User login
The Changing Landscape of Trauma Care, Part 2
Introduction
For decades, virtually all injury was treated with open operative surgery. Resuscitation was based on the belief that large-volume crystalloid infusion to raise blood pressure (BP) to normal was the optimal therapy. Advanced trauma life support teaching was that 2 L of crystalloid fluid should be the initial resuscitation for all trauma patients, and those who failed to respond should receive additional crystalloid fluid. Patients did not receive a blood transfusion until later in treatment, and fresh frozen plasma (FFP) and platelets were not given until 10 U of blood had been administered. Regardless of the fluid infused, the goal of initial resuscitation was to raise BP to a normal level. During the time I (TS) was chair of the emergency medicine department at the State University of New York’s Kings County Hospital, I remember administering liters of crystalloid fluid preoperatively, believing it was not safe to operate until the patient had been what we termed “adequately resuscitated.”
However, as early as 1918, Walter B. Cannon, MD, correctly observed that fluid therapy without hemostasis was not wise, and numerous animal studies since then also raised serious questions about this approach. This article points out the revolutionary changes in the thinking and practice of resuscitation that have occurred in the last 20 years. We now realize that raising BP to normal only perpetuates hemorrhage. Hypotension treated with additional volume resuscitation without surgical control of hemorrhage creates a cycle leading to dilution of clotting factors and red blood cells (RBCs), recurrent hypotension, and ultimately death.
The realization that early blood transfusion is probably the wisest course is a concept that has only been in clinical practice for less than 15 years. Major trauma centers now routinely keep type O negative blood in the ED refrigerators so that it is instantly available.
Our understanding of trauma coagulopathy also has changed dramatically. Once thought to be simply a consequence of hypotension and hypothermia, we now realize that coagulopathy following trauma is far more complicated and likely occurs in concert with the inflammatory response to serious injury. Regardless of its etiology, we have recognized that earlier administration of plasma and platelets following trauma prevents coagulopathy, and this approach is more beneficial than treating coagulopathy after it develops. There has been much debate about the optimal ratios of RBCs, plasma, and platelets, and the ideal ratio has yet to be determined. The idea that “one-size-fits-all” is almost certainly not the case: Different patients require different and more precise treatment strategies.
For years, we have relied on laboratory measurements of coagulation to guide transfusion therapy, but standard laboratory values often take over 30 minutes to obtain. In an extremely dynamic situation involving large-volume blood loss, this interval is too lax. A more personalized approach using rapidly available technology, such as thromboelastography (TEG), allows for real-time assessment of a multiplicity of coagulation dynamics and rapid correction of any abnormalities. Procoagulants such as factor 7A, prothrombin complex concentrate (PCC), and tranexamic acid (TXA) have a role. However, the data to support the use of these expensive agents is lacking. While they certainly can be life-saving, each of these components brings with it a risk of causing indiscriminate coagulation—even in areas of the body that are not injured. Moreover, their availability in nontrauma centers is either limited or not an option.
There is little question that our rapid advances in understanding resuscitation and transfusion practice has saved lives. Twenty years ago, intensive care units were populated by trauma patients who had received many liters of crystalloid fluid, and at least partly a consequence of the resuscitation experience, many had severe respiratory failure. Open abdomens were common and also a likely consequence of large-volume crystalloid use. While these problems have not entirely disappeared, they now occur much less frequently.
Standardizing trauma care has also helped enhance patient care a great deal. Most major trauma centers have a “massive transfusion” protocol which allows the blood bank to prepare coolers containing not only blood, but also plasma, platelets, and procoagulants. This practice obviates the need to order the components individually. Rapid access to technology such as TEG allows emergency physicians (EPs) and other trauma care professionals to precisely guide transfusion therapy, but this remains an area of intensely debated investigation. Hopefully, our understanding will continue to mature over the next few years.
Another area of trauma care that has rapidly evolved is the use of endovascular techniques for trauma hemostasis. The realization that we can obtain control of vascular injury without the need for a large open operation has revolutionized care. While endovascular techniques have been used for pelvic hemostasis since 1972, we now use it regularly in every body cavity. Splenic artery embolization was developed by our (TS) group in Brooklyn, New York in 1995, and its use has now expanded to other abdominal solid organ injuries.
Injuries to the thoracic aorta once required a thoracotomy, cardiopulmonary bypass, and open repair. Stent grafting is now the treatment of choice for these injuries, allowing for a minimally invasive solution, and permitting those with both aortic and many other injuries to receive care for all of these wounds much sooner than was possible in the past, when multiply injured patients were simply not considered candidates for early open repair.
Thoracotomy in the ED has been widely practiced for a variety of indications. While it is still the only available solution for injury to the heart and/or proximal pulmonary vasculature in a patient who is hemodynamically unstable and/or in extremis, other options now exist to obtain vascular inflow for patients bleeding in the abdomen or pelvis. The use of transfemoral balloons for aortic occlusion allows clinicians to temporize hemorrhage without a huge open operation, and resuscitative endovascular balloon occlusion of the aorta (REBOA), has only been available for the last several years. The exact indications, wisest strategy, length of time the balloon can be inflated, rate of complications, and who is the appropriate physician (eg, EP, intensivist, vascular surgeon) to insert it, all remain questions requiring resolution. Much more work is necessary to pursue the role that REBOA can have in the care of desperately injured trauma patients.
There has been a revolution in the care of severely injured patients. After 50 years of thinking that we knew the answers, we have come to realize that those answers were wrong. Newer resuscitation strategies, as well as new treatment strategies continue to evolve, allowing us to refine care of severely injured patients. Perhaps the one thing we have really learned is that we do not have all of the answers and that the discussion must continue if we are to do better at serving more trauma victims.
Damage Control Resuscitation
In the United States, trauma is the leading cause of death in patients younger than age 45 years and ranks as the fifth leading cause of death among all age groups. Hemorrhage remains the leading cause of preventable death in the trauma population,1 and one of the most important recent changes in our care of the injured patient is the manner in which we manage hemorrhage. As noted earlier, there has been a paradigm shift away from large-volume crystalloid resuscitation and toward what has been termed “damage control resuscitation” (DCR).2,3
The principles of the DCR strategy are aimed at preemptively treating the lethal triad of hypothermia, acidosis, and coagulopathy in conjunction with control of surgical bleeding using damage control surgery. The main principles of DCR include “permissive hypotension,” prevention of heat loss and/or active warming, minimizing the use of crystalloid infusions, and initiating resuscitation with blood products in a ratio that more closely resembles whole blood.2
Permissive Hypotension
Permissive hypotension, also referred to as hypotensive resuscitation, is not considered a goal or an endpoint, but rather a “bridge” to definitive surgical control of hemorrhage. The body’s initial response to injury involves vasoconstriction and early clot formation, a process facilitated by hypotension. The rationale for permissive hypotension is that attempting to drive the BP up to normal ranges may interfere with vasoconstriction, as well as physically disrupting this early clot, leading to increased bleeding and further hypotension.
This concept has been corroborated by many animal and human studies.3 In 1994, the landmark study by Bickell et al4 randomized patients with penetrating torso trauma and a systolic BP (SBP) of 90 mm Hg or lower to either immediate or delayed fluid resuscitation. Their study demonstrated that patients whose fluid resuscitation was delayed until they reached the operating room had improved outcomes. The study supported the long-time prehospital practice of the “scoop-and-run” strategy, especially in penetrating torso trauma.
In 2003, Sondeen et al5 used a swine model of aortic injury to find an inflection point for clot disruption and re-bleeding during volume resuscitation. They found the inflection point to be a mean arterial pressure (MAP) of 64 mm Hg and an SBP of 94 mm Hg, regardless of the size of the aortotomy. Using an animal model of hemorrhagic shock, Li et al6 demonstrated in 2011 that resuscitation to a MAP of 50 mm Hg was associated with a decreased amount of blood loss as well as with improved survival compared to patients who were resuscitated to a MAP of 80 mm Hg. However, they also showed that after a time period of more than 90 to 120 minutes, the lower MAP group had increased end organ damage and worse outcomes, emphasizing the importance of prompt surgical control of bleeding—regardless of preoperative resuscitation strategy.
Other studies, though, have not shown a clear benefit to permissive hypotension. A 2002 study by Dutton et al7 showed that titration of initial fluid to a lower SBP (70 mm Hg) did not affect mortality when compared to a target resuscitation MAP of more than 100 mm Hg. Further, in 2014, a plenary paper presented to the American Association for the Surgery of Trauma demonstrated that controlled resuscitation (CR) strategy was safe and feasible,8 but did not demonstrate a mortality benefit in the overall cohort, though patients with blunt trauma who received CR had improved survival at 24 hours.
The group at Ben Taub General Hospital in Houston, Texas recently performed a randomized controlled trial evaluating intraoperative hypotensive resuscitation strategies. Patients in hemorrhagic shock were randomized to either an intraoperative MAP goal of 50 mm Hg or 65 mm Hg.9,10 Preliminary results suggested that targeting a lower MAP resulted in fewer blood product transfusions, less fluid administration, less coagulopathy, and lower mortality in the early postoperative period. Additionally, they demonstrated a nonsignificant trend toward improved 30-day mortality in the lower MAP group.9 Moreover, in this study there was no increased morbidity associated with the hypotensive strategy,10 suggesting that the approach was safe. Unfortunately, the trial was stopped early due to slow enrollment.
Despite the overall promising results with permissive hypotension, it is important to remember that it is contraindicated in patients with known or suspected traumatic brain injury, as hypotension has been shown to be detrimental in this population.11
Hemostatic Resuscitation and Coagulopathy
Avoiding Aggressive Crystalloid Resuscitation. While the ideal MAP to target during DCR remains unclear, the potential harm caused by aggressive crystalloid resuscitation has become more evident. Infusing excessive amounts of crystalloid has been shown to be associated with increased ventilator days, multisystem organ failure, abdominal compartment syndrome, and surgical-site infections12—all of which have also been associated with systemic consequences of increased inflammation, including increased release of tumor necrosis factor-alpha and other proinflammatory cytokines.13
Rodent studies have demonstrated large-volume crystalloid administration and breakdown or “thinning” of the endothelial glycocalyx, which leads to increased capillary leak, third-spacing, and ultimately intravascular volume depletion.14,15 This mechanism has been linked to pulmonary complications, namely acute lung injury and acute respiratory distress syndrome. Enteric edema resulting from aggressive crystalloid resuscitation has also been associated with prolonged postoperative ileus, increased risk of anastomotic leak,13 and inability to achieve primary fascial closure.16 All of the aforementioned complications are reduced when employing a restrictive fluid resuscitation strategy.17
Aggressive crystalloid administration in hemorrhagic shock also leads to dilutional coagulopathy. Multiple animal and human studies have shown an association between increased crystalloid volumes in hemorrhaging patients and increasing coagulopathy, blood loss, and mortality. In 2004, Barak et al18 demonstrated that administration of a high volume of crystalloid fluid (>3 L) or colloid (500 mL) was associated with postoperative coagulopathy; whereas in 2017, Harada et al,19 at Cedars-Sinai Medical Center in New York, demonstrated over a 10-year period that decreased high-volume (>2 L) crystalloid resuscitation paralleled a decrease in mortality.
Massive Transfusion Protocols. Many trauma centers have shifted away from high-volume crystalloid resuscitation in favor of massive transfusion protocols (MTPs) utilizing standardized ratios that more closely mimic whole blood. The MTPs center on the principle of equal transfusion ratios of blood product as opposed to packed RBCs (PRBCs) alone. This means effecting a plasma-rich resuscitation and preemptive correction of coagulopathy with FFP and platelets in addition to PRBCs.
Data from a US Army combat support hospital have demonstrated improved survival with an FFP to PRBC ratio of more than 1:1.4,20 and civilian studies have produced similar findings.21-23 All of these studies also noted improved mortality with higher (>1:2) platelet to PRBC ratios.22,23 Although, the ideal ratio remains unknown, many MTPs aim for 1:1:1 ratio (6 U FFP to 6 packed platelets to 6 U PRBC), which most closely mimics whole blood.
The Pragmatic Randomized Optimal Platelet and Plasma Ratios trial was a recent large multicenter randomized trial that compared transfusion ratios of 1:1:1 and 1:1:2. The trial was unable to demonstrate a difference in mortality at either 24 hours or 30 days, though more patients in the 1:1:1 ratio group achieved hemostasis and fewer patients in this group died from exsanguination in the first 24 hours.24Prehospital PRBC Administration. A number of studies have looked at prehospital administration of PRBCs.25-27 Holcomb et al25 showed no overall survival advantage at 24 hours, but did demonstrate a negligible blood-product wastage. In 2015 Brown et al26 found an increase probability of 24-hour survival, decreased shock, and lower 24-hour PRBC requirements with pretrauma-center PRBC transfusion. That same year Brown et al27 demonstrated that prehospital PRBC transfusion in severely injured blunt trauma patients was associated with decreased 24-hour and 30-day mortality rates, and a lower risk of coagulopathy. Currently, the Prehospital Air Medical Plasma trial is enrolling patients to evaluate the prehospital administration of plasma.28 The primary endpoint of the study is 30-day mortality; the tentative completion date for the study is January 2018.
Tranexamic Acid. Another important development in the treatment of hemorrhagic shock in recent years has been the use of TXA, an antifibrinolytic agent which inhibits the conversion of plasminogen to plasmin. It has been shown to decrease mortality in both civilian and military trauma populations.29,30
The Clinical Randomization of an Antifibrinolytic in Significant Hemorrhage 2 trial was a large multicenter randomized trial, which showed a survival benefit among those who received TXA. The generalizability of the study has been questioned in the setting of modern urban trauma centers, as most of those enrolled in the study were from hospitals with no formal MTPs and a limited availability of blood products. Additionally, no laboratory measures of fibrinolysis were available.30
Most experts currently recommend TXA use as part of an MTP if there is evidence of hyperfibrinolysis on TEG or in severe hemorrhagic shock when the time from injury has been less than 3 hours, as studies have shown increased mortality when TXA was administered longer than 3 hours after injury.30
Viscoelastic Assays
An alternative approach to standardized ratio MTPs involves goal-directed hemostatic resuscitation using viscoelastic assays to guide transfusion of blood-product components. Both TEG and rotational thromboelastometry (ROTEM) are point-of-care tools for assessment of coagulation parameters of whole blood. Although they are not new technology, their use in trauma resuscitation is a relatively new concept.
While ROTEM is more commonly used in Europe, TEG is more popular and commonly used in the United States, though not exclusively.31,32
Thromboelastography
The TEG parameters most commonly used clinically are reaction time (R-time), kinetics time, angle, maximum amplitude (MA), and lysis at 30 minutes (LY30).
Reaction Time. The R-time is measured in minutes and represents the time to clot initiation, reflecting activity of coagulation factors. It is used in TEG-guided MTPs to trigger transfusion of FFP.31,32 The R-time is measured at the time the clot strength reaches an amplitude of 2 mm.31 The angle reflects the rate of rise of the amplitude of the TEG tracing, or the rate of increase in clot strength. Clinically, the angle represents fibrinogen concentration and function, and is used to trigger transfusion of cryoprecipitate or fibrinogen concentrate in MTPs.31,32
Maximum Amplitude. The MA is reached by the TEG tracing, or the maximum clot strength achieved. Although the MA has been shown to correlate with platelet count, it actually represents platelet count and function as well as fibrinogen activity, all of which contribute to clot strength. Clinically, MA is used to trigger platelet transfusion and/or administration of desmopressin in MTPs.31,32
Lysis at 30 Minutes. The LY30 is defined as the percent reduction in clot strength 30 minutes after reaching MA.31,32 Normal LY30 values are between 0% and 7.5%; however, these values have been challenged in recent studies, which have reported that an LY30 greater than 3% (termed hyperfibrinolysis) confers a significant increase in mortality and an increased likelihood of requiring massive transfusion.31,33 These findings have led to incorporation of this lower threshold as a trigger for administration of TXA during MTPs. Furthermore, an LY30 of less than 0.8% (described as fibrinolysis shutdown) has also been found to confer an increase in mortality,34 which has led many to advocate for goal-directed administration of TXA, rather than empiric administration, as these patients are more likely to be harmed than helped by such an intervention.31
Rapid Thromboelastography
Rapid TEG employs tissue factor to accelerate clot initiation and reaction time, providing an additional parameter which reflects coagulation factor activity: the activated clotting time (ACT).32
Activated Clotting Time. Historically used in cardiac surgery to measure anticoagulation during a cardiopulmonary bypass, ACT represents the same phase of coagulation as R-time, but is measured in seconds instead of minutes.31 The ACT has been found to correlate with prothrombin time/international normalized ratio (PT/INR), and accurately predicts the need for MTP.
Cotton et al35 found that an ACT of more than 128 seconds predicted patients requiring MTP, and an ACT lower than 105 seconds predicted those who required no transfusions in the first 24 hours after injury.35
The ACT can be used to trigger transfusion of FFP, but at certain thresholds, may also be used to trigger the early transfusion of cryoprecipitate and platelets.36 Moore et al36 found that an ACT over 140 seconds was able to predict an abnormal angle and MA. This had led to using this threshold as a trigger for early administration of cryoprecipitate and platelets, given this parameter is available within 5 minutes—long before the angle and MA have resulted.
Efficacy
The use of a TEG-guided strategy for MTP in trauma has shown great promise. In 2013, Tapia et al37 compared a historical cohort who received 1:1:1 MTP to a TEG-guided MTP and demonstrated improved mortality.In 2016, Gonzalez et al38 compared TEG-guided transfusion vs conventional coagulation tests (PT/INR, PTT, fibrinogen, platelets). The authors found a significant decrease in mortality and platelet and FFP transfusion when TEG-guided resuscitation is used.
Endovascular Techniques
The use of endovascular techniques in trauma continues to evolve. According to the National Trauma Data Bank, the use of endovascular therapies has increased from 1% of trauma cases in 2002 to 11% in 2008.39
Thoracic Endovascular Aortic Repair
Thoracic endovascular aortic repair (TEVAR) for blunt thoracic aortic injury has essentially replaced open surgical repair. (See Figures 1a and 1b for an example of a blunt traumatic aortic injury prior to and post-TEVAR placement.)

Transarterial Catheter Embolization
Endovascular treatments have also been used successfully in the management of injuries to aortic branch vessels and extremity vessels.42 Transarterial catheter embolization with coils, plugs, or gel foam is being employed with increasing frequency to achieve hemostasis in the pelvis and spleen.42 It may also be used as an adjunct to laparotomy and perihepatic packing in high-grade liver injuries, though it is associated with significant morbidity related to hepatic necrosis, bile leaks, and abscesses.43,44
Resuscitative Endovascular Balloon Occlusion of the Aorta
Most recently, REBOA has been used for noncompressible torso hemorrhage following trauma. This method involves percutaneous arterial cannulation of the common femoral artery and advancement of a balloon into the aorta, where it is then inflated at the desired level.

Once inflated, the balloon obstructs arterial inflow to the area of hemorrhage, curtailing blood loss, and increases proximal BP, improving coronary and cerebral perfusion. Multiple case reports and case series have described successful use of REBOA for hemorrhage control, including prehospital use by physicians in the United Kingdom. The largest series to date looked at 114 patients, of whom 46 had REBOA placement and 68 had open aortic occlusion through resuscitative thoracotomy.45 Those treated with REBOA were significantly more likely to achieve hemodynamic stability (defined as SBP >90 mm Hg for >5 minutes). Furthermore, the authors noted minimal complications from REBOA and no difference in time to successful aortic occlusion, regardless of technique. There was also no difference in mortality between the two groups. Despite the small number of studies in trauma patients, REBOA has been established as a viable alternative to open aortic occlusion. The prospective Aortic Occlusion for Resuscitation in Trauma and Acute Care Surgery registry established by the American Association for the Surgery of Trauma is continuing to enroll patients and will hopefully answer many of the current uncertainties regarding the use of REBOA.
Conclusion
Strategies and techniques for the care of the injured patient have changed significantly in the past few years. Damage control resuscitation includes three elements: damage control surgery, permissive hypotension, and blood-product resuscitation.
The goals of lowering MAP in hemorrhagic shock appear to be safe and make sense physiologically, but have yet to show clear mortality benefit. Avoidance of excessive crystalloid resuscitation and trends toward more physiological ratios of blood product resuscitation have shown better outcomes. While the ideal ratio of blood products in transfusion remains unknown, the use of a massive transfusion strategy is preferable to crystalloid fluids. The use of viscoelastic assays (TEG and ROTEM) have allowed for goal-directed blood product resuscitation and may improve outcomes when compared with prescribed resuscitation ratios.
Finally, endovascular techniques in trauma have been increasingly utilized over the past 15 years, making nonoperative management with angiographic embolization for solid organ injury common practice now in most trauma centers worldwide. Temporary aortic balloon occlusion with REBOA appears promising in many cases of noncompressible truncal hemorrhage until definitive hemostasis can be achieved, but studies are needed to determine its ultimate place in the care of the trauma patient.
1. Evans JA, van Wessem KJ, McDougall D, Lee KA, Lyons T, Balogh ZJ. Epidemiology of traumatic deaths: comprehensive population-based assessment. World J Surg. 2010;34(1):158-163. doi:10.1007/s00268-009-0266-1.
2. Bogert JN, Harvin JA, Cotton BA. Damage control resuscitation. J Intensive Care Med. 2016;31(3):177-186. doi:10.1177/0885066614558018.
3. Kaafarani HMA, Velmahos GC. Damage control resuscitation in trauma. Scand J Surg. 2014;103(2):81-88. doi:10.1177/1457496914524388.
4. Bickell WH, Wall MJ Jr, Pepe PE, et al. Immediate versus delayed fluid resuscitation for hypotensive patients with penetrating torso injuries. N Engl J Med. 1994;331(17):1105-1109. doi:10.1056/NEJM199410273311701.
5. Sondeen JL, Coppes VG, Holcomb JB. Blood pressure at which rebleeding occurs after resuscitation in swine with aortic injury. J Trauma. 2003;54(5 Suppl):S110-S117. doi:10.1097/01.TA.0000047220.81795.3D.
6. Li T, Zhu Y, Hu Y, et al. Ideal permissive hypotension to resuscitate uncontrolled hemorrhagic shock and the tolerance time in rats. Anesthesiology. 2011;114(1):111-119. doi:10.1097/ALN.0b013e3181fe3fe7.
7. Dutton RP, Mackenzie CF, Scalea TM. Hypotensive resuscitation during active hemorrhage: impact on in-hospital mortality. J Trauma. 2002;52(6):1141-1146.
8. Schreiber MA, Meier EN, Tisherman SA, et al; ROC Investigators. A controlled resuscitation strategy is feasible and safe in hypotensive trauma patients: results of a prospective randomized pilot trial. J Trauma Acute Care Surg. 2015;78(4):687-695. doi:10.1097/TA.0000000000000600.
9. Morrison CA, Carrick MM, Norman MA, et al. Hypotensive resuscitation strategy reduces transfusion requirements and severe postoperative coagulopathy in trauma patients with hemorrhagic shock: preliminary results of a randomized controlled trial. J Trauma. 2011;70(3):652-663. doi:10.1097/TA.0b013e31820e77ea.
10. Carrick MM, Morrison CA, Tapia NM, et al. Intraoperative hypotensive resuscitation for patients undergoing laparotomy or thoracotomy for trauma: Early termination of a randomized prospective clinical trial. J Trauma Acute Care Surg. 2016;80(6):886-896. doi:10.1097/TA.0000000000001044.
11. Chesnut RM, Marshall LF, Klauber MR, et al. The role of secondary brain injury in determining outcome from severe head injury. J Trauma. 1993;34(2):216-222.
12. Kasotakis G, Sideris A, Yang Y, et al. Inflammation and Host Response to Injury Investigators. Aggressive early crystalloid resuscitation adversely affects outcomes in adult blunt trauma patients: an analysis of the Glue Grant database. J Trauma Acute Care Surg. 2013;74(5):1215-1221; discussion 1221-1222. doi:10.1097/TA.0b013e3182826e13.
13. Cotton BA, Guy JS, Morris JA Jr, Abumrad NN. The cellular, metabolic, and systemic consequences of aggressive fluid resuscitation strategies. Shock. 2006;26(2):115-121. doi:10.1097/01.shk.0000209564.84822.f2.
14. Kozar RA, Peng Z, Zhang R, et al. Plasma restoration of endothelial glycocalyx in a rodent model of hemorrhagic shock. Anesth Analg. 2011;112(6):1289-1295. doi:10.1213/ANE.0b013e318210385c.
15. Torres LN, Sondeen JL, Ji L, Dubick MA, Filho IT. Evaluation of resuscitation fluids on endothelial glycocalyx, venular blood flow, and coagulation function after hemorrhagic shock in rats. J Trauma Acute Care Surg. 2013;75(5):759-766. doi:10.1097/TA.0b013e3182a92514.
16. Bradley M, Galvagno S, Dhanda A, et al. Damage control resuscitation protocol and the management of open abdomens in trauma patients. Am Surg. 2014;80(8):768-775.
17. Brandstrup B, Tønnesen H, Beier-Holgersen R, et al; Danish Study Group on Perioperative Fluid Therapy. Effects of intravenous fluid restriction on postoperative complications: comparison of two perioperative fluid regimens: a randomized assessor-blinded multicenter trial. Ann Surg. 2003;238(5):641-648. doi:10.1097/01.sla.0000094387.50865.23.
18. Barak M, Rudin M, Vofsi O, Droyan A, Katz Y. Fluid administration during abdominal surgery influences on coagulation in the postoperative period. Curr Surg. 2004;61(5):459-462. doi:10.1016/j.cursur.2004.02.002.
19. Harada MY, Ko A, Barmparas G, et al. 10-Year trend in crystalloid resuscitation: Reduced volume and lower mortality. Int J Surg. 2017;38:78-82. doi:10.1016/j.ijsu.2016.12.073.
20. Borgman MA, Spinella PC, Perkins JG, et al. The ratio of blood products transfused affects mortality in patients receiving massive transfusions at a combat support hospital. J Trauma. 2007;63(4):805-813. doi:10.1097/TA.0b013e3181271ba3.
21. Cotton BA, Gunter OL, Isbell J, et al. Damage control hematology: the impact of a trauma exsanguination protocol on survival and blood product utilization. J Trauma. 2008;64(5):1177-1782; discussion 1182-1183. doi:10.1097/TA.0b013e31816c5c80.
22. Holcomb JB, Wade CE, Michalek JE, et al. Increased plasma and platelet to red blood cell ratios improves outcome in 466 massively transfused civilian trauma patients. Ann Surg. 2008;248(3):447-458. doi:10.1097/SLA.0b013e318185a9ad.
23. Holcomb JB, del Junco DJ, Fox EE, et al; PROMMTT Study Group. The prospective, observational, multicenter, major trauma transfusion (PROMMTT) study: comparative effectiveness of a time-varying treatment with competing risks. JAMA Surg. 2013;148(2):127-136. doi:10.1001/2013.jamasurg.387.
24. Holcomb JB, Tilley BC, Baraniuk S, et al; PROPPR Study Group. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA. 2015;313(5):471-482. doi:10.1001/jama.2015.12.
25. Holcomb JB, Donathan DP, Cotton BA, et al. Prehospital transfusion of plasma and red blood cells in trauma patients. Prehosp Emerg Care. 2015;19(1):1-9. doi:10.3109/10903127.2014.923077.
26. Brown JB, Sperry JL, Fombona A, Billiar TR, Peitzman AB, Guyette FX. Pre-trauma center red blood cell transfusion is associated with improved early outcomes in air medical trauma patients. J Am Coll Surg. 2015;220(5):797-808. doi:10.1016/j.jamcollsurg.2015.01.006.
27. Brown JB, Cohen MJ, Minei JP, et al; Inflammation and the Host Response to Injury Investigators. Pretrauma center red blood cell transfusion is associated with reduced mortality and coagulopathy in severely injured patients with blunt trauma. Ann Surg. 2015;261(5):997-1005. doi:10.1097/SLA.0000000000000674.
28. Brown JB, Guyette FX, Neal MD, et al. Taking the blood bank to the field: the design and rationale of the Prehospital Air Medical Plasma (PAMPer) trial. Prehosp Emerg Care. 2015;19(3):343-350. doi:10.3109/10903127.2014.995851.
29. Morrison JJ, Dubose JJ, Rasmussen TE, Midwinter MJ. Military Application of Tranexamic Acid in Trauma Emergency Resuscitation (MATTERs) study. Arch Surg. 2012;147(2):113-119. doi:10.1001/archsurg.2011.287.
30. Shakur H, Roberts I, Bautista R, et al; CRASH-2 trial collaborators. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet. 2010;376(9734):23-32. doi:10.1016/S0140-6736(10)60835-5.
31. Gonzalez E, Moore EE, Moore HB. Management of trauma-induced coagulopathy with thrombelastography. Crit Care Clin. 2017;33(1):119-134. doi:10.1016/j.ccc.2016.09.002.
32. Abdelfattah K, Cripps MW. Thromboelastography and rotational thromboelastometry use in trauma. Int J Surg. 2016;33(Pt B):196-201. doi:10.1016/j.ijsu.2015.09.036.
33. Cotton BA, Harvin JA, Kostousouv V, et al. Hyperfibrinolysis at admission is an uncommon but highly lethal event associated with shock and prehospital fluid administration. J Trauma Acute Care Surg. 2012;73(2):365-370; discussion 370. doi:10.1097/TA.0b013e31825c1234.
34. Moore HB, Moore EE, Gonzalez E, et al. Hyperfibrinolysis, physiologic fibrinolysis, and fibrinolysis shutdown: the spectrum of postinjury fibrinolysis and relevance to antifibrinolytic therapy. J Trauma Acute Care Surg. 2014;77(6):811-817. doi:10.1097/TA.0000000000000341.
35. Cotton BA, Faz G, Hatch QM, et al. Rapid thrombelastography delivers real-time results that predict transfusion within 1 hour of admission. J Trauma. 2011;71(2):407-414; discussion 414-417. doi:10.1097/TA.0b013e31821e1bf0.
36. Moore HB, Moore EE, Chin TL, et al. Activated clotting time of thrombelastography (T-ACT) predicts early postinjury blood component transfusion beyond plasma. Surgery. 2014;156(3):564-569. doi:10.1016/j.surg.2014.04.017.
37. Tapia NM, Chang A, Norman M, et al. TEG-guided resuscitation is superior to standardized MTP resuscitation in massively transfused penetrating trauma patients. J Trauma Acute Care Surg. 2013;74(2):378-385; discussion 385-386. doi:10.1097/TA.0b013e31827e20e0.
38. Gonzalez E, Moore EE, Moore HB, et al. Goal-directed hemostatic resuscitation of trauma-induced coagulopathy: a pragmatic randomized clinical trial comparing a viscoelastic assay to conventional coagulation assays. Ann Surg. 2016;263(6):1051-1059. doi:10.1097/SLA.0000000000001608.
39. Avery LE, Stahlfeld KR, Corcos AC, et al. Evolving role of endovascular techniques for traumatic vascular injury: a changing landscape? J Trauma Acute Care Surg. 2012;72(1):41-46; discussion 46-47. doi:10.1097/TA.0b013e31823d0f03.
40. Demetriades D, Velmahos GC, Scalea TM, et al. Diagnosis and treatment of blunt thoracic aortic injuries: changing perspectives. J Trauma. 2008;64(6):1415-1418; discussion 1418-1419. doi:10.1097/TA.0b013e3181715e32.
41. Azizzadeh A, Ray HM, Dubose JJ, et al. Outcomes of endovascular repair for patients with blunt traumatic aortic injury. J Trauma Acute Care Surg. 2014;76(2):510-516. doi:10.1097/TA.0b013e3182aafe8c.
42. Brenner M, Hoehn M, Rasmussen TE. Endovascular therapy in trauma. Eur J Trauma Emerg Surg. 2014;40(6):671-678. doi:10.1007/s00068-014-0474-8.
43. Dabbs DN, Stein DM, Scalea TM. Major hepatic necrosis: a common complication after angioembolization for treatment of high-grade liver injuries. J Trauma. 2009;66(3):621-627; discussion 627-629. doi:10.1097/TA.0b013e31819919f2.
44. Letoublon C, Morra I, Chen Y, Monnin V, Voirin D, Arvieux C. Hepatic arterial embolization in the management of blunt hepatic trauma: indications and complications. J Trauma. 2011;70(5):1032-1036; discussion 1036-1037. doi:10.1097/TA.0b013e31820e7ca1.
45. DuBose JJ, Scalea TM, Brenner M, et al; AORTA Study Group. The AAST prospective Aortic Occlusion for Resuscitati on in Trauma and Acute Care Surgery (AORTA) registry: Data on contemporary utilization and outcomes of aortic occlusion and resuscitative balloon occlusion of the aorta (REBOA). J Trauma Acute Care Surg. 2016;81(3):409-419. doi:10.1097/TA.0000000000001079.
