User login
Children and Teens at Rising Risk for Diabetes
Diabetes is on the rise among children and teens in the U.S. Between 2002 and 2012, type 1 diabetes mellitus (T1DM) rose 1.8% annually and type 2 DM (T2DM) rose 4.8% annually, according to findings from the SEARCH for Diabetes in Youth study. The study involved 11,245 participants aged ≤ 19 years with T1 DM and 2,846 aged 10 to 19 years with T2DM.
Minority racial and ethnic groups saw the highest increases. The greatest rise (4.2%) in T1 DM was among Hispanics. The greatest increases of T2DM were in non-Hispanic blacks, Asians/Pacific Islanders, and Native Americans. The annual rate among Native American youth was 8.9%, followed by 8.5% among Asian Americans/Pacific Islanders, and 6.3% among non-Hispanic blacks. The researchers note that the results for Native Americans cannot be generalized to all Native American youth nationwide.
Across all ethnic/racial groups, T 1DM increased more each year in males (2.2%) than in females (1.4%). Type 2 DM, by contrast, increased twice as fast in girls as boys aged 10 to 19 years (6.2% vs 3.7%).
The study is the first to estimate trends in newly diagnosed cases of diabetes among people aged < 20 years from the 5 major racial and ethnic groups in the U.S.
Diabetes is on the rise among children and teens in the U.S. Between 2002 and 2012, type 1 diabetes mellitus (T1DM) rose 1.8% annually and type 2 DM (T2DM) rose 4.8% annually, according to findings from the SEARCH for Diabetes in Youth study. The study involved 11,245 participants aged ≤ 19 years with T1 DM and 2,846 aged 10 to 19 years with T2DM.
Minority racial and ethnic groups saw the highest increases. The greatest rise (4.2%) in T1 DM was among Hispanics. The greatest increases of T2DM were in non-Hispanic blacks, Asians/Pacific Islanders, and Native Americans. The annual rate among Native American youth was 8.9%, followed by 8.5% among Asian Americans/Pacific Islanders, and 6.3% among non-Hispanic blacks. The researchers note that the results for Native Americans cannot be generalized to all Native American youth nationwide.
Across all ethnic/racial groups, T 1DM increased more each year in males (2.2%) than in females (1.4%). Type 2 DM, by contrast, increased twice as fast in girls as boys aged 10 to 19 years (6.2% vs 3.7%).
The study is the first to estimate trends in newly diagnosed cases of diabetes among people aged < 20 years from the 5 major racial and ethnic groups in the U.S.
Diabetes is on the rise among children and teens in the U.S. Between 2002 and 2012, type 1 diabetes mellitus (T1DM) rose 1.8% annually and type 2 DM (T2DM) rose 4.8% annually, according to findings from the SEARCH for Diabetes in Youth study. The study involved 11,245 participants aged ≤ 19 years with T1 DM and 2,846 aged 10 to 19 years with T2DM.
Minority racial and ethnic groups saw the highest increases. The greatest rise (4.2%) in T1 DM was among Hispanics. The greatest increases of T2DM were in non-Hispanic blacks, Asians/Pacific Islanders, and Native Americans. The annual rate among Native American youth was 8.9%, followed by 8.5% among Asian Americans/Pacific Islanders, and 6.3% among non-Hispanic blacks. The researchers note that the results for Native Americans cannot be generalized to all Native American youth nationwide.
Across all ethnic/racial groups, T 1DM increased more each year in males (2.2%) than in females (1.4%). Type 2 DM, by contrast, increased twice as fast in girls as boys aged 10 to 19 years (6.2% vs 3.7%).
The study is the first to estimate trends in newly diagnosed cases of diabetes among people aged < 20 years from the 5 major racial and ethnic groups in the U.S.
Sunny Side's Up
A year ago, this 60-year-old man noticed an asymptomatic lesion on the dorsum of his right hand. When it grew in size over the course of a few months, he showed it to his primary care provider, who believed it to be a wart and froze it with liquid nitrogen. This reduced its size, but only temporarily. It has since been treated with topical and oral antibiotics to no avail.
The patient has had several basal cell carcinomas removed from his face, arms, and trunk in the past.
EXAMINATION
On the mid dorsum of the patient’s right hand is a 1.5-cm ovoid nodule with a smooth surface and very firm feel. It appears in the context of fully sun-exposed, sun-damaged skin. Several scars are noted in the area, consistent with his history of sun-caused skin cancers.

The lesion is removed by deep shave biopsy, and the base curetted. The entire lesion is sent to pathology.
What’s the diagnosis?
DISCUSSION
The pathology report shows a low-grade, well-differentiated squamous cell carcinoma (SCC)—in this case, a keratoacanthoma (KA). This common form of SCC is usually found on the sun-exposed skin of older patients. The lesions can range in size from 3 mm to 3 cm or larger and are usually round to oval and dome-like, with symmetrical architecture and, often, a central keratotic core. The differential includes cysts, warts, and seborrheic keratosis.
Histologically, KAs are composed of uniformly staining (blue) cells of similar size and shape (connoting relative benignancy), to which we apply the term well-differentiated. Poorly-differentiated cellular composition manifests with cells of different sizes, shapes, and colors; these characteristics suggest more aggressive malignancy.
Even though KAs are skin cancers, they are quite low-grade, which means they rarely metastasize; if left alone, they can resolve completely over time. However, their odd appearance and rapid growth are usually concerning enough to prompt their removal.
When suspected KAs are removed, it’s essential that the entire lesion be submitted for pathologic examination. This allows for the architecture of the entire lesion—its cellular composition and margins—to be evaluated. When only part of the lesion is removed for biopsy, the diagnosis will be “squamous cell carcinoma, well differentiated, without evidence of invasion.” In the minds of many dermatology providers, this diagnosis demands excision—but a KA lesion completely removed by shave biopsy is considered cured.
Histologic examination of these lesions is not always as straightforward as in this case. KAs can be poorly differentiated or demonstrate focal areas of invasion, which justifies excision with margins.
TAKE-HOME LEARNING POINTS
- Keratoacanthoma (KA) is an extremely common low-grade squamous cell carcinoma most often seen on directly sun-exposed skin (eg, hands, arms, face, ears) of older, sun-damaged patients.
- KA typically manifests as a round to oval, dome-like, firm nodule, often with a central keratotic core and a history of rapid growth.
- It’s important to remove these lesions in one piece (eg, by deep shave biopsy) because identification is based on architecture and cellular composition.
- The pathology report will show a well-differentiated squamous cell carcinoma with architecture consistent with KA.
- Although some believe that excision is necessary, a deep shave biopsy performed with clear margins is adequate treatment.
A year ago, this 60-year-old man noticed an asymptomatic lesion on the dorsum of his right hand. When it grew in size over the course of a few months, he showed it to his primary care provider, who believed it to be a wart and froze it with liquid nitrogen. This reduced its size, but only temporarily. It has since been treated with topical and oral antibiotics to no avail.
The patient has had several basal cell carcinomas removed from his face, arms, and trunk in the past.
EXAMINATION
On the mid dorsum of the patient’s right hand is a 1.5-cm ovoid nodule with a smooth surface and very firm feel. It appears in the context of fully sun-exposed, sun-damaged skin. Several scars are noted in the area, consistent with his history of sun-caused skin cancers.
The lesion is removed by deep shave biopsy, and the base curetted. The entire lesion is sent to pathology.
What’s the diagnosis?
DISCUSSION
The pathology report shows a low-grade, well-differentiated squamous cell carcinoma (SCC)—in this case, a keratoacanthoma (KA). This common form of SCC is usually found on the sun-exposed skin of older patients. The lesions can range in size from 3 mm to 3 cm or larger and are usually round to oval and dome-like, with symmetrical architecture and, often, a central keratotic core. The differential includes cysts, warts, and seborrheic keratosis.
Histologically, KAs are composed of uniformly staining (blue) cells of similar size and shape (connoting relative benignancy), to which we apply the term well-differentiated. Poorly-differentiated cellular composition manifests with cells of different sizes, shapes, and colors; these characteristics suggest more aggressive malignancy.
Even though KAs are skin cancers, they are quite low-grade, which means they rarely metastasize; if left alone, they can resolve completely over time. However, their odd appearance and rapid growth are usually concerning enough to prompt their removal.
When suspected KAs are removed, it’s essential that the entire lesion be submitted for pathologic examination. This allows for the architecture of the entire lesion—its cellular composition and margins—to be evaluated. When only part of the lesion is removed for biopsy, the diagnosis will be “squamous cell carcinoma, well differentiated, without evidence of invasion.” In the minds of many dermatology providers, this diagnosis demands excision—but a KA lesion completely removed by shave biopsy is considered cured.
Histologic examination of these lesions is not always as straightforward as in this case. KAs can be poorly differentiated or demonstrate focal areas of invasion, which justifies excision with margins.
TAKE-HOME LEARNING POINTS
- Keratoacanthoma (KA) is an extremely common low-grade squamous cell carcinoma most often seen on directly sun-exposed skin (eg, hands, arms, face, ears) of older, sun-damaged patients.
- KA typically manifests as a round to oval, dome-like, firm nodule, often with a central keratotic core and a history of rapid growth.
- It’s important to remove these lesions in one piece (eg, by deep shave biopsy) because identification is based on architecture and cellular composition.
- The pathology report will show a well-differentiated squamous cell carcinoma with architecture consistent with KA.
- Although some believe that excision is necessary, a deep shave biopsy performed with clear margins is adequate treatment.
A year ago, this 60-year-old man noticed an asymptomatic lesion on the dorsum of his right hand. When it grew in size over the course of a few months, he showed it to his primary care provider, who believed it to be a wart and froze it with liquid nitrogen. This reduced its size, but only temporarily. It has since been treated with topical and oral antibiotics to no avail.
The patient has had several basal cell carcinomas removed from his face, arms, and trunk in the past.
EXAMINATION
On the mid dorsum of the patient’s right hand is a 1.5-cm ovoid nodule with a smooth surface and very firm feel. It appears in the context of fully sun-exposed, sun-damaged skin. Several scars are noted in the area, consistent with his history of sun-caused skin cancers.
