User login
Topical tretinoin resolves inflammatory symptoms in rosacea, in small study
SYDNEY, AUSTRALIA – Treatment with topical tretinoin resulted in complete resolution of rosacea symptoms in a significant number of patients, in a small retrospective study presented at the annual meeting of the Australasian College of Dermatologists.
“As an intermediary step between topical antibiotics and oral isotretinoin, we propose that topical tretinoin may be effective in the management and reduction of rosacea symptoms,” Emily Forward, MD, of the University of Sydney, said at the meeting. There has been recent discussion regarding the use of low-dose isotretinoin in the treatment of rosacea, but safety with long-term use is an issue, she noted.

More than 80% of patients had complete or excellent resolution of papules and pustules, with only one patient showing no benefit. Of the patients with erythema as the primary feature of their rosacea, 42% achieved complete resolution, 33% achieved excellent resolution, 17% achieved a good response, and 8% showed no benefit, Dr. Forward reported.
Among patients with telangiectasia, 40% achieved complete resolution, while 37% of those with flushing achieved complete resolution.
Topical tretinoin should be considered among the treatment options for rosacea “as it is effective, well tolerated, and has synergistic benefits in the prevention of photoaging,” Dr. Forward said. The ideal patient candidate would be someone with inflammatory features such as papules, pustules, or erythema, she added.
No patients experienced worsening of their symptoms with treatment, although one patient stopped treatment because of adverse effects. Dr. Forward also stressed that tretinoin is a known teratogen, so it should not be used during pregnancy or breastfeeding, or in patients trying to conceive.
In an interview, Dr. Forward said that she and her associates were surprised at the degree of improvement with topical tretinoin, particularly for erythema symptoms. However, she said it was important to educate patients about how to use topical tretinoin. “You use a pea-sized amount, at night, and in the beginning we advise them to use it every second or third day, and if they tolerate it they can increase the amount,” she said.
No conflicts of interest were declared.
SYDNEY, AUSTRALIA – Treatment with topical tretinoin resulted in complete resolution of rosacea symptoms in a significant number of patients, in a small retrospective study presented at the annual meeting of the Australasian College of Dermatologists.
“As an intermediary step between topical antibiotics and oral isotretinoin, we propose that topical tretinoin may be effective in the management and reduction of rosacea symptoms,” Emily Forward, MD, of the University of Sydney, said at the meeting. There has been recent discussion regarding the use of low-dose isotretinoin in the treatment of rosacea, but safety with long-term use is an issue, she noted.
More than 80% of patients had complete or excellent resolution of papules and pustules, with only one patient showing no benefit. Of the patients with erythema as the primary feature of their rosacea, 42% achieved complete resolution, 33% achieved excellent resolution, 17% achieved a good response, and 8% showed no benefit, Dr. Forward reported.
Among patients with telangiectasia, 40% achieved complete resolution, while 37% of those with flushing achieved complete resolution.
Topical tretinoin should be considered among the treatment options for rosacea “as it is effective, well tolerated, and has synergistic benefits in the prevention of photoaging,” Dr. Forward said. The ideal patient candidate would be someone with inflammatory features such as papules, pustules, or erythema, she added.
No patients experienced worsening of their symptoms with treatment, although one patient stopped treatment because of adverse effects. Dr. Forward also stressed that tretinoin is a known teratogen, so it should not be used during pregnancy or breastfeeding, or in patients trying to conceive.
In an interview, Dr. Forward said that she and her associates were surprised at the degree of improvement with topical tretinoin, particularly for erythema symptoms. However, she said it was important to educate patients about how to use topical tretinoin. “You use a pea-sized amount, at night, and in the beginning we advise them to use it every second or third day, and if they tolerate it they can increase the amount,” she said.
No conflicts of interest were declared.
SYDNEY, AUSTRALIA – Treatment with topical tretinoin resulted in complete resolution of rosacea symptoms in a significant number of patients, in a small retrospective study presented at the annual meeting of the Australasian College of Dermatologists.
“As an intermediary step between topical antibiotics and oral isotretinoin, we propose that topical tretinoin may be effective in the management and reduction of rosacea symptoms,” Emily Forward, MD, of the University of Sydney, said at the meeting. There has been recent discussion regarding the use of low-dose isotretinoin in the treatment of rosacea, but safety with long-term use is an issue, she noted.
More than 80% of patients had complete or excellent resolution of papules and pustules, with only one patient showing no benefit. Of the patients with erythema as the primary feature of their rosacea, 42% achieved complete resolution, 33% achieved excellent resolution, 17% achieved a good response, and 8% showed no benefit, Dr. Forward reported.
Among patients with telangiectasia, 40% achieved complete resolution, while 37% of those with flushing achieved complete resolution.
Topical tretinoin should be considered among the treatment options for rosacea “as it is effective, well tolerated, and has synergistic benefits in the prevention of photoaging,” Dr. Forward said. The ideal patient candidate would be someone with inflammatory features such as papules, pustules, or erythema, she added.
No patients experienced worsening of their symptoms with treatment, although one patient stopped treatment because of adverse effects. Dr. Forward also stressed that tretinoin is a known teratogen, so it should not be used during pregnancy or breastfeeding, or in patients trying to conceive.
In an interview, Dr. Forward said that she and her associates were surprised at the degree of improvement with topical tretinoin, particularly for erythema symptoms. However, she said it was important to educate patients about how to use topical tretinoin. “You use a pea-sized amount, at night, and in the beginning we advise them to use it every second or third day, and if they tolerate it they can increase the amount,” she said.
No conflicts of interest were declared.
AT ACDASM 2017
Key clinical point: Topical tretinoin may be useful in treating erythema and the inflammatory symptoms of rosacea.
Major finding: More than 80% of patients with rosacea had complete or excellent resolution of papules and pustules with topical tretinoin 0.05%.
Data source: A retrospective study of 25 patients with mild to severe rosacea.
Disclosures: No conflicts of interest were declared.
My mundane genetic testing results
I’ve never been particularly curious about my genetic background. We have a pretty clear family history that I’m of central European and Russian descent, with my ancestors coming over in groups between 1900 and 1938.
Recently, my mother decided she wanted more genetic information on us, so she paid $99 for us to send saliva samples to a company that advertises such services.
A few weeks went by. You read about people who find out they have a genetic background that’s quite surprising. I began to wonder: Would there be some giant family history shocker when the results came in?

I’ve since spoken to others who had paid for this service and found most had the same experience. The test confirmed what was already well known, except for one friend whose results suggested a trace of Polynesian blood somewhere in his background. He believes this was likely artefactual, though he enjoys the idea that somewhere in history a Tongan warrior was blown off course at sea and somehow ended up in Odessa, Ukraine.
Of course, as I’ve now learned, that’s only the start of things. These days, I get emails advertising a more detailed panel (for an additional fee), looking for genetic markers for disease and more obscure traits. I also receive the occasional one from someone who, through the company’s anonymous servers, thinks they may be related to me.
I don’t answer either of those. I have no desire to expand my family circle beyond what it already is.
As for the disease testing? Not interested. Yes, some genetic tests may be helpful in making better choices, but the majority, at least to me, are still a work in progress. We deal with both false negatives and false positives in medicine. I routinely discourage my patients from spending money on unproven testing and treatments and have no desire to do the same myself. Maybe someday it will be worth the additional dollars, but I’m not convinced it’s there.
Money is, for better or worse, the driving force for all technologies, medical and otherwise. Maybe my $99 investment will help pay dividends down the road for someone, but today it only resulted in a shoulder shrug and chuckle at what I already knew.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
I’ve never been particularly curious about my genetic background. We have a pretty clear family history that I’m of central European and Russian descent, with my ancestors coming over in groups between 1900 and 1938.
Recently, my mother decided she wanted more genetic information on us, so she paid $99 for us to send saliva samples to a company that advertises such services.
A few weeks went by. You read about people who find out they have a genetic background that’s quite surprising. I began to wonder: Would there be some giant family history shocker when the results came in?
I’ve since spoken to others who had paid for this service and found most had the same experience. The test confirmed what was already well known, except for one friend whose results suggested a trace of Polynesian blood somewhere in his background. He believes this was likely artefactual, though he enjoys the idea that somewhere in history a Tongan warrior was blown off course at sea and somehow ended up in Odessa, Ukraine.
Of course, as I’ve now learned, that’s only the start of things. These days, I get emails advertising a more detailed panel (for an additional fee), looking for genetic markers for disease and more obscure traits. I also receive the occasional one from someone who, through the company’s anonymous servers, thinks they may be related to me.
I don’t answer either of those. I have no desire to expand my family circle beyond what it already is.
As for the disease testing? Not interested. Yes, some genetic tests may be helpful in making better choices, but the majority, at least to me, are still a work in progress. We deal with both false negatives and false positives in medicine. I routinely discourage my patients from spending money on unproven testing and treatments and have no desire to do the same myself. Maybe someday it will be worth the additional dollars, but I’m not convinced it’s there.
Money is, for better or worse, the driving force for all technologies, medical and otherwise. Maybe my $99 investment will help pay dividends down the road for someone, but today it only resulted in a shoulder shrug and chuckle at what I already knew.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
I’ve never been particularly curious about my genetic background. We have a pretty clear family history that I’m of central European and Russian descent, with my ancestors coming over in groups between 1900 and 1938.
Recently, my mother decided she wanted more genetic information on us, so she paid $99 for us to send saliva samples to a company that advertises such services.
A few weeks went by. You read about people who find out they have a genetic background that’s quite surprising. I began to wonder: Would there be some giant family history shocker when the results came in?
I’ve since spoken to others who had paid for this service and found most had the same experience. The test confirmed what was already well known, except for one friend whose results suggested a trace of Polynesian blood somewhere in his background. He believes this was likely artefactual, though he enjoys the idea that somewhere in history a Tongan warrior was blown off course at sea and somehow ended up in Odessa, Ukraine.
Of course, as I’ve now learned, that’s only the start of things. These days, I get emails advertising a more detailed panel (for an additional fee), looking for genetic markers for disease and more obscure traits. I also receive the occasional one from someone who, through the company’s anonymous servers, thinks they may be related to me.
I don’t answer either of those. I have no desire to expand my family circle beyond what it already is.
As for the disease testing? Not interested. Yes, some genetic tests may be helpful in making better choices, but the majority, at least to me, are still a work in progress. We deal with both false negatives and false positives in medicine. I routinely discourage my patients from spending money on unproven testing and treatments and have no desire to do the same myself. Maybe someday it will be worth the additional dollars, but I’m not convinced it’s there.
Money is, for better or worse, the driving force for all technologies, medical and otherwise. Maybe my $99 investment will help pay dividends down the road for someone, but today it only resulted in a shoulder shrug and chuckle at what I already knew.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
How to work with specialists in value-based care
The typical primary care physician has a patient base that consumes $10 million of health care a year. Yet the PCP receives only 6%-7% of those payments, with the rest of the costs resulting largely from the PCP’s referrals or lack of PCP care management of that patient.
The average PCP makes 1,000 referrals a year. Often, the referee specialist or facility not only does not coordinate with the PCP’s patient-centered medical home, they make their own downstream referrals.
A revolution in your compensation is underway. Under MACRA and other accountable care models, providers across the continuum of care are now being held responsible for the overall costs of those patients, not just their charges.
This is still hard to grasp, isn’t it? I was recently talking to a preeminent primary care physician who was an active member of an accountable care organization board of directors. I was fairly excited about the new impact this highly professional community leader could have on patients, now that he was in the PCP-driven ACO, not to mention his shared savings payment opportunities.
I was on a roll until he said, “But Bo, I’m already as efficient in treating patients as I can get.” He was still fighting the barriers you all face to do the best he could under the circumstances for the patients in his office each day.
Later, however, on a better day for me, we were working together on a cardiac care white paper. The physician leader told me, “I get it now – the biggest value-adding impact I might have is for the patient I don’t ever see.”
The above statistics show just what an opportunity you have in the new value care.
You can legally control referrals and patient care coordination with specialists. They don’t have to be in your ACO. You don’t even need to be in an ACO to take advantage of high-value referrals under the Medicare Merit-based Incentive Payment System (MIPS) program under MACRA. But how?
Let’s start by assuming the specialist you need to refer to is not in your ACO. You might be able to do this without an ACO, but it’s hard to get the critical mass of primary care physicians. If you’re under the Medicare Shared Savings Program or Next Gen initiative, there are important Stark Law and antikickback liability waivers that would benefit you by being in an ACO.
Otherwise, you should consider a high-value referral affiliation agreement.
If a critical mass of primary care physicians can access data that create a short list of high-value specialists, they can put them on the high-value specialist list. Specialists do not need to get part of the shared savings pool or other financial incentives – just referrals because of their high-quality and high-efficiency care. A superstar specialist or acute care or post–acute care facility may ultimately be invited into the ACO as a full participant.
The specialist/facility basically agrees to coordinate all care with the medical home and comanage that care with you. The agreement specifies that they will observe the care protocols of the ACO for that disease state. The provider will share data and agree to be monitored.
What is a high-value specialist/facility? The current common approach is to look at the insurance companies’ top tiers, but they are often too weighted to allowed charges. It’s really about being care coordinators and about readmission and complication rates.
For example, some bundled-payment specialists are selected solely based on the surgeons’ and anesthesiologists’ complication rates. If fees are mentioned at all, they are well down the list.
Of course, if the specialist is in the ACO with the primary care physician, this can be done internally.
How do you find value-added protocols involving specialists? I was lucky to be on a multiyear grant program whereby I worked with many primary care physicians and specialists to create white papers setting out high-value, practical initiatives. There are also guides for internists and family physicians. A condition of the grant was that they all can be accessed free of charge; they’re available at www.tac-consortium.org/resources.
This is a new day. Primary care is being asked to lead health care delivery today and be paid to do it. You are being rewarded or punished financially now based on the overall costs of your patients. You must have specialists and facilities coordinate with you in this new health care model. We have attempted to provide a road map to assist you on your journey.
Mr. Bobbitt is head of the health law group at the Smith Anderson law firm in Raleigh, N.C. He is president of Value Health Partners, LLC, a health care strategic consulting company. He has years of experience assisting physicians form integrated delivery systems. He has spoken and written nationally to primary care physicians on the strategies and practicalities of forming or joining ACOs. This article is meant to be educational and does not constitute legal advice. For additional information, readers may contact the author at [email protected] or 919-821-6612.
The typical primary care physician has a patient base that consumes $10 million of health care a year. Yet the PCP receives only 6%-7% of those payments, with the rest of the costs resulting largely from the PCP’s referrals or lack of PCP care management of that patient.
The average PCP makes 1,000 referrals a year. Often, the referee specialist or facility not only does not coordinate with the PCP’s patient-centered medical home, they make their own downstream referrals.
A revolution in your compensation is underway. Under MACRA and other accountable care models, providers across the continuum of care are now being held responsible for the overall costs of those patients, not just their charges.
This is still hard to grasp, isn’t it? I was recently talking to a preeminent primary care physician who was an active member of an accountable care organization board of directors. I was fairly excited about the new impact this highly professional community leader could have on patients, now that he was in the PCP-driven ACO, not to mention his shared savings payment opportunities.
I was on a roll until he said, “But Bo, I’m already as efficient in treating patients as I can get.” He was still fighting the barriers you all face to do the best he could under the circumstances for the patients in his office each day.
Later, however, on a better day for me, we were working together on a cardiac care white paper. The physician leader told me, “I get it now – the biggest value-adding impact I might have is for the patient I don’t ever see.”
The above statistics show just what an opportunity you have in the new value care.
You can legally control referrals and patient care coordination with specialists. They don’t have to be in your ACO. You don’t even need to be in an ACO to take advantage of high-value referrals under the Medicare Merit-based Incentive Payment System (MIPS) program under MACRA. But how?
Let’s start by assuming the specialist you need to refer to is not in your ACO. You might be able to do this without an ACO, but it’s hard to get the critical mass of primary care physicians. If you’re under the Medicare Shared Savings Program or Next Gen initiative, there are important Stark Law and antikickback liability waivers that would benefit you by being in an ACO.
Otherwise, you should consider a high-value referral affiliation agreement.
If a critical mass of primary care physicians can access data that create a short list of high-value specialists, they can put them on the high-value specialist list. Specialists do not need to get part of the shared savings pool or other financial incentives – just referrals because of their high-quality and high-efficiency care. A superstar specialist or acute care or post–acute care facility may ultimately be invited into the ACO as a full participant.
The specialist/facility basically agrees to coordinate all care with the medical home and comanage that care with you. The agreement specifies that they will observe the care protocols of the ACO for that disease state. The provider will share data and agree to be monitored.
What is a high-value specialist/facility? The current common approach is to look at the insurance companies’ top tiers, but they are often too weighted to allowed charges. It’s really about being care coordinators and about readmission and complication rates.
For example, some bundled-payment specialists are selected solely based on the surgeons’ and anesthesiologists’ complication rates. If fees are mentioned at all, they are well down the list.
Of course, if the specialist is in the ACO with the primary care physician, this can be done internally.
How do you find value-added protocols involving specialists? I was lucky to be on a multiyear grant program whereby I worked with many primary care physicians and specialists to create white papers setting out high-value, practical initiatives. There are also guides for internists and family physicians. A condition of the grant was that they all can be accessed free of charge; they’re available at www.tac-consortium.org/resources.
This is a new day. Primary care is being asked to lead health care delivery today and be paid to do it. You are being rewarded or punished financially now based on the overall costs of your patients. You must have specialists and facilities coordinate with you in this new health care model. We have attempted to provide a road map to assist you on your journey.
Mr. Bobbitt is head of the health law group at the Smith Anderson law firm in Raleigh, N.C. He is president of Value Health Partners, LLC, a health care strategic consulting company. He has years of experience assisting physicians form integrated delivery systems. He has spoken and written nationally to primary care physicians on the strategies and practicalities of forming or joining ACOs. This article is meant to be educational and does not constitute legal advice. For additional information, readers may contact the author at [email protected] or 919-821-6612.
The typical primary care physician has a patient base that consumes $10 million of health care a year. Yet the PCP receives only 6%-7% of those payments, with the rest of the costs resulting largely from the PCP’s referrals or lack of PCP care management of that patient.
The average PCP makes 1,000 referrals a year. Often, the referee specialist or facility not only does not coordinate with the PCP’s patient-centered medical home, they make their own downstream referrals.
A revolution in your compensation is underway. Under MACRA and other accountable care models, providers across the continuum of care are now being held responsible for the overall costs of those patients, not just their charges.
This is still hard to grasp, isn’t it? I was recently talking to a preeminent primary care physician who was an active member of an accountable care organization board of directors. I was fairly excited about the new impact this highly professional community leader could have on patients, now that he was in the PCP-driven ACO, not to mention his shared savings payment opportunities.
I was on a roll until he said, “But Bo, I’m already as efficient in treating patients as I can get.” He was still fighting the barriers you all face to do the best he could under the circumstances for the patients in his office each day.
Later, however, on a better day for me, we were working together on a cardiac care white paper. The physician leader told me, “I get it now – the biggest value-adding impact I might have is for the patient I don’t ever see.”
The above statistics show just what an opportunity you have in the new value care.
You can legally control referrals and patient care coordination with specialists. They don’t have to be in your ACO. You don’t even need to be in an ACO to take advantage of high-value referrals under the Medicare Merit-based Incentive Payment System (MIPS) program under MACRA. But how?
Let’s start by assuming the specialist you need to refer to is not in your ACO. You might be able to do this without an ACO, but it’s hard to get the critical mass of primary care physicians. If you’re under the Medicare Shared Savings Program or Next Gen initiative, there are important Stark Law and antikickback liability waivers that would benefit you by being in an ACO.
Otherwise, you should consider a high-value referral affiliation agreement.
If a critical mass of primary care physicians can access data that create a short list of high-value specialists, they can put them on the high-value specialist list. Specialists do not need to get part of the shared savings pool or other financial incentives – just referrals because of their high-quality and high-efficiency care. A superstar specialist or acute care or post–acute care facility may ultimately be invited into the ACO as a full participant.
The specialist/facility basically agrees to coordinate all care with the medical home and comanage that care with you. The agreement specifies that they will observe the care protocols of the ACO for that disease state. The provider will share data and agree to be monitored.
What is a high-value specialist/facility? The current common approach is to look at the insurance companies’ top tiers, but they are often too weighted to allowed charges. It’s really about being care coordinators and about readmission and complication rates.
For example, some bundled-payment specialists are selected solely based on the surgeons’ and anesthesiologists’ complication rates. If fees are mentioned at all, they are well down the list.
Of course, if the specialist is in the ACO with the primary care physician, this can be done internally.
How do you find value-added protocols involving specialists? I was lucky to be on a multiyear grant program whereby I worked with many primary care physicians and specialists to create white papers setting out high-value, practical initiatives. There are also guides for internists and family physicians. A condition of the grant was that they all can be accessed free of charge; they’re available at www.tac-consortium.org/resources.
This is a new day. Primary care is being asked to lead health care delivery today and be paid to do it. You are being rewarded or punished financially now based on the overall costs of your patients. You must have specialists and facilities coordinate with you in this new health care model. We have attempted to provide a road map to assist you on your journey.
Mr. Bobbitt is head of the health law group at the Smith Anderson law firm in Raleigh, N.C. He is president of Value Health Partners, LLC, a health care strategic consulting company. He has years of experience assisting physicians form integrated delivery systems. He has spoken and written nationally to primary care physicians on the strategies and practicalities of forming or joining ACOs. This article is meant to be educational and does not constitute legal advice. For additional information, readers may contact the author at [email protected] or 919-821-6612.
Illness-induced PTSD is common, understudied
SAN FRANCISCO – Posttraumatic stress disorder symptoms triggered by a life-threatening medical illness differ from the more common PTSD, the source of which is an external trauma such as an assault or natural disaster, according to Renee El-Gabalawy, PhD.
“This suggests implications for diagnostic classification. Maybe, in future editions of the DSM, we should think of this as a subtype of PTSD or potentially as a new diagnostic category, although it’s far too early to make any conclusions about that,” Dr. El-Gabalawy said at the annual conference of the Anxiety and Depression Association of America.

It’s estimated that PTSD occurs in 12%-25% of people who experience a life-threatening medical event.
“This is a fairly staggering proportion of people, and unfortunately this is a very overlooked area in the PTSD literature, almost all of which has been done in critical care units or oncology settings,” said Dr. El-Gabalawy, a psychologist at the University of Manitoba in Winnipeg.
She presented an analysis of data from the 2012-2013 National Epidemiologic Survey on Alcohol and Related Conditions, in which a nationally representative sample composed of 36,309 U.S. adults were interviewed face to face, with the current DSM-5 diagnostic criteria for PTSD being applied using the Alcohol Use Disorder and Association Disabilities Interview Schedule–5 (AUDADIS-5).
A total of 1,779 subjects (4.9%) indicated they had experienced physician-diagnosed PTSD during the previous year. Of those, 6.5% said their PTSD was triggered by an acute life-threatening medical event. The rest were attributed to nonmedical trauma.
There were sharp demographic differences between the two groups. Individuals with medical illness–induced PTSD were older – 35 years old at onset of their first episode, compared with age 23 in the others – with later onset of their PTSD. They were more likely to be men: 45.7% were male, compared with 31.8% for subjects with nonmedical PTSD. Comorbid depression was present in 25.4% of those with medical illness–induced PTSD, and comorbid panic disorder was present in 17%, significantly lower than the 37% and 24.5% rates in individuals with other triggers of PTSD.
Quality of life as measured by the Short Form-12 was similar in the two groups, after the investigators controlled for the number of medical conditions patients had.
Of people with medical illness–induced PTSD, 41% attributed their PTSD to a digestive disease, most often inflammatory bowel disease. In contrast, a digestive condition was present in 19.2% of subjects with nonmedical trauma as the source of their PTSD. Thus, a serious digestive disorder was associated with a 2.4-times increased risk of medical illness–induced PTSD in an analysis adjusted for socioeconomic factors and number of health conditions. Cancer, which was the trigger for 16.1% of cases of medical illness–induced PTSD and which had a prevalence of 5.8% in those with nonmedical sources of PTSD, was associated with a 2.64-times increased risk of medical illness–related PTSD.
“Those odds ratios are quite high for a population-based sample. This was a very dramatic effect,” Dr. El-Gabalawy commented.
The two groups of participants with PTSD had similar intensity of core PTSD symptom clusters with the exception of negative mood/cognition, which figured more prominently in those with medical illness–induced PTSD.
“This is very much in line with my clinical experience, that what’s really predominant in these folks are the maladaptive cognitions, their fear about their future health trajectory,” she said. “I tend to use cognitive processing therapy in these patients. It really taps into those maladaptive cognitions, and I’ve found that my patients are very receptive to this. Cognitive processing therapy might be more advantageous in this situation than prolonged exposure therapy .”
Dr. El-Gabalawy said she is a fan of the Enduring Somatic Threat model of medical illness–induced PTSD developed by Donald Edmondson, PhD, of Columbia University in New York (Soc Personal Psychol Compass. 2014 Mar 5;8[3]:118-34).
“It aligns with the literature and my own clinical experience,” she explained.
Dr. Edmondson’s model draws conceptual distinctions between medical illness–induced PTSD and other causes of PTSD. In medical illness–related PTSD, the trauma has a somatic source, the trauma tends to be chronic, and intrusive thoughts tend to be future oriented and highly cognitive in nature.
“It’s not uncommon that I’ll hear my patients with medical illness–induced PTSD say, ‘I’m really scared my disease is going to get worse.’ And behavioral avoidance is really difficult. Whereas, in the traditional conceptualization of PTSD, the intrusions are often past oriented and elicited by external triggers. Behavioral avoidance of those triggers is possible, but, in illness-related PTSD, arousal is keyed to internal triggers, often somatic in nature, such as heart palpitations,” according to the psychologist.
Her study was supported by the Canadian National Institutes of Health Research and the University of Manitoba. She reported having no financial conflicts.
SAN FRANCISCO – Posttraumatic stress disorder symptoms triggered by a life-threatening medical illness differ from the more common PTSD, the source of which is an external trauma such as an assault or natural disaster, according to Renee El-Gabalawy, PhD.
“This suggests implications for diagnostic classification. Maybe, in future editions of the DSM, we should think of this as a subtype of PTSD or potentially as a new diagnostic category, although it’s far too early to make any conclusions about that,” Dr. El-Gabalawy said at the annual conference of the Anxiety and Depression Association of America.
It’s estimated that PTSD occurs in 12%-25% of people who experience a life-threatening medical event.
“This is a fairly staggering proportion of people, and unfortunately this is a very overlooked area in the PTSD literature, almost all of which has been done in critical care units or oncology settings,” said Dr. El-Gabalawy, a psychologist at the University of Manitoba in Winnipeg.
She presented an analysis of data from the 2012-2013 National Epidemiologic Survey on Alcohol and Related Conditions, in which a nationally representative sample composed of 36,309 U.S. adults were interviewed face to face, with the current DSM-5 diagnostic criteria for PTSD being applied using the Alcohol Use Disorder and Association Disabilities Interview Schedule–5 (AUDADIS-5).
A total of 1,779 subjects (4.9%) indicated they had experienced physician-diagnosed PTSD during the previous year. Of those, 6.5% said their PTSD was triggered by an acute life-threatening medical event. The rest were attributed to nonmedical trauma.
There were sharp demographic differences between the two groups. Individuals with medical illness–induced PTSD were older – 35 years old at onset of their first episode, compared with age 23 in the others – with later onset of their PTSD. They were more likely to be men: 45.7% were male, compared with 31.8% for subjects with nonmedical PTSD. Comorbid depression was present in 25.4% of those with medical illness–induced PTSD, and comorbid panic disorder was present in 17%, significantly lower than the 37% and 24.5% rates in individuals with other triggers of PTSD.
Quality of life as measured by the Short Form-12 was similar in the two groups, after the investigators controlled for the number of medical conditions patients had.
Of people with medical illness–induced PTSD, 41% attributed their PTSD to a digestive disease, most often inflammatory bowel disease. In contrast, a digestive condition was present in 19.2% of subjects with nonmedical trauma as the source of their PTSD. Thus, a serious digestive disorder was associated with a 2.4-times increased risk of medical illness–induced PTSD in an analysis adjusted for socioeconomic factors and number of health conditions. Cancer, which was the trigger for 16.1% of cases of medical illness–induced PTSD and which had a prevalence of 5.8% in those with nonmedical sources of PTSD, was associated with a 2.64-times increased risk of medical illness–related PTSD.
“Those odds ratios are quite high for a population-based sample. This was a very dramatic effect,” Dr. El-Gabalawy commented.
The two groups of participants with PTSD had similar intensity of core PTSD symptom clusters with the exception of negative mood/cognition, which figured more prominently in those with medical illness–induced PTSD.
“This is very much in line with my clinical experience, that what’s really predominant in these folks are the maladaptive cognitions, their fear about their future health trajectory,” she said. “I tend to use cognitive processing therapy in these patients. It really taps into those maladaptive cognitions, and I’ve found that my patients are very receptive to this. Cognitive processing therapy might be more advantageous in this situation than prolonged exposure therapy .”
Dr. El-Gabalawy said she is a fan of the Enduring Somatic Threat model of medical illness–induced PTSD developed by Donald Edmondson, PhD, of Columbia University in New York (Soc Personal Psychol Compass. 2014 Mar 5;8[3]:118-34).
“It aligns with the literature and my own clinical experience,” she explained.
Dr. Edmondson’s model draws conceptual distinctions between medical illness–induced PTSD and other causes of PTSD. In medical illness–related PTSD, the trauma has a somatic source, the trauma tends to be chronic, and intrusive thoughts tend to be future oriented and highly cognitive in nature.
“It’s not uncommon that I’ll hear my patients with medical illness–induced PTSD say, ‘I’m really scared my disease is going to get worse.’ And behavioral avoidance is really difficult. Whereas, in the traditional conceptualization of PTSD, the intrusions are often past oriented and elicited by external triggers. Behavioral avoidance of those triggers is possible, but, in illness-related PTSD, arousal is keyed to internal triggers, often somatic in nature, such as heart palpitations,” according to the psychologist.
Her study was supported by the Canadian National Institutes of Health Research and the University of Manitoba. She reported having no financial conflicts.
SAN FRANCISCO – Posttraumatic stress disorder symptoms triggered by a life-threatening medical illness differ from the more common PTSD, the source of which is an external trauma such as an assault or natural disaster, according to Renee El-Gabalawy, PhD.
“This suggests implications for diagnostic classification. Maybe, in future editions of the DSM, we should think of this as a subtype of PTSD or potentially as a new diagnostic category, although it’s far too early to make any conclusions about that,” Dr. El-Gabalawy said at the annual conference of the Anxiety and Depression Association of America.
It’s estimated that PTSD occurs in 12%-25% of people who experience a life-threatening medical event.
“This is a fairly staggering proportion of people, and unfortunately this is a very overlooked area in the PTSD literature, almost all of which has been done in critical care units or oncology settings,” said Dr. El-Gabalawy, a psychologist at the University of Manitoba in Winnipeg.
She presented an analysis of data from the 2012-2013 National Epidemiologic Survey on Alcohol and Related Conditions, in which a nationally representative sample composed of 36,309 U.S. adults were interviewed face to face, with the current DSM-5 diagnostic criteria for PTSD being applied using the Alcohol Use Disorder and Association Disabilities Interview Schedule–5 (AUDADIS-5).
A total of 1,779 subjects (4.9%) indicated they had experienced physician-diagnosed PTSD during the previous year. Of those, 6.5% said their PTSD was triggered by an acute life-threatening medical event. The rest were attributed to nonmedical trauma.
There were sharp demographic differences between the two groups. Individuals with medical illness–induced PTSD were older – 35 years old at onset of their first episode, compared with age 23 in the others – with later onset of their PTSD. They were more likely to be men: 45.7% were male, compared with 31.8% for subjects with nonmedical PTSD. Comorbid depression was present in 25.4% of those with medical illness–induced PTSD, and comorbid panic disorder was present in 17%, significantly lower than the 37% and 24.5% rates in individuals with other triggers of PTSD.
Quality of life as measured by the Short Form-12 was similar in the two groups, after the investigators controlled for the number of medical conditions patients had.
Of people with medical illness–induced PTSD, 41% attributed their PTSD to a digestive disease, most often inflammatory bowel disease. In contrast, a digestive condition was present in 19.2% of subjects with nonmedical trauma as the source of their PTSD. Thus, a serious digestive disorder was associated with a 2.4-times increased risk of medical illness–induced PTSD in an analysis adjusted for socioeconomic factors and number of health conditions. Cancer, which was the trigger for 16.1% of cases of medical illness–induced PTSD and which had a prevalence of 5.8% in those with nonmedical sources of PTSD, was associated with a 2.64-times increased risk of medical illness–related PTSD.
“Those odds ratios are quite high for a population-based sample. This was a very dramatic effect,” Dr. El-Gabalawy commented.
The two groups of participants with PTSD had similar intensity of core PTSD symptom clusters with the exception of negative mood/cognition, which figured more prominently in those with medical illness–induced PTSD.
“This is very much in line with my clinical experience, that what’s really predominant in these folks are the maladaptive cognitions, their fear about their future health trajectory,” she said. “I tend to use cognitive processing therapy in these patients. It really taps into those maladaptive cognitions, and I’ve found that my patients are very receptive to this. Cognitive processing therapy might be more advantageous in this situation than prolonged exposure therapy .”
Dr. El-Gabalawy said she is a fan of the Enduring Somatic Threat model of medical illness–induced PTSD developed by Donald Edmondson, PhD, of Columbia University in New York (Soc Personal Psychol Compass. 2014 Mar 5;8[3]:118-34).
“It aligns with the literature and my own clinical experience,” she explained.
Dr. Edmondson’s model draws conceptual distinctions between medical illness–induced PTSD and other causes of PTSD. In medical illness–related PTSD, the trauma has a somatic source, the trauma tends to be chronic, and intrusive thoughts tend to be future oriented and highly cognitive in nature.
“It’s not uncommon that I’ll hear my patients with medical illness–induced PTSD say, ‘I’m really scared my disease is going to get worse.’ And behavioral avoidance is really difficult. Whereas, in the traditional conceptualization of PTSD, the intrusions are often past oriented and elicited by external triggers. Behavioral avoidance of those triggers is possible, but, in illness-related PTSD, arousal is keyed to internal triggers, often somatic in nature, such as heart palpitations,” according to the psychologist.
Her study was supported by the Canadian National Institutes of Health Research and the University of Manitoba. She reported having no financial conflicts.
AT ANXIETY AND DEPRESSION CONFERENCE 2017
Key clinical point:
Major finding: Individuals with PTSD and a serious digestive disease were 2.4-times more likely to have medical illness–induced PTSD than PTSD triggered by a nonmedical cause.
Data source: A cross-sectional study of a nationally representative sample of more than 36,000 U.S. adults, 4.9% of whom met DSM 5 criteria for PTSD.
Disclosures: The presenter reported no financial conflicts regarding her study, which was supported by the Canadian National Institutes of Health Research and the University of Manitoba.
Emergency department use by recently diagnosed cancer patients in California
In 2017 there will be nearly 1.7 million new cancer cases diagnosed, and over 600,000 cancer deaths in the Unites States.1 A 2013 Institute of Medicine report highlighted problems with the current quality of cancer care, including high costs and fragmentation of care.2 Other national reports have called for improvements in the overall quality of care and for reducing costly and possibly avoidable use of health services such as emergency department (ED) visits.3-6 Reduction of avoidable ED visits is often cited as a pathway to reduce costs by avoiding unnecessary tests and treatments that occur in the ED and subsequent hospital admissions.7,8
ED crowding, long waits, and unpredictable treatment environments can also make an ED visit an unpleasant experience for the patient. ED visits during cancer treatment can be particularly troubling and present health concerns for patients who are immunocompromised. In particular, cancer patients in the ED have been found to experience delays in the administration of analgesics, antiemetics, or antibiotics.9
Few studies have examined ED use or its associated predictors among cancer patients. Reports to date that have described ED use have focused on different cancers, which makes comparisons across studies difficult.10 Moreover, the time frames of interest and the type of event after which ED use is evaluated (ie, diagnosis or treatment) are inconsistent in the existing literature.10 Some studies quantifying ED use excluded patients admitted to the hospital after an ED visit.11,12 Taken together, these studies do not provide a clear overview of the extent of ED use by cancer patients or the amount of cancer-related care provided by EDs.
Patterns of ED use among cancer patients derived from large and generalizable samples may help inform providers about true risk factors for ED use. In addition, prioritizing new interventions and focusing future research on groups of patients who are at higher risk for preventable ED use could also improve overall care. To address these issues, accurate estimates of ED use among cancer patients are required.
To our knowledge, this is the first study to describe ED use across a range of cancers in a large population-based sample and to consider the timing of ED visits in relation to initial diagnosis. The findings could provide benchmark comparison data to inform future efforts to identify the subset of possibly preventable ED visits and to design interventions to address preventable ED use.
Material and methods
Data source
California’s Office of Statewide Health Planning and Development (OSHPD) manages the patient discharge dataset (PDD) and the emergency department use (EDU) dataset, providing a high-quality source of information on inpatient and ED use in the state.13 A principal diagnosis and up to 24 secondary diagnoses are recorded in OSHPD datasets. The EDU dataset was used to identify treat-and-release ED visits, and the PDD was used to identify hospitalizations initiated in the ED. The California Cancer Registry (CCR) obtains demographic and diagnosis information for every new invasive cancer diagnosed in California, and data collected by the registry are considered to be complete.14 CCR-OSPHD-linked data provide high-quality health care use information for cancer patients in California.15,16 Using an encrypted version of the social security number called the record linkage number (RLN), we linked the CCR records to the corresponding OSHPD files from 2009-2010.
Institutional review board approval for this study was obtained from the University of California, Davis, Human Subjects Committee and the State of California Committee for the Protection of Human Subjects.
Analysis
ED visits. Visits were included if they occurred on or up to 365 days after the date of cancer diagnosis recorded in the CCR. The visits were coded in mutually exclusive groups as occurring within 30, 31-180, and 181-365 days of diagnosis. Subsequently, we flagged each person as having any ED visit (Yes/No) within 180 days and within 365 days of diagnosis, and we tallied the total number of visits occurring within these time frames for each person.
Cancer type. We used relevant site and histology codes to classify cancer type into 24 mutually exclusive categories using the Surveillance Epidemiology and End Result‘s International Classification of Diseases for Oncology, 3rd Edition (ICD-O-3) Recode Definitions17-19 (Suppl Figure 1).
Individual-level variables. Sociodemographic information for each person was collected from the CCR including gender, age, race/ethnicity, marital status, health insurance status, rural residence, survival time in months, neighborhood socio-economic (SES) status based on the Yang index, and the American Joint Committee on Cancer (AJCC) stage.20,21
Data analysis
Demographic information was analyzed for the cohort using descriptive statistics (frequencies, proportions, means, standard deviations, and ranges) and evaluated for correlations. Fewer than 20 observations had missing data and we removed those observations from our analyses on an item-specific basis.
We tabulated ED visits by cancer type and time from diagnosis and then collapsed visit-level data by RLN to determine the number of ED visits for each person in the sample. The number of days from diagnosis to first ED visit was also tabulated. The cohort was stratified by cancer type and cumulative rates of ED visits were tabulated for individuals with ED visits within 0-180 and 0-365 days from diagnosis. To test the robustness of the findings adjusting for confounding factors known to impact ED use, we used logistic regression to model any ED use (Yes/No) as a function of age, gender, race/ethnicity, cancer stage, insurance status, marital status, urban residence, and Yang SES. After model estimation, we used the method of recycled predictions controlling for the confounding variables to compute the marginal probabilities of ED use by cancer type.22 To adjust for the possible impact of survival on ED use, we performed sensitivity analyses and estimated predicted probabilities adjusting for survival. Separate analyses were performed first adjusting for whether the patient died during the course of each month after diagnosis and then adjusting for whether or not the patient died within 180 days of diagnosis. All analyses were conducted using Stata 13.1.23
Results
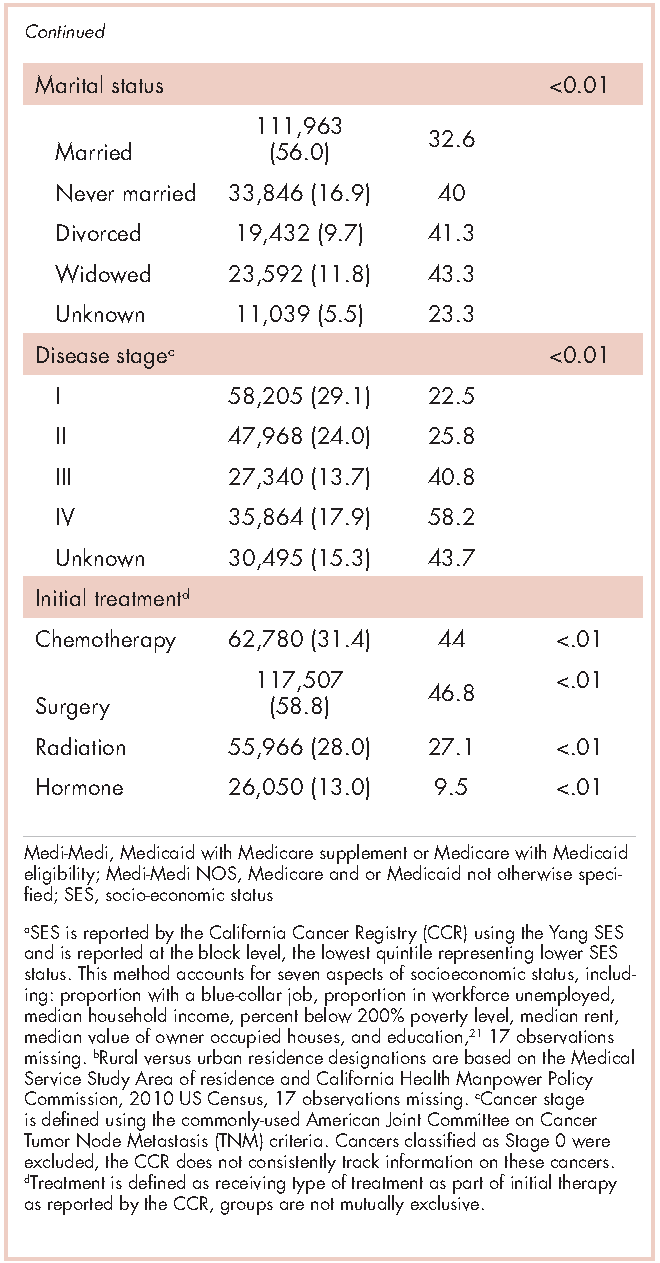
The CCR identified 222,087 adults with a new primary cancer diagnosis in 2009-2010. After excluding those with Stage 0 cancer (n = 21,154) and nonmelanoma skin cancer (n = 1,031), for whom data are inconsistently collected by CCR, a total of 199,872 individuals were included in the analytic sample. Of those patients (Table 1), most were white non-Hispanic (62%), women (51%), holders of private insurance (53%), married (56%), and urban residents (86%). Most were older than 50 years and had either Stage I or Stage II cancer. The most common cancer types were breast (17%), prostate (16%), lung (11%), and colon (9%; results not shown). In unadjusted comparisons, the incidence of ED use was significantly higher among those who were older, of non-Hispanic black race/ethnicity, uninsured, in the lowest SES group, widowed, or diagnosed with Stage IV cancer (Table 1).
ED visits
Within 365 days after initial cancer diagnosis, 87,025 cancer patients made a total of 197,886 ED visits (not shown in tables). Of those visits, 68% (n = 134,556) occurred within 180 days of diagnosis, with 22% (n = 43,535) occurring within the first 30 days and 46% (n = 91,027) occurring within 31-180 days after diagnosis (Figure). Given that most of the visits occurred within 180 days of diagnosis, we used that time frame in subsequent analyses. Among all ED visits within 180 days of diagnosis (Table 2), the largest proportions of visits were made by those with lung cancer (16%), breast cancer (11%), and colon cancer (10%).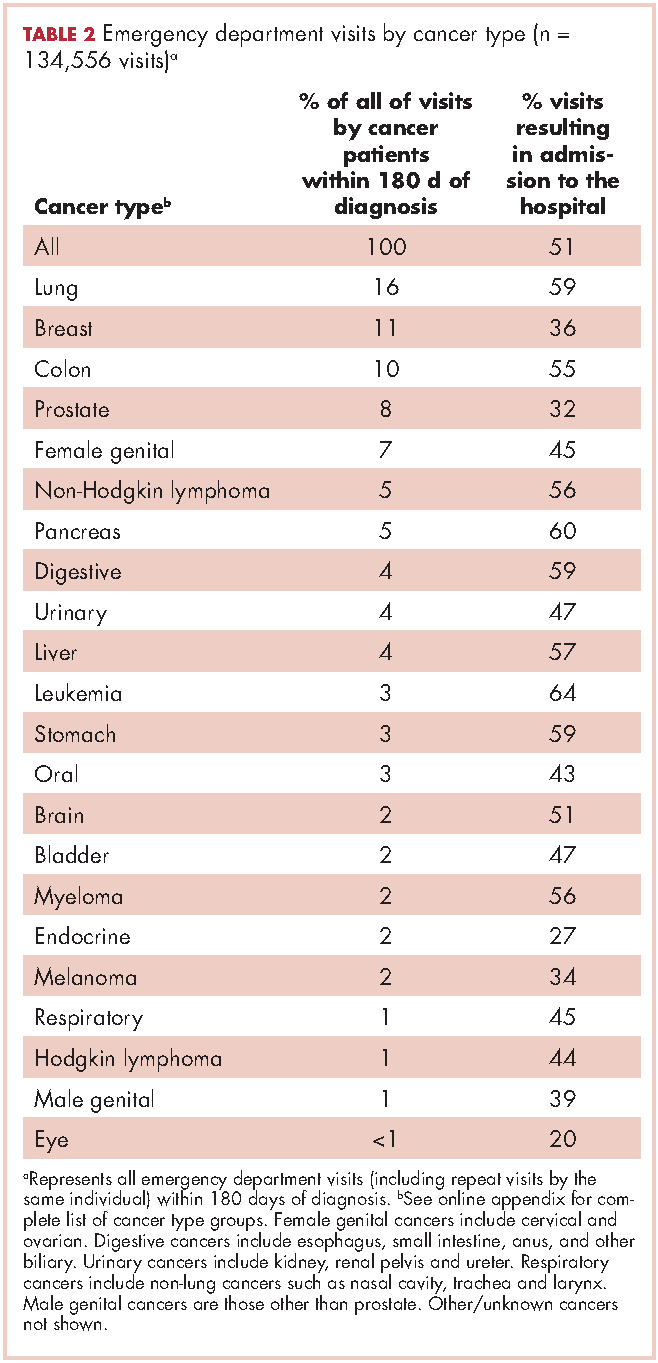
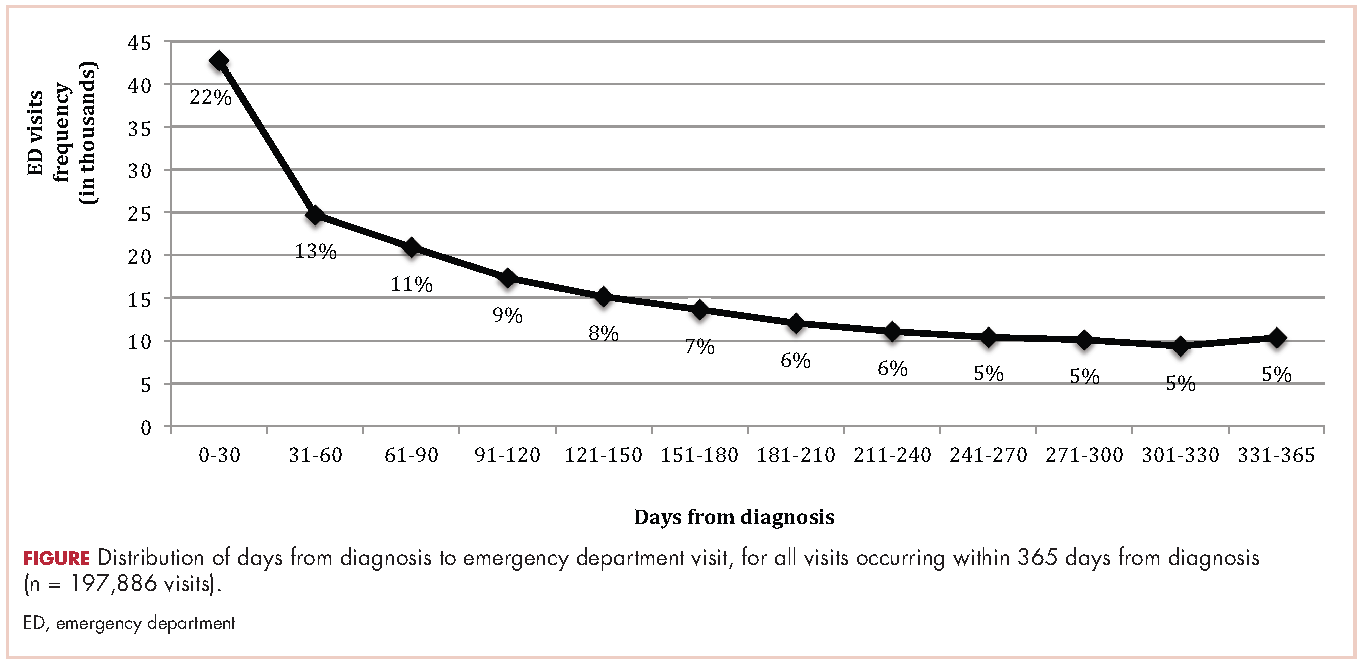
About 51% of visits resulted in admission to the hospital and 45% in discharge (Table 2). For some cancers (lung, colon, non-Hodgkin lymphoma, pancreatic, digestive, liver, stomach, leukemia, and myeloma) most of the visits resulted in admission to the hospital (Table 2). Among visits resulting in admission, the top three principal diagnoses were: septicemia (8%), cardiovascular problems (7%), and complications from surgery (5%) (not shown in tables). Among visits resulting in a discharge home, the three top principal diagnoses were abdominal pain (7%), cardiovascular problems (6%), and urinary, kidney, and bladder complaints other than a urinary tract infection (5%) (results not shown).
