User login
A Patient With Diabetes, Renal Disease, and Melanoma
Patients who are on hemodialysis and who have cancer present a “unique challenge,” say clinicians from Dartmouth-Hitchcock in New Hampshire.
Patients with end-stage renal disease (ESRD) are at risk of drug accumulation and toxicity. Many anticancer drugs and their metabolites are excreted by the kidney, but data to guide dose and schedule adjustments in renal dialysis are “scant,” the clinicians say. They cite a study that found 72% of dialysis patients receiving anticancer drugs needed dosage adjustments for at least 1 drug. The study researchers also found a significant number of chemotherapy drugs for which there were no available recommendations in dialysis patients.
However, a safe and effective alternative for these patients may exist. The clinicians report on a case—to the best of their knowledge, the first such—of a patient with metastatic melanoma who was successfully treated with pembrolizumab while on hemodialysis.
The patient, who had diabetes and ESRD, also had melanoma in his ear, which metastasized. After discussing his therapeutic options—including the limited data on available immunotherapy drugs—clinicians and the patient agreed to proceed with pembrolizumab, an IgG4-κ human antiprogrammed cell death protein 1 (PD-1) monoclonal antibody. Pembrolizumab has been shown to improve survival rates in patients with melanoma, although it had not been reported in patients with melanoma on dialysis.
The patient received pembrolizumab 2 mg/kg/dose, repeated every 3 weeks. After 1 dose, his abdominal pain and appetite improved. Serum lactate dehydrogenase dropped from 1,182 to 354 units/L. He continued on dialysis 3 times a week with stable serum creatinine levels. After 3 cycles, the computerized tomography scan showed the pulmonary nodules had resolved, and retroperitoneal lymphadenopathy was significantly reduced. After 10 cycles, he was in complete remission.
Pembrolizumab has distinct benefits for patients like theirs, the clinicians suggest. For one, the molecular weight of the antibody means it is not dialysable, so ultrafiltration (reducing drug exposure) is not the issue it might be. The drug can likely be given without regard to the timing of dialysis. Another benefit for these patients who are usually immunocompromised is that PD-1 antibodies “disrupt” the interactions that create an “immune-suppressive tumor microenvironment” and allow T-cell antitumor activity, the clinicians say.
Their report demonstrates that PD-1 antibodies can be effective in dialysis-dependent ESRD, they say, but add that further research into the induced immune response is warranted. In clinical trials, a small number of patients on pembrolizumab (0.4%) developed immune-mediated nephritis. That might not be as crucial for patients who are already on hemodialysis, the clinicians note, but they caution that the adverse effect (AE) could be a risk for patients with normal renal function or chronic kidney disease. However, their patient experienced no pembrolizumab-related AEs other than mild fatigue.
Source:
Chang R, Shirai K. BMJ Case Rep. 2016; pii: bcr2016216426.
doi: 10.1136/bcr-2016-216426.
Patients who are on hemodialysis and who have cancer present a “unique challenge,” say clinicians from Dartmouth-Hitchcock in New Hampshire.
Patients with end-stage renal disease (ESRD) are at risk of drug accumulation and toxicity. Many anticancer drugs and their metabolites are excreted by the kidney, but data to guide dose and schedule adjustments in renal dialysis are “scant,” the clinicians say. They cite a study that found 72% of dialysis patients receiving anticancer drugs needed dosage adjustments for at least 1 drug. The study researchers also found a significant number of chemotherapy drugs for which there were no available recommendations in dialysis patients.
However, a safe and effective alternative for these patients may exist. The clinicians report on a case—to the best of their knowledge, the first such—of a patient with metastatic melanoma who was successfully treated with pembrolizumab while on hemodialysis.
The patient, who had diabetes and ESRD, also had melanoma in his ear, which metastasized. After discussing his therapeutic options—including the limited data on available immunotherapy drugs—clinicians and the patient agreed to proceed with pembrolizumab, an IgG4-κ human antiprogrammed cell death protein 1 (PD-1) monoclonal antibody. Pembrolizumab has been shown to improve survival rates in patients with melanoma, although it had not been reported in patients with melanoma on dialysis.
The patient received pembrolizumab 2 mg/kg/dose, repeated every 3 weeks. After 1 dose, his abdominal pain and appetite improved. Serum lactate dehydrogenase dropped from 1,182 to 354 units/L. He continued on dialysis 3 times a week with stable serum creatinine levels. After 3 cycles, the computerized tomography scan showed the pulmonary nodules had resolved, and retroperitoneal lymphadenopathy was significantly reduced. After 10 cycles, he was in complete remission.
Pembrolizumab has distinct benefits for patients like theirs, the clinicians suggest. For one, the molecular weight of the antibody means it is not dialysable, so ultrafiltration (reducing drug exposure) is not the issue it might be. The drug can likely be given without regard to the timing of dialysis. Another benefit for these patients who are usually immunocompromised is that PD-1 antibodies “disrupt” the interactions that create an “immune-suppressive tumor microenvironment” and allow T-cell antitumor activity, the clinicians say.
Their report demonstrates that PD-1 antibodies can be effective in dialysis-dependent ESRD, they say, but add that further research into the induced immune response is warranted. In clinical trials, a small number of patients on pembrolizumab (0.4%) developed immune-mediated nephritis. That might not be as crucial for patients who are already on hemodialysis, the clinicians note, but they caution that the adverse effect (AE) could be a risk for patients with normal renal function or chronic kidney disease. However, their patient experienced no pembrolizumab-related AEs other than mild fatigue.
Source:
Chang R, Shirai K. BMJ Case Rep. 2016; pii: bcr2016216426.
doi: 10.1136/bcr-2016-216426.
Patients who are on hemodialysis and who have cancer present a “unique challenge,” say clinicians from Dartmouth-Hitchcock in New Hampshire.
Patients with end-stage renal disease (ESRD) are at risk of drug accumulation and toxicity. Many anticancer drugs and their metabolites are excreted by the kidney, but data to guide dose and schedule adjustments in renal dialysis are “scant,” the clinicians say. They cite a study that found 72% of dialysis patients receiving anticancer drugs needed dosage adjustments for at least 1 drug. The study researchers also found a significant number of chemotherapy drugs for which there were no available recommendations in dialysis patients.
However, a safe and effective alternative for these patients may exist. The clinicians report on a case—to the best of their knowledge, the first such—of a patient with metastatic melanoma who was successfully treated with pembrolizumab while on hemodialysis.
The patient, who had diabetes and ESRD, also had melanoma in his ear, which metastasized. After discussing his therapeutic options—including the limited data on available immunotherapy drugs—clinicians and the patient agreed to proceed with pembrolizumab, an IgG4-κ human antiprogrammed cell death protein 1 (PD-1) monoclonal antibody. Pembrolizumab has been shown to improve survival rates in patients with melanoma, although it had not been reported in patients with melanoma on dialysis.
The patient received pembrolizumab 2 mg/kg/dose, repeated every 3 weeks. After 1 dose, his abdominal pain and appetite improved. Serum lactate dehydrogenase dropped from 1,182 to 354 units/L. He continued on dialysis 3 times a week with stable serum creatinine levels. After 3 cycles, the computerized tomography scan showed the pulmonary nodules had resolved, and retroperitoneal lymphadenopathy was significantly reduced. After 10 cycles, he was in complete remission.
Pembrolizumab has distinct benefits for patients like theirs, the clinicians suggest. For one, the molecular weight of the antibody means it is not dialysable, so ultrafiltration (reducing drug exposure) is not the issue it might be. The drug can likely be given without regard to the timing of dialysis. Another benefit for these patients who are usually immunocompromised is that PD-1 antibodies “disrupt” the interactions that create an “immune-suppressive tumor microenvironment” and allow T-cell antitumor activity, the clinicians say.
Their report demonstrates that PD-1 antibodies can be effective in dialysis-dependent ESRD, they say, but add that further research into the induced immune response is warranted. In clinical trials, a small number of patients on pembrolizumab (0.4%) developed immune-mediated nephritis. That might not be as crucial for patients who are already on hemodialysis, the clinicians note, but they caution that the adverse effect (AE) could be a risk for patients with normal renal function or chronic kidney disease. However, their patient experienced no pembrolizumab-related AEs other than mild fatigue.
Source:
Chang R, Shirai K. BMJ Case Rep. 2016; pii: bcr2016216426.
doi: 10.1136/bcr-2016-216426.
High healthcare costs follow CCSs into adulthood

New research suggests survivors of childhood cancer can incur high out-of-pocket medical costs into adulthood and may forgo healthcare to lessen this financial burden.
The study showed that childhood cancer survivors (CCSs) were more likely than non-CCSs to have out-of-pocket medical costs that were at least 10% of their annual income.
And these high costs were associated with delaying care or skipping it altogether.
These findings were published in the Journal of Clinical Oncology.
“Survivors who reported spending a higher percentage of their income on out-of-pocket medical costs were not only more likely to report financial burden, they also were at risk for undertaking behaviors potentially detrimental to their health in order to save money,” said study author Ryan Nipp, MD, of Massachusetts General Hospital Cancer Center in Boston.
“While studies have identified associations between financial burden and patients’ treatment outcomes, quality of life, and even survival among adults with cancer, as far as we know, this is the first to report these associations in survivors of childhood cancer.”
For this research, Dr Nipp and his colleagues surveyed participants in the Childhood Cancer Survivor Study. This included adults who had been treated for childhood cancers between 1970 and 1986 along with a control group of siblings not affected by cancer.
In 2011 and 2012, participants were asked to provide information about their health insurance, the out-of-pocket healthcare costs they paid during the previous year, and sociodemographic information such as annual income and employment status.
The researchers also asked participants whether medical costs posed a financial burden and, if so, what measures they had taken to deal with that burden.
Study population
The researchers received complete responses from 580 CCSs and 173 of their siblings without a history of cancer. The most common cancer diagnosis was leukemia (33%), followed by Hodgkin lymphoma (14%), while non-Hodgkin lymphoma was less common (7%).
CCSs were a mean of 30.2 years from diagnosis. Use of chemotherapy (77%), radiation (66%), and surgery (81%) were common. Few patients had cancer recurrence (13%) or second cancers (5%).
There was no significant difference between CCSs and their siblings with regard to age at the time of the survey (P=0.071), household income (P=0.053), education (P=0.345), health insurance status (P=0.317), or having at least 1 hospitalization in the past year (P=0.270).
However, CCSs were significantly more likely than siblings to have chronic health conditions (P<0.001). Forty percent of CCSs had severe or life-threatening chronic conditions, compared to 17% of siblings.
Seventy-six percent of CCSs and 80% of siblings were employed. Twenty-nine percent of CCSs and 39% of siblings had household incomes exceeding $100,000. Twelve percent of CCSs and 5% of siblings had household incomes below $20,000.
Ninety-one percent of CCSs and 93% of siblings were insured. Most subjects in both groups (81% and 87%, respectively) had employer-sponsored insurance.
Results
CCSs were significantly more likely than their siblings to have out-of-pocket medical costs that were at least 10% of their annual income—10% and 3%, respectively (P<0.001).
Among CCSs, those with higher out-of-pocket costs (≥10% vs <10% of income) were more likely to have household incomes below $50,000 (odds ratio [OR]=5.5) and to report being hospitalized in the past year (OR=2.3).
CCSs with a higher percentage of their income spent on out-of-pocket costs were also more likely to:
- Have problems paying their medical bills (OR=8.8)
- Report inability to pay for basic costs of living such as food, heat, or rent (OR=6.1)
- Defer healthcare for a medical problem (OR=3.1)
- Skip a test, treatment, or follow-up (OR=2.1)
- Consider filing for bankruptcy (OR=6.4).
“A more comprehensive understanding of the relationship between high out-of-pocket medical costs and the adverse effects of increased financial burden on cancer survivors could be instrumental in helping us identify those at risk for higher costs to help us address their financial challenges and improve health outcomes,” Dr Nipp said.
“It could also help inform policy changes to help meet the unique needs of cancer survivors and improve our understanding of how both higher costs and resulting financial burden influence patients’ approach to their medical care and decision-making.” ![]()
New research suggests survivors of childhood cancer can incur high out-of-pocket medical costs into adulthood and may forgo healthcare to lessen this financial burden.
The study showed that childhood cancer survivors (CCSs) were more likely than non-CCSs to have out-of-pocket medical costs that were at least 10% of their annual income.
And these high costs were associated with delaying care or skipping it altogether.
These findings were published in the Journal of Clinical Oncology.
“Survivors who reported spending a higher percentage of their income on out-of-pocket medical costs were not only more likely to report financial burden, they also were at risk for undertaking behaviors potentially detrimental to their health in order to save money,” said study author Ryan Nipp, MD, of Massachusetts General Hospital Cancer Center in Boston.
“While studies have identified associations between financial burden and patients’ treatment outcomes, quality of life, and even survival among adults with cancer, as far as we know, this is the first to report these associations in survivors of childhood cancer.”
For this research, Dr Nipp and his colleagues surveyed participants in the Childhood Cancer Survivor Study. This included adults who had been treated for childhood cancers between 1970 and 1986 along with a control group of siblings not affected by cancer.
In 2011 and 2012, participants were asked to provide information about their health insurance, the out-of-pocket healthcare costs they paid during the previous year, and sociodemographic information such as annual income and employment status.
The researchers also asked participants whether medical costs posed a financial burden and, if so, what measures they had taken to deal with that burden.
Study population
The researchers received complete responses from 580 CCSs and 173 of their siblings without a history of cancer. The most common cancer diagnosis was leukemia (33%), followed by Hodgkin lymphoma (14%), while non-Hodgkin lymphoma was less common (7%).
CCSs were a mean of 30.2 years from diagnosis. Use of chemotherapy (77%), radiation (66%), and surgery (81%) were common. Few patients had cancer recurrence (13%) or second cancers (5%).
There was no significant difference between CCSs and their siblings with regard to age at the time of the survey (P=0.071), household income (P=0.053), education (P=0.345), health insurance status (P=0.317), or having at least 1 hospitalization in the past year (P=0.270).
However, CCSs were significantly more likely than siblings to have chronic health conditions (P<0.001). Forty percent of CCSs had severe or life-threatening chronic conditions, compared to 17% of siblings.
Seventy-six percent of CCSs and 80% of siblings were employed. Twenty-nine percent of CCSs and 39% of siblings had household incomes exceeding $100,000. Twelve percent of CCSs and 5% of siblings had household incomes below $20,000.
Ninety-one percent of CCSs and 93% of siblings were insured. Most subjects in both groups (81% and 87%, respectively) had employer-sponsored insurance.
Results
CCSs were significantly more likely than their siblings to have out-of-pocket medical costs that were at least 10% of their annual income—10% and 3%, respectively (P<0.001).
Among CCSs, those with higher out-of-pocket costs (≥10% vs <10% of income) were more likely to have household incomes below $50,000 (odds ratio [OR]=5.5) and to report being hospitalized in the past year (OR=2.3).
CCSs with a higher percentage of their income spent on out-of-pocket costs were also more likely to:
- Have problems paying their medical bills (OR=8.8)
- Report inability to pay for basic costs of living such as food, heat, or rent (OR=6.1)
- Defer healthcare for a medical problem (OR=3.1)
- Skip a test, treatment, or follow-up (OR=2.1)
- Consider filing for bankruptcy (OR=6.4).
“A more comprehensive understanding of the relationship between high out-of-pocket medical costs and the adverse effects of increased financial burden on cancer survivors could be instrumental in helping us identify those at risk for higher costs to help us address their financial challenges and improve health outcomes,” Dr Nipp said.
“It could also help inform policy changes to help meet the unique needs of cancer survivors and improve our understanding of how both higher costs and resulting financial burden influence patients’ approach to their medical care and decision-making.” ![]()
New research suggests survivors of childhood cancer can incur high out-of-pocket medical costs into adulthood and may forgo healthcare to lessen this financial burden.
The study showed that childhood cancer survivors (CCSs) were more likely than non-CCSs to have out-of-pocket medical costs that were at least 10% of their annual income.
And these high costs were associated with delaying care or skipping it altogether.
These findings were published in the Journal of Clinical Oncology.
“Survivors who reported spending a higher percentage of their income on out-of-pocket medical costs were not only more likely to report financial burden, they also were at risk for undertaking behaviors potentially detrimental to their health in order to save money,” said study author Ryan Nipp, MD, of Massachusetts General Hospital Cancer Center in Boston.
“While studies have identified associations between financial burden and patients’ treatment outcomes, quality of life, and even survival among adults with cancer, as far as we know, this is the first to report these associations in survivors of childhood cancer.”
For this research, Dr Nipp and his colleagues surveyed participants in the Childhood Cancer Survivor Study. This included adults who had been treated for childhood cancers between 1970 and 1986 along with a control group of siblings not affected by cancer.
