User login
What’s next for deep brain stimulation in OCD?
VIENNA – Even though deep brain stimulation has been used to treat obsessive-compulsive disorder in fewer than 300 patients worldwide, the therapy has had a huge impact on understanding of the disorder, Damiaan Denys, MD, PhD, said at the annual congress of the European College of Neuropsychopharmacology.
Indeed, the efficacy of deep brain stimulation (DBS) in the most severe, treatment-refractory cases of OCD casts doubt upon the fundamental construct clinicians have relied upon for decades to comprehend OCD: Namely, that affected patients first experience obsessions, which induce anxiety, which then stimulates compulsions, and engaging in those compulsions brings relief and reward, and then the whole cycle starts over again.

That construct may not actually be true.
“Deep brain stimulation will force us to rethink OCD,” predicted Dr. Denys, professor and head of psychiatry at the University of Amsterdam.
What does DBS change, and is there a temporal order? It varies, the psychiatrist said.
“In some patients, anxiety is the first thing that changes. In others, it starts with obsessions. Compulsions do decrease, but it’s difficult. It takes some time, and often we need supplemental cognitive-behavioral therapy (CBT). What changes, mainly, in my experience, is not symptoms, but something outside of OCD: namely, mood. Deep brain stimulation has a huge impact on mood. That’s an interesting finding, because it suggests the possibility that you can change a psychiatric disorder by changing symptoms that are thought of as being outside the disorder,” Dr. Denys observed.
“Another interesting thing is that deep brain stimulation changes things that are not even within psychiatry. When we ask our patients what has been the most profound impact deep brain stimulation has had on their lives, all of them say, ‘It increases my self-confidence.’ And that’s not something that is included in our scales. We assess patients using the HAM-D [Hamilton Rating Scale for Depression] and Y-BOCS [Yale-Brown Obsessive Compulsive Scale] and other measures, but there are still some very important aspects we are not taking into account and that have a profound effect on symptoms. In this case, using deep brain stimulation, we improve self-confidence and thereby change a whole chain of symptoms,” he continued.
Among the 60 highly refractory OCD patients treated by DBS by Dr. Denys and his colleagues at the Amsterdam center over the past 15 years, 15% were cured as a result.
“I purposely use the word ‘cured,’ because they don’t have obsessive-compulsive symptoms, anymore, which is, of course, extraordinary,” Dr. Denys noted.
An additional 35% of patients had a good response, defined as 60%-80% improvement on the Y-BOCS. Ten percent of patients were partial responders, with a 20%-40% improvement on the Y-BOCS. And 15% were nonresponders.
Closed-loop system coming
DBS entails implantation of electrodes deep within the brain to interrupt dysfunctional brain signals in local areas and across neural networks. This dysfunctional brain activity is expressed as symptoms. To date, DBS has been used in what’s called an open-loop system: Patients report the symptoms they’re experiencing and the clinician then adjusts the electrode settings in order to quell those symptoms. This approach is about to change. The technology has improved vastly since the early days, when the electrodes had four contact points. Now they have 64 contact points, and are capable of sending and receiving electrical signals.
“The next step in deep brain stimulation will be a closed-loop system. We will remove the clinician and the patient, and attempt to use a device capable of recording what happens in the brain and then changing electrical activity in response to the recordings. Our purpose will be to block these brain signals in advance of obsessions and compulsions so patients don’t have these symptoms. It’s technically possible. It has been done in Parkinson’s, and I think it’s the next step in psychiatry,” Dr. Denys said.
Indeed, he and his coinvestigators are planning formal studies of closed-loop DBS. For him, the prospect raises three key questions: What are the neural correlates of OCD symptoms? What about the ethics of implanting a device in the brain which by itself results in different life experiences? And will closed-loop DBS have superior efficacy, compared with open-loop DBS?
“How is the mind rooted in the brain, and how is the brain expressed in the mind? It’s the most fascinating question; it’s why we all love psychiatry, and up until now, there are no answers,” he observed.
Significant progress already has been made on the neural correlates question. Dr. Denys and his colleagues have found that when OCD patients with deep brain electrodes engage in cleaning compulsions, their local field potentials in the striatal area show peaks of roughly 9 Hz in the alpha range and in the beta/low gamma range. These patterns may represent compulsive behavior and likely could be useful in steering a closed-loop system. Also, the Dutch investigators have found that 3- to 8-Hz theta oscillations in local field potentials in the striatal area may represent a neural signature for anxiety and/or obsessions.
Using a closed-loop system, he continued, investigators plan to test two quite different hypotheses about the fundamental nature of OCD. One is that obsessions, anxiety, compulsions, and relief are each separately related to different brain areas. The other hypothesis is that one central brain stimulus drives the chain of symptoms that characterize OCD, and that by identifying and blocking that primary signal, the whole pathologic process can be stopped.
Current status of procedure
While Dr. Denys focused on the near future of DBS for OCD, another speaker at the session, Sina Kohl, PhD, addressed DBS for OCD as it exists today, particularly the who, how, and where.
The “who” is the relatively rare patient with truly refractory OCD after multiple drug trials of agents in different antidepressant classes, one of which should be clomipramine, as well as a failed course of CBT provided by an expert in CBT for OCD, of which there are relatively few. In a study led by investigators at Brown University, Providence, R.I., only 2 of 325 patients with OCD were deemed truly refractory (J Neuropsychiatry Clin Neurosci. 2014 Winter;26[1]:81-6). That sounds about right, according to Dr. Kohl, a psychologist at the University of Cologne, in Germany.

The “how” is to deliver DBS in conjunction with CBT. Response rates are higher at centers where that practice is routine, she added.
The “where” is an unsettled question. In Dr. Kohl’s meta-analysis of 25 published DBS studies, electrode placement in four different DBS target structures produced similar results: the nucleus accumbens, the anterior limb of the internal capsule, the ventral striatum, and the subthalamic nucleus. Stimulation of the inferior thalamic peduncle appeared to achieve better results, but this is a sketchy conclusion based upon two studies totaling just six patients (BMC Psychiatry. 2014 Aug 2;14:214. doi: 10.1186/s12888-014-0214-y).
Recently, Belgian investigators have reported particularly promising results – the best so far – for DBS targeting the bed nucleus of the stria terminalis (Mol Psychiatry. 2016 Sep;21[9]:1272-80).
Bilateral DBS appears to be more effective than unilateral.
Dr. Kohl and her colleagues in Cologne recently completed a study of DBS in 20 patients. She noted that it took 5 years to collect these 20 patients, underscoring the high bar that’s appropriate for resort to DBS, even though the therapy is approved for OCD by both the Food and Drug Administration and European regulatory authorities. Forty percent of the patients were DBS responders, with a mean 30% improvement in Y-BOCS scores. That’s a lower responder rate than in Dr. Denys’s and some other series, which Dr. Kohl attributed to the fact that in Germany, postimplantation CBT is not yet routine.
Asked about DBS side effects, the speakers agreed that they’re transient and fall off after initial stimulation parameters are changed.
“The most consistent and impressive side effect is that initially after surgical implantation of the electrodes and stimulation of the nucleus accumbens, patients experience 3 or 4 days of hypomania, which then disappears,” Dr. Denys said. “It causes a kind of imprinting, because even a decade later, patients ask us, ‘Could you bring back that really nice feeling?’ It’s 3 days of love, peace, and hypomania. It’s a side effect, but people like it.”
Dr. Denys and Dr. Kohl reported no financial conflicts of interest regarding their presentations.
VIENNA – Even though deep brain stimulation has been used to treat obsessive-compulsive disorder in fewer than 300 patients worldwide, the therapy has had a huge impact on understanding of the disorder, Damiaan Denys, MD, PhD, said at the annual congress of the European College of Neuropsychopharmacology.
Indeed, the efficacy of deep brain stimulation (DBS) in the most severe, treatment-refractory cases of OCD casts doubt upon the fundamental construct clinicians have relied upon for decades to comprehend OCD: Namely, that affected patients first experience obsessions, which induce anxiety, which then stimulates compulsions, and engaging in those compulsions brings relief and reward, and then the whole cycle starts over again.
That construct may not actually be true.
“Deep brain stimulation will force us to rethink OCD,” predicted Dr. Denys, professor and head of psychiatry at the University of Amsterdam.
What does DBS change, and is there a temporal order? It varies, the psychiatrist said.
“In some patients, anxiety is the first thing that changes. In others, it starts with obsessions. Compulsions do decrease, but it’s difficult. It takes some time, and often we need supplemental cognitive-behavioral therapy (CBT). What changes, mainly, in my experience, is not symptoms, but something outside of OCD: namely, mood. Deep brain stimulation has a huge impact on mood. That’s an interesting finding, because it suggests the possibility that you can change a psychiatric disorder by changing symptoms that are thought of as being outside the disorder,” Dr. Denys observed.
“Another interesting thing is that deep brain stimulation changes things that are not even within psychiatry. When we ask our patients what has been the most profound impact deep brain stimulation has had on their lives, all of them say, ‘It increases my self-confidence.’ And that’s not something that is included in our scales. We assess patients using the HAM-D [Hamilton Rating Scale for Depression] and Y-BOCS [Yale-Brown Obsessive Compulsive Scale] and other measures, but there are still some very important aspects we are not taking into account and that have a profound effect on symptoms. In this case, using deep brain stimulation, we improve self-confidence and thereby change a whole chain of symptoms,” he continued.
Among the 60 highly refractory OCD patients treated by DBS by Dr. Denys and his colleagues at the Amsterdam center over the past 15 years, 15% were cured as a result.
“I purposely use the word ‘cured,’ because they don’t have obsessive-compulsive symptoms, anymore, which is, of course, extraordinary,” Dr. Denys noted.
An additional 35% of patients had a good response, defined as 60%-80% improvement on the Y-BOCS. Ten percent of patients were partial responders, with a 20%-40% improvement on the Y-BOCS. And 15% were nonresponders.
Closed-loop system coming
DBS entails implantation of electrodes deep within the brain to interrupt dysfunctional brain signals in local areas and across neural networks. This dysfunctional brain activity is expressed as symptoms. To date, DBS has been used in what’s called an open-loop system: Patients report the symptoms they’re experiencing and the clinician then adjusts the electrode settings in order to quell those symptoms. This approach is about to change. The technology has improved vastly since the early days, when the electrodes had four contact points. Now they have 64 contact points, and are capable of sending and receiving electrical signals.
“The next step in deep brain stimulation will be a closed-loop system. We will remove the clinician and the patient, and attempt to use a device capable of recording what happens in the brain and then changing electrical activity in response to the recordings. Our purpose will be to block these brain signals in advance of obsessions and compulsions so patients don’t have these symptoms. It’s technically possible. It has been done in Parkinson’s, and I think it’s the next step in psychiatry,” Dr. Denys said.
Indeed, he and his coinvestigators are planning formal studies of closed-loop DBS. For him, the prospect raises three key questions: What are the neural correlates of OCD symptoms? What about the ethics of implanting a device in the brain which by itself results in different life experiences? And will closed-loop DBS have superior efficacy, compared with open-loop DBS?
“How is the mind rooted in the brain, and how is the brain expressed in the mind? It’s the most fascinating question; it’s why we all love psychiatry, and up until now, there are no answers,” he observed.
Significant progress already has been made on the neural correlates question. Dr. Denys and his colleagues have found that when OCD patients with deep brain electrodes engage in cleaning compulsions, their local field potentials in the striatal area show peaks of roughly 9 Hz in the alpha range and in the beta/low gamma range. These patterns may represent compulsive behavior and likely could be useful in steering a closed-loop system. Also, the Dutch investigators have found that 3- to 8-Hz theta oscillations in local field potentials in the striatal area may represent a neural signature for anxiety and/or obsessions.
Using a closed-loop system, he continued, investigators plan to test two quite different hypotheses about the fundamental nature of OCD. One is that obsessions, anxiety, compulsions, and relief are each separately related to different brain areas. The other hypothesis is that one central brain stimulus drives the chain of symptoms that characterize OCD, and that by identifying and blocking that primary signal, the whole pathologic process can be stopped.
Current status of procedure
While Dr. Denys focused on the near future of DBS for OCD, another speaker at the session, Sina Kohl, PhD, addressed DBS for OCD as it exists today, particularly the who, how, and where.
The “who” is the relatively rare patient with truly refractory OCD after multiple drug trials of agents in different antidepressant classes, one of which should be clomipramine, as well as a failed course of CBT provided by an expert in CBT for OCD, of which there are relatively few. In a study led by investigators at Brown University, Providence, R.I., only 2 of 325 patients with OCD were deemed truly refractory (J Neuropsychiatry Clin Neurosci. 2014 Winter;26[1]:81-6). That sounds about right, according to Dr. Kohl, a psychologist at the University of Cologne, in Germany.
The “how” is to deliver DBS in conjunction with CBT. Response rates are higher at centers where that practice is routine, she added.
The “where” is an unsettled question. In Dr. Kohl’s meta-analysis of 25 published DBS studies, electrode placement in four different DBS target structures produced similar results: the nucleus accumbens, the anterior limb of the internal capsule, the ventral striatum, and the subthalamic nucleus. Stimulation of the inferior thalamic peduncle appeared to achieve better results, but this is a sketchy conclusion based upon two studies totaling just six patients (BMC Psychiatry. 2014 Aug 2;14:214. doi: 10.1186/s12888-014-0214-y).
Recently, Belgian investigators have reported particularly promising results – the best so far – for DBS targeting the bed nucleus of the stria terminalis (Mol Psychiatry. 2016 Sep;21[9]:1272-80).
Bilateral DBS appears to be more effective than unilateral.
Dr. Kohl and her colleagues in Cologne recently completed a study of DBS in 20 patients. She noted that it took 5 years to collect these 20 patients, underscoring the high bar that’s appropriate for resort to DBS, even though the therapy is approved for OCD by both the Food and Drug Administration and European regulatory authorities. Forty percent of the patients were DBS responders, with a mean 30% improvement in Y-BOCS scores. That’s a lower responder rate than in Dr. Denys’s and some other series, which Dr. Kohl attributed to the fact that in Germany, postimplantation CBT is not yet routine.
Asked about DBS side effects, the speakers agreed that they’re transient and fall off after initial stimulation parameters are changed.
“The most consistent and impressive side effect is that initially after surgical implantation of the electrodes and stimulation of the nucleus accumbens, patients experience 3 or 4 days of hypomania, which then disappears,” Dr. Denys said. “It causes a kind of imprinting, because even a decade later, patients ask us, ‘Could you bring back that really nice feeling?’ It’s 3 days of love, peace, and hypomania. It’s a side effect, but people like it.”
Dr. Denys and Dr. Kohl reported no financial conflicts of interest regarding their presentations.
VIENNA – Even though deep brain stimulation has been used to treat obsessive-compulsive disorder in fewer than 300 patients worldwide, the therapy has had a huge impact on understanding of the disorder, Damiaan Denys, MD, PhD, said at the annual congress of the European College of Neuropsychopharmacology.
Indeed, the efficacy of deep brain stimulation (DBS) in the most severe, treatment-refractory cases of OCD casts doubt upon the fundamental construct clinicians have relied upon for decades to comprehend OCD: Namely, that affected patients first experience obsessions, which induce anxiety, which then stimulates compulsions, and engaging in those compulsions brings relief and reward, and then the whole cycle starts over again.
That construct may not actually be true.
“Deep brain stimulation will force us to rethink OCD,” predicted Dr. Denys, professor and head of psychiatry at the University of Amsterdam.
What does DBS change, and is there a temporal order? It varies, the psychiatrist said.
“In some patients, anxiety is the first thing that changes. In others, it starts with obsessions. Compulsions do decrease, but it’s difficult. It takes some time, and often we need supplemental cognitive-behavioral therapy (CBT). What changes, mainly, in my experience, is not symptoms, but something outside of OCD: namely, mood. Deep brain stimulation has a huge impact on mood. That’s an interesting finding, because it suggests the possibility that you can change a psychiatric disorder by changing symptoms that are thought of as being outside the disorder,” Dr. Denys observed.
“Another interesting thing is that deep brain stimulation changes things that are not even within psychiatry. When we ask our patients what has been the most profound impact deep brain stimulation has had on their lives, all of them say, ‘It increases my self-confidence.’ And that’s not something that is included in our scales. We assess patients using the HAM-D [Hamilton Rating Scale for Depression] and Y-BOCS [Yale-Brown Obsessive Compulsive Scale] and other measures, but there are still some very important aspects we are not taking into account and that have a profound effect on symptoms. In this case, using deep brain stimulation, we improve self-confidence and thereby change a whole chain of symptoms,” he continued.
Among the 60 highly refractory OCD patients treated by DBS by Dr. Denys and his colleagues at the Amsterdam center over the past 15 years, 15% were cured as a result.
“I purposely use the word ‘cured,’ because they don’t have obsessive-compulsive symptoms, anymore, which is, of course, extraordinary,” Dr. Denys noted.
An additional 35% of patients had a good response, defined as 60%-80% improvement on the Y-BOCS. Ten percent of patients were partial responders, with a 20%-40% improvement on the Y-BOCS. And 15% were nonresponders.
Closed-loop system coming
DBS entails implantation of electrodes deep within the brain to interrupt dysfunctional brain signals in local areas and across neural networks. This dysfunctional brain activity is expressed as symptoms. To date, DBS has been used in what’s called an open-loop system: Patients report the symptoms they’re experiencing and the clinician then adjusts the electrode settings in order to quell those symptoms. This approach is about to change. The technology has improved vastly since the early days, when the electrodes had four contact points. Now they have 64 contact points, and are capable of sending and receiving electrical signals.
“The next step in deep brain stimulation will be a closed-loop system. We will remove the clinician and the patient, and attempt to use a device capable of recording what happens in the brain and then changing electrical activity in response to the recordings. Our purpose will be to block these brain signals in advance of obsessions and compulsions so patients don’t have these symptoms. It’s technically possible. It has been done in Parkinson’s, and I think it’s the next step in psychiatry,” Dr. Denys said.
Indeed, he and his coinvestigators are planning formal studies of closed-loop DBS. For him, the prospect raises three key questions: What are the neural correlates of OCD symptoms? What about the ethics of implanting a device in the brain which by itself results in different life experiences? And will closed-loop DBS have superior efficacy, compared with open-loop DBS?
“How is the mind rooted in the brain, and how is the brain expressed in the mind? It’s the most fascinating question; it’s why we all love psychiatry, and up until now, there are no answers,” he observed.
Significant progress already has been made on the neural correlates question. Dr. Denys and his colleagues have found that when OCD patients with deep brain electrodes engage in cleaning compulsions, their local field potentials in the striatal area show peaks of roughly 9 Hz in the alpha range and in the beta/low gamma range. These patterns may represent compulsive behavior and likely could be useful in steering a closed-loop system. Also, the Dutch investigators have found that 3- to 8-Hz theta oscillations in local field potentials in the striatal area may represent a neural signature for anxiety and/or obsessions.
Using a closed-loop system, he continued, investigators plan to test two quite different hypotheses about the fundamental nature of OCD. One is that obsessions, anxiety, compulsions, and relief are each separately related to different brain areas. The other hypothesis is that one central brain stimulus drives the chain of symptoms that characterize OCD, and that by identifying and blocking that primary signal, the whole pathologic process can be stopped.
Current status of procedure
While Dr. Denys focused on the near future of DBS for OCD, another speaker at the session, Sina Kohl, PhD, addressed DBS for OCD as it exists today, particularly the who, how, and where.
The “who” is the relatively rare patient with truly refractory OCD after multiple drug trials of agents in different antidepressant classes, one of which should be clomipramine, as well as a failed course of CBT provided by an expert in CBT for OCD, of which there are relatively few. In a study led by investigators at Brown University, Providence, R.I., only 2 of 325 patients with OCD were deemed truly refractory (J Neuropsychiatry Clin Neurosci. 2014 Winter;26[1]:81-6). That sounds about right, according to Dr. Kohl, a psychologist at the University of Cologne, in Germany.
The “how” is to deliver DBS in conjunction with CBT. Response rates are higher at centers where that practice is routine, she added.
The “where” is an unsettled question. In Dr. Kohl’s meta-analysis of 25 published DBS studies, electrode placement in four different DBS target structures produced similar results: the nucleus accumbens, the anterior limb of the internal capsule, the ventral striatum, and the subthalamic nucleus. Stimulation of the inferior thalamic peduncle appeared to achieve better results, but this is a sketchy conclusion based upon two studies totaling just six patients (BMC Psychiatry. 2014 Aug 2;14:214. doi: 10.1186/s12888-014-0214-y).
Recently, Belgian investigators have reported particularly promising results – the best so far – for DBS targeting the bed nucleus of the stria terminalis (Mol Psychiatry. 2016 Sep;21[9]:1272-80).
Bilateral DBS appears to be more effective than unilateral.
Dr. Kohl and her colleagues in Cologne recently completed a study of DBS in 20 patients. She noted that it took 5 years to collect these 20 patients, underscoring the high bar that’s appropriate for resort to DBS, even though the therapy is approved for OCD by both the Food and Drug Administration and European regulatory authorities. Forty percent of the patients were DBS responders, with a mean 30% improvement in Y-BOCS scores. That’s a lower responder rate than in Dr. Denys’s and some other series, which Dr. Kohl attributed to the fact that in Germany, postimplantation CBT is not yet routine.
Asked about DBS side effects, the speakers agreed that they’re transient and fall off after initial stimulation parameters are changed.
“The most consistent and impressive side effect is that initially after surgical implantation of the electrodes and stimulation of the nucleus accumbens, patients experience 3 or 4 days of hypomania, which then disappears,” Dr. Denys said. “It causes a kind of imprinting, because even a decade later, patients ask us, ‘Could you bring back that really nice feeling?’ It’s 3 days of love, peace, and hypomania. It’s a side effect, but people like it.”
Dr. Denys and Dr. Kohl reported no financial conflicts of interest regarding their presentations.
Parental Feelings of Helplessness Predict Quality of Life in Children With Epilepsy
HOUSTON—The degree of helplessness that parents experience about their child’s epilepsy is significantly related to the child’s health-related quality of life, according to research presented at the 70th Annual Meeting of the American Epilepsy Society. “This relationship highlights the importance of taking into account a parent’s ability to cope … and should be an important target when developing interventions for families who have a child with epilepsy,” said Rachael McLaughlin, a research assistant in neuropsychology at Dell Children’s Comprehensive Epilepsy Program in Austin, and colleagues.

