User login
South African child has suppressed HIV without ART since infancy
A 9-year-old South African child treated with antiretroviral therapy (ART) for HIV infection for 40 weeks as an infant has been living with an undetectable level of the virus ever since without ART, researchers reported on July 24.
“To our knowledge, this is the first reported case of sustained control of HIV in a child enrolled in a randomized trial of ART interruption following treatment early in infancy,” said Avy Violari, MD, at the International AIDS Society Conference on HIV Pathogenesis and Treatment in Paris.
Starting with a high viral load at 9 weeks of age, the child’s treatment brought the viral load to undetectable levels and was halted after 40 weeks. Follow-up examinations and blood sampling over the next 8.5 years showed the child in good health, with only a small reservoir of virus in a tiny portion of immune cells, but a completely undetectable viral load by standard assays. The child’s immune system is healthy, and there are no symptoms of HIV infection.
“Further study is needed to learn how to induce long-term HIV remission in infected babies,” Anthony S. Fauci, MD, director of NIAID, said in an NIH news release about the case. “However, this new case strengthens our hope that by treating HIV-infected children for a brief period beginning in infancy, we may be able to spare them the burden of lifelong therapy and the health consequences of long-term immune activation typically associated with HIV disease.”
A 9-year-old South African child treated with antiretroviral therapy (ART) for HIV infection for 40 weeks as an infant has been living with an undetectable level of the virus ever since without ART, researchers reported on July 24.
“To our knowledge, this is the first reported case of sustained control of HIV in a child enrolled in a randomized trial of ART interruption following treatment early in infancy,” said Avy Violari, MD, at the International AIDS Society Conference on HIV Pathogenesis and Treatment in Paris.
Starting with a high viral load at 9 weeks of age, the child’s treatment brought the viral load to undetectable levels and was halted after 40 weeks. Follow-up examinations and blood sampling over the next 8.5 years showed the child in good health, with only a small reservoir of virus in a tiny portion of immune cells, but a completely undetectable viral load by standard assays. The child’s immune system is healthy, and there are no symptoms of HIV infection.
“Further study is needed to learn how to induce long-term HIV remission in infected babies,” Anthony S. Fauci, MD, director of NIAID, said in an NIH news release about the case. “However, this new case strengthens our hope that by treating HIV-infected children for a brief period beginning in infancy, we may be able to spare them the burden of lifelong therapy and the health consequences of long-term immune activation typically associated with HIV disease.”
A 9-year-old South African child treated with antiretroviral therapy (ART) for HIV infection for 40 weeks as an infant has been living with an undetectable level of the virus ever since without ART, researchers reported on July 24.
“To our knowledge, this is the first reported case of sustained control of HIV in a child enrolled in a randomized trial of ART interruption following treatment early in infancy,” said Avy Violari, MD, at the International AIDS Society Conference on HIV Pathogenesis and Treatment in Paris.
Starting with a high viral load at 9 weeks of age, the child’s treatment brought the viral load to undetectable levels and was halted after 40 weeks. Follow-up examinations and blood sampling over the next 8.5 years showed the child in good health, with only a small reservoir of virus in a tiny portion of immune cells, but a completely undetectable viral load by standard assays. The child’s immune system is healthy, and there are no symptoms of HIV infection.
“Further study is needed to learn how to induce long-term HIV remission in infected babies,” Anthony S. Fauci, MD, director of NIAID, said in an NIH news release about the case. “However, this new case strengthens our hope that by treating HIV-infected children for a brief period beginning in infancy, we may be able to spare them the burden of lifelong therapy and the health consequences of long-term immune activation typically associated with HIV disease.”
FROM IAS 2017
Solid organ transplantation contributes significantly to incidence of NHL among children and adolescents
Solid organ transplant recipients contribute a disproportionate fraction of pediatric non-Hodgkin lymphoma (NHL) cases, especially diffuse large B-cell lymphoma cases (DLBCL), according to an analysis of data drawn from transplant and cancer registries.
Investigators calculated that the incidence of NHL for the pediatric transplant population was 257 times higher than the general population, after analysis from the U.S. transplant registry and 16 cancer registries from around the country between 1990 and 2012. The incidence of NHL for the pediatric transplant population was 306 cases per 100,000 person-years (95% CI, 271-344), compared to 1.19 cases per 100,000 person-years (95% CI, 1.12-1.27) in the general population. Furthermore, transplant recipients made up a much larger proportion of general population DLBCL cases (7.62%; 95% CI, 6.35%-8.88%).
In the general population, the most common subtypes were DLBCL (25% of cases), Burkitt lymphoma (24%), and precursor cell lymphoblastic lymphoma (20%). Among NHLs diagnosed in transplant recipients, 65% were DLBCL, and 9% were Burkitt lymphoma, whereas there were no cases of precursor cell lymphoblastic lymphoma, reported Elizabeth L. Yanik, PhD, of Washington University, St. Louis, and her associates (Cancer. 2017 Jul 31. doi: 10.1002/cncr.30923).
The increased risk of NHL and other cancers for organ transplant recipients is primarily the result of the immunosuppressant medications administered after transplantation, leading to an increased risk of infection-related cancers, the authors said.
“NHL cases are largely attributable to EBV [Epstein-Barr virus] infections that occur while recipients are immunosuppressed, as evidenced by the high prevalence of EBV detectable in NHL tumors and particularly in cases diagnosed during the heavily immunosuppressed period early after transplantation,” wrote Dr. Yanik and her colleagues. “Patients who experience a primary EBV infection after transplantation are particularly susceptible because transplant recipients have a higher NHL risk if they are seronegative for EBV before transplantation.”
Among those at risk, Dr. Yanik and fellow investigators found transplant patients younger than 5 years were the most susceptible to NHL, making up 19.79% of those diagnosed with DLBCL.
The proportion of NHL in solid organ transplant recipients within the entire pediatric NHL population has risen over time, from 1.66% of the NHL population during 1990-1994 to 3.73% during 2010-2012.
“This trend is driven by the rising prevalence of transplant recipients in the general population and not by increases in NHL risk among transplant recipients,” noted Dr. Yanik and her coauthors. “The proportion of NHL diagnoses attributable to transplant recipients has grown over time, and it is likely that this population will be an important source of pediatric NHL cases in the future.”
This study was partially funded by the National Cancer Institute. One coauthor reported being an employee at GRAIL Inc. No other relevant financial disclosures were reported.
[email protected]
On Twitter @eaztweets
Solid organ transplant recipients contribute a disproportionate fraction of pediatric non-Hodgkin lymphoma (NHL) cases, especially diffuse large B-cell lymphoma cases (DLBCL), according to an analysis of data drawn from transplant and cancer registries.
Investigators calculated that the incidence of NHL for the pediatric transplant population was 257 times higher than the general population, after analysis from the U.S. transplant registry and 16 cancer registries from around the country between 1990 and 2012. The incidence of NHL for the pediatric transplant population was 306 cases per 100,000 person-years (95% CI, 271-344), compared to 1.19 cases per 100,000 person-years (95% CI, 1.12-1.27) in the general population. Furthermore, transplant recipients made up a much larger proportion of general population DLBCL cases (7.62%; 95% CI, 6.35%-8.88%).
In the general population, the most common subtypes were DLBCL (25% of cases), Burkitt lymphoma (24%), and precursor cell lymphoblastic lymphoma (20%). Among NHLs diagnosed in transplant recipients, 65% were DLBCL, and 9% were Burkitt lymphoma, whereas there were no cases of precursor cell lymphoblastic lymphoma, reported Elizabeth L. Yanik, PhD, of Washington University, St. Louis, and her associates (Cancer. 2017 Jul 31. doi: 10.1002/cncr.30923).
The increased risk of NHL and other cancers for organ transplant recipients is primarily the result of the immunosuppressant medications administered after transplantation, leading to an increased risk of infection-related cancers, the authors said.
“NHL cases are largely attributable to EBV [Epstein-Barr virus] infections that occur while recipients are immunosuppressed, as evidenced by the high prevalence of EBV detectable in NHL tumors and particularly in cases diagnosed during the heavily immunosuppressed period early after transplantation,” wrote Dr. Yanik and her colleagues. “Patients who experience a primary EBV infection after transplantation are particularly susceptible because transplant recipients have a higher NHL risk if they are seronegative for EBV before transplantation.”
Among those at risk, Dr. Yanik and fellow investigators found transplant patients younger than 5 years were the most susceptible to NHL, making up 19.79% of those diagnosed with DLBCL.
The proportion of NHL in solid organ transplant recipients within the entire pediatric NHL population has risen over time, from 1.66% of the NHL population during 1990-1994 to 3.73% during 2010-2012.
“This trend is driven by the rising prevalence of transplant recipients in the general population and not by increases in NHL risk among transplant recipients,” noted Dr. Yanik and her coauthors. “The proportion of NHL diagnoses attributable to transplant recipients has grown over time, and it is likely that this population will be an important source of pediatric NHL cases in the future.”
This study was partially funded by the National Cancer Institute. One coauthor reported being an employee at GRAIL Inc. No other relevant financial disclosures were reported.
[email protected]
On Twitter @eaztweets
Solid organ transplant recipients contribute a disproportionate fraction of pediatric non-Hodgkin lymphoma (NHL) cases, especially diffuse large B-cell lymphoma cases (DLBCL), according to an analysis of data drawn from transplant and cancer registries.
Investigators calculated that the incidence of NHL for the pediatric transplant population was 257 times higher than the general population, after analysis from the U.S. transplant registry and 16 cancer registries from around the country between 1990 and 2012. The incidence of NHL for the pediatric transplant population was 306 cases per 100,000 person-years (95% CI, 271-344), compared to 1.19 cases per 100,000 person-years (95% CI, 1.12-1.27) in the general population. Furthermore, transplant recipients made up a much larger proportion of general population DLBCL cases (7.62%; 95% CI, 6.35%-8.88%).
In the general population, the most common subtypes were DLBCL (25% of cases), Burkitt lymphoma (24%), and precursor cell lymphoblastic lymphoma (20%). Among NHLs diagnosed in transplant recipients, 65% were DLBCL, and 9% were Burkitt lymphoma, whereas there were no cases of precursor cell lymphoblastic lymphoma, reported Elizabeth L. Yanik, PhD, of Washington University, St. Louis, and her associates (Cancer. 2017 Jul 31. doi: 10.1002/cncr.30923).
The increased risk of NHL and other cancers for organ transplant recipients is primarily the result of the immunosuppressant medications administered after transplantation, leading to an increased risk of infection-related cancers, the authors said.
“NHL cases are largely attributable to EBV [Epstein-Barr virus] infections that occur while recipients are immunosuppressed, as evidenced by the high prevalence of EBV detectable in NHL tumors and particularly in cases diagnosed during the heavily immunosuppressed period early after transplantation,” wrote Dr. Yanik and her colleagues. “Patients who experience a primary EBV infection after transplantation are particularly susceptible because transplant recipients have a higher NHL risk if they are seronegative for EBV before transplantation.”
Among those at risk, Dr. Yanik and fellow investigators found transplant patients younger than 5 years were the most susceptible to NHL, making up 19.79% of those diagnosed with DLBCL.
The proportion of NHL in solid organ transplant recipients within the entire pediatric NHL population has risen over time, from 1.66% of the NHL population during 1990-1994 to 3.73% during 2010-2012.
“This trend is driven by the rising prevalence of transplant recipients in the general population and not by increases in NHL risk among transplant recipients,” noted Dr. Yanik and her coauthors. “The proportion of NHL diagnoses attributable to transplant recipients has grown over time, and it is likely that this population will be an important source of pediatric NHL cases in the future.”
This study was partially funded by the National Cancer Institute. One coauthor reported being an employee at GRAIL Inc. No other relevant financial disclosures were reported.
[email protected]
On Twitter @eaztweets
FROM CANCER
Key clinical point: Solid organ transplant recipients contribute a disproportionate fraction of pediatric NHL cases, especially DLBCL cases.
Major finding: Incidence of NHL among the pediatric transplant population was 257 times higher at 306 cases per 100,000 person-years (95% CI, 271-344), compared to 1.19 cases per 100,000 person-years (95% CI, 1.12-1.27) in the general pediatric population.
Data source: Retrospective cohort study of children and adolescents in the U.S. transplant registry and 16 cancer registries from around the country between 1990 and 2012.
Disclosures: This study was partially funded by the National Cancer Institute. One coauthor reported being an employee at GRAIL Inc. No other relevant financial disclosures were reported.
POLL: Draft classification criteria for SLE stir controversy
[polldaddy:9802068]
[polldaddy:9802068]
[polldaddy:9802068]
Thirdhand smoke shaping up as potential health hazard
DENVER – Thirdhand smoke – the persistent residue that collects on indoor surfaces where people have smoked – is “clearly” a potentially hazardous exposure, John M. Rogers, PhD, said at the annual meeting of the Teratology Society.
Everyone knows about the hazards of secondhand smoke, which have led to widespread bans on smoking in public spaces. Still, the Centers for Disease Control and Prevention estimates that 58 million nonsmokers in the United States are exposed to secondhand smoke on a regular basis. And where there is secondhand smoke, there is typically exposure to thirdhand smoke as well.
“If you walk into a hotel room you were told is a nonsmoking room and you take one breath and you know it’s not nonsmoking, that’s thirdhand smoke. Thirdhand smoke is all over the place where smokers have been,” explained Dr. Rogers, director of the toxicity assessment division at the Environmental Protection Agency in Research Triangle Park, N.C.

The main potential health risk is to young children, who ingest thirdhand smoke by the hand-to-mouth route and skin contact.
Thirdhand smoke is a much newer concept than secondhand smoke and has not yet actually been shown to pose a significant health risk. . But that is likely to change.
Thirdhand smoke has become an area of intensive research interest, with California leading the way. The Tobacco-Related Disease Research Program, a state agency funded by a tax on the sale of tobacco products, has created a research consortium on thirdhand smoke, with studies underway investigating thirdhand smoke’s precise chemical composition, cytotoxicity, genotoxicity, and true impact on public health (www.trdrp.org).
Concern regarding thirdhand smoke’s potential public health impact ramped up in response to a study in which investigators at the University of York, England, measured levels of various tobacco-specific nitrosamines, N-nitrosamines, and nicotine in house dust samples from the homes of smokers. The researchers estimated that years of early life exposure to these compounds at the levels they detected could result in one excess case of cancer per 1,000 exposed individuals (Environ Int. 2014 Oct;71:139-47).
