User login
Study links photosensitizing antihypertensives to SCC
PORTLAND, ORE. – Patients prescribed photosensitizing antihypertensive drugs had a 16% increase in risk of cutaneous squamous cell carcinoma (cSCC) in a large retrospective cohort study.
These drugs include alpha-2 receptor agonists and loop diuretics, potassium-sparing diuretics, thiazide diuretics, and combination diuretics, Katherine Levandoski said in an oral presentation at the annual meeting of the Society for Investigative Dermatology.
Furthermore, taking antihypertensive drugs of unknown photosensitizing potential conferred a 10% increase in risk of cSCC in the study, she added. Such medications include angiotensin–converting enzyme inhibitors, calcium channel blockers, and vasodilators, she said.
More than 50 million Americans take antihypertensive drugs, many of which are photosensitizing, noted Ms. Levandoski, a research assistant in the Patient Oriented Research on the Epidemiology of Skin Diseases Unit in the department of dermatology, Massachusetts General Hospital, and the department of population medicine, Harvard University, Boston. However, few studies have explored the oncogenic effects of exposure to these drugs, and those that have done so were subject to confounding, small sample sizes, missing data, lack of pathologic verification, and reliance on self-reported medication history, she added.
To help fill this knowledge gap, she and her associates studied 28,357 non-Hispanic whites diagnosed with hypertension and treated at Kaiser Permanente Northern California between 1997 and 2012. They limited the cohort to non-Hispanic whites because they represent the group with most cases of cSCC.
During follow-up, 3,010 patients were diagnosed with new-onset, pathologically verified cSCC, Ms. Levandoski said. Compared with nonusers of antihypertensives, users of photosensitizing antihypertensives had about a 16% increase in the rate of cSCC (hazard ratio, 1.16; 95% confidence interval, 1.06-1.27), even after accounting for age, sex, smoking, comorbidities, health care utilization, skin cancer history, length of health plan membership, and prior exposure to photosensitizing medications.
Strikingly, patients who used antihypertensives of unknown photosensitizing effect had a 10% increase in risk of incident cSCC (RR, 1.10; 95% CI, 1.02-1.19). Some antihypertensive drugs that are classified as unknown photosensitizers “may actually have photosensitizing properties,” Ms. Levandoski commented. Patients taking antihypertensives of known or unknown photosensitizing potential “should be educated on safe sun practices and may benefit from closer screening for cutaneous squamous cell carcinoma,” she added.
The risk of cSCC was not increased among users of nonphotosensitizing antihypertensives (HR, 0.99; 95% CI, 0.91-1.07), including alpha-blockers, beta-blockers, central agonists, and angiotensin receptor blockers, Ms. Levandoski reported.
Patients in the study cohort averaged aged 60 years (standard deviation, 10.6 years), and 56% were female. In all, 1,530 had never been prescribed antihypertensives, while about 17,000-19,000 had been prescribed unknown, known, or nonphotosensitizing antihypertensives.
The work was funded by the National Institutes of Health, a travel award from the Society for Investigative Dermatology, and a Massachusetts General Hospital Medical Student Award. Ms. Levandoski had no conflicts of interest.
PORTLAND, ORE. – Patients prescribed photosensitizing antihypertensive drugs had a 16% increase in risk of cutaneous squamous cell carcinoma (cSCC) in a large retrospective cohort study.
These drugs include alpha-2 receptor agonists and loop diuretics, potassium-sparing diuretics, thiazide diuretics, and combination diuretics, Katherine Levandoski said in an oral presentation at the annual meeting of the Society for Investigative Dermatology.
Furthermore, taking antihypertensive drugs of unknown photosensitizing potential conferred a 10% increase in risk of cSCC in the study, she added. Such medications include angiotensin–converting enzyme inhibitors, calcium channel blockers, and vasodilators, she said.
More than 50 million Americans take antihypertensive drugs, many of which are photosensitizing, noted Ms. Levandoski, a research assistant in the Patient Oriented Research on the Epidemiology of Skin Diseases Unit in the department of dermatology, Massachusetts General Hospital, and the department of population medicine, Harvard University, Boston. However, few studies have explored the oncogenic effects of exposure to these drugs, and those that have done so were subject to confounding, small sample sizes, missing data, lack of pathologic verification, and reliance on self-reported medication history, she added.
To help fill this knowledge gap, she and her associates studied 28,357 non-Hispanic whites diagnosed with hypertension and treated at Kaiser Permanente Northern California between 1997 and 2012. They limited the cohort to non-Hispanic whites because they represent the group with most cases of cSCC.
During follow-up, 3,010 patients were diagnosed with new-onset, pathologically verified cSCC, Ms. Levandoski said. Compared with nonusers of antihypertensives, users of photosensitizing antihypertensives had about a 16% increase in the rate of cSCC (hazard ratio, 1.16; 95% confidence interval, 1.06-1.27), even after accounting for age, sex, smoking, comorbidities, health care utilization, skin cancer history, length of health plan membership, and prior exposure to photosensitizing medications.
Strikingly, patients who used antihypertensives of unknown photosensitizing effect had a 10% increase in risk of incident cSCC (RR, 1.10; 95% CI, 1.02-1.19). Some antihypertensive drugs that are classified as unknown photosensitizers “may actually have photosensitizing properties,” Ms. Levandoski commented. Patients taking antihypertensives of known or unknown photosensitizing potential “should be educated on safe sun practices and may benefit from closer screening for cutaneous squamous cell carcinoma,” she added.
The risk of cSCC was not increased among users of nonphotosensitizing antihypertensives (HR, 0.99; 95% CI, 0.91-1.07), including alpha-blockers, beta-blockers, central agonists, and angiotensin receptor blockers, Ms. Levandoski reported.
Patients in the study cohort averaged aged 60 years (standard deviation, 10.6 years), and 56% were female. In all, 1,530 had never been prescribed antihypertensives, while about 17,000-19,000 had been prescribed unknown, known, or nonphotosensitizing antihypertensives.
The work was funded by the National Institutes of Health, a travel award from the Society for Investigative Dermatology, and a Massachusetts General Hospital Medical Student Award. Ms. Levandoski had no conflicts of interest.
PORTLAND, ORE. – Patients prescribed photosensitizing antihypertensive drugs had a 16% increase in risk of cutaneous squamous cell carcinoma (cSCC) in a large retrospective cohort study.
These drugs include alpha-2 receptor agonists and loop diuretics, potassium-sparing diuretics, thiazide diuretics, and combination diuretics, Katherine Levandoski said in an oral presentation at the annual meeting of the Society for Investigative Dermatology.
Furthermore, taking antihypertensive drugs of unknown photosensitizing potential conferred a 10% increase in risk of cSCC in the study, she added. Such medications include angiotensin–converting enzyme inhibitors, calcium channel blockers, and vasodilators, she said.
More than 50 million Americans take antihypertensive drugs, many of which are photosensitizing, noted Ms. Levandoski, a research assistant in the Patient Oriented Research on the Epidemiology of Skin Diseases Unit in the department of dermatology, Massachusetts General Hospital, and the department of population medicine, Harvard University, Boston. However, few studies have explored the oncogenic effects of exposure to these drugs, and those that have done so were subject to confounding, small sample sizes, missing data, lack of pathologic verification, and reliance on self-reported medication history, she added.
To help fill this knowledge gap, she and her associates studied 28,357 non-Hispanic whites diagnosed with hypertension and treated at Kaiser Permanente Northern California between 1997 and 2012. They limited the cohort to non-Hispanic whites because they represent the group with most cases of cSCC.
During follow-up, 3,010 patients were diagnosed with new-onset, pathologically verified cSCC, Ms. Levandoski said. Compared with nonusers of antihypertensives, users of photosensitizing antihypertensives had about a 16% increase in the rate of cSCC (hazard ratio, 1.16; 95% confidence interval, 1.06-1.27), even after accounting for age, sex, smoking, comorbidities, health care utilization, skin cancer history, length of health plan membership, and prior exposure to photosensitizing medications.
Strikingly, patients who used antihypertensives of unknown photosensitizing effect had a 10% increase in risk of incident cSCC (RR, 1.10; 95% CI, 1.02-1.19). Some antihypertensive drugs that are classified as unknown photosensitizers “may actually have photosensitizing properties,” Ms. Levandoski commented. Patients taking antihypertensives of known or unknown photosensitizing potential “should be educated on safe sun practices and may benefit from closer screening for cutaneous squamous cell carcinoma,” she added.
The risk of cSCC was not increased among users of nonphotosensitizing antihypertensives (HR, 0.99; 95% CI, 0.91-1.07), including alpha-blockers, beta-blockers, central agonists, and angiotensin receptor blockers, Ms. Levandoski reported.
Patients in the study cohort averaged aged 60 years (standard deviation, 10.6 years), and 56% were female. In all, 1,530 had never been prescribed antihypertensives, while about 17,000-19,000 had been prescribed unknown, known, or nonphotosensitizing antihypertensives.
The work was funded by the National Institutes of Health, a travel award from the Society for Investigative Dermatology, and a Massachusetts General Hospital Medical Student Award. Ms. Levandoski had no conflicts of interest.
AT SID 2017
Key clinical point: Consider skin cancer screening for patients who are taking antihypertensives of known or unknown photosensitizing potential.
Major finding: The risk of cutaneous squamous cell carcinoma associated with photosensitizing antihypertensives was about 16% .
Data source: A retrospective cohort study of 28,357 non-Hispanic whites with hypertension.
Disclosures: The work was funded by the National Institutes of Health, a travel award from the Society for Investigative Dermatology, and a Massachusetts General Hospital Medical Student Award. Ms. Levandoski had no conflicts of interest.
Pembrolizumab shows some activity against advanced endometrial cancer
In some women with heavily pretreated locally advanced or metastatic endometrial cancers, treatment with the immune checkpoint inhibitor pembrolizumab (Keytruda) produced partial, durable responses, investigators reported.
The women were participants in the KEYNOTE-028 trial, a multicohort, open-label phase Ib basket trial of patients with advanced solid tumors positive for programmed death ligand 1 (PD-L1). Of 24 women with PD-L1-positive endometrial tumors, 3 (13%) had confirmed partial responses, and 2 of these patients remained on therapy with a continued response at the time of data cutoff, reported Patrick A. Ott, MD, PhD, of Dana-Farber Cancer Institute, Boston, and his colleagues.
“[T]he results from the KEYNOTE-028 trial presented herein indicate that pembrolizumab shows promise as a treatment for advanced endometrial cancer, a disease for which current treatment options are limited,” they wrote (J Clin Oncol. 2017 May 10; doi: 10.1200/JCO.2017.72.5952).
Patients with advanced endometrial cancer have a poor prognosis, with 5-year survival rates below 50% for patients with lymph node metastases, and less than 20% for those with distant metastases or disease that has spread to the peritoneum, the investigators noted.
“Treatment options for patients presenting with advanced disease are limited, with no consensus on a standard regimen. Similarly, no optimal therapy for patients who experience progression during first-line therapy has been identified, with second-line chemotherapeutic options producing only modest activity,” they wrote.
