User login
This Month in CHEST: Editor’s picks

Giants in Chest Medicine:
Steven E. Weinberger, MD, FCCP
By Dr. J. Mandel
Editorial
Precision Medicine Urgency: The Case of Inhaled Corticosteroids in COPD By Drs. S. Suissa and P. Ernst
Original Research
Physician Assessment of Pretest Probability of Malignancy and Adherence With Guidelines for Pulmonary Nodule Evaluation By Dr. N. T. Tanner, et al.
The Long-Term Effect of Bacille Calmette-Guérin Vaccination on Tuberculin Skin Testing: A 55-Year Follow-Up Study By Dr. J. D. Mancuso, et al.
Clinical Characteristics of Pertussis-Associated Cough in Adults and Children: A Diagnostic Systematic Review and Meta-Analysis By Dr. A. Moore, et al.
Giants in Chest Medicine:
Steven E. Weinberger, MD, FCCP
By Dr. J. Mandel
Editorial
Precision Medicine Urgency: The Case of Inhaled Corticosteroids in COPD By Drs. S. Suissa and P. Ernst
Original Research
Physician Assessment of Pretest Probability of Malignancy and Adherence With Guidelines for Pulmonary Nodule Evaluation By Dr. N. T. Tanner, et al.
The Long-Term Effect of Bacille Calmette-Guérin Vaccination on Tuberculin Skin Testing: A 55-Year Follow-Up Study By Dr. J. D. Mancuso, et al.
Clinical Characteristics of Pertussis-Associated Cough in Adults and Children: A Diagnostic Systematic Review and Meta-Analysis By Dr. A. Moore, et al.
Giants in Chest Medicine:
Steven E. Weinberger, MD, FCCP
By Dr. J. Mandel
Editorial
Precision Medicine Urgency: The Case of Inhaled Corticosteroids in COPD By Drs. S. Suissa and P. Ernst
Original Research
Physician Assessment of Pretest Probability of Malignancy and Adherence With Guidelines for Pulmonary Nodule Evaluation By Dr. N. T. Tanner, et al.
The Long-Term Effect of Bacille Calmette-Guérin Vaccination on Tuberculin Skin Testing: A 55-Year Follow-Up Study By Dr. J. D. Mancuso, et al.
Clinical Characteristics of Pertussis-Associated Cough in Adults and Children: A Diagnostic Systematic Review and Meta-Analysis By Dr. A. Moore, et al.

CHEST Membership News
Introducing CHEST Participation Points
Everyday, you commit your time to helping patients. We recognize your dedication not only to your profession but to the CHEST community.
We’re happy to introduce CHEST Participation Points, designed to increase member recognition and reward you for participating and contributing to our diverse community. Wherever you are in your career, you can earn points for the things you do within the CHEST community.
Once you receive 50, 100, or 150 points, you can redeem your points for CHEST-branded apparel or discounts on courses and products.
Point accrual started on July 5, so you’ve already been earning points. If you are an FCCP, you began with 30 points awarded for becoming FCCP—that’s only 20 points away from the first tier of prizes. To accrue or redeem points, you must be an active member and current with your dues.
Log in to your CHEST account, and access Participation Points in the left column to see your points.
Start earning more points today! Learn more at chestnet.org/participationpoints.
Introducing CHEST Participation Points
Everyday, you commit your time to helping patients. We recognize your dedication not only to your profession but to the CHEST community.
We’re happy to introduce CHEST Participation Points, designed to increase member recognition and reward you for participating and contributing to our diverse community. Wherever you are in your career, you can earn points for the things you do within the CHEST community.
Once you receive 50, 100, or 150 points, you can redeem your points for CHEST-branded apparel or discounts on courses and products.
Point accrual started on July 5, so you’ve already been earning points. If you are an FCCP, you began with 30 points awarded for becoming FCCP—that’s only 20 points away from the first tier of prizes. To accrue or redeem points, you must be an active member and current with your dues.
Log in to your CHEST account, and access Participation Points in the left column to see your points.
Start earning more points today! Learn more at chestnet.org/participationpoints.
Introducing CHEST Participation Points
Everyday, you commit your time to helping patients. We recognize your dedication not only to your profession but to the CHEST community.
We’re happy to introduce CHEST Participation Points, designed to increase member recognition and reward you for participating and contributing to our diverse community. Wherever you are in your career, you can earn points for the things you do within the CHEST community.
Once you receive 50, 100, or 150 points, you can redeem your points for CHEST-branded apparel or discounts on courses and products.
Point accrual started on July 5, so you’ve already been earning points. If you are an FCCP, you began with 30 points awarded for becoming FCCP—that’s only 20 points away from the first tier of prizes. To accrue or redeem points, you must be an active member and current with your dues.
Log in to your CHEST account, and access Participation Points in the left column to see your points.
Start earning more points today! Learn more at chestnet.org/participationpoints.
CHEST ® journal — new online home
We are excited to share that the journal CHEST® has a new website with improved navigation, better search capabilities, alert sign-ups, and more multimedia elements. We are asking members to take a few minutes to activate their new account.
In order to maintain continuous access to the online journal, members will have to register for a free account and claim their subscription. If you go to chestjournal.org, CHEST members can then complete a 1- to 2-minute registration process.
CHEST members should have received an email with step-by-step instructions. Still have questions or need help? Contact Online Journal Support at 800/654-2452 (US and Canada) or +44 (0) 1865-843177 (Europe).
We are excited to share that the journal CHEST® has a new website with improved navigation, better search capabilities, alert sign-ups, and more multimedia elements. We are asking members to take a few minutes to activate their new account.
In order to maintain continuous access to the online journal, members will have to register for a free account and claim their subscription. If you go to chestjournal.org, CHEST members can then complete a 1- to 2-minute registration process.
CHEST members should have received an email with step-by-step instructions. Still have questions or need help? Contact Online Journal Support at 800/654-2452 (US and Canada) or +44 (0) 1865-843177 (Europe).
We are excited to share that the journal CHEST® has a new website with improved navigation, better search capabilities, alert sign-ups, and more multimedia elements. We are asking members to take a few minutes to activate their new account.
In order to maintain continuous access to the online journal, members will have to register for a free account and claim their subscription. If you go to chestjournal.org, CHEST members can then complete a 1- to 2-minute registration process.
CHEST members should have received an email with step-by-step instructions. Still have questions or need help? Contact Online Journal Support at 800/654-2452 (US and Canada) or +44 (0) 1865-843177 (Europe).
New Tools in Campaign to Fight Asthma
The Allergy & Asthma NetWork, the nation’s leading patient education and advocacy organization for people with allergy and asthma, has once again joined forces with the CHEST Foundation in an effort to empower patients suffering from severe asthma.
The campaign’s focus is to educate health-care providers, patients, parents of asthmatics, and the public about the most current treatment options for asthma, highlight the importance of referring to specialists to improve patient outcomes, and bring to light the role of the entire health-care team in the care of a patient with severe or difficult-to-control asthma.
Severity Assessment Tool
Available online and in print, the severity assessment tool was designed to help a patient, and the clinician, understand the severity of their asthma. Not only does the tool evaluate the severity of their condition, but it also helps the patient become more aware of their symptoms. The seven-question assessment includes questions on usage of quick-relief or rescue inhalers, visits to the ED/hospital, physical activity, controller medication, and quality of sleep.
Patient and Caregiver Testimonials
The campaign features several patient and caregiver testimonials that tell the stories of patients and parents of children with severe asthma.
“What we want people to understand, is that at the time of Ben’s passing, he was on a preventive med. He was going to the doctor routinely. We had actually just been to the asthma doctor. We were seeing somebody, had an action plan, and everybody knew what they had to do. Even with all of that, it still came to this. Benjamin still lost his life, and we never knew this was something that could happen,” stated Cristin Buckley, mother of Benjamin Buckley who was 7 years old at the time of his death. These testimonial videos will be used to raise awareness of the condition, and the importance of managing and monitoring symptoms.
Shared Decision Making Tool

Thank You to Our Supporters
The CHEST Foundation and Allergy and Asthma Network would like to thank our generous supporters, AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, and Novartis for making this campaign possible. It is through supporters, who are active participants in helping grow this campaign, that these important materials are able to have an impact on patient outcomes and create long-lasting social change.
To view the campaign materials, visit us at asthma.chestnet.org.
The Allergy & Asthma NetWork, the nation’s leading patient education and advocacy organization for people with allergy and asthma, has once again joined forces with the CHEST Foundation in an effort to empower patients suffering from severe asthma.
The campaign’s focus is to educate health-care providers, patients, parents of asthmatics, and the public about the most current treatment options for asthma, highlight the importance of referring to specialists to improve patient outcomes, and bring to light the role of the entire health-care team in the care of a patient with severe or difficult-to-control asthma.
Severity Assessment Tool
Available online and in print, the severity assessment tool was designed to help a patient, and the clinician, understand the severity of their asthma. Not only does the tool evaluate the severity of their condition, but it also helps the patient become more aware of their symptoms. The seven-question assessment includes questions on usage of quick-relief or rescue inhalers, visits to the ED/hospital, physical activity, controller medication, and quality of sleep.
Patient and Caregiver Testimonials
The campaign features several patient and caregiver testimonials that tell the stories of patients and parents of children with severe asthma.
“What we want people to understand, is that at the time of Ben’s passing, he was on a preventive med. He was going to the doctor routinely. We had actually just been to the asthma doctor. We were seeing somebody, had an action plan, and everybody knew what they had to do. Even with all of that, it still came to this. Benjamin still lost his life, and we never knew this was something that could happen,” stated Cristin Buckley, mother of Benjamin Buckley who was 7 years old at the time of his death. These testimonial videos will be used to raise awareness of the condition, and the importance of managing and monitoring symptoms.
Shared Decision Making Tool
Thank You to Our Supporters
The CHEST Foundation and Allergy and Asthma Network would like to thank our generous supporters, AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, and Novartis for making this campaign possible. It is through supporters, who are active participants in helping grow this campaign, that these important materials are able to have an impact on patient outcomes and create long-lasting social change.
To view the campaign materials, visit us at asthma.chestnet.org.
The Allergy & Asthma NetWork, the nation’s leading patient education and advocacy organization for people with allergy and asthma, has once again joined forces with the CHEST Foundation in an effort to empower patients suffering from severe asthma.
The campaign’s focus is to educate health-care providers, patients, parents of asthmatics, and the public about the most current treatment options for asthma, highlight the importance of referring to specialists to improve patient outcomes, and bring to light the role of the entire health-care team in the care of a patient with severe or difficult-to-control asthma.
Severity Assessment Tool
Available online and in print, the severity assessment tool was designed to help a patient, and the clinician, understand the severity of their asthma. Not only does the tool evaluate the severity of their condition, but it also helps the patient become more aware of their symptoms. The seven-question assessment includes questions on usage of quick-relief or rescue inhalers, visits to the ED/hospital, physical activity, controller medication, and quality of sleep.
Patient and Caregiver Testimonials
The campaign features several patient and caregiver testimonials that tell the stories of patients and parents of children with severe asthma.
“What we want people to understand, is that at the time of Ben’s passing, he was on a preventive med. He was going to the doctor routinely. We had actually just been to the asthma doctor. We were seeing somebody, had an action plan, and everybody knew what they had to do. Even with all of that, it still came to this. Benjamin still lost his life, and we never knew this was something that could happen,” stated Cristin Buckley, mother of Benjamin Buckley who was 7 years old at the time of his death. These testimonial videos will be used to raise awareness of the condition, and the importance of managing and monitoring symptoms.
Shared Decision Making Tool
Thank You to Our Supporters
The CHEST Foundation and Allergy and Asthma Network would like to thank our generous supporters, AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, and Novartis for making this campaign possible. It is through supporters, who are active participants in helping grow this campaign, that these important materials are able to have an impact on patient outcomes and create long-lasting social change.
