User login
Atherosclerosis severity in diabetes can be predicted by select biomarkers
in patient with type 2 diabetes, results from a long-term analysis of VA patients suggest.
Advanced glycation end products (AGEs) and oxidation products (OxPs) “can damage vascular cells by different mechanisms,” wrote researchers led by corresponding authors Aramesh Saremi, MD, and Peter D. Reaven, MD, and colleagues. The report appeared online Feb. 1 in Diabetes Care.
“One frequently reported pathway is AGE binding to their purported (and relatively promiscuous) receptors on cells, such as macrophages, vascular endothelial cells, and vascular smooth muscle cells, although this has not been consistent for all AGEs. Other mechanisms include, among others, binding to and altering the function of intracellular proteins, the activation of vascular NADPH [nicotinamide adenine dinucleotide phosphate] oxidase, and the uncoupling of endothelial nitric oxide synthase.”
Noting that data in the current medical literature are lacking with respect to long-term longitudinal associations between plasma levels of AGEs and OxPs on the extent of subclinical atherosclerosis in T2D patients, the researchers set out to determine whether baseline plasma levels of AGEs and OxPs are associated with the extent of carotid intima-media thickness (CIMT), coronary artery calcification (CAC), and abdominal aortic artery calcification (AAC) over an average of 10 years of follow-up in the VA Diabetes Trial (VADT). They also examined whether this relationship was altered by intervening improved glucose control (Diabetes Care 2017 Feb. 1. doi: 10.2337/dc16-1875]).
At baseline of the VADT, 411 study participants underwent plasma measurements of methylglyoxal hydroimidazolone, N epsilon–carboxymethyl lysine (CML), N epsilon–carboxyethyl lysine (CEL), 3-deoxyglucosone hydroimidazolone and glyoxal hydroimidazolone (G-H1), 2-aminoadipic acid (2-AAA), and methionine sulfoxide. The mean age of the study subjects was 58 years, 64% were non-Hispanic white, 96% were male, 69% had a history of hypertension, and they had diabetes for a mean of 11 years.
After a mean follow-up of 10 years, the 411 patients underwent ultrasound assessment of CIMT, and computed tomography scanning of CAC and AAC.
In risk factor–adjusted multivariable regression models, G-H1was associated with the extent of CIMT as well as with the extent of CAC (P = .01 for both associations). In addition, 2-AAA was strongly associated with the extent of CAC (P = .03 for continuous variables and P less than .01 for dichotomous variables), and CEL was strongly associated with the extent of AAC (P less than .01).
“These findings suggest that the effect of hyperglycemia and subsequent increased levels of AGEs and OxPs in patients with long-standing T2D may have long-lasting adverse effects on the development of macrovascular complications,” the researchers concluded. They acknowledged certain limitations of the analysis, including the fact that it was conducted in an older, primarily male population. Therefore, “extrapolation of the study findings to other populations must be done with caution,” they wrote. “This study also does not allow us to make a definite claim of causation between AGEs and OxPs with the extent of atherosclerosis.”
The Veterans Affairs Cooperative Studies Program of the U.S. Department of Veterans Affairs Office of Research and Development funded the study. Additional support was received from National Institutes of Health, the American Diabetes Association, and the National Institute of Diabetes and Digestive and Kidney Diseases. Two study authors, Scott Howell, MS, and Paul J. Beisswenger, MD, disclosed that they are affiliated with PreventAGE Healthcare. The other researchers reported having no financial disclosures.
[email protected]
For a long time the question about how glucose control relates to macrovascular disease has not been easy to answer. There are reports suggesting that glucose control correlates with macrovascular disease later in life, and others that do not make the association so convincing. This paper provides an important link between glucose control and the subsequent development of macrovascular disease. The link is by way of advanced glycation end products as well as oxidative byproducts and how they set the stage for macrovascular disease later on in life. It’s something that clinicians have postulated as being important, but to my knowledge this is one of the only studies to actually show the association.

The investigators used important endpoints like coronary artery calcification and carotid intima-media thickness. I was surprised at how well the relationship between glycosylated end products and oxidative products correlated, but there’s biologic plausibility; it makes sense. Glucose not only glycosates hemoglobin, but glycosates proteins throughout. This provides a logical, stepwise pathway for how the initial glycosylated protein will result years later in macrovascular disease as evidenced by the parameters that were used.
It was interesting to learn from this study that glycosylated end products and oxidative products interact with each other. That’s important, because as blood sugars rise acutely, both oxidative products and glycosylated products are produced. As a result of the oxidative stress that’s created by the sharp rise in blood sugar, endothelial function is affected and more glycosylated proteins are being formed.
Clinically, this study shows the importance of early, aggressive control of diabetes to not allow accumulation of both glycosylated end products and oxidative end products. It demonstrates that accumulation of these byproducts years later seems to relate strongly to macrovascular disease.
The study should be reproduced in younger, less sick patients. That may or may not further clarify the findings, but these findings need to be demonstrated in patients at a much earlier stage as well. We’ve been saying for a long time that early control of diabetes is so important because years later it makes a difference. This is a link to that rationale.
Paul S. Jellinger, MD, MACE, is professor of clinical medicine at the University of Miami Miller School of Medicine, Ft. Lauderdale. He is past president of the American Association of Clinical Endocrinologists (AACE) and past president of the American College of Endocrinology. Dr. Jellinger provided these comments in an interview.
For a long time the question about how glucose control relates to macrovascular disease has not been easy to answer. There are reports suggesting that glucose control correlates with macrovascular disease later in life, and others that do not make the association so convincing. This paper provides an important link between glucose control and the subsequent development of macrovascular disease. The link is by way of advanced glycation end products as well as oxidative byproducts and how they set the stage for macrovascular disease later on in life. It’s something that clinicians have postulated as being important, but to my knowledge this is one of the only studies to actually show the association.
The investigators used important endpoints like coronary artery calcification and carotid intima-media thickness. I was surprised at how well the relationship between glycosylated end products and oxidative products correlated, but there’s biologic plausibility; it makes sense. Glucose not only glycosates hemoglobin, but glycosates proteins throughout. This provides a logical, stepwise pathway for how the initial glycosylated protein will result years later in macrovascular disease as evidenced by the parameters that were used.
It was interesting to learn from this study that glycosylated end products and oxidative products interact with each other. That’s important, because as blood sugars rise acutely, both oxidative products and glycosylated products are produced. As a result of the oxidative stress that’s created by the sharp rise in blood sugar, endothelial function is affected and more glycosylated proteins are being formed.
Clinically, this study shows the importance of early, aggressive control of diabetes to not allow accumulation of both glycosylated end products and oxidative end products. It demonstrates that accumulation of these byproducts years later seems to relate strongly to macrovascular disease.
The study should be reproduced in younger, less sick patients. That may or may not further clarify the findings, but these findings need to be demonstrated in patients at a much earlier stage as well. We’ve been saying for a long time that early control of diabetes is so important because years later it makes a difference. This is a link to that rationale.
Paul S. Jellinger, MD, MACE, is professor of clinical medicine at the University of Miami Miller School of Medicine, Ft. Lauderdale. He is past president of the American Association of Clinical Endocrinologists (AACE) and past president of the American College of Endocrinology. Dr. Jellinger provided these comments in an interview.
For a long time the question about how glucose control relates to macrovascular disease has not been easy to answer. There are reports suggesting that glucose control correlates with macrovascular disease later in life, and others that do not make the association so convincing. This paper provides an important link between glucose control and the subsequent development of macrovascular disease. The link is by way of advanced glycation end products as well as oxidative byproducts and how they set the stage for macrovascular disease later on in life. It’s something that clinicians have postulated as being important, but to my knowledge this is one of the only studies to actually show the association.
The investigators used important endpoints like coronary artery calcification and carotid intima-media thickness. I was surprised at how well the relationship between glycosylated end products and oxidative products correlated, but there’s biologic plausibility; it makes sense. Glucose not only glycosates hemoglobin, but glycosates proteins throughout. This provides a logical, stepwise pathway for how the initial glycosylated protein will result years later in macrovascular disease as evidenced by the parameters that were used.
It was interesting to learn from this study that glycosylated end products and oxidative products interact with each other. That’s important, because as blood sugars rise acutely, both oxidative products and glycosylated products are produced. As a result of the oxidative stress that’s created by the sharp rise in blood sugar, endothelial function is affected and more glycosylated proteins are being formed.
Clinically, this study shows the importance of early, aggressive control of diabetes to not allow accumulation of both glycosylated end products and oxidative end products. It demonstrates that accumulation of these byproducts years later seems to relate strongly to macrovascular disease.
The study should be reproduced in younger, less sick patients. That may or may not further clarify the findings, but these findings need to be demonstrated in patients at a much earlier stage as well. We’ve been saying for a long time that early control of diabetes is so important because years later it makes a difference. This is a link to that rationale.
Paul S. Jellinger, MD, MACE, is professor of clinical medicine at the University of Miami Miller School of Medicine, Ft. Lauderdale. He is past president of the American Association of Clinical Endocrinologists (AACE) and past president of the American College of Endocrinology. Dr. Jellinger provided these comments in an interview.
in patient with type 2 diabetes, results from a long-term analysis of VA patients suggest.
Advanced glycation end products (AGEs) and oxidation products (OxPs) “can damage vascular cells by different mechanisms,” wrote researchers led by corresponding authors Aramesh Saremi, MD, and Peter D. Reaven, MD, and colleagues. The report appeared online Feb. 1 in Diabetes Care.
“One frequently reported pathway is AGE binding to their purported (and relatively promiscuous) receptors on cells, such as macrophages, vascular endothelial cells, and vascular smooth muscle cells, although this has not been consistent for all AGEs. Other mechanisms include, among others, binding to and altering the function of intracellular proteins, the activation of vascular NADPH [nicotinamide adenine dinucleotide phosphate] oxidase, and the uncoupling of endothelial nitric oxide synthase.”
Noting that data in the current medical literature are lacking with respect to long-term longitudinal associations between plasma levels of AGEs and OxPs on the extent of subclinical atherosclerosis in T2D patients, the researchers set out to determine whether baseline plasma levels of AGEs and OxPs are associated with the extent of carotid intima-media thickness (CIMT), coronary artery calcification (CAC), and abdominal aortic artery calcification (AAC) over an average of 10 years of follow-up in the VA Diabetes Trial (VADT). They also examined whether this relationship was altered by intervening improved glucose control (Diabetes Care 2017 Feb. 1. doi: 10.2337/dc16-1875]).
At baseline of the VADT, 411 study participants underwent plasma measurements of methylglyoxal hydroimidazolone, N epsilon–carboxymethyl lysine (CML), N epsilon–carboxyethyl lysine (CEL), 3-deoxyglucosone hydroimidazolone and glyoxal hydroimidazolone (G-H1), 2-aminoadipic acid (2-AAA), and methionine sulfoxide. The mean age of the study subjects was 58 years, 64% were non-Hispanic white, 96% were male, 69% had a history of hypertension, and they had diabetes for a mean of 11 years.
After a mean follow-up of 10 years, the 411 patients underwent ultrasound assessment of CIMT, and computed tomography scanning of CAC and AAC.
In risk factor–adjusted multivariable regression models, G-H1was associated with the extent of CIMT as well as with the extent of CAC (P = .01 for both associations). In addition, 2-AAA was strongly associated with the extent of CAC (P = .03 for continuous variables and P less than .01 for dichotomous variables), and CEL was strongly associated with the extent of AAC (P less than .01).
“These findings suggest that the effect of hyperglycemia and subsequent increased levels of AGEs and OxPs in patients with long-standing T2D may have long-lasting adverse effects on the development of macrovascular complications,” the researchers concluded. They acknowledged certain limitations of the analysis, including the fact that it was conducted in an older, primarily male population. Therefore, “extrapolation of the study findings to other populations must be done with caution,” they wrote. “This study also does not allow us to make a definite claim of causation between AGEs and OxPs with the extent of atherosclerosis.”
The Veterans Affairs Cooperative Studies Program of the U.S. Department of Veterans Affairs Office of Research and Development funded the study. Additional support was received from National Institutes of Health, the American Diabetes Association, and the National Institute of Diabetes and Digestive and Kidney Diseases. Two study authors, Scott Howell, MS, and Paul J. Beisswenger, MD, disclosed that they are affiliated with PreventAGE Healthcare. The other researchers reported having no financial disclosures.
[email protected]
in patient with type 2 diabetes, results from a long-term analysis of VA patients suggest.
Advanced glycation end products (AGEs) and oxidation products (OxPs) “can damage vascular cells by different mechanisms,” wrote researchers led by corresponding authors Aramesh Saremi, MD, and Peter D. Reaven, MD, and colleagues. The report appeared online Feb. 1 in Diabetes Care.
“One frequently reported pathway is AGE binding to their purported (and relatively promiscuous) receptors on cells, such as macrophages, vascular endothelial cells, and vascular smooth muscle cells, although this has not been consistent for all AGEs. Other mechanisms include, among others, binding to and altering the function of intracellular proteins, the activation of vascular NADPH [nicotinamide adenine dinucleotide phosphate] oxidase, and the uncoupling of endothelial nitric oxide synthase.”
Noting that data in the current medical literature are lacking with respect to long-term longitudinal associations between plasma levels of AGEs and OxPs on the extent of subclinical atherosclerosis in T2D patients, the researchers set out to determine whether baseline plasma levels of AGEs and OxPs are associated with the extent of carotid intima-media thickness (CIMT), coronary artery calcification (CAC), and abdominal aortic artery calcification (AAC) over an average of 10 years of follow-up in the VA Diabetes Trial (VADT). They also examined whether this relationship was altered by intervening improved glucose control (Diabetes Care 2017 Feb. 1. doi: 10.2337/dc16-1875]).
At baseline of the VADT, 411 study participants underwent plasma measurements of methylglyoxal hydroimidazolone, N epsilon–carboxymethyl lysine (CML), N epsilon–carboxyethyl lysine (CEL), 3-deoxyglucosone hydroimidazolone and glyoxal hydroimidazolone (G-H1), 2-aminoadipic acid (2-AAA), and methionine sulfoxide. The mean age of the study subjects was 58 years, 64% were non-Hispanic white, 96% were male, 69% had a history of hypertension, and they had diabetes for a mean of 11 years.
After a mean follow-up of 10 years, the 411 patients underwent ultrasound assessment of CIMT, and computed tomography scanning of CAC and AAC.
In risk factor–adjusted multivariable regression models, G-H1was associated with the extent of CIMT as well as with the extent of CAC (P = .01 for both associations). In addition, 2-AAA was strongly associated with the extent of CAC (P = .03 for continuous variables and P less than .01 for dichotomous variables), and CEL was strongly associated with the extent of AAC (P less than .01).