The lesion is removed by deep shave biopsy, and the base curetted. The entire lesion is sent to pathology.
What’s the diagnosis?
DISCUSSION
The pathology report shows a low-grade, well-differentiated squamous cell carcinoma (SCC)—in this case, a keratoacanthoma (KA). This common form of SCC is usually found on the sun-exposed skin of older patients. The lesions can range in size from 3 mm to 3 cm or larger and are usually round to oval and dome-like, with symmetrical architecture and, often, a central keratotic core. The differential includes cysts, warts, and seborrheic keratosis.
Histologically, KAs are composed of uniformly staining (blue) cells of similar size and shape (connoting relative benignancy), to which we apply the term well-differentiated. Poorly-differentiated cellular composition manifests with cells of different sizes, shapes, and colors; these characteristics suggest more aggressive malignancy.
Even though KAs are skin cancers, they are quite low-grade, which means they rarely metastasize; if left alone, they can resolve completely over time. However, their odd appearance and rapid growth are usually concerning enough to prompt their removal.
When suspected KAs are removed, it’s essential that the entire lesion be submitted for pathologic examination. This allows for the architecture of the entire lesion—its cellular composition and margins—to be evaluated. When only part of the lesion is removed for biopsy, the diagnosis will be “squamous cell carcinoma, well differentiated, without evidence of invasion.” In the minds of many dermatology providers, this diagnosis demands excision—but a KA lesion completely removed by shave biopsy is considered cured.
Histologic examination of these lesions is not always as straightforward as in this case. KAs can be poorly differentiated or demonstrate focal areas of invasion, which justifies excision with margins.
TAKE-HOME LEARNING POINTS
- Keratoacanthoma (KA) is an extremely common low-grade squamous cell carcinoma most often seen on directly sun-exposed skin (eg, hands, arms, face, ears) of older, sun-damaged patients.
- KA typically manifests as a round to oval, dome-like, firm nodule, often with a central keratotic core and a history of rapid growth.
- It’s important to remove these lesions in one piece (eg, by deep shave biopsy) because identification is based on architecture and cellular composition.
- The pathology report will show a well-differentiated squamous cell carcinoma with architecture consistent with KA.
- Although some believe that excision is necessary, a deep shave biopsy performed with clear margins is adequate treatment.
Single-cell analysis reveals TKI-resistant CML stem cells

Researchers say they’ve developed a technique for single-cell analysis that has revealed a population of treatment-resistant stem cells in patients with chronic myeloid leukemia (CML).
“It is increasingly recognized that tumors contain a variety of different cell types, including so-called cancer stem cells, that drive the growth and relapse of a patient’s cancer,” said study author Adam Mead, BM BCh, PhD, of University of Oxford in the UK.
“These cells can be very rare and extremely difficult to find after treatment as they become hidden within the normal tissue. We used a new genetic technique to identify and analyze single cancer stem cells in leukemia patients before and after treatment.”
Dr Mead and his colleagues detailed this research in Nature Medicine.
The team’s single-cell analysis technique combines high-sensitivity mutation detection with whole-transcriptome analysis.
The researchers used the method to analyze stem cells from patients with CML and found the cells to be heterogeneous.
In addition, the team was able to identify a subset of CML stem cells that proved resistant to treatment with tyrosine kinase inhibitors (TKIs).
“We found that, even in individual cases of leukemia, there are various types of cancer stem cell that respond differently to the treatment,” Dr Mead said.
“A small number of these cells are highly resistant to the treatment and are likely to be responsible for disease recurrence when the treatment is stopped. Our research allowed us uniquely to analyze these crucial cells that evade treatment so that we might learn how to more effectively eradicate them.”
The researchers said these TKI-resistant CML stem cells were “transcriptionally distinct” from normal hematopoietic stem cells.
The TKI-resistant cells were characterized by dysregulation of specific genes and pathways—TGF-β, TNF-α, JAK–STAT, CTNNB1, and NFKB1A—that could potentially be targeted to improve the treatment of CML.
The researchers also said their single-cell analysis technique can be used beyond CML.
“This technique could be adapted to analyze a range of different cancers to help predict both the likely response to treatment and the risk of the disease returning in the future,” Dr Mead said. “This should eventually enable treatment to be tailored to target each and every type of cancer stem cell that may be present.” ![]()
Researchers say they’ve developed a technique for single-cell analysis that has revealed a population of treatment-resistant stem cells in patients with chronic myeloid leukemia (CML).
“It is increasingly recognized that tumors contain a variety of different cell types, including so-called cancer stem cells, that drive the growth and relapse of a patient’s cancer,” said study author Adam Mead, BM BCh, PhD, of University of Oxford in the UK.
“These cells can be very rare and extremely difficult to find after treatment as they become hidden within the normal tissue. We used a new genetic technique to identify and analyze single cancer stem cells in leukemia patients before and after treatment.”
Dr Mead and his colleagues detailed this research in Nature Medicine.
The team’s single-cell analysis technique combines high-sensitivity mutation detection with whole-transcriptome analysis.
The researchers used the method to analyze stem cells from patients with CML and found the cells to be heterogeneous.
In addition, the team was able to identify a subset of CML stem cells that proved resistant to treatment with tyrosine kinase inhibitors (TKIs).
“We found that, even in individual cases of leukemia, there are various types of cancer stem cell that respond differently to the treatment,” Dr Mead said.
“A small number of these cells are highly resistant to the treatment and are likely to be responsible for disease recurrence when the treatment is stopped. Our research allowed us uniquely to analyze these crucial cells that evade treatment so that we might learn how to more effectively eradicate them.”
The researchers said these TKI-resistant CML stem cells were “transcriptionally distinct” from normal hematopoietic stem cells.
The TKI-resistant cells were characterized by dysregulation of specific genes and pathways—TGF-β, TNF-α, JAK–STAT, CTNNB1, and NFKB1A—that could potentially be targeted to improve the treatment of CML.
The researchers also said their single-cell analysis technique can be used beyond CML.
“This technique could be adapted to analyze a range of different cancers to help predict both the likely response to treatment and the risk of the disease returning in the future,” Dr Mead said. “This should eventually enable treatment to be tailored to target each and every type of cancer stem cell that may be present.” ![]()
Researchers say they’ve developed a technique for single-cell analysis that has revealed a population of treatment-resistant stem cells in patients with chronic myeloid leukemia (CML).
“It is increasingly recognized that tumors contain a variety of different cell types, including so-called cancer stem cells, that drive the growth and relapse of a patient’s cancer,” said study author Adam Mead, BM BCh, PhD, of University of Oxford in the UK.
“These cells can be very rare and extremely difficult to find after treatment as they become hidden within the normal tissue. We used a new genetic technique to identify and analyze single cancer stem cells in leukemia patients before and after treatment.”
Dr Mead and his colleagues detailed this research in Nature Medicine.
The team’s single-cell analysis technique combines high-sensitivity mutation detection with whole-transcriptome analysis.
The researchers used the method to analyze stem cells from patients with CML and found the cells to be heterogeneous.
In addition, the team was able to identify a subset of CML stem cells that proved resistant to treatment with tyrosine kinase inhibitors (TKIs).
“We found that, even in individual cases of leukemia, there are various types of cancer stem cell that respond differently to the treatment,” Dr Mead said.
“A small number of these cells are highly resistant to the treatment and are likely to be responsible for disease recurrence when the treatment is stopped. Our research allowed us uniquely to analyze these crucial cells that evade treatment so that we might learn how to more effectively eradicate them.”
The researchers said these TKI-resistant CML stem cells were “transcriptionally distinct” from normal hematopoietic stem cells.
The TKI-resistant cells were characterized by dysregulation of specific genes and pathways—TGF-β, TNF-α, JAK–STAT, CTNNB1, and NFKB1A—that could potentially be targeted to improve the treatment of CML.
The researchers also said their single-cell analysis technique can be used beyond CML.
“This technique could be adapted to analyze a range of different cancers to help predict both the likely response to treatment and the risk of the disease returning in the future,” Dr Mead said. “This should eventually enable treatment to be tailored to target each and every type of cancer stem cell that may be present.” ![]()
First generic version of clofarabine available in US

Clofarabine Injection, the first-to-market generic version of Sanofi Genzyme’s Clolar, is now available in the US.
The generic, a product of Fresenius Kabi, is available as a single dose vial containing 20 mg per 20 mL clofarabine.
Clofarabine is a purine nucleoside metabolic inhibitor indicated for the treatment of patients ages 1 to 21 with relapsed or refractory acute lymphoblastic leukemia (ALL) who received at least 2 prior treatment regimens.
Clolar was granted accelerated approval for this indication in the US in 2004.
The approval was based on response rates observed in ALL patients. There are no trials verifying that clofarabine confers improvement in survival or disease-related symptoms in ALL patients.
Clofarabine was assessed in a single-arm, phase 2 trial of 61 pediatric patients with relapsed/refractory ALL.
The patients’ median age was 12 (range, 1 to 20 years), and their median number of prior treatment regimens was 3 (range, 2 to 6).
The patients received clofarabine at 52 mg/m2 intravenously over 2 hours daily for 5 days, every 2 to 6 weeks.
The overall response rate was 30%. Seven patient achieved a complete response (CR), 5 had a CR without platelet recovery, and 6 patients had a partial response.
The median duration of CR in patients who did not go on to hematopoietic stem cell transplant was 6 weeks.
The most common grade 3 or higher adverse events were febrile neutropenia, anorexia, hypotension, and nausea.
These results were published in the Journal of Clinical Oncology in 2006. ![]()
Clofarabine Injection, the first-to-market generic version of Sanofi Genzyme’s Clolar, is now available in the US.
The generic, a product of Fresenius Kabi, is available as a single dose vial containing 20 mg per 20 mL clofarabine.
Clofarabine is a purine nucleoside metabolic inhibitor indicated for the treatment of patients ages 1 to 21 with relapsed or refractory acute lymphoblastic leukemia (ALL) who received at least 2 prior treatment regimens.
Clolar was granted accelerated approval for this indication in the US in 2004.
The approval was based on response rates observed in ALL patients. There are no trials verifying that clofarabine confers improvement in survival or disease-related symptoms in ALL patients.