Prior studies have found that psychosocial factors can play a role in the health-related quality of life of children with epilepsy. Parents’ adaptation to their child’s epilepsy is not often taken into account, however.
Mrs. McLaughlin and her research colleagues at Dell Children’s, William Schraegle, Nancy Nussbaum, PhD, and Jeffrey Titus, PhD, conducted a study using the Illness Cognition Questionnaire–Parent Version (ICQ). The ICQ assesses parents’ thoughts of helplessness, acceptance, and perceived benefits related to the illness. They studied how parental coping relates to a child’s internalizing psychopathology and health-related quality of life in the context of epilepsy.
The researchers analyzed data from 40 patients (23 females) who were seen for a neuropsychologic evaluation at a tertiary pediatric care epilepsy clinic. Parents completed the ICQ, Quality of Life Childhood Epilepsy (QOLCE) questionnaire, and the Behavior Assessment System for Children, Second Edition. They obtained family history of internalizing psychopathology, duration of epilepsy, and maternal education level from medical records. They assessed associations between parental, demographic, epilepsy-specific, behavioral, and functional variables. The researchers then assessed whether independent variables predicted quality of life.
Patients had an average age of 12 and average epilepsy duration of about six years. QOLCE was related to parental helplessness and acceptance on the ICQ, and to internalizing and externalizing psychopathology.
A simultaneous regression model found that parental helplessness and internalizing psychopathology predicted quality of life, with parental helplessness accounting for the most variance above and beyond all other variables. Analysis of variance found that parental helplessness significantly affected QOLCE. When divided by parental helplessness, patients whose parents had high levels of helplessness had significantly lower quality of life than patients whose parents had low levels of helplessness.
—Jake Remaly
HOUSTON—The degree of helplessness that parents experience about their child’s epilepsy is significantly related to the child’s health-related quality of life, according to research presented at the 70th Annual Meeting of the American Epilepsy Society. “This relationship highlights the importance of taking into account a parent’s ability to cope … and should be an important target when developing interventions for families who have a child with epilepsy,” said Rachael McLaughlin, a research assistant in neuropsychology at Dell Children’s Comprehensive Epilepsy Program in Austin, and colleagues.
Prior studies have found that psychosocial factors can play a role in the health-related quality of life of children with epilepsy. Parents’ adaptation to their child’s epilepsy is not often taken into account, however.
Mrs. McLaughlin and her research colleagues at Dell Children’s, William Schraegle, Nancy Nussbaum, PhD, and Jeffrey Titus, PhD, conducted a study using the Illness Cognition Questionnaire–Parent Version (ICQ). The ICQ assesses parents’ thoughts of helplessness, acceptance, and perceived benefits related to the illness. They studied how parental coping relates to a child’s internalizing psychopathology and health-related quality of life in the context of epilepsy.
The researchers analyzed data from 40 patients (23 females) who were seen for a neuropsychologic evaluation at a tertiary pediatric care epilepsy clinic. Parents completed the ICQ, Quality of Life Childhood Epilepsy (QOLCE) questionnaire, and the Behavior Assessment System for Children, Second Edition. They obtained family history of internalizing psychopathology, duration of epilepsy, and maternal education level from medical records. They assessed associations between parental, demographic, epilepsy-specific, behavioral, and functional variables. The researchers then assessed whether independent variables predicted quality of life.
Patients had an average age of 12 and average epilepsy duration of about six years. QOLCE was related to parental helplessness and acceptance on the ICQ, and to internalizing and externalizing psychopathology.
A simultaneous regression model found that parental helplessness and internalizing psychopathology predicted quality of life, with parental helplessness accounting for the most variance above and beyond all other variables. Analysis of variance found that parental helplessness significantly affected QOLCE. When divided by parental helplessness, patients whose parents had high levels of helplessness had significantly lower quality of life than patients whose parents had low levels of helplessness.
—Jake Remaly
HOUSTON—The degree of helplessness that parents experience about their child’s epilepsy is significantly related to the child’s health-related quality of life, according to research presented at the 70th Annual Meeting of the American Epilepsy Society. “This relationship highlights the importance of taking into account a parent’s ability to cope … and should be an important target when developing interventions for families who have a child with epilepsy,” said Rachael McLaughlin, a research assistant in neuropsychology at Dell Children’s Comprehensive Epilepsy Program in Austin, and colleagues.
Prior studies have found that psychosocial factors can play a role in the health-related quality of life of children with epilepsy. Parents’ adaptation to their child’s epilepsy is not often taken into account, however.
Mrs. McLaughlin and her research colleagues at Dell Children’s, William Schraegle, Nancy Nussbaum, PhD, and Jeffrey Titus, PhD, conducted a study using the Illness Cognition Questionnaire–Parent Version (ICQ). The ICQ assesses parents’ thoughts of helplessness, acceptance, and perceived benefits related to the illness. They studied how parental coping relates to a child’s internalizing psychopathology and health-related quality of life in the context of epilepsy.
The researchers analyzed data from 40 patients (23 females) who were seen for a neuropsychologic evaluation at a tertiary pediatric care epilepsy clinic. Parents completed the ICQ, Quality of Life Childhood Epilepsy (QOLCE) questionnaire, and the Behavior Assessment System for Children, Second Edition. They obtained family history of internalizing psychopathology, duration of epilepsy, and maternal education level from medical records. They assessed associations between parental, demographic, epilepsy-specific, behavioral, and functional variables. The researchers then assessed whether independent variables predicted quality of life.
Patients had an average age of 12 and average epilepsy duration of about six years. QOLCE was related to parental helplessness and acceptance on the ICQ, and to internalizing and externalizing psychopathology.
A simultaneous regression model found that parental helplessness and internalizing psychopathology predicted quality of life, with parental helplessness accounting for the most variance above and beyond all other variables. Analysis of variance found that parental helplessness significantly affected QOLCE. When divided by parental helplessness, patients whose parents had high levels of helplessness had significantly lower quality of life than patients whose parents had low levels of helplessness.
—Jake Remaly

Detecting Autoimmune Neural Antibodies Is Key to Preventing Misdiagnosis
LAS VEGAS—Advances in autoimmune neurology, a rapidly evolving field, have the potential to prevent misdiagnosis of many disorders, from multiple sclerosis to neurodegenerative diseases. The key: increased detection of autoimmune and paraneoplastic neural antibodies, said Sean J. Pittock, MD, at the American Academy of Neurology’s Fall 2016 Conference. The importance of autoimmune neurology “cannot be overstated because these are reversible, treatable conditions,” Dr. Pittock said. “If you miss them, you have done your patients a disservice.”