“These findings suggest that the effect of hyperglycemia and subsequent increased levels of AGEs and OxPs in patients with long-standing T2D may have long-lasting adverse effects on the development of macrovascular complications,” the researchers concluded. They acknowledged certain limitations of the analysis, including the fact that it was conducted in an older, primarily male population. Therefore, “extrapolation of the study findings to other populations must be done with caution,” they wrote. “This study also does not allow us to make a definite claim of causation between AGEs and OxPs with the extent of atherosclerosis.”
The Veterans Affairs Cooperative Studies Program of the U.S. Department of Veterans Affairs Office of Research and Development funded the study. Additional support was received from National Institutes of Health, the American Diabetes Association, and the National Institute of Diabetes and Digestive and Kidney Diseases. Two study authors, Scott Howell, MS, and Paul J. Beisswenger, MD, disclosed that they are affiliated with PreventAGE Healthcare. The other researchers reported having no financial disclosures.
[email protected]
Key clinical point:
Major finding: Glyoxal hydroimidazolone was associated with the extent of carotid intima-media thickness as well as with the extent of coronary artery calcification.
Data source: An analysis of 411 patients the VA Diabetes Trial.
Disclosures: The Veterans Affairs Cooperative Studies Program of the U.S. Department of Veterans Affairs Office of Research and Development funded the study. Additional support was received from National Institutes of Health, the American Diabetes Association, and the National Institute of Diabetes and Digestive and Kidney Diseases. Two study authors, Scott Howell, MS, and Paul J. Beisswenger, MD, disclosed that they are affiliated with PreventAGE Healthcare. The other researchers reported having no financial disclosures.

Liver transplantation largely effective in critically ill children
The use of advanced critical care in children and infants with liver failure is justified because orthotopic liver transplantation can be performed on the sickest children and achieve acceptable outcomes, results from a large analysis demonstrated.
“Hand in hand with improved care for critically ill children with liver failure, posttransplant critical care has made tremendous strides,” Abbas Rana, MD, wrote in a study published online in the Journal of the American College of Surgeons. “Our recipients have gotten sicker while our postoperative outcomes have improved. The question then becomes, have our operative skills and postoperative critical care management kept up with the abilities to keep sick children with liver failure alive? Just because transplantation is now possible in our sickest children, is it justified?”
To find out, Dr. Rana of the division of abdominal transplantation and hepatobiliary surgery at Baylor College of Medicine, Houston, and colleagues retrospectively analyzed United Network for Organ Sharing data from all orthotopic liver transplantation (OLT) recipients between Sept. 1, 1987, and June 30, 2015. The analysis paired the liver registry data with data collected by the Organ Procurement and Transplantation Network, and was limited to transplant recipients younger than age 18. The researchers followed a total of 13,723 recipients from date of transplant until either death or the date of last known follow-up (J Am Coll Surg. 2016 Dec 25. doi: 10.1016/j.jamcollsurg.2016.12.025).
In another part of the study, the researchers retrospectively reviewed the charts of 354 patients under 18 years of age who underwent OLT between March 1, 2002, and June 30, 2015, at Texas Children’s Hospital, including 65 who were admitted to the ICU at the time of transplantation.
In the analysis of national data, the researchers found that the rates of 1-year survival following OLT in children in the ICU improved from 60% in 1987 to 92% in 2013 (P less than .001). The rates of 1-year survival also improved for children on dialysis at the time of transplant (from 50% in 1995 to 95% in 2013; P less than .001) and for those dependent on a mechanical ventilator at the time of transplant (from 49% in 1994 to 94% in 2013; P less than .001). The significant risk factors were two previous transplants (hazard ratio, 4.2), one previous transplant (HR, 2.5), serum sodium greater than 150 mEq/L (HR, 2.0), dialysis or glomerular filtration rate less than 30 mL/min per 1.73 m2 (HR, 2.0), mechanical ventilator dependence (HR, 1.8), body weight under 6 kg (HR, 1.8), encephalopathy (HR, 1.8), and annual center volume of fewer than five cases (HR, 1.7).
In the experience at Texas Children’s Hospital, the researchers observed “preserved and successful patient survival outcomes” in many markers of acuity. For example, the 10-year survival rates for patients dependent on mechanical ventilation and dialysis were 85% and 96%, respectively, and reached 100% for those requiring therapeutic plasma exchange, molescular adsorbent recirculating system (MARS) liver dialysis, and vasopressors.
“Our collective ability to keep sick children alive with liver failure has improved considerably over the years,” the researchers wrote. “Keeping pace, this analysis demonstrates that the posttransplant outcomes have also improved dramatically. The survival outcomes are comparable to the general population, justifying the use of scarce donors. Although we cannot declare in absolute that no child should be left behind, we can demonstrate acceptable outcomes to date and urge the continual revisiting of our concepts of futility.
“We have learned throughout our experience that almost every child with end-stage liver disease and acute liver failure should be offered liver replacement, as long as the vasoactive medication and mechanical support are not maximized prior to the initiation of the OLT procedure. Every effort should be made to transplant our sickest children.”
This study was supported by the Cade R. Alpard Foundation. The researchers reported having no relevant financial disclosures.
The use of advanced critical care in children and infants with liver failure is justified because orthotopic liver transplantation can be performed on the sickest children and achieve acceptable outcomes, results from a large analysis demonstrated.
“Hand in hand with improved care for critically ill children with liver failure, posttransplant critical care has made tremendous strides,” Abbas Rana, MD, wrote in a study published online in the Journal of the American College of Surgeons. “Our recipients have gotten sicker while our postoperative outcomes have improved. The question then becomes, have our operative skills and postoperative critical care management kept up with the abilities to keep sick children with liver failure alive? Just because transplantation is now possible in our sickest children, is it justified?”
To find out, Dr. Rana of the division of abdominal transplantation and hepatobiliary surgery at Baylor College of Medicine, Houston, and colleagues retrospectively analyzed United Network for Organ Sharing data from all orthotopic liver transplantation (OLT) recipients between Sept. 1, 1987, and June 30, 2015. The analysis paired the liver registry data with data collected by the Organ Procurement and Transplantation Network, and was limited to transplant recipients younger than age 18. The researchers followed a total of 13,723 recipients from date of transplant until either death or the date of last known follow-up (J Am Coll Surg. 2016 Dec 25. doi: 10.1016/j.jamcollsurg.2016.12.025).
In another part of the study, the researchers retrospectively reviewed the charts of 354 patients under 18 years of age who underwent OLT between March 1, 2002, and June 30, 2015, at Texas Children’s Hospital, including 65 who were admitted to the ICU at the time of transplantation.
In the analysis of national data, the researchers found that the rates of 1-year survival following OLT in children in the ICU improved from 60% in 1987 to 92% in 2013 (P less than .001). The rates of 1-year survival also improved for children on dialysis at the time of transplant (from 50% in 1995 to 95% in 2013; P less than .001) and for those dependent on a mechanical ventilator at the time of transplant (from 49% in 1994 to 94% in 2013; P less than .001). The significant risk factors were two previous transplants (hazard ratio, 4.2), one previous transplant (HR, 2.5), serum sodium greater than 150 mEq/L (HR, 2.0), dialysis or glomerular filtration rate less than 30 mL/min per 1.73 m2 (HR, 2.0), mechanical ventilator dependence (HR, 1.8), body weight under 6 kg (HR, 1.8), encephalopathy (HR, 1.8), and annual center volume of fewer than five cases (HR, 1.7).
In the experience at Texas Children’s Hospital, the researchers observed “preserved and successful patient survival outcomes” in many markers of acuity. For example, the 10-year survival rates for patients dependent on mechanical ventilation and dialysis were 85% and 96%, respectively, and reached 100% for those requiring therapeutic plasma exchange, molescular adsorbent recirculating system (MARS) liver dialysis, and vasopressors.
“Our collective ability to keep sick children alive with liver failure has improved considerably over the years,” the researchers wrote. “Keeping pace, this analysis demonstrates that the posttransplant outcomes have also improved dramatically. The survival outcomes are comparable to the general population, justifying the use of scarce donors. Although we cannot declare in absolute that no child should be left behind, we can demonstrate acceptable outcomes to date and urge the continual revisiting of our concepts of futility.
“We have learned throughout our experience that almost every child with end-stage liver disease and acute liver failure should be offered liver replacement, as long as the vasoactive medication and mechanical support are not maximized prior to the initiation of the OLT procedure. Every effort should be made to transplant our sickest children.”
This study was supported by the Cade R. Alpard Foundation. The researchers reported having no relevant financial disclosures.
The use of advanced critical care in children and infants with liver failure is justified because orthotopic liver transplantation can be performed on the sickest children and achieve acceptable outcomes, results from a large analysis demonstrated.
“Hand in hand with improved care for critically ill children with liver failure, posttransplant critical care has made tremendous strides,” Abbas Rana, MD, wrote in a study published online in the Journal of the American College of Surgeons. “Our recipients have gotten sicker while our postoperative outcomes have improved. The question then becomes, have our operative skills and postoperative critical care management kept up with the abilities to keep sick children with liver failure alive? Just because transplantation is now possible in our sickest children, is it justified?”
To find out, Dr. Rana of the division of abdominal transplantation and hepatobiliary surgery at Baylor College of Medicine, Houston, and colleagues retrospectively analyzed United Network for Organ Sharing data from all orthotopic liver transplantation (OLT) recipients between Sept. 1, 1987, and June 30, 2015. The analysis paired the liver registry data with data collected by the Organ Procurement and Transplantation Network, and was limited to transplant recipients younger than age 18. The researchers followed a total of 13,723 recipients from date of transplant until either death or the date of last known follow-up (J Am Coll Surg. 2016 Dec 25. doi: 10.1016/j.jamcollsurg.2016.12.025).
In another part of the study, the researchers retrospectively reviewed the charts of 354 patients under 18 years of age who underwent OLT between March 1, 2002, and June 30, 2015, at Texas Children’s Hospital, including 65 who were admitted to the ICU at the time of transplantation.
In the analysis of national data, the researchers found that the rates of 1-year survival following OLT in children in the ICU improved from 60% in 1987 to 92% in 2013 (P less than .001). The rates of 1-year survival also improved for children on dialysis at the time of transplant (from 50% in 1995 to 95% in 2013; P less than .001) and for those dependent on a mechanical ventilator at the time of transplant (from 49% in 1994 to 94% in 2013; P less than .001). The significant risk factors were two previous transplants (hazard ratio, 4.2), one previous transplant (HR, 2.5), serum sodium greater than 150 mEq/L (HR, 2.0), dialysis or glomerular filtration rate less than 30 mL/min per 1.73 m2 (HR, 2.0), mechanical ventilator dependence (HR, 1.8), body weight under 6 kg (HR, 1.8), encephalopathy (HR, 1.8), and annual center volume of fewer than five cases (HR, 1.7).
In the experience at Texas Children’s Hospital, the researchers observed “preserved and successful patient survival outcomes” in many markers of acuity. For example, the 10-year survival rates for patients dependent on mechanical ventilation and dialysis were 85% and 96%, respectively, and reached 100% for those requiring therapeutic plasma exchange, molescular adsorbent recirculating system (MARS) liver dialysis, and vasopressors.
“Our collective ability to keep sick children alive with liver failure has improved considerably over the years,” the researchers wrote. “Keeping pace, this analysis demonstrates that the posttransplant outcomes have also improved dramatically. The survival outcomes are comparable to the general population, justifying the use of scarce donors. Although we cannot declare in absolute that no child should be left behind, we can demonstrate acceptable outcomes to date and urge the continual revisiting of our concepts of futility.
“We have learned throughout our experience that almost every child with end-stage liver disease and acute liver failure should be offered liver replacement, as long as the vasoactive medication and mechanical support are not maximized prior to the initiation of the OLT procedure. Every effort should be made to transplant our sickest children.”
This study was supported by the Cade R. Alpard Foundation. The researchers reported having no relevant financial disclosures.
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF SURGEONS
Key clinical point:
Major finding: The rates of 1-year survival following orthotopic liver transplantation in ICU children improved from 60% in 1987 to 92% in 2013 (P less than .001).
Data source: An analysis of 13,723 patients under the age of 18 years who underwent OLT between Sept. 1, 1987, and June 30, 2015.
Disclosures: The study was supported by the Cade R. Alpard Foundation. The researchers reported having no relevant financial disclosures.
How Will the Trump Travel Ban Impact the VA?
Rural health care and the VA seem to be on the front line in the battle over President Trump’s executive order halting travel to the U.S. for citizens of 7 Muslim-majority countries. The executive order, which has been temporarily suspended by a federal judge, could affect as many as 15,000 doctors and possibly even more pharmacists, nurses, and other health care providers. The ban also is expected to impact nearly 9,000 physicians from Iran and another 3,500 from Syria and 1,500 from Iraq.
The VA has long depended on foreign-born health care providers, and given the VA’s challenges in filling its more than 45,000 vacancies, this reliance was only expected to grow.. The Conrad 30 program allows J-1 medical doctors to apply for a waiver to the 2-year residence requirement upon completion of the J-1 exchange visitor program. Over the years, many international medical graduates (IMGs) earned permanent residence by agreeing to work 3 or 5 years in an underserved area or VA facility.
“The United States is facing a serious shortage of physicians. IMGs play an important role in U.S. health care, representing roughly 25% of the workforce,” AAMC (Association of American Medical Colleges) President and CEO Darrell G. Kirch, MD said in a statement. “In the last decade, Conrad 30 alone has directed nearly 10,000 physicians into rural and urban underserved communities. Impeding these U.S. immigration pathways jeopardizes critical access to high-quality physician care for our nation’s most vulnerable populations.”
Related: Medical Organizations Respond to Trump’s Immigration Order
According to an analysis of 2010 census data from the Migration Policy Institute (MPI), foreign-born workers make up 15% of the 1.1 million health care providers in the U.S., which includes a significant proportion of physicians and surgeons (27%) and a smaller, but still significant number of registered nurses (15%), technologists and technicians (12%), and therapists (10%). Among support personnel foreign-born workers range from 19% of health care support workers to 22% of nursing, psychiatric, and home health aides.
In a more recent analysis from the George Mason Institute for Immigration Research, 22% of nursing, psychiatric, and home health aides are foreign born.
Most of the foreign-born health care providers have acquired U.S. citizenship, according to MPI, “with naturalization rates ranging from 55% for those working as nursing, psychiatric, or home health aides to 78% for other health care practitioners and technical occupations. According to American Association of Family Practitioners (AAFP), 42% of office visits in rural America are with foreign-born physicians.