To view the campaign materials, visit us at asthma.chestnet.org.
CHEST Joint Congress in Basel, Switzerland
Members of CHEST leadership, faculty, and staff traveled to Basel, Switzerland, in June, to participate in the CHEST Joint Congress, which was co-hosted with the Swiss Respiratory Society, Schweizersche Gesellschaft Fur Pneumologie (SPG). Overall, there were approximately 1,100 total attendees, representing over 40 countries, who enjoyed the scientific program and gained valuable chest medicine knowledge.

The CHEST Joint Congress in Basel represented the second collaborative scientific conference endeavor with a third party, the first being the CHEST Conference held in Amsterdam May 6-9, COPD: Current Excellence and Future Development.
Members of CHEST leadership, faculty, and staff traveled to Basel, Switzerland, in June, to participate in the CHEST Joint Congress, which was co-hosted with the Swiss Respiratory Society, Schweizersche Gesellschaft Fur Pneumologie (SPG). Overall, there were approximately 1,100 total attendees, representing over 40 countries, who enjoyed the scientific program and gained valuable chest medicine knowledge.
The CHEST Joint Congress in Basel represented the second collaborative scientific conference endeavor with a third party, the first being the CHEST Conference held in Amsterdam May 6-9, COPD: Current Excellence and Future Development.
Members of CHEST leadership, faculty, and staff traveled to Basel, Switzerland, in June, to participate in the CHEST Joint Congress, which was co-hosted with the Swiss Respiratory Society, Schweizersche Gesellschaft Fur Pneumologie (SPG). Overall, there were approximately 1,100 total attendees, representing over 40 countries, who enjoyed the scientific program and gained valuable chest medicine knowledge.
The CHEST Joint Congress in Basel represented the second collaborative scientific conference endeavor with a third party, the first being the CHEST Conference held in Amsterdam May 6-9, COPD: Current Excellence and Future Development.
PHM17 session summary: Demonstrating teaching excellence with an educator’s portfolio
NASHVILLE, TENN. – Development of a medical educator’s portfolio is a necessary, but daunting, task for clinician educators when they enter the promotion process, according to an expert panel at Pediatric Hospital Medicine 2017, sponsored by the Society of Hospital Medicine, the American Academy of Pediatrics, and the Academic Pediatric Association.
Session
Promote yourself: Demonstrating teaching excellence with an educator’s portfolio
Presenters
Michael Ryan, MD, MEHP; Ashlie Tseng, MD; Jocelyn Schiller, MD; Rebecca Tenney-Soeiro, MD, MEd; Michele Long, MD; Corki Lehmann, MD, MEd; Amy Fleming, MD; and H. Barrett Fromme, MD, MHPE
Session summary
Development of an educator’s portfolio is a necessary, but daunting, task for clinician educators when they enter the promotion process. Each institution has its own specific requirements for the educator’s portfolio, but there are several general themes that should be considered for inclusion:
1. Develop an educational philosophy. This is a personal statement that frames the rest of the portfolio and describes how this philosophy is used by the educator in his/her approach to education.
2. Teaching. Include teaching activities that are both formal (i.e. lectures) and sessions that encourage more active participation (i.e. small group discussions). This can be accomplished by generating a teaching activities report, which helps to categorize these activities. This will not only demonstrate the volume of teaching experience, but also help to demonstrate the diversity of an educator’s teaching activities. In this section, an educator also should include teaching awards received.
3. Learner evaluations. A qualitative summary of comments will provide a narrative of the educator’s teaching skills. This section also may include summaries of annual reviews of teaching.
4. Curriculum development. Demonstrate the educator’s active engagement in the development of a novel curriculum or the improvement of a pre-existing curriculum and the successful outcomes of those improvements.
5. Mentoring and advising. Generating a list of advisees and highlighting their accomplishments reflects on the ability of the educator to guide and promote success in his/her learners.
6. Educational leadership and administration. This is a description of the past and present leadership roles that the educator has held, including courses or clerkships directed. This should allow the educator the opportunity to provide a narrative description of his/her involvement beyond what is typically stated on the curriculum vitae.
7. Professional development. The educator should develop a list of activities, including formal degree programs, certificate programs, and educational workshops, in which he/she has participated as a learner and have enhanced his/her skills as an educator.
8. Products of educational scholarship. Generate a list of education-related peer-reviewed publications authored, other educational products (such as a syllabus or curriculum) developed, and educational workshops that the educator was invited to give.
For clinician educators interested in developing an educator’s portfolio, there are several resources available, including the Academic Pediatric Association’s website and several MedEdPORTAL publications.

Key takeaways for Pediatric HM
• While each institution has its own specific requirements, there are general themes to consider including in an educator’s portfolio.
• Resources such as the Academic Pediatric Association’s website can help guide an educator in the development of his/her portfolio.
Dr. Player is a pediatric hospitalist at Children’s Hospital of Wisconsin and assistant professor at the Medical College of Wisconsin, Milwaukee.
NASHVILLE, TENN. – Development of a medical educator’s portfolio is a necessary, but daunting, task for clinician educators when they enter the promotion process, according to an expert panel at Pediatric Hospital Medicine 2017, sponsored by the Society of Hospital Medicine, the American Academy of Pediatrics, and the Academic Pediatric Association.
Session
Promote yourself: Demonstrating teaching excellence with an educator’s portfolio
Presenters
Michael Ryan, MD, MEHP; Ashlie Tseng, MD; Jocelyn Schiller, MD; Rebecca Tenney-Soeiro, MD, MEd; Michele Long, MD; Corki Lehmann, MD, MEd; Amy Fleming, MD; and H. Barrett Fromme, MD, MHPE
Session summary
Development of an educator’s portfolio is a necessary, but daunting, task for clinician educators when they enter the promotion process. Each institution has its own specific requirements for the educator’s portfolio, but there are several general themes that should be considered for inclusion:
1. Develop an educational philosophy. This is a personal statement that frames the rest of the portfolio and describes how this philosophy is used by the educator in his/her approach to education.
2. Teaching. Include teaching activities that are both formal (i.e. lectures) and sessions that encourage more active participation (i.e. small group discussions). This can be accomplished by generating a teaching activities report, which helps to categorize these activities. This will not only demonstrate the volume of teaching experience, but also help to demonstrate the diversity of an educator’s teaching activities. In this section, an educator also should include teaching awards received.
3. Learner evaluations. A qualitative summary of comments will provide a narrative of the educator’s teaching skills. This section also may include summaries of annual reviews of teaching.
4. Curriculum development. Demonstrate the educator’s active engagement in the development of a novel curriculum or the improvement of a pre-existing curriculum and the successful outcomes of those improvements.
5. Mentoring and advising. Generating a list of advisees and highlighting their accomplishments reflects on the ability of the educator to guide and promote success in his/her learners.
6. Educational leadership and administration. This is a description of the past and present leadership roles that the educator has held, including courses or clerkships directed. This should allow the educator the opportunity to provide a narrative description of his/her involvement beyond what is typically stated on the curriculum vitae.
7. Professional development. The educator should develop a list of activities, including formal degree programs, certificate programs, and educational workshops, in which he/she has participated as a learner and have enhanced his/her skills as an educator.
8. Products of educational scholarship. Generate a list of education-related peer-reviewed publications authored, other educational products (such as a syllabus or curriculum) developed, and educational workshops that the educator was invited to give.
For clinician educators interested in developing an educator’s portfolio, there are several resources available, including the Academic Pediatric Association’s website and several MedEdPORTAL publications.
Key takeaways for Pediatric HM
• While each institution has its own specific requirements, there are general themes to consider including in an educator’s portfolio.
• Resources such as the Academic Pediatric Association’s website can help guide an educator in the development of his/her portfolio.
Dr. Player is a pediatric hospitalist at Children’s Hospital of Wisconsin and assistant professor at the Medical College of Wisconsin, Milwaukee.
NASHVILLE, TENN. – Development of a medical educator’s portfolio is a necessary, but daunting, task for clinician educators when they enter the promotion process, according to an expert panel at Pediatric Hospital Medicine 2017, sponsored by the Society of Hospital Medicine, the American Academy of Pediatrics, and the Academic Pediatric Association.
Session
Promote yourself: Demonstrating teaching excellence with an educator’s portfolio
Presenters
Michael Ryan, MD, MEHP; Ashlie Tseng, MD; Jocelyn Schiller, MD; Rebecca Tenney-Soeiro, MD, MEd; Michele Long, MD; Corki Lehmann, MD, MEd; Amy Fleming, MD; and H. Barrett Fromme, MD, MHPE
Session summary
Development of an educator’s portfolio is a necessary, but daunting, task for clinician educators when they enter the promotion process. Each institution has its own specific requirements for the educator’s portfolio, but there are several general themes that should be considered for inclusion:
1. Develop an educational philosophy. This is a personal statement that frames the rest of the portfolio and describes how this philosophy is used by the educator in his/her approach to education.
2. Teaching. Include teaching activities that are both formal (i.e. lectures) and sessions that encourage more active participation (i.e. small group discussions). This can be accomplished by generating a teaching activities report, which helps to categorize these activities. This will not only demonstrate the volume of teaching experience, but also help to demonstrate the diversity of an educator’s teaching activities. In this section, an educator also should include teaching awards received.
3. Learner evaluations. A qualitative summary of comments will provide a narrative of the educator’s teaching skills. This section also may include summaries of annual reviews of teaching.
4. Curriculum development. Demonstrate the educator’s active engagement in the development of a novel curriculum or the improvement of a pre-existing curriculum and the successful outcomes of those improvements.
5. Mentoring and advising. Generating a list of advisees and highlighting their accomplishments reflects on the ability of the educator to guide and promote success in his/her learners.
6. Educational leadership and administration. This is a description of the past and present leadership roles that the educator has held, including courses or clerkships directed. This should allow the educator the opportunity to provide a narrative description of his/her involvement beyond what is typically stated on the curriculum vitae.
7. Professional development. The educator should develop a list of activities, including formal degree programs, certificate programs, and educational workshops, in which he/she has participated as a learner and have enhanced his/her skills as an educator.
8. Products of educational scholarship. Generate a list of education-related peer-reviewed publications authored, other educational products (such as a syllabus or curriculum) developed, and educational workshops that the educator was invited to give.
For clinician educators interested in developing an educator’s portfolio, there are several resources available, including the Academic Pediatric Association’s website and several MedEdPORTAL publications.
Key takeaways for Pediatric HM
• While each institution has its own specific requirements, there are general themes to consider including in an educator’s portfolio.
• Resources such as the Academic Pediatric Association’s website can help guide an educator in the development of his/her portfolio.
Dr. Player is a pediatric hospitalist at Children’s Hospital of Wisconsin and assistant professor at the Medical College of Wisconsin, Milwaukee.
At PHM 2017
Study aims to validate AAD criteria for diagnosing AD and create usable form
CHICAGO – A streamlined set of diagnostic criteria from the American Academy of Dermatology’s most recent consensus criteria for diagnosing atopic dermatitis (AD) produced a specificity of more than 95%, and was also highly sensitive, a prospective analysis found.
“Atopic dermatitis typically presents in childhood and is associated with a worsened quality of life, with severe itch and lack of sleep, and substantial health care costs due to therapeutic management and increased hospitalizations,” study author Jeremy Udkoff said at the World Congress of Pediatric Dermatology. “We also know that in order to treat the disease and to learn more about it, we have to have a good tool for diagnosing it. When it comes to clinical studies and research, we require a systematic and refined set of criteria.”

