User login
Artificial RBCs show promise in preclinical study

Photo by Aaron Logan
SAN DIEGO—Researchers have developed artificial red blood cells (RBCs) that appear able to emulate functions of natural red blood cells (RBCs), at least in rodents.
The artificial RBCs, known as ErythroMer, are designed to be freeze-dried, stored at ambient temperatures, and reconstituted with water when needed.
If ErythroMer proves safe and effective in humans, it could represent an alternative to blood transfusions that might be useful in situations where donated blood is difficult to obtain or store.
“There are currently no simple, practical means to bring transfusion to most trauma victims outside of hospitals,” said Allan Doctor, MD, of Washington University in Saint Louis, Missouri.
“ErythroMer would be a blood substitute that a medic can carry in his or her pack and literally take it out, add water, and inject it.”
Dr Doctor presented details on ErythroMer at the 2016 ASH Annual Meeting (abstract 1027).
Design
“Due to significant advances in synthetic chemistry and nanomedicine, we’re now able to encapsulate biologics with programmable polymers to generate nanoparticles that can emulate normal cellular physiology,” Dr Doctor noted.
With ErythroMer, he and his colleagues encapsulated human hemoglobin, methylene blue, and 2,3-DPG in an amphiphilic polymer shell. The polymer and its payload components, through microfluidization, self-assemble into toroids that are about one-fiftieth the size of human RBCs.
ErythroMer is designed to be pH-responsive, so that, in areas of high pH, 2,3-DPG is sequestered in the inner surface of the particle shell and does not bind to hemoglobin. In areas of low pH, 2,3-DPG is released from the shell and binds to hemoglobin, facilitating oxygen offloading. The role of methylene blue is to inhibit auto-oxidation of hemoglobin.
The last step in synthesis of the particle is crosslinking of the surface, which neutralizes the surface charge, stabilizes the particle, and generates a selective diffusion barrier to nitric oxide. The particle can be lyophilized for extended storage and later reconstituted.
Testing
Tests showed that ErythroMer matches the oxygen binding feature of human RBCs within 10%, a level researchers say should be sufficient to stabilize a bleeding patient until a blood transfusion can be obtained.
Experiments in mice showed that ErythroMer captures oxygen in the lungs and releases it to tissues in a pattern that is indistinguishable from that seen in a control group of mice injected with their own blood.
In rats, ErythroMer effectively resuscitated animals in shock following acute loss of 40% of their blood volume.
So far, tests suggest ErythroMer has overcome barriers that halted the development of previous blood substitutes.
However, Dr Doctor noted that ErythroMer does have its weaknesses. The particles are cleared rapidly from the bloodstream (in 3 to 7 hours), and hemoglobin sourcing presents a challenge. The researchers are now exploring the possibility of using recombinant hemoglobin genetically engineered in yeast.
The team hopes to further optimize ErythroMer’s shell, extend circulation time, confirm the efficacy of ErythroMer in a larger animal model (rabbits), evaluate the impact of the product on the coagulation and immune systems, and scale up production.
If further testing goes well, the researchers estimate that ErythroMer could be ready for use by field medics and emergency responders within 10 to 12 years.
ErythroMer development has been supported by the Children’s Discovery Institute at Washington University and St. Louis Children’s Hospital, the Skandalaris Center at Washington University, and the BioSTL Fundamentals Program.
This research was funded by the National Institute of General Medical Sciences; the National Heart, Lung, and Blood Institute; the National Institute of Child Health and Human Development, the US Department of Defense; the American Heart Association; Doris Duke Foundation; and Children’s Discovery Institute. ![]()
Photo by Aaron Logan
SAN DIEGO—Researchers have developed artificial red blood cells (RBCs) that appear able to emulate functions of natural red blood cells (RBCs), at least in rodents.
The artificial RBCs, known as ErythroMer, are designed to be freeze-dried, stored at ambient temperatures, and reconstituted with water when needed.
If ErythroMer proves safe and effective in humans, it could represent an alternative to blood transfusions that might be useful in situations where donated blood is difficult to obtain or store.
“There are currently no simple, practical means to bring transfusion to most trauma victims outside of hospitals,” said Allan Doctor, MD, of Washington University in Saint Louis, Missouri.
“ErythroMer would be a blood substitute that a medic can carry in his or her pack and literally take it out, add water, and inject it.”
Dr Doctor presented details on ErythroMer at the 2016 ASH Annual Meeting (abstract 1027).
Design
“Due to significant advances in synthetic chemistry and nanomedicine, we’re now able to encapsulate biologics with programmable polymers to generate nanoparticles that can emulate normal cellular physiology,” Dr Doctor noted.
With ErythroMer, he and his colleagues encapsulated human hemoglobin, methylene blue, and 2,3-DPG in an amphiphilic polymer shell. The polymer and its payload components, through microfluidization, self-assemble into toroids that are about one-fiftieth the size of human RBCs.
ErythroMer is designed to be pH-responsive, so that, in areas of high pH, 2,3-DPG is sequestered in the inner surface of the particle shell and does not bind to hemoglobin. In areas of low pH, 2,3-DPG is released from the shell and binds to hemoglobin, facilitating oxygen offloading. The role of methylene blue is to inhibit auto-oxidation of hemoglobin.
The last step in synthesis of the particle is crosslinking of the surface, which neutralizes the surface charge, stabilizes the particle, and generates a selective diffusion barrier to nitric oxide. The particle can be lyophilized for extended storage and later reconstituted.
Testing
Tests showed that ErythroMer matches the oxygen binding feature of human RBCs within 10%, a level researchers say should be sufficient to stabilize a bleeding patient until a blood transfusion can be obtained.
Experiments in mice showed that ErythroMer captures oxygen in the lungs and releases it to tissues in a pattern that is indistinguishable from that seen in a control group of mice injected with their own blood.
In rats, ErythroMer effectively resuscitated animals in shock following acute loss of 40% of their blood volume.
So far, tests suggest ErythroMer has overcome barriers that halted the development of previous blood substitutes.
However, Dr Doctor noted that ErythroMer does have its weaknesses. The particles are cleared rapidly from the bloodstream (in 3 to 7 hours), and hemoglobin sourcing presents a challenge. The researchers are now exploring the possibility of using recombinant hemoglobin genetically engineered in yeast.
The team hopes to further optimize ErythroMer’s shell, extend circulation time, confirm the efficacy of ErythroMer in a larger animal model (rabbits), evaluate the impact of the product on the coagulation and immune systems, and scale up production.
If further testing goes well, the researchers estimate that ErythroMer could be ready for use by field medics and emergency responders within 10 to 12 years.
ErythroMer development has been supported by the Children’s Discovery Institute at Washington University and St. Louis Children’s Hospital, the Skandalaris Center at Washington University, and the BioSTL Fundamentals Program.
This research was funded by the National Institute of General Medical Sciences; the National Heart, Lung, and Blood Institute; the National Institute of Child Health and Human Development, the US Department of Defense; the American Heart Association; Doris Duke Foundation; and Children’s Discovery Institute. ![]()
Photo by Aaron Logan
SAN DIEGO—Researchers have developed artificial red blood cells (RBCs) that appear able to emulate functions of natural red blood cells (RBCs), at least in rodents.
The artificial RBCs, known as ErythroMer, are designed to be freeze-dried, stored at ambient temperatures, and reconstituted with water when needed.
If ErythroMer proves safe and effective in humans, it could represent an alternative to blood transfusions that might be useful in situations where donated blood is difficult to obtain or store.
“There are currently no simple, practical means to bring transfusion to most trauma victims outside of hospitals,” said Allan Doctor, MD, of Washington University in Saint Louis, Missouri.
“ErythroMer would be a blood substitute that a medic can carry in his or her pack and literally take it out, add water, and inject it.”
Dr Doctor presented details on ErythroMer at the 2016 ASH Annual Meeting (abstract 1027).
Design
“Due to significant advances in synthetic chemistry and nanomedicine, we’re now able to encapsulate biologics with programmable polymers to generate nanoparticles that can emulate normal cellular physiology,” Dr Doctor noted.
With ErythroMer, he and his colleagues encapsulated human hemoglobin, methylene blue, and 2,3-DPG in an amphiphilic polymer shell. The polymer and its payload components, through microfluidization, self-assemble into toroids that are about one-fiftieth the size of human RBCs.
ErythroMer is designed to be pH-responsive, so that, in areas of high pH, 2,3-DPG is sequestered in the inner surface of the particle shell and does not bind to hemoglobin. In areas of low pH, 2,3-DPG is released from the shell and binds to hemoglobin, facilitating oxygen offloading. The role of methylene blue is to inhibit auto-oxidation of hemoglobin.
The last step in synthesis of the particle is crosslinking of the surface, which neutralizes the surface charge, stabilizes the particle, and generates a selective diffusion barrier to nitric oxide. The particle can be lyophilized for extended storage and later reconstituted.
Testing
Tests showed that ErythroMer matches the oxygen binding feature of human RBCs within 10%, a level researchers say should be sufficient to stabilize a bleeding patient until a blood transfusion can be obtained.
Experiments in mice showed that ErythroMer captures oxygen in the lungs and releases it to tissues in a pattern that is indistinguishable from that seen in a control group of mice injected with their own blood.
In rats, ErythroMer effectively resuscitated animals in shock following acute loss of 40% of their blood volume.
So far, tests suggest ErythroMer has overcome barriers that halted the development of previous blood substitutes.
However, Dr Doctor noted that ErythroMer does have its weaknesses. The particles are cleared rapidly from the bloodstream (in 3 to 7 hours), and hemoglobin sourcing presents a challenge. The researchers are now exploring the possibility of using recombinant hemoglobin genetically engineered in yeast.
The team hopes to further optimize ErythroMer’s shell, extend circulation time, confirm the efficacy of ErythroMer in a larger animal model (rabbits), evaluate the impact of the product on the coagulation and immune systems, and scale up production.
If further testing goes well, the researchers estimate that ErythroMer could be ready for use by field medics and emergency responders within 10 to 12 years.
ErythroMer development has been supported by the Children’s Discovery Institute at Washington University and St. Louis Children’s Hospital, the Skandalaris Center at Washington University, and the BioSTL Fundamentals Program.
This research was funded by the National Institute of General Medical Sciences; the National Heart, Lung, and Blood Institute; the National Institute of Child Health and Human Development, the US Department of Defense; the American Heart Association; Doris Duke Foundation; and Children’s Discovery Institute. ![]()
Celebrating 10 years of GI & Hepatology News
Our January 2017 issue marks the 10-year anniversary of GI & Hepatology News (GIHN), the official newspaper of the AGA Institute. In 2007, the AGA created the newspaper with the intent to communicate current news and emerging trends and technologies in GI. I am honored to serve as the third editor of GIHN, following in the esteemed footsteps of Charles J. Lightdale MD, AGAF, and Colin W. Howden MD, AGAF, who worked diligently to establish the publication’s credibility and quality.
The January 2007 issue of GIHN featured current AGA Institute President Timothy C. Wang, MD, AGAF, on its front page. At the time, he served as the chair of the AGA Future Trends Committee, which predicted emerging forces that would alter our practice, including that computed tomographic colonography would likely become an accepted CRC screening option in a few years (the full report of the committee was published in Gastroenterology 2008:134:597-616). For our 2017 10-year anniversary, we will feature a “Flashback” column, written by myself and our associate editors, that highlights and discusses the most impactful GIHN articles from each year of the previous decade.

John I. Allen, MD, MBA, AGAF
Editor in Chief
Our January 2017 issue marks the 10-year anniversary of GI & Hepatology News (GIHN), the official newspaper of the AGA Institute. In 2007, the AGA created the newspaper with the intent to communicate current news and emerging trends and technologies in GI. I am honored to serve as the third editor of GIHN, following in the esteemed footsteps of Charles J. Lightdale MD, AGAF, and Colin W. Howden MD, AGAF, who worked diligently to establish the publication’s credibility and quality.
The January 2007 issue of GIHN featured current AGA Institute President Timothy C. Wang, MD, AGAF, on its front page. At the time, he served as the chair of the AGA Future Trends Committee, which predicted emerging forces that would alter our practice, including that computed tomographic colonography would likely become an accepted CRC screening option in a few years (the full report of the committee was published in Gastroenterology 2008:134:597-616). For our 2017 10-year anniversary, we will feature a “Flashback” column, written by myself and our associate editors, that highlights and discusses the most impactful GIHN articles from each year of the previous decade.
John I. Allen, MD, MBA, AGAF
Editor in Chief
Our January 2017 issue marks the 10-year anniversary of GI & Hepatology News (GIHN), the official newspaper of the AGA Institute. In 2007, the AGA created the newspaper with the intent to communicate current news and emerging trends and technologies in GI. I am honored to serve as the third editor of GIHN, following in the esteemed footsteps of Charles J. Lightdale MD, AGAF, and Colin W. Howden MD, AGAF, who worked diligently to establish the publication’s credibility and quality.
The January 2007 issue of GIHN featured current AGA Institute President Timothy C. Wang, MD, AGAF, on its front page. At the time, he served as the chair of the AGA Future Trends Committee, which predicted emerging forces that would alter our practice, including that computed tomographic colonography would likely become an accepted CRC screening option in a few years (the full report of the committee was published in Gastroenterology 2008:134:597-616). For our 2017 10-year anniversary, we will feature a “Flashback” column, written by myself and our associate editors, that highlights and discusses the most impactful GIHN articles from each year of the previous decade.
John I. Allen, MD, MBA, AGAF
Editor in Chief

AGA Clinical Practice Update: Treatment for severe alcohol hepatitis challenging
Acute alcoholic hepatitis carries a high risk of mortality, yet only a minority of patients admitted to the hospital with the condition receive appropriate treatment, said the authors of an expert review.
Writing in the January 2017 issue of Clinical Gastroenterology and Hepatology, Mack C. Mitchell Jr., MD, of the University of Texas Southwestern Medical Center, Dallas, and Craig J. McClain, MD, of the University of Louisville (Ky.), described the challenges associated with treating acute alcoholic hepatitis and its consequences.
Acute alcohol hepatitis develops in heavy drinkers and presents with rapid onset of malaise, anorexia, tender hepatomegaly, and features of the systemic inflammatory response syndrome. Patients with alcoholic hepatitis also are at high risk of nutritional deficiency, infection, acute kidney injury, and multiorgan failure.
The two most widely used therapies are glucocorticoids – generally considered the standard of care for severe alcoholic hepatitis – and the phosphodiesterase inhibitor pentoxifylline (Clin Gastroenterol Hepatol. 2017. doi: 10.1016/j.cgh.2016.08.047).
“Although in its most severe form AH has a high short-term mortality rate if untreated, in 2011, only 28% of more than 1,600 patients admitted to U.S. hospitals were treated with glucocorticoids and 17% with pentoxifylline (PTX), suggesting a lack of widespread confidence in the two most frequently used therapies for AH,” the authors wrote.
Both drugs work by addressing the underlying inflammation that plays a key role in liver injury, but the evidence for both is mixed: A 2008 Cochrane systematic review of 15 trials concluded there was no benefit from glucocorticoids, largely because of substantial variability in bias across the trials, while two meta-analyses of pentoxifylline trials concluded that there were no differences in short-term mortality between those who received it and those who did not.
Some patients are unsuitable for glucocorticoids and others may develop resistance. There is also the possibility that, while glucocorticoids may improve short-term survival, the associated increase in infection risk removes that advantage at 90 days and 1 year after diagnosis. These infections, in turn, often precede the development of acute kidney injury and multiorgan failure.
The authors, however, did suggest that the approach of very high, short-term bursts of glucocorticoids to induce “immune paralysis” – an approach taken for lupus nephritis – might be considered.
They stressed that abstinence was the cornerstone of treatment for acute alcoholic hepatitis, with studies showing that patients with alcoholic hepatitis who resume heavy drinking have significantly worse outcomes than those who don’t.
“Although abstinence is important at all stages, it is particularly important to emphasize abstinence beyond 90 days when many patients are regaining normal functioning,” Dr. Mitchell and Dr. McClain wrote.
Infection, kidney injury, and malnutrition are all significant concerns in patients with acute alcoholic hepatitis.
With respect to infection, the authors said considerable suspicion is required to pick up bacterial and fungal infections, as patients may not always have a fever and an elevated white blood cell count is an unreliable indicator. Infection also can lead to acute kidney injury.
Malnutrition is not only common in patients with alcohol hepatitis, but it has a significant negative impact on recovery. All patients should be encouraged to meet nutritional goals as early as possible, but just how to achieve this is controversial, the authors stressed.
For example, one study suggested that enteral nutrition was as good as glucocorticoids in reducing 28-day mortality, while another found enteral nutrition via nasogastric tube – in addition to glucocorticoids – was no better than glucocorticoids alone. “Whether [nasogastric] tubes should be used to provide enteral nutrition is a subject of controversy,” the authors wrote. “Normal- to high-protein diets are safe and do not increase the risk of encephalopathy in patients with AH.”
No conflicts of interest were declared.
Acute alcoholic hepatitis carries a high risk of mortality, yet only a minority of patients admitted to the hospital with the condition receive appropriate treatment, said the authors of an expert review.
Writing in the January 2017 issue of Clinical Gastroenterology and Hepatology, Mack C. Mitchell Jr., MD, of the University of Texas Southwestern Medical Center, Dallas, and Craig J. McClain, MD, of the University of Louisville (Ky.), described the challenges associated with treating acute alcoholic hepatitis and its consequences.
Acute alcohol hepatitis develops in heavy drinkers and presents with rapid onset of malaise, anorexia, tender hepatomegaly, and features of the systemic inflammatory response syndrome. Patients with alcoholic hepatitis also are at high risk of nutritional deficiency, infection, acute kidney injury, and multiorgan failure.
The two most widely used therapies are glucocorticoids – generally considered the standard of care for severe alcoholic hepatitis – and the phosphodiesterase inhibitor pentoxifylline (Clin Gastroenterol Hepatol. 2017. doi: 10.1016/j.cgh.2016.08.047).
“Although in its most severe form AH has a high short-term mortality rate if untreated, in 2011, only 28% of more than 1,600 patients admitted to U.S. hospitals were treated with glucocorticoids and 17% with pentoxifylline (PTX), suggesting a lack of widespread confidence in the two most frequently used therapies for AH,” the authors wrote.
Both drugs work by addressing the underlying inflammation that plays a key role in liver injury, but the evidence for both is mixed: A 2008 Cochrane systematic review of 15 trials concluded there was no benefit from glucocorticoids, largely because of substantial variability in bias across the trials, while two meta-analyses of pentoxifylline trials concluded that there were no differences in short-term mortality between those who received it and those who did not.
Some patients are unsuitable for glucocorticoids and others may develop resistance. There is also the possibility that, while glucocorticoids may improve short-term survival, the associated increase in infection risk removes that advantage at 90 days and 1 year after diagnosis. These infections, in turn, often precede the development of acute kidney injury and multiorgan failure.
The authors, however, did suggest that the approach of very high, short-term bursts of glucocorticoids to induce “immune paralysis” – an approach taken for lupus nephritis – might be considered.
They stressed that abstinence was the cornerstone of treatment for acute alcoholic hepatitis, with studies showing that patients with alcoholic hepatitis who resume heavy drinking have significantly worse outcomes than those who don’t.
“Although abstinence is important at all stages, it is particularly important to emphasize abstinence beyond 90 days when many patients are regaining normal functioning,” Dr. Mitchell and Dr. McClain wrote.
Infection, kidney injury, and malnutrition are all significant concerns in patients with acute alcoholic hepatitis.
With respect to infection, the authors said considerable suspicion is required to pick up bacterial and fungal infections, as patients may not always have a fever and an elevated white blood cell count is an unreliable indicator. Infection also can lead to acute kidney injury.
Malnutrition is not only common in patients with alcohol hepatitis, but it has a significant negative impact on recovery. All patients should be encouraged to meet nutritional goals as early as possible, but just how to achieve this is controversial, the authors stressed.
For example, one study suggested that enteral nutrition was as good as glucocorticoids in reducing 28-day mortality, while another found enteral nutrition via nasogastric tube – in addition to glucocorticoids – was no better than glucocorticoids alone. “Whether [nasogastric] tubes should be used to provide enteral nutrition is a subject of controversy,” the authors wrote. “Normal- to high-protein diets are safe and do not increase the risk of encephalopathy in patients with AH.”
No conflicts of interest were declared.
Acute alcoholic hepatitis carries a high risk of mortality, yet only a minority of patients admitted to the hospital with the condition receive appropriate treatment, said the authors of an expert review.
Writing in the January 2017 issue of Clinical Gastroenterology and Hepatology, Mack C. Mitchell Jr., MD, of the University of Texas Southwestern Medical Center, Dallas, and Craig J. McClain, MD, of the University of Louisville (Ky.), described the challenges associated with treating acute alcoholic hepatitis and its consequences.
Acute alcohol hepatitis develops in heavy drinkers and presents with rapid onset of malaise, anorexia, tender hepatomegaly, and features of the systemic inflammatory response syndrome. Patients with alcoholic hepatitis also are at high risk of nutritional deficiency, infection, acute kidney injury, and multiorgan failure.
The two most widely used therapies are glucocorticoids – generally considered the standard of care for severe alcoholic hepatitis – and the phosphodiesterase inhibitor pentoxifylline (Clin Gastroenterol Hepatol. 2017. doi: 10.1016/j.cgh.2016.08.047).
“Although in its most severe form AH has a high short-term mortality rate if untreated, in 2011, only 28% of more than 1,600 patients admitted to U.S. hospitals were treated with glucocorticoids and 17% with pentoxifylline (PTX), suggesting a lack of widespread confidence in the two most frequently used therapies for AH,” the authors wrote.
Both drugs work by addressing the underlying inflammation that plays a key role in liver injury, but the evidence for both is mixed: A 2008 Cochrane systematic review of 15 trials concluded there was no benefit from glucocorticoids, largely because of substantial variability in bias across the trials, while two meta-analyses of pentoxifylline trials concluded that there were no differences in short-term mortality between those who received it and those who did not.
Some patients are unsuitable for glucocorticoids and others may develop resistance. There is also the possibility that, while glucocorticoids may improve short-term survival, the associated increase in infection risk removes that advantage at 90 days and 1 year after diagnosis. These infections, in turn, often precede the development of acute kidney injury and multiorgan failure.
The authors, however, did suggest that the approach of very high, short-term bursts of glucocorticoids to induce “immune paralysis” – an approach taken for lupus nephritis – might be considered.
They stressed that abstinence was the cornerstone of treatment for acute alcoholic hepatitis, with studies showing that patients with alcoholic hepatitis who resume heavy drinking have significantly worse outcomes than those who don’t.
“Although abstinence is important at all stages, it is particularly important to emphasize abstinence beyond 90 days when many patients are regaining normal functioning,” Dr. Mitchell and Dr. McClain wrote.
Infection, kidney injury, and malnutrition are all significant concerns in patients with acute alcoholic hepatitis.
With respect to infection, the authors said considerable suspicion is required to pick up bacterial and fungal infections, as patients may not always have a fever and an elevated white blood cell count is an unreliable indicator. Infection also can lead to acute kidney injury.
Malnutrition is not only common in patients with alcohol hepatitis, but it has a significant negative impact on recovery. All patients should be encouraged to meet nutritional goals as early as possible, but just how to achieve this is controversial, the authors stressed.
For example, one study suggested that enteral nutrition was as good as glucocorticoids in reducing 28-day mortality, while another found enteral nutrition via nasogastric tube – in addition to glucocorticoids – was no better than glucocorticoids alone. “Whether [nasogastric] tubes should be used to provide enteral nutrition is a subject of controversy,” the authors wrote. “Normal- to high-protein diets are safe and do not increase the risk of encephalopathy in patients with AH.”
No conflicts of interest were declared.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Treating depression after TBI

REIGNITE the desire: Tackle burnout in psychiatry
Burnout among psychiatric clinicians can lead to reduced job satisfaction, poorer quality of patient care, and depression.1 Signs of burnout include a feeling of cynicism (eg, negative attitudes toward patients), overwhelming exhaustion (eg, feeling depleted), and a sense of ineffectiveness (eg, reduced productivity).1 Workplace variables and other factors that could perpetuate burnout among psychiatrists include, but are not limited to:
- too much work
- chronic staff shortages
- working with difficult patients
- inability to meet self-imposed demands
- a lack of meaningful relationships with colleagues and supervisors.1,2
The mnemonic REIGNITE provides strategies to reduce the risk of burnout.1,3
Recognize your limits. Although saying “no” may be difficult for mental health clinicians, saying “yes” too often can be detrimental. Techniques for setting limits without alienating colleagues include:
- declining tasks (“I appreciate you thinking of me to do that, but I can’t complete it right now”)
- delaying an answer (“Let me ponder what you are asking”)
- delegating tasks (“I could really use your help”)
- avoid taking on too much (“I thought that I could do that extra task, but I realize that taking on the additional assignment isn’t going to work out”).
Expand your portfolio. Developing a diverse work portfolio (eg, teaching part-time) could diminish stagnation. Adding regenerative activities (eg, outdoor activities) could be restorative.
Itemize your priorities. Ask yourself what is important to you. Is it work? If so, can work be modified so it continues to be rewarding without resulting in burnout? If it isn’t work, then what is? Money? Family? Evaluating what is important and pursuing those priorities could increase overall life satisfaction.
Go after your passions. What do you like to do aside from work? Do you paint or play a musical instrument? Pursuing hobbies and interests can revitalize your spirit.
Now. We as a profession are notorious for saying to ourselves, “I will get to it (being happy) someday.” We delay happiness until we catch up with work, save enough money, and so on. This approach is unrealistic. It is better to live in the present because there are a finite number of days to seize the day. Focus your energy in the moment.
Interact. Isolating oneself will lead to burnout. If you are in solo practice, connect with other providers or get involved in community activities. If you work with other providers, interact with them in a meaningful manner (eg, don’t complain but rather air your concerns, accept honest feedback, be open to suggestions, and seek assistance; it is acceptable to admit that you can’t do everything).
Take time off and take care of yourself. Although that seems intuitive, psychiatrists, as a group, don’t do a good job of it. Waiting until you are burned out to take a vacation is counterproductive because you will be too drained to enjoy it. Taking care of your physical and mental health is equally important.
Enjoyment in and at work. We make a difference in our patients’ lives throughthe emotional connections we develop with them. By viewing what we do as fulfilling a higher calling, we can learn to enjoy what we do rather than feeling burdened by it. Advocating for better recognition—whether financial, institutional, or social—can create opportunities for personal satisfaction.
