User login
Tele-rheumatology reaches patients who lack access to care
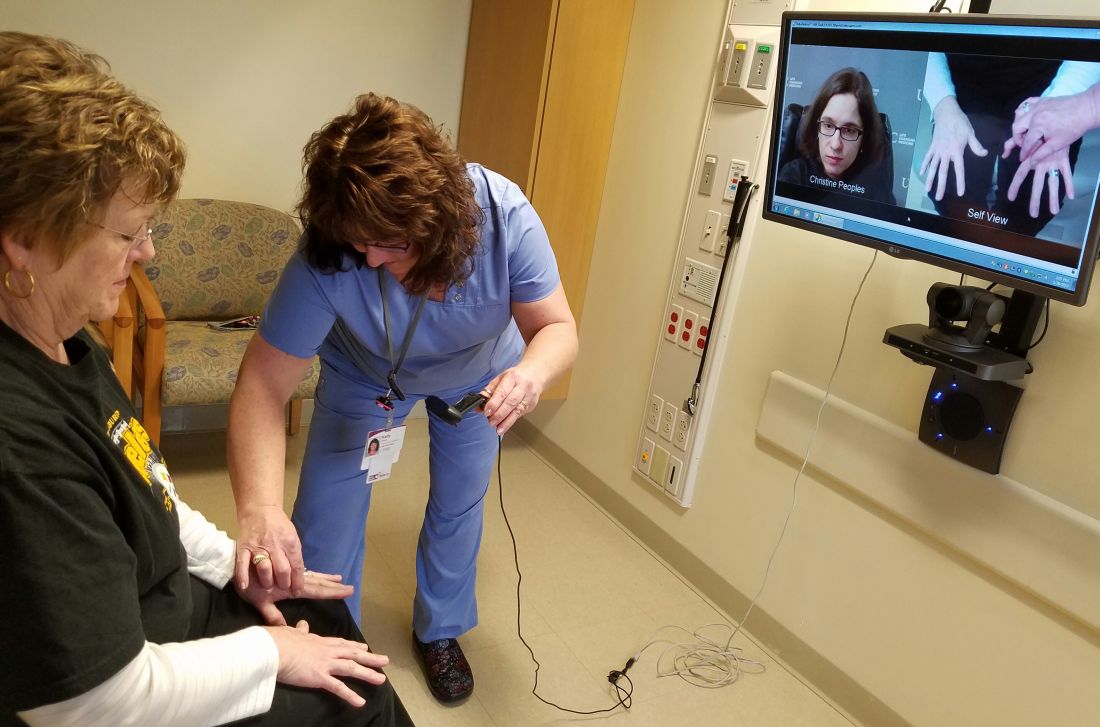
By the time Christine Peoples, MD, examined the patient, the middle-aged man had been treated inadequately for psoriatic arthritis for years.
Poor mobility made it difficult for him to travel and gaps in his care had exacerbated his condition. But a bidirectional video system at the University of Pittsburgh Medical Center (UPMC) allowed Dr. Peoples to assess the patient, correct his therapeutic regimen, and establish consistent follow up.
Without telemedicine, the patient would have likely continued to deteriorate, said Dr. Peoples, a rheumatologist with the UPMC Teleconsult Centers.
Across the country, more rheumatology practices are employing telemedicine to treat patients, particularly physicians at academic medicine centers that have the resources to launch comprehensive units. The technology is a way to bridge the shortage of rheumatologists in rural areas and reach patients who cannot access specialty care for rheumatic conditions, proponents say. A 2013 study in Arthritis & Rheumatism found that many towns and small cities with populations up to 50,000 have no practicing rheumatologists, with some patients having to travel more than 200 miles to get specialty care (2013 Dec;65[12]:3017-25).

“Telemedicine is expanding both in terms of the number of consultations and the breadth of services involved,” he said. “Tele-rheumatology was certainly not a big area to be looked at in years past, but it’s one more area that is starting to expand.”
Research is lacking regarding how prevalent tele-rheumatology has become in the United States and whether it’s as effective as face-to-face visits. In a recent analysis of 1,468 potentially eligible tele-rheumatology studies and literature, only 20 addressed direct provider to patient contact that influenced or had the potential to influence clinical care, according to a November 2016 review in Arthritis Care & Research (doi: 10.1002/acr.23153). Of the 20 studies, the majority of articles involved tele-rheumatology use in Europe or Great Britain, said study author John Allen McDougall, MD, a postdoctoral fellow at Yale University, New Haven, Conn. According to the literature, the most common condition treated through telemedicine was rheumatoid arthritis. Little information existed on telemedicine use for gout or the treatment of connective tissue diseases, Dr. McDougall said.

Increasing access, saving time
UPMC has operated a tele-rheumatology program for nearly 3 years that connects rural patients with their specialists. Patients present for video examination at teleconsult centers located in underserved areas within western and central Pennsylvania. On the other end of the screen, Dr. Peoples uses high-level audio and video technology to discuss the patient’s history, direct nurses, make diagnoses, answer questions, and prescribe treatment to patients.
“Patients comment about how much easier it is for them and what a difference this makes in their health care,” Dr. Peoples said. “[Tele-rheumatology also increases] the ability to have regular follow-up care. I see them every few months for their condition and manage it. You can really establish a long relationship with patients just as you would if you saw patients in person.”
Rheumatologists at Dartmouth–Hitchcock Medical Center (DHMC) in Lebanon, N.H., are achieving similar results.
The medical center’s tele-rheumatology unit began in 2011 with the aim of increasing specialty access for patients in northern New Hampshire. Dartmouth also employed a telemedicine clinic from 2012 to 2013 in southern Vermont while seeking to replace a well-established rheumatologist who had retired.
Between October 2011 and December 2014, 176 patients were seen via tele-rheumatology between the two sites over the course of 244 tele-rheumatology patient visits with Dartmouth providers, according to a study published in the June 2016 Seminars in Arthritis & Rheumatism (2016 Dec;46[3]:380-5). Patients lived on average 99 miles from DHMC and 22 miles from the remote clinic site, the study found. The top diagnosis was inflammatory arthritis.

For most of doctors and patients involved in the study, tele-rheumatology visits were positive and aided patient access, said Daniel Albert, MD, section chief of rheumatology at DHMC.
“There are some limitations, there’s no question about that,” he said. ... [However], right now there are way too few rheumatologists to care for patients with rheumatic disease. We’ve tried to improve that with [nurse practitioners and physician assistants], but this is another way of addressing that need.”
Pediatric rheumatologists are also using the benefits of telemedicine. Children’s Mercy in Kansas City, Mo., launched a pediatric rheumatology telemedicine clinic in 2014 that links children in Joplin, Mo., with rheumatologists at Children’s Mercy, about 160 miles away. Patients and their parents/guardians treated via telemedicine at the Joplin site missed less work and school and saved money on food, compared with traveling to Kansas City for treatment, according to a 2016 study published in Pediatric Rheumatology (doi: 10.1186/s12969-016-0116-2).
“This is much more convenient for patients,” said Elizabeth A. Kessler, MD, a pediatric rheumatologist at Children’s Mercy and coauthor of the study. “It saves them time and money. With the training of the tele-facilitator, I believe that we’re able to provide them with equivalent care compared with if they were in-person.”
Is tele-rheumatology right for all settings?
But tele-rheumatology may not be appropriate in all practice settings or the right method for all rheumatology patients, doctors say.
As the Dartmouth-Hitchcock study noted, patients with complex diseases or unclear underlying conditions do not make good telemedicine patients. In addition, opinions are mixed about whether physicians and patients should have an established relationship before telemedicine visits occur and what such visits should entail.
Anchorage, Alaska-based rheumatologist Elizabeth Ferucci, MD, uses telemedicine only for follow up. Like many rheumatologists in Alaska, she travels to field clinics in larger communities throughout the state twice a year to treat patients, said Dr. Ferucci, a rheumatologist for the Alaska Native Tribal Health Consortium. Because some patients need to be seen more than twice a year, Dr. Ferucci conducts follow-up visits every 2-3 months via video.

However, Dr. Ferucci notes that having a trained telemedicine presenter capable of performing a joint examination in the room with the patient is not always possible because of high staff turnover and the more than 200 villages served. A nurse or staff member remains in the room with the patient, but they are not always trained to perform joint exams.
At Dartmouth-Hitchcock, Dr. Albert’s practice accepts both initial and follow-up patient visits. He acknowledges that tele-rheumatology is best used for follow-up care, but explains that limiting the center’s telemedicine use would leave some patients without treatment.
“Because the constraints of getting to Dartmouth are so great, we don’t want to impose that restriction on patients,” Dr. Albert said. “However, in general, it is preferable to do an in-person consultation first to clarify the physical findings.”
Dartmouth has used both nurses and medical assistants as presenters during tele-rheumatology after developing a web-based video series to train presenters. Nurses at UPMC are also trained as telemedicine presenters, and telemedicine visits are only used for follow-up care, Dr. Peoples said.
At Children’s Mercy in Kansas City, telemedicine visits are restricted to follow-up care and include a nurse facilitator at the virtual site. However, the type of conditions and stages of disease examined through the technology has grown since the telemedicine services started, Dr. Kessler said.
“What we feel comfortable seeing has expanded since the time we initially started the clinic because some of the patients, when they were having flares, could not come to Kansas City for financial reasons,” she said, adding that she has since examined children with lupus, vasculitis, and active arthritis through telemedicine.
She stresses that in-person visits should depend on the patient, the circumstances, and the physician’s comfort level. ”You need to be realistic,” she said. “There are times when I’ve said to families, ‘I would like you to come to Kansas City because I’m a little unsure about your exam findings.’ There are definitely some patients who do more of a hybrid approach where they come to Kansas City and are also seen by telemedicine.”
Barriers slow telemedicine progress
Amid its benefits, challenges for tele-rheumatology, such as reimbursement and licensure obstacles, remain.
Commercial insurers pay for telemedicine at varying rates, making it difficult for doctors to consistently be paid, Dr. Peoples said.
“A lot of insurance companies do cover telemedicine visits, but still, a lot don’t,” she said. “I do think that in order to remain competitive, insurance companies overall are going to have to start covering these services.”
At least 32 states and the District of Columbia have parity laws that require commercial health insurers to cover services provided through telehealth to the same extent as those services are covered in person, according to an Aug. 15, 2016 Health Affairs policy brief. However, how private insurers pay and what services they cover vary widely.
Medicaid reimbursement for telehealth depends on individual state Medicaid programs. Only 9 states Medicaid programs reimburse for store-and-forward services, while at least 16 states have some sort of Medicaid reimbursement for remote patient monitoring, according to the Health Affairs report.
Medicare payment for telehealth is clouded in restrictions. The Centers for Medicare & Medicaid Services reimburses for synchronous communications only and does not cover store-and-forward services or remote patient monitoring for chronic diseases, except in Alaska and Hawaii. The patient must be present at an originating site for the visit and cannot be home to receive the services. In addition, CMS reimburses for telehealth only when the originating site is in a Health Professional Shortage Area or within a county outside a Metropolitan Statistical Area. In 2016, CMS expanded its coverage of telehealth to include several new codes including, end-stage renal disease–related services for dialysis, advance care planning services, and critical care consultations furnished via telehealth using new Medicare G-codes.
It’s worth noting that rheumatologists who are salaried employees of academic medical centers, as is the case for all rheumatologists interviewed here, don’t handle the billing for their telemedicine services, and so have not experienced the payment challenges that smaller practices may face.
Mr. Linkous of the American Telemedicine Association said that he believes that reimbursement for telemedicine will improve as health care heads toward value-based payment systems.
“In terms of reimbursement, with the move away from fee-based systems to value-based systems, it really opens the door because for many of the services, you no longer write out a bill with a code associated with it,” he said. “You are paid on the basis of keeping people well. That’s a great positive incentive for using telemedicine.”
Differing state licensure requirements also pose a challenge. The process to obtain licenses and navigate varying credentialing processes can be a headache for health providers and delay approval.
The barrier has led to model legislation by the Federation of State Medical Boards (FSMB) that aims to make it easier for telemedicine physicians to gain licenses in multiple states. Under the Interstate Medical Licensure Compact, physicians designate a member state as the state of principal licensure and select the other states in which they want to license. The state of principal licensure then verifies the physician’s eligibility and provides credential information to the interstate commission, which collects applicable fees and transmits the doctor’s information to the other states. Upon receipt in the additional states, the physician would be granted a license.
As of January, 18 states had enacted the compact legislation, and at least 4 states had introduced the legislation. In 2015, the Health Resources and Services Administration awarded the FSMB a grant to support establishment of the commission and aid with the compact’s infrastructure.
A new normal?
Dr. Albert notes that tele-rheumatology programs can succeed only with adequate resources, time, and staff.
“To set up Skype or Facetime is nothing; you just click on it,” he said. “To set up tele-rheumatology requires a fair amount of administrative substructure that has to be generated in terms of who’s going to do the documentation? Who is going to do the billing? Who is going to do the credentialing? Where is the credentialing going to be validated?”
The equipment required for telemedicine visits also requires a significant amount of time and regular maintenance, Dr. Peoples said. At UPMC for instance, the tele-rheumatology program has an information technology team that assists with technology glitches, changes, and updates, she said. That’s why it’s helpful to have the support of a large academic medical center with the sufficient resources to build a telemedicine program, she added.
In order for more practices to consider tele-rheumatology, more research about cost-effectiveness and best uses of the technology use would be useful, Dr. McDougall said.
“The main question that policy makers are going to want to answer is, ‘What’s the return on investment? Does this make sense for my practice?’ ” he said. “The methods reporting in tele-rheumatologist [literature] is lacking.” But regardless of barriers, telemedicine experts say the technology will likely continue to expand and transform the way rheumatologists are practicing and patients are receiving care.
“Tele-rheumatology will never replace an in-person exam,” Dr. Ferucci said. “But my vision is that it will be able to improve the quality of care for patients living in rural and remote locations, by allowing for more frequent visits and adjustment of medications, which are necessary to achieve the goal of treat-to-target for RA and other rheumatologic conditions.”
* This story was updated on 2/10/2017.
On Twitter @legal_med
By the time Christine Peoples, MD, examined the patient, the middle-aged man had been treated inadequately for psoriatic arthritis for years.
Poor mobility made it difficult for him to travel and gaps in his care had exacerbated his condition. But a bidirectional video system at the University of Pittsburgh Medical Center (UPMC) allowed Dr. Peoples to assess the patient, correct his therapeutic regimen, and establish consistent follow up.
Without telemedicine, the patient would have likely continued to deteriorate, said Dr. Peoples, a rheumatologist with the UPMC Teleconsult Centers.
Across the country, more rheumatology practices are employing telemedicine to treat patients, particularly physicians at academic medicine centers that have the resources to launch comprehensive units. The technology is a way to bridge the shortage of rheumatologists in rural areas and reach patients who cannot access specialty care for rheumatic conditions, proponents say. A 2013 study in Arthritis & Rheumatism found that many towns and small cities with populations up to 50,000 have no practicing rheumatologists, with some patients having to travel more than 200 miles to get specialty care (2013 Dec;65[12]:3017-25).
“Telemedicine is expanding both in terms of the number of consultations and the breadth of services involved,” he said. “Tele-rheumatology was certainly not a big area to be looked at in years past, but it’s one more area that is starting to expand.”
Research is lacking regarding how prevalent tele-rheumatology has become in the United States and whether it’s as effective as face-to-face visits. In a recent analysis of 1,468 potentially eligible tele-rheumatology studies and literature, only 20 addressed direct provider to patient contact that influenced or had the potential to influence clinical care, according to a November 2016 review in Arthritis Care & Research (doi: 10.1002/acr.23153). Of the 20 studies, the majority of articles involved tele-rheumatology use in Europe or Great Britain, said study author John Allen McDougall, MD, a postdoctoral fellow at Yale University, New Haven, Conn. According to the literature, the most common condition treated through telemedicine was rheumatoid arthritis. Little information existed on telemedicine use for gout or the treatment of connective tissue diseases, Dr. McDougall said.
Increasing access, saving time
UPMC has operated a tele-rheumatology program for nearly 3 years that connects rural patients with their specialists. Patients present for video examination at teleconsult centers located in underserved areas within western and central Pennsylvania. On the other end of the screen, Dr. Peoples uses high-level audio and video technology to discuss the patient’s history, direct nurses, make diagnoses, answer questions, and prescribe treatment to patients.
“Patients comment about how much easier it is for them and what a difference this makes in their health care,” Dr. Peoples said. “[Tele-rheumatology also increases] the ability to have regular follow-up care. I see them every few months for their condition and manage it. You can really establish a long relationship with patients just as you would if you saw patients in person.”
Rheumatologists at Dartmouth–Hitchcock Medical Center (DHMC) in Lebanon, N.H., are achieving similar results.
The medical center’s tele-rheumatology unit began in 2011 with the aim of increasing specialty access for patients in northern New Hampshire. Dartmouth also employed a telemedicine clinic from 2012 to 2013 in southern Vermont while seeking to replace a well-established rheumatologist who had retired.
Between October 2011 and December 2014, 176 patients were seen via tele-rheumatology between the two sites over the course of 244 tele-rheumatology patient visits with Dartmouth providers, according to a study published in the June 2016 Seminars in Arthritis & Rheumatism (2016 Dec;46[3]:380-5). Patients lived on average 99 miles from DHMC and 22 miles from the remote clinic site, the study found. The top diagnosis was inflammatory arthritis.
