User login
Psychiatry’s role rising in hospital care
BALTIMORE – A team of physicians and mental health experts at Johns Hopkins Hospital is trying something new: combining mental health services with medical ones. Hospital leadership hopes the experiment will pay off in shorter lengths of stay, lower readmission rates, and better overall patient care.
“We’re still collecting that data,” Melissa Richardson, the hospital’s director of care coordination, said in an interview. “We also will look at the impact on staffing ratios on the units. For example, has the number of patient observers gone down? Has the overall severity of certain cases on the unit been reduced by embedding mental health workers there?”
The medical-surgical mental health team debuted in April, and is separate from the hospital’s other psychiatric services. Comprised of a social worker, a nurse practitioner, a nurse care coordinator, and an attending psychiatrist, the team typically works a regular day shift, beginning each morning with patient chart reviews prepared by medical-surgical personnel. They discuss which patients will be seen by whom, since all team members are trained to do psychiatric evaluations.

Not all medical patients require psychiatric care, but according to the program’s clinical director and attending psychiatrist, Dr. Patrick T. Triplett, up to 38% of all medical admissions have a psychiatric comorbidity. Addressing those comorbidities while patients are in the hospital often leads to improved outcomes.
The team’s social worker connects patients with the appropriate outpatient mental health services in the community, and the team’s nurse care coordinator arranges any necessary transfers from the medical-surgical units to the inpatient psychiatric unit. Dr. Triplett and the psychiatric nurse practitioner are the only two team members who can diagnose and prescribe. Dr. Triplett’s time is billed as consultation services, and the hospital absorbs the cost of the rest of the team, according to Ms. Richardson.
‘Complex medically ill’ patients
As some procedures and medical treatments have shifted to the outpatient setting in recent years, and joint replacements or acute conditions such as myocardial infarctions can be managed successfully in shorter stays, more complicated patients, such as joint replacement patients who develop delirium, have been left on the medical-surgical unit, said Dr. Constantine G. Lyketsos, the Elizabeth Plank Althouse Professor at Johns Hopkins Bayview, Baltimore.
“Also, these days, up to 20% of our admissions are linked to opioids. Then, there are the chronically mentally ill. They tend to be a population with high rates of obesity, smoking, and diabetes, so they end up in the hospital with higher-level, more complicated conditions, that because of the disintegration of the mental health system, receive neither good psychiatric nor outpatient medical care,” Dr. Lyketsos, also chief of psychiatry at Johns Hopkins Bayview, said in an interview.
This surge in the number of complex medically ill patients has led to a growing number of hospitals nationwide calling upon psychiatrists for help in improving overall care. Hopkins is only the latest to join the ranks of other institutions such as Massachusetts General Hospital in Boston, State University of New York Downstate Medical Center in Brooklyn (N.Y.), Dartmouth-Hitchcock Medical Center in Lebanon, N.H., and New York–Presbyterian/Columbia University Medical Center in New York City.
The progenitor of this collaborative inpatient care model is Yale New Haven (Conn.) Hospital. The behavioral intervention team (BIT) at Yale New Haven includes nurses, social workers, and psychiatrists who proactively screen for and address behavioral barriers to care for medical patients with a co-occurring mental illness, said Dr. Hochang B. Lee, one of the psychiatrists who helped created the model in 2008. Dr. Lee is Yale’s psychological medicine section chief and director of the school’s Psychological Medicine Research Center. He also is an associate professor of psychiatry and an associate clinical professor of nursing.
“The goal was to create a proactive model of care, not the reactive one that is traditional consultation-liaison psychiatry,” Dr. Lee said in an interview. “Before the BIT model, medical teams often missed behavioral issues or made consultation requests too late in the course of hospitalization to avoid psychiatric crisis.”
LOS, costs reduced
A study

“The hospital is not making money off of us, but they’re losing less money because of us. That’s good!” Dr. Philip R. Muskin, chief of consultation-liaison psychiatry at New York–Presbyterian/Columbia, said in an interview.
Patients at New York Presbyterian have been comanaged since 2004 when, according to Dr. Muskin, a donor gift specifically intended for such a purpose was matched by the hospital’s department of medicine. The unspent money was enough to cover the cost of a consultation-liaison psychiatrist to round full time as an attending with the medical team.
“We were lucky at NYPCH, because someone gave us the gift to hire a full-time psychiatrist we could embed into the medical team,” Dr. Muskin said. “But there is no one right way to deliver collaborative care in the inpatient setting.”
The NYPCH program has grown to include a second full-time and one part-time psychiatrist, serving about half of all medical services at the campus, with plans to hire more. There is also a social worker to assist with outplacement services. Dr. Muskin said he is currently looking to hire a psychiatric nurse practitioner.
‘Nurses love us’

Restoration of staff morale is another benefit to this kind of practice, according to Maureen Lewis, an accredited psychiatric nurse practitioner who is part of the Hopkins integrated team.
“Med-surge nurses love us. Patients who come in with an overdose or who have any psychiatric conditions but need to have their medical comorbidities dealt with first, they are at their sickest with their psychiatric illnesses when they first arrive,” Ms. Lewis said. “It’s the med-surge nurses who have to care for them, but it’s not their comfort zone or their skill set. They like that we are helping them manage the patient.”
A decline in the number of assaults on the nursing staff also has been recorded since the Hopkins program began, Dr. Lyketsos said.
The psychiatrists themselves tend to be happier, too. “Knowing the cases before you even walk up on the unit is a huge benefit,” Dr. Triplett said. “To have to dive into an emergency all the time is just exhausting. It changes your relationship with the patient. When you’re not in that crisis mode, the patient isn’t a ‘problem’ anymore.”
A formal assessment of the entire medical-surgical staff satisfaction involved in the Hopkins program also is underway, said Ms. Richardson, the director of care coordination.
Having psychiatrists at the fore of this evolution in care provides more opportunities for training, too.
The comanagement model gives medical staff a chance to learn more not just about the direct care of complex behaviorally disordered patients, but also to understand their own emotions and states of mind as they interact with these patients. The teaching happens naturally as the team discusses patients while making rounds, Dr. Muskin said. “That’s really an integral part of what consult-liaison psychiatry is supposed to be about, anyway.”
By helping the medical staff reflect on their experiences, Dr. Muskin said, psychiatrists are helping to change the culture of inpatient medicine. “Once you change the culture, if you keep the things that brought it about in place, it doesn’t change back.”
Creating bridge services
A problem with this kind of inpatient collaborative care model is that hospitals that run them aren’t able to control all the variables associated with the cost of providing them.
The hope is that by spending more up front to identify and treat high-risk behavioral health patients, they won’t need readmission; but if the appropriate follow-up care can’t be found on the outpatient side, they could still end up back in the hospital, driving up readmission rates and possibly lengths of stay.
To address such contingencies, Hopkins has participated in state-sponsored partnerships for improving community care and is using monies from accountable care organization funding to create bridge services. The hospital system also has 14 primary care practices that have embedded psychiatric services, and the plan is to create a team to care for more complicated patients who need care 60-90 days after discharge.
For Dr. Lyketsos, however, fixing what he says is a broken mental health system isn’t up to the hospital alone. “We’re not going to be able to bring about real change without working with our legislators and our payers. It’s a complex problem that needs a complex solution. But there is a commonality of mind that we need to fix things, that patients need better care – and that this is a good place to start.”
On Twitter @whitneymcknight
CORRECTION, 6/28/16: A previous version of this article misstated Dr. Hochang B. Lee's name.
BALTIMORE – A team of physicians and mental health experts at Johns Hopkins Hospital is trying something new: combining mental health services with medical ones. Hospital leadership hopes the experiment will pay off in shorter lengths of stay, lower readmission rates, and better overall patient care.
“We’re still collecting that data,” Melissa Richardson, the hospital’s director of care coordination, said in an interview. “We also will look at the impact on staffing ratios on the units. For example, has the number of patient observers gone down? Has the overall severity of certain cases on the unit been reduced by embedding mental health workers there?”
The medical-surgical mental health team debuted in April, and is separate from the hospital’s other psychiatric services. Comprised of a social worker, a nurse practitioner, a nurse care coordinator, and an attending psychiatrist, the team typically works a regular day shift, beginning each morning with patient chart reviews prepared by medical-surgical personnel. They discuss which patients will be seen by whom, since all team members are trained to do psychiatric evaluations.
Not all medical patients require psychiatric care, but according to the program’s clinical director and attending psychiatrist, Dr. Patrick T. Triplett, up to 38% of all medical admissions have a psychiatric comorbidity. Addressing those comorbidities while patients are in the hospital often leads to improved outcomes.
The team’s social worker connects patients with the appropriate outpatient mental health services in the community, and the team’s nurse care coordinator arranges any necessary transfers from the medical-surgical units to the inpatient psychiatric unit. Dr. Triplett and the psychiatric nurse practitioner are the only two team members who can diagnose and prescribe. Dr. Triplett’s time is billed as consultation services, and the hospital absorbs the cost of the rest of the team, according to Ms. Richardson.
‘Complex medically ill’ patients
As some procedures and medical treatments have shifted to the outpatient setting in recent years, and joint replacements or acute conditions such as myocardial infarctions can be managed successfully in shorter stays, more complicated patients, such as joint replacement patients who develop delirium, have been left on the medical-surgical unit, said Dr. Constantine G. Lyketsos, the Elizabeth Plank Althouse Professor at Johns Hopkins Bayview, Baltimore.
“Also, these days, up to 20% of our admissions are linked to opioids. Then, there are the chronically mentally ill. They tend to be a population with high rates of obesity, smoking, and diabetes, so they end up in the hospital with higher-level, more complicated conditions, that because of the disintegration of the mental health system, receive neither good psychiatric nor outpatient medical care,” Dr. Lyketsos, also chief of psychiatry at Johns Hopkins Bayview, said in an interview.
This surge in the number of complex medically ill patients has led to a growing number of hospitals nationwide calling upon psychiatrists for help in improving overall care. Hopkins is only the latest to join the ranks of other institutions such as Massachusetts General Hospital in Boston, State University of New York Downstate Medical Center in Brooklyn (N.Y.), Dartmouth-Hitchcock Medical Center in Lebanon, N.H., and New York–Presbyterian/Columbia University Medical Center in New York City.
The progenitor of this collaborative inpatient care model is Yale New Haven (Conn.) Hospital. The behavioral intervention team (BIT) at Yale New Haven includes nurses, social workers, and psychiatrists who proactively screen for and address behavioral barriers to care for medical patients with a co-occurring mental illness, said Dr. Hochang B. Lee, one of the psychiatrists who helped created the model in 2008. Dr. Lee is Yale’s psychological medicine section chief and director of the school’s Psychological Medicine Research Center. He also is an associate professor of psychiatry and an associate clinical professor of nursing.
“The goal was to create a proactive model of care, not the reactive one that is traditional consultation-liaison psychiatry,” Dr. Lee said in an interview. “Before the BIT model, medical teams often missed behavioral issues or made consultation requests too late in the course of hospitalization to avoid psychiatric crisis.”
LOS, costs reduced
A study
“The hospital is not making money off of us, but they’re losing less money because of us. That’s good!” Dr. Philip R. Muskin, chief of consultation-liaison psychiatry at New York–Presbyterian/Columbia, said in an interview.
Patients at New York Presbyterian have been comanaged since 2004 when, according to Dr. Muskin, a donor gift specifically intended for such a purpose was matched by the hospital’s department of medicine. The unspent money was enough to cover the cost of a consultation-liaison psychiatrist to round full time as an attending with the medical team.
“We were lucky at NYPCH, because someone gave us the gift to hire a full-time psychiatrist we could embed into the medical team,” Dr. Muskin said. “But there is no one right way to deliver collaborative care in the inpatient setting.”
The NYPCH program has grown to include a second full-time and one part-time psychiatrist, serving about half of all medical services at the campus, with plans to hire more. There is also a social worker to assist with outplacement services. Dr. Muskin said he is currently looking to hire a psychiatric nurse practitioner.
‘Nurses love us’
Restoration of staff morale is another benefit to this kind of practice, according to Maureen Lewis, an accredited psychiatric nurse practitioner who is part of the Hopkins integrated team.
“Med-surge nurses love us. Patients who come in with an overdose or who have any psychiatric conditions but need to have their medical comorbidities dealt with first, they are at their sickest with their psychiatric illnesses when they first arrive,” Ms. Lewis said. “It’s the med-surge nurses who have to care for them, but it’s not their comfort zone or their skill set. They like that we are helping them manage the patient.”
A decline in the number of assaults on the nursing staff also has been recorded since the Hopkins program began, Dr. Lyketsos said.
The psychiatrists themselves tend to be happier, too. “Knowing the cases before you even walk up on the unit is a huge benefit,” Dr. Triplett said. “To have to dive into an emergency all the time is just exhausting. It changes your relationship with the patient. When you’re not in that crisis mode, the patient isn’t a ‘problem’ anymore.”
A formal assessment of the entire medical-surgical staff satisfaction involved in the Hopkins program also is underway, said Ms. Richardson, the director of care coordination.
Having psychiatrists at the fore of this evolution in care provides more opportunities for training, too.
The comanagement model gives medical staff a chance to learn more not just about the direct care of complex behaviorally disordered patients, but also to understand their own emotions and states of mind as they interact with these patients. The teaching happens naturally as the team discusses patients while making rounds, Dr. Muskin said. “That’s really an integral part of what consult-liaison psychiatry is supposed to be about, anyway.”
By helping the medical staff reflect on their experiences, Dr. Muskin said, psychiatrists are helping to change the culture of inpatient medicine. “Once you change the culture, if you keep the things that brought it about in place, it doesn’t change back.”
Creating bridge services
A problem with this kind of inpatient collaborative care model is that hospitals that run them aren’t able to control all the variables associated with the cost of providing them.
The hope is that by spending more up front to identify and treat high-risk behavioral health patients, they won’t need readmission; but if the appropriate follow-up care can’t be found on the outpatient side, they could still end up back in the hospital, driving up readmission rates and possibly lengths of stay.
To address such contingencies, Hopkins has participated in state-sponsored partnerships for improving community care and is using monies from accountable care organization funding to create bridge services. The hospital system also has 14 primary care practices that have embedded psychiatric services, and the plan is to create a team to care for more complicated patients who need care 60-90 days after discharge.
For Dr. Lyketsos, however, fixing what he says is a broken mental health system isn’t up to the hospital alone. “We’re not going to be able to bring about real change without working with our legislators and our payers. It’s a complex problem that needs a complex solution. But there is a commonality of mind that we need to fix things, that patients need better care – and that this is a good place to start.”
On Twitter @whitneymcknight
CORRECTION, 6/28/16: A previous version of this article misstated Dr. Hochang B. Lee's name.
BALTIMORE – A team of physicians and mental health experts at Johns Hopkins Hospital is trying something new: combining mental health services with medical ones. Hospital leadership hopes the experiment will pay off in shorter lengths of stay, lower readmission rates, and better overall patient care.
“We’re still collecting that data,” Melissa Richardson, the hospital’s director of care coordination, said in an interview. “We also will look at the impact on staffing ratios on the units. For example, has the number of patient observers gone down? Has the overall severity of certain cases on the unit been reduced by embedding mental health workers there?”
The medical-surgical mental health team debuted in April, and is separate from the hospital’s other psychiatric services. Comprised of a social worker, a nurse practitioner, a nurse care coordinator, and an attending psychiatrist, the team typically works a regular day shift, beginning each morning with patient chart reviews prepared by medical-surgical personnel. They discuss which patients will be seen by whom, since all team members are trained to do psychiatric evaluations.
Not all medical patients require psychiatric care, but according to the program’s clinical director and attending psychiatrist, Dr. Patrick T. Triplett, up to 38% of all medical admissions have a psychiatric comorbidity. Addressing those comorbidities while patients are in the hospital often leads to improved outcomes.
The team’s social worker connects patients with the appropriate outpatient mental health services in the community, and the team’s nurse care coordinator arranges any necessary transfers from the medical-surgical units to the inpatient psychiatric unit. Dr. Triplett and the psychiatric nurse practitioner are the only two team members who can diagnose and prescribe. Dr. Triplett’s time is billed as consultation services, and the hospital absorbs the cost of the rest of the team, according to Ms. Richardson.
‘Complex medically ill’ patients
As some procedures and medical treatments have shifted to the outpatient setting in recent years, and joint replacements or acute conditions such as myocardial infarctions can be managed successfully in shorter stays, more complicated patients, such as joint replacement patients who develop delirium, have been left on the medical-surgical unit, said Dr. Constantine G. Lyketsos, the Elizabeth Plank Althouse Professor at Johns Hopkins Bayview, Baltimore.
“Also, these days, up to 20% of our admissions are linked to opioids. Then, there are the chronically mentally ill. They tend to be a population with high rates of obesity, smoking, and diabetes, so they end up in the hospital with higher-level, more complicated conditions, that because of the disintegration of the mental health system, receive neither good psychiatric nor outpatient medical care,” Dr. Lyketsos, also chief of psychiatry at Johns Hopkins Bayview, said in an interview.
This surge in the number of complex medically ill patients has led to a growing number of hospitals nationwide calling upon psychiatrists for help in improving overall care. Hopkins is only the latest to join the ranks of other institutions such as Massachusetts General Hospital in Boston, State University of New York Downstate Medical Center in Brooklyn (N.Y.), Dartmouth-Hitchcock Medical Center in Lebanon, N.H., and New York–Presbyterian/Columbia University Medical Center in New York City.
The progenitor of this collaborative inpatient care model is Yale New Haven (Conn.) Hospital. The behavioral intervention team (BIT) at Yale New Haven includes nurses, social workers, and psychiatrists who proactively screen for and address behavioral barriers to care for medical patients with a co-occurring mental illness, said Dr. Hochang B. Lee, one of the psychiatrists who helped created the model in 2008. Dr. Lee is Yale’s psychological medicine section chief and director of the school’s Psychological Medicine Research Center. He also is an associate professor of psychiatry and an associate clinical professor of nursing.
“The goal was to create a proactive model of care, not the reactive one that is traditional consultation-liaison psychiatry,” Dr. Lee said in an interview. “Before the BIT model, medical teams often missed behavioral issues or made consultation requests too late in the course of hospitalization to avoid psychiatric crisis.”
LOS, costs reduced
A study
“The hospital is not making money off of us, but they’re losing less money because of us. That’s good!” Dr. Philip R. Muskin, chief of consultation-liaison psychiatry at New York–Presbyterian/Columbia, said in an interview.
Patients at New York Presbyterian have been comanaged since 2004 when, according to Dr. Muskin, a donor gift specifically intended for such a purpose was matched by the hospital’s department of medicine. The unspent money was enough to cover the cost of a consultation-liaison psychiatrist to round full time as an attending with the medical team.
“We were lucky at NYPCH, because someone gave us the gift to hire a full-time psychiatrist we could embed into the medical team,” Dr. Muskin said. “But there is no one right way to deliver collaborative care in the inpatient setting.”
The NYPCH program has grown to include a second full-time and one part-time psychiatrist, serving about half of all medical services at the campus, with plans to hire more. There is also a social worker to assist with outplacement services. Dr. Muskin said he is currently looking to hire a psychiatric nurse practitioner.
‘Nurses love us’
Restoration of staff morale is another benefit to this kind of practice, according to Maureen Lewis, an accredited psychiatric nurse practitioner who is part of the Hopkins integrated team.
“Med-surge nurses love us. Patients who come in with an overdose or who have any psychiatric conditions but need to have their medical comorbidities dealt with first, they are at their sickest with their psychiatric illnesses when they first arrive,” Ms. Lewis said. “It’s the med-surge nurses who have to care for them, but it’s not their comfort zone or their skill set. They like that we are helping them manage the patient.”
A decline in the number of assaults on the nursing staff also has been recorded since the Hopkins program began, Dr. Lyketsos said.
The psychiatrists themselves tend to be happier, too. “Knowing the cases before you even walk up on the unit is a huge benefit,” Dr. Triplett said. “To have to dive into an emergency all the time is just exhausting. It changes your relationship with the patient. When you’re not in that crisis mode, the patient isn’t a ‘problem’ anymore.”
A formal assessment of the entire medical-surgical staff satisfaction involved in the Hopkins program also is underway, said Ms. Richardson, the director of care coordination.
Having psychiatrists at the fore of this evolution in care provides more opportunities for training, too.
The comanagement model gives medical staff a chance to learn more not just about the direct care of complex behaviorally disordered patients, but also to understand their own emotions and states of mind as they interact with these patients. The teaching happens naturally as the team discusses patients while making rounds, Dr. Muskin said. “That’s really an integral part of what consult-liaison psychiatry is supposed to be about, anyway.”
By helping the medical staff reflect on their experiences, Dr. Muskin said, psychiatrists are helping to change the culture of inpatient medicine. “Once you change the culture, if you keep the things that brought it about in place, it doesn’t change back.”
Creating bridge services
A problem with this kind of inpatient collaborative care model is that hospitals that run them aren’t able to control all the variables associated with the cost of providing them.
The hope is that by spending more up front to identify and treat high-risk behavioral health patients, they won’t need readmission; but if the appropriate follow-up care can’t be found on the outpatient side, they could still end up back in the hospital, driving up readmission rates and possibly lengths of stay.
To address such contingencies, Hopkins has participated in state-sponsored partnerships for improving community care and is using monies from accountable care organization funding to create bridge services. The hospital system also has 14 primary care practices that have embedded psychiatric services, and the plan is to create a team to care for more complicated patients who need care 60-90 days after discharge.
For Dr. Lyketsos, however, fixing what he says is a broken mental health system isn’t up to the hospital alone. “We’re not going to be able to bring about real change without working with our legislators and our payers. It’s a complex problem that needs a complex solution. But there is a commonality of mind that we need to fix things, that patients need better care – and that this is a good place to start.”
On Twitter @whitneymcknight
CORRECTION, 6/28/16: A previous version of this article misstated Dr. Hochang B. Lee's name.

