User login
Need an add-on to metformin? Consider this
ILLUSTRATIVE CASE
A 58-year-old woman with type 2 diabetes mellitus (T2DM) and heart failure returns to your office for follow-up of her T2DM. She has been on the maximum dose of metformin alone for the past 6 months, but her HbA1c is now 7.8%. She is keen to avoid injections. What do you recommend next?
There is surprisingly little consensus about what to add to metformin for patients with T2DM who require a second agent to achieve their glycemic goal. Attainment of glycemic control earlier in the course of the disease may lead to reduced overall cardiovascular risk, so the choice of a second drug is an important one.2 While metformin is well established as initial pharmacotherapy because of its proven mortality benefit, wide availability, and low cost, no second-choice drug has amassed enough evidence of benefit to emerge as the add-on therapy of choice.
Furthermore, the professional societies and associations are of little assistance. Dual therapy recommendations from the American Diabetes Association (ADA) and the European Association for the Study of Diabetes do not denote a specific preference, and while the American Association of Clinical Endocrinologists/American College of Endocrinology do suggest a hierarchy of choices, it is based upon expert consensus recommendation.3,4
Sulfonylureas can cause hypoglycemia and weight gain
Options for add-on therapy include sulfonylureas, thiazolidines, dipeptidyl peptidase-4 (DPP-4) inhibitors, sodium glucose cotransporter 2 (SGLT2) inhibitors, glucagon-like peptide 1 (GLP-1) agonists, and insulin. Providers have frequently prescribed a sulfonylurea after metformin because such agents are low in cost, have long-term safety data, and are effective at lowering HbA1c. Sulfonylureas work by directly stimulating insulin secretion by pancreatic beta cells in a glucose-independent manner. But as a 2010 meta-analysis revealed, they carry significant risks of hypoglycemia (relative risk [RR]=4.57; 95% confidence interval [CI], 2.11-11.45) and weight gain (2.06 kg; 95% CI, 1.15-2.96) compared to placebo.5
DPP-4 inhibitors, on the other hand, work by inducing insulin secretion in a glucose-dependent manner through an incretin mechanism. Combined with metformin, they provide glucose control similar to that achieved with the combination of a sulfonylurea and metformin.6 DPP-4 inhibitors were initially found to be associated with fewer cardiovascular events and less hypoglycemia than sulfonylureas, but were subsequently linked to an increased risk of hospitalization for heart failure.7
This latest large observational study provides more evidence on the effects of DPP-4s when added to metformin.1
STUDY SUMMARY
DPP-4s as effective as sulfonylureas with no increased risks
This population-based observational cohort study compared DPP-4 inhibitors and sulfonylureas when added to metformin for the treatment of T2DM.1 Outcomes were all-cause mortality, major adverse cardiovascular events (MACEs; defined as hospitalization for ischemic stroke or myocardial infarction [MI]), and hospitalizations for either heart failure or hypoglycemia. Using the National Health Insurance Research Database in Taiwan, the study included data on over 70,000 patients ages 20 years and older with a diagnosis of T2DM. Individuals adherent to metformin were considered to be enrolled into the cohort on the day they began using either a DPP-4 inhibitor or a sulfonylurea, in addition to metformin.
The researchers collected additional data on the enrolled individuals regarding socioeconomic factors, urbanization, robustness of the local health care system, Charlson Comorbidity Index, adapted Diabetes Complications Severity Index, and other comorbidities and medications that could affect the outcomes of interest. Using these data, enrollees were matched by propensity score into 10,089 pairs consisting of a DPP-4 inhibitor user and a sulfonylurea user.
After a mean follow-up period of 2.8 years, the authors of the study used Cox regression analysis to evaluate the relative hazards of the outcomes. Subgroup analysis performed by age, sex, Charlson Comorbidity Index, hypertension, chronic kidney disease, hospitalization for heart failure, MI, and cerebrovascular disease yielded results similar to those of the primary analysis for each outcome. Additionally, similar results were obtained when the data were analyzed without propensity-score matching.
The researchers found that users of DPP-4 inhibitors—when compared to users of sulfonylureas—had a lower risk of all-cause mortality (366 vs 488 deaths; hazard ratio [HR]=0.63; 95% CI, 0.55-0.72; number needed to treat [NNT]=117), MACE (209 vs 282 events; HR=0.68; 95% CI, 0.55-0.83; NNT=191), ischemic stroke (144 vs 203 strokes; HR 0.64; 95% CI, 0.51-0.81; NNT=246), and hypoglycemia (89 vs 170 events; HR=0.43; 95% CI, 0.33-0.56; NNT=201). Further, there were no significant differences in either the number of MIs that occurred (69 vs 88 MIs; HR=0.75; 95% CI, 0.52-1.07) or in the number of hospitalizations for heart failure (100 vs 100 events; HR=0.78; 95% CI, 0.57-1.06) between users of DPP-4 inhibitors and those of sulfonylureas.
WHAT’S NEW
Lower risks of death, CV events, and hypoglycemia
This study found that when added to metformin, DPP-4 inhibitors were associated with lower risks for all-cause mortality, cardiovascular events, and hypoglycemia when compared to sulfonylureas. Additionally, DPP-4 inhibitors did not increase the risk of hospitalization for heart failure. A recent multicenter observational study of nearly 1.5 million patients on the effects of incretin-based treatments, including both DPP-4 inhibitors and GLP-1 agonists, similarly found no increased risk of hospitalization for heart failure, with DPP-4 inhibitors compared to other combinations of oral T2DM agents.8
CAVEATS
Did unmeasured confounders play a role?
Unmeasured confounders potentially bias all observational population cohort results. In this study, in particular, there may have been unmeasured, but significant, patient factors that providers used to choose diabetes medications. Also, the study did not evaluate diabetes control, although previous studies have shown similar glucose control between sulfonylureas and DPP-4 inhibitors when they were added to metformin.6
Another caveat is that the results from this study group may not be fully generalizable to other populations due to physiologic differences. People of Asian ancestry are at risk of developing T2DM at a lower body mass index than people of European ancestry, which could affect the outcomes of interest.9
Furthermore, the study did not evaluate outcomes based on whether patients were taking first-, second-, or third-generation sulfonylureas. Some sulfonylureas, such as glyburide, carry a higher risk of hypoglycemia, which could bias the results if a large number of patients were taking them.10
Lastly, the study only provides guidance when choosing between a sulfonylurea and a DPP-4 inhibitor for second-line pharmacotherapy. The GRADE trial, due to be completed in 2023, is comparing sulfonylureas, DPP-4 inhibitors, GLP-1 agonists, and insulin as add-on medications to metformin, and may provide more data on which to base treatment decisions.11
CHALLENGES TO IMPLEMENTATION
DPP-4s have a higher price tag than sulfonylureas
Sulfonylureas and DPP-4 inhibitors are both available as generic medications, but the cost of DPP-4 inhibitors remains significantly higher.12 Higher copays and deductibles could affect patient preference. Furthermore, for patients without health insurance, sulfonylureas are available on the discounted drug lists of many major retailers, while DPP-4 inhibitors are not.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Ou SM, Shih CJ, Chao PW, et al. Effects of clinical outcomes of adding dipeptidyl peptidase-4 inhibitors versus sulfonylureas to metformin therapy in patients with type 2 diabetes mellitus. Ann Intern Med. 2015;163:663-672.
2. Hayward RA, Reaven PD, Wiitala WL, et al. Follow-up of glycemic control and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;372:2197-2206.
3. American Diabetes Association. Approaches to glycemic treatment. Sec 7. In Standards of Medical Care in Diabetes—2016. Diabetes Care. 2016;39(Suppl. 1):S52-S59. Diabetes Care. 2016; 39:e88-e89.
4. Garber AJ, Abrahamson MJ, Barzilay JI, et al. Consensus Statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the Comprehensive Type 2 Diabetes 4. Management Algorithm—2016 Executive Summary. Endocr Pract. 2016;22:84-113.
5. Phung OJ, Scholle JM, Talwar M, et al. Effect of noninsulin antidiabetic drugs added to metformin therapy on glycemic control, weight gain, and hypoglycemia in type 2 diabetes. JAMA. 2010;303:1410-1418.
6. Gallwitz B, Rosenstock J, Rauch T, et al. 2-year efficacy and safety of linagliptin compared with glimepiride in patients with type 2 diabetes inadequately controlled on metformin: a randomised, double-blind, non-inferiority trial. Lancet. 2012;380:475-483.
7. Scirica BM, Bhatt DL, Braunwald E, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317-1326.
8. Filion KB, Azoulay L, Platt RW, et al. A multicenter observational study of incretin-based drugs and heart failure. N Engl J Med. 2016;374:1145-1154.
9. Chan JC, Malik V, Jia W, et al. Diabetes in Asia: epidemiology, risk factors, pathophysiology. JAMA. 2009;301:2129-2140.
10. Gangji AS, Cukierman T, Gerstein HC, et al. A systematic review and meta-analysis of hypoglycemia and cardiovascular events: a comparison of glyburide with other secretagogues and with insulin. Diabetes Care. 2007;30:389-394.
11. Nathan DM, Buse JB, Kahn SE, et al. Rationale and design of the glycemia reduction approaches in diabetes: a comparative effectiveness study (GRADE). Diabetes Care. 2013;36:2254-2261.
12. GoodRx. Gliptins. Available at: http://www.goodrx.com/gliptins. Accessed August 31, 2016.
ILLUSTRATIVE CASE
A 58-year-old woman with type 2 diabetes mellitus (T2DM) and heart failure returns to your office for follow-up of her T2DM. She has been on the maximum dose of metformin alone for the past 6 months, but her HbA1c is now 7.8%. She is keen to avoid injections. What do you recommend next?
There is surprisingly little consensus about what to add to metformin for patients with T2DM who require a second agent to achieve their glycemic goal. Attainment of glycemic control earlier in the course of the disease may lead to reduced overall cardiovascular risk, so the choice of a second drug is an important one.2 While metformin is well established as initial pharmacotherapy because of its proven mortality benefit, wide availability, and low cost, no second-choice drug has amassed enough evidence of benefit to emerge as the add-on therapy of choice.
Furthermore, the professional societies and associations are of little assistance. Dual therapy recommendations from the American Diabetes Association (ADA) and the European Association for the Study of Diabetes do not denote a specific preference, and while the American Association of Clinical Endocrinologists/American College of Endocrinology do suggest a hierarchy of choices, it is based upon expert consensus recommendation.3,4
Sulfonylureas can cause hypoglycemia and weight gain
Options for add-on therapy include sulfonylureas, thiazolidines, dipeptidyl peptidase-4 (DPP-4) inhibitors, sodium glucose cotransporter 2 (SGLT2) inhibitors, glucagon-like peptide 1 (GLP-1) agonists, and insulin. Providers have frequently prescribed a sulfonylurea after metformin because such agents are low in cost, have long-term safety data, and are effective at lowering HbA1c. Sulfonylureas work by directly stimulating insulin secretion by pancreatic beta cells in a glucose-independent manner. But as a 2010 meta-analysis revealed, they carry significant risks of hypoglycemia (relative risk [RR]=4.57; 95% confidence interval [CI], 2.11-11.45) and weight gain (2.06 kg; 95% CI, 1.15-2.96) compared to placebo.5
DPP-4 inhibitors, on the other hand, work by inducing insulin secretion in a glucose-dependent manner through an incretin mechanism. Combined with metformin, they provide glucose control similar to that achieved with the combination of a sulfonylurea and metformin.6 DPP-4 inhibitors were initially found to be associated with fewer cardiovascular events and less hypoglycemia than sulfonylureas, but were subsequently linked to an increased risk of hospitalization for heart failure.7
This latest large observational study provides more evidence on the effects of DPP-4s when added to metformin.1
STUDY SUMMARY
DPP-4s as effective as sulfonylureas with no increased risks
This population-based observational cohort study compared DPP-4 inhibitors and sulfonylureas when added to metformin for the treatment of T2DM.1 Outcomes were all-cause mortality, major adverse cardiovascular events (MACEs; defined as hospitalization for ischemic stroke or myocardial infarction [MI]), and hospitalizations for either heart failure or hypoglycemia. Using the National Health Insurance Research Database in Taiwan, the study included data on over 70,000 patients ages 20 years and older with a diagnosis of T2DM. Individuals adherent to metformin were considered to be enrolled into the cohort on the day they began using either a DPP-4 inhibitor or a sulfonylurea, in addition to metformin.
The researchers collected additional data on the enrolled individuals regarding socioeconomic factors, urbanization, robustness of the local health care system, Charlson Comorbidity Index, adapted Diabetes Complications Severity Index, and other comorbidities and medications that could affect the outcomes of interest. Using these data, enrollees were matched by propensity score into 10,089 pairs consisting of a DPP-4 inhibitor user and a sulfonylurea user.
After a mean follow-up period of 2.8 years, the authors of the study used Cox regression analysis to evaluate the relative hazards of the outcomes. Subgroup analysis performed by age, sex, Charlson Comorbidity Index, hypertension, chronic kidney disease, hospitalization for heart failure, MI, and cerebrovascular disease yielded results similar to those of the primary analysis for each outcome. Additionally, similar results were obtained when the data were analyzed without propensity-score matching.
The researchers found that users of DPP-4 inhibitors—when compared to users of sulfonylureas—had a lower risk of all-cause mortality (366 vs 488 deaths; hazard ratio [HR]=0.63; 95% CI, 0.55-0.72; number needed to treat [NNT]=117), MACE (209 vs 282 events; HR=0.68; 95% CI, 0.55-0.83; NNT=191), ischemic stroke (144 vs 203 strokes; HR 0.64; 95% CI, 0.51-0.81; NNT=246), and hypoglycemia (89 vs 170 events; HR=0.43; 95% CI, 0.33-0.56; NNT=201). Further, there were no significant differences in either the number of MIs that occurred (69 vs 88 MIs; HR=0.75; 95% CI, 0.52-1.07) or in the number of hospitalizations for heart failure (100 vs 100 events; HR=0.78; 95% CI, 0.57-1.06) between users of DPP-4 inhibitors and those of sulfonylureas.
WHAT’S NEW
Lower risks of death, CV events, and hypoglycemia
This study found that when added to metformin, DPP-4 inhibitors were associated with lower risks for all-cause mortality, cardiovascular events, and hypoglycemia when compared to sulfonylureas. Additionally, DPP-4 inhibitors did not increase the risk of hospitalization for heart failure. A recent multicenter observational study of nearly 1.5 million patients on the effects of incretin-based treatments, including both DPP-4 inhibitors and GLP-1 agonists, similarly found no increased risk of hospitalization for heart failure, with DPP-4 inhibitors compared to other combinations of oral T2DM agents.8
CAVEATS
Did unmeasured confounders play a role?
Unmeasured confounders potentially bias all observational population cohort results. In this study, in particular, there may have been unmeasured, but significant, patient factors that providers used to choose diabetes medications. Also, the study did not evaluate diabetes control, although previous studies have shown similar glucose control between sulfonylureas and DPP-4 inhibitors when they were added to metformin.6
Another caveat is that the results from this study group may not be fully generalizable to other populations due to physiologic differences. People of Asian ancestry are at risk of developing T2DM at a lower body mass index than people of European ancestry, which could affect the outcomes of interest.9
Furthermore, the study did not evaluate outcomes based on whether patients were taking first-, second-, or third-generation sulfonylureas. Some sulfonylureas, such as glyburide, carry a higher risk of hypoglycemia, which could bias the results if a large number of patients were taking them.10
Lastly, the study only provides guidance when choosing between a sulfonylurea and a DPP-4 inhibitor for second-line pharmacotherapy. The GRADE trial, due to be completed in 2023, is comparing sulfonylureas, DPP-4 inhibitors, GLP-1 agonists, and insulin as add-on medications to metformin, and may provide more data on which to base treatment decisions.11
CHALLENGES TO IMPLEMENTATION
DPP-4s have a higher price tag than sulfonylureas
Sulfonylureas and DPP-4 inhibitors are both available as generic medications, but the cost of DPP-4 inhibitors remains significantly higher.12 Higher copays and deductibles could affect patient preference. Furthermore, for patients without health insurance, sulfonylureas are available on the discounted drug lists of many major retailers, while DPP-4 inhibitors are not.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
ILLUSTRATIVE CASE
A 58-year-old woman with type 2 diabetes mellitus (T2DM) and heart failure returns to your office for follow-up of her T2DM. She has been on the maximum dose of metformin alone for the past 6 months, but her HbA1c is now 7.8%. She is keen to avoid injections. What do you recommend next?
There is surprisingly little consensus about what to add to metformin for patients with T2DM who require a second agent to achieve their glycemic goal. Attainment of glycemic control earlier in the course of the disease may lead to reduced overall cardiovascular risk, so the choice of a second drug is an important one.2 While metformin is well established as initial pharmacotherapy because of its proven mortality benefit, wide availability, and low cost, no second-choice drug has amassed enough evidence of benefit to emerge as the add-on therapy of choice.
Furthermore, the professional societies and associations are of little assistance. Dual therapy recommendations from the American Diabetes Association (ADA) and the European Association for the Study of Diabetes do not denote a specific preference, and while the American Association of Clinical Endocrinologists/American College of Endocrinology do suggest a hierarchy of choices, it is based upon expert consensus recommendation.3,4
Sulfonylureas can cause hypoglycemia and weight gain
Options for add-on therapy include sulfonylureas, thiazolidines, dipeptidyl peptidase-4 (DPP-4) inhibitors, sodium glucose cotransporter 2 (SGLT2) inhibitors, glucagon-like peptide 1 (GLP-1) agonists, and insulin. Providers have frequently prescribed a sulfonylurea after metformin because such agents are low in cost, have long-term safety data, and are effective at lowering HbA1c. Sulfonylureas work by directly stimulating insulin secretion by pancreatic beta cells in a glucose-independent manner. But as a 2010 meta-analysis revealed, they carry significant risks of hypoglycemia (relative risk [RR]=4.57; 95% confidence interval [CI], 2.11-11.45) and weight gain (2.06 kg; 95% CI, 1.15-2.96) compared to placebo.5
DPP-4 inhibitors, on the other hand, work by inducing insulin secretion in a glucose-dependent manner through an incretin mechanism. Combined with metformin, they provide glucose control similar to that achieved with the combination of a sulfonylurea and metformin.6 DPP-4 inhibitors were initially found to be associated with fewer cardiovascular events and less hypoglycemia than sulfonylureas, but were subsequently linked to an increased risk of hospitalization for heart failure.7
This latest large observational study provides more evidence on the effects of DPP-4s when added to metformin.1
STUDY SUMMARY
DPP-4s as effective as sulfonylureas with no increased risks
This population-based observational cohort study compared DPP-4 inhibitors and sulfonylureas when added to metformin for the treatment of T2DM.1 Outcomes were all-cause mortality, major adverse cardiovascular events (MACEs; defined as hospitalization for ischemic stroke or myocardial infarction [MI]), and hospitalizations for either heart failure or hypoglycemia. Using the National Health Insurance Research Database in Taiwan, the study included data on over 70,000 patients ages 20 years and older with a diagnosis of T2DM. Individuals adherent to metformin were considered to be enrolled into the cohort on the day they began using either a DPP-4 inhibitor or a sulfonylurea, in addition to metformin.
The researchers collected additional data on the enrolled individuals regarding socioeconomic factors, urbanization, robustness of the local health care system, Charlson Comorbidity Index, adapted Diabetes Complications Severity Index, and other comorbidities and medications that could affect the outcomes of interest. Using these data, enrollees were matched by propensity score into 10,089 pairs consisting of a DPP-4 inhibitor user and a sulfonylurea user.
After a mean follow-up period of 2.8 years, the authors of the study used Cox regression analysis to evaluate the relative hazards of the outcomes. Subgroup analysis performed by age, sex, Charlson Comorbidity Index, hypertension, chronic kidney disease, hospitalization for heart failure, MI, and cerebrovascular disease yielded results similar to those of the primary analysis for each outcome. Additionally, similar results were obtained when the data were analyzed without propensity-score matching.
The researchers found that users of DPP-4 inhibitors—when compared to users of sulfonylureas—had a lower risk of all-cause mortality (366 vs 488 deaths; hazard ratio [HR]=0.63; 95% CI, 0.55-0.72; number needed to treat [NNT]=117), MACE (209 vs 282 events; HR=0.68; 95% CI, 0.55-0.83; NNT=191), ischemic stroke (144 vs 203 strokes; HR 0.64; 95% CI, 0.51-0.81; NNT=246), and hypoglycemia (89 vs 170 events; HR=0.43; 95% CI, 0.33-0.56; NNT=201). Further, there were no significant differences in either the number of MIs that occurred (69 vs 88 MIs; HR=0.75; 95% CI, 0.52-1.07) or in the number of hospitalizations for heart failure (100 vs 100 events; HR=0.78; 95% CI, 0.57-1.06) between users of DPP-4 inhibitors and those of sulfonylureas.
WHAT’S NEW
Lower risks of death, CV events, and hypoglycemia
This study found that when added to metformin, DPP-4 inhibitors were associated with lower risks for all-cause mortality, cardiovascular events, and hypoglycemia when compared to sulfonylureas. Additionally, DPP-4 inhibitors did not increase the risk of hospitalization for heart failure. A recent multicenter observational study of nearly 1.5 million patients on the effects of incretin-based treatments, including both DPP-4 inhibitors and GLP-1 agonists, similarly found no increased risk of hospitalization for heart failure, with DPP-4 inhibitors compared to other combinations of oral T2DM agents.8
CAVEATS
Did unmeasured confounders play a role?
Unmeasured confounders potentially bias all observational population cohort results. In this study, in particular, there may have been unmeasured, but significant, patient factors that providers used to choose diabetes medications. Also, the study did not evaluate diabetes control, although previous studies have shown similar glucose control between sulfonylureas and DPP-4 inhibitors when they were added to metformin.6
Another caveat is that the results from this study group may not be fully generalizable to other populations due to physiologic differences. People of Asian ancestry are at risk of developing T2DM at a lower body mass index than people of European ancestry, which could affect the outcomes of interest.9
Furthermore, the study did not evaluate outcomes based on whether patients were taking first-, second-, or third-generation sulfonylureas. Some sulfonylureas, such as glyburide, carry a higher risk of hypoglycemia, which could bias the results if a large number of patients were taking them.10
Lastly, the study only provides guidance when choosing between a sulfonylurea and a DPP-4 inhibitor for second-line pharmacotherapy. The GRADE trial, due to be completed in 2023, is comparing sulfonylureas, DPP-4 inhibitors, GLP-1 agonists, and insulin as add-on medications to metformin, and may provide more data on which to base treatment decisions.11
CHALLENGES TO IMPLEMENTATION
DPP-4s have a higher price tag than sulfonylureas
Sulfonylureas and DPP-4 inhibitors are both available as generic medications, but the cost of DPP-4 inhibitors remains significantly higher.12 Higher copays and deductibles could affect patient preference. Furthermore, for patients without health insurance, sulfonylureas are available on the discounted drug lists of many major retailers, while DPP-4 inhibitors are not.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Ou SM, Shih CJ, Chao PW, et al. Effects of clinical outcomes of adding dipeptidyl peptidase-4 inhibitors versus sulfonylureas to metformin therapy in patients with type 2 diabetes mellitus. Ann Intern Med. 2015;163:663-672.
2. Hayward RA, Reaven PD, Wiitala WL, et al. Follow-up of glycemic control and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;372:2197-2206.
3. American Diabetes Association. Approaches to glycemic treatment. Sec 7. In Standards of Medical Care in Diabetes—2016. Diabetes Care. 2016;39(Suppl. 1):S52-S59. Diabetes Care. 2016; 39:e88-e89.
4. Garber AJ, Abrahamson MJ, Barzilay JI, et al. Consensus Statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the Comprehensive Type 2 Diabetes 4. Management Algorithm—2016 Executive Summary. Endocr Pract. 2016;22:84-113.
5. Phung OJ, Scholle JM, Talwar M, et al. Effect of noninsulin antidiabetic drugs added to metformin therapy on glycemic control, weight gain, and hypoglycemia in type 2 diabetes. JAMA. 2010;303:1410-1418.
6. Gallwitz B, Rosenstock J, Rauch T, et al. 2-year efficacy and safety of linagliptin compared with glimepiride in patients with type 2 diabetes inadequately controlled on metformin: a randomised, double-blind, non-inferiority trial. Lancet. 2012;380:475-483.
7. Scirica BM, Bhatt DL, Braunwald E, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317-1326.
8. Filion KB, Azoulay L, Platt RW, et al. A multicenter observational study of incretin-based drugs and heart failure. N Engl J Med. 2016;374:1145-1154.
9. Chan JC, Malik V, Jia W, et al. Diabetes in Asia: epidemiology, risk factors, pathophysiology. JAMA. 2009;301:2129-2140.
10. Gangji AS, Cukierman T, Gerstein HC, et al. A systematic review and meta-analysis of hypoglycemia and cardiovascular events: a comparison of glyburide with other secretagogues and with insulin. Diabetes Care. 2007;30:389-394.
11. Nathan DM, Buse JB, Kahn SE, et al. Rationale and design of the glycemia reduction approaches in diabetes: a comparative effectiveness study (GRADE). Diabetes Care. 2013;36:2254-2261.
12. GoodRx. Gliptins. Available at: http://www.goodrx.com/gliptins. Accessed August 31, 2016.
1. Ou SM, Shih CJ, Chao PW, et al. Effects of clinical outcomes of adding dipeptidyl peptidase-4 inhibitors versus sulfonylureas to metformin therapy in patients with type 2 diabetes mellitus. Ann Intern Med. 2015;163:663-672.
2. Hayward RA, Reaven PD, Wiitala WL, et al. Follow-up of glycemic control and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;372:2197-2206.
3. American Diabetes Association. Approaches to glycemic treatment. Sec 7. In Standards of Medical Care in Diabetes—2016. Diabetes Care. 2016;39(Suppl. 1):S52-S59. Diabetes Care. 2016; 39:e88-e89.
4. Garber AJ, Abrahamson MJ, Barzilay JI, et al. Consensus Statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the Comprehensive Type 2 Diabetes 4. Management Algorithm—2016 Executive Summary. Endocr Pract. 2016;22:84-113.
5. Phung OJ, Scholle JM, Talwar M, et al. Effect of noninsulin antidiabetic drugs added to metformin therapy on glycemic control, weight gain, and hypoglycemia in type 2 diabetes. JAMA. 2010;303:1410-1418.
6. Gallwitz B, Rosenstock J, Rauch T, et al. 2-year efficacy and safety of linagliptin compared with glimepiride in patients with type 2 diabetes inadequately controlled on metformin: a randomised, double-blind, non-inferiority trial. Lancet. 2012;380:475-483.
7. Scirica BM, Bhatt DL, Braunwald E, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317-1326.
8. Filion KB, Azoulay L, Platt RW, et al. A multicenter observational study of incretin-based drugs and heart failure. N Engl J Med. 2016;374:1145-1154.
9. Chan JC, Malik V, Jia W, et al. Diabetes in Asia: epidemiology, risk factors, pathophysiology. JAMA. 2009;301:2129-2140.
10. Gangji AS, Cukierman T, Gerstein HC, et al. A systematic review and meta-analysis of hypoglycemia and cardiovascular events: a comparison of glyburide with other secretagogues and with insulin. Diabetes Care. 2007;30:389-394.
11. Nathan DM, Buse JB, Kahn SE, et al. Rationale and design of the glycemia reduction approaches in diabetes: a comparative effectiveness study (GRADE). Diabetes Care. 2013;36:2254-2261.
12. GoodRx. Gliptins. Available at: http://www.goodrx.com/gliptins. Accessed August 31, 2016.
Copyright © 2017. The Family Physicians Inquiries Network. All rights reserved.
PRACTICE CHANGER
Consider a dipeptidyl peptidase-4 inhibitor before a sulfonylurea for patients with type 2 diabetes mellitus who require therapy in addition to metformin.
Ou SM, Shih CJ, Chao PW, et al. Effects of clinical outcomes of adding dipeptidyl peptidase-4 inhibitors versus sulfonylureas to metformin therapy in patients with type 2 diabetes mellitus. Ann Intern Med. 2015;163:663-672.1
STRENGTH OF RECOMMENDATION
B: Based on limited-quality, patient-oriented data from a high-quality, population-based cohort study.

Firm, non-tender mass in right breast • worsening, nonproductive cough • pleuritic pain • Dx?
THE CASE
A 44-year-old woman with a 15-year history of type 2 diabetes sought care for a firm, non-tender mass in the medial lower quadrant of her right breast. She hadn’t experienced any skin changes or axillary lymphadenopathy. The patient had immigrated to California from Afghanistan 22 years earlier, at which time she was briefly married to an Afghan man suffering from a chronic cough.
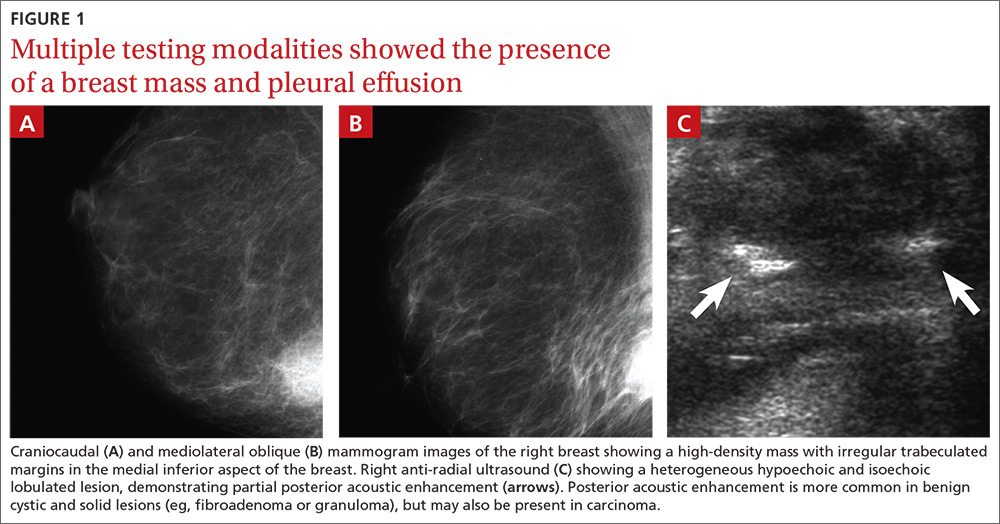
Mammography revealed a 3.5 x 4 x 4 cm lesion at the chest wall, which was highly suspicious for carcinoma (FIGURES 1A AND 1B). Sonography showed a heterogenous hypoechoic and isoechoic mass with posterior acoustic enhancement (FIGURE 1C). An excisional biopsy was performed.
One week postoperatively, the patient presented to the emergency department for a worsening nonproductive cough that intensified when supine, and was associated with subscapular pleuritic pain. She denied fever or weight loss. Biopsy results were pending.
THE DIAGNOSIS
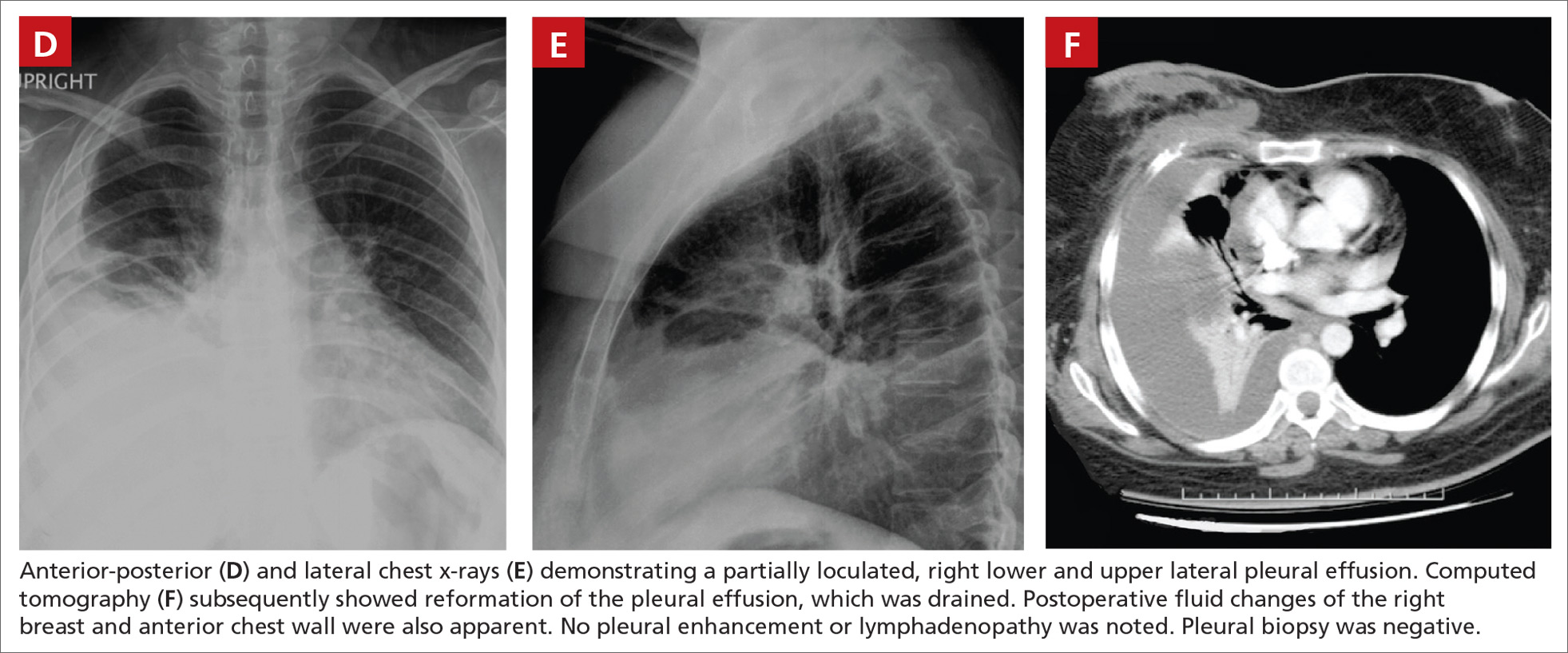
Chest x-rays revealed a large right pleural effusion that was presumed to be malignant (FIGURES 1D AND 1E). Thoracentesis yielded 1.5 liters of tea-colored exudate containing 2800 nucleated cells/mL—63% lymphocytes and 37% neutrophils—and a pleural fluid to serum protein ratio >0.5. Adenosine deaminase was <1 U/L. Fluid Gram stain, acid-fast bacillus (AFB) fluorescent antibody testing, AFB cultures, and cytology were negative. Computed tomography (CT) subsequently demonstrated recurrent effusion without hilar or mediastinal lymphadenopathy or pleural enhancement (FIGURE 1F).
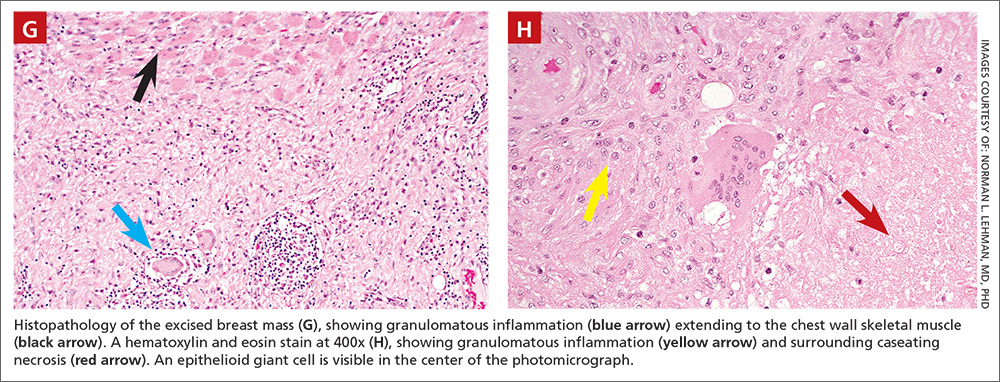
Histologically, the breast mass showed caseating granulomatous inflammation (FIGURES 1G AND 1H). An AFB stain was negative. Polymerase chain reaction (PCR) performed on DNA extracted from the formalin-fixed, paraffin-embedded biopsy material was positive for Mycobacterium tuberculosis.1 A CT-guided pleural biopsy showed only normal tissue. A follow-up tuberculin skin test (purified protein derivative [PPD]) yielded a 10-mm indurated reaction.
DISCUSSION
Granulomatous lesions, such as foreign body granuloma, idiopathic granulomatous mastitis (IGM), and sarcoidosis can mimic breast carcinoma.2,3 IGM is associated with elevated prolactin (eg, pregnancy or oral contraceptive use) and is usually subareolar.2 Infection, however, is also commonly subareolar. Sarcoidosis rarely exhibits unilateral pleural effusion and usually manifests with bilateral interstitial lung disease, hilar lymphadenopathy, and non-necrotizing granulomas.3,4
M tuberculosis and other granulomatous infections may also feign breast cancer.5-13 Breast TB, which is highly uncommon in the developed world, often demonstrates imaging similar to that which was seen in this case. Breast TB may appear nodular with ill-defined contours. Masses are sometimes attached to the chest wall and usually lack microcalcifications on mammography; they are also typically hypoechoic and heterogenous on ultrasound, often showing posterior enhancement.5,7,8 Like other breast infections, tuberculosis may show cutaneous sinus tract formation, which is seen in about one-third of patients.6,7 Alternatively, it may manifest as a diffuse mastitis with skin thickening and axillary lymphadenopathy.8
Primary breast TB without chest disease comprises up to 86% of mammary tuberculosis.6,7 Infection may occur via contamination of the skin or nipple.5-7 Lactation, pregnancy, and other causes of immunosuppression (especially human immunodeficiency virus) have been associated with an increased risk of breast infection.6-8 This patient was at risk for immunosuppression from longstanding diabetes.14
Many patients from TB-endemic areas have received the bacille Calmette-Guerin (BCG) vaccine and may exhibit equivocal or false-positive PPD results. Because interferon-gamma release assay TB blood tests (eg, QuantiFERON-TB Gold or T-SPOT.TB) are not affected by BCG, they are not associated with false-positive repeat testing results.15
Biopsy is necessary to rule out malignancy and diagnose breast TB
A pleural fluid to serum protein ratio >0.5 is consistent with infection, but also with sarcoidosis or malignancy.3,16 Elevated pleural fluid adenosine deaminase (>40 U/L) is sensitive, albeit nonspecific, for the presence of TB microorganisms. If a lymphocyte-dominant exudate is also present, however, its reliability greatly increases.16,17 Increased pleural fluid interferon-gamma is also sensitive and specific for TB pleurisy.18 Culture, along with drug sensitivity testing, should be performed on all unexplained pleural effusions.
A biopsy is often required to diagnose breast TB and should be performed on all suspicious lesions to exclude malignancy.5-7,9 AFB stains and cultures of aspirate fluids or tissue are often negative.7,9 PCR or other nucleic acid amplification tests of sputum, body fluids, or biopsy material may be positive in culture-negative cases and can rapidly confirm M tuberculosis infection.17,19 No testing modality offers 100% sensitivity or specificity; therefore, an additional confirmatory test is desirable.
Possible routes of transmission include activation of latent pulmonary tuberculosis and direct, lymphatic, or hematologic extension to the chest wall and breast.5-7 In this patient, we believe that activation of a latent breast granuloma may have resulted in a secondary or “sympathetic” pleural effusion, possibly triggered by surgical manipulation. This is compatible with her negative pleural adenosine deaminase result, negative culture, absence of pulmonary parenchymal disease, and negative pleural biopsy. Although we conducted a PubMed search, reviewing material as far back as 1966, we were unable to find a previous case of apparent sympathetic effusion associated with breast TB.
Our patient was treated with daily oral isoniazid, rifabutin, pyrazinamide, and ethambutol for 2 months, followed by isoniazid and rifabutin for 4 months. She has been disease-free for over 10 years.
THE TAKEAWAY
We describe a rare case of breast TB mimicking carcinoma that was associated with unilateral pleural effusion in a woman who had emigrated from Afghanistan. Patients at particular risk for breast TB include immigrants from endemic regions—especially parous females,6,7 those with a history of TB contacts, and those who are immunosuppressed.8 This case emphasizes the need for increased awareness of extrapulmonary TB by physicians in developed countries.
ACKNOWLEDGEMENTS
The authors thank Drs. Margie Scott, Harpreet Dhillon, Samir Vora, Todd Williams, Jeffrey Hawley, and Mr. Sergio Landeros. This report is dedicated to the memory of our friend and colleague in medicine, Dr. Jeanie Care Gillinta.
1. Bayer-Garner IB, Cox MD, Scott MA, et al. Mycobacteria other than Mycobacterium tuberculosis are not present in erythema induratum/nodular vasculitis: a case series and literature review of the clinical and histologic findings. J Cutan Pathol. 2005;32:220-226.
2. Verfaillie G, Breucq C, Sacre R, et al. Granulomatous lobular mastitis: a rare chronic inflammatory disease of the breast which can mimic breast carcinoma. Acta Chir Belg. 2006;106:222-224.
3. Fiorucci F, Conti V, Lucantoni G, et al. Sarcoidosis of the breast: a rare case report and a review. Eur Rev Med Pharmacol Sci. 2006;10:47-50.
4. Huggins JT, Doelken P, Sahn SA, et al. Pleural effusions in a series of 181 outpatients with sarcoidosis. Chest. 2006;129:1599-1604.
5. Zandrino F, Monetti F, Gandolfo N. Primary tuberculosis of the breast. A case report. Acta Radiol. 2000;41:61-63.
6. Khanna R, Prasanna GV, Gupta P, et al. Mammary tuberculosis: report on 52 cases. Postgrad Med J. 2002;78:422-424.
7. Harris SH, Khan MA, Khan R, et al. Mammary tuberculosis: analysis of thirty-eight patients. ANZ J Surg. 2006;76:234-237.
8. Meerkotter D, Spiegel K, Page-Shipp LS. Imaging of tuberculosis of the breast: 21 cases and a review of the literature. J Med Imaging Radiat Oncol. 2011;55:453-460.
9. Khodabakhshi B, Mehravar F. Breast tuberculosis in northeast Iran: review of 22 cases. BMC Womens Health. 2014;14:72.
10. Osborne BM. Granulomatous mastitis caused by histoplasma and mimicking inflammatory breast carcinoma. Hum Pathol. 1989;20:47-52.
11. Bocian JJ, Fahmy RN, Michas CA. A rare case of ‘coccidioidoma’ of the breast. Arch Pathol Lab Med. 1991;115:1064-1067.
12. Haddow LJ, Sahid F, Moosa MY. Cryptococcal breast abscess in an HIV-positive patient: arguments for reviewing the definition of immune reconstitution inflammatory syndrome. J Infect. 2008;57:82-84.
13. Lefkowitz M, Wear DJ. Cat-scratch disease masquerading as a solitary tumor of the breast. Arch Pathol Lab Med. 1989;113:473-475.
14. Ponce-De-Leon A, Garcia-Garcia Md Mde L, Garcia-Sancho MC, et al. Tuberculosis and diabetes in southern Mexico. Diabetes Care. 2004;27:1584-1590.
15. Mazurek GH, LoBue PA, Daley CL, et al. Comparison of a whole-blood interferon gamma assay with tuberculin skin testing for detecting latent Mycobacterium tuberculosis infection. JAMA. 2001;286:1740-1747.
16. Porcel JM, Light RW. Diagnostic approach to pleural effusion in adults. Am Fam Physician. 2006;73:1211-1220.
17. Burgess LJ, Maritz FJ, Le Roux I, et al. Combined use of pleural adenosine deaminase with lymphocyte/neutrophil ratio. Increased specificity for the diagnosis of tuberculous pleuritis. Chest. 1996;109:414-419.
18. Klimiuk J, Krenke R, Safianowska A, et al. Diagnostic performance of different pleural fluid biomarkers in tuberculous pleurisy. Adv Exp Med Biol. 2015;852:21-30.
19. Gopi A, Madhavan SM, Sharma SK, et al. Diagnosis and treatment of tuberculous pleural effusion in 2006. Chest. 2007;131:880-889.
THE CASE
A 44-year-old woman with a 15-year history of type 2 diabetes sought care for a firm, non-tender mass in the medial lower quadrant of her right breast. She hadn’t experienced any skin changes or axillary lymphadenopathy. The patient had immigrated to California from Afghanistan 22 years earlier, at which time she was briefly married to an Afghan man suffering from a chronic cough.
Mammography revealed a 3.5 x 4 x 4 cm lesion at the chest wall, which was highly suspicious for carcinoma (FIGURES 1A AND 1B). Sonography showed a heterogenous hypoechoic and isoechoic mass with posterior acoustic enhancement (FIGURE 1C). An excisional biopsy was performed.
One week postoperatively, the patient presented to the emergency department for a worsening nonproductive cough that intensified when supine, and was associated with subscapular pleuritic pain. She denied fever or weight loss. Biopsy results were pending.
THE DIAGNOSIS
Chest x-rays revealed a large right pleural effusion that was presumed to be malignant (FIGURES 1D AND 1E). Thoracentesis yielded 1.5 liters of tea-colored exudate containing 2800 nucleated cells/mL—63% lymphocytes and 37% neutrophils—and a pleural fluid to serum protein ratio >0.5. Adenosine deaminase was <1 U/L. Fluid Gram stain, acid-fast bacillus (AFB) fluorescent antibody testing, AFB cultures, and cytology were negative. Computed tomography (CT) subsequently demonstrated recurrent effusion without hilar or mediastinal lymphadenopathy or pleural enhancement (FIGURE 1F).
