User login
Noxious Nocebos in Dermatology
The medical dictum primum non nocere (first, do no harm) is a fundamental rule by which physicians have lived for centuries.1 Physicians are familiar with the term placebo (I shall please) and the placebo effect of improvement based on expectation of positive results; however, many are not familiar with the term nocebo (I shall harm) and the nocebo effect of lack of improvement or deterioration based on the expectation of negative results. The patient’s expectation of being pleased and/or being harmed may be on the conscious level and/or on one or more subconscious and unconscious levels.
Words can have as much of an impact on some patients as medications or procedures. Rudyard Kipling said, “Words are, of course, the most powerful drug used by mankind.” The words that a dermatologist chooses to use can have either a placebo or a nocebo effect on the patient. The purpose of this commentary is to elevate awareness that there are positive alternatives to unintended negative suggestions that are commonly used in dermatologic practice.
A search of PubMed articles indexed for MEDLINE and Scopus published from January 1966 through July 28, 2016, was conducted using the terms placebo or nocebo and cutaneous or skin. Prior publications in this area related specifically to dermatology include those of Poletti2 and Sonthalia et al.3
Patient expectations play an important role in both positive and negative treatment outcomes. Patient-physician communication can moderate these effects both positively and negatively.4 Nocebo effects can substantially reduce treatment efficacy and patient compliance. Patient expectations of negative results or side effects of a treatment or medication can be self-induced or can be induced by inappropriate physician-patient communication, drug information leaflets, influence of family or peers, or discovery of reported adverse effects through reading materials on the Internet.4 Expectation of negative effects can reduce patient adherence and compliance with treatment, reducing treatment efficacy. The psychosocial context around the patient and the treatment may change the neuronal biochemistry and circuitry in the patient’s brain, and the central and peripheral mechanisms activated by placebos and nocebos have been found to be the same as those activated by the medications, suggesting cognitive/affective enhancing or impeding of drug action.5
The subconscious and unconscious habitual automated parts of the brain hear words on the level of a 5- or 6-year-old child with literal unconscious cognitive interpretation of the words. These parts also do not connect words in a sentence with each other.5 For example, if the dermatologist or nurse says “This will not hurt,” the unconscious brain hears not and hurt but does not connect the two. On the other hand, if the dermatologist or nurse says “ You may experience some discomfort,” the unconscious brain hears comfort. Telling the patient “Don’t scratch” may be heard unconsciously as scratch. See the Table for suggested rephrasing of common nocebos used in dermatology. The conscious parts of the brain help determine cognitions influenced by associated unconscious memories, positive or negative. Both the conscious and unconscious parts of the brain influence affect or emotion.

When excess stress occurs, there is a natural shift downward from social communicative newer parasympathetic nervous system to fight or flight sympathetic nervous system, and possibly further shift to the freeze response of older parasympathetic nervous system dominance. Memories that are associated with a strong surge of norepinephrine tend to be much more strongly fixed in the memory than ordinary memories and frequently are associated with overwhelming traumatic experiences. When a threat is perceived, negative interpretations and perceptions generally win out over positive interpretations and perceptions. Unconscious fears generally prevail over conscious thoughts, and stronger emotions generally prevail over weaker emotions. Anxiety often is associated with rapid breathing and activation of the sympathetic nervous system. It can be countered by slow breathing to a rate of approximately 6 breaths per minute, helping to bring back more parasympathetic balance. Pacing a patient’s breathing to slow it and using a soothing tone of voice can help reduce patient anxiety. Reducing anxiety can decrease the patient’s tendency to jump to negative conclusions or have negative perceptions or emotions that can invoke the nocebo effect.
For the dermatologist, as for the patient, changing an old habit and creating a new habit requires repeating something differently and consistently 20 to 40 times. Becoming more conscious of the effects of language on the patient is an important part of the art of medicine. By carefully choosing words, intonation, and body language, it is possible to enhance the placebo effect and decrease the nocebo effect for the benefit of the patient. When describing possible adverse effects of treatments or medications, if the dermatologist says that most people do fine with the treatment but a few can experience the described adverse effect, it usually takes the edge off the potential suggested nocebo effect.
In conclusion, primum non nocere includes careful consideration and use of language, tone, and body language to maximize the placebo effect and minimize the nocebo effect.
- Hippocrates. Epidemics. Jones WHS, trans. Loeb Classical Library: Hippocrates. Vol 1. Cambridge, MA: Harvard University Press; 1923:164-165.
- Poletti ED. El efecto nocebo in dermatología. Dermatol Cosmet Quirg. 2007;5:74.
- Sonthalia S, Sahaya K, Arora R, et al. Nocebo effect in dermatology. Indian J Dermatol Venereol Leprol. 2015;81:242-250.
- Bingel U. Avoiding nocebo effects to optimize treatment outcomes. JAMA. 2014;312:693-694.
- Greenwald AG. New look 3: unconscious cognition reclaimed. Am Psychol. 1992;47:766-779.
- Porges SW. The Polyvagal Theory: Neurophysiological Foundations of Emotions, Attachment, Communication, and Self-regulation. New York, NY: W.W. Norton & Co; 2011.
The medical dictum primum non nocere (first, do no harm) is a fundamental rule by which physicians have lived for centuries.1 Physicians are familiar with the term placebo (I shall please) and the placebo effect of improvement based on expectation of positive results; however, many are not familiar with the term nocebo (I shall harm) and the nocebo effect of lack of improvement or deterioration based on the expectation of negative results. The patient’s expectation of being pleased and/or being harmed may be on the conscious level and/or on one or more subconscious and unconscious levels.
Words can have as much of an impact on some patients as medications or procedures. Rudyard Kipling said, “Words are, of course, the most powerful drug used by mankind.” The words that a dermatologist chooses to use can have either a placebo or a nocebo effect on the patient. The purpose of this commentary is to elevate awareness that there are positive alternatives to unintended negative suggestions that are commonly used in dermatologic practice.
A search of PubMed articles indexed for MEDLINE and Scopus published from January 1966 through July 28, 2016, was conducted using the terms placebo or nocebo and cutaneous or skin. Prior publications in this area related specifically to dermatology include those of Poletti2 and Sonthalia et al.3
Patient expectations play an important role in both positive and negative treatment outcomes. Patient-physician communication can moderate these effects both positively and negatively.4 Nocebo effects can substantially reduce treatment efficacy and patient compliance. Patient expectations of negative results or side effects of a treatment or medication can be self-induced or can be induced by inappropriate physician-patient communication, drug information leaflets, influence of family or peers, or discovery of reported adverse effects through reading materials on the Internet.4 Expectation of negative effects can reduce patient adherence and compliance with treatment, reducing treatment efficacy. The psychosocial context around the patient and the treatment may change the neuronal biochemistry and circuitry in the patient’s brain, and the central and peripheral mechanisms activated by placebos and nocebos have been found to be the same as those activated by the medications, suggesting cognitive/affective enhancing or impeding of drug action.5
The subconscious and unconscious habitual automated parts of the brain hear words on the level of a 5- or 6-year-old child with literal unconscious cognitive interpretation of the words. These parts also do not connect words in a sentence with each other.5 For example, if the dermatologist or nurse says “This will not hurt,” the unconscious brain hears not and hurt but does not connect the two. On the other hand, if the dermatologist or nurse says “ You may experience some discomfort,” the unconscious brain hears comfort. Telling the patient “Don’t scratch” may be heard unconsciously as scratch. See the Table for suggested rephrasing of common nocebos used in dermatology. The conscious parts of the brain help determine cognitions influenced by associated unconscious memories, positive or negative. Both the conscious and unconscious parts of the brain influence affect or emotion.
When excess stress occurs, there is a natural shift downward from social communicative newer parasympathetic nervous system to fight or flight sympathetic nervous system, and possibly further shift to the freeze response of older parasympathetic nervous system dominance. Memories that are associated with a strong surge of norepinephrine tend to be much more strongly fixed in the memory than ordinary memories and frequently are associated with overwhelming traumatic experiences. When a threat is perceived, negative interpretations and perceptions generally win out over positive interpretations and perceptions. Unconscious fears generally prevail over conscious thoughts, and stronger emotions generally prevail over weaker emotions. Anxiety often is associated with rapid breathing and activation of the sympathetic nervous system. It can be countered by slow breathing to a rate of approximately 6 breaths per minute, helping to bring back more parasympathetic balance. Pacing a patient’s breathing to slow it and using a soothing tone of voice can help reduce patient anxiety. Reducing anxiety can decrease the patient’s tendency to jump to negative conclusions or have negative perceptions or emotions that can invoke the nocebo effect.
For the dermatologist, as for the patient, changing an old habit and creating a new habit requires repeating something differently and consistently 20 to 40 times. Becoming more conscious of the effects of language on the patient is an important part of the art of medicine. By carefully choosing words, intonation, and body language, it is possible to enhance the placebo effect and decrease the nocebo effect for the benefit of the patient. When describing possible adverse effects of treatments or medications, if the dermatologist says that most people do fine with the treatment but a few can experience the described adverse effect, it usually takes the edge off the potential suggested nocebo effect.
In conclusion, primum non nocere includes careful consideration and use of language, tone, and body language to maximize the placebo effect and minimize the nocebo effect.