Additional antibiotics needed when implanting cryopreserved human aortic grafts
Infections of aortic prosthetic grafts can be a devastating complication, and while cryopreserved human allografts (CHA) can continue to possess antibacterial activity even after 5 years or more in storage, cardiac surgeons may want to apply additional antibiotic agents during implantation to boost bacterial resistance, investigators from Germany reported in the May issue of the Journal of Thoracic and Cardiovascular Surgery (2016;151:1251-9).
The researchers compared three different antibiotic regimens used in processing CHA aortic tissue and valves to determine the impact each can have on long-term bacterial resistance.
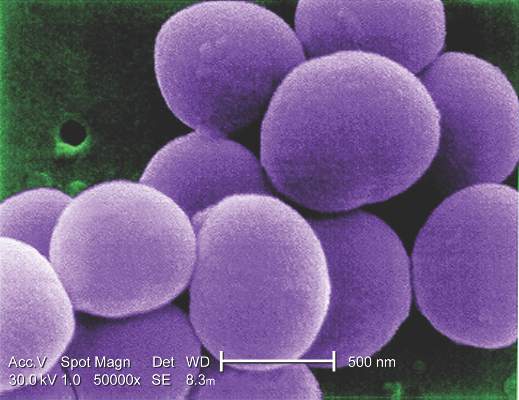
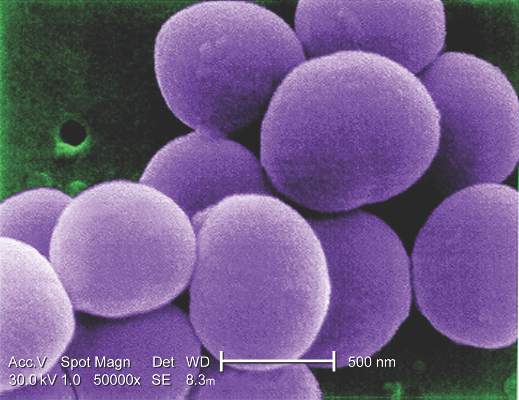
“Antibiotic combinations applied during CHA processing have a significant influence on their infection resistance,” said Dr. Viola Steffen of Hannover (Germany) Medical School and her colleagues. The average storage time of CHAs was 8.5 years, with the longest having been stored for 10 years.
The study involved microbiologic tests in vitro of three different antibiotic regimens used in processing CHA: gentamicin-piperacillin-vancomycin-metronidazole-amphotericin B (group A); gentamicin-piperacillin-flucloxacillin-metronidazole-amphotericin B (group B); and meropenem-vancomycin-tobramycin-colistin-amphotericin B (group C). The combinations are used to counteract Staphylococcus epidermidis and Staphylococcus aureus.
The study exposed pieces of 10 CHAs to different microbes and determined that regimen groups B and C were more effective than group A in eradicating gram-positive organisms. Specifically, group C was most resistant to Escherichia coli, whereas group B was most effective against Pseudomonas aeruginosa. Aortic tissue showed significantly less contamination with staphylococcal bacteria than valve grafts, the study reported.
Dr. Steffen and her colleagues said that tissue banks use antibiotic protocols during CHA processing, but they differ substantially. “Our results support the hypothesis that infection resistance of CHAs depends on the antibiotic pretreatment during processing and their residual activity,” they said.
The study had four key findings:
• The infection resistance of aortic wall and valve tissue differed significantly.
• In aortic wall specimens, group A specimens exhibited increased adherence of S. epidermidis, with vancomycin in group A and flucloxacillin in group B being the only differentiating agents between the two.
• Cryopreserved aortic vessels had a propensity toward reduced infection resistance against P. aeruginosa.
• Morphologic changes occurred in the microorganisms, especially rod-shaped E. coli, indicating that regional antibiotic release alters bacterial growth without eliminating all adherent bacteria.
Dr. Steffen and her colleagues noted that while previous studies determined that residual concentrations of antibiotics used in processing CHA heart valves and blood vessels are still present after they are prepared for implantation, they neither measured the antibacterial affect, clarified the period of cryopreservation nor differentiated between valve and vessel tissue (PLoS One. 2014;9:e112679; Transfus Med Hemother. 2011;38:379-86).
The Hannover researchers found that only a few gram-positive microorganisms adhered to aortic wall specimens, although they found “extensive adherence” of S. epidermidis in group A specimens. Valves, however, “were completely colonized” with both strains of staphylococcal bacteria, although the severity of contamination varied depending on the regimen used.
The findings raise some questions about the antibiotic properties of CHAs. “Because their infection resistance depends on the antibiotic combination selected during processing, further investigations concerning this treatment are necessary to improve the antimicrobial activity against frequent and highly virulent infection-causing bacteria,” Dr. Steffen and her colleagues said.
Dr. Steffen and her coauthors had no financial relationships to disclose.
Infections of aortic prosthetic grafts can be a devastating complication, and while cryopreserved human allografts (CHA) can continue to possess antibacterial activity even after 5 years or more in storage, cardiac surgeons may want to apply additional antibiotic agents during implantation to boost bacterial resistance, investigators from Germany reported in the May issue of the Journal of Thoracic and Cardiovascular Surgery (2016;151:1251-9).
The researchers compared three different antibiotic regimens used in processing CHA aortic tissue and valves to determine the impact each can have on long-term bacterial resistance.
“Antibiotic combinations applied during CHA processing have a significant influence on their infection resistance,” said Dr. Viola Steffen of Hannover (Germany) Medical School and her colleagues. The average storage time of CHAs was 8.5 years, with the longest having been stored for 10 years.
The study involved microbiologic tests in vitro of three different antibiotic regimens used in processing CHA: gentamicin-piperacillin-vancomycin-metronidazole-amphotericin B (group A); gentamicin-piperacillin-flucloxacillin-metronidazole-amphotericin B (group B); and meropenem-vancomycin-tobramycin-colistin-amphotericin B (group C). The combinations are used to counteract Staphylococcus epidermidis and Staphylococcus aureus.
The study exposed pieces of 10 CHAs to different microbes and determined that regimen groups B and C were more effective than group A in eradicating gram-positive organisms. Specifically, group C was most resistant to Escherichia coli, whereas group B was most effective against Pseudomonas aeruginosa. Aortic tissue showed significantly less contamination with staphylococcal bacteria than valve grafts, the study reported.
Dr. Steffen and her colleagues said that tissue banks use antibiotic protocols during CHA processing, but they differ substantially. “Our results support the hypothesis that infection resistance of CHAs depends on the antibiotic pretreatment during processing and their residual activity,” they said.
The study had four key findings:
• The infection resistance of aortic wall and valve tissue differed significantly.
• In aortic wall specimens, group A specimens exhibited increased adherence of S. epidermidis, with vancomycin in group A and flucloxacillin in group B being the only differentiating agents between the two.
• Cryopreserved aortic vessels had a propensity toward reduced infection resistance against P. aeruginosa.
• Morphologic changes occurred in the microorganisms, especially rod-shaped E. coli, indicating that regional antibiotic release alters bacterial growth without eliminating all adherent bacteria.
Dr. Steffen and her colleagues noted that while previous studies determined that residual concentrations of antibiotics used in processing CHA heart valves and blood vessels are still present after they are prepared for implantation, they neither measured the antibacterial affect, clarified the period of cryopreservation nor differentiated between valve and vessel tissue (PLoS One. 2014;9:e112679; Transfus Med Hemother. 2011;38:379-86).
The Hannover researchers found that only a few gram-positive microorganisms adhered to aortic wall specimens, although they found “extensive adherence” of S. epidermidis in group A specimens. Valves, however, “were completely colonized” with both strains of staphylococcal bacteria, although the severity of contamination varied depending on the regimen used.
The findings raise some questions about the antibiotic properties of CHAs. “Because their infection resistance depends on the antibiotic combination selected during processing, further investigations concerning this treatment are necessary to improve the antimicrobial activity against frequent and highly virulent infection-causing bacteria,” Dr. Steffen and her colleagues said.
Dr. Steffen and her coauthors had no financial relationships to disclose.
Infections of aortic prosthetic grafts can be a devastating complication, and while cryopreserved human allografts (CHA) can continue to possess antibacterial activity even after 5 years or more in storage, cardiac surgeons may want to apply additional antibiotic agents during implantation to boost bacterial resistance, investigators from Germany reported in the May issue of the Journal of Thoracic and Cardiovascular Surgery (2016;151:1251-9).
The researchers compared three different antibiotic regimens used in processing CHA aortic tissue and valves to determine the impact each can have on long-term bacterial resistance.
“Antibiotic combinations applied during CHA processing have a significant influence on their infection resistance,” said Dr. Viola Steffen of Hannover (Germany) Medical School and her colleagues. The average storage time of CHAs was 8.5 years, with the longest having been stored for 10 years.
The study involved microbiologic tests in vitro of three different antibiotic regimens used in processing CHA: gentamicin-piperacillin-vancomycin-metronidazole-amphotericin B (group A); gentamicin-piperacillin-flucloxacillin-metronidazole-amphotericin B (group B); and meropenem-vancomycin-tobramycin-colistin-amphotericin B (group C). The combinations are used to counteract Staphylococcus epidermidis and Staphylococcus aureus.
The study exposed pieces of 10 CHAs to different microbes and determined that regimen groups B and C were more effective than group A in eradicating gram-positive organisms. Specifically, group C was most resistant to Escherichia coli, whereas group B was most effective against Pseudomonas aeruginosa. Aortic tissue showed significantly less contamination with staphylococcal bacteria than valve grafts, the study reported.
Dr. Steffen and her colleagues said that tissue banks use antibiotic protocols during CHA processing, but they differ substantially. “Our results support the hypothesis that infection resistance of CHAs depends on the antibiotic pretreatment during processing and their residual activity,” they said.
The study had four key findings:
• The infection resistance of aortic wall and valve tissue differed significantly.
• In aortic wall specimens, group A specimens exhibited increased adherence of S. epidermidis, with vancomycin in group A and flucloxacillin in group B being the only differentiating agents between the two.
• Cryopreserved aortic vessels had a propensity toward reduced infection resistance against P. aeruginosa.
• Morphologic changes occurred in the microorganisms, especially rod-shaped E. coli, indicating that regional antibiotic release alters bacterial growth without eliminating all adherent bacteria.
Dr. Steffen and her colleagues noted that while previous studies determined that residual concentrations of antibiotics used in processing CHA heart valves and blood vessels are still present after they are prepared for implantation, they neither measured the antibacterial affect, clarified the period of cryopreservation nor differentiated between valve and vessel tissue (PLoS One. 2014;9:e112679; Transfus Med Hemother. 2011;38:379-86).
The Hannover researchers found that only a few gram-positive microorganisms adhered to aortic wall specimens, although they found “extensive adherence” of S. epidermidis in group A specimens. Valves, however, “were completely colonized” with both strains of staphylococcal bacteria, although the severity of contamination varied depending on the regimen used.
The findings raise some questions about the antibiotic properties of CHAs. “Because their infection resistance depends on the antibiotic combination selected during processing, further investigations concerning this treatment are necessary to improve the antimicrobial activity against frequent and highly virulent infection-causing bacteria,” Dr. Steffen and her colleagues said.
Dr. Steffen and her coauthors had no financial relationships to disclose.
FROM THE JOURNAL OF THORACIC AND CARDIOVASCULAR SURGERY
Key clinical point: The infection resistance of cryopreserved human allografts (CHAs) depends on antibiotic pretreatment during processing.
Major finding: Cardiac surgeons can recommend CHAs to patients either with active destructive infections or at high risk of reinfection, but they may want to apply additional antibiotic agents during implantation to enhance bacterial resistance.
Data source: Pieces of 10 CHAs were microbiologically tested in vitro and exposed to bacterial contamination and the number of attached bacteria quantified.
Disclosures: Dr. Steffen and her coauthors had no financial relationships to disclose.
Zika virus: The path to fetal infection
The question of how viruses can enter the intrauterine compartment and infect the fetus has long been a focus of research. It is of particular urgency today as the Zika virus spreads and causes perinatal infection that threatens the developing fetus with serious adverse outcomes such microcephaly and other brain anomalies, placental insufficiency, and fetal growth restriction.
We know that viruses can take a variety of routes to the fetal compartment, but we have also learned that the placenta has a robust level of inherent resistance to viruses. This resistance likely explains why we don’t see more viral infections in pregnancy.