Histologically, the breast mass showed caseating granulomatous inflammation (FIGURES 1G AND 1H). An AFB stain was negative. Polymerase chain reaction (PCR) performed on DNA extracted from the formalin-fixed, paraffin-embedded biopsy material was positive for Mycobacterium tuberculosis.1 A CT-guided pleural biopsy showed only normal tissue. A follow-up tuberculin skin test (purified protein derivative [PPD]) yielded a 10-mm indurated reaction.
DISCUSSION
Granulomatous lesions, such as foreign body granuloma, idiopathic granulomatous mastitis (IGM), and sarcoidosis can mimic breast carcinoma.2,3 IGM is associated with elevated prolactin (eg, pregnancy or oral contraceptive use) and is usually subareolar.2 Infection, however, is also commonly subareolar. Sarcoidosis rarely exhibits unilateral pleural effusion and usually manifests with bilateral interstitial lung disease, hilar lymphadenopathy, and non-necrotizing granulomas.3,4
M tuberculosis and other granulomatous infections may also feign breast cancer.5-13 Breast TB, which is highly uncommon in the developed world, often demonstrates imaging similar to that which was seen in this case. Breast TB may appear nodular with ill-defined contours. Masses are sometimes attached to the chest wall and usually lack microcalcifications on mammography; they are also typically hypoechoic and heterogenous on ultrasound, often showing posterior enhancement.5,7,8 Like other breast infections, tuberculosis may show cutaneous sinus tract formation, which is seen in about one-third of patients.6,7 Alternatively, it may manifest as a diffuse mastitis with skin thickening and axillary lymphadenopathy.8
Primary breast TB without chest disease comprises up to 86% of mammary tuberculosis.6,7 Infection may occur via contamination of the skin or nipple.5-7 Lactation, pregnancy, and other causes of immunosuppression (especially human immunodeficiency virus) have been associated with an increased risk of breast infection.6-8 This patient was at risk for immunosuppression from longstanding diabetes.14
Many patients from TB-endemic areas have received the bacille Calmette-Guerin (BCG) vaccine and may exhibit equivocal or false-positive PPD results. Because interferon-gamma release assay TB blood tests (eg, QuantiFERON-TB Gold or T-SPOT.TB) are not affected by BCG, they are not associated with false-positive repeat testing results.15
Biopsy is necessary to rule out malignancy and diagnose breast TB
A pleural fluid to serum protein ratio >0.5 is consistent with infection, but also with sarcoidosis or malignancy.3,16 Elevated pleural fluid adenosine deaminase (>40 U/L) is sensitive, albeit nonspecific, for the presence of TB microorganisms. If a lymphocyte-dominant exudate is also present, however, its reliability greatly increases.16,17 Increased pleural fluid interferon-gamma is also sensitive and specific for TB pleurisy.18 Culture, along with drug sensitivity testing, should be performed on all unexplained pleural effusions.
A biopsy is often required to diagnose breast TB and should be performed on all suspicious lesions to exclude malignancy.5-7,9 AFB stains and cultures of aspirate fluids or tissue are often negative.7,9 PCR or other nucleic acid amplification tests of sputum, body fluids, or biopsy material may be positive in culture-negative cases and can rapidly confirm M tuberculosis infection.17,19 No testing modality offers 100% sensitivity or specificity; therefore, an additional confirmatory test is desirable.
Possible routes of transmission include activation of latent pulmonary tuberculosis and direct, lymphatic, or hematologic extension to the chest wall and breast.5-7 In this patient, we believe that activation of a latent breast granuloma may have resulted in a secondary or “sympathetic” pleural effusion, possibly triggered by surgical manipulation. This is compatible with her negative pleural adenosine deaminase result, negative culture, absence of pulmonary parenchymal disease, and negative pleural biopsy. Although we conducted a PubMed search, reviewing material as far back as 1966, we were unable to find a previous case of apparent sympathetic effusion associated with breast TB.
Our patient was treated with daily oral isoniazid, rifabutin, pyrazinamide, and ethambutol for 2 months, followed by isoniazid and rifabutin for 4 months. She has been disease-free for over 10 years.
THE TAKEAWAY
We describe a rare case of breast TB mimicking carcinoma that was associated with unilateral pleural effusion in a woman who had emigrated from Afghanistan. Patients at particular risk for breast TB include immigrants from endemic regions—especially parous females,6,7 those with a history of TB contacts, and those who are immunosuppressed.8 This case emphasizes the need for increased awareness of extrapulmonary TB by physicians in developed countries.
ACKNOWLEDGEMENTS
The authors thank Drs. Margie Scott, Harpreet Dhillon, Samir Vora, Todd Williams, Jeffrey Hawley, and Mr. Sergio Landeros. This report is dedicated to the memory of our friend and colleague in medicine, Dr. Jeanie Care Gillinta.
THE CASE
A 44-year-old woman with a 15-year history of type 2 diabetes sought care for a firm, non-tender mass in the medial lower quadrant of her right breast. She hadn’t experienced any skin changes or axillary lymphadenopathy. The patient had immigrated to California from Afghanistan 22 years earlier, at which time she was briefly married to an Afghan man suffering from a chronic cough.
Mammography revealed a 3.5 x 4 x 4 cm lesion at the chest wall, which was highly suspicious for carcinoma (FIGURES 1A AND 1B). Sonography showed a heterogenous hypoechoic and isoechoic mass with posterior acoustic enhancement (FIGURE 1C). An excisional biopsy was performed.
One week postoperatively, the patient presented to the emergency department for a worsening nonproductive cough that intensified when supine, and was associated with subscapular pleuritic pain. She denied fever or weight loss. Biopsy results were pending.
THE DIAGNOSIS
Chest x-rays revealed a large right pleural effusion that was presumed to be malignant (FIGURES 1D AND 1E). Thoracentesis yielded 1.5 liters of tea-colored exudate containing 2800 nucleated cells/mL—63% lymphocytes and 37% neutrophils—and a pleural fluid to serum protein ratio >0.5. Adenosine deaminase was <1 U/L. Fluid Gram stain, acid-fast bacillus (AFB) fluorescent antibody testing, AFB cultures, and cytology were negative. Computed tomography (CT) subsequently demonstrated recurrent effusion without hilar or mediastinal lymphadenopathy or pleural enhancement (FIGURE 1F).
Histologically, the breast mass showed caseating granulomatous inflammation (FIGURES 1G AND 1H). An AFB stain was negative. Polymerase chain reaction (PCR) performed on DNA extracted from the formalin-fixed, paraffin-embedded biopsy material was positive for Mycobacterium tuberculosis.1 A CT-guided pleural biopsy showed only normal tissue. A follow-up tuberculin skin test (purified protein derivative [PPD]) yielded a 10-mm indurated reaction.
DISCUSSION
Granulomatous lesions, such as foreign body granuloma, idiopathic granulomatous mastitis (IGM), and sarcoidosis can mimic breast carcinoma.2,3 IGM is associated with elevated prolactin (eg, pregnancy or oral contraceptive use) and is usually subareolar.2 Infection, however, is also commonly subareolar. Sarcoidosis rarely exhibits unilateral pleural effusion and usually manifests with bilateral interstitial lung disease, hilar lymphadenopathy, and non-necrotizing granulomas.3,4
M tuberculosis and other granulomatous infections may also feign breast cancer.5-13 Breast TB, which is highly uncommon in the developed world, often demonstrates imaging similar to that which was seen in this case. Breast TB may appear nodular with ill-defined contours. Masses are sometimes attached to the chest wall and usually lack microcalcifications on mammography; they are also typically hypoechoic and heterogenous on ultrasound, often showing posterior enhancement.5,7,8 Like other breast infections, tuberculosis may show cutaneous sinus tract formation, which is seen in about one-third of patients.6,7 Alternatively, it may manifest as a diffuse mastitis with skin thickening and axillary lymphadenopathy.8
Primary breast TB without chest disease comprises up to 86% of mammary tuberculosis.6,7 Infection may occur via contamination of the skin or nipple.5-7 Lactation, pregnancy, and other causes of immunosuppression (especially human immunodeficiency virus) have been associated with an increased risk of breast infection.6-8 This patient was at risk for immunosuppression from longstanding diabetes.14
Many patients from TB-endemic areas have received the bacille Calmette-Guerin (BCG) vaccine and may exhibit equivocal or false-positive PPD results. Because interferon-gamma release assay TB blood tests (eg, QuantiFERON-TB Gold or T-SPOT.TB) are not affected by BCG, they are not associated with false-positive repeat testing results.15
Biopsy is necessary to rule out malignancy and diagnose breast TB
A pleural fluid to serum protein ratio >0.5 is consistent with infection, but also with sarcoidosis or malignancy.3,16 Elevated pleural fluid adenosine deaminase (>40 U/L) is sensitive, albeit nonspecific, for the presence of TB microorganisms. If a lymphocyte-dominant exudate is also present, however, its reliability greatly increases.16,17 Increased pleural fluid interferon-gamma is also sensitive and specific for TB pleurisy.18 Culture, along with drug sensitivity testing, should be performed on all unexplained pleural effusions.
A biopsy is often required to diagnose breast TB and should be performed on all suspicious lesions to exclude malignancy.5-7,9 AFB stains and cultures of aspirate fluids or tissue are often negative.7,9 PCR or other nucleic acid amplification tests of sputum, body fluids, or biopsy material may be positive in culture-negative cases and can rapidly confirm M tuberculosis infection.17,19 No testing modality offers 100% sensitivity or specificity; therefore, an additional confirmatory test is desirable.
Possible routes of transmission include activation of latent pulmonary tuberculosis and direct, lymphatic, or hematologic extension to the chest wall and breast.5-7 In this patient, we believe that activation of a latent breast granuloma may have resulted in a secondary or “sympathetic” pleural effusion, possibly triggered by surgical manipulation. This is compatible with her negative pleural adenosine deaminase result, negative culture, absence of pulmonary parenchymal disease, and negative pleural biopsy. Although we conducted a PubMed search, reviewing material as far back as 1966, we were unable to find a previous case of apparent sympathetic effusion associated with breast TB.
Our patient was treated with daily oral isoniazid, rifabutin, pyrazinamide, and ethambutol for 2 months, followed by isoniazid and rifabutin for 4 months. She has been disease-free for over 10 years.
THE TAKEAWAY
We describe a rare case of breast TB mimicking carcinoma that was associated with unilateral pleural effusion in a woman who had emigrated from Afghanistan. Patients at particular risk for breast TB include immigrants from endemic regions—especially parous females,6,7 those with a history of TB contacts, and those who are immunosuppressed.8 This case emphasizes the need for increased awareness of extrapulmonary TB by physicians in developed countries.
ACKNOWLEDGEMENTS
The authors thank Drs. Margie Scott, Harpreet Dhillon, Samir Vora, Todd Williams, Jeffrey Hawley, and Mr. Sergio Landeros. This report is dedicated to the memory of our friend and colleague in medicine, Dr. Jeanie Care Gillinta.
1. Bayer-Garner IB, Cox MD, Scott MA, et al. Mycobacteria other than Mycobacterium tuberculosis are not present in erythema induratum/nodular vasculitis: a case series and literature review of the clinical and histologic findings. J Cutan Pathol. 2005;32:220-226.
2. Verfaillie G, Breucq C, Sacre R, et al. Granulomatous lobular mastitis: a rare chronic inflammatory disease of the breast which can mimic breast carcinoma. Acta Chir Belg. 2006;106:222-224.
3. Fiorucci F, Conti V, Lucantoni G, et al. Sarcoidosis of the breast: a rare case report and a review. Eur Rev Med Pharmacol Sci. 2006;10:47-50.
4. Huggins JT, Doelken P, Sahn SA, et al. Pleural effusions in a series of 181 outpatients with sarcoidosis. Chest. 2006;129:1599-1604.
5. Zandrino F, Monetti F, Gandolfo N. Primary tuberculosis of the breast. A case report. Acta Radiol. 2000;41:61-63.
6. Khanna R, Prasanna GV, Gupta P, et al. Mammary tuberculosis: report on 52 cases. Postgrad Med J. 2002;78:422-424.
7. Harris SH, Khan MA, Khan R, et al. Mammary tuberculosis: analysis of thirty-eight patients. ANZ J Surg. 2006;76:234-237.
8. Meerkotter D, Spiegel K, Page-Shipp LS. Imaging of tuberculosis of the breast: 21 cases and a review of the literature. J Med Imaging Radiat Oncol. 2011;55:453-460.
9. Khodabakhshi B, Mehravar F. Breast tuberculosis in northeast Iran: review of 22 cases. BMC Womens Health. 2014;14:72.
10. Osborne BM. Granulomatous mastitis caused by histoplasma and mimicking inflammatory breast carcinoma. Hum Pathol. 1989;20:47-52.
11. Bocian JJ, Fahmy RN, Michas CA. A rare case of ‘coccidioidoma’ of the breast. Arch Pathol Lab Med. 1991;115:1064-1067.
12. Haddow LJ, Sahid F, Moosa MY. Cryptococcal breast abscess in an HIV-positive patient: arguments for reviewing the definition of immune reconstitution inflammatory syndrome. J Infect. 2008;57:82-84.
13. Lefkowitz M, Wear DJ. Cat-scratch disease masquerading as a solitary tumor of the breast. Arch Pathol Lab Med. 1989;113:473-475.
14. Ponce-De-Leon A, Garcia-Garcia Md Mde L, Garcia-Sancho MC, et al. Tuberculosis and diabetes in southern Mexico. Diabetes Care. 2004;27:1584-1590.
15. Mazurek GH, LoBue PA, Daley CL, et al. Comparison of a whole-blood interferon gamma assay with tuberculin skin testing for detecting latent Mycobacterium tuberculosis infection. JAMA. 2001;286:1740-1747.
16. Porcel JM, Light RW. Diagnostic approach to pleural effusion in adults. Am Fam Physician. 2006;73:1211-1220.
17. Burgess LJ, Maritz FJ, Le Roux I, et al. Combined use of pleural adenosine deaminase with lymphocyte/neutrophil ratio. Increased specificity for the diagnosis of tuberculous pleuritis. Chest. 1996;109:414-419.
18. Klimiuk J, Krenke R, Safianowska A, et al. Diagnostic performance of different pleural fluid biomarkers in tuberculous pleurisy. Adv Exp Med Biol. 2015;852:21-30.
19. Gopi A, Madhavan SM, Sharma SK, et al. Diagnosis and treatment of tuberculous pleural effusion in 2006. Chest. 2007;131:880-889.
1. Bayer-Garner IB, Cox MD, Scott MA, et al. Mycobacteria other than Mycobacterium tuberculosis are not present in erythema induratum/nodular vasculitis: a case series and literature review of the clinical and histologic findings. J Cutan Pathol. 2005;32:220-226.
2. Verfaillie G, Breucq C, Sacre R, et al. Granulomatous lobular mastitis: a rare chronic inflammatory disease of the breast which can mimic breast carcinoma. Acta Chir Belg. 2006;106:222-224.
3. Fiorucci F, Conti V, Lucantoni G, et al. Sarcoidosis of the breast: a rare case report and a review. Eur Rev Med Pharmacol Sci. 2006;10:47-50.
4. Huggins JT, Doelken P, Sahn SA, et al. Pleural effusions in a series of 181 outpatients with sarcoidosis. Chest. 2006;129:1599-1604.
5. Zandrino F, Monetti F, Gandolfo N. Primary tuberculosis of the breast. A case report. Acta Radiol. 2000;41:61-63.
6. Khanna R, Prasanna GV, Gupta P, et al. Mammary tuberculosis: report on 52 cases. Postgrad Med J. 2002;78:422-424.
7. Harris SH, Khan MA, Khan R, et al. Mammary tuberculosis: analysis of thirty-eight patients. ANZ J Surg. 2006;76:234-237.
8. Meerkotter D, Spiegel K, Page-Shipp LS. Imaging of tuberculosis of the breast: 21 cases and a review of the literature. J Med Imaging Radiat Oncol. 2011;55:453-460.
9. Khodabakhshi B, Mehravar F. Breast tuberculosis in northeast Iran: review of 22 cases. BMC Womens Health. 2014;14:72.
10. Osborne BM. Granulomatous mastitis caused by histoplasma and mimicking inflammatory breast carcinoma. Hum Pathol. 1989;20:47-52.
11. Bocian JJ, Fahmy RN, Michas CA. A rare case of ‘coccidioidoma’ of the breast. Arch Pathol Lab Med. 1991;115:1064-1067.
12. Haddow LJ, Sahid F, Moosa MY. Cryptococcal breast abscess in an HIV-positive patient: arguments for reviewing the definition of immune reconstitution inflammatory syndrome. J Infect. 2008;57:82-84.
13. Lefkowitz M, Wear DJ. Cat-scratch disease masquerading as a solitary tumor of the breast. Arch Pathol Lab Med. 1989;113:473-475.
14. Ponce-De-Leon A, Garcia-Garcia Md Mde L, Garcia-Sancho MC, et al. Tuberculosis and diabetes in southern Mexico. Diabetes Care. 2004;27:1584-1590.
15. Mazurek GH, LoBue PA, Daley CL, et al. Comparison of a whole-blood interferon gamma assay with tuberculin skin testing for detecting latent Mycobacterium tuberculosis infection. JAMA. 2001;286:1740-1747.
16. Porcel JM, Light RW. Diagnostic approach to pleural effusion in adults. Am Fam Physician. 2006;73:1211-1220.
17. Burgess LJ, Maritz FJ, Le Roux I, et al. Combined use of pleural adenosine deaminase with lymphocyte/neutrophil ratio. Increased specificity for the diagnosis of tuberculous pleuritis. Chest. 1996;109:414-419.
18. Klimiuk J, Krenke R, Safianowska A, et al. Diagnostic performance of different pleural fluid biomarkers in tuberculous pleurisy. Adv Exp Med Biol. 2015;852:21-30.
19. Gopi A, Madhavan SM, Sharma SK, et al. Diagnosis and treatment of tuberculous pleural effusion in 2006. Chest. 2007;131:880-889.

Malodorous discharge, redness, and crusting of the feet
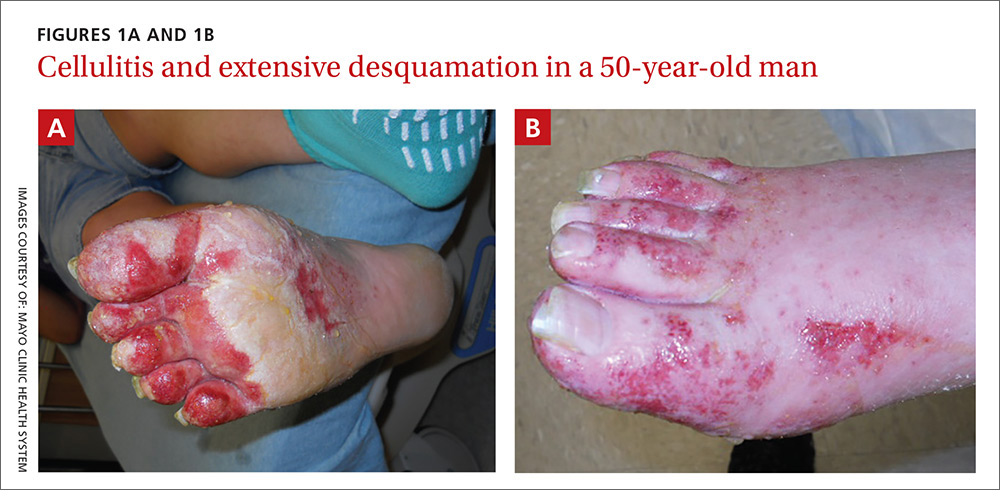
A 50-year-old man who worked in construction presented to a local urgent care facility complaining of 2 weeks of bilateral foot discomfort associated with local malodorous discharge, redness, and crusting. He had a past medical history of recurrent tinea pedis and had previously been treated with intravenous (IV) antibiotics for a severe episode of cellulitis. He denied recent fever, trauma, or swelling. Physical examination revealed extensive malodorous crusting of the interdigital webs of both feet, in addition to tenderness, erythema, and serous discharge. He was treated with topical clotrimazole and oral terbinafine for a presumed tinea pedis recurrence; cephalexin and triamcinolone were added a week later after minimal response. Two days later, he sought care at a local emergency department with progressive cellulitis, bullae formation, and extensive desquamation (FIGURES 1A AND 1B) and was hospitalized.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Gram-negative foot intertrigo
Plain x-rays and computed tomography imaging did not reveal evidence of abscess formation, subcutaneous gas/air formation, or osteomyelitis. Laboratory testing was normal. The patient was empirically started on cefepime, clindamycin, and ketoconazole. Wound therapy, consisting of normal saline washes, use of xeroform gauze to cover the wound, and frequent absorbent dressing changes, was also initiated. Wound cultures were obtained, which later grew Pseudomonas aeruginosa and Enterococcus faecalis. (P aeruginosa was the predominant organism.) We made the diagnosis of gram-negative foot intertrigo.
First described in 1973, researchers have found that P aeruginosa, as well as E faecalis and Staphylococcus aureus, are commonly associated with tinea pedis1 and toe web intertrigo.2 The infection usually involves the lateral 3 toe webs, and can present with malodorous discharge, itchy maceration, edema, and erythema of the surrounding tissue. Non-purulent lower extremity cellulitis is typically caused by beta-hemolytic streptococci residing in the toe webs, and is usually treated empirically with cephalexin or similar antibiotics.3 The presentation of foot intertrigo varies along a spectrum that includes tinea pedis, superinfected maceration, and infectious eczematoid dermatitis, all of which can encourage bacterial superinfections.
In this patient, it is likely that sweating (and consequent skin maceration), tinea pedis, and perhaps even friction between the affected area and the patient’s shoes led to skin ulceration. This then became colonized and infected with bacteria, including gram-negative varieties, which sometimes harbor at the edges of ulcerations. Addressing each of these factors is important to heal the infection.
Differential diagnosis includes other bacterial skin infections
The differential diagnosis includes erysipelas, cellulitis, lipodermatosclerosis, venous eczema, and burns. Erysipelas affects the superficial dermis with well demarcated borders, while cellulitis involves the subcutaneous fat.4 Both are more likely to be caused by beta-hemolytic streptococci, but at first may be difficult to differentiate from similar appearing gram-negative skin/soft tissue infection without microbiologic data.
Patients with lipodermatosclerosis and venous eczema often have a history of chronic venous insufficiency. Lipodermatosclerosis often presents with a subcutaneous panniculitis and hyperpigmentation; venous eczema is often associated with scaling of the involved areas. Although burns can become secondarily superinfected with bacteria, they can be differentiated from a primary bacterial infection by the history or presentation.
Gram-negative infections (especially those caused by Pseudomonas species) should be suspected if a toe web infection does not respond to empiric antimicrobial therapy with first- or second-generation cephalosporins, as their spectrum of antimicrobial activity does not include P aeruginosa. If the infection is severe, it may impact ambulation.2 Predisposing factors for gram-negative toe web infections include obesity, diabetes, moist environments, tight interdigital spaces, and recurrent tinea pedis.4
Administer appropriate wound care and start antimicrobial therapy
Foot intertrigo provides an easy portal of entry for pathogenic organisms.5 Therefore, it is important to modify risk factors from the outset to help prevent superinfection (as occurred with this patient) and other complications. Aggressive treatment of tinea pedis and use of compression stockings to reduce lymphedema are vital for prevention of recurrent infections.3
Physicians should be aware of likely, as well as unlikely, causative pathogens; “typical” skin and soft tissue infections that do not resolve may be due to atypical or gram-negative organisms, and typical first-line antibiotics will do nothing to eradicate them. Antimicrobial susceptibilities and a bacterial culture will steer you to the appropriate antimicrobial therapy. Debridement of the edge of the ulceration is necessary to remove any lingering bacteria.6 And appropriate wound care is paramount and should include the use of techniques to keep the affected areas dry, such as the use of astringent powders along with avoidance of damp shoes and socks.
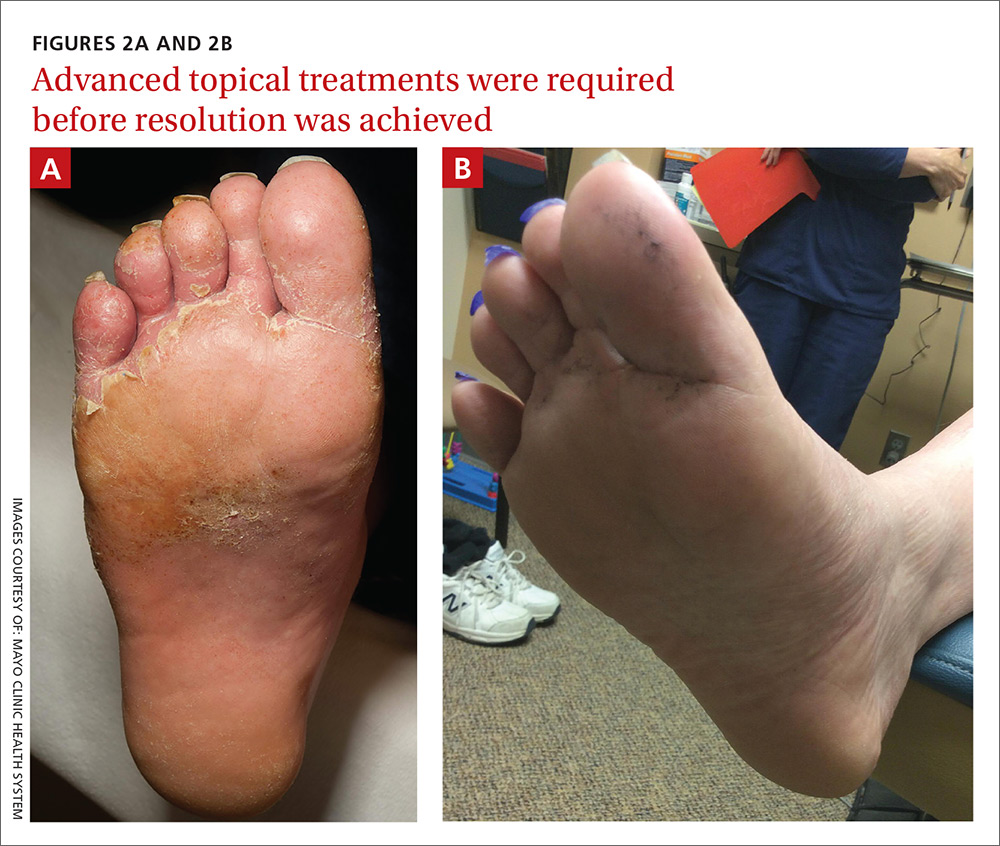
Our patient was switched from empiric therapy to culture-specific IV therapy with vancomycin 2 g every 12 hours, ciprofloxacin 400 mg every 12 hours, and fluconazole 400 mg daily. Two weeks later, he was discharged from the hospital on topical antifungals, oral fluconazole, and daily acetic acid soaks. He did, however, require further advanced topical treatments (miconazole 2% twice daily) due to recurrent flare-ups before complete resolution was achieved 6 weeks after presentation (FIGURES 2A AND 2B).
CORRESPONDENCE
Alberto Marcelin, MD, Department of Family Medicine, Mayo Clinic Health System, 1000 1st Dr NW, Austin, MN 55912; [email protected].
1. Westmoreland TA, Ross EV, Yeager JK. Pseudomonas toe web infections. Cutis. 1992;49:185-186.
2. Lin JY, Shih YL, Ho HC. Foot bacterial intertrigo mimicking interdigital tinea pedis. Chang Gung Med J. 2011;34:44-49.
3. Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis. 2014;59:e10-e52.
4. Hirschmann JV, Raugi GJ. Lower limb cellulitis and its mimics: part II. Conditions that simulate lower limb cellulitis. J Am Acad Dermatol. 2012;67:177, e1-e9; quiz 185-186.
5. Semel JD, Goldin H. Association of athlete’s foot with cellulitis of the lower extremities: diagnostic value of bacterial cultures of ipsilateral interdigital space samples. Clin Infect Dis. 1996;23:1162-1164.
6. Fangman W, Burton C. Hyperkeratotic rim of gram-negative toe web infections. Arch Dermatol. 2005;141:658.
A 50-year-old man who worked in construction presented to a local urgent care facility complaining of 2 weeks of bilateral foot discomfort associated with local malodorous discharge, redness, and crusting. He had a past medical history of recurrent tinea pedis and had previously been treated with intravenous (IV) antibiotics for a severe episode of cellulitis. He denied recent fever, trauma, or swelling. Physical examination revealed extensive malodorous crusting of the interdigital webs of both feet, in addition to tenderness, erythema, and serous discharge. He was treated with topical clotrimazole and oral terbinafine for a presumed tinea pedis recurrence; cephalexin and triamcinolone were added a week later after minimal response. Two days later, he sought care at a local emergency department with progressive cellulitis, bullae formation, and extensive desquamation (FIGURES 1A AND 1B) and was hospitalized.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Gram-negative foot intertrigo
Plain x-rays and computed tomography imaging did not reveal evidence of abscess formation, subcutaneous gas/air formation, or osteomyelitis. Laboratory testing was normal. The patient was empirically started on cefepime, clindamycin, and ketoconazole. Wound therapy, consisting of normal saline washes, use of xeroform gauze to cover the wound, and frequent absorbent dressing changes, was also initiated. Wound cultures were obtained, which later grew Pseudomonas aeruginosa and Enterococcus faecalis. (P aeruginosa was the predominant organism.) We made the diagnosis of gram-negative foot intertrigo.
First described in 1973, researchers have found that P aeruginosa, as well as E faecalis and Staphylococcus aureus, are commonly associated with tinea pedis1 and toe web intertrigo.2 The infection usually involves the lateral 3 toe webs, and can present with malodorous discharge, itchy maceration, edema, and erythema of the surrounding tissue. Non-purulent lower extremity cellulitis is typically caused by beta-hemolytic streptococci residing in the toe webs, and is usually treated empirically with cephalexin or similar antibiotics.3 The presentation of foot intertrigo varies along a spectrum that includes tinea pedis, superinfected maceration, and infectious eczematoid dermatitis, all of which can encourage bacterial superinfections.
In this patient, it is likely that sweating (and consequent skin maceration), tinea pedis, and perhaps even friction between the affected area and the patient’s shoes led to skin ulceration. This then became colonized and infected with bacteria, including gram-negative varieties, which sometimes harbor at the edges of ulcerations. Addressing each of these factors is important to heal the infection.
Differential diagnosis includes other bacterial skin infections
The differential diagnosis includes erysipelas, cellulitis, lipodermatosclerosis, venous eczema, and burns. Erysipelas affects the superficial dermis with well demarcated borders, while cellulitis involves the subcutaneous fat.4 Both are more likely to be caused by beta-hemolytic streptococci, but at first may be difficult to differentiate from similar appearing gram-negative skin/soft tissue infection without microbiologic data.
Patients with lipodermatosclerosis and venous eczema often have a history of chronic venous insufficiency. Lipodermatosclerosis often presents with a subcutaneous panniculitis and hyperpigmentation; venous eczema is often associated with scaling of the involved areas. Although burns can become secondarily superinfected with bacteria, they can be differentiated from a primary bacterial infection by the history or presentation.
Gram-negative infections (especially those caused by Pseudomonas species) should be suspected if a toe web infection does not respond to empiric antimicrobial therapy with first- or second-generation cephalosporins, as their spectrum of antimicrobial activity does not include P aeruginosa. If the infection is severe, it may impact ambulation.2 Predisposing factors for gram-negative toe web infections include obesity, diabetes, moist environments, tight interdigital spaces, and recurrent tinea pedis.4
Administer appropriate wound care and start antimicrobial therapy
Foot intertrigo provides an easy portal of entry for pathogenic organisms.5 Therefore, it is important to modify risk factors from the outset to help prevent superinfection (as occurred with this patient) and other complications. Aggressive treatment of tinea pedis and use of compression stockings to reduce lymphedema are vital for prevention of recurrent infections.3
Physicians should be aware of likely, as well as unlikely, causative pathogens; “typical” skin and soft tissue infections that do not resolve may be due to atypical or gram-negative organisms, and typical first-line antibiotics will do nothing to eradicate them. Antimicrobial susceptibilities and a bacterial culture will steer you to the appropriate antimicrobial therapy. Debridement of the edge of the ulceration is necessary to remove any lingering bacteria.6 And appropriate wound care is paramount and should include the use of techniques to keep the affected areas dry, such as the use of astringent powders along with avoidance of damp shoes and socks.
Our patient was switched from empiric therapy to culture-specific IV therapy with vancomycin 2 g every 12 hours, ciprofloxacin 400 mg every 12 hours, and fluconazole 400 mg daily. Two weeks later, he was discharged from the hospital on topical antifungals, oral fluconazole, and daily acetic acid soaks. He did, however, require further advanced topical treatments (miconazole 2% twice daily) due to recurrent flare-ups before complete resolution was achieved 6 weeks after presentation (FIGURES 2A AND 2B).
CORRESPONDENCE
Alberto Marcelin, MD, Department of Family Medicine, Mayo Clinic Health System, 1000 1st Dr NW, Austin, MN 55912; [email protected].
A 50-year-old man who worked in construction presented to a local urgent care facility complaining of 2 weeks of bilateral foot discomfort associated with local malodorous discharge, redness, and crusting. He had a past medical history of recurrent tinea pedis and had previously been treated with intravenous (IV) antibiotics for a severe episode of cellulitis. He denied recent fever, trauma, or swelling. Physical examination revealed extensive malodorous crusting of the interdigital webs of both feet, in addition to tenderness, erythema, and serous discharge. He was treated with topical clotrimazole and oral terbinafine for a presumed tinea pedis recurrence; cephalexin and triamcinolone were added a week later after minimal response. Two days later, he sought care at a local emergency department with progressive cellulitis, bullae formation, and extensive desquamation (FIGURES 1A AND 1B) and was hospitalized.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Gram-negative foot intertrigo
Plain x-rays and computed tomography imaging did not reveal evidence of abscess formation, subcutaneous gas/air formation, or osteomyelitis. Laboratory testing was normal. The patient was empirically started on cefepime, clindamycin, and ketoconazole. Wound therapy, consisting of normal saline washes, use of xeroform gauze to cover the wound, and frequent absorbent dressing changes, was also initiated. Wound cultures were obtained, which later grew Pseudomonas aeruginosa and Enterococcus faecalis. (P aeruginosa was the predominant organism.) We made the diagnosis of gram-negative foot intertrigo.
First described in 1973, researchers have found that P aeruginosa, as well as E faecalis and Staphylococcus aureus, are commonly associated with tinea pedis1 and toe web intertrigo.2 The infection usually involves the lateral 3 toe webs, and can present with malodorous discharge, itchy maceration, edema, and erythema of the surrounding tissue. Non-purulent lower extremity cellulitis is typically caused by beta-hemolytic streptococci residing in the toe webs, and is usually treated empirically with cephalexin or similar antibiotics.3 The presentation of foot intertrigo varies along a spectrum that includes tinea pedis, superinfected maceration, and infectious eczematoid dermatitis, all of which can encourage bacterial superinfections.
In this patient, it is likely that sweating (and consequent skin maceration), tinea pedis, and perhaps even friction between the affected area and the patient’s shoes led to skin ulceration. This then became colonized and infected with bacteria, including gram-negative varieties, which sometimes harbor at the edges of ulcerations. Addressing each of these factors is important to heal the infection.
Differential diagnosis includes other bacterial skin infections
The differential diagnosis includes erysipelas, cellulitis, lipodermatosclerosis, venous eczema, and burns. Erysipelas affects the superficial dermis with well demarcated borders, while cellulitis involves the subcutaneous fat.4 Both are more likely to be caused by beta-hemolytic streptococci, but at first may be difficult to differentiate from similar appearing gram-negative skin/soft tissue infection without microbiologic data.
Patients with lipodermatosclerosis and venous eczema often have a history of chronic venous insufficiency. Lipodermatosclerosis often presents with a subcutaneous panniculitis and hyperpigmentation; venous eczema is often associated with scaling of the involved areas. Although burns can become secondarily superinfected with bacteria, they can be differentiated from a primary bacterial infection by the history or presentation.
Gram-negative infections (especially those caused by Pseudomonas species) should be suspected if a toe web infection does not respond to empiric antimicrobial therapy with first- or second-generation cephalosporins, as their spectrum of antimicrobial activity does not include P aeruginosa. If the infection is severe, it may impact ambulation.2 Predisposing factors for gram-negative toe web infections include obesity, diabetes, moist environments, tight interdigital spaces, and recurrent tinea pedis.4
Administer appropriate wound care and start antimicrobial therapy
Foot intertrigo provides an easy portal of entry for pathogenic organisms.5 Therefore, it is important to modify risk factors from the outset to help prevent superinfection (as occurred with this patient) and other complications. Aggressive treatment of tinea pedis and use of compression stockings to reduce lymphedema are vital for prevention of recurrent infections.3
Physicians should be aware of likely, as well as unlikely, causative pathogens; “typical” skin and soft tissue infections that do not resolve may be due to atypical or gram-negative organisms, and typical first-line antibiotics will do nothing to eradicate them. Antimicrobial susceptibilities and a bacterial culture will steer you to the appropriate antimicrobial therapy. Debridement of the edge of the ulceration is necessary to remove any lingering bacteria.6 And appropriate wound care is paramount and should include the use of techniques to keep the affected areas dry, such as the use of astringent powders along with avoidance of damp shoes and socks.
Our patient was switched from empiric therapy to culture-specific IV therapy with vancomycin 2 g every 12 hours, ciprofloxacin 400 mg every 12 hours, and fluconazole 400 mg daily. Two weeks later, he was discharged from the hospital on topical antifungals, oral fluconazole, and daily acetic acid soaks. He did, however, require further advanced topical treatments (miconazole 2% twice daily) due to recurrent flare-ups before complete resolution was achieved 6 weeks after presentation (FIGURES 2A AND 2B).
CORRESPONDENCE
Alberto Marcelin, MD, Department of Family Medicine, Mayo Clinic Health System, 1000 1st Dr NW, Austin, MN 55912; [email protected].
1. Westmoreland TA, Ross EV, Yeager JK. Pseudomonas toe web infections. Cutis. 1992;49:185-186.
2. Lin JY, Shih YL, Ho HC. Foot bacterial intertrigo mimicking interdigital tinea pedis. Chang Gung Med J. 2011;34:44-49.
3. Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis. 2014;59:e10-e52.
4. Hirschmann JV, Raugi GJ. Lower limb cellulitis and its mimics: part II. Conditions that simulate lower limb cellulitis. J Am Acad Dermatol. 2012;67:177, e1-e9; quiz 185-186.
5. Semel JD, Goldin H. Association of athlete’s foot with cellulitis of the lower extremities: diagnostic value of bacterial cultures of ipsilateral interdigital space samples. Clin Infect Dis. 1996;23:1162-1164.
6. Fangman W, Burton C. Hyperkeratotic rim of gram-negative toe web infections. Arch Dermatol. 2005;141:658.
1. Westmoreland TA, Ross EV, Yeager JK. Pseudomonas toe web infections. Cutis. 1992;49:185-186.
2. Lin JY, Shih YL, Ho HC. Foot bacterial intertrigo mimicking interdigital tinea pedis. Chang Gung Med J. 2011;34:44-49.
3. Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis. 2014;59:e10-e52.
4. Hirschmann JV, Raugi GJ. Lower limb cellulitis and its mimics: part II. Conditions that simulate lower limb cellulitis. J Am Acad Dermatol. 2012;67:177, e1-e9; quiz 185-186.
5. Semel JD, Goldin H. Association of athlete’s foot with cellulitis of the lower extremities: diagnostic value of bacterial cultures of ipsilateral interdigital space samples. Clin Infect Dis. 1996;23:1162-1164.
6. Fangman W, Burton C. Hyperkeratotic rim of gram-negative toe web infections. Arch Dermatol. 2005;141:658.
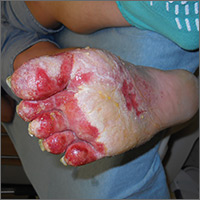
Rash on eyebrows and periumbilical region
An 8-year-old girl was brought to her family physician’s office (RU) because of a persistent rash on her lateral eyebrows and periumbilical region. The family indicated that she’d had the rash for more than 6 months. They also mentioned that the child had received a new pair of eyeglasses 8 months earlier. The child was otherwise in good health. The physical examination revealed erythematous scaling plaques near both lateral eyebrows and around the belly button (FIGURES 1 AND 2).
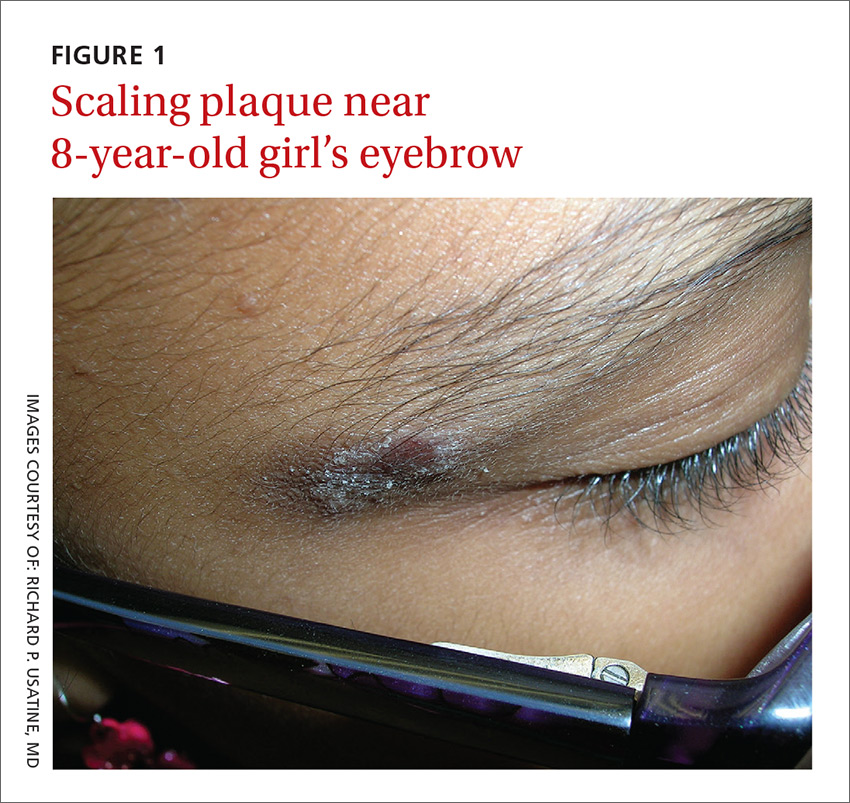
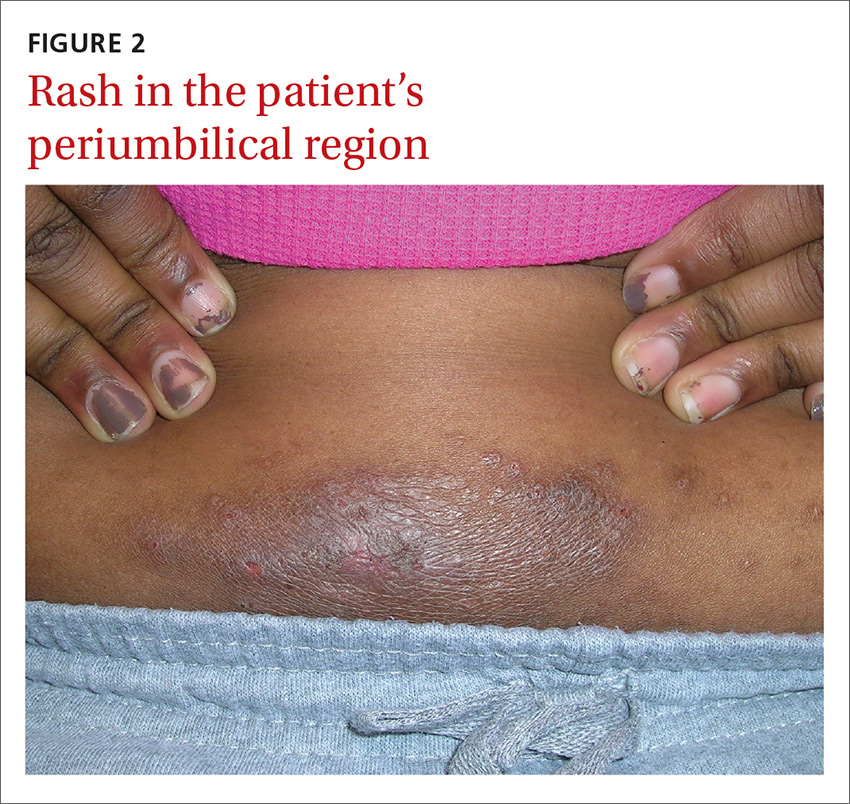
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Allergic contact dermatitis
We recognized that this was a case of allergic contact dermatitis (ACD), based on the clinical presentation. The distribution of the erythema, scale, and postinflammatory hyperpigmentation was highly suggestive of an ACD to nickel. In this case, the nickel in the patient’s eyeglasses and the snaps found on her pants were the culprits.
The lichenification of the plaque near the umbilicus suggested that the dermatitis was not acute and that the patient had likely been scratching the area due to itching. The plaque near the patient’s eye was actually in the shape of the metal on the inside of her glasses.