For most of doctors and patients involved in the study, tele-rheumatology visits were positive and aided patient access, said Daniel Albert, MD, section chief of rheumatology at DHMC.
“There are some limitations, there’s no question about that,” he said. ... [However], right now there are way too few rheumatologists to care for patients with rheumatic disease. We’ve tried to improve that with [nurse practitioners and physician assistants], but this is another way of addressing that need.”
Pediatric rheumatologists are also using the benefits of telemedicine. Children’s Mercy in Kansas City, Mo., launched a pediatric rheumatology telemedicine clinic in 2014 that links children in Joplin, Mo., with rheumatologists at Children’s Mercy, about 160 miles away. Patients and their parents/guardians treated via telemedicine at the Joplin site missed less work and school and saved money on food, compared with traveling to Kansas City for treatment, according to a 2016 study published in Pediatric Rheumatology (doi: 10.1186/s12969-016-0116-2).
“This is much more convenient for patients,” said Elizabeth A. Kessler, MD, a pediatric rheumatologist at Children’s Mercy and coauthor of the study. “It saves them time and money. With the training of the tele-facilitator, I believe that we’re able to provide them with equivalent care compared with if they were in-person.”
Is tele-rheumatology right for all settings?
But tele-rheumatology may not be appropriate in all practice settings or the right method for all rheumatology patients, doctors say.
As the Dartmouth-Hitchcock study noted, patients with complex diseases or unclear underlying conditions do not make good telemedicine patients. In addition, opinions are mixed about whether physicians and patients should have an established relationship before telemedicine visits occur and what such visits should entail.
Anchorage, Alaska-based rheumatologist Elizabeth Ferucci, MD, uses telemedicine only for follow up. Like many rheumatologists in Alaska, she travels to field clinics in larger communities throughout the state twice a year to treat patients, said Dr. Ferucci, a rheumatologist for the Alaska Native Tribal Health Consortium. Because some patients need to be seen more than twice a year, Dr. Ferucci conducts follow-up visits every 2-3 months via video.
However, Dr. Ferucci notes that having a trained telemedicine presenter capable of performing a joint examination in the room with the patient is not always possible because of high staff turnover and the more than 200 villages served. A nurse or staff member remains in the room with the patient, but they are not always trained to perform joint exams.
At Dartmouth-Hitchcock, Dr. Albert’s practice accepts both initial and follow-up patient visits. He acknowledges that tele-rheumatology is best used for follow-up care, but explains that limiting the center’s telemedicine use would leave some patients without treatment.
“Because the constraints of getting to Dartmouth are so great, we don’t want to impose that restriction on patients,” Dr. Albert said. “However, in general, it is preferable to do an in-person consultation first to clarify the physical findings.”
Dartmouth has used both nurses and medical assistants as presenters during tele-rheumatology after developing a web-based video series to train presenters. Nurses at UPMC are also trained as telemedicine presenters, and telemedicine visits are only used for follow-up care, Dr. Peoples said.
At Children’s Mercy in Kansas City, telemedicine visits are restricted to follow-up care and include a nurse facilitator at the virtual site. However, the type of conditions and stages of disease examined through the technology has grown since the telemedicine services started, Dr. Kessler said.
“What we feel comfortable seeing has expanded since the time we initially started the clinic because some of the patients, when they were having flares, could not come to Kansas City for financial reasons,” she said, adding that she has since examined children with lupus, vasculitis, and active arthritis through telemedicine.
She stresses that in-person visits should depend on the patient, the circumstances, and the physician’s comfort level. ”You need to be realistic,” she said. “There are times when I’ve said to families, ‘I would like you to come to Kansas City because I’m a little unsure about your exam findings.’ There are definitely some patients who do more of a hybrid approach where they come to Kansas City and are also seen by telemedicine.”
Barriers slow telemedicine progress
Amid its benefits, challenges for tele-rheumatology, such as reimbursement and licensure obstacles, remain.
Commercial insurers pay for telemedicine at varying rates, making it difficult for doctors to consistently be paid, Dr. Peoples said.
“A lot of insurance companies do cover telemedicine visits, but still, a lot don’t,” she said. “I do think that in order to remain competitive, insurance companies overall are going to have to start covering these services.”
At least 32 states and the District of Columbia have parity laws that require commercial health insurers to cover services provided through telehealth to the same extent as those services are covered in person, according to an Aug. 15, 2016 Health Affairs policy brief. However, how private insurers pay and what services they cover vary widely.
Medicaid reimbursement for telehealth depends on individual state Medicaid programs. Only 9 states Medicaid programs reimburse for store-and-forward services, while at least 16 states have some sort of Medicaid reimbursement for remote patient monitoring, according to the Health Affairs report.
Medicare payment for telehealth is clouded in restrictions. The Centers for Medicare & Medicaid Services reimburses for synchronous communications only and does not cover store-and-forward services or remote patient monitoring for chronic diseases, except in Alaska and Hawaii. The patient must be present at an originating site for the visit and cannot be home to receive the services. In addition, CMS reimburses for telehealth only when the originating site is in a Health Professional Shortage Area or within a county outside a Metropolitan Statistical Area. In 2016, CMS expanded its coverage of telehealth to include several new codes including, end-stage renal disease–related services for dialysis, advance care planning services, and critical care consultations furnished via telehealth using new Medicare G-codes.
It’s worth noting that rheumatologists who are salaried employees of academic medical centers, as is the case for all rheumatologists interviewed here, don’t handle the billing for their telemedicine services, and so have not experienced the payment challenges that smaller practices may face.
Mr. Linkous of the American Telemedicine Association said that he believes that reimbursement for telemedicine will improve as health care heads toward value-based payment systems.
“In terms of reimbursement, with the move away from fee-based systems to value-based systems, it really opens the door because for many of the services, you no longer write out a bill with a code associated with it,” he said. “You are paid on the basis of keeping people well. That’s a great positive incentive for using telemedicine.”
Differing state licensure requirements also pose a challenge. The process to obtain licenses and navigate varying credentialing processes can be a headache for health providers and delay approval.
The barrier has led to model legislation by the Federation of State Medical Boards (FSMB) that aims to make it easier for telemedicine physicians to gain licenses in multiple states. Under the Interstate Medical Licensure Compact, physicians designate a member state as the state of principal licensure and select the other states in which they want to license. The state of principal licensure then verifies the physician’s eligibility and provides credential information to the interstate commission, which collects applicable fees and transmits the doctor’s information to the other states. Upon receipt in the additional states, the physician would be granted a license.
As of January, 18 states had enacted the compact legislation, and at least 4 states had introduced the legislation. In 2015, the Health Resources and Services Administration awarded the FSMB a grant to support establishment of the commission and aid with the compact’s infrastructure.
A new normal?
Dr. Albert notes that tele-rheumatology programs can succeed only with adequate resources, time, and staff.
“To set up Skype or Facetime is nothing; you just click on it,” he said. “To set up tele-rheumatology requires a fair amount of administrative substructure that has to be generated in terms of who’s going to do the documentation? Who is going to do the billing? Who is going to do the credentialing? Where is the credentialing going to be validated?”
The equipment required for telemedicine visits also requires a significant amount of time and regular maintenance, Dr. Peoples said. At UPMC for instance, the tele-rheumatology program has an information technology team that assists with technology glitches, changes, and updates, she said. That’s why it’s helpful to have the support of a large academic medical center with the sufficient resources to build a telemedicine program, she added.
In order for more practices to consider tele-rheumatology, more research about cost-effectiveness and best uses of the technology use would be useful, Dr. McDougall said.
“The main question that policy makers are going to want to answer is, ‘What’s the return on investment? Does this make sense for my practice?’ ” he said. “The methods reporting in tele-rheumatologist [literature] is lacking.” But regardless of barriers, telemedicine experts say the technology will likely continue to expand and transform the way rheumatologists are practicing and patients are receiving care.
“Tele-rheumatology will never replace an in-person exam,” Dr. Ferucci said. “But my vision is that it will be able to improve the quality of care for patients living in rural and remote locations, by allowing for more frequent visits and adjustment of medications, which are necessary to achieve the goal of treat-to-target for RA and other rheumatologic conditions.”
* This story was updated on 2/10/2017.
On Twitter @legal_med
By the time Christine Peoples, MD, examined the patient, the middle-aged man had been treated inadequately for psoriatic arthritis for years.
Poor mobility made it difficult for him to travel and gaps in his care had exacerbated his condition. But a bidirectional video system at the University of Pittsburgh Medical Center (UPMC) allowed Dr. Peoples to assess the patient, correct his therapeutic regimen, and establish consistent follow up.
Without telemedicine, the patient would have likely continued to deteriorate, said Dr. Peoples, a rheumatologist with the UPMC Teleconsult Centers.
Across the country, more rheumatology practices are employing telemedicine to treat patients, particularly physicians at academic medicine centers that have the resources to launch comprehensive units. The technology is a way to bridge the shortage of rheumatologists in rural areas and reach patients who cannot access specialty care for rheumatic conditions, proponents say. A 2013 study in Arthritis & Rheumatism found that many towns and small cities with populations up to 50,000 have no practicing rheumatologists, with some patients having to travel more than 200 miles to get specialty care (2013 Dec;65[12]:3017-25).
“Telemedicine is expanding both in terms of the number of consultations and the breadth of services involved,” he said. “Tele-rheumatology was certainly not a big area to be looked at in years past, but it’s one more area that is starting to expand.”
Research is lacking regarding how prevalent tele-rheumatology has become in the United States and whether it’s as effective as face-to-face visits. In a recent analysis of 1,468 potentially eligible tele-rheumatology studies and literature, only 20 addressed direct provider to patient contact that influenced or had the potential to influence clinical care, according to a November 2016 review in Arthritis Care & Research (doi: 10.1002/acr.23153). Of the 20 studies, the majority of articles involved tele-rheumatology use in Europe or Great Britain, said study author John Allen McDougall, MD, a postdoctoral fellow at Yale University, New Haven, Conn. According to the literature, the most common condition treated through telemedicine was rheumatoid arthritis. Little information existed on telemedicine use for gout or the treatment of connective tissue diseases, Dr. McDougall said.
Increasing access, saving time
UPMC has operated a tele-rheumatology program for nearly 3 years that connects rural patients with their specialists. Patients present for video examination at teleconsult centers located in underserved areas within western and central Pennsylvania. On the other end of the screen, Dr. Peoples uses high-level audio and video technology to discuss the patient’s history, direct nurses, make diagnoses, answer questions, and prescribe treatment to patients.
“Patients comment about how much easier it is for them and what a difference this makes in their health care,” Dr. Peoples said. “[Tele-rheumatology also increases] the ability to have regular follow-up care. I see them every few months for their condition and manage it. You can really establish a long relationship with patients just as you would if you saw patients in person.”
Rheumatologists at Dartmouth–Hitchcock Medical Center (DHMC) in Lebanon, N.H., are achieving similar results.
The medical center’s tele-rheumatology unit began in 2011 with the aim of increasing specialty access for patients in northern New Hampshire. Dartmouth also employed a telemedicine clinic from 2012 to 2013 in southern Vermont while seeking to replace a well-established rheumatologist who had retired.
Between October 2011 and December 2014, 176 patients were seen via tele-rheumatology between the two sites over the course of 244 tele-rheumatology patient visits with Dartmouth providers, according to a study published in the June 2016 Seminars in Arthritis & Rheumatism (2016 Dec;46[3]:380-5). Patients lived on average 99 miles from DHMC and 22 miles from the remote clinic site, the study found. The top diagnosis was inflammatory arthritis.
For most of doctors and patients involved in the study, tele-rheumatology visits were positive and aided patient access, said Daniel Albert, MD, section chief of rheumatology at DHMC.
“There are some limitations, there’s no question about that,” he said. ... [However], right now there are way too few rheumatologists to care for patients with rheumatic disease. We’ve tried to improve that with [nurse practitioners and physician assistants], but this is another way of addressing that need.”
Pediatric rheumatologists are also using the benefits of telemedicine. Children’s Mercy in Kansas City, Mo., launched a pediatric rheumatology telemedicine clinic in 2014 that links children in Joplin, Mo., with rheumatologists at Children’s Mercy, about 160 miles away. Patients and their parents/guardians treated via telemedicine at the Joplin site missed less work and school and saved money on food, compared with traveling to Kansas City for treatment, according to a 2016 study published in Pediatric Rheumatology (doi: 10.1186/s12969-016-0116-2).
“This is much more convenient for patients,” said Elizabeth A. Kessler, MD, a pediatric rheumatologist at Children’s Mercy and coauthor of the study. “It saves them time and money. With the training of the tele-facilitator, I believe that we’re able to provide them with equivalent care compared with if they were in-person.”
Is tele-rheumatology right for all settings?
But tele-rheumatology may not be appropriate in all practice settings or the right method for all rheumatology patients, doctors say.
As the Dartmouth-Hitchcock study noted, patients with complex diseases or unclear underlying conditions do not make good telemedicine patients. In addition, opinions are mixed about whether physicians and patients should have an established relationship before telemedicine visits occur and what such visits should entail.
Anchorage, Alaska-based rheumatologist Elizabeth Ferucci, MD, uses telemedicine only for follow up. Like many rheumatologists in Alaska, she travels to field clinics in larger communities throughout the state twice a year to treat patients, said Dr. Ferucci, a rheumatologist for the Alaska Native Tribal Health Consortium. Because some patients need to be seen more than twice a year, Dr. Ferucci conducts follow-up visits every 2-3 months via video.
However, Dr. Ferucci notes that having a trained telemedicine presenter capable of performing a joint examination in the room with the patient is not always possible because of high staff turnover and the more than 200 villages served. A nurse or staff member remains in the room with the patient, but they are not always trained to perform joint exams.
At Dartmouth-Hitchcock, Dr. Albert’s practice accepts both initial and follow-up patient visits. He acknowledges that tele-rheumatology is best used for follow-up care, but explains that limiting the center’s telemedicine use would leave some patients without treatment.
“Because the constraints of getting to Dartmouth are so great, we don’t want to impose that restriction on patients,” Dr. Albert said. “However, in general, it is preferable to do an in-person consultation first to clarify the physical findings.”
Dartmouth has used both nurses and medical assistants as presenters during tele-rheumatology after developing a web-based video series to train presenters. Nurses at UPMC are also trained as telemedicine presenters, and telemedicine visits are only used for follow-up care, Dr. Peoples said.
At Children’s Mercy in Kansas City, telemedicine visits are restricted to follow-up care and include a nurse facilitator at the virtual site. However, the type of conditions and stages of disease examined through the technology has grown since the telemedicine services started, Dr. Kessler said.
“What we feel comfortable seeing has expanded since the time we initially started the clinic because some of the patients, when they were having flares, could not come to Kansas City for financial reasons,” she said, adding that she has since examined children with lupus, vasculitis, and active arthritis through telemedicine.
She stresses that in-person visits should depend on the patient, the circumstances, and the physician’s comfort level. ”You need to be realistic,” she said. “There are times when I’ve said to families, ‘I would like you to come to Kansas City because I’m a little unsure about your exam findings.’ There are definitely some patients who do more of a hybrid approach where they come to Kansas City and are also seen by telemedicine.”
Barriers slow telemedicine progress
Amid its benefits, challenges for tele-rheumatology, such as reimbursement and licensure obstacles, remain.
Commercial insurers pay for telemedicine at varying rates, making it difficult for doctors to consistently be paid, Dr. Peoples said.
“A lot of insurance companies do cover telemedicine visits, but still, a lot don’t,” she said. “I do think that in order to remain competitive, insurance companies overall are going to have to start covering these services.”
At least 32 states and the District of Columbia have parity laws that require commercial health insurers to cover services provided through telehealth to the same extent as those services are covered in person, according to an Aug. 15, 2016 Health Affairs policy brief. However, how private insurers pay and what services they cover vary widely.
Medicaid reimbursement for telehealth depends on individual state Medicaid programs. Only 9 states Medicaid programs reimburse for store-and-forward services, while at least 16 states have some sort of Medicaid reimbursement for remote patient monitoring, according to the Health Affairs report.
Medicare payment for telehealth is clouded in restrictions. The Centers for Medicare & Medicaid Services reimburses for synchronous communications only and does not cover store-and-forward services or remote patient monitoring for chronic diseases, except in Alaska and Hawaii. The patient must be present at an originating site for the visit and cannot be home to receive the services. In addition, CMS reimburses for telehealth only when the originating site is in a Health Professional Shortage Area or within a county outside a Metropolitan Statistical Area. In 2016, CMS expanded its coverage of telehealth to include several new codes including, end-stage renal disease–related services for dialysis, advance care planning services, and critical care consultations furnished via telehealth using new Medicare G-codes.
It’s worth noting that rheumatologists who are salaried employees of academic medical centers, as is the case for all rheumatologists interviewed here, don’t handle the billing for their telemedicine services, and so have not experienced the payment challenges that smaller practices may face.
Mr. Linkous of the American Telemedicine Association said that he believes that reimbursement for telemedicine will improve as health care heads toward value-based payment systems.
“In terms of reimbursement, with the move away from fee-based systems to value-based systems, it really opens the door because for many of the services, you no longer write out a bill with a code associated with it,” he said. “You are paid on the basis of keeping people well. That’s a great positive incentive for using telemedicine.”
Differing state licensure requirements also pose a challenge. The process to obtain licenses and navigate varying credentialing processes can be a headache for health providers and delay approval.
