User login
Current Concepts in Labral Repair and Refixation: Anatomical Approach to Labral Management
Take-Home Points
- Labral preservation is recommended when possible to ensure restoration of suction seal, stability, and contact pressure of the hip joint.
- Over 95% of labral tears can be addressed with primary repair.
- Consider using an accessory portal (ie, DALA) to allow for more anatomic placement of suture anchor.
- Mattress stitch when labrum >3 mm and looped stitch when labrum <3 mm.
- 10Control labral repair to avoid excessive inversion or eversion.
Arthroscopic labral repair and refixation have garnered much attention over the past several years. Restoration of suction seal and native labral function has been an evolving focus for achieving excellent results in hip preservation surgery.1-6 Given the superior results of labral repair, including level I evidence, repair or refixation should be pursued whenever possible.7 Authors have reported using several labral management techniques: débridement, labralization, looped suture fixation, base stitch fixation, inversion-eversion, and reconstruction.7-13 The optimal technique is yet to be determined. When possible, steps should be taken to repair the labrum to an anatomical position. Absolute indications for labral repair are a confirmed intra-articular diagnosis with symptomatic pain, joint space >2 mm with or without femoroacetabular impingement (FAI), labral tear or instability, and failed conservative management.9,11,12,14,15 More important, the surgeon must have a clear etiology of the pathologic cause of the tear and be aware of the limitations of the procedure. Labral repair is relatively contraindicated in end-stage arthritis and has failed when used alone in undiagnosed dysplasia or hip instability.16 In this article, we discuss indications for labral repair; describe Dr. Mather’s preoperative planning, labral repair technique, and postoperative care; and review published outcomes and future trends in labral repair.
Indications
At our institution, anatomical labral repair is the preferred procedure for most primary and revision hip arthroscopy procedures. We aim to restore the suction seal, re-create the contact of the labrum and the femoral head to facilitate proprioception, and restore normal stability of the labrum. Indications for primary repair are labrum width >3 mm, no more than 2 repairs, and ability to hold a suture. Our indications for reconstruction or débridement are stage 3 irreparable labral tear, calcified/cystic labrum, and multiple failed labral repairs or reconstructions. The decision to perform labral débridement or reconstruction is made on a case-by-case basis but is primarily influenced by the stability of the hip joint and the activity goals of the patient. If preoperative presentation and intraoperative examination suggest labral instability as a major component of the pathology, or if the patient wants to return to high-demand activity, we more strongly favor reconstruction over débridement. In our experience, with the technique described in this article, more than 95% of all primary labral tears can be addressed with repair.
Preoperative Planning
The goals in hip preservation surgery are to identify and address the underlying cause of the labral tear, whether it be FAI syndrome, trauma, labral instability, or all 3, and to re-create the anatomy and biomechanics of the acetabular labrum. For repair, we prefer an inversion-eversion technique with independent control of the labrum. Our initial work-up includes a thorough history and physical examination with baseline patient-reported outcome scores. Standard erect anteroposterior pelvis, Dunn lateral, and false-profile radiographs are obtained. Standard measurements of lateral center edge angle, anterior center edge angle, Tönnis angle, Tönnis grade, lateral joint space, and head extrusion indices are evaluated. Selective in-office ultrasound-guided injections are used to confirm an intra-articular source of pain. At our institution, noncontrast 3.0 Tesla magnetic resonance imaging (MRI) with volumetric interpolated breath-hold examination (VIBE) sequencing and 3-dimensional rendering is obtained for evaluation of labral and FAI morphology.17 All advanced imaging is performed without arthrogram or radiation exposure (Figures 1A-1C). 
With use of the radiographs and the MRI scans, we engage the patient in an informed discussion about the labral tear, FAI, and concomitant pathology. We discuss expected outcomes of conservative or operative management given the patient’s expected functional activities, and inform the patient that primary repair is indicated for many others in similar situations. The potential for possible labral reconstruction is discussed if the patient had prior intra-articular hip surgery, has a large calcified labrum or a cystic labrum, is an athlete with failed prior surgery, or is younger than 40 years.
Labral Repair Technique
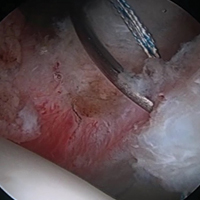
The patient is taken to the surgical suite, and a general anesthetic is administered. A peripheral nerve block is not routinely used. The patient’s feet are padded, and boots for the traction table are applied. The patient is carefully placed on a Hana table in modified supine position. Balanced traction is used to achieve proper joint distraction. The C-arm is used to verify proper distraction, assess hip stability, and achieve standard anterolateral (AL) portal placement. A midanterior portal (MAP) is created and an interportal capsulotomy is performed. Capsular suspension is performed with the InJector II Capsule Restoration System (Stryker Sports Medicine) and typically 4 or 5 high-strength No. 2 sutures (Zipline; Stryker Sports Medicine).19 Diagnostic arthroscopy is performed to identify the tear type, measure the labral width, determine the impingement area, and identify the intra-articular pathology. After the intra-articular pathology is addressed, a radiofrequency Ambient HIPVAC 50 Coblation Wand (Smith & Nephew) is used to expose the acetabular rim and subspine as indicated. Acetabuloplasty or subspine decompression is performed, and then a primary repair or refixation of the labrum is performed. We do not routinely detach the labrum for acetabular rim trimming. A crucial step here is to expose a bleeding surface to which the labrum can be repaired. If the rim is sclerotic, or the rim cannot be removed because of underlying low acetabular coverage, we prefer to obtain the bleeding surface with a microdrilling device (Stryker) that is routinely used for acetabular microfracture.
Labrum quality is used to determine which repair method to use. A hypertrophic labrum is debulked. The acetabular rim is seldom resected >3 mm, but, when it is, the newly exposed cartilage is removed. We have found that >3 mm of residual cartilage prevents refixation of the labrum directly to the bone and may interfere with anatomical positioning. When a labrum is <3 mm in width or will not hold a base technique, repair stability is the priority, and a looped method is used. A knotless anchor with No. 1 permanent suture designed for hip labral repair (CinchLock; Stryker) is our first-line anchor choice. A distal anterolateral accessory (DALA) portal is created with an outside-in technique, and anchors are drilled through this portal into zones 2 to 4 (Figures 2A-2E). 
A 2.4-mm drill guide is advanced through the DALA portal and placed in the appropriate position for drilling. We aim for 1 mm to 2 mm from the chondrolabral junction. Next, the probe is placed intra-articular and medial to the anchor insertion site, and the anchor is loaded and then inserted around the probe (Figures 3A-3E). 

The hip is then reduced. If indicated, a T-capsulotomy is performed for femoral osteochondroplasty. 
Postoperative Care
Patients are placed in a postoperative hip brace and use a continuous passive motion machine 6 hours a day for 2 weeks, and an ice machine. They maintain 30 lb of foot-flat weight-bearing for 3 weeks, and begin a standard labral repair protocol on postoperative days 3 to 7.
Discussion
Hip labral preservation has evolved over the past 10 years, and current options for labral management include excision, débridement, labralization, repair, and reconstruction.1-13 Labral excision was studied by Miozzari and colleagues,8 who postulated on the basis of animal models that the labrum may regenerate. In their series of 9 patients treated with surgical hip dislocation and labral excision at average 4-year follow-up, repeat magnetic resonance angiography revealed no regeneration of tissue—modified Harris Hip Score was 83. The hip scores were less than those of patients treated with the same procedure with repair, and the authors concluded that defining labral débridement versus excision in the literature, and treating patients with primary repair or reconstruction techniques, may lead to better results. Their study used a small sample and was limited to an open procedure. Arthroscopic labral débridement in isolation was also a poor option for treatment of a labral tear. In a 2-year follow-up of 59 isolated labral débridement procedures, Krych and colleagues9 found 47% combined poor results.
There is level I evidence of the importance of labral repair. In 2013, Krych and colleagues7 conducted a randomized control trial of 38 female patients who underwent hip arthroscopy for FAI. At time of surgery, patients were randomly assigned to either débridement or repair. At 1-year follow-up, activities of daily living and Sports specific Hip Outcome Scores were statistically significantly superior in the repair group. On a subjective scale, 94% vs 78% of patients reported normal or near normal hips in the repair versus débridement groups respectively. Ayeni and colleagues20 performed a systematic review of 6 studies in an attempt to develop labral management recommendations. Five of the studies (N = 490 patients total) had improved results with labral repair over reconstruction. Although the studies had a low level of evidence, they found a trend toward improved results with labral repair. These studies highlight the importance of labral preservation and proper FAI management.
Techniques for labrum repair have advanced as well—from a looped suture technique to a base stitch and knotless independent tensioning.11-13 Restoration of the hip labrum function as a suction seal, fluid circulator and anatomic capsular repair is paramount to excellent results and stresses the importance of performing an anatomic labral repair.1-6 Knotless anchor repair is not novel and has been previously described. Fry and Domb12 reported on a knotless labral repair technique that uses push-lock devices (Arthrex) that do not allow for independent tensioning. Inversion-eversion was introduced to the literature by Moreira and colleagues,13 who described an independent tensioning technique that uses speed-lock anchors (Smith & Nephew). Our technique differs in that it involves a DALA portal; labral reduction and tensioning with a probe assist to ensure the second pass of the base stitch is at the apex of the labrum; and use of No. 1 instead of No. 2 suture. Although seemingly subtle, these differences allow for proper anchor placement nearer the rim, additional support in achieving precise suture placement, and less disruption of small labra. These differences are particularly relevant for smaller labra.
Evaluating repair techniques on the basis of high-evidence literature is challenging. In a matched-cohort study of 220 patients, Jackson and colleagues21 compared 2 techniques: looped and base stitch. At 2-year follow-up, patients in both groups showed improvement, and there was no statistically significant difference in patient-reported outcome measures between the groups. Sawyer and colleagues22 studied the outcomes of 326 consecutive patients who underwent looped, pierced, or combined labral repair at an average 32-month follow-up. The groups’ revision rates were comparable, each group improved in postoperative patient-reported outcomes, and the pierced group had significantly higher preoperative scores on the Western Ontario and McMaster Universities Osteoarthritis Index. These studies described a base or pierce repair that did not differ from a looped repair, though the techniques did not allow for independent tensioning to re-create an anatomical inversion-eversion repair and may have altered the reported outcomes.
Our current technique uses independent tensioning of the repair to allow control of labrum inversion-eversion to give an anatomical repair with restoration of the suction seal. Preoperative planning, addressing the FAI appropriately, proper suture-passing technique, controlling the labrum in inversion-eversion fashion, and anatomical labral repair are the elements of Dr. Mather’s preferred method for preserving the native labrum and allowing it to assume its native function.
Future Directions
As our understanding of FAI and labral function evolves, labral preservation surgery continues to advance. With surgeons continually developing new techniques and following up on previous techniques, the ability to preserve the native hip with lasting procedures evolves as well. Proper identification of the underlying cause of the labral tear and proper anatomical repair are paramount to the success of FAI surgery.
Am J Orthop. 2017;46(1):42-48. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Philippon MJ, Nepple JJ, Campbell KJ, et al. The hip fluid seal—part I: the effect of an acetabular labral tear, repair, resection and reconstruction on hip fluid pressurization. Knee Surg Sports Traumatol Arthrosc. 2014;22(4):722-729.
2. Nepple JJ, Philippon MJ, Campbell KJ, et al. The hip fluid seal—part II: the effect of an acetabular labral tear, repair, resection and reconstruction on hip stability to distraction. Knee Surg Sports Traumatol Arthrosc. 2014;22(4):730-736.
3. Dwyer MK, Jones HL, Hogan MG, Field RE, McCarthy JC, Noble PC. The acetabular labrum regulates fluid circulation of the hip joint during functional activities. Am J Sports Med. 2014;42(4):812-819.
4. Greaves LL, Gilbart MK, Yung AC, Kozlowski, Wilson DR. Effect of acetabular labral tears, repair and resection on hip cartilage strain: a 7T MR study. J Biomech. 2010;43(5):858-863.
5. Freehill MT, Safran MR. The labrum of the hip: diagnosis and rationale for surgical correction. Clin Sports Med. 2011;30(2):293-315.
6. Myers CA, Register BC, Lertwanich P, et al. Role of the acetabular labrum and the iliofemoral ligament in hip stability: an in vitro biplane fluoroscopy study. Am J Sports Med. 2011;39(suppl):85S-91S.
7. Krych AJ, Thompson M, Knutson Z, Scoon J, Coleman SH. Arthroscopic labral repair versus selective labral debridement in female patients with femoroacetabular impingement: a prospective randomized study. Arthroscopy. 2013;29(1):46-53.
8. Miozzari HH, Celia M, Clark JM, Werlen S, Naal FD, Nötzli HP. No regeneration of the human acetabular labrum after excision to bone. Clin Orthop Relat Res. 2015;473(4):1349-1357.
9. Krych AJ, Kuzma SA, Kovachevich R, Hudgens JL, Stuart MJ, Levy BA. Modest mid-term outcomes after isolated arthroscopic debridement of acetabular labral tears. Knee Surg Sports Traumatol Arthrosc. 2014;22(4):763-767.
10. Matsuda DK. Arthroscopic labralization of the hip: an alternative to labral reconstruction. Arthrosc Tech. 2014;3(1):e131-e133.
11. Philippon MJ, Faucet SC, Briggs KK. Arthroscopic hip labral repair. Arthrosc Tech. 2013;2(2):e73-e76.
12. Fry D, Domb B. Labral base refixation in the hip: rationale and technique for an anatomic approach to labral repair. Arthroscopy. 2010;26(9 suppl):S81-S89.
13. Moreira B, Pascual-Garrido C, Chadayamurri V, Mei-Dan O. Eversion-inversion labral repair and reconstruction technique for optimal suction seal. Arthrosc Tech. 2015;4(6):e697-e700.
14. Mook WR, Briggs KK, Philippon MJ. Evidence and approach for management of labral deficiency: the role for labral reconstruction. Sports Med Arthrosc. 2015;23(4):205-212.
15. Gupta A, Suarez-Ahedo C, Redmond JM, et al. Best practices during hip arthroscopy: aggregate recommendations of high-volume surgeons. Arthroscopy. 2015;31(9):1722-1727.
16. Yeung M, Kowalczuk M, Simunovic N, Ayeni OR. Hip arthroscopy in the setting of hip dysplasia: a systematic review. Bone Joint Res. 2016;5(6):225-231.
17. Hash TW. Magnetic resonance imaging of the hip. In: Nho SJ, Leunig M, Larson CM, Bedi A, Kelly BT, eds. Hip Arthroscopy and Hip Joint Preservation Surgery, Vol. 1. New York, NY: Springer; 2015:65-113.
18. Sutter R, Zubler V, Hoffmann A, et al. Hip MRI: how useful is intraarticular contrast material for evaluating surgically proven lesions of the labrum and articular cartilage? AJR Am J Roentgenol. 2014;202(1):160-169.
19. Federer AE, Karas V, Nho S, Coleman SH, Mather RC 3rd. Capsular suspension technique for hip arthroscopy. Arthrosc Tech. 2015;4(4):e317-e322.
20. Ayeni OR, Adamich J, Farrokhyar F, et al. Surgical management of labral tears during femoroacetabular impingement surgery: a systematic review. Knee Surg Sports Traumatol Arthrosc. 2014;22(4):756-762.
21. Jackson TJ, Hammarstedt JE, Vemula SP, Domb BG. Acetabular labral base repair versus circumferential suture repair: a matched-paired comparison of clinical outcomes. Arthroscopy. 2015;31(9):1716-1721.
22. Sawyer GA, Briggs KK, Dornan GJ, Ommen ND, Philippon MJ. Clinical outcomes after arthroscopic hip labral repair using looped versus pierced suture techniques. Am J Sports Med. 2015;43(7):1683-1688.
Take-Home Points
- Labral preservation is recommended when possible to ensure restoration of suction seal, stability, and contact pressure of the hip joint.
- Over 95% of labral tears can be addressed with primary repair.
- Consider using an accessory portal (ie, DALA) to allow for more anatomic placement of suture anchor.
- Mattress stitch when labrum >3 mm and looped stitch when labrum <3 mm.
- 10Control labral repair to avoid excessive inversion or eversion.
Arthroscopic labral repair and refixation have garnered much attention over the past several years. Restoration of suction seal and native labral function has been an evolving focus for achieving excellent results in hip preservation surgery.1-6 Given the superior results of labral repair, including level I evidence, repair or refixation should be pursued whenever possible.7 Authors have reported using several labral management techniques: débridement, labralization, looped suture fixation, base stitch fixation, inversion-eversion, and reconstruction.7-13 The optimal technique is yet to be determined. When possible, steps should be taken to repair the labrum to an anatomical position. Absolute indications for labral repair are a confirmed intra-articular diagnosis with symptomatic pain, joint space >2 mm with or without femoroacetabular impingement (FAI), labral tear or instability, and failed conservative management.9,11,12,14,15 More important, the surgeon must have a clear etiology of the pathologic cause of the tear and be aware of the limitations of the procedure. Labral repair is relatively contraindicated in end-stage arthritis and has failed when used alone in undiagnosed dysplasia or hip instability.16 In this article, we discuss indications for labral repair; describe Dr. Mather’s preoperative planning, labral repair technique, and postoperative care; and review published outcomes and future trends in labral repair.
Indications
At our institution, anatomical labral repair is the preferred procedure for most primary and revision hip arthroscopy procedures. We aim to restore the suction seal, re-create the contact of the labrum and the femoral head to facilitate proprioception, and restore normal stability of the labrum. Indications for primary repair are labrum width >3 mm, no more than 2 repairs, and ability to hold a suture. Our indications for reconstruction or débridement are stage 3 irreparable labral tear, calcified/cystic labrum, and multiple failed labral repairs or reconstructions. The decision to perform labral débridement or reconstruction is made on a case-by-case basis but is primarily influenced by the stability of the hip joint and the activity goals of the patient. If preoperative presentation and intraoperative examination suggest labral instability as a major component of the pathology, or if the patient wants to return to high-demand activity, we more strongly favor reconstruction over débridement. In our experience, with the technique described in this article, more than 95% of all primary labral tears can be addressed with repair.
Preoperative Planning
The goals in hip preservation surgery are to identify and address the underlying cause of the labral tear, whether it be FAI syndrome, trauma, labral instability, or all 3, and to re-create the anatomy and biomechanics of the acetabular labrum. For repair, we prefer an inversion-eversion technique with independent control of the labrum. Our initial work-up includes a thorough history and physical examination with baseline patient-reported outcome scores. Standard erect anteroposterior pelvis, Dunn lateral, and false-profile radiographs are obtained. Standard measurements of lateral center edge angle, anterior center edge angle, Tönnis angle, Tönnis grade, lateral joint space, and head extrusion indices are evaluated. Selective in-office ultrasound-guided injections are used to confirm an intra-articular source of pain. At our institution, noncontrast 3.0 Tesla magnetic resonance imaging (MRI) with volumetric interpolated breath-hold examination (VIBE) sequencing and 3-dimensional rendering is obtained for evaluation of labral and FAI morphology.17 All advanced imaging is performed without arthrogram or radiation exposure (Figures 1A-1C).
With use of the radiographs and the MRI scans, we engage the patient in an informed discussion about the labral tear, FAI, and concomitant pathology. We discuss expected outcomes of conservative or operative management given the patient’s expected functional activities, and inform the patient that primary repair is indicated for many others in similar situations. The potential for possible labral reconstruction is discussed if the patient had prior intra-articular hip surgery, has a large calcified labrum or a cystic labrum, is an athlete with failed prior surgery, or is younger than 40 years.
Labral Repair Technique
The patient is taken to the surgical suite, and a general anesthetic is administered. A peripheral nerve block is not routinely used. The patient’s feet are padded, and boots for the traction table are applied. The patient is carefully placed on a Hana table in modified supine position. Balanced traction is used to achieve proper joint distraction. The C-arm is used to verify proper distraction, assess hip stability, and achieve standard anterolateral (AL) portal placement. A midanterior portal (MAP) is created and an interportal capsulotomy is performed. Capsular suspension is performed with the InJector II Capsule Restoration System (Stryker Sports Medicine) and typically 4 or 5 high-strength No. 2 sutures (Zipline; Stryker Sports Medicine).19 Diagnostic arthroscopy is performed to identify the tear type, measure the labral width, determine the impingement area, and identify the intra-articular pathology. After the intra-articular pathology is addressed, a radiofrequency Ambient HIPVAC 50 Coblation Wand (Smith & Nephew) is used to expose the acetabular rim and subspine as indicated. Acetabuloplasty or subspine decompression is performed, and then a primary repair or refixation of the labrum is performed. We do not routinely detach the labrum for acetabular rim trimming. A crucial step here is to expose a bleeding surface to which the labrum can be repaired. If the rim is sclerotic, or the rim cannot be removed because of underlying low acetabular coverage, we prefer to obtain the bleeding surface with a microdrilling device (Stryker) that is routinely used for acetabular microfracture.
Labrum quality is used to determine which repair method to use. A hypertrophic labrum is debulked. The acetabular rim is seldom resected >3 mm, but, when it is, the newly exposed cartilage is removed. We have found that >3 mm of residual cartilage prevents refixation of the labrum directly to the bone and may interfere with anatomical positioning. When a labrum is <3 mm in width or will not hold a base technique, repair stability is the priority, and a looped method is used. A knotless anchor with No. 1 permanent suture designed for hip labral repair (CinchLock; Stryker) is our first-line anchor choice. A distal anterolateral accessory (DALA) portal is created with an outside-in technique, and anchors are drilled through this portal into zones 2 to 4 (Figures 2A-2E).
A 2.4-mm drill guide is advanced through the DALA portal and placed in the appropriate position for drilling. We aim for 1 mm to 2 mm from the chondrolabral junction. Next, the probe is placed intra-articular and medial to the anchor insertion site, and the anchor is loaded and then inserted around the probe (Figures 3A-3E).
The hip is then reduced. If indicated, a T-capsulotomy is performed for femoral osteochondroplasty.
Postoperative Care
Patients are placed in a postoperative hip brace and use a continuous passive motion machine 6 hours a day for 2 weeks, and an ice machine. They maintain 30 lb of foot-flat weight-bearing for 3 weeks, and begin a standard labral repair protocol on postoperative days 3 to 7.
Discussion
Hip labral preservation has evolved over the past 10 years, and current options for labral management include excision, débridement, labralization, repair, and reconstruction.1-13 Labral excision was studied by Miozzari and colleagues,8 who postulated on the basis of animal models that the labrum may regenerate. In their series of 9 patients treated with surgical hip dislocation and labral excision at average 4-year follow-up, repeat magnetic resonance angiography revealed no regeneration of tissue—modified Harris Hip Score was 83. The hip scores were less than those of patients treated with the same procedure with repair, and the authors concluded that defining labral débridement versus excision in the literature, and treating patients with primary repair or reconstruction techniques, may lead to better results. Their study used a small sample and was limited to an open procedure. Arthroscopic labral débridement in isolation was also a poor option for treatment of a labral tear. In a 2-year follow-up of 59 isolated labral débridement procedures, Krych and colleagues9 found 47% combined poor results.
There is level I evidence of the importance of labral repair. In 2013, Krych and colleagues7 conducted a randomized control trial of 38 female patients who underwent hip arthroscopy for FAI. At time of surgery, patients were randomly assigned to either débridement or repair. At 1-year follow-up, activities of daily living and Sports specific Hip Outcome Scores were statistically significantly superior in the repair group. On a subjective scale, 94% vs 78% of patients reported normal or near normal hips in the repair versus débridement groups respectively. Ayeni and colleagues20 performed a systematic review of 6 studies in an attempt to develop labral management recommendations. Five of the studies (N = 490 patients total) had improved results with labral repair over reconstruction. Although the studies had a low level of evidence, they found a trend toward improved results with labral repair. These studies highlight the importance of labral preservation and proper FAI management.
Techniques for labrum repair have advanced as well—from a looped suture technique to a base stitch and knotless independent tensioning.11-13 Restoration of the hip labrum function as a suction seal, fluid circulator and anatomic capsular repair is paramount to excellent results and stresses the importance of performing an anatomic labral repair.1-6 Knotless anchor repair is not novel and has been previously described. Fry and Domb12 reported on a knotless labral repair technique that uses push-lock devices (Arthrex) that do not allow for independent tensioning. Inversion-eversion was introduced to the literature by Moreira and colleagues,13 who described an independent tensioning technique that uses speed-lock anchors (Smith & Nephew). Our technique differs in that it involves a DALA portal; labral reduction and tensioning with a probe assist to ensure the second pass of the base stitch is at the apex of the labrum; and use of No. 1 instead of No. 2 suture. Although seemingly subtle, these differences allow for proper anchor placement nearer the rim, additional support in achieving precise suture placement, and less disruption of small labra. These differences are particularly relevant for smaller labra.
Evaluating repair techniques on the basis of high-evidence literature is challenging. In a matched-cohort study of 220 patients, Jackson and colleagues21 compared 2 techniques: looped and base stitch. At 2-year follow-up, patients in both groups showed improvement, and there was no statistically significant difference in patient-reported outcome measures between the groups. Sawyer and colleagues22 studied the outcomes of 326 consecutive patients who underwent looped, pierced, or combined labral repair at an average 32-month follow-up. The groups’ revision rates were comparable, each group improved in postoperative patient-reported outcomes, and the pierced group had significantly higher preoperative scores on the Western Ontario and McMaster Universities Osteoarthritis Index. These studies described a base or pierce repair that did not differ from a looped repair, though the techniques did not allow for independent tensioning to re-create an anatomical inversion-eversion repair and may have altered the reported outcomes.
Our current technique uses independent tensioning of the repair to allow control of labrum inversion-eversion to give an anatomical repair with restoration of the suction seal. Preoperative planning, addressing the FAI appropriately, proper suture-passing technique, controlling the labrum in inversion-eversion fashion, and anatomical labral repair are the elements of Dr. Mather’s preferred method for preserving the native labrum and allowing it to assume its native function.
Future Directions
As our understanding of FAI and labral function evolves, labral preservation surgery continues to advance. With surgeons continually developing new techniques and following up on previous techniques, the ability to preserve the native hip with lasting procedures evolves as well. Proper identification of the underlying cause of the labral tear and proper anatomical repair are paramount to the success of FAI surgery.
Am J Orthop. 2017;46(1):42-48. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- Labral preservation is recommended when possible to ensure restoration of suction seal, stability, and contact pressure of the hip joint.
- Over 95% of labral tears can be addressed with primary repair.
- Consider using an accessory portal (ie, DALA) to allow for more anatomic placement of suture anchor.
- Mattress stitch when labrum >3 mm and looped stitch when labrum <3 mm.
- 10Control labral repair to avoid excessive inversion or eversion.
Arthroscopic labral repair and refixation have garnered much attention over the past several years. Restoration of suction seal and native labral function has been an evolving focus for achieving excellent results in hip preservation surgery.1-6 Given the superior results of labral repair, including level I evidence, repair or refixation should be pursued whenever possible.7 Authors have reported using several labral management techniques: débridement, labralization, looped suture fixation, base stitch fixation, inversion-eversion, and reconstruction.7-13 The optimal technique is yet to be determined. When possible, steps should be taken to repair the labrum to an anatomical position. Absolute indications for labral repair are a confirmed intra-articular diagnosis with symptomatic pain, joint space >2 mm with or without femoroacetabular impingement (FAI), labral tear or instability, and failed conservative management.9,11,12,14,15 More important, the surgeon must have a clear etiology of the pathologic cause of the tear and be aware of the limitations of the procedure. Labral repair is relatively contraindicated in end-stage arthritis and has failed when used alone in undiagnosed dysplasia or hip instability.16 In this article, we discuss indications for labral repair; describe Dr. Mather’s preoperative planning, labral repair technique, and postoperative care; and review published outcomes and future trends in labral repair.
Indications
At our institution, anatomical labral repair is the preferred procedure for most primary and revision hip arthroscopy procedures. We aim to restore the suction seal, re-create the contact of the labrum and the femoral head to facilitate proprioception, and restore normal stability of the labrum. Indications for primary repair are labrum width >3 mm, no more than 2 repairs, and ability to hold a suture. Our indications for reconstruction or débridement are stage 3 irreparable labral tear, calcified/cystic labrum, and multiple failed labral repairs or reconstructions. The decision to perform labral débridement or reconstruction is made on a case-by-case basis but is primarily influenced by the stability of the hip joint and the activity goals of the patient. If preoperative presentation and intraoperative examination suggest labral instability as a major component of the pathology, or if the patient wants to return to high-demand activity, we more strongly favor reconstruction over débridement. In our experience, with the technique described in this article, more than 95% of all primary labral tears can be addressed with repair.
Preoperative Planning
The goals in hip preservation surgery are to identify and address the underlying cause of the labral tear, whether it be FAI syndrome, trauma, labral instability, or all 3, and to re-create the anatomy and biomechanics of the acetabular labrum. For repair, we prefer an inversion-eversion technique with independent control of the labrum. Our initial work-up includes a thorough history and physical examination with baseline patient-reported outcome scores. Standard erect anteroposterior pelvis, Dunn lateral, and false-profile radiographs are obtained. Standard measurements of lateral center edge angle, anterior center edge angle, Tönnis angle, Tönnis grade, lateral joint space, and head extrusion indices are evaluated. Selective in-office ultrasound-guided injections are used to confirm an intra-articular source of pain. At our institution, noncontrast 3.0 Tesla magnetic resonance imaging (MRI) with volumetric interpolated breath-hold examination (VIBE) sequencing and 3-dimensional rendering is obtained for evaluation of labral and FAI morphology.17 All advanced imaging is performed without arthrogram or radiation exposure (Figures 1A-1C).