Most prevalent contact allergens. Patch testing data indicate that the 5 most prevalent contact allergens out of more than 3700 that are known are: nickel (14.3% of patients tested), fragrance mix (14%), the topical antibiotic neomycin (11.6%), balsam of Peru (used in some perfumes, toiletries, and pharmaceutical items) (10.4%), and the mercury-based vaccine preservative thimerosal (10.4%).1
ACD is a delayed-type hypersensitivity reaction in which a foreign substance comes into contact with the skin and is linked to skin proteins forming an antigen complex that leads to sensitization. When the epidermis is re-exposed to the antigen, the sensitized T cells initiate an inflammatory cascade, leading to the skin changes seen in ACD.2
Silverberg et al reported that in 30 children with a personal history of umbilical or wrist dermatitis or a family history of nickel ACD, 100% demonstrated a positive reaction to nickel sulfate.3 Nickel continues to be used (and unregulated) in a wide range of products, including costume jewelry, piercing posts, belt buckles, eyeglasses, and personal electronics (eg, tablets, cell phones, and laptop computers).
Making the diagnosis. Contact dermatitis can sometimes be diagnosed clinically with a good history and physical exam. However, there are many cases in which patch testing is needed to find the offending allergens or confirm the suspicion regarding a specific allergen. The only convenient and ready-to-use patch test in the United States is the T.R.U.E. test.
The differential includes other superficial skin infections
ACD characteristically presents with eczematoid plaques that are primarily in the area(s) of cutaneous contact with an allergen. The condition typically appears within a few days of exposure.
The differential diagnosis for ACD includes cutaneous candidiasis, impetigo, plaque psoriasis, and seborrheic dermatitis.
Cutaneous candidiasis is a superficial infection of the skin with a candida species. It can present as beefy red erythematous plaques on the buttocks, lower abdomen, thighs, or in intertriginous areas or oral commissures. A hallmark sign is pinpoint pustulovesicular satellite lesions.
Impetigo is a superficial bacterial skin infection that presents with edema, erythema, tenderness on palpation, and possible purulent drainage. It appears as honey-colored crusts with erythema and occurs most often on the face—especially around a child’s nose and mouth—but can occur anywhere on the head and body.
Plaque psoriasis presents as erythematous silver-scaled plaques on extensor surfaces, including the elbows and knees. Inverse psoriasis may present as erythema and maceration in intertriginous areas.
Seborrheic dermatitis appears as well-circumscribed greasy scale overlying erythematous skin. It is commonly found on the scalp, eyebrows, nasolabial folds, chest, face, and in the ear canals. It is thought to be an inflammatory reaction to Malassezia furfur.
Cool compresses, topical steroids can relieve symptoms
Patients with ACD should avoid the allergen that is causing the reaction. In cases of nickel ACD, the patient may cover the metal tab of their jeans with an iron-on patch or a few coats of clear nail polish. A better option is to buy jeans and pants that do not have nickel in the metal tab. (Levi’s has removed nickel from their pants.) Cool compresses can soothe the symptoms of acute cases of ACD.4 Calamine and colloidal oatmeal baths may help to dry and soothe acute, oozing lesions. Localized acute ACD lesions respond best to mid-potency (eg, 0.1% triamcinolone) to high-potency (eg, 0.05% clobetasol) topical steroids.4
On areas of thinner skin (eg, flexural surfaces, eyelids, face, anogenital region), lower-potency steroids such as desonide ointment can minimize the risk of skin atrophy.3,4 Be aware that some patients are actually allergic to topical steroids. This unfortunate situation can be diagnosed with patch testing.
We recommended that our patient get different glasses that were nickel-free. Fortunately, there are many frames for glasses that have no nickel in them. We also gave her advice on how to avoid the nickel that still exists in some pants. We gave her desonide 0.05% cream to apply to the affected area for symptomatic relief.
CORRESPONDENCE
Richard P. Usatine, MD, Skin clinic, University of Texas Health Science Center at San Antonio, 903 W. Martin, San Antonio, TX 78207; [email protected].
1. Krob HA, Fleischer AB Jr, D’Agostino R Jr, et al. Prevalence and relevance of contact dermatitis allergens: a meta-analysis of 15 years of published T.R.U.E. test data. J Am Acad Dermatol. 2004;51:349-353.
2. Usatine RP, Riojas M. Diagnosis and management of contact dermatitis. Am Fam Physician. 2010;82:249-255.
3. Silverberg NB, Licht J, Friedler S, et al. Nickel contact hypersensitivity in children. Pediatr Dermatol. 2002;19:110-113.
4. Beltrani VS, Bernstein IL, Cohen DE, et al. Contact dermatitis: a practice parameter. Ann Allergy Asthma Immunol. 2006;97:S1-S38.
An 8-year-old girl was brought to her family physician’s office (RU) because of a persistent rash on her lateral eyebrows and periumbilical region. The family indicated that she’d had the rash for more than 6 months. They also mentioned that the child had received a new pair of eyeglasses 8 months earlier. The child was otherwise in good health. The physical examination revealed erythematous scaling plaques near both lateral eyebrows and around the belly button (FIGURES 1 AND 2).
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Allergic contact dermatitis
We recognized that this was a case of allergic contact dermatitis (ACD), based on the clinical presentation. The distribution of the erythema, scale, and postinflammatory hyperpigmentation was highly suggestive of an ACD to nickel. In this case, the nickel in the patient’s eyeglasses and the snaps found on her pants were the culprits.
The lichenification of the plaque near the umbilicus suggested that the dermatitis was not acute and that the patient had likely been scratching the area due to itching. The plaque near the patient’s eye was actually in the shape of the metal on the inside of her glasses.
Most prevalent contact allergens. Patch testing data indicate that the 5 most prevalent contact allergens out of more than 3700 that are known are: nickel (14.3% of patients tested), fragrance mix (14%), the topical antibiotic neomycin (11.6%), balsam of Peru (used in some perfumes, toiletries, and pharmaceutical items) (10.4%), and the mercury-based vaccine preservative thimerosal (10.4%).1
ACD is a delayed-type hypersensitivity reaction in which a foreign substance comes into contact with the skin and is linked to skin proteins forming an antigen complex that leads to sensitization. When the epidermis is re-exposed to the antigen, the sensitized T cells initiate an inflammatory cascade, leading to the skin changes seen in ACD.2
Silverberg et al reported that in 30 children with a personal history of umbilical or wrist dermatitis or a family history of nickel ACD, 100% demonstrated a positive reaction to nickel sulfate.3 Nickel continues to be used (and unregulated) in a wide range of products, including costume jewelry, piercing posts, belt buckles, eyeglasses, and personal electronics (eg, tablets, cell phones, and laptop computers).
Making the diagnosis. Contact dermatitis can sometimes be diagnosed clinically with a good history and physical exam. However, there are many cases in which patch testing is needed to find the offending allergens or confirm the suspicion regarding a specific allergen. The only convenient and ready-to-use patch test in the United States is the T.R.U.E. test.
The differential includes other superficial skin infections
ACD characteristically presents with eczematoid plaques that are primarily in the area(s) of cutaneous contact with an allergen. The condition typically appears within a few days of exposure.
The differential diagnosis for ACD includes cutaneous candidiasis, impetigo, plaque psoriasis, and seborrheic dermatitis.
Cutaneous candidiasis is a superficial infection of the skin with a candida species. It can present as beefy red erythematous plaques on the buttocks, lower abdomen, thighs, or in intertriginous areas or oral commissures. A hallmark sign is pinpoint pustulovesicular satellite lesions.
Impetigo is a superficial bacterial skin infection that presents with edema, erythema, tenderness on palpation, and possible purulent drainage. It appears as honey-colored crusts with erythema and occurs most often on the face—especially around a child’s nose and mouth—but can occur anywhere on the head and body.
Plaque psoriasis presents as erythematous silver-scaled plaques on extensor surfaces, including the elbows and knees. Inverse psoriasis may present as erythema and maceration in intertriginous areas.
Seborrheic dermatitis appears as well-circumscribed greasy scale overlying erythematous skin. It is commonly found on the scalp, eyebrows, nasolabial folds, chest, face, and in the ear canals. It is thought to be an inflammatory reaction to Malassezia furfur.
Cool compresses, topical steroids can relieve symptoms
Patients with ACD should avoid the allergen that is causing the reaction. In cases of nickel ACD, the patient may cover the metal tab of their jeans with an iron-on patch or a few coats of clear nail polish. A better option is to buy jeans and pants that do not have nickel in the metal tab. (Levi’s has removed nickel from their pants.) Cool compresses can soothe the symptoms of acute cases of ACD.4 Calamine and colloidal oatmeal baths may help to dry and soothe acute, oozing lesions. Localized acute ACD lesions respond best to mid-potency (eg, 0.1% triamcinolone) to high-potency (eg, 0.05% clobetasol) topical steroids.4
On areas of thinner skin (eg, flexural surfaces, eyelids, face, anogenital region), lower-potency steroids such as desonide ointment can minimize the risk of skin atrophy.3,4 Be aware that some patients are actually allergic to topical steroids. This unfortunate situation can be diagnosed with patch testing.
We recommended that our patient get different glasses that were nickel-free. Fortunately, there are many frames for glasses that have no nickel in them. We also gave her advice on how to avoid the nickel that still exists in some pants. We gave her desonide 0.05% cream to apply to the affected area for symptomatic relief.
CORRESPONDENCE
Richard P. Usatine, MD, Skin clinic, University of Texas Health Science Center at San Antonio, 903 W. Martin, San Antonio, TX 78207; [email protected].
An 8-year-old girl was brought to her family physician’s office (RU) because of a persistent rash on her lateral eyebrows and periumbilical region. The family indicated that she’d had the rash for more than 6 months. They also mentioned that the child had received a new pair of eyeglasses 8 months earlier. The child was otherwise in good health. The physical examination revealed erythematous scaling plaques near both lateral eyebrows and around the belly button (FIGURES 1 AND 2).
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Allergic contact dermatitis
We recognized that this was a case of allergic contact dermatitis (ACD), based on the clinical presentation. The distribution of the erythema, scale, and postinflammatory hyperpigmentation was highly suggestive of an ACD to nickel. In this case, the nickel in the patient’s eyeglasses and the snaps found on her pants were the culprits.
The lichenification of the plaque near the umbilicus suggested that the dermatitis was not acute and that the patient had likely been scratching the area due to itching. The plaque near the patient’s eye was actually in the shape of the metal on the inside of her glasses.
Most prevalent contact allergens. Patch testing data indicate that the 5 most prevalent contact allergens out of more than 3700 that are known are: nickel (14.3% of patients tested), fragrance mix (14%), the topical antibiotic neomycin (11.6%), balsam of Peru (used in some perfumes, toiletries, and pharmaceutical items) (10.4%), and the mercury-based vaccine preservative thimerosal (10.4%).1
ACD is a delayed-type hypersensitivity reaction in which a foreign substance comes into contact with the skin and is linked to skin proteins forming an antigen complex that leads to sensitization. When the epidermis is re-exposed to the antigen, the sensitized T cells initiate an inflammatory cascade, leading to the skin changes seen in ACD.2
Silverberg et al reported that in 30 children with a personal history of umbilical or wrist dermatitis or a family history of nickel ACD, 100% demonstrated a positive reaction to nickel sulfate.3 Nickel continues to be used (and unregulated) in a wide range of products, including costume jewelry, piercing posts, belt buckles, eyeglasses, and personal electronics (eg, tablets, cell phones, and laptop computers).
Making the diagnosis. Contact dermatitis can sometimes be diagnosed clinically with a good history and physical exam. However, there are many cases in which patch testing is needed to find the offending allergens or confirm the suspicion regarding a specific allergen. The only convenient and ready-to-use patch test in the United States is the T.R.U.E. test.
The differential includes other superficial skin infections
ACD characteristically presents with eczematoid plaques that are primarily in the area(s) of cutaneous contact with an allergen. The condition typically appears within a few days of exposure.
The differential diagnosis for ACD includes cutaneous candidiasis, impetigo, plaque psoriasis, and seborrheic dermatitis.
Cutaneous candidiasis is a superficial infection of the skin with a candida species. It can present as beefy red erythematous plaques on the buttocks, lower abdomen, thighs, or in intertriginous areas or oral commissures. A hallmark sign is pinpoint pustulovesicular satellite lesions.
Impetigo is a superficial bacterial skin infection that presents with edema, erythema, tenderness on palpation, and possible purulent drainage. It appears as honey-colored crusts with erythema and occurs most often on the face—especially around a child’s nose and mouth—but can occur anywhere on the head and body.
Plaque psoriasis presents as erythematous silver-scaled plaques on extensor surfaces, including the elbows and knees. Inverse psoriasis may present as erythema and maceration in intertriginous areas.
Seborrheic dermatitis appears as well-circumscribed greasy scale overlying erythematous skin. It is commonly found on the scalp, eyebrows, nasolabial folds, chest, face, and in the ear canals. It is thought to be an inflammatory reaction to Malassezia furfur.
Cool compresses, topical steroids can relieve symptoms
Patients with ACD should avoid the allergen that is causing the reaction. In cases of nickel ACD, the patient may cover the metal tab of their jeans with an iron-on patch or a few coats of clear nail polish. A better option is to buy jeans and pants that do not have nickel in the metal tab. (Levi’s has removed nickel from their pants.) Cool compresses can soothe the symptoms of acute cases of ACD.4 Calamine and colloidal oatmeal baths may help to dry and soothe acute, oozing lesions. Localized acute ACD lesions respond best to mid-potency (eg, 0.1% triamcinolone) to high-potency (eg, 0.05% clobetasol) topical steroids.4
On areas of thinner skin (eg, flexural surfaces, eyelids, face, anogenital region), lower-potency steroids such as desonide ointment can minimize the risk of skin atrophy.3,4 Be aware that some patients are actually allergic to topical steroids. This unfortunate situation can be diagnosed with patch testing.
We recommended that our patient get different glasses that were nickel-free. Fortunately, there are many frames for glasses that have no nickel in them. We also gave her advice on how to avoid the nickel that still exists in some pants. We gave her desonide 0.05% cream to apply to the affected area for symptomatic relief.
CORRESPONDENCE
Richard P. Usatine, MD, Skin clinic, University of Texas Health Science Center at San Antonio, 903 W. Martin, San Antonio, TX 78207; [email protected].
1. Krob HA, Fleischer AB Jr, D’Agostino R Jr, et al. Prevalence and relevance of contact dermatitis allergens: a meta-analysis of 15 years of published T.R.U.E. test data. J Am Acad Dermatol. 2004;51:349-353.
2. Usatine RP, Riojas M. Diagnosis and management of contact dermatitis. Am Fam Physician. 2010;82:249-255.
3. Silverberg NB, Licht J, Friedler S, et al. Nickel contact hypersensitivity in children. Pediatr Dermatol. 2002;19:110-113.
4. Beltrani VS, Bernstein IL, Cohen DE, et al. Contact dermatitis: a practice parameter. Ann Allergy Asthma Immunol. 2006;97:S1-S38.
1. Krob HA, Fleischer AB Jr, D’Agostino R Jr, et al. Prevalence and relevance of contact dermatitis allergens: a meta-analysis of 15 years of published T.R.U.E. test data. J Am Acad Dermatol. 2004;51:349-353.
2. Usatine RP, Riojas M. Diagnosis and management of contact dermatitis. Am Fam Physician. 2010;82:249-255.
3. Silverberg NB, Licht J, Friedler S, et al. Nickel contact hypersensitivity in children. Pediatr Dermatol. 2002;19:110-113.
4. Beltrani VS, Bernstein IL, Cohen DE, et al. Contact dermatitis: a practice parameter. Ann Allergy Asthma Immunol. 2006;97:S1-S38.
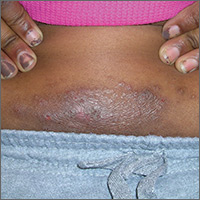
CPSTF: A lesser known, but valuable, resource for FPs
Family physicians have come to rely on the US Preventive Services Task Force (USPSTF) for rigorous, evidence-based recommendations on the use of clinical preventive services. Still, many such services reach too few individuals who need them. And that’s where the less well known Community Preventive Services Task Force comes in. The CPSTF makes recommendations regarding public health interventions and ways to increase the use of preventive services in the clinical setting—eg, means of improving childhood immunization rates or increasing screening for cervical, breast, and colon cancer.
To better understand how the CPSTF can serve as a resource to busy family physicians, it’s helpful to first understand a bit about the inner-workings of the CPSTF itself.
How CPSTF figures out what works
Formed in 1996, the CPSTF consists of 15 independent, nonfederal members with expertise in public health and preventive medicine, appointed by the Director of the Centers for Disease Control and Prevention (CDC). The Task Force makes recommendations and develops guidance on which community-based health promotion and disease-prevention interventions work and which do not, based on available scientific evidence. The Task Force uses an evidence-based methodology similar to that of the USPSTF—ie, assessing systematic reviews of the evidence and tying recommendations to the strength of the evidence. However, the Task Force has only 3 levels of recommendations: recommend for, recommend against, and insufficient evidence to recommend.
Three CPSTF meetings are held each year, and a representative from the American Academy of Family Physicians (AAFP) attends as a liaison, along with liaisons from other organizations with an interest in the methods and recommendations. The CDC provides the CPSTF with technical and administrative support. However, the recommendations developed do not undergo review or approval by the CDC and are the sole responsibility of the Task Force.
The recommendations made are contained in the Guide to Community Preventive Services, often called The Community Guide, which is available on the Task Force’s Web site at www.thecommunityguide.org/index.html. The topics on which the CPSTF currently has recommendations are listed in TABLE 1. (Since community-wide recommendations are rarely subjected to controlled clinical trials, methods of assessing and ranking other forms of evidence are required. To learn more about how the CPSTF approaches this, see: https://www.thecommunityguide.org/about/our-methodology.)

Improving immunization rates
The topic of immunizations is an example of how synergistic the CPSTF recommendations can be with those from clinical organizations. The Advisory Committee on Immunization Practices (ACIP) makes recommendations on the use of vaccines.1 The CPSTF has developed a set of recommendations on how to increase the uptake of vaccines to improve rates of immunization.2 Interventions they recommend include vaccine requirements for attendance at preschool, primary and secondary school, and college; patient reminder and recall systems; patient and family incentives and rewards; providing vaccines at Women, Infants, and Children clinics, schools, work sites, and homes; standing orders for vaccine administration; physician reminders; physician assessments and feedback; reducing out-of-pocket expenses for vaccines; and using immunization registries. Just as important, the CPSTF identifies interventions that lack hard evidence to support their effectiveness.
Cancer screening works, but patient buy-in lags
The USPSTF recommends screening for breast, cervical, and colorectal cancer. And yet, despite the proven effectiveness of these screening tests in decreasing cancer mortality, many people do not get screened. The CPSTF has developed a set of implementation recommendations that are proven to increase the uptake of recommended cancer screening tests.3 These include:
- sending reminders to patients when screening tests are due
- providing one-on-one or group educational sessions
- providing videos and printed materials that describe screening tests and recommendations
- offering testing at locations and times that are convenient for patients
- offering on-site translation, transportation, patient navigators, and other administrative services to facilitate screening
- assessing provider performance and providing feedback.
CPSTF’s range of resources
Resources provided by the CPSTF (TABLE 2) also include the following materials for physicians, patients, and policy makers:
- tools to assist communities in performing a community health assessment and in prioritizing health needs
- fact sheets on what works for specific populations or conditions. (One recently added fact sheet is a description of interventions to address the leading health problems that affect women.4)
- examples of how communities have used CPSTF recommendations to address a major health concern in their populations. (See “An immunization ‘success story’ from the field.”)

Tackling controversial social issues
Public health interventions are often politically charged, and the CPSTF at times makes recommendations that, while supported by evidence, raise objections from certain groups. One example is a recommendation for “comprehensive risk reduction interventions to promote behaviors that prevent or reduce the risk of pregnancy, human immunodeficiency virus (HIV), and other sexually transmitted infections (STIs).”5 These interventions may include a hierarchy of recommended behaviors that identifies abstinence as the best or preferred method, but also provides information about sexual risk reduction strategies. Abstinence-only education initiatives were rated as having insufficient evidence for effectiveness.6
Another example that falls in the controversial realm is a recommendation against “policies facilitating the transfer of juveniles from juvenile to adult criminal justice systems for the purpose of reducing violence, based on strong evidence that these laws and policies are associated with increased subsequent violent behavior among transferred youth.”7
And a third example is a recommendation for “the use of regulatory authority (eg, through licensing and zoning) to limit alcohol outlet density on the basis of sufficient evidence of a positive association between outlet density and excessive alcohol consumption and related harms.”8 The CPSTF also recommends increasing taxes on alcohol products to reduce excess alcohol consumption.9
SIDEBAR
An immunization “success story” from the field
Before 2009, the vaccination completion rates for 2-year-olds in Duval County, Florida, consistently ranked below the national target of 90%, with particularly low rates in Jacksonville. With the aim of improving vaccination rates—and not wanting to waste time “reinventing the wheel”—the Duval County Health Department (DCHD) turned to The Community Guide for interventions proven to work synergistically: system-based efforts (eg, client reminders, standing orders, clinic-based education) and community-based efforts (eg, staff outreach to clients, educational activities).
Checking the Florida Shots Registry, clinic staff identified infants and toddlers who were due for, or had missed, vaccinations. They sent monthly reminders to parents, urging them to make appointments. DCHD also provided parents with educational materials, vaccination schedules, and safety evidence to reinforce awareness of the need for immunizations.
At local clinics, DCHD trained staff to administer vaccines and established standing orders authorizing them to do so even in the absence of a physician or other approving practitioner.
DCHD also formed an immunization task force of community stakeholders that worked with hospitals to send nurses and physicians each week to immunize children at churches and other convenient locations.
Within one year, the rate of complete immunization for 2-year-olds rose from 75% to 90%—the national target. DCHD is now applying interventions from The Community Guide to discourage tobacco use and to prevent sexually transmitted infections.
Read the full story at: https://www.thecommunityguide.org/stories/good-shot-reaching-immunization-targets-duval-county.
Reducing health disparities
The CPSTF places a high priority on interventions that can reduce health disparities. Many of their topics of interest focus on interventions to reduce health inequities among racial and ethnic minorities and low-income populations. For instance, the Task Force recommends early childhood education, all-day kindergarten, and after-school academic programs as ways to improve health and decrease health disparities.10
Social determinants of health for individuals and populations are increasingly appreciated as issues to be addressed by physicians and health systems. The CPSTF can serve as a valuable evidence-based resource in these efforts, and their recommendations complement and build on those of other authoritative groups such as the USPSTF, ACIP, and AAFP.
1. Centers for Disease Control and Prevention. ACIP vaccine recommendations. Available at: http://www.cdc.gov/vaccines/hcp/acip-recs/index.html. Accessed December 6, 2016.
2. Community Preventive Services Task Force. The Community Guide. Vaccination. Available at: https://www.thecommunityguide.org/topic/vaccination. Accessed December 6, 2016.
3. Community Preventive Services Task Force. The Community Guide. Cancer prevention and control: cancer screening [fact sheet]. Available at: https://www.thecommunityguide.org/sites/default/files/assets/What-Works-Cancer-Screening-factsheet-and-insert.pdf. Accessed December 6, 2016.
4. Community Preventive Services Task Fo
5. Community Preventive Services Task Force. The Community Guide. HIV/AIDS, other STIs, and teen pregnancy: group-based comprehensive risk reduction interventions for adolescents. Available at: https://www.thecommunityguide.org/findings/hivaids-other-stis-and-teen-pregnancy-group-based-comprehensive-risk-reduction-interventions. Accessed December 6, 2016.
6. Community Preventive Services Task Force. The Community Guide. HIV/AIDS, STIs and pregnancy. Available at: https://www.thecommunityguide.org/topic/hivaids-stis-and-pregnancy. Accessed December 6, 2016.
7. Community Preventive Services Task Force. The Community Guide. Violence: policies facilitating the transfer of juveniles to adult justice systems. Available at: https://www.thecommunityguide.org/findings/violence-policies-facilitating-transfer-juveniles-adult-justice-systems. Accessed December 6, 2016.
8. Community Preventive Services Task Force. The Community Guide. Alcohol – excessive consumption: regulation of alcohol outlet density. Available at: https://www.thecommunityguide.org/findings/alcohol-excessive-consumption-regulation-alcohol-outlet-density. Accessed December 6, 2016.
9. Community Preventive Services Task Force. The Community Guide. Excessive alcohol consumption. Available at: https://www.thecommunityguide.org/topic/excessive-alcohol-consumption. Accessed December 6, 2016.
10. Community Preventive Services Task Force. The Community Guide. Health equity. Available at: https://www.thecommunityguide.org/topic/health-equity. Accessed December 6, 2016.
Family physicians have come to rely on the US Preventive Services Task Force (USPSTF) for rigorous, evidence-based recommendations on the use of clinical preventive services. Still, many such services reach too few individuals who need them. And that’s where the less well known Community Preventive Services Task Force comes in. The CPSTF makes recommendations regarding public health interventions and ways to increase the use of preventive services in the clinical setting—eg, means of improving childhood immunization rates or increasing screening for cervical, breast, and colon cancer.
To better understand how the CPSTF can serve as a resource to busy family physicians, it’s helpful to first understand a bit about the inner-workings of the CPSTF itself.
How CPSTF figures out what works
Formed in 1996, the CPSTF consists of 15 independent, nonfederal members with expertise in public health and preventive medicine, appointed by the Director of the Centers for Disease Control and Prevention (CDC). The Task Force makes recommendations and develops guidance on which community-based health promotion and disease-prevention interventions work and which do not, based on available scientific evidence. The Task Force uses an evidence-based methodology similar to that of the USPSTF—ie, assessing systematic reviews of the evidence and tying recommendations to the strength of the evidence. However, the Task Force has only 3 levels of recommendations: recommend for, recommend against, and insufficient evidence to recommend.
Three CPSTF meetings are held each year, and a representative from the American Academy of Family Physicians (AAFP) attends as a liaison, along with liaisons from other organizations with an interest in the methods and recommendations. The CDC provides the CPSTF with technical and administrative support. However, the recommendations developed do not undergo review or approval by the CDC and are the sole responsibility of the Task Force.
The recommendations made are contained in the Guide to Community Preventive Services, often called The Community Guide, which is available on the Task Force’s Web site at www.thecommunityguide.org/index.html. The topics on which the CPSTF currently has recommendations are listed in TABLE 1. (Since community-wide recommendations are rarely subjected to controlled clinical trials, methods of assessing and ranking other forms of evidence are required. To learn more about how the CPSTF approaches this, see: https://www.thecommunityguide.org/about/our-methodology.)
Improving immunization rates
The topic of immunizations is an example of how synergistic the CPSTF recommendations can be with those from clinical organizations. The Advisory Committee on Immunization Practices (ACIP) makes recommendations on the use of vaccines.1 The CPSTF has developed a set of recommendations on how to increase the uptake of vaccines to improve rates of immunization.2 Interventions they recommend include vaccine requirements for attendance at preschool, primary and secondary school, and college; patient reminder and recall systems; patient and family incentives and rewards; providing vaccines at Women, Infants, and Children clinics, schools, work sites, and homes; standing orders for vaccine administration; physician reminders; physician assessments and feedback; reducing out-of-pocket expenses for vaccines; and using immunization registries. Just as important, the CPSTF identifies interventions that lack hard evidence to support their effectiveness.
Cancer screening works, but patient buy-in lags
The USPSTF recommends screening for breast, cervical, and colorectal cancer. And yet, despite the proven effectiveness of these screening tests in decreasing cancer mortality, many people do not get screened. The CPSTF has developed a set of implementation recommendations that are proven to increase the uptake of recommended cancer screening tests.3 These include:
- sending reminders to patients when screening tests are due
- providing one-on-one or group educational sessions
- providing videos and printed materials that describe screening tests and recommendations
- offering testing at locations and times that are convenient for patients
- offering on-site translation, transportation, patient navigators, and other administrative services to facilitate screening
- assessing provider performance and providing feedback.
CPSTF’s range of resources
Resources provided by the CPSTF (TABLE 2) also include the following materials for physicians, patients, and policy makers:
- tools to assist communities in performing a community health assessment and in prioritizing health needs
- fact sheets on what works for specific populations or conditions. (One recently added fact sheet is a description of interventions to address the leading health problems that affect women.4)
- examples of how communities have used CPSTF recommendations to address a major health concern in their populations. (See “An immunization ‘success story’ from the field.”)
Tackling controversial social issues
Public health interventions are often politically charged, and the CPSTF at times makes recommendations that, while supported by evidence, raise objections from certain groups. One example is a recommendation for “comprehensive risk reduction interventions to promote behaviors that prevent or reduce the risk of pregnancy, human immunodeficiency virus (HIV), and other sexually transmitted infections (STIs).”5 These interventions may include a hierarchy of recommended behaviors that identifies abstinence as the best or preferred method, but also provides information about sexual risk reduction strategies. Abstinence-only education initiatives were rated as having insufficient evidence for effectiveness.6
Another example that falls in the controversial realm is a recommendation against “policies facilitating the transfer of juveniles from juvenile to adult criminal justice systems for the purpose of reducing violence, based on strong evidence that these laws and policies are associated with increased subsequent violent behavior among transferred youth.”7
And a third example is a recommendation for “the use of regulatory authority (eg, through licensing and zoning) to limit alcohol outlet density on the basis of sufficient evidence of a positive association between outlet density and excessive alcohol consumption and related harms.”8 The CPSTF also recommends increasing taxes on alcohol products to reduce excess alcohol consumption.9
SIDEBAR
An immunization “success story” from the field
Before 2009, the vaccination completion rates for 2-year-olds in Duval County, Florida, consistently ranked below the national target of 90%, with particularly low rates in Jacksonville. With the aim of improving vaccination rates—and not wanting to waste time “reinventing the wheel”—the Duval County Health Department (DCHD) turned to The Community Guide for interventions proven to work synergistically: system-based efforts (eg, client reminders, standing orders, clinic-based education) and community-based efforts (eg, staff outreach to clients, educational activities).
Checking the Florida Shots Registry, clinic staff identified infants and toddlers who were due for, or had missed, vaccinations. They sent monthly reminders to parents, urging them to make appointments. DCHD also provided parents with educational materials, vaccination schedules, and safety evidence to reinforce awareness of the need for immunizations.
At local clinics, DCHD trained staff to administer vaccines and established standing orders authorizing them to do so even in the absence of a physician or other approving practitioner.
DCHD also formed an immunization task force of community stakeholders that worked with hospitals to send nurses and physicians each week to immunize children at churches and other convenient locations.
Within one year, the rate of complete immunization for 2-year-olds rose from 75% to 90%—the national target. DCHD is now applying interventions from The Community Guide to discourage tobacco use and to prevent sexually transmitted infections.
Read the full story at: https://www.thecommunityguide.org/stories/good-shot-reaching-immunization-targets-duval-county.
Reducing health disparities
The CPSTF places a high priority on interventions that can reduce health disparities. Many of their topics of interest focus on interventions to reduce health inequities among racial and ethnic minorities and low-income populations. For instance, the Task Force recommends early childhood education, all-day kindergarten, and after-school academic programs as ways to improve health and decrease health disparities.10
Social determinants of health for individuals and populations are increasingly appreciated as issues to be addressed by physicians and health systems. The CPSTF can serve as a valuable evidence-based resource in these efforts, and their recommendations complement and build on those of other authoritative groups such as the USPSTF, ACIP, and AAFP.
Family physicians have come to rely on the US Preventive Services Task Force (USPSTF) for rigorous, evidence-based recommendations on the use of clinical preventive services. Still, many such services reach too few individuals who need them. And that’s where the less well known Community Preventive Services Task Force comes in. The CPSTF makes recommendations regarding public health interventions and ways to increase the use of preventive services in the clinical setting—eg, means of improving childhood immunization rates or increasing screening for cervical, breast, and colon cancer.
To better understand how the CPSTF can serve as a resource to busy family physicians, it’s helpful to first understand a bit about the inner-workings of the CPSTF itself.
How CPSTF figures out what works
Formed in 1996, the CPSTF consists of 15 independent, nonfederal members with expertise in public health and preventive medicine, appointed by the Director of the Centers for Disease Control and Prevention (CDC). The Task Force makes recommendations and develops guidance on which community-based health promotion and disease-prevention interventions work and which do not, based on available scientific evidence. The Task Force uses an evidence-based methodology similar to that of the USPSTF—ie, assessing systematic reviews of the evidence and tying recommendations to the strength of the evidence. However, the Task Force has only 3 levels of recommendations: recommend for, recommend against, and insufficient evidence to recommend.
Three CPSTF meetings are held each year, and a representative from the American Academy of Family Physicians (AAFP) attends as a liaison, along with liaisons from other organizations with an interest in the methods and recommendations. The CDC provides the CPSTF with technical and administrative support. However, the recommendations developed do not undergo review or approval by the CDC and are the sole responsibility of the Task Force.
The recommendations made are contained in the Guide to Community Preventive Services, often called The Community Guide, which is available on the Task Force’s Web site at www.thecommunityguide.org/index.html. The topics on which the CPSTF currently has recommendations are listed in TABLE 1. (Since community-wide recommendations are rarely subjected to controlled clinical trials, methods of assessing and ranking other forms of evidence are required. To learn more about how the CPSTF approaches this, see: https://www.thecommunityguide.org/about/our-methodology.)
Improving immunization rates
The topic of immunizations is an example of how synergistic the CPSTF recommendations can be with those from clinical organizations. The Advisory Committee on Immunization Practices (ACIP) makes recommendations on the use of vaccines.1 The CPSTF has developed a set of recommendations on how to increase the uptake of vaccines to improve rates of immunization.2 Interventions they recommend include vaccine requirements for attendance at preschool, primary and secondary school, and college; patient reminder and recall systems; patient and family incentives and rewards; providing vaccines at Women, Infants, and Children clinics, schools, work sites, and homes; standing orders for vaccine administration; physician reminders; physician assessments and feedback; reducing out-of-pocket expenses for vaccines; and using immunization registries. Just as important, the CPSTF identifies interventions that lack hard evidence to support their effectiveness.
Cancer screening works, but patient buy-in lags
The USPSTF recommends screening for breast, cervical, and colorectal cancer. And yet, despite the proven effectiveness of these screening tests in decreasing cancer mortality, many people do not get screened. The CPSTF has developed a set of implementation recommendations that are proven to increase the uptake of recommended cancer screening tests.3 These include:
- sending reminders to patients when screening tests are due
- providing one-on-one or group educational sessions
- providing videos and printed materials that describe screening tests and recommendations
- offering testing at locations and times that are convenient for patients
- offering on-site translation, transportation, patient navigators, and other administrative services to facilitate screening
- assessing provider performance and providing feedback.
CPSTF’s range of resources
Resources provided by the CPSTF (TABLE 2) also include the following materials for physicians, patients, and policy makers:
- tools to assist communities in performing a community health assessment and in prioritizing health needs
- fact sheets on what works for specific populations or conditions. (One recently added fact sheet is a description of interventions to address the leading health problems that affect women.4)
- examples of how communities have used CPSTF recommendations to address a major health concern in their populations. (See “An immunization ‘success story’ from the field.”)
Tackling controversial social issues
Public health interventions are often politically charged, and the CPSTF at times makes recommendations that, while supported by evidence, raise objections from certain groups. One example is a recommendation for “comprehensive risk reduction interventions to promote behaviors that prevent or reduce the risk of pregnancy, human immunodeficiency virus (HIV), and other sexually transmitted infections (STIs).”5 These interventions may include a hierarchy of recommended behaviors that identifies abstinence as the best or preferred method, but also provides information about sexual risk reduction strategies. Abstinence-only education initiatives were rated as having insufficient evidence for effectiveness.6
Another example that falls in the controversial realm is a recommendation against “policies facilitating the transfer of juveniles from juvenile to adult criminal justice systems for the purpose of reducing violence, based on strong evidence that these laws and policies are associated with increased subsequent violent behavior among transferred youth.”7
And a third example is a recommendation for “the use of regulatory authority (eg, through licensing and zoning) to limit alcohol outlet density on the basis of sufficient evidence of a positive association between outlet density and excessive alcohol consumption and related harms.”8 The CPSTF also recommends increasing taxes on alcohol products to reduce excess alcohol consumption.9
SIDEBAR
An immunization “success story” from the field
Before 2009, the vaccination completion rates for 2-year-olds in Duval County, Florida, consistently ranked below the national target of 90%, with particularly low rates in Jacksonville. With the aim of improving vaccination rates—and not wanting to waste time “reinventing the wheel”—the Duval County Health Department (DCHD) turned to The Community Guide for interventions proven to work synergistically: system-based efforts (eg, client reminders, standing orders, clinic-based education) and community-based efforts (eg, staff outreach to clients, educational activities).
Checking the Florida Shots Registry, clinic staff identified infants and toddlers who were due for, or had missed, vaccinations. They sent monthly reminders to parents, urging them to make appointments. DCHD also provided parents with educational materials, vaccination schedules, and safety evidence to reinforce awareness of the need for immunizations.
At local clinics, DCHD trained staff to administer vaccines and established standing orders authorizing them to do so even in the absence of a physician or other approving practitioner.
DCHD also formed an immunization task force of community stakeholders that worked with hospitals to send nurses and physicians each week to immunize children at churches and other convenient locations.
Within one year, the rate of complete immunization for 2-year-olds rose from 75% to 90%—the national target. DCHD is now applying interventions from The Community Guide to discourage tobacco use and to prevent sexually transmitted infections.
Read the full story at: https://www.thecommunityguide.org/stories/good-shot-reaching-immunization-targets-duval-county.
Reducing health disparities
The CPSTF places a high priority on interventions that can reduce health disparities. Many of their topics of interest focus on interventions to reduce health inequities among racial and ethnic minorities and low-income populations. For instance, the Task Force recommends early childhood education, all-day kindergarten, and after-school academic programs as ways to improve health and decrease health disparities.10
Social determinants of health for individuals and populations are increasingly appreciated as issues to be addressed by physicians and health systems. The CPSTF can serve as a valuable evidence-based resource in these efforts, and their recommendations complement and build on those of other authoritative groups such as the USPSTF, ACIP, and AAFP.
1. Centers for Disease Control and Prevention. ACIP vaccine recommendations. Available at: http://www.cdc.gov/vaccines/hcp/acip-recs/index.html. Accessed December 6, 2016.
2. Community Preventive Services Task Force. The Community Guide. Vaccination. Available at: https://www.thecommunityguide.org/topic/vaccination. Accessed December 6, 2016.
3. Community Preventive Services Task Force. The Community Guide. Cancer prevention and control: cancer screening [fact sheet]. Available at: https://www.thecommunityguide.org/sites/default/files/assets/What-Works-Cancer-Screening-factsheet-and-insert.pdf. Accessed December 6, 2016.
4. Community Preventive Services Task Fo
5. Community Preventive Services Task Force. The Community Guide. HIV/AIDS, other STIs, and teen pregnancy: group-based comprehensive risk reduction interventions for adolescents. Available at: https://www.thecommunityguide.org/findings/hivaids-other-stis-and-teen-pregnancy-group-based-comprehensive-risk-reduction-interventions. Accessed December 6, 2016.
6. Community Preventive Services Task Force. The Community Guide. HIV/AIDS, STIs and pregnancy. Available at: https://www.thecommunityguide.org/topic/hivaids-stis-and-pregnancy. Accessed December 6, 2016.
7. Community Preventive Services Task Force. The Community Guide. Violence: policies facilitating the transfer of juveniles to adult justice systems. Available at: https://www.thecommunityguide.org/findings/violence-policies-facilitating-transfer-juveniles-adult-justice-systems. Accessed December 6, 2016.
8. Community Preventive Services Task Force. The Community Guide. Alcohol – excessive consumption: regulation of alcohol outlet density. Available at: https://www.thecommunityguide.org/findings/alcohol-excessive-consumption-regulation-alcohol-outlet-density. Accessed December 6, 2016.
9. Community Preventive Services Task Force. The Community Guide. Excessive alcohol consumption. Available at: https://www.thecommunityguide.org/topic/excessive-alcohol-consumption. Accessed December 6, 2016.
10. Community Preventive Services Task Force. The Community Guide. Health equity. Available at: https://www.thecommunityguide.org/topic/health-equity. Accessed December 6, 2016.
1. Centers for Disease Control and Prevention. ACIP vaccine recommendations. Available at: http://www.cdc.gov/vaccines/hcp/acip-recs/index.html. Accessed December 6, 2016.
2. Community Preventive Services Task Force. The Community Guide. Vaccination. Available at: https://www.thecommunityguide.org/topic/vaccination. Accessed December 6, 2016.
3. Community Preventive Services Task Force. The Community Guide. Cancer prevention and control: cancer screening [fact sheet]. Available at: https://www.thecommunityguide.org/sites/default/files/assets/What-Works-Cancer-Screening-factsheet-and-insert.pdf. Accessed December 6, 2016.
4. Community Preventive Services Task Fo
5. Community Preventive Services Task Force. The Community Guide. HIV/AIDS, other STIs, and teen pregnancy: group-based comprehensive risk reduction interventions for adolescents. Available at: https://www.thecommunityguide.org/findings/hivaids-other-stis-and-teen-pregnancy-group-based-comprehensive-risk-reduction-interventions. Accessed December 6, 2016.
6. Community Preventive Services Task Force. The Community Guide. HIV/AIDS, STIs and pregnancy. Available at: https://www.thecommunityguide.org/topic/hivaids-stis-and-pregnancy. Accessed December 6, 2016.
7. Community Preventive Services Task Force. The Community Guide. Violence: policies facilitating the transfer of juveniles to adult justice systems. Available at: https://www.thecommunityguide.org/findings/violence-policies-facilitating-transfer-juveniles-adult-justice-systems. Accessed December 6, 2016.
8. Community Preventive Services Task Force. The Community Guide. Alcohol – excessive consumption: regulation of alcohol outlet density. Available at: https://www.thecommunityguide.org/findings/alcohol-excessive-consumption-regulation-alcohol-outlet-density. Accessed December 6, 2016.
9. Community Preventive Services Task Force. The Community Guide. Excessive alcohol consumption. Available at: https://www.thecommunityguide.org/topic/excessive-alcohol-consumption. Accessed December 6, 2016.
10. Community Preventive Services Task Force. The Community Guide. Health equity. Available at: https://www.thecommunityguide.org/topic/health-equity. Accessed December 6, 2016.
How in-office and ambulatory BP monitoring compare: A systematic review and meta-analysis
ABSTRACT
Purpose We performed a literature review and meta-analysis to ascertain the validity of office blood pressure (BP) measurement in a primary care setting, using ambulatory blood pressure measurement (ABPM) as a benchmark in the monitoring of hypertensive patients receiving treatment.
Methods We conducted a literature search for studies published up to December 2013 that included hypertensive patients receiving treatment in a primary care setting. We compared the mean office BP with readings obtained by ABPM. We summarized the diagnostic accuracy of office BP with respect to ABPM in terms of sensitivity, specificity, and positive and negative likelihood ratios (LR), with a 95% confidence interval (CI).
Results Only 12 studies met the inclusion criteria and contained data to calculate the differences between the means of office and ambulatory BP measurements. Five were suitable for calculating sensitivity, specificity, and likelihood ratios, and 4 contained sufficient extractable data for meta-analysis. Compared with ABPM (thresholds of 140/90 mm Hg for office BP; 130/80 mmHg for ABPM) in diagnosing uncontrolled BP, office BP measurement had a sensitivity of 81.9% (95% CI, 74.8%-87%) and specificity of 41.1% (95% CI, 35.1%-48.4%). Positive LR was 1.35 (95% CI, 1.32-1.38), and the negative LR was 0.44 (95% CI, 0.37-0.53).
Conclusion Likelihood ratios show that isolated BP measurement in the office does not confirm or rule out the presence of poor BP control. Likelihood of underestimating or overestimating BP control is high when relying on in-office BP measurement alone.
A growing body of evidence supports more frequent use of ambulatory blood pressure monitoring (ABPM) to confirm a diagnosis of hypertension1 and to monitor blood pressure (BP) response to treatment.2 The Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure has long accepted ABPM for diagnosis of hypertension,3 and many clinicians consider ABPM the reference standard for diagnosing true hypertension and for accurately assessing associated cardiovascular risk in adults, regardless of office BP readings.4 The US Preventive Services Task Force (USPSTF) recommends obtaining BP measurements outside the clinical setting to confirm a diagnosis of hypertension before starting treatment.5 The USPSTF also asserts that elevated 24-hour ambulatory systolic BP is consistently and significantly associated with stroke and other cardiovascular events independent of office BP readings and has greater predictive value than office monitoring.5 The USPSTF concludes that ABPM, because of its large evidence base, is the best confirmatory test for hypertension.6 The recommendation of the American Academy of Family Physicians is similar to that of the USPSTF.7
The challenge. Despite the considerable support for ABPM, this method of BP measurement is still not sufficiently integrated into primary care. And some guidelines, such as those of
But ABPM’s advantages are numerous. Ambulatory monitors, which can record BP for 24 hours, are typically programmed to take readings every 15 to 30 minutes, providing estimates of mean daytime and nighttime BP and revealing an individual’s circadian pattern of BP.8-10 Ambulatory BP values usually considered the uppermost limit of normal are 135/85 mm Hg (day), 120/70 mm Hg (night), and 130/80 mm Hg (24 hour).8
Office BP monitoring, usually performed manually by medical staff, has 2 main drawbacks: the well-known white-coat effect experienced by many patients, and the relatively small number of possible measurements. A more reliable in-office BP estimation of BP would require repeated measurements at each of several visits.