1. Maslach C, Leiter MP. Understanding the burnout experience: recent research and its implications for psychiatry. World Psychiatry. 2016;15(2):103-111.
2. Bressi C, Porcellana M, Gambini O, et al. Burnout among psychiatrists in Milan: a multicenter survey. Psychiatr Serv. 2009;60(7):985-988.
3. Bohnert P, O’Connell A. How to avoid burnout
Burnout among psychiatric clinicians can lead to reduced job satisfaction, poorer quality of patient care, and depression.1 Signs of burnout include a feeling of cynicism (eg, negative attitudes toward patients), overwhelming exhaustion (eg, feeling depleted), and a sense of ineffectiveness (eg, reduced productivity).1 Workplace variables and other factors that could perpetuate burnout among psychiatrists include, but are not limited to:
- too much work
- chronic staff shortages
- working with difficult patients
- inability to meet self-imposed demands
- a lack of meaningful relationships with colleagues and supervisors.1,2
The mnemonic REIGNITE provides strategies to reduce the risk of burnout.1,3
Recognize your limits. Although saying “no” may be difficult for mental health clinicians, saying “yes” too often can be detrimental. Techniques for setting limits without alienating colleagues include:
- declining tasks (“I appreciate you thinking of me to do that, but I can’t complete it right now”)
- delaying an answer (“Let me ponder what you are asking”)
- delegating tasks (“I could really use your help”)
- avoid taking on too much (“I thought that I could do that extra task, but I realize that taking on the additional assignment isn’t going to work out”).
Expand your portfolio. Developing a diverse work portfolio (eg, teaching part-time) could diminish stagnation. Adding regenerative activities (eg, outdoor activities) could be restorative.
Itemize your priorities. Ask yourself what is important to you. Is it work? If so, can work be modified so it continues to be rewarding without resulting in burnout? If it isn’t work, then what is? Money? Family? Evaluating what is important and pursuing those priorities could increase overall life satisfaction.
Go after your passions. What do you like to do aside from work? Do you paint or play a musical instrument? Pursuing hobbies and interests can revitalize your spirit.
Now. We as a profession are notorious for saying to ourselves, “I will get to it (being happy) someday.” We delay happiness until we catch up with work, save enough money, and so on. This approach is unrealistic. It is better to live in the present because there are a finite number of days to seize the day. Focus your energy in the moment.
Interact. Isolating oneself will lead to burnout. If you are in solo practice, connect with other providers or get involved in community activities. If you work with other providers, interact with them in a meaningful manner (eg, don’t complain but rather air your concerns, accept honest feedback, be open to suggestions, and seek assistance; it is acceptable to admit that you can’t do everything).
Take time off and take care of yourself. Although that seems intuitive, psychiatrists, as a group, don’t do a good job of it. Waiting until you are burned out to take a vacation is counterproductive because you will be too drained to enjoy it. Taking care of your physical and mental health is equally important.
Enjoyment in and at work. We make a difference in our patients’ lives throughthe emotional connections we develop with them. By viewing what we do as fulfilling a higher calling, we can learn to enjoy what we do rather than feeling burdened by it. Advocating for better recognition—whether financial, institutional, or social—can create opportunities for personal satisfaction.
Burnout among psychiatric clinicians can lead to reduced job satisfaction, poorer quality of patient care, and depression.1 Signs of burnout include a feeling of cynicism (eg, negative attitudes toward patients), overwhelming exhaustion (eg, feeling depleted), and a sense of ineffectiveness (eg, reduced productivity).1 Workplace variables and other factors that could perpetuate burnout among psychiatrists include, but are not limited to:
- too much work
- chronic staff shortages
- working with difficult patients
- inability to meet self-imposed demands
- a lack of meaningful relationships with colleagues and supervisors.1,2
The mnemonic REIGNITE provides strategies to reduce the risk of burnout.1,3
Recognize your limits. Although saying “no” may be difficult for mental health clinicians, saying “yes” too often can be detrimental. Techniques for setting limits without alienating colleagues include:
- declining tasks (“I appreciate you thinking of me to do that, but I can’t complete it right now”)
- delaying an answer (“Let me ponder what you are asking”)
- delegating tasks (“I could really use your help”)
- avoid taking on too much (“I thought that I could do that extra task, but I realize that taking on the additional assignment isn’t going to work out”).
Expand your portfolio. Developing a diverse work portfolio (eg, teaching part-time) could diminish stagnation. Adding regenerative activities (eg, outdoor activities) could be restorative.
Itemize your priorities. Ask yourself what is important to you. Is it work? If so, can work be modified so it continues to be rewarding without resulting in burnout? If it isn’t work, then what is? Money? Family? Evaluating what is important and pursuing those priorities could increase overall life satisfaction.
Go after your passions. What do you like to do aside from work? Do you paint or play a musical instrument? Pursuing hobbies and interests can revitalize your spirit.
Now. We as a profession are notorious for saying to ourselves, “I will get to it (being happy) someday.” We delay happiness until we catch up with work, save enough money, and so on. This approach is unrealistic. It is better to live in the present because there are a finite number of days to seize the day. Focus your energy in the moment.
Interact. Isolating oneself will lead to burnout. If you are in solo practice, connect with other providers or get involved in community activities. If you work with other providers, interact with them in a meaningful manner (eg, don’t complain but rather air your concerns, accept honest feedback, be open to suggestions, and seek assistance; it is acceptable to admit that you can’t do everything).
Take time off and take care of yourself. Although that seems intuitive, psychiatrists, as a group, don’t do a good job of it. Waiting until you are burned out to take a vacation is counterproductive because you will be too drained to enjoy it. Taking care of your physical and mental health is equally important.
Enjoyment in and at work. We make a difference in our patients’ lives throughthe emotional connections we develop with them. By viewing what we do as fulfilling a higher calling, we can learn to enjoy what we do rather than feeling burdened by it. Advocating for better recognition—whether financial, institutional, or social—can create opportunities for personal satisfaction.
1. Maslach C, Leiter MP. Understanding the burnout experience: recent research and its implications for psychiatry. World Psychiatry. 2016;15(2):103-111.
2. Bressi C, Porcellana M, Gambini O, et al. Burnout among psychiatrists in Milan: a multicenter survey. Psychiatr Serv. 2009;60(7):985-988.
3. Bohnert P, O’Connell A. How to avoid burnout
1. Maslach C, Leiter MP. Understanding the burnout experience: recent research and its implications for psychiatry. World Psychiatry. 2016;15(2):103-111.
2. Bressi C, Porcellana M, Gambini O, et al. Burnout among psychiatrists in Milan: a multicenter survey. Psychiatr Serv. 2009;60(7):985-988.
3. Bohnert P, O’Connell A. How to avoid burnout
Revisiting delirious mania; Correcting an error
Revisiting delirious mania
After treating a young woman with delirious mania, we were compelled to comment on the case report “Confused and nearly naked after going on spending sprees” (Cases That Test Your Skills,
A young woman with bipolar I disorder and mild intellectual disability was brought to our inpatient psychiatric unit after she disappeared from her home. Her family reported she was not compliant with her medications, and she recently showed deterioration marked by bizarre and violent behaviors for the previous month.
Although her presentation was consistent with earlier manic episodes, additional behaviors indicated an increase in severity. The patient was only oriented to name, was disrobing, had urinary and fecal incontinence, and showed purposeless hyperactivity such as continuously dancing in circles.
Because we thought she was experiencing a severe exacerbation of bipolar disorder, the patient was started on 4 different antipsychotic trials (typical and atypical) and 2 mood stabilizers, all of which did not produce adequate response. Even after augmentation with nightly long-acting benzodiazepines, the patient’s symptoms remained unchanged.
The patient received a diagnosis of delirious mania, with the underlying mechanism being severe catatonia. A literature search revealed electroconvulsive therapy (ECT) and benzodiazepines as first-line treatments, and discouraged use of typical antipsychotics because of an increased risk of neuroleptic malignant syndrome and malignant delirious mania.1 Because ECT was not available at our facility, we initiated benzodiazepines,
We agree it is prudent to rule out any medical illnesses that could cause delirium. Interestingly, in our patient a head CT revealed small calcifications suggestive of cysticercosis, which have been seen on imaging since age 13. We suggest that this finding contributed to her disinhibition, prolonged her recovery, and could explain why she did not respond adequately to medications.
Diagnosing and treating delirious mania in our patient was challenging. As mentioned by Davis et al, there is no classification of delirious mania in DSM-5. In addition, there are no large-scale studies to educate psychiatrists about the prevalence and appropriate treatment of this disorder.
Our treatment approach differed from that of Davis et al in that we chose scheduled benzodiazepines rather than antipsychotics to target the patient’s catatonia. However, both patients improved, prompting us to further question the mechanism behind this presentation.
We encourage the addition of delirious mania to the next edition of DSM. Without classification and establishment of this diagnosis, psychiatrists are unlikely to consider this serious and potentially fatal syndrome. Delirious mania is mysterious and rare and its inner workings are not fully elucidated.
Sabina Bera, MD MSc
PGY-2 Psychiatry Resident
Mohammed Molla, MD, DFAPA
Interim Joint Chair and Program Director
University of California Los Angeles-Kern
Psychiatry Training Program
Bakersfield, California
Reference
1. Jacobowski NL, Heckers S, Bobo WV. Delirious mania: detection, diagnosis, and clinical management in the acute setting. J Psychiatr Pract. 2013;19(1):15-28.
Correcting an error
In his informative guest editorial "
David A. Gorelick, MD, PhD
Professor of Psychiatry
Maryland Psychiatric Research Center
University of Maryland
Baltimore, Maryland
Revisiting delirious mania
After treating a young woman with delirious mania, we were compelled to comment on the case report “Confused and nearly naked after going on spending sprees” (Cases That Test Your Skills,
A young woman with bipolar I disorder and mild intellectual disability was brought to our inpatient psychiatric unit after she disappeared from her home. Her family reported she was not compliant with her medications, and she recently showed deterioration marked by bizarre and violent behaviors for the previous month.
Although her presentation was consistent with earlier manic episodes, additional behaviors indicated an increase in severity. The patient was only oriented to name, was disrobing, had urinary and fecal incontinence, and showed purposeless hyperactivity such as continuously dancing in circles.
Because we thought she was experiencing a severe exacerbation of bipolar disorder, the patient was started on 4 different antipsychotic trials (typical and atypical) and 2 mood stabilizers, all of which did not produce adequate response. Even after augmentation with nightly long-acting benzodiazepines, the patient’s symptoms remained unchanged.
The patient received a diagnosis of delirious mania, with the underlying mechanism being severe catatonia. A literature search revealed electroconvulsive therapy (ECT) and benzodiazepines as first-line treatments, and discouraged use of typical antipsychotics because of an increased risk of neuroleptic malignant syndrome and malignant delirious mania.1 Because ECT was not available at our facility, we initiated benzodiazepines,
We agree it is prudent to rule out any medical illnesses that could cause delirium. Interestingly, in our patient a head CT revealed small calcifications suggestive of cysticercosis, which have been seen on imaging since age 13. We suggest that this finding contributed to her disinhibition, prolonged her recovery, and could explain why she did not respond adequately to medications.
Diagnosing and treating delirious mania in our patient was challenging. As mentioned by Davis et al, there is no classification of delirious mania in DSM-5. In addition, there are no large-scale studies to educate psychiatrists about the prevalence and appropriate treatment of this disorder.
Our treatment approach differed from that of Davis et al in that we chose scheduled benzodiazepines rather than antipsychotics to target the patient’s catatonia. However, both patients improved, prompting us to further question the mechanism behind this presentation.
We encourage the addition of delirious mania to the next edition of DSM. Without classification and establishment of this diagnosis, psychiatrists are unlikely to consider this serious and potentially fatal syndrome. Delirious mania is mysterious and rare and its inner workings are not fully elucidated.
Sabina Bera, MD MSc
PGY-2 Psychiatry Resident
Mohammed Molla, MD, DFAPA
Interim Joint Chair and Program Director
University of California Los Angeles-Kern
Psychiatry Training Program
Bakersfield, California
Reference
1. Jacobowski NL, Heckers S, Bobo WV. Delirious mania: detection, diagnosis, and clinical management in the acute setting. J Psychiatr Pract. 2013;19(1):15-28.
Correcting an error
In his informative guest editorial "
David A. Gorelick, MD, PhD
Professor of Psychiatry
Maryland Psychiatric Research Center
University of Maryland
Baltimore, Maryland
Revisiting delirious mania
After treating a young woman with delirious mania, we were compelled to comment on the case report “Confused and nearly naked after going on spending sprees” (Cases That Test Your Skills,
A young woman with bipolar I disorder and mild intellectual disability was brought to our inpatient psychiatric unit after she disappeared from her home. Her family reported she was not compliant with her medications, and she recently showed deterioration marked by bizarre and violent behaviors for the previous month.
Although her presentation was consistent with earlier manic episodes, additional behaviors indicated an increase in severity. The patient was only oriented to name, was disrobing, had urinary and fecal incontinence, and showed purposeless hyperactivity such as continuously dancing in circles.
Because we thought she was experiencing a severe exacerbation of bipolar disorder, the patient was started on 4 different antipsychotic trials (typical and atypical) and 2 mood stabilizers, all of which did not produce adequate response. Even after augmentation with nightly long-acting benzodiazepines, the patient’s symptoms remained unchanged.
The patient received a diagnosis of delirious mania, with the underlying mechanism being severe catatonia. A literature search revealed electroconvulsive therapy (ECT) and benzodiazepines as first-line treatments, and discouraged use of typical antipsychotics because of an increased risk of neuroleptic malignant syndrome and malignant delirious mania.1 Because ECT was not available at our facility, we initiated benzodiazepines,
We agree it is prudent to rule out any medical illnesses that could cause delirium. Interestingly, in our patient a head CT revealed small calcifications suggestive of cysticercosis, which have been seen on imaging since age 13. We suggest that this finding contributed to her disinhibition, prolonged her recovery, and could explain why she did not respond adequately to medications.
Diagnosing and treating delirious mania in our patient was challenging. As mentioned by Davis et al, there is no classification of delirious mania in DSM-5. In addition, there are no large-scale studies to educate psychiatrists about the prevalence and appropriate treatment of this disorder.
Our treatment approach differed from that of Davis et al in that we chose scheduled benzodiazepines rather than antipsychotics to target the patient’s catatonia. However, both patients improved, prompting us to further question the mechanism behind this presentation.
We encourage the addition of delirious mania to the next edition of DSM. Without classification and establishment of this diagnosis, psychiatrists are unlikely to consider this serious and potentially fatal syndrome. Delirious mania is mysterious and rare and its inner workings are not fully elucidated.
Sabina Bera, MD MSc
PGY-2 Psychiatry Resident
Mohammed Molla, MD, DFAPA
Interim Joint Chair and Program Director
University of California Los Angeles-Kern
Psychiatry Training Program
Bakersfield, California
Reference
1. Jacobowski NL, Heckers S, Bobo WV. Delirious mania: detection, diagnosis, and clinical management in the acute setting. J Psychiatr Pract. 2013;19(1):15-28.
Correcting an error
In his informative guest editorial "
David A. Gorelick, MD, PhD
Professor of Psychiatry
Maryland Psychiatric Research Center
University of Maryland
Baltimore, Maryland

Worsening agitation and hallucinations: Could it be PTSD?
CASE Confusion, hallucinations
Mr. G, age 57, is brought to the emergency department (ED) from a hospice care facility for worsening agitation and psychosis over 2 days. His wife, who accompanies him, describes a 2-month onset of “confusion” with occasional visual hallucinations. She says that at baseline Mr. G was alert and oriented and able to engage appropriately in conversations. The hospice facility administered emergency medications, including unknown dosages of haloperidol and chlorpromazine, the morning before transfer to the ED.
Mr. G has a history of posttraumatic stress disorder (PTSD), anxiety, and depression that has been managed for 6 years with several trials of antidepressant monotherapy, including fluoxetine, citalopram, mirtazapine, bupropion, and augmentation using aripiprazole, risperidone, topiramate, and zolpidem. At the time of this hospital presentation, his symptoms are controlled on clonazepam, 2 mg/d, and trazodone, 50 mg/d. For his pain attributed to non-small cell lung cancer (NSCLC), he receives methadone, 25 mg, 6 times a day, and hydromorphone, 8 mg, every 4 hours as needed, for breakthrough pain. Mr. G underwent a right upper lobectomy 5 years ago and neurosurgery with a right suboccipital craniectomy for right-sided cerebellar metastatic tumor measuring 2 × 1 × 0.6 cm, along with chemotherapy and radiation for metastasis in the brain 1 year ago. His last chemotherapy session was 3 months ago.
In the ED, Mr. G is sedated and oriented only to person and his wife. He is observed mumbling incoherently. Abnormal vital signs and laboratory findings are elevated pulse, 97 beats per minute; mild anemia, 13.5 g/dL hemoglobin and 40.8% hematocrit; an elevated glucose of 136 mg/dL; and small amounts of blood, trace ketones, and hyaline casts in urinalysis. Vital signs, laboratory resu
In addition to psychotropic and pain medication, Mr. G is taking cyclobenzaprine, 5 mg, every 6 hours as needed, for muscle spasms; docusate, 200 mg/d; enoxaparin, 100 mg/1mL, every 12 hours; folic acid, 1 mg/d; gabapentin, 600 mg, 3 times daily; lidocaine ointment, twice daily as needed, for pain; omeprazole, 80 mg/d; ondansetron, 4 mg, every 8 hours as needed, for nausea; and tamsulosin, 0.4 mg/d.
What is your differential diagnosis for Mr. G?
a) brain metastases
b) infection
c) PTSD
d) polypharmacy
e) benzodiazepine withdrawal
The authors’ observations
Altered mental status (AMS), or acute confusional state, describes an individual who fails to interact with environmental stimuli in an appropriate, anticipated manner. The disturbance usually is acute and transient.1 Often providers struggle to obtain relevant facts about a patient’s history of illness and must use laboratory and diagnostic data to determine the underlying cause of the patient’s disorientation.
Mental status includes 2 components: arousal and awareness. Arousal refers to a person’s wakeful state and how an individual responds to his (her) surroundings. Impairment in arousal can result in variable states including lethargy, drowsiness, and even coma. Awareness, on the other hand, is an individual’s perception of his environment, including orientation to surroundings, executive functioning, and memory. Although arousal level is controlled by the reticular activating system of the brainstem, awareness of consciousness is mediated at the cortical level. Mr. G experienced increased arousal and AMS with a clear change in behavior from his baseline. With increasing frequency of hallucinations and agitated behaviors, several tests must be ordered to determine the etiology of his altered mentation (Table 1).
Which test would you order next?
a) urine drug screen (UDS)
b) chest CT with pulmonary embolism protocol
c) CT of the head
d) blood cultures
e) chest radiography
EVALUATION Awake, still confused
The ED physician orders a UDS, non-contrasted CT of the head, and chest radiography for preliminary workup investigating the cause of Mr. G’s AMS. UDS is negative for illicit substances. The non-contrasted CT of the head shows a stable, right cerebellar hemisphere lesion from a prior lung metastasis. Mr. G’s chest radiography reading describes an ill-defined opacity at the left lung base.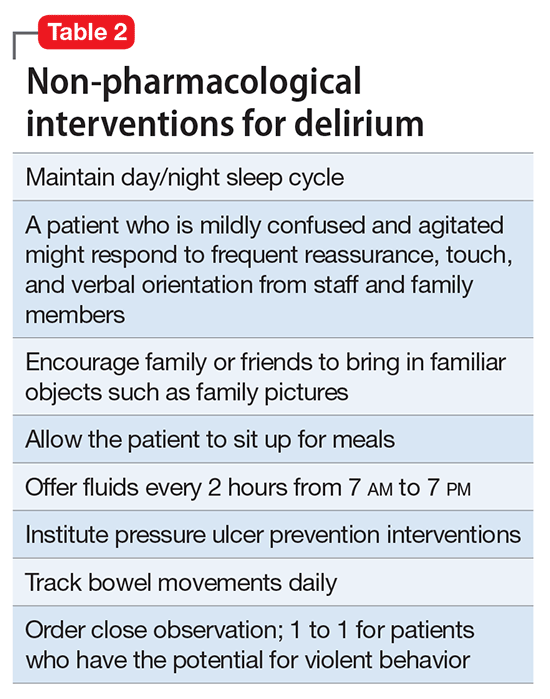
Mr. G is admitted to the medical service and is started on dexamethasone, 8 mg/d, for his NSCLC with brain metastasis. Clonazepam is continued to prevent benzodiazepine withdrawal. The psychiatry and palliative care teams are consulted to determine if Mr. G’s PTSD symptoms and/or opioids are contributing to his AMS and psychosis. After evaluation, the psychiatry team recommends decreasing clonazepam to 0.5 mg, twice daily, starting olanzapine, 5 mg, every 12 hours, for agitation and psychosis involving auditory and visual hallucinations as well as paranoid themes related to food contamination, and using non-pharmacologic interventions for delirium treatment (Table 2). In a prospective, randomized controlled trial of olanzapine vs haloperidol, clinical improvement in delirious states was seen in individuals who received either antipsychotic medication; however, haloperidol was associated with extrapyramidal side effects. Therefore, olanzapine is a safe alternative to haloperidol in delirious patients.2
The psychiatry consult service suspects delirium due to polypharmacy or Mr. G’s metastatic brain lesion. However, other collaborating treatment teams feel that Mr. G’s presentation was precipitated by an exacerbation of PTSD symptoms because of the observed psychotic themes, in addition to metabolic encephalopathy. Acute stress disorder can present with emotional numbing, depersonalization, reduced awareness of surroundings, or dissociative amnesia. However, Mr. G has not experienced PTSD symptoms involving mental status changes with fluctuating orientation in the past nor has he displayed persistent dissociation during outpatient psychiatric care. Therefore, it is unlikely that PTSD is the primary cause of his hospital admission.
The palliative care team recommends switching Mr. G’s pain medications to methadone, 20 mg, every 6 hours, to reduce possibility that opioids are contributing to his delirious state. Mr. G’s medical providers report that the chest radiography is suspicious for pneumonia and start him on levofloxacin, 500 mg/d.
The authors’ observations
DSM-5 criteria for delirium has 4 components:
- disturbance in attention and awareness
- change in cognition
- the disturbance develops over a short period of time
- there is evidence that the disturbance is a direct consequence of a medical condition, medication, or substance, or more than 1 cause.3
Mr. G presented with multi-factorial delirium, and as a result, all underlying contributions, including infection, polypharmacy, brain metastasis, and steroids needed to be considered. Treating delirium requires investigating the underlying cause and keeping the patient safe in the process (Figure). Mr. G was agitated at presentation; therefore, low-dosage olanzapine was initiated to address the imbalance between the cholinergic and dopaminergic systems in the CNS, which are thought to be the mechanism behind delirious presentations.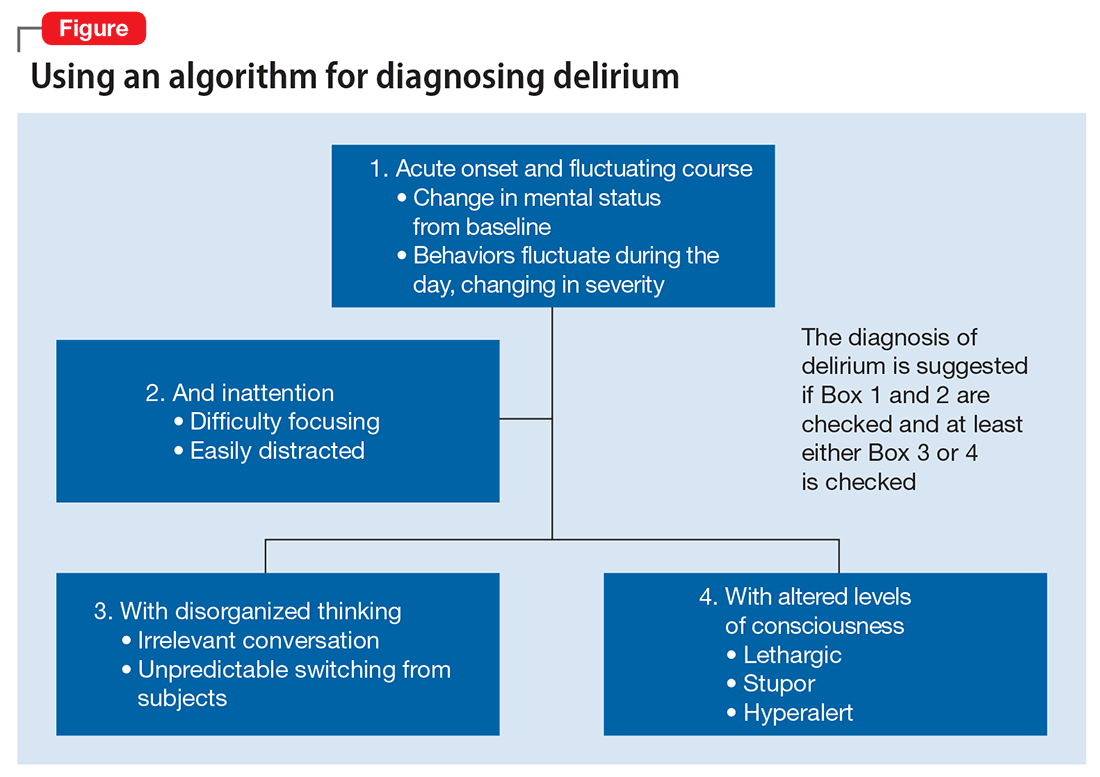
In Mr. G’s case, methadone was lowered, with continual monitoring and evaluation for his comfort. Infections, specifically urinary tract infections and pneumonia, can cause delirium states and must be treated with appropriate antibiotics. Metastatic tumors have been known to precipitate changes in mental status and can be ruled out via imaging. In Mr. G’s case, his metastatic lesion remained stable from prior radiographic studies.
TREATMENT Delirium resolves
Mr. G slowly responds to multi-modal treatment including decreased opioids and benzodiazepines and the use of low-dosage antipsychotics. He begins to return to baseline with antibiotic administration. By hospital day 5, Mr. G is alert and oriented. He notes resolution of his auditory and visual hallucinations and denies any persistent paranoia or delusions. The medical team observes Mr. G is having difficulty swallowing with meals, and orders a speech therapy evaluation. After assessment, the team suspects that aspiration pneumonia could have precipitated Mr. G’s initial decline and recommends a mechanic diet with thin liquids to reduce the risk of future aspiration.