In 2011 and 2012, participants were asked to provide information about their health insurance, the out-of-pocket healthcare costs they paid during the previous year, and sociodemographic information such as annual income and employment status.
The researchers also asked participants whether medical costs posed a financial burden and, if so, what measures they had taken to deal with that burden.
Study population
The researchers received complete responses from 580 CCSs and 173 of their siblings without a history of cancer. The most common cancer diagnosis was leukemia (33%), followed by Hodgkin lymphoma (14%), while non-Hodgkin lymphoma was less common (7%).
CCSs were a mean of 30.2 years from diagnosis. Use of chemotherapy (77%), radiation (66%), and surgery (81%) were common. Few patients had cancer recurrence (13%) or second cancers (5%).
There was no significant difference between CCSs and their siblings with regard to age at the time of the survey (P=0.071), household income (P=0.053), education (P=0.345), health insurance status (P=0.317), or having at least 1 hospitalization in the past year (P=0.270).
However, CCSs were significantly more likely than siblings to have chronic health conditions (P<0.001). Forty percent of CCSs had severe or life-threatening chronic conditions, compared to 17% of siblings.
Seventy-six percent of CCSs and 80% of siblings were employed. Twenty-nine percent of CCSs and 39% of siblings had household incomes exceeding $100,000. Twelve percent of CCSs and 5% of siblings had household incomes below $20,000.
Ninety-one percent of CCSs and 93% of siblings were insured. Most subjects in both groups (81% and 87%, respectively) had employer-sponsored insurance.
Results
CCSs were significantly more likely than their siblings to have out-of-pocket medical costs that were at least 10% of their annual income—10% and 3%, respectively (P<0.001).
Among CCSs, those with higher out-of-pocket costs (≥10% vs <10% of income) were more likely to have household incomes below $50,000 (odds ratio [OR]=5.5) and to report being hospitalized in the past year (OR=2.3).
CCSs with a higher percentage of their income spent on out-of-pocket costs were also more likely to:
- Have problems paying their medical bills (OR=8.8)
- Report inability to pay for basic costs of living such as food, heat, or rent (OR=6.1)
- Defer healthcare for a medical problem (OR=3.1)
- Skip a test, treatment, or follow-up (OR=2.1)
- Consider filing for bankruptcy (OR=6.4).
“A more comprehensive understanding of the relationship between high out-of-pocket medical costs and the adverse effects of increased financial burden on cancer survivors could be instrumental in helping us identify those at risk for higher costs to help us address their financial challenges and improve health outcomes,” Dr Nipp said.
“It could also help inform policy changes to help meet the unique needs of cancer survivors and improve our understanding of how both higher costs and resulting financial burden influence patients’ approach to their medical care and decision-making.” ![]()
Vitamin C regulates HSCs, curbs AML development

Researchers have described a molecular mechanism that could help explain the link between low vitamin C (ascorbate) levels and acute myeloid leukemia (AML).
The team found that mice with low levels of ascorbate in their blood experience a notable increase in hematopoietic stem cell (HSC) frequency and function.
This, in turn, accelerates AML development, partly by inhibiting the tumor suppressor Tet2.
Sean Morrison, PhD, of the University of Texas Southwestern Medical Center in Dallas, and his colleagues reported these findings in Nature.
“We have known for a while that people with lower levels of ascorbate (vitamin C) are at increased cancer risk, but we haven’t fully understood why,” Dr Morrison said. “Our research provides part of the explanation, at least for the blood-forming system.”
Dr Morrison and his colleagues developed a technique for analyzing the metabolic profiles of rare cell populations and used it to compare HSCs to restricted hematopoietic progenitors.
The researchers found that each hematopoietic cell type had a “distinct metabolic signature,” and both human and mouse HSCs had “unusually high” levels of ascorbate, which decreased as the cells differentiated.
To determine if ascorbate is important for HSC function, the team studied mice that lacked gulonolactone oxidase (Gulo), an enzyme mice use to synthesize their own ascorbate. Loss of the enzyme requires Gulo-deficient mice to obtain ascorbate exclusively through their diet like humans do.
So when the researchers fed the mice a standard diet, which contains little ascorbate, the animals’ ascorbate levels were depleted.
The team expected ascorbate depletion might lead to loss of HSC function, but they found the opposite was true. HSCs actually gained function, and this increased the incidence of AML in the mice.
This increase is partly tied to how ascorbate affects Tet2. The researchers found that ascorbate depletion can limit Tet2 function in tissues in a way that increases the risk of AML.
In addition, ascorbate depletion cooperated with Flt3 internal tandem duplication mutations to accelerate leukemogenesis. But the researchers were able to suppress leukemogenesis by feeding animals higher levels of ascorbate.
“Stem cells use ascorbate to regulate the abundance of certain chemical modifications on DNA, which are part of the epigenome,” said study author Michalis Agathocleous, PhD, of the University of Texas Southwestern Medical Center.
“So when stem cells don’t receive enough vitamin C, the epigenome can become damaged in a way that increases stem cell function but also increases the risk of leukemia.”
The researchers said further studies are needed to better understand the potential clinical implications of these findings.
However, the findings may have implications for older patients with clonal hematopoiesis. This condition increases a person’s risk of developing leukemia, but it is not well understood why certain patients develop leukemia and others do not. The results of this study might offer an explanation.
“One of the most common mutations in patients with clonal hematopoiesis is a loss of one copy of TET2,” Dr Morrison said. “Our results suggest patients with clonal hematopoiesis and a TET2 mutation should be particularly careful to get 100% of their daily vitamin C requirement. Because these patients only have one good copy of TET2 left, they need to maximize the residual TET2 tumor-suppressor activity to protect themselves from cancer.” ![]()
Researchers have described a molecular mechanism that could help explain the link between low vitamin C (ascorbate) levels and acute myeloid leukemia (AML).
The team found that mice with low levels of ascorbate in their blood experience a notable increase in hematopoietic stem cell (HSC) frequency and function.
This, in turn, accelerates AML development, partly by inhibiting the tumor suppressor Tet2.
Sean Morrison, PhD, of the University of Texas Southwestern Medical Center in Dallas, and his colleagues reported these findings in Nature.
“We have known for a while that people with lower levels of ascorbate (vitamin C) are at increased cancer risk, but we haven’t fully understood why,” Dr Morrison said. “Our research provides part of the explanation, at least for the blood-forming system.”
Dr Morrison and his colleagues developed a technique for analyzing the metabolic profiles of rare cell populations and used it to compare HSCs to restricted hematopoietic progenitors.
The researchers found that each hematopoietic cell type had a “distinct metabolic signature,” and both human and mouse HSCs had “unusually high” levels of ascorbate, which decreased as the cells differentiated.
To determine if ascorbate is important for HSC function, the team studied mice that lacked gulonolactone oxidase (Gulo), an enzyme mice use to synthesize their own ascorbate. Loss of the enzyme requires Gulo-deficient mice to obtain ascorbate exclusively through their diet like humans do.
So when the researchers fed the mice a standard diet, which contains little ascorbate, the animals’ ascorbate levels were depleted.
The team expected ascorbate depletion might lead to loss of HSC function, but they found the opposite was true. HSCs actually gained function, and this increased the incidence of AML in the mice.
This increase is partly tied to how ascorbate affects Tet2. The researchers found that ascorbate depletion can limit Tet2 function in tissues in a way that increases the risk of AML.
In addition, ascorbate depletion cooperated with Flt3 internal tandem duplication mutations to accelerate leukemogenesis. But the researchers were able to suppress leukemogenesis by feeding animals higher levels of ascorbate.
“Stem cells use ascorbate to regulate the abundance of certain chemical modifications on DNA, which are part of the epigenome,” said study author Michalis Agathocleous, PhD, of the University of Texas Southwestern Medical Center.
“So when stem cells don’t receive enough vitamin C, the epigenome can become damaged in a way that increases stem cell function but also increases the risk of leukemia.”
The researchers said further studies are needed to better understand the potential clinical implications of these findings.
However, the findings may have implications for older patients with clonal hematopoiesis. This condition increases a person’s risk of developing leukemia, but it is not well understood why certain patients develop leukemia and others do not. The results of this study might offer an explanation.
“One of the most common mutations in patients with clonal hematopoiesis is a loss of one copy of TET2,” Dr Morrison said. “Our results suggest patients with clonal hematopoiesis and a TET2 mutation should be particularly careful to get 100% of their daily vitamin C requirement. Because these patients only have one good copy of TET2 left, they need to maximize the residual TET2 tumor-suppressor activity to protect themselves from cancer.” ![]()
Researchers have described a molecular mechanism that could help explain the link between low vitamin C (ascorbate) levels and acute myeloid leukemia (AML).
The team found that mice with low levels of ascorbate in their blood experience a notable increase in hematopoietic stem cell (HSC) frequency and function.
This, in turn, accelerates AML development, partly by inhibiting the tumor suppressor Tet2.
Sean Morrison, PhD, of the University of Texas Southwestern Medical Center in Dallas, and his colleagues reported these findings in Nature.
“We have known for a while that people with lower levels of ascorbate (vitamin C) are at increased cancer risk, but we haven’t fully understood why,” Dr Morrison said. “Our research provides part of the explanation, at least for the blood-forming system.”
Dr Morrison and his colleagues developed a technique for analyzing the metabolic profiles of rare cell populations and used it to compare HSCs to restricted hematopoietic progenitors.
The researchers found that each hematopoietic cell type had a “distinct metabolic signature,” and both human and mouse HSCs had “unusually high” levels of ascorbate, which decreased as the cells differentiated.
To determine if ascorbate is important for HSC function, the team studied mice that lacked gulonolactone oxidase (Gulo), an enzyme mice use to synthesize their own ascorbate. Loss of the enzyme requires Gulo-deficient mice to obtain ascorbate exclusively through their diet like humans do.
So when the researchers fed the mice a standard diet, which contains little ascorbate, the animals’ ascorbate levels were depleted.
The team expected ascorbate depletion might lead to loss of HSC function, but they found the opposite was true. HSCs actually gained function, and this increased the incidence of AML in the mice.
This increase is partly tied to how ascorbate affects Tet2. The researchers found that ascorbate depletion can limit Tet2 function in tissues in a way that increases the risk of AML.
In addition, ascorbate depletion cooperated with Flt3 internal tandem duplication mutations to accelerate leukemogenesis. But the researchers were able to suppress leukemogenesis by feeding animals higher levels of ascorbate.
“Stem cells use ascorbate to regulate the abundance of certain chemical modifications on DNA, which are part of the epigenome,” said study author Michalis Agathocleous, PhD, of the University of Texas Southwestern Medical Center.
“So when stem cells don’t receive enough vitamin C, the epigenome can become damaged in a way that increases stem cell function but also increases the risk of leukemia.”
The researchers said further studies are needed to better understand the potential clinical implications of these findings.
However, the findings may have implications for older patients with clonal hematopoiesis. This condition increases a person’s risk of developing leukemia, but it is not well understood why certain patients develop leukemia and others do not. The results of this study might offer an explanation.
“One of the most common mutations in patients with clonal hematopoiesis is a loss of one copy of TET2,” Dr Morrison said. “Our results suggest patients with clonal hematopoiesis and a TET2 mutation should be particularly careful to get 100% of their daily vitamin C requirement. Because these patients only have one good copy of TET2 left, they need to maximize the residual TET2 tumor-suppressor activity to protect themselves from cancer.” ![]()
FDA approves new strengths of hemophilia therapy
The US Food and Drug Administration (FDA) has approved new product strengths for the factor VIII therapy simoctocog alfa (NUWIQ®).
Simoctocog alfa is the first B-domain-deleted recombinant factor VIII product derived from a human cell line—not chemically modified or fused with another protein—designed to treat hemophilia A.
Simoctocog alfa is FDA-approved to treat adults and children with hemophilia A. This includes on-demand treatment and control of bleeding episodes, routine prophylaxis to reduce the frequency of bleeding episodes, and perioperative management of bleeding.
Now, the FDA has approved single-dose simoctocog alfa vial strengths of 2500, 3000, and 4000 International Units (IU), which will be available for order in the US starting September 2017.
These new vial strengths will be provided in addition to the already available strengths of 250, 500, 1000, and 2000 IU.
“The new vial options will benefit patients, physicians, and healthcare professionals by providing greater treatment flexibility and convenience,” said Flemming Nielsen, president of Octapharma USA, makers of simoctocog alfa.
According to Octapharma, the additional vial strengths offer benefits beyond potentially reducing the number of vials used per patient. The new vial sizes may benefit heavier patients who could use fewer product vials and, in some cases, just one vial.
More vial options will increase dosing flexibility by allowing physicians to select various vial combinations to align closer to the prescribed dose. The new vial sizes could be particularly beneficial to patients using a pharmacokinetic (PK)-guided, personalized prophylaxis approach.
Results of Octapharma’s clinical trial on the PK-guided dosing with simoctocog alfa were published in Haemophilia in April.
This study, known as GENA-21 or NuPreviq, enrolled 66 previously treated adults with severe hemophilia A. Patients were originally started on infusions 3 times a week or every other day. Subsequent dosing intervals were then determined based on individual PK data.
The median dosing interval with PK-guided prophylaxis was 3.5 days, and 57% of patients were able to decrease infusions to twice-weekly or less.
The median weekly prophylaxis dose was reduced by 7.2%, from 100.0 IU kg−1 with standard prophylaxis to 92.8 IU kg−1 during the last 2 months of personalized prophylaxis.
For all bleeds, the mean annualized bleeding rate (ABR) during personalized prophylaxis was 1.45, and the median was 0 (interquartile range, [IQR]: 0, 1.9).
For spontaneous bleeds, the mean ABR was 0.79, and the median was 0 (IQR: 0, 0). For joint bleeds, the mean ABR was 0.91, and the median was 0 (IQR: 0, 0).
None of the patients developed FVIII inhibitors. There were no treatment-related serious or severe adverse events, clinically significant abnormalities in laboratory parameters, or cases of thromboembolism.
The ongoing trial GENA-21b is designed to confirm the results of GENA-21. ![]()
The US Food and Drug Administration (FDA) has approved new product strengths for the factor VIII therapy simoctocog alfa (NUWIQ®).
Simoctocog alfa is the first B-domain-deleted recombinant factor VIII product derived from a human cell line—not chemically modified or fused with another protein—designed to treat hemophilia A.
Simoctocog alfa is FDA-approved to treat adults and children with hemophilia A. This includes on-demand treatment and control of bleeding episodes, routine prophylaxis to reduce the frequency of bleeding episodes, and perioperative management of bleeding.
Now, the FDA has approved single-dose simoctocog alfa vial strengths of 2500, 3000, and 4000 International Units (IU), which will be available for order in the US starting September 2017.
These new vial strengths will be provided in addition to the already available strengths of 250, 500, 1000, and 2000 IU.
“The new vial options will benefit patients, physicians, and healthcare professionals by providing greater treatment flexibility and convenience,” said Flemming Nielsen, president of Octapharma USA, makers of simoctocog alfa.
According to Octapharma, the additional vial strengths offer benefits beyond potentially reducing the number of vials used per patient. The new vial sizes may benefit heavier patients who could use fewer product vials and, in some cases, just one vial.
More vial options will increase dosing flexibility by allowing physicians to select various vial combinations to align closer to the prescribed dose. The new vial sizes could be particularly beneficial to patients using a pharmacokinetic (PK)-guided, personalized prophylaxis approach.
Results of Octapharma’s clinical trial on the PK-guided dosing with simoctocog alfa were published in Haemophilia in April.
This study, known as GENA-21 or NuPreviq, enrolled 66 previously treated adults with severe hemophilia A. Patients were originally started on infusions 3 times a week or every other day. Subsequent dosing intervals were then determined based on individual PK data.
The median dosing interval with PK-guided prophylaxis was 3.5 days, and 57% of patients were able to decrease infusions to twice-weekly or less.
The median weekly prophylaxis dose was reduced by 7.2%, from 100.0 IU kg−1 with standard prophylaxis to 92.8 IU kg−1 during the last 2 months of personalized prophylaxis.
For all bleeds, the mean annualized bleeding rate (ABR) during personalized prophylaxis was 1.45, and the median was 0 (interquartile range, [IQR]: 0, 1.9).
For spontaneous bleeds, the mean ABR was 0.79, and the median was 0 (IQR: 0, 0). For joint bleeds, the mean ABR was 0.91, and the median was 0 (IQR: 0, 0).
None of the patients developed FVIII inhibitors. There were no treatment-related serious or severe adverse events, clinically significant abnormalities in laboratory parameters, or cases of thromboembolism.
The ongoing trial GENA-21b is designed to confirm the results of GENA-21. ![]()
The US Food and Drug Administration (FDA) has approved new product strengths for the factor VIII therapy simoctocog alfa (NUWIQ®).