By comparing ABPM and office measurements, 4 clinical findings are possible: isolated clinic or office (white-coat) hypertension (ICH); isolated ambulatory (masked) hypertension (IAH); consistent normotension; or sustained hypertension. With ICH, BP is high in the office and normal with ABPM. With IAH, BP is normal in the office and high with ABPM. With consistent normotension and sustained hypertension, BP readings with both types of measurement agree.8,9
In patients being treated for hypertension, ICH leads to an overestimation of uncontrolled BP and may result in overtreatment. The cardiovascular risk, although controversial, is usually lower than in patients diagnosed with sustained hypertension.11 IAH leads to an underestimation of uncontrolled BP and may result in undertreatment; its associated cardiovascular risk is similar to that of sustained hypertension.12
Our research objective. We recently published a study conducted with 137 hypertensive patients in a primary care center.13 Our conclusion was that in-office measurement of BP had insufficient clinical validity to be recommended as a sole method of monitoring BP control. In accurately classifying BP as controlled or uncontrolled, clinic measurement agreed with 24h-ABPM in just 64.2% of cases.13
In our present study, we performed a literature review and meta-analysis to ascertain the validity of office BP measurement in a primary care setting, using ABPM as a benchmark in the monitoring of hypertensive patients receiving treatment.
METHODS
Most published studies comparing conventional office BP measurement with ABPM have been conducted with patients not taking antihypertensive medication. We excluded these studies and conducted a literature search for studies published up to December 2013 that included hypertensive patients receiving treatment in a primary care setting.
We searched Medline (from 1950 onward) and the Cochrane Database of Systematic Reviews. For the Medline search, we combined keywords for office BP, hypertension, and ambulatory BP with keywords for outpatient setting and primary care, using the following syntax: (((“clinic blood pressure” OR “office blood pressure” OR “casual blood pressure”))) AND (“hypertension” AND ((((“24-h ambulatory blood pressure”) OR “24 h ambulatory blood pressure”) OR “24 hour ambulatory blood pressure”) OR “blood pressure monitoring, ambulatory”[Mesh]) AND ((((((“outpatient setting”) OR “primary care”) OR “family care”) OR “family physician”) OR “family practice”) OR “general practice”)). We chose studies published in English and reviewed the titles and abstracts of identified articles.
With the aim of identifying additional candidate studies, we reviewed the reference lists of eligible primary studies, narrative reviews, and systematic reviews. The studies were generally of good quality and used appropriate statistical methods. Only primary studies qualified for meta-analysis.
Inclusion and exclusion criteria
Acceptable studies had to be conducted in a primary care setting with patients being treated for hypertension, and had to provide data comparing office BP measurement with ABPM. We excluded studies in which participants were treated in the hospital, were untreated, or had not been diagnosed with hypertension.
The quality of the studies included in the meta-analysis was judged by 2 independent observers according to the following criteria: the clear classification and initial comparison of both measurements; explicit and defined diagnostic criteria; compliance with the inclusion/exclusion criteria; and clear and precise definition of outcome variables.
Data extraction
We extracted the following data from each included study: study population, number of patients included, age, gender distribution, number of measurements (ambulatory and office BP), equipment validation, mean office and ambulatory BP, and the period of ambulatory BP measurement. We included adult patients of all ages, and we compared the mean office BP with those obtained by ABPM in hypertensive patients.
STATISTICAL ANALYSIS
For each study, we summarized the diagnostic accuracy of office BP with respect to ABPM in terms of sensitivity, specificity, and positive and negative likelihood ratios (LRs), with the 95% confidence interval (CI), if available. If these rates were not directly reported in the original papers, we used the published data to calculate them.
We used the R v2.15.1 software with the “mada” package for meta-analysis.14 Although a bivariate approach is preferred for the meta-analysis of diagnostic accuracy, it cannot be recommended if the number of primary studies to pool is too small,14 as happened in our case. Therefore, we used a univariate approach and pooled summary statistics for positive LR, negative LR, and the diagnostic odds ratio (DOR) with their 95% confidence intervals. We used the DerSimonian-Laird method to perform a random-effect meta-analysis. To explore heterogeneity between the studies, we used the Cochran’s Q heterogeneity test, I2 index, and Galbraith and L’Abbé plots.
RESULTS
Our search identified 237 studies, only 12 of which met the inclusion criteria and contained data to calculate the differences between the means of office and ambulatory BP measurements (TABLES 1 AND 2).15-26 Of these 12 studies, 5 were suitable for calculating sensitivity, specificity, and LR (TABLE 3),16,18,22,24,26 and 4 contained sufficient extractable data for meta-analysis. The study by Little et al18 was not included in the meta-analysis, as the number of true-positive, true-negative, false-positive, and false-negative results could not be deduced from published data.
The studies differed in sample size (40-31,530), patient ages (mean, 55-72.8 years), sex (percentage of men, 31%-52.9%), and number of measurements for office BP (1-9) and ABPM (32-96) (TABLE 1),15-26 as well as in daytime and nighttime periods for ABPM and BP thresholds, and in differences between the mean office and ambulatory BPs (TABLE 2).15-26
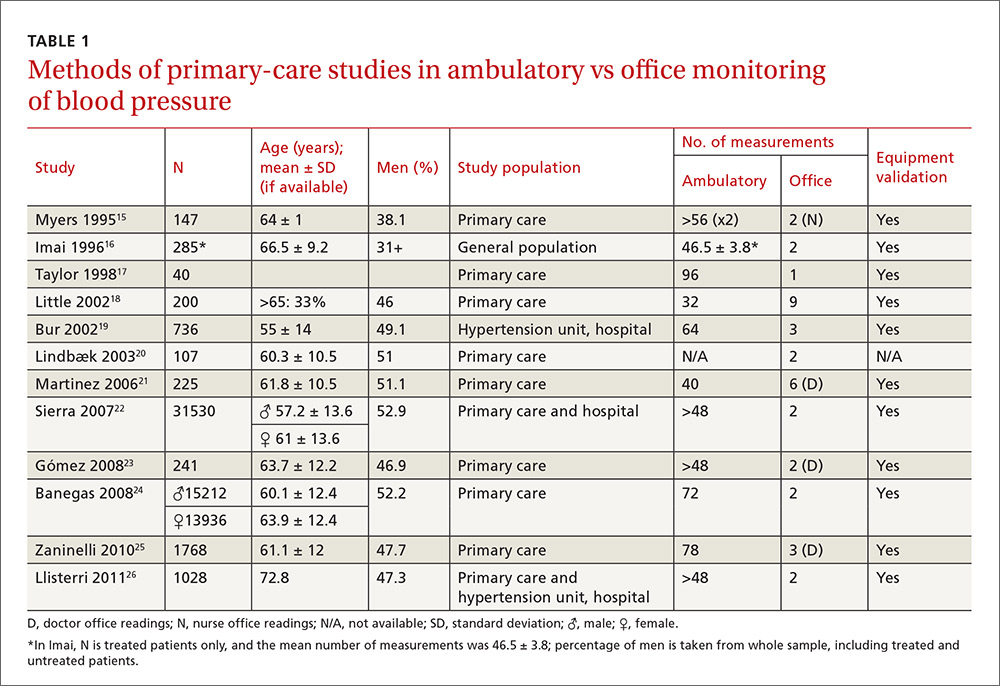
In general, the mean office BP measurements were higher than those obtained with ABPM in any period—from 5/0 mm Hg to 27.4/10.1 mm Hg in the day, and from 7.9/6.3 mm Hg to 31.2/13.7 mm Hg over 24 hours (TABLE 2).15-26
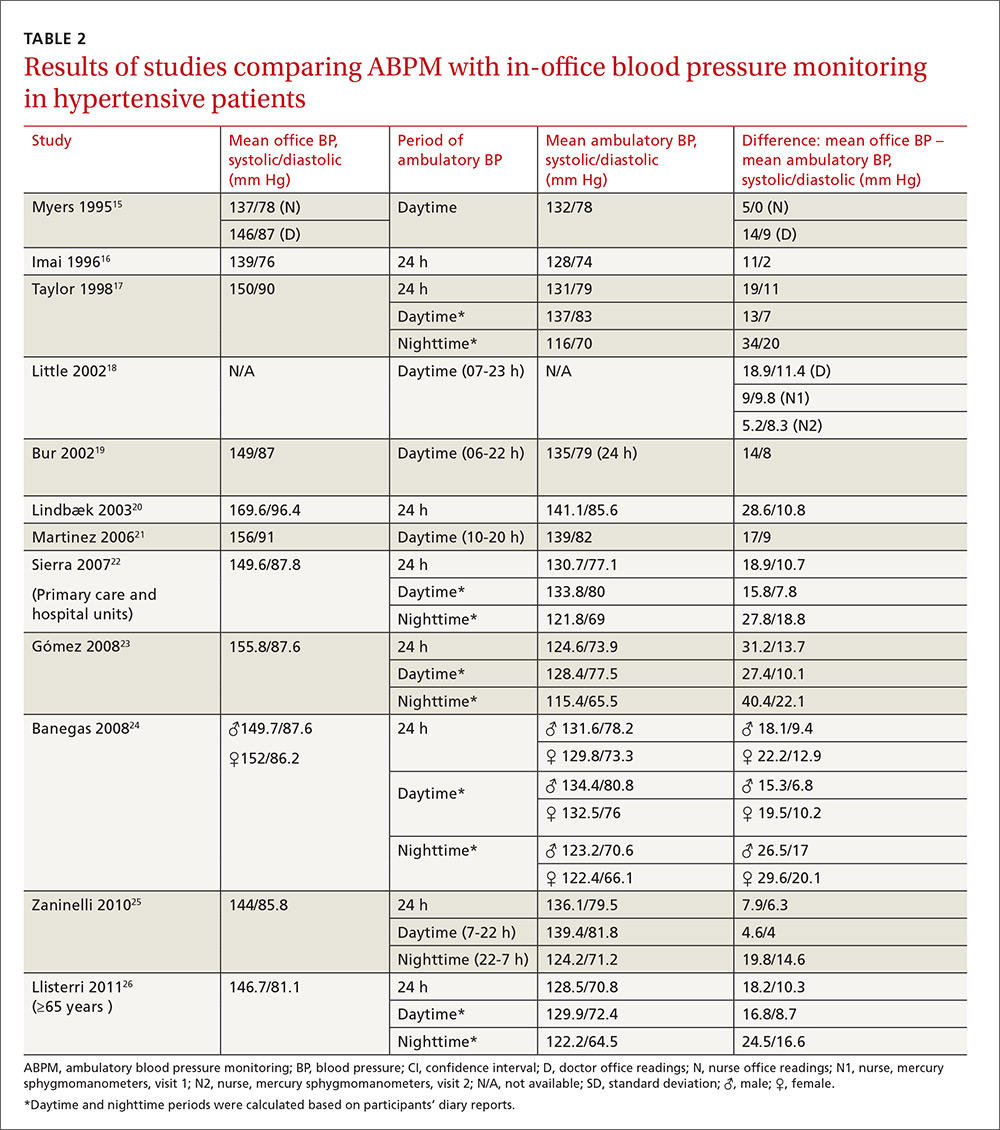
Compared with ABPM in diagnosing uncontrolled BP, office BP measurement had a sensitivity of 55.7% to 91.2% and a specificity of 25.8% to 61.8% (depending on whether the measure was carried out by the doctor or nurse18); positive LR ranged from 1.2 to 1.4, and negative LR from 0.3 to 0.72 (TABLE 3).16,18,22,24,26
For meta-analysis, we pooled studies with the same thresholds (140/90 mm Hg for office BP; 130/80 mm Hg for ABPM), with diagnostic accuracy of office BP expressed as pooled positive and negative LR, and as pooled DOR. The meta-analysis revealed that the pooled positive LR was 1.35 (95% CI, 1.32-1.38), and the pooled negative LR was 0.44 (95% CI, 0.37-0.53). The pooled DOR was 3.47 (95% CI, 3.02-3.98). Sensitivity was 81.9% (95% CI, 74.8%-87%) and specificity was 41.1% (95% CI, 35.1%-48.4%).
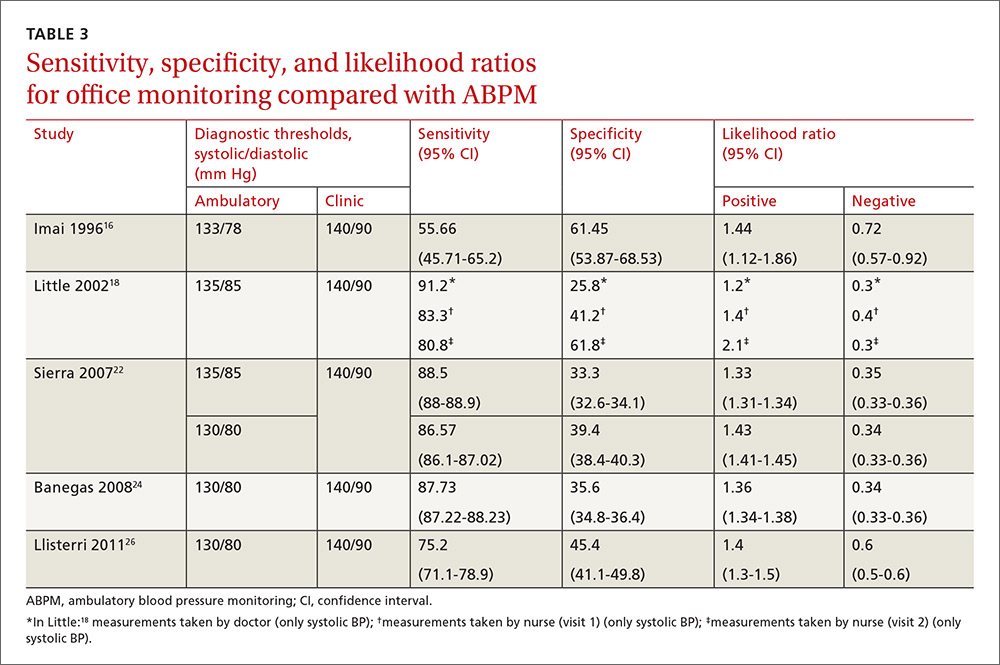
One study16 had a slightly different ambulatory diagnostic threshold (133/78 mm Hg), so we excluded it from a second meta-analysis. Results after the exclusion did not change significantly: positive LR was 1.39 (95% CI, 1.34-1.45); negative LR was 0.38 (95% CI, 0.33-0.44); and DOR was 3.77 (95% CI, 3.31-4.43).
In conclusion, the use of office-based BP readings in the outpatient clinic does not correlate well with ABPM. Therefore, caution must be used when making management decisions based solely on in-office readings of BP.
DISCUSSION
The European Society of Hypertension still regards office BP measurement as the gold standard in screening for, diagnosing, and managing hypertension. As previously mentioned, though, office measurements are usually handled by medical staff and can be compromised by the white-coat effect and a small number of measurements. The USPSTF now considers ABPM the reference standard in primary care to diagnose hypertension in adults, to corroborate or contradict office-based determinations of elevated BP (whether based on single or repeated-interval measurements), and to avoid overtreatment of individuals displaying elevated office BP yet proven normotensive by ABPM.4,7 The recommendation of the American Academy of Family Physicians is similar to that of the USPSTF.7 Therefore, evidence supports ABPM as the reference standard for confirming elevated office BP screening results to avoid misdiagnosis and overtreatment of individuals with isolated clinic hypertension.7
How office measurements stack up against ABPM
Checking the validity of decisions in clinical practice is extremely important for patient management. One of the tools used for decision-making is an estimate of the LR. We used the LR to assess the value of office BP measurement in determining controlled or uncontrolled BP. A high LR (eg, >10) indicates that the office BP can be used to rule in the disease (uncontrolled BP) with a high probability, while a low LR (eg, <0.1) could rule it out. An LR of around one indicates that the office BP measurement cannot rule the diagnosis of uncontrolled BP in or out.27 In our meta-analysis, the positive LR is 1.35 and negative LR is 0.44. Therefore, in treated hypertensive patients, an indication of uncontrolled BP as measured in the clinic does not confirm a diagnosis of uncontrolled BP (as judged by the reference standard of ABPM). On the other hand, the negative LR means that normal office BP does not rule out uncontrolled BP, which may be detected with ABPM. Consequently, the measurement of BP in the office does not change the degree of (un)certainty of adequate control of BP. This knowledge is important, to avoid overtreatment of white coat hypertension and undertreatment of masked cases.
As previously mentioned, we reported similar results in a study designed to determine the validity of office BP measurement in a primary care setting compared with ABPM.13 In that paper, the level of agreement between both methods was poor, indicating that clinic measurements could not be recommended as a single method of BP control in hypertensive patients.
The use of ABPM in diagnosing hypertension is likely to increase as a consequence of some guideline updates.2 Our study emphasizes the importance of their use in the control of hypertensive patients.
Another published meta-analysis1 investigated the validity of office BP for the diagnosis of hypertension in untreated patients, with diagnostic thresholds for arterial hypertension set at 140/90 mm Hg for office measurement, and 135/85 mm Hg for ABPM. In that paper, the sensitivity of office BP was 74.6% (95% CI, 60.7-84.8) and the specificity was 74.6% (95% CI, 47.9-90.4).
In our present study carried out with hypertensive patients receiving treatment, we obtained a slightly higher sensitivity value of 81.9% (within the CI of this meta-analysis) and a lower specificity of 41.1%. Therefore, the discordance between office BP and ABPM seems to be similar for the diagnosis of hypertension and the classification of hypertension as being well or poorly controlled. This confirms the low validity of the office BP, both for diagnosis and monitoring of hypertensive patients.
Strengths of our study. The study focused on (treated) hypertensive patients in a primary care setting, where hypertension is most often managed. It confirms that ABPM is indispensable to a good clinical practice.
Limitations of our study are those inherent to meta-analyses. The main weakness of our study is the paucity of data available regarding the utility of ABPM for monitoring BP control with treatment in a primary care setting. Other limitations are the variability in BP thresholds used, the number of measurements performed, and the ambulatory BP devices used. These differences could contribute to the observed heterogeneity.
Application of our results must take into account that we included only those studies performed in a primary care setting with treated hypertensive patients.
Moreover, this study was not designed to evaluate the consequences of over- and undertreatment of blood pressure, nor to address the accuracy of automated blood pressure machines or newer health and fitness devices.
Implications for practice, policy, or future research. Alternative monitoring methods are home BP self-measurement and automated 30-minute clinic BP measurement.28 However, ABPM provides us with unique information about the BP pattern (dipping or non-dipping), BP variability, and mean nighttime BP. This paper establishes that the measurement of BP in the office is not an accurate method to monitor BP control. ABPM should be incorporated in usual clinical practice in primary care. Although the consequences of ambulatory monitoring are not the focus of this study, we acknowledge that the decision to incorporate ABPM in clinical practice depends on the availability of ambulatory devices, proper training of health care workers, and a cost-effectiveness analysis of its use.
CORRESPONDENCE
Sergio Reino-González, MD, PhD, Adormideras Primary Health Center, Poligono de Adormideras s/n. 15002 A Coruña, Spain; [email protected].
1. Hodgkinson J, Mant J, Martin U, et al. Relative effectiveness of clinic and home blood pressure monitoring compared with ambulatory blood pressure monitoring in diagnosis of hypertension: systematic review. BMJ. 2011;342:d3621.
2. National Institute for Health and Clinical Excellence. Hypertension in adults: diagnosis and management. Available at: http://www.nice.org.uk/guidance/CG127. Accessed November 15, 2016.
3. Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension. 2003;42:1206-1252.
4. Hermida RC, Smolensky MH, Ayala DE, et al. Ambulatory Blood Pressure Monitoring (ABPM) as the reference standard for diagnosis of hypertension and assessment of vascular risk in adults. Chronobiol Int. 2015;32:1329-1342.
5. Siu AL; U.S. Preventive Services Task Force. Screening for high blood pressure in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2015;163:778-786.
6. Piper MA, Evans CV
7. American Academy of Family Physicians. Hypertension. Available at: www.aafp.org/patient-care/clinical-recommendations/all/hypertension.html. Accessed February 10, 2016.
8. Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC Practice Guidelines for the Management of Arterial Hypertension. Blood Press. 2013;23:3-16.
9. Marin R, de la Sierra A, Armario P, et al. 2005 Spanish guidelines in diagnosis and treatment of arterial hypertension. Medicina Clínica. 2005;125:24-34.
10. Fagard RH, Celis H, Thijs L, et al. Daytime and nighttime blood pressure as predictors of death and cause-specific cardiovascular events in hypertension. Hypertension. 2008;51:55-61.
11. Sega R, Trocino G, Lanzarotti A, et al. Alterations of cardiac structure in patients with isolated office, ambulatory, or home hypertension: Data from the general population (Pressione Arteriose Monitorate E Loro Associazioni [PAMELA] Study). Circulation. 2001;104:1385-1392.
12. Verberk WJ, Kessels AG, de Leeuw PW. Prevalence, causes, and consequences of masked hypertension: a meta-analysis. Am J Hypertens. 2008;21:969-975.
13. Reino-González S, Pita-Fernández S, Cibiriain-Sola M, et al. Validity of clinic blood pressure compared to ambulatory monitoring in hypertensive patients in a primary care setting. Blood Press. 2015;24:111-118.
14. Doebler P, Holling H. Meta-analysis of diagnostic accuracy with mada. Available at: https://cran.r-project.org/web/packages/mada/vignettes/mada.pdf. Accessed October 5, 2015.
15. Myers MG, Oh PI, Reeves RA, et al. Prevalence of white coat effect in treated hypertensive patients in the community. Am J Hypertens. 1995;8:591-597.
16. Imai Y, Tsuji I, Nagai K, et al. Ambulatory blood pressure monitoring in evaluating the prevalence of hypertension in adults in Ohasama, a rural Japanese community. Hypertens Res. 1996;19:207-212.
17. Taylor RS, Stockman J, Kernick D, et al. Ambulatory blood pressure monitoring for hypertension in general practice. J R Soc Med. 1998;91:301-304.
18. Little P, Barnett J, Barnsley L, et al. Comparison of agreement between different measures of blood pressure in primary care and daytime ambulatory blood pressure. BMJ. 2002;325:254.
19. Bur A, Herkner H, Vlcek M, et al. Classification of blood pressure levels by ambulatory blood pressure in hypertension. Hypertension. 2002;40:817-822.
20. Lindbaek M, Sandvik E, Liodden K, et al. Predictors for the white coat effect in general practice patients with suspected and treated hypertension. Br J Gen Pract. 2003;53:790-793.
21. Martínez MA, Sancho T, García P, et al. Home blood pressure in poorly controlled hypertension: relationship with ambulatory blood pressure and organ damage. Blood Press Monit. 2006;11:207-213.
22. Sierra BC, de la Sierra IA, Sobrino J, et al. Monitorización ambulatoria de la presión arterial (MAPA): características clínicas de 31.530 pacientes. Medicina Clínica. 2007;129:1-5.
23. Gómez MA, García L, Sánchez Á, et al. Agreement and disagreement between different methods of measuring blood pressure. Hipertensión (Madr). 2008;25:231-239.
24. Banegas JR, Segura J, De la Sierra A, et al. Gender differences in office and ambulatory control of hypertension. Am J Med. 2008;121:1078-1084.
25. Zaninelli A, Parati G, Cricelli C, et al. Office and 24-h ambulatory blood pressure control by treatment in general practice: the ‘Monitoraggio della pressione ARteriosa nella medicina TErritoriale’ study. J Hypertens. 2010;28:910-917.
26. Llisterri JL, Morillas P, Pallarés V, et al. Differences in the degree of control of arterial hypertension according to the measurement procedure of blood pressure in patients ≥ 65 years. FAPRES study. Rev Clin Esp. 2011;211:76-84.
27. Straus SE, Richardson WS, Glasziou P, et al. Evidence-Based Medicine: How to practice and teach it. 4th ed. Edinburgh, Scotland: Churchill Livingstone; 2010.
28. Van der Wel MC, Buunk IE, van Weel C, et al. A novel approach to office blood pressure measurement: 30-minute office blood pressure vs daytime ambulatory blood pressure. Ann Fam Med. 2011;9:128-135.
ABSTRACT
Purpose We performed a literature review and meta-analysis to ascertain the validity of office blood pressure (BP) measurement in a primary care setting, using ambulatory blood pressure measurement (ABPM) as a benchmark in the monitoring of hypertensive patients receiving treatment.
Methods We conducted a literature search for studies published up to December 2013 that included hypertensive patients receiving treatment in a primary care setting. We compared the mean office BP with readings obtained by ABPM. We summarized the diagnostic accuracy of office BP with respect to ABPM in terms of sensitivity, specificity, and positive and negative likelihood ratios (LR), with a 95% confidence interval (CI).
Results Only 12 studies met the inclusion criteria and contained data to calculate the differences between the means of office and ambulatory BP measurements. Five were suitable for calculating sensitivity, specificity, and likelihood ratios, and 4 contained sufficient extractable data for meta-analysis. Compared with ABPM (thresholds of 140/90 mm Hg for office BP; 130/80 mmHg for ABPM) in diagnosing uncontrolled BP, office BP measurement had a sensitivity of 81.9% (95% CI, 74.8%-87%) and specificity of 41.1% (95% CI, 35.1%-48.4%). Positive LR was 1.35 (95% CI, 1.32-1.38), and the negative LR was 0.44 (95% CI, 0.37-0.53).
Conclusion Likelihood ratios show that isolated BP measurement in the office does not confirm or rule out the presence of poor BP control. Likelihood of underestimating or overestimating BP control is high when relying on in-office BP measurement alone.
A growing body of evidence supports more frequent use of ambulatory blood pressure monitoring (ABPM) to confirm a diagnosis of hypertension1 and to monitor blood pressure (BP) response to treatment.2 The Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure has long accepted ABPM for diagnosis of hypertension,3 and many clinicians consider ABPM the reference standard for diagnosing true hypertension and for accurately assessing associated cardiovascular risk in adults, regardless of office BP readings.4 The US Preventive Services Task Force (USPSTF) recommends obtaining BP measurements outside the clinical setting to confirm a diagnosis of hypertension before starting treatment.5 The USPSTF also asserts that elevated 24-hour ambulatory systolic BP is consistently and significantly associated with stroke and other cardiovascular events independent of office BP readings and has greater predictive value than office monitoring.5 The USPSTF concludes that ABPM, because of its large evidence base, is the best confirmatory test for hypertension.6 The recommendation of the American Academy of Family Physicians is similar to that of the USPSTF.7
The challenge. Despite the considerable support for ABPM, this method of BP measurement is still not sufficiently integrated into primary care. And some guidelines, such as those of
But ABPM’s advantages are numerous. Ambulatory monitors, which can record BP for 24 hours, are typically programmed to take readings every 15 to 30 minutes, providing estimates of mean daytime and nighttime BP and revealing an individual’s circadian pattern of BP.8-10 Ambulatory BP values usually considered the uppermost limit of normal are 135/85 mm Hg (day), 120/70 mm Hg (night), and 130/80 mm Hg (24 hour).8
Office BP monitoring, usually performed manually by medical staff, has 2 main drawbacks: the well-known white-coat effect experienced by many patients, and the relatively small number of possible measurements. A more reliable in-office BP estimation of BP would require repeated measurements at each of several visits.
By comparing ABPM and office measurements, 4 clinical findings are possible: isolated clinic or office (white-coat) hypertension (ICH); isolated ambulatory (masked) hypertension (IAH); consistent normotension; or sustained hypertension. With ICH, BP is high in the office and normal with ABPM. With IAH, BP is normal in the office and high with ABPM. With consistent normotension and sustained hypertension, BP readings with both types of measurement agree.8,9
In patients being treated for hypertension, ICH leads to an overestimation of uncontrolled BP and may result in overtreatment. The cardiovascular risk, although controversial, is usually lower than in patients diagnosed with sustained hypertension.11 IAH leads to an underestimation of uncontrolled BP and may result in undertreatment; its associated cardiovascular risk is similar to that of sustained hypertension.12
Our research objective. We recently published a study conducted with 137 hypertensive patients in a primary care center.13 Our conclusion was that in-office measurement of BP had insufficient clinical validity to be recommended as a sole method of monitoring BP control. In accurately classifying BP as controlled or uncontrolled, clinic measurement agreed with 24h-ABPM in just 64.2% of cases.13
In our present study, we performed a literature review and meta-analysis to ascertain the validity of office BP measurement in a primary care setting, using ABPM as a benchmark in the monitoring of hypertensive patients receiving treatment.
METHODS
Most published studies comparing conventional office BP measurement with ABPM have been conducted with patients not taking antihypertensive medication. We excluded these studies and conducted a literature search for studies published up to December 2013 that included hypertensive patients receiving treatment in a primary care setting.
We searched Medline (from 1950 onward) and the Cochrane Database of Systematic Reviews. For the Medline search, we combined keywords for office BP, hypertension, and ambulatory BP with keywords for outpatient setting and primary care, using the following syntax: (((“clinic blood pressure” OR “office blood pressure” OR “casual blood pressure”))) AND (“hypertension” AND ((((“24-h ambulatory blood pressure”) OR “24 h ambulatory blood pressure”) OR “24 hour ambulatory blood pressure”) OR “blood pressure monitoring, ambulatory”[Mesh]) AND ((((((“outpatient setting”) OR “primary care”) OR “family care”) OR “family physician”) OR “family practice”) OR “general practice”)). We chose studies published in English and reviewed the titles and abstracts of identified articles.
With the aim of identifying additional candidate studies, we reviewed the reference lists of eligible primary studies, narrative reviews, and systematic reviews. The studies were generally of good quality and used appropriate statistical methods. Only primary studies qualified for meta-analysis.
Inclusion and exclusion criteria
Acceptable studies had to be conducted in a primary care setting with patients being treated for hypertension, and had to provide data comparing office BP measurement with ABPM. We excluded studies in which participants were treated in the hospital, were untreated, or had not been diagnosed with hypertension.
The quality of the studies included in the meta-analysis was judged by 2 independent observers according to the following criteria: the clear classification and initial comparison of both measurements; explicit and defined diagnostic criteria; compliance with the inclusion/exclusion criteria; and clear and precise definition of outcome variables.
Data extraction
We extracted the following data from each included study: study population, number of patients included, age, gender distribution, number of measurements (ambulatory and office BP), equipment validation, mean office and ambulatory BP, and the period of ambulatory BP measurement. We included adult patients of all ages, and we compared the mean office BP with those obtained by ABPM in hypertensive patients.
STATISTICAL ANALYSIS
For each study, we summarized the diagnostic accuracy of office BP with respect to ABPM in terms of sensitivity, specificity, and positive and negative likelihood ratios (LRs), with the 95% confidence interval (CI), if available. If these rates were not directly reported in the original papers, we used the published data to calculate them.
We used the R v2.15.1 software with the “mada” package for meta-analysis.14 Although a bivariate approach is preferred for the meta-analysis of diagnostic accuracy, it cannot be recommended if the number of primary studies to pool is too small,14 as happened in our case. Therefore, we used a univariate approach and pooled summary statistics for positive LR, negative LR, and the diagnostic odds ratio (DOR) with their 95% confidence intervals. We used the DerSimonian-Laird method to perform a random-effect meta-analysis. To explore heterogeneity between the studies, we used the Cochran’s Q heterogeneity test, I2 index, and Galbraith and L’Abbé plots.
RESULTS
Our search identified 237 studies, only 12 of which met the inclusion criteria and contained data to calculate the differences between the means of office and ambulatory BP measurements (TABLES 1 AND 2).15-26 Of these 12 studies, 5 were suitable for calculating sensitivity, specificity, and LR (TABLE 3),16,18,22,24,26 and 4 contained sufficient extractable data for meta-analysis. The study by Little et al18 was not included in the meta-analysis, as the number of true-positive, true-negative, false-positive, and false-negative results could not be deduced from published data.
The studies differed in sample size (40-31,530), patient ages (mean, 55-72.8 years), sex (percentage of men, 31%-52.9%), and number of measurements for office BP (1-9) and ABPM (32-96) (TABLE 1),15-26 as well as in daytime and nighttime periods for ABPM and BP thresholds, and in differences between the mean office and ambulatory BPs (TABLE 2).15-26
In general, the mean office BP measurements were higher than those obtained with ABPM in any period—from 5/0 mm Hg to 27.4/10.1 mm Hg in the day, and from 7.9/6.3 mm Hg to 31.2/13.7 mm Hg over 24 hours (TABLE 2).15-26
Compared with ABPM in diagnosing uncontrolled BP, office BP measurement had a sensitivity of 55.7% to 91.2% and a specificity of 25.8% to 61.8% (depending on whether the measure was carried out by the doctor or nurse18); positive LR ranged from 1.2 to 1.4, and negative LR from 0.3 to 0.72 (TABLE 3).16,18,22,24,26
For meta-analysis, we pooled studies with the same thresholds (140/90 mm Hg for office BP; 130/80 mm Hg for ABPM), with diagnostic accuracy of office BP expressed as pooled positive and negative LR, and as pooled DOR. The meta-analysis revealed that the pooled positive LR was 1.35 (95% CI, 1.32-1.38), and the pooled negative LR was 0.44 (95% CI, 0.37-0.53). The pooled DOR was 3.47 (95% CI, 3.02-3.98). Sensitivity was 81.9% (95% CI, 74.8%-87%) and specificity was 41.1% (95% CI, 35.1%-48.4%).
One study16 had a slightly different ambulatory diagnostic threshold (133/78 mm Hg), so we excluded it from a second meta-analysis. Results after the exclusion did not change significantly: positive LR was 1.39 (95% CI, 1.34-1.45); negative LR was 0.38 (95% CI, 0.33-0.44); and DOR was 3.77 (95% CI, 3.31-4.43).
In conclusion, the use of office-based BP readings in the outpatient clinic does not correlate well with ABPM. Therefore, caution must be used when making management decisions based solely on in-office readings of BP.
DISCUSSION
The European Society of Hypertension still regards office BP measurement as the gold standard in screening for, diagnosing, and managing hypertension. As previously mentioned, though, office measurements are usually handled by medical staff and can be compromised by the white-coat effect and a small number of measurements. The USPSTF now considers ABPM the reference standard in primary care to diagnose hypertension in adults, to corroborate or contradict office-based determinations of elevated BP (whether based on single or repeated-interval measurements), and to avoid overtreatment of individuals displaying elevated office BP yet proven normotensive by ABPM.4,7 The recommendation of the American Academy of Family Physicians is similar to that of the USPSTF.7 Therefore, evidence supports ABPM as the reference standard for confirming elevated office BP screening results to avoid misdiagnosis and overtreatment of individuals with isolated clinic hypertension.7
How office measurements stack up against ABPM
Checking the validity of decisions in clinical practice is extremely important for patient management. One of the tools used for decision-making is an estimate of the LR. We used the LR to assess the value of office BP measurement in determining controlled or uncontrolled BP. A high LR (eg, >10) indicates that the office BP can be used to rule in the disease (uncontrolled BP) with a high probability, while a low LR (eg, <0.1) could rule it out. An LR of around one indicates that the office BP measurement cannot rule the diagnosis of uncontrolled BP in or out.27 In our meta-analysis, the positive LR is 1.35 and negative LR is 0.44. Therefore, in treated hypertensive patients, an indication of uncontrolled BP as measured in the clinic does not confirm a diagnosis of uncontrolled BP (as judged by the reference standard of ABPM). On the other hand, the negative LR means that normal office BP does not rule out uncontrolled BP, which may be detected with ABPM. Consequently, the measurement of BP in the office does not change the degree of (un)certainty of adequate control of BP. This knowledge is important, to avoid overtreatment of white coat hypertension and undertreatment of masked cases.
As previously mentioned, we reported similar results in a study designed to determine the validity of office BP measurement in a primary care setting compared with ABPM.13 In that paper, the level of agreement between both methods was poor, indicating that clinic measurements could not be recommended as a single method of BP control in hypertensive patients.
The use of ABPM in diagnosing hypertension is likely to increase as a consequence of some guideline updates.2 Our study emphasizes the importance of their use in the control of hypertensive patients.
Another published meta-analysis1 investigated the validity of office BP for the diagnosis of hypertension in untreated patients, with diagnostic thresholds for arterial hypertension set at 140/90 mm Hg for office measurement, and 135/85 mm Hg for ABPM. In that paper, the sensitivity of office BP was 74.6% (95% CI, 60.7-84.8) and the specificity was 74.6% (95% CI, 47.9-90.4).
In our present study carried out with hypertensive patients receiving treatment, we obtained a slightly higher sensitivity value of 81.9% (within the CI of this meta-analysis) and a lower specificity of 41.1%. Therefore, the discordance between office BP and ABPM seems to be similar for the diagnosis of hypertension and the classification of hypertension as being well or poorly controlled. This confirms the low validity of the office BP, both for diagnosis and monitoring of hypertensive patients.
Strengths of our study. The study focused on (treated) hypertensive patients in a primary care setting, where hypertension is most often managed. It confirms that ABPM is indispensable to a good clinical practice.
Limitations of our study are those inherent to meta-analyses. The main weakness of our study is the paucity of data available regarding the utility of ABPM for monitoring BP control with treatment in a primary care setting. Other limitations are the variability in BP thresholds used, the number of measurements performed, and the ambulatory BP devices used. These differences could contribute to the observed heterogeneity.
Application of our results must take into account that we included only those studies performed in a primary care setting with treated hypertensive patients.
Moreover, this study was not designed to evaluate the consequences of over- and undertreatment of blood pressure, nor to address the accuracy of automated blood pressure machines or newer health and fitness devices.
Implications for practice, policy, or future research. Alternative monitoring methods are home BP self-measurement and automated 30-minute clinic BP measurement.28 However, ABPM provides us with unique information about the BP pattern (dipping or non-dipping), BP variability, and mean nighttime BP. This paper establishes that the measurement of BP in the office is not an accurate method to monitor BP control. ABPM should be incorporated in usual clinical practice in primary care. Although the consequences of ambulatory monitoring are not the focus of this study, we acknowledge that the decision to incorporate ABPM in clinical practice depends on the availability of ambulatory devices, proper training of health care workers, and a cost-effectiveness analysis of its use.
CORRESPONDENCE
Sergio Reino-González, MD, PhD, Adormideras Primary Health Center, Poligono de Adormideras s/n. 15002 A Coruña, Spain; [email protected].
ABSTRACT
Purpose We performed a literature review and meta-analysis to ascertain the validity of office blood pressure (BP) measurement in a primary care setting, using ambulatory blood pressure measurement (ABPM) as a benchmark in the monitoring of hypertensive patients receiving treatment.
Methods We conducted a literature search for studies published up to December 2013 that included hypertensive patients receiving treatment in a primary care setting. We compared the mean office BP with readings obtained by ABPM. We summarized the diagnostic accuracy of office BP with respect to ABPM in terms of sensitivity, specificity, and positive and negative likelihood ratios (LR), with a 95% confidence interval (CI).
Results Only 12 studies met the inclusion criteria and contained data to calculate the differences between the means of office and ambulatory BP measurements. Five were suitable for calculating sensitivity, specificity, and likelihood ratios, and 4 contained sufficient extractable data for meta-analysis. Compared with ABPM (thresholds of 140/90 mm Hg for office BP; 130/80 mmHg for ABPM) in diagnosing uncontrolled BP, office BP measurement had a sensitivity of 81.9% (95% CI, 74.8%-87%) and specificity of 41.1% (95% CI, 35.1%-48.4%). Positive LR was 1.35 (95% CI, 1.32-1.38), and the negative LR was 0.44 (95% CI, 0.37-0.53).
Conclusion Likelihood ratios show that isolated BP measurement in the office does not confirm or rule out the presence of poor BP control. Likelihood of underestimating or overestimating BP control is high when relying on in-office BP measurement alone.
A growing body of evidence supports more frequent use of ambulatory blood pressure monitoring (ABPM) to confirm a diagnosis of hypertension1 and to monitor blood pressure (BP) response to treatment.2 The Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure has long accepted ABPM for diagnosis of hypertension,3 and many clinicians consider ABPM the reference standard for diagnosing true hypertension and for accurately assessing associated cardiovascular risk in adults, regardless of office BP readings.4 The US Preventive Services Task Force (USPSTF) recommends obtaining BP measurements outside the clinical setting to confirm a diagnosis of hypertension before starting treatment.5 The USPSTF also asserts that elevated 24-hour ambulatory systolic BP is consistently and significantly associated with stroke and other cardiovascular events independent of office BP readings and has greater predictive value than office monitoring.5 The USPSTF concludes that ABPM, because of its large evidence base, is the best confirmatory test for hypertension.6 The recommendation of the American Academy of Family Physicians is similar to that of the USPSTF.7
The challenge. Despite the considerable support for ABPM, this method of BP measurement is still not sufficiently integrated into primary care. And some guidelines, such as those of
But ABPM’s advantages are numerous. Ambulatory monitors, which can record BP for 24 hours, are typically programmed to take readings every 15 to 30 minutes, providing estimates of mean daytime and nighttime BP and revealing an individual’s circadian pattern of BP.8-10 Ambulatory BP values usually considered the uppermost limit of normal are 135/85 mm Hg (day), 120/70 mm Hg (night), and 130/80 mm Hg (24 hour).8
Office BP monitoring, usually performed manually by medical staff, has 2 main drawbacks: the well-known white-coat effect experienced by many patients, and the relatively small number of possible measurements. A more reliable in-office BP estimation of BP would require repeated measurements at each of several visits.
By comparing ABPM and office measurements, 4 clinical findings are possible: isolated clinic or office (white-coat) hypertension (ICH); isolated ambulatory (masked) hypertension (IAH); consistent normotension; or sustained hypertension. With ICH, BP is high in the office and normal with ABPM. With IAH, BP is normal in the office and high with ABPM. With consistent normotension and sustained hypertension, BP readings with both types of measurement agree.8,9
In patients being treated for hypertension, ICH leads to an overestimation of uncontrolled BP and may result in overtreatment. The cardiovascular risk, although controversial, is usually lower than in patients diagnosed with sustained hypertension.11 IAH leads to an underestimation of uncontrolled BP and may result in undertreatment; its associated cardiovascular risk is similar to that of sustained hypertension.12
Our research objective. We recently published a study conducted with 137 hypertensive patients in a primary care center.13 Our conclusion was that in-office measurement of BP had insufficient clinical validity to be recommended as a sole method of monitoring BP control. In accurately classifying BP as controlled or uncontrolled, clinic measurement agreed with 24h-ABPM in just 64.2% of cases.13
In our present study, we performed a literature review and meta-analysis to ascertain the validity of office BP measurement in a primary care setting, using ABPM as a benchmark in the monitoring of hypertensive patients receiving treatment.
METHODS
Most published studies comparing conventional office BP measurement with ABPM have been conducted with patients not taking antihypertensive medication. We excluded these studies and conducted a literature search for studies published up to December 2013 that included hypertensive patients receiving treatment in a primary care setting.
We searched Medline (from 1950 onward) and the Cochrane Database of Systematic Reviews. For the Medline search, we combined keywords for office BP, hypertension, and ambulatory BP with keywords for outpatient setting and primary care, using the following syntax: (((“clinic blood pressure” OR “office blood pressure” OR “casual blood pressure”))) AND (“hypertension” AND ((((“24-h ambulatory blood pressure”) OR “24 h ambulatory blood pressure”) OR “24 hour ambulatory blood pressure”) OR “blood pressure monitoring, ambulatory”[Mesh]) AND ((((((“outpatient setting”) OR “primary care”) OR “family care”) OR “family physician”) OR “family practice”) OR “general practice”)). We chose studies published in English and reviewed the titles and abstracts of identified articles.
With the aim of identifying additional candidate studies, we reviewed the reference lists of eligible primary studies, narrative reviews, and systematic reviews. The studies were generally of good quality and used appropriate statistical methods. Only primary studies qualified for meta-analysis.
Inclusion and exclusion criteria
Acceptable studies had to be conducted in a primary care setting with patients being treated for hypertension, and had to provide data comparing office BP measurement with ABPM. We excluded studies in which participants were treated in the hospital, were untreated, or had not been diagnosed with hypertension.
The quality of the studies included in the meta-analysis was judged by 2 independent observers according to the following criteria: the clear classification and initial comparison of both measurements; explicit and defined diagnostic criteria; compliance with the inclusion/exclusion criteria; and clear and precise definition of outcome variables.
Data extraction
We extracted the following data from each included study: study population, number of patients included, age, gender distribution, number of measurements (ambulatory and office BP), equipment validation, mean office and ambulatory BP, and the period of ambulatory BP measurement. We included adult patients of all ages, and we compared the mean office BP with those obtained by ABPM in hypertensive patients.
STATISTICAL ANALYSIS
For each study, we summarized the diagnostic accuracy of office BP with respect to ABPM in terms of sensitivity, specificity, and positive and negative likelihood ratios (LRs), with the 95% confidence interval (CI), if available. If these rates were not directly reported in the original papers, we used the published data to calculate them.
We used the R v2.15.1 software with the “mada” package for meta-analysis.14 Although a bivariate approach is preferred for the meta-analysis of diagnostic accuracy, it cannot be recommended if the number of primary studies to pool is too small,14 as happened in our case. Therefore, we used a univariate approach and pooled summary statistics for positive LR, negative LR, and the diagnostic odds ratio (DOR) with their 95% confidence intervals. We used the DerSimonian-Laird method to perform a random-effect meta-analysis. To explore heterogeneity between the studies, we used the Cochran’s Q heterogeneity test, I2 index, and Galbraith and L’Abbé plots.