Mr. G is discharged home in his wife’s care with home hospice to continue end-of-life care. His medication regimen includes olanzapine, 10 mg/d, to continue until his next outpatient appointment, trazodone, 50 mg/d, for depression and PTSD symptoms, and clonazepam is decreased to 0.5 mg, at bedtime, for anxiety.
The authors’ observations
Mr. G’s case highlights the importance of fully evaluating all common underlying causes of delirium. The etiology of delirium is more likely to be missed in medically complex patients or in patients with a history of psychiatric illness. Palliative care patients have several risk factors for delirium, such as benzodiazepine or opioid treatment, dementia, and organic diseases such as brain metastasis.6 A recent study assessed the frequency of delirium in cancer patients admitted to an inpatient palliative unit and found that 71% of individuals had a diagnosis of delirium at admission and 26% developed delirium afterward.7 Despite the increased likelihood of developing delirium, more than one-half of palliative patients have delirium that is missed by their primary providers.8 Similarly, patients with documented psychiatric illness were approximately 2.5 times more likely to have overlooked delirium compared with patients without psychiatric illness.9
Risk and prevention
Patients with risk factors for delirium—which includes sedative and narcotic usage, advanced cancer, older age, prolonged hospital stays, surgical procedures, and/or cognitive impairment—should receive interventions to prevent delirium. However, if symptoms of AMS are present, providers should perform a complete workup for underlying causes of delirium. Remembering that individuals with delirium have an impaired ability to voice symptoms, such as dyspnea, dysuria, and headache, clinicians should have a high index of suspicion for delirium in patients at heightened risk.10
Perhaps most important, teams treating patients at high risk for delirium should employ preventive measures to reduce the development of delirium. Although more studies are needed to clarify the role of drug therapies for preventing delirium, there is strong evidence for several non-pharmacotherapeutic interventions including:
- frequent orientation activities
- early mobilization
- maintaining healthy sleep–wake cycles
- minimizing the use of psychoactive drugs and frequently reviewing the medication regimen
- allowing use of eyeglasses and hearing aids
- treating volume depletion.10
These preventive measures are important when treating delirium, such as minimizing Mr. G’s use of benzodiazepine and opioids—medications known to contribute to iatrogenic delirium.
A delirium diagnosis portends grave adverse outcomes. Research has shown significant associations with morbidity and mortality, financial and emotional burden, and prolonged hospitalizations. Often, symptoms of delirium persist for months and patients do not recover completely. However, studies have found that when underlying causes are treated effectively, delirium is more likely to be reversible.11
The prompt diagnosis of delirium with good interdisciplinary communication can reduce the risk of these adverse outcomes.12 Consultation-liaison psychiatrists are well positioned to facilitate the diagnoses of delirium and play a role in educating other health care providers of the importance of prevention, early symptom recognition, full workup, and effective treatment of its underlying causes.
1. Posner JB, Saper CB, Schiff ND, et al. Plum and Posner’s diagnosis of stupor and coma. New York, NY: Oxford University Press; 2007.
2. Skrobik YK, Bergeron N, Dumont M, et al. Olanzapine vs haldoperidol: treating delirium in a critical care setting. Intensive Care Med. 2004;30(3):444-449.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Lonergan E, Luxenberg J, Areosa Sastre A, et al. Benzodiazepines for delirium. Cochrane Database Syst Rev. 2009;(1):CD006379. doi: 10.1002/14651858.CD006379.pub2.
5. Vella-Brincat J, Macleod AD. Adverse effects of opioids on the central nervous system of palliative care patients. J Pain Palliat Care Pharmacother. 2007;21(1):15-25.
6. Grassi L, Caraceni A, Mitchell AJ, et al. Management of delirium in palliative care: a review. Curr Psychiatry Rep. 2015;17(3):550.
7. de la Cruz M, Ransing V, Yennu S, et al. The frequency, characteristics, and outcomes among cancer patients with delirium admitted to an acute palliative care unit. Oncologist. 2015;20(12):1425-1431.
8. de la Cruz, M, Fan J, Yennu S, et al. The frequency of missed delirium in patients referred to palliative care in a comprehensive cancer center. Support Care Cancer. 2015;23(8):2427-2433.
9. Swigart SE, Kishi Y, Thurber S, et al. Misdiagnosed delirium in patient referrals to a university-based hospital psychiatry department. Psychosomatics. 2008;49(2):104-108.
10. Inouye SK, Bogardus ST Jr, Charpentier PA, et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med. 1999;340(9):669-676.
11. Dasgupta M, Hillier LM. Factors associated with prolonged delirium: a systematic review. Int Psychogeriatr. 2010;22(3):373-394.
12. Detweiler MB, Kenneth A, Bader G, et al. Can improved intra- and inter-team communication reduce missed delirium? Psychiatr Q. 2014;85(2):211-224.
CASE Confusion, hallucinations
Mr. G, age 57, is brought to the emergency department (ED) from a hospice care facility for worsening agitation and psychosis over 2 days. His wife, who accompanies him, describes a 2-month onset of “confusion” with occasional visual hallucinations. She says that at baseline Mr. G was alert and oriented and able to engage appropriately in conversations. The hospice facility administered emergency medications, including unknown dosages of haloperidol and chlorpromazine, the morning before transfer to the ED.
Mr. G has a history of posttraumatic stress disorder (PTSD), anxiety, and depression that has been managed for 6 years with several trials of antidepressant monotherapy, including fluoxetine, citalopram, mirtazapine, bupropion, and augmentation using aripiprazole, risperidone, topiramate, and zolpidem. At the time of this hospital presentation, his symptoms are controlled on clonazepam, 2 mg/d, and trazodone, 50 mg/d. For his pain attributed to non-small cell lung cancer (NSCLC), he receives methadone, 25 mg, 6 times a day, and hydromorphone, 8 mg, every 4 hours as needed, for breakthrough pain. Mr. G underwent a right upper lobectomy 5 years ago and neurosurgery with a right suboccipital craniectomy for right-sided cerebellar metastatic tumor measuring 2 × 1 × 0.6 cm, along with chemotherapy and radiation for metastasis in the brain 1 year ago. His last chemotherapy session was 3 months ago.
In the ED, Mr. G is sedated and oriented only to person and his wife. He is observed mumbling incoherently. Abnormal vital signs and laboratory findings are elevated pulse, 97 beats per minute; mild anemia, 13.5 g/dL hemoglobin and 40.8% hematocrit; an elevated glucose of 136 mg/dL; and small amounts of blood, trace ketones, and hyaline casts in urinalysis. Vital signs, laboratory resu
In addition to psychotropic and pain medication, Mr. G is taking cyclobenzaprine, 5 mg, every 6 hours as needed, for muscle spasms; docusate, 200 mg/d; enoxaparin, 100 mg/1mL, every 12 hours; folic acid, 1 mg/d; gabapentin, 600 mg, 3 times daily; lidocaine ointment, twice daily as needed, for pain; omeprazole, 80 mg/d; ondansetron, 4 mg, every 8 hours as needed, for nausea; and tamsulosin, 0.4 mg/d.
What is your differential diagnosis for Mr. G?
a) brain metastases
b) infection
c) PTSD
d) polypharmacy
e) benzodiazepine withdrawal
The authors’ observations
Altered mental status (AMS), or acute confusional state, describes an individual who fails to interact with environmental stimuli in an appropriate, anticipated manner. The disturbance usually is acute and transient.1 Often providers struggle to obtain relevant facts about a patient’s history of illness and must use laboratory and diagnostic data to determine the underlying cause of the patient’s disorientation.
Mental status includes 2 components: arousal and awareness. Arousal refers to a person’s wakeful state and how an individual responds to his (her) surroundings. Impairment in arousal can result in variable states including lethargy, drowsiness, and even coma. Awareness, on the other hand, is an individual’s perception of his environment, including orientation to surroundings, executive functioning, and memory. Although arousal level is controlled by the reticular activating system of the brainstem, awareness of consciousness is mediated at the cortical level. Mr. G experienced increased arousal and AMS with a clear change in behavior from his baseline. With increasing frequency of hallucinations and agitated behaviors, several tests must be ordered to determine the etiology of his altered mentation (Table 1).
Which test would you order next?
a) urine drug screen (UDS)
b) chest CT with pulmonary embolism protocol
c) CT of the head
d) blood cultures
e) chest radiography
EVALUATION Awake, still confused
The ED physician orders a UDS, non-contrasted CT of the head, and chest radiography for preliminary workup investigating the cause of Mr. G’s AMS. UDS is negative for illicit substances. The non-contrasted CT of the head shows a stable, right cerebellar hemisphere lesion from a prior lung metastasis. Mr. G’s chest radiography reading describes an ill-defined opacity at the left lung base.
Mr. G is admitted to the medical service and is started on dexamethasone, 8 mg/d, for his NSCLC with brain metastasis. Clonazepam is continued to prevent benzodiazepine withdrawal. The psychiatry and palliative care teams are consulted to determine if Mr. G’s PTSD symptoms and/or opioids are contributing to his AMS and psychosis. After evaluation, the psychiatry team recommends decreasing clonazepam to 0.5 mg, twice daily, starting olanzapine, 5 mg, every 12 hours, for agitation and psychosis involving auditory and visual hallucinations as well as paranoid themes related to food contamination, and using non-pharmacologic interventions for delirium treatment (Table 2). In a prospective, randomized controlled trial of olanzapine vs haloperidol, clinical improvement in delirious states was seen in individuals who received either antipsychotic medication; however, haloperidol was associated with extrapyramidal side effects. Therefore, olanzapine is a safe alternative to haloperidol in delirious patients.2
The psychiatry consult service suspects delirium due to polypharmacy or Mr. G’s metastatic brain lesion. However, other collaborating treatment teams feel that Mr. G’s presentation was precipitated by an exacerbation of PTSD symptoms because of the observed psychotic themes, in addition to metabolic encephalopathy. Acute stress disorder can present with emotional numbing, depersonalization, reduced awareness of surroundings, or dissociative amnesia. However, Mr. G has not experienced PTSD symptoms involving mental status changes with fluctuating orientation in the past nor has he displayed persistent dissociation during outpatient psychiatric care. Therefore, it is unlikely that PTSD is the primary cause of his hospital admission.
The palliative care team recommends switching Mr. G’s pain medications to methadone, 20 mg, every 6 hours, to reduce possibility that opioids are contributing to his delirious state. Mr. G’s medical providers report that the chest radiography is suspicious for pneumonia and start him on levofloxacin, 500 mg/d.
The authors’ observations
DSM-5 criteria for delirium has 4 components:
- disturbance in attention and awareness
- change in cognition
- the disturbance develops over a short period of time
- there is evidence that the disturbance is a direct consequence of a medical condition, medication, or substance, or more than 1 cause.3
Mr. G presented with multi-factorial delirium, and as a result, all underlying contributions, including infection, polypharmacy, brain metastasis, and steroids needed to be considered. Treating delirium requires investigating the underlying cause and keeping the patient safe in the process (Figure). Mr. G was agitated at presentation; therefore, low-dosage olanzapine was initiated to address the imbalance between the cholinergic and dopaminergic systems in the CNS, which are thought to be the mechanism behind delirious presentations.
In Mr. G’s case, methadone was lowered, with continual monitoring and evaluation for his comfort. Infections, specifically urinary tract infections and pneumonia, can cause delirium states and must be treated with appropriate antibiotics. Metastatic tumors have been known to precipitate changes in mental status and can be ruled out via imaging. In Mr. G’s case, his metastatic lesion remained stable from prior radiographic studies.
TREATMENT Delirium resolves
Mr. G slowly responds to multi-modal treatment including decreased opioids and benzodiazepines and the use of low-dosage antipsychotics. He begins to return to baseline with antibiotic administration. By hospital day 5, Mr. G is alert and oriented. He notes resolution of his auditory and visual hallucinations and denies any persistent paranoia or delusions. The medical team observes Mr. G is having difficulty swallowing with meals, and orders a speech therapy evaluation. After assessment, the team suspects that aspiration pneumonia could have precipitated Mr. G’s initial decline and recommends a mechanic diet with thin liquids to reduce the risk of future aspiration.
Mr. G is discharged home in his wife’s care with home hospice to continue end-of-life care. His medication regimen includes olanzapine, 10 mg/d, to continue until his next outpatient appointment, trazodone, 50 mg/d, for depression and PTSD symptoms, and clonazepam is decreased to 0.5 mg, at bedtime, for anxiety.
The authors’ observations
Mr. G’s case highlights the importance of fully evaluating all common underlying causes of delirium. The etiology of delirium is more likely to be missed in medically complex patients or in patients with a history of psychiatric illness. Palliative care patients have several risk factors for delirium, such as benzodiazepine or opioid treatment, dementia, and organic diseases such as brain metastasis.6 A recent study assessed the frequency of delirium in cancer patients admitted to an inpatient palliative unit and found that 71% of individuals had a diagnosis of delirium at admission and 26% developed delirium afterward.7 Despite the increased likelihood of developing delirium, more than one-half of palliative patients have delirium that is missed by their primary providers.8 Similarly, patients with documented psychiatric illness were approximately 2.5 times more likely to have overlooked delirium compared with patients without psychiatric illness.9
Risk and prevention
Patients with risk factors for delirium—which includes sedative and narcotic usage, advanced cancer, older age, prolonged hospital stays, surgical procedures, and/or cognitive impairment—should receive interventions to prevent delirium. However, if symptoms of AMS are present, providers should perform a complete workup for underlying causes of delirium. Remembering that individuals with delirium have an impaired ability to voice symptoms, such as dyspnea, dysuria, and headache, clinicians should have a high index of suspicion for delirium in patients at heightened risk.10
Perhaps most important, teams treating patients at high risk for delirium should employ preventive measures to reduce the development of delirium. Although more studies are needed to clarify the role of drug therapies for preventing delirium, there is strong evidence for several non-pharmacotherapeutic interventions including:
- frequent orientation activities
- early mobilization
- maintaining healthy sleep–wake cycles
- minimizing the use of psychoactive drugs and frequently reviewing the medication regimen
- allowing use of eyeglasses and hearing aids
- treating volume depletion.10
These preventive measures are important when treating delirium, such as minimizing Mr. G’s use of benzodiazepine and opioids—medications known to contribute to iatrogenic delirium.
A delirium diagnosis portends grave adverse outcomes. Research has shown significant associations with morbidity and mortality, financial and emotional burden, and prolonged hospitalizations. Often, symptoms of delirium persist for months and patients do not recover completely. However, studies have found that when underlying causes are treated effectively, delirium is more likely to be reversible.11
The prompt diagnosis of delirium with good interdisciplinary communication can reduce the risk of these adverse outcomes.12 Consultation-liaison psychiatrists are well positioned to facilitate the diagnoses of delirium and play a role in educating other health care providers of the importance of prevention, early symptom recognition, full workup, and effective treatment of its underlying causes.
CASE Confusion, hallucinations
Mr. G, age 57, is brought to the emergency department (ED) from a hospice care facility for worsening agitation and psychosis over 2 days. His wife, who accompanies him, describes a 2-month onset of “confusion” with occasional visual hallucinations. She says that at baseline Mr. G was alert and oriented and able to engage appropriately in conversations. The hospice facility administered emergency medications, including unknown dosages of haloperidol and chlorpromazine, the morning before transfer to the ED.
Mr. G has a history of posttraumatic stress disorder (PTSD), anxiety, and depression that has been managed for 6 years with several trials of antidepressant monotherapy, including fluoxetine, citalopram, mirtazapine, bupropion, and augmentation using aripiprazole, risperidone, topiramate, and zolpidem. At the time of this hospital presentation, his symptoms are controlled on clonazepam, 2 mg/d, and trazodone, 50 mg/d. For his pain attributed to non-small cell lung cancer (NSCLC), he receives methadone, 25 mg, 6 times a day, and hydromorphone, 8 mg, every 4 hours as needed, for breakthrough pain. Mr. G underwent a right upper lobectomy 5 years ago and neurosurgery with a right suboccipital craniectomy for right-sided cerebellar metastatic tumor measuring 2 × 1 × 0.6 cm, along with chemotherapy and radiation for metastasis in the brain 1 year ago. His last chemotherapy session was 3 months ago.
In the ED, Mr. G is sedated and oriented only to person and his wife. He is observed mumbling incoherently. Abnormal vital signs and laboratory findings are elevated pulse, 97 beats per minute; mild anemia, 13.5 g/dL hemoglobin and 40.8% hematocrit; an elevated glucose of 136 mg/dL; and small amounts of blood, trace ketones, and hyaline casts in urinalysis. Vital signs, laboratory resu
In addition to psychotropic and pain medication, Mr. G is taking cyclobenzaprine, 5 mg, every 6 hours as needed, for muscle spasms; docusate, 200 mg/d; enoxaparin, 100 mg/1mL, every 12 hours; folic acid, 1 mg/d; gabapentin, 600 mg, 3 times daily; lidocaine ointment, twice daily as needed, for pain; omeprazole, 80 mg/d; ondansetron, 4 mg, every 8 hours as needed, for nausea; and tamsulosin, 0.4 mg/d.
What is your differential diagnosis for Mr. G?
a) brain metastases
b) infection
c) PTSD
d) polypharmacy
e) benzodiazepine withdrawal
The authors’ observations
Altered mental status (AMS), or acute confusional state, describes an individual who fails to interact with environmental stimuli in an appropriate, anticipated manner. The disturbance usually is acute and transient.1 Often providers struggle to obtain relevant facts about a patient’s history of illness and must use laboratory and diagnostic data to determine the underlying cause of the patient’s disorientation.
Mental status includes 2 components: arousal and awareness. Arousal refers to a person’s wakeful state and how an individual responds to his (her) surroundings. Impairment in arousal can result in variable states including lethargy, drowsiness, and even coma. Awareness, on the other hand, is an individual’s perception of his environment, including orientation to surroundings, executive functioning, and memory. Although arousal level is controlled by the reticular activating system of the brainstem, awareness of consciousness is mediated at the cortical level. Mr. G experienced increased arousal and AMS with a clear change in behavior from his baseline. With increasing frequency of hallucinations and agitated behaviors, several tests must be ordered to determine the etiology of his altered mentation (Table 1).
Which test would you order next?
a) urine drug screen (UDS)
b) chest CT with pulmonary embolism protocol
c) CT of the head
d) blood cultures
e) chest radiography
EVALUATION Awake, still confused
The ED physician orders a UDS, non-contrasted CT of the head, and chest radiography for preliminary workup investigating the cause of Mr. G’s AMS. UDS is negative for illicit substances. The non-contrasted CT of the head shows a stable, right cerebellar hemisphere lesion from a prior lung metastasis. Mr. G’s chest radiography reading describes an ill-defined opacity at the left lung base.
Mr. G is admitted to the medical service and is started on dexamethasone, 8 mg/d, for his NSCLC with brain metastasis. Clonazepam is continued to prevent benzodiazepine withdrawal. The psychiatry and palliative care teams are consulted to determine if Mr. G’s PTSD symptoms and/or opioids are contributing to his AMS and psychosis. After evaluation, the psychiatry team recommends decreasing clonazepam to 0.5 mg, twice daily, starting olanzapine, 5 mg, every 12 hours, for agitation and psychosis involving auditory and visual hallucinations as well as paranoid themes related to food contamination, and using non-pharmacologic interventions for delirium treatment (Table 2). In a prospective, randomized controlled trial of olanzapine vs haloperidol, clinical improvement in delirious states was seen in individuals who received either antipsychotic medication; however, haloperidol was associated with extrapyramidal side effects. Therefore, olanzapine is a safe alternative to haloperidol in delirious patients.2
The psychiatry consult service suspects delirium due to polypharmacy or Mr. G’s metastatic brain lesion. However, other collaborating treatment teams feel that Mr. G’s presentation was precipitated by an exacerbation of PTSD symptoms because of the observed psychotic themes, in addition to metabolic encephalopathy. Acute stress disorder can present with emotional numbing, depersonalization, reduced awareness of surroundings, or dissociative amnesia. However, Mr. G has not experienced PTSD symptoms involving mental status changes with fluctuating orientation in the past nor has he displayed persistent dissociation during outpatient psychiatric care. Therefore, it is unlikely that PTSD is the primary cause of his hospital admission.
The palliative care team recommends switching Mr. G’s pain medications to methadone, 20 mg, every 6 hours, to reduce possibility that opioids are contributing to his delirious state. Mr. G’s medical providers report that the chest radiography is suspicious for pneumonia and start him on levofloxacin, 500 mg/d.
The authors’ observations
DSM-5 criteria for delirium has 4 components:
- disturbance in attention and awareness
- change in cognition
- the disturbance develops over a short period of time
- there is evidence that the disturbance is a direct consequence of a medical condition, medication, or substance, or more than 1 cause.3
Mr. G presented with multi-factorial delirium, and as a result, all underlying contributions, including infection, polypharmacy, brain metastasis, and steroids needed to be considered. Treating delirium requires investigating the underlying cause and keeping the patient safe in the process (Figure). Mr. G was agitated at presentation; therefore, low-dosage olanzapine was initiated to address the imbalance between the cholinergic and dopaminergic systems in the CNS, which are thought to be the mechanism behind delirious presentations.
In Mr. G’s case, methadone was lowered, with continual monitoring and evaluation for his comfort. Infections, specifically urinary tract infections and pneumonia, can cause delirium states and must be treated with appropriate antibiotics. Metastatic tumors have been known to precipitate changes in mental status and can be ruled out via imaging. In Mr. G’s case, his metastatic lesion remained stable from prior radiographic studies.
TREATMENT Delirium resolves
Mr. G slowly responds to multi-modal treatment including decreased opioids and benzodiazepines and the use of low-dosage antipsychotics. He begins to return to baseline with antibiotic administration. By hospital day 5, Mr. G is alert and oriented. He notes resolution of his auditory and visual hallucinations and denies any persistent paranoia or delusions. The medical team observes Mr. G is having difficulty swallowing with meals, and orders a speech therapy evaluation. After assessment, the team suspects that aspiration pneumonia could have precipitated Mr. G’s initial decline and recommends a mechanic diet with thin liquids to reduce the risk of future aspiration.
Mr. G is discharged home in his wife’s care with home hospice to continue end-of-life care. His medication regimen includes olanzapine, 10 mg/d, to continue until his next outpatient appointment, trazodone, 50 mg/d, for depression and PTSD symptoms, and clonazepam is decreased to 0.5 mg, at bedtime, for anxiety.
The authors’ observations
Mr. G’s case highlights the importance of fully evaluating all common underlying causes of delirium. The etiology of delirium is more likely to be missed in medically complex patients or in patients with a history of psychiatric illness. Palliative care patients have several risk factors for delirium, such as benzodiazepine or opioid treatment, dementia, and organic diseases such as brain metastasis.6 A recent study assessed the frequency of delirium in cancer patients admitted to an inpatient palliative unit and found that 71% of individuals had a diagnosis of delirium at admission and 26% developed delirium afterward.7 Despite the increased likelihood of developing delirium, more than one-half of palliative patients have delirium that is missed by their primary providers.8 Similarly, patients with documented psychiatric illness were approximately 2.5 times more likely to have overlooked delirium compared with patients without psychiatric illness.9
Risk and prevention
Patients with risk factors for delirium—which includes sedative and narcotic usage, advanced cancer, older age, prolonged hospital stays, surgical procedures, and/or cognitive impairment—should receive interventions to prevent delirium. However, if symptoms of AMS are present, providers should perform a complete workup for underlying causes of delirium. Remembering that individuals with delirium have an impaired ability to voice symptoms, such as dyspnea, dysuria, and headache, clinicians should have a high index of suspicion for delirium in patients at heightened risk.10
Perhaps most important, teams treating patients at high risk for delirium should employ preventive measures to reduce the development of delirium. Although more studies are needed to clarify the role of drug therapies for preventing delirium, there is strong evidence for several non-pharmacotherapeutic interventions including:
- frequent orientation activities
- early mobilization
- maintaining healthy sleep–wake cycles
- minimizing the use of psychoactive drugs and frequently reviewing the medication regimen
- allowing use of eyeglasses and hearing aids
- treating volume depletion.10
These preventive measures are important when treating delirium, such as minimizing Mr. G’s use of benzodiazepine and opioids—medications known to contribute to iatrogenic delirium.
A delirium diagnosis portends grave adverse outcomes. Research has shown significant associations with morbidity and mortality, financial and emotional burden, and prolonged hospitalizations. Often, symptoms of delirium persist for months and patients do not recover completely. However, studies have found that when underlying causes are treated effectively, delirium is more likely to be reversible.11
The prompt diagnosis of delirium with good interdisciplinary communication can reduce the risk of these adverse outcomes.12 Consultation-liaison psychiatrists are well positioned to facilitate the diagnoses of delirium and play a role in educating other health care providers of the importance of prevention, early symptom recognition, full workup, and effective treatment of its underlying causes.
1. Posner JB, Saper CB, Schiff ND, et al. Plum and Posner’s diagnosis of stupor and coma. New York, NY: Oxford University Press; 2007.
2. Skrobik YK, Bergeron N, Dumont M, et al. Olanzapine vs haldoperidol: treating delirium in a critical care setting. Intensive Care Med. 2004;30(3):444-449.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Lonergan E, Luxenberg J, Areosa Sastre A, et al. Benzodiazepines for delirium. Cochrane Database Syst Rev. 2009;(1):CD006379. doi: 10.1002/14651858.CD006379.pub2.
5. Vella-Brincat J, Macleod AD. Adverse effects of opioids on the central nervous system of palliative care patients. J Pain Palliat Care Pharmacother. 2007;21(1):15-25.
6. Grassi L, Caraceni A, Mitchell AJ, et al. Management of delirium in palliative care: a review. Curr Psychiatry Rep. 2015;17(3):550.
7. de la Cruz M, Ransing V, Yennu S, et al. The frequency, characteristics, and outcomes among cancer patients with delirium admitted to an acute palliative care unit. Oncologist. 2015;20(12):1425-1431.
8. de la Cruz, M, Fan J, Yennu S, et al. The frequency of missed delirium in patients referred to palliative care in a comprehensive cancer center. Support Care Cancer. 2015;23(8):2427-2433.