Introduction
For decades, virtually all injury was treated with open operative surgery. Resuscitation was based on the belief that large-volume crystalloid infusion to raise blood pressure (BP) to normal was the optimal therapy. Advanced trauma life support teaching was that 2 L of crystalloid fluid should be the initial resuscitation for all trauma patients, and those who failed to respond should receive additional crystalloid fluid. Patients did not receive a blood transfusion until later in treatment, and fresh frozen plasma (FFP) and platelets were not given until 10 U of blood had been administered. Regardless of the fluid infused, the goal of initial resuscitation was to raise BP to a normal level. During the time I (TS) was chair of the emergency medicine department at the State University of New York’s Kings County Hospital, I remember administering liters of crystalloid fluid preoperatively, believing it was not safe to operate until the patient had been what we termed “adequately resuscitated.”
However, as early as 1918, Walter B. Cannon, MD, correctly observed that fluid therapy without hemostasis was not wise, and numerous animal studies since then also raised serious questions about this approach. This article points out the revolutionary changes in the thinking and practice of resuscitation that have occurred in the last 20 years. We now realize that raising BP to normal only perpetuates hemorrhage. Hypotension treated with additional volume resuscitation without surgical control of hemorrhage creates a cycle leading to dilution of clotting factors and red blood cells (RBCs), recurrent hypotension, and ultimately death.
The realization that early blood transfusion is probably the wisest course is a concept that has only been in clinical practice for less than 15 years. Major trauma centers now routinely keep type O negative blood in the ED refrigerators so that it is instantly available.
Our understanding of trauma coagulopathy also has changed dramatically. Once thought to be simply a consequence of hypotension and hypothermia, we now realize that coagulopathy following trauma is far more complicated and likely occurs in concert with the inflammatory response to serious injury. Regardless of its etiology, we have recognized that earlier administration of plasma and platelets following trauma prevents coagulopathy, and this approach is more beneficial than treating coagulopathy after it develops. There has been much debate about the optimal ratios of RBCs, plasma, and platelets, and the ideal ratio has yet to be determined. The idea that “one-size-fits-all” is almost certainly not the case: Different patients require different and more precise treatment strategies.
For years, we have relied on laboratory measurements of coagulation to guide transfusion therapy, but standard laboratory values often take over 30 minutes to obtain. In an extremely dynamic situation involving large-volume blood loss, this interval is too lax. A more personalized approach using rapidly available technology, such as thromboelastography (TEG), allows for real-time assessment of a multiplicity of coagulation dynamics and rapid correction of any abnormalities. Procoagulants such as factor 7A, prothrombin complex concentrate (PCC), and tranexamic acid (TXA) have a role. However, the data to support the use of these expensive agents is lacking. While they certainly can be life-saving, each of these components brings with it a risk of causing indiscriminate coagulation—even in areas of the body that are not injured. Moreover, their availability in nontrauma centers is either limited or not an option.
There is little question that our rapid advances in understanding resuscitation and transfusion practice has saved lives. Twenty years ago, intensive care units were populated by trauma patients who had received many liters of crystalloid fluid, and at least partly a consequence of the resuscitation experience, many had severe respiratory failure. Open abdomens were common and also a likely consequence of large-volume crystalloid use. While these problems have not entirely disappeared, they now occur much less frequently.
Standardizing trauma care has also helped enhance patient care a great deal. Most major trauma centers have a “massive transfusion” protocol which allows the blood bank to prepare coolers containing not only blood, but also plasma, platelets, and procoagulants. This practice obviates the need to order the components individually. Rapid access to technology such as TEG allows emergency physicians (EPs) and other trauma care professionals to precisely guide transfusion therapy, but this remains an area of intensely debated investigation. Hopefully, our understanding will continue to mature over the next few years.
Another area of trauma care that has rapidly evolved is the use of endovascular techniques for trauma hemostasis. The realization that we can obtain control of vascular injury without the need for a large open operation has revolutionized care. While endovascular techniques have been used for pelvic hemostasis since 1972, we now use it regularly in every body cavity. Splenic artery embolization was developed by our (TS) group in Brooklyn, New York in 1995, and its use has now expanded to other abdominal solid organ injuries.
Injuries to the thoracic aorta once required a thoracotomy, cardiopulmonary bypass, and open repair. Stent grafting is now the treatment of choice for these injuries, allowing for a minimally invasive solution, and permitting those with both aortic and many other injuries to receive care for all of these wounds much sooner than was possible in the past, when multiply injured patients were simply not considered candidates for early open repair.
Thoracotomy in the ED has been widely practiced for a variety of indications. While it is still the only available solution for injury to the heart and/or proximal pulmonary vasculature in a patient who is hemodynamically unstable and/or in extremis, other options now exist to obtain vascular inflow for patients bleeding in the abdomen or pelvis. The use of transfemoral balloons for aortic occlusion allows clinicians to temporize hemorrhage without a huge open operation, and resuscitative endovascular balloon occlusion of the aorta (REBOA), has only been available for the last several years. The exact indications, wisest strategy, length of time the balloon can be inflated, rate of complications, and who is the appropriate physician (eg, EP, intensivist, vascular surgeon) to insert it, all remain questions requiring resolution. Much more work is necessary to pursue the role that REBOA can have in the care of desperately injured trauma patients.
There has been a revolution in the care of severely injured patients. After 50 years of thinking that we knew the answers, we have come to realize that those answers were wrong. Newer resuscitation strategies, as well as new treatment strategies continue to evolve, allowing us to refine care of severely injured patients. Perhaps the one thing we have really learned is that we do not have all of the answers and that the discussion must continue if we are to do better at serving more trauma victims.
Damage Control Resuscitation
In the United States, trauma is the leading cause of death in patients younger than age 45 years and ranks as the fifth leading cause of death among all age groups. Hemorrhage remains the leading cause of preventable death in the trauma population,1 and one of the most important recent changes in our care of the injured patient is the manner in which we manage hemorrhage. As noted earlier, there has been a paradigm shift away from large-volume crystalloid resuscitation and toward what has been termed “damage control resuscitation” (DCR).2,3
The principles of the DCR strategy are aimed at preemptively treating the lethal triad of hypothermia, acidosis, and coagulopathy in conjunction with control of surgical bleeding using damage control surgery. The main principles of DCR include “permissive hypotension,” prevention of heat loss and/or active warming, minimizing the use of crystalloid infusions, and initiating resuscitation with blood products in a ratio that more closely resembles whole blood.2
Permissive Hypotension
Permissive hypotension, also referred to as hypotensive resuscitation, is not considered a goal or an endpoint, but rather a “bridge” to definitive surgical control of hemorrhage. The body’s initial response to injury involves vasoconstriction and early clot formation, a process facilitated by hypotension. The rationale for permissive hypotension is that attempting to drive the BP up to normal ranges may interfere with vasoconstriction, as well as physically disrupting this early clot, leading to increased bleeding and further hypotension.
This concept has been corroborated by many animal and human studies.3 In 1994, the landmark study by Bickell et al4 randomized patients with penetrating torso trauma and a systolic BP (SBP) of 90 mm Hg or lower to either immediate or delayed fluid resuscitation. Their study demonstrated that patients whose fluid resuscitation was delayed until they reached the operating room had improved outcomes. The study supported the long-time prehospital practice of the “scoop-and-run” strategy, especially in penetrating torso trauma.
In 2003, Sondeen et al5 used a swine model of aortic injury to find an inflection point for clot disruption and re-bleeding during volume resuscitation. They found the inflection point to be a mean arterial pressure (MAP) of 64 mm Hg and an SBP of 94 mm Hg, regardless of the size of the aortotomy. Using an animal model of hemorrhagic shock, Li et al6 demonstrated in 2011 that resuscitation to a MAP of 50 mm Hg was associated with a decreased amount of blood loss as well as with improved survival compared to patients who were resuscitated to a MAP of 80 mm Hg. However, they also showed that after a time period of more than 90 to 120 minutes, the lower MAP group had increased end organ damage and worse outcomes, emphasizing the importance of prompt surgical control of bleeding—regardless of preoperative resuscitation strategy.
Other studies, though, have not shown a clear benefit to permissive hypotension. A 2002 study by Dutton et al7 showed that titration of initial fluid to a lower SBP (70 mm Hg) did not affect mortality when compared to a target resuscitation MAP of more than 100 mm Hg. Further, in 2014, a plenary paper presented to the American Association for the Surgery of Trauma demonstrated that controlled resuscitation (CR) strategy was safe and feasible,8 but did not demonstrate a mortality benefit in the overall cohort, though patients with blunt trauma who received CR had improved survival at 24 hours.
The group at Ben Taub General Hospital in Houston, Texas recently performed a randomized controlled trial evaluating intraoperative hypotensive resuscitation strategies. Patients in hemorrhagic shock were randomized to either an intraoperative MAP goal of 50 mm Hg or 65 mm Hg.9,10 Preliminary results suggested that targeting a lower MAP resulted in fewer blood product transfusions, less fluid administration, less coagulopathy, and lower mortality in the early postoperative period. Additionally, they demonstrated a nonsignificant trend toward improved 30-day mortality in the lower MAP group.9 Moreover, in this study there was no increased morbidity associated with the hypotensive strategy,10 suggesting that the approach was safe. Unfortunately, the trial was stopped early due to slow enrollment.
Despite the overall promising results with permissive hypotension, it is important to remember that it is contraindicated in patients with known or suspected traumatic brain injury, as hypotension has been shown to be detrimental in this population.11
Hemostatic Resuscitation and Coagulopathy
Avoiding Aggressive Crystalloid Resuscitation. While the ideal MAP to target during DCR remains unclear, the potential harm caused by aggressive crystalloid resuscitation has become more evident. Infusing excessive amounts of crystalloid has been shown to be associated with increased ventilator days, multisystem organ failure, abdominal compartment syndrome, and surgical-site infections12—all of which have also been associated with systemic consequences of increased inflammation, including increased release of tumor necrosis factor-alpha and other proinflammatory cytokines.13
Rodent studies have demonstrated large-volume crystalloid administration and breakdown or “thinning” of the endothelial glycocalyx, which leads to increased capillary leak, third-spacing, and ultimately intravascular volume depletion.14,15 This mechanism has been linked to pulmonary complications, namely acute lung injury and acute respiratory distress syndrome. Enteric edema resulting from aggressive crystalloid resuscitation has also been associated with prolonged postoperative ileus, increased risk of anastomotic leak,13 and inability to achieve primary fascial closure.16 All of the aforementioned complications are reduced when employing a restrictive fluid resuscitation strategy.17
Aggressive crystalloid administration in hemorrhagic shock also leads to dilutional coagulopathy. Multiple animal and human studies have shown an association between increased crystalloid volumes in hemorrhaging patients and increasing coagulopathy, blood loss, and mortality. In 2004, Barak et al18 demonstrated that administration of a high volume of crystalloid fluid (>3 L) or colloid (500 mL) was associated with postoperative coagulopathy; whereas in 2017, Harada et al,19 at Cedars-Sinai Medical Center in New York, demonstrated over a 10-year period that decreased high-volume (>2 L) crystalloid resuscitation paralleled a decrease in mortality.
Massive Transfusion Protocols. Many trauma centers have shifted away from high-volume crystalloid resuscitation in favor of massive transfusion protocols (MTPs) utilizing standardized ratios that more closely mimic whole blood. The MTPs center on the principle of equal transfusion ratios of blood product as opposed to packed RBCs (PRBCs) alone. This means effecting a plasma-rich resuscitation and preemptive correction of coagulopathy with FFP and platelets in addition to PRBCs.
Data from a US Army combat support hospital have demonstrated improved survival with an FFP to PRBC ratio of more than 1:1.4,20 and civilian studies have produced similar findings.21-23 All of these studies also noted improved mortality with higher (>1:2) platelet to PRBC ratios.22,23 Although, the ideal ratio remains unknown, many MTPs aim for 1:1:1 ratio (6 U FFP to 6 packed platelets to 6 U PRBC), which most closely mimics whole blood.
The Pragmatic Randomized Optimal Platelet and Plasma Ratios trial was a recent large multicenter randomized trial that compared transfusion ratios of 1:1:1 and 1:1:2. The trial was unable to demonstrate a difference in mortality at either 24 hours or 30 days, though more patients in the 1:1:1 ratio group achieved hemostasis and fewer patients in this group died from exsanguination in the first 24 hours.24Prehospital PRBC Administration. A number of studies have looked at prehospital administration of PRBCs.25-27 Holcomb et al25 showed no overall survival advantage at 24 hours, but did demonstrate a negligible blood-product wastage. In 2015 Brown et al26 found an increase probability of 24-hour survival, decreased shock, and lower 24-hour PRBC requirements with pretrauma-center PRBC transfusion. That same year Brown et al27 demonstrated that prehospital PRBC transfusion in severely injured blunt trauma patients was associated with decreased 24-hour and 30-day mortality rates, and a lower risk of coagulopathy. Currently, the Prehospital Air Medical Plasma trial is enrolling patients to evaluate the prehospital administration of plasma.28 The primary endpoint of the study is 30-day mortality; the tentative completion date for the study is January 2018.
Tranexamic Acid. Another important development in the treatment of hemorrhagic shock in recent years has been the use of TXA, an antifibrinolytic agent which inhibits the conversion of plasminogen to plasmin. It has been shown to decrease mortality in both civilian and military trauma populations.29,30
The Clinical Randomization of an Antifibrinolytic in Significant Hemorrhage 2 trial was a large multicenter randomized trial, which showed a survival benefit among those who received TXA. The generalizability of the study has been questioned in the setting of modern urban trauma centers, as most of those enrolled in the study were from hospitals with no formal MTPs and a limited availability of blood products. Additionally, no laboratory measures of fibrinolysis were available.30
Most experts currently recommend TXA use as part of an MTP if there is evidence of hyperfibrinolysis on TEG or in severe hemorrhagic shock when the time from injury has been less than 3 hours, as studies have shown increased mortality when TXA was administered longer than 3 hours after injury.30
Viscoelastic Assays
An alternative approach to standardized ratio MTPs involves goal-directed hemostatic resuscitation using viscoelastic assays to guide transfusion of blood-product components. Both TEG and rotational thromboelastometry (ROTEM) are point-of-care tools for assessment of coagulation parameters of whole blood. Although they are not new technology, their use in trauma resuscitation is a relatively new concept.
While ROTEM is more commonly used in Europe, TEG is more popular and commonly used in the United States, though not exclusively.31,32
Thromboelastography
The TEG parameters most commonly used clinically are reaction time (R-time), kinetics time, angle, maximum amplitude (MA), and lysis at 30 minutes (LY30).
Reaction Time. The R-time is measured in minutes and represents the time to clot initiation, reflecting activity of coagulation factors. It is used in TEG-guided MTPs to trigger transfusion of FFP.31,32 The R-time is measured at the time the clot strength reaches an amplitude of 2 mm.31 The angle reflects the rate of rise of the amplitude of the TEG tracing, or the rate of increase in clot strength. Clinically, the angle represents fibrinogen concentration and function, and is used to trigger transfusion of cryoprecipitate or fibrinogen concentrate in MTPs.31,32
Maximum Amplitude. The MA is reached by the TEG tracing, or the maximum clot strength achieved. Although the MA has been shown to correlate with platelet count, it actually represents platelet count and function as well as fibrinogen activity, all of which contribute to clot strength. Clinically, MA is used to trigger platelet transfusion and/or administration of desmopressin in MTPs.31,32
Lysis at 30 Minutes. The LY30 is defined as the percent reduction in clot strength 30 minutes after reaching MA.31,32 Normal LY30 values are between 0% and 7.5%; however, these values have been challenged in recent studies, which have reported that an LY30 greater than 3% (termed hyperfibrinolysis) confers a significant increase in mortality and an increased likelihood of requiring massive transfusion.31,33 These findings have led to incorporation of this lower threshold as a trigger for administration of TXA during MTPs. Furthermore, an LY30 of less than 0.8% (described as fibrinolysis shutdown) has also been found to confer an increase in mortality,34 which has led many to advocate for goal-directed administration of TXA, rather than empiric administration, as these patients are more likely to be harmed than helped by such an intervention.31
Rapid Thromboelastography
Rapid TEG employs tissue factor to accelerate clot initiation and reaction time, providing an additional parameter which reflects coagulation factor activity: the activated clotting time (ACT).32
Activated Clotting Time. Historically used in cardiac surgery to measure anticoagulation during a cardiopulmonary bypass, ACT represents the same phase of coagulation as R-time, but is measured in seconds instead of minutes.31 The ACT has been found to correlate with prothrombin time/international normalized ratio (PT/INR), and accurately predicts the need for MTP.
Cotton et al35 found that an ACT of more than 128 seconds predicted patients requiring MTP, and an ACT lower than 105 seconds predicted those who required no transfusions in the first 24 hours after injury.35
The ACT can be used to trigger transfusion of FFP, but at certain thresholds, may also be used to trigger the early transfusion of cryoprecipitate and platelets.36 Moore et al36 found that an ACT over 140 seconds was able to predict an abnormal angle and MA. This had led to using this threshold as a trigger for early administration of cryoprecipitate and platelets, given this parameter is available within 5 minutes—long before the angle and MA have resulted.
Efficacy
The use of a TEG-guided strategy for MTP in trauma has shown great promise. In 2013, Tapia et al37 compared a historical cohort who received 1:1:1 MTP to a TEG-guided MTP and demonstrated improved mortality.In 2016, Gonzalez et al38 compared TEG-guided transfusion vs conventional coagulation tests (PT/INR, PTT, fibrinogen, platelets). The authors found a significant decrease in mortality and platelet and FFP transfusion when TEG-guided resuscitation is used.
Endovascular Techniques
The use of endovascular techniques in trauma continues to evolve. According to the National Trauma Data Bank, the use of endovascular therapies has increased from 1% of trauma cases in 2002 to 11% in 2008.39
Thoracic Endovascular Aortic Repair
Thoracic endovascular aortic repair (TEVAR) for blunt thoracic aortic injury has essentially replaced open surgical repair. (See Figures 1a and 1b for an example of a blunt traumatic aortic injury prior to and post-TEVAR placement.)
Transarterial Catheter Embolization
Endovascular treatments have also been used successfully in the management of injuries to aortic branch vessels and extremity vessels.42 Transarterial catheter embolization with coils, plugs, or gel foam is being employed with increasing frequency to achieve hemostasis in the pelvis and spleen.42 It may also be used as an adjunct to laparotomy and perihepatic packing in high-grade liver injuries, though it is associated with significant morbidity related to hepatic necrosis, bile leaks, and abscesses.43,44
Resuscitative Endovascular Balloon Occlusion of the Aorta
Most recently, REBOA has been used for noncompressible torso hemorrhage following trauma. This method involves percutaneous arterial cannulation of the common femoral artery and advancement of a balloon into the aorta, where it is then inflated at the desired level.
Once inflated, the balloon obstructs arterial inflow to the area of hemorrhage, curtailing blood loss, and increases proximal BP, improving coronary and cerebral perfusion. Multiple case reports and case series have described successful use of REBOA for hemorrhage control, including prehospital use by physicians in the United Kingdom. The largest series to date looked at 114 patients, of whom 46 had REBOA placement and 68 had open aortic occlusion through resuscitative thoracotomy.45 Those treated with REBOA were significantly more likely to achieve hemodynamic stability (defined as SBP >90 mm Hg for >5 minutes). Furthermore, the authors noted minimal complications from REBOA and no difference in time to successful aortic occlusion, regardless of technique. There was also no difference in mortality between the two groups. Despite the small number of studies in trauma patients, REBOA has been established as a viable alternative to open aortic occlusion. The prospective Aortic Occlusion for Resuscitation in Trauma and Acute Care Surgery registry established by the American Association for the Surgery of Trauma is continuing to enroll patients and will hopefully answer many of the current uncertainties regarding the use of REBOA.
Conclusion
Strategies and techniques for the care of the injured patient have changed significantly in the past few years. Damage control resuscitation includes three elements: damage control surgery, permissive hypotension, and blood-product resuscitation.
The goals of lowering MAP in hemorrhagic shock appear to be safe and make sense physiologically, but have yet to show clear mortality benefit. Avoidance of excessive crystalloid resuscitation and trends toward more physiological ratios of blood product resuscitation have shown better outcomes. While the ideal ratio of blood products in transfusion remains unknown, the use of a massive transfusion strategy is preferable to crystalloid fluids. The use of viscoelastic assays (TEG and ROTEM) have allowed for goal-directed blood product resuscitation and may improve outcomes when compared with prescribed resuscitation ratios.
Finally, endovascular techniques in trauma have been increasingly utilized over the past 15 years, making nonoperative management with angiographic embolization for solid organ injury common practice now in most trauma centers worldwide. Temporary aortic balloon occlusion with REBOA appears promising in many cases of noncompressible truncal hemorrhage until definitive hemostasis can be achieved, but studies are needed to determine its ultimate place in the care of the trauma patient.
Introduction
For decades, virtually all injury was treated with open operative surgery. Resuscitation was based on the belief that large-volume crystalloid infusion to raise blood pressure (BP) to normal was the optimal therapy. Advanced trauma life support teaching was that 2 L of crystalloid fluid should be the initial resuscitation for all trauma patients, and those who failed to respond should receive additional crystalloid fluid. Patients did not receive a blood transfusion until later in treatment, and fresh frozen plasma (FFP) and platelets were not given until 10 U of blood had been administered. Regardless of the fluid infused, the goal of initial resuscitation was to raise BP to a normal level. During the time I (TS) was chair of the emergency medicine department at the State University of New York’s Kings County Hospital, I remember administering liters of crystalloid fluid preoperatively, believing it was not safe to operate until the patient had been what we termed “adequately resuscitated.”
However, as early as 1918, Walter B. Cannon, MD, correctly observed that fluid therapy without hemostasis was not wise, and numerous animal studies since then also raised serious questions about this approach. This article points out the revolutionary changes in the thinking and practice of resuscitation that have occurred in the last 20 years. We now realize that raising BP to normal only perpetuates hemorrhage. Hypotension treated with additional volume resuscitation without surgical control of hemorrhage creates a cycle leading to dilution of clotting factors and red blood cells (RBCs), recurrent hypotension, and ultimately death.
The realization that early blood transfusion is probably the wisest course is a concept that has only been in clinical practice for less than 15 years. Major trauma centers now routinely keep type O negative blood in the ED refrigerators so that it is instantly available.
Our understanding of trauma coagulopathy also has changed dramatically. Once thought to be simply a consequence of hypotension and hypothermia, we now realize that coagulopathy following trauma is far more complicated and likely occurs in concert with the inflammatory response to serious injury. Regardless of its etiology, we have recognized that earlier administration of plasma and platelets following trauma prevents coagulopathy, and this approach is more beneficial than treating coagulopathy after it develops. There has been much debate about the optimal ratios of RBCs, plasma, and platelets, and the ideal ratio has yet to be determined. The idea that “one-size-fits-all” is almost certainly not the case: Different patients require different and more precise treatment strategies.
For years, we have relied on laboratory measurements of coagulation to guide transfusion therapy, but standard laboratory values often take over 30 minutes to obtain. In an extremely dynamic situation involving large-volume blood loss, this interval is too lax. A more personalized approach using rapidly available technology, such as thromboelastography (TEG), allows for real-time assessment of a multiplicity of coagulation dynamics and rapid correction of any abnormalities. Procoagulants such as factor 7A, prothrombin complex concentrate (PCC), and tranexamic acid (TXA) have a role. However, the data to support the use of these expensive agents is lacking. While they certainly can be life-saving, each of these components brings with it a risk of causing indiscriminate coagulation—even in areas of the body that are not injured. Moreover, their availability in nontrauma centers is either limited or not an option.
There is little question that our rapid advances in understanding resuscitation and transfusion practice has saved lives. Twenty years ago, intensive care units were populated by trauma patients who had received many liters of crystalloid fluid, and at least partly a consequence of the resuscitation experience, many had severe respiratory failure. Open abdomens were common and also a likely consequence of large-volume crystalloid use. While these problems have not entirely disappeared, they now occur much less frequently.
Standardizing trauma care has also helped enhance patient care a great deal. Most major trauma centers have a “massive transfusion” protocol which allows the blood bank to prepare coolers containing not only blood, but also plasma, platelets, and procoagulants. This practice obviates the need to order the components individually. Rapid access to technology such as TEG allows emergency physicians (EPs) and other trauma care professionals to precisely guide transfusion therapy, but this remains an area of intensely debated investigation. Hopefully, our understanding will continue to mature over the next few years.
Another area of trauma care that has rapidly evolved is the use of endovascular techniques for trauma hemostasis. The realization that we can obtain control of vascular injury without the need for a large open operation has revolutionized care. While endovascular techniques have been used for pelvic hemostasis since 1972, we now use it regularly in every body cavity. Splenic artery embolization was developed by our (TS) group in Brooklyn, New York in 1995, and its use has now expanded to other abdominal solid organ injuries.
Injuries to the thoracic aorta once required a thoracotomy, cardiopulmonary bypass, and open repair. Stent grafting is now the treatment of choice for these injuries, allowing for a minimally invasive solution, and permitting those with both aortic and many other injuries to receive care for all of these wounds much sooner than was possible in the past, when multiply injured patients were simply not considered candidates for early open repair.
Thoracotomy in the ED has been widely practiced for a variety of indications. While it is still the only available solution for injury to the heart and/or proximal pulmonary vasculature in a patient who is hemodynamically unstable and/or in extremis, other options now exist to obtain vascular inflow for patients bleeding in the abdomen or pelvis. The use of transfemoral balloons for aortic occlusion allows clinicians to temporize hemorrhage without a huge open operation, and resuscitative endovascular balloon occlusion of the aorta (REBOA), has only been available for the last several years. The exact indications, wisest strategy, length of time the balloon can be inflated, rate of complications, and who is the appropriate physician (eg, EP, intensivist, vascular surgeon) to insert it, all remain questions requiring resolution. Much more work is necessary to pursue the role that REBOA can have in the care of desperately injured trauma patients.
There has been a revolution in the care of severely injured patients. After 50 years of thinking that we knew the answers, we have come to realize that those answers were wrong. Newer resuscitation strategies, as well as new treatment strategies continue to evolve, allowing us to refine care of severely injured patients. Perhaps the one thing we have really learned is that we do not have all of the answers and that the discussion must continue if we are to do better at serving more trauma victims.
Damage Control Resuscitation
In the United States, trauma is the leading cause of death in patients younger than age 45 years and ranks as the fifth leading cause of death among all age groups. Hemorrhage remains the leading cause of preventable death in the trauma population,1 and one of the most important recent changes in our care of the injured patient is the manner in which we manage hemorrhage. As noted earlier, there has been a paradigm shift away from large-volume crystalloid resuscitation and toward what has been termed “damage control resuscitation” (DCR).2,3
The principles of the DCR strategy are aimed at preemptively treating the lethal triad of hypothermia, acidosis, and coagulopathy in conjunction with control of surgical bleeding using damage control surgery. The main principles of DCR include “permissive hypotension,” prevention of heat loss and/or active warming, minimizing the use of crystalloid infusions, and initiating resuscitation with blood products in a ratio that more closely resembles whole blood.2
Permissive Hypotension
Permissive hypotension, also referred to as hypotensive resuscitation, is not considered a goal or an endpoint, but rather a “bridge” to definitive surgical control of hemorrhage. The body’s initial response to injury involves vasoconstriction and early clot formation, a process facilitated by hypotension. The rationale for permissive hypotension is that attempting to drive the BP up to normal ranges may interfere with vasoconstriction, as well as physically disrupting this early clot, leading to increased bleeding and further hypotension.
This concept has been corroborated by many animal and human studies.3 In 1994, the landmark study by Bickell et al4 randomized patients with penetrating torso trauma and a systolic BP (SBP) of 90 mm Hg or lower to either immediate or delayed fluid resuscitation. Their study demonstrated that patients whose fluid resuscitation was delayed until they reached the operating room had improved outcomes. The study supported the long-time prehospital practice of the “scoop-and-run” strategy, especially in penetrating torso trauma.
In 2003, Sondeen et al5 used a swine model of aortic injury to find an inflection point for clot disruption and re-bleeding during volume resuscitation. They found the inflection point to be a mean arterial pressure (MAP) of 64 mm Hg and an SBP of 94 mm Hg, regardless of the size of the aortotomy. Using an animal model of hemorrhagic shock, Li et al6 demonstrated in 2011 that resuscitation to a MAP of 50 mm Hg was associated with a decreased amount of blood loss as well as with improved survival compared to patients who were resuscitated to a MAP of 80 mm Hg. However, they also showed that after a time period of more than 90 to 120 minutes, the lower MAP group had increased end organ damage and worse outcomes, emphasizing the importance of prompt surgical control of bleeding—regardless of preoperative resuscitation strategy.
Other studies, though, have not shown a clear benefit to permissive hypotension. A 2002 study by Dutton et al7 showed that titration of initial fluid to a lower SBP (70 mm Hg) did not affect mortality when compared to a target resuscitation MAP of more than 100 mm Hg. Further, in 2014, a plenary paper presented to the American Association for the Surgery of Trauma demonstrated that controlled resuscitation (CR) strategy was safe and feasible,8 but did not demonstrate a mortality benefit in the overall cohort, though patients with blunt trauma who received CR had improved survival at 24 hours.
The group at Ben Taub General Hospital in Houston, Texas recently performed a randomized controlled trial evaluating intraoperative hypotensive resuscitation strategies. Patients in hemorrhagic shock were randomized to either an intraoperative MAP goal of 50 mm Hg or 65 mm Hg.9,10 Preliminary results suggested that targeting a lower MAP resulted in fewer blood product transfusions, less fluid administration, less coagulopathy, and lower mortality in the early postoperative period. Additionally, they demonstrated a nonsignificant trend toward improved 30-day mortality in the lower MAP group.9 Moreover, in this study there was no increased morbidity associated with the hypotensive strategy,10 suggesting that the approach was safe. Unfortunately, the trial was stopped early due to slow enrollment.
Despite the overall promising results with permissive hypotension, it is important to remember that it is contraindicated in patients with known or suspected traumatic brain injury, as hypotension has been shown to be detrimental in this population.11
Hemostatic Resuscitation and Coagulopathy
Avoiding Aggressive Crystalloid Resuscitation. While the ideal MAP to target during DCR remains unclear, the potential harm caused by aggressive crystalloid resuscitation has become more evident. Infusing excessive amounts of crystalloid has been shown to be associated with increased ventilator days, multisystem organ failure, abdominal compartment syndrome, and surgical-site infections12—all of which have also been associated with systemic consequences of increased inflammation, including increased release of tumor necrosis factor-alpha and other proinflammatory cytokines.13
Rodent studies have demonstrated large-volume crystalloid administration and breakdown or “thinning” of the endothelial glycocalyx, which leads to increased capillary leak, third-spacing, and ultimately intravascular volume depletion.14,15 This mechanism has been linked to pulmonary complications, namely acute lung injury and acute respiratory distress syndrome. Enteric edema resulting from aggressive crystalloid resuscitation has also been associated with prolonged postoperative ileus, increased risk of anastomotic leak,13 and inability to achieve primary fascial closure.16 All of the aforementioned complications are reduced when employing a restrictive fluid resuscitation strategy.17
Aggressive crystalloid administration in hemorrhagic shock also leads to dilutional coagulopathy. Multiple animal and human studies have shown an association between increased crystalloid volumes in hemorrhaging patients and increasing coagulopathy, blood loss, and mortality. In 2004, Barak et al18 demonstrated that administration of a high volume of crystalloid fluid (>3 L) or colloid (500 mL) was associated with postoperative coagulopathy; whereas in 2017, Harada et al,19 at Cedars-Sinai Medical Center in New York, demonstrated over a 10-year period that decreased high-volume (>2 L) crystalloid resuscitation paralleled a decrease in mortality.
Massive Transfusion Protocols. Many trauma centers have shifted away from high-volume crystalloid resuscitation in favor of massive transfusion protocols (MTPs) utilizing standardized ratios that more closely mimic whole blood. The MTPs center on the principle of equal transfusion ratios of blood product as opposed to packed RBCs (PRBCs) alone. This means effecting a plasma-rich resuscitation and preemptive correction of coagulopathy with FFP and platelets in addition to PRBCs.
Data from a US Army combat support hospital have demonstrated improved survival with an FFP to PRBC ratio of more than 1:1.4,20 and civilian studies have produced similar findings.21-23 All of these studies also noted improved mortality with higher (>1:2) platelet to PRBC ratios.22,23 Although, the ideal ratio remains unknown, many MTPs aim for 1:1:1 ratio (6 U FFP to 6 packed platelets to 6 U PRBC), which most closely mimics whole blood.