Individuals
The cumulative incidence of at least one ED visit was 35% (n = 70,813) within 180 days after diagnosis (Table 3). Visit rates varied by cancer type: individuals with pancreatic (62%), brain (60%), and lung (55%) cancers had the highest cumulative incidences of ED use within 180 days of diagnosis (Table 3). Those with melanoma (14%), prostate (17%), and eye (18%) cancers had the lowest cumulative incidences of ED visits (Table 3).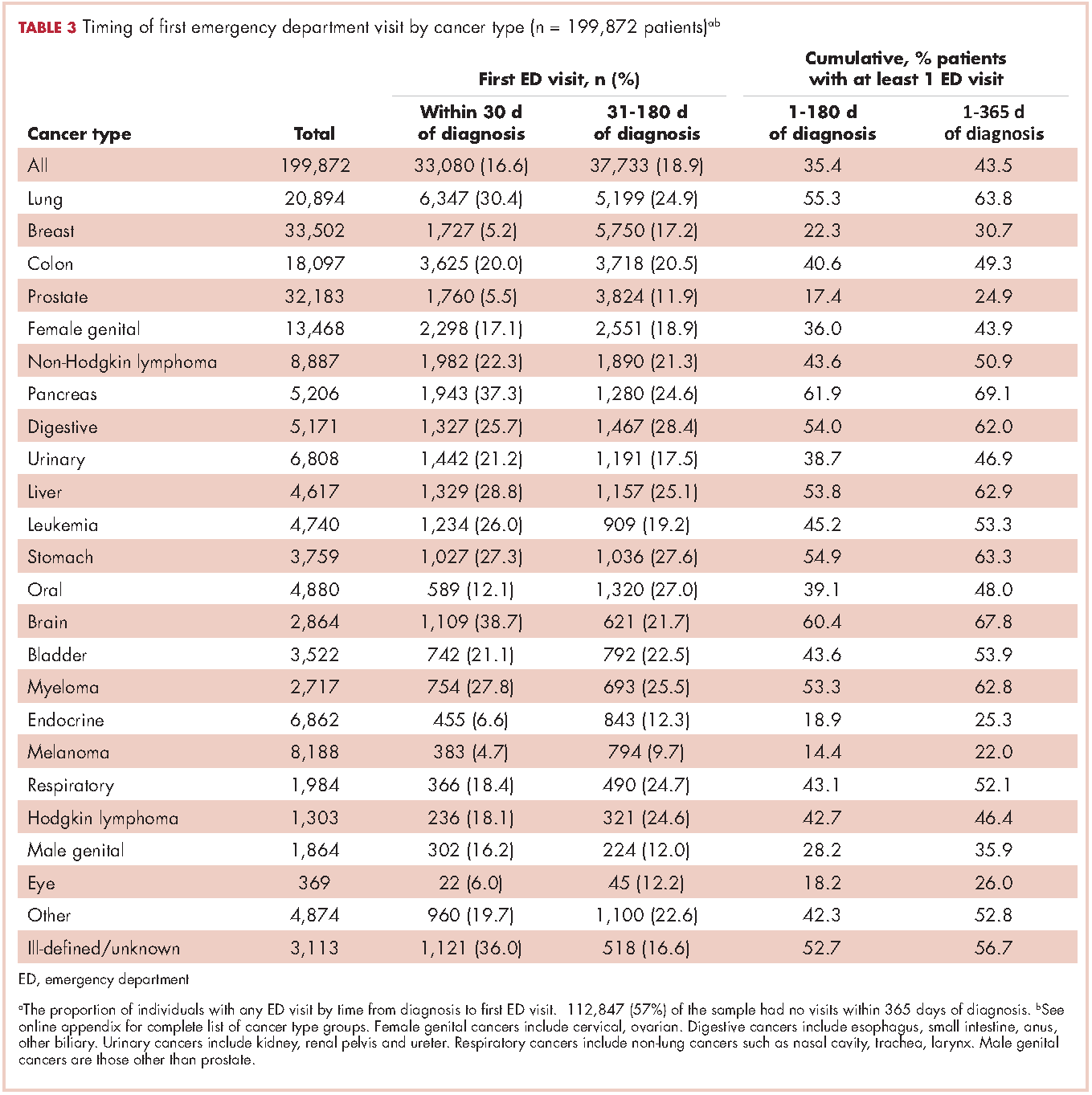
Recycled predictions from logistic regression models, accounting for potential confounding factors, yielded substantively similar results for the cumulative incidence of ED use across cancer types (Table 4). Results did not differ substantially after accounting for survival. Differences in the predicted probability of an ED visits adjusting for death within 180 days of diagnosis were noted to be 2% or greater from estimates reported in Table 4 for only four cancers. Estimates of having any ED visits for those with lung cancer decreased from 46% to 44% (95% CI: 43.0-44.4%), pancreatic cancer from 53% to 49% (95% CI: 48-51%), liver cancer from 51% to 47% (95% CI: 49-53%), and those with eye cancers increased from 20% to 22% (95% CI: 18-26%) (not shown in tables).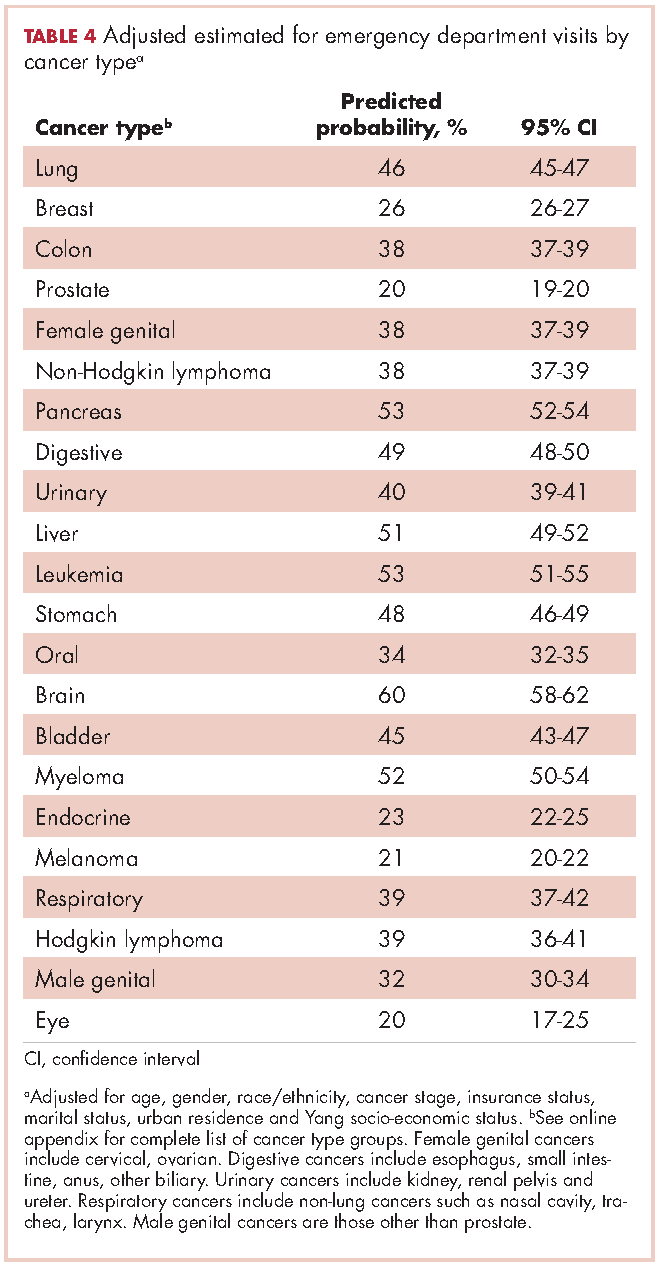
For patients with certain cancers (eg, lung, pancreas, leukemia) the proportion of individuals with an initial ED visit was highest in the first 30 days after diagnosis (Table 3). For individuals with other cancers (eg, breast, prostate, melanoma) the proportion of individuals with an initial ED visit increased by more than 5% during the 31-181–day time period. Those with the remaining cancers had less than 5% change in cumulative ED use between the two time periods.
The number of visits per person ranged from 0-44 during the first 180 days after diagnosis (results not shown in tables). Of all patients diagnosed with cancer, 20% (n = 39,429) had one ED visit, 8% (n = 16,238) had two visits, and 7% (n = 14,760) had three or more visits. Of those patients having at least one ED visit within 180 days of diagnosis, 44% (n = 31,080) had two or more visits and 21% (n = 14,760) had 3 or more visits.
Discussion
This study extends previous research by describing ED use for more than 20 cancer types by time from diagnosis in a large, heterogeneous and population-based sample of recently diagnosed adults in California. We found that 16% of newly diagnosed individuals with cancer used the ED within 30 days of diagnosis, 35% within 6 months of diagnosis, and 44% within 1 year of diagnosis. These findings suggest that ED use by cancer patients is more than double that of the US general population and is higher than previously estimated for cancer patients.10,24 In 2010, about 21% of the US population visited the ED, compared with 44% of cancer patients in the same time period.24 Although persons with greater medical need, such as those with cancer, inevitably require more health services, new approaches are needed to explore the extent to which some of these visits by cancer patients could be prevented by providing care in other settings.
Few studies have examined ED use by cancer patients, but previous findings suggest that 1%-12% of cancer patients use the ED within 30 days of diagnosis, and 15%-25% use the ED within a year of diagnosis.10,25,26 One study did report higher rates of ED use by cancer patients, but attributed the increased use to changes in Medicaid copayments.27 The finding that ED use is higher among cancer patients than previously considered is important for several reasons. First, high rates of ED use may reflect excessive fragmentation in cancer care, or patients’ inability to access providers when acute concerns arise. Furthermore, providers and policymakers may be particularly interested in populations with high ED use because reducing potentially preventable ED use is often cited as one of the goals of care coordination and alternative health care model programs.2,28,29
The number of newly diagnosed cancer patients with multiple ED visits is also substantial. We found 15% of recently diagnosed cancer patients had two or more ED visits within 180 days of diagnosis, compared with 8% of the general US population having two or more ED visits in all of 2010.24 Among cancer patients with at least one ED visit, 44% visited more than once. Repeat visits may represent worsening health status, continued unmet health needs, or new complications that might have been prevented or treated in other health care settings. In addition, there may be opportunities to identify cancer patients at risk for multiple visits at their initial ED visit. A better understanding of the reasons for ED visits and the factors driving unmet need – such as inadequate patient education, limited access to specialty services, or failure to admit a patient to resolve a problem appropriately (eg, pain, infection) – may help to identify which visits are potentially preventable. Ultimately, failure to adequately describe the number of cancer patients that visit the ED and the number of times they visit may result in a lost opportunity for improvement in care, the patient experience and cost reduction in cancer care.
The distinction between cancer types that account for the most ED visits and cancer types with the highest cumulative incidences of ED use is informative. For instance, lung cancer patients accounted for the largest number of ED visits and over half of those with lung cancer visited the ED within 180 days of diagnosis. However, although more than 60% of individuals with pancreatic cancer visited an ED within 180 days of diagnosis; they accounted for only 5% of all ED visits by cancer patients during the same time period. This in part reflects the relative frequency of these cancers. However, prostate cancer, which has a high incidence rate, represents about 8% of all ED visits by cancer patients, yet only 17% of all prostate cancer patients visit the ED within 180 days of diagnosis.
One approach to reduce the absolute number of ED visits by cancer patients would be to target the most frequent users of the ED such as lung, breast, prostate, and colon cancer patients. These cancers are the most common in the general population, so proportionate reduction in ED visits in these groups would have a large overall impact on ED use. Alternatively, patients with cancers that have high rates of ED use could be targeted with interventions to better address their needs. Additional studies of ED use among cancer patients, including understudied cancers, are needed to determine whether care provided in the ED could be provided in alternate clinical settings. Such research can also support training of emergency department staff to manage the full range of cancer-related conditions presenting to the ED.
Another approach to identifying potentially avoidable ED visits is to explore visits that result in admission to the hospital compared with those that result in discharge from the ED. In some circumstances, visits that result in discharge home may not have been true medical emergencies, and therefore might have been preventable. It is also true that even an acute problem requiring admission may have been preventable with timely outpatient management. While we found that 45% of visits ended in discharge home, over half of cancer patients who visit the ED are admitted to the hospital. This is higher than admission rates from the ED for the U.S. population overall (11%-15%),30-32and even higher than the estimated rate of individuals with chronic conditions, such as diabetes (42%), who visit the ED and then are subsequently admitted to the hospital.33
Relatively high rates of admissions may indicate that cancer patients seeking care in the ED require increased medical attention; however, it is possible that other explanations exist. For instance, ED physicians may be uncomfortable with complex cancer cases and may admit patients to be evaluated by a specialist. As such, it is possible that some of these admissions could have been appropriate for outpatient follow up. It is also possible that patients are referred to the ED for admission to the hospital. In these situations the ED visit may be entirely preventable through a direct admission process, although such processes are not available at all institutions and may vary by the admitting provider. For instance, if a hospitalist is overseeing the hospital stay, they may prefer the admission to occur through the ED, whereas a primary care provider or oncologist may be more likely to facilitate a direct admission. Future research could address the extent to which admissions from the ED may be avoidable by examining reasons for and length of admission following an ED visit. While this study found top reasons for admission (principal diagnosis) to be septicemia, cardiovascular complaints and complications from surgery, cumulatively these diagnosis accounted for less than 20% of admissions from the ED. Examining frequent diagnoses by cancer type will also provide insight into potentially avoidable ED use, which may vary by disease course and treatment regimen.
The distribution of days from diagnosis to the first ED visit also varied by cancer type. This variation is likely attributable, at least in part, to differences in condition-specific treatment regimens, severity of illness, and stage at diagnosis. For example, patients with ED visits within 30 days of diagnosis may be those with advanced stage cancers who are at higher risk of complications, or they may be visiting the ED for post-surgical problems. Likewise, individuals who incur visits during later time periods may be undergoing longer treatment regimens. Further research is warranted to explore site-specific predictors of ED use and high-risk periods, accounting for cancer treatment and the timing of treatments.
In summary, ED use among cancer patients is substantial and higher than previously reported. Most ED visits occur within the first 180 days after diagnosis, suggesting focus on the first 30 days after hospital discharge may be misguided. Time frames for ED measurement in future research should be selected with careful attention to cancer-specific periods within which most ED use occurs and the outcomes of interest. Furthermore, better models identifying cancer-specific predictors of ED use, which account for treatment and comorbidities, will facilitate the development of interventions focused on high-risk segments of this population. Research is needed to explore cancer-specific reasons for ED visits and which ED admission diagnoses may be potentially preventable.
Limitations
The limitations of this study include those common to use of administrative and registry data and the CCR and OSHPD data in particular. While CCR data are known to be complete with respect to demographic and cancer information, treatment data is less robust and specific treatment dates are not available.14,34 As a result, we were unable to analyze ED use in relation to receipt of outpatient treatment. As we included all ED visits on or up to a year after the day of diagnosis, it is possible that our analysis includes diagnoses that occurred in conjunction with an ED visits. However, it is unlikely a reporting hospital would report a cancer diagnosis to the CCR without a corresponding hospital admission. Therefore, we assume such cases to be rare.
Lastly, California had lower prevalence of health insurance coverage and higher market penetration by health maintenance organizations, relative to the national average, which may limit the generalizability of the results to other states.35 At the same time, CCR-OSHPD linked data offer the advantage of providing complete data to enumerate ED visits among patients whether they were discharged home or subsequently admitted to hospital.
1. American Cancer Society. Facts & Figures 2017. https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2017.html. Published 2017. Accessed March 16, 2017.
2. Levit L, Balogh E, Nass S, Ganz PA. Delivering high-quality cancer care: charting a new course for a system in crisis. https://www.nap.edu/read/18359/chapter/1. Published 2013. Accessed March 16, 2017.
3. Readmissions Reduction Program (HRRP). CMS website. https://www.cms.gov/medicare/medicare-fee-for-service-payment/acuteinpatientpps/readmissions-reduction-program.html. Updated April 8, 2016. Accessed March 16, 2017.
4. Erikson C, Salsberg E, Forte G, Bruinooge S, Goldstein M. Future supply and demand for oncologists: challenges to assuring access to oncology services. J Clin Oncol. 2007;3(2):79-86.
5. Guadagnolo B, Dohan D, Raich P. Metrics for evaluating patient navigation during cancer diagnosis and treatment: crafting a policy-relevant research agenda for patient navigation in cancer care. Cancer. 2011;117(15 Suppl):3565-3574.
6. Medicare Patient Access to Cancer Treatment Act of 2013, H.R.2869, 113th Cong.(2013). https://www.congress.gov/bill/113th-congress/house-bill/2869/text?format=txt. Introduced July 31, 2013; latest action, referred to the Subcommittee on Health, August 2, 2013. Accessed March 16, 2017.
7. Smulowitz P, Honigman L, Landon B. A novel approach to identifying targets for cost reduction in the emergency department. Ann Emerg Med. 2013;61(3):293-300.
8. Agrawal S, Conway P. Integrating emergency care into a patient- and outcome-centered health care system. Ann Emerg Med. 2013;61(3):301-302.
9. Swenson K, Rose M, Ritz L, Murray CL, Adlis S. Recognition and evaluation of oncology-related symptoms in the emergency department. Ann Emerg Med.1995;26(1):12-17.
10. Lash R, Bell J, Reed S, et al. A systematic review of emergency department use among cancer patients. Cancer Nurs. 2017;40(2):135-144.
11. Sanoff H, Carpenter W, Freburger J, et al. Comparison of adverse events during 5-fluorouracil versus 5-fluorouracil/oxaliplatin adjuvant chemotherapy for stage III colon cancer: a population-based analysis. Cancer. 2012;118(17):4309-4320.
12. Hansen D, Fox J, Gross C, Bruun J. Hospital readmissions and emergency department visits following laparoscopic and open colon resection for cancer. Dis Colon Rectum. 2013;56(9):1053-1061.
13. Office of Statewide Health Planning and Development. Hospital Data Products. http://www.oshpd.ca.gov/HID/DataFlow/HospData.html. Last updated September 6, 2016. Accessed March 16, 2017.
14. California Cancer Registry. Overview. http://www.ccrcal.org/Inside_CCR/About_Us.shtml. Published 2009. Accessed April 15, 2015.
15. Patel M, Ma Y, Mitchell B, Rhoads K. How do differences in treatment impact racial and ethnic disparities in acute myeloid leukemia? Cancer Epidemiol Biomarkers Prev. 2015;24(2):344-349.
16. Parikh-Patel A, White R, Allen M, Cress R. Risk of cancer among rheumatoid arthritis patients in California. Cancer Causes Control. 2009;20(6):1001-1010.
17. National Cancer Institute: Surveillance, Epidemiology and End Results Program. Site recode ICD-O-3/WHO 2008 definition. https://seer.cancer.gov/siterecode/icdo3_dwhoheme/. Published 2008. Accessed March 16, 2017.
18. Washington State Cancer Registry. Cancer Codes Used in Reports. https://fortress. wa.gov/doh/wscr/WSCR/CancerCode.mvc/CancerCode. Data updated, January 2016; report updated, March 2016. Accessed March 16, 2017.
19. National Cancer Institute: Surveillance, Epidemiology and End Results Program. ICD-O-3 SEER Site/Histology Validation List. https://seer.cancer.gov/icd-o-3/. Published 2012, updated September 2015. Accessed March 16, 2017.
20. American Joint Committee on Cancer. What is Cancer Staging? https://cancerstaging.org/references-tools/Pages/What-is-Cancer-Staging.aspx. Published 2010. Accessed March 16, 2017.
21. Yang J S, Harrati A, Clarke C, Keegan T, Gomez S. Cancer Prevention Institute of California. Developing an area based socioeconomic measures from American Community Survey data. http://www.cpic.org/files/PDF/Research_Files/Reports/CPIC_ACS_SES_Index_Documentation_3-102014.pdf. Published March 10, 2014. Accessed March 16, 2017.
22. Basu A, Rathouz P. Estimating marginal and incremental effects on health outcomes using flexible link and variance function models. Biostatistics. 2005;6(1):93-109.
23. Stata Statistical Software [computer program]. Version 13 College Station, TX: StataCorp LP. 2013.
24. National Center for Health Statistics. Health, United States, 2013 – with a special feature on prescription drugs. https://www.cdc.gov/nchs/data/hus/hus13.pdf. Updated May 2014. Accessed March 16, 2017.
25. Goyal R, Wheeler S, Kohler R, et al. Health care utilization from chemotherapy-related adverse events among low-income breast cancer patients: effect of enrollment in a medical home program. N C Med J. 2014;75(4):231-238.
26. Hassett M, O’Malley A, Pakes J, Newhouse J, Earle C. Frequency and cost of chemotherapy-related serious adverse effects in a population sample of women with breast cancer. J Natl Cancer Inst. 2006;98(16):1108-1117.
27. Subramanian S. Impact of Medicaid copayments on patients with cancer: lessons for Medicaid expansion under health reform. Med Care. 2011;49(9):842-847.
28. Coyle Y, Miller A, Paulson R. Model for the cost-efficient delivery of continuous quality cancer care: a hospital and private-practice collaboration. Proc (Bayl Univ Med Cent). 2013;26(2):95-99.
29. Agency for Healthcare Research and Quality. 2011 National Healthcare Disparities Report. https://archive.ahrq.gov/research/findings/nhqrdr/nhdr11/index.html. Last reviewed October 2014. Accessed March 16, 2017.
30. Healthcare Cost and Utilization Project. Introduction to the HCUP Nationwide Emergency Department Sample (NEDS) 2010. https://www.hcup-us.ahrq.gov/db/nation/neds/NEDS_Introduction_2010.jsp. Issued November 2012, updated November 2015. Accessed March 16, 2017.
31. Healthcare Cost and Utilization Project. Introduction to the HCUP Nationwide Emergency Department Sample (NEDS) 2013. https://www.hcup-us.ahrq.gov/db/nation/neds/NEDS_Introduction_2013.jsp. Published November 2015. Accessed March 16, 2017.
32. Weiss AJ, Wier LM, Stocks C, Blanchard J. Overview of emergency department visits in the United States, 2011. Statistical Brief #174. https://www.hcup-us.ahrq.gov/reports/statbriefs/sb174-Emergency-Department-Visits-Overview.pdf. Published June 2014. Accessed March 16, 2017.
33. Washington R, Andrews R, Mutter, R. Emergency department visits for adults with diabetes, 2010. Statistical Brief #167. https://www.hcup-us.ahrq.gov/reports/statbriefs/sb167.jsp. Published November 2013. Accessed March 16, 2017.
34. Penberthy L, Petkov V, McClish D, et al. The value of billing data from oncology practice to supplement treatment information for cancer surveillance. Journal of registry management. 2014;41(2):57-64.
35. National Center for Health Statistics. Health, United States, 2014 – with a special feature on adults aged 55-64. https://www.cdc.gov/nchs/data/hus/hus14.pdf. Published May 2015. Accessed March 16, 2017.
In 2017 there will be nearly 1.7 million new cancer cases diagnosed, and over 600,000 cancer deaths in the Unites States.1 A 2013 Institute of Medicine report highlighted problems with the current quality of cancer care, including high costs and fragmentation of care.2 Other national reports have called for improvements in the overall quality of care and for reducing costly and possibly avoidable use of health services such as emergency department (ED) visits.3-6 Reduction of avoidable ED visits is often cited as a pathway to reduce costs by avoiding unnecessary tests and treatments that occur in the ED and subsequent hospital admissions.7,8
ED crowding, long waits, and unpredictable treatment environments can also make an ED visit an unpleasant experience for the patient. ED visits during cancer treatment can be particularly troubling and present health concerns for patients who are immunocompromised. In particular, cancer patients in the ED have been found to experience delays in the administration of analgesics, antiemetics, or antibiotics.9
Few studies have examined ED use or its associated predictors among cancer patients. Reports to date that have described ED use have focused on different cancers, which makes comparisons across studies difficult.10 Moreover, the time frames of interest and the type of event after which ED use is evaluated (ie, diagnosis or treatment) are inconsistent in the existing literature.10 Some studies quantifying ED use excluded patients admitted to the hospital after an ED visit.11,12 Taken together, these studies do not provide a clear overview of the extent of ED use by cancer patients or the amount of cancer-related care provided by EDs.
Patterns of ED use among cancer patients derived from large and generalizable samples may help inform providers about true risk factors for ED use. In addition, prioritizing new interventions and focusing future research on groups of patients who are at higher risk for preventable ED use could also improve overall care. To address these issues, accurate estimates of ED use among cancer patients are required.
To our knowledge, this is the first study to describe ED use across a range of cancers in a large population-based sample and to consider the timing of ED visits in relation to initial diagnosis. The findings could provide benchmark comparison data to inform future efforts to identify the subset of possibly preventable ED visits and to design interventions to address preventable ED use.
Material and methods
Data source
California’s Office of Statewide Health Planning and Development (OSHPD) manages the patient discharge dataset (PDD) and the emergency department use (EDU) dataset, providing a high-quality source of information on inpatient and ED use in the state.13 A principal diagnosis and up to 24 secondary diagnoses are recorded in OSHPD datasets. The EDU dataset was used to identify treat-and-release ED visits, and the PDD was used to identify hospitalizations initiated in the ED. The California Cancer Registry (CCR) obtains demographic and diagnosis information for every new invasive cancer diagnosed in California, and data collected by the registry are considered to be complete.14 CCR-OSPHD-linked data provide high-quality health care use information for cancer patients in California.15,16 Using an encrypted version of the social security number called the record linkage number (RLN), we linked the CCR records to the corresponding OSHPD files from 2009-2010.
Institutional review board approval for this study was obtained from the University of California, Davis, Human Subjects Committee and the State of California Committee for the Protection of Human Subjects.
Analysis
ED visits. Visits were included if they occurred on or up to 365 days after the date of cancer diagnosis recorded in the CCR. The visits were coded in mutually exclusive groups as occurring within 30, 31-180, and 181-365 days of diagnosis. Subsequently, we flagged each person as having any ED visit (Yes/No) within 180 days and within 365 days of diagnosis, and we tallied the total number of visits occurring within these time frames for each person.
Cancer type. We used relevant site and histology codes to classify cancer type into 24 mutually exclusive categories using the Surveillance Epidemiology and End Result‘s International Classification of Diseases for Oncology, 3rd Edition (ICD-O-3) Recode Definitions17-19 (Suppl Figure 1).
Individual-level variables. Sociodemographic information for each person was collected from the CCR including gender, age, race/ethnicity, marital status, health insurance status, rural residence, survival time in months, neighborhood socio-economic (SES) status based on the Yang index, and the American Joint Committee on Cancer (AJCC) stage.20,21
Data analysis
Demographic information was analyzed for the cohort using descriptive statistics (frequencies, proportions, means, standard deviations, and ranges) and evaluated for correlations. Fewer than 20 observations had missing data and we removed those observations from our analyses on an item-specific basis.
We tabulated ED visits by cancer type and time from diagnosis and then collapsed visit-level data by RLN to determine the number of ED visits for each person in the sample. The number of days from diagnosis to first ED visit was also tabulated. The cohort was stratified by cancer type and cumulative rates of ED visits were tabulated for individuals with ED visits within 0-180 and 0-365 days from diagnosis. To test the robustness of the findings adjusting for confounding factors known to impact ED use, we used logistic regression to model any ED use (Yes/No) as a function of age, gender, race/ethnicity, cancer stage, insurance status, marital status, urban residence, and Yang SES. After model estimation, we used the method of recycled predictions controlling for the confounding variables to compute the marginal probabilities of ED use by cancer type.22 To adjust for the possible impact of survival on ED use, we performed sensitivity analyses and estimated predicted probabilities adjusting for survival. Separate analyses were performed first adjusting for whether the patient died during the course of each month after diagnosis and then adjusting for whether or not the patient died within 180 days of diagnosis. All analyses were conducted using Stata 13.1.23
Results
The CCR identified 222,087 adults with a new primary cancer diagnosis in 2009-2010. After excluding those with Stage 0 cancer (n = 21,154) and nonmelanoma skin cancer (n = 1,031), for whom data are inconsistently collected by CCR, a total of 199,872 individuals were included in the analytic sample. Of those patients (Table 1), most were white non-Hispanic (62%), women (51%), holders of private insurance (53%), married (56%), and urban residents (86%). Most were older than 50 years and had either Stage I or Stage II cancer. The most common cancer types were breast (17%), prostate (16%), lung (11%), and colon (9%; results not shown). In unadjusted comparisons, the incidence of ED use was significantly higher among those who were older, of non-Hispanic black race/ethnicity, uninsured, in the lowest SES group, widowed, or diagnosed with Stage IV cancer (Table 1).
ED visits
Within 365 days after initial cancer diagnosis, 87,025 cancer patients made a total of 197,886 ED visits (not shown in tables). Of those visits, 68% (n = 134,556) occurred within 180 days of diagnosis, with 22% (n = 43,535) occurring within the first 30 days and 46% (n = 91,027) occurring within 31-180 days after diagnosis (Figure). Given that most of the visits occurred within 180 days of diagnosis, we used that time frame in subsequent analyses. Among all ED visits within 180 days of diagnosis (Table 2), the largest proportions of visits were made by those with lung cancer (16%), breast cancer (11%), and colon cancer (10%).
About 51% of visits resulted in admission to the hospital and 45% in discharge (Table 2). For some cancers (lung, colon, non-Hodgkin lymphoma, pancreatic, digestive, liver, stomach, leukemia, and myeloma) most of the visits resulted in admission to the hospital (Table 2). Among visits resulting in admission, the top three principal diagnoses were: septicemia (8%), cardiovascular problems (7%), and complications from surgery (5%) (not shown in tables). Among visits resulting in a discharge home, the three top principal diagnoses were abdominal pain (7%), cardiovascular problems (6%), and urinary, kidney, and bladder complaints other than a urinary tract infection (5%) (results not shown).
Individuals
The cumulative incidence of at least one ED visit was 35% (n = 70,813) within 180 days after diagnosis (Table 3). Visit rates varied by cancer type: individuals with pancreatic (62%), brain (60%), and lung (55%) cancers had the highest cumulative incidences of ED use within 180 days of diagnosis (Table 3). Those with melanoma (14%), prostate (17%), and eye (18%) cancers had the lowest cumulative incidences of ED visits (Table 3).
Recycled predictions from logistic regression models, accounting for potential confounding factors, yielded substantively similar results for the cumulative incidence of ED use across cancer types (Table 4). Results did not differ substantially after accounting for survival. Differences in the predicted probability of an ED visits adjusting for death within 180 days of diagnosis were noted to be 2% or greater from estimates reported in Table 4 for only four cancers. Estimates of having any ED visits for those with lung cancer decreased from 46% to 44% (95% CI: 43.0-44.4%), pancreatic cancer from 53% to 49% (95% CI: 48-51%), liver cancer from 51% to 47% (95% CI: 49-53%), and those with eye cancers increased from 20% to 22% (95% CI: 18-26%) (not shown in tables).
For patients with certain cancers (eg, lung, pancreas, leukemia) the proportion of individuals with an initial ED visit was highest in the first 30 days after diagnosis (Table 3). For individuals with other cancers (eg, breast, prostate, melanoma) the proportion of individuals with an initial ED visit increased by more than 5% during the 31-181–day time period. Those with the remaining cancers had less than 5% change in cumulative ED use between the two time periods.
The number of visits per person ranged from 0-44 during the first 180 days after diagnosis (results not shown in tables). Of all patients diagnosed with cancer, 20% (n = 39,429) had one ED visit, 8% (n = 16,238) had two visits, and 7% (n = 14,760) had three or more visits. Of those patients having at least one ED visit within 180 days of diagnosis, 44% (n = 31,080) had two or more visits and 21% (n = 14,760) had 3 or more visits.
Discussion
This study extends previous research by describing ED use for more than 20 cancer types by time from diagnosis in a large, heterogeneous and population-based sample of recently diagnosed adults in California. We found that 16% of newly diagnosed individuals with cancer used the ED within 30 days of diagnosis, 35% within 6 months of diagnosis, and 44% within 1 year of diagnosis. These findings suggest that ED use by cancer patients is more than double that of the US general population and is higher than previously estimated for cancer patients.10,24 In 2010, about 21% of the US population visited the ED, compared with 44% of cancer patients in the same time period.24 Although persons with greater medical need, such as those with cancer, inevitably require more health services, new approaches are needed to explore the extent to which some of these visits by cancer patients could be prevented by providing care in other settings.
Few studies have examined ED use by cancer patients, but previous findings suggest that 1%-12% of cancer patients use the ED within 30 days of diagnosis, and 15%-25% use the ED within a year of diagnosis.10,25,26 One study did report higher rates of ED use by cancer patients, but attributed the increased use to changes in Medicaid copayments.27 The finding that ED use is higher among cancer patients than previously considered is important for several reasons. First, high rates of ED use may reflect excessive fragmentation in cancer care, or patients’ inability to access providers when acute concerns arise. Furthermore, providers and policymakers may be particularly interested in populations with high ED use because reducing potentially preventable ED use is often cited as one of the goals of care coordination and alternative health care model programs.2,28,29
The number of newly diagnosed cancer patients with multiple ED visits is also substantial. We found 15% of recently diagnosed cancer patients had two or more ED visits within 180 days of diagnosis, compared with 8% of the general US population having two or more ED visits in all of 2010.24 Among cancer patients with at least one ED visit, 44% visited more than once. Repeat visits may represent worsening health status, continued unmet health needs, or new complications that might have been prevented or treated in other health care settings. In addition, there may be opportunities to identify cancer patients at risk for multiple visits at their initial ED visit. A better understanding of the reasons for ED visits and the factors driving unmet need – such as inadequate patient education, limited access to specialty services, or failure to admit a patient to resolve a problem appropriately (eg, pain, infection) – may help to identify which visits are potentially preventable. Ultimately, failure to adequately describe the number of cancer patients that visit the ED and the number of times they visit may result in a lost opportunity for improvement in care, the patient experience and cost reduction in cancer care.
The distinction between cancer types that account for the most ED visits and cancer types with the highest cumulative incidences of ED use is informative. For instance, lung cancer patients accounted for the largest number of ED visits and over half of those with lung cancer visited the ED within 180 days of diagnosis. However, although more than 60% of individuals with pancreatic cancer visited an ED within 180 days of diagnosis; they accounted for only 5% of all ED visits by cancer patients during the same time period. This in part reflects the relative frequency of these cancers. However, prostate cancer, which has a high incidence rate, represents about 8% of all ED visits by cancer patients, yet only 17% of all prostate cancer patients visit the ED within 180 days of diagnosis.
One approach to reduce the absolute number of ED visits by cancer patients would be to target the most frequent users of the ED such as lung, breast, prostate, and colon cancer patients. These cancers are the most common in the general population, so proportionate reduction in ED visits in these groups would have a large overall impact on ED use. Alternatively, patients with cancers that have high rates of ED use could be targeted with interventions to better address their needs. Additional studies of ED use among cancer patients, including understudied cancers, are needed to determine whether care provided in the ED could be provided in alternate clinical settings. Such research can also support training of emergency department staff to manage the full range of cancer-related conditions presenting to the ED.
Another approach to identifying potentially avoidable ED visits is to explore visits that result in admission to the hospital compared with those that result in discharge from the ED. In some circumstances, visits that result in discharge home may not have been true medical emergencies, and therefore might have been preventable. It is also true that even an acute problem requiring admission may have been preventable with timely outpatient management. While we found that 45% of visits ended in discharge home, over half of cancer patients who visit the ED are admitted to the hospital. This is higher than admission rates from the ED for the U.S. population overall (11%-15%),30-32and even higher than the estimated rate of individuals with chronic conditions, such as diabetes (42%), who visit the ED and then are subsequently admitted to the hospital.33
Relatively high rates of admissions may indicate that cancer patients seeking care in the ED require increased medical attention; however, it is possible that other explanations exist. For instance, ED physicians may be uncomfortable with complex cancer cases and may admit patients to be evaluated by a specialist. As such, it is possible that some of these admissions could have been appropriate for outpatient follow up. It is also possible that patients are referred to the ED for admission to the hospital. In these situations the ED visit may be entirely preventable through a direct admission process, although such processes are not available at all institutions and may vary by the admitting provider. For instance, if a hospitalist is overseeing the hospital stay, they may prefer the admission to occur through the ED, whereas a primary care provider or oncologist may be more likely to facilitate a direct admission. Future research could address the extent to which admissions from the ED may be avoidable by examining reasons for and length of admission following an ED visit. While this study found top reasons for admission (principal diagnosis) to be septicemia, cardiovascular complaints and complications from surgery, cumulatively these diagnosis accounted for less than 20% of admissions from the ED. Examining frequent diagnoses by cancer type will also provide insight into potentially avoidable ED use, which may vary by disease course and treatment regimen.
The distribution of days from diagnosis to the first ED visit also varied by cancer type. This variation is likely attributable, at least in part, to differences in condition-specific treatment regimens, severity of illness, and stage at diagnosis. For example, patients with ED visits within 30 days of diagnosis may be those with advanced stage cancers who are at higher risk of complications, or they may be visiting the ED for post-surgical problems. Likewise, individuals who incur visits during later time periods may be undergoing longer treatment regimens. Further research is warranted to explore site-specific predictors of ED use and high-risk periods, accounting for cancer treatment and the timing of treatments.
In summary, ED use among cancer patients is substantial and higher than previously reported. Most ED visits occur within the first 180 days after diagnosis, suggesting focus on the first 30 days after hospital discharge may be misguided. Time frames for ED measurement in future research should be selected with careful attention to cancer-specific periods within which most ED use occurs and the outcomes of interest. Furthermore, better models identifying cancer-specific predictors of ED use, which account for treatment and comorbidities, will facilitate the development of interventions focused on high-risk segments of this population. Research is needed to explore cancer-specific reasons for ED visits and which ED admission diagnoses may be potentially preventable.
Limitations
The limitations of this study include those common to use of administrative and registry data and the CCR and OSHPD data in particular. While CCR data are known to be complete with respect to demographic and cancer information, treatment data is less robust and specific treatment dates are not available.14,34 As a result, we were unable to analyze ED use in relation to receipt of outpatient treatment. As we included all ED visits on or up to a year after the day of diagnosis, it is possible that our analysis includes diagnoses that occurred in conjunction with an ED visits. However, it is unlikely a reporting hospital would report a cancer diagnosis to the CCR without a corresponding hospital admission. Therefore, we assume such cases to be rare.
Lastly, California had lower prevalence of health insurance coverage and higher market penetration by health maintenance organizations, relative to the national average, which may limit the generalizability of the results to other states.35 At the same time, CCR-OSHPD linked data offer the advantage of providing complete data to enumerate ED visits among patients whether they were discharged home or subsequently admitted to hospital.
In 2017 there will be nearly 1.7 million new cancer cases diagnosed, and over 600,000 cancer deaths in the Unites States.1 A 2013 Institute of Medicine report highlighted problems with the current quality of cancer care, including high costs and fragmentation of care.2 Other national reports have called for improvements in the overall quality of care and for reducing costly and possibly avoidable use of health services such as emergency department (ED) visits.3-6 Reduction of avoidable ED visits is often cited as a pathway to reduce costs by avoiding unnecessary tests and treatments that occur in the ED and subsequent hospital admissions.7,8
ED crowding, long waits, and unpredictable treatment environments can also make an ED visit an unpleasant experience for the patient. ED visits during cancer treatment can be particularly troubling and present health concerns for patients who are immunocompromised. In particular, cancer patients in the ED have been found to experience delays in the administration of analgesics, antiemetics, or antibiotics.9
Few studies have examined ED use or its associated predictors among cancer patients. Reports to date that have described ED use have focused on different cancers, which makes comparisons across studies difficult.10 Moreover, the time frames of interest and the type of event after which ED use is evaluated (ie, diagnosis or treatment) are inconsistent in the existing literature.10 Some studies quantifying ED use excluded patients admitted to the hospital after an ED visit.11,12 Taken together, these studies do not provide a clear overview of the extent of ED use by cancer patients or the amount of cancer-related care provided by EDs.
Patterns of ED use among cancer patients derived from large and generalizable samples may help inform providers about true risk factors for ED use. In addition, prioritizing new interventions and focusing future research on groups of patients who are at higher risk for preventable ED use could also improve overall care. To address these issues, accurate estimates of ED use among cancer patients are required.
To our knowledge, this is the first study to describe ED use across a range of cancers in a large population-based sample and to consider the timing of ED visits in relation to initial diagnosis. The findings could provide benchmark comparison data to inform future efforts to identify the subset of possibly preventable ED visits and to design interventions to address preventable ED use.
Material and methods
Data source
California’s Office of Statewide Health Planning and Development (OSHPD) manages the patient discharge dataset (PDD) and the emergency department use (EDU) dataset, providing a high-quality source of information on inpatient and ED use in the state.13 A principal diagnosis and up to 24 secondary diagnoses are recorded in OSHPD datasets. The EDU dataset was used to identify treat-and-release ED visits, and the PDD was used to identify hospitalizations initiated in the ED. The California Cancer Registry (CCR) obtains demographic and diagnosis information for every new invasive cancer diagnosed in California, and data collected by the registry are considered to be complete.14 CCR-OSPHD-linked data provide high-quality health care use information for cancer patients in California.15,16 Using an encrypted version of the social security number called the record linkage number (RLN), we linked the CCR records to the corresponding OSHPD files from 2009-2010.
Institutional review board approval for this study was obtained from the University of California, Davis, Human Subjects Committee and the State of California Committee for the Protection of Human Subjects.
Analysis
ED visits. Visits were included if they occurred on or up to 365 days after the date of cancer diagnosis recorded in the CCR. The visits were coded in mutually exclusive groups as occurring within 30, 31-180, and 181-365 days of diagnosis. Subsequently, we flagged each person as having any ED visit (Yes/No) within 180 days and within 365 days of diagnosis, and we tallied the total number of visits occurring within these time frames for each person.
Cancer type. We used relevant site and histology codes to classify cancer type into 24 mutually exclusive categories using the Surveillance Epidemiology and End Result‘s International Classification of Diseases for Oncology, 3rd Edition (ICD-O-3) Recode Definitions17-19 (Suppl Figure 1).
Individual-level variables. Sociodemographic information for each person was collected from the CCR including gender, age, race/ethnicity, marital status, health insurance status, rural residence, survival time in months, neighborhood socio-economic (SES) status based on the Yang index, and the American Joint Committee on Cancer (AJCC) stage.20,21
Data analysis
Demographic information was analyzed for the cohort using descriptive statistics (frequencies, proportions, means, standard deviations, and ranges) and evaluated for correlations. Fewer than 20 observations had missing data and we removed those observations from our analyses on an item-specific basis.
We tabulated ED visits by cancer type and time from diagnosis and then collapsed visit-level data by RLN to determine the number of ED visits for each person in the sample. The number of days from diagnosis to first ED visit was also tabulated. The cohort was stratified by cancer type and cumulative rates of ED visits were tabulated for individuals with ED visits within 0-180 and 0-365 days from diagnosis. To test the robustness of the findings adjusting for confounding factors known to impact ED use, we used logistic regression to model any ED use (Yes/No) as a function of age, gender, race/ethnicity, cancer stage, insurance status, marital status, urban residence, and Yang SES. After model estimation, we used the method of recycled predictions controlling for the confounding variables to compute the marginal probabilities of ED use by cancer type.22 To adjust for the possible impact of survival on ED use, we performed sensitivity analyses and estimated predicted probabilities adjusting for survival. Separate analyses were performed first adjusting for whether the patient died during the course of each month after diagnosis and then adjusting for whether or not the patient died within 180 days of diagnosis. All analyses were conducted using Stata 13.1.23
Results
The CCR identified 222,087 adults with a new primary cancer diagnosis in 2009-2010. After excluding those with Stage 0 cancer (n = 21,154) and nonmelanoma skin cancer (n = 1,031), for whom data are inconsistently collected by CCR, a total of 199,872 individuals were included in the analytic sample. Of those patients (Table 1), most were white non-Hispanic (62%), women (51%), holders of private insurance (53%), married (56%), and urban residents (86%). Most were older than 50 years and had either Stage I or Stage II cancer. The most common cancer types were breast (17%), prostate (16%), lung (11%), and colon (9%; results not shown). In unadjusted comparisons, the incidence of ED use was significantly higher among those who were older, of non-Hispanic black race/ethnicity, uninsured, in the lowest SES group, widowed, or diagnosed with Stage IV cancer (Table 1).
ED visits
Within 365 days after initial cancer diagnosis, 87,025 cancer patients made a total of 197,886 ED visits (not shown in tables). Of those visits, 68% (n = 134,556) occurred within 180 days of diagnosis, with 22% (n = 43,535) occurring within the first 30 days and 46% (n = 91,027) occurring within 31-180 days after diagnosis (Figure). Given that most of the visits occurred within 180 days of diagnosis, we used that time frame in subsequent analyses. Among all ED visits within 180 days of diagnosis (Table 2), the largest proportions of visits were made by those with lung cancer (16%), breast cancer (11%), and colon cancer (10%).
About 51% of visits resulted in admission to the hospital and 45% in discharge (Table 2). For some cancers (lung, colon, non-Hodgkin lymphoma, pancreatic, digestive, liver, stomach, leukemia, and myeloma) most of the visits resulted in admission to the hospital (Table 2). Among visits resulting in admission, the top three principal diagnoses were: septicemia (8%), cardiovascular problems (7%), and complications from surgery (5%) (not shown in tables). Among visits resulting in a discharge home, the three top principal diagnoses were abdominal pain (7%), cardiovascular problems (6%), and urinary, kidney, and bladder complaints other than a urinary tract infection (5%) (results not shown).
Individuals
The cumulative incidence of at least one ED visit was 35% (n = 70,813) within 180 days after diagnosis (Table 3). Visit rates varied by cancer type: individuals with pancreatic (62%), brain (60%), and lung (55%) cancers had the highest cumulative incidences of ED use within 180 days of diagnosis (Table 3). Those with melanoma (14%), prostate (17%), and eye (18%) cancers had the lowest cumulative incidences of ED visits (Table 3).
Recycled predictions from logistic regression models, accounting for potential confounding factors, yielded substantively similar results for the cumulative incidence of ED use across cancer types (Table 4). Results did not differ substantially after accounting for survival. Differences in the predicted probability of an ED visits adjusting for death within 180 days of diagnosis were noted to be 2% or greater from estimates reported in Table 4 for only four cancers. Estimates of having any ED visits for those with lung cancer decreased from 46% to 44% (95% CI: 43.0-44.4%), pancreatic cancer from 53% to 49% (95% CI: 48-51%), liver cancer from 51% to 47% (95% CI: 49-53%), and those with eye cancers increased from 20% to 22% (95% CI: 18-26%) (not shown in tables).
For patients with certain cancers (eg, lung, pancreas, leukemia) the proportion of individuals with an initial ED visit was highest in the first 30 days after diagnosis (Table 3). For individuals with other cancers (eg, breast, prostate, melanoma) the proportion of individuals with an initial ED visit increased by more than 5% during the 31-181–day time period. Those with the remaining cancers had less than 5% change in cumulative ED use between the two time periods.
The number of visits per person ranged from 0-44 during the first 180 days after diagnosis (results not shown in tables). Of all patients diagnosed with cancer, 20% (n = 39,429) had one ED visit, 8% (n = 16,238) had two visits, and 7% (n = 14,760) had three or more visits. Of those patients having at least one ED visit within 180 days of diagnosis, 44% (n = 31,080) had two or more visits and 21% (n = 14,760) had 3 or more visits.
Discussion
This study extends previous research by describing ED use for more than 20 cancer types by time from diagnosis in a large, heterogeneous and population-based sample of recently diagnosed adults in California. We found that 16% of newly diagnosed individuals with cancer used the ED within 30 days of diagnosis, 35% within 6 months of diagnosis, and 44% within 1 year of diagnosis. These findings suggest that ED use by cancer patients is more than double that of the US general population and is higher than previously estimated for cancer patients.10,24 In 2010, about 21% of the US population visited the ED, compared with 44% of cancer patients in the same time period.24 Although persons with greater medical need, such as those with cancer, inevitably require more health services, new approaches are needed to explore the extent to which some of these visits by cancer patients could be prevented by providing care in other settings.
Few studies have examined ED use by cancer patients, but previous findings suggest that 1%-12% of cancer patients use the ED within 30 days of diagnosis, and 15%-25% use the ED within a year of diagnosis.10,25,26 One study did report higher rates of ED use by cancer patients, but attributed the increased use to changes in Medicaid copayments.27 The finding that ED use is higher among cancer patients than previously considered is important for several reasons. First, high rates of ED use may reflect excessive fragmentation in cancer care, or patients’ inability to access providers when acute concerns arise. Furthermore, providers and policymakers may be particularly interested in populations with high ED use because reducing potentially preventable ED use is often cited as one of the goals of care coordination and alternative health care model programs.2,28,29
The number of newly diagnosed cancer patients with multiple ED visits is also substantial. We found 15% of recently diagnosed cancer patients had two or more ED visits within 180 days of diagnosis, compared with 8% of the general US population having two or more ED visits in all of 2010.24 Among cancer patients with at least one ED visit, 44% visited more than once. Repeat visits may represent worsening health status, continued unmet health needs, or new complications that might have been prevented or treated in other health care settings. In addition, there may be opportunities to identify cancer patients at risk for multiple visits at their initial ED visit. A better understanding of the reasons for ED visits and the factors driving unmet need – such as inadequate patient education, limited access to specialty services, or failure to admit a patient to resolve a problem appropriately (eg, pain, infection) – may help to identify which visits are potentially preventable. Ultimately, failure to adequately describe the number of cancer patients that visit the ED and the number of times they visit may result in a lost opportunity for improvement in care, the patient experience and cost reduction in cancer care.
The distinction between cancer types that account for the most ED visits and cancer types with the highest cumulative incidences of ED use is informative. For instance, lung cancer patients accounted for the largest number of ED visits and over half of those with lung cancer visited the ED within 180 days of diagnosis. However, although more than 60% of individuals with pancreatic cancer visited an ED within 180 days of diagnosis; they accounted for only 5% of all ED visits by cancer patients during the same time period. This in part reflects the relative frequency of these cancers. However, prostate cancer, which has a high incidence rate, represents about 8% of all ED visits by cancer patients, yet only 17% of all prostate cancer patients visit the ED within 180 days of diagnosis.
One approach to reduce the absolute number of ED visits by cancer patients would be to target the most frequent users of the ED such as lung, breast, prostate, and colon cancer patients. These cancers are the most common in the general population, so proportionate reduction in ED visits in these groups would have a large overall impact on ED use. Alternatively, patients with cancers that have high rates of ED use could be targeted with interventions to better address their needs. Additional studies of ED use among cancer patients, including understudied cancers, are needed to determine whether care provided in the ED could be provided in alternate clinical settings. Such research can also support training of emergency department staff to manage the full range of cancer-related conditions presenting to the ED.
Another approach to identifying potentially avoidable ED visits is to explore visits that result in admission to the hospital compared with those that result in discharge from the ED. In some circumstances, visits that result in discharge home may not have been true medical emergencies, and therefore might have been preventable. It is also true that even an acute problem requiring admission may have been preventable with timely outpatient management. While we found that 45% of visits ended in discharge home, over half of cancer patients who visit the ED are admitted to the hospital. This is higher than admission rates from the ED for the U.S. population overall (11%-15%),30-32and even higher than the estimated rate of individuals with chronic conditions, such as diabetes (42%), who visit the ED and then are subsequently admitted to the hospital.33
Relatively high rates of admissions may indicate that cancer patients seeking care in the ED require increased medical attention; however, it is possible that other explanations exist. For instance, ED physicians may be uncomfortable with complex cancer cases and may admit patients to be evaluated by a specialist. As such, it is possible that some of these admissions could have been appropriate for outpatient follow up. It is also possible that patients are referred to the ED for admission to the hospital. In these situations the ED visit may be entirely preventable through a direct admission process, although such processes are not available at all institutions and may vary by the admitting provider. For instance, if a hospitalist is overseeing the hospital stay, they may prefer the admission to occur through the ED, whereas a primary care provider or oncologist may be more likely to facilitate a direct admission. Future research could address the extent to which admissions from the ED may be avoidable by examining reasons for and length of admission following an ED visit. While this study found top reasons for admission (principal diagnosis) to be septicemia, cardiovascular complaints and complications from surgery, cumulatively these diagnosis accounted for less than 20% of admissions from the ED. Examining frequent diagnoses by cancer type will also provide insight into potentially avoidable ED use, which may vary by disease course and treatment regimen.
The distribution of days from diagnosis to the first ED visit also varied by cancer type. This variation is likely attributable, at least in part, to differences in condition-specific treatment regimens, severity of illness, and stage at diagnosis. For example, patients with ED visits within 30 days of diagnosis may be those with advanced stage cancers who are at higher risk of complications, or they may be visiting the ED for post-surgical problems. Likewise, individuals who incur visits during later time periods may be undergoing longer treatment regimens. Further research is warranted to explore site-specific predictors of ED use and high-risk periods, accounting for cancer treatment and the timing of treatments.
In summary, ED use among cancer patients is substantial and higher than previously reported. Most ED visits occur within the first 180 days after diagnosis, suggesting focus on the first 30 days after hospital discharge may be misguided. Time frames for ED measurement in future research should be selected with careful attention to cancer-specific periods within which most ED use occurs and the outcomes of interest. Furthermore, better models identifying cancer-specific predictors of ED use, which account for treatment and comorbidities, will facilitate the development of interventions focused on high-risk segments of this population. Research is needed to explore cancer-specific reasons for ED visits and which ED admission diagnoses may be potentially preventable.
Limitations
The limitations of this study include those common to use of administrative and registry data and the CCR and OSHPD data in particular. While CCR data are known to be complete with respect to demographic and cancer information, treatment data is less robust and specific treatment dates are not available.14,34 As a result, we were unable to analyze ED use in relation to receipt of outpatient treatment. As we included all ED visits on or up to a year after the day of diagnosis, it is possible that our analysis includes diagnoses that occurred in conjunction with an ED visits. However, it is unlikely a reporting hospital would report a cancer diagnosis to the CCR without a corresponding hospital admission. Therefore, we assume such cases to be rare.
Lastly, California had lower prevalence of health insurance coverage and higher market penetration by health maintenance organizations, relative to the national average, which may limit the generalizability of the results to other states.35 At the same time, CCR-OSHPD linked data offer the advantage of providing complete data to enumerate ED visits among patients whether they were discharged home or subsequently admitted to hospital.
1. American Cancer Society. Facts & Figures 2017. https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2017.html. Published 2017. Accessed March 16, 2017.
2. Levit L, Balogh E, Nass S, Ganz PA. Delivering high-quality cancer care: charting a new course for a system in crisis. https://www.nap.edu/read/18359/chapter/1. Published 2013. Accessed March 16, 2017.