The next set of commonly used criteria to appear were created by the U.K. working party, for which researchers used logistic regression to systematically create a minimum set of effective criteria for AD (Br J Dermatol. 1994;131[3]:383-96). For these guidelines, meeting a diagnosis of AD requires an itchy skin condition, followed by three or more of the following: a history of flexural involvement; a personal history of asthma or hay fever; a history of general dry skin in the last year; visible flexural eczema, and onset under the age of 2 years. “A subsequent validation trial found [the U.K. working party criteria] to have a low sensitivity, which as you can imagine, could be a very large problem,” he said (Arch Dermatol. 1999;135[5]:514-6).
In 2001, the AAD consensus conference created revised hierarchical criteria known as the AAD consensus criteria (J Am Acad Dermatol. 2003;49[6]:1088-95). “These were initially created for more of a gestalt-type picture of AD in the clinic, but because it flows so well, it’s currently being used in about one-third of clinical trials,” Mr. Udkoff said. “However, [the AAD criteria] have not been validated, so we didn’t know its sensitivity or specificity. In addition, we didn’t have a ‘checkbox’ form that tells us how many of each of the criteria are required to make the diagnosis. We didn’t know how many ‘essential,’ ‘important,’ or ‘associated’ features we need to make this diagnosis.”
For the current study, he and his associates set out to determine how many “essential,” “important,” and “associated” criteria are necessary to make the AAD consensus criteria work. They also set out to create a usable checkbox form, validate the criteria, and compare it to the Hanifin-Rajka (HR) and U.K. criteria. To accomplish this, they created a questionnaire comprised of HR, U.K., and AAD criteria, examined the criteria on 60 subjects with and without AD, and compared the diagnostic features of each of those criteria against a gold standard dermatology diagnosis from one of seven pediatric dermatologists. Next, they ranked all 56 possible AAD criterion combinations based on their overall sensitivity and specificity, and chose the most predictive combination. “Once we had the optimal set of criteria, we validated it on a new cohort to determine its sensitivity and specificity, and compared it with the classic HR and U.K. criteria,” Mr. Udkoff explained.
Overall, the researchers evaluated findings from 100 subjects: 58 with AD, and 42 controls. Those with AD were about 3 years younger, compared with controls (a mean age of 5 years vs. about 8 years, respectively). About 40% of patients were Hispanic and about 30% were white. Mr. Udkoff and his associates confirmed the hierarchical structure of the AAD criteria and found that individual “essential” AAD criteria of pruritus, typical AD pattern, and chronic/relapsing course each had a sensitivity that exceeded 96%. This was followed by the “important” criteria of early age of onset, atopy, and xerosis, which had a sensitivity that ranged between 88% and 95%, while the associated criteria had a sensitivity that ranged between 50% and 85%.
Next, the researchers systematically tested all combinations of the AAD criteria and found that three “essential” AAD criteria, two or more of the “important” criteria, and one or more of the “associated” criteria were optimal in diagnosing AD. Mr. Udkoff noted that the findings can be translated into a simple “3-2-1 rule” that “is both practical and pragmatic,” he said. Using this rule, sensitivity was 91.4% and specificity was 95.2%.
Currently, the researchers are working to validate this criteria in different subgroups of patients. To date, they have found that children younger than 1.5 years get one bonus “essential” criteria for being an infant, so for that population a 2-2-1 rule would apply.
Mr. Udkoff reported that the research was supported by a training grant from the National Institutes of Health. He reported having no financial disclosures.
CHICAGO – A streamlined set of diagnostic criteria from the American Academy of Dermatology’s most recent consensus criteria for diagnosing atopic dermatitis (AD) produced a specificity of more than 95%, and was also highly sensitive, a prospective analysis found.
“Atopic dermatitis typically presents in childhood and is associated with a worsened quality of life, with severe itch and lack of sleep, and substantial health care costs due to therapeutic management and increased hospitalizations,” study author Jeremy Udkoff said at the World Congress of Pediatric Dermatology. “We also know that in order to treat the disease and to learn more about it, we have to have a good tool for diagnosing it. When it comes to clinical studies and research, we require a systematic and refined set of criteria.”
The next set of commonly used criteria to appear were created by the U.K. working party, for which researchers used logistic regression to systematically create a minimum set of effective criteria for AD (Br J Dermatol. 1994;131[3]:383-96). For these guidelines, meeting a diagnosis of AD requires an itchy skin condition, followed by three or more of the following: a history of flexural involvement; a personal history of asthma or hay fever; a history of general dry skin in the last year; visible flexural eczema, and onset under the age of 2 years. “A subsequent validation trial found [the U.K. working party criteria] to have a low sensitivity, which as you can imagine, could be a very large problem,” he said (Arch Dermatol. 1999;135[5]:514-6).
In 2001, the AAD consensus conference created revised hierarchical criteria known as the AAD consensus criteria (J Am Acad Dermatol. 2003;49[6]:1088-95). “These were initially created for more of a gestalt-type picture of AD in the clinic, but because it flows so well, it’s currently being used in about one-third of clinical trials,” Mr. Udkoff said. “However, [the AAD criteria] have not been validated, so we didn’t know its sensitivity or specificity. In addition, we didn’t have a ‘checkbox’ form that tells us how many of each of the criteria are required to make the diagnosis. We didn’t know how many ‘essential,’ ‘important,’ or ‘associated’ features we need to make this diagnosis.”
For the current study, he and his associates set out to determine how many “essential,” “important,” and “associated” criteria are necessary to make the AAD consensus criteria work. They also set out to create a usable checkbox form, validate the criteria, and compare it to the Hanifin-Rajka (HR) and U.K. criteria. To accomplish this, they created a questionnaire comprised of HR, U.K., and AAD criteria, examined the criteria on 60 subjects with and without AD, and compared the diagnostic features of each of those criteria against a gold standard dermatology diagnosis from one of seven pediatric dermatologists. Next, they ranked all 56 possible AAD criterion combinations based on their overall sensitivity and specificity, and chose the most predictive combination. “Once we had the optimal set of criteria, we validated it on a new cohort to determine its sensitivity and specificity, and compared it with the classic HR and U.K. criteria,” Mr. Udkoff explained.
Overall, the researchers evaluated findings from 100 subjects: 58 with AD, and 42 controls. Those with AD were about 3 years younger, compared with controls (a mean age of 5 years vs. about 8 years, respectively). About 40% of patients were Hispanic and about 30% were white. Mr. Udkoff and his associates confirmed the hierarchical structure of the AAD criteria and found that individual “essential” AAD criteria of pruritus, typical AD pattern, and chronic/relapsing course each had a sensitivity that exceeded 96%. This was followed by the “important” criteria of early age of onset, atopy, and xerosis, which had a sensitivity that ranged between 88% and 95%, while the associated criteria had a sensitivity that ranged between 50% and 85%.
Next, the researchers systematically tested all combinations of the AAD criteria and found that three “essential” AAD criteria, two or more of the “important” criteria, and one or more of the “associated” criteria were optimal in diagnosing AD. Mr. Udkoff noted that the findings can be translated into a simple “3-2-1 rule” that “is both practical and pragmatic,” he said. Using this rule, sensitivity was 91.4% and specificity was 95.2%.
Currently, the researchers are working to validate this criteria in different subgroups of patients. To date, they have found that children younger than 1.5 years get one bonus “essential” criteria for being an infant, so for that population a 2-2-1 rule would apply.
Mr. Udkoff reported that the research was supported by a training grant from the National Institutes of Health. He reported having no financial disclosures.
CHICAGO – A streamlined set of diagnostic criteria from the American Academy of Dermatology’s most recent consensus criteria for diagnosing atopic dermatitis (AD) produced a specificity of more than 95%, and was also highly sensitive, a prospective analysis found.
“Atopic dermatitis typically presents in childhood and is associated with a worsened quality of life, with severe itch and lack of sleep, and substantial health care costs due to therapeutic management and increased hospitalizations,” study author Jeremy Udkoff said at the World Congress of Pediatric Dermatology. “We also know that in order to treat the disease and to learn more about it, we have to have a good tool for diagnosing it. When it comes to clinical studies and research, we require a systematic and refined set of criteria.”
The next set of commonly used criteria to appear were created by the U.K. working party, for which researchers used logistic regression to systematically create a minimum set of effective criteria for AD (Br J Dermatol. 1994;131[3]:383-96). For these guidelines, meeting a diagnosis of AD requires an itchy skin condition, followed by three or more of the following: a history of flexural involvement; a personal history of asthma or hay fever; a history of general dry skin in the last year; visible flexural eczema, and onset under the age of 2 years. “A subsequent validation trial found [the U.K. working party criteria] to have a low sensitivity, which as you can imagine, could be a very large problem,” he said (Arch Dermatol. 1999;135[5]:514-6).
In 2001, the AAD consensus conference created revised hierarchical criteria known as the AAD consensus criteria (J Am Acad Dermatol. 2003;49[6]:1088-95). “These were initially created for more of a gestalt-type picture of AD in the clinic, but because it flows so well, it’s currently being used in about one-third of clinical trials,” Mr. Udkoff said. “However, [the AAD criteria] have not been validated, so we didn’t know its sensitivity or specificity. In addition, we didn’t have a ‘checkbox’ form that tells us how many of each of the criteria are required to make the diagnosis. We didn’t know how many ‘essential,’ ‘important,’ or ‘associated’ features we need to make this diagnosis.”
For the current study, he and his associates set out to determine how many “essential,” “important,” and “associated” criteria are necessary to make the AAD consensus criteria work. They also set out to create a usable checkbox form, validate the criteria, and compare it to the Hanifin-Rajka (HR) and U.K. criteria. To accomplish this, they created a questionnaire comprised of HR, U.K., and AAD criteria, examined the criteria on 60 subjects with and without AD, and compared the diagnostic features of each of those criteria against a gold standard dermatology diagnosis from one of seven pediatric dermatologists. Next, they ranked all 56 possible AAD criterion combinations based on their overall sensitivity and specificity, and chose the most predictive combination. “Once we had the optimal set of criteria, we validated it on a new cohort to determine its sensitivity and specificity, and compared it with the classic HR and U.K. criteria,” Mr. Udkoff explained.
Overall, the researchers evaluated findings from 100 subjects: 58 with AD, and 42 controls. Those with AD were about 3 years younger, compared with controls (a mean age of 5 years vs. about 8 years, respectively). About 40% of patients were Hispanic and about 30% were white. Mr. Udkoff and his associates confirmed the hierarchical structure of the AAD criteria and found that individual “essential” AAD criteria of pruritus, typical AD pattern, and chronic/relapsing course each had a sensitivity that exceeded 96%. This was followed by the “important” criteria of early age of onset, atopy, and xerosis, which had a sensitivity that ranged between 88% and 95%, while the associated criteria had a sensitivity that ranged between 50% and 85%.
Next, the researchers systematically tested all combinations of the AAD criteria and found that three “essential” AAD criteria, two or more of the “important” criteria, and one or more of the “associated” criteria were optimal in diagnosing AD. Mr. Udkoff noted that the findings can be translated into a simple “3-2-1 rule” that “is both practical and pragmatic,” he said. Using this rule, sensitivity was 91.4% and specificity was 95.2%.
Currently, the researchers are working to validate this criteria in different subgroups of patients. To date, they have found that children younger than 1.5 years get one bonus “essential” criteria for being an infant, so for that population a 2-2-1 rule would apply.
Mr. Udkoff reported that the research was supported by a training grant from the National Institutes of Health. He reported having no financial disclosures.
AT WCPD 2017
Key clinical point:
Major finding: The “important” AD criteria of early age of onset, atopy, and xerosis had a sensitivity that ranged between 88% and 95%.
Data source: An analysis of optimal AAD criteria for AD that included 58 patients with AD and 42 controls.
Disclosures: Mr. Udkoff reported that the research was supported by a training grant from the National Institutes of Health. He reported having no financial disclosures.