“Many family physicians are IMGs, who have completed all or part of their education and training in the U.S.,” AAFP President John Meigs, Jr, MD, FAAFP, wrote in a letter to President Trump. “They are professionals who dedicate their careers to the service of their patients in communities large and small, urban and rural. In fact, 20% of our membership and over 25% of family medicine residents [comprise] IMGs. The AAFP applauds and supports wholly the contributions of these individual family physicians to their patients and communities and we celebrate their diversity.”
Rural health care and the VA seem to be on the front line in the battle over President Trump’s executive order halting travel to the U.S. for citizens of 7 Muslim-majority countries. The executive order, which has been temporarily suspended by a federal judge, could affect as many as 15,000 doctors and possibly even more pharmacists, nurses, and other health care providers. The ban also is expected to impact nearly 9,000 physicians from Iran and another 3,500 from Syria and 1,500 from Iraq.
The VA has long depended on foreign-born health care providers, and given the VA’s challenges in filling its more than 45,000 vacancies, this reliance was only expected to grow.. The Conrad 30 program allows J-1 medical doctors to apply for a waiver to the 2-year residence requirement upon completion of the J-1 exchange visitor program. Over the years, many international medical graduates (IMGs) earned permanent residence by agreeing to work 3 or 5 years in an underserved area or VA facility.
“The United States is facing a serious shortage of physicians. IMGs play an important role in U.S. health care, representing roughly 25% of the workforce,” AAMC (Association of American Medical Colleges) President and CEO Darrell G. Kirch, MD said in a statement. “In the last decade, Conrad 30 alone has directed nearly 10,000 physicians into rural and urban underserved communities. Impeding these U.S. immigration pathways jeopardizes critical access to high-quality physician care for our nation’s most vulnerable populations.”
Related: Medical Organizations Respond to Trump’s Immigration Order
According to an analysis of 2010 census data from the Migration Policy Institute (MPI), foreign-born workers make up 15% of the 1.1 million health care providers in the U.S., which includes a significant proportion of physicians and surgeons (27%) and a smaller, but still significant number of registered nurses (15%), technologists and technicians (12%), and therapists (10%). Among support personnel foreign-born workers range from 19% of health care support workers to 22% of nursing, psychiatric, and home health aides.
In a more recent analysis from the George Mason Institute for Immigration Research, 22% of nursing, psychiatric, and home health aides are foreign born.
Most of the foreign-born health care providers have acquired U.S. citizenship, according to MPI, “with naturalization rates ranging from 55% for those working as nursing, psychiatric, or home health aides to 78% for other health care practitioners and technical occupations. According to American Association of Family Practitioners (AAFP), 42% of office visits in rural America are with foreign-born physicians.
“Many family physicians are IMGs, who have completed all or part of their education and training in the U.S.,” AAFP President John Meigs, Jr, MD, FAAFP, wrote in a letter to President Trump. “They are professionals who dedicate their careers to the service of their patients in communities large and small, urban and rural. In fact, 20% of our membership and over 25% of family medicine residents [comprise] IMGs. The AAFP applauds and supports wholly the contributions of these individual family physicians to their patients and communities and we celebrate their diversity.”
Rural health care and the VA seem to be on the front line in the battle over President Trump’s executive order halting travel to the U.S. for citizens of 7 Muslim-majority countries. The executive order, which has been temporarily suspended by a federal judge, could affect as many as 15,000 doctors and possibly even more pharmacists, nurses, and other health care providers. The ban also is expected to impact nearly 9,000 physicians from Iran and another 3,500 from Syria and 1,500 from Iraq.
The VA has long depended on foreign-born health care providers, and given the VA’s challenges in filling its more than 45,000 vacancies, this reliance was only expected to grow.. The Conrad 30 program allows J-1 medical doctors to apply for a waiver to the 2-year residence requirement upon completion of the J-1 exchange visitor program. Over the years, many international medical graduates (IMGs) earned permanent residence by agreeing to work 3 or 5 years in an underserved area or VA facility.
“The United States is facing a serious shortage of physicians. IMGs play an important role in U.S. health care, representing roughly 25% of the workforce,” AAMC (Association of American Medical Colleges) President and CEO Darrell G. Kirch, MD said in a statement. “In the last decade, Conrad 30 alone has directed nearly 10,000 physicians into rural and urban underserved communities. Impeding these U.S. immigration pathways jeopardizes critical access to high-quality physician care for our nation’s most vulnerable populations.”
Related: Medical Organizations Respond to Trump’s Immigration Order
According to an analysis of 2010 census data from the Migration Policy Institute (MPI), foreign-born workers make up 15% of the 1.1 million health care providers in the U.S., which includes a significant proportion of physicians and surgeons (27%) and a smaller, but still significant number of registered nurses (15%), technologists and technicians (12%), and therapists (10%). Among support personnel foreign-born workers range from 19% of health care support workers to 22% of nursing, psychiatric, and home health aides.
In a more recent analysis from the George Mason Institute for Immigration Research, 22% of nursing, psychiatric, and home health aides are foreign born.
Most of the foreign-born health care providers have acquired U.S. citizenship, according to MPI, “with naturalization rates ranging from 55% for those working as nursing, psychiatric, or home health aides to 78% for other health care practitioners and technical occupations. According to American Association of Family Practitioners (AAFP), 42% of office visits in rural America are with foreign-born physicians.
“Many family physicians are IMGs, who have completed all or part of their education and training in the U.S.,” AAFP President John Meigs, Jr, MD, FAAFP, wrote in a letter to President Trump. “They are professionals who dedicate their careers to the service of their patients in communities large and small, urban and rural. In fact, 20% of our membership and over 25% of family medicine residents [comprise] IMGs. The AAFP applauds and supports wholly the contributions of these individual family physicians to their patients and communities and we celebrate their diversity.”
Widespread Poikilodermatous Dermatomyositis Associated With Chronic Lymphocytic Leukemia
To the Editor:
Dermatomyositis represents a rare idiopathic inflammatory process presenting with cutaneous lesions and muscular weakness. It often represents a paraneoplastic syndrome. We report the case of a 62-year-old man with a history of total-body poikiloderma and a recent diagnosis of chronic lymphocytic leukemia (CLL). Despite lacking typical features of the disease, a diagnosis of dermatomyositis was made. Our patient may represent a distinct poikilodermatous variant of dermatomyositis, sharing the generalized distribution of the erythrodermic subtype.
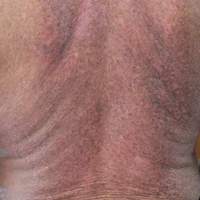
A 62-year-old man presented with pruritic poikiloderma involving the neck, arms, legs, abdomen, chest, and back of 2 years’ duration (Figure). He also experienced dysphagia and weakness of the legs. The rash was previously treated by other dermatologists with a combination of high-potency topical steroids and topical tacrolimus 0.1% without success. His history was notable for CLL, which had been diagnosed by a dermatologist 6 months prior to the current presentation. Prior to his visit to the dermatologist, the patient had received 6 chemotherapeutic sessions with a combination of rituximab and cyclophosphamide for the treatment of CLL. The rash did not improve with chemotherapy.

Repeat biopsies of affected regions only demonstrated features of mild interface dermatitis. Direct immunofluorescence studies showed scattered colloid body fluorescence for IgM. Because of bilateral weakness of the legs, a muscle biopsy was taken, which demonstrated severe atrophy and interstitial fibrosis, with neurogenic abnormalities detected in areas of lesser atrophy via abnormal muscle fiber–type grouping. Metabolic panel showed elevated muscle enzymes in the blood: creatine kinase, 243 U/L (reference range, 10–225 U/L); serum aldolase, 16 U/L (reference range, ≤8.1 U/L); lactate dehydrogenase, 314 U/L (reference range, 60–200 U/L). An autoimmune panel was negative for Jo-1, Scl-70, U1 ribonucleoprotein, DNA, desmoglein 1 and 3, and antiacetylcholine receptor antibodies. An elevated erythrocyte sedimentation rate was measured at 16 mm/h (reference range, 0–10 mm/h). Given these findings, the lesions were confirmed as a widespread poikilodermatous variant of dermatomyositis.
The patient was placed on a daily 50-mg dose of prednisone, which produced rapid improvement in scaling and erythema. Creatine kinase and serum aldolase levels normalized and motor strength increased. After 1 week the prednisone dosage was reduced to a daily 30-mg dose, and then 20 mg a week later. The skin lesions completely resolved within 4 to 5 months and the patient is currently on a prednisone dose of 5 mg, alternating with 2.5 mg of prednisone and rituximab infusion every 2 months.
Dermatomyositis is a rare entity with an incidence of approximately 0.5 to 1 per 100,000 individuals.1 It presents with a characteristic rash composed of Gottron papules; pathognomonic flat violaceous papules on the dorsal interphalangeal joints, elbows, or knees; and a heliotrope rash, a violaceous erythema involving the eyelids. Poikiloderma frequently is reported to present in a shawl-like distribution, encompassing the shoulders, arms, and upper back.1,2 Dermatomyositis of the poikilodermatous type can present in nonphotoexposed areas and photoexposed areas. The unusual feature is the total-body involvement, which is analogous to erythroderma.3
Our case may represent a distinct poikilodermatous manifestation sharing the distribution of the erythrodermic subtype. We believe that the skin lesions may have represented a paraneoplastic event presenting prior to diagnosis with CLL. Dermatomyositis has a strong association with cancer, with patients 3 times more likely to develop internal malignancy.4 Association is strongest for non-Hodgkin lymphoma, as well as ovarian, lung, colorectal, pancreatic, and gastric cancer. When associated with malignancy, symptoms of dermatomyositis or myositis typically precede the discovery of malignancy by an average of 1.9 years.5 Dermatomyositis has been previously reported to present as a paraneoplastic manifestation of CLL.6 One case has been reported of a patient with CLL who developed leukemia cutis presenting with poikiloderma in the characteristic dermatomyositis shawl-like distribution.7 The lack of dermal infiltration with leukemic cells in our patient, however, makes a paraneoplastic etiology much more likely.
Our patient’s rash did not initially improve with treatment of CLL, but dermatomyositis associated with hematological malignancy may precede, occur simultaneously, or follow the diagnosis of malignancy.8 Additionally, symptoms of dermatomyositis do not always parallel the course of hematological malignancy outcome. However, rituximab has been used as a treatment of dermatomyositis and may have contributed some synergistic effect in combination with prednisone in our patient.9
- Dourmishev LA, Dourmishev AL, Schwartz RA. Dermatomyositis: cutaneous manifestations of its variants. Int J Dermatol. 2002;41:625-630.
- Kovacs SO, Kovacs SC. Dermatomyositis. J Am Acad Dermatol. 1998;39:899-920; quiz 921-992.
- Liu ZH, Wang XD. Acute-onset adult dermatomyositis presenting with erythroderma and diplopia. Clin Exp Dermatol. 2007;32:751-752.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Bohan A, Peter JB, Bowman RL, et al. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore). 1977;56:255-286.
- Ishida T, Aikawa K, Tamura T, et al. Chronic lymphocytic leukemia associated with nephrotic syndrome and dermatomyositis. Intern Med. 1995;34:15-17.
- Nousari HC, Kimyai-Asadi A, Huang CH, et al. T-cell chronic lymphocytic leukemia mimicking dermatomyositis. Int J Dermatol. 2000;39:144-146.
- Marie I, Guillevin L, Menard JF, et al. Hematological malignancy associated with polymyositis and dermatomyositis. Autoimmun Rev. 2012;11:615-620.
- Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52:601-607.
To the Editor:
Dermatomyositis represents a rare idiopathic inflammatory process presenting with cutaneous lesions and muscular weakness. It often represents a paraneoplastic syndrome. We report the case of a 62-year-old man with a history of total-body poikiloderma and a recent diagnosis of chronic lymphocytic leukemia (CLL). Despite lacking typical features of the disease, a diagnosis of dermatomyositis was made. Our patient may represent a distinct poikilodermatous variant of dermatomyositis, sharing the generalized distribution of the erythrodermic subtype.
A 62-year-old man presented with pruritic poikiloderma involving the neck, arms, legs, abdomen, chest, and back of 2 years’ duration (Figure). He also experienced dysphagia and weakness of the legs. The rash was previously treated by other dermatologists with a combination of high-potency topical steroids and topical tacrolimus 0.1% without success. His history was notable for CLL, which had been diagnosed by a dermatologist 6 months prior to the current presentation. Prior to his visit to the dermatologist, the patient had received 6 chemotherapeutic sessions with a combination of rituximab and cyclophosphamide for the treatment of CLL. The rash did not improve with chemotherapy.
Repeat biopsies of affected regions only demonstrated features of mild interface dermatitis. Direct immunofluorescence studies showed scattered colloid body fluorescence for IgM. Because of bilateral weakness of the legs, a muscle biopsy was taken, which demonstrated severe atrophy and interstitial fibrosis, with neurogenic abnormalities detected in areas of lesser atrophy via abnormal muscle fiber–type grouping. Metabolic panel showed elevated muscle enzymes in the blood: creatine kinase, 243 U/L (reference range, 10–225 U/L); serum aldolase, 16 U/L (reference range, ≤8.1 U/L); lactate dehydrogenase, 314 U/L (reference range, 60–200 U/L). An autoimmune panel was negative for Jo-1, Scl-70, U1 ribonucleoprotein, DNA, desmoglein 1 and 3, and antiacetylcholine receptor antibodies. An elevated erythrocyte sedimentation rate was measured at 16 mm/h (reference range, 0–10 mm/h). Given these findings, the lesions were confirmed as a widespread poikilodermatous variant of dermatomyositis.
The patient was placed on a daily 50-mg dose of prednisone, which produced rapid improvement in scaling and erythema. Creatine kinase and serum aldolase levels normalized and motor strength increased. After 1 week the prednisone dosage was reduced to a daily 30-mg dose, and then 20 mg a week later. The skin lesions completely resolved within 4 to 5 months and the patient is currently on a prednisone dose of 5 mg, alternating with 2.5 mg of prednisone and rituximab infusion every 2 months.
Dermatomyositis is a rare entity with an incidence of approximately 0.5 to 1 per 100,000 individuals.1 It presents with a characteristic rash composed of Gottron papules; pathognomonic flat violaceous papules on the dorsal interphalangeal joints, elbows, or knees; and a heliotrope rash, a violaceous erythema involving the eyelids. Poikiloderma frequently is reported to present in a shawl-like distribution, encompassing the shoulders, arms, and upper back.1,2 Dermatomyositis of the poikilodermatous type can present in nonphotoexposed areas and photoexposed areas. The unusual feature is the total-body involvement, which is analogous to erythroderma.3
Our case may represent a distinct poikilodermatous manifestation sharing the distribution of the erythrodermic subtype. We believe that the skin lesions may have represented a paraneoplastic event presenting prior to diagnosis with CLL. Dermatomyositis has a strong association with cancer, with patients 3 times more likely to develop internal malignancy.4 Association is strongest for non-Hodgkin lymphoma, as well as ovarian, lung, colorectal, pancreatic, and gastric cancer. When associated with malignancy, symptoms of dermatomyositis or myositis typically precede the discovery of malignancy by an average of 1.9 years.5 Dermatomyositis has been previously reported to present as a paraneoplastic manifestation of CLL.6 One case has been reported of a patient with CLL who developed leukemia cutis presenting with poikiloderma in the characteristic dermatomyositis shawl-like distribution.7 The lack of dermal infiltration with leukemic cells in our patient, however, makes a paraneoplastic etiology much more likely.