Recent studies performed at our institution suggest that placental trophoblasts – the placenta’s primary line of defense – have inherent resistance to viruses such as Zika. It appears, therefore, that the Zika virus invades the intrauterine cavity by crossing the trophoblasts, perhaps earlier in pregnancy and prior to the development of full trophoblast resistance, by entering through breaks in this outer layer, or by utilizing alternative pathways to access the fetal compartment.
Further study of the placenta and its various cell types and mechanisms of viral defense will be critical for designing therapeutic strategies for preventing perinatal infections.
Various routes and affinities
Viruses have long been known to affect mothers and their unborn children. The rubella virus, for instance, posed a significant threat to the fetus until a vaccine program was introduced almost 50 years ago. Cytomegalovirus (CMV), on the other hand, continues be passed from mothers to their unborn children. While not as threatening as rubella once was, it can in some cases cause severe defects.
One might expect viruses to infect the placenta and then secondarily infect the fetus. While this may indeed occur, direct placental infection is not the only route by which viruses may enter the intrauterine compartment. Some viruses may be carried by macrophages or other immune cells through the placenta and into the fetal compartment, while others colonize the uterine cavity prior to conception, ready to proliferate during pregnancy.

In still other cases, viruses may be inadvertently introduced during medical procedures such as amniocentesis or transmitted through transvaginal ascending infection, most likely after rupture of the membranes. Viruses may also be transported through infected sperm (this appears to be one of the Zika virus’s modes of transportation), and as is the case with HIV and herpes simplex viruses, transmission sometimes occurs during delivery.
When we investigate whether or not the fetus is protected against particular viruses, we must therefore think about the multifaceted mechanisms by which viruses may be transmitted. With respect to the placenta specifically, we seek to understand how viruses enter the placenta, and how the placenta resists the propagation of some viruses while allowing other viruses to gain entry to the intrauterine compartment.
An additional consideration – one that is of utmost importance in the case of Zika – is whether viruses have any special affinity for particular fetal tissues. Some viruses, like CMV, infect multiple types of fetal tissue. The Zika virus, on the other hand, appears to target neuronal tissue in the fetus. In May, investigators of two studies reported that a strain of the Zika virus efficiently infected human cortical neural progenitor cells (Cell Stem Cell. 2016 May 5;18[5]:587-90), and that Zika infection of mice early in pregnancy resulted in infection of the placenta and of the fetal brain (Cell. 2016 May 19;165[5]:1081-91).
Interestingly, other flaviviruses such as the dengue and chikungunya viruses have not been associated with microcephaly or other congenital disorders. This suggests that the Zika virus employs unique mechanisms to infect or bypass the placental barrier and, in turn, to cause neuronal-focused damage.
Placental passage
The villous trophoblasts, cells that are bathed in maternal blood, form the placenta’s first line of defense. Viruses, including the Zika virus, must cross or somehow bypass this initial barrier before crossing the placental basement membrane and endothelial cells, if they are to potentially invade the intrauterine cavity and infect the fetal brain and other tissues.
Research has demonstrated that cells of various types of tissue may express certain proteins, such as AXL, MER and TYRO3. While not yet proven, these proteins may mediate the entry of viruses such as Zika, enabling them to cross the placental trophoblast layer. These proteins are indeed expressed in trophoblasts, especially in early pregnancy, but we do not yet know if the proteins actually aid Zika’s passage through the placenta.
Another mechanism that has been postulated in the case of Zika infection is antibody-dependent enhancement, a process by which a current infection is enhanced by prior infection with another virus from the same family. Some experts believe that pre-existing immunity to the dengue virus – another member of the flavivirus family that has been endemic in Brazil – may be enhancing the spread of Zika infection as antibodies against dengue cross-react with the Zika virus.
While antibody-dependent enhancement has been shown to occur and to advance infection in various body systems, it has not been proven to affect the placenta. Until we learn more, we must simply appreciate that the presence of antibodies from another member of a family of viruses does not necessarily confer resistance. Instead, it may enable new infections to advance.
One might view pregnancy as a time of immune compromise, but we have shown in our laboratories that trophoblasts in fact have inherent resistance to a number of viruses. In a recent study, we found that trophoblasts are refractory to direct infection with the Zika virus. We isolated trophoblast cells from healthy full-term human placentas, cultured these cells for several days, and infected them with the Zika virus. We then measured viral replication and compared the infectivity of these cells with the infectivity of human brain microvascular endothelial cells – nontrophoblast cells that served as a control.
Our findings were extremely interesting to us: The trophoblast cells appeared to be significantly more resistant to the Zika virus than the nontrophoblast cells.
We learned, moreover, that this resistance was mediated by a particular interferon released by the trophoblast cells – type III interferon IFN1 – and that this type III interferon appeared to protect not only the trophoblasts but the nontrophoblast cells as well. It acted in both an autocrine and a paracrine manner to protect cells from the Zika virus. When we blocked the antiviral signaling of this interferon, resistance to the virus was attenuated.
These findings suggest that while Zika appears able to cross through the placenta and infect the fetus, the mechanism does not involve direct infection of the trophoblasts, at least in the later stages of pregnancy. The virus must either evade the type III interferon antiviral signals generated by the trophoblasts or somehow bypass these cells to cross the placenta (Cell Host Microbe. 2016 May 11;19[5]:705-12).
Interestingly, the Cell study mentioned above, in which Zika infection of mice early in pregnancy infected placental cells and the brain, also showed reduced Zika presence in the mouse mononuclear trophoblasts and syncytiotrophoblasts, in areas of the placenta analogous to the human villi.
Some experts have suggested, based the study of other viruses, that the Zika virus is better able to infect the placenta when the infection occurs early in the first trimester or the second trimester. It is indeed possible – and makes intuitive sense – that first-trimester trophoblasts confer less resistance and a lower level of protection than the mature trophoblasts we studied. At this point, however, we cannot say with certainty whether or not the placenta is more or less permissive to Zika infection at different points in pregnancy.
Interestingly, investigators who prospectively followed a small cohort of pregnant women in Brazil with suspected Zika infection identified abnormalities in fetuses of women who were infected at various points of their pregnancies, even in the third trimester. Fetuses infected in the first trimester had findings suggestive of pathologic change during embryogenesis, but central nervous system abnormalities were seen in fetuses infected as late as 27 weeks of gestation, the investigators said (N Engl J Med. 2016 Mar 4. doi: 10.1056/NEJMoa1602412).
The interferon-conferred resistance demonstrated in our recent study is one of two mechanisms we’ve identified by which placental trophoblasts orchestrate resistance to viral infection. In earlier research, we found that resistance can be conferred to nontrophoblast cells by the delivery of micro-RNAs. These micro-RNAs (C19MC miRNAs) are uniquely expressed in the placenta and packaged within trophoblast-derived nanovesicles called exosomes. The nanovesicles can latch onto other cells in the vicinity of the trophoblasts, attenuating viral replication in these recipient cells.
This earlier in-vitro study involved a panel of diverse and unrelated viruses, including coxsackievirus B3, poliovirus, vesicular stomatitis virus, and human cytomegalovirus (Proc Natl Acad Sci U S A. 2013 Jul 16;110[29]:12048-53). It did not include the Zika virus, but our ongoing preliminary research suggests that the same mechanisms might be active against Zika.
Furthering research
Research at our institution and in other laboratories has shed light on various ways in which the fetus is protected from viruses, but we must learn more in order to understand how particular viruses, such as Zika, are able to reach the fetal compartment and cause particular birth defects.
We must further investigate the role and importance of antibody-dependent enhancement, and we must continue to study the placenta and its various cell types. Continuing efforts to better elucidate the placenta’s defense mechanisms and to identify cell types that are more or less resistant to the Zika virus – and understand their differences – may lead us to potential therapeutic strategies.
Dr. Sadovsky is scientific director of the Magee-Womens Research Institute, Elsie Hilliard Hillman Chair of Women’s Health Research, and professor of ob.gyn., reproductive sciences, microbiology, and molecular genetics at the University of Pittsburgh. Dr. Coyne is associate professor of microbiology and molecular genetics, and ob.gyn. and reproductive sciences, at the University of Pittsburgh.* Their research addressed in this Master Class was supported by grants from the National Institutes of Health, State of Pennsylvania Formula Research Funds, and Burroughs Wellcome Fund.
*Correction, 7/05/2016: An earlier version of this article misstated Dr. Coyne's academic title.
The question of how viruses can enter the intrauterine compartment and infect the fetus has long been a focus of research. It is of particular urgency today as the Zika virus spreads and causes perinatal infection that threatens the developing fetus with serious adverse outcomes such microcephaly and other brain anomalies, placental insufficiency, and fetal growth restriction.
We know that viruses can take a variety of routes to the fetal compartment, but we have also learned that the placenta has a robust level of inherent resistance to viruses. This resistance likely explains why we don’t see more viral infections in pregnancy.
Recent studies performed at our institution suggest that placental trophoblasts – the placenta’s primary line of defense – have inherent resistance to viruses such as Zika. It appears, therefore, that the Zika virus invades the intrauterine cavity by crossing the trophoblasts, perhaps earlier in pregnancy and prior to the development of full trophoblast resistance, by entering through breaks in this outer layer, or by utilizing alternative pathways to access the fetal compartment.
Further study of the placenta and its various cell types and mechanisms of viral defense will be critical for designing therapeutic strategies for preventing perinatal infections.
Various routes and affinities
Viruses have long been known to affect mothers and their unborn children. The rubella virus, for instance, posed a significant threat to the fetus until a vaccine program was introduced almost 50 years ago. Cytomegalovirus (CMV), on the other hand, continues be passed from mothers to their unborn children. While not as threatening as rubella once was, it can in some cases cause severe defects.
One might expect viruses to infect the placenta and then secondarily infect the fetus. While this may indeed occur, direct placental infection is not the only route by which viruses may enter the intrauterine compartment. Some viruses may be carried by macrophages or other immune cells through the placenta and into the fetal compartment, while others colonize the uterine cavity prior to conception, ready to proliferate during pregnancy.
In still other cases, viruses may be inadvertently introduced during medical procedures such as amniocentesis or transmitted through transvaginal ascending infection, most likely after rupture of the membranes. Viruses may also be transported through infected sperm (this appears to be one of the Zika virus’s modes of transportation), and as is the case with HIV and herpes simplex viruses, transmission sometimes occurs during delivery.
When we investigate whether or not the fetus is protected against particular viruses, we must therefore think about the multifaceted mechanisms by which viruses may be transmitted. With respect to the placenta specifically, we seek to understand how viruses enter the placenta, and how the placenta resists the propagation of some viruses while allowing other viruses to gain entry to the intrauterine compartment.
An additional consideration – one that is of utmost importance in the case of Zika – is whether viruses have any special affinity for particular fetal tissues. Some viruses, like CMV, infect multiple types of fetal tissue. The Zika virus, on the other hand, appears to target neuronal tissue in the fetus. In May, investigators of two studies reported that a strain of the Zika virus efficiently infected human cortical neural progenitor cells (Cell Stem Cell. 2016 May 5;18[5]:587-90), and that Zika infection of mice early in pregnancy resulted in infection of the placenta and of the fetal brain (Cell. 2016 May 19;165[5]:1081-91).
Interestingly, other flaviviruses such as the dengue and chikungunya viruses have not been associated with microcephaly or other congenital disorders. This suggests that the Zika virus employs unique mechanisms to infect or bypass the placental barrier and, in turn, to cause neuronal-focused damage.
Placental passage
The villous trophoblasts, cells that are bathed in maternal blood, form the placenta’s first line of defense. Viruses, including the Zika virus, must cross or somehow bypass this initial barrier before crossing the placental basement membrane and endothelial cells, if they are to potentially invade the intrauterine cavity and infect the fetal brain and other tissues.
Research has demonstrated that cells of various types of tissue may express certain proteins, such as AXL, MER and TYRO3. While not yet proven, these proteins may mediate the entry of viruses such as Zika, enabling them to cross the placental trophoblast layer. These proteins are indeed expressed in trophoblasts, especially in early pregnancy, but we do not yet know if the proteins actually aid Zika’s passage through the placenta.
Another mechanism that has been postulated in the case of Zika infection is antibody-dependent enhancement, a process by which a current infection is enhanced by prior infection with another virus from the same family. Some experts believe that pre-existing immunity to the dengue virus – another member of the flavivirus family that has been endemic in Brazil – may be enhancing the spread of Zika infection as antibodies against dengue cross-react with the Zika virus.
While antibody-dependent enhancement has been shown to occur and to advance infection in various body systems, it has not been proven to affect the placenta. Until we learn more, we must simply appreciate that the presence of antibodies from another member of a family of viruses does not necessarily confer resistance. Instead, it may enable new infections to advance.
One might view pregnancy as a time of immune compromise, but we have shown in our laboratories that trophoblasts in fact have inherent resistance to a number of viruses. In a recent study, we found that trophoblasts are refractory to direct infection with the Zika virus. We isolated trophoblast cells from healthy full-term human placentas, cultured these cells for several days, and infected them with the Zika virus. We then measured viral replication and compared the infectivity of these cells with the infectivity of human brain microvascular endothelial cells – nontrophoblast cells that served as a control.
Our findings were extremely interesting to us: The trophoblast cells appeared to be significantly more resistant to the Zika virus than the nontrophoblast cells.
We learned, moreover, that this resistance was mediated by a particular interferon released by the trophoblast cells – type III interferon IFN1 – and that this type III interferon appeared to protect not only the trophoblasts but the nontrophoblast cells as well. It acted in both an autocrine and a paracrine manner to protect cells from the Zika virus. When we blocked the antiviral signaling of this interferon, resistance to the virus was attenuated.
These findings suggest that while Zika appears able to cross through the placenta and infect the fetus, the mechanism does not involve direct infection of the trophoblasts, at least in the later stages of pregnancy. The virus must either evade the type III interferon antiviral signals generated by the trophoblasts or somehow bypass these cells to cross the placenta (Cell Host Microbe. 2016 May 11;19[5]:705-12).
Interestingly, the Cell study mentioned above, in which Zika infection of mice early in pregnancy infected placental cells and the brain, also showed reduced Zika presence in the mouse mononuclear trophoblasts and syncytiotrophoblasts, in areas of the placenta analogous to the human villi.
Some experts have suggested, based the study of other viruses, that the Zika virus is better able to infect the placenta when the infection occurs early in the first trimester or the second trimester. It is indeed possible – and makes intuitive sense – that first-trimester trophoblasts confer less resistance and a lower level of protection than the mature trophoblasts we studied. At this point, however, we cannot say with certainty whether or not the placenta is more or less permissive to Zika infection at different points in pregnancy.
Interestingly, investigators who prospectively followed a small cohort of pregnant women in Brazil with suspected Zika infection identified abnormalities in fetuses of women who were infected at various points of their pregnancies, even in the third trimester. Fetuses infected in the first trimester had findings suggestive of pathologic change during embryogenesis, but central nervous system abnormalities were seen in fetuses infected as late as 27 weeks of gestation, the investigators said (N Engl J Med. 2016 Mar 4. doi: 10.1056/NEJMoa1602412).
The interferon-conferred resistance demonstrated in our recent study is one of two mechanisms we’ve identified by which placental trophoblasts orchestrate resistance to viral infection. In earlier research, we found that resistance can be conferred to nontrophoblast cells by the delivery of micro-RNAs. These micro-RNAs (C19MC miRNAs) are uniquely expressed in the placenta and packaged within trophoblast-derived nanovesicles called exosomes. The nanovesicles can latch onto other cells in the vicinity of the trophoblasts, attenuating viral replication in these recipient cells.
This earlier in-vitro study involved a panel of diverse and unrelated viruses, including coxsackievirus B3, poliovirus, vesicular stomatitis virus, and human cytomegalovirus (Proc Natl Acad Sci U S A. 2013 Jul 16;110[29]:12048-53). It did not include the Zika virus, but our ongoing preliminary research suggests that the same mechanisms might be active against Zika.
Furthering research
Research at our institution and in other laboratories has shed light on various ways in which the fetus is protected from viruses, but we must learn more in order to understand how particular viruses, such as Zika, are able to reach the fetal compartment and cause particular birth defects.
We must further investigate the role and importance of antibody-dependent enhancement, and we must continue to study the placenta and its various cell types. Continuing efforts to better elucidate the placenta’s defense mechanisms and to identify cell types that are more or less resistant to the Zika virus – and understand their differences – may lead us to potential therapeutic strategies.
Dr. Sadovsky is scientific director of the Magee-Womens Research Institute, Elsie Hilliard Hillman Chair of Women’s Health Research, and professor of ob.gyn., reproductive sciences, microbiology, and molecular genetics at the University of Pittsburgh. Dr. Coyne is associate professor of microbiology and molecular genetics, and ob.gyn. and reproductive sciences, at the University of Pittsburgh.* Their research addressed in this Master Class was supported by grants from the National Institutes of Health, State of Pennsylvania Formula Research Funds, and Burroughs Wellcome Fund.
*Correction, 7/05/2016: An earlier version of this article misstated Dr. Coyne's academic title.
The question of how viruses can enter the intrauterine compartment and infect the fetus has long been a focus of research. It is of particular urgency today as the Zika virus spreads and causes perinatal infection that threatens the developing fetus with serious adverse outcomes such microcephaly and other brain anomalies, placental insufficiency, and fetal growth restriction.
We know that viruses can take a variety of routes to the fetal compartment, but we have also learned that the placenta has a robust level of inherent resistance to viruses. This resistance likely explains why we don’t see more viral infections in pregnancy.
Recent studies performed at our institution suggest that placental trophoblasts – the placenta’s primary line of defense – have inherent resistance to viruses such as Zika. It appears, therefore, that the Zika virus invades the intrauterine cavity by crossing the trophoblasts, perhaps earlier in pregnancy and prior to the development of full trophoblast resistance, by entering through breaks in this outer layer, or by utilizing alternative pathways to access the fetal compartment.
Further study of the placenta and its various cell types and mechanisms of viral defense will be critical for designing therapeutic strategies for preventing perinatal infections.
Various routes and affinities
Viruses have long been known to affect mothers and their unborn children. The rubella virus, for instance, posed a significant threat to the fetus until a vaccine program was introduced almost 50 years ago. Cytomegalovirus (CMV), on the other hand, continues be passed from mothers to their unborn children. While not as threatening as rubella once was, it can in some cases cause severe defects.
One might expect viruses to infect the placenta and then secondarily infect the fetus. While this may indeed occur, direct placental infection is not the only route by which viruses may enter the intrauterine compartment. Some viruses may be carried by macrophages or other immune cells through the placenta and into the fetal compartment, while others colonize the uterine cavity prior to conception, ready to proliferate during pregnancy.
In still other cases, viruses may be inadvertently introduced during medical procedures such as amniocentesis or transmitted through transvaginal ascending infection, most likely after rupture of the membranes. Viruses may also be transported through infected sperm (this appears to be one of the Zika virus’s modes of transportation), and as is the case with HIV and herpes simplex viruses, transmission sometimes occurs during delivery.
When we investigate whether or not the fetus is protected against particular viruses, we must therefore think about the multifaceted mechanisms by which viruses may be transmitted. With respect to the placenta specifically, we seek to understand how viruses enter the placenta, and how the placenta resists the propagation of some viruses while allowing other viruses to gain entry to the intrauterine compartment.
An additional consideration – one that is of utmost importance in the case of Zika – is whether viruses have any special affinity for particular fetal tissues. Some viruses, like CMV, infect multiple types of fetal tissue. The Zika virus, on the other hand, appears to target neuronal tissue in the fetus. In May, investigators of two studies reported that a strain of the Zika virus efficiently infected human cortical neural progenitor cells (Cell Stem Cell. 2016 May 5;18[5]:587-90), and that Zika infection of mice early in pregnancy resulted in infection of the placenta and of the fetal brain (Cell. 2016 May 19;165[5]:1081-91).
Interestingly, other flaviviruses such as the dengue and chikungunya viruses have not been associated with microcephaly or other congenital disorders. This suggests that the Zika virus employs unique mechanisms to infect or bypass the placental barrier and, in turn, to cause neuronal-focused damage.
Placental passage
The villous trophoblasts, cells that are bathed in maternal blood, form the placenta’s first line of defense. Viruses, including the Zika virus, must cross or somehow bypass this initial barrier before crossing the placental basement membrane and endothelial cells, if they are to potentially invade the intrauterine cavity and infect the fetal brain and other tissues.
Research has demonstrated that cells of various types of tissue may express certain proteins, such as AXL, MER and TYRO3. While not yet proven, these proteins may mediate the entry of viruses such as Zika, enabling them to cross the placental trophoblast layer. These proteins are indeed expressed in trophoblasts, especially in early pregnancy, but we do not yet know if the proteins actually aid Zika’s passage through the placenta.
Another mechanism that has been postulated in the case of Zika infection is antibody-dependent enhancement, a process by which a current infection is enhanced by prior infection with another virus from the same family. Some experts believe that pre-existing immunity to the dengue virus – another member of the flavivirus family that has been endemic in Brazil – may be enhancing the spread of Zika infection as antibodies against dengue cross-react with the Zika virus.
While antibody-dependent enhancement has been shown to occur and to advance infection in various body systems, it has not been proven to affect the placenta. Until we learn more, we must simply appreciate that the presence of antibodies from another member of a family of viruses does not necessarily confer resistance. Instead, it may enable new infections to advance.
One might view pregnancy as a time of immune compromise, but we have shown in our laboratories that trophoblasts in fact have inherent resistance to a number of viruses. In a recent study, we found that trophoblasts are refractory to direct infection with the Zika virus. We isolated trophoblast cells from healthy full-term human placentas, cultured these cells for several days, and infected them with the Zika virus. We then measured viral replication and compared the infectivity of these cells with the infectivity of human brain microvascular endothelial cells – nontrophoblast cells that served as a control.
Our findings were extremely interesting to us: The trophoblast cells appeared to be significantly more resistant to the Zika virus than the nontrophoblast cells.
We learned, moreover, that this resistance was mediated by a particular interferon released by the trophoblast cells – type III interferon IFN1 – and that this type III interferon appeared to protect not only the trophoblasts but the nontrophoblast cells as well. It acted in both an autocrine and a paracrine manner to protect cells from the Zika virus. When we blocked the antiviral signaling of this interferon, resistance to the virus was attenuated.
These findings suggest that while Zika appears able to cross through the placenta and infect the fetus, the mechanism does not involve direct infection of the trophoblasts, at least in the later stages of pregnancy. The virus must either evade the type III interferon antiviral signals generated by the trophoblasts or somehow bypass these cells to cross the placenta (Cell Host Microbe. 2016 May 11;19[5]:705-12).
Interestingly, the Cell study mentioned above, in which Zika infection of mice early in pregnancy infected placental cells and the brain, also showed reduced Zika presence in the mouse mononuclear trophoblasts and syncytiotrophoblasts, in areas of the placenta analogous to the human villi.
Some experts have suggested, based the study of other viruses, that the Zika virus is better able to infect the placenta when the infection occurs early in the first trimester or the second trimester. It is indeed possible – and makes intuitive sense – that first-trimester trophoblasts confer less resistance and a lower level of protection than the mature trophoblasts we studied. At this point, however, we cannot say with certainty whether or not the placenta is more or less permissive to Zika infection at different points in pregnancy.
Interestingly, investigators who prospectively followed a small cohort of pregnant women in Brazil with suspected Zika infection identified abnormalities in fetuses of women who were infected at various points of their pregnancies, even in the third trimester. Fetuses infected in the first trimester had findings suggestive of pathologic change during embryogenesis, but central nervous system abnormalities were seen in fetuses infected as late as 27 weeks of gestation, the investigators said (N Engl J Med. 2016 Mar 4. doi: 10.1056/NEJMoa1602412).
The interferon-conferred resistance demonstrated in our recent study is one of two mechanisms we’ve identified by which placental trophoblasts orchestrate resistance to viral infection. In earlier research, we found that resistance can be conferred to nontrophoblast cells by the delivery of micro-RNAs. These micro-RNAs (C19MC miRNAs) are uniquely expressed in the placenta and packaged within trophoblast-derived nanovesicles called exosomes. The nanovesicles can latch onto other cells in the vicinity of the trophoblasts, attenuating viral replication in these recipient cells.
This earlier in-vitro study involved a panel of diverse and unrelated viruses, including coxsackievirus B3, poliovirus, vesicular stomatitis virus, and human cytomegalovirus (Proc Natl Acad Sci U S A. 2013 Jul 16;110[29]:12048-53). It did not include the Zika virus, but our ongoing preliminary research suggests that the same mechanisms might be active against Zika.
Furthering research
Research at our institution and in other laboratories has shed light on various ways in which the fetus is protected from viruses, but we must learn more in order to understand how particular viruses, such as Zika, are able to reach the fetal compartment and cause particular birth defects.
We must further investigate the role and importance of antibody-dependent enhancement, and we must continue to study the placenta and its various cell types. Continuing efforts to better elucidate the placenta’s defense mechanisms and to identify cell types that are more or less resistant to the Zika virus – and understand their differences – may lead us to potential therapeutic strategies.
Dr. Sadovsky is scientific director of the Magee-Womens Research Institute, Elsie Hilliard Hillman Chair of Women’s Health Research, and professor of ob.gyn., reproductive sciences, microbiology, and molecular genetics at the University of Pittsburgh. Dr. Coyne is associate professor of microbiology and molecular genetics, and ob.gyn. and reproductive sciences, at the University of Pittsburgh.* Their research addressed in this Master Class was supported by grants from the National Institutes of Health, State of Pennsylvania Formula Research Funds, and Burroughs Wellcome Fund.
*Correction, 7/05/2016: An earlier version of this article misstated Dr. Coyne's academic title.