The barrier has led to model legislation by the Federation of State Medical Boards (FSMB) that aims to make it easier for telemedicine physicians to gain licenses in multiple states. Under the Interstate Medical Licensure Compact, physicians designate a member state as the state of principal licensure and select the other states in which they want to license. The state of principal licensure then verifies the physician’s eligibility and provides credential information to the interstate commission, which collects applicable fees and transmits the doctor’s information to the other states. Upon receipt in the additional states, the physician would be granted a license.
As of January, 18 states had enacted the compact legislation, and at least 4 states had introduced the legislation. In 2015, the Health Resources and Services Administration awarded the FSMB a grant to support establishment of the commission and aid with the compact’s infrastructure.
A new normal?
Dr. Albert notes that tele-rheumatology programs can succeed only with adequate resources, time, and staff.
“To set up Skype or Facetime is nothing; you just click on it,” he said. “To set up tele-rheumatology requires a fair amount of administrative substructure that has to be generated in terms of who’s going to do the documentation? Who is going to do the billing? Who is going to do the credentialing? Where is the credentialing going to be validated?”
The equipment required for telemedicine visits also requires a significant amount of time and regular maintenance, Dr. Peoples said. At UPMC for instance, the tele-rheumatology program has an information technology team that assists with technology glitches, changes, and updates, she said. That’s why it’s helpful to have the support of a large academic medical center with the sufficient resources to build a telemedicine program, she added.
In order for more practices to consider tele-rheumatology, more research about cost-effectiveness and best uses of the technology use would be useful, Dr. McDougall said.
“The main question that policy makers are going to want to answer is, ‘What’s the return on investment? Does this make sense for my practice?’ ” he said. “The methods reporting in tele-rheumatologist [literature] is lacking.” But regardless of barriers, telemedicine experts say the technology will likely continue to expand and transform the way rheumatologists are practicing and patients are receiving care.
“Tele-rheumatology will never replace an in-person exam,” Dr. Ferucci said. “But my vision is that it will be able to improve the quality of care for patients living in rural and remote locations, by allowing for more frequent visits and adjustment of medications, which are necessary to achieve the goal of treat-to-target for RA and other rheumatologic conditions.”
* This story was updated on 2/10/2017.
On Twitter @legal_med
Recent FDA Boxed Warnings
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. You can search these and other label changes in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential bene t from the drug that it is essential that it be considered in assessing the risks and bene ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted.
QUINOLONE:
- Edited and updated warning September 2016
WARNING: SERIOUS ADVERSE REACTIONS INCLUDING TENDINITIS, TENDON RUPTURE, PERIPHERAL NEUROPATHY, CENTRAL NERVOUS SYSTEM EFFECTS AND EXACERBATION OF MYASTHENIA GRAVIS
Fluoroquinolones have been associated with disabling and potentially irreversible serious adverse reactions that have occurred together including:
- Tendinitis and tendon rupture
- Peripheral neuropathy
- Central nervous system effects
Discontinue immediately and avoid the use of fluoroquinolones in patients who experience any of these serious adverse reactions. Fluoroquinolones may exacerbate muscle weakness in patients with myasthenia gravis. Avoid quinolones in patients with known history of myasthenia gravis. Because fluoroquinolones
have been associated with serious adverse reactions, reserve quinolones for use in patients who have no alternative treatment options for the following
indications:
Avelox (moxifloxacin hydrochloride): Avelox in sodium chloride 0.8% in plastic container; moxifloxacin hydrochloride; Cipro in dextrose 5% in plastic container):
Acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis.
Cipro (ciprofloxacin; ciprofloxacin hydrochloride): Acute exacerbation of chronic bronchitis, acute uncomplicated cystitis, and acute sinusitis.
Cipro XR; Noroxin (norfloxacin): Uncomplicated urinary tract infections.
Factive (gemifloxacin mesylate): Acute bacterial exacerbation of chronic bronchitis.
Levaquin (levofloxacin): Uncomplicated urinary tract infection, acute bacterial exacerbation of chronic bronchitis, and acute bacterial sinusitis.
KRYSTEXXA (PEGLOTICASE):
- Added section to warning September 2016
WARNING: ANAPHYLAXIS AND INFUSION REACTIONS; G6PD DEFICIENCY ASSOCIATED HEMOLYSIS AND METHEMOGLOBINEMIA (Title Updated)
Addition of: Screen patients at risk for G6PD deficiency prior to starting Krystexxa. Hemolysis and methemoglobinemia have been reported with Krystexxa in patients with G6PD deficiency. Do not administer Krystexxa to patients with G6PD deficiency.
PLAVIX (CLOPIDOGREL BISULFATE):
- Edited and updated warning September 2016
WARNING: DIMINISHED ANTIPLATELET EFFECT IN PATIENTS WITH TWO LOSS-OF-FUNCTION ALLELES OF THE CYP2C19 GENE
The effectiveness of Plavix results from its antiplatelet activity, which is dependent on its conversion to an active metabolite by the cytochrome P450 (CYP) system, principally CYP2C19. Plavix at recommended doses forms less of the active metabolite and so has a reduced effect on platelet activity in patients who are homozygous for nonfunctional alleles of the CYP2C19 gene, (termed “CYP2C19 poor metabolizers”). Tests are available to identify patients who are CYP2C19 poor metabolizers. Consider use of another platelet P2Y12 inhibitor in patients identified as CYP2C19 poor metabolizers.
SYNJARDY (EMPAGLIFLOZIN; METFORMIN HYDROCHLORIDE):
- Edited and updated warning September 2016
Postmarketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin-associated lactic acidosis is often subtle, accompanied only by nonspecific symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metforminassociated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/pyruvate ratio; and metformin plasma levels generally > 5 mcg/mL.
Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (e.g., carbonic anhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (e.g., acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Steps to reduce the risk of and manage metformin-associated lactic acidosis in these high-risk groups are provided in the full prescribing information.
If metformin-associated lactic acidosis is suspected, immediately discontinue Synjardy and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
ZYDELIG (IDELALISIB)
- Edited and updated warning September 2016
WARNING: FATAL AND SERIOUS TOXICITIES: HEPATIC, SEVERE DIARRHEA, COLITIS, PNEUMONITIS, INFECTIONS, AND INTESTINAL PERFORATION
- Fatal and/or serious hepatotoxicity occurred in 11 % to 18% of Zydelig-treated patients. Monitor hepatic function prior to and during treatment. Interrupt and then reduce or discontinue Zydelig as recommended.
- Fatal and/or serious and severe diarrhea or colitis occurred in 14% to 19% of Zydelig-treated patients. Monitor for the development of severe diarrhea or colitis. Interrupt and then reduce or discontinue Zydelig as recommended.
- Fatal and/or serious pneumonitis occurred in 4% of Zydelig-treated patients. Monitor for pulmonary symptoms and bilateral interstitial infiltrates. Interrupt or discontinue Zydelig as recommended.
- Fatal and/or serious infections occurred in 21% to 36% of Zydelig-treated patients. Monitor for signs and symptoms of infection. Interrupt Zydelig if infection is suspected.
- Fatal and serious intestinal perforation can occur in Zydelig-treated patients across clinical trials. Discontinue Zydelig for intestinal perforation.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. You can search these and other label changes in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential bene t from the drug that it is essential that it be considered in assessing the risks and bene ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted.
QUINOLONE:
- Edited and updated warning September 2016
WARNING: SERIOUS ADVERSE REACTIONS INCLUDING TENDINITIS, TENDON RUPTURE, PERIPHERAL NEUROPATHY, CENTRAL NERVOUS SYSTEM EFFECTS AND EXACERBATION OF MYASTHENIA GRAVIS
Fluoroquinolones have been associated with disabling and potentially irreversible serious adverse reactions that have occurred together including:
- Tendinitis and tendon rupture
- Peripheral neuropathy
- Central nervous system effects
Discontinue immediately and avoid the use of fluoroquinolones in patients who experience any of these serious adverse reactions. Fluoroquinolones may exacerbate muscle weakness in patients with myasthenia gravis. Avoid quinolones in patients with known history of myasthenia gravis. Because fluoroquinolones
have been associated with serious adverse reactions, reserve quinolones for use in patients who have no alternative treatment options for the following
indications:
Avelox (moxifloxacin hydrochloride): Avelox in sodium chloride 0.8% in plastic container; moxifloxacin hydrochloride; Cipro in dextrose 5% in plastic container):
Acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis.
Cipro (ciprofloxacin; ciprofloxacin hydrochloride): Acute exacerbation of chronic bronchitis, acute uncomplicated cystitis, and acute sinusitis.
Cipro XR; Noroxin (norfloxacin): Uncomplicated urinary tract infections.
Factive (gemifloxacin mesylate): Acute bacterial exacerbation of chronic bronchitis.
Levaquin (levofloxacin): Uncomplicated urinary tract infection, acute bacterial exacerbation of chronic bronchitis, and acute bacterial sinusitis.
KRYSTEXXA (PEGLOTICASE):
- Added section to warning September 2016
WARNING: ANAPHYLAXIS AND INFUSION REACTIONS; G6PD DEFICIENCY ASSOCIATED HEMOLYSIS AND METHEMOGLOBINEMIA (Title Updated)
Addition of: Screen patients at risk for G6PD deficiency prior to starting Krystexxa. Hemolysis and methemoglobinemia have been reported with Krystexxa in patients with G6PD deficiency. Do not administer Krystexxa to patients with G6PD deficiency.
PLAVIX (CLOPIDOGREL BISULFATE):
- Edited and updated warning September 2016
WARNING: DIMINISHED ANTIPLATELET EFFECT IN PATIENTS WITH TWO LOSS-OF-FUNCTION ALLELES OF THE CYP2C19 GENE
The effectiveness of Plavix results from its antiplatelet activity, which is dependent on its conversion to an active metabolite by the cytochrome P450 (CYP) system, principally CYP2C19. Plavix at recommended doses forms less of the active metabolite and so has a reduced effect on platelet activity in patients who are homozygous for nonfunctional alleles of the CYP2C19 gene, (termed “CYP2C19 poor metabolizers”). Tests are available to identify patients who are CYP2C19 poor metabolizers. Consider use of another platelet P2Y12 inhibitor in patients identified as CYP2C19 poor metabolizers.
SYNJARDY (EMPAGLIFLOZIN; METFORMIN HYDROCHLORIDE):
- Edited and updated warning September 2016
Postmarketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin-associated lactic acidosis is often subtle, accompanied only by nonspecific symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metforminassociated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/pyruvate ratio; and metformin plasma levels generally > 5 mcg/mL.
Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (e.g., carbonic anhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (e.g., acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Steps to reduce the risk of and manage metformin-associated lactic acidosis in these high-risk groups are provided in the full prescribing information.
If metformin-associated lactic acidosis is suspected, immediately discontinue Synjardy and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
ZYDELIG (IDELALISIB)
- Edited and updated warning September 2016
WARNING: FATAL AND SERIOUS TOXICITIES: HEPATIC, SEVERE DIARRHEA, COLITIS, PNEUMONITIS, INFECTIONS, AND INTESTINAL PERFORATION
- Fatal and/or serious hepatotoxicity occurred in 11 % to 18% of Zydelig-treated patients. Monitor hepatic function prior to and during treatment. Interrupt and then reduce or discontinue Zydelig as recommended.
- Fatal and/or serious and severe diarrhea or colitis occurred in 14% to 19% of Zydelig-treated patients. Monitor for the development of severe diarrhea or colitis. Interrupt and then reduce or discontinue Zydelig as recommended.
- Fatal and/or serious pneumonitis occurred in 4% of Zydelig-treated patients. Monitor for pulmonary symptoms and bilateral interstitial infiltrates. Interrupt or discontinue Zydelig as recommended.
- Fatal and/or serious infections occurred in 21% to 36% of Zydelig-treated patients. Monitor for signs and symptoms of infection. Interrupt Zydelig if infection is suspected.
- Fatal and serious intestinal perforation can occur in Zydelig-treated patients across clinical trials. Discontinue Zydelig for intestinal perforation.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. You can search these and other label changes in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential bene t from the drug that it is essential that it be considered in assessing the risks and bene ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted.
QUINOLONE:
- Edited and updated warning September 2016
WARNING: SERIOUS ADVERSE REACTIONS INCLUDING TENDINITIS, TENDON RUPTURE, PERIPHERAL NEUROPATHY, CENTRAL NERVOUS SYSTEM EFFECTS AND EXACERBATION OF MYASTHENIA GRAVIS
Fluoroquinolones have been associated with disabling and potentially irreversible serious adverse reactions that have occurred together including:
- Tendinitis and tendon rupture
- Peripheral neuropathy
- Central nervous system effects
Discontinue immediately and avoid the use of fluoroquinolones in patients who experience any of these serious adverse reactions. Fluoroquinolones may exacerbate muscle weakness in patients with myasthenia gravis. Avoid quinolones in patients with known history of myasthenia gravis. Because fluoroquinolones
have been associated with serious adverse reactions, reserve quinolones for use in patients who have no alternative treatment options for the following
indications:
Avelox (moxifloxacin hydrochloride): Avelox in sodium chloride 0.8% in plastic container; moxifloxacin hydrochloride; Cipro in dextrose 5% in plastic container):
Acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis.
Cipro (ciprofloxacin; ciprofloxacin hydrochloride): Acute exacerbation of chronic bronchitis, acute uncomplicated cystitis, and acute sinusitis.
Cipro XR; Noroxin (norfloxacin): Uncomplicated urinary tract infections.
Factive (gemifloxacin mesylate): Acute bacterial exacerbation of chronic bronchitis.
Levaquin (levofloxacin): Uncomplicated urinary tract infection, acute bacterial exacerbation of chronic bronchitis, and acute bacterial sinusitis.
KRYSTEXXA (PEGLOTICASE):
- Added section to warning September 2016
WARNING: ANAPHYLAXIS AND INFUSION REACTIONS; G6PD DEFICIENCY ASSOCIATED HEMOLYSIS AND METHEMOGLOBINEMIA (Title Updated)
Addition of: Screen patients at risk for G6PD deficiency prior to starting Krystexxa. Hemolysis and methemoglobinemia have been reported with Krystexxa in patients with G6PD deficiency. Do not administer Krystexxa to patients with G6PD deficiency.
PLAVIX (CLOPIDOGREL BISULFATE):
- Edited and updated warning September 2016
WARNING: DIMINISHED ANTIPLATELET EFFECT IN PATIENTS WITH TWO LOSS-OF-FUNCTION ALLELES OF THE CYP2C19 GENE
The effectiveness of Plavix results from its antiplatelet activity, which is dependent on its conversion to an active metabolite by the cytochrome P450 (CYP) system, principally CYP2C19. Plavix at recommended doses forms less of the active metabolite and so has a reduced effect on platelet activity in patients who are homozygous for nonfunctional alleles of the CYP2C19 gene, (termed “CYP2C19 poor metabolizers”). Tests are available to identify patients who are CYP2C19 poor metabolizers. Consider use of another platelet P2Y12 inhibitor in patients identified as CYP2C19 poor metabolizers.
SYNJARDY (EMPAGLIFLOZIN; METFORMIN HYDROCHLORIDE):
- Edited and updated warning September 2016
Postmarketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin-associated lactic acidosis is often subtle, accompanied only by nonspecific symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metforminassociated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/pyruvate ratio; and metformin plasma levels generally > 5 mcg/mL.
Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (e.g., carbonic anhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (e.g., acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Steps to reduce the risk of and manage metformin-associated lactic acidosis in these high-risk groups are provided in the full prescribing information.
If metformin-associated lactic acidosis is suspected, immediately discontinue Synjardy and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
ZYDELIG (IDELALISIB)
- Edited and updated warning September 2016
WARNING: FATAL AND SERIOUS TOXICITIES: HEPATIC, SEVERE DIARRHEA, COLITIS, PNEUMONITIS, INFECTIONS, AND INTESTINAL PERFORATION
- Fatal and/or serious hepatotoxicity occurred in 11 % to 18% of Zydelig-treated patients. Monitor hepatic function prior to and during treatment. Interrupt and then reduce or discontinue Zydelig as recommended.
- Fatal and/or serious and severe diarrhea or colitis occurred in 14% to 19% of Zydelig-treated patients. Monitor for the development of severe diarrhea or colitis. Interrupt and then reduce or discontinue Zydelig as recommended.
- Fatal and/or serious pneumonitis occurred in 4% of Zydelig-treated patients. Monitor for pulmonary symptoms and bilateral interstitial infiltrates. Interrupt or discontinue Zydelig as recommended.
- Fatal and/or serious infections occurred in 21% to 36% of Zydelig-treated patients. Monitor for signs and symptoms of infection. Interrupt Zydelig if infection is suspected.
- Fatal and serious intestinal perforation can occur in Zydelig-treated patients across clinical trials. Discontinue Zydelig for intestinal perforation.
Hearing Loss Is Less Common in Adults
The number of older Americans is growing, but the number of those with hearing loss is declining, according to data from the National Health and Nutrition Examination Survey. Researchers compared 2 time periods (1999-2004 and 2011-2012) and found overall annual prevalence of hearing loss dropped from 16% to 14% in 1999-2004, to 28 million adults, then dropped further to 27.7 million in 2011-2012.
Age was the greatest predictor of hearing loss; people in the oldest age group surveyed (aged 60 to 69) had the most loss. Although not included in the study, people aged ≥ 70 years have the highest prevalence of hearing loss of any age group, the authors say. Men of all ages were twice as likely as women to have hearing loss. Non-Hispanic white adults were more likely to have hearing loss than were adults in other ethnic groups. Non-Hispanic black adults had the lowest risk.