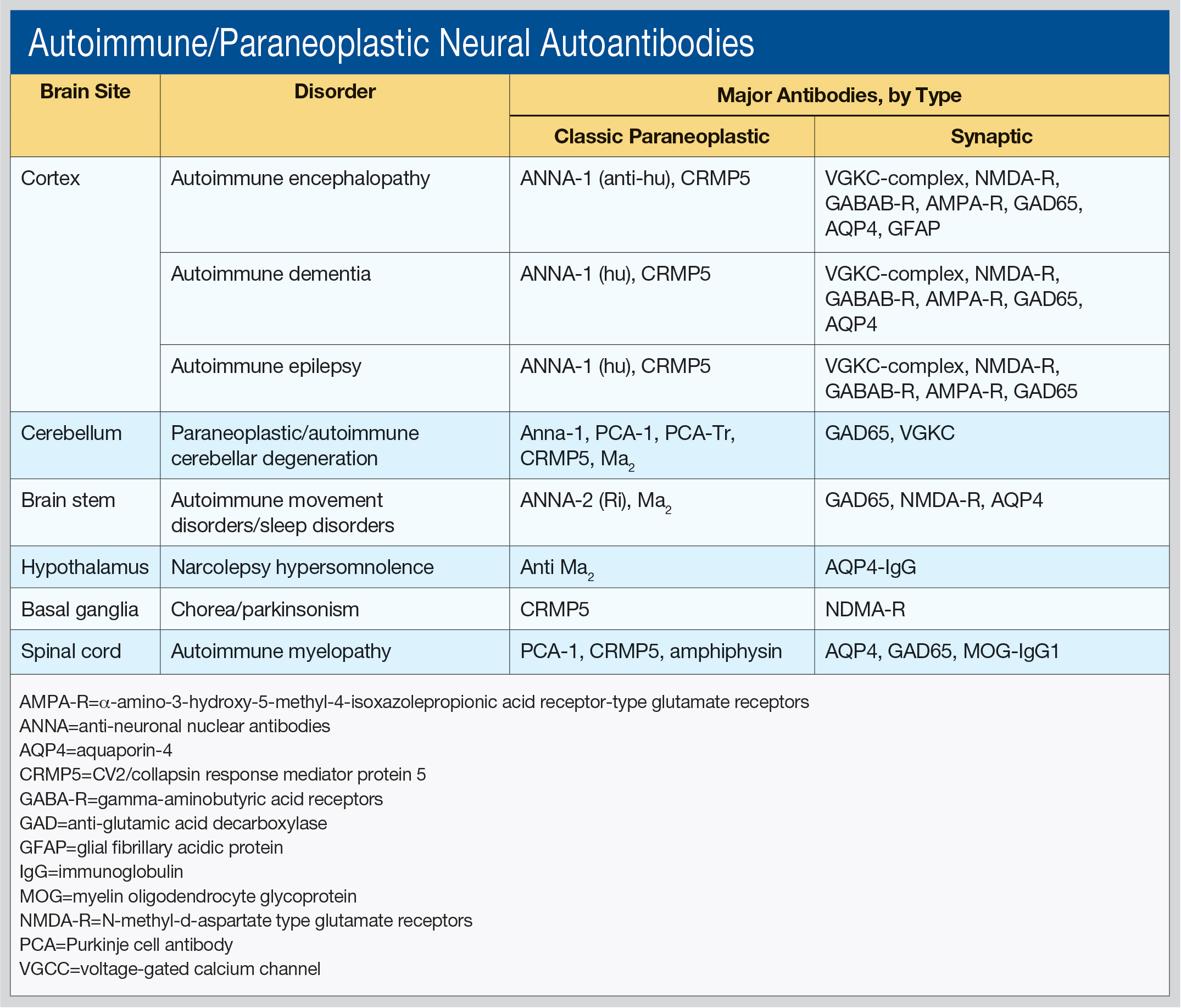
Dr. Pittock, Professor of Neurology, Director of the Neuroimmunology Laboratory, and Director of the Center for MS and Autoimmune Neurology at the Mayo Clinic in Rochester, Minnesota, said he works primarily “in the field of discovering novel antibodies that make sense to the clinician.” He outlined these antibodies by brain site (cortex, cerebellum, brainstem, hypothalamus, basal ganglia, and spinal cord), as well as associated disorders and type of antibodies (see table).
“Different antibodies tell you what cancers you should be looking for,” he said. The classic paraneoplastic antibodies have oncological associations with small cell and aerodigestive carcinomas, breast and gynecologic adenocarcinomas, Hodgkin lymphoma, and thymoma. The synaptic antibodies are associated with prostate, lung, thymic, and endometrial carcinomas, as well as with teratoma.
In his laboratory, Dr. Pittock uses a complex algorithm to test for 20 types of neural antibodies using diverse methodologies, from indirect immunofluorescence to cell-based and immunoprecipitation assays. Results are usually available within six to eight days. “We are moving towards a movement disorder evaluation, a CNS hyperexcitability evaluation, and a demyelinating disease evaluation,” he said.
“In paraneoplastic disorders, the tumors are often difficult to find,” Dr. Pittock said. “The immune system is having a dramatic impact on the tumor, and so these tumors are very small, so you may miss them.”
Dr. Pittock provided an overview of how clinicians might approach diagnosing autoimmune disorders. “For children, obviously, anaplastic diseases are rare, but if you have a patient with opsoclonus myoclonus, 50% of those children will have a cancer. For adolescents and young females, you always think about teratoma. If it is a young man, you really need to think about testicular tumors.”
The type of cancer defines what type of investigation is appropriate, he said. For example, due to low resolution, PET scans are not as useful for thymoma, teratoma, or testicular or gastrointestinal tumors. On the other hand, PET scans can help evaluate equivocal findings, such as pulmonary nodules or lymph nodes. With the exception of Medicare, however, PET scans are not generally covered by insurance. Medicare covers PET scanning under ICD-9 code V71.1, “observation for suspected malignant neoplasm,” and ICD-10, Z12, “encounter for screening for malignant neoplasm”; Z12.9, “site unspecified.”
At the Mayo Clinic, Dr. Pittock said they see five patients per week with NMDA-receptor encephalitis. To diagnose a possible autoimmune neurologic disorder, he asked that clinicians “please consider using the concept of immunotherapy as kind of a diagnostic test.” To do so, the patient should be treated acutely with IV methylprednisolone, IVIg, or plasma exchanges. If the patient improves, continue acute IV therapy and taper oral prednisone; other options include oral azathioprine or mycophenolate mofetil. If the patient does not improve, consider alternative acute therapy or no further therapy.
In one study that he and his colleagues conducted, 81% of patients who had failed antiepileptic drugs (AED) and were having daily seizures were treated with such a diagnostic test. Of these, 62% responded and 34% of patients became seizure-free. In contrast, Dr. Pittock said, patients treated with a third AED in a trial rarely experience meaningful improvement.
An autoimmune condition may also have a neuropsychiatric presentation. Dr. Pittock urged attendees to watch a recent film based on the 2013 book, “Brain on Fire: My Month of Madness,” by Susannah Cahalan, who went from psychosis to catatonia before a physician determined she had brain inflammation due to an autoimmune reaction.
“Many of these patients previously would have ended up in a psychiatric institution or in an ICU setting where, potentially, in the setting of intractable disease, they may have had their machine turned off, they may have died, when they have the potential to make a full recovery and, sometimes, spontaneously improve. The take-home message for this one is, if you want to investigate a patient with this, test spinal fluid.
“Autoimmune neurology is here to stay. It is a new field; it is very exciting; it has just been recognized by the American Academy of Neurology as having its own section,” Dr. Pittock said, and invited those interested to join.
Dr. Pittock has received research support from Alexion Pharmaceuticals, the Guthy-Jackson Charitable Foundation, and the National Institutes of Health.
—Debra Hughes
Suggested Reading
Linnoila J, Pittock SJ. Autoantibody-associated central nervous system neurologic disorders. Semin Neurol. 2016;36(4):382-396.
Toledano M, Britton JW, McKeon A, et al. Utility of an immunotherapy trial in evaluating patients with presumed autoimmune epilepsy. Neurology. 2014;82(18):1578-1586.
LAS VEGAS—Advances in autoimmune neurology, a rapidly evolving field, have the potential to prevent misdiagnosis of many disorders, from multiple sclerosis to neurodegenerative diseases. The key: increased detection of autoimmune and paraneoplastic neural antibodies, said Sean J. Pittock, MD, at the American Academy of Neurology’s Fall 2016 Conference. The importance of autoimmune neurology “cannot be overstated because these are reversible, treatable conditions,” Dr. Pittock said. “If you miss them, you have done your patients a disservice.”
Dr. Pittock, Professor of Neurology, Director of the Neuroimmunology Laboratory, and Director of the Center for MS and Autoimmune Neurology at the Mayo Clinic in Rochester, Minnesota, said he works primarily “in the field of discovering novel antibodies that make sense to the clinician.” He outlined these antibodies by brain site (cortex, cerebellum, brainstem, hypothalamus, basal ganglia, and spinal cord), as well as associated disorders and type of antibodies (see table).
“Different antibodies tell you what cancers you should be looking for,” he said. The classic paraneoplastic antibodies have oncological associations with small cell and aerodigestive carcinomas, breast and gynecologic adenocarcinomas, Hodgkin lymphoma, and thymoma. The synaptic antibodies are associated with prostate, lung, thymic, and endometrial carcinomas, as well as with teratoma.
In his laboratory, Dr. Pittock uses a complex algorithm to test for 20 types of neural antibodies using diverse methodologies, from indirect immunofluorescence to cell-based and immunoprecipitation assays. Results are usually available within six to eight days. “We are moving towards a movement disorder evaluation, a CNS hyperexcitability evaluation, and a demyelinating disease evaluation,” he said.
“In paraneoplastic disorders, the tumors are often difficult to find,” Dr. Pittock said. “The immune system is having a dramatic impact on the tumor, and so these tumors are very small, so you may miss them.”
Dr. Pittock provided an overview of how clinicians might approach diagnosing autoimmune disorders. “For children, obviously, anaplastic diseases are rare, but if you have a patient with opsoclonus myoclonus, 50% of those children will have a cancer. For adolescents and young females, you always think about teratoma. If it is a young man, you really need to think about testicular tumors.”
The type of cancer defines what type of investigation is appropriate, he said. For example, due to low resolution, PET scans are not as useful for thymoma, teratoma, or testicular or gastrointestinal tumors. On the other hand, PET scans can help evaluate equivocal findings, such as pulmonary nodules or lymph nodes. With the exception of Medicare, however, PET scans are not generally covered by insurance. Medicare covers PET scanning under ICD-9 code V71.1, “observation for suspected malignant neoplasm,” and ICD-10, Z12, “encounter for screening for malignant neoplasm”; Z12.9, “site unspecified.”
At the Mayo Clinic, Dr. Pittock said they see five patients per week with NMDA-receptor encephalitis. To diagnose a possible autoimmune neurologic disorder, he asked that clinicians “please consider using the concept of immunotherapy as kind of a diagnostic test.” To do so, the patient should be treated acutely with IV methylprednisolone, IVIg, or plasma exchanges. If the patient improves, continue acute IV therapy and taper oral prednisone; other options include oral azathioprine or mycophenolate mofetil. If the patient does not improve, consider alternative acute therapy or no further therapy.
In one study that he and his colleagues conducted, 81% of patients who had failed antiepileptic drugs (AED) and were having daily seizures were treated with such a diagnostic test. Of these, 62% responded and 34% of patients became seizure-free. In contrast, Dr. Pittock said, patients treated with a third AED in a trial rarely experience meaningful improvement.
An autoimmune condition may also have a neuropsychiatric presentation. Dr. Pittock urged attendees to watch a recent film based on the 2013 book, “Brain on Fire: My Month of Madness,” by Susannah Cahalan, who went from psychosis to catatonia before a physician determined she had brain inflammation due to an autoimmune reaction.
“Many of these patients previously would have ended up in a psychiatric institution or in an ICU setting where, potentially, in the setting of intractable disease, they may have had their machine turned off, they may have died, when they have the potential to make a full recovery and, sometimes, spontaneously improve. The take-home message for this one is, if you want to investigate a patient with this, test spinal fluid.
“Autoimmune neurology is here to stay. It is a new field; it is very exciting; it has just been recognized by the American Academy of Neurology as having its own section,” Dr. Pittock said, and invited those interested to join.
Dr. Pittock has received research support from Alexion Pharmaceuticals, the Guthy-Jackson Charitable Foundation, and the National Institutes of Health.
—Debra Hughes
Suggested Reading
Linnoila J, Pittock SJ. Autoantibody-associated central nervous system neurologic disorders. Semin Neurol. 2016;36(4):382-396.
Toledano M, Britton JW, McKeon A, et al. Utility of an immunotherapy trial in evaluating patients with presumed autoimmune epilepsy. Neurology. 2014;82(18):1578-1586.
LAS VEGAS—Advances in autoimmune neurology, a rapidly evolving field, have the potential to prevent misdiagnosis of many disorders, from multiple sclerosis to neurodegenerative diseases. The key: increased detection of autoimmune and paraneoplastic neural antibodies, said Sean J. Pittock, MD, at the American Academy of Neurology’s Fall 2016 Conference. The importance of autoimmune neurology “cannot be overstated because these are reversible, treatable conditions,” Dr. Pittock said. “If you miss them, you have done your patients a disservice.”
Dr. Pittock, Professor of Neurology, Director of the Neuroimmunology Laboratory, and Director of the Center for MS and Autoimmune Neurology at the Mayo Clinic in Rochester, Minnesota, said he works primarily “in the field of discovering novel antibodies that make sense to the clinician.” He outlined these antibodies by brain site (cortex, cerebellum, brainstem, hypothalamus, basal ganglia, and spinal cord), as well as associated disorders and type of antibodies (see table).
“Different antibodies tell you what cancers you should be looking for,” he said. The classic paraneoplastic antibodies have oncological associations with small cell and aerodigestive carcinomas, breast and gynecologic adenocarcinomas, Hodgkin lymphoma, and thymoma. The synaptic antibodies are associated with prostate, lung, thymic, and endometrial carcinomas, as well as with teratoma.
In his laboratory, Dr. Pittock uses a complex algorithm to test for 20 types of neural antibodies using diverse methodologies, from indirect immunofluorescence to cell-based and immunoprecipitation assays. Results are usually available within six to eight days. “We are moving towards a movement disorder evaluation, a CNS hyperexcitability evaluation, and a demyelinating disease evaluation,” he said.
“In paraneoplastic disorders, the tumors are often difficult to find,” Dr. Pittock said. “The immune system is having a dramatic impact on the tumor, and so these tumors are very small, so you may miss them.”
Dr. Pittock provided an overview of how clinicians might approach diagnosing autoimmune disorders. “For children, obviously, anaplastic diseases are rare, but if you have a patient with opsoclonus myoclonus, 50% of those children will have a cancer. For adolescents and young females, you always think about teratoma. If it is a young man, you really need to think about testicular tumors.”
The type of cancer defines what type of investigation is appropriate, he said. For example, due to low resolution, PET scans are not as useful for thymoma, teratoma, or testicular or gastrointestinal tumors. On the other hand, PET scans can help evaluate equivocal findings, such as pulmonary nodules or lymph nodes. With the exception of Medicare, however, PET scans are not generally covered by insurance. Medicare covers PET scanning under ICD-9 code V71.1, “observation for suspected malignant neoplasm,” and ICD-10, Z12, “encounter for screening for malignant neoplasm”; Z12.9, “site unspecified.”
At the Mayo Clinic, Dr. Pittock said they see five patients per week with NMDA-receptor encephalitis. To diagnose a possible autoimmune neurologic disorder, he asked that clinicians “please consider using the concept of immunotherapy as kind of a diagnostic test.” To do so, the patient should be treated acutely with IV methylprednisolone, IVIg, or plasma exchanges. If the patient improves, continue acute IV therapy and taper oral prednisone; other options include oral azathioprine or mycophenolate mofetil. If the patient does not improve, consider alternative acute therapy or no further therapy.
In one study that he and his colleagues conducted, 81% of patients who had failed antiepileptic drugs (AED) and were having daily seizures were treated with such a diagnostic test. Of these, 62% responded and 34% of patients became seizure-free. In contrast, Dr. Pittock said, patients treated with a third AED in a trial rarely experience meaningful improvement.
An autoimmune condition may also have a neuropsychiatric presentation. Dr. Pittock urged attendees to watch a recent film based on the 2013 book, “Brain on Fire: My Month of Madness,” by Susannah Cahalan, who went from psychosis to catatonia before a physician determined she had brain inflammation due to an autoimmune reaction.
“Many of these patients previously would have ended up in a psychiatric institution or in an ICU setting where, potentially, in the setting of intractable disease, they may have had their machine turned off, they may have died, when they have the potential to make a full recovery and, sometimes, spontaneously improve. The take-home message for this one is, if you want to investigate a patient with this, test spinal fluid.
“Autoimmune neurology is here to stay. It is a new field; it is very exciting; it has just been recognized by the American Academy of Neurology as having its own section,” Dr. Pittock said, and invited those interested to join.
Dr. Pittock has received research support from Alexion Pharmaceuticals, the Guthy-Jackson Charitable Foundation, and the National Institutes of Health.
—Debra Hughes
Suggested Reading
Linnoila J, Pittock SJ. Autoantibody-associated central nervous system neurologic disorders. Semin Neurol. 2016;36(4):382-396.
Toledano M, Britton JW, McKeon A, et al. Utility of an immunotherapy trial in evaluating patients with presumed autoimmune epilepsy. Neurology. 2014;82(18):1578-1586.

Gene Mutation Linked to Early Onset of Parkinson’s Disease
A defect in a gene that produces dopamine in nigrostriatal cells appears to accelerate the onset of Parkinson’s disease, according to a study published in the February issue of Neurobiology of Aging. The effect is particularly pronounced for people younger than 50.
Auriel Willette, PhD, an Assistant Professor in the Departments of Food Science and Human Nutrition and Psychology at Iowa State University in Ames, and Joseph Webb, a graduate research assistant also at Iowa State, found on average that Caucasians with one mutated version of the gene—guanosine triphosphate cyclohydrolase-1 (GCH1)—had a 23% increased risk for Parkinson’s disease and developed symptoms five years earlier than those without the gene mutation.