In addition to his update on thirdhand smoke, Dr. Rogers also touched on other recent tobacco-related developments, including a determination by the Food and Drug Administration that there has been no decline in tobacco use in the last 5 years in adolescents and young adults. While cigarette smoking by young people decreased, this was offset by a large increase in the use of electronic cigarettes and a smaller rise in the use of hookah tobacco. Indeed, e-cigarette use is now about double that of cigarettes among youth.
Also of concern is evidence of a striking socioeconomic disparity in smoking prevalence: Low-education, low-income Americans have far higher tobacco use rates.
“That’s pretty alarming,” he said. “I think a lot of people in this audience probably don’t see a lot of smoking these days, but it’s still around.”
Dr. Rogers drew attention to updated evidence reviews on the reproductive and developmental effects of smoking contained in the U.S. Surgeon General’s voluminous 2014 report on the health consequences of smoking. The report concluded that there is now sufficient evidence to infer a causal relationship between maternal smoking in pregnancy, ectopic pregnancy, and orofacial clefts. The available evidence is “suggestive but not sufficient” to infer causality between maternal smoking in pregnancy and atrial septal defects, clubfoot, gastroschisis, and attention-deficit/hyperactivity disorder and other disruptive behavior disorders.
Dr. Rogers reported having no financial disclosures related to his presentation, which he noted did not necessarily reflect the views and policies of the EPA.
DENVER – Thirdhand smoke – the persistent residue that collects on indoor surfaces where people have smoked – is “clearly” a potentially hazardous exposure, John M. Rogers, PhD, said at the annual meeting of the Teratology Society.
Everyone knows about the hazards of secondhand smoke, which have led to widespread bans on smoking in public spaces. Still, the Centers for Disease Control and Prevention estimates that 58 million nonsmokers in the United States are exposed to secondhand smoke on a regular basis. And where there is secondhand smoke, there is typically exposure to thirdhand smoke as well.
“If you walk into a hotel room you were told is a nonsmoking room and you take one breath and you know it’s not nonsmoking, that’s thirdhand smoke. Thirdhand smoke is all over the place where smokers have been,” explained Dr. Rogers, director of the toxicity assessment division at the Environmental Protection Agency in Research Triangle Park, N.C.
The main potential health risk is to young children, who ingest thirdhand smoke by the hand-to-mouth route and skin contact.
Thirdhand smoke is a much newer concept than secondhand smoke and has not yet actually been shown to pose a significant health risk. . But that is likely to change.
Thirdhand smoke has become an area of intensive research interest, with California leading the way. The Tobacco-Related Disease Research Program, a state agency funded by a tax on the sale of tobacco products, has created a research consortium on thirdhand smoke, with studies underway investigating thirdhand smoke’s precise chemical composition, cytotoxicity, genotoxicity, and true impact on public health (www.trdrp.org).
Concern regarding thirdhand smoke’s potential public health impact ramped up in response to a study in which investigators at the University of York, England, measured levels of various tobacco-specific nitrosamines, N-nitrosamines, and nicotine in house dust samples from the homes of smokers. The researchers estimated that years of early life exposure to these compounds at the levels they detected could result in one excess case of cancer per 1,000 exposed individuals (Environ Int. 2014 Oct;71:139-47).
In addition to his update on thirdhand smoke, Dr. Rogers also touched on other recent tobacco-related developments, including a determination by the Food and Drug Administration that there has been no decline in tobacco use in the last 5 years in adolescents and young adults. While cigarette smoking by young people decreased, this was offset by a large increase in the use of electronic cigarettes and a smaller rise in the use of hookah tobacco. Indeed, e-cigarette use is now about double that of cigarettes among youth.
Also of concern is evidence of a striking socioeconomic disparity in smoking prevalence: Low-education, low-income Americans have far higher tobacco use rates.
“That’s pretty alarming,” he said. “I think a lot of people in this audience probably don’t see a lot of smoking these days, but it’s still around.”
Dr. Rogers drew attention to updated evidence reviews on the reproductive and developmental effects of smoking contained in the U.S. Surgeon General’s voluminous 2014 report on the health consequences of smoking. The report concluded that there is now sufficient evidence to infer a causal relationship between maternal smoking in pregnancy, ectopic pregnancy, and orofacial clefts. The available evidence is “suggestive but not sufficient” to infer causality between maternal smoking in pregnancy and atrial septal defects, clubfoot, gastroschisis, and attention-deficit/hyperactivity disorder and other disruptive behavior disorders.
Dr. Rogers reported having no financial disclosures related to his presentation, which he noted did not necessarily reflect the views and policies of the EPA.
DENVER – Thirdhand smoke – the persistent residue that collects on indoor surfaces where people have smoked – is “clearly” a potentially hazardous exposure, John M. Rogers, PhD, said at the annual meeting of the Teratology Society.
Everyone knows about the hazards of secondhand smoke, which have led to widespread bans on smoking in public spaces. Still, the Centers for Disease Control and Prevention estimates that 58 million nonsmokers in the United States are exposed to secondhand smoke on a regular basis. And where there is secondhand smoke, there is typically exposure to thirdhand smoke as well.
“If you walk into a hotel room you were told is a nonsmoking room and you take one breath and you know it’s not nonsmoking, that’s thirdhand smoke. Thirdhand smoke is all over the place where smokers have been,” explained Dr. Rogers, director of the toxicity assessment division at the Environmental Protection Agency in Research Triangle Park, N.C.
The main potential health risk is to young children, who ingest thirdhand smoke by the hand-to-mouth route and skin contact.
Thirdhand smoke is a much newer concept than secondhand smoke and has not yet actually been shown to pose a significant health risk. . But that is likely to change.
Thirdhand smoke has become an area of intensive research interest, with California leading the way. The Tobacco-Related Disease Research Program, a state agency funded by a tax on the sale of tobacco products, has created a research consortium on thirdhand smoke, with studies underway investigating thirdhand smoke’s precise chemical composition, cytotoxicity, genotoxicity, and true impact on public health (www.trdrp.org).
Concern regarding thirdhand smoke’s potential public health impact ramped up in response to a study in which investigators at the University of York, England, measured levels of various tobacco-specific nitrosamines, N-nitrosamines, and nicotine in house dust samples from the homes of smokers. The researchers estimated that years of early life exposure to these compounds at the levels they detected could result in one excess case of cancer per 1,000 exposed individuals (Environ Int. 2014 Oct;71:139-47).
In addition to his update on thirdhand smoke, Dr. Rogers also touched on other recent tobacco-related developments, including a determination by the Food and Drug Administration that there has been no decline in tobacco use in the last 5 years in adolescents and young adults. While cigarette smoking by young people decreased, this was offset by a large increase in the use of electronic cigarettes and a smaller rise in the use of hookah tobacco. Indeed, e-cigarette use is now about double that of cigarettes among youth.
Also of concern is evidence of a striking socioeconomic disparity in smoking prevalence: Low-education, low-income Americans have far higher tobacco use rates.
“That’s pretty alarming,” he said. “I think a lot of people in this audience probably don’t see a lot of smoking these days, but it’s still around.”
Dr. Rogers drew attention to updated evidence reviews on the reproductive and developmental effects of smoking contained in the U.S. Surgeon General’s voluminous 2014 report on the health consequences of smoking. The report concluded that there is now sufficient evidence to infer a causal relationship between maternal smoking in pregnancy, ectopic pregnancy, and orofacial clefts. The available evidence is “suggestive but not sufficient” to infer causality between maternal smoking in pregnancy and atrial septal defects, clubfoot, gastroschisis, and attention-deficit/hyperactivity disorder and other disruptive behavior disorders.
Dr. Rogers reported having no financial disclosures related to his presentation, which he noted did not necessarily reflect the views and policies of the EPA.
EXPERT ANALYSIS FROM TERATOLOGY SOCIETY 2017
New and Noteworthy Information—August 2017
Brain Training Shows Little Benefit
Commercial brain training with Lumosity has no effect on decision making or cognitive function beyond practice effects on training tasks, according to a study published online ahead of print July 10 in the Journal of Neuroscience. Researchers tested whether training executive cognitive function could influence choice behavior and brain responses. In a randomized controlled trial, 128 young adults (71 male) participated in 10 weeks of training with either a commercial web-based cognitive training program or web-based video games that do not specifically target executive function or adapt the level of difficulty throughout training. The participants also completed a series of cognitive tests that were not part of the training. Although both groups showed improvement, commercial brain training did not lead to more improvement than online video games did.
Kable JW, Caulfield MK, Falcone M, et al. No effect of commercial cognitive training on neural activity during decision-making. J Neurosci. 2017 Jul 10 [Epub ahead of print].
Sense of Purpose Linked to Better Sleep
A higher level of meaning and purpose in life among older adults is associated with better sleep quality and appears to protect against symptoms of sleep apnea and restless legs syndrome (RLS), according to a study published online ahead of print July 10 in Sleep Science and Practice. Included in this study were 825 nondemented older African Americans (n = 428) and whites (n = 397), from the Minority Aging Research Study and the Rush Memory and Aging Project. Participants completed a 32-item questionnaire assessing sleep quality and symptoms of sleep apnea, RLS, and REM sleep behavior disorder. Longitudinal follow-up data indicated that higher levels of purpose in life were associated with lower risk of sleep apnea at baseline, one-year follow-up, and two-year follow-up, and with reduced RLS symptoms at one-year and two-year follow-up.
Turner AD, Smith CE, Ong JC. Is purpose in life associated with less sleep disturbance in older adults? Sleep Sci Pract. 2017 July 10 [Epub ahead of print].
Can Breastfeeding Reduce MS Risk in Mothers?
Mothers who breastfeed longer may be at lower subsequent risk of developing multiple sclerosis (MS), according to a study published online ahead of print July 12 in Neurology. Researchers recruited women with newly diagnosed MS or clinically isolated syndrome (CIS) and matched controls into the MS Sunshine Study from the membership of Kaiser Permanente Southern California. An in-person questionnaire was administered to collect behavioral and biologic factors to calculate ovulatory years. Among women who had live births, a cumulative duration of breastfeeding for 15 months or more was associated with a reduced risk of MS and CIS (adjusted odds ratio, 0.47). Being age 15 or older at menarche also was associated with a lower risk of MS and CIS (adjusted odds ratio, 0.56).
Langer-Gould A, Smith JB, Hellwig K, et al. Breastfeeding, ovulatory years, and risk of multiple sclerosis. Neurology. 2017 July 12 [Epub ahead of print].
Does Added Weight Increase Survival After Stroke?
People who are overweight or mildly obese survive strokes at a higher rate, compared with people of normal body weight, according to a study published June 24 in the Journal of the American Heart Association. Participants from the Framingham Heart Study were followed for as long as 10 years, with BMI measured prior to their strokes. Researchers compared all-cause mortality in participants stratified by prestroke weight. Separate analyses were performed for ischemic stroke and all stroke and for age-, sex-, and BMI category-matched stroke-free controls. There were 782 stroke cases and 2,346 controls. The association of reduced mortality with BMI of 25 or higher, compared with BMI of 18.5 to less than 25, was pronounced among ischemic stroke cases, but diminished with inclusion of hemorrhagic strokes.
Aparicio HJ, Himali JJ, Beiser AS, et al. Overweight, obesity, and survival after stroke in the Framingham Heart Study. J Am Heart Assoc. 2017;6(6).
Poor Sleep Linked to CSF Biomarkers
Self-reported poor sleep is associated with greater Alzheimer’s disease-related pathology in cognitively healthy adults at risk for Alzheimer’s disease, according to a study published online ahead of print July 5 in Neurology. Researchers investigated the relationship between sleep quality and CSF Alzheimer’s disease biomarkers in a cohort enriched for parental history of sporadic Alzheimer’s disease. In all, 101 participants completed sleep assessments and CSF collection and were cognitively normal. CSF was assayed for biomarkers of amyloid metabolism and plaques, tau pathology, neuronal and axonal degeneration, neuroinflammation and astroglial activation, and synaptic dysfunction and degeneration. Worse subjective sleep quality, more sleep problems, and daytime somnolence were associated with greater Alzheimer’s disease pathology, indicated by lower CSF Aβ42/Aβ40 and higher t-tau/Aβ42, p-tau/Aβ42, MCP-1/Aβ42, and YKL-40/Aβ42.
Sprecher KE, Koscik RL, Carlsson CM, et al. Poor sleep is associated with CSF biomarkers of amyloid pathology in cognitively normal adults. Neurology. 2017 Jul 5 [Epub ahead of print].
Is There a Link Between Parkinson’s Disease and Melanoma?
Melanoma and Parkinson’s disease may be associated, according to a study published in the July issue of Mayo Clinic Proceedings. For phase I of the Rochester Epidemiology Project, investigators used records to identify patients with Parkinson’s disease and match three controls per case. During phase II of this study, all Rochester Epidemiology Project cases of melanoma were identified, with one control per case. Investigators used a Cox proportional hazards model to assess the risk of developing Parkinson’s disease after the index date in cases versus controls, and performed Kaplan-Meier analysis to determine the 35-year cumulative risk of Parkinson’s disease. Patients with Parkinson’s disease had a 3.8-fold increased likelihood of having preexisting melanoma, compared with controls. Patients with melanoma had a 4.2-fold increased risk of developing Parkinson’s disease.
Dalvin LA, Damento GM, Yawn BP, et al. Parkinson disease and melanoma: confirming and reexamining an association. Mayo Clin Proc. 2017;92(7):1070-1079.
Zolpidem Treats Various Neurologic Disorders
A systematic review shows that zolpidem can treat various neurologic disorders, most often related to movement disorders and disorders of consciousness, according to a literature review published online ahead of print June 26 in JAMA Neurology. The investigators searched for English-language articles, published by March 20, 2015, that examined the use of zolpidem for noninsomnia neurologic disorders. Searched databases included PubMed, Scopus, Web of Science Core Collection, the Cochrane Library, EMBASE, CENTRAL, and clinicaltrials.gov. In all, 67 articles were eligible for full manuscript review. Thirty-one studies treated movement disorders, 22 treated disorders of consciousness, and 14 treated other neurologic conditions. The effects of zolpidem were wide ranging and generally lasted for one to four hours before the participant returned to baseline. Sedation was the most common adverse effect.
Bomalaski MN, Claflin ES, Townsend W, Peterson MD. Zolpidem for the treatment of neurologic disorders: a systematic review. JAMA Neurol. 2017 Jun 26 [Epub ahead of print].