The Pragmatic Randomized Optimal Platelet and Plasma Ratios trial was a recent large multicenter randomized trial that compared transfusion ratios of 1:1:1 and 1:1:2. The trial was unable to demonstrate a difference in mortality at either 24 hours or 30 days, though more patients in the 1:1:1 ratio group achieved hemostasis and fewer patients in this group died from exsanguination in the first 24 hours.24Prehospital PRBC Administration. A number of studies have looked at prehospital administration of PRBCs.25-27 Holcomb et al25 showed no overall survival advantage at 24 hours, but did demonstrate a negligible blood-product wastage. In 2015 Brown et al26 found an increase probability of 24-hour survival, decreased shock, and lower 24-hour PRBC requirements with pretrauma-center PRBC transfusion. That same year Brown et al27 demonstrated that prehospital PRBC transfusion in severely injured blunt trauma patients was associated with decreased 24-hour and 30-day mortality rates, and a lower risk of coagulopathy. Currently, the Prehospital Air Medical Plasma trial is enrolling patients to evaluate the prehospital administration of plasma.28 The primary endpoint of the study is 30-day mortality; the tentative completion date for the study is January 2018.
Tranexamic Acid. Another important development in the treatment of hemorrhagic shock in recent years has been the use of TXA, an antifibrinolytic agent which inhibits the conversion of plasminogen to plasmin. It has been shown to decrease mortality in both civilian and military trauma populations.29,30
The Clinical Randomization of an Antifibrinolytic in Significant Hemorrhage 2 trial was a large multicenter randomized trial, which showed a survival benefit among those who received TXA. The generalizability of the study has been questioned in the setting of modern urban trauma centers, as most of those enrolled in the study were from hospitals with no formal MTPs and a limited availability of blood products. Additionally, no laboratory measures of fibrinolysis were available.30
Most experts currently recommend TXA use as part of an MTP if there is evidence of hyperfibrinolysis on TEG or in severe hemorrhagic shock when the time from injury has been less than 3 hours, as studies have shown increased mortality when TXA was administered longer than 3 hours after injury.30
Viscoelastic Assays
An alternative approach to standardized ratio MTPs involves goal-directed hemostatic resuscitation using viscoelastic assays to guide transfusion of blood-product components. Both TEG and rotational thromboelastometry (ROTEM) are point-of-care tools for assessment of coagulation parameters of whole blood. Although they are not new technology, their use in trauma resuscitation is a relatively new concept.
While ROTEM is more commonly used in Europe, TEG is more popular and commonly used in the United States, though not exclusively.31,32
Thromboelastography
The TEG parameters most commonly used clinically are reaction time (R-time), kinetics time, angle, maximum amplitude (MA), and lysis at 30 minutes (LY30).
Reaction Time. The R-time is measured in minutes and represents the time to clot initiation, reflecting activity of coagulation factors. It is used in TEG-guided MTPs to trigger transfusion of FFP.31,32 The R-time is measured at the time the clot strength reaches an amplitude of 2 mm.31 The angle reflects the rate of rise of the amplitude of the TEG tracing, or the rate of increase in clot strength. Clinically, the angle represents fibrinogen concentration and function, and is used to trigger transfusion of cryoprecipitate or fibrinogen concentrate in MTPs.31,32
Maximum Amplitude. The MA is reached by the TEG tracing, or the maximum clot strength achieved. Although the MA has been shown to correlate with platelet count, it actually represents platelet count and function as well as fibrinogen activity, all of which contribute to clot strength. Clinically, MA is used to trigger platelet transfusion and/or administration of desmopressin in MTPs.31,32
Lysis at 30 Minutes. The LY30 is defined as the percent reduction in clot strength 30 minutes after reaching MA.31,32 Normal LY30 values are between 0% and 7.5%; however, these values have been challenged in recent studies, which have reported that an LY30 greater than 3% (termed hyperfibrinolysis) confers a significant increase in mortality and an increased likelihood of requiring massive transfusion.31,33 These findings have led to incorporation of this lower threshold as a trigger for administration of TXA during MTPs. Furthermore, an LY30 of less than 0.8% (described as fibrinolysis shutdown) has also been found to confer an increase in mortality,34 which has led many to advocate for goal-directed administration of TXA, rather than empiric administration, as these patients are more likely to be harmed than helped by such an intervention.31
Rapid Thromboelastography
Rapid TEG employs tissue factor to accelerate clot initiation and reaction time, providing an additional parameter which reflects coagulation factor activity: the activated clotting time (ACT).32
Activated Clotting Time. Historically used in cardiac surgery to measure anticoagulation during a cardiopulmonary bypass, ACT represents the same phase of coagulation as R-time, but is measured in seconds instead of minutes.31 The ACT has been found to correlate with prothrombin time/international normalized ratio (PT/INR), and accurately predicts the need for MTP.
Cotton et al35 found that an ACT of more than 128 seconds predicted patients requiring MTP, and an ACT lower than 105 seconds predicted those who required no transfusions in the first 24 hours after injury.35
The ACT can be used to trigger transfusion of FFP, but at certain thresholds, may also be used to trigger the early transfusion of cryoprecipitate and platelets.36 Moore et al36 found that an ACT over 140 seconds was able to predict an abnormal angle and MA. This had led to using this threshold as a trigger for early administration of cryoprecipitate and platelets, given this parameter is available within 5 minutes—long before the angle and MA have resulted.
Efficacy
The use of a TEG-guided strategy for MTP in trauma has shown great promise. In 2013, Tapia et al37 compared a historical cohort who received 1:1:1 MTP to a TEG-guided MTP and demonstrated improved mortality.In 2016, Gonzalez et al38 compared TEG-guided transfusion vs conventional coagulation tests (PT/INR, PTT, fibrinogen, platelets). The authors found a significant decrease in mortality and platelet and FFP transfusion when TEG-guided resuscitation is used.
Endovascular Techniques
The use of endovascular techniques in trauma continues to evolve. According to the National Trauma Data Bank, the use of endovascular therapies has increased from 1% of trauma cases in 2002 to 11% in 2008.39
Thoracic Endovascular Aortic Repair
Thoracic endovascular aortic repair (TEVAR) for blunt thoracic aortic injury has essentially replaced open surgical repair. (See Figures 1a and 1b for an example of a blunt traumatic aortic injury prior to and post-TEVAR placement.)
Transarterial Catheter Embolization
Endovascular treatments have also been used successfully in the management of injuries to aortic branch vessels and extremity vessels.42 Transarterial catheter embolization with coils, plugs, or gel foam is being employed with increasing frequency to achieve hemostasis in the pelvis and spleen.42 It may also be used as an adjunct to laparotomy and perihepatic packing in high-grade liver injuries, though it is associated with significant morbidity related to hepatic necrosis, bile leaks, and abscesses.43,44
Resuscitative Endovascular Balloon Occlusion of the Aorta
Most recently, REBOA has been used for noncompressible torso hemorrhage following trauma. This method involves percutaneous arterial cannulation of the common femoral artery and advancement of a balloon into the aorta, where it is then inflated at the desired level.
Once inflated, the balloon obstructs arterial inflow to the area of hemorrhage, curtailing blood loss, and increases proximal BP, improving coronary and cerebral perfusion. Multiple case reports and case series have described successful use of REBOA for hemorrhage control, including prehospital use by physicians in the United Kingdom. The largest series to date looked at 114 patients, of whom 46 had REBOA placement and 68 had open aortic occlusion through resuscitative thoracotomy.45 Those treated with REBOA were significantly more likely to achieve hemodynamic stability (defined as SBP >90 mm Hg for >5 minutes). Furthermore, the authors noted minimal complications from REBOA and no difference in time to successful aortic occlusion, regardless of technique. There was also no difference in mortality between the two groups. Despite the small number of studies in trauma patients, REBOA has been established as a viable alternative to open aortic occlusion. The prospective Aortic Occlusion for Resuscitation in Trauma and Acute Care Surgery registry established by the American Association for the Surgery of Trauma is continuing to enroll patients and will hopefully answer many of the current uncertainties regarding the use of REBOA.
Conclusion
Strategies and techniques for the care of the injured patient have changed significantly in the past few years. Damage control resuscitation includes three elements: damage control surgery, permissive hypotension, and blood-product resuscitation.
The goals of lowering MAP in hemorrhagic shock appear to be safe and make sense physiologically, but have yet to show clear mortality benefit. Avoidance of excessive crystalloid resuscitation and trends toward more physiological ratios of blood product resuscitation have shown better outcomes. While the ideal ratio of blood products in transfusion remains unknown, the use of a massive transfusion strategy is preferable to crystalloid fluids. The use of viscoelastic assays (TEG and ROTEM) have allowed for goal-directed blood product resuscitation and may improve outcomes when compared with prescribed resuscitation ratios.
Finally, endovascular techniques in trauma have been increasingly utilized over the past 15 years, making nonoperative management with angiographic embolization for solid organ injury common practice now in most trauma centers worldwide. Temporary aortic balloon occlusion with REBOA appears promising in many cases of noncompressible truncal hemorrhage until definitive hemostasis can be achieved, but studies are needed to determine its ultimate place in the care of the trauma patient.
1. Evans JA, van Wessem KJ, McDougall D, Lee KA, Lyons T, Balogh ZJ. Epidemiology of traumatic deaths: comprehensive population-based assessment. World J Surg. 2010;34(1):158-163. doi:10.1007/s00268-009-0266-1.
2. Bogert JN, Harvin JA, Cotton BA. Damage control resuscitation. J Intensive Care Med. 2016;31(3):177-186. doi:10.1177/0885066614558018.
3. Kaafarani HMA, Velmahos GC. Damage control resuscitation in trauma. Scand J Surg. 2014;103(2):81-88. doi:10.1177/1457496914524388.
4. Bickell WH, Wall MJ Jr, Pepe PE, et al. Immediate versus delayed fluid resuscitation for hypotensive patients with penetrating torso injuries. N Engl J Med. 1994;331(17):1105-1109. doi:10.1056/NEJM199410273311701.
5. Sondeen JL, Coppes VG, Holcomb JB. Blood pressure at which rebleeding occurs after resuscitation in swine with aortic injury. J Trauma. 2003;54(5 Suppl):S110-S117. doi:10.1097/01.TA.0000047220.81795.3D.
6. Li T, Zhu Y, Hu Y, et al. Ideal permissive hypotension to resuscitate uncontrolled hemorrhagic shock and the tolerance time in rats. Anesthesiology. 2011;114(1):111-119. doi:10.1097/ALN.0b013e3181fe3fe7.
7. Dutton RP, Mackenzie CF, Scalea TM. Hypotensive resuscitation during active hemorrhage: impact on in-hospital mortality. J Trauma. 2002;52(6):1141-1146.
8. Schreiber MA, Meier EN, Tisherman SA, et al; ROC Investigators. A controlled resuscitation strategy is feasible and safe in hypotensive trauma patients: results of a prospective randomized pilot trial. J Trauma Acute Care Surg. 2015;78(4):687-695. doi:10.1097/TA.0000000000000600.
9. Morrison CA, Carrick MM, Norman MA, et al. Hypotensive resuscitation strategy reduces transfusion requirements and severe postoperative coagulopathy in trauma patients with hemorrhagic shock: preliminary results of a randomized controlled trial. J Trauma. 2011;70(3):652-663. doi:10.1097/TA.0b013e31820e77ea.
10. Carrick MM, Morrison CA, Tapia NM, et al. Intraoperative hypotensive resuscitation for patients undergoing laparotomy or thoracotomy for trauma: Early termination of a randomized prospective clinical trial. J Trauma Acute Care Surg. 2016;80(6):886-896. doi:10.1097/TA.0000000000001044.
11. Chesnut RM, Marshall LF, Klauber MR, et al. The role of secondary brain injury in determining outcome from severe head injury. J Trauma. 1993;34(2):216-222.
12. Kasotakis G, Sideris A, Yang Y, et al. Inflammation and Host Response to Injury Investigators. Aggressive early crystalloid resuscitation adversely affects outcomes in adult blunt trauma patients: an analysis of the Glue Grant database. J Trauma Acute Care Surg. 2013;74(5):1215-1221; discussion 1221-1222. doi:10.1097/TA.0b013e3182826e13.
13. Cotton BA, Guy JS, Morris JA Jr, Abumrad NN. The cellular, metabolic, and systemic consequences of aggressive fluid resuscitation strategies. Shock. 2006;26(2):115-121. doi:10.1097/01.shk.0000209564.84822.f2.
14. Kozar RA, Peng Z, Zhang R, et al. Plasma restoration of endothelial glycocalyx in a rodent model of hemorrhagic shock. Anesth Analg. 2011;112(6):1289-1295. doi:10.1213/ANE.0b013e318210385c.
15. Torres LN, Sondeen JL, Ji L, Dubick MA, Filho IT. Evaluation of resuscitation fluids on endothelial glycocalyx, venular blood flow, and coagulation function after hemorrhagic shock in rats. J Trauma Acute Care Surg. 2013;75(5):759-766. doi:10.1097/TA.0b013e3182a92514.
16. Bradley M, Galvagno S, Dhanda A, et al. Damage control resuscitation protocol and the management of open abdomens in trauma patients. Am Surg. 2014;80(8):768-775.
17. Brandstrup B, Tønnesen H, Beier-Holgersen R, et al; Danish Study Group on Perioperative Fluid Therapy. Effects of intravenous fluid restriction on postoperative complications: comparison of two perioperative fluid regimens: a randomized assessor-blinded multicenter trial. Ann Surg. 2003;238(5):641-648. doi:10.1097/01.sla.0000094387.50865.23.
18. Barak M, Rudin M, Vofsi O, Droyan A, Katz Y. Fluid administration during abdominal surgery influences on coagulation in the postoperative period. Curr Surg. 2004;61(5):459-462. doi:10.1016/j.cursur.2004.02.002.
19. Harada MY, Ko A, Barmparas G, et al. 10-Year trend in crystalloid resuscitation: Reduced volume and lower mortality. Int J Surg. 2017;38:78-82. doi:10.1016/j.ijsu.2016.12.073.
20. Borgman MA, Spinella PC, Perkins JG, et al. The ratio of blood products transfused affects mortality in patients receiving massive transfusions at a combat support hospital. J Trauma. 2007;63(4):805-813. doi:10.1097/TA.0b013e3181271ba3.
21. Cotton BA, Gunter OL, Isbell J, et al. Damage control hematology: the impact of a trauma exsanguination protocol on survival and blood product utilization. J Trauma. 2008;64(5):1177-1782; discussion 1182-1183. doi:10.1097/TA.0b013e31816c5c80.
22. Holcomb JB, Wade CE, Michalek JE, et al. Increased plasma and platelet to red blood cell ratios improves outcome in 466 massively transfused civilian trauma patients. Ann Surg. 2008;248(3):447-458. doi:10.1097/SLA.0b013e318185a9ad.
23. Holcomb JB, del Junco DJ, Fox EE, et al; PROMMTT Study Group. The prospective, observational, multicenter, major trauma transfusion (PROMMTT) study: comparative effectiveness of a time-varying treatment with competing risks. JAMA Surg. 2013;148(2):127-136. doi:10.1001/2013.jamasurg.387.
24. Holcomb JB, Tilley BC, Baraniuk S, et al; PROPPR Study Group. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA. 2015;313(5):471-482. doi:10.1001/jama.2015.12.
25. Holcomb JB, Donathan DP, Cotton BA, et al. Prehospital transfusion of plasma and red blood cells in trauma patients. Prehosp Emerg Care. 2015;19(1):1-9. doi:10.3109/10903127.2014.923077.
26. Brown JB, Sperry JL, Fombona A, Billiar TR, Peitzman AB, Guyette FX. Pre-trauma center red blood cell transfusion is associated with improved early outcomes in air medical trauma patients. J Am Coll Surg. 2015;220(5):797-808. doi:10.1016/j.jamcollsurg.2015.01.006.
27. Brown JB, Cohen MJ, Minei JP, et al; Inflammation and the Host Response to Injury Investigators. Pretrauma center red blood cell transfusion is associated with reduced mortality and coagulopathy in severely injured patients with blunt trauma. Ann Surg. 2015;261(5):997-1005. doi:10.1097/SLA.0000000000000674.
28. Brown JB, Guyette FX, Neal MD, et al. Taking the blood bank to the field: the design and rationale of the Prehospital Air Medical Plasma (PAMPer) trial. Prehosp Emerg Care. 2015;19(3):343-350. doi:10.3109/10903127.2014.995851.
29. Morrison JJ, Dubose JJ, Rasmussen TE, Midwinter MJ. Military Application of Tranexamic Acid in Trauma Emergency Resuscitation (MATTERs) study. Arch Surg. 2012;147(2):113-119. doi:10.1001/archsurg.2011.287.
30. Shakur H, Roberts I, Bautista R, et al; CRASH-2 trial collaborators. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet. 2010;376(9734):23-32. doi:10.1016/S0140-6736(10)60835-5.
31. Gonzalez E, Moore EE, Moore HB. Management of trauma-induced coagulopathy with thrombelastography. Crit Care Clin. 2017;33(1):119-134. doi:10.1016/j.ccc.2016.09.002.
32. Abdelfattah K, Cripps MW. Thromboelastography and rotational thromboelastometry use in trauma. Int J Surg. 2016;33(Pt B):196-201. doi:10.1016/j.ijsu.2015.09.036.
33. Cotton BA, Harvin JA, Kostousouv V, et al. Hyperfibrinolysis at admission is an uncommon but highly lethal event associated with shock and prehospital fluid administration. J Trauma Acute Care Surg. 2012;73(2):365-370; discussion 370. doi:10.1097/TA.0b013e31825c1234.
34. Moore HB, Moore EE, Gonzalez E, et al. Hyperfibrinolysis, physiologic fibrinolysis, and fibrinolysis shutdown: the spectrum of postinjury fibrinolysis and relevance to antifibrinolytic therapy. J Trauma Acute Care Surg. 2014;77(6):811-817. doi:10.1097/TA.0000000000000341.
35. Cotton BA, Faz G, Hatch QM, et al. Rapid thrombelastography delivers real-time results that predict transfusion within 1 hour of admission. J Trauma. 2011;71(2):407-414; discussion 414-417. doi:10.1097/TA.0b013e31821e1bf0.
36. Moore HB, Moore EE, Chin TL, et al. Activated clotting time of thrombelastography (T-ACT) predicts early postinjury blood component transfusion beyond plasma. Surgery. 2014;156(3):564-569. doi:10.1016/j.surg.2014.04.017.
37. Tapia NM, Chang A, Norman M, et al. TEG-guided resuscitation is superior to standardized MTP resuscitation in massively transfused penetrating trauma patients. J Trauma Acute Care Surg. 2013;74(2):378-385; discussion 385-386. doi:10.1097/TA.0b013e31827e20e0.
38. Gonzalez E, Moore EE, Moore HB, et al. Goal-directed hemostatic resuscitation of trauma-induced coagulopathy: a pragmatic randomized clinical trial comparing a viscoelastic assay to conventional coagulation assays. Ann Surg. 2016;263(6):1051-1059. doi:10.1097/SLA.0000000000001608.
39. Avery LE, Stahlfeld KR, Corcos AC, et al. Evolving role of endovascular techniques for traumatic vascular injury: a changing landscape? J Trauma Acute Care Surg. 2012;72(1):41-46; discussion 46-47. doi:10.1097/TA.0b013e31823d0f03.
40. Demetriades D, Velmahos GC, Scalea TM, et al. Diagnosis and treatment of blunt thoracic aortic injuries: changing perspectives. J Trauma. 2008;64(6):1415-1418; discussion 1418-1419. doi:10.1097/TA.0b013e3181715e32.
41. Azizzadeh A, Ray HM, Dubose JJ, et al. Outcomes of endovascular repair for patients with blunt traumatic aortic injury. J Trauma Acute Care Surg. 2014;76(2):510-516. doi:10.1097/TA.0b013e3182aafe8c.
42. Brenner M, Hoehn M, Rasmussen TE. Endovascular therapy in trauma. Eur J Trauma Emerg Surg. 2014;40(6):671-678. doi:10.1007/s00068-014-0474-8.
43. Dabbs DN, Stein DM, Scalea TM. Major hepatic necrosis: a common complication after angioembolization for treatment of high-grade liver injuries. J Trauma. 2009;66(3):621-627; discussion 627-629. doi:10.1097/TA.0b013e31819919f2.
44. Letoublon C, Morra I, Chen Y, Monnin V, Voirin D, Arvieux C. Hepatic arterial embolization in the management of blunt hepatic trauma: indications and complications. J Trauma. 2011;70(5):1032-1036; discussion 1036-1037. doi:10.1097/TA.0b013e31820e7ca1.
45. DuBose JJ, Scalea TM, Brenner M, et al; AORTA Study Group. The AAST prospective Aortic Occlusion for Resuscitati on in Trauma and Acute Care Surgery (AORTA) registry: Data on contemporary utilization and outcomes of aortic occlusion and resuscitative balloon occlusion of the aorta (REBOA). J Trauma Acute Care Surg. 2016;81(3):409-419. doi:10.1097/TA.0000000000001079.
1. Evans JA, van Wessem KJ, McDougall D, Lee KA, Lyons T, Balogh ZJ. Epidemiology of traumatic deaths: comprehensive population-based assessment. World J Surg. 2010;34(1):158-163. doi:10.1007/s00268-009-0266-1.
2. Bogert JN, Harvin JA, Cotton BA. Damage control resuscitation. J Intensive Care Med. 2016;31(3):177-186. doi:10.1177/0885066614558018.
3. Kaafarani HMA, Velmahos GC. Damage control resuscitation in trauma. Scand J Surg. 2014;103(2):81-88. doi:10.1177/1457496914524388.
4. Bickell WH, Wall MJ Jr, Pepe PE, et al. Immediate versus delayed fluid resuscitation for hypotensive patients with penetrating torso injuries. N Engl J Med. 1994;331(17):1105-1109. doi:10.1056/NEJM199410273311701.
5. Sondeen JL, Coppes VG, Holcomb JB. Blood pressure at which rebleeding occurs after resuscitation in swine with aortic injury. J Trauma. 2003;54(5 Suppl):S110-S117. doi:10.1097/01.TA.0000047220.81795.3D.
6. Li T, Zhu Y, Hu Y, et al. Ideal permissive hypotension to resuscitate uncontrolled hemorrhagic shock and the tolerance time in rats. Anesthesiology. 2011;114(1):111-119. doi:10.1097/ALN.0b013e3181fe3fe7.
7. Dutton RP, Mackenzie CF, Scalea TM. Hypotensive resuscitation during active hemorrhage: impact on in-hospital mortality. J Trauma. 2002;52(6):1141-1146.
8. Schreiber MA, Meier EN, Tisherman SA, et al; ROC Investigators. A controlled resuscitation strategy is feasible and safe in hypotensive trauma patients: results of a prospective randomized pilot trial. J Trauma Acute Care Surg. 2015;78(4):687-695. doi:10.1097/TA.0000000000000600.
9. Morrison CA, Carrick MM, Norman MA, et al. Hypotensive resuscitation strategy reduces transfusion requirements and severe postoperative coagulopathy in trauma patients with hemorrhagic shock: preliminary results of a randomized controlled trial. J Trauma. 2011;70(3):652-663. doi:10.1097/TA.0b013e31820e77ea.
10. Carrick MM, Morrison CA, Tapia NM, et al. Intraoperative hypotensive resuscitation for patients undergoing laparotomy or thoracotomy for trauma: Early termination of a randomized prospective clinical trial. J Trauma Acute Care Surg. 2016;80(6):886-896. doi:10.1097/TA.0000000000001044.
11. Chesnut RM, Marshall LF, Klauber MR, et al. The role of secondary brain injury in determining outcome from severe head injury. J Trauma. 1993;34(2):216-222.
12. Kasotakis G, Sideris A, Yang Y, et al. Inflammation and Host Response to Injury Investigators. Aggressive early crystalloid resuscitation adversely affects outcomes in adult blunt trauma patients: an analysis of the Glue Grant database. J Trauma Acute Care Surg. 2013;74(5):1215-1221; discussion 1221-1222. doi:10.1097/TA.0b013e3182826e13.
13. Cotton BA, Guy JS, Morris JA Jr, Abumrad NN. The cellular, metabolic, and systemic consequences of aggressive fluid resuscitation strategies. Shock. 2006;26(2):115-121. doi:10.1097/01.shk.0000209564.84822.f2.
14. Kozar RA, Peng Z, Zhang R, et al. Plasma restoration of endothelial glycocalyx in a rodent model of hemorrhagic shock. Anesth Analg. 2011;112(6):1289-1295. doi:10.1213/ANE.0b013e318210385c.
15. Torres LN, Sondeen JL, Ji L, Dubick MA, Filho IT. Evaluation of resuscitation fluids on endothelial glycocalyx, venular blood flow, and coagulation function after hemorrhagic shock in rats. J Trauma Acute Care Surg. 2013;75(5):759-766. doi:10.1097/TA.0b013e3182a92514.
16. Bradley M, Galvagno S, Dhanda A, et al. Damage control resuscitation protocol and the management of open abdomens in trauma patients. Am Surg. 2014;80(8):768-775.
17. Brandstrup B, Tønnesen H, Beier-Holgersen R, et al; Danish Study Group on Perioperative Fluid Therapy. Effects of intravenous fluid restriction on postoperative complications: comparison of two perioperative fluid regimens: a randomized assessor-blinded multicenter trial. Ann Surg. 2003;238(5):641-648. doi:10.1097/01.sla.0000094387.50865.23.
18. Barak M, Rudin M, Vofsi O, Droyan A, Katz Y. Fluid administration during abdominal surgery influences on coagulation in the postoperative period. Curr Surg. 2004;61(5):459-462. doi:10.1016/j.cursur.2004.02.002.
19. Harada MY, Ko A, Barmparas G, et al. 10-Year trend in crystalloid resuscitation: Reduced volume and lower mortality. Int J Surg. 2017;38:78-82. doi:10.1016/j.ijsu.2016.12.073.
20. Borgman MA, Spinella PC, Perkins JG, et al. The ratio of blood products transfused affects mortality in patients receiving massive transfusions at a combat support hospital. J Trauma. 2007;63(4):805-813. doi:10.1097/TA.0b013e3181271ba3.
21. Cotton BA, Gunter OL, Isbell J, et al. Damage control hematology: the impact of a trauma exsanguination protocol on survival and blood product utilization. J Trauma. 2008;64(5):1177-1782; discussion 1182-1183. doi:10.1097/TA.0b013e31816c5c80.
22. Holcomb JB, Wade CE, Michalek JE, et al. Increased plasma and platelet to red blood cell ratios improves outcome in 466 massively transfused civilian trauma patients. Ann Surg. 2008;248(3):447-458. doi:10.1097/SLA.0b013e318185a9ad.
23. Holcomb JB, del Junco DJ, Fox EE, et al; PROMMTT Study Group. The prospective, observational, multicenter, major trauma transfusion (PROMMTT) study: comparative effectiveness of a time-varying treatment with competing risks. JAMA Surg. 2013;148(2):127-136. doi:10.1001/2013.jamasurg.387.
24. Holcomb JB, Tilley BC, Baraniuk S, et al; PROPPR Study Group. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA. 2015;313(5):471-482. doi:10.1001/jama.2015.12.
25. Holcomb JB, Donathan DP, Cotton BA, et al. Prehospital transfusion of plasma and red blood cells in trauma patients. Prehosp Emerg Care. 2015;19(1):1-9. doi:10.3109/10903127.2014.923077.
26. Brown JB, Sperry JL, Fombona A, Billiar TR, Peitzman AB, Guyette FX. Pre-trauma center red blood cell transfusion is associated with improved early outcomes in air medical trauma patients. J Am Coll Surg. 2015;220(5):797-808. doi:10.1016/j.jamcollsurg.2015.01.006.
27. Brown JB, Cohen MJ, Minei JP, et al; Inflammation and the Host Response to Injury Investigators. Pretrauma center red blood cell transfusion is associated with reduced mortality and coagulopathy in severely injured patients with blunt trauma. Ann Surg. 2015;261(5):997-1005. doi:10.1097/SLA.0000000000000674.
28. Brown JB, Guyette FX, Neal MD, et al. Taking the blood bank to the field: the design and rationale of the Prehospital Air Medical Plasma (PAMPer) trial. Prehosp Emerg Care. 2015;19(3):343-350. doi:10.3109/10903127.2014.995851.
29. Morrison JJ, Dubose JJ, Rasmussen TE, Midwinter MJ. Military Application of Tranexamic Acid in Trauma Emergency Resuscitation (MATTERs) study. Arch Surg. 2012;147(2):113-119. doi:10.1001/archsurg.2011.287.
30. Shakur H, Roberts I, Bautista R, et al; CRASH-2 trial collaborators. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet. 2010;376(9734):23-32. doi:10.1016/S0140-6736(10)60835-5.
31. Gonzalez E, Moore EE, Moore HB. Management of trauma-induced coagulopathy with thrombelastography. Crit Care Clin. 2017;33(1):119-134. doi:10.1016/j.ccc.2016.09.002.
32. Abdelfattah K, Cripps MW. Thromboelastography and rotational thromboelastometry use in trauma. Int J Surg. 2016;33(Pt B):196-201. doi:10.1016/j.ijsu.2015.09.036.
33. Cotton BA, Harvin JA, Kostousouv V, et al. Hyperfibrinolysis at admission is an uncommon but highly lethal event associated with shock and prehospital fluid administration. J Trauma Acute Care Surg. 2012;73(2):365-370; discussion 370. doi:10.1097/TA.0b013e31825c1234.
34. Moore HB, Moore EE, Gonzalez E, et al. Hyperfibrinolysis, physiologic fibrinolysis, and fibrinolysis shutdown: the spectrum of postinjury fibrinolysis and relevance to antifibrinolytic therapy. J Trauma Acute Care Surg. 2014;77(6):811-817. doi:10.1097/TA.0000000000000341.
35. Cotton BA, Faz G, Hatch QM, et al. Rapid thrombelastography delivers real-time results that predict transfusion within 1 hour of admission. J Trauma. 2011;71(2):407-414; discussion 414-417. doi:10.1097/TA.0b013e31821e1bf0.
36. Moore HB, Moore EE, Chin TL, et al. Activated clotting time of thrombelastography (T-ACT) predicts early postinjury blood component transfusion beyond plasma. Surgery. 2014;156(3):564-569. doi:10.1016/j.surg.2014.04.017.
37. Tapia NM, Chang A, Norman M, et al. TEG-guided resuscitation is superior to standardized MTP resuscitation in massively transfused penetrating trauma patients. J Trauma Acute Care Surg. 2013;74(2):378-385; discussion 385-386. doi:10.1097/TA.0b013e31827e20e0.
38. Gonzalez E, Moore EE, Moore HB, et al. Goal-directed hemostatic resuscitation of trauma-induced coagulopathy: a pragmatic randomized clinical trial comparing a viscoelastic assay to conventional coagulation assays. Ann Surg. 2016;263(6):1051-1059. doi:10.1097/SLA.0000000000001608.
39. Avery LE, Stahlfeld KR, Corcos AC, et al. Evolving role of endovascular techniques for traumatic vascular injury: a changing landscape? J Trauma Acute Care Surg. 2012;72(1):41-46; discussion 46-47. doi:10.1097/TA.0b013e31823d0f03.
40. Demetriades D, Velmahos GC, Scalea TM, et al. Diagnosis and treatment of blunt thoracic aortic injuries: changing perspectives. J Trauma. 2008;64(6):1415-1418; discussion 1418-1419. doi:10.1097/TA.0b013e3181715e32.
41. Azizzadeh A, Ray HM, Dubose JJ, et al. Outcomes of endovascular repair for patients with blunt traumatic aortic injury. J Trauma Acute Care Surg. 2014;76(2):510-516. doi:10.1097/TA.0b013e3182aafe8c.
42. Brenner M, Hoehn M, Rasmussen TE. Endovascular therapy in trauma. Eur J Trauma Emerg Surg. 2014;40(6):671-678. doi:10.1007/s00068-014-0474-8.
43. Dabbs DN, Stein DM, Scalea TM. Major hepatic necrosis: a common complication after angioembolization for treatment of high-grade liver injuries. J Trauma. 2009;66(3):621-627; discussion 627-629. doi:10.1097/TA.0b013e31819919f2.
44. Letoublon C, Morra I, Chen Y, Monnin V, Voirin D, Arvieux C. Hepatic arterial embolization in the management of blunt hepatic trauma: indications and complications. J Trauma. 2011;70(5):1032-1036; discussion 1036-1037. doi:10.1097/TA.0b013e31820e7ca1.
45. DuBose JJ, Scalea TM, Brenner M, et al; AORTA Study Group. The AAST prospective Aortic Occlusion for Resuscitati on in Trauma and Acute Care Surgery (AORTA) registry: Data on contemporary utilization and outcomes of aortic occlusion and resuscitative balloon occlusion of the aorta (REBOA). J Trauma Acute Care Surg. 2016;81(3):409-419. doi:10.1097/TA.0000000000001079.