However, young-to-middle-aged adults with the mutation had a 45% increased risk of developing Parkinson’s disease. Researchers said that the presence of the mutated gene in older adults had minimal effect.
The Potential for Personalized Medicine
It is widely known that rigidity and loss of muscle function associated with Parkinson’s disease are linked to a depletion of dopamine in the substantia nigra. Taking a holistic approach to their study, Dr. Willette and Mr. Webb sought to better understand how the GCH1 gene affects the course of Parkinson’s disease and certain outcomes such as motor skills and anxiety.
The study is the first to look at these biological markers, as well as the first to examine how the gene’s impact on dopamine production specifically affects Caucasian populations. Previous studies have focused primarily on Chinese and Taiwanese populations, said Dr. Willette. The findings have the potential to help personalize medical care for people with a family history of Parkinson’s disease, he said, in a way similar to testing for the BRCA gene in women at risk for breast cancer.
“We want to have a more comprehensive understanding of what these genes related to Parkinson’s disease are doing at different points in someone’s lifetime,” Dr. Willette said. “Then, with genetic testing we can determine the risk for illness based on someone’s age, gender, weight, and other intervening factors.”
The Impact of Age
Data for the study were collected through the Parkinson’s Progression Markers Initiative, a public–private partnership sponsored by the Michael J. Fox Foundation for Parkinson’s Research. The initiative evaluates people with the disease to develop new and better treatments. The Iowa State study included 289 treatment-naïve people recently diagnosed with Parkinson’s disease and 233 healthy controls.
The researchers analyzed anxiety and motor function using the Unified Parkinson’s Disease Rating Scale (UPDRS). They found that those with polymorphisms at rs11158026 coding for the GCH1 enzyme, regardless of age, were more anxious and struggled more with daily activities. The defective gene was not as strong of a predictor of developing Parkinson’s disease in people older than 50, however.
“As we age, we make progressively less dopamine, and this effect strongly outweighs the genetic influences from the ‘bad version’ of this gene,” said Mr. Webb. “Simply by aging, our dopamine production decreases to the point that the effects from a mutation in this gene are not noticeable in older adults, but make a big difference in younger populations.”
The researchers noted that it is also important to pay attention to blood cholesterol levels. Cholesterol is directly related to the ability to produce dopamine. High low-density lipoprotein (LDL) is an established risk factor for Parkinson’s disease, Dr. Willette said. Their study shows that carriers of the mutated GCH1 gene had higher cholesterol than noncarriers, regardless of age.
Suggested Reading
Webb J, Willette AA. Aging modifies the effect of GCH1 RS11158026 on DAT uptake and Parkinson’s disease clinical severity. Neurobiol Aging. 2017;50:39-46.
A defect in a gene that produces dopamine in nigrostriatal cells appears to accelerate the onset of Parkinson’s disease, according to a study published in the February issue of Neurobiology of Aging. The effect is particularly pronounced for people younger than 50.
Auriel Willette, PhD, an Assistant Professor in the Departments of Food Science and Human Nutrition and Psychology at Iowa State University in Ames, and Joseph Webb, a graduate research assistant also at Iowa State, found on average that Caucasians with one mutated version of the gene—guanosine triphosphate cyclohydrolase-1 (GCH1)—had a 23% increased risk for Parkinson’s disease and developed symptoms five years earlier than those without the gene mutation.
However, young-to-middle-aged adults with the mutation had a 45% increased risk of developing Parkinson’s disease. Researchers said that the presence of the mutated gene in older adults had minimal effect.
The Potential for Personalized Medicine
It is widely known that rigidity and loss of muscle function associated with Parkinson’s disease are linked to a depletion of dopamine in the substantia nigra. Taking a holistic approach to their study, Dr. Willette and Mr. Webb sought to better understand how the GCH1 gene affects the course of Parkinson’s disease and certain outcomes such as motor skills and anxiety.
The study is the first to look at these biological markers, as well as the first to examine how the gene’s impact on dopamine production specifically affects Caucasian populations. Previous studies have focused primarily on Chinese and Taiwanese populations, said Dr. Willette. The findings have the potential to help personalize medical care for people with a family history of Parkinson’s disease, he said, in a way similar to testing for the BRCA gene in women at risk for breast cancer.
“We want to have a more comprehensive understanding of what these genes related to Parkinson’s disease are doing at different points in someone’s lifetime,” Dr. Willette said. “Then, with genetic testing we can determine the risk for illness based on someone’s age, gender, weight, and other intervening factors.”
The Impact of Age
Data for the study were collected through the Parkinson’s Progression Markers Initiative, a public–private partnership sponsored by the Michael J. Fox Foundation for Parkinson’s Research. The initiative evaluates people with the disease to develop new and better treatments. The Iowa State study included 289 treatment-naïve people recently diagnosed with Parkinson’s disease and 233 healthy controls.
The researchers analyzed anxiety and motor function using the Unified Parkinson’s Disease Rating Scale (UPDRS). They found that those with polymorphisms at rs11158026 coding for the GCH1 enzyme, regardless of age, were more anxious and struggled more with daily activities. The defective gene was not as strong of a predictor of developing Parkinson’s disease in people older than 50, however.
“As we age, we make progressively less dopamine, and this effect strongly outweighs the genetic influences from the ‘bad version’ of this gene,” said Mr. Webb. “Simply by aging, our dopamine production decreases to the point that the effects from a mutation in this gene are not noticeable in older adults, but make a big difference in younger populations.”
The researchers noted that it is also important to pay attention to blood cholesterol levels. Cholesterol is directly related to the ability to produce dopamine. High low-density lipoprotein (LDL) is an established risk factor for Parkinson’s disease, Dr. Willette said. Their study shows that carriers of the mutated GCH1 gene had higher cholesterol than noncarriers, regardless of age.
Suggested Reading
Webb J, Willette AA. Aging modifies the effect of GCH1 RS11158026 on DAT uptake and Parkinson’s disease clinical severity. Neurobiol Aging. 2017;50:39-46.
A defect in a gene that produces dopamine in nigrostriatal cells appears to accelerate the onset of Parkinson’s disease, according to a study published in the February issue of Neurobiology of Aging. The effect is particularly pronounced for people younger than 50.
Auriel Willette, PhD, an Assistant Professor in the Departments of Food Science and Human Nutrition and Psychology at Iowa State University in Ames, and Joseph Webb, a graduate research assistant also at Iowa State, found on average that Caucasians with one mutated version of the gene—guanosine triphosphate cyclohydrolase-1 (GCH1)—had a 23% increased risk for Parkinson’s disease and developed symptoms five years earlier than those without the gene mutation.
However, young-to-middle-aged adults with the mutation had a 45% increased risk of developing Parkinson’s disease. Researchers said that the presence of the mutated gene in older adults had minimal effect.
The Potential for Personalized Medicine
It is widely known that rigidity and loss of muscle function associated with Parkinson’s disease are linked to a depletion of dopamine in the substantia nigra. Taking a holistic approach to their study, Dr. Willette and Mr. Webb sought to better understand how the GCH1 gene affects the course of Parkinson’s disease and certain outcomes such as motor skills and anxiety.
The study is the first to look at these biological markers, as well as the first to examine how the gene’s impact on dopamine production specifically affects Caucasian populations. Previous studies have focused primarily on Chinese and Taiwanese populations, said Dr. Willette. The findings have the potential to help personalize medical care for people with a family history of Parkinson’s disease, he said, in a way similar to testing for the BRCA gene in women at risk for breast cancer.
“We want to have a more comprehensive understanding of what these genes related to Parkinson’s disease are doing at different points in someone’s lifetime,” Dr. Willette said. “Then, with genetic testing we can determine the risk for illness based on someone’s age, gender, weight, and other intervening factors.”
The Impact of Age
Data for the study were collected through the Parkinson’s Progression Markers Initiative, a public–private partnership sponsored by the Michael J. Fox Foundation for Parkinson’s Research. The initiative evaluates people with the disease to develop new and better treatments. The Iowa State study included 289 treatment-naïve people recently diagnosed with Parkinson’s disease and 233 healthy controls.
The researchers analyzed anxiety and motor function using the Unified Parkinson’s Disease Rating Scale (UPDRS). They found that those with polymorphisms at rs11158026 coding for the GCH1 enzyme, regardless of age, were more anxious and struggled more with daily activities. The defective gene was not as strong of a predictor of developing Parkinson’s disease in people older than 50, however.
“As we age, we make progressively less dopamine, and this effect strongly outweighs the genetic influences from the ‘bad version’ of this gene,” said Mr. Webb. “Simply by aging, our dopamine production decreases to the point that the effects from a mutation in this gene are not noticeable in older adults, but make a big difference in younger populations.”
The researchers noted that it is also important to pay attention to blood cholesterol levels. Cholesterol is directly related to the ability to produce dopamine. High low-density lipoprotein (LDL) is an established risk factor for Parkinson’s disease, Dr. Willette said. Their study shows that carriers of the mutated GCH1 gene had higher cholesterol than noncarriers, regardless of age.
Suggested Reading
Webb J, Willette AA. Aging modifies the effect of GCH1 RS11158026 on DAT uptake and Parkinson’s disease clinical severity. Neurobiol Aging. 2017;50:39-46.

Why Do Seizures Sometimes Continue After Surgery?
Roughly one out of every two patients with drug-resistant temporal lobe epilepsy will not become completely seizure-free after temporal lobe surgery. The reasons for this remain unclear and are most likely due to multiple factors. Preoperative automated fiber quantification (AFQ), however, may predict postoperative seizure outcome in patients with temporal lobe epilepsy, according to a study published online ahead of print November 15, 2016, in Brain.
“We have identified three important factors that contribute to persistent postoperative seizures: diffusion abnormalities of the ipsilateral dorsal fornix outside the future margins of resection, diffusion abnormalities of the contralateral parahippocampal white matter bundle, and insufficient resection of the uncinate fasciculus,” said lead author Simon S. Keller, MSc, PhD, and colleagues. Dr. Keller is a Lecturer in Molecular and Clinical Pharmacology at the University of Liverpool in the United Kingdom. “These results may have the potential to be developed into imaging prognostic markers of postoperative outcome and provide new insights for why some patients with temporal lobe epilepsy continue to experience postoperative seizures.”

Sensitive Imaging Technology
MRI techniques such as quantitative volumetric imaging have provided limited insight into what causes recurrent seizures after temporal lobe surgery. AFQ is a diffusion tensor imaging (DTI) tractography technique that permits a comprehensive analysis of tissue characteristics along the length of white matter tract bundles. This technique may allow for a more sensitive measure of neuroanatomic white matter alterations in patients with neurologic disorders than whole-tract approaches.
Dr. Keller and colleagues conducted a comprehensive DTI study to evaluate the local tissue physical characteristics of preoperative temporal lobe white matter tracts by applying DTI and AFQ in patients with temporal lobe epilepsy who underwent surgical treatment and postoperative follow-up. The primary goal of their research was to identify preoperative diffusion markers of postoperative seizure outcome. Their secondary goal was to determine whether the extent of resection of the temporal lobe tract bundles was associated with seizure outcome.
Forty-three patients with mesial temporal lobe epilepsy associated with hippocampal sclerosis and 44 healthy controls were included in the study. Patients underwent preoperative imaging, amygdalohippocampectomy, and postoperative assessment using the International League Against Epilepsy seizure outcome scale. The fimbria-fornix, parahippocampal, white matter bundle, and uncinate fasciculus were reconstructed from preoperative imaging. In addition, scalar diffusion metrics were calculated along the length of each tract.
Results revealed that 51.2% of patients had a completely seizure-free outcome, and 48.8% of patients had persistent postoperative seizures. More men were rendered seizure-free, relative to women. Compared to controls, both patient groups showed strong and significant diffusion abnormalities along the length of the uncinate bilaterally, the ipsilateral parahippocampal white matter bundle, and the ipsilateral fimbria-fornix in regions located within the medial temporal lobe.
However, only patients with persistent postoperative seizures showed evidence of significant pathology of tract sections located in the ipsilateral dorsal fornix and in the contralateral parahippocampal white matter bundle. Using receiver operating characteristic curves, diffusion characteristics of these regions could project individual patient outcomes with 84% sensitivity and 89% specificity.
Pathologic changes in the dorsal fornix were observed beyond the margins of resection. In addition, contralateral parahippocampal changes may suggest a bitemporal disorder in some patients. Diffusion characteristics of the ipsilateral uncinate could potentially classify patients from controls with a sensitivity of 98%.
By coregistering the preoperative fiber maps to postoperative surgical lacuna maps, Dr. Keller and colleagues observed that the extent of the surgical uncinate resection was significantly greater in patients who were rendered seizure-free, suggesting that a smaller surgical resection of the uncinate may represent insufficient disconnection of an anterior temporal epileptogenic network.
“An important future step will be to perform a pragmatic prospective study of consecutive patients with consideration of these new findings,” said Dr. Keller and colleagues.
—Erica Tricarico
Suggested Reading
Keller SS, Glenn RG, Weber B, et al. Preoperative automated fibre quantification predicts postoperative seizure outcome in temporal lobe epilepsy. Brain. 2016 Nov 15 [Epub ahead of print].
Roughly one out of every two patients with drug-resistant temporal lobe epilepsy will not become completely seizure-free after temporal lobe surgery. The reasons for this remain unclear and are most likely due to multiple factors. Preoperative automated fiber quantification (AFQ), however, may predict postoperative seizure outcome in patients with temporal lobe epilepsy, according to a study published online ahead of print November 15, 2016, in Brain.
“We have identified three important factors that contribute to persistent postoperative seizures: diffusion abnormalities of the ipsilateral dorsal fornix outside the future margins of resection, diffusion abnormalities of the contralateral parahippocampal white matter bundle, and insufficient resection of the uncinate fasciculus,” said lead author Simon S. Keller, MSc, PhD, and colleagues. Dr. Keller is a Lecturer in Molecular and Clinical Pharmacology at the University of Liverpool in the United Kingdom. “These results may have the potential to be developed into imaging prognostic markers of postoperative outcome and provide new insights for why some patients with temporal lobe epilepsy continue to experience postoperative seizures.”
Sensitive Imaging Technology
MRI techniques such as quantitative volumetric imaging have provided limited insight into what causes recurrent seizures after temporal lobe surgery. AFQ is a diffusion tensor imaging (DTI) tractography technique that permits a comprehensive analysis of tissue characteristics along the length of white matter tract bundles. This technique may allow for a more sensitive measure of neuroanatomic white matter alterations in patients with neurologic disorders than whole-tract approaches.
Dr. Keller and colleagues conducted a comprehensive DTI study to evaluate the local tissue physical characteristics of preoperative temporal lobe white matter tracts by applying DTI and AFQ in patients with temporal lobe epilepsy who underwent surgical treatment and postoperative follow-up. The primary goal of their research was to identify preoperative diffusion markers of postoperative seizure outcome. Their secondary goal was to determine whether the extent of resection of the temporal lobe tract bundles was associated with seizure outcome.
Forty-three patients with mesial temporal lobe epilepsy associated with hippocampal sclerosis and 44 healthy controls were included in the study. Patients underwent preoperative imaging, amygdalohippocampectomy, and postoperative assessment using the International League Against Epilepsy seizure outcome scale. The fimbria-fornix, parahippocampal, white matter bundle, and uncinate fasciculus were reconstructed from preoperative imaging. In addition, scalar diffusion metrics were calculated along the length of each tract.
Results revealed that 51.2% of patients had a completely seizure-free outcome, and 48.8% of patients had persistent postoperative seizures. More men were rendered seizure-free, relative to women. Compared to controls, both patient groups showed strong and significant diffusion abnormalities along the length of the uncinate bilaterally, the ipsilateral parahippocampal white matter bundle, and the ipsilateral fimbria-fornix in regions located within the medial temporal lobe.
However, only patients with persistent postoperative seizures showed evidence of significant pathology of tract sections located in the ipsilateral dorsal fornix and in the contralateral parahippocampal white matter bundle. Using receiver operating characteristic curves, diffusion characteristics of these regions could project individual patient outcomes with 84% sensitivity and 89% specificity.
Pathologic changes in the dorsal fornix were observed beyond the margins of resection. In addition, contralateral parahippocampal changes may suggest a bitemporal disorder in some patients. Diffusion characteristics of the ipsilateral uncinate could potentially classify patients from controls with a sensitivity of 98%.
By coregistering the preoperative fiber maps to postoperative surgical lacuna maps, Dr. Keller and colleagues observed that the extent of the surgical uncinate resection was significantly greater in patients who were rendered seizure-free, suggesting that a smaller surgical resection of the uncinate may represent insufficient disconnection of an anterior temporal epileptogenic network.
“An important future step will be to perform a pragmatic prospective study of consecutive patients with consideration of these new findings,” said Dr. Keller and colleagues.
—Erica Tricarico
Suggested Reading
Keller SS, Glenn RG, Weber B, et al. Preoperative automated fibre quantification predicts postoperative seizure outcome in temporal lobe epilepsy. Brain. 2016 Nov 15 [Epub ahead of print].
Roughly one out of every two patients with drug-resistant temporal lobe epilepsy will not become completely seizure-free after temporal lobe surgery. The reasons for this remain unclear and are most likely due to multiple factors. Preoperative automated fiber quantification (AFQ), however, may predict postoperative seizure outcome in patients with temporal lobe epilepsy, according to a study published online ahead of print November 15, 2016, in Brain.
“We have identified three important factors that contribute to persistent postoperative seizures: diffusion abnormalities of the ipsilateral dorsal fornix outside the future margins of resection, diffusion abnormalities of the contralateral parahippocampal white matter bundle, and insufficient resection of the uncinate fasciculus,” said lead author Simon S. Keller, MSc, PhD, and colleagues. Dr. Keller is a Lecturer in Molecular and Clinical Pharmacology at the University of Liverpool in the United Kingdom. “These results may have the potential to be developed into imaging prognostic markers of postoperative outcome and provide new insights for why some patients with temporal lobe epilepsy continue to experience postoperative seizures.”
Sensitive Imaging Technology
MRI techniques such as quantitative volumetric imaging have provided limited insight into what causes recurrent seizures after temporal lobe surgery. AFQ is a diffusion tensor imaging (DTI) tractography technique that permits a comprehensive analysis of tissue characteristics along the length of white matter tract bundles. This technique may allow for a more sensitive measure of neuroanatomic white matter alterations in patients with neurologic disorders than whole-tract approaches.
Dr. Keller and colleagues conducted a comprehensive DTI study to evaluate the local tissue physical characteristics of preoperative temporal lobe white matter tracts by applying DTI and AFQ in patients with temporal lobe epilepsy who underwent surgical treatment and postoperative follow-up. The primary goal of their research was to identify preoperative diffusion markers of postoperative seizure outcome. Their secondary goal was to determine whether the extent of resection of the temporal lobe tract bundles was associated with seizure outcome.
Forty-three patients with mesial temporal lobe epilepsy associated with hippocampal sclerosis and 44 healthy controls were included in the study. Patients underwent preoperative imaging, amygdalohippocampectomy, and postoperative assessment using the International League Against Epilepsy seizure outcome scale. The fimbria-fornix, parahippocampal, white matter bundle, and uncinate fasciculus were reconstructed from preoperative imaging. In addition, scalar diffusion metrics were calculated along the length of each tract.
Results revealed that 51.2% of patients had a completely seizure-free outcome, and 48.8% of patients had persistent postoperative seizures. More men were rendered seizure-free, relative to women. Compared to controls, both patient groups showed strong and significant diffusion abnormalities along the length of the uncinate bilaterally, the ipsilateral parahippocampal white matter bundle, and the ipsilateral fimbria-fornix in regions located within the medial temporal lobe.
However, only patients with persistent postoperative seizures showed evidence of significant pathology of tract sections located in the ipsilateral dorsal fornix and in the contralateral parahippocampal white matter bundle. Using receiver operating characteristic curves, diffusion characteristics of these regions could project individual patient outcomes with 84% sensitivity and 89% specificity.
Pathologic changes in the dorsal fornix were observed beyond the margins of resection. In addition, contralateral parahippocampal changes may suggest a bitemporal disorder in some patients. Diffusion characteristics of the ipsilateral uncinate could potentially classify patients from controls with a sensitivity of 98%.
By coregistering the preoperative fiber maps to postoperative surgical lacuna maps, Dr. Keller and colleagues observed that the extent of the surgical uncinate resection was significantly greater in patients who were rendered seizure-free, suggesting that a smaller surgical resection of the uncinate may represent insufficient disconnection of an anterior temporal epileptogenic network.
“An important future step will be to perform a pragmatic prospective study of consecutive patients with consideration of these new findings,” said Dr. Keller and colleagues.
—Erica Tricarico
Suggested Reading
Keller SS, Glenn RG, Weber B, et al. Preoperative automated fibre quantification predicts postoperative seizure outcome in temporal lobe epilepsy. Brain. 2016 Nov 15 [Epub ahead of print].