Colored Light Triggers Responses in Migraineurs
Lights trigger more changes in autonomic functions and negative emotions during migraine than in control subjects, and the association between light and positive emotions is stronger in control subjects than in migraineurs, according to a study published online ahead of print June 26 in the Proceedings of the National Academy of Sciences. Researchers showed different colored lights to 81 migraineurs and 17 people who had never had a migraine. The effects of light and color were tested three times. Investigators found that all colors of light triggered unpleasant physiologic sensations in patients with migraines, during and between attacks. Additionally, migraineurs reported intense emotional responses such as anger, nervousness, hopelessness, sadness, depression, anxiety, and fear when exposed to all light colors except green.
Noseda R, Lee AJ, Nir RR, et al. Neural mechanism for hypothalamic-mediated autonomic responses to light during migraine. Proc Natl Acad Sci. 2017 Jun 26 [Epub ahead of print].
TBI May Not Hasten Cognitive Decline
Having a history of traumatic brain injury (TBI) with loss of consciousness does not affect the rate of cognitive change over time for people with normal cognition or people with Alzheimer’s disease, according to a study published online ahead of print June 22 in the Journal of Alzheimer’s Disease. Researchers compared performance on cognitive tests over time for 432 participants with normal cognition and 274 participants with probable Alzheimer’s disease. They matched participants with a history of TBI with loss of consciousness to an equal number of demographically and clinically similar participants without a history of TBI. Mixed-effects regressions showed that a history of TBI with loss of consciousness did not affect rates of cognitive change in APOE ε4 carriers and noncarriers.
Tripodis Y, Alosco ML, Zirogiannis N, et al. The effect of traumatic brain injury history with loss of consciousness on rate of cognitive decline among older adults with normal cognition and Alzheimer’s disease dementia. J Alzheimers Dis. 2017 Jun 22 [Epub ahead of print].
Visual Changes in Parkinson’s Disease
Visual system alterations can be detected in early stages of Parkinson’s disease, and the entire intracranial visual system can be involved, according to a study published online ahead of print July 11 in Radiology. Twenty patients with newly diagnosed Parkinson’s disease and 20 age-matched control subjects were studied. Researchers used diffusion-weighted imaging to assess white matter changes and voxel-based morphometry (VBM) to investigate concentration changes of gray and white matter. In patients with Parkinson’s disease, significant alterations were found in optic radiation connectivity distribution, with decreased lateral geniculate nucleus V2 density, a significant increase in optic radiation mean diffusivity, and a significant reduction in white matter concentration. VBM analysis also showed a significant reduction in visual cortical volumes.
Arrigo A, Calamuneri A, Milardi D, et al. Visual system involvement in patients with newly diagnosed Parkinson disease. Radiology. 2017 Jul 11 [Epub ahead of print].
—Kimberly Williams
Brain Training Shows Little Benefit
Commercial brain training with Lumosity has no effect on decision making or cognitive function beyond practice effects on training tasks, according to a study published online ahead of print July 10 in the Journal of Neuroscience. Researchers tested whether training executive cognitive function could influence choice behavior and brain responses. In a randomized controlled trial, 128 young adults (71 male) participated in 10 weeks of training with either a commercial web-based cognitive training program or web-based video games that do not specifically target executive function or adapt the level of difficulty throughout training. The participants also completed a series of cognitive tests that were not part of the training. Although both groups showed improvement, commercial brain training did not lead to more improvement than online video games did.
Kable JW, Caulfield MK, Falcone M, et al. No effect of commercial cognitive training on neural activity during decision-making. J Neurosci. 2017 Jul 10 [Epub ahead of print].
Sense of Purpose Linked to Better Sleep
A higher level of meaning and purpose in life among older adults is associated with better sleep quality and appears to protect against symptoms of sleep apnea and restless legs syndrome (RLS), according to a study published online ahead of print July 10 in Sleep Science and Practice. Included in this study were 825 nondemented older African Americans (n = 428) and whites (n = 397), from the Minority Aging Research Study and the Rush Memory and Aging Project. Participants completed a 32-item questionnaire assessing sleep quality and symptoms of sleep apnea, RLS, and REM sleep behavior disorder. Longitudinal follow-up data indicated that higher levels of purpose in life were associated with lower risk of sleep apnea at baseline, one-year follow-up, and two-year follow-up, and with reduced RLS symptoms at one-year and two-year follow-up.
Turner AD, Smith CE, Ong JC. Is purpose in life associated with less sleep disturbance in older adults? Sleep Sci Pract. 2017 July 10 [Epub ahead of print].
Can Breastfeeding Reduce MS Risk in Mothers?
Mothers who breastfeed longer may be at lower subsequent risk of developing multiple sclerosis (MS), according to a study published online ahead of print July 12 in Neurology. Researchers recruited women with newly diagnosed MS or clinically isolated syndrome (CIS) and matched controls into the MS Sunshine Study from the membership of Kaiser Permanente Southern California. An in-person questionnaire was administered to collect behavioral and biologic factors to calculate ovulatory years. Among women who had live births, a cumulative duration of breastfeeding for 15 months or more was associated with a reduced risk of MS and CIS (adjusted odds ratio, 0.47). Being age 15 or older at menarche also was associated with a lower risk of MS and CIS (adjusted odds ratio, 0.56).
Langer-Gould A, Smith JB, Hellwig K, et al. Breastfeeding, ovulatory years, and risk of multiple sclerosis. Neurology. 2017 July 12 [Epub ahead of print].
Does Added Weight Increase Survival After Stroke?
People who are overweight or mildly obese survive strokes at a higher rate, compared with people of normal body weight, according to a study published June 24 in the Journal of the American Heart Association. Participants from the Framingham Heart Study were followed for as long as 10 years, with BMI measured prior to their strokes. Researchers compared all-cause mortality in participants stratified by prestroke weight. Separate analyses were performed for ischemic stroke and all stroke and for age-, sex-, and BMI category-matched stroke-free controls. There were 782 stroke cases and 2,346 controls. The association of reduced mortality with BMI of 25 or higher, compared with BMI of 18.5 to less than 25, was pronounced among ischemic stroke cases, but diminished with inclusion of hemorrhagic strokes.
Aparicio HJ, Himali JJ, Beiser AS, et al. Overweight, obesity, and survival after stroke in the Framingham Heart Study. J Am Heart Assoc. 2017;6(6).
Poor Sleep Linked to CSF Biomarkers
Self-reported poor sleep is associated with greater Alzheimer’s disease-related pathology in cognitively healthy adults at risk for Alzheimer’s disease, according to a study published online ahead of print July 5 in Neurology. Researchers investigated the relationship between sleep quality and CSF Alzheimer’s disease biomarkers in a cohort enriched for parental history of sporadic Alzheimer’s disease. In all, 101 participants completed sleep assessments and CSF collection and were cognitively normal. CSF was assayed for biomarkers of amyloid metabolism and plaques, tau pathology, neuronal and axonal degeneration, neuroinflammation and astroglial activation, and synaptic dysfunction and degeneration. Worse subjective sleep quality, more sleep problems, and daytime somnolence were associated with greater Alzheimer’s disease pathology, indicated by lower CSF Aβ42/Aβ40 and higher t-tau/Aβ42, p-tau/Aβ42, MCP-1/Aβ42, and YKL-40/Aβ42.
Sprecher KE, Koscik RL, Carlsson CM, et al. Poor sleep is associated with CSF biomarkers of amyloid pathology in cognitively normal adults. Neurology. 2017 Jul 5 [Epub ahead of print].
Is There a Link Between Parkinson’s Disease and Melanoma?
Melanoma and Parkinson’s disease may be associated, according to a study published in the July issue of Mayo Clinic Proceedings. For phase I of the Rochester Epidemiology Project, investigators used records to identify patients with Parkinson’s disease and match three controls per case. During phase II of this study, all Rochester Epidemiology Project cases of melanoma were identified, with one control per case. Investigators used a Cox proportional hazards model to assess the risk of developing Parkinson’s disease after the index date in cases versus controls, and performed Kaplan-Meier analysis to determine the 35-year cumulative risk of Parkinson’s disease. Patients with Parkinson’s disease had a 3.8-fold increased likelihood of having preexisting melanoma, compared with controls. Patients with melanoma had a 4.2-fold increased risk of developing Parkinson’s disease.
Dalvin LA, Damento GM, Yawn BP, et al. Parkinson disease and melanoma: confirming and reexamining an association. Mayo Clin Proc. 2017;92(7):1070-1079.
Zolpidem Treats Various Neurologic Disorders
A systematic review shows that zolpidem can treat various neurologic disorders, most often related to movement disorders and disorders of consciousness, according to a literature review published online ahead of print June 26 in JAMA Neurology. The investigators searched for English-language articles, published by March 20, 2015, that examined the use of zolpidem for noninsomnia neurologic disorders. Searched databases included PubMed, Scopus, Web of Science Core Collection, the Cochrane Library, EMBASE, CENTRAL, and clinicaltrials.gov. In all, 67 articles were eligible for full manuscript review. Thirty-one studies treated movement disorders, 22 treated disorders of consciousness, and 14 treated other neurologic conditions. The effects of zolpidem were wide ranging and generally lasted for one to four hours before the participant returned to baseline. Sedation was the most common adverse effect.
Bomalaski MN, Claflin ES, Townsend W, Peterson MD. Zolpidem for the treatment of neurologic disorders: a systematic review. JAMA Neurol. 2017 Jun 26 [Epub ahead of print].
Colored Light Triggers Responses in Migraineurs
Lights trigger more changes in autonomic functions and negative emotions during migraine than in control subjects, and the association between light and positive emotions is stronger in control subjects than in migraineurs, according to a study published online ahead of print June 26 in the Proceedings of the National Academy of Sciences. Researchers showed different colored lights to 81 migraineurs and 17 people who had never had a migraine. The effects of light and color were tested three times. Investigators found that all colors of light triggered unpleasant physiologic sensations in patients with migraines, during and between attacks. Additionally, migraineurs reported intense emotional responses such as anger, nervousness, hopelessness, sadness, depression, anxiety, and fear when exposed to all light colors except green.
Noseda R, Lee AJ, Nir RR, et al. Neural mechanism for hypothalamic-mediated autonomic responses to light during migraine. Proc Natl Acad Sci. 2017 Jun 26 [Epub ahead of print].
TBI May Not Hasten Cognitive Decline
Having a history of traumatic brain injury (TBI) with loss of consciousness does not affect the rate of cognitive change over time for people with normal cognition or people with Alzheimer’s disease, according to a study published online ahead of print June 22 in the Journal of Alzheimer’s Disease. Researchers compared performance on cognitive tests over time for 432 participants with normal cognition and 274 participants with probable Alzheimer’s disease. They matched participants with a history of TBI with loss of consciousness to an equal number of demographically and clinically similar participants without a history of TBI. Mixed-effects regressions showed that a history of TBI with loss of consciousness did not affect rates of cognitive change in APOE ε4 carriers and noncarriers.
Tripodis Y, Alosco ML, Zirogiannis N, et al. The effect of traumatic brain injury history with loss of consciousness on rate of cognitive decline among older adults with normal cognition and Alzheimer’s disease dementia. J Alzheimers Dis. 2017 Jun 22 [Epub ahead of print].
Visual Changes in Parkinson’s Disease
Visual system alterations can be detected in early stages of Parkinson’s disease, and the entire intracranial visual system can be involved, according to a study published online ahead of print July 11 in Radiology. Twenty patients with newly diagnosed Parkinson’s disease and 20 age-matched control subjects were studied. Researchers used diffusion-weighted imaging to assess white matter changes and voxel-based morphometry (VBM) to investigate concentration changes of gray and white matter. In patients with Parkinson’s disease, significant alterations were found in optic radiation connectivity distribution, with decreased lateral geniculate nucleus V2 density, a significant increase in optic radiation mean diffusivity, and a significant reduction in white matter concentration. VBM analysis also showed a significant reduction in visual cortical volumes.
Arrigo A, Calamuneri A, Milardi D, et al. Visual system involvement in patients with newly diagnosed Parkinson disease. Radiology. 2017 Jul 11 [Epub ahead of print].
—Kimberly Williams
Brain Training Shows Little Benefit
Commercial brain training with Lumosity has no effect on decision making or cognitive function beyond practice effects on training tasks, according to a study published online ahead of print July 10 in the Journal of Neuroscience. Researchers tested whether training executive cognitive function could influence choice behavior and brain responses. In a randomized controlled trial, 128 young adults (71 male) participated in 10 weeks of training with either a commercial web-based cognitive training program or web-based video games that do not specifically target executive function or adapt the level of difficulty throughout training. The participants also completed a series of cognitive tests that were not part of the training. Although both groups showed improvement, commercial brain training did not lead to more improvement than online video games did.
Kable JW, Caulfield MK, Falcone M, et al. No effect of commercial cognitive training on neural activity during decision-making. J Neurosci. 2017 Jul 10 [Epub ahead of print].
Sense of Purpose Linked to Better Sleep
A higher level of meaning and purpose in life among older adults is associated with better sleep quality and appears to protect against symptoms of sleep apnea and restless legs syndrome (RLS), according to a study published online ahead of print July 10 in Sleep Science and Practice. Included in this study were 825 nondemented older African Americans (n = 428) and whites (n = 397), from the Minority Aging Research Study and the Rush Memory and Aging Project. Participants completed a 32-item questionnaire assessing sleep quality and symptoms of sleep apnea, RLS, and REM sleep behavior disorder. Longitudinal follow-up data indicated that higher levels of purpose in life were associated with lower risk of sleep apnea at baseline, one-year follow-up, and two-year follow-up, and with reduced RLS symptoms at one-year and two-year follow-up.
Turner AD, Smith CE, Ong JC. Is purpose in life associated with less sleep disturbance in older adults? Sleep Sci Pract. 2017 July 10 [Epub ahead of print].
Can Breastfeeding Reduce MS Risk in Mothers?
Mothers who breastfeed longer may be at lower subsequent risk of developing multiple sclerosis (MS), according to a study published online ahead of print July 12 in Neurology. Researchers recruited women with newly diagnosed MS or clinically isolated syndrome (CIS) and matched controls into the MS Sunshine Study from the membership of Kaiser Permanente Southern California. An in-person questionnaire was administered to collect behavioral and biologic factors to calculate ovulatory years. Among women who had live births, a cumulative duration of breastfeeding for 15 months or more was associated with a reduced risk of MS and CIS (adjusted odds ratio, 0.47). Being age 15 or older at menarche also was associated with a lower risk of MS and CIS (adjusted odds ratio, 0.56).
Langer-Gould A, Smith JB, Hellwig K, et al. Breastfeeding, ovulatory years, and risk of multiple sclerosis. Neurology. 2017 July 12 [Epub ahead of print].
Does Added Weight Increase Survival After Stroke?