3. Readmissions Reduction Program (HRRP). CMS website. https://www.cms.gov/medicare/medicare-fee-for-service-payment/acuteinpatientpps/readmissions-reduction-program.html. Updated April 8, 2016. Accessed March 16, 2017.
4. Erikson C, Salsberg E, Forte G, Bruinooge S, Goldstein M. Future supply and demand for oncologists: challenges to assuring access to oncology services. J Clin Oncol. 2007;3(2):79-86.
5. Guadagnolo B, Dohan D, Raich P. Metrics for evaluating patient navigation during cancer diagnosis and treatment: crafting a policy-relevant research agenda for patient navigation in cancer care. Cancer. 2011;117(15 Suppl):3565-3574.
6. Medicare Patient Access to Cancer Treatment Act of 2013, H.R.2869, 113th Cong.(2013). https://www.congress.gov/bill/113th-congress/house-bill/2869/text?format=txt. Introduced July 31, 2013; latest action, referred to the Subcommittee on Health, August 2, 2013. Accessed March 16, 2017.
7. Smulowitz P, Honigman L, Landon B. A novel approach to identifying targets for cost reduction in the emergency department. Ann Emerg Med. 2013;61(3):293-300.
8. Agrawal S, Conway P. Integrating emergency care into a patient- and outcome-centered health care system. Ann Emerg Med. 2013;61(3):301-302.
9. Swenson K, Rose M, Ritz L, Murray CL, Adlis S. Recognition and evaluation of oncology-related symptoms in the emergency department. Ann Emerg Med.1995;26(1):12-17.
10. Lash R, Bell J, Reed S, et al. A systematic review of emergency department use among cancer patients. Cancer Nurs. 2017;40(2):135-144.
11. Sanoff H, Carpenter W, Freburger J, et al. Comparison of adverse events during 5-fluorouracil versus 5-fluorouracil/oxaliplatin adjuvant chemotherapy for stage III colon cancer: a population-based analysis. Cancer. 2012;118(17):4309-4320.
12. Hansen D, Fox J, Gross C, Bruun J. Hospital readmissions and emergency department visits following laparoscopic and open colon resection for cancer. Dis Colon Rectum. 2013;56(9):1053-1061.
13. Office of Statewide Health Planning and Development. Hospital Data Products. http://www.oshpd.ca.gov/HID/DataFlow/HospData.html. Last updated September 6, 2016. Accessed March 16, 2017.
14. California Cancer Registry. Overview. http://www.ccrcal.org/Inside_CCR/About_Us.shtml. Published 2009. Accessed April 15, 2015.
15. Patel M, Ma Y, Mitchell B, Rhoads K. How do differences in treatment impact racial and ethnic disparities in acute myeloid leukemia? Cancer Epidemiol Biomarkers Prev. 2015;24(2):344-349.
16. Parikh-Patel A, White R, Allen M, Cress R. Risk of cancer among rheumatoid arthritis patients in California. Cancer Causes Control. 2009;20(6):1001-1010.
17. National Cancer Institute: Surveillance, Epidemiology and End Results Program. Site recode ICD-O-3/WHO 2008 definition. https://seer.cancer.gov/siterecode/icdo3_dwhoheme/. Published 2008. Accessed March 16, 2017.
18. Washington State Cancer Registry. Cancer Codes Used in Reports. https://fortress. wa.gov/doh/wscr/WSCR/CancerCode.mvc/CancerCode. Data updated, January 2016; report updated, March 2016. Accessed March 16, 2017.
19. National Cancer Institute: Surveillance, Epidemiology and End Results Program. ICD-O-3 SEER Site/Histology Validation List. https://seer.cancer.gov/icd-o-3/. Published 2012, updated September 2015. Accessed March 16, 2017.
20. American Joint Committee on Cancer. What is Cancer Staging? https://cancerstaging.org/references-tools/Pages/What-is-Cancer-Staging.aspx. Published 2010. Accessed March 16, 2017.
21. Yang J S, Harrati A, Clarke C, Keegan T, Gomez S. Cancer Prevention Institute of California. Developing an area based socioeconomic measures from American Community Survey data. http://www.cpic.org/files/PDF/Research_Files/Reports/CPIC_ACS_SES_Index_Documentation_3-102014.pdf. Published March 10, 2014. Accessed March 16, 2017.
22. Basu A, Rathouz P. Estimating marginal and incremental effects on health outcomes using flexible link and variance function models. Biostatistics. 2005;6(1):93-109.
23. Stata Statistical Software [computer program]. Version 13 College Station, TX: StataCorp LP. 2013.
24. National Center for Health Statistics. Health, United States, 2013 – with a special feature on prescription drugs. https://www.cdc.gov/nchs/data/hus/hus13.pdf. Updated May 2014. Accessed March 16, 2017.
25. Goyal R, Wheeler S, Kohler R, et al. Health care utilization from chemotherapy-related adverse events among low-income breast cancer patients: effect of enrollment in a medical home program. N C Med J. 2014;75(4):231-238.
26. Hassett M, O’Malley A, Pakes J, Newhouse J, Earle C. Frequency and cost of chemotherapy-related serious adverse effects in a population sample of women with breast cancer. J Natl Cancer Inst. 2006;98(16):1108-1117.
27. Subramanian S. Impact of Medicaid copayments on patients with cancer: lessons for Medicaid expansion under health reform. Med Care. 2011;49(9):842-847.
28. Coyle Y, Miller A, Paulson R. Model for the cost-efficient delivery of continuous quality cancer care: a hospital and private-practice collaboration. Proc (Bayl Univ Med Cent). 2013;26(2):95-99.
29. Agency for Healthcare Research and Quality. 2011 National Healthcare Disparities Report. https://archive.ahrq.gov/research/findings/nhqrdr/nhdr11/index.html. Last reviewed October 2014. Accessed March 16, 2017.
30. Healthcare Cost and Utilization Project. Introduction to the HCUP Nationwide Emergency Department Sample (NEDS) 2010. https://www.hcup-us.ahrq.gov/db/nation/neds/NEDS_Introduction_2010.jsp. Issued November 2012, updated November 2015. Accessed March 16, 2017.
31. Healthcare Cost and Utilization Project. Introduction to the HCUP Nationwide Emergency Department Sample (NEDS) 2013. https://www.hcup-us.ahrq.gov/db/nation/neds/NEDS_Introduction_2013.jsp. Published November 2015. Accessed March 16, 2017.
32. Weiss AJ, Wier LM, Stocks C, Blanchard J. Overview of emergency department visits in the United States, 2011. Statistical Brief #174. https://www.hcup-us.ahrq.gov/reports/statbriefs/sb174-Emergency-Department-Visits-Overview.pdf. Published June 2014. Accessed March 16, 2017.
33. Washington R, Andrews R, Mutter, R. Emergency department visits for adults with diabetes, 2010. Statistical Brief #167. https://www.hcup-us.ahrq.gov/reports/statbriefs/sb167.jsp. Published November 2013. Accessed March 16, 2017.
34. Penberthy L, Petkov V, McClish D, et al. The value of billing data from oncology practice to supplement treatment information for cancer surveillance. Journal of registry management. 2014;41(2):57-64.
35. National Center for Health Statistics. Health, United States, 2014 – with a special feature on adults aged 55-64. https://www.cdc.gov/nchs/data/hus/hus14.pdf. Published May 2015. Accessed March 16, 2017.
1. American Cancer Society. Facts & Figures 2017. https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2017.html. Published 2017. Accessed March 16, 2017.
2. Levit L, Balogh E, Nass S, Ganz PA. Delivering high-quality cancer care: charting a new course for a system in crisis. https://www.nap.edu/read/18359/chapter/1. Published 2013. Accessed March 16, 2017.
3. Readmissions Reduction Program (HRRP). CMS website. https://www.cms.gov/medicare/medicare-fee-for-service-payment/acuteinpatientpps/readmissions-reduction-program.html. Updated April 8, 2016. Accessed March 16, 2017.
4. Erikson C, Salsberg E, Forte G, Bruinooge S, Goldstein M. Future supply and demand for oncologists: challenges to assuring access to oncology services. J Clin Oncol. 2007;3(2):79-86.
5. Guadagnolo B, Dohan D, Raich P. Metrics for evaluating patient navigation during cancer diagnosis and treatment: crafting a policy-relevant research agenda for patient navigation in cancer care. Cancer. 2011;117(15 Suppl):3565-3574.
6. Medicare Patient Access to Cancer Treatment Act of 2013, H.R.2869, 113th Cong.(2013). https://www.congress.gov/bill/113th-congress/house-bill/2869/text?format=txt. Introduced July 31, 2013; latest action, referred to the Subcommittee on Health, August 2, 2013. Accessed March 16, 2017.
7. Smulowitz P, Honigman L, Landon B. A novel approach to identifying targets for cost reduction in the emergency department. Ann Emerg Med. 2013;61(3):293-300.
8. Agrawal S, Conway P. Integrating emergency care into a patient- and outcome-centered health care system. Ann Emerg Med. 2013;61(3):301-302.
9. Swenson K, Rose M, Ritz L, Murray CL, Adlis S. Recognition and evaluation of oncology-related symptoms in the emergency department. Ann Emerg Med.1995;26(1):12-17.
10. Lash R, Bell J, Reed S, et al. A systematic review of emergency department use among cancer patients. Cancer Nurs. 2017;40(2):135-144.
11. Sanoff H, Carpenter W, Freburger J, et al. Comparison of adverse events during 5-fluorouracil versus 5-fluorouracil/oxaliplatin adjuvant chemotherapy for stage III colon cancer: a population-based analysis. Cancer. 2012;118(17):4309-4320.
12. Hansen D, Fox J, Gross C, Bruun J. Hospital readmissions and emergency department visits following laparoscopic and open colon resection for cancer. Dis Colon Rectum. 2013;56(9):1053-1061.
13. Office of Statewide Health Planning and Development. Hospital Data Products. http://www.oshpd.ca.gov/HID/DataFlow/HospData.html. Last updated September 6, 2016. Accessed March 16, 2017.
14. California Cancer Registry. Overview. http://www.ccrcal.org/Inside_CCR/About_Us.shtml. Published 2009. Accessed April 15, 2015.
15. Patel M, Ma Y, Mitchell B, Rhoads K. How do differences in treatment impact racial and ethnic disparities in acute myeloid leukemia? Cancer Epidemiol Biomarkers Prev. 2015;24(2):344-349.
16. Parikh-Patel A, White R, Allen M, Cress R. Risk of cancer among rheumatoid arthritis patients in California. Cancer Causes Control. 2009;20(6):1001-1010.
17. National Cancer Institute: Surveillance, Epidemiology and End Results Program. Site recode ICD-O-3/WHO 2008 definition. https://seer.cancer.gov/siterecode/icdo3_dwhoheme/. Published 2008. Accessed March 16, 2017.
18. Washington State Cancer Registry. Cancer Codes Used in Reports. https://fortress. wa.gov/doh/wscr/WSCR/CancerCode.mvc/CancerCode. Data updated, January 2016; report updated, March 2016. Accessed March 16, 2017.
19. National Cancer Institute: Surveillance, Epidemiology and End Results Program. ICD-O-3 SEER Site/Histology Validation List. https://seer.cancer.gov/icd-o-3/. Published 2012, updated September 2015. Accessed March 16, 2017.
20. American Joint Committee on Cancer. What is Cancer Staging? https://cancerstaging.org/references-tools/Pages/What-is-Cancer-Staging.aspx. Published 2010. Accessed March 16, 2017.
21. Yang J S, Harrati A, Clarke C, Keegan T, Gomez S. Cancer Prevention Institute of California. Developing an area based socioeconomic measures from American Community Survey data. http://www.cpic.org/files/PDF/Research_Files/Reports/CPIC_ACS_SES_Index_Documentation_3-102014.pdf. Published March 10, 2014. Accessed March 16, 2017.
22. Basu A, Rathouz P. Estimating marginal and incremental effects on health outcomes using flexible link and variance function models. Biostatistics. 2005;6(1):93-109.
23. Stata Statistical Software [computer program]. Version 13 College Station, TX: StataCorp LP. 2013.
24. National Center for Health Statistics. Health, United States, 2013 – with a special feature on prescription drugs. https://www.cdc.gov/nchs/data/hus/hus13.pdf. Updated May 2014. Accessed March 16, 2017.
25. Goyal R, Wheeler S, Kohler R, et al. Health care utilization from chemotherapy-related adverse events among low-income breast cancer patients: effect of enrollment in a medical home program. N C Med J. 2014;75(4):231-238.
26. Hassett M, O’Malley A, Pakes J, Newhouse J, Earle C. Frequency and cost of chemotherapy-related serious adverse effects in a population sample of women with breast cancer. J Natl Cancer Inst. 2006;98(16):1108-1117.
27. Subramanian S. Impact of Medicaid copayments on patients with cancer: lessons for Medicaid expansion under health reform. Med Care. 2011;49(9):842-847.
28. Coyle Y, Miller A, Paulson R. Model for the cost-efficient delivery of continuous quality cancer care: a hospital and private-practice collaboration. Proc (Bayl Univ Med Cent). 2013;26(2):95-99.
29. Agency for Healthcare Research and Quality. 2011 National Healthcare Disparities Report. https://archive.ahrq.gov/research/findings/nhqrdr/nhdr11/index.html. Last reviewed October 2014. Accessed March 16, 2017.
30. Healthcare Cost and Utilization Project. Introduction to the HCUP Nationwide Emergency Department Sample (NEDS) 2010. https://www.hcup-us.ahrq.gov/db/nation/neds/NEDS_Introduction_2010.jsp. Issued November 2012, updated November 2015. Accessed March 16, 2017.
31. Healthcare Cost and Utilization Project. Introduction to the HCUP Nationwide Emergency Department Sample (NEDS) 2013. https://www.hcup-us.ahrq.gov/db/nation/neds/NEDS_Introduction_2013.jsp. Published November 2015. Accessed March 16, 2017.
32. Weiss AJ, Wier LM, Stocks C, Blanchard J. Overview of emergency department visits in the United States, 2011. Statistical Brief #174. https://www.hcup-us.ahrq.gov/reports/statbriefs/sb174-Emergency-Department-Visits-Overview.pdf. Published June 2014. Accessed March 16, 2017.
33. Washington R, Andrews R, Mutter, R. Emergency department visits for adults with diabetes, 2010. Statistical Brief #167. https://www.hcup-us.ahrq.gov/reports/statbriefs/sb167.jsp. Published November 2013. Accessed March 16, 2017.
34. Penberthy L, Petkov V, McClish D, et al. The value of billing data from oncology practice to supplement treatment information for cancer surveillance. Journal of registry management. 2014;41(2):57-64.
35. National Center for Health Statistics. Health, United States, 2014 – with a special feature on adults aged 55-64. https://www.cdc.gov/nchs/data/hus/hus14.pdf. Published May 2015. Accessed March 16, 2017.
Prevention and treatment options for mTOR inhibitor-associated stomatitis
Mammalian target of rapamycin (mTOR), a serine–threonine protein kinase, operates in the phosphoinositide 3-kinase (PI3K)–protein kinase B (AKT)–mTOR signal transduction pathway regulating both normal and cancer cellular processes, including cell growth, proliferation, motility, survival, and protein and lipid synthesis.1 Genetic alterations affecting this pathway, including mutations in receptor tyrosine kinases PI3K and AKT, occur frequently in human cancers,2 supporting the rationale to develop drugs that target pathway components, such as mTOR inhibitors.
Two mTOR inhibitors are currently approved by the US Food and Drug Administration for cancer treatment: temsirolimus, for advanced renal cell carcinoma (RCC; approved 2007)3 and everolimus, for advanced RCC (approved 2009), advanced pancreatic neuroendocrine tumors (pNET; approved 2011), and hormone receptor-positive (HR-positive), human epidermal growth factor receptor-2 (HER2)-negative advanced breast cancer (approved 2012).4 Another mTOR inhibitor, sirolimus, is approved for use as an immunosuppressive agent and prophylactic against organ rejection after kidney transplant.5
Stomatitis, inflammation of the oral mucosa with contributing factors of genetic predisposition, nutritional deficiencies, infections, and immunological or hematologic dysfunction,6 occurs frequently as a side effect associated with mTOR inhibitor treatment.7-9 Left untreated or managed unsatisfactorily, mTOR inhibitor-associated stomatitis (mIAS) may cause patients discomfort and trouble with maintaining adequate nutritional intake and proper oral hygiene, as well as strict adherence to cancer treatment. It is therefore important for health care providers of cancer patients receiving mTOR inhibitor treatment to be knowledgeable about this side effect. The purpose of the present systematic review of published literature is to provide a better understanding of the differential diagnosis of mIAS, the pathophysiology of mIAS, preventive strategies for patients initiating mTOR inhibitor treatment, and treatment options available to manage mIAS.
Method
The PubMed database was searched with the terms mTOR inhibitor and stomatitis (no date restriction); 79 articles were retrieved, and all abstracts were reviewed to select those relevant to the aims of this review article. To understand future directions for management and prevention of mIAS, a search of clinicaltrials.gov was performed with the terms temsirolimus everolimus stomatitis yielding 12 clinical trials, of which 4 were excluded: 1 trial was terminated due to slow accrual, the status of 1 trial had not been verified in >2 years, and 2 studies focused on efficacy outcomes. A search of the American Society of Clinical Oncology (ASCO) meeting abstracts database was performed to assess the availability of clinical trial data; the search was limited to 2011-2016 and terms were stomatitis in the title and mTOR in the abstract or title. Seven abstracts were retrieved; 2 discussed stomatitis prevention (1 as a “trial-in-progress” and 1 presented results of the trial); the other 5 abstracts presented meta-analyses or reviews of previous clinical studies to assess the risk, incidence, management, and resolution of mIAS.
Review findings
Incidence of mIAS in patients treated for cancer
Two recent meta-analyses quantified the rate of mIAS in patients receiving mTOR inhibitors. Shameem and colleagues10 identified 9 randomized studies of everolimus (8 phase 3, 1 phase 2) and 2 of temsirolimus (1 each phase 2 and 3) involving a total of 4752 patients with a variety of tumor types including angiomyolipoma, breast, gastric, giant cell astrocytoma, pNET, and RCC. Patients received everolimus monotherapy (n = 1,075) or in combination with exemestane (n = 485), tamoxifen (n = 54), letrozole (n = 137), or octreotide (n = 216). Temsirolimus was administered as monotherapy (n = 208) or in combination with interferon
(n = 210) or letrozole (n = 550). The incidence of all-grade stomatitis in the 11 studies ranged from 11%-63%, and the overall incidence of any grade stomatitis was 33.5% (95% confidence interval [CI], 21.9%-47.6%). The concurrent use of a second agent may have confounded these findings because, for example, stomatitis has been reported in pooled analyses and in postmarketing experience with letrozole.11
Rugo and colleagues12 evaluated the incidence of stomatitis in 1455 patients participating in 5 phase 3 randomized clinical trials of everolimus in breast cancer, carcinoid tumor, pNET, and RCC. Patients received everolimus monotherapy
(n = 478) or in combination with exemestane (n = 482), trastuzumab plus vinorelbine (n = 280), or octreotide
(n = 215). The incidence of stomatitis in patients receiving everolimus was 59%-71%, compared with 19%-29% in 1,071 patients of the comparator arms (placebo, and placebo–trastuzumab–vinorelbine). The overall incidence of any grade stomatitis was 67%; most events were mild (grade 1/2); 9% of stomatitis events were moderate to severe (grade 3/4).
Differential clinical presentation of mIAS and severity
Oral mucositis is a common significant adverse event (AE) that occurs in patients with cancer who receive standard chemotherapy regimens and/or radiation therapy,13 so it is important to recognize that the clinical presentation of mIAS differs from that of oral mucositis (Table 1, Figure 14,15).16 mIAS shares some similarities with aphthous ulcers (also referred to as canker sores), a common oral condition with varied causes related to systemic disorders, gastrointestinal disorders, and infections, among others .17 In general, mIAS ulcers develop with a median onset of 10 days (range, 4-25 days) after initiation of mTOR inhibitor treatment and resolve in about 1-3 weeks after dose interruption/reduction of everolimus.16,18,19 mIAS ulcers appear as distinct, oval lesions with a central gray area surrounded by peripheral erythema. They are usually localized to the movable mucosa of the mouth and oropharynx. Although mIAS lesions are usually small, they are quite painful and may cluster.
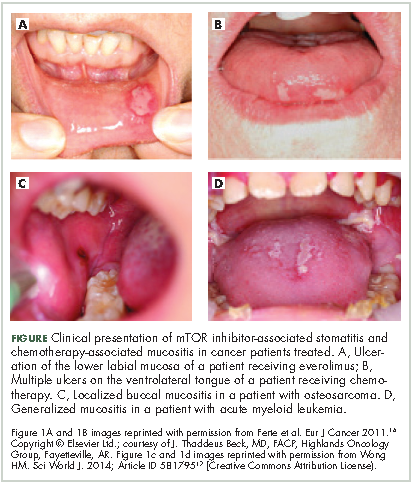
Differential diagnosis of mIAS should be made based on physical examination and medical history, with consideration given to appearance of lesions (number, size, and location), current infection status, and current medications. Specific diagnostic testing should be conducted to confirm a coexisting or alternative cause of oral lesions.17
Although there are many different scales for grading mIAS severity, the most commonly used are the National Cancer Institute Common Terminology Criteria for Adverse Events (based on patient function, symptoms, and intervention needs) and the World Health Organization oral mucositis scales (based on symptoms, clinical presentation, and interference with patient function).20-22 These scales distinguish between mild lesions (grade 1/2) and moderate to severe lesions (grade 3/4) that cause significant pain or interfere with oral intake.
Pathophysiology of mIAS
The pathophysiology mIAS is incompletely understood. The ubiquitous role of the PI3K-AKT-mTOR pathway in regulating broad cellular functions suggests that mTOR inhibition is likely to have wide-ranging effects on many biological processes. It is not known whether disruption of one or more processes – or upsetting the balance of mTOR activities – underlies the formation of mIAS.
Differences between mIAS and oral mucositis, including clinical presentation and concomitant toxicities,16,23 suggest that the two types of oral lesions are fundamentally distinct. This distinction is supported by animal studies in which mTOR inhibition was found to almost completely prevent the appearance of oral mucositis in irradiated mice. The protective effect of mTOR inhibition is mediated through suppression of oxidative stress generated by radiation therapy.24
Although mIAS and recurrent aphthous ulcers share some similarities, it is not clear whether they also share a common pathophysiology. Recent studies suggest that patients with recurrent aphthous ulcers have immune dysfunction that leads to excessive immune response to normally innocuous substrates in the oral mucosa.25 mTOR inhibition can have proinflammatory activity by promoting autophagy, a process that stimulates antigen presentation and activation of T cells that produce proinflammatory cytokines.26 It is interesting to note that the incidence of stomatitis in patients receiving sirolimus after kidney transplant is relatively low, 3%-20%.5 Sirolimus is administered in combination with other immunosuppressants, namely cyclosporine and corticosteroids, so it suggests that concomitant use of a steroid-based regimen may have a preventive or therapeutic effect. However, posttransplant sirolimus is typically administered at relatively low doses, which might account in part for the lower incidence of mIAS observed. Ongoing clinical studies of steroid-based mouthwashes in patients receiving everolimus should shed light on this.
Other study findings have shown that inhibition of the PI3K-AKT-mTOR signaling pathway affects skin wound healing,27,28 which raises the possibility that mIAS may stem from a diminished capacity to repair physical injuries to the oral mucosa. More research is needed to elucidate the pathophysiology of mIAS.
Preventive measures for patients initiating mTOR inhibitor treatment
There are preventive measures for mIAS that have not yet been backed up with evidence-based findings, although several clinical studies that are underway aim to address this gap (Table 2). The hypotheses about the pathophysiology of mIAS suggest that certain preventive and therapeutic interventions might be effective against mIAS. For example, two studies are evaluating the use of steroid-based mouthwashes in patients receiving everolimus, based on the hypothesis that mIAS may arise from an inflammatory process; another study will evaluate a mucoadhesive oral wound rinse, based on the hypothesis that wound protection might prevent mIAS. Glutamine suspension is also under evaluation as it is understood to have wound-preventative and tissue-repair properties, and another study is focused on dentist-guided oral management. Recent results of one of these trials (SWISH),29 reported that preventative care with a dexamethasone mouthwash 3-4 times a day significantly minimized or prevented the incidence of all grades of stomatitis in women receiving everolimus plus exemestane therapy for advanced/metastatic breast cancer compared with the incidence of stomatitis observed in a previously published phase 3 trial (BOLERO-2)30,31 of everolimus plus exemestane in the same patient population. Results from several other studies are expected soon.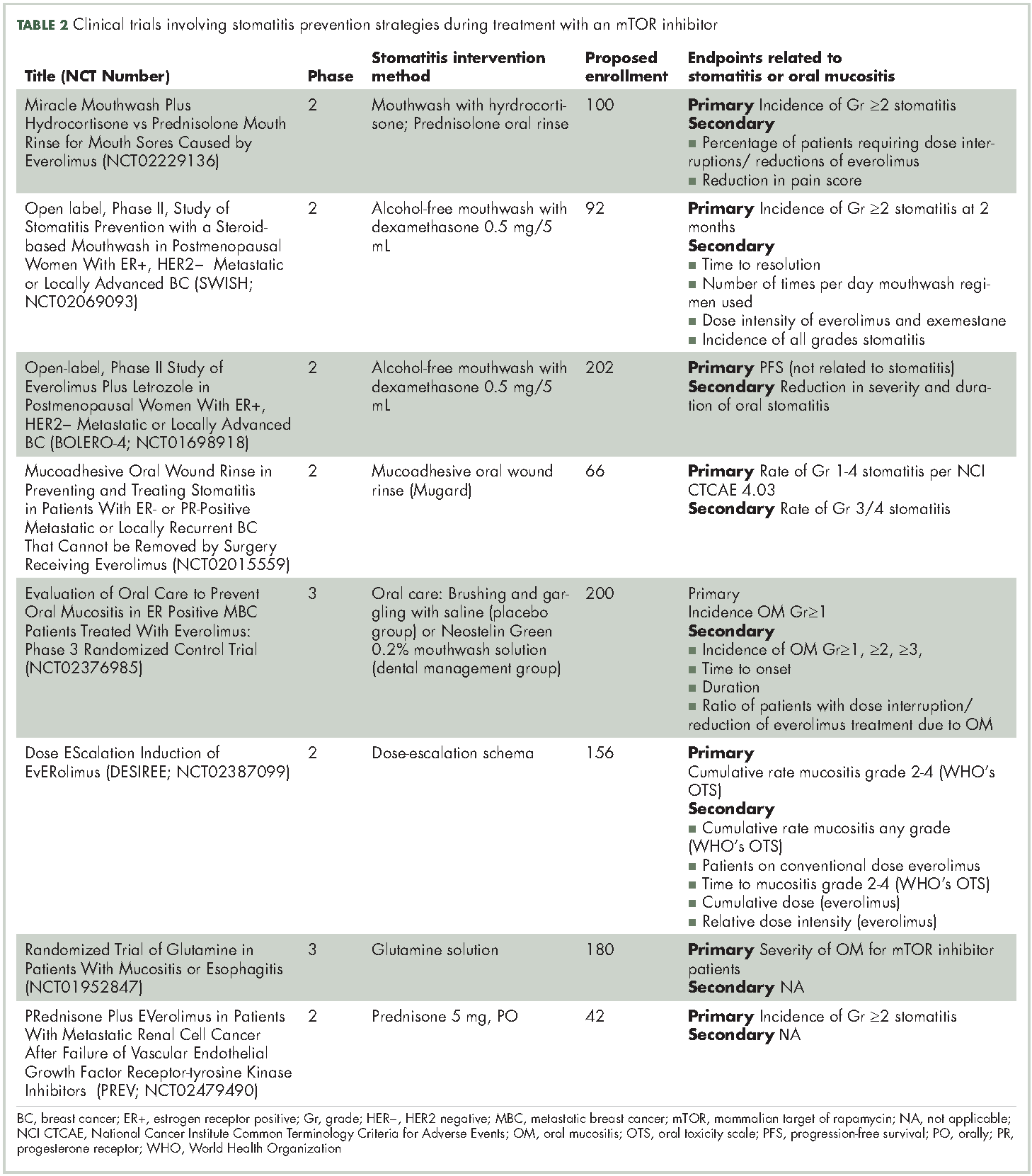
Current approaches to mIAS prevention are based largely on clinical experience with chemotherapy- or radiation-induced oral mucositis (Table 3).13,32 Preventive measures use three main strategies: establish and maintain good routine oral care; modify diet to avoid potentially damaging foods; and improve patient education about mIAS. In regard to patient education, numerous studies have reported that establishing an institutional protocol for oral care helped reduce the incidence of chemotherapy- or radiation-induced oral mucositis.33-40 An ongoing clinical study that will randomize patients to receive oral care education from oral surgeons or instruction on brushing only (NCT02376985) is investigating whether having an oral care protocol holds for patients with mIAS. The hypothesis is that focusing attention on oral care and educating patients to recognize the onset of mIAS facilitates early detection and promotes early intervention.
Therapeutic measures for patients with mIAS
Therapeutic measures for mIAS are based largely on experience with chemotherapy- or radiation-induced oral mucositis or recurrent aphthous ulcers (Table 3) and vary in part by the severity of lesions. Treatments for mild mIAS aim to ameliorate symptoms (eg, topical analgesics for pain), protect the oral mucosa (eg, mucoadhesive gels or viscous solutions that coat the oral cavity), prevent potential sequelae (eg, prophylactic antibiotics to avoid secondary infections), and reduce inflammation/immune response (eg, steroid-based mouth rinses, topical steroids, or topical anti-inflammatory agents). Treatments for mild mIAS are generally local rather than systemic.
Treatment options for moderate to severe mIAS include systemic approaches that generally carry increased risk of AEs and, therefore, should be reserved for patients with multiple lesions, uncontrolled or poorly controlled pain, or greatly diminished oral food intake (Table 3).41 When mIAS cannot be controlled with the interventions described, the dose of the mTOR inhibitor can be reduced with the recognition that dose modification of anticancer therapy may affect disease outcomes.29 The experience of reduction or interruption of treatment with everolimus in the BOLERO-2 trial as a strategy for management of AEs is discussed in a recent review.29 Prescribing information for both temsirolimus and everolimus specify that grade 3 AEs be treated with temporary dose interruption, and with resolution (temsirolimus: grade ≤2; everolimus: grade ≤1), treatment may be resumed at lower doses (temsirolimus: reduce by 5 mg/week; no lower than 15 mg/week; everolimus: reduce by half the previously administered dose).3,4 Grade 4 events due to treatment with temsirolimus may also be treated with dose interruption/reduction; the everolimus prescribing information advises treatment discontinuation for grade 4 stomatitis.
Summary and discussion
mTOR inhibitors can be effective treatments for patients with advanced cancer, specifically for advanced RCC, advanced pNET, and HR+, HER2-negative advanced breast cancer. Although mIAS may occur in many patients, it is usually grade 1 or 2 in severity. mIAS has an early onset, usually within the first 2 weeks of treatment16,19,42 and a relatively rapid resolution, usually within 3 weeks.16,19 Thus, most cases of mIAS are self-limiting.
The relatively recent emergence of mIAS poses short-term challenges regarding diagnosis, assessment, prevention, and treatment. Several clinical studies are underway to evaluate a range of interventions for their preventive and therapeutic efficacy in mIAS. Furthermore, our growing understanding of the underlying pathophysiology of mIAS can guide how mIAS is managed and what interventions patients receive.
Although mIAS is believed to differ from chemotherapy- or radiation-induced oral mucositis and aphthous ulcers, much can be learned from the treatment of both of these. Several strategies have been proposed to limit the occurrence of mIAS (Table 3). First, establish an oral care protocol. Educate patients who are initiating treatment with an mTOR inhibitor on implementation of the oral care protocol and emphasize adherence. Second, educate patients on the symptoms and timing of mIAS. Patients may hesitate to report mild symptoms or assume they are innocuous, so be clear that reporting all symptoms is important to allow timely clinical evaluation. Early recognition of mIAS facilitates early intervention and can prevent dose modification and interruption. Third, implement the preventive and treatment measures described. Many of the preventive measures can be incorporated into an oral care protocol.
The advent of mTOR inhibitors has clinically benefited many patients with cancer. Although side effects, like mIAS, may develop during treatment, they should not be considered insurmountable. Through education, vigilance, and aggressive management, health care providers and patients can work together to help patients maintain their quality of life while continuing to optimally address their disease.
Acknowledgment
The authors thank Anna Lau, PhD, and Patricia Segarini, PhD, of Percolation Communications LLC, for their editorial assistance. Funding for manuscript development was provided by Novartis Pharmaceuticals Corp.
1. Lauring J, Park BH, Wolff AC. The phosphoinositide-3-kinase-Akt-mTOR pathway as a therapeutic target in breast cancer. J Natl Compr Canc Netw. 2013;11:670-678.
2. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140-156.
3. Torisel (temsirolimus) [prescribing information]. Philadelphia, PA: Wyeth Pharmaceuticals; 2014.
4. Afinitor (everolimus) [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2015.
5. Rapamune (sirolimus) [prescribing information]. Philadelphia, PA: Wyeth Pharmaceuticals; 2012.
6. Peterson DE, Boers-Doets CB, Bensadoun RJ, Herrstedt J, ESMO Guidelines Committee. Management of oral and gastrointestinal mucosal injury: ESMO Clinical Practice Guidelines for diagnosis, treatment, and follow-up. Ann Oncol. 2015;26 Suppl 5:v139-151.
7. Hidalgo M, Buckner JC, Erlichman C, et al. A phase I and pharmacokinetic study of temsirolimus (CCI-779) administered intravenously daily for 5 days every 2 weeks to patients with advanced cancer. Clin Cancer Res. 2006;12:5755-5763.
8. Martins F, de Oliveira MA, Wang Q, et al. A review of oral toxicity associated with mTOR inhibitor therapy in cancer patients. Oral Oncol. 2013;49:293-298.
9. O’Donnell A, Faivre S, Burris HA, 3rd, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J Clin Oncol. 2008;26:1588-1595.
10. Shameem R, Lacouture M, Wu S. Incidence and risk of high-grade stomatitis with mTOR inhibitors in cancer patients. Cancer Invest. 2015;33:70-77.
11. Femara (letrozole) [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
12. Rugo HS, Hortobagyi GN, Yao J, et al. Meta-analysis of stomatitis in clinical studies of everolimus: incidence and relationship with efficacy. Ann Oncol. 2016;27:519-525.
13. Keefe DM, Schubert MM, Elting LS, et al. Updated clinical practice guidelines for the prevention and treatment of mucositis. Cancer. 2007;109:820-831.
14. Sonis S, Treister N, Chawla S, Demetri G, Haluska F. Preliminary characterization of oral lesions associated with inhibitors of mammalian target of rapamycin in cancer patients. Cancer. 2010;116:210-215.
15. Scully C. Clinical practice. Aphthous ulceration. N Engl J Med. 2006;355:165-172.
16. Ferte C, Paci A, Zizi M, et al. Natural history, management and pharmacokinetics of everolimus-induced-oral ulcers: insights into compliance issues. Eur J Cancer. 2011;47:2249-2255.
17. Wong HM. Oral complications and management strategies for patients undergoing cancer therapy ScienceWorldJournal. 2014;581795.
18. de Oliveira MA, Martins EMF, Wang Q, et al. Clinical presentation and management of mTOR inhibitor-associated stomatitis. Oral Oncol. 2011;47:998-1003.
19. Rugo HS, Pritchard KI, Gnant M, et al. Incidence and time course of everolimus-related adverse events in postmenopausal women with hormone receptor-positive advanced breast cancer: insights from BOLERO-2. Ann Oncol. 2014;25:808-815.
20. National Cancer Institute. Cancer Therapy Evaluation Program. Common Terminology Criteria for Adverse Events v3.0 (CTCAE). http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed February 13, 2017.
21. National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) v4.03. http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf. Accessed February 13, 2017.
22. World Health Organization. WHO Handbook for Reporting Results of Cancer Treatment. Geneva, Switzerland: World Health Organization (WHO Offset Publication No. 48); 1979.
23. Epstein JB, Thariat J, Bensadoun RJ, et al. Oral complications of cancer and cancer therapy: from cancer treatment to survivorship. CA Cancer J Clin. 2012;62:400-422.
24. Iglesias-Bartolome R, Patel V, Cotrim A, et al. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell. 2012;11:401-414.
25. Lewkowicz N, Lewkowicz P, Dzitko K, et al. Dysfunction of CD4+CD25high T regulatory cells in patients with recurrent aphthous stomatitis. J Oral Pathol Med. 2008;37:454-461.
26. Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767-777.
27. Jin Y, Tymen SD, Chen D, et al. MicroRNA-99 family targets AKT/mTOR signaling pathway in dermal wound healing. PLoS One. 2013;8:e64434.
28. Rosselli-Murai LK, Almeida LO, Zagni C, et al. Periostin responds to mechanical stress and tension by activating the MTOR signaling pathway. PLoS One. 2013;8:e83580.
29. Rugo HS. Dosing and safety implications for oncologists when administering everolimus to patients with hormone receptor-positive breast cancer. Clin Breast Cancer. 2016;16:18-22.
30. Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520-529.
31. Yardley DA, Noguchi S, Pritchard KI, et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther. 2013;30:870-884.
32. Rubenstein EB, Peterson DE, Schubert M, et al. Clinical practice guidelines for the prevention and treatment of cancer therapy-induced oral and gastrointestinal mucositis. Cancer. 2004;100:2026-2046.
33. Borowski B, Benhamou E, Pico JL, Laplanche A, Margainaud JP, Hayat M. Prevention of oral mucositis in patients treated with high-dose chemotherapy and bone marrow transplantation: a randomised controlled trial comparing two protocols of dental care. Eur J Cancer B Oral Oncol. 1994;30B:93-97.
34. Cheng KK, Molassiotis A, Chang AM, Wai WC, Cheung SS. Evaluation of an oral care protocol intervention in the prevention of chemotherapy-induced oral mucositis in paediatric cancer patients. Eur J Cancer. 2001;37:2056-2063.
35. Dudjak LA. Mouth care for mucositis due to radiation therapy. Cancer Nurs. 1987;10:131-140.
36. Graham KM, Pecoraro DA, Ventura M, Meyer CC. Reducing the incidence of stomatitis using a quality assessment and improvement approach. Cancer Nurs. 1993;16:117-122.
37. Kenny SA. Effect of two oral care protocols on the incidence of stomatitis in hematology patients. Cancer Nurs. 1990;13:345-353.
38. Larson PJ, Miaskowski C, MacPhail L, et al. The PRO-SELF Mouth Aware program: an effective approach for reducing chemotherapy-induced mucositis. Cancer Nurs. 1998;21:263-268.
39. Levy-Polack MP, Sebelli P, Polack NL. Incidence of oral complications and application of a preventive protocol in children with acute leukemia. Spec Care Dentist. 1998;18:189-193.
40. Yeager KA, Webster J, Crain M, Kasow J, McGuire DB. Implementation of an oral care standard for leukemia and transplantation patients. Cancer Nurs. 2000;23:40-47; quiz 47-48.
41. Pilotte AP, Hohos MB, Polson KM, Huftalen TM, Treister N. Managing stomatitis in patients treated with mammalian target of rapamycin inhibitors. Clin J Oncol Nurs. 2011;15:E83-89.
42. Gomez-Fernandez C, Garden BC, Wu S, Feldman DR, Lacouture ME. The risk of skin rash and stomatitis with the mammalian target of rapamycin inhibitor temsirolimus: a systematic review of the literature and meta-analysis. Eur J Cancer. 2012;48:340-346.
43. Bonnaure-Mallet M, Bunetel L, Tricot-Doleux S, Guerin J, Bergeron C, LeGall E. Oral complications during treatment of malignant diseases in childhood: effects of tooth brushing. Eur J Cancer. 1998;34:1588-1591.
44. Chuang P, Langone AJ. Clobetasol ameliorates aphthous ulceration in renal transplant patients on sirolimus. Am J Transplant. 2007;7:714-717.
45. Femiano F, Buonaiuto C, Gombos F, Lanza A, Cirillo N. Pilot study on recurrent aphthous stomatitis (RAS): a randomized placebo-controlled trial for the comparative therapeutic effects of systemic prednisone and systemic montelukast in subjects unresponsive to topical therapy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2010;109:402-407.
Mammalian target of rapamycin (mTOR), a serine–threonine protein kinase, operates in the phosphoinositide 3-kinase (PI3K)–protein kinase B (AKT)–mTOR signal transduction pathway regulating both normal and cancer cellular processes, including cell growth, proliferation, motility, survival, and protein and lipid synthesis.1 Genetic alterations affecting this pathway, including mutations in receptor tyrosine kinases PI3K and AKT, occur frequently in human cancers,2 supporting the rationale to develop drugs that target pathway components, such as mTOR inhibitors.
Two mTOR inhibitors are currently approved by the US Food and Drug Administration for cancer treatment: temsirolimus, for advanced renal cell carcinoma (RCC; approved 2007)3 and everolimus, for advanced RCC (approved 2009), advanced pancreatic neuroendocrine tumors (pNET; approved 2011), and hormone receptor-positive (HR-positive), human epidermal growth factor receptor-2 (HER2)-negative advanced breast cancer (approved 2012).4 Another mTOR inhibitor, sirolimus, is approved for use as an immunosuppressive agent and prophylactic against organ rejection after kidney transplant.5
Stomatitis, inflammation of the oral mucosa with contributing factors of genetic predisposition, nutritional deficiencies, infections, and immunological or hematologic dysfunction,6 occurs frequently as a side effect associated with mTOR inhibitor treatment.7-9 Left untreated or managed unsatisfactorily, mTOR inhibitor-associated stomatitis (mIAS) may cause patients discomfort and trouble with maintaining adequate nutritional intake and proper oral hygiene, as well as strict adherence to cancer treatment. It is therefore important for health care providers of cancer patients receiving mTOR inhibitor treatment to be knowledgeable about this side effect. The purpose of the present systematic review of published literature is to provide a better understanding of the differential diagnosis of mIAS, the pathophysiology of mIAS, preventive strategies for patients initiating mTOR inhibitor treatment, and treatment options available to manage mIAS.
Method
The PubMed database was searched with the terms mTOR inhibitor and stomatitis (no date restriction); 79 articles were retrieved, and all abstracts were reviewed to select those relevant to the aims of this review article. To understand future directions for management and prevention of mIAS, a search of clinicaltrials.gov was performed with the terms temsirolimus everolimus stomatitis yielding 12 clinical trials, of which 4 were excluded: 1 trial was terminated due to slow accrual, the status of 1 trial had not been verified in >2 years, and 2 studies focused on efficacy outcomes. A search of the American Society of Clinical Oncology (ASCO) meeting abstracts database was performed to assess the availability of clinical trial data; the search was limited to 2011-2016 and terms were stomatitis in the title and mTOR in the abstract or title. Seven abstracts were retrieved; 2 discussed stomatitis prevention (1 as a “trial-in-progress” and 1 presented results of the trial); the other 5 abstracts presented meta-analyses or reviews of previous clinical studies to assess the risk, incidence, management, and resolution of mIAS.
Review findings
Incidence of mIAS in patients treated for cancer
Two recent meta-analyses quantified the rate of mIAS in patients receiving mTOR inhibitors. Shameem and colleagues10 identified 9 randomized studies of everolimus (8 phase 3, 1 phase 2) and 2 of temsirolimus (1 each phase 2 and 3) involving a total of 4752 patients with a variety of tumor types including angiomyolipoma, breast, gastric, giant cell astrocytoma, pNET, and RCC. Patients received everolimus monotherapy (n = 1,075) or in combination with exemestane (n = 485), tamoxifen (n = 54), letrozole (n = 137), or octreotide (n = 216). Temsirolimus was administered as monotherapy (n = 208) or in combination with interferon
(n = 210) or letrozole (n = 550). The incidence of all-grade stomatitis in the 11 studies ranged from 11%-63%, and the overall incidence of any grade stomatitis was 33.5% (95% confidence interval [CI], 21.9%-47.6%). The concurrent use of a second agent may have confounded these findings because, for example, stomatitis has been reported in pooled analyses and in postmarketing experience with letrozole.11
Rugo and colleagues12 evaluated the incidence of stomatitis in 1455 patients participating in 5 phase 3 randomized clinical trials of everolimus in breast cancer, carcinoid tumor, pNET, and RCC. Patients received everolimus monotherapy
(n = 478) or in combination with exemestane (n = 482), trastuzumab plus vinorelbine (n = 280), or octreotide
(n = 215). The incidence of stomatitis in patients receiving everolimus was 59%-71%, compared with 19%-29% in 1,071 patients of the comparator arms (placebo, and placebo–trastuzumab–vinorelbine). The overall incidence of any grade stomatitis was 67%; most events were mild (grade 1/2); 9% of stomatitis events were moderate to severe (grade 3/4).
Differential clinical presentation of mIAS and severity
Oral mucositis is a common significant adverse event (AE) that occurs in patients with cancer who receive standard chemotherapy regimens and/or radiation therapy,13 so it is important to recognize that the clinical presentation of mIAS differs from that of oral mucositis (Table 1, Figure 14,15).16 mIAS shares some similarities with aphthous ulcers (also referred to as canker sores), a common oral condition with varied causes related to systemic disorders, gastrointestinal disorders, and infections, among others .17 In general, mIAS ulcers develop with a median onset of 10 days (range, 4-25 days) after initiation of mTOR inhibitor treatment and resolve in about 1-3 weeks after dose interruption/reduction of everolimus.16,18,19 mIAS ulcers appear as distinct, oval lesions with a central gray area surrounded by peripheral erythema. They are usually localized to the movable mucosa of the mouth and oropharynx. Although mIAS lesions are usually small, they are quite painful and may cluster.
Differential diagnosis of mIAS should be made based on physical examination and medical history, with consideration given to appearance of lesions (number, size, and location), current infection status, and current medications. Specific diagnostic testing should be conducted to confirm a coexisting or alternative cause of oral lesions.17
Although there are many different scales for grading mIAS severity, the most commonly used are the National Cancer Institute Common Terminology Criteria for Adverse Events (based on patient function, symptoms, and intervention needs) and the World Health Organization oral mucositis scales (based on symptoms, clinical presentation, and interference with patient function).20-22 These scales distinguish between mild lesions (grade 1/2) and moderate to severe lesions (grade 3/4) that cause significant pain or interfere with oral intake.
Pathophysiology of mIAS
The pathophysiology mIAS is incompletely understood. The ubiquitous role of the PI3K-AKT-mTOR pathway in regulating broad cellular functions suggests that mTOR inhibition is likely to have wide-ranging effects on many biological processes. It is not known whether disruption of one or more processes – or upsetting the balance of mTOR activities – underlies the formation of mIAS.
Differences between mIAS and oral mucositis, including clinical presentation and concomitant toxicities,16,23 suggest that the two types of oral lesions are fundamentally distinct. This distinction is supported by animal studies in which mTOR inhibition was found to almost completely prevent the appearance of oral mucositis in irradiated mice. The protective effect of mTOR inhibition is mediated through suppression of oxidative stress generated by radiation therapy.24
Although mIAS and recurrent aphthous ulcers share some similarities, it is not clear whether they also share a common pathophysiology. Recent studies suggest that patients with recurrent aphthous ulcers have immune dysfunction that leads to excessive immune response to normally innocuous substrates in the oral mucosa.25 mTOR inhibition can have proinflammatory activity by promoting autophagy, a process that stimulates antigen presentation and activation of T cells that produce proinflammatory cytokines.26 It is interesting to note that the incidence of stomatitis in patients receiving sirolimus after kidney transplant is relatively low, 3%-20%.5 Sirolimus is administered in combination with other immunosuppressants, namely cyclosporine and corticosteroids, so it suggests that concomitant use of a steroid-based regimen may have a preventive or therapeutic effect. However, posttransplant sirolimus is typically administered at relatively low doses, which might account in part for the lower incidence of mIAS observed. Ongoing clinical studies of steroid-based mouthwashes in patients receiving everolimus should shed light on this.
Other study findings have shown that inhibition of the PI3K-AKT-mTOR signaling pathway affects skin wound healing,27,28 which raises the possibility that mIAS may stem from a diminished capacity to repair physical injuries to the oral mucosa. More research is needed to elucidate the pathophysiology of mIAS.
Preventive measures for patients initiating mTOR inhibitor treatment
There are preventive measures for mIAS that have not yet been backed up with evidence-based findings, although several clinical studies that are underway aim to address this gap (Table 2). The hypotheses about the pathophysiology of mIAS suggest that certain preventive and therapeutic interventions might be effective against mIAS. For example, two studies are evaluating the use of steroid-based mouthwashes in patients receiving everolimus, based on the hypothesis that mIAS may arise from an inflammatory process; another study will evaluate a mucoadhesive oral wound rinse, based on the hypothesis that wound protection might prevent mIAS. Glutamine suspension is also under evaluation as it is understood to have wound-preventative and tissue-repair properties, and another study is focused on dentist-guided oral management. Recent results of one of these trials (SWISH),29 reported that preventative care with a dexamethasone mouthwash 3-4 times a day significantly minimized or prevented the incidence of all grades of stomatitis in women receiving everolimus plus exemestane therapy for advanced/metastatic breast cancer compared with the incidence of stomatitis observed in a previously published phase 3 trial (BOLERO-2)30,31 of everolimus plus exemestane in the same patient population. Results from several other studies are expected soon.
Current approaches to mIAS prevention are based largely on clinical experience with chemotherapy- or radiation-induced oral mucositis (Table 3).13,32 Preventive measures use three main strategies: establish and maintain good routine oral care; modify diet to avoid potentially damaging foods; and improve patient education about mIAS. In regard to patient education, numerous studies have reported that establishing an institutional protocol for oral care helped reduce the incidence of chemotherapy- or radiation-induced oral mucositis.33-40 An ongoing clinical study that will randomize patients to receive oral care education from oral surgeons or instruction on brushing only (NCT02376985) is investigating whether having an oral care protocol holds for patients with mIAS. The hypothesis is that focusing attention on oral care and educating patients to recognize the onset of mIAS facilitates early detection and promotes early intervention.
Therapeutic measures for patients with mIAS
Therapeutic measures for mIAS are based largely on experience with chemotherapy- or radiation-induced oral mucositis or recurrent aphthous ulcers (Table 3) and vary in part by the severity of lesions. Treatments for mild mIAS aim to ameliorate symptoms (eg, topical analgesics for pain), protect the oral mucosa (eg, mucoadhesive gels or viscous solutions that coat the oral cavity), prevent potential sequelae (eg, prophylactic antibiotics to avoid secondary infections), and reduce inflammation/immune response (eg, steroid-based mouth rinses, topical steroids, or topical anti-inflammatory agents). Treatments for mild mIAS are generally local rather than systemic.
Treatment options for moderate to severe mIAS include systemic approaches that generally carry increased risk of AEs and, therefore, should be reserved for patients with multiple lesions, uncontrolled or poorly controlled pain, or greatly diminished oral food intake (Table 3).41 When mIAS cannot be controlled with the interventions described, the dose of the mTOR inhibitor can be reduced with the recognition that dose modification of anticancer therapy may affect disease outcomes.29 The experience of reduction or interruption of treatment with everolimus in the BOLERO-2 trial as a strategy for management of AEs is discussed in a recent review.29 Prescribing information for both temsirolimus and everolimus specify that grade 3 AEs be treated with temporary dose interruption, and with resolution (temsirolimus: grade ≤2; everolimus: grade ≤1), treatment may be resumed at lower doses (temsirolimus: reduce by 5 mg/week; no lower than 15 mg/week; everolimus: reduce by half the previously administered dose).3,4 Grade 4 events due to treatment with temsirolimus may also be treated with dose interruption/reduction; the everolimus prescribing information advises treatment discontinuation for grade 4 stomatitis.
Summary and discussion
mTOR inhibitors can be effective treatments for patients with advanced cancer, specifically for advanced RCC, advanced pNET, and HR+, HER2-negative advanced breast cancer. Although mIAS may occur in many patients, it is usually grade 1 or 2 in severity. mIAS has an early onset, usually within the first 2 weeks of treatment16,19,42 and a relatively rapid resolution, usually within 3 weeks.16,19 Thus, most cases of mIAS are self-limiting.
