User login
How Does Cannabidiol Interact With Antiepileptic Drugs?
HOUSTON—Cannabidiol (CBD) interacts significantly with clobazam, rufinamide, topiramate, zonisamide, and eslicarbazepine, researchers said at the 70th Annual Meeting of the American Epilepsy Society. The study results underscore the importance of monitoring levels of antiepileptic drugs (AEDs) during treatment with CBD. “In the future, these data will need to be correlated with reported side effects or laboratory abnormalities to determine whether they are clinically significant,” said Jerzy P. Szaflarski, MD, PhD, Director of the University of Alabama at Birmingham Epilepsy Center.

Dr. Szaflarski and colleagues monitored serum AED levels during active titration of pharmaceutical grade CBD in patients with refractory epilepsy who were enrolled in an open-label safety study. The study was intended to investigate CBD as a potential adjunctive therapy. As part of the study protocol, researchers checked serum AED levels frequently to identify interactions between CBD and AEDs. Based on previous data and anecdotal observations, Dr. Szaflarski and colleagues hypothesized that they would find interactions between CBD and clobazam and valproate.
Participants received an initial CBD dose of 5 mg/kg/day. The dose was increased by 5 mg/kg/day every two weeks, provided that tolerability was maintained, to a maximum of 50 mg/kg/day. Baseline AED levels were drawn, and AEDs were required to have been at a stable dose for one month before enrollment. The researchers obtained AED levels at almost all study visits during dose titration and maintenance. AED doses were adjusted at the investigators’ discretion if an adverse effect, laboratory abnormality, or drug level change was considered related to a potential interaction between CBD and the AED. The researchers frequently adjusted doses of clobazam and valproate because of complaints of sedation and alteration in liver function tests, respectively. At the time of Dr. Szaflarski’s analysis, 81 participants were enrolled in the study (39 adults and 42 children). There were sufficient data to analyze potential interactions between CBD and 19 AEDs.
The researchers found increases in serum levels of topiramate, rufinamide, and desmethylclobazam (an active metabolite of clobazam), and a decrease in levels of clobazam, with increasing CBD dose in the pediatric and adult arms. In addition, they noted significant increases in serum levels of zonisamide and eslicarbazepine with increasing CBD dose in the adult arm. Dr. Szaflarski and colleagues observed no significant interactions between CBD and the other AEDs investigated, which included valproate, levetiracetam, lacosamide, and perampanel.
—Erik Greb
HOUSTON—Cannabidiol (CBD) interacts significantly with clobazam, rufinamide, topiramate, zonisamide, and eslicarbazepine, researchers said at the 70th Annual Meeting of the American Epilepsy Society. The study results underscore the importance of monitoring levels of antiepileptic drugs (AEDs) during treatment with CBD. “In the future, these data will need to be correlated with reported side effects or laboratory abnormalities to determine whether they are clinically significant,” said Jerzy P. Szaflarski, MD, PhD, Director of the University of Alabama at Birmingham Epilepsy Center.
Dr. Szaflarski and colleagues monitored serum AED levels during active titration of pharmaceutical grade CBD in patients with refractory epilepsy who were enrolled in an open-label safety study. The study was intended to investigate CBD as a potential adjunctive therapy. As part of the study protocol, researchers checked serum AED levels frequently to identify interactions between CBD and AEDs. Based on previous data and anecdotal observations, Dr. Szaflarski and colleagues hypothesized that they would find interactions between CBD and clobazam and valproate.
Participants received an initial CBD dose of 5 mg/kg/day. The dose was increased by 5 mg/kg/day every two weeks, provided that tolerability was maintained, to a maximum of 50 mg/kg/day. Baseline AED levels were drawn, and AEDs were required to have been at a stable dose for one month before enrollment. The researchers obtained AED levels at almost all study visits during dose titration and maintenance. AED doses were adjusted at the investigators’ discretion if an adverse effect, laboratory abnormality, or drug level change was considered related to a potential interaction between CBD and the AED. The researchers frequently adjusted doses of clobazam and valproate because of complaints of sedation and alteration in liver function tests, respectively. At the time of Dr. Szaflarski’s analysis, 81 participants were enrolled in the study (39 adults and 42 children). There were sufficient data to analyze potential interactions between CBD and 19 AEDs.
The researchers found increases in serum levels of topiramate, rufinamide, and desmethylclobazam (an active metabolite of clobazam), and a decrease in levels of clobazam, with increasing CBD dose in the pediatric and adult arms. In addition, they noted significant increases in serum levels of zonisamide and eslicarbazepine with increasing CBD dose in the adult arm. Dr. Szaflarski and colleagues observed no significant interactions between CBD and the other AEDs investigated, which included valproate, levetiracetam, lacosamide, and perampanel.
—Erik Greb
HOUSTON—Cannabidiol (CBD) interacts significantly with clobazam, rufinamide, topiramate, zonisamide, and eslicarbazepine, researchers said at the 70th Annual Meeting of the American Epilepsy Society. The study results underscore the importance of monitoring levels of antiepileptic drugs (AEDs) during treatment with CBD. “In the future, these data will need to be correlated with reported side effects or laboratory abnormalities to determine whether they are clinically significant,” said Jerzy P. Szaflarski, MD, PhD, Director of the University of Alabama at Birmingham Epilepsy Center.
Dr. Szaflarski and colleagues monitored serum AED levels during active titration of pharmaceutical grade CBD in patients with refractory epilepsy who were enrolled in an open-label safety study. The study was intended to investigate CBD as a potential adjunctive therapy. As part of the study protocol, researchers checked serum AED levels frequently to identify interactions between CBD and AEDs. Based on previous data and anecdotal observations, Dr. Szaflarski and colleagues hypothesized that they would find interactions between CBD and clobazam and valproate.
Participants received an initial CBD dose of 5 mg/kg/day. The dose was increased by 5 mg/kg/day every two weeks, provided that tolerability was maintained, to a maximum of 50 mg/kg/day. Baseline AED levels were drawn, and AEDs were required to have been at a stable dose for one month before enrollment. The researchers obtained AED levels at almost all study visits during dose titration and maintenance. AED doses were adjusted at the investigators’ discretion if an adverse effect, laboratory abnormality, or drug level change was considered related to a potential interaction between CBD and the AED. The researchers frequently adjusted doses of clobazam and valproate because of complaints of sedation and alteration in liver function tests, respectively. At the time of Dr. Szaflarski’s analysis, 81 participants were enrolled in the study (39 adults and 42 children). There were sufficient data to analyze potential interactions between CBD and 19 AEDs.
The researchers found increases in serum levels of topiramate, rufinamide, and desmethylclobazam (an active metabolite of clobazam), and a decrease in levels of clobazam, with increasing CBD dose in the pediatric and adult arms. In addition, they noted significant increases in serum levels of zonisamide and eslicarbazepine with increasing CBD dose in the adult arm. Dr. Szaflarski and colleagues observed no significant interactions between CBD and the other AEDs investigated, which included valproate, levetiracetam, lacosamide, and perampanel.
—Erik Greb

VIDEO: Protein-rich diet can help manage type 2 diabetes, NAFLD
Patients with type 2 diabetes should be put on diets rich in either animal or plant protein to reduce not only liver fat, but insulin resistance and hepatic necroinflammation markers as well, according to a study published in the February issue of Gastroenterology (doi: 10.1053/j.gastro.2016.10.007).
“High-protein diets have shown variable and sometimes even favorable effects on glucose metabolism and insulin sensitivity in people with type 2 diabetes and it is unclear which metabolic pathways are involved,” wrote the authors of the study, led by Mariya Markova, MD, of the German Institute of Human Nutrition Potsdam-Rehbrücke in Nuthetal, Germany.
SOURCE: American Gastroenterological Association
Obesity and insulin resistance have long been linked to liver fat, with excessive amounts of the latter causing nonalcoholic fatty liver disease (NAFLD), with a significant risk of nonalcoholic steatohepatitis (NASH) developing as well. Compounding this issue, at least in the United States, are widespread dietary and nutritional habits that promote consumption of animal protein, carbohydrates, and saturated fats. This “hypercaloric Western style diet,” as the authors call it, exacerbates the accumulation of fat deposits in the liver and complicates the health of patients across the country, regardless of weight.
“Remarkably, diets restricted in methionine were shown to prevent the development of insulin resistance and of the metabolic syndrome in animal models [so] the type of protein may elicit different metabolic responses depending on the amino acid composition,” Dr. Markova and her coinvestigators noted. “It is therefore hypothesized that high-plant-protein diets exert favorable effects on hepatic fat content and metabolic responses as compared to high intake of animal protein rich in BCAA [branched-chain amino acids] and methionine,” both of which can be found in suitably low levels via plant protein.
Dr. Markova and her team devised a prospective, randomized, open-label clinical trial involving 44 patients with type 2 diabetes and NAFLD, all of whom were recruited at the department of clinical nutrition of the German Institute of Human Nutrition Potsdam-Rehbrücke between June 2013 and March 2015. Subjects were randomized into one of two cohorts, each of which were assigned a diet rich in either animal protein (AP) or plant protein (PP) for a period of 6 weeks. Median body mass index in the AP cohort was 31.0 ± 0.8, and was 29.4 ± 1.0 in the PP cohort.
The AP cohort diet consisted mainly of meat and dairy products, while legumes constituted the bulk of the PP cohort diet. Both diets were isocaloric and had the same macronutrient makeup: 30% protein, 40% carbohydrates, and 30% fat. Seven subjects dropped out prior to completion of the study; of the 37 that remained all the way through – 19 in the AP cohort, 18 in the PP cohort – the age range was 49-78 years. Subjects maintained the same physical exercise regimens throughout the study that they had beforehand, and were asked not to alter them. Hemoglobin A1c levels ranged from 5.8% to 8.8% at baseline, and evaluations were carried out at fasting levels for each subject.
Patients in both cohorts saw significant decreases in intrahepatic fat content by the end of the trial period. Those in the AP cohort saw decreases of 48.0% (P = .0002), while those in the PP cohort saw a decrease of 35.7% (P = .001). Perhaps most importantly, the reductions in both cohorts were not correlated to body weight. In addition, levels of fibroblast growth factor 21 (FGF21), which has been shown to be a predictive marker of NAFLD, decreased by nearly 50% for both AP and PP cohorts (P less than .0002 for both).
“Despite the elevated intake and postprandial uptake of methionine and BCAA in the AP group, there was no indication of negative effects of these components,” the authors stated in the study. “The origin of protein – animal or plant – did not play a major role. Both high-protein diets unexpectedly induced strong reductions of FGF21, which was associated with metabolic improvements and the decrease of IHL.”
Despite these findings, however, the 6-week time span used here is not sufficient to determine just how viable this diet may be in the long term, according to the authors. Further studies will be needed, and will need to take place over longer periods of time, to “show the durability of the responses and eventual adverse effects of the diets.” Furthermore, different age groups must be examined to find out if the benefits observed by Dr. Markova and her coinvestigators were somehow related to the age of these subjects.
The study was funded by grants from German Federal Ministry of Food and Agriculture and German Center for Diabetes Research. Dr. Markova and her coauthors did not report any financial disclosures.
Human studies to assess the effects of isocaloric macronutrient substitution are fraught with difficulty. If one macronutrient is increased, what happens to the others? If you observe an effect, is it the phenomenon you were seeking due to the macronutrient you altered, or an epiphenomenon due to changes in the others?
Markova et al. attempted to study a 6-week “isocaloric” increase of animal vs. plant protein (from 17% to 30% of calories as protein). However, a decrease of percent fat from 41% to 30%, and a reduction in carbohydrate from 42% to 40% occurred commensurately. This brings up three concerns. First, despite the diet’s being “isocaloric,” weight and body mass index decreased by 2 kg and 0.8 kg/m2, respectively. Reductions in intrahepatic, visceral, and subcutaneous fat, and an increase in lean body mass were noted. So was the diet isocaloric? Protein reduces plasma ghrelin levels and is more satiating. Furthermore, metabolism of protein to ATP is inefficient compared to that of carbohydrate or fat. The authors say only that calories were “unrestricted.” These issues do not engender “isocaloric” confidence.

Lastly, the type of carbohydrate was not controlled for. Fructose is significantly more lipogenic than glucose. Yet they were lumped together as “carbohydrate,” and were uncontrolled. So what macronutrient really caused the reduction in liver fat? These methodological issues detract from the author’s message, and this study must be considered preliminary.
Robert H. Lustig, MD, MSL, is in the division of pediatric endocrinology, UCSF Benioff Children’s Hospital, San Francisco; member, UCSF Institute for Health Policy Studies. Dr. Lustig declared no conflicts of interest.
Human studies to assess the effects of isocaloric macronutrient substitution are fraught with difficulty. If one macronutrient is increased, what happens to the others? If you observe an effect, is it the phenomenon you were seeking due to the macronutrient you altered, or an epiphenomenon due to changes in the others?
Markova et al. attempted to study a 6-week “isocaloric” increase of animal vs. plant protein (from 17% to 30% of calories as protein). However, a decrease of percent fat from 41% to 30%, and a reduction in carbohydrate from 42% to 40% occurred commensurately. This brings up three concerns. First, despite the diet’s being “isocaloric,” weight and body mass index decreased by 2 kg and 0.8 kg/m2, respectively. Reductions in intrahepatic, visceral, and subcutaneous fat, and an increase in lean body mass were noted. So was the diet isocaloric? Protein reduces plasma ghrelin levels and is more satiating. Furthermore, metabolism of protein to ATP is inefficient compared to that of carbohydrate or fat. The authors say only that calories were “unrestricted.” These issues do not engender “isocaloric” confidence.
Lastly, the type of carbohydrate was not controlled for. Fructose is significantly more lipogenic than glucose. Yet they were lumped together as “carbohydrate,” and were uncontrolled. So what macronutrient really caused the reduction in liver fat? These methodological issues detract from the author’s message, and this study must be considered preliminary.
Robert H. Lustig, MD, MSL, is in the division of pediatric endocrinology, UCSF Benioff Children’s Hospital, San Francisco; member, UCSF Institute for Health Policy Studies. Dr. Lustig declared no conflicts of interest.
Human studies to assess the effects of isocaloric macronutrient substitution are fraught with difficulty. If one macronutrient is increased, what happens to the others? If you observe an effect, is it the phenomenon you were seeking due to the macronutrient you altered, or an epiphenomenon due to changes in the others?
Markova et al. attempted to study a 6-week “isocaloric” increase of animal vs. plant protein (from 17% to 30% of calories as protein). However, a decrease of percent fat from 41% to 30%, and a reduction in carbohydrate from 42% to 40% occurred commensurately. This brings up three concerns. First, despite the diet’s being “isocaloric,” weight and body mass index decreased by 2 kg and 0.8 kg/m2, respectively. Reductions in intrahepatic, visceral, and subcutaneous fat, and an increase in lean body mass were noted. So was the diet isocaloric? Protein reduces plasma ghrelin levels and is more satiating. Furthermore, metabolism of protein to ATP is inefficient compared to that of carbohydrate or fat. The authors say only that calories were “unrestricted.” These issues do not engender “isocaloric” confidence.