Zika virus challenges ob.gyn. practice
Viral illnesses in pregnancy are not unheard of. When a patient presents with symptoms, we often think of an influenza type of infection that will be cleared within a short period of time and with few negative consequences for the developing fetus. Other infections that can occur include TORCH – Toxoplasmosis, Other (syphilis, varicella-zoster, parvovirus B19), Rubella, Cytomegalovirus (CMV), and Herpes – infections, but these are also relatively common.
Rarely do we in the United States consider a gravida’s vulnerability to tropical infectious diseases such as dengue, chikungunya, and now Zika virus. With the popularity and ease of international travel, and the potential for women’s exposure to more exotic diseases, the practice of ob.gyn. must undergo a significant transition in perspective. It is vital for us to understand these illnesses because of their potency and reported injury to both the mother and baby, for several reasons.

First, there is the public health concern. As of June 16, 2016, the Pan American Health Organization of the World Health Organization, reported 39 countries and territories in the Americas with confirmed cases of Zika virus, with 21 of those countries having confirmed cases in pregnant women.
As of June 9, 2016, the Centers for Disease Control and Prevention reported that 234 pregnant women in the United States have laboratory evidence of possible Zika infection, along with 189 pregnant women living in U.S. territories. Since the current outbreak, which began in July 2015 in Brazil, seven countries – accounting for more than 1,600 cases – have reported babies with congenital syndrome associated with Zika virus, the majority of which have been in Brazil. With the Summer Olympics in Rio starting in August 2016, the potential spread of Zika virus is dizzying.
Second, there is the counseling and management concern. Without a treatment or vaccine available, ob.gyns. must stay current on the latest research and findings to inform their patients of the risks associated with travel to an area with confirmed, or areas at risk for developing, Zika virus transmission.
Third, there is a diagnostic concern. Women who have visited areas with Zika virus, or who have had intimate contact with someone who has traveled to these areas, must be diagnosed and then counseled immediately.
We have devoted this Master Class to a discussion of Zika virus and the work being conducted in the United States to understand this disease. We have invited Dr. Yoel Sadovsky, an expert on placental development and trophoblast function, and his colleague, Carolyn Coyne, Ph.D., a leading researcher on host-virus interactions, to address this important topic.
Dr. Reece, who specializes in maternal-fetal medicine, is vice president for medical affairs at the University of Maryland, Baltimore, as well as the John Z. and Akiko K. Bowers Distinguished Professor and dean of the school of medicine. Dr. Reece said he had no relevant financial disclosures. He is the medical editor of this column. Contact him at [email protected].
Viral illnesses in pregnancy are not unheard of. When a patient presents with symptoms, we often think of an influenza type of infection that will be cleared within a short period of time and with few negative consequences for the developing fetus. Other infections that can occur include TORCH – Toxoplasmosis, Other (syphilis, varicella-zoster, parvovirus B19), Rubella, Cytomegalovirus (CMV), and Herpes – infections, but these are also relatively common.
Rarely do we in the United States consider a gravida’s vulnerability to tropical infectious diseases such as dengue, chikungunya, and now Zika virus. With the popularity and ease of international travel, and the potential for women’s exposure to more exotic diseases, the practice of ob.gyn. must undergo a significant transition in perspective. It is vital for us to understand these illnesses because of their potency and reported injury to both the mother and baby, for several reasons.
First, there is the public health concern. As of June 16, 2016, the Pan American Health Organization of the World Health Organization, reported 39 countries and territories in the Americas with confirmed cases of Zika virus, with 21 of those countries having confirmed cases in pregnant women.
As of June 9, 2016, the Centers for Disease Control and Prevention reported that 234 pregnant women in the United States have laboratory evidence of possible Zika infection, along with 189 pregnant women living in U.S. territories. Since the current outbreak, which began in July 2015 in Brazil, seven countries – accounting for more than 1,600 cases – have reported babies with congenital syndrome associated with Zika virus, the majority of which have been in Brazil. With the Summer Olympics in Rio starting in August 2016, the potential spread of Zika virus is dizzying.
Second, there is the counseling and management concern. Without a treatment or vaccine available, ob.gyns. must stay current on the latest research and findings to inform their patients of the risks associated with travel to an area with confirmed, or areas at risk for developing, Zika virus transmission.
Third, there is a diagnostic concern. Women who have visited areas with Zika virus, or who have had intimate contact with someone who has traveled to these areas, must be diagnosed and then counseled immediately.
We have devoted this Master Class to a discussion of Zika virus and the work being conducted in the United States to understand this disease. We have invited Dr. Yoel Sadovsky, an expert on placental development and trophoblast function, and his colleague, Carolyn Coyne, Ph.D., a leading researcher on host-virus interactions, to address this important topic.
Dr. Reece, who specializes in maternal-fetal medicine, is vice president for medical affairs at the University of Maryland, Baltimore, as well as the John Z. and Akiko K. Bowers Distinguished Professor and dean of the school of medicine. Dr. Reece said he had no relevant financial disclosures. He is the medical editor of this column. Contact him at [email protected].
Viral illnesses in pregnancy are not unheard of. When a patient presents with symptoms, we often think of an influenza type of infection that will be cleared within a short period of time and with few negative consequences for the developing fetus. Other infections that can occur include TORCH – Toxoplasmosis, Other (syphilis, varicella-zoster, parvovirus B19), Rubella, Cytomegalovirus (CMV), and Herpes – infections, but these are also relatively common.
Rarely do we in the United States consider a gravida’s vulnerability to tropical infectious diseases such as dengue, chikungunya, and now Zika virus. With the popularity and ease of international travel, and the potential for women’s exposure to more exotic diseases, the practice of ob.gyn. must undergo a significant transition in perspective. It is vital for us to understand these illnesses because of their potency and reported injury to both the mother and baby, for several reasons.
First, there is the public health concern. As of June 16, 2016, the Pan American Health Organization of the World Health Organization, reported 39 countries and territories in the Americas with confirmed cases of Zika virus, with 21 of those countries having confirmed cases in pregnant women.
As of June 9, 2016, the Centers for Disease Control and Prevention reported that 234 pregnant women in the United States have laboratory evidence of possible Zika infection, along with 189 pregnant women living in U.S. territories. Since the current outbreak, which began in July 2015 in Brazil, seven countries – accounting for more than 1,600 cases – have reported babies with congenital syndrome associated with Zika virus, the majority of which have been in Brazil. With the Summer Olympics in Rio starting in August 2016, the potential spread of Zika virus is dizzying.
Second, there is the counseling and management concern. Without a treatment or vaccine available, ob.gyns. must stay current on the latest research and findings to inform their patients of the risks associated with travel to an area with confirmed, or areas at risk for developing, Zika virus transmission.
Third, there is a diagnostic concern. Women who have visited areas with Zika virus, or who have had intimate contact with someone who has traveled to these areas, must be diagnosed and then counseled immediately.
We have devoted this Master Class to a discussion of Zika virus and the work being conducted in the United States to understand this disease. We have invited Dr. Yoel Sadovsky, an expert on placental development and trophoblast function, and his colleague, Carolyn Coyne, Ph.D., a leading researcher on host-virus interactions, to address this important topic.
Dr. Reece, who specializes in maternal-fetal medicine, is vice president for medical affairs at the University of Maryland, Baltimore, as well as the John Z. and Akiko K. Bowers Distinguished Professor and dean of the school of medicine. Dr. Reece said he had no relevant financial disclosures. He is the medical editor of this column. Contact him at [email protected].