Clofarabine was assessed in a single-arm, phase 2 trial of 61 pediatric patients with relapsed/refractory ALL.
The patients’ median age was 12 (range, 1 to 20 years), and their median number of prior treatment regimens was 3 (range, 2 to 6).
The patients received clofarabine at 52 mg/m2 intravenously over 2 hours daily for 5 days, every 2 to 6 weeks.
The overall response rate was 30%. Seven patient achieved a complete response (CR), 5 had a CR without platelet recovery, and 6 patients had a partial response.
The median duration of CR in patients who did not go on to hematopoietic stem cell transplant was 6 weeks.
The most common grade 3 or higher adverse events were febrile neutropenia, anorexia, hypotension, and nausea.
These results were published in the Journal of Clinical Oncology in 2006. ![]()
Clofarabine Injection, the first-to-market generic version of Sanofi Genzyme’s Clolar, is now available in the US.
The generic, a product of Fresenius Kabi, is available as a single dose vial containing 20 mg per 20 mL clofarabine.
Clofarabine is a purine nucleoside metabolic inhibitor indicated for the treatment of patients ages 1 to 21 with relapsed or refractory acute lymphoblastic leukemia (ALL) who received at least 2 prior treatment regimens.
Clolar was granted accelerated approval for this indication in the US in 2004.
The approval was based on response rates observed in ALL patients. There are no trials verifying that clofarabine confers improvement in survival or disease-related symptoms in ALL patients.
Clofarabine was assessed in a single-arm, phase 2 trial of 61 pediatric patients with relapsed/refractory ALL.
The patients’ median age was 12 (range, 1 to 20 years), and their median number of prior treatment regimens was 3 (range, 2 to 6).
The patients received clofarabine at 52 mg/m2 intravenously over 2 hours daily for 5 days, every 2 to 6 weeks.
The overall response rate was 30%. Seven patient achieved a complete response (CR), 5 had a CR without platelet recovery, and 6 patients had a partial response.
The median duration of CR in patients who did not go on to hematopoietic stem cell transplant was 6 weeks.
The most common grade 3 or higher adverse events were febrile neutropenia, anorexia, hypotension, and nausea.
These results were published in the Journal of Clinical Oncology in 2006. ![]()
FDA grants priority review to NDA for copanlisib

The US Food and Drug Administration (FDA) has granted priority review to the new drug application (NDA) for copanlisib, an intravenous PI3K inhibitor.
The NDA is for copanlisib as a treatment for patients with relapsed or refractory follicular lymphoma (FL) who have received at least 2 prior therapies.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The application for copanlisib is supported by data from the CHRONOS-1 trial. This phase 2 trial enrolled 141 patients with relapsed/refractory, indolent non-Hodgkin lymphoma. Most of these patients had FL (n=104).
In all patients, copanlisib produced an objective response rate of 59.2%, with a complete response rate of 12%. The median duration of response exceeded 98 weeks.
In the FL subset, copanlisib produced an overall response rate of 58.7%, with a complete response rate of 14.4%. The median duration of response exceeded 52 weeks.
In the entire cohort, there were 3 deaths considered related to copanlisib.
The most common treatment-related adverse events were transient hyperglycemia (all grades: 49%/grade 3-4: 40%) and hypertension (all grades: 29%/grade 3: 23%).
“Patients with relapsed or refractory follicular lymphoma have a poor prognosis, and new treatment options which are well tolerated and effective are needed to prolong progression-free survival and improve quality of life for these patients,” said Martin Dreyling, MD, a professor at the University of Munich Hospital (Grosshadern) in Germany and lead investigator of the CHRONOS-1 study.
“Based on the CHRONOS-1 results, where copanlisib showed durable efficacy with a manageable and distinct safety profile, the compound may have the potential to address this unmet medical need.”
Data from CHRONOS-1 were presented at the AACR Annual Meeting 2017.
Data from the FL subset of the trial are scheduled to be presented at the 2017 ASCO Annual Meeting in June.
Copanlisib is being developed by Bayer. The compound has fast track and orphan drug designations from the FDA.
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The FDA’s fast track program is designed to facilitate the development and expedite the review of products intended to treat or prevent serious or life-threatening conditions and address unmet medical need.
Through the fast track program, a product may be eligible for priority review. In addition, the company developing the product may be allowed to submit sections of the NDA or biologic license application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings and written communications with the FDA. ![]()
The US Food and Drug Administration (FDA) has granted priority review to the new drug application (NDA) for copanlisib, an intravenous PI3K inhibitor.
The NDA is for copanlisib as a treatment for patients with relapsed or refractory follicular lymphoma (FL) who have received at least 2 prior therapies.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The application for copanlisib is supported by data from the CHRONOS-1 trial. This phase 2 trial enrolled 141 patients with relapsed/refractory, indolent non-Hodgkin lymphoma. Most of these patients had FL (n=104).
In all patients, copanlisib produced an objective response rate of 59.2%, with a complete response rate of 12%. The median duration of response exceeded 98 weeks.
In the FL subset, copanlisib produced an overall response rate of 58.7%, with a complete response rate of 14.4%. The median duration of response exceeded 52 weeks.
In the entire cohort, there were 3 deaths considered related to copanlisib.
The most common treatment-related adverse events were transient hyperglycemia (all grades: 49%/grade 3-4: 40%) and hypertension (all grades: 29%/grade 3: 23%).
“Patients with relapsed or refractory follicular lymphoma have a poor prognosis, and new treatment options which are well tolerated and effective are needed to prolong progression-free survival and improve quality of life for these patients,” said Martin Dreyling, MD, a professor at the University of Munich Hospital (Grosshadern) in Germany and lead investigator of the CHRONOS-1 study.
“Based on the CHRONOS-1 results, where copanlisib showed durable efficacy with a manageable and distinct safety profile, the compound may have the potential to address this unmet medical need.”
Data from CHRONOS-1 were presented at the AACR Annual Meeting 2017.
Data from the FL subset of the trial are scheduled to be presented at the 2017 ASCO Annual Meeting in June.
Copanlisib is being developed by Bayer. The compound has fast track and orphan drug designations from the FDA.
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The FDA’s fast track program is designed to facilitate the development and expedite the review of products intended to treat or prevent serious or life-threatening conditions and address unmet medical need.
Through the fast track program, a product may be eligible for priority review. In addition, the company developing the product may be allowed to submit sections of the NDA or biologic license application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings and written communications with the FDA. ![]()
The US Food and Drug Administration (FDA) has granted priority review to the new drug application (NDA) for copanlisib, an intravenous PI3K inhibitor.
The NDA is for copanlisib as a treatment for patients with relapsed or refractory follicular lymphoma (FL) who have received at least 2 prior therapies.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The application for copanlisib is supported by data from the CHRONOS-1 trial. This phase 2 trial enrolled 141 patients with relapsed/refractory, indolent non-Hodgkin lymphoma. Most of these patients had FL (n=104).
In all patients, copanlisib produced an objective response rate of 59.2%, with a complete response rate of 12%. The median duration of response exceeded 98 weeks.
In the FL subset, copanlisib produced an overall response rate of 58.7%, with a complete response rate of 14.4%. The median duration of response exceeded 52 weeks.
In the entire cohort, there were 3 deaths considered related to copanlisib.
The most common treatment-related adverse events were transient hyperglycemia (all grades: 49%/grade 3-4: 40%) and hypertension (all grades: 29%/grade 3: 23%).
“Patients with relapsed or refractory follicular lymphoma have a poor prognosis, and new treatment options which are well tolerated and effective are needed to prolong progression-free survival and improve quality of life for these patients,” said Martin Dreyling, MD, a professor at the University of Munich Hospital (Grosshadern) in Germany and lead investigator of the CHRONOS-1 study.
“Based on the CHRONOS-1 results, where copanlisib showed durable efficacy with a manageable and distinct safety profile, the compound may have the potential to address this unmet medical need.”
Data from CHRONOS-1 were presented at the AACR Annual Meeting 2017.
Data from the FL subset of the trial are scheduled to be presented at the 2017 ASCO Annual Meeting in June.
Copanlisib is being developed by Bayer. The compound has fast track and orphan drug designations from the FDA.
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The FDA’s fast track program is designed to facilitate the development and expedite the review of products intended to treat or prevent serious or life-threatening conditions and address unmet medical need.
Through the fast track program, a product may be eligible for priority review. In addition, the company developing the product may be allowed to submit sections of the NDA or biologic license application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings and written communications with the FDA. ![]()
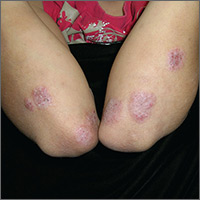
Itchy rash on forearms
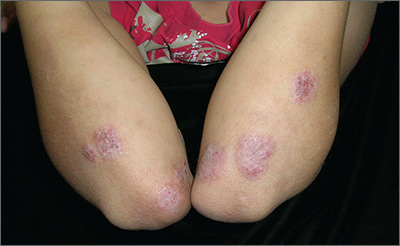
The FP strongly suspected that this was a case of nummular eczema, based on the round shape of the plaques, but the location of the lesions suggested psoriasis. The FP also considered tinea corporis with psoriasis in the differential.
The FP checked the patient's scalp, nails, and umbilicus for other signs of psoriasis and found none. He also performed a potassium hydroxide (KOH) preparation, which was negative for hyphae and fungal elements. (See a video on how to perform a KOH preparation here: http://www.mdedge.com/jfponline/article/100603/dermatology/koh-preparation.) To be sure that this wasn’t psoriasis, the FP also performed a punch biopsy. (The pathology subsequently came back positive for nummular eczema.) Ultimately, the yellow crusting, along with the round shape of the plaques, supported a diagnosis of nummular eczema. (“Nummus” is Latin for “coin.”)
Treatment for nummular eczema typically includes clobetasol, an ultra-high-potency corticosteroid. (The patient’s lack of response to the over-the-counter [1%] hydrocortisone was not unusual for nummular eczema because it is a low-potency steroid.) The FP in this case prescribed 0.05% clobetasol ointment to be applied twice daily to the lesions until the follow-up appointment 10 days later. At follow-up, the patient reported that the itching had almost completely resolved and the lesions were looking much better. The stitch from the biopsy was removed and the patient was told to continue using the clobetasol until the lesions completely resolved.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Wah Y, Usatine R. Eczema. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP strongly suspected that this was a case of nummular eczema, based on the round shape of the plaques, but the location of the lesions suggested psoriasis. The FP also considered tinea corporis with psoriasis in the differential.