With use of the radiographs and the MRI scans, we engage the patient in an informed discussion about the labral tear, FAI, and concomitant pathology. We discuss expected outcomes of conservative or operative management given the patient’s expected functional activities, and inform the patient that primary repair is indicated for many others in similar situations. The potential for possible labral reconstruction is discussed if the patient had prior intra-articular hip surgery, has a large calcified labrum or a cystic labrum, is an athlete with failed prior surgery, or is younger than 40 years.
Labral Repair Technique
The patient is taken to the surgical suite, and a general anesthetic is administered. A peripheral nerve block is not routinely used. The patient’s feet are padded, and boots for the traction table are applied. The patient is carefully placed on a Hana table in modified supine position. Balanced traction is used to achieve proper joint distraction. The C-arm is used to verify proper distraction, assess hip stability, and achieve standard anterolateral (AL) portal placement. A midanterior portal (MAP) is created and an interportal capsulotomy is performed. Capsular suspension is performed with the InJector II Capsule Restoration System (Stryker Sports Medicine) and typically 4 or 5 high-strength No. 2 sutures (Zipline; Stryker Sports Medicine).19 Diagnostic arthroscopy is performed to identify the tear type, measure the labral width, determine the impingement area, and identify the intra-articular pathology. After the intra-articular pathology is addressed, a radiofrequency Ambient HIPVAC 50 Coblation Wand (Smith & Nephew) is used to expose the acetabular rim and subspine as indicated. Acetabuloplasty or subspine decompression is performed, and then a primary repair or refixation of the labrum is performed. We do not routinely detach the labrum for acetabular rim trimming. A crucial step here is to expose a bleeding surface to which the labrum can be repaired. If the rim is sclerotic, or the rim cannot be removed because of underlying low acetabular coverage, we prefer to obtain the bleeding surface with a microdrilling device (Stryker) that is routinely used for acetabular microfracture.
Labrum quality is used to determine which repair method to use. A hypertrophic labrum is debulked. The acetabular rim is seldom resected >3 mm, but, when it is, the newly exposed cartilage is removed. We have found that >3 mm of residual cartilage prevents refixation of the labrum directly to the bone and may interfere with anatomical positioning. When a labrum is <3 mm in width or will not hold a base technique, repair stability is the priority, and a looped method is used. A knotless anchor with No. 1 permanent suture designed for hip labral repair (CinchLock; Stryker) is our first-line anchor choice. A distal anterolateral accessory (DALA) portal is created with an outside-in technique, and anchors are drilled through this portal into zones 2 to 4 (Figures 2A-2E).
A 2.4-mm drill guide is advanced through the DALA portal and placed in the appropriate position for drilling. We aim for 1 mm to 2 mm from the chondrolabral junction. Next, the probe is placed intra-articular and medial to the anchor insertion site, and the anchor is loaded and then inserted around the probe (Figures 3A-3E).
The hip is then reduced. If indicated, a T-capsulotomy is performed for femoral osteochondroplasty.
Postoperative Care
Patients are placed in a postoperative hip brace and use a continuous passive motion machine 6 hours a day for 2 weeks, and an ice machine. They maintain 30 lb of foot-flat weight-bearing for 3 weeks, and begin a standard labral repair protocol on postoperative days 3 to 7.
Discussion
Hip labral preservation has evolved over the past 10 years, and current options for labral management include excision, débridement, labralization, repair, and reconstruction.1-13 Labral excision was studied by Miozzari and colleagues,8 who postulated on the basis of animal models that the labrum may regenerate. In their series of 9 patients treated with surgical hip dislocation and labral excision at average 4-year follow-up, repeat magnetic resonance angiography revealed no regeneration of tissue—modified Harris Hip Score was 83. The hip scores were less than those of patients treated with the same procedure with repair, and the authors concluded that defining labral débridement versus excision in the literature, and treating patients with primary repair or reconstruction techniques, may lead to better results. Their study used a small sample and was limited to an open procedure. Arthroscopic labral débridement in isolation was also a poor option for treatment of a labral tear. In a 2-year follow-up of 59 isolated labral débridement procedures, Krych and colleagues9 found 47% combined poor results.
There is level I evidence of the importance of labral repair. In 2013, Krych and colleagues7 conducted a randomized control trial of 38 female patients who underwent hip arthroscopy for FAI. At time of surgery, patients were randomly assigned to either débridement or repair. At 1-year follow-up, activities of daily living and Sports specific Hip Outcome Scores were statistically significantly superior in the repair group. On a subjective scale, 94% vs 78% of patients reported normal or near normal hips in the repair versus débridement groups respectively. Ayeni and colleagues20 performed a systematic review of 6 studies in an attempt to develop labral management recommendations. Five of the studies (N = 490 patients total) had improved results with labral repair over reconstruction. Although the studies had a low level of evidence, they found a trend toward improved results with labral repair. These studies highlight the importance of labral preservation and proper FAI management.
Techniques for labrum repair have advanced as well—from a looped suture technique to a base stitch and knotless independent tensioning.11-13 Restoration of the hip labrum function as a suction seal, fluid circulator and anatomic capsular repair is paramount to excellent results and stresses the importance of performing an anatomic labral repair.1-6 Knotless anchor repair is not novel and has been previously described. Fry and Domb12 reported on a knotless labral repair technique that uses push-lock devices (Arthrex) that do not allow for independent tensioning. Inversion-eversion was introduced to the literature by Moreira and colleagues,13 who described an independent tensioning technique that uses speed-lock anchors (Smith & Nephew). Our technique differs in that it involves a DALA portal; labral reduction and tensioning with a probe assist to ensure the second pass of the base stitch is at the apex of the labrum; and use of No. 1 instead of No. 2 suture. Although seemingly subtle, these differences allow for proper anchor placement nearer the rim, additional support in achieving precise suture placement, and less disruption of small labra. These differences are particularly relevant for smaller labra.
Evaluating repair techniques on the basis of high-evidence literature is challenging. In a matched-cohort study of 220 patients, Jackson and colleagues21 compared 2 techniques: looped and base stitch. At 2-year follow-up, patients in both groups showed improvement, and there was no statistically significant difference in patient-reported outcome measures between the groups. Sawyer and colleagues22 studied the outcomes of 326 consecutive patients who underwent looped, pierced, or combined labral repair at an average 32-month follow-up. The groups’ revision rates were comparable, each group improved in postoperative patient-reported outcomes, and the pierced group had significantly higher preoperative scores on the Western Ontario and McMaster Universities Osteoarthritis Index. These studies described a base or pierce repair that did not differ from a looped repair, though the techniques did not allow for independent tensioning to re-create an anatomical inversion-eversion repair and may have altered the reported outcomes.
Our current technique uses independent tensioning of the repair to allow control of labrum inversion-eversion to give an anatomical repair with restoration of the suction seal. Preoperative planning, addressing the FAI appropriately, proper suture-passing technique, controlling the labrum in inversion-eversion fashion, and anatomical labral repair are the elements of Dr. Mather’s preferred method for preserving the native labrum and allowing it to assume its native function.
Future Directions
As our understanding of FAI and labral function evolves, labral preservation surgery continues to advance. With surgeons continually developing new techniques and following up on previous techniques, the ability to preserve the native hip with lasting procedures evolves as well. Proper identification of the underlying cause of the labral tear and proper anatomical repair are paramount to the success of FAI surgery.
Am J Orthop. 2017;46(1):42-48. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Philippon MJ, Nepple JJ, Campbell KJ, et al. The hip fluid seal—part I: the effect of an acetabular labral tear, repair, resection and reconstruction on hip fluid pressurization. Knee Surg Sports Traumatol Arthrosc. 2014;22(4):722-729.
2. Nepple JJ, Philippon MJ, Campbell KJ, et al. The hip fluid seal—part II: the effect of an acetabular labral tear, repair, resection and reconstruction on hip stability to distraction. Knee Surg Sports Traumatol Arthrosc. 2014;22(4):730-736.
3. Dwyer MK, Jones HL, Hogan MG, Field RE, McCarthy JC, Noble PC. The acetabular labrum regulates fluid circulation of the hip joint during functional activities. Am J Sports Med. 2014;42(4):812-819.
4. Greaves LL, Gilbart MK, Yung AC, Kozlowski, Wilson DR. Effect of acetabular labral tears, repair and resection on hip cartilage strain: a 7T MR study. J Biomech. 2010;43(5):858-863.
5. Freehill MT, Safran MR. The labrum of the hip: diagnosis and rationale for surgical correction. Clin Sports Med. 2011;30(2):293-315.
6. Myers CA, Register BC, Lertwanich P, et al. Role of the acetabular labrum and the iliofemoral ligament in hip stability: an in vitro biplane fluoroscopy study. Am J Sports Med. 2011;39(suppl):85S-91S.
7. Krych AJ, Thompson M, Knutson Z, Scoon J, Coleman SH. Arthroscopic labral repair versus selective labral debridement in female patients with femoroacetabular impingement: a prospective randomized study. Arthroscopy. 2013;29(1):46-53.
8. Miozzari HH, Celia M, Clark JM, Werlen S, Naal FD, Nötzli HP. No regeneration of the human acetabular labrum after excision to bone. Clin Orthop Relat Res. 2015;473(4):1349-1357.
9. Krych AJ, Kuzma SA, Kovachevich R, Hudgens JL, Stuart MJ, Levy BA. Modest mid-term outcomes after isolated arthroscopic debridement of acetabular labral tears. Knee Surg Sports Traumatol Arthrosc. 2014;22(4):763-767.
10. Matsuda DK. Arthroscopic labralization of the hip: an alternative to labral reconstruction. Arthrosc Tech. 2014;3(1):e131-e133.
11. Philippon MJ, Faucet SC, Briggs KK. Arthroscopic hip labral repair. Arthrosc Tech. 2013;2(2):e73-e76.
12. Fry D, Domb B. Labral base refixation in the hip: rationale and technique for an anatomic approach to labral repair. Arthroscopy. 2010;26(9 suppl):S81-S89.
13. Moreira B, Pascual-Garrido C, Chadayamurri V, Mei-Dan O. Eversion-inversion labral repair and reconstruction technique for optimal suction seal. Arthrosc Tech. 2015;4(6):e697-e700.
14. Mook WR, Briggs KK, Philippon MJ. Evidence and approach for management of labral deficiency: the role for labral reconstruction. Sports Med Arthrosc. 2015;23(4):205-212.
15. Gupta A, Suarez-Ahedo C, Redmond JM, et al. Best practices during hip arthroscopy: aggregate recommendations of high-volume surgeons. Arthroscopy. 2015;31(9):1722-1727.
16. Yeung M, Kowalczuk M, Simunovic N, Ayeni OR. Hip arthroscopy in the setting of hip dysplasia: a systematic review. Bone Joint Res. 2016;5(6):225-231.
17. Hash TW. Magnetic resonance imaging of the hip. In: Nho SJ, Leunig M, Larson CM, Bedi A, Kelly BT, eds. Hip Arthroscopy and Hip Joint Preservation Surgery, Vol. 1. New York, NY: Springer; 2015:65-113.
18. Sutter R, Zubler V, Hoffmann A, et al. Hip MRI: how useful is intraarticular contrast material for evaluating surgically proven lesions of the labrum and articular cartilage? AJR Am J Roentgenol. 2014;202(1):160-169.
19. Federer AE, Karas V, Nho S, Coleman SH, Mather RC 3rd. Capsular suspension technique for hip arthroscopy. Arthrosc Tech. 2015;4(4):e317-e322.
20. Ayeni OR, Adamich J, Farrokhyar F, et al. Surgical management of labral tears during femoroacetabular impingement surgery: a systematic review. Knee Surg Sports Traumatol Arthrosc. 2014;22(4):756-762.
21. Jackson TJ, Hammarstedt JE, Vemula SP, Domb BG. Acetabular labral base repair versus circumferential suture repair: a matched-paired comparison of clinical outcomes. Arthroscopy. 2015;31(9):1716-1721.
22. Sawyer GA, Briggs KK, Dornan GJ, Ommen ND, Philippon MJ. Clinical outcomes after arthroscopic hip labral repair using looped versus pierced suture techniques. Am J Sports Med. 2015;43(7):1683-1688.
1. Philippon MJ, Nepple JJ, Campbell KJ, et al. The hip fluid seal—part I: the effect of an acetabular labral tear, repair, resection and reconstruction on hip fluid pressurization. Knee Surg Sports Traumatol Arthrosc. 2014;22(4):722-729.
2. Nepple JJ, Philippon MJ, Campbell KJ, et al. The hip fluid seal—part II: the effect of an acetabular labral tear, repair, resection and reconstruction on hip stability to distraction. Knee Surg Sports Traumatol Arthrosc. 2014;22(4):730-736.
3. Dwyer MK, Jones HL, Hogan MG, Field RE, McCarthy JC, Noble PC. The acetabular labrum regulates fluid circulation of the hip joint during functional activities. Am J Sports Med. 2014;42(4):812-819.
4. Greaves LL, Gilbart MK, Yung AC, Kozlowski, Wilson DR. Effect of acetabular labral tears, repair and resection on hip cartilage strain: a 7T MR study. J Biomech. 2010;43(5):858-863.
5. Freehill MT, Safran MR. The labrum of the hip: diagnosis and rationale for surgical correction. Clin Sports Med. 2011;30(2):293-315.
6. Myers CA, Register BC, Lertwanich P, et al. Role of the acetabular labrum and the iliofemoral ligament in hip stability: an in vitro biplane fluoroscopy study. Am J Sports Med. 2011;39(suppl):85S-91S.
7. Krych AJ, Thompson M, Knutson Z, Scoon J, Coleman SH. Arthroscopic labral repair versus selective labral debridement in female patients with femoroacetabular impingement: a prospective randomized study. Arthroscopy. 2013;29(1):46-53.
8. Miozzari HH, Celia M, Clark JM, Werlen S, Naal FD, Nötzli HP. No regeneration of the human acetabular labrum after excision to bone. Clin Orthop Relat Res. 2015;473(4):1349-1357.
9. Krych AJ, Kuzma SA, Kovachevich R, Hudgens JL, Stuart MJ, Levy BA. Modest mid-term outcomes after isolated arthroscopic debridement of acetabular labral tears. Knee Surg Sports Traumatol Arthrosc. 2014;22(4):763-767.
10. Matsuda DK. Arthroscopic labralization of the hip: an alternative to labral reconstruction. Arthrosc Tech. 2014;3(1):e131-e133.
11. Philippon MJ, Faucet SC, Briggs KK. Arthroscopic hip labral repair. Arthrosc Tech. 2013;2(2):e73-e76.
12. Fry D, Domb B. Labral base refixation in the hip: rationale and technique for an anatomic approach to labral repair. Arthroscopy. 2010;26(9 suppl):S81-S89.
13. Moreira B, Pascual-Garrido C, Chadayamurri V, Mei-Dan O. Eversion-inversion labral repair and reconstruction technique for optimal suction seal. Arthrosc Tech. 2015;4(6):e697-e700.
14. Mook WR, Briggs KK, Philippon MJ. Evidence and approach for management of labral deficiency: the role for labral reconstruction. Sports Med Arthrosc. 2015;23(4):205-212.
15. Gupta A, Suarez-Ahedo C, Redmond JM, et al. Best practices during hip arthroscopy: aggregate recommendations of high-volume surgeons. Arthroscopy. 2015;31(9):1722-1727.
16. Yeung M, Kowalczuk M, Simunovic N, Ayeni OR. Hip arthroscopy in the setting of hip dysplasia: a systematic review. Bone Joint Res. 2016;5(6):225-231.
17. Hash TW. Magnetic resonance imaging of the hip. In: Nho SJ, Leunig M, Larson CM, Bedi A, Kelly BT, eds. Hip Arthroscopy and Hip Joint Preservation Surgery, Vol. 1. New York, NY: Springer; 2015:65-113.
18. Sutter R, Zubler V, Hoffmann A, et al. Hip MRI: how useful is intraarticular contrast material for evaluating surgically proven lesions of the labrum and articular cartilage? AJR Am J Roentgenol. 2014;202(1):160-169.
19. Federer AE, Karas V, Nho S, Coleman SH, Mather RC 3rd. Capsular suspension technique for hip arthroscopy. Arthrosc Tech. 2015;4(4):e317-e322.
20. Ayeni OR, Adamich J, Farrokhyar F, et al. Surgical management of labral tears during femoroacetabular impingement surgery: a systematic review. Knee Surg Sports Traumatol Arthrosc. 2014;22(4):756-762.
21. Jackson TJ, Hammarstedt JE, Vemula SP, Domb BG. Acetabular labral base repair versus circumferential suture repair: a matched-paired comparison of clinical outcomes. Arthroscopy. 2015;31(9):1716-1721.
22. Sawyer GA, Briggs KK, Dornan GJ, Ommen ND, Philippon MJ. Clinical outcomes after arthroscopic hip labral repair using looped versus pierced suture techniques. Am J Sports Med. 2015;43(7):1683-1688.
New and Noteworthy Information—January 2017
Students who played varsity high school football between 1956 and 1970 do not have an increased risk of neurodegenerative diseases, compared with athletes engaged in other varsity sports, according to a study published online ahead of print December 12, 2016, in Mayo Clinic Proceedings. Researchers identified 296 male varsity football players in public high schools in Rochester, Minnesota, and 190 male varsity swimmers, wrestlers, and basketball players. Using records from the Rochester Epidemiology Project, investigators ascertained the incidence of late-life neurodegenerative diseases. Football players had an increased risk of medically documented head trauma, especially if they played football for more than one year. Compared with other athletes, football players did not have an increased risk of neurodegenerative disease overall, nor an increased risk of dementia, parkinsonism, or amyotrophic lateral sclerosis.
Antipsychotic drug use is associated with a 60% increased risk of mortality among persons with Alzheimer's disease, according to a study published online ahead of print December 5, 2016, in the Journal of Alzheimer's Disease. Researchers examined data from the MEDALZ study for 70,718 people who were newly diagnosed with Alzheimer's disease in Finland from 2005 to 2011. Death, excluding death from cancer, was extracted from the Causes of Death Register. Incident antipsychotic use was compared with time without antipsychotics using Cox proportional hazard models. The absolute difference in mortality rate was 4.58 deaths per 100 person-years. The risk of mortality was increased from the first days of antipsychotic use and attenuated gradually. Antipsychotic polypharmacy was associated with an almost doubled risk of mortality, compared with monotherapy.
A disruption of structural connections in a brain network contributes to cognitive deficits in patients with Parkinson's disease, according to a study published online ahead of print December 7, 2016, in Radiology. The structural brain connectomes of 170 patients with Parkinson's disease and 41 healthy controls were obtained with deterministic diffusion-tensor tractography. Patients with Parkinson's disease and mild cognitive impairment (MCI) had global network alterations, compared with controls and patients with Parkinson's disease without MCI. Relative to controls, patients with Parkinson's disease and MCI had a large basal ganglia and frontoparietal network with decreased fractional anisotropy in the right hemisphere and a subnetwork with increased mean diffusivity involving similar regions bilaterally. Compared with patients with Parkinson's disease without MCI, people with Parkinson's disease and MCI had networks with decreased fractional anisotropy.
A proposed diagnostic algorithm for sporadic Creutzfeldt-Jakob disease combines CSF and olfactory mucosa real-time quaking-induced conversion testing to provide approximately 100% sensitivity and specificity in the clinical phase of the disease, according to a study published online ahead of print December 12, 2016, in JAMA Neurology. Among the 86 patients included in this analysis, 61 patients with sporadic Creutzfeldt-Jakob disease had positive real-time quaking-induced conversion findings using olfactory mucosa, CSF samples, or both, for an overall real-time quaking-induced conversion sensitivity of 100%. All patients with a final diagnosis of nonprion disease had negative real-time quaking-induced conversion findings, for 100% specificity. Of eight symptomatic patients with various mutations causing Creutzfeldt-Jakob disease or Gerstmann-Sträussler-Scheinker syndrome, six had positive and two had negative real-time quaking-induced conversion findings, for a sensitivity of 75%.
CSF autotaxin may be a useful biomarker of dysmetabolism for examining risk for and outcomes of Alzheimer's disease, according to research published December 1, 2016, in the Journal of Alzheimer's Disease. Investigators studied 287 participants in the Alzheimer's Disease Neuroimaging Initiative, including 86 cognitively normal participants, 135 participants with mild cognitive impairment (MCI), and 66 participants with Alzheimer's disease. Autotaxin levels were significantly higher in patients with MCI and those with Alzheimer's disease. Each point increase in log-based autotaxin corresponded to a 3.5- to 5-times higher likelihood of having MCI and Alzheimer's disease, respectively. Higher autotaxin in Alzheimer's disease predicted hypometabolism in the medial temporal lobe and prefrontal cortex, and worse performance on executive function and memory factors. Autotaxin was associated with decreased cortical thickness in prefrontal cortex areas.
Marital history is significantly associated with survival after stroke, according to a study published December 14, 2016, in the Journal of the American Heart Association. Data from a nationally representative sample of 2,351 older adults who experienced a stroke were used to examine whether and to what extent current marital status and past marital losses were associated with risks of dying after the onset of disease. Results showed that the risks of dying following a stroke were significantly higher among people who were never married, remarried, divorced, and widowed, relative to those who remained continuously married. Researchers also found that having multiple marital losses was especially detrimental to survival, regardless of current marital status and accounting for multiple socioeconomic, psychosocial, behavioral, and physiologic risk factors.
Prefrontal brain activity levels during a cognitively demanding walking condition predict falls in high-functioning senior citizens, according to a study published online ahead of print December 7, 2016, in Neurology. Researchers examined 166 people with a mean age of 75 with functional near-infrared spectroscopy during motor, cognitive, and combined motor and cognitive tasks. Incident falls were prospectively assessed during a 50-month study period. During a mean follow-up of 33.9 months, 116 falls occurred. Higher levels of prefrontal cortical activation during the dual-task walking condition predicted falls. Neither behavioral outcomes on the dual task nor brain activation patterns on the single tasks predicted falls in this high-functioning sample. The results remained robust after accounting for multiple confounders, cognitive status, slow gait, previous falls, and frailty.
Localized brain injury and repair, indicated by higher translocator protein 18 kDa signal and white matter changes, may be associated with National Football League (NFL) play, according to a study published online ahead of print November 28, 2016, in JAMA Neurology. This cross-sectional, case-control study included young active or former NFL players recruited from across the United States and 16 age-, sex-, highest educational level-, and BMI-matched control participants. Researchers used [11C]DPA-713 PET data and other imaging data from 12 active or former NFL players and 11 matched control participants. The NFL players showed higher total distribution volume in eight of 12 brain regions examined. Investigators also observed limited change in white matter fractional anisotropy and mean diffusivity in 13 players, compared with 15 control participants.
Exposure to maternal rheumatoid arthritis is associated with an increased risk of childhood epilepsy, while exposure to paternal rheumatoid arthritis is not, according to a study published December 13, 2016, in Neurology. Researchers performed a nationwide cohort study of 1,917,723 people that were born between 1977 and 2008. Compared with unexposed children, children exposed to maternal rheumatoid arthritis had an increased risk of early and late childhood epilepsy, while children exposed to maternal rheumatoid arthritis had no increased risk of epilepsy in adolescence and adulthood. Paternal rheumatoid arthritis was not associated with an overall risk of epilepsy in the offspring or at any age. Children exposed to maternal rheumatoid arthritis in utero had a more pronounced increased risk of early childhood epilepsy than children of mothers who were diagnosed with rheumatoid arthritis after childbirth.
Having surgery may be linked to developing Guillain-Barré syndrome for people with cancer or autoimmune disorders, according to a study published online ahead of print November 23, 2016, in Neurology Clinical Practice. Researchers retrospectively reviewed consecutive patients diagnosed with Guillain-Barré syndrome within eight weeks of a surgical procedure between January 1995 and June 2014. Of the 208 people treated for Guillain-Barré syndrome, 31 people developed the syndrome within eight weeks of having a surgical procedure. People who had had cancer within the previous six months were seven times more likely to develop Guillain-Barré syndrome after surgery than people who had not had cancer. People who had pre-existing autoimmune disorders were five times more likely to develop Guillain-Barré syndrome after surgery than those without autoimmune disorders.
Patients with Parkinson's disease and orthostatic hypotension have transient, posture-mediated changes in cognition, according to a study published online ahead of print November 30, 2016, in Neurology. To investigate the relation between orthostatic hypotension and posture-mediated cognitive impairment in Parkinson disease, researchers used a cross-sectional and within-group design. Participants included 18 patients with Parkinson's disease and orthostatic hypotension, 19 patients with Parkinson's disease but without orthostatic hypotension, and 18 healthy controls. Participants underwent neuropsychologic tests in the supine and upright-tilted positions. When relative performances were compared with each other, postural changes had no significant impact on participants with Parkinson's disease but without orthostatic hypotension, compared with the control group. Participants with Parkinson's disease and orthostatic hypotension, however, were more susceptible to posture-related impairment on several tests.
Low concentrations of neonatal vitamin D are associated with an increased risk of multiple sclerosis (MS), according to a study published online ahead of print November 30, 2016, in Neurology. Researchers conducted a matched case-control study. Dried blood spots samples from 521 patients with MS were identified in the Danish Newborn Screening Biobank. For every patient with MS, one to two controls with the same sex and birth date were retrieved from the Biobank. Lower levels of 25-hydroxyvitamin D in neonates were associated with an increased risk of MS. In the analysis by quintiles, MS risk was highest among individuals in the bottom quintile and lowest among those in the top quintile of 25-hydroxyvitamin D, with an odds ratio for top versus bottom of 0.53.
Children exposed to valproate in the womb are at an increased risk of having a malformation at birth, and the dose of valproate that the child is exposed to determines the level of risk, according to a study published November 7, 2016, in the Cochrane Database of Systematic Reviews. Researchers analyzed 50 studies, with 31 contributing to a meta-analysis. Children exposed to valproate were at a higher risk of malformation, compared with children born to women without epilepsy and to women with untreated epilepsy. Investigators found significantly higher rates of specific malformations associating phenobarbital exposure with cardiac malformations and valproate exposure with neural tube, cardiac, orofacial, craniofacial, skeletal, and limb malformations, compared with other antiepileptic drugs. Dose of exposure mediated the risk of malformation following valproate exposure.
—Kimberly Williams
Students who played varsity high school football between 1956 and 1970 do not have an increased risk of neurodegenerative diseases, compared with athletes engaged in other varsity sports, according to a study published online ahead of print December 12, 2016, in Mayo Clinic Proceedings. Researchers identified 296 male varsity football players in public high schools in Rochester, Minnesota, and 190 male varsity swimmers, wrestlers, and basketball players. Using records from the Rochester Epidemiology Project, investigators ascertained the incidence of late-life neurodegenerative diseases. Football players had an increased risk of medically documented head trauma, especially if they played football for more than one year. Compared with other athletes, football players did not have an increased risk of neurodegenerative disease overall, nor an increased risk of dementia, parkinsonism, or amyotrophic lateral sclerosis.
Antipsychotic drug use is associated with a 60% increased risk of mortality among persons with Alzheimer's disease, according to a study published online ahead of print December 5, 2016, in the Journal of Alzheimer's Disease. Researchers examined data from the MEDALZ study for 70,718 people who were newly diagnosed with Alzheimer's disease in Finland from 2005 to 2011. Death, excluding death from cancer, was extracted from the Causes of Death Register. Incident antipsychotic use was compared with time without antipsychotics using Cox proportional hazard models. The absolute difference in mortality rate was 4.58 deaths per 100 person-years. The risk of mortality was increased from the first days of antipsychotic use and attenuated gradually. Antipsychotic polypharmacy was associated with an almost doubled risk of mortality, compared with monotherapy.
A disruption of structural connections in a brain network contributes to cognitive deficits in patients with Parkinson's disease, according to a study published online ahead of print December 7, 2016, in Radiology. The structural brain connectomes of 170 patients with Parkinson's disease and 41 healthy controls were obtained with deterministic diffusion-tensor tractography. Patients with Parkinson's disease and mild cognitive impairment (MCI) had global network alterations, compared with controls and patients with Parkinson's disease without MCI. Relative to controls, patients with Parkinson's disease and MCI had a large basal ganglia and frontoparietal network with decreased fractional anisotropy in the right hemisphere and a subnetwork with increased mean diffusivity involving similar regions bilaterally. Compared with patients with Parkinson's disease without MCI, people with Parkinson's disease and MCI had networks with decreased fractional anisotropy.
A proposed diagnostic algorithm for sporadic Creutzfeldt-Jakob disease combines CSF and olfactory mucosa real-time quaking-induced conversion testing to provide approximately 100% sensitivity and specificity in the clinical phase of the disease, according to a study published online ahead of print December 12, 2016, in JAMA Neurology. Among the 86 patients included in this analysis, 61 patients with sporadic Creutzfeldt-Jakob disease had positive real-time quaking-induced conversion findings using olfactory mucosa, CSF samples, or both, for an overall real-time quaking-induced conversion sensitivity of 100%. All patients with a final diagnosis of nonprion disease had negative real-time quaking-induced conversion findings, for 100% specificity. Of eight symptomatic patients with various mutations causing Creutzfeldt-Jakob disease or Gerstmann-Sträussler-Scheinker syndrome, six had positive and two had negative real-time quaking-induced conversion findings, for a sensitivity of 75%.