Simoctocog alfa is the first B-domain-deleted recombinant factor VIII product derived from a human cell line—not chemically modified or fused with another protein—designed to treat hemophilia A.
Simoctocog alfa is FDA-approved to treat adults and children with hemophilia A. This includes on-demand treatment and control of bleeding episodes, routine prophylaxis to reduce the frequency of bleeding episodes, and perioperative management of bleeding.
Now, the FDA has approved single-dose simoctocog alfa vial strengths of 2500, 3000, and 4000 International Units (IU), which will be available for order in the US starting September 2017.
These new vial strengths will be provided in addition to the already available strengths of 250, 500, 1000, and 2000 IU.
“The new vial options will benefit patients, physicians, and healthcare professionals by providing greater treatment flexibility and convenience,” said Flemming Nielsen, president of Octapharma USA, makers of simoctocog alfa.
According to Octapharma, the additional vial strengths offer benefits beyond potentially reducing the number of vials used per patient. The new vial sizes may benefit heavier patients who could use fewer product vials and, in some cases, just one vial.
More vial options will increase dosing flexibility by allowing physicians to select various vial combinations to align closer to the prescribed dose. The new vial sizes could be particularly beneficial to patients using a pharmacokinetic (PK)-guided, personalized prophylaxis approach.
Results of Octapharma’s clinical trial on the PK-guided dosing with simoctocog alfa were published in Haemophilia in April.
This study, known as GENA-21 or NuPreviq, enrolled 66 previously treated adults with severe hemophilia A. Patients were originally started on infusions 3 times a week or every other day. Subsequent dosing intervals were then determined based on individual PK data.
The median dosing interval with PK-guided prophylaxis was 3.5 days, and 57% of patients were able to decrease infusions to twice-weekly or less.
The median weekly prophylaxis dose was reduced by 7.2%, from 100.0 IU kg−1 with standard prophylaxis to 92.8 IU kg−1 during the last 2 months of personalized prophylaxis.
For all bleeds, the mean annualized bleeding rate (ABR) during personalized prophylaxis was 1.45, and the median was 0 (interquartile range, [IQR]: 0, 1.9).
For spontaneous bleeds, the mean ABR was 0.79, and the median was 0 (IQR: 0, 0). For joint bleeds, the mean ABR was 0.91, and the median was 0 (IQR: 0, 0).
None of the patients developed FVIII inhibitors. There were no treatment-related serious or severe adverse events, clinically significant abnormalities in laboratory parameters, or cases of thromboembolism.
The ongoing trial GENA-21b is designed to confirm the results of GENA-21. ![]()
Worsening rash
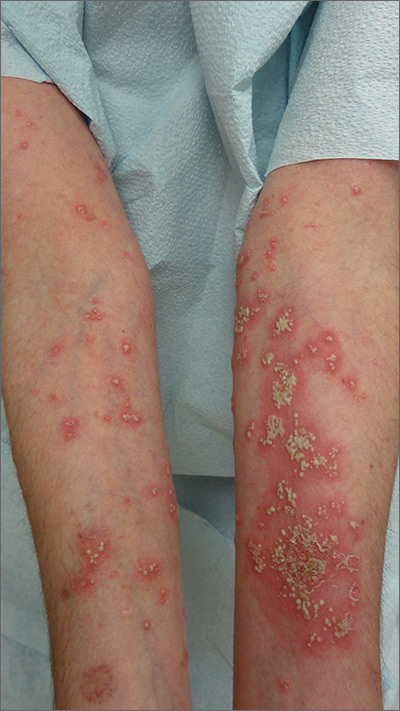
The FP suspected pustular psoriasis, which was confirmed by a dermatologist the patient saw the following day. The dermatologist confirmed this diagnosis by performing a 4-mm punch biopsy, which included an area of new pustules on the patient’s arm. When a rash is extensive, it’s best to biopsy new lesions from the upper body, rather than old lesions below the waist.
Pustular psoriasis can lead to confluent pustules, a condition in which the skin peels off in sheets, resulting in dehydration and a risk of sepsis. This is why you shouldn’t prescribe oral prednisone for any rash before you know that it’s not psoriasis. While oral prednisone may be an effective treatment for many types of dermatitis, it can exacerbate psoriasis (as it did with this patient), and is therefore not an effective or safe treatment for it.
Knowing that pustular psoriasis can become a dermatologic emergency, the FP prescribed oral cyclosporine for the most rapid result. The patient’s blood pressure and kidney function were normal, so it was only necessary for the FP to order baseline laboratory tests. At the follow-up appointment 2 days later, the patient’s condition was already improving and there were no new pustules. Her vital signs remained stable and the pathology report confirmed pustular psoriasis.
The dermatologist planned to transition the patient from cyclosporine to acitretin, determining it was safe to prescribe an oral retinoid. The patient’s hysterectomy 2 years earlier also eliminated concerns about acitretin’s teratogenic potency. The dermatologist prescribed acitretin with directions for the patient to begin taking it in one week, tapering off the cyclosporine once the pustular psoriasis was significantly better.
After a month, the patient was off of cyclosporine completely. Her skin was clear while using daily acitretin. Monitoring of lab tests did not show any adverse effects of the patient using these 2 potentially toxic medications.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Psoriasis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 878-895.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP suspected pustular psoriasis, which was confirmed by a dermatologist the patient saw the following day. The dermatologist confirmed this diagnosis by performing a 4-mm punch biopsy, which included an area of new pustules on the patient’s arm. When a rash is extensive, it’s best to biopsy new lesions from the upper body, rather than old lesions below the waist.
Pustular psoriasis can lead to confluent pustules, a condition in which the skin peels off in sheets, resulting in dehydration and a risk of sepsis. This is why you shouldn’t prescribe oral prednisone for any rash before you know that it’s not psoriasis. While oral prednisone may be an effective treatment for many types of dermatitis, it can exacerbate psoriasis (as it did with this patient), and is therefore not an effective or safe treatment for it.
Knowing that pustular psoriasis can become a dermatologic emergency, the FP prescribed oral cyclosporine for the most rapid result. The patient’s blood pressure and kidney function were normal, so it was only necessary for the FP to order baseline laboratory tests. At the follow-up appointment 2 days later, the patient’s condition was already improving and there were no new pustules. Her vital signs remained stable and the pathology report confirmed pustular psoriasis.
The dermatologist planned to transition the patient from cyclosporine to acitretin, determining it was safe to prescribe an oral retinoid. The patient’s hysterectomy 2 years earlier also eliminated concerns about acitretin’s teratogenic potency. The dermatologist prescribed acitretin with directions for the patient to begin taking it in one week, tapering off the cyclosporine once the pustular psoriasis was significantly better.
After a month, the patient was off of cyclosporine completely. Her skin was clear while using daily acitretin. Monitoring of lab tests did not show any adverse effects of the patient using these 2 potentially toxic medications.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Psoriasis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 878-895.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP suspected pustular psoriasis, which was confirmed by a dermatologist the patient saw the following day. The dermatologist confirmed this diagnosis by performing a 4-mm punch biopsy, which included an area of new pustules on the patient’s arm. When a rash is extensive, it’s best to biopsy new lesions from the upper body, rather than old lesions below the waist.
Pustular psoriasis can lead to confluent pustules, a condition in which the skin peels off in sheets, resulting in dehydration and a risk of sepsis. This is why you shouldn’t prescribe oral prednisone for any rash before you know that it’s not psoriasis. While oral prednisone may be an effective treatment for many types of dermatitis, it can exacerbate psoriasis (as it did with this patient), and is therefore not an effective or safe treatment for it.
Knowing that pustular psoriasis can become a dermatologic emergency, the FP prescribed oral cyclosporine for the most rapid result. The patient’s blood pressure and kidney function were normal, so it was only necessary for the FP to order baseline laboratory tests. At the follow-up appointment 2 days later, the patient’s condition was already improving and there were no new pustules. Her vital signs remained stable and the pathology report confirmed pustular psoriasis.
The dermatologist planned to transition the patient from cyclosporine to acitretin, determining it was safe to prescribe an oral retinoid. The patient’s hysterectomy 2 years earlier also eliminated concerns about acitretin’s teratogenic potency. The dermatologist prescribed acitretin with directions for the patient to begin taking it in one week, tapering off the cyclosporine once the pustular psoriasis was significantly better.
After a month, the patient was off of cyclosporine completely. Her skin was clear while using daily acitretin. Monitoring of lab tests did not show any adverse effects of the patient using these 2 potentially toxic medications.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Psoriasis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 878-895.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
Epidemiology of meningitis and encephalitis in the United States
Clinical Question: What is the epidemiology of meningitis and encephalitis in adults in the United States?
Background: Previous epidemiologic studies have been smaller with less clinical information available and without steroid usage rates.
Study Design: A retrospective database review.
Setting: The Premier HealthCare Database, including hospitals of all types and sizes.

Synopsis: Of patients aged 18 or older, 26,429 were included with a primary or secondary discharge diagnosis of meningitis or encephalitis from 2011-2014. Enterovirus was the most common infectious cause (51%), followed by unknown etiology (19%), bacterial (14%), herpetic (8%), fungal (3%), and arboviruses (1%). Of patients, 4.2% had HIV.
Steroids were given on the first day of antibiotics in 25.9%. The only statistical mortality benefit was found with steroid use in pneumococcal meningitis (6.7% vs. 12.5%; P = .0245), with a trend toward increased mortality for steroids in fungal meningitis.
Of patients, 87.2% were admitted through the ED, though 22.5% of lumbar punctures were done after admission and 77.4% were discharged home.
Bottom Line: Enterovirus was the most common cause of adult meningoencephalitis, and patients with pneumococcal meningitis who received steroids had decreased mortality.
Citation: Hasbun R, Ning R, Balada-Llasat JM, Chung J, Duff S, Bozzette S, et al. Meningitis and encephalitis in the United States from 2011-2014. Published online, Apr 17, 2017. Clin Infect Dis. 2017. doi: 10.1093/cid/cix319.
Dr. Hall is an assistant professor in the University of Kentucky division of hospital medicine and pediatrics.
Clinical Question: What is the epidemiology of meningitis and encephalitis in adults in the United States?
Background: Previous epidemiologic studies have been smaller with less clinical information available and without steroid usage rates.
Study Design: A retrospective database review.
Setting: The Premier HealthCare Database, including hospitals of all types and sizes.
Synopsis: Of patients aged 18 or older, 26,429 were included with a primary or secondary discharge diagnosis of meningitis or encephalitis from 2011-2014. Enterovirus was the most common infectious cause (51%), followed by unknown etiology (19%), bacterial (14%), herpetic (8%), fungal (3%), and arboviruses (1%). Of patients, 4.2% had HIV.
Steroids were given on the first day of antibiotics in 25.9%. The only statistical mortality benefit was found with steroid use in pneumococcal meningitis (6.7% vs. 12.5%; P = .0245), with a trend toward increased mortality for steroids in fungal meningitis.
Of patients, 87.2% were admitted through the ED, though 22.5% of lumbar punctures were done after admission and 77.4% were discharged home.
Bottom Line: Enterovirus was the most common cause of adult meningoencephalitis, and patients with pneumococcal meningitis who received steroids had decreased mortality.
Citation: Hasbun R, Ning R, Balada-Llasat JM, Chung J, Duff S, Bozzette S, et al. Meningitis and encephalitis in the United States from 2011-2014. Published online, Apr 17, 2017. Clin Infect Dis. 2017. doi: 10.1093/cid/cix319.
Dr. Hall is an assistant professor in the University of Kentucky division of hospital medicine and pediatrics.
Clinical Question: What is the epidemiology of meningitis and encephalitis in adults in the United States?
Background: Previous epidemiologic studies have been smaller with less clinical information available and without steroid usage rates.
Study Design: A retrospective database review.
Setting: The Premier HealthCare Database, including hospitals of all types and sizes.
Synopsis: Of patients aged 18 or older, 26,429 were included with a primary or secondary discharge diagnosis of meningitis or encephalitis from 2011-2014. Enterovirus was the most common infectious cause (51%), followed by unknown etiology (19%), bacterial (14%), herpetic (8%), fungal (3%), and arboviruses (1%). Of patients, 4.2% had HIV.
Steroids were given on the first day of antibiotics in 25.9%. The only statistical mortality benefit was found with steroid use in pneumococcal meningitis (6.7% vs. 12.5%; P = .0245), with a trend toward increased mortality for steroids in fungal meningitis.
Of patients, 87.2% were admitted through the ED, though 22.5% of lumbar punctures were done after admission and 77.4% were discharged home.
Bottom Line: Enterovirus was the most common cause of adult meningoencephalitis, and patients with pneumococcal meningitis who received steroids had decreased mortality.
Citation: Hasbun R, Ning R, Balada-Llasat JM, Chung J, Duff S, Bozzette S, et al. Meningitis and encephalitis in the United States from 2011-2014. Published online, Apr 17, 2017. Clin Infect Dis. 2017. doi: 10.1093/cid/cix319.
Dr. Hall is an assistant professor in the University of Kentucky division of hospital medicine and pediatrics.
Remediation for surgical trainees may lower attrition
Remediation programs and program director attitudes can make the difference in attrition rates among general surgery residents, according to a survey-based study.
A study by the Association of American Medical Colleges, projects a shortage of 29,000 general surgeons by 2030. Some residency programs are taking steps lower program dropout rates, which has been reported as high as 26% in some programs, according to Alexander Schwed, MD, general surgeon at Harbor–University of California, Los Angeles Medical Center.

Dr. Schwed and his colleagues conducted a survey of 21 general surgery residency program directors. In those programs, the overall attrition rate was found to be much lower than expected – 8.8% over a 5-year period (JAMA Surg. 2017 Aug 16. doi: 10.1001/jamasurg.2017.2656).
The survey showed that programs that implemented resident remediation had lower attrition rates, (21.0% vs 6.8%; P less than .001).
“The association between increased use of remediation by residency programs and low rates of resident attrition is novel,” the investigators wrote. “Nevertheless, based on our findings, high-attrition programs could lower their attrition rates through the increased use of resident remediation and increased focus on resident education.”
Both high- and low-attrition programs selected to participate in the study showed relatively similar median numbers of residents, with low-attrition programs reporting a median of 28 participants per year, and high-attrition programs reporting with 35.
Other similarities between low- and high-attrition programs include percentage of female and minority residents, median of 33.3% and 39.8% respectively, and the number of cases performed by first-, second-, and third-year residents.
The other difference between the six low-attrition programs and the five high-attrition programs was the attitude of the program directors regarding their role in the training of residents, according to researchers.
Investigators asked directors a series of questions using a Likert scale with 1 representing “strongly disagree” and 4 representing “strongly agree.”
Program directors from high- and low-attrition programs tended to agree strongly (scoring 3.8 and 3.2, respectively) with the statement that one of their main roles as a program leader was to “redirect residents who should not be surgeons.”
When asked whether “some degree of resident attrition is a necessary phenomenon,” directors from low-attrition programs scored 2.2, while those from high-attrition programs indicated stronger agreement with an overall score of 3.2.
Directors from programs with high dropout rates were also more likely to consider a 6% dropout rate to be too low, compared with directors from low-attrition programs who thought it was too high.
“When we recruit residents, we are very careful to recruit those who seem to buy into our mission, our vision, and our ideals and fit in well with our culture,” said Sharmila Dissanaike, MD, FACS, department of surgery chair at Texas Tech University Health Sciences Center, Lubbock, in an interview. “We emphasize teamwork, collegiality, and an ‘all for one and one for all’ type of mentality.”
This kind of recruitment includes having current residents be a part of the process, Dr. Dissanaike explained, and encouraging current and potential residents to have an informal dinner to get to know one another better.
For the department of surgery at Texas Tech, the collaborative culture combined with a remediation program has resulted in a drop in attrition from 20% down to 7% in recent years, Dr. Dissanaike said. In addition, the current success of her program can be partly attributed to a recent decision to maintain the number of incoming residents at five, she said.*
Larger programs can achieve similar improvement, she noted and the rising demand for surgeons makes it essential to find a solution that incorporates the benefits of both types of programs.
“We need more surgeons, we need more Graduate Medical Education spots, we need more training spots for general surgeons,” said Dr. Dissanaike. “I think within those large programs we need to find ways to structure smaller groups, maybe little pods, to help support residents so they don’t get lost.”
Dr. Schwed and his colleagues expressed concern that institutional barriers, such as the focus on test scores, may impede directors from embracing remediation.
“Greater emphasis on the written and oral General Surgery Qualifying Examination pass rates, which are now publicly posted and used by residency review committees, will likely exert pressure on program directors, who may fear that attempting to remediate a resident with poor medical knowledge may affect their program’s 5-year board pass rates,” the investigators wrote. “Our study suggests that such fears may be unfounded because programs with high levels of remediation and low attrition had similar board pass rates as those with high attrition.”