Lastly, the type of carbohydrate was not controlled for. Fructose is significantly more lipogenic than glucose. Yet they were lumped together as “carbohydrate,” and were uncontrolled. So what macronutrient really caused the reduction in liver fat? These methodological issues detract from the author’s message, and this study must be considered preliminary.
Robert H. Lustig, MD, MSL, is in the division of pediatric endocrinology, UCSF Benioff Children’s Hospital, San Francisco; member, UCSF Institute for Health Policy Studies. Dr. Lustig declared no conflicts of interest.
Patients with type 2 diabetes should be put on diets rich in either animal or plant protein to reduce not only liver fat, but insulin resistance and hepatic necroinflammation markers as well, according to a study published in the February issue of Gastroenterology (doi: 10.1053/j.gastro.2016.10.007).
“High-protein diets have shown variable and sometimes even favorable effects on glucose metabolism and insulin sensitivity in people with type 2 diabetes and it is unclear which metabolic pathways are involved,” wrote the authors of the study, led by Mariya Markova, MD, of the German Institute of Human Nutrition Potsdam-Rehbrücke in Nuthetal, Germany.
SOURCE: American Gastroenterological Association
Obesity and insulin resistance have long been linked to liver fat, with excessive amounts of the latter causing nonalcoholic fatty liver disease (NAFLD), with a significant risk of nonalcoholic steatohepatitis (NASH) developing as well. Compounding this issue, at least in the United States, are widespread dietary and nutritional habits that promote consumption of animal protein, carbohydrates, and saturated fats. This “hypercaloric Western style diet,” as the authors call it, exacerbates the accumulation of fat deposits in the liver and complicates the health of patients across the country, regardless of weight.
“Remarkably, diets restricted in methionine were shown to prevent the development of insulin resistance and of the metabolic syndrome in animal models [so] the type of protein may elicit different metabolic responses depending on the amino acid composition,” Dr. Markova and her coinvestigators noted. “It is therefore hypothesized that high-plant-protein diets exert favorable effects on hepatic fat content and metabolic responses as compared to high intake of animal protein rich in BCAA [branched-chain amino acids] and methionine,” both of which can be found in suitably low levels via plant protein.
Dr. Markova and her team devised a prospective, randomized, open-label clinical trial involving 44 patients with type 2 diabetes and NAFLD, all of whom were recruited at the department of clinical nutrition of the German Institute of Human Nutrition Potsdam-Rehbrücke between June 2013 and March 2015. Subjects were randomized into one of two cohorts, each of which were assigned a diet rich in either animal protein (AP) or plant protein (PP) for a period of 6 weeks. Median body mass index in the AP cohort was 31.0 ± 0.8, and was 29.4 ± 1.0 in the PP cohort.
The AP cohort diet consisted mainly of meat and dairy products, while legumes constituted the bulk of the PP cohort diet. Both diets were isocaloric and had the same macronutrient makeup: 30% protein, 40% carbohydrates, and 30% fat. Seven subjects dropped out prior to completion of the study; of the 37 that remained all the way through – 19 in the AP cohort, 18 in the PP cohort – the age range was 49-78 years. Subjects maintained the same physical exercise regimens throughout the study that they had beforehand, and were asked not to alter them. Hemoglobin A1c levels ranged from 5.8% to 8.8% at baseline, and evaluations were carried out at fasting levels for each subject.
Patients in both cohorts saw significant decreases in intrahepatic fat content by the end of the trial period. Those in the AP cohort saw decreases of 48.0% (P = .0002), while those in the PP cohort saw a decrease of 35.7% (P = .001). Perhaps most importantly, the reductions in both cohorts were not correlated to body weight. In addition, levels of fibroblast growth factor 21 (FGF21), which has been shown to be a predictive marker of NAFLD, decreased by nearly 50% for both AP and PP cohorts (P less than .0002 for both).
“Despite the elevated intake and postprandial uptake of methionine and BCAA in the AP group, there was no indication of negative effects of these components,” the authors stated in the study. “The origin of protein – animal or plant – did not play a major role. Both high-protein diets unexpectedly induced strong reductions of FGF21, which was associated with metabolic improvements and the decrease of IHL.”
Despite these findings, however, the 6-week time span used here is not sufficient to determine just how viable this diet may be in the long term, according to the authors. Further studies will be needed, and will need to take place over longer periods of time, to “show the durability of the responses and eventual adverse effects of the diets.” Furthermore, different age groups must be examined to find out if the benefits observed by Dr. Markova and her coinvestigators were somehow related to the age of these subjects.
The study was funded by grants from German Federal Ministry of Food and Agriculture and German Center for Diabetes Research. Dr. Markova and her coauthors did not report any financial disclosures.
Patients with type 2 diabetes should be put on diets rich in either animal or plant protein to reduce not only liver fat, but insulin resistance and hepatic necroinflammation markers as well, according to a study published in the February issue of Gastroenterology (doi: 10.1053/j.gastro.2016.10.007).
“High-protein diets have shown variable and sometimes even favorable effects on glucose metabolism and insulin sensitivity in people with type 2 diabetes and it is unclear which metabolic pathways are involved,” wrote the authors of the study, led by Mariya Markova, MD, of the German Institute of Human Nutrition Potsdam-Rehbrücke in Nuthetal, Germany.
SOURCE: American Gastroenterological Association
Obesity and insulin resistance have long been linked to liver fat, with excessive amounts of the latter causing nonalcoholic fatty liver disease (NAFLD), with a significant risk of nonalcoholic steatohepatitis (NASH) developing as well. Compounding this issue, at least in the United States, are widespread dietary and nutritional habits that promote consumption of animal protein, carbohydrates, and saturated fats. This “hypercaloric Western style diet,” as the authors call it, exacerbates the accumulation of fat deposits in the liver and complicates the health of patients across the country, regardless of weight.
“Remarkably, diets restricted in methionine were shown to prevent the development of insulin resistance and of the metabolic syndrome in animal models [so] the type of protein may elicit different metabolic responses depending on the amino acid composition,” Dr. Markova and her coinvestigators noted. “It is therefore hypothesized that high-plant-protein diets exert favorable effects on hepatic fat content and metabolic responses as compared to high intake of animal protein rich in BCAA [branched-chain amino acids] and methionine,” both of which can be found in suitably low levels via plant protein.
Dr. Markova and her team devised a prospective, randomized, open-label clinical trial involving 44 patients with type 2 diabetes and NAFLD, all of whom were recruited at the department of clinical nutrition of the German Institute of Human Nutrition Potsdam-Rehbrücke between June 2013 and March 2015. Subjects were randomized into one of two cohorts, each of which were assigned a diet rich in either animal protein (AP) or plant protein (PP) for a period of 6 weeks. Median body mass index in the AP cohort was 31.0 ± 0.8, and was 29.4 ± 1.0 in the PP cohort.
The AP cohort diet consisted mainly of meat and dairy products, while legumes constituted the bulk of the PP cohort diet. Both diets were isocaloric and had the same macronutrient makeup: 30% protein, 40% carbohydrates, and 30% fat. Seven subjects dropped out prior to completion of the study; of the 37 that remained all the way through – 19 in the AP cohort, 18 in the PP cohort – the age range was 49-78 years. Subjects maintained the same physical exercise regimens throughout the study that they had beforehand, and were asked not to alter them. Hemoglobin A1c levels ranged from 5.8% to 8.8% at baseline, and evaluations were carried out at fasting levels for each subject.
Patients in both cohorts saw significant decreases in intrahepatic fat content by the end of the trial period. Those in the AP cohort saw decreases of 48.0% (P = .0002), while those in the PP cohort saw a decrease of 35.7% (P = .001). Perhaps most importantly, the reductions in both cohorts were not correlated to body weight. In addition, levels of fibroblast growth factor 21 (FGF21), which has been shown to be a predictive marker of NAFLD, decreased by nearly 50% for both AP and PP cohorts (P less than .0002 for both).
“Despite the elevated intake and postprandial uptake of methionine and BCAA in the AP group, there was no indication of negative effects of these components,” the authors stated in the study. “The origin of protein – animal or plant – did not play a major role. Both high-protein diets unexpectedly induced strong reductions of FGF21, which was associated with metabolic improvements and the decrease of IHL.”
Despite these findings, however, the 6-week time span used here is not sufficient to determine just how viable this diet may be in the long term, according to the authors. Further studies will be needed, and will need to take place over longer periods of time, to “show the durability of the responses and eventual adverse effects of the diets.” Furthermore, different age groups must be examined to find out if the benefits observed by Dr. Markova and her coinvestigators were somehow related to the age of these subjects.
The study was funded by grants from German Federal Ministry of Food and Agriculture and German Center for Diabetes Research. Dr. Markova and her coauthors did not report any financial disclosures.
FROM GASTROENTEROLOGY
Key clinical point:
Major finding: Animal- and plant-protein diets reduced liver fat for type 2 diabetes patients by 36%-48% over the course of 6 months (P = .0002 and P = .001, respectively).
Data source: Prospective study of 37 type 2 diabetes patients from June 2013 to March 2015.
Disclosures: The German Federal Ministry of Food and Agriculture and German Center for Diabetes Research supported the study. The authors did not report any financial disclosures.

Papillary Transitional Cell Bladder Carcinoma and Systematized Epidermal Nevus Syndrome
Epidermal nevi can occur in isolation or in association with internal abnormalities. Epidermal nevus syndrome (ENS) is a heterogeneous group of neurocutaneous disorders characterized by mosaicism and epidermal nevi found in association with various systemic abnormalities.1-4 There are many possible associated systemic findings, including abnormalities of the central nervous, musculoskeletal, renal, and hematologic systems. Epidermal nevi have been associated with internal malignancies. We present the case of a patient with epidermal nevi associated with papillary transitional cell bladder carcinoma. According to a PubMed search of articles indexed for MEDLINE using the search terms transitional cell bladder carcinoma and epidermal nevus, there have only been 4 other cases of transitional cell bladder carcinoma and ENS reported in the literature,5-8 2 of which were reports of papillary transitional cell bladder carcinoma.5,6
Case Report
A 29-year-old woman presented to our clinic with a rash that had been present since 3 years of age. The emergency department consulted dermatology for evaluation of what was believed to be contact dermatitis; however, upon questioning the patient, it was revealed that the rash was chronic and persistent.
The rash was nonpruritic and was located on the face, hands (Figure 1), chest, buttocks, thighs, legs, and back (Figure 2). Although asymptomatic, the appearance of the skin caused the patient some emotional distress. As a child she had been evaluated by a dermatologist and a biopsy was performed, but she did not recall the results or have any records. She had been prescribed an oral medication by the dermatologist, but treatment was terminated early due to nausea. The skin lesions did not improve with the short course of treatment.