People who are overweight or mildly obese survive strokes at a higher rate, compared with people of normal body weight, according to a study published June 24 in the Journal of the American Heart Association. Participants from the Framingham Heart Study were followed for as long as 10 years, with BMI measured prior to their strokes. Researchers compared all-cause mortality in participants stratified by prestroke weight. Separate analyses were performed for ischemic stroke and all stroke and for age-, sex-, and BMI category-matched stroke-free controls. There were 782 stroke cases and 2,346 controls. The association of reduced mortality with BMI of 25 or higher, compared with BMI of 18.5 to less than 25, was pronounced among ischemic stroke cases, but diminished with inclusion of hemorrhagic strokes.
Aparicio HJ, Himali JJ, Beiser AS, et al. Overweight, obesity, and survival after stroke in the Framingham Heart Study. J Am Heart Assoc. 2017;6(6).
Poor Sleep Linked to CSF Biomarkers
Self-reported poor sleep is associated with greater Alzheimer’s disease-related pathology in cognitively healthy adults at risk for Alzheimer’s disease, according to a study published online ahead of print July 5 in Neurology. Researchers investigated the relationship between sleep quality and CSF Alzheimer’s disease biomarkers in a cohort enriched for parental history of sporadic Alzheimer’s disease. In all, 101 participants completed sleep assessments and CSF collection and were cognitively normal. CSF was assayed for biomarkers of amyloid metabolism and plaques, tau pathology, neuronal and axonal degeneration, neuroinflammation and astroglial activation, and synaptic dysfunction and degeneration. Worse subjective sleep quality, more sleep problems, and daytime somnolence were associated with greater Alzheimer’s disease pathology, indicated by lower CSF Aβ42/Aβ40 and higher t-tau/Aβ42, p-tau/Aβ42, MCP-1/Aβ42, and YKL-40/Aβ42.
Sprecher KE, Koscik RL, Carlsson CM, et al. Poor sleep is associated with CSF biomarkers of amyloid pathology in cognitively normal adults. Neurology. 2017 Jul 5 [Epub ahead of print].
Is There a Link Between Parkinson’s Disease and Melanoma?
Melanoma and Parkinson’s disease may be associated, according to a study published in the July issue of Mayo Clinic Proceedings. For phase I of the Rochester Epidemiology Project, investigators used records to identify patients with Parkinson’s disease and match three controls per case. During phase II of this study, all Rochester Epidemiology Project cases of melanoma were identified, with one control per case. Investigators used a Cox proportional hazards model to assess the risk of developing Parkinson’s disease after the index date in cases versus controls, and performed Kaplan-Meier analysis to determine the 35-year cumulative risk of Parkinson’s disease. Patients with Parkinson’s disease had a 3.8-fold increased likelihood of having preexisting melanoma, compared with controls. Patients with melanoma had a 4.2-fold increased risk of developing Parkinson’s disease.
Dalvin LA, Damento GM, Yawn BP, et al. Parkinson disease and melanoma: confirming and reexamining an association. Mayo Clin Proc. 2017;92(7):1070-1079.
Zolpidem Treats Various Neurologic Disorders
A systematic review shows that zolpidem can treat various neurologic disorders, most often related to movement disorders and disorders of consciousness, according to a literature review published online ahead of print June 26 in JAMA Neurology. The investigators searched for English-language articles, published by March 20, 2015, that examined the use of zolpidem for noninsomnia neurologic disorders. Searched databases included PubMed, Scopus, Web of Science Core Collection, the Cochrane Library, EMBASE, CENTRAL, and clinicaltrials.gov. In all, 67 articles were eligible for full manuscript review. Thirty-one studies treated movement disorders, 22 treated disorders of consciousness, and 14 treated other neurologic conditions. The effects of zolpidem were wide ranging and generally lasted for one to four hours before the participant returned to baseline. Sedation was the most common adverse effect.
Bomalaski MN, Claflin ES, Townsend W, Peterson MD. Zolpidem for the treatment of neurologic disorders: a systematic review. JAMA Neurol. 2017 Jun 26 [Epub ahead of print].
Colored Light Triggers Responses in Migraineurs
Lights trigger more changes in autonomic functions and negative emotions during migraine than in control subjects, and the association between light and positive emotions is stronger in control subjects than in migraineurs, according to a study published online ahead of print June 26 in the Proceedings of the National Academy of Sciences. Researchers showed different colored lights to 81 migraineurs and 17 people who had never had a migraine. The effects of light and color were tested three times. Investigators found that all colors of light triggered unpleasant physiologic sensations in patients with migraines, during and between attacks. Additionally, migraineurs reported intense emotional responses such as anger, nervousness, hopelessness, sadness, depression, anxiety, and fear when exposed to all light colors except green.
Noseda R, Lee AJ, Nir RR, et al. Neural mechanism for hypothalamic-mediated autonomic responses to light during migraine. Proc Natl Acad Sci. 2017 Jun 26 [Epub ahead of print].
TBI May Not Hasten Cognitive Decline
Having a history of traumatic brain injury (TBI) with loss of consciousness does not affect the rate of cognitive change over time for people with normal cognition or people with Alzheimer’s disease, according to a study published online ahead of print June 22 in the Journal of Alzheimer’s Disease. Researchers compared performance on cognitive tests over time for 432 participants with normal cognition and 274 participants with probable Alzheimer’s disease. They matched participants with a history of TBI with loss of consciousness to an equal number of demographically and clinically similar participants without a history of TBI. Mixed-effects regressions showed that a history of TBI with loss of consciousness did not affect rates of cognitive change in APOE ε4 carriers and noncarriers.
Tripodis Y, Alosco ML, Zirogiannis N, et al. The effect of traumatic brain injury history with loss of consciousness on rate of cognitive decline among older adults with normal cognition and Alzheimer’s disease dementia. J Alzheimers Dis. 2017 Jun 22 [Epub ahead of print].
Visual Changes in Parkinson’s Disease
Visual system alterations can be detected in early stages of Parkinson’s disease, and the entire intracranial visual system can be involved, according to a study published online ahead of print July 11 in Radiology. Twenty patients with newly diagnosed Parkinson’s disease and 20 age-matched control subjects were studied. Researchers used diffusion-weighted imaging to assess white matter changes and voxel-based morphometry (VBM) to investigate concentration changes of gray and white matter. In patients with Parkinson’s disease, significant alterations were found in optic radiation connectivity distribution, with decreased lateral geniculate nucleus V2 density, a significant increase in optic radiation mean diffusivity, and a significant reduction in white matter concentration. VBM analysis also showed a significant reduction in visual cortical volumes.
Arrigo A, Calamuneri A, Milardi D, et al. Visual system involvement in patients with newly diagnosed Parkinson disease. Radiology. 2017 Jul 11 [Epub ahead of print].
—Kimberly Williams

LIFSCREEN data support broader cancer screening in Li-Fraumeni syndrome
Broader cancer screening of individuals with Li-Fraumeni syndrome (LFS), with or without whole-body magnetic resonance imaging, has a good diagnostic yield and identifies a wide range of cancers, according to a preliminary analysis of the ongoing LIFSCREEN phase 3, randomized, controlled trial.
Investigators led by Olivier Caron, MD, chair of the oncogenetics committee, department of medical oncology, at the Gustave Roussy University Hospital in Villejuif, France, enrolled in the trial 107 individuals from 75 families carrying a TP53 mutation, a genetic aberration commonly present in LFS that confers heightened risk of a variety of malignancies.
Participants had a median age at baseline of 32.9 years, with a range from 5 to 67 years. Fully 98% had a family history of cancer, and 48% had a personal history of cancer.
The participants were assigned to 5 years of standard screening – annual clinical examination, abdomen and pelvis ultrasound, brain MRI, complete blood cell count, and, for women older than 20 years, breast ultrasound and MRI – or intensive screening, entailing the addition of diffusion whole-body MRI.
At the time of the preliminary analysis, 15 patients had undergone only one round of screening; 35, two rounds; 19, three rounds; 24, four rounds; and 7, five rounds, Dr. Caron and associates reported in a research letter (JAMA Oncol. 2017; Aug 3 doi: 10.1001/jamaoncol.2017.1358).
Collectively, this amounted to 226.4 person-years of follow-up.
Screening with either trial strategy (with or without whole-body MRI) led to diagnosis of 23 new primary cancers in 20 patients. Nearly half of the total (12 cancers) were detected at the first round. Patients had a median age of 39.8 at the new cancer diagnosis, with a range from 6 to 70 years.
Of the new cancers, 10 belonged to the core LFS spectrum of breast cancer, sarcoma, and brain tumors. However, the other 13 were outside that spectrum, for example, lung adenocarcinomas, all seen in never or light smokers, and leukemias. Screening also detected three relapses of previous cancers.
Analyses further showed that prior cancer diagnosis was not a reliable marker for risk of new primaries. Although 12 of the patients with a screening-detected new primary had a personal cancer history, 8 did not (P = .22).
“The proportion and diversity of off–core LFS spectrum cancers detected in TP53 mutation carriers as reported by others give growing evidence of a broader LFS spectrum, in agreement with the permissive role of TP53 mutations,” write Dr. Caron and colleagues, who report having no relevant disclosures. “Our observations seem to support recent moves toward broader cancer screening in TP53 mutation carriers.”
The investigators continue to collect data in LIFSCREEN and plan to undertake main analysis later this year. “Our final analysis will help to determine the benefits and drawbacks (mostly related to false-positive test results) of whole-body MRI in TP53 mutation carrier surveillance,” they conclude. “Studies focused on TP53 mutation penetrance, using methods limiting selection bias, are required to refine cancer risks to improve TP53 mutation carrier management.”
Broader cancer screening of individuals with Li-Fraumeni syndrome (LFS), with or without whole-body magnetic resonance imaging, has a good diagnostic yield and identifies a wide range of cancers, according to a preliminary analysis of the ongoing LIFSCREEN phase 3, randomized, controlled trial.
Investigators led by Olivier Caron, MD, chair of the oncogenetics committee, department of medical oncology, at the Gustave Roussy University Hospital in Villejuif, France, enrolled in the trial 107 individuals from 75 families carrying a TP53 mutation, a genetic aberration commonly present in LFS that confers heightened risk of a variety of malignancies.
Participants had a median age at baseline of 32.9 years, with a range from 5 to 67 years. Fully 98% had a family history of cancer, and 48% had a personal history of cancer.
The participants were assigned to 5 years of standard screening – annual clinical examination, abdomen and pelvis ultrasound, brain MRI, complete blood cell count, and, for women older than 20 years, breast ultrasound and MRI – or intensive screening, entailing the addition of diffusion whole-body MRI.
At the time of the preliminary analysis, 15 patients had undergone only one round of screening; 35, two rounds; 19, three rounds; 24, four rounds; and 7, five rounds, Dr. Caron and associates reported in a research letter (JAMA Oncol. 2017; Aug 3 doi: 10.1001/jamaoncol.2017.1358).
Collectively, this amounted to 226.4 person-years of follow-up.
Screening with either trial strategy (with or without whole-body MRI) led to diagnosis of 23 new primary cancers in 20 patients. Nearly half of the total (12 cancers) were detected at the first round. Patients had a median age of 39.8 at the new cancer diagnosis, with a range from 6 to 70 years.
Of the new cancers, 10 belonged to the core LFS spectrum of breast cancer, sarcoma, and brain tumors. However, the other 13 were outside that spectrum, for example, lung adenocarcinomas, all seen in never or light smokers, and leukemias. Screening also detected three relapses of previous cancers.
Analyses further showed that prior cancer diagnosis was not a reliable marker for risk of new primaries. Although 12 of the patients with a screening-detected new primary had a personal cancer history, 8 did not (P = .22).
“The proportion and diversity of off–core LFS spectrum cancers detected in TP53 mutation carriers as reported by others give growing evidence of a broader LFS spectrum, in agreement with the permissive role of TP53 mutations,” write Dr. Caron and colleagues, who report having no relevant disclosures. “Our observations seem to support recent moves toward broader cancer screening in TP53 mutation carriers.”
The investigators continue to collect data in LIFSCREEN and plan to undertake main analysis later this year. “Our final analysis will help to determine the benefits and drawbacks (mostly related to false-positive test results) of whole-body MRI in TP53 mutation carrier surveillance,” they conclude. “Studies focused on TP53 mutation penetrance, using methods limiting selection bias, are required to refine cancer risks to improve TP53 mutation carrier management.”
Broader cancer screening of individuals with Li-Fraumeni syndrome (LFS), with or without whole-body magnetic resonance imaging, has a good diagnostic yield and identifies a wide range of cancers, according to a preliminary analysis of the ongoing LIFSCREEN phase 3, randomized, controlled trial.
Investigators led by Olivier Caron, MD, chair of the oncogenetics committee, department of medical oncology, at the Gustave Roussy University Hospital in Villejuif, France, enrolled in the trial 107 individuals from 75 families carrying a TP53 mutation, a genetic aberration commonly present in LFS that confers heightened risk of a variety of malignancies.
Participants had a median age at baseline of 32.9 years, with a range from 5 to 67 years. Fully 98% had a family history of cancer, and 48% had a personal history of cancer.
The participants were assigned to 5 years of standard screening – annual clinical examination, abdomen and pelvis ultrasound, brain MRI, complete blood cell count, and, for women older than 20 years, breast ultrasound and MRI – or intensive screening, entailing the addition of diffusion whole-body MRI.
At the time of the preliminary analysis, 15 patients had undergone only one round of screening; 35, two rounds; 19, three rounds; 24, four rounds; and 7, five rounds, Dr. Caron and associates reported in a research letter (JAMA Oncol. 2017; Aug 3 doi: 10.1001/jamaoncol.2017.1358).
Collectively, this amounted to 226.4 person-years of follow-up.
Screening with either trial strategy (with or without whole-body MRI) led to diagnosis of 23 new primary cancers in 20 patients. Nearly half of the total (12 cancers) were detected at the first round. Patients had a median age of 39.8 at the new cancer diagnosis, with a range from 6 to 70 years.
Of the new cancers, 10 belonged to the core LFS spectrum of breast cancer, sarcoma, and brain tumors. However, the other 13 were outside that spectrum, for example, lung adenocarcinomas, all seen in never or light smokers, and leukemias. Screening also detected three relapses of previous cancers.
Analyses further showed that prior cancer diagnosis was not a reliable marker for risk of new primaries. Although 12 of the patients with a screening-detected new primary had a personal cancer history, 8 did not (P = .22).
“The proportion and diversity of off–core LFS spectrum cancers detected in TP53 mutation carriers as reported by others give growing evidence of a broader LFS spectrum, in agreement with the permissive role of TP53 mutations,” write Dr. Caron and colleagues, who report having no relevant disclosures. “Our observations seem to support recent moves toward broader cancer screening in TP53 mutation carriers.”