PD-L1 was shown to be expressed on 83% of primary endometrial tumors and 100% of metastatic endometrial cancers in one study (Cancer Immunol Immunother. 2014;63[6]:545-57), suggesting that could be a viable target in this difficult-to-treat disease, noted Dr. Ott and his colleagues.
In their study, 24 women with locally advanced or metastatic PD-L1–positive endometrial cancer who had experienced progression after standard therapy were treated with pembrolizumab 10 mg/kg every 2 weeks for up to 24 months or until disease progression or unacceptable toxicity. As noted, 3 of the 24 patients met the primary efficacy endpoint of objective response rate by Response Evaluation Criteria in Solid Tumors (RECIST; version 1.1) with a partial response, with the median duration of response (a secondary endpoint) not reached at the time of data cutoff on Feb. 17, 2016.
Three other patients (13%) had stable disease, with a median duration of 24.6 weeks.
Safety, also a secondary endpoint, was comparable to that seen in other studies with pembrolizumab. Four patients had grade 3 treatment-related adverse events (AEs). There were no grade 4 AEs of any kind, no immune-mediated AEs of any grade, and no treatment-related deaths.
One of the patients who had a partial response was found through genomic profiling to have a polymerase E, or POLE mutation, and this patient had a rapid improvement after starting on pembrolizumab, with the response lasting longer than 14 months.
“These exceptional results support the theory that the presence of POLE mutations may aid in identifying patients for whom pembrolizumab may be particularly effective, a concept that should be investigated further,” wrote Dr. Ott and his colleagues.
Merck Sharp & Dohme sponsored the study. Dr. Ott and multiple coauthors disclosed institution research funding, honoraria, consulting, or advising with the company, and three are Merck employees..
In some women with heavily pretreated locally advanced or metastatic endometrial cancers, treatment with the immune checkpoint inhibitor pembrolizumab (Keytruda) produced partial, durable responses, investigators reported.
The women were participants in the KEYNOTE-028 trial, a multicohort, open-label phase Ib basket trial of patients with advanced solid tumors positive for programmed death ligand 1 (PD-L1). Of 24 women with PD-L1-positive endometrial tumors, 3 (13%) had confirmed partial responses, and 2 of these patients remained on therapy with a continued response at the time of data cutoff, reported Patrick A. Ott, MD, PhD, of Dana-Farber Cancer Institute, Boston, and his colleagues.
“[T]he results from the KEYNOTE-028 trial presented herein indicate that pembrolizumab shows promise as a treatment for advanced endometrial cancer, a disease for which current treatment options are limited,” they wrote (J Clin Oncol. 2017 May 10; doi: 10.1200/JCO.2017.72.5952).
Patients with advanced endometrial cancer have a poor prognosis, with 5-year survival rates below 50% for patients with lymph node metastases, and less than 20% for those with distant metastases or disease that has spread to the peritoneum, the investigators noted.
“Treatment options for patients presenting with advanced disease are limited, with no consensus on a standard regimen. Similarly, no optimal therapy for patients who experience progression during first-line therapy has been identified, with second-line chemotherapeutic options producing only modest activity,” they wrote.
PD-L1 was shown to be expressed on 83% of primary endometrial tumors and 100% of metastatic endometrial cancers in one study (Cancer Immunol Immunother. 2014;63[6]:545-57), suggesting that could be a viable target in this difficult-to-treat disease, noted Dr. Ott and his colleagues.
In their study, 24 women with locally advanced or metastatic PD-L1–positive endometrial cancer who had experienced progression after standard therapy were treated with pembrolizumab 10 mg/kg every 2 weeks for up to 24 months or until disease progression or unacceptable toxicity. As noted, 3 of the 24 patients met the primary efficacy endpoint of objective response rate by Response Evaluation Criteria in Solid Tumors (RECIST; version 1.1) with a partial response, with the median duration of response (a secondary endpoint) not reached at the time of data cutoff on Feb. 17, 2016.
Three other patients (13%) had stable disease, with a median duration of 24.6 weeks.
Safety, also a secondary endpoint, was comparable to that seen in other studies with pembrolizumab. Four patients had grade 3 treatment-related adverse events (AEs). There were no grade 4 AEs of any kind, no immune-mediated AEs of any grade, and no treatment-related deaths.
One of the patients who had a partial response was found through genomic profiling to have a polymerase E, or POLE mutation, and this patient had a rapid improvement after starting on pembrolizumab, with the response lasting longer than 14 months.
“These exceptional results support the theory that the presence of POLE mutations may aid in identifying patients for whom pembrolizumab may be particularly effective, a concept that should be investigated further,” wrote Dr. Ott and his colleagues.
Merck Sharp & Dohme sponsored the study. Dr. Ott and multiple coauthors disclosed institution research funding, honoraria, consulting, or advising with the company, and three are Merck employees..
In some women with heavily pretreated locally advanced or metastatic endometrial cancers, treatment with the immune checkpoint inhibitor pembrolizumab (Keytruda) produced partial, durable responses, investigators reported.
The women were participants in the KEYNOTE-028 trial, a multicohort, open-label phase Ib basket trial of patients with advanced solid tumors positive for programmed death ligand 1 (PD-L1). Of 24 women with PD-L1-positive endometrial tumors, 3 (13%) had confirmed partial responses, and 2 of these patients remained on therapy with a continued response at the time of data cutoff, reported Patrick A. Ott, MD, PhD, of Dana-Farber Cancer Institute, Boston, and his colleagues.
“[T]he results from the KEYNOTE-028 trial presented herein indicate that pembrolizumab shows promise as a treatment for advanced endometrial cancer, a disease for which current treatment options are limited,” they wrote (J Clin Oncol. 2017 May 10; doi: 10.1200/JCO.2017.72.5952).
Patients with advanced endometrial cancer have a poor prognosis, with 5-year survival rates below 50% for patients with lymph node metastases, and less than 20% for those with distant metastases or disease that has spread to the peritoneum, the investigators noted.
“Treatment options for patients presenting with advanced disease are limited, with no consensus on a standard regimen. Similarly, no optimal therapy for patients who experience progression during first-line therapy has been identified, with second-line chemotherapeutic options producing only modest activity,” they wrote.
PD-L1 was shown to be expressed on 83% of primary endometrial tumors and 100% of metastatic endometrial cancers in one study (Cancer Immunol Immunother. 2014;63[6]:545-57), suggesting that could be a viable target in this difficult-to-treat disease, noted Dr. Ott and his colleagues.
In their study, 24 women with locally advanced or metastatic PD-L1–positive endometrial cancer who had experienced progression after standard therapy were treated with pembrolizumab 10 mg/kg every 2 weeks for up to 24 months or until disease progression or unacceptable toxicity. As noted, 3 of the 24 patients met the primary efficacy endpoint of objective response rate by Response Evaluation Criteria in Solid Tumors (RECIST; version 1.1) with a partial response, with the median duration of response (a secondary endpoint) not reached at the time of data cutoff on Feb. 17, 2016.
Three other patients (13%) had stable disease, with a median duration of 24.6 weeks.
Safety, also a secondary endpoint, was comparable to that seen in other studies with pembrolizumab. Four patients had grade 3 treatment-related adverse events (AEs). There were no grade 4 AEs of any kind, no immune-mediated AEs of any grade, and no treatment-related deaths.
One of the patients who had a partial response was found through genomic profiling to have a polymerase E, or POLE mutation, and this patient had a rapid improvement after starting on pembrolizumab, with the response lasting longer than 14 months.
“These exceptional results support the theory that the presence of POLE mutations may aid in identifying patients for whom pembrolizumab may be particularly effective, a concept that should be investigated further,” wrote Dr. Ott and his colleagues.
Merck Sharp & Dohme sponsored the study. Dr. Ott and multiple coauthors disclosed institution research funding, honoraria, consulting, or advising with the company, and three are Merck employees..
Key clinical point: The anti-PD-L1 checkpoint inhibitor had some activity against locally advanced or metastatic endometrial cancer.
Major finding: Three of 24 patients had a partial response, and three had stable disease.
Data source: Cohort of patients with heavily pretreated advanced endometrial cancer in the multicohort, open-label, phase Ib basket trial KEYNOTE 028.
Disclosures: Merck Sharp & Dohme sponsored the study. Dr. Ott and multiple coauthors disclosed institution research funding, honoraria, consulting, or advising with the company, and three are Merck employees.
Are you prepared for MACRA?
MACRA (Medicare Access and CHIP Reauthorization Act of 2015) replaces the flawed sustainable growth rate (SGR) formula and significantly changes the way Medicare pays physicians.
Many things about the Affordable Care Act (ACA; Obamacare) are likely to change under the new administration, but MACRA and the commitment to cost-effective, value-based care is here to stay. MACRA is separate from the ACA. Congress overwhelmingly passed MACRA legislation with bipartisan support. MACRA will eventually transition physicians toward more value-based payments.
It is important to understand MACRA to ensure you are doing everything required under the current rules in 2017. Ignore MACRA in 2017 and you will face an automatic reduction of 4% to your payments under Medicare in 2019.
AGA offers educational webinars and videos to help you prepare. Visit gastro.org/MACRA to learn more.
MACRA (Medicare Access and CHIP Reauthorization Act of 2015) replaces the flawed sustainable growth rate (SGR) formula and significantly changes the way Medicare pays physicians.
Many things about the Affordable Care Act (ACA; Obamacare) are likely to change under the new administration, but MACRA and the commitment to cost-effective, value-based care is here to stay. MACRA is separate from the ACA. Congress overwhelmingly passed MACRA legislation with bipartisan support. MACRA will eventually transition physicians toward more value-based payments.
It is important to understand MACRA to ensure you are doing everything required under the current rules in 2017. Ignore MACRA in 2017 and you will face an automatic reduction of 4% to your payments under Medicare in 2019.
AGA offers educational webinars and videos to help you prepare. Visit gastro.org/MACRA to learn more.
MACRA (Medicare Access and CHIP Reauthorization Act of 2015) replaces the flawed sustainable growth rate (SGR) formula and significantly changes the way Medicare pays physicians.
Many things about the Affordable Care Act (ACA; Obamacare) are likely to change under the new administration, but MACRA and the commitment to cost-effective, value-based care is here to stay. MACRA is separate from the ACA. Congress overwhelmingly passed MACRA legislation with bipartisan support. MACRA will eventually transition physicians toward more value-based payments.
It is important to understand MACRA to ensure you are doing everything required under the current rules in 2017. Ignore MACRA in 2017 and you will face an automatic reduction of 4% to your payments under Medicare in 2019.
AGA offers educational webinars and videos to help you prepare. Visit gastro.org/MACRA to learn more.
BILCAP: adjuvant capecitabine boosts overall survival of biliary tract cancers
Patients who have undergone complete resection of biliary tract cancers live longer if they receive the oral chemotherapy agent capecitabine instead of simple observation, according to findings of the phase III randomized controlled BILCAP trial.
“The only curative treatment [for these cancers] is surgical resection, but even in that circumstance, most patients will ultimately succumb to the disease,” lead study author John N. Primrose, MD, professor of surgery at the University of Southampton (England), said in a presscast leading up to the annual meeting of the American Society of Clinical Oncology.