RESULTS
Our search identified 237 studies, only 12 of which met the inclusion criteria and contained data to calculate the differences between the means of office and ambulatory BP measurements (TABLES 1 AND 2).15-26 Of these 12 studies, 5 were suitable for calculating sensitivity, specificity, and LR (TABLE 3),16,18,22,24,26 and 4 contained sufficient extractable data for meta-analysis. The study by Little et al18 was not included in the meta-analysis, as the number of true-positive, true-negative, false-positive, and false-negative results could not be deduced from published data.
The studies differed in sample size (40-31,530), patient ages (mean, 55-72.8 years), sex (percentage of men, 31%-52.9%), and number of measurements for office BP (1-9) and ABPM (32-96) (TABLE 1),15-26 as well as in daytime and nighttime periods for ABPM and BP thresholds, and in differences between the mean office and ambulatory BPs (TABLE 2).15-26
In general, the mean office BP measurements were higher than those obtained with ABPM in any period—from 5/0 mm Hg to 27.4/10.1 mm Hg in the day, and from 7.9/6.3 mm Hg to 31.2/13.7 mm Hg over 24 hours (TABLE 2).15-26
Compared with ABPM in diagnosing uncontrolled BP, office BP measurement had a sensitivity of 55.7% to 91.2% and a specificity of 25.8% to 61.8% (depending on whether the measure was carried out by the doctor or nurse18); positive LR ranged from 1.2 to 1.4, and negative LR from 0.3 to 0.72 (TABLE 3).16,18,22,24,26
For meta-analysis, we pooled studies with the same thresholds (140/90 mm Hg for office BP; 130/80 mm Hg for ABPM), with diagnostic accuracy of office BP expressed as pooled positive and negative LR, and as pooled DOR. The meta-analysis revealed that the pooled positive LR was 1.35 (95% CI, 1.32-1.38), and the pooled negative LR was 0.44 (95% CI, 0.37-0.53). The pooled DOR was 3.47 (95% CI, 3.02-3.98). Sensitivity was 81.9% (95% CI, 74.8%-87%) and specificity was 41.1% (95% CI, 35.1%-48.4%).
One study16 had a slightly different ambulatory diagnostic threshold (133/78 mm Hg), so we excluded it from a second meta-analysis. Results after the exclusion did not change significantly: positive LR was 1.39 (95% CI, 1.34-1.45); negative LR was 0.38 (95% CI, 0.33-0.44); and DOR was 3.77 (95% CI, 3.31-4.43).
In conclusion, the use of office-based BP readings in the outpatient clinic does not correlate well with ABPM. Therefore, caution must be used when making management decisions based solely on in-office readings of BP.
DISCUSSION
The European Society of Hypertension still regards office BP measurement as the gold standard in screening for, diagnosing, and managing hypertension. As previously mentioned, though, office measurements are usually handled by medical staff and can be compromised by the white-coat effect and a small number of measurements. The USPSTF now considers ABPM the reference standard in primary care to diagnose hypertension in adults, to corroborate or contradict office-based determinations of elevated BP (whether based on single or repeated-interval measurements), and to avoid overtreatment of individuals displaying elevated office BP yet proven normotensive by ABPM.4,7 The recommendation of the American Academy of Family Physicians is similar to that of the USPSTF.7 Therefore, evidence supports ABPM as the reference standard for confirming elevated office BP screening results to avoid misdiagnosis and overtreatment of individuals with isolated clinic hypertension.7
How office measurements stack up against ABPM
Checking the validity of decisions in clinical practice is extremely important for patient management. One of the tools used for decision-making is an estimate of the LR. We used the LR to assess the value of office BP measurement in determining controlled or uncontrolled BP. A high LR (eg, >10) indicates that the office BP can be used to rule in the disease (uncontrolled BP) with a high probability, while a low LR (eg, <0.1) could rule it out. An LR of around one indicates that the office BP measurement cannot rule the diagnosis of uncontrolled BP in or out.27 In our meta-analysis, the positive LR is 1.35 and negative LR is 0.44. Therefore, in treated hypertensive patients, an indication of uncontrolled BP as measured in the clinic does not confirm a diagnosis of uncontrolled BP (as judged by the reference standard of ABPM). On the other hand, the negative LR means that normal office BP does not rule out uncontrolled BP, which may be detected with ABPM. Consequently, the measurement of BP in the office does not change the degree of (un)certainty of adequate control of BP. This knowledge is important, to avoid overtreatment of white coat hypertension and undertreatment of masked cases.
As previously mentioned, we reported similar results in a study designed to determine the validity of office BP measurement in a primary care setting compared with ABPM.13 In that paper, the level of agreement between both methods was poor, indicating that clinic measurements could not be recommended as a single method of BP control in hypertensive patients.
The use of ABPM in diagnosing hypertension is likely to increase as a consequence of some guideline updates.2 Our study emphasizes the importance of their use in the control of hypertensive patients.
Another published meta-analysis1 investigated the validity of office BP for the diagnosis of hypertension in untreated patients, with diagnostic thresholds for arterial hypertension set at 140/90 mm Hg for office measurement, and 135/85 mm Hg for ABPM. In that paper, the sensitivity of office BP was 74.6% (95% CI, 60.7-84.8) and the specificity was 74.6% (95% CI, 47.9-90.4).
In our present study carried out with hypertensive patients receiving treatment, we obtained a slightly higher sensitivity value of 81.9% (within the CI of this meta-analysis) and a lower specificity of 41.1%. Therefore, the discordance between office BP and ABPM seems to be similar for the diagnosis of hypertension and the classification of hypertension as being well or poorly controlled. This confirms the low validity of the office BP, both for diagnosis and monitoring of hypertensive patients.
Strengths of our study. The study focused on (treated) hypertensive patients in a primary care setting, where hypertension is most often managed. It confirms that ABPM is indispensable to a good clinical practice.
Limitations of our study are those inherent to meta-analyses. The main weakness of our study is the paucity of data available regarding the utility of ABPM for monitoring BP control with treatment in a primary care setting. Other limitations are the variability in BP thresholds used, the number of measurements performed, and the ambulatory BP devices used. These differences could contribute to the observed heterogeneity.
Application of our results must take into account that we included only those studies performed in a primary care setting with treated hypertensive patients.
Moreover, this study was not designed to evaluate the consequences of over- and undertreatment of blood pressure, nor to address the accuracy of automated blood pressure machines or newer health and fitness devices.
Implications for practice, policy, or future research. Alternative monitoring methods are home BP self-measurement and automated 30-minute clinic BP measurement.28 However, ABPM provides us with unique information about the BP pattern (dipping or non-dipping), BP variability, and mean nighttime BP. This paper establishes that the measurement of BP in the office is not an accurate method to monitor BP control. ABPM should be incorporated in usual clinical practice in primary care. Although the consequences of ambulatory monitoring are not the focus of this study, we acknowledge that the decision to incorporate ABPM in clinical practice depends on the availability of ambulatory devices, proper training of health care workers, and a cost-effectiveness analysis of its use.
CORRESPONDENCE
Sergio Reino-González, MD, PhD, Adormideras Primary Health Center, Poligono de Adormideras s/n. 15002 A Coruña, Spain; [email protected].
1. Hodgkinson J, Mant J, Martin U, et al. Relative effectiveness of clinic and home blood pressure monitoring compared with ambulatory blood pressure monitoring in diagnosis of hypertension: systematic review. BMJ. 2011;342:d3621.
2. National Institute for Health and Clinical Excellence. Hypertension in adults: diagnosis and management. Available at: http://www.nice.org.uk/guidance/CG127. Accessed November 15, 2016.
3. Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension. 2003;42:1206-1252.
4. Hermida RC, Smolensky MH, Ayala DE, et al. Ambulatory Blood Pressure Monitoring (ABPM) as the reference standard for diagnosis of hypertension and assessment of vascular risk in adults. Chronobiol Int. 2015;32:1329-1342.
5. Siu AL; U.S. Preventive Services Task Force. Screening for high blood pressure in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2015;163:778-786.
6. Piper MA, Evans CV
7. American Academy of Family Physicians. Hypertension. Available at: www.aafp.org/patient-care/clinical-recommendations/all/hypertension.html. Accessed February 10, 2016.
8. Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC Practice Guidelines for the Management of Arterial Hypertension. Blood Press. 2013;23:3-16.
9. Marin R, de la Sierra A, Armario P, et al. 2005 Spanish guidelines in diagnosis and treatment of arterial hypertension. Medicina Clínica. 2005;125:24-34.
10. Fagard RH, Celis H, Thijs L, et al. Daytime and nighttime blood pressure as predictors of death and cause-specific cardiovascular events in hypertension. Hypertension. 2008;51:55-61.
11. Sega R, Trocino G, Lanzarotti A, et al. Alterations of cardiac structure in patients with isolated office, ambulatory, or home hypertension: Data from the general population (Pressione Arteriose Monitorate E Loro Associazioni [PAMELA] Study). Circulation. 2001;104:1385-1392.
12. Verberk WJ, Kessels AG, de Leeuw PW. Prevalence, causes, and consequences of masked hypertension: a meta-analysis. Am J Hypertens. 2008;21:969-975.
13. Reino-González S, Pita-Fernández S, Cibiriain-Sola M, et al. Validity of clinic blood pressure compared to ambulatory monitoring in hypertensive patients in a primary care setting. Blood Press. 2015;24:111-118.
14. Doebler P, Holling H. Meta-analysis of diagnostic accuracy with mada. Available at: https://cran.r-project.org/web/packages/mada/vignettes/mada.pdf. Accessed October 5, 2015.
15. Myers MG, Oh PI, Reeves RA, et al. Prevalence of white coat effect in treated hypertensive patients in the community. Am J Hypertens. 1995;8:591-597.
16. Imai Y, Tsuji I, Nagai K, et al. Ambulatory blood pressure monitoring in evaluating the prevalence of hypertension in adults in Ohasama, a rural Japanese community. Hypertens Res. 1996;19:207-212.
17. Taylor RS, Stockman J, Kernick D, et al. Ambulatory blood pressure monitoring for hypertension in general practice. J R Soc Med. 1998;91:301-304.
18. Little P, Barnett J, Barnsley L, et al. Comparison of agreement between different measures of blood pressure in primary care and daytime ambulatory blood pressure. BMJ. 2002;325:254.
19. Bur A, Herkner H, Vlcek M, et al. Classification of blood pressure levels by ambulatory blood pressure in hypertension. Hypertension. 2002;40:817-822.
20. Lindbaek M, Sandvik E, Liodden K, et al. Predictors for the white coat effect in general practice patients with suspected and treated hypertension. Br J Gen Pract. 2003;53:790-793.
21. Martínez MA, Sancho T, García P, et al. Home blood pressure in poorly controlled hypertension: relationship with ambulatory blood pressure and organ damage. Blood Press Monit. 2006;11:207-213.
22. Sierra BC, de la Sierra IA, Sobrino J, et al. Monitorización ambulatoria de la presión arterial (MAPA): características clínicas de 31.530 pacientes. Medicina Clínica. 2007;129:1-5.
23. Gómez MA, García L, Sánchez Á, et al. Agreement and disagreement between different methods of measuring blood pressure. Hipertensión (Madr). 2008;25:231-239.
24. Banegas JR, Segura J, De la Sierra A, et al. Gender differences in office and ambulatory control of hypertension. Am J Med. 2008;121:1078-1084.
25. Zaninelli A, Parati G, Cricelli C, et al. Office and 24-h ambulatory blood pressure control by treatment in general practice: the ‘Monitoraggio della pressione ARteriosa nella medicina TErritoriale’ study. J Hypertens. 2010;28:910-917.
26. Llisterri JL, Morillas P, Pallarés V, et al. Differences in the degree of control of arterial hypertension according to the measurement procedure of blood pressure in patients ≥ 65 years. FAPRES study. Rev Clin Esp. 2011;211:76-84.
27. Straus SE, Richardson WS, Glasziou P, et al. Evidence-Based Medicine: How to practice and teach it. 4th ed. Edinburgh, Scotland: Churchill Livingstone; 2010.
28. Van der Wel MC, Buunk IE, van Weel C, et al. A novel approach to office blood pressure measurement: 30-minute office blood pressure vs daytime ambulatory blood pressure. Ann Fam Med. 2011;9:128-135.
1. Hodgkinson J, Mant J, Martin U, et al. Relative effectiveness of clinic and home blood pressure monitoring compared with ambulatory blood pressure monitoring in diagnosis of hypertension: systematic review. BMJ. 2011;342:d3621.
2. National Institute for Health and Clinical Excellence. Hypertension in adults: diagnosis and management. Available at: http://www.nice.org.uk/guidance/CG127. Accessed November 15, 2016.
3. Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension. 2003;42:1206-1252.
4. Hermida RC, Smolensky MH, Ayala DE, et al. Ambulatory Blood Pressure Monitoring (ABPM) as the reference standard for diagnosis of hypertension and assessment of vascular risk in adults. Chronobiol Int. 2015;32:1329-1342.
5. Siu AL; U.S. Preventive Services Task Force. Screening for high blood pressure in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2015;163:778-786.
6. Piper MA, Evans CV
7. American Academy of Family Physicians. Hypertension. Available at: www.aafp.org/patient-care/clinical-recommendations/all/hypertension.html. Accessed February 10, 2016.
8. Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC Practice Guidelines for the Management of Arterial Hypertension. Blood Press. 2013;23:3-16.
9. Marin R, de la Sierra A, Armario P, et al. 2005 Spanish guidelines in diagnosis and treatment of arterial hypertension. Medicina Clínica. 2005;125:24-34.
10. Fagard RH, Celis H, Thijs L, et al. Daytime and nighttime blood pressure as predictors of death and cause-specific cardiovascular events in hypertension. Hypertension. 2008;51:55-61.
11. Sega R, Trocino G, Lanzarotti A, et al. Alterations of cardiac structure in patients with isolated office, ambulatory, or home hypertension: Data from the general population (Pressione Arteriose Monitorate E Loro Associazioni [PAMELA] Study). Circulation. 2001;104:1385-1392.
12. Verberk WJ, Kessels AG, de Leeuw PW. Prevalence, causes, and consequences of masked hypertension: a meta-analysis. Am J Hypertens. 2008;21:969-975.
13. Reino-González S, Pita-Fernández S, Cibiriain-Sola M, et al. Validity of clinic blood pressure compared to ambulatory monitoring in hypertensive patients in a primary care setting. Blood Press. 2015;24:111-118.
14. Doebler P, Holling H. Meta-analysis of diagnostic accuracy with mada. Available at: https://cran.r-project.org/web/packages/mada/vignettes/mada.pdf. Accessed October 5, 2015.
15. Myers MG, Oh PI, Reeves RA, et al. Prevalence of white coat effect in treated hypertensive patients in the community. Am J Hypertens. 1995;8:591-597.
16. Imai Y, Tsuji I, Nagai K, et al. Ambulatory blood pressure monitoring in evaluating the prevalence of hypertension in adults in Ohasama, a rural Japanese community. Hypertens Res. 1996;19:207-212.
17. Taylor RS, Stockman J, Kernick D, et al. Ambulatory blood pressure monitoring for hypertension in general practice. J R Soc Med. 1998;91:301-304.
18. Little P, Barnett J, Barnsley L, et al. Comparison of agreement between different measures of blood pressure in primary care and daytime ambulatory blood pressure. BMJ. 2002;325:254.
19. Bur A, Herkner H, Vlcek M, et al. Classification of blood pressure levels by ambulatory blood pressure in hypertension. Hypertension. 2002;40:817-822.
20. Lindbaek M, Sandvik E, Liodden K, et al. Predictors for the white coat effect in general practice patients with suspected and treated hypertension. Br J Gen Pract. 2003;53:790-793.
21. Martínez MA, Sancho T, García P, et al. Home blood pressure in poorly controlled hypertension: relationship with ambulatory blood pressure and organ damage. Blood Press Monit. 2006;11:207-213.
22. Sierra BC, de la Sierra IA, Sobrino J, et al. Monitorización ambulatoria de la presión arterial (MAPA): características clínicas de 31.530 pacientes. Medicina Clínica. 2007;129:1-5.
23. Gómez MA, García L, Sánchez Á, et al. Agreement and disagreement between different methods of measuring blood pressure. Hipertensión (Madr). 2008;25:231-239.
24. Banegas JR, Segura J, De la Sierra A, et al. Gender differences in office and ambulatory control of hypertension. Am J Med. 2008;121:1078-1084.
25. Zaninelli A, Parati G, Cricelli C, et al. Office and 24-h ambulatory blood pressure control by treatment in general practice: the ‘Monitoraggio della pressione ARteriosa nella medicina TErritoriale’ study. J Hypertens. 2010;28:910-917.
26. Llisterri JL, Morillas P, Pallarés V, et al. Differences in the degree of control of arterial hypertension according to the measurement procedure of blood pressure in patients ≥ 65 years. FAPRES study. Rev Clin Esp. 2011;211:76-84.
27. Straus SE, Richardson WS, Glasziou P, et al. Evidence-Based Medicine: How to practice and teach it. 4th ed. Edinburgh, Scotland: Churchill Livingstone; 2010.
28. Van der Wel MC, Buunk IE, van Weel C, et al. A novel approach to office blood pressure measurement: 30-minute office blood pressure vs daytime ambulatory blood pressure. Ann Fam Med. 2011;9:128-135.

Is diabetes distress on your radar screen?
Managing diabetes is a complex undertaking, with an extensive regimen of self-care—including regular exercise, meal planning, blood glucose monitoring, medication scheduling, and multiple visits—that is critically linked to glycemic control and the prevention of complications. Incorporating all of these elements into daily life can be daunting.1-3
In fact, nearly half of US adults with diabetes fail to meet the recommended targets.4 This leads to frustration, which often manifests in psychosocial problems that further hamper efforts to manage the disease.5-10 The most notable is a psychosocial disorder known as diabetes distress, which affects close to 45% of those with diabetes.11,12
It is important to note that diabetes distress is not a psychiatric disorder;13 rather, it is a broad affective reaction to the stress of living with this chronic and complex disease.14,15 By negatively affecting adherence to a self-care regimen, diabetes distress contributes to worsening glycemic control and increasing morbidity.16-18
Recognizing that about 80% of those with diabetes are treated in primary care settings,19 we wrote this review to call your attention to diabetes distress, alert you to brief screening tools that can easily be incorporated into clinic visits, and offer guidance in matching proposed interventions to the aspects of diabetes self-management that cause patients the greatest distress.
Diabetes distress: What it is, what it’s not
For patients with type 2 diabetes, diabetes distress centers around 4 main issues:
- frustration with the demands of self-care;
- apprehension about the future and the possibility of developing serious complications;
- concern about both the quality and the cost of required medical care; and
- perceived lack of support from family and/or friends.11,12,20
As mentioned earlier, diabetes distress is not a psychiatric condition and should not be confused with major depressive disorder (MDD). Here’s help in telling the difference.
For starters, a diagnosis of depression is symptom-based.13 MDD requires the presence of at least 5 of the 9 symptoms defined by the Diagnostic and Statistical Manual of Mental Disorders, Fifth ed. (DSM-5)—eg, persistent feelings of worthlessness or guilt, sleep disturbances, lack of interest in normal activities—for at least 2 weeks.21 What’s more, the diagnostic criteria for MDD do not specify a cause or disease process. Nor do they distinguish between a pathological response and an expected reaction to a stressful life event.22 Further, depression measures reflect symptoms (eg, hyperglycemia), as well as stressful experiences resulting from diabetes self-care, which may contribute to the high rate of false positives or incorrect diagnoses of MDD and missed diagnoses of diabetes distress.23
Unlike MDD, diabetes distress has a specific cause—diabetes—and can best be understood as an emotional response to a demanding health condition.13 And, because the source of the problem is identified, diabetes distress can be treated with specific interventions targeting the areas causing the highest levels of stress.
When a psychiatric condition and diabetes distress overlap
MDD, anxiety disorders, and diabetes distress are all common in patients with diabetes,24 and the co-occurrence of a psychiatric disorder and diabetes distress is high.25 Thus, it is important not only to identify cases of diabetes distress but also to consider comorbid depression and/or anxiety in patients with diabetes distress.
More often, though, it is the other way around, according to the Distress and Depression in Diabetes (3D) study. The researchers recently found that 84% of patients with moderate or high diabetes distress did not fulfill the criteria for MDD, but that 67% of diabetes patients with MDD also had moderate or high diabetes distress.13,15,17,25
The data highlight the importance of screening patients with a dual diagnosis of diabetes and MDD for diabetes distress. Keep in mind that individuals diagnosed with both diabetes distress and a comorbid psychiatric condition may require more complex and intensive treatment than those with either diabetes distress or MDD alone.25
Screening for diabetes distress
Diabetes distress can be easily assessed using one of several patient-reported outcome measures. Six validated measures, ranging in length from one to 28 questions, are designed for use in primary care (TABLE).26-30 Some of the measures are easily accessible online; others require subscription to MEDLINE.
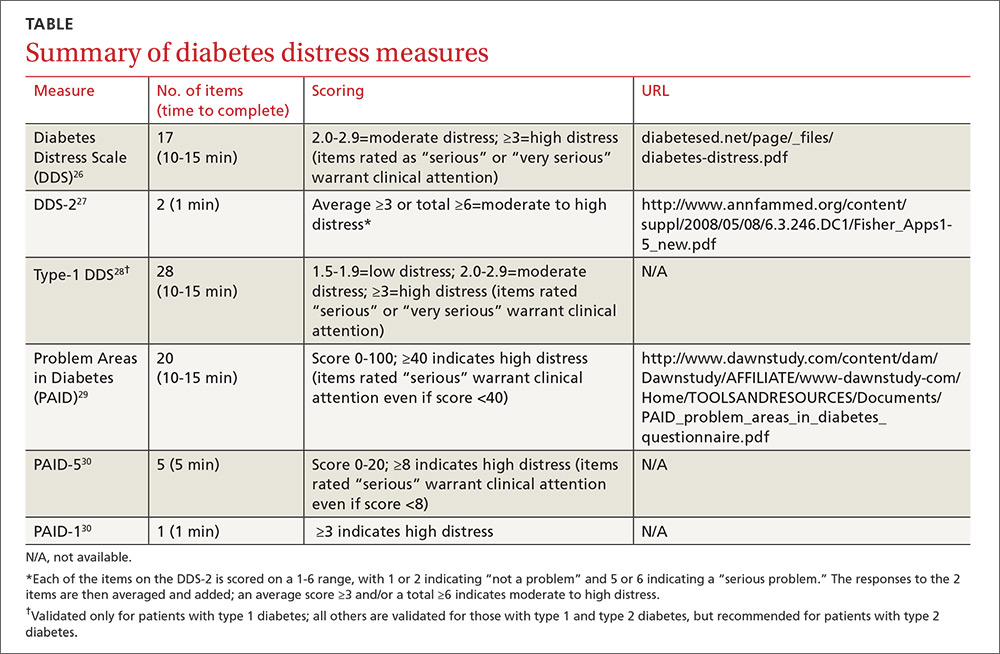
Problem Areas in Diabetes (PAID): There are 3 versions of PAID—a 20-item screen assessing a broad range of feelings related to living with diabetes and its treatment, a 5-item version (PAID-5) with high rates of sensitivity (95%) and specificity (89%), and a single-item test (PAID-1) that is highly correlated with the longer version.26,27
Diabetes Distress Scale (DDS): This tool is available in a 17-item measure assessing diabetes distress as it relates to the emotional burden, physician-related distress, regimen-related distress, and interpersonal distress.28 DDS is also available in a short form (DDS-2) with 2 items29 and a 28-item scale specifically for patients with type 1 diabetes.30 T1-DDS, the only diabetes distress measure focused on this particular patient population, assesses the 7 sources of distress found to be common among adults with type 1 diabetes: powerlessness, negative social perceptions, physician distress, friend/family distress, hypoglycemia distress, management distress, and eating distress.
Studies have shown that not only do those with type 1 diabetes experience different stressors compared with their type 2 counterparts, but that they tend to experience distress differently. For patients with type 1 diabetes, for example, powerlessness ranked as the highest source of distress, followed by eating distress and hypoglycemia distress. These sources of distress differ from the regimen distress, emotional burden, interpersonal distress, and physician distress identified by those with type 2 diabetes.30
How to respond to diabetes distress
Diabetes distress is easier to identify than to successfully treat. Few validated treatments for diabetes distress exist and, to our knowledge, only 2 studies have assessed interventions aimed at reduction of such distress.31,32
The REDEEM trial31 recruited adults with type 2 diabetes and diabetes distress to participate in a 12-month randomized controlled trial (RCT). The trial had 3 arms, comparing the effectiveness of a computer-assisted self-management (CASM) program alone, a CASM program plus in-person diabetes distress-specific problem-solving therapy, and a computer-assisted minimally supportive intervention. The main outcomes included diabetes distress (using the DDS scale and subscales), along with self-management behaviors and HbA1c.
Participants in all 3 arms showed significant reductions in total diabetes distress and improvements in self-management behaviors, with no significant differences among the groups. No differences in HbA1c were found. However, those in the CASM program plus distress-specific therapy arm showed a larger reduction in regimen distress compared with the other 2 groups.31
The DIAMOS trial32 recruited adults who had type 1 or type 2 diabetes, diabetes distress, and subclinical depressive symptoms for a 2-arm RCT. One group underwent cognitive behavioral interventions, while the controls had standard group-based diabetes education. The main outcomes included diabetes distress (measured via the PAID scale), depressive symptoms, well-being, diabetes self-care, diabetes acceptance, satisfaction with diabetes treatment, HbA1c, and subclinical inflammation.
The intervention group showed greater improvement in diabetes distress and depressive symptoms compared with the control group, but no differences in well-being, self-care, treatment satisfaction, HbA1c, or subclinical inflammation were observed.32
Both studies support the use of problem-solving therapy and cognitive behavioral interventions for patients with diabetes distress. Future research should evaluate the effectiveness of these interventions in the primary care setting.
What else to offer when challenges mount?
Diabetes is a progressive disease, and most patients experience multiple challenges over time. These typically include complications and comorbidities, physical limitations, polypharmacy, hypoglycemia, and cognitive impairment, as well as changes in everything from medication and lifestyle to insurance coverage and social support.33,34 All increase the risk for diabetes distress, as well as related psychiatric conditions.
Aging and diabetes are independent risk factors for cognitive impairment, for example, and the presence of both increases this risk.35 What’s more, diabetes alone is associated with poorer executive function,36-38 the higher-level cognitive processes that allow individuals to engage in independent, purposeful, and flexible goal-related behaviors. Both poor cognitive function and impaired executive function interfere with the ability to perform self-care behaviors such as adjusting insulin doses, drawing insulin into a syringe, or dialing an insulin dose with an insulin pen.39 This in turn can lead to frustration and increase the likelihood of moderate to high diabetes distress.
Assessing diabetes distress in patients with cognitive impairment, poor executive functioning, or other psychological limitations is particularly difficult, however, as no diabetes distress measures take such deficits into account. Thus, primary care physicians without expertise in neuropsychology should consider referring patients with such problems to specialists for assessment.
The progressive nature of diabetes also highlights the need for primary care physicians to periodically screen for diabetes distress and engage in ongoing discussions about what type of care is best for individual patients, and why. When developing or updating treatment plans and making recommendations, it is crucial to consider the impact the treatment would likely have on the patient’s physical and mental health and to explicitly inquire about and acknowledge his or her values and preferences for care.40-44
It is also important to remain aware of socioeconomic changes—in employment, insurance coverage, and living situations, for example—which are not addressed in the screening tools.
Moderate to high diabetes distress scores, as well as individual items patients identify as “very serious” problems, represent clinical red flags that should be the focus of careful discussion during a medical visit. Patients with moderate to high distress should be referred to a therapist trained in cognitive behavioral therapy or problem-solving therapy. Physicians who lack access to such resources can incorporate cognitive behavioral and problem-solving techniques into patient discussion. (See “Directing help where it’s most needed.”) All patients should be referred to a certified diabetes educator—a key component of diabetes care.45,46
SIDEBAR
Directing help where it's most needed
CASE 1 ›
Conduct a behavioral experiment
Fred J, a 67-year-old diagnosed with type 2 diabetes 6 years ago, comes in for a diabetes check-up. He is a new patient who recently retired from his job as a contractor and was referred by a colleague. In response to a question about his diabetes management, Mr. J tells you he’s having a hard time.
“I get down on myself,” the patient says. “I take my medications every day at the exact same time, but when I test my sugar, it’s 260 or 280. I know I did this to myself. If only I weighed less, ate better, or exercised more.”
At other times, “I think, 'Why bother?'” Mr. J adds. “I feel like there’s nothing I can do to make it better.”
The DDS-2 screen you gave Mr. J bears out his high level of distress and his fear of complications. He tells you about an aunt who “had diabetes like me and had to go on dialysis, then died 2 years later.” When you ask what he fears most, Mr. J says he worries about kidney failure. “I don’t want to go on dialysis,” he insists.
You take the opportunity to point out that nephropathy is not inevitable and that he can perform self-care behaviors now that will prevent or delay kidney complications.
You also decide to try a cognitive behavioral technique in an attempt to change his thought process. You ask Mr. J to agree to a week-long behavioral experiment to examine the effect of walking for 30 minutes each day.
He agrees. You advise him to write down his predictions before he begins the experiment and then to keep a log, checking and recording his glucose levels before and after each walk. You schedule a follow-up visit to discuss the results, hoping that a reduction in blood glucose levels will convince Mr. J that exercise is beneficial to his diabetes.
CASE 2 ›
Identify the problem; brainstorm with the patient
Susan T, a 46-year-old with a husband and 2 teenage children, comes in for her 3-month diabetes check-up. At her last visit, she expressed concerns about her family’s lack of cooperation as she struggled to change her diet. This time, she appears frustrated and distraught.
Your nurse administered the PAID-5 while Ms. T was in the waiting room and entered her score—8, indicating high diabetes distress—in the electronic medical record. You ask Ms. T what’s happening, knowing that encouraging her to verbalize her feelings is a way to increase her trust and help alleviate her concerns.
You also try the following problem-solving technique:
Define the problem. Ms. T is having a hard time maintaining a healthy diet. Her husband and children refuse to eat the healthy meals she prepares and want her to cook separate dinners for them.
Identify challenges. The patient works full-time and does not have the time or energy to cook separate meals. In addition, she is upset by her family’s lack of support in her efforts to control her disease.
Brainstorm multiple solutions:
1) Ms. T can prepare all of her own meals for the work week on Sunday, then cook for the others when she returns from work.
2) Her husband and children can make their own dinner if they do not want to eat the healthier meals she prepares.
3) The patient can join a diabetes support group where she will meet, and possibly learn from, other patients who may be struggling with diabetes self-care.
4) Ms. T can ask her husband and children to come to her next diabetes check-up so they can learn about the importance of family support in diabetes management directly from you.
5) The patient’s family can receive information about a healthy diabetes diet from a certified diabetes educator.
Decide on appropriate solutions. The patient agrees to try and prepare her weekday meals on Sunday so that she is not tempted to eat less healthy options. She also agrees to bring her family to her next diabetes check-up and to diabetes education classes.
CORRESPONDENCE
Elizabeth A. Beverly, PhD, Department of Family Medicine, Ohio University Heritage College of Osteopathic Medicine, 35 W. Green Drive, Athens, OH 45701; [email protected].
1. Gafarian CT, Heiby EM, Blair P, et al. The diabetes time management questionnaire. Diabetes Educator. 1999;25:585-592.
2. Wdowik MJ, Kendall PA, Harris MA. College students with diabetes: using focus groups and interviews to determine psychosocial issues and barriers to control. Diabetes Educator. 1997;23:558-562.
3. Rubin RR. Psychological issues and treatment for people with diabetes. J Clin Psych. 2001;57:457-478.
4. Ali MK, Bullard KM, Gregg EW. Achievement of goals in US diabetes care, 1999-2010. New Engl J Med. 2013;369:287-288.
5. Lloyd CE, Smith J, Weinger K. Stress and diabetes: Review of the links. Diabetes Spectrum. 2005;18:121-127.
6. Weinger K. Psychosocial issues and self-care. Am J Nurs. 2007;107(6 suppl): S34-S38.
7. Weinger K, Jacobson AM. Psychosocial and quality of life correlates of glycemic control during intensive treatment of type 1 diabetes. Patient Education Counseling. 2001;42:123-131.
8. Albright TL, Parchman M, Burge SK. Predictors of self-care behavior in adults with type 2 diabetes: an RRNeST study. Fam Med. 2001;33:354-360.
9. Gonzalez JS, Safren SA, Cagliero E, et al. Depression, self-care, and medication adherence in type 2 diabetes: relationships across the full range of symptom severity. Diabetes Care. 2007;30:2222-2227.
10. Gonzalez JS, Safren SA, Delahanty LM, et al. Symptoms of depression prospectively predict poorer self-care in patients with Type 2 diabetes. Diabetic Med. 2008;25:1102-1107.
11. Nicolucci A, Kovacs Burns K, Holt RI, et al. Diabetes Attitudes, Wishes and Needs second study (DAWN2): cross-national benchmarking of diabetes-related psychosocial outcomes for people with diabetes. Diabetic Med. 2013;30:767-777.
12. Fisher L, Hessler DM, Polonsky W, et al. When is diabetes distress clinically meaningful?: establishing cut points for the Diabetes Distress Scale. Diabetes Care. 2012;35:259-264.
13. Fisher L, Gonzalez JS, Polonsky WH. The confusing tale of depression and distress in patients with diabetes: a call for greater clarity and precision. Diabetic Med. 2014;31:764-772.
14. Fisher L, Mullan JT, Skaff MM, et al. Predicting diabetes distress in patients with Type 2 diabetes: a longitudinal study. Diabetic Med. 2009;26:622-627.
15. Fisher L, Skaff MM, Mullan JT, et al. Clinical depression versus distress among patients with type 2 diabetes: not just a question of semantics. Diabetes Care. 2007;30:542-548.
16. Gonzalez JS, Delahanty LM, Safren SA, et al. Differentiating symptoms of depression from diabetes-specific distress: relationships with self-care in type 2 diabetes. Diabetologia. 2008;51:2822-1825.
17. Fisher L, Mullan JT, Arean P, et al. Diabetes distress but not clinical depression or depressive symptoms is associated with glycemic control in both cross-sectional and longitudinal analyses. Diabetes Care. 2010;33:23-28.
18. Fisher EB, Thorpe CT, Devellis BM, et al. Healthy coping, negative emotions, and diabetes management: a systematic review and appraisal. Diabetes Educator. 2007;33:1080-1103; 1104-1086.
19. Peterson KA, Radosevich DM, O’Connor PJ, et al. Improving diabetes care in practice: findings from the TRANSLATE trial. Diabetes Care. 2008;31:2238-2243.
20. Fisher L, Glasgow RE, Strycker LA. The relationship between diabetes distress and clinical depression with glycemic control among patients with type 2 diabetes. Diabetes Care. 2010;33:1034-1036.
21. Cole J, McGuffin P, Farmer AE. The classification of depression: are we still confused? Br J Psychiatr. 2008;192:83-85.
22. Wakefield JC. The concept of mental disorder. On the boundary between biological facts and social values. Am Psychologist. 1992;47:373-388.
23. Fisher L, Gonzalez JS, Polonsky WH. The confusing tale of depression and distress in patients with diabetes: a call for greater clarity and precision. Diabetic Med. 2014;31:764-772.
24. Ciechanowski PS, Katon WJ, Russo JE. Depression and diabetes: impact of depressive symptoms on adherence, function, and costs. Arch Intern Med. 2000;160:3278-3285.
25. Fisher L, Skaff MM, Mullan JT, et al. A longitudinal study of affective and anxiety disorders, depressive affect and diabetes distress in adults with Type 2 diabetes. Diabetic Med. 2008;25:1096-1101.
26. Polonsky WH, Anderson BJ, Lohrer PA, et al. Assessment of diabetes-related distress. Diabetes Care. 1995;18:754-760.
27. McGuire BE, Morrison TG, Hermanns N, et al. Short-form measures of diabetes-related emotional distress: the Problem Areas in Diabetes Scale (PAID)-5 and PAID-1. Diabetologia. 2010;53:66-69.
28. Polonsky WH, Fisher L, Earles J, et al. Assessing psychosocial distress in diabetes: development of the diabetes distress scale. Diabetes Care. 2005;28:626-631.
29. Fisher L, Glasgow RE, Mullan JT, et al. Development of a brief diabetes distress screening instrument. Ann Fam Med. 2008;6:246-252.
30. Fisher L, Polonsky WH, Hessler DM, et al. Understanding the sources of diabetes distress in adults with type 1 diabetes. J Diabetes Complications. 2015;29:572-577.
31. Fisher L, Hessler D, Glasgow RE, et al. REDEEM: a pragmatic trial to reduce diabetes distress. Diabetes Care. 2013;36:2551-2558.
32. Hermanns N, Schmitt A, Gahr A, et al. The effect of a Diabetes-Specific Cognitive Behavioral Treatment Program (DIAMOS) for patients with diabetes and subclinical depression: results of a randomized controlled trial. Diabetes Care. 2015;38:551-560.
33. Weinger K, Beverly EA, Smaldone A. Diabetes self-care and the older adult. Western J Nurs Res. 2014;36:1272-1298.
34. Beverly EA, Ritholz MD, Shepherd C, et al. The psychosocial challenges and care of older adults with diabetes: “can’t do what I used to do; can’t be who I once was.” Curr Diabetes Rep. 2016;16:48.
35. Lu FP, Lin KP, Kuo HK. Diabetes and the risk of multi-system aging phenotypes: a systematic review and meta-analysis. PloS One. 2009;4:e4144.
36. Thabit H, Kyaw TT, McDermott J, et al. Executive function and diabetes mellitus—a stone left unturned? Curr Diabetes Rev. 2012;8:109-115.
37. McNally K, Rohan J, Pendley JS, et al. Executive functioning, treatment adherence, and glycemic control in children with type 1 diabetes. Diabetes Care. 2010;33:1159-1162.
38. Rucker JL, McDowd JM, Kluding PM. Executive function and type 2 diabetes: putting the pieces together. Phys Ther. 2012;92:454-462.
39. Kirkman MS, Briscoe VJ, Clark N, et al. Diabetes in older adults. Diabetes Care. 2012;35:2650-2664.
40. Durso SC. Using clinical guidelines designed for older adults with diabetes mellitus and complex health status. JAMA. 2006;295:1935-1940.
41. Oftedal B, Karlsen B, Bru E. Life values and self-regulation behaviours among adults with type 2 diabetes. J Clin Nurs. 2010;19:2548-2556.
42. Morrow AS, Haidet P, Skinner J, et al. Integrating diabetes self-management with the health goals of older adults: a qualitative exploration. Patient Education Counseling. 2008;72:418-423.
43. Huang ES, Gorawara-Bhat R, Chin MH. Self-reported goals of older patients with type 2 diabetes mellitus. J Am Geriatr Soc. 2005;53:306-311.
44. Beverly EA, Wray LA, LaCoe CL, et al. Listening to older adults’ values and preferences for Type 2 diabetes care: a qualitative study. Diabetes Spectrum. 2014;27:44-49.
45. American Association of Diabetes Educators. Why refer for diabetes education? American Association of Diabetes Educators. Available at: https://www.diabeteseducator.org/practice/provider-resources/why-refer-for-diabetes-education. Accessed August 15, 2016.
46. Ismail K, Winkley K, Rabe-Hesketh S. Systematic review and meta-analysis of randomised controlled trials of psychological interventions to improve glycaemic control in patients with type 2 diabetes. Lancet. 2004;363:1589-1597.
Managing diabetes is a complex undertaking, with an extensive regimen of self-care—including regular exercise, meal planning, blood glucose monitoring, medication scheduling, and multiple visits—that is critically linked to glycemic control and the prevention of complications. Incorporating all of these elements into daily life can be daunting.1-3
In fact, nearly half of US adults with diabetes fail to meet the recommended targets.4 This leads to frustration, which often manifests in psychosocial problems that further hamper efforts to manage the disease.5-10 The most notable is a psychosocial disorder known as diabetes distress, which affects close to 45% of those with diabetes.11,12
It is important to note that diabetes distress is not a psychiatric disorder;13 rather, it is a broad affective reaction to the stress of living with this chronic and complex disease.14,15 By negatively affecting adherence to a self-care regimen, diabetes distress contributes to worsening glycemic control and increasing morbidity.16-18
Recognizing that about 80% of those with diabetes are treated in primary care settings,19 we wrote this review to call your attention to diabetes distress, alert you to brief screening tools that can easily be incorporated into clinic visits, and offer guidance in matching proposed interventions to the aspects of diabetes self-management that cause patients the greatest distress.
Diabetes distress: What it is, what it’s not
For patients with type 2 diabetes, diabetes distress centers around 4 main issues:
- frustration with the demands of self-care;
- apprehension about the future and the possibility of developing serious complications;
- concern about both the quality and the cost of required medical care; and
- perceived lack of support from family and/or friends.11,12,20
As mentioned earlier, diabetes distress is not a psychiatric condition and should not be confused with major depressive disorder (MDD). Here’s help in telling the difference.
For starters, a diagnosis of depression is symptom-based.13 MDD requires the presence of at least 5 of the 9 symptoms defined by the Diagnostic and Statistical Manual of Mental Disorders, Fifth ed. (DSM-5)—eg, persistent feelings of worthlessness or guilt, sleep disturbances, lack of interest in normal activities—for at least 2 weeks.21 What’s more, the diagnostic criteria for MDD do not specify a cause or disease process. Nor do they distinguish between a pathological response and an expected reaction to a stressful life event.22 Further, depression measures reflect symptoms (eg, hyperglycemia), as well as stressful experiences resulting from diabetes self-care, which may contribute to the high rate of false positives or incorrect diagnoses of MDD and missed diagnoses of diabetes distress.23
Unlike MDD, diabetes distress has a specific cause—diabetes—and can best be understood as an emotional response to a demanding health condition.13 And, because the source of the problem is identified, diabetes distress can be treated with specific interventions targeting the areas causing the highest levels of stress.
When a psychiatric condition and diabetes distress overlap
MDD, anxiety disorders, and diabetes distress are all common in patients with diabetes,24 and the co-occurrence of a psychiatric disorder and diabetes distress is high.25 Thus, it is important not only to identify cases of diabetes distress but also to consider comorbid depression and/or anxiety in patients with diabetes distress.
More often, though, it is the other way around, according to the Distress and Depression in Diabetes (3D) study. The researchers recently found that 84% of patients with moderate or high diabetes distress did not fulfill the criteria for MDD, but that 67% of diabetes patients with MDD also had moderate or high diabetes distress.13,15,17,25
The data highlight the importance of screening patients with a dual diagnosis of diabetes and MDD for diabetes distress. Keep in mind that individuals diagnosed with both diabetes distress and a comorbid psychiatric condition may require more complex and intensive treatment than those with either diabetes distress or MDD alone.25
Screening for diabetes distress
Diabetes distress can be easily assessed using one of several patient-reported outcome measures. Six validated measures, ranging in length from one to 28 questions, are designed for use in primary care (TABLE).26-30 Some of the measures are easily accessible online; others require subscription to MEDLINE.
Problem Areas in Diabetes (PAID): There are 3 versions of PAID—a 20-item screen assessing a broad range of feelings related to living with diabetes and its treatment, a 5-item version (PAID-5) with high rates of sensitivity (95%) and specificity (89%), and a single-item test (PAID-1) that is highly correlated with the longer version.26,27
Diabetes Distress Scale (DDS): This tool is available in a 17-item measure assessing diabetes distress as it relates to the emotional burden, physician-related distress, regimen-related distress, and interpersonal distress.28 DDS is also available in a short form (DDS-2) with 2 items29 and a 28-item scale specifically for patients with type 1 diabetes.30 T1-DDS, the only diabetes distress measure focused on this particular patient population, assesses the 7 sources of distress found to be common among adults with type 1 diabetes: powerlessness, negative social perceptions, physician distress, friend/family distress, hypoglycemia distress, management distress, and eating distress.
Studies have shown that not only do those with type 1 diabetes experience different stressors compared with their type 2 counterparts, but that they tend to experience distress differently. For patients with type 1 diabetes, for example, powerlessness ranked as the highest source of distress, followed by eating distress and hypoglycemia distress. These sources of distress differ from the regimen distress, emotional burden, interpersonal distress, and physician distress identified by those with type 2 diabetes.30
How to respond to diabetes distress
Diabetes distress is easier to identify than to successfully treat. Few validated treatments for diabetes distress exist and, to our knowledge, only 2 studies have assessed interventions aimed at reduction of such distress.31,32
The REDEEM trial31 recruited adults with type 2 diabetes and diabetes distress to participate in a 12-month randomized controlled trial (RCT). The trial had 3 arms, comparing the effectiveness of a computer-assisted self-management (CASM) program alone, a CASM program plus in-person diabetes distress-specific problem-solving therapy, and a computer-assisted minimally supportive intervention. The main outcomes included diabetes distress (using the DDS scale and subscales), along with self-management behaviors and HbA1c.
Participants in all 3 arms showed significant reductions in total diabetes distress and improvements in self-management behaviors, with no significant differences among the groups. No differences in HbA1c were found. However, those in the CASM program plus distress-specific therapy arm showed a larger reduction in regimen distress compared with the other 2 groups.31
The DIAMOS trial32 recruited adults who had type 1 or type 2 diabetes, diabetes distress, and subclinical depressive symptoms for a 2-arm RCT. One group underwent cognitive behavioral interventions, while the controls had standard group-based diabetes education. The main outcomes included diabetes distress (measured via the PAID scale), depressive symptoms, well-being, diabetes self-care, diabetes acceptance, satisfaction with diabetes treatment, HbA1c, and subclinical inflammation.