Dr. Schwed and his coinvestigators acknowledged that the programs studied may not be representative of U.S. residencies and selection bias may have affected the findings.
Researchers reported no relevant financial disclosures.
*Correction, 10/26/17: An earlier version of this article misstated the number of incoming residents in the program.
[email protected]
On Twitter @eaztweets
Remediation programs and program director attitudes can make the difference in attrition rates among general surgery residents, according to a survey-based study.
A study by the Association of American Medical Colleges, projects a shortage of 29,000 general surgeons by 2030. Some residency programs are taking steps lower program dropout rates, which has been reported as high as 26% in some programs, according to Alexander Schwed, MD, general surgeon at Harbor–University of California, Los Angeles Medical Center.
Dr. Schwed and his colleagues conducted a survey of 21 general surgery residency program directors. In those programs, the overall attrition rate was found to be much lower than expected – 8.8% over a 5-year period (JAMA Surg. 2017 Aug 16. doi: 10.1001/jamasurg.2017.2656).
The survey showed that programs that implemented resident remediation had lower attrition rates, (21.0% vs 6.8%; P less than .001).
“The association between increased use of remediation by residency programs and low rates of resident attrition is novel,” the investigators wrote. “Nevertheless, based on our findings, high-attrition programs could lower their attrition rates through the increased use of resident remediation and increased focus on resident education.”
Both high- and low-attrition programs selected to participate in the study showed relatively similar median numbers of residents, with low-attrition programs reporting a median of 28 participants per year, and high-attrition programs reporting with 35.
Other similarities between low- and high-attrition programs include percentage of female and minority residents, median of 33.3% and 39.8% respectively, and the number of cases performed by first-, second-, and third-year residents.
The other difference between the six low-attrition programs and the five high-attrition programs was the attitude of the program directors regarding their role in the training of residents, according to researchers.
Investigators asked directors a series of questions using a Likert scale with 1 representing “strongly disagree” and 4 representing “strongly agree.”
Program directors from high- and low-attrition programs tended to agree strongly (scoring 3.8 and 3.2, respectively) with the statement that one of their main roles as a program leader was to “redirect residents who should not be surgeons.”
When asked whether “some degree of resident attrition is a necessary phenomenon,” directors from low-attrition programs scored 2.2, while those from high-attrition programs indicated stronger agreement with an overall score of 3.2.
Directors from programs with high dropout rates were also more likely to consider a 6% dropout rate to be too low, compared with directors from low-attrition programs who thought it was too high.
“When we recruit residents, we are very careful to recruit those who seem to buy into our mission, our vision, and our ideals and fit in well with our culture,” said Sharmila Dissanaike, MD, FACS, department of surgery chair at Texas Tech University Health Sciences Center, Lubbock, in an interview. “We emphasize teamwork, collegiality, and an ‘all for one and one for all’ type of mentality.”
This kind of recruitment includes having current residents be a part of the process, Dr. Dissanaike explained, and encouraging current and potential residents to have an informal dinner to get to know one another better.
For the department of surgery at Texas Tech, the collaborative culture combined with a remediation program has resulted in a drop in attrition from 20% down to 7% in recent years, Dr. Dissanaike said. In addition, the current success of her program can be partly attributed to a recent decision to maintain the number of incoming residents at five, she said.*
Larger programs can achieve similar improvement, she noted and the rising demand for surgeons makes it essential to find a solution that incorporates the benefits of both types of programs.
“We need more surgeons, we need more Graduate Medical Education spots, we need more training spots for general surgeons,” said Dr. Dissanaike. “I think within those large programs we need to find ways to structure smaller groups, maybe little pods, to help support residents so they don’t get lost.”
Dr. Schwed and his colleagues expressed concern that institutional barriers, such as the focus on test scores, may impede directors from embracing remediation.
“Greater emphasis on the written and oral General Surgery Qualifying Examination pass rates, which are now publicly posted and used by residency review committees, will likely exert pressure on program directors, who may fear that attempting to remediate a resident with poor medical knowledge may affect their program’s 5-year board pass rates,” the investigators wrote. “Our study suggests that such fears may be unfounded because programs with high levels of remediation and low attrition had similar board pass rates as those with high attrition.”
Dr. Schwed and his coinvestigators acknowledged that the programs studied may not be representative of U.S. residencies and selection bias may have affected the findings.
Researchers reported no relevant financial disclosures.
*Correction, 10/26/17: An earlier version of this article misstated the number of incoming residents in the program.
[email protected]
On Twitter @eaztweets
Remediation programs and program director attitudes can make the difference in attrition rates among general surgery residents, according to a survey-based study.
A study by the Association of American Medical Colleges, projects a shortage of 29,000 general surgeons by 2030. Some residency programs are taking steps lower program dropout rates, which has been reported as high as 26% in some programs, according to Alexander Schwed, MD, general surgeon at Harbor–University of California, Los Angeles Medical Center.
Dr. Schwed and his colleagues conducted a survey of 21 general surgery residency program directors. In those programs, the overall attrition rate was found to be much lower than expected – 8.8% over a 5-year period (JAMA Surg. 2017 Aug 16. doi: 10.1001/jamasurg.2017.2656).
The survey showed that programs that implemented resident remediation had lower attrition rates, (21.0% vs 6.8%; P less than .001).
“The association between increased use of remediation by residency programs and low rates of resident attrition is novel,” the investigators wrote. “Nevertheless, based on our findings, high-attrition programs could lower their attrition rates through the increased use of resident remediation and increased focus on resident education.”
Both high- and low-attrition programs selected to participate in the study showed relatively similar median numbers of residents, with low-attrition programs reporting a median of 28 participants per year, and high-attrition programs reporting with 35.
Other similarities between low- and high-attrition programs include percentage of female and minority residents, median of 33.3% and 39.8% respectively, and the number of cases performed by first-, second-, and third-year residents.
The other difference between the six low-attrition programs and the five high-attrition programs was the attitude of the program directors regarding their role in the training of residents, according to researchers.
Investigators asked directors a series of questions using a Likert scale with 1 representing “strongly disagree” and 4 representing “strongly agree.”
Program directors from high- and low-attrition programs tended to agree strongly (scoring 3.8 and 3.2, respectively) with the statement that one of their main roles as a program leader was to “redirect residents who should not be surgeons.”
When asked whether “some degree of resident attrition is a necessary phenomenon,” directors from low-attrition programs scored 2.2, while those from high-attrition programs indicated stronger agreement with an overall score of 3.2.
Directors from programs with high dropout rates were also more likely to consider a 6% dropout rate to be too low, compared with directors from low-attrition programs who thought it was too high.
“When we recruit residents, we are very careful to recruit those who seem to buy into our mission, our vision, and our ideals and fit in well with our culture,” said Sharmila Dissanaike, MD, FACS, department of surgery chair at Texas Tech University Health Sciences Center, Lubbock, in an interview. “We emphasize teamwork, collegiality, and an ‘all for one and one for all’ type of mentality.”
This kind of recruitment includes having current residents be a part of the process, Dr. Dissanaike explained, and encouraging current and potential residents to have an informal dinner to get to know one another better.
For the department of surgery at Texas Tech, the collaborative culture combined with a remediation program has resulted in a drop in attrition from 20% down to 7% in recent years, Dr. Dissanaike said. In addition, the current success of her program can be partly attributed to a recent decision to maintain the number of incoming residents at five, she said.*
Larger programs can achieve similar improvement, she noted and the rising demand for surgeons makes it essential to find a solution that incorporates the benefits of both types of programs.
“We need more surgeons, we need more Graduate Medical Education spots, we need more training spots for general surgeons,” said Dr. Dissanaike. “I think within those large programs we need to find ways to structure smaller groups, maybe little pods, to help support residents so they don’t get lost.”
Dr. Schwed and his colleagues expressed concern that institutional barriers, such as the focus on test scores, may impede directors from embracing remediation.
“Greater emphasis on the written and oral General Surgery Qualifying Examination pass rates, which are now publicly posted and used by residency review committees, will likely exert pressure on program directors, who may fear that attempting to remediate a resident with poor medical knowledge may affect their program’s 5-year board pass rates,” the investigators wrote. “Our study suggests that such fears may be unfounded because programs with high levels of remediation and low attrition had similar board pass rates as those with high attrition.”
Dr. Schwed and his coinvestigators acknowledged that the programs studied may not be representative of U.S. residencies and selection bias may have affected the findings.
Researchers reported no relevant financial disclosures.
*Correction, 10/26/17: An earlier version of this article misstated the number of incoming residents in the program.
[email protected]
On Twitter @eaztweets
FROM JAMA SURGERY
Key clinical point:
Major finding: Of the 21 programs surveyed, there was an average attrition rate of 8.8% over 5 years.
Data source: Survey of 21 general surgery residency program directors between July 2010 and June 2015.
Disclosures: Investigators report no relevant financial disclosures.
Practicing medicine for all, regardless of differences
I’m a doctor, specifically a neurologist.
I’m also a father with three kids.
I’m also a small business owner and part of the American economy. My practice is small, but provides jobs to two awesome women and in doing so allows them to have insurance, raise their families with job security, own homes, and contribute to the economy. I’m not required to by law, but I provide both with insurance coverage and a retirement plan. I pay my taxes on time and to the penny.
I’m a third-generation American, and a first-generation native Arizonan.
I’m a Phoenix Suns, ASU Sun Devils, and Creighton Bluejays basketball fan.
And, somewhere in all of the above, I’m Jewish.
I’ve never understood hate very well. To me, people are people. I’ve never treated patients differently based on race, religion, political beliefs, or pretty much any other factor. That’s part of my job, and I wouldn’t have it any other way.
For the same reason, I don’t understand anti-Semitism. I’ve never ripped anyone off and try very hard to practice ethical medicine, doing what’s right for patients and not for my pocketbook. Some could even argue that this approach has cost me financially over time.
My first direct experience with hate was in 1975, when my family moved from central Phoenix to the suburbs. When we were building our house and meeting future neighbors, one lady circulated a petition to keep Jews out of the neighborhood. A few weeks later, when the school year started, her kids looked me and my sister over and asked us where our horns were.
I don’t encounter it, at least not directly, as much anymore. Perhaps one to two times a year someone will call my office to make an appointment and will ask what my religion is. My secretary tells them that we don’t discuss this professionally here.
But it never goes away entirely. There are always those looking to blame anyone who is slightly different from them for economic and social changes, perhaps because it’s easier than actually working together to solve things. Or because they find it a welcome distraction from the real issues facing our society.
The recent events in Charlottesville are frightening to all of us, regardless of religion, who are trying to get along in everyday life. All I’ve ever wanted is to be able to work and raise my family in peace, yet we’re faced with a stark reminder of those who see this as a threat. Worse, their fires are stoked by seeming indifference (at best) and overt support (at worst) at the highest level of our government – one founded on freedom of religion.
Hate is hate, whether it’s ISIS, the Westboro Baptist Church, KKK, Kahane Chai, or the modern interpretations of Nazism lurking in Europe and America. Although they’ve always been there, today the Internet has given them a larger voice. People whom I’ve never done anything to consider me an enemy.
My kids’ school is a block from a mosque, so I drive by it all the time. It’s an attractive, well-maintained building. Sometimes I see younger kids running around out in the yard, or others playing basketball on a court between buildings. Its proximity has never bothered me. But, like myself, I know those inside are hated by others who don’t even know them. Like me, all they’ve done is raise kids, work, and pay taxes.
There have always been, and will always be, bad people in all religions, races, and ethnic groups. This is the nature of humans. But the association of hating all because of a few is very troubling. I believe the majority of people are good and that none are born hating others.
I try hard to run a blind practice: treating all patients as equal, and giving them the best care I can, regardless of who they are, what they believe, or where they’re from.
Unfortunately, too many people seem to find it easier to slap labels on anyone who doesn’t look or think like them, and decide that’s all they need to hate them and avoid looking at the person inside.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
I’m a doctor, specifically a neurologist.
I’m also a father with three kids.
I’m also a small business owner and part of the American economy. My practice is small, but provides jobs to two awesome women and in doing so allows them to have insurance, raise their families with job security, own homes, and contribute to the economy. I’m not required to by law, but I provide both with insurance coverage and a retirement plan. I pay my taxes on time and to the penny.
I’m a third-generation American, and a first-generation native Arizonan.
I’m a Phoenix Suns, ASU Sun Devils, and Creighton Bluejays basketball fan.
And, somewhere in all of the above, I’m Jewish.
I’ve never understood hate very well. To me, people are people. I’ve never treated patients differently based on race, religion, political beliefs, or pretty much any other factor. That’s part of my job, and I wouldn’t have it any other way.
For the same reason, I don’t understand anti-Semitism. I’ve never ripped anyone off and try very hard to practice ethical medicine, doing what’s right for patients and not for my pocketbook. Some could even argue that this approach has cost me financially over time.
My first direct experience with hate was in 1975, when my family moved from central Phoenix to the suburbs. When we were building our house and meeting future neighbors, one lady circulated a petition to keep Jews out of the neighborhood. A few weeks later, when the school year started, her kids looked me and my sister over and asked us where our horns were.
I don’t encounter it, at least not directly, as much anymore. Perhaps one to two times a year someone will call my office to make an appointment and will ask what my religion is. My secretary tells them that we don’t discuss this professionally here.
But it never goes away entirely. There are always those looking to blame anyone who is slightly different from them for economic and social changes, perhaps because it’s easier than actually working together to solve things. Or because they find it a welcome distraction from the real issues facing our society.
The recent events in Charlottesville are frightening to all of us, regardless of religion, who are trying to get along in everyday life. All I’ve ever wanted is to be able to work and raise my family in peace, yet we’re faced with a stark reminder of those who see this as a threat. Worse, their fires are stoked by seeming indifference (at best) and overt support (at worst) at the highest level of our government – one founded on freedom of religion.
Hate is hate, whether it’s ISIS, the Westboro Baptist Church, KKK, Kahane Chai, or the modern interpretations of Nazism lurking in Europe and America. Although they’ve always been there, today the Internet has given them a larger voice. People whom I’ve never done anything to consider me an enemy.
My kids’ school is a block from a mosque, so I drive by it all the time. It’s an attractive, well-maintained building. Sometimes I see younger kids running around out in the yard, or others playing basketball on a court between buildings. Its proximity has never bothered me. But, like myself, I know those inside are hated by others who don’t even know them. Like me, all they’ve done is raise kids, work, and pay taxes.
There have always been, and will always be, bad people in all religions, races, and ethnic groups. This is the nature of humans. But the association of hating all because of a few is very troubling. I believe the majority of people are good and that none are born hating others.
I try hard to run a blind practice: treating all patients as equal, and giving them the best care I can, regardless of who they are, what they believe, or where they’re from.
Unfortunately, too many people seem to find it easier to slap labels on anyone who doesn’t look or think like them, and decide that’s all they need to hate them and avoid looking at the person inside.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
I’m a doctor, specifically a neurologist.
I’m also a father with three kids.
I’m also a small business owner and part of the American economy. My practice is small, but provides jobs to two awesome women and in doing so allows them to have insurance, raise their families with job security, own homes, and contribute to the economy. I’m not required to by law, but I provide both with insurance coverage and a retirement plan. I pay my taxes on time and to the penny.
I’m a third-generation American, and a first-generation native Arizonan.
I’m a Phoenix Suns, ASU Sun Devils, and Creighton Bluejays basketball fan.
And, somewhere in all of the above, I’m Jewish.
I’ve never understood hate very well. To me, people are people. I’ve never treated patients differently based on race, religion, political beliefs, or pretty much any other factor. That’s part of my job, and I wouldn’t have it any other way.
For the same reason, I don’t understand anti-Semitism. I’ve never ripped anyone off and try very hard to practice ethical medicine, doing what’s right for patients and not for my pocketbook. Some could even argue that this approach has cost me financially over time.
My first direct experience with hate was in 1975, when my family moved from central Phoenix to the suburbs. When we were building our house and meeting future neighbors, one lady circulated a petition to keep Jews out of the neighborhood. A few weeks later, when the school year started, her kids looked me and my sister over and asked us where our horns were.
I don’t encounter it, at least not directly, as much anymore. Perhaps one to two times a year someone will call my office to make an appointment and will ask what my religion is. My secretary tells them that we don’t discuss this professionally here.
But it never goes away entirely. There are always those looking to blame anyone who is slightly different from them for economic and social changes, perhaps because it’s easier than actually working together to solve things. Or because they find it a welcome distraction from the real issues facing our society.
The recent events in Charlottesville are frightening to all of us, regardless of religion, who are trying to get along in everyday life. All I’ve ever wanted is to be able to work and raise my family in peace, yet we’re faced with a stark reminder of those who see this as a threat. Worse, their fires are stoked by seeming indifference (at best) and overt support (at worst) at the highest level of our government – one founded on freedom of religion.