The FP checked the patient's scalp, nails, and umbilicus for other signs of psoriasis and found none. He also performed a potassium hydroxide (KOH) preparation, which was negative for hyphae and fungal elements. (See a video on how to perform a KOH preparation here: http://www.mdedge.com/jfponline/article/100603/dermatology/koh-preparation.) To be sure that this wasn’t psoriasis, the FP also performed a punch biopsy. (The pathology subsequently came back positive for nummular eczema.) Ultimately, the yellow crusting, along with the round shape of the plaques, supported a diagnosis of nummular eczema. (“Nummus” is Latin for “coin.”)
Treatment for nummular eczema typically includes clobetasol, an ultra-high-potency corticosteroid. (The patient’s lack of response to the over-the-counter [1%] hydrocortisone was not unusual for nummular eczema because it is a low-potency steroid.) The FP in this case prescribed 0.05% clobetasol ointment to be applied twice daily to the lesions until the follow-up appointment 10 days later. At follow-up, the patient reported that the itching had almost completely resolved and the lesions were looking much better. The stitch from the biopsy was removed and the patient was told to continue using the clobetasol until the lesions completely resolved.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Wah Y, Usatine R. Eczema. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP strongly suspected that this was a case of nummular eczema, based on the round shape of the plaques, but the location of the lesions suggested psoriasis. The FP also considered tinea corporis with psoriasis in the differential.
The FP checked the patient's scalp, nails, and umbilicus for other signs of psoriasis and found none. He also performed a potassium hydroxide (KOH) preparation, which was negative for hyphae and fungal elements. (See a video on how to perform a KOH preparation here: http://www.mdedge.com/jfponline/article/100603/dermatology/koh-preparation.) To be sure that this wasn’t psoriasis, the FP also performed a punch biopsy. (The pathology subsequently came back positive for nummular eczema.) Ultimately, the yellow crusting, along with the round shape of the plaques, supported a diagnosis of nummular eczema. (“Nummus” is Latin for “coin.”)
Treatment for nummular eczema typically includes clobetasol, an ultra-high-potency corticosteroid. (The patient’s lack of response to the over-the-counter [1%] hydrocortisone was not unusual for nummular eczema because it is a low-potency steroid.) The FP in this case prescribed 0.05% clobetasol ointment to be applied twice daily to the lesions until the follow-up appointment 10 days later. At follow-up, the patient reported that the itching had almost completely resolved and the lesions were looking much better. The stitch from the biopsy was removed and the patient was told to continue using the clobetasol until the lesions completely resolved.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Wah Y, Usatine R. Eczema. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
Alternating therapy in renal cell carcinoma fails to show an advantage
There was no efficacy or safety advantage for alternating everolimus with pazopanib over pazopanib alone in patients with metastatic or locally advanced clear cell renal cell carcinoma (ccRCC), according to a newly published randomized trial.
The study hypothesis was that alternating the two drugs would improve outcomes and reduce toxicity, but differences between arms for the major outcomes were not clinically significant, according to results of a multicenter trial led by Geert A. Cirkel, MD, of the department of medical oncology, University Medical Center, Utrecht, the Netherlands.
Investigators randomized 101 patients with histologically confirmed ccRCC to receive 8 weeks of pazopanib in a daily dose of 800 mg alternated with 8 weeks of everolimus in a daily dose of 10 mg or 800 mg per day of continuous pazopanib. Patients remained on either regimen until disease progression.
Median time until first progression or death was 7.4 months for the experimental alternating arm versus 9.4 months for the control arm of continuous single-agent pazopanib (P = .37), Dr. Cirkel and associates reported (JAMA Onc. 2017 Apr 1. doi: 10.1001/jamaoncol.2016.5202).
Progression-free survival after starting on a second-line therapy was 20.2 months for the alternating treatment vs. 14.5 months for the control, but the confidence intervals were wide, and the difference was not significant (P = .86).
There was no apparent toxicity or tolerability advantage from alternating therapy. Nearly 40% of patients in both arms required pazopanib dose reductions, while 14% in the alternating arm also required an everolimus dose reduction. The incidence of serious adverse events possibly related to treatment was comparable between arms.
Quality of life was measured with several tools, including the Functional Assessment of Cancer Therapy – Kidney Symptom Index Disease-Related Symptoms (FKSI-DRS), but no significant differences between treatment arms were observed in any measure.
Current guidelines recommend pazopanib, which is a tyrosine kinase inhibitor of the vascular endothelial growth factor receptor, as a first-line therapy in ccRCC. Everolimus, an inhibitor of mammalian target of rapamycin, is recommended in the second-line setting. Noting that resistance to pazopanib has been shown to be reversible after a period of withdrawal in experimental studies, the authors had speculated an on-off strategy with everolimus might better preserve the efficacy of pazopanib, providing longer periods of disease control. They had also hypothesized that the cumulative adverse events might be less if the drugs were sequenced, allowing recovery from each set of drug-specific adverse events.
Several potential explanations were offered for the lack of improved efficacy from alternating everolimus with pazopanib. For one, the improved activity of pazopanib after withdrawal in experimental models was observed after drug-free periods. The authors questioned whether a period of tumor regrowth may be needed in order to overcome pazopanib resistance.
The study may still have supported the use of an alternating regimen if the alternating therapy had led to a significantly improved quality of life, but the authors found none, a outcome that they characterized as unexpected. They concluded that there are no apparent advantages for the alternating regimen of pazopanib and everolimus relative to pazopanib alone.
There was no efficacy or safety advantage for alternating everolimus with pazopanib over pazopanib alone in patients with metastatic or locally advanced clear cell renal cell carcinoma (ccRCC), according to a newly published randomized trial.
The study hypothesis was that alternating the two drugs would improve outcomes and reduce toxicity, but differences between arms for the major outcomes were not clinically significant, according to results of a multicenter trial led by Geert A. Cirkel, MD, of the department of medical oncology, University Medical Center, Utrecht, the Netherlands.
Investigators randomized 101 patients with histologically confirmed ccRCC to receive 8 weeks of pazopanib in a daily dose of 800 mg alternated with 8 weeks of everolimus in a daily dose of 10 mg or 800 mg per day of continuous pazopanib. Patients remained on either regimen until disease progression.
Median time until first progression or death was 7.4 months for the experimental alternating arm versus 9.4 months for the control arm of continuous single-agent pazopanib (P = .37), Dr. Cirkel and associates reported (JAMA Onc. 2017 Apr 1. doi: 10.1001/jamaoncol.2016.5202).
Progression-free survival after starting on a second-line therapy was 20.2 months for the alternating treatment vs. 14.5 months for the control, but the confidence intervals were wide, and the difference was not significant (P = .86).
There was no apparent toxicity or tolerability advantage from alternating therapy. Nearly 40% of patients in both arms required pazopanib dose reductions, while 14% in the alternating arm also required an everolimus dose reduction. The incidence of serious adverse events possibly related to treatment was comparable between arms.
Quality of life was measured with several tools, including the Functional Assessment of Cancer Therapy – Kidney Symptom Index Disease-Related Symptoms (FKSI-DRS), but no significant differences between treatment arms were observed in any measure.
Current guidelines recommend pazopanib, which is a tyrosine kinase inhibitor of the vascular endothelial growth factor receptor, as a first-line therapy in ccRCC. Everolimus, an inhibitor of mammalian target of rapamycin, is recommended in the second-line setting. Noting that resistance to pazopanib has been shown to be reversible after a period of withdrawal in experimental studies, the authors had speculated an on-off strategy with everolimus might better preserve the efficacy of pazopanib, providing longer periods of disease control. They had also hypothesized that the cumulative adverse events might be less if the drugs were sequenced, allowing recovery from each set of drug-specific adverse events.
Several potential explanations were offered for the lack of improved efficacy from alternating everolimus with pazopanib. For one, the improved activity of pazopanib after withdrawal in experimental models was observed after drug-free periods. The authors questioned whether a period of tumor regrowth may be needed in order to overcome pazopanib resistance.
The study may still have supported the use of an alternating regimen if the alternating therapy had led to a significantly improved quality of life, but the authors found none, a outcome that they characterized as unexpected. They concluded that there are no apparent advantages for the alternating regimen of pazopanib and everolimus relative to pazopanib alone.
There was no efficacy or safety advantage for alternating everolimus with pazopanib over pazopanib alone in patients with metastatic or locally advanced clear cell renal cell carcinoma (ccRCC), according to a newly published randomized trial.
The study hypothesis was that alternating the two drugs would improve outcomes and reduce toxicity, but differences between arms for the major outcomes were not clinically significant, according to results of a multicenter trial led by Geert A. Cirkel, MD, of the department of medical oncology, University Medical Center, Utrecht, the Netherlands.
Investigators randomized 101 patients with histologically confirmed ccRCC to receive 8 weeks of pazopanib in a daily dose of 800 mg alternated with 8 weeks of everolimus in a daily dose of 10 mg or 800 mg per day of continuous pazopanib. Patients remained on either regimen until disease progression.
Median time until first progression or death was 7.4 months for the experimental alternating arm versus 9.4 months for the control arm of continuous single-agent pazopanib (P = .37), Dr. Cirkel and associates reported (JAMA Onc. 2017 Apr 1. doi: 10.1001/jamaoncol.2016.5202).
Progression-free survival after starting on a second-line therapy was 20.2 months for the alternating treatment vs. 14.5 months for the control, but the confidence intervals were wide, and the difference was not significant (P = .86).
There was no apparent toxicity or tolerability advantage from alternating therapy. Nearly 40% of patients in both arms required pazopanib dose reductions, while 14% in the alternating arm also required an everolimus dose reduction. The incidence of serious adverse events possibly related to treatment was comparable between arms.
Quality of life was measured with several tools, including the Functional Assessment of Cancer Therapy – Kidney Symptom Index Disease-Related Symptoms (FKSI-DRS), but no significant differences between treatment arms were observed in any measure.