Results of the trial showed that compared with observation, capecitabine prolonged survival by a nonsignificant 15 months in the intention-to-treat population but by a significant 17 months in the per-protocol population. The drug had modest toxicity consistent with past experience and little impact on quality of life.
“On this basis, we believe that capecitabine should now become the standard of care for patients following curative resection of biliary tract cancer,” Dr. Primrose maintained.
The trial took place in a U.K. population, noted ASCO President Daniel F. Hayes, MD, clinical director of the breast oncology program and Stuart B. Padnos Professor in Breast Cancer Research at the University of Michigan Comprehensive Cancer Center in Ann Arbor.
“This is a cancer that is much more common in Asia than it is in the western world, and I think that will be one of the questions that will be raised, as to whether these [results] apply to patients from Asia with the same cancer,” he said. “Otherwise, this is an impressive study, an enormous amount of work, and a very important finding.”
Study coauthor John A. Bridgewater, PhD, a professor at University College Hospital in London, said that he was not concerned that the trial missed its primary endpoint.
“It would of course have been much nicer if it had been significant, but I don’t think there is any doubt that there is a genuine effect here,” he maintained, agreeing that capecitabine should be standard of care going forward.
Other chemotherapies have made their way into similar adjuvant trials since BILCAP began, including the combination of cisplatin and gemcitabine being tested in the randomized ACTICCA-1 trial, Dr. Bridgewater acknowledged. “We’ve been discussing the possible permutations if BILCAP turned out to be positive with [those investigators], and that study, cisplatin-gemcitabine compared to surveillance, will now be modified to cisplatin-gemcitabine versus capecitabine. We came to that agreement some time ago.”
The BILCAP investigators are undertaking biomarker analyses of patients’ tumors. “The genotype of a bile duct cancer that will do well with fluoropyrimidine [such as capecitabine] is unknown, and that is exactly what we’ll be looking at when we look at the material. That surely will be one of the most important questions,” he said.
At present, there is no evidence to suggest that biliary tract cancers arising in Asian populations, which are mainly due to chronic infection with liver flukes, will differ in their response to capecitabine, according to Dr. Bridgewater.
“Certainly, you’ll be able to see in the clinical subgroup analyses, the long and the short of it is that it’s actually very difficult to distinguish, certainly on clinical grounds, any group that benefits more than other groups,” he said. “So the short answer is, there shouldn’t be any difference. But do we really know? Not yet.”
Study details
Patients enrolled in BILCAP had macroscopically completely resected cholangiocarcinoma or gallbladder cancer (including liver and pancreatic resection, as appropriate). They were randomized evenly to eight cycles of capecitabine (Xeloda) at a conventional dose or observation (Capecitabine is approved by the Food and Drug Administration for treatment of breast and colorectal cancers).
With more than 80% of patients having at least 3 years of follow-up, median overall survival in the intention-to-treat population – the trial’s primary endpoint – was 51 months with capecitabine and 36 months with observation (hazard ratio [HR], 0.81; P = .097), Dr. Primrose reported. The benefit became significant in a sensitivity analysis that adjusted for prognostic factors (HR, 0.70; P = .007).
In addition, median overall survival in the per-protocol population was significantly longer with capecitabine, at 53 months, than with observation, at 36 months (HR, 0.75; P = .028).
Median recurrence-free survival in the intention-to-treat population was 25 months for the capecitabine group and 18 months for the observation group.
“The toxicity associated with chemotherapy was relatively modest and in fact very similar to what has been observed in other studies,” Dr. Primrose said. The predominant grade 3 or 4 toxicity seen with capecitabine was plantar-palmar erythema, which occurred in 20.7% of patients who received the drug. There were no capecitabine-related deaths.
“Our quality of life analysis showed that there was very little difference in quality of life related to chemotherapy over those who did not have chemotherapy,” he added.
Dr. Primrose reported that he had no disclosures. Dr. Bridgewater disclosed ties with Merck Serono, Servier, Roche, Celgene, and MSD Oncology.
Patients who have undergone complete resection of biliary tract cancers live longer if they receive the oral chemotherapy agent capecitabine instead of simple observation, according to findings of the phase III randomized controlled BILCAP trial.
“The only curative treatment [for these cancers] is surgical resection, but even in that circumstance, most patients will ultimately succumb to the disease,” lead study author John N. Primrose, MD, professor of surgery at the University of Southampton (England), said in a presscast leading up to the annual meeting of the American Society of Clinical Oncology.
Results of the trial showed that compared with observation, capecitabine prolonged survival by a nonsignificant 15 months in the intention-to-treat population but by a significant 17 months in the per-protocol population. The drug had modest toxicity consistent with past experience and little impact on quality of life.
“On this basis, we believe that capecitabine should now become the standard of care for patients following curative resection of biliary tract cancer,” Dr. Primrose maintained.
The trial took place in a U.K. population, noted ASCO President Daniel F. Hayes, MD, clinical director of the breast oncology program and Stuart B. Padnos Professor in Breast Cancer Research at the University of Michigan Comprehensive Cancer Center in Ann Arbor.
“This is a cancer that is much more common in Asia than it is in the western world, and I think that will be one of the questions that will be raised, as to whether these [results] apply to patients from Asia with the same cancer,” he said. “Otherwise, this is an impressive study, an enormous amount of work, and a very important finding.”
Study coauthor John A. Bridgewater, PhD, a professor at University College Hospital in London, said that he was not concerned that the trial missed its primary endpoint.
“It would of course have been much nicer if it had been significant, but I don’t think there is any doubt that there is a genuine effect here,” he maintained, agreeing that capecitabine should be standard of care going forward.
Other chemotherapies have made their way into similar adjuvant trials since BILCAP began, including the combination of cisplatin and gemcitabine being tested in the randomized ACTICCA-1 trial, Dr. Bridgewater acknowledged. “We’ve been discussing the possible permutations if BILCAP turned out to be positive with [those investigators], and that study, cisplatin-gemcitabine compared to surveillance, will now be modified to cisplatin-gemcitabine versus capecitabine. We came to that agreement some time ago.”
The BILCAP investigators are undertaking biomarker analyses of patients’ tumors. “The genotype of a bile duct cancer that will do well with fluoropyrimidine [such as capecitabine] is unknown, and that is exactly what we’ll be looking at when we look at the material. That surely will be one of the most important questions,” he said.
At present, there is no evidence to suggest that biliary tract cancers arising in Asian populations, which are mainly due to chronic infection with liver flukes, will differ in their response to capecitabine, according to Dr. Bridgewater.
“Certainly, you’ll be able to see in the clinical subgroup analyses, the long and the short of it is that it’s actually very difficult to distinguish, certainly on clinical grounds, any group that benefits more than other groups,” he said. “So the short answer is, there shouldn’t be any difference. But do we really know? Not yet.”
Study details
Patients enrolled in BILCAP had macroscopically completely resected cholangiocarcinoma or gallbladder cancer (including liver and pancreatic resection, as appropriate). They were randomized evenly to eight cycles of capecitabine (Xeloda) at a conventional dose or observation (Capecitabine is approved by the Food and Drug Administration for treatment of breast and colorectal cancers).
With more than 80% of patients having at least 3 years of follow-up, median overall survival in the intention-to-treat population – the trial’s primary endpoint – was 51 months with capecitabine and 36 months with observation (hazard ratio [HR], 0.81; P = .097), Dr. Primrose reported. The benefit became significant in a sensitivity analysis that adjusted for prognostic factors (HR, 0.70; P = .007).
In addition, median overall survival in the per-protocol population was significantly longer with capecitabine, at 53 months, than with observation, at 36 months (HR, 0.75; P = .028).
Median recurrence-free survival in the intention-to-treat population was 25 months for the capecitabine group and 18 months for the observation group.
“The toxicity associated with chemotherapy was relatively modest and in fact very similar to what has been observed in other studies,” Dr. Primrose said. The predominant grade 3 or 4 toxicity seen with capecitabine was plantar-palmar erythema, which occurred in 20.7% of patients who received the drug. There were no capecitabine-related deaths.
“Our quality of life analysis showed that there was very little difference in quality of life related to chemotherapy over those who did not have chemotherapy,” he added.
Dr. Primrose reported that he had no disclosures. Dr. Bridgewater disclosed ties with Merck Serono, Servier, Roche, Celgene, and MSD Oncology.
Patients who have undergone complete resection of biliary tract cancers live longer if they receive the oral chemotherapy agent capecitabine instead of simple observation, according to findings of the phase III randomized controlled BILCAP trial.
“The only curative treatment [for these cancers] is surgical resection, but even in that circumstance, most patients will ultimately succumb to the disease,” lead study author John N. Primrose, MD, professor of surgery at the University of Southampton (England), said in a presscast leading up to the annual meeting of the American Society of Clinical Oncology.
Results of the trial showed that compared with observation, capecitabine prolonged survival by a nonsignificant 15 months in the intention-to-treat population but by a significant 17 months in the per-protocol population. The drug had modest toxicity consistent with past experience and little impact on quality of life.
“On this basis, we believe that capecitabine should now become the standard of care for patients following curative resection of biliary tract cancer,” Dr. Primrose maintained.
The trial took place in a U.K. population, noted ASCO President Daniel F. Hayes, MD, clinical director of the breast oncology program and Stuart B. Padnos Professor in Breast Cancer Research at the University of Michigan Comprehensive Cancer Center in Ann Arbor.
“This is a cancer that is much more common in Asia than it is in the western world, and I think that will be one of the questions that will be raised, as to whether these [results] apply to patients from Asia with the same cancer,” he said. “Otherwise, this is an impressive study, an enormous amount of work, and a very important finding.”
Study coauthor John A. Bridgewater, PhD, a professor at University College Hospital in London, said that he was not concerned that the trial missed its primary endpoint.
“It would of course have been much nicer if it had been significant, but I don’t think there is any doubt that there is a genuine effect here,” he maintained, agreeing that capecitabine should be standard of care going forward.
Other chemotherapies have made their way into similar adjuvant trials since BILCAP began, including the combination of cisplatin and gemcitabine being tested in the randomized ACTICCA-1 trial, Dr. Bridgewater acknowledged. “We’ve been discussing the possible permutations if BILCAP turned out to be positive with [those investigators], and that study, cisplatin-gemcitabine compared to surveillance, will now be modified to cisplatin-gemcitabine versus capecitabine. We came to that agreement some time ago.”
The BILCAP investigators are undertaking biomarker analyses of patients’ tumors. “The genotype of a bile duct cancer that will do well with fluoropyrimidine [such as capecitabine] is unknown, and that is exactly what we’ll be looking at when we look at the material. That surely will be one of the most important questions,” he said.
At present, there is no evidence to suggest that biliary tract cancers arising in Asian populations, which are mainly due to chronic infection with liver flukes, will differ in their response to capecitabine, according to Dr. Bridgewater.
“Certainly, you’ll be able to see in the clinical subgroup analyses, the long and the short of it is that it’s actually very difficult to distinguish, certainly on clinical grounds, any group that benefits more than other groups,” he said. “So the short answer is, there shouldn’t be any difference. But do we really know? Not yet.”
