User login
Refining SLE cardiovascular risk estimation
WASHINGTON – Red blood cell distribution width provides a novel tool for cardiovascular risk stratification in patients with systemic lupus erythematosus (SLE), Chang H. Kim, MD, reported at the annual meeting of the American College of Cardiology.
In a retrospective cohort study of nearly 70,000 patients with SLE, the 10-year rate of major adverse cardiovascular events (MACE) rose stepwise according to quintile of red cell distribution width (RDW) from 5.3% in patients with an RDW of 13.2% or less to 38.6% in those with an RDW of 15.8% or greater, according to Dr. Kim of Case Western Reserve University in Cleveland.
He utilized the Explorys database to determine the 10-year cumulative incidence of MACE – defined as acute MI, heart failure, or cerebrovascular accident – during 2007-2016 in 69,920 patients with SLE and 14,825,240 controls. Explorys is an 8-year-old Cleveland-based company that maintains a health care database incorporating 26 health care systems across the United States with nearly 50 million patients. It is part of IBM Watson Health.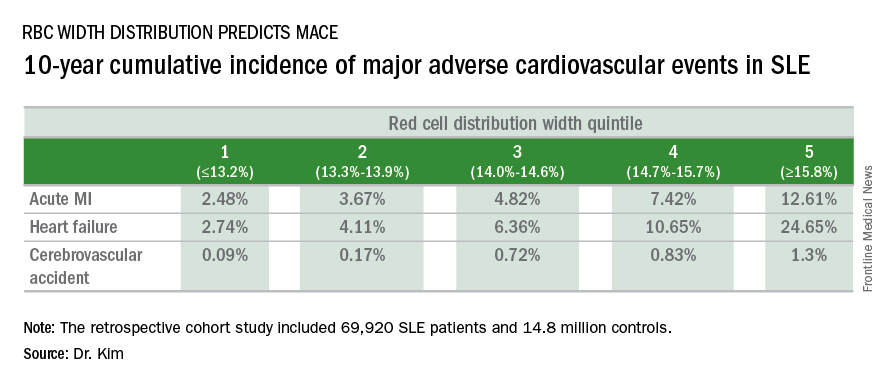
The MACE rate in patients with SLE displayed a graded increase in association with RDW quintile as measured in a routine CBC. (See table.) MACE rates were significantly higher in male than female SLE patients, but the graded relationship between RDW quintile and 10-year incidence of MACE persisted after adjustment for gender and the presence of anemia.
A graded association between RDW quintile and MACE also was noted in the control group of nearly 15 million individuals, but the absolute incidence of MACE in the non-SLE controls was far lower.
Dr. Kim reported having no financial conflicts regarding this unfunded study.
WASHINGTON – Red blood cell distribution width provides a novel tool for cardiovascular risk stratification in patients with systemic lupus erythematosus (SLE), Chang H. Kim, MD, reported at the annual meeting of the American College of Cardiology.
In a retrospective cohort study of nearly 70,000 patients with SLE, the 10-year rate of major adverse cardiovascular events (MACE) rose stepwise according to quintile of red cell distribution width (RDW) from 5.3% in patients with an RDW of 13.2% or less to 38.6% in those with an RDW of 15.8% or greater, according to Dr. Kim of Case Western Reserve University in Cleveland.
He utilized the Explorys database to determine the 10-year cumulative incidence of MACE – defined as acute MI, heart failure, or cerebrovascular accident – during 2007-2016 in 69,920 patients with SLE and 14,825,240 controls. Explorys is an 8-year-old Cleveland-based company that maintains a health care database incorporating 26 health care systems across the United States with nearly 50 million patients. It is part of IBM Watson Health.
The MACE rate in patients with SLE displayed a graded increase in association with RDW quintile as measured in a routine CBC. (See table.) MACE rates were significantly higher in male than female SLE patients, but the graded relationship between RDW quintile and 10-year incidence of MACE persisted after adjustment for gender and the presence of anemia.
A graded association between RDW quintile and MACE also was noted in the control group of nearly 15 million individuals, but the absolute incidence of MACE in the non-SLE controls was far lower.
Dr. Kim reported having no financial conflicts regarding this unfunded study.
WASHINGTON – Red blood cell distribution width provides a novel tool for cardiovascular risk stratification in patients with systemic lupus erythematosus (SLE), Chang H. Kim, MD, reported at the annual meeting of the American College of Cardiology.
In a retrospective cohort study of nearly 70,000 patients with SLE, the 10-year rate of major adverse cardiovascular events (MACE) rose stepwise according to quintile of red cell distribution width (RDW) from 5.3% in patients with an RDW of 13.2% or less to 38.6% in those with an RDW of 15.8% or greater, according to Dr. Kim of Case Western Reserve University in Cleveland.
He utilized the Explorys database to determine the 10-year cumulative incidence of MACE – defined as acute MI, heart failure, or cerebrovascular accident – during 2007-2016 in 69,920 patients with SLE and 14,825,240 controls. Explorys is an 8-year-old Cleveland-based company that maintains a health care database incorporating 26 health care systems across the United States with nearly 50 million patients. It is part of IBM Watson Health.
The MACE rate in patients with SLE displayed a graded increase in association with RDW quintile as measured in a routine CBC. (See table.) MACE rates were significantly higher in male than female SLE patients, but the graded relationship between RDW quintile and 10-year incidence of MACE persisted after adjustment for gender and the presence of anemia.
A graded association between RDW quintile and MACE also was noted in the control group of nearly 15 million individuals, but the absolute incidence of MACE in the non-SLE controls was far lower.
Dr. Kim reported having no financial conflicts regarding this unfunded study.
AT ACC 2017
Key clinical point:
Major finding: Systemic lupus erythematosus patients in the top quintile of RBC distribution width had a 10-year incidence of major adverse cardiovascular events of 39%.
Data source: This retrospective cohort study included nearly 70,000 SLE patients and 14.8 million controls.
Disclosures: The presenter reported no financial conflicts with regard to this unfunded study.
Parents rate telepediatric experience as better than office visits
Three-quarters of parents who have used telemedicine services for their children say the experience was better than an in-person office visit, according to a new study.
The analysis, released April 23, 2017, by Nemours Children’s Health System operating in five states, surveyed 500 child caregivers between February 15 and 20 about their awareness and usage of telemedicine services for their children aged 17 years and younger (mean age 10 years). Of caregivers surveyed, 15% had tried telemedicine services for their children, while another 61% reported they plan to use telemedicine in the next year for their children’s medical care, according to the study. Eight percent of caregivers did not plan to use telemedicine, and 31% of those surveyed were unsure.
Of child caregivers who have used telemedicine, 75% of reported their telemedicine experience was better than an in-person visit, according to the survey. Convenience, after-hours accessibility, and immediacy were the top three reasons caregivers sought an online visit for their child. Compared with a similar survey conducted by Nemours in 2014, parents’ use of telemedicine for their children has grown by 125% and awareness of the practice has increased by 88%, the study found.
The overall takeaway is that parents are using telemedicine because of its ease and accessibility and they’re having satisfactory experiences with the technology, said Shayan Vyas, MD, a pediatrician based in Nemours’ Orlando location and director of telemedicine for Nemours Children’s Hospital.

“Parents are citing telemedicine because of convenience and that’s true for other industries that have gone mobile,” Dr. Vyas said in an interview. “We no longer hail a cab at the intersection, we use our smartphones. We now use Amazon and other companies to order diapers and other everyday items. ... More and more, patients are driving telemedicine more than the systems or the providers.”
Caregivers surveyed said they are most willing to use telehealth services for cold and flu (58%), pink eye (51%), and rashes (48%) as well as well-child visits (41%). However, parents and caregivers were hesitant to use telemedicine for chronic conditions and reported they would likely never consider telehealth services for diabetes (53%), asthma (43%), ear pain (37%), and ADHD (36%).
The findings are expected because there are limitations for providers when it comes to treating some conditions and chronic diseases with telemedicine, Dr. Vyas said. He also stressed that telemedicine should not be used to replace the medical home for children.
“We’re working really hard to ensure there’s great pediatric care online and that the medical home stays intact,” he said. “We don’t want telemedicine to become the wild wild west of retail clinics where children are getting care from providers they don’t know or who are not in touch with their primary care world.”
Nemours has incorporated telemedicine throughout its health system with direct-to-consumer care for acute, chronic, and postsurgical appointments. The telehealth program, called Nemours CareConnect, is a 24/7, on-demand pediatric service that provides access to Nemours pediatricians through smartphones, tablets, or computers. In addition, Nemours CareConnect is used to bring pediatric specialists into affiliated community hospitals.
“At Nemours, we’ve seen how telemedicine can positively impact patients’ lives,” Dr. Vyas said. “The overwhelmingly positive response we’ve seen from parents who are early adopters of telemedicine really reinforces the feasibility of online doctor visits and sets the stage for real change in the way health care is delivered.”
[email protected]
On Twitter @legal_med
Three-quarters of parents who have used telemedicine services for their children say the experience was better than an in-person office visit, according to a new study.
The analysis, released April 23, 2017, by Nemours Children’s Health System operating in five states, surveyed 500 child caregivers between February 15 and 20 about their awareness and usage of telemedicine services for their children aged 17 years and younger (mean age 10 years). Of caregivers surveyed, 15% had tried telemedicine services for their children, while another 61% reported they plan to use telemedicine in the next year for their children’s medical care, according to the study. Eight percent of caregivers did not plan to use telemedicine, and 31% of those surveyed were unsure.
Of child caregivers who have used telemedicine, 75% of reported their telemedicine experience was better than an in-person visit, according to the survey. Convenience, after-hours accessibility, and immediacy were the top three reasons caregivers sought an online visit for their child. Compared with a similar survey conducted by Nemours in 2014, parents’ use of telemedicine for their children has grown by 125% and awareness of the practice has increased by 88%, the study found.
The overall takeaway is that parents are using telemedicine because of its ease and accessibility and they’re having satisfactory experiences with the technology, said Shayan Vyas, MD, a pediatrician based in Nemours’ Orlando location and director of telemedicine for Nemours Children’s Hospital.
“Parents are citing telemedicine because of convenience and that’s true for other industries that have gone mobile,” Dr. Vyas said in an interview. “We no longer hail a cab at the intersection, we use our smartphones. We now use Amazon and other companies to order diapers and other everyday items. ... More and more, patients are driving telemedicine more than the systems or the providers.”
Caregivers surveyed said they are most willing to use telehealth services for cold and flu (58%), pink eye (51%), and rashes (48%) as well as well-child visits (41%). However, parents and caregivers were hesitant to use telemedicine for chronic conditions and reported they would likely never consider telehealth services for diabetes (53%), asthma (43%), ear pain (37%), and ADHD (36%).
The findings are expected because there are limitations for providers when it comes to treating some conditions and chronic diseases with telemedicine, Dr. Vyas said. He also stressed that telemedicine should not be used to replace the medical home for children.
“We’re working really hard to ensure there’s great pediatric care online and that the medical home stays intact,” he said. “We don’t want telemedicine to become the wild wild west of retail clinics where children are getting care from providers they don’t know or who are not in touch with their primary care world.”
Nemours has incorporated telemedicine throughout its health system with direct-to-consumer care for acute, chronic, and postsurgical appointments. The telehealth program, called Nemours CareConnect, is a 24/7, on-demand pediatric service that provides access to Nemours pediatricians through smartphones, tablets, or computers. In addition, Nemours CareConnect is used to bring pediatric specialists into affiliated community hospitals.
“At Nemours, we’ve seen how telemedicine can positively impact patients’ lives,” Dr. Vyas said. “The overwhelmingly positive response we’ve seen from parents who are early adopters of telemedicine really reinforces the feasibility of online doctor visits and sets the stage for real change in the way health care is delivered.”
[email protected]
On Twitter @legal_med
Three-quarters of parents who have used telemedicine services for their children say the experience was better than an in-person office visit, according to a new study.
The analysis, released April 23, 2017, by Nemours Children’s Health System operating in five states, surveyed 500 child caregivers between February 15 and 20 about their awareness and usage of telemedicine services for their children aged 17 years and younger (mean age 10 years). Of caregivers surveyed, 15% had tried telemedicine services for their children, while another 61% reported they plan to use telemedicine in the next year for their children’s medical care, according to the study. Eight percent of caregivers did not plan to use telemedicine, and 31% of those surveyed were unsure.
Of child caregivers who have used telemedicine, 75% of reported their telemedicine experience was better than an in-person visit, according to the survey. Convenience, after-hours accessibility, and immediacy were the top three reasons caregivers sought an online visit for their child. Compared with a similar survey conducted by Nemours in 2014, parents’ use of telemedicine for their children has grown by 125% and awareness of the practice has increased by 88%, the study found.
The overall takeaway is that parents are using telemedicine because of its ease and accessibility and they’re having satisfactory experiences with the technology, said Shayan Vyas, MD, a pediatrician based in Nemours’ Orlando location and director of telemedicine for Nemours Children’s Hospital.
“Parents are citing telemedicine because of convenience and that’s true for other industries that have gone mobile,” Dr. Vyas said in an interview. “We no longer hail a cab at the intersection, we use our smartphones. We now use Amazon and other companies to order diapers and other everyday items. ... More and more, patients are driving telemedicine more than the systems or the providers.”
Caregivers surveyed said they are most willing to use telehealth services for cold and flu (58%), pink eye (51%), and rashes (48%) as well as well-child visits (41%). However, parents and caregivers were hesitant to use telemedicine for chronic conditions and reported they would likely never consider telehealth services for diabetes (53%), asthma (43%), ear pain (37%), and ADHD (36%).
The findings are expected because there are limitations for providers when it comes to treating some conditions and chronic diseases with telemedicine, Dr. Vyas said. He also stressed that telemedicine should not be used to replace the medical home for children.
“We’re working really hard to ensure there’s great pediatric care online and that the medical home stays intact,” he said. “We don’t want telemedicine to become the wild wild west of retail clinics where children are getting care from providers they don’t know or who are not in touch with their primary care world.”
Nemours has incorporated telemedicine throughout its health system with direct-to-consumer care for acute, chronic, and postsurgical appointments. The telehealth program, called Nemours CareConnect, is a 24/7, on-demand pediatric service that provides access to Nemours pediatricians through smartphones, tablets, or computers. In addition, Nemours CareConnect is used to bring pediatric specialists into affiliated community hospitals.
“At Nemours, we’ve seen how telemedicine can positively impact patients’ lives,” Dr. Vyas said. “The overwhelmingly positive response we’ve seen from parents who are early adopters of telemedicine really reinforces the feasibility of online doctor visits and sets the stage for real change in the way health care is delivered.”
[email protected]
On Twitter @legal_med
Parkinson’s patients’ quality of life improves after deep brain stimulation
BOSTON– Early results from an industry-funded registry of Parkinson’s disease patients who underwent deep brain stimulation (DBS) reveal that their quality-of-life scores grew by 22% at 6 months.
Patients, their caregivers, and their clinicians all overwhelmingly reported improvement.
The study, which examined two types of technology that are not approved in the United States, aims to fill a gap in DBS research: How do patients fare in normal conditions – “real life” – outside of clinical trials?