Affordable Care: Back to the Future?
In the days before this issue of Emergency Medicine (EM) went to press, the United States Senate tried unsuccessfully, first to repeal and replace the Affordable Care Act (ACA), then to repeal key provisions of ACA without a replacement bill. Despite having a majority in both the Senate and House of Representatives as well as a Republican President, after 7 years of vowing to repeal “Obama Care,” Republicans have still not been able to fulfill that vow.
When ACA was signed into law in March 2010 (See “Springing Forward,” April 2010 EM), we wrote “though the new law will undoubtedly be challenged, tested, modified, refined, used—and probably abused—it will not be repealed. As was the case with Medicare and Medicaid previously, this will change everything in subtle and not-so-subtle ways.” (For a discussion of how the healthcare industry has managed to co-opt and abuse ACA, see the recently published book An American Sickness by Elisabeth Rosenthal, who was an emergency physician [EP] in our department before becoming a senior science and healthcare reporter for the New York Times.)
But the failure of ACA to deliver on many of its promises, its uncertain financial future, and the lack of improvements to ACA since 2010, directly or indirectly affects every American. Predictably, for those in need of care who cannot find a physician to accept their insurance or schedule a timely appointment, the ED remains the safety net for obtaining care.
After the constitutionality of ACA was upheld by the Supreme Court in June 2012 (See “Our National Pastime,” July 2012 EM), we noted that “ACA contains no provisions for increasing the number of healthcare providers [and] if 24 million more Americans now have access to affordable health insurance, but there are no new providers, who will they go to for care?” Seven years after passage of ACA, the answer to this question has been provided by published studies confirming that even more insured Americans are now seeking care in EDs than before “affordable care” became available. At the same time, urgent care centers, freestanding EDs, and “convenient care” centers, have sprung up and proliferated throughout the country, while in many states, nurse practitioners, physician assistants, and now emergency medical technicians and paramedics have sought and received authorization to evaluate and treat patients independent of physician supervision and oversight. Telemedicine or “telehealth” is the latest attempt to stretch the available supply of physicians to manage patients remotely, in the hope of obviating the need for an ED visit.
But none of these measures completely addresses a basic weakness of ACA: There are not enough physicians, including EPs, in this country to care for everyone entitled to healthcare; at the same time, there is a generation of highly qualified, highly motivated young men and women seeking entrance to medical school who will never get the opportunity to become fine physicians because there are not enough places for them. The solution to these problems seems obvious and the funds needed to finance it would be well spent, though the benefits of increasing the number of medical school places would not be realized for 4 to 8 years after they are made available.
In the meantime, we leave you with the solution President George W. Bush offered to a Cleveland audience on July 10, 2007 (See “Dream On,” March 2008 EM): “people have access to healthcare in America. After all, you just go to an emergency room.”
In the days before this issue of Emergency Medicine (EM) went to press, the United States Senate tried unsuccessfully, first to repeal and replace the Affordable Care Act (ACA), then to repeal key provisions of ACA without a replacement bill. Despite having a majority in both the Senate and House of Representatives as well as a Republican President, after 7 years of vowing to repeal “Obama Care,” Republicans have still not been able to fulfill that vow.
When ACA was signed into law in March 2010 (See “Springing Forward,” April 2010 EM), we wrote “though the new law will undoubtedly be challenged, tested, modified, refined, used—and probably abused—it will not be repealed. As was the case with Medicare and Medicaid previously, this will change everything in subtle and not-so-subtle ways.” (For a discussion of how the healthcare industry has managed to co-opt and abuse ACA, see the recently published book An American Sickness by Elisabeth Rosenthal, who was an emergency physician [EP] in our department before becoming a senior science and healthcare reporter for the New York Times.)
But the failure of ACA to deliver on many of its promises, its uncertain financial future, and the lack of improvements to ACA since 2010, directly or indirectly affects every American. Predictably, for those in need of care who cannot find a physician to accept their insurance or schedule a timely appointment, the ED remains the safety net for obtaining care.
After the constitutionality of ACA was upheld by the Supreme Court in June 2012 (See “Our National Pastime,” July 2012 EM), we noted that “ACA contains no provisions for increasing the number of healthcare providers [and] if 24 million more Americans now have access to affordable health insurance, but there are no new providers, who will they go to for care?” Seven years after passage of ACA, the answer to this question has been provided by published studies confirming that even more insured Americans are now seeking care in EDs than before “affordable care” became available. At the same time, urgent care centers, freestanding EDs, and “convenient care” centers, have sprung up and proliferated throughout the country, while in many states, nurse practitioners, physician assistants, and now emergency medical technicians and paramedics have sought and received authorization to evaluate and treat patients independent of physician supervision and oversight. Telemedicine or “telehealth” is the latest attempt to stretch the available supply of physicians to manage patients remotely, in the hope of obviating the need for an ED visit.
But none of these measures completely addresses a basic weakness of ACA: There are not enough physicians, including EPs, in this country to care for everyone entitled to healthcare; at the same time, there is a generation of highly qualified, highly motivated young men and women seeking entrance to medical school who will never get the opportunity to become fine physicians because there are not enough places for them. The solution to these problems seems obvious and the funds needed to finance it would be well spent, though the benefits of increasing the number of medical school places would not be realized for 4 to 8 years after they are made available.
In the meantime, we leave you with the solution President George W. Bush offered to a Cleveland audience on July 10, 2007 (See “Dream On,” March 2008 EM): “people have access to healthcare in America. After all, you just go to an emergency room.”
In the days before this issue of Emergency Medicine (EM) went to press, the United States Senate tried unsuccessfully, first to repeal and replace the Affordable Care Act (ACA), then to repeal key provisions of ACA without a replacement bill. Despite having a majority in both the Senate and House of Representatives as well as a Republican President, after 7 years of vowing to repeal “Obama Care,” Republicans have still not been able to fulfill that vow.
When ACA was signed into law in March 2010 (See “Springing Forward,” April 2010 EM), we wrote “though the new law will undoubtedly be challenged, tested, modified, refined, used—and probably abused—it will not be repealed. As was the case with Medicare and Medicaid previously, this will change everything in subtle and not-so-subtle ways.” (For a discussion of how the healthcare industry has managed to co-opt and abuse ACA, see the recently published book An American Sickness by Elisabeth Rosenthal, who was an emergency physician [EP] in our department before becoming a senior science and healthcare reporter for the New York Times.)
But the failure of ACA to deliver on many of its promises, its uncertain financial future, and the lack of improvements to ACA since 2010, directly or indirectly affects every American. Predictably, for those in need of care who cannot find a physician to accept their insurance or schedule a timely appointment, the ED remains the safety net for obtaining care.
After the constitutionality of ACA was upheld by the Supreme Court in June 2012 (See “Our National Pastime,” July 2012 EM), we noted that “ACA contains no provisions for increasing the number of healthcare providers [and] if 24 million more Americans now have access to affordable health insurance, but there are no new providers, who will they go to for care?” Seven years after passage of ACA, the answer to this question has been provided by published studies confirming that even more insured Americans are now seeking care in EDs than before “affordable care” became available. At the same time, urgent care centers, freestanding EDs, and “convenient care” centers, have sprung up and proliferated throughout the country, while in many states, nurse practitioners, physician assistants, and now emergency medical technicians and paramedics have sought and received authorization to evaluate and treat patients independent of physician supervision and oversight. Telemedicine or “telehealth” is the latest attempt to stretch the available supply of physicians to manage patients remotely, in the hope of obviating the need for an ED visit.
But none of these measures completely addresses a basic weakness of ACA: There are not enough physicians, including EPs, in this country to care for everyone entitled to healthcare; at the same time, there is a generation of highly qualified, highly motivated young men and women seeking entrance to medical school who will never get the opportunity to become fine physicians because there are not enough places for them. The solution to these problems seems obvious and the funds needed to finance it would be well spent, though the benefits of increasing the number of medical school places would not be realized for 4 to 8 years after they are made available.
In the meantime, we leave you with the solution President George W. Bush offered to a Cleveland audience on July 10, 2007 (See “Dream On,” March 2008 EM): “people have access to healthcare in America. After all, you just go to an emergency room.”

Approach to the Multitrauma Patient With Sternoclavicular Joint Dislocation
Case
A 28-year-old woman was brought to the ED by emergency medical services for evaluation of injuries sustained from a high-speed, rollover motor vehicle collision (MVC), during which she was partially ejected from the passenger front seat. The patient did not experience any loss of consciousness following the MVC. At presentation, she was oriented to place and time, and actively complained of bilateral clavicular pain (which she noted was worse on her right side) as well as right forearm pain.
The patient’s vital signs upon arrival were: heart rate, 94 beats/min; respiratory rate, 24 breaths/min; blood pressure, 107/84 mm Hg; and temperature,
The focused assessment with sonography for trauma examination was negative. In addition to the bilateral clavicular and right forearm pain, the patient also had tenderness bilaterally over the sternoclavicular joints (SCJ) and the right midclavicle, though there were no obvious deformities. Laboratory studies were within normal limits, with the exception of an elevated white blood cell count of 21 x 109/L.
Initial plain radiographs showed a normal chest X-ray (CXR) and right forearm fracture.

Orthopedic services were contacted and, with cardiothoracic surgery services readily available, the patient underwent an open reduction and internal fixation of the unstable SCJ. After surgical intervention, the patient experienced motor and sensory dysfunction, and a magnetic resonance imaging (MRI) study of the upper right extremity was ordered, which revealed brachial plexus injury secondary to hematoma and soft-tissue contusion in the right supraclavicular region. The patient remained in the hospital until postoperative day 3, at which time she was discharged home.
Three weeks after discharge, the patient followed up with the orthopedist for further surgical intervention of the comminuted distal forearm fracture. At that time, she had no further neurological or motor deficits from the upper extremity hematoma. However, 4 months after the MVC, she presented to the ED for evaluation of continued right shoulder pain. During this visit, X-ray studies confirmed posttraumatic arthritis; otherwise, the workup was negative for any further sequela or injury.
Discussion
Sternoclavicular joint dislocation is a rare traumatic injury, accounting for less than 3% of shoulder girdle injuries.1-4 Dislocations of the SCJ, which may be anterior or posterior, superior or inferior, are typically due to an MVC or athletic injury involving great force.5
Although an injury involving mediastinal compression such as a posterior SCJ dislocation can be fatal if not treated early, these dislocations are typically not detected in a multitrauma patient until the secondary survey.6 A missed diagnosis of posterior SCJ dislocation carries a mortality rate of 3% to 4% due to the potential for retrosternal injuries.4,6 Unfortunately, SCJ dislocations can be an easily overlooked injury in the multitrauma patient, as plain radiographs are difficult to interpret and physical examination findings other than tenderness may not always be present.5-7
Anatomy
The SCJ is comprised of the manubrium’s clavicular notch, the medial end of the clavicle, and the cartilage of the first rib.3 The capsular ligaments cover the anterior and posterior parts of the joint and offer stability along with the costoclavicular and interclavicular ligaments. Since the surrounding ligaments provide strong support to the joint, an incredible amount of force is needed for a dislocation to occur.
The clavicle is the first long bone in the body to ossify, and it does so in utero; however, the epiphysis of the medial clavicle is the last to ossify, and it does not fuse with the body of the clavicle until the early-to-mid-third decade of life. Since younger patients are therefore more prone to physeal fracture than joint dislocation, it is imperative to keep this as part of the differential in patients younger than age 25 years.1,3,8-10
Primary Survey
The emergency physician (EP) should approach the multitrauma patient in the usual fashion, ie, by first performing the primary survey. However, there may be some signs present in this early examination to indicate a posterior SCJ dislocation, including painful range of motion at the joint, inability to move the shoulder joint, hoarseness, dyspnea, dysphagia, neurovascular compromise of the arm, or frank hemodynamic instability.6-8 After the first survey is completed, if the EP has a high level of suspicion for SCJ dislocation, it is essential to perform a thorough secondary survey to confirm the diagnosis.
Secondary Survey
Anterior SCJ dislocations might be easier to detect clinically during the secondary survey, as the patient might have a deformity of the clavicle and swelling.8 However, posterior and superior SCJ dislocations might be more subtle during examination, and may only exhibit tenderness and limited range of motion.
Imaging Studies
Computed Tomography. Since overlying structures often make it difficult to interpret simple radiographs, advanced imaging studies such as CT are often needed for diagnosis. A CT angiogram (CTA) may be considered if there is concern for vascular injury and compromise of the limb, as this modality is more accurate in evaluating vasculature such as the subclavian artery.4,10
Special Radiographic Views. If advanced imaging is not immediately available or if the patient is not in a suitable condition to leave the ED, an alternative is to obtain a serendipity view X-ray. Described in 2009 by Wirth and Rockwood,11 the serendipity view is obtained with the patient in the supine position and the X-ray beam tilted to a 40-degree cephalic angle centered through the manubrium. This view permits comparison of both clavicles without overlying structures. The usefulness of serendipity view X-ray, however, is limited, as it does not allow for differentiation of sprains.
Other plain radiographic views, such as the Hobbs, Heinig, and Kattan views, have also been described to evaluate for SCJ dislocation, but these views are often not feasible or easily obtained in an emergency setting with an acutely injured patient.6,9,12
Magnetic Resonance Imaging. Though CT is typically the advanced initial imaging modality of choice for assessing the presence of an SCJ dislocation, additional studies using MRI are indicated for patients in whom there is a concern of physeal injury.1Ultrasound. Point-of-care ultrasound has become an important tool in the EP’s armamentarium, and can easily be employed to diagnose a posterior SCJ dislocation, as well as confirm the reduction. The method described by Bengtzen and Petering13 involves placing a linear array probe in the long axis to the clavicle and scanning until the clavicle and sternum are identified by finding the hyperechoic areas. The hypoechoic area in between the clavicle and sternum is the SCJ space. An ultrasound of the unaffected side can be useful for comparison purposes.6,13
Management
Posterior SCJ dislocations are considered a true emergency because of the potential structures associated with this type of injury. Concomitant injuries requiring immediate intervention include mediastinal compression, pneumothorax, laceration of the superior vena cava, tracheal erosion, esophageal injury, and brachial plexus compression and injury. Moreover, an unstable patient with an SCJ dislocation may have a lacerated thoracic vessel and need immediate thoracotomy.6
Anterior Reduction. Prior to any attempts at reduction, it is imperative to consult with orthopedic and cardiothoracic surgery services. However, if the patient’s dislocation is causing limb or life compromise, then the EP should attempt closed reduction in the ED.1,3 One reduction technique is to place the patient in the supine position with a towel rolled up between his or her shoulders. The EP then extends and abducts the affected arm using a traction-countertraction approach.
Another technique is to have an assistant either pushing posteriorly or pulling anteriorly on the medial clavicle, while the EP performs lateral traction. An audible “snap” sound might be heard with successful reduction. If the assistant is unable to grasp the medial clavicle, then a towel clip should be used percutaneously to grasp it. If the joint remains reduced, the limb is immobilized with a figure-of-8 bandage1,8
Further treatment options for complete SCJ dislocation include operative and nonoperative management. Posterior Reduction. While anterior dislocations are often managed conservatively with closed reduction and nonoperative treatment, posterior dislocations can often be reduced using either an open or closed approach.1-3,8,9,14 If a posterior SCJ dislocation is reduced using a closed approach, it is more likely to be stable after reduction when compared to anterior SCJ dislocation reduction.
An attempt of closed reduction of posterior SCJ dislocation is often recommended before open approach is attempted, if it occurred within 48 hours and there are no signs of mediastinal compression.9 Some authors however, prefer immediate surgical approach and treatment of all complete dislocations due to better visualization of other structures such as the meniscus and potentially damaged fibrocartilage, which if untreated can result in decreased mobility and pain.14,15
Conclusion
Although rare, posterior SCJ dislocations can be fatal when they are not diagnosed early. The EP must keep the possibility of an SCJ dislocation in mind based on the mechanism of injury—usually direct force to the joint such as occurs in an MVC or a lateral compression of the shoulder. There are clues during the primary survey that might point in the direction of an SCJ dislocation.
If the patient is hemodynamically unstable, immediate reduction is warranted and the possibility of a thoracotomy must be considered. Thirty percent of all posterior SCJ dislocations will have concomitant life-threatening injuries involving structures such as the esophagus, great vessels, and trachea.
Since sternoclavicular dislocation is often difficult to detect on CXR, the gold standard for diagnosis is CT or MRI. While the serendipity view X-ray can facilitate the evaluation of the SCJ, its value is limited. Other available plain radiographs are the Hobbs, Heinig, and Kattan views, but bedside ultrasound is often more useful and allows for faster evaluation and without ionizing radiation. Orthopedic services should be immediately consulted, and cardiothoracic surgery should readily available.
1. Groh GI, Wirth MA. Management of traumatic sternoclavicular joint injuries. J Am Acad Orthop Surg. 2011;19(1):1-7.
2. Glass ER, Thompson JD, Cole PA, Gause TM 2nd, Altman GT. Treatment of sternoclavicular joint dislocations: a systematic review of 251 dislocations in 24 case series. J Trauma. 2011;70(5):1294-1298. doi:10.1097/TA.0b013e3182092c7b.
3. Van Tongel A, De Wilde L. Sternoclavicular joint injuries: a literature review. Muscles Ligaments Tendons J. 2012;1(3):100-105.
4. Morell DJ, Thygarajan DS. Sternoclavicular joint dislocation and its management: A review of the literature. World J Orthop. 2016;7(4):244-250. doi:10.5312/wjo.v7.i4.244.
5. de Jong KP, Sukul DM. Anterior sternoclavicular dislocation: a long-term follow-up study. J Orthop Trauma. 1990;4(4):420-423.
6. Fenig M, Lowman R, Thompson BP, Shayne PH. Fatal posterior sternoclavicular joint dislocation due to occult trauma. Am J Emerg Med. 2010;28(3):385.e5-e8. doi:10.1016/j.ajem.2009.05.011.
7. Mirza AH, Alam K, Ali A. Posterior sternoclavicular dislocation in a rugby player as a cause of silent vascular compromise: a case report. Br J Sports Med. 2005;39(5):e28.
8. Roepke C, Kleiner M, Jhun P, Bright A, Herbert M. Chest pain bounce-back: posterior sternoclavicular dislocation. Annals Emerg Med. 2015;66(5):559-561. doi:10.1016/j.annemergmed.2015.09.015.
9. Laffosse JM, Espié A, Bonnevialle N, et al. Posterior dislocation of sternoclavicular joint and epiphyseal disruption of the medial clavicle with posterior displacement in sports participants. J Bone Joint Surg Br. 2010;92(1):103-109. doi:10.1302/0301-620X.92B1.22186.
10. Salvatore JE. Sternoclavicular joint dislocation. Clin Orthop Relat Res. 1968;58:51-55.
11. Wirth MA, Rockwood CA. Disorders of the sternoclavicular joint. In: Rockwood CA, Matsen FA, Wirth MA, Lippitt SB, eds. The Shoulder. 4th ed. Philadelphia, PA: Saunders; 2009:527-560.
12. Yang JS, Bogunovic L, Brophy RH, Wright RW, Scott R, Matava M. A case of posterior sternoclavicular dislocation in a professional American football player. Sports Health. 2013;7(4):318-325. doi:10.1177/1941738113502153.
13. Bengtzen RR, Petering RC. Point-of-care ultrasound diagnosis of posterior sternoclavicular joint dislocation. J Emerg Med. 2017;52(4):513-515. doi:10.1016/j.jemermed.2016.11.001.
14. Ferrandez L, Yubero J, Usabiaga J, No L, Martin F. Sternoclavicular dislocation. Treatment and complications. Ital J Orthop Traumatol. 1988;14(3):349-355.
15. Bicos J, Nicholson GP. Treatment and results of sternoclavicular joint injuries. Clin Sports Med. 2003;22(2):359-370.
Case
A 28-year-old woman was brought to the ED by emergency medical services for evaluation of injuries sustained from a high-speed, rollover motor vehicle collision (MVC), during which she was partially ejected from the passenger front seat. The patient did not experience any loss of consciousness following the MVC. At presentation, she was oriented to place and time, and actively complained of bilateral clavicular pain (which she noted was worse on her right side) as well as right forearm pain.
The patient’s vital signs upon arrival were: heart rate, 94 beats/min; respiratory rate, 24 breaths/min; blood pressure, 107/84 mm Hg; and temperature,
The focused assessment with sonography for trauma examination was negative. In addition to the bilateral clavicular and right forearm pain, the patient also had tenderness bilaterally over the sternoclavicular joints (SCJ) and the right midclavicle, though there were no obvious deformities. Laboratory studies were within normal limits, with the exception of an elevated white blood cell count of 21 x 109/L.
Initial plain radiographs showed a normal chest X-ray (CXR) and right forearm fracture.
Orthopedic services were contacted and, with cardiothoracic surgery services readily available, the patient underwent an open reduction and internal fixation of the unstable SCJ. After surgical intervention, the patient experienced motor and sensory dysfunction, and a magnetic resonance imaging (MRI) study of the upper right extremity was ordered, which revealed brachial plexus injury secondary to hematoma and soft-tissue contusion in the right supraclavicular region. The patient remained in the hospital until postoperative day 3, at which time she was discharged home.
Three weeks after discharge, the patient followed up with the orthopedist for further surgical intervention of the comminuted distal forearm fracture. At that time, she had no further neurological or motor deficits from the upper extremity hematoma. However, 4 months after the MVC, she presented to the ED for evaluation of continued right shoulder pain. During this visit, X-ray studies confirmed posttraumatic arthritis; otherwise, the workup was negative for any further sequela or injury.
Discussion
Sternoclavicular joint dislocation is a rare traumatic injury, accounting for less than 3% of shoulder girdle injuries.1-4 Dislocations of the SCJ, which may be anterior or posterior, superior or inferior, are typically due to an MVC or athletic injury involving great force.5
Although an injury involving mediastinal compression such as a posterior SCJ dislocation can be fatal if not treated early, these dislocations are typically not detected in a multitrauma patient until the secondary survey.6 A missed diagnosis of posterior SCJ dislocation carries a mortality rate of 3% to 4% due to the potential for retrosternal injuries.4,6 Unfortunately, SCJ dislocations can be an easily overlooked injury in the multitrauma patient, as plain radiographs are difficult to interpret and physical examination findings other than tenderness may not always be present.5-7
Anatomy
The SCJ is comprised of the manubrium’s clavicular notch, the medial end of the clavicle, and the cartilage of the first rib.3 The capsular ligaments cover the anterior and posterior parts of the joint and offer stability along with the costoclavicular and interclavicular ligaments. Since the surrounding ligaments provide strong support to the joint, an incredible amount of force is needed for a dislocation to occur.
The clavicle is the first long bone in the body to ossify, and it does so in utero; however, the epiphysis of the medial clavicle is the last to ossify, and it does not fuse with the body of the clavicle until the early-to-mid-third decade of life. Since younger patients are therefore more prone to physeal fracture than joint dislocation, it is imperative to keep this as part of the differential in patients younger than age 25 years.1,3,8-10
Primary Survey
The emergency physician (EP) should approach the multitrauma patient in the usual fashion, ie, by first performing the primary survey. However, there may be some signs present in this early examination to indicate a posterior SCJ dislocation, including painful range of motion at the joint, inability to move the shoulder joint, hoarseness, dyspnea, dysphagia, neurovascular compromise of the arm, or frank hemodynamic instability.6-8 After the first survey is completed, if the EP has a high level of suspicion for SCJ dislocation, it is essential to perform a thorough secondary survey to confirm the diagnosis.
Secondary Survey
Anterior SCJ dislocations might be easier to detect clinically during the secondary survey, as the patient might have a deformity of the clavicle and swelling.8 However, posterior and superior SCJ dislocations might be more subtle during examination, and may only exhibit tenderness and limited range of motion.
Imaging Studies
Computed Tomography. Since overlying structures often make it difficult to interpret simple radiographs, advanced imaging studies such as CT are often needed for diagnosis. A CT angiogram (CTA) may be considered if there is concern for vascular injury and compromise of the limb, as this modality is more accurate in evaluating vasculature such as the subclavian artery.4,10
Special Radiographic Views. If advanced imaging is not immediately available or if the patient is not in a suitable condition to leave the ED, an alternative is to obtain a serendipity view X-ray. Described in 2009 by Wirth and Rockwood,11 the serendipity view is obtained with the patient in the supine position and the X-ray beam tilted to a 40-degree cephalic angle centered through the manubrium. This view permits comparison of both clavicles without overlying structures. The usefulness of serendipity view X-ray, however, is limited, as it does not allow for differentiation of sprains.
Other plain radiographic views, such as the Hobbs, Heinig, and Kattan views, have also been described to evaluate for SCJ dislocation, but these views are often not feasible or easily obtained in an emergency setting with an acutely injured patient.6,9,12
Magnetic Resonance Imaging. Though CT is typically the advanced initial imaging modality of choice for assessing the presence of an SCJ dislocation, additional studies using MRI are indicated for patients in whom there is a concern of physeal injury.1Ultrasound. Point-of-care ultrasound has become an important tool in the EP’s armamentarium, and can easily be employed to diagnose a posterior SCJ dislocation, as well as confirm the reduction. The method described by Bengtzen and Petering13 involves placing a linear array probe in the long axis to the clavicle and scanning until the clavicle and sternum are identified by finding the hyperechoic areas. The hypoechoic area in between the clavicle and sternum is the SCJ space. An ultrasound of the unaffected side can be useful for comparison purposes.6,13
Management
Posterior SCJ dislocations are considered a true emergency because of the potential structures associated with this type of injury. Concomitant injuries requiring immediate intervention include mediastinal compression, pneumothorax, laceration of the superior vena cava, tracheal erosion, esophageal injury, and brachial plexus compression and injury. Moreover, an unstable patient with an SCJ dislocation may have a lacerated thoracic vessel and need immediate thoracotomy.6
Anterior Reduction. Prior to any attempts at reduction, it is imperative to consult with orthopedic and cardiothoracic surgery services. However, if the patient’s dislocation is causing limb or life compromise, then the EP should attempt closed reduction in the ED.1,3 One reduction technique is to place the patient in the supine position with a towel rolled up between his or her shoulders. The EP then extends and abducts the affected arm using a traction-countertraction approach.
Another technique is to have an assistant either pushing posteriorly or pulling anteriorly on the medial clavicle, while the EP performs lateral traction. An audible “snap” sound might be heard with successful reduction. If the assistant is unable to grasp the medial clavicle, then a towel clip should be used percutaneously to grasp it. If the joint remains reduced, the limb is immobilized with a figure-of-8 bandage1,8
Further treatment options for complete SCJ dislocation include operative and nonoperative management. Posterior Reduction. While anterior dislocations are often managed conservatively with closed reduction and nonoperative treatment, posterior dislocations can often be reduced using either an open or closed approach.1-3,8,9,14 If a posterior SCJ dislocation is reduced using a closed approach, it is more likely to be stable after reduction when compared to anterior SCJ dislocation reduction.
An attempt of closed reduction of posterior SCJ dislocation is often recommended before open approach is attempted, if it occurred within 48 hours and there are no signs of mediastinal compression.9 Some authors however, prefer immediate surgical approach and treatment of all complete dislocations due to better visualization of other structures such as the meniscus and potentially damaged fibrocartilage, which if untreated can result in decreased mobility and pain.14,15
Conclusion
Although rare, posterior SCJ dislocations can be fatal when they are not diagnosed early. The EP must keep the possibility of an SCJ dislocation in mind based on the mechanism of injury—usually direct force to the joint such as occurs in an MVC or a lateral compression of the shoulder. There are clues during the primary survey that might point in the direction of an SCJ dislocation.
If the patient is hemodynamically unstable, immediate reduction is warranted and the possibility of a thoracotomy must be considered. Thirty percent of all posterior SCJ dislocations will have concomitant life-threatening injuries involving structures such as the esophagus, great vessels, and trachea.
Since sternoclavicular dislocation is often difficult to detect on CXR, the gold standard for diagnosis is CT or MRI. While the serendipity view X-ray can facilitate the evaluation of the SCJ, its value is limited. Other available plain radiographs are the Hobbs, Heinig, and Kattan views, but bedside ultrasound is often more useful and allows for faster evaluation and without ionizing radiation. Orthopedic services should be immediately consulted, and cardiothoracic surgery should readily available.
Case
A 28-year-old woman was brought to the ED by emergency medical services for evaluation of injuries sustained from a high-speed, rollover motor vehicle collision (MVC), during which she was partially ejected from the passenger front seat. The patient did not experience any loss of consciousness following the MVC. At presentation, she was oriented to place and time, and actively complained of bilateral clavicular pain (which she noted was worse on her right side) as well as right forearm pain.
The patient’s vital signs upon arrival were: heart rate, 94 beats/min; respiratory rate, 24 breaths/min; blood pressure, 107/84 mm Hg; and temperature,
The focused assessment with sonography for trauma examination was negative. In addition to the bilateral clavicular and right forearm pain, the patient also had tenderness bilaterally over the sternoclavicular joints (SCJ) and the right midclavicle, though there were no obvious deformities. Laboratory studies were within normal limits, with the exception of an elevated white blood cell count of 21 x 109/L.
Initial plain radiographs showed a normal chest X-ray (CXR) and right forearm fracture.
Orthopedic services were contacted and, with cardiothoracic surgery services readily available, the patient underwent an open reduction and internal fixation of the unstable SCJ. After surgical intervention, the patient experienced motor and sensory dysfunction, and a magnetic resonance imaging (MRI) study of the upper right extremity was ordered, which revealed brachial plexus injury secondary to hematoma and soft-tissue contusion in the right supraclavicular region. The patient remained in the hospital until postoperative day 3, at which time she was discharged home.
Three weeks after discharge, the patient followed up with the orthopedist for further surgical intervention of the comminuted distal forearm fracture. At that time, she had no further neurological or motor deficits from the upper extremity hematoma. However, 4 months after the MVC, she presented to the ED for evaluation of continued right shoulder pain. During this visit, X-ray studies confirmed posttraumatic arthritis; otherwise, the workup was negative for any further sequela or injury.
Discussion
Sternoclavicular joint dislocation is a rare traumatic injury, accounting for less than 3% of shoulder girdle injuries.1-4 Dislocations of the SCJ, which may be anterior or posterior, superior or inferior, are typically due to an MVC or athletic injury involving great force.5
Although an injury involving mediastinal compression such as a posterior SCJ dislocation can be fatal if not treated early, these dislocations are typically not detected in a multitrauma patient until the secondary survey.6 A missed diagnosis of posterior SCJ dislocation carries a mortality rate of 3% to 4% due to the potential for retrosternal injuries.4,6 Unfortunately, SCJ dislocations can be an easily overlooked injury in the multitrauma patient, as plain radiographs are difficult to interpret and physical examination findings other than tenderness may not always be present.5-7
Anatomy
The SCJ is comprised of the manubrium’s clavicular notch, the medial end of the clavicle, and the cartilage of the first rib.3 The capsular ligaments cover the anterior and posterior parts of the joint and offer stability along with the costoclavicular and interclavicular ligaments. Since the surrounding ligaments provide strong support to the joint, an incredible amount of force is needed for a dislocation to occur.
The clavicle is the first long bone in the body to ossify, and it does so in utero; however, the epiphysis of the medial clavicle is the last to ossify, and it does not fuse with the body of the clavicle until the early-to-mid-third decade of life. Since younger patients are therefore more prone to physeal fracture than joint dislocation, it is imperative to keep this as part of the differential in patients younger than age 25 years.1,3,8-10
Primary Survey
The emergency physician (EP) should approach the multitrauma patient in the usual fashion, ie, by first performing the primary survey. However, there may be some signs present in this early examination to indicate a posterior SCJ dislocation, including painful range of motion at the joint, inability to move the shoulder joint, hoarseness, dyspnea, dysphagia, neurovascular compromise of the arm, or frank hemodynamic instability.6-8 After the first survey is completed, if the EP has a high level of suspicion for SCJ dislocation, it is essential to perform a thorough secondary survey to confirm the diagnosis.
Secondary Survey
Anterior SCJ dislocations might be easier to detect clinically during the secondary survey, as the patient might have a deformity of the clavicle and swelling.8 However, posterior and superior SCJ dislocations might be more subtle during examination, and may only exhibit tenderness and limited range of motion.
Imaging Studies
Computed Tomography. Since overlying structures often make it difficult to interpret simple radiographs, advanced imaging studies such as CT are often needed for diagnosis. A CT angiogram (CTA) may be considered if there is concern for vascular injury and compromise of the limb, as this modality is more accurate in evaluating vasculature such as the subclavian artery.4,10
Special Radiographic Views. If advanced imaging is not immediately available or if the patient is not in a suitable condition to leave the ED, an alternative is to obtain a serendipity view X-ray. Described in 2009 by Wirth and Rockwood,11 the serendipity view is obtained with the patient in the supine position and the X-ray beam tilted to a 40-degree cephalic angle centered through the manubrium. This view permits comparison of both clavicles without overlying structures. The usefulness of serendipity view X-ray, however, is limited, as it does not allow for differentiation of sprains.