Study details
Patients enrolled in BILCAP had macroscopically completely resected cholangiocarcinoma or gallbladder cancer (including liver and pancreatic resection, as appropriate). They were randomized evenly to eight cycles of capecitabine (Xeloda) at a conventional dose or observation (Capecitabine is approved by the Food and Drug Administration for treatment of breast and colorectal cancers).
With more than 80% of patients having at least 3 years of follow-up, median overall survival in the intention-to-treat population – the trial’s primary endpoint – was 51 months with capecitabine and 36 months with observation (hazard ratio [HR], 0.81; P = .097), Dr. Primrose reported. The benefit became significant in a sensitivity analysis that adjusted for prognostic factors (HR, 0.70; P = .007).
In addition, median overall survival in the per-protocol population was significantly longer with capecitabine, at 53 months, than with observation, at 36 months (HR, 0.75; P = .028).
Median recurrence-free survival in the intention-to-treat population was 25 months for the capecitabine group and 18 months for the observation group.
“The toxicity associated with chemotherapy was relatively modest and in fact very similar to what has been observed in other studies,” Dr. Primrose said. The predominant grade 3 or 4 toxicity seen with capecitabine was plantar-palmar erythema, which occurred in 20.7% of patients who received the drug. There were no capecitabine-related deaths.
“Our quality of life analysis showed that there was very little difference in quality of life related to chemotherapy over those who did not have chemotherapy,” he added.
Dr. Primrose reported that he had no disclosures. Dr. Bridgewater disclosed ties with Merck Serono, Servier, Roche, Celgene, and MSD Oncology.
FROM THE 2017 ASCO ANNUAL MEETING
Key clinical point:
Major finding: Compared with observation, capecitabine prolonged median overall survival by 15 months in the intention-to-treat population (P = .097) and by 17 months in the per-protocol population (P = .028).
Data source: BILCAP, a phase III randomized controlled trial among 447 patients who had undergone complete resection of biliary tract cancers.
Disclosures: Dr. Primrose reported that he had no disclosures. Dr. Bridgewater disclosed ties with Merck Serono, Servier, Roche, Celgene, and MSD Oncology.
DDSEP® 8 Quick quiz - June 2017 Question 2
Answer: E
Critique: Children with early-onset inflammatory bowel disease tend to present with colonic disease. Many of them eventually develop signs and symptoms consistent with Crohn’s disease as they get older. They are often diagnosed as “indeterminant” colitis or even ulcerative colitis but the diagnosis often changes. The colitis does not usually improve with age and these early-onset patients often follow a complicated course. This boy may eventually require enteral tube feedings or even a colectomy — however that cannot be predicted at this time. Provided his disease is well managed, he should reach his expected mid-parental height.
References
1. Oliva-Hemker M., Hutfless S., Al Kazzi E., et al. Clinical presentation and five-year therapeutic management of very early-onset inflammatory bowel disease in a large North American cohort. J Pediatr. 2015;167:527-32.
2. Aloi M., Lionetti P., Barabino A., et al. Phenotype and disease course of early-onset pediatric inflammatory bowel disease. Inflamm Bowel Dis. 2014;20:597-605.
3. Mamula P., Markowitz J.E., Baldassano R.N. Inflammatory bowel disease in early childhood and adolescence: special considerations. Gastroenterol Clin North Am. 2003;32(3):967–95, viii.
Answer: E
Critique: Children with early-onset inflammatory bowel disease tend to present with colonic disease. Many of them eventually develop signs and symptoms consistent with Crohn’s disease as they get older. They are often diagnosed as “indeterminant” colitis or even ulcerative colitis but the diagnosis often changes. The colitis does not usually improve with age and these early-onset patients often follow a complicated course. This boy may eventually require enteral tube feedings or even a colectomy — however that cannot be predicted at this time. Provided his disease is well managed, he should reach his expected mid-parental height.
References
1. Oliva-Hemker M., Hutfless S., Al Kazzi E., et al. Clinical presentation and five-year therapeutic management of very early-onset inflammatory bowel disease in a large North American cohort. J Pediatr. 2015;167:527-32.
2. Aloi M., Lionetti P., Barabino A., et al. Phenotype and disease course of early-onset pediatric inflammatory bowel disease. Inflamm Bowel Dis. 2014;20:597-605.
3. Mamula P., Markowitz J.E., Baldassano R.N. Inflammatory bowel disease in early childhood and adolescence: special considerations. Gastroenterol Clin North Am. 2003;32(3):967–95, viii.
Answer: E
Critique: Children with early-onset inflammatory bowel disease tend to present with colonic disease. Many of them eventually develop signs and symptoms consistent with Crohn’s disease as they get older. They are often diagnosed as “indeterminant” colitis or even ulcerative colitis but the diagnosis often changes. The colitis does not usually improve with age and these early-onset patients often follow a complicated course. This boy may eventually require enteral tube feedings or even a colectomy — however that cannot be predicted at this time. Provided his disease is well managed, he should reach his expected mid-parental height.
References
1. Oliva-Hemker M., Hutfless S., Al Kazzi E., et al. Clinical presentation and five-year therapeutic management of very early-onset inflammatory bowel disease in a large North American cohort. J Pediatr. 2015;167:527-32.
2. Aloi M., Lionetti P., Barabino A., et al. Phenotype and disease course of early-onset pediatric inflammatory bowel disease. Inflamm Bowel Dis. 2014;20:597-605.
3. Mamula P., Markowitz J.E., Baldassano R.N. Inflammatory bowel disease in early childhood and adolescence: special considerations. Gastroenterol Clin North Am. 2003;32(3):967–95, viii.
The family of a 7-year-old boy that has been followed for colitis for the past 2 years presents with many questions and concerns. He was diagnosed with colitis at age 4 when he presented with several weeks of bloody diarrhea. He is doing well on a maintenance regimen of mesalamine but the family would like to discuss what the future may hold.
DDSEP® 8 Quick quiz - June 2017 Question 1
Answer: D
Objective: Recognize that spontaneous bacterial peritonitis may present in an asymptomatic manner.
Discussion: It is important to recognize that patients with spontaneous bacterial peritonitis may present in various ways and may not exhibit classic abdominal pain or fevers. Patients may have atypical or no overt symptoms at all. With direct inoculation of ascitic fluid into culture bottles at bedside, cultures may identify bacteria in up to 40%-50% of cases.
Patients who survive an episode of SBP have a very high risk of recurrence (70%) within the first year of the index episode. It is therefore essential that patients recovering from SBP be started on prophylactic therapy prior to hospital discharge. Nonabsorbable (or poorly absorbable) antibiotics are most effective for such prophylaxis by selectively eliminating gram-negative organisms in the gut.
These agents reduce the rate of SBP recurrence to around 15–20%. Nosocomial infections respond poorly (~40% of cases) to 3rd generation cephalosporins. Those who have been in the hospital and received antibiotics within the past 90 days should receive extended-spectrum antibiotics.
References
1. Rimola A., Garcia-Tsao G., Navasa M., et al. Diagnosis, treatment and prophylaxis of spontaneous bacterial peri- tonitis: a consensus document. J Hepatol. 2000;32:142-53.
2. Tandon P., Garcia-Tsao G. Bacterial infections, sepsis, and multiorgan failure in cirrhosis. Semin Liver Dis. 2008;28(1):26-42.
Answer: D
Objective: Recognize that spontaneous bacterial peritonitis may present in an asymptomatic manner.
Discussion: It is important to recognize that patients with spontaneous bacterial peritonitis may present in various ways and may not exhibit classic abdominal pain or fevers. Patients may have atypical or no overt symptoms at all. With direct inoculation of ascitic fluid into culture bottles at bedside, cultures may identify bacteria in up to 40%-50% of cases.
Patients who survive an episode of SBP have a very high risk of recurrence (70%) within the first year of the index episode. It is therefore essential that patients recovering from SBP be started on prophylactic therapy prior to hospital discharge. Nonabsorbable (or poorly absorbable) antibiotics are most effective for such prophylaxis by selectively eliminating gram-negative organisms in the gut.
These agents reduce the rate of SBP recurrence to around 15–20%. Nosocomial infections respond poorly (~40% of cases) to 3rd generation cephalosporins. Those who have been in the hospital and received antibiotics within the past 90 days should receive extended-spectrum antibiotics.
References
1. Rimola A., Garcia-Tsao G., Navasa M., et al. Diagnosis, treatment and prophylaxis of spontaneous bacterial peri- tonitis: a consensus document. J Hepatol. 2000;32:142-53.
2. Tandon P., Garcia-Tsao G. Bacterial infections, sepsis, and multiorgan failure in cirrhosis. Semin Liver Dis. 2008;28(1):26-42.
Answer: D
Objective: Recognize that spontaneous bacterial peritonitis may present in an asymptomatic manner.
Discussion: It is important to recognize that patients with spontaneous bacterial peritonitis may present in various ways and may not exhibit classic abdominal pain or fevers. Patients may have atypical or no overt symptoms at all. With direct inoculation of ascitic fluid into culture bottles at bedside, cultures may identify bacteria in up to 40%-50% of cases.
Patients who survive an episode of SBP have a very high risk of recurrence (70%) within the first year of the index episode. It is therefore essential that patients recovering from SBP be started on prophylactic therapy prior to hospital discharge. Nonabsorbable (or poorly absorbable) antibiotics are most effective for such prophylaxis by selectively eliminating gram-negative organisms in the gut.
These agents reduce the rate of SBP recurrence to around 15–20%. Nosocomial infections respond poorly (~40% of cases) to 3rd generation cephalosporins. Those who have been in the hospital and received antibiotics within the past 90 days should receive extended-spectrum antibiotics.
References
1. Rimola A., Garcia-Tsao G., Navasa M., et al. Diagnosis, treatment and prophylaxis of spontaneous bacterial peri- tonitis: a consensus document. J Hepatol. 2000;32:142-53.
2. Tandon P., Garcia-Tsao G. Bacterial infections, sepsis, and multiorgan failure in cirrhosis. Semin Liver Dis. 2008;28(1):26-42.
A 56-year old man with a history of decompensated cirrhosis due to hepatitis C, complicated by ascites, presents with abdominal distension. A therapeutic paracentesis is performed, and is positive for spontaneous bacterial peritonitis.
2018 AGA Fellows Program now accepting applications
The application period for the 2018 AGA Fellows Program is now open. The program recognizes members whose accomplishments demonstrate personal commitment to the field of gastroenterology with the distinction of fellowship.
AGA Fellows receive this honor from AGA for their superior professional achievement in clinical private or academic practice and in basic or clinical research.
AGA Fellows receive:
- The privilege of using the designation “AGAF” in professional activities.
- An official certificate and pin denoting your status.
- A listing on the AGA website.
And more.
Apply today to join this international community of excellence.
Find more information, including the list of benefits and criteria for fellowship, at www.gastro.org/fellowship. The deadline for application submissions is Monday, July 31, 2017.
The application period for the 2018 AGA Fellows Program is now open. The program recognizes members whose accomplishments demonstrate personal commitment to the field of gastroenterology with the distinction of fellowship.
AGA Fellows receive this honor from AGA for their superior professional achievement in clinical private or academic practice and in basic or clinical research.