The intervention group showed greater improvement in diabetes distress and depressive symptoms compared with the control group, but no differences in well-being, self-care, treatment satisfaction, HbA1c, or subclinical inflammation were observed.32
Both studies support the use of problem-solving therapy and cognitive behavioral interventions for patients with diabetes distress. Future research should evaluate the effectiveness of these interventions in the primary care setting.
What else to offer when challenges mount?
Diabetes is a progressive disease, and most patients experience multiple challenges over time. These typically include complications and comorbidities, physical limitations, polypharmacy, hypoglycemia, and cognitive impairment, as well as changes in everything from medication and lifestyle to insurance coverage and social support.33,34 All increase the risk for diabetes distress, as well as related psychiatric conditions.
Aging and diabetes are independent risk factors for cognitive impairment, for example, and the presence of both increases this risk.35 What’s more, diabetes alone is associated with poorer executive function,36-38 the higher-level cognitive processes that allow individuals to engage in independent, purposeful, and flexible goal-related behaviors. Both poor cognitive function and impaired executive function interfere with the ability to perform self-care behaviors such as adjusting insulin doses, drawing insulin into a syringe, or dialing an insulin dose with an insulin pen.39 This in turn can lead to frustration and increase the likelihood of moderate to high diabetes distress.
Assessing diabetes distress in patients with cognitive impairment, poor executive functioning, or other psychological limitations is particularly difficult, however, as no diabetes distress measures take such deficits into account. Thus, primary care physicians without expertise in neuropsychology should consider referring patients with such problems to specialists for assessment.
The progressive nature of diabetes also highlights the need for primary care physicians to periodically screen for diabetes distress and engage in ongoing discussions about what type of care is best for individual patients, and why. When developing or updating treatment plans and making recommendations, it is crucial to consider the impact the treatment would likely have on the patient’s physical and mental health and to explicitly inquire about and acknowledge his or her values and preferences for care.40-44
It is also important to remain aware of socioeconomic changes—in employment, insurance coverage, and living situations, for example—which are not addressed in the screening tools.
Moderate to high diabetes distress scores, as well as individual items patients identify as “very serious” problems, represent clinical red flags that should be the focus of careful discussion during a medical visit. Patients with moderate to high distress should be referred to a therapist trained in cognitive behavioral therapy or problem-solving therapy. Physicians who lack access to such resources can incorporate cognitive behavioral and problem-solving techniques into patient discussion. (See “Directing help where it’s most needed.”) All patients should be referred to a certified diabetes educator—a key component of diabetes care.45,46
SIDEBAR
Directing help where it's most needed
CASE 1 ›
Conduct a behavioral experiment
Fred J, a 67-year-old diagnosed with type 2 diabetes 6 years ago, comes in for a diabetes check-up. He is a new patient who recently retired from his job as a contractor and was referred by a colleague. In response to a question about his diabetes management, Mr. J tells you he’s having a hard time.
“I get down on myself,” the patient says. “I take my medications every day at the exact same time, but when I test my sugar, it’s 260 or 280. I know I did this to myself. If only I weighed less, ate better, or exercised more.”
At other times, “I think, 'Why bother?'” Mr. J adds. “I feel like there’s nothing I can do to make it better.”
The DDS-2 screen you gave Mr. J bears out his high level of distress and his fear of complications. He tells you about an aunt who “had diabetes like me and had to go on dialysis, then died 2 years later.” When you ask what he fears most, Mr. J says he worries about kidney failure. “I don’t want to go on dialysis,” he insists.
You take the opportunity to point out that nephropathy is not inevitable and that he can perform self-care behaviors now that will prevent or delay kidney complications.
You also decide to try a cognitive behavioral technique in an attempt to change his thought process. You ask Mr. J to agree to a week-long behavioral experiment to examine the effect of walking for 30 minutes each day.
He agrees. You advise him to write down his predictions before he begins the experiment and then to keep a log, checking and recording his glucose levels before and after each walk. You schedule a follow-up visit to discuss the results, hoping that a reduction in blood glucose levels will convince Mr. J that exercise is beneficial to his diabetes.
CASE 2 ›
Identify the problem; brainstorm with the patient
Susan T, a 46-year-old with a husband and 2 teenage children, comes in for her 3-month diabetes check-up. At her last visit, she expressed concerns about her family’s lack of cooperation as she struggled to change her diet. This time, she appears frustrated and distraught.
Your nurse administered the PAID-5 while Ms. T was in the waiting room and entered her score—8, indicating high diabetes distress—in the electronic medical record. You ask Ms. T what’s happening, knowing that encouraging her to verbalize her feelings is a way to increase her trust and help alleviate her concerns.
You also try the following problem-solving technique:
Define the problem. Ms. T is having a hard time maintaining a healthy diet. Her husband and children refuse to eat the healthy meals she prepares and want her to cook separate dinners for them.
Identify challenges. The patient works full-time and does not have the time or energy to cook separate meals. In addition, she is upset by her family’s lack of support in her efforts to control her disease.
Brainstorm multiple solutions:
1) Ms. T can prepare all of her own meals for the work week on Sunday, then cook for the others when she returns from work.
2) Her husband and children can make their own dinner if they do not want to eat the healthier meals she prepares.
3) The patient can join a diabetes support group where she will meet, and possibly learn from, other patients who may be struggling with diabetes self-care.
4) Ms. T can ask her husband and children to come to her next diabetes check-up so they can learn about the importance of family support in diabetes management directly from you.
5) The patient’s family can receive information about a healthy diabetes diet from a certified diabetes educator.
Decide on appropriate solutions. The patient agrees to try and prepare her weekday meals on Sunday so that she is not tempted to eat less healthy options. She also agrees to bring her family to her next diabetes check-up and to diabetes education classes.
CORRESPONDENCE
Elizabeth A. Beverly, PhD, Department of Family Medicine, Ohio University Heritage College of Osteopathic Medicine, 35 W. Green Drive, Athens, OH 45701; [email protected].
Managing diabetes is a complex undertaking, with an extensive regimen of self-care—including regular exercise, meal planning, blood glucose monitoring, medication scheduling, and multiple visits—that is critically linked to glycemic control and the prevention of complications. Incorporating all of these elements into daily life can be daunting.1-3
In fact, nearly half of US adults with diabetes fail to meet the recommended targets.4 This leads to frustration, which often manifests in psychosocial problems that further hamper efforts to manage the disease.5-10 The most notable is a psychosocial disorder known as diabetes distress, which affects close to 45% of those with diabetes.11,12
It is important to note that diabetes distress is not a psychiatric disorder;13 rather, it is a broad affective reaction to the stress of living with this chronic and complex disease.14,15 By negatively affecting adherence to a self-care regimen, diabetes distress contributes to worsening glycemic control and increasing morbidity.16-18
Recognizing that about 80% of those with diabetes are treated in primary care settings,19 we wrote this review to call your attention to diabetes distress, alert you to brief screening tools that can easily be incorporated into clinic visits, and offer guidance in matching proposed interventions to the aspects of diabetes self-management that cause patients the greatest distress.
Diabetes distress: What it is, what it’s not
For patients with type 2 diabetes, diabetes distress centers around 4 main issues:
- frustration with the demands of self-care;
- apprehension about the future and the possibility of developing serious complications;
- concern about both the quality and the cost of required medical care; and
- perceived lack of support from family and/or friends.11,12,20
As mentioned earlier, diabetes distress is not a psychiatric condition and should not be confused with major depressive disorder (MDD). Here’s help in telling the difference.
For starters, a diagnosis of depression is symptom-based.13 MDD requires the presence of at least 5 of the 9 symptoms defined by the Diagnostic and Statistical Manual of Mental Disorders, Fifth ed. (DSM-5)—eg, persistent feelings of worthlessness or guilt, sleep disturbances, lack of interest in normal activities—for at least 2 weeks.21 What’s more, the diagnostic criteria for MDD do not specify a cause or disease process. Nor do they distinguish between a pathological response and an expected reaction to a stressful life event.22 Further, depression measures reflect symptoms (eg, hyperglycemia), as well as stressful experiences resulting from diabetes self-care, which may contribute to the high rate of false positives or incorrect diagnoses of MDD and missed diagnoses of diabetes distress.23
Unlike MDD, diabetes distress has a specific cause—diabetes—and can best be understood as an emotional response to a demanding health condition.13 And, because the source of the problem is identified, diabetes distress can be treated with specific interventions targeting the areas causing the highest levels of stress.
When a psychiatric condition and diabetes distress overlap
MDD, anxiety disorders, and diabetes distress are all common in patients with diabetes,24 and the co-occurrence of a psychiatric disorder and diabetes distress is high.25 Thus, it is important not only to identify cases of diabetes distress but also to consider comorbid depression and/or anxiety in patients with diabetes distress.
More often, though, it is the other way around, according to the Distress and Depression in Diabetes (3D) study. The researchers recently found that 84% of patients with moderate or high diabetes distress did not fulfill the criteria for MDD, but that 67% of diabetes patients with MDD also had moderate or high diabetes distress.13,15,17,25
The data highlight the importance of screening patients with a dual diagnosis of diabetes and MDD for diabetes distress. Keep in mind that individuals diagnosed with both diabetes distress and a comorbid psychiatric condition may require more complex and intensive treatment than those with either diabetes distress or MDD alone.25
Screening for diabetes distress
Diabetes distress can be easily assessed using one of several patient-reported outcome measures. Six validated measures, ranging in length from one to 28 questions, are designed for use in primary care (TABLE).26-30 Some of the measures are easily accessible online; others require subscription to MEDLINE.
Problem Areas in Diabetes (PAID): There are 3 versions of PAID—a 20-item screen assessing a broad range of feelings related to living with diabetes and its treatment, a 5-item version (PAID-5) with high rates of sensitivity (95%) and specificity (89%), and a single-item test (PAID-1) that is highly correlated with the longer version.26,27
Diabetes Distress Scale (DDS): This tool is available in a 17-item measure assessing diabetes distress as it relates to the emotional burden, physician-related distress, regimen-related distress, and interpersonal distress.28 DDS is also available in a short form (DDS-2) with 2 items29 and a 28-item scale specifically for patients with type 1 diabetes.30 T1-DDS, the only diabetes distress measure focused on this particular patient population, assesses the 7 sources of distress found to be common among adults with type 1 diabetes: powerlessness, negative social perceptions, physician distress, friend/family distress, hypoglycemia distress, management distress, and eating distress.
Studies have shown that not only do those with type 1 diabetes experience different stressors compared with their type 2 counterparts, but that they tend to experience distress differently. For patients with type 1 diabetes, for example, powerlessness ranked as the highest source of distress, followed by eating distress and hypoglycemia distress. These sources of distress differ from the regimen distress, emotional burden, interpersonal distress, and physician distress identified by those with type 2 diabetes.30
How to respond to diabetes distress
Diabetes distress is easier to identify than to successfully treat. Few validated treatments for diabetes distress exist and, to our knowledge, only 2 studies have assessed interventions aimed at reduction of such distress.31,32
The REDEEM trial31 recruited adults with type 2 diabetes and diabetes distress to participate in a 12-month randomized controlled trial (RCT). The trial had 3 arms, comparing the effectiveness of a computer-assisted self-management (CASM) program alone, a CASM program plus in-person diabetes distress-specific problem-solving therapy, and a computer-assisted minimally supportive intervention. The main outcomes included diabetes distress (using the DDS scale and subscales), along with self-management behaviors and HbA1c.
Participants in all 3 arms showed significant reductions in total diabetes distress and improvements in self-management behaviors, with no significant differences among the groups. No differences in HbA1c were found. However, those in the CASM program plus distress-specific therapy arm showed a larger reduction in regimen distress compared with the other 2 groups.31
The DIAMOS trial32 recruited adults who had type 1 or type 2 diabetes, diabetes distress, and subclinical depressive symptoms for a 2-arm RCT. One group underwent cognitive behavioral interventions, while the controls had standard group-based diabetes education. The main outcomes included diabetes distress (measured via the PAID scale), depressive symptoms, well-being, diabetes self-care, diabetes acceptance, satisfaction with diabetes treatment, HbA1c, and subclinical inflammation.
The intervention group showed greater improvement in diabetes distress and depressive symptoms compared with the control group, but no differences in well-being, self-care, treatment satisfaction, HbA1c, or subclinical inflammation were observed.32
Both studies support the use of problem-solving therapy and cognitive behavioral interventions for patients with diabetes distress. Future research should evaluate the effectiveness of these interventions in the primary care setting.
What else to offer when challenges mount?
Diabetes is a progressive disease, and most patients experience multiple challenges over time. These typically include complications and comorbidities, physical limitations, polypharmacy, hypoglycemia, and cognitive impairment, as well as changes in everything from medication and lifestyle to insurance coverage and social support.33,34 All increase the risk for diabetes distress, as well as related psychiatric conditions.
Aging and diabetes are independent risk factors for cognitive impairment, for example, and the presence of both increases this risk.35 What’s more, diabetes alone is associated with poorer executive function,36-38 the higher-level cognitive processes that allow individuals to engage in independent, purposeful, and flexible goal-related behaviors. Both poor cognitive function and impaired executive function interfere with the ability to perform self-care behaviors such as adjusting insulin doses, drawing insulin into a syringe, or dialing an insulin dose with an insulin pen.39 This in turn can lead to frustration and increase the likelihood of moderate to high diabetes distress.
Assessing diabetes distress in patients with cognitive impairment, poor executive functioning, or other psychological limitations is particularly difficult, however, as no diabetes distress measures take such deficits into account. Thus, primary care physicians without expertise in neuropsychology should consider referring patients with such problems to specialists for assessment.
The progressive nature of diabetes also highlights the need for primary care physicians to periodically screen for diabetes distress and engage in ongoing discussions about what type of care is best for individual patients, and why. When developing or updating treatment plans and making recommendations, it is crucial to consider the impact the treatment would likely have on the patient’s physical and mental health and to explicitly inquire about and acknowledge his or her values and preferences for care.40-44
It is also important to remain aware of socioeconomic changes—in employment, insurance coverage, and living situations, for example—which are not addressed in the screening tools.
Moderate to high diabetes distress scores, as well as individual items patients identify as “very serious” problems, represent clinical red flags that should be the focus of careful discussion during a medical visit. Patients with moderate to high distress should be referred to a therapist trained in cognitive behavioral therapy or problem-solving therapy. Physicians who lack access to such resources can incorporate cognitive behavioral and problem-solving techniques into patient discussion. (See “Directing help where it’s most needed.”) All patients should be referred to a certified diabetes educator—a key component of diabetes care.45,46
SIDEBAR
Directing help where it's most needed
CASE 1 ›
Conduct a behavioral experiment
Fred J, a 67-year-old diagnosed with type 2 diabetes 6 years ago, comes in for a diabetes check-up. He is a new patient who recently retired from his job as a contractor and was referred by a colleague. In response to a question about his diabetes management, Mr. J tells you he’s having a hard time.
“I get down on myself,” the patient says. “I take my medications every day at the exact same time, but when I test my sugar, it’s 260 or 280. I know I did this to myself. If only I weighed less, ate better, or exercised more.”
At other times, “I think, 'Why bother?'” Mr. J adds. “I feel like there’s nothing I can do to make it better.”
The DDS-2 screen you gave Mr. J bears out his high level of distress and his fear of complications. He tells you about an aunt who “had diabetes like me and had to go on dialysis, then died 2 years later.” When you ask what he fears most, Mr. J says he worries about kidney failure. “I don’t want to go on dialysis,” he insists.
You take the opportunity to point out that nephropathy is not inevitable and that he can perform self-care behaviors now that will prevent or delay kidney complications.
You also decide to try a cognitive behavioral technique in an attempt to change his thought process. You ask Mr. J to agree to a week-long behavioral experiment to examine the effect of walking for 30 minutes each day.
He agrees. You advise him to write down his predictions before he begins the experiment and then to keep a log, checking and recording his glucose levels before and after each walk. You schedule a follow-up visit to discuss the results, hoping that a reduction in blood glucose levels will convince Mr. J that exercise is beneficial to his diabetes.
CASE 2 ›
Identify the problem; brainstorm with the patient
Susan T, a 46-year-old with a husband and 2 teenage children, comes in for her 3-month diabetes check-up. At her last visit, she expressed concerns about her family’s lack of cooperation as she struggled to change her diet. This time, she appears frustrated and distraught.
Your nurse administered the PAID-5 while Ms. T was in the waiting room and entered her score—8, indicating high diabetes distress—in the electronic medical record. You ask Ms. T what’s happening, knowing that encouraging her to verbalize her feelings is a way to increase her trust and help alleviate her concerns.
You also try the following problem-solving technique:
Define the problem. Ms. T is having a hard time maintaining a healthy diet. Her husband and children refuse to eat the healthy meals she prepares and want her to cook separate dinners for them.
Identify challenges. The patient works full-time and does not have the time or energy to cook separate meals. In addition, she is upset by her family’s lack of support in her efforts to control her disease.
Brainstorm multiple solutions:
1) Ms. T can prepare all of her own meals for the work week on Sunday, then cook for the others when she returns from work.
2) Her husband and children can make their own dinner if they do not want to eat the healthier meals she prepares.
3) The patient can join a diabetes support group where she will meet, and possibly learn from, other patients who may be struggling with diabetes self-care.
4) Ms. T can ask her husband and children to come to her next diabetes check-up so they can learn about the importance of family support in diabetes management directly from you.
5) The patient’s family can receive information about a healthy diabetes diet from a certified diabetes educator.
Decide on appropriate solutions. The patient agrees to try and prepare her weekday meals on Sunday so that she is not tempted to eat less healthy options. She also agrees to bring her family to her next diabetes check-up and to diabetes education classes.
CORRESPONDENCE
Elizabeth A. Beverly, PhD, Department of Family Medicine, Ohio University Heritage College of Osteopathic Medicine, 35 W. Green Drive, Athens, OH 45701; [email protected].
1. Gafarian CT, Heiby EM, Blair P, et al. The diabetes time management questionnaire. Diabetes Educator. 1999;25:585-592.
2. Wdowik MJ, Kendall PA, Harris MA. College students with diabetes: using focus groups and interviews to determine psychosocial issues and barriers to control. Diabetes Educator. 1997;23:558-562.
3. Rubin RR. Psychological issues and treatment for people with diabetes. J Clin Psych. 2001;57:457-478.
4. Ali MK, Bullard KM, Gregg EW. Achievement of goals in US diabetes care, 1999-2010. New Engl J Med. 2013;369:287-288.
5. Lloyd CE, Smith J, Weinger K. Stress and diabetes: Review of the links. Diabetes Spectrum. 2005;18:121-127.
6. Weinger K. Psychosocial issues and self-care. Am J Nurs. 2007;107(6 suppl): S34-S38.
7. Weinger K, Jacobson AM. Psychosocial and quality of life correlates of glycemic control during intensive treatment of type 1 diabetes. Patient Education Counseling. 2001;42:123-131.
8. Albright TL, Parchman M, Burge SK. Predictors of self-care behavior in adults with type 2 diabetes: an RRNeST study. Fam Med. 2001;33:354-360.
9. Gonzalez JS, Safren SA, Cagliero E, et al. Depression, self-care, and medication adherence in type 2 diabetes: relationships across the full range of symptom severity. Diabetes Care. 2007;30:2222-2227.
10. Gonzalez JS, Safren SA, Delahanty LM, et al. Symptoms of depression prospectively predict poorer self-care in patients with Type 2 diabetes. Diabetic Med. 2008;25:1102-1107.
11. Nicolucci A, Kovacs Burns K, Holt RI, et al. Diabetes Attitudes, Wishes and Needs second study (DAWN2): cross-national benchmarking of diabetes-related psychosocial outcomes for people with diabetes. Diabetic Med. 2013;30:767-777.
12. Fisher L, Hessler DM, Polonsky W, et al. When is diabetes distress clinically meaningful?: establishing cut points for the Diabetes Distress Scale. Diabetes Care. 2012;35:259-264.
13. Fisher L, Gonzalez JS, Polonsky WH. The confusing tale of depression and distress in patients with diabetes: a call for greater clarity and precision. Diabetic Med. 2014;31:764-772.
14. Fisher L, Mullan JT, Skaff MM, et al. Predicting diabetes distress in patients with Type 2 diabetes: a longitudinal study. Diabetic Med. 2009;26:622-627.
15. Fisher L, Skaff MM, Mullan JT, et al. Clinical depression versus distress among patients with type 2 diabetes: not just a question of semantics. Diabetes Care. 2007;30:542-548.
16. Gonzalez JS, Delahanty LM, Safren SA, et al. Differentiating symptoms of depression from diabetes-specific distress: relationships with self-care in type 2 diabetes. Diabetologia. 2008;51:2822-1825.
17. Fisher L, Mullan JT, Arean P, et al. Diabetes distress but not clinical depression or depressive symptoms is associated with glycemic control in both cross-sectional and longitudinal analyses. Diabetes Care. 2010;33:23-28.
18. Fisher EB, Thorpe CT, Devellis BM, et al. Healthy coping, negative emotions, and diabetes management: a systematic review and appraisal. Diabetes Educator. 2007;33:1080-1103; 1104-1086.
19. Peterson KA, Radosevich DM, O’Connor PJ, et al. Improving diabetes care in practice: findings from the TRANSLATE trial. Diabetes Care. 2008;31:2238-2243.
20. Fisher L, Glasgow RE, Strycker LA. The relationship between diabetes distress and clinical depression with glycemic control among patients with type 2 diabetes. Diabetes Care. 2010;33:1034-1036.
21. Cole J, McGuffin P, Farmer AE. The classification of depression: are we still confused? Br J Psychiatr. 2008;192:83-85.
22. Wakefield JC. The concept of mental disorder. On the boundary between biological facts and social values. Am Psychologist. 1992;47:373-388.
23. Fisher L, Gonzalez JS, Polonsky WH. The confusing tale of depression and distress in patients with diabetes: a call for greater clarity and precision. Diabetic Med. 2014;31:764-772.
24. Ciechanowski PS, Katon WJ, Russo JE. Depression and diabetes: impact of depressive symptoms on adherence, function, and costs. Arch Intern Med. 2000;160:3278-3285.
25. Fisher L, Skaff MM, Mullan JT, et al. A longitudinal study of affective and anxiety disorders, depressive affect and diabetes distress in adults with Type 2 diabetes. Diabetic Med. 2008;25:1096-1101.
26. Polonsky WH, Anderson BJ, Lohrer PA, et al. Assessment of diabetes-related distress. Diabetes Care. 1995;18:754-760.
27. McGuire BE, Morrison TG, Hermanns N, et al. Short-form measures of diabetes-related emotional distress: the Problem Areas in Diabetes Scale (PAID)-5 and PAID-1. Diabetologia. 2010;53:66-69.
28. Polonsky WH, Fisher L, Earles J, et al. Assessing psychosocial distress in diabetes: development of the diabetes distress scale. Diabetes Care. 2005;28:626-631.
29. Fisher L, Glasgow RE, Mullan JT, et al. Development of a brief diabetes distress screening instrument. Ann Fam Med. 2008;6:246-252.
30. Fisher L, Polonsky WH, Hessler DM, et al. Understanding the sources of diabetes distress in adults with type 1 diabetes. J Diabetes Complications. 2015;29:572-577.
31. Fisher L, Hessler D, Glasgow RE, et al. REDEEM: a pragmatic trial to reduce diabetes distress. Diabetes Care. 2013;36:2551-2558.
32. Hermanns N, Schmitt A, Gahr A, et al. The effect of a Diabetes-Specific Cognitive Behavioral Treatment Program (DIAMOS) for patients with diabetes and subclinical depression: results of a randomized controlled trial. Diabetes Care. 2015;38:551-560.
33. Weinger K, Beverly EA, Smaldone A. Diabetes self-care and the older adult. Western J Nurs Res. 2014;36:1272-1298.
34. Beverly EA, Ritholz MD, Shepherd C, et al. The psychosocial challenges and care of older adults with diabetes: “can’t do what I used to do; can’t be who I once was.” Curr Diabetes Rep. 2016;16:48.
35. Lu FP, Lin KP, Kuo HK. Diabetes and the risk of multi-system aging phenotypes: a systematic review and meta-analysis. PloS One. 2009;4:e4144.
36. Thabit H, Kyaw TT, McDermott J, et al. Executive function and diabetes mellitus—a stone left unturned? Curr Diabetes Rev. 2012;8:109-115.
37. McNally K, Rohan J, Pendley JS, et al. Executive functioning, treatment adherence, and glycemic control in children with type 1 diabetes. Diabetes Care. 2010;33:1159-1162.
38. Rucker JL, McDowd JM, Kluding PM. Executive function and type 2 diabetes: putting the pieces together. Phys Ther. 2012;92:454-462.
39. Kirkman MS, Briscoe VJ, Clark N, et al. Diabetes in older adults. Diabetes Care. 2012;35:2650-2664.
40. Durso SC. Using clinical guidelines designed for older adults with diabetes mellitus and complex health status. JAMA. 2006;295:1935-1940.
41. Oftedal B, Karlsen B, Bru E. Life values and self-regulation behaviours among adults with type 2 diabetes. J Clin Nurs. 2010;19:2548-2556.
42. Morrow AS, Haidet P, Skinner J, et al. Integrating diabetes self-management with the health goals of older adults: a qualitative exploration. Patient Education Counseling. 2008;72:418-423.
43. Huang ES, Gorawara-Bhat R, Chin MH. Self-reported goals of older patients with type 2 diabetes mellitus. J Am Geriatr Soc. 2005;53:306-311.
44. Beverly EA, Wray LA, LaCoe CL, et al. Listening to older adults’ values and preferences for Type 2 diabetes care: a qualitative study. Diabetes Spectrum. 2014;27:44-49.
45. American Association of Diabetes Educators. Why refer for diabetes education? American Association of Diabetes Educators. Available at: https://www.diabeteseducator.org/practice/provider-resources/why-refer-for-diabetes-education. Accessed August 15, 2016.
46. Ismail K, Winkley K, Rabe-Hesketh S. Systematic review and meta-analysis of randomised controlled trials of psychological interventions to improve glycaemic control in patients with type 2 diabetes. Lancet. 2004;363:1589-1597.
1. Gafarian CT, Heiby EM, Blair P, et al. The diabetes time management questionnaire. Diabetes Educator. 1999;25:585-592.
2. Wdowik MJ, Kendall PA, Harris MA. College students with diabetes: using focus groups and interviews to determine psychosocial issues and barriers to control. Diabetes Educator. 1997;23:558-562.
3. Rubin RR. Psychological issues and treatment for people with diabetes. J Clin Psych. 2001;57:457-478.
4. Ali MK, Bullard KM, Gregg EW. Achievement of goals in US diabetes care, 1999-2010. New Engl J Med. 2013;369:287-288.
5. Lloyd CE, Smith J, Weinger K. Stress and diabetes: Review of the links. Diabetes Spectrum. 2005;18:121-127.
6. Weinger K. Psychosocial issues and self-care. Am J Nurs. 2007;107(6 suppl): S34-S38.
7. Weinger K, Jacobson AM. Psychosocial and quality of life correlates of glycemic control during intensive treatment of type 1 diabetes. Patient Education Counseling. 2001;42:123-131.
8. Albright TL, Parchman M, Burge SK. Predictors of self-care behavior in adults with type 2 diabetes: an RRNeST study. Fam Med. 2001;33:354-360.
9. Gonzalez JS, Safren SA, Cagliero E, et al. Depression, self-care, and medication adherence in type 2 diabetes: relationships across the full range of symptom severity. Diabetes Care. 2007;30:2222-2227.
10. Gonzalez JS, Safren SA, Delahanty LM, et al. Symptoms of depression prospectively predict poorer self-care in patients with Type 2 diabetes. Diabetic Med. 2008;25:1102-1107.
11. Nicolucci A, Kovacs Burns K, Holt RI, et al. Diabetes Attitudes, Wishes and Needs second study (DAWN2): cross-national benchmarking of diabetes-related psychosocial outcomes for people with diabetes. Diabetic Med. 2013;30:767-777.
12. Fisher L, Hessler DM, Polonsky W, et al. When is diabetes distress clinically meaningful?: establishing cut points for the Diabetes Distress Scale. Diabetes Care. 2012;35:259-264.
13. Fisher L, Gonzalez JS, Polonsky WH. The confusing tale of depression and distress in patients with diabetes: a call for greater clarity and precision. Diabetic Med. 2014;31:764-772.
14. Fisher L, Mullan JT, Skaff MM, et al. Predicting diabetes distress in patients with Type 2 diabetes: a longitudinal study. Diabetic Med. 2009;26:622-627.
15. Fisher L, Skaff MM, Mullan JT, et al. Clinical depression versus distress among patients with type 2 diabetes: not just a question of semantics. Diabetes Care. 2007;30:542-548.
16. Gonzalez JS, Delahanty LM, Safren SA, et al. Differentiating symptoms of depression from diabetes-specific distress: relationships with self-care in type 2 diabetes. Diabetologia. 2008;51:2822-1825.
17. Fisher L, Mullan JT, Arean P, et al. Diabetes distress but not clinical depression or depressive symptoms is associated with glycemic control in both cross-sectional and longitudinal analyses. Diabetes Care. 2010;33:23-28.
18. Fisher EB, Thorpe CT, Devellis BM, et al. Healthy coping, negative emotions, and diabetes management: a systematic review and appraisal. Diabetes Educator. 2007;33:1080-1103; 1104-1086.
19. Peterson KA, Radosevich DM, O’Connor PJ, et al. Improving diabetes care in practice: findings from the TRANSLATE trial. Diabetes Care. 2008;31:2238-2243.
20. Fisher L, Glasgow RE, Strycker LA. The relationship between diabetes distress and clinical depression with glycemic control among patients with type 2 diabetes. Diabetes Care. 2010;33:1034-1036.
21. Cole J, McGuffin P, Farmer AE. The classification of depression: are we still confused? Br J Psychiatr. 2008;192:83-85.
22. Wakefield JC. The concept of mental disorder. On the boundary between biological facts and social values. Am Psychologist. 1992;47:373-388.
23. Fisher L, Gonzalez JS, Polonsky WH. The confusing tale of depression and distress in patients with diabetes: a call for greater clarity and precision. Diabetic Med. 2014;31:764-772.
24. Ciechanowski PS, Katon WJ, Russo JE. Depression and diabetes: impact of depressive symptoms on adherence, function, and costs. Arch Intern Med. 2000;160:3278-3285.
25. Fisher L, Skaff MM, Mullan JT, et al. A longitudinal study of affective and anxiety disorders, depressive affect and diabetes distress in adults with Type 2 diabetes. Diabetic Med. 2008;25:1096-1101.
26. Polonsky WH, Anderson BJ, Lohrer PA, et al. Assessment of diabetes-related distress. Diabetes Care. 1995;18:754-760.
27. McGuire BE, Morrison TG, Hermanns N, et al. Short-form measures of diabetes-related emotional distress: the Problem Areas in Diabetes Scale (PAID)-5 and PAID-1. Diabetologia. 2010;53:66-69.
28. Polonsky WH, Fisher L, Earles J, et al. Assessing psychosocial distress in diabetes: development of the diabetes distress scale. Diabetes Care. 2005;28:626-631.
29. Fisher L, Glasgow RE, Mullan JT, et al. Development of a brief diabetes distress screening instrument. Ann Fam Med. 2008;6:246-252.
30. Fisher L, Polonsky WH, Hessler DM, et al. Understanding the sources of diabetes distress in adults with type 1 diabetes. J Diabetes Complications. 2015;29:572-577.
31. Fisher L, Hessler D, Glasgow RE, et al. REDEEM: a pragmatic trial to reduce diabetes distress. Diabetes Care. 2013;36:2551-2558.
32. Hermanns N, Schmitt A, Gahr A, et al. The effect of a Diabetes-Specific Cognitive Behavioral Treatment Program (DIAMOS) for patients with diabetes and subclinical depression: results of a randomized controlled trial. Diabetes Care. 2015;38:551-560.
33. Weinger K, Beverly EA, Smaldone A. Diabetes self-care and the older adult. Western J Nurs Res. 2014;36:1272-1298.
34. Beverly EA, Ritholz MD, Shepherd C, et al. The psychosocial challenges and care of older adults with diabetes: “can’t do what I used to do; can’t be who I once was.” Curr Diabetes Rep. 2016;16:48.
35. Lu FP, Lin KP, Kuo HK. Diabetes and the risk of multi-system aging phenotypes: a systematic review and meta-analysis. PloS One. 2009;4:e4144.
36. Thabit H, Kyaw TT, McDermott J, et al. Executive function and diabetes mellitus—a stone left unturned? Curr Diabetes Rev. 2012;8:109-115.
37. McNally K, Rohan J, Pendley JS, et al. Executive functioning, treatment adherence, and glycemic control in children with type 1 diabetes. Diabetes Care. 2010;33:1159-1162.
38. Rucker JL, McDowd JM, Kluding PM. Executive function and type 2 diabetes: putting the pieces together. Phys Ther. 2012;92:454-462.
39. Kirkman MS, Briscoe VJ, Clark N, et al. Diabetes in older adults. Diabetes Care. 2012;35:2650-2664.
40. Durso SC. Using clinical guidelines designed for older adults with diabetes mellitus and complex health status. JAMA. 2006;295:1935-1940.
41. Oftedal B, Karlsen B, Bru E. Life values and self-regulation behaviours among adults with type 2 diabetes. J Clin Nurs. 2010;19:2548-2556.
42. Morrow AS, Haidet P, Skinner J, et al. Integrating diabetes self-management with the health goals of older adults: a qualitative exploration. Patient Education Counseling. 2008;72:418-423.
43. Huang ES, Gorawara-Bhat R, Chin MH. Self-reported goals of older patients with type 2 diabetes mellitus. J Am Geriatr Soc. 2005;53:306-311.
44. Beverly EA, Wray LA, LaCoe CL, et al. Listening to older adults’ values and preferences for Type 2 diabetes care: a qualitative study. Diabetes Spectrum. 2014;27:44-49.
45. American Association of Diabetes Educators. Why refer for diabetes education? American Association of Diabetes Educators. Available at: https://www.diabeteseducator.org/practice/provider-resources/why-refer-for-diabetes-education. Accessed August 15, 2016.
46. Ismail K, Winkley K, Rabe-Hesketh S. Systematic review and meta-analysis of randomised controlled trials of psychological interventions to improve glycaemic control in patients with type 2 diabetes. Lancet. 2004;363:1589-1597.
PRACTICE RECOMMENDATIONS
› Educate patients about diabetes distress, explaining that diabetes is manageable and that neither complications nor diabetes distress is inevitable. C
› Empower patients to take an active role in self-management of diabetes, encouraging them to express their concerns and ask open-ended questions. A
› Support shared decision-making by inquiring about patients’ values and treatment preferences, presenting options, and reviewing the risks and benefits of each. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Atrial fibrillation: Effective strategies using the latest tools
Atrial fibrillation (AF)—the most common supraventricular tachycardia—affects as many as 6.1 million adults in the United States.1 It is associated with a 5-fold increased risk of stroke,2 a 3-fold increased risk of heart failure (HF),3 and about a 2-fold increased risk of dementia4 and mortality.2 The prevalence of AF increases with maturity, from 2% in people <65 years of age to 9% in those ≥65 years,5 and that prevalence is expected to double over the next 25 years as the population ages.1
The primary goals of treatment are to alleviate symptoms and prevent thromboembolism. Strokes related to AF are more likely to result in severe disability or death when compared with those unrelated to AF.6 And yet anticoagulation remains underutilized.7
The net clinical benefit of oral anticoagulation appears to be greatest in patients with the highest risk of bleeding, since these patients are also at the highest risk for stroke.8 Patients at increased risk of stroke are more likely to receive oral anticoagulation; however, for unknown reasons, more than half of people with the highest risk of stroke are not prescribed these important anti-blood-clotting medications.7 One theory is that physicians may be relying on their gut rather than objective risk scores, and underuse of validated schemata leads to poor estimation of risk.
For example, results from the ORBIT-AF (Outcomes Registry for Better Informed Treatment of Atrial Fibrillation) trial, which involved over 10,000 people with AF, found that although 72% (n=7251) had high-risk CHADS2 scores (≥2), only 16% were assessed as having a high risk of stroke by physicians.9 Along the same lines, a recent study of Canadian primary care physicians showed that stroke risk and bleeding risk were not evaluated with validated tools in 58% and 81% of patients, respectively, leading to both significant underestimation and overestimation of risk.10
This review provides the tools to identify when anticoagulation is indicated, reports the advantages and disadvantages of the currently available anticoagulants, and discusses the selection and implementation of rate- vs rhythm-control strategies. But first, a word about the etiology, classification, and diagnosis of AF.
AF: The result of any number of cardiac and non-cardiac causes
AF is characterized by uncoordinated activation of the atria, which results in ineffective atrial contractions and an irregular, often rapid, ventricular response. It is the ultimate clinical manifestation of multiple diseases that alter atrial tissue through inflammation, fibrosis, or hypertrophy.5 The most common causes are hypertension, coronary artery disease, HF, cardiomyopathies, and valvular heart disease, all of which stimulate the renin-angiotensin-aldosterone system, leading to increased susceptibility to arrhythmia.5 Atrial ectopic tachycardia, Wolff-Parkinson-White (WPW) syndrome, and atrioventricular (AV) nodal reentrant tachycardia also may precipitate AF.5 In these cases, AF usually resolves after catheter ablation (CA) of the primary arrhythmia.11 Unrecognized AF may trigger atrial flutter, and more than 80% of patients who undergo radiofrequency ablation for atrial flutter experience AF at some point in the subsequent 5 years.12
Non-cardiac causes of AF include sleep apnea, obesity, hyperthyroidism, drugs, electrocution, pneumonia, and pulmonary embolism.5 An association between binge drinking and AF (“holiday heart syndrome”) has long been recognized. The evidence now suggests that alcohol increases the risk of AF in a dose-dependent manner with intakes of ≥1 drink per day (12 g per drink).13
Classification schema no longer includes “lone AF”
AF is classified in terms of the duration of episodes:5
- Paroxysmal AF is characterized by brief episodes that terminate spontaneously or with intervention within 7 days of onset. These episodes recur with variable frequency.
- Persistent AF refers to AF that is continuously sustained for more than 7 days.
- Longstanding persistent AF refers to continuous AF that lasts longer than 12 months.
- Permanent AF is not an inherent pathophysiologic attribute of AF, but rather an acceptance of AF where the patient and physician abandon further efforts to restore and/or maintain sinus rhythm.
- Nonvalvular AF occurs in the absence of a valve replacement (mechanical or bioprosthetic), rheumatic mitral stenosis, or mitral valve repair.
Although paroxysmal and persistent AF may occur in the same individual, the distinction is still clinically relevant, as outcomes of certain therapies, such as CA, are superior in patients with paroxysmal AF.14 With a more complete understanding of AF pathophysiology, guidelines now discourage use of the potentially confusing term “lone AF,” which has historically been applied to younger patients with no known clinical risk factors or echocardiographic abnormalities. As a result, therapeutic decisions are no longer based on this nomenclature, according to the 2014 AF practice guideline from the American College of Cardiology (ACC)/American Heart Association (AHA)/Heart Rhythm Society (HRS).5
Patient complaints—or incidental findings—can prompt a Dx
Fatigue is the most common symptom of AF. Other signs and symptoms include palpitations, dyspnea, HF, hypotension, syncope, chest pain, and stroke. Some patients are asymptomatic, and AF is an incidental finding when an irregular pulse is discovered during a physical examination. The diagnosis is confirmed by electrocardiogram (EKG), telemetry, Holter monitor, event recorder, or an implanted electrocardiographic recording device. A chest x-ray, serum electrolyte levels, a complete blood count, thyroid testing, and renal and hepatic function testing are recommended. Transthoracic echocardiography to measure cardiac function, detect underlying structural heart disease, and evaluate atrial size is essential.5
An electrophysiologic (EP) study may be needed for diagnosis or treatment if another arrhythmia is present. Aberrant conduction may cause AF to present as a wide complex tachycardia and be mislabeled as ventricular tachycardia. The presence of delta waves is an indication for an EP study targeting the WPW accessory pathway. Transesophageal echocardiography (TEE) is the most sensitive and specific test for left atrial thrombi. If you are considering a TEE for a patient with AF of unknown, or >48 hours’, duration who has not been anticoagulated in the preceding 3 weeks, obtain it before performing cardioversion because of the risk of embolism.5
Stroke prevention
The ACC/AHA/HRS AF guideline recommends basing anticoagulation decisions on thromboembolic risk, regardless of AF pattern (paroxysmal, persistent, or permanent) (Class I recommendation).5 For patients with nonvalvular AF and atrial flutter, the guideline recommends using the Birmingham 2009 schema (CHA2DS2-VASc score) (TABLE 115-18) to estimate thromboembolic risk.5,15 CHA2DS2-VASc improves on the older CHADS2 score by significantly reducing the number of patients categorized as having intermediate risk and better identifying truly low-risk patients who are unlikely to benefit from anticoagulation.16,17,19
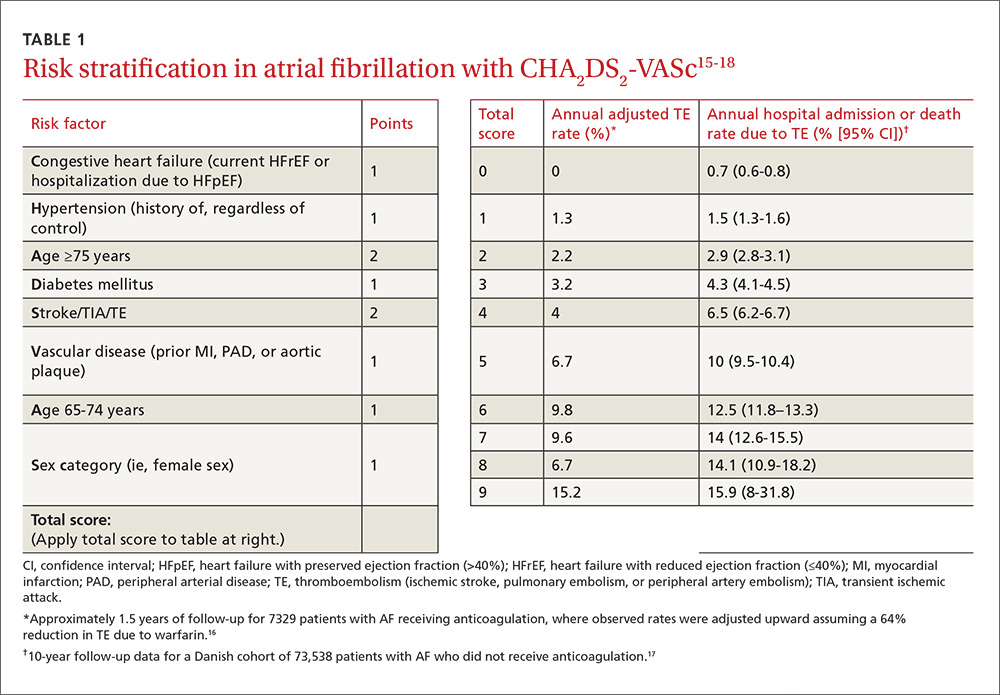
Men with a CHA2DS2-VASc score of zero and women with a score of one do not need anticoagulation.5,20 Discuss the risks and benefits of oral anticoagulation with men who have a score of one. In these intermediate-risk men, antiplatelet therapy with aspirin and/or clopidogrel may be reasonable, especially if there is an indication other than stroke prevention (eg, post-myocardial infarction). Oral anticoagulation is strongly recommended for all patients with a CHA2DS2-VASc score of 2 or higher.5,18,21,22
Anticoagulant considerations: Warfarin vs DOACs
Warfarin was the gold standard for stroke prevention in nonvalvular AF until the direct oral anticoagulants (DOACs) became available in 2010. Guidelines in the United States and the United Kingdom recommend shared decision-making to help patients with AF who do not have a specific indication for warfarin choose between warfarin and the DOACs.5,21 Canadian and European guidelines recommend DOACs as the first-line option for anticoagulation and reserve warfarin for patients who have contraindications to, or are unable to afford, DOACs.18,22 All current guidelines recommend continuing warfarin in patients who are stable, well controlled, and satisfied with warfarin therapy and the monitoring and dietary restrictions it entails.
DOACs are as effective as warfarin. All of the DOACs are approved for stroke prevention based on individual phase III non-inferiority trials in which they were compared to warfarin.23-26 In addition, a meta-analysis of these 4 trials involving a total of 71,683 patients (mean age 70-73 years; median follow-up, 1.8-2.8 years) evaluated the benefits and risks of the 4 DOACs against the former gold standard.27
Higher doses of the DOACs (dabigatran 150 mg BID, rivaroxaban 20 mg/d, edoxaban 60 mg/d, and apixaban 5 mg BID) reduced the rates of stroke or systemic embolism (relative risk [RR]=0.81; 95% confidence interval [CI], 0.73-0.91; P<.0001; number needed to treat [NNT]=147), hemorrhagic stroke (RR=0.49; 95% CI, 0.38-0.64; P<.0001; NNT=219), and all-cause mortality (RR=0.90; 95% CI, 0.85-0.95; P=.0003; NNT=128), compared with warfarin.27 It is important to note that while lower doses of some DOACs (dabigatran 110 mg BID and edoxaban 30 mg/d) were not as effective at preventing ischemic stroke when compared with warfarin (RR=1.3; 95% CI, 1-1.6; P=.045), they still significantly reduced hemorrhagic stroke (RR=0.33; 95% CI, 0.23-0.46; P<.0001) and all-cause mortality (RR=0.89; 95% CI, 0.83-0.96; P=.003).