The investigators continue to collect data in LIFSCREEN and plan to undertake main analysis later this year. “Our final analysis will help to determine the benefits and drawbacks (mostly related to false-positive test results) of whole-body MRI in TP53 mutation carrier surveillance,” they conclude. “Studies focused on TP53 mutation penetrance, using methods limiting selection bias, are required to refine cancer risks to improve TP53 mutation carrier management.”
FROM JAMA ONCOLOGY
Key clinical point:
Major finding: A total of 23 new primary cancers were diagnosed in 20 patients; more than half were outside the core spectrum of Li-Fraumeni syndrome.
Data source: A preliminary analysis of a phase 3, randomized, controlled trial comparing standard and intensive screening among 107 individuals with Li-Fraumeni syndrome carrying a TP53 mutation (LIFSCREEN trial).
Disclosures: The investigators report having no relevant disclosures. The trial was funded by the French Ligue Contre le Cancer.
AADE: New standards for diabetes self-management programs
New standards for diabetes self-management education and support outline 10 key evidence-based standards for services that meet the Medicare diabetes self-management training regulations, although they do not guarantee coverage. The standards, produced by the American Association of Diabetes Educators in association with the American Diabetes Association, are an update to a similar document produced in 2014. The 2017 revision of the standards is the first to combine support and education to reflect the value of ongoing counsel for improved diabetes self-care, according to an accompanying statement (Diab Care. 2017 Jul 28. doi: 10.2337/dci17-0025).
The new document is full of good recommendations, but they do not cover some important areas. “I don’t disagree with any of them,” said Richard Hellman, MD, clinical professor of medicine at the University of Missouri–Kansas City School of Medicine (UMKC) and associate program director of the UMKC Endocrine Fellowship. But he pointed out that the standards did not include any mention of integrating patient care with other teams. “I think that’s unfortunate. Certainly in our small diabetes practice, we have certified diabetes educators, and all of our patients see them at some point. I hope in subsequent documents they’ll talk about that more,” said Dr. Hellman.

The standards focus on a sort of nuts-and-bolts approach and may be directed toward health care providers who operate in areas with relatively few resources to turn to for help. “It seems it’s answering what to do if you don’t have backup and support, and perhaps that is what it’s for, but the standards should be good in any setting, whether totally integrated or separate. I do think in the future they need to address that large group of people in an integrated setting, and also talk about people with behavioral health expertise. Both are very important,” said Dr. Hellman.
One recommendation he praised in particular was the call for oversight from a quality coordinator. The document calls for the quality coordinator to ensure that the standards are properly implemented, including evidence-based practice, service design, evaluation, and quality improvement. That’s a key consideration because many patients may have disabilities that interfere with comprehension, such as hearing loss or cognitive dysfunction. Such impediments may prevent patients from learning key skills, such as properly checking blood glucose. “That can have a profound effect on diabetes control,” said Dr. Hellman.
He pointed out that quality control can play a wider role in medicine. “Checking your own work isn’t something people always like to do, but it’s really essential. If you think you’re giving high quality care, why don’t you just check your work to see that it’s getting the results that you thought it would?” said Dr. Hellman.
The paper disclosed no sources of funding or conflict of interest information. Dr. Hellman reported having no financial disclosures.
From AADE: 10 standards
Diabetes self-management education and support service providers should:
• Adopt a mission statement and goals.
• Adopt ongoing input from stakeholders and experts to improve quality and participation.
• Analyze the needs of the communities they serve to ensure the best design, delivery method, and use of resources to meet their needs.
• Employ a quality coordinator to oversee services. This individual should be responsible for evidence-based practice, service design, evaluation, and continuous quality improvement.
• Include at least one registered nurse, registered dietitian nutritionist, or pharmacist with training and experience related to DSMES, or a health care professional with a certificate as a diabetes educator (CDE) or Board Certification in Advanced Diabetes Management (BC-ADM).
• Employ a curriculum that follows current evidence and practice guidelines, including a means for evaluating outcomes. The specific elements of the curriculum required will depend on the individual participant’s needs.
• Identify the needs of individual participants and be led by the participant, supported by diabetes self-management education and support team members. They should cooperatively develop an individualized diabetes self-management education and support plan.
• Provide options and resources for ongoing support that participants can choose.
• Monitor participants’ progress toward self-management goals and other outcomes.
• Have their quality control coordinators measure the impact and effectiveness of the diabetes self-management education and support services and determine potential improvements by systematically evaluating process and outcome data.
New standards for diabetes self-management education and support outline 10 key evidence-based standards for services that meet the Medicare diabetes self-management training regulations, although they do not guarantee coverage. The standards, produced by the American Association of Diabetes Educators in association with the American Diabetes Association, are an update to a similar document produced in 2014. The 2017 revision of the standards is the first to combine support and education to reflect the value of ongoing counsel for improved diabetes self-care, according to an accompanying statement (Diab Care. 2017 Jul 28. doi: 10.2337/dci17-0025).
The new document is full of good recommendations, but they do not cover some important areas. “I don’t disagree with any of them,” said Richard Hellman, MD, clinical professor of medicine at the University of Missouri–Kansas City School of Medicine (UMKC) and associate program director of the UMKC Endocrine Fellowship. But he pointed out that the standards did not include any mention of integrating patient care with other teams. “I think that’s unfortunate. Certainly in our small diabetes practice, we have certified diabetes educators, and all of our patients see them at some point. I hope in subsequent documents they’ll talk about that more,” said Dr. Hellman.
The standards focus on a sort of nuts-and-bolts approach and may be directed toward health care providers who operate in areas with relatively few resources to turn to for help. “It seems it’s answering what to do if you don’t have backup and support, and perhaps that is what it’s for, but the standards should be good in any setting, whether totally integrated or separate. I do think in the future they need to address that large group of people in an integrated setting, and also talk about people with behavioral health expertise. Both are very important,” said Dr. Hellman.
One recommendation he praised in particular was the call for oversight from a quality coordinator. The document calls for the quality coordinator to ensure that the standards are properly implemented, including evidence-based practice, service design, evaluation, and quality improvement. That’s a key consideration because many patients may have disabilities that interfere with comprehension, such as hearing loss or cognitive dysfunction. Such impediments may prevent patients from learning key skills, such as properly checking blood glucose. “That can have a profound effect on diabetes control,” said Dr. Hellman.
He pointed out that quality control can play a wider role in medicine. “Checking your own work isn’t something people always like to do, but it’s really essential. If you think you’re giving high quality care, why don’t you just check your work to see that it’s getting the results that you thought it would?” said Dr. Hellman.
The paper disclosed no sources of funding or conflict of interest information. Dr. Hellman reported having no financial disclosures.
From AADE: 10 standards
Diabetes self-management education and support service providers should:
• Adopt a mission statement and goals.
• Adopt ongoing input from stakeholders and experts to improve quality and participation.
• Analyze the needs of the communities they serve to ensure the best design, delivery method, and use of resources to meet their needs.
• Employ a quality coordinator to oversee services. This individual should be responsible for evidence-based practice, service design, evaluation, and continuous quality improvement.
• Include at least one registered nurse, registered dietitian nutritionist, or pharmacist with training and experience related to DSMES, or a health care professional with a certificate as a diabetes educator (CDE) or Board Certification in Advanced Diabetes Management (BC-ADM).
• Employ a curriculum that follows current evidence and practice guidelines, including a means for evaluating outcomes. The specific elements of the curriculum required will depend on the individual participant’s needs.
• Identify the needs of individual participants and be led by the participant, supported by diabetes self-management education and support team members. They should cooperatively develop an individualized diabetes self-management education and support plan.
• Provide options and resources for ongoing support that participants can choose.
• Monitor participants’ progress toward self-management goals and other outcomes.
• Have their quality control coordinators measure the impact and effectiveness of the diabetes self-management education and support services and determine potential improvements by systematically evaluating process and outcome data.
New standards for diabetes self-management education and support outline 10 key evidence-based standards for services that meet the Medicare diabetes self-management training regulations, although they do not guarantee coverage. The standards, produced by the American Association of Diabetes Educators in association with the American Diabetes Association, are an update to a similar document produced in 2014. The 2017 revision of the standards is the first to combine support and education to reflect the value of ongoing counsel for improved diabetes self-care, according to an accompanying statement (Diab Care. 2017 Jul 28. doi: 10.2337/dci17-0025).
The new document is full of good recommendations, but they do not cover some important areas. “I don’t disagree with any of them,” said Richard Hellman, MD, clinical professor of medicine at the University of Missouri–Kansas City School of Medicine (UMKC) and associate program director of the UMKC Endocrine Fellowship. But he pointed out that the standards did not include any mention of integrating patient care with other teams. “I think that’s unfortunate. Certainly in our small diabetes practice, we have certified diabetes educators, and all of our patients see them at some point. I hope in subsequent documents they’ll talk about that more,” said Dr. Hellman.
The standards focus on a sort of nuts-and-bolts approach and may be directed toward health care providers who operate in areas with relatively few resources to turn to for help. “It seems it’s answering what to do if you don’t have backup and support, and perhaps that is what it’s for, but the standards should be good in any setting, whether totally integrated or separate. I do think in the future they need to address that large group of people in an integrated setting, and also talk about people with behavioral health expertise. Both are very important,” said Dr. Hellman.
One recommendation he praised in particular was the call for oversight from a quality coordinator. The document calls for the quality coordinator to ensure that the standards are properly implemented, including evidence-based practice, service design, evaluation, and quality improvement. That’s a key consideration because many patients may have disabilities that interfere with comprehension, such as hearing loss or cognitive dysfunction. Such impediments may prevent patients from learning key skills, such as properly checking blood glucose. “That can have a profound effect on diabetes control,” said Dr. Hellman.
He pointed out that quality control can play a wider role in medicine. “Checking your own work isn’t something people always like to do, but it’s really essential. If you think you’re giving high quality care, why don’t you just check your work to see that it’s getting the results that you thought it would?” said Dr. Hellman.
The paper disclosed no sources of funding or conflict of interest information. Dr. Hellman reported having no financial disclosures.
From AADE: 10 standards
Diabetes self-management education and support service providers should:
• Adopt a mission statement and goals.
• Adopt ongoing input from stakeholders and experts to improve quality and participation.
• Analyze the needs of the communities they serve to ensure the best design, delivery method, and use of resources to meet their needs.
• Employ a quality coordinator to oversee services. This individual should be responsible for evidence-based practice, service design, evaluation, and continuous quality improvement.
• Include at least one registered nurse, registered dietitian nutritionist, or pharmacist with training and experience related to DSMES, or a health care professional with a certificate as a diabetes educator (CDE) or Board Certification in Advanced Diabetes Management (BC-ADM).
• Employ a curriculum that follows current evidence and practice guidelines, including a means for evaluating outcomes. The specific elements of the curriculum required will depend on the individual participant’s needs.
• Identify the needs of individual participants and be led by the participant, supported by diabetes self-management education and support team members. They should cooperatively develop an individualized diabetes self-management education and support plan.
• Provide options and resources for ongoing support that participants can choose.
• Monitor participants’ progress toward self-management goals and other outcomes.
• Have their quality control coordinators measure the impact and effectiveness of the diabetes self-management education and support services and determine potential improvements by systematically evaluating process and outcome data.
FROM DIABETES CARE
Inflammatory bowel disease rate higher among urban residents
Children born in urban areas are more likely to develop inflammatory bowel disease (IBD) when they grow up than are children born in rural areas, a Canadian study showed.
With rising rates of IBD in developing nations and urbanized areas, the investigators interpreted these findings as a positive step toward further understanding, and eventually eliminating, the risk of developing IBD.
The retrospective, population-based study gathered a total of 45,567 IBD patients: 6,662 living in rural residences and 38,905 living in urban residences in Nova Scotia, Ontario, Alberta, and Manitoba, Canada.
Patients in rural areas were on average older than urban patients (average age, 43 years vs. 40 years). Rural patients were also, on average, diagnosed later than were urban patients, with an average age at diagnosis of 42 years, compared with 38 years for urban residents.
The IBD incidence rate among urban patients was 33.16/100,000 (95% CI, 27.24-39.08), compared with 30.72/100,000 (95% CI, 23.81-37.64) among rural residents (Am J Gastroenterol. 2017 Jul 25. doi: 10.1038/ajg.2017.208).
Exposure to these environments while growing up was especially significant, with the lowest rate among children younger than 10 years in rural areas (incidence rate ratio, 0.58; 95% CI, 0.43-0.73), followed by adolescents between 10 and 17.9 years (IRR, 0.72; 95% CI, 0.64-0.81), according to investigators.
The incidence rate of IBD among rural children stayed consistent from birth through age 5 years, which may be evidence that development of IBD later in life is correlated with patients’ time in rural areas, the investigators reported.
Although Dr. Benchimol and his coauthors could not point to the exact reason for these results, they said factors such as diet and early exposure to animals, which may help develop useful bacteria that could help fight IBD development, are possible explanations.
“The mechanism by which rurality protects against IBD is uncertain, and may include dietary and lifestyle factors, environmental exposures, or segregation of individuals with different genetic risk profiles,” the investigators wrote. “These effects may be stronger in children because their gut microbiome is in evolution and may be vulnerable to changes in the first 2 years of life.”
This study was limited by certain classification factors, such as what constitutes an urban or rural area, which may have affected the outcomes. A lack of information on the effects of confounding factors, particularly ethnicity, genotype, phenotype, disease severity, or family history also limited this study, the investigators said.
The Janssen Future Leaders in IBD Program funded the study. Investigators reported receiving financial support from or holding leadership positions in the Canadian Institutes of Health Research, the Canadian Child Health Clinician Scientist program, and the Nova Scotia Health Research Foundation.
[email protected]
On Twitter @eaztweets
Children born in urban areas are more likely to develop inflammatory bowel disease (IBD) when they grow up than are children born in rural areas, a Canadian study showed.
With rising rates of IBD in developing nations and urbanized areas, the investigators interpreted these findings as a positive step toward further understanding, and eventually eliminating, the risk of developing IBD.
The retrospective, population-based study gathered a total of 45,567 IBD patients: 6,662 living in rural residences and 38,905 living in urban residences in Nova Scotia, Ontario, Alberta, and Manitoba, Canada.
Patients in rural areas were on average older than urban patients (average age, 43 years vs. 40 years). Rural patients were also, on average, diagnosed later than were urban patients, with an average age at diagnosis of 42 years, compared with 38 years for urban residents.