While DBS has been widely used for many years, “much of the available information is from controlled trials that usually select the best possible patients – relatively young and in the condition to go through a clinical trial. There’s less information about people who may not be in the best possible shape,” said Michele Tagliati, MD, professor and director of movement disorders at Cedars-Sinai Medical Center in Los Angeles. He did not take part in the study but is familiar with its findings.
The study’s lead author, Günther Deuschl, MD, PhD, of University Hospital Schleswig-Holstein, released preliminary findings at the annual meeting of the American Academy of Neurology.
The researchers have enrolled 203 patients who were treated with Boston Scientific’s Vercise DBS system, which is not approved for use in the United States. The researchers plan to track patients for 3 years.
“This is the first such industry-sponsored registry, which addresses a need in the field to track DBS practice and outcomes across multiple centers,” said Mark Richardson, MD, PhD, associate professor and director of epilepsy and movement disorders surgery at the University of Pittsburgh Medical Center. He did not take part in the study but is familiar with its findings.
The average age of participants is 59 years, which Dr. Deuschl said is a bit younger than many other studies, and 69% are male. Eighty-five serious adverse events were reported in 52 patients; 57 were not linked to the procedure. One patient died of a surgery-related hematoma.
At 6 months, patients reported a 22% improvement (P less than .0001) on the Parkinson’s Disease Questionnaire (PDQ-39), Dr. Deuschl said, and that level was sustained at 1 year.
That level of improvement is significant, Dr. Richardson said in an interview.
“Previous randomized, controlled trials have shown that patients who are candidates for DBS but who continue medical management alone are most likely to have no improvement at all on any quality of life measures,” he said. In addition, “6 months is fairly short, and some patients likely have not reached stable stimulation programming and effectiveness.”
The researchers also reported that more than 90% of patients, their clinicians, and their caregivers reported improvement.
This kind of study is valuable in light of skepticism about DBS, which is used to treat patients who do not respond to medication, Dr. Tagliati noted.
“Despite 15 years of [Food and Drug Administration] approval, there is still some form of resistance out there in referring patients with Parkinson’s at the right time when they can still fully benefit from the procedure,” he said. “Registries have this potential great benefit in terms of awareness and reassuring people.”
As for the high rating of improvement, Dr. Tagliati said it reflects what he sees in the clinic.
“Barring complications, we have very substantial satisfaction in our patients, definitely in the short term after DBS,” he said. “Over the long term, the picture is more difficult to read.”
Dr. Deuschl said researchers would like to add hundreds more patients to the study, and they hope to gain data about the differences between the results from standard and directional-lead DBS systems.
Boston Scientific funded the study, and some of the authors work for the company.
Dr. Richardson reported receiving research grant funding from Medtronic. Dr. Tagliati reported receiving funding from Abbott, Boston Scientific, Medtronic, and all DBS manufacturers, and his clinic is an investigational center for the Vercise DBS system. Dr. Deuschl reported receiving consulting fees from Boston Scientific, grant funding from Medtronic, lecture fees from Almirall and Novartis, and royalties from Thieme Publishers.
BOSTON– Early results from an industry-funded registry of Parkinson’s disease patients who underwent deep brain stimulation (DBS) reveal that their quality-of-life scores grew by 22% at 6 months.
Patients, their caregivers, and their clinicians all overwhelmingly reported improvement.
The study, which examined two types of technology that are not approved in the United States, aims to fill a gap in DBS research: How do patients fare in normal conditions – “real life” – outside of clinical trials?
While DBS has been widely used for many years, “much of the available information is from controlled trials that usually select the best possible patients – relatively young and in the condition to go through a clinical trial. There’s less information about people who may not be in the best possible shape,” said Michele Tagliati, MD, professor and director of movement disorders at Cedars-Sinai Medical Center in Los Angeles. He did not take part in the study but is familiar with its findings.
The study’s lead author, Günther Deuschl, MD, PhD, of University Hospital Schleswig-Holstein, released preliminary findings at the annual meeting of the American Academy of Neurology.
The researchers have enrolled 203 patients who were treated with Boston Scientific’s Vercise DBS system, which is not approved for use in the United States. The researchers plan to track patients for 3 years.
“This is the first such industry-sponsored registry, which addresses a need in the field to track DBS practice and outcomes across multiple centers,” said Mark Richardson, MD, PhD, associate professor and director of epilepsy and movement disorders surgery at the University of Pittsburgh Medical Center. He did not take part in the study but is familiar with its findings.
The average age of participants is 59 years, which Dr. Deuschl said is a bit younger than many other studies, and 69% are male. Eighty-five serious adverse events were reported in 52 patients; 57 were not linked to the procedure. One patient died of a surgery-related hematoma.
At 6 months, patients reported a 22% improvement (P less than .0001) on the Parkinson’s Disease Questionnaire (PDQ-39), Dr. Deuschl said, and that level was sustained at 1 year.
That level of improvement is significant, Dr. Richardson said in an interview.
“Previous randomized, controlled trials have shown that patients who are candidates for DBS but who continue medical management alone are most likely to have no improvement at all on any quality of life measures,” he said. In addition, “6 months is fairly short, and some patients likely have not reached stable stimulation programming and effectiveness.”
The researchers also reported that more than 90% of patients, their clinicians, and their caregivers reported improvement.
This kind of study is valuable in light of skepticism about DBS, which is used to treat patients who do not respond to medication, Dr. Tagliati noted.
“Despite 15 years of [Food and Drug Administration] approval, there is still some form of resistance out there in referring patients with Parkinson’s at the right time when they can still fully benefit from the procedure,” he said. “Registries have this potential great benefit in terms of awareness and reassuring people.”
As for the high rating of improvement, Dr. Tagliati said it reflects what he sees in the clinic.
“Barring complications, we have very substantial satisfaction in our patients, definitely in the short term after DBS,” he said. “Over the long term, the picture is more difficult to read.”
Dr. Deuschl said researchers would like to add hundreds more patients to the study, and they hope to gain data about the differences between the results from standard and directional-lead DBS systems.
Boston Scientific funded the study, and some of the authors work for the company.
Dr. Richardson reported receiving research grant funding from Medtronic. Dr. Tagliati reported receiving funding from Abbott, Boston Scientific, Medtronic, and all DBS manufacturers, and his clinic is an investigational center for the Vercise DBS system. Dr. Deuschl reported receiving consulting fees from Boston Scientific, grant funding from Medtronic, lecture fees from Almirall and Novartis, and royalties from Thieme Publishers.
BOSTON– Early results from an industry-funded registry of Parkinson’s disease patients who underwent deep brain stimulation (DBS) reveal that their quality-of-life scores grew by 22% at 6 months.
Patients, their caregivers, and their clinicians all overwhelmingly reported improvement.
The study, which examined two types of technology that are not approved in the United States, aims to fill a gap in DBS research: How do patients fare in normal conditions – “real life” – outside of clinical trials?
While DBS has been widely used for many years, “much of the available information is from controlled trials that usually select the best possible patients – relatively young and in the condition to go through a clinical trial. There’s less information about people who may not be in the best possible shape,” said Michele Tagliati, MD, professor and director of movement disorders at Cedars-Sinai Medical Center in Los Angeles. He did not take part in the study but is familiar with its findings.
The study’s lead author, Günther Deuschl, MD, PhD, of University Hospital Schleswig-Holstein, released preliminary findings at the annual meeting of the American Academy of Neurology.
The researchers have enrolled 203 patients who were treated with Boston Scientific’s Vercise DBS system, which is not approved for use in the United States. The researchers plan to track patients for 3 years.
“This is the first such industry-sponsored registry, which addresses a need in the field to track DBS practice and outcomes across multiple centers,” said Mark Richardson, MD, PhD, associate professor and director of epilepsy and movement disorders surgery at the University of Pittsburgh Medical Center. He did not take part in the study but is familiar with its findings.
The average age of participants is 59 years, which Dr. Deuschl said is a bit younger than many other studies, and 69% are male. Eighty-five serious adverse events were reported in 52 patients; 57 were not linked to the procedure. One patient died of a surgery-related hematoma.
At 6 months, patients reported a 22% improvement (P less than .0001) on the Parkinson’s Disease Questionnaire (PDQ-39), Dr. Deuschl said, and that level was sustained at 1 year.
That level of improvement is significant, Dr. Richardson said in an interview.
“Previous randomized, controlled trials have shown that patients who are candidates for DBS but who continue medical management alone are most likely to have no improvement at all on any quality of life measures,” he said. In addition, “6 months is fairly short, and some patients likely have not reached stable stimulation programming and effectiveness.”
The researchers also reported that more than 90% of patients, their clinicians, and their caregivers reported improvement.
This kind of study is valuable in light of skepticism about DBS, which is used to treat patients who do not respond to medication, Dr. Tagliati noted.
“Despite 15 years of [Food and Drug Administration] approval, there is still some form of resistance out there in referring patients with Parkinson’s at the right time when they can still fully benefit from the procedure,” he said. “Registries have this potential great benefit in terms of awareness and reassuring people.”
As for the high rating of improvement, Dr. Tagliati said it reflects what he sees in the clinic.
“Barring complications, we have very substantial satisfaction in our patients, definitely in the short term after DBS,” he said. “Over the long term, the picture is more difficult to read.”
Dr. Deuschl said researchers would like to add hundreds more patients to the study, and they hope to gain data about the differences between the results from standard and directional-lead DBS systems.
Boston Scientific funded the study, and some of the authors work for the company.
Dr. Richardson reported receiving research grant funding from Medtronic. Dr. Tagliati reported receiving funding from Abbott, Boston Scientific, Medtronic, and all DBS manufacturers, and his clinic is an investigational center for the Vercise DBS system. Dr. Deuschl reported receiving consulting fees from Boston Scientific, grant funding from Medtronic, lecture fees from Almirall and Novartis, and royalties from Thieme Publishers.
Flow Diverters Successfully Treat Small, Unruptured Aneurysms
HOUSTON—A flow-diverter device can treat small to medium-sized unruptured aneurysms safely and effectively, according to research presented at the International Stroke Conference 2017. The device has a high rate of aneurysm occlusion and is associated with no aneurysm rupture or recurrence at one year. The treatment also entails low rates of morbidity and mortality at one year.
One flow diverter is the Pipeline embolization device. The Pipeline device is inserted into the blood vessel and incorporates a mesh that covers the mouth of the aneurysm. The mesh allows the inner lining of the blood vessel to grow and patch the aneurysm from the inside. In April 2011, the Pipeline embolization device was approved in the United States for the treatment of aneurysms larger than 10 mm on the carotid artery. Many physicians have used the device to treat smaller aneurysms, but no investigators had examined its safety and efficacy in this indication.
Examining an Off-Label Use of the Device
Ricardo Hanel, MD, PhD, Director of Baptist Neurological Institute in Jacksonville, Florida, and colleagues conducted a prospective, multicenter study to evaluate the safety and efficacy of the Pipeline device in the treatment of small to medium-sized (ie, 12 mm or smaller), wide-necked, unruptured aneurysms on the internal carotid artery or vertebral artery. Eligible patients were between ages 22 and 80 and had an aneurysm neck that was 4 mm or larger. Patients with subarachnoid hemorrhage in the previous 30 days and those who had received an intracranial implant in the area of the target aneurysm within the previous 12 weeks were excluded.

The primary efficacy end point was complete aneurysm occlusion without significant parent artery stenosis at one year post procedure. The secondary efficacy end point was device deployment success rate. The primary safety end point was major stroke in the area supplied by the treated artery or neurologic death at one year post treatment. Secondary safety end points included major stroke or neurologic death at 30 days post treatment. Follow-up was conducted at 30 days, six months, one year, two years, and three years.
Dr. Hanel and colleagues enrolled 141 patients into the trial. The population’s mean age was approximately 55, and about 88% of the population was female. Participants had few, if any, symptoms. Approximately 48% of the population had hypertension, 38% had hyperlipidemia, and 44% were current smokers or had smoked within the previous 10 years.
Most Patients Had Complete Occlusion
Mean aneurysm size at baseline was 5 mm. About 84% of the aneurysms were smaller than 7 mm, and 16% were between 7 mm and 12 mm. Approximately 97% of aneurysms were saccular; 12% of saccular aneurysms involved a side branch, and 84% involved a sidewall, but no branch. About 4% of aneurysms were fusiform. About 95% of aneurysms were on the carotid artery. The two most common patient risk factors for treatment were patient preference (63%) and hypertension (48%).
The device was deployed to the target site successfully in about 99% of patients. Mean procedure time was approximately 80 minutes. The mean number of Pipeline devices required to treat each aneurysm was one. Ten patients received multiple Pipeline devices.
The investigators achieved complete occlusion without significant stenosis or retreatment at one year in approximately 84% of patients. This end point was documented by angiogram and adjudicated by an independent core laboratory. The reasons for treatment failure included residual aneurysm (8% of all patients), residual neck (6%), stenosis greater than 50% (1%), and aneurysm retreatment (2%).
At 30 days, three safety events had occurred in two patients. Both patients had a major stroke, and one of them died. The 30-day safety event rate thus was about 1%. One other safety event, a stroke, occurred at 169 days after treatment. The rate of major stroke and death at one year was approximately 2%. Two-year and three-year data are forthcoming.
The study was not designed to define which patients should be treated with a flow diverter, which is “a much broader and harder question,” said Dr. Hanel. Neurologists treat between 20% and 25% of patients with aneurysms smaller than 7 mm. The decision to treat is based on risk factors such as medical history and aneurysm location.
Aneurysms like those in the study “are difficult to treat with current armamentarium devices,” said Ralph L. Sacco, MD, Professor and Olemberg Chair of Neurology at the University of Miami. The current study indicates that the Pipeline device is safe and that these aneurysms can be treated, he added.
Dr. Hanel is a consultant for Medtronic, the manufacturer of the Pipeline device and the funder of the study.
—Erik Greb
Suggested Reading
Becske T, Potts MB, Shapiro M, et al. Pipeline for uncoilable or failed aneurysms: 3-year follow-up results. J Neurosurg. 2016 Oct 14:1-8. [Epub ahead of print].
Brinjikji W, Murad MH, Lanzino G, et al. Endovascular treatment of intracranial aneurysms with flow diverters: a meta-analysis. Stroke. 2013;44(2):442-447.
HOUSTON—A flow-diverter device can treat small to medium-sized unruptured aneurysms safely and effectively, according to research presented at the International Stroke Conference 2017. The device has a high rate of aneurysm occlusion and is associated with no aneurysm rupture or recurrence at one year. The treatment also entails low rates of morbidity and mortality at one year.
One flow diverter is the Pipeline embolization device. The Pipeline device is inserted into the blood vessel and incorporates a mesh that covers the mouth of the aneurysm. The mesh allows the inner lining of the blood vessel to grow and patch the aneurysm from the inside. In April 2011, the Pipeline embolization device was approved in the United States for the treatment of aneurysms larger than 10 mm on the carotid artery. Many physicians have used the device to treat smaller aneurysms, but no investigators had examined its safety and efficacy in this indication.