ATO enables anthracycline reduction in pediatric APL

Consolidation therapy that includes arsenic trioxide (ATO) can decrease anthracycline dosing by about 40% in children and young adults with acute promyelocytic leukemia (APL), according to new research.
And it can accomplish this without compromising survival in standard-risk patients.
Outcomes for high-risk patients compared favorably to other pediatric APL trials, the research indicated.
Investigators compared ATO consolidation in the AAML0631 trial to the historic control trial AIDA0493 and reported the results in the Journal of Clinical Oncology.
The AAML0631 phase 3 trial, conducted by the Children’s Oncology Group, compared newly diagnosed pediatric APL patients receiving ATO consolidation to the benchmark of event-free survival (EFS) in standard-risk (SR) patients established by the AIDA0493 trial.
AIDA0493 enrolled patients between January 1993 and June 2000. The protocol involved treatment with all-trans retinoic acid (ATRA), anthracyclines, and high-dose cytarabine. The trial resulted in overall survival (OS) of approximately 90%.
AAML0631
AAML063 investigators defined SR as a white blood cell count (WBC) at presentation less than 10,000 cells/μL. They defined high risk (HR) as a WBC count of 10,000 cells/μL or more.
AAML0631 patients had to be at least 2 years old and younger than 22, and their de novo APL had to be confirmed by PML-RARα polymerase chain reaction.
The patients could have had no prior leukemia treatment, except for steroids, hydroxyurea, or leukapheresis.
AAML0631 did not exclude patients based on organ function or performance status. AIDA0493, however, excluded patients with performance status of 4 or liver function tests greater than 3 times the upper limit of normal.
Patients were excluded from AAML0631 if they had preexisting prolonged QT syndrome because of the risk of QT interval prolongation with ATO.
AAML0631 treatment protocol
All patients received ATRA during induction, each consolidation course, and maintenance.
Induction therapy consisted of ATRA and idarubicin.
All patients received 2 cycles of ATO during the first consolidation. SR patients received an additional 2 consolidation courses, and HR patients received 3 consolidation courses that included high-dose cytarabine and anthracycline.
Maintenance therapy consisted of ATRA, oral methotrexate, and 6-mercaptopurine for 2 years.
Patients also received prophylactic treatment with intrathecal cytarabine.
Patient demographics
Investigators enrolled 108 patients between March 2009 and November 2012, of which 101 (66 SR and 35 HR) were evaluable.
Patients were a median age of 15.04 years (range, 2.01 – 21.34), 56% were female, 80% were white, 10% black, 2% Native American, 3% Asian, and 5% unknown.
Three quarters of the patients had an ECOG score of 0 or 1, median WBC counts of 3.8 x 1000 cells/uL (range, 0.4 – 173.8), and median platelet counts of 21.5 x 1000/uL (range, 3 – 198).
Almost two-thirds of patients (63%) had the classic translocation (15;17), and 37% had an additional 1 or more cytogenetic abnormalities.
The SR patients in AAML0631 had similar characteristics to the patients in AIDA0493 except for the distribution of performance status scores and differences in racial/ethnic diversity.
Efficacy
After a median follow-up of 3.73 years (range, 0.003 – 5.97), the 3-year overall survival (OS) was 94% ± 5% and the 3-year EFS was 91% ± 6%.
For SR patients, the OS was 98% ± 3% and the EFS 95% ± 5%.
For HR patients, the OS was 86% ± 12% and the EFS was 83% ± 13%.
SR patients had a 2-year EFS of 97%. This compared with 91% for patients in the AIDA0493 trial, which means that therapy with ATO was not inferior to therapy in the historic comparator trial (P=0.93).
And these results were achieved with a cumulative anthracycline dosing of idarubicin at 51 mg/m2 (SR) and 61 mg/m2 (HR) and mitoxantrone at 20 mg/m2.
This compared with the AIDA0493 cumulative anthracycline dosing of 80 mg/m2 of idarubicin and 50 mg/m2 of mitoxantrone.
The cumulative daunorubicin equivalent in the AAML0631 trial was 335 mg/m2 (SR) and 385 mg/m2 (HR) compared with 600 mg/m2 in the AIDA 0493 trial.
Toxicity
The percentage of patients with adverse events varied according to treatment cycle and was highest during induction and high-dose cytarabine-containing courses.
The most common adverse events were fever/neutropenia and infection.
Differentiation syndrome occurred in 20% of patients during induction, 31% in HR patients and 13% in SR patients. ATRA was held for 15 of these patients during induction. It was subsequently re-started at a lower dose and increased to the full dose.
QTc interval prolongation of grade 1 or 2 occurred in 16% (n=15) and 12% (n=11) during the ATO cycles.
One patient experienced grade 3 QTc interval prolongation during ATO consolidation. There were no grade 4 or 5 events for this toxicity.
One event of grade 1 ventricular arrhythmia and 1 event of grade 1 left ventricular systolic dysfunction occurred during ATO consolidation.
Two off-therapy cardiac events have been reported: a grade 1 QTc interval prolongation and a grade 2 ventricular arrhythmia.
No cardiac deaths have occurred, and liver toxicity was minimal during ATO cycles.
The investigators believe the favorable results of this study provide a new benchmark for outcomes in pediatric APL.
The Children’s Oncology Group is currently accruing pediatric APL patients to further investigate similar treatment approaches. ![]()
Consolidation therapy that includes arsenic trioxide (ATO) can decrease anthracycline dosing by about 40% in children and young adults with acute promyelocytic leukemia (APL), according to new research.
And it can accomplish this without compromising survival in standard-risk patients.
Outcomes for high-risk patients compared favorably to other pediatric APL trials, the research indicated.
Investigators compared ATO consolidation in the AAML0631 trial to the historic control trial AIDA0493 and reported the results in the Journal of Clinical Oncology.
The AAML0631 phase 3 trial, conducted by the Children’s Oncology Group, compared newly diagnosed pediatric APL patients receiving ATO consolidation to the benchmark of event-free survival (EFS) in standard-risk (SR) patients established by the AIDA0493 trial.
AIDA0493 enrolled patients between January 1993 and June 2000. The protocol involved treatment with all-trans retinoic acid (ATRA), anthracyclines, and high-dose cytarabine. The trial resulted in overall survival (OS) of approximately 90%.
AAML0631
AAML063 investigators defined SR as a white blood cell count (WBC) at presentation less than 10,000 cells/μL. They defined high risk (HR) as a WBC count of 10,000 cells/μL or more.
AAML0631 patients had to be at least 2 years old and younger than 22, and their de novo APL had to be confirmed by PML-RARα polymerase chain reaction.
The patients could have had no prior leukemia treatment, except for steroids, hydroxyurea, or leukapheresis.
AAML0631 did not exclude patients based on organ function or performance status. AIDA0493, however, excluded patients with performance status of 4 or liver function tests greater than 3 times the upper limit of normal.
Patients were excluded from AAML0631 if they had preexisting prolonged QT syndrome because of the risk of QT interval prolongation with ATO.
AAML0631 treatment protocol
All patients received ATRA during induction, each consolidation course, and maintenance.
Induction therapy consisted of ATRA and idarubicin.
All patients received 2 cycles of ATO during the first consolidation. SR patients received an additional 2 consolidation courses, and HR patients received 3 consolidation courses that included high-dose cytarabine and anthracycline.
Maintenance therapy consisted of ATRA, oral methotrexate, and 6-mercaptopurine for 2 years.
Patients also received prophylactic treatment with intrathecal cytarabine.
Patient demographics
Investigators enrolled 108 patients between March 2009 and November 2012, of which 101 (66 SR and 35 HR) were evaluable.
Patients were a median age of 15.04 years (range, 2.01 – 21.34), 56% were female, 80% were white, 10% black, 2% Native American, 3% Asian, and 5% unknown.
Three quarters of the patients had an ECOG score of 0 or 1, median WBC counts of 3.8 x 1000 cells/uL (range, 0.4 – 173.8), and median platelet counts of 21.5 x 1000/uL (range, 3 – 198).
Almost two-thirds of patients (63%) had the classic translocation (15;17), and 37% had an additional 1 or more cytogenetic abnormalities.
The SR patients in AAML0631 had similar characteristics to the patients in AIDA0493 except for the distribution of performance status scores and differences in racial/ethnic diversity.
Efficacy
After a median follow-up of 3.73 years (range, 0.003 – 5.97), the 3-year overall survival (OS) was 94% ± 5% and the 3-year EFS was 91% ± 6%.
For SR patients, the OS was 98% ± 3% and the EFS 95% ± 5%.
For HR patients, the OS was 86% ± 12% and the EFS was 83% ± 13%.
SR patients had a 2-year EFS of 97%. This compared with 91% for patients in the AIDA0493 trial, which means that therapy with ATO was not inferior to therapy in the historic comparator trial (P=0.93).
And these results were achieved with a cumulative anthracycline dosing of idarubicin at 51 mg/m2 (SR) and 61 mg/m2 (HR) and mitoxantrone at 20 mg/m2.
This compared with the AIDA0493 cumulative anthracycline dosing of 80 mg/m2 of idarubicin and 50 mg/m2 of mitoxantrone.
The cumulative daunorubicin equivalent in the AAML0631 trial was 335 mg/m2 (SR) and 385 mg/m2 (HR) compared with 600 mg/m2 in the AIDA 0493 trial.
Toxicity
The percentage of patients with adverse events varied according to treatment cycle and was highest during induction and high-dose cytarabine-containing courses.
The most common adverse events were fever/neutropenia and infection.
Differentiation syndrome occurred in 20% of patients during induction, 31% in HR patients and 13% in SR patients. ATRA was held for 15 of these patients during induction. It was subsequently re-started at a lower dose and increased to the full dose.
QTc interval prolongation of grade 1 or 2 occurred in 16% (n=15) and 12% (n=11) during the ATO cycles.
One patient experienced grade 3 QTc interval prolongation during ATO consolidation. There were no grade 4 or 5 events for this toxicity.
One event of grade 1 ventricular arrhythmia and 1 event of grade 1 left ventricular systolic dysfunction occurred during ATO consolidation.
Two off-therapy cardiac events have been reported: a grade 1 QTc interval prolongation and a grade 2 ventricular arrhythmia.
No cardiac deaths have occurred, and liver toxicity was minimal during ATO cycles.
The investigators believe the favorable results of this study provide a new benchmark for outcomes in pediatric APL.
The Children’s Oncology Group is currently accruing pediatric APL patients to further investigate similar treatment approaches. ![]()
Consolidation therapy that includes arsenic trioxide (ATO) can decrease anthracycline dosing by about 40% in children and young adults with acute promyelocytic leukemia (APL), according to new research.
And it can accomplish this without compromising survival in standard-risk patients.
Outcomes for high-risk patients compared favorably to other pediatric APL trials, the research indicated.
Investigators compared ATO consolidation in the AAML0631 trial to the historic control trial AIDA0493 and reported the results in the Journal of Clinical Oncology.
The AAML0631 phase 3 trial, conducted by the Children’s Oncology Group, compared newly diagnosed pediatric APL patients receiving ATO consolidation to the benchmark of event-free survival (EFS) in standard-risk (SR) patients established by the AIDA0493 trial.
AIDA0493 enrolled patients between January 1993 and June 2000. The protocol involved treatment with all-trans retinoic acid (ATRA), anthracyclines, and high-dose cytarabine. The trial resulted in overall survival (OS) of approximately 90%.
AAML0631
AAML063 investigators defined SR as a white blood cell count (WBC) at presentation less than 10,000 cells/μL. They defined high risk (HR) as a WBC count of 10,000 cells/μL or more.