The researchers don’t know why hearing loss is becoming less prevalent but suggest reasons include fewer manufacturing jobs, increased use of hearing protectors, less smoking, and better medical care to manage risk factors associated with hearing loss. They did find that lower education level and heavy use of firearms were associated with hearing loss.
“Our findings show a promising trend of better hearing among adults that spans more than half a century,” says Howard Hoffman, MA, first author on the paper and director of the National Institute on Deafness and Other Communication Disorders Epidemiology and Statistics Program. “The decline in hearing loss rates among adults aged < 70 years suggests that age-related hearing loss may be delayed until later in life. This is good news because for those who do develop hearing loss, they will have experienced more quality years of life with better hearing than earlier generations."
The number of older Americans is growing, but the number of those with hearing loss is declining, according to data from the National Health and Nutrition Examination Survey. Researchers compared 2 time periods (1999-2004 and 2011-2012) and found overall annual prevalence of hearing loss dropped from 16% to 14% in 1999-2004, to 28 million adults, then dropped further to 27.7 million in 2011-2012.
Age was the greatest predictor of hearing loss; people in the oldest age group surveyed (aged 60 to 69) had the most loss. Although not included in the study, people aged ≥ 70 years have the highest prevalence of hearing loss of any age group, the authors say. Men of all ages were twice as likely as women to have hearing loss. Non-Hispanic white adults were more likely to have hearing loss than were adults in other ethnic groups. Non-Hispanic black adults had the lowest risk.
The researchers don’t know why hearing loss is becoming less prevalent but suggest reasons include fewer manufacturing jobs, increased use of hearing protectors, less smoking, and better medical care to manage risk factors associated with hearing loss. They did find that lower education level and heavy use of firearms were associated with hearing loss.
“Our findings show a promising trend of better hearing among adults that spans more than half a century,” says Howard Hoffman, MA, first author on the paper and director of the National Institute on Deafness and Other Communication Disorders Epidemiology and Statistics Program. “The decline in hearing loss rates among adults aged < 70 years suggests that age-related hearing loss may be delayed until later in life. This is good news because for those who do develop hearing loss, they will have experienced more quality years of life with better hearing than earlier generations."
The number of older Americans is growing, but the number of those with hearing loss is declining, according to data from the National Health and Nutrition Examination Survey. Researchers compared 2 time periods (1999-2004 and 2011-2012) and found overall annual prevalence of hearing loss dropped from 16% to 14% in 1999-2004, to 28 million adults, then dropped further to 27.7 million in 2011-2012.
Age was the greatest predictor of hearing loss; people in the oldest age group surveyed (aged 60 to 69) had the most loss. Although not included in the study, people aged ≥ 70 years have the highest prevalence of hearing loss of any age group, the authors say. Men of all ages were twice as likely as women to have hearing loss. Non-Hispanic white adults were more likely to have hearing loss than were adults in other ethnic groups. Non-Hispanic black adults had the lowest risk.
The researchers don’t know why hearing loss is becoming less prevalent but suggest reasons include fewer manufacturing jobs, increased use of hearing protectors, less smoking, and better medical care to manage risk factors associated with hearing loss. They did find that lower education level and heavy use of firearms were associated with hearing loss.
“Our findings show a promising trend of better hearing among adults that spans more than half a century,” says Howard Hoffman, MA, first author on the paper and director of the National Institute on Deafness and Other Communication Disorders Epidemiology and Statistics Program. “The decline in hearing loss rates among adults aged < 70 years suggests that age-related hearing loss may be delayed until later in life. This is good news because for those who do develop hearing loss, they will have experienced more quality years of life with better hearing than earlier generations."
Third-hand smoke affects blood cell development in mice

and Antoine Snijders analyze
blood cells collected from mice
exposed to third-hand smoke.
Photo courtesy of
Marilyn Chung/Berkeley Lab
Exposure to third-hand smoke leads to biological effects on weight and blood cell development, according to preclinical research published in Scientific Reports.
Researchers found that newborn mice housed with smoke-treated cloths for 3 weeks weighed significantly less than mice in a control group.
Moreover, newborn and adult mice exposed to third-hand smoke experienced persistent changes in blood cell counts.
The blood cell count changes are associated with inflammatory and allergic reactions upon exposure to third-hand smoke, the researchers said.
For this study, the team set out to characterize the biological effects of exposure to third-hand smoke by placing 5-square-centimeter pieces of smoke-contaminated cotton cloth in cages with mice.
The researchers then compared smoke-exposed mice to control mice. The team assessed changes to body weight and the hematopoietic system after 3 weeks of exposure (or no exposure) for mice belonging to 2 age groups: birth to 3 weeks (neonatal) and 12 to 15 weeks (young adult).
The results showed that smoke exposure temporarily inhibited weight gain in the neonatal mice. There was no effect on weight gain in the young adult mice.
In addition, smoke exposure produced changes in blood cell populations that persisted over time and were evident in mice from both age groups.
In general, there were lower levels of platelets and specific types of white blood cells in the smoke-exposed mice.
For example, neonatal mice exposed to third-hand smoke had higher levels of eosinophils, female mice had higher levels of neutrophils, males had higher levels of basophils, and all mice had higher levels of B cells.
“Those are all types of white blood cells associated with inflammation and allergic reactions,” said study author Jian-Hua Mao, PhD, of Lawrence Berkeley National Laboratory in Berkeley, California.
“And the effects on blood cell count persisted even after exposure ended. Changes remained at least 14 weeks after exposure ended for the neonatal group and 2 weeks after it ended for the adults.”
The researchers pointed out that they did not study whether the observed biological changes led to specific diseases or other health outcomes, but other studies suggest links to adverse health effects.
“Third-hand smoke is an underappreciated risk factor in health,” said study author Antoine Snijders, PhD, of Lawrence Berkeley National Laboratory.
“It’s clear that more and bigger studies are needed, particularly in humans, so we can support policy decisions on third-hand smoke.” ![]()
and Antoine Snijders analyze
blood cells collected from mice
exposed to third-hand smoke.
Photo courtesy of
Marilyn Chung/Berkeley Lab
Exposure to third-hand smoke leads to biological effects on weight and blood cell development, according to preclinical research published in Scientific Reports.
Researchers found that newborn mice housed with smoke-treated cloths for 3 weeks weighed significantly less than mice in a control group.
Moreover, newborn and adult mice exposed to third-hand smoke experienced persistent changes in blood cell counts.
The blood cell count changes are associated with inflammatory and allergic reactions upon exposure to third-hand smoke, the researchers said.
For this study, the team set out to characterize the biological effects of exposure to third-hand smoke by placing 5-square-centimeter pieces of smoke-contaminated cotton cloth in cages with mice.
The researchers then compared smoke-exposed mice to control mice. The team assessed changes to body weight and the hematopoietic system after 3 weeks of exposure (or no exposure) for mice belonging to 2 age groups: birth to 3 weeks (neonatal) and 12 to 15 weeks (young adult).
The results showed that smoke exposure temporarily inhibited weight gain in the neonatal mice. There was no effect on weight gain in the young adult mice.
In addition, smoke exposure produced changes in blood cell populations that persisted over time and were evident in mice from both age groups.
In general, there were lower levels of platelets and specific types of white blood cells in the smoke-exposed mice.
For example, neonatal mice exposed to third-hand smoke had higher levels of eosinophils, female mice had higher levels of neutrophils, males had higher levels of basophils, and all mice had higher levels of B cells.
“Those are all types of white blood cells associated with inflammation and allergic reactions,” said study author Jian-Hua Mao, PhD, of Lawrence Berkeley National Laboratory in Berkeley, California.
“And the effects on blood cell count persisted even after exposure ended. Changes remained at least 14 weeks after exposure ended for the neonatal group and 2 weeks after it ended for the adults.”
The researchers pointed out that they did not study whether the observed biological changes led to specific diseases or other health outcomes, but other studies suggest links to adverse health effects.
“Third-hand smoke is an underappreciated risk factor in health,” said study author Antoine Snijders, PhD, of Lawrence Berkeley National Laboratory.
“It’s clear that more and bigger studies are needed, particularly in humans, so we can support policy decisions on third-hand smoke.” ![]()
and Antoine Snijders analyze
blood cells collected from mice
exposed to third-hand smoke.
Photo courtesy of
Marilyn Chung/Berkeley Lab
Exposure to third-hand smoke leads to biological effects on weight and blood cell development, according to preclinical research published in Scientific Reports.
Researchers found that newborn mice housed with smoke-treated cloths for 3 weeks weighed significantly less than mice in a control group.
Moreover, newborn and adult mice exposed to third-hand smoke experienced persistent changes in blood cell counts.
The blood cell count changes are associated with inflammatory and allergic reactions upon exposure to third-hand smoke, the researchers said.
For this study, the team set out to characterize the biological effects of exposure to third-hand smoke by placing 5-square-centimeter pieces of smoke-contaminated cotton cloth in cages with mice.
The researchers then compared smoke-exposed mice to control mice. The team assessed changes to body weight and the hematopoietic system after 3 weeks of exposure (or no exposure) for mice belonging to 2 age groups: birth to 3 weeks (neonatal) and 12 to 15 weeks (young adult).
The results showed that smoke exposure temporarily inhibited weight gain in the neonatal mice. There was no effect on weight gain in the young adult mice.
In addition, smoke exposure produced changes in blood cell populations that persisted over time and were evident in mice from both age groups.
In general, there were lower levels of platelets and specific types of white blood cells in the smoke-exposed mice.
For example, neonatal mice exposed to third-hand smoke had higher levels of eosinophils, female mice had higher levels of neutrophils, males had higher levels of basophils, and all mice had higher levels of B cells.
“Those are all types of white blood cells associated with inflammation and allergic reactions,” said study author Jian-Hua Mao, PhD, of Lawrence Berkeley National Laboratory in Berkeley, California.
“And the effects on blood cell count persisted even after exposure ended. Changes remained at least 14 weeks after exposure ended for the neonatal group and 2 weeks after it ended for the adults.”
The researchers pointed out that they did not study whether the observed biological changes led to specific diseases or other health outcomes, but other studies suggest links to adverse health effects.
“Third-hand smoke is an underappreciated risk factor in health,” said study author Antoine Snijders, PhD, of Lawrence Berkeley National Laboratory.
“It’s clear that more and bigger studies are needed, particularly in humans, so we can support policy decisions on third-hand smoke.” ![]()
NCCN releases patient guidelines for WM/LPL
The National Comprehensive Cancer Network® (NCCN) has published a set of
guidelines for patients with
Waldenström’s macroglobulinemia/ lymphoplasmacytic lymphoma (WM/LPL).
This resource describes what WM is and how it develops,
explains testing for WM, and provides information on the treatment of
primary, relapsed, and refractory WM.
NCCN Guidelines for Patients are adaptations of the
NCCN Clinical Practice Guidelines in Oncology.
The NCCN also publishes Quick Guide™ sheets, which are 1-page summaries of key points in the patient guidelines.
Both patient resources are available free of charge at NCCN.org/patients as well as on the NCCN Patient Guides for Cancer mobile app.
“The treatment approach to patients with Waldenström’s macroglobulinemia has significantly changed in the recent years with better understanding of the disease biology and its natural history and availability of new drugs, allowing for a more individualized approach,” said Shaji Kumar, MD, of the Mayo Clinic in Rochester, Minnesota.
“The revised guidelines reflect these changes and will be a valuable guide for patients in shared decision-making with their oncologists.”
NCCN Guidelines for Patients are based on the same clinical practice guidelines used by healthcare professionals to determine the best way to treat a patient with cancer.
Each resource features expert guidance from US cancer centers designed to help people living with cancer talk to their physicians about the best treatment options for their disease.
The Guidelines for Patients and Quick Guide sheets are written in plain language and include patient-friendly elements, such as “questions to ask your doctor,” a glossary of terms, and medical illustrations of anatomy, tests, and treatment.
NCCN currently offers NCCN Guidelines for Patients covering the following topics: brain, breast, colon esophageal, kidney, non-small cell lung, ovarian, pancreatic, prostate, and stomach cancers; acute lymphoblastic leukemia; adolescents and young adults with cancer; chronic lymphocytic leukemia; chronic myelogenous leukemia; Hodgkin lymphoma; lung cancer screening; malignant pleural mesothelioma; melanoma; multiple myeloma; nausea and vomiting; non-Hodgkin lymphomas; soft tissue sarcoma; and WM/LPL. ![]()
The National Comprehensive Cancer Network® (NCCN) has published a set of
guidelines for patients with
Waldenström’s macroglobulinemia/ lymphoplasmacytic lymphoma (WM/LPL).
This resource describes what WM is and how it develops,
explains testing for WM, and provides information on the treatment of
primary, relapsed, and refractory WM.
NCCN Guidelines for Patients are adaptations of the
NCCN Clinical Practice Guidelines in Oncology.
The NCCN also publishes Quick Guide™ sheets, which are 1-page summaries of key points in the patient guidelines.
Both patient resources are available free of charge at NCCN.org/patients as well as on the NCCN Patient Guides for Cancer mobile app.
“The treatment approach to patients with Waldenström’s macroglobulinemia has significantly changed in the recent years with better understanding of the disease biology and its natural history and availability of new drugs, allowing for a more individualized approach,” said Shaji Kumar, MD, of the Mayo Clinic in Rochester, Minnesota.
“The revised guidelines reflect these changes and will be a valuable guide for patients in shared decision-making with their oncologists.”
NCCN Guidelines for Patients are based on the same clinical practice guidelines used by healthcare professionals to determine the best way to treat a patient with cancer.
Each resource features expert guidance from US cancer centers designed to help people living with cancer talk to their physicians about the best treatment options for their disease.
The Guidelines for Patients and Quick Guide sheets are written in plain language and include patient-friendly elements, such as “questions to ask your doctor,” a glossary of terms, and medical illustrations of anatomy, tests, and treatment.
NCCN currently offers NCCN Guidelines for Patients covering the following topics: brain, breast, colon esophageal, kidney, non-small cell lung, ovarian, pancreatic, prostate, and stomach cancers; acute lymphoblastic leukemia; adolescents and young adults with cancer; chronic lymphocytic leukemia; chronic myelogenous leukemia; Hodgkin lymphoma; lung cancer screening; malignant pleural mesothelioma; melanoma; multiple myeloma; nausea and vomiting; non-Hodgkin lymphomas; soft tissue sarcoma; and WM/LPL. ![]()
The National Comprehensive Cancer Network® (NCCN) has published a set of
guidelines for patients with
Waldenström’s macroglobulinemia/ lymphoplasmacytic lymphoma (WM/LPL).
This resource describes what WM is and how it develops,
explains testing for WM, and provides information on the treatment of
primary, relapsed, and refractory WM.
NCCN Guidelines for Patients are adaptations of the
NCCN Clinical Practice Guidelines in Oncology.
The NCCN also publishes Quick Guide™ sheets, which are 1-page summaries of key points in the patient guidelines.
Both patient resources are available free of charge at NCCN.org/patients as well as on the NCCN Patient Guides for Cancer mobile app.
“The treatment approach to patients with Waldenström’s macroglobulinemia has significantly changed in the recent years with better understanding of the disease biology and its natural history and availability of new drugs, allowing for a more individualized approach,” said Shaji Kumar, MD, of the Mayo Clinic in Rochester, Minnesota.
“The revised guidelines reflect these changes and will be a valuable guide for patients in shared decision-making with their oncologists.”
NCCN Guidelines for Patients are based on the same clinical practice guidelines used by healthcare professionals to determine the best way to treat a patient with cancer.
Each resource features expert guidance from US cancer centers designed to help people living with cancer talk to their physicians about the best treatment options for their disease.
The Guidelines for Patients and Quick Guide sheets are written in plain language and include patient-friendly elements, such as “questions to ask your doctor,” a glossary of terms, and medical illustrations of anatomy, tests, and treatment.
NCCN currently offers NCCN Guidelines for Patients covering the following topics: brain, breast, colon esophageal, kidney, non-small cell lung, ovarian, pancreatic, prostate, and stomach cancers; acute lymphoblastic leukemia; adolescents and young adults with cancer; chronic lymphocytic leukemia; chronic myelogenous leukemia; Hodgkin lymphoma; lung cancer screening; malignant pleural mesothelioma; melanoma; multiple myeloma; nausea and vomiting; non-Hodgkin lymphomas; soft tissue sarcoma; and WM/LPL. ![]()
Azathioprine may increase risk of MDS, AML

Results of a large, retrospective study suggest that taking azathioprine, a drug commonly used to treat autoimmune disease, may increase a person’s risk of developing myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML).
Researchers analyzed data on more than 40,000 patients with 27 common autoimmune diseases and found that azathioprine use was significantly associated with an increased risk of MDS and AML.
“Similar associations were already documented in case reports and case series but have never been evaluated in a broad spectrum of autoimmune diseases in that many patients and in context of individual medications,” said study author Raoul Tibes, MD, PhD, of the Mayo Clinic in Phoenix, Arizona.
“Interestingly, there was no association with length of time on therapy and resulting myeloid neoplasm.”
Dr Tibes and his colleagues reported these findings in JAMA Oncology.
The researchers reviewed data on 40,011 patients with primary autoimmune disorders, such as lupus and rheumatoid arthritis, who were seen at 2 centers from January 1, 2004, to December 31, 2014.
There were 311 patients with MDS or AML, but only 86 met strict inclusion criteria. Fifty-five patients had MDS, 21 had de novo AML, and 10 had AML and a history of MDS.