9. Swigart SE, Kishi Y, Thurber S, et al. Misdiagnosed delirium in patient referrals to a university-based hospital psychiatry department. Psychosomatics. 2008;49(2):104-108.
10. Inouye SK, Bogardus ST Jr, Charpentier PA, et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med. 1999;340(9):669-676.
11. Dasgupta M, Hillier LM. Factors associated with prolonged delirium: a systematic review. Int Psychogeriatr. 2010;22(3):373-394.
12. Detweiler MB, Kenneth A, Bader G, et al. Can improved intra- and inter-team communication reduce missed delirium? Psychiatr Q. 2014;85(2):211-224.
1. Posner JB, Saper CB, Schiff ND, et al. Plum and Posner’s diagnosis of stupor and coma. New York, NY: Oxford University Press; 2007.
2. Skrobik YK, Bergeron N, Dumont M, et al. Olanzapine vs haldoperidol: treating delirium in a critical care setting. Intensive Care Med. 2004;30(3):444-449.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Lonergan E, Luxenberg J, Areosa Sastre A, et al. Benzodiazepines for delirium. Cochrane Database Syst Rev. 2009;(1):CD006379. doi: 10.1002/14651858.CD006379.pub2.
5. Vella-Brincat J, Macleod AD. Adverse effects of opioids on the central nervous system of palliative care patients. J Pain Palliat Care Pharmacother. 2007;21(1):15-25.
6. Grassi L, Caraceni A, Mitchell AJ, et al. Management of delirium in palliative care: a review. Curr Psychiatry Rep. 2015;17(3):550.
7. de la Cruz M, Ransing V, Yennu S, et al. The frequency, characteristics, and outcomes among cancer patients with delirium admitted to an acute palliative care unit. Oncologist. 2015;20(12):1425-1431.
8. de la Cruz, M, Fan J, Yennu S, et al. The frequency of missed delirium in patients referred to palliative care in a comprehensive cancer center. Support Care Cancer. 2015;23(8):2427-2433.
9. Swigart SE, Kishi Y, Thurber S, et al. Misdiagnosed delirium in patient referrals to a university-based hospital psychiatry department. Psychosomatics. 2008;49(2):104-108.
10. Inouye SK, Bogardus ST Jr, Charpentier PA, et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med. 1999;340(9):669-676.
11. Dasgupta M, Hillier LM. Factors associated with prolonged delirium: a systematic review. Int Psychogeriatr. 2010;22(3):373-394.
12. Detweiler MB, Kenneth A, Bader G, et al. Can improved intra- and inter-team communication reduce missed delirium? Psychiatr Q. 2014;85(2):211-224.
Alcohol-use disorders after bariatric surgery: The case for targeted group therapy
Maladaptive alcohol use has emerged as a risk for a subset of individuals who have undergone weight loss surgery (WLS); studies report they are vulnerable to consuming alcohol in greater quantities or more frequently.1,2 Estimates of the prevalence of “high-risk” or “hazardous” alcohol use after WLS range from 4% to 28%,3,4 while the prevalence of alcohol use meeting DSM-IV-TR5 criteria for alcohol use disorders (AUDs) hovers around 10%.6
Heavy alcohol users or patients who have active AUD at the time of WLS are at greater risk for continuation of these problems after surgery.2,6 For patients with a long-remitted history of AUD, the evidence regarding risk for post-WLS relapse is lacking, and some evidence suggests they may have better weight loss outcomes after WLS.7
However, approximately two-third of cases of post-WLS alcohol problems occur in patients who have had no history of such problems before surgery.5,8,9 Reported prevalence rates of new-onset alcohol problems range from 3% to 18%,6,9 with the modal finding being approximately 7% to 8%. New-onset alcohol problems appear to occur at a considerable latency after surgery. One study found little risk at 1 year post-surgery, but a significant increase in AUD symptoms at 2 years.6 Another study identified 3 years post-surgery as a high-risk time point,8 and yet another reported a linear increase in the risk for developing alcohol problems for at least 10 years after WLS.10
This article describes a group treatment protocol developed specifically for patients with post-WLS substance use disorder (SUD), and explores:
- risk factors and causal mechanisms of post-WLS AUDs
- weight stigma and emotional stressors
- the role of specialized treatment
- group treatment based on the Health at Every Size® (HAES)-oriented, trauma-informed and fat acceptance framework.
Post-WLS patients with alcohol problems may be a distinct phenotype among people with substance abuse issues. For this reason, they may have a need to address their experiences and issues specific to WLS as part of their alcohol treatment.
Etiology
Risk factors. Empirical findings have identified few predictors or risk factors for post-WLS SUD. These patients are more likely to be male and of a younger age.6 Notably, the vast majority of individuals reporting post-WLS alcohol problems have undergone Roux-en-Y gastric bypass (RYGB), rather than other WLS procedures, such as the laparoscopic adjustable gastric band,6,11 suggesting some physiological mechanism specific to RYGB.
Other potential predictors of postoperative alcohol problems include a pre-operative history of depression, generalized anxiety disorder, smoking, and/or recreational drug use.3,6 Likewise, patients with depression or anxiety disorder symptoms after surgery also may be at higher risk for postoperative alcohol problems.4 The evidence of an association between postoperative weight outcomes and post-WLS alcohol problems is mixed.3,12 Interestingly, patients who had no personal history of substance abuse but who have a family history may have a higher risk of new-onset alcohol problems after surgery.9,12
Causal mechanisms. The etiology of post-WLS alcohol problems is not well understood. If anything, epidemiological data suggest that larger-bodied individuals tend to consume lower levels of alcohol and have lower rates of AUD than individuals in the general population with thinner bodies.13 However, an association has been found between a family history of SUD, but not a personal history, and being large.14 This suggests a shared etiological pathway between addiction and being “overweight,” of which the onset of AUD after RYGB may be a manifestation.
Human and animal studies have shown that WLS may affect alcohol use differently in specific subgroups. Studies have shown that wild-type rats greatly increase their consumption of, or operant responding for, alcohol after RYGB,15 while genetically “alcohol-preferring” rats decrease consumption of, or responding for, alcohol after RYGB.16 A human study likewise found some patients decreased alcohol use or experienced improvement of or remission of AUD symptoms after WLS.4 Combined with the finding that a family history of substance abuse is related to risk for post-operative AUD, these data suggest a potential genetic vulnerability or protection in some individuals.
Turning to potential psychosocial explanations, the lay media has popularized the concepts of “addiction transfer,” or “transfer addiction,”12 with the implication that some patients, who had a preoperative history of “food addiction,” transfer that “addiction” after surgery to substances of abuse.
However, the “addiction transfer” model has a number of flaws:
- it is stigmatizing, because it assumes the patient possesses an innate, chronic, and inalterable pathology
- it relies upon the validity of the controversial construct of “food addiction,” a construct of mixed scientific evidence.17
Further, our knowledge of post-WLS SUD argues against “addiction transfer.” As noted, postoperative alcohol problems are more likely to develop years after surgery, rather than in the first few months afterward when eating is most significantly curtailed. Additionally, post-WLS alcohol problems are significantly more likely to occur after RYGB than other procedures, whereas the “addiction transfer” model would hypothesize that all WLS patients would be at equal risk for postoperative “addiction transfer,” because their eating is similarly affected after surgery.
Links to RYGB. Some clues to physiological mechanisms underlying alcohol problems after RYGB have been identified. After surgery, many RYGB patients report a quicker effect from a smaller amount of alcohol than was the case pre-surgery.18 Studies have demonstrated a number of changes in the pharmacodynamics of alcohol after RYGB not seen in other WLS procedures19:
- a much faster time to peak blood (or breath) alcohol content (BAC)
- significantly higher peak BAC
- a precipitous initial decline in perceived intoxication.18,20
Anatomical features of RYGB may explain such changes.8 However, an increased response to both IV alcohol and IV morphine after RYGB21,22 in rodents suggests that gastrointestinal tract changes are not solely responsible for changes in alcohol use. Emerging research reports that WLS has been found to cause alterations in brain reward pathways,23 which may be an additional contributor to changes in alcohol misuse after surgery.
However, even combined, pharmacokinetic and neurobiological factors cannot entirely explain new-onset alcohol problems after WLS; if they could, one would expect to see a much higher prevalence of this complication. Some psychosocial factors are likely involved as well.
Emotional stressors. One possibility involves a mismatch between post-WLS stressors and coping skills. After WLS, these patients face a multitude of challenges inherent in adjusting to changes in lifestyle, weight, body image, and social functioning, which most individuals would find daunting. These challenges become even more acute in the absence of appropriate psychoeducation, preparation, and intervention from qualified professionals. Individuals who lack effective and adaptive coping skills and supports may have a particularly heightened vulnerability to increased alcohol use in the setting of post-surgery changes in brain reward circuits and pharmacodynamics in alcohol metabolism. For example, one patient reported that her spouse’s pressure to “do something about her weight” was a significant factor in her decision to undergo surgery, but that her spouse was blaming and unsupportive when post-WLS complications developed. The patient believed that these experiences helped fuel development of her post-RYGB alcohol abuse.
Specialized treatment
The number of patients experiencing post-WLS alcohol problems likely will continue to grow, given that the risk of onset of has been shown increase over years. Already, post-WLS patients are proportionally overrepresented among substance abuse treatment populations.24 Empirically, however, we do not know yet if these patients need a different type of addiction treatment than patients who have not had WLS.
Some evidence suggests that post-WLS patients with alcohol problems may be a distinct phenotype within the general population with alcohol problems, as their presentations differ in several ways, including their demographics, alcohol use patterns, and premorbid functioning. A number of studies have found that, despite their increased pharmacodynamic sensitivity to alcohol, people with post-WLS AUDs actually consume a larger amount of alcohol on both typical and maximum drinking days than other individuals with AUDs.24 Additionally, although the median age of onset for AUD is around age 20,25 patients presenting with new-onset, post-WLS alcohol problems are usually in their late 30s, or even 40s or 50s. Further, many of these patients were quite high functioning before their alcohol problems, and are unlikely to identify with the cultural stereotype of a person with AUD (eg, homeless, unemployed), which may hamper or delay their own willingness to accept that they have a problem. These phenotypic differences suggest that post-WLS patients may require substance abuse treatment approaches tailored to their unique presentation. There are additional factors specific to the experiences of being larger-bodied and WLS that also may need to be addressed in specialized treatment for post-WLS addiction patients.
Weight stigma. By definition, patients who have undergone WLS have spent a significant portion of their lives inhabiting larger bodies, an experience that, in our culture, can produce adverse psychosocial effects. Compared with the general population, patients seeking WLS exhibit psychological distress equivalent to psychiatric patients.26 Weight stigma or weight bias—negative judgments directed toward people in larger bodies—is pervasive and continues to increase.27 Further, evidence suggests that, unlike almost all other stigmatized groups, people in larger bodies tend to internalize this stigma, holding an unfavorable attitude toward their own social group.28 Weight stigma impacts the well-being of people all along the weight spectrum, affecting many domains including educational achievements and classroom experiences, job opportunities, salaries, and medical care.27 Weight stigma increases the likelihood of bullying, teasing, and harassment for both adults and children.27 Weight bias has been associated with any number of adverse psychosocial effects, including symptoms of depression, anxiety, and eating pathology; poor body image; and a decrease in healthy self-care behaviors.29-33
Weight stigma makes it more difficult for people to enjoy physical activities, nourish their bodies, and manage stress, which contributes to poorer health outcomes and lower quality of life.33,34 For example, one study showed that, regardless of actual body mass index, people experiencing weight stigma have significantly increased risk of developing an illness or dying.35
Factors specific to WLS. WLS may lead to significant changes in eating habits, and some patients experience a sense of loss, particularly if eating represented one of their primary coping strategies—this may represent a heightened emotional vulnerability for developing AUD.
The fairly rapid and substantial weight loss that WLS produces can lead to sweeping changes in lifestyle, body image, and functional factors for many individuals. Patients often report profound changes, both positive and negative, in their relationships and interactions not only with people in their support network, but also with strangers.36
After the first year or 2 post-WLS, it is fairly common for patients to regain some weight, sometimes in significant amounts.37 This can lead to a sense of “failure.” Life stressors, including difficulties in important relationships, can further add to patients’ vulnerability. For example, one patient noticed that when she was at her thinnest after WLS, drivers were more likely to stop for her when she crossed the street, which pleased but also angered her because they hadn’t extended the same courtesy before WLS. After she regained a significant amount of weight, she began to notice drivers stopping for her less and less frequently. This took her back to her previous feelings of being ignored but now with the certainty that she would be treated better if she were thinner.
Patients also may experience ambivalence about changes in their body size. One might expect that body image would improve after weight loss, but the evidence is mixed.38 Although there is some evidence that body image improves in the short term after WLS,38 other research indicates that body image does not improve with weight loss.39 However, the evidence is clear that the appearance of excess skin after weight loss worsens some patients’ body image.40
To date, there has been no research examining treatment modalities for this population. Because experiences common to individuals who have had WLS could play a role in the development of AUD after surgery, it is intuitive that it would be important to address these factors when designing a treatment plan for post-WLS substance abuse.
Group treatment approach
In 2013, in response to the increase in rates of post-WLS addictions presenting to West End Clinic, an outpatient dual-diagnosis (addiction and psychiatry) service at Massachusetts General Hospital, a specialized treatment group was developed. Nine patients have enrolled since October 2013.
The Post-WLS Addictions Group (PWAG) was designed to be HAES-oriented, trauma-informed, and run within a fat acceptance framework. The HAES model prioritizes a weight-neutral approach that sees health and well-being as multifaceted. This approach directs both patient and clinician to focus on improving health behaviors and reducing internalized weight bias, while building a supportive community that buffers against external cultural weight bias.41
Trauma-informed care42 emphasizes the principles of safety, trustworthiness, and transparency; peer support; collaboration and mutuality; empowerment; and awareness of cultural, historical, and gender issues. In the context of PWAG, weight stigma is conceptualized as a traumatic experience.43 The fat acceptance approach promotes a culture that accepts people of every size with dignity and equality in all aspects of life.44
Self-care emphasis. The HAES model encourages patients to allow their bodies to determine what weight to settle at, and to focus on sustainable health-enhancing behaviors rather than weight loss. Patients who asked about the PWAG were told that this group would not explicitly support, or even encourage, continued pursuit of weight loss per se, but instead would assist patients with relapse prevention, mindful eating, improving self-care, and ongoing stress management. Moving away from a focus on weight loss and toward improvement of self-care skills allowed patients to focus on behaviors and outcomes over which they had more direct control and were more likely to yield immediate benefits.
All of the PWAG group members were in early recovery from an SUD, with a minimum of 4 weeks of abstinence; all had at least 1 co-occurring mental health diagnosis. A licensed independent clinical social worker (LICSW) and a physician familiar with bariatric surgery ran the sessions. The group met weekly for 1 hour. The 8 weekly sessions included both psychoeducation and discussion, with each session covering different topics (Table). The first 20 minutes of each session were devoted to an educational presentation; the remaining 40 minutes for reflection and discussion. In sessions 2 through 8, participants were asked about any recent use or cravings, and problem-solving techniques were employed as needed.
The PWAG group leader herself is a large person who modeled fat acceptance and follows the HAES approach; she led the group using both this experience and her specialized clinical training. As is the case with other addictions recovery treatment modalities, clinicians with lived experience may add a valuable component to both the program design and patient experience.
After the first 8 sessions, all members expressed interest in continuing as an ongoing relapse prevention and HAES support group, and they reported that meeting regularly was very helpful. The group continued with the LICSW alone, who continued to share HAES-oriented and fat acceptance information and resources that group members requested specifically. Over time, new members joined following an individual orientation session with the group leader, and the group has revisited each of the psychoeducational topics repeatedly, though not in a formally structured way.
Process and observations. Participants described high levels of excitement and hopefulness about being in a group with other WLS patients who had developed SUDs. They had a particular interest in reviewing medical/anatomical information about WLS and understanding more about the potential reasons for the elevated risk for developing SUD following WLS. Discussions regarding weight stigma proved to be quite emotional; most participants reported that this material readily related to their own experiences with weight stigma, but they had never discussed these ideas before.
Participants explored the role that grief, loss, guilt, and shame had in the decision to have WLS, the development of SUDs, weight regain or medical complications from the surgery or from substance abuse, career and relationship changes, and worsened body image. Another theme that emerged was the various reasons that prompted the members have WLS that they may not have been conscious of, or willing to discuss with others, such as pressure from a spouse, fears of remaining single due to their size, and a desire to finally “fit in.”
Repeatedly, group members expressed how satisfied and emotionally validated they felt being with people with similar experiences. Most of them had felt alone. They reported a belief that “everyone else” who had WLS was doing well, and that they were the exceptions. Such beliefs and emotions increased the risk of relapse and decreased participants’ ability to develop more positive coping strategies and self-care skills.
Participants reported that feeling less alone, understanding how stigma impacts health and well-being, and focusing on the general benefits of good self-care rather than the pursuit of weight loss were particularly helpful. The HAES and fat acceptance approaches have given group members new ways to think about their bodies and decreased shame. Several group members reported that if they had learned about the HAES approach prior to having a WLS, they might have made a different decision about having surgery, or at least might have been better prepared to handle the emotional and psychological challenges after WLS.
Although evidence for post-WLS addictions is fairly robust, causal mechanisms are not well understood, and research identifying specific risk factors is lacking. Because post-WLS patients with addictions seem to represent a specific phenotype, specialized treatment might be indicated. Future research will be needed to determine optimal treatment approaches for post-WLS addictions. However, a number of aspects are likely to be important. For example, it is likely that unaddressed experiences of weight stigma contribute to challenges, including substance abuse, after WLS; therefore, clinicians involved in the care of individuals presenting with post-WLS SUD should be knowledgeable about weight stigma and how to address it. Because of the specific nature of post-WLS addictions, patients often feel alone and isolated, and seem to benefit from the specialized group setting. We note that the PWAG group leader is herself a large person who models fat acceptance and follows the HAES approach, and therefore led the group using this experience and her specialized clinical training. As with other addiction recovery treatment modalities, clinicians who have lived the experience can add a valuable component to the program design and patient experience.
2. Lent MR, Hayes SM, Wood GC, et al. Smoking and alcohol use in gastric bypass patients. Eat Behav. 2013;14(4):460-463.
4. Wee CC, Mukamal KJ, Huskey KW, et al. High-risk alcohol use after weight loss surgery. Surg Obes Relat Dis. 2014;10(3):508-513.
5. Diagnostic and statistical manual of mental disorders, 4th, text rev. Washington, DC: American Psychiatric Association; 2000.
6. King WC, Chen JY, Mitchell JE, et al. Prevalence of alcohol use disorders before and after bariatric surgery. JAMA. 2012;307(23):2516-2525.
9. Ivezaj V, Saules KK, Schuh LM. New-onset substance use disorder after gastric bypass surgery: rates and associated characteristics. Obes Surg. 2014;24(11):1975-1980.
10. Svensson PA, Anveden Å, Romeo S, et al. Alcohol consumption and alcohol problems after bariatric surgery in the Swedish obese subjects study. Obesity. 2013;21(12):2444-2451.
11. Ostlund MP, Backman O, Marsk R, et al. Increased admission for alcohol dependence after gastric bypass surgery compared with restrictive bariatric surgery. JAMA Surg. 2013;148(4):374-377.
13. Gearhardt AN, Corbin WR. Body mass index and alcohol consumption: family history of alcoholism as a moderator. Psychol Addict Behav. 2009;23(2):216-225.
14. Grucza RA, Krueger RF, Racette SB, et al. The emerging link between alcoholism risk and obesity in the United States. Arch Gen Psychiatry. 2010;67(12):1301-1308.
15. Davis JF, Tracy AL, Schurdak JD, et al. Roux en y gastric bypass increases ethanol intake in the rat. Obes Surg. 2013;23(7):920-930.
16. Davis JF, Schurdak JD, Magrisso IJ, et al. Gastric bypass surgery attenuates ethanol consumption in ethanol-preferring rats. Biol Psychiatry. 2012;72(5):354-360.
18. Pepino MY, Okunade AL, Eagon JC, et al. Effect of Roux-en-Y gastric bypass surgery: converting 2 alcoholic drinks to 4. JAMA Surg. 2015
19. Changchien EM, Woodard GA, Hernandez-Boussard T, et al. Normal alcohol metabolism after gastric banding and sleeve gastrectomy: a case-cross-over trial. J Am Coll Surg. 2012;215(4):475-479.
22. Polston JE, Pritchett CE, Tomasko JM, et al. Roux-en-Y gastric bypass increases intravenous ethanol self-administration in dietary obese rats. PLoS ONE. 2013;8(12):e83741. doi: 10.1371/journal.pone.0083741.
26. Higgs ML, Wade T, Cescato M, et al. Differences between treatment seekers in an obese population: medical intervention vs. dietary restriction. J Behav Med. 1997;20(4):391-405.
35. Sutin AR, Stephan Y, Terracciano A. Weight discrimination and risk of mortality. Psychol Sci. 2015;26(11):1803-1811.
36. Sogg S, Gorman MJ. Interpersonal changes and challenges after weight loss surgery. Prim Psychiatry. 2008;15(8):61-66.
37. Yanos BR, Saules KK, Schuh LM, et al. Predictors of lowest weight and long-term weight regain among Roux-en-Y gastric bypass patients. Obes Surg. 2015;25(8):1364-1370.
40. van der Beek E, Te Riele W, Specken TF, et al. The impact of reconstructive procedures following bariatric surgery on patient well-being and quality of life. Obes Surg. 2010;20(1):36-41.
Maladaptive alcohol use has emerged as a risk for a subset of individuals who have undergone weight loss surgery (WLS); studies report they are vulnerable to consuming alcohol in greater quantities or more frequently.1,2 Estimates of the prevalence of “high-risk” or “hazardous” alcohol use after WLS range from 4% to 28%,3,4 while the prevalence of alcohol use meeting DSM-IV-TR5 criteria for alcohol use disorders (AUDs) hovers around 10%.6
Heavy alcohol users or patients who have active AUD at the time of WLS are at greater risk for continuation of these problems after surgery.2,6 For patients with a long-remitted history of AUD, the evidence regarding risk for post-WLS relapse is lacking, and some evidence suggests they may have better weight loss outcomes after WLS.7
However, approximately two-third of cases of post-WLS alcohol problems occur in patients who have had no history of such problems before surgery.5,8,9 Reported prevalence rates of new-onset alcohol problems range from 3% to 18%,6,9 with the modal finding being approximately 7% to 8%. New-onset alcohol problems appear to occur at a considerable latency after surgery. One study found little risk at 1 year post-surgery, but a significant increase in AUD symptoms at 2 years.6 Another study identified 3 years post-surgery as a high-risk time point,8 and yet another reported a linear increase in the risk for developing alcohol problems for at least 10 years after WLS.10
This article describes a group treatment protocol developed specifically for patients with post-WLS substance use disorder (SUD), and explores:
- risk factors and causal mechanisms of post-WLS AUDs
- weight stigma and emotional stressors
- the role of specialized treatment
- group treatment based on the Health at Every Size® (HAES)-oriented, trauma-informed and fat acceptance framework.
Post-WLS patients with alcohol problems may be a distinct phenotype among people with substance abuse issues. For this reason, they may have a need to address their experiences and issues specific to WLS as part of their alcohol treatment.
Etiology
Risk factors. Empirical findings have identified few predictors or risk factors for post-WLS SUD. These patients are more likely to be male and of a younger age.6 Notably, the vast majority of individuals reporting post-WLS alcohol problems have undergone Roux-en-Y gastric bypass (RYGB), rather than other WLS procedures, such as the laparoscopic adjustable gastric band,6,11 suggesting some physiological mechanism specific to RYGB.
Other potential predictors of postoperative alcohol problems include a pre-operative history of depression, generalized anxiety disorder, smoking, and/or recreational drug use.3,6 Likewise, patients with depression or anxiety disorder symptoms after surgery also may be at higher risk for postoperative alcohol problems.4 The evidence of an association between postoperative weight outcomes and post-WLS alcohol problems is mixed.3,12 Interestingly, patients who had no personal history of substance abuse but who have a family history may have a higher risk of new-onset alcohol problems after surgery.9,12
Causal mechanisms. The etiology of post-WLS alcohol problems is not well understood. If anything, epidemiological data suggest that larger-bodied individuals tend to consume lower levels of alcohol and have lower rates of AUD than individuals in the general population with thinner bodies.13 However, an association has been found between a family history of SUD, but not a personal history, and being large.14 This suggests a shared etiological pathway between addiction and being “overweight,” of which the onset of AUD after RYGB may be a manifestation.
Human and animal studies have shown that WLS may affect alcohol use differently in specific subgroups. Studies have shown that wild-type rats greatly increase their consumption of, or operant responding for, alcohol after RYGB,15 while genetically “alcohol-preferring” rats decrease consumption of, or responding for, alcohol after RYGB.16 A human study likewise found some patients decreased alcohol use or experienced improvement of or remission of AUD symptoms after WLS.4 Combined with the finding that a family history of substance abuse is related to risk for post-operative AUD, these data suggest a potential genetic vulnerability or protection in some individuals.
Turning to potential psychosocial explanations, the lay media has popularized the concepts of “addiction transfer,” or “transfer addiction,”12 with the implication that some patients, who had a preoperative history of “food addiction,” transfer that “addiction” after surgery to substances of abuse.
However, the “addiction transfer” model has a number of flaws:
- it is stigmatizing, because it assumes the patient possesses an innate, chronic, and inalterable pathology
- it relies upon the validity of the controversial construct of “food addiction,” a construct of mixed scientific evidence.17
Further, our knowledge of post-WLS SUD argues against “addiction transfer.” As noted, postoperative alcohol problems are more likely to develop years after surgery, rather than in the first few months afterward when eating is most significantly curtailed. Additionally, post-WLS alcohol problems are significantly more likely to occur after RYGB than other procedures, whereas the “addiction transfer” model would hypothesize that all WLS patients would be at equal risk for postoperative “addiction transfer,” because their eating is similarly affected after surgery.