Does congenital cardiac surgery training need a makeover?
Trainees in congenital cardiac surgery fellowship programs are doing more operations since the programs became accredited in 2007, but no clear parameters have emerged to determine if certification has improved the quality of training, according to an evaluation of fellowship training programs published in the June issue of the Journal of Thoracic and Cardiovascular Surgery (2016 Jun;151:1488-95).
Overall, the training has become standardized, the fellows’ operative experience is “robust,” and fellows are mostly satisfied since the Accreditation Council of Graduate Medical Education (ACGME) recognized congenital cardiac surgery as a fellowship in 2007, lead study author Dr. Brian Kogon of Emory University, Atlanta, said.
However, Dr. Kogon and his colleagues also found some shortcomings in fellowship training. They received survey responses from 36 of 44 fellows in 12 accredited programs nationwide. To determine if fellows were meeting minimum case requirements, they also reviewed operative logs of 38 of the 44 fellows. They compared their findings to a study of congenital cardiac surgery fellowship programs they did pre-ACGME accreditation (J Thorac Cardiovasc Surg. 2006 Dec;132:1280). “The number of operations performed by the fellows during their training was underwhelming, and most of the fellows were dissatisfied with their operative experience,” Dr. Kogon and his colleagues wrote in the earlier study.
The study found that all fellows achieved the minimum number of 75 total cases the standards require for graduation, with a median of 136; and the minimum standard of 36 specific qualifying cases with a median of 63. However, seven did not meet the minimum of five complex neonate cases. Among other types of operations for which fellows failed to meet the minimum cases were atrioventricular septal defect repair, arch reconstruction including coarctation procedures and systemic-to-pulmonary artery shunt procedures.
The comparative lack of adult cardiac surgery operations was also considered a potential problem, the authors noted, pointing out that “the number of adults who have congenital heart disease now exceeds the number of children who have the disease, and many of these patients will require an operation.”
Another shortcoming the study found was a drop-off in international fellowships since 2007. “This change places us at risk of becoming intellectually isolated and losing international relationships that are critical to the future of our specialty,” Dr. Kogon and his colleagues wrote. Graduated fellows also acknowledged dissatisfaction with their lack of exposure to neonate surgery.
The study also determined the following demographics of the fellows: 83% are men and the median age at graduation was 40 years, with a range of 35-48 years. Only 25% of graduates participated in nonsurgical rotations such as cardiac catheterization and echocardiography.
“Although the operative experience seems to be much more robust, and this finding has been corroborated in other surgical disciplines after the advent of ACGME accreditation, comparing training before and after the accreditation process came into existence is difficult,” Dr. Kogon and his colleagues said.
The study also noted that the Thoracic Surgery Directors Association developed a congenital curriculum for congenital cardiothoracic surgery fellows, but only 28% used that curriculum and only 61% used any formal curriculum. “Unfortunately, regardless of the curriculum, only 50% of the graduates found it helpful,” Dr. Kogon and his colleagues said.
And regardless of the curriculum, only half of the graduates have passed the written qualifying and oral certifying examinations after completing their fellowship. “Although the curriculum is quite robust, the latter statistic suggests that we need either more emphasis on education by the program directors or a better and/or different curriculum,” Dr. Kogon and his colleagues said. However, they added that “after training, former fellows have adequate case volumes and mixes and seem to be thriving in the field.”
Dr. Kogon and his study coauthors had no financial disclosures.
In his invited commentary, Dr. Charles D. Fraser Jr. of Texas Children’s Hospital, Baylor University, Houston, called the study findings that only 50% of congenital cardiac surgery fellowship graduates had passed the congenital examination “quite disturbing” and the demographic data and surgical and nonsurgical experience of the trainees “thought provoking” (J Thorac Cardiovasc Surg. 2016;151:1496-7)
“Is the bar too high or too low?” Dr. Fraser asked. He suggested the fellowship training system for congenital cardiac surgeons may be a work in progress. “For one, having a median age of 40 years for graduates is unacceptable,” he said. For half of trainees to not pass the examination “at this advanced age is tragic.” That 25% of fellows participate in nonsurgical rotations “also is concerning.”

|
Dr. Charles D. Fraser |
A challenge is that after fellows complete their training in general and cardiothoracic surgery, opportunities to operate on newborns in a new fellowship setting are extremely limited, Dr. Fraser said. “To expect someone to be able to perform complex newborn heart surgery with excellent outcomes in a brand-new environment after just learning how to perform adult cardiac surgery is unrealistic,” he said.
Dr. Fraser said 1 formal year of training for congenital cardiac surgery fellows may not be enough. “Our colleagues in general pediatric surgery have a 2-year fellowship, and our specialty is every bit as complex as theirs,” he said. The basic American Board of Thoracic Surgery thoracic fellowship should have more latitude in its congenital heart surgery rotations, including exposure to pediatrics, neonatal/pediatric critical care, and the nonsurgical rotations the study referred to. Congenital heart surgery fellowships should also embrace adult congenital heart surgery with a more formalized experience requirement, he said.
“As a specialty, we owe it to our fine young surgeon candidates to offer the most robust and fair pathway to success while never compromising on the public trust and patient well-being,” Dr. Fraser said.
Dr. Fraser is chief of the division of congenital heart surgery at Baylor and codirector of the Texas Children’s Heart Center. He had no financial relationships to disclose.
In his invited commentary, Dr. Charles D. Fraser Jr. of Texas Children’s Hospital, Baylor University, Houston, called the study findings that only 50% of congenital cardiac surgery fellowship graduates had passed the congenital examination “quite disturbing” and the demographic data and surgical and nonsurgical experience of the trainees “thought provoking” (J Thorac Cardiovasc Surg. 2016;151:1496-7)
“Is the bar too high or too low?” Dr. Fraser asked. He suggested the fellowship training system for congenital cardiac surgeons may be a work in progress. “For one, having a median age of 40 years for graduates is unacceptable,” he said. For half of trainees to not pass the examination “at this advanced age is tragic.” That 25% of fellows participate in nonsurgical rotations “also is concerning.”
|
|
Dr. Charles D. Fraser |
A challenge is that after fellows complete their training in general and cardiothoracic surgery, opportunities to operate on newborns in a new fellowship setting are extremely limited, Dr. Fraser said. “To expect someone to be able to perform complex newborn heart surgery with excellent outcomes in a brand-new environment after just learning how to perform adult cardiac surgery is unrealistic,” he said.
Dr. Fraser said 1 formal year of training for congenital cardiac surgery fellows may not be enough. “Our colleagues in general pediatric surgery have a 2-year fellowship, and our specialty is every bit as complex as theirs,” he said. The basic American Board of Thoracic Surgery thoracic fellowship should have more latitude in its congenital heart surgery rotations, including exposure to pediatrics, neonatal/pediatric critical care, and the nonsurgical rotations the study referred to. Congenital heart surgery fellowships should also embrace adult congenital heart surgery with a more formalized experience requirement, he said.
“As a specialty, we owe it to our fine young surgeon candidates to offer the most robust and fair pathway to success while never compromising on the public trust and patient well-being,” Dr. Fraser said.
Dr. Fraser is chief of the division of congenital heart surgery at Baylor and codirector of the Texas Children’s Heart Center. He had no financial relationships to disclose.
In his invited commentary, Dr. Charles D. Fraser Jr. of Texas Children’s Hospital, Baylor University, Houston, called the study findings that only 50% of congenital cardiac surgery fellowship graduates had passed the congenital examination “quite disturbing” and the demographic data and surgical and nonsurgical experience of the trainees “thought provoking” (J Thorac Cardiovasc Surg. 2016;151:1496-7)
“Is the bar too high or too low?” Dr. Fraser asked. He suggested the fellowship training system for congenital cardiac surgeons may be a work in progress. “For one, having a median age of 40 years for graduates is unacceptable,” he said. For half of trainees to not pass the examination “at this advanced age is tragic.” That 25% of fellows participate in nonsurgical rotations “also is concerning.”
|
|
Dr. Charles D. Fraser |
A challenge is that after fellows complete their training in general and cardiothoracic surgery, opportunities to operate on newborns in a new fellowship setting are extremely limited, Dr. Fraser said. “To expect someone to be able to perform complex newborn heart surgery with excellent outcomes in a brand-new environment after just learning how to perform adult cardiac surgery is unrealistic,” he said.
Dr. Fraser said 1 formal year of training for congenital cardiac surgery fellows may not be enough. “Our colleagues in general pediatric surgery have a 2-year fellowship, and our specialty is every bit as complex as theirs,” he said. The basic American Board of Thoracic Surgery thoracic fellowship should have more latitude in its congenital heart surgery rotations, including exposure to pediatrics, neonatal/pediatric critical care, and the nonsurgical rotations the study referred to. Congenital heart surgery fellowships should also embrace adult congenital heart surgery with a more formalized experience requirement, he said.
“As a specialty, we owe it to our fine young surgeon candidates to offer the most robust and fair pathway to success while never compromising on the public trust and patient well-being,” Dr. Fraser said.
Dr. Fraser is chief of the division of congenital heart surgery at Baylor and codirector of the Texas Children’s Heart Center. He had no financial relationships to disclose.
Trainees in congenital cardiac surgery fellowship programs are doing more operations since the programs became accredited in 2007, but no clear parameters have emerged to determine if certification has improved the quality of training, according to an evaluation of fellowship training programs published in the June issue of the Journal of Thoracic and Cardiovascular Surgery (2016 Jun;151:1488-95).
Overall, the training has become standardized, the fellows’ operative experience is “robust,” and fellows are mostly satisfied since the Accreditation Council of Graduate Medical Education (ACGME) recognized congenital cardiac surgery as a fellowship in 2007, lead study author Dr. Brian Kogon of Emory University, Atlanta, said.
However, Dr. Kogon and his colleagues also found some shortcomings in fellowship training. They received survey responses from 36 of 44 fellows in 12 accredited programs nationwide. To determine if fellows were meeting minimum case requirements, they also reviewed operative logs of 38 of the 44 fellows. They compared their findings to a study of congenital cardiac surgery fellowship programs they did pre-ACGME accreditation (J Thorac Cardiovasc Surg. 2006 Dec;132:1280). “The number of operations performed by the fellows during their training was underwhelming, and most of the fellows were dissatisfied with their operative experience,” Dr. Kogon and his colleagues wrote in the earlier study.
The study found that all fellows achieved the minimum number of 75 total cases the standards require for graduation, with a median of 136; and the minimum standard of 36 specific qualifying cases with a median of 63. However, seven did not meet the minimum of five complex neonate cases. Among other types of operations for which fellows failed to meet the minimum cases were atrioventricular septal defect repair, arch reconstruction including coarctation procedures and systemic-to-pulmonary artery shunt procedures.
The comparative lack of adult cardiac surgery operations was also considered a potential problem, the authors noted, pointing out that “the number of adults who have congenital heart disease now exceeds the number of children who have the disease, and many of these patients will require an operation.”
Another shortcoming the study found was a drop-off in international fellowships since 2007. “This change places us at risk of becoming intellectually isolated and losing international relationships that are critical to the future of our specialty,” Dr. Kogon and his colleagues wrote. Graduated fellows also acknowledged dissatisfaction with their lack of exposure to neonate surgery.
The study also determined the following demographics of the fellows: 83% are men and the median age at graduation was 40 years, with a range of 35-48 years. Only 25% of graduates participated in nonsurgical rotations such as cardiac catheterization and echocardiography.
“Although the operative experience seems to be much more robust, and this finding has been corroborated in other surgical disciplines after the advent of ACGME accreditation, comparing training before and after the accreditation process came into existence is difficult,” Dr. Kogon and his colleagues said.
The study also noted that the Thoracic Surgery Directors Association developed a congenital curriculum for congenital cardiothoracic surgery fellows, but only 28% used that curriculum and only 61% used any formal curriculum. “Unfortunately, regardless of the curriculum, only 50% of the graduates found it helpful,” Dr. Kogon and his colleagues said.
And regardless of the curriculum, only half of the graduates have passed the written qualifying and oral certifying examinations after completing their fellowship. “Although the curriculum is quite robust, the latter statistic suggests that we need either more emphasis on education by the program directors or a better and/or different curriculum,” Dr. Kogon and his colleagues said. However, they added that “after training, former fellows have adequate case volumes and mixes and seem to be thriving in the field.”
Dr. Kogon and his study coauthors had no financial disclosures.
Trainees in congenital cardiac surgery fellowship programs are doing more operations since the programs became accredited in 2007, but no clear parameters have emerged to determine if certification has improved the quality of training, according to an evaluation of fellowship training programs published in the June issue of the Journal of Thoracic and Cardiovascular Surgery (2016 Jun;151:1488-95).
Overall, the training has become standardized, the fellows’ operative experience is “robust,” and fellows are mostly satisfied since the Accreditation Council of Graduate Medical Education (ACGME) recognized congenital cardiac surgery as a fellowship in 2007, lead study author Dr. Brian Kogon of Emory University, Atlanta, said.
However, Dr. Kogon and his colleagues also found some shortcomings in fellowship training. They received survey responses from 36 of 44 fellows in 12 accredited programs nationwide. To determine if fellows were meeting minimum case requirements, they also reviewed operative logs of 38 of the 44 fellows. They compared their findings to a study of congenital cardiac surgery fellowship programs they did pre-ACGME accreditation (J Thorac Cardiovasc Surg. 2006 Dec;132:1280). “The number of operations performed by the fellows during their training was underwhelming, and most of the fellows were dissatisfied with their operative experience,” Dr. Kogon and his colleagues wrote in the earlier study.
The study found that all fellows achieved the minimum number of 75 total cases the standards require for graduation, with a median of 136; and the minimum standard of 36 specific qualifying cases with a median of 63. However, seven did not meet the minimum of five complex neonate cases. Among other types of operations for which fellows failed to meet the minimum cases were atrioventricular septal defect repair, arch reconstruction including coarctation procedures and systemic-to-pulmonary artery shunt procedures.
The comparative lack of adult cardiac surgery operations was also considered a potential problem, the authors noted, pointing out that “the number of adults who have congenital heart disease now exceeds the number of children who have the disease, and many of these patients will require an operation.”
Another shortcoming the study found was a drop-off in international fellowships since 2007. “This change places us at risk of becoming intellectually isolated and losing international relationships that are critical to the future of our specialty,” Dr. Kogon and his colleagues wrote. Graduated fellows also acknowledged dissatisfaction with their lack of exposure to neonate surgery.
The study also determined the following demographics of the fellows: 83% are men and the median age at graduation was 40 years, with a range of 35-48 years. Only 25% of graduates participated in nonsurgical rotations such as cardiac catheterization and echocardiography.
“Although the operative experience seems to be much more robust, and this finding has been corroborated in other surgical disciplines after the advent of ACGME accreditation, comparing training before and after the accreditation process came into existence is difficult,” Dr. Kogon and his colleagues said.
The study also noted that the Thoracic Surgery Directors Association developed a congenital curriculum for congenital cardiothoracic surgery fellows, but only 28% used that curriculum and only 61% used any formal curriculum. “Unfortunately, regardless of the curriculum, only 50% of the graduates found it helpful,” Dr. Kogon and his colleagues said.
And regardless of the curriculum, only half of the graduates have passed the written qualifying and oral certifying examinations after completing their fellowship. “Although the curriculum is quite robust, the latter statistic suggests that we need either more emphasis on education by the program directors or a better and/or different curriculum,” Dr. Kogon and his colleagues said. However, they added that “after training, former fellows have adequate case volumes and mixes and seem to be thriving in the field.”
Dr. Kogon and his study coauthors had no financial disclosures.
FROM THE JOURNAL OF THORACIC AND CARDIOVASCULAR SURGERY
Key clinical point: Since congenital cardiac fellowship programs became accredited in 2007, training requirements have been standardized and the surgical experience robust.
Major finding: Recent graduates of fellowship programs are thriving in practice, but shortcomings with existing fellowship training exist, including only 50% gaining certification by passing the written and oral exams.
Data source: The study drew on survey responses from 36 of 44 fellows in 12 accredited programs and a review of operative logs of 38 of the 44 fellows.
Disclosures: Dr. Kogon and his study coauthors had no financial disclosures.

Helmets trump face mask for noninvasive ventilation
Treating acute respiratory distress syndrome patients with helmets instead of face masks significantly reduced intubation rates and 90-day mortality rates, based on data from a randomized trial of 83 adults published in JAMA.
Acute respiratory distress syndrome (ARDS) can be treated using a face mask, but the mask is often inadequate, forcing patients to undergo intubation that can lead to additional problems including discomfort, delirium, and muscle deterioration, wrote Tianna Hicklin, Ph.D., in a column on the NIH website.