The relatively recent emergence of mIAS poses short-term challenges regarding diagnosis, assessment, prevention, and treatment. Several clinical studies are underway to evaluate a range of interventions for their preventive and therapeutic efficacy in mIAS. Furthermore, our growing understanding of the underlying pathophysiology of mIAS can guide how mIAS is managed and what interventions patients receive.
Although mIAS is believed to differ from chemotherapy- or radiation-induced oral mucositis and aphthous ulcers, much can be learned from the treatment of both of these. Several strategies have been proposed to limit the occurrence of mIAS (Table 3). First, establish an oral care protocol. Educate patients who are initiating treatment with an mTOR inhibitor on implementation of the oral care protocol and emphasize adherence. Second, educate patients on the symptoms and timing of mIAS. Patients may hesitate to report mild symptoms or assume they are innocuous, so be clear that reporting all symptoms is important to allow timely clinical evaluation. Early recognition of mIAS facilitates early intervention and can prevent dose modification and interruption. Third, implement the preventive and treatment measures described. Many of the preventive measures can be incorporated into an oral care protocol.
The advent of mTOR inhibitors has clinically benefited many patients with cancer. Although side effects, like mIAS, may develop during treatment, they should not be considered insurmountable. Through education, vigilance, and aggressive management, health care providers and patients can work together to help patients maintain their quality of life while continuing to optimally address their disease.
Acknowledgment
The authors thank Anna Lau, PhD, and Patricia Segarini, PhD, of Percolation Communications LLC, for their editorial assistance. Funding for manuscript development was provided by Novartis Pharmaceuticals Corp.
Mammalian target of rapamycin (mTOR), a serine–threonine protein kinase, operates in the phosphoinositide 3-kinase (PI3K)–protein kinase B (AKT)–mTOR signal transduction pathway regulating both normal and cancer cellular processes, including cell growth, proliferation, motility, survival, and protein and lipid synthesis.1 Genetic alterations affecting this pathway, including mutations in receptor tyrosine kinases PI3K and AKT, occur frequently in human cancers,2 supporting the rationale to develop drugs that target pathway components, such as mTOR inhibitors.
Two mTOR inhibitors are currently approved by the US Food and Drug Administration for cancer treatment: temsirolimus, for advanced renal cell carcinoma (RCC; approved 2007)3 and everolimus, for advanced RCC (approved 2009), advanced pancreatic neuroendocrine tumors (pNET; approved 2011), and hormone receptor-positive (HR-positive), human epidermal growth factor receptor-2 (HER2)-negative advanced breast cancer (approved 2012).4 Another mTOR inhibitor, sirolimus, is approved for use as an immunosuppressive agent and prophylactic against organ rejection after kidney transplant.5
Stomatitis, inflammation of the oral mucosa with contributing factors of genetic predisposition, nutritional deficiencies, infections, and immunological or hematologic dysfunction,6 occurs frequently as a side effect associated with mTOR inhibitor treatment.7-9 Left untreated or managed unsatisfactorily, mTOR inhibitor-associated stomatitis (mIAS) may cause patients discomfort and trouble with maintaining adequate nutritional intake and proper oral hygiene, as well as strict adherence to cancer treatment. It is therefore important for health care providers of cancer patients receiving mTOR inhibitor treatment to be knowledgeable about this side effect. The purpose of the present systematic review of published literature is to provide a better understanding of the differential diagnosis of mIAS, the pathophysiology of mIAS, preventive strategies for patients initiating mTOR inhibitor treatment, and treatment options available to manage mIAS.
Method
The PubMed database was searched with the terms mTOR inhibitor and stomatitis (no date restriction); 79 articles were retrieved, and all abstracts were reviewed to select those relevant to the aims of this review article. To understand future directions for management and prevention of mIAS, a search of clinicaltrials.gov was performed with the terms temsirolimus everolimus stomatitis yielding 12 clinical trials, of which 4 were excluded: 1 trial was terminated due to slow accrual, the status of 1 trial had not been verified in >2 years, and 2 studies focused on efficacy outcomes. A search of the American Society of Clinical Oncology (ASCO) meeting abstracts database was performed to assess the availability of clinical trial data; the search was limited to 2011-2016 and terms were stomatitis in the title and mTOR in the abstract or title. Seven abstracts were retrieved; 2 discussed stomatitis prevention (1 as a “trial-in-progress” and 1 presented results of the trial); the other 5 abstracts presented meta-analyses or reviews of previous clinical studies to assess the risk, incidence, management, and resolution of mIAS.
Review findings
Incidence of mIAS in patients treated for cancer
Two recent meta-analyses quantified the rate of mIAS in patients receiving mTOR inhibitors. Shameem and colleagues10 identified 9 randomized studies of everolimus (8 phase 3, 1 phase 2) and 2 of temsirolimus (1 each phase 2 and 3) involving a total of 4752 patients with a variety of tumor types including angiomyolipoma, breast, gastric, giant cell astrocytoma, pNET, and RCC. Patients received everolimus monotherapy (n = 1,075) or in combination with exemestane (n = 485), tamoxifen (n = 54), letrozole (n = 137), or octreotide (n = 216). Temsirolimus was administered as monotherapy (n = 208) or in combination with interferon
(n = 210) or letrozole (n = 550). The incidence of all-grade stomatitis in the 11 studies ranged from 11%-63%, and the overall incidence of any grade stomatitis was 33.5% (95% confidence interval [CI], 21.9%-47.6%). The concurrent use of a second agent may have confounded these findings because, for example, stomatitis has been reported in pooled analyses and in postmarketing experience with letrozole.11
Rugo and colleagues12 evaluated the incidence of stomatitis in 1455 patients participating in 5 phase 3 randomized clinical trials of everolimus in breast cancer, carcinoid tumor, pNET, and RCC. Patients received everolimus monotherapy
(n = 478) or in combination with exemestane (n = 482), trastuzumab plus vinorelbine (n = 280), or octreotide
(n = 215). The incidence of stomatitis in patients receiving everolimus was 59%-71%, compared with 19%-29% in 1,071 patients of the comparator arms (placebo, and placebo–trastuzumab–vinorelbine). The overall incidence of any grade stomatitis was 67%; most events were mild (grade 1/2); 9% of stomatitis events were moderate to severe (grade 3/4).
Differential clinical presentation of mIAS and severity
Oral mucositis is a common significant adverse event (AE) that occurs in patients with cancer who receive standard chemotherapy regimens and/or radiation therapy,13 so it is important to recognize that the clinical presentation of mIAS differs from that of oral mucositis (Table 1, Figure 14,15).16 mIAS shares some similarities with aphthous ulcers (also referred to as canker sores), a common oral condition with varied causes related to systemic disorders, gastrointestinal disorders, and infections, among others .17 In general, mIAS ulcers develop with a median onset of 10 days (range, 4-25 days) after initiation of mTOR inhibitor treatment and resolve in about 1-3 weeks after dose interruption/reduction of everolimus.16,18,19 mIAS ulcers appear as distinct, oval lesions with a central gray area surrounded by peripheral erythema. They are usually localized to the movable mucosa of the mouth and oropharynx. Although mIAS lesions are usually small, they are quite painful and may cluster.
Differential diagnosis of mIAS should be made based on physical examination and medical history, with consideration given to appearance of lesions (number, size, and location), current infection status, and current medications. Specific diagnostic testing should be conducted to confirm a coexisting or alternative cause of oral lesions.17
Although there are many different scales for grading mIAS severity, the most commonly used are the National Cancer Institute Common Terminology Criteria for Adverse Events (based on patient function, symptoms, and intervention needs) and the World Health Organization oral mucositis scales (based on symptoms, clinical presentation, and interference with patient function).20-22 These scales distinguish between mild lesions (grade 1/2) and moderate to severe lesions (grade 3/4) that cause significant pain or interfere with oral intake.
Pathophysiology of mIAS
The pathophysiology mIAS is incompletely understood. The ubiquitous role of the PI3K-AKT-mTOR pathway in regulating broad cellular functions suggests that mTOR inhibition is likely to have wide-ranging effects on many biological processes. It is not known whether disruption of one or more processes – or upsetting the balance of mTOR activities – underlies the formation of mIAS.
Differences between mIAS and oral mucositis, including clinical presentation and concomitant toxicities,16,23 suggest that the two types of oral lesions are fundamentally distinct. This distinction is supported by animal studies in which mTOR inhibition was found to almost completely prevent the appearance of oral mucositis in irradiated mice. The protective effect of mTOR inhibition is mediated through suppression of oxidative stress generated by radiation therapy.24
Although mIAS and recurrent aphthous ulcers share some similarities, it is not clear whether they also share a common pathophysiology. Recent studies suggest that patients with recurrent aphthous ulcers have immune dysfunction that leads to excessive immune response to normally innocuous substrates in the oral mucosa.25 mTOR inhibition can have proinflammatory activity by promoting autophagy, a process that stimulates antigen presentation and activation of T cells that produce proinflammatory cytokines.26 It is interesting to note that the incidence of stomatitis in patients receiving sirolimus after kidney transplant is relatively low, 3%-20%.5 Sirolimus is administered in combination with other immunosuppressants, namely cyclosporine and corticosteroids, so it suggests that concomitant use of a steroid-based regimen may have a preventive or therapeutic effect. However, posttransplant sirolimus is typically administered at relatively low doses, which might account in part for the lower incidence of mIAS observed. Ongoing clinical studies of steroid-based mouthwashes in patients receiving everolimus should shed light on this.
Other study findings have shown that inhibition of the PI3K-AKT-mTOR signaling pathway affects skin wound healing,27,28 which raises the possibility that mIAS may stem from a diminished capacity to repair physical injuries to the oral mucosa. More research is needed to elucidate the pathophysiology of mIAS.
Preventive measures for patients initiating mTOR inhibitor treatment
There are preventive measures for mIAS that have not yet been backed up with evidence-based findings, although several clinical studies that are underway aim to address this gap (Table 2). The hypotheses about the pathophysiology of mIAS suggest that certain preventive and therapeutic interventions might be effective against mIAS. For example, two studies are evaluating the use of steroid-based mouthwashes in patients receiving everolimus, based on the hypothesis that mIAS may arise from an inflammatory process; another study will evaluate a mucoadhesive oral wound rinse, based on the hypothesis that wound protection might prevent mIAS. Glutamine suspension is also under evaluation as it is understood to have wound-preventative and tissue-repair properties, and another study is focused on dentist-guided oral management. Recent results of one of these trials (SWISH),29 reported that preventative care with a dexamethasone mouthwash 3-4 times a day significantly minimized or prevented the incidence of all grades of stomatitis in women receiving everolimus plus exemestane therapy for advanced/metastatic breast cancer compared with the incidence of stomatitis observed in a previously published phase 3 trial (BOLERO-2)30,31 of everolimus plus exemestane in the same patient population. Results from several other studies are expected soon.
Current approaches to mIAS prevention are based largely on clinical experience with chemotherapy- or radiation-induced oral mucositis (Table 3).13,32 Preventive measures use three main strategies: establish and maintain good routine oral care; modify diet to avoid potentially damaging foods; and improve patient education about mIAS. In regard to patient education, numerous studies have reported that establishing an institutional protocol for oral care helped reduce the incidence of chemotherapy- or radiation-induced oral mucositis.33-40 An ongoing clinical study that will randomize patients to receive oral care education from oral surgeons or instruction on brushing only (NCT02376985) is investigating whether having an oral care protocol holds for patients with mIAS. The hypothesis is that focusing attention on oral care and educating patients to recognize the onset of mIAS facilitates early detection and promotes early intervention.
Therapeutic measures for patients with mIAS
Therapeutic measures for mIAS are based largely on experience with chemotherapy- or radiation-induced oral mucositis or recurrent aphthous ulcers (Table 3) and vary in part by the severity of lesions. Treatments for mild mIAS aim to ameliorate symptoms (eg, topical analgesics for pain), protect the oral mucosa (eg, mucoadhesive gels or viscous solutions that coat the oral cavity), prevent potential sequelae (eg, prophylactic antibiotics to avoid secondary infections), and reduce inflammation/immune response (eg, steroid-based mouth rinses, topical steroids, or topical anti-inflammatory agents). Treatments for mild mIAS are generally local rather than systemic.
Treatment options for moderate to severe mIAS include systemic approaches that generally carry increased risk of AEs and, therefore, should be reserved for patients with multiple lesions, uncontrolled or poorly controlled pain, or greatly diminished oral food intake (Table 3).41 When mIAS cannot be controlled with the interventions described, the dose of the mTOR inhibitor can be reduced with the recognition that dose modification of anticancer therapy may affect disease outcomes.29 The experience of reduction or interruption of treatment with everolimus in the BOLERO-2 trial as a strategy for management of AEs is discussed in a recent review.29 Prescribing information for both temsirolimus and everolimus specify that grade 3 AEs be treated with temporary dose interruption, and with resolution (temsirolimus: grade ≤2; everolimus: grade ≤1), treatment may be resumed at lower doses (temsirolimus: reduce by 5 mg/week; no lower than 15 mg/week; everolimus: reduce by half the previously administered dose).3,4 Grade 4 events due to treatment with temsirolimus may also be treated with dose interruption/reduction; the everolimus prescribing information advises treatment discontinuation for grade 4 stomatitis.
Summary and discussion
mTOR inhibitors can be effective treatments for patients with advanced cancer, specifically for advanced RCC, advanced pNET, and HR+, HER2-negative advanced breast cancer. Although mIAS may occur in many patients, it is usually grade 1 or 2 in severity. mIAS has an early onset, usually within the first 2 weeks of treatment16,19,42 and a relatively rapid resolution, usually within 3 weeks.16,19 Thus, most cases of mIAS are self-limiting.
The relatively recent emergence of mIAS poses short-term challenges regarding diagnosis, assessment, prevention, and treatment. Several clinical studies are underway to evaluate a range of interventions for their preventive and therapeutic efficacy in mIAS. Furthermore, our growing understanding of the underlying pathophysiology of mIAS can guide how mIAS is managed and what interventions patients receive.
Although mIAS is believed to differ from chemotherapy- or radiation-induced oral mucositis and aphthous ulcers, much can be learned from the treatment of both of these. Several strategies have been proposed to limit the occurrence of mIAS (Table 3). First, establish an oral care protocol. Educate patients who are initiating treatment with an mTOR inhibitor on implementation of the oral care protocol and emphasize adherence. Second, educate patients on the symptoms and timing of mIAS. Patients may hesitate to report mild symptoms or assume they are innocuous, so be clear that reporting all symptoms is important to allow timely clinical evaluation. Early recognition of mIAS facilitates early intervention and can prevent dose modification and interruption. Third, implement the preventive and treatment measures described. Many of the preventive measures can be incorporated into an oral care protocol.
The advent of mTOR inhibitors has clinically benefited many patients with cancer. Although side effects, like mIAS, may develop during treatment, they should not be considered insurmountable. Through education, vigilance, and aggressive management, health care providers and patients can work together to help patients maintain their quality of life while continuing to optimally address their disease.
Acknowledgment
The authors thank Anna Lau, PhD, and Patricia Segarini, PhD, of Percolation Communications LLC, for their editorial assistance. Funding for manuscript development was provided by Novartis Pharmaceuticals Corp.
1. Lauring J, Park BH, Wolff AC. The phosphoinositide-3-kinase-Akt-mTOR pathway as a therapeutic target in breast cancer. J Natl Compr Canc Netw. 2013;11:670-678.
2. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140-156.
3. Torisel (temsirolimus) [prescribing information]. Philadelphia, PA: Wyeth Pharmaceuticals; 2014.
4. Afinitor (everolimus) [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2015.
5. Rapamune (sirolimus) [prescribing information]. Philadelphia, PA: Wyeth Pharmaceuticals; 2012.
6. Peterson DE, Boers-Doets CB, Bensadoun RJ, Herrstedt J, ESMO Guidelines Committee. Management of oral and gastrointestinal mucosal injury: ESMO Clinical Practice Guidelines for diagnosis, treatment, and follow-up. Ann Oncol. 2015;26 Suppl 5:v139-151.
7. Hidalgo M, Buckner JC, Erlichman C, et al. A phase I and pharmacokinetic study of temsirolimus (CCI-779) administered intravenously daily for 5 days every 2 weeks to patients with advanced cancer. Clin Cancer Res. 2006;12:5755-5763.
8. Martins F, de Oliveira MA, Wang Q, et al. A review of oral toxicity associated with mTOR inhibitor therapy in cancer patients. Oral Oncol. 2013;49:293-298.
9. O’Donnell A, Faivre S, Burris HA, 3rd, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J Clin Oncol. 2008;26:1588-1595.
10. Shameem R, Lacouture M, Wu S. Incidence and risk of high-grade stomatitis with mTOR inhibitors in cancer patients. Cancer Invest. 2015;33:70-77.
11. Femara (letrozole) [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
12. Rugo HS, Hortobagyi GN, Yao J, et al. Meta-analysis of stomatitis in clinical studies of everolimus: incidence and relationship with efficacy. Ann Oncol. 2016;27:519-525.
13. Keefe DM, Schubert MM, Elting LS, et al. Updated clinical practice guidelines for the prevention and treatment of mucositis. Cancer. 2007;109:820-831.
14. Sonis S, Treister N, Chawla S, Demetri G, Haluska F. Preliminary characterization of oral lesions associated with inhibitors of mammalian target of rapamycin in cancer patients. Cancer. 2010;116:210-215.
15. Scully C. Clinical practice. Aphthous ulceration. N Engl J Med. 2006;355:165-172.
16. Ferte C, Paci A, Zizi M, et al. Natural history, management and pharmacokinetics of everolimus-induced-oral ulcers: insights into compliance issues. Eur J Cancer. 2011;47:2249-2255.
17. Wong HM. Oral complications and management strategies for patients undergoing cancer therapy ScienceWorldJournal. 2014;581795.
18. de Oliveira MA, Martins EMF, Wang Q, et al. Clinical presentation and management of mTOR inhibitor-associated stomatitis. Oral Oncol. 2011;47:998-1003.
19. Rugo HS, Pritchard KI, Gnant M, et al. Incidence and time course of everolimus-related adverse events in postmenopausal women with hormone receptor-positive advanced breast cancer: insights from BOLERO-2. Ann Oncol. 2014;25:808-815.
20. National Cancer Institute. Cancer Therapy Evaluation Program. Common Terminology Criteria for Adverse Events v3.0 (CTCAE). http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed February 13, 2017.
21. National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) v4.03. http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf. Accessed February 13, 2017.
22. World Health Organization. WHO Handbook for Reporting Results of Cancer Treatment. Geneva, Switzerland: World Health Organization (WHO Offset Publication No. 48); 1979.
23. Epstein JB, Thariat J, Bensadoun RJ, et al. Oral complications of cancer and cancer therapy: from cancer treatment to survivorship. CA Cancer J Clin. 2012;62:400-422.
24. Iglesias-Bartolome R, Patel V, Cotrim A, et al. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell. 2012;11:401-414.
25. Lewkowicz N, Lewkowicz P, Dzitko K, et al. Dysfunction of CD4+CD25high T regulatory cells in patients with recurrent aphthous stomatitis. J Oral Pathol Med. 2008;37:454-461.
26. Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767-777.
27. Jin Y, Tymen SD, Chen D, et al. MicroRNA-99 family targets AKT/mTOR signaling pathway in dermal wound healing. PLoS One. 2013;8:e64434.
28. Rosselli-Murai LK, Almeida LO, Zagni C, et al. Periostin responds to mechanical stress and tension by activating the MTOR signaling pathway. PLoS One. 2013;8:e83580.
29. Rugo HS. Dosing and safety implications for oncologists when administering everolimus to patients with hormone receptor-positive breast cancer. Clin Breast Cancer. 2016;16:18-22.
30. Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520-529.
31. Yardley DA, Noguchi S, Pritchard KI, et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther. 2013;30:870-884.
32. Rubenstein EB, Peterson DE, Schubert M, et al. Clinical practice guidelines for the prevention and treatment of cancer therapy-induced oral and gastrointestinal mucositis. Cancer. 2004;100:2026-2046.
33. Borowski B, Benhamou E, Pico JL, Laplanche A, Margainaud JP, Hayat M. Prevention of oral mucositis in patients treated with high-dose chemotherapy and bone marrow transplantation: a randomised controlled trial comparing two protocols of dental care. Eur J Cancer B Oral Oncol. 1994;30B:93-97.
34. Cheng KK, Molassiotis A, Chang AM, Wai WC, Cheung SS. Evaluation of an oral care protocol intervention in the prevention of chemotherapy-induced oral mucositis in paediatric cancer patients. Eur J Cancer. 2001;37:2056-2063.
35. Dudjak LA. Mouth care for mucositis due to radiation therapy. Cancer Nurs. 1987;10:131-140.
36. Graham KM, Pecoraro DA, Ventura M, Meyer CC. Reducing the incidence of stomatitis using a quality assessment and improvement approach. Cancer Nurs. 1993;16:117-122.
37. Kenny SA. Effect of two oral care protocols on the incidence of stomatitis in hematology patients. Cancer Nurs. 1990;13:345-353.
38. Larson PJ, Miaskowski C, MacPhail L, et al. The PRO-SELF Mouth Aware program: an effective approach for reducing chemotherapy-induced mucositis. Cancer Nurs. 1998;21:263-268.
39. Levy-Polack MP, Sebelli P, Polack NL. Incidence of oral complications and application of a preventive protocol in children with acute leukemia. Spec Care Dentist. 1998;18:189-193.
40. Yeager KA, Webster J, Crain M, Kasow J, McGuire DB. Implementation of an oral care standard for leukemia and transplantation patients. Cancer Nurs. 2000;23:40-47; quiz 47-48.
41. Pilotte AP, Hohos MB, Polson KM, Huftalen TM, Treister N. Managing stomatitis in patients treated with mammalian target of rapamycin inhibitors. Clin J Oncol Nurs. 2011;15:E83-89.
42. Gomez-Fernandez C, Garden BC, Wu S, Feldman DR, Lacouture ME. The risk of skin rash and stomatitis with the mammalian target of rapamycin inhibitor temsirolimus: a systematic review of the literature and meta-analysis. Eur J Cancer. 2012;48:340-346.
43. Bonnaure-Mallet M, Bunetel L, Tricot-Doleux S, Guerin J, Bergeron C, LeGall E. Oral complications during treatment of malignant diseases in childhood: effects of tooth brushing. Eur J Cancer. 1998;34:1588-1591.
44. Chuang P, Langone AJ. Clobetasol ameliorates aphthous ulceration in renal transplant patients on sirolimus. Am J Transplant. 2007;7:714-717.
45. Femiano F, Buonaiuto C, Gombos F, Lanza A, Cirillo N. Pilot study on recurrent aphthous stomatitis (RAS): a randomized placebo-controlled trial for the comparative therapeutic effects of systemic prednisone and systemic montelukast in subjects unresponsive to topical therapy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2010;109:402-407.
1. Lauring J, Park BH, Wolff AC. The phosphoinositide-3-kinase-Akt-mTOR pathway as a therapeutic target in breast cancer. J Natl Compr Canc Netw. 2013;11:670-678.
2. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140-156.
3. Torisel (temsirolimus) [prescribing information]. Philadelphia, PA: Wyeth Pharmaceuticals; 2014.
4. Afinitor (everolimus) [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2015.
5. Rapamune (sirolimus) [prescribing information]. Philadelphia, PA: Wyeth Pharmaceuticals; 2012.
6. Peterson DE, Boers-Doets CB, Bensadoun RJ, Herrstedt J, ESMO Guidelines Committee. Management of oral and gastrointestinal mucosal injury: ESMO Clinical Practice Guidelines for diagnosis, treatment, and follow-up. Ann Oncol. 2015;26 Suppl 5:v139-151.
7. Hidalgo M, Buckner JC, Erlichman C, et al. A phase I and pharmacokinetic study of temsirolimus (CCI-779) administered intravenously daily for 5 days every 2 weeks to patients with advanced cancer. Clin Cancer Res. 2006;12:5755-5763.
8. Martins F, de Oliveira MA, Wang Q, et al. A review of oral toxicity associated with mTOR inhibitor therapy in cancer patients. Oral Oncol. 2013;49:293-298.
9. O’Donnell A, Faivre S, Burris HA, 3rd, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J Clin Oncol. 2008;26:1588-1595.
10. Shameem R, Lacouture M, Wu S. Incidence and risk of high-grade stomatitis with mTOR inhibitors in cancer patients. Cancer Invest. 2015;33:70-77.
11. Femara (letrozole) [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
12. Rugo HS, Hortobagyi GN, Yao J, et al. Meta-analysis of stomatitis in clinical studies of everolimus: incidence and relationship with efficacy. Ann Oncol. 2016;27:519-525.
13. Keefe DM, Schubert MM, Elting LS, et al. Updated clinical practice guidelines for the prevention and treatment of mucositis. Cancer. 2007;109:820-831.
14. Sonis S, Treister N, Chawla S, Demetri G, Haluska F. Preliminary characterization of oral lesions associated with inhibitors of mammalian target of rapamycin in cancer patients. Cancer. 2010;116:210-215.
15. Scully C. Clinical practice. Aphthous ulceration. N Engl J Med. 2006;355:165-172.
16. Ferte C, Paci A, Zizi M, et al. Natural history, management and pharmacokinetics of everolimus-induced-oral ulcers: insights into compliance issues. Eur J Cancer. 2011;47:2249-2255.
17. Wong HM. Oral complications and management strategies for patients undergoing cancer therapy ScienceWorldJournal. 2014;581795.
18. de Oliveira MA, Martins EMF, Wang Q, et al. Clinical presentation and management of mTOR inhibitor-associated stomatitis. Oral Oncol. 2011;47:998-1003.
19. Rugo HS, Pritchard KI, Gnant M, et al. Incidence and time course of everolimus-related adverse events in postmenopausal women with hormone receptor-positive advanced breast cancer: insights from BOLERO-2. Ann Oncol. 2014;25:808-815.
20. National Cancer Institute. Cancer Therapy Evaluation Program. Common Terminology Criteria for Adverse Events v3.0 (CTCAE). http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed February 13, 2017.
21. National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) v4.03. http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf. Accessed February 13, 2017.
22. World Health Organization. WHO Handbook for Reporting Results of Cancer Treatment. Geneva, Switzerland: World Health Organization (WHO Offset Publication No. 48); 1979.
23. Epstein JB, Thariat J, Bensadoun RJ, et al. Oral complications of cancer and cancer therapy: from cancer treatment to survivorship. CA Cancer J Clin. 2012;62:400-422.
24. Iglesias-Bartolome R, Patel V, Cotrim A, et al. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell. 2012;11:401-414.
25. Lewkowicz N, Lewkowicz P, Dzitko K, et al. Dysfunction of CD4+CD25high T regulatory cells in patients with recurrent aphthous stomatitis. J Oral Pathol Med. 2008;37:454-461.
26. Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767-777.
27. Jin Y, Tymen SD, Chen D, et al. MicroRNA-99 family targets AKT/mTOR signaling pathway in dermal wound healing. PLoS One. 2013;8:e64434.
28. Rosselli-Murai LK, Almeida LO, Zagni C, et al. Periostin responds to mechanical stress and tension by activating the MTOR signaling pathway. PLoS One. 2013;8:e83580.
29. Rugo HS. Dosing and safety implications for oncologists when administering everolimus to patients with hormone receptor-positive breast cancer. Clin Breast Cancer. 2016;16:18-22.
30. Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520-529.
31. Yardley DA, Noguchi S, Pritchard KI, et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther. 2013;30:870-884.
32. Rubenstein EB, Peterson DE, Schubert M, et al. Clinical practice guidelines for the prevention and treatment of cancer therapy-induced oral and gastrointestinal mucositis. Cancer. 2004;100:2026-2046.
33. Borowski B, Benhamou E, Pico JL, Laplanche A, Margainaud JP, Hayat M. Prevention of oral mucositis in patients treated with high-dose chemotherapy and bone marrow transplantation: a randomised controlled trial comparing two protocols of dental care. Eur J Cancer B Oral Oncol. 1994;30B:93-97.
34. Cheng KK, Molassiotis A, Chang AM, Wai WC, Cheung SS. Evaluation of an oral care protocol intervention in the prevention of chemotherapy-induced oral mucositis in paediatric cancer patients. Eur J Cancer. 2001;37:2056-2063.
35. Dudjak LA. Mouth care for mucositis due to radiation therapy. Cancer Nurs. 1987;10:131-140.
36. Graham KM, Pecoraro DA, Ventura M, Meyer CC. Reducing the incidence of stomatitis using a quality assessment and improvement approach. Cancer Nurs. 1993;16:117-122.
37. Kenny SA. Effect of two oral care protocols on the incidence of stomatitis in hematology patients. Cancer Nurs. 1990;13:345-353.
38. Larson PJ, Miaskowski C, MacPhail L, et al. The PRO-SELF Mouth Aware program: an effective approach for reducing chemotherapy-induced mucositis. Cancer Nurs. 1998;21:263-268.
39. Levy-Polack MP, Sebelli P, Polack NL. Incidence of oral complications and application of a preventive protocol in children with acute leukemia. Spec Care Dentist. 1998;18:189-193.
40. Yeager KA, Webster J, Crain M, Kasow J, McGuire DB. Implementation of an oral care standard for leukemia and transplantation patients. Cancer Nurs. 2000;23:40-47; quiz 47-48.
41. Pilotte AP, Hohos MB, Polson KM, Huftalen TM, Treister N. Managing stomatitis in patients treated with mammalian target of rapamycin inhibitors. Clin J Oncol Nurs. 2011;15:E83-89.
42. Gomez-Fernandez C, Garden BC, Wu S, Feldman DR, Lacouture ME. The risk of skin rash and stomatitis with the mammalian target of rapamycin inhibitor temsirolimus: a systematic review of the literature and meta-analysis. Eur J Cancer. 2012;48:340-346.
43. Bonnaure-Mallet M, Bunetel L, Tricot-Doleux S, Guerin J, Bergeron C, LeGall E. Oral complications during treatment of malignant diseases in childhood: effects of tooth brushing. Eur J Cancer. 1998;34:1588-1591.
44. Chuang P, Langone AJ. Clobetasol ameliorates aphthous ulceration in renal transplant patients on sirolimus. Am J Transplant. 2007;7:714-717.
45. Femiano F, Buonaiuto C, Gombos F, Lanza A, Cirillo N. Pilot study on recurrent aphthous stomatitis (RAS): a randomized placebo-controlled trial for the comparative therapeutic effects of systemic prednisone and systemic montelukast in subjects unresponsive to topical therapy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2010;109:402-407.
APF530 for nausea and vomiting prevention following cisplatin: phase 3 MAGIC trial analysis
Despite available antiemetic therapies, chemotherapy-induced nausea and vomiting (CINV) following highly emetogenic chemotherapy (HEC), particularly in the delayed phase (>24-120 h after chemotherapy), continues to impair patient quality of life and chemotherapy compliance.1 Cisplatin-based chemotherapy, classified as HEC at any dose,2 is widely used to treat cancers such as non–small-cell and small
5-hydroxytryptamine type 3 (5-HT3) receptor antagonists (RAs; eg, granisetron, ondansetron, dolasetron, and palonosetron) have been the cornerstone of CINV therapy for decades and remain an integral part of contemporary antiemetic treatment regimens. Most current antiemetic guidelines for HEC recommend a 3-drug regimen, comprising a 5-HT3 RA, a neurokinin 1 (NK-1) RA, and a corticosteroid (dexamethasone).2,6,7 A regimen of olanzapine (antipsychotic), palonosetron (5-HT3 RA), and dexamethasone (corticosteroid) has been recommended as an alternative option. Recently, the oral fixed-dose combination of netupitant and palonosetron (NEPA) was approved and has shown efficacy in the cisplatin setting.8,9 However, the administration of oral medication to patients experiencing CINV and those with head and neck cancer may be difficult.10 Alternative antiemetic treatments that provide CINV control into the delayed phase and with a convenient route of administration, are needed.
APF530 is a novel extended-release granisetron formulation that provides sustained release of therapeutic concentrations for ≥5 days. The Biochronomer tri(ethylene glycol) poly(orthoester) (TEG-POE) vehicle releases granisetron slowly by polymer hydrolysis after it has been injected subcutaneously (SC) into the abdomen or upper arm.11,12 In 2016, the US Food and Drug Administration approved APF530 in combination with other antiemetics for the prevention of acute and delayed nausea and vomiting associated with initial and repeat courses of moderately emetogenic chemotherapy (MEC) or anthracycline plus cyclophosphamide (AC) combination chemotherapy regimens based on data from 2 pivotal phase 3 trials.13
A phase 3 trial demonstrated noninferiority of APF530 (500 mg, SC) to palonosetron (0.25 mg, intravenously [IV]), each with dexamethasone (corticosteroid), in the control of acute-phase CINV after MEC or HEC, and delayed-phase CINV after MEC (classified by Hesketh criteria).14,15 Furthermore, APF530 provided sustained CINV control over multiple cycles of chemotherapy.16 Numerically higher complete response (CR: no emesis, no rescue medication use) rates were observed with APF530, compared with palonosetron, in the delayed phase after HEC (APF530 500 mg, 67.1%; palonosetron 0.25 mg, 64.3%).15
A reanalysis of study endpoints by newer emetogenicity classification guidelines from the American Society of Clinical Oncology (ASCO)7 maintained overall study conclusions.17 Notably, the numerically higher CR rates with APF530 in the delayed phase following HEC were enhanced (APF530 500 mg, 55.8%; palonosetron 0.25 mg, 50.5%), suggesting a need for further examination in this setting. The subsequent APF530 phase 3 MAGIC trial (Modified Absorption of Granisetron In the prevention of CINV; NCT02106494), compared APF530 (500 mg, SC) with ondansetron (0.15 mg/kg, IV), each with fosaprepitant (NK-1 RA) and dexamethasone in patients receiving HEC. The primary endpoint was met: the APF530 regimen demonstrated superior delayed-phase CR compared with the ondansetron regimen (64.7% vs 56.6%; 95% confidence interval [CI]:
1.7-14.4; P = .014; 8.0% absolute improvement).18
APF530 also demonstrated a significant benefit over ondansetron for other endpoints including nausea control, rescue medication use, and satisfaction with antiemetic therapy.18 APF530 is the first and only 5-HT3 RA to demonstrate superiority over another in a phase 3 efficacy trial using a guideline-recommended 3-drug regimen for both arms.
A prespecified MAGIC trial analysis of the primary endpoint by intent to receive cisplatin (≥50 mg/m2, Yes/No) demonstrated a pronounced treatment benefit in terms of delayed-phase CR rates with the APF530 regimen among patients in the cisplatin (≥50 mg/m2, Yes) stratum (CR: 65.3% vs 54.7%; 95% CI: -1.4-22.7; 10.6% absolute improvement).18 These results are compelling, since cisplatin represents a particularly emetogenic class of chemotherapy; a more in-depth analysis of additional MAGIC trial endpoints for these patients would be of clinical interest, and is presented here. Efficacy endpoints in this analysis include CR in the overall and acute phases, complete control (CC) and total response (TR) rates, rescue medication use, nausea frequency, and safety.
Methods
Study design and patients
The MAGIC trial was a prospective, randomized, multicenter, placebo-controlled, double-blind, double-dummy phase 3 trial conducted at 77 centers across the United States. The study protocol was reviewed and approved by the institutional review board at each participating center, and conducted according to the Declaration of Helsinki. The study design, previously presented in detail,18 is reviewed briefly here.
Eligible men and women were 18-80 years of age with histologically or cytologically confirmed malignancy (cancer type information was not captured) and were entering the first cycle of their single-day HEC treatment (defined by ASCO 2011 emetogenicity criteria).7 Patients had Eastern Cooperative Oncology Group Performance Status (ECOG-PS) of 0 or 1, no history or presence of significant cardiac disease or QT interval prolongation, and adequate bone marrow, kidney, and liver function. All patients provided written informed consent.
Procedures
Patients were stratified by planned receipt of the cisplatin regimen ≥50 mg/m2 (Yes/No), randomized 1:1 to receive APF530 500 mg SC (granisetron 10 mg) or ondansetron 0.15 mg/kg IV (up to a maximum of 16 mg as a single dose) on day 1 (Figure 1). The APF530 arm received the ondansetron saline placebo, and the ondansetron arm received the APF530 SC placebo containing the TEG-POE vehicle. All patients were scheduled to receive fosaprepitant 150 mg IV and dexamethasone 12 mg IV on day 1, then oral dexamethasone 8 mg once daily on day 2 and 8 mg twice daily on days 3 and 4. Rescue medication was allowed at the investigator’s discretion.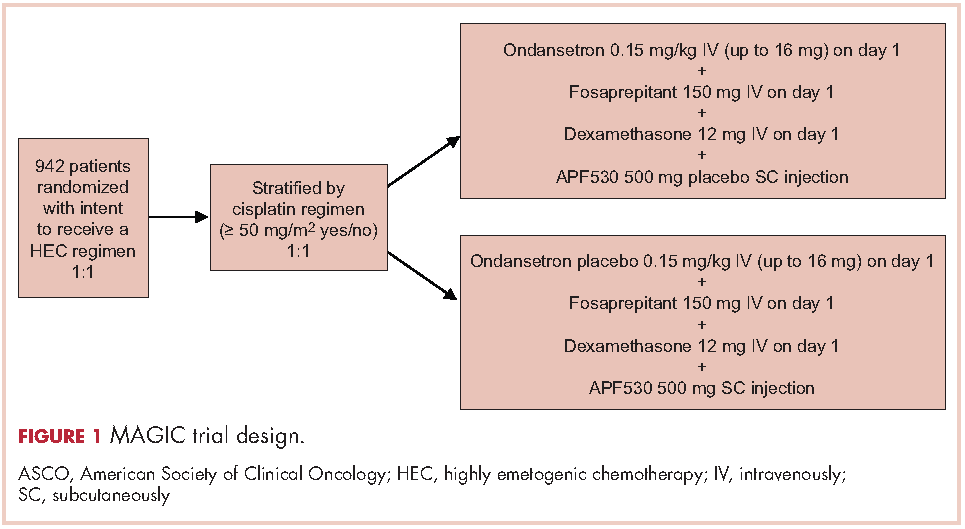
Outcomes
The primary objective of the trial was to demonstrate the superiority of APF530 500 mg SC compared with ondansetron 0.15 mg/kg IV, as part of the current guideline-recommended 3-drug regimen, in preventing delayed-phase CINV after HEC. The primary endpoint was delayed-phase (24-120 h) CR (no emetic episodes [vomit or retch] and no rescue medication use). In addition, a prespecified analysis of delayed-phase CR by randomization strata (planned use of cisplatin) was performed.
Secondary and other endpoints included overall-phase CR
(0-120 h); delayed-, overall-, and acute-phase complete control (CC: CR and no more than mild nausea); delayed-, overall-, and acute-phase total response (TR; CR and no nausea); and rescue medication use. A post hoc analysis of nausea severity was also conducted. Safety assessments included treatment-emergent adverse events (TEAEs), injection-site reactions (ISRs), laboratory parameters, and vital signs. TEAEs were assessed by type, duration, severity, and relationship to study drug. ISR timing and severity were captured in patient diaries.
Statistical analysis
All efficacy analyses were conducted using the modified intent-to-treat population (mITT; randomized patients who received study drug and a HEC regimen and had post-baseline efficacy data). Safety assessments were performed on the safety population (randomized patients who received study drug).
This analysis conducted on the subgroup of patients with intent to receive cisplatin (cisplatin randomization stratum, ≥50 mg/m2, Yes) was exploratory and was not powered to detect treatment differences. Preplanned analyses compared CR, CC, and TR rates across treatment arms using 95% CIs.
Post hoc analyses of time to first rescue medication use, proportion of patients with rescue medication use, and less frequent nausea were performed. All P values were calculated using the Cochran-Mantel-Haenszel chi square test. Rescue medication use results were based on observed data, without imputation for missing results (ie, calculated from the number of patients with a response). Further analyses of efficacy endpoints CR, CC, and TR in the subset of female patients in the cisplatin randomization stratum were performed. Safety assessments were summarized descriptively.
Results
A total of 942 patients were randomized across 77 US centers during March 31, 2014 and May 15, 2015 (471 APF530, 471 ondansetron). Among those, 264 had intent to receive cisplatin and were included in the cisplatin randomization stratum (≥50 mg/m2, Yes) (Figure 2). A total of 256 patients in the cisplatin stratum received study drug and were included in the safety population (126 APF530, 130 ondansetron); 252 patients were included in the mITT population (124 APF530, 128 ondansetron).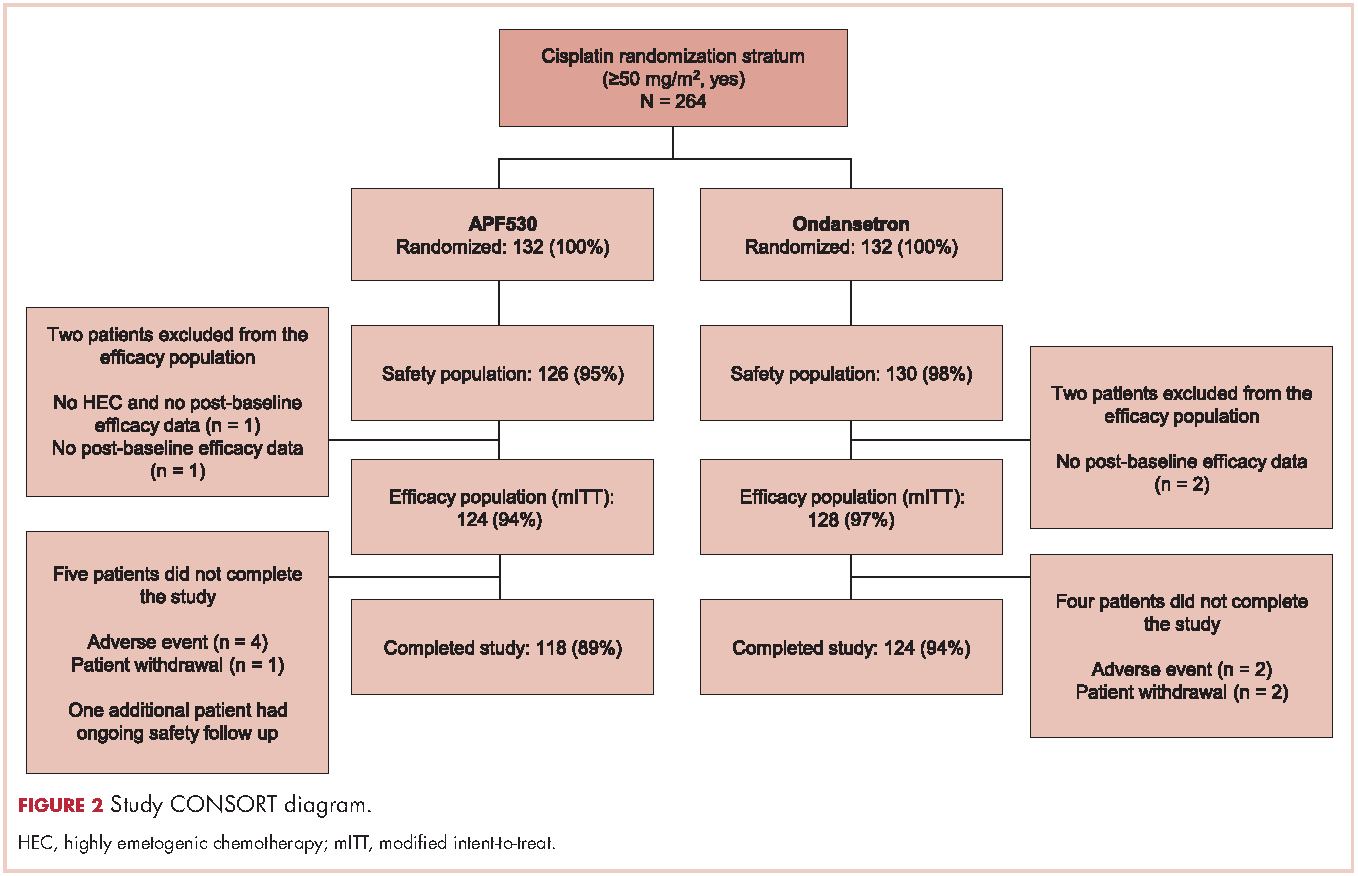
Baseline demographics were generally balanced between treatment arms (Table 1). The proportion of female patients was 41.1% (51/124) and 48.4% (62/128) in the APF530 and ondansetron arms, respectively. The majority of patients had an ECOG PS of 0 (57.3% [71/124] APF530; 60.2% [77/128] ondansetron). The most common cisplatin-based chemotherapy regimen in both treatment arms was cisplatin and gemcitabine (25.0% [31/124] APF530; 28.9% [37/128] ondansetron) (Suppl Table 1). Two patients in the APF530 arm and 3 patients in the ondansetron arm either received a lower cisplatin dose (<50 mg/m2) or did not go on to receive cisplatin as intended at randomization (Suppl Table 1). As previously reported, in the cisplatin stratum (Table 2), delayed-phase CR was numerically higher in the APF530 arm versus the ondansetron arm, with a corresponding treatment difference of 10.6% (65.3% [81/124] APF530; 54.7% [70/128] ondansetron; 95% CI [-1.4, 22.7]; P = .085). Although the CI contains 0, the result is consistent with the significant benefit observed in the overall study population (64.7% [291/450] APF530; 56.6% [256/452] ondansetron; 95% CI [1.7, 14.4];
P = .014).18 This more in-depth analysis found similar trends favoring the APF530 over the ondansetron regimen across overall- and acute-phase CR (Table 2).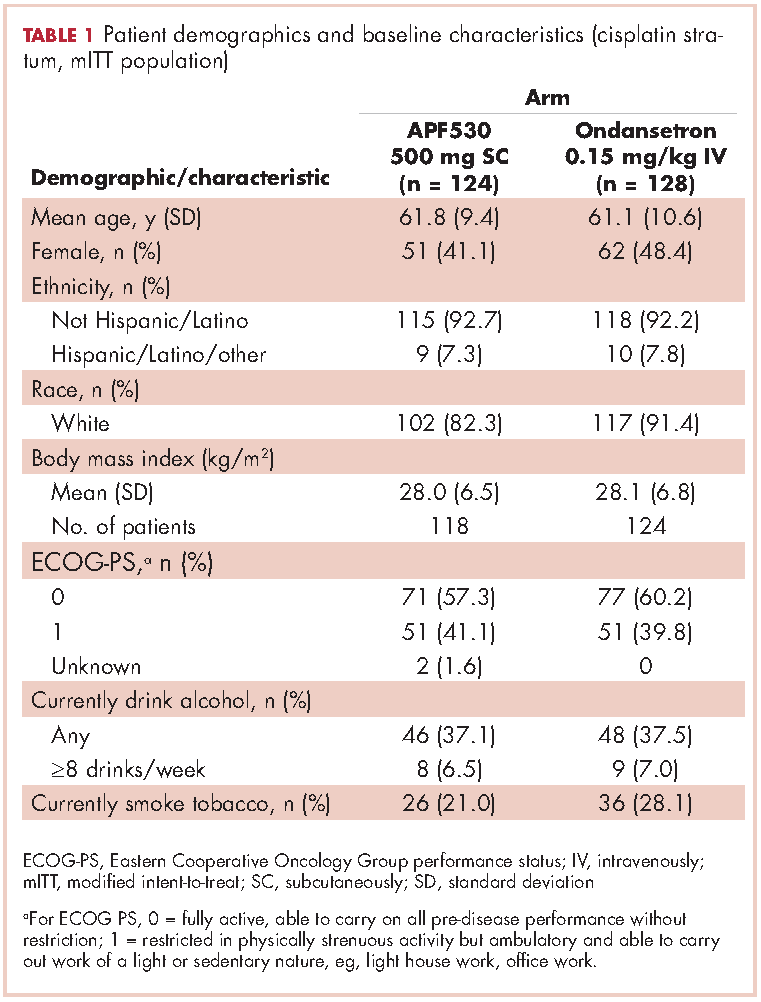
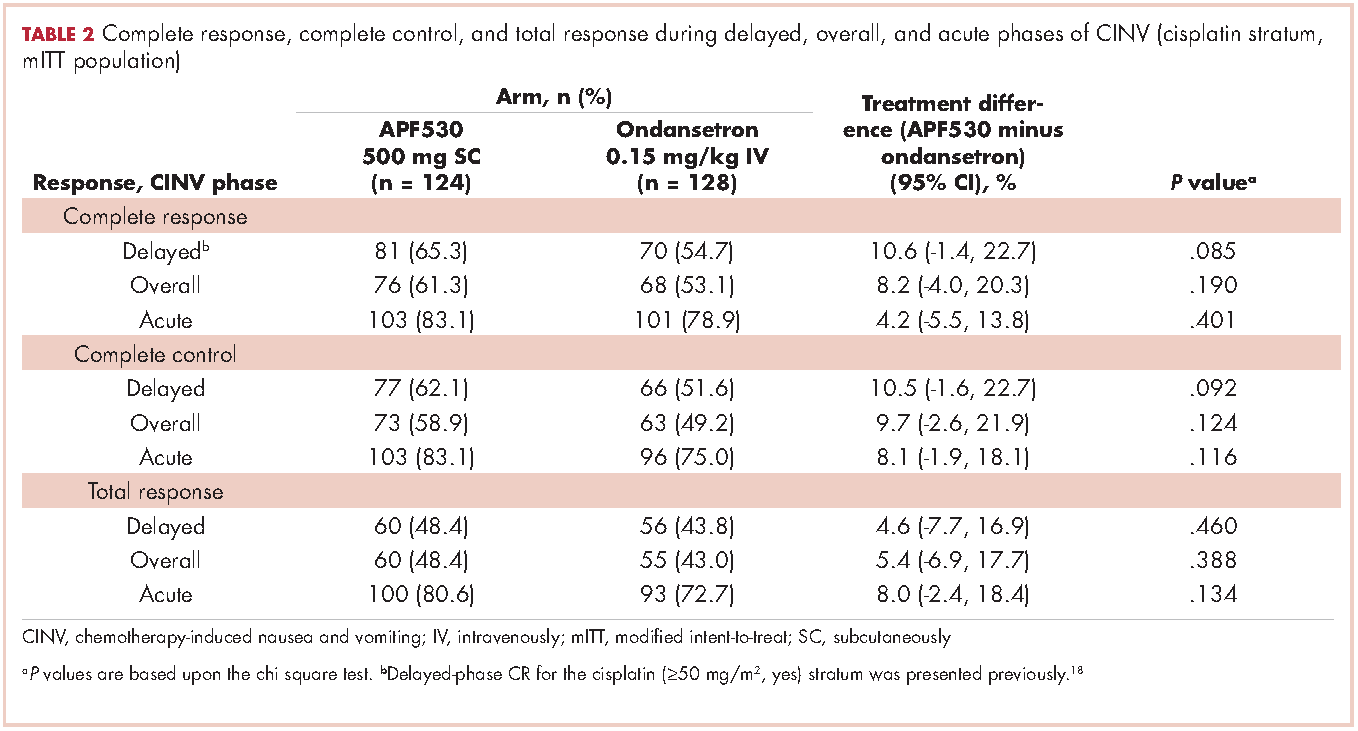
A significantly greater proportion of patients in the APF530 arm, compared with the ondansetron arm, reported no rescue medication use during the delayed phase (74.4% [90/121] APF530; 62.6% [77/123] ondansetron; P = .048). Trends in favor of APF530 were observed in the overall phase (71.1% [86/121] APF530; 61.8% [76/123] ondansetron; P = .125) and acute phase (86.9% [106/122] APF530; 81.9% [104/127] ondansetron; P = .278). Time to first rescue medication use was consistently longer in the APF530 arm, compared with the ondansetron arm, although not statistically significantly (P = .150) (Figure 3).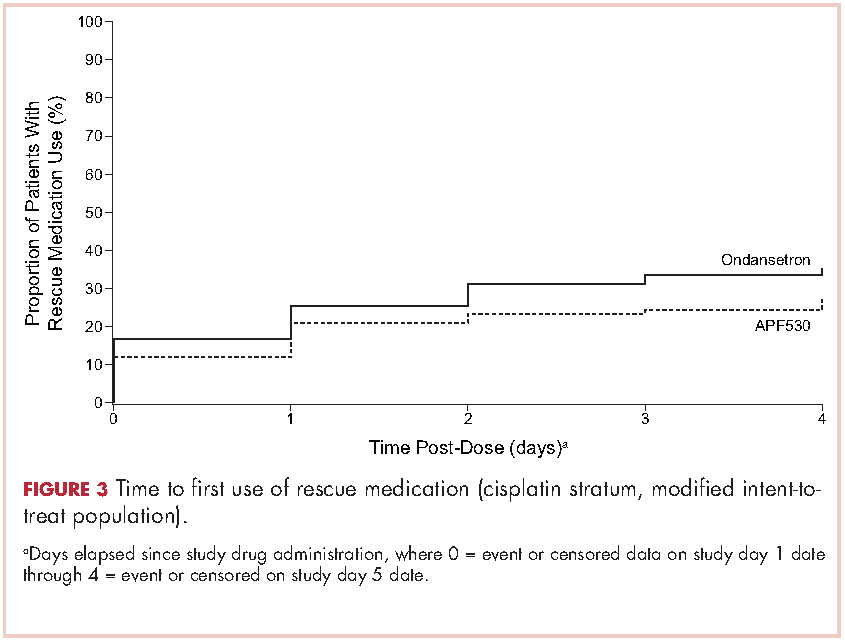
The APF530 regimen was generally well tolerated in the cisplatin subgroup, and no new safety signals were identified (Table 3). Most patients experienced at least one TEAE. Excluding ISRs, TEAE incidences were 72.2% and 66.9% in the APF530 and ondansetron arms, respectively; most common were constipation, fatigue, nausea, diarrhea, dehydration, and headache. Excluding ISRs, the most common treatment-related TEAEs in the APF530 and ondansetron arms were constipation (2.4% and 2.3%, respectively and headache (3.2% and 4.6%).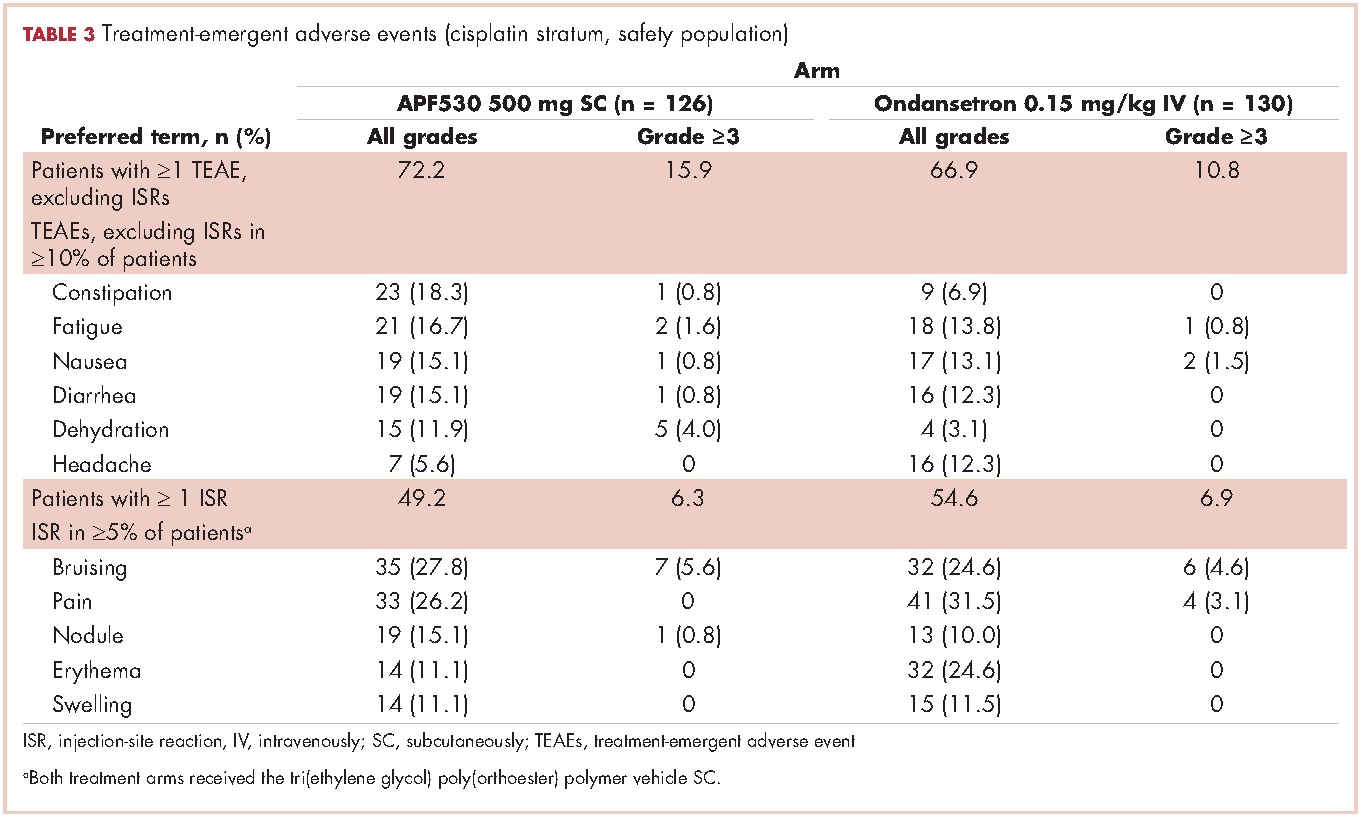
Discussion
The MAGIC trial is the first phase 3 efficacy trial in the prevention of CINV in patients receiving HEC using the current guideline-recommended 3-drug antiemetic regimen in both treatment arms.18 Ondansetron was chosen as the appropriate 5-HT3 RA comparator because no other 5-HT3 RA has shown superiority to ondansetron in delayed-phase CINV following HEC. Furthermore, ondansetron is indicated for prevention of nausea and vomiting associated with initial and repeat courses of chemotherapy, including high-dose cisplatin.19 The MAGIC trial primary endpoint was met for the overall study population; in the context of a 3-drug regimen, APF530 demonstrated superior control of delayed-phase CINV following HEC compared with standard-of-care ondansetron.18 As reported previously, significant benefits were also observed with the APF530 regimen over the ondansetron regimen in terms of rescue medication use, patient satisfaction with antiemetic therapy, and nausea frequency in the overall study population.18
Cisplatin is generally regarded as one of the most emetogenic chemotherapeutic agents. For this reason, cisplatin is often evaluated separately in clinical trials, and was a stratification factor in the MAGIC trial. Consistent with the previously reported significant results,18 trends in the cisplatin stratum analysis favored the APF530 regimen, compared with the ondansetron regimen, in delayed- and overall-phase CR (treatment difference: 10.6%).18 Numerical trends presented here favoring the APF530 regimen over the ondansetron regimen were observed in CC and TR, two more stringent measures of CINV control that account for incidence of nausea. Furthermore, among women in the cisplatin stratum, a population at increased risk for CINV, the numerically higher CR, CC, and TR persisted in the APF530 arm, compared with the ondansetron arm.