The medical dictum primum non nocere (first, do no harm) is a fundamental rule by which physicians have lived for centuries.1 Physicians are familiar with the term placebo (I shall please) and the placebo effect of improvement based on expectation of positive results; however, many are not familiar with the term nocebo (I shall harm) and the nocebo effect of lack of improvement or deterioration based on the expectation of negative results. The patient’s expectation of being pleased and/or being harmed may be on the conscious level and/or on one or more subconscious and unconscious levels.
Words can have as much of an impact on some patients as medications or procedures. Rudyard Kipling said, “Words are, of course, the most powerful drug used by mankind.” The words that a dermatologist chooses to use can have either a placebo or a nocebo effect on the patient. The purpose of this commentary is to elevate awareness that there are positive alternatives to unintended negative suggestions that are commonly used in dermatologic practice.
A search of PubMed articles indexed for MEDLINE and Scopus published from January 1966 through July 28, 2016, was conducted using the terms placebo or nocebo and cutaneous or skin. Prior publications in this area related specifically to dermatology include those of Poletti2 and Sonthalia et al.3
Patient expectations play an important role in both positive and negative treatment outcomes. Patient-physician communication can moderate these effects both positively and negatively.4 Nocebo effects can substantially reduce treatment efficacy and patient compliance. Patient expectations of negative results or side effects of a treatment or medication can be self-induced or can be induced by inappropriate physician-patient communication, drug information leaflets, influence of family or peers, or discovery of reported adverse effects through reading materials on the Internet.4 Expectation of negative effects can reduce patient adherence and compliance with treatment, reducing treatment efficacy. The psychosocial context around the patient and the treatment may change the neuronal biochemistry and circuitry in the patient’s brain, and the central and peripheral mechanisms activated by placebos and nocebos have been found to be the same as those activated by the medications, suggesting cognitive/affective enhancing or impeding of drug action.5
The subconscious and unconscious habitual automated parts of the brain hear words on the level of a 5- or 6-year-old child with literal unconscious cognitive interpretation of the words. These parts also do not connect words in a sentence with each other.5 For example, if the dermatologist or nurse says “This will not hurt,” the unconscious brain hears not and hurt but does not connect the two. On the other hand, if the dermatologist or nurse says “ You may experience some discomfort,” the unconscious brain hears comfort. Telling the patient “Don’t scratch” may be heard unconsciously as scratch. See the Table for suggested rephrasing of common nocebos used in dermatology. The conscious parts of the brain help determine cognitions influenced by associated unconscious memories, positive or negative. Both the conscious and unconscious parts of the brain influence affect or emotion.
When excess stress occurs, there is a natural shift downward from social communicative newer parasympathetic nervous system to fight or flight sympathetic nervous system, and possibly further shift to the freeze response of older parasympathetic nervous system dominance. Memories that are associated with a strong surge of norepinephrine tend to be much more strongly fixed in the memory than ordinary memories and frequently are associated with overwhelming traumatic experiences. When a threat is perceived, negative interpretations and perceptions generally win out over positive interpretations and perceptions. Unconscious fears generally prevail over conscious thoughts, and stronger emotions generally prevail over weaker emotions. Anxiety often is associated with rapid breathing and activation of the sympathetic nervous system. It can be countered by slow breathing to a rate of approximately 6 breaths per minute, helping to bring back more parasympathetic balance. Pacing a patient’s breathing to slow it and using a soothing tone of voice can help reduce patient anxiety. Reducing anxiety can decrease the patient’s tendency to jump to negative conclusions or have negative perceptions or emotions that can invoke the nocebo effect.
For the dermatologist, as for the patient, changing an old habit and creating a new habit requires repeating something differently and consistently 20 to 40 times. Becoming more conscious of the effects of language on the patient is an important part of the art of medicine. By carefully choosing words, intonation, and body language, it is possible to enhance the placebo effect and decrease the nocebo effect for the benefit of the patient. When describing possible adverse effects of treatments or medications, if the dermatologist says that most people do fine with the treatment but a few can experience the described adverse effect, it usually takes the edge off the potential suggested nocebo effect.
In conclusion, primum non nocere includes careful consideration and use of language, tone, and body language to maximize the placebo effect and minimize the nocebo effect.
- Hippocrates. Epidemics. Jones WHS, trans. Loeb Classical Library: Hippocrates. Vol 1. Cambridge, MA: Harvard University Press; 1923:164-165.
- Poletti ED. El efecto nocebo in dermatología. Dermatol Cosmet Quirg. 2007;5:74.
- Sonthalia S, Sahaya K, Arora R, et al. Nocebo effect in dermatology. Indian J Dermatol Venereol Leprol. 2015;81:242-250.
- Bingel U. Avoiding nocebo effects to optimize treatment outcomes. JAMA. 2014;312:693-694.
- Greenwald AG. New look 3: unconscious cognition reclaimed. Am Psychol. 1992;47:766-779.
- Porges SW. The Polyvagal Theory: Neurophysiological Foundations of Emotions, Attachment, Communication, and Self-regulation. New York, NY: W.W. Norton & Co; 2011.
- Hippocrates. Epidemics. Jones WHS, trans. Loeb Classical Library: Hippocrates. Vol 1. Cambridge, MA: Harvard University Press; 1923:164-165.
- Poletti ED. El efecto nocebo in dermatología. Dermatol Cosmet Quirg. 2007;5:74.
- Sonthalia S, Sahaya K, Arora R, et al. Nocebo effect in dermatology. Indian J Dermatol Venereol Leprol. 2015;81:242-250.
- Bingel U. Avoiding nocebo effects to optimize treatment outcomes. JAMA. 2014;312:693-694.
- Greenwald AG. New look 3: unconscious cognition reclaimed. Am Psychol. 1992;47:766-779.
- Porges SW. The Polyvagal Theory: Neurophysiological Foundations of Emotions, Attachment, Communication, and Self-regulation. New York, NY: W.W. Norton & Co; 2011.

Presumed Serum Sickness Following Thymoglobulin Treatment of Acute Cellular Rejection of a Cardiac Allograft
Serum sickness was first described by von Pirquet and Schick1 as a constellation of signs and symptoms displayed in patients receiving equine serum as an antitoxin for the treatment of scarlet fever and diphtheria. Serum sickness is an immune complex–mediated hypersensitivity reaction that can be clinically diagnosed in patients who present with fever, rash, and polyarthralgia or polyarthritis following exposure to heterologous serum proteins.2,3 Symptom onset typically occurs within 1 to 2 weeks of first exposure to the serum, and resolution frequently occurs with discontinuation of the offending agent. Other symptoms may include malaise, gastrointestinal tract concerns, headache, blurred vision, or lymphadenopathy.4 Proteinuria, hematuria, and a transient decrease in creatinine clearance also have been reported in serum sickness.4
Serum sickness is caused by a type III immune complex–mediated hypersensitivity reaction to heterologous rabbit or equine serum proteins. Nonhuman proteins present in antithymocyte globulin (ATG) stimulate the production of IgG, IgM, IgA, and IgE antibodies.2-4 If the resultant immune complexes overwhelm the mononuclear phagocyte system, these complexes are deposited in blood vessels and tissues, which leads to complement activation and the production of complement fragments such as C3a and C5a.5 C3a is an anaphylatoxin that causes mast cell degranulation and the consequent formation of urticarial lesions. C5a is a neutrophil chemoattractant that promotes inflammation at the site of complement deposition.
Serum sickness–like reactions may occur days to weeks following administration of certain drugs, such as cefaclor or penicillin. Although the symptoms and timing of serum sickness–like reactions are similar to serum sickness, they are not caused by an immune complex–mediated mechanism and are believed to be secondary to an idiosyncratic delayed drug reaction.6
Thymoglobulin, a type of ATG, is a polyclonal antibody generated in rabbits that targets numerous human epitopes, including cell surface markers on T cells (CD2, CD3, CD4, CD8), B cells (CD21, CD19, CD40), and adhesion molecules (CD6, CD25, CD44, CD45, and the integrin LFA-1 [lymphocyte function-associated antigen-1]).7,8 Thymoglobulin has proven efficacy in the setting of cardiac transplantation.9-11 Although calcineurin inhibitors form the foundation in the armamentarium of immunosuppressive agents in cardiac transplantation, their nephrotoxicity has limited their unrestrained use in patients.9 By delaying the need for calcineurin inhibitors, thymoglobulin preserves greater renal function without increasing the risk for acute rejection.9,10 Akin to its use in the patient presented in this case report, thymoglobulin also is used in the treatment of acute cellular rejection in heart transplant recipients with signs of heart failure.11
Case Report
A 35-year-old man with a history of familial cardiomyopathy who underwent orthotopic heart transplantation presented with grade 3R acute cellular rejection. The patient’s immunosuppressive regimen consisted of thymoglobulin 150 mg once daily, tacrolimus 2.5 mg twice daily, hydrocortisone 100 mg once daily, and mycophenolate mofetil 1000 mg twice daily. On day 7 of thymoglobulin treatment, the dermatology department was consulted to evaluate a pruritic eruption. The patient reported that he noticed redness of the palms and soles, as well as redness accentuated in the axilla, groin, and other skin creases 2 days prior. The patient also reported symmetric bilateral hand pain that had started 1 day following rash onset. He denied fever and remained afebrile throughout his hospitalization.