“The proposed mechanism for improved efficacy of these helmets is the preservation of applied pressures and avoidance of air leak. If the helmets indeed do allow clinicians both to be able to increase airway pressures above levels they typically would with mask-NIV and maintain those pressures without unpredictable system leak, that would be of great physiologic importance for patients with acute respiratory failure,” said Dr. Eric J. Gartman.
To explore the potential of noninvasive ventilation (NIV) using a helmet instead of a mask, both of which allow patients to remain awake in the ICU, the researchers randomized 83 adults with ARDS to treatment with either a helmet (44 patients) or a face mask (39 patients).
Intubation incidence was 18% in patients treated with helmets, compared with 62% for those treated with face masks. In addition, patients in the helmet group had significantly more ventilator-free days than the mask group (28 vs. 13; P less than .001), and both hospital mortality and 90-day mortality rates were significantly lower in the helmet group compared to the mask group (27% vs. 49%; P = .04 and 34% vs. 56%; P = .02, respectively)
Adverse event rates were rare, and similar between the helmet and mask groups. Three interface-related skin ulcers occurred in each group; nose ulcers in 7% of the mask group and neck ulcers in 7% of the helmet group.
The study population included adults aged 18 years and older who were admitted to the ICU at the University of Chicago between September 2012 and September 2015 and required face mask NIV. The average age of the patients was 58 years in the helmet group and 61 years in the mask group; there were no significant demographic differences between the groups. The most common causes of acute respiratory failure in both groups were pneumonia and pneumonia caused by immunosuppression.
The study results were limited by several factors including a lack of blinding, the need for a learning curve for clinicians using the helmet, and the potential for patient-ventilator dyssynchrony in the helmet group, noted Dr. Bhakti K. Patel of the University of Chicago and her colleagues (JAMA 2016;315:2435-41). Multicenter studies are needed to support the findings, they added. However, the findings “affirm the far-reaching benefits of spontaneous yet highly supported ventilation in an awake, animated patient over invasive medical ventilation via endotracheal tube,” they wrote. “These findings warrant further investigation of helmet NIV for patients with ARDS and other types of AHRF [acute hypoxemic respiratory failure], particularly with attention to long-term outcomes.”
The researchers had no financial conflicts to disclose.
“While the results of this study are very impressive, it is a single-center study, and obviously a larger multicenter trial (with all types of institutions included – not just large academic centers) would be helpful to elucidate the benefit of this technique and support a change in standard of care in the use of NIV. A change to this system would be a very large culture shift in NIV and would mean a significant amount of training (physician, nursing, respiratory care), purchasing the helmets, and ensuring that it is implemented properly and safely. As stated by the authors, this fact is similar to the change the occurred originally with NIV – but if their results reflect a true benefit over FM-NIV [face mask-noninvasive ventilation], such a large change would certainly be worth the effort,” Dr. Gartman said.
In a related study published in the Journal of Cardiothoracic and Vascular Anesthesia, patients treated with noninvasive positive pressure ventilation (NPPV) through helmets had significantly lower heart rates, lower average arterial pressure, and improved left ventricular ejection fraction at the end of treatment, compared with patients treated with ventilation masks and controls. Dr. Yi Yang of Capital Medical University in Beijing, China, and colleagues conducted the prospective study of 75 adults experiencing hypoxemia within 24 hours of extubation after Stanford type A aortic dissection. The participants were divided into three 25-patient groups. The control group was treated with high-flux inhalation of oxygen via a Venturi mask, another group was treated with NPPV via a mask, and the third group was treated with NPPV via a helmet. (J Card Vasc Anesth. 2016. http://dx.doi.org/10.1053/j.jvca.2016.03.129).
The study was funded by China’s public welfare industry of health.
“Several key clinical messages can be gained from the study by Patel et al.,” wrote Dr. Jeremy R. Beitler, Dr. Robert L. Owens, and Dr. Atul Malhotra, in an accompanying editorial (JAMA 2016 Jun; 315:2401-3.).
“The helmet interface has unique advantages and disadvantages that may influence efficacy of noninvasive positive pressure ventilation (NIV) depending on patient and disease characteristics,” they said. “External validation of the findings by Patel et al. and clarification of appropriate eligibility criteria, optimal ventilator settings, and potential mechanisms of effect are needed before clinicians could consider an expanded role for helmet NIV in routine management. Regardless, it is increasingly clear that there may be an important albeit underinvestigated role for some form of high-level noninvasive respiratory support to prevent intubation, and perhaps mortality, in acute hypoxemic respiratory failure,” they noted.
“In future studies, reporting of interruptions to wearing the prescribed NIV interface continuously, leak severity, biomarkers of lung injury, and sedative administration would help delineate potential mechanisms [of action],” they added.
Dr. Beitler, Dr. Owens, and Dr. Malhotra are all affiliated with the division of pulmonary and critical care medicine at the University of California, San Diego. They had no financial conflicts to disclose.
“Several key clinical messages can be gained from the study by Patel et al.,” wrote Dr. Jeremy R. Beitler, Dr. Robert L. Owens, and Dr. Atul Malhotra, in an accompanying editorial (JAMA 2016 Jun; 315:2401-3.).
“The helmet interface has unique advantages and disadvantages that may influence efficacy of noninvasive positive pressure ventilation (NIV) depending on patient and disease characteristics,” they said. “External validation of the findings by Patel et al. and clarification of appropriate eligibility criteria, optimal ventilator settings, and potential mechanisms of effect are needed before clinicians could consider an expanded role for helmet NIV in routine management. Regardless, it is increasingly clear that there may be an important albeit underinvestigated role for some form of high-level noninvasive respiratory support to prevent intubation, and perhaps mortality, in acute hypoxemic respiratory failure,” they noted.
“In future studies, reporting of interruptions to wearing the prescribed NIV interface continuously, leak severity, biomarkers of lung injury, and sedative administration would help delineate potential mechanisms [of action],” they added.
Dr. Beitler, Dr. Owens, and Dr. Malhotra are all affiliated with the division of pulmonary and critical care medicine at the University of California, San Diego. They had no financial conflicts to disclose.
“Several key clinical messages can be gained from the study by Patel et al.,” wrote Dr. Jeremy R. Beitler, Dr. Robert L. Owens, and Dr. Atul Malhotra, in an accompanying editorial (JAMA 2016 Jun; 315:2401-3.).
“The helmet interface has unique advantages and disadvantages that may influence efficacy of noninvasive positive pressure ventilation (NIV) depending on patient and disease characteristics,” they said. “External validation of the findings by Patel et al. and clarification of appropriate eligibility criteria, optimal ventilator settings, and potential mechanisms of effect are needed before clinicians could consider an expanded role for helmet NIV in routine management. Regardless, it is increasingly clear that there may be an important albeit underinvestigated role for some form of high-level noninvasive respiratory support to prevent intubation, and perhaps mortality, in acute hypoxemic respiratory failure,” they noted.
“In future studies, reporting of interruptions to wearing the prescribed NIV interface continuously, leak severity, biomarkers of lung injury, and sedative administration would help delineate potential mechanisms [of action],” they added.
Dr. Beitler, Dr. Owens, and Dr. Malhotra are all affiliated with the division of pulmonary and critical care medicine at the University of California, San Diego. They had no financial conflicts to disclose.
Treating acute respiratory distress syndrome patients with helmets instead of face masks significantly reduced intubation rates and 90-day mortality rates, based on data from a randomized trial of 83 adults published in JAMA.
Acute respiratory distress syndrome (ARDS) can be treated using a face mask, but the mask is often inadequate, forcing patients to undergo intubation that can lead to additional problems including discomfort, delirium, and muscle deterioration, wrote Tianna Hicklin, Ph.D., in a column on the NIH website.
“The proposed mechanism for improved efficacy of these helmets is the preservation of applied pressures and avoidance of air leak. If the helmets indeed do allow clinicians both to be able to increase airway pressures above levels they typically would with mask-NIV and maintain those pressures without unpredictable system leak, that would be of great physiologic importance for patients with acute respiratory failure,” said Dr. Eric J. Gartman.
To explore the potential of noninvasive ventilation (NIV) using a helmet instead of a mask, both of which allow patients to remain awake in the ICU, the researchers randomized 83 adults with ARDS to treatment with either a helmet (44 patients) or a face mask (39 patients).
Intubation incidence was 18% in patients treated with helmets, compared with 62% for those treated with face masks. In addition, patients in the helmet group had significantly more ventilator-free days than the mask group (28 vs. 13; P less than .001), and both hospital mortality and 90-day mortality rates were significantly lower in the helmet group compared to the mask group (27% vs. 49%; P = .04 and 34% vs. 56%; P = .02, respectively)
Adverse event rates were rare, and similar between the helmet and mask groups. Three interface-related skin ulcers occurred in each group; nose ulcers in 7% of the mask group and neck ulcers in 7% of the helmet group.
The study population included adults aged 18 years and older who were admitted to the ICU at the University of Chicago between September 2012 and September 2015 and required face mask NIV. The average age of the patients was 58 years in the helmet group and 61 years in the mask group; there were no significant demographic differences between the groups. The most common causes of acute respiratory failure in both groups were pneumonia and pneumonia caused by immunosuppression.
The study results were limited by several factors including a lack of blinding, the need for a learning curve for clinicians using the helmet, and the potential for patient-ventilator dyssynchrony in the helmet group, noted Dr. Bhakti K. Patel of the University of Chicago and her colleagues (JAMA 2016;315:2435-41). Multicenter studies are needed to support the findings, they added. However, the findings “affirm the far-reaching benefits of spontaneous yet highly supported ventilation in an awake, animated patient over invasive medical ventilation via endotracheal tube,” they wrote. “These findings warrant further investigation of helmet NIV for patients with ARDS and other types of AHRF [acute hypoxemic respiratory failure], particularly with attention to long-term outcomes.”
The researchers had no financial conflicts to disclose.
“While the results of this study are very impressive, it is a single-center study, and obviously a larger multicenter trial (with all types of institutions included – not just large academic centers) would be helpful to elucidate the benefit of this technique and support a change in standard of care in the use of NIV. A change to this system would be a very large culture shift in NIV and would mean a significant amount of training (physician, nursing, respiratory care), purchasing the helmets, and ensuring that it is implemented properly and safely. As stated by the authors, this fact is similar to the change the occurred originally with NIV – but if their results reflect a true benefit over FM-NIV [face mask-noninvasive ventilation], such a large change would certainly be worth the effort,” Dr. Gartman said.
In a related study published in the Journal of Cardiothoracic and Vascular Anesthesia, patients treated with noninvasive positive pressure ventilation (NPPV) through helmets had significantly lower heart rates, lower average arterial pressure, and improved left ventricular ejection fraction at the end of treatment, compared with patients treated with ventilation masks and controls. Dr. Yi Yang of Capital Medical University in Beijing, China, and colleagues conducted the prospective study of 75 adults experiencing hypoxemia within 24 hours of extubation after Stanford type A aortic dissection. The participants were divided into three 25-patient groups. The control group was treated with high-flux inhalation of oxygen via a Venturi mask, another group was treated with NPPV via a mask, and the third group was treated with NPPV via a helmet. (J Card Vasc Anesth. 2016. http://dx.doi.org/10.1053/j.jvca.2016.03.129).
The study was funded by China’s public welfare industry of health.
Treating acute respiratory distress syndrome patients with helmets instead of face masks significantly reduced intubation rates and 90-day mortality rates, based on data from a randomized trial of 83 adults published in JAMA.
Acute respiratory distress syndrome (ARDS) can be treated using a face mask, but the mask is often inadequate, forcing patients to undergo intubation that can lead to additional problems including discomfort, delirium, and muscle deterioration, wrote Tianna Hicklin, Ph.D., in a column on the NIH website.
“The proposed mechanism for improved efficacy of these helmets is the preservation of applied pressures and avoidance of air leak. If the helmets indeed do allow clinicians both to be able to increase airway pressures above levels they typically would with mask-NIV and maintain those pressures without unpredictable system leak, that would be of great physiologic importance for patients with acute respiratory failure,” said Dr. Eric J. Gartman.
To explore the potential of noninvasive ventilation (NIV) using a helmet instead of a mask, both of which allow patients to remain awake in the ICU, the researchers randomized 83 adults with ARDS to treatment with either a helmet (44 patients) or a face mask (39 patients).
Intubation incidence was 18% in patients treated with helmets, compared with 62% for those treated with face masks. In addition, patients in the helmet group had significantly more ventilator-free days than the mask group (28 vs. 13; P less than .001), and both hospital mortality and 90-day mortality rates were significantly lower in the helmet group compared to the mask group (27% vs. 49%; P = .04 and 34% vs. 56%; P = .02, respectively)
Adverse event rates were rare, and similar between the helmet and mask groups. Three interface-related skin ulcers occurred in each group; nose ulcers in 7% of the mask group and neck ulcers in 7% of the helmet group.
The study population included adults aged 18 years and older who were admitted to the ICU at the University of Chicago between September 2012 and September 2015 and required face mask NIV. The average age of the patients was 58 years in the helmet group and 61 years in the mask group; there were no significant demographic differences between the groups. The most common causes of acute respiratory failure in both groups were pneumonia and pneumonia caused by immunosuppression.
The study results were limited by several factors including a lack of blinding, the need for a learning curve for clinicians using the helmet, and the potential for patient-ventilator dyssynchrony in the helmet group, noted Dr. Bhakti K. Patel of the University of Chicago and her colleagues (JAMA 2016;315:2435-41). Multicenter studies are needed to support the findings, they added. However, the findings “affirm the far-reaching benefits of spontaneous yet highly supported ventilation in an awake, animated patient over invasive medical ventilation via endotracheal tube,” they wrote. “These findings warrant further investigation of helmet NIV for patients with ARDS and other types of AHRF [acute hypoxemic respiratory failure], particularly with attention to long-term outcomes.”
The researchers had no financial conflicts to disclose.
“While the results of this study are very impressive, it is a single-center study, and obviously a larger multicenter trial (with all types of institutions included – not just large academic centers) would be helpful to elucidate the benefit of this technique and support a change in standard of care in the use of NIV. A change to this system would be a very large culture shift in NIV and would mean a significant amount of training (physician, nursing, respiratory care), purchasing the helmets, and ensuring that it is implemented properly and safely. As stated by the authors, this fact is similar to the change the occurred originally with NIV – but if their results reflect a true benefit over FM-NIV [face mask-noninvasive ventilation], such a large change would certainly be worth the effort,” Dr. Gartman said.
In a related study published in the Journal of Cardiothoracic and Vascular Anesthesia, patients treated with noninvasive positive pressure ventilation (NPPV) through helmets had significantly lower heart rates, lower average arterial pressure, and improved left ventricular ejection fraction at the end of treatment, compared with patients treated with ventilation masks and controls. Dr. Yi Yang of Capital Medical University in Beijing, China, and colleagues conducted the prospective study of 75 adults experiencing hypoxemia within 24 hours of extubation after Stanford type A aortic dissection. The participants were divided into three 25-patient groups. The control group was treated with high-flux inhalation of oxygen via a Venturi mask, another group was treated with NPPV via a mask, and the third group was treated with NPPV via a helmet. (J Card Vasc Anesth. 2016. http://dx.doi.org/10.1053/j.jvca.2016.03.129).
The study was funded by China’s public welfare industry of health.
FROM JAMA
Key clinical point: Patients with ARDS who were treated with noninvasive continuous positive airway pressure ventilation via a helmet needed significantly less intubation and had significantly higher survival rates through 90 days than did those treated with noninvasive ventilation via face masks.
Major finding: Intubation incidence was 18% in patients treated with helmets vs. 62% for those treated with face masks; 90-day survival rates for the helmet and mask groups were 66% and 44%, respectively.
Data source: A randomized, single center study of 83 patients with ARDS.
Disclosures: The researchers had no financial conflicts to disclose. The study was funded in part by the National Heart, Lung, and Blood Institute at the National Institutes of Health.

Perform urine culture if UTI suspected despite absence of pyuria
Urine cultures should be performed in all children clinically suspected of having a urinary tract infection (UTI), results of a study in Pediatrics suggest.
That’s because pyuria may be absent in some children with certain uropathogens.
“Clinicians rely heavily on the degree of pyuria when making a presumptive diagnosis of UTI [but] lack of pyuria on an initial urinalysis may result in delayed diagnosis and delayed antimicrobial therapy,” explained Dr. Nader Shaikh of the University of Pittsburgh and his associates. “We hypothesized, based on some preliminary data in adults, that gram-positive organisms would cause less inflammation of the urinary tract and consequently cause less pyuria on urinalysis than infections caused by gram-negative organisms, in which pyuria is observed in the vast majority of cases.”