The APF530 regimen was generally well tolerated in the cisplatin stratum, and no new safety signals were identified. The most common TEAEs were ISRs, mostly mild or moderate and resolving by study end. The double-dummy design resulted in ISRs in the ondansetron arm due to TEG-POE vehicle as the dummy APF530 injection. Transient ISRs have been observed with other agents administered SC, and are expected.20,21 Excluding ISRs, TEAEs were generally consistent with those observed for the 5-HT3 RA class.22
This analysis of patients randomized to receive cisplatin-based HEC in the MAGIC trial is exploratory and was not sufficiently powered to detect between-arm differences. Five total patients did not go on to receive cisplatin ≥50 mg/m2 as intended at randomization (2 APF530, 3 ondansetron); however, this is not uncommon in large clinical trials and represents less than 2% of patients in this analysis.
Recent phase 3 studies in patients receiving cisplatin-based HEC showed significant improvement in CINV prevention with the current guideline-recommended 3-drug regimen over the traditional 2-drug regimen (5-HT3 RA + dexamethasone).8,23 Results presented here, in a similar population receiving cisplatin-based HEC, suggest that in the context of a 3-drug antiemetic regimen in both treatment arms, APF530 provides additional benefit in CINV prevention compared with the standard of care, ondansetron. Furthermore, a recent phase 3 trial in patients receiving cisplatin or AC-based HEC demonstrated significant improvement in nausea when olanzapine was added to a traditional 3-drug regimen of a 5-HT3 RA, NK-1 RA, and dexamethasone.24 These compelling data support the addition of olanzapine as a fourth agent to the CINV treatment regimen to provide further control of nausea, which has been one of the more difficult components of CINV to control to date.
APF530 is the only 5-HT3 RA to demonstrate superiority over another as part of the guideline-recommended regimen in a 3-drug versus 3-drug phase 3 efficacy trial examining antiemetic efficacy following HEC. Results from the MAGIC trial, this exploratory analysis, and previous studies in MEC and HEC provide clinically meaningful benefits in preventing both acute- and delayed-phase CINV following guideline-specified MEC or HEC regimens. Consequently, APF530 was approved for use in combination with other antiemetics for prevention of acute and delayed nausea and vomiting associated with initial and repeat courses of MEC or AC combination chemotherapy regimens.13 Both the superior control of delayed-phase CINV following HEC demonstrated by the MAGIC trial18 and the consistent trends in the cisplatin stratum indicate a particular benefit for high-risk patients receiving high doses of cisplatin.
Acknowledgments
Joanna K Sandilos Rega, PhD, of SciStrategy Communications provided medical writing assistance, supported by Heron Therapeutics Inc, the maker of the study drug.
1. Hilarius DL, Kloeg PH, van der Wall E, van den Heuvel JJ, Gundy CM, Aaronson NK. Chemotherapy-induced nausea and vomiting in daily clinical practice: a community hospital-based study. Support Care Cancer. 2012;20:107-117.
2. NCCN clinical practice guidelines in oncology: antiemesis—version 1.2017. https://www.nccn.org/professionals/physician_gls/f_guidelines.asp#antiemesis. Accessed March 1, 2017.
3. Platinol (cisplatin for injection, USP) [prescribing information]. Princeton, NJ: Bristol-Myers Squibb Company; 2010. http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/018057s079lbl.pdf. Updated May 2010. Accessed September 12, 2016.
4. Pollera CF, Giannarelli D. Prognostic factors influencing cisplatin-induced emesis. Definition and validation of a predictive logistic model. Cancer. 1989;64:1117-1122.
5. Roila F, Boschetti E, Tonato M, et al. Predictive factors of delayed emesis in cisplatin-treated patients and antiemetic activity and tolerability of metoclopramide or dexamethasone. A randomized single-blind study. Am J Clin Oncol. 1991;14:238-242.
6. Roila F, Molassiotis A, Herrstedt J, et al. 2016 MASCC and ESMO guideline update for the prevention of chemotherapy- and radiotherapy-induced nausea and vomiting and of nausea and vomiting in advanced cancer patients. Ann Oncol. 2016;27(suppl 5):v119-v133.
7. Basch E, Prestrud AA, Hesketh PJ, et al. Antiemetics: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol. 2011;29:4189-4198.
8. Hesketh PJ, Rossi G, Rizzi G, et al. Efficacy and safety of NEPA, an oral combination of netupitant and palonosetron, for prevention of chemotherapy-induced nausea and vomiting following highly emetogenic chemotherapy: a randomized dose-ranging pivotal study. Ann Oncol. 2014;25:1340-1346.
9. Akynzeo (netupitant and palonosetron capsules) [prescribing information]. Woodcliff, NJ: Eisai; 2015. http://www.akynzeo.com. Revised April 2015. Accessed September 12, 2016.
10. Tsukahara K, Nakamura K, Motohashi R, et al. Antiemetic therapy of fosaprepitant, palonosetron, and dexamethasone combined with cisplatin-based chemotherapy for head and neck carcinomas. Acta Otolaryngol. 2014;134:1198-1204.
11. Ottoboni. Biochronomer technology and the development of APF530, a sustained release formulation of granisetron. J Exp Pharmacol. 2014;6:15-21.
12. Gabrail N, Yanagihara R, Spaczynski M, et al. Pharmacokinetics, safety, and efficacy of APF530 (extended-release granisetron) in patients receiving moderately or highly emetogenic chemotherapy: results of two phase II trials. Cancer Manag Res. 2015;7:83-92.
13. Sustol (granisetron) extended-release injection, for subcutaneous use [prescribing information]. Redwood City, CA; Heron Therapeutics; 2016. http://sustol.com/hcp/healthcare-professionals. Updated August 2016. Accessed September 12, 2016.
14. Hesketh PJ, Kris MG, Grunberg SM, et al. Proposal for classifying the acute emetogenicity of cancer chemotherapy. J Clin Oncol. 1997;15:103-109.
15. Raftopoulos H, Cooper W, O’Boyle E, Gabrail N, Boccia R, Gralla RJ. Comparison of an extended-release formulation of granisetron (APF530) versus palonosetron for the prevention of chemotherapy-induced nausea and vomiting associated with moderately or highly emetogenic chemotherapy: results of a prospective, randomized, double-blind, noninferiority phase 3 trial. Support Care Cancer. 2015;23:723-732.
16. Boccia RV, Cooper W, O’Boyle E. Sustained antiemetic responses with APF530 (sustained-release granisetron) during multiple cycles of emetogenic chemotherapy. J Community Support Oncol. 2015;13:38-46.
17. Raftopoulos H, Boccia R, Cooper W, O’Boyle E, Gralla RJ. Slow-release granisetron (APF530) versus palonosetron for chemotherapy-induced nausea/vomiting: analysis by American Society of Clinical Oncology emetogenicity criteria. Future Oncol. 2015;11:2541-2551.
18. Schnadig ID, Agajanian R, Dakhil C, et al. APF530 (Granisetron injection extended-release) in a three-drug regimen for delayed CINV in highly emetogenic chemotherapy. Future Oncol. 2016;12:1469-1481.
19. Zofran (ondansetron hydrochloride) injection for intravenous use. Research Triangle Park, NC: GlaxoSmithKline; 2014. http://www.pharma.us.novartis.com/product/pi/pdf/zofran_inj.pdf. Revised September 2014. Accessed September 12, 2016.
20. Eligard (luprolide acetate) kit for subcutaneous use [prescribing information]. Fort Collins, CO: Tolmar Pharmaceuticals Inc; 2017. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=b78d1919-9dee-44fa-90f9-e0a26d32481d. Revised January 2017. Accessed March 1, 2017.
21. Sandostatin LAR Depot (octreotide acetate for injectable suspension) [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2016. https://www.pharma.us.novartis.com/product/pi/pdf/sandostatin_lar.pdf. Revised July 2016. Accessed September 12, 2016.
22. Navari RM. Management of chemotherapy-induced nausea and vomiting: focus on newer agents and new uses for older agents. Drugs. 2013;73:249-262.
23. Rapoport BL, Chasen MR, Gridelli C, et al. Safety and efficacy of rolapitant for prevention of chemotherapy-induced nausea and vomiting after administration of cisplatin-based highly emetogenic chemotherapy in patients with cancer: two randomised, active-controlled, double-blind, phase 3 trials. Lancet Oncol. 2015;16:1079-1089.
24. Navari RM, Qin R, Ruddy KJ, et al. Olanzapine for the prevention of chemotherapy-induced nausea and vomiting. N Engl J Med. 2016;375:134-142.
Despite available antiemetic therapies, chemotherapy-induced nausea and vomiting (CINV) following highly emetogenic chemotherapy (HEC), particularly in the delayed phase (>24-120 h after chemotherapy), continues to impair patient quality of life and chemotherapy compliance.1 Cisplatin-based chemotherapy, classified as HEC at any dose,2 is widely used to treat cancers such as non–small-cell and small
5-hydroxytryptamine type 3 (5-HT3) receptor antagonists (RAs; eg, granisetron, ondansetron, dolasetron, and palonosetron) have been the cornerstone of CINV therapy for decades and remain an integral part of contemporary antiemetic treatment regimens. Most current antiemetic guidelines for HEC recommend a 3-drug regimen, comprising a 5-HT3 RA, a neurokinin 1 (NK-1) RA, and a corticosteroid (dexamethasone).2,6,7 A regimen of olanzapine (antipsychotic), palonosetron (5-HT3 RA), and dexamethasone (corticosteroid) has been recommended as an alternative option. Recently, the oral fixed-dose combination of netupitant and palonosetron (NEPA) was approved and has shown efficacy in the cisplatin setting.8,9 However, the administration of oral medication to patients experiencing CINV and those with head and neck cancer may be difficult.10 Alternative antiemetic treatments that provide CINV control into the delayed phase and with a convenient route of administration, are needed.
APF530 is a novel extended-release granisetron formulation that provides sustained release of therapeutic concentrations for ≥5 days. The Biochronomer tri(ethylene glycol) poly(orthoester) (TEG-POE) vehicle releases granisetron slowly by polymer hydrolysis after it has been injected subcutaneously (SC) into the abdomen or upper arm.11,12 In 2016, the US Food and Drug Administration approved APF530 in combination with other antiemetics for the prevention of acute and delayed nausea and vomiting associated with initial and repeat courses of moderately emetogenic chemotherapy (MEC) or anthracycline plus cyclophosphamide (AC) combination chemotherapy regimens based on data from 2 pivotal phase 3 trials.13
A phase 3 trial demonstrated noninferiority of APF530 (500 mg, SC) to palonosetron (0.25 mg, intravenously [IV]), each with dexamethasone (corticosteroid), in the control of acute-phase CINV after MEC or HEC, and delayed-phase CINV after MEC (classified by Hesketh criteria).14,15 Furthermore, APF530 provided sustained CINV control over multiple cycles of chemotherapy.16 Numerically higher complete response (CR: no emesis, no rescue medication use) rates were observed with APF530, compared with palonosetron, in the delayed phase after HEC (APF530 500 mg, 67.1%; palonosetron 0.25 mg, 64.3%).15
A reanalysis of study endpoints by newer emetogenicity classification guidelines from the American Society of Clinical Oncology (ASCO)7 maintained overall study conclusions.17 Notably, the numerically higher CR rates with APF530 in the delayed phase following HEC were enhanced (APF530 500 mg, 55.8%; palonosetron 0.25 mg, 50.5%), suggesting a need for further examination in this setting. The subsequent APF530 phase 3 MAGIC trial (Modified Absorption of Granisetron In the prevention of CINV; NCT02106494), compared APF530 (500 mg, SC) with ondansetron (0.15 mg/kg, IV), each with fosaprepitant (NK-1 RA) and dexamethasone in patients receiving HEC. The primary endpoint was met: the APF530 regimen demonstrated superior delayed-phase CR compared with the ondansetron regimen (64.7% vs 56.6%; 95% confidence interval [CI]:
1.7-14.4; P = .014; 8.0% absolute improvement).18
APF530 also demonstrated a significant benefit over ondansetron for other endpoints including nausea control, rescue medication use, and satisfaction with antiemetic therapy.18 APF530 is the first and only 5-HT3 RA to demonstrate superiority over another in a phase 3 efficacy trial using a guideline-recommended 3-drug regimen for both arms.
A prespecified MAGIC trial analysis of the primary endpoint by intent to receive cisplatin (≥50 mg/m2, Yes/No) demonstrated a pronounced treatment benefit in terms of delayed-phase CR rates with the APF530 regimen among patients in the cisplatin (≥50 mg/m2, Yes) stratum (CR: 65.3% vs 54.7%; 95% CI: -1.4-22.7; 10.6% absolute improvement).18 These results are compelling, since cisplatin represents a particularly emetogenic class of chemotherapy; a more in-depth analysis of additional MAGIC trial endpoints for these patients would be of clinical interest, and is presented here. Efficacy endpoints in this analysis include CR in the overall and acute phases, complete control (CC) and total response (TR) rates, rescue medication use, nausea frequency, and safety.
Methods
Study design and patients
The MAGIC trial was a prospective, randomized, multicenter, placebo-controlled, double-blind, double-dummy phase 3 trial conducted at 77 centers across the United States. The study protocol was reviewed and approved by the institutional review board at each participating center, and conducted according to the Declaration of Helsinki. The study design, previously presented in detail,18 is reviewed briefly here.
Eligible men and women were 18-80 years of age with histologically or cytologically confirmed malignancy (cancer type information was not captured) and were entering the first cycle of their single-day HEC treatment (defined by ASCO 2011 emetogenicity criteria).7 Patients had Eastern Cooperative Oncology Group Performance Status (ECOG-PS) of 0 or 1, no history or presence of significant cardiac disease or QT interval prolongation, and adequate bone marrow, kidney, and liver function. All patients provided written informed consent.
Procedures
Patients were stratified by planned receipt of the cisplatin regimen ≥50 mg/m2 (Yes/No), randomized 1:1 to receive APF530 500 mg SC (granisetron 10 mg) or ondansetron 0.15 mg/kg IV (up to a maximum of 16 mg as a single dose) on day 1 (Figure 1). The APF530 arm received the ondansetron saline placebo, and the ondansetron arm received the APF530 SC placebo containing the TEG-POE vehicle. All patients were scheduled to receive fosaprepitant 150 mg IV and dexamethasone 12 mg IV on day 1, then oral dexamethasone 8 mg once daily on day 2 and 8 mg twice daily on days 3 and 4. Rescue medication was allowed at the investigator’s discretion.
Outcomes
The primary objective of the trial was to demonstrate the superiority of APF530 500 mg SC compared with ondansetron 0.15 mg/kg IV, as part of the current guideline-recommended 3-drug regimen, in preventing delayed-phase CINV after HEC. The primary endpoint was delayed-phase (24-120 h) CR (no emetic episodes [vomit or retch] and no rescue medication use). In addition, a prespecified analysis of delayed-phase CR by randomization strata (planned use of cisplatin) was performed.
Secondary and other endpoints included overall-phase CR
(0-120 h); delayed-, overall-, and acute-phase complete control (CC: CR and no more than mild nausea); delayed-, overall-, and acute-phase total response (TR; CR and no nausea); and rescue medication use. A post hoc analysis of nausea severity was also conducted. Safety assessments included treatment-emergent adverse events (TEAEs), injection-site reactions (ISRs), laboratory parameters, and vital signs. TEAEs were assessed by type, duration, severity, and relationship to study drug. ISR timing and severity were captured in patient diaries.
Statistical analysis
All efficacy analyses were conducted using the modified intent-to-treat population (mITT; randomized patients who received study drug and a HEC regimen and had post-baseline efficacy data). Safety assessments were performed on the safety population (randomized patients who received study drug).
This analysis conducted on the subgroup of patients with intent to receive cisplatin (cisplatin randomization stratum, ≥50 mg/m2, Yes) was exploratory and was not powered to detect treatment differences. Preplanned analyses compared CR, CC, and TR rates across treatment arms using 95% CIs.
Post hoc analyses of time to first rescue medication use, proportion of patients with rescue medication use, and less frequent nausea were performed. All P values were calculated using the Cochran-Mantel-Haenszel chi square test. Rescue medication use results were based on observed data, without imputation for missing results (ie, calculated from the number of patients with a response). Further analyses of efficacy endpoints CR, CC, and TR in the subset of female patients in the cisplatin randomization stratum were performed. Safety assessments were summarized descriptively.
Results
A total of 942 patients were randomized across 77 US centers during March 31, 2014 and May 15, 2015 (471 APF530, 471 ondansetron). Among those, 264 had intent to receive cisplatin and were included in the cisplatin randomization stratum (≥50 mg/m2, Yes) (Figure 2). A total of 256 patients in the cisplatin stratum received study drug and were included in the safety population (126 APF530, 130 ondansetron); 252 patients were included in the mITT population (124 APF530, 128 ondansetron).
Baseline demographics were generally balanced between treatment arms (Table 1). The proportion of female patients was 41.1% (51/124) and 48.4% (62/128) in the APF530 and ondansetron arms, respectively. The majority of patients had an ECOG PS of 0 (57.3% [71/124] APF530; 60.2% [77/128] ondansetron). The most common cisplatin-based chemotherapy regimen in both treatment arms was cisplatin and gemcitabine (25.0% [31/124] APF530; 28.9% [37/128] ondansetron) (Suppl Table 1). Two patients in the APF530 arm and 3 patients in the ondansetron arm either received a lower cisplatin dose (<50 mg/m2) or did not go on to receive cisplatin as intended at randomization (Suppl Table 1). As previously reported, in the cisplatin stratum (Table 2), delayed-phase CR was numerically higher in the APF530 arm versus the ondansetron arm, with a corresponding treatment difference of 10.6% (65.3% [81/124] APF530; 54.7% [70/128] ondansetron; 95% CI [-1.4, 22.7]; P = .085). Although the CI contains 0, the result is consistent with the significant benefit observed in the overall study population (64.7% [291/450] APF530; 56.6% [256/452] ondansetron; 95% CI [1.7, 14.4];
P = .014).18 This more in-depth analysis found similar trends favoring the APF530 over the ondansetron regimen across overall- and acute-phase CR (Table 2).
A significantly greater proportion of patients in the APF530 arm, compared with the ondansetron arm, reported no rescue medication use during the delayed phase (74.4% [90/121] APF530; 62.6% [77/123] ondansetron; P = .048). Trends in favor of APF530 were observed in the overall phase (71.1% [86/121] APF530; 61.8% [76/123] ondansetron; P = .125) and acute phase (86.9% [106/122] APF530; 81.9% [104/127] ondansetron; P = .278). Time to first rescue medication use was consistently longer in the APF530 arm, compared with the ondansetron arm, although not statistically significantly (P = .150) (Figure 3).
The APF530 regimen was generally well tolerated in the cisplatin subgroup, and no new safety signals were identified (Table 3). Most patients experienced at least one TEAE. Excluding ISRs, TEAE incidences were 72.2% and 66.9% in the APF530 and ondansetron arms, respectively; most common were constipation, fatigue, nausea, diarrhea, dehydration, and headache. Excluding ISRs, the most common treatment-related TEAEs in the APF530 and ondansetron arms were constipation (2.4% and 2.3%, respectively and headache (3.2% and 4.6%).
Discussion
The MAGIC trial is the first phase 3 efficacy trial in the prevention of CINV in patients receiving HEC using the current guideline-recommended 3-drug antiemetic regimen in both treatment arms.18 Ondansetron was chosen as the appropriate 5-HT3 RA comparator because no other 5-HT3 RA has shown superiority to ondansetron in delayed-phase CINV following HEC. Furthermore, ondansetron is indicated for prevention of nausea and vomiting associated with initial and repeat courses of chemotherapy, including high-dose cisplatin.19 The MAGIC trial primary endpoint was met for the overall study population; in the context of a 3-drug regimen, APF530 demonstrated superior control of delayed-phase CINV following HEC compared with standard-of-care ondansetron.18 As reported previously, significant benefits were also observed with the APF530 regimen over the ondansetron regimen in terms of rescue medication use, patient satisfaction with antiemetic therapy, and nausea frequency in the overall study population.18
Cisplatin is generally regarded as one of the most emetogenic chemotherapeutic agents. For this reason, cisplatin is often evaluated separately in clinical trials, and was a stratification factor in the MAGIC trial. Consistent with the previously reported significant results,18 trends in the cisplatin stratum analysis favored the APF530 regimen, compared with the ondansetron regimen, in delayed- and overall-phase CR (treatment difference: 10.6%).18 Numerical trends presented here favoring the APF530 regimen over the ondansetron regimen were observed in CC and TR, two more stringent measures of CINV control that account for incidence of nausea. Furthermore, among women in the cisplatin stratum, a population at increased risk for CINV, the numerically higher CR, CC, and TR persisted in the APF530 arm, compared with the ondansetron arm.
The APF530 regimen was generally well tolerated in the cisplatin stratum, and no new safety signals were identified. The most common TEAEs were ISRs, mostly mild or moderate and resolving by study end. The double-dummy design resulted in ISRs in the ondansetron arm due to TEG-POE vehicle as the dummy APF530 injection. Transient ISRs have been observed with other agents administered SC, and are expected.20,21 Excluding ISRs, TEAEs were generally consistent with those observed for the 5-HT3 RA class.22
This analysis of patients randomized to receive cisplatin-based HEC in the MAGIC trial is exploratory and was not sufficiently powered to detect between-arm differences. Five total patients did not go on to receive cisplatin ≥50 mg/m2 as intended at randomization (2 APF530, 3 ondansetron); however, this is not uncommon in large clinical trials and represents less than 2% of patients in this analysis.
Recent phase 3 studies in patients receiving cisplatin-based HEC showed significant improvement in CINV prevention with the current guideline-recommended 3-drug regimen over the traditional 2-drug regimen (5-HT3 RA + dexamethasone).8,23 Results presented here, in a similar population receiving cisplatin-based HEC, suggest that in the context of a 3-drug antiemetic regimen in both treatment arms, APF530 provides additional benefit in CINV prevention compared with the standard of care, ondansetron. Furthermore, a recent phase 3 trial in patients receiving cisplatin or AC-based HEC demonstrated significant improvement in nausea when olanzapine was added to a traditional 3-drug regimen of a 5-HT3 RA, NK-1 RA, and dexamethasone.24 These compelling data support the addition of olanzapine as a fourth agent to the CINV treatment regimen to provide further control of nausea, which has been one of the more difficult components of CINV to control to date.
APF530 is the only 5-HT3 RA to demonstrate superiority over another as part of the guideline-recommended regimen in a 3-drug versus 3-drug phase 3 efficacy trial examining antiemetic efficacy following HEC. Results from the MAGIC trial, this exploratory analysis, and previous studies in MEC and HEC provide clinically meaningful benefits in preventing both acute- and delayed-phase CINV following guideline-specified MEC or HEC regimens. Consequently, APF530 was approved for use in combination with other antiemetics for prevention of acute and delayed nausea and vomiting associated with initial and repeat courses of MEC or AC combination chemotherapy regimens.13 Both the superior control of delayed-phase CINV following HEC demonstrated by the MAGIC trial18 and the consistent trends in the cisplatin stratum indicate a particular benefit for high-risk patients receiving high doses of cisplatin.
Acknowledgments
Joanna K Sandilos Rega, PhD, of SciStrategy Communications provided medical writing assistance, supported by Heron Therapeutics Inc, the maker of the study drug.
Despite available antiemetic therapies, chemotherapy-induced nausea and vomiting (CINV) following highly emetogenic chemotherapy (HEC), particularly in the delayed phase (>24-120 h after chemotherapy), continues to impair patient quality of life and chemotherapy compliance.1 Cisplatin-based chemotherapy, classified as HEC at any dose,2 is widely used to treat cancers such as non–small-cell and small
5-hydroxytryptamine type 3 (5-HT3) receptor antagonists (RAs; eg, granisetron, ondansetron, dolasetron, and palonosetron) have been the cornerstone of CINV therapy for decades and remain an integral part of contemporary antiemetic treatment regimens. Most current antiemetic guidelines for HEC recommend a 3-drug regimen, comprising a 5-HT3 RA, a neurokinin 1 (NK-1) RA, and a corticosteroid (dexamethasone).2,6,7 A regimen of olanzapine (antipsychotic), palonosetron (5-HT3 RA), and dexamethasone (corticosteroid) has been recommended as an alternative option. Recently, the oral fixed-dose combination of netupitant and palonosetron (NEPA) was approved and has shown efficacy in the cisplatin setting.8,9 However, the administration of oral medication to patients experiencing CINV and those with head and neck cancer may be difficult.10 Alternative antiemetic treatments that provide CINV control into the delayed phase and with a convenient route of administration, are needed.
APF530 is a novel extended-release granisetron formulation that provides sustained release of therapeutic concentrations for ≥5 days. The Biochronomer tri(ethylene glycol) poly(orthoester) (TEG-POE) vehicle releases granisetron slowly by polymer hydrolysis after it has been injected subcutaneously (SC) into the abdomen or upper arm.11,12 In 2016, the US Food and Drug Administration approved APF530 in combination with other antiemetics for the prevention of acute and delayed nausea and vomiting associated with initial and repeat courses of moderately emetogenic chemotherapy (MEC) or anthracycline plus cyclophosphamide (AC) combination chemotherapy regimens based on data from 2 pivotal phase 3 trials.13
A phase 3 trial demonstrated noninferiority of APF530 (500 mg, SC) to palonosetron (0.25 mg, intravenously [IV]), each with dexamethasone (corticosteroid), in the control of acute-phase CINV after MEC or HEC, and delayed-phase CINV after MEC (classified by Hesketh criteria).14,15 Furthermore, APF530 provided sustained CINV control over multiple cycles of chemotherapy.16 Numerically higher complete response (CR: no emesis, no rescue medication use) rates were observed with APF530, compared with palonosetron, in the delayed phase after HEC (APF530 500 mg, 67.1%; palonosetron 0.25 mg, 64.3%).15
A reanalysis of study endpoints by newer emetogenicity classification guidelines from the American Society of Clinical Oncology (ASCO)7 maintained overall study conclusions.17 Notably, the numerically higher CR rates with APF530 in the delayed phase following HEC were enhanced (APF530 500 mg, 55.8%; palonosetron 0.25 mg, 50.5%), suggesting a need for further examination in this setting. The subsequent APF530 phase 3 MAGIC trial (Modified Absorption of Granisetron In the prevention of CINV; NCT02106494), compared APF530 (500 mg, SC) with ondansetron (0.15 mg/kg, IV), each with fosaprepitant (NK-1 RA) and dexamethasone in patients receiving HEC. The primary endpoint was met: the APF530 regimen demonstrated superior delayed-phase CR compared with the ondansetron regimen (64.7% vs 56.6%; 95% confidence interval [CI]:
1.7-14.4; P = .014; 8.0% absolute improvement).18
APF530 also demonstrated a significant benefit over ondansetron for other endpoints including nausea control, rescue medication use, and satisfaction with antiemetic therapy.18 APF530 is the first and only 5-HT3 RA to demonstrate superiority over another in a phase 3 efficacy trial using a guideline-recommended 3-drug regimen for both arms.
A prespecified MAGIC trial analysis of the primary endpoint by intent to receive cisplatin (≥50 mg/m2, Yes/No) demonstrated a pronounced treatment benefit in terms of delayed-phase CR rates with the APF530 regimen among patients in the cisplatin (≥50 mg/m2, Yes) stratum (CR: 65.3% vs 54.7%; 95% CI: -1.4-22.7; 10.6% absolute improvement).18 These results are compelling, since cisplatin represents a particularly emetogenic class of chemotherapy; a more in-depth analysis of additional MAGIC trial endpoints for these patients would be of clinical interest, and is presented here. Efficacy endpoints in this analysis include CR in the overall and acute phases, complete control (CC) and total response (TR) rates, rescue medication use, nausea frequency, and safety.
Methods
Study design and patients
The MAGIC trial was a prospective, randomized, multicenter, placebo-controlled, double-blind, double-dummy phase 3 trial conducted at 77 centers across the United States. The study protocol was reviewed and approved by the institutional review board at each participating center, and conducted according to the Declaration of Helsinki. The study design, previously presented in detail,18 is reviewed briefly here.
Eligible men and women were 18-80 years of age with histologically or cytologically confirmed malignancy (cancer type information was not captured) and were entering the first cycle of their single-day HEC treatment (defined by ASCO 2011 emetogenicity criteria).7 Patients had Eastern Cooperative Oncology Group Performance Status (ECOG-PS) of 0 or 1, no history or presence of significant cardiac disease or QT interval prolongation, and adequate bone marrow, kidney, and liver function. All patients provided written informed consent.
Procedures
Patients were stratified by planned receipt of the cisplatin regimen ≥50 mg/m2 (Yes/No), randomized 1:1 to receive APF530 500 mg SC (granisetron 10 mg) or ondansetron 0.15 mg/kg IV (up to a maximum of 16 mg as a single dose) on day 1 (Figure 1). The APF530 arm received the ondansetron saline placebo, and the ondansetron arm received the APF530 SC placebo containing the TEG-POE vehicle. All patients were scheduled to receive fosaprepitant 150 mg IV and dexamethasone 12 mg IV on day 1, then oral dexamethasone 8 mg once daily on day 2 and 8 mg twice daily on days 3 and 4. Rescue medication was allowed at the investigator’s discretion.
Outcomes
The primary objective of the trial was to demonstrate the superiority of APF530 500 mg SC compared with ondansetron 0.15 mg/kg IV, as part of the current guideline-recommended 3-drug regimen, in preventing delayed-phase CINV after HEC. The primary endpoint was delayed-phase (24-120 h) CR (no emetic episodes [vomit or retch] and no rescue medication use). In addition, a prespecified analysis of delayed-phase CR by randomization strata (planned use of cisplatin) was performed.
Secondary and other endpoints included overall-phase CR
(0-120 h); delayed-, overall-, and acute-phase complete control (CC: CR and no more than mild nausea); delayed-, overall-, and acute-phase total response (TR; CR and no nausea); and rescue medication use. A post hoc analysis of nausea severity was also conducted. Safety assessments included treatment-emergent adverse events (TEAEs), injection-site reactions (ISRs), laboratory parameters, and vital signs. TEAEs were assessed by type, duration, severity, and relationship to study drug. ISR timing and severity were captured in patient diaries.
Statistical analysis
All efficacy analyses were conducted using the modified intent-to-treat population (mITT; randomized patients who received study drug and a HEC regimen and had post-baseline efficacy data). Safety assessments were performed on the safety population (randomized patients who received study drug).
This analysis conducted on the subgroup of patients with intent to receive cisplatin (cisplatin randomization stratum, ≥50 mg/m2, Yes) was exploratory and was not powered to detect treatment differences. Preplanned analyses compared CR, CC, and TR rates across treatment arms using 95% CIs.
Post hoc analyses of time to first rescue medication use, proportion of patients with rescue medication use, and less frequent nausea were performed. All P values were calculated using the Cochran-Mantel-Haenszel chi square test. Rescue medication use results were based on observed data, without imputation for missing results (ie, calculated from the number of patients with a response). Further analyses of efficacy endpoints CR, CC, and TR in the subset of female patients in the cisplatin randomization stratum were performed. Safety assessments were summarized descriptively.
Results
A total of 942 patients were randomized across 77 US centers during March 31, 2014 and May 15, 2015 (471 APF530, 471 ondansetron). Among those, 264 had intent to receive cisplatin and were included in the cisplatin randomization stratum (≥50 mg/m2, Yes) (Figure 2). A total of 256 patients in the cisplatin stratum received study drug and were included in the safety population (126 APF530, 130 ondansetron); 252 patients were included in the mITT population (124 APF530, 128 ondansetron).
Baseline demographics were generally balanced between treatment arms (Table 1). The proportion of female patients was 41.1% (51/124) and 48.4% (62/128) in the APF530 and ondansetron arms, respectively. The majority of patients had an ECOG PS of 0 (57.3% [71/124] APF530; 60.2% [77/128] ondansetron). The most common cisplatin-based chemotherapy regimen in both treatment arms was cisplatin and gemcitabine (25.0% [31/124] APF530; 28.9% [37/128] ondansetron) (Suppl Table 1). Two patients in the APF530 arm and 3 patients in the ondansetron arm either received a lower cisplatin dose (<50 mg/m2) or did not go on to receive cisplatin as intended at randomization (Suppl Table 1). As previously reported, in the cisplatin stratum (Table 2), delayed-phase CR was numerically higher in the APF530 arm versus the ondansetron arm, with a corresponding treatment difference of 10.6% (65.3% [81/124] APF530; 54.7% [70/128] ondansetron; 95% CI [-1.4, 22.7]; P = .085). Although the CI contains 0, the result is consistent with the significant benefit observed in the overall study population (64.7% [291/450] APF530; 56.6% [256/452] ondansetron; 95% CI [1.7, 14.4];
P = .014).18 This more in-depth analysis found similar trends favoring the APF530 over the ondansetron regimen across overall- and acute-phase CR (Table 2).
A significantly greater proportion of patients in the APF530 arm, compared with the ondansetron arm, reported no rescue medication use during the delayed phase (74.4% [90/121] APF530; 62.6% [77/123] ondansetron; P = .048). Trends in favor of APF530 were observed in the overall phase (71.1% [86/121] APF530; 61.8% [76/123] ondansetron; P = .125) and acute phase (86.9% [106/122] APF530; 81.9% [104/127] ondansetron; P = .278). Time to first rescue medication use was consistently longer in the APF530 arm, compared with the ondansetron arm, although not statistically significantly (P = .150) (Figure 3).
The APF530 regimen was generally well tolerated in the cisplatin subgroup, and no new safety signals were identified (Table 3). Most patients experienced at least one TEAE. Excluding ISRs, TEAE incidences were 72.2% and 66.9% in the APF530 and ondansetron arms, respectively; most common were constipation, fatigue, nausea, diarrhea, dehydration, and headache. Excluding ISRs, the most common treatment-related TEAEs in the APF530 and ondansetron arms were constipation (2.4% and 2.3%, respectively and headache (3.2% and 4.6%).
Discussion
The MAGIC trial is the first phase 3 efficacy trial in the prevention of CINV in patients receiving HEC using the current guideline-recommended 3-drug antiemetic regimen in both treatment arms.18 Ondansetron was chosen as the appropriate 5-HT3 RA comparator because no other 5-HT3 RA has shown superiority to ondansetron in delayed-phase CINV following HEC. Furthermore, ondansetron is indicated for prevention of nausea and vomiting associated with initial and repeat courses of chemotherapy, including high-dose cisplatin.19 The MAGIC trial primary endpoint was met for the overall study population; in the context of a 3-drug regimen, APF530 demonstrated superior control of delayed-phase CINV following HEC compared with standard-of-care ondansetron.18 As reported previously, significant benefits were also observed with the APF530 regimen over the ondansetron regimen in terms of rescue medication use, patient satisfaction with antiemetic therapy, and nausea frequency in the overall study population.18
Cisplatin is generally regarded as one of the most emetogenic chemotherapeutic agents. For this reason, cisplatin is often evaluated separately in clinical trials, and was a stratification factor in the MAGIC trial. Consistent with the previously reported significant results,18 trends in the cisplatin stratum analysis favored the APF530 regimen, compared with the ondansetron regimen, in delayed- and overall-phase CR (treatment difference: 10.6%).18 Numerical trends presented here favoring the APF530 regimen over the ondansetron regimen were observed in CC and TR, two more stringent measures of CINV control that account for incidence of nausea. Furthermore, among women in the cisplatin stratum, a population at increased risk for CINV, the numerically higher CR, CC, and TR persisted in the APF530 arm, compared with the ondansetron arm.
The APF530 regimen was generally well tolerated in the cisplatin stratum, and no new safety signals were identified. The most common TEAEs were ISRs, mostly mild or moderate and resolving by study end. The double-dummy design resulted in ISRs in the ondansetron arm due to TEG-POE vehicle as the dummy APF530 injection. Transient ISRs have been observed with other agents administered SC, and are expected.20,21 Excluding ISRs, TEAEs were generally consistent with those observed for the 5-HT3 RA class.22
This analysis of patients randomized to receive cisplatin-based HEC in the MAGIC trial is exploratory and was not sufficiently powered to detect between-arm differences. Five total patients did not go on to receive cisplatin ≥50 mg/m2 as intended at randomization (2 APF530, 3 ondansetron); however, this is not uncommon in large clinical trials and represents less than 2% of patients in this analysis.
Recent phase 3 studies in patients receiving cisplatin-based HEC showed significant improvement in CINV prevention with the current guideline-recommended 3-drug regimen over the traditional 2-drug regimen (5-HT3 RA + dexamethasone).8,23 Results presented here, in a similar population receiving cisplatin-based HEC, suggest that in the context of a 3-drug antiemetic regimen in both treatment arms, APF530 provides additional benefit in CINV prevention compared with the standard of care, ondansetron. Furthermore, a recent phase 3 trial in patients receiving cisplatin or AC-based HEC demonstrated significant improvement in nausea when olanzapine was added to a traditional 3-drug regimen of a 5-HT3 RA, NK-1 RA, and dexamethasone.24 These compelling data support the addition of olanzapine as a fourth agent to the CINV treatment regimen to provide further control of nausea, which has been one of the more difficult components of CINV to control to date.
APF530 is the only 5-HT3 RA to demonstrate superiority over another as part of the guideline-recommended regimen in a 3-drug versus 3-drug phase 3 efficacy trial examining antiemetic efficacy following HEC. Results from the MAGIC trial, this exploratory analysis, and previous studies in MEC and HEC provide clinically meaningful benefits in preventing both acute- and delayed-phase CINV following guideline-specified MEC or HEC regimens. Consequently, APF530 was approved for use in combination with other antiemetics for prevention of acute and delayed nausea and vomiting associated with initial and repeat courses of MEC or AC combination chemotherapy regimens.13 Both the superior control of delayed-phase CINV following HEC demonstrated by the MAGIC trial18 and the consistent trends in the cisplatin stratum indicate a particular benefit for high-risk patients receiving high doses of cisplatin.
Acknowledgments
Joanna K Sandilos Rega, PhD, of SciStrategy Communications provided medical writing assistance, supported by Heron Therapeutics Inc, the maker of the study drug.
1. Hilarius DL, Kloeg PH, van der Wall E, van den Heuvel JJ, Gundy CM, Aaronson NK. Chemotherapy-induced nausea and vomiting in daily clinical practice: a community hospital-based study. Support Care Cancer. 2012;20:107-117.
2. NCCN clinical practice guidelines in oncology: antiemesis—version 1.2017. https://www.nccn.org/professionals/physician_gls/f_guidelines.asp#antiemesis. Accessed March 1, 2017.
3. Platinol (cisplatin for injection, USP) [prescribing information]. Princeton, NJ: Bristol-Myers Squibb Company; 2010. http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/018057s079lbl.pdf. Updated May 2010. Accessed September 12, 2016.
4. Pollera CF, Giannarelli D. Prognostic factors influencing cisplatin-induced emesis. Definition and validation of a predictive logistic model. Cancer. 1989;64:1117-1122.
5. Roila F, Boschetti E, Tonato M, et al. Predictive factors of delayed emesis in cisplatin-treated patients and antiemetic activity and tolerability of metoclopramide or dexamethasone. A randomized single-blind study. Am J Clin Oncol. 1991;14:238-242.
6. Roila F, Molassiotis A, Herrstedt J, et al. 2016 MASCC and ESMO guideline update for the prevention of chemotherapy- and radiotherapy-induced nausea and vomiting and of nausea and vomiting in advanced cancer patients. Ann Oncol. 2016;27(suppl 5):v119-v133.
7. Basch E, Prestrud AA, Hesketh PJ, et al. Antiemetics: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol. 2011;29:4189-4198.
8. Hesketh PJ, Rossi G, Rizzi G, et al. Efficacy and safety of NEPA, an oral combination of netupitant and palonosetron, for prevention of chemotherapy-induced nausea and vomiting following highly emetogenic chemotherapy: a randomized dose-ranging pivotal study. Ann Oncol. 2014;25:1340-1346.
9. Akynzeo (netupitant and palonosetron capsules) [prescribing information]. Woodcliff, NJ: Eisai; 2015. http://www.akynzeo.com. Revised April 2015. Accessed September 12, 2016.
10. Tsukahara K, Nakamura K, Motohashi R, et al. Antiemetic therapy of fosaprepitant, palonosetron, and dexamethasone combined with cisplatin-based chemotherapy for head and neck carcinomas. Acta Otolaryngol. 2014;134:1198-1204.
11. Ottoboni. Biochronomer technology and the development of APF530, a sustained release formulation of granisetron. J Exp Pharmacol. 2014;6:15-21.
12. Gabrail N, Yanagihara R, Spaczynski M, et al. Pharmacokinetics, safety, and efficacy of APF530 (extended-release granisetron) in patients receiving moderately or highly emetogenic chemotherapy: results of two phase II trials. Cancer Manag Res. 2015;7:83-92.
13. Sustol (granisetron) extended-release injection, for subcutaneous use [prescribing information]. Redwood City, CA; Heron Therapeutics; 2016. http://sustol.com/hcp/healthcare-professionals. Updated August 2016. Accessed September 12, 2016.
14. Hesketh PJ, Kris MG, Grunberg SM, et al. Proposal for classifying the acute emetogenicity of cancer chemotherapy. J Clin Oncol. 1997;15:103-109.
15. Raftopoulos H, Cooper W, O’Boyle E, Gabrail N, Boccia R, Gralla RJ. Comparison of an extended-release formulation of granisetron (APF530) versus palonosetron for the prevention of chemotherapy-induced nausea and vomiting associated with moderately or highly emetogenic chemotherapy: results of a prospective, randomized, double-blind, noninferiority phase 3 trial. Support Care Cancer. 2015;23:723-732.
16. Boccia RV, Cooper W, O’Boyle E. Sustained antiemetic responses with APF530 (sustained-release granisetron) during multiple cycles of emetogenic chemotherapy. J Community Support Oncol. 2015;13:38-46.
17. Raftopoulos H, Boccia R, Cooper W, O’Boyle E, Gralla RJ. Slow-release granisetron (APF530) versus palonosetron for chemotherapy-induced nausea/vomiting: analysis by American Society of Clinical Oncology emetogenicity criteria. Future Oncol. 2015;11:2541-2551.
18. Schnadig ID, Agajanian R, Dakhil C, et al. APF530 (Granisetron injection extended-release) in a three-drug regimen for delayed CINV in highly emetogenic chemotherapy. Future Oncol. 2016;12:1469-1481.
19. Zofran (ondansetron hydrochloride) injection for intravenous use. Research Triangle Park, NC: GlaxoSmithKline; 2014. http://www.pharma.us.novartis.com/product/pi/pdf/zofran_inj.pdf. Revised September 2014. Accessed September 12, 2016.
20. Eligard (luprolide acetate) kit for subcutaneous use [prescribing information]. Fort Collins, CO: Tolmar Pharmaceuticals Inc; 2017. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=b78d1919-9dee-44fa-90f9-e0a26d32481d. Revised January 2017. Accessed March 1, 2017.
21. Sandostatin LAR Depot (octreotide acetate for injectable suspension) [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2016. https://www.pharma.us.novartis.com/product/pi/pdf/sandostatin_lar.pdf. Revised July 2016. Accessed September 12, 2016.
22. Navari RM. Management of chemotherapy-induced nausea and vomiting: focus on newer agents and new uses for older agents. Drugs. 2013;73:249-262.
23. Rapoport BL, Chasen MR, Gridelli C, et al. Safety and efficacy of rolapitant for prevention of chemotherapy-induced nausea and vomiting after administration of cisplatin-based highly emetogenic chemotherapy in patients with cancer: two randomised, active-controlled, double-blind, phase 3 trials. Lancet Oncol. 2015;16:1079-1089.
24. Navari RM, Qin R, Ruddy KJ, et al. Olanzapine for the prevention of chemotherapy-induced nausea and vomiting. N Engl J Med. 2016;375:134-142.
1. Hilarius DL, Kloeg PH, van der Wall E, van den Heuvel JJ, Gundy CM, Aaronson NK. Chemotherapy-induced nausea and vomiting in daily clinical practice: a community hospital-based study. Support Care Cancer. 2012;20:107-117.
2. NCCN clinical practice guidelines in oncology: antiemesis—version 1.2017. https://www.nccn.org/professionals/physician_gls/f_guidelines.asp#antiemesis. Accessed March 1, 2017.
3. Platinol (cisplatin for injection, USP) [prescribing information]. Princeton, NJ: Bristol-Myers Squibb Company; 2010. http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/018057s079lbl.pdf. Updated May 2010. Accessed September 12, 2016.
4. Pollera CF, Giannarelli D. Prognostic factors influencing cisplatin-induced emesis. Definition and validation of a predictive logistic model. Cancer. 1989;64:1117-1122.
5. Roila F, Boschetti E, Tonato M, et al. Predictive factors of delayed emesis in cisplatin-treated patients and antiemetic activity and tolerability of metoclopramide or dexamethasone. A randomized single-blind study. Am J Clin Oncol. 1991;14:238-242.
6. Roila F, Molassiotis A, Herrstedt J, et al. 2016 MASCC and ESMO guideline update for the prevention of chemotherapy- and radiotherapy-induced nausea and vomiting and of nausea and vomiting in advanced cancer patients. Ann Oncol. 2016;27(suppl 5):v119-v133.
7. Basch E, Prestrud AA, Hesketh PJ, et al. Antiemetics: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol. 2011;29:4189-4198.
8. Hesketh PJ, Rossi G, Rizzi G, et al. Efficacy and safety of NEPA, an oral combination of netupitant and palonosetron, for prevention of chemotherapy-induced nausea and vomiting following highly emetogenic chemotherapy: a randomized dose-ranging pivotal study. Ann Oncol. 2014;25:1340-1346.
9. Akynzeo (netupitant and palonosetron capsules) [prescribing information]. Woodcliff, NJ: Eisai; 2015. http://www.akynzeo.com. Revised April 2015. Accessed September 12, 2016.
10. Tsukahara K, Nakamura K, Motohashi R, et al. Antiemetic therapy of fosaprepitant, palonosetron, and dexamethasone combined with cisplatin-based chemotherapy for head and neck carcinomas. Acta Otolaryngol. 2014;134:1198-1204.