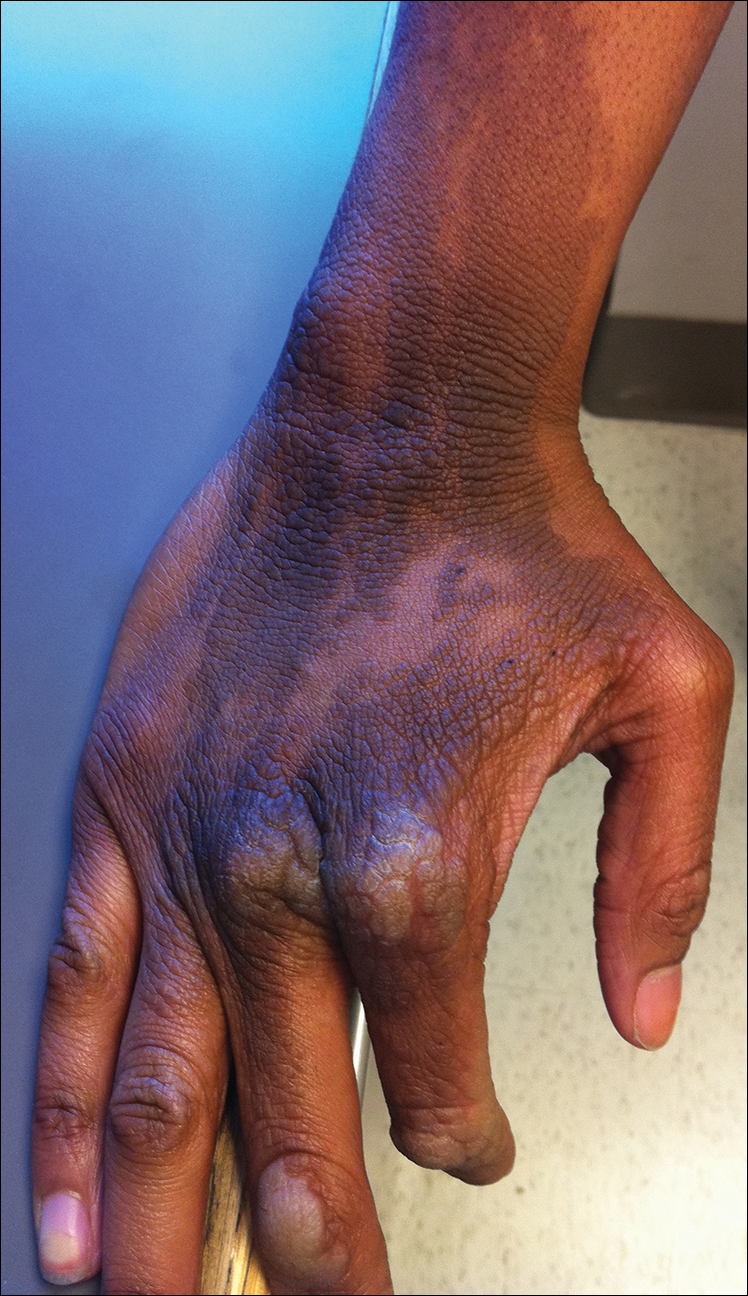
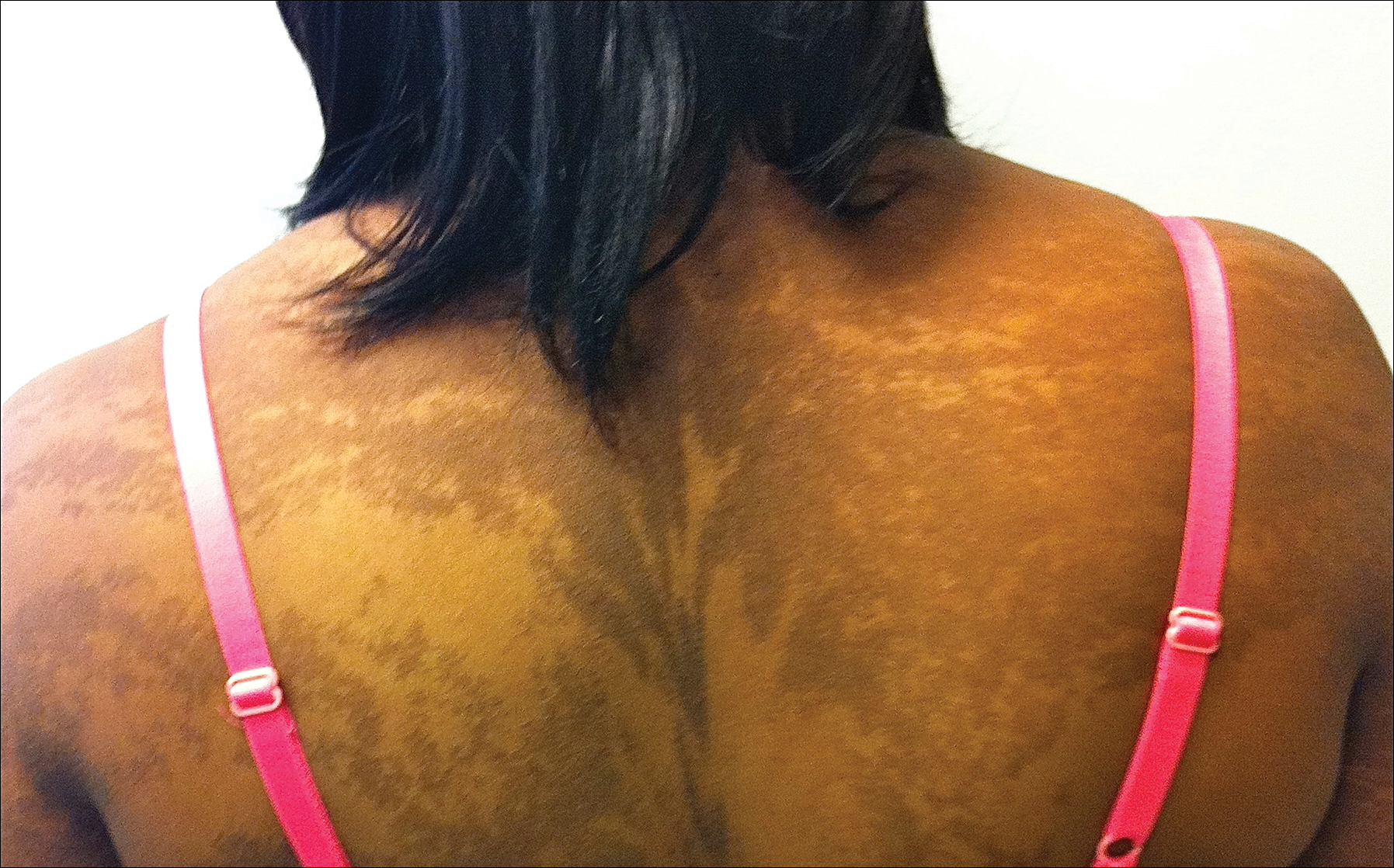
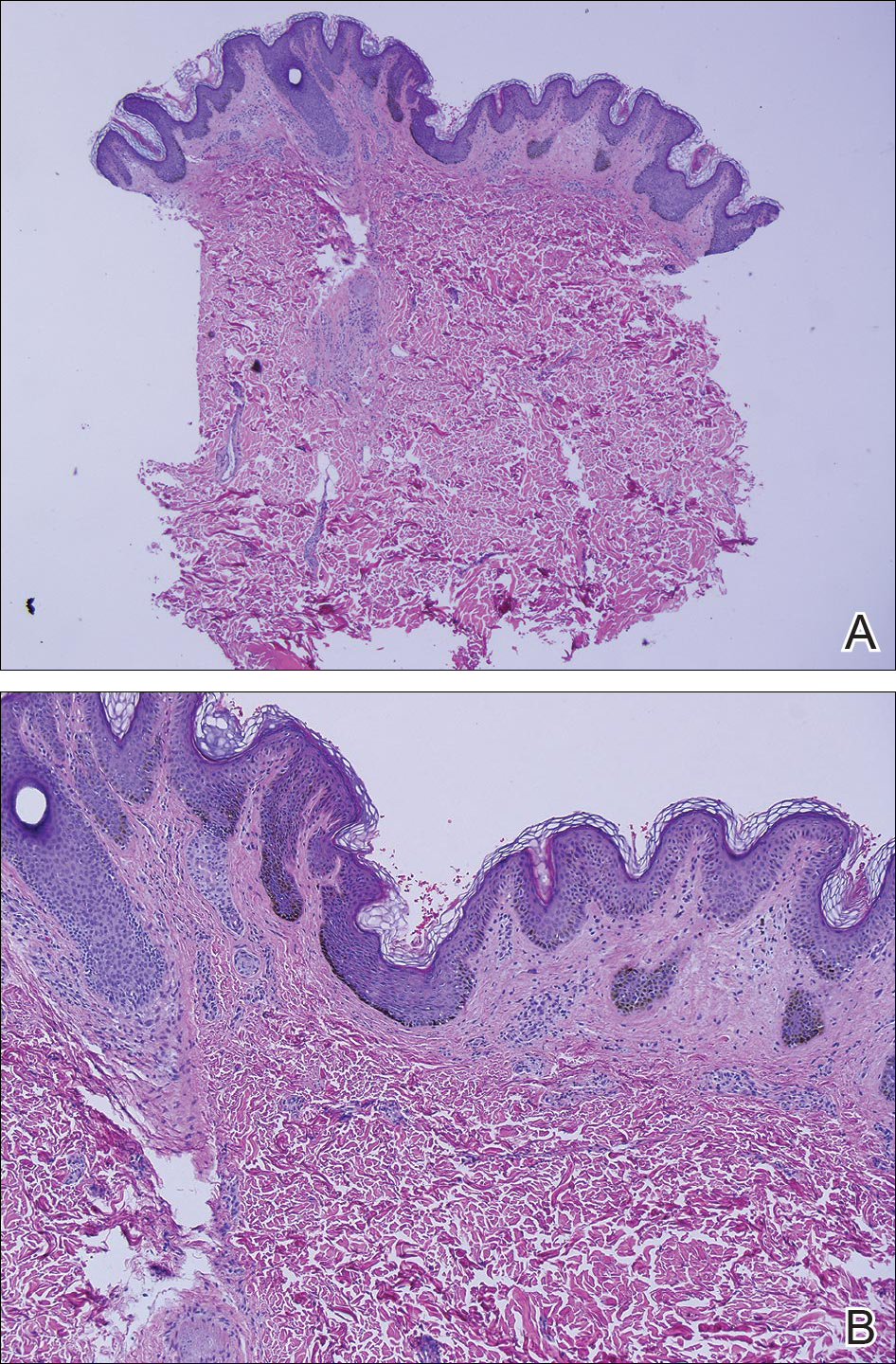
Eighteen months prior to presentation to our clinic, the patient was discovered to have hematuria on routine examination by her primary care physician. At that time, the patient underwent a workup for hematuria and a mass was discovered in the bladder via cystoscopy. A diagnosis of low-grade papillary transitional cell bladder carcinoma was made, and she underwent a partial cystectomy. No radiation or chemotherapy was required. The remainder of her medical history was only remarkable for asthma, which was well controlled with albuterol. On examination, generalized, hyperpigmented, reticulated patches, macules, and hyperpigmented verrucous plaques were distributed along the Blaschko lines, sparing the face. No limb abnormalities or dental or nail abnormalities were noted. Examination of the axillary and cervical lymph nodes was unremarkable, and no neurological abnormalities were noted. A 3-mm punch biopsy of the mid upper back was performed. Histopathology revealed papillomatous, nonorganoid, nonepidermolytic hyperplasia of the epidermis with elongated rete ridges (Figure 3), which was diagnosed as a nonorganoid nonepidermolytic epidermal nevus.
Comment
Epidermal nevus syndrome is a group of disorders characterized by both local or systematized epidermal nevi and systemic findings. Solomon et al4 first coined the term epidermal nevus syndrome more than 40 years ago; however, since then there has been confusion about how to define ENS. Epidermal nevus syndrome has been considered an umbrella term that includes more specific syndromes involving epidermal nevi, such as Proteus syndrome and Schimmelpenning syndrome; conversely, it also has been considered a term for those who do not meet the criteria for more specific syndromes.1,9 Happle1 discussed that the genetic variations found in ENS warrant recognition. Simply put, ENS is a heterogeneous group of syndromes that are similar in that they involve epidermal nevi and internal abnormalities but are genetically distinct. The list of definitive ENSs, as suggested by Happle1 and others, will likely continue to grow.3,5
The exact pathomechanism of ENS is unknown, but the clinical presentation most likely represents a lethal disorder mitigated by mosaicism.2,9 Gene defects vary depending on the specific ENS. For instance, the phosphatase and tensin homolog gene, PTEN, mutations have been associated with type 2 segmental Cowden disease. Fibroblast growth factor receptor 3, FGFR3, mutations have been linked to Garcia-Hafner-Happle syndrome.3FGFR3 mutations have been found in nonepidermolytic epidermal nevi, and some suggest that the majority of epidermal nevi exhibit mutations in FGFR3.5,10,11 On the other hand, other gene defects have not been elucidated, such as in Schimmelpenning syndrome.3
Clinically, ENS may involve nonepidermolytic verrucous nevi, sebaceous nevi, organoid nevi, linear Cowden nevi, and woolly hair nevi. Lesions may be flesh-colored, pink, yellow, or hyperpigmented plaques in a blaschkoid distribution and may be localized or systematized. Nevi typically are present at birth or develop within the first year of life.9,12,13 Other cutaneous findings may be noted apart from epidermal nevi, including melanocytic nevi, aplasia cutis congenita, and hemangiomas.13,14
Extracutaneous findings include central nervous system, skeletal, ocular, cardiac, and genitourinary defects, which are often observed in these patients.3,9,13,14 Central nervous system findings are seen in 50% to 70% of cases, with seizures and mental retardation among the most common.13-15 Genitourinary abnormalities associated with epidermal nevi, including horseshoe kidney, cystic kidney, duplicated collecting system, testicular and paratesticular tumors, and hypospadias have been documented in the literature.16 Our patient had a history of papillary transitional cell bladder carcinoma, which is rare for a patient younger than 30 years. The overall median age of diagnosis of bladder cancer is 65 years, and it is more common in men than in women.17 Transitional cell carcinomas account for approximately 90% of all bladder cancers in the United States. Other common types of bladder cancer include squamous cell carcinoma, adenocarcinoma, and rhabdomyosarcoma.16 Typically, transitional cell carcinoma is associated with smoking, exposure to aniline dyes, cyclophosphamide, and living in industrialized areas.16,17 Individuals who work with textiles, dyes, leather, tires, rubber, and/or petroleum; painters; truck drivers; drill press operators; and hairdressers are at an increased risk for development of bladder cancer.16
Interestingly, it has been shown in some studies that papillary transitional cell bladder carcinoma frequently is associated with FGFR3 mutations, which may be the missing link in the rare finding of papillary transitional cell bladder carcinoma and epidermal nevi.5,18,19 In addition, PTEN mutations also have been identified in low-grade papillary transitional cell carcinomas of the bladder, another gene linked to an ENS with type 2 segmental Cowden disease.3,20
Histopathologically, epidermal nevi have 10 different descriptions. Our patient had a nonorganoid nonepidermolytic epidermal nevus characterized by hyperkeratosis, acanthosis, papillomatosis, and elongated rete ridges. Focal acantholysis and epidermolytic hyperkeratosis also is seen in some epidermal nevi but was not seen in this case.9,21
Simple epidermal nevi occur in approximately 1 in 1000 newborns; however, when a child presents with multiple or systematized epidermal nevi, investigation should be undertaken for other possible associations.13,14 Of note, there have been several cases of squamous cell, verrucous, basal cell, and adnexal carcinomas arising in linear epidermal nevi.22-24
Epidermal nevi can be difficult to treat. Some patients are troubled by the appearance of these nevi, especially those with systematized disease. Unfortunately, for patients with multiple nevi or systematized disease, there are no consistently effective treatment options; however, there are case reports25,26 in the literature citing improvement or cure of epidermal nevi with full-thickness excision, continuous and pulsed CO2 laser, pulsed dye laser, and erbium-doped YAG laser.25 Other therapies that have been purported to help improve epidermal nevi are topical and oral retinoids, corticosteroids, topical 5-fluorouracil, anthralin, and podophyllin.26
Conclusion
Transitional cell bladder carcinoma is rare in patients in the third decade of life and younger. Given the age of our patient and her concomitant lack of risk factors, such as older age, history of smoking, and exposure to certain chemicals (eg, aniline dyes) and medications (eg, cyclophosphamide), it is more likely that the finding of papillary transitional cell bladder carcinoma and ENS are related. A clear genetic link between ENS and transitional cell papillary bladder carcinoma has yet to be elucidated, but the FGFR3 gene is promising.
- Happle R. What is a nevus? a proposed definition of a common medical term. Dermatology. 1995;191:1-5.
- Gonzalez ME, Jabbari A, Tlougan BE, et al. Epidermal nevus. Dermatol Online J. 2010;16:12.
- Happle R. The group of epidermal nevus syndromes. part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22.
- Solomon LM, Fretzin DF, Dewald RL. The epidermal nevus syndrome. Arch Dermatol. 1968;97:273-285.
- Flosadottir E, Bjarnason B. A non-epidermolytic epidermal nevus of a soft, papillomatous type with transitional cell cancer of the bladder: a case report and review of non-cutaneous cancers associated with epidermal naevi. Acta Derm Venerol. 2008;88:173-175.
- Rosenthal D, Fretzin DF. Epidermal nevus syndrome: report of association with transitional cell carcinoma of the bladder. Pediatr Dermatol. 1986;3:455-458.
- Garcia de Jalon A, Azua-Romea J, Trivez MA, et al. Epidermal naevus syndrome (Solomon’s syndrome) associated with bladder cancer in a 20-year-old female. Scand J Urol Nephrol. 2004;38:85-87.
- Rongioletti F, Rebora A. Epidermal nevus with transitional cell carcinomas of the urinary tract. J Am Acad Dermatol. 1991;25:856-858.
- Moss C. Mosacism and linear lesions. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012:943-962.
- Hafner C, van Oers JM, Vogt T, et al. Mosaicisim of activating FGFR3 mutations in human skin causes epidermal nevi. J Clin Invest. 2006;116:2201-2207.
- Bygum A, Fagerberg CR, Clemmensen OJ, et al. Systemic epidermal nevus with involvement of the oral mucosa due to FGFR3 mutation. BMC Med Genet. 2011;12:79.