AGA Fellows receive:
- The privilege of using the designation “AGAF” in professional activities.
- An official certificate and pin denoting your status.
- A listing on the AGA website.
And more.
Apply today to join this international community of excellence.
Find more information, including the list of benefits and criteria for fellowship, at www.gastro.org/fellowship. The deadline for application submissions is Monday, July 31, 2017.
The application period for the 2018 AGA Fellows Program is now open. The program recognizes members whose accomplishments demonstrate personal commitment to the field of gastroenterology with the distinction of fellowship.
AGA Fellows receive this honor from AGA for their superior professional achievement in clinical private or academic practice and in basic or clinical research.
AGA Fellows receive:
- The privilege of using the designation “AGAF” in professional activities.
- An official certificate and pin denoting your status.
- A listing on the AGA website.
And more.
Apply today to join this international community of excellence.
Find more information, including the list of benefits and criteria for fellowship, at www.gastro.org/fellowship. The deadline for application submissions is Monday, July 31, 2017.
Dual-task walking test may be effective dementia predictor
Poor performance in dual-task gait testing was significantly associated with dementia progression in patients with mild cognitive impairment (MCI), according to a small prospective study.
The dementia prediction model could provide clinicians with a minimally invasive, low-cost analysis tool for patients with MCI and a compass to help guide decisions for further testing, according to Manuel Montero-Odasso, MD, PhD, an associate professor at the University of Western Ontario, London, and his colleagues.
In a study of 112 patients with MCI who were part of the Gait and Brain Study, investigators measured patients’ walking speed while only focusing on walking and then again while walking and counting backward from 100 by ones, counting backward from 100 by 7, or naming animals. The percentage difference between patients’ single-test walking speed and the dual-task speed was the dual-task gait cost. Patients received a biannual follow-up over a 6-year period.
Investigators found that higher gait costs in the dual gait test involving either counting backward by ones or naming animals were associated with a significantly increased risk of progression to dementia (JAMA Neurol. 2017 May 15. doi: 10.1001/jamaneurol.2017.0643).
“Our sensitivity analysis showed that dual-task gait was comparable with cognitive testing to predict incident dementia,” said Dr. Montero-Odasso. “Clinicians may use dual-task gait testing in screening patients with MCI who could benefit the most from additional testing, optimizing recommendations for imaging, spinal fluid examinations, and genetic testing.”
Patients were an average of 75 years old, with an even distribution of men and women. Patients had an average of five comorbidities, most common among them hypertension (60.4%) and history of smoking (56.1%). Of the 112 participants, 24% progressed to dementia, an incidence rate of 121 per 1,000 person-years. A total of 39% of the study participants carried an APOE e4 allele.
Investigators confirmed the presence of MCI by evaluating patients’ levels of cognitive challenges, impairment in memory, executive function, attention, language, certain living activities, and an absence of dementia using criteria from the Diagnostic and Statistical Manual of Mental Disorders, fourth edition.
When comparing the three dual-task gait tests, researchers found that higher gait cost when asking patients to count backward by ones or name animals presented a 3.8 times (P = .003) and 2.4 times (P = .04), respectively, increased risk of dementia progression. Gait cost when counting from 100 by sevens wasn’t significantly associated with dementia risk.
The researchers said that they still do not understand completely the relationship between walking velocity and cognitive function, although they hypothesized the connection may have to do with shared networks in the brain.
“What does seem clear is that executive demands used for gait and for the selected cognitive tasks may share a similar pathogenic mechanism at the brain level,” according to Dr. Montero-Odasso and his fellow investigators. “Episodic memory, a cognitive domain that was affected in all of our participants who progressed to dementia, relies on frontal-hippocampal circuits that are also central for gait control.”
The study was limited by the small population size, causing investigators to conclude that their findings are only generalizable at a clinic-based level. The investigators also noted the need for cross-validation in other MCI cohorts.
The model would be accessible for clinicians and researchers alike when identifying high-risk patients, designing further testing strategies, or planning primary prevention or intervention studies by more easily selecting patients with a greater risk of dementia progression, Dr. Montero-Odasso and his peers asserted.
The Canadian Institutes of Health Research funded the study. One of the authors reported having been a part-time employee of Pfizer and owning employee stock options. The investigators reported no other relevant financial disclosures.
[email protected]
On Twitter @eaztweets
Poor performance in dual-task gait testing was significantly associated with dementia progression in patients with mild cognitive impairment (MCI), according to a small prospective study.
The dementia prediction model could provide clinicians with a minimally invasive, low-cost analysis tool for patients with MCI and a compass to help guide decisions for further testing, according to Manuel Montero-Odasso, MD, PhD, an associate professor at the University of Western Ontario, London, and his colleagues.
In a study of 112 patients with MCI who were part of the Gait and Brain Study, investigators measured patients’ walking speed while only focusing on walking and then again while walking and counting backward from 100 by ones, counting backward from 100 by 7, or naming animals. The percentage difference between patients’ single-test walking speed and the dual-task speed was the dual-task gait cost. Patients received a biannual follow-up over a 6-year period.
Investigators found that higher gait costs in the dual gait test involving either counting backward by ones or naming animals were associated with a significantly increased risk of progression to dementia (JAMA Neurol. 2017 May 15. doi: 10.1001/jamaneurol.2017.0643).
“Our sensitivity analysis showed that dual-task gait was comparable with cognitive testing to predict incident dementia,” said Dr. Montero-Odasso. “Clinicians may use dual-task gait testing in screening patients with MCI who could benefit the most from additional testing, optimizing recommendations for imaging, spinal fluid examinations, and genetic testing.”
Patients were an average of 75 years old, with an even distribution of men and women. Patients had an average of five comorbidities, most common among them hypertension (60.4%) and history of smoking (56.1%). Of the 112 participants, 24% progressed to dementia, an incidence rate of 121 per 1,000 person-years. A total of 39% of the study participants carried an APOE e4 allele.
Investigators confirmed the presence of MCI by evaluating patients’ levels of cognitive challenges, impairment in memory, executive function, attention, language, certain living activities, and an absence of dementia using criteria from the Diagnostic and Statistical Manual of Mental Disorders, fourth edition.
When comparing the three dual-task gait tests, researchers found that higher gait cost when asking patients to count backward by ones or name animals presented a 3.8 times (P = .003) and 2.4 times (P = .04), respectively, increased risk of dementia progression. Gait cost when counting from 100 by sevens wasn’t significantly associated with dementia risk.
The researchers said that they still do not understand completely the relationship between walking velocity and cognitive function, although they hypothesized the connection may have to do with shared networks in the brain.
“What does seem clear is that executive demands used for gait and for the selected cognitive tasks may share a similar pathogenic mechanism at the brain level,” according to Dr. Montero-Odasso and his fellow investigators. “Episodic memory, a cognitive domain that was affected in all of our participants who progressed to dementia, relies on frontal-hippocampal circuits that are also central for gait control.”
The study was limited by the small population size, causing investigators to conclude that their findings are only generalizable at a clinic-based level. The investigators also noted the need for cross-validation in other MCI cohorts.
The model would be accessible for clinicians and researchers alike when identifying high-risk patients, designing further testing strategies, or planning primary prevention or intervention studies by more easily selecting patients with a greater risk of dementia progression, Dr. Montero-Odasso and his peers asserted.
The Canadian Institutes of Health Research funded the study. One of the authors reported having been a part-time employee of Pfizer and owning employee stock options. The investigators reported no other relevant financial disclosures.
[email protected]
On Twitter @eaztweets
Poor performance in dual-task gait testing was significantly associated with dementia progression in patients with mild cognitive impairment (MCI), according to a small prospective study.
The dementia prediction model could provide clinicians with a minimally invasive, low-cost analysis tool for patients with MCI and a compass to help guide decisions for further testing, according to Manuel Montero-Odasso, MD, PhD, an associate professor at the University of Western Ontario, London, and his colleagues.
In a study of 112 patients with MCI who were part of the Gait and Brain Study, investigators measured patients’ walking speed while only focusing on walking and then again while walking and counting backward from 100 by ones, counting backward from 100 by 7, or naming animals. The percentage difference between patients’ single-test walking speed and the dual-task speed was the dual-task gait cost. Patients received a biannual follow-up over a 6-year period.
Investigators found that higher gait costs in the dual gait test involving either counting backward by ones or naming animals were associated with a significantly increased risk of progression to dementia (JAMA Neurol. 2017 May 15. doi: 10.1001/jamaneurol.2017.0643).
“Our sensitivity analysis showed that dual-task gait was comparable with cognitive testing to predict incident dementia,” said Dr. Montero-Odasso. “Clinicians may use dual-task gait testing in screening patients with MCI who could benefit the most from additional testing, optimizing recommendations for imaging, spinal fluid examinations, and genetic testing.”
Patients were an average of 75 years old, with an even distribution of men and women. Patients had an average of five comorbidities, most common among them hypertension (60.4%) and history of smoking (56.1%). Of the 112 participants, 24% progressed to dementia, an incidence rate of 121 per 1,000 person-years. A total of 39% of the study participants carried an APOE e4 allele.
Investigators confirmed the presence of MCI by evaluating patients’ levels of cognitive challenges, impairment in memory, executive function, attention, language, certain living activities, and an absence of dementia using criteria from the Diagnostic and Statistical Manual of Mental Disorders, fourth edition.
When comparing the three dual-task gait tests, researchers found that higher gait cost when asking patients to count backward by ones or name animals presented a 3.8 times (P = .003) and 2.4 times (P = .04), respectively, increased risk of dementia progression. Gait cost when counting from 100 by sevens wasn’t significantly associated with dementia risk.
The researchers said that they still do not understand completely the relationship between walking velocity and cognitive function, although they hypothesized the connection may have to do with shared networks in the brain.
“What does seem clear is that executive demands used for gait and for the selected cognitive tasks may share a similar pathogenic mechanism at the brain level,” according to Dr. Montero-Odasso and his fellow investigators. “Episodic memory, a cognitive domain that was affected in all of our participants who progressed to dementia, relies on frontal-hippocampal circuits that are also central for gait control.”
The study was limited by the small population size, causing investigators to conclude that their findings are only generalizable at a clinic-based level. The investigators also noted the need for cross-validation in other MCI cohorts.
The model would be accessible for clinicians and researchers alike when identifying high-risk patients, designing further testing strategies, or planning primary prevention or intervention studies by more easily selecting patients with a greater risk of dementia progression, Dr. Montero-Odasso and his peers asserted.
The Canadian Institutes of Health Research funded the study. One of the authors reported having been a part-time employee of Pfizer and owning employee stock options. The investigators reported no other relevant financial disclosures.
[email protected]
On Twitter @eaztweets
FROM JAMA NEUROLOGY
Key clinical point:
Major finding: Greater changes in the walking speeds of patients as they counted backward (hazard ratio, 3.79; P = .003) or named animals (HR, 2.41; P = .04) were associated with increased risk of progression to dementia.
Data source: A prospective cohort study of 122 MCI patients enrolled in the Gait and Brain Study, with follow-up reports gathered between July 2007 and March 2016.