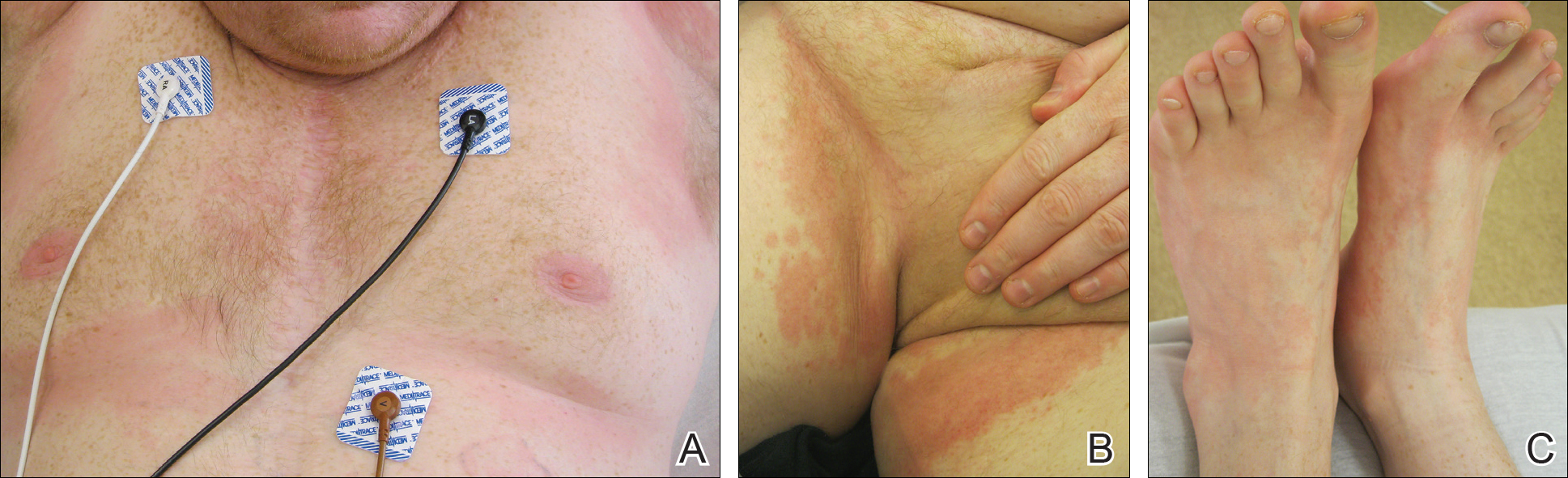
On physical examination, the patient displayed a blanching, erythematous, edematous, evanescent macular rash with some areas of wheal formation symmetrically distributed in the bilateral axillae, inframammary folds, and groin (Figure, A and B). The palms and soles were tender with diffuse blanching erythema. The eruption was accentuated at the lateral and medial borders of both feet (Figure, C). There was concern that the patient may have a form of serum sickness with a blunted incomplete response due to his concomitant use of immunosuppressive agents. Shortly after evaluation, the patient left the hospital against medical advice before the recommended evaluation and systemic workup could be implemented.
The patient returned for an outpatient appointment approximately 1 week later. Medical records indicated that the patient’s skin eruption had resolved. Tests for antithymoglobulin antibodies at this visit were negative. The antithymoglobulin antibody enzyme-linked immunosorbent assay has a diagnostic sensitivity of 86%12 and large interlaboratory variability.13 Given the presence of other features of serum sickness, a false-negative result was considered by dermatology. Nonetheless, one must consider other differential diagnoses, including a simple cutaneous adverse drug eruption or viral exanthem that might have in fact been causative.
Comment
We present an atypical case of possible serum sickness in a heart transplant recipient following thymoglobulin treatment of acute cellular rejection of the cardiac allograft. Serum sickness is a clinical diagnosis supported by laboratory data. Some authors have suggested major and minor diagnostic criteria to aid with the diagnosis.7 Major diagnostic criteria include onset more than 7 days after the initial thymoglobulin administration, persistent high fevers (temperature, >38.4°C), persistent arthritis/arthralgia, and positive heterologous antibodies on enzyme-linked immunosorbent assay. Minor diagnostic criteria include rash, acute renal failure, trismus, and low serum complement (C3 and C4).
The variable cutaneous presentations of serum sickness are important to recognize in the process of making the correct diagnosis. Rash is frequently reported in serum sickness, with some studies displaying rates of up to 93%.4,14 The skin findings are most frequently described as urticarial or serpiginous macular lesions.3 Other variations of the eruption exist, and morbilliform eruptions or a combination of morbilliform and urticarial eruptions have been reported.3 It is important to judge cutaneous eruptions of serum sickness within the context of the potential cytopenia in a patient being treated with ATG. As such, purpuric eruptions have been attributed to serum sickness in thrombocytopenic patients receiving ATG for bone marrow failure.14
Usually, cutaneous eruptions of serum sickness initially are identified in the groin, axilla, and periumbilical region, and then they proceed to include the trunk and extremities. Erythema of the palms and soles frequently is described as well as a linear accentuation of the rash along the lateral and medial borders of the feet and hands at the margin of the plantar or palmar skin, respectively.14 The mucous membranes frequently are spared in serum sickness.
Despite the lack of evidence-based guidelines, case series and literature reviews have suggested a treatment regimen for serum sickness,7,15-18 calling for immediate withdrawal of the offending agent. Antihistamines may be added to control pruritus and rash. Patients with high fever, a progressive rash, or severe arthralgia have benefited from short courses of oral16,18 or intravenous7,17 glucocorticoids. The extent of the eruption in our patient was concerning, particularly because he was already receiving systemic corticosteroids in conjunction with other immunosuppressives, which may have explained his lack of fever.
Because our patient satisfied some diagnostic criteria for serum sickness and failed to satisfy others, our team was faced with the challenge of balancing the risks of possible serum sickness with the risks of the potential for progressive cardiac rejection from the withdrawal of thymoglobulin.7 There is some evidence in the literature for the use of therapeutic plasma exchange (TPE) for the treatment of serum sickness if the offending agent could not be discontinued. Tanriover et al19 presented a case series of 5 renal transplant recipients treated with thymoglobulin who developed serum sickness. The diagnosis of serum sickness was made clinically and augmented by the presence of antiheterologous antibodies. All 5 patients had persistent symptoms of serum sickness despite 2 days of glucocorticoid treatment. Interestingly, 3 patients had complete resolution of all symptoms after a single TPE treatment, and 2 patients achieved resolution of fever and arthritis after 2 consecutive days of TPE treatments.19 Because plasmapheresis is used to treat cardiac allograft rejection in patients showing signs of heart failure,11 the employment of TPE in these patients may have dual beneficial effects of concurrently treating serum sickness and allograft rejection.
Given the patient’s noncompliance and leaving the hospital against medical advice, a full workup was not able to be pursued in this case, though fortunately the eruption and his other symptoms had resolved by the time he was seen for outpatient follow-up 1 week later. Noncompliance with immunosuppressive therapy is a considerable risk factor for morbidity and mortality following heart transplantation. These patients have more transplant coronary artery disease and substantially shorter clinical event-free time.20 Our patient demonstrates the need for proactive compliance-enhancing interventions in heart transplant patients who experience allograft rejection.
- von Pirquet C, Schick B. Serum Sickness. Schick B, trans-ed. Baltimore, MD; Williams & Wilkins; 1951.
- Vincent C, Revillard JP. Antibody response to horse gamma-globulin in recipients of renal allografts: relationship with transplant crises and transplant survival. Transplantation. 1977;24:141-147.
- Lawley TJ, Bielory L, Gascon P, et al. A prospective clinical and immunologic analysis of patients with serum sickness. N Engl J Med. 1984;311:1407-1413.
- Bielory L, Gascon P, Lawley TJ, et al. Human serum sickness: a prospective analysis of 35 patients treated with equine anti-thymocyte globulin for bone marrow failure. Medicine (Baltimore). 1988;67:40-57.
- Chen M, Daha MR, Kallenberg CG. The complement system in systemic autoimmune disease. J Autoimmun. 2010;34:J276-J286.
- Knowles SR, Uetrecht J, Shear NH. Idiosyncratic drug reactions: the reactive metabolite syndromes. Lancet. 2000;356:1587-1591.
- Lundquist AL, Chari RS, Wood JH, et al. Serum sickness following rabbit antithymocyte-globulin induction in a liver transplant recipient: case report and literature review. Liver Transpl. 2007;13:647-650.
- Bourdage JS, Hamlin DM. Comparative polyclonal antithymocyte globulin and antilymphocyte/antilymphoblast globulin anti-CD antigen analysis by flow cytometry. Transplantation. 1995;59:1194-1200.
- Zuckermann AO, Aliabadi AZ. Calcineurin-inhibitor minimization protocols in heart transplantation. Transpl Int. 2009;22:78-89.
- Cantarovich M, Giannetti N, Barkun J, et al. Antithymocyte globulin induction allows a prolonged delay in the initiation of cyclosporine in heart transplant patients with postoperative renal dysfunction. Transplantation. 2004;78:779-781.
- Patel JK, Kittleson M, Kobashigawa JA. Cardiac allograft rejection. Surgeon. 2010;9:160-167.
- Tatum AH, Bollinger RR, Sanfilippo F. Rapid serologic diagnosis of serum sickness from antithymocyte globulin therapy using enzyme immunoassay. Transplantation. 1984;38:582-586.