Examining an Off-Label Use of the Device
Ricardo Hanel, MD, PhD, Director of Baptist Neurological Institute in Jacksonville, Florida, and colleagues conducted a prospective, multicenter study to evaluate the safety and efficacy of the Pipeline device in the treatment of small to medium-sized (ie, 12 mm or smaller), wide-necked, unruptured aneurysms on the internal carotid artery or vertebral artery. Eligible patients were between ages 22 and 80 and had an aneurysm neck that was 4 mm or larger. Patients with subarachnoid hemorrhage in the previous 30 days and those who had received an intracranial implant in the area of the target aneurysm within the previous 12 weeks were excluded.
The primary efficacy end point was complete aneurysm occlusion without significant parent artery stenosis at one year post procedure. The secondary efficacy end point was device deployment success rate. The primary safety end point was major stroke in the area supplied by the treated artery or neurologic death at one year post treatment. Secondary safety end points included major stroke or neurologic death at 30 days post treatment. Follow-up was conducted at 30 days, six months, one year, two years, and three years.
Dr. Hanel and colleagues enrolled 141 patients into the trial. The population’s mean age was approximately 55, and about 88% of the population was female. Participants had few, if any, symptoms. Approximately 48% of the population had hypertension, 38% had hyperlipidemia, and 44% were current smokers or had smoked within the previous 10 years.
Most Patients Had Complete Occlusion
Mean aneurysm size at baseline was 5 mm. About 84% of the aneurysms were smaller than 7 mm, and 16% were between 7 mm and 12 mm. Approximately 97% of aneurysms were saccular; 12% of saccular aneurysms involved a side branch, and 84% involved a sidewall, but no branch. About 4% of aneurysms were fusiform. About 95% of aneurysms were on the carotid artery. The two most common patient risk factors for treatment were patient preference (63%) and hypertension (48%).
The device was deployed to the target site successfully in about 99% of patients. Mean procedure time was approximately 80 minutes. The mean number of Pipeline devices required to treat each aneurysm was one. Ten patients received multiple Pipeline devices.
The investigators achieved complete occlusion without significant stenosis or retreatment at one year in approximately 84% of patients. This end point was documented by angiogram and adjudicated by an independent core laboratory. The reasons for treatment failure included residual aneurysm (8% of all patients), residual neck (6%), stenosis greater than 50% (1%), and aneurysm retreatment (2%).
At 30 days, three safety events had occurred in two patients. Both patients had a major stroke, and one of them died. The 30-day safety event rate thus was about 1%. One other safety event, a stroke, occurred at 169 days after treatment. The rate of major stroke and death at one year was approximately 2%. Two-year and three-year data are forthcoming.
The study was not designed to define which patients should be treated with a flow diverter, which is “a much broader and harder question,” said Dr. Hanel. Neurologists treat between 20% and 25% of patients with aneurysms smaller than 7 mm. The decision to treat is based on risk factors such as medical history and aneurysm location.
Aneurysms like those in the study “are difficult to treat with current armamentarium devices,” said Ralph L. Sacco, MD, Professor and Olemberg Chair of Neurology at the University of Miami. The current study indicates that the Pipeline device is safe and that these aneurysms can be treated, he added.
Dr. Hanel is a consultant for Medtronic, the manufacturer of the Pipeline device and the funder of the study.
—Erik Greb
Suggested Reading
Becske T, Potts MB, Shapiro M, et al. Pipeline for uncoilable or failed aneurysms: 3-year follow-up results. J Neurosurg. 2016 Oct 14:1-8. [Epub ahead of print].
Brinjikji W, Murad MH, Lanzino G, et al. Endovascular treatment of intracranial aneurysms with flow diverters: a meta-analysis. Stroke. 2013;44(2):442-447.
HOUSTON—A flow-diverter device can treat small to medium-sized unruptured aneurysms safely and effectively, according to research presented at the International Stroke Conference 2017. The device has a high rate of aneurysm occlusion and is associated with no aneurysm rupture or recurrence at one year. The treatment also entails low rates of morbidity and mortality at one year.
One flow diverter is the Pipeline embolization device. The Pipeline device is inserted into the blood vessel and incorporates a mesh that covers the mouth of the aneurysm. The mesh allows the inner lining of the blood vessel to grow and patch the aneurysm from the inside. In April 2011, the Pipeline embolization device was approved in the United States for the treatment of aneurysms larger than 10 mm on the carotid artery. Many physicians have used the device to treat smaller aneurysms, but no investigators had examined its safety and efficacy in this indication.
Examining an Off-Label Use of the Device
Ricardo Hanel, MD, PhD, Director of Baptist Neurological Institute in Jacksonville, Florida, and colleagues conducted a prospective, multicenter study to evaluate the safety and efficacy of the Pipeline device in the treatment of small to medium-sized (ie, 12 mm or smaller), wide-necked, unruptured aneurysms on the internal carotid artery or vertebral artery. Eligible patients were between ages 22 and 80 and had an aneurysm neck that was 4 mm or larger. Patients with subarachnoid hemorrhage in the previous 30 days and those who had received an intracranial implant in the area of the target aneurysm within the previous 12 weeks were excluded.
The primary efficacy end point was complete aneurysm occlusion without significant parent artery stenosis at one year post procedure. The secondary efficacy end point was device deployment success rate. The primary safety end point was major stroke in the area supplied by the treated artery or neurologic death at one year post treatment. Secondary safety end points included major stroke or neurologic death at 30 days post treatment. Follow-up was conducted at 30 days, six months, one year, two years, and three years.
Dr. Hanel and colleagues enrolled 141 patients into the trial. The population’s mean age was approximately 55, and about 88% of the population was female. Participants had few, if any, symptoms. Approximately 48% of the population had hypertension, 38% had hyperlipidemia, and 44% were current smokers or had smoked within the previous 10 years.
Most Patients Had Complete Occlusion
Mean aneurysm size at baseline was 5 mm. About 84% of the aneurysms were smaller than 7 mm, and 16% were between 7 mm and 12 mm. Approximately 97% of aneurysms were saccular; 12% of saccular aneurysms involved a side branch, and 84% involved a sidewall, but no branch. About 4% of aneurysms were fusiform. About 95% of aneurysms were on the carotid artery. The two most common patient risk factors for treatment were patient preference (63%) and hypertension (48%).
The device was deployed to the target site successfully in about 99% of patients. Mean procedure time was approximately 80 minutes. The mean number of Pipeline devices required to treat each aneurysm was one. Ten patients received multiple Pipeline devices.
The investigators achieved complete occlusion without significant stenosis or retreatment at one year in approximately 84% of patients. This end point was documented by angiogram and adjudicated by an independent core laboratory. The reasons for treatment failure included residual aneurysm (8% of all patients), residual neck (6%), stenosis greater than 50% (1%), and aneurysm retreatment (2%).
At 30 days, three safety events had occurred in two patients. Both patients had a major stroke, and one of them died. The 30-day safety event rate thus was about 1%. One other safety event, a stroke, occurred at 169 days after treatment. The rate of major stroke and death at one year was approximately 2%. Two-year and three-year data are forthcoming.
The study was not designed to define which patients should be treated with a flow diverter, which is “a much broader and harder question,” said Dr. Hanel. Neurologists treat between 20% and 25% of patients with aneurysms smaller than 7 mm. The decision to treat is based on risk factors such as medical history and aneurysm location.
Aneurysms like those in the study “are difficult to treat with current armamentarium devices,” said Ralph L. Sacco, MD, Professor and Olemberg Chair of Neurology at the University of Miami. The current study indicates that the Pipeline device is safe and that these aneurysms can be treated, he added.
Dr. Hanel is a consultant for Medtronic, the manufacturer of the Pipeline device and the funder of the study.
—Erik Greb
Suggested Reading
Becske T, Potts MB, Shapiro M, et al. Pipeline for uncoilable or failed aneurysms: 3-year follow-up results. J Neurosurg. 2016 Oct 14:1-8. [Epub ahead of print].
Brinjikji W, Murad MH, Lanzino G, et al. Endovascular treatment of intracranial aneurysms with flow diverters: a meta-analysis. Stroke. 2013;44(2):442-447.
Can Environmental Toxicants Cause Parkinson’s Disease?
MIAMI—Accumulating evidence suggests that exposure to certain toxicants may increase the risk of Parkinson’s disease, according to an overview presented at the First Pan American Parkinson’s Disease and Movement Disorders Congress. Researchers seek to learn more about these chemicals and to investigate interventions that could reduce the risks that they present.

Synthetic Heroin and Parkinsonism
In 1983, several cases prompted researchers to think about whether toxicants could cause Parkinson’s disease. A 39-year-old man in California presented to an emergency room with visual hallucinations, jerking of limbs, generalized slowing, and difficulty walking. He had no prior medical history, neurologic history, or family history of neurologic disease. At around the same time, a woman and two men from the same area developed young-onset subacute parkinsonism. James Tetrud, MD, and J. William Langston, MD, the neurologists who examined these patients, learned that they were all IV narcotic addicts. Between two and six weeks before presentation, the patients had injected a synthetic heroin that they had obtained from the same supplier. The toxicant in the synthetic heroin that had induced the parkinsonism was identified as 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP). All of these patients responded to levodopa.
Herbicides and Insecticides
In 2009, Dr. Tanner and colleagues conducted a case–control study to investigate whether specific occupations or toxicant exposures were associated with parkinsonism. They found that 2,4-Dichlorophenoxyacetic acid (2,4-D) was associated with a greater than twofold increased risk of Parkinson’s disease. This chemical was introduced as an herbicide in 1945 and is found in more than 1,500 products, including Agent Orange, which the US military used as a defoliant in Vietnam. Parkinson’s disease is considered to be service-connected in certain US military veterans who served in Vietnam. Currently, 2,4-D is used on lawns, golf courses, and large farms.
The authors also found that exposure to paraquat, another herbicide, nearly doubled the risk of Parkinson’s disease. In people with a homozygous deletion of GSTT1, a gene that encodes an enzyme important to xenobiotic metabolism, exposure to paraquat increased the risk of Parkinson’s disease 11-fold. In addition, exposure to rotenone, an insecticide and piscicide, increased the risk of Parkinson’s disease by more than two times.
Persistent Organic Pollutants
Exposure to persistent organic pollutants such as polychlorinated biphenyls (PCBs) and organochlorine pesticides can also increase the risk of developing Parkinson’s disease. A study by Becker et al in 2000 found an elevated prevalence of Parkinson’s disease in Greenland that may have resulted from exposure to PCBs.
This research prompted Dr. Tanner and colleagues to conduct a case–control study of Alaska natives. The investigators examined the food, diet, occupation, toxicant exposure, blood, and DNA of 69 people with Parkinson’s disease and 179 controls in the Alaska native health system. They found higher blood levels of hexachlorobenzene and PCBs in people with Parkinson’s disease, compared with healthy controls. The blood levels approximately doubled the risk of Parkinson’s disease.
In the Agricultural Health Study, investigators found a similar association between serum PCB level and risk of Parkinson’s disease. Furthermore, people with a particular variant of the efflux transporter gene, which protects cells from exogenous chemicals, and people who also had high serum PCB levels had as much as a 12-fold increased risk of Parkinson’s disease. When Dr. Tanner and colleagues reexamined the data from the Alaska native population, they found that this gene variant had a similar effect. People with a low-risk genotype did not have a greatly increased risk of Parkinson’s disease, even after high exposure to PCBs.
In the 1980s, Hawaiian pineapple farmers sprayed organochlorine pesticides on plants that later were fed to dairy cows. More recently, the Honolulu Asia Aging Study, a prospective cohort study, suggested that milk consumption was associated with increased risk of parkinsonism. In addition, G. Webster Ross, MD, and colleagues analyzed postmortem data and found that nonsmokers who consumed high amounts of milk had low neuron density. Other research has found that brain organochlorine levels were associated with Lewy pathology.
Solvents
Solvents are another class of chemicals that has been associated with Parkinson’s disease. In a 2008 study of 30 industrial coworkers with Parkinson’s disease, Gash et al found that trichloroethylene, a solvent used in many industrial processes such as dry cleaning, was a risk factor for parkinsonism. In a twin study, Dr. Tanner and colleagues found that chlorinated solvents were associated with an increased risk of Parkinson’s disease.
From 1953 to 1985, the water at the Marine Corps base at Camp Lejeune in North Carolina was contaminated with trichloroethylene and perchloroethylene. In 2015, the Institute of Medicine found that among people who had lived at Camp Lejeune, Parkinson’s disease may have resulted from drinking the contaminated water.
Researchers have also noted a higher prevalence of Parkinson’s disease in areas with high traffic. This finding might result from exposure to metals, such as manganese, and gasoline fuels. In addition, exposure to particulate matter may also increase the risk of developing the disease.
Can Parkinson’s Disease Be Prevented?
“Purely genetic Parkinson’s [disease] is very rare, and purely environmental Parkinson’s [disease] is rare. It is most likely that the combined effects of genes and the environment, for most people, are the cause of Parkinson’s disease,” said Dr. Tanner. Preventive measures such as wearing gloves during pesticide application can protect against the disease. “We can change the environment. We can identify genes…. We can make a difference.”
—Erica Tricarico
Suggested Reading
Abbot RD, Ross GW, Petrovitch H, et al. Midlife milk consumption and substantia nigra neuron density at death. Neurology. 2016;86(6):512-519.
Goldman SM, Kamel F, Ross GW, et al. Genetic modification of the association of paraquat and Parkinson’s disease. Mov Disord. 2012;27(13):1652-1658.
Gordon PH, Mehal JM, Holman RC, et al. Parkinson’s disease among American Indians and Alaska natives: a nationwide prevalence study. Mov Disord. 2012;27(11): 1456-1459.
Petrovitch H, Ross GW, Abbott RD, et al. Plantation work and risk of Parkinson disease in a population-based longitudinal study. Arch Neurol. 2002;59(11):1787-1792.
Tanner CM, Ross GW, Jewell SA, et al. Occupation and risk of parkinsonism: a multicenter case-control study. Arch Neurol. 2009;66(9):1106-1113.
MIAMI—Accumulating evidence suggests that exposure to certain toxicants may increase the risk of Parkinson’s disease, according to an overview presented at the First Pan American Parkinson’s Disease and Movement Disorders Congress. Researchers seek to learn more about these chemicals and to investigate interventions that could reduce the risks that they present.
Synthetic Heroin and Parkinsonism
In 1983, several cases prompted researchers to think about whether toxicants could cause Parkinson’s disease. A 39-year-old man in California presented to an emergency room with visual hallucinations, jerking of limbs, generalized slowing, and difficulty walking. He had no prior medical history, neurologic history, or family history of neurologic disease. At around the same time, a woman and two men from the same area developed young-onset subacute parkinsonism. James Tetrud, MD, and J. William Langston, MD, the neurologists who examined these patients, learned that they were all IV narcotic addicts. Between two and six weeks before presentation, the patients had injected a synthetic heroin that they had obtained from the same supplier. The toxicant in the synthetic heroin that had induced the parkinsonism was identified as 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP). All of these patients responded to levodopa.