The researchers collected detailed data on each patient’s drug exposures, treatment duration, and disease characteristics and compared this information to data from patients with autoimmune disorders who did not have MDS or AML.
This revealed that use of azathioprine sodium was more frequent in cases than controls, and azathioprine was significantly associated with an increased risk of MDS and AML. The odds ratio was 7.05 (P<0.001).
Other agents used showed a similar trend, but the results were not statistically significant. The odds ratios were 3.58 for cyclophosphamide and 2.73 for mitoxantrone hydrochloride.
The researchers said that, while these results are intriguing, they should not change or replace the clinical judgments, monitoring, and current standard treatments for patients with autoimmune diseases.
Despite its large size, this study had limitations, including its retrospective nature, the fact that many different autoimmune diseases were analyzed, and that the researchers only looked at cases of MDS and AML.
No definitive causal association was made between taking a particular drug and MDS or AML. The number of patients with autoimmune disease developing MDS or AML is still low overall, and no prediction for individual patients can be concluded from the study.
The researchers plan to perform molecular investigations into the genetic susceptibility for therapy-related myeloid neoplasms as the next phase of this research. ![]()
Results of a large, retrospective study suggest that taking azathioprine, a drug commonly used to treat autoimmune disease, may increase a person’s risk of developing myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML).
Researchers analyzed data on more than 40,000 patients with 27 common autoimmune diseases and found that azathioprine use was significantly associated with an increased risk of MDS and AML.
“Similar associations were already documented in case reports and case series but have never been evaluated in a broad spectrum of autoimmune diseases in that many patients and in context of individual medications,” said study author Raoul Tibes, MD, PhD, of the Mayo Clinic in Phoenix, Arizona.
“Interestingly, there was no association with length of time on therapy and resulting myeloid neoplasm.”
Dr Tibes and his colleagues reported these findings in JAMA Oncology.
The researchers reviewed data on 40,011 patients with primary autoimmune disorders, such as lupus and rheumatoid arthritis, who were seen at 2 centers from January 1, 2004, to December 31, 2014.
There were 311 patients with MDS or AML, but only 86 met strict inclusion criteria. Fifty-five patients had MDS, 21 had de novo AML, and 10 had AML and a history of MDS.
The researchers collected detailed data on each patient’s drug exposures, treatment duration, and disease characteristics and compared this information to data from patients with autoimmune disorders who did not have MDS or AML.
This revealed that use of azathioprine sodium was more frequent in cases than controls, and azathioprine was significantly associated with an increased risk of MDS and AML. The odds ratio was 7.05 (P<0.001).
Other agents used showed a similar trend, but the results were not statistically significant. The odds ratios were 3.58 for cyclophosphamide and 2.73 for mitoxantrone hydrochloride.
The researchers said that, while these results are intriguing, they should not change or replace the clinical judgments, monitoring, and current standard treatments for patients with autoimmune diseases.
Despite its large size, this study had limitations, including its retrospective nature, the fact that many different autoimmune diseases were analyzed, and that the researchers only looked at cases of MDS and AML.
No definitive causal association was made between taking a particular drug and MDS or AML. The number of patients with autoimmune disease developing MDS or AML is still low overall, and no prediction for individual patients can be concluded from the study.
The researchers plan to perform molecular investigations into the genetic susceptibility for therapy-related myeloid neoplasms as the next phase of this research. ![]()
Results of a large, retrospective study suggest that taking azathioprine, a drug commonly used to treat autoimmune disease, may increase a person’s risk of developing myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML).
Researchers analyzed data on more than 40,000 patients with 27 common autoimmune diseases and found that azathioprine use was significantly associated with an increased risk of MDS and AML.
“Similar associations were already documented in case reports and case series but have never been evaluated in a broad spectrum of autoimmune diseases in that many patients and in context of individual medications,” said study author Raoul Tibes, MD, PhD, of the Mayo Clinic in Phoenix, Arizona.
“Interestingly, there was no association with length of time on therapy and resulting myeloid neoplasm.”
Dr Tibes and his colleagues reported these findings in JAMA Oncology.
The researchers reviewed data on 40,011 patients with primary autoimmune disorders, such as lupus and rheumatoid arthritis, who were seen at 2 centers from January 1, 2004, to December 31, 2014.
There were 311 patients with MDS or AML, but only 86 met strict inclusion criteria. Fifty-five patients had MDS, 21 had de novo AML, and 10 had AML and a history of MDS.
The researchers collected detailed data on each patient’s drug exposures, treatment duration, and disease characteristics and compared this information to data from patients with autoimmune disorders who did not have MDS or AML.
This revealed that use of azathioprine sodium was more frequent in cases than controls, and azathioprine was significantly associated with an increased risk of MDS and AML. The odds ratio was 7.05 (P<0.001).
Other agents used showed a similar trend, but the results were not statistically significant. The odds ratios were 3.58 for cyclophosphamide and 2.73 for mitoxantrone hydrochloride.
The researchers said that, while these results are intriguing, they should not change or replace the clinical judgments, monitoring, and current standard treatments for patients with autoimmune diseases.
Despite its large size, this study had limitations, including its retrospective nature, the fact that many different autoimmune diseases were analyzed, and that the researchers only looked at cases of MDS and AML.
No definitive causal association was made between taking a particular drug and MDS or AML. The number of patients with autoimmune disease developing MDS or AML is still low overall, and no prediction for individual patients can be concluded from the study.
The researchers plan to perform molecular investigations into the genetic susceptibility for therapy-related myeloid neoplasms as the next phase of this research. ![]()
Adverse event reporting in second-dose varicella vaccination shows no surprises
No new or unexpected adverse events were reported in association with second-dose varicella vaccination in children age 4-18 years during 2006-2014.
During the study period of 2006-2014, 14,641 reports regarding second-dose varicella vaccinations were made to the Vaccine Adverse Event Reporting System, according to John Su, MD, PhD, and his associates. Of these reports, only 3% were serious adverse events (AE), with the most common nonserious AE being injection-site reaction, which occurred in 48% of patients age 4-6 years reporting AEs, and in 38% of patients age 7-18 years reporting AEs.
Of the 494 serious AEs reported, pyrexia was the most common in children age 4-6 years, and headache and vomiting were the most common in children age 7-18 years. A total of seven deaths from various causes were reported, though no causal relation to vaccination was seen, and significant chronic medical problems were common in these children.
“The safety data on second-dose varicella vaccination are reassuring,” the investigators noted. “Reported AEs after second-dose varicella vaccination were mild, self limiting, and similar in reported frequency to AEs after first-dose vaccination, with no new or unexpected safety concerns.”
Find the full study in Pediatrics (2017 Feb 7. doi: 1542/peds.2016-2536).
No new or unexpected adverse events were reported in association with second-dose varicella vaccination in children age 4-18 years during 2006-2014.
During the study period of 2006-2014, 14,641 reports regarding second-dose varicella vaccinations were made to the Vaccine Adverse Event Reporting System, according to John Su, MD, PhD, and his associates. Of these reports, only 3% were serious adverse events (AE), with the most common nonserious AE being injection-site reaction, which occurred in 48% of patients age 4-6 years reporting AEs, and in 38% of patients age 7-18 years reporting AEs.
Of the 494 serious AEs reported, pyrexia was the most common in children age 4-6 years, and headache and vomiting were the most common in children age 7-18 years. A total of seven deaths from various causes were reported, though no causal relation to vaccination was seen, and significant chronic medical problems were common in these children.
“The safety data on second-dose varicella vaccination are reassuring,” the investigators noted. “Reported AEs after second-dose varicella vaccination were mild, self limiting, and similar in reported frequency to AEs after first-dose vaccination, with no new or unexpected safety concerns.”
Find the full study in Pediatrics (2017 Feb 7. doi: 1542/peds.2016-2536).
No new or unexpected adverse events were reported in association with second-dose varicella vaccination in children age 4-18 years during 2006-2014.
During the study period of 2006-2014, 14,641 reports regarding second-dose varicella vaccinations were made to the Vaccine Adverse Event Reporting System, according to John Su, MD, PhD, and his associates. Of these reports, only 3% were serious adverse events (AE), with the most common nonserious AE being injection-site reaction, which occurred in 48% of patients age 4-6 years reporting AEs, and in 38% of patients age 7-18 years reporting AEs.
Of the 494 serious AEs reported, pyrexia was the most common in children age 4-6 years, and headache and vomiting were the most common in children age 7-18 years. A total of seven deaths from various causes were reported, though no causal relation to vaccination was seen, and significant chronic medical problems were common in these children.
“The safety data on second-dose varicella vaccination are reassuring,” the investigators noted. “Reported AEs after second-dose varicella vaccination were mild, self limiting, and similar in reported frequency to AEs after first-dose vaccination, with no new or unexpected safety concerns.”
Find the full study in Pediatrics (2017 Feb 7. doi: 1542/peds.2016-2536).
Hypertriglyceridemia: Identifying Secondary Causes
Screening for cardiovascular (CV) risk often includes a routine serum fasting lipid profile. However, with the focus on LDL cholesterol, triglyceride measurement is frequently overlooked. Yet this element of the lipid profile is particularly important, given its strong association with not only atherosclerotic coronary heart disease but also pancreatitis.
Hypertriglyceridemia is defined as a serum triglyceride level that exceeds 150 mg/dL. In the US, an estimated 25% of patients have hypertriglyceridemia.1 Of these, 33.1% have “borderline high” triglyceride levels (150 to 199 mg/dL), 17.8% have “high” levels (200 to 499 mg/dL), and 1.7% have “very high” levels (> 500 mg/dL).1,2
Most of the time, hypertriglyceridemia is caused (or at least exacerbated) by underlying etiology. The best way to identify and manage these secondary causes is through a systematic approach.
CONSIDER THE EVIDENCE
For mild to moderately elevated (borderline high) triglyceride levels, our reflex reaction may be to recommend a triglyceride-lowering medication, such as fenofibrate. But this may not be the best answer. Although there is increasing evidence of an independent association between elevated triglyceride levels and CV risk, it remains unclear whether targeting them specifically can reduce that risk.3
In well-designed, peer-reviewed clinical trials, statins have been shown to reduce CV risk in patients with known cardiovascular disease (CVD) and those at high risk for CVD, as well as in primary prevention. However, these trials also suggest that significant residual CV risk remains after statin therapy.4
Several trials have attempted to prove residual risk reduction following combination therapy including statins—with inconclusive results:
ACCORD: Fenofibrate showed no overall macrovascular benefit when added to a statin in patients with type 2 diabetes and a triglyceride level < 204 mg/dL.3,5
AIM-HIGH: There was a 25% reduction in triglyceride levels when niacin was added to a regimen of a statin +/- ezetimibe, with an aggressive LDL treatment target (40 to 80 mg/dL). But the study was stopped early due to the lack of expected reduction in CVD events.4,6
JELIS: A reduction in major CV events was seen with 1,800 mg/d of eicosapentaenoic acid (EPA) supplementation plus a low-dose statin, compared to statin monotherapy. However, there was minimal change in triglyceride levels, leading the researchers to hypothesize that multiple mechanisms—such as decreasing oxidative stress, platelet aggregation, plaque formation and stabilization—contributed to the outcome.4,7
Informed by the JELIS results, the much-anticipated REDUCE-IT trial is currently in progress to address the lingering question of whether combination therapy can reduce residual CV risk. In this trial, EPA omega-3 fatty acid is being added to the regimen of statin-treated patients with persistently elevated triglycerides. Results are expected in 2017 to 2018.8
Remember that a triglyceride level of 150 mg/dL is a parameter—it does not represent a therapeutic target. There is insufficient evidence that treating to this level improves CV risk beyond LDL target recommendations.7
The National Lipid Association Expert Panel’s consensus view is that non-HDL is a better primary target than triglycerides alone or LDL. Using non-HDL as a target for intervention also simplifies the management of patients with high triglycerides (200 to 499 mg/dL). The non-HDL goal is considered to be 30 mg/dL greater than the LDL target. For patients with diabetes and those with CVD, the individualized non-HDL targets are 130 mg/dL and 100 mg/dL, respectively.9
REVIEW THE MEDICATION LIST
Several commonly used medications, including ß-blockers and thiazide diuretics, can increase triglyceride levels.10 Other medications with exacerbating effects on triglycerides include corticosteroids, retrovirals, immunosuppressants, retinoids, and some antipsychotics.10 Bile acid sequestrants (eg, colesevelam) should be avoided in patients with elevated triglycerides (> 200 mg/dL).7
In women, oral estrogen (ie, menopausal hormone replacement and oral birth control) can greatly exacerbate triglyceride levels, making transdermal delivery a better option. Tamoxifen, the hormonal medication used for breast cancer prophylaxis, can also increase triglyceride levels.11
LOOK FOR UNDERLYING CONDITIONS
Among those to consider: Hypothyroidism is common and easily ruled out by a simple blood test. Nephrotic syndrome should be ruled out, particularly in patients with concomitant renal dysfunction and peripheral edema, by checking a random urine protein-to-creatinine ratio or 24-hour urine for protein. Other factors that should be explored because of their potential effect on lipid metabolism include obesity and excessive intake of sugary beverages (ie, soda, fruit juice) and alcohol.11
High triglyceride levels occurring with low HDL are characteristic of insulin resistance and concerning for metabolic syndrome and/or polycystic ovarian syndrome.3,12 Often, patients will have underlying prediabetes (fasting glucose ≥ 100 mg/dL or random glucose ≥ 140 mg/dL with an A1C > 5.7%13) or covert type 2 diabetes. Another underdiagnosed but very common condition, obstructive sleep apnea, can greatly affect insulin sensitivity and has been associated with lipid abnormalities and metabolic syndrome.14
EXAMINE YOUR PATIENT
The physical exam is an essential component of assessment for patients with high triglycerides. As discussed, elevated triglycerides and low HDL are hallmarks for insulin resistance. As triglyceride levels are affected by obesity and body fat distribution, measuring BMI and assessing for visceral adiposity are an important part of the physical exam.4
The physical exam may also yield dermatologic clues, such as skin tags or acanthosis nigricans, a dark, velvety lesion usually found on the posterior and lateral neck creases, axillae, groin, and elbows.13 In rare cases—usually those with genetic involvement from a familial lipid metabolism disorder—patients may exhibit xanthomas. These cutaneous, lipid-rich lesions can appear as flat, yellowish plaques on various parts of the body, such as the eyelids (xanthelasma) or tendons of the hands, feet, and heels. Widespread, eruptive xanthomas, which manifest as pruritic pink papules with creamy centers, are associated with severe emergent triglyceride elevation and pancreatitis.10
CONSIDER NONPHARMACOLOGIC MANAGEMENT
In mild to moderate hypertriglyceridemia, intensive lifestyle changes are considered firstline therapy. Weight loss is recommended in obese patients; a 5% to 10% reduction in body weight can lower triglycerides by 20%.15
A quick 24-hour diet recall, including beverages, is helpful for identifying key issues. The goal should be to reduce carbohydrates—in particular, simple, high glycemic index, processed foods—as well as total and saturated fats. A substantial problem in our population is the consumption of high-fructose beverages and fruit juices. Referral to a dietitian can be very helpful, not only for initial meal planning but also for continuing counseling on successful long-term weight loss and maintenance.
Exercise is also very helpful for improving lipid parameters. A daily minimum of 30 to 60 minutes of intermittent aerobic exercise or mild resistance exercise has been shown to reduce triglyceride levels.10
PRESCRIBE APPROPRIATELY
The most important indication for treatment of hypertriglyceridemia is reduction of CVD risk. However, in patients with very high triglyceride levels (> 500 mg/dL), the goal is to decrease risk for life-threatening pancreatitis.15 Lipid-lowering medications and dietary restrictions should be promptly employed.
There are medications, as discussed earlier, that specifically lower triglycerides. Fibrates offer the most robust decrease, with a 20% to 50% reduction in triglyceride levels. Fenofibrate is considered a safer option when used in combination with a statin, due to the risk for significant muscle toxicity with gemfibrozil. There is some evidence that adding a fibrate may actually increase risk for pancreatitis; since this risk is otherwise low in patients with mild to moderate triglyceride elevation, the addition of a fibrate to their regimen should be avoided.3
Statins are the drug of choice when CV risk reduction is the goal (for patients with hypertriglyceridemia < 500 mg/dL). In addition to lowering LDL, statins can reduce triglycerides by 7% to 30%, depending on the dose.15
Other triglyceride-lowering medications include omega-3 fatty acids and niacin preparations. Prescription-strength omega-3 fatty acids have been found to lower serum triglyceride levels by 50% or more; the newest preparation, icosapent ethyl, demonstrated up to 45% reduction without significant effect on LDL levels.3 (Other preparations have been shown to substantially increase LDL in many cases.) Niacin (1,500 to 2,000 mg/d) can decrease triglycerides by 15% to 25%. However, it is no longer recommended for CV risk reduction; recent data indicate it may increase stroke risk when used in combination with statins.3,10 In April 2016, the FDA revoked its approval of the co-administration of niacin and fenofibrate with statin therapy, due to a lack of CV benefit.16
Other secondline options to consider for patients with insulin resistance or diabetes are metformin and pioglitazone. These medications have been shown to improve insulin sensitivity and decrease LDL and triglycerides in patients with prediabetes. Pioglitazone has proven beneficial in the treatment of steatohepatitis.17 Insulin is an excellent rapid triglyceride-lowering agent for patients with diabetes. It is important to reinforce that reduction of glucose is a key component in reduction of triglyceride levels.3
CONCLUSION
Hypertriglyceridemia is a complex condition that requires individualized and comprehensive management strategies. Clinicians must be able to identify and address secondary causes. Treatment options should be tailored to decrease CV and pancreatitis risk, and medication recommendations should be evidenced based and carefully selected to mitigate potential adverse effects. Patients should receive education and lifestyle management support to help motivate and equip them to employ strategies to improve their health.