Hate is hate, whether it’s ISIS, the Westboro Baptist Church, KKK, Kahane Chai, or the modern interpretations of Nazism lurking in Europe and America. Although they’ve always been there, today the Internet has given them a larger voice. People whom I’ve never done anything to consider me an enemy.
My kids’ school is a block from a mosque, so I drive by it all the time. It’s an attractive, well-maintained building. Sometimes I see younger kids running around out in the yard, or others playing basketball on a court between buildings. Its proximity has never bothered me. But, like myself, I know those inside are hated by others who don’t even know them. Like me, all they’ve done is raise kids, work, and pay taxes.
There have always been, and will always be, bad people in all religions, races, and ethnic groups. This is the nature of humans. But the association of hating all because of a few is very troubling. I believe the majority of people are good and that none are born hating others.
I try hard to run a blind practice: treating all patients as equal, and giving them the best care I can, regardless of who they are, what they believe, or where they’re from.
Unfortunately, too many people seem to find it easier to slap labels on anyone who doesn’t look or think like them, and decide that’s all they need to hate them and avoid looking at the person inside.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Fueling the Alzheimer’s brain with fat
LONDON – A 3-month diet comprised of 70% fat improved cognition in Alzheimer’s disease patients better than any anti-amyloid drug that has ever been tested.
In a small pilot study, Alzheimer’s patients who followed the University of Kansas’s ketogenic diet program improved an average of 4 points on one of the most important cognitive assessments in dementia care, the Alzheimer’s Disease Assessment Scale–cognitive domain (ADAS-cog). Not only was this gain statistically significant, but it reached a level that clinical trialists believe to be clinically meaningful, and it was similar to the gains that won Food and Drug Administration approval for donepezil in 1996, according to Russell Swerdlow, MD, director of the University of Kansas Alzheimer’s Disease Center in Fairway.

To put the results in perspective, donepezil was approved on a 4-point spread between the active and placebo arm over 3 months, said Dr. Swerdlow, who is also the Gene and Marge Sweeney Professor of Neurology at the university. Part of this difference was driven by a 2-point decline in the placebo group. Relative to its baseline, the treatment group improved, on average, by about 2 points.
But in the Ketogenic Diet Retention and Feasibility Trail (KDRAFT), also 3 months long, patients’ ADAS-cog scores didn’t decline at all. Everyone who stayed with the diet and kept on their baseline medications improved, although to varying degrees.
KDRAFT was very small, with just 10 patients completing the intervention, and lacked a comparator group, so the results should be interpreted extremely cautiously, Dr. Swerdlow said in an interview. “We have to very careful about overinterpreting these findings. It’s a pilot study, and a small group, so we don’t know how genuine the finding is. But if it is true, it’s a big deal.”
Diet and dementia
Emerging evidence suggests that modifying diet can help prevent Alzheimer’s and may even help AD patients think and function better. But this research has largely focused on the heart-healthy diets already proven successful in preventing and treating hypertension, diabetes, and cardiovascular disease. Most notably, the Mediterranean-DASH Intervention for Neurodegenerative Delay (MIND) diet cut the risk of AD by up to 53% (Alzheimers Dement. 2015 Sep;11[9]:1007-14) and also slowed aging-related cognitive decline (Alzheimers Dement. 2015 Sep; 11[9]:1015-22).
MIND is a combination of the low-salt, plant-focused DASH diet, and the heart-healthy Mediterranean diet. It is a moderate-fat plan, with a ratio of 33% fat, 38% carbohydrates, and 26% protein. Ideally, only 3% of the fat should be saturated, so MIND draws on olive oil, nuts, and other foods with monounsaturated fats, largely eschewing animal fats. It’s generally considered fairly easy to follow, since it allows a wide variety of whole grains, beans, nuts, fruits, vegetables, salads, fish, and poultry. Butter, red meat, fried foods, full-fat dairy, and fast foods are strict no-nos.
A ketogenic diet, however, turns MIND on its head. With a 70% fat, 20% protein, 10% carbohydrate ratio, a typical ketogenic diet nearly eliminates most fruits, and virtually all starchy vegetables, beans, and grains. It does, however, incorporate a large amount of fat from many sources, including olive oil, butter, cream, eggs, nuts, all kinds of meat, and fish. For a ketogenic diet, Dr. Swerdlow said, the ratio of fat to protein and carbs is more critical than the source of the fat.
MIND was designed to prevent the cardiovascular and endocrine disorders than predispose to dementia over the long term. But a ketogenic diet for patients with Alzheimer’s acutely manipulates the brain’s energy metabolism system, forcing it to use ketone bodies instead of glucose for fuel.
In normal energy metabolism, carbohydrates provide a ready supply of glucose, the brain’s primary fuel. When carbs are limited or absent, serum insulin decreases and glucagon increases. This promotes lipolysis. Ketones (primarily beta-hydroxybutyrate and acetoacetate) are formed in the liver from the newly released fatty acids, and released into the circulation, including into the brain during times of decreased glucose availability – a state characteristic of Alzheimer’s disease.
Induced ketogenesis trial
Inducing ketosis through diet seems to help correct the normal, age-related decline in the brain’s ability to use glucose, said Stephen Cunnane, PhD, who also presented ketogenic intervention results at AAIC. “Cognitively normal, healthy older adults experience a 10% reduction in the brain’s ability to metabolize glucose compared to healthy young people,” he said in an interview. But this decline accelerates as Alzheimer’s hits. Those with early AD have a 20% decrement in glucose utilization, compared with healthy elders.

Changes in brain glucose metabolism can develop years before any cognitive symptoms manifest and seem to increase the risk of Alzheimer’s, said Dr. Cunnane of Sherbrooke University, Que.
“We propose that this vicious cycle of presymptomatic glucose hypometabolism causes chronic brain energy deprivation, and might contribute to deteriorating neuronal function. That could cause a further decrease in the demand for glucose, leading to cognitive decline.”
“What doesn’t change, though, is the brain’s ability to take up ketone bodies,” he said. If anything, the brain appears to use ketones more efficiently as AD becomes established. “It’s almost like the brain is trying to rescue itself. If those cells were dead, they would not be able to take up ketones. Because they do, we think they are instead starving because of their inability to use glucose and that maybe we can rescue them with ketones before they die.”
At AAIC, Dr. Cunnane reported interim results of an investigation of induced ketogenesis in patients with mild cognitive impairment (MCI). The 6-month BENEFIC trial comprises 50 patients, randomized to either a daily nutritional supplement with 30 g medium chain triglycerides (MCT) in a unflavored, nondairy emulsion, or a fat-equivalent placebo drink. When consumed, the liver very quickly converts MCT fatty acids into ketone bodies, which then circulate throughout the body, including passing the blood-brain barrier.
All of the participants in the BENEFIC trial underwent brain PET scanning for both glucose and ketone uptake. Early results clearly showed that the MCI brains took up just as much acetoacetate as did the brains of cognitively normal young adults. And although the study wasn’t powered for a full cognitive assessment, Dr. Cunnane did present 6-month data on three measures in the MCI group: trail making time, verbal fluency, and the Boston Naming Test. In the active group on MCT, scores on all three measures improved “modestly” in direct correlation with brain ketone uptake. In the placebo group, scores remained unchanged.
“We don’t have enough people in the study to make any definitive statement about cognition, but it’s nice to see the trend going in the right direction, Dr. Cunnane said. “I really think of this as a dose-finding study and a chance to demonstrate the safety and tolerability of a liquid MCT supplement in people with MCI. Our next study will use a 45 g per day supplement of MCT.”
Details of the KDRAFT study
The BENEFIC study looked only at the effects of an MCT supplement, which may not deliver all the metabolic benefits of a ketogenic diet. KDRAFT, however, employed both, and assessed not only cognitive outcomes and adverse effects, but the practical matter of whether AD patients and their caregivers could implement the diet and stick to it.
Couples recruited into the trial met with a dietitian who explained the importance of sticking with the strict fat:carb:protein ratio. It’s not easy to stay in that zone, Dr. Swerdlow said, and the MCT supplement really helps there.
“Adding the MCT, which is typically done for the ketogenic diet in epilepsy, increases the fat intake so you can tolerate a bit more carbohydrate and still remain in ketosis. MCT therefore makes it easier to successfully do the diet, if we define success by time in ketosis. Ultimately, it is an iterative diet. You check your urine, and if you are in ketosis, you are doing well. If you are not in ketosis, you have to increase your fat intake, decrease your carb intake, or both.”
The study comprised 15 patients (7 with very mild AD, 4 with mild, and 4 with moderate disease). All patients were instructed to remain on their current medications for Alzheimer’s disease for the duration of the study if they were taking any. All of the patients with moderate AD and one with very mild AD dropped out of the study within the first month, citing caregiver burden. The supplement was in the form of an oil, not an emulsion like the BENEFIC supplement, and it caused diarrhea and nausea in five subjects, although none discontinued because of that.
“We found that a slow titration of the oil could deal with the GI issues. Rather, the primary deal-breaker seemed to be the stress of planning the menus and preparing the meals.”
One patient discontinued his cholinesterase inhibitor during the study, for unknown reasons. His cognitive scores declined, but was still included in the diet-compliant analysis.
The diet didn’t affect weight, blood pressure, insulin sensitivity or resistance, or glucose level, but the intervention was short-lived. Nor were there any significant changes in high-density, low-density, or total cholesterol. Liver enzymes were stable, too.
“The only thing that changed was that they really did increase their fat and decrease their carb intake,” Dr. Swerdlow said. Daily fat jumped from 91 g to 167 g, and carbs dropped from 201 g to 46 g.
Almost everyone who stuck with the diet achieved and maintained ketosis during the study, although with varying degrees of success. “Many only had a trace amount of urinary ketones,” Dr. Swerdlow said. The investigators tracked serum beta hydroxybutyrate levels every month as well, and those measures also confirmed ketosis in the group as a whole, although some patients fluctuated in and out of the state.
The cognitive changes were striking, he said. In the 10-patient analysis, ADAS-cog scores improved by an average of 4.1 points. The results were better when Dr. Swerdlow excluded the patient who stopped his cholinesterase inhibitor medication. In that nine-patient group, the ADAS-cog improved an average of 5.3 points.
While urging caution over the small sample size and lack of a control comparator, Dr. Swerdlow expressed deep satisfaction over the outcomes. A clinician as well as a researcher, he is accustomed to the slow but inexorable decline of AD patients.
“I’m going to try to relate the impression you get in the clinic with these scores,” he said. “Very rarely, but sometimes, with a cholinesterase inhibitor in patients, we’ll see something like a 7-point change. That’s a fantastic response, an improvement you can see across the room. A change of 2 points really doesn’t look that much different, although caregivers will tell you there is a subtle change, maybe a little more focus. The average we got in our 10 subjects was a 4-point improvement. That’s impressive. And a 5-point change is like rolling the clock back by a year.”
The improvements didn’t last, though. A 1-month washout period followed the intervention. By the end, both ADAS-cog and Mini-Mental State Examination scores had returned to their baseline levels. At the end of the study, a few of the patients and their partners expressed their intent to resume the diet, but the investigators do not know whether this indeed happened. Still, the results are encouraging enough that, like Dr. Cunnane, Dr. Swerdlow hopes to conduct a larger, longer study – one that would include a control group.
Future investigations of the ketogenic diet in AD might do well to also include an exercise component, both researchers mentioned. In addition to starvation, ketogenic dieting, and MCT supplementation, exercise is an effective way to induce ketogenesis.
“Exercise produces ketones, but most importantly, it increases the capacity of the brain to use ketones,” Dr. Cunnane said. The connection may help explain some of the cognitive benefits seen in exercise trials in patients with MCI and AD.
“This raises the possibility that if in fact exercise benefits the brain, ketone bodies may mediate some of that effect,” Dr. Swerdlow said. “Could exercise potentiate the ketosis from the diet? That is possible, and maybe using these interventions in conjunction would be synergistic. At this point, we are just happy to show the diet is feasible, if even for a limited period.”
Implementing KDRAFT: Research team dishes the skinny on fats
The KDRAFT study diet is surprisingly flexible despite its strict ratio of fat to protein and carbohydrate, according to the University of Kansas research team that implemented it. It only took a few counseling sessions to get most study participants enthusiastically embracing the new eating plan, even one so radically different from the way they were accustomed to eating.
“We focused mainly on the macronutrient makeup,” said Matthew Taylor, PhD, who supervised the diet study on a day-to-day basis. Instead of distributing a rigid diet plan, with prespecified meals and snacks, “We talked more in general about foods they could have and foods they couldn’t have.”
“When people think ‘ketogenic,’ they think bacon, eggs, oil, butter and cream, and may have an automatic negative connotation that this is unhealthy eating,” Dr. Taylor said in an interview. “But yes, eggs were in there and, because a lot of people really like bacon, there was bacon, too!”
The educational sessions did include teaching about healthy and unhealthy fats, and Dr. Taylor “tried to steer people toward the healthier ones, like olive oil, avocados, and nuts. But I didn’t say, ‘Eat this one and not that one.’ If it took melting butter on vegetables to get to that fat ratio, I was not as concerned about where the fat came from as about getting there and maintaining ketosis.”
KDRAFT also had a twist that’s becoming more common among ketogenic eating plans: lots of vegetables. Dr. Taylor asked participants to concentrate on nonstarchy vegetables and forgo potatoes, corn, beans, and lima beans, although some people did enjoy peas occasionally.
“We used to be think we had to restrict vegetables or people would go out of ketosis more easily. But that doesn’t seem to be true. We focused a lot on eating vegetables, and everyone increased their vegetable intake dramatically. We actually tried to use vegetables as a vehicle for fat. For example, people would roast Brussels sprouts or broccoli in olive oil and then put melted butter on it. It was pretty much, ‘Eat all the vegetables you can and put fat on them.’”
Fruits are full of sugar, so they are not liberally used in most ketogenic diets, but KDRAFT did allow one type: berries, and blueberries in particular. “We had people eating a couple of small handfuls of berries throughout the day and still being able to maintain ketosis. We did severely cut back on the amount and type of fruit people could have, but berries seemed to work well.”
Whipping cream had a place, too. “It fit really well in the diet, because it’s basically all fat,” Dr. Taylor said. “It’s used more often in pediatric ketogenic diets as a milk substitute. One thing our subjects liked to do was use it to make a sweet snack. All it takes is a packet of [stevia] sweetener and some vanilla. Then you whip and freeze it and it’s like an ice cream dessert.”
After the initial drop-outs, the remaining study pairs embraced the intervention enthusiastically.
“When the study partner took the diet on too, we had our best success. One of our last pairs had an entire family join in – children, grandchildren, everyone decided to follow the diet. That is a very helpful piece to this. It’s difficult to always say, ‘Here’s our normal food and here’s the keto food over here.’”
The dropouts occurred very early. These study pairs, all of whom included patients with moderate Alzheimer’s, never embraced the plan at all, and this is a telling point, Dr. Taylor noted.
“When you get to a level of dementia there are so many other things in the caregiving process that taking on big behavioral changes is very difficult.”
Although the study showed that the diet wasn’t practical for sicker patients at home, it still might be beneficial in other settings, said Debra Sullivan, PhD, RD. Dr. Sullivan chairs the department of dietetics and nutrition at the University of Kansas Medical Center and holds the Midwest Dairy Council Endowed Professorship in Clinical Nutrition.
“I think that we might be able to create a version of the diet that could be used in an institutional setting for our more advanced patients,” she said. “But there’s no denying that this can be challenging. It’s a big change from the way the typical American eats.”
None of the KDRAFT participants experienced any lipid changes, for either better or worse. The 3-month intervention was long enough to have picked up such changes if they were in the offing, said principal investigator Russell Swerdlow, MD. While there are mixed data on ketogenic diets’ atherogenic effects, many people respond positively, with improved cholesterol.
“Much of what it comes down to is, are you in a catabolic or anabolic states? Are you building up or tearing down? Excessive cholesterol is a sign of being overfed and laying down energy supplies. You take in carbon and turn it into cholesterol. But if you can trick your body into a catabolic state – essentially make it think it’s starving, which a ketogenic diet does – then you have consistently low insulin levels, and you don’t turn on the cholesterol synthesis pathway. You may increase your cholesterol intake through diet, but you’re not synthesizing it in your body, and that synthesis is what really drives your cholesterol level. If you’re not overeating, your body’s production goes down.”
Brain Energy and Memory (BEAM) study
Dr. Swerdlow isn’t the only clinician researcher looking at how a ketogenic diet might influence cognition. Suzanne Craft, PhD, well known for her investigations of the role of insulin signaling and therapy in AD, is running a ketogenic diet trial as well.