Links to RYGB. Some clues to physiological mechanisms underlying alcohol problems after RYGB have been identified. After surgery, many RYGB patients report a quicker effect from a smaller amount of alcohol than was the case pre-surgery.18 Studies have demonstrated a number of changes in the pharmacodynamics of alcohol after RYGB not seen in other WLS procedures19:
- a much faster time to peak blood (or breath) alcohol content (BAC)
- significantly higher peak BAC
- a precipitous initial decline in perceived intoxication.18,20
Anatomical features of RYGB may explain such changes.8 However, an increased response to both IV alcohol and IV morphine after RYGB21,22 in rodents suggests that gastrointestinal tract changes are not solely responsible for changes in alcohol use. Emerging research reports that WLS has been found to cause alterations in brain reward pathways,23 which may be an additional contributor to changes in alcohol misuse after surgery.
However, even combined, pharmacokinetic and neurobiological factors cannot entirely explain new-onset alcohol problems after WLS; if they could, one would expect to see a much higher prevalence of this complication. Some psychosocial factors are likely involved as well.
Emotional stressors. One possibility involves a mismatch between post-WLS stressors and coping skills. After WLS, these patients face a multitude of challenges inherent in adjusting to changes in lifestyle, weight, body image, and social functioning, which most individuals would find daunting. These challenges become even more acute in the absence of appropriate psychoeducation, preparation, and intervention from qualified professionals. Individuals who lack effective and adaptive coping skills and supports may have a particularly heightened vulnerability to increased alcohol use in the setting of post-surgery changes in brain reward circuits and pharmacodynamics in alcohol metabolism. For example, one patient reported that her spouse’s pressure to “do something about her weight” was a significant factor in her decision to undergo surgery, but that her spouse was blaming and unsupportive when post-WLS complications developed. The patient believed that these experiences helped fuel development of her post-RYGB alcohol abuse.
Specialized treatment
The number of patients experiencing post-WLS alcohol problems likely will continue to grow, given that the risk of onset of has been shown increase over years. Already, post-WLS patients are proportionally overrepresented among substance abuse treatment populations.24 Empirically, however, we do not know yet if these patients need a different type of addiction treatment than patients who have not had WLS.
Some evidence suggests that post-WLS patients with alcohol problems may be a distinct phenotype within the general population with alcohol problems, as their presentations differ in several ways, including their demographics, alcohol use patterns, and premorbid functioning. A number of studies have found that, despite their increased pharmacodynamic sensitivity to alcohol, people with post-WLS AUDs actually consume a larger amount of alcohol on both typical and maximum drinking days than other individuals with AUDs.24 Additionally, although the median age of onset for AUD is around age 20,25 patients presenting with new-onset, post-WLS alcohol problems are usually in their late 30s, or even 40s or 50s. Further, many of these patients were quite high functioning before their alcohol problems, and are unlikely to identify with the cultural stereotype of a person with AUD (eg, homeless, unemployed), which may hamper or delay their own willingness to accept that they have a problem. These phenotypic differences suggest that post-WLS patients may require substance abuse treatment approaches tailored to their unique presentation. There are additional factors specific to the experiences of being larger-bodied and WLS that also may need to be addressed in specialized treatment for post-WLS addiction patients.
Weight stigma. By definition, patients who have undergone WLS have spent a significant portion of their lives inhabiting larger bodies, an experience that, in our culture, can produce adverse psychosocial effects. Compared with the general population, patients seeking WLS exhibit psychological distress equivalent to psychiatric patients.26 Weight stigma or weight bias—negative judgments directed toward people in larger bodies—is pervasive and continues to increase.27 Further, evidence suggests that, unlike almost all other stigmatized groups, people in larger bodies tend to internalize this stigma, holding an unfavorable attitude toward their own social group.28 Weight stigma impacts the well-being of people all along the weight spectrum, affecting many domains including educational achievements and classroom experiences, job opportunities, salaries, and medical care.27 Weight stigma increases the likelihood of bullying, teasing, and harassment for both adults and children.27 Weight bias has been associated with any number of adverse psychosocial effects, including symptoms of depression, anxiety, and eating pathology; poor body image; and a decrease in healthy self-care behaviors.29-33
Weight stigma makes it more difficult for people to enjoy physical activities, nourish their bodies, and manage stress, which contributes to poorer health outcomes and lower quality of life.33,34 For example, one study showed that, regardless of actual body mass index, people experiencing weight stigma have significantly increased risk of developing an illness or dying.35
Factors specific to WLS. WLS may lead to significant changes in eating habits, and some patients experience a sense of loss, particularly if eating represented one of their primary coping strategies—this may represent a heightened emotional vulnerability for developing AUD.
The fairly rapid and substantial weight loss that WLS produces can lead to sweeping changes in lifestyle, body image, and functional factors for many individuals. Patients often report profound changes, both positive and negative, in their relationships and interactions not only with people in their support network, but also with strangers.36
After the first year or 2 post-WLS, it is fairly common for patients to regain some weight, sometimes in significant amounts.37 This can lead to a sense of “failure.” Life stressors, including difficulties in important relationships, can further add to patients’ vulnerability. For example, one patient noticed that when she was at her thinnest after WLS, drivers were more likely to stop for her when she crossed the street, which pleased but also angered her because they hadn’t extended the same courtesy before WLS. After she regained a significant amount of weight, she began to notice drivers stopping for her less and less frequently. This took her back to her previous feelings of being ignored but now with the certainty that she would be treated better if she were thinner.
Patients also may experience ambivalence about changes in their body size. One might expect that body image would improve after weight loss, but the evidence is mixed.38 Although there is some evidence that body image improves in the short term after WLS,38 other research indicates that body image does not improve with weight loss.39 However, the evidence is clear that the appearance of excess skin after weight loss worsens some patients’ body image.40
To date, there has been no research examining treatment modalities for this population. Because experiences common to individuals who have had WLS could play a role in the development of AUD after surgery, it is intuitive that it would be important to address these factors when designing a treatment plan for post-WLS substance abuse.
Group treatment approach
In 2013, in response to the increase in rates of post-WLS addictions presenting to West End Clinic, an outpatient dual-diagnosis (addiction and psychiatry) service at Massachusetts General Hospital, a specialized treatment group was developed. Nine patients have enrolled since October 2013.
The Post-WLS Addictions Group (PWAG) was designed to be HAES-oriented, trauma-informed, and run within a fat acceptance framework. The HAES model prioritizes a weight-neutral approach that sees health and well-being as multifaceted. This approach directs both patient and clinician to focus on improving health behaviors and reducing internalized weight bias, while building a supportive community that buffers against external cultural weight bias.41
Trauma-informed care42 emphasizes the principles of safety, trustworthiness, and transparency; peer support; collaboration and mutuality; empowerment; and awareness of cultural, historical, and gender issues. In the context of PWAG, weight stigma is conceptualized as a traumatic experience.43 The fat acceptance approach promotes a culture that accepts people of every size with dignity and equality in all aspects of life.44
Self-care emphasis. The HAES model encourages patients to allow their bodies to determine what weight to settle at, and to focus on sustainable health-enhancing behaviors rather than weight loss. Patients who asked about the PWAG were told that this group would not explicitly support, or even encourage, continued pursuit of weight loss per se, but instead would assist patients with relapse prevention, mindful eating, improving self-care, and ongoing stress management. Moving away from a focus on weight loss and toward improvement of self-care skills allowed patients to focus on behaviors and outcomes over which they had more direct control and were more likely to yield immediate benefits.
All of the PWAG group members were in early recovery from an SUD, with a minimum of 4 weeks of abstinence; all had at least 1 co-occurring mental health diagnosis. A licensed independent clinical social worker (LICSW) and a physician familiar with bariatric surgery ran the sessions. The group met weekly for 1 hour. The 8 weekly sessions included both psychoeducation and discussion, with each session covering different topics (Table). The first 20 minutes of each session were devoted to an educational presentation; the remaining 40 minutes for reflection and discussion. In sessions 2 through 8, participants were asked about any recent use or cravings, and problem-solving techniques were employed as needed.
The PWAG group leader herself is a large person who modeled fat acceptance and follows the HAES approach; she led the group using both this experience and her specialized clinical training. As is the case with other addictions recovery treatment modalities, clinicians with lived experience may add a valuable component to both the program design and patient experience.
After the first 8 sessions, all members expressed interest in continuing as an ongoing relapse prevention and HAES support group, and they reported that meeting regularly was very helpful. The group continued with the LICSW alone, who continued to share HAES-oriented and fat acceptance information and resources that group members requested specifically. Over time, new members joined following an individual orientation session with the group leader, and the group has revisited each of the psychoeducational topics repeatedly, though not in a formally structured way.
Process and observations. Participants described high levels of excitement and hopefulness about being in a group with other WLS patients who had developed SUDs. They had a particular interest in reviewing medical/anatomical information about WLS and understanding more about the potential reasons for the elevated risk for developing SUD following WLS. Discussions regarding weight stigma proved to be quite emotional; most participants reported that this material readily related to their own experiences with weight stigma, but they had never discussed these ideas before.
Participants explored the role that grief, loss, guilt, and shame had in the decision to have WLS, the development of SUDs, weight regain or medical complications from the surgery or from substance abuse, career and relationship changes, and worsened body image. Another theme that emerged was the various reasons that prompted the members have WLS that they may not have been conscious of, or willing to discuss with others, such as pressure from a spouse, fears of remaining single due to their size, and a desire to finally “fit in.”
Repeatedly, group members expressed how satisfied and emotionally validated they felt being with people with similar experiences. Most of them had felt alone. They reported a belief that “everyone else” who had WLS was doing well, and that they were the exceptions. Such beliefs and emotions increased the risk of relapse and decreased participants’ ability to develop more positive coping strategies and self-care skills.
Participants reported that feeling less alone, understanding how stigma impacts health and well-being, and focusing on the general benefits of good self-care rather than the pursuit of weight loss were particularly helpful. The HAES and fat acceptance approaches have given group members new ways to think about their bodies and decreased shame. Several group members reported that if they had learned about the HAES approach prior to having a WLS, they might have made a different decision about having surgery, or at least might have been better prepared to handle the emotional and psychological challenges after WLS.
Although evidence for post-WLS addictions is fairly robust, causal mechanisms are not well understood, and research identifying specific risk factors is lacking. Because post-WLS patients with addictions seem to represent a specific phenotype, specialized treatment might be indicated. Future research will be needed to determine optimal treatment approaches for post-WLS addictions. However, a number of aspects are likely to be important. For example, it is likely that unaddressed experiences of weight stigma contribute to challenges, including substance abuse, after WLS; therefore, clinicians involved in the care of individuals presenting with post-WLS SUD should be knowledgeable about weight stigma and how to address it. Because of the specific nature of post-WLS addictions, patients often feel alone and isolated, and seem to benefit from the specialized group setting. We note that the PWAG group leader is herself a large person who models fat acceptance and follows the HAES approach, and therefore led the group using this experience and her specialized clinical training. As with other addiction recovery treatment modalities, clinicians who have lived the experience can add a valuable component to the program design and patient experience.
Maladaptive alcohol use has emerged as a risk for a subset of individuals who have undergone weight loss surgery (WLS); studies report they are vulnerable to consuming alcohol in greater quantities or more frequently.1,2 Estimates of the prevalence of “high-risk” or “hazardous” alcohol use after WLS range from 4% to 28%,3,4 while the prevalence of alcohol use meeting DSM-IV-TR5 criteria for alcohol use disorders (AUDs) hovers around 10%.6
Heavy alcohol users or patients who have active AUD at the time of WLS are at greater risk for continuation of these problems after surgery.2,6 For patients with a long-remitted history of AUD, the evidence regarding risk for post-WLS relapse is lacking, and some evidence suggests they may have better weight loss outcomes after WLS.7
However, approximately two-third of cases of post-WLS alcohol problems occur in patients who have had no history of such problems before surgery.5,8,9 Reported prevalence rates of new-onset alcohol problems range from 3% to 18%,6,9 with the modal finding being approximately 7% to 8%. New-onset alcohol problems appear to occur at a considerable latency after surgery. One study found little risk at 1 year post-surgery, but a significant increase in AUD symptoms at 2 years.6 Another study identified 3 years post-surgery as a high-risk time point,8 and yet another reported a linear increase in the risk for developing alcohol problems for at least 10 years after WLS.10
This article describes a group treatment protocol developed specifically for patients with post-WLS substance use disorder (SUD), and explores:
- risk factors and causal mechanisms of post-WLS AUDs
- weight stigma and emotional stressors
- the role of specialized treatment
- group treatment based on the Health at Every Size® (HAES)-oriented, trauma-informed and fat acceptance framework.
Post-WLS patients with alcohol problems may be a distinct phenotype among people with substance abuse issues. For this reason, they may have a need to address their experiences and issues specific to WLS as part of their alcohol treatment.
Etiology
Risk factors. Empirical findings have identified few predictors or risk factors for post-WLS SUD. These patients are more likely to be male and of a younger age.6 Notably, the vast majority of individuals reporting post-WLS alcohol problems have undergone Roux-en-Y gastric bypass (RYGB), rather than other WLS procedures, such as the laparoscopic adjustable gastric band,6,11 suggesting some physiological mechanism specific to RYGB.
Other potential predictors of postoperative alcohol problems include a pre-operative history of depression, generalized anxiety disorder, smoking, and/or recreational drug use.3,6 Likewise, patients with depression or anxiety disorder symptoms after surgery also may be at higher risk for postoperative alcohol problems.4 The evidence of an association between postoperative weight outcomes and post-WLS alcohol problems is mixed.3,12 Interestingly, patients who had no personal history of substance abuse but who have a family history may have a higher risk of new-onset alcohol problems after surgery.9,12
Causal mechanisms. The etiology of post-WLS alcohol problems is not well understood. If anything, epidemiological data suggest that larger-bodied individuals tend to consume lower levels of alcohol and have lower rates of AUD than individuals in the general population with thinner bodies.13 However, an association has been found between a family history of SUD, but not a personal history, and being large.14 This suggests a shared etiological pathway between addiction and being “overweight,” of which the onset of AUD after RYGB may be a manifestation.
Human and animal studies have shown that WLS may affect alcohol use differently in specific subgroups. Studies have shown that wild-type rats greatly increase their consumption of, or operant responding for, alcohol after RYGB,15 while genetically “alcohol-preferring” rats decrease consumption of, or responding for, alcohol after RYGB.16 A human study likewise found some patients decreased alcohol use or experienced improvement of or remission of AUD symptoms after WLS.4 Combined with the finding that a family history of substance abuse is related to risk for post-operative AUD, these data suggest a potential genetic vulnerability or protection in some individuals.
Turning to potential psychosocial explanations, the lay media has popularized the concepts of “addiction transfer,” or “transfer addiction,”12 with the implication that some patients, who had a preoperative history of “food addiction,” transfer that “addiction” after surgery to substances of abuse.
However, the “addiction transfer” model has a number of flaws:
- it is stigmatizing, because it assumes the patient possesses an innate, chronic, and inalterable pathology
- it relies upon the validity of the controversial construct of “food addiction,” a construct of mixed scientific evidence.17
Further, our knowledge of post-WLS SUD argues against “addiction transfer.” As noted, postoperative alcohol problems are more likely to develop years after surgery, rather than in the first few months afterward when eating is most significantly curtailed. Additionally, post-WLS alcohol problems are significantly more likely to occur after RYGB than other procedures, whereas the “addiction transfer” model would hypothesize that all WLS patients would be at equal risk for postoperative “addiction transfer,” because their eating is similarly affected after surgery.
Links to RYGB. Some clues to physiological mechanisms underlying alcohol problems after RYGB have been identified. After surgery, many RYGB patients report a quicker effect from a smaller amount of alcohol than was the case pre-surgery.18 Studies have demonstrated a number of changes in the pharmacodynamics of alcohol after RYGB not seen in other WLS procedures19:
- a much faster time to peak blood (or breath) alcohol content (BAC)
- significantly higher peak BAC
- a precipitous initial decline in perceived intoxication.18,20
Anatomical features of RYGB may explain such changes.8 However, an increased response to both IV alcohol and IV morphine after RYGB21,22 in rodents suggests that gastrointestinal tract changes are not solely responsible for changes in alcohol use. Emerging research reports that WLS has been found to cause alterations in brain reward pathways,23 which may be an additional contributor to changes in alcohol misuse after surgery.
However, even combined, pharmacokinetic and neurobiological factors cannot entirely explain new-onset alcohol problems after WLS; if they could, one would expect to see a much higher prevalence of this complication. Some psychosocial factors are likely involved as well.
Emotional stressors. One possibility involves a mismatch between post-WLS stressors and coping skills. After WLS, these patients face a multitude of challenges inherent in adjusting to changes in lifestyle, weight, body image, and social functioning, which most individuals would find daunting. These challenges become even more acute in the absence of appropriate psychoeducation, preparation, and intervention from qualified professionals. Individuals who lack effective and adaptive coping skills and supports may have a particularly heightened vulnerability to increased alcohol use in the setting of post-surgery changes in brain reward circuits and pharmacodynamics in alcohol metabolism. For example, one patient reported that her spouse’s pressure to “do something about her weight” was a significant factor in her decision to undergo surgery, but that her spouse was blaming and unsupportive when post-WLS complications developed. The patient believed that these experiences helped fuel development of her post-RYGB alcohol abuse.
Specialized treatment
The number of patients experiencing post-WLS alcohol problems likely will continue to grow, given that the risk of onset of has been shown increase over years. Already, post-WLS patients are proportionally overrepresented among substance abuse treatment populations.24 Empirically, however, we do not know yet if these patients need a different type of addiction treatment than patients who have not had WLS.
Some evidence suggests that post-WLS patients with alcohol problems may be a distinct phenotype within the general population with alcohol problems, as their presentations differ in several ways, including their demographics, alcohol use patterns, and premorbid functioning. A number of studies have found that, despite their increased pharmacodynamic sensitivity to alcohol, people with post-WLS AUDs actually consume a larger amount of alcohol on both typical and maximum drinking days than other individuals with AUDs.24 Additionally, although the median age of onset for AUD is around age 20,25 patients presenting with new-onset, post-WLS alcohol problems are usually in their late 30s, or even 40s or 50s. Further, many of these patients were quite high functioning before their alcohol problems, and are unlikely to identify with the cultural stereotype of a person with AUD (eg, homeless, unemployed), which may hamper or delay their own willingness to accept that they have a problem. These phenotypic differences suggest that post-WLS patients may require substance abuse treatment approaches tailored to their unique presentation. There are additional factors specific to the experiences of being larger-bodied and WLS that also may need to be addressed in specialized treatment for post-WLS addiction patients.
Weight stigma. By definition, patients who have undergone WLS have spent a significant portion of their lives inhabiting larger bodies, an experience that, in our culture, can produce adverse psychosocial effects. Compared with the general population, patients seeking WLS exhibit psychological distress equivalent to psychiatric patients.26 Weight stigma or weight bias—negative judgments directed toward people in larger bodies—is pervasive and continues to increase.27 Further, evidence suggests that, unlike almost all other stigmatized groups, people in larger bodies tend to internalize this stigma, holding an unfavorable attitude toward their own social group.28 Weight stigma impacts the well-being of people all along the weight spectrum, affecting many domains including educational achievements and classroom experiences, job opportunities, salaries, and medical care.27 Weight stigma increases the likelihood of bullying, teasing, and harassment for both adults and children.27 Weight bias has been associated with any number of adverse psychosocial effects, including symptoms of depression, anxiety, and eating pathology; poor body image; and a decrease in healthy self-care behaviors.29-33
Weight stigma makes it more difficult for people to enjoy physical activities, nourish their bodies, and manage stress, which contributes to poorer health outcomes and lower quality of life.33,34 For example, one study showed that, regardless of actual body mass index, people experiencing weight stigma have significantly increased risk of developing an illness or dying.35
Factors specific to WLS. WLS may lead to significant changes in eating habits, and some patients experience a sense of loss, particularly if eating represented one of their primary coping strategies—this may represent a heightened emotional vulnerability for developing AUD.
The fairly rapid and substantial weight loss that WLS produces can lead to sweeping changes in lifestyle, body image, and functional factors for many individuals. Patients often report profound changes, both positive and negative, in their relationships and interactions not only with people in their support network, but also with strangers.36
After the first year or 2 post-WLS, it is fairly common for patients to regain some weight, sometimes in significant amounts.37 This can lead to a sense of “failure.” Life stressors, including difficulties in important relationships, can further add to patients’ vulnerability. For example, one patient noticed that when she was at her thinnest after WLS, drivers were more likely to stop for her when she crossed the street, which pleased but also angered her because they hadn’t extended the same courtesy before WLS. After she regained a significant amount of weight, she began to notice drivers stopping for her less and less frequently. This took her back to her previous feelings of being ignored but now with the certainty that she would be treated better if she were thinner.
Patients also may experience ambivalence about changes in their body size. One might expect that body image would improve after weight loss, but the evidence is mixed.38 Although there is some evidence that body image improves in the short term after WLS,38 other research indicates that body image does not improve with weight loss.39 However, the evidence is clear that the appearance of excess skin after weight loss worsens some patients’ body image.40
To date, there has been no research examining treatment modalities for this population. Because experiences common to individuals who have had WLS could play a role in the development of AUD after surgery, it is intuitive that it would be important to address these factors when designing a treatment plan for post-WLS substance abuse.
Group treatment approach
In 2013, in response to the increase in rates of post-WLS addictions presenting to West End Clinic, an outpatient dual-diagnosis (addiction and psychiatry) service at Massachusetts General Hospital, a specialized treatment group was developed. Nine patients have enrolled since October 2013.
The Post-WLS Addictions Group (PWAG) was designed to be HAES-oriented, trauma-informed, and run within a fat acceptance framework. The HAES model prioritizes a weight-neutral approach that sees health and well-being as multifaceted. This approach directs both patient and clinician to focus on improving health behaviors and reducing internalized weight bias, while building a supportive community that buffers against external cultural weight bias.41
Trauma-informed care42 emphasizes the principles of safety, trustworthiness, and transparency; peer support; collaboration and mutuality; empowerment; and awareness of cultural, historical, and gender issues. In the context of PWAG, weight stigma is conceptualized as a traumatic experience.43 The fat acceptance approach promotes a culture that accepts people of every size with dignity and equality in all aspects of life.44
Self-care emphasis. The HAES model encourages patients to allow their bodies to determine what weight to settle at, and to focus on sustainable health-enhancing behaviors rather than weight loss. Patients who asked about the PWAG were told that this group would not explicitly support, or even encourage, continued pursuit of weight loss per se, but instead would assist patients with relapse prevention, mindful eating, improving self-care, and ongoing stress management. Moving away from a focus on weight loss and toward improvement of self-care skills allowed patients to focus on behaviors and outcomes over which they had more direct control and were more likely to yield immediate benefits.
All of the PWAG group members were in early recovery from an SUD, with a minimum of 4 weeks of abstinence; all had at least 1 co-occurring mental health diagnosis. A licensed independent clinical social worker (LICSW) and a physician familiar with bariatric surgery ran the sessions. The group met weekly for 1 hour. The 8 weekly sessions included both psychoeducation and discussion, with each session covering different topics (Table). The first 20 minutes of each session were devoted to an educational presentation; the remaining 40 minutes for reflection and discussion. In sessions 2 through 8, participants were asked about any recent use or cravings, and problem-solving techniques were employed as needed.
The PWAG group leader herself is a large person who modeled fat acceptance and follows the HAES approach; she led the group using both this experience and her specialized clinical training. As is the case with other addictions recovery treatment modalities, clinicians with lived experience may add a valuable component to both the program design and patient experience.
After the first 8 sessions, all members expressed interest in continuing as an ongoing relapse prevention and HAES support group, and they reported that meeting regularly was very helpful. The group continued with the LICSW alone, who continued to share HAES-oriented and fat acceptance information and resources that group members requested specifically. Over time, new members joined following an individual orientation session with the group leader, and the group has revisited each of the psychoeducational topics repeatedly, though not in a formally structured way.
Process and observations. Participants described high levels of excitement and hopefulness about being in a group with other WLS patients who had developed SUDs. They had a particular interest in reviewing medical/anatomical information about WLS and understanding more about the potential reasons for the elevated risk for developing SUD following WLS. Discussions regarding weight stigma proved to be quite emotional; most participants reported that this material readily related to their own experiences with weight stigma, but they had never discussed these ideas before.
Participants explored the role that grief, loss, guilt, and shame had in the decision to have WLS, the development of SUDs, weight regain or medical complications from the surgery or from substance abuse, career and relationship changes, and worsened body image. Another theme that emerged was the various reasons that prompted the members have WLS that they may not have been conscious of, or willing to discuss with others, such as pressure from a spouse, fears of remaining single due to their size, and a desire to finally “fit in.”
Repeatedly, group members expressed how satisfied and emotionally validated they felt being with people with similar experiences. Most of them had felt alone. They reported a belief that “everyone else” who had WLS was doing well, and that they were the exceptions. Such beliefs and emotions increased the risk of relapse and decreased participants’ ability to develop more positive coping strategies and self-care skills.
Participants reported that feeling less alone, understanding how stigma impacts health and well-being, and focusing on the general benefits of good self-care rather than the pursuit of weight loss were particularly helpful. The HAES and fat acceptance approaches have given group members new ways to think about their bodies and decreased shame. Several group members reported that if they had learned about the HAES approach prior to having a WLS, they might have made a different decision about having surgery, or at least might have been better prepared to handle the emotional and psychological challenges after WLS.
Although evidence for post-WLS addictions is fairly robust, causal mechanisms are not well understood, and research identifying specific risk factors is lacking. Because post-WLS patients with addictions seem to represent a specific phenotype, specialized treatment might be indicated. Future research will be needed to determine optimal treatment approaches for post-WLS addictions. However, a number of aspects are likely to be important. For example, it is likely that unaddressed experiences of weight stigma contribute to challenges, including substance abuse, after WLS; therefore, clinicians involved in the care of individuals presenting with post-WLS SUD should be knowledgeable about weight stigma and how to address it. Because of the specific nature of post-WLS addictions, patients often feel alone and isolated, and seem to benefit from the specialized group setting. We note that the PWAG group leader is herself a large person who models fat acceptance and follows the HAES approach, and therefore led the group using this experience and her specialized clinical training. As with other addiction recovery treatment modalities, clinicians who have lived the experience can add a valuable component to the program design and patient experience.
2. Lent MR, Hayes SM, Wood GC, et al. Smoking and alcohol use in gastric bypass patients. Eat Behav. 2013;14(4):460-463.
4. Wee CC, Mukamal KJ, Huskey KW, et al. High-risk alcohol use after weight loss surgery. Surg Obes Relat Dis. 2014;10(3):508-513.