Current guidelines recommend pazopanib, which is a tyrosine kinase inhibitor of the vascular endothelial growth factor receptor, as a first-line therapy in ccRCC. Everolimus, an inhibitor of mammalian target of rapamycin, is recommended in the second-line setting. Noting that resistance to pazopanib has been shown to be reversible after a period of withdrawal in experimental studies, the authors had speculated an on-off strategy with everolimus might better preserve the efficacy of pazopanib, providing longer periods of disease control. They had also hypothesized that the cumulative adverse events might be less if the drugs were sequenced, allowing recovery from each set of drug-specific adverse events.
Several potential explanations were offered for the lack of improved efficacy from alternating everolimus with pazopanib. For one, the improved activity of pazopanib after withdrawal in experimental models was observed after drug-free periods. The authors questioned whether a period of tumor regrowth may be needed in order to overcome pazopanib resistance.
The study may still have supported the use of an alternating regimen if the alternating therapy had led to a significantly improved quality of life, but the authors found none, a outcome that they characterized as unexpected. They concluded that there are no apparent advantages for the alternating regimen of pazopanib and everolimus relative to pazopanib alone.
FROM JAMA ONCOLOGY
Key clinical point:
Major finding: The median time to progression or death was 7.4 months for the combination versus 9.4 months for pazopanib alone (P = .37).
Data source: Randomized, multicenter controlled trial.
Disclosures: The principal investigator Dr. Cirkel reports travel expenses from Novartis, which, along with GlaxoSmithKline, provided funding for this study.
More early-stage cancer diagnosis since ACA implementation
Implementation of the Affordable Care Act (ACA) has been associated with a shift toward earlier stage at diagnosis for common screenable cancers, finds an analysis of nearly 273,000 patients reported in a presscast leading up to the annual meeting of the American Society of Clinical Oncology.
“Extensive evidence has shown that people without insurance are more likely to be diagnosed at later stage, especially for the cancers that can be detected earlier through screening or symptoms,” said lead study author Xuesong Han, PhD, strategic director of health policy and health care delivery research at the American Cancer Society in Atlanta. “In 2014, two major components of the Affordable Care Act – Medicaid expansion and marketplace exchange – were implemented. As a result, insurance coverage has substantially increased for nonelderly Americans.”
Study findings showed that, for four of five screenable cancers – breast and cervical cancer in women and lung and colorectal cancer in both sexes combined – the proportion of cancers that were stage I at diagnosis, and hence most curable, increased by an absolute 1% or so after the ACA was implemented. Prostate cancer was the outlier: the value for this malignancy decreased by 1%.
“The increases for the first four cancers were consistent with our hypothesis, with more people gaining insurance and access to screening services or access to physicians to detect early symptoms,” Dr. Han summarized. “But what about prostate cancer? We think [that pattern] may reflect the recent USPSTF recommendations against routine prostate cancer screening.”
“We think that this is an important study,” commented ASCO president-elect Bruce E. Johnson, MD, who is also chief clinical research officer and an institute physician at the Dana-Farber Cancer Institute in Boston. “Obviously, the changes are not enormous; they are not dramatic. But … because the uptake of screening is relatively slow, this is certainly consistent with the idea that, by doing additional screening, you can potentially find more stage I patients, and, the earlier the stage, the more likely one is to be cured.”
“The other important thing is that ASCO strongly supports the relative ease of access to screening capabilities, and that’s one of the characteristics of the Affordable Care Act, that most of the cancer screening is covered,” he further stated. “Whatever form our health care takes over the next several years, we advocate for patients to have early access to screening, which can identify cancers at an earlier stage in their more curable forms.”
Study details
For the study, the investigators used the National Cancer Database – which captures 70% of newly diagnosed cases in the United States – to identify patients younger than 65 who were eligible for cancer screening and who received a diagnosis of any of the five screenable cancers in 2013 or 2014. They compared stage distribution before ACA implementation (first nine months of 2013) and afterward (last nine months of 2014).
Analyses were based on data from 121,402 female breast cancer patients aged 40-64 years, 39,418 colorectal cancer patients aged 50-64 years, 11,190 cervical cancer patients aged 21-64 years, 59,210 prostate cancer patients aged 50-64 years, and 41,436 lung cancer patients aged 55-64 years.
Results showed that the proportion of cancers that were stage I at diagnosis increased after ACA implementation from 47.8% to 48.9% for breast cancer (adjusted prevalence ratio, 1.02) and from 47.3% to 48.8% for cervical cancer (APR, 1.02) in women, and from 16.6% to 17.7% for lung cancer (APR, 1.07) and from 22.8% to 23.7% for colorectal cancer (APR, 1.04) in men and women combined, Dr. Han reported.
Prostate cancer was the exception, with the proportion of cases that were stage I at diagnosis falling from 18.5% to 17.2% (APR, 0.93).
In a stratified analysis, the significant downshift in lung and colorectal cancer stage were seen only in states that had actually adopted the Medicaid expansion component of the ACA, which covers low-income individuals, according to Dr. Han. The downshift in female breast cancer stage and upshift in prostate cancer stage occurred regardless of whether states had done so.
Implementation of the Affordable Care Act (ACA) has been associated with a shift toward earlier stage at diagnosis for common screenable cancers, finds an analysis of nearly 273,000 patients reported in a presscast leading up to the annual meeting of the American Society of Clinical Oncology.
“Extensive evidence has shown that people without insurance are more likely to be diagnosed at later stage, especially for the cancers that can be detected earlier through screening or symptoms,” said lead study author Xuesong Han, PhD, strategic director of health policy and health care delivery research at the American Cancer Society in Atlanta. “In 2014, two major components of the Affordable Care Act – Medicaid expansion and marketplace exchange – were implemented. As a result, insurance coverage has substantially increased for nonelderly Americans.”
Study findings showed that, for four of five screenable cancers – breast and cervical cancer in women and lung and colorectal cancer in both sexes combined – the proportion of cancers that were stage I at diagnosis, and hence most curable, increased by an absolute 1% or so after the ACA was implemented. Prostate cancer was the outlier: the value for this malignancy decreased by 1%.
“The increases for the first four cancers were consistent with our hypothesis, with more people gaining insurance and access to screening services or access to physicians to detect early symptoms,” Dr. Han summarized. “But what about prostate cancer? We think [that pattern] may reflect the recent USPSTF recommendations against routine prostate cancer screening.”
“We think that this is an important study,” commented ASCO president-elect Bruce E. Johnson, MD, who is also chief clinical research officer and an institute physician at the Dana-Farber Cancer Institute in Boston. “Obviously, the changes are not enormous; they are not dramatic. But … because the uptake of screening is relatively slow, this is certainly consistent with the idea that, by doing additional screening, you can potentially find more stage I patients, and, the earlier the stage, the more likely one is to be cured.”
“The other important thing is that ASCO strongly supports the relative ease of access to screening capabilities, and that’s one of the characteristics of the Affordable Care Act, that most of the cancer screening is covered,” he further stated. “Whatever form our health care takes over the next several years, we advocate for patients to have early access to screening, which can identify cancers at an earlier stage in their more curable forms.”
Study details
For the study, the investigators used the National Cancer Database – which captures 70% of newly diagnosed cases in the United States – to identify patients younger than 65 who were eligible for cancer screening and who received a diagnosis of any of the five screenable cancers in 2013 or 2014. They compared stage distribution before ACA implementation (first nine months of 2013) and afterward (last nine months of 2014).
Analyses were based on data from 121,402 female breast cancer patients aged 40-64 years, 39,418 colorectal cancer patients aged 50-64 years, 11,190 cervical cancer patients aged 21-64 years, 59,210 prostate cancer patients aged 50-64 years, and 41,436 lung cancer patients aged 55-64 years.
Results showed that the proportion of cancers that were stage I at diagnosis increased after ACA implementation from 47.8% to 48.9% for breast cancer (adjusted prevalence ratio, 1.02) and from 47.3% to 48.8% for cervical cancer (APR, 1.02) in women, and from 16.6% to 17.7% for lung cancer (APR, 1.07) and from 22.8% to 23.7% for colorectal cancer (APR, 1.04) in men and women combined, Dr. Han reported.
Prostate cancer was the exception, with the proportion of cases that were stage I at diagnosis falling from 18.5% to 17.2% (APR, 0.93).
In a stratified analysis, the significant downshift in lung and colorectal cancer stage were seen only in states that had actually adopted the Medicaid expansion component of the ACA, which covers low-income individuals, according to Dr. Han. The downshift in female breast cancer stage and upshift in prostate cancer stage occurred regardless of whether states had done so.
Implementation of the Affordable Care Act (ACA) has been associated with a shift toward earlier stage at diagnosis for common screenable cancers, finds an analysis of nearly 273,000 patients reported in a presscast leading up to the annual meeting of the American Society of Clinical Oncology.
“Extensive evidence has shown that people without insurance are more likely to be diagnosed at later stage, especially for the cancers that can be detected earlier through screening or symptoms,” said lead study author Xuesong Han, PhD, strategic director of health policy and health care delivery research at the American Cancer Society in Atlanta. “In 2014, two major components of the Affordable Care Act – Medicaid expansion and marketplace exchange – were implemented. As a result, insurance coverage has substantially increased for nonelderly Americans.”
Study findings showed that, for four of five screenable cancers – breast and cervical cancer in women and lung and colorectal cancer in both sexes combined – the proportion of cancers that were stage I at diagnosis, and hence most curable, increased by an absolute 1% or so after the ACA was implemented. Prostate cancer was the outlier: the value for this malignancy decreased by 1%.
“The increases for the first four cancers were consistent with our hypothesis, with more people gaining insurance and access to screening services or access to physicians to detect early symptoms,” Dr. Han summarized. “But what about prostate cancer? We think [that pattern] may reflect the recent USPSTF recommendations against routine prostate cancer screening.”
“We think that this is an important study,” commented ASCO president-elect Bruce E. Johnson, MD, who is also chief clinical research officer and an institute physician at the Dana-Farber Cancer Institute in Boston. “Obviously, the changes are not enormous; they are not dramatic. But … because the uptake of screening is relatively slow, this is certainly consistent with the idea that, by doing additional screening, you can potentially find more stage I patients, and, the earlier the stage, the more likely one is to be cured.”