- Happle R. Linear Cowden nevus: a new distinct epidermal nevus. Eur J Dermatol. 2007;17:133-136.
- Vujevich JJ, Mancini AJ. The epidermal nevus syndromes: multisystem disorders. J Am Acad Dermatol. 2004;50:957-961.
- Solomon L, Esterly N. Epidermal and other congenital organoid nevi. Curr Probl Pediatr. 1975;6:1-56.
- Grebe TA, Rimsa ME, Richter SF, et al. Further delineation of the epidermal nevus syndrome: two cases with new findings and literature review. Am J Med Genet. 1993;47:24-30.
- Lamm DL, Torti FM. Bladder cancer, 1996. Ca Cancer J Clin. 1996;46:93-112.
- Metts MC, Metts JC, Milito SJ, et al. Bladder cancer: a review of diagnosis and management. J Natl Med Assoc. 2000;92:285-294.
- Kimura T, Suzuki H, Ohashi T, et al. The incidence of thanatophoric dysplasia mutations in FGFR3 gene is higher in low-grade or superficial bladder carcinomas. Cancer. 2001;92:2555-2561.
- Cappellen D, DeOliveira C, Ricol D, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18-20.
- Knowles MA, Platt FM, Ross RL, et al. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 2009;28:305-316.
- Luzar B, Calonje E, Bastian B. Tumors of the surface epithelium. In: Calonje JE, Breen T, McKee PH, eds. McKee’s Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012:1076-1149.
- Masood Q, Narayan D. Squamous cell carcinoma in a linear epidermal nevus. J Plast Reconstr Aesthet Surg. 2009;62:693-694.
- Cramer SF, Mandel MA, Hauler R, et al. Squamous cell carcinoma arising in a linear epidermal nevus. Arch Dermatol. 1981;117:222-224.
- Affleck AG, Leach IJ, Varma S. Two squamous cell carcinomas arising in a linear epidermal nevus in a 28-year-old female. Clin Exp Dermatol. 2005;30:382-384.
- Alam M, Arndt KA. A method for pulsed carbon dioxide laser treatment of epidermal nevi. J Am Acad Dermatol. 2002;46:554-556.
- Requena L, Requena C, Cockerell CJ. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012:1809-1810.
Epidermal nevi can occur in isolation or in association with internal abnormalities. Epidermal nevus syndrome (ENS) is a heterogeneous group of neurocutaneous disorders characterized by mosaicism and epidermal nevi found in association with various systemic abnormalities.1-4 There are many possible associated systemic findings, including abnormalities of the central nervous, musculoskeletal, renal, and hematologic systems. Epidermal nevi have been associated with internal malignancies. We present the case of a patient with epidermal nevi associated with papillary transitional cell bladder carcinoma. According to a PubMed search of articles indexed for MEDLINE using the search terms transitional cell bladder carcinoma and epidermal nevus, there have only been 4 other cases of transitional cell bladder carcinoma and ENS reported in the literature,5-8 2 of which were reports of papillary transitional cell bladder carcinoma.5,6
Case Report
A 29-year-old woman presented to our clinic with a rash that had been present since 3 years of age. The emergency department consulted dermatology for evaluation of what was believed to be contact dermatitis; however, upon questioning the patient, it was revealed that the rash was chronic and persistent.
The rash was nonpruritic and was located on the face, hands (Figure 1), chest, buttocks, thighs, legs, and back (Figure 2). Although asymptomatic, the appearance of the skin caused the patient some emotional distress. As a child she had been evaluated by a dermatologist and a biopsy was performed, but she did not recall the results or have any records. She had been prescribed an oral medication by the dermatologist, but treatment was terminated early due to nausea. The skin lesions did not improve with the short course of treatment.
Eighteen months prior to presentation to our clinic, the patient was discovered to have hematuria on routine examination by her primary care physician. At that time, the patient underwent a workup for hematuria and a mass was discovered in the bladder via cystoscopy. A diagnosis of low-grade papillary transitional cell bladder carcinoma was made, and she underwent a partial cystectomy. No radiation or chemotherapy was required. The remainder of her medical history was only remarkable for asthma, which was well controlled with albuterol. On examination, generalized, hyperpigmented, reticulated patches, macules, and hyperpigmented verrucous plaques were distributed along the Blaschko lines, sparing the face. No limb abnormalities or dental or nail abnormalities were noted. Examination of the axillary and cervical lymph nodes was unremarkable, and no neurological abnormalities were noted. A 3-mm punch biopsy of the mid upper back was performed. Histopathology revealed papillomatous, nonorganoid, nonepidermolytic hyperplasia of the epidermis with elongated rete ridges (Figure 3), which was diagnosed as a nonorganoid nonepidermolytic epidermal nevus.
Comment
Epidermal nevus syndrome is a group of disorders characterized by both local or systematized epidermal nevi and systemic findings. Solomon et al4 first coined the term epidermal nevus syndrome more than 40 years ago; however, since then there has been confusion about how to define ENS. Epidermal nevus syndrome has been considered an umbrella term that includes more specific syndromes involving epidermal nevi, such as Proteus syndrome and Schimmelpenning syndrome; conversely, it also has been considered a term for those who do not meet the criteria for more specific syndromes.1,9 Happle1 discussed that the genetic variations found in ENS warrant recognition. Simply put, ENS is a heterogeneous group of syndromes that are similar in that they involve epidermal nevi and internal abnormalities but are genetically distinct. The list of definitive ENSs, as suggested by Happle1 and others, will likely continue to grow.3,5
The exact pathomechanism of ENS is unknown, but the clinical presentation most likely represents a lethal disorder mitigated by mosaicism.2,9 Gene defects vary depending on the specific ENS. For instance, the phosphatase and tensin homolog gene, PTEN, mutations have been associated with type 2 segmental Cowden disease. Fibroblast growth factor receptor 3, FGFR3, mutations have been linked to Garcia-Hafner-Happle syndrome.3FGFR3 mutations have been found in nonepidermolytic epidermal nevi, and some suggest that the majority of epidermal nevi exhibit mutations in FGFR3.5,10,11 On the other hand, other gene defects have not been elucidated, such as in Schimmelpenning syndrome.3
Clinically, ENS may involve nonepidermolytic verrucous nevi, sebaceous nevi, organoid nevi, linear Cowden nevi, and woolly hair nevi. Lesions may be flesh-colored, pink, yellow, or hyperpigmented plaques in a blaschkoid distribution and may be localized or systematized. Nevi typically are present at birth or develop within the first year of life.9,12,13 Other cutaneous findings may be noted apart from epidermal nevi, including melanocytic nevi, aplasia cutis congenita, and hemangiomas.13,14
Extracutaneous findings include central nervous system, skeletal, ocular, cardiac, and genitourinary defects, which are often observed in these patients.3,9,13,14 Central nervous system findings are seen in 50% to 70% of cases, with seizures and mental retardation among the most common.13-15 Genitourinary abnormalities associated with epidermal nevi, including horseshoe kidney, cystic kidney, duplicated collecting system, testicular and paratesticular tumors, and hypospadias have been documented in the literature.16 Our patient had a history of papillary transitional cell bladder carcinoma, which is rare for a patient younger than 30 years. The overall median age of diagnosis of bladder cancer is 65 years, and it is more common in men than in women.17 Transitional cell carcinomas account for approximately 90% of all bladder cancers in the United States. Other common types of bladder cancer include squamous cell carcinoma, adenocarcinoma, and rhabdomyosarcoma.16 Typically, transitional cell carcinoma is associated with smoking, exposure to aniline dyes, cyclophosphamide, and living in industrialized areas.16,17 Individuals who work with textiles, dyes, leather, tires, rubber, and/or petroleum; painters; truck drivers; drill press operators; and hairdressers are at an increased risk for development of bladder cancer.16
Interestingly, it has been shown in some studies that papillary transitional cell bladder carcinoma frequently is associated with FGFR3 mutations, which may be the missing link in the rare finding of papillary transitional cell bladder carcinoma and epidermal nevi.5,18,19 In addition, PTEN mutations also have been identified in low-grade papillary transitional cell carcinomas of the bladder, another gene linked to an ENS with type 2 segmental Cowden disease.3,20
Histopathologically, epidermal nevi have 10 different descriptions. Our patient had a nonorganoid nonepidermolytic epidermal nevus characterized by hyperkeratosis, acanthosis, papillomatosis, and elongated rete ridges. Focal acantholysis and epidermolytic hyperkeratosis also is seen in some epidermal nevi but was not seen in this case.9,21
Simple epidermal nevi occur in approximately 1 in 1000 newborns; however, when a child presents with multiple or systematized epidermal nevi, investigation should be undertaken for other possible associations.13,14 Of note, there have been several cases of squamous cell, verrucous, basal cell, and adnexal carcinomas arising in linear epidermal nevi.22-24
Epidermal nevi can be difficult to treat. Some patients are troubled by the appearance of these nevi, especially those with systematized disease. Unfortunately, for patients with multiple nevi or systematized disease, there are no consistently effective treatment options; however, there are case reports25,26 in the literature citing improvement or cure of epidermal nevi with full-thickness excision, continuous and pulsed CO2 laser, pulsed dye laser, and erbium-doped YAG laser.25 Other therapies that have been purported to help improve epidermal nevi are topical and oral retinoids, corticosteroids, topical 5-fluorouracil, anthralin, and podophyllin.26
Conclusion
Transitional cell bladder carcinoma is rare in patients in the third decade of life and younger. Given the age of our patient and her concomitant lack of risk factors, such as older age, history of smoking, and exposure to certain chemicals (eg, aniline dyes) and medications (eg, cyclophosphamide), it is more likely that the finding of papillary transitional cell bladder carcinoma and ENS are related. A clear genetic link between ENS and transitional cell papillary bladder carcinoma has yet to be elucidated, but the FGFR3 gene is promising.
Epidermal nevi can occur in isolation or in association with internal abnormalities. Epidermal nevus syndrome (ENS) is a heterogeneous group of neurocutaneous disorders characterized by mosaicism and epidermal nevi found in association with various systemic abnormalities.1-4 There are many possible associated systemic findings, including abnormalities of the central nervous, musculoskeletal, renal, and hematologic systems. Epidermal nevi have been associated with internal malignancies. We present the case of a patient with epidermal nevi associated with papillary transitional cell bladder carcinoma. According to a PubMed search of articles indexed for MEDLINE using the search terms transitional cell bladder carcinoma and epidermal nevus, there have only been 4 other cases of transitional cell bladder carcinoma and ENS reported in the literature,5-8 2 of which were reports of papillary transitional cell bladder carcinoma.5,6
Case Report
A 29-year-old woman presented to our clinic with a rash that had been present since 3 years of age. The emergency department consulted dermatology for evaluation of what was believed to be contact dermatitis; however, upon questioning the patient, it was revealed that the rash was chronic and persistent.
The rash was nonpruritic and was located on the face, hands (Figure 1), chest, buttocks, thighs, legs, and back (Figure 2). Although asymptomatic, the appearance of the skin caused the patient some emotional distress. As a child she had been evaluated by a dermatologist and a biopsy was performed, but she did not recall the results or have any records. She had been prescribed an oral medication by the dermatologist, but treatment was terminated early due to nausea. The skin lesions did not improve with the short course of treatment.
Eighteen months prior to presentation to our clinic, the patient was discovered to have hematuria on routine examination by her primary care physician. At that time, the patient underwent a workup for hematuria and a mass was discovered in the bladder via cystoscopy. A diagnosis of low-grade papillary transitional cell bladder carcinoma was made, and she underwent a partial cystectomy. No radiation or chemotherapy was required. The remainder of her medical history was only remarkable for asthma, which was well controlled with albuterol. On examination, generalized, hyperpigmented, reticulated patches, macules, and hyperpigmented verrucous plaques were distributed along the Blaschko lines, sparing the face. No limb abnormalities or dental or nail abnormalities were noted. Examination of the axillary and cervical lymph nodes was unremarkable, and no neurological abnormalities were noted. A 3-mm punch biopsy of the mid upper back was performed. Histopathology revealed papillomatous, nonorganoid, nonepidermolytic hyperplasia of the epidermis with elongated rete ridges (Figure 3), which was diagnosed as a nonorganoid nonepidermolytic epidermal nevus.
Comment
Epidermal nevus syndrome is a group of disorders characterized by both local or systematized epidermal nevi and systemic findings. Solomon et al4 first coined the term epidermal nevus syndrome more than 40 years ago; however, since then there has been confusion about how to define ENS. Epidermal nevus syndrome has been considered an umbrella term that includes more specific syndromes involving epidermal nevi, such as Proteus syndrome and Schimmelpenning syndrome; conversely, it also has been considered a term for those who do not meet the criteria for more specific syndromes.1,9 Happle1 discussed that the genetic variations found in ENS warrant recognition. Simply put, ENS is a heterogeneous group of syndromes that are similar in that they involve epidermal nevi and internal abnormalities but are genetically distinct. The list of definitive ENSs, as suggested by Happle1 and others, will likely continue to grow.3,5
The exact pathomechanism of ENS is unknown, but the clinical presentation most likely represents a lethal disorder mitigated by mosaicism.2,9 Gene defects vary depending on the specific ENS. For instance, the phosphatase and tensin homolog gene, PTEN, mutations have been associated with type 2 segmental Cowden disease. Fibroblast growth factor receptor 3, FGFR3, mutations have been linked to Garcia-Hafner-Happle syndrome.3FGFR3 mutations have been found in nonepidermolytic epidermal nevi, and some suggest that the majority of epidermal nevi exhibit mutations in FGFR3.5,10,11 On the other hand, other gene defects have not been elucidated, such as in Schimmelpenning syndrome.3
Clinically, ENS may involve nonepidermolytic verrucous nevi, sebaceous nevi, organoid nevi, linear Cowden nevi, and woolly hair nevi. Lesions may be flesh-colored, pink, yellow, or hyperpigmented plaques in a blaschkoid distribution and may be localized or systematized. Nevi typically are present at birth or develop within the first year of life.9,12,13 Other cutaneous findings may be noted apart from epidermal nevi, including melanocytic nevi, aplasia cutis congenita, and hemangiomas.13,14
Extracutaneous findings include central nervous system, skeletal, ocular, cardiac, and genitourinary defects, which are often observed in these patients.3,9,13,14 Central nervous system findings are seen in 50% to 70% of cases, with seizures and mental retardation among the most common.13-15 Genitourinary abnormalities associated with epidermal nevi, including horseshoe kidney, cystic kidney, duplicated collecting system, testicular and paratesticular tumors, and hypospadias have been documented in the literature.16 Our patient had a history of papillary transitional cell bladder carcinoma, which is rare for a patient younger than 30 years. The overall median age of diagnosis of bladder cancer is 65 years, and it is more common in men than in women.17 Transitional cell carcinomas account for approximately 90% of all bladder cancers in the United States. Other common types of bladder cancer include squamous cell carcinoma, adenocarcinoma, and rhabdomyosarcoma.16 Typically, transitional cell carcinoma is associated with smoking, exposure to aniline dyes, cyclophosphamide, and living in industrialized areas.16,17 Individuals who work with textiles, dyes, leather, tires, rubber, and/or petroleum; painters; truck drivers; drill press operators; and hairdressers are at an increased risk for development of bladder cancer.16
Interestingly, it has been shown in some studies that papillary transitional cell bladder carcinoma frequently is associated with FGFR3 mutations, which may be the missing link in the rare finding of papillary transitional cell bladder carcinoma and epidermal nevi.5,18,19 In addition, PTEN mutations also have been identified in low-grade papillary transitional cell carcinomas of the bladder, another gene linked to an ENS with type 2 segmental Cowden disease.3,20
Histopathologically, epidermal nevi have 10 different descriptions. Our patient had a nonorganoid nonepidermolytic epidermal nevus characterized by hyperkeratosis, acanthosis, papillomatosis, and elongated rete ridges. Focal acantholysis and epidermolytic hyperkeratosis also is seen in some epidermal nevi but was not seen in this case.9,21
Simple epidermal nevi occur in approximately 1 in 1000 newborns; however, when a child presents with multiple or systematized epidermal nevi, investigation should be undertaken for other possible associations.13,14 Of note, there have been several cases of squamous cell, verrucous, basal cell, and adnexal carcinomas arising in linear epidermal nevi.22-24
Epidermal nevi can be difficult to treat. Some patients are troubled by the appearance of these nevi, especially those with systematized disease. Unfortunately, for patients with multiple nevi or systematized disease, there are no consistently effective treatment options; however, there are case reports25,26 in the literature citing improvement or cure of epidermal nevi with full-thickness excision, continuous and pulsed CO2 laser, pulsed dye laser, and erbium-doped YAG laser.25 Other therapies that have been purported to help improve epidermal nevi are topical and oral retinoids, corticosteroids, topical 5-fluorouracil, anthralin, and podophyllin.26
Conclusion
Transitional cell bladder carcinoma is rare in patients in the third decade of life and younger. Given the age of our patient and her concomitant lack of risk factors, such as older age, history of smoking, and exposure to certain chemicals (eg, aniline dyes) and medications (eg, cyclophosphamide), it is more likely that the finding of papillary transitional cell bladder carcinoma and ENS are related. A clear genetic link between ENS and transitional cell papillary bladder carcinoma has yet to be elucidated, but the FGFR3 gene is promising.