CSF autotaxin may be a useful biomarker of dysmetabolism for examining risk for and outcomes of Alzheimer's disease, according to research published December 1, 2016, in the Journal of Alzheimer's Disease. Investigators studied 287 participants in the Alzheimer's Disease Neuroimaging Initiative, including 86 cognitively normal participants, 135 participants with mild cognitive impairment (MCI), and 66 participants with Alzheimer's disease. Autotaxin levels were significantly higher in patients with MCI and those with Alzheimer's disease. Each point increase in log-based autotaxin corresponded to a 3.5- to 5-times higher likelihood of having MCI and Alzheimer's disease, respectively. Higher autotaxin in Alzheimer's disease predicted hypometabolism in the medial temporal lobe and prefrontal cortex, and worse performance on executive function and memory factors. Autotaxin was associated with decreased cortical thickness in prefrontal cortex areas.
Marital history is significantly associated with survival after stroke, according to a study published December 14, 2016, in the Journal of the American Heart Association. Data from a nationally representative sample of 2,351 older adults who experienced a stroke were used to examine whether and to what extent current marital status and past marital losses were associated with risks of dying after the onset of disease. Results showed that the risks of dying following a stroke were significantly higher among people who were never married, remarried, divorced, and widowed, relative to those who remained continuously married. Researchers also found that having multiple marital losses was especially detrimental to survival, regardless of current marital status and accounting for multiple socioeconomic, psychosocial, behavioral, and physiologic risk factors.
Prefrontal brain activity levels during a cognitively demanding walking condition predict falls in high-functioning senior citizens, according to a study published online ahead of print December 7, 2016, in Neurology. Researchers examined 166 people with a mean age of 75 with functional near-infrared spectroscopy during motor, cognitive, and combined motor and cognitive tasks. Incident falls were prospectively assessed during a 50-month study period. During a mean follow-up of 33.9 months, 116 falls occurred. Higher levels of prefrontal cortical activation during the dual-task walking condition predicted falls. Neither behavioral outcomes on the dual task nor brain activation patterns on the single tasks predicted falls in this high-functioning sample. The results remained robust after accounting for multiple confounders, cognitive status, slow gait, previous falls, and frailty.
Localized brain injury and repair, indicated by higher translocator protein 18 kDa signal and white matter changes, may be associated with National Football League (NFL) play, according to a study published online ahead of print November 28, 2016, in JAMA Neurology. This cross-sectional, case-control study included young active or former NFL players recruited from across the United States and 16 age-, sex-, highest educational level-, and BMI-matched control participants. Researchers used [11C]DPA-713 PET data and other imaging data from 12 active or former NFL players and 11 matched control participants. The NFL players showed higher total distribution volume in eight of 12 brain regions examined. Investigators also observed limited change in white matter fractional anisotropy and mean diffusivity in 13 players, compared with 15 control participants.
Exposure to maternal rheumatoid arthritis is associated with an increased risk of childhood epilepsy, while exposure to paternal rheumatoid arthritis is not, according to a study published December 13, 2016, in Neurology. Researchers performed a nationwide cohort study of 1,917,723 people that were born between 1977 and 2008. Compared with unexposed children, children exposed to maternal rheumatoid arthritis had an increased risk of early and late childhood epilepsy, while children exposed to maternal rheumatoid arthritis had no increased risk of epilepsy in adolescence and adulthood. Paternal rheumatoid arthritis was not associated with an overall risk of epilepsy in the offspring or at any age. Children exposed to maternal rheumatoid arthritis in utero had a more pronounced increased risk of early childhood epilepsy than children of mothers who were diagnosed with rheumatoid arthritis after childbirth.
Having surgery may be linked to developing Guillain-Barré syndrome for people with cancer or autoimmune disorders, according to a study published online ahead of print November 23, 2016, in Neurology Clinical Practice. Researchers retrospectively reviewed consecutive patients diagnosed with Guillain-Barré syndrome within eight weeks of a surgical procedure between January 1995 and June 2014. Of the 208 people treated for Guillain-Barré syndrome, 31 people developed the syndrome within eight weeks of having a surgical procedure. People who had had cancer within the previous six months were seven times more likely to develop Guillain-Barré syndrome after surgery than people who had not had cancer. People who had pre-existing autoimmune disorders were five times more likely to develop Guillain-Barré syndrome after surgery than those without autoimmune disorders.
Patients with Parkinson's disease and orthostatic hypotension have transient, posture-mediated changes in cognition, according to a study published online ahead of print November 30, 2016, in Neurology. To investigate the relation between orthostatic hypotension and posture-mediated cognitive impairment in Parkinson disease, researchers used a cross-sectional and within-group design. Participants included 18 patients with Parkinson's disease and orthostatic hypotension, 19 patients with Parkinson's disease but without orthostatic hypotension, and 18 healthy controls. Participants underwent neuropsychologic tests in the supine and upright-tilted positions. When relative performances were compared with each other, postural changes had no significant impact on participants with Parkinson's disease but without orthostatic hypotension, compared with the control group. Participants with Parkinson's disease and orthostatic hypotension, however, were more susceptible to posture-related impairment on several tests.
Low concentrations of neonatal vitamin D are associated with an increased risk of multiple sclerosis (MS), according to a study published online ahead of print November 30, 2016, in Neurology. Researchers conducted a matched case-control study. Dried blood spots samples from 521 patients with MS were identified in the Danish Newborn Screening Biobank. For every patient with MS, one to two controls with the same sex and birth date were retrieved from the Biobank. Lower levels of 25-hydroxyvitamin D in neonates were associated with an increased risk of MS. In the analysis by quintiles, MS risk was highest among individuals in the bottom quintile and lowest among those in the top quintile of 25-hydroxyvitamin D, with an odds ratio for top versus bottom of 0.53.
Children exposed to valproate in the womb are at an increased risk of having a malformation at birth, and the dose of valproate that the child is exposed to determines the level of risk, according to a study published November 7, 2016, in the Cochrane Database of Systematic Reviews. Researchers analyzed 50 studies, with 31 contributing to a meta-analysis. Children exposed to valproate were at a higher risk of malformation, compared with children born to women without epilepsy and to women with untreated epilepsy. Investigators found significantly higher rates of specific malformations associating phenobarbital exposure with cardiac malformations and valproate exposure with neural tube, cardiac, orofacial, craniofacial, skeletal, and limb malformations, compared with other antiepileptic drugs. Dose of exposure mediated the risk of malformation following valproate exposure.
—Kimberly Williams
Students who played varsity high school football between 1956 and 1970 do not have an increased risk of neurodegenerative diseases, compared with athletes engaged in other varsity sports, according to a study published online ahead of print December 12, 2016, in Mayo Clinic Proceedings. Researchers identified 296 male varsity football players in public high schools in Rochester, Minnesota, and 190 male varsity swimmers, wrestlers, and basketball players. Using records from the Rochester Epidemiology Project, investigators ascertained the incidence of late-life neurodegenerative diseases. Football players had an increased risk of medically documented head trauma, especially if they played football for more than one year. Compared with other athletes, football players did not have an increased risk of neurodegenerative disease overall, nor an increased risk of dementia, parkinsonism, or amyotrophic lateral sclerosis.
Antipsychotic drug use is associated with a 60% increased risk of mortality among persons with Alzheimer's disease, according to a study published online ahead of print December 5, 2016, in the Journal of Alzheimer's Disease. Researchers examined data from the MEDALZ study for 70,718 people who were newly diagnosed with Alzheimer's disease in Finland from 2005 to 2011. Death, excluding death from cancer, was extracted from the Causes of Death Register. Incident antipsychotic use was compared with time without antipsychotics using Cox proportional hazard models. The absolute difference in mortality rate was 4.58 deaths per 100 person-years. The risk of mortality was increased from the first days of antipsychotic use and attenuated gradually. Antipsychotic polypharmacy was associated with an almost doubled risk of mortality, compared with monotherapy.
A disruption of structural connections in a brain network contributes to cognitive deficits in patients with Parkinson's disease, according to a study published online ahead of print December 7, 2016, in Radiology. The structural brain connectomes of 170 patients with Parkinson's disease and 41 healthy controls were obtained with deterministic diffusion-tensor tractography. Patients with Parkinson's disease and mild cognitive impairment (MCI) had global network alterations, compared with controls and patients with Parkinson's disease without MCI. Relative to controls, patients with Parkinson's disease and MCI had a large basal ganglia and frontoparietal network with decreased fractional anisotropy in the right hemisphere and a subnetwork with increased mean diffusivity involving similar regions bilaterally. Compared with patients with Parkinson's disease without MCI, people with Parkinson's disease and MCI had networks with decreased fractional anisotropy.
A proposed diagnostic algorithm for sporadic Creutzfeldt-Jakob disease combines CSF and olfactory mucosa real-time quaking-induced conversion testing to provide approximately 100% sensitivity and specificity in the clinical phase of the disease, according to a study published online ahead of print December 12, 2016, in JAMA Neurology. Among the 86 patients included in this analysis, 61 patients with sporadic Creutzfeldt-Jakob disease had positive real-time quaking-induced conversion findings using olfactory mucosa, CSF samples, or both, for an overall real-time quaking-induced conversion sensitivity of 100%. All patients with a final diagnosis of nonprion disease had negative real-time quaking-induced conversion findings, for 100% specificity. Of eight symptomatic patients with various mutations causing Creutzfeldt-Jakob disease or Gerstmann-Sträussler-Scheinker syndrome, six had positive and two had negative real-time quaking-induced conversion findings, for a sensitivity of 75%.
CSF autotaxin may be a useful biomarker of dysmetabolism for examining risk for and outcomes of Alzheimer's disease, according to research published December 1, 2016, in the Journal of Alzheimer's Disease. Investigators studied 287 participants in the Alzheimer's Disease Neuroimaging Initiative, including 86 cognitively normal participants, 135 participants with mild cognitive impairment (MCI), and 66 participants with Alzheimer's disease. Autotaxin levels were significantly higher in patients with MCI and those with Alzheimer's disease. Each point increase in log-based autotaxin corresponded to a 3.5- to 5-times higher likelihood of having MCI and Alzheimer's disease, respectively. Higher autotaxin in Alzheimer's disease predicted hypometabolism in the medial temporal lobe and prefrontal cortex, and worse performance on executive function and memory factors. Autotaxin was associated with decreased cortical thickness in prefrontal cortex areas.
Marital history is significantly associated with survival after stroke, according to a study published December 14, 2016, in the Journal of the American Heart Association. Data from a nationally representative sample of 2,351 older adults who experienced a stroke were used to examine whether and to what extent current marital status and past marital losses were associated with risks of dying after the onset of disease. Results showed that the risks of dying following a stroke were significantly higher among people who were never married, remarried, divorced, and widowed, relative to those who remained continuously married. Researchers also found that having multiple marital losses was especially detrimental to survival, regardless of current marital status and accounting for multiple socioeconomic, psychosocial, behavioral, and physiologic risk factors.
Prefrontal brain activity levels during a cognitively demanding walking condition predict falls in high-functioning senior citizens, according to a study published online ahead of print December 7, 2016, in Neurology. Researchers examined 166 people with a mean age of 75 with functional near-infrared spectroscopy during motor, cognitive, and combined motor and cognitive tasks. Incident falls were prospectively assessed during a 50-month study period. During a mean follow-up of 33.9 months, 116 falls occurred. Higher levels of prefrontal cortical activation during the dual-task walking condition predicted falls. Neither behavioral outcomes on the dual task nor brain activation patterns on the single tasks predicted falls in this high-functioning sample. The results remained robust after accounting for multiple confounders, cognitive status, slow gait, previous falls, and frailty.
Localized brain injury and repair, indicated by higher translocator protein 18 kDa signal and white matter changes, may be associated with National Football League (NFL) play, according to a study published online ahead of print November 28, 2016, in JAMA Neurology. This cross-sectional, case-control study included young active or former NFL players recruited from across the United States and 16 age-, sex-, highest educational level-, and BMI-matched control participants. Researchers used [11C]DPA-713 PET data and other imaging data from 12 active or former NFL players and 11 matched control participants. The NFL players showed higher total distribution volume in eight of 12 brain regions examined. Investigators also observed limited change in white matter fractional anisotropy and mean diffusivity in 13 players, compared with 15 control participants.
Exposure to maternal rheumatoid arthritis is associated with an increased risk of childhood epilepsy, while exposure to paternal rheumatoid arthritis is not, according to a study published December 13, 2016, in Neurology. Researchers performed a nationwide cohort study of 1,917,723 people that were born between 1977 and 2008. Compared with unexposed children, children exposed to maternal rheumatoid arthritis had an increased risk of early and late childhood epilepsy, while children exposed to maternal rheumatoid arthritis had no increased risk of epilepsy in adolescence and adulthood. Paternal rheumatoid arthritis was not associated with an overall risk of epilepsy in the offspring or at any age. Children exposed to maternal rheumatoid arthritis in utero had a more pronounced increased risk of early childhood epilepsy than children of mothers who were diagnosed with rheumatoid arthritis after childbirth.
Having surgery may be linked to developing Guillain-Barré syndrome for people with cancer or autoimmune disorders, according to a study published online ahead of print November 23, 2016, in Neurology Clinical Practice. Researchers retrospectively reviewed consecutive patients diagnosed with Guillain-Barré syndrome within eight weeks of a surgical procedure between January 1995 and June 2014. Of the 208 people treated for Guillain-Barré syndrome, 31 people developed the syndrome within eight weeks of having a surgical procedure. People who had had cancer within the previous six months were seven times more likely to develop Guillain-Barré syndrome after surgery than people who had not had cancer. People who had pre-existing autoimmune disorders were five times more likely to develop Guillain-Barré syndrome after surgery than those without autoimmune disorders.
Patients with Parkinson's disease and orthostatic hypotension have transient, posture-mediated changes in cognition, according to a study published online ahead of print November 30, 2016, in Neurology. To investigate the relation between orthostatic hypotension and posture-mediated cognitive impairment in Parkinson disease, researchers used a cross-sectional and within-group design. Participants included 18 patients with Parkinson's disease and orthostatic hypotension, 19 patients with Parkinson's disease but without orthostatic hypotension, and 18 healthy controls. Participants underwent neuropsychologic tests in the supine and upright-tilted positions. When relative performances were compared with each other, postural changes had no significant impact on participants with Parkinson's disease but without orthostatic hypotension, compared with the control group. Participants with Parkinson's disease and orthostatic hypotension, however, were more susceptible to posture-related impairment on several tests.
Low concentrations of neonatal vitamin D are associated with an increased risk of multiple sclerosis (MS), according to a study published online ahead of print November 30, 2016, in Neurology. Researchers conducted a matched case-control study. Dried blood spots samples from 521 patients with MS were identified in the Danish Newborn Screening Biobank. For every patient with MS, one to two controls with the same sex and birth date were retrieved from the Biobank. Lower levels of 25-hydroxyvitamin D in neonates were associated with an increased risk of MS. In the analysis by quintiles, MS risk was highest among individuals in the bottom quintile and lowest among those in the top quintile of 25-hydroxyvitamin D, with an odds ratio for top versus bottom of 0.53.
Children exposed to valproate in the womb are at an increased risk of having a malformation at birth, and the dose of valproate that the child is exposed to determines the level of risk, according to a study published November 7, 2016, in the Cochrane Database of Systematic Reviews. Researchers analyzed 50 studies, with 31 contributing to a meta-analysis. Children exposed to valproate were at a higher risk of malformation, compared with children born to women without epilepsy and to women with untreated epilepsy. Investigators found significantly higher rates of specific malformations associating phenobarbital exposure with cardiac malformations and valproate exposure with neural tube, cardiac, orofacial, craniofacial, skeletal, and limb malformations, compared with other antiepileptic drugs. Dose of exposure mediated the risk of malformation following valproate exposure.
—Kimberly Williams

Current Techniques in Treating Femoroacetabular Impingement: Capsular Repair and Plication
Take-Home Points
- Hip capsule provides static stabilization for the hip joint.
- Capsular management must weigh visualization to address underlying osseous deformity but also repair/plication of the capsule to maintain biomechanical characteristics.
- T-capsulotomy provides optimal visualization with a small interportal incision with a vertical incision along the femoral neck.
- Extensile interportal capsulotomy is the most widely used capsulotomy and size may vary depending on capsular and patient characteristics.
- Orthopedic surgeons should be equipped to employ either technique depending on the patients individual hip pathomorphology.
Hip arthroscopy has emerged as a common surgical treatment for a number of hip pathologies. Surgical treatment strategies, including management of the hip capsule, have evolved. Whereas earlier hip arthroscopies often involved capsulectomy or capsulotomy without repair, more recently capsular closure has been considered an important step in restoring the anatomy of the hip joint and preventing microinstability or gross macroinstability.
The anatomy of the hip joint includes both static and dynamic stabilizers designed to maintain a functioning articulation. The osseous articulation of the femoral head and acetabulum is the first static stabilizer, with variations in offset, version, and inclination of the acetabulum and the proximal femur. The joint capsule consists of 3 ligaments—iliofemoral, pubofemoral, and ischiofemoral—that converge to form the zona orbicularis. Other soft-tissue structures, such as the articular cartilage, the labrum, the transverse acetabular ligament, the pulvinar, and the ligamentum teres, also provide static constraint.1 The surrounding musculature provides the hip joint with dynamic stability, which contributes to overall maintenance of proper joint kinematics.
Management of the hip capsule has evolved as our understanding of hip pathology and biomechanics has matured. Initial articles on using hip arthroscopy to treat labral tears described improvement in clinical outcomes,2 but the cases involved limited focal capsulotomy. Not until the idea of femoroacetabular impingement (FAI) was introduced were extensive capsulotomies and capsulectomies performed to address the underlying osseous deformities and emulate open techniques. Soon after our ability to access osseous pathomorphology improved with enhanced visualization and comprehensive resection, cases of hip instability after hip arthroscopy surfaced.3-5 Although frank dislocation after hip arthroscopy is rare and largely underreported, it is a catastrophic complication. In addition, focal capsular defects were also described in cases of failed hip arthroscopy and thought to lead to microinstability of the hip.6 Iatrogenic microinstability is thought to be more common, but it is also underrecognized as a cause of failure of hip arthroscopy.7Microinstability is a pathologic condition that can affect hip function. In cases of recurrent pain and unimproved functional status after surgery, microinstability should be considered. In an imaging study of capsule integrity, McCormick and colleagues6 found that 78% of patients who underwent revision arthroscopic surgery after hip arthroscopic surgery for FAI showed evidence of capsular and iliofemoral defects on magnetic resonance angiography. Frank and colleagues8 reported that, though all patients showed preoperative-to-postoperative improvement on outcome measures, those who underwent complete repair of their T-capsulotomy (vs repair of only its longitudinal portion) had superior outcomes, particularly increased sport-specific activity.
For patients undergoing hip arthroscopy, several predisposing factors can increase the risk of postoperative instability. Patient-related hip instability factors include generalized ligamentous laxity, supraphysiologic athletics (eg, dance), and borderline or true hip dysplasia. Surgeon-related factors include overaggressive acetabular rim resection, excessive labral débridement, and lack of capsular repair.5,9 Although there are multiple techniques for accessing the hip joint and addressing capsular closure at the end of surgery,9-14 we think capsular closure is an important aspect of the case.
Surgical Technique
For a demonstration of this technique, click here to see the video that accompanies this article. The patient is moved to a traction table and placed in the supine position. Induction of general anesthesia with muscle relaxation allows for atraumatic axial traction. The anesthetized patient is assessed for passive motion and ligamentous laxity. Well-padded boots are applied, and a well-padded perineal post is used for positioning. Gentle traction is applied to the contralateral limb, and axial traction is applied through the surgical limb with the hip abducted and minimally flexed. The leg is then adducted and neutrally extended, inducing a transverse vector cantilever moment to the proximal femur. The foot is internally rotated to optimize femoral neck length on an anteroposterior radiograph. The circulating nursing staff notes the onset of hip distraction in order to ensure safe traction duration.
Bony landmarks are marked with a sterile marking pen. Under fluoroscopic guidance, an anterolateral (AL) portal is established 1 cm proximal and 1 cm anterior to the AL tip of the greater trochanter. Standard cannulation allows for intra-articular visualization with a 70° arthroscope. A needle is used to localize placement of a modified anterior portal. After cannulation, the arthroscope is placed in the modified anterior portal to confirm safe entry of the portal without labral violation. An arthroscopic scalpel (Samurai Blade; Stryker Sports Medicine) is used to make a transverse interportal capsulotomy 8 mm to 10 mm from the labrum and extending from 12 to 2 o’clock; length is 2 cm to 4 cm, depending on the extent of the intra-articular injury (Figure 1A). 
The acetabular rim is trimmed with a 5.0-mm arthroscopic burr. Distal AL accessory (DALA) portal placement (4-6 cm distal to and in line with the AL portal) allows for suture anchor–based labral refixation. Generally, 2 to 4 anchors (1.4-mm NanoTack Anatomic Labrum Restoration System; Stryker Sports Medicine) are placed as near the articular cartilage as possible without penetration (Figure 1B). On completion of labral refixation, traction is released, and the hip is flexed to 20° to 30°.
T-Capsulotomy
Pericapsular fatty tissue is débrided with an arthroscopic shaver to visualize the interval between the iliocapsularis and gluteus minimus muscles. An arthroscopic scalpel is used, through a 5.0-mm cannula in the DALA portal, to extend the capsulotomy longitudinally and perpendicular to the interportal capsulotomy (Figure 1C). The T-capsulotomy is performed along the length of the femoral neck distally to the capsular reflection at the intertrochanteric line. The arthroscopic burr is used to perform a femoral osteochondroplasty between the lateral synovial folds (12 o’clock) and the medial synovial folds (6 o’clock). Dynamic examination and fluoroscopic imaging confirm that the entire cam deformity has been excised and that there is no evidence of impingement.
Although various suture-shuttling or tissue-penetrating/retrieving devices may be used, we recommend whichever device is appropriate for closing the capsule in its entirety. With the arthroscope in the modified anterior portal, an 8.25-mm × 90-mm cannula is placed in the AL portal, and an 8.25-mm × 110-mm cannula in the DALA portal. These portals will facilitate suture passage.
The vertical limb of the T-capsulotomy is closed with 2 to 4 side-to-side sutures, and the interportal capsulotomy limb with 2 or 3 sutures. Capsular closure begins with the distal portion of the longitudinal limb at the base of the iliofemoral ligament (IFL). A crescent tissue penetrating device (Slingshot; Stryker Sports Medicine) is loaded with high-strength No. 2 suture (Zipline; Stryker Sports Medicine) and placed through the AL portal to sharply pierce the lateral leaflet of the IFL (Figure 1D). The No. 2 suture is shuttled into the intra-articular side of the capsule (Figure 1E). Through the DALA portal, the penetrating device is used to pierce the medial leaflet to retrieve the free suture (Figure 1F). Next, the looped suture retriever is used to pull the suture from the AL portal to the DALA portal so the suture can be tied. We prefer to tie each suture individually after it is passed, but all of the sutures can be passed first, and then tied. As successive suture placement and knot tying inherently tighten the capsule, successive visualization requires more precision. Each subsequent suture is similarly passed, about 1 cm proximal to the previous stitch.
After closure of the vertical limb of the T-capsulotomy, we prefer to close the interportal capsulotomy with the InJector II Capsule Restoration System (Stryker Sports Medicine), a device that allows for closure through a single cannula lateral to medial. This device is passed through the AL cannula in order to bring the suture end through the proximal IFL attached to the acetabulum (Figure 1G). The device is removed from the cannula, and the other suture end is placed in the device and passed through the distal IFL (Figure 1H). The stitch is then tensioned and tied. Likewise, closure of the medial IFL involves passing the InJector through the DALA cannula and bringing the first suture end through the proximal IFL attached to the acetabulum. The Injector is removed from the cannula, and the other suture end is placed in the device and passed through the distal IFL. The stitch is then tensioned and tied with the hip in neutral extension. Generally, 2 or 3 stitches are used to close the interportal capsulotomy. Complete capsular closure is confirmed by the inability to visualize the underlying femoral head/neck and by probing the anterior capsule to ensure proper tension (Figure 1I).
Extensile Interportal Capsulotomy
An alternative to T-capsulotomy is interportal capsulotomy. Just as with T-capsulotomy closure, multiple different suture passing devices can be used. Good visualization for accessing the peripheral compartment generally is achieved by making the interportal capsulotomy 4 cm to 6 cm longer than the horizontal limb of the T-capsulotomy (Figures 2A, 2B). Capsular closure usually begins with the medial portion of the interportal capsulotomy. With the arthroscope in the AL portal, the 8.25-mm × 90-mm cannula is placed in the midanterior portal (MAP), and an 8.25-mm × 110-mm cannula is placed in the DALA portal. 
Ligamentous laxity determines degree of capsular closure. The capsular leaflets can be closed end to end if there is little concern for laxity and instability. If there is more concern for capsular laxity, a larger bite of the capsular tissue can be taken to allow for a greater degree of plication. Further, the interportal capsule can be tightened by alternately advancing the location where sutures are passed through the capsule. Specifically, the sutures are passed such that larger bites of the distal capsule are taken, increasing the tightness of the capsule in external rotation.9
Rehabilitation
After surgery, hip extension and external rotation are limited to decrease stress on the capsular closure. The patient is placed into a hip orthosis with 0° to 90° of flexion and a night abduction pillow to limit hip external rotation. Crutch-assisted gait with 20 lb of foot-flat weight-bearing is maintained the first 3 weeks. Continuous passive motion and use of a stationary bicycle are recommended for the first 3 weeks, and then the patient slowly progresses to muscle strengthening, including core and proximal motor control. Closed-chain exercises are begun 6 weeks after surgery. Treadmill running may start at 12 weeks, with the goal of returning to sport at 4 to 6 months.
Discussion
Capsular closure during hip arthroscopy restores the normal anatomy of the IFL and therefore restores the biomechanical characteristics of the hip joint. Scientific studies have found that capsular repair or plication after hip arthroscopy restores normal hip translation, rotation, and strain. Clinical studies have also demonstrated a lower revision rate and more rapid return to athletic activity. Capsular closure, however, is technically challenging and increases operative time, but gross instability and microinstability can be avoided with meticulous closure/plication.
Am J Orthop. 2017;46(1):49-54. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Boykin RE, Anz AW, Bushnell BD, Kocher MS, Stubbs AJ, Philippon MJ. Hip instability. J Am Acad Orthop Surg. 2011;19(6):340-349.
2. Byrd JW, Jones KS. Hip arthroscopy for labral pathology: prospective analysis with 10-year follow-up. Arthroscopy. 2009;25(4):365-368.
3. Benali Y, Katthagen BD. Hip subluxation as a complication of arthroscopic debridement. Arthroscopy. 2009;25(4):405-407.
4. Matsuda DK. Acute iatrogenic dislocation following hip impingement arthroscopic surgery. Arthroscopy. 2009;25(4):400-404.
5. Ranawat AS, McClincy M, Sekiya JK. Anterior dislocation of the hip after arthroscopy in a patient with capsular laxity of the hip. A case report. J Bone Joint Surg Am. 2009;91(1):192-197.
6. McCormick F, Slikker W 3rd, Harris JD, et al. Evidence of capsular defect following hip arthroscopy. Knee Surg Sports Traumatol Arthrosc. 2014;22(4):902-905.
7. Wylie JD, Beckmann JT, Maak TG, Aoki SK. Arthroscopic capsular repair for symptomatic hip instability after previous hip arthroscopic surgery. Am J Sports Med. 2016;44(1):39-45.
8. Frank RM, Lee S, Bush-Joseph CA, Kelly BT, Salata MJ, Nho SJ. Improved outcomes after hip arthroscopic surgery in patients undergoing T-capsulotomy with complete repair versus partial repair for femoroacetabular impingement: a comparative matched-pair analysis. Am J Sports Med. 2014;42(11):2634-2642.
9. Domb BG, Philippon MJ, Giordano BD. Arthroscopic capsulotomy, capsular repair, and capsular plication of the hip: relation to atraumatic instability. Arthroscopy. 2013;29(1):162-173.
10. Asopa V, Singh PJ. The intracapsular atraumatic arthroscopic technique for closure of the hip capsule. Arthrosc Tech. 2014;3(2):e245-e247.
11. Camp CL, Reardon PJ, Levy BA, Krych AJ. A simple technique for capsular repair after hip arthroscopy. Arthrosc Tech. 2015;4(6):e737-e740.
12. Chow RM, Engasser WM, Krych AJ, Levy BA. Arthroscopic capsular repair in the treatment of femoroacetabular impingement. Arthrosc Tech. 2014;3(1):e27-e30.
13. Harris JD, Slikker W 3rd, Gupta AK, McCormick FM, Nho SJ. Routine complete capsular closure during hip arthroscopy. Arthrosc Tech. 2013;2(2):e89-e94.
14. Kuhns BD, Weber AE, Levy DM, et al. Capsular management in hip arthroscopy: an anatomic, biomechanical, and technical review. Front Surg. 2016;3:13.