AAML0631 patients had to be at least 2 years old and younger than 22, and their de novo APL had to be confirmed by PML-RARα polymerase chain reaction.
The patients could have had no prior leukemia treatment, except for steroids, hydroxyurea, or leukapheresis.
AAML0631 did not exclude patients based on organ function or performance status. AIDA0493, however, excluded patients with performance status of 4 or liver function tests greater than 3 times the upper limit of normal.
Patients were excluded from AAML0631 if they had preexisting prolonged QT syndrome because of the risk of QT interval prolongation with ATO.
AAML0631 treatment protocol
All patients received ATRA during induction, each consolidation course, and maintenance.
Induction therapy consisted of ATRA and idarubicin.
All patients received 2 cycles of ATO during the first consolidation. SR patients received an additional 2 consolidation courses, and HR patients received 3 consolidation courses that included high-dose cytarabine and anthracycline.
Maintenance therapy consisted of ATRA, oral methotrexate, and 6-mercaptopurine for 2 years.
Patients also received prophylactic treatment with intrathecal cytarabine.
Patient demographics
Investigators enrolled 108 patients between March 2009 and November 2012, of which 101 (66 SR and 35 HR) were evaluable.
Patients were a median age of 15.04 years (range, 2.01 – 21.34), 56% were female, 80% were white, 10% black, 2% Native American, 3% Asian, and 5% unknown.
Three quarters of the patients had an ECOG score of 0 or 1, median WBC counts of 3.8 x 1000 cells/uL (range, 0.4 – 173.8), and median platelet counts of 21.5 x 1000/uL (range, 3 – 198).
Almost two-thirds of patients (63%) had the classic translocation (15;17), and 37% had an additional 1 or more cytogenetic abnormalities.
The SR patients in AAML0631 had similar characteristics to the patients in AIDA0493 except for the distribution of performance status scores and differences in racial/ethnic diversity.
Efficacy
After a median follow-up of 3.73 years (range, 0.003 – 5.97), the 3-year overall survival (OS) was 94% ± 5% and the 3-year EFS was 91% ± 6%.
For SR patients, the OS was 98% ± 3% and the EFS 95% ± 5%.
For HR patients, the OS was 86% ± 12% and the EFS was 83% ± 13%.
SR patients had a 2-year EFS of 97%. This compared with 91% for patients in the AIDA0493 trial, which means that therapy with ATO was not inferior to therapy in the historic comparator trial (P=0.93).
And these results were achieved with a cumulative anthracycline dosing of idarubicin at 51 mg/m2 (SR) and 61 mg/m2 (HR) and mitoxantrone at 20 mg/m2.
This compared with the AIDA0493 cumulative anthracycline dosing of 80 mg/m2 of idarubicin and 50 mg/m2 of mitoxantrone.
The cumulative daunorubicin equivalent in the AAML0631 trial was 335 mg/m2 (SR) and 385 mg/m2 (HR) compared with 600 mg/m2 in the AIDA 0493 trial.
Toxicity
The percentage of patients with adverse events varied according to treatment cycle and was highest during induction and high-dose cytarabine-containing courses.
The most common adverse events were fever/neutropenia and infection.
Differentiation syndrome occurred in 20% of patients during induction, 31% in HR patients and 13% in SR patients. ATRA was held for 15 of these patients during induction. It was subsequently re-started at a lower dose and increased to the full dose.
QTc interval prolongation of grade 1 or 2 occurred in 16% (n=15) and 12% (n=11) during the ATO cycles.
One patient experienced grade 3 QTc interval prolongation during ATO consolidation. There were no grade 4 or 5 events for this toxicity.
One event of grade 1 ventricular arrhythmia and 1 event of grade 1 left ventricular systolic dysfunction occurred during ATO consolidation.
Two off-therapy cardiac events have been reported: a grade 1 QTc interval prolongation and a grade 2 ventricular arrhythmia.
No cardiac deaths have occurred, and liver toxicity was minimal during ATO cycles.
The investigators believe the favorable results of this study provide a new benchmark for outcomes in pediatric APL.
The Children’s Oncology Group is currently accruing pediatric APL patients to further investigate similar treatment approaches. ![]()
Incontinentia Pigmenti: Do You Know the Signs?
IN THIS ARTICLE
- Presenting stages
- Diagnostic criteria
- Management of IP
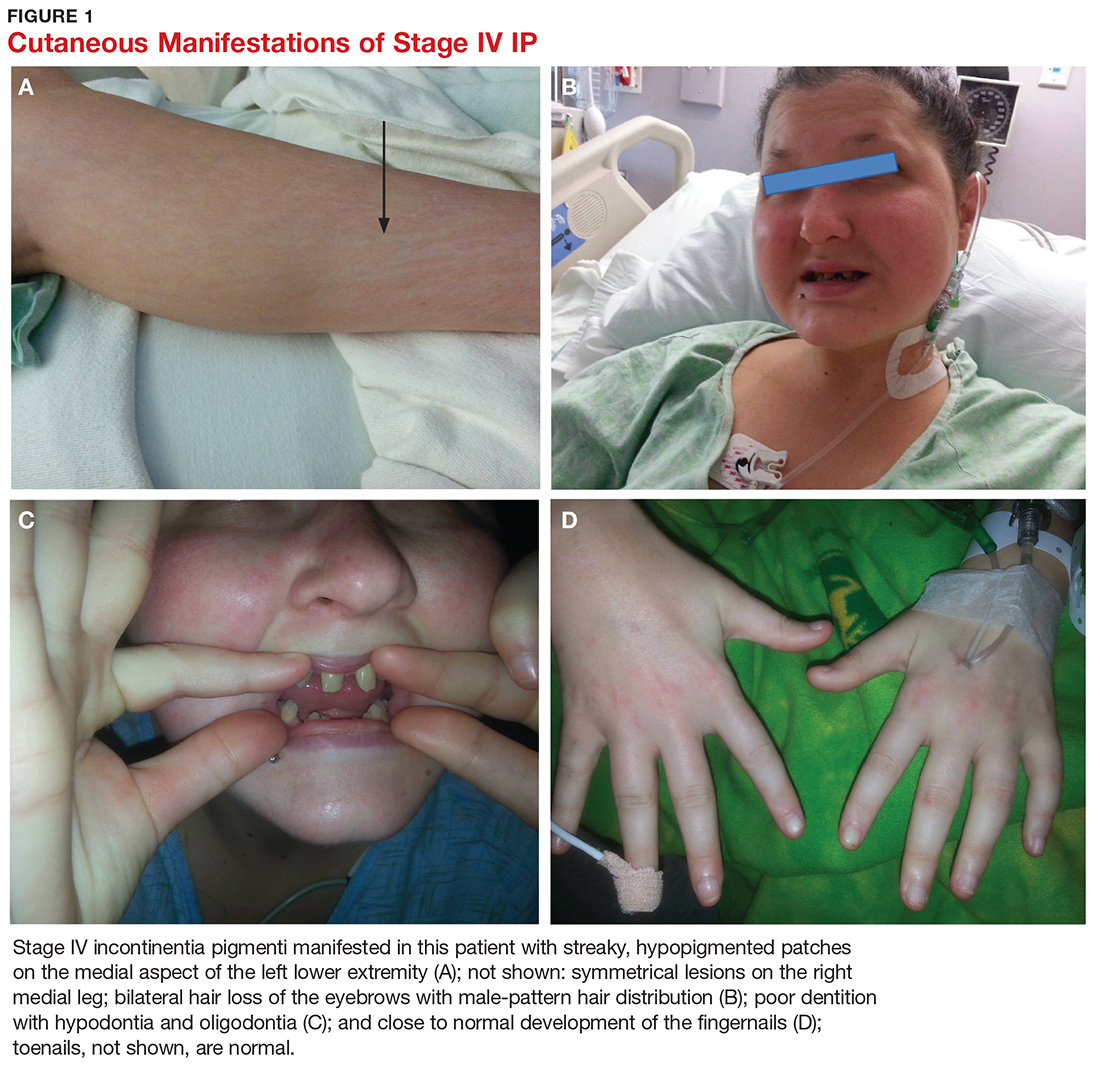
A 21-year-old woman with type 1 diabetes is admitted for recurrent diabetic ketoacidosis. Physical exam reveals hypopigmented, linear, streaky patches on the medial aspects of the bilateral lower legs (Figure 1A). The patient denies tenderness, pruritus, or paresthesia. There is obvious symmetrical hair loss on the lateral aspects of the eyebrows, as well as slightly wooly male-pattern hair distribution with patchy alopecia on the vertex of the head (Figure 1B). She has very poor dentition with hypodontia and malformed teeth (Figure 1C). Her fingernails and toenails appear normal, with no visible atrophy (Figure 1D). What explains her condition?
Incontinentia pigmenti (IP), also known as Bloch-Sulzberger syndrome, is a rare, X-linked dominant genodermatosis involving the cutaneous, ophthalmic, neurologic, and dental systems.1-3 It results from X-inactivation due to mutations in the NF-kappaB essential modulator (NEMO) gene with deletion of exons 4-10 in most cases. The NEMO gene encodes a regulatory component of the IkappaB kinase complex required to activate the NF-kappa B pathway, which is important for many immune, inflammatory, and apoptotic processes.4-6 This deletional mutation is typically lethal in normal 46,XY male karyotypes. Male fetuses with this mutation usually die in utero, making the reported cases predominantly female.4,7
The estimated incidence of IP is between 1/10,000 and 1/100,000.4 Due to the rarity of the condition, IP may be underrecognized and underdiagnosed.
CLINICAL PRESENTATION
Characteristic skin lesions of IP begin to develop at birth or in utero, in an evolving pattern that consists of four stages:
- The vesicular stage (stage I) is characterized by linear erythematous papules and blisters that manifest in newborns.
- The verrucous stage (stage II) begins as the blisters start to heal—usually after several weeks—and is distinguished by hyperkeratotic warty papules in linear or swirling distribution. This stage resolves on its own within months.
- The hyperpigmentation stage (stage III) is when swirling macules or patches develop. This hallmark stage of IP tends to remain static until adolescence.