- Happle R. What is a nevus? a proposed definition of a common medical term. Dermatology. 1995;191:1-5.
- Gonzalez ME, Jabbari A, Tlougan BE, et al. Epidermal nevus. Dermatol Online J. 2010;16:12.
- Happle R. The group of epidermal nevus syndromes. part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22.
- Solomon LM, Fretzin DF, Dewald RL. The epidermal nevus syndrome. Arch Dermatol. 1968;97:273-285.
- Flosadottir E, Bjarnason B. A non-epidermolytic epidermal nevus of a soft, papillomatous type with transitional cell cancer of the bladder: a case report and review of non-cutaneous cancers associated with epidermal naevi. Acta Derm Venerol. 2008;88:173-175.
- Rosenthal D, Fretzin DF. Epidermal nevus syndrome: report of association with transitional cell carcinoma of the bladder. Pediatr Dermatol. 1986;3:455-458.
- Garcia de Jalon A, Azua-Romea J, Trivez MA, et al. Epidermal naevus syndrome (Solomon’s syndrome) associated with bladder cancer in a 20-year-old female. Scand J Urol Nephrol. 2004;38:85-87.
- Rongioletti F, Rebora A. Epidermal nevus with transitional cell carcinomas of the urinary tract. J Am Acad Dermatol. 1991;25:856-858.
- Moss C. Mosacism and linear lesions. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012:943-962.
- Hafner C, van Oers JM, Vogt T, et al. Mosaicisim of activating FGFR3 mutations in human skin causes epidermal nevi. J Clin Invest. 2006;116:2201-2207.
- Bygum A, Fagerberg CR, Clemmensen OJ, et al. Systemic epidermal nevus with involvement of the oral mucosa due to FGFR3 mutation. BMC Med Genet. 2011;12:79.
- Happle R. Linear Cowden nevus: a new distinct epidermal nevus. Eur J Dermatol. 2007;17:133-136.
- Vujevich JJ, Mancini AJ. The epidermal nevus syndromes: multisystem disorders. J Am Acad Dermatol. 2004;50:957-961.
- Solomon L, Esterly N. Epidermal and other congenital organoid nevi. Curr Probl Pediatr. 1975;6:1-56.
- Grebe TA, Rimsa ME, Richter SF, et al. Further delineation of the epidermal nevus syndrome: two cases with new findings and literature review. Am J Med Genet. 1993;47:24-30.
- Lamm DL, Torti FM. Bladder cancer, 1996. Ca Cancer J Clin. 1996;46:93-112.
- Metts MC, Metts JC, Milito SJ, et al. Bladder cancer: a review of diagnosis and management. J Natl Med Assoc. 2000;92:285-294.
- Kimura T, Suzuki H, Ohashi T, et al. The incidence of thanatophoric dysplasia mutations in FGFR3 gene is higher in low-grade or superficial bladder carcinomas. Cancer. 2001;92:2555-2561.
- Cappellen D, DeOliveira C, Ricol D, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18-20.
- Knowles MA, Platt FM, Ross RL, et al. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 2009;28:305-316.
- Luzar B, Calonje E, Bastian B. Tumors of the surface epithelium. In: Calonje JE, Breen T, McKee PH, eds. McKee’s Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012:1076-1149.
- Masood Q, Narayan D. Squamous cell carcinoma in a linear epidermal nevus. J Plast Reconstr Aesthet Surg. 2009;62:693-694.
- Cramer SF, Mandel MA, Hauler R, et al. Squamous cell carcinoma arising in a linear epidermal nevus. Arch Dermatol. 1981;117:222-224.
- Affleck AG, Leach IJ, Varma S. Two squamous cell carcinomas arising in a linear epidermal nevus in a 28-year-old female. Clin Exp Dermatol. 2005;30:382-384.
- Alam M, Arndt KA. A method for pulsed carbon dioxide laser treatment of epidermal nevi. J Am Acad Dermatol. 2002;46:554-556.
- Requena L, Requena C, Cockerell CJ. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012:1809-1810.
- Happle R. What is a nevus? a proposed definition of a common medical term. Dermatology. 1995;191:1-5.
- Gonzalez ME, Jabbari A, Tlougan BE, et al. Epidermal nevus. Dermatol Online J. 2010;16:12.
- Happle R. The group of epidermal nevus syndromes. part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22.
- Solomon LM, Fretzin DF, Dewald RL. The epidermal nevus syndrome. Arch Dermatol. 1968;97:273-285.
- Flosadottir E, Bjarnason B. A non-epidermolytic epidermal nevus of a soft, papillomatous type with transitional cell cancer of the bladder: a case report and review of non-cutaneous cancers associated with epidermal naevi. Acta Derm Venerol. 2008;88:173-175.
- Rosenthal D, Fretzin DF. Epidermal nevus syndrome: report of association with transitional cell carcinoma of the bladder. Pediatr Dermatol. 1986;3:455-458.
- Garcia de Jalon A, Azua-Romea J, Trivez MA, et al. Epidermal naevus syndrome (Solomon’s syndrome) associated with bladder cancer in a 20-year-old female. Scand J Urol Nephrol. 2004;38:85-87.
- Rongioletti F, Rebora A. Epidermal nevus with transitional cell carcinomas of the urinary tract. J Am Acad Dermatol. 1991;25:856-858.
- Moss C. Mosacism and linear lesions. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012:943-962.
- Hafner C, van Oers JM, Vogt T, et al. Mosaicisim of activating FGFR3 mutations in human skin causes epidermal nevi. J Clin Invest. 2006;116:2201-2207.
- Bygum A, Fagerberg CR, Clemmensen OJ, et al. Systemic epidermal nevus with involvement of the oral mucosa due to FGFR3 mutation. BMC Med Genet. 2011;12:79.
- Happle R. Linear Cowden nevus: a new distinct epidermal nevus. Eur J Dermatol. 2007;17:133-136.
- Vujevich JJ, Mancini AJ. The epidermal nevus syndromes: multisystem disorders. J Am Acad Dermatol. 2004;50:957-961.
- Solomon L, Esterly N. Epidermal and other congenital organoid nevi. Curr Probl Pediatr. 1975;6:1-56.
- Grebe TA, Rimsa ME, Richter SF, et al. Further delineation of the epidermal nevus syndrome: two cases with new findings and literature review. Am J Med Genet. 1993;47:24-30.
- Lamm DL, Torti FM. Bladder cancer, 1996. Ca Cancer J Clin. 1996;46:93-112.
- Metts MC, Metts JC, Milito SJ, et al. Bladder cancer: a review of diagnosis and management. J Natl Med Assoc. 2000;92:285-294.
- Kimura T, Suzuki H, Ohashi T, et al. The incidence of thanatophoric dysplasia mutations in FGFR3 gene is higher in low-grade or superficial bladder carcinomas. Cancer. 2001;92:2555-2561.
- Cappellen D, DeOliveira C, Ricol D, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18-20.
- Knowles MA, Platt FM, Ross RL, et al. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 2009;28:305-316.
- Luzar B, Calonje E, Bastian B. Tumors of the surface epithelium. In: Calonje JE, Breen T, McKee PH, eds. McKee’s Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012:1076-1149.
- Masood Q, Narayan D. Squamous cell carcinoma in a linear epidermal nevus. J Plast Reconstr Aesthet Surg. 2009;62:693-694.
- Cramer SF, Mandel MA, Hauler R, et al. Squamous cell carcinoma arising in a linear epidermal nevus. Arch Dermatol. 1981;117:222-224.
- Affleck AG, Leach IJ, Varma S. Two squamous cell carcinomas arising in a linear epidermal nevus in a 28-year-old female. Clin Exp Dermatol. 2005;30:382-384.
- Alam M, Arndt KA. A method for pulsed carbon dioxide laser treatment of epidermal nevi. J Am Acad Dermatol. 2002;46:554-556.
- Requena L, Requena C, Cockerell CJ. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012:1809-1810.
Practice Points
- Epidermal nevi are common benign cutaneous neoplasms.
- Extensive systematized epidermal nevi can be a sign of internal disease.
Team characterizes therapy-resistant ALL cells

Image by Vashi Donsk
Researchers say they have characterized a subpopulation of leukemia cells that are responsible for relapse in acute lymphoblastic leukemia (ALL).
The team identified these dormant, therapy-resistant cells in mouse models of ALL and found that removing the cells from their environment makes them sensitive to treatment.
The researchers believe these findings could pave the way to better relapse prevention in patients with ALL.
“Previously, the biological principles responsible for a relapse in leukemia were not fully understood,” said study author Irmela Jeremias, MD, PhD, of Helmholtz Zentrum München in Munich, Germany.
“Our new approach is to isolate dormant cells, which gives us the first possibility of developing therapies that switch off these cells.”
Dr Jeremias and colleagues described this approach in Cancer Cell.
First, the researchers created mouse models recapitulating minimal residual disease (MRD) and relapse in ALL patients.
The team then used genetic engineering and proliferation-sensitive dyes to isolate and characterize relapse-inducing cells.
This revealed a subpopulation of leukemia cells that exhibited long-term dormancy, treatment resistance, and stemness. These cells were similar to primary ALL cells isolated from pediatric and adult patients with MRD.
However, the dormant leukemia cells found in the mice changed once they were removed from the in vivo environment. They began to proliferate and became sensitive to ex vivo treatment with chemotherapy drugs.
“[T]hese cells, once they have been dissolved out of their surroundings, are indeed susceptible to therapy and react well to therapeutics,” said study author Erbey Özdemir, a doctoral candidate at Helmholtz Zentrum München.
The researchers therefore believe that therapeutic strategies aimed at dissociating dormant leukemia cells from their protective niche might prevent relapse in ALL patients.
“This has brought us a small step closer to the global goal of preventing disease relapse in patients suffering from leukemia,” Dr Jeremias said. “It might serve as basis for new therapies that destroy resistant leukemia cells before they induce relapse.” ![]()
Image by Vashi Donsk
Researchers say they have characterized a subpopulation of leukemia cells that are responsible for relapse in acute lymphoblastic leukemia (ALL).
The team identified these dormant, therapy-resistant cells in mouse models of ALL and found that removing the cells from their environment makes them sensitive to treatment.
The researchers believe these findings could pave the way to better relapse prevention in patients with ALL.
“Previously, the biological principles responsible for a relapse in leukemia were not fully understood,” said study author Irmela Jeremias, MD, PhD, of Helmholtz Zentrum München in Munich, Germany.
“Our new approach is to isolate dormant cells, which gives us the first possibility of developing therapies that switch off these cells.”
Dr Jeremias and colleagues described this approach in Cancer Cell.
First, the researchers created mouse models recapitulating minimal residual disease (MRD) and relapse in ALL patients.
The team then used genetic engineering and proliferation-sensitive dyes to isolate and characterize relapse-inducing cells.
This revealed a subpopulation of leukemia cells that exhibited long-term dormancy, treatment resistance, and stemness. These cells were similar to primary ALL cells isolated from pediatric and adult patients with MRD.
However, the dormant leukemia cells found in the mice changed once they were removed from the in vivo environment. They began to proliferate and became sensitive to ex vivo treatment with chemotherapy drugs.
“[T]hese cells, once they have been dissolved out of their surroundings, are indeed susceptible to therapy and react well to therapeutics,” said study author Erbey Özdemir, a doctoral candidate at Helmholtz Zentrum München.
The researchers therefore believe that therapeutic strategies aimed at dissociating dormant leukemia cells from their protective niche might prevent relapse in ALL patients.
“This has brought us a small step closer to the global goal of preventing disease relapse in patients suffering from leukemia,” Dr Jeremias said. “It might serve as basis for new therapies that destroy resistant leukemia cells before they induce relapse.” ![]()
Image by Vashi Donsk
Researchers say they have characterized a subpopulation of leukemia cells that are responsible for relapse in acute lymphoblastic leukemia (ALL).
The team identified these dormant, therapy-resistant cells in mouse models of ALL and found that removing the cells from their environment makes them sensitive to treatment.
The researchers believe these findings could pave the way to better relapse prevention in patients with ALL.
“Previously, the biological principles responsible for a relapse in leukemia were not fully understood,” said study author Irmela Jeremias, MD, PhD, of Helmholtz Zentrum München in Munich, Germany.
“Our new approach is to isolate dormant cells, which gives us the first possibility of developing therapies that switch off these cells.”