Of course, the biggest concern is bleeding. In that same meta-analysis, the difference in major bleeding events with DOACs vs warfarin was not statistically significant (RR=0.86; 95% CI, 0.73-1; P=.06). While DOACs likely lower rates of intracranial hemorrhage (RR=0.48; 95% CI, 0.39-0.59; P<.0001; NNT=132), they seem to increase the risk of gastrointestinal (GI) bleeding (RR=1.3; 95% CI, 1-1.6; P=.043; number needed to harm [NNH]=185).27
There was significant heterogeneity in the GI bleeding outcome, however. When compared with warfarin, GI bleeding was increased by dabigatran 150 mg BID (RR=1.5; 95% CI, 1.2-1.9; P<.001) and edoxaban 60 mg/d (HR=1.2; 95% CI, 1.02-1.5; P=.03), but there were no significant differences for dabigatran 110 mg BID or apixaban 5 mg BID.23,25,26
On the other hand, edoxaban 30 mg/d had a lower risk of GI bleeding when compared with warfarin (HR=0.67; 95% CI, 0.53-0.83; P<.001).25 Without head-to-head trials, it is impossible to know if one DOAC is superior to another. Apixaban 5 mg BID appears to offer the best overall balance between efficacy and safety. Other DOACs may be better options for patients who have specific concerns regarding efficacy or safety.28,29
Convenience, interactions, and cost may be the deciding factors. Since all DOACs are fairly comparable in efficacy and safety, other factors such as convenience, interactions with other medications, and cost should be considered when deciding on a medication for an individual patient (TABLE 230,31). The DOACs require no lab monitoring or dose titration, and all 4 have fewer potential drug interactions than warfarin.30 Due to their relatively short half-lives, strict adherence is critical; DOACs are not suitable for patients who frequently miss doses.5 (For more information on starting or switching to DOACs, see, “Is a novel anticoagulant right for your patient?” J Fam Pract. 2014;63:22-28.)

A word about DOACs and renal impairment. Another concern with DOACs is their reliance on renal metabolism and excretion. A meta-analysis of the 4 phase III trials of the DOACs, this time involving 58,338 patients, evaluated DOAC efficacy and safety compared to warfarin in the presence of kidney dysfunction.32 Renal function was categorized as normal (estimated glomerular filtration rate [eGFR] >80 mL/min/1.73 m2), mildly impaired (eGFR 50-80 mL/min/1.73 m2), or moderately impaired (eGFR <50 mL/min/1.73m2). Compared with warfarin, DOACs lowered stroke risk in patients with mild (RR=0.71; 95% CI, 0.62-0.81) or moderate (RR=0.79; 95% CI, 0.66-0.94) renal impairment. DOACs also reduced major bleeding compared to warfarin in patients with mild (RR=0.88; 95% CI, 0.80-0.97) or moderate (RR=0.80; 95% CI, 0.66-0.94) renal impairment. How the DOACs fare in patients with severe renal dysfunction could not be determined because such patients were excluded from the trials.
Keep in mind that the DOACs require dose adjustment at different levels of renal impairment (TABLE 230,31), and warfarin remains the only recommended treatment for patients with severe renal impairment, according to both AHA/ACC/HRS and European Society of Cardiology guidelines.5,18
Tools to help assess patients’ bleeding risk
Of the available scoring mechanisms to identify risk factors for bleeding, 3 have been specifically validated in AF populations (ie, ATRIA,33 HEMORR2HAGES,34 and HAS-BLED35). Of the 3, HAS-BLED is superior,36 the most practical, and recommended by expert guidelines.18,21,22 Additionally, HAS-BLED has good correlation with intracranial hemorrhage risk. The HAS-BLED score ranges from 0 to 9 points with one point assigned for each of the following:35
- Hypertension–uncontrolled with systolic BP >160 mm Hg
- Abnormal liver function–cirrhosis, bilirubin >2× normal, or liver enzymes >3× normal
- Abnormal renal function–dialysis, transplant, or serum creatinine >2.26 mg/dL
- Stroke history–including lacunar infarcts
- Bleeding predisposition–history of major bleeding due to any cause
- Labile international normalized ratio (INR)–time in therapeutic range <60%
- Elderly–age >65 years
- Drug–antiplatelet agents, including nonsteroidal anti-inflammatory drugs
- Alcohol usage–>8 drinks per week.
Patients with a HAS-BLED score ≥3 warrant additional monitoring and attempts to reduce bleeding risk by addressing modifiable risk factors. Bleeding risk scores should not be used to exclude patients from anticoagulation therapy.5 In fact, the British National Institute for Health and Clinical Excellence (NICE) guidelines state that anticoagulation should not be withheld solely due to fall risk.21
Also, anticoagulation with warfarin should not be permanently discontinued because of a single GI bleed, since restarting warfarin is associated with decreased risks of thromboembolism and mortality and a statistically insignificant increase in recurrent GI bleeding.37 Restarting DOAC therapy following a GI bleed has not been evaluated in clinical trials; however, it may be reasonable to use one of the DOAC doses with a lower risk of GI bleeding (dabigatran 110 mg BID, apixaban 5 mg BID, or edoxaban 30 mg/d) in patients who have experienced a GI bleed on warfarin or another DOAC.18,22
An online calculator is available that uses CHA2DS2-VASc and HAS-BLED scores to determine an individual’s risk/benefit profile with the various anticoagulation strategies available (http://www.sparctool.com). Consider percutaneous left atrial appendage occlusion if the risks of anticoagulation truly exceed the benefits.38
Rate control vs rhythm control
Most patients who present with AF require immediate ventricular rate control to reduce symptoms. In the acute setting, this can be accomplished with intravenous (IV) beta-blockers or IV calcium channel antagonists.5,39 If the patient is hemodynamically unstable, urgent direct-current cardioversion is the preferred treatment strategy and should not be delayed pending anticoagulation. IV amiodarone can be used in the ICU patient who does not require cardioversion, but is unable to tolerate beta-blockers or calcium channel antagonists.40 Once the patient is stable, long-term treatment focuses on ventricular rate control or restoration and maintenance of sinus rhythm.
The AFFIRM (Atrial Fibrillation Follow-up Investigation of Rhythm Management) trial enrolled 4060 patients (mean age 70 years, mean follow-up 3.5 years) with paroxysmal and persistent AF and randomized them to either pharmacologic rate control or rhythm control.41 No significant differences were found in all-cause mortality or in the composite secondary endpoint of death, ischemic stroke, anoxic encephalopathy, major bleeding, or cardiac arrest. In addition, no significant differences emerged in quality of life or global functional status. The number of patients requiring hospitalization during follow-up was significantly lower in the rate-control group vs the rhythm-control group (73% vs 80%; P<.001). Anticoagulation was encouraged but not mandated in the rhythm-control group after 4 weeks in sinus rhythm, and there was a trend toward higher mortality in the rhythm-control group (27% vs 26%; P=.08).
Patients <65 years were excluded from the AFFIRM trial. When younger patients experience significant symptoms, early referral to Cardiology should be considered to discuss the long-term benefits and risks of a rhythm-control strategy. Regardless of age, when patients remain symptomatic despite rate- or rhythm-control management, the strategy should be changed.5
Rate-control targets and options
Target heart rates should be individualized. The 2014 ACC/AHA/HRS guideline recommends a resting target heart rate <80 beats per minute (bpm) in symptomatic patients.5 In patients with permanent AF who remain asymptomatic at higher resting heart rates, a more lenient rate-control strategy (resting heart rate <110 bpm) has demonstrated outcomes equivalent to those of a more strict approach (resting heart rate <80 bpm and heart rate during moderate exercise <110 bpm).42 Pharmacologic rate-control options include beta-blockers, non-dihydropyridine calcium channel antagonists, and digoxin (TABLE 35). Digoxin is associated with increased all-cause mortality in patients with AF regardless of HF status (HR=1.4; 95% CI, 1.2-1.6, P=.0001).43 Digoxin should be reserved for patients who are sedentary or have inadequate control with first-line medications.5
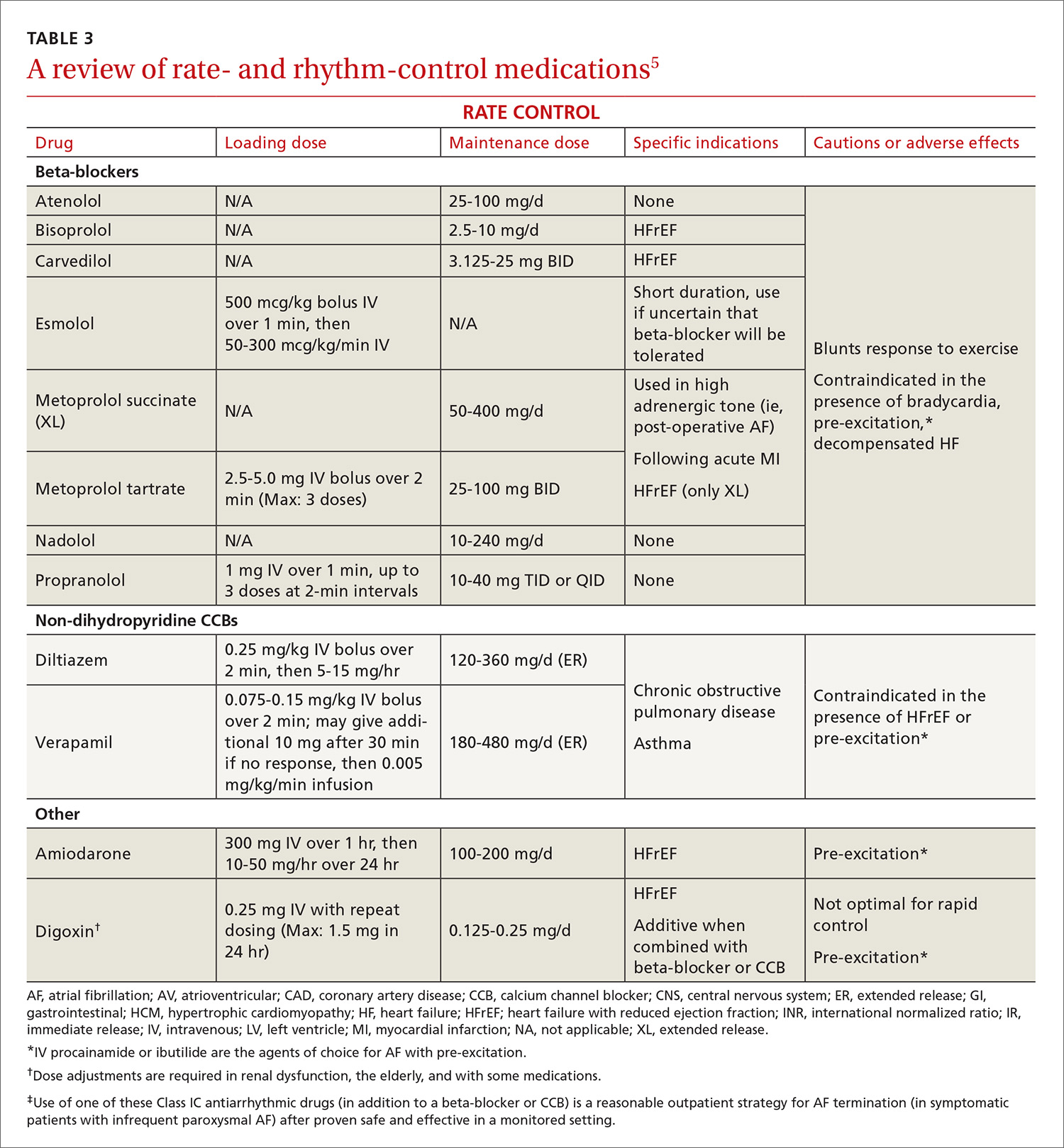
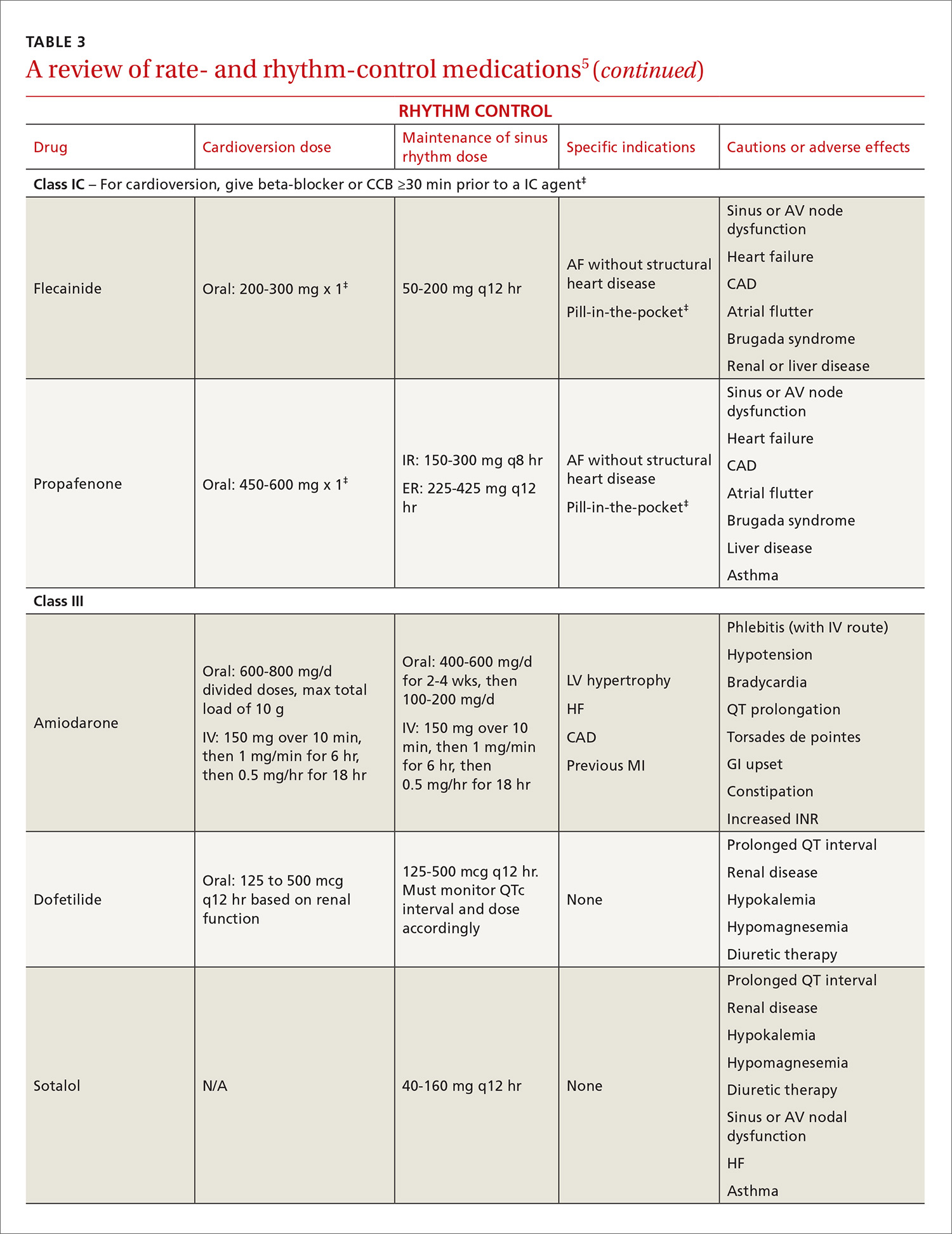
Indications for rhythm control
The NICE guidelines, which are consistent with the ACC/AHA/HRS guidelines, recommend rate control as the first-line strategy for AF management, except in people:21
- whose AF has a reversible cause
- who have HF believed to be primarily caused by AF
- with new-onset AF
- with atrial flutter that is considered suitable for an ablation strategy to restore sinus rhythm
- for whom a rhythm-control strategy would be more suitable based on clinical judgment.
In addition, patients who continue to experience symptomatic AF despite an adequate trial of rate control should be offered rhythm control.5
Pharmacologic rhythm-control strategies. Antiarrhythmic drugs can be used for chemical cardioversion, reduction of paroxysms, and long-term maintenance of sinus rhythm. The most commonly used antiarrhythmic drugs are Class IC and Class III agents (TABLE 3).5 Tailored drug selection for each patient is key. Patients with left atrial diameters >4.5 cm are less likely to remain in sinus rhythm, and patients with left ventricular hypertrophy are at increased risk for proarrhythmic adverse effects.44 Patients with paroxysmal AF may be candidates for a “pill-in-the-pocket” strategy using propafenone or flecainide.5
AF frequently progresses from paroxysmal to persistent and can subsequently result in electrical and structural remodeling that becomes irreversible over time.45 The patient with uncontrolled symptoms despite attempts at rate control and rhythm control should be promptly referred to an electrophysiologist.
Surgical interventions for rate or rhythm control
Electrophysiology interventions include AV nodal ablation with pacemaker placement for rate control, or catheter-directed ablation (radiofrequency or cryotherapy) for rhythm control. CA appears to be more effective than pharmacologic rhythm control.46,47 Treatment with CA is indicated for symptomatic paroxysmal AF when a rhythm-control strategy is desired and the AF is refractory to, or the patient is intolerant of, at least one class I or III antiarrhythmic medication.5 With these same caveats, CA is a reasonable strategy for symptomatic persistent AF.
Consider more invasive interventions, such as an atrial maze procedure, when patients require cardiac surgery for another indication. Patients with an increased risk of thromboembolism (based on CHA2DS2-VASc) remain at high risk even after successful ablation.48 As a result, some guidelines recommend continued long-term anticoagulation following CA.18,22
CORRESPONDENCE
Philip Dooley, MD, University of Kansas School of Medicine–Wichita Family Medicine Residency at Via Christi, 707 North Emporia, Wichita, KS 67207; [email protected].
ACKNOWLEDGMENTS
We thank Professor Anne Walling, MB, ChB, FFPHM, Department of Family and Community Medicine, University of Kansas School of Medicine–Wichita for her suggestions and critical review of an earlier version of this manuscript.
1. Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults. National implications for Rhythm Management and Stroke Prevention: The AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA. 2001;285:2370-2375.
2. Kannel WB, Wolf PA, Benjamin EJ, et al. Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: population-based estimates. Am J Cardiol. 1998;82:2N-9N.
3. Krahn AD, Manfreda J, Tate RB, et al. The natural history of atrial fibrillation: incidence, risk factors, and prognosis in the Manitoba follow-up study. Am J Med. 1995;98:476-484.
4. Ott A, Breteler MMB, de Bruyne MC, et al. Atrial fibrillation and dementia in a population-based study: The Rotterdam Study. Stroke. 1997;28:316-321.
5. January CT, Wann L, Alpert JS, et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2014;64:e1-e76.
6. Lin HJ, Wolf PA, Kelly-Hayes M, et al. Stroke severity in atrial fibrillation. Stroke. 1996;27:1760-1764.
7. Hsu JC, Maddox TM, Kennedy KF, et al. Oral anticoagulant therapy prescription in patients with atrial fibrillation across the spectrum of stroke risk: insights from the NCDR PINNACLE registry. JAMA Cardiol. 2016;1:55-62.
8. Olesen JB, Lip GY, Lindhardsen J, et al. Risks of thromboembolism and bleeding with thromboprophylaxis in patients with atrial fibrillation: a net clinical benefit analysis using a ‘real world’ nationwide cohort study. Thromb Haemost. 2011;106:739-749.
9. Steinberg BA, Kim S, Thomas L, et al. Lack of concordance between empirical scores and physician assessments of stroke and bleeding risk in atrial fibrillation: results from the Outcomes Registry for Better Informed Treatment of Atrial Fibrillation (ORBIT-AF) registry. Circulation. 2014;129:2005-2012.
10. Angaran P, Dorian P, Tan MK, et al. The risk stratification and stroke prevention therapy care gap in Canadian atrial fibrillation patients. Can J Cardiol. 2016;32:336-343.
11. Waldo AL, Feld GK. Inter-relationships of atrial fibrillation and atrial flutter: mechanisms and clinical implications. J Am Coll Cardiol. 2008;51:779-786.
12. Ellis K, Wazni O, Marrouche N, et al. Incidence of atrial fibrillation post-cavotricuspid isthmus ablation in patients with typical atrial flutter: left-atrial size as an independent predictor of atrial fibrillation recurrence. J Cardiovasc Electrophysiol. 2007;18:799-802.
13. Larsson SC, Drca N, Wolk A. Alcohol consumption and risk of atrial fibrillation: a prospective study and dose-response meta-analysis. J Am Coll Cardiol. 2014;64:281-289.
14. Calkins H, Kuck KH, Cappato R, et al. 2012 HRS/EHRA/ECAS expert consensus statement on catheter and surgical ablation of atrial fibrillation: recommendations for patient selection, procedural techniques, patient management and follow-up, definitions, endpoints, and research trial design. J Interv Card Electrophysiol. 2012;33:171-257.
15. Lip GY, Nieuwlaat R, Pisters R, et al. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: the Euro Heart Survey on Atrial Fibrillation. Chest. 2010;137:263-272.
16. Lip GYH, Frison L, Halperin JL, et al. Identifying patients at high risk for stroke despite anticoagulation: a comparison of contemporary stroke risk stratification schemes in an anticoagulated atrial fibrillation cohort. Stroke. 2010;41:2731-2738.
17. Olesen JB, Lip GYH, Hansen ML, et al. Validation of risk stratification schemes for predicting stroke and thromboembolism in patients with atrial fibrillation: nationwide cohort study. BMJ. 2011;342:d124.
18. Camm AJ, Lip GYH, De Caterina R, et al. 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J. 2012;33:2719-2747.
19. Olesen JB, Torp-Pedersen C, Hansen ML, et al. The value of the CHA2DS2-VASc score for refining stroke risk stratification in patients with atrial fibrillation with a CHADS2 score 0-1: a nationwide cohort study. Thromb Haemost. 2012;107:1172-1179.
20. Friberg L, Benson L, Rosenqvist M, et al. Assessment of female sex as a risk factor in atrial fibrillation in Sweden: nationwide retrospective cohort study. BMJ. 2012;344:e3522.
21. National Institute for Health and Clinical Excellence (NICE). Atrial fibrillation: the management of atrial fibrillation [CG180]. 2014. Available at: https://www.nice.org.uk/guidance/cg180. Accessed July 31, 2016.
22. Verma A, Cairns JA, Mitchell LB, et al. 2014 focused update of the Canadian Cardiovascular Society Guidelines for the management of atrial fibrillation. Can J Cardiol. 2014;30:1114-1130.
23. Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361:1139-1151.
24. Patel MR, Mahaffey KW, Garg J, et al. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365:883-891.
25. Giugliano RP, Ruff CT, Braunwald E, et al. Edoxaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2013;369:2093-2104.
26. Granger CB, Alexander JH, McMurray JJV, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365:981-992.
27. Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383:955-962.
28. Morimoto T, Crawford B, Wada K, et al. Comparative efficacy and safety of novel oral anticoagulants in patients with atrial fibrillation: a network meta-analysis with the adjustment for the possible bias from open label studies. J Cardiol. 2015;66:466-474.
29. Verdecchia P, Angeli F, Bartolini C, et al. Safety and efficacy of non-vitamin K oral anticoagulants in non-valvular atrial fibrillation: a Bayesian meta-analysis approach. Expert Opin Drug Saf. 2015;14:7-20.
30. Micromedex® 2.0 (electronic version). Truven Health Analytics, Greenwood Village, Colorado, USA. Available at: http://www.micromedexsolutions.com. Accessed August 18, 2016.
31. GoodRx. Available at: https://www.goodrx.com. Accessed August 18, 2016.
32. Del-Carpio Munoz F, Gharacholou SM, Munger TM, et al. Meta-analysis of renal function on the safety and efficacy of novel oral anticoagulants for atrial fibrillation. Am J Cardiol. 2016;117:69-75.
33. Fang MC, Go AS, Chang Y, et al. A new risk scheme to predict warfarin-associated hemorrhage: the ATRIA (Anticoagulation and Risk Factors in Atrial Fibrillation) Study. J Am Coll Cardiol. 2011;58:395-401.
34. Gage BF, Yan Y, Milligan PE, et al. Clinical classification schemes for predicting hemorrhage: results from the National Registry of Atrial Fibrillation (NRAF). Am Heart J. 2006;151:713-719.
35. Pisters R, Lane DA, Nieuwlaat R, et al. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest. 2010;138:1093-1100.
36. Zhu W, He W, Guo L, et al. The HAS-BLED Score for predicting major bleeding risk in anticoagulated patients with atrial fibrillation: a systematic review and meta-analysis. Clin Cardiol. 2015;38:555-561.
37. Chai-Adisaksopha C, Hillis C, Monreal M, et al. Thromboembolic events, recurrent bleeding and mortality after resuming anticoagulant following gastrointestinal bleeding. A meta-analysis. Thromb Haemost. 2015;114:819-825.
38. Xu H, Xie X, Wang B, et al. Efficacy and safety of percutaneous left atrial appendage occlusion for stroke prevention in nonvalvular atrial fibrillation: a meta-analysis of contemporary studies. Heart Lung Circ. 2016;25:1107-1117.
39. Siu CW, Lau CP, Lee WL, et al. Intravenous diltiazem is superior to intravenous amiodarone or digoxin for achieving ventricular rate control in patients with acute uncomplicated atrial fibrillation. Crit Care Med. 2009;37:2174-2179.
40. Clemo HF, Wood MA, Gilligan DM, et al. Intravenous amiodarone for acute heart rate control in the critically ill patient with atrial tachyarrhythmias. Am J Cardiol. 1998;81:594-598.
41. The Atrial Fibrillation Follow-Up Investigation of Rhythm Management (AFFIRM) Investigators. A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med. 2002;347:1825-1833.
42. Van Gelder IC, Groenveld HF, Crijns HJGM, et al. Lenient versus strict rate control in patients with atrial fibrillation. N Engl J Med. 2010;362:1363-1373.
43. Wang ZQ, Zhang R, Chen MT, et al. Digoxin is associated with increased all-cause mortality in patients with atrial fibrillation regardless of concomitant heart failure: a meta-analysis. J Cardiovasc Pharmacol. 2015;66:270-275.
44. Olshansky B, Heller EN, Mitchell LB, et al. Are transthoracic echocardiographic parameters associated with atrial fibrillation recurrence or stroke? Results from the Atrial Fibrillation Follow-Up Investigation of Rhythm Management (AFFIRM) study. J Am Coll Cardiol. 2005;45:2026-2033.
45. de Vos CB, Pisters R, Nieuwlaat R, et al. Progression from paroxysmal to persistent atrial fibrillation: clinical correlates and prognosis. J Am Coll Cardiol. 2010;55:725-731.
46. Cheng X, Li X, He Y, et al. Catheter ablation versus anti-arrhythmic drug therapy for the management of atrial fibrillation: a meta-analysis. J Interv Card Electrophysiol. 2014;41:267-272.
47. Di Biase L, Mohanty P, Mohanty S, et al. Ablation versus amiodarone for treatment of persistent atrial fibrillation in patients with congestive heart failure and an implanted device: results from the AATAC multicenter randomized trial. Circulation. 2016;133:1637-1644.
48.
Atrial fibrillation (AF)—the most common supraventricular tachycardia—affects as many as 6.1 million adults in the United States.1 It is associated with a 5-fold increased risk of stroke,2 a 3-fold increased risk of heart failure (HF),3 and about a 2-fold increased risk of dementia4 and mortality.2 The prevalence of AF increases with maturity, from 2% in people <65 years of age to 9% in those ≥65 years,5 and that prevalence is expected to double over the next 25 years as the population ages.1
The primary goals of treatment are to alleviate symptoms and prevent thromboembolism. Strokes related to AF are more likely to result in severe disability or death when compared with those unrelated to AF.6 And yet anticoagulation remains underutilized.7
The net clinical benefit of oral anticoagulation appears to be greatest in patients with the highest risk of bleeding, since these patients are also at the highest risk for stroke.8 Patients at increased risk of stroke are more likely to receive oral anticoagulation; however, for unknown reasons, more than half of people with the highest risk of stroke are not prescribed these important anti-blood-clotting medications.7 One theory is that physicians may be relying on their gut rather than objective risk scores, and underuse of validated schemata leads to poor estimation of risk.
For example, results from the ORBIT-AF (Outcomes Registry for Better Informed Treatment of Atrial Fibrillation) trial, which involved over 10,000 people with AF, found that although 72% (n=7251) had high-risk CHADS2 scores (≥2), only 16% were assessed as having a high risk of stroke by physicians.9 Along the same lines, a recent study of Canadian primary care physicians showed that stroke risk and bleeding risk were not evaluated with validated tools in 58% and 81% of patients, respectively, leading to both significant underestimation and overestimation of risk.10
This review provides the tools to identify when anticoagulation is indicated, reports the advantages and disadvantages of the currently available anticoagulants, and discusses the selection and implementation of rate- vs rhythm-control strategies. But first, a word about the etiology, classification, and diagnosis of AF.
AF: The result of any number of cardiac and non-cardiac causes
AF is characterized by uncoordinated activation of the atria, which results in ineffective atrial contractions and an irregular, often rapid, ventricular response. It is the ultimate clinical manifestation of multiple diseases that alter atrial tissue through inflammation, fibrosis, or hypertrophy.5 The most common causes are hypertension, coronary artery disease, HF, cardiomyopathies, and valvular heart disease, all of which stimulate the renin-angiotensin-aldosterone system, leading to increased susceptibility to arrhythmia.5 Atrial ectopic tachycardia, Wolff-Parkinson-White (WPW) syndrome, and atrioventricular (AV) nodal reentrant tachycardia also may precipitate AF.5 In these cases, AF usually resolves after catheter ablation (CA) of the primary arrhythmia.11 Unrecognized AF may trigger atrial flutter, and more than 80% of patients who undergo radiofrequency ablation for atrial flutter experience AF at some point in the subsequent 5 years.12
Non-cardiac causes of AF include sleep apnea, obesity, hyperthyroidism, drugs, electrocution, pneumonia, and pulmonary embolism.5 An association between binge drinking and AF (“holiday heart syndrome”) has long been recognized. The evidence now suggests that alcohol increases the risk of AF in a dose-dependent manner with intakes of ≥1 drink per day (12 g per drink).13
Classification schema no longer includes “lone AF”
AF is classified in terms of the duration of episodes:5
- Paroxysmal AF is characterized by brief episodes that terminate spontaneously or with intervention within 7 days of onset. These episodes recur with variable frequency.
- Persistent AF refers to AF that is continuously sustained for more than 7 days.
- Longstanding persistent AF refers to continuous AF that lasts longer than 12 months.
- Permanent AF is not an inherent pathophysiologic attribute of AF, but rather an acceptance of AF where the patient and physician abandon further efforts to restore and/or maintain sinus rhythm.
- Nonvalvular AF occurs in the absence of a valve replacement (mechanical or bioprosthetic), rheumatic mitral stenosis, or mitral valve repair.
Although paroxysmal and persistent AF may occur in the same individual, the distinction is still clinically relevant, as outcomes of certain therapies, such as CA, are superior in patients with paroxysmal AF.14 With a more complete understanding of AF pathophysiology, guidelines now discourage use of the potentially confusing term “lone AF,” which has historically been applied to younger patients with no known clinical risk factors or echocardiographic abnormalities. As a result, therapeutic decisions are no longer based on this nomenclature, according to the 2014 AF practice guideline from the American College of Cardiology (ACC)/American Heart Association (AHA)/Heart Rhythm Society (HRS).5
Patient complaints—or incidental findings—can prompt a Dx
Fatigue is the most common symptom of AF. Other signs and symptoms include palpitations, dyspnea, HF, hypotension, syncope, chest pain, and stroke. Some patients are asymptomatic, and AF is an incidental finding when an irregular pulse is discovered during a physical examination. The diagnosis is confirmed by electrocardiogram (EKG), telemetry, Holter monitor, event recorder, or an implanted electrocardiographic recording device. A chest x-ray, serum electrolyte levels, a complete blood count, thyroid testing, and renal and hepatic function testing are recommended. Transthoracic echocardiography to measure cardiac function, detect underlying structural heart disease, and evaluate atrial size is essential.5
An electrophysiologic (EP) study may be needed for diagnosis or treatment if another arrhythmia is present. Aberrant conduction may cause AF to present as a wide complex tachycardia and be mislabeled as ventricular tachycardia. The presence of delta waves is an indication for an EP study targeting the WPW accessory pathway. Transesophageal echocardiography (TEE) is the most sensitive and specific test for left atrial thrombi. If you are considering a TEE for a patient with AF of unknown, or >48 hours’, duration who has not been anticoagulated in the preceding 3 weeks, obtain it before performing cardioversion because of the risk of embolism.5
Stroke prevention
The ACC/AHA/HRS AF guideline recommends basing anticoagulation decisions on thromboembolic risk, regardless of AF pattern (paroxysmal, persistent, or permanent) (Class I recommendation).5 For patients with nonvalvular AF and atrial flutter, the guideline recommends using the Birmingham 2009 schema (CHA2DS2-VASc score) (TABLE 115-18) to estimate thromboembolic risk.5,15 CHA2DS2-VASc improves on the older CHADS2 score by significantly reducing the number of patients categorized as having intermediate risk and better identifying truly low-risk patients who are unlikely to benefit from anticoagulation.16,17,19
Men with a CHA2DS2-VASc score of zero and women with a score of one do not need anticoagulation.5,20 Discuss the risks and benefits of oral anticoagulation with men who have a score of one. In these intermediate-risk men, antiplatelet therapy with aspirin and/or clopidogrel may be reasonable, especially if there is an indication other than stroke prevention (eg, post-myocardial infarction). Oral anticoagulation is strongly recommended for all patients with a CHA2DS2-VASc score of 2 or higher.5,18,21,22
Anticoagulant considerations: Warfarin vs DOACs
Warfarin was the gold standard for stroke prevention in nonvalvular AF until the direct oral anticoagulants (DOACs) became available in 2010. Guidelines in the United States and the United Kingdom recommend shared decision-making to help patients with AF who do not have a specific indication for warfarin choose between warfarin and the DOACs.5,21 Canadian and European guidelines recommend DOACs as the first-line option for anticoagulation and reserve warfarin for patients who have contraindications to, or are unable to afford, DOACs.18,22 All current guidelines recommend continuing warfarin in patients who are stable, well controlled, and satisfied with warfarin therapy and the monitoring and dietary restrictions it entails.
DOACs are as effective as warfarin. All of the DOACs are approved for stroke prevention based on individual phase III non-inferiority trials in which they were compared to warfarin.23-26 In addition, a meta-analysis of these 4 trials involving a total of 71,683 patients (mean age 70-73 years; median follow-up, 1.8-2.8 years) evaluated the benefits and risks of the 4 DOACs against the former gold standard.27
Higher doses of the DOACs (dabigatran 150 mg BID, rivaroxaban 20 mg/d, edoxaban 60 mg/d, and apixaban 5 mg BID) reduced the rates of stroke or systemic embolism (relative risk [RR]=0.81; 95% confidence interval [CI], 0.73-0.91; P<.0001; number needed to treat [NNT]=147), hemorrhagic stroke (RR=0.49; 95% CI, 0.38-0.64; P<.0001; NNT=219), and all-cause mortality (RR=0.90; 95% CI, 0.85-0.95; P=.0003; NNT=128), compared with warfarin.27 It is important to note that while lower doses of some DOACs (dabigatran 110 mg BID and edoxaban 30 mg/d) were not as effective at preventing ischemic stroke when compared with warfarin (RR=1.3; 95% CI, 1-1.6; P=.045), they still significantly reduced hemorrhagic stroke (RR=0.33; 95% CI, 0.23-0.46; P<.0001) and all-cause mortality (RR=0.89; 95% CI, 0.83-0.96; P=.003).
Of course, the biggest concern is bleeding. In that same meta-analysis, the difference in major bleeding events with DOACs vs warfarin was not statistically significant (RR=0.86; 95% CI, 0.73-1; P=.06). While DOACs likely lower rates of intracranial hemorrhage (RR=0.48; 95% CI, 0.39-0.59; P<.0001; NNT=132), they seem to increase the risk of gastrointestinal (GI) bleeding (RR=1.3; 95% CI, 1-1.6; P=.043; number needed to harm [NNH]=185).27
There was significant heterogeneity in the GI bleeding outcome, however. When compared with warfarin, GI bleeding was increased by dabigatran 150 mg BID (RR=1.5; 95% CI, 1.2-1.9; P<.001) and edoxaban 60 mg/d (HR=1.2; 95% CI, 1.02-1.5; P=.03), but there were no significant differences for dabigatran 110 mg BID or apixaban 5 mg BID.23,25,26
On the other hand, edoxaban 30 mg/d had a lower risk of GI bleeding when compared with warfarin (HR=0.67; 95% CI, 0.53-0.83; P<.001).25 Without head-to-head trials, it is impossible to know if one DOAC is superior to another. Apixaban 5 mg BID appears to offer the best overall balance between efficacy and safety. Other DOACs may be better options for patients who have specific concerns regarding efficacy or safety.28,29
Convenience, interactions, and cost may be the deciding factors. Since all DOACs are fairly comparable in efficacy and safety, other factors such as convenience, interactions with other medications, and cost should be considered when deciding on a medication for an individual patient (TABLE 230,31). The DOACs require no lab monitoring or dose titration, and all 4 have fewer potential drug interactions than warfarin.30 Due to their relatively short half-lives, strict adherence is critical; DOACs are not suitable for patients who frequently miss doses.5 (For more information on starting or switching to DOACs, see, “Is a novel anticoagulant right for your patient?” J Fam Pract. 2014;63:22-28.)
A word about DOACs and renal impairment. Another concern with DOACs is their reliance on renal metabolism and excretion. A meta-analysis of the 4 phase III trials of the DOACs, this time involving 58,338 patients, evaluated DOAC efficacy and safety compared to warfarin in the presence of kidney dysfunction.32 Renal function was categorized as normal (estimated glomerular filtration rate [eGFR] >80 mL/min/1.73 m2), mildly impaired (eGFR 50-80 mL/min/1.73 m2), or moderately impaired (eGFR <50 mL/min/1.73m2). Compared with warfarin, DOACs lowered stroke risk in patients with mild (RR=0.71; 95% CI, 0.62-0.81) or moderate (RR=0.79; 95% CI, 0.66-0.94) renal impairment. DOACs also reduced major bleeding compared to warfarin in patients with mild (RR=0.88; 95% CI, 0.80-0.97) or moderate (RR=0.80; 95% CI, 0.66-0.94) renal impairment. How the DOACs fare in patients with severe renal dysfunction could not be determined because such patients were excluded from the trials.
Keep in mind that the DOACs require dose adjustment at different levels of renal impairment (TABLE 230,31), and warfarin remains the only recommended treatment for patients with severe renal impairment, according to both AHA/ACC/HRS and European Society of Cardiology guidelines.5,18
Tools to help assess patients’ bleeding risk
Of the available scoring mechanisms to identify risk factors for bleeding, 3 have been specifically validated in AF populations (ie, ATRIA,33 HEMORR2HAGES,34 and HAS-BLED35). Of the 3, HAS-BLED is superior,36 the most practical, and recommended by expert guidelines.18,21,22 Additionally, HAS-BLED has good correlation with intracranial hemorrhage risk. The HAS-BLED score ranges from 0 to 9 points with one point assigned for each of the following:35
- Hypertension–uncontrolled with systolic BP >160 mm Hg
- Abnormal liver function–cirrhosis, bilirubin >2× normal, or liver enzymes >3× normal
- Abnormal renal function–dialysis, transplant, or serum creatinine >2.26 mg/dL
- Stroke history–including lacunar infarcts
- Bleeding predisposition–history of major bleeding due to any cause
- Labile international normalized ratio (INR)–time in therapeutic range <60%
- Elderly–age >65 years
- Drug–antiplatelet agents, including nonsteroidal anti-inflammatory drugs
- Alcohol usage–>8 drinks per week.
Patients with a HAS-BLED score ≥3 warrant additional monitoring and attempts to reduce bleeding risk by addressing modifiable risk factors. Bleeding risk scores should not be used to exclude patients from anticoagulation therapy.5 In fact, the British National Institute for Health and Clinical Excellence (NICE) guidelines state that anticoagulation should not be withheld solely due to fall risk.21
Also, anticoagulation with warfarin should not be permanently discontinued because of a single GI bleed, since restarting warfarin is associated with decreased risks of thromboembolism and mortality and a statistically insignificant increase in recurrent GI bleeding.37 Restarting DOAC therapy following a GI bleed has not been evaluated in clinical trials; however, it may be reasonable to use one of the DOAC doses with a lower risk of GI bleeding (dabigatran 110 mg BID, apixaban 5 mg BID, or edoxaban 30 mg/d) in patients who have experienced a GI bleed on warfarin or another DOAC.18,22
An online calculator is available that uses CHA2DS2-VASc and HAS-BLED scores to determine an individual’s risk/benefit profile with the various anticoagulation strategies available (http://www.sparctool.com). Consider percutaneous left atrial appendage occlusion if the risks of anticoagulation truly exceed the benefits.38
Rate control vs rhythm control
Most patients who present with AF require immediate ventricular rate control to reduce symptoms. In the acute setting, this can be accomplished with intravenous (IV) beta-blockers or IV calcium channel antagonists.5,39 If the patient is hemodynamically unstable, urgent direct-current cardioversion is the preferred treatment strategy and should not be delayed pending anticoagulation. IV amiodarone can be used in the ICU patient who does not require cardioversion, but is unable to tolerate beta-blockers or calcium channel antagonists.40 Once the patient is stable, long-term treatment focuses on ventricular rate control or restoration and maintenance of sinus rhythm.
The AFFIRM (Atrial Fibrillation Follow-up Investigation of Rhythm Management) trial enrolled 4060 patients (mean age 70 years, mean follow-up 3.5 years) with paroxysmal and persistent AF and randomized them to either pharmacologic rate control or rhythm control.41 No significant differences were found in all-cause mortality or in the composite secondary endpoint of death, ischemic stroke, anoxic encephalopathy, major bleeding, or cardiac arrest. In addition, no significant differences emerged in quality of life or global functional status. The number of patients requiring hospitalization during follow-up was significantly lower in the rate-control group vs the rhythm-control group (73% vs 80%; P<.001). Anticoagulation was encouraged but not mandated in the rhythm-control group after 4 weeks in sinus rhythm, and there was a trend toward higher mortality in the rhythm-control group (27% vs 26%; P=.08).
Patients <65 years were excluded from the AFFIRM trial. When younger patients experience significant symptoms, early referral to Cardiology should be considered to discuss the long-term benefits and risks of a rhythm-control strategy. Regardless of age, when patients remain symptomatic despite rate- or rhythm-control management, the strategy should be changed.5
Rate-control targets and options
Target heart rates should be individualized. The 2014 ACC/AHA/HRS guideline recommends a resting target heart rate <80 beats per minute (bpm) in symptomatic patients.5 In patients with permanent AF who remain asymptomatic at higher resting heart rates, a more lenient rate-control strategy (resting heart rate <110 bpm) has demonstrated outcomes equivalent to those of a more strict approach (resting heart rate <80 bpm and heart rate during moderate exercise <110 bpm).42 Pharmacologic rate-control options include beta-blockers, non-dihydropyridine calcium channel antagonists, and digoxin (TABLE 35). Digoxin is associated with increased all-cause mortality in patients with AF regardless of HF status (HR=1.4; 95% CI, 1.2-1.6, P=.0001).43 Digoxin should be reserved for patients who are sedentary or have inadequate control with first-line medications.5
Indications for rhythm control
The NICE guidelines, which are consistent with the ACC/AHA/HRS guidelines, recommend rate control as the first-line strategy for AF management, except in people:21
- whose AF has a reversible cause
- who have HF believed to be primarily caused by AF
- with new-onset AF
- with atrial flutter that is considered suitable for an ablation strategy to restore sinus rhythm
- for whom a rhythm-control strategy would be more suitable based on clinical judgment.
In addition, patients who continue to experience symptomatic AF despite an adequate trial of rate control should be offered rhythm control.5
Pharmacologic rhythm-control strategies. Antiarrhythmic drugs can be used for chemical cardioversion, reduction of paroxysms, and long-term maintenance of sinus rhythm. The most commonly used antiarrhythmic drugs are Class IC and Class III agents (TABLE 3).5 Tailored drug selection for each patient is key. Patients with left atrial diameters >4.5 cm are less likely to remain in sinus rhythm, and patients with left ventricular hypertrophy are at increased risk for proarrhythmic adverse effects.44 Patients with paroxysmal AF may be candidates for a “pill-in-the-pocket” strategy using propafenone or flecainide.5
AF frequently progresses from paroxysmal to persistent and can subsequently result in electrical and structural remodeling that becomes irreversible over time.45 The patient with uncontrolled symptoms despite attempts at rate control and rhythm control should be promptly referred to an electrophysiologist.
Surgical interventions for rate or rhythm control
Electrophysiology interventions include AV nodal ablation with pacemaker placement for rate control, or catheter-directed ablation (radiofrequency or cryotherapy) for rhythm control. CA appears to be more effective than pharmacologic rhythm control.46,47 Treatment with CA is indicated for symptomatic paroxysmal AF when a rhythm-control strategy is desired and the AF is refractory to, or the patient is intolerant of, at least one class I or III antiarrhythmic medication.5 With these same caveats, CA is a reasonable strategy for symptomatic persistent AF.