5. Diagnostic and statistical manual of mental disorders, 4th, text rev. Washington, DC: American Psychiatric Association; 2000.
6. King WC, Chen JY, Mitchell JE, et al. Prevalence of alcohol use disorders before and after bariatric surgery. JAMA. 2012;307(23):2516-2525.
9. Ivezaj V, Saules KK, Schuh LM. New-onset substance use disorder after gastric bypass surgery: rates and associated characteristics. Obes Surg. 2014;24(11):1975-1980.
10. Svensson PA, Anveden Å, Romeo S, et al. Alcohol consumption and alcohol problems after bariatric surgery in the Swedish obese subjects study. Obesity. 2013;21(12):2444-2451.
11. Ostlund MP, Backman O, Marsk R, et al. Increased admission for alcohol dependence after gastric bypass surgery compared with restrictive bariatric surgery. JAMA Surg. 2013;148(4):374-377.
13. Gearhardt AN, Corbin WR. Body mass index and alcohol consumption: family history of alcoholism as a moderator. Psychol Addict Behav. 2009;23(2):216-225.
14. Grucza RA, Krueger RF, Racette SB, et al. The emerging link between alcoholism risk and obesity in the United States. Arch Gen Psychiatry. 2010;67(12):1301-1308.
15. Davis JF, Tracy AL, Schurdak JD, et al. Roux en y gastric bypass increases ethanol intake in the rat. Obes Surg. 2013;23(7):920-930.
16. Davis JF, Schurdak JD, Magrisso IJ, et al. Gastric bypass surgery attenuates ethanol consumption in ethanol-preferring rats. Biol Psychiatry. 2012;72(5):354-360.
18. Pepino MY, Okunade AL, Eagon JC, et al. Effect of Roux-en-Y gastric bypass surgery: converting 2 alcoholic drinks to 4. JAMA Surg. 2015
19. Changchien EM, Woodard GA, Hernandez-Boussard T, et al. Normal alcohol metabolism after gastric banding and sleeve gastrectomy: a case-cross-over trial. J Am Coll Surg. 2012;215(4):475-479.
22. Polston JE, Pritchett CE, Tomasko JM, et al. Roux-en-Y gastric bypass increases intravenous ethanol self-administration in dietary obese rats. PLoS ONE. 2013;8(12):e83741. doi: 10.1371/journal.pone.0083741.
26. Higgs ML, Wade T, Cescato M, et al. Differences between treatment seekers in an obese population: medical intervention vs. dietary restriction. J Behav Med. 1997;20(4):391-405.
35. Sutin AR, Stephan Y, Terracciano A. Weight discrimination and risk of mortality. Psychol Sci. 2015;26(11):1803-1811.
36. Sogg S, Gorman MJ. Interpersonal changes and challenges after weight loss surgery. Prim Psychiatry. 2008;15(8):61-66.
37. Yanos BR, Saules KK, Schuh LM, et al. Predictors of lowest weight and long-term weight regain among Roux-en-Y gastric bypass patients. Obes Surg. 2015;25(8):1364-1370.
40. van der Beek E, Te Riele W, Specken TF, et al. The impact of reconstructive procedures following bariatric surgery on patient well-being and quality of life. Obes Surg. 2010;20(1):36-41.
2. Lent MR, Hayes SM, Wood GC, et al. Smoking and alcohol use in gastric bypass patients. Eat Behav. 2013;14(4):460-463.
4. Wee CC, Mukamal KJ, Huskey KW, et al. High-risk alcohol use after weight loss surgery. Surg Obes Relat Dis. 2014;10(3):508-513.
5. Diagnostic and statistical manual of mental disorders, 4th, text rev. Washington, DC: American Psychiatric Association; 2000.
6. King WC, Chen JY, Mitchell JE, et al. Prevalence of alcohol use disorders before and after bariatric surgery. JAMA. 2012;307(23):2516-2525.
9. Ivezaj V, Saules KK, Schuh LM. New-onset substance use disorder after gastric bypass surgery: rates and associated characteristics. Obes Surg. 2014;24(11):1975-1980.
10. Svensson PA, Anveden Å, Romeo S, et al. Alcohol consumption and alcohol problems after bariatric surgery in the Swedish obese subjects study. Obesity. 2013;21(12):2444-2451.
11. Ostlund MP, Backman O, Marsk R, et al. Increased admission for alcohol dependence after gastric bypass surgery compared with restrictive bariatric surgery. JAMA Surg. 2013;148(4):374-377.
13. Gearhardt AN, Corbin WR. Body mass index and alcohol consumption: family history of alcoholism as a moderator. Psychol Addict Behav. 2009;23(2):216-225.
14. Grucza RA, Krueger RF, Racette SB, et al. The emerging link between alcoholism risk and obesity in the United States. Arch Gen Psychiatry. 2010;67(12):1301-1308.
15. Davis JF, Tracy AL, Schurdak JD, et al. Roux en y gastric bypass increases ethanol intake in the rat. Obes Surg. 2013;23(7):920-930.
16. Davis JF, Schurdak JD, Magrisso IJ, et al. Gastric bypass surgery attenuates ethanol consumption in ethanol-preferring rats. Biol Psychiatry. 2012;72(5):354-360.
18. Pepino MY, Okunade AL, Eagon JC, et al. Effect of Roux-en-Y gastric bypass surgery: converting 2 alcoholic drinks to 4. JAMA Surg. 2015
19. Changchien EM, Woodard GA, Hernandez-Boussard T, et al. Normal alcohol metabolism after gastric banding and sleeve gastrectomy: a case-cross-over trial. J Am Coll Surg. 2012;215(4):475-479.
22. Polston JE, Pritchett CE, Tomasko JM, et al. Roux-en-Y gastric bypass increases intravenous ethanol self-administration in dietary obese rats. PLoS ONE. 2013;8(12):e83741. doi: 10.1371/journal.pone.0083741.
26. Higgs ML, Wade T, Cescato M, et al. Differences between treatment seekers in an obese population: medical intervention vs. dietary restriction. J Behav Med. 1997;20(4):391-405.
35. Sutin AR, Stephan Y, Terracciano A. Weight discrimination and risk of mortality. Psychol Sci. 2015;26(11):1803-1811.
36. Sogg S, Gorman MJ. Interpersonal changes and challenges after weight loss surgery. Prim Psychiatry. 2008;15(8):61-66.
37. Yanos BR, Saules KK, Schuh LM, et al. Predictors of lowest weight and long-term weight regain among Roux-en-Y gastric bypass patients. Obes Surg. 2015;25(8):1364-1370.
40. van der Beek E, Te Riele W, Specken TF, et al. The impact of reconstructive procedures following bariatric surgery on patient well-being and quality of life. Obes Surg. 2010;20(1):36-41.

Evaluating the risk of sexually transmitted infections in mentally ill patients
Sexually transmitted infections (STIs) continue to be a significant public health problem with potentially serious complications.1 The incidence of new STIs, including viral STIs, in the United States is estimated at 19 million cases per year.2Chlamydia trachomatis remains the most common bacterial STI with an estimated annual incidence of 2.8 million cases in the United States and 50 million worldwide. Second in prevalence is gonococcal infection. Herpes simplex virus is one of the most common viral STIs, but the incidence of human papillomavirus virus (HPV), which is associated with cervical cancer, has steadily increased worldwide.3 Young persons age 15 to 24 are at the highest risk of acquiring new STIs with almost 50% of new cases reported among this age group.4
STIs can have serious complications and sequelae. For example, 20% to 40% of women who have chlamydia infections and 10% to 20% of women who have gonococcal infections develop pelvic inflammatory disease (PID),2 which increases the risk for ectopic pregnancy, infertility, and chronic pelvic pain.
Patients with mental illness are at high risk of acquiring STIs. In the United States, the prevalence of HIV among patients with psychiatric illness is 10 to 20 times higher than in the general population.4,5 Factors contributing to increased vulnerability to STIs among psychiatric patients include:
- impaired autonomy
- increased impulsivity
- increased susceptibility to coerced sex.6


Furthermore, a higher incidence of poverty, placement in risky environments, and overall poor health and medical care also contribute to the high prevalence of STIs and their complications in this population (Table 1). Because of risk factors specific to psychiatric illness, standard STI prevention interventions are not always successful and novel and innovative behavioral approaches are necessary.7
Case Abdominal pain and fever
Ms. K, age 25, has a history of bipolar disorder treated with lithium and presents to the community psychiatrist with lower abdominal pain. She recently recovered from a manic episode and has started to reintegrate with the community mental health team. She refuses to see her primary care physician and is adamant that she wishes to see her psychiatrist, who is the only doctor she has rapport with.
Ms. K reports lower abdominal pain for 3 or 4 days and fever for 1 day. The pain is dull in character. She denies diarrhea, vomiting, or urinary symptoms, but on further questioning describes new-onset, foul-smelling vaginal discharge without vaginal bleeding. Her menstrual cycle usually is regular, but her last menstrual period occurred 2 months ago. Her medical history includes an appendectomy at age 10 and she is a current cigarette smoker. Chart notes taken during her manic episode describe high-risk behavior, including having unprotected sexual intercourse with several partners. On examination, she is febrile and tachycardic with a tender lower abdomen.
Diagnosing STIs


To diagnose an STI, first a clinician must consider its likelihood. Taking a thorough sexual history allows assessment of the need for further investigation and provides an opportunity to discuss risk reduction. In accordance with recent guidelines,8 all health care providers are encouraged to consider the sexual history a routine aspect of the clinical encounter. The Centers for Disease Control and Prevention’s (CDC’s) “Five Ps” approach (Table 2) is an excellent tool for guiding investigation and counseling.9
The Figure provides health care providers with an algorithm to guide testing for STIs among psychiatric patients. Note that chlamydia, gonorrhea, syphilis, chancroid, viral hepatitis, and HIV must be reported to state public health agencies and the CDC.
Modern laboratory techniques make diagnosing STIs easier. Analysis of urine or serum reduces the need for invasive sampling. If swabs are required for diagnosis, patient self-collection of urethral, vulvovaginal, rectal, or pharyngeal specimens is as accurate as clinician collected samples and is better tolerated.8 Because of variation in diagnostic assays, we recommend contacting the laboratory before sending non-standard samples to ensure accurate collection and analysis.

Guidelines for preventing and screening for STIs
There are no prevention guidelines for STIs specific to the psychiatric population, although there is a clear need for focused intervention in this vulnerable patient group.10 Rates of STI screening generally are low in the psychiatric setting,11 which results in a considerable burden of disease. All psychiatric patients should be encouraged to engage with STI screening programs that are in line with national guidelines. In the inpatient psychiatric or medical environment, clinicians have a responsibility to ensure that STI screening is considered for each patient.
Patients with mental illness should be assumed to be sexually active, even if they do not volunteer this information to clinicians. Employ a low threshold for recommending safer sex practices including condom use. Encourage women to develop a relationship with a family practitioner, internist, or gynecologist. Advise men who have sex with men (MSM) to visit a doctor regularly for screening of HIV and rectal, anal, and oral STIs as behavior and symptoms dictate.
There is general agreement about STI screening among the United States Preventive Services Task Force (USPSTF), CDC, American Academy of Family Physicians, American Academy of Pediatrics, and American College of Obstetricians and Gynecologists. USPSTF guidelines are summarized in Table 3.12
In addition to these guidelines, the CDC suggests that all adults and adolescents be tested at least once for HIV.13 The CDC also recommends annual testing of MSM for HIV, syphilis, chlamydia, and gonorrhea. In MSM who have multiple partners or who have sex while using illicit drugs, testing should occur more frequently, such as every 3 to 6 months.14
HPV. Routine HPV screening is not recommended; however, 2 vaccines are available to prevent oncogenic HPV (types 16 and 18). All females age 13 to 26 should receive 3 doses of HPV vaccine over a 6-month period. The quadrivalent vaccine (Gardasil) also protects against HPV types 6 and 11, which cause 90% of genital warts and is preferred when available. Males age 9 to 26 also can receive the vaccine, although ideally it should be administered before sexual activity begins.15 Women still should attend routine cervical cancer screening even if they have the vaccine because 30% of cervical cancers are not caused by HPV 16/18. However, this means that 70% of cervical cancers are associated with HPV 16/18, making screening and the vaccine an important public health initiative. There also is a link between HPV and oral cancers.
Treating STIs among mentally ill individuals
Treatment of STIs among mentally ill individuals is important to prevent medical complications and to reduce transmission. Here are a few additional questions to keep in mind when treating a patient with psychiatric illness:
Does the patient have a primary psychiatric disorder, or is the patient’s current psychiatric presentation a result of the infection?
Some STIs can manifest with psychiatric symptoms—for example, neurosyphilis and HIV-associated neurocognitive disorders—and pose a diagnostic challenge. Obtaining a longitudinal history of the patient’s mental health, age of onset, and family history can help clarify the cause.
Are there any psychiatric adverse effects of STI treatment?
Most drugs used for treating common STIs are not known to cause psychiatric adverse effects (See the American Psychiatric Association16 and Sockalingham et al17 for a thorough discussion of HIV and hepatitis C treatment). The exception is fluoroquinolones, which could be prescribed for PID if cephalosporin therapy is not feasible. CNS effects of fluoroquinolones include insomnia, restlessness, confusion, and, in rare cases, mania and psychosis.
What are possible medication interactions to keep in mind when treating a psychiatric patient?
Nonsteroidal anti-inflammatory drugs (NSAIDs), other than sulindac, could increase serum lithium levels. Although NSAIDs are not contraindicated in patients taking lithium, other pain relievers, such as acetaminophen, may be preferred as a first-line choice.
Carbamazepine could lower serum levels of doxycycline.18
Azithromycin and other macrolides, as well as fluoroquinolones, could have QTc prolonging effects and has been associated with torsades de pointes.19 Several psychiatric medications, in particular, atypical antipsychotics, also could prolong the QTc interval. This could be a consideration in patients with underlying long QT intervals at baseline or a family history of sudden cardiac death.
Psychiatric patients might refuse or not adhere to their medication. Refusals could be the result of grandiose delusions (“I don’t need treatment”) or paranoia (“The doctor is trying to poison me”). Consider 1-time doses of antibiotics that can be given in the clinic for uncomplicated infections when adherence is an issue. Because psychiatric patients are at higher risk for acquiring STIs, education and counseling—especially substance abuse counseling—are vital as both primary and secondary prevention strategies. Treatment of STIs should be accompanied by referrals to the social work team or a therapist when appropriate.
Finally, as with any proposed treatment, it is important to consider whether the patient has capacity to consent to or refuse treatment. To assess for capacity, a patient must be able to:
- communicate a choice
- understand the relevant information
- appreciate the medical consequences of the decision
- demonstrate the ability to reason about treatment choices.20
Case continued
In the emergency department, Ms. K’s vital signs are: temperature 39.5°C; pulse 110 beats per minute; blood pressure 96/67 mm Hg; and breathing 20 respirations per minute. She complains of nausea and has 2 episodes of emesis. She allows clinicians to perform a complete physical examination, including pelvic exam. Her cervix is inflamed, and she is noted to have adnexal and cervical motion tenderness.
Labs and imaging confirm a diagnosis of PID due to gonorrhea and she is admitted to the hospital for IV antibiotics. She continues to experience nausea and vomiting, but also complains of dizziness and diarrhea. Her speech is slurred and a coarse tremor is noticed in her hands. Renal function tests show slight impairment, probably due to dehydration. A pregnancy test is negative.
Lithium is held. Her nausea, vomiting, and diarrhea resolve quickly, and Ms. K asks to leave. When she is told that she is not ready for discharge, Ms. K becomes upset and rips out her IV yelling, “I don’t need treatment from you guys!” A psychiatry consult is called to assess for her capacity to refuse treatment. The team determines that she has capacity, but she becomes agreeable to remaining in the hospital after a phone conversation with her community mental health team.
Ms. K improves with antibiotic treatment. HIV and syphilis serology tests are negative. Before discharge, both the community psychiatrist and her primary care physicians are informed her lithium was held during hospitalization and restarted before discharge. Ms. K also is educated about the signs and symptoms of lithium toxicity, as well as common STIs.
Clinical considerations
- Physicians should have a low threshold of suspicion for PID in a sexually active young woman who presents with abdominal pain and shuffling gait, which is a natural attempt to reduce cervical irritation and is associated with PID.
- Ask about sexual history and symptoms of STIs.
- Rule out STIs in men presenting with urinary tract infections.
- If chlamydia is diagnosed, treatment for gonorrhea also is essential, and vice versa.
- Always think about HIV and hepatatis B and C in a patient with a STI.
- Treatment with single-dose medications can be effective.
- Risk of STIs is higher during episodes of mania or psychosis.
- Consider hospitalization if medically indicated or if you suspect non-adherence to therapy. It is important to remember that all kinds of systemic infections—including PID—can result in dehydration and alter renal metabolism leading to lithium accumulation.
- Mentally ill patients might require placement under involuntary commitment if they are found to be a danger to themselves or others. It is important to liaise with both the community psychiatry team and primary care physician both during hospitalization and before discharge to ensure a smooth transition.
1. Fenton KA, Lowndes CM. Recent trends in the epidemiology of sexually transmitted infections in the European Union. Sex Transm Infect. 2004;80(4):255-263.
2. Trigg BG, Kerndt PR, Aynalem G. Sexually transmitted infections and pelvic inflammatory disease in women. Med Clin North Am. 2008;92(5):1083-1113, x.
3. Frenkl TL, Potts J. Sexually transmitted infections. Urol Clin North Am. 2008;35(1):33-46; vi.
4. Weinstock H, Berman S, Cates W Jr. Sexually transmitted diseases among American youth: incidence and prevalence estimates, 2000. Perspect Sex Reprod Health. 2004;36(1):6-10.
5. Rosenberg SD, Goodman LA, Osher FC, et al. Prevalence of HIV, hepatitis B, and hepatitis C in people with severe mental illness. Am J Public Health. 2001;91(1):31-37.
6. King C, Feldman J, Waithaka Y, et al. Sexual risk behaviors and sexually transmitted infection prevalence in an outpatient psychiatry clinic. Sex Transm Dis. 2008;35(10):877-882.
7. Erbelding EJ, Hutton HE, Zenilman JM, et al. The prevalence of psychiatric disorders in sexually transmitted disease clinic patients and their association with sexually transmitted disease risk. Sex Transm Dis. 2004;31(1):8-12.
8. Freeman AH, Bernstein KT, Kohn RP, et al. Evaluation of self-collected versus clinician-collected swabs for the detection of Chlamydia trachomatis and Neisseria gonorrhoeae pharyngeal infection among men who have sex with men. Sex Transm Dis. 2011;38(11):1036-1039.
9. Workowski KA, Berman S; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2010. MMWR Recomm Rep. 2010;59(RR-12):1-110.
10. Rein DB, Anderson LA, Irwin KL. Mental health disorders and sexually transmitted diseases in a privately insured population. Am J Manag Care. 2004;10(12):917-924.
11. Rothbard AB, Blank MB, Staab JP, et al. Previously undetected metabolic syndromes and infectious diseases among psychiatric inpatients. Psychiatr Serv. 2009;60(4):534-537.
12. Meyers D, Wolff T, Gregory K, et al. USPSTF recommendations for STI screening. Am Fam Physician. 2008;77(6):819-824.
13. Branson BM, Handsfield HH, Lampe MA, et al; Centers for Disease Control and Prevention (CDC). Revised recommendations for HIV testing of adults, adolescents, and pregnant women in health-care settings. MMWR Recomm Rep. 2006;55(RR-14):1-17; quiz CE1-CE 4.
14. Centers for Disease Control and Prevention. Incidence, prevalence, and cost of sexually transmitted infections in the United States. https://npin.cdc.gov/publication/incidence-prevalence-and-cost-sexually-transmitted-infections-united-states. Published February 2013. Accessed December 12, 2016.
15. Centers for Disease Control and Prevention (CDC). Recommendations on the use of quadrivalent human papillomavirus vaccine in males—Advisory Committee on Immunization Practices (ACIP), 2011. MMWR Morb Mortal Wkly Rep. 2011;60(50):1705-1708.
16. American Psychiatric Association. HIV psychiatry. https://www.psychiatry.org/psychiatrists/practice/professional-interests/hiv-psychiatry. Accessed December 13, 2016.
17. Sockalingam S, Sheehan K, Feld JJ, et al. Psychiatric care during hepatitis C treatment: the changing role of psychiatrists in the era of direct-acting antivirals. Am J Psychiatry. 2015;172(6):512-516.
18. Neuvonen PJ, Pentikäinen PJ, Gothoni G. Inhibition of iron absorption by tetracycline. Br J Clin Pharmacol. 1975;2(1):94-96.
19. Sears SP, Getz TW, Austin CO, et al. Incidence of sustained ventricular tachycardia in patients with prolonged QTc after the administration of azithromycin: a retrospective study. Drugs Real World Outcomes. 2016;3:99-105.
20. Appelbaum PS. Clinical practice. Assessment of patients’ competence to consent to treatment. N Engl J Med. 2007;357(18):1834-1840.
Sexually transmitted infections (STIs) continue to be a significant public health problem with potentially serious complications.1 The incidence of new STIs, including viral STIs, in the United States is estimated at 19 million cases per year.2Chlamydia trachomatis remains the most common bacterial STI with an estimated annual incidence of 2.8 million cases in the United States and 50 million worldwide. Second in prevalence is gonococcal infection. Herpes simplex virus is one of the most common viral STIs, but the incidence of human papillomavirus virus (HPV), which is associated with cervical cancer, has steadily increased worldwide.3 Young persons age 15 to 24 are at the highest risk of acquiring new STIs with almost 50% of new cases reported among this age group.4
STIs can have serious complications and sequelae. For example, 20% to 40% of women who have chlamydia infections and 10% to 20% of women who have gonococcal infections develop pelvic inflammatory disease (PID),2 which increases the risk for ectopic pregnancy, infertility, and chronic pelvic pain.
Patients with mental illness are at high risk of acquiring STIs. In the United States, the prevalence of HIV among patients with psychiatric illness is 10 to 20 times higher than in the general population.4,5 Factors contributing to increased vulnerability to STIs among psychiatric patients include:
- impaired autonomy
- increased impulsivity
- increased susceptibility to coerced sex.6
Furthermore, a higher incidence of poverty, placement in risky environments, and overall poor health and medical care also contribute to the high prevalence of STIs and their complications in this population (Table 1). Because of risk factors specific to psychiatric illness, standard STI prevention interventions are not always successful and novel and innovative behavioral approaches are necessary.7
Case Abdominal pain and fever
Ms. K, age 25, has a history of bipolar disorder treated with lithium and presents to the community psychiatrist with lower abdominal pain. She recently recovered from a manic episode and has started to reintegrate with the community mental health team. She refuses to see her primary care physician and is adamant that she wishes to see her psychiatrist, who is the only doctor she has rapport with.
Ms. K reports lower abdominal pain for 3 or 4 days and fever for 1 day. The pain is dull in character. She denies diarrhea, vomiting, or urinary symptoms, but on further questioning describes new-onset, foul-smelling vaginal discharge without vaginal bleeding. Her menstrual cycle usually is regular, but her last menstrual period occurred 2 months ago. Her medical history includes an appendectomy at age 10 and she is a current cigarette smoker. Chart notes taken during her manic episode describe high-risk behavior, including having unprotected sexual intercourse with several partners. On examination, she is febrile and tachycardic with a tender lower abdomen.
Diagnosing STIs
To diagnose an STI, first a clinician must consider its likelihood. Taking a thorough sexual history allows assessment of the need for further investigation and provides an opportunity to discuss risk reduction. In accordance with recent guidelines,8 all health care providers are encouraged to consider the sexual history a routine aspect of the clinical encounter. The Centers for Disease Control and Prevention’s (CDC’s) “Five Ps” approach (Table 2) is an excellent tool for guiding investigation and counseling.9
The Figure provides health care providers with an algorithm to guide testing for STIs among psychiatric patients. Note that chlamydia, gonorrhea, syphilis, chancroid, viral hepatitis, and HIV must be reported to state public health agencies and the CDC.
Modern laboratory techniques make diagnosing STIs easier. Analysis of urine or serum reduces the need for invasive sampling. If swabs are required for diagnosis, patient self-collection of urethral, vulvovaginal, rectal, or pharyngeal specimens is as accurate as clinician collected samples and is better tolerated.8 Because of variation in diagnostic assays, we recommend contacting the laboratory before sending non-standard samples to ensure accurate collection and analysis.
Guidelines for preventing and screening for STIs
There are no prevention guidelines for STIs specific to the psychiatric population, although there is a clear need for focused intervention in this vulnerable patient group.10 Rates of STI screening generally are low in the psychiatric setting,11 which results in a considerable burden of disease. All psychiatric patients should be encouraged to engage with STI screening programs that are in line with national guidelines. In the inpatient psychiatric or medical environment, clinicians have a responsibility to ensure that STI screening is considered for each patient.
Patients with mental illness should be assumed to be sexually active, even if they do not volunteer this information to clinicians. Employ a low threshold for recommending safer sex practices including condom use. Encourage women to develop a relationship with a family practitioner, internist, or gynecologist. Advise men who have sex with men (MSM) to visit a doctor regularly for screening of HIV and rectal, anal, and oral STIs as behavior and symptoms dictate.
There is general agreement about STI screening among the United States Preventive Services Task Force (USPSTF), CDC, American Academy of Family Physicians, American Academy of Pediatrics, and American College of Obstetricians and Gynecologists. USPSTF guidelines are summarized in Table 3.12
In addition to these guidelines, the CDC suggests that all adults and adolescents be tested at least once for HIV.13 The CDC also recommends annual testing of MSM for HIV, syphilis, chlamydia, and gonorrhea. In MSM who have multiple partners or who have sex while using illicit drugs, testing should occur more frequently, such as every 3 to 6 months.14
HPV. Routine HPV screening is not recommended; however, 2 vaccines are available to prevent oncogenic HPV (types 16 and 18). All females age 13 to 26 should receive 3 doses of HPV vaccine over a 6-month period. The quadrivalent vaccine (Gardasil) also protects against HPV types 6 and 11, which cause 90% of genital warts and is preferred when available. Males age 9 to 26 also can receive the vaccine, although ideally it should be administered before sexual activity begins.15 Women still should attend routine cervical cancer screening even if they have the vaccine because 30% of cervical cancers are not caused by HPV 16/18. However, this means that 70% of cervical cancers are associated with HPV 16/18, making screening and the vaccine an important public health initiative. There also is a link between HPV and oral cancers.