1. CDC. Trends in elevated triglyceride in adults: United States, 2001-2012. www.cdc.gov/nchs/data/databriefs/db198.pdf. Accessed December 27, 2016.
2. Maki KC, Bays HE, Dicklin MR. Treatment options for the management of hypertriglyceridemia: strategies based on the best-available evidence. J Clin Lipidol. 2012;6(5):413-426.
3. Rosenson RS. Approach to the patient with hypertriglyceridemia. www.uptodate.com/contents/approach-to-the-patient-with-hypertriglyceridemia. Accessed December 28, 2016.
4. Talayero BG, Sacks FM. The role of triglycerides in atherosclerosis. Curr Cardiol Rep. 2011;13(6): 544-552.
5. Ginsberg HN, Elam MB, Lovato LC, et al; ACCORD Study Group. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010; 362(17):1563-1574.
6. AIM-HIGH Investigators. The role of niacin in raising high-density lipoprotein cholesterol to reduce cardiovascular events in patients with atherosclerotic cardiovascular disease and optimally treated low-density lipoprotein cholesterol. Rationale and study design. The Atherothrombosis Intervention in Metabolic syndrome with low HDL/high triglycerides: impact on Global Health outcomes (AIM-HIGH). Am Heart J. 2011;161(3):471-477.
7. Miller M, Stone NJ, Ballantyne C, et al. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123(20):2292-2333.
8. Borow KM, Nelson JR, Mason RP. Biologic plausibility, cellular effects, and molecular mechanisms of eicosapentaenoic acid (EPA) in atherosclerosis. Atherosclerosis. 2015;242(1):357-366.
9. Jacobson TA, Ito MK, Maki KC, et al. National Lipid Association recommendations for patient-centered management of dyslipidemia. J Clin Lipidol. 2015;9(2):129-169.
10. Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012; 97(9):2969-2989.
11. Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 pt B):2889-2934.
12. Amini L, Sadeghi MR, Oskuie F, Maleki H. Lipid profile in women with polycystic ovary syndrome. Crescent J Med Biol Sci. 2014;1(4):147-150.
13. Mantzoros C. Insulin resistance: definition and clinical spectrum. www.uptodate.com/contents/insulin-resistance-definition-and-clinical-spectrum. Accessed December 28, 2016.
14. Lin M, Lin H, Lee P, et al. Beneficial effect of continuous positive airway pressure on lipid profiles in obstructive sleep apnea: a meta-analysis. Sleep Breath. 2015;19(3):809-817.
15. Kaur J. A comprehensive review on metabolic syndrome. Cardiol Res Pract. 2014;2014:943162.
16. FDA. Withdrawal of approval of indications related to the coadministration with statins in applications for niacin extended-release tablets and fenofibric acid delayed-release capsules. https://s3.amazonaws.com/public-inspection.federalregister.gov/2016-08887.pdf. Accessed December 28, 2016.
17. Mazza A, Fruci B, Garinis GA, et al. The role of metformin in the management of NAFLD. Exp Diabetes Res. 2012;2012: 716404.
Clinician Reviews in partnership with

Carrie Bowling practices at the South Georgia Medical Center Diabetes Management Center and Valdosta Specialty Clinic, Endocrinology, in Valdosta, Georgia. Joyce Ross is President of theNational Lipid Association and Past President of the Preventive Cardiovascular Nurses Association.
Ms Bowling has no disclosures relevant to the content of this article. Ms Ross is on the Speakers' Bureau for Sanofi/Regeneron, AstraZeneca, Abbott/AbbVie, Amarin, and Amgen; she is also a consultant for Amarin.
Clinician Reviews in partnership with
Carrie Bowling practices at the South Georgia Medical Center Diabetes Management Center and Valdosta Specialty Clinic, Endocrinology, in Valdosta, Georgia. Joyce Ross is President of theNational Lipid Association and Past President of the Preventive Cardiovascular Nurses Association.
Ms Bowling has no disclosures relevant to the content of this article. Ms Ross is on the Speakers' Bureau for Sanofi/Regeneron, AstraZeneca, Abbott/AbbVie, Amarin, and Amgen; she is also a consultant for Amarin.
Clinician Reviews in partnership with
Carrie Bowling practices at the South Georgia Medical Center Diabetes Management Center and Valdosta Specialty Clinic, Endocrinology, in Valdosta, Georgia. Joyce Ross is President of theNational Lipid Association and Past President of the Preventive Cardiovascular Nurses Association.
Ms Bowling has no disclosures relevant to the content of this article. Ms Ross is on the Speakers' Bureau for Sanofi/Regeneron, AstraZeneca, Abbott/AbbVie, Amarin, and Amgen; she is also a consultant for Amarin.
Screening for cardiovascular (CV) risk often includes a routine serum fasting lipid profile. However, with the focus on LDL cholesterol, triglyceride measurement is frequently overlooked. Yet this element of the lipid profile is particularly important, given its strong association with not only atherosclerotic coronary heart disease but also pancreatitis.
Hypertriglyceridemia is defined as a serum triglyceride level that exceeds 150 mg/dL. In the US, an estimated 25% of patients have hypertriglyceridemia.1 Of these, 33.1% have “borderline high” triglyceride levels (150 to 199 mg/dL), 17.8% have “high” levels (200 to 499 mg/dL), and 1.7% have “very high” levels (> 500 mg/dL).1,2
Most of the time, hypertriglyceridemia is caused (or at least exacerbated) by underlying etiology. The best way to identify and manage these secondary causes is through a systematic approach.
CONSIDER THE EVIDENCE
For mild to moderately elevated (borderline high) triglyceride levels, our reflex reaction may be to recommend a triglyceride-lowering medication, such as fenofibrate. But this may not be the best answer. Although there is increasing evidence of an independent association between elevated triglyceride levels and CV risk, it remains unclear whether targeting them specifically can reduce that risk.3
In well-designed, peer-reviewed clinical trials, statins have been shown to reduce CV risk in patients with known cardiovascular disease (CVD) and those at high risk for CVD, as well as in primary prevention. However, these trials also suggest that significant residual CV risk remains after statin therapy.4
Several trials have attempted to prove residual risk reduction following combination therapy including statins—with inconclusive results:
ACCORD: Fenofibrate showed no overall macrovascular benefit when added to a statin in patients with type 2 diabetes and a triglyceride level < 204 mg/dL.3,5
AIM-HIGH: There was a 25% reduction in triglyceride levels when niacin was added to a regimen of a statin +/- ezetimibe, with an aggressive LDL treatment target (40 to 80 mg/dL). But the study was stopped early due to the lack of expected reduction in CVD events.4,6
JELIS: A reduction in major CV events was seen with 1,800 mg/d of eicosapentaenoic acid (EPA) supplementation plus a low-dose statin, compared to statin monotherapy. However, there was minimal change in triglyceride levels, leading the researchers to hypothesize that multiple mechanisms—such as decreasing oxidative stress, platelet aggregation, plaque formation and stabilization—contributed to the outcome.4,7
Informed by the JELIS results, the much-anticipated REDUCE-IT trial is currently in progress to address the lingering question of whether combination therapy can reduce residual CV risk. In this trial, EPA omega-3 fatty acid is being added to the regimen of statin-treated patients with persistently elevated triglycerides. Results are expected in 2017 to 2018.8
Remember that a triglyceride level of 150 mg/dL is a parameter—it does not represent a therapeutic target. There is insufficient evidence that treating to this level improves CV risk beyond LDL target recommendations.7
The National Lipid Association Expert Panel’s consensus view is that non-HDL is a better primary target than triglycerides alone or LDL. Using non-HDL as a target for intervention also simplifies the management of patients with high triglycerides (200 to 499 mg/dL). The non-HDL goal is considered to be 30 mg/dL greater than the LDL target. For patients with diabetes and those with CVD, the individualized non-HDL targets are 130 mg/dL and 100 mg/dL, respectively.9
REVIEW THE MEDICATION LIST
Several commonly used medications, including ß-blockers and thiazide diuretics, can increase triglyceride levels.10 Other medications with exacerbating effects on triglycerides include corticosteroids, retrovirals, immunosuppressants, retinoids, and some antipsychotics.10 Bile acid sequestrants (eg, colesevelam) should be avoided in patients with elevated triglycerides (> 200 mg/dL).7
In women, oral estrogen (ie, menopausal hormone replacement and oral birth control) can greatly exacerbate triglyceride levels, making transdermal delivery a better option. Tamoxifen, the hormonal medication used for breast cancer prophylaxis, can also increase triglyceride levels.11
LOOK FOR UNDERLYING CONDITIONS
Among those to consider: Hypothyroidism is common and easily ruled out by a simple blood test. Nephrotic syndrome should be ruled out, particularly in patients with concomitant renal dysfunction and peripheral edema, by checking a random urine protein-to-creatinine ratio or 24-hour urine for protein. Other factors that should be explored because of their potential effect on lipid metabolism include obesity and excessive intake of sugary beverages (ie, soda, fruit juice) and alcohol.11
High triglyceride levels occurring with low HDL are characteristic of insulin resistance and concerning for metabolic syndrome and/or polycystic ovarian syndrome.3,12 Often, patients will have underlying prediabetes (fasting glucose ≥ 100 mg/dL or random glucose ≥ 140 mg/dL with an A1C > 5.7%13) or covert type 2 diabetes. Another underdiagnosed but very common condition, obstructive sleep apnea, can greatly affect insulin sensitivity and has been associated with lipid abnormalities and metabolic syndrome.14
EXAMINE YOUR PATIENT
The physical exam is an essential component of assessment for patients with high triglycerides. As discussed, elevated triglycerides and low HDL are hallmarks for insulin resistance. As triglyceride levels are affected by obesity and body fat distribution, measuring BMI and assessing for visceral adiposity are an important part of the physical exam.4
The physical exam may also yield dermatologic clues, such as skin tags or acanthosis nigricans, a dark, velvety lesion usually found on the posterior and lateral neck creases, axillae, groin, and elbows.13 In rare cases—usually those with genetic involvement from a familial lipid metabolism disorder—patients may exhibit xanthomas. These cutaneous, lipid-rich lesions can appear as flat, yellowish plaques on various parts of the body, such as the eyelids (xanthelasma) or tendons of the hands, feet, and heels. Widespread, eruptive xanthomas, which manifest as pruritic pink papules with creamy centers, are associated with severe emergent triglyceride elevation and pancreatitis.10
CONSIDER NONPHARMACOLOGIC MANAGEMENT
In mild to moderate hypertriglyceridemia, intensive lifestyle changes are considered firstline therapy. Weight loss is recommended in obese patients; a 5% to 10% reduction in body weight can lower triglycerides by 20%.15
A quick 24-hour diet recall, including beverages, is helpful for identifying key issues. The goal should be to reduce carbohydrates—in particular, simple, high glycemic index, processed foods—as well as total and saturated fats. A substantial problem in our population is the consumption of high-fructose beverages and fruit juices. Referral to a dietitian can be very helpful, not only for initial meal planning but also for continuing counseling on successful long-term weight loss and maintenance.
Exercise is also very helpful for improving lipid parameters. A daily minimum of 30 to 60 minutes of intermittent aerobic exercise or mild resistance exercise has been shown to reduce triglyceride levels.10
PRESCRIBE APPROPRIATELY
The most important indication for treatment of hypertriglyceridemia is reduction of CVD risk. However, in patients with very high triglyceride levels (> 500 mg/dL), the goal is to decrease risk for life-threatening pancreatitis.15 Lipid-lowering medications and dietary restrictions should be promptly employed.
There are medications, as discussed earlier, that specifically lower triglycerides. Fibrates offer the most robust decrease, with a 20% to 50% reduction in triglyceride levels. Fenofibrate is considered a safer option when used in combination with a statin, due to the risk for significant muscle toxicity with gemfibrozil. There is some evidence that adding a fibrate may actually increase risk for pancreatitis; since this risk is otherwise low in patients with mild to moderate triglyceride elevation, the addition of a fibrate to their regimen should be avoided.3
Statins are the drug of choice when CV risk reduction is the goal (for patients with hypertriglyceridemia < 500 mg/dL). In addition to lowering LDL, statins can reduce triglycerides by 7% to 30%, depending on the dose.15
Other triglyceride-lowering medications include omega-3 fatty acids and niacin preparations. Prescription-strength omega-3 fatty acids have been found to lower serum triglyceride levels by 50% or more; the newest preparation, icosapent ethyl, demonstrated up to 45% reduction without significant effect on LDL levels.3 (Other preparations have been shown to substantially increase LDL in many cases.) Niacin (1,500 to 2,000 mg/d) can decrease triglycerides by 15% to 25%. However, it is no longer recommended for CV risk reduction; recent data indicate it may increase stroke risk when used in combination with statins.3,10 In April 2016, the FDA revoked its approval of the co-administration of niacin and fenofibrate with statin therapy, due to a lack of CV benefit.16
Other secondline options to consider for patients with insulin resistance or diabetes are metformin and pioglitazone. These medications have been shown to improve insulin sensitivity and decrease LDL and triglycerides in patients with prediabetes. Pioglitazone has proven beneficial in the treatment of steatohepatitis.17 Insulin is an excellent rapid triglyceride-lowering agent for patients with diabetes. It is important to reinforce that reduction of glucose is a key component in reduction of triglyceride levels.3
CONCLUSION
Hypertriglyceridemia is a complex condition that requires individualized and comprehensive management strategies. Clinicians must be able to identify and address secondary causes. Treatment options should be tailored to decrease CV and pancreatitis risk, and medication recommendations should be evidenced based and carefully selected to mitigate potential adverse effects. Patients should receive education and lifestyle management support to help motivate and equip them to employ strategies to improve their health.
Screening for cardiovascular (CV) risk often includes a routine serum fasting lipid profile. However, with the focus on LDL cholesterol, triglyceride measurement is frequently overlooked. Yet this element of the lipid profile is particularly important, given its strong association with not only atherosclerotic coronary heart disease but also pancreatitis.
Hypertriglyceridemia is defined as a serum triglyceride level that exceeds 150 mg/dL. In the US, an estimated 25% of patients have hypertriglyceridemia.1 Of these, 33.1% have “borderline high” triglyceride levels (150 to 199 mg/dL), 17.8% have “high” levels (200 to 499 mg/dL), and 1.7% have “very high” levels (> 500 mg/dL).1,2
Most of the time, hypertriglyceridemia is caused (or at least exacerbated) by underlying etiology. The best way to identify and manage these secondary causes is through a systematic approach.