Our patient’s rash did not initially improve with treatment of CLL, but dermatomyositis associated with hematological malignancy may precede, occur simultaneously, or follow the diagnosis of malignancy.8 Additionally, symptoms of dermatomyositis do not always parallel the course of hematological malignancy outcome. However, rituximab has been used as a treatment of dermatomyositis and may have contributed some synergistic effect in combination with prednisone in our patient.9
To the Editor:
Dermatomyositis represents a rare idiopathic inflammatory process presenting with cutaneous lesions and muscular weakness. It often represents a paraneoplastic syndrome. We report the case of a 62-year-old man with a history of total-body poikiloderma and a recent diagnosis of chronic lymphocytic leukemia (CLL). Despite lacking typical features of the disease, a diagnosis of dermatomyositis was made. Our patient may represent a distinct poikilodermatous variant of dermatomyositis, sharing the generalized distribution of the erythrodermic subtype.
A 62-year-old man presented with pruritic poikiloderma involving the neck, arms, legs, abdomen, chest, and back of 2 years’ duration (Figure). He also experienced dysphagia and weakness of the legs. The rash was previously treated by other dermatologists with a combination of high-potency topical steroids and topical tacrolimus 0.1% without success. His history was notable for CLL, which had been diagnosed by a dermatologist 6 months prior to the current presentation. Prior to his visit to the dermatologist, the patient had received 6 chemotherapeutic sessions with a combination of rituximab and cyclophosphamide for the treatment of CLL. The rash did not improve with chemotherapy.
Repeat biopsies of affected regions only demonstrated features of mild interface dermatitis. Direct immunofluorescence studies showed scattered colloid body fluorescence for IgM. Because of bilateral weakness of the legs, a muscle biopsy was taken, which demonstrated severe atrophy and interstitial fibrosis, with neurogenic abnormalities detected in areas of lesser atrophy via abnormal muscle fiber–type grouping. Metabolic panel showed elevated muscle enzymes in the blood: creatine kinase, 243 U/L (reference range, 10–225 U/L); serum aldolase, 16 U/L (reference range, ≤8.1 U/L); lactate dehydrogenase, 314 U/L (reference range, 60–200 U/L). An autoimmune panel was negative for Jo-1, Scl-70, U1 ribonucleoprotein, DNA, desmoglein 1 and 3, and antiacetylcholine receptor antibodies. An elevated erythrocyte sedimentation rate was measured at 16 mm/h (reference range, 0–10 mm/h). Given these findings, the lesions were confirmed as a widespread poikilodermatous variant of dermatomyositis.
The patient was placed on a daily 50-mg dose of prednisone, which produced rapid improvement in scaling and erythema. Creatine kinase and serum aldolase levels normalized and motor strength increased. After 1 week the prednisone dosage was reduced to a daily 30-mg dose, and then 20 mg a week later. The skin lesions completely resolved within 4 to 5 months and the patient is currently on a prednisone dose of 5 mg, alternating with 2.5 mg of prednisone and rituximab infusion every 2 months.
Dermatomyositis is a rare entity with an incidence of approximately 0.5 to 1 per 100,000 individuals.1 It presents with a characteristic rash composed of Gottron papules; pathognomonic flat violaceous papules on the dorsal interphalangeal joints, elbows, or knees; and a heliotrope rash, a violaceous erythema involving the eyelids. Poikiloderma frequently is reported to present in a shawl-like distribution, encompassing the shoulders, arms, and upper back.1,2 Dermatomyositis of the poikilodermatous type can present in nonphotoexposed areas and photoexposed areas. The unusual feature is the total-body involvement, which is analogous to erythroderma.3
Our case may represent a distinct poikilodermatous manifestation sharing the distribution of the erythrodermic subtype. We believe that the skin lesions may have represented a paraneoplastic event presenting prior to diagnosis with CLL. Dermatomyositis has a strong association with cancer, with patients 3 times more likely to develop internal malignancy.4 Association is strongest for non-Hodgkin lymphoma, as well as ovarian, lung, colorectal, pancreatic, and gastric cancer. When associated with malignancy, symptoms of dermatomyositis or myositis typically precede the discovery of malignancy by an average of 1.9 years.5 Dermatomyositis has been previously reported to present as a paraneoplastic manifestation of CLL.6 One case has been reported of a patient with CLL who developed leukemia cutis presenting with poikiloderma in the characteristic dermatomyositis shawl-like distribution.7 The lack of dermal infiltration with leukemic cells in our patient, however, makes a paraneoplastic etiology much more likely.
Our patient’s rash did not initially improve with treatment of CLL, but dermatomyositis associated with hematological malignancy may precede, occur simultaneously, or follow the diagnosis of malignancy.8 Additionally, symptoms of dermatomyositis do not always parallel the course of hematological malignancy outcome. However, rituximab has been used as a treatment of dermatomyositis and may have contributed some synergistic effect in combination with prednisone in our patient.9
- Dourmishev LA, Dourmishev AL, Schwartz RA. Dermatomyositis: cutaneous manifestations of its variants. Int J Dermatol. 2002;41:625-630.
- Kovacs SO, Kovacs SC. Dermatomyositis. J Am Acad Dermatol. 1998;39:899-920; quiz 921-992.
- Liu ZH, Wang XD. Acute-onset adult dermatomyositis presenting with erythroderma and diplopia. Clin Exp Dermatol. 2007;32:751-752.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Bohan A, Peter JB, Bowman RL, et al. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore). 1977;56:255-286.
- Ishida T, Aikawa K, Tamura T, et al. Chronic lymphocytic leukemia associated with nephrotic syndrome and dermatomyositis. Intern Med. 1995;34:15-17.
- Nousari HC, Kimyai-Asadi A, Huang CH, et al. T-cell chronic lymphocytic leukemia mimicking dermatomyositis. Int J Dermatol. 2000;39:144-146.
- Marie I, Guillevin L, Menard JF, et al. Hematological malignancy associated with polymyositis and dermatomyositis. Autoimmun Rev. 2012;11:615-620.
- Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52:601-607.
- Dourmishev LA, Dourmishev AL, Schwartz RA. Dermatomyositis: cutaneous manifestations of its variants. Int J Dermatol. 2002;41:625-630.
- Kovacs SO, Kovacs SC. Dermatomyositis. J Am Acad Dermatol. 1998;39:899-920; quiz 921-992.
- Liu ZH, Wang XD. Acute-onset adult dermatomyositis presenting with erythroderma and diplopia. Clin Exp Dermatol. 2007;32:751-752.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Bohan A, Peter JB, Bowman RL, et al. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore). 1977;56:255-286.
- Ishida T, Aikawa K, Tamura T, et al. Chronic lymphocytic leukemia associated with nephrotic syndrome and dermatomyositis. Intern Med. 1995;34:15-17.
- Nousari HC, Kimyai-Asadi A, Huang CH, et al. T-cell chronic lymphocytic leukemia mimicking dermatomyositis. Int J Dermatol. 2000;39:144-146.
- Marie I, Guillevin L, Menard JF, et al. Hematological malignancy associated with polymyositis and dermatomyositis. Autoimmun Rev. 2012;11:615-620.
- Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52:601-607.
Practice Points
- Poikiloderma, even with an unusual clinical presentation, can be a useful clinical clue for the diagnosis of dermatomyositis or other collagen vascular disease.
- Dermatomyositis can be paraneoplastic and though often associated with epithelial malignancies and solid tumors can also be associated with leukemias.
Are Breast Cancer Patients Satisfied With Their Care?
Japan has a universal health care system with low copays and short wait times for appointments, including those with specialists. Yet patient satisfaction scores are low compared with those of other countries. Researchers from Juntendo Urayasu Hospital, a university hospital in a Tokyo suburb, conducted a study of 214 patients with breast cancer to find out which aspects of radiation oncology care might affect patient satisfaction. The survey included questions about overall treatment, time from diagnosis to treatment start, wait times in the hospital, and length of consultations.
In general, levels of satisfaction were high. However, wait time was significantly negatively associated with both overall satisfaction and satisfaction with the radiation oncologist. Wait time was just under an hour for an average 11-minute consultation. Although this was longer than the “notorious” Japanese situation of a “3 hours wait and 3 minutes consultation,” the researchers say, “We expect that an international audience will appreciate that even 11 minutes is an exceptionally short duration for a consultation visit with a specialist in radiation oncology.”
They note, though, a reasonable caveat. Anyone can walk into their hospital and, for an additional fee, see a specialist on the day they want, which can lead to extended wait times from sheer congestion. Their hospital’s chief breast cancer surgeon sees 60 to 70 patients a day; the radiation oncologist treats 500 to 600 patients a year without resident or trainee support. This situation is typical of Japanese university hospitals, the researchers add.
Related: Breast Cancer Treatment Among Rural and Urban Women at the Veterans Health Administration
Importantly, for Japanese patients, the researchers also included questions to measure patients’ opinions about sharing how they felt with their physicians. The level of sharing correlated with satisfaction, but the researchers point out that in Japan sharing feelings remains “challenging.” Their findings suggest, they say, that if this were improved, patients’ satisfaction might increase.
Japan has a universal health care system with low copays and short wait times for appointments, including those with specialists. Yet patient satisfaction scores are low compared with those of other countries. Researchers from Juntendo Urayasu Hospital, a university hospital in a Tokyo suburb, conducted a study of 214 patients with breast cancer to find out which aspects of radiation oncology care might affect patient satisfaction. The survey included questions about overall treatment, time from diagnosis to treatment start, wait times in the hospital, and length of consultations.
In general, levels of satisfaction were high. However, wait time was significantly negatively associated with both overall satisfaction and satisfaction with the radiation oncologist. Wait time was just under an hour for an average 11-minute consultation. Although this was longer than the “notorious” Japanese situation of a “3 hours wait and 3 minutes consultation,” the researchers say, “We expect that an international audience will appreciate that even 11 minutes is an exceptionally short duration for a consultation visit with a specialist in radiation oncology.”
They note, though, a reasonable caveat. Anyone can walk into their hospital and, for an additional fee, see a specialist on the day they want, which can lead to extended wait times from sheer congestion. Their hospital’s chief breast cancer surgeon sees 60 to 70 patients a day; the radiation oncologist treats 500 to 600 patients a year without resident or trainee support. This situation is typical of Japanese university hospitals, the researchers add.
Related: Breast Cancer Treatment Among Rural and Urban Women at the Veterans Health Administration
Importantly, for Japanese patients, the researchers also included questions to measure patients’ opinions about sharing how they felt with their physicians. The level of sharing correlated with satisfaction, but the researchers point out that in Japan sharing feelings remains “challenging.” Their findings suggest, they say, that if this were improved, patients’ satisfaction might increase.
Japan has a universal health care system with low copays and short wait times for appointments, including those with specialists. Yet patient satisfaction scores are low compared with those of other countries. Researchers from Juntendo Urayasu Hospital, a university hospital in a Tokyo suburb, conducted a study of 214 patients with breast cancer to find out which aspects of radiation oncology care might affect patient satisfaction. The survey included questions about overall treatment, time from diagnosis to treatment start, wait times in the hospital, and length of consultations.
In general, levels of satisfaction were high. However, wait time was significantly negatively associated with both overall satisfaction and satisfaction with the radiation oncologist. Wait time was just under an hour for an average 11-minute consultation. Although this was longer than the “notorious” Japanese situation of a “3 hours wait and 3 minutes consultation,” the researchers say, “We expect that an international audience will appreciate that even 11 minutes is an exceptionally short duration for a consultation visit with a specialist in radiation oncology.”
They note, though, a reasonable caveat. Anyone can walk into their hospital and, for an additional fee, see a specialist on the day they want, which can lead to extended wait times from sheer congestion. Their hospital’s chief breast cancer surgeon sees 60 to 70 patients a day; the radiation oncologist treats 500 to 600 patients a year without resident or trainee support. This situation is typical of Japanese university hospitals, the researchers add.
Related: Breast Cancer Treatment Among Rural and Urban Women at the Veterans Health Administration
Importantly, for Japanese patients, the researchers also included questions to measure patients’ opinions about sharing how they felt with their physicians. The level of sharing correlated with satisfaction, but the researchers point out that in Japan sharing feelings remains “challenging.” Their findings suggest, they say, that if this were improved, patients’ satisfaction might increase.
Intrathecal hydromorphone boosts post-op pain control
LAS VEGAS – Intrathecal hydromorphone, administered alone or with lidocaine, effectively controlled pain and decreased postoperative opioid use after colorectal surgery in a retrospective study.
The technique was so effective that 28% of patients required no postoperative opioids at all, Amit Merchea, MD, said at the Association for Academic Surgery/Society of University Surgeons Academic Surgical Congress.
The intrathecal analgesic was part of an enhanced recovery pathway (ERP) for patients undergoing elective colorectal surgery at the Mayo Clinic, Jacksonville, Fla., where Dr. Merchea practices colorectal surgery.
Morphine has been the gold standard for this approach, he said. Dr. Merchea and his colleagues investigated the use of hydromorphone in 601 patients who underwent open or minimally invasive colorectal surgery at the Mayo Clinic from 2012 to 2013.
The patients were a median of 52 years old. The surgical approach was almost evenly split between open and laparoscopic. The median length of hospital stay was 3 days. All received intrathecal hydromorphone either alone (91%) or with a local anesthetic (9%).
Everyone was on the same presurgical and postsurgical pain control regimen, which consisted of celecoxib, gabapentin, and acetaminophen before surgery, followed by nonsteroidal anti-inflammatories and acetaminophen, with oxycodone as needed, after surgery.
Overall, the procedure was well tolerated, with seven cases of pruritus requiring Nubain (nalbuphine), one case of respiratory depression that required naloxone, and one postdural headache that required a patch. The rate of ileus was 16%.
At 4 hours, the median pain score was 3 on a 1- to 10-point scale. At 24 hours, it was a median of 4. By 48 hours, the median pain score was 6. This increase is to be expected as the hydrocodone exists in the intrathecal space for up to 36 hours, Dr. Merchea noted.
The median total oral morphine equivalent (OME) was 24; 170 patients (28%) needed no opioid medications after surgery.