“The other important thing is that ASCO strongly supports the relative ease of access to screening capabilities, and that’s one of the characteristics of the Affordable Care Act, that most of the cancer screening is covered,” he further stated. “Whatever form our health care takes over the next several years, we advocate for patients to have early access to screening, which can identify cancers at an earlier stage in their more curable forms.”
Study details
For the study, the investigators used the National Cancer Database – which captures 70% of newly diagnosed cases in the United States – to identify patients younger than 65 who were eligible for cancer screening and who received a diagnosis of any of the five screenable cancers in 2013 or 2014. They compared stage distribution before ACA implementation (first nine months of 2013) and afterward (last nine months of 2014).
Analyses were based on data from 121,402 female breast cancer patients aged 40-64 years, 39,418 colorectal cancer patients aged 50-64 years, 11,190 cervical cancer patients aged 21-64 years, 59,210 prostate cancer patients aged 50-64 years, and 41,436 lung cancer patients aged 55-64 years.
Results showed that the proportion of cancers that were stage I at diagnosis increased after ACA implementation from 47.8% to 48.9% for breast cancer (adjusted prevalence ratio, 1.02) and from 47.3% to 48.8% for cervical cancer (APR, 1.02) in women, and from 16.6% to 17.7% for lung cancer (APR, 1.07) and from 22.8% to 23.7% for colorectal cancer (APR, 1.04) in men and women combined, Dr. Han reported.
Prostate cancer was the exception, with the proportion of cases that were stage I at diagnosis falling from 18.5% to 17.2% (APR, 0.93).
In a stratified analysis, the significant downshift in lung and colorectal cancer stage were seen only in states that had actually adopted the Medicaid expansion component of the ACA, which covers low-income individuals, according to Dr. Han. The downshift in female breast cancer stage and upshift in prostate cancer stage occurred regardless of whether states had done so.
FROM THE 2017 ASCO ANNUAL MEETING
Key clinical point:
Major finding: The proportion of cancers that were stage I when diagnosed increased by about 1% after ACA implementation for breast, cervical, lung, and colorectal cancer, while it decreased by 1% for prostate cancer.
Data source: A cohort study of 272,656 patients with these five cancers from the National Cancer Database.
Disclosures: Dr. Han reported that she had no disclosures.
Mepolizumab proves effective for eosinophilic granulomatosis with polyangiitis
Adding mepolizumab to standard-of-care glucocorticoids with or without immunosuppressive agents can induce remission in many patients who have eosinophilic granulomatosis with polyangiitis (EGPA), according to a report published online May 18 in the New England Journal of Medicine.
EGPA, a rare disorder characterized by asthma, sinusitis, pulmonary infiltrates, neuropathy, and eosinophilic vasculitis in at least one end-organ, frequently relapses despite glucocorticoid therapy or fails to respond adequately to the treatment. Patients have elevated levels of the cytokine interleukin-5, which regulates eosinophil maturation, differentiation, and proliferation. Neutralizing this cytokine is thought to be a potential therapeutic approach, said Michael E. Wechsler, MD, of National Jewish Health, Denver, and his associates.
Proof-of-concept studies have demonstrated the efficacy of subcutaneous mepolizumab, an anti–interleukin-5 monoclonal antibody, in EGPA, so Dr. Wechsler and his colleagues assessed the safety and efficacy of a 1-year course of mepolizumab (300 mg) as add-on therapy in a double-blind, randomized, phase III trial, which involved 136 adults treated at 31 academic medical centers in nine countries. The study was sponsored by GlaxoSmithKline and the National Institute of Allergy and Infectious Diseases.
The first of two primary efficacy endpoints was the total accrued weeks of remission. A total of 28% of the mepolizumab group achieved remission for at least 24 weeks, compared with only 3% of the placebo group, for an odds ratio of 5.91.
The second primary efficacy endpoint was the proportion of patients in remission at both week 36 and week 48. Again, significantly more patients in the mepolizumab group (32%) than in the placebo group (3%) met this end point (OR, 16.74).
Mepolizumab also proved superior to placebo regarding numerous secondary endpoints, the investigators said (N Engl J Med. 2017 May 18. doi: 10.1056/NEJMoa1702079). More patients who received active treatment achieved remission within the first 6 months of treatment and remained in remission for a full year (19% vs. 1%; OR, 19.65). The time to first relapse was significantly longer for mepolizumab, with only 56% of that group experiencing a relapse within 1 year, compared with 82% of the placebo group. The annualized relapse rate was half as high with mepolizumab (1.14) as with placebo (2.27).
In addition, patients in the mepolizumab group were more likely to reduce their doses of glucocorticoids (OR, 0.20) or discontinue the drugs altogether (18% vs. 3% taking placebo).
Mepolizumab was most effective among the 79 patients who had a high absolute eosinophil count (150 or more cells per cubic millimeter) at baseline. In this subgroup, 33% of patients taking mepolizumab achieved remission for 6 months or more, compared with none of the patients taking placebo (OR, 26.1).
Although the effectiveness of mepolizumab in this difficult-to-treat population was noteworthy, only about half of the patients given the active treatment achieved remission as defined by the study protocol. It is unclear why the drug was not effective in the other half of patients. One possible reason is that some manifestations of the disorder are not driven by eosinophils. Another is that nonresponsive patients may have sustained longstanding, irreversible vasculitic damage that is no longer amenable to anti–interleukin-5 therapy.
Alternatively, it’s possible that mepolizumab reduced eosinophils in the blood but not those in the body tissues of nonresponsive patients or that the patients who didn’t respond well simply required a higher dose of the drug, Dr. Wechsler and his associates said.
The NIAID is now supporting a study of blood, urine, sputum, and tissue samples from some of these participants “to address questions related to disease risk and pathological features, as well as response to treatment,” they added.
Many authors reported receiving payments from pharmaceutical companies, including several from GlaxoSmithKline. Four authors are employees of the company.
The study by Michael E. Wechsler, MD, and his associates can be considered proof of concept. Now, researchers must turn to identifying biomarkers that predict the success or failure of mepolizumab in patients.
Researchers must also elucidate the fate of eosinophils in the tissues, especially in vasculitic lesions, after treatment with mepolizumab. And they should address possible synergistic activity when the drug is given together with immunosuppressants such as azathioprine and cyclophosphamide.
In addition, future studies should include patients who have organ-threatening or life-threatening eosinophilic granulomatosis with polyangiitis, who were excluded from this trial but who are most in need of novel treatments.
Ratko Djukanovic, MD, is with the University of Southampton (England) and the National Institute for Health Research Southampton Biomedical Research Centre. Paul M. O’Byrne, MD, is with the Firestone Institute for Respiratory Health within St. Joseph’s Healthcare and McMaster University in Hamilton, Ont. Dr. Djukanovic and Dr. O’Byrne both reported financial relationships with pharmaceutical companies outside their editorial. They made these remarks in an editorial accompanying Dr. Wechsler and colleagues’ report (N Engl J Med. 2017 May 18. doi: 10.1056/NEJMe1704402).
The study by Michael E. Wechsler, MD, and his associates can be considered proof of concept. Now, researchers must turn to identifying biomarkers that predict the success or failure of mepolizumab in patients.
Researchers must also elucidate the fate of eosinophils in the tissues, especially in vasculitic lesions, after treatment with mepolizumab. And they should address possible synergistic activity when the drug is given together with immunosuppressants such as azathioprine and cyclophosphamide.
In addition, future studies should include patients who have organ-threatening or life-threatening eosinophilic granulomatosis with polyangiitis, who were excluded from this trial but who are most in need of novel treatments.
Ratko Djukanovic, MD, is with the University of Southampton (England) and the National Institute for Health Research Southampton Biomedical Research Centre. Paul M. O’Byrne, MD, is with the Firestone Institute for Respiratory Health within St. Joseph’s Healthcare and McMaster University in Hamilton, Ont. Dr. Djukanovic and Dr. O’Byrne both reported financial relationships with pharmaceutical companies outside their editorial. They made these remarks in an editorial accompanying Dr. Wechsler and colleagues’ report (N Engl J Med. 2017 May 18. doi: 10.1056/NEJMe1704402).
The study by Michael E. Wechsler, MD, and his associates can be considered proof of concept. Now, researchers must turn to identifying biomarkers that predict the success or failure of mepolizumab in patients.
Researchers must also elucidate the fate of eosinophils in the tissues, especially in vasculitic lesions, after treatment with mepolizumab. And they should address possible synergistic activity when the drug is given together with immunosuppressants such as azathioprine and cyclophosphamide.
In addition, future studies should include patients who have organ-threatening or life-threatening eosinophilic granulomatosis with polyangiitis, who were excluded from this trial but who are most in need of novel treatments.
Ratko Djukanovic, MD, is with the University of Southampton (England) and the National Institute for Health Research Southampton Biomedical Research Centre. Paul M. O’Byrne, MD, is with the Firestone Institute for Respiratory Health within St. Joseph’s Healthcare and McMaster University in Hamilton, Ont. Dr. Djukanovic and Dr. O’Byrne both reported financial relationships with pharmaceutical companies outside their editorial. They made these remarks in an editorial accompanying Dr. Wechsler and colleagues’ report (N Engl J Med. 2017 May 18. doi: 10.1056/NEJMe1704402).
Adding mepolizumab to standard-of-care glucocorticoids with or without immunosuppressive agents can induce remission in many patients who have eosinophilic granulomatosis with polyangiitis (EGPA), according to a report published online May 18 in the New England Journal of Medicine.
EGPA, a rare disorder characterized by asthma, sinusitis, pulmonary infiltrates, neuropathy, and eosinophilic vasculitis in at least one end-organ, frequently relapses despite glucocorticoid therapy or fails to respond adequately to the treatment. Patients have elevated levels of the cytokine interleukin-5, which regulates eosinophil maturation, differentiation, and proliferation. Neutralizing this cytokine is thought to be a potential therapeutic approach, said Michael E. Wechsler, MD, of National Jewish Health, Denver, and his associates.