Disclosures: The Canadian Institutes of Health Research funded the study. One of the authors reported having been a part-time employee of Pfizer and owning employee stock options. The investigators reported no other relevant financial disclosures.
Systemic Interferon Alfa Injections for the Treatment of a Giant Orf
Orf, also known as ecthyma contagiosum, is a common viral zoonotic infection caused by a parapoxvirus. It is widespread among small ruminants such as sheep and goats, and it can be transmitted to humans by close contact with infected animals or contaminated fomites. It usually manifests as vesiculoulcerative lesions or nodules on the inoculation sites, mostly on the hands, but other sites such as the head and scalp occasionally may be involved.1 We report the case of an orf that proliferated dramatically and became giant after total excision. It was successfully treated with systemic interferon alfa-2a injections and imiquimod cream.
Case Report
A 68-year-old man presented with a rapidly enlarging mass on the left hand that developed 4 weeks prior after close contact with a freshly slaughtered sheep during an Islamic holiday in Turkey. His medical history was remarkable for chronic lymphocytic leukemia (CLL), which was diagnosed one year prior. The patient had been treated with systemic prednisolone and cyclophosphamide therapies, but his disease was in remission at the current presentation and he currently was not receiving any treatment. On physical examination, a 2-cm, exophytic, pinkish gray, weeping nodule was observed on the proximal aspect of the right thumb. Based on the clinical findings and typical anamnesis, a diagnosis of an orf was concluded. It was decided to monitor the patient without any intervention; however, because the lesion did not resolve and remained stable, he was referred to a plastic surgeon for surgical removal after 6 weeks of follow-up.
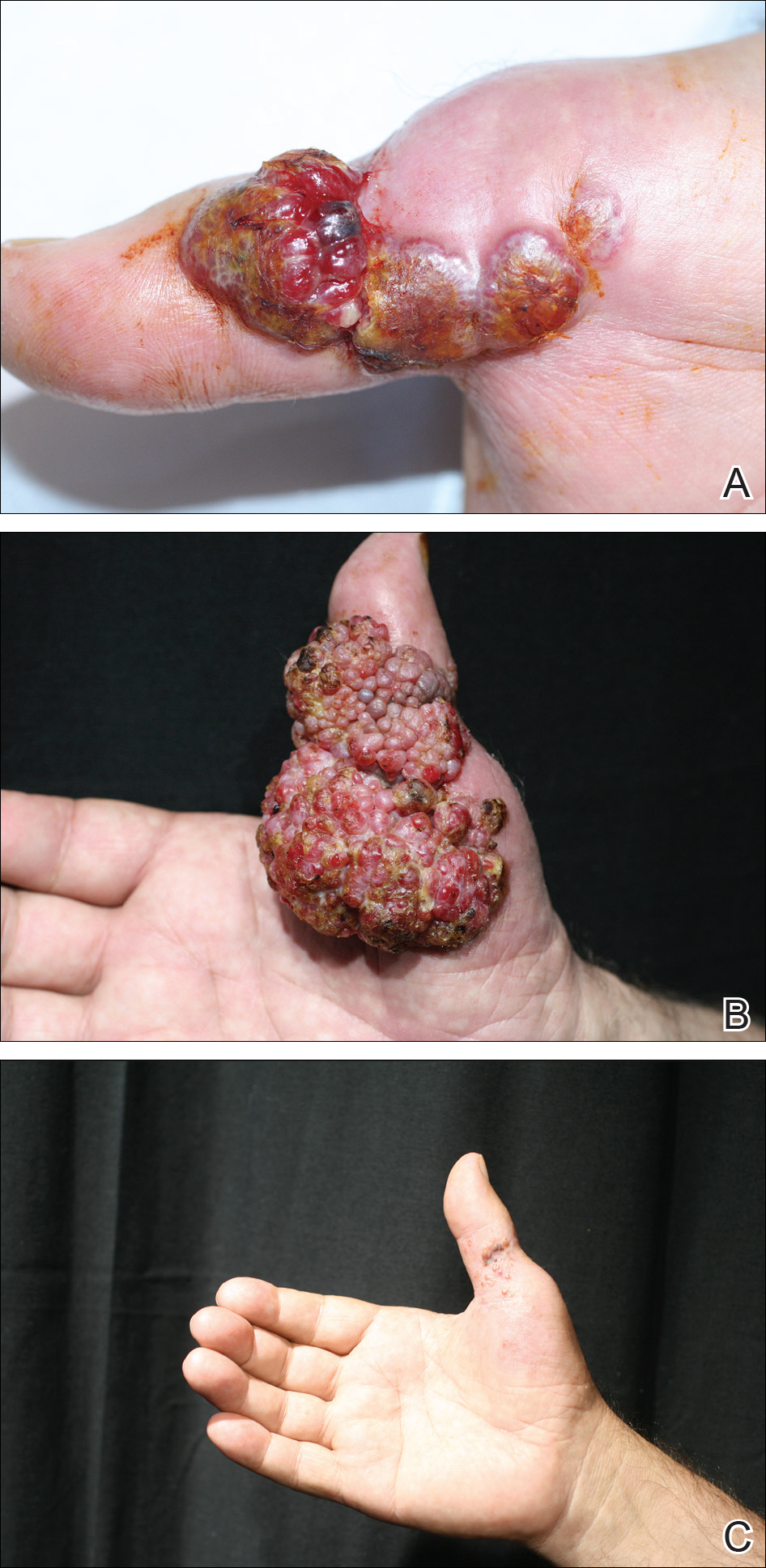
Histopathologic examination of the excision specimen revealed pseudoepitheliomatous hyperplasia, massive capillary proliferation, and viral cytopathic changes in keratinocytes characterized by ballooning degeneration and eosinophilic cytoplasmic inclusions, which was consistent with the clinical diagnosis of an orf. Unfortunately, the lesion relapsed rapidly following excision (Figure, A). Treatment with oral valacyclovir (1 g 3 times daily) and imiquimod cream 5% (3 times weekly) was initiated. However, this treatment was unsuccessful and was discontinued after 6 weeks, as the lesion kept growing, reaching a diameter of approximately 5 cm and becoming lobulated on the surface (Figure, B). Combination therapy was started with imiquimod cream 5% (3 times weekly) and intralesional interferon alfa-2a injections (3 million IU twice weekly). The injections were so painful that the patient refused further therapy after only 2 injections. The therapy was switched from intralesional to systemic subcutaneous injections of interferon alfa-2a (3 million IU twice weekly) with concomitant imiquimod cream 5% 3 times weekly. This treatment was well tolerated by our patient with no notable side effects, except for mild fever on the night of each injection. Three weeks after the commencement of systemic injections, remarkable healing of the lesions with reduced size and exudation was noted. The frequency of injections was decreased to once weekly, which was then discontinued after 6 weeks when the lesion totally resolved (Figure, C). At 12 months’ follow-up, there were no signs of relapse.
Comment
Orf is an occupational disease that usually develops in farmers, butchers, and veterinarians; however, epidemic outbreaks of human orf are commonly observed in Turkey after the feast of sacrifice, as many individuals have close contact with the animals during sacrification.2 In Turkey, orf is well recognized by dermatologists, and clinical diagnosis usually is not difficult.
Human orf has a self-limited course in which lesions spontaneously resolve in 4 to 8 weeks; however, in immunocompromised patients, such as our patient with CLL, orf lesions may be persistent, atypical, and giant, requiring early and effective treatment. Treatment options for giant orf tumors in immunocompromised individuals include surgical excision,3 cryotherapy, topical imiquimod,4,5 topical or intralesional cidofovir,6 and intralesional interferon alfa injections.7 According to our clinical observations, surgical interventions for treatment of orfs usually cause a delay in the natural healing process; however, because surgical excision is a recommended treatment option for exophytic and recalcitrant orfs, we decided to treat our patient with surgical excision, which resulted in rapid recurrence and massive proliferation. A similar case of giant orf that was aggravated after surgery has been reported.8 In light of these cases, it is our opinion that treatment options other than surgery may be reasonable.
Chronic lymphocytic leukemia may show features of both humoral and cell-mediated deficiency. Patients are known to be prone to viral infections such as varicella-zoster virus, herpes simplex virus, cytomegalovirus, and human papillomavirus. A giant orf infection on the background of CLL also has been described.9
Interferons were first discovered in 1957 and named after their ability to interfere with viral replication. They represent a family of cytokines that has an essential role in the innate immune response to virus infections. Because of their antiviral properties, recombinant forms of interferon alfa are widely used with success in the treatment of chronic hepatitis B and hepatitis C virus infections. A few other antiviral clinical applications of interferon alfa include infections caused by human herpesvirus 8 (the etiological agent in Kaposi sarcoma) and human papillomatosis virus (the etiological agent in juvenile laryngeal papillomatosis and condyloma acuminatum).10
In a report by Ran et al,7 intralesional interferon alfa injections were successfully used for treatment of giant orf lesions in an immunocompromised patient. As a result, we started treating the patient with intralesional interferon alfa-2a, but it was not well tolerated by our patient, as it was quite painful. We then decided to continue the therapy with systemic interferon alfa-2a injections, as we believed that it was a good option due to its antiviral, antiproliferative, and antiangiogenic properties. With the experimental combined therapy of systemic interferon alfa-2a and topical imiquimod, our patient achieved a complete response in 9 weeks (3 weeks of twice weekly injections and then 6 weeks of once weekly injections) and had no relapses during 12 months of follow-up.
Conclusion
We present a rare case of a giant orf treated with systemic interferon alfa-2a injections. Because intralesional injections are quite painful, systemic subcutaneous injections of interferon might be a good and safe alternative for recalcitrant orf lesions in immunocompromised patients. However, more studies and reports are needed to confirm its effectiveness and safety.
The 9th Cosmetic Surgery Forum will be held November 29-December 2, 2017, in Las Vegas, Nevada. Get more information at www.cosmeticsurgeryforum.com.
- Gurel MS, Ozardali I, Bitiren M, et al. Giant orf on the nose. Eur J Dermatol. 2002;12:183-185.
- Uzel M, Sasmaz S, Bakaris S, et al. A viral infection of the hand commonly seen after the feast of sacrifice: human orf (orf of the hand). Epidemiol Infect. 2005;133:653-657.
- Ballanger F, Barbarot S, Mollat C, et al. Two giant orf lesions in a heart/lung transplant patient. Eur J Dermatol. 2006;16:284-286.
- Zaharia D, Kanitakis J, Pouteil-Noble C, et al. Rapidly growing orf in a renal transplant recipient: favourable outcome with reduction of immunosuppression and imiquimod. Transpl Int. 2010;23:E62-E64.
- Lederman ER, Green GM, DeGroot HE, et al. Progressive ORF virus infection in a patient with lymphoma: successful treatment using imiquimod. Clin Infect Dis. 2007;44:e100-e103.
- Geerinck K, Lukito G, Snoeck R, et al. A case of human orf in an immunocompromised patient treated successfully with cidofovir cream. J Med Virol. 2001;64:543-549.
- Ran M, Lee M, Gong J, et al. Oral acyclovir and intralesional interferon injections for treatment of giant pyogenic granuloma–like lesions in an immunocompromised patient with human orf. JAMA Dermatol. 2015;151:1032-1034.