Herbicides and Insecticides
In 2009, Dr. Tanner and colleagues conducted a case–control study to investigate whether specific occupations or toxicant exposures were associated with parkinsonism. They found that 2,4-Dichlorophenoxyacetic acid (2,4-D) was associated with a greater than twofold increased risk of Parkinson’s disease. This chemical was introduced as an herbicide in 1945 and is found in more than 1,500 products, including Agent Orange, which the US military used as a defoliant in Vietnam. Parkinson’s disease is considered to be service-connected in certain US military veterans who served in Vietnam. Currently, 2,4-D is used on lawns, golf courses, and large farms.
The authors also found that exposure to paraquat, another herbicide, nearly doubled the risk of Parkinson’s disease. In people with a homozygous deletion of GSTT1, a gene that encodes an enzyme important to xenobiotic metabolism, exposure to paraquat increased the risk of Parkinson’s disease 11-fold. In addition, exposure to rotenone, an insecticide and piscicide, increased the risk of Parkinson’s disease by more than two times.
Persistent Organic Pollutants
Exposure to persistent organic pollutants such as polychlorinated biphenyls (PCBs) and organochlorine pesticides can also increase the risk of developing Parkinson’s disease. A study by Becker et al in 2000 found an elevated prevalence of Parkinson’s disease in Greenland that may have resulted from exposure to PCBs.
This research prompted Dr. Tanner and colleagues to conduct a case–control study of Alaska natives. The investigators examined the food, diet, occupation, toxicant exposure, blood, and DNA of 69 people with Parkinson’s disease and 179 controls in the Alaska native health system. They found higher blood levels of hexachlorobenzene and PCBs in people with Parkinson’s disease, compared with healthy controls. The blood levels approximately doubled the risk of Parkinson’s disease.
In the Agricultural Health Study, investigators found a similar association between serum PCB level and risk of Parkinson’s disease. Furthermore, people with a particular variant of the efflux transporter gene, which protects cells from exogenous chemicals, and people who also had high serum PCB levels had as much as a 12-fold increased risk of Parkinson’s disease. When Dr. Tanner and colleagues reexamined the data from the Alaska native population, they found that this gene variant had a similar effect. People with a low-risk genotype did not have a greatly increased risk of Parkinson’s disease, even after high exposure to PCBs.
In the 1980s, Hawaiian pineapple farmers sprayed organochlorine pesticides on plants that later were fed to dairy cows. More recently, the Honolulu Asia Aging Study, a prospective cohort study, suggested that milk consumption was associated with increased risk of parkinsonism. In addition, G. Webster Ross, MD, and colleagues analyzed postmortem data and found that nonsmokers who consumed high amounts of milk had low neuron density. Other research has found that brain organochlorine levels were associated with Lewy pathology.
Solvents
Solvents are another class of chemicals that has been associated with Parkinson’s disease. In a 2008 study of 30 industrial coworkers with Parkinson’s disease, Gash et al found that trichloroethylene, a solvent used in many industrial processes such as dry cleaning, was a risk factor for parkinsonism. In a twin study, Dr. Tanner and colleagues found that chlorinated solvents were associated with an increased risk of Parkinson’s disease.
From 1953 to 1985, the water at the Marine Corps base at Camp Lejeune in North Carolina was contaminated with trichloroethylene and perchloroethylene. In 2015, the Institute of Medicine found that among people who had lived at Camp Lejeune, Parkinson’s disease may have resulted from drinking the contaminated water.
Researchers have also noted a higher prevalence of Parkinson’s disease in areas with high traffic. This finding might result from exposure to metals, such as manganese, and gasoline fuels. In addition, exposure to particulate matter may also increase the risk of developing the disease.
Can Parkinson’s Disease Be Prevented?
“Purely genetic Parkinson’s [disease] is very rare, and purely environmental Parkinson’s [disease] is rare. It is most likely that the combined effects of genes and the environment, for most people, are the cause of Parkinson’s disease,” said Dr. Tanner. Preventive measures such as wearing gloves during pesticide application can protect against the disease. “We can change the environment. We can identify genes…. We can make a difference.”
—Erica Tricarico
Suggested Reading
Abbot RD, Ross GW, Petrovitch H, et al. Midlife milk consumption and substantia nigra neuron density at death. Neurology. 2016;86(6):512-519.
Goldman SM, Kamel F, Ross GW, et al. Genetic modification of the association of paraquat and Parkinson’s disease. Mov Disord. 2012;27(13):1652-1658.
Gordon PH, Mehal JM, Holman RC, et al. Parkinson’s disease among American Indians and Alaska natives: a nationwide prevalence study. Mov Disord. 2012;27(11): 1456-1459.
Petrovitch H, Ross GW, Abbott RD, et al. Plantation work and risk of Parkinson disease in a population-based longitudinal study. Arch Neurol. 2002;59(11):1787-1792.
Tanner CM, Ross GW, Jewell SA, et al. Occupation and risk of parkinsonism: a multicenter case-control study. Arch Neurol. 2009;66(9):1106-1113.
MIAMI—Accumulating evidence suggests that exposure to certain toxicants may increase the risk of Parkinson’s disease, according to an overview presented at the First Pan American Parkinson’s Disease and Movement Disorders Congress. Researchers seek to learn more about these chemicals and to investigate interventions that could reduce the risks that they present.
Synthetic Heroin and Parkinsonism
In 1983, several cases prompted researchers to think about whether toxicants could cause Parkinson’s disease. A 39-year-old man in California presented to an emergency room with visual hallucinations, jerking of limbs, generalized slowing, and difficulty walking. He had no prior medical history, neurologic history, or family history of neurologic disease. At around the same time, a woman and two men from the same area developed young-onset subacute parkinsonism. James Tetrud, MD, and J. William Langston, MD, the neurologists who examined these patients, learned that they were all IV narcotic addicts. Between two and six weeks before presentation, the patients had injected a synthetic heroin that they had obtained from the same supplier. The toxicant in the synthetic heroin that had induced the parkinsonism was identified as 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP). All of these patients responded to levodopa.
Herbicides and Insecticides
In 2009, Dr. Tanner and colleagues conducted a case–control study to investigate whether specific occupations or toxicant exposures were associated with parkinsonism. They found that 2,4-Dichlorophenoxyacetic acid (2,4-D) was associated with a greater than twofold increased risk of Parkinson’s disease. This chemical was introduced as an herbicide in 1945 and is found in more than 1,500 products, including Agent Orange, which the US military used as a defoliant in Vietnam. Parkinson’s disease is considered to be service-connected in certain US military veterans who served in Vietnam. Currently, 2,4-D is used on lawns, golf courses, and large farms.
The authors also found that exposure to paraquat, another herbicide, nearly doubled the risk of Parkinson’s disease. In people with a homozygous deletion of GSTT1, a gene that encodes an enzyme important to xenobiotic metabolism, exposure to paraquat increased the risk of Parkinson’s disease 11-fold. In addition, exposure to rotenone, an insecticide and piscicide, increased the risk of Parkinson’s disease by more than two times.
Persistent Organic Pollutants
Exposure to persistent organic pollutants such as polychlorinated biphenyls (PCBs) and organochlorine pesticides can also increase the risk of developing Parkinson’s disease. A study by Becker et al in 2000 found an elevated prevalence of Parkinson’s disease in Greenland that may have resulted from exposure to PCBs.
This research prompted Dr. Tanner and colleagues to conduct a case–control study of Alaska natives. The investigators examined the food, diet, occupation, toxicant exposure, blood, and DNA of 69 people with Parkinson’s disease and 179 controls in the Alaska native health system. They found higher blood levels of hexachlorobenzene and PCBs in people with Parkinson’s disease, compared with healthy controls. The blood levels approximately doubled the risk of Parkinson’s disease.
In the Agricultural Health Study, investigators found a similar association between serum PCB level and risk of Parkinson’s disease. Furthermore, people with a particular variant of the efflux transporter gene, which protects cells from exogenous chemicals, and people who also had high serum PCB levels had as much as a 12-fold increased risk of Parkinson’s disease. When Dr. Tanner and colleagues reexamined the data from the Alaska native population, they found that this gene variant had a similar effect. People with a low-risk genotype did not have a greatly increased risk of Parkinson’s disease, even after high exposure to PCBs.
In the 1980s, Hawaiian pineapple farmers sprayed organochlorine pesticides on plants that later were fed to dairy cows. More recently, the Honolulu Asia Aging Study, a prospective cohort study, suggested that milk consumption was associated with increased risk of parkinsonism. In addition, G. Webster Ross, MD, and colleagues analyzed postmortem data and found that nonsmokers who consumed high amounts of milk had low neuron density. Other research has found that brain organochlorine levels were associated with Lewy pathology.
Solvents
Solvents are another class of chemicals that has been associated with Parkinson’s disease. In a 2008 study of 30 industrial coworkers with Parkinson’s disease, Gash et al found that trichloroethylene, a solvent used in many industrial processes such as dry cleaning, was a risk factor for parkinsonism. In a twin study, Dr. Tanner and colleagues found that chlorinated solvents were associated with an increased risk of Parkinson’s disease.
From 1953 to 1985, the water at the Marine Corps base at Camp Lejeune in North Carolina was contaminated with trichloroethylene and perchloroethylene. In 2015, the Institute of Medicine found that among people who had lived at Camp Lejeune, Parkinson’s disease may have resulted from drinking the contaminated water.
Researchers have also noted a higher prevalence of Parkinson’s disease in areas with high traffic. This finding might result from exposure to metals, such as manganese, and gasoline fuels. In addition, exposure to particulate matter may also increase the risk of developing the disease.
Can Parkinson’s Disease Be Prevented?
“Purely genetic Parkinson’s [disease] is very rare, and purely environmental Parkinson’s [disease] is rare. It is most likely that the combined effects of genes and the environment, for most people, are the cause of Parkinson’s disease,” said Dr. Tanner. Preventive measures such as wearing gloves during pesticide application can protect against the disease. “We can change the environment. We can identify genes…. We can make a difference.”
—Erica Tricarico
Suggested Reading
Abbot RD, Ross GW, Petrovitch H, et al. Midlife milk consumption and substantia nigra neuron density at death. Neurology. 2016;86(6):512-519.
Goldman SM, Kamel F, Ross GW, et al. Genetic modification of the association of paraquat and Parkinson’s disease. Mov Disord. 2012;27(13):1652-1658.
Gordon PH, Mehal JM, Holman RC, et al. Parkinson’s disease among American Indians and Alaska natives: a nationwide prevalence study. Mov Disord. 2012;27(11): 1456-1459.
Petrovitch H, Ross GW, Abbott RD, et al. Plantation work and risk of Parkinson disease in a population-based longitudinal study. Arch Neurol. 2002;59(11):1787-1792.
Tanner CM, Ross GW, Jewell SA, et al. Occupation and risk of parkinsonism: a multicenter case-control study. Arch Neurol. 2009;66(9):1106-1113.

Just the FRAX: National fracture risks estimated
Adults aged 40 years and over have a mean 10-year risk of 5.3% for major osteoporotic fracture, according to the National Center for Health Statistics.
That risk includes a 10-year probability of 0.5% for hip fracture, based on estimates using the FRAX algorithm. These nationally representative estimates, the first to use the FRAX algorithm in this population, are based on 2013-2014 data from the National Health and Nutrition Examination Survey and show that the adjusted mean risks for those aged 50 years and over are 7.4% for major osteoporotic fracture and 0.9% for hip fracture, the NCHS reported.
The risk estimates in this analysis agree in large part with guidelines from the National Osteoporosis Foundation, which “use elevated FRAX scores in combination with low bone mass to define treatment eligibility,” the investigators said. FRAX-based findings, they noted, may provide “a more global evaluation of fracture risk than can be obtained from estimates based on [bone mineral density] alone.”
Adults aged 40 years and over have a mean 10-year risk of 5.3% for major osteoporotic fracture, according to the National Center for Health Statistics.
That risk includes a 10-year probability of 0.5% for hip fracture, based on estimates using the FRAX algorithm. These nationally representative estimates, the first to use the FRAX algorithm in this population, are based on 2013-2014 data from the National Health and Nutrition Examination Survey and show that the adjusted mean risks for those aged 50 years and over are 7.4% for major osteoporotic fracture and 0.9% for hip fracture, the NCHS reported.
The risk estimates in this analysis agree in large part with guidelines from the National Osteoporosis Foundation, which “use elevated FRAX scores in combination with low bone mass to define treatment eligibility,” the investigators said. FRAX-based findings, they noted, may provide “a more global evaluation of fracture risk than can be obtained from estimates based on [bone mineral density] alone.”
Adults aged 40 years and over have a mean 10-year risk of 5.3% for major osteoporotic fracture, according to the National Center for Health Statistics.
That risk includes a 10-year probability of 0.5% for hip fracture, based on estimates using the FRAX algorithm. These nationally representative estimates, the first to use the FRAX algorithm in this population, are based on 2013-2014 data from the National Health and Nutrition Examination Survey and show that the adjusted mean risks for those aged 50 years and over are 7.4% for major osteoporotic fracture and 0.9% for hip fracture, the NCHS reported.
The risk estimates in this analysis agree in large part with guidelines from the National Osteoporosis Foundation, which “use elevated FRAX scores in combination with low bone mass to define treatment eligibility,” the investigators said. FRAX-based findings, they noted, may provide “a more global evaluation of fracture risk than can be obtained from estimates based on [bone mineral density] alone.”
Lenalidomide maintains posttransplant remissions in myeloma
NEW YORK – Recent trial results have shown the importance of treating patients with multiple myeloma with the immunomodulator lenalidomide for maintaining negative minimal residual disease and sustained complete responses following autologous stem cell transplantation, experts said at a conference held by Imedex.
“How do you keep the therapy pedal to the metal over time? Lenalidomide is easy to deliver, convenient, and improves progression-free survival and overall survival,” said Paul G. Richardson, MD, professor of medicine at Harvard University and clinical program leader of the Multiple Myeloma Center at the Dana-Farber Cancer Institute in Boston. “Lenalidomide maintenance is standard of care. It provides a platform on which you can reliably add new agents for maintenance of remission following autologous stem cell transplantation,” in patients with multiple myeloma.

In a meta-analysis of three randomized, controlled trials with 1,208 patients with multiple myeloma who had undergone induction and autologous stem cell transplant, the overall mortality rate dropped by 0.75 (95% CI, 0.63-0.90) among patients maintained on lenalidomide, compared with placebo (J Clin Oncol. 2016;34:suppl;abstract 8001).
A third study that Dr. Richardson cited showed the difficulty of improving on lenalidomide. The BMT CTN 0702 (Stem Cell Transplant With Lenalidomide Maintenance in Patients With Multiple Myeloma) trial randomized 758 multiple myeloma patients to three different autologous stem cell transplant regimens, each followed by lenalidomide maintenance. One arm followed the transplant with four cycles of consolidation therapy with lenalidomide, dexamethasone, and bortezomib (Velcade); one arm used two tandem transplantations; and the third arm used a single transplantation. All three arms had similar rates of progression-free survival and overall survival during follow-up (Blood. 2016 Dec 6;LBA-1). The results showed that “lenalidomide maintenance is an equalizer,” Dr. Richardson said.