- The hypopigmentation stage (stage IV) manifests with faded streaky patches, which may be subtly atrophic. This final stage usually develops in the second or third decade of life.2,3
All these cutaneous lesions follow Blaschko lines—invisible lines believed to result from embryonic cell migration that become visible with the manifestation of cutaneous or mucous lesions.6
Other associated cutaneous findings include patchy alopecia, nail dystrophy, and oral/dental anomalies such as hypodontia, oligodontia, and tooth deformities. In addition, ophthalmologic involvement can result in strabismus, cataracts, and retinal vascular changes that can lead to blindness. Central nervous system manifestations include seizures, cognitive impairment, and spastic paralysis.3
DIFFERENTIAL DIAGNOSIS
Because IP is uncommon, it may be easily overlooked or misdiagnosed as another, similar cutaneous manifestation. Cutaneous sarcoidosis, for example, is a skin lesion of noncaseating granuloma. It can present as patches, papules, ulcers, scars, ichthyosis, and alopecia. The development of cutaneous sarcoidosis can be idiopathic or iatrogenic, particularly in patients using anti-TNF therapy. The diagnosis is made clinically and can be confirmed pathologically.8
Stage I IP can also be confused with neonatal herpes simplex virus-1 (HSV-1) infection, given the similarities in vesicular morphology and linear distribution. The diagnosis of HSV-1 can be made based on history, physical exam, and pathology. Given the serious sequelae of neonatal HSV-1 infection, antiviral therapy should not be delayed until confirmation of the diagnosis in infants with vesicular eruptions.9
Erythema multiforme (EM) is another dermatologic condition frequently encountered in children and young adults. Its characteristic round target lesion usually has two rings surrounding the dusky-appearing central zone. Atypical lesions can be bullous or crusty, mimicking the appearance of stage I or II IP. EM is usually a self-limiting condition, but specific treatment may be required if the infectious agent is identified.10
Vitiligo, the development of white patches due to the loss of melanocytes, is another item in the differential. Although it most commonly involves the skin, the hair may also be affected. The diagnosis is made clinically and can be confirmed with skin biopsy if needed.11
DIAGNOSIS
Diagnostic criteria for IP have been proposed, with family history playing a role (see Table).2,3,12 Results of a case-study series indicate that 28% of patients with IP have a family history involving at least one first-degree female relative. IP was considered “sporadic” in 62% of cases studied.3

Without a family history of IP, at least one major criterion must be present to support the diagnosis. These include
- Neonatal rash (erythema, vesicles)
- Linear, atrophic, hairless lesions
- Hyperpigmentation (mainly on trunk, following Blaschko lines)
In a patient with a family history of IP, the presence of any major criterion strongly supports the diagnosis. These, as well as minor criteria, are outlined in the Table.2,3,12
In stages I and II of IP, pathologic features include spongiotic dermatitis with characteristic eosinophils and large dyskeratotic cells.3,13 In stage IV, skin biopsies may reveal slight atrophy and scattered apoptotic cells in the epidermis and epidermal hypopigmentation due to reduced melanocytes. The dermis typically appears thickened and is absent hair follicles and sweat glands.14 In a 2014 update, these pathologic features were proposed to be included in the major diagnostic criteria.12
TREATMENT/MANAGEMENT
Treatment of IP is centered on the involved organ systems. For cutaneous lesions, treatment is not usually necessary unless inflammation persists. In such cases, topical steroids or tacrolimus have been used with some success.15,16 In the vesicular stage, the patient should be monitored for bacterial infection, with appropriate prevention or treatment as necessary.
With other involved systems—such as dental, ophthalmologic, or neurologic (eg, seizures or other encephalopathy) anomalies—consultation and follow-up with the relevant specialist is warranted.
In this case, the patient denied family history of IP. She did have a history of infantile cataract and seizure. Her presenting signs were typical of stage IV IP: hypopigmented streaky patches on the skin of the lower legs, dental abnormalities, somewhat wooly hair, alopecia on the head, and loss of hair on the lateral aspects of the eyebrows. The uniqueness of this case is that the patient also had type 1 diabetes, a condition with a strong genetic predisposition. However, there is no evidence supporting an association between IP and either type of diabetes.
CONCLUSION
Although rare, when IP does occur, its manifestations are vast and severe enough to significantly reduce quality of life for patients; when it occurs in males, it is usually lethal. This genetic disorder can affect multiple body systems, making knowledge of its symptoms essential for proper diagnosis. Because its characteristic stages may be present at birth or in infancy, early identification and diagnosis of IP can help guide treatment intervention.
1. Roberts AP. Incontinentia pigmenti (Bloch-Sulzberger). Br Med. J. 1958;1(5079):1106-1107.
2. Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-Sulzberger syndrome). J Med Genet. 1993;30(1):53-59.
3. Hadj-Rabia S, Froidevaux N, Bodak D, et al. Clinical study of 40 cases of incontinentia pigmenti. Arch Dermatol. 2003; 139(9):1163-1170.
4. Smahi A, Courtois G, Vabres P, et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. Nature. 2000;405(6785):466-472.
5. Aradhya S, Courtois G, Rajkovic A, et al. Atypical forms of incontinentia pigmenti in male individuals result from mutations of a cytosine tract in exon 10 of NEMO (IKK-gamma). Am J Hum Genet. 2001;68(3):765-771.
6. Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89(1):26-36.
7. Kenwrick S, Woffendin H, Jakins T, et al. Survival of male patients with incontinentia pigmenti carrying a lethal mutation can be explained by somatic mosaicism or Klinefelter Syndrome. Am J Hum Genet. 2001;69(6):1210-1217.
8. Katta R. Cutaneous sarcoidosis: a dermatologic masquerader. Am Fam Physician. 2002;65(8):1581-1584.
9. Faloyin M, Levitt J, Bercowitz E, et al. All that is vesicular is not herpes: incontinentia pigmenti masquerading as herpes simplex virus in a newborn. Pediatrics. 2004;114(2):e270-272.
10. Siedner-Weintraub Y, Gross I, David A, et al. Paediatric erythema multiforme: epidemiological, clinical and laboratory characteristics. Acta Derm Venereol. 2016 Nov 10. doi: 10.2340/00015555-2569.
11. Gawkrodger DJ, Ormerod AD, Shaw L, et al. Guideline for the diagnosis and management of vitiligo. Br J Dermatol. 2008;159(5):1051-1076.
12. Minic´ S, Trpinac D, Obradovic´ M. Incontinentia pigmenti diagnostic criteria update. Clin Genet. 2014;85(6):536-542.
13. Jean-Baptiste S, O’Toole EA, Chen M, et al. Expression of eotaxin, an eosinophil-selective chemokine, parallels eosinophil accumulation in the vesiculobullous stage of incontinentia pigmenti. Clin Exp Immunol. 2002;127(3):470-478.
14. Hadj-Rabia S, Rimella A, Smahi A, et al. Clinical and histologic features of incontinentia pigmenti in adults with nuclear factor-κ B essential modulator gene mutations. J Am Acad Dermatol. 2011;64(3):508-515.
15. Kaya TI, Tursen U, Ikizoglu G. Therapeutic use of topical corticosteroids in the vesiculobullous lesions of incontinentia pigmenti. Clin Exp Dermatol. 2009;34(8):e611-613.
16. Jessup CJ, Morgan SC, Cohen LM, Viders DE. Incontinentia pigmenti: treatment of IP with topical tacrolimus. J Drugs Dermatol. 2009;8(10):944-946.
IN THIS ARTICLE
- Presenting stages
- Diagnostic criteria
- Management of IP
A 21-year-old woman with type 1 diabetes is admitted for recurrent diabetic ketoacidosis. Physical exam reveals hypopigmented, linear, streaky patches on the medial aspects of the bilateral lower legs (Figure 1A). The patient denies tenderness, pruritus, or paresthesia. There is obvious symmetrical hair loss on the lateral aspects of the eyebrows, as well as slightly wooly male-pattern hair distribution with patchy alopecia on the vertex of the head (Figure 1B). She has very poor dentition with hypodontia and malformed teeth (Figure 1C). Her fingernails and toenails appear normal, with no visible atrophy (Figure 1D). What explains her condition?
Incontinentia pigmenti (IP), also known as Bloch-Sulzberger syndrome, is a rare, X-linked dominant genodermatosis involving the cutaneous, ophthalmic, neurologic, and dental systems.1-3 It results from X-inactivation due to mutations in the NF-kappaB essential modulator (NEMO) gene with deletion of exons 4-10 in most cases. The NEMO gene encodes a regulatory component of the IkappaB kinase complex required to activate the NF-kappa B pathway, which is important for many immune, inflammatory, and apoptotic processes.4-6 This deletional mutation is typically lethal in normal 46,XY male karyotypes. Male fetuses with this mutation usually die in utero, making the reported cases predominantly female.4,7
The estimated incidence of IP is between 1/10,000 and 1/100,000.4 Due to the rarity of the condition, IP may be underrecognized and underdiagnosed.
CLINICAL PRESENTATION
Characteristic skin lesions of IP begin to develop at birth or in utero, in an evolving pattern that consists of four stages:
- The vesicular stage (stage I) is characterized by linear erythematous papules and blisters that manifest in newborns.
- The verrucous stage (stage II) begins as the blisters start to heal—usually after several weeks—and is distinguished by hyperkeratotic warty papules in linear or swirling distribution. This stage resolves on its own within months.
- The hyperpigmentation stage (stage III) is when swirling macules or patches develop. This hallmark stage of IP tends to remain static until adolescence.
- The hypopigmentation stage (stage IV) manifests with faded streaky patches, which may be subtly atrophic. This final stage usually develops in the second or third decade of life.2,3
All these cutaneous lesions follow Blaschko lines—invisible lines believed to result from embryonic cell migration that become visible with the manifestation of cutaneous or mucous lesions.6
Other associated cutaneous findings include patchy alopecia, nail dystrophy, and oral/dental anomalies such as hypodontia, oligodontia, and tooth deformities. In addition, ophthalmologic involvement can result in strabismus, cataracts, and retinal vascular changes that can lead to blindness. Central nervous system manifestations include seizures, cognitive impairment, and spastic paralysis.3
DIFFERENTIAL DIAGNOSIS
Because IP is uncommon, it may be easily overlooked or misdiagnosed as another, similar cutaneous manifestation. Cutaneous sarcoidosis, for example, is a skin lesion of noncaseating granuloma. It can present as patches, papules, ulcers, scars, ichthyosis, and alopecia. The development of cutaneous sarcoidosis can be idiopathic or iatrogenic, particularly in patients using anti-TNF therapy. The diagnosis is made clinically and can be confirmed pathologically.8
Stage I IP can also be confused with neonatal herpes simplex virus-1 (HSV-1) infection, given the similarities in vesicular morphology and linear distribution. The diagnosis of HSV-1 can be made based on history, physical exam, and pathology. Given the serious sequelae of neonatal HSV-1 infection, antiviral therapy should not be delayed until confirmation of the diagnosis in infants with vesicular eruptions.9
Erythema multiforme (EM) is another dermatologic condition frequently encountered in children and young adults. Its characteristic round target lesion usually has two rings surrounding the dusky-appearing central zone. Atypical lesions can be bullous or crusty, mimicking the appearance of stage I or II IP. EM is usually a self-limiting condition, but specific treatment may be required if the infectious agent is identified.10
Vitiligo, the development of white patches due to the loss of melanocytes, is another item in the differential. Although it most commonly involves the skin, the hair may also be affected. The diagnosis is made clinically and can be confirmed with skin biopsy if needed.11
DIAGNOSIS
Diagnostic criteria for IP have been proposed, with family history playing a role (see Table).2,3,12 Results of a case-study series indicate that 28% of patients with IP have a family history involving at least one first-degree female relative. IP was considered “sporadic” in 62% of cases studied.3
Without a family history of IP, at least one major criterion must be present to support the diagnosis. These include
- Neonatal rash (erythema, vesicles)
- Linear, atrophic, hairless lesions
- Hyperpigmentation (mainly on trunk, following Blaschko lines)
In a patient with a family history of IP, the presence of any major criterion strongly supports the diagnosis. These, as well as minor criteria, are outlined in the Table.2,3,12
In stages I and II of IP, pathologic features include spongiotic dermatitis with characteristic eosinophils and large dyskeratotic cells.3,13 In stage IV, skin biopsies may reveal slight atrophy and scattered apoptotic cells in the epidermis and epidermal hypopigmentation due to reduced melanocytes. The dermis typically appears thickened and is absent hair follicles and sweat glands.14 In a 2014 update, these pathologic features were proposed to be included in the major diagnostic criteria.12
TREATMENT/MANAGEMENT
Treatment of IP is centered on the involved organ systems. For cutaneous lesions, treatment is not usually necessary unless inflammation persists. In such cases, topical steroids or tacrolimus have been used with some success.15,16 In the vesicular stage, the patient should be monitored for bacterial infection, with appropriate prevention or treatment as necessary.
With other involved systems—such as dental, ophthalmologic, or neurologic (eg, seizures or other encephalopathy) anomalies—consultation and follow-up with the relevant specialist is warranted.
In this case, the patient denied family history of IP. She did have a history of infantile cataract and seizure. Her presenting signs were typical of stage IV IP: hypopigmented streaky patches on the skin of the lower legs, dental abnormalities, somewhat wooly hair, alopecia on the head, and loss of hair on the lateral aspects of the eyebrows. The uniqueness of this case is that the patient also had type 1 diabetes, a condition with a strong genetic predisposition. However, there is no evidence supporting an association between IP and either type of diabetes.