Take-Home Points
- Hip capsule provides static stabilization for the hip joint.
- Capsular management must weigh visualization to address underlying osseous deformity but also repair/plication of the capsule to maintain biomechanical characteristics.
- T-capsulotomy provides optimal visualization with a small interportal incision with a vertical incision along the femoral neck.
- Extensile interportal capsulotomy is the most widely used capsulotomy and size may vary depending on capsular and patient characteristics.
- Orthopedic surgeons should be equipped to employ either technique depending on the patients individual hip pathomorphology.
Hip arthroscopy has emerged as a common surgical treatment for a number of hip pathologies. Surgical treatment strategies, including management of the hip capsule, have evolved. Whereas earlier hip arthroscopies often involved capsulectomy or capsulotomy without repair, more recently capsular closure has been considered an important step in restoring the anatomy of the hip joint and preventing microinstability or gross macroinstability.
The anatomy of the hip joint includes both static and dynamic stabilizers designed to maintain a functioning articulation. The osseous articulation of the femoral head and acetabulum is the first static stabilizer, with variations in offset, version, and inclination of the acetabulum and the proximal femur. The joint capsule consists of 3 ligaments—iliofemoral, pubofemoral, and ischiofemoral—that converge to form the zona orbicularis. Other soft-tissue structures, such as the articular cartilage, the labrum, the transverse acetabular ligament, the pulvinar, and the ligamentum teres, also provide static constraint.1 The surrounding musculature provides the hip joint with dynamic stability, which contributes to overall maintenance of proper joint kinematics.
Management of the hip capsule has evolved as our understanding of hip pathology and biomechanics has matured. Initial articles on using hip arthroscopy to treat labral tears described improvement in clinical outcomes,2 but the cases involved limited focal capsulotomy. Not until the idea of femoroacetabular impingement (FAI) was introduced were extensive capsulotomies and capsulectomies performed to address the underlying osseous deformities and emulate open techniques. Soon after our ability to access osseous pathomorphology improved with enhanced visualization and comprehensive resection, cases of hip instability after hip arthroscopy surfaced.3-5 Although frank dislocation after hip arthroscopy is rare and largely underreported, it is a catastrophic complication. In addition, focal capsular defects were also described in cases of failed hip arthroscopy and thought to lead to microinstability of the hip.6 Iatrogenic microinstability is thought to be more common, but it is also underrecognized as a cause of failure of hip arthroscopy.7Microinstability is a pathologic condition that can affect hip function. In cases of recurrent pain and unimproved functional status after surgery, microinstability should be considered. In an imaging study of capsule integrity, McCormick and colleagues6 found that 78% of patients who underwent revision arthroscopic surgery after hip arthroscopic surgery for FAI showed evidence of capsular and iliofemoral defects on magnetic resonance angiography. Frank and colleagues8 reported that, though all patients showed preoperative-to-postoperative improvement on outcome measures, those who underwent complete repair of their T-capsulotomy (vs repair of only its longitudinal portion) had superior outcomes, particularly increased sport-specific activity.
For patients undergoing hip arthroscopy, several predisposing factors can increase the risk of postoperative instability. Patient-related hip instability factors include generalized ligamentous laxity, supraphysiologic athletics (eg, dance), and borderline or true hip dysplasia. Surgeon-related factors include overaggressive acetabular rim resection, excessive labral débridement, and lack of capsular repair.5,9 Although there are multiple techniques for accessing the hip joint and addressing capsular closure at the end of surgery,9-14 we think capsular closure is an important aspect of the case.
Surgical Technique
For a demonstration of this technique, click here to see the video that accompanies this article. The patient is moved to a traction table and placed in the supine position. Induction of general anesthesia with muscle relaxation allows for atraumatic axial traction. The anesthetized patient is assessed for passive motion and ligamentous laxity. Well-padded boots are applied, and a well-padded perineal post is used for positioning. Gentle traction is applied to the contralateral limb, and axial traction is applied through the surgical limb with the hip abducted and minimally flexed. The leg is then adducted and neutrally extended, inducing a transverse vector cantilever moment to the proximal femur. The foot is internally rotated to optimize femoral neck length on an anteroposterior radiograph. The circulating nursing staff notes the onset of hip distraction in order to ensure safe traction duration.
Bony landmarks are marked with a sterile marking pen. Under fluoroscopic guidance, an anterolateral (AL) portal is established 1 cm proximal and 1 cm anterior to the AL tip of the greater trochanter. Standard cannulation allows for intra-articular visualization with a 70° arthroscope. A needle is used to localize placement of a modified anterior portal. After cannulation, the arthroscope is placed in the modified anterior portal to confirm safe entry of the portal without labral violation. An arthroscopic scalpel (Samurai Blade; Stryker Sports Medicine) is used to make a transverse interportal capsulotomy 8 mm to 10 mm from the labrum and extending from 12 to 2 o’clock; length is 2 cm to 4 cm, depending on the extent of the intra-articular injury (Figure 1A).
The acetabular rim is trimmed with a 5.0-mm arthroscopic burr. Distal AL accessory (DALA) portal placement (4-6 cm distal to and in line with the AL portal) allows for suture anchor–based labral refixation. Generally, 2 to 4 anchors (1.4-mm NanoTack Anatomic Labrum Restoration System; Stryker Sports Medicine) are placed as near the articular cartilage as possible without penetration (Figure 1B). On completion of labral refixation, traction is released, and the hip is flexed to 20° to 30°.
T-Capsulotomy
Pericapsular fatty tissue is débrided with an arthroscopic shaver to visualize the interval between the iliocapsularis and gluteus minimus muscles. An arthroscopic scalpel is used, through a 5.0-mm cannula in the DALA portal, to extend the capsulotomy longitudinally and perpendicular to the interportal capsulotomy (Figure 1C). The T-capsulotomy is performed along the length of the femoral neck distally to the capsular reflection at the intertrochanteric line. The arthroscopic burr is used to perform a femoral osteochondroplasty between the lateral synovial folds (12 o’clock) and the medial synovial folds (6 o’clock). Dynamic examination and fluoroscopic imaging confirm that the entire cam deformity has been excised and that there is no evidence of impingement.
Although various suture-shuttling or tissue-penetrating/retrieving devices may be used, we recommend whichever device is appropriate for closing the capsule in its entirety. With the arthroscope in the modified anterior portal, an 8.25-mm × 90-mm cannula is placed in the AL portal, and an 8.25-mm × 110-mm cannula in the DALA portal. These portals will facilitate suture passage.
The vertical limb of the T-capsulotomy is closed with 2 to 4 side-to-side sutures, and the interportal capsulotomy limb with 2 or 3 sutures. Capsular closure begins with the distal portion of the longitudinal limb at the base of the iliofemoral ligament (IFL). A crescent tissue penetrating device (Slingshot; Stryker Sports Medicine) is loaded with high-strength No. 2 suture (Zipline; Stryker Sports Medicine) and placed through the AL portal to sharply pierce the lateral leaflet of the IFL (Figure 1D). The No. 2 suture is shuttled into the intra-articular side of the capsule (Figure 1E). Through the DALA portal, the penetrating device is used to pierce the medial leaflet to retrieve the free suture (Figure 1F). Next, the looped suture retriever is used to pull the suture from the AL portal to the DALA portal so the suture can be tied. We prefer to tie each suture individually after it is passed, but all of the sutures can be passed first, and then tied. As successive suture placement and knot tying inherently tighten the capsule, successive visualization requires more precision. Each subsequent suture is similarly passed, about 1 cm proximal to the previous stitch.
After closure of the vertical limb of the T-capsulotomy, we prefer to close the interportal capsulotomy with the InJector II Capsule Restoration System (Stryker Sports Medicine), a device that allows for closure through a single cannula lateral to medial. This device is passed through the AL cannula in order to bring the suture end through the proximal IFL attached to the acetabulum (Figure 1G). The device is removed from the cannula, and the other suture end is placed in the device and passed through the distal IFL (Figure 1H). The stitch is then tensioned and tied. Likewise, closure of the medial IFL involves passing the InJector through the DALA cannula and bringing the first suture end through the proximal IFL attached to the acetabulum. The Injector is removed from the cannula, and the other suture end is placed in the device and passed through the distal IFL. The stitch is then tensioned and tied with the hip in neutral extension. Generally, 2 or 3 stitches are used to close the interportal capsulotomy. Complete capsular closure is confirmed by the inability to visualize the underlying femoral head/neck and by probing the anterior capsule to ensure proper tension (Figure 1I).
Extensile Interportal Capsulotomy
An alternative to T-capsulotomy is interportal capsulotomy. Just as with T-capsulotomy closure, multiple different suture passing devices can be used. Good visualization for accessing the peripheral compartment generally is achieved by making the interportal capsulotomy 4 cm to 6 cm longer than the horizontal limb of the T-capsulotomy (Figures 2A, 2B). Capsular closure usually begins with the medial portion of the interportal capsulotomy. With the arthroscope in the AL portal, the 8.25-mm × 90-mm cannula is placed in the midanterior portal (MAP), and an 8.25-mm × 110-mm cannula is placed in the DALA portal.
Ligamentous laxity determines degree of capsular closure. The capsular leaflets can be closed end to end if there is little concern for laxity and instability. If there is more concern for capsular laxity, a larger bite of the capsular tissue can be taken to allow for a greater degree of plication. Further, the interportal capsule can be tightened by alternately advancing the location where sutures are passed through the capsule. Specifically, the sutures are passed such that larger bites of the distal capsule are taken, increasing the tightness of the capsule in external rotation.9
Rehabilitation
After surgery, hip extension and external rotation are limited to decrease stress on the capsular closure. The patient is placed into a hip orthosis with 0° to 90° of flexion and a night abduction pillow to limit hip external rotation. Crutch-assisted gait with 20 lb of foot-flat weight-bearing is maintained the first 3 weeks. Continuous passive motion and use of a stationary bicycle are recommended for the first 3 weeks, and then the patient slowly progresses to muscle strengthening, including core and proximal motor control. Closed-chain exercises are begun 6 weeks after surgery. Treadmill running may start at 12 weeks, with the goal of returning to sport at 4 to 6 months.
Discussion
Capsular closure during hip arthroscopy restores the normal anatomy of the IFL and therefore restores the biomechanical characteristics of the hip joint. Scientific studies have found that capsular repair or plication after hip arthroscopy restores normal hip translation, rotation, and strain. Clinical studies have also demonstrated a lower revision rate and more rapid return to athletic activity. Capsular closure, however, is technically challenging and increases operative time, but gross instability and microinstability can be avoided with meticulous closure/plication.
Am J Orthop. 2017;46(1):49-54. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- Hip capsule provides static stabilization for the hip joint.
- Capsular management must weigh visualization to address underlying osseous deformity but also repair/plication of the capsule to maintain biomechanical characteristics.
- T-capsulotomy provides optimal visualization with a small interportal incision with a vertical incision along the femoral neck.
- Extensile interportal capsulotomy is the most widely used capsulotomy and size may vary depending on capsular and patient characteristics.
- Orthopedic surgeons should be equipped to employ either technique depending on the patients individual hip pathomorphology.
Hip arthroscopy has emerged as a common surgical treatment for a number of hip pathologies. Surgical treatment strategies, including management of the hip capsule, have evolved. Whereas earlier hip arthroscopies often involved capsulectomy or capsulotomy without repair, more recently capsular closure has been considered an important step in restoring the anatomy of the hip joint and preventing microinstability or gross macroinstability.
The anatomy of the hip joint includes both static and dynamic stabilizers designed to maintain a functioning articulation. The osseous articulation of the femoral head and acetabulum is the first static stabilizer, with variations in offset, version, and inclination of the acetabulum and the proximal femur. The joint capsule consists of 3 ligaments—iliofemoral, pubofemoral, and ischiofemoral—that converge to form the zona orbicularis. Other soft-tissue structures, such as the articular cartilage, the labrum, the transverse acetabular ligament, the pulvinar, and the ligamentum teres, also provide static constraint.1 The surrounding musculature provides the hip joint with dynamic stability, which contributes to overall maintenance of proper joint kinematics.
Management of the hip capsule has evolved as our understanding of hip pathology and biomechanics has matured. Initial articles on using hip arthroscopy to treat labral tears described improvement in clinical outcomes,2 but the cases involved limited focal capsulotomy. Not until the idea of femoroacetabular impingement (FAI) was introduced were extensive capsulotomies and capsulectomies performed to address the underlying osseous deformities and emulate open techniques. Soon after our ability to access osseous pathomorphology improved with enhanced visualization and comprehensive resection, cases of hip instability after hip arthroscopy surfaced.3-5 Although frank dislocation after hip arthroscopy is rare and largely underreported, it is a catastrophic complication. In addition, focal capsular defects were also described in cases of failed hip arthroscopy and thought to lead to microinstability of the hip.6 Iatrogenic microinstability is thought to be more common, but it is also underrecognized as a cause of failure of hip arthroscopy.7Microinstability is a pathologic condition that can affect hip function. In cases of recurrent pain and unimproved functional status after surgery, microinstability should be considered. In an imaging study of capsule integrity, McCormick and colleagues6 found that 78% of patients who underwent revision arthroscopic surgery after hip arthroscopic surgery for FAI showed evidence of capsular and iliofemoral defects on magnetic resonance angiography. Frank and colleagues8 reported that, though all patients showed preoperative-to-postoperative improvement on outcome measures, those who underwent complete repair of their T-capsulotomy (vs repair of only its longitudinal portion) had superior outcomes, particularly increased sport-specific activity.
For patients undergoing hip arthroscopy, several predisposing factors can increase the risk of postoperative instability. Patient-related hip instability factors include generalized ligamentous laxity, supraphysiologic athletics (eg, dance), and borderline or true hip dysplasia. Surgeon-related factors include overaggressive acetabular rim resection, excessive labral débridement, and lack of capsular repair.5,9 Although there are multiple techniques for accessing the hip joint and addressing capsular closure at the end of surgery,9-14 we think capsular closure is an important aspect of the case.
Surgical Technique
For a demonstration of this technique, click here to see the video that accompanies this article. The patient is moved to a traction table and placed in the supine position. Induction of general anesthesia with muscle relaxation allows for atraumatic axial traction. The anesthetized patient is assessed for passive motion and ligamentous laxity. Well-padded boots are applied, and a well-padded perineal post is used for positioning. Gentle traction is applied to the contralateral limb, and axial traction is applied through the surgical limb with the hip abducted and minimally flexed. The leg is then adducted and neutrally extended, inducing a transverse vector cantilever moment to the proximal femur. The foot is internally rotated to optimize femoral neck length on an anteroposterior radiograph. The circulating nursing staff notes the onset of hip distraction in order to ensure safe traction duration.
Bony landmarks are marked with a sterile marking pen. Under fluoroscopic guidance, an anterolateral (AL) portal is established 1 cm proximal and 1 cm anterior to the AL tip of the greater trochanter. Standard cannulation allows for intra-articular visualization with a 70° arthroscope. A needle is used to localize placement of a modified anterior portal. After cannulation, the arthroscope is placed in the modified anterior portal to confirm safe entry of the portal without labral violation. An arthroscopic scalpel (Samurai Blade; Stryker Sports Medicine) is used to make a transverse interportal capsulotomy 8 mm to 10 mm from the labrum and extending from 12 to 2 o’clock; length is 2 cm to 4 cm, depending on the extent of the intra-articular injury (Figure 1A).
The acetabular rim is trimmed with a 5.0-mm arthroscopic burr. Distal AL accessory (DALA) portal placement (4-6 cm distal to and in line with the AL portal) allows for suture anchor–based labral refixation. Generally, 2 to 4 anchors (1.4-mm NanoTack Anatomic Labrum Restoration System; Stryker Sports Medicine) are placed as near the articular cartilage as possible without penetration (Figure 1B). On completion of labral refixation, traction is released, and the hip is flexed to 20° to 30°.
T-Capsulotomy
Pericapsular fatty tissue is débrided with an arthroscopic shaver to visualize the interval between the iliocapsularis and gluteus minimus muscles. An arthroscopic scalpel is used, through a 5.0-mm cannula in the DALA portal, to extend the capsulotomy longitudinally and perpendicular to the interportal capsulotomy (Figure 1C). The T-capsulotomy is performed along the length of the femoral neck distally to the capsular reflection at the intertrochanteric line. The arthroscopic burr is used to perform a femoral osteochondroplasty between the lateral synovial folds (12 o’clock) and the medial synovial folds (6 o’clock). Dynamic examination and fluoroscopic imaging confirm that the entire cam deformity has been excised and that there is no evidence of impingement.
Although various suture-shuttling or tissue-penetrating/retrieving devices may be used, we recommend whichever device is appropriate for closing the capsule in its entirety. With the arthroscope in the modified anterior portal, an 8.25-mm × 90-mm cannula is placed in the AL portal, and an 8.25-mm × 110-mm cannula in the DALA portal. These portals will facilitate suture passage.
The vertical limb of the T-capsulotomy is closed with 2 to 4 side-to-side sutures, and the interportal capsulotomy limb with 2 or 3 sutures. Capsular closure begins with the distal portion of the longitudinal limb at the base of the iliofemoral ligament (IFL). A crescent tissue penetrating device (Slingshot; Stryker Sports Medicine) is loaded with high-strength No. 2 suture (Zipline; Stryker Sports Medicine) and placed through the AL portal to sharply pierce the lateral leaflet of the IFL (Figure 1D). The No. 2 suture is shuttled into the intra-articular side of the capsule (Figure 1E). Through the DALA portal, the penetrating device is used to pierce the medial leaflet to retrieve the free suture (Figure 1F). Next, the looped suture retriever is used to pull the suture from the AL portal to the DALA portal so the suture can be tied. We prefer to tie each suture individually after it is passed, but all of the sutures can be passed first, and then tied. As successive suture placement and knot tying inherently tighten the capsule, successive visualization requires more precision. Each subsequent suture is similarly passed, about 1 cm proximal to the previous stitch.
After closure of the vertical limb of the T-capsulotomy, we prefer to close the interportal capsulotomy with the InJector II Capsule Restoration System (Stryker Sports Medicine), a device that allows for closure through a single cannula lateral to medial. This device is passed through the AL cannula in order to bring the suture end through the proximal IFL attached to the acetabulum (Figure 1G). The device is removed from the cannula, and the other suture end is placed in the device and passed through the distal IFL (Figure 1H). The stitch is then tensioned and tied. Likewise, closure of the medial IFL involves passing the InJector through the DALA cannula and bringing the first suture end through the proximal IFL attached to the acetabulum. The Injector is removed from the cannula, and the other suture end is placed in the device and passed through the distal IFL. The stitch is then tensioned and tied with the hip in neutral extension. Generally, 2 or 3 stitches are used to close the interportal capsulotomy. Complete capsular closure is confirmed by the inability to visualize the underlying femoral head/neck and by probing the anterior capsule to ensure proper tension (Figure 1I).
Extensile Interportal Capsulotomy
An alternative to T-capsulotomy is interportal capsulotomy. Just as with T-capsulotomy closure, multiple different suture passing devices can be used. Good visualization for accessing the peripheral compartment generally is achieved by making the interportal capsulotomy 4 cm to 6 cm longer than the horizontal limb of the T-capsulotomy (Figures 2A, 2B). Capsular closure usually begins with the medial portion of the interportal capsulotomy. With the arthroscope in the AL portal, the 8.25-mm × 90-mm cannula is placed in the midanterior portal (MAP), and an 8.25-mm × 110-mm cannula is placed in the DALA portal.
Ligamentous laxity determines degree of capsular closure. The capsular leaflets can be closed end to end if there is little concern for laxity and instability. If there is more concern for capsular laxity, a larger bite of the capsular tissue can be taken to allow for a greater degree of plication. Further, the interportal capsule can be tightened by alternately advancing the location where sutures are passed through the capsule. Specifically, the sutures are passed such that larger bites of the distal capsule are taken, increasing the tightness of the capsule in external rotation.9
Rehabilitation
After surgery, hip extension and external rotation are limited to decrease stress on the capsular closure. The patient is placed into a hip orthosis with 0° to 90° of flexion and a night abduction pillow to limit hip external rotation. Crutch-assisted gait with 20 lb of foot-flat weight-bearing is maintained the first 3 weeks. Continuous passive motion and use of a stationary bicycle are recommended for the first 3 weeks, and then the patient slowly progresses to muscle strengthening, including core and proximal motor control. Closed-chain exercises are begun 6 weeks after surgery. Treadmill running may start at 12 weeks, with the goal of returning to sport at 4 to 6 months.
Discussion
Capsular closure during hip arthroscopy restores the normal anatomy of the IFL and therefore restores the biomechanical characteristics of the hip joint. Scientific studies have found that capsular repair or plication after hip arthroscopy restores normal hip translation, rotation, and strain. Clinical studies have also demonstrated a lower revision rate and more rapid return to athletic activity. Capsular closure, however, is technically challenging and increases operative time, but gross instability and microinstability can be avoided with meticulous closure/plication.
Am J Orthop. 2017;46(1):49-54. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Boykin RE, Anz AW, Bushnell BD, Kocher MS, Stubbs AJ, Philippon MJ. Hip instability. J Am Acad Orthop Surg. 2011;19(6):340-349.
2. Byrd JW, Jones KS. Hip arthroscopy for labral pathology: prospective analysis with 10-year follow-up. Arthroscopy. 2009;25(4):365-368.
3. Benali Y, Katthagen BD. Hip subluxation as a complication of arthroscopic debridement. Arthroscopy. 2009;25(4):405-407.
4. Matsuda DK. Acute iatrogenic dislocation following hip impingement arthroscopic surgery. Arthroscopy. 2009;25(4):400-404.
5. Ranawat AS, McClincy M, Sekiya JK. Anterior dislocation of the hip after arthroscopy in a patient with capsular laxity of the hip. A case report. J Bone Joint Surg Am. 2009;91(1):192-197.
6. McCormick F, Slikker W 3rd, Harris JD, et al. Evidence of capsular defect following hip arthroscopy. Knee Surg Sports Traumatol Arthrosc. 2014;22(4):902-905.
7. Wylie JD, Beckmann JT, Maak TG, Aoki SK. Arthroscopic capsular repair for symptomatic hip instability after previous hip arthroscopic surgery. Am J Sports Med. 2016;44(1):39-45.
8. Frank RM, Lee S, Bush-Joseph CA, Kelly BT, Salata MJ, Nho SJ. Improved outcomes after hip arthroscopic surgery in patients undergoing T-capsulotomy with complete repair versus partial repair for femoroacetabular impingement: a comparative matched-pair analysis. Am J Sports Med. 2014;42(11):2634-2642.
9. Domb BG, Philippon MJ, Giordano BD. Arthroscopic capsulotomy, capsular repair, and capsular plication of the hip: relation to atraumatic instability. Arthroscopy. 2013;29(1):162-173.
10. Asopa V, Singh PJ. The intracapsular atraumatic arthroscopic technique for closure of the hip capsule. Arthrosc Tech. 2014;3(2):e245-e247.
11. Camp CL, Reardon PJ, Levy BA, Krych AJ. A simple technique for capsular repair after hip arthroscopy. Arthrosc Tech. 2015;4(6):e737-e740.
12. Chow RM, Engasser WM, Krych AJ, Levy BA. Arthroscopic capsular repair in the treatment of femoroacetabular impingement. Arthrosc Tech. 2014;3(1):e27-e30.
13. Harris JD, Slikker W 3rd, Gupta AK, McCormick FM, Nho SJ. Routine complete capsular closure during hip arthroscopy. Arthrosc Tech. 2013;2(2):e89-e94.
14. Kuhns BD, Weber AE, Levy DM, et al. Capsular management in hip arthroscopy: an anatomic, biomechanical, and technical review. Front Surg. 2016;3:13.
1. Boykin RE, Anz AW, Bushnell BD, Kocher MS, Stubbs AJ, Philippon MJ. Hip instability. J Am Acad Orthop Surg. 2011;19(6):340-349.
2. Byrd JW, Jones KS. Hip arthroscopy for labral pathology: prospective analysis with 10-year follow-up. Arthroscopy. 2009;25(4):365-368.
3. Benali Y, Katthagen BD. Hip subluxation as a complication of arthroscopic debridement. Arthroscopy. 2009;25(4):405-407.
4. Matsuda DK. Acute iatrogenic dislocation following hip impingement arthroscopic surgery. Arthroscopy. 2009;25(4):400-404.
5. Ranawat AS, McClincy M, Sekiya JK. Anterior dislocation of the hip after arthroscopy in a patient with capsular laxity of the hip. A case report. J Bone Joint Surg Am. 2009;91(1):192-197.
6. McCormick F, Slikker W 3rd, Harris JD, et al. Evidence of capsular defect following hip arthroscopy. Knee Surg Sports Traumatol Arthrosc. 2014;22(4):902-905.
7. Wylie JD, Beckmann JT, Maak TG, Aoki SK. Arthroscopic capsular repair for symptomatic hip instability after previous hip arthroscopic surgery. Am J Sports Med. 2016;44(1):39-45.
8. Frank RM, Lee S, Bush-Joseph CA, Kelly BT, Salata MJ, Nho SJ. Improved outcomes after hip arthroscopic surgery in patients undergoing T-capsulotomy with complete repair versus partial repair for femoroacetabular impingement: a comparative matched-pair analysis. Am J Sports Med. 2014;42(11):2634-2642.
9. Domb BG, Philippon MJ, Giordano BD. Arthroscopic capsulotomy, capsular repair, and capsular plication of the hip: relation to atraumatic instability. Arthroscopy. 2013;29(1):162-173.
10. Asopa V, Singh PJ. The intracapsular atraumatic arthroscopic technique for closure of the hip capsule. Arthrosc Tech. 2014;3(2):e245-e247.
11. Camp CL, Reardon PJ, Levy BA, Krych AJ. A simple technique for capsular repair after hip arthroscopy. Arthrosc Tech. 2015;4(6):e737-e740.
12. Chow RM, Engasser WM, Krych AJ, Levy BA. Arthroscopic capsular repair in the treatment of femoroacetabular impingement. Arthrosc Tech. 2014;3(1):e27-e30.
13. Harris JD, Slikker W 3rd, Gupta AK, McCormick FM, Nho SJ. Routine complete capsular closure during hip arthroscopy. Arthrosc Tech. 2013;2(2):e89-e94.
14. Kuhns BD, Weber AE, Levy DM, et al. Capsular management in hip arthroscopy: an anatomic, biomechanical, and technical review. Front Surg. 2016;3:13.
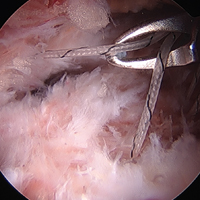
Relapsing Polychondritis With Meningoencephalitis
Relapsing polychondritis (RP) is an autoimmune disease affecting cartilaginous structures such as the ears, respiratory passages, joints, and cardiovascular system.1,2 In rare cases, the systemic effects of this autoimmune process can cause central nervous system (CNS) involvement such as meningoencephalitis (ME).3 In 2011, Wang et al4 described 4 cases of RP with ME and reviewed 24 cases from the literature. We present a case of a man with RP-associated ME that was responsive to steroid treatment. We also provide an updated review of the literature.
Case Report
A 44-year-old man developed gradually worsening bilateral ear pain, headaches, and seizures. He was briefly hospitalized and discharged with levetiracetam and quetiapine. However, his mental status continued to deteriorate and he was subsequently hospitalized 3 months later with confusion, hallucinations, and seizures.
On physical examination the patient was disoriented and unable to form cohesive sentences. He had bilateral tenderness, erythema, and edema of the auricles, which notably spared the lobules (Figure 1). The conjunctivae were injected bilaterally, and joint involvement included bilateral knee tenderness and swelling. Neurologic examination revealed questionable meningeal signs but no motor or sensory deficits. An extensive laboratory workup for the etiology of his altered mental status was unremarkable, except for a mildly elevated white blood cell count in the cerebrospinal fluid with predominantly lymphocytes. No infectious etiologies were identified on laboratory testing, and rheumatologic markers were negative including antinuclear antibody, rheumatoid factor, and anti–Sjögren syndrome antigen A/Sjögren syndrome antigen B. Magnetic resonance imaging revealed nonspecific findings of bilateral T2 hyperdensities in the subcortical white matter; however, cerebral angiography revealed no evidence of vasculitis. A biopsy of the right antihelix revealed prominent perichondritis and a neutrophilic inflammatory infiltrate with several lymphocytes and histiocytes (Figure 2). There was degeneration of the cartilaginous tissue with evidence of pyknotic nuclei, eosinophilia, and vacuolization of the chondrocytes. He was diagnosed with RP on the basis of clinical and histologic inflammation of the auricular cartilage, polyarthritis, and ocular inflammation.
The patient was treated with high-dose immunosuppression with methylprednisolone (1000 mg intravenous once daily for 5 days) and cyclophosphamide (one dose at 500 mg/m2), which resulted in remarkable improvement in his mental status, auricular inflammation, and knee pain. After 31 days of hospitalization the patient was discharged with a course of oral prednisone (starting at 60 mg/d, then tapered over the following 2 months) and monthly cyclophosphamide infusions (5 months total; starting at 500 mg/m2, then uptitrated to goal of 1000 mg/m2). Maintenance suppression was achieved with azathioprine (starting at 50 mg daily, then uptitrated to 100 mg daily), which was continued without any evidence of relapsed disease through his last outpatient visit 1 year after the diagnosis.