Proof-of-concept studies have demonstrated the efficacy of subcutaneous mepolizumab, an anti–interleukin-5 monoclonal antibody, in EGPA, so Dr. Wechsler and his colleagues assessed the safety and efficacy of a 1-year course of mepolizumab (300 mg) as add-on therapy in a double-blind, randomized, phase III trial, which involved 136 adults treated at 31 academic medical centers in nine countries. The study was sponsored by GlaxoSmithKline and the National Institute of Allergy and Infectious Diseases.
The first of two primary efficacy endpoints was the total accrued weeks of remission. A total of 28% of the mepolizumab group achieved remission for at least 24 weeks, compared with only 3% of the placebo group, for an odds ratio of 5.91.
The second primary efficacy endpoint was the proportion of patients in remission at both week 36 and week 48. Again, significantly more patients in the mepolizumab group (32%) than in the placebo group (3%) met this end point (OR, 16.74).
Mepolizumab also proved superior to placebo regarding numerous secondary endpoints, the investigators said (N Engl J Med. 2017 May 18. doi: 10.1056/NEJMoa1702079). More patients who received active treatment achieved remission within the first 6 months of treatment and remained in remission for a full year (19% vs. 1%; OR, 19.65). The time to first relapse was significantly longer for mepolizumab, with only 56% of that group experiencing a relapse within 1 year, compared with 82% of the placebo group. The annualized relapse rate was half as high with mepolizumab (1.14) as with placebo (2.27).
In addition, patients in the mepolizumab group were more likely to reduce their doses of glucocorticoids (OR, 0.20) or discontinue the drugs altogether (18% vs. 3% taking placebo).
Mepolizumab was most effective among the 79 patients who had a high absolute eosinophil count (150 or more cells per cubic millimeter) at baseline. In this subgroup, 33% of patients taking mepolizumab achieved remission for 6 months or more, compared with none of the patients taking placebo (OR, 26.1).
Although the effectiveness of mepolizumab in this difficult-to-treat population was noteworthy, only about half of the patients given the active treatment achieved remission as defined by the study protocol. It is unclear why the drug was not effective in the other half of patients. One possible reason is that some manifestations of the disorder are not driven by eosinophils. Another is that nonresponsive patients may have sustained longstanding, irreversible vasculitic damage that is no longer amenable to anti–interleukin-5 therapy.
Alternatively, it’s possible that mepolizumab reduced eosinophils in the blood but not those in the body tissues of nonresponsive patients or that the patients who didn’t respond well simply required a higher dose of the drug, Dr. Wechsler and his associates said.
The NIAID is now supporting a study of blood, urine, sputum, and tissue samples from some of these participants “to address questions related to disease risk and pathological features, as well as response to treatment,” they added.
Many authors reported receiving payments from pharmaceutical companies, including several from GlaxoSmithKline. Four authors are employees of the company.
Adding mepolizumab to standard-of-care glucocorticoids with or without immunosuppressive agents can induce remission in many patients who have eosinophilic granulomatosis with polyangiitis (EGPA), according to a report published online May 18 in the New England Journal of Medicine.
EGPA, a rare disorder characterized by asthma, sinusitis, pulmonary infiltrates, neuropathy, and eosinophilic vasculitis in at least one end-organ, frequently relapses despite glucocorticoid therapy or fails to respond adequately to the treatment. Patients have elevated levels of the cytokine interleukin-5, which regulates eosinophil maturation, differentiation, and proliferation. Neutralizing this cytokine is thought to be a potential therapeutic approach, said Michael E. Wechsler, MD, of National Jewish Health, Denver, and his associates.
Proof-of-concept studies have demonstrated the efficacy of subcutaneous mepolizumab, an anti–interleukin-5 monoclonal antibody, in EGPA, so Dr. Wechsler and his colleagues assessed the safety and efficacy of a 1-year course of mepolizumab (300 mg) as add-on therapy in a double-blind, randomized, phase III trial, which involved 136 adults treated at 31 academic medical centers in nine countries. The study was sponsored by GlaxoSmithKline and the National Institute of Allergy and Infectious Diseases.
The first of two primary efficacy endpoints was the total accrued weeks of remission. A total of 28% of the mepolizumab group achieved remission for at least 24 weeks, compared with only 3% of the placebo group, for an odds ratio of 5.91.
The second primary efficacy endpoint was the proportion of patients in remission at both week 36 and week 48. Again, significantly more patients in the mepolizumab group (32%) than in the placebo group (3%) met this end point (OR, 16.74).
Mepolizumab also proved superior to placebo regarding numerous secondary endpoints, the investigators said (N Engl J Med. 2017 May 18. doi: 10.1056/NEJMoa1702079). More patients who received active treatment achieved remission within the first 6 months of treatment and remained in remission for a full year (19% vs. 1%; OR, 19.65). The time to first relapse was significantly longer for mepolizumab, with only 56% of that group experiencing a relapse within 1 year, compared with 82% of the placebo group. The annualized relapse rate was half as high with mepolizumab (1.14) as with placebo (2.27).
In addition, patients in the mepolizumab group were more likely to reduce their doses of glucocorticoids (OR, 0.20) or discontinue the drugs altogether (18% vs. 3% taking placebo).
Mepolizumab was most effective among the 79 patients who had a high absolute eosinophil count (150 or more cells per cubic millimeter) at baseline. In this subgroup, 33% of patients taking mepolizumab achieved remission for 6 months or more, compared with none of the patients taking placebo (OR, 26.1).
Although the effectiveness of mepolizumab in this difficult-to-treat population was noteworthy, only about half of the patients given the active treatment achieved remission as defined by the study protocol. It is unclear why the drug was not effective in the other half of patients. One possible reason is that some manifestations of the disorder are not driven by eosinophils. Another is that nonresponsive patients may have sustained longstanding, irreversible vasculitic damage that is no longer amenable to anti–interleukin-5 therapy.
Alternatively, it’s possible that mepolizumab reduced eosinophils in the blood but not those in the body tissues of nonresponsive patients or that the patients who didn’t respond well simply required a higher dose of the drug, Dr. Wechsler and his associates said.
The NIAID is now supporting a study of blood, urine, sputum, and tissue samples from some of these participants “to address questions related to disease risk and pathological features, as well as response to treatment,” they added.
Many authors reported receiving payments from pharmaceutical companies, including several from GlaxoSmithKline. Four authors are employees of the company.
FROM NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point:
Major finding: Of the mepolizumab group, 28% achieved remission for at least 24 weeks, compared with only 3% of the placebo group (OR, 5.91).
Data source: An international double-blind randomized placebo-controlled phase III trial involving 136 adults treated for 1 year.
Disclosures: This study was supported by GlaxoSmithKline and the National Institute of Allergy and Infectious Diseases. Many authors reported receiving payments from pharmaceutical companies, including several from GlaxoSmithKline. Four authors are employees of the company.
Imatinib cuts mast cells in severe asthma
Imatinib decreased airway mast-cell counts and airway hyperresponsiveness in adults with asthma, who were not responding well to maximal therapy, according to a report published online May 17 in the New England Journal of Medicine.
Imatinib is an inhibitor of the stem-cell factor receptor KIT, which is essential for mast-cell development and survival in bodily tissues. This study’s findings suggest that KIT-dependent processes and mast cells contribute to the pathobiology of severe asthma.
The researchers undertook this study because imatinib is known to reduce bone-marrow mast cells and tryptase levels in chronic myeloid leukemia and to reduce serum tryptase in patients with pulmonary hypertension. Tryptase is a marker of mast-cell burden and activation when detected in extracellular fluids, and it is elevated in the bronchoalveolar lavage fluid from patients with uncontrolled asthma.
To examine whether imatinib would decrease mast-cell counts and activation in the airways of adults with severe, refractory asthma, the investigators performed the randomized double-blind proof-of-principle trial at seven academic centers across the United States over the course of 5 years. A total of 62 patients were assigned to 24 weeks of either oral imatinib (32 participants) or a matching placebo (30 participants). Fifty patients, 24 in the imatinib group and 26 in the placebo group, completed the trial.
The primary outcome measure was the change in airway hyperresponsiveness at 6 months, as measured by the increase in the concentration of methacholine that causes significant bronchoconstriction (PC20). Imatinib decreased airway hyperresponsiveness to a greater degree than did placebo. Imatinib increased PC20 by a mean of 1.20 doubling doses at 3 months and by a mean of 1.73 doubling doses at 6 months, compared with 0.03 and 1.07, respectively, for placebo.
The small improvement in the placebo group is consistent with a phenomenon reported in other studies, in which patients show a delayed improvement in airway hyperresponsiveness for several months after they started inhaled glucocorticoids, Dr. Cahill and her associates noted (N Engl J Med. 2017 May 18. doi: 10.1056/NEJMoa1613125).
Imatinib also reduced mast-cell activity as measured by serum and airway levels of tryptase. Serum tryptase decreased by 43% in the imatinib group, compared with a 12% decline in the placebo group. And tryptase levels in bronchoalveolar lavage fluid tended to decrease in the imatinib group but to increase in the placebo group.
Imatinib also increased mean forced expiratory volume in 1 second (FEV1).
“Although the increase in FEV1 may not seem substantial, it suggests that mast-cell–dependent processes contribute to airway obstruction in these patients despite high-dose, anti-inflammatory glucocorticoid therapy. The near–50-mL difference in the change in baseline FEV1 between the imatinib and placebo groups is small, but it is likely to be important in light of the population we studied,” Dr. Cahill and her associates wrote.
In addition, exploratory analyses showed that the reduction in airway hyperresponsiveness with imatinib “negatively correlated with baseline blood eosinophil counts, and baseline numbers of neutrophils in bronchoalveolar lavage fluid were strongly correlated with increases in FEV1. Together, these findings support a role for mast cells in noneosinophilic asthma. Since almost half of the patients with severe asthma have neutrophilic airway inflammation, we speculate that KIT inhibition might represent an important approach to treatment for this group,” they said.
This study was supported by the National Heart, Lung, and Blood Institute, the National Institute of Allergy and Infectious Diseases, the Vinik family, and the Kaye family; Novartis provided imatinib free of charge. The authors’ financial disclosures are available at www.nejm.org.
As Cahill et al. noted, these data suggest that the role of mast cells should be studied further, but cautiously.