Other plain radiographic views, such as the Hobbs, Heinig, and Kattan views, have also been described to evaluate for SCJ dislocation, but these views are often not feasible or easily obtained in an emergency setting with an acutely injured patient.6,9,12
Magnetic Resonance Imaging. Though CT is typically the advanced initial imaging modality of choice for assessing the presence of an SCJ dislocation, additional studies using MRI are indicated for patients in whom there is a concern of physeal injury.1Ultrasound. Point-of-care ultrasound has become an important tool in the EP’s armamentarium, and can easily be employed to diagnose a posterior SCJ dislocation, as well as confirm the reduction. The method described by Bengtzen and Petering13 involves placing a linear array probe in the long axis to the clavicle and scanning until the clavicle and sternum are identified by finding the hyperechoic areas. The hypoechoic area in between the clavicle and sternum is the SCJ space. An ultrasound of the unaffected side can be useful for comparison purposes.6,13
Management
Posterior SCJ dislocations are considered a true emergency because of the potential structures associated with this type of injury. Concomitant injuries requiring immediate intervention include mediastinal compression, pneumothorax, laceration of the superior vena cava, tracheal erosion, esophageal injury, and brachial plexus compression and injury. Moreover, an unstable patient with an SCJ dislocation may have a lacerated thoracic vessel and need immediate thoracotomy.6
Anterior Reduction. Prior to any attempts at reduction, it is imperative to consult with orthopedic and cardiothoracic surgery services. However, if the patient’s dislocation is causing limb or life compromise, then the EP should attempt closed reduction in the ED.1,3 One reduction technique is to place the patient in the supine position with a towel rolled up between his or her shoulders. The EP then extends and abducts the affected arm using a traction-countertraction approach.
Another technique is to have an assistant either pushing posteriorly or pulling anteriorly on the medial clavicle, while the EP performs lateral traction. An audible “snap” sound might be heard with successful reduction. If the assistant is unable to grasp the medial clavicle, then a towel clip should be used percutaneously to grasp it. If the joint remains reduced, the limb is immobilized with a figure-of-8 bandage1,8
Further treatment options for complete SCJ dislocation include operative and nonoperative management. Posterior Reduction. While anterior dislocations are often managed conservatively with closed reduction and nonoperative treatment, posterior dislocations can often be reduced using either an open or closed approach.1-3,8,9,14 If a posterior SCJ dislocation is reduced using a closed approach, it is more likely to be stable after reduction when compared to anterior SCJ dislocation reduction.
An attempt of closed reduction of posterior SCJ dislocation is often recommended before open approach is attempted, if it occurred within 48 hours and there are no signs of mediastinal compression.9 Some authors however, prefer immediate surgical approach and treatment of all complete dislocations due to better visualization of other structures such as the meniscus and potentially damaged fibrocartilage, which if untreated can result in decreased mobility and pain.14,15
Conclusion
Although rare, posterior SCJ dislocations can be fatal when they are not diagnosed early. The EP must keep the possibility of an SCJ dislocation in mind based on the mechanism of injury—usually direct force to the joint such as occurs in an MVC or a lateral compression of the shoulder. There are clues during the primary survey that might point in the direction of an SCJ dislocation.
If the patient is hemodynamically unstable, immediate reduction is warranted and the possibility of a thoracotomy must be considered. Thirty percent of all posterior SCJ dislocations will have concomitant life-threatening injuries involving structures such as the esophagus, great vessels, and trachea.
Since sternoclavicular dislocation is often difficult to detect on CXR, the gold standard for diagnosis is CT or MRI. While the serendipity view X-ray can facilitate the evaluation of the SCJ, its value is limited. Other available plain radiographs are the Hobbs, Heinig, and Kattan views, but bedside ultrasound is often more useful and allows for faster evaluation and without ionizing radiation. Orthopedic services should be immediately consulted, and cardiothoracic surgery should readily available.
1. Groh GI, Wirth MA. Management of traumatic sternoclavicular joint injuries. J Am Acad Orthop Surg. 2011;19(1):1-7.
2. Glass ER, Thompson JD, Cole PA, Gause TM 2nd, Altman GT. Treatment of sternoclavicular joint dislocations: a systematic review of 251 dislocations in 24 case series. J Trauma. 2011;70(5):1294-1298. doi:10.1097/TA.0b013e3182092c7b.
3. Van Tongel A, De Wilde L. Sternoclavicular joint injuries: a literature review. Muscles Ligaments Tendons J. 2012;1(3):100-105.
4. Morell DJ, Thygarajan DS. Sternoclavicular joint dislocation and its management: A review of the literature. World J Orthop. 2016;7(4):244-250. doi:10.5312/wjo.v7.i4.244.
5. de Jong KP, Sukul DM. Anterior sternoclavicular dislocation: a long-term follow-up study. J Orthop Trauma. 1990;4(4):420-423.
6. Fenig M, Lowman R, Thompson BP, Shayne PH. Fatal posterior sternoclavicular joint dislocation due to occult trauma. Am J Emerg Med. 2010;28(3):385.e5-e8. doi:10.1016/j.ajem.2009.05.011.
7. Mirza AH, Alam K, Ali A. Posterior sternoclavicular dislocation in a rugby player as a cause of silent vascular compromise: a case report. Br J Sports Med. 2005;39(5):e28.
8. Roepke C, Kleiner M, Jhun P, Bright A, Herbert M. Chest pain bounce-back: posterior sternoclavicular dislocation. Annals Emerg Med. 2015;66(5):559-561. doi:10.1016/j.annemergmed.2015.09.015.
9. Laffosse JM, Espié A, Bonnevialle N, et al. Posterior dislocation of sternoclavicular joint and epiphyseal disruption of the medial clavicle with posterior displacement in sports participants. J Bone Joint Surg Br. 2010;92(1):103-109. doi:10.1302/0301-620X.92B1.22186.
10. Salvatore JE. Sternoclavicular joint dislocation. Clin Orthop Relat Res. 1968;58:51-55.
11. Wirth MA, Rockwood CA. Disorders of the sternoclavicular joint. In: Rockwood CA, Matsen FA, Wirth MA, Lippitt SB, eds. The Shoulder. 4th ed. Philadelphia, PA: Saunders; 2009:527-560.
12. Yang JS, Bogunovic L, Brophy RH, Wright RW, Scott R, Matava M. A case of posterior sternoclavicular dislocation in a professional American football player. Sports Health. 2013;7(4):318-325. doi:10.1177/1941738113502153.
13. Bengtzen RR, Petering RC. Point-of-care ultrasound diagnosis of posterior sternoclavicular joint dislocation. J Emerg Med. 2017;52(4):513-515. doi:10.1016/j.jemermed.2016.11.001.
14. Ferrandez L, Yubero J, Usabiaga J, No L, Martin F. Sternoclavicular dislocation. Treatment and complications. Ital J Orthop Traumatol. 1988;14(3):349-355.
15. Bicos J, Nicholson GP. Treatment and results of sternoclavicular joint injuries. Clin Sports Med. 2003;22(2):359-370.
1. Groh GI, Wirth MA. Management of traumatic sternoclavicular joint injuries. J Am Acad Orthop Surg. 2011;19(1):1-7.
2. Glass ER, Thompson JD, Cole PA, Gause TM 2nd, Altman GT. Treatment of sternoclavicular joint dislocations: a systematic review of 251 dislocations in 24 case series. J Trauma. 2011;70(5):1294-1298. doi:10.1097/TA.0b013e3182092c7b.
3. Van Tongel A, De Wilde L. Sternoclavicular joint injuries: a literature review. Muscles Ligaments Tendons J. 2012;1(3):100-105.
4. Morell DJ, Thygarajan DS. Sternoclavicular joint dislocation and its management: A review of the literature. World J Orthop. 2016;7(4):244-250. doi:10.5312/wjo.v7.i4.244.
5. de Jong KP, Sukul DM. Anterior sternoclavicular dislocation: a long-term follow-up study. J Orthop Trauma. 1990;4(4):420-423.
6. Fenig M, Lowman R, Thompson BP, Shayne PH. Fatal posterior sternoclavicular joint dislocation due to occult trauma. Am J Emerg Med. 2010;28(3):385.e5-e8. doi:10.1016/j.ajem.2009.05.011.
7. Mirza AH, Alam K, Ali A. Posterior sternoclavicular dislocation in a rugby player as a cause of silent vascular compromise: a case report. Br J Sports Med. 2005;39(5):e28.
8. Roepke C, Kleiner M, Jhun P, Bright A, Herbert M. Chest pain bounce-back: posterior sternoclavicular dislocation. Annals Emerg Med. 2015;66(5):559-561. doi:10.1016/j.annemergmed.2015.09.015.
9. Laffosse JM, Espié A, Bonnevialle N, et al. Posterior dislocation of sternoclavicular joint and epiphyseal disruption of the medial clavicle with posterior displacement in sports participants. J Bone Joint Surg Br. 2010;92(1):103-109. doi:10.1302/0301-620X.92B1.22186.
10. Salvatore JE. Sternoclavicular joint dislocation. Clin Orthop Relat Res. 1968;58:51-55.
11. Wirth MA, Rockwood CA. Disorders of the sternoclavicular joint. In: Rockwood CA, Matsen FA, Wirth MA, Lippitt SB, eds. The Shoulder. 4th ed. Philadelphia, PA: Saunders; 2009:527-560.
12. Yang JS, Bogunovic L, Brophy RH, Wright RW, Scott R, Matava M. A case of posterior sternoclavicular dislocation in a professional American football player. Sports Health. 2013;7(4):318-325. doi:10.1177/1941738113502153.
13. Bengtzen RR, Petering RC. Point-of-care ultrasound diagnosis of posterior sternoclavicular joint dislocation. J Emerg Med. 2017;52(4):513-515. doi:10.1016/j.jemermed.2016.11.001.
14. Ferrandez L, Yubero J, Usabiaga J, No L, Martin F. Sternoclavicular dislocation. Treatment and complications. Ital J Orthop Traumatol. 1988;14(3):349-355.
15. Bicos J, Nicholson GP. Treatment and results of sternoclavicular joint injuries. Clin Sports Med. 2003;22(2):359-370.

Malpractice Counsel: Never Too Young to Have a Heart Attack
Case
A 21-year-old woman presented to the ED for evaluation of severe chest pain radiating to her left arm, and associated shortness of breath, nausea, and vomiting. She stated that the pain started 2 hours earlier while she was resting and had become progressively worse. She denied any history of similar symptoms. The patient denied fever, chills, or cough. She stated that she was otherwise in good health and did not take any medication on a regular basis. Regarding her social history, she admitted to smoking one pack of cigarettes a day and drinking alcohol on occasion.
On physical examination, the patient appeared uncomfortable. Her vital signs were: blood pressure, 136/86 mm Hg; heart rate, 102 beats/min; respiratory rate, 22 breaths/min; and temperature, 98.60F. Oxygen saturation was 97% on room air. The head, eyes, ears, nose, and throat examination was unremarkable. Auscultation of the lungs revealed clear breath sounds bilaterally. The heart examination revealed tachycardia, but with regular rhythm and without murmurs, rubs, or gallops. The abdomen was soft and nontender. No lower extremity examination was documented.
The patient was seen by a physician assistant (PA) in the ED. An electrocardiogram (ECG), complete blood count (CBC), basic metabolic profile (BMP), troponin level, chest X-ray (CXR), and urine pregnancy test were ordered. The patient was given intravenous (IV) fluids and prochlorperazine 10 mg IV. The ECG and CXR were interpreted as normal. The urine pregnancy test was negative, and the remaining blood test results were within normal limits.
The PA believed the patient suffered from gastroenteritis, coupled with anxiety. He discharged the patient home with instructions to drink clear liquids for 24 hours, and take the prescribed prochlorperazine tablets as needed for continued nausea and vomiting.
At home, the patient continued to experience increasingly severe chest pain, shortness of breath, and vomiting. The next morning, she could no longer tolerate the pain and returned to the same ED via emergency medical services.
The patient’s history and physical examination remained unchanged from her presentation 16 hours earlier. At this ED visit, the patient was seen by an emergency physician (EP) who, concerned the patient had suffered an ischemic coronary event, ordered repeat ECG, CBC, BMP, and troponin evaluation. The EP also contacted cardiology services, but the cardiologist did not see the patient for several hours. When the cardiologist evaluated the patient and interpreted the ECG, he was concerned for an ST-segment elevation myocardial infarction (STEMI), and activated the catheterization lab.
Unfortunately, the patient had significant myocardial damage, with a resulting ejection fraction of only 10%. She was judged to be a candidate for heart transplantation, and received a left ventricular assist device (LVAD) as a bridge until a suitable donor heart could be identified. One month after implantation of the LVAD, the patient experienced an ischemic stroke that resulted in dense left-side weakness, leaving her confined to bed.
The patient sued the PA, the EP, the hospital, and the cardiologist for failing to identify and treat the acute STEMI in a timely manner. The plaintiff claimed the STEMI began at her first presentation to the ED, and that it should have been diagnosed and treated at that time. The plaintiff further argued that she should at least have been monitored and undergone repeat testing (ie, ECG and troponin level evaluation) at the first visit, stating that if she had received proper treatment, she would not have required an LVAD and therefore would not have had a stroke. The patient also alleged that at the second ED visit, there was a significant time delay before she was taken to the catheterization lab, which resulted in additional myocardial injury.
The defendants argued the patient was appropriately evaluated and treated at the first presentation, and that there was no evidence to suggest an MI. The EP argued that the delay in the patient’s care at the second visit was not his fault. All of the parties involved negotiated a settlement in the amount of $6 million in favor of the plaintiff.
Discussion
Myocardial infarction in adults younger than age 45 years is relatively rare, comprising only 2% to 10% of all MIs.1,2 The percentage of MI in patients younger than age 25 years must be even smaller, but no good data are available. In fact, age 40 years and younger is usually an exclusion criteria in many of the multicenter studies involving MI. Women are relatively spared from coronary artery disease (CAD) before menopause, thanks to the cardioprotective effects of estrogen. Young women who do experience an MI usually will have cardiovascular risk factors, especially smoking.
Risk Factors for MI in Young Patients
Cigarette Smoking. When examining common risk factors in young patients who had an MI (defined as patients younger than age 45 years), cigarette smoking is the most common risk factor.1,2 Between 76% and 91% of young patients with an MI are smokers, compared to only 40% incidence in older patients.1 It is thought that cigarette smoking produces endothelial dysfunction and can precipitate coronary spasm.1
Nonatherosclerotic Etiology. Interestingly, several studies of MI in young patients found a higher incidence of nonatherosclerotic causes of MI in women compared to men.2 One explanation for this finding is that women experience vasospastic syndromes and hypercoagulable states, secondary to oral contraceptive use or hereditary coagulation disorders.2 It has also been shown that young women have more active platelets following an MI and experience plaque erosions, rather than the plaque ruptures that occur in men and older women.2,3
Hyperlipidemia. Hyperlipidemia is an additional risk factor for MI in the younger adult patient population. In one study of young patients who had an MI, hyperlipidemia was the most important risk factor, in the absence of other obvious risk factors.1,4 In fact, some researchers think hyperlipidemia may be a more reliable predictor of MI in patients aged 30 to 39 years than in older patients.1,5 Unfortunately, many of these young adults are not aware that they have hyperlipidemia until they experience a complication such as an acute coronary syndrome. With respect to the patient in this case, it is not clear from the published report whether or not she had hyperlipidemia.
Family History. Another risk factor for MI in younger patients is a positive family history of CAD in a first-degree relative younger than age 55 years.1 Siblings of a young patient who experienced an MI have up to a 10-fold increase for developing CAD.1 It is currently not known why a positive family history increases the risk of MI in younger patients, but it may be related to inherited disorders of lipid metabolism, blood coagulation, or other genetic factors.1
Drug Abuse. Finally, drug abuse must be considered in young patients presenting with an MI. The use of cocaine, methamphetamine, marijuana, and K2 (synthetic marijuana) have all been associated with MI, especially in young patients,6-9 who typically do not have cardiac risk factors and do not show evidence of atherosclerotic disease on cardiac catheterization. As for the patient in this case, we do not know if she used any illicit drugs prior to presentation.
Summary
This case underscores the importance of not excluding MI in the differential diagnosis based simply on age or sex. While MI is uncommon in a 21-year-old woman, it can and does occur. In young patients presenting with chest pain, it is important to obtain a thorough history, including smoking, family history of MI, hyperlipidemia, and illicit drug use. While MI may be low on the differential diagnosis, it still needs to be considered.
1. Choudhury L, Marsh JD. Myocardial infarction in young patients. Am J Med. 1999;107(3):254-261.
2. Lawesson SS, Stenestrand U, Lagerqvist B, Wallentin L, Swahn E. Gender perspective on risk factors, coronary lesions and long-term outcome in young patients with ST-elevation myocardial infarction. Heart. 2010;96(6):453-459. doi:10.1136/hrt.2009.175463.
3. Burke AP, Farb A, Malcom G,Virmani R. Effect of menopause on plaque morphologic characteristics in coronary atherosclerosis. Am Heart J. 2001;141(2 Suppl):S58-S62.
4. Tomono S, Ohshima S, Murata K. The risk factors for ischemic heart disease in young adults. Jpn Circ J. 1990;54(4):436-441.
5. Gofman JW, Young W, Tandy R. Ischemic heart disease, atherosclerosis, and longevity. Circulation. 1966;34(4):679-697.
6. Zimmerman JL. Cocaine intoxication. Crit Care Clin. 2012;28(4):517-525. doi:10.1016/j.ccc.2012.07.003.
7. Hawley LA, Auten JD, Matteucci MJ, et al. Cardiac complications of adult methamphetamine exposures. J Emerg Med. 2013;45(6):821-827. doi:10.1016/j.jemermed.2013.04.061.
8. Bachs L, Mørland H. Acute cardiovascular fatalities following cannabis use. Forensic Sci Int. 2001;124(2-3):200-203.
9. Mir A, Obafemi A, Young A, Kane C. Myocardial infarction associated with use of the synthetic cannabinoid K2. Pediatrics. 2011;128(6):e1622-e1627. doi:10.1542 peds.2010-3823.
Case
A 21-year-old woman presented to the ED for evaluation of severe chest pain radiating to her left arm, and associated shortness of breath, nausea, and vomiting. She stated that the pain started 2 hours earlier while she was resting and had become progressively worse. She denied any history of similar symptoms. The patient denied fever, chills, or cough. She stated that she was otherwise in good health and did not take any medication on a regular basis. Regarding her social history, she admitted to smoking one pack of cigarettes a day and drinking alcohol on occasion.
On physical examination, the patient appeared uncomfortable. Her vital signs were: blood pressure, 136/86 mm Hg; heart rate, 102 beats/min; respiratory rate, 22 breaths/min; and temperature, 98.60F. Oxygen saturation was 97% on room air. The head, eyes, ears, nose, and throat examination was unremarkable. Auscultation of the lungs revealed clear breath sounds bilaterally. The heart examination revealed tachycardia, but with regular rhythm and without murmurs, rubs, or gallops. The abdomen was soft and nontender. No lower extremity examination was documented.
The patient was seen by a physician assistant (PA) in the ED. An electrocardiogram (ECG), complete blood count (CBC), basic metabolic profile (BMP), troponin level, chest X-ray (CXR), and urine pregnancy test were ordered. The patient was given intravenous (IV) fluids and prochlorperazine 10 mg IV. The ECG and CXR were interpreted as normal. The urine pregnancy test was negative, and the remaining blood test results were within normal limits.
The PA believed the patient suffered from gastroenteritis, coupled with anxiety. He discharged the patient home with instructions to drink clear liquids for 24 hours, and take the prescribed prochlorperazine tablets as needed for continued nausea and vomiting.
At home, the patient continued to experience increasingly severe chest pain, shortness of breath, and vomiting. The next morning, she could no longer tolerate the pain and returned to the same ED via emergency medical services.
The patient’s history and physical examination remained unchanged from her presentation 16 hours earlier. At this ED visit, the patient was seen by an emergency physician (EP) who, concerned the patient had suffered an ischemic coronary event, ordered repeat ECG, CBC, BMP, and troponin evaluation. The EP also contacted cardiology services, but the cardiologist did not see the patient for several hours. When the cardiologist evaluated the patient and interpreted the ECG, he was concerned for an ST-segment elevation myocardial infarction (STEMI), and activated the catheterization lab.
Unfortunately, the patient had significant myocardial damage, with a resulting ejection fraction of only 10%. She was judged to be a candidate for heart transplantation, and received a left ventricular assist device (LVAD) as a bridge until a suitable donor heart could be identified. One month after implantation of the LVAD, the patient experienced an ischemic stroke that resulted in dense left-side weakness, leaving her confined to bed.
The patient sued the PA, the EP, the hospital, and the cardiologist for failing to identify and treat the acute STEMI in a timely manner. The plaintiff claimed the STEMI began at her first presentation to the ED, and that it should have been diagnosed and treated at that time. The plaintiff further argued that she should at least have been monitored and undergone repeat testing (ie, ECG and troponin level evaluation) at the first visit, stating that if she had received proper treatment, she would not have required an LVAD and therefore would not have had a stroke. The patient also alleged that at the second ED visit, there was a significant time delay before she was taken to the catheterization lab, which resulted in additional myocardial injury.
The defendants argued the patient was appropriately evaluated and treated at the first presentation, and that there was no evidence to suggest an MI. The EP argued that the delay in the patient’s care at the second visit was not his fault. All of the parties involved negotiated a settlement in the amount of $6 million in favor of the plaintiff.
Discussion
Myocardial infarction in adults younger than age 45 years is relatively rare, comprising only 2% to 10% of all MIs.1,2 The percentage of MI in patients younger than age 25 years must be even smaller, but no good data are available. In fact, age 40 years and younger is usually an exclusion criteria in many of the multicenter studies involving MI. Women are relatively spared from coronary artery disease (CAD) before menopause, thanks to the cardioprotective effects of estrogen. Young women who do experience an MI usually will have cardiovascular risk factors, especially smoking.
Risk Factors for MI in Young Patients
Cigarette Smoking. When examining common risk factors in young patients who had an MI (defined as patients younger than age 45 years), cigarette smoking is the most common risk factor.1,2 Between 76% and 91% of young patients with an MI are smokers, compared to only 40% incidence in older patients.1 It is thought that cigarette smoking produces endothelial dysfunction and can precipitate coronary spasm.1
Nonatherosclerotic Etiology. Interestingly, several studies of MI in young patients found a higher incidence of nonatherosclerotic causes of MI in women compared to men.2 One explanation for this finding is that women experience vasospastic syndromes and hypercoagulable states, secondary to oral contraceptive use or hereditary coagulation disorders.2 It has also been shown that young women have more active platelets following an MI and experience plaque erosions, rather than the plaque ruptures that occur in men and older women.2,3
Hyperlipidemia. Hyperlipidemia is an additional risk factor for MI in the younger adult patient population. In one study of young patients who had an MI, hyperlipidemia was the most important risk factor, in the absence of other obvious risk factors.1,4 In fact, some researchers think hyperlipidemia may be a more reliable predictor of MI in patients aged 30 to 39 years than in older patients.1,5 Unfortunately, many of these young adults are not aware that they have hyperlipidemia until they experience a complication such as an acute coronary syndrome. With respect to the patient in this case, it is not clear from the published report whether or not she had hyperlipidemia.
Family History. Another risk factor for MI in younger patients is a positive family history of CAD in a first-degree relative younger than age 55 years.1 Siblings of a young patient who experienced an MI have up to a 10-fold increase for developing CAD.1 It is currently not known why a positive family history increases the risk of MI in younger patients, but it may be related to inherited disorders of lipid metabolism, blood coagulation, or other genetic factors.1
Drug Abuse. Finally, drug abuse must be considered in young patients presenting with an MI. The use of cocaine, methamphetamine, marijuana, and K2 (synthetic marijuana) have all been associated with MI, especially in young patients,6-9 who typically do not have cardiac risk factors and do not show evidence of atherosclerotic disease on cardiac catheterization. As for the patient in this case, we do not know if she used any illicit drugs prior to presentation.
Summary
This case underscores the importance of not excluding MI in the differential diagnosis based simply on age or sex. While MI is uncommon in a 21-year-old woman, it can and does occur. In young patients presenting with chest pain, it is important to obtain a thorough history, including smoking, family history of MI, hyperlipidemia, and illicit drug use. While MI may be low on the differential diagnosis, it still needs to be considered.
Case
A 21-year-old woman presented to the ED for evaluation of severe chest pain radiating to her left arm, and associated shortness of breath, nausea, and vomiting. She stated that the pain started 2 hours earlier while she was resting and had become progressively worse. She denied any history of similar symptoms. The patient denied fever, chills, or cough. She stated that she was otherwise in good health and did not take any medication on a regular basis. Regarding her social history, she admitted to smoking one pack of cigarettes a day and drinking alcohol on occasion.
On physical examination, the patient appeared uncomfortable. Her vital signs were: blood pressure, 136/86 mm Hg; heart rate, 102 beats/min; respiratory rate, 22 breaths/min; and temperature, 98.60F. Oxygen saturation was 97% on room air. The head, eyes, ears, nose, and throat examination was unremarkable. Auscultation of the lungs revealed clear breath sounds bilaterally. The heart examination revealed tachycardia, but with regular rhythm and without murmurs, rubs, or gallops. The abdomen was soft and nontender. No lower extremity examination was documented.
The patient was seen by a physician assistant (PA) in the ED. An electrocardiogram (ECG), complete blood count (CBC), basic metabolic profile (BMP), troponin level, chest X-ray (CXR), and urine pregnancy test were ordered. The patient was given intravenous (IV) fluids and prochlorperazine 10 mg IV. The ECG and CXR were interpreted as normal. The urine pregnancy test was negative, and the remaining blood test results were within normal limits.
The PA believed the patient suffered from gastroenteritis, coupled with anxiety. He discharged the patient home with instructions to drink clear liquids for 24 hours, and take the prescribed prochlorperazine tablets as needed for continued nausea and vomiting.
At home, the patient continued to experience increasingly severe chest pain, shortness of breath, and vomiting. The next morning, she could no longer tolerate the pain and returned to the same ED via emergency medical services.
The patient’s history and physical examination remained unchanged from her presentation 16 hours earlier. At this ED visit, the patient was seen by an emergency physician (EP) who, concerned the patient had suffered an ischemic coronary event, ordered repeat ECG, CBC, BMP, and troponin evaluation. The EP also contacted cardiology services, but the cardiologist did not see the patient for several hours. When the cardiologist evaluated the patient and interpreted the ECG, he was concerned for an ST-segment elevation myocardial infarction (STEMI), and activated the catheterization lab.
Unfortunately, the patient had significant myocardial damage, with a resulting ejection fraction of only 10%. She was judged to be a candidate for heart transplantation, and received a left ventricular assist device (LVAD) as a bridge until a suitable donor heart could be identified. One month after implantation of the LVAD, the patient experienced an ischemic stroke that resulted in dense left-side weakness, leaving her confined to bed.
The patient sued the PA, the EP, the hospital, and the cardiologist for failing to identify and treat the acute STEMI in a timely manner. The plaintiff claimed the STEMI began at her first presentation to the ED, and that it should have been diagnosed and treated at that time. The plaintiff further argued that she should at least have been monitored and undergone repeat testing (ie, ECG and troponin level evaluation) at the first visit, stating that if she had received proper treatment, she would not have required an LVAD and therefore would not have had a stroke. The patient also alleged that at the second ED visit, there was a significant time delay before she was taken to the catheterization lab, which resulted in additional myocardial injury.
The defendants argued the patient was appropriately evaluated and treated at the first presentation, and that there was no evidence to suggest an MI. The EP argued that the delay in the patient’s care at the second visit was not his fault. All of the parties involved negotiated a settlement in the amount of $6 million in favor of the plaintiff.
Discussion
Myocardial infarction in adults younger than age 45 years is relatively rare, comprising only 2% to 10% of all MIs.1,2 The percentage of MI in patients younger than age 25 years must be even smaller, but no good data are available. In fact, age 40 years and younger is usually an exclusion criteria in many of the multicenter studies involving MI. Women are relatively spared from coronary artery disease (CAD) before menopause, thanks to the cardioprotective effects of estrogen. Young women who do experience an MI usually will have cardiovascular risk factors, especially smoking.
Risk Factors for MI in Young Patients
Cigarette Smoking. When examining common risk factors in young patients who had an MI (defined as patients younger than age 45 years), cigarette smoking is the most common risk factor.1,2 Between 76% and 91% of young patients with an MI are smokers, compared to only 40% incidence in older patients.1 It is thought that cigarette smoking produces endothelial dysfunction and can precipitate coronary spasm.1
Nonatherosclerotic Etiology. Interestingly, several studies of MI in young patients found a higher incidence of nonatherosclerotic causes of MI in women compared to men.2 One explanation for this finding is that women experience vasospastic syndromes and hypercoagulable states, secondary to oral contraceptive use or hereditary coagulation disorders.2 It has also been shown that young women have more active platelets following an MI and experience plaque erosions, rather than the plaque ruptures that occur in men and older women.2,3
Hyperlipidemia. Hyperlipidemia is an additional risk factor for MI in the younger adult patient population. In one study of young patients who had an MI, hyperlipidemia was the most important risk factor, in the absence of other obvious risk factors.1,4 In fact, some researchers think hyperlipidemia may be a more reliable predictor of MI in patients aged 30 to 39 years than in older patients.1,5 Unfortunately, many of these young adults are not aware that they have hyperlipidemia until they experience a complication such as an acute coronary syndrome. With respect to the patient in this case, it is not clear from the published report whether or not she had hyperlipidemia.
Family History. Another risk factor for MI in younger patients is a positive family history of CAD in a first-degree relative younger than age 55 years.1 Siblings of a young patient who experienced an MI have up to a 10-fold increase for developing CAD.1 It is currently not known why a positive family history increases the risk of MI in younger patients, but it may be related to inherited disorders of lipid metabolism, blood coagulation, or other genetic factors.1
Drug Abuse. Finally, drug abuse must be considered in young patients presenting with an MI. The use of cocaine, methamphetamine, marijuana, and K2 (synthetic marijuana) have all been associated with MI, especially in young patients,6-9 who typically do not have cardiac risk factors and do not show evidence of atherosclerotic disease on cardiac catheterization. As for the patient in this case, we do not know if she used any illicit drugs prior to presentation.
Summary
This case underscores the importance of not excluding MI in the differential diagnosis based simply on age or sex. While MI is uncommon in a 21-year-old woman, it can and does occur. In young patients presenting with chest pain, it is important to obtain a thorough history, including smoking, family history of MI, hyperlipidemia, and illicit drug use. While MI may be low on the differential diagnosis, it still needs to be considered.
1. Choudhury L, Marsh JD. Myocardial infarction in young patients. Am J Med. 1999;107(3):254-261.
2. Lawesson SS, Stenestrand U, Lagerqvist B, Wallentin L, Swahn E. Gender perspective on risk factors, coronary lesions and long-term outcome in young patients with ST-elevation myocardial infarction. Heart. 2010;96(6):453-459. doi:10.1136/hrt.2009.175463.
3. Burke AP, Farb A, Malcom G,Virmani R. Effect of menopause on plaque morphologic characteristics in coronary atherosclerosis. Am Heart J. 2001;141(2 Suppl):S58-S62.
4. Tomono S, Ohshima S, Murata K. The risk factors for ischemic heart disease in young adults. Jpn Circ J. 1990;54(4):436-441.
5. Gofman JW, Young W, Tandy R. Ischemic heart disease, atherosclerosis, and longevity. Circulation. 1966;34(4):679-697.
6. Zimmerman JL. Cocaine intoxication. Crit Care Clin. 2012;28(4):517-525. doi:10.1016/j.ccc.2012.07.003.
7. Hawley LA, Auten JD, Matteucci MJ, et al. Cardiac complications of adult methamphetamine exposures. J Emerg Med. 2013;45(6):821-827. doi:10.1016/j.jemermed.2013.04.061.
8. Bachs L, Mørland H. Acute cardiovascular fatalities following cannabis use. Forensic Sci Int. 2001;124(2-3):200-203.
9. Mir A, Obafemi A, Young A, Kane C. Myocardial infarction associated with use of the synthetic cannabinoid K2. Pediatrics. 2011;128(6):e1622-e1627. doi:10.1542 peds.2010-3823.
1. Choudhury L, Marsh JD. Myocardial infarction in young patients. Am J Med. 1999;107(3):254-261.
2. Lawesson SS, Stenestrand U, Lagerqvist B, Wallentin L, Swahn E. Gender perspective on risk factors, coronary lesions and long-term outcome in young patients with ST-elevation myocardial infarction. Heart. 2010;96(6):453-459. doi:10.1136/hrt.2009.175463.