Treating STIs among mentally ill individuals
Treatment of STIs among mentally ill individuals is important to prevent medical complications and to reduce transmission. Here are a few additional questions to keep in mind when treating a patient with psychiatric illness:
Does the patient have a primary psychiatric disorder, or is the patient’s current psychiatric presentation a result of the infection?
Some STIs can manifest with psychiatric symptoms—for example, neurosyphilis and HIV-associated neurocognitive disorders—and pose a diagnostic challenge. Obtaining a longitudinal history of the patient’s mental health, age of onset, and family history can help clarify the cause.
Are there any psychiatric adverse effects of STI treatment?
Most drugs used for treating common STIs are not known to cause psychiatric adverse effects (See the American Psychiatric Association16 and Sockalingham et al17 for a thorough discussion of HIV and hepatitis C treatment). The exception is fluoroquinolones, which could be prescribed for PID if cephalosporin therapy is not feasible. CNS effects of fluoroquinolones include insomnia, restlessness, confusion, and, in rare cases, mania and psychosis.
What are possible medication interactions to keep in mind when treating a psychiatric patient?
Nonsteroidal anti-inflammatory drugs (NSAIDs), other than sulindac, could increase serum lithium levels. Although NSAIDs are not contraindicated in patients taking lithium, other pain relievers, such as acetaminophen, may be preferred as a first-line choice.
Carbamazepine could lower serum levels of doxycycline.18
Azithromycin and other macrolides, as well as fluoroquinolones, could have QTc prolonging effects and has been associated with torsades de pointes.19 Several psychiatric medications, in particular, atypical antipsychotics, also could prolong the QTc interval. This could be a consideration in patients with underlying long QT intervals at baseline or a family history of sudden cardiac death.
Psychiatric patients might refuse or not adhere to their medication. Refusals could be the result of grandiose delusions (“I don’t need treatment”) or paranoia (“The doctor is trying to poison me”). Consider 1-time doses of antibiotics that can be given in the clinic for uncomplicated infections when adherence is an issue. Because psychiatric patients are at higher risk for acquiring STIs, education and counseling—especially substance abuse counseling—are vital as both primary and secondary prevention strategies. Treatment of STIs should be accompanied by referrals to the social work team or a therapist when appropriate.
Finally, as with any proposed treatment, it is important to consider whether the patient has capacity to consent to or refuse treatment. To assess for capacity, a patient must be able to:
- communicate a choice
- understand the relevant information
- appreciate the medical consequences of the decision
- demonstrate the ability to reason about treatment choices.20
Case continued
In the emergency department, Ms. K’s vital signs are: temperature 39.5°C; pulse 110 beats per minute; blood pressure 96/67 mm Hg; and breathing 20 respirations per minute. She complains of nausea and has 2 episodes of emesis. She allows clinicians to perform a complete physical examination, including pelvic exam. Her cervix is inflamed, and she is noted to have adnexal and cervical motion tenderness.
Labs and imaging confirm a diagnosis of PID due to gonorrhea and she is admitted to the hospital for IV antibiotics. She continues to experience nausea and vomiting, but also complains of dizziness and diarrhea. Her speech is slurred and a coarse tremor is noticed in her hands. Renal function tests show slight impairment, probably due to dehydration. A pregnancy test is negative.
Lithium is held. Her nausea, vomiting, and diarrhea resolve quickly, and Ms. K asks to leave. When she is told that she is not ready for discharge, Ms. K becomes upset and rips out her IV yelling, “I don’t need treatment from you guys!” A psychiatry consult is called to assess for her capacity to refuse treatment. The team determines that she has capacity, but she becomes agreeable to remaining in the hospital after a phone conversation with her community mental health team.
Ms. K improves with antibiotic treatment. HIV and syphilis serology tests are negative. Before discharge, both the community psychiatrist and her primary care physicians are informed her lithium was held during hospitalization and restarted before discharge. Ms. K also is educated about the signs and symptoms of lithium toxicity, as well as common STIs.
Clinical considerations
- Physicians should have a low threshold of suspicion for PID in a sexually active young woman who presents with abdominal pain and shuffling gait, which is a natural attempt to reduce cervical irritation and is associated with PID.
- Ask about sexual history and symptoms of STIs.
- Rule out STIs in men presenting with urinary tract infections.
- If chlamydia is diagnosed, treatment for gonorrhea also is essential, and vice versa.
- Always think about HIV and hepatatis B and C in a patient with a STI.
- Treatment with single-dose medications can be effective.
- Risk of STIs is higher during episodes of mania or psychosis.
- Consider hospitalization if medically indicated or if you suspect non-adherence to therapy. It is important to remember that all kinds of systemic infections—including PID—can result in dehydration and alter renal metabolism leading to lithium accumulation.
- Mentally ill patients might require placement under involuntary commitment if they are found to be a danger to themselves or others. It is important to liaise with both the community psychiatry team and primary care physician both during hospitalization and before discharge to ensure a smooth transition.
Sexually transmitted infections (STIs) continue to be a significant public health problem with potentially serious complications.1 The incidence of new STIs, including viral STIs, in the United States is estimated at 19 million cases per year.2Chlamydia trachomatis remains the most common bacterial STI with an estimated annual incidence of 2.8 million cases in the United States and 50 million worldwide. Second in prevalence is gonococcal infection. Herpes simplex virus is one of the most common viral STIs, but the incidence of human papillomavirus virus (HPV), which is associated with cervical cancer, has steadily increased worldwide.3 Young persons age 15 to 24 are at the highest risk of acquiring new STIs with almost 50% of new cases reported among this age group.4
STIs can have serious complications and sequelae. For example, 20% to 40% of women who have chlamydia infections and 10% to 20% of women who have gonococcal infections develop pelvic inflammatory disease (PID),2 which increases the risk for ectopic pregnancy, infertility, and chronic pelvic pain.
Patients with mental illness are at high risk of acquiring STIs. In the United States, the prevalence of HIV among patients with psychiatric illness is 10 to 20 times higher than in the general population.4,5 Factors contributing to increased vulnerability to STIs among psychiatric patients include:
- impaired autonomy
- increased impulsivity
- increased susceptibility to coerced sex.6
Furthermore, a higher incidence of poverty, placement in risky environments, and overall poor health and medical care also contribute to the high prevalence of STIs and their complications in this population (Table 1). Because of risk factors specific to psychiatric illness, standard STI prevention interventions are not always successful and novel and innovative behavioral approaches are necessary.7
Case Abdominal pain and fever
Ms. K, age 25, has a history of bipolar disorder treated with lithium and presents to the community psychiatrist with lower abdominal pain. She recently recovered from a manic episode and has started to reintegrate with the community mental health team. She refuses to see her primary care physician and is adamant that she wishes to see her psychiatrist, who is the only doctor she has rapport with.
Ms. K reports lower abdominal pain for 3 or 4 days and fever for 1 day. The pain is dull in character. She denies diarrhea, vomiting, or urinary symptoms, but on further questioning describes new-onset, foul-smelling vaginal discharge without vaginal bleeding. Her menstrual cycle usually is regular, but her last menstrual period occurred 2 months ago. Her medical history includes an appendectomy at age 10 and she is a current cigarette smoker. Chart notes taken during her manic episode describe high-risk behavior, including having unprotected sexual intercourse with several partners. On examination, she is febrile and tachycardic with a tender lower abdomen.
Diagnosing STIs
To diagnose an STI, first a clinician must consider its likelihood. Taking a thorough sexual history allows assessment of the need for further investigation and provides an opportunity to discuss risk reduction. In accordance with recent guidelines,8 all health care providers are encouraged to consider the sexual history a routine aspect of the clinical encounter. The Centers for Disease Control and Prevention’s (CDC’s) “Five Ps” approach (Table 2) is an excellent tool for guiding investigation and counseling.9
The Figure provides health care providers with an algorithm to guide testing for STIs among psychiatric patients. Note that chlamydia, gonorrhea, syphilis, chancroid, viral hepatitis, and HIV must be reported to state public health agencies and the CDC.
Modern laboratory techniques make diagnosing STIs easier. Analysis of urine or serum reduces the need for invasive sampling. If swabs are required for diagnosis, patient self-collection of urethral, vulvovaginal, rectal, or pharyngeal specimens is as accurate as clinician collected samples and is better tolerated.8 Because of variation in diagnostic assays, we recommend contacting the laboratory before sending non-standard samples to ensure accurate collection and analysis.
Guidelines for preventing and screening for STIs
There are no prevention guidelines for STIs specific to the psychiatric population, although there is a clear need for focused intervention in this vulnerable patient group.10 Rates of STI screening generally are low in the psychiatric setting,11 which results in a considerable burden of disease. All psychiatric patients should be encouraged to engage with STI screening programs that are in line with national guidelines. In the inpatient psychiatric or medical environment, clinicians have a responsibility to ensure that STI screening is considered for each patient.
Patients with mental illness should be assumed to be sexually active, even if they do not volunteer this information to clinicians. Employ a low threshold for recommending safer sex practices including condom use. Encourage women to develop a relationship with a family practitioner, internist, or gynecologist. Advise men who have sex with men (MSM) to visit a doctor regularly for screening of HIV and rectal, anal, and oral STIs as behavior and symptoms dictate.
There is general agreement about STI screening among the United States Preventive Services Task Force (USPSTF), CDC, American Academy of Family Physicians, American Academy of Pediatrics, and American College of Obstetricians and Gynecologists. USPSTF guidelines are summarized in Table 3.12
In addition to these guidelines, the CDC suggests that all adults and adolescents be tested at least once for HIV.13 The CDC also recommends annual testing of MSM for HIV, syphilis, chlamydia, and gonorrhea. In MSM who have multiple partners or who have sex while using illicit drugs, testing should occur more frequently, such as every 3 to 6 months.14
HPV. Routine HPV screening is not recommended; however, 2 vaccines are available to prevent oncogenic HPV (types 16 and 18). All females age 13 to 26 should receive 3 doses of HPV vaccine over a 6-month period. The quadrivalent vaccine (Gardasil) also protects against HPV types 6 and 11, which cause 90% of genital warts and is preferred when available. Males age 9 to 26 also can receive the vaccine, although ideally it should be administered before sexual activity begins.15 Women still should attend routine cervical cancer screening even if they have the vaccine because 30% of cervical cancers are not caused by HPV 16/18. However, this means that 70% of cervical cancers are associated with HPV 16/18, making screening and the vaccine an important public health initiative. There also is a link between HPV and oral cancers.
Treating STIs among mentally ill individuals
Treatment of STIs among mentally ill individuals is important to prevent medical complications and to reduce transmission. Here are a few additional questions to keep in mind when treating a patient with psychiatric illness:
Does the patient have a primary psychiatric disorder, or is the patient’s current psychiatric presentation a result of the infection?
Some STIs can manifest with psychiatric symptoms—for example, neurosyphilis and HIV-associated neurocognitive disorders—and pose a diagnostic challenge. Obtaining a longitudinal history of the patient’s mental health, age of onset, and family history can help clarify the cause.
Are there any psychiatric adverse effects of STI treatment?
Most drugs used for treating common STIs are not known to cause psychiatric adverse effects (See the American Psychiatric Association16 and Sockalingham et al17 for a thorough discussion of HIV and hepatitis C treatment). The exception is fluoroquinolones, which could be prescribed for PID if cephalosporin therapy is not feasible. CNS effects of fluoroquinolones include insomnia, restlessness, confusion, and, in rare cases, mania and psychosis.
What are possible medication interactions to keep in mind when treating a psychiatric patient?
Nonsteroidal anti-inflammatory drugs (NSAIDs), other than sulindac, could increase serum lithium levels. Although NSAIDs are not contraindicated in patients taking lithium, other pain relievers, such as acetaminophen, may be preferred as a first-line choice.
Carbamazepine could lower serum levels of doxycycline.18
Azithromycin and other macrolides, as well as fluoroquinolones, could have QTc prolonging effects and has been associated with torsades de pointes.19 Several psychiatric medications, in particular, atypical antipsychotics, also could prolong the QTc interval. This could be a consideration in patients with underlying long QT intervals at baseline or a family history of sudden cardiac death.
Psychiatric patients might refuse or not adhere to their medication. Refusals could be the result of grandiose delusions (“I don’t need treatment”) or paranoia (“The doctor is trying to poison me”). Consider 1-time doses of antibiotics that can be given in the clinic for uncomplicated infections when adherence is an issue. Because psychiatric patients are at higher risk for acquiring STIs, education and counseling—especially substance abuse counseling—are vital as both primary and secondary prevention strategies. Treatment of STIs should be accompanied by referrals to the social work team or a therapist when appropriate.
Finally, as with any proposed treatment, it is important to consider whether the patient has capacity to consent to or refuse treatment. To assess for capacity, a patient must be able to:
- communicate a choice
- understand the relevant information
- appreciate the medical consequences of the decision
- demonstrate the ability to reason about treatment choices.20
Case continued
In the emergency department, Ms. K’s vital signs are: temperature 39.5°C; pulse 110 beats per minute; blood pressure 96/67 mm Hg; and breathing 20 respirations per minute. She complains of nausea and has 2 episodes of emesis. She allows clinicians to perform a complete physical examination, including pelvic exam. Her cervix is inflamed, and she is noted to have adnexal and cervical motion tenderness.
Labs and imaging confirm a diagnosis of PID due to gonorrhea and she is admitted to the hospital for IV antibiotics. She continues to experience nausea and vomiting, but also complains of dizziness and diarrhea. Her speech is slurred and a coarse tremor is noticed in her hands. Renal function tests show slight impairment, probably due to dehydration. A pregnancy test is negative.
Lithium is held. Her nausea, vomiting, and diarrhea resolve quickly, and Ms. K asks to leave. When she is told that she is not ready for discharge, Ms. K becomes upset and rips out her IV yelling, “I don’t need treatment from you guys!” A psychiatry consult is called to assess for her capacity to refuse treatment. The team determines that she has capacity, but she becomes agreeable to remaining in the hospital after a phone conversation with her community mental health team.
Ms. K improves with antibiotic treatment. HIV and syphilis serology tests are negative. Before discharge, both the community psychiatrist and her primary care physicians are informed her lithium was held during hospitalization and restarted before discharge. Ms. K also is educated about the signs and symptoms of lithium toxicity, as well as common STIs.
Clinical considerations
- Physicians should have a low threshold of suspicion for PID in a sexually active young woman who presents with abdominal pain and shuffling gait, which is a natural attempt to reduce cervical irritation and is associated with PID.
- Ask about sexual history and symptoms of STIs.
- Rule out STIs in men presenting with urinary tract infections.
- If chlamydia is diagnosed, treatment for gonorrhea also is essential, and vice versa.
- Always think about HIV and hepatatis B and C in a patient with a STI.
- Treatment with single-dose medications can be effective.
- Risk of STIs is higher during episodes of mania or psychosis.
- Consider hospitalization if medically indicated or if you suspect non-adherence to therapy. It is important to remember that all kinds of systemic infections—including PID—can result in dehydration and alter renal metabolism leading to lithium accumulation.
- Mentally ill patients might require placement under involuntary commitment if they are found to be a danger to themselves or others. It is important to liaise with both the community psychiatry team and primary care physician both during hospitalization and before discharge to ensure a smooth transition.
1. Fenton KA, Lowndes CM. Recent trends in the epidemiology of sexually transmitted infections in the European Union. Sex Transm Infect. 2004;80(4):255-263.
2. Trigg BG, Kerndt PR, Aynalem G. Sexually transmitted infections and pelvic inflammatory disease in women. Med Clin North Am. 2008;92(5):1083-1113, x.
3. Frenkl TL, Potts J. Sexually transmitted infections. Urol Clin North Am. 2008;35(1):33-46; vi.
4. Weinstock H, Berman S, Cates W Jr. Sexually transmitted diseases among American youth: incidence and prevalence estimates, 2000. Perspect Sex Reprod Health. 2004;36(1):6-10.
5. Rosenberg SD, Goodman LA, Osher FC, et al. Prevalence of HIV, hepatitis B, and hepatitis C in people with severe mental illness. Am J Public Health. 2001;91(1):31-37.
6. King C, Feldman J, Waithaka Y, et al. Sexual risk behaviors and sexually transmitted infection prevalence in an outpatient psychiatry clinic. Sex Transm Dis. 2008;35(10):877-882.
7. Erbelding EJ, Hutton HE, Zenilman JM, et al. The prevalence of psychiatric disorders in sexually transmitted disease clinic patients and their association with sexually transmitted disease risk. Sex Transm Dis. 2004;31(1):8-12.
8. Freeman AH, Bernstein KT, Kohn RP, et al. Evaluation of self-collected versus clinician-collected swabs for the detection of Chlamydia trachomatis and Neisseria gonorrhoeae pharyngeal infection among men who have sex with men. Sex Transm Dis. 2011;38(11):1036-1039.
9. Workowski KA, Berman S; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2010. MMWR Recomm Rep. 2010;59(RR-12):1-110.
10. Rein DB, Anderson LA, Irwin KL. Mental health disorders and sexually transmitted diseases in a privately insured population. Am J Manag Care. 2004;10(12):917-924.
11. Rothbard AB, Blank MB, Staab JP, et al. Previously undetected metabolic syndromes and infectious diseases among psychiatric inpatients. Psychiatr Serv. 2009;60(4):534-537.
12. Meyers D, Wolff T, Gregory K, et al. USPSTF recommendations for STI screening. Am Fam Physician. 2008;77(6):819-824.
13. Branson BM, Handsfield HH, Lampe MA, et al; Centers for Disease Control and Prevention (CDC). Revised recommendations for HIV testing of adults, adolescents, and pregnant women in health-care settings. MMWR Recomm Rep. 2006;55(RR-14):1-17; quiz CE1-CE 4.
14. Centers for Disease Control and Prevention. Incidence, prevalence, and cost of sexually transmitted infections in the United States. https://npin.cdc.gov/publication/incidence-prevalence-and-cost-sexually-transmitted-infections-united-states. Published February 2013. Accessed December 12, 2016.
15. Centers for Disease Control and Prevention (CDC). Recommendations on the use of quadrivalent human papillomavirus vaccine in males—Advisory Committee on Immunization Practices (ACIP), 2011. MMWR Morb Mortal Wkly Rep. 2011;60(50):1705-1708.
16. American Psychiatric Association. HIV psychiatry. https://www.psychiatry.org/psychiatrists/practice/professional-interests/hiv-psychiatry. Accessed December 13, 2016.
17. Sockalingam S, Sheehan K, Feld JJ, et al. Psychiatric care during hepatitis C treatment: the changing role of psychiatrists in the era of direct-acting antivirals. Am J Psychiatry. 2015;172(6):512-516.
18. Neuvonen PJ, Pentikäinen PJ, Gothoni G. Inhibition of iron absorption by tetracycline. Br J Clin Pharmacol. 1975;2(1):94-96.
19. Sears SP, Getz TW, Austin CO, et al. Incidence of sustained ventricular tachycardia in patients with prolonged QTc after the administration of azithromycin: a retrospective study. Drugs Real World Outcomes. 2016;3:99-105.
20. Appelbaum PS. Clinical practice. Assessment of patients’ competence to consent to treatment. N Engl J Med. 2007;357(18):1834-1840.
1. Fenton KA, Lowndes CM. Recent trends in the epidemiology of sexually transmitted infections in the European Union. Sex Transm Infect. 2004;80(4):255-263.
2. Trigg BG, Kerndt PR, Aynalem G. Sexually transmitted infections and pelvic inflammatory disease in women. Med Clin North Am. 2008;92(5):1083-1113, x.
3. Frenkl TL, Potts J. Sexually transmitted infections. Urol Clin North Am. 2008;35(1):33-46; vi.
4. Weinstock H, Berman S, Cates W Jr. Sexually transmitted diseases among American youth: incidence and prevalence estimates, 2000. Perspect Sex Reprod Health. 2004;36(1):6-10.
5. Rosenberg SD, Goodman LA, Osher FC, et al. Prevalence of HIV, hepatitis B, and hepatitis C in people with severe mental illness. Am J Public Health. 2001;91(1):31-37.
6. King C, Feldman J, Waithaka Y, et al. Sexual risk behaviors and sexually transmitted infection prevalence in an outpatient psychiatry clinic. Sex Transm Dis. 2008;35(10):877-882.
7. Erbelding EJ, Hutton HE, Zenilman JM, et al. The prevalence of psychiatric disorders in sexually transmitted disease clinic patients and their association with sexually transmitted disease risk. Sex Transm Dis. 2004;31(1):8-12.
8. Freeman AH, Bernstein KT, Kohn RP, et al. Evaluation of self-collected versus clinician-collected swabs for the detection of Chlamydia trachomatis and Neisseria gonorrhoeae pharyngeal infection among men who have sex with men. Sex Transm Dis. 2011;38(11):1036-1039.
9. Workowski KA, Berman S; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2010. MMWR Recomm Rep. 2010;59(RR-12):1-110.
10. Rein DB, Anderson LA, Irwin KL. Mental health disorders and sexually transmitted diseases in a privately insured population. Am J Manag Care. 2004;10(12):917-924.
11. Rothbard AB, Blank MB, Staab JP, et al. Previously undetected metabolic syndromes and infectious diseases among psychiatric inpatients. Psychiatr Serv. 2009;60(4):534-537.
12. Meyers D, Wolff T, Gregory K, et al. USPSTF recommendations for STI screening. Am Fam Physician. 2008;77(6):819-824.
13. Branson BM, Handsfield HH, Lampe MA, et al; Centers for Disease Control and Prevention (CDC). Revised recommendations for HIV testing of adults, adolescents, and pregnant women in health-care settings. MMWR Recomm Rep. 2006;55(RR-14):1-17; quiz CE1-CE 4.
14. Centers for Disease Control and Prevention. Incidence, prevalence, and cost of sexually transmitted infections in the United States. https://npin.cdc.gov/publication/incidence-prevalence-and-cost-sexually-transmitted-infections-united-states. Published February 2013. Accessed December 12, 2016.
15. Centers for Disease Control and Prevention (CDC). Recommendations on the use of quadrivalent human papillomavirus vaccine in males—Advisory Committee on Immunization Practices (ACIP), 2011. MMWR Morb Mortal Wkly Rep. 2011;60(50):1705-1708.
16. American Psychiatric Association. HIV psychiatry. https://www.psychiatry.org/psychiatrists/practice/professional-interests/hiv-psychiatry. Accessed December 13, 2016.
17. Sockalingam S, Sheehan K, Feld JJ, et al. Psychiatric care during hepatitis C treatment: the changing role of psychiatrists in the era of direct-acting antivirals. Am J Psychiatry. 2015;172(6):512-516.
18. Neuvonen PJ, Pentikäinen PJ, Gothoni G. Inhibition of iron absorption by tetracycline. Br J Clin Pharmacol. 1975;2(1):94-96.
19. Sears SP, Getz TW, Austin CO, et al. Incidence of sustained ventricular tachycardia in patients with prolonged QTc after the administration of azithromycin: a retrospective study. Drugs Real World Outcomes. 2016;3:99-105.
20. Appelbaum PS. Clinical practice. Assessment of patients’ competence to consent to treatment. N Engl J Med. 2007;357(18):1834-1840.

Maddening therapies: How hallucinogens morphed into novel treatments
Snake venom is deadly but is being used to treat some cancers,1 because it produces contortrostatin, a protein that “paralyzes” cancer cells and prevents them from migrating. Venoms from spiders are being investigated as a treatment to slow the progression of muscular dystrophy by preventing muscle cells from deteriorating. Venom from tarantulas can relieve chronic pain, and those from centipedes help rodents tolerate thermal, chemical, or acid pain. Scorpion venom can cause cancer cells to glow under a flashlight, enabling surgeons to locate and remove them. Anemones toxin could be used to treat autoimmune diseases, such as rheumatoid arthritis, multiple sclerosis, and lupus.
Vaccines are an excellent example of how deadly pathogens can be transformed into life-saving therapies. Billions of people have been protected from polio, smallpox, tetanus, diphtheria, measles, mumps, rubella, influenza, pneumococcus, hepatitis A and B, rabies, shingles, typhoid, meningitis, or cholera. Turning killers into saviors is one of the most remarkable miracles of medical research.2
The mind-boggling transformation of mind-altering drugs
In psychiatry, psychedelic drugs have been repurposed into useful therapies for mental illness. As recently as a decade ago, psychiatric practitioners—physicians and nurse practitioners—regarded hallucinogens as dangerous, “must-avoid” drugs of abuse that could trigger or exacerbate serious psychiatric disorders. Then, thanks to ongoing research, the psychedelic “caterpillars” transformed into therapeutic “butterflies,” and the despised drugs of abuse became welcome adjuncts for treating some stubborn psychopathologies. Such paradoxical developments are emblematic of how one can always find a silver lining.
Consider the following transformations of various psychedelics and hallucinogens—also called “entheogens”—into novel pharmacotherapies. Note that in most cases, the application of these mind-altering drugs into useful medications is still a work in progress.
LSD
Lysergic acid diethylamide (LSD) was used extensively for treating mood disorders in the pre-antidepressant era, before it was prohibited in the late 1960s. A review of 19 studies—many uncontrolled—concluded that approximately 80% of patients improved, according to the treating physicians.3 However, research on LSD was halted for several decades after it became illegal, and resumed in 2010. Neuropsychiatrists and neuroscience researchers are now employing advanced techniques, such as neuroimaging, molecular pharmacology, and connectomics, to study its therapeutic effects.4 LSD is not only being used for treatment-resistant depression but also anxiety, alcoholism, autism, and even schizophrenia. However, despite its potential uses for treating alcoholism and anxiety, enhancing creativity, or caring for terminally ill patients, using LSD requires expertise, caution, and adherence to ethical standards.5
In healthy individuals, the effects of LSD include visual hallucinations, audiovisual synesthesia, depersonalization and derealization, and a sense of well-being, happiness, closeness to others, and trust.
Biologic effects include increased heart rate and blood pressure, elevated temperature, dilated pupils, and increased serum cortisol, prolactin, oxytocin, and epinephrine. All effects subside within 3 days.6
Psilocybin
Psilocybin, a component of some mushrooms that is known for its use during rituals in some cultures, has been discovered to have antidepressant, anxiolytic, and anti-addictive effects.7 Recent controlled studies at Johns Hopkins University reported that a single dose of psilocybin can relieve anxiety or depression for up to 6 months, which, if replicated, could lead to a remarkable paradigm shift in treating mood and anxiety disorders, especially if patients do not respond to standard antidepressants.3 Other emerging uses of both psilocybin and LSD are in treating addictions8 where psychiatry is desperately looking for innovative new therapies.