As noted on clinicaltrials.gov, the 24-week Brain Energy and Memory (BEAM) study aimed to recruit 25 subjects in two cohorts: adults with mild memory complaints, and cognitively normal adults with prediabetes. A comparator group of healthy controls will contribute cognitive assessments, blood and stool sample collection, neuroimaging, and lumbar puncture at baseline.
Both active groups will be randomized to 6 weeks of either a low-fat, high-carbohydrate diet, with carbs making up 50%-60% of daily caloric intake, or a modified ketogenic-Mediterranean Diet with carbs comprising less than 10% of daily caloric intake.
BEAM’s primary outcome will be changes in the AD cerebrospinal fluid biomarkers beta-amyloid and tau. Secondary endpoints include cognitive assessments, brain ketone uptake on PET scanning, and insulin sensitivity.
Dr. Cunnane has no financial interest in the MCT emulsion, which was supplied by Abitec. He reported conference travel support from Abitec, Nisshin OilliO, and Pruvit. He also reported receiving research project funding from Nestlé and Bulletproof.
Dr. Swerdlow had no financial disclosures.
[email protected]
On Twitter @alz_gal
In Alzheimer’s disease (AD), there are early significant deficits in glucose utilization that become increasingly severe as disease progresses.
Most reports from early-onset AD animal models find that these energy deficits are largely due to defects in mitochondrial complex IV and V, and possibly related to mitochondrial fusion and fission regulators. Animal models of tauopathy demonstrate Complex I deficits.
In AD-vulnerable brain regions with early glucose utilization deficits, surviving neurons show large reductions in mitochondrial complex I, IV, and V gene expression and proteins. These changes appear sufficient to contribute to cognitive deficits. These are not shared by nondemented individuals, even in the presences of AD pathology.
The precise causes of reduced glucose utilization in AD are unknown, but may reflect these mitochondrial deficits, as well as defective insulin signaling. These changes lead to adenosine triphosphate deficits and disruptions in the balance of NAD+/NADH, both of which are already altered by normal aging.
However, because metabolism is coupled to synaptic activity, it is difficult to ascertain whether these “bioenergetic” deficits are simply secondary to progressive neuron and synapse loss or a contributing factor to neuron and synapse loss and cognitive deficits.
One of the best ways to discern the contribution of bioenergetics deficits is to treat them. Many animal models and some small trials appear to show possible benefits from supplements directed at improving energy metabolism.
In the context of these known deficits in Alzheimer’s, the new positive results with ketogenic diet reported by Dr. Swerdlow should not be ignored despite the small sample size and open-label design with the diet. The impressive 4-5 point increase in ADAS-cog that they saw is not easily achieved, and the rapid loss with washout suggests a real benefit with a large effect size.
Similarly, despite the study’s limitations with dose and size, Dr. Cunnane’s imaging of ketone body uptake and its correlation with cognitive improvement suggests that ameliorating energy deficits can be a real target capable of producing substantial short-term benefits for patients with Alzheimer’s.
Given the rapid results and large effect size, this is an area that needs to see more trials.
Gregory Cole, PhD , is a professor of neurology at the University of California, Los Angeles, and interim director of the Mary S. Easton Alzheimer Center. He had no relevant financial disclosures.
In Alzheimer’s disease (AD), there are early significant deficits in glucose utilization that become increasingly severe as disease progresses.
Most reports from early-onset AD animal models find that these energy deficits are largely due to defects in mitochondrial complex IV and V, and possibly related to mitochondrial fusion and fission regulators. Animal models of tauopathy demonstrate Complex I deficits.
In AD-vulnerable brain regions with early glucose utilization deficits, surviving neurons show large reductions in mitochondrial complex I, IV, and V gene expression and proteins. These changes appear sufficient to contribute to cognitive deficits. These are not shared by nondemented individuals, even in the presences of AD pathology.
The precise causes of reduced glucose utilization in AD are unknown, but may reflect these mitochondrial deficits, as well as defective insulin signaling. These changes lead to adenosine triphosphate deficits and disruptions in the balance of NAD+/NADH, both of which are already altered by normal aging.
However, because metabolism is coupled to synaptic activity, it is difficult to ascertain whether these “bioenergetic” deficits are simply secondary to progressive neuron and synapse loss or a contributing factor to neuron and synapse loss and cognitive deficits.
One of the best ways to discern the contribution of bioenergetics deficits is to treat them. Many animal models and some small trials appear to show possible benefits from supplements directed at improving energy metabolism.
In the context of these known deficits in Alzheimer’s, the new positive results with ketogenic diet reported by Dr. Swerdlow should not be ignored despite the small sample size and open-label design with the diet. The impressive 4-5 point increase in ADAS-cog that they saw is not easily achieved, and the rapid loss with washout suggests a real benefit with a large effect size.
Similarly, despite the study’s limitations with dose and size, Dr. Cunnane’s imaging of ketone body uptake and its correlation with cognitive improvement suggests that ameliorating energy deficits can be a real target capable of producing substantial short-term benefits for patients with Alzheimer’s.
Given the rapid results and large effect size, this is an area that needs to see more trials.
Gregory Cole, PhD , is a professor of neurology at the University of California, Los Angeles, and interim director of the Mary S. Easton Alzheimer Center. He had no relevant financial disclosures.
In Alzheimer’s disease (AD), there are early significant deficits in glucose utilization that become increasingly severe as disease progresses.
Most reports from early-onset AD animal models find that these energy deficits are largely due to defects in mitochondrial complex IV and V, and possibly related to mitochondrial fusion and fission regulators. Animal models of tauopathy demonstrate Complex I deficits.
In AD-vulnerable brain regions with early glucose utilization deficits, surviving neurons show large reductions in mitochondrial complex I, IV, and V gene expression and proteins. These changes appear sufficient to contribute to cognitive deficits. These are not shared by nondemented individuals, even in the presences of AD pathology.
The precise causes of reduced glucose utilization in AD are unknown, but may reflect these mitochondrial deficits, as well as defective insulin signaling. These changes lead to adenosine triphosphate deficits and disruptions in the balance of NAD+/NADH, both of which are already altered by normal aging.
However, because metabolism is coupled to synaptic activity, it is difficult to ascertain whether these “bioenergetic” deficits are simply secondary to progressive neuron and synapse loss or a contributing factor to neuron and synapse loss and cognitive deficits.
One of the best ways to discern the contribution of bioenergetics deficits is to treat them. Many animal models and some small trials appear to show possible benefits from supplements directed at improving energy metabolism.
In the context of these known deficits in Alzheimer’s, the new positive results with ketogenic diet reported by Dr. Swerdlow should not be ignored despite the small sample size and open-label design with the diet. The impressive 4-5 point increase in ADAS-cog that they saw is not easily achieved, and the rapid loss with washout suggests a real benefit with a large effect size.
Similarly, despite the study’s limitations with dose and size, Dr. Cunnane’s imaging of ketone body uptake and its correlation with cognitive improvement suggests that ameliorating energy deficits can be a real target capable of producing substantial short-term benefits for patients with Alzheimer’s.
Given the rapid results and large effect size, this is an area that needs to see more trials.
Gregory Cole, PhD , is a professor of neurology at the University of California, Los Angeles, and interim director of the Mary S. Easton Alzheimer Center. He had no relevant financial disclosures.
LONDON – A 3-month diet comprised of 70% fat improved cognition in Alzheimer’s disease patients better than any anti-amyloid drug that has ever been tested.
In a small pilot study, Alzheimer’s patients who followed the University of Kansas’s ketogenic diet program improved an average of 4 points on one of the most important cognitive assessments in dementia care, the Alzheimer’s Disease Assessment Scale–cognitive domain (ADAS-cog). Not only was this gain statistically significant, but it reached a level that clinical trialists believe to be clinically meaningful, and it was similar to the gains that won Food and Drug Administration approval for donepezil in 1996, according to Russell Swerdlow, MD, director of the University of Kansas Alzheimer’s Disease Center in Fairway.
To put the results in perspective, donepezil was approved on a 4-point spread between the active and placebo arm over 3 months, said Dr. Swerdlow, who is also the Gene and Marge Sweeney Professor of Neurology at the university. Part of this difference was driven by a 2-point decline in the placebo group. Relative to its baseline, the treatment group improved, on average, by about 2 points.
But in the Ketogenic Diet Retention and Feasibility Trail (KDRAFT), also 3 months long, patients’ ADAS-cog scores didn’t decline at all. Everyone who stayed with the diet and kept on their baseline medications improved, although to varying degrees.
KDRAFT was very small, with just 10 patients completing the intervention, and lacked a comparator group, so the results should be interpreted extremely cautiously, Dr. Swerdlow said in an interview. “We have to very careful about overinterpreting these findings. It’s a pilot study, and a small group, so we don’t know how genuine the finding is. But if it is true, it’s a big deal.”
Diet and dementia
Emerging evidence suggests that modifying diet can help prevent Alzheimer’s and may even help AD patients think and function better. But this research has largely focused on the heart-healthy diets already proven successful in preventing and treating hypertension, diabetes, and cardiovascular disease. Most notably, the Mediterranean-DASH Intervention for Neurodegenerative Delay (MIND) diet cut the risk of AD by up to 53% (Alzheimers Dement. 2015 Sep;11[9]:1007-14) and also slowed aging-related cognitive decline (Alzheimers Dement. 2015 Sep; 11[9]:1015-22).
MIND is a combination of the low-salt, plant-focused DASH diet, and the heart-healthy Mediterranean diet. It is a moderate-fat plan, with a ratio of 33% fat, 38% carbohydrates, and 26% protein. Ideally, only 3% of the fat should be saturated, so MIND draws on olive oil, nuts, and other foods with monounsaturated fats, largely eschewing animal fats. It’s generally considered fairly easy to follow, since it allows a wide variety of whole grains, beans, nuts, fruits, vegetables, salads, fish, and poultry. Butter, red meat, fried foods, full-fat dairy, and fast foods are strict no-nos.
A ketogenic diet, however, turns MIND on its head. With a 70% fat, 20% protein, 10% carbohydrate ratio, a typical ketogenic diet nearly eliminates most fruits, and virtually all starchy vegetables, beans, and grains. It does, however, incorporate a large amount of fat from many sources, including olive oil, butter, cream, eggs, nuts, all kinds of meat, and fish. For a ketogenic diet, Dr. Swerdlow said, the ratio of fat to protein and carbs is more critical than the source of the fat.
MIND was designed to prevent the cardiovascular and endocrine disorders than predispose to dementia over the long term. But a ketogenic diet for patients with Alzheimer’s acutely manipulates the brain’s energy metabolism system, forcing it to use ketone bodies instead of glucose for fuel.
In normal energy metabolism, carbohydrates provide a ready supply of glucose, the brain’s primary fuel. When carbs are limited or absent, serum insulin decreases and glucagon increases. This promotes lipolysis. Ketones (primarily beta-hydroxybutyrate and acetoacetate) are formed in the liver from the newly released fatty acids, and released into the circulation, including into the brain during times of decreased glucose availability – a state characteristic of Alzheimer’s disease.
Induced ketogenesis trial
Inducing ketosis through diet seems to help correct the normal, age-related decline in the brain’s ability to use glucose, said Stephen Cunnane, PhD, who also presented ketogenic intervention results at AAIC. “Cognitively normal, healthy older adults experience a 10% reduction in the brain’s ability to metabolize glucose compared to healthy young people,” he said in an interview. But this decline accelerates as Alzheimer’s hits. Those with early AD have a 20% decrement in glucose utilization, compared with healthy elders.
Changes in brain glucose metabolism can develop years before any cognitive symptoms manifest and seem to increase the risk of Alzheimer’s, said Dr. Cunnane of Sherbrooke University, Que.
“We propose that this vicious cycle of presymptomatic glucose hypometabolism causes chronic brain energy deprivation, and might contribute to deteriorating neuronal function. That could cause a further decrease in the demand for glucose, leading to cognitive decline.”
“What doesn’t change, though, is the brain’s ability to take up ketone bodies,” he said. If anything, the brain appears to use ketones more efficiently as AD becomes established. “It’s almost like the brain is trying to rescue itself. If those cells were dead, they would not be able to take up ketones. Because they do, we think they are instead starving because of their inability to use glucose and that maybe we can rescue them with ketones before they die.”
At AAIC, Dr. Cunnane reported interim results of an investigation of induced ketogenesis in patients with mild cognitive impairment (MCI). The 6-month BENEFIC trial comprises 50 patients, randomized to either a daily nutritional supplement with 30 g medium chain triglycerides (MCT) in a unflavored, nondairy emulsion, or a fat-equivalent placebo drink. When consumed, the liver very quickly converts MCT fatty acids into ketone bodies, which then circulate throughout the body, including passing the blood-brain barrier.
All of the participants in the BENEFIC trial underwent brain PET scanning for both glucose and ketone uptake. Early results clearly showed that the MCI brains took up just as much acetoacetate as did the brains of cognitively normal young adults. And although the study wasn’t powered for a full cognitive assessment, Dr. Cunnane did present 6-month data on three measures in the MCI group: trail making time, verbal fluency, and the Boston Naming Test. In the active group on MCT, scores on all three measures improved “modestly” in direct correlation with brain ketone uptake. In the placebo group, scores remained unchanged.
“We don’t have enough people in the study to make any definitive statement about cognition, but it’s nice to see the trend going in the right direction, Dr. Cunnane said. “I really think of this as a dose-finding study and a chance to demonstrate the safety and tolerability of a liquid MCT supplement in people with MCI. Our next study will use a 45 g per day supplement of MCT.”
Details of the KDRAFT study
The BENEFIC study looked only at the effects of an MCT supplement, which may not deliver all the metabolic benefits of a ketogenic diet. KDRAFT, however, employed both, and assessed not only cognitive outcomes and adverse effects, but the practical matter of whether AD patients and their caregivers could implement the diet and stick to it.
Couples recruited into the trial met with a dietitian who explained the importance of sticking with the strict fat:carb:protein ratio. It’s not easy to stay in that zone, Dr. Swerdlow said, and the MCT supplement really helps there.
“Adding the MCT, which is typically done for the ketogenic diet in epilepsy, increases the fat intake so you can tolerate a bit more carbohydrate and still remain in ketosis. MCT therefore makes it easier to successfully do the diet, if we define success by time in ketosis. Ultimately, it is an iterative diet. You check your urine, and if you are in ketosis, you are doing well. If you are not in ketosis, you have to increase your fat intake, decrease your carb intake, or both.”
The study comprised 15 patients (7 with very mild AD, 4 with mild, and 4 with moderate disease). All patients were instructed to remain on their current medications for Alzheimer’s disease for the duration of the study if they were taking any. All of the patients with moderate AD and one with very mild AD dropped out of the study within the first month, citing caregiver burden. The supplement was in the form of an oil, not an emulsion like the BENEFIC supplement, and it caused diarrhea and nausea in five subjects, although none discontinued because of that.
“We found that a slow titration of the oil could deal with the GI issues. Rather, the primary deal-breaker seemed to be the stress of planning the menus and preparing the meals.”
One patient discontinued his cholinesterase inhibitor during the study, for unknown reasons. His cognitive scores declined, but was still included in the diet-compliant analysis.
The diet didn’t affect weight, blood pressure, insulin sensitivity or resistance, or glucose level, but the intervention was short-lived. Nor were there any significant changes in high-density, low-density, or total cholesterol. Liver enzymes were stable, too.
“The only thing that changed was that they really did increase their fat and decrease their carb intake,” Dr. Swerdlow said. Daily fat jumped from 91 g to 167 g, and carbs dropped from 201 g to 46 g.
Almost everyone who stuck with the diet achieved and maintained ketosis during the study, although with varying degrees of success. “Many only had a trace amount of urinary ketones,” Dr. Swerdlow said. The investigators tracked serum beta hydroxybutyrate levels every month as well, and those measures also confirmed ketosis in the group as a whole, although some patients fluctuated in and out of the state.
The cognitive changes were striking, he said. In the 10-patient analysis, ADAS-cog scores improved by an average of 4.1 points. The results were better when Dr. Swerdlow excluded the patient who stopped his cholinesterase inhibitor medication. In that nine-patient group, the ADAS-cog improved an average of 5.3 points.
While urging caution over the small sample size and lack of a control comparator, Dr. Swerdlow expressed deep satisfaction over the outcomes. A clinician as well as a researcher, he is accustomed to the slow but inexorable decline of AD patients.
“I’m going to try to relate the impression you get in the clinic with these scores,” he said. “Very rarely, but sometimes, with a cholinesterase inhibitor in patients, we’ll see something like a 7-point change. That’s a fantastic response, an improvement you can see across the room. A change of 2 points really doesn’t look that much different, although caregivers will tell you there is a subtle change, maybe a little more focus. The average we got in our 10 subjects was a 4-point improvement. That’s impressive. And a 5-point change is like rolling the clock back by a year.”