CONSIDER THE EVIDENCE
For mild to moderately elevated (borderline high) triglyceride levels, our reflex reaction may be to recommend a triglyceride-lowering medication, such as fenofibrate. But this may not be the best answer. Although there is increasing evidence of an independent association between elevated triglyceride levels and CV risk, it remains unclear whether targeting them specifically can reduce that risk.3
In well-designed, peer-reviewed clinical trials, statins have been shown to reduce CV risk in patients with known cardiovascular disease (CVD) and those at high risk for CVD, as well as in primary prevention. However, these trials also suggest that significant residual CV risk remains after statin therapy.4
Several trials have attempted to prove residual risk reduction following combination therapy including statins—with inconclusive results:
ACCORD: Fenofibrate showed no overall macrovascular benefit when added to a statin in patients with type 2 diabetes and a triglyceride level < 204 mg/dL.3,5
AIM-HIGH: There was a 25% reduction in triglyceride levels when niacin was added to a regimen of a statin +/- ezetimibe, with an aggressive LDL treatment target (40 to 80 mg/dL). But the study was stopped early due to the lack of expected reduction in CVD events.4,6
JELIS: A reduction in major CV events was seen with 1,800 mg/d of eicosapentaenoic acid (EPA) supplementation plus a low-dose statin, compared to statin monotherapy. However, there was minimal change in triglyceride levels, leading the researchers to hypothesize that multiple mechanisms—such as decreasing oxidative stress, platelet aggregation, plaque formation and stabilization—contributed to the outcome.4,7
Informed by the JELIS results, the much-anticipated REDUCE-IT trial is currently in progress to address the lingering question of whether combination therapy can reduce residual CV risk. In this trial, EPA omega-3 fatty acid is being added to the regimen of statin-treated patients with persistently elevated triglycerides. Results are expected in 2017 to 2018.8
Remember that a triglyceride level of 150 mg/dL is a parameter—it does not represent a therapeutic target. There is insufficient evidence that treating to this level improves CV risk beyond LDL target recommendations.7
The National Lipid Association Expert Panel’s consensus view is that non-HDL is a better primary target than triglycerides alone or LDL. Using non-HDL as a target for intervention also simplifies the management of patients with high triglycerides (200 to 499 mg/dL). The non-HDL goal is considered to be 30 mg/dL greater than the LDL target. For patients with diabetes and those with CVD, the individualized non-HDL targets are 130 mg/dL and 100 mg/dL, respectively.9
REVIEW THE MEDICATION LIST
Several commonly used medications, including ß-blockers and thiazide diuretics, can increase triglyceride levels.10 Other medications with exacerbating effects on triglycerides include corticosteroids, retrovirals, immunosuppressants, retinoids, and some antipsychotics.10 Bile acid sequestrants (eg, colesevelam) should be avoided in patients with elevated triglycerides (> 200 mg/dL).7
In women, oral estrogen (ie, menopausal hormone replacement and oral birth control) can greatly exacerbate triglyceride levels, making transdermal delivery a better option. Tamoxifen, the hormonal medication used for breast cancer prophylaxis, can also increase triglyceride levels.11
LOOK FOR UNDERLYING CONDITIONS
Among those to consider: Hypothyroidism is common and easily ruled out by a simple blood test. Nephrotic syndrome should be ruled out, particularly in patients with concomitant renal dysfunction and peripheral edema, by checking a random urine protein-to-creatinine ratio or 24-hour urine for protein. Other factors that should be explored because of their potential effect on lipid metabolism include obesity and excessive intake of sugary beverages (ie, soda, fruit juice) and alcohol.11
High triglyceride levels occurring with low HDL are characteristic of insulin resistance and concerning for metabolic syndrome and/or polycystic ovarian syndrome.3,12 Often, patients will have underlying prediabetes (fasting glucose ≥ 100 mg/dL or random glucose ≥ 140 mg/dL with an A1C > 5.7%13) or covert type 2 diabetes. Another underdiagnosed but very common condition, obstructive sleep apnea, can greatly affect insulin sensitivity and has been associated with lipid abnormalities and metabolic syndrome.14
EXAMINE YOUR PATIENT
The physical exam is an essential component of assessment for patients with high triglycerides. As discussed, elevated triglycerides and low HDL are hallmarks for insulin resistance. As triglyceride levels are affected by obesity and body fat distribution, measuring BMI and assessing for visceral adiposity are an important part of the physical exam.4
The physical exam may also yield dermatologic clues, such as skin tags or acanthosis nigricans, a dark, velvety lesion usually found on the posterior and lateral neck creases, axillae, groin, and elbows.13 In rare cases—usually those with genetic involvement from a familial lipid metabolism disorder—patients may exhibit xanthomas. These cutaneous, lipid-rich lesions can appear as flat, yellowish plaques on various parts of the body, such as the eyelids (xanthelasma) or tendons of the hands, feet, and heels. Widespread, eruptive xanthomas, which manifest as pruritic pink papules with creamy centers, are associated with severe emergent triglyceride elevation and pancreatitis.10
CONSIDER NONPHARMACOLOGIC MANAGEMENT
In mild to moderate hypertriglyceridemia, intensive lifestyle changes are considered firstline therapy. Weight loss is recommended in obese patients; a 5% to 10% reduction in body weight can lower triglycerides by 20%.15
A quick 24-hour diet recall, including beverages, is helpful for identifying key issues. The goal should be to reduce carbohydrates—in particular, simple, high glycemic index, processed foods—as well as total and saturated fats. A substantial problem in our population is the consumption of high-fructose beverages and fruit juices. Referral to a dietitian can be very helpful, not only for initial meal planning but also for continuing counseling on successful long-term weight loss and maintenance.
Exercise is also very helpful for improving lipid parameters. A daily minimum of 30 to 60 minutes of intermittent aerobic exercise or mild resistance exercise has been shown to reduce triglyceride levels.10
PRESCRIBE APPROPRIATELY
The most important indication for treatment of hypertriglyceridemia is reduction of CVD risk. However, in patients with very high triglyceride levels (> 500 mg/dL), the goal is to decrease risk for life-threatening pancreatitis.15 Lipid-lowering medications and dietary restrictions should be promptly employed.
There are medications, as discussed earlier, that specifically lower triglycerides. Fibrates offer the most robust decrease, with a 20% to 50% reduction in triglyceride levels. Fenofibrate is considered a safer option when used in combination with a statin, due to the risk for significant muscle toxicity with gemfibrozil. There is some evidence that adding a fibrate may actually increase risk for pancreatitis; since this risk is otherwise low in patients with mild to moderate triglyceride elevation, the addition of a fibrate to their regimen should be avoided.3
Statins are the drug of choice when CV risk reduction is the goal (for patients with hypertriglyceridemia < 500 mg/dL). In addition to lowering LDL, statins can reduce triglycerides by 7% to 30%, depending on the dose.15
Other triglyceride-lowering medications include omega-3 fatty acids and niacin preparations. Prescription-strength omega-3 fatty acids have been found to lower serum triglyceride levels by 50% or more; the newest preparation, icosapent ethyl, demonstrated up to 45% reduction without significant effect on LDL levels.3 (Other preparations have been shown to substantially increase LDL in many cases.) Niacin (1,500 to 2,000 mg/d) can decrease triglycerides by 15% to 25%. However, it is no longer recommended for CV risk reduction; recent data indicate it may increase stroke risk when used in combination with statins.3,10 In April 2016, the FDA revoked its approval of the co-administration of niacin and fenofibrate with statin therapy, due to a lack of CV benefit.16
Other secondline options to consider for patients with insulin resistance or diabetes are metformin and pioglitazone. These medications have been shown to improve insulin sensitivity and decrease LDL and triglycerides in patients with prediabetes. Pioglitazone has proven beneficial in the treatment of steatohepatitis.17 Insulin is an excellent rapid triglyceride-lowering agent for patients with diabetes. It is important to reinforce that reduction of glucose is a key component in reduction of triglyceride levels.3
CONCLUSION
Hypertriglyceridemia is a complex condition that requires individualized and comprehensive management strategies. Clinicians must be able to identify and address secondary causes. Treatment options should be tailored to decrease CV and pancreatitis risk, and medication recommendations should be evidenced based and carefully selected to mitigate potential adverse effects. Patients should receive education and lifestyle management support to help motivate and equip them to employ strategies to improve their health.
1. CDC. Trends in elevated triglyceride in adults: United States, 2001-2012. www.cdc.gov/nchs/data/databriefs/db198.pdf. Accessed December 27, 2016.
2. Maki KC, Bays HE, Dicklin MR. Treatment options for the management of hypertriglyceridemia: strategies based on the best-available evidence. J Clin Lipidol. 2012;6(5):413-426.
3. Rosenson RS. Approach to the patient with hypertriglyceridemia. www.uptodate.com/contents/approach-to-the-patient-with-hypertriglyceridemia. Accessed December 28, 2016.
4. Talayero BG, Sacks FM. The role of triglycerides in atherosclerosis. Curr Cardiol Rep. 2011;13(6): 544-552.
5. Ginsberg HN, Elam MB, Lovato LC, et al; ACCORD Study Group. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010; 362(17):1563-1574.
6. AIM-HIGH Investigators. The role of niacin in raising high-density lipoprotein cholesterol to reduce cardiovascular events in patients with atherosclerotic cardiovascular disease and optimally treated low-density lipoprotein cholesterol. Rationale and study design. The Atherothrombosis Intervention in Metabolic syndrome with low HDL/high triglycerides: impact on Global Health outcomes (AIM-HIGH). Am Heart J. 2011;161(3):471-477.
7. Miller M, Stone NJ, Ballantyne C, et al. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123(20):2292-2333.
8. Borow KM, Nelson JR, Mason RP. Biologic plausibility, cellular effects, and molecular mechanisms of eicosapentaenoic acid (EPA) in atherosclerosis. Atherosclerosis. 2015;242(1):357-366.
9. Jacobson TA, Ito MK, Maki KC, et al. National Lipid Association recommendations for patient-centered management of dyslipidemia. J Clin Lipidol. 2015;9(2):129-169.
10. Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012; 97(9):2969-2989.
11. Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 pt B):2889-2934.
12. Amini L, Sadeghi MR, Oskuie F, Maleki H. Lipid profile in women with polycystic ovary syndrome. Crescent J Med Biol Sci. 2014;1(4):147-150.
13. Mantzoros C. Insulin resistance: definition and clinical spectrum. www.uptodate.com/contents/insulin-resistance-definition-and-clinical-spectrum. Accessed December 28, 2016.
14. Lin M, Lin H, Lee P, et al. Beneficial effect of continuous positive airway pressure on lipid profiles in obstructive sleep apnea: a meta-analysis. Sleep Breath. 2015;19(3):809-817.
15. Kaur J. A comprehensive review on metabolic syndrome. Cardiol Res Pract. 2014;2014:943162.
16. FDA. Withdrawal of approval of indications related to the coadministration with statins in applications for niacin extended-release tablets and fenofibric acid delayed-release capsules. https://s3.amazonaws.com/public-inspection.federalregister.gov/2016-08887.pdf. Accessed December 28, 2016.
17. Mazza A, Fruci B, Garinis GA, et al. The role of metformin in the management of NAFLD. Exp Diabetes Res. 2012;2012: 716404.
1. CDC. Trends in elevated triglyceride in adults: United States, 2001-2012. www.cdc.gov/nchs/data/databriefs/db198.pdf. Accessed December 27, 2016.
2. Maki KC, Bays HE, Dicklin MR. Treatment options for the management of hypertriglyceridemia: strategies based on the best-available evidence. J Clin Lipidol. 2012;6(5):413-426.
3. Rosenson RS. Approach to the patient with hypertriglyceridemia. www.uptodate.com/contents/approach-to-the-patient-with-hypertriglyceridemia. Accessed December 28, 2016.
4. Talayero BG, Sacks FM. The role of triglycerides in atherosclerosis. Curr Cardiol Rep. 2011;13(6): 544-552.
5. Ginsberg HN, Elam MB, Lovato LC, et al; ACCORD Study Group. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010; 362(17):1563-1574.
6. AIM-HIGH Investigators. The role of niacin in raising high-density lipoprotein cholesterol to reduce cardiovascular events in patients with atherosclerotic cardiovascular disease and optimally treated low-density lipoprotein cholesterol. Rationale and study design. The Atherothrombosis Intervention in Metabolic syndrome with low HDL/high triglycerides: impact on Global Health outcomes (AIM-HIGH). Am Heart J. 2011;161(3):471-477.
7. Miller M, Stone NJ, Ballantyne C, et al. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123(20):2292-2333.
8. Borow KM, Nelson JR, Mason RP. Biologic plausibility, cellular effects, and molecular mechanisms of eicosapentaenoic acid (EPA) in atherosclerosis. Atherosclerosis. 2015;242(1):357-366.
9. Jacobson TA, Ito MK, Maki KC, et al. National Lipid Association recommendations for patient-centered management of dyslipidemia. J Clin Lipidol. 2015;9(2):129-169.
10. Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012; 97(9):2969-2989.
11. Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 pt B):2889-2934.
12. Amini L, Sadeghi MR, Oskuie F, Maleki H. Lipid profile in women with polycystic ovary syndrome. Crescent J Med Biol Sci. 2014;1(4):147-150.
13. Mantzoros C. Insulin resistance: definition and clinical spectrum. www.uptodate.com/contents/insulin-resistance-definition-and-clinical-spectrum. Accessed December 28, 2016.
14. Lin M, Lin H, Lee P, et al. Beneficial effect of continuous positive airway pressure on lipid profiles in obstructive sleep apnea: a meta-analysis. Sleep Breath. 2015;19(3):809-817.
15. Kaur J. A comprehensive review on metabolic syndrome. Cardiol Res Pract. 2014;2014:943162.
16. FDA. Withdrawal of approval of indications related to the coadministration with statins in applications for niacin extended-release tablets and fenofibric acid delayed-release capsules. https://s3.amazonaws.com/public-inspection.federalregister.gov/2016-08887.pdf. Accessed December 28, 2016.
17. Mazza A, Fruci B, Garinis GA, et al. The role of metformin in the management of NAFLD. Exp Diabetes Res. 2012;2012: 716404.
Scholar grants help future hospitalists explore career pathways
Editor’s note: Each month, SHM puts the spotlight on some of our most active members who are making substantial contributions to hospital medicine. Log on to www.hospitalmedicine.org/getinvolved for more information on how you can lend your expertise to help SHM improve the care of hospitalized patients.
This month, The Hospitalist spotlights Ernie L. Esquivel, MD, FACP, FHM, the clerkship director, medicine, and assistant professor of clinical medicine in the Division of General Internal Medicine at the Weill Cornell Medical College in New York City. Dr. Esquivel is involved with SHM’s Physicians in Training Committee, and has spearheaded the creation of the Student Hospitalist Scholar Grant program.
What inspired you to become a hospitalist?
I became a hospitalist serendipitously. At a critical juncture in my life about 8 years ago (when a change in career direction became necessary), I chanced upon a locum tenens position in a small community hospital in Lansdale, Pa., as a hospitalist. Having been a primary care track resident who subsequently chose to specialize in nephrology, I rediscovered my generalist inclinations during this job. I fell in love with the fast pace of the hospitalist’s work, the complexity of delivering care and the diversity of diseases, and of personal life stories on the general medicine wards and the ICU.

Subsequently, I worked as an intensivist in Philadelphia for a year before joining the Academic Hospital Medicine Division at Weill Cornell in New York City. At Cornell, I have managed to cultivate my passion for medical education, especially for working with and mentoring students and residents, while continuing to care for patients on the general medicine wards. As the medicine clerkship director, I have had the privilege of creating an innovative curriculum that I hope prepares medical students for the challenges in, and the richness of, encounters in the practice of inpatient medicine.
How and why did you become a member of SHM and the Physicians in Training Committee (PIT)?
I joined SHM 6 years ago as I started to explore my career options more deeply. In 2010, I attended the Academic Hospitalist Academy, and that really offered me a closer look at the different ways in which SHM could help me advance. I went to my first SHM annual meeting 5 years ago; it motivated me to become involved in committee work. Because of my interest in medical education, I volunteered for the PIT Committee and it has given me the opportunity to work closely with other hospitalists around the country, and develop programming specifically targeted toward future hospitalists.
What is the PIT Committee working on?
The committee has continued to find ways for increased engagement of residents and students in SHM. Dr. Brian Kwan, an academic hospitalist at UC San Diego, and I have been developing a travel grant program for resident trainees and hospital medicine fellows to attend the annual meeting. By offering them a stipend to defray the costs of travel if their quality improvement innovation or research project is accepted, we hope that the annual meeting can become a venue for them to highlight their work, while becoming exposed to the many activities and opportunities offered by our society. In addition, it could be a way for them to network with other future hospitalists and established future mentors.
What prompted you to lead the creation of the Student Hospitalist Scholar Grant summer program?
Before I became a hospitalist, I spent about 7 years in research, studying renal genetics. I have always been fascinated by science and asked how I can help to advance our knowledge. As a hospitalist, it became clear to me early on that there are many questions that one can pose about the clinical work we do, the way we practice medicine, or ways to innovate education, and that there are many academic hospitalists who engage in advancing the field. I spearheaded this program because I would like students to see the field of hospital medicine as one in which they can develop a future career in academic medicine, not only by caring for patients, but also by involving themselves in research questions or QI projects.
Do you have any specific advice for students and residents interested in hospital medicine? In what ways can early-career hospitalists utilize SHM resources to leverage their careers?
The decision to pursue a career as a hospitalist will open up many more questions in the future, because there are so many opportunities available. I would suggest that trainees ask in which ways they see themselves growing in the future – clinical research, medical education, QI/patient safety, operations, and hospital leadership are the main avenues. When I interview future faculty, I always pose the same question to each of them: “Every year you are allocated X amount of money that you can use for CME, etc. How are you going to use this money to improve your skills in any particular area?” The ability of candidates to answer this question reflects for me their preparedness to develop themselves as career hospitalists and their willingness to contribute to their group or division in an innovative manner.
The reality is that as one gets older, most will find it difficult to sustain a 26-week/year schedule. So find ways for your energies, in whichever area, to be noticed and developed toward a position of leadership in the hospital or medical school.
As you take care of patients in the hospital or consider your education and training, identify ways in which things can be done better. Invariably, someone in the Society of Hospital Medicine is interested in the same issue(s). Explore your ideas, share them at the meeting, talk to people, go to the SHM website and identify what resources are already available.
If SHM will be your future academic home, volunteer to engage in activities at the chapter or national levels. Our society is really dedicated to identifying ways to welcome you into our exciting and continually evolving field.
Felicia Steele is SHM’s communications coordinator.
Editor’s note: Each month, SHM puts the spotlight on some of our most active members who are making substantial contributions to hospital medicine. Log on to www.hospitalmedicine.org/getinvolved for more information on how you can lend your expertise to help SHM improve the care of hospitalized patients.
This month, The Hospitalist spotlights Ernie L. Esquivel, MD, FACP, FHM, the clerkship director, medicine, and assistant professor of clinical medicine in the Division of General Internal Medicine at the Weill Cornell Medical College in New York City. Dr. Esquivel is involved with SHM’s Physicians in Training Committee, and has spearheaded the creation of the Student Hospitalist Scholar Grant program.
What inspired you to become a hospitalist?
I became a hospitalist serendipitously. At a critical juncture in my life about 8 years ago (when a change in career direction became necessary), I chanced upon a locum tenens position in a small community hospital in Lansdale, Pa., as a hospitalist. Having been a primary care track resident who subsequently chose to specialize in nephrology, I rediscovered my generalist inclinations during this job. I fell in love with the fast pace of the hospitalist’s work, the complexity of delivering care and the diversity of diseases, and of personal life stories on the general medicine wards and the ICU.
Subsequently, I worked as an intensivist in Philadelphia for a year before joining the Academic Hospital Medicine Division at Weill Cornell in New York City. At Cornell, I have managed to cultivate my passion for medical education, especially for working with and mentoring students and residents, while continuing to care for patients on the general medicine wards. As the medicine clerkship director, I have had the privilege of creating an innovative curriculum that I hope prepares medical students for the challenges in, and the richness of, encounters in the practice of inpatient medicine.