He also presented outcomes by infusion composition. There was no difference in the rate of ileus among those who had hydromorphone alone and those who had it with lidocaine. The length of stay was 3 vs. 3.5 days, respectively. The only significant difference in pain scores was the 48-hour maximum, which was a median of 7 in the combination group and 6 in the hydromorphone-only group.
The combination group, however, required more postoperative opioids (33.8 vs. 22.5 OMEs). Significantly more patients in the hydromorphone-only group were able to go without any postoperative opioids (30% vs. 15%).
Dr. Merchea also broke down the results by hydromorphone dosage, but there were no significant differences in ileus rate, length of stay, or pain scores correlated with dosage. However, those who received higher doses were significantly more likely to need more postoperative opioids than those who had lower doses.
Session moderator Peter Muscarella, MD, of Montefiore Medical Center, New York, asked whether the intrathecal infusion was associated with hypotension. “Some of these procedures with epidural analgesics intraoperatively, we have seen shifts in blood pressure that result in excess fluid administration, sometimes leading to tissue complications.”
Dr. Merchea said hypotension was not an outcome of this trial, but that he has looked at it before. “We have previously reported that epidural analgesia was associated with a 15% occurrence of hypotension, but it had no clinical impact and didn’t warrant giving any additional fluids.”
Dr. Merchea had no relevant financial disclosures.
[email protected]
On Twitter @Alz_Gal
LAS VEGAS – Intrathecal hydromorphone, administered alone or with lidocaine, effectively controlled pain and decreased postoperative opioid use after colorectal surgery in a retrospective study.
The technique was so effective that 28% of patients required no postoperative opioids at all, Amit Merchea, MD, said at the Association for Academic Surgery/Society of University Surgeons Academic Surgical Congress.
The intrathecal analgesic was part of an enhanced recovery pathway (ERP) for patients undergoing elective colorectal surgery at the Mayo Clinic, Jacksonville, Fla., where Dr. Merchea practices colorectal surgery.
Morphine has been the gold standard for this approach, he said. Dr. Merchea and his colleagues investigated the use of hydromorphone in 601 patients who underwent open or minimally invasive colorectal surgery at the Mayo Clinic from 2012 to 2013.
The patients were a median of 52 years old. The surgical approach was almost evenly split between open and laparoscopic. The median length of hospital stay was 3 days. All received intrathecal hydromorphone either alone (91%) or with a local anesthetic (9%).
Everyone was on the same presurgical and postsurgical pain control regimen, which consisted of celecoxib, gabapentin, and acetaminophen before surgery, followed by nonsteroidal anti-inflammatories and acetaminophen, with oxycodone as needed, after surgery.
Overall, the procedure was well tolerated, with seven cases of pruritus requiring Nubain (nalbuphine), one case of respiratory depression that required naloxone, and one postdural headache that required a patch. The rate of ileus was 16%.
At 4 hours, the median pain score was 3 on a 1- to 10-point scale. At 24 hours, it was a median of 4. By 48 hours, the median pain score was 6. This increase is to be expected as the hydrocodone exists in the intrathecal space for up to 36 hours, Dr. Merchea noted.
The median total oral morphine equivalent (OME) was 24; 170 patients (28%) needed no opioid medications after surgery.
He also presented outcomes by infusion composition. There was no difference in the rate of ileus among those who had hydromorphone alone and those who had it with lidocaine. The length of stay was 3 vs. 3.5 days, respectively. The only significant difference in pain scores was the 48-hour maximum, which was a median of 7 in the combination group and 6 in the hydromorphone-only group.
The combination group, however, required more postoperative opioids (33.8 vs. 22.5 OMEs). Significantly more patients in the hydromorphone-only group were able to go without any postoperative opioids (30% vs. 15%).
Dr. Merchea also broke down the results by hydromorphone dosage, but there were no significant differences in ileus rate, length of stay, or pain scores correlated with dosage. However, those who received higher doses were significantly more likely to need more postoperative opioids than those who had lower doses.
Session moderator Peter Muscarella, MD, of Montefiore Medical Center, New York, asked whether the intrathecal infusion was associated with hypotension. “Some of these procedures with epidural analgesics intraoperatively, we have seen shifts in blood pressure that result in excess fluid administration, sometimes leading to tissue complications.”
Dr. Merchea said hypotension was not an outcome of this trial, but that he has looked at it before. “We have previously reported that epidural analgesia was associated with a 15% occurrence of hypotension, but it had no clinical impact and didn’t warrant giving any additional fluids.”
Dr. Merchea had no relevant financial disclosures.
[email protected]
On Twitter @Alz_Gal
LAS VEGAS – Intrathecal hydromorphone, administered alone or with lidocaine, effectively controlled pain and decreased postoperative opioid use after colorectal surgery in a retrospective study.
The technique was so effective that 28% of patients required no postoperative opioids at all, Amit Merchea, MD, said at the Association for Academic Surgery/Society of University Surgeons Academic Surgical Congress.
The intrathecal analgesic was part of an enhanced recovery pathway (ERP) for patients undergoing elective colorectal surgery at the Mayo Clinic, Jacksonville, Fla., where Dr. Merchea practices colorectal surgery.
Morphine has been the gold standard for this approach, he said. Dr. Merchea and his colleagues investigated the use of hydromorphone in 601 patients who underwent open or minimally invasive colorectal surgery at the Mayo Clinic from 2012 to 2013.
The patients were a median of 52 years old. The surgical approach was almost evenly split between open and laparoscopic. The median length of hospital stay was 3 days. All received intrathecal hydromorphone either alone (91%) or with a local anesthetic (9%).
Everyone was on the same presurgical and postsurgical pain control regimen, which consisted of celecoxib, gabapentin, and acetaminophen before surgery, followed by nonsteroidal anti-inflammatories and acetaminophen, with oxycodone as needed, after surgery.
Overall, the procedure was well tolerated, with seven cases of pruritus requiring Nubain (nalbuphine), one case of respiratory depression that required naloxone, and one postdural headache that required a patch. The rate of ileus was 16%.
At 4 hours, the median pain score was 3 on a 1- to 10-point scale. At 24 hours, it was a median of 4. By 48 hours, the median pain score was 6. This increase is to be expected as the hydrocodone exists in the intrathecal space for up to 36 hours, Dr. Merchea noted.
The median total oral morphine equivalent (OME) was 24; 170 patients (28%) needed no opioid medications after surgery.
He also presented outcomes by infusion composition. There was no difference in the rate of ileus among those who had hydromorphone alone and those who had it with lidocaine. The length of stay was 3 vs. 3.5 days, respectively. The only significant difference in pain scores was the 48-hour maximum, which was a median of 7 in the combination group and 6 in the hydromorphone-only group.
The combination group, however, required more postoperative opioids (33.8 vs. 22.5 OMEs). Significantly more patients in the hydromorphone-only group were able to go without any postoperative opioids (30% vs. 15%).
Dr. Merchea also broke down the results by hydromorphone dosage, but there were no significant differences in ileus rate, length of stay, or pain scores correlated with dosage. However, those who received higher doses were significantly more likely to need more postoperative opioids than those who had lower doses.
Session moderator Peter Muscarella, MD, of Montefiore Medical Center, New York, asked whether the intrathecal infusion was associated with hypotension. “Some of these procedures with epidural analgesics intraoperatively, we have seen shifts in blood pressure that result in excess fluid administration, sometimes leading to tissue complications.”
Dr. Merchea said hypotension was not an outcome of this trial, but that he has looked at it before. “We have previously reported that epidural analgesia was associated with a 15% occurrence of hypotension, but it had no clinical impact and didn’t warrant giving any additional fluids.”
Dr. Merchea had no relevant financial disclosures.
[email protected]
On Twitter @Alz_Gal
AT THE ACADEMIC SURGICAL CONGRESS
Key clinical point:
Major finding: About a quarter of patients (28%) needed no postoperative opioids.
Data source: A retrospective study of 601 patients.
Disclosures: Dr. Merchea had no relevant financial disclosures.
Out to lunch
I’m sure there are folks here in town who wondered how I could keep up a professional pace that often included being on call 2 nights a week and working every third weekend. Even when I was in my early 50s, people asked me if I was getting ready to retire. I hope it wasn’t because I appeared unhappy or looked 15 years older than I was. I suspect that some parents who didn’t know me well predicted that my career would have ended far short of 40 years.
One of the secrets of what appeared to be my superhuman stamina was that almost every day at noon I was out to lunch. That doesn’t mean that I always took time to eat lunch. In fact, I must admit that more often than not my midday diet consisted of several handfuls of cashews or an energy bar eaten on the fly.

The feeling of invigoration and renewal that came in its wake fueled my commitment to my habit of lunchtime outdoor activity. Although to some people it may be counterintuitive, the physical activity energized me. The second half of my workday was no more fatiguing than the morning. However, if some thoughtless hospital or practice administrator scheduled a noon meeting, the rest of my day was a grump fest.
A recent study has demonstrated just how powerful lunchtime exercise can be in improving worker attitude and mood, even if the activity is just going for a walk. (“Changes in work affect in response to lunchtime walking in previously physically inactive employees: A randomized trial” (Scand J Med Sci Sports. 2015 Dec;25[6]:778-87). There have been other studies that have pointed to the value of an activity break, but these investigators collected real-time reports from subjects using their cell phones. “Lunchtime walks improved enthusiasm, relaxation, and nervousness at work,” the researchers noted.
The problem comes in getting employees to take that first step toward developing a lunchtime activity habit. A few, usually women, have discovered the value for themselves and enjoy the social interaction as much as they do the affect-improving aspects of the activity and change of scene. I have tried to encourage lunchtime walking in the workplace with several strategies, including small monetary rewards, prizes, and contests between groups of workers. One year we even bought umbrellas to encourage employees to walk even if it was raining. But without a vigorous and persistent support system, inertia wins, and only those who have discovered the benefits of lunchtime activity for themselves persist.
You may be asking yourself how I managed to find time in my schedule for that hour of lunchtime activity; actually it was usually an hour and half to include a shower. The answer is that I built my schedule around it, and that meant getting to the office earlier and working later. But in my mind that was a small price to pay for the benefits I received. The other secret to my apparent stamina was that I lived a 5-minute bike ride from both hospitals and my office. Don’t underestimate the toll your commute is taking on your life and happiness.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.”
I’m sure there are folks here in town who wondered how I could keep up a professional pace that often included being on call 2 nights a week and working every third weekend. Even when I was in my early 50s, people asked me if I was getting ready to retire. I hope it wasn’t because I appeared unhappy or looked 15 years older than I was. I suspect that some parents who didn’t know me well predicted that my career would have ended far short of 40 years.
One of the secrets of what appeared to be my superhuman stamina was that almost every day at noon I was out to lunch. That doesn’t mean that I always took time to eat lunch. In fact, I must admit that more often than not my midday diet consisted of several handfuls of cashews or an energy bar eaten on the fly.
The feeling of invigoration and renewal that came in its wake fueled my commitment to my habit of lunchtime outdoor activity. Although to some people it may be counterintuitive, the physical activity energized me. The second half of my workday was no more fatiguing than the morning. However, if some thoughtless hospital or practice administrator scheduled a noon meeting, the rest of my day was a grump fest.
A recent study has demonstrated just how powerful lunchtime exercise can be in improving worker attitude and mood, even if the activity is just going for a walk. (“Changes in work affect in response to lunchtime walking in previously physically inactive employees: A randomized trial” (Scand J Med Sci Sports. 2015 Dec;25[6]:778-87). There have been other studies that have pointed to the value of an activity break, but these investigators collected real-time reports from subjects using their cell phones. “Lunchtime walks improved enthusiasm, relaxation, and nervousness at work,” the researchers noted.
The problem comes in getting employees to take that first step toward developing a lunchtime activity habit. A few, usually women, have discovered the value for themselves and enjoy the social interaction as much as they do the affect-improving aspects of the activity and change of scene. I have tried to encourage lunchtime walking in the workplace with several strategies, including small monetary rewards, prizes, and contests between groups of workers. One year we even bought umbrellas to encourage employees to walk even if it was raining. But without a vigorous and persistent support system, inertia wins, and only those who have discovered the benefits of lunchtime activity for themselves persist.
You may be asking yourself how I managed to find time in my schedule for that hour of lunchtime activity; actually it was usually an hour and half to include a shower. The answer is that I built my schedule around it, and that meant getting to the office earlier and working later. But in my mind that was a small price to pay for the benefits I received. The other secret to my apparent stamina was that I lived a 5-minute bike ride from both hospitals and my office. Don’t underestimate the toll your commute is taking on your life and happiness.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.”
I’m sure there are folks here in town who wondered how I could keep up a professional pace that often included being on call 2 nights a week and working every third weekend. Even when I was in my early 50s, people asked me if I was getting ready to retire. I hope it wasn’t because I appeared unhappy or looked 15 years older than I was. I suspect that some parents who didn’t know me well predicted that my career would have ended far short of 40 years.
One of the secrets of what appeared to be my superhuman stamina was that almost every day at noon I was out to lunch. That doesn’t mean that I always took time to eat lunch. In fact, I must admit that more often than not my midday diet consisted of several handfuls of cashews or an energy bar eaten on the fly.
The feeling of invigoration and renewal that came in its wake fueled my commitment to my habit of lunchtime outdoor activity. Although to some people it may be counterintuitive, the physical activity energized me. The second half of my workday was no more fatiguing than the morning. However, if some thoughtless hospital or practice administrator scheduled a noon meeting, the rest of my day was a grump fest.
A recent study has demonstrated just how powerful lunchtime exercise can be in improving worker attitude and mood, even if the activity is just going for a walk. (“Changes in work affect in response to lunchtime walking in previously physically inactive employees: A randomized trial” (Scand J Med Sci Sports. 2015 Dec;25[6]:778-87). There have been other studies that have pointed to the value of an activity break, but these investigators collected real-time reports from subjects using their cell phones. “Lunchtime walks improved enthusiasm, relaxation, and nervousness at work,” the researchers noted.
The problem comes in getting employees to take that first step toward developing a lunchtime activity habit. A few, usually women, have discovered the value for themselves and enjoy the social interaction as much as they do the affect-improving aspects of the activity and change of scene. I have tried to encourage lunchtime walking in the workplace with several strategies, including small monetary rewards, prizes, and contests between groups of workers. One year we even bought umbrellas to encourage employees to walk even if it was raining. But without a vigorous and persistent support system, inertia wins, and only those who have discovered the benefits of lunchtime activity for themselves persist.
You may be asking yourself how I managed to find time in my schedule for that hour of lunchtime activity; actually it was usually an hour and half to include a shower. The answer is that I built my schedule around it, and that meant getting to the office earlier and working later. But in my mind that was a small price to pay for the benefits I received. The other secret to my apparent stamina was that I lived a 5-minute bike ride from both hospitals and my office. Don’t underestimate the toll your commute is taking on your life and happiness.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.”