The improvements didn’t last, though. A 1-month washout period followed the intervention. By the end, both ADAS-cog and Mini-Mental State Examination scores had returned to their baseline levels. At the end of the study, a few of the patients and their partners expressed their intent to resume the diet, but the investigators do not know whether this indeed happened. Still, the results are encouraging enough that, like Dr. Cunnane, Dr. Swerdlow hopes to conduct a larger, longer study – one that would include a control group.
Future investigations of the ketogenic diet in AD might do well to also include an exercise component, both researchers mentioned. In addition to starvation, ketogenic dieting, and MCT supplementation, exercise is an effective way to induce ketogenesis.
“Exercise produces ketones, but most importantly, it increases the capacity of the brain to use ketones,” Dr. Cunnane said. The connection may help explain some of the cognitive benefits seen in exercise trials in patients with MCI and AD.
“This raises the possibility that if in fact exercise benefits the brain, ketone bodies may mediate some of that effect,” Dr. Swerdlow said. “Could exercise potentiate the ketosis from the diet? That is possible, and maybe using these interventions in conjunction would be synergistic. At this point, we are just happy to show the diet is feasible, if even for a limited period.”
Implementing KDRAFT: Research team dishes the skinny on fats
The KDRAFT study diet is surprisingly flexible despite its strict ratio of fat to protein and carbohydrate, according to the University of Kansas research team that implemented it. It only took a few counseling sessions to get most study participants enthusiastically embracing the new eating plan, even one so radically different from the way they were accustomed to eating.
“We focused mainly on the macronutrient makeup,” said Matthew Taylor, PhD, who supervised the diet study on a day-to-day basis. Instead of distributing a rigid diet plan, with prespecified meals and snacks, “We talked more in general about foods they could have and foods they couldn’t have.”
“When people think ‘ketogenic,’ they think bacon, eggs, oil, butter and cream, and may have an automatic negative connotation that this is unhealthy eating,” Dr. Taylor said in an interview. “But yes, eggs were in there and, because a lot of people really like bacon, there was bacon, too!”
The educational sessions did include teaching about healthy and unhealthy fats, and Dr. Taylor “tried to steer people toward the healthier ones, like olive oil, avocados, and nuts. But I didn’t say, ‘Eat this one and not that one.’ If it took melting butter on vegetables to get to that fat ratio, I was not as concerned about where the fat came from as about getting there and maintaining ketosis.”
KDRAFT also had a twist that’s becoming more common among ketogenic eating plans: lots of vegetables. Dr. Taylor asked participants to concentrate on nonstarchy vegetables and forgo potatoes, corn, beans, and lima beans, although some people did enjoy peas occasionally.
“We used to be think we had to restrict vegetables or people would go out of ketosis more easily. But that doesn’t seem to be true. We focused a lot on eating vegetables, and everyone increased their vegetable intake dramatically. We actually tried to use vegetables as a vehicle for fat. For example, people would roast Brussels sprouts or broccoli in olive oil and then put melted butter on it. It was pretty much, ‘Eat all the vegetables you can and put fat on them.’”
Fruits are full of sugar, so they are not liberally used in most ketogenic diets, but KDRAFT did allow one type: berries, and blueberries in particular. “We had people eating a couple of small handfuls of berries throughout the day and still being able to maintain ketosis. We did severely cut back on the amount and type of fruit people could have, but berries seemed to work well.”
Whipping cream had a place, too. “It fit really well in the diet, because it’s basically all fat,” Dr. Taylor said. “It’s used more often in pediatric ketogenic diets as a milk substitute. One thing our subjects liked to do was use it to make a sweet snack. All it takes is a packet of [stevia] sweetener and some vanilla. Then you whip and freeze it and it’s like an ice cream dessert.”
After the initial drop-outs, the remaining study pairs embraced the intervention enthusiastically.
“When the study partner took the diet on too, we had our best success. One of our last pairs had an entire family join in – children, grandchildren, everyone decided to follow the diet. That is a very helpful piece to this. It’s difficult to always say, ‘Here’s our normal food and here’s the keto food over here.’”
The dropouts occurred very early. These study pairs, all of whom included patients with moderate Alzheimer’s, never embraced the plan at all, and this is a telling point, Dr. Taylor noted.
“When you get to a level of dementia there are so many other things in the caregiving process that taking on big behavioral changes is very difficult.”
Although the study showed that the diet wasn’t practical for sicker patients at home, it still might be beneficial in other settings, said Debra Sullivan, PhD, RD. Dr. Sullivan chairs the department of dietetics and nutrition at the University of Kansas Medical Center and holds the Midwest Dairy Council Endowed Professorship in Clinical Nutrition.
“I think that we might be able to create a version of the diet that could be used in an institutional setting for our more advanced patients,” she said. “But there’s no denying that this can be challenging. It’s a big change from the way the typical American eats.”
None of the KDRAFT participants experienced any lipid changes, for either better or worse. The 3-month intervention was long enough to have picked up such changes if they were in the offing, said principal investigator Russell Swerdlow, MD. While there are mixed data on ketogenic diets’ atherogenic effects, many people respond positively, with improved cholesterol.
“Much of what it comes down to is, are you in a catabolic or anabolic states? Are you building up or tearing down? Excessive cholesterol is a sign of being overfed and laying down energy supplies. You take in carbon and turn it into cholesterol. But if you can trick your body into a catabolic state – essentially make it think it’s starving, which a ketogenic diet does – then you have consistently low insulin levels, and you don’t turn on the cholesterol synthesis pathway. You may increase your cholesterol intake through diet, but you’re not synthesizing it in your body, and that synthesis is what really drives your cholesterol level. If you’re not overeating, your body’s production goes down.”
Brain Energy and Memory (BEAM) study
Dr. Swerdlow isn’t the only clinician researcher looking at how a ketogenic diet might influence cognition. Suzanne Craft, PhD, well known for her investigations of the role of insulin signaling and therapy in AD, is running a ketogenic diet trial as well.
As noted on clinicaltrials.gov, the 24-week Brain Energy and Memory (BEAM) study aimed to recruit 25 subjects in two cohorts: adults with mild memory complaints, and cognitively normal adults with prediabetes. A comparator group of healthy controls will contribute cognitive assessments, blood and stool sample collection, neuroimaging, and lumbar puncture at baseline.
Both active groups will be randomized to 6 weeks of either a low-fat, high-carbohydrate diet, with carbs making up 50%-60% of daily caloric intake, or a modified ketogenic-Mediterranean Diet with carbs comprising less than 10% of daily caloric intake.
BEAM’s primary outcome will be changes in the AD cerebrospinal fluid biomarkers beta-amyloid and tau. Secondary endpoints include cognitive assessments, brain ketone uptake on PET scanning, and insulin sensitivity.
Dr. Cunnane has no financial interest in the MCT emulsion, which was supplied by Abitec. He reported conference travel support from Abitec, Nisshin OilliO, and Pruvit. He also reported receiving research project funding from Nestlé and Bulletproof.
Dr. Swerdlow had no financial disclosures.
[email protected]
On Twitter @alz_gal
LONDON – A 3-month diet comprised of 70% fat improved cognition in Alzheimer’s disease patients better than any anti-amyloid drug that has ever been tested.
In a small pilot study, Alzheimer’s patients who followed the University of Kansas’s ketogenic diet program improved an average of 4 points on one of the most important cognitive assessments in dementia care, the Alzheimer’s Disease Assessment Scale–cognitive domain (ADAS-cog). Not only was this gain statistically significant, but it reached a level that clinical trialists believe to be clinically meaningful, and it was similar to the gains that won Food and Drug Administration approval for donepezil in 1996, according to Russell Swerdlow, MD, director of the University of Kansas Alzheimer’s Disease Center in Fairway.
To put the results in perspective, donepezil was approved on a 4-point spread between the active and placebo arm over 3 months, said Dr. Swerdlow, who is also the Gene and Marge Sweeney Professor of Neurology at the university. Part of this difference was driven by a 2-point decline in the placebo group. Relative to its baseline, the treatment group improved, on average, by about 2 points.
But in the Ketogenic Diet Retention and Feasibility Trail (KDRAFT), also 3 months long, patients’ ADAS-cog scores didn’t decline at all. Everyone who stayed with the diet and kept on their baseline medications improved, although to varying degrees.
KDRAFT was very small, with just 10 patients completing the intervention, and lacked a comparator group, so the results should be interpreted extremely cautiously, Dr. Swerdlow said in an interview. “We have to very careful about overinterpreting these findings. It’s a pilot study, and a small group, so we don’t know how genuine the finding is. But if it is true, it’s a big deal.”
Diet and dementia
Emerging evidence suggests that modifying diet can help prevent Alzheimer’s and may even help AD patients think and function better. But this research has largely focused on the heart-healthy diets already proven successful in preventing and treating hypertension, diabetes, and cardiovascular disease. Most notably, the Mediterranean-DASH Intervention for Neurodegenerative Delay (MIND) diet cut the risk of AD by up to 53% (Alzheimers Dement. 2015 Sep;11[9]:1007-14) and also slowed aging-related cognitive decline (Alzheimers Dement. 2015 Sep; 11[9]:1015-22).
MIND is a combination of the low-salt, plant-focused DASH diet, and the heart-healthy Mediterranean diet. It is a moderate-fat plan, with a ratio of 33% fat, 38% carbohydrates, and 26% protein. Ideally, only 3% of the fat should be saturated, so MIND draws on olive oil, nuts, and other foods with monounsaturated fats, largely eschewing animal fats. It’s generally considered fairly easy to follow, since it allows a wide variety of whole grains, beans, nuts, fruits, vegetables, salads, fish, and poultry. Butter, red meat, fried foods, full-fat dairy, and fast foods are strict no-nos.
A ketogenic diet, however, turns MIND on its head. With a 70% fat, 20% protein, 10% carbohydrate ratio, a typical ketogenic diet nearly eliminates most fruits, and virtually all starchy vegetables, beans, and grains. It does, however, incorporate a large amount of fat from many sources, including olive oil, butter, cream, eggs, nuts, all kinds of meat, and fish. For a ketogenic diet, Dr. Swerdlow said, the ratio of fat to protein and carbs is more critical than the source of the fat.
MIND was designed to prevent the cardiovascular and endocrine disorders than predispose to dementia over the long term. But a ketogenic diet for patients with Alzheimer’s acutely manipulates the brain’s energy metabolism system, forcing it to use ketone bodies instead of glucose for fuel.
In normal energy metabolism, carbohydrates provide a ready supply of glucose, the brain’s primary fuel. When carbs are limited or absent, serum insulin decreases and glucagon increases. This promotes lipolysis. Ketones (primarily beta-hydroxybutyrate and acetoacetate) are formed in the liver from the newly released fatty acids, and released into the circulation, including into the brain during times of decreased glucose availability – a state characteristic of Alzheimer’s disease.
Induced ketogenesis trial
Inducing ketosis through diet seems to help correct the normal, age-related decline in the brain’s ability to use glucose, said Stephen Cunnane, PhD, who also presented ketogenic intervention results at AAIC. “Cognitively normal, healthy older adults experience a 10% reduction in the brain’s ability to metabolize glucose compared to healthy young people,” he said in an interview. But this decline accelerates as Alzheimer’s hits. Those with early AD have a 20% decrement in glucose utilization, compared with healthy elders.
Changes in brain glucose metabolism can develop years before any cognitive symptoms manifest and seem to increase the risk of Alzheimer’s, said Dr. Cunnane of Sherbrooke University, Que.
“We propose that this vicious cycle of presymptomatic glucose hypometabolism causes chronic brain energy deprivation, and might contribute to deteriorating neuronal function. That could cause a further decrease in the demand for glucose, leading to cognitive decline.”
“What doesn’t change, though, is the brain’s ability to take up ketone bodies,” he said. If anything, the brain appears to use ketones more efficiently as AD becomes established. “It’s almost like the brain is trying to rescue itself. If those cells were dead, they would not be able to take up ketones. Because they do, we think they are instead starving because of their inability to use glucose and that maybe we can rescue them with ketones before they die.”
At AAIC, Dr. Cunnane reported interim results of an investigation of induced ketogenesis in patients with mild cognitive impairment (MCI). The 6-month BENEFIC trial comprises 50 patients, randomized to either a daily nutritional supplement with 30 g medium chain triglycerides (MCT) in a unflavored, nondairy emulsion, or a fat-equivalent placebo drink. When consumed, the liver very quickly converts MCT fatty acids into ketone bodies, which then circulate throughout the body, including passing the blood-brain barrier.
All of the participants in the BENEFIC trial underwent brain PET scanning for both glucose and ketone uptake. Early results clearly showed that the MCI brains took up just as much acetoacetate as did the brains of cognitively normal young adults. And although the study wasn’t powered for a full cognitive assessment, Dr. Cunnane did present 6-month data on three measures in the MCI group: trail making time, verbal fluency, and the Boston Naming Test. In the active group on MCT, scores on all three measures improved “modestly” in direct correlation with brain ketone uptake. In the placebo group, scores remained unchanged.
“We don’t have enough people in the study to make any definitive statement about cognition, but it’s nice to see the trend going in the right direction, Dr. Cunnane said. “I really think of this as a dose-finding study and a chance to demonstrate the safety and tolerability of a liquid MCT supplement in people with MCI. Our next study will use a 45 g per day supplement of MCT.”
Details of the KDRAFT study
The BENEFIC study looked only at the effects of an MCT supplement, which may not deliver all the metabolic benefits of a ketogenic diet. KDRAFT, however, employed both, and assessed not only cognitive outcomes and adverse effects, but the practical matter of whether AD patients and their caregivers could implement the diet and stick to it.
Couples recruited into the trial met with a dietitian who explained the importance of sticking with the strict fat:carb:protein ratio. It’s not easy to stay in that zone, Dr. Swerdlow said, and the MCT supplement really helps there.
“Adding the MCT, which is typically done for the ketogenic diet in epilepsy, increases the fat intake so you can tolerate a bit more carbohydrate and still remain in ketosis. MCT therefore makes it easier to successfully do the diet, if we define success by time in ketosis. Ultimately, it is an iterative diet. You check your urine, and if you are in ketosis, you are doing well. If you are not in ketosis, you have to increase your fat intake, decrease your carb intake, or both.”
The study comprised 15 patients (7 with very mild AD, 4 with mild, and 4 with moderate disease). All patients were instructed to remain on their current medications for Alzheimer’s disease for the duration of the study if they were taking any. All of the patients with moderate AD and one with very mild AD dropped out of the study within the first month, citing caregiver burden. The supplement was in the form of an oil, not an emulsion like the BENEFIC supplement, and it caused diarrhea and nausea in five subjects, although none discontinued because of that.
“We found that a slow titration of the oil could deal with the GI issues. Rather, the primary deal-breaker seemed to be the stress of planning the menus and preparing the meals.”
One patient discontinued his cholinesterase inhibitor during the study, for unknown reasons. His cognitive scores declined, but was still included in the diet-compliant analysis.
The diet didn’t affect weight, blood pressure, insulin sensitivity or resistance, or glucose level, but the intervention was short-lived. Nor were there any significant changes in high-density, low-density, or total cholesterol. Liver enzymes were stable, too.
“The only thing that changed was that they really did increase their fat and decrease their carb intake,” Dr. Swerdlow said. Daily fat jumped from 91 g to 167 g, and carbs dropped from 201 g to 46 g.
Almost everyone who stuck with the diet achieved and maintained ketosis during the study, although with varying degrees of success. “Many only had a trace amount of urinary ketones,” Dr. Swerdlow said. The investigators tracked serum beta hydroxybutyrate levels every month as well, and those measures also confirmed ketosis in the group as a whole, although some patients fluctuated in and out of the state.
The cognitive changes were striking, he said. In the 10-patient analysis, ADAS-cog scores improved by an average of 4.1 points. The results were better when Dr. Swerdlow excluded the patient who stopped his cholinesterase inhibitor medication. In that nine-patient group, the ADAS-cog improved an average of 5.3 points.
While urging caution over the small sample size and lack of a control comparator, Dr. Swerdlow expressed deep satisfaction over the outcomes. A clinician as well as a researcher, he is accustomed to the slow but inexorable decline of AD patients.
“I’m going to try to relate the impression you get in the clinic with these scores,” he said. “Very rarely, but sometimes, with a cholinesterase inhibitor in patients, we’ll see something like a 7-point change. That’s a fantastic response, an improvement you can see across the room. A change of 2 points really doesn’t look that much different, although caregivers will tell you there is a subtle change, maybe a little more focus. The average we got in our 10 subjects was a 4-point improvement. That’s impressive. And a 5-point change is like rolling the clock back by a year.”