She cited a U.S., multicenter, phase II study that followed 66 newly diagnosed patients with multiple myeloma who proceeded through induction, autologous stem cell transplantation, and multiple cycles of consolidation therapy with a regimen of lenalidomide, the proteasome inhibitor carfilzomib (Kyprolis), and dexamethasone. Fifty patients went through 18 cycles of this consolidation regimen and showed an 84% rate of stringent complete response, “the first time we’ve seen such results,” said Dr. Lentzsch, director of the Multiple Myeloma and Amyloidosis Program at Columbia University in New York. “This translated into an excellent” progression-free survival of 86% after 3 years and an overall survival of 95% after 3 years (Blood. 2016 Dec 5;Abstract 675). “I’m pretty impressed” by the results, she added.
An advantage of the combined regimen used in this study is that it is “relatively well tolerated,” as well as effective for keeping patients in remission, Dr. Lentzsch said. However, she highlighted that this was a small study, so its treatment implications are limited for the time being. “For high-risk patients, use the full combination. For everyone else, we need to wait for results from a randomized, controlled trial,” she advised.
Dr. Richardson has been a consultant to Celgene, the company that markets lenalidomide, and is also a consultant to Genmab, Janssen, Novartis, Oncopeptides, and Takeda and has received research funding from Celgene and Takeda. Dr. Lentzsch has been a consultant to Amgen, Bristol-Myers Squibb, and Caelum Biosciences.
[email protected]
On Twitter @mitchelzoler
NEW YORK – Recent trial results have shown the importance of treating patients with multiple myeloma with the immunomodulator lenalidomide for maintaining negative minimal residual disease and sustained complete responses following autologous stem cell transplantation, experts said at a conference held by Imedex.
“How do you keep the therapy pedal to the metal over time? Lenalidomide is easy to deliver, convenient, and improves progression-free survival and overall survival,” said Paul G. Richardson, MD, professor of medicine at Harvard University and clinical program leader of the Multiple Myeloma Center at the Dana-Farber Cancer Institute in Boston. “Lenalidomide maintenance is standard of care. It provides a platform on which you can reliably add new agents for maintenance of remission following autologous stem cell transplantation,” in patients with multiple myeloma.
In a meta-analysis of three randomized, controlled trials with 1,208 patients with multiple myeloma who had undergone induction and autologous stem cell transplant, the overall mortality rate dropped by 0.75 (95% CI, 0.63-0.90) among patients maintained on lenalidomide, compared with placebo (J Clin Oncol. 2016;34:suppl;abstract 8001).
A third study that Dr. Richardson cited showed the difficulty of improving on lenalidomide. The BMT CTN 0702 (Stem Cell Transplant With Lenalidomide Maintenance in Patients With Multiple Myeloma) trial randomized 758 multiple myeloma patients to three different autologous stem cell transplant regimens, each followed by lenalidomide maintenance. One arm followed the transplant with four cycles of consolidation therapy with lenalidomide, dexamethasone, and bortezomib (Velcade); one arm used two tandem transplantations; and the third arm used a single transplantation. All three arms had similar rates of progression-free survival and overall survival during follow-up (Blood. 2016 Dec 6;LBA-1). The results showed that “lenalidomide maintenance is an equalizer,” Dr. Richardson said.
She cited a U.S., multicenter, phase II study that followed 66 newly diagnosed patients with multiple myeloma who proceeded through induction, autologous stem cell transplantation, and multiple cycles of consolidation therapy with a regimen of lenalidomide, the proteasome inhibitor carfilzomib (Kyprolis), and dexamethasone. Fifty patients went through 18 cycles of this consolidation regimen and showed an 84% rate of stringent complete response, “the first time we’ve seen such results,” said Dr. Lentzsch, director of the Multiple Myeloma and Amyloidosis Program at Columbia University in New York. “This translated into an excellent” progression-free survival of 86% after 3 years and an overall survival of 95% after 3 years (Blood. 2016 Dec 5;Abstract 675). “I’m pretty impressed” by the results, she added.
An advantage of the combined regimen used in this study is that it is “relatively well tolerated,” as well as effective for keeping patients in remission, Dr. Lentzsch said. However, she highlighted that this was a small study, so its treatment implications are limited for the time being. “For high-risk patients, use the full combination. For everyone else, we need to wait for results from a randomized, controlled trial,” she advised.
Dr. Richardson has been a consultant to Celgene, the company that markets lenalidomide, and is also a consultant to Genmab, Janssen, Novartis, Oncopeptides, and Takeda and has received research funding from Celgene and Takeda. Dr. Lentzsch has been a consultant to Amgen, Bristol-Myers Squibb, and Caelum Biosciences.
[email protected]
On Twitter @mitchelzoler
NEW YORK – Recent trial results have shown the importance of treating patients with multiple myeloma with the immunomodulator lenalidomide for maintaining negative minimal residual disease and sustained complete responses following autologous stem cell transplantation, experts said at a conference held by Imedex.
“How do you keep the therapy pedal to the metal over time? Lenalidomide is easy to deliver, convenient, and improves progression-free survival and overall survival,” said Paul G. Richardson, MD, professor of medicine at Harvard University and clinical program leader of the Multiple Myeloma Center at the Dana-Farber Cancer Institute in Boston. “Lenalidomide maintenance is standard of care. It provides a platform on which you can reliably add new agents for maintenance of remission following autologous stem cell transplantation,” in patients with multiple myeloma.
In a meta-analysis of three randomized, controlled trials with 1,208 patients with multiple myeloma who had undergone induction and autologous stem cell transplant, the overall mortality rate dropped by 0.75 (95% CI, 0.63-0.90) among patients maintained on lenalidomide, compared with placebo (J Clin Oncol. 2016;34:suppl;abstract 8001).
A third study that Dr. Richardson cited showed the difficulty of improving on lenalidomide. The BMT CTN 0702 (Stem Cell Transplant With Lenalidomide Maintenance in Patients With Multiple Myeloma) trial randomized 758 multiple myeloma patients to three different autologous stem cell transplant regimens, each followed by lenalidomide maintenance. One arm followed the transplant with four cycles of consolidation therapy with lenalidomide, dexamethasone, and bortezomib (Velcade); one arm used two tandem transplantations; and the third arm used a single transplantation. All three arms had similar rates of progression-free survival and overall survival during follow-up (Blood. 2016 Dec 6;LBA-1). The results showed that “lenalidomide maintenance is an equalizer,” Dr. Richardson said.
She cited a U.S., multicenter, phase II study that followed 66 newly diagnosed patients with multiple myeloma who proceeded through induction, autologous stem cell transplantation, and multiple cycles of consolidation therapy with a regimen of lenalidomide, the proteasome inhibitor carfilzomib (Kyprolis), and dexamethasone. Fifty patients went through 18 cycles of this consolidation regimen and showed an 84% rate of stringent complete response, “the first time we’ve seen such results,” said Dr. Lentzsch, director of the Multiple Myeloma and Amyloidosis Program at Columbia University in New York. “This translated into an excellent” progression-free survival of 86% after 3 years and an overall survival of 95% after 3 years (Blood. 2016 Dec 5;Abstract 675). “I’m pretty impressed” by the results, she added.
An advantage of the combined regimen used in this study is that it is “relatively well tolerated,” as well as effective for keeping patients in remission, Dr. Lentzsch said. However, she highlighted that this was a small study, so its treatment implications are limited for the time being. “For high-risk patients, use the full combination. For everyone else, we need to wait for results from a randomized, controlled trial,” she advised.
Dr. Richardson has been a consultant to Celgene, the company that markets lenalidomide, and is also a consultant to Genmab, Janssen, Novartis, Oncopeptides, and Takeda and has received research funding from Celgene and Takeda. Dr. Lentzsch has been a consultant to Amgen, Bristol-Myers Squibb, and Caelum Biosciences.
[email protected]
On Twitter @mitchelzoler
VIDEO: Study confirms uneven access to liver cancer treatment at VA hospitals
Only 25% of Veterans Affairs (VA) patients with potentially curable (Barcelona Clinic Liver Cancer stage 0/A) hepatocellular carcinoma received resection, transplantation, or ablative therapy, according to the results of a national retrospective cohort study published in the June issue of Gastroenterology (doi: 10.1053/j.gastro.2017.02.040).
Furthermore, 13% of the fittest (ECOG performance status 1-2) patients received no active treatment for their hepatocellular carcinoma, Marina Serper, MD, of Corporal Michael J. Crescenz VA Medical Center, Philadelphia, and Tamar H. Taddei, MD, of VA New York Harbor Health Care System, Brooklyn, New York, wrote with their associates in Gastroenterology.
Source: American Gastroenterological Association
“Delivery of curative therapies conferred the highest survival benefit, and notable geographic and specialist variation was observed in the delivery of active treatment,” they added. “Future studies should further evaluate modifiable health system and provider-specific barriers to delivering high quality, multidisciplinary care in hepatocellular carcinoma [in order] to optimize patient outcomes.”
Hepatocellular carcinoma ranks second worldwide and fifth in the United States as a cause of cancer mortality. Gastroenterologists, hepatologists, medical oncologists, or surgeons may take primary responsibility for treatment in community settings, but little is known about how provider and health system factors affect outcomes or the likelihood of receiving active treatments, such as liver transplantation, resection, ablative or transarterial therapy, sorafenib, systemic chemotherapy, or radiation. Accordingly, the researchers reviewed medical records and demographic data from all 3,988 U.S. patients diagnosed with hepatocellular carcinoma between 2008 and 2010 who received care at 128 Veterans Affairs centers. Patients were followed through the end of 2014. Data were from the Veterans Outcomes and Costs Associated with Liver Disease (VOCAL) cohort study (Gastroenterology. 2017 Mar 7. doi: 10.1053/j.gastro.2017.02.040).
After diagnosis, most (54%) patients only underwent transarterial palliative therapy, and 24% received no cancer treatment, the researchers reported. Being treated at an academically affiliated VA hospital nearly doubled the odds of receiving active therapy (odds ratio, 1.97; 95% confidence interval, 1.6 to 2.4; P less than .001), even after the researchers controlled for race, Charlson-Deyo comorbidity, and presenting Barcelona Clinic Liver Cancer stage. Evaluation by multiple specialists also significantly increased the odds of active treatment (OR, 1.60; 95% CI, 1.15 to 2.21; P = .005), but review by a multidisciplinary tumor board did not (OR, 1.19; P = .1).
Receipt of active therapy also varied significantly by region. Compared with patients in the Northeastern United States, those in the mid-South were significantly less likely to receive active therapy (HR, 0.62; 95% CI, 0.44-0.85). Patients in the Southeast, Central, and Western United States also were less likely to receive active treatment than were those in the Northeast, but 95% CIs for these hazard ratios were nonsignificant. Virtual tumor boards could help overcome diagnostic and treatment delays, but costs, care coordination, patient factors, and compensation issues are major barriers against implementation, the investigators noted.
Overall survival was associated with active treatment of hepatocellular carcinoma, including liver transplantation (hazard ratio, 0.22; 95% CI, 0.16-0.31), liver resection (HR, 0.38; 95% CI, 0.28-0.52), ablative therapy (HR, 0.63; 95% CI, 0.52-0.76), and transarterial therapy (HR, 0.83; 95% CI, 0.74-0.92). Reduced mortality was associated with seeing a hepatologist (HR, 0.7), medical oncologist (HR, 0.82), or surgeon (HR, 0.79) within 30 days of diagnosis (P less than .001 for each). Undergoing review by a multidisciplinary tumor board was associated with significantly reduced mortality (HR, 0.83; P less than .001), said the researchers.
“Findings from the VOCAL cohort of predominantly older males with significant medical comorbidities are important in light of the aging U.S. population and a nearly 70% expected increase in cancer among older adults,” they wrote. Together, the results indicate that access to multidisciplinary and expert care “is critical for optimizing treatment choices and for maximizing survival, but that such access is non-uniform,” they noted. “Detailed national VA clinical and administrative data are a unique resource that may be tapped to facilitate development of a parsimonious set of evidence-based, patient-centered, liver cancer–specific quality measures,” they emphasized. Quality measures based on timeliness, receipt of appropriate care, survival, or patient-reported outcomes “could be applicable both within and outside the VA system.”
The study was funded by unrestricted grants from Bayer Healthcare Pharmaceuticals and the VA HIV, Hepatitis and Public Health Pathogens Programs. The investigators had no conflicts.
The treatment of hepatocellular carcinoma (HCC) can be challenging because of the presence of underlying chronic liver disease and cirrhosis in the majority of patients. The study by Dr. Serper and colleagues evaluated the care of patients diagnosed with HCC in the Veterans Affairs (VA) system between 2008 and 2010. There are important aspects of this study worth highlighting.
First, 36% of patients presented with early-stage HCC and clearly had a better overall survival. This highlights the need for surveillance of patients with cirrhosis not only in the VA but also in other health systems. Second, only a minority of patients with early-stage HCC received curative interventions. In order to improve outcomes, patients with early stage disease should receive appropriate curative interventions. Third, gastroenterologists saw a large number of patients with HCC in the VA system, but, unfortunately, this led to less receipt of active therapy and a trend for a worse all-cause mortality, compared with hepatologists and other specialties. It is critical that gastroenterologists refer patients to specialties more adept at treating HCC in order to achieve better outcomes.
Lastly, only 34% of patients with HCC were managed via a multidisciplinary tumor conference. Importantly, these patients had an increased probability of receipt of active treatment and a 17% reduction in all-cause mortality. Our group has shown that a multidisciplinary approach to treating HCC improves overall survival. It is critical that medical centers develop a multidisciplinary treatment approach to HCC.
Jorge A. Marrero, MD, MS, AGAF, is professor of medicine and medical director for liver transplantation at UT Southwestern Medical Center Dallas. He has no conflicts of interest to report regarding this manuscript or commentary.
The treatment of hepatocellular carcinoma (HCC) can be challenging because of the presence of underlying chronic liver disease and cirrhosis in the majority of patients. The study by Dr. Serper and colleagues evaluated the care of patients diagnosed with HCC in the Veterans Affairs (VA) system between 2008 and 2010. There are important aspects of this study worth highlighting.
First, 36% of patients presented with early-stage HCC and clearly had a better overall survival. This highlights the need for surveillance of patients with cirrhosis not only in the VA but also in other health systems. Second, only a minority of patients with early-stage HCC received curative interventions. In order to improve outcomes, patients with early stage disease should receive appropriate curative interventions. Third, gastroenterologists saw a large number of patients with HCC in the VA system, but, unfortunately, this led to less receipt of active therapy and a trend for a worse all-cause mortality, compared with hepatologists and other specialties. It is critical that gastroenterologists refer patients to specialties more adept at treating HCC in order to achieve better outcomes.
Lastly, only 34% of patients with HCC were managed via a multidisciplinary tumor conference. Importantly, these patients had an increased probability of receipt of active treatment and a 17% reduction in all-cause mortality. Our group has shown that a multidisciplinary approach to treating HCC improves overall survival. It is critical that medical centers develop a multidisciplinary treatment approach to HCC.