The IBD incidence rate among urban patients was 33.16/100,000 (95% CI, 27.24-39.08), compared with 30.72/100,000 (95% CI, 23.81-37.64) among rural residents (Am J Gastroenterol. 2017 Jul 25. doi: 10.1038/ajg.2017.208).
Exposure to these environments while growing up was especially significant, with the lowest rate among children younger than 10 years in rural areas (incidence rate ratio, 0.58; 95% CI, 0.43-0.73), followed by adolescents between 10 and 17.9 years (IRR, 0.72; 95% CI, 0.64-0.81), according to investigators.
The incidence rate of IBD among rural children stayed consistent from birth through age 5 years, which may be evidence that development of IBD later in life is correlated with patients’ time in rural areas, the investigators reported.
Although Dr. Benchimol and his coauthors could not point to the exact reason for these results, they said factors such as diet and early exposure to animals, which may help develop useful bacteria that could help fight IBD development, are possible explanations.
“The mechanism by which rurality protects against IBD is uncertain, and may include dietary and lifestyle factors, environmental exposures, or segregation of individuals with different genetic risk profiles,” the investigators wrote. “These effects may be stronger in children because their gut microbiome is in evolution and may be vulnerable to changes in the first 2 years of life.”
This study was limited by certain classification factors, such as what constitutes an urban or rural area, which may have affected the outcomes. A lack of information on the effects of confounding factors, particularly ethnicity, genotype, phenotype, disease severity, or family history also limited this study, the investigators said.
The Janssen Future Leaders in IBD Program funded the study. Investigators reported receiving financial support from or holding leadership positions in the Canadian Institutes of Health Research, the Canadian Child Health Clinician Scientist program, and the Nova Scotia Health Research Foundation.
[email protected]
On Twitter @eaztweets
Children born in urban areas are more likely to develop inflammatory bowel disease (IBD) when they grow up than are children born in rural areas, a Canadian study showed.
With rising rates of IBD in developing nations and urbanized areas, the investigators interpreted these findings as a positive step toward further understanding, and eventually eliminating, the risk of developing IBD.
The retrospective, population-based study gathered a total of 45,567 IBD patients: 6,662 living in rural residences and 38,905 living in urban residences in Nova Scotia, Ontario, Alberta, and Manitoba, Canada.
Patients in rural areas were on average older than urban patients (average age, 43 years vs. 40 years). Rural patients were also, on average, diagnosed later than were urban patients, with an average age at diagnosis of 42 years, compared with 38 years for urban residents.
The IBD incidence rate among urban patients was 33.16/100,000 (95% CI, 27.24-39.08), compared with 30.72/100,000 (95% CI, 23.81-37.64) among rural residents (Am J Gastroenterol. 2017 Jul 25. doi: 10.1038/ajg.2017.208).
Exposure to these environments while growing up was especially significant, with the lowest rate among children younger than 10 years in rural areas (incidence rate ratio, 0.58; 95% CI, 0.43-0.73), followed by adolescents between 10 and 17.9 years (IRR, 0.72; 95% CI, 0.64-0.81), according to investigators.
The incidence rate of IBD among rural children stayed consistent from birth through age 5 years, which may be evidence that development of IBD later in life is correlated with patients’ time in rural areas, the investigators reported.
Although Dr. Benchimol and his coauthors could not point to the exact reason for these results, they said factors such as diet and early exposure to animals, which may help develop useful bacteria that could help fight IBD development, are possible explanations.
“The mechanism by which rurality protects against IBD is uncertain, and may include dietary and lifestyle factors, environmental exposures, or segregation of individuals with different genetic risk profiles,” the investigators wrote. “These effects may be stronger in children because their gut microbiome is in evolution and may be vulnerable to changes in the first 2 years of life.”
This study was limited by certain classification factors, such as what constitutes an urban or rural area, which may have affected the outcomes. A lack of information on the effects of confounding factors, particularly ethnicity, genotype, phenotype, disease severity, or family history also limited this study, the investigators said.
The Janssen Future Leaders in IBD Program funded the study. Investigators reported receiving financial support from or holding leadership positions in the Canadian Institutes of Health Research, the Canadian Child Health Clinician Scientist program, and the Nova Scotia Health Research Foundation.
[email protected]
On Twitter @eaztweets
FROM THE AMERICAN JOURNAL OF GASTROENTEROLOGY
Key clinical point:
Major finding: The incidence of IBD among urban residents was 33.16/100,000 (95% CI, 27.24-39.08), compared with 30.72/100,000 (95% CI, 23.81-37.64) among rural residents.
Data source: A population-based, retrospective analysis of residents among four Canadian provinces between 1999 and 2010.
Disclosures: The Janssen Future Leaders in IBD Program sponsored the study. Investigators reported receiving financial support from or holding leadership positions in the Canadian Institutes of Health Research, the Canadian Child Health Clinician Scientist program, and the Nova Scotia Health Research Foundation.
Ob.gyns. split on cosmetic genital surgery
Ob.gyns. appear divided on whether it is appropriate to perform genital surgery for cosmetic reasons, according to the results of a recent online reader poll.
In a poll conducted by Ob.Gyn. News, 48.7% of respondents said it is appropriate to perform gynecologic procedures, such as labiaplasty, for cosmetic reasons, while 51.3% said it is not appropriate.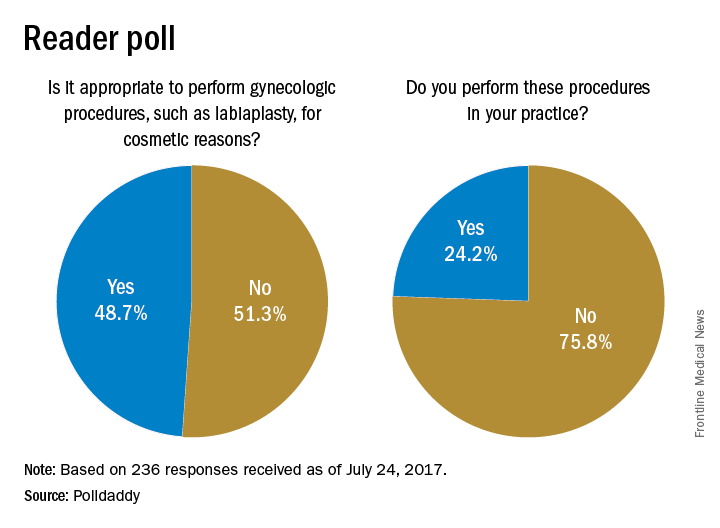
Results are based on 236 reader responses from June 29 to July 24, 2017.
[email protected]
On Twitter @maryellenny
Ob.gyns. appear divided on whether it is appropriate to perform genital surgery for cosmetic reasons, according to the results of a recent online reader poll.
In a poll conducted by Ob.Gyn. News, 48.7% of respondents said it is appropriate to perform gynecologic procedures, such as labiaplasty, for cosmetic reasons, while 51.3% said it is not appropriate.
Results are based on 236 reader responses from June 29 to July 24, 2017.
[email protected]
On Twitter @maryellenny
Ob.gyns. appear divided on whether it is appropriate to perform genital surgery for cosmetic reasons, according to the results of a recent online reader poll.
In a poll conducted by Ob.Gyn. News, 48.7% of respondents said it is appropriate to perform gynecologic procedures, such as labiaplasty, for cosmetic reasons, while 51.3% said it is not appropriate.
Results are based on 236 reader responses from June 29 to July 24, 2017.
[email protected]
On Twitter @maryellenny
Pruritic Eruption on the Chest
The Diagnosis: Grover Disease
Grover disease (also known as transient acantholytic dermatosis) was first described by Ralph W. Grover in 1970 as an idiopathic, acquired, monomorphous, papulovesicular eruption. Although originally characterized by solely transient acantholytic dermatosis, over time the term Grover disease has been expanded to include persistent acantholytic dermatoses. Grover disease chiefly affects white adults older than 40 years and is more prevalent in males than females. Cases generally are self-limited but correlate with age, as older adults are more likely to have prolonged eruptions.1
Grover disease typically erupts with discrete, erythematous, edematous, acneform, red-brown or flesh-colored papules, papulovesicles, or keratotic papules that primarily are seen on the trunk and anterior portion of the chest. As the rash spreads, it can erupt on the neck and thighs. The etiology of Grover disease is unknown, but many factors have been associated with the condition in a limited number of patients, including exposure to UV radiation, excessive heat or sweating, use of sulfadoxine-pyrimethamine and recombinant human IL-4, and infection with Malassezia furfur and Demodex folliculorum.1 Grover disease also has been associated with other conditions such as asteatotic eczema, allergic contact dermatitis, and atopic dermatitis.2
Histologically, Grover disease (Figure 1) is an acantholytic process that can exhibit dyskeratosis (corps ronds and grains). Foci often are small and multiple foci are seen on shave biopsy. There also may be spongiotic changes when associated with an eczematous element. A perivascular lymphohistiocytic infiltrate with eosinophils usually is seen.3 Basket weave keratin may be seen; however, as the lesions cause pruritus, erosions and ulcerations often are present.4
Grover disease has multiple histologic variants that may resemble Darier disease, Hailey-Hailey disease, pemphigus foliaceus, pemphigus vulgaris, and spongiotic dermatitis and can present in combination.5
The variant of Grover disease that has a Darier-like pattern is difficult to distinguish from Darier disease, an autosomal-dominant-inherited disorder classified by small papules that emerge in seborrheic areas during childhood and adolescence. Histologically, Darier disease (Figure 2) shows broad areas of dyskeratosis and acantholysis that lead to suprabasal cleavage. Follicular extension may be present. In addition, there often is prominent vertical parakeratosis in Darier disease.6 Histologic features that favor Darier disease over the Darier-like variant of Grover disease include a broad focus of acanthotic dyskeratosis with follicular extension; the presence of a hyperkeratotic stratum corneum; and a lack of spongiosis and eosinophils, which are notably absent in Darier disease but may be present in Grover disease.4
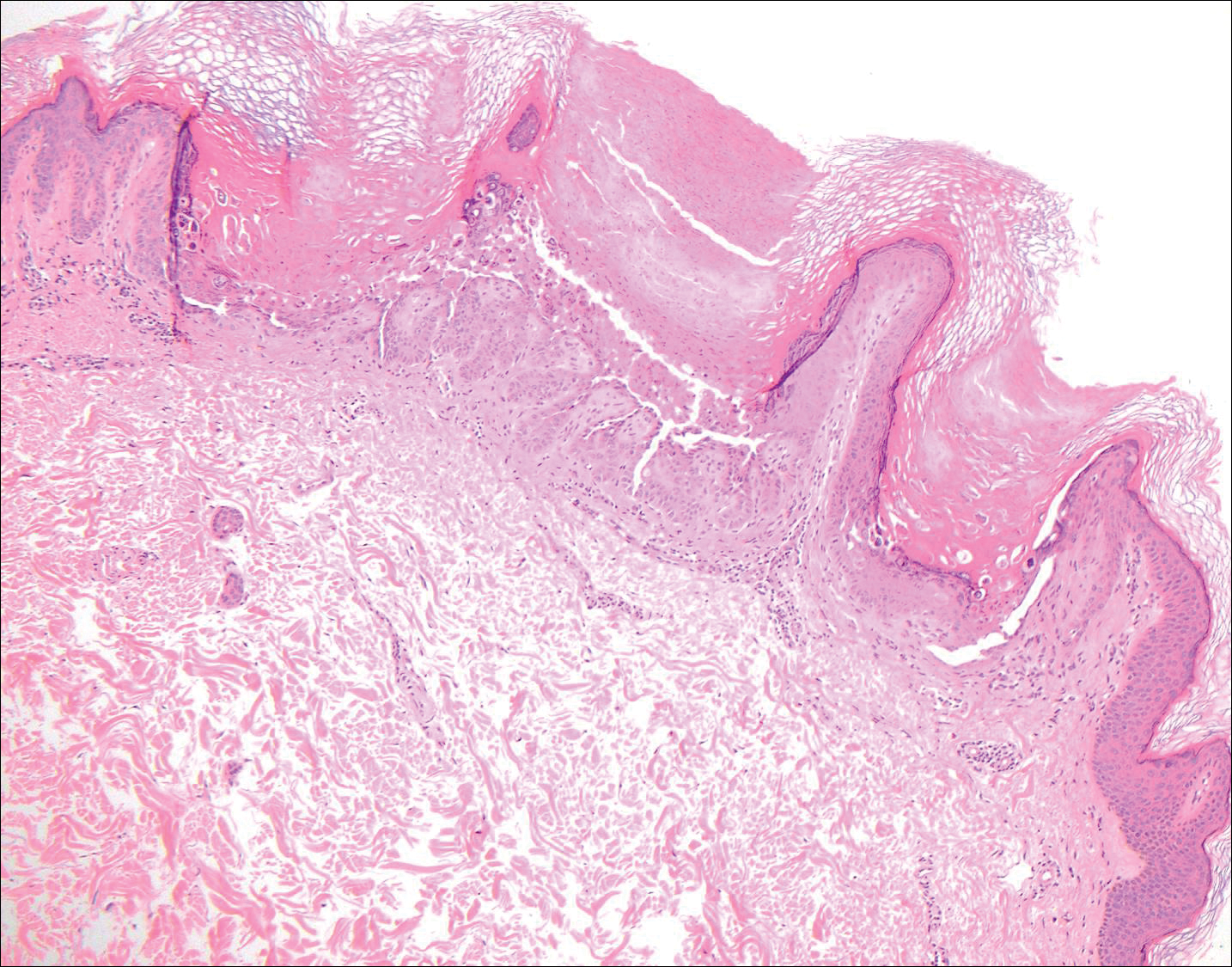
Another variant of Grover disease has a Hailey-Hailey-like pattern, which is characterized by Hailey-Hailey disease's dilapidated brick wall appearance or the diffuse suprabasal acantholysis of all epidermal layers without notable dyskeratosis.4 Hailey-Hailey disease, also known as familial benign pemphigus, is an autosomal-dominant disorder that presents with erythematous vesicular plaques in flexural areas. The plaques progress to flaccid bullae with rupture and crusting and spread peripherally.7 Pathology shows suprabasilar clefts and numerous acantholytic cells (Figure 3). Dyskeratotic keratinocytes are rare with infrequent corps ronds and rare grains. The epidermis also is less hyperplastic in Grover disease than in Hailey-Hailey disease.1
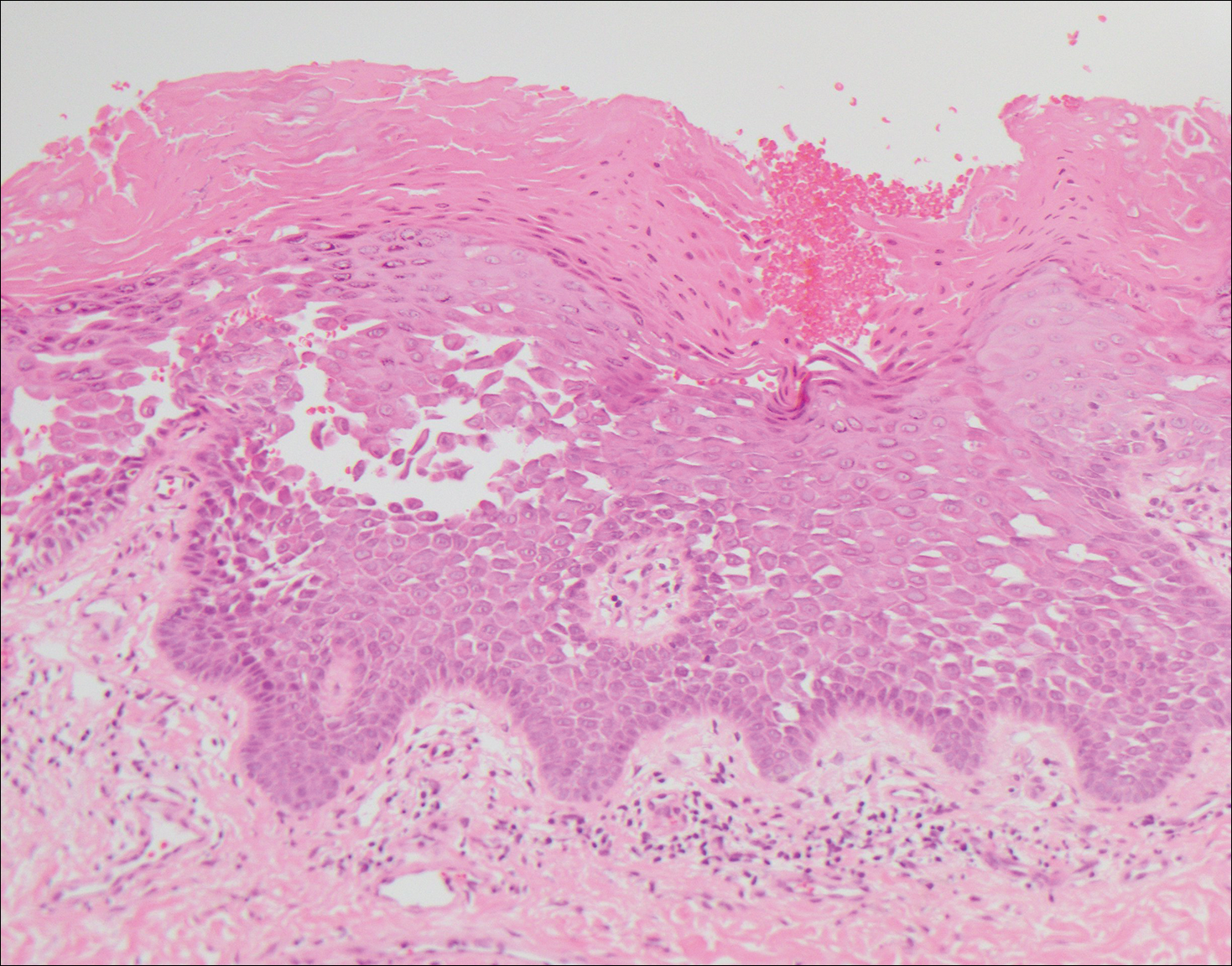
Grover disease also may present histologically with a pemphiguslike pattern, mimicking pemphigus foliaceus and pemphigus vulgaris; however, direct immunofluorescence studies are negative in Grover disease.