- Key SJ, Catania J, Mustafa SF, et al. Unusual presentation of human giant orf (ecthyma contagiosum). J Craniofac Surg. 2007;18:1076-1078.
- Hunskaar S. Giant orf in a patient with chronic lymphocytic leukaemia. Br J Dermatol. 1986;114:631-634.
- Friedman RM. Clinical uses of interferons. Br J Clin Pharmacol. 2008;65:158-162.
Orf, also known as ecthyma contagiosum, is a common viral zoonotic infection caused by a parapoxvirus. It is widespread among small ruminants such as sheep and goats, and it can be transmitted to humans by close contact with infected animals or contaminated fomites. It usually manifests as vesiculoulcerative lesions or nodules on the inoculation sites, mostly on the hands, but other sites such as the head and scalp occasionally may be involved.1 We report the case of an orf that proliferated dramatically and became giant after total excision. It was successfully treated with systemic interferon alfa-2a injections and imiquimod cream.
Case Report
A 68-year-old man presented with a rapidly enlarging mass on the left hand that developed 4 weeks prior after close contact with a freshly slaughtered sheep during an Islamic holiday in Turkey. His medical history was remarkable for chronic lymphocytic leukemia (CLL), which was diagnosed one year prior. The patient had been treated with systemic prednisolone and cyclophosphamide therapies, but his disease was in remission at the current presentation and he currently was not receiving any treatment. On physical examination, a 2-cm, exophytic, pinkish gray, weeping nodule was observed on the proximal aspect of the right thumb. Based on the clinical findings and typical anamnesis, a diagnosis of an orf was concluded. It was decided to monitor the patient without any intervention; however, because the lesion did not resolve and remained stable, he was referred to a plastic surgeon for surgical removal after 6 weeks of follow-up.
Histopathologic examination of the excision specimen revealed pseudoepitheliomatous hyperplasia, massive capillary proliferation, and viral cytopathic changes in keratinocytes characterized by ballooning degeneration and eosinophilic cytoplasmic inclusions, which was consistent with the clinical diagnosis of an orf. Unfortunately, the lesion relapsed rapidly following excision (Figure, A). Treatment with oral valacyclovir (1 g 3 times daily) and imiquimod cream 5% (3 times weekly) was initiated. However, this treatment was unsuccessful and was discontinued after 6 weeks, as the lesion kept growing, reaching a diameter of approximately 5 cm and becoming lobulated on the surface (Figure, B). Combination therapy was started with imiquimod cream 5% (3 times weekly) and intralesional interferon alfa-2a injections (3 million IU twice weekly). The injections were so painful that the patient refused further therapy after only 2 injections. The therapy was switched from intralesional to systemic subcutaneous injections of interferon alfa-2a (3 million IU twice weekly) with concomitant imiquimod cream 5% 3 times weekly. This treatment was well tolerated by our patient with no notable side effects, except for mild fever on the night of each injection. Three weeks after the commencement of systemic injections, remarkable healing of the lesions with reduced size and exudation was noted. The frequency of injections was decreased to once weekly, which was then discontinued after 6 weeks when the lesion totally resolved (Figure, C). At 12 months’ follow-up, there were no signs of relapse.
Comment
Orf is an occupational disease that usually develops in farmers, butchers, and veterinarians; however, epidemic outbreaks of human orf are commonly observed in Turkey after the feast of sacrifice, as many individuals have close contact with the animals during sacrification.2 In Turkey, orf is well recognized by dermatologists, and clinical diagnosis usually is not difficult.
Human orf has a self-limited course in which lesions spontaneously resolve in 4 to 8 weeks; however, in immunocompromised patients, such as our patient with CLL, orf lesions may be persistent, atypical, and giant, requiring early and effective treatment. Treatment options for giant orf tumors in immunocompromised individuals include surgical excision,3 cryotherapy, topical imiquimod,4,5 topical or intralesional cidofovir,6 and intralesional interferon alfa injections.7 According to our clinical observations, surgical interventions for treatment of orfs usually cause a delay in the natural healing process; however, because surgical excision is a recommended treatment option for exophytic and recalcitrant orfs, we decided to treat our patient with surgical excision, which resulted in rapid recurrence and massive proliferation. A similar case of giant orf that was aggravated after surgery has been reported.8 In light of these cases, it is our opinion that treatment options other than surgery may be reasonable.
Chronic lymphocytic leukemia may show features of both humoral and cell-mediated deficiency. Patients are known to be prone to viral infections such as varicella-zoster virus, herpes simplex virus, cytomegalovirus, and human papillomavirus. A giant orf infection on the background of CLL also has been described.9
Interferons were first discovered in 1957 and named after their ability to interfere with viral replication. They represent a family of cytokines that has an essential role in the innate immune response to virus infections. Because of their antiviral properties, recombinant forms of interferon alfa are widely used with success in the treatment of chronic hepatitis B and hepatitis C virus infections. A few other antiviral clinical applications of interferon alfa include infections caused by human herpesvirus 8 (the etiological agent in Kaposi sarcoma) and human papillomatosis virus (the etiological agent in juvenile laryngeal papillomatosis and condyloma acuminatum).10
In a report by Ran et al,7 intralesional interferon alfa injections were successfully used for treatment of giant orf lesions in an immunocompromised patient. As a result, we started treating the patient with intralesional interferon alfa-2a, but it was not well tolerated by our patient, as it was quite painful. We then decided to continue the therapy with systemic interferon alfa-2a injections, as we believed that it was a good option due to its antiviral, antiproliferative, and antiangiogenic properties. With the experimental combined therapy of systemic interferon alfa-2a and topical imiquimod, our patient achieved a complete response in 9 weeks (3 weeks of twice weekly injections and then 6 weeks of once weekly injections) and had no relapses during 12 months of follow-up.
Conclusion
We present a rare case of a giant orf treated with systemic interferon alfa-2a injections. Because intralesional injections are quite painful, systemic subcutaneous injections of interferon might be a good and safe alternative for recalcitrant orf lesions in immunocompromised patients. However, more studies and reports are needed to confirm its effectiveness and safety.
The 9th Cosmetic Surgery Forum will be held November 29-December 2, 2017, in Las Vegas, Nevada. Get more information at www.cosmeticsurgeryforum.com.
Orf, also known as ecthyma contagiosum, is a common viral zoonotic infection caused by a parapoxvirus. It is widespread among small ruminants such as sheep and goats, and it can be transmitted to humans by close contact with infected animals or contaminated fomites. It usually manifests as vesiculoulcerative lesions or nodules on the inoculation sites, mostly on the hands, but other sites such as the head and scalp occasionally may be involved.1 We report the case of an orf that proliferated dramatically and became giant after total excision. It was successfully treated with systemic interferon alfa-2a injections and imiquimod cream.
Case Report
A 68-year-old man presented with a rapidly enlarging mass on the left hand that developed 4 weeks prior after close contact with a freshly slaughtered sheep during an Islamic holiday in Turkey. His medical history was remarkable for chronic lymphocytic leukemia (CLL), which was diagnosed one year prior. The patient had been treated with systemic prednisolone and cyclophosphamide therapies, but his disease was in remission at the current presentation and he currently was not receiving any treatment. On physical examination, a 2-cm, exophytic, pinkish gray, weeping nodule was observed on the proximal aspect of the right thumb. Based on the clinical findings and typical anamnesis, a diagnosis of an orf was concluded. It was decided to monitor the patient without any intervention; however, because the lesion did not resolve and remained stable, he was referred to a plastic surgeon for surgical removal after 6 weeks of follow-up.
Histopathologic examination of the excision specimen revealed pseudoepitheliomatous hyperplasia, massive capillary proliferation, and viral cytopathic changes in keratinocytes characterized by ballooning degeneration and eosinophilic cytoplasmic inclusions, which was consistent with the clinical diagnosis of an orf. Unfortunately, the lesion relapsed rapidly following excision (Figure, A). Treatment with oral valacyclovir (1 g 3 times daily) and imiquimod cream 5% (3 times weekly) was initiated. However, this treatment was unsuccessful and was discontinued after 6 weeks, as the lesion kept growing, reaching a diameter of approximately 5 cm and becoming lobulated on the surface (Figure, B). Combination therapy was started with imiquimod cream 5% (3 times weekly) and intralesional interferon alfa-2a injections (3 million IU twice weekly). The injections were so painful that the patient refused further therapy after only 2 injections. The therapy was switched from intralesional to systemic subcutaneous injections of interferon alfa-2a (3 million IU twice weekly) with concomitant imiquimod cream 5% 3 times weekly. This treatment was well tolerated by our patient with no notable side effects, except for mild fever on the night of each injection. Three weeks after the commencement of systemic injections, remarkable healing of the lesions with reduced size and exudation was noted. The frequency of injections was decreased to once weekly, which was then discontinued after 6 weeks when the lesion totally resolved (Figure, C). At 12 months’ follow-up, there were no signs of relapse.
Comment
Orf is an occupational disease that usually develops in farmers, butchers, and veterinarians; however, epidemic outbreaks of human orf are commonly observed in Turkey after the feast of sacrifice, as many individuals have close contact with the animals during sacrification.2 In Turkey, orf is well recognized by dermatologists, and clinical diagnosis usually is not difficult.
Human orf has a self-limited course in which lesions spontaneously resolve in 4 to 8 weeks; however, in immunocompromised patients, such as our patient with CLL, orf lesions may be persistent, atypical, and giant, requiring early and effective treatment. Treatment options for giant orf tumors in immunocompromised individuals include surgical excision,3 cryotherapy, topical imiquimod,4,5 topical or intralesional cidofovir,6 and intralesional interferon alfa injections.7 According to our clinical observations, surgical interventions for treatment of orfs usually cause a delay in the natural healing process; however, because surgical excision is a recommended treatment option for exophytic and recalcitrant orfs, we decided to treat our patient with surgical excision, which resulted in rapid recurrence and massive proliferation. A similar case of giant orf that was aggravated after surgery has been reported.8 In light of these cases, it is our opinion that treatment options other than surgery may be reasonable.
Chronic lymphocytic leukemia may show features of both humoral and cell-mediated deficiency. Patients are known to be prone to viral infections such as varicella-zoster virus, herpes simplex virus, cytomegalovirus, and human papillomavirus. A giant orf infection on the background of CLL also has been described.9
Interferons were first discovered in 1957 and named after their ability to interfere with viral replication. They represent a family of cytokines that has an essential role in the innate immune response to virus infections. Because of their antiviral properties, recombinant forms of interferon alfa are widely used with success in the treatment of chronic hepatitis B and hepatitis C virus infections. A few other antiviral clinical applications of interferon alfa include infections caused by human herpesvirus 8 (the etiological agent in Kaposi sarcoma) and human papillomatosis virus (the etiological agent in juvenile laryngeal papillomatosis and condyloma acuminatum).10
In a report by Ran et al,7 intralesional interferon alfa injections were successfully used for treatment of giant orf lesions in an immunocompromised patient. As a result, we started treating the patient with intralesional interferon alfa-2a, but it was not well tolerated by our patient, as it was quite painful. We then decided to continue the therapy with systemic interferon alfa-2a injections, as we believed that it was a good option due to its antiviral, antiproliferative, and antiangiogenic properties. With the experimental combined therapy of systemic interferon alfa-2a and topical imiquimod, our patient achieved a complete response in 9 weeks (3 weeks of twice weekly injections and then 6 weeks of once weekly injections) and had no relapses during 12 months of follow-up.