Jorge A. Marrero, MD, MS, AGAF, is professor of medicine and medical director for liver transplantation at UT Southwestern Medical Center Dallas. He has no conflicts of interest to report regarding this manuscript or commentary.
The treatment of hepatocellular carcinoma (HCC) can be challenging because of the presence of underlying chronic liver disease and cirrhosis in the majority of patients. The study by Dr. Serper and colleagues evaluated the care of patients diagnosed with HCC in the Veterans Affairs (VA) system between 2008 and 2010. There are important aspects of this study worth highlighting.
First, 36% of patients presented with early-stage HCC and clearly had a better overall survival. This highlights the need for surveillance of patients with cirrhosis not only in the VA but also in other health systems. Second, only a minority of patients with early-stage HCC received curative interventions. In order to improve outcomes, patients with early stage disease should receive appropriate curative interventions. Third, gastroenterologists saw a large number of patients with HCC in the VA system, but, unfortunately, this led to less receipt of active therapy and a trend for a worse all-cause mortality, compared with hepatologists and other specialties. It is critical that gastroenterologists refer patients to specialties more adept at treating HCC in order to achieve better outcomes.
Lastly, only 34% of patients with HCC were managed via a multidisciplinary tumor conference. Importantly, these patients had an increased probability of receipt of active treatment and a 17% reduction in all-cause mortality. Our group has shown that a multidisciplinary approach to treating HCC improves overall survival. It is critical that medical centers develop a multidisciplinary treatment approach to HCC.
Jorge A. Marrero, MD, MS, AGAF, is professor of medicine and medical director for liver transplantation at UT Southwestern Medical Center Dallas. He has no conflicts of interest to report regarding this manuscript or commentary.
Only 25% of Veterans Affairs (VA) patients with potentially curable (Barcelona Clinic Liver Cancer stage 0/A) hepatocellular carcinoma received resection, transplantation, or ablative therapy, according to the results of a national retrospective cohort study published in the June issue of Gastroenterology (doi: 10.1053/j.gastro.2017.02.040).
Furthermore, 13% of the fittest (ECOG performance status 1-2) patients received no active treatment for their hepatocellular carcinoma, Marina Serper, MD, of Corporal Michael J. Crescenz VA Medical Center, Philadelphia, and Tamar H. Taddei, MD, of VA New York Harbor Health Care System, Brooklyn, New York, wrote with their associates in Gastroenterology.
Source: American Gastroenterological Association
“Delivery of curative therapies conferred the highest survival benefit, and notable geographic and specialist variation was observed in the delivery of active treatment,” they added. “Future studies should further evaluate modifiable health system and provider-specific barriers to delivering high quality, multidisciplinary care in hepatocellular carcinoma [in order] to optimize patient outcomes.”
Hepatocellular carcinoma ranks second worldwide and fifth in the United States as a cause of cancer mortality. Gastroenterologists, hepatologists, medical oncologists, or surgeons may take primary responsibility for treatment in community settings, but little is known about how provider and health system factors affect outcomes or the likelihood of receiving active treatments, such as liver transplantation, resection, ablative or transarterial therapy, sorafenib, systemic chemotherapy, or radiation. Accordingly, the researchers reviewed medical records and demographic data from all 3,988 U.S. patients diagnosed with hepatocellular carcinoma between 2008 and 2010 who received care at 128 Veterans Affairs centers. Patients were followed through the end of 2014. Data were from the Veterans Outcomes and Costs Associated with Liver Disease (VOCAL) cohort study (Gastroenterology. 2017 Mar 7. doi: 10.1053/j.gastro.2017.02.040).
After diagnosis, most (54%) patients only underwent transarterial palliative therapy, and 24% received no cancer treatment, the researchers reported. Being treated at an academically affiliated VA hospital nearly doubled the odds of receiving active therapy (odds ratio, 1.97; 95% confidence interval, 1.6 to 2.4; P less than .001), even after the researchers controlled for race, Charlson-Deyo comorbidity, and presenting Barcelona Clinic Liver Cancer stage. Evaluation by multiple specialists also significantly increased the odds of active treatment (OR, 1.60; 95% CI, 1.15 to 2.21; P = .005), but review by a multidisciplinary tumor board did not (OR, 1.19; P = .1).
Receipt of active therapy also varied significantly by region. Compared with patients in the Northeastern United States, those in the mid-South were significantly less likely to receive active therapy (HR, 0.62; 95% CI, 0.44-0.85). Patients in the Southeast, Central, and Western United States also were less likely to receive active treatment than were those in the Northeast, but 95% CIs for these hazard ratios were nonsignificant. Virtual tumor boards could help overcome diagnostic and treatment delays, but costs, care coordination, patient factors, and compensation issues are major barriers against implementation, the investigators noted.
Overall survival was associated with active treatment of hepatocellular carcinoma, including liver transplantation (hazard ratio, 0.22; 95% CI, 0.16-0.31), liver resection (HR, 0.38; 95% CI, 0.28-0.52), ablative therapy (HR, 0.63; 95% CI, 0.52-0.76), and transarterial therapy (HR, 0.83; 95% CI, 0.74-0.92). Reduced mortality was associated with seeing a hepatologist (HR, 0.7), medical oncologist (HR, 0.82), or surgeon (HR, 0.79) within 30 days of diagnosis (P less than .001 for each). Undergoing review by a multidisciplinary tumor board was associated with significantly reduced mortality (HR, 0.83; P less than .001), said the researchers.
“Findings from the VOCAL cohort of predominantly older males with significant medical comorbidities are important in light of the aging U.S. population and a nearly 70% expected increase in cancer among older adults,” they wrote. Together, the results indicate that access to multidisciplinary and expert care “is critical for optimizing treatment choices and for maximizing survival, but that such access is non-uniform,” they noted. “Detailed national VA clinical and administrative data are a unique resource that may be tapped to facilitate development of a parsimonious set of evidence-based, patient-centered, liver cancer–specific quality measures,” they emphasized. Quality measures based on timeliness, receipt of appropriate care, survival, or patient-reported outcomes “could be applicable both within and outside the VA system.”
The study was funded by unrestricted grants from Bayer Healthcare Pharmaceuticals and the VA HIV, Hepatitis and Public Health Pathogens Programs. The investigators had no conflicts.
Only 25% of Veterans Affairs (VA) patients with potentially curable (Barcelona Clinic Liver Cancer stage 0/A) hepatocellular carcinoma received resection, transplantation, or ablative therapy, according to the results of a national retrospective cohort study published in the June issue of Gastroenterology (doi: 10.1053/j.gastro.2017.02.040).
Furthermore, 13% of the fittest (ECOG performance status 1-2) patients received no active treatment for their hepatocellular carcinoma, Marina Serper, MD, of Corporal Michael J. Crescenz VA Medical Center, Philadelphia, and Tamar H. Taddei, MD, of VA New York Harbor Health Care System, Brooklyn, New York, wrote with their associates in Gastroenterology.
Source: American Gastroenterological Association
“Delivery of curative therapies conferred the highest survival benefit, and notable geographic and specialist variation was observed in the delivery of active treatment,” they added. “Future studies should further evaluate modifiable health system and provider-specific barriers to delivering high quality, multidisciplinary care in hepatocellular carcinoma [in order] to optimize patient outcomes.”
Hepatocellular carcinoma ranks second worldwide and fifth in the United States as a cause of cancer mortality. Gastroenterologists, hepatologists, medical oncologists, or surgeons may take primary responsibility for treatment in community settings, but little is known about how provider and health system factors affect outcomes or the likelihood of receiving active treatments, such as liver transplantation, resection, ablative or transarterial therapy, sorafenib, systemic chemotherapy, or radiation. Accordingly, the researchers reviewed medical records and demographic data from all 3,988 U.S. patients diagnosed with hepatocellular carcinoma between 2008 and 2010 who received care at 128 Veterans Affairs centers. Patients were followed through the end of 2014. Data were from the Veterans Outcomes and Costs Associated with Liver Disease (VOCAL) cohort study (Gastroenterology. 2017 Mar 7. doi: 10.1053/j.gastro.2017.02.040).
After diagnosis, most (54%) patients only underwent transarterial palliative therapy, and 24% received no cancer treatment, the researchers reported. Being treated at an academically affiliated VA hospital nearly doubled the odds of receiving active therapy (odds ratio, 1.97; 95% confidence interval, 1.6 to 2.4; P less than .001), even after the researchers controlled for race, Charlson-Deyo comorbidity, and presenting Barcelona Clinic Liver Cancer stage. Evaluation by multiple specialists also significantly increased the odds of active treatment (OR, 1.60; 95% CI, 1.15 to 2.21; P = .005), but review by a multidisciplinary tumor board did not (OR, 1.19; P = .1).
Receipt of active therapy also varied significantly by region. Compared with patients in the Northeastern United States, those in the mid-South were significantly less likely to receive active therapy (HR, 0.62; 95% CI, 0.44-0.85). Patients in the Southeast, Central, and Western United States also were less likely to receive active treatment than were those in the Northeast, but 95% CIs for these hazard ratios were nonsignificant. Virtual tumor boards could help overcome diagnostic and treatment delays, but costs, care coordination, patient factors, and compensation issues are major barriers against implementation, the investigators noted.
Overall survival was associated with active treatment of hepatocellular carcinoma, including liver transplantation (hazard ratio, 0.22; 95% CI, 0.16-0.31), liver resection (HR, 0.38; 95% CI, 0.28-0.52), ablative therapy (HR, 0.63; 95% CI, 0.52-0.76), and transarterial therapy (HR, 0.83; 95% CI, 0.74-0.92). Reduced mortality was associated with seeing a hepatologist (HR, 0.7), medical oncologist (HR, 0.82), or surgeon (HR, 0.79) within 30 days of diagnosis (P less than .001 for each). Undergoing review by a multidisciplinary tumor board was associated with significantly reduced mortality (HR, 0.83; P less than .001), said the researchers.
“Findings from the VOCAL cohort of predominantly older males with significant medical comorbidities are important in light of the aging U.S. population and a nearly 70% expected increase in cancer among older adults,” they wrote. Together, the results indicate that access to multidisciplinary and expert care “is critical for optimizing treatment choices and for maximizing survival, but that such access is non-uniform,” they noted. “Detailed national VA clinical and administrative data are a unique resource that may be tapped to facilitate development of a parsimonious set of evidence-based, patient-centered, liver cancer–specific quality measures,” they emphasized. Quality measures based on timeliness, receipt of appropriate care, survival, or patient-reported outcomes “could be applicable both within and outside the VA system.”
The study was funded by unrestricted grants from Bayer Healthcare Pharmaceuticals and the VA HIV, Hepatitis and Public Health Pathogens Programs. The investigators had no conflicts.
FROM GASTROENTEROLOGY
Key clinical point: Undertreatment of hepatocellular carcinoma was common within the Veterans Affairs system, and varied by geographic region.
Major finding: Only 25% of Barcelona Clinic Liver Cancer stage 0/A patients received potentially curative therapies. Those in the mid-South were significantly less likely to receive active treatment than were those in the Northeast (HR, 0.62; 95% CI, 0.44-0.85). In an adjusted model, treatment at an academically affiliated VA hospital nearly doubled the odds of receiving active therapy (odds ratio, 1.97; P less than .001).
Data source: A national, retrospective cohort study of all 3,988 patients who were diagnosed with hepatocellular carcinoma between 2008 and 2010 and received care through Veterans Affairs.
Disclosures: The study was funded by unrestricted grants from Bayer Healthcare Pharmaceuticals and the VA HIV, Hepatitis and Public Health Pathogens Programs. The investigators had no conflicts.
Behavioral Medicine Approaches to Migraine

Multiaxial Assessment
All workups for headache evaluate frequency, intensity, duration, and disability. From a behavioral perspective, Dr. Baskin recommended considering a few more factors—adherence to therapy, stress-related issues, comorbid psychiatric disorders, and factors that transform migraine from episodic to chronic.
“When you talk about adherence, one of the basics of this is readiness to change,” said Dr. Baskin. “Is the patient motivated for treatment? That doesn’t necessarily happen immediately.” For example, a patient who is motivated to have an abortive agent might not be motivated to pursue prevention, lose weight, exercise, or make lifestyle changes. “Over time, that might change,” Dr. Baskin noted. Patient motivation may evolve with a greater understanding of the goals of treatment.
“Has the patient adhered to past therapy regimens? If the answer is no, then the answer will most likely be no again,” Dr. Baskin said. If medication overuse was a problem, it may be again. Whenever possible, ask open-ended questions, such as “how do you decide when to take an acute medication?” Dr. Baskin advised.
Stress and Migraine
High levels of daily stress, even daily stressors that are not catastrophic, can transform migraine from episodic to chronic. “Patients with depression show a greater effect of stress,” Dr. Baskin said. Similarly, victims of trauma or childhood maltreatment can have more disabling headaches that may be more likely to transform into daily headaches. “Many patients do not have the coping skills necessary to manage stress or recurrent headache,” Dr. Baskin said. One of the goals of behavioral therapy for migraine is to increase patients’ self-efficacy and give them the tools and the confidence to manage headaches and stressors.
Psychiatric Comorbidity
Psychiatric comorbidity may complicate differential diagnosis, and nonadherence to medication is greater in people with mood disorders and anxiety disorders. These patients may show a reduced response to pharmacologic and behavioral treatments for headache. Psychiatric comorbidities also can contribute to migraine chronification. “However, if you add a small behavioral component to your headache program, patients with psychiatric comorbidities seem to respond relatively well,” Dr. Baskin said.
Migraineurs have a two- to threefold greater prevalence of depression. There is a bidirectional relationship between migraine and depression in population studies. The relationship between migraine and depression is greater in clinic populations, in chronic migraine, and in medication overuse headache.
There is a significant relationship between migraine and anxiety disorders. Anxiety disorders entail a sense of danger, fear, or worry. “All patients with anxiety disorder have physical symptoms, and they often show avoidance behaviors,” said Dr. Baskin. For example, in generalized anxiety disorder, the object of avoidance is probably the fear of uncertainty. People worry excessively, thinking they are avoiding uncertainty. “Avoidance learning is huge in anxiety,” said Dr. Baskin. People fear unexpected events and perceive things as more unmanageable, dangerous, or threatening than they objectively are. “We see that frequently in migraineurs with anxiety disorders,” he said. Dr. Baskin noted that anxious patients tend to be sensitive to medication side effects and somatic sensations in general.
Patients with headache and anxiety often develop strong fear reactions. They identify a warning signal for headache, real or imagined, and may treat their fear, which they perceive as a headache prodrome, with a medication. “There is a preemptive strike with medicine treating a sensation that may or may not develop into headache,” Dr. Baskin said. That medication reduces their emotional distress and prevents the migraine, a powerful avoidance learning paradigm. That cycle can be a major part of medication overuse headache.
Anxiety disorders are much more prevalent than depression in migraineurs. They are associated with greater long-term persistence of headache, greater headache-related disability, and reduced satisfaction with acute therapy. “Across all emotional disorders, anxiety is the driver of distress,” Dr. Baskin said. “When you add anxiety to any disorder, it becomes much more problematic.”