Pemphigus foliaceus is an autoimmune disorder caused by autoantibodies to desmoglein 1, which are present on the surfaces of keratinocytes, and is characterized by scaly crusts and blisters.8 Histologically, pemphigus foliaceus (Figure 4) shows a superficial epidermal blistering process. The acantholysis may be subtle and is commonly localized to the stratum granulosum, extending into the stratum corneum. Complete loss of the stratum corneum can be seen, resulting in only scattered acantholytic cells. Spongiosis also may be seen. The dermis shows a perivascular infiltrate that often contains eosinophils. Pemphigus foliaceus is confirmed by direct immunofluorescence.9
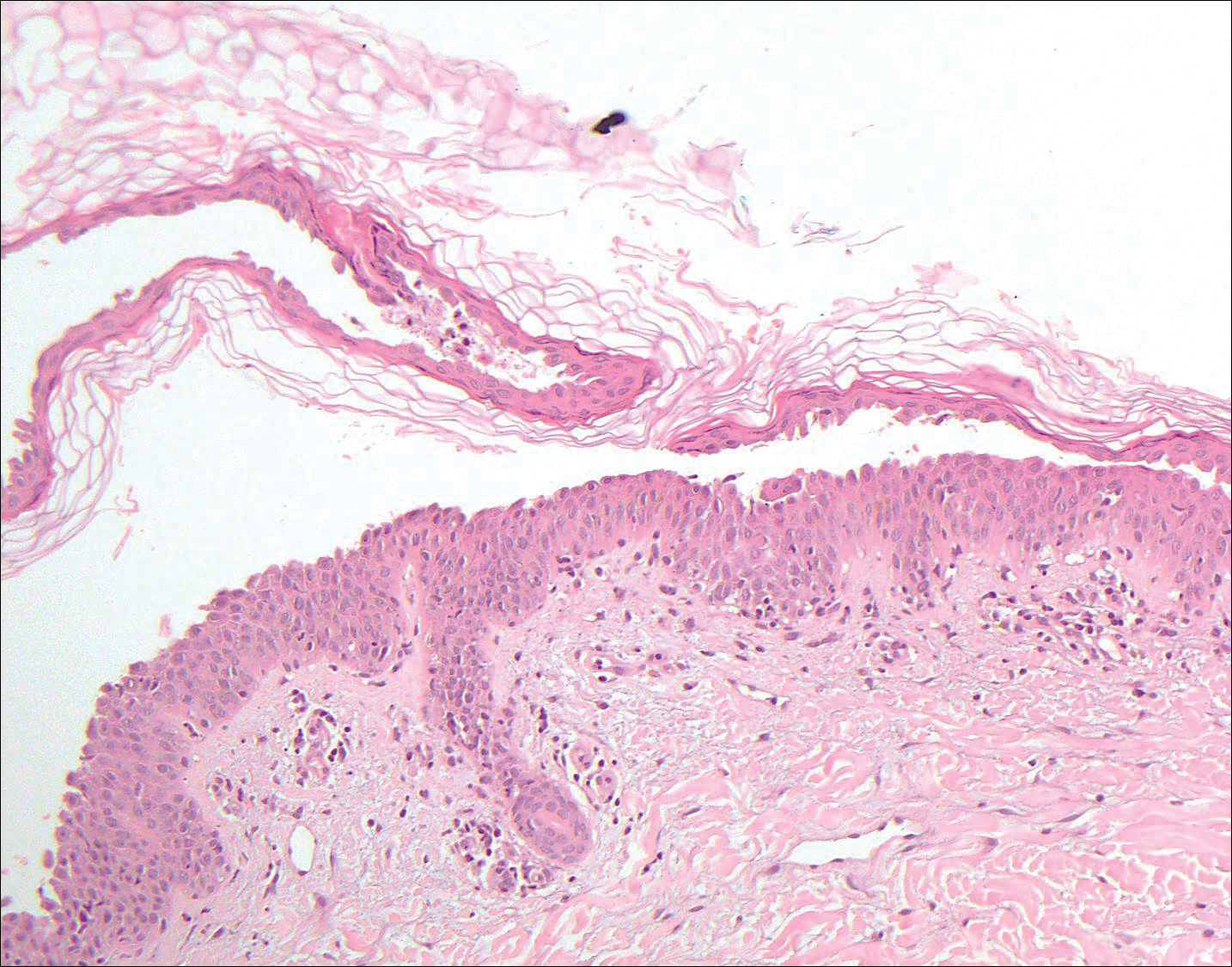
Pemphigus vulgaris is an autoimmune blistering disorder that is characterized by IgG autoantibodies to desmoglein 3, a component of desmosomes that are involved in keratinocyte-to-keratinocyte adhesion. Clinically, patients present with flaccid fragile blisters on the skin and mucous membranes that rupture easily, leading to painful erosions.10 Intraepidermal blisters are seen histologically (Figure 5) with the loss of cohesion (acantholysis) seen classically in the lower portions of the epidermis where desmoglein 3 is most prominent. When only the basal layer remains, the histology has been likened to a tombstone row.11 Extension of the blister along the adnexa is common. The underlying dermis shows a perivascular infiltrate with eosinophils. Early lesions may show only eosinophilic spongiosis. Direct immunofluorescence studies show IgG and C3 in an intercellular pattern that resembles a fish net or chicken wire.4,11
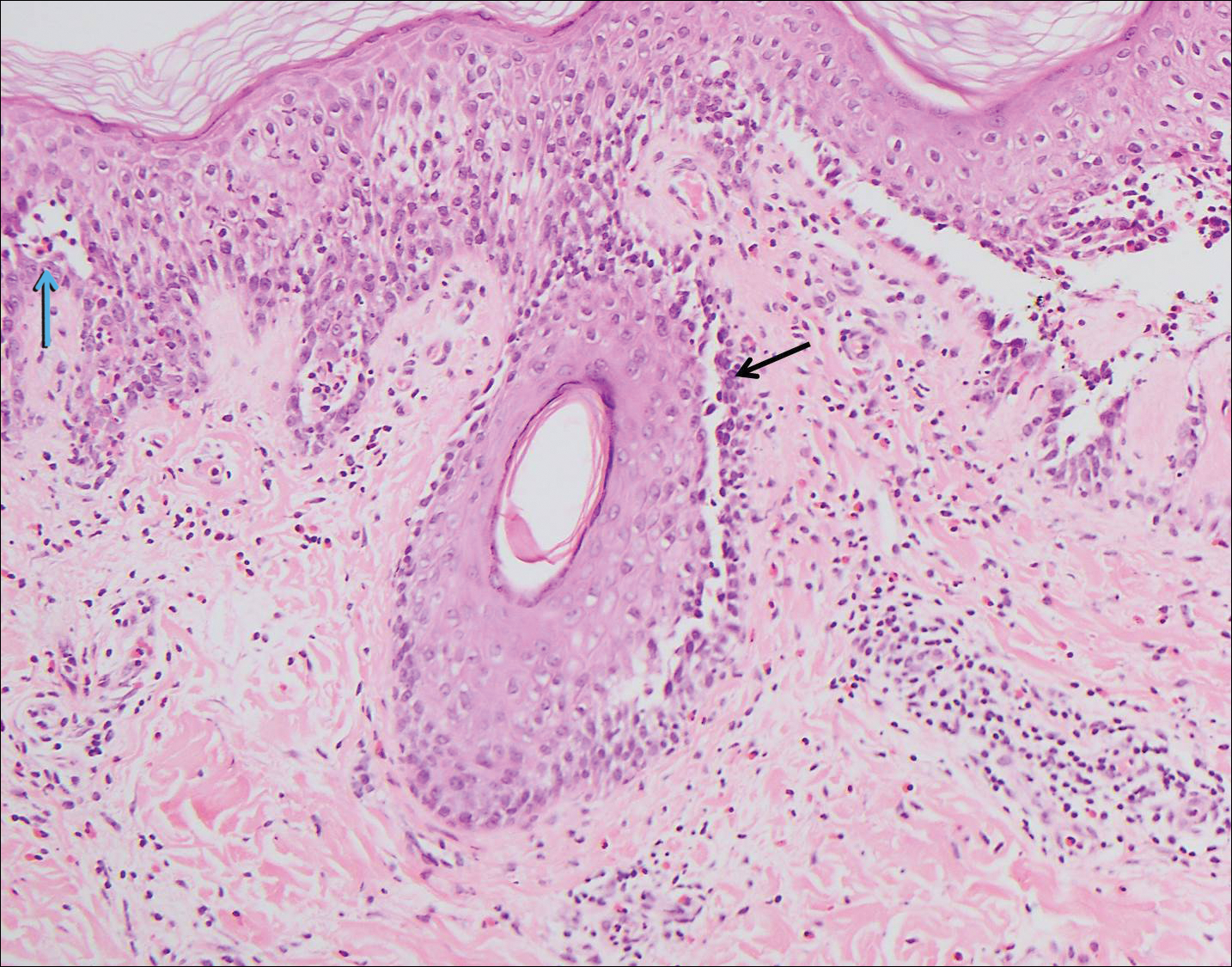
The spongioticlike pattern of Grover disease is marked by epidermal edema with separation of the keratinocytes and the revelation of their intracellular bridges,4 which manifests as vesiculation in the stratum corneum or upper layers of the epidermis.12
Grover disease is self-limited and may spontaneously resolve; however, the disease may be responsive to topical and systemic steroids. Additionally, avoidance of aggravating factors such as sunlight, heat, and sweating can improve symptoms.2
- Parsons JM. Transient acantholytic dermatosis (Grover's disease): a global perspective. J Am Acad Dermatol. 1996;35(5, pt 1):653-666; quiz 667-670.
- Quirk CJ, Heenan PJ. Grover's disease: 34 years on. Australas J Dermatol. 2004;45:83-86.
- Davis MD, Dinneen AM, Landa N, et al. Grover's disease: clinicopathologic review of 72 cases. Mayo Clin Proc. 1999;74:229-234.
- Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133:1490-1494.
- Chalet M, Grover R, Ackerman AB. Transient acantholytic dermatosis: a reevaluation. Arch Dermatol. 1977;133:431-435.
- Takagi A, Kamijo M, Ikeda S. Darier disease. J Dermatol. 2016;43:275-279.
- Engin B, Kutlubay Z, Celik U, et al. Hailey-Hailey disease: a fold (intertriginous) dermatosis. Clin Dermatol. 2015;33:452-455.
- de Sena Nogueira Maehara L, Huizinga J, Jonkman MF. Rituximab therapy in pemphigus foliaceus: report of 12 cases and review of recent literature [published online March 31, 2015]. Br J Dermatol. 2015;172:1420-1423.
- James KA, Culton DA, Diaz LA. Diagnosis and clinical features of pemphigus foliaceus. Dermatol Clin. 2011;29:405-412.