Comment
Auricular inflammation is a hallmark of RP and is present in 83% to 95% of patients.1,3 The affected ears can appear erythematous to violaceous with tender edema of the auricle that spares the lobules where no cartilage is present. The inflammation can extend into the ear canal and cause hearing loss, tinnitus, and vertigo. Histologically, RP can present with a nonspecific leukocytoclastic vasculitis and inflammatory destruction of the cartilage. Therefore, diagnosis of RP is reliant mainly on clinical characteristics rather than pathologic findings. In 1976, McAdam et al5 established diagnostic criteria for RP based on the presence of common clinical manifestations (eg, auricular chondritis, seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation). Michet et al6 later proposed major and minor criteria to classify and diagnose RP based on clinical manifestations. Diagnosis of our patient was confirmed by the presence of auricular chondritis, polyarthritis, and ocular inflammation. Diagnosing RP can be difficult because it has many systemic manifestations that can evoke a broad differential diagnosis. The time to diagnosis in our patient was 3 months, but the mean delay in diagnosis for patients with RP and ME is 2.9 years.4
The etiology of RP remains unclear, but current evidence supports an immune-mediated process directed toward proteins found in cartilage. Animal studies have suggested that RP may be driven by antibodies to matrillin 1 and type II collagen. There also may be a familial association with HLA-DR4 and genetic predisposition to autoimmune diseases in individuals affected by RP.1,3 The pathogenesis of CNS involvement in RP is thought to be due to a localized small vessel vasculitis.7,8 In our patient, however, cerebral angiography was negative for vasculitis, and thus our case may represent another mechanism for CNS involvement. There have been cases of encephalitis in RP caused by pathways other than CNS vasculitis. Kashihara et al9 reported a case of RP with encephalitis associated with antiglutamate receptor antibodies found in the cerebrospinal fluid and blood.
Treatment of RP has been based on pathophysiological considerations rather than empiric data due to its rarity. Relapsing polychondritis has been responsive to steroid treatment in reported cases as well as in our patient; however, in cases in which RP did not respond to steroids, infliximab may be effective for RP with ME.10 Further research regarding the treatment outcomes of RP with ME may be warranted.
Although rare, additional cases of RP with ME have been reported (Table). Wang et al4 described a series of 28 patients with RP and ME from 1960 to 2010. A PubMed search of articles indexed for MEDLINE that were published in the English-language literature from 2010 to 2016 was performed using the search terms relapsing polychondritis and nervous system. Including our patient, RP with ME was reported in 17 additional cases since Wang et al4 published their findings. These cases involved adults ranging in age from 44 to 73 years who were mainly men (14/17 [82%]). All of the patients presented with bilateral auricular chondritis, except for a case of unilateral ear involvement reported by Storey et al.11 Common neurologic manifestations included fever, headache, and altered mental status. Motor symptoms ranged from dysarthria and agraphia12 to hemiparesis.13 The mechanism of CNS involvement in RP was not identified in most cases; however, Mattiassich et al14 documented cerebral vasculitis in their patient, and Niwa et al16 found diffuse cerebral vasculitis on autopsy. Eleven of 17 (65%) cases responded to steroid treatment. Of the 6 cases in which RP did not respond to steroids, 2 patients died despite high-dose steroid treatment,11,20 2 responded to infliximab,10,15 1 responded to tocilizumab,21 and 1 was lost to follow-up after initial treatment failure.20
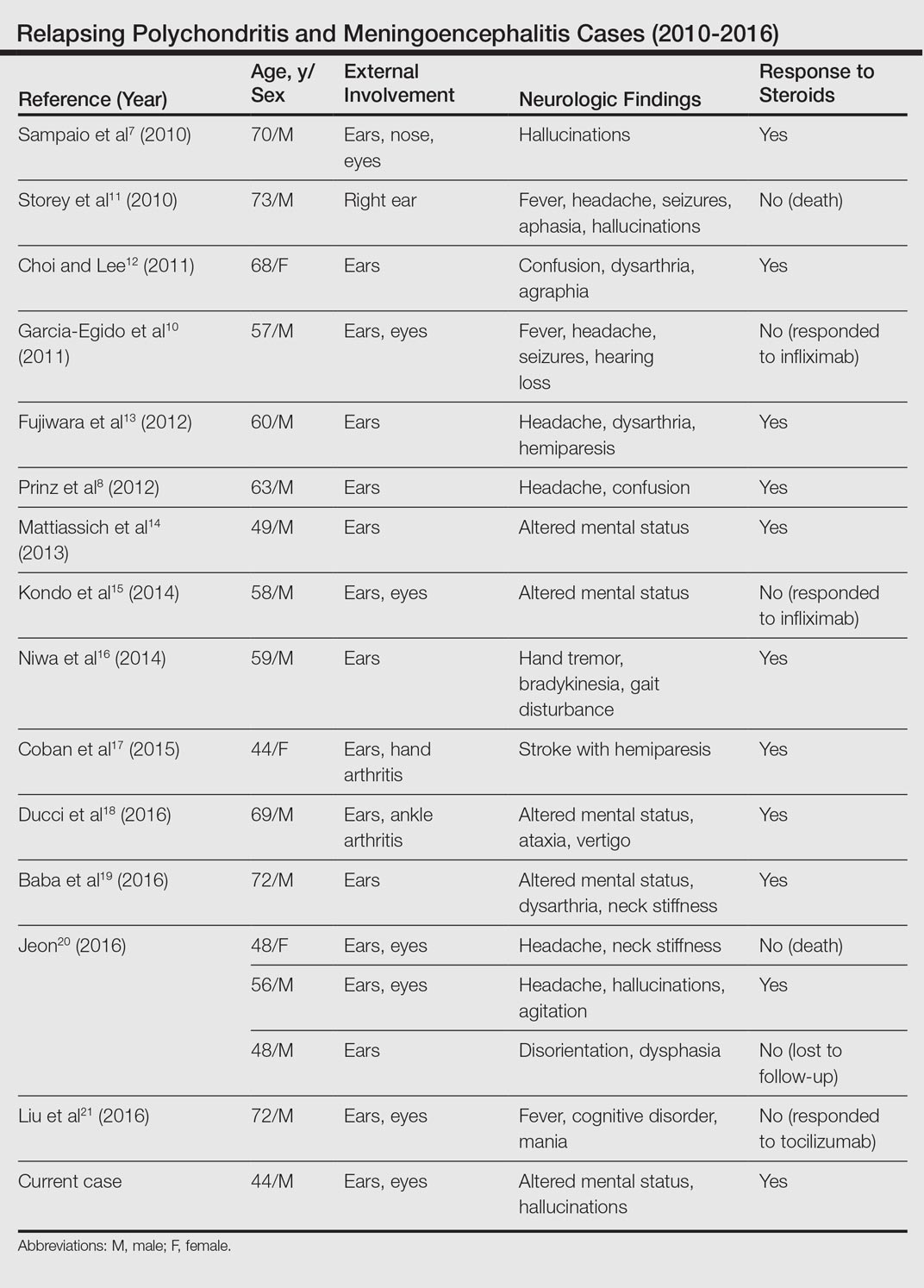
Conclusion
Although rare, RP should not be overlooked in the inpatient setting due to its potential for life-threatening systemic effects. Early diagnosis of this condition may be of benefit to this select population of patients, and further research regarding the prognosis, mechanisms, and treatment of RP may be necessary in the future.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis: a 2013 update. Autoimmun Rev. 2014;13:90-95.
- Ostrowski RA, Takagishi T, Robinson J. Rheumatoid arthritis, spondyloarthropathies, and relapsing polychondritis. Handb Clin Neurol. 2014;119:449-461.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Wang ZJ, Pu CQ, Wang ZJ, et al. Meningoencephalitis or meningitis in relapsing polychondritis: four case reports and a literature review. J Clin Neurosci. 2011;18:1608-1615.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet C, McKenna C, Luthra H, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Sampaio L, Silva L, Mariz E, et al. CNS involvement in relapsing polychondritis. Joint Bone Spine. 2010;77:619-620.
- Prinz S, Dafotakis M, Schneider RK, et al. The red puffy ear sign—a clinical sign to diagnose a rare cause of meningoencephalitis. Fortschr Neurol Psychiatr. 2012;80:463-467.
- Kashihara K, Kawada S, Takahashi Y. Autoantibodies to glutamate receptor GluR2 in a patient with limic encephalitis associated with relapsing polychondritis. J Neurol Sci. 2009;287:275-277.
- Garcia-Egido A, Gutierrez C, de la Fuente C, et al. Relapsing polychondritis-associated meningitis and encephalitis: response to infliximab. Rheumatology (Oxford). 2011;50:1721-1723.
- Storey K, Matej R, Rusina R. Unusual association of seronegative, nonparaneoplastic limbic encephalitis and relapsing polychondritis in a patient with history of thymectomy for myasthemia: a case study. J Neurol. 2010;258:159-161.
- Choi HJ, Lee HJ. Relapsing polychondritis with encephalitis. J Clin Rheum. 2011;6:329-331.
- Fujiwara S, Zenke K, Iwata S, et al. Relapsing polychondritis presenting as encephalitis. No Shinkei Geka. 2012;40:247-253.
- Mattiassich G, Egger M, Semlitsch G, et al. Occurrence of relapsing polychondritis with a rising cANCA titre in a cANCA-positive systemic and cerebral vasculitis patient [published online February 5, 2013]. BMJ Case Rep. doi:10.1136/bcr-2013-008717.
- Kondo T, Fukuta M, Takemoto A, et al. Limbic encephalitis associated with relapsing polychondritis responded to infliximab and maintained its condition without recurrence after discontinuation: a case report and review of the literature. Nagoya J Med Sci. 2014;76:361-368.
- Niwa A, Okamoto Y, Kondo T, et al. Perivasculitic pancencephalitis with relapsing polychondritis: an autopsy case report and review of previous cases. Intern Med. 2014;53:1191-1195.
- Coban EK, Xanmemmedoy E, Colak M, et al. A rare complication of a rare disease; stroke due to relapsing polychondritis. Ideggyogy Sz. 2015;68:429-432.
- Ducci R, Germiniani F, Czecko L, et al. Relapsing polychondritis and lymphocytic meningitis with varied neurological symptoms [published online February 5, 2016]. Rev Bras Reumatol. doi:10.1016/j.rbr.2015.09.005.
- Baba T, Kanno S, Shijo T, et al. Callosal disconnection syndrome associated with relapsing polychondritis. Intern Med. 2016;55:1191-1193.
- Jeon C. Relapsing polychondritis with central nervous system involvement: experience of three different cases in a single center. J Korean Med. 2016;31:1846-1850.
- Liu L, Liu S, Guan W, et al. Efficacy of tocilizumab for psychiatric symptoms associated with relapsing polychondritis: the first case report and review of the literature. Rheumatol Int. 2016;36:1185-1189.
Relapsing polychondritis (RP) is an autoimmune disease affecting cartilaginous structures such as the ears, respiratory passages, joints, and cardiovascular system.1,2 In rare cases, the systemic effects of this autoimmune process can cause central nervous system (CNS) involvement such as meningoencephalitis (ME).3 In 2011, Wang et al4 described 4 cases of RP with ME and reviewed 24 cases from the literature. We present a case of a man with RP-associated ME that was responsive to steroid treatment. We also provide an updated review of the literature.
Case Report
A 44-year-old man developed gradually worsening bilateral ear pain, headaches, and seizures. He was briefly hospitalized and discharged with levetiracetam and quetiapine. However, his mental status continued to deteriorate and he was subsequently hospitalized 3 months later with confusion, hallucinations, and seizures.
On physical examination the patient was disoriented and unable to form cohesive sentences. He had bilateral tenderness, erythema, and edema of the auricles, which notably spared the lobules (Figure 1). The conjunctivae were injected bilaterally, and joint involvement included bilateral knee tenderness and swelling. Neurologic examination revealed questionable meningeal signs but no motor or sensory deficits. An extensive laboratory workup for the etiology of his altered mental status was unremarkable, except for a mildly elevated white blood cell count in the cerebrospinal fluid with predominantly lymphocytes. No infectious etiologies were identified on laboratory testing, and rheumatologic markers were negative including antinuclear antibody, rheumatoid factor, and anti–Sjögren syndrome antigen A/Sjögren syndrome antigen B. Magnetic resonance imaging revealed nonspecific findings of bilateral T2 hyperdensities in the subcortical white matter; however, cerebral angiography revealed no evidence of vasculitis. A biopsy of the right antihelix revealed prominent perichondritis and a neutrophilic inflammatory infiltrate with several lymphocytes and histiocytes (Figure 2). There was degeneration of the cartilaginous tissue with evidence of pyknotic nuclei, eosinophilia, and vacuolization of the chondrocytes. He was diagnosed with RP on the basis of clinical and histologic inflammation of the auricular cartilage, polyarthritis, and ocular inflammation.
The patient was treated with high-dose immunosuppression with methylprednisolone (1000 mg intravenous once daily for 5 days) and cyclophosphamide (one dose at 500 mg/m2), which resulted in remarkable improvement in his mental status, auricular inflammation, and knee pain. After 31 days of hospitalization the patient was discharged with a course of oral prednisone (starting at 60 mg/d, then tapered over the following 2 months) and monthly cyclophosphamide infusions (5 months total; starting at 500 mg/m2, then uptitrated to goal of 1000 mg/m2). Maintenance suppression was achieved with azathioprine (starting at 50 mg daily, then uptitrated to 100 mg daily), which was continued without any evidence of relapsed disease through his last outpatient visit 1 year after the diagnosis.
Comment
Auricular inflammation is a hallmark of RP and is present in 83% to 95% of patients.1,3 The affected ears can appear erythematous to violaceous with tender edema of the auricle that spares the lobules where no cartilage is present. The inflammation can extend into the ear canal and cause hearing loss, tinnitus, and vertigo. Histologically, RP can present with a nonspecific leukocytoclastic vasculitis and inflammatory destruction of the cartilage. Therefore, diagnosis of RP is reliant mainly on clinical characteristics rather than pathologic findings. In 1976, McAdam et al5 established diagnostic criteria for RP based on the presence of common clinical manifestations (eg, auricular chondritis, seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation). Michet et al6 later proposed major and minor criteria to classify and diagnose RP based on clinical manifestations. Diagnosis of our patient was confirmed by the presence of auricular chondritis, polyarthritis, and ocular inflammation. Diagnosing RP can be difficult because it has many systemic manifestations that can evoke a broad differential diagnosis. The time to diagnosis in our patient was 3 months, but the mean delay in diagnosis for patients with RP and ME is 2.9 years.4
The etiology of RP remains unclear, but current evidence supports an immune-mediated process directed toward proteins found in cartilage. Animal studies have suggested that RP may be driven by antibodies to matrillin 1 and type II collagen. There also may be a familial association with HLA-DR4 and genetic predisposition to autoimmune diseases in individuals affected by RP.1,3 The pathogenesis of CNS involvement in RP is thought to be due to a localized small vessel vasculitis.7,8 In our patient, however, cerebral angiography was negative for vasculitis, and thus our case may represent another mechanism for CNS involvement. There have been cases of encephalitis in RP caused by pathways other than CNS vasculitis. Kashihara et al9 reported a case of RP with encephalitis associated with antiglutamate receptor antibodies found in the cerebrospinal fluid and blood.
Treatment of RP has been based on pathophysiological considerations rather than empiric data due to its rarity. Relapsing polychondritis has been responsive to steroid treatment in reported cases as well as in our patient; however, in cases in which RP did not respond to steroids, infliximab may be effective for RP with ME.10 Further research regarding the treatment outcomes of RP with ME may be warranted.
Although rare, additional cases of RP with ME have been reported (Table). Wang et al4 described a series of 28 patients with RP and ME from 1960 to 2010. A PubMed search of articles indexed for MEDLINE that were published in the English-language literature from 2010 to 2016 was performed using the search terms relapsing polychondritis and nervous system. Including our patient, RP with ME was reported in 17 additional cases since Wang et al4 published their findings. These cases involved adults ranging in age from 44 to 73 years who were mainly men (14/17 [82%]). All of the patients presented with bilateral auricular chondritis, except for a case of unilateral ear involvement reported by Storey et al.11 Common neurologic manifestations included fever, headache, and altered mental status. Motor symptoms ranged from dysarthria and agraphia12 to hemiparesis.13 The mechanism of CNS involvement in RP was not identified in most cases; however, Mattiassich et al14 documented cerebral vasculitis in their patient, and Niwa et al16 found diffuse cerebral vasculitis on autopsy. Eleven of 17 (65%) cases responded to steroid treatment. Of the 6 cases in which RP did not respond to steroids, 2 patients died despite high-dose steroid treatment,11,20 2 responded to infliximab,10,15 1 responded to tocilizumab,21 and 1 was lost to follow-up after initial treatment failure.20
Conclusion
Although rare, RP should not be overlooked in the inpatient setting due to its potential for life-threatening systemic effects. Early diagnosis of this condition may be of benefit to this select population of patients, and further research regarding the prognosis, mechanisms, and treatment of RP may be necessary in the future.
Relapsing polychondritis (RP) is an autoimmune disease affecting cartilaginous structures such as the ears, respiratory passages, joints, and cardiovascular system.1,2 In rare cases, the systemic effects of this autoimmune process can cause central nervous system (CNS) involvement such as meningoencephalitis (ME).3 In 2011, Wang et al4 described 4 cases of RP with ME and reviewed 24 cases from the literature. We present a case of a man with RP-associated ME that was responsive to steroid treatment. We also provide an updated review of the literature.
Case Report
A 44-year-old man developed gradually worsening bilateral ear pain, headaches, and seizures. He was briefly hospitalized and discharged with levetiracetam and quetiapine. However, his mental status continued to deteriorate and he was subsequently hospitalized 3 months later with confusion, hallucinations, and seizures.
On physical examination the patient was disoriented and unable to form cohesive sentences. He had bilateral tenderness, erythema, and edema of the auricles, which notably spared the lobules (Figure 1). The conjunctivae were injected bilaterally, and joint involvement included bilateral knee tenderness and swelling. Neurologic examination revealed questionable meningeal signs but no motor or sensory deficits. An extensive laboratory workup for the etiology of his altered mental status was unremarkable, except for a mildly elevated white blood cell count in the cerebrospinal fluid with predominantly lymphocytes. No infectious etiologies were identified on laboratory testing, and rheumatologic markers were negative including antinuclear antibody, rheumatoid factor, and anti–Sjögren syndrome antigen A/Sjögren syndrome antigen B. Magnetic resonance imaging revealed nonspecific findings of bilateral T2 hyperdensities in the subcortical white matter; however, cerebral angiography revealed no evidence of vasculitis. A biopsy of the right antihelix revealed prominent perichondritis and a neutrophilic inflammatory infiltrate with several lymphocytes and histiocytes (Figure 2). There was degeneration of the cartilaginous tissue with evidence of pyknotic nuclei, eosinophilia, and vacuolization of the chondrocytes. He was diagnosed with RP on the basis of clinical and histologic inflammation of the auricular cartilage, polyarthritis, and ocular inflammation.
The patient was treated with high-dose immunosuppression with methylprednisolone (1000 mg intravenous once daily for 5 days) and cyclophosphamide (one dose at 500 mg/m2), which resulted in remarkable improvement in his mental status, auricular inflammation, and knee pain. After 31 days of hospitalization the patient was discharged with a course of oral prednisone (starting at 60 mg/d, then tapered over the following 2 months) and monthly cyclophosphamide infusions (5 months total; starting at 500 mg/m2, then uptitrated to goal of 1000 mg/m2). Maintenance suppression was achieved with azathioprine (starting at 50 mg daily, then uptitrated to 100 mg daily), which was continued without any evidence of relapsed disease through his last outpatient visit 1 year after the diagnosis.
Comment
Auricular inflammation is a hallmark of RP and is present in 83% to 95% of patients.1,3 The affected ears can appear erythematous to violaceous with tender edema of the auricle that spares the lobules where no cartilage is present. The inflammation can extend into the ear canal and cause hearing loss, tinnitus, and vertigo. Histologically, RP can present with a nonspecific leukocytoclastic vasculitis and inflammatory destruction of the cartilage. Therefore, diagnosis of RP is reliant mainly on clinical characteristics rather than pathologic findings. In 1976, McAdam et al5 established diagnostic criteria for RP based on the presence of common clinical manifestations (eg, auricular chondritis, seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation). Michet et al6 later proposed major and minor criteria to classify and diagnose RP based on clinical manifestations. Diagnosis of our patient was confirmed by the presence of auricular chondritis, polyarthritis, and ocular inflammation. Diagnosing RP can be difficult because it has many systemic manifestations that can evoke a broad differential diagnosis. The time to diagnosis in our patient was 3 months, but the mean delay in diagnosis for patients with RP and ME is 2.9 years.4
The etiology of RP remains unclear, but current evidence supports an immune-mediated process directed toward proteins found in cartilage. Animal studies have suggested that RP may be driven by antibodies to matrillin 1 and type II collagen. There also may be a familial association with HLA-DR4 and genetic predisposition to autoimmune diseases in individuals affected by RP.1,3 The pathogenesis of CNS involvement in RP is thought to be due to a localized small vessel vasculitis.7,8 In our patient, however, cerebral angiography was negative for vasculitis, and thus our case may represent another mechanism for CNS involvement. There have been cases of encephalitis in RP caused by pathways other than CNS vasculitis. Kashihara et al9 reported a case of RP with encephalitis associated with antiglutamate receptor antibodies found in the cerebrospinal fluid and blood.
Treatment of RP has been based on pathophysiological considerations rather than empiric data due to its rarity. Relapsing polychondritis has been responsive to steroid treatment in reported cases as well as in our patient; however, in cases in which RP did not respond to steroids, infliximab may be effective for RP with ME.10 Further research regarding the treatment outcomes of RP with ME may be warranted.
Although rare, additional cases of RP with ME have been reported (Table). Wang et al4 described a series of 28 patients with RP and ME from 1960 to 2010. A PubMed search of articles indexed for MEDLINE that were published in the English-language literature from 2010 to 2016 was performed using the search terms relapsing polychondritis and nervous system. Including our patient, RP with ME was reported in 17 additional cases since Wang et al4 published their findings. These cases involved adults ranging in age from 44 to 73 years who were mainly men (14/17 [82%]). All of the patients presented with bilateral auricular chondritis, except for a case of unilateral ear involvement reported by Storey et al.11 Common neurologic manifestations included fever, headache, and altered mental status. Motor symptoms ranged from dysarthria and agraphia12 to hemiparesis.13 The mechanism of CNS involvement in RP was not identified in most cases; however, Mattiassich et al14 documented cerebral vasculitis in their patient, and Niwa et al16 found diffuse cerebral vasculitis on autopsy. Eleven of 17 (65%) cases responded to steroid treatment. Of the 6 cases in which RP did not respond to steroids, 2 patients died despite high-dose steroid treatment,11,20 2 responded to infliximab,10,15 1 responded to tocilizumab,21 and 1 was lost to follow-up after initial treatment failure.20
Conclusion
Although rare, RP should not be overlooked in the inpatient setting due to its potential for life-threatening systemic effects. Early diagnosis of this condition may be of benefit to this select population of patients, and further research regarding the prognosis, mechanisms, and treatment of RP may be necessary in the future.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis: a 2013 update. Autoimmun Rev. 2014;13:90-95.
- Ostrowski RA, Takagishi T, Robinson J. Rheumatoid arthritis, spondyloarthropathies, and relapsing polychondritis. Handb Clin Neurol. 2014;119:449-461.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Wang ZJ, Pu CQ, Wang ZJ, et al. Meningoencephalitis or meningitis in relapsing polychondritis: four case reports and a literature review. J Clin Neurosci. 2011;18:1608-1615.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet C, McKenna C, Luthra H, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Sampaio L, Silva L, Mariz E, et al. CNS involvement in relapsing polychondritis. Joint Bone Spine. 2010;77:619-620.
- Prinz S, Dafotakis M, Schneider RK, et al. The red puffy ear sign—a clinical sign to diagnose a rare cause of meningoencephalitis. Fortschr Neurol Psychiatr. 2012;80:463-467.
- Kashihara K, Kawada S, Takahashi Y. Autoantibodies to glutamate receptor GluR2 in a patient with limic encephalitis associated with relapsing polychondritis. J Neurol Sci. 2009;287:275-277.
- Garcia-Egido A, Gutierrez C, de la Fuente C, et al. Relapsing polychondritis-associated meningitis and encephalitis: response to infliximab. Rheumatology (Oxford). 2011;50:1721-1723.
- Storey K, Matej R, Rusina R. Unusual association of seronegative, nonparaneoplastic limbic encephalitis and relapsing polychondritis in a patient with history of thymectomy for myasthemia: a case study. J Neurol. 2010;258:159-161.
- Choi HJ, Lee HJ. Relapsing polychondritis with encephalitis. J Clin Rheum. 2011;6:329-331.
- Fujiwara S, Zenke K, Iwata S, et al. Relapsing polychondritis presenting as encephalitis. No Shinkei Geka. 2012;40:247-253.
- Mattiassich G, Egger M, Semlitsch G, et al. Occurrence of relapsing polychondritis with a rising cANCA titre in a cANCA-positive systemic and cerebral vasculitis patient [published online February 5, 2013]. BMJ Case Rep. doi:10.1136/bcr-2013-008717.
- Kondo T, Fukuta M, Takemoto A, et al. Limbic encephalitis associated with relapsing polychondritis responded to infliximab and maintained its condition without recurrence after discontinuation: a case report and review of the literature. Nagoya J Med Sci. 2014;76:361-368.
- Niwa A, Okamoto Y, Kondo T, et al. Perivasculitic pancencephalitis with relapsing polychondritis: an autopsy case report and review of previous cases. Intern Med. 2014;53:1191-1195.
- Coban EK, Xanmemmedoy E, Colak M, et al. A rare complication of a rare disease; stroke due to relapsing polychondritis. Ideggyogy Sz. 2015;68:429-432.
- Ducci R, Germiniani F, Czecko L, et al. Relapsing polychondritis and lymphocytic meningitis with varied neurological symptoms [published online February 5, 2016]. Rev Bras Reumatol. doi:10.1016/j.rbr.2015.09.005.
- Baba T, Kanno S, Shijo T, et al. Callosal disconnection syndrome associated with relapsing polychondritis. Intern Med. 2016;55:1191-1193.
- Jeon C. Relapsing polychondritis with central nervous system involvement: experience of three different cases in a single center. J Korean Med. 2016;31:1846-1850.
- Liu L, Liu S, Guan W, et al. Efficacy of tocilizumab for psychiatric symptoms associated with relapsing polychondritis: the first case report and review of the literature. Rheumatol Int. 2016;36:1185-1189.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis: a 2013 update. Autoimmun Rev. 2014;13:90-95.
- Ostrowski RA, Takagishi T, Robinson J. Rheumatoid arthritis, spondyloarthropathies, and relapsing polychondritis. Handb Clin Neurol. 2014;119:449-461.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Wang ZJ, Pu CQ, Wang ZJ, et al. Meningoencephalitis or meningitis in relapsing polychondritis: four case reports and a literature review. J Clin Neurosci. 2011;18:1608-1615.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet C, McKenna C, Luthra H, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Sampaio L, Silva L, Mariz E, et al. CNS involvement in relapsing polychondritis. Joint Bone Spine. 2010;77:619-620.
- Prinz S, Dafotakis M, Schneider RK, et al. The red puffy ear sign—a clinical sign to diagnose a rare cause of meningoencephalitis. Fortschr Neurol Psychiatr. 2012;80:463-467.
- Kashihara K, Kawada S, Takahashi Y. Autoantibodies to glutamate receptor GluR2 in a patient with limic encephalitis associated with relapsing polychondritis. J Neurol Sci. 2009;287:275-277.
- Garcia-Egido A, Gutierrez C, de la Fuente C, et al. Relapsing polychondritis-associated meningitis and encephalitis: response to infliximab. Rheumatology (Oxford). 2011;50:1721-1723.
- Storey K, Matej R, Rusina R. Unusual association of seronegative, nonparaneoplastic limbic encephalitis and relapsing polychondritis in a patient with history of thymectomy for myasthemia: a case study. J Neurol. 2010;258:159-161.
- Choi HJ, Lee HJ. Relapsing polychondritis with encephalitis. J Clin Rheum. 2011;6:329-331.
- Fujiwara S, Zenke K, Iwata S, et al. Relapsing polychondritis presenting as encephalitis. No Shinkei Geka. 2012;40:247-253.
- Mattiassich G, Egger M, Semlitsch G, et al. Occurrence of relapsing polychondritis with a rising cANCA titre in a cANCA-positive systemic and cerebral vasculitis patient [published online February 5, 2013]. BMJ Case Rep. doi:10.1136/bcr-2013-008717.
- Kondo T, Fukuta M, Takemoto A, et al. Limbic encephalitis associated with relapsing polychondritis responded to infliximab and maintained its condition without recurrence after discontinuation: a case report and review of the literature. Nagoya J Med Sci. 2014;76:361-368.
- Niwa A, Okamoto Y, Kondo T, et al. Perivasculitic pancencephalitis with relapsing polychondritis: an autopsy case report and review of previous cases. Intern Med. 2014;53:1191-1195.
- Coban EK, Xanmemmedoy E, Colak M, et al. A rare complication of a rare disease; stroke due to relapsing polychondritis. Ideggyogy Sz. 2015;68:429-432.