The concept of interoceptive awareness—an individual’s sensitivity to bodily signals—is important in people with panic disorder. Like people with panic disorder, some migraineurs often have high interoceptive awareness. Anxiety sensitivity—fear that benign physical sensations will have harmful or catastrophic consequences—also may be a factor in panic disorder, as well as migraine. As in panic disorder, highly anxious migraineurs may develop hypervigilance to somatic sensations and conditioned fear to these internal sensations.
Dr. Baskin recommends screening patients for psychiatric comorbidity. He recommends two questions from the Patient Health Questionnaire-9 screener—“Do you have little interest or pleasure in doing things?” and “Are you feeling down, depressed, or hopeless?”—and two additional anxiety questions—“Are you feeling anxious, nervous, or on edge?” and “Are you unable to stop or control worrying?” “With just those four questions, you can capture a good number of people with anxiety or mood issues,” Dr. Baskin said.
Behavioral Therapy
“Our behavioral medicine program is time-limited and goal-oriented,” said Dr. Baskin. “We try to get people to develop self-efficacy and personal responsibility. We monitor and maximize adherence to medications and help patients to regulate their routine activities, including going to bed, getting up, and exercising on a consistent schedule. We offer biofeedback for self-regulation, relaxation and coping skills training utilizing a cognitive behavioral model, and we treat psychiatric issues,” said Dr. Baskin. Many of those things can be done by clinicians who are not behavioral clinicians, he said. “You do not necessarily have to refer all these people. Schedule frequent revisits for complicated or difficult patients. Do not overwhelm patients with too much information. Simplify jargon, provide written instructions, and make sure the patient understands the plan.”
An important component of behavioral treatment is teaching relaxation exercises, which can reduce muscle tension and autonomic arousal. There are many types of relaxation strategies. Dr. Baskin recommended breathing pacer apps, which are designed to encourage slower abdominal breathing. The goal is to gradually reduce the breathing rate to six breaths per minute for five- to 10-minute practice sessions. “If you can teach people diaphragmatic breathing—it takes about 30 seconds to begin the discussion—it is helpful.” Dr. Baskin recommended having patients do it three to five times per day for a few minutes at a time.
“We deliver relaxation training alone, sometimes with biofeedback, and teach it as a self-regulation coping skill. We try to get people to develop an internal locus of control so they can manage some of their own physiology, relax muscles, and learn a nonspecific low-arousal response and use it as a coping skill to apply in different situations,” Dr. Baskin said.
Cognitive behavioral therapy is another tool. It gives people an opportunity to modify distress-related thoughts and to examine their personal danger cognitions, their sense of threat, and the negative predictions that they may have. Dr. Baskin uses cognitive behavioral therapy to help patients develop an action plan based on their prescribed strategy to treat an acute migraine attack as well as manage concomitant emotional reactivity and maintain functionality in the presence of a significant headache disorder.
Trigger Management
Historically, migraineurs have avoided headache triggers. The down side to that strategy is that they can unnecessarily restrict themselves. Studies suggest that avoiding triggers may lead to sensitization to those triggers. Gradual exposure coping models are being developed. “Cope, do not avoid,” Dr. Baskin said.
Biofeedback
Relaxation training, EMG biofeedback, thermal biofeedback, and cognitive behavioral therapy show grade A but modest efficacy. There is recent evidence that combining behavioral therapy with preventive pharmacologic treatment improves outcomes. The behavioral section of the American Headache Society will soon be reviewing the most recent evidence on behavioral interventions in migraine.
Sleep Hygiene
Three main messages regarding sleep are to adopt a routine, consistent bedtime and wake up time, avoid all non–sleep-related activities at bedtime, and employ relaxation strategies to reduce sleep onset latency. In addition, patients should not eat or drink a lot of fluid too close to bedtime, not exercise in the evening, and avoid napping. A patient should be advised that if he or she cannot sleep, the best solution is to get out of bed, go to another room in the house, engage in a relaxing activity, and go back to bed when he or she gets tired. With these strategies, clinicians have converted chronic migraine to episodic migraine for a significant number of patients.
Combination Treatment of Migraine and Psychiatric Disorder
Many doctors support the idea that one drug should treat migraine and associated conditions whenever possible. The idea is to use one agent to treat migraine and associated conditions (“two-fer”) This strategy is simpler and may entail lower cost, fewer adverse events, and fewer potential drug interactions. “It makes sense on one level, however, there’s a risk of treating only one condition optimally or treating none of them optimally,” Dr. Baskin said. “It is important to treat both disorders the way you think they should be treated. Sometimes two drugs are better than one. You want to treat both conditions effectively.”
When treating anxiety disorders with selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs), prescribers should know that people with anxiety are exceptionally sensitive to side effects. Also, anxiety disorders often require higher doses than treating depression. “You should start dosing incredibly low and titrate slowly,” Dr. Baskin said.
—Glenn S. Williams
Multiaxial Assessment
All workups for headache evaluate frequency, intensity, duration, and disability. From a behavioral perspective, Dr. Baskin recommended considering a few more factors—adherence to therapy, stress-related issues, comorbid psychiatric disorders, and factors that transform migraine from episodic to chronic.
“When you talk about adherence, one of the basics of this is readiness to change,” said Dr. Baskin. “Is the patient motivated for treatment? That doesn’t necessarily happen immediately.” For example, a patient who is motivated to have an abortive agent might not be motivated to pursue prevention, lose weight, exercise, or make lifestyle changes. “Over time, that might change,” Dr. Baskin noted. Patient motivation may evolve with a greater understanding of the goals of treatment.
“Has the patient adhered to past therapy regimens? If the answer is no, then the answer will most likely be no again,” Dr. Baskin said. If medication overuse was a problem, it may be again. Whenever possible, ask open-ended questions, such as “how do you decide when to take an acute medication?” Dr. Baskin advised.
Stress and Migraine
High levels of daily stress, even daily stressors that are not catastrophic, can transform migraine from episodic to chronic. “Patients with depression show a greater effect of stress,” Dr. Baskin said. Similarly, victims of trauma or childhood maltreatment can have more disabling headaches that may be more likely to transform into daily headaches. “Many patients do not have the coping skills necessary to manage stress or recurrent headache,” Dr. Baskin said. One of the goals of behavioral therapy for migraine is to increase patients’ self-efficacy and give them the tools and the confidence to manage headaches and stressors.
Psychiatric Comorbidity
Psychiatric comorbidity may complicate differential diagnosis, and nonadherence to medication is greater in people with mood disorders and anxiety disorders. These patients may show a reduced response to pharmacologic and behavioral treatments for headache. Psychiatric comorbidities also can contribute to migraine chronification. “However, if you add a small behavioral component to your headache program, patients with psychiatric comorbidities seem to respond relatively well,” Dr. Baskin said.
Migraineurs have a two- to threefold greater prevalence of depression. There is a bidirectional relationship between migraine and depression in population studies. The relationship between migraine and depression is greater in clinic populations, in chronic migraine, and in medication overuse headache.
There is a significant relationship between migraine and anxiety disorders. Anxiety disorders entail a sense of danger, fear, or worry. “All patients with anxiety disorder have physical symptoms, and they often show avoidance behaviors,” said Dr. Baskin. For example, in generalized anxiety disorder, the object of avoidance is probably the fear of uncertainty. People worry excessively, thinking they are avoiding uncertainty. “Avoidance learning is huge in anxiety,” said Dr. Baskin. People fear unexpected events and perceive things as more unmanageable, dangerous, or threatening than they objectively are. “We see that frequently in migraineurs with anxiety disorders,” he said. Dr. Baskin noted that anxious patients tend to be sensitive to medication side effects and somatic sensations in general.
Patients with headache and anxiety often develop strong fear reactions. They identify a warning signal for headache, real or imagined, and may treat their fear, which they perceive as a headache prodrome, with a medication. “There is a preemptive strike with medicine treating a sensation that may or may not develop into headache,” Dr. Baskin said. That medication reduces their emotional distress and prevents the migraine, a powerful avoidance learning paradigm. That cycle can be a major part of medication overuse headache.
Anxiety disorders are much more prevalent than depression in migraineurs. They are associated with greater long-term persistence of headache, greater headache-related disability, and reduced satisfaction with acute therapy. “Across all emotional disorders, anxiety is the driver of distress,” Dr. Baskin said. “When you add anxiety to any disorder, it becomes much more problematic.”
The concept of interoceptive awareness—an individual’s sensitivity to bodily signals—is important in people with panic disorder. Like people with panic disorder, some migraineurs often have high interoceptive awareness. Anxiety sensitivity—fear that benign physical sensations will have harmful or catastrophic consequences—also may be a factor in panic disorder, as well as migraine. As in panic disorder, highly anxious migraineurs may develop hypervigilance to somatic sensations and conditioned fear to these internal sensations.
Dr. Baskin recommends screening patients for psychiatric comorbidity. He recommends two questions from the Patient Health Questionnaire-9 screener—“Do you have little interest or pleasure in doing things?” and “Are you feeling down, depressed, or hopeless?”—and two additional anxiety questions—“Are you feeling anxious, nervous, or on edge?” and “Are you unable to stop or control worrying?” “With just those four questions, you can capture a good number of people with anxiety or mood issues,” Dr. Baskin said.
Behavioral Therapy
“Our behavioral medicine program is time-limited and goal-oriented,” said Dr. Baskin. “We try to get people to develop self-efficacy and personal responsibility. We monitor and maximize adherence to medications and help patients to regulate their routine activities, including going to bed, getting up, and exercising on a consistent schedule. We offer biofeedback for self-regulation, relaxation and coping skills training utilizing a cognitive behavioral model, and we treat psychiatric issues,” said Dr. Baskin. Many of those things can be done by clinicians who are not behavioral clinicians, he said. “You do not necessarily have to refer all these people. Schedule frequent revisits for complicated or difficult patients. Do not overwhelm patients with too much information. Simplify jargon, provide written instructions, and make sure the patient understands the plan.”
An important component of behavioral treatment is teaching relaxation exercises, which can reduce muscle tension and autonomic arousal. There are many types of relaxation strategies. Dr. Baskin recommended breathing pacer apps, which are designed to encourage slower abdominal breathing. The goal is to gradually reduce the breathing rate to six breaths per minute for five- to 10-minute practice sessions. “If you can teach people diaphragmatic breathing—it takes about 30 seconds to begin the discussion—it is helpful.” Dr. Baskin recommended having patients do it three to five times per day for a few minutes at a time.
“We deliver relaxation training alone, sometimes with biofeedback, and teach it as a self-regulation coping skill. We try to get people to develop an internal locus of control so they can manage some of their own physiology, relax muscles, and learn a nonspecific low-arousal response and use it as a coping skill to apply in different situations,” Dr. Baskin said.
Cognitive behavioral therapy is another tool. It gives people an opportunity to modify distress-related thoughts and to examine their personal danger cognitions, their sense of threat, and the negative predictions that they may have. Dr. Baskin uses cognitive behavioral therapy to help patients develop an action plan based on their prescribed strategy to treat an acute migraine attack as well as manage concomitant emotional reactivity and maintain functionality in the presence of a significant headache disorder.
Trigger Management
Historically, migraineurs have avoided headache triggers. The down side to that strategy is that they can unnecessarily restrict themselves. Studies suggest that avoiding triggers may lead to sensitization to those triggers. Gradual exposure coping models are being developed. “Cope, do not avoid,” Dr. Baskin said.
Biofeedback
Relaxation training, EMG biofeedback, thermal biofeedback, and cognitive behavioral therapy show grade A but modest efficacy. There is recent evidence that combining behavioral therapy with preventive pharmacologic treatment improves outcomes. The behavioral section of the American Headache Society will soon be reviewing the most recent evidence on behavioral interventions in migraine.
Sleep Hygiene
Three main messages regarding sleep are to adopt a routine, consistent bedtime and wake up time, avoid all non–sleep-related activities at bedtime, and employ relaxation strategies to reduce sleep onset latency. In addition, patients should not eat or drink a lot of fluid too close to bedtime, not exercise in the evening, and avoid napping. A patient should be advised that if he or she cannot sleep, the best solution is to get out of bed, go to another room in the house, engage in a relaxing activity, and go back to bed when he or she gets tired. With these strategies, clinicians have converted chronic migraine to episodic migraine for a significant number of patients.
Combination Treatment of Migraine and Psychiatric Disorder
Many doctors support the idea that one drug should treat migraine and associated conditions whenever possible. The idea is to use one agent to treat migraine and associated conditions (“two-fer”) This strategy is simpler and may entail lower cost, fewer adverse events, and fewer potential drug interactions. “It makes sense on one level, however, there’s a risk of treating only one condition optimally or treating none of them optimally,” Dr. Baskin said. “It is important to treat both disorders the way you think they should be treated. Sometimes two drugs are better than one. You want to treat both conditions effectively.”
When treating anxiety disorders with selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs), prescribers should know that people with anxiety are exceptionally sensitive to side effects. Also, anxiety disorders often require higher doses than treating depression. “You should start dosing incredibly low and titrate slowly,” Dr. Baskin said.
—Glenn S. Williams
Multiaxial Assessment
All workups for headache evaluate frequency, intensity, duration, and disability. From a behavioral perspective, Dr. Baskin recommended considering a few more factors—adherence to therapy, stress-related issues, comorbid psychiatric disorders, and factors that transform migraine from episodic to chronic.
“When you talk about adherence, one of the basics of this is readiness to change,” said Dr. Baskin. “Is the patient motivated for treatment? That doesn’t necessarily happen immediately.” For example, a patient who is motivated to have an abortive agent might not be motivated to pursue prevention, lose weight, exercise, or make lifestyle changes. “Over time, that might change,” Dr. Baskin noted. Patient motivation may evolve with a greater understanding of the goals of treatment.
“Has the patient adhered to past therapy regimens? If the answer is no, then the answer will most likely be no again,” Dr. Baskin said. If medication overuse was a problem, it may be again. Whenever possible, ask open-ended questions, such as “how do you decide when to take an acute medication?” Dr. Baskin advised.
Stress and Migraine
High levels of daily stress, even daily stressors that are not catastrophic, can transform migraine from episodic to chronic. “Patients with depression show a greater effect of stress,” Dr. Baskin said. Similarly, victims of trauma or childhood maltreatment can have more disabling headaches that may be more likely to transform into daily headaches. “Many patients do not have the coping skills necessary to manage stress or recurrent headache,” Dr. Baskin said. One of the goals of behavioral therapy for migraine is to increase patients’ self-efficacy and give them the tools and the confidence to manage headaches and stressors.
Psychiatric Comorbidity
Psychiatric comorbidity may complicate differential diagnosis, and nonadherence to medication is greater in people with mood disorders and anxiety disorders. These patients may show a reduced response to pharmacologic and behavioral treatments for headache. Psychiatric comorbidities also can contribute to migraine chronification. “However, if you add a small behavioral component to your headache program, patients with psychiatric comorbidities seem to respond relatively well,” Dr. Baskin said.