Racial disparities not seen in child asthma hospitalizations
A study of racial disparities in the hospitalization outcomes of children with asthma in the Medicaid system has found no significant differences in outcomes such as revisit and readmission rates, a study published online Dec. 26 shows.
Researchers examined the outcomes for 11,079 matched pairs of black and white children from the same state, admitted for asthma during a nearly 2-year period across 33 states. The black and white patients were matched on clinical characteristics (Pediatrics. 2016 Dec 26. doi: 10.1542/peds.2016-1221).
However, the study did find that ICU use was significantly higher among black patients, compared with white patients in four states (22.2% vs. 17.5%, P less than .001). Only 23 deaths were recorded among the 22,158 patients – 12 among black patients and 13 among white; a difference that was not significant.
“Because the number of children in Medicaid continues to increase due to the Affordable Care Act, it will be important to keep monitoring for potential racial disparities in hospitalization treatment styles and patient outcomes,” wrote Dr. Silber, of the Children’s Hospital of Philadelphia, and his coauthors. “Because our study was large, including more than 11,000 pairs of patients, we did see some statistically significant differences between black and white Medicaid patients in ICU use and [length of stay], but in most cases, such differences were small in any economic or clinical sense.”
The authors did note some key limitations of study, including a reliance on retrospective Medicaid claims from billing records – which they said may have led to false positives or negatives – and an absence of data on household smoking status and controller medication compliance, both of which could influence readmission and revisit risk.
Dr. Silber and his coauthors also pointed out that children were matched within the state, not within the hospital. “If black children went to worse hospitals than whites, we may not have seen these outcome differences if whites were matched to blacks always within the same hospital.”
The study was supported by the Agency for Healthcare Research and Quality. No conflicts of interest were declared.
The “most disturbing disparities in asthma care” are tied to the practices that limit or deny both outpatient and community care to pediatric Medicaid patients, wrote Lisa D. Young, MD, and Jay G. Berry, MD, MPH, in an accompanying editorial (Pediatrics 2016 Dec 26. doi: 10.1542/peds.2016-3485). “Although we are pleased that Silber and [his] colleagues’ work reports parity in hospital use for children with asthma by race, it will remain important to shine light on other disparities in children with asthma until those disparities are eliminated.”
Among the reasons that such care is either limited or denied is that in some states, Medicaid payments do not underwrite “the cost of the time and effort intensive health services required to optimize the health of the children. Moreover, some states recurrently threaten or enact reductions in funding for their Medicaid program. These legislative actions undoubtedly deincentivize outpatient pediatric providers to care for children with Medicaid,” they wrote.
Dr. Young is affiliated with the Pediatric Clinic and the East Alabama Medical Center, Auburn; Dr. Berry is with the division of general pediatrics at Boston Children’s Hospital and with Harvard Medical School, Boston. No conflicts of interest were declared.
The “most disturbing disparities in asthma care” are tied to the practices that limit or deny both outpatient and community care to pediatric Medicaid patients, wrote Lisa D. Young, MD, and Jay G. Berry, MD, MPH, in an accompanying editorial (Pediatrics 2016 Dec 26. doi: 10.1542/peds.2016-3485). “Although we are pleased that Silber and [his] colleagues’ work reports parity in hospital use for children with asthma by race, it will remain important to shine light on other disparities in children with asthma until those disparities are eliminated.”
Among the reasons that such care is either limited or denied is that in some states, Medicaid payments do not underwrite “the cost of the time and effort intensive health services required to optimize the health of the children. Moreover, some states recurrently threaten or enact reductions in funding for their Medicaid program. These legislative actions undoubtedly deincentivize outpatient pediatric providers to care for children with Medicaid,” they wrote.
Dr. Young is affiliated with the Pediatric Clinic and the East Alabama Medical Center, Auburn; Dr. Berry is with the division of general pediatrics at Boston Children’s Hospital and with Harvard Medical School, Boston. No conflicts of interest were declared.
The “most disturbing disparities in asthma care” are tied to the practices that limit or deny both outpatient and community care to pediatric Medicaid patients, wrote Lisa D. Young, MD, and Jay G. Berry, MD, MPH, in an accompanying editorial (Pediatrics 2016 Dec 26. doi: 10.1542/peds.2016-3485). “Although we are pleased that Silber and [his] colleagues’ work reports parity in hospital use for children with asthma by race, it will remain important to shine light on other disparities in children with asthma until those disparities are eliminated.”
Among the reasons that such care is either limited or denied is that in some states, Medicaid payments do not underwrite “the cost of the time and effort intensive health services required to optimize the health of the children. Moreover, some states recurrently threaten or enact reductions in funding for their Medicaid program. These legislative actions undoubtedly deincentivize outpatient pediatric providers to care for children with Medicaid,” they wrote.
Dr. Young is affiliated with the Pediatric Clinic and the East Alabama Medical Center, Auburn; Dr. Berry is with the division of general pediatrics at Boston Children’s Hospital and with Harvard Medical School, Boston. No conflicts of interest were declared.
A study of racial disparities in the hospitalization outcomes of children with asthma in the Medicaid system has found no significant differences in outcomes such as revisit and readmission rates, a study published online Dec. 26 shows.
Researchers examined the outcomes for 11,079 matched pairs of black and white children from the same state, admitted for asthma during a nearly 2-year period across 33 states. The black and white patients were matched on clinical characteristics (Pediatrics. 2016 Dec 26. doi: 10.1542/peds.2016-1221).
However, the study did find that ICU use was significantly higher among black patients, compared with white patients in four states (22.2% vs. 17.5%, P less than .001). Only 23 deaths were recorded among the 22,158 patients – 12 among black patients and 13 among white; a difference that was not significant.
“Because the number of children in Medicaid continues to increase due to the Affordable Care Act, it will be important to keep monitoring for potential racial disparities in hospitalization treatment styles and patient outcomes,” wrote Dr. Silber, of the Children’s Hospital of Philadelphia, and his coauthors. “Because our study was large, including more than 11,000 pairs of patients, we did see some statistically significant differences between black and white Medicaid patients in ICU use and [length of stay], but in most cases, such differences were small in any economic or clinical sense.”
The authors did note some key limitations of study, including a reliance on retrospective Medicaid claims from billing records – which they said may have led to false positives or negatives – and an absence of data on household smoking status and controller medication compliance, both of which could influence readmission and revisit risk.
Dr. Silber and his coauthors also pointed out that children were matched within the state, not within the hospital. “If black children went to worse hospitals than whites, we may not have seen these outcome differences if whites were matched to blacks always within the same hospital.”
The study was supported by the Agency for Healthcare Research and Quality. No conflicts of interest were declared.
A study of racial disparities in the hospitalization outcomes of children with asthma in the Medicaid system has found no significant differences in outcomes such as revisit and readmission rates, a study published online Dec. 26 shows.
Researchers examined the outcomes for 11,079 matched pairs of black and white children from the same state, admitted for asthma during a nearly 2-year period across 33 states. The black and white patients were matched on clinical characteristics (Pediatrics. 2016 Dec 26. doi: 10.1542/peds.2016-1221).
However, the study did find that ICU use was significantly higher among black patients, compared with white patients in four states (22.2% vs. 17.5%, P less than .001). Only 23 deaths were recorded among the 22,158 patients – 12 among black patients and 13 among white; a difference that was not significant.
“Because the number of children in Medicaid continues to increase due to the Affordable Care Act, it will be important to keep monitoring for potential racial disparities in hospitalization treatment styles and patient outcomes,” wrote Dr. Silber, of the Children’s Hospital of Philadelphia, and his coauthors. “Because our study was large, including more than 11,000 pairs of patients, we did see some statistically significant differences between black and white Medicaid patients in ICU use and [length of stay], but in most cases, such differences were small in any economic or clinical sense.”
The authors did note some key limitations of study, including a reliance on retrospective Medicaid claims from billing records – which they said may have led to false positives or negatives – and an absence of data on household smoking status and controller medication compliance, both of which could influence readmission and revisit risk.
Dr. Silber and his coauthors also pointed out that children were matched within the state, not within the hospital. “If black children went to worse hospitals than whites, we may not have seen these outcome differences if whites were matched to blacks always within the same hospital.”
The study was supported by the Agency for Healthcare Research and Quality. No conflicts of interest were declared.
Key clinical point: No significant differences in revisit or readmission rates were found between black and white children admitted to hospital for asthma.
Major finding: Black and white children hospitalized for asthma across 33 states show no significant differences in revisit or readmission rates or length of stay.
Data source: Retrospective matched cohort study in 11,079 matched pairs of black and white children from within same state.
Disclosures: The study was supported by the Agency for Healthcare Research and Quality. No conflicts of interest were declared.
Regenerative medicine is likely game changer for cardiovascular disease
NEW ORLEANS – Regenerative medicine has much to offer the cardiovascular field, although there is still a way to go before it is ready for routine clinical application, according to Andre Terzic, MD, PhD, director of the Mayo Clinic Center for Regenerative Medicine and a professor in Cardiovascular Diseases Research at the Mayo Clinic, in Rochester, Minn.
Trials testing a variety of stem cell approaches in patients with conditions such as acute myocardial infarction, peripheral arterial disease, and dilated cardiomyopathy are providing important lessons about this novel strategy and showing its potential, he said in a state of the science talk at the American Heart Association scientific sessions.
Impact on therapeutic approaches and goals
“This is a true paradigm shift in how we are approaching patients from a more traditional fighting of disease, whether it’s in the vascular or cardiac arena, to ultimately really where regenerative medicine is driving: rebuilding vascular and heart health,” Dr. Terzic maintained.