- Ducci R, Germiniani F, Czecko L, et al. Relapsing polychondritis and lymphocytic meningitis with varied neurological symptoms [published online February 5, 2016]. Rev Bras Reumatol. doi:10.1016/j.rbr.2015.09.005.
- Baba T, Kanno S, Shijo T, et al. Callosal disconnection syndrome associated with relapsing polychondritis. Intern Med. 2016;55:1191-1193.
- Jeon C. Relapsing polychondritis with central nervous system involvement: experience of three different cases in a single center. J Korean Med. 2016;31:1846-1850.
- Liu L, Liu S, Guan W, et al. Efficacy of tocilizumab for psychiatric symptoms associated with relapsing polychondritis: the first case report and review of the literature. Rheumatol Int. 2016;36:1185-1189.
Practice Points
- Meningoencephalitis (ME) is a potentially rare complication of relapsing polychondritis (RP).
- Treatment of ME due to RP can include high-dose steroids and biologics.
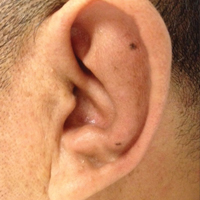
Clinicians Should Retain the Ability to Choose a Pathologist
As employers search for ways to reduce the cost of providing health care to their employees, there is a growing trend toward narrowed provider networks and exclusive laboratory contracts. In the case of clinical pathology, some of these choices make sense from the employer’s perspective. A complete blood cell count or comprehensive metabolic panel is done on a machine and the result is much the same regardless of the laboratory. So why not have all laboratory tests performed by the lowest bidder?
Laboratories vary in quality and anatomic pathology services are different from blood tests. Each slide must be interpreted by a physician and skill in the interpretation of skin specimens varies widely. Dermatopathology was one of the first subspecialties to be recognized within pathology, as it requires a high level of expertise. Clinicopathological correlation often is key to the accurate interpretation of a specimen. The stakes are high, and a delay in diagnosis of melanoma remains one of the most serious errors in medicine and one of the most common causes for litigation in dermatology.
The accurate interpretation of skin biopsy specimens becomes especially difficult when inadequate or misleading clinical information accompanies the specimen. A study of 589 biopsies submitted by primary care physicians and reported by general pathologists demonstrated a 6.5% error rate. False-negative errors were the most common, but false-positives also were observed.1 A study of pigmented lesions referred to the University of California, San Francisco, demonstrated a discordance rate of 14.3%.2 The degree of discordance would be expected to vary based on the range of diagnoses included in each study.
Board-certified dermatopathologists have varying areas of expertise and there is notable subjectivity in the interpretation of biopsy specimens. In the case of problematic pigmented lesions such as atypical Spitz nevi, there can be low interobserver agreement even among the experts in categorizing lesions as malignant versus nonmalignant (κ=0.30).3 The low concordance among expert dermatopathologists demonstrates that light microscopic features alone often are inadequate for diagnosis. Advanced studies, including immunohistochemical stains, can help to clarify the diagnosis. In the case of atypical Spitz tumors, the contribution of special stains to the final diagnosis is statistically similar to that of hematoxylin and eosin sections and age, suggesting that nothing alone is sufficiently reliable to establish a definitive diagnosis in every case.4 Although helpful, these studies are costly, and savings obtained by sending cases to the lowest bidder can evaporate quickly. Costs are higher when factoring in molecular studies, which can run upwards of $3000 per slide; the cost of litigation related to incorrect diagnoses; or the human costs of an incorrect diagnosis.
As a group, dermatopathologists are highly skilled in the interpretation of skin specimens, but challenging lesions are common in the routine practice of dermatopathology. A study of 1249 pigmented melanocytic lesions demonstrated substantial agreement among expert dermatopathologists for less problematic lesions, though agreement was greater for patients 40 years and older (κ=0.67) than for younger patients (κ=0.49). Agreement was lower for patients with atypical mole syndrome (κ=0.31).5 These discrepancies occur despite the fact that there is good interobserver reproducibility for grading of individual histological features such as asymmetry, circumscription, irregular confluent nests, single melanocytes predominating, absence of maturation, suprabasal melanocytes, symmetrical melanin, deep melanin, cytological atypia, mitoses, dermal lymphocytic infiltrate, and necrosis.6 These results indicate that accurate diagnoses cannot be reliably established simply by grading a list of histological features. Accurate diagnosis requires complex pattern recognition and integration of findings. Conflicting criteria often are present and an accurate interpretation requires considerable judgment as to which features are significant and which are not.
Separation of sebaceous adenoma, sebaceoma, and well-differentiated sebaceous carcinoma is another challenging area, and interobserver consensus can be as low as 11%,7 which suggests notable subjectivity in the criteria for diagnosis of nonmelanocytic tumors and emphasizes the importance of communication between the dermatopathologist and clinician when determining how to manage an ambiguous lesion. The interpretation of inflammatory skin diseases, alopecia, and lymphoid proliferations also can be problematic, and expert consultation often is required.
All dermatologists receive substantial training in dermatopathology, which puts them in an excellent position to interpret ambiguous findings in the context of the clinical presentation. Sometimes the dermatologist who has seen the clinical presentation can be in the best position to make the diagnosis. All clinicians must be wary of bias and an objective set of eyes often can be helpful. Communication is crucial to ensure appropriate care for each patient, and policies that restrict the choice of pathologist can be damaging.
- Trotter MJ, Bruecks AK. Interpretation of skin biopsies by general pathologists: diagnostic discrepancy rate measured by blinded review. Arch Pathol Lab Med. 2003;127:1489-1492.
- Shoo BA, Sagebiel RW, Kashani-Sabet M. Discordance in the histopathologic diagnosis of melanoma at a melanoma referral center [published online March 19, 2010]. J Am Acad Dermatol. 2010;62:751-756.
- Gerami P, Busam K, Cochran A, et al. Histomorphologic assessment and interobserver diagnostic reproducibility of atypical spitzoid melanocytic neoplasms with long-term follow-up. Am J Surg Pathol. 2014;38:934-940.
- Puri PK, Ferringer TC, Tyler WB, et al. Statistical analysis of the concordance of immunohistochemical stains with the final diagnosis in spitzoid neoplasms. Am J Dermatopathol. 2011;33:72-77.
- Braun RP, Gutkowicz-Krusin D, Rabinovitz H, et al. Agreement of dermatopathologists in the evaluation of clinically difficult melanocytic lesions: how golden is the ‘gold standard’? Dermatology. 2012;224:51-58.
- Urso C, Rongioletti F, Innocenzi D, et al. Interobserver reproducibility of histological features in cutaneous malignant melanoma. J Clin Pathol. 2005;58:1194-1198.
- Harvey NT, Budgeon CA, Leecy T, et al. Interobserver variability in the diagnosis of circumscribed sebaceous neoplasms of the skin. Pathology. 2013;45:581-586.
As employers search for ways to reduce the cost of providing health care to their employees, there is a growing trend toward narrowed provider networks and exclusive laboratory contracts. In the case of clinical pathology, some of these choices make sense from the employer’s perspective. A complete blood cell count or comprehensive metabolic panel is done on a machine and the result is much the same regardless of the laboratory. So why not have all laboratory tests performed by the lowest bidder?
Laboratories vary in quality and anatomic pathology services are different from blood tests. Each slide must be interpreted by a physician and skill in the interpretation of skin specimens varies widely. Dermatopathology was one of the first subspecialties to be recognized within pathology, as it requires a high level of expertise. Clinicopathological correlation often is key to the accurate interpretation of a specimen. The stakes are high, and a delay in diagnosis of melanoma remains one of the most serious errors in medicine and one of the most common causes for litigation in dermatology.
The accurate interpretation of skin biopsy specimens becomes especially difficult when inadequate or misleading clinical information accompanies the specimen. A study of 589 biopsies submitted by primary care physicians and reported by general pathologists demonstrated a 6.5% error rate. False-negative errors were the most common, but false-positives also were observed.1 A study of pigmented lesions referred to the University of California, San Francisco, demonstrated a discordance rate of 14.3%.2 The degree of discordance would be expected to vary based on the range of diagnoses included in each study.
Board-certified dermatopathologists have varying areas of expertise and there is notable subjectivity in the interpretation of biopsy specimens. In the case of problematic pigmented lesions such as atypical Spitz nevi, there can be low interobserver agreement even among the experts in categorizing lesions as malignant versus nonmalignant (κ=0.30).3 The low concordance among expert dermatopathologists demonstrates that light microscopic features alone often are inadequate for diagnosis. Advanced studies, including immunohistochemical stains, can help to clarify the diagnosis. In the case of atypical Spitz tumors, the contribution of special stains to the final diagnosis is statistically similar to that of hematoxylin and eosin sections and age, suggesting that nothing alone is sufficiently reliable to establish a definitive diagnosis in every case.4 Although helpful, these studies are costly, and savings obtained by sending cases to the lowest bidder can evaporate quickly. Costs are higher when factoring in molecular studies, which can run upwards of $3000 per slide; the cost of litigation related to incorrect diagnoses; or the human costs of an incorrect diagnosis.
As a group, dermatopathologists are highly skilled in the interpretation of skin specimens, but challenging lesions are common in the routine practice of dermatopathology. A study of 1249 pigmented melanocytic lesions demonstrated substantial agreement among expert dermatopathologists for less problematic lesions, though agreement was greater for patients 40 years and older (κ=0.67) than for younger patients (κ=0.49). Agreement was lower for patients with atypical mole syndrome (κ=0.31).5 These discrepancies occur despite the fact that there is good interobserver reproducibility for grading of individual histological features such as asymmetry, circumscription, irregular confluent nests, single melanocytes predominating, absence of maturation, suprabasal melanocytes, symmetrical melanin, deep melanin, cytological atypia, mitoses, dermal lymphocytic infiltrate, and necrosis.6 These results indicate that accurate diagnoses cannot be reliably established simply by grading a list of histological features. Accurate diagnosis requires complex pattern recognition and integration of findings. Conflicting criteria often are present and an accurate interpretation requires considerable judgment as to which features are significant and which are not.
Separation of sebaceous adenoma, sebaceoma, and well-differentiated sebaceous carcinoma is another challenging area, and interobserver consensus can be as low as 11%,7 which suggests notable subjectivity in the criteria for diagnosis of nonmelanocytic tumors and emphasizes the importance of communication between the dermatopathologist and clinician when determining how to manage an ambiguous lesion. The interpretation of inflammatory skin diseases, alopecia, and lymphoid proliferations also can be problematic, and expert consultation often is required.
All dermatologists receive substantial training in dermatopathology, which puts them in an excellent position to interpret ambiguous findings in the context of the clinical presentation. Sometimes the dermatologist who has seen the clinical presentation can be in the best position to make the diagnosis. All clinicians must be wary of bias and an objective set of eyes often can be helpful. Communication is crucial to ensure appropriate care for each patient, and policies that restrict the choice of pathologist can be damaging.
As employers search for ways to reduce the cost of providing health care to their employees, there is a growing trend toward narrowed provider networks and exclusive laboratory contracts. In the case of clinical pathology, some of these choices make sense from the employer’s perspective. A complete blood cell count or comprehensive metabolic panel is done on a machine and the result is much the same regardless of the laboratory. So why not have all laboratory tests performed by the lowest bidder?
Laboratories vary in quality and anatomic pathology services are different from blood tests. Each slide must be interpreted by a physician and skill in the interpretation of skin specimens varies widely. Dermatopathology was one of the first subspecialties to be recognized within pathology, as it requires a high level of expertise. Clinicopathological correlation often is key to the accurate interpretation of a specimen. The stakes are high, and a delay in diagnosis of melanoma remains one of the most serious errors in medicine and one of the most common causes for litigation in dermatology.
The accurate interpretation of skin biopsy specimens becomes especially difficult when inadequate or misleading clinical information accompanies the specimen. A study of 589 biopsies submitted by primary care physicians and reported by general pathologists demonstrated a 6.5% error rate. False-negative errors were the most common, but false-positives also were observed.1 A study of pigmented lesions referred to the University of California, San Francisco, demonstrated a discordance rate of 14.3%.2 The degree of discordance would be expected to vary based on the range of diagnoses included in each study.
Board-certified dermatopathologists have varying areas of expertise and there is notable subjectivity in the interpretation of biopsy specimens. In the case of problematic pigmented lesions such as atypical Spitz nevi, there can be low interobserver agreement even among the experts in categorizing lesions as malignant versus nonmalignant (κ=0.30).3 The low concordance among expert dermatopathologists demonstrates that light microscopic features alone often are inadequate for diagnosis. Advanced studies, including immunohistochemical stains, can help to clarify the diagnosis. In the case of atypical Spitz tumors, the contribution of special stains to the final diagnosis is statistically similar to that of hematoxylin and eosin sections and age, suggesting that nothing alone is sufficiently reliable to establish a definitive diagnosis in every case.4 Although helpful, these studies are costly, and savings obtained by sending cases to the lowest bidder can evaporate quickly. Costs are higher when factoring in molecular studies, which can run upwards of $3000 per slide; the cost of litigation related to incorrect diagnoses; or the human costs of an incorrect diagnosis.
As a group, dermatopathologists are highly skilled in the interpretation of skin specimens, but challenging lesions are common in the routine practice of dermatopathology. A study of 1249 pigmented melanocytic lesions demonstrated substantial agreement among expert dermatopathologists for less problematic lesions, though agreement was greater for patients 40 years and older (κ=0.67) than for younger patients (κ=0.49). Agreement was lower for patients with atypical mole syndrome (κ=0.31).5 These discrepancies occur despite the fact that there is good interobserver reproducibility for grading of individual histological features such as asymmetry, circumscription, irregular confluent nests, single melanocytes predominating, absence of maturation, suprabasal melanocytes, symmetrical melanin, deep melanin, cytological atypia, mitoses, dermal lymphocytic infiltrate, and necrosis.6 These results indicate that accurate diagnoses cannot be reliably established simply by grading a list of histological features. Accurate diagnosis requires complex pattern recognition and integration of findings. Conflicting criteria often are present and an accurate interpretation requires considerable judgment as to which features are significant and which are not.
Separation of sebaceous adenoma, sebaceoma, and well-differentiated sebaceous carcinoma is another challenging area, and interobserver consensus can be as low as 11%,7 which suggests notable subjectivity in the criteria for diagnosis of nonmelanocytic tumors and emphasizes the importance of communication between the dermatopathologist and clinician when determining how to manage an ambiguous lesion. The interpretation of inflammatory skin diseases, alopecia, and lymphoid proliferations also can be problematic, and expert consultation often is required.
All dermatologists receive substantial training in dermatopathology, which puts them in an excellent position to interpret ambiguous findings in the context of the clinical presentation. Sometimes the dermatologist who has seen the clinical presentation can be in the best position to make the diagnosis. All clinicians must be wary of bias and an objective set of eyes often can be helpful. Communication is crucial to ensure appropriate care for each patient, and policies that restrict the choice of pathologist can be damaging.
- Trotter MJ, Bruecks AK. Interpretation of skin biopsies by general pathologists: diagnostic discrepancy rate measured by blinded review. Arch Pathol Lab Med. 2003;127:1489-1492.
- Shoo BA, Sagebiel RW, Kashani-Sabet M. Discordance in the histopathologic diagnosis of melanoma at a melanoma referral center [published online March 19, 2010]. J Am Acad Dermatol. 2010;62:751-756.
- Gerami P, Busam K, Cochran A, et al. Histomorphologic assessment and interobserver diagnostic reproducibility of atypical spitzoid melanocytic neoplasms with long-term follow-up. Am J Surg Pathol. 2014;38:934-940.
- Puri PK, Ferringer TC, Tyler WB, et al. Statistical analysis of the concordance of immunohistochemical stains with the final diagnosis in spitzoid neoplasms. Am J Dermatopathol. 2011;33:72-77.
- Braun RP, Gutkowicz-Krusin D, Rabinovitz H, et al. Agreement of dermatopathologists in the evaluation of clinically difficult melanocytic lesions: how golden is the ‘gold standard’? Dermatology. 2012;224:51-58.
- Urso C, Rongioletti F, Innocenzi D, et al. Interobserver reproducibility of histological features in cutaneous malignant melanoma. J Clin Pathol. 2005;58:1194-1198.
- Harvey NT, Budgeon CA, Leecy T, et al. Interobserver variability in the diagnosis of circumscribed sebaceous neoplasms of the skin. Pathology. 2013;45:581-586.
- Trotter MJ, Bruecks AK. Interpretation of skin biopsies by general pathologists: diagnostic discrepancy rate measured by blinded review. Arch Pathol Lab Med. 2003;127:1489-1492.
- Shoo BA, Sagebiel RW, Kashani-Sabet M. Discordance in the histopathologic diagnosis of melanoma at a melanoma referral center [published online March 19, 2010]. J Am Acad Dermatol. 2010;62:751-756.
- Gerami P, Busam K, Cochran A, et al. Histomorphologic assessment and interobserver diagnostic reproducibility of atypical spitzoid melanocytic neoplasms with long-term follow-up. Am J Surg Pathol. 2014;38:934-940.
- Puri PK, Ferringer TC, Tyler WB, et al. Statistical analysis of the concordance of immunohistochemical stains with the final diagnosis in spitzoid neoplasms. Am J Dermatopathol. 2011;33:72-77.
- Braun RP, Gutkowicz-Krusin D, Rabinovitz H, et al. Agreement of dermatopathologists in the evaluation of clinically difficult melanocytic lesions: how golden is the ‘gold standard’? Dermatology. 2012;224:51-58.
- Urso C, Rongioletti F, Innocenzi D, et al. Interobserver reproducibility of histological features in cutaneous malignant melanoma. J Clin Pathol. 2005;58:1194-1198.
- Harvey NT, Budgeon CA, Leecy T, et al. Interobserver variability in the diagnosis of circumscribed sebaceous neoplasms of the skin. Pathology. 2013;45:581-586.

French psychiatrist condemned for society’s deficiency
On Dec. 14, 2016, a French psychiatrist was sentenced to an 18-month suspended prison sentence. Lekhraj Gujadhur, MD, was the supervisor of unit 101 at the Psychiatric Hospital Center of Saint-Egrève in France. In November 2008, he had approved the nonsupervised release of a schizophrenia patient, Jean-Pierre Guillaud, to outside of his unit but within the hospital facility. Mr. Guillaud, while outside supervision, escaped. He subsequently purchased a large knife and murdered a 26-year-old student, Luc Meunier, Le Monde reported.1
This is reminiscent of a similar case in 2012 in Marseille, where a psychiatrist received a suspended prison sentence after his patient committed murder. That prior case was later dismissed in appellate court. In my opinion, both trials point to a failure in psychiatry’s responsibility to educate the public in our limitations and roles. They also highlight the necessary discourse that society should have on the role of mental illness when it comes to crime.

Although I appreciate society’s concern about such crimes, I think that displacement of our anger onto Dr. Gujadhur is misguided, and instead, allows us to forget to look at our own poor judgment. Dr. Gujadhur, other psychiatrists, and mental hospitals do not have the responsibility to enact sentences for crimes; the legal system does. Law enforcement and prosecutors had numerous opportunities to charge and commit Mr. Guillaud over the years but chose not to do so, instead permitting him to stay within society under the care of the mental health system.
Asking Dr. Gujadhur to primarily focus on becoming an agent of the law, instead of treating his patient, is unfair. Schizophrenia, and in particular paranoia, are greatly worsened by social isolation. Confining Mr. Guillaud would be countertherapeutic and possibly lead to his suicide. Would Dr. Gujadhur have been responsible for the suicide? Mental health providers have to understand and support the psychological functioning of their patients. Creating a dual agency blurs and effaces the doctor-patient relationship, already so fragile in the treatment of paranoid patients.
The publicity of such cases, and of Mr. Guillaud’s mental illness, seems to go against current mental health research. Recent work has suggested that mental illness is not a significant risk factor for violence but rather a risk factor for being the victim of violence. Certainly, some patients with mental illness commit acts of violence, but studies suggest that this is mostly independent of their mental illness (Law Hum Behav. 2014 Oct;38[5];439-49).2 Our overemphasis on the mental status of criminals belittles their crimes and suggests that psychiatrists are responsible for the failings of our legal system.
As a supervising psychiatrist at one of the largest jail systems in America, I am familiar with the challenges in such cases. All of my patients are facing legal charges, and many suffer from severe mental illness like schizophrenia. As their treating psychiatrist, I am not asked to also sentence them for the charges they are facing. Simply working for the sheriff makes my ability to gain the trust of my patients much more difficult. Conspiring with the city or district attorney in an attempt to protect society would obliterate any chance at rapport building.
Working in corrections, I am deeply familiar with the current debate on the solitary confinement of our mentally ill offenders. Ironically, in that context, society has blamed the legal system for socially isolating our mentally ill offenders, especially ones with severe mental illness.3 In our jail, we meet regularly and discuss in an interdisciplinary fashion the role and consequences of social isolation. During our weekly sessions, a case involving stabbing someone 2 years prior would not have justified the punishment of social isolation and constant monitoring.
As a field, psychiatry must educate society on its ability to create a therapeutic environment and its ability to provide risk assessment of violence. We must also remind others of the impossibility of doing both simultaneously. Decisions on removing patients’ right to freedom can be informed by the mental health perspective but should be left to the courts. Society’s need to find a target after such tragedies is understandable, but blaming the treating psychiatrists will not help past or future victims.
References
1. Le psychiatre d’un schizophrène meurtrier condamné pour homicide involontaire, Le Monde, Dec. 14, 2016.
2. How often and how consistently do symptoms directly precede criminal behavior among offenders with mental illness? (Law Hum Behav. 2014 Oct;38[5]:439-49).
3. How to fix solitary confinement in American prisons, Los Angeles Times, Oct. 17, 2016.
Dr. Badre is a supervising psychiatric contractor at the San Diego Central Jail. He also holds teaching positions at the University of California, San Diego, and the University of San Diego. He teaches on medical education, psychopharmacology, ethics in psychiatry, and correctional care. He mentors several residents on projects, including reduction in the use of solitary confinement of patients with mental illness, reduction in the use of involuntary treatment of the mentally ill, and examination of the mentally ill offender.
On Dec. 14, 2016, a French psychiatrist was sentenced to an 18-month suspended prison sentence. Lekhraj Gujadhur, MD, was the supervisor of unit 101 at the Psychiatric Hospital Center of Saint-Egrève in France. In November 2008, he had approved the nonsupervised release of a schizophrenia patient, Jean-Pierre Guillaud, to outside of his unit but within the hospital facility. Mr. Guillaud, while outside supervision, escaped. He subsequently purchased a large knife and murdered a 26-year-old student, Luc Meunier, Le Monde reported.1
This is reminiscent of a similar case in 2012 in Marseille, where a psychiatrist received a suspended prison sentence after his patient committed murder. That prior case was later dismissed in appellate court. In my opinion, both trials point to a failure in psychiatry’s responsibility to educate the public in our limitations and roles. They also highlight the necessary discourse that society should have on the role of mental illness when it comes to crime.
Although I appreciate society’s concern about such crimes, I think that displacement of our anger onto Dr. Gujadhur is misguided, and instead, allows us to forget to look at our own poor judgment. Dr. Gujadhur, other psychiatrists, and mental hospitals do not have the responsibility to enact sentences for crimes; the legal system does. Law enforcement and prosecutors had numerous opportunities to charge and commit Mr. Guillaud over the years but chose not to do so, instead permitting him to stay within society under the care of the mental health system.
Asking Dr. Gujadhur to primarily focus on becoming an agent of the law, instead of treating his patient, is unfair. Schizophrenia, and in particular paranoia, are greatly worsened by social isolation. Confining Mr. Guillaud would be countertherapeutic and possibly lead to his suicide. Would Dr. Gujadhur have been responsible for the suicide? Mental health providers have to understand and support the psychological functioning of their patients. Creating a dual agency blurs and effaces the doctor-patient relationship, already so fragile in the treatment of paranoid patients.
The publicity of such cases, and of Mr. Guillaud’s mental illness, seems to go against current mental health research. Recent work has suggested that mental illness is not a significant risk factor for violence but rather a risk factor for being the victim of violence. Certainly, some patients with mental illness commit acts of violence, but studies suggest that this is mostly independent of their mental illness (Law Hum Behav. 2014 Oct;38[5];439-49).2 Our overemphasis on the mental status of criminals belittles their crimes and suggests that psychiatrists are responsible for the failings of our legal system.
As a supervising psychiatrist at one of the largest jail systems in America, I am familiar with the challenges in such cases. All of my patients are facing legal charges, and many suffer from severe mental illness like schizophrenia. As their treating psychiatrist, I am not asked to also sentence them for the charges they are facing. Simply working for the sheriff makes my ability to gain the trust of my patients much more difficult. Conspiring with the city or district attorney in an attempt to protect society would obliterate any chance at rapport building.
Working in corrections, I am deeply familiar with the current debate on the solitary confinement of our mentally ill offenders. Ironically, in that context, society has blamed the legal system for socially isolating our mentally ill offenders, especially ones with severe mental illness.3 In our jail, we meet regularly and discuss in an interdisciplinary fashion the role and consequences of social isolation. During our weekly sessions, a case involving stabbing someone 2 years prior would not have justified the punishment of social isolation and constant monitoring.
As a field, psychiatry must educate society on its ability to create a therapeutic environment and its ability to provide risk assessment of violence. We must also remind others of the impossibility of doing both simultaneously. Decisions on removing patients’ right to freedom can be informed by the mental health perspective but should be left to the courts. Society’s need to find a target after such tragedies is understandable, but blaming the treating psychiatrists will not help past or future victims.
References
1. Le psychiatre d’un schizophrène meurtrier condamné pour homicide involontaire, Le Monde, Dec. 14, 2016.
2. How often and how consistently do symptoms directly precede criminal behavior among offenders with mental illness? (Law Hum Behav. 2014 Oct;38[5]:439-49).
3. How to fix solitary confinement in American prisons, Los Angeles Times, Oct. 17, 2016.
Dr. Badre is a supervising psychiatric contractor at the San Diego Central Jail. He also holds teaching positions at the University of California, San Diego, and the University of San Diego. He teaches on medical education, psychopharmacology, ethics in psychiatry, and correctional care. He mentors several residents on projects, including reduction in the use of solitary confinement of patients with mental illness, reduction in the use of involuntary treatment of the mentally ill, and examination of the mentally ill offender.
On Dec. 14, 2016, a French psychiatrist was sentenced to an 18-month suspended prison sentence. Lekhraj Gujadhur, MD, was the supervisor of unit 101 at the Psychiatric Hospital Center of Saint-Egrève in France. In November 2008, he had approved the nonsupervised release of a schizophrenia patient, Jean-Pierre Guillaud, to outside of his unit but within the hospital facility. Mr. Guillaud, while outside supervision, escaped. He subsequently purchased a large knife and murdered a 26-year-old student, Luc Meunier, Le Monde reported.1
This is reminiscent of a similar case in 2012 in Marseille, where a psychiatrist received a suspended prison sentence after his patient committed murder. That prior case was later dismissed in appellate court. In my opinion, both trials point to a failure in psychiatry’s responsibility to educate the public in our limitations and roles. They also highlight the necessary discourse that society should have on the role of mental illness when it comes to crime.
Although I appreciate society’s concern about such crimes, I think that displacement of our anger onto Dr. Gujadhur is misguided, and instead, allows us to forget to look at our own poor judgment. Dr. Gujadhur, other psychiatrists, and mental hospitals do not have the responsibility to enact sentences for crimes; the legal system does. Law enforcement and prosecutors had numerous opportunities to charge and commit Mr. Guillaud over the years but chose not to do so, instead permitting him to stay within society under the care of the mental health system.
Asking Dr. Gujadhur to primarily focus on becoming an agent of the law, instead of treating his patient, is unfair. Schizophrenia, and in particular paranoia, are greatly worsened by social isolation. Confining Mr. Guillaud would be countertherapeutic and possibly lead to his suicide. Would Dr. Gujadhur have been responsible for the suicide? Mental health providers have to understand and support the psychological functioning of their patients. Creating a dual agency blurs and effaces the doctor-patient relationship, already so fragile in the treatment of paranoid patients.
The publicity of such cases, and of Mr. Guillaud’s mental illness, seems to go against current mental health research. Recent work has suggested that mental illness is not a significant risk factor for violence but rather a risk factor for being the victim of violence. Certainly, some patients with mental illness commit acts of violence, but studies suggest that this is mostly independent of their mental illness (Law Hum Behav. 2014 Oct;38[5];439-49).2 Our overemphasis on the mental status of criminals belittles their crimes and suggests that psychiatrists are responsible for the failings of our legal system.
As a supervising psychiatrist at one of the largest jail systems in America, I am familiar with the challenges in such cases. All of my patients are facing legal charges, and many suffer from severe mental illness like schizophrenia. As their treating psychiatrist, I am not asked to also sentence them for the charges they are facing. Simply working for the sheriff makes my ability to gain the trust of my patients much more difficult. Conspiring with the city or district attorney in an attempt to protect society would obliterate any chance at rapport building.
Working in corrections, I am deeply familiar with the current debate on the solitary confinement of our mentally ill offenders. Ironically, in that context, society has blamed the legal system for socially isolating our mentally ill offenders, especially ones with severe mental illness.3 In our jail, we meet regularly and discuss in an interdisciplinary fashion the role and consequences of social isolation. During our weekly sessions, a case involving stabbing someone 2 years prior would not have justified the punishment of social isolation and constant monitoring.