Dr Jeremias and colleagues described this approach in Cancer Cell.
First, the researchers created mouse models recapitulating minimal residual disease (MRD) and relapse in ALL patients.
The team then used genetic engineering and proliferation-sensitive dyes to isolate and characterize relapse-inducing cells.
This revealed a subpopulation of leukemia cells that exhibited long-term dormancy, treatment resistance, and stemness. These cells were similar to primary ALL cells isolated from pediatric and adult patients with MRD.
However, the dormant leukemia cells found in the mice changed once they were removed from the in vivo environment. They began to proliferate and became sensitive to ex vivo treatment with chemotherapy drugs.
“[T]hese cells, once they have been dissolved out of their surroundings, are indeed susceptible to therapy and react well to therapeutics,” said study author Erbey Özdemir, a doctoral candidate at Helmholtz Zentrum München.
The researchers therefore believe that therapeutic strategies aimed at dissociating dormant leukemia cells from their protective niche might prevent relapse in ALL patients.
“This has brought us a small step closer to the global goal of preventing disease relapse in patients suffering from leukemia,” Dr Jeremias said. “It might serve as basis for new therapies that destroy resistant leukemia cells before they induce relapse.” ![]()
Ryan White Program Announces New Funding Grants
“The Ryan White HIV/AIDS Program plays an instrumental role in the United States’ public health response to HIV,” said HHS Secretary Sylvia Burwell, announcing nearly $2.3 billion in grants to the program to ease access to HIV/AIDS care and medications.
The program provides primary medical care, drug assistance, education and training, and a number of other essential support services to more than half a million people—more than50% of those living with diagnosed HIV infection in the U..S. The services are crucial in “preserving health, extending life expectancy, and reducing HIV transmission,” said HRSA Acting Administrator Jim Macrae. “In 2014, more than 80% of Ryan White HIV/AIDS Program clients who received HIV medical care were retained in care, and more than 81% of program clients who received HIV medical care were virally suppressed.”
About $627.8 million was awarded to 24 metropolitan areas and 28 transitional grant areas with the highest number of people living with HIV and AIDS or those experiencing increases in HIV and AIDS cases and emerging care needs. Another approximate $1.3 billion was awarded to 59 states and territories for core medical and support services and for the AIDS Drug Assistance Program.
Sixteen states received Emerging Community grants based on the number of AIDS cases over the most recent 5-year period. Thirty-two states and territories were awarded $10.4 million in Part B Minority AIDS Initiative grants.
Local community-based organizations and other groups across the country also were awarded funding to provide family-centered comprehensive care for women and children; technical assistance, clinical training, and oral health services; and education and training for health care professionals. Grant money will support the demonstration and evaluation of innovative models of care delivery for hard-to-reach populations as well as efforts to reduce new HIV infections.
“The Ryan White HIV/AIDS Program plays an instrumental role in the United States’ public health response to HIV,” said HHS Secretary Sylvia Burwell, announcing nearly $2.3 billion in grants to the program to ease access to HIV/AIDS care and medications.
The program provides primary medical care, drug assistance, education and training, and a number of other essential support services to more than half a million people—more than50% of those living with diagnosed HIV infection in the U..S. The services are crucial in “preserving health, extending life expectancy, and reducing HIV transmission,” said HRSA Acting Administrator Jim Macrae. “In 2014, more than 80% of Ryan White HIV/AIDS Program clients who received HIV medical care were retained in care, and more than 81% of program clients who received HIV medical care were virally suppressed.”
About $627.8 million was awarded to 24 metropolitan areas and 28 transitional grant areas with the highest number of people living with HIV and AIDS or those experiencing increases in HIV and AIDS cases and emerging care needs. Another approximate $1.3 billion was awarded to 59 states and territories for core medical and support services and for the AIDS Drug Assistance Program.
Sixteen states received Emerging Community grants based on the number of AIDS cases over the most recent 5-year period. Thirty-two states and territories were awarded $10.4 million in Part B Minority AIDS Initiative grants.
Local community-based organizations and other groups across the country also were awarded funding to provide family-centered comprehensive care for women and children; technical assistance, clinical training, and oral health services; and education and training for health care professionals. Grant money will support the demonstration and evaluation of innovative models of care delivery for hard-to-reach populations as well as efforts to reduce new HIV infections.
“The Ryan White HIV/AIDS Program plays an instrumental role in the United States’ public health response to HIV,” said HHS Secretary Sylvia Burwell, announcing nearly $2.3 billion in grants to the program to ease access to HIV/AIDS care and medications.
The program provides primary medical care, drug assistance, education and training, and a number of other essential support services to more than half a million people—more than50% of those living with diagnosed HIV infection in the U..S. The services are crucial in “preserving health, extending life expectancy, and reducing HIV transmission,” said HRSA Acting Administrator Jim Macrae. “In 2014, more than 80% of Ryan White HIV/AIDS Program clients who received HIV medical care were retained in care, and more than 81% of program clients who received HIV medical care were virally suppressed.”
About $627.8 million was awarded to 24 metropolitan areas and 28 transitional grant areas with the highest number of people living with HIV and AIDS or those experiencing increases in HIV and AIDS cases and emerging care needs. Another approximate $1.3 billion was awarded to 59 states and territories for core medical and support services and for the AIDS Drug Assistance Program.
Sixteen states received Emerging Community grants based on the number of AIDS cases over the most recent 5-year period. Thirty-two states and territories were awarded $10.4 million in Part B Minority AIDS Initiative grants.
Local community-based organizations and other groups across the country also were awarded funding to provide family-centered comprehensive care for women and children; technical assistance, clinical training, and oral health services; and education and training for health care professionals. Grant money will support the demonstration and evaluation of innovative models of care delivery for hard-to-reach populations as well as efforts to reduce new HIV infections.
Food Security Can Help Reduce Cardiovascular Risk Factors
Food insecurity has been linked to hypertension, diabetes, elevated cholesterol, and obesity—all cardiovascular risk factors and dangerous for pregnant women and infants. Researchers from Massachusetts General Hospital theorized that enrolling pregnant women in a program to ensure their access to food banks and other resources could help reduce their risks.
The researchers conducted a retrospective analysis of 1,295 women who visited the obstetrics clinic at a community health center. Of those, 145 (11%) were referred to Food for Families, which connects patients to resources such as the Supplemental Nutrition Assistance Program (SNAP) and the Special Supplemental Nutrition Program for Women, Infants, and Children (WIC).
Two-thirds of referred women enrolled in Food for Families. A majority rated their health as good, very good, or excellent. Most had never used a free meal program, soup kitchen, or food pantry, although 43% were eligible for SNAP, and 87% were enrolled in WIC.
The primary outcomes measured were trends in blood pressure (BP) and blood glucose during pregnancy. Blood pressure numbers trended “modestly better” for women in the intervention program. They had a better systolic BP (0.2015 mm Hg/wk lower) and diastolic BP (0.1049 mm Hg/wk lower) than those who were not referred. The researchers found no differences in blood glucose trend.
The findings suggest that programs to reduce food insecurity can improve cardiovascular health in pregnant women, the researchers say. If so, screening for food insecurity in obstetric care may be a useful tool—particularly if the next step is to get patients the food they need
Food insecurity has been linked to hypertension, diabetes, elevated cholesterol, and obesity—all cardiovascular risk factors and dangerous for pregnant women and infants. Researchers from Massachusetts General Hospital theorized that enrolling pregnant women in a program to ensure their access to food banks and other resources could help reduce their risks.
The researchers conducted a retrospective analysis of 1,295 women who visited the obstetrics clinic at a community health center. Of those, 145 (11%) were referred to Food for Families, which connects patients to resources such as the Supplemental Nutrition Assistance Program (SNAP) and the Special Supplemental Nutrition Program for Women, Infants, and Children (WIC).
Two-thirds of referred women enrolled in Food for Families. A majority rated their health as good, very good, or excellent. Most had never used a free meal program, soup kitchen, or food pantry, although 43% were eligible for SNAP, and 87% were enrolled in WIC.
The primary outcomes measured were trends in blood pressure (BP) and blood glucose during pregnancy. Blood pressure numbers trended “modestly better” for women in the intervention program. They had a better systolic BP (0.2015 mm Hg/wk lower) and diastolic BP (0.1049 mm Hg/wk lower) than those who were not referred. The researchers found no differences in blood glucose trend.
The findings suggest that programs to reduce food insecurity can improve cardiovascular health in pregnant women, the researchers say. If so, screening for food insecurity in obstetric care may be a useful tool—particularly if the next step is to get patients the food they need
Food insecurity has been linked to hypertension, diabetes, elevated cholesterol, and obesity—all cardiovascular risk factors and dangerous for pregnant women and infants. Researchers from Massachusetts General Hospital theorized that enrolling pregnant women in a program to ensure their access to food banks and other resources could help reduce their risks.
The researchers conducted a retrospective analysis of 1,295 women who visited the obstetrics clinic at a community health center. Of those, 145 (11%) were referred to Food for Families, which connects patients to resources such as the Supplemental Nutrition Assistance Program (SNAP) and the Special Supplemental Nutrition Program for Women, Infants, and Children (WIC).
Two-thirds of referred women enrolled in Food for Families. A majority rated their health as good, very good, or excellent. Most had never used a free meal program, soup kitchen, or food pantry, although 43% were eligible for SNAP, and 87% were enrolled in WIC.
The primary outcomes measured were trends in blood pressure (BP) and blood glucose during pregnancy. Blood pressure numbers trended “modestly better” for women in the intervention program. They had a better systolic BP (0.2015 mm Hg/wk lower) and diastolic BP (0.1049 mm Hg/wk lower) than those who were not referred. The researchers found no differences in blood glucose trend.
The findings suggest that programs to reduce food insecurity can improve cardiovascular health in pregnant women, the researchers say. If so, screening for food insecurity in obstetric care may be a useful tool—particularly if the next step is to get patients the food they need
Pain Management: How About Holistic?
I recently received the letter and instruction card for prescribing narcotic analgesics from US Surgeon General Vice Admiral Vivek H. Murthy, MD, MBA. While I agree in principle with the movement to improve pain management, I feel there is a lot being overlooked in this crusade and would like to suggest alternative evidence-based methods that don’t involve prescription narcotics.
I have extensive training in energy therapy, guided imagery, and ozonotherapy. Healing touch is one well-researched energy therapy that has been shown to reduce pain. I have performed and published research demonstrating its efficacy (see http://healingbeyondborders.org/index.php/research-integrative-health/research); patients’ functional abilities improve, and they are able to decrease or eliminate use of pain medication. Providers from many disciplines, including MDs, DOs, NPs, and DCs, practice healing touch. Training for healing touch is available worldwide. The certification process is similar to masters-level education, including both classroom and hands-on clinical practice experience. My practice uses healing touch for patients, and I teach classes using the international curriculum.
In addition to the research published on the efficacy of guided imagery (another method of pain relief therapy), I have personally witnessed and been part of several successful examples in my clinical practice. In the burn unit, Dr. Jean Achterberg Lawlis and I used guided imagery to relieve pain in patients with third- and fourth-degree burns over 70% or more of their body. We performed tanking and dressing changes without narcotic pain medications; patients were comfortable during treatment and slept peacefully after. In another instance, a 23-year-old man presented with major chest and spine injuries after a motorcycle accident. Morphine (100 mg IV) did nothing to relieve his pain. But guided imagery of racing his stock car around a racetrack eliminated any need for narcotics during dressing changes. I’ve also worked with women prenatally, teaching guided imagery for smooth, successful deliveries without pain medications or epidural.
Ozonotherapy has an extensive international evidence base, and many studies show that it relieves pain without the need for narcotics (see http://aaot.us/?page=Literature). I have seen many cases of chronic pain relieved by major autohemolytic therapy and prolozone injection therapies. Here, too, patients are able to decrease and eventually stop their narcotic medications. Some patients are able to avoid joint replacement surgery, achieving improved function and comfort without the adverse effects of steroids.
An effective way to release muscle tension and relieve pain from injury (eg, low back pain, plantar fasciitis, whiplash, carpal tunnel) is through massage therapy. Providers who refer patients to massage practitioners can avoid narcotic medication prescriptions by addressing the problem that is causing the pain. Chiropractic care is a standard care for low back pain; it can also resolve problems that cause migraines, trigeminal neuralgia, and Bell’s palsy without narcotics, steroids, or the sedating muscle relaxants and seizure medications. Yet several veterans in my community were denied chiropractic care until they had tried narcotics and physical therapy (which involved a four-hour roundtrip car ride, no less). Oh, and in the meantime, they were prescribed an additional narcotic!
By focusing only on narcotics, we miss out on other options to treat pain. If we overlook the full range of evidence, then the “evidence-based” mantra isn’t truthful, nor is it useful. To follow the pledge to “do no harm,” we must treat the causes of pain. Of the Surgeon General, I request: Please don’t just send us a teaching card on how to prescribe narcotics. Get providers involved in seeking continuing education credits in therapies that help us avoid prescribing them in the first place.

Susan Peck, PhD, GNP-BC, APNP, FAAO, APT, CHTP/I
Eau Claire, WI
I recently received the letter and instruction card for prescribing narcotic analgesics from US Surgeon General Vice Admiral Vivek H. Murthy, MD, MBA. While I agree in principle with the movement to improve pain management, I feel there is a lot being overlooked in this crusade and would like to suggest alternative evidence-based methods that don’t involve prescription narcotics.
I have extensive training in energy therapy, guided imagery, and ozonotherapy. Healing touch is one well-researched energy therapy that has been shown to reduce pain. I have performed and published research demonstrating its efficacy (see http://healingbeyondborders.org/index.php/research-integrative-health/research); patients’ functional abilities improve, and they are able to decrease or eliminate use of pain medication. Providers from many disciplines, including MDs, DOs, NPs, and DCs, practice healing touch. Training for healing touch is available worldwide. The certification process is similar to masters-level education, including both classroom and hands-on clinical practice experience. My practice uses healing touch for patients, and I teach classes using the international curriculum.
In addition to the research published on the efficacy of guided imagery (another method of pain relief therapy), I have personally witnessed and been part of several successful examples in my clinical practice. In the burn unit, Dr. Jean Achterberg Lawlis and I used guided imagery to relieve pain in patients with third- and fourth-degree burns over 70% or more of their body. We performed tanking and dressing changes without narcotic pain medications; patients were comfortable during treatment and slept peacefully after. In another instance, a 23-year-old man presented with major chest and spine injuries after a motorcycle accident. Morphine (100 mg IV) did nothing to relieve his pain. But guided imagery of racing his stock car around a racetrack eliminated any need for narcotics during dressing changes. I’ve also worked with women prenatally, teaching guided imagery for smooth, successful deliveries without pain medications or epidural.