Migraineurs have a two- to threefold greater prevalence of depression. There is a bidirectional relationship between migraine and depression in population studies. The relationship between migraine and depression is greater in clinic populations, in chronic migraine, and in medication overuse headache.
There is a significant relationship between migraine and anxiety disorders. Anxiety disorders entail a sense of danger, fear, or worry. “All patients with anxiety disorder have physical symptoms, and they often show avoidance behaviors,” said Dr. Baskin. For example, in generalized anxiety disorder, the object of avoidance is probably the fear of uncertainty. People worry excessively, thinking they are avoiding uncertainty. “Avoidance learning is huge in anxiety,” said Dr. Baskin. People fear unexpected events and perceive things as more unmanageable, dangerous, or threatening than they objectively are. “We see that frequently in migraineurs with anxiety disorders,” he said. Dr. Baskin noted that anxious patients tend to be sensitive to medication side effects and somatic sensations in general.
Patients with headache and anxiety often develop strong fear reactions. They identify a warning signal for headache, real or imagined, and may treat their fear, which they perceive as a headache prodrome, with a medication. “There is a preemptive strike with medicine treating a sensation that may or may not develop into headache,” Dr. Baskin said. That medication reduces their emotional distress and prevents the migraine, a powerful avoidance learning paradigm. That cycle can be a major part of medication overuse headache.
Anxiety disorders are much more prevalent than depression in migraineurs. They are associated with greater long-term persistence of headache, greater headache-related disability, and reduced satisfaction with acute therapy. “Across all emotional disorders, anxiety is the driver of distress,” Dr. Baskin said. “When you add anxiety to any disorder, it becomes much more problematic.”
The concept of interoceptive awareness—an individual’s sensitivity to bodily signals—is important in people with panic disorder. Like people with panic disorder, some migraineurs often have high interoceptive awareness. Anxiety sensitivity—fear that benign physical sensations will have harmful or catastrophic consequences—also may be a factor in panic disorder, as well as migraine. As in panic disorder, highly anxious migraineurs may develop hypervigilance to somatic sensations and conditioned fear to these internal sensations.
Dr. Baskin recommends screening patients for psychiatric comorbidity. He recommends two questions from the Patient Health Questionnaire-9 screener—“Do you have little interest or pleasure in doing things?” and “Are you feeling down, depressed, or hopeless?”—and two additional anxiety questions—“Are you feeling anxious, nervous, or on edge?” and “Are you unable to stop or control worrying?” “With just those four questions, you can capture a good number of people with anxiety or mood issues,” Dr. Baskin said.
Behavioral Therapy
“Our behavioral medicine program is time-limited and goal-oriented,” said Dr. Baskin. “We try to get people to develop self-efficacy and personal responsibility. We monitor and maximize adherence to medications and help patients to regulate their routine activities, including going to bed, getting up, and exercising on a consistent schedule. We offer biofeedback for self-regulation, relaxation and coping skills training utilizing a cognitive behavioral model, and we treat psychiatric issues,” said Dr. Baskin. Many of those things can be done by clinicians who are not behavioral clinicians, he said. “You do not necessarily have to refer all these people. Schedule frequent revisits for complicated or difficult patients. Do not overwhelm patients with too much information. Simplify jargon, provide written instructions, and make sure the patient understands the plan.”
An important component of behavioral treatment is teaching relaxation exercises, which can reduce muscle tension and autonomic arousal. There are many types of relaxation strategies. Dr. Baskin recommended breathing pacer apps, which are designed to encourage slower abdominal breathing. The goal is to gradually reduce the breathing rate to six breaths per minute for five- to 10-minute practice sessions. “If you can teach people diaphragmatic breathing—it takes about 30 seconds to begin the discussion—it is helpful.” Dr. Baskin recommended having patients do it three to five times per day for a few minutes at a time.
“We deliver relaxation training alone, sometimes with biofeedback, and teach it as a self-regulation coping skill. We try to get people to develop an internal locus of control so they can manage some of their own physiology, relax muscles, and learn a nonspecific low-arousal response and use it as a coping skill to apply in different situations,” Dr. Baskin said.
Cognitive behavioral therapy is another tool. It gives people an opportunity to modify distress-related thoughts and to examine their personal danger cognitions, their sense of threat, and the negative predictions that they may have. Dr. Baskin uses cognitive behavioral therapy to help patients develop an action plan based on their prescribed strategy to treat an acute migraine attack as well as manage concomitant emotional reactivity and maintain functionality in the presence of a significant headache disorder.
Trigger Management
Historically, migraineurs have avoided headache triggers. The down side to that strategy is that they can unnecessarily restrict themselves. Studies suggest that avoiding triggers may lead to sensitization to those triggers. Gradual exposure coping models are being developed. “Cope, do not avoid,” Dr. Baskin said.
Biofeedback
Relaxation training, EMG biofeedback, thermal biofeedback, and cognitive behavioral therapy show grade A but modest efficacy. There is recent evidence that combining behavioral therapy with preventive pharmacologic treatment improves outcomes. The behavioral section of the American Headache Society will soon be reviewing the most recent evidence on behavioral interventions in migraine.
Sleep Hygiene
Three main messages regarding sleep are to adopt a routine, consistent bedtime and wake up time, avoid all non–sleep-related activities at bedtime, and employ relaxation strategies to reduce sleep onset latency. In addition, patients should not eat or drink a lot of fluid too close to bedtime, not exercise in the evening, and avoid napping. A patient should be advised that if he or she cannot sleep, the best solution is to get out of bed, go to another room in the house, engage in a relaxing activity, and go back to bed when he or she gets tired. With these strategies, clinicians have converted chronic migraine to episodic migraine for a significant number of patients.
Combination Treatment of Migraine and Psychiatric Disorder
Many doctors support the idea that one drug should treat migraine and associated conditions whenever possible. The idea is to use one agent to treat migraine and associated conditions (“two-fer”) This strategy is simpler and may entail lower cost, fewer adverse events, and fewer potential drug interactions. “It makes sense on one level, however, there’s a risk of treating only one condition optimally or treating none of them optimally,” Dr. Baskin said. “It is important to treat both disorders the way you think they should be treated. Sometimes two drugs are better than one. You want to treat both conditions effectively.”
When treating anxiety disorders with selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs), prescribers should know that people with anxiety are exceptionally sensitive to side effects. Also, anxiety disorders often require higher doses than treating depression. “You should start dosing incredibly low and titrate slowly,” Dr. Baskin said.
—Glenn S. Williams
Early-Onset Parkinson’s Disease Psychosis May Be Linked to Amyloid Pathology
Patients with newly diagnosed Parkinson’s disease and reduced CSF amyloid β1–42 may be more likely to develop Parkinson’s disease psychosis within three to four years, according to a study published in the April issue of the Journal of Neurology, Neurosurgery and Psychiatry. The type of psychotic symptom (eg, illusions or formed hallucinations) may indicate a specific profile of visual, cognitive, cortical, and hippocampal involvement in Parkinson’s disease, said the authors.

“Our finding may suggest [that] early-onset Parkinson’s disease psychosis is a biomarker for the subsequent development of Alzheimer pathology,” said Dominic H. Ffytche, MD, PhD, of the Department of Old Age Psychiatry and Dementia at King’s College London.
Previous studies have indicated that visual hallucinations and illusions may be caused by specific cognitive and higher visual function deficits. Patients who develop these symptoms early in the disease course have greater rates of cognitive decline and progression to dementia, said the authors. No studies have investigated whether cognitive and higher visual function deficits are found before the onset of Parkinson’s disease, however.
Dr. Ffytche and colleagues examined the profile of cognitive, biomarker, and other risk factors before the onset of illusions and hallucinations in the Parkinson’s Progression Markers Initiative (PPMI) data set.
The PPMI is an observational multicenter study of newly diagnosed, untreated patients and healthy controls. It includes standardized, clinical, imaging, CSF, and cognitive assessments at three-month intervals during the first year and at six-month intervals in subsequent years. Assessment tools include the Montreal Cognitive Assessment, the Benton Judgement of Line Orientation, and the Unified Parkinson’s Disease Rating Scale.
Dr. Ffytche’s group included 195 healthy controls and 423 patients with Parkinson’s disease in its analysis. They compared baseline assessments in patients who developed illusions or hallucinations within three to four years of follow-up and in patients who did not develop these symptoms.
Of all patients with Parkinson’s disease, 115 people reported psychotic symptoms (predominantly illusions) at a median time of 19.5 months. At baseline, these patients had reduced CSF amyloid β1–42, lower olfaction scores, higher depression scores, and increased REM sleep behavior disorder symptoms, compared with patients without early-onset Parkinson’s disease psychosis. No differences in cognitive, higher visual, or structural imaging measures were observed, however. Dr. Ffytche and colleagues also found a subset of 21 participants with early-onset formed hallucinations who had reduced higher visual function at baseline; cortical thinning in the parietal, occipital, and frontal cortex; and reduced hippocampal volume.
One limitation of the study was its lack of phenomenologic detail about the symptoms of Parkinson’s disease psychosis. In addition, information was collected about symptoms during the week before each given assessment, thus some symptoms that occurred between assessments were missed.
—Erica Tricarico
Suggested Reading
Ffytche DH, Pereira JB, Ballard C, et al. Risk factors for early psychosis in PD: insights from the Parkinson’s Progression Markers Initiative. J Neurol Neurosurg Psychiatry. 2017;88(4):325-331.
Patients with newly diagnosed Parkinson’s disease and reduced CSF amyloid β1–42 may be more likely to develop Parkinson’s disease psychosis within three to four years, according to a study published in the April issue of the Journal of Neurology, Neurosurgery and Psychiatry. The type of psychotic symptom (eg, illusions or formed hallucinations) may indicate a specific profile of visual, cognitive, cortical, and hippocampal involvement in Parkinson’s disease, said the authors.
“Our finding may suggest [that] early-onset Parkinson’s disease psychosis is a biomarker for the subsequent development of Alzheimer pathology,” said Dominic H. Ffytche, MD, PhD, of the Department of Old Age Psychiatry and Dementia at King’s College London.
Previous studies have indicated that visual hallucinations and illusions may be caused by specific cognitive and higher visual function deficits. Patients who develop these symptoms early in the disease course have greater rates of cognitive decline and progression to dementia, said the authors. No studies have investigated whether cognitive and higher visual function deficits are found before the onset of Parkinson’s disease, however.
Dr. Ffytche and colleagues examined the profile of cognitive, biomarker, and other risk factors before the onset of illusions and hallucinations in the Parkinson’s Progression Markers Initiative (PPMI) data set.
The PPMI is an observational multicenter study of newly diagnosed, untreated patients and healthy controls. It includes standardized, clinical, imaging, CSF, and cognitive assessments at three-month intervals during the first year and at six-month intervals in subsequent years. Assessment tools include the Montreal Cognitive Assessment, the Benton Judgement of Line Orientation, and the Unified Parkinson’s Disease Rating Scale.
Dr. Ffytche’s group included 195 healthy controls and 423 patients with Parkinson’s disease in its analysis. They compared baseline assessments in patients who developed illusions or hallucinations within three to four years of follow-up and in patients who did not develop these symptoms.
Of all patients with Parkinson’s disease, 115 people reported psychotic symptoms (predominantly illusions) at a median time of 19.5 months. At baseline, these patients had reduced CSF amyloid β1–42, lower olfaction scores, higher depression scores, and increased REM sleep behavior disorder symptoms, compared with patients without early-onset Parkinson’s disease psychosis. No differences in cognitive, higher visual, or structural imaging measures were observed, however. Dr. Ffytche and colleagues also found a subset of 21 participants with early-onset formed hallucinations who had reduced higher visual function at baseline; cortical thinning in the parietal, occipital, and frontal cortex; and reduced hippocampal volume.
One limitation of the study was its lack of phenomenologic detail about the symptoms of Parkinson’s disease psychosis. In addition, information was collected about symptoms during the week before each given assessment, thus some symptoms that occurred between assessments were missed.
—Erica Tricarico
Suggested Reading
Ffytche DH, Pereira JB, Ballard C, et al. Risk factors for early psychosis in PD: insights from the Parkinson’s Progression Markers Initiative. J Neurol Neurosurg Psychiatry. 2017;88(4):325-331.
Patients with newly diagnosed Parkinson’s disease and reduced CSF amyloid β1–42 may be more likely to develop Parkinson’s disease psychosis within three to four years, according to a study published in the April issue of the Journal of Neurology, Neurosurgery and Psychiatry. The type of psychotic symptom (eg, illusions or formed hallucinations) may indicate a specific profile of visual, cognitive, cortical, and hippocampal involvement in Parkinson’s disease, said the authors.
“Our finding may suggest [that] early-onset Parkinson’s disease psychosis is a biomarker for the subsequent development of Alzheimer pathology,” said Dominic H. Ffytche, MD, PhD, of the Department of Old Age Psychiatry and Dementia at King’s College London.
Previous studies have indicated that visual hallucinations and illusions may be caused by specific cognitive and higher visual function deficits. Patients who develop these symptoms early in the disease course have greater rates of cognitive decline and progression to dementia, said the authors. No studies have investigated whether cognitive and higher visual function deficits are found before the onset of Parkinson’s disease, however.
Dr. Ffytche and colleagues examined the profile of cognitive, biomarker, and other risk factors before the onset of illusions and hallucinations in the Parkinson’s Progression Markers Initiative (PPMI) data set.
The PPMI is an observational multicenter study of newly diagnosed, untreated patients and healthy controls. It includes standardized, clinical, imaging, CSF, and cognitive assessments at three-month intervals during the first year and at six-month intervals in subsequent years. Assessment tools include the Montreal Cognitive Assessment, the Benton Judgement of Line Orientation, and the Unified Parkinson’s Disease Rating Scale.
Dr. Ffytche’s group included 195 healthy controls and 423 patients with Parkinson’s disease in its analysis. They compared baseline assessments in patients who developed illusions or hallucinations within three to four years of follow-up and in patients who did not develop these symptoms.
Of all patients with Parkinson’s disease, 115 people reported psychotic symptoms (predominantly illusions) at a median time of 19.5 months. At baseline, these patients had reduced CSF amyloid β1–42, lower olfaction scores, higher depression scores, and increased REM sleep behavior disorder symptoms, compared with patients without early-onset Parkinson’s disease psychosis. No differences in cognitive, higher visual, or structural imaging measures were observed, however. Dr. Ffytche and colleagues also found a subset of 21 participants with early-onset formed hallucinations who had reduced higher visual function at baseline; cortical thinning in the parietal, occipital, and frontal cortex; and reduced hippocampal volume.
One limitation of the study was its lack of phenomenologic detail about the symptoms of Parkinson’s disease psychosis. In addition, information was collected about symptoms during the week before each given assessment, thus some symptoms that occurred between assessments were missed.
—Erica Tricarico
Suggested Reading
Ffytche DH, Pereira JB, Ballard C, et al. Risk factors for early psychosis in PD: insights from the Parkinson’s Progression Markers Initiative. J Neurol Neurosurg Psychiatry. 2017;88(4):325-331.