Consider more invasive interventions, such as an atrial maze procedure, when patients require cardiac surgery for another indication. Patients with an increased risk of thromboembolism (based on CHA2DS2-VASc) remain at high risk even after successful ablation.48 As a result, some guidelines recommend continued long-term anticoagulation following CA.18,22
CORRESPONDENCE
Philip Dooley, MD, University of Kansas School of Medicine–Wichita Family Medicine Residency at Via Christi, 707 North Emporia, Wichita, KS 67207; [email protected].
ACKNOWLEDGMENTS
We thank Professor Anne Walling, MB, ChB, FFPHM, Department of Family and Community Medicine, University of Kansas School of Medicine–Wichita for her suggestions and critical review of an earlier version of this manuscript.
Atrial fibrillation (AF)—the most common supraventricular tachycardia—affects as many as 6.1 million adults in the United States.1 It is associated with a 5-fold increased risk of stroke,2 a 3-fold increased risk of heart failure (HF),3 and about a 2-fold increased risk of dementia4 and mortality.2 The prevalence of AF increases with maturity, from 2% in people <65 years of age to 9% in those ≥65 years,5 and that prevalence is expected to double over the next 25 years as the population ages.1
The primary goals of treatment are to alleviate symptoms and prevent thromboembolism. Strokes related to AF are more likely to result in severe disability or death when compared with those unrelated to AF.6 And yet anticoagulation remains underutilized.7
The net clinical benefit of oral anticoagulation appears to be greatest in patients with the highest risk of bleeding, since these patients are also at the highest risk for stroke.8 Patients at increased risk of stroke are more likely to receive oral anticoagulation; however, for unknown reasons, more than half of people with the highest risk of stroke are not prescribed these important anti-blood-clotting medications.7 One theory is that physicians may be relying on their gut rather than objective risk scores, and underuse of validated schemata leads to poor estimation of risk.
For example, results from the ORBIT-AF (Outcomes Registry for Better Informed Treatment of Atrial Fibrillation) trial, which involved over 10,000 people with AF, found that although 72% (n=7251) had high-risk CHADS2 scores (≥2), only 16% were assessed as having a high risk of stroke by physicians.9 Along the same lines, a recent study of Canadian primary care physicians showed that stroke risk and bleeding risk were not evaluated with validated tools in 58% and 81% of patients, respectively, leading to both significant underestimation and overestimation of risk.10
This review provides the tools to identify when anticoagulation is indicated, reports the advantages and disadvantages of the currently available anticoagulants, and discusses the selection and implementation of rate- vs rhythm-control strategies. But first, a word about the etiology, classification, and diagnosis of AF.
AF: The result of any number of cardiac and non-cardiac causes
AF is characterized by uncoordinated activation of the atria, which results in ineffective atrial contractions and an irregular, often rapid, ventricular response. It is the ultimate clinical manifestation of multiple diseases that alter atrial tissue through inflammation, fibrosis, or hypertrophy.5 The most common causes are hypertension, coronary artery disease, HF, cardiomyopathies, and valvular heart disease, all of which stimulate the renin-angiotensin-aldosterone system, leading to increased susceptibility to arrhythmia.5 Atrial ectopic tachycardia, Wolff-Parkinson-White (WPW) syndrome, and atrioventricular (AV) nodal reentrant tachycardia also may precipitate AF.5 In these cases, AF usually resolves after catheter ablation (CA) of the primary arrhythmia.11 Unrecognized AF may trigger atrial flutter, and more than 80% of patients who undergo radiofrequency ablation for atrial flutter experience AF at some point in the subsequent 5 years.12
Non-cardiac causes of AF include sleep apnea, obesity, hyperthyroidism, drugs, electrocution, pneumonia, and pulmonary embolism.5 An association between binge drinking and AF (“holiday heart syndrome”) has long been recognized. The evidence now suggests that alcohol increases the risk of AF in a dose-dependent manner with intakes of ≥1 drink per day (12 g per drink).13
Classification schema no longer includes “lone AF”
AF is classified in terms of the duration of episodes:5
- Paroxysmal AF is characterized by brief episodes that terminate spontaneously or with intervention within 7 days of onset. These episodes recur with variable frequency.
- Persistent AF refers to AF that is continuously sustained for more than 7 days.
- Longstanding persistent AF refers to continuous AF that lasts longer than 12 months.
- Permanent AF is not an inherent pathophysiologic attribute of AF, but rather an acceptance of AF where the patient and physician abandon further efforts to restore and/or maintain sinus rhythm.
- Nonvalvular AF occurs in the absence of a valve replacement (mechanical or bioprosthetic), rheumatic mitral stenosis, or mitral valve repair.
Although paroxysmal and persistent AF may occur in the same individual, the distinction is still clinically relevant, as outcomes of certain therapies, such as CA, are superior in patients with paroxysmal AF.14 With a more complete understanding of AF pathophysiology, guidelines now discourage use of the potentially confusing term “lone AF,” which has historically been applied to younger patients with no known clinical risk factors or echocardiographic abnormalities. As a result, therapeutic decisions are no longer based on this nomenclature, according to the 2014 AF practice guideline from the American College of Cardiology (ACC)/American Heart Association (AHA)/Heart Rhythm Society (HRS).5
Patient complaints—or incidental findings—can prompt a Dx
Fatigue is the most common symptom of AF. Other signs and symptoms include palpitations, dyspnea, HF, hypotension, syncope, chest pain, and stroke. Some patients are asymptomatic, and AF is an incidental finding when an irregular pulse is discovered during a physical examination. The diagnosis is confirmed by electrocardiogram (EKG), telemetry, Holter monitor, event recorder, or an implanted electrocardiographic recording device. A chest x-ray, serum electrolyte levels, a complete blood count, thyroid testing, and renal and hepatic function testing are recommended. Transthoracic echocardiography to measure cardiac function, detect underlying structural heart disease, and evaluate atrial size is essential.5
An electrophysiologic (EP) study may be needed for diagnosis or treatment if another arrhythmia is present. Aberrant conduction may cause AF to present as a wide complex tachycardia and be mislabeled as ventricular tachycardia. The presence of delta waves is an indication for an EP study targeting the WPW accessory pathway. Transesophageal echocardiography (TEE) is the most sensitive and specific test for left atrial thrombi. If you are considering a TEE for a patient with AF of unknown, or >48 hours’, duration who has not been anticoagulated in the preceding 3 weeks, obtain it before performing cardioversion because of the risk of embolism.5
Stroke prevention
The ACC/AHA/HRS AF guideline recommends basing anticoagulation decisions on thromboembolic risk, regardless of AF pattern (paroxysmal, persistent, or permanent) (Class I recommendation).5 For patients with nonvalvular AF and atrial flutter, the guideline recommends using the Birmingham 2009 schema (CHA2DS2-VASc score) (TABLE 115-18) to estimate thromboembolic risk.5,15 CHA2DS2-VASc improves on the older CHADS2 score by significantly reducing the number of patients categorized as having intermediate risk and better identifying truly low-risk patients who are unlikely to benefit from anticoagulation.16,17,19
Men with a CHA2DS2-VASc score of zero and women with a score of one do not need anticoagulation.5,20 Discuss the risks and benefits of oral anticoagulation with men who have a score of one. In these intermediate-risk men, antiplatelet therapy with aspirin and/or clopidogrel may be reasonable, especially if there is an indication other than stroke prevention (eg, post-myocardial infarction). Oral anticoagulation is strongly recommended for all patients with a CHA2DS2-VASc score of 2 or higher.5,18,21,22
Anticoagulant considerations: Warfarin vs DOACs
Warfarin was the gold standard for stroke prevention in nonvalvular AF until the direct oral anticoagulants (DOACs) became available in 2010. Guidelines in the United States and the United Kingdom recommend shared decision-making to help patients with AF who do not have a specific indication for warfarin choose between warfarin and the DOACs.5,21 Canadian and European guidelines recommend DOACs as the first-line option for anticoagulation and reserve warfarin for patients who have contraindications to, or are unable to afford, DOACs.18,22 All current guidelines recommend continuing warfarin in patients who are stable, well controlled, and satisfied with warfarin therapy and the monitoring and dietary restrictions it entails.
DOACs are as effective as warfarin. All of the DOACs are approved for stroke prevention based on individual phase III non-inferiority trials in which they were compared to warfarin.23-26 In addition, a meta-analysis of these 4 trials involving a total of 71,683 patients (mean age 70-73 years; median follow-up, 1.8-2.8 years) evaluated the benefits and risks of the 4 DOACs against the former gold standard.27
Higher doses of the DOACs (dabigatran 150 mg BID, rivaroxaban 20 mg/d, edoxaban 60 mg/d, and apixaban 5 mg BID) reduced the rates of stroke or systemic embolism (relative risk [RR]=0.81; 95% confidence interval [CI], 0.73-0.91; P<.0001; number needed to treat [NNT]=147), hemorrhagic stroke (RR=0.49; 95% CI, 0.38-0.64; P<.0001; NNT=219), and all-cause mortality (RR=0.90; 95% CI, 0.85-0.95; P=.0003; NNT=128), compared with warfarin.27 It is important to note that while lower doses of some DOACs (dabigatran 110 mg BID and edoxaban 30 mg/d) were not as effective at preventing ischemic stroke when compared with warfarin (RR=1.3; 95% CI, 1-1.6; P=.045), they still significantly reduced hemorrhagic stroke (RR=0.33; 95% CI, 0.23-0.46; P<.0001) and all-cause mortality (RR=0.89; 95% CI, 0.83-0.96; P=.003).
Of course, the biggest concern is bleeding. In that same meta-analysis, the difference in major bleeding events with DOACs vs warfarin was not statistically significant (RR=0.86; 95% CI, 0.73-1; P=.06). While DOACs likely lower rates of intracranial hemorrhage (RR=0.48; 95% CI, 0.39-0.59; P<.0001; NNT=132), they seem to increase the risk of gastrointestinal (GI) bleeding (RR=1.3; 95% CI, 1-1.6; P=.043; number needed to harm [NNH]=185).27
There was significant heterogeneity in the GI bleeding outcome, however. When compared with warfarin, GI bleeding was increased by dabigatran 150 mg BID (RR=1.5; 95% CI, 1.2-1.9; P<.001) and edoxaban 60 mg/d (HR=1.2; 95% CI, 1.02-1.5; P=.03), but there were no significant differences for dabigatran 110 mg BID or apixaban 5 mg BID.23,25,26
On the other hand, edoxaban 30 mg/d had a lower risk of GI bleeding when compared with warfarin (HR=0.67; 95% CI, 0.53-0.83; P<.001).25 Without head-to-head trials, it is impossible to know if one DOAC is superior to another. Apixaban 5 mg BID appears to offer the best overall balance between efficacy and safety. Other DOACs may be better options for patients who have specific concerns regarding efficacy or safety.28,29
Convenience, interactions, and cost may be the deciding factors. Since all DOACs are fairly comparable in efficacy and safety, other factors such as convenience, interactions with other medications, and cost should be considered when deciding on a medication for an individual patient (TABLE 230,31). The DOACs require no lab monitoring or dose titration, and all 4 have fewer potential drug interactions than warfarin.30 Due to their relatively short half-lives, strict adherence is critical; DOACs are not suitable for patients who frequently miss doses.5 (For more information on starting or switching to DOACs, see, “Is a novel anticoagulant right for your patient?” J Fam Pract. 2014;63:22-28.)
A word about DOACs and renal impairment. Another concern with DOACs is their reliance on renal metabolism and excretion. A meta-analysis of the 4 phase III trials of the DOACs, this time involving 58,338 patients, evaluated DOAC efficacy and safety compared to warfarin in the presence of kidney dysfunction.32 Renal function was categorized as normal (estimated glomerular filtration rate [eGFR] >80 mL/min/1.73 m2), mildly impaired (eGFR 50-80 mL/min/1.73 m2), or moderately impaired (eGFR <50 mL/min/1.73m2). Compared with warfarin, DOACs lowered stroke risk in patients with mild (RR=0.71; 95% CI, 0.62-0.81) or moderate (RR=0.79; 95% CI, 0.66-0.94) renal impairment. DOACs also reduced major bleeding compared to warfarin in patients with mild (RR=0.88; 95% CI, 0.80-0.97) or moderate (RR=0.80; 95% CI, 0.66-0.94) renal impairment. How the DOACs fare in patients with severe renal dysfunction could not be determined because such patients were excluded from the trials.
Keep in mind that the DOACs require dose adjustment at different levels of renal impairment (TABLE 230,31), and warfarin remains the only recommended treatment for patients with severe renal impairment, according to both AHA/ACC/HRS and European Society of Cardiology guidelines.5,18
Tools to help assess patients’ bleeding risk
Of the available scoring mechanisms to identify risk factors for bleeding, 3 have been specifically validated in AF populations (ie, ATRIA,33 HEMORR2HAGES,34 and HAS-BLED35). Of the 3, HAS-BLED is superior,36 the most practical, and recommended by expert guidelines.18,21,22 Additionally, HAS-BLED has good correlation with intracranial hemorrhage risk. The HAS-BLED score ranges from 0 to 9 points with one point assigned for each of the following:35
- Hypertension–uncontrolled with systolic BP >160 mm Hg
- Abnormal liver function–cirrhosis, bilirubin >2× normal, or liver enzymes >3× normal
- Abnormal renal function–dialysis, transplant, or serum creatinine >2.26 mg/dL
- Stroke history–including lacunar infarcts
- Bleeding predisposition–history of major bleeding due to any cause
- Labile international normalized ratio (INR)–time in therapeutic range <60%
- Elderly–age >65 years
- Drug–antiplatelet agents, including nonsteroidal anti-inflammatory drugs
- Alcohol usage–>8 drinks per week.
Patients with a HAS-BLED score ≥3 warrant additional monitoring and attempts to reduce bleeding risk by addressing modifiable risk factors. Bleeding risk scores should not be used to exclude patients from anticoagulation therapy.5 In fact, the British National Institute for Health and Clinical Excellence (NICE) guidelines state that anticoagulation should not be withheld solely due to fall risk.21
Also, anticoagulation with warfarin should not be permanently discontinued because of a single GI bleed, since restarting warfarin is associated with decreased risks of thromboembolism and mortality and a statistically insignificant increase in recurrent GI bleeding.37 Restarting DOAC therapy following a GI bleed has not been evaluated in clinical trials; however, it may be reasonable to use one of the DOAC doses with a lower risk of GI bleeding (dabigatran 110 mg BID, apixaban 5 mg BID, or edoxaban 30 mg/d) in patients who have experienced a GI bleed on warfarin or another DOAC.18,22
An online calculator is available that uses CHA2DS2-VASc and HAS-BLED scores to determine an individual’s risk/benefit profile with the various anticoagulation strategies available (http://www.sparctool.com). Consider percutaneous left atrial appendage occlusion if the risks of anticoagulation truly exceed the benefits.38
Rate control vs rhythm control
Most patients who present with AF require immediate ventricular rate control to reduce symptoms. In the acute setting, this can be accomplished with intravenous (IV) beta-blockers or IV calcium channel antagonists.5,39 If the patient is hemodynamically unstable, urgent direct-current cardioversion is the preferred treatment strategy and should not be delayed pending anticoagulation. IV amiodarone can be used in the ICU patient who does not require cardioversion, but is unable to tolerate beta-blockers or calcium channel antagonists.40 Once the patient is stable, long-term treatment focuses on ventricular rate control or restoration and maintenance of sinus rhythm.
The AFFIRM (Atrial Fibrillation Follow-up Investigation of Rhythm Management) trial enrolled 4060 patients (mean age 70 years, mean follow-up 3.5 years) with paroxysmal and persistent AF and randomized them to either pharmacologic rate control or rhythm control.41 No significant differences were found in all-cause mortality or in the composite secondary endpoint of death, ischemic stroke, anoxic encephalopathy, major bleeding, or cardiac arrest. In addition, no significant differences emerged in quality of life or global functional status. The number of patients requiring hospitalization during follow-up was significantly lower in the rate-control group vs the rhythm-control group (73% vs 80%; P<.001). Anticoagulation was encouraged but not mandated in the rhythm-control group after 4 weeks in sinus rhythm, and there was a trend toward higher mortality in the rhythm-control group (27% vs 26%; P=.08).
Patients <65 years were excluded from the AFFIRM trial. When younger patients experience significant symptoms, early referral to Cardiology should be considered to discuss the long-term benefits and risks of a rhythm-control strategy. Regardless of age, when patients remain symptomatic despite rate- or rhythm-control management, the strategy should be changed.5
Rate-control targets and options
Target heart rates should be individualized. The 2014 ACC/AHA/HRS guideline recommends a resting target heart rate <80 beats per minute (bpm) in symptomatic patients.5 In patients with permanent AF who remain asymptomatic at higher resting heart rates, a more lenient rate-control strategy (resting heart rate <110 bpm) has demonstrated outcomes equivalent to those of a more strict approach (resting heart rate <80 bpm and heart rate during moderate exercise <110 bpm).42 Pharmacologic rate-control options include beta-blockers, non-dihydropyridine calcium channel antagonists, and digoxin (TABLE 35). Digoxin is associated with increased all-cause mortality in patients with AF regardless of HF status (HR=1.4; 95% CI, 1.2-1.6, P=.0001).43 Digoxin should be reserved for patients who are sedentary or have inadequate control with first-line medications.5
Indications for rhythm control
The NICE guidelines, which are consistent with the ACC/AHA/HRS guidelines, recommend rate control as the first-line strategy for AF management, except in people:21
- whose AF has a reversible cause
- who have HF believed to be primarily caused by AF
- with new-onset AF
- with atrial flutter that is considered suitable for an ablation strategy to restore sinus rhythm
- for whom a rhythm-control strategy would be more suitable based on clinical judgment.
In addition, patients who continue to experience symptomatic AF despite an adequate trial of rate control should be offered rhythm control.5
Pharmacologic rhythm-control strategies. Antiarrhythmic drugs can be used for chemical cardioversion, reduction of paroxysms, and long-term maintenance of sinus rhythm. The most commonly used antiarrhythmic drugs are Class IC and Class III agents (TABLE 3).5 Tailored drug selection for each patient is key. Patients with left atrial diameters >4.5 cm are less likely to remain in sinus rhythm, and patients with left ventricular hypertrophy are at increased risk for proarrhythmic adverse effects.44 Patients with paroxysmal AF may be candidates for a “pill-in-the-pocket” strategy using propafenone or flecainide.5
AF frequently progresses from paroxysmal to persistent and can subsequently result in electrical and structural remodeling that becomes irreversible over time.45 The patient with uncontrolled symptoms despite attempts at rate control and rhythm control should be promptly referred to an electrophysiologist.
Surgical interventions for rate or rhythm control
Electrophysiology interventions include AV nodal ablation with pacemaker placement for rate control, or catheter-directed ablation (radiofrequency or cryotherapy) for rhythm control. CA appears to be more effective than pharmacologic rhythm control.46,47 Treatment with CA is indicated for symptomatic paroxysmal AF when a rhythm-control strategy is desired and the AF is refractory to, or the patient is intolerant of, at least one class I or III antiarrhythmic medication.5 With these same caveats, CA is a reasonable strategy for symptomatic persistent AF.
Consider more invasive interventions, such as an atrial maze procedure, when patients require cardiac surgery for another indication. Patients with an increased risk of thromboembolism (based on CHA2DS2-VASc) remain at high risk even after successful ablation.48 As a result, some guidelines recommend continued long-term anticoagulation following CA.18,22
CORRESPONDENCE
Philip Dooley, MD, University of Kansas School of Medicine–Wichita Family Medicine Residency at Via Christi, 707 North Emporia, Wichita, KS 67207; [email protected].
ACKNOWLEDGMENTS
We thank Professor Anne Walling, MB, ChB, FFPHM, Department of Family and Community Medicine, University of Kansas School of Medicine–Wichita for her suggestions and critical review of an earlier version of this manuscript.
1. Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults. National implications for Rhythm Management and Stroke Prevention: The AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA. 2001;285:2370-2375.
2. Kannel WB, Wolf PA, Benjamin EJ, et al. Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: population-based estimates. Am J Cardiol. 1998;82:2N-9N.
3. Krahn AD, Manfreda J, Tate RB, et al. The natural history of atrial fibrillation: incidence, risk factors, and prognosis in the Manitoba follow-up study. Am J Med. 1995;98:476-484.
4. Ott A, Breteler MMB, de Bruyne MC, et al. Atrial fibrillation and dementia in a population-based study: The Rotterdam Study. Stroke. 1997;28:316-321.
5. January CT, Wann L, Alpert JS, et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2014;64:e1-e76.
6. Lin HJ, Wolf PA, Kelly-Hayes M, et al. Stroke severity in atrial fibrillation. Stroke. 1996;27:1760-1764.
7. Hsu JC, Maddox TM, Kennedy KF, et al. Oral anticoagulant therapy prescription in patients with atrial fibrillation across the spectrum of stroke risk: insights from the NCDR PINNACLE registry. JAMA Cardiol. 2016;1:55-62.
8. Olesen JB, Lip GY, Lindhardsen J, et al. Risks of thromboembolism and bleeding with thromboprophylaxis in patients with atrial fibrillation: a net clinical benefit analysis using a ‘real world’ nationwide cohort study. Thromb Haemost. 2011;106:739-749.
9. Steinberg BA, Kim S, Thomas L, et al. Lack of concordance between empirical scores and physician assessments of stroke and bleeding risk in atrial fibrillation: results from the Outcomes Registry for Better Informed Treatment of Atrial Fibrillation (ORBIT-AF) registry. Circulation. 2014;129:2005-2012.
10. Angaran P, Dorian P, Tan MK, et al. The risk stratification and stroke prevention therapy care gap in Canadian atrial fibrillation patients. Can J Cardiol. 2016;32:336-343.
11. Waldo AL, Feld GK. Inter-relationships of atrial fibrillation and atrial flutter: mechanisms and clinical implications. J Am Coll Cardiol. 2008;51:779-786.
12. Ellis K, Wazni O, Marrouche N, et al. Incidence of atrial fibrillation post-cavotricuspid isthmus ablation in patients with typical atrial flutter: left-atrial size as an independent predictor of atrial fibrillation recurrence. J Cardiovasc Electrophysiol. 2007;18:799-802.
13. Larsson SC, Drca N, Wolk A. Alcohol consumption and risk of atrial fibrillation: a prospective study and dose-response meta-analysis. J Am Coll Cardiol. 2014;64:281-289.
14. Calkins H, Kuck KH, Cappato R, et al. 2012 HRS/EHRA/ECAS expert consensus statement on catheter and surgical ablation of atrial fibrillation: recommendations for patient selection, procedural techniques, patient management and follow-up, definitions, endpoints, and research trial design. J Interv Card Electrophysiol. 2012;33:171-257.
15. Lip GY, Nieuwlaat R, Pisters R, et al. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: the Euro Heart Survey on Atrial Fibrillation. Chest. 2010;137:263-272.
16. Lip GYH, Frison L, Halperin JL, et al. Identifying patients at high risk for stroke despite anticoagulation: a comparison of contemporary stroke risk stratification schemes in an anticoagulated atrial fibrillation cohort. Stroke. 2010;41:2731-2738.
17. Olesen JB, Lip GYH, Hansen ML, et al. Validation of risk stratification schemes for predicting stroke and thromboembolism in patients with atrial fibrillation: nationwide cohort study. BMJ. 2011;342:d124.
18. Camm AJ, Lip GYH, De Caterina R, et al. 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J. 2012;33:2719-2747.
19. Olesen JB, Torp-Pedersen C, Hansen ML, et al. The value of the CHA2DS2-VASc score for refining stroke risk stratification in patients with atrial fibrillation with a CHADS2 score 0-1: a nationwide cohort study. Thromb Haemost. 2012;107:1172-1179.
20. Friberg L, Benson L, Rosenqvist M, et al. Assessment of female sex as a risk factor in atrial fibrillation in Sweden: nationwide retrospective cohort study. BMJ. 2012;344:e3522.
21. National Institute for Health and Clinical Excellence (NICE). Atrial fibrillation: the management of atrial fibrillation [CG180]. 2014. Available at: https://www.nice.org.uk/guidance/cg180. Accessed July 31, 2016.
22. Verma A, Cairns JA, Mitchell LB, et al. 2014 focused update of the Canadian Cardiovascular Society Guidelines for the management of atrial fibrillation. Can J Cardiol. 2014;30:1114-1130.
23. Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361:1139-1151.
24. Patel MR, Mahaffey KW, Garg J, et al. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365:883-891.
25. Giugliano RP, Ruff CT, Braunwald E, et al. Edoxaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2013;369:2093-2104.
26. Granger CB, Alexander JH, McMurray JJV, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365:981-992.
27. Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383:955-962.
28. Morimoto T, Crawford B, Wada K, et al. Comparative efficacy and safety of novel oral anticoagulants in patients with atrial fibrillation: a network meta-analysis with the adjustment for the possible bias from open label studies. J Cardiol. 2015;66:466-474.
29. Verdecchia P, Angeli F, Bartolini C, et al. Safety and efficacy of non-vitamin K oral anticoagulants in non-valvular atrial fibrillation: a Bayesian meta-analysis approach. Expert Opin Drug Saf. 2015;14:7-20.
30. Micromedex® 2.0 (electronic version). Truven Health Analytics, Greenwood Village, Colorado, USA. Available at: http://www.micromedexsolutions.com. Accessed August 18, 2016.
31. GoodRx. Available at: https://www.goodrx.com. Accessed August 18, 2016.
32. Del-Carpio Munoz F, Gharacholou SM, Munger TM, et al. Meta-analysis of renal function on the safety and efficacy of novel oral anticoagulants for atrial fibrillation. Am J Cardiol. 2016;117:69-75.
33. Fang MC, Go AS, Chang Y, et al. A new risk scheme to predict warfarin-associated hemorrhage: the ATRIA (Anticoagulation and Risk Factors in Atrial Fibrillation) Study. J Am Coll Cardiol. 2011;58:395-401.
34. Gage BF, Yan Y, Milligan PE, et al. Clinical classification schemes for predicting hemorrhage: results from the National Registry of Atrial Fibrillation (NRAF). Am Heart J. 2006;151:713-719.
35. Pisters R, Lane DA, Nieuwlaat R, et al. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest. 2010;138:1093-1100.
36. Zhu W, He W, Guo L, et al. The HAS-BLED Score for predicting major bleeding risk in anticoagulated patients with atrial fibrillation: a systematic review and meta-analysis. Clin Cardiol. 2015;38:555-561.
37. Chai-Adisaksopha C, Hillis C, Monreal M, et al. Thromboembolic events, recurrent bleeding and mortality after resuming anticoagulant following gastrointestinal bleeding. A meta-analysis. Thromb Haemost. 2015;114:819-825.
38. Xu H, Xie X, Wang B, et al. Efficacy and safety of percutaneous left atrial appendage occlusion for stroke prevention in nonvalvular atrial fibrillation: a meta-analysis of contemporary studies. Heart Lung Circ. 2016;25:1107-1117.
39. Siu CW, Lau CP, Lee WL, et al. Intravenous diltiazem is superior to intravenous amiodarone or digoxin for achieving ventricular rate control in patients with acute uncomplicated atrial fibrillation. Crit Care Med. 2009;37:2174-2179.
40. Clemo HF, Wood MA, Gilligan DM, et al. Intravenous amiodarone for acute heart rate control in the critically ill patient with atrial tachyarrhythmias. Am J Cardiol. 1998;81:594-598.
41. The Atrial Fibrillation Follow-Up Investigation of Rhythm Management (AFFIRM) Investigators. A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med. 2002;347:1825-1833.
42. Van Gelder IC, Groenveld HF, Crijns HJGM, et al. Lenient versus strict rate control in patients with atrial fibrillation. N Engl J Med. 2010;362:1363-1373.
43. Wang ZQ, Zhang R, Chen MT, et al. Digoxin is associated with increased all-cause mortality in patients with atrial fibrillation regardless of concomitant heart failure: a meta-analysis. J Cardiovasc Pharmacol. 2015;66:270-275.
44. Olshansky B, Heller EN, Mitchell LB, et al. Are transthoracic echocardiographic parameters associated with atrial fibrillation recurrence or stroke? Results from the Atrial Fibrillation Follow-Up Investigation of Rhythm Management (AFFIRM) study. J Am Coll Cardiol. 2005;45:2026-2033.
45. de Vos CB, Pisters R, Nieuwlaat R, et al. Progression from paroxysmal to persistent atrial fibrillation: clinical correlates and prognosis. J Am Coll Cardiol. 2010;55:725-731.
46. Cheng X, Li X, He Y, et al. Catheter ablation versus anti-arrhythmic drug therapy for the management of atrial fibrillation: a meta-analysis. J Interv Card Electrophysiol. 2014;41:267-272.
47. Di Biase L, Mohanty P, Mohanty S, et al. Ablation versus amiodarone for treatment of persistent atrial fibrillation in patients with congestive heart failure and an implanted device: results from the AATAC multicenter randomized trial. Circulation. 2016;133:1637-1644.
48.
1. Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults. National implications for Rhythm Management and Stroke Prevention: The AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA. 2001;285:2370-2375.
2. Kannel WB, Wolf PA, Benjamin EJ, et al. Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: population-based estimates. Am J Cardiol. 1998;82:2N-9N.
3. Krahn AD, Manfreda J, Tate RB, et al. The natural history of atrial fibrillation: incidence, risk factors, and prognosis in the Manitoba follow-up study. Am J Med. 1995;98:476-484.
4. Ott A, Breteler MMB, de Bruyne MC, et al. Atrial fibrillation and dementia in a population-based study: The Rotterdam Study. Stroke. 1997;28:316-321.
5. January CT, Wann L, Alpert JS, et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2014;64:e1-e76.
6. Lin HJ, Wolf PA, Kelly-Hayes M, et al. Stroke severity in atrial fibrillation. Stroke. 1996;27:1760-1764.
7. Hsu JC, Maddox TM, Kennedy KF, et al. Oral anticoagulant therapy prescription in patients with atrial fibrillation across the spectrum of stroke risk: insights from the NCDR PINNACLE registry. JAMA Cardiol. 2016;1:55-62.
8. Olesen JB, Lip GY, Lindhardsen J, et al. Risks of thromboembolism and bleeding with thromboprophylaxis in patients with atrial fibrillation: a net clinical benefit analysis using a ‘real world’ nationwide cohort study. Thromb Haemost. 2011;106:739-749.
9. Steinberg BA, Kim S, Thomas L, et al. Lack of concordance between empirical scores and physician assessments of stroke and bleeding risk in atrial fibrillation: results from the Outcomes Registry for Better Informed Treatment of Atrial Fibrillation (ORBIT-AF) registry. Circulation. 2014;129:2005-2012.
10. Angaran P, Dorian P, Tan MK, et al. The risk stratification and stroke prevention therapy care gap in Canadian atrial fibrillation patients. Can J Cardiol. 2016;32:336-343.
11. Waldo AL, Feld GK. Inter-relationships of atrial fibrillation and atrial flutter: mechanisms and clinical implications. J Am Coll Cardiol. 2008;51:779-786.
12. Ellis K, Wazni O, Marrouche N, et al. Incidence of atrial fibrillation post-cavotricuspid isthmus ablation in patients with typical atrial flutter: left-atrial size as an independent predictor of atrial fibrillation recurrence. J Cardiovasc Electrophysiol. 2007;18:799-802.
13. Larsson SC, Drca N, Wolk A. Alcohol consumption and risk of atrial fibrillation: a prospective study and dose-response meta-analysis. J Am Coll Cardiol. 2014;64:281-289.
14. Calkins H, Kuck KH, Cappato R, et al. 2012 HRS/EHRA/ECAS expert consensus statement on catheter and surgical ablation of atrial fibrillation: recommendations for patient selection, procedural techniques, patient management and follow-up, definitions, endpoints, and research trial design. J Interv Card Electrophysiol. 2012;33:171-257.
15. Lip GY, Nieuwlaat R, Pisters R, et al. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: the Euro Heart Survey on Atrial Fibrillation. Chest. 2010;137:263-272.
16. Lip GYH, Frison L, Halperin JL, et al. Identifying patients at high risk for stroke despite anticoagulation: a comparison of contemporary stroke risk stratification schemes in an anticoagulated atrial fibrillation cohort. Stroke. 2010;41:2731-2738.
17. Olesen JB, Lip GYH, Hansen ML, et al. Validation of risk stratification schemes for predicting stroke and thromboembolism in patients with atrial fibrillation: nationwide cohort study. BMJ. 2011;342:d124.
18. Camm AJ, Lip GYH, De Caterina R, et al. 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J. 2012;33:2719-2747.
19. Olesen JB, Torp-Pedersen C, Hansen ML, et al. The value of the CHA2DS2-VASc score for refining stroke risk stratification in patients with atrial fibrillation with a CHADS2 score 0-1: a nationwide cohort study. Thromb Haemost. 2012;107:1172-1179.
20. Friberg L, Benson L, Rosenqvist M, et al. Assessment of female sex as a risk factor in atrial fibrillation in Sweden: nationwide retrospective cohort study. BMJ. 2012;344:e3522.
21. National Institute for Health and Clinical Excellence (NICE). Atrial fibrillation: the management of atrial fibrillation [CG180]. 2014. Available at: https://www.nice.org.uk/guidance/cg180. Accessed July 31, 2016.
22. Verma A, Cairns JA, Mitchell LB, et al. 2014 focused update of the Canadian Cardiovascular Society Guidelines for the management of atrial fibrillation. Can J Cardiol. 2014;30:1114-1130.
23. Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361:1139-1151.
24. Patel MR, Mahaffey KW, Garg J, et al. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365:883-891.
25. Giugliano RP, Ruff CT, Braunwald E, et al. Edoxaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2013;369:2093-2104.
26. Granger CB, Alexander JH, McMurray JJV, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365:981-992.
27. Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383:955-962.
28. Morimoto T, Crawford B, Wada K, et al. Comparative efficacy and safety of novel oral anticoagulants in patients with atrial fibrillation: a network meta-analysis with the adjustment for the possible bias from open label studies. J Cardiol. 2015;66:466-474.
29. Verdecchia P, Angeli F, Bartolini C, et al. Safety and efficacy of non-vitamin K oral anticoagulants in non-valvular atrial fibrillation: a Bayesian meta-analysis approach. Expert Opin Drug Saf. 2015;14:7-20.
30. Micromedex® 2.0 (electronic version). Truven Health Analytics, Greenwood Village, Colorado, USA. Available at: http://www.micromedexsolutions.com. Accessed August 18, 2016.
31. GoodRx. Available at: https://www.goodrx.com. Accessed August 18, 2016.
32. Del-Carpio Munoz F, Gharacholou SM, Munger TM, et al. Meta-analysis of renal function on the safety and efficacy of novel oral anticoagulants for atrial fibrillation. Am J Cardiol. 2016;117:69-75.
33. Fang MC, Go AS, Chang Y, et al. A new risk scheme to predict warfarin-associated hemorrhage: the ATRIA (Anticoagulation and Risk Factors in Atrial Fibrillation) Study. J Am Coll Cardiol. 2011;58:395-401.
34. Gage BF, Yan Y, Milligan PE, et al. Clinical classification schemes for predicting hemorrhage: results from the National Registry of Atrial Fibrillation (NRAF). Am Heart J. 2006;151:713-719.
35. Pisters R, Lane DA, Nieuwlaat R, et al. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest. 2010;138:1093-1100.
36. Zhu W, He W, Guo L, et al. The HAS-BLED Score for predicting major bleeding risk in anticoagulated patients with atrial fibrillation: a systematic review and meta-analysis. Clin Cardiol. 2015;38:555-561.
37. Chai-Adisaksopha C, Hillis C, Monreal M, et al. Thromboembolic events, recurrent bleeding and mortality after resuming anticoagulant following gastrointestinal bleeding. A meta-analysis. Thromb Haemost. 2015;114:819-825.
38. Xu H, Xie X, Wang B, et al. Efficacy and safety of percutaneous left atrial appendage occlusion for stroke prevention in nonvalvular atrial fibrillation: a meta-analysis of contemporary studies. Heart Lung Circ. 2016;25:1107-1117.
39. Siu CW, Lau CP, Lee WL, et al. Intravenous diltiazem is superior to intravenous amiodarone or digoxin for achieving ventricular rate control in patients with acute uncomplicated atrial fibrillation. Crit Care Med. 2009;37:2174-2179.
40. Clemo HF, Wood MA, Gilligan DM, et al. Intravenous amiodarone for acute heart rate control in the critically ill patient with atrial tachyarrhythmias. Am J Cardiol. 1998;81:594-598.
41. The Atrial Fibrillation Follow-Up Investigation of Rhythm Management (AFFIRM) Investigators. A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med. 2002;347:1825-1833.
42. Van Gelder IC, Groenveld HF, Crijns HJGM, et al. Lenient versus strict rate control in patients with atrial fibrillation. N Engl J Med. 2010;362:1363-1373.
43. Wang ZQ, Zhang R, Chen MT, et al. Digoxin is associated with increased all-cause mortality in patients with atrial fibrillation regardless of concomitant heart failure: a meta-analysis. J Cardiovasc Pharmacol. 2015;66:270-275.
44. Olshansky B, Heller EN, Mitchell LB, et al. Are transthoracic echocardiographic parameters associated with atrial fibrillation recurrence or stroke? Results from the Atrial Fibrillation Follow-Up Investigation of Rhythm Management (AFFIRM) study. J Am Coll Cardiol. 2005;45:2026-2033.
45. de Vos CB, Pisters R, Nieuwlaat R, et al. Progression from paroxysmal to persistent atrial fibrillation: clinical correlates and prognosis. J Am Coll Cardiol. 2010;55:725-731.
46. Cheng X, Li X, He Y, et al. Catheter ablation versus anti-arrhythmic drug therapy for the management of atrial fibrillation: a meta-analysis. J Interv Card Electrophysiol. 2014;41:267-272.
47. Di Biase L, Mohanty P, Mohanty S, et al. Ablation versus amiodarone for treatment of persistent atrial fibrillation in patients with congestive heart failure and an implanted device: results from the AATAC multicenter randomized trial. Circulation. 2016;133:1637-1644.
48.
PRACTICE RECOMMENDATIONS
› Use the CHA2DS2-VASc score to assess the risk of thromboembolism, including ischemic stroke. A
› Consider prescribing a direct oral anticoagulant (DOAC) instead of warfarin for patients with nonvalvular atrial fibrillation (AF) because they are superior at preventing strokes and lowering all-cause mortality in this population. B
› Do not use a DOAC in patients with mechanical heart valves, hemodynamically significant mitral stenosis, or severe chronic kidney disease (estimated glomerular filtration rate [eGFR] <30 mL/min/1.73 m2). A
› Pursue a rate-control strategy for most patients with AF, although rhythm control may be preferable for younger (<65 years) symptomatic patients. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
How to provide effective pain treatment
![]()
Chief, UW Division of Pain Medicine;
Clinical Professor, Department of Medicine and
Department of Anesthesiology & Pain Medicine
University of Washington, Seattle
![]()
Chief, UW Division of Pain Medicine;
Clinical Professor, Department of Medicine and
Department of Anesthesiology & Pain Medicine
University of Washington, Seattle
![]()
Chief, UW Division of Pain Medicine;
Clinical Professor, Department of Medicine and
Department of Anesthesiology & Pain Medicine
University of Washington, Seattle
Drugs may be effective against hematologic, other cancers

Image courtesy of PNAS
A diabetes medication and an antihypertensive drug may prove effective in the treatment of hematologic malignancies and other cancers, according to preclinical research published in Science Advances.
Past research has shown that metformin, a drug used to treat type 2 diabetes, has anticancer properties.
However, the usual therapeutic dose is too low to effectively fight cancer, and higher doses of metformin could be too toxic.
With the current study, researchers found that the antihypertensive drug syrosingopine enhances the anticancer efficacy of metformin without harming normal blood cells.
The team screened over a thousand drugs to find one that could boost metformin’s efficacy against cancers.
They identified syrosingopine and tested it in combination with metformin—at concentrations substantially below the drugs’ therapeutic thresholds—on a range of cancer cell lines and in mouse models of liver cancer.
Thirty-five of the 43 cell lines tested were susceptible to both syrosingopine and metformin. This included leukemia, lymphoma, and multiple myeloma cell lines.
In addition, the mice given a short course of syrosingopine and metformin experienced a reduction in the number of visible liver tumors.
The researchers also tested syrosingopine and metformin in peripheral blasts from 12 patients with acute myeloid leukemia and a patient with blast crisis chronic myeloid leukemia. All 13 samples responded to the treatment.
On the other hand, syrosingopine and metformin did not affect peripheral blood cells from healthy subjects.
“[A]lmost all tumor cells were killed by this cocktail and at doses that are actually not toxic to normal cells,” said study author Don Benjamin, of the University of Basel in Switzerland.
“And the effect was exclusively confined to cancer cells, as the blood cells from healthy donors were insensitive to the treatment.”
The researchers believe metformin functions by lowering blood glucose levels for cancer cells, starving them of essential nutrients needed for their survival. However, it is not clear how syrosingopine works in conjunction with metformin.
The team emphasized the need for more research evaluating the drugs in combination.
“We have been able to show that the 2 known drugs lead to more profound effects on cancer cell proliferation than each drug alone,” Dr Benjamin said. “The data from this study support the development of combination approaches for the treatment of cancer patients.” ![]()
Image courtesy of PNAS
A diabetes medication and an antihypertensive drug may prove effective in the treatment of hematologic malignancies and other cancers, according to preclinical research published in Science Advances.
Past research has shown that metformin, a drug used to treat type 2 diabetes, has anticancer properties.
However, the usual therapeutic dose is too low to effectively fight cancer, and higher doses of metformin could be too toxic.
With the current study, researchers found that the antihypertensive drug syrosingopine enhances the anticancer efficacy of metformin without harming normal blood cells.
The team screened over a thousand drugs to find one that could boost metformin’s efficacy against cancers.
They identified syrosingopine and tested it in combination with metformin—at concentrations substantially below the drugs’ therapeutic thresholds—on a range of cancer cell lines and in mouse models of liver cancer.
Thirty-five of the 43 cell lines tested were susceptible to both syrosingopine and metformin. This included leukemia, lymphoma, and multiple myeloma cell lines.
In addition, the mice given a short course of syrosingopine and metformin experienced a reduction in the number of visible liver tumors.
The researchers also tested syrosingopine and metformin in peripheral blasts from 12 patients with acute myeloid leukemia and a patient with blast crisis chronic myeloid leukemia. All 13 samples responded to the treatment.
On the other hand, syrosingopine and metformin did not affect peripheral blood cells from healthy subjects.
“[A]lmost all tumor cells were killed by this cocktail and at doses that are actually not toxic to normal cells,” said study author Don Benjamin, of the University of Basel in Switzerland.
“And the effect was exclusively confined to cancer cells, as the blood cells from healthy donors were insensitive to the treatment.”
The researchers believe metformin functions by lowering blood glucose levels for cancer cells, starving them of essential nutrients needed for their survival. However, it is not clear how syrosingopine works in conjunction with metformin.
The team emphasized the need for more research evaluating the drugs in combination.
“We have been able to show that the 2 known drugs lead to more profound effects on cancer cell proliferation than each drug alone,” Dr Benjamin said. “The data from this study support the development of combination approaches for the treatment of cancer patients.” ![]()
Image courtesy of PNAS
A diabetes medication and an antihypertensive drug may prove effective in the treatment of hematologic malignancies and other cancers, according to preclinical research published in Science Advances.
Past research has shown that metformin, a drug used to treat type 2 diabetes, has anticancer properties.
However, the usual therapeutic dose is too low to effectively fight cancer, and higher doses of metformin could be too toxic.
With the current study, researchers found that the antihypertensive drug syrosingopine enhances the anticancer efficacy of metformin without harming normal blood cells.
The team screened over a thousand drugs to find one that could boost metformin’s efficacy against cancers.
They identified syrosingopine and tested it in combination with metformin—at concentrations substantially below the drugs’ therapeutic thresholds—on a range of cancer cell lines and in mouse models of liver cancer.
Thirty-five of the 43 cell lines tested were susceptible to both syrosingopine and metformin. This included leukemia, lymphoma, and multiple myeloma cell lines.
In addition, the mice given a short course of syrosingopine and metformin experienced a reduction in the number of visible liver tumors.
The researchers also tested syrosingopine and metformin in peripheral blasts from 12 patients with acute myeloid leukemia and a patient with blast crisis chronic myeloid leukemia. All 13 samples responded to the treatment.
On the other hand, syrosingopine and metformin did not affect peripheral blood cells from healthy subjects.
“[A]lmost all tumor cells were killed by this cocktail and at doses that are actually not toxic to normal cells,” said study author Don Benjamin, of the University of Basel in Switzerland.
“And the effect was exclusively confined to cancer cells, as the blood cells from healthy donors were insensitive to the treatment.”
The researchers believe metformin functions by lowering blood glucose levels for cancer cells, starving them of essential nutrients needed for their survival. However, it is not clear how syrosingopine works in conjunction with metformin.
The team emphasized the need for more research evaluating the drugs in combination.
“We have been able to show that the 2 known drugs lead to more profound effects on cancer cell proliferation than each drug alone,” Dr Benjamin said. “The data from this study support the development of combination approaches for the treatment of cancer patients.” ![]()