Leaders in the field have put forth an associated value proposition, estimating that a decade from now, regenerative medicine will account for about 10% of all health care delivered globally, according to Dr. Terzic. And research presented at the meeting is a major step in that direction.
The potential applications in cardiovascular medicine are numerous, he proposed. Regenerative medicine might be used in prophylactic strategies, for protection against chronic disease, and in bridging strategies, to delay transplantation in patients with end organ damage. It might also be used in combination approaches, to augment the efficacy of primary therapy, and even to generate new organs for patients who have run out of options.
The specific therapeutic goals depend on the stage of disease. For example, the goal is to boost cardioprotection and limit inflammation in acute myocardial infarction; to augment myocardial regenerative reserve and improve survival in subacute disease; and to enhance the pro-reparative environment and restore structure and function in the setting of chronic cardiomyopathy.
Bringing regenerative medicine to the clinic
Ultimately, the health care field is striving to bring the science of regenerative medicine into the clinic, and turn the research pipeline into a clinical service line, Dr. Terzic said.
As might be expected, the whole process begins with identification of an unmet need in patients, which in turn generates a specific practice advancement goal. In the cardiovascular arena, the process has largely been driven by smaller collaborative groups and champions in the field, but is increasingly garnering new support from the business world and national networks.
“The importance at the moment is the experiences that are being built allow for a true iterative process in harnessing this new knowledge and essentially developing even more refined and more effective solutions as we look into the future,” Dr. Terzic commented.
“The cardiovascular field is clearly not alone, but is working in tandem with many other fields that are evolving rapidly in this area,” he added. “And I think the collective experience will be one of the main drivers as we build this shared vision for a practice not only for regenerative solutions, but for practice-integrated regenerative medicine, ultimately, as one of the models of care in the decade to come.”
A blueprint for moving forward
Dr. Terzic and his colleagues at the Mayo Clinic have developed a framework for advancing the field that is being used at their institution but could also be used elsewhere (Eur Heart J. 2016;37:1089-90).
“What we have historically been doing for the last decade or so is really building this discovery knowledge, and then moving it into translation and ultimately application,” he explained. “We call this a blueprint, a blueprint that has helped us all collectively get where we are today.”
For each of these three domains, investigators strive to build out the concept, starting with the specific regenerative technology or product, proceeding to its manufacture or production, and eventually devising ways to deliver it to patients through a clinical-grade supply chain available at the point of care.
“There is a new evolving concept of truly building a regenerative medicine care model,” summarized Dr. Terzic, who disclosed no relevant conflicts of interest. “Today, we are not fully there. We are really speaking about technological and translational readiness. But ultimately, the goal is to ensure clinical readiness.”
NEW ORLEANS – Regenerative medicine has much to offer the cardiovascular field, although there is still a way to go before it is ready for routine clinical application, according to Andre Terzic, MD, PhD, director of the Mayo Clinic Center for Regenerative Medicine and a professor in Cardiovascular Diseases Research at the Mayo Clinic, in Rochester, Minn.
Trials testing a variety of stem cell approaches in patients with conditions such as acute myocardial infarction, peripheral arterial disease, and dilated cardiomyopathy are providing important lessons about this novel strategy and showing its potential, he said in a state of the science talk at the American Heart Association scientific sessions.
Impact on therapeutic approaches and goals
“This is a true paradigm shift in how we are approaching patients from a more traditional fighting of disease, whether it’s in the vascular or cardiac arena, to ultimately really where regenerative medicine is driving: rebuilding vascular and heart health,” Dr. Terzic maintained.
Leaders in the field have put forth an associated value proposition, estimating that a decade from now, regenerative medicine will account for about 10% of all health care delivered globally, according to Dr. Terzic. And research presented at the meeting is a major step in that direction.
The potential applications in cardiovascular medicine are numerous, he proposed. Regenerative medicine might be used in prophylactic strategies, for protection against chronic disease, and in bridging strategies, to delay transplantation in patients with end organ damage. It might also be used in combination approaches, to augment the efficacy of primary therapy, and even to generate new organs for patients who have run out of options.
The specific therapeutic goals depend on the stage of disease. For example, the goal is to boost cardioprotection and limit inflammation in acute myocardial infarction; to augment myocardial regenerative reserve and improve survival in subacute disease; and to enhance the pro-reparative environment and restore structure and function in the setting of chronic cardiomyopathy.
Bringing regenerative medicine to the clinic
Ultimately, the health care field is striving to bring the science of regenerative medicine into the clinic, and turn the research pipeline into a clinical service line, Dr. Terzic said.
As might be expected, the whole process begins with identification of an unmet need in patients, which in turn generates a specific practice advancement goal. In the cardiovascular arena, the process has largely been driven by smaller collaborative groups and champions in the field, but is increasingly garnering new support from the business world and national networks.
“The importance at the moment is the experiences that are being built allow for a true iterative process in harnessing this new knowledge and essentially developing even more refined and more effective solutions as we look into the future,” Dr. Terzic commented.
“The cardiovascular field is clearly not alone, but is working in tandem with many other fields that are evolving rapidly in this area,” he added. “And I think the collective experience will be one of the main drivers as we build this shared vision for a practice not only for regenerative solutions, but for practice-integrated regenerative medicine, ultimately, as one of the models of care in the decade to come.”
A blueprint for moving forward
Dr. Terzic and his colleagues at the Mayo Clinic have developed a framework for advancing the field that is being used at their institution but could also be used elsewhere (Eur Heart J. 2016;37:1089-90).
“What we have historically been doing for the last decade or so is really building this discovery knowledge, and then moving it into translation and ultimately application,” he explained. “We call this a blueprint, a blueprint that has helped us all collectively get where we are today.”
For each of these three domains, investigators strive to build out the concept, starting with the specific regenerative technology or product, proceeding to its manufacture or production, and eventually devising ways to deliver it to patients through a clinical-grade supply chain available at the point of care.
“There is a new evolving concept of truly building a regenerative medicine care model,” summarized Dr. Terzic, who disclosed no relevant conflicts of interest. “Today, we are not fully there. We are really speaking about technological and translational readiness. But ultimately, the goal is to ensure clinical readiness.”
NEW ORLEANS – Regenerative medicine has much to offer the cardiovascular field, although there is still a way to go before it is ready for routine clinical application, according to Andre Terzic, MD, PhD, director of the Mayo Clinic Center for Regenerative Medicine and a professor in Cardiovascular Diseases Research at the Mayo Clinic, in Rochester, Minn.
Trials testing a variety of stem cell approaches in patients with conditions such as acute myocardial infarction, peripheral arterial disease, and dilated cardiomyopathy are providing important lessons about this novel strategy and showing its potential, he said in a state of the science talk at the American Heart Association scientific sessions.
Impact on therapeutic approaches and goals
“This is a true paradigm shift in how we are approaching patients from a more traditional fighting of disease, whether it’s in the vascular or cardiac arena, to ultimately really where regenerative medicine is driving: rebuilding vascular and heart health,” Dr. Terzic maintained.
Leaders in the field have put forth an associated value proposition, estimating that a decade from now, regenerative medicine will account for about 10% of all health care delivered globally, according to Dr. Terzic. And research presented at the meeting is a major step in that direction.
The potential applications in cardiovascular medicine are numerous, he proposed. Regenerative medicine might be used in prophylactic strategies, for protection against chronic disease, and in bridging strategies, to delay transplantation in patients with end organ damage. It might also be used in combination approaches, to augment the efficacy of primary therapy, and even to generate new organs for patients who have run out of options.
The specific therapeutic goals depend on the stage of disease. For example, the goal is to boost cardioprotection and limit inflammation in acute myocardial infarction; to augment myocardial regenerative reserve and improve survival in subacute disease; and to enhance the pro-reparative environment and restore structure and function in the setting of chronic cardiomyopathy.
Bringing regenerative medicine to the clinic
Ultimately, the health care field is striving to bring the science of regenerative medicine into the clinic, and turn the research pipeline into a clinical service line, Dr. Terzic said.
As might be expected, the whole process begins with identification of an unmet need in patients, which in turn generates a specific practice advancement goal. In the cardiovascular arena, the process has largely been driven by smaller collaborative groups and champions in the field, but is increasingly garnering new support from the business world and national networks.
“The importance at the moment is the experiences that are being built allow for a true iterative process in harnessing this new knowledge and essentially developing even more refined and more effective solutions as we look into the future,” Dr. Terzic commented.
“The cardiovascular field is clearly not alone, but is working in tandem with many other fields that are evolving rapidly in this area,” he added. “And I think the collective experience will be one of the main drivers as we build this shared vision for a practice not only for regenerative solutions, but for practice-integrated regenerative medicine, ultimately, as one of the models of care in the decade to come.”
A blueprint for moving forward
Dr. Terzic and his colleagues at the Mayo Clinic have developed a framework for advancing the field that is being used at their institution but could also be used elsewhere (Eur Heart J. 2016;37:1089-90).
“What we have historically been doing for the last decade or so is really building this discovery knowledge, and then moving it into translation and ultimately application,” he explained. “We call this a blueprint, a blueprint that has helped us all collectively get where we are today.”
For each of these three domains, investigators strive to build out the concept, starting with the specific regenerative technology or product, proceeding to its manufacture or production, and eventually devising ways to deliver it to patients through a clinical-grade supply chain available at the point of care.
“There is a new evolving concept of truly building a regenerative medicine care model,” summarized Dr. Terzic, who disclosed no relevant conflicts of interest. “Today, we are not fully there. We are really speaking about technological and translational readiness. But ultimately, the goal is to ensure clinical readiness.”
Key clinical point:
Major finding: Trials testing a variety of stem cell approaches in patients with conditions such as acute MI, PAD, and dilated cardiomyopathy are providing important lessons.
Data source: Expert analysis of current trials by Dr. Terzic.
Disclosures: Dr. Terzic disclosed that he had no relevant conflicts of interest.
Options for Acute Stroke Evolving Rapidly With New Standards for Optimal Management
LAS VEGAS—Recent changes in the labeling of t-PA, along with FDA approval of a clot retrieval device, are increasing the pressure for clinicians to consider aggressive treatment of acute stroke in a broader range of patients, according to an update at the American Academy of Neurology’s Fall 2016 Conference. These changes have been shifting the orientation so that t-PA is not just an option, but is now widely regarded as a benchmark for quality of care at centers where acute stroke is treated.
Offering patients with acute stroke this lytic agent “has also become an incredibly important legal benchmark, as there is reportedly more litigation ... in regard to missing opportunities to administer t-PA rather than as a result of complications of t-PA administration,” said Natalia S. Rost, MD, Director of Acute Stroke Services at Massachusetts General Hospital in Boston. Delivering the update on acute stroke, Dr. Rost said it is important that clinicians adjust to these label changes to increase the proportion of eligible patients who receive t-PA, which she characterized as “the only FDA-approved medication shown to improve stroke outcomes.”

Reconsidering Contraindications
The major risk of t-PA is potentially life-threatening bleeding events, which is the basis for most contraindications and the source of hesitation for many clinicians concerned about an iatrogenic complication. However, more than a decade of experience has demonstrated that the benefit-to-risk ratio favors treatment in many patients originally listed as contraindicated. A reevaluation of the evidence by an American Heart Association/American Stroke Association (AHA/ASA) joint committee contributed to the 2015 decision to adjust the labeling.
“For example, the history of prior stroke as a contraindication has been removed entirely,” Dr. Rost said. Other examples singled out by Dr. Rost include patients who had a seizure at stroke onset, patients with mild stroke, patients with severe stroke, patients with early signs of infarction on CT scan, and patients with diabetes.
In other cases, absolute contraindications have been relisted as warnings. These include elderly age, history of gastrointestinal bleeding, and pregnancy. Severe and difficult-to-control hypertension and history of subarachnoid hemorrhage remain contraindications, but Dr. Rost noted that the specifics have been removed, permitting some discretion in how these terms are defined.
While loosening of the contraindications for t-PA is designed to increase the proportion of patients who receive this potentially life-saving therapy, the relisting of the indications may also bring more rigor to clinical practice. A review by Demaerschalk et al included a survey of clinicians that showed wide variability in practice. The survey showed that less than 40% of clinicians reported that they were willing to administer t-PA to patients who had had a myocardial infarction in the previous three months; less than 20% reported a willingness to administer t-PA to patients who had had major surgery in the previous 14 days.
Stroke specialists have long considered t-PA to be the standard of care for patients with stroke presenting within three hours of symptoms, but the AHA/ASA review of safety data has shifted the orientation. Dr. Rost said that clinicians should no longer consider t-PA among other options in patients presenting within three hours of symptom onset, but rather “assume t-PA treatment unless contraindicated.”
Intra-Arterial Thrombolysis
There have also been recent changes in regard to the indications of intra-arterial thrombolysis devices in conjunction with t-PA. Various mechanical devices for removing clots associated with stroke (ie, mechanical thrombectomy) have been available for more than 10 years, but were not initially supported by evidence that they improved functional outcomes. This situation has changed, according to Dr. Rost. She gave credit to stent retrievers, the most recent evolution in mechanical devices, which provide “almost an immediate path to reperfusion.” She called the technology instrumental to the clinical benefits now being seen.
Although the first multicenter trial to demonstrate an improvement in functional independence, MR CLEAN, was published in December 2014, there have been several subsequent trials, including ESCAPE, EXTEND-IA, and SWIFT-PRIME, all showing similar benefits of mechanical thrombectomy when conducted under standardized patient selection criteria.
More data are likely to be coming, according to Dr. Rost, but the AHA/ASA guidelines for intra-arterial thrombolysis have already been revised. According to the 2015 update published in Stroke, patients should be considered for endovascular therapy with a stent retriever if they have an occlusion in the internal carotid artery or proximal middle cerebral artery, age of at least 18, modified Rankin Scale score no higher than 1, NIH Stroke Scale (NIHSS) score of at least 6, and an Alberta Stroke Program Early CT Score (ASPECTS) of at least 6.