Ibrutinib, palbociclib yield durable complete responses in pretreated mantle cell lymphoma
SAN DIEGO – A “mechanism-based” combination of ibrutinib and palbociclib was reasonably well tolerated and induced complete responses in 44% of patients with previously treated mantle cell lymphoma, Peter Martin, MD, reported at the annual meeting of the American Society of Hematology.
Fully 67% of patients remained alive and progression-free after a median of 11 months of follow-up, and no responders progressed during this phase I trial, added Dr. Martin of Weill Cornell Medical College in New York. These rates “appear better than those reported in studies of single-agent ibrutinib, although the number of patients was very small,” he acknowledged. Most patients tolerated therapy, although 25% developed dose-limiting toxicities or stopped treatment because of adverse effects. Based on these results, the investigators are studying biomarkers for resistance and are planning a phase II, multicenter trial to evaluate time to progression.
Single-agent ibrutinib (Imbruvica) has shown promise in mantle cell lymphoma, but treatment failure affects about half of patients within 1 year, Dr. Martin noted. The CDK4/6 inhibitor palbociclib (Ibrance) induces prolonged arrest early in the G1 phase of the cell cycle, which overcame ibrutinib resistance in mantle cell lymphoma cell lines in a prior study (Cancer Discov. 2014;4[9]:1022-35).
To test the maximum tolerated dose of combination therapy, Dr. Martin and his associates enrolled 20 adults with previously treated mantle cell lymphoma who were naive to ibrutinib and CD4/6 inhibitors. The patients had received a median of one and up to five prior lines of therapy, and six (30%) were refractory to their most recent therapy. They received ibrutinib daily and palbociclib on the first 21 days of each 28-day treatment cycle. Dosing began at one of five levels, ranging from 280 mg ibrutinib/75 mg palbociclib to 560 mg ibrutinib/125 mg palbociclib. Doses were escalated based on a standard phase I 3+3 design.
Among 18 patients evaluated, 12 (67%) responded to treatment, and 8 (44%) had a complete response. Median time to complete response was three cycles. The most common grade 1-2 adverse events were diarrhea, fatigue, rash, and bruising. Three patients (15%) developed dose-limiting toxicities. These included one case of grade 4 thrombocytopenia at 420 mg ibrutinib/100 mg palbociclib and two cases of grade 3 rash at 560 mg ibrutinib/125 mg palbociclib. The grade 3 rashes led to dose reductions, and six patients needed dose interruptions. Also, four patients stopped treatment because of disease progression, two did so because of elevated liver enzymes or prolonged cytopenia, and one did so to undergo allogeneic stem cell transplantation.
The National Cancer Institute sponsored the study. Dr. Martin disclosed ties to Janssen, which makes ibrutinib, and to Celgene, Gilead, Novartis, Acerta, and Teva. Senior author John P. Leonard, MD, and one of 10 coinvestigators disclosed ties to several pharmaceutical companies.
SAN DIEGO – A “mechanism-based” combination of ibrutinib and palbociclib was reasonably well tolerated and induced complete responses in 44% of patients with previously treated mantle cell lymphoma, Peter Martin, MD, reported at the annual meeting of the American Society of Hematology.
Fully 67% of patients remained alive and progression-free after a median of 11 months of follow-up, and no responders progressed during this phase I trial, added Dr. Martin of Weill Cornell Medical College in New York. These rates “appear better than those reported in studies of single-agent ibrutinib, although the number of patients was very small,” he acknowledged. Most patients tolerated therapy, although 25% developed dose-limiting toxicities or stopped treatment because of adverse effects. Based on these results, the investigators are studying biomarkers for resistance and are planning a phase II, multicenter trial to evaluate time to progression.
Single-agent ibrutinib (Imbruvica) has shown promise in mantle cell lymphoma, but treatment failure affects about half of patients within 1 year, Dr. Martin noted. The CDK4/6 inhibitor palbociclib (Ibrance) induces prolonged arrest early in the G1 phase of the cell cycle, which overcame ibrutinib resistance in mantle cell lymphoma cell lines in a prior study (Cancer Discov. 2014;4[9]:1022-35).
To test the maximum tolerated dose of combination therapy, Dr. Martin and his associates enrolled 20 adults with previously treated mantle cell lymphoma who were naive to ibrutinib and CD4/6 inhibitors. The patients had received a median of one and up to five prior lines of therapy, and six (30%) were refractory to their most recent therapy. They received ibrutinib daily and palbociclib on the first 21 days of each 28-day treatment cycle. Dosing began at one of five levels, ranging from 280 mg ibrutinib/75 mg palbociclib to 560 mg ibrutinib/125 mg palbociclib. Doses were escalated based on a standard phase I 3+3 design.
Among 18 patients evaluated, 12 (67%) responded to treatment, and 8 (44%) had a complete response. Median time to complete response was three cycles. The most common grade 1-2 adverse events were diarrhea, fatigue, rash, and bruising. Three patients (15%) developed dose-limiting toxicities. These included one case of grade 4 thrombocytopenia at 420 mg ibrutinib/100 mg palbociclib and two cases of grade 3 rash at 560 mg ibrutinib/125 mg palbociclib. The grade 3 rashes led to dose reductions, and six patients needed dose interruptions. Also, four patients stopped treatment because of disease progression, two did so because of elevated liver enzymes or prolonged cytopenia, and one did so to undergo allogeneic stem cell transplantation.
The National Cancer Institute sponsored the study. Dr. Martin disclosed ties to Janssen, which makes ibrutinib, and to Celgene, Gilead, Novartis, Acerta, and Teva. Senior author John P. Leonard, MD, and one of 10 coinvestigators disclosed ties to several pharmaceutical companies.
SAN DIEGO – A “mechanism-based” combination of ibrutinib and palbociclib was reasonably well tolerated and induced complete responses in 44% of patients with previously treated mantle cell lymphoma, Peter Martin, MD, reported at the annual meeting of the American Society of Hematology.
Fully 67% of patients remained alive and progression-free after a median of 11 months of follow-up, and no responders progressed during this phase I trial, added Dr. Martin of Weill Cornell Medical College in New York. These rates “appear better than those reported in studies of single-agent ibrutinib, although the number of patients was very small,” he acknowledged. Most patients tolerated therapy, although 25% developed dose-limiting toxicities or stopped treatment because of adverse effects. Based on these results, the investigators are studying biomarkers for resistance and are planning a phase II, multicenter trial to evaluate time to progression.
Single-agent ibrutinib (Imbruvica) has shown promise in mantle cell lymphoma, but treatment failure affects about half of patients within 1 year, Dr. Martin noted. The CDK4/6 inhibitor palbociclib (Ibrance) induces prolonged arrest early in the G1 phase of the cell cycle, which overcame ibrutinib resistance in mantle cell lymphoma cell lines in a prior study (Cancer Discov. 2014;4[9]:1022-35).
To test the maximum tolerated dose of combination therapy, Dr. Martin and his associates enrolled 20 adults with previously treated mantle cell lymphoma who were naive to ibrutinib and CD4/6 inhibitors. The patients had received a median of one and up to five prior lines of therapy, and six (30%) were refractory to their most recent therapy. They received ibrutinib daily and palbociclib on the first 21 days of each 28-day treatment cycle. Dosing began at one of five levels, ranging from 280 mg ibrutinib/75 mg palbociclib to 560 mg ibrutinib/125 mg palbociclib. Doses were escalated based on a standard phase I 3+3 design.
Among 18 patients evaluated, 12 (67%) responded to treatment, and 8 (44%) had a complete response. Median time to complete response was three cycles. The most common grade 1-2 adverse events were diarrhea, fatigue, rash, and bruising. Three patients (15%) developed dose-limiting toxicities. These included one case of grade 4 thrombocytopenia at 420 mg ibrutinib/100 mg palbociclib and two cases of grade 3 rash at 560 mg ibrutinib/125 mg palbociclib. The grade 3 rashes led to dose reductions, and six patients needed dose interruptions. Also, four patients stopped treatment because of disease progression, two did so because of elevated liver enzymes or prolonged cytopenia, and one did so to undergo allogeneic stem cell transplantation.
The National Cancer Institute sponsored the study. Dr. Martin disclosed ties to Janssen, which makes ibrutinib, and to Celgene, Gilead, Novartis, Acerta, and Teva. Senior author John P. Leonard, MD, and one of 10 coinvestigators disclosed ties to several pharmaceutical companies.
AT ASH 2016
Key clinical point: Combination therapy with ibrutinib and palbociclib was generally well tolerated and induced complete responses in patients with pretreated mantle cell lymphoma.
Major finding: A total of 44% of patients had complete responses, and 67% remained alive and progression-free after a median of 11 months of follow-up. Severe rashes occurred at the highest dose studied (420 mg ibrutinib/100 mg palbociclib).
Data source: A phase I trial of 20 patients with previously treated mantle cell lymphoma.
Disclosures: The National Cancer Institute sponsored the study. Dr. Martin disclosed ties to Janssen, which makes ibrutinib, and to Celgene, Gilead, Novartis, Acerta, and Teva. Senior author John P. Leonard, MD, and one of 10 coinvestigators disclosed ties to several pharmaceutical companies.
HHS pick Price made ‘brazen’ trades while committee was under scrutiny
Health and Human Services secretary nominee Tom Price showed little restraint in his personal stock trading during the 3 years that federal investigators were bearing down on a key House committee on which the Republican congressman served, a review of his financial disclosures shows.
Rep. Price (Ga.) made dozens of health industry stock trades during a 3-year investigation by the Securities and Exchange Commission that focused on the Ways and Means Committee, according to financial disclosure records he filed with the House of Representatives. The investigation was considered the first test of a law passed to ban members of Congress and their staffs from trading stock based on insider information.
Rep. Price, who is a retired orthopedic surgeon, was never a target of the federal investigation, which scrutinized a top Ways and Means staffer, and no charges were brought. But ethics experts say Rep. Price’s personal trading, even during the thick of federal pressure on his committee, shows he was unconcerned about financial investments that could create an appearance of impropriety.
“He should have known better,” Richard Painter, former White House chief ethics attorney under President George W. Bush and a professor at the University of Minnesota Law School said of Rep. Price’s conduct during the SEC inquiry.
As Rep. Price awaits a Senate vote on his confirmation, Senate Democrats and a number of watchdog groups have asked the SEC to investigate whether Rep. Price engaged in insider trading with some of his trades in health care companies. Rep. Price has said he abided by all ethics rules, although he acknowledged to the Senate Finance Committee that he did not consult the House Ethics Committee on trades that have now become controversial.
The SEC’s inquiry began in 2013, as it battled Ways and Means for documents to develop its case.
A few weeks ago, the day before President Donald Trump’s inauguration, the SEC quietly dropped its pursuit of committee documents without explanation, according to federal court records. No charges were brought against the staffer, Brian Sutter, who is now a health care lobbyist. Sutter’s lawyer declined to comment.
Craig Holman, government affairs lobbyist with Public Citizen, described Rep. Price’s volume of stock trades during the SEC inquiry as “brazen,” given the congressman’s access to nonpublic information affecting the companies’ fortunes.
“The public is seeing this and they really don’t like it,” said Holman, whose watchdog group recently filed complaints about Rep. Price’s stock trading with both the SEC and the Office of Congressional Ethics.
Trump administration officials and Rep. Price have dismissed questions that news reports and lawmakers have raised about stock trades coinciding with official actions to help certain companies, saying Rep. Price’s brokers chose the stocks independently and all of his conduct was transparent.
After acknowledging that he asked his broker to buy stock in an Australian drug company, he told the Senate Finance Committee that he did not direct his broker to make other trades.
“To the best of my knowledge, I have not undertaken such actions,” he wrote in response to finance committee questions. “I have abided by and adhered to all ethics and conflict of interest rules applicable to me.”
An analysis of Rep. Price’s trades shows that he bought health stocks in 2007, the first year Congress financial disclosures are posted online. In 2011, the first year Rep. Price sat on the health subcommittee, he traded no health-related stocks, according to his financial disclosures filed with Congress.
That same year, members were facing public criticism because of a book detailing how they could use inside information and a “60 Minutes” investigation focused on how members and staff could legally use inside information to gain from their own stock trades.
In 2012, President Barack Obama signed the Stop Trading on Congressional Knowledge Act to rein in insider trading by members and require more disclosure. Public watchdog groups suggested at the time that the law would curb the practice.
That year, after his 1-year break in health care trades, Rep. Price resumed investing in health care companies.
Along with investments in technology, financial services, and retail stocks, he also bought and sold stock in companies that could be impacted by actions of his subcommittee, which has a role in determining rates the government pays under the Medicare program.
Health care firms spend heavily to influence members of Congress, lobbying on health matters, funding political campaigns, and seeking favor with Medicare officials who decide how much the program will pay for certain drugs and devices. The Food and Drug Administration holds similar power, approving or putting conditions on drug and device use.
Beyond his personal investments in health care companies, Rep. Price has also advocated their interests in letters to officials and proposed laws, government records show.
In 2012, disclosure records show Rep. Price sold stock in several drug firms, including more than $110,000 worth of Amgen stock. Amgen’s stock price had steadily climbed out of a recession-level slump, but Rep. Price’s sale came a few weeks before the company pleaded guilty to illegally marketing an anemia drug.
By 2013, the health subcommittee was at the center of a major conflict between Medicare, which sets Medicare Advantage rates, and the insurance industry. Medicare issued a notice early that year announcing its intention to reduce Medicare Advantage rates by 2.3 percent as part of a major cost-cutting initiative.
That prompted fierce lobbying by the health insurance industry. Members of Congress, including Rep. Price, wrote a letter to Marilyn Tavenner, then acting administrator for the Centers for Medicare & Medicaid Services, protesting the rate cut, saying the decrease would “disadvantage vulnerable beneficiaries with multiple chronic conditions.”
Ultimately, Medicare decided not to cut rates but instead, to increase them. Yet an hour before Medicare announced the change, a Height Securities analyst fired off a “flash” report to 200 clients that touched off a surge of trading.
The analyst’s report said a political deal was hatched on Capitol Hill to prevent the cuts as a condition for moving forward on Tavenner’s confirmation. Medicare officials increased rates by nearly 4 percent, a change that would positively impact the bottom lines of health insurance companies.
The SEC began looking for the leak’s source, and within weeks, FBI agents began interviewing staffers at the Ways and Means Committee, court records show.
They discovered communications between Sutter and a health care lobbyist. The HHS Inspector General also began a probe, and federal prosecutors briefly examined the matter as well.