11. Ottoboni. Biochronomer technology and the development of APF530, a sustained release formulation of granisetron. J Exp Pharmacol. 2014;6:15-21.
12. Gabrail N, Yanagihara R, Spaczynski M, et al. Pharmacokinetics, safety, and efficacy of APF530 (extended-release granisetron) in patients receiving moderately or highly emetogenic chemotherapy: results of two phase II trials. Cancer Manag Res. 2015;7:83-92.
13. Sustol (granisetron) extended-release injection, for subcutaneous use [prescribing information]. Redwood City, CA; Heron Therapeutics; 2016. http://sustol.com/hcp/healthcare-professionals. Updated August 2016. Accessed September 12, 2016.
14. Hesketh PJ, Kris MG, Grunberg SM, et al. Proposal for classifying the acute emetogenicity of cancer chemotherapy. J Clin Oncol. 1997;15:103-109.
15. Raftopoulos H, Cooper W, O’Boyle E, Gabrail N, Boccia R, Gralla RJ. Comparison of an extended-release formulation of granisetron (APF530) versus palonosetron for the prevention of chemotherapy-induced nausea and vomiting associated with moderately or highly emetogenic chemotherapy: results of a prospective, randomized, double-blind, noninferiority phase 3 trial. Support Care Cancer. 2015;23:723-732.
16. Boccia RV, Cooper W, O’Boyle E. Sustained antiemetic responses with APF530 (sustained-release granisetron) during multiple cycles of emetogenic chemotherapy. J Community Support Oncol. 2015;13:38-46.
17. Raftopoulos H, Boccia R, Cooper W, O’Boyle E, Gralla RJ. Slow-release granisetron (APF530) versus palonosetron for chemotherapy-induced nausea/vomiting: analysis by American Society of Clinical Oncology emetogenicity criteria. Future Oncol. 2015;11:2541-2551.
18. Schnadig ID, Agajanian R, Dakhil C, et al. APF530 (Granisetron injection extended-release) in a three-drug regimen for delayed CINV in highly emetogenic chemotherapy. Future Oncol. 2016;12:1469-1481.
19. Zofran (ondansetron hydrochloride) injection for intravenous use. Research Triangle Park, NC: GlaxoSmithKline; 2014. http://www.pharma.us.novartis.com/product/pi/pdf/zofran_inj.pdf. Revised September 2014. Accessed September 12, 2016.
20. Eligard (luprolide acetate) kit for subcutaneous use [prescribing information]. Fort Collins, CO: Tolmar Pharmaceuticals Inc; 2017. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=b78d1919-9dee-44fa-90f9-e0a26d32481d. Revised January 2017. Accessed March 1, 2017.
21. Sandostatin LAR Depot (octreotide acetate for injectable suspension) [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2016. https://www.pharma.us.novartis.com/product/pi/pdf/sandostatin_lar.pdf. Revised July 2016. Accessed September 12, 2016.
22. Navari RM. Management of chemotherapy-induced nausea and vomiting: focus on newer agents and new uses for older agents. Drugs. 2013;73:249-262.
23. Rapoport BL, Chasen MR, Gridelli C, et al. Safety and efficacy of rolapitant for prevention of chemotherapy-induced nausea and vomiting after administration of cisplatin-based highly emetogenic chemotherapy in patients with cancer: two randomised, active-controlled, double-blind, phase 3 trials. Lancet Oncol. 2015;16:1079-1089.
24. Navari RM, Qin R, Ruddy KJ, et al. Olanzapine for the prevention of chemotherapy-induced nausea and vomiting. N Engl J Med. 2016;375:134-142.
Patterns of care with regard to whole-brain radiotherapy technique and delivery among academic centers in the United States
Despite the recent advances in systemic therapy, metastatic spread to the brain continues to be the most common neurologic complication of many cancers. The clinical incidence of brain metastases varies with primary cancer diagnosis, with estimates ranging from 1.2%-19.8%.1,2 Metastatic spread to the brain is even more prevalent at autopsy, with evidence of intracranial tumor being found in 26% of patients in some series.3 It is possible that the clinical incidence of metastatic disease to the brain will continue to increase as newer therapeutic agents improve survival and imaging techniques continue to improve.
The management of brain metastases has changed rapidly as technological improvements have made treatment increasingly safe and efficacious. Traditionally, treatment consisted of radiotherapy to the whole brain, with or without surgical resection.4,5 More recently, stereotactic radiosurgery (SRS) has been adopted on the basis of evidence that it is safe and efficacious alone or in combination with radiotherapy to the whole brain.6 Further evidence is emerging that neurocognitive outcomes are improved when whole-brain radiotherapy (WBRT) is omitted, which possibly contributes to improved patient quality of life.7 Taking into account this and other data, the American Society for Radiation Oncology’s Choosing Wisely campaign now recommends not routinely adding WBRT to radiosurgery in patients with limited brain metastases.8
Despite this recommendation, many patients continue to benefit from WBRT, and it remains a common treatment in radiation oncology clinics across the US for several reasons. Many patients present with multiple brain metastases and are ineligible for radiosurgery. Even for technically eligible patients, WBRT has been shown to improve local control and decrease the rate of distant brain failure over radiosurgery alone.6 With higher rates of subsequent failures, patients receiving radiosurgery alone must adhere to more rigorous follow-up and imaging schedules, which can be difficult for many rural patients who have to travel long distances to centers. Furthermore, there is some suggestion that this decreased failure rate may result in improved survival in highly selected patients with excellent disease and performance status.9 Controversies exist, however, and strong institutional biases persist, contributing to significant differences in practice. We surveyed academic radiation oncologists and in an effort to identify and describe practice patterns in the delivery of WBRT at academic centers.
Methods
We conducted a thorough review of available literature on radiation for brain metastases and based on our findings, devised a survey 19 questions to ascertain practice patterns and treatment delivery among US academic physicians (Table 1). After obtaining institutional review board approval to do the study, we sent the survey to program coordinators at radiation oncology programs that are accredited by the Accreditation Council for Graduate Medical Education. We instructed coordinators to e-mail the survey to their practicing resident and attending physicians. The surveys were created using SurveyMonkey software. We obtained informed consent from the providers. A total of 3 follow-up e-mails were sent to each recipient of the survey to solicit responses, similar to the Dillman Total Design Survey Method.10
SPSS version 22.0 was used to analyze the data in an exploratory fashion. Statistical methods were used to assess the association of demographic data with SRS and WBRT delivery and treatment technique items when the analyses involved percentages that included the Pearson chi-square statistic and the chi-square test for linear trend. When the analysis focused on ranking data, the Kruskal-Wallis test, Mann-Whitney U test, the Jonckheere-Terpstra and the Kendall tau-b rank correlation were used as appropriate. If there were small sample sizes within some groups, then exact significant levels were assessed. Statistical significance was set by convention at P < .05.
Results
We received 95 responses of which 87 were considered complete for analysis. Forty-seven percent of the 87 respondents were not board-certified, and the remainder had passed their radiobiology and physics boards exams. A majority of respondents (70%, 61 of 87) were physicians who had been in practice for ≤5 years. Fifty-four percent of respondents were located in the Northeast US, 22% in the South, 14% in the West, and 10% in the Midwest and Hawaii (Table 2).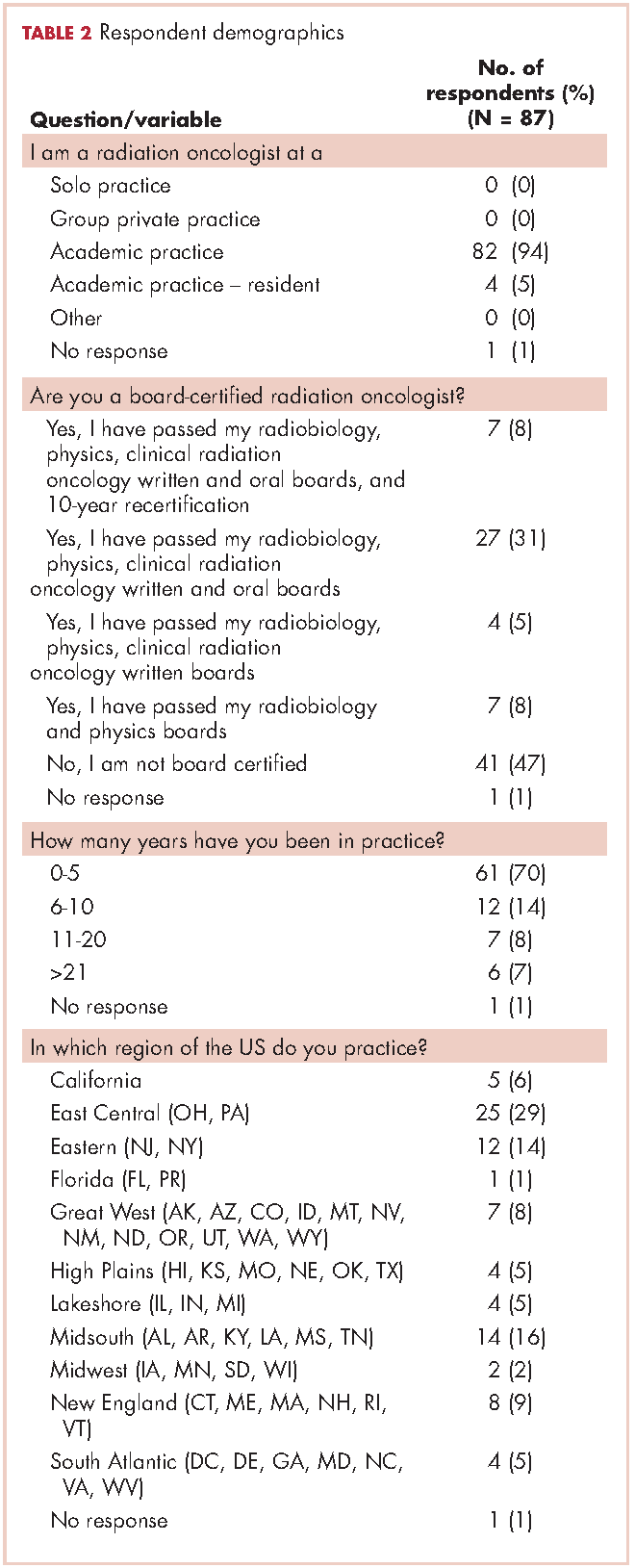
We used the chi-square test for linear trends to assess for a relationship between years of practice and whether respondents deviated from their typical method of WBRT therapy when treating more radioresistant tumors (melanoma, renal cell carcinoma). Respondents were classified by years in practice: 0-5, 6-10, 11-20, and >21 years. The results showed a linear association, with those in practice for longer periods more likely to use SRS alone, P = .027 (Figure 1).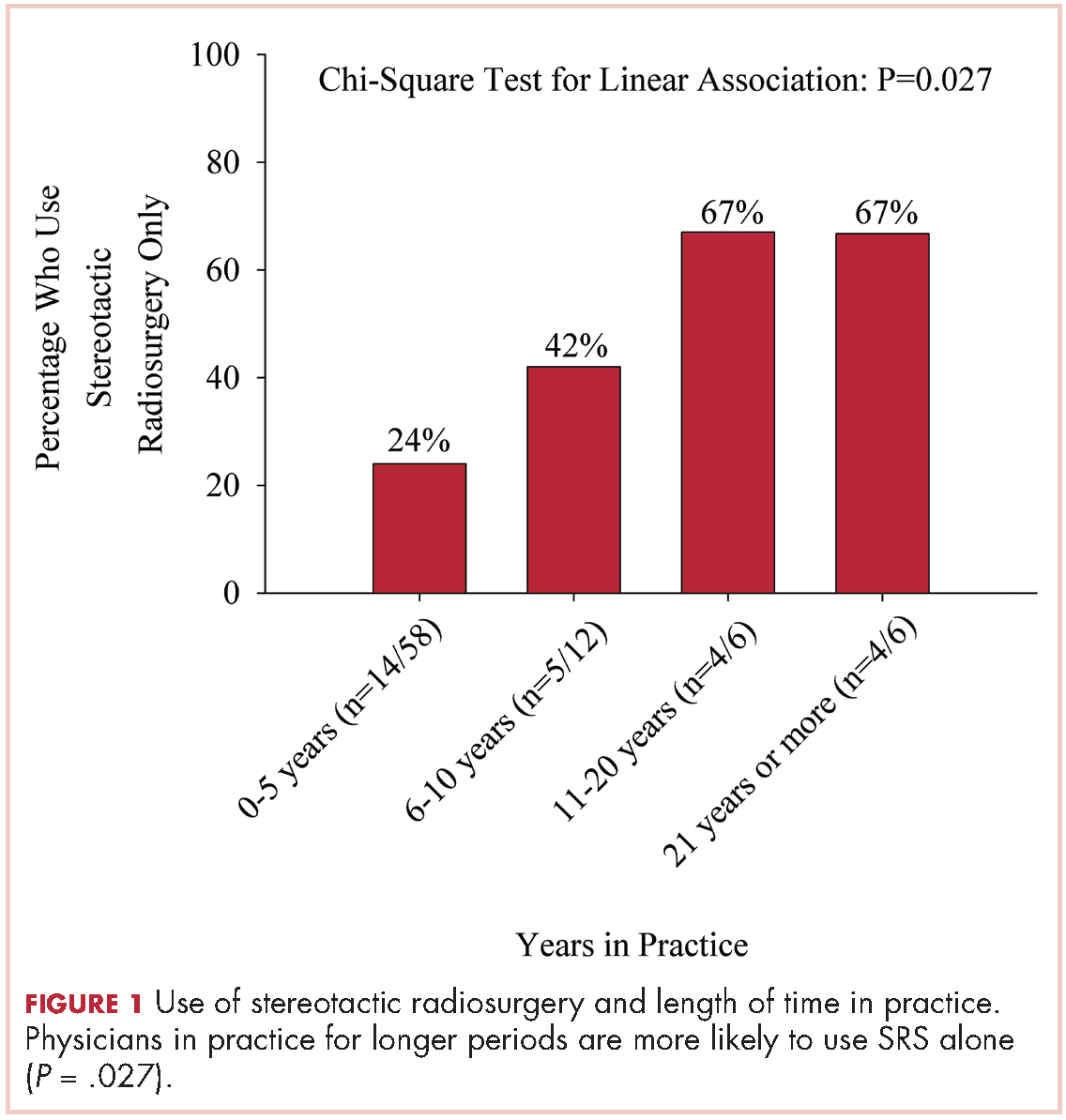
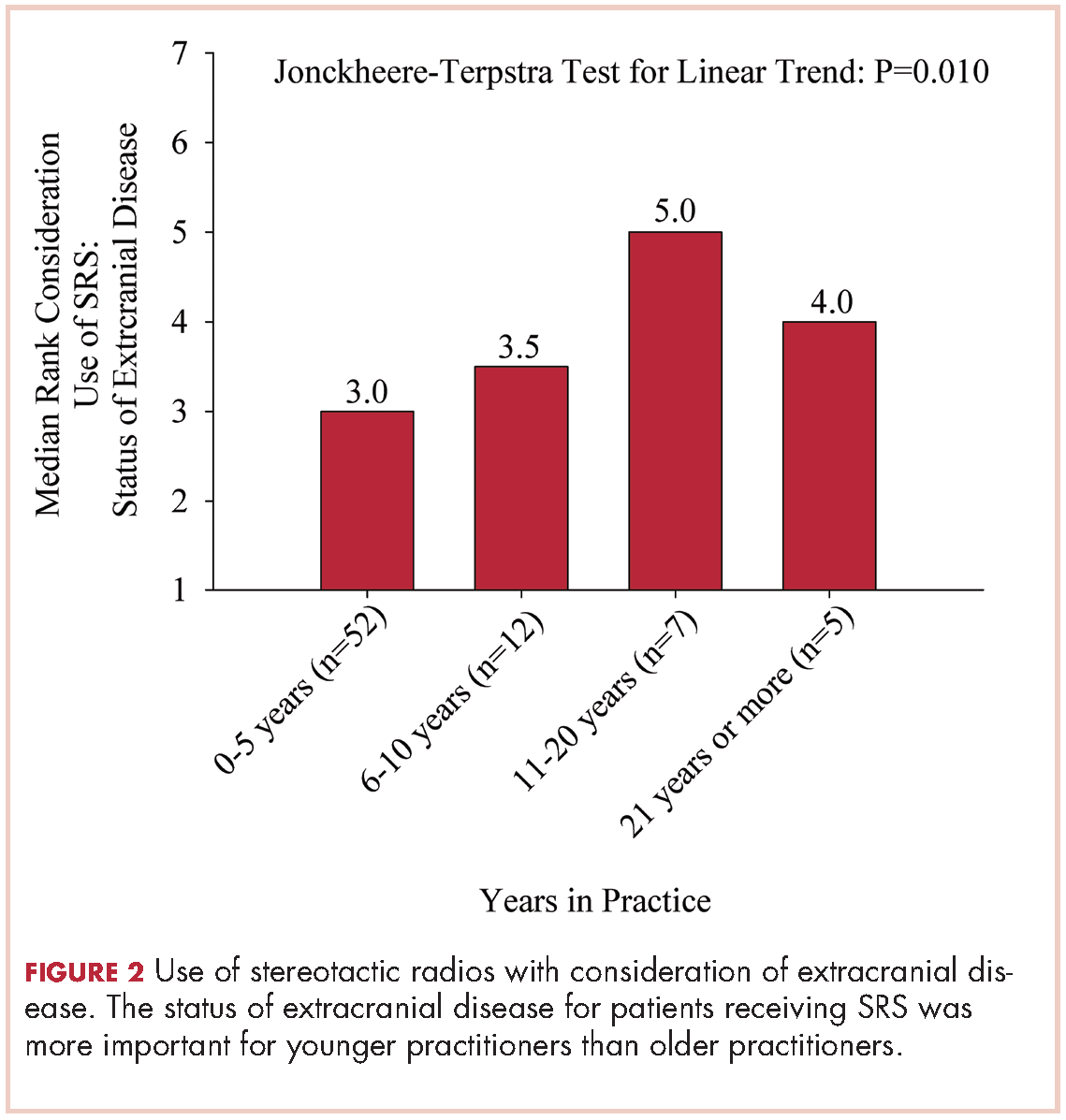
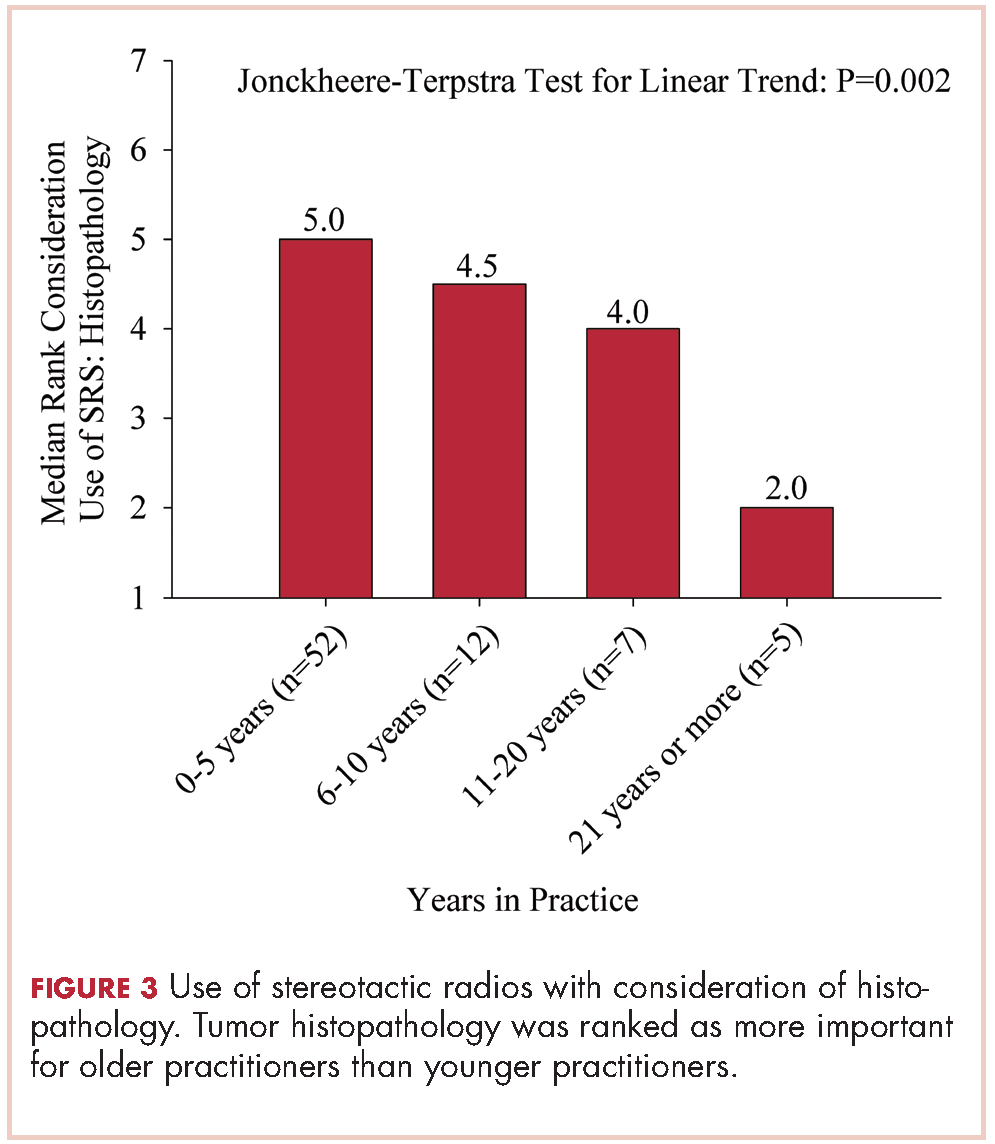
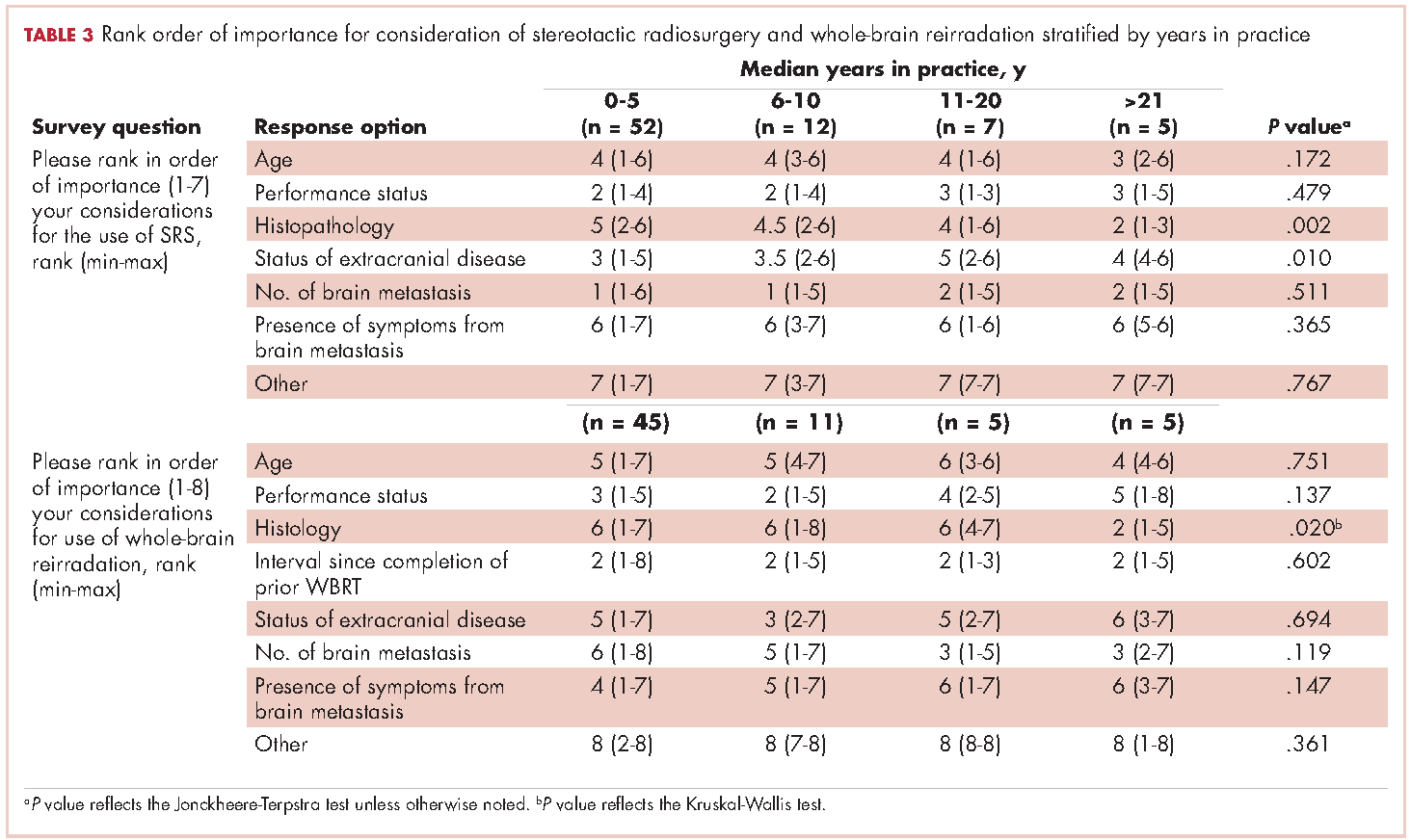
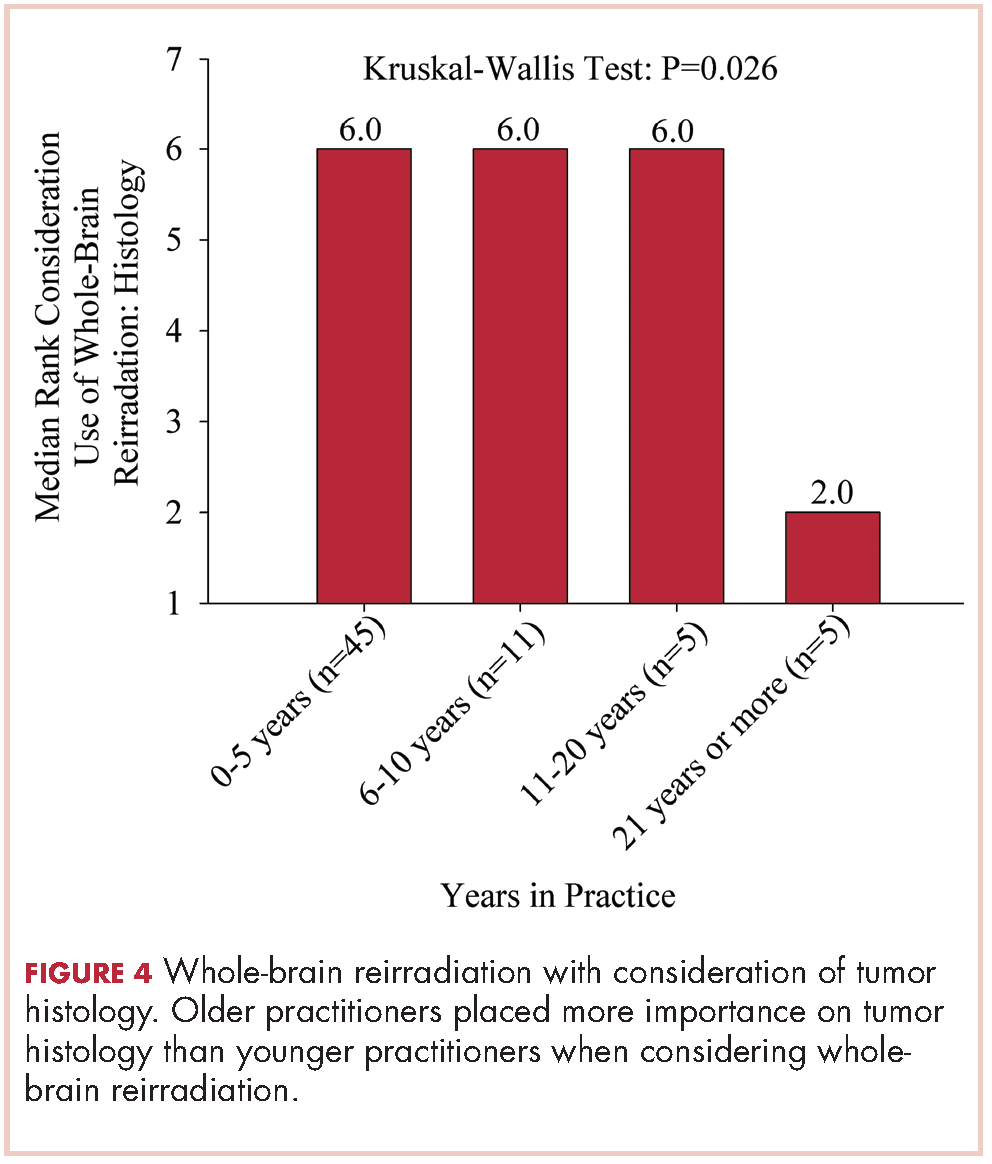
Discussion
The incidence of brain metastases is increasing because of improvements in diagnostic imaging techniques and advancements in systemic therapy control of extracranial disease but not of intracranial disease or metastasis, because therapies do not cross the blood-brain barrier.11,12 Brain metastases are the most common type of brain tumor. Given that most chemotherapeutic agents cannot cross the blood-brain barrier, radiotherapy is considered a means of treatment and of controlling brain metastases. Early data from the 1950s13 and 1960s14 have suggested clinical improvement with brain radiation, making radiotherapy the cornerstone for treatment of brain metastases.
The Radiation Therapy Oncology Group (RTOG) has evaluated several fractionation schedules, with 5 schemas evaluated by the RTOG 6901 and 7361 studies: 30 Gy in 10 fractions, 30 Gy in 15 fractions, 40 Gy in 15 fractions, 40 Gy in 20 fractions, and 20 Gy in 5 fractions. The combined results from these two trials showed that outcomes were similar for patients treated with a shorter regimen than for those treated with a more protracted schedule. In our study, respondents reported that they most frequently treated brain metastases to a total dose of 30 Gy in 10 fractions. Given the results of the aforementioned RTOG trials and practice patterns among academic physicians, we recommend all practitioners consider a shorter hypofractioned course when treating brain metastases with WBRT. This will also reduce delays for patients who are likely to benefit greatly from earlier enrollment into hospice care, because protracted radiation schedules typically are not covered while a patient is in hospice.
Pharmacologic management for patients with brain metastases is important for symptomatic improvement. Glucocorticoids are important for palliation of symptoms from edema and increased intracranial pressure.15 However, steroids have a multitude of side effects and their use in asymptomatic patients is unnecessary. Improvements in imaging and detection11 have allowed us to find smaller and asymptomatic brain tumors. In our survey, it was promising to see a change in former practice patterns, with only 8% of academic practitioners regularly prescribing steroids to all of their patients receiving whole-brain radiation.
Diminished cognitive function and short-term memory loss are troublesome side effects of WBRT. As cancer patients live longer, such cognitive dysfunction will become more than just a nuisance. The RTOG has investigated the use of prophylactic memantine for patients receiving whole-brain radiation to determine if it would aid in the preservation of cognition. It found that patients who received memantine did better and had delayed time to cognitive decline and a reduced rate of memory decline, executive function, and processing speed.16 In our study, about a third of practitioners prescribed memantine and it was reserved for patients who had an otherwise favorable prognosis.
The RTOG has also investigated adjusting treatment technique for patients who receive WBRT. RTOG 0933 was a phase 2 trial that evaluated hippocampal avoidance during deliverance of WBRT with intensity-modulated radiation therapy (IMRT). Results showed that avoiding the hippocampus during WBRT was associated with improved memory preservation and patient quality of life.17 In a survey of practicing radiation oncologists in the US, most reported that they did not use memantine or IMRT for hippocampal sparing when delivering whole-brain radiation.18 Given the positive results of RTOG 0933 and 0614, the NRG Oncology research organization is conducting a phase 3 randomized trial that compares memantine use for patients receiving whole-brain radiation with or without hippocampal sparing to determine if patients will have reduced cognitive decline. All patients receiving WBRT should be considered for enrolment on this trial if they are eligible.
The delivery of brain radiation has continued to change, especially with the introduction of SRS. Recent publication of a meta-analysis of three phase 3 trials evaluating SRS with or without WBRT for 1-4 brain metastases showed that patients aged 50 years or younger experienced a survival benefit with SRS, and the omission of whole-brain radiation did not affect distant brain relapse rates. 19 The authors recommended that for this population, SRS alone is the preferred treatment. In our study, physicians who had been in practice for a longer time were more likely to treat using SRS alone. The results showed a linear association, with those in practice for a longer time being more likely to use SRS alone compared with those practicing for a shorter time (P = .027). Accordingly, 67% of respondents (8 of 12) who had been in practice for 11 or more years used SRS alone, whereas 24% (14 of 58) who had practiced for 0-5 years and 42% (5 of 12) who had practice from 6-10 years used SRS alone (Figure 1). When treating with SRS, younger practitioners placed more importance on the status of extracranial disease, whereas older practitioners placed more importance on tumor histopathology.
The use of repeat whole-brain reirradiation is more controversial among practitioners.20-22 Son and colleagues evaluated patients who needed whole-brain reirradiation after intracranial disease progression.22 The authors noted that patients with stable extracranial disease benefited from reirradiation. In our study, we found that when considering whole-brain reirradiation, older practitioners placed more importance on tumor histology than other factors.
As far as we know, this is the first study evaluating the practices and patterns of care with regard to the delivery of brain radiation in academic centers in the US. We found that time in practice was the most significant predictor of treatment technique and delivery. We also found that older practitioners place more importance on tumor histopathology compared with younger practitioners. A limitation of this study is that we had contact information only for program coordinators at ACGME-accredited programs. As such, we were not able to assess practice patterns among community practitioners. In addition, it seemed that residents and junior faculty were more likely to respond to this survey, likely because of the dissemination pattern. Given the evolution and diversity of treatment regimens for brain metastases, we believe that patients with brain metastases should be managed individually using a multidisciplinary approach.
1. Barnholtz-Sloan JS, Sloan AE, Davis FG, Vigneau FD, Lai P, Sawaya RE. Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. J Clin Oncol. 2004;22(14):2865-2872.
2. Schouten LJ, Rutten J, Huveneers HA, Twijnstra A. Incidence of brain metastases in a cohort of patients with carcinoma of the breast, colon, kidney, and lung and melanoma. Cancer. 2002;94(10):2698-2705.
3. Takakura K. Metastatic tumors of the central nervous system. Tokyo: Igaku-Shoin; 1982.
4. Patchell RA, Tibbs PA, Regine WF, et al. Postoperative radiotherapy in the treatment of single metastases to the brain: a randomized trial. JAMA. 1998;280(17):1485-1489.
5. Patchell RA, Tibbs PA, Walsh JW, et al. A randomized trial of surgery in the treatment of single metastases to the brain. New Engl J Med. 1990;322(8):494-500.
6. Sahgal A, Aoyama H, Kocher M, et al. Phase 3 trials of stereotactic radiosurgery with or without whole-brain radiation therapy for 1 to 4 brain metastases: individual patient data meta-analysis. Int J Radiat Oncol Biol Phys. 2015;91(4):710-717.
7. Chang EL, Wefel JS, Hess KR, et al. Neurocognition in patients with brain metastases treated with radiosurgery or radiosurgery plus whole-brain irradiation: a randomised controlled trial. Lancet Oncol. 2009;10(11):1037-1044.
8. Choosing Wisely [ASTRO]. Don’t routinely add adjuvant whole-brain radiation therapy to stereotactic radiosurgery for limited brain metastases. http://www.choosingwisely.org/clinician-lists/american-society-radiation-oncology-adjunct-whole-brain-radiation-therapy/. Updated June 21, 2016. Accessed November 10, 2016.
9. Aoyama H, Tago M, Shirato H, Japanese Radiation Oncology Study Group I. Stereotactic radiosurgery with or without whole-brain radiotherapy for brain metastases: secondary analysis of the JROSG 99-1 Randomized Clinical Trial. JAMA Oncol. 2015;1(4):457-464.
10. Hoddinott SN, Bass MJ. The Dillman total design survey method. Can Fam Physician. 1986;32:2366-2368.
11. Nayak L, Lee EQ, Wen PY. Epidemiology of brain metastases. Curr Oncol Rep. 2012;14(1):48-54.
12. Gavrilovic IT, Posner JB. Brain metastases: epidemiology and pathophysiology. J Neurooncol. 2005;75(1):5-14.
13. Chao JH, Phillips R, Nickson JJ. Roentgen-ray therapy of cerebral metastases. Cancer. 1954;7(4):682-689.
14. Nieder C, Niewald M, Schnabel K. Treatment of brain metastases from hypernephroma. Urol Int. 1996;57(1):17-20.
15. Ryken TC, McDermott M, Robinson PD, et al. The role of steroids in the management of brain metastases: a systematic review and evidence-based clinical practice guideline. J Neurooncol. 2010;96(1):103-114.
16. Brown PD, Pugh S, Laack NN, et al. Memantine for the prevention of cognitive dysfunction in patients receiving whole-brain radiotherapy: a randomized, double-blind, placebo-controlled trial. Neuro Oncol. 2013;15(10):1429-1437.
17. Gondi V, Pugh SL, Tome WA, et al. Preservation of memory with conformal avoidance of the hippocampal neural stem-cell compartment during whole-brain radiotherapy for brain metastases (RTOG 0933): a phase II multi-institutional trial. J Clin Oncol. 2014;32(34):3810-3816.
18. Slade AN, Stanic S. The impact of RTOG 0614 and RTOG 0933 trials in routine clinical practice: The US Survey of Utilization of Memantine and IMRT planning for hippocampus sparing in patients receiving whole-brain radiotherapy for brain metastases. Contemp Clin Trials. 2016;47:74-77.
19. Sahgal A, Aoyama H, Kocher M, et al. Phase 3 trials of stereotactic radiosurgery with or without whole-brain radiation therapy for 1 to 4 brain metastases: individual patient data meta-analysis. International journal of radiation oncology, biology, physics. 2015;91(4):710-717.
20. Hazuka MB, Kinzie JJ. Brain metastases: results and effects of re-irradiation. Int J Radiat Oncol Biol Phys. 1988;15(2):433-437.
21. Sadikov E, Bezjak A, Yi QL, et al. Value of whole-brain re-irradiation for brain metastases — single centre experience. Clin Oncol (R Coll Radiol). 2007;19(7):532-538.
22. Son CH, Jimenez R, Niemierko A, Loeffler JS, Oh KS, Shih HA. outcomes after whole-brain reirradiation in patients with brain metastases. Int J Radiat Oncol Biol Phys. 2012;82(2):e167-e172.
Despite the recent advances in systemic therapy, metastatic spread to the brain continues to be the most common neurologic complication of many cancers. The clinical incidence of brain metastases varies with primary cancer diagnosis, with estimates ranging from 1.2%-19.8%.1,2 Metastatic spread to the brain is even more prevalent at autopsy, with evidence of intracranial tumor being found in 26% of patients in some series.3 It is possible that the clinical incidence of metastatic disease to the brain will continue to increase as newer therapeutic agents improve survival and imaging techniques continue to improve.
The management of brain metastases has changed rapidly as technological improvements have made treatment increasingly safe and efficacious. Traditionally, treatment consisted of radiotherapy to the whole brain, with or without surgical resection.4,5 More recently, stereotactic radiosurgery (SRS) has been adopted on the basis of evidence that it is safe and efficacious alone or in combination with radiotherapy to the whole brain.6 Further evidence is emerging that neurocognitive outcomes are improved when whole-brain radiotherapy (WBRT) is omitted, which possibly contributes to improved patient quality of life.7 Taking into account this and other data, the American Society for Radiation Oncology’s Choosing Wisely campaign now recommends not routinely adding WBRT to radiosurgery in patients with limited brain metastases.8
Despite this recommendation, many patients continue to benefit from WBRT, and it remains a common treatment in radiation oncology clinics across the US for several reasons. Many patients present with multiple brain metastases and are ineligible for radiosurgery. Even for technically eligible patients, WBRT has been shown to improve local control and decrease the rate of distant brain failure over radiosurgery alone.6 With higher rates of subsequent failures, patients receiving radiosurgery alone must adhere to more rigorous follow-up and imaging schedules, which can be difficult for many rural patients who have to travel long distances to centers. Furthermore, there is some suggestion that this decreased failure rate may result in improved survival in highly selected patients with excellent disease and performance status.9 Controversies exist, however, and strong institutional biases persist, contributing to significant differences in practice. We surveyed academic radiation oncologists and in an effort to identify and describe practice patterns in the delivery of WBRT at academic centers.
Methods
We conducted a thorough review of available literature on radiation for brain metastases and based on our findings, devised a survey 19 questions to ascertain practice patterns and treatment delivery among US academic physicians (Table 1). After obtaining institutional review board approval to do the study, we sent the survey to program coordinators at radiation oncology programs that are accredited by the Accreditation Council for Graduate Medical Education. We instructed coordinators to e-mail the survey to their practicing resident and attending physicians. The surveys were created using SurveyMonkey software. We obtained informed consent from the providers. A total of 3 follow-up e-mails were sent to each recipient of the survey to solicit responses, similar to the Dillman Total Design Survey Method.10
SPSS version 22.0 was used to analyze the data in an exploratory fashion. Statistical methods were used to assess the association of demographic data with SRS and WBRT delivery and treatment technique items when the analyses involved percentages that included the Pearson chi-square statistic and the chi-square test for linear trend. When the analysis focused on ranking data, the Kruskal-Wallis test, Mann-Whitney U test, the Jonckheere-Terpstra and the Kendall tau-b rank correlation were used as appropriate. If there were small sample sizes within some groups, then exact significant levels were assessed. Statistical significance was set by convention at P < .05.
Results
We received 95 responses of which 87 were considered complete for analysis. Forty-seven percent of the 87 respondents were not board-certified, and the remainder had passed their radiobiology and physics boards exams. A majority of respondents (70%, 61 of 87) were physicians who had been in practice for ≤5 years. Fifty-four percent of respondents were located in the Northeast US, 22% in the South, 14% in the West, and 10% in the Midwest and Hawaii (Table 2).
We used the chi-square test for linear trends to assess for a relationship between years of practice and whether respondents deviated from their typical method of WBRT therapy when treating more radioresistant tumors (melanoma, renal cell carcinoma). Respondents were classified by years in practice: 0-5, 6-10, 11-20, and >21 years. The results showed a linear association, with those in practice for longer periods more likely to use SRS alone, P = .027 (Figure 1).
Discussion
The incidence of brain metastases is increasing because of improvements in diagnostic imaging techniques and advancements in systemic therapy control of extracranial disease but not of intracranial disease or metastasis, because therapies do not cross the blood-brain barrier.11,12 Brain metastases are the most common type of brain tumor. Given that most chemotherapeutic agents cannot cross the blood-brain barrier, radiotherapy is considered a means of treatment and of controlling brain metastases. Early data from the 1950s13 and 1960s14 have suggested clinical improvement with brain radiation, making radiotherapy the cornerstone for treatment of brain metastases.
The Radiation Therapy Oncology Group (RTOG) has evaluated several fractionation schedules, with 5 schemas evaluated by the RTOG 6901 and 7361 studies: 30 Gy in 10 fractions, 30 Gy in 15 fractions, 40 Gy in 15 fractions, 40 Gy in 20 fractions, and 20 Gy in 5 fractions. The combined results from these two trials showed that outcomes were similar for patients treated with a shorter regimen than for those treated with a more protracted schedule. In our study, respondents reported that they most frequently treated brain metastases to a total dose of 30 Gy in 10 fractions. Given the results of the aforementioned RTOG trials and practice patterns among academic physicians, we recommend all practitioners consider a shorter hypofractioned course when treating brain metastases with WBRT. This will also reduce delays for patients who are likely to benefit greatly from earlier enrollment into hospice care, because protracted radiation schedules typically are not covered while a patient is in hospice.
Pharmacologic management for patients with brain metastases is important for symptomatic improvement. Glucocorticoids are important for palliation of symptoms from edema and increased intracranial pressure.15 However, steroids have a multitude of side effects and their use in asymptomatic patients is unnecessary. Improvements in imaging and detection11 have allowed us to find smaller and asymptomatic brain tumors. In our survey, it was promising to see a change in former practice patterns, with only 8% of academic practitioners regularly prescribing steroids to all of their patients receiving whole-brain radiation.
Diminished cognitive function and short-term memory loss are troublesome side effects of WBRT. As cancer patients live longer, such cognitive dysfunction will become more than just a nuisance. The RTOG has investigated the use of prophylactic memantine for patients receiving whole-brain radiation to determine if it would aid in the preservation of cognition. It found that patients who received memantine did better and had delayed time to cognitive decline and a reduced rate of memory decline, executive function, and processing speed.16 In our study, about a third of practitioners prescribed memantine and it was reserved for patients who had an otherwise favorable prognosis.
The RTOG has also investigated adjusting treatment technique for patients who receive WBRT. RTOG 0933 was a phase 2 trial that evaluated hippocampal avoidance during deliverance of WBRT with intensity-modulated radiation therapy (IMRT). Results showed that avoiding the hippocampus during WBRT was associated with improved memory preservation and patient quality of life.17 In a survey of practicing radiation oncologists in the US, most reported that they did not use memantine or IMRT for hippocampal sparing when delivering whole-brain radiation.18 Given the positive results of RTOG 0933 and 0614, the NRG Oncology research organization is conducting a phase 3 randomized trial that compares memantine use for patients receiving whole-brain radiation with or without hippocampal sparing to determine if patients will have reduced cognitive decline. All patients receiving WBRT should be considered for enrolment on this trial if they are eligible.
The delivery of brain radiation has continued to change, especially with the introduction of SRS. Recent publication of a meta-analysis of three phase 3 trials evaluating SRS with or without WBRT for 1-4 brain metastases showed that patients aged 50 years or younger experienced a survival benefit with SRS, and the omission of whole-brain radiation did not affect distant brain relapse rates. 19 The authors recommended that for this population, SRS alone is the preferred treatment. In our study, physicians who had been in practice for a longer time were more likely to treat using SRS alone. The results showed a linear association, with those in practice for a longer time being more likely to use SRS alone compared with those practicing for a shorter time (P = .027). Accordingly, 67% of respondents (8 of 12) who had been in practice for 11 or more years used SRS alone, whereas 24% (14 of 58) who had practiced for 0-5 years and 42% (5 of 12) who had practice from 6-10 years used SRS alone (Figure 1). When treating with SRS, younger practitioners placed more importance on the status of extracranial disease, whereas older practitioners placed more importance on tumor histopathology.
The use of repeat whole-brain reirradiation is more controversial among practitioners.20-22 Son and colleagues evaluated patients who needed whole-brain reirradiation after intracranial disease progression.22 The authors noted that patients with stable extracranial disease benefited from reirradiation. In our study, we found that when considering whole-brain reirradiation, older practitioners placed more importance on tumor histology than other factors.
As far as we know, this is the first study evaluating the practices and patterns of care with regard to the delivery of brain radiation in academic centers in the US. We found that time in practice was the most significant predictor of treatment technique and delivery. We also found that older practitioners place more importance on tumor histopathology compared with younger practitioners. A limitation of this study is that we had contact information only for program coordinators at ACGME-accredited programs. As such, we were not able to assess practice patterns among community practitioners. In addition, it seemed that residents and junior faculty were more likely to respond to this survey, likely because of the dissemination pattern. Given the evolution and diversity of treatment regimens for brain metastases, we believe that patients with brain metastases should be managed individually using a multidisciplinary approach.
Despite the recent advances in systemic therapy, metastatic spread to the brain continues to be the most common neurologic complication of many cancers. The clinical incidence of brain metastases varies with primary cancer diagnosis, with estimates ranging from 1.2%-19.8%.1,2 Metastatic spread to the brain is even more prevalent at autopsy, with evidence of intracranial tumor being found in 26% of patients in some series.3 It is possible that the clinical incidence of metastatic disease to the brain will continue to increase as newer therapeutic agents improve survival and imaging techniques continue to improve.
The management of brain metastases has changed rapidly as technological improvements have made treatment increasingly safe and efficacious. Traditionally, treatment consisted of radiotherapy to the whole brain, with or without surgical resection.4,5 More recently, stereotactic radiosurgery (SRS) has been adopted on the basis of evidence that it is safe and efficacious alone or in combination with radiotherapy to the whole brain.6 Further evidence is emerging that neurocognitive outcomes are improved when whole-brain radiotherapy (WBRT) is omitted, which possibly contributes to improved patient quality of life.7 Taking into account this and other data, the American Society for Radiation Oncology’s Choosing Wisely campaign now recommends not routinely adding WBRT to radiosurgery in patients with limited brain metastases.8
Despite this recommendation, many patients continue to benefit from WBRT, and it remains a common treatment in radiation oncology clinics across the US for several reasons. Many patients present with multiple brain metastases and are ineligible for radiosurgery. Even for technically eligible patients, WBRT has been shown to improve local control and decrease the rate of distant brain failure over radiosurgery alone.6 With higher rates of subsequent failures, patients receiving radiosurgery alone must adhere to more rigorous follow-up and imaging schedules, which can be difficult for many rural patients who have to travel long distances to centers. Furthermore, there is some suggestion that this decreased failure rate may result in improved survival in highly selected patients with excellent disease and performance status.9 Controversies exist, however, and strong institutional biases persist, contributing to significant differences in practice. We surveyed academic radiation oncologists and in an effort to identify and describe practice patterns in the delivery of WBRT at academic centers.
Methods
We conducted a thorough review of available literature on radiation for brain metastases and based on our findings, devised a survey 19 questions to ascertain practice patterns and treatment delivery among US academic physicians (Table 1). After obtaining institutional review board approval to do the study, we sent the survey to program coordinators at radiation oncology programs that are accredited by the Accreditation Council for Graduate Medical Education. We instructed coordinators to e-mail the survey to their practicing resident and attending physicians. The surveys were created using SurveyMonkey software. We obtained informed consent from the providers. A total of 3 follow-up e-mails were sent to each recipient of the survey to solicit responses, similar to the Dillman Total Design Survey Method.10
SPSS version 22.0 was used to analyze the data in an exploratory fashion. Statistical methods were used to assess the association of demographic data with SRS and WBRT delivery and treatment technique items when the analyses involved percentages that included the Pearson chi-square statistic and the chi-square test for linear trend. When the analysis focused on ranking data, the Kruskal-Wallis test, Mann-Whitney U test, the Jonckheere-Terpstra and the Kendall tau-b rank correlation were used as appropriate. If there were small sample sizes within some groups, then exact significant levels were assessed. Statistical significance was set by convention at P < .05.
Results
We received 95 responses of which 87 were considered complete for analysis. Forty-seven percent of the 87 respondents were not board-certified, and the remainder had passed their radiobiology and physics boards exams. A majority of respondents (70%, 61 of 87) were physicians who had been in practice for ≤5 years. Fifty-four percent of respondents were located in the Northeast US, 22% in the South, 14% in the West, and 10% in the Midwest and Hawaii (Table 2).
We used the chi-square test for linear trends to assess for a relationship between years of practice and whether respondents deviated from their typical method of WBRT therapy when treating more radioresistant tumors (melanoma, renal cell carcinoma). Respondents were classified by years in practice: 0-5, 6-10, 11-20, and >21 years. The results showed a linear association, with those in practice for longer periods more likely to use SRS alone, P = .027 (Figure 1).
Discussion
The incidence of brain metastases is increasing because of improvements in diagnostic imaging techniques and advancements in systemic therapy control of extracranial disease but not of intracranial disease or metastasis, because therapies do not cross the blood-brain barrier.11,12 Brain metastases are the most common type of brain tumor. Given that most chemotherapeutic agents cannot cross the blood-brain barrier, radiotherapy is considered a means of treatment and of controlling brain metastases. Early data from the 1950s13 and 1960s14 have suggested clinical improvement with brain radiation, making radiotherapy the cornerstone for treatment of brain metastases.