3. Burke AP, Farb A, Malcom G,Virmani R. Effect of menopause on plaque morphologic characteristics in coronary atherosclerosis. Am Heart J. 2001;141(2 Suppl):S58-S62.
4. Tomono S, Ohshima S, Murata K. The risk factors for ischemic heart disease in young adults. Jpn Circ J. 1990;54(4):436-441.
5. Gofman JW, Young W, Tandy R. Ischemic heart disease, atherosclerosis, and longevity. Circulation. 1966;34(4):679-697.
6. Zimmerman JL. Cocaine intoxication. Crit Care Clin. 2012;28(4):517-525. doi:10.1016/j.ccc.2012.07.003.
7. Hawley LA, Auten JD, Matteucci MJ, et al. Cardiac complications of adult methamphetamine exposures. J Emerg Med. 2013;45(6):821-827. doi:10.1016/j.jemermed.2013.04.061.
8. Bachs L, Mørland H. Acute cardiovascular fatalities following cannabis use. Forensic Sci Int. 2001;124(2-3):200-203.
9. Mir A, Obafemi A, Young A, Kane C. Myocardial infarction associated with use of the synthetic cannabinoid K2. Pediatrics. 2011;128(6):e1622-e1627. doi:10.1542 peds.2010-3823.

Emergency Imaging: Severe Left Testicular Swelling
A 32-year-old man presented to the ED with acute onset of left testicular swelling and pain. He described the pain as severe, radiating to his lower back and lower abdomen. Regarding his medical history, the patient stated he had experienced similar episodes of significant testicular swelling in the past, for which he was treated with antibiotics.

Physical examination revealed mild enlargement of the left testis with tenderness to palpation. The right testis was normal in appearance and nontender. An ultrasound study of the testicles was ordered; representative images are shown (Figures 1a-1c).
What is the diagnosis?
The transverse image of both testes demonstrated an enlarged left testicle compared to the right testicle (Figure 2a). On color-flow Doppler ultrasound, spots of color within the testicle were noted within the right testicle only. The lack of blood flow was confirmed on the sagittal image of the left testicle, which also revealed a small hydrocele (white arrows, Figure 2b). A sagittal color Doppler image of the normal right testicle showed color flow (white arrows, Figure 2c) and normal vascular waveforms (red arrow, Figure 2c) within the testis, but no hydrocele, confirming the diagnosis of left testicular torsion. The Doppler ultrasound of the right testicle (white arrows, Figure 2c) further confirmed a normal right testicle but no evidence of flow in the left testicle. These findings were further consistent with the presence of left testicular torsion.
Answer
Testicular Torsion
Testicular torsion is a urological emergency that results from a twisting of the spermatic cord, cutting off arterial flow to, and venous drainage from, the affected testis. There are two types of testicular torsion depending on which side of the tunica vaginalis (the serous membrane pouch covering the testes) the torsion occurs: extra vaginal, seen mainly in newborns; and intravaginal, which can occur at any age, but is more common in adolescents.

“Bell clapper deformity” is a predisposing congenital condition resulting from intravaginal torsion of the testis in which the tunica vaginalis joins high on the spermatic cord, leaving the testis free to rotate.1 Testicular torsion most commonly occurs in young males, with an estimated incidence of 4.5 cases per 100,000 patients between ages 1 and 25 years.2
Clinical Presentation
Patients with testicular torsion typically experience a sudden onset of severe unilateral pain often accompanied by nausea and vomiting, which can occur spontaneously or after vigorous physical activity or trauma. Associated complaints may include urinary symptoms and/or fever.3 The affected testis may lie transversely in the scrotum and be retracted, although physical examination is often nonspecific and unreliable. Since an absence of the cremasteric reflex is neither sensitive nor specific in determining the need for surgical intervention, further diagnostic testing is required.4
Doppler Ultrasound
Ultrasound utilizing color and spectral Doppler techniques is the imaging test of choice to evaluate for testicular torsion, and has a reported sensitivity of 82% to 89%, and a specificity of 98% to 100%.5,6 Ultrasound findings include enlargement and decreased echogenicity of the affected testicle due to edema. Scrotal wall thickening and a small hydrocele also may be seen. Doppler imaging also typically demonstrates absence of flow, though hyperemia and increased flow may be present early in the disease process.
It is important to note that torsion may be intermittent; therefore, imaging studies can appear normal during periods of intermittent perfusion. If there is incomplete torsion and some arterial flow persists in the affected testis, comparison of the two testes using transverse views is very useful in making the diagnosis.7
With respect to the differential diagnoses, ultrasound imaging studies are also useful in diagnosing other conditions associated with testicular pain, including torsion of the appendix testis, epididymitis, orchitis, trauma, varicocele, and tumors.
Treatment
Rapid diagnosis of testicular torsion is important, as delay in diagnosis may lead to irreversible damage and loss of the testicle. Infertility can result even with a normal contralateral testis.8 When surgical intervention is performed within 6 hours from onset of torsion, salvage of the testicle has been reported to be 90% to 100%, but only 50% and 10% at 12 and 24 hours, respectively.3 The patient in this case was taken immediately for emergent surgical detorsion, and the left testicle was salvaged.
1. Caesar RE, Kaplan GW. Incidence of the bell-clapper deformity in an autopsy series. Urology. 1994;44 (1):114-116.
2. Mansbach JM, Forbes P, Peters C. Testicular torsion and risk factors for orchiectomy. Arch Pediatr Adolesc Med. 2005;159(12):1167-1171. doi:10.1001/archpedi.159.12.1167.
3. Sharp VJ, Kieran K, Arlen AM. Testicular torsion: diagnosis, evaluation, and management. Am Fam Physician. 2013;88(12):835-840.
4. Mellick LB. Torsion of the testicle: It is time to stop tossing the dice. Pediatr Emerg Care. 2012;28:80Y86. doi:10.1097/PEC.0b013e31823f5ed9.
5. Baker LA, Sigman D, Mathews RI, Benson J, Docimo SG. An analysis of clinical outcomes using color doppler testicular ultrasound for testicular torsion. Pediatrics. 2000;105(3 Pt 1):604-607.
6. Burks DD, Markey BJ, Burkhard TK, Balsara ZN, Haluszka MM, Canning DA. Suspected testicular torsion and ischemia: evaluation with color Doppler sonography. Radiology. 1990;175(3):815-821. doi:10.1148/radiology.175.3.2188301.
7. Aso C, Enríquez G, Fité M, et al. Gray-scale and color doppler sonography of scrotal disorders in children: an update. Radiographics. 2005;25(5):1197-1214. doi:10.1148/rg.255045109.
8. Hadziselimovic F, Geneto R, Emmons LR. Increased apoptosis in the contralateral testes of patients with testicular torsion as a factor for infertility. J Urol. 1998;160(3 Pt 2):1158-1160.
A 32-year-old man presented to the ED with acute onset of left testicular swelling and pain. He described the pain as severe, radiating to his lower back and lower abdomen. Regarding his medical history, the patient stated he had experienced similar episodes of significant testicular swelling in the past, for which he was treated with antibiotics.
Physical examination revealed mild enlargement of the left testis with tenderness to palpation. The right testis was normal in appearance and nontender. An ultrasound study of the testicles was ordered; representative images are shown (Figures 1a-1c).
What is the diagnosis?
The transverse image of both testes demonstrated an enlarged left testicle compared to the right testicle (Figure 2a). On color-flow Doppler ultrasound, spots of color within the testicle were noted within the right testicle only. The lack of blood flow was confirmed on the sagittal image of the left testicle, which also revealed a small hydrocele (white arrows, Figure 2b). A sagittal color Doppler image of the normal right testicle showed color flow (white arrows, Figure 2c) and normal vascular waveforms (red arrow, Figure 2c) within the testis, but no hydrocele, confirming the diagnosis of left testicular torsion. The Doppler ultrasound of the right testicle (white arrows, Figure 2c) further confirmed a normal right testicle but no evidence of flow in the left testicle. These findings were further consistent with the presence of left testicular torsion.
Answer
Testicular Torsion
Testicular torsion is a urological emergency that results from a twisting of the spermatic cord, cutting off arterial flow to, and venous drainage from, the affected testis. There are two types of testicular torsion depending on which side of the tunica vaginalis (the serous membrane pouch covering the testes) the torsion occurs: extra vaginal, seen mainly in newborns; and intravaginal, which can occur at any age, but is more common in adolescents.
“Bell clapper deformity” is a predisposing congenital condition resulting from intravaginal torsion of the testis in which the tunica vaginalis joins high on the spermatic cord, leaving the testis free to rotate.1 Testicular torsion most commonly occurs in young males, with an estimated incidence of 4.5 cases per 100,000 patients between ages 1 and 25 years.2
Clinical Presentation
Patients with testicular torsion typically experience a sudden onset of severe unilateral pain often accompanied by nausea and vomiting, which can occur spontaneously or after vigorous physical activity or trauma. Associated complaints may include urinary symptoms and/or fever.3 The affected testis may lie transversely in the scrotum and be retracted, although physical examination is often nonspecific and unreliable. Since an absence of the cremasteric reflex is neither sensitive nor specific in determining the need for surgical intervention, further diagnostic testing is required.4
Doppler Ultrasound
Ultrasound utilizing color and spectral Doppler techniques is the imaging test of choice to evaluate for testicular torsion, and has a reported sensitivity of 82% to 89%, and a specificity of 98% to 100%.5,6 Ultrasound findings include enlargement and decreased echogenicity of the affected testicle due to edema. Scrotal wall thickening and a small hydrocele also may be seen. Doppler imaging also typically demonstrates absence of flow, though hyperemia and increased flow may be present early in the disease process.
It is important to note that torsion may be intermittent; therefore, imaging studies can appear normal during periods of intermittent perfusion. If there is incomplete torsion and some arterial flow persists in the affected testis, comparison of the two testes using transverse views is very useful in making the diagnosis.7
With respect to the differential diagnoses, ultrasound imaging studies are also useful in diagnosing other conditions associated with testicular pain, including torsion of the appendix testis, epididymitis, orchitis, trauma, varicocele, and tumors.
Treatment
Rapid diagnosis of testicular torsion is important, as delay in diagnosis may lead to irreversible damage and loss of the testicle. Infertility can result even with a normal contralateral testis.8 When surgical intervention is performed within 6 hours from onset of torsion, salvage of the testicle has been reported to be 90% to 100%, but only 50% and 10% at 12 and 24 hours, respectively.3 The patient in this case was taken immediately for emergent surgical detorsion, and the left testicle was salvaged.
A 32-year-old man presented to the ED with acute onset of left testicular swelling and pain. He described the pain as severe, radiating to his lower back and lower abdomen. Regarding his medical history, the patient stated he had experienced similar episodes of significant testicular swelling in the past, for which he was treated with antibiotics.
Physical examination revealed mild enlargement of the left testis with tenderness to palpation. The right testis was normal in appearance and nontender. An ultrasound study of the testicles was ordered; representative images are shown (Figures 1a-1c).
What is the diagnosis?
The transverse image of both testes demonstrated an enlarged left testicle compared to the right testicle (Figure 2a). On color-flow Doppler ultrasound, spots of color within the testicle were noted within the right testicle only. The lack of blood flow was confirmed on the sagittal image of the left testicle, which also revealed a small hydrocele (white arrows, Figure 2b). A sagittal color Doppler image of the normal right testicle showed color flow (white arrows, Figure 2c) and normal vascular waveforms (red arrow, Figure 2c) within the testis, but no hydrocele, confirming the diagnosis of left testicular torsion. The Doppler ultrasound of the right testicle (white arrows, Figure 2c) further confirmed a normal right testicle but no evidence of flow in the left testicle. These findings were further consistent with the presence of left testicular torsion.
Answer
Testicular Torsion
Testicular torsion is a urological emergency that results from a twisting of the spermatic cord, cutting off arterial flow to, and venous drainage from, the affected testis. There are two types of testicular torsion depending on which side of the tunica vaginalis (the serous membrane pouch covering the testes) the torsion occurs: extra vaginal, seen mainly in newborns; and intravaginal, which can occur at any age, but is more common in adolescents.
“Bell clapper deformity” is a predisposing congenital condition resulting from intravaginal torsion of the testis in which the tunica vaginalis joins high on the spermatic cord, leaving the testis free to rotate.1 Testicular torsion most commonly occurs in young males, with an estimated incidence of 4.5 cases per 100,000 patients between ages 1 and 25 years.2
Clinical Presentation
Patients with testicular torsion typically experience a sudden onset of severe unilateral pain often accompanied by nausea and vomiting, which can occur spontaneously or after vigorous physical activity or trauma. Associated complaints may include urinary symptoms and/or fever.3 The affected testis may lie transversely in the scrotum and be retracted, although physical examination is often nonspecific and unreliable. Since an absence of the cremasteric reflex is neither sensitive nor specific in determining the need for surgical intervention, further diagnostic testing is required.4
Doppler Ultrasound
Ultrasound utilizing color and spectral Doppler techniques is the imaging test of choice to evaluate for testicular torsion, and has a reported sensitivity of 82% to 89%, and a specificity of 98% to 100%.5,6 Ultrasound findings include enlargement and decreased echogenicity of the affected testicle due to edema. Scrotal wall thickening and a small hydrocele also may be seen. Doppler imaging also typically demonstrates absence of flow, though hyperemia and increased flow may be present early in the disease process.
It is important to note that torsion may be intermittent; therefore, imaging studies can appear normal during periods of intermittent perfusion. If there is incomplete torsion and some arterial flow persists in the affected testis, comparison of the two testes using transverse views is very useful in making the diagnosis.7
With respect to the differential diagnoses, ultrasound imaging studies are also useful in diagnosing other conditions associated with testicular pain, including torsion of the appendix testis, epididymitis, orchitis, trauma, varicocele, and tumors.
Treatment
Rapid diagnosis of testicular torsion is important, as delay in diagnosis may lead to irreversible damage and loss of the testicle. Infertility can result even with a normal contralateral testis.8 When surgical intervention is performed within 6 hours from onset of torsion, salvage of the testicle has been reported to be 90% to 100%, but only 50% and 10% at 12 and 24 hours, respectively.3 The patient in this case was taken immediately for emergent surgical detorsion, and the left testicle was salvaged.
1. Caesar RE, Kaplan GW. Incidence of the bell-clapper deformity in an autopsy series. Urology. 1994;44 (1):114-116.
2. Mansbach JM, Forbes P, Peters C. Testicular torsion and risk factors for orchiectomy. Arch Pediatr Adolesc Med. 2005;159(12):1167-1171. doi:10.1001/archpedi.159.12.1167.
3. Sharp VJ, Kieran K, Arlen AM. Testicular torsion: diagnosis, evaluation, and management. Am Fam Physician. 2013;88(12):835-840.
4. Mellick LB. Torsion of the testicle: It is time to stop tossing the dice. Pediatr Emerg Care. 2012;28:80Y86. doi:10.1097/PEC.0b013e31823f5ed9.
5. Baker LA, Sigman D, Mathews RI, Benson J, Docimo SG. An analysis of clinical outcomes using color doppler testicular ultrasound for testicular torsion. Pediatrics. 2000;105(3 Pt 1):604-607.
6. Burks DD, Markey BJ, Burkhard TK, Balsara ZN, Haluszka MM, Canning DA. Suspected testicular torsion and ischemia: evaluation with color Doppler sonography. Radiology. 1990;175(3):815-821. doi:10.1148/radiology.175.3.2188301.
7. Aso C, Enríquez G, Fité M, et al. Gray-scale and color doppler sonography of scrotal disorders in children: an update. Radiographics. 2005;25(5):1197-1214. doi:10.1148/rg.255045109.
8. Hadziselimovic F, Geneto R, Emmons LR. Increased apoptosis in the contralateral testes of patients with testicular torsion as a factor for infertility. J Urol. 1998;160(3 Pt 2):1158-1160.
1. Caesar RE, Kaplan GW. Incidence of the bell-clapper deformity in an autopsy series. Urology. 1994;44 (1):114-116.
2. Mansbach JM, Forbes P, Peters C. Testicular torsion and risk factors for orchiectomy. Arch Pediatr Adolesc Med. 2005;159(12):1167-1171. doi:10.1001/archpedi.159.12.1167.
3. Sharp VJ, Kieran K, Arlen AM. Testicular torsion: diagnosis, evaluation, and management. Am Fam Physician. 2013;88(12):835-840.
4. Mellick LB. Torsion of the testicle: It is time to stop tossing the dice. Pediatr Emerg Care. 2012;28:80Y86. doi:10.1097/PEC.0b013e31823f5ed9.
5. Baker LA, Sigman D, Mathews RI, Benson J, Docimo SG. An analysis of clinical outcomes using color doppler testicular ultrasound for testicular torsion. Pediatrics. 2000;105(3 Pt 1):604-607.
6. Burks DD, Markey BJ, Burkhard TK, Balsara ZN, Haluszka MM, Canning DA. Suspected testicular torsion and ischemia: evaluation with color Doppler sonography. Radiology. 1990;175(3):815-821. doi:10.1148/radiology.175.3.2188301.
7. Aso C, Enríquez G, Fité M, et al. Gray-scale and color doppler sonography of scrotal disorders in children: an update. Radiographics. 2005;25(5):1197-1214. doi:10.1148/rg.255045109.
8. Hadziselimovic F, Geneto R, Emmons LR. Increased apoptosis in the contralateral testes of patients with testicular torsion as a factor for infertility. J Urol. 1998;160(3 Pt 2):1158-1160.
Focused on value-based care: Harry Cho, MD
Education and service have always been important for Harry Cho, MD, who recently joined the editorial advisory board of The Hospitalist.
From joining AmeriCorps as a fresh faced college graduate, to his ongoing work as assistant professor of medicine and director of quality, safety, and value for the division of hospital medicine at Mount Sinai in New York, and as senior fellow at the Lown Institute, Dr. Cho has found a passion in helping others learn.

When not teaching or working with patients, Dr. Cho is committed to improving value-based medicine, a path that has lead him to create the High Value Chair Initiatives, a program dedicated to offering clinicians resources on how to reduce wasteful testing and harmful practices.
Dr. Cho said he is excited to contribute as one of eight new members of The Hospitalist editorial advisory board in 2017 and took time to tell us more about himself in a recent interview.
Q: Why did you choose medicine as a career?
A: Right after I finished undergrad at Cornell, I spent the summer and the following year doing AmeriCorps, which is service learning work, and I worked in the inner city of Philadelphia. I worked on after-school programs and weekend programs for inner city youth and I loved it. I was organizing and developing these programs, and I thought it was fantastic. The one thing that I thought was lacking, and I think what really drove me to get into medicine, was that at the end of the day, although I felt really connected with all the kids, I felt like I was a role model, like I was a mentor, and we had a really good connection, but I wanted something a little bit more concrete on improving outcomes. I knew we made connections, but I really wanted to know more – such as, did we reduce the dropout rate in high school for these students? I think that’s why medicine was really interesting.
Q: How did you end up in hospital medicine?
A: I think it’s a lot of things. I love the acuity, I love playing the quarterback in a place where a lot of things are going back and forth and you have to coordinate with others. You have to make sure you see the patient from top to bottom, the whole picture, and I love that part. I also love the action and the communication and the teamwork aspect of it.
Q: What part of being a hospitalist do you like the most?
A: I love the education on a daily basis: the morning rounds where you walk around for an hour or two with your team, and you teach them at the bedside, and these little pearls come up along the way. My career is positioned more within quality, value improvement, and safety, so I think that participating in the education process is really helpful. I think hospital medicine has taken over that spirit in the hospital setting, and I love that.
Q: Which part do you like the least?
A: I think we’re in a unique time right now. Burnout is getting a little tougher to beat. People are getting a bit more tired, and I don’t think we have a good solution to solve this. With quality improvement and the electronic medical record system, a lot of us are expected to do more. I still get queries from clinical documentation saying, “I need you to document this for billing purposes” or “I need you to document this for increasing the expected length of stay,” and doctors are not quite at the point where they can balance these requirements in an effective way. There tends to be an emphasis on “one more click,” one more thing to document, just one more thing to do on the checklist. It’s getting more complex.
Q: What is the most rewarding part of your work?
A: Larger scale accomplishments. When you give a talk, or teach a group of residents during morning rounds, and they look at you with wonder because you have this teaching pearl they’ve never heard before, and they think you’re a great attending – that’s very instant gratification, but there's more to be done beyond that. I’ve been co-directing in the Right Care educator program, and we have a High Value Care curriculum that we’ve been implementing across the country, and we’ve just finished our second year. There are around 60 programs involved, and it’s a great feeling. You’re not seeing actual people face to face after they’ve been taught, and you’re not getting that instant gratification. But just knowing what one of those chief residents who has implemented the program is feeling, and extrapolating across the number of programs this year alone, that makes me feel good.
Q: Outside of hospital work, what else are you interested in?
A: High-value care is my central aim right now. I want to expand it, and I want to do things on a national scale. We formed a High Value Care committee and I’m hoping to create new guidelines to reduce overuse, overtesting, and Choosing Wisely. Outside of medicine, I like photography. Nothing professional, but I love taking pictures, especially nature and travel. Back in the day, I used to do a lot of running and martial arts too.
Q: Where do you see yourself in 10 years?
A: I’m not sure if I will go the chief medical officer or chief quality officer route. That’s probably where I see myself. I definitely want to continue making bigger changes on a national scale, like implementing the overuse educator program across the country.
Q: What do you see as the future of hospital medicine?
A: Value-based health care is always going to get bigger as the cost of health care and the cost of overuse rises, and we start to see a lot of harms outlined in research. We’re going to be on top of it much more, because the hospital setting is complex and continues to change.
Education and service have always been important for Harry Cho, MD, who recently joined the editorial advisory board of The Hospitalist.
From joining AmeriCorps as a fresh faced college graduate, to his ongoing work as assistant professor of medicine and director of quality, safety, and value for the division of hospital medicine at Mount Sinai in New York, and as senior fellow at the Lown Institute, Dr. Cho has found a passion in helping others learn.
When not teaching or working with patients, Dr. Cho is committed to improving value-based medicine, a path that has lead him to create the High Value Chair Initiatives, a program dedicated to offering clinicians resources on how to reduce wasteful testing and harmful practices.
Dr. Cho said he is excited to contribute as one of eight new members of The Hospitalist editorial advisory board in 2017 and took time to tell us more about himself in a recent interview.
Q: Why did you choose medicine as a career?
A: Right after I finished undergrad at Cornell, I spent the summer and the following year doing AmeriCorps, which is service learning work, and I worked in the inner city of Philadelphia. I worked on after-school programs and weekend programs for inner city youth and I loved it. I was organizing and developing these programs, and I thought it was fantastic. The one thing that I thought was lacking, and I think what really drove me to get into medicine, was that at the end of the day, although I felt really connected with all the kids, I felt like I was a role model, like I was a mentor, and we had a really good connection, but I wanted something a little bit more concrete on improving outcomes. I knew we made connections, but I really wanted to know more – such as, did we reduce the dropout rate in high school for these students? I think that’s why medicine was really interesting.
Q: How did you end up in hospital medicine?
A: I think it’s a lot of things. I love the acuity, I love playing the quarterback in a place where a lot of things are going back and forth and you have to coordinate with others. You have to make sure you see the patient from top to bottom, the whole picture, and I love that part. I also love the action and the communication and the teamwork aspect of it.
Q: What part of being a hospitalist do you like the most?
A: I love the education on a daily basis: the morning rounds where you walk around for an hour or two with your team, and you teach them at the bedside, and these little pearls come up along the way. My career is positioned more within quality, value improvement, and safety, so I think that participating in the education process is really helpful. I think hospital medicine has taken over that spirit in the hospital setting, and I love that.
Q: Which part do you like the least?
A: I think we’re in a unique time right now. Burnout is getting a little tougher to beat. People are getting a bit more tired, and I don’t think we have a good solution to solve this. With quality improvement and the electronic medical record system, a lot of us are expected to do more. I still get queries from clinical documentation saying, “I need you to document this for billing purposes” or “I need you to document this for increasing the expected length of stay,” and doctors are not quite at the point where they can balance these requirements in an effective way. There tends to be an emphasis on “one more click,” one more thing to document, just one more thing to do on the checklist. It’s getting more complex.
Q: What is the most rewarding part of your work?
A: Larger scale accomplishments. When you give a talk, or teach a group of residents during morning rounds, and they look at you with wonder because you have this teaching pearl they’ve never heard before, and they think you’re a great attending – that’s very instant gratification, but there's more to be done beyond that. I’ve been co-directing in the Right Care educator program, and we have a High Value Care curriculum that we’ve been implementing across the country, and we’ve just finished our second year. There are around 60 programs involved, and it’s a great feeling. You’re not seeing actual people face to face after they’ve been taught, and you’re not getting that instant gratification. But just knowing what one of those chief residents who has implemented the program is feeling, and extrapolating across the number of programs this year alone, that makes me feel good.
Q: Outside of hospital work, what else are you interested in?
A: High-value care is my central aim right now. I want to expand it, and I want to do things on a national scale. We formed a High Value Care committee and I’m hoping to create new guidelines to reduce overuse, overtesting, and Choosing Wisely. Outside of medicine, I like photography. Nothing professional, but I love taking pictures, especially nature and travel. Back in the day, I used to do a lot of running and martial arts too.
Q: Where do you see yourself in 10 years?
A: I’m not sure if I will go the chief medical officer or chief quality officer route. That’s probably where I see myself. I definitely want to continue making bigger changes on a national scale, like implementing the overuse educator program across the country.
Q: What do you see as the future of hospital medicine?
A: Value-based health care is always going to get bigger as the cost of health care and the cost of overuse rises, and we start to see a lot of harms outlined in research. We’re going to be on top of it much more, because the hospital setting is complex and continues to change.
Education and service have always been important for Harry Cho, MD, who recently joined the editorial advisory board of The Hospitalist.
From joining AmeriCorps as a fresh faced college graduate, to his ongoing work as assistant professor of medicine and director of quality, safety, and value for the division of hospital medicine at Mount Sinai in New York, and as senior fellow at the Lown Institute, Dr. Cho has found a passion in helping others learn.
When not teaching or working with patients, Dr. Cho is committed to improving value-based medicine, a path that has lead him to create the High Value Chair Initiatives, a program dedicated to offering clinicians resources on how to reduce wasteful testing and harmful practices.
Dr. Cho said he is excited to contribute as one of eight new members of The Hospitalist editorial advisory board in 2017 and took time to tell us more about himself in a recent interview.
Q: Why did you choose medicine as a career?
A: Right after I finished undergrad at Cornell, I spent the summer and the following year doing AmeriCorps, which is service learning work, and I worked in the inner city of Philadelphia. I worked on after-school programs and weekend programs for inner city youth and I loved it. I was organizing and developing these programs, and I thought it was fantastic. The one thing that I thought was lacking, and I think what really drove me to get into medicine, was that at the end of the day, although I felt really connected with all the kids, I felt like I was a role model, like I was a mentor, and we had a really good connection, but I wanted something a little bit more concrete on improving outcomes. I knew we made connections, but I really wanted to know more – such as, did we reduce the dropout rate in high school for these students? I think that’s why medicine was really interesting.
Q: How did you end up in hospital medicine?
A: I think it’s a lot of things. I love the acuity, I love playing the quarterback in a place where a lot of things are going back and forth and you have to coordinate with others. You have to make sure you see the patient from top to bottom, the whole picture, and I love that part. I also love the action and the communication and the teamwork aspect of it.
Q: What part of being a hospitalist do you like the most?
A: I love the education on a daily basis: the morning rounds where you walk around for an hour or two with your team, and you teach them at the bedside, and these little pearls come up along the way. My career is positioned more within quality, value improvement, and safety, so I think that participating in the education process is really helpful. I think hospital medicine has taken over that spirit in the hospital setting, and I love that.
Q: Which part do you like the least?
A: I think we’re in a unique time right now. Burnout is getting a little tougher to beat. People are getting a bit more tired, and I don’t think we have a good solution to solve this. With quality improvement and the electronic medical record system, a lot of us are expected to do more. I still get queries from clinical documentation saying, “I need you to document this for billing purposes” or “I need you to document this for increasing the expected length of stay,” and doctors are not quite at the point where they can balance these requirements in an effective way. There tends to be an emphasis on “one more click,” one more thing to document, just one more thing to do on the checklist. It’s getting more complex.
Q: What is the most rewarding part of your work?
A: Larger scale accomplishments. When you give a talk, or teach a group of residents during morning rounds, and they look at you with wonder because you have this teaching pearl they’ve never heard before, and they think you’re a great attending – that’s very instant gratification, but there's more to be done beyond that. I’ve been co-directing in the Right Care educator program, and we have a High Value Care curriculum that we’ve been implementing across the country, and we’ve just finished our second year. There are around 60 programs involved, and it’s a great feeling. You’re not seeing actual people face to face after they’ve been taught, and you’re not getting that instant gratification. But just knowing what one of those chief residents who has implemented the program is feeling, and extrapolating across the number of programs this year alone, that makes me feel good.
Q: Outside of hospital work, what else are you interested in?
A: High-value care is my central aim right now. I want to expand it, and I want to do things on a national scale. We formed a High Value Care committee and I’m hoping to create new guidelines to reduce overuse, overtesting, and Choosing Wisely. Outside of medicine, I like photography. Nothing professional, but I love taking pictures, especially nature and travel. Back in the day, I used to do a lot of running and martial arts too.
Q: Where do you see yourself in 10 years?
A: I’m not sure if I will go the chief medical officer or chief quality officer route. That’s probably where I see myself. I definitely want to continue making bigger changes on a national scale, like implementing the overuse educator program across the country.
Q: What do you see as the future of hospital medicine?
A: Value-based health care is always going to get bigger as the cost of health care and the cost of overuse rises, and we start to see a lot of harms outlined in research. We’re going to be on top of it much more, because the hospital setting is complex and continues to change.
Parkinsonism and Vitamin C Deficiency
Vitamin C (ascorbic acid) deficiency is known to affect brain function and is associated with parkinsonism.1 In 1752, James Lind, MD, described emotional and behavioral changes that herald the onset of scurvy and precede hemorrhagic findings.2 The World Health Organization (WHO) today refers to this stage as latent scurvy.3 The 2 case studies that follow present examples of patients with vitamin C deficiencies whose parkinsonism responded robustly to vitamin C replacement. These cases suggest that vitamin C deficiency may be a treatable cause of parkinsonism.
Case 1
Mr. A, a 60-year-old white male, was admitted to the Medicine Service for alcohol detoxification. The patient had a history of alcohol dependence, alcohol withdrawal seizures, tobacco dependence, and hyperlipidemia. He took no medications as an outpatient. On admission Mr. A’s body mass index (BMI) was 27.2. An initial examination revealed a marked resting tremor of the patient’s right hand with cogwheeling, which had not been present in examinations conducted in the previous 3 years. Mr. A had no prior history of a tremor. He had no cerebellar findings and no evidence of asterixis or of tremulousness associated with high-output cardiac states, such as de Musset sign.
Mr. A reported he had experienced the tremor for a month and that it had been worsening. He also was having difficulty using his dominant right hand, for routine daily activities. Mr. A was oriented, and his short-term memory was intact. He was ill-appearing, irritable with psychomotor slowing, and did not wish to rise from his bed. He had no gingival or periungual bleeding and did not bruise easily. He had no corkscrew hairs. The patient was started on no medications known to cause extrapyramidal symptoms (EPS).
In the hospital, the tremor persisted unabated for 2 days. On the third day, Mr. A was started on 1,000 mg vitamin C IV twice daily. He received a total of 2,000 mg IV that day, but the IV fell out, and he refused its replacement. Several hours later, Mr. A stated that he felt much better, got out of bed, and asked to go outside to smoke. The author noted complete resolution of the right hand tremor and cogwheeling 20 hours after starting the vitamin C IV. Mr. A refused a repeat serum vitamin C assay.
Laboratory studies initially revealed that Mr. A had hyponatremia with a serum sodium of 121 mmol/L (normal range: 133 to 145 mmol/L) as well as hypokalemia with a serum potassium of 3.2 mmol/L (normal range: 3.5 to 5.0 mmol/L). He was hypoosmolar, with a serum osmolality of 276 mOsm/kg (normal range: 278 to 305 mOsm/kg). His vitamin C level was low at 0.2 mg/dL (normal range: 0.4 to 2.0 mg/dL). Mr. A also had a serum vitamin C level drawn 2 years prior that showed no symptoms of EPS, and at that time, the reading was 0.7 mg/dL. At admission to Medicine Services, Mr. A had a serum alcohol level of 211 mg/dL. Neuroimaging revealed diffuse cerebral and cerebellar volume loss.
Normal laboratory results included serum levels of vitamin B12, red cell folate, homocysteine, methylmalonic acid, free and total carnitine, alkaline phosphatase, manganese, and zinc. A urine drug screen was negative.