As the case unfolded, Rep. Price bought more health care-related stocks, according to his financial disclosures. He has testified that his broker directed all of the trades, except for his investments in Innate Immunotherapeutics, an Australian company partly owned by Rep. Chris Collins (R-N.Y.), according to Collins’ disclosures. An HHS spokesman said Monday that Rep. Price held three broker-directed accounts.
Ethics experts have said that Rep. Price should have further distanced himself by placing his assets in a blind trust.
On April 30, 2013, Rep. Price bought $2,093 worth of stocks in Incyte, a company that develops cancer drugs; $2,076 in Onyx Pharmaceuticals, a drug maker that would soon merge with a larger drug firm; and $2,097 in Parexel International, a consultancy that helps drugs and devices win FDA approval, according to the financial disclosure records.
The same day, Rep. Price shed shares of Express Scripts, a drug management firm, and Danaher, which makes products hospitals and doctor’s offices using for testing and diagnostics. In August of that year, he bought a $2,429 stake in Jazz Pharmaceuticals, which makes sleep and cancer drugs.
On May 6, 2014, the SEC served its first subpoena for the Ways and Means Committee documents. The committee launched a vigorous fight, appealing a federal district judge’s ruling that it should comply with the SEC subpoena.
Rep. Price continued his health stock trades, including $1,000 to $15,000 in drug firms Amgen, Biogen, Bristol-Myers Squibb, Eli Lilly, and Pfizer. He also bought stock in Aetna, a major health insurer, and Athenahealth, which sells electronic medical record and medical billing software. In 2016, he also increased his investment in Innate Immunotherapeutics.
The purchase became controversial because both he and Collins bought stock in a private placement at a discounted price.
“You’re asking for trouble if you have access to nonpublic information about the health care industry and you’re buying and selling health care stocks,” Painter said.
Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation.
Health and Human Services secretary nominee Tom Price showed little restraint in his personal stock trading during the 3 years that federal investigators were bearing down on a key House committee on which the Republican congressman served, a review of his financial disclosures shows.
Rep. Price (Ga.) made dozens of health industry stock trades during a 3-year investigation by the Securities and Exchange Commission that focused on the Ways and Means Committee, according to financial disclosure records he filed with the House of Representatives. The investigation was considered the first test of a law passed to ban members of Congress and their staffs from trading stock based on insider information.
Rep. Price, who is a retired orthopedic surgeon, was never a target of the federal investigation, which scrutinized a top Ways and Means staffer, and no charges were brought. But ethics experts say Rep. Price’s personal trading, even during the thick of federal pressure on his committee, shows he was unconcerned about financial investments that could create an appearance of impropriety.
“He should have known better,” Richard Painter, former White House chief ethics attorney under President George W. Bush and a professor at the University of Minnesota Law School said of Rep. Price’s conduct during the SEC inquiry.
As Rep. Price awaits a Senate vote on his confirmation, Senate Democrats and a number of watchdog groups have asked the SEC to investigate whether Rep. Price engaged in insider trading with some of his trades in health care companies. Rep. Price has said he abided by all ethics rules, although he acknowledged to the Senate Finance Committee that he did not consult the House Ethics Committee on trades that have now become controversial.
The SEC’s inquiry began in 2013, as it battled Ways and Means for documents to develop its case.
A few weeks ago, the day before President Donald Trump’s inauguration, the SEC quietly dropped its pursuit of committee documents without explanation, according to federal court records. No charges were brought against the staffer, Brian Sutter, who is now a health care lobbyist. Sutter’s lawyer declined to comment.
Craig Holman, government affairs lobbyist with Public Citizen, described Rep. Price’s volume of stock trades during the SEC inquiry as “brazen,” given the congressman’s access to nonpublic information affecting the companies’ fortunes.
“The public is seeing this and they really don’t like it,” said Holman, whose watchdog group recently filed complaints about Rep. Price’s stock trading with both the SEC and the Office of Congressional Ethics.
Trump administration officials and Rep. Price have dismissed questions that news reports and lawmakers have raised about stock trades coinciding with official actions to help certain companies, saying Rep. Price’s brokers chose the stocks independently and all of his conduct was transparent.
After acknowledging that he asked his broker to buy stock in an Australian drug company, he told the Senate Finance Committee that he did not direct his broker to make other trades.
“To the best of my knowledge, I have not undertaken such actions,” he wrote in response to finance committee questions. “I have abided by and adhered to all ethics and conflict of interest rules applicable to me.”
An analysis of Rep. Price’s trades shows that he bought health stocks in 2007, the first year Congress financial disclosures are posted online. In 2011, the first year Rep. Price sat on the health subcommittee, he traded no health-related stocks, according to his financial disclosures filed with Congress.
That same year, members were facing public criticism because of a book detailing how they could use inside information and a “60 Minutes” investigation focused on how members and staff could legally use inside information to gain from their own stock trades.
In 2012, President Barack Obama signed the Stop Trading on Congressional Knowledge Act to rein in insider trading by members and require more disclosure. Public watchdog groups suggested at the time that the law would curb the practice.
That year, after his 1-year break in health care trades, Rep. Price resumed investing in health care companies.
Along with investments in technology, financial services, and retail stocks, he also bought and sold stock in companies that could be impacted by actions of his subcommittee, which has a role in determining rates the government pays under the Medicare program.
Health care firms spend heavily to influence members of Congress, lobbying on health matters, funding political campaigns, and seeking favor with Medicare officials who decide how much the program will pay for certain drugs and devices. The Food and Drug Administration holds similar power, approving or putting conditions on drug and device use.
Beyond his personal investments in health care companies, Rep. Price has also advocated their interests in letters to officials and proposed laws, government records show.
In 2012, disclosure records show Rep. Price sold stock in several drug firms, including more than $110,000 worth of Amgen stock. Amgen’s stock price had steadily climbed out of a recession-level slump, but Rep. Price’s sale came a few weeks before the company pleaded guilty to illegally marketing an anemia drug.
By 2013, the health subcommittee was at the center of a major conflict between Medicare, which sets Medicare Advantage rates, and the insurance industry. Medicare issued a notice early that year announcing its intention to reduce Medicare Advantage rates by 2.3 percent as part of a major cost-cutting initiative.
That prompted fierce lobbying by the health insurance industry. Members of Congress, including Rep. Price, wrote a letter to Marilyn Tavenner, then acting administrator for the Centers for Medicare & Medicaid Services, protesting the rate cut, saying the decrease would “disadvantage vulnerable beneficiaries with multiple chronic conditions.”
Ultimately, Medicare decided not to cut rates but instead, to increase them. Yet an hour before Medicare announced the change, a Height Securities analyst fired off a “flash” report to 200 clients that touched off a surge of trading.
The analyst’s report said a political deal was hatched on Capitol Hill to prevent the cuts as a condition for moving forward on Tavenner’s confirmation. Medicare officials increased rates by nearly 4 percent, a change that would positively impact the bottom lines of health insurance companies.
The SEC began looking for the leak’s source, and within weeks, FBI agents began interviewing staffers at the Ways and Means Committee, court records show.
They discovered communications between Sutter and a health care lobbyist. The HHS Inspector General also began a probe, and federal prosecutors briefly examined the matter as well.
As the case unfolded, Rep. Price bought more health care-related stocks, according to his financial disclosures. He has testified that his broker directed all of the trades, except for his investments in Innate Immunotherapeutics, an Australian company partly owned by Rep. Chris Collins (R-N.Y.), according to Collins’ disclosures. An HHS spokesman said Monday that Rep. Price held three broker-directed accounts.
Ethics experts have said that Rep. Price should have further distanced himself by placing his assets in a blind trust.
On April 30, 2013, Rep. Price bought $2,093 worth of stocks in Incyte, a company that develops cancer drugs; $2,076 in Onyx Pharmaceuticals, a drug maker that would soon merge with a larger drug firm; and $2,097 in Parexel International, a consultancy that helps drugs and devices win FDA approval, according to the financial disclosure records.
The same day, Rep. Price shed shares of Express Scripts, a drug management firm, and Danaher, which makes products hospitals and doctor’s offices using for testing and diagnostics. In August of that year, he bought a $2,429 stake in Jazz Pharmaceuticals, which makes sleep and cancer drugs.
On May 6, 2014, the SEC served its first subpoena for the Ways and Means Committee documents. The committee launched a vigorous fight, appealing a federal district judge’s ruling that it should comply with the SEC subpoena.
Rep. Price continued his health stock trades, including $1,000 to $15,000 in drug firms Amgen, Biogen, Bristol-Myers Squibb, Eli Lilly, and Pfizer. He also bought stock in Aetna, a major health insurer, and Athenahealth, which sells electronic medical record and medical billing software. In 2016, he also increased his investment in Innate Immunotherapeutics.
The purchase became controversial because both he and Collins bought stock in a private placement at a discounted price.
“You’re asking for trouble if you have access to nonpublic information about the health care industry and you’re buying and selling health care stocks,” Painter said.
Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation.
Health and Human Services secretary nominee Tom Price showed little restraint in his personal stock trading during the 3 years that federal investigators were bearing down on a key House committee on which the Republican congressman served, a review of his financial disclosures shows.
Rep. Price (Ga.) made dozens of health industry stock trades during a 3-year investigation by the Securities and Exchange Commission that focused on the Ways and Means Committee, according to financial disclosure records he filed with the House of Representatives. The investigation was considered the first test of a law passed to ban members of Congress and their staffs from trading stock based on insider information.
Rep. Price, who is a retired orthopedic surgeon, was never a target of the federal investigation, which scrutinized a top Ways and Means staffer, and no charges were brought. But ethics experts say Rep. Price’s personal trading, even during the thick of federal pressure on his committee, shows he was unconcerned about financial investments that could create an appearance of impropriety.
“He should have known better,” Richard Painter, former White House chief ethics attorney under President George W. Bush and a professor at the University of Minnesota Law School said of Rep. Price’s conduct during the SEC inquiry.
As Rep. Price awaits a Senate vote on his confirmation, Senate Democrats and a number of watchdog groups have asked the SEC to investigate whether Rep. Price engaged in insider trading with some of his trades in health care companies. Rep. Price has said he abided by all ethics rules, although he acknowledged to the Senate Finance Committee that he did not consult the House Ethics Committee on trades that have now become controversial.
The SEC’s inquiry began in 2013, as it battled Ways and Means for documents to develop its case.
A few weeks ago, the day before President Donald Trump’s inauguration, the SEC quietly dropped its pursuit of committee documents without explanation, according to federal court records. No charges were brought against the staffer, Brian Sutter, who is now a health care lobbyist. Sutter’s lawyer declined to comment.
Craig Holman, government affairs lobbyist with Public Citizen, described Rep. Price’s volume of stock trades during the SEC inquiry as “brazen,” given the congressman’s access to nonpublic information affecting the companies’ fortunes.
“The public is seeing this and they really don’t like it,” said Holman, whose watchdog group recently filed complaints about Rep. Price’s stock trading with both the SEC and the Office of Congressional Ethics.
Trump administration officials and Rep. Price have dismissed questions that news reports and lawmakers have raised about stock trades coinciding with official actions to help certain companies, saying Rep. Price’s brokers chose the stocks independently and all of his conduct was transparent.
After acknowledging that he asked his broker to buy stock in an Australian drug company, he told the Senate Finance Committee that he did not direct his broker to make other trades.
“To the best of my knowledge, I have not undertaken such actions,” he wrote in response to finance committee questions. “I have abided by and adhered to all ethics and conflict of interest rules applicable to me.”
An analysis of Rep. Price’s trades shows that he bought health stocks in 2007, the first year Congress financial disclosures are posted online. In 2011, the first year Rep. Price sat on the health subcommittee, he traded no health-related stocks, according to his financial disclosures filed with Congress.
That same year, members were facing public criticism because of a book detailing how they could use inside information and a “60 Minutes” investigation focused on how members and staff could legally use inside information to gain from their own stock trades.
In 2012, President Barack Obama signed the Stop Trading on Congressional Knowledge Act to rein in insider trading by members and require more disclosure. Public watchdog groups suggested at the time that the law would curb the practice.
That year, after his 1-year break in health care trades, Rep. Price resumed investing in health care companies.
Along with investments in technology, financial services, and retail stocks, he also bought and sold stock in companies that could be impacted by actions of his subcommittee, which has a role in determining rates the government pays under the Medicare program.
Health care firms spend heavily to influence members of Congress, lobbying on health matters, funding political campaigns, and seeking favor with Medicare officials who decide how much the program will pay for certain drugs and devices. The Food and Drug Administration holds similar power, approving or putting conditions on drug and device use.
Beyond his personal investments in health care companies, Rep. Price has also advocated their interests in letters to officials and proposed laws, government records show.
In 2012, disclosure records show Rep. Price sold stock in several drug firms, including more than $110,000 worth of Amgen stock. Amgen’s stock price had steadily climbed out of a recession-level slump, but Rep. Price’s sale came a few weeks before the company pleaded guilty to illegally marketing an anemia drug.
By 2013, the health subcommittee was at the center of a major conflict between Medicare, which sets Medicare Advantage rates, and the insurance industry. Medicare issued a notice early that year announcing its intention to reduce Medicare Advantage rates by 2.3 percent as part of a major cost-cutting initiative.
That prompted fierce lobbying by the health insurance industry. Members of Congress, including Rep. Price, wrote a letter to Marilyn Tavenner, then acting administrator for the Centers for Medicare & Medicaid Services, protesting the rate cut, saying the decrease would “disadvantage vulnerable beneficiaries with multiple chronic conditions.”
Ultimately, Medicare decided not to cut rates but instead, to increase them. Yet an hour before Medicare announced the change, a Height Securities analyst fired off a “flash” report to 200 clients that touched off a surge of trading.
The analyst’s report said a political deal was hatched on Capitol Hill to prevent the cuts as a condition for moving forward on Tavenner’s confirmation. Medicare officials increased rates by nearly 4 percent, a change that would positively impact the bottom lines of health insurance companies.
The SEC began looking for the leak’s source, and within weeks, FBI agents began interviewing staffers at the Ways and Means Committee, court records show.
They discovered communications between Sutter and a health care lobbyist. The HHS Inspector General also began a probe, and federal prosecutors briefly examined the matter as well.
As the case unfolded, Rep. Price bought more health care-related stocks, according to his financial disclosures. He has testified that his broker directed all of the trades, except for his investments in Innate Immunotherapeutics, an Australian company partly owned by Rep. Chris Collins (R-N.Y.), according to Collins’ disclosures. An HHS spokesman said Monday that Rep. Price held three broker-directed accounts.
Ethics experts have said that Rep. Price should have further distanced himself by placing his assets in a blind trust.