CONCLUSION
Although rare, when IP does occur, its manifestations are vast and severe enough to significantly reduce quality of life for patients; when it occurs in males, it is usually lethal. This genetic disorder can affect multiple body systems, making knowledge of its symptoms essential for proper diagnosis. Because its characteristic stages may be present at birth or in infancy, early identification and diagnosis of IP can help guide treatment intervention.
IN THIS ARTICLE
- Presenting stages
- Diagnostic criteria
- Management of IP
A 21-year-old woman with type 1 diabetes is admitted for recurrent diabetic ketoacidosis. Physical exam reveals hypopigmented, linear, streaky patches on the medial aspects of the bilateral lower legs (Figure 1A). The patient denies tenderness, pruritus, or paresthesia. There is obvious symmetrical hair loss on the lateral aspects of the eyebrows, as well as slightly wooly male-pattern hair distribution with patchy alopecia on the vertex of the head (Figure 1B). She has very poor dentition with hypodontia and malformed teeth (Figure 1C). Her fingernails and toenails appear normal, with no visible atrophy (Figure 1D). What explains her condition?
Incontinentia pigmenti (IP), also known as Bloch-Sulzberger syndrome, is a rare, X-linked dominant genodermatosis involving the cutaneous, ophthalmic, neurologic, and dental systems.1-3 It results from X-inactivation due to mutations in the NF-kappaB essential modulator (NEMO) gene with deletion of exons 4-10 in most cases. The NEMO gene encodes a regulatory component of the IkappaB kinase complex required to activate the NF-kappa B pathway, which is important for many immune, inflammatory, and apoptotic processes.4-6 This deletional mutation is typically lethal in normal 46,XY male karyotypes. Male fetuses with this mutation usually die in utero, making the reported cases predominantly female.4,7
The estimated incidence of IP is between 1/10,000 and 1/100,000.4 Due to the rarity of the condition, IP may be underrecognized and underdiagnosed.
CLINICAL PRESENTATION
Characteristic skin lesions of IP begin to develop at birth or in utero, in an evolving pattern that consists of four stages:
- The vesicular stage (stage I) is characterized by linear erythematous papules and blisters that manifest in newborns.
- The verrucous stage (stage II) begins as the blisters start to heal—usually after several weeks—and is distinguished by hyperkeratotic warty papules in linear or swirling distribution. This stage resolves on its own within months.
- The hyperpigmentation stage (stage III) is when swirling macules or patches develop. This hallmark stage of IP tends to remain static until adolescence.
- The hypopigmentation stage (stage IV) manifests with faded streaky patches, which may be subtly atrophic. This final stage usually develops in the second or third decade of life.2,3
All these cutaneous lesions follow Blaschko lines—invisible lines believed to result from embryonic cell migration that become visible with the manifestation of cutaneous or mucous lesions.6
Other associated cutaneous findings include patchy alopecia, nail dystrophy, and oral/dental anomalies such as hypodontia, oligodontia, and tooth deformities. In addition, ophthalmologic involvement can result in strabismus, cataracts, and retinal vascular changes that can lead to blindness. Central nervous system manifestations include seizures, cognitive impairment, and spastic paralysis.3
DIFFERENTIAL DIAGNOSIS
Because IP is uncommon, it may be easily overlooked or misdiagnosed as another, similar cutaneous manifestation. Cutaneous sarcoidosis, for example, is a skin lesion of noncaseating granuloma. It can present as patches, papules, ulcers, scars, ichthyosis, and alopecia. The development of cutaneous sarcoidosis can be idiopathic or iatrogenic, particularly in patients using anti-TNF therapy. The diagnosis is made clinically and can be confirmed pathologically.8
Stage I IP can also be confused with neonatal herpes simplex virus-1 (HSV-1) infection, given the similarities in vesicular morphology and linear distribution. The diagnosis of HSV-1 can be made based on history, physical exam, and pathology. Given the serious sequelae of neonatal HSV-1 infection, antiviral therapy should not be delayed until confirmation of the diagnosis in infants with vesicular eruptions.9
Erythema multiforme (EM) is another dermatologic condition frequently encountered in children and young adults. Its characteristic round target lesion usually has two rings surrounding the dusky-appearing central zone. Atypical lesions can be bullous or crusty, mimicking the appearance of stage I or II IP. EM is usually a self-limiting condition, but specific treatment may be required if the infectious agent is identified.10
Vitiligo, the development of white patches due to the loss of melanocytes, is another item in the differential. Although it most commonly involves the skin, the hair may also be affected. The diagnosis is made clinically and can be confirmed with skin biopsy if needed.11
DIAGNOSIS
Diagnostic criteria for IP have been proposed, with family history playing a role (see Table).2,3,12 Results of a case-study series indicate that 28% of patients with IP have a family history involving at least one first-degree female relative. IP was considered “sporadic” in 62% of cases studied.3
Without a family history of IP, at least one major criterion must be present to support the diagnosis. These include
- Neonatal rash (erythema, vesicles)
- Linear, atrophic, hairless lesions
- Hyperpigmentation (mainly on trunk, following Blaschko lines)
In a patient with a family history of IP, the presence of any major criterion strongly supports the diagnosis. These, as well as minor criteria, are outlined in the Table.2,3,12
In stages I and II of IP, pathologic features include spongiotic dermatitis with characteristic eosinophils and large dyskeratotic cells.3,13 In stage IV, skin biopsies may reveal slight atrophy and scattered apoptotic cells in the epidermis and epidermal hypopigmentation due to reduced melanocytes. The dermis typically appears thickened and is absent hair follicles and sweat glands.14 In a 2014 update, these pathologic features were proposed to be included in the major diagnostic criteria.12
TREATMENT/MANAGEMENT
Treatment of IP is centered on the involved organ systems. For cutaneous lesions, treatment is not usually necessary unless inflammation persists. In such cases, topical steroids or tacrolimus have been used with some success.15,16 In the vesicular stage, the patient should be monitored for bacterial infection, with appropriate prevention or treatment as necessary.
With other involved systems—such as dental, ophthalmologic, or neurologic (eg, seizures or other encephalopathy) anomalies—consultation and follow-up with the relevant specialist is warranted.
In this case, the patient denied family history of IP. She did have a history of infantile cataract and seizure. Her presenting signs were typical of stage IV IP: hypopigmented streaky patches on the skin of the lower legs, dental abnormalities, somewhat wooly hair, alopecia on the head, and loss of hair on the lateral aspects of the eyebrows. The uniqueness of this case is that the patient also had type 1 diabetes, a condition with a strong genetic predisposition. However, there is no evidence supporting an association between IP and either type of diabetes.
CONCLUSION
Although rare, when IP does occur, its manifestations are vast and severe enough to significantly reduce quality of life for patients; when it occurs in males, it is usually lethal. This genetic disorder can affect multiple body systems, making knowledge of its symptoms essential for proper diagnosis. Because its characteristic stages may be present at birth or in infancy, early identification and diagnosis of IP can help guide treatment intervention.
1. Roberts AP. Incontinentia pigmenti (Bloch-Sulzberger). Br Med. J. 1958;1(5079):1106-1107.
2. Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-Sulzberger syndrome). J Med Genet. 1993;30(1):53-59.
3. Hadj-Rabia S, Froidevaux N, Bodak D, et al. Clinical study of 40 cases of incontinentia pigmenti. Arch Dermatol. 2003; 139(9):1163-1170.
4. Smahi A, Courtois G, Vabres P, et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. Nature. 2000;405(6785):466-472.
5. Aradhya S, Courtois G, Rajkovic A, et al. Atypical forms of incontinentia pigmenti in male individuals result from mutations of a cytosine tract in exon 10 of NEMO (IKK-gamma). Am J Hum Genet. 2001;68(3):765-771.
6. Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89(1):26-36.
7. Kenwrick S, Woffendin H, Jakins T, et al. Survival of male patients with incontinentia pigmenti carrying a lethal mutation can be explained by somatic mosaicism or Klinefelter Syndrome. Am J Hum Genet. 2001;69(6):1210-1217.
8. Katta R. Cutaneous sarcoidosis: a dermatologic masquerader. Am Fam Physician. 2002;65(8):1581-1584.
9. Faloyin M, Levitt J, Bercowitz E, et al. All that is vesicular is not herpes: incontinentia pigmenti masquerading as herpes simplex virus in a newborn. Pediatrics. 2004;114(2):e270-272.
10. Siedner-Weintraub Y, Gross I, David A, et al. Paediatric erythema multiforme: epidemiological, clinical and laboratory characteristics. Acta Derm Venereol. 2016 Nov 10. doi: 10.2340/00015555-2569.
11. Gawkrodger DJ, Ormerod AD, Shaw L, et al. Guideline for the diagnosis and management of vitiligo. Br J Dermatol. 2008;159(5):1051-1076.
12. Minic´ S, Trpinac D, Obradovic´ M. Incontinentia pigmenti diagnostic criteria update. Clin Genet. 2014;85(6):536-542.
13. Jean-Baptiste S, O’Toole EA, Chen M, et al. Expression of eotaxin, an eosinophil-selective chemokine, parallels eosinophil accumulation in the vesiculobullous stage of incontinentia pigmenti. Clin Exp Immunol. 2002;127(3):470-478.
14. Hadj-Rabia S, Rimella A, Smahi A, et al. Clinical and histologic features of incontinentia pigmenti in adults with nuclear factor-κ B essential modulator gene mutations. J Am Acad Dermatol. 2011;64(3):508-515.
15. Kaya TI, Tursen U, Ikizoglu G. Therapeutic use of topical corticosteroids in the vesiculobullous lesions of incontinentia pigmenti. Clin Exp Dermatol. 2009;34(8):e611-613.
16. Jessup CJ, Morgan SC, Cohen LM, Viders DE. Incontinentia pigmenti: treatment of IP with topical tacrolimus. J Drugs Dermatol. 2009;8(10):944-946.
1. Roberts AP. Incontinentia pigmenti (Bloch-Sulzberger). Br Med. J. 1958;1(5079):1106-1107.
2. Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-Sulzberger syndrome). J Med Genet. 1993;30(1):53-59.
3. Hadj-Rabia S, Froidevaux N, Bodak D, et al. Clinical study of 40 cases of incontinentia pigmenti. Arch Dermatol. 2003; 139(9):1163-1170.
4. Smahi A, Courtois G, Vabres P, et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. Nature. 2000;405(6785):466-472.
5. Aradhya S, Courtois G, Rajkovic A, et al. Atypical forms of incontinentia pigmenti in male individuals result from mutations of a cytosine tract in exon 10 of NEMO (IKK-gamma). Am J Hum Genet. 2001;68(3):765-771.
6. Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89(1):26-36.
7. Kenwrick S, Woffendin H, Jakins T, et al. Survival of male patients with incontinentia pigmenti carrying a lethal mutation can be explained by somatic mosaicism or Klinefelter Syndrome. Am J Hum Genet. 2001;69(6):1210-1217.
8. Katta R. Cutaneous sarcoidosis: a dermatologic masquerader. Am Fam Physician. 2002;65(8):1581-1584.
9. Faloyin M, Levitt J, Bercowitz E, et al. All that is vesicular is not herpes: incontinentia pigmenti masquerading as herpes simplex virus in a newborn. Pediatrics. 2004;114(2):e270-272.
10. Siedner-Weintraub Y, Gross I, David A, et al. Paediatric erythema multiforme: epidemiological, clinical and laboratory characteristics. Acta Derm Venereol. 2016 Nov 10. doi: 10.2340/00015555-2569.