Ozonotherapy has an extensive international evidence base, and many studies show that it relieves pain without the need for narcotics (see http://aaot.us/?page=Literature). I have seen many cases of chronic pain relieved by major autohemolytic therapy and prolozone injection therapies. Here, too, patients are able to decrease and eventually stop their narcotic medications. Some patients are able to avoid joint replacement surgery, achieving improved function and comfort without the adverse effects of steroids.
An effective way to release muscle tension and relieve pain from injury (eg, low back pain, plantar fasciitis, whiplash, carpal tunnel) is through massage therapy. Providers who refer patients to massage practitioners can avoid narcotic medication prescriptions by addressing the problem that is causing the pain. Chiropractic care is a standard care for low back pain; it can also resolve problems that cause migraines, trigeminal neuralgia, and Bell’s palsy without narcotics, steroids, or the sedating muscle relaxants and seizure medications. Yet several veterans in my community were denied chiropractic care until they had tried narcotics and physical therapy (which involved a four-hour roundtrip car ride, no less). Oh, and in the meantime, they were prescribed an additional narcotic!
By focusing only on narcotics, we miss out on other options to treat pain. If we overlook the full range of evidence, then the “evidence-based” mantra isn’t truthful, nor is it useful. To follow the pledge to “do no harm,” we must treat the causes of pain. Of the Surgeon General, I request: Please don’t just send us a teaching card on how to prescribe narcotics. Get providers involved in seeking continuing education credits in therapies that help us avoid prescribing them in the first place.
Susan Peck, PhD, GNP-BC, APNP, FAAO, APT, CHTP/I
Eau Claire, WI
I recently received the letter and instruction card for prescribing narcotic analgesics from US Surgeon General Vice Admiral Vivek H. Murthy, MD, MBA. While I agree in principle with the movement to improve pain management, I feel there is a lot being overlooked in this crusade and would like to suggest alternative evidence-based methods that don’t involve prescription narcotics.
I have extensive training in energy therapy, guided imagery, and ozonotherapy. Healing touch is one well-researched energy therapy that has been shown to reduce pain. I have performed and published research demonstrating its efficacy (see http://healingbeyondborders.org/index.php/research-integrative-health/research); patients’ functional abilities improve, and they are able to decrease or eliminate use of pain medication. Providers from many disciplines, including MDs, DOs, NPs, and DCs, practice healing touch. Training for healing touch is available worldwide. The certification process is similar to masters-level education, including both classroom and hands-on clinical practice experience. My practice uses healing touch for patients, and I teach classes using the international curriculum.
In addition to the research published on the efficacy of guided imagery (another method of pain relief therapy), I have personally witnessed and been part of several successful examples in my clinical practice. In the burn unit, Dr. Jean Achterberg Lawlis and I used guided imagery to relieve pain in patients with third- and fourth-degree burns over 70% or more of their body. We performed tanking and dressing changes without narcotic pain medications; patients were comfortable during treatment and slept peacefully after. In another instance, a 23-year-old man presented with major chest and spine injuries after a motorcycle accident. Morphine (100 mg IV) did nothing to relieve his pain. But guided imagery of racing his stock car around a racetrack eliminated any need for narcotics during dressing changes. I’ve also worked with women prenatally, teaching guided imagery for smooth, successful deliveries without pain medications or epidural.
Ozonotherapy has an extensive international evidence base, and many studies show that it relieves pain without the need for narcotics (see http://aaot.us/?page=Literature). I have seen many cases of chronic pain relieved by major autohemolytic therapy and prolozone injection therapies. Here, too, patients are able to decrease and eventually stop their narcotic medications. Some patients are able to avoid joint replacement surgery, achieving improved function and comfort without the adverse effects of steroids.
An effective way to release muscle tension and relieve pain from injury (eg, low back pain, plantar fasciitis, whiplash, carpal tunnel) is through massage therapy. Providers who refer patients to massage practitioners can avoid narcotic medication prescriptions by addressing the problem that is causing the pain. Chiropractic care is a standard care for low back pain; it can also resolve problems that cause migraines, trigeminal neuralgia, and Bell’s palsy without narcotics, steroids, or the sedating muscle relaxants and seizure medications. Yet several veterans in my community were denied chiropractic care until they had tried narcotics and physical therapy (which involved a four-hour roundtrip car ride, no less). Oh, and in the meantime, they were prescribed an additional narcotic!
By focusing only on narcotics, we miss out on other options to treat pain. If we overlook the full range of evidence, then the “evidence-based” mantra isn’t truthful, nor is it useful. To follow the pledge to “do no harm,” we must treat the causes of pain. Of the Surgeon General, I request: Please don’t just send us a teaching card on how to prescribe narcotics. Get providers involved in seeking continuing education credits in therapies that help us avoid prescribing them in the first place.
Susan Peck, PhD, GNP-BC, APNP, FAAO, APT, CHTP/I
Eau Claire, WI
Statins in adults with CVD risk: The latest from the USPSTF

Do pedometers increase activity and improve health outcomes?
EVIDENCE SUMMARY
A systematic review and meta-analysis identified 26 studies evaluating activity and health outcomes with the use of pedometers.1 The studies included 8 RCTs and 18 observational studies with 2767 patients (mean body mass index [BMI]: 30 kg/m2; mean age: 49 years; 85% women). The studies ranged from 3 to 104 weeks. From the RCT data, patients using pedometers had an increase of 2491 steps per day (about one mile) more than control group patients (8 trials, n=305; 95% confidence interval [CI], 1098-3885 steps/day; P<.001).
Across all of the observational studies, pedometer users had a 26.9% increase from their baseline physical activity (P=.001). When data from all of the studies were combined, the researchers found a decrease from baseline BMI (18 studies, n=562; mean difference [MD]=0.38 kg/m2; 95% CI, 0.05-0.72; P=.03) and a decrease in systolic BP (12 studies, n=468; MD=3.8 mm Hg; 95% CI, 1.7-5.9 mm Hg; P<.001). No statistically significant change was noted in cholesterol or fasting glucose levels. Weaknesses of this review include the heterogeneity of the interventions, relatively small study sizes, and short study durations.
A systematic review and meta-analysis of 11 RCTs (N=1258) evaluated pedometer effects in overweight patients with type 2 diabetes.2 (One RCT was included in the above meta-analysis.) Studies ran from 6 to 48 weeks, and mean enrollment BMI (where reported) was 30 kg/m2 or more in at least one treatment arm. Compared to controls, patients using pedometers had greater reductions in weight (weighted mean difference [WMD]= -0.65 kg; 95% CI, -1.12 to -0.17 kg) and BMI (WMD= -0.15 kg/m2; 95% CI, -0.29 to -0.02 kg/m2). The effect persisted in the subset of studies in which the intervention and control groups both received dietary counseling (WMD weight= -0.86 kg; 95% CI, -1.45 to -0.27 kg; WMD BMI= -0.30 kg/m2; 95% CI, -0.50 to -0.10 kg/m2). Study quality was low to moderate, and 5 studies used per-protocol analysis instead of intention-to-treat analysis.
Pedometer use benefits patients with musculoskeletal diseases, too
A systematic review and meta-analysis examined the use of pedometers in patients with musculoskeletal diseases.3 It included 7 RCTs lasting 4 weeks to one year with 484 adults, 40 to 82 years of age, with musculoskeletal disorders (eg, back pain, knee pain, hip pain). (One RCT was also included in the diabetes meta-analysis.) Pedometer use resulted in a mean increase in physical activity of 1950 steps per day above baseline (range=818-2829 steps/day; P<.05). The authors noted that 4 of the 7 studies also demonstrated significant improvement in pain scores and physical function. BMI data were not tracked in this review.
Pedometers increase walking in older patients
A RCT compared the effects of pedometer-based activity prescriptions with standard time-based activity prescriptions in 330 patients ≥65 years of age with baseline low activity levels.4 All patients received an initial physician visit followed by 3 telephone counseling sessions encouraging increased activity. The pedometer group was counseled on increasing steps (without specific targets), while the standard activity prescription group received time-related activity goals.
At one year, “leisure walking” had increased more for the pedometer group than for the standard group (mean 50 minutes/week vs 28 minutes/week; P=.03), although both groups equally increased their amount of “total activity.”
1. Bravata DM, Smith-Spangler C, Sundaram V, et al. Using pedometers to increase physical activity and improve health: a systematic review. JAMA. 2007;298:2296-2304.
2. Cai X, Qiu SH, Yin H, et al. Pedometer intervention and weight loss in overweight and obese adults with type 2 diabetes: a meta-analysis. Diabet Med. 2016;33:1035-1044.
3. Mansi S, Milosavljevic S, Baxter GD, et al. A systematic review of studies using pedometers as an intervention for musculoskeletal diseases. BMC Musculoskeletal Disorders. 2014;15:231.
4. Kolt GS, Schofield GM, Kerse N, et al. Healthy Steps trial: pedometer-based advice and physical activity for low-active older adults. Ann Fam Med. 2012;10:206-212.
EVIDENCE SUMMARY
A systematic review and meta-analysis identified 26 studies evaluating activity and health outcomes with the use of pedometers.1 The studies included 8 RCTs and 18 observational studies with 2767 patients (mean body mass index [BMI]: 30 kg/m2; mean age: 49 years; 85% women). The studies ranged from 3 to 104 weeks. From the RCT data, patients using pedometers had an increase of 2491 steps per day (about one mile) more than control group patients (8 trials, n=305; 95% confidence interval [CI], 1098-3885 steps/day; P<.001).
Across all of the observational studies, pedometer users had a 26.9% increase from their baseline physical activity (P=.001). When data from all of the studies were combined, the researchers found a decrease from baseline BMI (18 studies, n=562; mean difference [MD]=0.38 kg/m2; 95% CI, 0.05-0.72; P=.03) and a decrease in systolic BP (12 studies, n=468; MD=3.8 mm Hg; 95% CI, 1.7-5.9 mm Hg; P<.001). No statistically significant change was noted in cholesterol or fasting glucose levels. Weaknesses of this review include the heterogeneity of the interventions, relatively small study sizes, and short study durations.
A systematic review and meta-analysis of 11 RCTs (N=1258) evaluated pedometer effects in overweight patients with type 2 diabetes.2 (One RCT was included in the above meta-analysis.) Studies ran from 6 to 48 weeks, and mean enrollment BMI (where reported) was 30 kg/m2 or more in at least one treatment arm. Compared to controls, patients using pedometers had greater reductions in weight (weighted mean difference [WMD]= -0.65 kg; 95% CI, -1.12 to -0.17 kg) and BMI (WMD= -0.15 kg/m2; 95% CI, -0.29 to -0.02 kg/m2). The effect persisted in the subset of studies in which the intervention and control groups both received dietary counseling (WMD weight= -0.86 kg; 95% CI, -1.45 to -0.27 kg; WMD BMI= -0.30 kg/m2; 95% CI, -0.50 to -0.10 kg/m2). Study quality was low to moderate, and 5 studies used per-protocol analysis instead of intention-to-treat analysis.
Pedometer use benefits patients with musculoskeletal diseases, too
A systematic review and meta-analysis examined the use of pedometers in patients with musculoskeletal diseases.3 It included 7 RCTs lasting 4 weeks to one year with 484 adults, 40 to 82 years of age, with musculoskeletal disorders (eg, back pain, knee pain, hip pain). (One RCT was also included in the diabetes meta-analysis.) Pedometer use resulted in a mean increase in physical activity of 1950 steps per day above baseline (range=818-2829 steps/day; P<.05). The authors noted that 4 of the 7 studies also demonstrated significant improvement in pain scores and physical function. BMI data were not tracked in this review.
Pedometers increase walking in older patients
A RCT compared the effects of pedometer-based activity prescriptions with standard time-based activity prescriptions in 330 patients ≥65 years of age with baseline low activity levels.4 All patients received an initial physician visit followed by 3 telephone counseling sessions encouraging increased activity. The pedometer group was counseled on increasing steps (without specific targets), while the standard activity prescription group received time-related activity goals.
At one year, “leisure walking” had increased more for the pedometer group than for the standard group (mean 50 minutes/week vs 28 minutes/week; P=.03), although both groups equally increased their amount of “total activity.”
EVIDENCE SUMMARY
A systematic review and meta-analysis identified 26 studies evaluating activity and health outcomes with the use of pedometers.1 The studies included 8 RCTs and 18 observational studies with 2767 patients (mean body mass index [BMI]: 30 kg/m2; mean age: 49 years; 85% women). The studies ranged from 3 to 104 weeks. From the RCT data, patients using pedometers had an increase of 2491 steps per day (about one mile) more than control group patients (8 trials, n=305; 95% confidence interval [CI], 1098-3885 steps/day; P<.001).
Across all of the observational studies, pedometer users had a 26.9% increase from their baseline physical activity (P=.001). When data from all of the studies were combined, the researchers found a decrease from baseline BMI (18 studies, n=562; mean difference [MD]=0.38 kg/m2; 95% CI, 0.05-0.72; P=.03) and a decrease in systolic BP (12 studies, n=468; MD=3.8 mm Hg; 95% CI, 1.7-5.9 mm Hg; P<.001). No statistically significant change was noted in cholesterol or fasting glucose levels. Weaknesses of this review include the heterogeneity of the interventions, relatively small study sizes, and short study durations.
A systematic review and meta-analysis of 11 RCTs (N=1258) evaluated pedometer effects in overweight patients with type 2 diabetes.2 (One RCT was included in the above meta-analysis.) Studies ran from 6 to 48 weeks, and mean enrollment BMI (where reported) was 30 kg/m2 or more in at least one treatment arm. Compared to controls, patients using pedometers had greater reductions in weight (weighted mean difference [WMD]= -0.65 kg; 95% CI, -1.12 to -0.17 kg) and BMI (WMD= -0.15 kg/m2; 95% CI, -0.29 to -0.02 kg/m2). The effect persisted in the subset of studies in which the intervention and control groups both received dietary counseling (WMD weight= -0.86 kg; 95% CI, -1.45 to -0.27 kg; WMD BMI= -0.30 kg/m2; 95% CI, -0.50 to -0.10 kg/m2). Study quality was low to moderate, and 5 studies used per-protocol analysis instead of intention-to-treat analysis.
Pedometer use benefits patients with musculoskeletal diseases, too
A systematic review and meta-analysis examined the use of pedometers in patients with musculoskeletal diseases.3 It included 7 RCTs lasting 4 weeks to one year with 484 adults, 40 to 82 years of age, with musculoskeletal disorders (eg, back pain, knee pain, hip pain). (One RCT was also included in the diabetes meta-analysis.) Pedometer use resulted in a mean increase in physical activity of 1950 steps per day above baseline (range=818-2829 steps/day; P<.05). The authors noted that 4 of the 7 studies also demonstrated significant improvement in pain scores and physical function. BMI data were not tracked in this review.