- Kimball JA, Pescovitz MD, Book BK, et al. Reduced human IgG anti-ATGAM antibody formation in renal transplant recipients receiving mycophenolate mofetil. Transplantation. 1995;60:1379-1383.
- Bielory L, Yancey KB, Young NS, et al. Cutaneous manifestations of serum sickness in patients receiving antithymocyte globulin. J Am Acad Dermatol. 1985;13:411-417.
- Joubert GI, Hadad K, Matsui D, et al. Selection of treatment of cefaclor-associated urticarial, serum sickness-like reactions and erythema multiforme by emergency pediatricians: lack of a uniform standard of care. Can J Clin Pharmacol. 1999;6:197-201.
- Clark BM, Kotti GH, Shah AD, et al. Severe serum sickness reaction to oral and intramuscular penicillin. Pharmacotherapy. 2006;26:705-708.
- Finger E, Scheinberg M. Development of serum sickness-like symptoms after rituximab infusion in two patients with severe hypergammaglobulinemia. J Clin Rheumatol. 2007;13:94-95.
- Tatum AJ, Ditto AM, Patterson R. Severe serum sickness-like reaction to oral penicillin drugs: three case reports. Ann Allergy Asthma Immunol. 2001;86:330-334.
- Tanriover B, Chuang P, Fishbach B, et al. Polyclonal antibody-induced serum sickness in renal transplant recipients: treatment with therapeutic plasma exchange. Transplantation. 2005;80:279-281.
- Dobbels F, De Geest S, van Cleemput J, et al. Effect of late medication non-compliance on outcome after heart transplantation: a 5-year follow-up. J Heart Lung Transplant. 2004;23:1245-1251.
Serum sickness was first described by von Pirquet and Schick1 as a constellation of signs and symptoms displayed in patients receiving equine serum as an antitoxin for the treatment of scarlet fever and diphtheria. Serum sickness is an immune complex–mediated hypersensitivity reaction that can be clinically diagnosed in patients who present with fever, rash, and polyarthralgia or polyarthritis following exposure to heterologous serum proteins.2,3 Symptom onset typically occurs within 1 to 2 weeks of first exposure to the serum, and resolution frequently occurs with discontinuation of the offending agent. Other symptoms may include malaise, gastrointestinal tract concerns, headache, blurred vision, or lymphadenopathy.4 Proteinuria, hematuria, and a transient decrease in creatinine clearance also have been reported in serum sickness.4
Serum sickness is caused by a type III immune complex–mediated hypersensitivity reaction to heterologous rabbit or equine serum proteins. Nonhuman proteins present in antithymocyte globulin (ATG) stimulate the production of IgG, IgM, IgA, and IgE antibodies.2-4 If the resultant immune complexes overwhelm the mononuclear phagocyte system, these complexes are deposited in blood vessels and tissues, which leads to complement activation and the production of complement fragments such as C3a and C5a.5 C3a is an anaphylatoxin that causes mast cell degranulation and the consequent formation of urticarial lesions. C5a is a neutrophil chemoattractant that promotes inflammation at the site of complement deposition.
Serum sickness–like reactions may occur days to weeks following administration of certain drugs, such as cefaclor or penicillin. Although the symptoms and timing of serum sickness–like reactions are similar to serum sickness, they are not caused by an immune complex–mediated mechanism and are believed to be secondary to an idiosyncratic delayed drug reaction.6
Thymoglobulin, a type of ATG, is a polyclonal antibody generated in rabbits that targets numerous human epitopes, including cell surface markers on T cells (CD2, CD3, CD4, CD8), B cells (CD21, CD19, CD40), and adhesion molecules (CD6, CD25, CD44, CD45, and the integrin LFA-1 [lymphocyte function-associated antigen-1]).7,8 Thymoglobulin has proven efficacy in the setting of cardiac transplantation.9-11 Although calcineurin inhibitors form the foundation in the armamentarium of immunosuppressive agents in cardiac transplantation, their nephrotoxicity has limited their unrestrained use in patients.9 By delaying the need for calcineurin inhibitors, thymoglobulin preserves greater renal function without increasing the risk for acute rejection.9,10 Akin to its use in the patient presented in this case report, thymoglobulin also is used in the treatment of acute cellular rejection in heart transplant recipients with signs of heart failure.11
Case Report
A 35-year-old man with a history of familial cardiomyopathy who underwent orthotopic heart transplantation presented with grade 3R acute cellular rejection. The patient’s immunosuppressive regimen consisted of thymoglobulin 150 mg once daily, tacrolimus 2.5 mg twice daily, hydrocortisone 100 mg once daily, and mycophenolate mofetil 1000 mg twice daily. On day 7 of thymoglobulin treatment, the dermatology department was consulted to evaluate a pruritic eruption. The patient reported that he noticed redness of the palms and soles, as well as redness accentuated in the axilla, groin, and other skin creases 2 days prior. The patient also reported symmetric bilateral hand pain that had started 1 day following rash onset. He denied fever and remained afebrile throughout his hospitalization.
On physical examination, the patient displayed a blanching, erythematous, edematous, evanescent macular rash with some areas of wheal formation symmetrically distributed in the bilateral axillae, inframammary folds, and groin (Figure, A and B). The palms and soles were tender with diffuse blanching erythema. The eruption was accentuated at the lateral and medial borders of both feet (Figure, C). There was concern that the patient may have a form of serum sickness with a blunted incomplete response due to his concomitant use of immunosuppressive agents. Shortly after evaluation, the patient left the hospital against medical advice before the recommended evaluation and systemic workup could be implemented.
The patient returned for an outpatient appointment approximately 1 week later. Medical records indicated that the patient’s skin eruption had resolved. Tests for antithymoglobulin antibodies at this visit were negative. The antithymoglobulin antibody enzyme-linked immunosorbent assay has a diagnostic sensitivity of 86%12 and large interlaboratory variability.13 Given the presence of other features of serum sickness, a false-negative result was considered by dermatology. Nonetheless, one must consider other differential diagnoses, including a simple cutaneous adverse drug eruption or viral exanthem that might have in fact been causative.
Comment
We present an atypical case of possible serum sickness in a heart transplant recipient following thymoglobulin treatment of acute cellular rejection of the cardiac allograft. Serum sickness is a clinical diagnosis supported by laboratory data. Some authors have suggested major and minor diagnostic criteria to aid with the diagnosis.7 Major diagnostic criteria include onset more than 7 days after the initial thymoglobulin administration, persistent high fevers (temperature, >38.4°C), persistent arthritis/arthralgia, and positive heterologous antibodies on enzyme-linked immunosorbent assay. Minor diagnostic criteria include rash, acute renal failure, trismus, and low serum complement (C3 and C4).
The variable cutaneous presentations of serum sickness are important to recognize in the process of making the correct diagnosis. Rash is frequently reported in serum sickness, with some studies displaying rates of up to 93%.4,14 The skin findings are most frequently described as urticarial or serpiginous macular lesions.3 Other variations of the eruption exist, and morbilliform eruptions or a combination of morbilliform and urticarial eruptions have been reported.3 It is important to judge cutaneous eruptions of serum sickness within the context of the potential cytopenia in a patient being treated with ATG. As such, purpuric eruptions have been attributed to serum sickness in thrombocytopenic patients receiving ATG for bone marrow failure.14
Usually, cutaneous eruptions of serum sickness initially are identified in the groin, axilla, and periumbilical region, and then they proceed to include the trunk and extremities. Erythema of the palms and soles frequently is described as well as a linear accentuation of the rash along the lateral and medial borders of the feet and hands at the margin of the plantar or palmar skin, respectively.14 The mucous membranes frequently are spared in serum sickness.
Despite the lack of evidence-based guidelines, case series and literature reviews have suggested a treatment regimen for serum sickness,7,15-18 calling for immediate withdrawal of the offending agent. Antihistamines may be added to control pruritus and rash. Patients with high fever, a progressive rash, or severe arthralgia have benefited from short courses of oral16,18 or intravenous7,17 glucocorticoids. The extent of the eruption in our patient was concerning, particularly because he was already receiving systemic corticosteroids in conjunction with other immunosuppressives, which may have explained his lack of fever.
Because our patient satisfied some diagnostic criteria for serum sickness and failed to satisfy others, our team was faced with the challenge of balancing the risks of possible serum sickness with the risks of the potential for progressive cardiac rejection from the withdrawal of thymoglobulin.7 There is some evidence in the literature for the use of therapeutic plasma exchange (TPE) for the treatment of serum sickness if the offending agent could not be discontinued. Tanriover et al19 presented a case series of 5 renal transplant recipients treated with thymoglobulin who developed serum sickness. The diagnosis of serum sickness was made clinically and augmented by the presence of antiheterologous antibodies. All 5 patients had persistent symptoms of serum sickness despite 2 days of glucocorticoid treatment. Interestingly, 3 patients had complete resolution of all symptoms after a single TPE treatment, and 2 patients achieved resolution of fever and arthritis after 2 consecutive days of TPE treatments.19 Because plasmapheresis is used to treat cardiac allograft rejection in patients showing signs of heart failure,11 the employment of TPE in these patients may have dual beneficial effects of concurrently treating serum sickness and allograft rejection.
Given the patient’s noncompliance and leaving the hospital against medical advice, a full workup was not able to be pursued in this case, though fortunately the eruption and his other symptoms had resolved by the time he was seen for outpatient follow-up 1 week later. Noncompliance with immunosuppressive therapy is a considerable risk factor for morbidity and mortality following heart transplantation. These patients have more transplant coronary artery disease and substantially shorter clinical event-free time.20 Our patient demonstrates the need for proactive compliance-enhancing interventions in heart transplant patients who experience allograft rejection.