Case 2
Mr. B, a 69-year-old black male, was admitted to the hospital for depression complicated by alcohol dependence. He also had tobacco dependence, type 2 diabetes mellitus, hypertension, and gout. The patient’s BMI at admission was 16.1. Mr. B appeared ill, was worried about his health, and remained recumbent unless asked to move. He reported that his right hand had begun to shake at rest in the month prior to admission. The tremor made it difficult for him to drink. He pointed out stains on his hospital gurney from an attempt to drink orange juice prior to being assessed.
A physical examination revealed a distinct resting tremor with cogwheeling of the right hand; there was no other evidence of EPS, nor was there evidence of cognitive, cerebellar, or skin abnormalities, such as hemorrhages or corkscrew hairs. Asterixis was absent as was evidence of a high-output cardiac state that might produce a tremor, such as de Musset sign. A serum vitamin C level was obtained and returned at 0.0 mg/dL. A head computed tomography scan obtained the next day revealed mild cerebellar volume loss. A serum alkaline phosphatase level was elevated slightly at 136 U/L (normal range: 42 to 113 U/L). Normal serum values were returned for zinc, vitamins B12 and folate, rapid plasma reagin, sodium, and serum osmolality. A urine drug screen was negative, and serum alcohol level was < 5.0 mg/dL.
Mr. B took no medications expected to cause EPS. He received no micronutrient replacement until the day after admission when he began receiving oral vitamin C 1,000 mg twice a day. After receiving 3 doses, Mr. B’s resting tremor and cogwheeling completely resolved. He noticed he had stopped shaking and could now drink without spilling fluids. He also got out of bed and began interacting with others. Mr. B said he felt he was “doing well.” A repeat serum vitamin C level was 0.2 mg/dL on that day. The improvement was sustained over 3 days, and Mr. B was discharged to home.
Discussion
Both Mr. A and Mr. B presented with a typical picture of latent scurvy and the additional finding of parkinsonism. These cases are important for 2 reasons. First, the swift and full response of these patients’ parkinsonism to vitamin C replacement underscores the importance of considering a vitamin C deficiency when confronted with EPS. And second, both patients lacked signs of bleeding or of impaired collagen synthesis. This differs from the classic presentation of scurvy as a disorder primarily of collagen metabolism.4
Lind described the onset of scurvy as one in which striking emotional and behavior changes developed and later were followed by abnormal bleeding and even death.2 These early changes also were recognized by Shapter in 1847.5 Furthermore, the evidence that exists about the time-course of scurvy’s development suggests that neuropsychiatric findings precede the hemorrhagic.6 Indeed, classic skin findings, such as petechiae or corkscrew hairs, may develop years after the onset of neuropsychiatric changes.7,8
Despite WHO characterizing it as latent scurvy, the distinct syndromal presentation of hypovitaminosis C with parkinsonism along with the rapid response to vitamin C replacement argues for its recognition as a distinct clinical entity and not just a prelude to the hemorrhagic state. To assist in recognizing neuropsychiatric scurvy, the author suggests the operationalized approach described in Table 1.9

Pathophysiology
Vitamin C has an intimate role in the normal functioning of the basal ganglia. It is involved in the synthesis of catechecholamines, the regulation of the release and postsynaptic activities of various neurotransmitters, and managing the oxyradical toxicity of aerobic metabolism. Table 2 outlines some of the normal brain functions of vitamin C and the potential consequences of inadequate central vitamin C.9,10 Risk factors for vitamin C deficiency include those affecting the uptake, response to, and elimination of this vitamin (Table 3).11-14
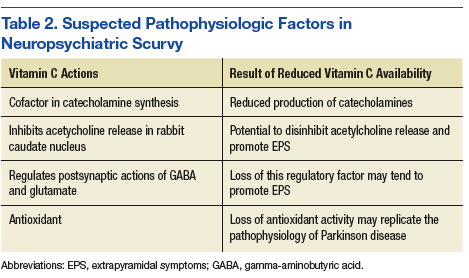
The potential role of alcohol use by both patients also warrants mention. Current data suggest a nonlinear relationship between alcohol use and neurotoxicity. Epidemiologic data show that moderate alcohol consumption protects against the development of such neurodegenerative processes as Parkinson disease and Alzheimer disease.15,16 But the cases here reflect excessive use of alcohol. In this situation, a variety of progressive insults, such as those caused by oxyradical toxicity as well as malnutrition may foster the development of basal ganglia dysfunction.17
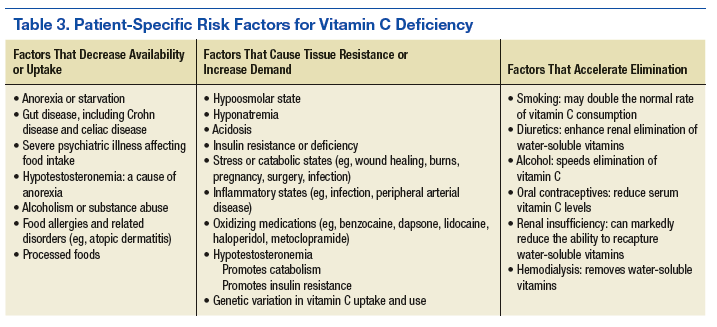
Measuring Deficiency
A deficiency of vitamin C may be determined in several ways. The most frequently used laboratory measure of vitamin C status is the serum vitamin C level. This level is included in the WHO’s recommendations for diagnosis.3 However, this assay is limited because when facing total body depletion, the kidneys may restrict the elimination of vitamin C and tend to maintain serum vitamin C levels even as target tissue levels fall. An interesting example of this is the 0.2 mg/dL value that each patient registered. In Mr. A’s case, this reflected a systemic deficit of vitamin C, while in Mr. B’s case it correlated with the onset of effective repletion of body’s stores.
A fall in urinary output of vitamin C is another marker of hypovitaminosis C. When available, this laboratory test can be used with the serum level to assess total body stores of vitamin C. Lymphocytes, neutrophils, and platelets also store vitamin C. These target tissues tend to saturate when the oral intake ranges between 100 mg to 200 mg a day. This is the same point at which serum vitamin C levels peak and level off in normal, healthy adults.18,19 Once again, the limited availability of target-tissue assays puts these studies out of reach for most clinicians.
No evaluation is complete without some assurance of what the disease is not. Deficiencies of biotin, zinc, folate, and B12 all may affect the function of the basal ganglia.20 The biotin deficiencies literature is particularly robust. Biotin deficiencies affecting basal ganglia function are best known as inherited disorders of metabolism.21 Manganese intoxication also may present as a movement disorder.22
Treatment
Treatment of neuropsychiatric scurvy has relied on IV administration of vitamin C. Although the bioavailability of oral vitamin C among healthy adult volunteers is nearly complete up to about 200 mg a day, a patient with neuropsychiatric scurvy may need substantially more than that amount to accommodate total body deficiencies and increased demands.23 The IV route allows serum vitamin C levels up to 100 times higher than by the oral route.24 Mr. B is, in fact, the first person reported in the literature with neuropsychiatric scurvy to respond to oral vitamin C replacement alone. Once repletion of vitamin C is complete, it is useful to consider a maintenance replacement dose based on a patient’s risk factors and needs.
A healthy adult should ingest about 120 mg of vitamin C daily. Smokers and pregnant women may require more, but this recommendation was intended to address their needs as well.25 Many commercial multivitamins use the old recommended daily allowance of 60 mg, so it may be safest to recommend specifically a vitamin C tablet with at least 120 mg when ordering vitamin C replacement.
Tight control of the serum vitamin C concentration means that little additional vitamin C will be taken up by the gut beyond 200 mg orally a day, which helps minimize any concerns about long-term toxicity. It takes several weeks to deplete vitamin C from the human body when vitamin C is removed from the diet, so a patient with a previously treated deficiency of vitamin C should wait a month before a repeat serum vitamin C level measurement.
The half life of vitamin C is normally ≤ 2 hours. When renal function is intact, vitamin C in excess of immediate need is lost through renal filtration. Toxicity is rare under these conditions.26 When vitamin C toxicity has been reported, it has occurred in the setting of prolonged supplementation, usually when a patient already experienced a renal injury. The main toxicities attributed to vitamin C are oxalate crystal formation with subsequent renal injury and exacerbation of glucose 6-phosphate dehydrogenase deficiency (G6PD).24
Oxalate formation due to vitamin C replacement is uncommon, but patients with preexisting calcium oxalate stones may be at risk for further stone formation when they receive additional vitamin C.27 This is most likely to occur when treatment with parenteral vitamin C is prolonged, which is not typical for patients with neuropsychiatric scurvy who tend to respond rapidly to vitamin C replenishment. Reports of acute hemolytic episodes among patient with G6PD deficiency receiving vitamin C exist, although these cases are rare.28 Furthermore, some authors advocate for the use of ascorbic acid to treat methemoglobinemia associated with G6PD deficiency, when methylene blue is not available.29 It may be reasonable to begin treatment with oral vitamin C for patients with NPS and G6PD deficiency. This is equivalent to a low-dose form of vitamin C replacement and may help avoid the theoretically pro-oxidant effects of larger, IV doses of vitamin C.30
Conclusion
The recent discovery of movement disorders in scurvy has enlarged the picture of vitamin C deficiency. The cases here demonstrate how hypovitaminosis C with central nervous system manifestations may be identified and treated. This relationship fits well within the established basic science and clinical framework for scurvy, and the clinical implications for scurvy remain in many ways unchanged. First, malnutrition must be considered even when a patient’s habitus suggests he is well fed. Also, it is more likely to see scurvy without all of the classic findings than an end-stage case of the disease.31 In the right clinical setting, it is reasonable to think of a vitamin C deficiency before the patient develops bleeding gums and corkscrew hairs. And as is typical of vitamin deficiencies, the treatment of a vitamin C deficiency usually results in swift improvement. Finally, for those who treat movement disorders or who prescribe agents such as antipsychotics that may cause movement disorders, it is important to recognize vitamin C deficiency as another potential explanation for EPS.
1. Ide K, Yamada H, Umegaki K, et al. Lymphocyte vitamin C levels as potential biomarker for progression of Parkinson’s disease. Nutrition. 2015;31(2):406-408.
2. Lind J. The diagnostics, or symptoms. A Treatise on the Scurvy, in Three Parts. 3rd ed. London: S. Crowder, D. Wilson and G. Nicholls, T. Cadell, T. Becket and Co., G. Pearch, and Woodfall; 1772:98-129.
3. World Health Organization. Scurvy and its prevention and control in major emergencies. http://whqlibdoc.who.int/hq/1999/WHO_NHD_99.11.pdf. Published 1999. Accessed July 6, 2017.
4. Sasseville D. Scurvy: curse and cure in New France. JAMA Dermatol. 2015;151(4):431.
5. Shapter T. On the recent occurrence of scurvy in Exeter and the neighbourhood. Prov Med Surg J. 1847;11(11):281-285.
6. Kinsman RA, Hood J. Some behavioral effects of ascorbic acid deficiency. Am J Clin Nutr. 1971;24(4):455-464.
7. DeSantis J. Scurvy and psychiatric symptoms. Perspect Psychiatr Care. 1993;29(1):18-22.
8. Walter JF. Scurvy resulting from a self-imposed diet. West J Med. 1979;130(2):177-179.
9. Brown TM. Neuropsychiatric scurvy. Psychosomatics. 2015;56(1):12-20.
10. Feuerstein TJ, Weinheimer G, Lang G, Ginap T, Rossner R. Inhibition by ascorbic acid of NMDA-evoked acetylcholine release in rabbit caudate nucleus. Naunyn Schmiedebergs Arch Pharmacol. 1993;348(5):549-551.
11. Kim J, Kwon J, Noh G, Lee SS. The effects of elimination diet on nutritional status in subjects with atopic dermatitis. Nutr Res Pract. 2013;7(6):488-494.
12. Langlois M, Duprez D, Delanghe J, De Buyzere M, Clement DL. Serum vitamin C concentration is low in peripheral arterial disease and is associated with inflammation and severity of atherosclerosis. Circulation. 2001;103(14):1863-1868.
13. Nappe TM, Pacelli AM, Katz K. An atypical case of methemoglobinemia due to self-administered benzocaine. Case Rep Emerg Med. 2015;2015:670979.
14. Wright AD, Stevens E, Ali M, Carroll DW, Brown TM. The neuropsychiatry of scurvy. Psychosomatics. 2014;55(2):179-185.
15. Bate C, Williams A. Ethanol protects cultured neurons against amyloid-β and α-synuclein-induced synapse damage. Neuropharmacology. 2011;61(8):1406-1412.
16. Vasanthi HR, Parameswari RP, DeLeiris J, Das DK. Health benefits of wine and alcohol from neuroprotection to heart health. Front Biosci (Elite Ed). 2012;4:1505-1512.
17. Vaglini F, Viaggi C, Piro V, et al. Acetaldehyde and parkinsonism: role of CYP450 2E1. Front Behav Neurosci. 2013;7:71.
18. Levine M, Wang Y, Padayatty SJ, Morrow J. A new recommended dietary allowance of vitamin C for healthy young women. Proc Natl Acad Sci USA. 2001;98(17):9842-9846.
19. Levine M, Padayatty SJ, Espey MG. Vitamin C: a concentration-function approach yields pharmacology and therapeutic discoveries. Adv Nutr. 2011;2(2):78-88.
20. Quiroga MJ, Carroll DW, Brown TM. Ascorbate- and zinc-responsive parkinsonism. Ann Pharmacother. 2014;48(11):1515-1520.
21. Tabarki B, Al-Shafi S, Al-Shahwan S, et al. Biotin-responsive basal ganglia disease revisited: clinical, radiologic, and genetic findings. Neurology. 2013;80(3):261-267.
22. Tuschl K, Mills PB, Clayton PT. Manganese and the brain. Int Rev Neurobiol. 2013;110:277-312.
23. Levine M, Conry-Cantilena C, Wang Y, et al. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci USA. 1996;93(8):3704-3709.
24. Wilson MK, Baguley BC, Wall C, Jameson MB, Findlay MP. Review of high-dose intravenous vitamin C as an anticancer agent. Asia Pac J Clin Oncol. 2014;10(1):22-37.
25. Carr AC, Frei B. Toward a new recommended dietary allowance for vitamin C based on antioxidant and health effects in humans. Am J Clin Nutr. 1999;69(6):1086-1107.
26. Nielsen TK, Højgaard M, Andersen JT, Poulsen HE, Lykkesfeldt J, Mikines KJ. Elimination of ascorbic acid after high-dose infusion in prostate cancer patients: a pharmacokinetic evaluation. Basic Clin Pharmacol Toxicol. 2015;116(4):343-348.
27. Baxmann AC, De O G Mendonça C, Heilberg IP. Effect of vitamin C supplements on urinary oxalate and pH in calcium stone-forming patients. Kidney Int. 2003;63(3):1066-1071.
28. Huang YC, Chang TK, Fu YC, Jan SL. C for colored urine: acute hemolysis induced by high-dose ascorbic acid. Clin Toxicol (Phila). 2014;52(9):984.
29. Rino PB, Scolnik D, Fustiñana A, Mitelpunkt A, Glatstein M. Ascorbic acid for the treatment of methemoglobinemia: the experience of a large tertiary care pediatric hospital. Am J Ther. 2014;21(4):240-243.
30. Du J, Cullen JJ, Buettner GR. Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim Biophys Acta. 2012;1826(2):443-457.
31. Fouron JC, Chicoine L. Le scorbut: aspects particuliers de l’association rachitisme-scorbut. Can Med Assoc J. 1962;86(26):1191-1196.
Vitamin C (ascorbic acid) deficiency is known to affect brain function and is associated with parkinsonism.1 In 1752, James Lind, MD, described emotional and behavioral changes that herald the onset of scurvy and precede hemorrhagic findings.2 The World Health Organization (WHO) today refers to this stage as latent scurvy.3 The 2 case studies that follow present examples of patients with vitamin C deficiencies whose parkinsonism responded robustly to vitamin C replacement. These cases suggest that vitamin C deficiency may be a treatable cause of parkinsonism.
Case 1
Mr. A, a 60-year-old white male, was admitted to the Medicine Service for alcohol detoxification. The patient had a history of alcohol dependence, alcohol withdrawal seizures, tobacco dependence, and hyperlipidemia. He took no medications as an outpatient. On admission Mr. A’s body mass index (BMI) was 27.2. An initial examination revealed a marked resting tremor of the patient’s right hand with cogwheeling, which had not been present in examinations conducted in the previous 3 years. Mr. A had no prior history of a tremor. He had no cerebellar findings and no evidence of asterixis or of tremulousness associated with high-output cardiac states, such as de Musset sign.
Mr. A reported he had experienced the tremor for a month and that it had been worsening. He also was having difficulty using his dominant right hand, for routine daily activities. Mr. A was oriented, and his short-term memory was intact. He was ill-appearing, irritable with psychomotor slowing, and did not wish to rise from his bed. He had no gingival or periungual bleeding and did not bruise easily. He had no corkscrew hairs. The patient was started on no medications known to cause extrapyramidal symptoms (EPS).
In the hospital, the tremor persisted unabated for 2 days. On the third day, Mr. A was started on 1,000 mg vitamin C IV twice daily. He received a total of 2,000 mg IV that day, but the IV fell out, and he refused its replacement. Several hours later, Mr. A stated that he felt much better, got out of bed, and asked to go outside to smoke. The author noted complete resolution of the right hand tremor and cogwheeling 20 hours after starting the vitamin C IV. Mr. A refused a repeat serum vitamin C assay.
Laboratory studies initially revealed that Mr. A had hyponatremia with a serum sodium of 121 mmol/L (normal range: 133 to 145 mmol/L) as well as hypokalemia with a serum potassium of 3.2 mmol/L (normal range: 3.5 to 5.0 mmol/L). He was hypoosmolar, with a serum osmolality of 276 mOsm/kg (normal range: 278 to 305 mOsm/kg). His vitamin C level was low at 0.2 mg/dL (normal range: 0.4 to 2.0 mg/dL). Mr. A also had a serum vitamin C level drawn 2 years prior that showed no symptoms of EPS, and at that time, the reading was 0.7 mg/dL. At admission to Medicine Services, Mr. A had a serum alcohol level of 211 mg/dL. Neuroimaging revealed diffuse cerebral and cerebellar volume loss.
Normal laboratory results included serum levels of vitamin B12, red cell folate, homocysteine, methylmalonic acid, free and total carnitine, alkaline phosphatase, manganese, and zinc. A urine drug screen was negative.
Case 2
Mr. B, a 69-year-old black male, was admitted to the hospital for depression complicated by alcohol dependence. He also had tobacco dependence, type 2 diabetes mellitus, hypertension, and gout. The patient’s BMI at admission was 16.1. Mr. B appeared ill, was worried about his health, and remained recumbent unless asked to move. He reported that his right hand had begun to shake at rest in the month prior to admission. The tremor made it difficult for him to drink. He pointed out stains on his hospital gurney from an attempt to drink orange juice prior to being assessed.
A physical examination revealed a distinct resting tremor with cogwheeling of the right hand; there was no other evidence of EPS, nor was there evidence of cognitive, cerebellar, or skin abnormalities, such as hemorrhages or corkscrew hairs. Asterixis was absent as was evidence of a high-output cardiac state that might produce a tremor, such as de Musset sign. A serum vitamin C level was obtained and returned at 0.0 mg/dL. A head computed tomography scan obtained the next day revealed mild cerebellar volume loss. A serum alkaline phosphatase level was elevated slightly at 136 U/L (normal range: 42 to 113 U/L). Normal serum values were returned for zinc, vitamins B12 and folate, rapid plasma reagin, sodium, and serum osmolality. A urine drug screen was negative, and serum alcohol level was < 5.0 mg/dL.
Mr. B took no medications expected to cause EPS. He received no micronutrient replacement until the day after admission when he began receiving oral vitamin C 1,000 mg twice a day. After receiving 3 doses, Mr. B’s resting tremor and cogwheeling completely resolved. He noticed he had stopped shaking and could now drink without spilling fluids. He also got out of bed and began interacting with others. Mr. B said he felt he was “doing well.” A repeat serum vitamin C level was 0.2 mg/dL on that day. The improvement was sustained over 3 days, and Mr. B was discharged to home.
Discussion
Both Mr. A and Mr. B presented with a typical picture of latent scurvy and the additional finding of parkinsonism. These cases are important for 2 reasons. First, the swift and full response of these patients’ parkinsonism to vitamin C replacement underscores the importance of considering a vitamin C deficiency when confronted with EPS. And second, both patients lacked signs of bleeding or of impaired collagen synthesis. This differs from the classic presentation of scurvy as a disorder primarily of collagen metabolism.4
Lind described the onset of scurvy as one in which striking emotional and behavior changes developed and later were followed by abnormal bleeding and even death.2 These early changes also were recognized by Shapter in 1847.5 Furthermore, the evidence that exists about the time-course of scurvy’s development suggests that neuropsychiatric findings precede the hemorrhagic.6 Indeed, classic skin findings, such as petechiae or corkscrew hairs, may develop years after the onset of neuropsychiatric changes.7,8
Despite WHO characterizing it as latent scurvy, the distinct syndromal presentation of hypovitaminosis C with parkinsonism along with the rapid response to vitamin C replacement argues for its recognition as a distinct clinical entity and not just a prelude to the hemorrhagic state. To assist in recognizing neuropsychiatric scurvy, the author suggests the operationalized approach described in Table 1.9
Pathophysiology
Vitamin C has an intimate role in the normal functioning of the basal ganglia. It is involved in the synthesis of catechecholamines, the regulation of the release and postsynaptic activities of various neurotransmitters, and managing the oxyradical toxicity of aerobic metabolism. Table 2 outlines some of the normal brain functions of vitamin C and the potential consequences of inadequate central vitamin C.9,10 Risk factors for vitamin C deficiency include those affecting the uptake, response to, and elimination of this vitamin (Table 3).11-14
The potential role of alcohol use by both patients also warrants mention. Current data suggest a nonlinear relationship between alcohol use and neurotoxicity. Epidemiologic data show that moderate alcohol consumption protects against the development of such neurodegenerative processes as Parkinson disease and Alzheimer disease.15,16 But the cases here reflect excessive use of alcohol. In this situation, a variety of progressive insults, such as those caused by oxyradical toxicity as well as malnutrition may foster the development of basal ganglia dysfunction.17
Measuring Deficiency
A deficiency of vitamin C may be determined in several ways. The most frequently used laboratory measure of vitamin C status is the serum vitamin C level. This level is included in the WHO’s recommendations for diagnosis.3 However, this assay is limited because when facing total body depletion, the kidneys may restrict the elimination of vitamin C and tend to maintain serum vitamin C levels even as target tissue levels fall. An interesting example of this is the 0.2 mg/dL value that each patient registered. In Mr. A’s case, this reflected a systemic deficit of vitamin C, while in Mr. B’s case it correlated with the onset of effective repletion of body’s stores.
A fall in urinary output of vitamin C is another marker of hypovitaminosis C. When available, this laboratory test can be used with the serum level to assess total body stores of vitamin C. Lymphocytes, neutrophils, and platelets also store vitamin C. These target tissues tend to saturate when the oral intake ranges between 100 mg to 200 mg a day. This is the same point at which serum vitamin C levels peak and level off in normal, healthy adults.18,19 Once again, the limited availability of target-tissue assays puts these studies out of reach for most clinicians.
No evaluation is complete without some assurance of what the disease is not. Deficiencies of biotin, zinc, folate, and B12 all may affect the function of the basal ganglia.20 The biotin deficiencies literature is particularly robust. Biotin deficiencies affecting basal ganglia function are best known as inherited disorders of metabolism.21 Manganese intoxication also may present as a movement disorder.22
Treatment
Treatment of neuropsychiatric scurvy has relied on IV administration of vitamin C. Although the bioavailability of oral vitamin C among healthy adult volunteers is nearly complete up to about 200 mg a day, a patient with neuropsychiatric scurvy may need substantially more than that amount to accommodate total body deficiencies and increased demands.23 The IV route allows serum vitamin C levels up to 100 times higher than by the oral route.24 Mr. B is, in fact, the first person reported in the literature with neuropsychiatric scurvy to respond to oral vitamin C replacement alone. Once repletion of vitamin C is complete, it is useful to consider a maintenance replacement dose based on a patient’s risk factors and needs.
A healthy adult should ingest about 120 mg of vitamin C daily. Smokers and pregnant women may require more, but this recommendation was intended to address their needs as well.25 Many commercial multivitamins use the old recommended daily allowance of 60 mg, so it may be safest to recommend specifically a vitamin C tablet with at least 120 mg when ordering vitamin C replacement.
Tight control of the serum vitamin C concentration means that little additional vitamin C will be taken up by the gut beyond 200 mg orally a day, which helps minimize any concerns about long-term toxicity. It takes several weeks to deplete vitamin C from the human body when vitamin C is removed from the diet, so a patient with a previously treated deficiency of vitamin C should wait a month before a repeat serum vitamin C level measurement.
The half life of vitamin C is normally ≤ 2 hours. When renal function is intact, vitamin C in excess of immediate need is lost through renal filtration. Toxicity is rare under these conditions.26 When vitamin C toxicity has been reported, it has occurred in the setting of prolonged supplementation, usually when a patient already experienced a renal injury. The main toxicities attributed to vitamin C are oxalate crystal formation with subsequent renal injury and exacerbation of glucose 6-phosphate dehydrogenase deficiency (G6PD).24
Oxalate formation due to vitamin C replacement is uncommon, but patients with preexisting calcium oxalate stones may be at risk for further stone formation when they receive additional vitamin C.27 This is most likely to occur when treatment with parenteral vitamin C is prolonged, which is not typical for patients with neuropsychiatric scurvy who tend to respond rapidly to vitamin C replenishment. Reports of acute hemolytic episodes among patient with G6PD deficiency receiving vitamin C exist, although these cases are rare.28 Furthermore, some authors advocate for the use of ascorbic acid to treat methemoglobinemia associated with G6PD deficiency, when methylene blue is not available.29 It may be reasonable to begin treatment with oral vitamin C for patients with NPS and G6PD deficiency. This is equivalent to a low-dose form of vitamin C replacement and may help avoid the theoretically pro-oxidant effects of larger, IV doses of vitamin C.30
Conclusion
The recent discovery of movement disorders in scurvy has enlarged the picture of vitamin C deficiency. The cases here demonstrate how hypovitaminosis C with central nervous system manifestations may be identified and treated. This relationship fits well within the established basic science and clinical framework for scurvy, and the clinical implications for scurvy remain in many ways unchanged. First, malnutrition must be considered even when a patient’s habitus suggests he is well fed. Also, it is more likely to see scurvy without all of the classic findings than an end-stage case of the disease.31 In the right clinical setting, it is reasonable to think of a vitamin C deficiency before the patient develops bleeding gums and corkscrew hairs. And as is typical of vitamin deficiencies, the treatment of a vitamin C deficiency usually results in swift improvement. Finally, for those who treat movement disorders or who prescribe agents such as antipsychotics that may cause movement disorders, it is important to recognize vitamin C deficiency as another potential explanation for EPS.
Vitamin C (ascorbic acid) deficiency is known to affect brain function and is associated with parkinsonism.1 In 1752, James Lind, MD, described emotional and behavioral changes that herald the onset of scurvy and precede hemorrhagic findings.2 The World Health Organization (WHO) today refers to this stage as latent scurvy.3 The 2 case studies that follow present examples of patients with vitamin C deficiencies whose parkinsonism responded robustly to vitamin C replacement. These cases suggest that vitamin C deficiency may be a treatable cause of parkinsonism.
Case 1
Mr. A, a 60-year-old white male, was admitted to the Medicine Service for alcohol detoxification. The patient had a history of alcohol dependence, alcohol withdrawal seizures, tobacco dependence, and hyperlipidemia. He took no medications as an outpatient. On admission Mr. A’s body mass index (BMI) was 27.2. An initial examination revealed a marked resting tremor of the patient’s right hand with cogwheeling, which had not been present in examinations conducted in the previous 3 years. Mr. A had no prior history of a tremor. He had no cerebellar findings and no evidence of asterixis or of tremulousness associated with high-output cardiac states, such as de Musset sign.
Mr. A reported he had experienced the tremor for a month and that it had been worsening. He also was having difficulty using his dominant right hand, for routine daily activities. Mr. A was oriented, and his short-term memory was intact. He was ill-appearing, irritable with psychomotor slowing, and did not wish to rise from his bed. He had no gingival or periungual bleeding and did not bruise easily. He had no corkscrew hairs. The patient was started on no medications known to cause extrapyramidal symptoms (EPS).
In the hospital, the tremor persisted unabated for 2 days. On the third day, Mr. A was started on 1,000 mg vitamin C IV twice daily. He received a total of 2,000 mg IV that day, but the IV fell out, and he refused its replacement. Several hours later, Mr. A stated that he felt much better, got out of bed, and asked to go outside to smoke. The author noted complete resolution of the right hand tremor and cogwheeling 20 hours after starting the vitamin C IV. Mr. A refused a repeat serum vitamin C assay.
Laboratory studies initially revealed that Mr. A had hyponatremia with a serum sodium of 121 mmol/L (normal range: 133 to 145 mmol/L) as well as hypokalemia with a serum potassium of 3.2 mmol/L (normal range: 3.5 to 5.0 mmol/L). He was hypoosmolar, with a serum osmolality of 276 mOsm/kg (normal range: 278 to 305 mOsm/kg). His vitamin C level was low at 0.2 mg/dL (normal range: 0.4 to 2.0 mg/dL). Mr. A also had a serum vitamin C level drawn 2 years prior that showed no symptoms of EPS, and at that time, the reading was 0.7 mg/dL. At admission to Medicine Services, Mr. A had a serum alcohol level of 211 mg/dL. Neuroimaging revealed diffuse cerebral and cerebellar volume loss.
Normal laboratory results included serum levels of vitamin B12, red cell folate, homocysteine, methylmalonic acid, free and total carnitine, alkaline phosphatase, manganese, and zinc. A urine drug screen was negative.
Case 2
Mr. B, a 69-year-old black male, was admitted to the hospital for depression complicated by alcohol dependence. He also had tobacco dependence, type 2 diabetes mellitus, hypertension, and gout. The patient’s BMI at admission was 16.1. Mr. B appeared ill, was worried about his health, and remained recumbent unless asked to move. He reported that his right hand had begun to shake at rest in the month prior to admission. The tremor made it difficult for him to drink. He pointed out stains on his hospital gurney from an attempt to drink orange juice prior to being assessed.
A physical examination revealed a distinct resting tremor with cogwheeling of the right hand; there was no other evidence of EPS, nor was there evidence of cognitive, cerebellar, or skin abnormalities, such as hemorrhages or corkscrew hairs. Asterixis was absent as was evidence of a high-output cardiac state that might produce a tremor, such as de Musset sign. A serum vitamin C level was obtained and returned at 0.0 mg/dL. A head computed tomography scan obtained the next day revealed mild cerebellar volume loss. A serum alkaline phosphatase level was elevated slightly at 136 U/L (normal range: 42 to 113 U/L). Normal serum values were returned for zinc, vitamins B12 and folate, rapid plasma reagin, sodium, and serum osmolality. A urine drug screen was negative, and serum alcohol level was < 5.0 mg/dL.
Mr. B took no medications expected to cause EPS. He received no micronutrient replacement until the day after admission when he began receiving oral vitamin C 1,000 mg twice a day. After receiving 3 doses, Mr. B’s resting tremor and cogwheeling completely resolved. He noticed he had stopped shaking and could now drink without spilling fluids. He also got out of bed and began interacting with others. Mr. B said he felt he was “doing well.” A repeat serum vitamin C level was 0.2 mg/dL on that day. The improvement was sustained over 3 days, and Mr. B was discharged to home.