- Black M, Mignogna MD, Scully C. Number II. pemphigus vulgaris. Oral Dis. 2005;11:119-130.
- Madke B, Doshi B, Khopkar U, et al. Appearances in dermatopathology: the diagnostic and the deceptive. Indian J Dermatol Venerol Leprol. 2013;79:338-348.
- Motaparthi K. Pseudoherpetic transient acantholytic dermatosis (Grover disease): case series and review of the literature [published online February 16, 2017]. J Cutan Pathol. 2017;44:486-489.
The Diagnosis: Grover Disease
Grover disease (also known as transient acantholytic dermatosis) was first described by Ralph W. Grover in 1970 as an idiopathic, acquired, monomorphous, papulovesicular eruption. Although originally characterized by solely transient acantholytic dermatosis, over time the term Grover disease has been expanded to include persistent acantholytic dermatoses. Grover disease chiefly affects white adults older than 40 years and is more prevalent in males than females. Cases generally are self-limited but correlate with age, as older adults are more likely to have prolonged eruptions.1
Grover disease typically erupts with discrete, erythematous, edematous, acneform, red-brown or flesh-colored papules, papulovesicles, or keratotic papules that primarily are seen on the trunk and anterior portion of the chest. As the rash spreads, it can erupt on the neck and thighs. The etiology of Grover disease is unknown, but many factors have been associated with the condition in a limited number of patients, including exposure to UV radiation, excessive heat or sweating, use of sulfadoxine-pyrimethamine and recombinant human IL-4, and infection with Malassezia furfur and Demodex folliculorum.1 Grover disease also has been associated with other conditions such as asteatotic eczema, allergic contact dermatitis, and atopic dermatitis.2
Histologically, Grover disease (Figure 1) is an acantholytic process that can exhibit dyskeratosis (corps ronds and grains). Foci often are small and multiple foci are seen on shave biopsy. There also may be spongiotic changes when associated with an eczematous element. A perivascular lymphohistiocytic infiltrate with eosinophils usually is seen.3 Basket weave keratin may be seen; however, as the lesions cause pruritus, erosions and ulcerations often are present.4
Grover disease has multiple histologic variants that may resemble Darier disease, Hailey-Hailey disease, pemphigus foliaceus, pemphigus vulgaris, and spongiotic dermatitis and can present in combination.5
The variant of Grover disease that has a Darier-like pattern is difficult to distinguish from Darier disease, an autosomal-dominant-inherited disorder classified by small papules that emerge in seborrheic areas during childhood and adolescence. Histologically, Darier disease (Figure 2) shows broad areas of dyskeratosis and acantholysis that lead to suprabasal cleavage. Follicular extension may be present. In addition, there often is prominent vertical parakeratosis in Darier disease.6 Histologic features that favor Darier disease over the Darier-like variant of Grover disease include a broad focus of acanthotic dyskeratosis with follicular extension; the presence of a hyperkeratotic stratum corneum; and a lack of spongiosis and eosinophils, which are notably absent in Darier disease but may be present in Grover disease.4
Another variant of Grover disease has a Hailey-Hailey-like pattern, which is characterized by Hailey-Hailey disease's dilapidated brick wall appearance or the diffuse suprabasal acantholysis of all epidermal layers without notable dyskeratosis.4 Hailey-Hailey disease, also known as familial benign pemphigus, is an autosomal-dominant disorder that presents with erythematous vesicular plaques in flexural areas. The plaques progress to flaccid bullae with rupture and crusting and spread peripherally.7 Pathology shows suprabasilar clefts and numerous acantholytic cells (Figure 3). Dyskeratotic keratinocytes are rare with infrequent corps ronds and rare grains. The epidermis also is less hyperplastic in Grover disease than in Hailey-Hailey disease.1
Grover disease also may present histologically with a pemphiguslike pattern, mimicking pemphigus foliaceus and pemphigus vulgaris; however, direct immunofluorescence studies are negative in Grover disease.
Pemphigus foliaceus is an autoimmune disorder caused by autoantibodies to desmoglein 1, which are present on the surfaces of keratinocytes, and is characterized by scaly crusts and blisters.8 Histologically, pemphigus foliaceus (Figure 4) shows a superficial epidermal blistering process. The acantholysis may be subtle and is commonly localized to the stratum granulosum, extending into the stratum corneum. Complete loss of the stratum corneum can be seen, resulting in only scattered acantholytic cells. Spongiosis also may be seen. The dermis shows a perivascular infiltrate that often contains eosinophils. Pemphigus foliaceus is confirmed by direct immunofluorescence.9
Pemphigus vulgaris is an autoimmune blistering disorder that is characterized by IgG autoantibodies to desmoglein 3, a component of desmosomes that are involved in keratinocyte-to-keratinocyte adhesion. Clinically, patients present with flaccid fragile blisters on the skin and mucous membranes that rupture easily, leading to painful erosions.10 Intraepidermal blisters are seen histologically (Figure 5) with the loss of cohesion (acantholysis) seen classically in the lower portions of the epidermis where desmoglein 3 is most prominent. When only the basal layer remains, the histology has been likened to a tombstone row.11 Extension of the blister along the adnexa is common. The underlying dermis shows a perivascular infiltrate with eosinophils. Early lesions may show only eosinophilic spongiosis. Direct immunofluorescence studies show IgG and C3 in an intercellular pattern that resembles a fish net or chicken wire.4,11
The spongioticlike pattern of Grover disease is marked by epidermal edema with separation of the keratinocytes and the revelation of their intracellular bridges,4 which manifests as vesiculation in the stratum corneum or upper layers of the epidermis.12
Grover disease is self-limited and may spontaneously resolve; however, the disease may be responsive to topical and systemic steroids. Additionally, avoidance of aggravating factors such as sunlight, heat, and sweating can improve symptoms.2
The Diagnosis: Grover Disease
Grover disease (also known as transient acantholytic dermatosis) was first described by Ralph W. Grover in 1970 as an idiopathic, acquired, monomorphous, papulovesicular eruption. Although originally characterized by solely transient acantholytic dermatosis, over time the term Grover disease has been expanded to include persistent acantholytic dermatoses. Grover disease chiefly affects white adults older than 40 years and is more prevalent in males than females. Cases generally are self-limited but correlate with age, as older adults are more likely to have prolonged eruptions.1
Grover disease typically erupts with discrete, erythematous, edematous, acneform, red-brown or flesh-colored papules, papulovesicles, or keratotic papules that primarily are seen on the trunk and anterior portion of the chest. As the rash spreads, it can erupt on the neck and thighs. The etiology of Grover disease is unknown, but many factors have been associated with the condition in a limited number of patients, including exposure to UV radiation, excessive heat or sweating, use of sulfadoxine-pyrimethamine and recombinant human IL-4, and infection with Malassezia furfur and Demodex folliculorum.1 Grover disease also has been associated with other conditions such as asteatotic eczema, allergic contact dermatitis, and atopic dermatitis.2
Histologically, Grover disease (Figure 1) is an acantholytic process that can exhibit dyskeratosis (corps ronds and grains). Foci often are small and multiple foci are seen on shave biopsy. There also may be spongiotic changes when associated with an eczematous element. A perivascular lymphohistiocytic infiltrate with eosinophils usually is seen.3 Basket weave keratin may be seen; however, as the lesions cause pruritus, erosions and ulcerations often are present.4
Grover disease has multiple histologic variants that may resemble Darier disease, Hailey-Hailey disease, pemphigus foliaceus, pemphigus vulgaris, and spongiotic dermatitis and can present in combination.5
The variant of Grover disease that has a Darier-like pattern is difficult to distinguish from Darier disease, an autosomal-dominant-inherited disorder classified by small papules that emerge in seborrheic areas during childhood and adolescence. Histologically, Darier disease (Figure 2) shows broad areas of dyskeratosis and acantholysis that lead to suprabasal cleavage. Follicular extension may be present. In addition, there often is prominent vertical parakeratosis in Darier disease.6 Histologic features that favor Darier disease over the Darier-like variant of Grover disease include a broad focus of acanthotic dyskeratosis with follicular extension; the presence of a hyperkeratotic stratum corneum; and a lack of spongiosis and eosinophils, which are notably absent in Darier disease but may be present in Grover disease.4
Another variant of Grover disease has a Hailey-Hailey-like pattern, which is characterized by Hailey-Hailey disease's dilapidated brick wall appearance or the diffuse suprabasal acantholysis of all epidermal layers without notable dyskeratosis.4 Hailey-Hailey disease, also known as familial benign pemphigus, is an autosomal-dominant disorder that presents with erythematous vesicular plaques in flexural areas. The plaques progress to flaccid bullae with rupture and crusting and spread peripherally.7 Pathology shows suprabasilar clefts and numerous acantholytic cells (Figure 3). Dyskeratotic keratinocytes are rare with infrequent corps ronds and rare grains. The epidermis also is less hyperplastic in Grover disease than in Hailey-Hailey disease.1
Grover disease also may present histologically with a pemphiguslike pattern, mimicking pemphigus foliaceus and pemphigus vulgaris; however, direct immunofluorescence studies are negative in Grover disease.
Pemphigus foliaceus is an autoimmune disorder caused by autoantibodies to desmoglein 1, which are present on the surfaces of keratinocytes, and is characterized by scaly crusts and blisters.8 Histologically, pemphigus foliaceus (Figure 4) shows a superficial epidermal blistering process. The acantholysis may be subtle and is commonly localized to the stratum granulosum, extending into the stratum corneum. Complete loss of the stratum corneum can be seen, resulting in only scattered acantholytic cells. Spongiosis also may be seen. The dermis shows a perivascular infiltrate that often contains eosinophils. Pemphigus foliaceus is confirmed by direct immunofluorescence.9
Pemphigus vulgaris is an autoimmune blistering disorder that is characterized by IgG autoantibodies to desmoglein 3, a component of desmosomes that are involved in keratinocyte-to-keratinocyte adhesion. Clinically, patients present with flaccid fragile blisters on the skin and mucous membranes that rupture easily, leading to painful erosions.10 Intraepidermal blisters are seen histologically (Figure 5) with the loss of cohesion (acantholysis) seen classically in the lower portions of the epidermis where desmoglein 3 is most prominent. When only the basal layer remains, the histology has been likened to a tombstone row.11 Extension of the blister along the adnexa is common. The underlying dermis shows a perivascular infiltrate with eosinophils. Early lesions may show only eosinophilic spongiosis. Direct immunofluorescence studies show IgG and C3 in an intercellular pattern that resembles a fish net or chicken wire.4,11
The spongioticlike pattern of Grover disease is marked by epidermal edema with separation of the keratinocytes and the revelation of their intracellular bridges,4 which manifests as vesiculation in the stratum corneum or upper layers of the epidermis.12
Grover disease is self-limited and may spontaneously resolve; however, the disease may be responsive to topical and systemic steroids. Additionally, avoidance of aggravating factors such as sunlight, heat, and sweating can improve symptoms.2
- Parsons JM. Transient acantholytic dermatosis (Grover's disease): a global perspective. J Am Acad Dermatol. 1996;35(5, pt 1):653-666; quiz 667-670.
- Quirk CJ, Heenan PJ. Grover's disease: 34 years on. Australas J Dermatol. 2004;45:83-86.
- Davis MD, Dinneen AM, Landa N, et al. Grover's disease: clinicopathologic review of 72 cases. Mayo Clin Proc. 1999;74:229-234.
- Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133:1490-1494.
- Chalet M, Grover R, Ackerman AB. Transient acantholytic dermatosis: a reevaluation. Arch Dermatol. 1977;133:431-435.
- Takagi A, Kamijo M, Ikeda S. Darier disease. J Dermatol. 2016;43:275-279.
- Engin B, Kutlubay Z, Celik U, et al. Hailey-Hailey disease: a fold (intertriginous) dermatosis. Clin Dermatol. 2015;33:452-455.
- de Sena Nogueira Maehara L, Huizinga J, Jonkman MF. Rituximab therapy in pemphigus foliaceus: report of 12 cases and review of recent literature [published online March 31, 2015]. Br J Dermatol. 2015;172:1420-1423.
- James KA, Culton DA, Diaz LA. Diagnosis and clinical features of pemphigus foliaceus. Dermatol Clin. 2011;29:405-412.
- Black M, Mignogna MD, Scully C. Number II. pemphigus vulgaris. Oral Dis. 2005;11:119-130.
- Madke B, Doshi B, Khopkar U, et al. Appearances in dermatopathology: the diagnostic and the deceptive. Indian J Dermatol Venerol Leprol. 2013;79:338-348.
- Motaparthi K. Pseudoherpetic transient acantholytic dermatosis (Grover disease): case series and review of the literature [published online February 16, 2017]. J Cutan Pathol. 2017;44:486-489.
- Parsons JM. Transient acantholytic dermatosis (Grover's disease): a global perspective. J Am Acad Dermatol. 1996;35(5, pt 1):653-666; quiz 667-670.
- Quirk CJ, Heenan PJ. Grover's disease: 34 years on. Australas J Dermatol. 2004;45:83-86.
- Davis MD, Dinneen AM, Landa N, et al. Grover's disease: clinicopathologic review of 72 cases. Mayo Clin Proc. 1999;74:229-234.
- Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133:1490-1494.
- Chalet M, Grover R, Ackerman AB. Transient acantholytic dermatosis: a reevaluation. Arch Dermatol. 1977;133:431-435.
- Takagi A, Kamijo M, Ikeda S. Darier disease. J Dermatol. 2016;43:275-279.
- Engin B, Kutlubay Z, Celik U, et al. Hailey-Hailey disease: a fold (intertriginous) dermatosis. Clin Dermatol. 2015;33:452-455.
- de Sena Nogueira Maehara L, Huizinga J, Jonkman MF. Rituximab therapy in pemphigus foliaceus: report of 12 cases and review of recent literature [published online March 31, 2015]. Br J Dermatol. 2015;172:1420-1423.
- James KA, Culton DA, Diaz LA. Diagnosis and clinical features of pemphigus foliaceus. Dermatol Clin. 2011;29:405-412.
- Black M, Mignogna MD, Scully C. Number II. pemphigus vulgaris. Oral Dis. 2005;11:119-130.
- Madke B, Doshi B, Khopkar U, et al. Appearances in dermatopathology: the diagnostic and the deceptive. Indian J Dermatol Venerol Leprol. 2013;79:338-348.
- Motaparthi K. Pseudoherpetic transient acantholytic dermatosis (Grover disease): case series and review of the literature [published online February 16, 2017]. J Cutan Pathol. 2017;44:486-489.
A 55-year-old man presented with small, erythematous, nonfollicular, pruritic papules on the mid chest.