Serum sickness was first described by von Pirquet and Schick1 as a constellation of signs and symptoms displayed in patients receiving equine serum as an antitoxin for the treatment of scarlet fever and diphtheria. Serum sickness is an immune complex–mediated hypersensitivity reaction that can be clinically diagnosed in patients who present with fever, rash, and polyarthralgia or polyarthritis following exposure to heterologous serum proteins.2,3 Symptom onset typically occurs within 1 to 2 weeks of first exposure to the serum, and resolution frequently occurs with discontinuation of the offending agent. Other symptoms may include malaise, gastrointestinal tract concerns, headache, blurred vision, or lymphadenopathy.4 Proteinuria, hematuria, and a transient decrease in creatinine clearance also have been reported in serum sickness.4
Serum sickness is caused by a type III immune complex–mediated hypersensitivity reaction to heterologous rabbit or equine serum proteins. Nonhuman proteins present in antithymocyte globulin (ATG) stimulate the production of IgG, IgM, IgA, and IgE antibodies.2-4 If the resultant immune complexes overwhelm the mononuclear phagocyte system, these complexes are deposited in blood vessels and tissues, which leads to complement activation and the production of complement fragments such as C3a and C5a.5 C3a is an anaphylatoxin that causes mast cell degranulation and the consequent formation of urticarial lesions. C5a is a neutrophil chemoattractant that promotes inflammation at the site of complement deposition.
Serum sickness–like reactions may occur days to weeks following administration of certain drugs, such as cefaclor or penicillin. Although the symptoms and timing of serum sickness–like reactions are similar to serum sickness, they are not caused by an immune complex–mediated mechanism and are believed to be secondary to an idiosyncratic delayed drug reaction.6
Thymoglobulin, a type of ATG, is a polyclonal antibody generated in rabbits that targets numerous human epitopes, including cell surface markers on T cells (CD2, CD3, CD4, CD8), B cells (CD21, CD19, CD40), and adhesion molecules (CD6, CD25, CD44, CD45, and the integrin LFA-1 [lymphocyte function-associated antigen-1]).7,8 Thymoglobulin has proven efficacy in the setting of cardiac transplantation.9-11 Although calcineurin inhibitors form the foundation in the armamentarium of immunosuppressive agents in cardiac transplantation, their nephrotoxicity has limited their unrestrained use in patients.9 By delaying the need for calcineurin inhibitors, thymoglobulin preserves greater renal function without increasing the risk for acute rejection.9,10 Akin to its use in the patient presented in this case report, thymoglobulin also is used in the treatment of acute cellular rejection in heart transplant recipients with signs of heart failure.11
Case Report
A 35-year-old man with a history of familial cardiomyopathy who underwent orthotopic heart transplantation presented with grade 3R acute cellular rejection. The patient’s immunosuppressive regimen consisted of thymoglobulin 150 mg once daily, tacrolimus 2.5 mg twice daily, hydrocortisone 100 mg once daily, and mycophenolate mofetil 1000 mg twice daily. On day 7 of thymoglobulin treatment, the dermatology department was consulted to evaluate a pruritic eruption. The patient reported that he noticed redness of the palms and soles, as well as redness accentuated in the axilla, groin, and other skin creases 2 days prior. The patient also reported symmetric bilateral hand pain that had started 1 day following rash onset. He denied fever and remained afebrile throughout his hospitalization.
On physical examination, the patient displayed a blanching, erythematous, edematous, evanescent macular rash with some areas of wheal formation symmetrically distributed in the bilateral axillae, inframammary folds, and groin (Figure, A and B). The palms and soles were tender with diffuse blanching erythema. The eruption was accentuated at the lateral and medial borders of both feet (Figure, C). There was concern that the patient may have a form of serum sickness with a blunted incomplete response due to his concomitant use of immunosuppressive agents. Shortly after evaluation, the patient left the hospital against medical advice before the recommended evaluation and systemic workup could be implemented.
The patient returned for an outpatient appointment approximately 1 week later. Medical records indicated that the patient’s skin eruption had resolved. Tests for antithymoglobulin antibodies at this visit were negative. The antithymoglobulin antibody enzyme-linked immunosorbent assay has a diagnostic sensitivity of 86%12 and large interlaboratory variability.13 Given the presence of other features of serum sickness, a false-negative result was considered by dermatology. Nonetheless, one must consider other differential diagnoses, including a simple cutaneous adverse drug eruption or viral exanthem that might have in fact been causative.
Comment
We present an atypical case of possible serum sickness in a heart transplant recipient following thymoglobulin treatment of acute cellular rejection of the cardiac allograft. Serum sickness is a clinical diagnosis supported by laboratory data. Some authors have suggested major and minor diagnostic criteria to aid with the diagnosis.7 Major diagnostic criteria include onset more than 7 days after the initial thymoglobulin administration, persistent high fevers (temperature, >38.4°C), persistent arthritis/arthralgia, and positive heterologous antibodies on enzyme-linked immunosorbent assay. Minor diagnostic criteria include rash, acute renal failure, trismus, and low serum complement (C3 and C4).
The variable cutaneous presentations of serum sickness are important to recognize in the process of making the correct diagnosis. Rash is frequently reported in serum sickness, with some studies displaying rates of up to 93%.4,14 The skin findings are most frequently described as urticarial or serpiginous macular lesions.3 Other variations of the eruption exist, and morbilliform eruptions or a combination of morbilliform and urticarial eruptions have been reported.3 It is important to judge cutaneous eruptions of serum sickness within the context of the potential cytopenia in a patient being treated with ATG. As such, purpuric eruptions have been attributed to serum sickness in thrombocytopenic patients receiving ATG for bone marrow failure.14
Usually, cutaneous eruptions of serum sickness initially are identified in the groin, axilla, and periumbilical region, and then they proceed to include the trunk and extremities. Erythema of the palms and soles frequently is described as well as a linear accentuation of the rash along the lateral and medial borders of the feet and hands at the margin of the plantar or palmar skin, respectively.14 The mucous membranes frequently are spared in serum sickness.
Despite the lack of evidence-based guidelines, case series and literature reviews have suggested a treatment regimen for serum sickness,7,15-18 calling for immediate withdrawal of the offending agent. Antihistamines may be added to control pruritus and rash. Patients with high fever, a progressive rash, or severe arthralgia have benefited from short courses of oral16,18 or intravenous7,17 glucocorticoids. The extent of the eruption in our patient was concerning, particularly because he was already receiving systemic corticosteroids in conjunction with other immunosuppressives, which may have explained his lack of fever.
Because our patient satisfied some diagnostic criteria for serum sickness and failed to satisfy others, our team was faced with the challenge of balancing the risks of possible serum sickness with the risks of the potential for progressive cardiac rejection from the withdrawal of thymoglobulin.7 There is some evidence in the literature for the use of therapeutic plasma exchange (TPE) for the treatment of serum sickness if the offending agent could not be discontinued. Tanriover et al19 presented a case series of 5 renal transplant recipients treated with thymoglobulin who developed serum sickness. The diagnosis of serum sickness was made clinically and augmented by the presence of antiheterologous antibodies. All 5 patients had persistent symptoms of serum sickness despite 2 days of glucocorticoid treatment. Interestingly, 3 patients had complete resolution of all symptoms after a single TPE treatment, and 2 patients achieved resolution of fever and arthritis after 2 consecutive days of TPE treatments.19 Because plasmapheresis is used to treat cardiac allograft rejection in patients showing signs of heart failure,11 the employment of TPE in these patients may have dual beneficial effects of concurrently treating serum sickness and allograft rejection.
Given the patient’s noncompliance and leaving the hospital against medical advice, a full workup was not able to be pursued in this case, though fortunately the eruption and his other symptoms had resolved by the time he was seen for outpatient follow-up 1 week later. Noncompliance with immunosuppressive therapy is a considerable risk factor for morbidity and mortality following heart transplantation. These patients have more transplant coronary artery disease and substantially shorter clinical event-free time.20 Our patient demonstrates the need for proactive compliance-enhancing interventions in heart transplant patients who experience allograft rejection.
- von Pirquet C, Schick B. Serum Sickness. Schick B, trans-ed. Baltimore, MD; Williams & Wilkins; 1951.
- Vincent C, Revillard JP. Antibody response to horse gamma-globulin in recipients of renal allografts: relationship with transplant crises and transplant survival. Transplantation. 1977;24:141-147.
- Lawley TJ, Bielory L, Gascon P, et al. A prospective clinical and immunologic analysis of patients with serum sickness. N Engl J Med. 1984;311:1407-1413.
- Bielory L, Gascon P, Lawley TJ, et al. Human serum sickness: a prospective analysis of 35 patients treated with equine anti-thymocyte globulin for bone marrow failure. Medicine (Baltimore). 1988;67:40-57.
- Chen M, Daha MR, Kallenberg CG. The complement system in systemic autoimmune disease. J Autoimmun. 2010;34:J276-J286.
- Knowles SR, Uetrecht J, Shear NH. Idiosyncratic drug reactions: the reactive metabolite syndromes. Lancet. 2000;356:1587-1591.