It is not yet time to target mast cells in patients with asthma. Evolution has given us these cells for a reason. They appear to assist in host defense against parasites and play a role in other innate and adaptive immune responses. And they likely have other beneficial effects that haven’t been discovered yet.
However, in the unfortunate patients in whom mast cells can be strongly implicated as contributing to disease, either reducing their numbers or suppressing their function may confer more benefit than harm. This is particularly true in the case of asthma, where mast cells can be targeted locally, in the airways.
Stephen J. Galli, MD, is the Mary Hewitt Loveless, MD, Professor in the school of medicine, and is at the Sean N. Parker Center for Allergy and Asthma Research at Stanford (Calif.) University. His financial disclosures are available at www.nejm.org. Dr. Galli made these remarks in an editorial accompanying Dr. Cahill’s report (N Engl J Med. 2017 May 18. doi: 10.1056/NEJMe1702653).
As Cahill et al. noted, these data suggest that the role of mast cells should be studied further, but cautiously.
It is not yet time to target mast cells in patients with asthma. Evolution has given us these cells for a reason. They appear to assist in host defense against parasites and play a role in other innate and adaptive immune responses. And they likely have other beneficial effects that haven’t been discovered yet.
However, in the unfortunate patients in whom mast cells can be strongly implicated as contributing to disease, either reducing their numbers or suppressing their function may confer more benefit than harm. This is particularly true in the case of asthma, where mast cells can be targeted locally, in the airways.
Stephen J. Galli, MD, is the Mary Hewitt Loveless, MD, Professor in the school of medicine, and is at the Sean N. Parker Center for Allergy and Asthma Research at Stanford (Calif.) University. His financial disclosures are available at www.nejm.org. Dr. Galli made these remarks in an editorial accompanying Dr. Cahill’s report (N Engl J Med. 2017 May 18. doi: 10.1056/NEJMe1702653).
As Cahill et al. noted, these data suggest that the role of mast cells should be studied further, but cautiously.
It is not yet time to target mast cells in patients with asthma. Evolution has given us these cells for a reason. They appear to assist in host defense against parasites and play a role in other innate and adaptive immune responses. And they likely have other beneficial effects that haven’t been discovered yet.
However, in the unfortunate patients in whom mast cells can be strongly implicated as contributing to disease, either reducing their numbers or suppressing their function may confer more benefit than harm. This is particularly true in the case of asthma, where mast cells can be targeted locally, in the airways.
Stephen J. Galli, MD, is the Mary Hewitt Loveless, MD, Professor in the school of medicine, and is at the Sean N. Parker Center for Allergy and Asthma Research at Stanford (Calif.) University. His financial disclosures are available at www.nejm.org. Dr. Galli made these remarks in an editorial accompanying Dr. Cahill’s report (N Engl J Med. 2017 May 18. doi: 10.1056/NEJMe1702653).
Imatinib decreased airway mast-cell counts and airway hyperresponsiveness in adults with asthma, who were not responding well to maximal therapy, according to a report published online May 17 in the New England Journal of Medicine.
Imatinib is an inhibitor of the stem-cell factor receptor KIT, which is essential for mast-cell development and survival in bodily tissues. This study’s findings suggest that KIT-dependent processes and mast cells contribute to the pathobiology of severe asthma.
The researchers undertook this study because imatinib is known to reduce bone-marrow mast cells and tryptase levels in chronic myeloid leukemia and to reduce serum tryptase in patients with pulmonary hypertension. Tryptase is a marker of mast-cell burden and activation when detected in extracellular fluids, and it is elevated in the bronchoalveolar lavage fluid from patients with uncontrolled asthma.
To examine whether imatinib would decrease mast-cell counts and activation in the airways of adults with severe, refractory asthma, the investigators performed the randomized double-blind proof-of-principle trial at seven academic centers across the United States over the course of 5 years. A total of 62 patients were assigned to 24 weeks of either oral imatinib (32 participants) or a matching placebo (30 participants). Fifty patients, 24 in the imatinib group and 26 in the placebo group, completed the trial.
The primary outcome measure was the change in airway hyperresponsiveness at 6 months, as measured by the increase in the concentration of methacholine that causes significant bronchoconstriction (PC20). Imatinib decreased airway hyperresponsiveness to a greater degree than did placebo. Imatinib increased PC20 by a mean of 1.20 doubling doses at 3 months and by a mean of 1.73 doubling doses at 6 months, compared with 0.03 and 1.07, respectively, for placebo.
The small improvement in the placebo group is consistent with a phenomenon reported in other studies, in which patients show a delayed improvement in airway hyperresponsiveness for several months after they started inhaled glucocorticoids, Dr. Cahill and her associates noted (N Engl J Med. 2017 May 18. doi: 10.1056/NEJMoa1613125).
Imatinib also reduced mast-cell activity as measured by serum and airway levels of tryptase. Serum tryptase decreased by 43% in the imatinib group, compared with a 12% decline in the placebo group. And tryptase levels in bronchoalveolar lavage fluid tended to decrease in the imatinib group but to increase in the placebo group.
Imatinib also increased mean forced expiratory volume in 1 second (FEV1).
“Although the increase in FEV1 may not seem substantial, it suggests that mast-cell–dependent processes contribute to airway obstruction in these patients despite high-dose, anti-inflammatory glucocorticoid therapy. The near–50-mL difference in the change in baseline FEV1 between the imatinib and placebo groups is small, but it is likely to be important in light of the population we studied,” Dr. Cahill and her associates wrote.
In addition, exploratory analyses showed that the reduction in airway hyperresponsiveness with imatinib “negatively correlated with baseline blood eosinophil counts, and baseline numbers of neutrophils in bronchoalveolar lavage fluid were strongly correlated with increases in FEV1. Together, these findings support a role for mast cells in noneosinophilic asthma. Since almost half of the patients with severe asthma have neutrophilic airway inflammation, we speculate that KIT inhibition might represent an important approach to treatment for this group,” they said.
This study was supported by the National Heart, Lung, and Blood Institute, the National Institute of Allergy and Infectious Diseases, the Vinik family, and the Kaye family; Novartis provided imatinib free of charge. The authors’ financial disclosures are available at www.nejm.org.
Imatinib decreased airway mast-cell counts and airway hyperresponsiveness in adults with asthma, who were not responding well to maximal therapy, according to a report published online May 17 in the New England Journal of Medicine.
Imatinib is an inhibitor of the stem-cell factor receptor KIT, which is essential for mast-cell development and survival in bodily tissues. This study’s findings suggest that KIT-dependent processes and mast cells contribute to the pathobiology of severe asthma.
The researchers undertook this study because imatinib is known to reduce bone-marrow mast cells and tryptase levels in chronic myeloid leukemia and to reduce serum tryptase in patients with pulmonary hypertension. Tryptase is a marker of mast-cell burden and activation when detected in extracellular fluids, and it is elevated in the bronchoalveolar lavage fluid from patients with uncontrolled asthma.
To examine whether imatinib would decrease mast-cell counts and activation in the airways of adults with severe, refractory asthma, the investigators performed the randomized double-blind proof-of-principle trial at seven academic centers across the United States over the course of 5 years. A total of 62 patients were assigned to 24 weeks of either oral imatinib (32 participants) or a matching placebo (30 participants). Fifty patients, 24 in the imatinib group and 26 in the placebo group, completed the trial.
The primary outcome measure was the change in airway hyperresponsiveness at 6 months, as measured by the increase in the concentration of methacholine that causes significant bronchoconstriction (PC20). Imatinib decreased airway hyperresponsiveness to a greater degree than did placebo. Imatinib increased PC20 by a mean of 1.20 doubling doses at 3 months and by a mean of 1.73 doubling doses at 6 months, compared with 0.03 and 1.07, respectively, for placebo.
The small improvement in the placebo group is consistent with a phenomenon reported in other studies, in which patients show a delayed improvement in airway hyperresponsiveness for several months after they started inhaled glucocorticoids, Dr. Cahill and her associates noted (N Engl J Med. 2017 May 18. doi: 10.1056/NEJMoa1613125).
Imatinib also reduced mast-cell activity as measured by serum and airway levels of tryptase. Serum tryptase decreased by 43% in the imatinib group, compared with a 12% decline in the placebo group. And tryptase levels in bronchoalveolar lavage fluid tended to decrease in the imatinib group but to increase in the placebo group.
Imatinib also increased mean forced expiratory volume in 1 second (FEV1).
“Although the increase in FEV1 may not seem substantial, it suggests that mast-cell–dependent processes contribute to airway obstruction in these patients despite high-dose, anti-inflammatory glucocorticoid therapy. The near–50-mL difference in the change in baseline FEV1 between the imatinib and placebo groups is small, but it is likely to be important in light of the population we studied,” Dr. Cahill and her associates wrote.
In addition, exploratory analyses showed that the reduction in airway hyperresponsiveness with imatinib “negatively correlated with baseline blood eosinophil counts, and baseline numbers of neutrophils in bronchoalveolar lavage fluid were strongly correlated with increases in FEV1. Together, these findings support a role for mast cells in noneosinophilic asthma. Since almost half of the patients with severe asthma have neutrophilic airway inflammation, we speculate that KIT inhibition might represent an important approach to treatment for this group,” they said.
This study was supported by the National Heart, Lung, and Blood Institute, the National Institute of Allergy and Infectious Diseases, the Vinik family, and the Kaye family; Novartis provided imatinib free of charge. The authors’ financial disclosures are available at www.nejm.org.
Key clinical point: Imatinib, a KIT inhibitor, reduced mast cell counts and airway hyperresponsiveness in severe asthma.
Major finding: Imatinib increased PC20 by a mean of 1.20 doubling doses at 3 months and by a mean of 1.73 doubling doses at 6 months, compared with 0.03 and 1.07, respectively, for placebo.
Data source: A randomized, double-blind, placebo-controlled proof-of-principle trial involving 62 adults treated for 24 weeks.
Disclosures: This study was supported by the National Heart, Lung, and Blood Institute, the National Institute of Allergy and Infectious Diseases, the Vinik family, and the Kaye family; Novartis provided imatinib free of charge. The researchers’ financial disclosures are available at www.nejm.org.