How and why did you become a member of SHM and the Physicians in Training Committee (PIT)?
I joined SHM 6 years ago as I started to explore my career options more deeply. In 2010, I attended the Academic Hospitalist Academy, and that really offered me a closer look at the different ways in which SHM could help me advance. I went to my first SHM annual meeting 5 years ago; it motivated me to become involved in committee work. Because of my interest in medical education, I volunteered for the PIT Committee and it has given me the opportunity to work closely with other hospitalists around the country, and develop programming specifically targeted toward future hospitalists.
What is the PIT Committee working on?
The committee has continued to find ways for increased engagement of residents and students in SHM. Dr. Brian Kwan, an academic hospitalist at UC San Diego, and I have been developing a travel grant program for resident trainees and hospital medicine fellows to attend the annual meeting. By offering them a stipend to defray the costs of travel if their quality improvement innovation or research project is accepted, we hope that the annual meeting can become a venue for them to highlight their work, while becoming exposed to the many activities and opportunities offered by our society. In addition, it could be a way for them to network with other future hospitalists and established future mentors.
What prompted you to lead the creation of the Student Hospitalist Scholar Grant summer program?
Before I became a hospitalist, I spent about 7 years in research, studying renal genetics. I have always been fascinated by science and asked how I can help to advance our knowledge. As a hospitalist, it became clear to me early on that there are many questions that one can pose about the clinical work we do, the way we practice medicine, or ways to innovate education, and that there are many academic hospitalists who engage in advancing the field. I spearheaded this program because I would like students to see the field of hospital medicine as one in which they can develop a future career in academic medicine, not only by caring for patients, but also by involving themselves in research questions or QI projects.
Do you have any specific advice for students and residents interested in hospital medicine? In what ways can early-career hospitalists utilize SHM resources to leverage their careers?
The decision to pursue a career as a hospitalist will open up many more questions in the future, because there are so many opportunities available. I would suggest that trainees ask in which ways they see themselves growing in the future – clinical research, medical education, QI/patient safety, operations, and hospital leadership are the main avenues. When I interview future faculty, I always pose the same question to each of them: “Every year you are allocated X amount of money that you can use for CME, etc. How are you going to use this money to improve your skills in any particular area?” The ability of candidates to answer this question reflects for me their preparedness to develop themselves as career hospitalists and their willingness to contribute to their group or division in an innovative manner.
The reality is that as one gets older, most will find it difficult to sustain a 26-week/year schedule. So find ways for your energies, in whichever area, to be noticed and developed toward a position of leadership in the hospital or medical school.
As you take care of patients in the hospital or consider your education and training, identify ways in which things can be done better. Invariably, someone in the Society of Hospital Medicine is interested in the same issue(s). Explore your ideas, share them at the meeting, talk to people, go to the SHM website and identify what resources are already available.
If SHM will be your future academic home, volunteer to engage in activities at the chapter or national levels. Our society is really dedicated to identifying ways to welcome you into our exciting and continually evolving field.
Felicia Steele is SHM’s communications coordinator.
Editor’s note: Each month, SHM puts the spotlight on some of our most active members who are making substantial contributions to hospital medicine. Log on to www.hospitalmedicine.org/getinvolved for more information on how you can lend your expertise to help SHM improve the care of hospitalized patients.
This month, The Hospitalist spotlights Ernie L. Esquivel, MD, FACP, FHM, the clerkship director, medicine, and assistant professor of clinical medicine in the Division of General Internal Medicine at the Weill Cornell Medical College in New York City. Dr. Esquivel is involved with SHM’s Physicians in Training Committee, and has spearheaded the creation of the Student Hospitalist Scholar Grant program.
What inspired you to become a hospitalist?
I became a hospitalist serendipitously. At a critical juncture in my life about 8 years ago (when a change in career direction became necessary), I chanced upon a locum tenens position in a small community hospital in Lansdale, Pa., as a hospitalist. Having been a primary care track resident who subsequently chose to specialize in nephrology, I rediscovered my generalist inclinations during this job. I fell in love with the fast pace of the hospitalist’s work, the complexity of delivering care and the diversity of diseases, and of personal life stories on the general medicine wards and the ICU.
Subsequently, I worked as an intensivist in Philadelphia for a year before joining the Academic Hospital Medicine Division at Weill Cornell in New York City. At Cornell, I have managed to cultivate my passion for medical education, especially for working with and mentoring students and residents, while continuing to care for patients on the general medicine wards. As the medicine clerkship director, I have had the privilege of creating an innovative curriculum that I hope prepares medical students for the challenges in, and the richness of, encounters in the practice of inpatient medicine.
How and why did you become a member of SHM and the Physicians in Training Committee (PIT)?
I joined SHM 6 years ago as I started to explore my career options more deeply. In 2010, I attended the Academic Hospitalist Academy, and that really offered me a closer look at the different ways in which SHM could help me advance. I went to my first SHM annual meeting 5 years ago; it motivated me to become involved in committee work. Because of my interest in medical education, I volunteered for the PIT Committee and it has given me the opportunity to work closely with other hospitalists around the country, and develop programming specifically targeted toward future hospitalists.
What is the PIT Committee working on?
The committee has continued to find ways for increased engagement of residents and students in SHM. Dr. Brian Kwan, an academic hospitalist at UC San Diego, and I have been developing a travel grant program for resident trainees and hospital medicine fellows to attend the annual meeting. By offering them a stipend to defray the costs of travel if their quality improvement innovation or research project is accepted, we hope that the annual meeting can become a venue for them to highlight their work, while becoming exposed to the many activities and opportunities offered by our society. In addition, it could be a way for them to network with other future hospitalists and established future mentors.
What prompted you to lead the creation of the Student Hospitalist Scholar Grant summer program?
Before I became a hospitalist, I spent about 7 years in research, studying renal genetics. I have always been fascinated by science and asked how I can help to advance our knowledge. As a hospitalist, it became clear to me early on that there are many questions that one can pose about the clinical work we do, the way we practice medicine, or ways to innovate education, and that there are many academic hospitalists who engage in advancing the field. I spearheaded this program because I would like students to see the field of hospital medicine as one in which they can develop a future career in academic medicine, not only by caring for patients, but also by involving themselves in research questions or QI projects.
Do you have any specific advice for students and residents interested in hospital medicine? In what ways can early-career hospitalists utilize SHM resources to leverage their careers?
The decision to pursue a career as a hospitalist will open up many more questions in the future, because there are so many opportunities available. I would suggest that trainees ask in which ways they see themselves growing in the future – clinical research, medical education, QI/patient safety, operations, and hospital leadership are the main avenues. When I interview future faculty, I always pose the same question to each of them: “Every year you are allocated X amount of money that you can use for CME, etc. How are you going to use this money to improve your skills in any particular area?” The ability of candidates to answer this question reflects for me their preparedness to develop themselves as career hospitalists and their willingness to contribute to their group or division in an innovative manner.
The reality is that as one gets older, most will find it difficult to sustain a 26-week/year schedule. So find ways for your energies, in whichever area, to be noticed and developed toward a position of leadership in the hospital or medical school.
As you take care of patients in the hospital or consider your education and training, identify ways in which things can be done better. Invariably, someone in the Society of Hospital Medicine is interested in the same issue(s). Explore your ideas, share them at the meeting, talk to people, go to the SHM website and identify what resources are already available.
If SHM will be your future academic home, volunteer to engage in activities at the chapter or national levels. Our society is really dedicated to identifying ways to welcome you into our exciting and continually evolving field.
Felicia Steele is SHM’s communications coordinator.
Postdischarge antibiotics for complicated pneumonia
Clinical question: Are oral antibiotics as effective as intravenous (IV) antibiotics in the treatment of complicated pneumonia after discharge to home?
Background: Pneumonia is the most common illness among hospitalized children and adolescents (excluding neonates). Among children admitted with community acquired pneumonia, 15% may develop a complicated pneumonia (one with a pleural effusion or empyema). Treatment for these complicated pneumonias may include a variety of invasive procedures, such as video-assisted thorascopic surgery or chest tube placement.
Typically, a long course of antibiotics is prescribed on discharge, which may be oral or parenterally administered via a peripherally inserted central catheter (PICC). Previous studies have shown that oral antibiotics are equivalent to parenteral antibiotics for outpatient treatment of osteomyelitis. However, little evidence exists comparing the effectiveness of the two routes in treating complicated pneumonia.

The rate of PICC complications in complicated pneumonia also has not been well studied.
Study design: Retrospective cohort study.
Setting: Thirty-eight children’s hospitals affiliated with the Children’s Hospital Association.
Synopsis: Over 4 years, 7,820 encounters were identified with 2,123 patients ultimately being included in the cohort. Inclusion criteria were age 2 months to 18 years, and discharge diagnoses of pneumonia and pleural effusion. The authors excluded patients with chronic medical conditions, length of stay (LOS) less than 4 and more than 14 days, patients transferred to or from other institutions, and patients receiving no antibiotics on hospital day 1. The final criteria attempted to avoid inclusion of patients with nosocomial pneumonia. After application of these criteria, individual patient records were reviewed.
Patients were categorized as PICC or oral antibiotics based upon antibiotic route at their initial discharge. Treatment failure was defined as an ED revisit or rehospitalization that led to a change in antibiotic, lengthening of antibiotic course, or pleural drainage. Records were searched for evidence of PICC complications, adverse drug reactions, and other illness-related revisits. Patients in the PICC arm and oral arm were matched by age, race, insurance, LOS, positive vs. negative blood culture, ICU admission, and timing and type of pleural drainage.
Fifty-seven patients had treatment failure (2.7%). In matched analysis, there was no difference in treatment failure between PICC and oral routes (PICC treatment failure OR, 1.26 95% CI, 0.54-2.94). PICC complications were found in 7.1% of patients. Patients with PICC had significantly higher rates of adverse drug reactions (OR, 19.1 95% CI, 4.2-87.3) and illness-related revisits (OR 3.27 95% CI, 1.65-6.48), and all revisits (OR, 4.71 95% CI, 2.97-7.46).
PICC use varied markedly across geographic regions and institutions, with rates varying from less than 10% of cases to approximately 70%. Of geographic regions, the Mid-Atlantic used PICCs least often while the East North Central used them the most.
Bottom line: Treatment failure with both oral and PICC treatment of complicated pneumonia occur at the same rate, and are uncommon. Patients with PICCs had an increased rate of complications, including adverse drug reactions and revisits.
Citation: Shah SS, Srivastava R, Wu S, et al. Intravenous versus oral antibiotics for postdischarge treatment of complicated pneumonia. Pediatrics. 2016;138(6):e20161692. doi: 10.1542/peds.2016-1692.
Dr. Stubblefield is a pediatric hospitalist at Nemours/Alfred I. duPont Hospital for Children in Wilmington, Del., and clinical assistant professor of pediatrics at Sidney Kimmel Medical College at Thomas Jefferson University in Philadelphia.
Clinical question: Are oral antibiotics as effective as intravenous (IV) antibiotics in the treatment of complicated pneumonia after discharge to home?
Background: Pneumonia is the most common illness among hospitalized children and adolescents (excluding neonates). Among children admitted with community acquired pneumonia, 15% may develop a complicated pneumonia (one with a pleural effusion or empyema). Treatment for these complicated pneumonias may include a variety of invasive procedures, such as video-assisted thorascopic surgery or chest tube placement.
Typically, a long course of antibiotics is prescribed on discharge, which may be oral or parenterally administered via a peripherally inserted central catheter (PICC). Previous studies have shown that oral antibiotics are equivalent to parenteral antibiotics for outpatient treatment of osteomyelitis. However, little evidence exists comparing the effectiveness of the two routes in treating complicated pneumonia.
The rate of PICC complications in complicated pneumonia also has not been well studied.
Study design: Retrospective cohort study.
Setting: Thirty-eight children’s hospitals affiliated with the Children’s Hospital Association.
Synopsis: Over 4 years, 7,820 encounters were identified with 2,123 patients ultimately being included in the cohort. Inclusion criteria were age 2 months to 18 years, and discharge diagnoses of pneumonia and pleural effusion. The authors excluded patients with chronic medical conditions, length of stay (LOS) less than 4 and more than 14 days, patients transferred to or from other institutions, and patients receiving no antibiotics on hospital day 1. The final criteria attempted to avoid inclusion of patients with nosocomial pneumonia. After application of these criteria, individual patient records were reviewed.
Patients were categorized as PICC or oral antibiotics based upon antibiotic route at their initial discharge. Treatment failure was defined as an ED revisit or rehospitalization that led to a change in antibiotic, lengthening of antibiotic course, or pleural drainage. Records were searched for evidence of PICC complications, adverse drug reactions, and other illness-related revisits. Patients in the PICC arm and oral arm were matched by age, race, insurance, LOS, positive vs. negative blood culture, ICU admission, and timing and type of pleural drainage.
Fifty-seven patients had treatment failure (2.7%). In matched analysis, there was no difference in treatment failure between PICC and oral routes (PICC treatment failure OR, 1.26 95% CI, 0.54-2.94). PICC complications were found in 7.1% of patients. Patients with PICC had significantly higher rates of adverse drug reactions (OR, 19.1 95% CI, 4.2-87.3) and illness-related revisits (OR 3.27 95% CI, 1.65-6.48), and all revisits (OR, 4.71 95% CI, 2.97-7.46).
PICC use varied markedly across geographic regions and institutions, with rates varying from less than 10% of cases to approximately 70%. Of geographic regions, the Mid-Atlantic used PICCs least often while the East North Central used them the most.
Bottom line: Treatment failure with both oral and PICC treatment of complicated pneumonia occur at the same rate, and are uncommon. Patients with PICCs had an increased rate of complications, including adverse drug reactions and revisits.
Citation: Shah SS, Srivastava R, Wu S, et al. Intravenous versus oral antibiotics for postdischarge treatment of complicated pneumonia. Pediatrics. 2016;138(6):e20161692. doi: 10.1542/peds.2016-1692.
Dr. Stubblefield is a pediatric hospitalist at Nemours/Alfred I. duPont Hospital for Children in Wilmington, Del., and clinical assistant professor of pediatrics at Sidney Kimmel Medical College at Thomas Jefferson University in Philadelphia.
Clinical question: Are oral antibiotics as effective as intravenous (IV) antibiotics in the treatment of complicated pneumonia after discharge to home?
Background: Pneumonia is the most common illness among hospitalized children and adolescents (excluding neonates). Among children admitted with community acquired pneumonia, 15% may develop a complicated pneumonia (one with a pleural effusion or empyema). Treatment for these complicated pneumonias may include a variety of invasive procedures, such as video-assisted thorascopic surgery or chest tube placement.
Typically, a long course of antibiotics is prescribed on discharge, which may be oral or parenterally administered via a peripherally inserted central catheter (PICC). Previous studies have shown that oral antibiotics are equivalent to parenteral antibiotics for outpatient treatment of osteomyelitis. However, little evidence exists comparing the effectiveness of the two routes in treating complicated pneumonia.
The rate of PICC complications in complicated pneumonia also has not been well studied.
Study design: Retrospective cohort study.
Setting: Thirty-eight children’s hospitals affiliated with the Children’s Hospital Association.
Synopsis: Over 4 years, 7,820 encounters were identified with 2,123 patients ultimately being included in the cohort. Inclusion criteria were age 2 months to 18 years, and discharge diagnoses of pneumonia and pleural effusion. The authors excluded patients with chronic medical conditions, length of stay (LOS) less than 4 and more than 14 days, patients transferred to or from other institutions, and patients receiving no antibiotics on hospital day 1. The final criteria attempted to avoid inclusion of patients with nosocomial pneumonia. After application of these criteria, individual patient records were reviewed.
Patients were categorized as PICC or oral antibiotics based upon antibiotic route at their initial discharge. Treatment failure was defined as an ED revisit or rehospitalization that led to a change in antibiotic, lengthening of antibiotic course, or pleural drainage. Records were searched for evidence of PICC complications, adverse drug reactions, and other illness-related revisits. Patients in the PICC arm and oral arm were matched by age, race, insurance, LOS, positive vs. negative blood culture, ICU admission, and timing and type of pleural drainage.
Fifty-seven patients had treatment failure (2.7%). In matched analysis, there was no difference in treatment failure between PICC and oral routes (PICC treatment failure OR, 1.26 95% CI, 0.54-2.94). PICC complications were found in 7.1% of patients. Patients with PICC had significantly higher rates of adverse drug reactions (OR, 19.1 95% CI, 4.2-87.3) and illness-related revisits (OR 3.27 95% CI, 1.65-6.48), and all revisits (OR, 4.71 95% CI, 2.97-7.46).
PICC use varied markedly across geographic regions and institutions, with rates varying from less than 10% of cases to approximately 70%. Of geographic regions, the Mid-Atlantic used PICCs least often while the East North Central used them the most.
Bottom line: Treatment failure with both oral and PICC treatment of complicated pneumonia occur at the same rate, and are uncommon. Patients with PICCs had an increased rate of complications, including adverse drug reactions and revisits.
Citation: Shah SS, Srivastava R, Wu S, et al. Intravenous versus oral antibiotics for postdischarge treatment of complicated pneumonia. Pediatrics. 2016;138(6):e20161692. doi: 10.1542/peds.2016-1692.
Dr. Stubblefield is a pediatric hospitalist at Nemours/Alfred I. duPont Hospital for Children in Wilmington, Del., and clinical assistant professor of pediatrics at Sidney Kimmel Medical College at Thomas Jefferson University in Philadelphia.