- Lundquist AL, Chari RS, Wood JH, et al. Serum sickness following rabbit antithymocyte-globulin induction in a liver transplant recipient: case report and literature review. Liver Transpl. 2007;13:647-650.
- Bourdage JS, Hamlin DM. Comparative polyclonal antithymocyte globulin and antilymphocyte/antilymphoblast globulin anti-CD antigen analysis by flow cytometry. Transplantation. 1995;59:1194-1200.
- Zuckermann AO, Aliabadi AZ. Calcineurin-inhibitor minimization protocols in heart transplantation. Transpl Int. 2009;22:78-89.
- Cantarovich M, Giannetti N, Barkun J, et al. Antithymocyte globulin induction allows a prolonged delay in the initiation of cyclosporine in heart transplant patients with postoperative renal dysfunction. Transplantation. 2004;78:779-781.
- Patel JK, Kittleson M, Kobashigawa JA. Cardiac allograft rejection. Surgeon. 2010;9:160-167.
- Tatum AH, Bollinger RR, Sanfilippo F. Rapid serologic diagnosis of serum sickness from antithymocyte globulin therapy using enzyme immunoassay. Transplantation. 1984;38:582-586.
- Kimball JA, Pescovitz MD, Book BK, et al. Reduced human IgG anti-ATGAM antibody formation in renal transplant recipients receiving mycophenolate mofetil. Transplantation. 1995;60:1379-1383.
- Bielory L, Yancey KB, Young NS, et al. Cutaneous manifestations of serum sickness in patients receiving antithymocyte globulin. J Am Acad Dermatol. 1985;13:411-417.
- Joubert GI, Hadad K, Matsui D, et al. Selection of treatment of cefaclor-associated urticarial, serum sickness-like reactions and erythema multiforme by emergency pediatricians: lack of a uniform standard of care. Can J Clin Pharmacol. 1999;6:197-201.
- Clark BM, Kotti GH, Shah AD, et al. Severe serum sickness reaction to oral and intramuscular penicillin. Pharmacotherapy. 2006;26:705-708.
- Finger E, Scheinberg M. Development of serum sickness-like symptoms after rituximab infusion in two patients with severe hypergammaglobulinemia. J Clin Rheumatol. 2007;13:94-95.
- Tatum AJ, Ditto AM, Patterson R. Severe serum sickness-like reaction to oral penicillin drugs: three case reports. Ann Allergy Asthma Immunol. 2001;86:330-334.
- Tanriover B, Chuang P, Fishbach B, et al. Polyclonal antibody-induced serum sickness in renal transplant recipients: treatment with therapeutic plasma exchange. Transplantation. 2005;80:279-281.
- Dobbels F, De Geest S, van Cleemput J, et al. Effect of late medication non-compliance on outcome after heart transplantation: a 5-year follow-up. J Heart Lung Transplant. 2004;23:1245-1251.
- von Pirquet C, Schick B. Serum Sickness. Schick B, trans-ed. Baltimore, MD; Williams & Wilkins; 1951.
- Vincent C, Revillard JP. Antibody response to horse gamma-globulin in recipients of renal allografts: relationship with transplant crises and transplant survival. Transplantation. 1977;24:141-147.
- Lawley TJ, Bielory L, Gascon P, et al. A prospective clinical and immunologic analysis of patients with serum sickness. N Engl J Med. 1984;311:1407-1413.
- Bielory L, Gascon P, Lawley TJ, et al. Human serum sickness: a prospective analysis of 35 patients treated with equine anti-thymocyte globulin for bone marrow failure. Medicine (Baltimore). 1988;67:40-57.
- Chen M, Daha MR, Kallenberg CG. The complement system in systemic autoimmune disease. J Autoimmun. 2010;34:J276-J286.
- Knowles SR, Uetrecht J, Shear NH. Idiosyncratic drug reactions: the reactive metabolite syndromes. Lancet. 2000;356:1587-1591.
- Lundquist AL, Chari RS, Wood JH, et al. Serum sickness following rabbit antithymocyte-globulin induction in a liver transplant recipient: case report and literature review. Liver Transpl. 2007;13:647-650.
- Bourdage JS, Hamlin DM. Comparative polyclonal antithymocyte globulin and antilymphocyte/antilymphoblast globulin anti-CD antigen analysis by flow cytometry. Transplantation. 1995;59:1194-1200.
- Zuckermann AO, Aliabadi AZ. Calcineurin-inhibitor minimization protocols in heart transplantation. Transpl Int. 2009;22:78-89.
- Cantarovich M, Giannetti N, Barkun J, et al. Antithymocyte globulin induction allows a prolonged delay in the initiation of cyclosporine in heart transplant patients with postoperative renal dysfunction. Transplantation. 2004;78:779-781.
- Patel JK, Kittleson M, Kobashigawa JA. Cardiac allograft rejection. Surgeon. 2010;9:160-167.
- Tatum AH, Bollinger RR, Sanfilippo F. Rapid serologic diagnosis of serum sickness from antithymocyte globulin therapy using enzyme immunoassay. Transplantation. 1984;38:582-586.
- Kimball JA, Pescovitz MD, Book BK, et al. Reduced human IgG anti-ATGAM antibody formation in renal transplant recipients receiving mycophenolate mofetil. Transplantation. 1995;60:1379-1383.
- Bielory L, Yancey KB, Young NS, et al. Cutaneous manifestations of serum sickness in patients receiving antithymocyte globulin. J Am Acad Dermatol. 1985;13:411-417.
- Joubert GI, Hadad K, Matsui D, et al. Selection of treatment of cefaclor-associated urticarial, serum sickness-like reactions and erythema multiforme by emergency pediatricians: lack of a uniform standard of care. Can J Clin Pharmacol. 1999;6:197-201.
- Clark BM, Kotti GH, Shah AD, et al. Severe serum sickness reaction to oral and intramuscular penicillin. Pharmacotherapy. 2006;26:705-708.
- Finger E, Scheinberg M. Development of serum sickness-like symptoms after rituximab infusion in two patients with severe hypergammaglobulinemia. J Clin Rheumatol. 2007;13:94-95.
- Tatum AJ, Ditto AM, Patterson R. Severe serum sickness-like reaction to oral penicillin drugs: three case reports. Ann Allergy Asthma Immunol. 2001;86:330-334.
- Tanriover B, Chuang P, Fishbach B, et al. Polyclonal antibody-induced serum sickness in renal transplant recipients: treatment with therapeutic plasma exchange. Transplantation. 2005;80:279-281.
- Dobbels F, De Geest S, van Cleemput J, et al. Effect of late medication non-compliance on outcome after heart transplantation: a 5-year follow-up. J Heart Lung Transplant. 2004;23:1245-1251.
Practice Points
- Serum sickness can be seen in patients treated with thymoglobulin to prevent transplant rejection.
- Serum sickness can display multiple cutaneous manifestation, thus making it an important entity for dermatologists.
Acronymic Despair: MACRA, MIPS, and Me
The year is moving ahead, and we are in the first year with a new president and a new administration. There have been multiple attempts to defund, revoke, or otherwise eliminate the Patient Protection and Affordable Care Act. As a physician, you may be asking, “What should I be doing for MACRA (Medicare Access and CHIP Reauthorization Act of 2015) and MIPS (Merit-Based Incentive Payments System)?”
RELATED VIDEO: Update on Coding Changes: Report From the Mount Sinai Fall Symposium
What is MACRA?
Of course, there is no such thing as a free lunch. The less pleasant side of MACRA is the Quality Payment Program under which providers will be paid based on the quality and effectiveness of the care provided; physician assistants, nurse practitioners, clinical nurse specialists, and certified registered nurse anesthetists also will be under the new system in addition to physicians. We are to be paid based on value, not volume. Heady stuff. The devil, as always, is in the details, as the factors we will be measured against are diverse. Having an electronic medical record (EMR) can make capturing data for some of these measures a bit less onerous. If you do not have an EMR, the cost of transitioning to one, especially if you are a small solo practice or approaching the end of your career, may outweigh the benefits.
RELATED VIDEO: Update on Coding Changes: Report From the Mount Sinai Fall Symposium
What is MIPS?
A small group of providers, most likely those in large multispecialty groups or academic settings, will instead participate in advanced Alternative Payment Models that will provide a lump sum bonus payment of 5% of their Medicare charges from 2019 to 2024.
For those taking the more common MIPS pathway, beginning in 2019 you can see a penalty of up to 4% on your Medicare payments if you do nothing and a bonus of up to 4% if you do it all. This rate will increase to a 5% penalty or a reward of up to 5% in 2020, 7% in 2021, and 9% in 2022. The penalty is a result of nonparticipation, while complete participation might get you to the maximum bonus. Of course, the bonus pool is limited, and if everyone does it all, the bonus would be much less, assuming the program is not changed or eliminated by the current administration. At the time of writing this column, Senate Majority Leader Mitch McConnell (R-KY) has failed multiple times to pass a Patient Protection and Affordable Care Act repeal bill following rebellions in his own party.4
So what do you, dear colleague, need to do right now, or at least before the end of the calendar year? You could do it all and try to grab the brass ring 4% bonus for 2019, putting time, effort, and expense into going after what could be an elusive reward. Or you could simply avoid the penalty and go back to work knowing you have locked in normal payments (whatever that will be!) for 2019. We are both doing the latter, and so might you, especially if you have not done anything yet this year.