Dr. Shaikh and his coinvestigators looked at patients under the age of 18 years admitted to the Children’s Hospital of Pittsburgh at the University of Pittsburgh Medical Center between 2007 and 2013 with a UTI. Of 46,158 relevant hospital visits over the course of the study period, 1,181 children were ultimately selected for inclusion. Urine samples were tested for the presence of uropathogens, followed by analysis for the presence of pyuria, defined as having 5 or more white blood cells per high-powered field or at least 10 white blood cells per mm3 of urine (Pediatrics. 2016. doi: 10.1542/peds.2016-0087).
Only 150 subjects (13%) did not have pyuria; the remaining 1,031 (87%) of subjects did. Escherichia coli was found in 999 of the 1,181 (85%) subjects included, of which 892 (89%) had pyuria. Additionally, of the 27 children found to have Staphylococcus saprophyticus, all had pyuria. High rates of pyuria also were found in children whose urine had Proteus species and Enterobacter species uropathogens: 25 of 31 (81%) and 13 of 15 (87%), respectively.
However, the presence of three other pathogens suggested a lower association between pyuria and UTI. Pseudomonas aeruginosa was found in 13 children, of which 8 (62%) had pyuria; similarly, those with Enterococcus species uropathogens had a 54% chance of having pyuria (19 of 35) and those with Klebsiella species had a 74% chance (34 of 46). The takeaway, therefore, is that pyuria may not always be present in cases of UTI, especially if these pathogens are the cause of it; in fact, “the odds of pyuria were three to five times lower” with these pathogens, compared with E. coli.
“The most recent American Academy of Pediatrics guideline suggests that pyuria should be present when diagnosing a UTI [but] only 90% of children with UTI exhibit pyuria even when the urine specimen is collected by bladder catheterization or suprapubic aspiration,” Dr. Shaikh and his associates found, adding that the “findings of our study offer an additional explanation for the absence of pyuria – some uropathogens may not elicit a strong host inflammatory response – [and] suggest that bedside biomarkers that are more sensitive and specific than pyuria are needed to improve the accuracy of early diagnosis.”
There was no outside funding involved with this study. Dr. Shaikh and his coauthors did not report any relevant financial disclosures.
Shaikh et al. demonstrate that certain urinary pathogens fail to reliably elicit pyuria. In 1,181 children where both a urine culture and concomitant urinalysis were performed, only 87% of the time was pyuria found in the setting of a positive culture. The authors further found that Enterococcus, Klebsiella, and Pseudomonas species were less likely to elicit pyuria or a positive leukocyte esterase test despite causing a urinary tract infection.
So how does the urinalysis help? More pointedly, why do a urinalysis? The report by Shaikh et al. agrees with a previously published meta-analysis that reveals pyuria to be absent in at least 10% of urines that culture positive. For a condition as common as UTI, this is too high a false-negative rate. Shaikh et al. conclude that a urine culture should be obtained in all children suspected of UTI. Indeed, the AAP guidelines are consistent with this statement. The AAP guidelines recognize that a clinician may have a low level of suspicion for UTI and may choose not to treat. However, given recent analyses of the utility of urinalysis and the report by Shaikh et al., it is difficult to see how a negative urinalysis might reassure a clinician if there are signs of a UTI. Hence, if one is considering treating for presumptive UTI, a culture is needed. If one is considering waiting and not treating, but suspects a UTI, a culture is still needed. Shaikh et al. conclude that new biomarkers are needed if we really want help with the “point of care” testing. Although we wait for new biomarkers, we should recognize the limitations of a negative urinalysis and still get that urine culture.
Dr. Aaron Friedman is the former dean of the University of Minnesota Medical School, Minneapolis. These comments are excerpted from a commentary accompanying Dr. Shaikh and his associates’ report (Pediatrics. 2016. doi: 10.1542/peds.2016-1247). Dr. Friedman did not report any relevant financial disclosures or sources of external funding.
Shaikh et al. demonstrate that certain urinary pathogens fail to reliably elicit pyuria. In 1,181 children where both a urine culture and concomitant urinalysis were performed, only 87% of the time was pyuria found in the setting of a positive culture. The authors further found that Enterococcus, Klebsiella, and Pseudomonas species were less likely to elicit pyuria or a positive leukocyte esterase test despite causing a urinary tract infection.
So how does the urinalysis help? More pointedly, why do a urinalysis? The report by Shaikh et al. agrees with a previously published meta-analysis that reveals pyuria to be absent in at least 10% of urines that culture positive. For a condition as common as UTI, this is too high a false-negative rate. Shaikh et al. conclude that a urine culture should be obtained in all children suspected of UTI. Indeed, the AAP guidelines are consistent with this statement. The AAP guidelines recognize that a clinician may have a low level of suspicion for UTI and may choose not to treat. However, given recent analyses of the utility of urinalysis and the report by Shaikh et al., it is difficult to see how a negative urinalysis might reassure a clinician if there are signs of a UTI. Hence, if one is considering treating for presumptive UTI, a culture is needed. If one is considering waiting and not treating, but suspects a UTI, a culture is still needed. Shaikh et al. conclude that new biomarkers are needed if we really want help with the “point of care” testing. Although we wait for new biomarkers, we should recognize the limitations of a negative urinalysis and still get that urine culture.
Dr. Aaron Friedman is the former dean of the University of Minnesota Medical School, Minneapolis. These comments are excerpted from a commentary accompanying Dr. Shaikh and his associates’ report (Pediatrics. 2016. doi: 10.1542/peds.2016-1247). Dr. Friedman did not report any relevant financial disclosures or sources of external funding.
Shaikh et al. demonstrate that certain urinary pathogens fail to reliably elicit pyuria. In 1,181 children where both a urine culture and concomitant urinalysis were performed, only 87% of the time was pyuria found in the setting of a positive culture. The authors further found that Enterococcus, Klebsiella, and Pseudomonas species were less likely to elicit pyuria or a positive leukocyte esterase test despite causing a urinary tract infection.
So how does the urinalysis help? More pointedly, why do a urinalysis? The report by Shaikh et al. agrees with a previously published meta-analysis that reveals pyuria to be absent in at least 10% of urines that culture positive. For a condition as common as UTI, this is too high a false-negative rate. Shaikh et al. conclude that a urine culture should be obtained in all children suspected of UTI. Indeed, the AAP guidelines are consistent with this statement. The AAP guidelines recognize that a clinician may have a low level of suspicion for UTI and may choose not to treat. However, given recent analyses of the utility of urinalysis and the report by Shaikh et al., it is difficult to see how a negative urinalysis might reassure a clinician if there are signs of a UTI. Hence, if one is considering treating for presumptive UTI, a culture is needed. If one is considering waiting and not treating, but suspects a UTI, a culture is still needed. Shaikh et al. conclude that new biomarkers are needed if we really want help with the “point of care” testing. Although we wait for new biomarkers, we should recognize the limitations of a negative urinalysis and still get that urine culture.
Dr. Aaron Friedman is the former dean of the University of Minnesota Medical School, Minneapolis. These comments are excerpted from a commentary accompanying Dr. Shaikh and his associates’ report (Pediatrics. 2016. doi: 10.1542/peds.2016-1247). Dr. Friedman did not report any relevant financial disclosures or sources of external funding.
Urine cultures should be performed in all children clinically suspected of having a urinary tract infection (UTI), results of a study in Pediatrics suggest.
That’s because pyuria may be absent in some children with certain uropathogens.
“Clinicians rely heavily on the degree of pyuria when making a presumptive diagnosis of UTI [but] lack of pyuria on an initial urinalysis may result in delayed diagnosis and delayed antimicrobial therapy,” explained Dr. Nader Shaikh of the University of Pittsburgh and his associates. “We hypothesized, based on some preliminary data in adults, that gram-positive organisms would cause less inflammation of the urinary tract and consequently cause less pyuria on urinalysis than infections caused by gram-negative organisms, in which pyuria is observed in the vast majority of cases.”
Dr. Shaikh and his coinvestigators looked at patients under the age of 18 years admitted to the Children’s Hospital of Pittsburgh at the University of Pittsburgh Medical Center between 2007 and 2013 with a UTI. Of 46,158 relevant hospital visits over the course of the study period, 1,181 children were ultimately selected for inclusion. Urine samples were tested for the presence of uropathogens, followed by analysis for the presence of pyuria, defined as having 5 or more white blood cells per high-powered field or at least 10 white blood cells per mm3 of urine (Pediatrics. 2016. doi: 10.1542/peds.2016-0087).
Only 150 subjects (13%) did not have pyuria; the remaining 1,031 (87%) of subjects did. Escherichia coli was found in 999 of the 1,181 (85%) subjects included, of which 892 (89%) had pyuria. Additionally, of the 27 children found to have Staphylococcus saprophyticus, all had pyuria. High rates of pyuria also were found in children whose urine had Proteus species and Enterobacter species uropathogens: 25 of 31 (81%) and 13 of 15 (87%), respectively.
However, the presence of three other pathogens suggested a lower association between pyuria and UTI. Pseudomonas aeruginosa was found in 13 children, of which 8 (62%) had pyuria; similarly, those with Enterococcus species uropathogens had a 54% chance of having pyuria (19 of 35) and those with Klebsiella species had a 74% chance (34 of 46). The takeaway, therefore, is that pyuria may not always be present in cases of UTI, especially if these pathogens are the cause of it; in fact, “the odds of pyuria were three to five times lower” with these pathogens, compared with E. coli.
“The most recent American Academy of Pediatrics guideline suggests that pyuria should be present when diagnosing a UTI [but] only 90% of children with UTI exhibit pyuria even when the urine specimen is collected by bladder catheterization or suprapubic aspiration,” Dr. Shaikh and his associates found, adding that the “findings of our study offer an additional explanation for the absence of pyuria – some uropathogens may not elicit a strong host inflammatory response – [and] suggest that bedside biomarkers that are more sensitive and specific than pyuria are needed to improve the accuracy of early diagnosis.”
There was no outside funding involved with this study. Dr. Shaikh and his coauthors did not report any relevant financial disclosures.
Urine cultures should be performed in all children clinically suspected of having a urinary tract infection (UTI), results of a study in Pediatrics suggest.
That’s because pyuria may be absent in some children with certain uropathogens.
“Clinicians rely heavily on the degree of pyuria when making a presumptive diagnosis of UTI [but] lack of pyuria on an initial urinalysis may result in delayed diagnosis and delayed antimicrobial therapy,” explained Dr. Nader Shaikh of the University of Pittsburgh and his associates. “We hypothesized, based on some preliminary data in adults, that gram-positive organisms would cause less inflammation of the urinary tract and consequently cause less pyuria on urinalysis than infections caused by gram-negative organisms, in which pyuria is observed in the vast majority of cases.”
Dr. Shaikh and his coinvestigators looked at patients under the age of 18 years admitted to the Children’s Hospital of Pittsburgh at the University of Pittsburgh Medical Center between 2007 and 2013 with a UTI. Of 46,158 relevant hospital visits over the course of the study period, 1,181 children were ultimately selected for inclusion. Urine samples were tested for the presence of uropathogens, followed by analysis for the presence of pyuria, defined as having 5 or more white blood cells per high-powered field or at least 10 white blood cells per mm3 of urine (Pediatrics. 2016. doi: 10.1542/peds.2016-0087).
Only 150 subjects (13%) did not have pyuria; the remaining 1,031 (87%) of subjects did. Escherichia coli was found in 999 of the 1,181 (85%) subjects included, of which 892 (89%) had pyuria. Additionally, of the 27 children found to have Staphylococcus saprophyticus, all had pyuria. High rates of pyuria also were found in children whose urine had Proteus species and Enterobacter species uropathogens: 25 of 31 (81%) and 13 of 15 (87%), respectively.
However, the presence of three other pathogens suggested a lower association between pyuria and UTI. Pseudomonas aeruginosa was found in 13 children, of which 8 (62%) had pyuria; similarly, those with Enterococcus species uropathogens had a 54% chance of having pyuria (19 of 35) and those with Klebsiella species had a 74% chance (34 of 46). The takeaway, therefore, is that pyuria may not always be present in cases of UTI, especially if these pathogens are the cause of it; in fact, “the odds of pyuria were three to five times lower” with these pathogens, compared with E. coli.
“The most recent American Academy of Pediatrics guideline suggests that pyuria should be present when diagnosing a UTI [but] only 90% of children with UTI exhibit pyuria even when the urine specimen is collected by bladder catheterization or suprapubic aspiration,” Dr. Shaikh and his associates found, adding that the “findings of our study offer an additional explanation for the absence of pyuria – some uropathogens may not elicit a strong host inflammatory response – [and] suggest that bedside biomarkers that are more sensitive and specific than pyuria are needed to improve the accuracy of early diagnosis.”
There was no outside funding involved with this study. Dr. Shaikh and his coauthors did not report any relevant financial disclosures.
FROM PEDIATRICS
Key clinical point: Certain pathogens found in urine, such as E. coli, are more reliable than others at indicating a child’s risk for pyuria.
Major finding: 89% of subjects with E. coli in their urinary tract also had pyuria, compared with 62% of those with P. aeruginosa, 54.3% of those with a member of the Enterococcus species, and 74% of those with a member of the Klebsiella species found in their urinary tract.
Data source: Retrospective review of 1,181 children diagnosed with UTIs between 2007 and 2013 at the Children’s Hospital of Pittsburgh at UPMC.
Disclosures: The study had no external funding. Dr. Shaikh and his coauthors did not report any relevant financial disclosures.