Discussion
Both Mr. A and Mr. B presented with a typical picture of latent scurvy and the additional finding of parkinsonism. These cases are important for 2 reasons. First, the swift and full response of these patients’ parkinsonism to vitamin C replacement underscores the importance of considering a vitamin C deficiency when confronted with EPS. And second, both patients lacked signs of bleeding or of impaired collagen synthesis. This differs from the classic presentation of scurvy as a disorder primarily of collagen metabolism.4
Lind described the onset of scurvy as one in which striking emotional and behavior changes developed and later were followed by abnormal bleeding and even death.2 These early changes also were recognized by Shapter in 1847.5 Furthermore, the evidence that exists about the time-course of scurvy’s development suggests that neuropsychiatric findings precede the hemorrhagic.6 Indeed, classic skin findings, such as petechiae or corkscrew hairs, may develop years after the onset of neuropsychiatric changes.7,8
Despite WHO characterizing it as latent scurvy, the distinct syndromal presentation of hypovitaminosis C with parkinsonism along with the rapid response to vitamin C replacement argues for its recognition as a distinct clinical entity and not just a prelude to the hemorrhagic state. To assist in recognizing neuropsychiatric scurvy, the author suggests the operationalized approach described in Table 1.9
Pathophysiology
Vitamin C has an intimate role in the normal functioning of the basal ganglia. It is involved in the synthesis of catechecholamines, the regulation of the release and postsynaptic activities of various neurotransmitters, and managing the oxyradical toxicity of aerobic metabolism. Table 2 outlines some of the normal brain functions of vitamin C and the potential consequences of inadequate central vitamin C.9,10 Risk factors for vitamin C deficiency include those affecting the uptake, response to, and elimination of this vitamin (Table 3).11-14
The potential role of alcohol use by both patients also warrants mention. Current data suggest a nonlinear relationship between alcohol use and neurotoxicity. Epidemiologic data show that moderate alcohol consumption protects against the development of such neurodegenerative processes as Parkinson disease and Alzheimer disease.15,16 But the cases here reflect excessive use of alcohol. In this situation, a variety of progressive insults, such as those caused by oxyradical toxicity as well as malnutrition may foster the development of basal ganglia dysfunction.17
Measuring Deficiency
A deficiency of vitamin C may be determined in several ways. The most frequently used laboratory measure of vitamin C status is the serum vitamin C level. This level is included in the WHO’s recommendations for diagnosis.3 However, this assay is limited because when facing total body depletion, the kidneys may restrict the elimination of vitamin C and tend to maintain serum vitamin C levels even as target tissue levels fall. An interesting example of this is the 0.2 mg/dL value that each patient registered. In Mr. A’s case, this reflected a systemic deficit of vitamin C, while in Mr. B’s case it correlated with the onset of effective repletion of body’s stores.
A fall in urinary output of vitamin C is another marker of hypovitaminosis C. When available, this laboratory test can be used with the serum level to assess total body stores of vitamin C. Lymphocytes, neutrophils, and platelets also store vitamin C. These target tissues tend to saturate when the oral intake ranges between 100 mg to 200 mg a day. This is the same point at which serum vitamin C levels peak and level off in normal, healthy adults.18,19 Once again, the limited availability of target-tissue assays puts these studies out of reach for most clinicians.
No evaluation is complete without some assurance of what the disease is not. Deficiencies of biotin, zinc, folate, and B12 all may affect the function of the basal ganglia.20 The biotin deficiencies literature is particularly robust. Biotin deficiencies affecting basal ganglia function are best known as inherited disorders of metabolism.21 Manganese intoxication also may present as a movement disorder.22
Treatment
Treatment of neuropsychiatric scurvy has relied on IV administration of vitamin C. Although the bioavailability of oral vitamin C among healthy adult volunteers is nearly complete up to about 200 mg a day, a patient with neuropsychiatric scurvy may need substantially more than that amount to accommodate total body deficiencies and increased demands.23 The IV route allows serum vitamin C levels up to 100 times higher than by the oral route.24 Mr. B is, in fact, the first person reported in the literature with neuropsychiatric scurvy to respond to oral vitamin C replacement alone. Once repletion of vitamin C is complete, it is useful to consider a maintenance replacement dose based on a patient’s risk factors and needs.
A healthy adult should ingest about 120 mg of vitamin C daily. Smokers and pregnant women may require more, but this recommendation was intended to address their needs as well.25 Many commercial multivitamins use the old recommended daily allowance of 60 mg, so it may be safest to recommend specifically a vitamin C tablet with at least 120 mg when ordering vitamin C replacement.
Tight control of the serum vitamin C concentration means that little additional vitamin C will be taken up by the gut beyond 200 mg orally a day, which helps minimize any concerns about long-term toxicity. It takes several weeks to deplete vitamin C from the human body when vitamin C is removed from the diet, so a patient with a previously treated deficiency of vitamin C should wait a month before a repeat serum vitamin C level measurement.
The half life of vitamin C is normally ≤ 2 hours. When renal function is intact, vitamin C in excess of immediate need is lost through renal filtration. Toxicity is rare under these conditions.26 When vitamin C toxicity has been reported, it has occurred in the setting of prolonged supplementation, usually when a patient already experienced a renal injury. The main toxicities attributed to vitamin C are oxalate crystal formation with subsequent renal injury and exacerbation of glucose 6-phosphate dehydrogenase deficiency (G6PD).24
Oxalate formation due to vitamin C replacement is uncommon, but patients with preexisting calcium oxalate stones may be at risk for further stone formation when they receive additional vitamin C.27 This is most likely to occur when treatment with parenteral vitamin C is prolonged, which is not typical for patients with neuropsychiatric scurvy who tend to respond rapidly to vitamin C replenishment. Reports of acute hemolytic episodes among patient with G6PD deficiency receiving vitamin C exist, although these cases are rare.28 Furthermore, some authors advocate for the use of ascorbic acid to treat methemoglobinemia associated with G6PD deficiency, when methylene blue is not available.29 It may be reasonable to begin treatment with oral vitamin C for patients with NPS and G6PD deficiency. This is equivalent to a low-dose form of vitamin C replacement and may help avoid the theoretically pro-oxidant effects of larger, IV doses of vitamin C.30
Conclusion
The recent discovery of movement disorders in scurvy has enlarged the picture of vitamin C deficiency. The cases here demonstrate how hypovitaminosis C with central nervous system manifestations may be identified and treated. This relationship fits well within the established basic science and clinical framework for scurvy, and the clinical implications for scurvy remain in many ways unchanged. First, malnutrition must be considered even when a patient’s habitus suggests he is well fed. Also, it is more likely to see scurvy without all of the classic findings than an end-stage case of the disease.31 In the right clinical setting, it is reasonable to think of a vitamin C deficiency before the patient develops bleeding gums and corkscrew hairs. And as is typical of vitamin deficiencies, the treatment of a vitamin C deficiency usually results in swift improvement. Finally, for those who treat movement disorders or who prescribe agents such as antipsychotics that may cause movement disorders, it is important to recognize vitamin C deficiency as another potential explanation for EPS.
1. Ide K, Yamada H, Umegaki K, et al. Lymphocyte vitamin C levels as potential biomarker for progression of Parkinson’s disease. Nutrition. 2015;31(2):406-408.
2. Lind J. The diagnostics, or symptoms. A Treatise on the Scurvy, in Three Parts. 3rd ed. London: S. Crowder, D. Wilson and G. Nicholls, T. Cadell, T. Becket and Co., G. Pearch, and Woodfall; 1772:98-129.
3. World Health Organization. Scurvy and its prevention and control in major emergencies. http://whqlibdoc.who.int/hq/1999/WHO_NHD_99.11.pdf. Published 1999. Accessed July 6, 2017.
4. Sasseville D. Scurvy: curse and cure in New France. JAMA Dermatol. 2015;151(4):431.
5. Shapter T. On the recent occurrence of scurvy in Exeter and the neighbourhood. Prov Med Surg J. 1847;11(11):281-285.
6. Kinsman RA, Hood J. Some behavioral effects of ascorbic acid deficiency. Am J Clin Nutr. 1971;24(4):455-464.
7. DeSantis J. Scurvy and psychiatric symptoms. Perspect Psychiatr Care. 1993;29(1):18-22.
8. Walter JF. Scurvy resulting from a self-imposed diet. West J Med. 1979;130(2):177-179.
9. Brown TM. Neuropsychiatric scurvy. Psychosomatics. 2015;56(1):12-20.
10. Feuerstein TJ, Weinheimer G, Lang G, Ginap T, Rossner R. Inhibition by ascorbic acid of NMDA-evoked acetylcholine release in rabbit caudate nucleus. Naunyn Schmiedebergs Arch Pharmacol. 1993;348(5):549-551.
11. Kim J, Kwon J, Noh G, Lee SS. The effects of elimination diet on nutritional status in subjects with atopic dermatitis. Nutr Res Pract. 2013;7(6):488-494.
12. Langlois M, Duprez D, Delanghe J, De Buyzere M, Clement DL. Serum vitamin C concentration is low in peripheral arterial disease and is associated with inflammation and severity of atherosclerosis. Circulation. 2001;103(14):1863-1868.
13. Nappe TM, Pacelli AM, Katz K. An atypical case of methemoglobinemia due to self-administered benzocaine. Case Rep Emerg Med. 2015;2015:670979.
14. Wright AD, Stevens E, Ali M, Carroll DW, Brown TM. The neuropsychiatry of scurvy. Psychosomatics. 2014;55(2):179-185.
15. Bate C, Williams A. Ethanol protects cultured neurons against amyloid-β and α-synuclein-induced synapse damage. Neuropharmacology. 2011;61(8):1406-1412.
16. Vasanthi HR, Parameswari RP, DeLeiris J, Das DK. Health benefits of wine and alcohol from neuroprotection to heart health. Front Biosci (Elite Ed). 2012;4:1505-1512.
17. Vaglini F, Viaggi C, Piro V, et al. Acetaldehyde and parkinsonism: role of CYP450 2E1. Front Behav Neurosci. 2013;7:71.
18. Levine M, Wang Y, Padayatty SJ, Morrow J. A new recommended dietary allowance of vitamin C for healthy young women. Proc Natl Acad Sci USA. 2001;98(17):9842-9846.
19. Levine M, Padayatty SJ, Espey MG. Vitamin C: a concentration-function approach yields pharmacology and therapeutic discoveries. Adv Nutr. 2011;2(2):78-88.
20. Quiroga MJ, Carroll DW, Brown TM. Ascorbate- and zinc-responsive parkinsonism. Ann Pharmacother. 2014;48(11):1515-1520.
21. Tabarki B, Al-Shafi S, Al-Shahwan S, et al. Biotin-responsive basal ganglia disease revisited: clinical, radiologic, and genetic findings. Neurology. 2013;80(3):261-267.
22. Tuschl K, Mills PB, Clayton PT. Manganese and the brain. Int Rev Neurobiol. 2013;110:277-312.
23. Levine M, Conry-Cantilena C, Wang Y, et al. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci USA. 1996;93(8):3704-3709.
24. Wilson MK, Baguley BC, Wall C, Jameson MB, Findlay MP. Review of high-dose intravenous vitamin C as an anticancer agent. Asia Pac J Clin Oncol. 2014;10(1):22-37.
25. Carr AC, Frei B. Toward a new recommended dietary allowance for vitamin C based on antioxidant and health effects in humans. Am J Clin Nutr. 1999;69(6):1086-1107.
26. Nielsen TK, Højgaard M, Andersen JT, Poulsen HE, Lykkesfeldt J, Mikines KJ. Elimination of ascorbic acid after high-dose infusion in prostate cancer patients: a pharmacokinetic evaluation. Basic Clin Pharmacol Toxicol. 2015;116(4):343-348.
27. Baxmann AC, De O G Mendonça C, Heilberg IP. Effect of vitamin C supplements on urinary oxalate and pH in calcium stone-forming patients. Kidney Int. 2003;63(3):1066-1071.
28. Huang YC, Chang TK, Fu YC, Jan SL. C for colored urine: acute hemolysis induced by high-dose ascorbic acid. Clin Toxicol (Phila). 2014;52(9):984.
29. Rino PB, Scolnik D, Fustiñana A, Mitelpunkt A, Glatstein M. Ascorbic acid for the treatment of methemoglobinemia: the experience of a large tertiary care pediatric hospital. Am J Ther. 2014;21(4):240-243.
30. Du J, Cullen JJ, Buettner GR. Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim Biophys Acta. 2012;1826(2):443-457.
31. Fouron JC, Chicoine L. Le scorbut: aspects particuliers de l’association rachitisme-scorbut. Can Med Assoc J. 1962;86(26):1191-1196.
1. Ide K, Yamada H, Umegaki K, et al. Lymphocyte vitamin C levels as potential biomarker for progression of Parkinson’s disease. Nutrition. 2015;31(2):406-408.
2. Lind J. The diagnostics, or symptoms. A Treatise on the Scurvy, in Three Parts. 3rd ed. London: S. Crowder, D. Wilson and G. Nicholls, T. Cadell, T. Becket and Co., G. Pearch, and Woodfall; 1772:98-129.
3. World Health Organization. Scurvy and its prevention and control in major emergencies. http://whqlibdoc.who.int/hq/1999/WHO_NHD_99.11.pdf. Published 1999. Accessed July 6, 2017.
4. Sasseville D. Scurvy: curse and cure in New France. JAMA Dermatol. 2015;151(4):431.
5. Shapter T. On the recent occurrence of scurvy in Exeter and the neighbourhood. Prov Med Surg J. 1847;11(11):281-285.
6. Kinsman RA, Hood J. Some behavioral effects of ascorbic acid deficiency. Am J Clin Nutr. 1971;24(4):455-464.
7. DeSantis J. Scurvy and psychiatric symptoms. Perspect Psychiatr Care. 1993;29(1):18-22.
8. Walter JF. Scurvy resulting from a self-imposed diet. West J Med. 1979;130(2):177-179.
9. Brown TM. Neuropsychiatric scurvy. Psychosomatics. 2015;56(1):12-20.
10. Feuerstein TJ, Weinheimer G, Lang G, Ginap T, Rossner R. Inhibition by ascorbic acid of NMDA-evoked acetylcholine release in rabbit caudate nucleus. Naunyn Schmiedebergs Arch Pharmacol. 1993;348(5):549-551.
11. Kim J, Kwon J, Noh G, Lee SS. The effects of elimination diet on nutritional status in subjects with atopic dermatitis. Nutr Res Pract. 2013;7(6):488-494.
12. Langlois M, Duprez D, Delanghe J, De Buyzere M, Clement DL. Serum vitamin C concentration is low in peripheral arterial disease and is associated with inflammation and severity of atherosclerosis. Circulation. 2001;103(14):1863-1868.
13. Nappe TM, Pacelli AM, Katz K. An atypical case of methemoglobinemia due to self-administered benzocaine. Case Rep Emerg Med. 2015;2015:670979.
14. Wright AD, Stevens E, Ali M, Carroll DW, Brown TM. The neuropsychiatry of scurvy. Psychosomatics. 2014;55(2):179-185.
15. Bate C, Williams A. Ethanol protects cultured neurons against amyloid-β and α-synuclein-induced synapse damage. Neuropharmacology. 2011;61(8):1406-1412.
16. Vasanthi HR, Parameswari RP, DeLeiris J, Das DK. Health benefits of wine and alcohol from neuroprotection to heart health. Front Biosci (Elite Ed). 2012;4:1505-1512.
17. Vaglini F, Viaggi C, Piro V, et al. Acetaldehyde and parkinsonism: role of CYP450 2E1. Front Behav Neurosci. 2013;7:71.
18. Levine M, Wang Y, Padayatty SJ, Morrow J. A new recommended dietary allowance of vitamin C for healthy young women. Proc Natl Acad Sci USA. 2001;98(17):9842-9846.
19. Levine M, Padayatty SJ, Espey MG. Vitamin C: a concentration-function approach yields pharmacology and therapeutic discoveries. Adv Nutr. 2011;2(2):78-88.
20. Quiroga MJ, Carroll DW, Brown TM. Ascorbate- and zinc-responsive parkinsonism. Ann Pharmacother. 2014;48(11):1515-1520.
21. Tabarki B, Al-Shafi S, Al-Shahwan S, et al. Biotin-responsive basal ganglia disease revisited: clinical, radiologic, and genetic findings. Neurology. 2013;80(3):261-267.
22. Tuschl K, Mills PB, Clayton PT. Manganese and the brain. Int Rev Neurobiol. 2013;110:277-312.
23. Levine M, Conry-Cantilena C, Wang Y, et al. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci USA. 1996;93(8):3704-3709.
24. Wilson MK, Baguley BC, Wall C, Jameson MB, Findlay MP. Review of high-dose intravenous vitamin C as an anticancer agent. Asia Pac J Clin Oncol. 2014;10(1):22-37.
25. Carr AC, Frei B. Toward a new recommended dietary allowance for vitamin C based on antioxidant and health effects in humans. Am J Clin Nutr. 1999;69(6):1086-1107.
26. Nielsen TK, Højgaard M, Andersen JT, Poulsen HE, Lykkesfeldt J, Mikines KJ. Elimination of ascorbic acid after high-dose infusion in prostate cancer patients: a pharmacokinetic evaluation. Basic Clin Pharmacol Toxicol. 2015;116(4):343-348.
27. Baxmann AC, De O G Mendonça C, Heilberg IP. Effect of vitamin C supplements on urinary oxalate and pH in calcium stone-forming patients. Kidney Int. 2003;63(3):1066-1071.
28. Huang YC, Chang TK, Fu YC, Jan SL. C for colored urine: acute hemolysis induced by high-dose ascorbic acid. Clin Toxicol (Phila). 2014;52(9):984.
29. Rino PB, Scolnik D, Fustiñana A, Mitelpunkt A, Glatstein M. Ascorbic acid for the treatment of methemoglobinemia: the experience of a large tertiary care pediatric hospital. Am J Ther. 2014;21(4):240-243.
30. Du J, Cullen JJ, Buettner GR. Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim Biophys Acta. 2012;1826(2):443-457.
31. Fouron JC, Chicoine L. Le scorbut: aspects particuliers de l’association rachitisme-scorbut. Can Med Assoc J. 1962;86(26):1191-1196.
Testing the Limits of Dual Antiplatelet Treatment for PCI Patients
What’s the right duration of dual antiplatelet therapy (DAPT) for patients who have had drug-eluting stents implanted? According to researchers from Sungkyunkwan University in Seoul, Korea, long-term clinical outcomes are similar whether patients receive therapy for less than or longer than 12 months.
The researchers conducted a retrospective study of 512 patients who had undergone percutaneous coronary intervention (PCI) for coronary chronic total occlusion (CTO) and were event free at 12 months. They separated the patients into 2 groups: 199 received aspirin and clopidogrel for ≤ 12 months, and 313 for > 12 months. The primary outcome was major adverse cardiac and cerebrovascular event (MACCE) during follow-up. Median follow-up was 64 months.
Related: Statins for the Physically Fit: Do They Help or Hurt?
No significant differences were seen between the groups in the incidence of MACCE: 21.6% of the patients in the ≤ 12-month group and 17.6% of the patients in the > 12-month group developed MACCE. After propensity matching, moderate or severe bleeding rates were also similar (1.6% in the shorter duration group and 2.2% in the longer duration group).
The researchers note that previously published data showed that longer duration DAPT was not associated with improved clinical outcomes in patients with CTO, although other subsets of complex PCI, such as longer stent length, bifurcation stenting, or multiple stents showed better clinical outcomes. To the best of their knowledge, the researchers add, theirs is the first study to directly compare DAPT durations in patients with CTO-PCI.
Related: A Heart Failure Management Program Using Shared Medical Appointments
Source:
Lee SH, Yang JH, Choi SH, et al. PLoS One. 2017;12(5):e0176737.
doi: 10.1371/journal.pone.0176737.
What’s the right duration of dual antiplatelet therapy (DAPT) for patients who have had drug-eluting stents implanted? According to researchers from Sungkyunkwan University in Seoul, Korea, long-term clinical outcomes are similar whether patients receive therapy for less than or longer than 12 months.
The researchers conducted a retrospective study of 512 patients who had undergone percutaneous coronary intervention (PCI) for coronary chronic total occlusion (CTO) and were event free at 12 months. They separated the patients into 2 groups: 199 received aspirin and clopidogrel for ≤ 12 months, and 313 for > 12 months. The primary outcome was major adverse cardiac and cerebrovascular event (MACCE) during follow-up. Median follow-up was 64 months.
Related: Statins for the Physically Fit: Do They Help or Hurt?
No significant differences were seen between the groups in the incidence of MACCE: 21.6% of the patients in the ≤ 12-month group and 17.6% of the patients in the > 12-month group developed MACCE. After propensity matching, moderate or severe bleeding rates were also similar (1.6% in the shorter duration group and 2.2% in the longer duration group).
The researchers note that previously published data showed that longer duration DAPT was not associated with improved clinical outcomes in patients with CTO, although other subsets of complex PCI, such as longer stent length, bifurcation stenting, or multiple stents showed better clinical outcomes. To the best of their knowledge, the researchers add, theirs is the first study to directly compare DAPT durations in patients with CTO-PCI.
Related: A Heart Failure Management Program Using Shared Medical Appointments
Source:
Lee SH, Yang JH, Choi SH, et al. PLoS One. 2017;12(5):e0176737.
doi: 10.1371/journal.pone.0176737.
What’s the right duration of dual antiplatelet therapy (DAPT) for patients who have had drug-eluting stents implanted? According to researchers from Sungkyunkwan University in Seoul, Korea, long-term clinical outcomes are similar whether patients receive therapy for less than or longer than 12 months.
The researchers conducted a retrospective study of 512 patients who had undergone percutaneous coronary intervention (PCI) for coronary chronic total occlusion (CTO) and were event free at 12 months. They separated the patients into 2 groups: 199 received aspirin and clopidogrel for ≤ 12 months, and 313 for > 12 months. The primary outcome was major adverse cardiac and cerebrovascular event (MACCE) during follow-up. Median follow-up was 64 months.
Related: Statins for the Physically Fit: Do They Help or Hurt?
No significant differences were seen between the groups in the incidence of MACCE: 21.6% of the patients in the ≤ 12-month group and 17.6% of the patients in the > 12-month group developed MACCE. After propensity matching, moderate or severe bleeding rates were also similar (1.6% in the shorter duration group and 2.2% in the longer duration group).
The researchers note that previously published data showed that longer duration DAPT was not associated with improved clinical outcomes in patients with CTO, although other subsets of complex PCI, such as longer stent length, bifurcation stenting, or multiple stents showed better clinical outcomes. To the best of their knowledge, the researchers add, theirs is the first study to directly compare DAPT durations in patients with CTO-PCI.
Related: A Heart Failure Management Program Using Shared Medical Appointments
Source:
Lee SH, Yang JH, Choi SH, et al. PLoS One. 2017;12(5):e0176737.
doi: 10.1371/journal.pone.0176737.
Sleep Apnea on the Rise Among Veterans
Male veterans who reported mild-to-moderate psychological distress in the previous year had 61% higher odds of experiencing sleep apnea, according to a California State University study. Those with serious psychological distress had 138% higher odds. The average prevalence of sleep apnea was 5.9%, but the proportions rose from 3.7% in 2005 to 8.1% in 2014.
The researchers analyzed data from the 2005-2014 National Survey on Drug Use and Health. They cite other research that found the age-adjusted prevalence of sleep apnea among U.S. veterans increased almost 6-fold from 2000 to 2010. They also point to an evaluation of veterans of Operation Enduring Freedom, Operation Iraqi Freedom, and Operation New Dawn that found that 69.2% of 159 veterans screened were at high risk for obstructive sleep apnea.
An even stronger risk factor was asthma. Veterans with a past-year diagnosis of asthma had 256% higher odds of experiencing sleep apnea than among those without asthma. The researchers note that men and women may be asymptomatic when they are recruited but develop asthma due to environmental factors during deployment. The researchers urge more and better screening regardless of whether the service member had asthma symptoms during recruitment.
Their study is unique, the researchers say, in that it demonstrates a putative relationship between sleep apnea and mental illness. They suggest multidisciplinary interventions, including peer-support strategies to improve veterans’ mental health and community-based resources to help improve access to health care. Above all, the researchers urge more rigorous screening of sleep apnea and better sleep apnea treatment for veterans.
Male veterans who reported mild-to-moderate psychological distress in the previous year had 61% higher odds of experiencing sleep apnea, according to a California State University study. Those with serious psychological distress had 138% higher odds. The average prevalence of sleep apnea was 5.9%, but the proportions rose from 3.7% in 2005 to 8.1% in 2014.
The researchers analyzed data from the 2005-2014 National Survey on Drug Use and Health. They cite other research that found the age-adjusted prevalence of sleep apnea among U.S. veterans increased almost 6-fold from 2000 to 2010. They also point to an evaluation of veterans of Operation Enduring Freedom, Operation Iraqi Freedom, and Operation New Dawn that found that 69.2% of 159 veterans screened were at high risk for obstructive sleep apnea.
An even stronger risk factor was asthma. Veterans with a past-year diagnosis of asthma had 256% higher odds of experiencing sleep apnea than among those without asthma. The researchers note that men and women may be asymptomatic when they are recruited but develop asthma due to environmental factors during deployment. The researchers urge more and better screening regardless of whether the service member had asthma symptoms during recruitment.
Their study is unique, the researchers say, in that it demonstrates a putative relationship between sleep apnea and mental illness. They suggest multidisciplinary interventions, including peer-support strategies to improve veterans’ mental health and community-based resources to help improve access to health care. Above all, the researchers urge more rigorous screening of sleep apnea and better sleep apnea treatment for veterans.
Male veterans who reported mild-to-moderate psychological distress in the previous year had 61% higher odds of experiencing sleep apnea, according to a California State University study. Those with serious psychological distress had 138% higher odds. The average prevalence of sleep apnea was 5.9%, but the proportions rose from 3.7% in 2005 to 8.1% in 2014.
The researchers analyzed data from the 2005-2014 National Survey on Drug Use and Health. They cite other research that found the age-adjusted prevalence of sleep apnea among U.S. veterans increased almost 6-fold from 2000 to 2010. They also point to an evaluation of veterans of Operation Enduring Freedom, Operation Iraqi Freedom, and Operation New Dawn that found that 69.2% of 159 veterans screened were at high risk for obstructive sleep apnea.
An even stronger risk factor was asthma. Veterans with a past-year diagnosis of asthma had 256% higher odds of experiencing sleep apnea than among those without asthma. The researchers note that men and women may be asymptomatic when they are recruited but develop asthma due to environmental factors during deployment. The researchers urge more and better screening regardless of whether the service member had asthma symptoms during recruitment.
Their study is unique, the researchers say, in that it demonstrates a putative relationship between sleep apnea and mental illness. They suggest multidisciplinary interventions, including peer-support strategies to improve veterans’ mental health and community-based resources to help improve access to health care. Above all, the researchers urge more rigorous screening of sleep apnea and better sleep apnea treatment for veterans.
Glucose metabolism deemed key to platelet survival
New research published in Cell Reports suggests platelets are highly reliant on their ability to metabolize glucose.
Researchers studied mice lacking proteins that platelets use to import glucose—glucose transporter (GLUT) 1 and GLUT3.
These mice had lower platelet counts and their platelets had shorter life spans than platelets in wild-type mice.
“We found that glucose metabolism is very critical across the entire life cycle of platelets—from production, to what they do in the body, to how they get cleared from the body,” said E. Dale Abel, MBBS, PhD, of the University of Iowa in Iowa City.
For this study, Dr Abel and his colleagues studied genetically engineered mouse models that were missing GLUT1 and GLUT3 or GLUT3 alone and observed how platelet formation, function, and clearance were affected.
Mice missing glucose transporter proteins did produce platelets, and the platelets’ mitochondria metabolized other substances in place of glucose. However, the mice had platelet counts that were lower than normal.
The researchers were able to pinpoint 2 causes for the low platelet count in mice lacking GLUT1 and GLUT3—fewer platelets being produced and increased clearance of platelets.
The team tested megakaryocytes’ ability to generate new platelets by depleting the blood of platelets and observing the subsequent recovery, which was lower than normal.
They also tested the megakaryocytes in culture, stimulating them to create new platelets, and found the generation of new platelets was defective.
“Clearly, we show that there’s an obligate need for glucose to bud platelets off from the bone marrow,” Dr Abel said.
In addition, the team observed that young platelets functioned normally, even in the absence of glucose. But as they aged, the platelets were cleared from the circulation earlier than normal because they were being destroyed.
“We identified a new mechanism of necrosis by which the absence of glucose leads to the cleavage of a protein called calpain, which marks them for this necrotic pathway,” Dr Abel said. “If we treated the animals with a calpain inhibitor, we could reduce the increased platelet clearance.”
The team also sought to determine whether platelets could exist and function when mitochondria metabolism is halted.
They injected the mice with oligomycin, which inhibits mitochondrial metabolism. In the mice deficient in GLUT1 and GLUT3, platelet counts dropped to 0 within about 30 minutes. The same effect did not occur in wild-type mice.
“This work defined a very important role for metabolism in how platelets leave the bone marrow, how they come into circulation, and how they stay in circulation,” Dr Abel said. “It could even have implications for why platelets have to be used within a certain period of time when they’re donated at the blood bank—the fresher the better.” ![]()
New research published in Cell Reports suggests platelets are highly reliant on their ability to metabolize glucose.
Researchers studied mice lacking proteins that platelets use to import glucose—glucose transporter (GLUT) 1 and GLUT3.
These mice had lower platelet counts and their platelets had shorter life spans than platelets in wild-type mice.
“We found that glucose metabolism is very critical across the entire life cycle of platelets—from production, to what they do in the body, to how they get cleared from the body,” said E. Dale Abel, MBBS, PhD, of the University of Iowa in Iowa City.
For this study, Dr Abel and his colleagues studied genetically engineered mouse models that were missing GLUT1 and GLUT3 or GLUT3 alone and observed how platelet formation, function, and clearance were affected.
Mice missing glucose transporter proteins did produce platelets, and the platelets’ mitochondria metabolized other substances in place of glucose. However, the mice had platelet counts that were lower than normal.
The researchers were able to pinpoint 2 causes for the low platelet count in mice lacking GLUT1 and GLUT3—fewer platelets being produced and increased clearance of platelets.
The team tested megakaryocytes’ ability to generate new platelets by depleting the blood of platelets and observing the subsequent recovery, which was lower than normal.
They also tested the megakaryocytes in culture, stimulating them to create new platelets, and found the generation of new platelets was defective.
“Clearly, we show that there’s an obligate need for glucose to bud platelets off from the bone marrow,” Dr Abel said.
In addition, the team observed that young platelets functioned normally, even in the absence of glucose. But as they aged, the platelets were cleared from the circulation earlier than normal because they were being destroyed.
“We identified a new mechanism of necrosis by which the absence of glucose leads to the cleavage of a protein called calpain, which marks them for this necrotic pathway,” Dr Abel said. “If we treated the animals with a calpain inhibitor, we could reduce the increased platelet clearance.”
The team also sought to determine whether platelets could exist and function when mitochondria metabolism is halted.
They injected the mice with oligomycin, which inhibits mitochondrial metabolism. In the mice deficient in GLUT1 and GLUT3, platelet counts dropped to 0 within about 30 minutes. The same effect did not occur in wild-type mice.
“This work defined a very important role for metabolism in how platelets leave the bone marrow, how they come into circulation, and how they stay in circulation,” Dr Abel said. “It could even have implications for why platelets have to be used within a certain period of time when they’re donated at the blood bank—the fresher the better.” ![]()
New research published in Cell Reports suggests platelets are highly reliant on their ability to metabolize glucose.
Researchers studied mice lacking proteins that platelets use to import glucose—glucose transporter (GLUT) 1 and GLUT3.
These mice had lower platelet counts and their platelets had shorter life spans than platelets in wild-type mice.
“We found that glucose metabolism is very critical across the entire life cycle of platelets—from production, to what they do in the body, to how they get cleared from the body,” said E. Dale Abel, MBBS, PhD, of the University of Iowa in Iowa City.
For this study, Dr Abel and his colleagues studied genetically engineered mouse models that were missing GLUT1 and GLUT3 or GLUT3 alone and observed how platelet formation, function, and clearance were affected.
Mice missing glucose transporter proteins did produce platelets, and the platelets’ mitochondria metabolized other substances in place of glucose. However, the mice had platelet counts that were lower than normal.
The researchers were able to pinpoint 2 causes for the low platelet count in mice lacking GLUT1 and GLUT3—fewer platelets being produced and increased clearance of platelets.
The team tested megakaryocytes’ ability to generate new platelets by depleting the blood of platelets and observing the subsequent recovery, which was lower than normal.
They also tested the megakaryocytes in culture, stimulating them to create new platelets, and found the generation of new platelets was defective.
“Clearly, we show that there’s an obligate need for glucose to bud platelets off from the bone marrow,” Dr Abel said.
In addition, the team observed that young platelets functioned normally, even in the absence of glucose. But as they aged, the platelets were cleared from the circulation earlier than normal because they were being destroyed.
“We identified a new mechanism of necrosis by which the absence of glucose leads to the cleavage of a protein called calpain, which marks them for this necrotic pathway,” Dr Abel said. “If we treated the animals with a calpain inhibitor, we could reduce the increased platelet clearance.”
The team also sought to determine whether platelets could exist and function when mitochondria metabolism is halted.
They injected the mice with oligomycin, which inhibits mitochondrial metabolism. In the mice deficient in GLUT1 and GLUT3, platelet counts dropped to 0 within about 30 minutes. The same effect did not occur in wild-type mice.
“This work defined a very important role for metabolism in how platelets leave the bone marrow, how they come into circulation, and how they stay in circulation,” Dr Abel said. “It could even have implications for why platelets have to be used within a certain period of time when they’re donated at the blood bank—the fresher the better.” ![]()