MIPS Made Merry
To learn what you need to do or can do, pay a visit to the Quality Payment Program website (https://qpp.cms.gov/) where you can look yourself up with your national provider identifier number and find out what system you are under. Unless you are part of a large enterprise, you are likely under MIPS, but it never hurts to check.
It will then give you the options for reporting as an individual or a group. Either way, you can send in quality data through your routine Medicare claims process, which is our suggested route; no registry, no EMR, just an extra line on a claim form. You can review the complete list of quality measures that are available on the Quality Payment Program website (https://qpp.cms.gov/mips/quality-measures). There are 271 measures to read through and ponder, but by now you already have a headache, so take the following advice:
- Filter with the “Data Submission Method” by checking off “Claims,” which gives you 74 choices.
- Filter further with the “Specialty Measure Set”by checking off “Dermatology,” which gives you 4 choices.
- The top choice and probably the easiest one to get your staff to help with is “Documentation of Current Medications in the Medical Record,” which if you click on it further identifies it as “Quality ID: 130,” the official name of this measure.
You can see the MIPS program information in all its bureaucratic glory on the Quality Payment Program website (https://qpp.cms.gov/resources/education); click on “Quality Measure Specifications” to download a 250 MB zip file that contains information on all the measures in detail. The Measure #130 (Documentation of Current Medications in the Medical Record) file indicates that the clinician must use a G code (G8427) to report that current medications have been documented. The measure reads: “Eligible clinician attests to documenting, updating or reviewing a patient’s current medications using all immediate resources available on the date of encounter. This list must include ALL known prescriptions, over-the counters, herbals, and vitamin/mineral/dietary (nutritional) supplements AND must contain the medications’ name, dosages, frequency and route of administration.”5
You likely already confirm current medications with patients in some form or other, so simply look at the list of medications and supplements with all their dosages, frequencies, and routes of administration and sign the sheet of paper your practice likely already uses as an extra way of confirming that you have reviewed it. You report code G8427 as you would any Current Procedural Terminology code and link it to any International Classification of Diseases, Tenth Revision, code in your claim along with any evaluation and management and/or procedure codes that you would otherwise report for that encounter.
Some clearinghouses will not accept $0 charges, so we recommend you place a $0.01 charge for G8427 and write it off later. Upon receiving your explanation of benefits, you should notice 2 remark codes relating to the G8427 line: CO-246 and N620. Both of these codes indicate that the Centers for Medicare & Medicaid Services acknowledge your quality submission. To avoid that 4% penalty in 2019, you only need to do it once, but doing it a few times until you get back an explanation of benefits acknowledging it may help you sleep better.
Conclusion
Although the future of the Patient Protection and Affordable Care Act is still unclear, one thing is for sure: MACRA and MIPS are here to stay. Avoid the 4% penalty in 2019 and take good care of your patients and, if eligible, make donations to the American Academy of Dermatology Association Political Action Committee (skinPAC). It is going to be a wild ride.
- MACRA: delivery system reform, Medicare payment reform. Centers for Medicare & Medicaid Services website. https://www.cms.gov/Medicare/Quality-Initiatives-Patient-Assessment-Instruments/Value-Based-Programs/MACRA-MIPS-and-APMs/MACRA-MIPS-and-APMs.html. Updated June 26, 2016. Accessed August 1, 2017.
- MACRA tools and resources. American Academy of Dermatology website. https://www.aad.org/practicecenter/coding-and-reimbursement/macra. Accessed August 1, 2017.
- Balanced Budget Act of 1997. Senate and House of Representatives of the United States of America. https://www.gpo.gov/fdsys/pkg/PLAW-105publ33/html/PLAW-105publ33.htm. Accessed August 1, 2017.
- Bruni F. The misery of Mitch McConnell. New York Times. June 28, 2017. https://www.nytimes.com/2017/06/28/opinion/mitch-mcconnell-health-care-misery.html. Accessed August 1, 2017.
- American Medical Association. Measure #130 (NQF 0419): Documentation of Current Medications in the Medical Record-National Quality Strategy Domain: Patient Safety. Published November 15, 2016. Accessed August 18, 2016.
The year is moving ahead, and we are in the first year with a new president and a new administration. There have been multiple attempts to defund, revoke, or otherwise eliminate the Patient Protection and Affordable Care Act. As a physician, you may be asking, “What should I be doing for MACRA (Medicare Access and CHIP Reauthorization Act of 2015) and MIPS (Merit-Based Incentive Payments System)?”
RELATED VIDEO: Update on Coding Changes: Report From the Mount Sinai Fall Symposium
What is MACRA?
Of course, there is no such thing as a free lunch. The less pleasant side of MACRA is the Quality Payment Program under which providers will be paid based on the quality and effectiveness of the care provided; physician assistants, nurse practitioners, clinical nurse specialists, and certified registered nurse anesthetists also will be under the new system in addition to physicians. We are to be paid based on value, not volume. Heady stuff. The devil, as always, is in the details, as the factors we will be measured against are diverse. Having an electronic medical record (EMR) can make capturing data for some of these measures a bit less onerous. If you do not have an EMR, the cost of transitioning to one, especially if you are a small solo practice or approaching the end of your career, may outweigh the benefits.
RELATED VIDEO: Update on Coding Changes: Report From the Mount Sinai Fall Symposium
What is MIPS?
A small group of providers, most likely those in large multispecialty groups or academic settings, will instead participate in advanced Alternative Payment Models that will provide a lump sum bonus payment of 5% of their Medicare charges from 2019 to 2024.
For those taking the more common MIPS pathway, beginning in 2019 you can see a penalty of up to 4% on your Medicare payments if you do nothing and a bonus of up to 4% if you do it all. This rate will increase to a 5% penalty or a reward of up to 5% in 2020, 7% in 2021, and 9% in 2022. The penalty is a result of nonparticipation, while complete participation might get you to the maximum bonus. Of course, the bonus pool is limited, and if everyone does it all, the bonus would be much less, assuming the program is not changed or eliminated by the current administration. At the time of writing this column, Senate Majority Leader Mitch McConnell (R-KY) has failed multiple times to pass a Patient Protection and Affordable Care Act repeal bill following rebellions in his own party.4
So what do you, dear colleague, need to do right now, or at least before the end of the calendar year? You could do it all and try to grab the brass ring 4% bonus for 2019, putting time, effort, and expense into going after what could be an elusive reward. Or you could simply avoid the penalty and go back to work knowing you have locked in normal payments (whatever that will be!) for 2019. We are both doing the latter, and so might you, especially if you have not done anything yet this year.
MIPS Made Merry
To learn what you need to do or can do, pay a visit to the Quality Payment Program website (https://qpp.cms.gov/) where you can look yourself up with your national provider identifier number and find out what system you are under. Unless you are part of a large enterprise, you are likely under MIPS, but it never hurts to check.
It will then give you the options for reporting as an individual or a group. Either way, you can send in quality data through your routine Medicare claims process, which is our suggested route; no registry, no EMR, just an extra line on a claim form. You can review the complete list of quality measures that are available on the Quality Payment Program website (https://qpp.cms.gov/mips/quality-measures). There are 271 measures to read through and ponder, but by now you already have a headache, so take the following advice:
- Filter with the “Data Submission Method” by checking off “Claims,” which gives you 74 choices.
- Filter further with the “Specialty Measure Set”by checking off “Dermatology,” which gives you 4 choices.
- The top choice and probably the easiest one to get your staff to help with is “Documentation of Current Medications in the Medical Record,” which if you click on it further identifies it as “Quality ID: 130,” the official name of this measure.
You can see the MIPS program information in all its bureaucratic glory on the Quality Payment Program website (https://qpp.cms.gov/resources/education); click on “Quality Measure Specifications” to download a 250 MB zip file that contains information on all the measures in detail. The Measure #130 (Documentation of Current Medications in the Medical Record) file indicates that the clinician must use a G code (G8427) to report that current medications have been documented. The measure reads: “Eligible clinician attests to documenting, updating or reviewing a patient’s current medications using all immediate resources available on the date of encounter. This list must include ALL known prescriptions, over-the counters, herbals, and vitamin/mineral/dietary (nutritional) supplements AND must contain the medications’ name, dosages, frequency and route of administration.”5
You likely already confirm current medications with patients in some form or other, so simply look at the list of medications and supplements with all their dosages, frequencies, and routes of administration and sign the sheet of paper your practice likely already uses as an extra way of confirming that you have reviewed it. You report code G8427 as you would any Current Procedural Terminology code and link it to any International Classification of Diseases, Tenth Revision, code in your claim along with any evaluation and management and/or procedure codes that you would otherwise report for that encounter.
Some clearinghouses will not accept $0 charges, so we recommend you place a $0.01 charge for G8427 and write it off later. Upon receiving your explanation of benefits, you should notice 2 remark codes relating to the G8427 line: CO-246 and N620. Both of these codes indicate that the Centers for Medicare & Medicaid Services acknowledge your quality submission. To avoid that 4% penalty in 2019, you only need to do it once, but doing it a few times until you get back an explanation of benefits acknowledging it may help you sleep better.
Conclusion
Although the future of the Patient Protection and Affordable Care Act is still unclear, one thing is for sure: MACRA and MIPS are here to stay. Avoid the 4% penalty in 2019 and take good care of your patients and, if eligible, make donations to the American Academy of Dermatology Association Political Action Committee (skinPAC). It is going to be a wild ride.