On April 30, 2013, Rep. Price bought $2,093 worth of stocks in Incyte, a company that develops cancer drugs; $2,076 in Onyx Pharmaceuticals, a drug maker that would soon merge with a larger drug firm; and $2,097 in Parexel International, a consultancy that helps drugs and devices win FDA approval, according to the financial disclosure records.
The same day, Rep. Price shed shares of Express Scripts, a drug management firm, and Danaher, which makes products hospitals and doctor’s offices using for testing and diagnostics. In August of that year, he bought a $2,429 stake in Jazz Pharmaceuticals, which makes sleep and cancer drugs.
On May 6, 2014, the SEC served its first subpoena for the Ways and Means Committee documents. The committee launched a vigorous fight, appealing a federal district judge’s ruling that it should comply with the SEC subpoena.
Rep. Price continued his health stock trades, including $1,000 to $15,000 in drug firms Amgen, Biogen, Bristol-Myers Squibb, Eli Lilly, and Pfizer. He also bought stock in Aetna, a major health insurer, and Athenahealth, which sells electronic medical record and medical billing software. In 2016, he also increased his investment in Innate Immunotherapeutics.
The purchase became controversial because both he and Collins bought stock in a private placement at a discounted price.
“You’re asking for trouble if you have access to nonpublic information about the health care industry and you’re buying and selling health care stocks,” Painter said.
Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation.
Heart disease risk soars in young adults with coronary calcium
Younger adults who have any calcium deposited in their coronary arteries, even a small amount, are at increased risk for adverse coronary heart disease (CHD) outcomes and death, finds an analysis of the Coronary Artery Risk Development in Young Adults (CARDIA) Study.
There’s no evidence, however, that treating such patients would make a difference in outcomes, John Jeffrey Carr, MD, reported in JAMA Cardiology on Feb. 8.
In the prospective, community-based, cohort study, 5,115 black and white adults underwent coronary computed tomographic (CT) imaging between the ages of 32 and 46 years, and had a mean follow-up of 12.5 years.
Compared with counterparts not having any coronary artery calcium (CAC), those having at least some had a 5.0-fold increased risk of CHD events and a 1.6-fold increased risk of death (JAMA Cardiol. 2017 Feb 8; doi: 10.1001/jamacardio.2016.5493).
Estimates suggested that identification of individuals at elevated risk for developing CAC could inform a selective CT screening strategy whereby the number of younger adults screened could be reduced by half, and the number needing to be imaged to find one person with CAC could be reduced from 3.5 to 2.2.
“The finding that CAC present by ages 32-46 years is associated with increased risk of premature CHD and death emphasizes the need for reduction of risk factors and primordial prevention beginning in early life,” wrote Dr. Carr, professor radiology at Vanderbilt University in Nashville, Tenn.
“Whether any kind of general screening for CAC is warranted needs further study, although we suggest that a strategy in which all individuals aged 32 to 46 years are screened is not indicated. Rather, a more targeted approach based on measuring risk factors in early adult life to predict individuals at high risk for developing CAC in whom the CT scan would have the greatest value can be considered,” they propose.
Study details
Participants were recruited to CARDIA when aged 18-30 years, and they underwent CAC measurement at 15, 20, and 25 years after recruitment. Incident events were ascertained starting from the time of the year-15 scan.
At that year-15 scan, 10.2% of participants were found to have CAC. The geometric mean Agatston score was 21.6.
In adjusted analyses, participants with any CAC had sharply higher risks of CHD events (hazard ratio, 5.0), as well as cardiovascular disease events (HR, 3.0). The risk of CHD events increased with CAC score, with hazard ratios of 2.6, 5.8, and 9.8 for individuals with scores of 1-19, 20-99, and 100 or more, respectively.
In addition, participants with any CAC had an elevated adjusted risk of all-cause mortality (HR, 1.6). This risk similarly rose with score but was significant for those having a score of 100 or greater only (hazard ratio, 3.7); the large majority of deaths in this group were deemed to be from CHD events.
The model that the investigators developed predicted the probability of CAC by ages 32-56 years based on risk factors assessed 7 years apart, between the ages of 18 and 38 years.
When stratified by this model, 4.2% of study participants falling into the lowest-risk decile had CAC, compared with 67.8% of those falling into the highest-risk decile.
Analyses suggested that if screening were restricted to those participants having an above-median risk score, fully 77.3% of all those with coronary calcium and 95.5% of all those with CHD events would be identified. Moreover, these yields would be obtained while reducing the number of individuals recommended to be screened by 50.0%.
Several challenges will need to be addressed before computed tomographic (CT) screening of younger adults for coronary artery calcium is ready for prime time.
First, such screenings must be efficient, and the investigator’s new model seems to be a step in this direction.
The model should be further validated in other populations as well as across younger populations (i.e., during the first CAC test, when the age of the cohort was aged 32-46 years) to help substantiate whether testing of younger individuals is efficient or if waiting to screen those who are older than 40-45 years may be preferable.
Second, even if coronary calcium is detected in young adults, individuals’ risk may not be sufficiently elevated to justify long-term statin therapy.
Finally, there are no data in this context to show that intervening with statins improves cardiovascular outcomes. The absence of such data, and consequently the fact that treatment is often not started until later in life, is owing to the economic and ethical considerations of performing a trial that would take decades to conduct.
In the meantime, the study’s findings have implications for care in younger adults who are found to have coronary calcium incidentally and underscore the importance of primordial prevention.
Future studies will be needed to refine our approaches to better select appropriate candidates for CAC testing, even more so in younger than in older individuals.
Ron Blankstein, MD, of Harvard University, Boston, and Philip Greenland, MD, of Northwestern University, Chicago, made these comments in an accompanying editorial (JAMA Cardiol. 2017 Feb 8; doi: 10.1001/jamacardio.2016.5552). They reported having no relevant financial disclosures.
Several challenges will need to be addressed before computed tomographic (CT) screening of younger adults for coronary artery calcium is ready for prime time.
First, such screenings must be efficient, and the investigator’s new model seems to be a step in this direction.
The model should be further validated in other populations as well as across younger populations (i.e., during the first CAC test, when the age of the cohort was aged 32-46 years) to help substantiate whether testing of younger individuals is efficient or if waiting to screen those who are older than 40-45 years may be preferable.
Second, even if coronary calcium is detected in young adults, individuals’ risk may not be sufficiently elevated to justify long-term statin therapy.
Finally, there are no data in this context to show that intervening with statins improves cardiovascular outcomes. The absence of such data, and consequently the fact that treatment is often not started until later in life, is owing to the economic and ethical considerations of performing a trial that would take decades to conduct.
In the meantime, the study’s findings have implications for care in younger adults who are found to have coronary calcium incidentally and underscore the importance of primordial prevention.
Future studies will be needed to refine our approaches to better select appropriate candidates for CAC testing, even more so in younger than in older individuals.
Ron Blankstein, MD, of Harvard University, Boston, and Philip Greenland, MD, of Northwestern University, Chicago, made these comments in an accompanying editorial (JAMA Cardiol. 2017 Feb 8; doi: 10.1001/jamacardio.2016.5552). They reported having no relevant financial disclosures.
Several challenges will need to be addressed before computed tomographic (CT) screening of younger adults for coronary artery calcium is ready for prime time.
First, such screenings must be efficient, and the investigator’s new model seems to be a step in this direction.
The model should be further validated in other populations as well as across younger populations (i.e., during the first CAC test, when the age of the cohort was aged 32-46 years) to help substantiate whether testing of younger individuals is efficient or if waiting to screen those who are older than 40-45 years may be preferable.
Second, even if coronary calcium is detected in young adults, individuals’ risk may not be sufficiently elevated to justify long-term statin therapy.
Finally, there are no data in this context to show that intervening with statins improves cardiovascular outcomes. The absence of such data, and consequently the fact that treatment is often not started until later in life, is owing to the economic and ethical considerations of performing a trial that would take decades to conduct.
In the meantime, the study’s findings have implications for care in younger adults who are found to have coronary calcium incidentally and underscore the importance of primordial prevention.
Future studies will be needed to refine our approaches to better select appropriate candidates for CAC testing, even more so in younger than in older individuals.
Ron Blankstein, MD, of Harvard University, Boston, and Philip Greenland, MD, of Northwestern University, Chicago, made these comments in an accompanying editorial (JAMA Cardiol. 2017 Feb 8; doi: 10.1001/jamacardio.2016.5552). They reported having no relevant financial disclosures.
Younger adults who have any calcium deposited in their coronary arteries, even a small amount, are at increased risk for adverse coronary heart disease (CHD) outcomes and death, finds an analysis of the Coronary Artery Risk Development in Young Adults (CARDIA) Study.
There’s no evidence, however, that treating such patients would make a difference in outcomes, John Jeffrey Carr, MD, reported in JAMA Cardiology on Feb. 8.
In the prospective, community-based, cohort study, 5,115 black and white adults underwent coronary computed tomographic (CT) imaging between the ages of 32 and 46 years, and had a mean follow-up of 12.5 years.
Compared with counterparts not having any coronary artery calcium (CAC), those having at least some had a 5.0-fold increased risk of CHD events and a 1.6-fold increased risk of death (JAMA Cardiol. 2017 Feb 8; doi: 10.1001/jamacardio.2016.5493).
Estimates suggested that identification of individuals at elevated risk for developing CAC could inform a selective CT screening strategy whereby the number of younger adults screened could be reduced by half, and the number needing to be imaged to find one person with CAC could be reduced from 3.5 to 2.2.
“The finding that CAC present by ages 32-46 years is associated with increased risk of premature CHD and death emphasizes the need for reduction of risk factors and primordial prevention beginning in early life,” wrote Dr. Carr, professor radiology at Vanderbilt University in Nashville, Tenn.
“Whether any kind of general screening for CAC is warranted needs further study, although we suggest that a strategy in which all individuals aged 32 to 46 years are screened is not indicated. Rather, a more targeted approach based on measuring risk factors in early adult life to predict individuals at high risk for developing CAC in whom the CT scan would have the greatest value can be considered,” they propose.
Study details
Participants were recruited to CARDIA when aged 18-30 years, and they underwent CAC measurement at 15, 20, and 25 years after recruitment. Incident events were ascertained starting from the time of the year-15 scan.
At that year-15 scan, 10.2% of participants were found to have CAC. The geometric mean Agatston score was 21.6.
In adjusted analyses, participants with any CAC had sharply higher risks of CHD events (hazard ratio, 5.0), as well as cardiovascular disease events (HR, 3.0). The risk of CHD events increased with CAC score, with hazard ratios of 2.6, 5.8, and 9.8 for individuals with scores of 1-19, 20-99, and 100 or more, respectively.
In addition, participants with any CAC had an elevated adjusted risk of all-cause mortality (HR, 1.6). This risk similarly rose with score but was significant for those having a score of 100 or greater only (hazard ratio, 3.7); the large majority of deaths in this group were deemed to be from CHD events.
The model that the investigators developed predicted the probability of CAC by ages 32-56 years based on risk factors assessed 7 years apart, between the ages of 18 and 38 years.
When stratified by this model, 4.2% of study participants falling into the lowest-risk decile had CAC, compared with 67.8% of those falling into the highest-risk decile.
Analyses suggested that if screening were restricted to those participants having an above-median risk score, fully 77.3% of all those with coronary calcium and 95.5% of all those with CHD events would be identified. Moreover, these yields would be obtained while reducing the number of individuals recommended to be screened by 50.0%.
Younger adults who have any calcium deposited in their coronary arteries, even a small amount, are at increased risk for adverse coronary heart disease (CHD) outcomes and death, finds an analysis of the Coronary Artery Risk Development in Young Adults (CARDIA) Study.
There’s no evidence, however, that treating such patients would make a difference in outcomes, John Jeffrey Carr, MD, reported in JAMA Cardiology on Feb. 8.
In the prospective, community-based, cohort study, 5,115 black and white adults underwent coronary computed tomographic (CT) imaging between the ages of 32 and 46 years, and had a mean follow-up of 12.5 years.
Compared with counterparts not having any coronary artery calcium (CAC), those having at least some had a 5.0-fold increased risk of CHD events and a 1.6-fold increased risk of death (JAMA Cardiol. 2017 Feb 8; doi: 10.1001/jamacardio.2016.5493).
Estimates suggested that identification of individuals at elevated risk for developing CAC could inform a selective CT screening strategy whereby the number of younger adults screened could be reduced by half, and the number needing to be imaged to find one person with CAC could be reduced from 3.5 to 2.2.
“The finding that CAC present by ages 32-46 years is associated with increased risk of premature CHD and death emphasizes the need for reduction of risk factors and primordial prevention beginning in early life,” wrote Dr. Carr, professor radiology at Vanderbilt University in Nashville, Tenn.
“Whether any kind of general screening for CAC is warranted needs further study, although we suggest that a strategy in which all individuals aged 32 to 46 years are screened is not indicated. Rather, a more targeted approach based on measuring risk factors in early adult life to predict individuals at high risk for developing CAC in whom the CT scan would have the greatest value can be considered,” they propose.
Study details
Participants were recruited to CARDIA when aged 18-30 years, and they underwent CAC measurement at 15, 20, and 25 years after recruitment. Incident events were ascertained starting from the time of the year-15 scan.
At that year-15 scan, 10.2% of participants were found to have CAC. The geometric mean Agatston score was 21.6.
In adjusted analyses, participants with any CAC had sharply higher risks of CHD events (hazard ratio, 5.0), as well as cardiovascular disease events (HR, 3.0). The risk of CHD events increased with CAC score, with hazard ratios of 2.6, 5.8, and 9.8 for individuals with scores of 1-19, 20-99, and 100 or more, respectively.
In addition, participants with any CAC had an elevated adjusted risk of all-cause mortality (HR, 1.6). This risk similarly rose with score but was significant for those having a score of 100 or greater only (hazard ratio, 3.7); the large majority of deaths in this group were deemed to be from CHD events.
The model that the investigators developed predicted the probability of CAC by ages 32-56 years based on risk factors assessed 7 years apart, between the ages of 18 and 38 years.
When stratified by this model, 4.2% of study participants falling into the lowest-risk decile had CAC, compared with 67.8% of those falling into the highest-risk decile.
Analyses suggested that if screening were restricted to those participants having an above-median risk score, fully 77.3% of all those with coronary calcium and 95.5% of all those with CHD events would be identified. Moreover, these yields would be obtained while reducing the number of individuals recommended to be screened by 50.0%.
FROM JAMA CARDIOLOGY
Key clinical point:
Major finding: Individuals having any versus no coronary artery calcium when aged 32-46 years had elevated risks of CHD events (HR, 5.0) and death (HR, 1.6) by the age of 58 years.
Data source: A prospective community-based cohort study of 5,115 black and white adults (CARDIA Study).
Disclosures: Dr. Carr disclosed that he had no relevant conflicts of interest.