11. Gawkrodger DJ, Ormerod AD, Shaw L, et al. Guideline for the diagnosis and management of vitiligo. Br J Dermatol. 2008;159(5):1051-1076.
12. Minic´ S, Trpinac D, Obradovic´ M. Incontinentia pigmenti diagnostic criteria update. Clin Genet. 2014;85(6):536-542.
13. Jean-Baptiste S, O’Toole EA, Chen M, et al. Expression of eotaxin, an eosinophil-selective chemokine, parallels eosinophil accumulation in the vesiculobullous stage of incontinentia pigmenti. Clin Exp Immunol. 2002;127(3):470-478.
14. Hadj-Rabia S, Rimella A, Smahi A, et al. Clinical and histologic features of incontinentia pigmenti in adults with nuclear factor-κ B essential modulator gene mutations. J Am Acad Dermatol. 2011;64(3):508-515.
15. Kaya TI, Tursen U, Ikizoglu G. Therapeutic use of topical corticosteroids in the vesiculobullous lesions of incontinentia pigmenti. Clin Exp Dermatol. 2009;34(8):e611-613.
16. Jessup CJ, Morgan SC, Cohen LM, Viders DE. Incontinentia pigmenti: treatment of IP with topical tacrolimus. J Drugs Dermatol. 2009;8(10):944-946.
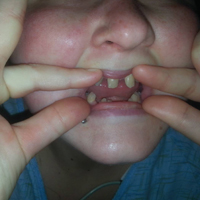
Program reduces transfusions in leukemia, HSCT patients

New research suggests a patient blood management (PBM) program can safely reduce transfusion use in patients with acute leukemia and those undergoing hematopoietic stem cell transplant (HSCT).
The program significantly reduced the use of red blood cell (RBC) and platelet transfusions without increasing morbidity or mortality in patients who were receiving intensive chemotherapy to treat acute leukemia and in patients receiving an allogeneic or autologous HSCT.
“There has been a long-standing belief among hematologists that patients with leukemia undergoing chemotherapy should have a transfusion of red blood cells if their hemoglobin level drops below about 9 g/dL to help avoid adverse outcomes,” said study author Michael Leahy, MB ChB, a consultant hematologist at the University of Western Australia in Perth.
“Findings in this real-world, non-clinical trial setting challenge that belief.”
Dr Leahy and his colleagues published their findings in Transfusion.
The researchers said the PBM program used in this study was built around the “3 pillars” concept of PBM, which are:
- Optimize the patient’s RBC mass
- Minimize blood loss
- Harness and optimize the patient’s physiologic anemia reserve.
No specific transfusion thresholds were established. However, the hospitals did adopt a single-unit RBC transfusion policy for symptomatic anemic patients who were not actively bleeding.
Results
The study included 695 admissions to 2 major hospitals in Western Australia. Patients were admitted between July 2010 and December 2014 for treatment of acute leukemia or for autologous or allogeneic HSCT.
During this time, the patients received 3384 RBC units and 3639 units of platelets.
The mean number of platelet units transfused per hospital admission decreased 35% from baseline to the end of the study period, from 6.3 to 4.1 units (P<0.001).
The mean number of RBC units transfused decreased 39%, from 6.1 to 3.7 (P<0.001). Meanwhile, the use of single-unit RBC transfusions increased from 39% to 67% (P<0.001).
And the mean hemoglobin level prior to RBC transfusion decreased from 8.0 g/dL to 6.8 g/dL (P<0.001).
“This study suggests that patients undergoing chemotherapy with hematological disease may tolerate much lower levels of hemoglobin than previously thought,” said Shannon Farmer, an adjunct research fellow at the University of Western Australia.
“The transfusion threshold, the hemoglobin value at which a transfusion is given, dropped significantly from 8.0 g/dL at the beginning of the study to 6.8 g/dL at the end. This was associated with significant reductions in transfusion and substantial costs savings without evidence of harm to the patients. In fact, it was associated with a trend toward improved survival.”
The reduction in blood products over the study period resulted in a cost savings of AU$694,886 (US$654,007)—AU$389,537 (US$364,177) for RBCs and AU$305,349 (US$289,830) for platelets.
There were no significant changes over the study period in length of hospital stay, serious bleeding events, or in-hospital mortality.
There was a non-significant reduction in the mean length of hospital stay, from 24.5 days to 22.6 days (P=0.338). The difference was still not significant after the researchers adjusted for patient age, patient group, and comorbidities (incident rate ratio=0.88; 95% CI, 0.75-1.04).
The rate of serious bleeding increased over the study period, from 5.3% to 7.0% (P=0.582). After adjustment, the odds ratio was 1.14 (95% CI, 0.38-3.44; P=0.811).
There was a non-significant decrease in in-hospital mortality, from 5.3% to 2.0% (P=0.218). After adjustment, the odds ratio was 0.31 (95% CI, 0.06-1.56; P=0.154).
Based on these results, the researchers concluded that PBM programs could have a substantial impact in this patient population, reducing blood utilization and healthcare costs. ![]()
New research suggests a patient blood management (PBM) program can safely reduce transfusion use in patients with acute leukemia and those undergoing hematopoietic stem cell transplant (HSCT).
The program significantly reduced the use of red blood cell (RBC) and platelet transfusions without increasing morbidity or mortality in patients who were receiving intensive chemotherapy to treat acute leukemia and in patients receiving an allogeneic or autologous HSCT.
“There has been a long-standing belief among hematologists that patients with leukemia undergoing chemotherapy should have a transfusion of red blood cells if their hemoglobin level drops below about 9 g/dL to help avoid adverse outcomes,” said study author Michael Leahy, MB ChB, a consultant hematologist at the University of Western Australia in Perth.
“Findings in this real-world, non-clinical trial setting challenge that belief.”
Dr Leahy and his colleagues published their findings in Transfusion.
The researchers said the PBM program used in this study was built around the “3 pillars” concept of PBM, which are:
- Optimize the patient’s RBC mass
- Minimize blood loss
- Harness and optimize the patient’s physiologic anemia reserve.
No specific transfusion thresholds were established. However, the hospitals did adopt a single-unit RBC transfusion policy for symptomatic anemic patients who were not actively bleeding.
Results
The study included 695 admissions to 2 major hospitals in Western Australia. Patients were admitted between July 2010 and December 2014 for treatment of acute leukemia or for autologous or allogeneic HSCT.
During this time, the patients received 3384 RBC units and 3639 units of platelets.
The mean number of platelet units transfused per hospital admission decreased 35% from baseline to the end of the study period, from 6.3 to 4.1 units (P<0.001).
The mean number of RBC units transfused decreased 39%, from 6.1 to 3.7 (P<0.001). Meanwhile, the use of single-unit RBC transfusions increased from 39% to 67% (P<0.001).
And the mean hemoglobin level prior to RBC transfusion decreased from 8.0 g/dL to 6.8 g/dL (P<0.001).
“This study suggests that patients undergoing chemotherapy with hematological disease may tolerate much lower levels of hemoglobin than previously thought,” said Shannon Farmer, an adjunct research fellow at the University of Western Australia.
“The transfusion threshold, the hemoglobin value at which a transfusion is given, dropped significantly from 8.0 g/dL at the beginning of the study to 6.8 g/dL at the end. This was associated with significant reductions in transfusion and substantial costs savings without evidence of harm to the patients. In fact, it was associated with a trend toward improved survival.”
The reduction in blood products over the study period resulted in a cost savings of AU$694,886 (US$654,007)—AU$389,537 (US$364,177) for RBCs and AU$305,349 (US$289,830) for platelets.
There were no significant changes over the study period in length of hospital stay, serious bleeding events, or in-hospital mortality.
There was a non-significant reduction in the mean length of hospital stay, from 24.5 days to 22.6 days (P=0.338). The difference was still not significant after the researchers adjusted for patient age, patient group, and comorbidities (incident rate ratio=0.88; 95% CI, 0.75-1.04).
The rate of serious bleeding increased over the study period, from 5.3% to 7.0% (P=0.582). After adjustment, the odds ratio was 1.14 (95% CI, 0.38-3.44; P=0.811).
There was a non-significant decrease in in-hospital mortality, from 5.3% to 2.0% (P=0.218). After adjustment, the odds ratio was 0.31 (95% CI, 0.06-1.56; P=0.154).
Based on these results, the researchers concluded that PBM programs could have a substantial impact in this patient population, reducing blood utilization and healthcare costs. ![]()
New research suggests a patient blood management (PBM) program can safely reduce transfusion use in patients with acute leukemia and those undergoing hematopoietic stem cell transplant (HSCT).
The program significantly reduced the use of red blood cell (RBC) and platelet transfusions without increasing morbidity or mortality in patients who were receiving intensive chemotherapy to treat acute leukemia and in patients receiving an allogeneic or autologous HSCT.
“There has been a long-standing belief among hematologists that patients with leukemia undergoing chemotherapy should have a transfusion of red blood cells if their hemoglobin level drops below about 9 g/dL to help avoid adverse outcomes,” said study author Michael Leahy, MB ChB, a consultant hematologist at the University of Western Australia in Perth.
“Findings in this real-world, non-clinical trial setting challenge that belief.”
Dr Leahy and his colleagues published their findings in Transfusion.
The researchers said the PBM program used in this study was built around the “3 pillars” concept of PBM, which are:
- Optimize the patient’s RBC mass
- Minimize blood loss
- Harness and optimize the patient’s physiologic anemia reserve.
No specific transfusion thresholds were established. However, the hospitals did adopt a single-unit RBC transfusion policy for symptomatic anemic patients who were not actively bleeding.
Results
The study included 695 admissions to 2 major hospitals in Western Australia. Patients were admitted between July 2010 and December 2014 for treatment of acute leukemia or for autologous or allogeneic HSCT.
During this time, the patients received 3384 RBC units and 3639 units of platelets.
The mean number of platelet units transfused per hospital admission decreased 35% from baseline to the end of the study period, from 6.3 to 4.1 units (P<0.001).
The mean number of RBC units transfused decreased 39%, from 6.1 to 3.7 (P<0.001). Meanwhile, the use of single-unit RBC transfusions increased from 39% to 67% (P<0.001).
And the mean hemoglobin level prior to RBC transfusion decreased from 8.0 g/dL to 6.8 g/dL (P<0.001).
“This study suggests that patients undergoing chemotherapy with hematological disease may tolerate much lower levels of hemoglobin than previously thought,” said Shannon Farmer, an adjunct research fellow at the University of Western Australia.
“The transfusion threshold, the hemoglobin value at which a transfusion is given, dropped significantly from 8.0 g/dL at the beginning of the study to 6.8 g/dL at the end. This was associated with significant reductions in transfusion and substantial costs savings without evidence of harm to the patients. In fact, it was associated with a trend toward improved survival.”
The reduction in blood products over the study period resulted in a cost savings of AU$694,886 (US$654,007)—AU$389,537 (US$364,177) for RBCs and AU$305,349 (US$289,830) for platelets.
There were no significant changes over the study period in length of hospital stay, serious bleeding events, or in-hospital mortality.
There was a non-significant reduction in the mean length of hospital stay, from 24.5 days to 22.6 days (P=0.338). The difference was still not significant after the researchers adjusted for patient age, patient group, and comorbidities (incident rate ratio=0.88; 95% CI, 0.75-1.04).
The rate of serious bleeding increased over the study period, from 5.3% to 7.0% (P=0.582). After adjustment, the odds ratio was 1.14 (95% CI, 0.38-3.44; P=0.811).
There was a non-significant decrease in in-hospital mortality, from 5.3% to 2.0% (P=0.218). After adjustment, the odds ratio was 0.31 (95% CI, 0.06-1.56; P=0.154).
Based on these results, the researchers concluded that PBM programs could have a substantial impact in this patient population, reducing blood utilization and healthcare costs. ![]()