Ecstasy
MDMA (3,4-methylenedioxymethamphetamine), also known as ecstasy, is widely regarded as a harmful party drug that produces euphoria, but not hallucinations. However, it has emerged as a useful treatment for posttraumatic stress disorder (PTSD). In one study of female sexual abuse victims, 80% of the patients who received MDMA with psychotherapy no longer met diagnostic criteria for PTSD after 2 months.9 Other studies showed no effects. Despite persistent skepticisms by many, the Multidisciplinary Association for Psychedelics Studies organization is investing millions of dollars into studying MDMA for PTSD in several countries.9,10 One hurdle is that it is difficult to conduct truly blind studies with psychedelic drugs because of their profound effects. MDMA releases cortisol, oxytocin—which are known to facilitate psychotherapy—and testosterone, but the debate about the risk–benefit ratio will continue.11 MDMA also is being studied for treating social anxiety in adults with autism.12
Ketamine
Ketamine is a weaker cousin of the potent psychotogenic phencyclidine (approximately one-fiftieth the potency) and is a well-known drug of abuse that causes dissociation and hallucinations. It is used as an anesthetic in veterinary medicine and in children undergoing surgical procedures. Until recently, its only use in psychiatry has been as an anesthetic during electroconvulsive therapy. However, over the past few years, IV ketamine has been in the spotlight as a breakthrough, rapid-onset antidepressant and anti-suicidal agent in several controlled studies.13 This drug is revolutionizing the management of treatment-resistant depression and suicidal ideation and generating new insights into the neurobiology of depression.
Cannabis
Last, but certainly not least, is marijuana, which is more widely used than all the other psychedelics combined, and is currently at the center of a national debate about its legalization. Although the director of the National Institute on Drug Abuse highlighted the many risk of marijuana,14 studies have pointed to the myriad medical uses of Cannabis.15,16 An editorial in Nature Medicine recently urged that regulators reconsider the tight constraints on marijuana research.17 Some of the medical applications of marijuana include:
- psychiatry (anxiety, PTSD)
- neurology (severe epilepsy, tremors in Parkinson’s disease, traumatic brain injury, pain of multiple sclerosis, muscle spasms, and progression of Alzheimer’s disease)
- oncology (nausea and pain of chemotherapy, reduction of metastasis)
- ophthalmology (decrease of intraocular pressure in glaucoma)
- autoimmune disorders (rheumatoid arthritis, Crohn’s disease, lupus).
However, as a schizophrenia researcher, I am wary about marijuana’s high risk of triggering psychosis in young adults with a family history of schizophrenia spectrum disorders.18
The above are examples of how psychiatry is finally recognizing the therapeutic value inherent in traditionally “evil” street drugs that we euphemistically refer to as “recreational drugs.” Even methamphetamine, the universally condemned and clearly harmful drug, was recently reported to be neuroprotective at low dosages!19 Could our field have suffered from a blind eye to the benefits of these hallucinogens and ignored the possibility that some persons with addiction who use these “recreational drugs” may have been self-medicating to alleviate their un-diagnosed psychiatric disorder? We need to reconceptualize the pejorative term “mind-altering drug” because of its implicitly negative connotation. After all, alteration may indicate a favorable, not just a deleterious, outcome.
1. Vyas VK, Brahmbhatt K, Bhatt H, et al. Therapeutic potential of snake venom in cancer therapy: current perspectives. Asian Pac J Trop Biomed. 2013;3(2):156-162.
2. Loehr J. The vaccine answer book: 200 essential answers to help you make the right decisions for your child. Naperville, IL: Sourcebooks Inc; 2009.
3. Rucker JJ, Jelen LA, Flynn S, et al. Psychedelics in the treatment of unipolar mood disorders: a systematic review. J Psychopharmacol. 2016;30(12):1220-1229.
4. Mucke HA. From psychiatry to flower power and back again: the amazing story of lysergic acid diethylamide [published online July 8, 2016]. Assay Drug Dev Technol. doi: 10.1089/adt.2016.747.
5. Das S, Barnwal P, Ramasamy A, et al. Lysergic acid diethylamide: a drug of ‘use’? Ther Advances Pychopharmacol. 2016;6(3):214-228.
6. Schmid Y, Enzler F, Gasser P, et al. Acute effects of lysergic acid diethylamide in healthy subjects. Biol Psychiatry. 2015;78(8):544-553.
7. Dos Santos RG, Osório FL, Crippa JA, et al. Antidepressive, anxiolytic, and antiaddictive effects of ayahuasca, psilocybin and lysergic acid diethylamide (LSD): a systematic review of clinical trials published in the last 25 years. Ther Adv Psychopharmacol. 2016;6(3):193-213.
8. Bogenschutz MP. Studying the effects of classic hallucinogens in the treatment of alcoholism: rationale, methodology, and current research with psilocybin. Curr Drug Abuse Rev. 2013;6(1):17-29.
9. Kupferschmidt K. Can ecstasy treat the agony of PTSD? Science. 2014;345:22-23.
10. Sessa B. MDMA and PTSD treatment: PTSD: from novel pathophysiology to innovative therapeutics [published online July 6, 2016]. Neurosci Lett. doi: 10.1016/j.neulet.2016.07.004.
11. Parrott AC. The potential dangers of using MDMA for psychotherapy. J Psychoactive Drugs. 2014;46(1):37-43.
12. Danforth AL, Struble CM, Yazar-Klosinski B, et al. MDMA-assisted therapy: a new treatment model for social anxiety in autistic adults. Prog Neuropsychopharmacol Biol Psychiatry. 2016;64:237-249.
13. Feifel D. Breaking sad: unleashing the breakthrough potential of ketamine’s rapid antidepressant effects [published online November 26, 2016]. Drug Dev Res. doi: 10.1002/ddr.21347.
14. Volkow ND, Baler RD, Compton WM, et al. Adverse health effects of marijuana use. N Engl J Med. 2014;370(23):2219-2227.
15. Murnion B. Medicinal cannabis. Aust Prescr. 2015;38(6):212-215.
16. Borgelt LM, Franson KL, Nussbaum AM, et al. The pharmacologic and clinical effects of medical cannabis. Pharmacotherapy. 2013;33(2):195-209.
17. Release the strains. Nat Med. 2015;21(9):963.
18. Moore TH, Zammit S, Lingford-Hughes A, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet. 2007;370(9584):319-328.
19. Rau T, Ziemniak J, Poulsen D, et al. The neuroprotective potential of low-dose methamphetamine in preclinical models of stroke and traumatic brain injury. Prog Neuropsychopharmacol Biol Psychiatry. 2016;64:231-236.
Snake venom is deadly but is being used to treat some cancers,1 because it produces contortrostatin, a protein that “paralyzes” cancer cells and prevents them from migrating. Venoms from spiders are being investigated as a treatment to slow the progression of muscular dystrophy by preventing muscle cells from deteriorating. Venom from tarantulas can relieve chronic pain, and those from centipedes help rodents tolerate thermal, chemical, or acid pain. Scorpion venom can cause cancer cells to glow under a flashlight, enabling surgeons to locate and remove them. Anemones toxin could be used to treat autoimmune diseases, such as rheumatoid arthritis, multiple sclerosis, and lupus.
Vaccines are an excellent example of how deadly pathogens can be transformed into life-saving therapies. Billions of people have been protected from polio, smallpox, tetanus, diphtheria, measles, mumps, rubella, influenza, pneumococcus, hepatitis A and B, rabies, shingles, typhoid, meningitis, or cholera. Turning killers into saviors is one of the most remarkable miracles of medical research.2
The mind-boggling transformation of mind-altering drugs
In psychiatry, psychedelic drugs have been repurposed into useful therapies for mental illness. As recently as a decade ago, psychiatric practitioners—physicians and nurse practitioners—regarded hallucinogens as dangerous, “must-avoid” drugs of abuse that could trigger or exacerbate serious psychiatric disorders. Then, thanks to ongoing research, the psychedelic “caterpillars” transformed into therapeutic “butterflies,” and the despised drugs of abuse became welcome adjuncts for treating some stubborn psychopathologies. Such paradoxical developments are emblematic of how one can always find a silver lining.
Consider the following transformations of various psychedelics and hallucinogens—also called “entheogens”—into novel pharmacotherapies. Note that in most cases, the application of these mind-altering drugs into useful medications is still a work in progress.
LSD
Lysergic acid diethylamide (LSD) was used extensively for treating mood disorders in the pre-antidepressant era, before it was prohibited in the late 1960s. A review of 19 studies—many uncontrolled—concluded that approximately 80% of patients improved, according to the treating physicians.3 However, research on LSD was halted for several decades after it became illegal, and resumed in 2010. Neuropsychiatrists and neuroscience researchers are now employing advanced techniques, such as neuroimaging, molecular pharmacology, and connectomics, to study its therapeutic effects.4 LSD is not only being used for treatment-resistant depression but also anxiety, alcoholism, autism, and even schizophrenia. However, despite its potential uses for treating alcoholism and anxiety, enhancing creativity, or caring for terminally ill patients, using LSD requires expertise, caution, and adherence to ethical standards.5
In healthy individuals, the effects of LSD include visual hallucinations, audiovisual synesthesia, depersonalization and derealization, and a sense of well-being, happiness, closeness to others, and trust.
Biologic effects include increased heart rate and blood pressure, elevated temperature, dilated pupils, and increased serum cortisol, prolactin, oxytocin, and epinephrine. All effects subside within 3 days.6
Psilocybin
Psilocybin, a component of some mushrooms that is known for its use during rituals in some cultures, has been discovered to have antidepressant, anxiolytic, and anti-addictive effects.7 Recent controlled studies at Johns Hopkins University reported that a single dose of psilocybin can relieve anxiety or depression for up to 6 months, which, if replicated, could lead to a remarkable paradigm shift in treating mood and anxiety disorders, especially if patients do not respond to standard antidepressants.3 Other emerging uses of both psilocybin and LSD are in treating addictions8 where psychiatry is desperately looking for innovative new therapies.
Ecstasy
MDMA (3,4-methylenedioxymethamphetamine), also known as ecstasy, is widely regarded as a harmful party drug that produces euphoria, but not hallucinations. However, it has emerged as a useful treatment for posttraumatic stress disorder (PTSD). In one study of female sexual abuse victims, 80% of the patients who received MDMA with psychotherapy no longer met diagnostic criteria for PTSD after 2 months.9 Other studies showed no effects. Despite persistent skepticisms by many, the Multidisciplinary Association for Psychedelics Studies organization is investing millions of dollars into studying MDMA for PTSD in several countries.9,10 One hurdle is that it is difficult to conduct truly blind studies with psychedelic drugs because of their profound effects. MDMA releases cortisol, oxytocin—which are known to facilitate psychotherapy—and testosterone, but the debate about the risk–benefit ratio will continue.11 MDMA also is being studied for treating social anxiety in adults with autism.12
Ketamine
Ketamine is a weaker cousin of the potent psychotogenic phencyclidine (approximately one-fiftieth the potency) and is a well-known drug of abuse that causes dissociation and hallucinations. It is used as an anesthetic in veterinary medicine and in children undergoing surgical procedures. Until recently, its only use in psychiatry has been as an anesthetic during electroconvulsive therapy. However, over the past few years, IV ketamine has been in the spotlight as a breakthrough, rapid-onset antidepressant and anti-suicidal agent in several controlled studies.13 This drug is revolutionizing the management of treatment-resistant depression and suicidal ideation and generating new insights into the neurobiology of depression.
Cannabis
Last, but certainly not least, is marijuana, which is more widely used than all the other psychedelics combined, and is currently at the center of a national debate about its legalization. Although the director of the National Institute on Drug Abuse highlighted the many risk of marijuana,14 studies have pointed to the myriad medical uses of Cannabis.15,16 An editorial in Nature Medicine recently urged that regulators reconsider the tight constraints on marijuana research.17 Some of the medical applications of marijuana include:
- psychiatry (anxiety, PTSD)
- neurology (severe epilepsy, tremors in Parkinson’s disease, traumatic brain injury, pain of multiple sclerosis, muscle spasms, and progression of Alzheimer’s disease)
- oncology (nausea and pain of chemotherapy, reduction of metastasis)
- ophthalmology (decrease of intraocular pressure in glaucoma)
- autoimmune disorders (rheumatoid arthritis, Crohn’s disease, lupus).
However, as a schizophrenia researcher, I am wary about marijuana’s high risk of triggering psychosis in young adults with a family history of schizophrenia spectrum disorders.18
The above are examples of how psychiatry is finally recognizing the therapeutic value inherent in traditionally “evil” street drugs that we euphemistically refer to as “recreational drugs.” Even methamphetamine, the universally condemned and clearly harmful drug, was recently reported to be neuroprotective at low dosages!19 Could our field have suffered from a blind eye to the benefits of these hallucinogens and ignored the possibility that some persons with addiction who use these “recreational drugs” may have been self-medicating to alleviate their un-diagnosed psychiatric disorder? We need to reconceptualize the pejorative term “mind-altering drug” because of its implicitly negative connotation. After all, alteration may indicate a favorable, not just a deleterious, outcome.
Snake venom is deadly but is being used to treat some cancers,1 because it produces contortrostatin, a protein that “paralyzes” cancer cells and prevents them from migrating. Venoms from spiders are being investigated as a treatment to slow the progression of muscular dystrophy by preventing muscle cells from deteriorating. Venom from tarantulas can relieve chronic pain, and those from centipedes help rodents tolerate thermal, chemical, or acid pain. Scorpion venom can cause cancer cells to glow under a flashlight, enabling surgeons to locate and remove them. Anemones toxin could be used to treat autoimmune diseases, such as rheumatoid arthritis, multiple sclerosis, and lupus.
Vaccines are an excellent example of how deadly pathogens can be transformed into life-saving therapies. Billions of people have been protected from polio, smallpox, tetanus, diphtheria, measles, mumps, rubella, influenza, pneumococcus, hepatitis A and B, rabies, shingles, typhoid, meningitis, or cholera. Turning killers into saviors is one of the most remarkable miracles of medical research.2
The mind-boggling transformation of mind-altering drugs
In psychiatry, psychedelic drugs have been repurposed into useful therapies for mental illness. As recently as a decade ago, psychiatric practitioners—physicians and nurse practitioners—regarded hallucinogens as dangerous, “must-avoid” drugs of abuse that could trigger or exacerbate serious psychiatric disorders. Then, thanks to ongoing research, the psychedelic “caterpillars” transformed into therapeutic “butterflies,” and the despised drugs of abuse became welcome adjuncts for treating some stubborn psychopathologies. Such paradoxical developments are emblematic of how one can always find a silver lining.
Consider the following transformations of various psychedelics and hallucinogens—also called “entheogens”—into novel pharmacotherapies. Note that in most cases, the application of these mind-altering drugs into useful medications is still a work in progress.
LSD
Lysergic acid diethylamide (LSD) was used extensively for treating mood disorders in the pre-antidepressant era, before it was prohibited in the late 1960s. A review of 19 studies—many uncontrolled—concluded that approximately 80% of patients improved, according to the treating physicians.3 However, research on LSD was halted for several decades after it became illegal, and resumed in 2010. Neuropsychiatrists and neuroscience researchers are now employing advanced techniques, such as neuroimaging, molecular pharmacology, and connectomics, to study its therapeutic effects.4 LSD is not only being used for treatment-resistant depression but also anxiety, alcoholism, autism, and even schizophrenia. However, despite its potential uses for treating alcoholism and anxiety, enhancing creativity, or caring for terminally ill patients, using LSD requires expertise, caution, and adherence to ethical standards.5
In healthy individuals, the effects of LSD include visual hallucinations, audiovisual synesthesia, depersonalization and derealization, and a sense of well-being, happiness, closeness to others, and trust.
Biologic effects include increased heart rate and blood pressure, elevated temperature, dilated pupils, and increased serum cortisol, prolactin, oxytocin, and epinephrine. All effects subside within 3 days.6
Psilocybin
Psilocybin, a component of some mushrooms that is known for its use during rituals in some cultures, has been discovered to have antidepressant, anxiolytic, and anti-addictive effects.7 Recent controlled studies at Johns Hopkins University reported that a single dose of psilocybin can relieve anxiety or depression for up to 6 months, which, if replicated, could lead to a remarkable paradigm shift in treating mood and anxiety disorders, especially if patients do not respond to standard antidepressants.3 Other emerging uses of both psilocybin and LSD are in treating addictions8 where psychiatry is desperately looking for innovative new therapies.
Ecstasy
MDMA (3,4-methylenedioxymethamphetamine), also known as ecstasy, is widely regarded as a harmful party drug that produces euphoria, but not hallucinations. However, it has emerged as a useful treatment for posttraumatic stress disorder (PTSD). In one study of female sexual abuse victims, 80% of the patients who received MDMA with psychotherapy no longer met diagnostic criteria for PTSD after 2 months.9 Other studies showed no effects. Despite persistent skepticisms by many, the Multidisciplinary Association for Psychedelics Studies organization is investing millions of dollars into studying MDMA for PTSD in several countries.9,10 One hurdle is that it is difficult to conduct truly blind studies with psychedelic drugs because of their profound effects. MDMA releases cortisol, oxytocin—which are known to facilitate psychotherapy—and testosterone, but the debate about the risk–benefit ratio will continue.11 MDMA also is being studied for treating social anxiety in adults with autism.12
Ketamine
Ketamine is a weaker cousin of the potent psychotogenic phencyclidine (approximately one-fiftieth the potency) and is a well-known drug of abuse that causes dissociation and hallucinations. It is used as an anesthetic in veterinary medicine and in children undergoing surgical procedures. Until recently, its only use in psychiatry has been as an anesthetic during electroconvulsive therapy. However, over the past few years, IV ketamine has been in the spotlight as a breakthrough, rapid-onset antidepressant and anti-suicidal agent in several controlled studies.13 This drug is revolutionizing the management of treatment-resistant depression and suicidal ideation and generating new insights into the neurobiology of depression.
Cannabis
Last, but certainly not least, is marijuana, which is more widely used than all the other psychedelics combined, and is currently at the center of a national debate about its legalization. Although the director of the National Institute on Drug Abuse highlighted the many risk of marijuana,14 studies have pointed to the myriad medical uses of Cannabis.15,16 An editorial in Nature Medicine recently urged that regulators reconsider the tight constraints on marijuana research.17 Some of the medical applications of marijuana include:
- psychiatry (anxiety, PTSD)
- neurology (severe epilepsy, tremors in Parkinson’s disease, traumatic brain injury, pain of multiple sclerosis, muscle spasms, and progression of Alzheimer’s disease)
- oncology (nausea and pain of chemotherapy, reduction of metastasis)
- ophthalmology (decrease of intraocular pressure in glaucoma)
- autoimmune disorders (rheumatoid arthritis, Crohn’s disease, lupus).
However, as a schizophrenia researcher, I am wary about marijuana’s high risk of triggering psychosis in young adults with a family history of schizophrenia spectrum disorders.18
The above are examples of how psychiatry is finally recognizing the therapeutic value inherent in traditionally “evil” street drugs that we euphemistically refer to as “recreational drugs.” Even methamphetamine, the universally condemned and clearly harmful drug, was recently reported to be neuroprotective at low dosages!19 Could our field have suffered from a blind eye to the benefits of these hallucinogens and ignored the possibility that some persons with addiction who use these “recreational drugs” may have been self-medicating to alleviate their un-diagnosed psychiatric disorder? We need to reconceptualize the pejorative term “mind-altering drug” because of its implicitly negative connotation. After all, alteration may indicate a favorable, not just a deleterious, outcome.
1. Vyas VK, Brahmbhatt K, Bhatt H, et al. Therapeutic potential of snake venom in cancer therapy: current perspectives. Asian Pac J Trop Biomed. 2013;3(2):156-162.
2. Loehr J. The vaccine answer book: 200 essential answers to help you make the right decisions for your child. Naperville, IL: Sourcebooks Inc; 2009.
3. Rucker JJ, Jelen LA, Flynn S, et al. Psychedelics in the treatment of unipolar mood disorders: a systematic review. J Psychopharmacol. 2016;30(12):1220-1229.
4. Mucke HA. From psychiatry to flower power and back again: the amazing story of lysergic acid diethylamide [published online July 8, 2016]. Assay Drug Dev Technol. doi: 10.1089/adt.2016.747.
5. Das S, Barnwal P, Ramasamy A, et al. Lysergic acid diethylamide: a drug of ‘use’? Ther Advances Pychopharmacol. 2016;6(3):214-228.
6. Schmid Y, Enzler F, Gasser P, et al. Acute effects of lysergic acid diethylamide in healthy subjects. Biol Psychiatry. 2015;78(8):544-553.
7. Dos Santos RG, Osório FL, Crippa JA, et al. Antidepressive, anxiolytic, and antiaddictive effects of ayahuasca, psilocybin and lysergic acid diethylamide (LSD): a systematic review of clinical trials published in the last 25 years. Ther Adv Psychopharmacol. 2016;6(3):193-213.
8. Bogenschutz MP. Studying the effects of classic hallucinogens in the treatment of alcoholism: rationale, methodology, and current research with psilocybin. Curr Drug Abuse Rev. 2013;6(1):17-29.
9. Kupferschmidt K. Can ecstasy treat the agony of PTSD? Science. 2014;345:22-23.
10. Sessa B. MDMA and PTSD treatment: PTSD: from novel pathophysiology to innovative therapeutics [published online July 6, 2016]. Neurosci Lett. doi: 10.1016/j.neulet.2016.07.004.
11. Parrott AC. The potential dangers of using MDMA for psychotherapy. J Psychoactive Drugs. 2014;46(1):37-43.
12. Danforth AL, Struble CM, Yazar-Klosinski B, et al. MDMA-assisted therapy: a new treatment model for social anxiety in autistic adults. Prog Neuropsychopharmacol Biol Psychiatry. 2016;64:237-249.
13. Feifel D. Breaking sad: unleashing the breakthrough potential of ketamine’s rapid antidepressant effects [published online November 26, 2016]. Drug Dev Res. doi: 10.1002/ddr.21347.
14. Volkow ND, Baler RD, Compton WM, et al. Adverse health effects of marijuana use. N Engl J Med. 2014;370(23):2219-2227.
15. Murnion B. Medicinal cannabis. Aust Prescr. 2015;38(6):212-215.
16. Borgelt LM, Franson KL, Nussbaum AM, et al. The pharmacologic and clinical effects of medical cannabis. Pharmacotherapy. 2013;33(2):195-209.
17. Release the strains. Nat Med. 2015;21(9):963.
18. Moore TH, Zammit S, Lingford-Hughes A, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet. 2007;370(9584):319-328.
19. Rau T, Ziemniak J, Poulsen D, et al. The neuroprotective potential of low-dose methamphetamine in preclinical models of stroke and traumatic brain injury. Prog Neuropsychopharmacol Biol Psychiatry. 2016;64:231-236.
1. Vyas VK, Brahmbhatt K, Bhatt H, et al. Therapeutic potential of snake venom in cancer therapy: current perspectives. Asian Pac J Trop Biomed. 2013;3(2):156-162.
2. Loehr J. The vaccine answer book: 200 essential answers to help you make the right decisions for your child. Naperville, IL: Sourcebooks Inc; 2009.
3. Rucker JJ, Jelen LA, Flynn S, et al. Psychedelics in the treatment of unipolar mood disorders: a systematic review. J Psychopharmacol. 2016;30(12):1220-1229.
4. Mucke HA. From psychiatry to flower power and back again: the amazing story of lysergic acid diethylamide [published online July 8, 2016]. Assay Drug Dev Technol. doi: 10.1089/adt.2016.747.
5. Das S, Barnwal P, Ramasamy A, et al. Lysergic acid diethylamide: a drug of ‘use’? Ther Advances Pychopharmacol. 2016;6(3):214-228.
6. Schmid Y, Enzler F, Gasser P, et al. Acute effects of lysergic acid diethylamide in healthy subjects. Biol Psychiatry. 2015;78(8):544-553.
7. Dos Santos RG, Osório FL, Crippa JA, et al. Antidepressive, anxiolytic, and antiaddictive effects of ayahuasca, psilocybin and lysergic acid diethylamide (LSD): a systematic review of clinical trials published in the last 25 years. Ther Adv Psychopharmacol. 2016;6(3):193-213.
8. Bogenschutz MP. Studying the effects of classic hallucinogens in the treatment of alcoholism: rationale, methodology, and current research with psilocybin. Curr Drug Abuse Rev. 2013;6(1):17-29.
9. Kupferschmidt K. Can ecstasy treat the agony of PTSD? Science. 2014;345:22-23.
10. Sessa B. MDMA and PTSD treatment: PTSD: from novel pathophysiology to innovative therapeutics [published online July 6, 2016]. Neurosci Lett. doi: 10.1016/j.neulet.2016.07.004.
11. Parrott AC. The potential dangers of using MDMA for psychotherapy. J Psychoactive Drugs. 2014;46(1):37-43.
12. Danforth AL, Struble CM, Yazar-Klosinski B, et al. MDMA-assisted therapy: a new treatment model for social anxiety in autistic adults. Prog Neuropsychopharmacol Biol Psychiatry. 2016;64:237-249.
13. Feifel D. Breaking sad: unleashing the breakthrough potential of ketamine’s rapid antidepressant effects [published online November 26, 2016]. Drug Dev Res. doi: 10.1002/ddr.21347.
14. Volkow ND, Baler RD, Compton WM, et al. Adverse health effects of marijuana use. N Engl J Med. 2014;370(23):2219-2227.
15. Murnion B. Medicinal cannabis. Aust Prescr. 2015;38(6):212-215.
16. Borgelt LM, Franson KL, Nussbaum AM, et al. The pharmacologic and clinical effects of medical cannabis. Pharmacotherapy. 2013;33(2):195-209.
17. Release the strains. Nat Med. 2015;21(9):963.
18. Moore TH, Zammit S, Lingford-Hughes A, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet. 2007;370(9584):319-328.
19. Rau T, Ziemniak J, Poulsen D, et al. The neuroprotective potential of low-dose methamphetamine in preclinical models of stroke and traumatic brain injury. Prog Neuropsychopharmacol Biol Psychiatry. 2016;64:231-236.