As a field, psychiatry must educate society on its ability to create a therapeutic environment and its ability to provide risk assessment of violence. We must also remind others of the impossibility of doing both simultaneously. Decisions on removing patients’ right to freedom can be informed by the mental health perspective but should be left to the courts. Society’s need to find a target after such tragedies is understandable, but blaming the treating psychiatrists will not help past or future victims.
References
1. Le psychiatre d’un schizophrène meurtrier condamné pour homicide involontaire, Le Monde, Dec. 14, 2016.
2. How often and how consistently do symptoms directly precede criminal behavior among offenders with mental illness? (Law Hum Behav. 2014 Oct;38[5]:439-49).
3. How to fix solitary confinement in American prisons, Los Angeles Times, Oct. 17, 2016.
Dr. Badre is a supervising psychiatric contractor at the San Diego Central Jail. He also holds teaching positions at the University of California, San Diego, and the University of San Diego. He teaches on medical education, psychopharmacology, ethics in psychiatry, and correctional care. He mentors several residents on projects, including reduction in the use of solitary confinement of patients with mental illness, reduction in the use of involuntary treatment of the mentally ill, and examination of the mentally ill offender.
Endoscopy during pregnancy increases risk of preterm, SGA birth
Women who undergo an endoscopy during pregnancy are increasing the chances that their baby will be born preterm, or be small for gestational age (SGA), according to research published in the February issue of Gastroenterology (doi: 10.1053/j.gastro.2016.10.016).
“Research in pregnancy outcome in women undergoing endoscopy during pregnancy is scarce,” wrote the authors, led by Jonas F. Ludvigsson, MD, of the Karolinska Institutet in Stockholm, adding that there are nine studies with original data on a total of 379 pregnant women undergoing endoscopy; two of these studies examined pregnancy outcome in upper endoscopy (n = 143), two examined pregnancy outcome in sigmoidoscopy or colonoscopy (n = 116), and four examined pregnancy outcome in endoscopic retrograde cholangiopancreatography (n = 120).
Additionally, the authors noted that, to their knowledge, there are no studies that offer data on the relative risk of endoscopy during pregnancy, and none that followed up subjects after birth. Of the few studies that do exist, a handful conclude that endoscopy during pregnancy is actually safe, but do not include data on stillbirths and neonatal deaths that did not occur immediately after patients underwent endoscopy, which could compromise that data.
To address the lack of reliable research on the effect of endoscopy on pregnancy, Dr. Ludvigsson and his coinvestigators launched a nationwide study of pregnancies in Sweden that occurred between 1992 and 2011, all of which were registered in the Swedish Medical Birth Registry and the Swedish Patient Registry. The databases revealed 2,025 upper endoscopies, 1,109 lower endoscopies, and 58 endoscopic retrograde cholangiopancreatographies, for a total of 3,052 pregnancies exposed to endoscopy over that time period.
The primary endpoint of the study was the frequency of preterm birth and stillbirth in this population. To measure this, the investigators used adjusted relative risk (ARR), calculated via Poisson regression by using data on 1,589,173 pregnancies that were not exposed to endoscopy as reference.
“Stillbirth is recorded from 22 completed gestational weeks since mid-2008, and before that from gestational week 28. Gestational age was determined using ultrasound, and when ultrasound data were missing, we used the first day of the last menstrual period for pregnancy start,” the authors wrote.
The results showed that mothers who had any kind of endoscopy during pregnancy were more likely to experience a preterm birth or give birth to a baby who was SGA, with the ARR being 1.54 (95% confidence interval, 1.36-1.75) and 1.30 (95% CI, 1.07-1.57), respectively. However, the risk of other adverse effects, such as stillbirth or congenital malformation, was not significant: Stillbirth ARR was 1.45 (95% CI, 0.87-2.40) and congenital malformation ARR was 1.00 (95% CI, 0.83-1.20).
Women who were exposed to endoscopy during pregnancy were more likely to have a preterm birth, compared with women who had endoscopy 1 year before or after pregnancy, but were not more highly predisposed to SGA, stillbirth, or congenital malformations. Additionally, when data on multiple pregnancies carried by the same mother were compared, no correlation was found between endoscopy and gestational age or birth weight, if the mother was exposed to endoscopy during only one of the pregnancies.
“Earlier recommendations suggest that endoscopy should only be performed during pregnancy if there are strong indications, and if so, not during the second trimester, [but] our study shows that endoscopy is unlikely to have a more than marginal influence on pregnancy outcome independently of trimester,” the authors concluded. “Neither does it seem that sigmoidoscopy is preferable to a full colonoscopy in the pregnant woman.”
Regarding the latter conclusion, the authors clarified that “it is possible that in women with particularly severe gastrointestinal disease where endoscopy is inevitable, the physician will prefer a sigmoidoscopy rather than a full colonoscopy, and under such circumstances the sigmoidoscopy will signal a more severe disease.”
The investigators also noted that their study had several limitations, including not knowing the length of time each endoscopy took, the sedatives and bowel preparations that were used, the patient’s position during the procedure, and the indication that prompted the endoscopy in the first place.
The study was funded by grants from the Swedish Society of Medicine and the Stockholm County Council, and the Swedish Research Council. Dr. Ludvigsson and his coauthors did not report any relevant financial disclosures.
Women who undergo an endoscopy during pregnancy are increasing the chances that their baby will be born preterm, or be small for gestational age (SGA), according to research published in the February issue of Gastroenterology (doi: 10.1053/j.gastro.2016.10.016).
“Research in pregnancy outcome in women undergoing endoscopy during pregnancy is scarce,” wrote the authors, led by Jonas F. Ludvigsson, MD, of the Karolinska Institutet in Stockholm, adding that there are nine studies with original data on a total of 379 pregnant women undergoing endoscopy; two of these studies examined pregnancy outcome in upper endoscopy (n = 143), two examined pregnancy outcome in sigmoidoscopy or colonoscopy (n = 116), and four examined pregnancy outcome in endoscopic retrograde cholangiopancreatography (n = 120).
Additionally, the authors noted that, to their knowledge, there are no studies that offer data on the relative risk of endoscopy during pregnancy, and none that followed up subjects after birth. Of the few studies that do exist, a handful conclude that endoscopy during pregnancy is actually safe, but do not include data on stillbirths and neonatal deaths that did not occur immediately after patients underwent endoscopy, which could compromise that data.
To address the lack of reliable research on the effect of endoscopy on pregnancy, Dr. Ludvigsson and his coinvestigators launched a nationwide study of pregnancies in Sweden that occurred between 1992 and 2011, all of which were registered in the Swedish Medical Birth Registry and the Swedish Patient Registry. The databases revealed 2,025 upper endoscopies, 1,109 lower endoscopies, and 58 endoscopic retrograde cholangiopancreatographies, for a total of 3,052 pregnancies exposed to endoscopy over that time period.
The primary endpoint of the study was the frequency of preterm birth and stillbirth in this population. To measure this, the investigators used adjusted relative risk (ARR), calculated via Poisson regression by using data on 1,589,173 pregnancies that were not exposed to endoscopy as reference.
“Stillbirth is recorded from 22 completed gestational weeks since mid-2008, and before that from gestational week 28. Gestational age was determined using ultrasound, and when ultrasound data were missing, we used the first day of the last menstrual period for pregnancy start,” the authors wrote.
The results showed that mothers who had any kind of endoscopy during pregnancy were more likely to experience a preterm birth or give birth to a baby who was SGA, with the ARR being 1.54 (95% confidence interval, 1.36-1.75) and 1.30 (95% CI, 1.07-1.57), respectively. However, the risk of other adverse effects, such as stillbirth or congenital malformation, was not significant: Stillbirth ARR was 1.45 (95% CI, 0.87-2.40) and congenital malformation ARR was 1.00 (95% CI, 0.83-1.20).
Women who were exposed to endoscopy during pregnancy were more likely to have a preterm birth, compared with women who had endoscopy 1 year before or after pregnancy, but were not more highly predisposed to SGA, stillbirth, or congenital malformations. Additionally, when data on multiple pregnancies carried by the same mother were compared, no correlation was found between endoscopy and gestational age or birth weight, if the mother was exposed to endoscopy during only one of the pregnancies.
“Earlier recommendations suggest that endoscopy should only be performed during pregnancy if there are strong indications, and if so, not during the second trimester, [but] our study shows that endoscopy is unlikely to have a more than marginal influence on pregnancy outcome independently of trimester,” the authors concluded. “Neither does it seem that sigmoidoscopy is preferable to a full colonoscopy in the pregnant woman.”
Regarding the latter conclusion, the authors clarified that “it is possible that in women with particularly severe gastrointestinal disease where endoscopy is inevitable, the physician will prefer a sigmoidoscopy rather than a full colonoscopy, and under such circumstances the sigmoidoscopy will signal a more severe disease.”
The investigators also noted that their study had several limitations, including not knowing the length of time each endoscopy took, the sedatives and bowel preparations that were used, the patient’s position during the procedure, and the indication that prompted the endoscopy in the first place.
The study was funded by grants from the Swedish Society of Medicine and the Stockholm County Council, and the Swedish Research Council. Dr. Ludvigsson and his coauthors did not report any relevant financial disclosures.
Women who undergo an endoscopy during pregnancy are increasing the chances that their baby will be born preterm, or be small for gestational age (SGA), according to research published in the February issue of Gastroenterology (doi: 10.1053/j.gastro.2016.10.016).
“Research in pregnancy outcome in women undergoing endoscopy during pregnancy is scarce,” wrote the authors, led by Jonas F. Ludvigsson, MD, of the Karolinska Institutet in Stockholm, adding that there are nine studies with original data on a total of 379 pregnant women undergoing endoscopy; two of these studies examined pregnancy outcome in upper endoscopy (n = 143), two examined pregnancy outcome in sigmoidoscopy or colonoscopy (n = 116), and four examined pregnancy outcome in endoscopic retrograde cholangiopancreatography (n = 120).
Additionally, the authors noted that, to their knowledge, there are no studies that offer data on the relative risk of endoscopy during pregnancy, and none that followed up subjects after birth. Of the few studies that do exist, a handful conclude that endoscopy during pregnancy is actually safe, but do not include data on stillbirths and neonatal deaths that did not occur immediately after patients underwent endoscopy, which could compromise that data.
To address the lack of reliable research on the effect of endoscopy on pregnancy, Dr. Ludvigsson and his coinvestigators launched a nationwide study of pregnancies in Sweden that occurred between 1992 and 2011, all of which were registered in the Swedish Medical Birth Registry and the Swedish Patient Registry. The databases revealed 2,025 upper endoscopies, 1,109 lower endoscopies, and 58 endoscopic retrograde cholangiopancreatographies, for a total of 3,052 pregnancies exposed to endoscopy over that time period.
The primary endpoint of the study was the frequency of preterm birth and stillbirth in this population. To measure this, the investigators used adjusted relative risk (ARR), calculated via Poisson regression by using data on 1,589,173 pregnancies that were not exposed to endoscopy as reference.
“Stillbirth is recorded from 22 completed gestational weeks since mid-2008, and before that from gestational week 28. Gestational age was determined using ultrasound, and when ultrasound data were missing, we used the first day of the last menstrual period for pregnancy start,” the authors wrote.
The results showed that mothers who had any kind of endoscopy during pregnancy were more likely to experience a preterm birth or give birth to a baby who was SGA, with the ARR being 1.54 (95% confidence interval, 1.36-1.75) and 1.30 (95% CI, 1.07-1.57), respectively. However, the risk of other adverse effects, such as stillbirth or congenital malformation, was not significant: Stillbirth ARR was 1.45 (95% CI, 0.87-2.40) and congenital malformation ARR was 1.00 (95% CI, 0.83-1.20).
Women who were exposed to endoscopy during pregnancy were more likely to have a preterm birth, compared with women who had endoscopy 1 year before or after pregnancy, but were not more highly predisposed to SGA, stillbirth, or congenital malformations. Additionally, when data on multiple pregnancies carried by the same mother were compared, no correlation was found between endoscopy and gestational age or birth weight, if the mother was exposed to endoscopy during only one of the pregnancies.
“Earlier recommendations suggest that endoscopy should only be performed during pregnancy if there are strong indications, and if so, not during the second trimester, [but] our study shows that endoscopy is unlikely to have a more than marginal influence on pregnancy outcome independently of trimester,” the authors concluded. “Neither does it seem that sigmoidoscopy is preferable to a full colonoscopy in the pregnant woman.”
Regarding the latter conclusion, the authors clarified that “it is possible that in women with particularly severe gastrointestinal disease where endoscopy is inevitable, the physician will prefer a sigmoidoscopy rather than a full colonoscopy, and under such circumstances the sigmoidoscopy will signal a more severe disease.”
The investigators also noted that their study had several limitations, including not knowing the length of time each endoscopy took, the sedatives and bowel preparations that were used, the patient’s position during the procedure, and the indication that prompted the endoscopy in the first place.
The study was funded by grants from the Swedish Society of Medicine and the Stockholm County Council, and the Swedish Research Council. Dr. Ludvigsson and his coauthors did not report any relevant financial disclosures.
FROM GASTROENTEROLOGY
Key clinical point:
Major finding: The adjusted relative risk of preterm birth was 1.54 (95% CI, 1.36-1.75) and was 1.30 (95% CI, 1.07-1.57) for SGA.
Data source: A population-based cohort study of 3,052 pregnancies in Sweden exposed to endoscopy from 1992 through 2011.
Disclosures: The study was funded by the Swedish Society of Medicine and the Stockholm County Council, and the Swedish Research Council. The authors did not report any relevant financial disclosures.
Recognizing Congenital Zika Syndrome
VANCOUVER—Infants infected with Zika virus in utero may develop a syndrome characterized by brain volume loss, intracerebral calcifications, and spasticity. They may develop dyskinesia or seizures after several months, and a subset of children has severe arthrogryposis.
And although microcephaly at birth is common, infants may have a normal head size at birth, but develop postnatal microcephaly or other neurologic symptoms at six months, according to research described at the 45th Annual Meeting of the Child Neurology Society.
“The spectrum of congenital Zika syndrome is expanding as we come to understand it better,” said William B. Dobyns, MD, Professor of Pediatrics at the University of Washington in Seattle and a faculty member at the Center for Integrative Brain Research at Seattle Children’s Research Institute. “We all need to stop calling this microcephaly. This is much more than that. This is the congenital Zika syndrome.”

Zika virus is trophic for neural stem cells, and the first reports of microcephaly associated with prenatal Zika virus infection came from Brazil in January 2016. In the US, mosquitoes that transmit Zika virus, Aedes aegypti and albopictus, are present year round in Florida and seasonally in about a quarter of the states. “It is pretty clear that it will be coming.… We need to take precautions until treatments or preventives are available,” he said. In addition, child neurologists need to be able to recognize congenital Zika syndrome. “It is entirely possible for us to do so,” Dr. Dobyns said. “You do not even need viral titers in the more classically affected children.”
A Case Series of 57 Children
Dr. Dobyns worked with André Pessoa, MD, a child neurologist at Hospital Infantil Albert Sabin in Fortaleza, Brazil, and other neurologists in the region to compile data on a series of 57 children with congenital microcephaly and presumed or proven Zika exposure of the mothers during pregnancy. Microcephaly was defined as occipitofrontal head circumference of at least two standard deviations below the mean.
About half of the children had a bony protuberance of the occipital bone, known as an occipital shelf, Dr. Dobyns said. This feature occurs when the fetal brain, instead of growing and pushing out the skull plates, is severely injured and shrinks. The frontal and parietal bones, but not the occipital bone, collapse over the injured brain.
Almost all of the children had prominent calcifications in the brain. Unlike in children infected with cytomegalovirus, periventricular calcifications are the exception in children infected with Zika virus. Researchers observed subcortical or cortical calcifications on CT in 51 of the 57 children infected with Zika virus and basal ganglia calcifications in 33 of the 57 children.
Furthermore, calcifications with Zika virus infection tend to be diffuse and bilateral, whereas calcifications with cytomegalovirus infection tend to be patchy, Dr. Dobyns said.
All patients had the same general pattern of enlarged extra-axial space, ventriculomegaly, or both, indicating brain volume loss.
About 20% of patients had severe arthrogryposis multiplex congenita, and all of these children had abnormally positioned proximal joints.
Twenty of the children underwent brain MRI. MRI showed an abnormal cortex in all 20 children. The patients appear to have a diffuse cortical malformation that is most consistent with polymicrogyria, Dr. Dobyns said.
Nearly 20% of children in the series had microcephaly between two and three standard deviations below the mean. But 81% had microcephaly of three or more standard deviations below the mean. The mean occipitofrontal head circumference was four standard deviations below the mean.
Neurologic features included spasticity in 94% of the children and severe irritability or tremor in 64% of the children. About 20% had seizures after several months. Some patients had eye abnormalities, including optic nerve pallor, macular atrophy, and strabismus.
“The exam is characteristic,” Dr. Dobyns said. “They all develop a dyskinesia later in the first year of life. They have spastic quadriparesis. They frequently have tremors at birth. They feed poorly. They tend to be irritable and scream all the time. They are starting to have seizures as they get past six months of age.”
As in other studies, data from this series suggest that children whose mothers have a symptomatic illness or are infected earlier in pregnancy may be at higher risk of congenital Zika syndrome.
Infants Without Microcephaly at Birth
Dr. Dobyns presented preliminary data from children who were exposed to Zika virus but did not have microcephaly at birth. These children had most of the same features on exam as children with microcephaly, although the features tended to be less severe. The children started to have seizures after several months. When their head size was measured at six months or older, it fell below the second percentile, meaning that these children had postnatal microcephaly. The children did not have congenital contractures, Dr. Dobyns said.
Vanessa van der Linden, MD, a pediatric neurologist at the Association for Assistance of Disabled Children in Recife, Brazil, Dr. Pessoa, Dr. Dobyns, and colleagues on November 22, 2016, published a description of 13 infants who had evidence of congenital Zika infection but did not have microcephaly at birth. Their report was published online in the CDC’s Morbidity and Mortality Weekly Report. The researchers found that head growth decelerated in all 13 of the infants by as early as age 5 months, and 11 of the infants had microcephaly. The findings suggest that infants exposed to Zika virus prenatally should receive comprehensive medical and developmental follow-up, even in the absence of microcephaly at birth, the investigators said.
That infants with prenatal Zika infection may develop postnatal microcephaly is not surprising, Dr. Dobyns said. Microcephaly, however, remains only one possible symptom of congenital Zika syndrome. “It is a pattern of features seen clinically, on CT scans, and behaviorally that will mark this syndrome,” he said.
—Jake Remaly
Suggested Reading
Moore CA, Staples JE, Dobyns WB, et al. Characterizing the pattern of anomalies in congenital Zika syndrome for pediatric clinicians. JAMA Pediatr. 2016 Nov 3 [Epub ahead of print].
van der Linden V, Pessoa A, Dobyns W, et al. Description of 13 infants born during October 2015–January 2016 with congenital Zika virus infection without microcephaly at birth — Brazil. MMWR Morb Mortal Wkly Rep. 22 Nov 2016 [Epub ahead of print].
VANCOUVER—Infants infected with Zika virus in utero may develop a syndrome characterized by brain volume loss, intracerebral calcifications, and spasticity. They may develop dyskinesia or seizures after several months, and a subset of children has severe arthrogryposis.
And although microcephaly at birth is common, infants may have a normal head size at birth, but develop postnatal microcephaly or other neurologic symptoms at six months, according to research described at the 45th Annual Meeting of the Child Neurology Society.
“The spectrum of congenital Zika syndrome is expanding as we come to understand it better,” said William B. Dobyns, MD, Professor of Pediatrics at the University of Washington in Seattle and a faculty member at the Center for Integrative Brain Research at Seattle Children’s Research Institute. “We all need to stop calling this microcephaly. This is much more than that. This is the congenital Zika syndrome.”
Zika virus is trophic for neural stem cells, and the first reports of microcephaly associated with prenatal Zika virus infection came from Brazil in January 2016. In the US, mosquitoes that transmit Zika virus, Aedes aegypti and albopictus, are present year round in Florida and seasonally in about a quarter of the states. “It is pretty clear that it will be coming.… We need to take precautions until treatments or preventives are available,” he said. In addition, child neurologists need to be able to recognize congenital Zika syndrome. “It is entirely possible for us to do so,” Dr. Dobyns said. “You do not even need viral titers in the more classically affected children.”
A Case Series of 57 Children
Dr. Dobyns worked with André Pessoa, MD, a child neurologist at Hospital Infantil Albert Sabin in Fortaleza, Brazil, and other neurologists in the region to compile data on a series of 57 children with congenital microcephaly and presumed or proven Zika exposure of the mothers during pregnancy. Microcephaly was defined as occipitofrontal head circumference of at least two standard deviations below the mean.
About half of the children had a bony protuberance of the occipital bone, known as an occipital shelf, Dr. Dobyns said. This feature occurs when the fetal brain, instead of growing and pushing out the skull plates, is severely injured and shrinks. The frontal and parietal bones, but not the occipital bone, collapse over the injured brain.
Almost all of the children had prominent calcifications in the brain. Unlike in children infected with cytomegalovirus, periventricular calcifications are the exception in children infected with Zika virus. Researchers observed subcortical or cortical calcifications on CT in 51 of the 57 children infected with Zika virus and basal ganglia calcifications in 33 of the 57 children.
Furthermore, calcifications with Zika virus infection tend to be diffuse and bilateral, whereas calcifications with cytomegalovirus infection tend to be patchy, Dr. Dobyns said.
All patients had the same general pattern of enlarged extra-axial space, ventriculomegaly, or both, indicating brain volume loss.
About 20% of patients had severe arthrogryposis multiplex congenita, and all of these children had abnormally positioned proximal joints.
Twenty of the children underwent brain MRI. MRI showed an abnormal cortex in all 20 children. The patients appear to have a diffuse cortical malformation that is most consistent with polymicrogyria, Dr. Dobyns said.
Nearly 20% of children in the series had microcephaly between two and three standard deviations below the mean. But 81% had microcephaly of three or more standard deviations below the mean. The mean occipitofrontal head circumference was four standard deviations below the mean.
Neurologic features included spasticity in 94% of the children and severe irritability or tremor in 64% of the children. About 20% had seizures after several months. Some patients had eye abnormalities, including optic nerve pallor, macular atrophy, and strabismus.
“The exam is characteristic,” Dr. Dobyns said. “They all develop a dyskinesia later in the first year of life. They have spastic quadriparesis. They frequently have tremors at birth. They feed poorly. They tend to be irritable and scream all the time. They are starting to have seizures as they get past six months of age.”
As in other studies, data from this series suggest that children whose mothers have a symptomatic illness or are infected earlier in pregnancy may be at higher risk of congenital Zika syndrome.
Infants Without Microcephaly at Birth
Dr. Dobyns presented preliminary data from children who were exposed to Zika virus but did not have microcephaly at birth. These children had most of the same features on exam as children with microcephaly, although the features tended to be less severe. The children started to have seizures after several months. When their head size was measured at six months or older, it fell below the second percentile, meaning that these children had postnatal microcephaly. The children did not have congenital contractures, Dr. Dobyns said.
Vanessa van der Linden, MD, a pediatric neurologist at the Association for Assistance of Disabled Children in Recife, Brazil, Dr. Pessoa, Dr. Dobyns, and colleagues on November 22, 2016, published a description of 13 infants who had evidence of congenital Zika infection but did not have microcephaly at birth. Their report was published online in the CDC’s Morbidity and Mortality Weekly Report. The researchers found that head growth decelerated in all 13 of the infants by as early as age 5 months, and 11 of the infants had microcephaly. The findings suggest that infants exposed to Zika virus prenatally should receive comprehensive medical and developmental follow-up, even in the absence of microcephaly at birth, the investigators said.
That infants with prenatal Zika infection may develop postnatal microcephaly is not surprising, Dr. Dobyns said. Microcephaly, however, remains only one possible symptom of congenital Zika syndrome. “It is a pattern of features seen clinically, on CT scans, and behaviorally that will mark this syndrome,” he said.
—Jake Remaly
Suggested Reading
Moore CA, Staples JE, Dobyns WB, et al. Characterizing the pattern of anomalies in congenital Zika syndrome for pediatric clinicians. JAMA Pediatr. 2016 Nov 3 [Epub ahead of print].
van der Linden V, Pessoa A, Dobyns W, et al. Description of 13 infants born during October 2015–January 2016 with congenital Zika virus infection without microcephaly at birth — Brazil. MMWR Morb Mortal Wkly Rep. 22 Nov 2016 [Epub ahead of print].
VANCOUVER—Infants infected with Zika virus in utero may develop a syndrome characterized by brain volume loss, intracerebral calcifications, and spasticity. They may develop dyskinesia or seizures after several months, and a subset of children has severe arthrogryposis.
And although microcephaly at birth is common, infants may have a normal head size at birth, but develop postnatal microcephaly or other neurologic symptoms at six months, according to research described at the 45th Annual Meeting of the Child Neurology Society.
“The spectrum of congenital Zika syndrome is expanding as we come to understand it better,” said William B. Dobyns, MD, Professor of Pediatrics at the University of Washington in Seattle and a faculty member at the Center for Integrative Brain Research at Seattle Children’s Research Institute. “We all need to stop calling this microcephaly. This is much more than that. This is the congenital Zika syndrome.”
Zika virus is trophic for neural stem cells, and the first reports of microcephaly associated with prenatal Zika virus infection came from Brazil in January 2016. In the US, mosquitoes that transmit Zika virus, Aedes aegypti and albopictus, are present year round in Florida and seasonally in about a quarter of the states. “It is pretty clear that it will be coming.… We need to take precautions until treatments or preventives are available,” he said. In addition, child neurologists need to be able to recognize congenital Zika syndrome. “It is entirely possible for us to do so,” Dr. Dobyns said. “You do not even need viral titers in the more classically affected children.”
A Case Series of 57 Children
Dr. Dobyns worked with André Pessoa, MD, a child neurologist at Hospital Infantil Albert Sabin in Fortaleza, Brazil, and other neurologists in the region to compile data on a series of 57 children with congenital microcephaly and presumed or proven Zika exposure of the mothers during pregnancy. Microcephaly was defined as occipitofrontal head circumference of at least two standard deviations below the mean.
About half of the children had a bony protuberance of the occipital bone, known as an occipital shelf, Dr. Dobyns said. This feature occurs when the fetal brain, instead of growing and pushing out the skull plates, is severely injured and shrinks. The frontal and parietal bones, but not the occipital bone, collapse over the injured brain.
Almost all of the children had prominent calcifications in the brain. Unlike in children infected with cytomegalovirus, periventricular calcifications are the exception in children infected with Zika virus. Researchers observed subcortical or cortical calcifications on CT in 51 of the 57 children infected with Zika virus and basal ganglia calcifications in 33 of the 57 children.
Furthermore, calcifications with Zika virus infection tend to be diffuse and bilateral, whereas calcifications with cytomegalovirus infection tend to be patchy, Dr. Dobyns said.
All patients had the same general pattern of enlarged extra-axial space, ventriculomegaly, or both, indicating brain volume loss.
About 20% of patients had severe arthrogryposis multiplex congenita, and all of these children had abnormally positioned proximal joints.
Twenty of the children underwent brain MRI. MRI showed an abnormal cortex in all 20 children. The patients appear to have a diffuse cortical malformation that is most consistent with polymicrogyria, Dr. Dobyns said.
Nearly 20% of children in the series had microcephaly between two and three standard deviations below the mean. But 81% had microcephaly of three or more standard deviations below the mean. The mean occipitofrontal head circumference was four standard deviations below the mean.
Neurologic features included spasticity in 94% of the children and severe irritability or tremor in 64% of the children. About 20% had seizures after several months. Some patients had eye abnormalities, including optic nerve pallor, macular atrophy, and strabismus.
“The exam is characteristic,” Dr. Dobyns said. “They all develop a dyskinesia later in the first year of life. They have spastic quadriparesis. They frequently have tremors at birth. They feed poorly. They tend to be irritable and scream all the time. They are starting to have seizures as they get past six months of age.”
As in other studies, data from this series suggest that children whose mothers have a symptomatic illness or are infected earlier in pregnancy may be at higher risk of congenital Zika syndrome.
Infants Without Microcephaly at Birth
Dr. Dobyns presented preliminary data from children who were exposed to Zika virus but did not have microcephaly at birth. These children had most of the same features on exam as children with microcephaly, although the features tended to be less severe. The children started to have seizures after several months. When their head size was measured at six months or older, it fell below the second percentile, meaning that these children had postnatal microcephaly. The children did not have congenital contractures, Dr. Dobyns said.
Vanessa van der Linden, MD, a pediatric neurologist at the Association for Assistance of Disabled Children in Recife, Brazil, Dr. Pessoa, Dr. Dobyns, and colleagues on November 22, 2016, published a description of 13 infants who had evidence of congenital Zika infection but did not have microcephaly at birth. Their report was published online in the CDC’s Morbidity and Mortality Weekly Report. The researchers found that head growth decelerated in all 13 of the infants by as early as age 5 months, and 11 of the infants had microcephaly. The findings suggest that infants exposed to Zika virus prenatally should receive comprehensive medical and developmental follow-up, even in the absence of microcephaly at birth, the investigators said.