The Radiation Therapy Oncology Group (RTOG) has evaluated several fractionation schedules, with 5 schemas evaluated by the RTOG 6901 and 7361 studies: 30 Gy in 10 fractions, 30 Gy in 15 fractions, 40 Gy in 15 fractions, 40 Gy in 20 fractions, and 20 Gy in 5 fractions. The combined results from these two trials showed that outcomes were similar for patients treated with a shorter regimen than for those treated with a more protracted schedule. In our study, respondents reported that they most frequently treated brain metastases to a total dose of 30 Gy in 10 fractions. Given the results of the aforementioned RTOG trials and practice patterns among academic physicians, we recommend all practitioners consider a shorter hypofractioned course when treating brain metastases with WBRT. This will also reduce delays for patients who are likely to benefit greatly from earlier enrollment into hospice care, because protracted radiation schedules typically are not covered while a patient is in hospice.
Pharmacologic management for patients with brain metastases is important for symptomatic improvement. Glucocorticoids are important for palliation of symptoms from edema and increased intracranial pressure.15 However, steroids have a multitude of side effects and their use in asymptomatic patients is unnecessary. Improvements in imaging and detection11 have allowed us to find smaller and asymptomatic brain tumors. In our survey, it was promising to see a change in former practice patterns, with only 8% of academic practitioners regularly prescribing steroids to all of their patients receiving whole-brain radiation.
Diminished cognitive function and short-term memory loss are troublesome side effects of WBRT. As cancer patients live longer, such cognitive dysfunction will become more than just a nuisance. The RTOG has investigated the use of prophylactic memantine for patients receiving whole-brain radiation to determine if it would aid in the preservation of cognition. It found that patients who received memantine did better and had delayed time to cognitive decline and a reduced rate of memory decline, executive function, and processing speed.16 In our study, about a third of practitioners prescribed memantine and it was reserved for patients who had an otherwise favorable prognosis.
The RTOG has also investigated adjusting treatment technique for patients who receive WBRT. RTOG 0933 was a phase 2 trial that evaluated hippocampal avoidance during deliverance of WBRT with intensity-modulated radiation therapy (IMRT). Results showed that avoiding the hippocampus during WBRT was associated with improved memory preservation and patient quality of life.17 In a survey of practicing radiation oncologists in the US, most reported that they did not use memantine or IMRT for hippocampal sparing when delivering whole-brain radiation.18 Given the positive results of RTOG 0933 and 0614, the NRG Oncology research organization is conducting a phase 3 randomized trial that compares memantine use for patients receiving whole-brain radiation with or without hippocampal sparing to determine if patients will have reduced cognitive decline. All patients receiving WBRT should be considered for enrolment on this trial if they are eligible.
The delivery of brain radiation has continued to change, especially with the introduction of SRS. Recent publication of a meta-analysis of three phase 3 trials evaluating SRS with or without WBRT for 1-4 brain metastases showed that patients aged 50 years or younger experienced a survival benefit with SRS, and the omission of whole-brain radiation did not affect distant brain relapse rates. 19 The authors recommended that for this population, SRS alone is the preferred treatment. In our study, physicians who had been in practice for a longer time were more likely to treat using SRS alone. The results showed a linear association, with those in practice for a longer time being more likely to use SRS alone compared with those practicing for a shorter time (P = .027). Accordingly, 67% of respondents (8 of 12) who had been in practice for 11 or more years used SRS alone, whereas 24% (14 of 58) who had practiced for 0-5 years and 42% (5 of 12) who had practice from 6-10 years used SRS alone (Figure 1). When treating with SRS, younger practitioners placed more importance on the status of extracranial disease, whereas older practitioners placed more importance on tumor histopathology.
The use of repeat whole-brain reirradiation is more controversial among practitioners.20-22 Son and colleagues evaluated patients who needed whole-brain reirradiation after intracranial disease progression.22 The authors noted that patients with stable extracranial disease benefited from reirradiation. In our study, we found that when considering whole-brain reirradiation, older practitioners placed more importance on tumor histology than other factors.
As far as we know, this is the first study evaluating the practices and patterns of care with regard to the delivery of brain radiation in academic centers in the US. We found that time in practice was the most significant predictor of treatment technique and delivery. We also found that older practitioners place more importance on tumor histopathology compared with younger practitioners. A limitation of this study is that we had contact information only for program coordinators at ACGME-accredited programs. As such, we were not able to assess practice patterns among community practitioners. In addition, it seemed that residents and junior faculty were more likely to respond to this survey, likely because of the dissemination pattern. Given the evolution and diversity of treatment regimens for brain metastases, we believe that patients with brain metastases should be managed individually using a multidisciplinary approach.
1. Barnholtz-Sloan JS, Sloan AE, Davis FG, Vigneau FD, Lai P, Sawaya RE. Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. J Clin Oncol. 2004;22(14):2865-2872.
2. Schouten LJ, Rutten J, Huveneers HA, Twijnstra A. Incidence of brain metastases in a cohort of patients with carcinoma of the breast, colon, kidney, and lung and melanoma. Cancer. 2002;94(10):2698-2705.
3. Takakura K. Metastatic tumors of the central nervous system. Tokyo: Igaku-Shoin; 1982.
4. Patchell RA, Tibbs PA, Regine WF, et al. Postoperative radiotherapy in the treatment of single metastases to the brain: a randomized trial. JAMA. 1998;280(17):1485-1489.
5. Patchell RA, Tibbs PA, Walsh JW, et al. A randomized trial of surgery in the treatment of single metastases to the brain. New Engl J Med. 1990;322(8):494-500.
6. Sahgal A, Aoyama H, Kocher M, et al. Phase 3 trials of stereotactic radiosurgery with or without whole-brain radiation therapy for 1 to 4 brain metastases: individual patient data meta-analysis. Int J Radiat Oncol Biol Phys. 2015;91(4):710-717.
7. Chang EL, Wefel JS, Hess KR, et al. Neurocognition in patients with brain metastases treated with radiosurgery or radiosurgery plus whole-brain irradiation: a randomised controlled trial. Lancet Oncol. 2009;10(11):1037-1044.
8. Choosing Wisely [ASTRO]. Don’t routinely add adjuvant whole-brain radiation therapy to stereotactic radiosurgery for limited brain metastases. http://www.choosingwisely.org/clinician-lists/american-society-radiation-oncology-adjunct-whole-brain-radiation-therapy/. Updated June 21, 2016. Accessed November 10, 2016.
9. Aoyama H, Tago M, Shirato H, Japanese Radiation Oncology Study Group I. Stereotactic radiosurgery with or without whole-brain radiotherapy for brain metastases: secondary analysis of the JROSG 99-1 Randomized Clinical Trial. JAMA Oncol. 2015;1(4):457-464.
10. Hoddinott SN, Bass MJ. The Dillman total design survey method. Can Fam Physician. 1986;32:2366-2368.
11. Nayak L, Lee EQ, Wen PY. Epidemiology of brain metastases. Curr Oncol Rep. 2012;14(1):48-54.
12. Gavrilovic IT, Posner JB. Brain metastases: epidemiology and pathophysiology. J Neurooncol. 2005;75(1):5-14.
13. Chao JH, Phillips R, Nickson JJ. Roentgen-ray therapy of cerebral metastases. Cancer. 1954;7(4):682-689.
14. Nieder C, Niewald M, Schnabel K. Treatment of brain metastases from hypernephroma. Urol Int. 1996;57(1):17-20.
15. Ryken TC, McDermott M, Robinson PD, et al. The role of steroids in the management of brain metastases: a systematic review and evidence-based clinical practice guideline. J Neurooncol. 2010;96(1):103-114.
16. Brown PD, Pugh S, Laack NN, et al. Memantine for the prevention of cognitive dysfunction in patients receiving whole-brain radiotherapy: a randomized, double-blind, placebo-controlled trial. Neuro Oncol. 2013;15(10):1429-1437.
17. Gondi V, Pugh SL, Tome WA, et al. Preservation of memory with conformal avoidance of the hippocampal neural stem-cell compartment during whole-brain radiotherapy for brain metastases (RTOG 0933): a phase II multi-institutional trial. J Clin Oncol. 2014;32(34):3810-3816.
18. Slade AN, Stanic S. The impact of RTOG 0614 and RTOG 0933 trials in routine clinical practice: The US Survey of Utilization of Memantine and IMRT planning for hippocampus sparing in patients receiving whole-brain radiotherapy for brain metastases. Contemp Clin Trials. 2016;47:74-77.
19. Sahgal A, Aoyama H, Kocher M, et al. Phase 3 trials of stereotactic radiosurgery with or without whole-brain radiation therapy for 1 to 4 brain metastases: individual patient data meta-analysis. International journal of radiation oncology, biology, physics. 2015;91(4):710-717.
20. Hazuka MB, Kinzie JJ. Brain metastases: results and effects of re-irradiation. Int J Radiat Oncol Biol Phys. 1988;15(2):433-437.
21. Sadikov E, Bezjak A, Yi QL, et al. Value of whole-brain re-irradiation for brain metastases — single centre experience. Clin Oncol (R Coll Radiol). 2007;19(7):532-538.
22. Son CH, Jimenez R, Niemierko A, Loeffler JS, Oh KS, Shih HA. outcomes after whole-brain reirradiation in patients with brain metastases. Int J Radiat Oncol Biol Phys. 2012;82(2):e167-e172.
1. Barnholtz-Sloan JS, Sloan AE, Davis FG, Vigneau FD, Lai P, Sawaya RE. Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. J Clin Oncol. 2004;22(14):2865-2872.
2. Schouten LJ, Rutten J, Huveneers HA, Twijnstra A. Incidence of brain metastases in a cohort of patients with carcinoma of the breast, colon, kidney, and lung and melanoma. Cancer. 2002;94(10):2698-2705.
3. Takakura K. Metastatic tumors of the central nervous system. Tokyo: Igaku-Shoin; 1982.
4. Patchell RA, Tibbs PA, Regine WF, et al. Postoperative radiotherapy in the treatment of single metastases to the brain: a randomized trial. JAMA. 1998;280(17):1485-1489.
5. Patchell RA, Tibbs PA, Walsh JW, et al. A randomized trial of surgery in the treatment of single metastases to the brain. New Engl J Med. 1990;322(8):494-500.
6. Sahgal A, Aoyama H, Kocher M, et al. Phase 3 trials of stereotactic radiosurgery with or without whole-brain radiation therapy for 1 to 4 brain metastases: individual patient data meta-analysis. Int J Radiat Oncol Biol Phys. 2015;91(4):710-717.
7. Chang EL, Wefel JS, Hess KR, et al. Neurocognition in patients with brain metastases treated with radiosurgery or radiosurgery plus whole-brain irradiation: a randomised controlled trial. Lancet Oncol. 2009;10(11):1037-1044.
8. Choosing Wisely [ASTRO]. Don’t routinely add adjuvant whole-brain radiation therapy to stereotactic radiosurgery for limited brain metastases. http://www.choosingwisely.org/clinician-lists/american-society-radiation-oncology-adjunct-whole-brain-radiation-therapy/. Updated June 21, 2016. Accessed November 10, 2016.
9. Aoyama H, Tago M, Shirato H, Japanese Radiation Oncology Study Group I. Stereotactic radiosurgery with or without whole-brain radiotherapy for brain metastases: secondary analysis of the JROSG 99-1 Randomized Clinical Trial. JAMA Oncol. 2015;1(4):457-464.
10. Hoddinott SN, Bass MJ. The Dillman total design survey method. Can Fam Physician. 1986;32:2366-2368.
11. Nayak L, Lee EQ, Wen PY. Epidemiology of brain metastases. Curr Oncol Rep. 2012;14(1):48-54.
12. Gavrilovic IT, Posner JB. Brain metastases: epidemiology and pathophysiology. J Neurooncol. 2005;75(1):5-14.
13. Chao JH, Phillips R, Nickson JJ. Roentgen-ray therapy of cerebral metastases. Cancer. 1954;7(4):682-689.
14. Nieder C, Niewald M, Schnabel K. Treatment of brain metastases from hypernephroma. Urol Int. 1996;57(1):17-20.
15. Ryken TC, McDermott M, Robinson PD, et al. The role of steroids in the management of brain metastases: a systematic review and evidence-based clinical practice guideline. J Neurooncol. 2010;96(1):103-114.
16. Brown PD, Pugh S, Laack NN, et al. Memantine for the prevention of cognitive dysfunction in patients receiving whole-brain radiotherapy: a randomized, double-blind, placebo-controlled trial. Neuro Oncol. 2013;15(10):1429-1437.
17. Gondi V, Pugh SL, Tome WA, et al. Preservation of memory with conformal avoidance of the hippocampal neural stem-cell compartment during whole-brain radiotherapy for brain metastases (RTOG 0933): a phase II multi-institutional trial. J Clin Oncol. 2014;32(34):3810-3816.
18. Slade AN, Stanic S. The impact of RTOG 0614 and RTOG 0933 trials in routine clinical practice: The US Survey of Utilization of Memantine and IMRT planning for hippocampus sparing in patients receiving whole-brain radiotherapy for brain metastases. Contemp Clin Trials. 2016;47:74-77.
19. Sahgal A, Aoyama H, Kocher M, et al. Phase 3 trials of stereotactic radiosurgery with or without whole-brain radiation therapy for 1 to 4 brain metastases: individual patient data meta-analysis. International journal of radiation oncology, biology, physics. 2015;91(4):710-717.
20. Hazuka MB, Kinzie JJ. Brain metastases: results and effects of re-irradiation. Int J Radiat Oncol Biol Phys. 1988;15(2):433-437.
21. Sadikov E, Bezjak A, Yi QL, et al. Value of whole-brain re-irradiation for brain metastases — single centre experience. Clin Oncol (R Coll Radiol). 2007;19(7):532-538.
22. Son CH, Jimenez R, Niemierko A, Loeffler JS, Oh KS, Shih HA. outcomes after whole-brain reirradiation in patients with brain metastases. Int J Radiat Oncol Biol Phys. 2012;82(2):e167-e172.
Distress management in cancer patients in Puerto Rico
A comprehensive, patient-centered approach is required to accomplish cancer best standards of care.1 This approach reflects the holistic conceptualization of health in which the physical, emotional, and social dimensions of the human being are considered when providing medical care. As a result, to look after all patient needs, interdisciplinary and well-coordinated interventions are recommended. Cancer patients should be provided not only with diagnostic, treatment, and follow-up clinical service, but also with the supportive assistance that may positively influence all aspects of their health.
To appraise physical, social, emotional and spiritual issues and to develop supportive interventional action plans, the National Comprehensive Cancer Network (NCCN) recommends screening all cancer patients for distress.2 In particular, screening the emotional component of distress occupies a prominent place in this process because it is now recognized as the sixth vital sign in oncology.3 Even though the influence of emotional distress over cancer mortality rates and disease progression is still under scrutiny,4 its plausible implications over treatment compliance have been pointed out. Patients with higher levels of emotional distress show lower adherence to treatment and poorer health outcomes.5 Furthermore, prevalence rates of emotional distress in cancer patients from ambulatory settings6 and oncology surgical units have been studied and have provided justification for distress management.7 Studies have shown low ability among oncologists to identify patients in distress and oncologists’ tendency to judge distress higher than the patients themselves.8 As a consequence, to achieve systematic distress evaluations and appropriate referrals for care, guidelines for distress management should be implemented in clinical settings. It is recommended that tests are conducted to find brief screening instruments and procedures to assure accurate interventions according to patient specific needs.
This article presents the process of implementing a distress management program at HIMA-San Pablo Oncologic Hospital in Caguas, Puerto Rico, with particular emphasis on the management of emotional distress, which has been defined as the feeling of suffering that cancer patients may experience after diagnosis. In addition, we have included data from a pilot study that was completed for content validation of the Patient Health Questionnaire (PHQ-9) to estimate depression levels in Puerto Rican cancer patients.
Methods
HIMA-San Pablo operates a group of privately owned hospitals in Puerto Rico. It established a cancer center in Caguas in 2007, recruiting a multispecialty medical faculty to provide cancer care and bone marrow transplants for adult and pediatric patients. The cancer center, currently named HIMA-San Pablo Oncologic Hospital (HSPOH), is a hospital within a hospital licensed by the Puerto Rico Department of Health. In 2007, a cancer committee was established as the steering committee to ensure the delivery of cancer care according to best standards of care. The committee took responsibility for developing all activities needed to achieve the American College of Surgeons’ Commission on Cancer (CoC) accreditation under the category of Comprehensive Community Cancer Center. The committee established a psychosocial team to develop a protocol for the delivery of distress management for adult patients. (The psychosocial needs of pediatric patients are assessed through other procedures.)
To develop the protocol, principles of input-output model of research and quality analysis in health care were applied.9 The input-output model, with its origin in engineering, helped map systematic activities to transform empirical data on cancer psychosocial care into operational procedures. Focus was given to data gathering (input), information organization and analysis (throughput), and the schematization of emotional distress management care (output).
The input phase
In the input phase, elements of psychosocial care and operational definitions related to distress management in general were identified through literature review (Table 1). Basic parameters for distress management were clarified, resulting in a conceptual framework based in four remarks: First, according to NCCN, distress is a multifactorial unpleasant emotional experience of a psychological, social, and spiritual nature. It may interfere with the ability to cope effectively with cancer, its symptoms and its treatment. Its intensity may fluctuate from feelings of suffering and fear to incapacitating manifestations of anxiety and depression2 and its severity may hamper patient quality of life and treatment compliance.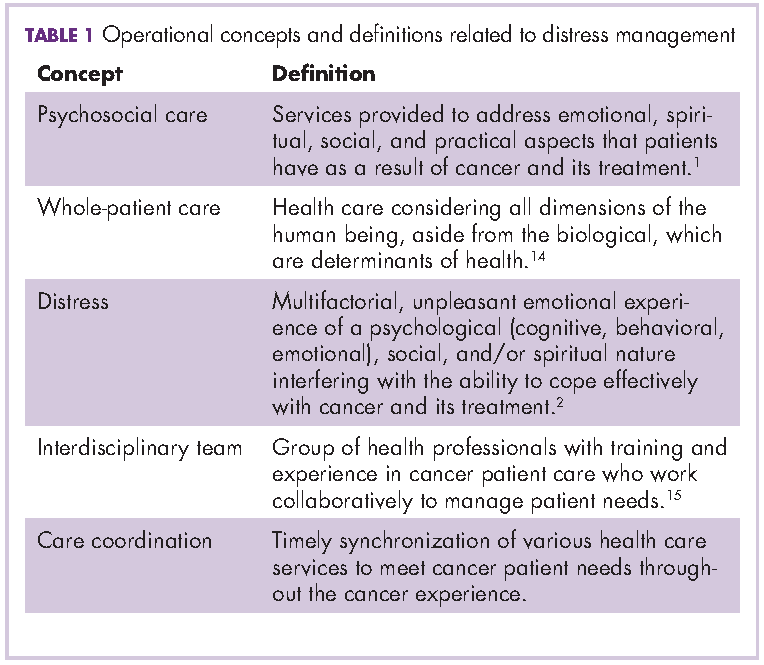
Second, distress management requires the intervention of an interdisciplinary team with both medical and allied health professionals. This may include mental health specialists and other professionals with training and experience in cancer-related issues, who work with reciprocal channels of communication for the exchange of patient information.
Third, NCCN recommends using the Distress Thermometer for patient initial distress screening.10-12 It consists of a numeric scale ranging from 0 (no distress) to 10 (severe distress) in which patients classify their level of distress. The numeric scale is followed by a section in which patients identify areas of practical, familiar, emotional, spiritual/ religious, and physical concerns. Based on responses, interviews may follow to set distress management interventions.
Fourth, screening and assessment are different but sequential and complementary stages of distress management. Screening is viewed as a rapid strategy to identify cancer patients in distress. Assessment looks out for a broader appraisal and documentation of factors with repercussions over patient distress level and resiliency capability.13 In many instances, the patient’s emotional distress is better understood in the assessment phase.
The throughput phase
Within the throughput phase of information organization and analysis, an inventory of health professionals and other in-house consultants needed for distress management was completed. Roles and procedures for information sharing were determined, and we established collaborative agreements with professionals in the community who could contribute to distress management. Members of the psychosocial team held workshops to discuss elements of NCCN guidelines for distress management and to create an action plan for the implementation of the protocol. Data analyses were performed to create a demographic profile of the oncology population at the hospital and assess patient willingness to receive emotional support services,16 which led to the implementation of support group meetings at which additional substantive information was collected about issues affecting cancer patients’ emotions.
The NCCN Distress Thermometer for measuring distress was translated to Spanish. Its format was adapted, and it was identified as a distress screening tool (DST), which we named Distress Assessment Tool for Oncology Patients (Figure 1). The instrument helps for rapid screening of patient needs and proper determination of initial interventions. In addition, psychometric properties of several instruments were reviewed for instances when patient emotional distress could not be clearly determined. We decided to proceed with the validation of the 9-item Patient Health Questionnaire (PHQ-9) to estimate patient depression level. A proposal for content validation of the PHQ-9 was approved by the University of Puerto Rico institutional review board, and patients were recruited to participate in the pilot study.
The PHQ-9. The PHQ-9 is a self-report version of the PRIME-MD instrument developed to assess mental disorders in clinical settings. It is based on DSM-IV diagnostic criteria.17 The PHQ-9 is the depression module with nine depression symptoms to check off if they become the cause of emotional impairment. Respondents categorized depression symptoms in four frequency degrees representing numeric values: 0 (not at all), 1 (several days), 2 (more than half the days), 3 (nearly every day). Measures of depression severity are subsequently determined in a Likert-type scale according to numeric calculations of responses: 0-4 (none severe depression), 5-9 (mild), 10-14 (moderate), 15-19 (moderately severe), and 20-27 (severe-major depression).
The instrument is widely used because of its validity in small and large populations. It showed adequate reliability and validity in a small sample of head and neck cancer patients, with a Cronbach’s alpha of 0.80 and a correlation coefficient of 0.71.18 Similarly, it showed good performance in identifying major depression in 4264 cancer outpatients, with sensitivity of 93%, specificity of 81%, and a positive predictive value (PPV) of 25% and negative predictive value (NPV) of 99%.19 Even when administered on a touch screen computer, the instrument showed valid data of depression from patients in treatment.20
The Beck Depression Inventory. We used the Beck Depression Inventory (BDI-II) Spanish version as the gold standard measure for the validation study. It is a 22-items inventory that measures attitudes and symptoms of depression.21 It can be administered in 10 minutes and has shown good psychometric measures when administered in Spain and Puerto Rico.22, 23
The pilot study. In all, 44 cancer patients who were receiving outpatient treatment at the radiotherapy unit agreed to participate in the study. The participants signed a consent form after the confidentiality protection measures and the main objectives of the study had been explained to them. Patients were interviewed individually during November and December 2012, with the Spanish versions of the PHQ-9 and BDI-II administered by one of two interviewers. At the beginning of each interview, the patient was asked 10 questions so that we could gather demographic data and confirm participant eligibility: aged 21 years or older, born and raised in Puerto Rico, being a Spanish speaker, and having a primary cancer diagnosis with no previous disease. Three patients were excluded from the sample because they either had cancer previously or had a recurrence or metastasis. The final sample consisted of 41 outpatients (N = 41).
Data analysis for demographics was completed with STATA v.12 software. Measures of central tendency and dispersion as well as PHQ-9 internal consistency analysis were made through Cronbach alpha with SPSS.
From a total of 41 patients surveyed, 22 (54%) were men and 19 (46%) were women, with an overall median age of 61 years. Among the men, 15 (68%) had a prostate cancer diagnosis and among women, 9 (47.4%) had a breast cancer diagnosis. In regard to health insurance, 19 (46%) had Medicare or Veterans/federal insurance coverage, and 13 (32%) had Reforma, the Puerto Rican government health insurance program partially funded by Medicaid funds. In addition, 8 participants (20%) were unemployed or disabled. As previously stated, all of the patients were in ambulatory care. Only 3 (7%) were participating in support groups.
Of all the respondents, 16 (39%) reported some level of depression. In particular, 2 (5%) showed severe-major depression, 4 (10%) moderately severe depression, and 10 (24%) moderate depression. Of those with depression, 8 (50%) were women, 8 (50%) were men. All 6 of the patients with head and neck cancer showed moderate or moderately severe depression (Table 2).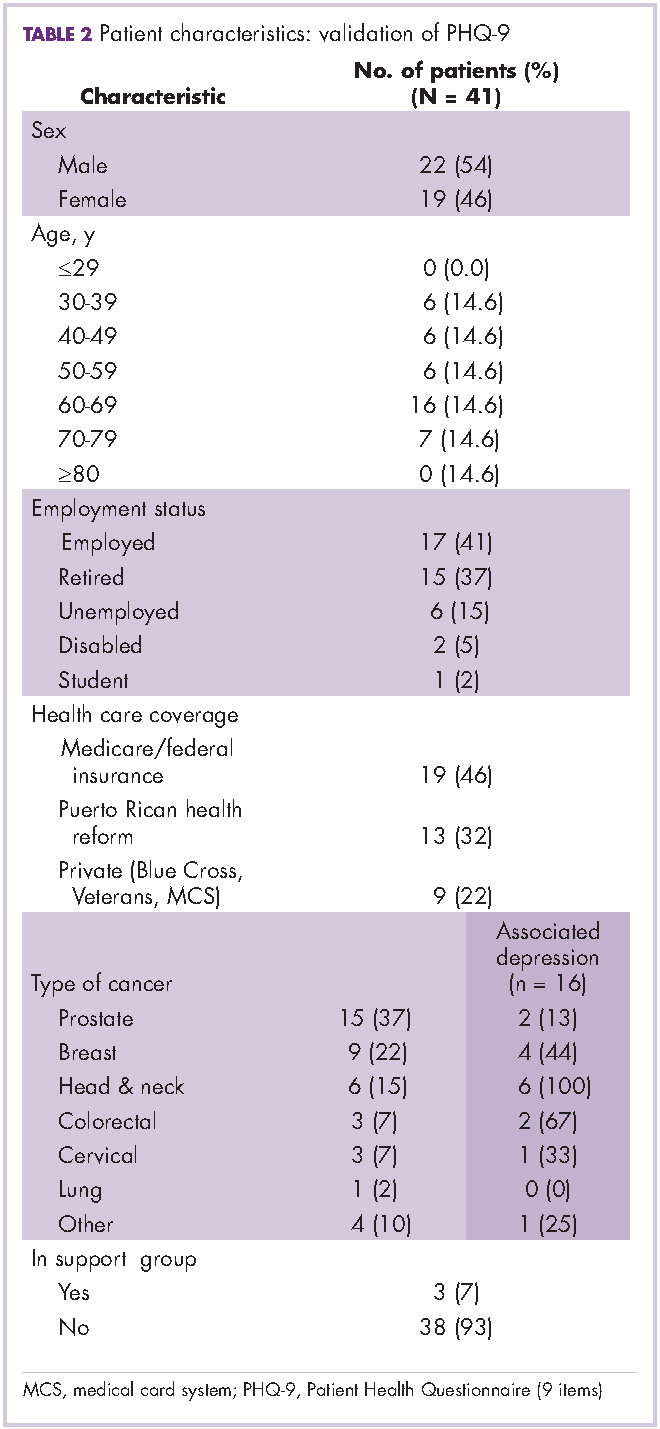
When respondent PHQ-9 scoring reflected moderate to severe depression (>10), a letter was sent to the patient’s radio-oncologist for referral to counseling and clinical psychological evaluation. All participants had access to the support group program, to a radiotherapy education program meeting weekly, and written information about their cancer diagnosis and treatment. They also were interviewed by the psychosocial coordinator or patient navigator for further assessment.
The output phase
In the output phase, a graphic representing the process of emotional assessment at the institution was created and then modified. PHQ-9 was added to the process when it was found suitable to assess level of depression contributing to the identification of patients requiring psychological and psychiatric assistance which by other means would be missed. PHQ-9 was useful in the busy clinical setting as it was completed, scored and interpreted in minutes. It showed the potential for routine evaluations when looking to identify improvement or deterioration in depression levels thus helping to monitor responses to treatment and providing insights for follow up interventions. As stated by NCCN guidelines, distress should be monitored, documented and managed at all stages of the cancer continuum.
Results and discussion
The protocol for distress management at HSPOH is based on the 2013 NCCN guidelines. Cancer patients are screened for levels of distress in all settings (inpatients and outpatients). Screening is held with the DST Spanish translation at the moment of diagnosis or as soon as possible after a diagnosis is made. Screening for distress is also done before or after surgery, in recurrence or progression, and when clinically indicated. Patients are informed that distress management is an essential part of their care and are encouraged to provide information so that we can make a proper need assessment.
Patients are screened by the psychosocial coordinator or patient navigator who administers the DST followed by in-depth interviews for additional appraisal. An action plan is designed based on patient needs, which include their intervention and the intervention of other members of the psychosocial team from the institution and/or from the community. Additional in-house health professionals contributing in distress management include, but are not limited to: physicians; clinical psychologists; health educators; social workers; dietitians; chaplains; and physical, respiratory, speech, and/or swallow therapists. Follow-up and rescreening sessions are scheduled to assure coordination of services between those health professionals as well as to secure continuity of distress management during all stages of the cancer continuum.
The results of the DST are filed in patient medical records. Members of the psychosocial team also document their interventions in the patient medical record, which helps in the exchange of information among the cancer care team. The psychosocial team meets once a month – or as required for extraordinary cases – to review and discuss the cases, determine the best options for distress management, and identify areas for psychosocial care improvement. Those findings and the results of distress management in patient level of satisfaction are then reported and discussed quarterly by the psychosocial coordinator and the cancer committee.
Figure 2 shows in what phase of emotional distress assessment the PHQ-9 was included. Patients reporting four or more of the six areas of concern related to emotional distress in the DST (Figure 1) are automatically referred to a mental health specialist. But when patients report three areas of concern with no clear data on their specific level of depression, PHQ-9 is administered to differentiate those who need a mental health specialist from those who could be adequately supported by health education and support group interventions. In this way detrimental outcomes such as duplicity and over or underuse of services and resources are reduced. In addition, it is recognized that using an interview after the administration of the DST to determine distress management actions does not always provide enough information about a patient’s emotional circumstances and previous comorbidities. Patient responses during interviews may be influenced by the patient’s level of literacy, verbal comprehension, and communication style,24 so emotional distress can go unrecognized during interviews, resulting in delays for treatment and supportive care.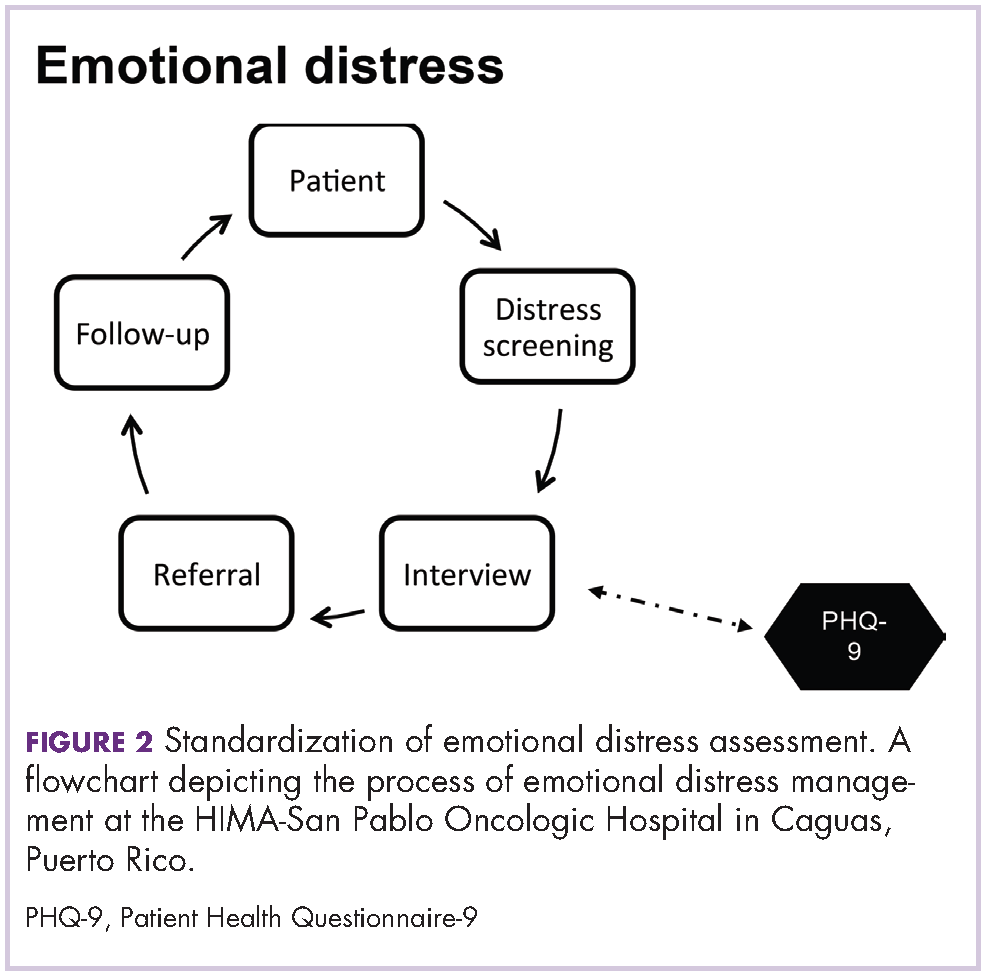
National guidelines in oncology consider such socio-ecological models emphasizing the delivery of patient-centered, interdisciplinary, and evidence-based care. That does not mean that institutions should apply protocols of psychosocial care as previously developed, but that they should test, review, adapt, and improve them during the implementation of the care. In fact, NCCN encourages conducting trials to examine protocols, screening instruments, and models of intervention to determine applicability to particular settings.2
Findings from a study by NCCN member institutions to evaluate progress of implementing distress management guidelines found that 53% (n = 8) of respondent institutions conducted routine distress screening. Of those, 37.5% (3) relied only on interviews. That finding is of concern because if interviews are not standardized and have not been systematically evaluated, then their sensitivity and specificity in identifying distressed patients is unknown.26 Accordingly, the process described in this article and the PHQ-9 validation was an effort to standardize emotional distress management, and was underlined as an achievement during the CoC accreditation visit to the cancer center in December 2013. The hospital was accredited as a comprehensive community cancer center with gold commendations, becoming the first privately owned hospital in Puerto Rico to achieve the accreditation.
1. Commission on Cancer, American College of Surgeons. Cancer Programs Standards 2012: Ensuring Patient-Centered Care. Version 1.2.1. https://www.facs.org/~/media/files/quality%20programs/cancer/coc/programstandards2012.ashx. Published 2012. Accessed March 5, 2013.
2. National Comprehensive Cancer Network clinical practice guidelines in oncology (NCCN guidelines): Distress management. Version I. 2012. https://www.nccn.org/professionals/physician_gls/f_guidelines.asp#supportive. Accessed March 5, 2013.
3. Bultz BD, Groff SL. Screening for distress, the 6th vital sign in oncology: from theory to practice: http://www.oncologyex.com/issue/2009/vol8_no1/8_comment2_1.html. Published February 2009. Accessed February 16, 2017.
4. Satin JR, Linden W, Phillips MJ. Depression as a predictor of disease progression and mortality in cancer patients: a meta-analysis. Cancer. 2009;115:5349-5361.
5. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Int Med. 2000;160:2101-2107.
6. Jadoon NA, Munir W, Shahzad MA, Choudhry ZS. Assessment of depression and anxiety in adult cancer outpatients: a cross sectional study. BMC Cancer. 2010;10:594.
7. Fisher D, Wedel B. Anxiety and depression disorders in cancer patients: incidence, diagnosis and therapy. Mag Eur Med Oncol. 2012;5:52-54.
8. Sollner W, DeVries A, Steixner E, et al. How successful are oncologists in identifying patient distress, perceived social support, and in need for psychosocial counselling? Br J Cancer. 2001;84:179-185.
9. Scott RD, Solomon SL, McGowan JE. Applying economic principles to health care: special issue. Emerg Infect Dis. 2001;7:282-285.
10. Adler NE, Page AEK. A model for delivering psychosocial health services. In: Cancer care for the whole patient: meeting psychosocial health needs. Washington, DC: National Academies Press (US); 2008.
11. Holland JC, Alici Y. Management of distress in cancer patients. J Support Oncol. 2010;8:4-12.
12. Jacobsen PB, Donovan KA, Trask PC, et al. Screening for psychologic distress in ambulatory cancer patients. Cancer. 2005;103:1494-1502.
13. Maihoff SE. Assessment. In Washington CM, Leaver D, eds. Principles and practice of radiation therapy. St Louis, MO: Mosby Elsevier; 2004:243-264.
14. National Academy of Sciences. Adler NE, Page AEK, eds. Cancer care for the whole patient: meeting psychosocial health needs. https://www.ncbi.nlm.nih.gov/books/NBK4015/. Published 2008. Accessed February 22, 2012.
15. Nancarrow SA, Booth A, Ariss
16. Baker-Glenn EA, Park B, Granger L, Symonds P, Mitchell AJ. Desire for psychological support in cancer patients with depression or distress: validation of a simple help question. Psychooncology. 2011;20:525-531.
17. Kroenke K, Spitzer RL, Williams JBW. The PHQ-9: Validity of a brief depression severity measure. J Gen Intern Med. 2001;16:606-613.
18. Omoro SA, Fann JR, Weymuller EA, Macharia IM, Yueh B. Swahili translation and validation of the Patient Health Questionnaire-9 depression scale in the Kenyan head and neck cancer patient population. Int J Psychiatry Med. 2006;36:367-381.
19. Thekkumpurath P, Walker J, Butcher I, et al. Screening for major depression on cancer outpatients: the diagnostic accuracy of the 9-item Patient Health Questionnaire. Cancer. 2011;117:218-227.
20. Fann JR, Berry DL, Wolpin S, et al. Depression screening using the Patient Health Questionnaire-9 administered on a touch screen computer. Psychooncology. 2009;18:14-22.
21. Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry. 1961;4:651-571.
22. Sanz J, Perdigón AL, Vázquez C. The Spanish adaptation of Beck’s Depression Inventory–II (BDI-II): psychometric properties in the general population. Clínica y Salud. 2003;14:249-280.
23. Bonilla J, Bernal G, Santos A, Santos D. A revised Spanish version of the Beck Depression Inventory: psychometric properties with a Puerto Rican sample of college students. J Clin Psychol. 2004;60:119-130.
24. Alcántara C, Gone JP. Multicultural issues in the clinical interview and diagnostic process. In Leong FTL, ed. APA handbook of multicultural psychology. Vol 2. Applications and training. Washington, DC: American Psychological Association; 2014:153-163.
25. Sharma M, Romas JA. Theoretical foundations of health education and health promotion. 2nd ed. Burlington, MA: Jones & Barlett Learning; 2012.
26. Jacobsen PB, Ransom S. Implementation of NCCN distress management guidelines by member institutions. J Natl Compr Canc Netw. 2007;5:99-103.
A comprehensive, patient-centered approach is required to accomplish cancer best standards of care.1 This approach reflects the holistic conceptualization of health in which the physical, emotional, and social dimensions of the human being are considered when providing medical care. As a result, to look after all patient needs, interdisciplinary and well-coordinated interventions are recommended. Cancer patients should be provided not only with diagnostic, treatment, and follow-up clinical service, but also with the supportive assistance that may positively influence all aspects of their health.
To appraise physical, social, emotional and spiritual issues and to develop supportive interventional action plans, the National Comprehensive Cancer Network (NCCN) recommends screening all cancer patients for distress.2 In particular, screening the emotional component of distress occupies a prominent place in this process because it is now recognized as the sixth vital sign in oncology.3 Even though the influence of emotional distress over cancer mortality rates and disease progression is still under scrutiny,4 its plausible implications over treatment compliance have been pointed out. Patients with higher levels of emotional distress show lower adherence to treatment and poorer health outcomes.5 Furthermore, prevalence rates of emotional distress in cancer patients from ambulatory settings6 and oncology surgical units have been studied and have provided justification for distress management.7 Studies have shown low ability among oncologists to identify patients in distress and oncologists’ tendency to judge distress higher than the patients themselves.8 As a consequence, to achieve systematic distress evaluations and appropriate referrals for care, guidelines for distress management should be implemented in clinical settings. It is recommended that tests are conducted to find brief screening instruments and procedures to assure accurate interventions according to patient specific needs.
This article presents the process of implementing a distress management program at HIMA-San Pablo Oncologic Hospital in Caguas, Puerto Rico, with particular emphasis on the management of emotional distress, which has been defined as the feeling of suffering that cancer patients may experience after diagnosis. In addition, we have included data from a pilot study that was completed for content validation of the Patient Health Questionnaire (PHQ-9) to estimate depression levels in Puerto Rican cancer patients.
Methods
HIMA-San Pablo operates a group of privately owned hospitals in Puerto Rico. It established a cancer center in Caguas in 2007, recruiting a multispecialty medical faculty to provide cancer care and bone marrow transplants for adult and pediatric patients. The cancer center, currently named HIMA-San Pablo Oncologic Hospital (HSPOH), is a hospital within a hospital licensed by the Puerto Rico Department of Health. In 2007, a cancer committee was established as the steering committee to ensure the delivery of cancer care according to best standards of care. The committee took responsibility for developing all activities needed to achieve the American College of Surgeons’ Commission on Cancer (CoC) accreditation under the category of Comprehensive Community Cancer Center. The committee established a psychosocial team to develop a protocol for the delivery of distress management for adult patients. (The psychosocial needs of pediatric patients are assessed through other procedures.)
To develop the protocol, principles of input-output model of research and quality analysis in health care were applied.9 The input-output model, with its origin in engineering, helped map systematic activities to transform empirical data on cancer psychosocial care into operational procedures. Focus was given to data gathering (input), information organization and analysis (throughput), and the schematization of emotional distress management care (output).
The input phase
In the input phase, elements of psychosocial care and operational definitions related to distress management in general were identified through literature review (Table 1). Basic parameters for distress management were clarified, resulting in a conceptual framework based in four remarks: First, according to NCCN, distress is a multifactorial unpleasant emotional experience of a psychological, social, and spiritual nature. It may interfere with the ability to cope effectively with cancer, its symptoms and its treatment. Its intensity may fluctuate from feelings of suffering and fear to incapacitating manifestations of anxiety and depression2 and its severity may hamper patient quality of life and treatment compliance.
Second, distress management requires the intervention of an interdisciplinary team with both medical and allied health professionals. This may include mental health specialists and other professionals with training and experience in cancer-related issues, who work with reciprocal channels of communication for the exchange of patient information.
Third, NCCN recommends using the Distress Thermometer for patient initial distress screening.10-12 It consists of a numeric scale ranging from 0 (no distress) to 10 (severe distress) in which patients classify their level of distress. The numeric scale is followed by a section in which patients identify areas of practical, familiar, emotional, spiritual/ religious, and physical concerns. Based on responses, interviews may follow to set distress management interventions.
Fourth, screening and assessment are different but sequential and complementary stages of distress management. Screening is viewed as a rapid strategy to identify cancer patients in distress. Assessment looks out for a broader appraisal and documentation of factors with repercussions over patient distress level and resiliency capability.13 In many instances, the patient’s emotional distress is better understood in the assessment phase.
The throughput phase
Within the throughput phase of information organization and analysis, an inventory of health professionals and other in-house consultants needed for distress management was completed. Roles and procedures for information sharing were determined, and we established collaborative agreements with professionals in the community who could contribute to distress management. Members of the psychosocial team held workshops to discuss elements of NCCN guidelines for distress management and to create an action plan for the implementation of the protocol. Data analyses were performed to create a demographic profile of the oncology population at the hospital and assess patient willingness to receive emotional support services,16 which led to the implementation of support group meetings at which additional substantive information was collected about issues affecting cancer patients’ emotions.
The NCCN Distress Thermometer for measuring distress was translated to Spanish. Its format was adapted, and it was identified as a distress screening tool (DST), which we named Distress Assessment Tool for Oncology Patients (Figure 1). The instrument helps for rapid screening of patient needs and proper determination of initial interventions. In addition, psychometric properties of several instruments were reviewed for instances when patient emotional distress could not be clearly determined. We decided to proceed with the validation of the 9-item Patient Health Questionnaire (PHQ-9) to estimate patient depression level. A proposal for content validation of the PHQ-9 was approved by the University of Puerto Rico institutional review board, and patients were recruited to participate in the pilot study.
The PHQ-9. The PHQ-9 is a self-report version of the PRIME-MD instrument developed to assess mental disorders in clinical settings. It is based on DSM-IV diagnostic criteria.17 The PHQ-9 is the depression module with nine depression symptoms to check off if they become the cause of emotional impairment. Respondents categorized depression symptoms in four frequency degrees representing numeric values: 0 (not at all), 1 (several days), 2 (more than half the days), 3 (nearly every day). Measures of depression severity are subsequently determined in a Likert-type scale according to numeric calculations of responses: 0-4 (none severe depression), 5-9 (mild), 10-14 (moderate), 15-19 (moderately severe), and 20-27 (severe-major depression).
The instrument is widely used because of its validity in small and large populations. It showed adequate reliability and validity in a small sample of head and neck cancer patients, with a Cronbach’s alpha of 0.80 and a correlation coefficient of 0.71.18 Similarly, it showed good performance in identifying major depression in 4264 cancer outpatients, with sensitivity of 93%, specificity of 81%, and a positive predictive value (PPV) of 25% and negative predictive value (NPV) of 99%.19 Even when administered on a touch screen computer, the instrument showed valid data of depression from patients in treatment.20
The Beck Depression Inventory. We used the Beck Depression Inventory (BDI-II) Spanish version as the gold standard measure for the validation study. It is a 22-items inventory that measures attitudes and symptoms of depression.21 It can be administered in 10 minutes and has shown good psychometric measures when administered in Spain and Puerto Rico.22, 23
The pilot study. In all, 44 cancer patients who were receiving outpatient treatment at the radiotherapy unit agreed to participate in the study. The participants signed a consent form after the confidentiality protection measures and the main objectives of the study had been explained to them. Patients were interviewed individually during November and December 2012, with the Spanish versions of the PHQ-9 and BDI-II administered by one of two interviewers. At the beginning of each interview, the patient was asked 10 questions so that we could gather demographic data and confirm participant eligibility: aged 21 years or older, born and raised in Puerto Rico, being a Spanish speaker, and having a primary cancer diagnosis with no previous disease. Three patients were excluded from the sample because they either had cancer previously or had a recurrence or metastasis. The final sample consisted of 41 outpatients (N = 41).
Data analysis for demographics was completed with STATA v.12 software. Measures of central tendency and dispersion as well as PHQ-9 internal consistency analysis were made through Cronbach alpha with SPSS.
From a total of 41 patients surveyed, 22 (54%) were men and 19 (46%) were women, with an overall median age of 61 years. Among the men, 15 (68%) had a prostate cancer diagnosis and among women, 9 (47.4%) had a breast cancer diagnosis. In regard to health insurance, 19 (46%) had Medicare or Veterans/federal insurance coverage, and 13 (32%) had Reforma, the Puerto Rican government health insurance program partially funded by Medicaid funds. In addition, 8 participants (20%) were unemployed or disabled. As previously stated, all of the patients were in ambulatory care. Only 3 (7%) were participating in support groups.
Of all the respondents, 16 (39%) reported some level of depression. In particular, 2 (5%) showed severe-major depression, 4 (10%) moderately severe depression, and 10 (24%) moderate depression. Of those with depression, 8 (50%) were women, 8 (50%) were men. All 6 of the patients with head and neck cancer showed moderate or moderately severe depression (Table 2).
When respondent PHQ-9 scoring reflected moderate to severe depression (>10), a letter was sent to the patient’s radio-oncologist for referral to counseling and clinical psychological evaluation. All participants had access to the support group program, to a radiotherapy education program meeting weekly, and written information about their cancer diagnosis and treatment. They also were interviewed by the psychosocial coordinator or patient navigator for further assessment.
The output phase
In the output phase, a graphic representing the process of emotional assessment at the institution was created and then modified. PHQ-9 was added to the process when it was found suitable to assess level of depression contributing to the identification of patients requiring psychological and psychiatric assistance which by other means would be missed. PHQ-9 was useful in the busy clinical setting as it was completed, scored and interpreted in minutes. It showed the potential for routine evaluations when looking to identify improvement or deterioration in depression levels thus helping to monitor responses to treatment and providing insights for follow up interventions. As stated by NCCN guidelines, distress should be monitored, documented and managed at all stages of the cancer continuum.
Results and discussion
The protocol for distress management at HSPOH is based on the 2013 NCCN guidelines. Cancer patients are screened for levels of distress in all settings (inpatients and outpatients). Screening is held with the DST Spanish translation at the moment of diagnosis or as soon as possible after a diagnosis is made. Screening for distress is also done before or after surgery, in recurrence or progression, and when clinically indicated. Patients are informed that distress management is an essential part of their care and are encouraged to provide information so that we can make a proper need assessment.
Patients are screened by the psychosocial coordinator or patient navigator who administers the DST followed by in-depth interviews for additional appraisal. An action plan is designed based on patient needs, which include their intervention and the intervention of other members of the psychosocial team from the institution and/or from the community. Additional in-house health professionals contributing in distress management include, but are not limited to: physicians; clinical psychologists; health educators; social workers; dietitians; chaplains; and physical, respiratory, speech, and/or swallow therapists. Follow-up and rescreening sessions are scheduled to assure coordination of services between those health professionals as well as to secure continuity of distress management during all stages of the cancer continuum.
The results of the DST are filed in patient medical records. Members of the psychosocial team also document their interventions in the patient medical record, which helps in the exchange of information among the cancer care team. The psychosocial team meets once a month – or as required for extraordinary cases – to review and discuss the cases, determine the best options for distress management, and identify areas for psychosocial care improvement. Those findings and the results of distress management in patient level of satisfaction are then reported and discussed quarterly by the psychosocial coordinator and the cancer committee.
Figure 2 shows in what phase of emotional distress assessment the PHQ-9 was included. Patients reporting four or more of the six areas of concern related to emotional distress in the DST (Figure 1) are automatically referred to a mental health specialist. But when patients report three areas of concern with no clear data on their specific level of depression, PHQ-9 is administered to differentiate those who need a mental health specialist from those who could be adequately supported by health education and support group interventions. In this way detrimental outcomes such as duplicity and over or underuse of services and resources are reduced. In addition, it is recognized that using an interview after the administration of the DST to determine distress management actions does not always provide enough information about a patient’s emotional circumstances and previous comorbidities. Patient responses during interviews may be influenced by the patient’s level of literacy, verbal comprehension, and communication style,24 so emotional distress can go unrecognized during interviews, resulting in delays for treatment and supportive care.
National guidelines in oncology consider such socio-ecological models emphasizing the delivery of patient-centered, interdisciplinary, and evidence-based care. That does not mean that institutions should apply protocols of psychosocial care as previously developed, but that they should test, review, adapt, and improve them during the implementation of the care. In fact, NCCN encourages conducting trials to examine protocols, screening instruments, and models of intervention to determine applicability to particular settings.2
Findings from a study by NCCN member institutions to evaluate progress of implementing distress management guidelines found that 53% (n = 8) of respondent institutions conducted routine distress screening. Of those, 37.5% (3) relied only on interviews. That finding is of concern because if interviews are not standardized and have not been systematically evaluated, then their sensitivity and specificity in identifying distressed patients is unknown.26 Accordingly, the process described in this article and the PHQ-9 validation was an effort to standardize emotional distress management, and was underlined as an achievement during the CoC accreditation visit to the cancer center in December 2013. The hospital was accredited as a comprehensive community cancer center with gold commendations, becoming the first privately owned hospital in Puerto Rico to achieve the accreditation.
A comprehensive, patient-centered approach is required to accomplish cancer best standards of care.1 This approach reflects the holistic conceptualization of health in which the physical, emotional, and social dimensions of the human being are considered when providing medical care. As a result, to look after all patient needs, interdisciplinary and well-coordinated interventions are recommended. Cancer patients should be provided not only with diagnostic, treatment, and follow-up clinical service, but also with the supportive assistance that may positively influence all aspects of their health.
To appraise physical, social, emotional and spiritual issues and to develop supportive interventional action plans, the National Comprehensive Cancer Network (NCCN) recommends screening all cancer patients for distress.2 In particular, screening the emotional component of distress occupies a prominent place in this process because it is now recognized as the sixth vital sign in oncology.3 Even though the influence of emotional distress over cancer mortality rates and disease progression is still under scrutiny,4 its plausible implications over treatment compliance have been pointed out. Patients with higher levels of emotional distress show lower adherence to treatment and poorer health outcomes.5 Furthermore, prevalence rates of emotional distress in cancer patients from ambulatory settings6 and oncology surgical units have been studied and have provided justification for distress management.7 Studies have shown low ability among oncologists to identify patients in distress and oncologists’ tendency to judge distress higher than the patients themselves.8 As a consequence, to achieve systematic distress evaluations and appropriate referrals for care, guidelines for distress management should be implemented in clinical settings. It is recommended that tests are conducted to find brief screening instruments and procedures to assure accurate interventions according to patient specific needs.