Conclusion
We present a rare case of a giant orf treated with systemic interferon alfa-2a injections. Because intralesional injections are quite painful, systemic subcutaneous injections of interferon might be a good and safe alternative for recalcitrant orf lesions in immunocompromised patients. However, more studies and reports are needed to confirm its effectiveness and safety.
The 9th Cosmetic Surgery Forum will be held November 29-December 2, 2017, in Las Vegas, Nevada. Get more information at www.cosmeticsurgeryforum.com.
- Gurel MS, Ozardali I, Bitiren M, et al. Giant orf on the nose. Eur J Dermatol. 2002;12:183-185.
- Uzel M, Sasmaz S, Bakaris S, et al. A viral infection of the hand commonly seen after the feast of sacrifice: human orf (orf of the hand). Epidemiol Infect. 2005;133:653-657.
- Ballanger F, Barbarot S, Mollat C, et al. Two giant orf lesions in a heart/lung transplant patient. Eur J Dermatol. 2006;16:284-286.
- Zaharia D, Kanitakis J, Pouteil-Noble C, et al. Rapidly growing orf in a renal transplant recipient: favourable outcome with reduction of immunosuppression and imiquimod. Transpl Int. 2010;23:E62-E64.
- Lederman ER, Green GM, DeGroot HE, et al. Progressive ORF virus infection in a patient with lymphoma: successful treatment using imiquimod. Clin Infect Dis. 2007;44:e100-e103.
- Geerinck K, Lukito G, Snoeck R, et al. A case of human orf in an immunocompromised patient treated successfully with cidofovir cream. J Med Virol. 2001;64:543-549.
- Ran M, Lee M, Gong J, et al. Oral acyclovir and intralesional interferon injections for treatment of giant pyogenic granuloma–like lesions in an immunocompromised patient with human orf. JAMA Dermatol. 2015;151:1032-1034.
- Key SJ, Catania J, Mustafa SF, et al. Unusual presentation of human giant orf (ecthyma contagiosum). J Craniofac Surg. 2007;18:1076-1078.
- Hunskaar S. Giant orf in a patient with chronic lymphocytic leukaemia. Br J Dermatol. 1986;114:631-634.
- Friedman RM. Clinical uses of interferons. Br J Clin Pharmacol. 2008;65:158-162.
- Gurel MS, Ozardali I, Bitiren M, et al. Giant orf on the nose. Eur J Dermatol. 2002;12:183-185.
- Uzel M, Sasmaz S, Bakaris S, et al. A viral infection of the hand commonly seen after the feast of sacrifice: human orf (orf of the hand). Epidemiol Infect. 2005;133:653-657.
- Ballanger F, Barbarot S, Mollat C, et al. Two giant orf lesions in a heart/lung transplant patient. Eur J Dermatol. 2006;16:284-286.
- Zaharia D, Kanitakis J, Pouteil-Noble C, et al. Rapidly growing orf in a renal transplant recipient: favourable outcome with reduction of immunosuppression and imiquimod. Transpl Int. 2010;23:E62-E64.
- Lederman ER, Green GM, DeGroot HE, et al. Progressive ORF virus infection in a patient with lymphoma: successful treatment using imiquimod. Clin Infect Dis. 2007;44:e100-e103.
- Geerinck K, Lukito G, Snoeck R, et al. A case of human orf in an immunocompromised patient treated successfully with cidofovir cream. J Med Virol. 2001;64:543-549.
- Ran M, Lee M, Gong J, et al. Oral acyclovir and intralesional interferon injections for treatment of giant pyogenic granuloma–like lesions in an immunocompromised patient with human orf. JAMA Dermatol. 2015;151:1032-1034.
- Key SJ, Catania J, Mustafa SF, et al. Unusual presentation of human giant orf (ecthyma contagiosum). J Craniofac Surg. 2007;18:1076-1078.
- Hunskaar S. Giant orf in a patient with chronic lymphocytic leukaemia. Br J Dermatol. 1986;114:631-634.
- Friedman RM. Clinical uses of interferons. Br J Clin Pharmacol. 2008;65:158-162.
Resident Pearl
- Human orf lesions spontaneously resolve in 4 to 8 weeks; however, in immunocompromised patients, orf lesions may be persistent, atypical, and giant. We observed that surgical interventions for treatment of orfs cause a delay in the natural healing process, and other treatment options such as subcutaneous interferon alfa-2a may be used.
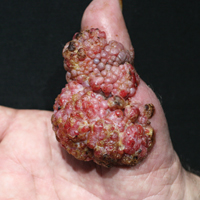
Few states fully support HCV prevention, treatment
The prevalence of hepatitis C virus (HCV) varies considerably by state, and the same can be said for the state laws and policies attempting to decrease that prevalence, according to an assessment by the Centers for Disease Control and Prevention.
In 2015, incidence of acute HCV infection exceeded the national average of 0.8 per 100,000 population in 17 states, including seven with rates that at least doubled it, the report noted. New HCV infections have increased in recent years despite curative therapies “and known preventive measures to interrupt transmission.”
The U.S. incidence of HCV jumped by 294% from 2010 to 2015, and “this increase in acute cases of HCV is largely attributed to injection drug use,” the CDC investigators said. Since state laws and policies affect access to HCV preventive and treatment measures, the researchers reviewed laws related to access to clean needles and policies on Medicaid fee-for-service treatment.
Only three states – Massachusetts, New Mexico, and Washington – had a comprehensive (all three were considered “more comprehensive”) set of prevention laws and a permissive treatment policy, the investigators said, while also noting that two of the three – Massachusetts and New Mexico – were among the states with acute HCV rates that were at least twice the national average.
“Although the costs of HCV therapies have raised budgetary issues for state Medicaid programs in the past, the costs of HCV treatment have declined in recent years, increasing the cost-effectiveness of treatment, particularly among persons who inject drugs and who might serve as an ongoing source of transmission to others,” the report concluded.
The analysis examined three types of laws on access to clean needles and syringes: authorization of exchange programs, the scope of drug paraphernalia laws, and retail sale of needles and syringes. Each law was assessed for five elements, including authorization of syringe exchange statewide or in selected jurisdictions and exemption of needles or syringes from the definition of drug paraphernalia.
For the accompanying map (see “Acute hepatitis C infection incidence rates, 2015: State vs. national”), each state’s acute HCV incidence rate for 2015 was divided by the national rate to determine the incidence rate ratio, with data unavailable for 10 states.
AGA Resource
The AGA HCV Clinical Service Line offers tools to help you become more efficient, understand quality standards and improve the process of care for patients. Read more at http://www.gastro.org/patient-care/conditions-diseases/hepatitis-c.
The prevalence of hepatitis C virus (HCV) varies considerably by state, and the same can be said for the state laws and policies attempting to decrease that prevalence, according to an assessment by the Centers for Disease Control and Prevention.
In 2015, incidence of acute HCV infection exceeded the national average of 0.8 per 100,000 population in 17 states, including seven with rates that at least doubled it, the report noted. New HCV infections have increased in recent years despite curative therapies “and known preventive measures to interrupt transmission.”
The U.S. incidence of HCV jumped by 294% from 2010 to 2015, and “this increase in acute cases of HCV is largely attributed to injection drug use,” the CDC investigators said. Since state laws and policies affect access to HCV preventive and treatment measures, the researchers reviewed laws related to access to clean needles and policies on Medicaid fee-for-service treatment.
Only three states – Massachusetts, New Mexico, and Washington – had a comprehensive (all three were considered “more comprehensive”) set of prevention laws and a permissive treatment policy, the investigators said, while also noting that two of the three – Massachusetts and New Mexico – were among the states with acute HCV rates that were at least twice the national average.
“Although the costs of HCV therapies have raised budgetary issues for state Medicaid programs in the past, the costs of HCV treatment have declined in recent years, increasing the cost-effectiveness of treatment, particularly among persons who inject drugs and who might serve as an ongoing source of transmission to others,” the report concluded.
The analysis examined three types of laws on access to clean needles and syringes: authorization of exchange programs, the scope of drug paraphernalia laws, and retail sale of needles and syringes. Each law was assessed for five elements, including authorization of syringe exchange statewide or in selected jurisdictions and exemption of needles or syringes from the definition of drug paraphernalia.
For the accompanying map (see “Acute hepatitis C infection incidence rates, 2015: State vs. national”), each state’s acute HCV incidence rate for 2015 was divided by the national rate to determine the incidence rate ratio, with data unavailable for 10 states.
AGA Resource
The AGA HCV Clinical Service Line offers tools to help you become more efficient, understand quality standards and improve the process of care for patients. Read more at http://www.gastro.org/patient-care/conditions-diseases/hepatitis-c.
The prevalence of hepatitis C virus (HCV) varies considerably by state, and the same can be said for the state laws and policies attempting to decrease that prevalence, according to an assessment by the Centers for Disease Control and Prevention.
In 2015, incidence of acute HCV infection exceeded the national average of 0.8 per 100,000 population in 17 states, including seven with rates that at least doubled it, the report noted. New HCV infections have increased in recent years despite curative therapies “and known preventive measures to interrupt transmission.”
The U.S. incidence of HCV jumped by 294% from 2010 to 2015, and “this increase in acute cases of HCV is largely attributed to injection drug use,” the CDC investigators said. Since state laws and policies affect access to HCV preventive and treatment measures, the researchers reviewed laws related to access to clean needles and policies on Medicaid fee-for-service treatment.
Only three states – Massachusetts, New Mexico, and Washington – had a comprehensive (all three were considered “more comprehensive”) set of prevention laws and a permissive treatment policy, the investigators said, while also noting that two of the three – Massachusetts and New Mexico – were among the states with acute HCV rates that were at least twice the national average.
“Although the costs of HCV therapies have raised budgetary issues for state Medicaid programs in the past, the costs of HCV treatment have declined in recent years, increasing the cost-effectiveness of treatment, particularly among persons who inject drugs and who might serve as an ongoing source of transmission to others,” the report concluded.
The analysis examined three types of laws on access to clean needles and syringes: authorization of exchange programs, the scope of drug paraphernalia laws, and retail sale of needles and syringes. Each law was assessed for five elements, including authorization of syringe exchange statewide or in selected jurisdictions and exemption of needles or syringes from the definition of drug paraphernalia.
For the accompanying map (see “Acute hepatitis C infection incidence rates, 2015: State vs. national”), each state’s acute HCV incidence rate for 2015 was divided by the national rate to determine the incidence rate ratio, with data unavailable for 10 states.
AGA Resource
The AGA HCV Clinical Service Line offers tools to help you become more efficient, understand quality standards and improve the process of care for patients. Read more at http://www.gastro.org/patient-care/conditions-diseases/hepatitis-c.
FROM MMWR