That infants with prenatal Zika infection may develop postnatal microcephaly is not surprising, Dr. Dobyns said. Microcephaly, however, remains only one possible symptom of congenital Zika syndrome. “It is a pattern of features seen clinically, on CT scans, and behaviorally that will mark this syndrome,” he said.
—Jake Remaly
Suggested Reading
Moore CA, Staples JE, Dobyns WB, et al. Characterizing the pattern of anomalies in congenital Zika syndrome for pediatric clinicians. JAMA Pediatr. 2016 Nov 3 [Epub ahead of print].
van der Linden V, Pessoa A, Dobyns W, et al. Description of 13 infants born during October 2015–January 2016 with congenital Zika virus infection without microcephaly at birth — Brazil. MMWR Morb Mortal Wkly Rep. 22 Nov 2016 [Epub ahead of print].

IV Ketamine May Be Effective as Subacute Treatment for Refractory Chronic Migraine
Ketamine may help to treat pain in patients with refractory chronic migraine, according to a case series published in the December 2016 Journal of Headache and Pain. IV ketamine treatment was associated with short-term improvement in pain severity in six of six patients with refractory chronic migraine.
“This study highlights the need for further research regarding new treatment options for patients who suffer daily consequences of refractory migraine and have failed many abortive and preventive medications,” said Clinton Lauritsen, MD, a Headache Fellow at Thomas Jefferson University Hospital in Philadelphia.

Ketamine is a dissociative anesthetic that acts on glutamate binding sites at the N-methyl-D-aspartate (NMDA) receptor, as well as at opioid, monoaminergic, cholinergic, nicotinic, and muscarinic receptors. IV ketamine was previously studied in several refractory pain conditions, including complex regional pain. While intranasal ketamine reduced the severity of migraine aura in a small randomized trial, the use of IV ketamine has only been reported in case series. Krusz et al showed improvement in pain scores in patients who used IV ketamine for refractory migraine; few side effects were reported.
Inpatient IV Ketamine
To further investigate the effect of IV ketamine in patients with intractable migraine, Dr. Lauritsen and colleagues conducted a retrospective chart review study. The researchers identified six patients with refractory chronic migraine admitted to Mount Sinai Beth Israel Hospital in New York from 2010 through 2014 for treatment with continuous IV ketamine.
Patients were given a starting dose of 0.1 mg/kg/h that was increased by 0.1 mg/kg/h every three to four hours as tolerated until the target pain score of 3 out of 10 was achieved and maintained for at least eight hours. Subsequently, the infusion was decreased by 0.2 mg/kg/h every three to four hours until the infusion rate reached 0 mg/kg/h.
The dose of ketamine was increased until maximum response was achieved or undesirable side effects, including psychomimetic and dysphoric effects, developed. Researchers used the Visual Analogue Score (VAS) at admission and during follow-up. VAS scores at different ketamine infusion rates were assessed from nursing and infusion records. Pain response was defined as a reduction in the initial VAS to a score of 3 or less. In addition, researchers attempted to contact patients for a telephone follow-up; however, they were only able to reach two of the six patients. During the telephone interview, researchers administered a questionnaire.
Pain Relief Achieved
Results from the data revealed a median age of 36.5 years; 83% of the patients were women. All of the patients were Caucasian, and the median age of migraine onset was 17. The median duration of the disease was 17 years. The mean number of failed acute migraine treatments was 18, and the mean number of failed preventive medications was 25. Pre-treatment pain scores ranged from 9 to 10.
In this small case series, all six patients with refractory migraine met
Overall, IV ketamine relieved pain in patients with chronic migraine without substantial adverse effects. “However, future study of this benefit on short-term headache relief needs to be conducted in a placebo-controlled fashion,” said Dr. Lauritsen.
“It is biologically plausible that ketamine could be an effective treatment for intractable headache,” the researchers said. “Ketamine is an antagonist at NMDA receptors, blocking the excitatory action of glutamate, a neurotransmitter long implicated in the pathophysiology of migraine. Glutamate has been … implicated in induction of cortical spreading depression [and] activation of trigeminal nociceptive neurons [and may] play a role in central sensitization.”
—Erica Tricarico
Suggested Reading
Lauritsen C, Mazuera S, Lipton RB, Ashina S. Intravenous ketamine for subacute treatment of refractory chronic migraine: a case series. J Headache Pain. 2016;17(1)106-110.
Ketamine may help to treat pain in patients with refractory chronic migraine, according to a case series published in the December 2016 Journal of Headache and Pain. IV ketamine treatment was associated with short-term improvement in pain severity in six of six patients with refractory chronic migraine.
“This study highlights the need for further research regarding new treatment options for patients who suffer daily consequences of refractory migraine and have failed many abortive and preventive medications,” said Clinton Lauritsen, MD, a Headache Fellow at Thomas Jefferson University Hospital in Philadelphia.
Ketamine is a dissociative anesthetic that acts on glutamate binding sites at the N-methyl-D-aspartate (NMDA) receptor, as well as at opioid, monoaminergic, cholinergic, nicotinic, and muscarinic receptors. IV ketamine was previously studied in several refractory pain conditions, including complex regional pain. While intranasal ketamine reduced the severity of migraine aura in a small randomized trial, the use of IV ketamine has only been reported in case series. Krusz et al showed improvement in pain scores in patients who used IV ketamine for refractory migraine; few side effects were reported.
Inpatient IV Ketamine
To further investigate the effect of IV ketamine in patients with intractable migraine, Dr. Lauritsen and colleagues conducted a retrospective chart review study. The researchers identified six patients with refractory chronic migraine admitted to Mount Sinai Beth Israel Hospital in New York from 2010 through 2014 for treatment with continuous IV ketamine.
Patients were given a starting dose of 0.1 mg/kg/h that was increased by 0.1 mg/kg/h every three to four hours as tolerated until the target pain score of 3 out of 10 was achieved and maintained for at least eight hours. Subsequently, the infusion was decreased by 0.2 mg/kg/h every three to four hours until the infusion rate reached 0 mg/kg/h.
The dose of ketamine was increased until maximum response was achieved or undesirable side effects, including psychomimetic and dysphoric effects, developed. Researchers used the Visual Analogue Score (VAS) at admission and during follow-up. VAS scores at different ketamine infusion rates were assessed from nursing and infusion records. Pain response was defined as a reduction in the initial VAS to a score of 3 or less. In addition, researchers attempted to contact patients for a telephone follow-up; however, they were only able to reach two of the six patients. During the telephone interview, researchers administered a questionnaire.
Pain Relief Achieved
Results from the data revealed a median age of 36.5 years; 83% of the patients were women. All of the patients were Caucasian, and the median age of migraine onset was 17. The median duration of the disease was 17 years. The mean number of failed acute migraine treatments was 18, and the mean number of failed preventive medications was 25. Pre-treatment pain scores ranged from 9 to 10.
In this small case series, all six patients with refractory migraine met
Overall, IV ketamine relieved pain in patients with chronic migraine without substantial adverse effects. “However, future study of this benefit on short-term headache relief needs to be conducted in a placebo-controlled fashion,” said Dr. Lauritsen.
“It is biologically plausible that ketamine could be an effective treatment for intractable headache,” the researchers said. “Ketamine is an antagonist at NMDA receptors, blocking the excitatory action of glutamate, a neurotransmitter long implicated in the pathophysiology of migraine. Glutamate has been … implicated in induction of cortical spreading depression [and] activation of trigeminal nociceptive neurons [and may] play a role in central sensitization.”
—Erica Tricarico
Suggested Reading
Lauritsen C, Mazuera S, Lipton RB, Ashina S. Intravenous ketamine for subacute treatment of refractory chronic migraine: a case series. J Headache Pain. 2016;17(1)106-110.
Ketamine may help to treat pain in patients with refractory chronic migraine, according to a case series published in the December 2016 Journal of Headache and Pain. IV ketamine treatment was associated with short-term improvement in pain severity in six of six patients with refractory chronic migraine.
“This study highlights the need for further research regarding new treatment options for patients who suffer daily consequences of refractory migraine and have failed many abortive and preventive medications,” said Clinton Lauritsen, MD, a Headache Fellow at Thomas Jefferson University Hospital in Philadelphia.
Ketamine is a dissociative anesthetic that acts on glutamate binding sites at the N-methyl-D-aspartate (NMDA) receptor, as well as at opioid, monoaminergic, cholinergic, nicotinic, and muscarinic receptors. IV ketamine was previously studied in several refractory pain conditions, including complex regional pain. While intranasal ketamine reduced the severity of migraine aura in a small randomized trial, the use of IV ketamine has only been reported in case series. Krusz et al showed improvement in pain scores in patients who used IV ketamine for refractory migraine; few side effects were reported.
Inpatient IV Ketamine
To further investigate the effect of IV ketamine in patients with intractable migraine, Dr. Lauritsen and colleagues conducted a retrospective chart review study. The researchers identified six patients with refractory chronic migraine admitted to Mount Sinai Beth Israel Hospital in New York from 2010 through 2014 for treatment with continuous IV ketamine.
Patients were given a starting dose of 0.1 mg/kg/h that was increased by 0.1 mg/kg/h every three to four hours as tolerated until the target pain score of 3 out of 10 was achieved and maintained for at least eight hours. Subsequently, the infusion was decreased by 0.2 mg/kg/h every three to four hours until the infusion rate reached 0 mg/kg/h.
The dose of ketamine was increased until maximum response was achieved or undesirable side effects, including psychomimetic and dysphoric effects, developed. Researchers used the Visual Analogue Score (VAS) at admission and during follow-up. VAS scores at different ketamine infusion rates were assessed from nursing and infusion records. Pain response was defined as a reduction in the initial VAS to a score of 3 or less. In addition, researchers attempted to contact patients for a telephone follow-up; however, they were only able to reach two of the six patients. During the telephone interview, researchers administered a questionnaire.
Pain Relief Achieved
Results from the data revealed a median age of 36.5 years; 83% of the patients were women. All of the patients were Caucasian, and the median age of migraine onset was 17. The median duration of the disease was 17 years. The mean number of failed acute migraine treatments was 18, and the mean number of failed preventive medications was 25. Pre-treatment pain scores ranged from 9 to 10.
In this small case series, all six patients with refractory migraine met
Overall, IV ketamine relieved pain in patients with chronic migraine without substantial adverse effects. “However, future study of this benefit on short-term headache relief needs to be conducted in a placebo-controlled fashion,” said Dr. Lauritsen.
“It is biologically plausible that ketamine could be an effective treatment for intractable headache,” the researchers said. “Ketamine is an antagonist at NMDA receptors, blocking the excitatory action of glutamate, a neurotransmitter long implicated in the pathophysiology of migraine. Glutamate has been … implicated in induction of cortical spreading depression [and] activation of trigeminal nociceptive neurons [and may] play a role in central sensitization.”
—Erica Tricarico
Suggested Reading
Lauritsen C, Mazuera S, Lipton RB, Ashina S. Intravenous ketamine for subacute treatment of refractory chronic migraine: a case series. J Headache Pain. 2016;17(1)106-110.

Propofol safety similar to that of traditional sedatives used in endoscopy
For doctors performing gastrointestinal endoscopic procedures, use of propofol as a sedative instead of the more commonly used drugs carries about the same risk of causing cardiopulmonary adverse events, according to a study published in the February issue of Clinical Gastroenterology and Hepatology (doi: 10.1016/j.cgh.2016.07.013).
“Because of its popularity, propofol is being used for both simple endoscopic procedures such as esophagogastroduodenoscopy and colonoscopy, and advanced endoscopic procedures [but] despite the widespread use of propofol, significant concerns remain regarding its safety profile,” according to the authors of the study, led by Vaibhav Wadhwa, MD, of Fairview Hospital in Cleveland.
The use of propofol as a sedative in gastrointestinal endoscopic procedures has increased in recent years, but because of an increasing number of advanced and, therefore, more complicated procedures being performed, the safety of sedatives has come into question because of their more prolonged use. Before use of propofol became prevalent, the more traditionally used sedative was a combination of benzodiazepine with an opioid. While still used today, this combination has seen a dramatic decline in usage because of its longer recovery time and lower rates of satisfaction among both patients and doctors, according to the authors. Combinations including midazolam, meperidine, pethidine, remifentanil, and fentanyl have also been used.
To compare the safety of propofol and a more traditional sedative combination, Dr. Wadhwa and his coauthors conducted a meta-analysis of published studies in the Medline (Ovid), EMBASE, and the Cochrane controlled trials registry databases. All searches were for research conducted through September of 2014, with the Medline database search starting in 1960, and the EMBASE and Cochrane searches starting in 1980, yielding a total of 2,117 studies eligible for inclusion.
Of those, 1,568 remained after duplicates were removed, then 136 were screened after removal of those deemed irrelevant or otherwise unsuitable. From those 136, 83 were excluded for various reasons – because they featured either ineligible populations, or were retrospective studies, single-arm studies, or conference abstracts – leaving 53 full-text articles to be evaluated for inclusion in the study. Of those, 27 were deemed eligible and were ultimately included.
“The primary outcomes measured were cardiopulmonary complications such as hypoxia, if oxygen saturation decreased to less than 90%; hypotension, if systolic blood pressure decreased to less than 90 mm Hg; arrhythmias, including bradycardia, supraventricular and ventricular arrhythmias, and ectopy,” Dr. Wadhwa and his coauthors wrote. “A subgroup analysis also was performed to assess studies in which sedation was directed by gastroenterologists and was compared with nongastroenterologists.” Apnea was not measured because of the lack of studies that assessed it qualitatively.
Pooled odds ratios were used to measure and compare results. The 27 included studies featured data on a total of 2,518 patients. Traditional sedatives were used on 1,194 of these subjects, while the remaining 1,324 received propofol. Regarding hypoxia, 26 of the 27 studies addressed this, of which 13 concluded that propofol was safer and 9 found that traditional sedatives were safer, with a pooled OR for propofol of 0.82 (95% confidence interval [CI] 0.63-1.07).
Twenty-five studies examined hypotension, of which 9 favored propofol and 10 favored traditional sedatives, for an OR of 0.92 (95% CI, 0.64-1.32). Of the 20 studies that included arrhythmia, 8 favored propofol and 7 favored traditional sedatives, for an OR of 1.07 (95% CI, 0.68-1.68).
“Our results showed that propofol sedation for gastrointestinal endoscopic procedures, whether simple or advanced, did not increase the cardiopulmonary adverse event rate when compared with traditional sedative agents,” the authors concluded.
In terms of the risk of developing any of the aforementioned complications, of the 20 relevant studies, 9 found propofol to be safer versus 6 that found traditional sedatives to be the better option, yielding an overall OR of 0.77 (95% CI, 0.56-1.07) for propofol. For the subanalysis regarding which type of clinician administered each sedative, 25 studies contained relevant data, of which 9 studies reported gastroenterologists administering sedatives, 5 studies reported endoscopy nurses administering sedatives under the supervision of the gastroenterologist, and 11 studies reported either an anesthesiologist, intensive care unit physician, or critical care physician administering sedatives.
“Gastroenterologist-directed sedation with propofol was noninferior to nongastroenterologist sedation,” Dr. Wadhwa and his coinvestigators wrote. “The risk of complications was similar to [that of traditional sedatives] both during simple and advanced endoscopic procedures.”
While the authors point to the sheer size of the study population as a huge strength of these results, they also note that because this is a study-level analysis rather than one conducted on an individual level, there is an inherent limitation to this study. Furthermore, variations from study to study in how propofol was administered to each patient may have caused heterogeneity with the findings of the meta-analysis. A large clinical trial would be the next logical step to affirm what this analysis has found.
“Because it may not be feasible to perform such a study, this meta-analysis should provide a rough idea of the possible associations,” the authors wrote. “However, the difference in complications between propofol and other agents might not be clinically relevant owing to the lack of any serious complications such as intubations or deaths in the studies used in this meta-analysis.”
No funding source was reported for this study. Dr. Wadhwa and his coauthors reported no relevant financial disclosures.
The use of propofol-mediated sedation and, in particular, anesthetist-directed sedation has become a hot-button item in the landscape of gastrointestinal endoscopy by virtue of its overall cost. Some experts place the cost of this at over $1.1 billion annually. Recent studies stemming from a large administrative database question the safety of propofol-mediated sedation when compared to the standard combination of a benzodiazepine and opioid. Still other studies have found that anesthesiologist-directed sedation did not improve the rate of polyp detection or polypectomy. Given these findings, our research group decided to embark upon a meta-analysis to further study the safety profile of propofol when compared to the combination of a benzodiazepine and opioid. We found that when compared to the traditional sedation agents, the pooled odds ratio of propofol-mediated sedation was not associated with a safety benefit in terms of the development of hypoxia or hypotension. We also found that the safety profile of propofol-mediated sedation was equivalent whether it was administered by a gastroenterologist or nongastroenterologist.

John Vargo, MD, MPH, is the department chair of gastroenterology and hepatology at Cleveland Clinic as well as vice chairman of Cleveland Clinic’s Digestive Disease Institute.
The use of propofol-mediated sedation and, in particular, anesthetist-directed sedation has become a hot-button item in the landscape of gastrointestinal endoscopy by virtue of its overall cost. Some experts place the cost of this at over $1.1 billion annually. Recent studies stemming from a large administrative database question the safety of propofol-mediated sedation when compared to the standard combination of a benzodiazepine and opioid. Still other studies have found that anesthesiologist-directed sedation did not improve the rate of polyp detection or polypectomy. Given these findings, our research group decided to embark upon a meta-analysis to further study the safety profile of propofol when compared to the combination of a benzodiazepine and opioid. We found that when compared to the traditional sedation agents, the pooled odds ratio of propofol-mediated sedation was not associated with a safety benefit in terms of the development of hypoxia or hypotension. We also found that the safety profile of propofol-mediated sedation was equivalent whether it was administered by a gastroenterologist or nongastroenterologist.
John Vargo, MD, MPH, is the department chair of gastroenterology and hepatology at Cleveland Clinic as well as vice chairman of Cleveland Clinic’s Digestive Disease Institute.
The use of propofol-mediated sedation and, in particular, anesthetist-directed sedation has become a hot-button item in the landscape of gastrointestinal endoscopy by virtue of its overall cost. Some experts place the cost of this at over $1.1 billion annually. Recent studies stemming from a large administrative database question the safety of propofol-mediated sedation when compared to the standard combination of a benzodiazepine and opioid. Still other studies have found that anesthesiologist-directed sedation did not improve the rate of polyp detection or polypectomy. Given these findings, our research group decided to embark upon a meta-analysis to further study the safety profile of propofol when compared to the combination of a benzodiazepine and opioid. We found that when compared to the traditional sedation agents, the pooled odds ratio of propofol-mediated sedation was not associated with a safety benefit in terms of the development of hypoxia or hypotension. We also found that the safety profile of propofol-mediated sedation was equivalent whether it was administered by a gastroenterologist or nongastroenterologist.
John Vargo, MD, MPH, is the department chair of gastroenterology and hepatology at Cleveland Clinic as well as vice chairman of Cleveland Clinic’s Digestive Disease Institute.
For doctors performing gastrointestinal endoscopic procedures, use of propofol as a sedative instead of the more commonly used drugs carries about the same risk of causing cardiopulmonary adverse events, according to a study published in the February issue of Clinical Gastroenterology and Hepatology (doi: 10.1016/j.cgh.2016.07.013).
“Because of its popularity, propofol is being used for both simple endoscopic procedures such as esophagogastroduodenoscopy and colonoscopy, and advanced endoscopic procedures [but] despite the widespread use of propofol, significant concerns remain regarding its safety profile,” according to the authors of the study, led by Vaibhav Wadhwa, MD, of Fairview Hospital in Cleveland.
The use of propofol as a sedative in gastrointestinal endoscopic procedures has increased in recent years, but because of an increasing number of advanced and, therefore, more complicated procedures being performed, the safety of sedatives has come into question because of their more prolonged use. Before use of propofol became prevalent, the more traditionally used sedative was a combination of benzodiazepine with an opioid. While still used today, this combination has seen a dramatic decline in usage because of its longer recovery time and lower rates of satisfaction among both patients and doctors, according to the authors. Combinations including midazolam, meperidine, pethidine, remifentanil, and fentanyl have also been used.
To compare the safety of propofol and a more traditional sedative combination, Dr. Wadhwa and his coauthors conducted a meta-analysis of published studies in the Medline (Ovid), EMBASE, and the Cochrane controlled trials registry databases. All searches were for research conducted through September of 2014, with the Medline database search starting in 1960, and the EMBASE and Cochrane searches starting in 1980, yielding a total of 2,117 studies eligible for inclusion.
Of those, 1,568 remained after duplicates were removed, then 136 were screened after removal of those deemed irrelevant or otherwise unsuitable. From those 136, 83 were excluded for various reasons – because they featured either ineligible populations, or were retrospective studies, single-arm studies, or conference abstracts – leaving 53 full-text articles to be evaluated for inclusion in the study. Of those, 27 were deemed eligible and were ultimately included.
“The primary outcomes measured were cardiopulmonary complications such as hypoxia, if oxygen saturation decreased to less than 90%; hypotension, if systolic blood pressure decreased to less than 90 mm Hg; arrhythmias, including bradycardia, supraventricular and ventricular arrhythmias, and ectopy,” Dr. Wadhwa and his coauthors wrote. “A subgroup analysis also was performed to assess studies in which sedation was directed by gastroenterologists and was compared with nongastroenterologists.” Apnea was not measured because of the lack of studies that assessed it qualitatively.
Pooled odds ratios were used to measure and compare results. The 27 included studies featured data on a total of 2,518 patients. Traditional sedatives were used on 1,194 of these subjects, while the remaining 1,324 received propofol. Regarding hypoxia, 26 of the 27 studies addressed this, of which 13 concluded that propofol was safer and 9 found that traditional sedatives were safer, with a pooled OR for propofol of 0.82 (95% confidence interval [CI] 0.63-1.07).
Twenty-five studies examined hypotension, of which 9 favored propofol and 10 favored traditional sedatives, for an OR of 0.92 (95% CI, 0.64-1.32). Of the 20 studies that included arrhythmia, 8 favored propofol and 7 favored traditional sedatives, for an OR of 1.07 (95% CI, 0.68-1.68).
“Our results showed that propofol sedation for gastrointestinal endoscopic procedures, whether simple or advanced, did not increase the cardiopulmonary adverse event rate when compared with traditional sedative agents,” the authors concluded.
In terms of the risk of developing any of the aforementioned complications, of the 20 relevant studies, 9 found propofol to be safer versus 6 that found traditional sedatives to be the better option, yielding an overall OR of 0.77 (95% CI, 0.56-1.07) for propofol. For the subanalysis regarding which type of clinician administered each sedative, 25 studies contained relevant data, of which 9 studies reported gastroenterologists administering sedatives, 5 studies reported endoscopy nurses administering sedatives under the supervision of the gastroenterologist, and 11 studies reported either an anesthesiologist, intensive care unit physician, or critical care physician administering sedatives.
“Gastroenterologist-directed sedation with propofol was noninferior to nongastroenterologist sedation,” Dr. Wadhwa and his coinvestigators wrote. “The risk of complications was similar to [that of traditional sedatives] both during simple and advanced endoscopic procedures.”
While the authors point to the sheer size of the study population as a huge strength of these results, they also note that because this is a study-level analysis rather than one conducted on an individual level, there is an inherent limitation to this study. Furthermore, variations from study to study in how propofol was administered to each patient may have caused heterogeneity with the findings of the meta-analysis. A large clinical trial would be the next logical step to affirm what this analysis has found.
“Because it may not be feasible to perform such a study, this meta-analysis should provide a rough idea of the possible associations,” the authors wrote. “However, the difference in complications between propofol and other agents might not be clinically relevant owing to the lack of any serious complications such as intubations or deaths in the studies used in this meta-analysis.”
No funding source was reported for this study. Dr. Wadhwa and his coauthors reported no relevant financial disclosures.
For doctors performing gastrointestinal endoscopic procedures, use of propofol as a sedative instead of the more commonly used drugs carries about the same risk of causing cardiopulmonary adverse events, according to a study published in the February issue of Clinical Gastroenterology and Hepatology (doi: 10.1016/j.cgh.2016.07.013).
“Because of its popularity, propofol is being used for both simple endoscopic procedures such as esophagogastroduodenoscopy and colonoscopy, and advanced endoscopic procedures [but] despite the widespread use of propofol, significant concerns remain regarding its safety profile,” according to the authors of the study, led by Vaibhav Wadhwa, MD, of Fairview Hospital in Cleveland.
The use of propofol as a sedative in gastrointestinal endoscopic procedures has increased in recent years, but because of an increasing number of advanced and, therefore, more complicated procedures being performed, the safety of sedatives has come into question because of their more prolonged use. Before use of propofol became prevalent, the more traditionally used sedative was a combination of benzodiazepine with an opioid. While still used today, this combination has seen a dramatic decline in usage because of its longer recovery time and lower rates of satisfaction among both patients and doctors, according to the authors. Combinations including midazolam, meperidine, pethidine, remifentanil, and fentanyl have also been used.
To compare the safety of propofol and a more traditional sedative combination, Dr. Wadhwa and his coauthors conducted a meta-analysis of published studies in the Medline (Ovid), EMBASE, and the Cochrane controlled trials registry databases. All searches were for research conducted through September of 2014, with the Medline database search starting in 1960, and the EMBASE and Cochrane searches starting in 1980, yielding a total of 2,117 studies eligible for inclusion.
Of those, 1,568 remained after duplicates were removed, then 136 were screened after removal of those deemed irrelevant or otherwise unsuitable. From those 136, 83 were excluded for various reasons – because they featured either ineligible populations, or were retrospective studies, single-arm studies, or conference abstracts – leaving 53 full-text articles to be evaluated for inclusion in the study. Of those, 27 were deemed eligible and were ultimately included.
“The primary outcomes measured were cardiopulmonary complications such as hypoxia, if oxygen saturation decreased to less than 90%; hypotension, if systolic blood pressure decreased to less than 90 mm Hg; arrhythmias, including bradycardia, supraventricular and ventricular arrhythmias, and ectopy,” Dr. Wadhwa and his coauthors wrote. “A subgroup analysis also was performed to assess studies in which sedation was directed by gastroenterologists and was compared with nongastroenterologists.” Apnea was not measured because of the lack of studies that assessed it qualitatively.
Pooled odds ratios were used to measure and compare results. The 27 included studies featured data on a total of 2,518 patients. Traditional sedatives were used on 1,194 of these subjects, while the remaining 1,324 received propofol. Regarding hypoxia, 26 of the 27 studies addressed this, of which 13 concluded that propofol was safer and 9 found that traditional sedatives were safer, with a pooled OR for propofol of 0.82 (95% confidence interval [CI] 0.63-1.07).
Twenty-five studies examined hypotension, of which 9 favored propofol and 10 favored traditional sedatives, for an OR of 0.92 (95% CI, 0.64-1.32). Of the 20 studies that included arrhythmia, 8 favored propofol and 7 favored traditional sedatives, for an OR of 1.07 (95% CI, 0.68-1.68).
“Our results showed that propofol sedation for gastrointestinal endoscopic procedures, whether simple or advanced, did not increase the cardiopulmonary adverse event rate when compared with traditional sedative agents,” the authors concluded.
In terms of the risk of developing any of the aforementioned complications, of the 20 relevant studies, 9 found propofol to be safer versus 6 that found traditional sedatives to be the better option, yielding an overall OR of 0.77 (95% CI, 0.56-1.07) for propofol. For the subanalysis regarding which type of clinician administered each sedative, 25 studies contained relevant data, of which 9 studies reported gastroenterologists administering sedatives, 5 studies reported endoscopy nurses administering sedatives under the supervision of the gastroenterologist, and 11 studies reported either an anesthesiologist, intensive care unit physician, or critical care physician administering sedatives.
“Gastroenterologist-directed sedation with propofol was noninferior to nongastroenterologist sedation,” Dr. Wadhwa and his coinvestigators wrote. “The risk of complications was similar to [that of traditional sedatives] both during simple and advanced endoscopic procedures.”
While the authors point to the sheer size of the study population as a huge strength of these results, they also note that because this is a study-level analysis rather than one conducted on an individual level, there is an inherent limitation to this study. Furthermore, variations from study to study in how propofol was administered to each patient may have caused heterogeneity with the findings of the meta-analysis. A large clinical trial would be the next logical step to affirm what this analysis has found.
“Because it may not be feasible to perform such a study, this meta-analysis should provide a rough idea of the possible associations,” the authors wrote. “However, the difference in complications between propofol and other agents might not be clinically relevant owing to the lack of any serious complications such as intubations or deaths in the studies used in this meta-analysis.”
No funding source was reported for this study. Dr. Wadhwa and his coauthors reported no relevant financial disclosures.
CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Key clinical point:
Major finding: Pooled odds ratio for propofol was 0.82 for hypoxia (95% CI, 0.63-1.07), 0.92 for hypotension (95% CI, 0.64-1.32), and 0.86 (95% CI, 0.56-1.34) for complication rate in advanced endoscopic procedures; subjects who received propofol were 39% less likely to have complications than were those who received traditional sedatives.
Data source: Retrospective meta-analysis of 27 studies involving 2,518 patients from 1966 through 2014.
Disclosures: The authors reported no relevant financial disclosures.