This article presents the process of implementing a distress management program at HIMA-San Pablo Oncologic Hospital in Caguas, Puerto Rico, with particular emphasis on the management of emotional distress, which has been defined as the feeling of suffering that cancer patients may experience after diagnosis. In addition, we have included data from a pilot study that was completed for content validation of the Patient Health Questionnaire (PHQ-9) to estimate depression levels in Puerto Rican cancer patients.
Methods
HIMA-San Pablo operates a group of privately owned hospitals in Puerto Rico. It established a cancer center in Caguas in 2007, recruiting a multispecialty medical faculty to provide cancer care and bone marrow transplants for adult and pediatric patients. The cancer center, currently named HIMA-San Pablo Oncologic Hospital (HSPOH), is a hospital within a hospital licensed by the Puerto Rico Department of Health. In 2007, a cancer committee was established as the steering committee to ensure the delivery of cancer care according to best standards of care. The committee took responsibility for developing all activities needed to achieve the American College of Surgeons’ Commission on Cancer (CoC) accreditation under the category of Comprehensive Community Cancer Center. The committee established a psychosocial team to develop a protocol for the delivery of distress management for adult patients. (The psychosocial needs of pediatric patients are assessed through other procedures.)
To develop the protocol, principles of input-output model of research and quality analysis in health care were applied.9 The input-output model, with its origin in engineering, helped map systematic activities to transform empirical data on cancer psychosocial care into operational procedures. Focus was given to data gathering (input), information organization and analysis (throughput), and the schematization of emotional distress management care (output).
The input phase
In the input phase, elements of psychosocial care and operational definitions related to distress management in general were identified through literature review (Table 1). Basic parameters for distress management were clarified, resulting in a conceptual framework based in four remarks: First, according to NCCN, distress is a multifactorial unpleasant emotional experience of a psychological, social, and spiritual nature. It may interfere with the ability to cope effectively with cancer, its symptoms and its treatment. Its intensity may fluctuate from feelings of suffering and fear to incapacitating manifestations of anxiety and depression2 and its severity may hamper patient quality of life and treatment compliance.
Second, distress management requires the intervention of an interdisciplinary team with both medical and allied health professionals. This may include mental health specialists and other professionals with training and experience in cancer-related issues, who work with reciprocal channels of communication for the exchange of patient information.
Third, NCCN recommends using the Distress Thermometer for patient initial distress screening.10-12 It consists of a numeric scale ranging from 0 (no distress) to 10 (severe distress) in which patients classify their level of distress. The numeric scale is followed by a section in which patients identify areas of practical, familiar, emotional, spiritual/ religious, and physical concerns. Based on responses, interviews may follow to set distress management interventions.
Fourth, screening and assessment are different but sequential and complementary stages of distress management. Screening is viewed as a rapid strategy to identify cancer patients in distress. Assessment looks out for a broader appraisal and documentation of factors with repercussions over patient distress level and resiliency capability.13 In many instances, the patient’s emotional distress is better understood in the assessment phase.
The throughput phase
Within the throughput phase of information organization and analysis, an inventory of health professionals and other in-house consultants needed for distress management was completed. Roles and procedures for information sharing were determined, and we established collaborative agreements with professionals in the community who could contribute to distress management. Members of the psychosocial team held workshops to discuss elements of NCCN guidelines for distress management and to create an action plan for the implementation of the protocol. Data analyses were performed to create a demographic profile of the oncology population at the hospital and assess patient willingness to receive emotional support services,16 which led to the implementation of support group meetings at which additional substantive information was collected about issues affecting cancer patients’ emotions.
The NCCN Distress Thermometer for measuring distress was translated to Spanish. Its format was adapted, and it was identified as a distress screening tool (DST), which we named Distress Assessment Tool for Oncology Patients (Figure 1). The instrument helps for rapid screening of patient needs and proper determination of initial interventions. In addition, psychometric properties of several instruments were reviewed for instances when patient emotional distress could not be clearly determined. We decided to proceed with the validation of the 9-item Patient Health Questionnaire (PHQ-9) to estimate patient depression level. A proposal for content validation of the PHQ-9 was approved by the University of Puerto Rico institutional review board, and patients were recruited to participate in the pilot study.
The PHQ-9. The PHQ-9 is a self-report version of the PRIME-MD instrument developed to assess mental disorders in clinical settings. It is based on DSM-IV diagnostic criteria.17 The PHQ-9 is the depression module with nine depression symptoms to check off if they become the cause of emotional impairment. Respondents categorized depression symptoms in four frequency degrees representing numeric values: 0 (not at all), 1 (several days), 2 (more than half the days), 3 (nearly every day). Measures of depression severity are subsequently determined in a Likert-type scale according to numeric calculations of responses: 0-4 (none severe depression), 5-9 (mild), 10-14 (moderate), 15-19 (moderately severe), and 20-27 (severe-major depression).
The instrument is widely used because of its validity in small and large populations. It showed adequate reliability and validity in a small sample of head and neck cancer patients, with a Cronbach’s alpha of 0.80 and a correlation coefficient of 0.71.18 Similarly, it showed good performance in identifying major depression in 4264 cancer outpatients, with sensitivity of 93%, specificity of 81%, and a positive predictive value (PPV) of 25% and negative predictive value (NPV) of 99%.19 Even when administered on a touch screen computer, the instrument showed valid data of depression from patients in treatment.20
The Beck Depression Inventory. We used the Beck Depression Inventory (BDI-II) Spanish version as the gold standard measure for the validation study. It is a 22-items inventory that measures attitudes and symptoms of depression.21 It can be administered in 10 minutes and has shown good psychometric measures when administered in Spain and Puerto Rico.22, 23
The pilot study. In all, 44 cancer patients who were receiving outpatient treatment at the radiotherapy unit agreed to participate in the study. The participants signed a consent form after the confidentiality protection measures and the main objectives of the study had been explained to them. Patients were interviewed individually during November and December 2012, with the Spanish versions of the PHQ-9 and BDI-II administered by one of two interviewers. At the beginning of each interview, the patient was asked 10 questions so that we could gather demographic data and confirm participant eligibility: aged 21 years or older, born and raised in Puerto Rico, being a Spanish speaker, and having a primary cancer diagnosis with no previous disease. Three patients were excluded from the sample because they either had cancer previously or had a recurrence or metastasis. The final sample consisted of 41 outpatients (N = 41).
Data analysis for demographics was completed with STATA v.12 software. Measures of central tendency and dispersion as well as PHQ-9 internal consistency analysis were made through Cronbach alpha with SPSS.
From a total of 41 patients surveyed, 22 (54%) were men and 19 (46%) were women, with an overall median age of 61 years. Among the men, 15 (68%) had a prostate cancer diagnosis and among women, 9 (47.4%) had a breast cancer diagnosis. In regard to health insurance, 19 (46%) had Medicare or Veterans/federal insurance coverage, and 13 (32%) had Reforma, the Puerto Rican government health insurance program partially funded by Medicaid funds. In addition, 8 participants (20%) were unemployed or disabled. As previously stated, all of the patients were in ambulatory care. Only 3 (7%) were participating in support groups.
Of all the respondents, 16 (39%) reported some level of depression. In particular, 2 (5%) showed severe-major depression, 4 (10%) moderately severe depression, and 10 (24%) moderate depression. Of those with depression, 8 (50%) were women, 8 (50%) were men. All 6 of the patients with head and neck cancer showed moderate or moderately severe depression (Table 2).
When respondent PHQ-9 scoring reflected moderate to severe depression (>10), a letter was sent to the patient’s radio-oncologist for referral to counseling and clinical psychological evaluation. All participants had access to the support group program, to a radiotherapy education program meeting weekly, and written information about their cancer diagnosis and treatment. They also were interviewed by the psychosocial coordinator or patient navigator for further assessment.
The output phase
In the output phase, a graphic representing the process of emotional assessment at the institution was created and then modified. PHQ-9 was added to the process when it was found suitable to assess level of depression contributing to the identification of patients requiring psychological and psychiatric assistance which by other means would be missed. PHQ-9 was useful in the busy clinical setting as it was completed, scored and interpreted in minutes. It showed the potential for routine evaluations when looking to identify improvement or deterioration in depression levels thus helping to monitor responses to treatment and providing insights for follow up interventions. As stated by NCCN guidelines, distress should be monitored, documented and managed at all stages of the cancer continuum.
Results and discussion
The protocol for distress management at HSPOH is based on the 2013 NCCN guidelines. Cancer patients are screened for levels of distress in all settings (inpatients and outpatients). Screening is held with the DST Spanish translation at the moment of diagnosis or as soon as possible after a diagnosis is made. Screening for distress is also done before or after surgery, in recurrence or progression, and when clinically indicated. Patients are informed that distress management is an essential part of their care and are encouraged to provide information so that we can make a proper need assessment.
Patients are screened by the psychosocial coordinator or patient navigator who administers the DST followed by in-depth interviews for additional appraisal. An action plan is designed based on patient needs, which include their intervention and the intervention of other members of the psychosocial team from the institution and/or from the community. Additional in-house health professionals contributing in distress management include, but are not limited to: physicians; clinical psychologists; health educators; social workers; dietitians; chaplains; and physical, respiratory, speech, and/or swallow therapists. Follow-up and rescreening sessions are scheduled to assure coordination of services between those health professionals as well as to secure continuity of distress management during all stages of the cancer continuum.
The results of the DST are filed in patient medical records. Members of the psychosocial team also document their interventions in the patient medical record, which helps in the exchange of information among the cancer care team. The psychosocial team meets once a month – or as required for extraordinary cases – to review and discuss the cases, determine the best options for distress management, and identify areas for psychosocial care improvement. Those findings and the results of distress management in patient level of satisfaction are then reported and discussed quarterly by the psychosocial coordinator and the cancer committee.
Figure 2 shows in what phase of emotional distress assessment the PHQ-9 was included. Patients reporting four or more of the six areas of concern related to emotional distress in the DST (Figure 1) are automatically referred to a mental health specialist. But when patients report three areas of concern with no clear data on their specific level of depression, PHQ-9 is administered to differentiate those who need a mental health specialist from those who could be adequately supported by health education and support group interventions. In this way detrimental outcomes such as duplicity and over or underuse of services and resources are reduced. In addition, it is recognized that using an interview after the administration of the DST to determine distress management actions does not always provide enough information about a patient’s emotional circumstances and previous comorbidities. Patient responses during interviews may be influenced by the patient’s level of literacy, verbal comprehension, and communication style,24 so emotional distress can go unrecognized during interviews, resulting in delays for treatment and supportive care.
National guidelines in oncology consider such socio-ecological models emphasizing the delivery of patient-centered, interdisciplinary, and evidence-based care. That does not mean that institutions should apply protocols of psychosocial care as previously developed, but that they should test, review, adapt, and improve them during the implementation of the care. In fact, NCCN encourages conducting trials to examine protocols, screening instruments, and models of intervention to determine applicability to particular settings.2
Findings from a study by NCCN member institutions to evaluate progress of implementing distress management guidelines found that 53% (n = 8) of respondent institutions conducted routine distress screening. Of those, 37.5% (3) relied only on interviews. That finding is of concern because if interviews are not standardized and have not been systematically evaluated, then their sensitivity and specificity in identifying distressed patients is unknown.26 Accordingly, the process described in this article and the PHQ-9 validation was an effort to standardize emotional distress management, and was underlined as an achievement during the CoC accreditation visit to the cancer center in December 2013. The hospital was accredited as a comprehensive community cancer center with gold commendations, becoming the first privately owned hospital in Puerto Rico to achieve the accreditation.
1. Commission on Cancer, American College of Surgeons. Cancer Programs Standards 2012: Ensuring Patient-Centered Care. Version 1.2.1. https://www.facs.org/~/media/files/quality%20programs/cancer/coc/programstandards2012.ashx. Published 2012. Accessed March 5, 2013.
2. National Comprehensive Cancer Network clinical practice guidelines in oncology (NCCN guidelines): Distress management. Version I. 2012. https://www.nccn.org/professionals/physician_gls/f_guidelines.asp#supportive. Accessed March 5, 2013.
3. Bultz BD, Groff SL. Screening for distress, the 6th vital sign in oncology: from theory to practice: http://www.oncologyex.com/issue/2009/vol8_no1/8_comment2_1.html. Published February 2009. Accessed February 16, 2017.
4. Satin JR, Linden W, Phillips MJ. Depression as a predictor of disease progression and mortality in cancer patients: a meta-analysis. Cancer. 2009;115:5349-5361.
5. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Int Med. 2000;160:2101-2107.
6. Jadoon NA, Munir W, Shahzad MA, Choudhry ZS. Assessment of depression and anxiety in adult cancer outpatients: a cross sectional study. BMC Cancer. 2010;10:594.
7. Fisher D, Wedel B. Anxiety and depression disorders in cancer patients: incidence, diagnosis and therapy. Mag Eur Med Oncol. 2012;5:52-54.
8. Sollner W, DeVries A, Steixner E, et al. How successful are oncologists in identifying patient distress, perceived social support, and in need for psychosocial counselling? Br J Cancer. 2001;84:179-185.
9. Scott RD, Solomon SL, McGowan JE. Applying economic principles to health care: special issue. Emerg Infect Dis. 2001;7:282-285.
10. Adler NE, Page AEK. A model for delivering psychosocial health services. In: Cancer care for the whole patient: meeting psychosocial health needs. Washington, DC: National Academies Press (US); 2008.
11. Holland JC, Alici Y. Management of distress in cancer patients. J Support Oncol. 2010;8:4-12.
12. Jacobsen PB, Donovan KA, Trask PC, et al. Screening for psychologic distress in ambulatory cancer patients. Cancer. 2005;103:1494-1502.
13. Maihoff SE. Assessment. In Washington CM, Leaver D, eds. Principles and practice of radiation therapy. St Louis, MO: Mosby Elsevier; 2004:243-264.
14. National Academy of Sciences. Adler NE, Page AEK, eds. Cancer care for the whole patient: meeting psychosocial health needs. https://www.ncbi.nlm.nih.gov/books/NBK4015/. Published 2008. Accessed February 22, 2012.
15. Nancarrow SA, Booth A, Ariss
16. Baker-Glenn EA, Park B, Granger L, Symonds P, Mitchell AJ. Desire for psychological support in cancer patients with depression or distress: validation of a simple help question. Psychooncology. 2011;20:525-531.
17. Kroenke K, Spitzer RL, Williams JBW. The PHQ-9: Validity of a brief depression severity measure. J Gen Intern Med. 2001;16:606-613.
18. Omoro SA, Fann JR, Weymuller EA, Macharia IM, Yueh B. Swahili translation and validation of the Patient Health Questionnaire-9 depression scale in the Kenyan head and neck cancer patient population. Int J Psychiatry Med. 2006;36:367-381.
19. Thekkumpurath P, Walker J, Butcher I, et al. Screening for major depression on cancer outpatients: the diagnostic accuracy of the 9-item Patient Health Questionnaire. Cancer. 2011;117:218-227.
20. Fann JR, Berry DL, Wolpin S, et al. Depression screening using the Patient Health Questionnaire-9 administered on a touch screen computer. Psychooncology. 2009;18:14-22.
21. Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry. 1961;4:651-571.
22. Sanz J, Perdigón AL, Vázquez C. The Spanish adaptation of Beck’s Depression Inventory–II (BDI-II): psychometric properties in the general population. Clínica y Salud. 2003;14:249-280.
23. Bonilla J, Bernal G, Santos A, Santos D. A revised Spanish version of the Beck Depression Inventory: psychometric properties with a Puerto Rican sample of college students. J Clin Psychol. 2004;60:119-130.
24. Alcántara C, Gone JP. Multicultural issues in the clinical interview and diagnostic process. In Leong FTL, ed. APA handbook of multicultural psychology. Vol 2. Applications and training. Washington, DC: American Psychological Association; 2014:153-163.
25. Sharma M, Romas JA. Theoretical foundations of health education and health promotion. 2nd ed. Burlington, MA: Jones & Barlett Learning; 2012.
26. Jacobsen PB, Ransom S. Implementation of NCCN distress management guidelines by member institutions. J Natl Compr Canc Netw. 2007;5:99-103.
1. Commission on Cancer, American College of Surgeons. Cancer Programs Standards 2012: Ensuring Patient-Centered Care. Version 1.2.1. https://www.facs.org/~/media/files/quality%20programs/cancer/coc/programstandards2012.ashx. Published 2012. Accessed March 5, 2013.
2. National Comprehensive Cancer Network clinical practice guidelines in oncology (NCCN guidelines): Distress management. Version I. 2012. https://www.nccn.org/professionals/physician_gls/f_guidelines.asp#supportive. Accessed March 5, 2013.
3. Bultz BD, Groff SL. Screening for distress, the 6th vital sign in oncology: from theory to practice: http://www.oncologyex.com/issue/2009/vol8_no1/8_comment2_1.html. Published February 2009. Accessed February 16, 2017.
4. Satin JR, Linden W, Phillips MJ. Depression as a predictor of disease progression and mortality in cancer patients: a meta-analysis. Cancer. 2009;115:5349-5361.
5. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Int Med. 2000;160:2101-2107.
6. Jadoon NA, Munir W, Shahzad MA, Choudhry ZS. Assessment of depression and anxiety in adult cancer outpatients: a cross sectional study. BMC Cancer. 2010;10:594.
7. Fisher D, Wedel B. Anxiety and depression disorders in cancer patients: incidence, diagnosis and therapy. Mag Eur Med Oncol. 2012;5:52-54.
8. Sollner W, DeVries A, Steixner E, et al. How successful are oncologists in identifying patient distress, perceived social support, and in need for psychosocial counselling? Br J Cancer. 2001;84:179-185.
9. Scott RD, Solomon SL, McGowan JE. Applying economic principles to health care: special issue. Emerg Infect Dis. 2001;7:282-285.
10. Adler NE, Page AEK. A model for delivering psychosocial health services. In: Cancer care for the whole patient: meeting psychosocial health needs. Washington, DC: National Academies Press (US); 2008.
11. Holland JC, Alici Y. Management of distress in cancer patients. J Support Oncol. 2010;8:4-12.
12. Jacobsen PB, Donovan KA, Trask PC, et al. Screening for psychologic distress in ambulatory cancer patients. Cancer. 2005;103:1494-1502.
13. Maihoff SE. Assessment. In Washington CM, Leaver D, eds. Principles and practice of radiation therapy. St Louis, MO: Mosby Elsevier; 2004:243-264.
14. National Academy of Sciences. Adler NE, Page AEK, eds. Cancer care for the whole patient: meeting psychosocial health needs. https://www.ncbi.nlm.nih.gov/books/NBK4015/. Published 2008. Accessed February 22, 2012.
15. Nancarrow SA, Booth A, Ariss
16. Baker-Glenn EA, Park B, Granger L, Symonds P, Mitchell AJ. Desire for psychological support in cancer patients with depression or distress: validation of a simple help question. Psychooncology. 2011;20:525-531.
17. Kroenke K, Spitzer RL, Williams JBW. The PHQ-9: Validity of a brief depression severity measure. J Gen Intern Med. 2001;16:606-613.
18. Omoro SA, Fann JR, Weymuller EA, Macharia IM, Yueh B. Swahili translation and validation of the Patient Health Questionnaire-9 depression scale in the Kenyan head and neck cancer patient population. Int J Psychiatry Med. 2006;36:367-381.
19. Thekkumpurath P, Walker J, Butcher I, et al. Screening for major depression on cancer outpatients: the diagnostic accuracy of the 9-item Patient Health Questionnaire. Cancer. 2011;117:218-227.
20. Fann JR, Berry DL, Wolpin S, et al. Depression screening using the Patient Health Questionnaire-9 administered on a touch screen computer. Psychooncology. 2009;18:14-22.
21. Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry. 1961;4:651-571.
22. Sanz J, Perdigón AL, Vázquez C. The Spanish adaptation of Beck’s Depression Inventory–II (BDI-II): psychometric properties in the general population. Clínica y Salud. 2003;14:249-280.
23. Bonilla J, Bernal G, Santos A, Santos D. A revised Spanish version of the Beck Depression Inventory: psychometric properties with a Puerto Rican sample of college students. J Clin Psychol. 2004;60:119-130.
24. Alcántara C, Gone JP. Multicultural issues in the clinical interview and diagnostic process. In Leong FTL, ed. APA handbook of multicultural psychology. Vol 2. Applications and training. Washington, DC: American Psychological Association; 2014:153-163.
25. Sharma M, Romas JA. Theoretical foundations of health education and health promotion. 2nd ed. Burlington, MA: Jones & Barlett Learning; 2012.
26. Jacobsen PB, Ransom S. Implementation of NCCN distress management guidelines by member institutions. J Natl Compr Canc Netw. 2007;5:99-103.
Electronic Health Record Implementation Is Associated With a Negligible Change in Outpatient Volume and Billing
Take-Home Points
- With EHR implementation there are small changes in the level of billing coding.
- Although these changes may be statistically significant they are relatively minor.
- In the general internal medicine department, level 4 coding increased by 1.2% while level 3 coding decreased by 0.5%.
- In the orthopedics department, level 4 coding increased by 3.3% while level 3 coding decreased by 3.1%.
- Reports in the lay media regarding dramatic up-coding after EHR implementation may be misleading.
The Health Information Technology for Economic and Clinical Health (HITECH) Act, which was signed into law in 2009, mandated that hospitals that care for Medicare patients either begin using electronic health records (EHRs) or pay a nontrivial penalty.1 By now, the majority of orthopedic surgeons have implemented EHRs in their practices.2 Despite ongoing debate in the orthopedic literature,3 EHRs are expected to improve coordination of care, reduce duplicate testing, and reduce costs over the long term as healthcare insurance coverage is extended to millions more Americans.
In early coverage, however, media reported that EHR implementation at some hospitals was correlated with substantial increases in Medicare payments.4 Journalists suggested the billion dollars more paid by Medicare to hospitals in 2010 than in 2005 were partly attributable to up-coding facilitated by EHRs.5 The secretary of the Department of Health and Human Services (DHHS) and the attorney general of the Department of Justice also weighed in on this controversy by expressing their concerns in a letter to the presidents of 5 hospital associations.6 The inspector general of DHHS also published a report critical of Medicare officials’ oversight of EHRs.7Responding to the critical reception of EHR implementations, investigators studied the validity of the early reports and anecdotes. Some initial reports cited the emergency department (ED) as an area at high risk for using the convenience of EHRs to up-code visits.5 The DHHS Office of the Inspector General noted that, between 2001 and 2010, the proportion of claims for lower reimbursement categories of American Medical Association Current Procedural Terminology (CPT) codes decreased while the proportion for higher-paid billing codes increased for all visit types.8 Addressing these concerns, the American Hospital Association9 issued a brief that noted that any observed coding increases were more likely attributable to more ED use by Medicare patients and increased average illness severity. In a thoughtful perspective, Pitts10 conceded that, though utilization and illness severity may explain part of the trend, the trend may also be related to technological innovations and changes in culture and practice style in the ED.
Because these studies and reports variously suggested that EHR implementation affects patient volume and up-coding, and because none of the reports specifically addressed orthopedics, we conducted a study to determine whether any significant up-coding or change in patient volumes occurred around the time of EHR implementation in ambulatory practices at our academic medical center. In a recent national study, Adler-Milstein and Jha11 compared billing data of hospitals that adopted EHRs and hospitals that did not. Although both groups showed increased billing trends, the increases were not significantly different between the EHR adopters and nonadopters. To more effectively control for the confounding differences between groups of EHR adopters and nonadopters, we studied individual departments during EHR implementation at our institution.
Methods
In 2011, our academic medical center began the transition to EHRs (Epic). We examined our center’s trends in patient volumes and billing coding around the time of the transition in the outpatient practice of the general internal medicine (GIM) department (EHR transition, October 2011) and the outpatient practice of the orthopedics department (EHR transition, March 2012). These departments were chosen because they are representative of a GIM practice and a subspecialty practice, and because a recent study found that GIM practitioners and orthopedic surgeons were among those specialists who used EHRs the most.12
After this study was approved by our Human Investigations Committee, we began using CPT codes to identify all outpatient visits (new, consultation, and return) on a monthly basis. We compared the volume of patient visits and the billing coding level in the GIM and orthopedics departments before and after EHR implementation. Pearson χ2 test was used when appropriate, and statistical analyses were performed with SPSS for Windows Version 16.0.
Results
In the GIM department, mean monthly volume of patient visits in the 12 months before EHR implementation was similar to that in the 12 months afterward (613 vs 587; P = .439). Even when normalized for changes in provider availability (maternity leave), the decrease in volume of patient visits after EHR implementation in the GIM department was not significant (6.9%; P = .107). Likewise, in the orthopedics department, mean monthly volume of patient visits in the 17 months before EHR implementation was similar to that in the 7 months afterward (2157 vs 2317; P = .156). In fact, patient volumes remained constant during the EHR transition (Figure 1).
EHR implementation brought small changes in billing coding levels. In the GIM department, the largest change was a 1.2% increase in level 4 billing coding—an increase accompanied by a 0.5% decrease in level 3 coding. 
Discussion
It is remarkable that the volumes of patient visits in the GIM and orthopedics departments at our academic center were not affected by EHR implementation. 
Rather than reduce scheduling during the EHR transition, surgeons in our practice either added or lengthened clinic sessions, and the level of ancillary staffing was adjusted accordingly. As staffing costs at any given time are multifactorial and vary widely, estimating the cost of these staffing changes during the EHR transition is difficult. We should note that extending ancillary staff hours during the transition very likely increased costs, and it is unclear whether they were higher or lower than the costs that would have been incurred had we reduced scheduling or tried some combination of these strategies.
Although billing coding levels changed with EHR implementation, the changes were small. In the GIM department, level 4 CPT coded visits as percentages of all visits increased to 59.5% from 58.3%, and level 5 visits increased to 6.2% from 6.0%; in the orthopedics department, level 4 visits increased to 40.2% from 37.1%, and level 5 visits increased to 5.5% from 3.8% (Table). The 1.2% and 0.2% absolute increases in level 4 and level 5 visits in the GIM department represent 2.1% and 3.3% relative increases in level 4 and level 5 visits, and the 3.3% and 1.7% absolute increases in the orthopedics department represent 8.4% and 44.7% relative increases in level 4 and level 5 visits after EHR implementation.
Although the absolute increases in level 4 and level 5 visits were relatively minor, popular media have raised the alarm about 43% and 82% relative increases in level 5 visits after EHR implementation in some hospitals’ EDs.4 Although our orthopedics department showed a 44.7% relative increase in level 5 visits after EHR implementation, this represented an increase of only 1.7% of patient visits overall. Our findings therefore indicate that lay media reports could be misleading. Nevertheless, the small changes we found were statistically significant.
One explanation for these small changes is that EHRs facilitate better documentation of services provided. Therefore, what seem to be billing coding changes could be more accurate reports of high-level care that is the same as before. In addition, because of meaningful use mandates that coincided with the requirement to implement EHRs, additional data elements are now being consistently collected and reviewed (these may not necessarily have been collected and reviewed before). In some patient encounters, these additional data elements may have contributed to higher levels of service, and this effect could be especially apparent in EDs.
Some have suggested a potential for large-scale up-coding during EHR transitions. Others have contended that coding level increases are a consequence of a time-intensive data entry process, collection and review of additional data, and more accurate reporting of services already being provided. We are not convinced that large coding changes are attributable solely to EHR implementation, as the changes at our center have been relatively small.
Nevertheless, minor coding level changes could translate to large changes in healthcare costs when scaled nationally. Although causes may be innocuous, any increases in national healthcare costs are concerning in our time of limited budgets and scrutinized healthcare utilization.
This study had its limitations. First, including billing data from only 2 departments at a single center may limit the generalizability of findings. However, we specifically selected a GIM department and a specialty (orthopedics) department in an attempt to capture a representative sample of practices. Another limitation is that we investigated billing codes over only 2 years, around the implementation of EHRs in these departments, and therefore may have captured only short-term changes. However, as patient volumes and billing are subject to many factors, including staffing changes (eg, new partners, new hires, retirements, other departures), we attempted to limit the effect of confounding variables by limiting the period of analysis.
Overall, changes in patient volume and coded level of service during EHR implementation at our institution were relatively small. Although the trend toward higher billing coding levels was statistically significant, these 0.2% and 1.7% increases in level 5 coding hardly deserve the negative attention from lay media. These small increases are unlikely caused by intentional up-coding, and more likely reflect better documentation of an already high level of care. We hope these findings allay the concern that up-coding increased dramatically with EHR implementation.
Am J Orthop. 2017;46(3):E172-E176. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Centers for Medicare & Medicaid Services. Electronic health records (EHR) incentive programs. http://www.cms.gov/Regulations-and-Guidance/Legislation/EHRIncentivePrograms. Accessed February 5, 2015.
2. American Academy of Orthopaedic Surgeons Practice Management Committee. EMR: A Primer for Orthopaedic Surgeons. 2nd ed. Rosemont, IL: American Academy of Orthopaedic Surgeons; 2010.
3. Ries MD. Electronic medical records: friends or foes? Clin Orthop Relat Res. 2014;472(1):16-21.
4. Abelson R. Medicare is faulted on shift to electronic records. New York Times. November 29, 2012;B1. http://www.nytimes.com/2012/11/29/business/medicare-is-faulted-in-electronic-medical-records-conversion.html. Accessed February 5, 2015.
5. Abelson R, Creswell J, Palmer G. Medicare bills rise as records turn electronic. New York Times. September 22, 2012;A1. http://www.nytimes.com/2012/09/22/business/medicare-billing-rises-at-hospitals-with-electronic-records.html. Accessed February 5, 2015.
6. Carlson J. Warning bell. Potential for fraud through use of EHRs draws federal scrutiny. Mod Healthc. 2012;42(40):8-9.
7. Levinson DR. Early assessment finds that CMS faces obstacles in overseeing the Medicare EHR Incentive Program. Dept of Health and Human Services, Office of Inspector General website. https://oig.hss.gov/oei/reports/oei-05-11-00250.pdf. Publication OEI-05-11-00250. Published November 2012. Accessed February 5, 2015.
8. Levinson DR. Coding trends of Medicare evaluation and management services. Dept of Health and Human Services, Office of Inspector General website. https://oig.hhs.gov/oei/reports/oei-04-10-00180.pdf. Publication OEI-04-10-00180. Published May 2012. Accessed February 5, 2015.
9. American Hospital Association. Sicker, more complex patients are driving up intensity of ED care [issue brief]. http://www.aha.org/content/13/13issuebrief-ed.pdf. Published May 2, 2013. Accessed February 5, 2015.
10. Pitts SR. Higher-complexity ED billing codes—sicker patients, more intensive practice, or improper payments? N Engl J Med. 2012;367(26):2465-2467.
11. Adler-Milstein J, Jha AK. No evidence found that hospitals are using new electronic health records to increase Medicare reimbursements. Health Aff (Millwood). 2014;33(7):1271-1277.
12. Kokkonen EW, Davis SA, Lin HC, Dabade TS, Feldman SR, Fleischer AB Jr. Use of electronic medical records differs by specialty and office settings. J Am Med Inform Assoc. 2013;20(e1):e33-e38.
13. Samaan ZM, Klein MD, Mansour ME, DeWitt TG. The impact of the electronic health record on an academic pediatric primary care center. J Ambul Care Manage. 2009;32(3):180-187.
Take-Home Points
- With EHR implementation there are small changes in the level of billing coding.
- Although these changes may be statistically significant they are relatively minor.
- In the general internal medicine department, level 4 coding increased by 1.2% while level 3 coding decreased by 0.5%.
- In the orthopedics department, level 4 coding increased by 3.3% while level 3 coding decreased by 3.1%.
- Reports in the lay media regarding dramatic up-coding after EHR implementation may be misleading.
The Health Information Technology for Economic and Clinical Health (HITECH) Act, which was signed into law in 2009, mandated that hospitals that care for Medicare patients either begin using electronic health records (EHRs) or pay a nontrivial penalty.1 By now, the majority of orthopedic surgeons have implemented EHRs in their practices.2 Despite ongoing debate in the orthopedic literature,3 EHRs are expected to improve coordination of care, reduce duplicate testing, and reduce costs over the long term as healthcare insurance coverage is extended to millions more Americans.
In early coverage, however, media reported that EHR implementation at some hospitals was correlated with substantial increases in Medicare payments.4 Journalists suggested the billion dollars more paid by Medicare to hospitals in 2010 than in 2005 were partly attributable to up-coding facilitated by EHRs.5 The secretary of the Department of Health and Human Services (DHHS) and the attorney general of the Department of Justice also weighed in on this controversy by expressing their concerns in a letter to the presidents of 5 hospital associations.6 The inspector general of DHHS also published a report critical of Medicare officials’ oversight of EHRs.7Responding to the critical reception of EHR implementations, investigators studied the validity of the early reports and anecdotes. Some initial reports cited the emergency department (ED) as an area at high risk for using the convenience of EHRs to up-code visits.5 The DHHS Office of the Inspector General noted that, between 2001 and 2010, the proportion of claims for lower reimbursement categories of American Medical Association Current Procedural Terminology (CPT) codes decreased while the proportion for higher-paid billing codes increased for all visit types.8 Addressing these concerns, the American Hospital Association9 issued a brief that noted that any observed coding increases were more likely attributable to more ED use by Medicare patients and increased average illness severity. In a thoughtful perspective, Pitts10 conceded that, though utilization and illness severity may explain part of the trend, the trend may also be related to technological innovations and changes in culture and practice style in the ED.
Because these studies and reports variously suggested that EHR implementation affects patient volume and up-coding, and because none of the reports specifically addressed orthopedics, we conducted a study to determine whether any significant up-coding or change in patient volumes occurred around the time of EHR implementation in ambulatory practices at our academic medical center. In a recent national study, Adler-Milstein and Jha11 compared billing data of hospitals that adopted EHRs and hospitals that did not. Although both groups showed increased billing trends, the increases were not significantly different between the EHR adopters and nonadopters. To more effectively control for the confounding differences between groups of EHR adopters and nonadopters, we studied individual departments during EHR implementation at our institution.
Methods
In 2011, our academic medical center began the transition to EHRs (Epic). We examined our center’s trends in patient volumes and billing coding around the time of the transition in the outpatient practice of the general internal medicine (GIM) department (EHR transition, October 2011) and the outpatient practice of the orthopedics department (EHR transition, March 2012). These departments were chosen because they are representative of a GIM practice and a subspecialty practice, and because a recent study found that GIM practitioners and orthopedic surgeons were among those specialists who used EHRs the most.12
After this study was approved by our Human Investigations Committee, we began using CPT codes to identify all outpatient visits (new, consultation, and return) on a monthly basis. We compared the volume of patient visits and the billing coding level in the GIM and orthopedics departments before and after EHR implementation. Pearson χ2 test was used when appropriate, and statistical analyses were performed with SPSS for Windows Version 16.0.
Results
In the GIM department, mean monthly volume of patient visits in the 12 months before EHR implementation was similar to that in the 12 months afterward (613 vs 587; P = .439). Even when normalized for changes in provider availability (maternity leave), the decrease in volume of patient visits after EHR implementation in the GIM department was not significant (6.9%; P = .107). Likewise, in the orthopedics department, mean monthly volume of patient visits in the 17 months before EHR implementation was similar to that in the 7 months afterward (2157 vs 2317; P = .156). In fact, patient volumes remained constant during the EHR transition (Figure 1).
EHR implementation brought small changes in billing coding levels. In the GIM department, the largest change was a 1.2% increase in level 4 billing coding—an increase accompanied by a 0.5% decrease in level 3 coding.
Discussion
It is remarkable that the volumes of patient visits in the GIM and orthopedics departments at our academic center were not affected by EHR implementation.
Rather than reduce scheduling during the EHR transition, surgeons in our practice either added or lengthened clinic sessions, and the level of ancillary staffing was adjusted accordingly. As staffing costs at any given time are multifactorial and vary widely, estimating the cost of these staffing changes during the EHR transition is difficult. We should note that extending ancillary staff hours during the transition very likely increased costs, and it is unclear whether they were higher or lower than the costs that would have been incurred had we reduced scheduling or tried some combination of these strategies.
Although billing coding levels changed with EHR implementation, the changes were small. In the GIM department, level 4 CPT coded visits as percentages of all visits increased to 59.5% from 58.3%, and level 5 visits increased to 6.2% from 6.0%; in the orthopedics department, level 4 visits increased to 40.2% from 37.1%, and level 5 visits increased to 5.5% from 3.8% (Table). The 1.2% and 0.2% absolute increases in level 4 and level 5 visits in the GIM department represent 2.1% and 3.3% relative increases in level 4 and level 5 visits, and the 3.3% and 1.7% absolute increases in the orthopedics department represent 8.4% and 44.7% relative increases in level 4 and level 5 visits after EHR implementation.
Although the absolute increases in level 4 and level 5 visits were relatively minor, popular media have raised the alarm about 43% and 82% relative increases in level 5 visits after EHR implementation in some hospitals’ EDs.4 Although our orthopedics department showed a 44.7% relative increase in level 5 visits after EHR implementation, this represented an increase of only 1.7% of patient visits overall. Our findings therefore indicate that lay media reports could be misleading. Nevertheless, the small changes we found were statistically significant.
One explanation for these small changes is that EHRs facilitate better documentation of services provided. Therefore, what seem to be billing coding changes could be more accurate reports of high-level care that is the same as before. In addition, because of meaningful use mandates that coincided with the requirement to implement EHRs, additional data elements are now being consistently collected and reviewed (these may not necessarily have been collected and reviewed before). In some patient encounters, these additional data elements may have contributed to higher levels of service, and this effect could be especially apparent in EDs.
Some have suggested a potential for large-scale up-coding during EHR transitions. Others have contended that coding level increases are a consequence of a time-intensive data entry process, collection and review of additional data, and more accurate reporting of services already being provided. We are not convinced that large coding changes are attributable solely to EHR implementation, as the changes at our center have been relatively small.
Nevertheless, minor coding level changes could translate to large changes in healthcare costs when scaled nationally. Although causes may be innocuous, any increases in national healthcare costs are concerning in our time of limited budgets and scrutinized healthcare utilization.
This study had its limitations. First, including billing data from only 2 departments at a single center may limit the generalizability of findings. However, we specifically selected a GIM department and a specialty (orthopedics) department in an attempt to capture a representative sample of practices. Another limitation is that we investigated billing codes over only 2 years, around the implementation of EHRs in these departments, and therefore may have captured only short-term changes. However, as patient volumes and billing are subject to many factors, including staffing changes (eg, new partners, new hires, retirements, other departures), we attempted to limit the effect of confounding variables by limiting the period of analysis.
Overall, changes in patient volume and coded level of service during EHR implementation at our institution were relatively small. Although the trend toward higher billing coding levels was statistically significant, these 0.2% and 1.7% increases in level 5 coding hardly deserve the negative attention from lay media. These small increases are unlikely caused by intentional up-coding, and more likely reflect better documentation of an already high level of care. We hope these findings allay the concern that up-coding increased dramatically with EHR implementation.
Am J Orthop. 2017;46(3):E172-E176. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- With EHR implementation there are small changes in the level of billing coding.
- Although these changes may be statistically significant they are relatively minor.
- In the general internal medicine department, level 4 coding increased by 1.2% while level 3 coding decreased by 0.5%.
- In the orthopedics department, level 4 coding increased by 3.3% while level 3 coding decreased by 3.1%.
- Reports in the lay media regarding dramatic up-coding after EHR implementation may be misleading.
The Health Information Technology for Economic and Clinical Health (HITECH) Act, which was signed into law in 2009, mandated that hospitals that care for Medicare patients either begin using electronic health records (EHRs) or pay a nontrivial penalty.1 By now, the majority of orthopedic surgeons have implemented EHRs in their practices.2 Despite ongoing debate in the orthopedic literature,3 EHRs are expected to improve coordination of care, reduce duplicate testing, and reduce costs over the long term as healthcare insurance coverage is extended to millions more Americans.
In early coverage, however, media reported that EHR implementation at some hospitals was correlated with substantial increases in Medicare payments.4 Journalists suggested the billion dollars more paid by Medicare to hospitals in 2010 than in 2005 were partly attributable to up-coding facilitated by EHRs.5 The secretary of the Department of Health and Human Services (DHHS) and the attorney general of the Department of Justice also weighed in on this controversy by expressing their concerns in a letter to the presidents of 5 hospital associations.6 The inspector general of DHHS also published a report critical of Medicare officials’ oversight of EHRs.7Responding to the critical reception of EHR implementations, investigators studied the validity of the early reports and anecdotes. Some initial reports cited the emergency department (ED) as an area at high risk for using the convenience of EHRs to up-code visits.5 The DHHS Office of the Inspector General noted that, between 2001 and 2010, the proportion of claims for lower reimbursement categories of American Medical Association Current Procedural Terminology (CPT) codes decreased while the proportion for higher-paid billing codes increased for all visit types.8 Addressing these concerns, the American Hospital Association9 issued a brief that noted that any observed coding increases were more likely attributable to more ED use by Medicare patients and increased average illness severity. In a thoughtful perspective, Pitts10 conceded that, though utilization and illness severity may explain part of the trend, the trend may also be related to technological innovations and changes in culture and practice style in the ED.
Because these studies and reports variously suggested that EHR implementation affects patient volume and up-coding, and because none of the reports specifically addressed orthopedics, we conducted a study to determine whether any significant up-coding or change in patient volumes occurred around the time of EHR implementation in ambulatory practices at our academic medical center. In a recent national study, Adler-Milstein and Jha11 compared billing data of hospitals that adopted EHRs and hospitals that did not. Although both groups showed increased billing trends, the increases were not significantly different between the EHR adopters and nonadopters. To more effectively control for the confounding differences between groups of EHR adopters and nonadopters, we studied individual departments during EHR implementation at our institution.
Methods
In 2011, our academic medical center began the transition to EHRs (Epic). We examined our center’s trends in patient volumes and billing coding around the time of the transition in the outpatient practice of the general internal medicine (GIM) department (EHR transition, October 2011) and the outpatient practice of the orthopedics department (EHR transition, March 2012). These departments were chosen because they are representative of a GIM practice and a subspecialty practice, and because a recent study found that GIM practitioners and orthopedic surgeons were among those specialists who used EHRs the most.12
After this study was approved by our Human Investigations Committee, we began using CPT codes to identify all outpatient visits (new, consultation, and return) on a monthly basis. We compared the volume of patient visits and the billing coding level in the GIM and orthopedics departments before and after EHR implementation. Pearson χ2 test was used when appropriate, and statistical analyses were performed with SPSS for Windows Version 16.0.
Results
In the GIM department, mean monthly volume of patient visits in the 12 months before EHR implementation was similar to that in the 12 months afterward (613 vs 587; P = .439). Even when normalized for changes in provider availability (maternity leave), the decrease in volume of patient visits after EHR implementation in the GIM department was not significant (6.9%; P = .107). Likewise, in the orthopedics department, mean monthly volume of patient visits in the 17 months before EHR implementation was similar to that in the 7 months afterward (2157 vs 2317; P = .156). In fact, patient volumes remained constant during the EHR transition (Figure 1).
EHR implementation brought small changes in billing coding levels. In the GIM department, the largest change was a 1.2% increase in level 4 billing coding—an increase accompanied by a 0.5% decrease in level 3 coding.
Discussion
It is remarkable that the volumes of patient visits in the GIM and orthopedics departments at our academic center were not affected by EHR implementation.
Rather than reduce scheduling during the EHR transition, surgeons in our practice either added or lengthened clinic sessions, and the level of ancillary staffing was adjusted accordingly. As staffing costs at any given time are multifactorial and vary widely, estimating the cost of these staffing changes during the EHR transition is difficult. We should note that extending ancillary staff hours during the transition very likely increased costs, and it is unclear whether they were higher or lower than the costs that would have been incurred had we reduced scheduling or tried some combination of these strategies.
Although billing coding levels changed with EHR implementation, the changes were small. In the GIM department, level 4 CPT coded visits as percentages of all visits increased to 59.5% from 58.3%, and level 5 visits increased to 6.2% from 6.0%; in the orthopedics department, level 4 visits increased to 40.2% from 37.1%, and level 5 visits increased to 5.5% from 3.8% (Table). The 1.2% and 0.2% absolute increases in level 4 and level 5 visits in the GIM department represent 2.1% and 3.3% relative increases in level 4 and level 5 visits, and the 3.3% and 1.7% absolute increases in the orthopedics department represent 8.4% and 44.7% relative increases in level 4 and level 5 visits after EHR implementation.
Although the absolute increases in level 4 and level 5 visits were relatively minor, popular media have raised the alarm about 43% and 82% relative increases in level 5 visits after EHR implementation in some hospitals’ EDs.4 Although our orthopedics department showed a 44.7% relative increase in level 5 visits after EHR implementation, this represented an increase of only 1.7% of patient visits overall. Our findings therefore indicate that lay media reports could be misleading. Nevertheless, the small changes we found were statistically significant.
One explanation for these small changes is that EHRs facilitate better documentation of services provided. Therefore, what seem to be billing coding changes could be more accurate reports of high-level care that is the same as before. In addition, because of meaningful use mandates that coincided with the requirement to implement EHRs, additional data elements are now being consistently collected and reviewed (these may not necessarily have been collected and reviewed before). In some patient encounters, these additional data elements may have contributed to higher levels of service, and this effect could be especially apparent in EDs.
Some have suggested a potential for large-scale up-coding during EHR transitions. Others have contended that coding level increases are a consequence of a time-intensive data entry process, collection and review of additional data, and more accurate reporting of services already being provided. We are not convinced that large coding changes are attributable solely to EHR implementation, as the changes at our center have been relatively small.
Nevertheless, minor coding level changes could translate to large changes in healthcare costs when scaled nationally. Although causes may be innocuous, any increases in national healthcare costs are concerning in our time of limited budgets and scrutinized healthcare utilization.
This study had its limitations. First, including billing data from only 2 departments at a single center may limit the generalizability of findings. However, we specifically selected a GIM department and a specialty (orthopedics) department in an attempt to capture a representative sample of practices. Another limitation is that we investigated billing codes over only 2 years, around the implementation of EHRs in these departments, and therefore may have captured only short-term changes. However, as patient volumes and billing are subject to many factors, including staffing changes (eg, new partners, new hires, retirements, other departures), we attempted to limit the effect of confounding variables by limiting the period of analysis.
Overall, changes in patient volume and coded level of service during EHR implementation at our institution were relatively small. Although the trend toward higher billing coding levels was statistically significant, these 0.2% and 1.7% increases in level 5 coding hardly deserve the negative attention from lay media. These small increases are unlikely caused by intentional up-coding, and more likely reflect better documentation of an already high level of care. We hope these findings allay the concern that up-coding increased dramatically with EHR implementation.
Am J Orthop. 2017;46(3):E172-E176. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Centers for Medicare & Medicaid Services. Electronic health records (EHR) incentive programs. http://www.cms.gov/Regulations-and-Guidance/Legislation/EHRIncentivePrograms. Accessed February 5, 2015.
2. American Academy of Orthopaedic Surgeons Practice Management Committee. EMR: A Primer for Orthopaedic Surgeons. 2nd ed. Rosemont, IL: American Academy of Orthopaedic Surgeons; 2010.
3. Ries MD. Electronic medical records: friends or foes? Clin Orthop Relat Res. 2014;472(1):16-21.
4. Abelson R. Medicare is faulted on shift to electronic records. New York Times. November 29, 2012;B1. http://www.nytimes.com/2012/11/29/business/medicare-is-faulted-in-electronic-medical-records-conversion.html. Accessed February 5, 2015.
5. Abelson R, Creswell J, Palmer G. Medicare bills rise as records turn electronic. New York Times. September 22, 2012;A1. http://www.nytimes.com/2012/09/22/business/medicare-billing-rises-at-hospitals-with-electronic-records.html. Accessed February 5, 2015.
6. Carlson J. Warning bell. Potential for fraud through use of EHRs draws federal scrutiny. Mod Healthc. 2012;42(40):8-9.
7. Levinson DR. Early assessment finds that CMS faces obstacles in overseeing the Medicare EHR Incentive Program. Dept of Health and Human Services, Office of Inspector General website. https://oig.hss.gov/oei/reports/oei-05-11-00250.pdf. Publication OEI-05-11-00250. Published November 2012. Accessed February 5, 2015.
8. Levinson DR. Coding trends of Medicare evaluation and management services. Dept of Health and Human Services, Office of Inspector General website. https://oig.hhs.gov/oei/reports/oei-04-10-00180.pdf. Publication OEI-04-10-00180. Published May 2012. Accessed February 5, 2015.
9. American Hospital Association. Sicker, more complex patients are driving up intensity of ED care [issue brief]. http://www.aha.org/content/13/13issuebrief-ed.pdf. Published May 2, 2013. Accessed February 5, 2015.
10. Pitts SR. Higher-complexity ED billing codes—sicker patients, more intensive practice, or improper payments? N Engl J Med. 2012;367(26):2465-2467.
11. Adler-Milstein J, Jha AK. No evidence found that hospitals are using new electronic health records to increase Medicare reimbursements. Health Aff (Millwood). 2014;33(7):1271-1277.
12. Kokkonen EW, Davis SA, Lin HC, Dabade TS, Feldman SR, Fleischer AB Jr. Use of electronic medical records differs by specialty and office settings. J Am Med Inform Assoc. 2013;20(e1):e33-e38.
13. Samaan ZM, Klein MD, Mansour ME, DeWitt TG. The impact of the electronic health record on an academic pediatric primary care center. J Ambul Care Manage. 2009;32(3):180-187.
1. Centers for Medicare & Medicaid Services. Electronic health records (EHR) incentive programs. http://www.cms.gov/Regulations-and-Guidance/Legislation/EHRIncentivePrograms. Accessed February 5, 2015.
2. American Academy of Orthopaedic Surgeons Practice Management Committee. EMR: A Primer for Orthopaedic Surgeons. 2nd ed. Rosemont, IL: American Academy of Orthopaedic Surgeons; 2010.
3. Ries MD. Electronic medical records: friends or foes? Clin Orthop Relat Res. 2014;472(1):16-21.
4. Abelson R. Medicare is faulted on shift to electronic records. New York Times. November 29, 2012;B1. http://www.nytimes.com/2012/11/29/business/medicare-is-faulted-in-electronic-medical-records-conversion.html. Accessed February 5, 2015.
5. Abelson R, Creswell J, Palmer G. Medicare bills rise as records turn electronic. New York Times. September 22, 2012;A1. http://www.nytimes.com/2012/09/22/business/medicare-billing-rises-at-hospitals-with-electronic-records.html. Accessed February 5, 2015.
6. Carlson J. Warning bell. Potential for fraud through use of EHRs draws federal scrutiny. Mod Healthc. 2012;42(40):8-9.
7. Levinson DR. Early assessment finds that CMS faces obstacles in overseeing the Medicare EHR Incentive Program. Dept of Health and Human Services, Office of Inspector General website. https://oig.hss.gov/oei/reports/oei-05-11-00250.pdf. Publication OEI-05-11-00250. Published November 2012. Accessed February 5, 2015.
8. Levinson DR. Coding trends of Medicare evaluation and management services. Dept of Health and Human Services, Office of Inspector General website. https://oig.hhs.gov/oei/reports/oei-04-10-00180.pdf. Publication OEI-04-10-00180. Published May 2012. Accessed February 5, 2015.
9. American Hospital Association. Sicker, more complex patients are driving up intensity of ED care [issue brief]. http://www.aha.org/content/13/13issuebrief-ed.pdf. Published May 2, 2013. Accessed February 5, 2015.
10. Pitts SR. Higher-complexity ED billing codes—sicker patients, more intensive practice, or improper payments? N Engl J Med. 2012;367(26):2465-2467.
11. Adler-Milstein J, Jha AK. No evidence found that hospitals are using new electronic health records to increase Medicare reimbursements. Health Aff (Millwood). 2014;33(7):1271-1277.
12. Kokkonen EW, Davis SA, Lin HC, Dabade TS, Feldman SR, Fleischer AB Jr. Use of electronic medical records differs by specialty and office settings. J Am Med Inform Assoc. 2013;20(e1):e33-e38.
13. Samaan ZM, Klein MD, Mansour ME, DeWitt TG. The impact of the electronic health record on an academic pediatric primary care center. J Ambul Care Manage. 2009;32(3):180-187.