These guidelines further specify that groin puncture for this procedure must be initiated within six hours of symptom onset. The update highlights that all patients presenting within 4.5 hours of symptom onset should be considered for treatment with t-PA prior to consideration of mechanical thrombectomy.
On September 2, 2016, the “FDA for the first time approved a mechanical device for the indication of improvement in functional outcome,” said Dr. Rost. The approval included devices that were used strictly within six hours of symptom onset and only following treatment with t-PA in the first three hours of stroke. The new labeling is specific for the Trevo ProVue and Trevo XP ProVue clot retrieval devices, but Dr. Rost predicted that many of the other devices using similar technology will eventually receive the same approval.
Stroke Science Continues to Evolve
The adage that time is brain in acute stroke is well known, but it was recently reinforced in a meta-analysis of thrombectomy trials that demonstrated that most benefit from stroke treatments is achieved if reperfusion occurs within seven hours of symptom onset. Citing data from Saver et al, Dr. Rost reiterated the importance of adhering to recommendations for early administration of the most effective therapies to improve functional outcomes.
Although clinicians should know and adhere to guidelines to deliver acute stroke treatments to patients “proven to benefit from these therapies,” Dr. Rost expects the guidelines to continue to evolve with new studies and more experience, particularly with mechanical endovascular devices. “Just as we have recently seen with t-PA, we may in time be able to relax the parameters of who is treated [with these devices],” Dr. Rost said.
Dr. Rost reported financial relationships with BioTelemetry, Daiichi Sankyo, Genzyme, and Omniox.
—Theodore Bosworth
Suggested Reading
Berkhemer OA, Fransen PS, Beumer D, et al. A randomized trial of intraarterial treatment for acute ischemic stroke. N Engl J Med. 2015;372(1):11-20.
Demaerschalk BM, Kleindorfer Do, Adeoye OM, et al. Scientific rationale for the inclusion and exlusion criteria for intravenous alteplase in acute ischemeic stroke: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2016;47(2):581-641.
Powers WJ, Derdeyn CP, Biller J, et al. 2015 American Heart Association/American Stroke Association focused update of the 2013 guidelines for the early management of patients with acute ischemic stroke regarding endovascular treatment: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2015;46(10):3020-3035.
Saver JL, Goyal M, van der Lugt A, et al. Time to treatment with endovascular thrombectomy and outcomes from ischemic stroke: a meta-analysis. JAMA. 2016;316(12):1279-1288.
LAS VEGAS—Recent changes in the labeling of t-PA, along with FDA approval of a clot retrieval device, are increasing the pressure for clinicians to consider aggressive treatment of acute stroke in a broader range of patients, according to an update at the American Academy of Neurology’s Fall 2016 Conference. These changes have been shifting the orientation so that t-PA is not just an option, but is now widely regarded as a benchmark for quality of care at centers where acute stroke is treated.
Offering patients with acute stroke this lytic agent “has also become an incredibly important legal benchmark, as there is reportedly more litigation ... in regard to missing opportunities to administer t-PA rather than as a result of complications of t-PA administration,” said Natalia S. Rost, MD, Director of Acute Stroke Services at Massachusetts General Hospital in Boston. Delivering the update on acute stroke, Dr. Rost said it is important that clinicians adjust to these label changes to increase the proportion of eligible patients who receive t-PA, which she characterized as “the only FDA-approved medication shown to improve stroke outcomes.”
Reconsidering Contraindications
The major risk of t-PA is potentially life-threatening bleeding events, which is the basis for most contraindications and the source of hesitation for many clinicians concerned about an iatrogenic complication. However, more than a decade of experience has demonstrated that the benefit-to-risk ratio favors treatment in many patients originally listed as contraindicated. A reevaluation of the evidence by an American Heart Association/American Stroke Association (AHA/ASA) joint committee contributed to the 2015 decision to adjust the labeling.
“For example, the history of prior stroke as a contraindication has been removed entirely,” Dr. Rost said. Other examples singled out by Dr. Rost include patients who had a seizure at stroke onset, patients with mild stroke, patients with severe stroke, patients with early signs of infarction on CT scan, and patients with diabetes.
In other cases, absolute contraindications have been relisted as warnings. These include elderly age, history of gastrointestinal bleeding, and pregnancy. Severe and difficult-to-control hypertension and history of subarachnoid hemorrhage remain contraindications, but Dr. Rost noted that the specifics have been removed, permitting some discretion in how these terms are defined.
While loosening of the contraindications for t-PA is designed to increase the proportion of patients who receive this potentially life-saving therapy, the relisting of the indications may also bring more rigor to clinical practice. A review by Demaerschalk et al included a survey of clinicians that showed wide variability in practice. The survey showed that less than 40% of clinicians reported that they were willing to administer t-PA to patients who had had a myocardial infarction in the previous three months; less than 20% reported a willingness to administer t-PA to patients who had had major surgery in the previous 14 days.
Stroke specialists have long considered t-PA to be the standard of care for patients with stroke presenting within three hours of symptoms, but the AHA/ASA review of safety data has shifted the orientation. Dr. Rost said that clinicians should no longer consider t-PA among other options in patients presenting within three hours of symptom onset, but rather “assume t-PA treatment unless contraindicated.”
Intra-Arterial Thrombolysis
There have also been recent changes in regard to the indications of intra-arterial thrombolysis devices in conjunction with t-PA. Various mechanical devices for removing clots associated with stroke (ie, mechanical thrombectomy) have been available for more than 10 years, but were not initially supported by evidence that they improved functional outcomes. This situation has changed, according to Dr. Rost. She gave credit to stent retrievers, the most recent evolution in mechanical devices, which provide “almost an immediate path to reperfusion.” She called the technology instrumental to the clinical benefits now being seen.
Although the first multicenter trial to demonstrate an improvement in functional independence, MR CLEAN, was published in December 2014, there have been several subsequent trials, including ESCAPE, EXTEND-IA, and SWIFT-PRIME, all showing similar benefits of mechanical thrombectomy when conducted under standardized patient selection criteria.
More data are likely to be coming, according to Dr. Rost, but the AHA/ASA guidelines for intra-arterial thrombolysis have already been revised. According to the 2015 update published in Stroke, patients should be considered for endovascular therapy with a stent retriever if they have an occlusion in the internal carotid artery or proximal middle cerebral artery, age of at least 18, modified Rankin Scale score no higher than 1, NIH Stroke Scale (NIHSS) score of at least 6, and an Alberta Stroke Program Early CT Score (ASPECTS) of at least 6.
These guidelines further specify that groin puncture for this procedure must be initiated within six hours of symptom onset. The update highlights that all patients presenting within 4.5 hours of symptom onset should be considered for treatment with t-PA prior to consideration of mechanical thrombectomy.
On September 2, 2016, the “FDA for the first time approved a mechanical device for the indication of improvement in functional outcome,” said Dr. Rost. The approval included devices that were used strictly within six hours of symptom onset and only following treatment with t-PA in the first three hours of stroke. The new labeling is specific for the Trevo ProVue and Trevo XP ProVue clot retrieval devices, but Dr. Rost predicted that many of the other devices using similar technology will eventually receive the same approval.
Stroke Science Continues to Evolve
The adage that time is brain in acute stroke is well known, but it was recently reinforced in a meta-analysis of thrombectomy trials that demonstrated that most benefit from stroke treatments is achieved if reperfusion occurs within seven hours of symptom onset. Citing data from Saver et al, Dr. Rost reiterated the importance of adhering to recommendations for early administration of the most effective therapies to improve functional outcomes.
Although clinicians should know and adhere to guidelines to deliver acute stroke treatments to patients “proven to benefit from these therapies,” Dr. Rost expects the guidelines to continue to evolve with new studies and more experience, particularly with mechanical endovascular devices. “Just as we have recently seen with t-PA, we may in time be able to relax the parameters of who is treated [with these devices],” Dr. Rost said.
Dr. Rost reported financial relationships with BioTelemetry, Daiichi Sankyo, Genzyme, and Omniox.
—Theodore Bosworth
Suggested Reading
Berkhemer OA, Fransen PS, Beumer D, et al. A randomized trial of intraarterial treatment for acute ischemic stroke. N Engl J Med. 2015;372(1):11-20.
Demaerschalk BM, Kleindorfer Do, Adeoye OM, et al. Scientific rationale for the inclusion and exlusion criteria for intravenous alteplase in acute ischemeic stroke: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2016;47(2):581-641.
Powers WJ, Derdeyn CP, Biller J, et al. 2015 American Heart Association/American Stroke Association focused update of the 2013 guidelines for the early management of patients with acute ischemic stroke regarding endovascular treatment: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2015;46(10):3020-3035.
Saver JL, Goyal M, van der Lugt A, et al. Time to treatment with endovascular thrombectomy and outcomes from ischemic stroke: a meta-analysis. JAMA. 2016;316(12):1279-1288.
LAS VEGAS—Recent changes in the labeling of t-PA, along with FDA approval of a clot retrieval device, are increasing the pressure for clinicians to consider aggressive treatment of acute stroke in a broader range of patients, according to an update at the American Academy of Neurology’s Fall 2016 Conference. These changes have been shifting the orientation so that t-PA is not just an option, but is now widely regarded as a benchmark for quality of care at centers where acute stroke is treated.
Offering patients with acute stroke this lytic agent “has also become an incredibly important legal benchmark, as there is reportedly more litigation ... in regard to missing opportunities to administer t-PA rather than as a result of complications of t-PA administration,” said Natalia S. Rost, MD, Director of Acute Stroke Services at Massachusetts General Hospital in Boston. Delivering the update on acute stroke, Dr. Rost said it is important that clinicians adjust to these label changes to increase the proportion of eligible patients who receive t-PA, which she characterized as “the only FDA-approved medication shown to improve stroke outcomes.”
Reconsidering Contraindications
The major risk of t-PA is potentially life-threatening bleeding events, which is the basis for most contraindications and the source of hesitation for many clinicians concerned about an iatrogenic complication. However, more than a decade of experience has demonstrated that the benefit-to-risk ratio favors treatment in many patients originally listed as contraindicated. A reevaluation of the evidence by an American Heart Association/American Stroke Association (AHA/ASA) joint committee contributed to the 2015 decision to adjust the labeling.
“For example, the history of prior stroke as a contraindication has been removed entirely,” Dr. Rost said. Other examples singled out by Dr. Rost include patients who had a seizure at stroke onset, patients with mild stroke, patients with severe stroke, patients with early signs of infarction on CT scan, and patients with diabetes.
In other cases, absolute contraindications have been relisted as warnings. These include elderly age, history of gastrointestinal bleeding, and pregnancy. Severe and difficult-to-control hypertension and history of subarachnoid hemorrhage remain contraindications, but Dr. Rost noted that the specifics have been removed, permitting some discretion in how these terms are defined.
While loosening of the contraindications for t-PA is designed to increase the proportion of patients who receive this potentially life-saving therapy, the relisting of the indications may also bring more rigor to clinical practice. A review by Demaerschalk et al included a survey of clinicians that showed wide variability in practice. The survey showed that less than 40% of clinicians reported that they were willing to administer t-PA to patients who had had a myocardial infarction in the previous three months; less than 20% reported a willingness to administer t-PA to patients who had had major surgery in the previous 14 days.
Stroke specialists have long considered t-PA to be the standard of care for patients with stroke presenting within three hours of symptoms, but the AHA/ASA review of safety data has shifted the orientation. Dr. Rost said that clinicians should no longer consider t-PA among other options in patients presenting within three hours of symptom onset, but rather “assume t-PA treatment unless contraindicated.”
Intra-Arterial Thrombolysis
There have also been recent changes in regard to the indications of intra-arterial thrombolysis devices in conjunction with t-PA. Various mechanical devices for removing clots associated with stroke (ie, mechanical thrombectomy) have been available for more than 10 years, but were not initially supported by evidence that they improved functional outcomes. This situation has changed, according to Dr. Rost. She gave credit to stent retrievers, the most recent evolution in mechanical devices, which provide “almost an immediate path to reperfusion.” She called the technology instrumental to the clinical benefits now being seen.
Although the first multicenter trial to demonstrate an improvement in functional independence, MR CLEAN, was published in December 2014, there have been several subsequent trials, including ESCAPE, EXTEND-IA, and SWIFT-PRIME, all showing similar benefits of mechanical thrombectomy when conducted under standardized patient selection criteria.
More data are likely to be coming, according to Dr. Rost, but the AHA/ASA guidelines for intra-arterial thrombolysis have already been revised. According to the 2015 update published in Stroke, patients should be considered for endovascular therapy with a stent retriever if they have an occlusion in the internal carotid artery or proximal middle cerebral artery, age of at least 18, modified Rankin Scale score no higher than 1, NIH Stroke Scale (NIHSS) score of at least 6, and an Alberta Stroke Program Early CT Score (ASPECTS) of at least 6.
These guidelines further specify that groin puncture for this procedure must be initiated within six hours of symptom onset. The update highlights that all patients presenting within 4.5 hours of symptom onset should be considered for treatment with t-PA prior to consideration of mechanical thrombectomy.
On September 2, 2016, the “FDA for the first time approved a mechanical device for the indication of improvement in functional outcome,” said Dr. Rost. The approval included devices that were used strictly within six hours of symptom onset and only following treatment with t-PA in the first three hours of stroke. The new labeling is specific for the Trevo ProVue and Trevo XP ProVue clot retrieval devices, but Dr. Rost predicted that many of the other devices using similar technology will eventually receive the same approval.
Stroke Science Continues to Evolve
The adage that time is brain in acute stroke is well known, but it was recently reinforced in a meta-analysis of thrombectomy trials that demonstrated that most benefit from stroke treatments is achieved if reperfusion occurs within seven hours of symptom onset. Citing data from Saver et al, Dr. Rost reiterated the importance of adhering to recommendations for early administration of the most effective therapies to improve functional outcomes.
Although clinicians should know and adhere to guidelines to deliver acute stroke treatments to patients “proven to benefit from these therapies,” Dr. Rost expects the guidelines to continue to evolve with new studies and more experience, particularly with mechanical endovascular devices. “Just as we have recently seen with t-PA, we may in time be able to relax the parameters of who is treated [with these devices],” Dr. Rost said.
Dr. Rost reported financial relationships with BioTelemetry, Daiichi Sankyo, Genzyme, and Omniox.
—Theodore Bosworth
Suggested Reading
Berkhemer OA, Fransen PS, Beumer D, et al. A randomized trial of intraarterial treatment for acute ischemic stroke. N Engl J Med. 2015;372(1):11-20.
Demaerschalk BM, Kleindorfer Do, Adeoye OM, et al. Scientific rationale for the inclusion and exlusion criteria for intravenous alteplase in acute ischemeic stroke: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2016;47(2):581-641.
Powers WJ, Derdeyn CP, Biller J, et al. 2015 American Heart Association/American Stroke Association focused update of the 2013 guidelines for the early management of patients with acute ischemic stroke regarding endovascular treatment: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2015;46(10):3020-3035.
Saver JL, Goyal M, van der Lugt A, et al. Time to treatment with endovascular thrombectomy and outcomes from ischemic stroke: a meta-analysis. JAMA. 2016;316(12):1279-1288.