LETTER: Emory Hospital Medicine’s Growth Sparks Establishment of NP, PA Career Track
Due to many reasons, the healthcare paradigm has shifted, dictating alternative staffing models to manage the burgeoning inpatient census of hospital-based physicians. Herein, we will briefly describe the Emory University Division of Hospital Medicine (EDHM) approach to utilizing advanced practice providers (APPs) in the care of inpatients and summarize key components of the program that improve sustainability for providers.
The EDHM in Atlanta matriculated APPs into its service in 2004. Currently, there are 22 APPs across all Emory HM sites. The largest group is at Emory University Hospital Midtown (EUHM).
At EUHM, the addition of a renal service created concern for increased workload for the physicians. APPs were recruited to bridge the gap in 2011. Initially, the role was ill-defined, but over time, with physician and administrative leadership buy-in and support, the role has evolved. Currently at EUHM, APPs are practicing in other HM services, allowing them to practice near or at the top of their scope of practice. The 12 hospitalist APPs at EUHM practice in four roles: nocturnist, frontline provider in the observation unit, dedicated renal service, and generalist on an overflow team.
Along with the rapid growth of APPs on the service came the need for structured leadership, improved onboarding procedures, competency maintenance, advocacy, and professional development activities. Essentially, we needed to create a career track parallel to that of the physicians without compromising the portion of our scopes of our practice that overlap (i.e., patient care) while supporting our regulatory differences.
The professional development plans incorporated findings from APP exit interviews at the University of Maryland Medical Center highlighting the following retention issues:1
- Length of time for credentialing
- Role clarity
- Inadequate clinical orientation
- Feelings of clinical incompetence
- Feelings of isolation
With the instillation of APP leadership, the team created a comprehensive APP program. The Hospital Medicine APP program at EUHM includes the following components:
- APP representation at monthly clinical operation meetings and quarterly education council meetings to ensure that APP competency and regulatory issues are always represented.
- Orientation personally tailored to the APP’s level of clinical expertise, with a post-orientation meeting with leadership and remediation, if needed.
- APP incentives to teach NP or PA students, conduct in-services, join committees, or participate in other leadership opportunities.
- APPs invited to attend and/or present at all divisional small and large group learning opportunities (e.g., Grand Rounds, Lunch and Learn, Journal Club).
- APPs allocated time and space to meet and discuss practice issues.
- Newly developed Mini-Hospitalist Academy, which offers monthly workshops to all hospitalist physicians and APPs, from novice to expert.
- Dedicated APP Ongoing Professional Performance Evaluation (OPPE) program.
- In addition to the annual monetary support offered for educational opportunities, the division offers an annual Faculty Development Award. This award is by application for eligible educational opportunities; APPs are welcome to apply and have consistently been awarded support to pursue myriad opportunities.
This successful APP-physician collaboration is driven by a committed group of professionals who are sensitive to the shifting healthcare paradigm. Our APPs and physicians are constantly adapting their practice so that our collaboration is safe, evidence-based, and professionally fulfilling. TH
Yvonne Brown, DNP, MSN, ACNP-C, FNP-C, nurse practitioner, lead advanced practice provider, Division of Hospital Medicine, Emory Healthcare, Emory University Hospital Midtown, Atlanta
Reference
1. Bahouth MN, Esposito-Herr MB. Orientation program for hospital-based nurse practitioners. AACN Adv Crit Care. 2009;20(1):82-90.
Due to many reasons, the healthcare paradigm has shifted, dictating alternative staffing models to manage the burgeoning inpatient census of hospital-based physicians. Herein, we will briefly describe the Emory University Division of Hospital Medicine (EDHM) approach to utilizing advanced practice providers (APPs) in the care of inpatients and summarize key components of the program that improve sustainability for providers.
The EDHM in Atlanta matriculated APPs into its service in 2004. Currently, there are 22 APPs across all Emory HM sites. The largest group is at Emory University Hospital Midtown (EUHM).
At EUHM, the addition of a renal service created concern for increased workload for the physicians. APPs were recruited to bridge the gap in 2011. Initially, the role was ill-defined, but over time, with physician and administrative leadership buy-in and support, the role has evolved. Currently at EUHM, APPs are practicing in other HM services, allowing them to practice near or at the top of their scope of practice. The 12 hospitalist APPs at EUHM practice in four roles: nocturnist, frontline provider in the observation unit, dedicated renal service, and generalist on an overflow team.
Along with the rapid growth of APPs on the service came the need for structured leadership, improved onboarding procedures, competency maintenance, advocacy, and professional development activities. Essentially, we needed to create a career track parallel to that of the physicians without compromising the portion of our scopes of our practice that overlap (i.e., patient care) while supporting our regulatory differences.
The professional development plans incorporated findings from APP exit interviews at the University of Maryland Medical Center highlighting the following retention issues:1
- Length of time for credentialing
- Role clarity
- Inadequate clinical orientation
- Feelings of clinical incompetence
- Feelings of isolation
With the instillation of APP leadership, the team created a comprehensive APP program. The Hospital Medicine APP program at EUHM includes the following components:
- APP representation at monthly clinical operation meetings and quarterly education council meetings to ensure that APP competency and regulatory issues are always represented.
- Orientation personally tailored to the APP’s level of clinical expertise, with a post-orientation meeting with leadership and remediation, if needed.
- APP incentives to teach NP or PA students, conduct in-services, join committees, or participate in other leadership opportunities.
- APPs invited to attend and/or present at all divisional small and large group learning opportunities (e.g., Grand Rounds, Lunch and Learn, Journal Club).
- APPs allocated time and space to meet and discuss practice issues.
- Newly developed Mini-Hospitalist Academy, which offers monthly workshops to all hospitalist physicians and APPs, from novice to expert.
- Dedicated APP Ongoing Professional Performance Evaluation (OPPE) program.
- In addition to the annual monetary support offered for educational opportunities, the division offers an annual Faculty Development Award. This award is by application for eligible educational opportunities; APPs are welcome to apply and have consistently been awarded support to pursue myriad opportunities.
This successful APP-physician collaboration is driven by a committed group of professionals who are sensitive to the shifting healthcare paradigm. Our APPs and physicians are constantly adapting their practice so that our collaboration is safe, evidence-based, and professionally fulfilling. TH
Yvonne Brown, DNP, MSN, ACNP-C, FNP-C, nurse practitioner, lead advanced practice provider, Division of Hospital Medicine, Emory Healthcare, Emory University Hospital Midtown, Atlanta
Reference
1. Bahouth MN, Esposito-Herr MB. Orientation program for hospital-based nurse practitioners. AACN Adv Crit Care. 2009;20(1):82-90.
Due to many reasons, the healthcare paradigm has shifted, dictating alternative staffing models to manage the burgeoning inpatient census of hospital-based physicians. Herein, we will briefly describe the Emory University Division of Hospital Medicine (EDHM) approach to utilizing advanced practice providers (APPs) in the care of inpatients and summarize key components of the program that improve sustainability for providers.
The EDHM in Atlanta matriculated APPs into its service in 2004. Currently, there are 22 APPs across all Emory HM sites. The largest group is at Emory University Hospital Midtown (EUHM).
At EUHM, the addition of a renal service created concern for increased workload for the physicians. APPs were recruited to bridge the gap in 2011. Initially, the role was ill-defined, but over time, with physician and administrative leadership buy-in and support, the role has evolved. Currently at EUHM, APPs are practicing in other HM services, allowing them to practice near or at the top of their scope of practice. The 12 hospitalist APPs at EUHM practice in four roles: nocturnist, frontline provider in the observation unit, dedicated renal service, and generalist on an overflow team.
Along with the rapid growth of APPs on the service came the need for structured leadership, improved onboarding procedures, competency maintenance, advocacy, and professional development activities. Essentially, we needed to create a career track parallel to that of the physicians without compromising the portion of our scopes of our practice that overlap (i.e., patient care) while supporting our regulatory differences.
The professional development plans incorporated findings from APP exit interviews at the University of Maryland Medical Center highlighting the following retention issues:1
- Length of time for credentialing
- Role clarity
- Inadequate clinical orientation
- Feelings of clinical incompetence
- Feelings of isolation
With the instillation of APP leadership, the team created a comprehensive APP program. The Hospital Medicine APP program at EUHM includes the following components:
- APP representation at monthly clinical operation meetings and quarterly education council meetings to ensure that APP competency and regulatory issues are always represented.
- Orientation personally tailored to the APP’s level of clinical expertise, with a post-orientation meeting with leadership and remediation, if needed.
- APP incentives to teach NP or PA students, conduct in-services, join committees, or participate in other leadership opportunities.
- APPs invited to attend and/or present at all divisional small and large group learning opportunities (e.g., Grand Rounds, Lunch and Learn, Journal Club).
- APPs allocated time and space to meet and discuss practice issues.
- Newly developed Mini-Hospitalist Academy, which offers monthly workshops to all hospitalist physicians and APPs, from novice to expert.
- Dedicated APP Ongoing Professional Performance Evaluation (OPPE) program.
- In addition to the annual monetary support offered for educational opportunities, the division offers an annual Faculty Development Award. This award is by application for eligible educational opportunities; APPs are welcome to apply and have consistently been awarded support to pursue myriad opportunities.
This successful APP-physician collaboration is driven by a committed group of professionals who are sensitive to the shifting healthcare paradigm. Our APPs and physicians are constantly adapting their practice so that our collaboration is safe, evidence-based, and professionally fulfilling. TH
Yvonne Brown, DNP, MSN, ACNP-C, FNP-C, nurse practitioner, lead advanced practice provider, Division of Hospital Medicine, Emory Healthcare, Emory University Hospital Midtown, Atlanta
Reference
1. Bahouth MN, Esposito-Herr MB. Orientation program for hospital-based nurse practitioners. AACN Adv Crit Care. 2009;20(1):82-90.
Site of major bleeding differs with NOACs and VKAs

COPENHAGEN—Results of a new study suggest that vitamin K antagonists (VKAs) and non-VKA oral anticoagulants (NOACs) can both confer an increased risk of major bleeding, but the type of bleeding tends to differ with the type of anticoagulant.
The research showed that patients taking VKAs were more likely to be hospitalized for intracranial hemorrhage (ICH), while patients taking NOACs were more likely to be hospitalized for gastrointestinal (GI) bleeding.
In addition, the 30-day mortality rate was significantly lower in patients who were taking NOACs than in those who were taking VKAs. However, there was no significant difference between the 2 treatment groups when the researchers assessed mortality according to the type of major bleeding.
Laura Franco, MD, of the University of Perugia in Italy, presented these findings at the 21st Congress of the European Hematology Association (abstract S139*).
For this study, Dr Franco and her colleagues wanted to evaluate the real-life clinical presentation, management, and outcome of major bleeding in patients on anticoagulants.
So the team analyzed a cohort of patients treated at 9 Italian hospitals and a group of patients enrolled in Germany’s Dresden NOAC registry.
Between September 2013 and May 2016, there were 874 patients who were hospitalized for major bleeding—220 of whom who were taking NOACs and 654 of whom were taking VKAs.
Overall, 44% (n=386) of patients were hospitalized for an ICH, and 30% (n=261) were hospitalized for GI bleeding.
The incidence of ICH was 22% (n=48) among patients who received NOACs and 52% (n=338) among patients who received VKAs. The odds ratio was 0.26 (95% CI, 0.18-0.37; P<0.001).
The incidence of GI major bleeding was 46% (n=102) in the NOAC group and 24% (n=159) in the VKA group. The odds ratio was 2.69 (95% CI, 1.95-3.70; P<0.001).
The incidence of 30-day mortality was significantly higher in the VKA group than the NOAC group—17% and 10%, respectively. The adjusted hazard ratio (aHR) was 0.59 (95% CI, 0.36-0.94, P=0.012).
However, when the researchers looked at 30-day mortality according to the site of major bleeding, they observed no significant differences between the NOAC and VKA groups.
The incidence of 30-day mortality among patients with ICH was 25% in both the NOAC and VKA groups (aHR=1.07; 95% CI, 0.57-1.97). And the incidence of 30-day mortality among patients with GI bleeding was 7% in the NOAC group and 10% in the VKA group (aHR=0.66; 95% CI, 0.26-1.68). ![]()
*Data in the abstract differ from data presented at the meeting.
COPENHAGEN—Results of a new study suggest that vitamin K antagonists (VKAs) and non-VKA oral anticoagulants (NOACs) can both confer an increased risk of major bleeding, but the type of bleeding tends to differ with the type of anticoagulant.
The research showed that patients taking VKAs were more likely to be hospitalized for intracranial hemorrhage (ICH), while patients taking NOACs were more likely to be hospitalized for gastrointestinal (GI) bleeding.
In addition, the 30-day mortality rate was significantly lower in patients who were taking NOACs than in those who were taking VKAs. However, there was no significant difference between the 2 treatment groups when the researchers assessed mortality according to the type of major bleeding.
Laura Franco, MD, of the University of Perugia in Italy, presented these findings at the 21st Congress of the European Hematology Association (abstract S139*).
For this study, Dr Franco and her colleagues wanted to evaluate the real-life clinical presentation, management, and outcome of major bleeding in patients on anticoagulants.
So the team analyzed a cohort of patients treated at 9 Italian hospitals and a group of patients enrolled in Germany’s Dresden NOAC registry.
Between September 2013 and May 2016, there were 874 patients who were hospitalized for major bleeding—220 of whom who were taking NOACs and 654 of whom were taking VKAs.
Overall, 44% (n=386) of patients were hospitalized for an ICH, and 30% (n=261) were hospitalized for GI bleeding.
The incidence of ICH was 22% (n=48) among patients who received NOACs and 52% (n=338) among patients who received VKAs. The odds ratio was 0.26 (95% CI, 0.18-0.37; P<0.001).
The incidence of GI major bleeding was 46% (n=102) in the NOAC group and 24% (n=159) in the VKA group. The odds ratio was 2.69 (95% CI, 1.95-3.70; P<0.001).
The incidence of 30-day mortality was significantly higher in the VKA group than the NOAC group—17% and 10%, respectively. The adjusted hazard ratio (aHR) was 0.59 (95% CI, 0.36-0.94, P=0.012).
However, when the researchers looked at 30-day mortality according to the site of major bleeding, they observed no significant differences between the NOAC and VKA groups.
The incidence of 30-day mortality among patients with ICH was 25% in both the NOAC and VKA groups (aHR=1.07; 95% CI, 0.57-1.97). And the incidence of 30-day mortality among patients with GI bleeding was 7% in the NOAC group and 10% in the VKA group (aHR=0.66; 95% CI, 0.26-1.68). ![]()
*Data in the abstract differ from data presented at the meeting.
COPENHAGEN—Results of a new study suggest that vitamin K antagonists (VKAs) and non-VKA oral anticoagulants (NOACs) can both confer an increased risk of major bleeding, but the type of bleeding tends to differ with the type of anticoagulant.
The research showed that patients taking VKAs were more likely to be hospitalized for intracranial hemorrhage (ICH), while patients taking NOACs were more likely to be hospitalized for gastrointestinal (GI) bleeding.
In addition, the 30-day mortality rate was significantly lower in patients who were taking NOACs than in those who were taking VKAs. However, there was no significant difference between the 2 treatment groups when the researchers assessed mortality according to the type of major bleeding.
Laura Franco, MD, of the University of Perugia in Italy, presented these findings at the 21st Congress of the European Hematology Association (abstract S139*).
For this study, Dr Franco and her colleagues wanted to evaluate the real-life clinical presentation, management, and outcome of major bleeding in patients on anticoagulants.
So the team analyzed a cohort of patients treated at 9 Italian hospitals and a group of patients enrolled in Germany’s Dresden NOAC registry.
Between September 2013 and May 2016, there were 874 patients who were hospitalized for major bleeding—220 of whom who were taking NOACs and 654 of whom were taking VKAs.
Overall, 44% (n=386) of patients were hospitalized for an ICH, and 30% (n=261) were hospitalized for GI bleeding.
The incidence of ICH was 22% (n=48) among patients who received NOACs and 52% (n=338) among patients who received VKAs. The odds ratio was 0.26 (95% CI, 0.18-0.37; P<0.001).
The incidence of GI major bleeding was 46% (n=102) in the NOAC group and 24% (n=159) in the VKA group. The odds ratio was 2.69 (95% CI, 1.95-3.70; P<0.001).
The incidence of 30-day mortality was significantly higher in the VKA group than the NOAC group—17% and 10%, respectively. The adjusted hazard ratio (aHR) was 0.59 (95% CI, 0.36-0.94, P=0.012).
However, when the researchers looked at 30-day mortality according to the site of major bleeding, they observed no significant differences between the NOAC and VKA groups.
The incidence of 30-day mortality among patients with ICH was 25% in both the NOAC and VKA groups (aHR=1.07; 95% CI, 0.57-1.97). And the incidence of 30-day mortality among patients with GI bleeding was 7% in the NOAC group and 10% in the VKA group (aHR=0.66; 95% CI, 0.26-1.68). ![]()
*Data in the abstract differ from data presented at the meeting.
How CML cells respond to stress

Image by Difu Wu
Researchers have used a tiny force probe to compare how healthy hematopoietic cells and cancerous ones respond to stress.
They found that cells harvested from the bone marrow of 5 patients with chronic myeloid leukemia (CML) appeared much stiffer than comparable samples taken from 5 healthy volunteers.
In addition, the researchers were able to identify areas of localized brittle failure events in the CML cells.
“What makes this work so exciting to us is not simply seeing a difference between the stiffness of healthy and cancerous cells but observing that the cancerous cells also lost their dynamic ductility and behaved as more breakable objects,” said study author Françoise Argoul, PhD, of the French National Centre for Research (CNRS) in Lyon, France.
Dr Argoul and her colleagues described this work in Physical Biology.
The researchers believe the mechanical signatures obtained by squeezing or deforming cells could potentially assist physicians in determining the presence of CML and other hematologic malignancies.
The mechanical data might also provide clues as to how long the cells have been affected by the cancer.
“We would like to construct a hematopoietic cancer cell chart where the loss of cell mechanical functions could be graded, depending on the leukemia and its stage of evolution,” Dr Argoul said.
Thinking about how the technique might be applied in a hospital setting, she added that biopsy needles could, in principle, be adapted to allow local sensing of internal soft tissue structures.
However, before the researchers can even progress to testing cells inside the body and preparing for clinical trials, they must first build up sufficient information from their measurements on isolated cells under a range of conditions in the lab. ![]()
Image by Difu Wu
Researchers have used a tiny force probe to compare how healthy hematopoietic cells and cancerous ones respond to stress.
They found that cells harvested from the bone marrow of 5 patients with chronic myeloid leukemia (CML) appeared much stiffer than comparable samples taken from 5 healthy volunteers.
In addition, the researchers were able to identify areas of localized brittle failure events in the CML cells.
“What makes this work so exciting to us is not simply seeing a difference between the stiffness of healthy and cancerous cells but observing that the cancerous cells also lost their dynamic ductility and behaved as more breakable objects,” said study author Françoise Argoul, PhD, of the French National Centre for Research (CNRS) in Lyon, France.
Dr Argoul and her colleagues described this work in Physical Biology.
The researchers believe the mechanical signatures obtained by squeezing or deforming cells could potentially assist physicians in determining the presence of CML and other hematologic malignancies.
The mechanical data might also provide clues as to how long the cells have been affected by the cancer.
“We would like to construct a hematopoietic cancer cell chart where the loss of cell mechanical functions could be graded, depending on the leukemia and its stage of evolution,” Dr Argoul said.
Thinking about how the technique might be applied in a hospital setting, she added that biopsy needles could, in principle, be adapted to allow local sensing of internal soft tissue structures.
However, before the researchers can even progress to testing cells inside the body and preparing for clinical trials, they must first build up sufficient information from their measurements on isolated cells under a range of conditions in the lab. ![]()
Image by Difu Wu
Researchers have used a tiny force probe to compare how healthy hematopoietic cells and cancerous ones respond to stress.
They found that cells harvested from the bone marrow of 5 patients with chronic myeloid leukemia (CML) appeared much stiffer than comparable samples taken from 5 healthy volunteers.
In addition, the researchers were able to identify areas of localized brittle failure events in the CML cells.
“What makes this work so exciting to us is not simply seeing a difference between the stiffness of healthy and cancerous cells but observing that the cancerous cells also lost their dynamic ductility and behaved as more breakable objects,” said study author Françoise Argoul, PhD, of the French National Centre for Research (CNRS) in Lyon, France.
Dr Argoul and her colleagues described this work in Physical Biology.
The researchers believe the mechanical signatures obtained by squeezing or deforming cells could potentially assist physicians in determining the presence of CML and other hematologic malignancies.
The mechanical data might also provide clues as to how long the cells have been affected by the cancer.
“We would like to construct a hematopoietic cancer cell chart where the loss of cell mechanical functions could be graded, depending on the leukemia and its stage of evolution,” Dr Argoul said.
Thinking about how the technique might be applied in a hospital setting, she added that biopsy needles could, in principle, be adapted to allow local sensing of internal soft tissue structures.
However, before the researchers can even progress to testing cells inside the body and preparing for clinical trials, they must first build up sufficient information from their measurements on isolated cells under a range of conditions in the lab. ![]()