The improvements didn’t last, though. A 1-month washout period followed the intervention. By the end, both ADAS-cog and Mini-Mental State Examination scores had returned to their baseline levels. At the end of the study, a few of the patients and their partners expressed their intent to resume the diet, but the investigators do not know whether this indeed happened. Still, the results are encouraging enough that, like Dr. Cunnane, Dr. Swerdlow hopes to conduct a larger, longer study – one that would include a control group.
Future investigations of the ketogenic diet in AD might do well to also include an exercise component, both researchers mentioned. In addition to starvation, ketogenic dieting, and MCT supplementation, exercise is an effective way to induce ketogenesis.
“Exercise produces ketones, but most importantly, it increases the capacity of the brain to use ketones,” Dr. Cunnane said. The connection may help explain some of the cognitive benefits seen in exercise trials in patients with MCI and AD.
“This raises the possibility that if in fact exercise benefits the brain, ketone bodies may mediate some of that effect,” Dr. Swerdlow said. “Could exercise potentiate the ketosis from the diet? That is possible, and maybe using these interventions in conjunction would be synergistic. At this point, we are just happy to show the diet is feasible, if even for a limited period.”
Implementing KDRAFT: Research team dishes the skinny on fats
The KDRAFT study diet is surprisingly flexible despite its strict ratio of fat to protein and carbohydrate, according to the University of Kansas research team that implemented it. It only took a few counseling sessions to get most study participants enthusiastically embracing the new eating plan, even one so radically different from the way they were accustomed to eating.
“We focused mainly on the macronutrient makeup,” said Matthew Taylor, PhD, who supervised the diet study on a day-to-day basis. Instead of distributing a rigid diet plan, with prespecified meals and snacks, “We talked more in general about foods they could have and foods they couldn’t have.”
“When people think ‘ketogenic,’ they think bacon, eggs, oil, butter and cream, and may have an automatic negative connotation that this is unhealthy eating,” Dr. Taylor said in an interview. “But yes, eggs were in there and, because a lot of people really like bacon, there was bacon, too!”
The educational sessions did include teaching about healthy and unhealthy fats, and Dr. Taylor “tried to steer people toward the healthier ones, like olive oil, avocados, and nuts. But I didn’t say, ‘Eat this one and not that one.’ If it took melting butter on vegetables to get to that fat ratio, I was not as concerned about where the fat came from as about getting there and maintaining ketosis.”
KDRAFT also had a twist that’s becoming more common among ketogenic eating plans: lots of vegetables. Dr. Taylor asked participants to concentrate on nonstarchy vegetables and forgo potatoes, corn, beans, and lima beans, although some people did enjoy peas occasionally.
“We used to be think we had to restrict vegetables or people would go out of ketosis more easily. But that doesn’t seem to be true. We focused a lot on eating vegetables, and everyone increased their vegetable intake dramatically. We actually tried to use vegetables as a vehicle for fat. For example, people would roast Brussels sprouts or broccoli in olive oil and then put melted butter on it. It was pretty much, ‘Eat all the vegetables you can and put fat on them.’”
Fruits are full of sugar, so they are not liberally used in most ketogenic diets, but KDRAFT did allow one type: berries, and blueberries in particular. “We had people eating a couple of small handfuls of berries throughout the day and still being able to maintain ketosis. We did severely cut back on the amount and type of fruit people could have, but berries seemed to work well.”
Whipping cream had a place, too. “It fit really well in the diet, because it’s basically all fat,” Dr. Taylor said. “It’s used more often in pediatric ketogenic diets as a milk substitute. One thing our subjects liked to do was use it to make a sweet snack. All it takes is a packet of [stevia] sweetener and some vanilla. Then you whip and freeze it and it’s like an ice cream dessert.”
After the initial drop-outs, the remaining study pairs embraced the intervention enthusiastically.
“When the study partner took the diet on too, we had our best success. One of our last pairs had an entire family join in – children, grandchildren, everyone decided to follow the diet. That is a very helpful piece to this. It’s difficult to always say, ‘Here’s our normal food and here’s the keto food over here.’”
The dropouts occurred very early. These study pairs, all of whom included patients with moderate Alzheimer’s, never embraced the plan at all, and this is a telling point, Dr. Taylor noted.
“When you get to a level of dementia there are so many other things in the caregiving process that taking on big behavioral changes is very difficult.”
Although the study showed that the diet wasn’t practical for sicker patients at home, it still might be beneficial in other settings, said Debra Sullivan, PhD, RD. Dr. Sullivan chairs the department of dietetics and nutrition at the University of Kansas Medical Center and holds the Midwest Dairy Council Endowed Professorship in Clinical Nutrition.
“I think that we might be able to create a version of the diet that could be used in an institutional setting for our more advanced patients,” she said. “But there’s no denying that this can be challenging. It’s a big change from the way the typical American eats.”
None of the KDRAFT participants experienced any lipid changes, for either better or worse. The 3-month intervention was long enough to have picked up such changes if they were in the offing, said principal investigator Russell Swerdlow, MD. While there are mixed data on ketogenic diets’ atherogenic effects, many people respond positively, with improved cholesterol.
“Much of what it comes down to is, are you in a catabolic or anabolic states? Are you building up or tearing down? Excessive cholesterol is a sign of being overfed and laying down energy supplies. You take in carbon and turn it into cholesterol. But if you can trick your body into a catabolic state – essentially make it think it’s starving, which a ketogenic diet does – then you have consistently low insulin levels, and you don’t turn on the cholesterol synthesis pathway. You may increase your cholesterol intake through diet, but you’re not synthesizing it in your body, and that synthesis is what really drives your cholesterol level. If you’re not overeating, your body’s production goes down.”
Brain Energy and Memory (BEAM) study
Dr. Swerdlow isn’t the only clinician researcher looking at how a ketogenic diet might influence cognition. Suzanne Craft, PhD, well known for her investigations of the role of insulin signaling and therapy in AD, is running a ketogenic diet trial as well.
As noted on clinicaltrials.gov, the 24-week Brain Energy and Memory (BEAM) study aimed to recruit 25 subjects in two cohorts: adults with mild memory complaints, and cognitively normal adults with prediabetes. A comparator group of healthy controls will contribute cognitive assessments, blood and stool sample collection, neuroimaging, and lumbar puncture at baseline.
Both active groups will be randomized to 6 weeks of either a low-fat, high-carbohydrate diet, with carbs making up 50%-60% of daily caloric intake, or a modified ketogenic-Mediterranean Diet with carbs comprising less than 10% of daily caloric intake.
BEAM’s primary outcome will be changes in the AD cerebrospinal fluid biomarkers beta-amyloid and tau. Secondary endpoints include cognitive assessments, brain ketone uptake on PET scanning, and insulin sensitivity.
Dr. Cunnane has no financial interest in the MCT emulsion, which was supplied by Abitec. He reported conference travel support from Abitec, Nisshin OilliO, and Pruvit. He also reported receiving research project funding from Nestlé and Bulletproof.
Dr. Swerdlow had no financial disclosures.
[email protected]
On Twitter @alz_gal
AT AAIC 2017

Expert shares tips for spotting allergic contact dermatitis in children
CHICAGO – If severe eczema persists in a pediatric patient despite your best treatment efforts, think allergic contact dermatitis.
“Or, if your eczema patients tell you that they have a cream that’s making things worse, you should think about a contact allergen,” Catalina Matiz, MD, said at the World Congress of Pediatric Dermatology.
Allergic contact dermatitis (ACD) is a type IV delayed-type hypersensitivity reaction to haptens that come into contact with the skin. Poison ivy is a common plant-based culprit, while nickel is the most common metal allergen in adults and children. “The skin barrier also plays a role,” said Dr. Matiz of the department of dermatology at Rady Children’s Hospital–San Diego, and the University of California, San Diego. “Compared with adults, children have a thinner stratum corneum, and some haptens can penetrate the skin. Some studies suggest that patients with atopic dermatitis may have increased rates of allergic sensitization, and filaggrin mutations have been found in patients with atopic dermatitis and in patients with ACD to nickel. Filaggrin helps to aggregate the cytoskeletal proteins that form the cornified cell envelope. Without filaggrin, the skin barrier is defective.”

The top 10 pediatric allergens found in personal hygiene products across five studies in the medical literature include neomycin, balsam of Peru, fragrance mix, benzalkonium chloride, lanolin, cocamidopropyl betaine, formaldehyde, methylchloroisothiazolinone/methylisothiazolinone (MCI/MI), propylene glycol, and corticosteroids. Dr. Matiz makes it practice to patch test as a last resort. “I always try to get a history, try to improve their symptoms, and have them start avoidance first, following the preemptive avoidance list,” she said (Expert Rev Clin Immunol. 2016;12[5]:551-61).
The T.R.U.E. test includes 35 allergens. “The T.R.U.E test is a good tool, which can capture up to 70% of relevant reactions in children with the inconvenience that some of the allergens in the test are not that relevant in children, and it’s not yet [Food and Drug Administration] approved to use in children,” she noted. The comprehensive chamber test allows you to select from unlimited number of allergens, “but that’s difficult. You have to have specialized staff to help you make the cells.”
A list of the minimum 20 allergens you should test for in children and the recommended supplemental allergens depending on history and locations of their dermatitis can be found in the following article: Curr Allergy Asthma Rep 2014;14[6]:444. “I always tell patients when they come for consultations to bring in everything they’re using: their shampoos, creams, and medications, because we want to see what they’re exposed to, so we can select the right allergens and also test their own products,” Dr. Matiz said. She recommends avoiding testing for strong sensitizers such as paraphenylenediamine, in children younger than 12 years of age who don’t have a history of exposure.
Testing tips for children younger than age 5 include decreasing concentrations to half for nickel, formaldehyde, and rubber accelerators. “Don’t test for paraphenylenediamine unless there is high suspicion,” she said. “Consider removing patches by 24 hours in the very young.”
The best antidote to contact dermatitis is avoidance of the known trigger. “You want to spend a lot of time with patients and parents on this,” she advised. “Give a list of safe products to use from the American Contact Dermatitis Society’s Contact Allergen Management Program [www.contactderm.org], and provide handouts about the location and history of positive allergens [www.truetest.com].” And, she added, “make a plan of treatment and follow-up in 6 weeks.”
Dr. Matiz disclosed that she is a subinvestigator in the Clinical Evaluation of T.R.U.E Test Panel 3.3 in Children and Adolescents study.
CHICAGO – If severe eczema persists in a pediatric patient despite your best treatment efforts, think allergic contact dermatitis.
“Or, if your eczema patients tell you that they have a cream that’s making things worse, you should think about a contact allergen,” Catalina Matiz, MD, said at the World Congress of Pediatric Dermatology.
Allergic contact dermatitis (ACD) is a type IV delayed-type hypersensitivity reaction to haptens that come into contact with the skin. Poison ivy is a common plant-based culprit, while nickel is the most common metal allergen in adults and children. “The skin barrier also plays a role,” said Dr. Matiz of the department of dermatology at Rady Children’s Hospital–San Diego, and the University of California, San Diego. “Compared with adults, children have a thinner stratum corneum, and some haptens can penetrate the skin. Some studies suggest that patients with atopic dermatitis may have increased rates of allergic sensitization, and filaggrin mutations have been found in patients with atopic dermatitis and in patients with ACD to nickel. Filaggrin helps to aggregate the cytoskeletal proteins that form the cornified cell envelope. Without filaggrin, the skin barrier is defective.”
The top 10 pediatric allergens found in personal hygiene products across five studies in the medical literature include neomycin, balsam of Peru, fragrance mix, benzalkonium chloride, lanolin, cocamidopropyl betaine, formaldehyde, methylchloroisothiazolinone/methylisothiazolinone (MCI/MI), propylene glycol, and corticosteroids. Dr. Matiz makes it practice to patch test as a last resort. “I always try to get a history, try to improve their symptoms, and have them start avoidance first, following the preemptive avoidance list,” she said (Expert Rev Clin Immunol. 2016;12[5]:551-61).
The T.R.U.E. test includes 35 allergens. “The T.R.U.E test is a good tool, which can capture up to 70% of relevant reactions in children with the inconvenience that some of the allergens in the test are not that relevant in children, and it’s not yet [Food and Drug Administration] approved to use in children,” she noted. The comprehensive chamber test allows you to select from unlimited number of allergens, “but that’s difficult. You have to have specialized staff to help you make the cells.”
A list of the minimum 20 allergens you should test for in children and the recommended supplemental allergens depending on history and locations of their dermatitis can be found in the following article: Curr Allergy Asthma Rep 2014;14[6]:444. “I always tell patients when they come for consultations to bring in everything they’re using: their shampoos, creams, and medications, because we want to see what they’re exposed to, so we can select the right allergens and also test their own products,” Dr. Matiz said. She recommends avoiding testing for strong sensitizers such as paraphenylenediamine, in children younger than 12 years of age who don’t have a history of exposure.
Testing tips for children younger than age 5 include decreasing concentrations to half for nickel, formaldehyde, and rubber accelerators. “Don’t test for paraphenylenediamine unless there is high suspicion,” she said. “Consider removing patches by 24 hours in the very young.”
The best antidote to contact dermatitis is avoidance of the known trigger. “You want to spend a lot of time with patients and parents on this,” she advised. “Give a list of safe products to use from the American Contact Dermatitis Society’s Contact Allergen Management Program [www.contactderm.org], and provide handouts about the location and history of positive allergens [www.truetest.com].” And, she added, “make a plan of treatment and follow-up in 6 weeks.”
Dr. Matiz disclosed that she is a subinvestigator in the Clinical Evaluation of T.R.U.E Test Panel 3.3 in Children and Adolescents study.
CHICAGO – If severe eczema persists in a pediatric patient despite your best treatment efforts, think allergic contact dermatitis.
“Or, if your eczema patients tell you that they have a cream that’s making things worse, you should think about a contact allergen,” Catalina Matiz, MD, said at the World Congress of Pediatric Dermatology.
Allergic contact dermatitis (ACD) is a type IV delayed-type hypersensitivity reaction to haptens that come into contact with the skin. Poison ivy is a common plant-based culprit, while nickel is the most common metal allergen in adults and children. “The skin barrier also plays a role,” said Dr. Matiz of the department of dermatology at Rady Children’s Hospital–San Diego, and the University of California, San Diego. “Compared with adults, children have a thinner stratum corneum, and some haptens can penetrate the skin. Some studies suggest that patients with atopic dermatitis may have increased rates of allergic sensitization, and filaggrin mutations have been found in patients with atopic dermatitis and in patients with ACD to nickel. Filaggrin helps to aggregate the cytoskeletal proteins that form the cornified cell envelope. Without filaggrin, the skin barrier is defective.”
The top 10 pediatric allergens found in personal hygiene products across five studies in the medical literature include neomycin, balsam of Peru, fragrance mix, benzalkonium chloride, lanolin, cocamidopropyl betaine, formaldehyde, methylchloroisothiazolinone/methylisothiazolinone (MCI/MI), propylene glycol, and corticosteroids. Dr. Matiz makes it practice to patch test as a last resort. “I always try to get a history, try to improve their symptoms, and have them start avoidance first, following the preemptive avoidance list,” she said (Expert Rev Clin Immunol. 2016;12[5]:551-61).
The T.R.U.E. test includes 35 allergens. “The T.R.U.E test is a good tool, which can capture up to 70% of relevant reactions in children with the inconvenience that some of the allergens in the test are not that relevant in children, and it’s not yet [Food and Drug Administration] approved to use in children,” she noted. The comprehensive chamber test allows you to select from unlimited number of allergens, “but that’s difficult. You have to have specialized staff to help you make the cells.”
A list of the minimum 20 allergens you should test for in children and the recommended supplemental allergens depending on history and locations of their dermatitis can be found in the following article: Curr Allergy Asthma Rep 2014;14[6]:444. “I always tell patients when they come for consultations to bring in everything they’re using: their shampoos, creams, and medications, because we want to see what they’re exposed to, so we can select the right allergens and also test their own products,” Dr. Matiz said. She recommends avoiding testing for strong sensitizers such as paraphenylenediamine, in children younger than 12 years of age who don’t have a history of exposure.
Testing tips for children younger than age 5 include decreasing concentrations to half for nickel, formaldehyde, and rubber accelerators. “Don’t test for paraphenylenediamine unless there is high suspicion,” she said. “Consider removing patches by 24 hours in the very young.”
The best antidote to contact dermatitis is avoidance of the known trigger. “You want to spend a lot of time with patients and parents on this,” she advised. “Give a list of safe products to use from the American Contact Dermatitis Society’s Contact Allergen Management Program [www.contactderm.org], and provide handouts about the location and history of positive allergens [www.truetest.com].” And, she added, “make a plan of treatment and follow-up in 6 weeks.”
Dr. Matiz disclosed that she is a subinvestigator in the Clinical Evaluation of T.R.U.E Test Panel 3.3 in Children and Adolescents study.
AT WCPD 2017