Which Factors Predict Response to Acute Migraine Treatment?
Demographic variables, headache features, comorbidity, and treatment factors may predict inadequate response to acute migraine treatment at two hours and at 24 hours, according to research published in the November issue of Headache. Similar factors may predict which patients with an adequate response at two hours will have an inadequate response at 24 hours.
The data suggest substantial unmet acute treatment needs at the population level, said Richard B. Lipton, MD, Edwin S. Lowe Chair in Neurology at the Albert Einstein College of Medicine in Bronx, New York, and colleagues. “A good response at two hours was associated with doing well at 24 hours. This [result] highlights the importance of initial response to treatment in overall patient outcomes.”

An Analysis of AMPP Data
Much of the literature intended to identify predictors of response to migraine treatment focuses on outcomes of individual attacks. Study populations generally are limited, furthermore, to the select group of patients willing to participate in trials. A thorough evaluation of unmet acute treatment needs requires a more representative sample population, as well as information about long-term responses to multiple attacks, said Dr. Lipton.
He and his colleagues examined data from the American Migraine Prevalence and Prevention (AMPP) Study to identify factors that predict the success or failure of acute treatment at two hours and at 24 hours. The investigators focused on the 2006 AMPP survey, which included the Migraine Treatment Optimization Questionnaire (mTOQ). Eligible participants met criteria for episodic migraine, used acute pharmacologic treatment for migraine, and provided the necessary data for the researchers’ analysis.
In all, 14,520 people responded to the 2006 survey, 10,006 of whom met International Classification of Headache Disorders-3 beta criteria for migraine. Dr. Lipton’s group examined two questions from the mTOQ to assess pain-response outcomes following acute treatment. The first question asked whether the respondent was pain-free within two hours of treatment for most attacks. The second asked whether one dose of medication usually relieved the respondent’s headache and kept it away for at least 24 hours.
In all, 8,233 people responded to both questions. Patients who responded “never,” “rarely,” or “less than half the time” to the first or second question were considered to have an inadequate two-hour pain-free response or an inadequate 24-hour pain relief response, respectively. A response of “half the time or more” was defined as an adequate response. In addition, the researchers defined a 24-hour sustained pain-free response as an adequate response to both questions. Participants with an adequate two-hour response and an inadequate 24-hour response were considered to have recurrence. To identify outcome predictors, Dr. Lipton and colleagues conducted logistic regression analyses.
Most Participants Had Inadequate Response
Most participants (56.0%) reported inadequate two-hour pain-free response to usual acute treatment, and 53.6% of respondents reported inadequate 24-hour pain relief. Of the 44.0% of people with adequate two-hour pain-free response, 74.3% reported sustained pain relief at 24 hours.
The significant predictors of inadequate two-hour pain-free response were greater pain intensity, cutaneous allodynia, depression, higher BMI, and higher average monthly headache day frequency. Factors that protected against an inadequate two-hour pain-free response included using a preventive medication for migraine, female gender, and being married.
Factors that predicted inadequate 24-hour pain relief included greater feelings of depression, cutaneous allodynia, greater monthly headache day frequency, greater headache pain intensity, overuse of acute medication, lack of health insurance, being a smoker, and being unmarried. Predictors of inadequate 24-hour sustained pain-free response were greater monthly headache day frequency, cutaneous allodynia, meeting criteria for depression, acute medication overuse, and migraine symptom severity.
A Need for Treatment Optimization
Previous studies have found an association between high BMI and severe and progressive forms of migraine. This association “may reflect a proinflammatory state in obesity that renders treatment less effective,” said Dr. Lipton.
The authors’ finding of an association between depression and inadequate response is consistent with previous research suggesting that depression is a risk factor for headache progression. Preventive migraine medications were protective against this outcome, however.
A possible explanation for smokers’ higher likelihood of having inadequate 24-hour pain relief is that “smoking may alter drug metabolism and shorten the duration of action of selected acute treatments,” said Dr. Lipton. In addition, the association between monthly headache days and inadequate 24-hour pain relief “may reflect the fact that more frequent attacks may be associated with prolonged activation of neuronal networks involved in pain processing during attacks, which may lead to lowering the threshold for subsequent attacks.”
One limitation of the current study is its reliance on self-reported data, said the authors. The questionnaire that the researchers used is limited by recall bias, recency effects, and the risk that the preceding month did not represent the individual’s usual experience. Nonetheless, mTOQ items have demonstrated high reliability and validity. Other study limitations include the retrospective design, the high proportion of participants who used more than one acute treatment, and the fact that the data are 10 years old.
On the other hand, the study examined a large, representative sample of the US population. It also included various validated measures to diagnose migraine and to assess headache-related disability, allodynia, and depression.
“These results show that unmet needs remain, and the expansion of therapeutic options for episodic migraine is needed, as well as optimizing treatment by carefully designing comprehensive treatment plans with existing acute therapies with various doses, routes of administration, preventive and interventional treatment approaches, behavioral therapies, neuromodulators, and other empirically validated approaches to achieve optimized treatment,” Dr. Lipton concluded.
—Erik Greb
Suggested Reading
Lipton RB, Munjal S, Buse DC, et al. Predicting inadequate response to acute migraine medication: results from the American Migraine Prevalence and Prevention (AMPP) Study. Headache. 2016;56(10):1635-1648.
Demographic variables, headache features, comorbidity, and treatment factors may predict inadequate response to acute migraine treatment at two hours and at 24 hours, according to research published in the November issue of Headache. Similar factors may predict which patients with an adequate response at two hours will have an inadequate response at 24 hours.
The data suggest substantial unmet acute treatment needs at the population level, said Richard B. Lipton, MD, Edwin S. Lowe Chair in Neurology at the Albert Einstein College of Medicine in Bronx, New York, and colleagues. “A good response at two hours was associated with doing well at 24 hours. This [result] highlights the importance of initial response to treatment in overall patient outcomes.”
An Analysis of AMPP Data
Much of the literature intended to identify predictors of response to migraine treatment focuses on outcomes of individual attacks. Study populations generally are limited, furthermore, to the select group of patients willing to participate in trials. A thorough evaluation of unmet acute treatment needs requires a more representative sample population, as well as information about long-term responses to multiple attacks, said Dr. Lipton.
He and his colleagues examined data from the American Migraine Prevalence and Prevention (AMPP) Study to identify factors that predict the success or failure of acute treatment at two hours and at 24 hours. The investigators focused on the 2006 AMPP survey, which included the Migraine Treatment Optimization Questionnaire (mTOQ). Eligible participants met criteria for episodic migraine, used acute pharmacologic treatment for migraine, and provided the necessary data for the researchers’ analysis.
In all, 14,520 people responded to the 2006 survey, 10,006 of whom met International Classification of Headache Disorders-3 beta criteria for migraine. Dr. Lipton’s group examined two questions from the mTOQ to assess pain-response outcomes following acute treatment. The first question asked whether the respondent was pain-free within two hours of treatment for most attacks. The second asked whether one dose of medication usually relieved the respondent’s headache and kept it away for at least 24 hours.
In all, 8,233 people responded to both questions. Patients who responded “never,” “rarely,” or “less than half the time” to the first or second question were considered to have an inadequate two-hour pain-free response or an inadequate 24-hour pain relief response, respectively. A response of “half the time or more” was defined as an adequate response. In addition, the researchers defined a 24-hour sustained pain-free response as an adequate response to both questions. Participants with an adequate two-hour response and an inadequate 24-hour response were considered to have recurrence. To identify outcome predictors, Dr. Lipton and colleagues conducted logistic regression analyses.
Most Participants Had Inadequate Response
Most participants (56.0%) reported inadequate two-hour pain-free response to usual acute treatment, and 53.6% of respondents reported inadequate 24-hour pain relief. Of the 44.0% of people with adequate two-hour pain-free response, 74.3% reported sustained pain relief at 24 hours.
The significant predictors of inadequate two-hour pain-free response were greater pain intensity, cutaneous allodynia, depression, higher BMI, and higher average monthly headache day frequency. Factors that protected against an inadequate two-hour pain-free response included using a preventive medication for migraine, female gender, and being married.
Factors that predicted inadequate 24-hour pain relief included greater feelings of depression, cutaneous allodynia, greater monthly headache day frequency, greater headache pain intensity, overuse of acute medication, lack of health insurance, being a smoker, and being unmarried. Predictors of inadequate 24-hour sustained pain-free response were greater monthly headache day frequency, cutaneous allodynia, meeting criteria for depression, acute medication overuse, and migraine symptom severity.
A Need for Treatment Optimization
Previous studies have found an association between high BMI and severe and progressive forms of migraine. This association “may reflect a proinflammatory state in obesity that renders treatment less effective,” said Dr. Lipton.
The authors’ finding of an association between depression and inadequate response is consistent with previous research suggesting that depression is a risk factor for headache progression. Preventive migraine medications were protective against this outcome, however.
A possible explanation for smokers’ higher likelihood of having inadequate 24-hour pain relief is that “smoking may alter drug metabolism and shorten the duration of action of selected acute treatments,” said Dr. Lipton. In addition, the association between monthly headache days and inadequate 24-hour pain relief “may reflect the fact that more frequent attacks may be associated with prolonged activation of neuronal networks involved in pain processing during attacks, which may lead to lowering the threshold for subsequent attacks.”
One limitation of the current study is its reliance on self-reported data, said the authors. The questionnaire that the researchers used is limited by recall bias, recency effects, and the risk that the preceding month did not represent the individual’s usual experience. Nonetheless, mTOQ items have demonstrated high reliability and validity. Other study limitations include the retrospective design, the high proportion of participants who used more than one acute treatment, and the fact that the data are 10 years old.
On the other hand, the study examined a large, representative sample of the US population. It also included various validated measures to diagnose migraine and to assess headache-related disability, allodynia, and depression.
“These results show that unmet needs remain, and the expansion of therapeutic options for episodic migraine is needed, as well as optimizing treatment by carefully designing comprehensive treatment plans with existing acute therapies with various doses, routes of administration, preventive and interventional treatment approaches, behavioral therapies, neuromodulators, and other empirically validated approaches to achieve optimized treatment,” Dr. Lipton concluded.
—Erik Greb
Suggested Reading
Lipton RB, Munjal S, Buse DC, et al. Predicting inadequate response to acute migraine medication: results from the American Migraine Prevalence and Prevention (AMPP) Study. Headache. 2016;56(10):1635-1648.
Demographic variables, headache features, comorbidity, and treatment factors may predict inadequate response to acute migraine treatment at two hours and at 24 hours, according to research published in the November issue of Headache. Similar factors may predict which patients with an adequate response at two hours will have an inadequate response at 24 hours.
The data suggest substantial unmet acute treatment needs at the population level, said Richard B. Lipton, MD, Edwin S. Lowe Chair in Neurology at the Albert Einstein College of Medicine in Bronx, New York, and colleagues. “A good response at two hours was associated with doing well at 24 hours. This [result] highlights the importance of initial response to treatment in overall patient outcomes.”
An Analysis of AMPP Data
Much of the literature intended to identify predictors of response to migraine treatment focuses on outcomes of individual attacks. Study populations generally are limited, furthermore, to the select group of patients willing to participate in trials. A thorough evaluation of unmet acute treatment needs requires a more representative sample population, as well as information about long-term responses to multiple attacks, said Dr. Lipton.
He and his colleagues examined data from the American Migraine Prevalence and Prevention (AMPP) Study to identify factors that predict the success or failure of acute treatment at two hours and at 24 hours. The investigators focused on the 2006 AMPP survey, which included the Migraine Treatment Optimization Questionnaire (mTOQ). Eligible participants met criteria for episodic migraine, used acute pharmacologic treatment for migraine, and provided the necessary data for the researchers’ analysis.
In all, 14,520 people responded to the 2006 survey, 10,006 of whom met International Classification of Headache Disorders-3 beta criteria for migraine. Dr. Lipton’s group examined two questions from the mTOQ to assess pain-response outcomes following acute treatment. The first question asked whether the respondent was pain-free within two hours of treatment for most attacks. The second asked whether one dose of medication usually relieved the respondent’s headache and kept it away for at least 24 hours.
In all, 8,233 people responded to both questions. Patients who responded “never,” “rarely,” or “less than half the time” to the first or second question were considered to have an inadequate two-hour pain-free response or an inadequate 24-hour pain relief response, respectively. A response of “half the time or more” was defined as an adequate response. In addition, the researchers defined a 24-hour sustained pain-free response as an adequate response to both questions. Participants with an adequate two-hour response and an inadequate 24-hour response were considered to have recurrence. To identify outcome predictors, Dr. Lipton and colleagues conducted logistic regression analyses.
Most Participants Had Inadequate Response
Most participants (56.0%) reported inadequate two-hour pain-free response to usual acute treatment, and 53.6% of respondents reported inadequate 24-hour pain relief. Of the 44.0% of people with adequate two-hour pain-free response, 74.3% reported sustained pain relief at 24 hours.
The significant predictors of inadequate two-hour pain-free response were greater pain intensity, cutaneous allodynia, depression, higher BMI, and higher average monthly headache day frequency. Factors that protected against an inadequate two-hour pain-free response included using a preventive medication for migraine, female gender, and being married.
Factors that predicted inadequate 24-hour pain relief included greater feelings of depression, cutaneous allodynia, greater monthly headache day frequency, greater headache pain intensity, overuse of acute medication, lack of health insurance, being a smoker, and being unmarried. Predictors of inadequate 24-hour sustained pain-free response were greater monthly headache day frequency, cutaneous allodynia, meeting criteria for depression, acute medication overuse, and migraine symptom severity.
A Need for Treatment Optimization
Previous studies have found an association between high BMI and severe and progressive forms of migraine. This association “may reflect a proinflammatory state in obesity that renders treatment less effective,” said Dr. Lipton.
The authors’ finding of an association between depression and inadequate response is consistent with previous research suggesting that depression is a risk factor for headache progression. Preventive migraine medications were protective against this outcome, however.
A possible explanation for smokers’ higher likelihood of having inadequate 24-hour pain relief is that “smoking may alter drug metabolism and shorten the duration of action of selected acute treatments,” said Dr. Lipton. In addition, the association between monthly headache days and inadequate 24-hour pain relief “may reflect the fact that more frequent attacks may be associated with prolonged activation of neuronal networks involved in pain processing during attacks, which may lead to lowering the threshold for subsequent attacks.”
One limitation of the current study is its reliance on self-reported data, said the authors. The questionnaire that the researchers used is limited by recall bias, recency effects, and the risk that the preceding month did not represent the individual’s usual experience. Nonetheless, mTOQ items have demonstrated high reliability and validity. Other study limitations include the retrospective design, the high proportion of participants who used more than one acute treatment, and the fact that the data are 10 years old.
On the other hand, the study examined a large, representative sample of the US population. It also included various validated measures to diagnose migraine and to assess headache-related disability, allodynia, and depression.
“These results show that unmet needs remain, and the expansion of therapeutic options for episodic migraine is needed, as well as optimizing treatment by carefully designing comprehensive treatment plans with existing acute therapies with various doses, routes of administration, preventive and interventional treatment approaches, behavioral therapies, neuromodulators, and other empirically validated approaches to achieve optimized treatment,” Dr. Lipton concluded.
—Erik Greb
Suggested Reading
Lipton RB, Munjal S, Buse DC, et al. Predicting inadequate response to acute migraine medication: results from the American Migraine Prevalence and Prevention (AMPP) Study. Headache. 2016;56(10):1635-1648.

Rituximab May Have Advantages Over Fingolimod and Natalizumab in MS
LONDON—As a first- or second-line agent for relapsing-remitting multiple sclerosis (MS), rituximab demonstrates superior drug survival (ie, the proportion of patients remaining on a
Rituximab Is Not Indicated for MS
Rituximab, a chimeric mouse–human anti-CD20 monoclonal antibody, is not approved as an MS treatment, but researchers have studied it for this indication. Naismith et al found that rituximab effectively treated breakthrough disease in patients with relapsing-remitting MS.
In a retrospective, observational study, Fredrik Piehl, MD, Professor of Neuroimmunology at Karolinska Institutet in Stockholm, and colleagues examined outcomes in patients with MS who switched from natalizumab to rituximab or fingolimod as part of a risk-management strategy. Compared with patients who switched to fingolimod, patients who switched to rituximab had lower risks of clinical relapses and adverse events.

These results prompted Dr. Piehl and colleagues to conduct a study to compare the baseline characteristics and outcomes of patients with relapsing-remitting MS starting rituximab, fingolimod, or natalizumab. The investigators used data from the Swedish Neuroregistry, which contains information on approximately 80% of all Swedish patients with MS. They included 4,244 patients in their final analysis and examined only the first period for each therapy and patient.
Rituximab Reduced Likelihood of Relapse
Approximately 14% of Swedish patients with relapsing-remitting MS received natalizumab, 10% received fingolimod, and 29% received rituximab. Patients who initiated therapy with natalizumab tended to be younger, have a higher Expanded Disability Status Scale (EDSS) score, and have more active disease at baseline, compared with the other participants.
Drug survival was greater among all patients who ever received rituximab, compared with all patients who ever received fingolimod or natalizumab. When the researchers examined the drugs as first-line agents, drug survival was greater with rituximab than with the other therapies. They found the same result when they analyzed the treatments as second-line agents. Among patients who switched from natalizumab, drug survival was greater in patients who initiated rituximab, compared with those who initiated fingolimod.
Relapse-free survival was most likely among patients receiving rituximab, compared with patients receiving fingolimod or natalizumab. When the researchers analyzed the drugs as first-line agents, they found that relapse-free survival was more likely with rituximab than with the other therapies, but the difference was small. The difference in relapse-free survival was greater, however, when the researchers analyzed the three drugs as second-line therapies.
“The strengths of this study are that it is nationwide and [that] it is a real-world population, including patients with various comorbidities,” said Dr. Piehl. The study results were susceptible to hidden confounding by indication, however. Another weakness of the study was its retrospective, observational design, said Dr. Piehl.
—Erik Greb
Suggested Reading
Naismith RT, Piccio L, Lyons JA, et al. Rituximab add-on therapy for breakthrough relapsing multiple sclerosis: a 52-week phase II trial. Neurology. 2010;74(23):1860-1867.
Salzer J, Svenningsson R, Alping P, et al. Rituximab in multiple sclerosis: A retrospective observational study on safety and efficacy. Neurology. 2016 Oct 19 [Epub ahead of print].
LONDON—As a first- or second-line agent for relapsing-remitting multiple sclerosis (MS), rituximab demonstrates superior drug survival (ie, the proportion of patients remaining on a
Rituximab Is Not Indicated for MS
Rituximab, a chimeric mouse–human anti-CD20 monoclonal antibody, is not approved as an MS treatment, but researchers have studied it for this indication. Naismith et al found that rituximab effectively treated breakthrough disease in patients with relapsing-remitting MS.
In a retrospective, observational study, Fredrik Piehl, MD, Professor of Neuroimmunology at Karolinska Institutet in Stockholm, and colleagues examined outcomes in patients with MS who switched from natalizumab to rituximab or fingolimod as part of a risk-management strategy. Compared with patients who switched to fingolimod, patients who switched to rituximab had lower risks of clinical relapses and adverse events.
These results prompted Dr. Piehl and colleagues to conduct a study to compare the baseline characteristics and outcomes of patients with relapsing-remitting MS starting rituximab, fingolimod, or natalizumab. The investigators used data from the Swedish Neuroregistry, which contains information on approximately 80% of all Swedish patients with MS. They included 4,244 patients in their final analysis and examined only the first period for each therapy and patient.
Rituximab Reduced Likelihood of Relapse
Approximately 14% of Swedish patients with relapsing-remitting MS received natalizumab, 10% received fingolimod, and 29% received rituximab. Patients who initiated therapy with natalizumab tended to be younger, have a higher Expanded Disability Status Scale (EDSS) score, and have more active disease at baseline, compared with the other participants.
Drug survival was greater among all patients who ever received rituximab, compared with all patients who ever received fingolimod or natalizumab. When the researchers examined the drugs as first-line agents, drug survival was greater with rituximab than with the other therapies. They found the same result when they analyzed the treatments as second-line agents. Among patients who switched from natalizumab, drug survival was greater in patients who initiated rituximab, compared with those who initiated fingolimod.
Relapse-free survival was most likely among patients receiving rituximab, compared with patients receiving fingolimod or natalizumab. When the researchers analyzed the drugs as first-line agents, they found that relapse-free survival was more likely with rituximab than with the other therapies, but the difference was small. The difference in relapse-free survival was greater, however, when the researchers analyzed the three drugs as second-line therapies.
“The strengths of this study are that it is nationwide and [that] it is a real-world population, including patients with various comorbidities,” said Dr. Piehl. The study results were susceptible to hidden confounding by indication, however. Another weakness of the study was its retrospective, observational design, said Dr. Piehl.
—Erik Greb
Suggested Reading
Naismith RT, Piccio L, Lyons JA, et al. Rituximab add-on therapy for breakthrough relapsing multiple sclerosis: a 52-week phase II trial. Neurology. 2010;74(23):1860-1867.
Salzer J, Svenningsson R, Alping P, et al. Rituximab in multiple sclerosis: A retrospective observational study on safety and efficacy. Neurology. 2016 Oct 19 [Epub ahead of print].
LONDON—As a first- or second-line agent for relapsing-remitting multiple sclerosis (MS), rituximab demonstrates superior drug survival (ie, the proportion of patients remaining on a
Rituximab Is Not Indicated for MS
Rituximab, a chimeric mouse–human anti-CD20 monoclonal antibody, is not approved as an MS treatment, but researchers have studied it for this indication. Naismith et al found that rituximab effectively treated breakthrough disease in patients with relapsing-remitting MS.
In a retrospective, observational study, Fredrik Piehl, MD, Professor of Neuroimmunology at Karolinska Institutet in Stockholm, and colleagues examined outcomes in patients with MS who switched from natalizumab to rituximab or fingolimod as part of a risk-management strategy. Compared with patients who switched to fingolimod, patients who switched to rituximab had lower risks of clinical relapses and adverse events.
These results prompted Dr. Piehl and colleagues to conduct a study to compare the baseline characteristics and outcomes of patients with relapsing-remitting MS starting rituximab, fingolimod, or natalizumab. The investigators used data from the Swedish Neuroregistry, which contains information on approximately 80% of all Swedish patients with MS. They included 4,244 patients in their final analysis and examined only the first period for each therapy and patient.
Rituximab Reduced Likelihood of Relapse
Approximately 14% of Swedish patients with relapsing-remitting MS received natalizumab, 10% received fingolimod, and 29% received rituximab. Patients who initiated therapy with natalizumab tended to be younger, have a higher Expanded Disability Status Scale (EDSS) score, and have more active disease at baseline, compared with the other participants.
Drug survival was greater among all patients who ever received rituximab, compared with all patients who ever received fingolimod or natalizumab. When the researchers examined the drugs as first-line agents, drug survival was greater with rituximab than with the other therapies. They found the same result when they analyzed the treatments as second-line agents. Among patients who switched from natalizumab, drug survival was greater in patients who initiated rituximab, compared with those who initiated fingolimod.
Relapse-free survival was most likely among patients receiving rituximab, compared with patients receiving fingolimod or natalizumab. When the researchers analyzed the drugs as first-line agents, they found that relapse-free survival was more likely with rituximab than with the other therapies, but the difference was small. The difference in relapse-free survival was greater, however, when the researchers analyzed the three drugs as second-line therapies.
“The strengths of this study are that it is nationwide and [that] it is a real-world population, including patients with various comorbidities,” said Dr. Piehl. The study results were susceptible to hidden confounding by indication, however. Another weakness of the study was its retrospective, observational design, said Dr. Piehl.
—Erik Greb
Suggested Reading
Naismith RT, Piccio L, Lyons JA, et al. Rituximab add-on therapy for breakthrough relapsing multiple sclerosis: a 52-week phase II trial. Neurology. 2010;74(23):1860-1867.
Salzer J, Svenningsson R, Alping P, et al. Rituximab in multiple sclerosis: A retrospective observational study on safety and efficacy. Neurology. 2016 Oct 19 [Epub ahead of print].