Pedometers increase walking in older patients
A RCT compared the effects of pedometer-based activity prescriptions with standard time-based activity prescriptions in 330 patients ≥65 years of age with baseline low activity levels.4 All patients received an initial physician visit followed by 3 telephone counseling sessions encouraging increased activity. The pedometer group was counseled on increasing steps (without specific targets), while the standard activity prescription group received time-related activity goals.
At one year, “leisure walking” had increased more for the pedometer group than for the standard group (mean 50 minutes/week vs 28 minutes/week; P=.03), although both groups equally increased their amount of “total activity.”
1. Bravata DM, Smith-Spangler C, Sundaram V, et al. Using pedometers to increase physical activity and improve health: a systematic review. JAMA. 2007;298:2296-2304.
2. Cai X, Qiu SH, Yin H, et al. Pedometer intervention and weight loss in overweight and obese adults with type 2 diabetes: a meta-analysis. Diabet Med. 2016;33:1035-1044.
3. Mansi S, Milosavljevic S, Baxter GD, et al. A systematic review of studies using pedometers as an intervention for musculoskeletal diseases. BMC Musculoskeletal Disorders. 2014;15:231.
4. Kolt GS, Schofield GM, Kerse N, et al. Healthy Steps trial: pedometer-based advice and physical activity for low-active older adults. Ann Fam Med. 2012;10:206-212.
1. Bravata DM, Smith-Spangler C, Sundaram V, et al. Using pedometers to increase physical activity and improve health: a systematic review. JAMA. 2007;298:2296-2304.
2. Cai X, Qiu SH, Yin H, et al. Pedometer intervention and weight loss in overweight and obese adults with type 2 diabetes: a meta-analysis. Diabet Med. 2016;33:1035-1044.
3. Mansi S, Milosavljevic S, Baxter GD, et al. A systematic review of studies using pedometers as an intervention for musculoskeletal diseases. BMC Musculoskeletal Disorders. 2014;15:231.
4. Kolt GS, Schofield GM, Kerse N, et al. Healthy Steps trial: pedometer-based advice and physical activity for low-active older adults. Ann Fam Med. 2012;10:206-212.
Evidence-based answers from the Family Physicians Inquiries Network
EVIDENCE-BASED ANSWER:
Yes. In overweight and obese patients, exercise interventions using a pedometer increase steps by about a mile per day over the same interventions without access to pedometer information (strength of recommendation [SOR]: A, meta-analysis of randomized controlled trials [RCTs]) and are associated with a modest 4 mm Hg reduction in systolic blood pressure (BP) over baseline (SOR: B, meta-analysis of RCTs and cohort studies). In overweight patients with diabetes, pedometer use with nutritional counseling is associated with 0.86 kg greater weight loss than nutritional counseling alone (SOR: B, meta-analysis of lower quality RCTs).
Pedometers increase activity in patients with various musculoskeletal conditions and may help reduce pain (SOR: B, meta-analysis of RCTs with heterogeneous outcomes). In low-activity elderly patients, pedometers do not appear to increase total activity when added to an exercise program, but they do appear to increase walking (SOR: B, RCT).
There is no evidence concerning the impact of pedometers on cardiovascular outcomes.

Staring down the opioid epidemic
Nearly 80 people die every day in America from an opioid overdose.1 At the same time, sales of prescription painkillers have increased 4-fold since 1999.2 My own medical assistant was given an unsolicited prescription for 40 oxycodone after a wisdom tooth extraction.
Meanwhile, about 80% of the country’s 2 million opioid-dependent patients are not receiving the treatment they need.3,4 In Vermont, for example, more than 500 patients are on waiting lists to receive buprenorphine (the partial opioid agonist used to treat opioid addiction)—a wait that for many of them will last for more than a year and may cost them their life.5
Buprenorphine makes good sense. Fortunately, buprenorphine can reverse opioid cravings within minutes. Medication-assisted treatment with buprenorphine derivatives allows patients to lead normal, productive, and stable lives. Every dollar invested in treating opioid addiction saves society $7 in drug-related crime and criminal justice costs.6 In addition, 50% to 80% of opioid-dependent patients remain opioid-free for 12 months while taking buprenorphine.7
Steps we can take. As family physicians (FPs), we are frequently overwhelmed by regulatory concerns, overhead expenses, and providing meaningful use data to third-party payers. And we sometimes take the easy route of simply prescribing or refilling scheduled drugs. Instead, we should educate ourselves and our patients about alternative therapeutic interventions for pain control and addiction.
To that end, I encourage all FPs to take the 8-hour online course provided by the American Society of Addiction Medicine to obtain a US Drug Enforcement Administration waiver for prescribing buprenorphine (available at: http://www.asam.org/education/live-online-cme/buprenorphine-course). It costs less than $200 and successful completion of this CME program allows FPs to deliver office-based opioid dependency interventions as per the Drug Addiction Treatment Act of 2000.
Right now, monthly patient censuses indicate that there are about 3234 buprenorphine prescribers providing care for 245,016 opioid-dependent patients, and fewer than 20% of those prescribers are FPs.8 We need to change that. We have an opportunity to invest in the future of these high-risk patients. Let’s not let them down.
1. Democratic staff of the senate committee on finance. Dying waiting for treatment: the opioid use disorder treatment gap and the need for funding. October 10, 2016. Available at: https://www.finance.senate.gov/imo/media/doc/101116%20Opioid%20Treatment%20Gap%20Report%20Final.pdf. Accessed December 14, 2016.
2. Centers for Disease Control and Prevention. Vital signs: overdoses of prescription opioid pain relievers—United States, 1999-2008. MMWR Morb Mortal Wkly Rep. 2011;60:1487-1492.
3. Saloner B, Karthikeyan S. Changes in substance abuse treatment use among individuals with opioid use disorders in the United States. JAMA. 2015;314:1515-1517.
4. Substance Abuse and Mental Health Services Administration. Opioids. Available at: https://www.samhsa.gov/atod/opioids. Accessed December 14, 2016.
5. Vestal C. Waiting lists grow for medicine to fight opioid addiction. Stateline. February 11, 2016. Available at: http://www.pewtrusts.org/en/research-and-analysis/blogs/stateline/2016/02/11/waiting-lists-grow-for-medicine-to-fight-opioid-addiction. Accessed December 14, 2016.
6. National Institute on Drug Abuse. Principles of drug addiction treatment: a research-based guide (third edition). Is drug addiction treatment worth its cost? Available at: https://www.drugabuse.gov/publications/principles-drug-addiction-treatment-research-based-guide-third-edition/frequently-asked-questions/drug-addiction-treatment-worth-its-cost. Accessed December 14, 2016.
7. Kleber HD. Pharmacologic treatments for opioid dependence: detoxification and maintenance options. Dialogues Clin Neurosci. 2007; 9:455-470.
8. Stein BD, Sorbero MJ, Dick AW, et al. Physician capacity to treat opioid use disorder with buprenorphine-assisted treatment. JAMA. 2016;316:1211-1212.

Unger Primary Care Concierge Medical Group, Rancho Cucamonga, Calif. Dr. Unger is a member of JFP’s editorial board. He reported no potential conflict of interest relevant to this article.
Unger Primary Care Concierge Medical Group, Rancho Cucamonga, Calif. Dr. Unger is a member of JFP’s editorial board. He reported no potential conflict of interest relevant to this article.
Unger Primary Care Concierge Medical Group, Rancho Cucamonga, Calif. Dr. Unger is a member of JFP’s editorial board. He reported no potential conflict of interest relevant to this article.
Nearly 80 people die every day in America from an opioid overdose.1 At the same time, sales of prescription painkillers have increased 4-fold since 1999.2 My own medical assistant was given an unsolicited prescription for 40 oxycodone after a wisdom tooth extraction.
Meanwhile, about 80% of the country’s 2 million opioid-dependent patients are not receiving the treatment they need.3,4 In Vermont, for example, more than 500 patients are on waiting lists to receive buprenorphine (the partial opioid agonist used to treat opioid addiction)—a wait that for many of them will last for more than a year and may cost them their life.5
Buprenorphine makes good sense. Fortunately, buprenorphine can reverse opioid cravings within minutes. Medication-assisted treatment with buprenorphine derivatives allows patients to lead normal, productive, and stable lives. Every dollar invested in treating opioid addiction saves society $7 in drug-related crime and criminal justice costs.6 In addition, 50% to 80% of opioid-dependent patients remain opioid-free for 12 months while taking buprenorphine.7
Steps we can take. As family physicians (FPs), we are frequently overwhelmed by regulatory concerns, overhead expenses, and providing meaningful use data to third-party payers. And we sometimes take the easy route of simply prescribing or refilling scheduled drugs. Instead, we should educate ourselves and our patients about alternative therapeutic interventions for pain control and addiction.
To that end, I encourage all FPs to take the 8-hour online course provided by the American Society of Addiction Medicine to obtain a US Drug Enforcement Administration waiver for prescribing buprenorphine (available at: http://www.asam.org/education/live-online-cme/buprenorphine-course). It costs less than $200 and successful completion of this CME program allows FPs to deliver office-based opioid dependency interventions as per the Drug Addiction Treatment Act of 2000.
Right now, monthly patient censuses indicate that there are about 3234 buprenorphine prescribers providing care for 245,016 opioid-dependent patients, and fewer than 20% of those prescribers are FPs.8 We need to change that. We have an opportunity to invest in the future of these high-risk patients. Let’s not let them down.
Nearly 80 people die every day in America from an opioid overdose.1 At the same time, sales of prescription painkillers have increased 4-fold since 1999.2 My own medical assistant was given an unsolicited prescription for 40 oxycodone after a wisdom tooth extraction.
Meanwhile, about 80% of the country’s 2 million opioid-dependent patients are not receiving the treatment they need.3,4 In Vermont, for example, more than 500 patients are on waiting lists to receive buprenorphine (the partial opioid agonist used to treat opioid addiction)—a wait that for many of them will last for more than a year and may cost them their life.5
Buprenorphine makes good sense. Fortunately, buprenorphine can reverse opioid cravings within minutes. Medication-assisted treatment with buprenorphine derivatives allows patients to lead normal, productive, and stable lives. Every dollar invested in treating opioid addiction saves society $7 in drug-related crime and criminal justice costs.6 In addition, 50% to 80% of opioid-dependent patients remain opioid-free for 12 months while taking buprenorphine.7
Steps we can take. As family physicians (FPs), we are frequently overwhelmed by regulatory concerns, overhead expenses, and providing meaningful use data to third-party payers. And we sometimes take the easy route of simply prescribing or refilling scheduled drugs. Instead, we should educate ourselves and our patients about alternative therapeutic interventions for pain control and addiction.
To that end, I encourage all FPs to take the 8-hour online course provided by the American Society of Addiction Medicine to obtain a US Drug Enforcement Administration waiver for prescribing buprenorphine (available at: http://www.asam.org/education/live-online-cme/buprenorphine-course). It costs less than $200 and successful completion of this CME program allows FPs to deliver office-based opioid dependency interventions as per the Drug Addiction Treatment Act of 2000.
Right now, monthly patient censuses indicate that there are about 3234 buprenorphine prescribers providing care for 245,016 opioid-dependent patients, and fewer than 20% of those prescribers are FPs.8 We need to change that. We have an opportunity to invest in the future of these high-risk patients. Let’s not let them down.
1. Democratic staff of the senate committee on finance. Dying waiting for treatment: the opioid use disorder treatment gap and the need for funding. October 10, 2016. Available at: https://www.finance.senate.gov/imo/media/doc/101116%20Opioid%20Treatment%20Gap%20Report%20Final.pdf. Accessed December 14, 2016.
2. Centers for Disease Control and Prevention. Vital signs: overdoses of prescription opioid pain relievers—United States, 1999-2008. MMWR Morb Mortal Wkly Rep. 2011;60:1487-1492.
3. Saloner B, Karthikeyan S. Changes in substance abuse treatment use among individuals with opioid use disorders in the United States. JAMA. 2015;314:1515-1517.
4. Substance Abuse and Mental Health Services Administration. Opioids. Available at: https://www.samhsa.gov/atod/opioids. Accessed December 14, 2016.
5. Vestal C. Waiting lists grow for medicine to fight opioid addiction. Stateline. February 11, 2016. Available at: http://www.pewtrusts.org/en/research-and-analysis/blogs/stateline/2016/02/11/waiting-lists-grow-for-medicine-to-fight-opioid-addiction. Accessed December 14, 2016.
6. National Institute on Drug Abuse. Principles of drug addiction treatment: a research-based guide (third edition). Is drug addiction treatment worth its cost? Available at: https://www.drugabuse.gov/publications/principles-drug-addiction-treatment-research-based-guide-third-edition/frequently-asked-questions/drug-addiction-treatment-worth-its-cost. Accessed December 14, 2016.
7. Kleber HD. Pharmacologic treatments for opioid dependence: detoxification and maintenance options. Dialogues Clin Neurosci. 2007; 9:455-470.
8. Stein BD, Sorbero MJ, Dick AW, et al. Physician capacity to treat opioid use disorder with buprenorphine-assisted treatment. JAMA. 2016;316:1211-1212.
1. Democratic staff of the senate committee on finance. Dying waiting for treatment: the opioid use disorder treatment gap and the need for funding. October 10, 2016. Available at: https://www.finance.senate.gov/imo/media/doc/101116%20Opioid%20Treatment%20Gap%20Report%20Final.pdf. Accessed December 14, 2016.
2. Centers for Disease Control and Prevention. Vital signs: overdoses of prescription opioid pain relievers—United States, 1999-2008. MMWR Morb Mortal Wkly Rep. 2011;60:1487-1492.
3. Saloner B, Karthikeyan S. Changes in substance abuse treatment use among individuals with opioid use disorders in the United States. JAMA. 2015;314:1515-1517.
4. Substance Abuse and Mental Health Services Administration. Opioids. Available at: https://www.samhsa.gov/atod/opioids. Accessed December 14, 2016.
5. Vestal C. Waiting lists grow for medicine to fight opioid addiction. Stateline. February 11, 2016. Available at: http://www.pewtrusts.org/en/research-and-analysis/blogs/stateline/2016/02/11/waiting-lists-grow-for-medicine-to-fight-opioid-addiction. Accessed December 14, 2016.
6. National Institute on Drug Abuse. Principles of drug addiction treatment: a research-based guide (third edition). Is drug addiction treatment worth its cost? Available at: https://www.drugabuse.gov/publications/principles-drug-addiction-treatment-research-based-guide-third-edition/frequently-asked-questions/drug-addiction-treatment-worth-its-cost. Accessed December 14, 2016.
7. Kleber HD. Pharmacologic treatments for opioid dependence: detoxification and maintenance options. Dialogues Clin Neurosci. 2007; 9:455-470.
8. Stein BD, Sorbero MJ, Dick AW, et al. Physician capacity to treat opioid use disorder with buprenorphine-assisted treatment. JAMA. 2016;316:1211-1212.