The year is moving ahead, and we are in the first year with a new president and a new administration. There have been multiple attempts to defund, revoke, or otherwise eliminate the Patient Protection and Affordable Care Act. As a physician, you may be asking, “What should I be doing for MACRA (Medicare Access and CHIP Reauthorization Act of 2015) and MIPS (Merit-Based Incentive Payments System)?”
RELATED VIDEO: Update on Coding Changes: Report From the Mount Sinai Fall Symposium
What is MACRA?
Of course, there is no such thing as a free lunch. The less pleasant side of MACRA is the Quality Payment Program under which providers will be paid based on the quality and effectiveness of the care provided; physician assistants, nurse practitioners, clinical nurse specialists, and certified registered nurse anesthetists also will be under the new system in addition to physicians. We are to be paid based on value, not volume. Heady stuff. The devil, as always, is in the details, as the factors we will be measured against are diverse. Having an electronic medical record (EMR) can make capturing data for some of these measures a bit less onerous. If you do not have an EMR, the cost of transitioning to one, especially if you are a small solo practice or approaching the end of your career, may outweigh the benefits.
RELATED VIDEO: Update on Coding Changes: Report From the Mount Sinai Fall Symposium
What is MIPS?
A small group of providers, most likely those in large multispecialty groups or academic settings, will instead participate in advanced Alternative Payment Models that will provide a lump sum bonus payment of 5% of their Medicare charges from 2019 to 2024.
For those taking the more common MIPS pathway, beginning in 2019 you can see a penalty of up to 4% on your Medicare payments if you do nothing and a bonus of up to 4% if you do it all. This rate will increase to a 5% penalty or a reward of up to 5% in 2020, 7% in 2021, and 9% in 2022. The penalty is a result of nonparticipation, while complete participation might get you to the maximum bonus. Of course, the bonus pool is limited, and if everyone does it all, the bonus would be much less, assuming the program is not changed or eliminated by the current administration. At the time of writing this column, Senate Majority Leader Mitch McConnell (R-KY) has failed multiple times to pass a Patient Protection and Affordable Care Act repeal bill following rebellions in his own party.4
So what do you, dear colleague, need to do right now, or at least before the end of the calendar year? You could do it all and try to grab the brass ring 4% bonus for 2019, putting time, effort, and expense into going after what could be an elusive reward. Or you could simply avoid the penalty and go back to work knowing you have locked in normal payments (whatever that will be!) for 2019. We are both doing the latter, and so might you, especially if you have not done anything yet this year.
MIPS Made Merry
To learn what you need to do or can do, pay a visit to the Quality Payment Program website (https://qpp.cms.gov/) where you can look yourself up with your national provider identifier number and find out what system you are under. Unless you are part of a large enterprise, you are likely under MIPS, but it never hurts to check.
It will then give you the options for reporting as an individual or a group. Either way, you can send in quality data through your routine Medicare claims process, which is our suggested route; no registry, no EMR, just an extra line on a claim form. You can review the complete list of quality measures that are available on the Quality Payment Program website (https://qpp.cms.gov/mips/quality-measures). There are 271 measures to read through and ponder, but by now you already have a headache, so take the following advice:
- Filter with the “Data Submission Method” by checking off “Claims,” which gives you 74 choices.
- Filter further with the “Specialty Measure Set”by checking off “Dermatology,” which gives you 4 choices.
- The top choice and probably the easiest one to get your staff to help with is “Documentation of Current Medications in the Medical Record,” which if you click on it further identifies it as “Quality ID: 130,” the official name of this measure.
You can see the MIPS program information in all its bureaucratic glory on the Quality Payment Program website (https://qpp.cms.gov/resources/education); click on “Quality Measure Specifications” to download a 250 MB zip file that contains information on all the measures in detail. The Measure #130 (Documentation of Current Medications in the Medical Record) file indicates that the clinician must use a G code (G8427) to report that current medications have been documented. The measure reads: “Eligible clinician attests to documenting, updating or reviewing a patient’s current medications using all immediate resources available on the date of encounter. This list must include ALL known prescriptions, over-the counters, herbals, and vitamin/mineral/dietary (nutritional) supplements AND must contain the medications’ name, dosages, frequency and route of administration.”5
You likely already confirm current medications with patients in some form or other, so simply look at the list of medications and supplements with all their dosages, frequencies, and routes of administration and sign the sheet of paper your practice likely already uses as an extra way of confirming that you have reviewed it. You report code G8427 as you would any Current Procedural Terminology code and link it to any International Classification of Diseases, Tenth Revision, code in your claim along with any evaluation and management and/or procedure codes that you would otherwise report for that encounter.
Some clearinghouses will not accept $0 charges, so we recommend you place a $0.01 charge for G8427 and write it off later. Upon receiving your explanation of benefits, you should notice 2 remark codes relating to the G8427 line: CO-246 and N620. Both of these codes indicate that the Centers for Medicare & Medicaid Services acknowledge your quality submission. To avoid that 4% penalty in 2019, you only need to do it once, but doing it a few times until you get back an explanation of benefits acknowledging it may help you sleep better.
Conclusion
Although the future of the Patient Protection and Affordable Care Act is still unclear, one thing is for sure: MACRA and MIPS are here to stay. Avoid the 4% penalty in 2019 and take good care of your patients and, if eligible, make donations to the American Academy of Dermatology Association Political Action Committee (skinPAC). It is going to be a wild ride.
- MACRA: delivery system reform, Medicare payment reform. Centers for Medicare & Medicaid Services website. https://www.cms.gov/Medicare/Quality-Initiatives-Patient-Assessment-Instruments/Value-Based-Programs/MACRA-MIPS-and-APMs/MACRA-MIPS-and-APMs.html. Updated June 26, 2016. Accessed August 1, 2017.
- MACRA tools and resources. American Academy of Dermatology website. https://www.aad.org/practicecenter/coding-and-reimbursement/macra. Accessed August 1, 2017.
- Balanced Budget Act of 1997. Senate and House of Representatives of the United States of America. https://www.gpo.gov/fdsys/pkg/PLAW-105publ33/html/PLAW-105publ33.htm. Accessed August 1, 2017.
- Bruni F. The misery of Mitch McConnell. New York Times. June 28, 2017. https://www.nytimes.com/2017/06/28/opinion/mitch-mcconnell-health-care-misery.html. Accessed August 1, 2017.
- American Medical Association. Measure #130 (NQF 0419): Documentation of Current Medications in the Medical Record-National Quality Strategy Domain: Patient Safety. Published November 15, 2016. Accessed August 18, 2016.
- MACRA: delivery system reform, Medicare payment reform. Centers for Medicare & Medicaid Services website. https://www.cms.gov/Medicare/Quality-Initiatives-Patient-Assessment-Instruments/Value-Based-Programs/MACRA-MIPS-and-APMs/MACRA-MIPS-and-APMs.html. Updated June 26, 2016. Accessed August 1, 2017.
- MACRA tools and resources. American Academy of Dermatology website. https://www.aad.org/practicecenter/coding-and-reimbursement/macra. Accessed August 1, 2017.
- Balanced Budget Act of 1997. Senate and House of Representatives of the United States of America. https://www.gpo.gov/fdsys/pkg/PLAW-105publ33/html/PLAW-105publ33.htm. Accessed August 1, 2017.
- Bruni F. The misery of Mitch McConnell. New York Times. June 28, 2017. https://www.nytimes.com/2017/06/28/opinion/mitch-mcconnell-health-care-misery.html. Accessed August 1, 2017.
- American Medical Association. Measure #130 (NQF 0419): Documentation of Current Medications in the Medical Record-National Quality Strategy Domain: Patient Safety. Published November 15, 2016. Accessed August 18, 2016.
Practice Points
- MACRA (Medicare Access and CHIP Reauthorization Act of 2015) and MIPS (Merit-Based Incentive Payments System) need not ruin your life this year.
- A single measure can eliminate your downside risk for 2019.

Atopic Dermatitis Prevention and Treatment
Atopic dermatitis (AD) is a disease that finally is coming of age in dermatology research. New topical agents and systemic biologic agents offer patients with AD other options for medical management. This article provides a practical review of prevention strategies and treatment guidelines for AD.
PREVENTION
Prevention strategies for AD have been largely unsuccessful in the past, which may relate to factors such as prenatal triggers.1 However, some newer interventional studies have shown some promise in AD prevention in specific settings. For example, a randomized trial of infants in the United States and United Kingdom at high risk for AD (ie, family history of atopy) reported that the AD risk was reduced by 50% when patients were treated with at least once-daily application of full-body emollients for 6 months (beginning by 3 weeks of life).2 The strategy of daily application of emollients for avoidance of AD in infants with a family history of AD is reasonable but may not offer lifetime prevention, and the benefit in children not from AD families is unknown.
Other trials to prevent AD have included usage of dust avoidance and dust covers for mattresses. This strategy showed modest benefit in reducing the incidence of atopic diatheses in the first year3 but did not gain endorsement by the most recent guidelines of the American Academy of Dermatology (AAD).4
Prenatal and postnatal (maternal and child) supplementation of Lactobacillus rhamnosus has shown promise in prevention.5 The exact regimen likely makes an impact on efficacy. An early study showed the usage of probiotics (eg, Lactobacillus reuteri) prenatally in pregnant women and postnatally in infants resulted in no reduction in occurrence of AD and possible reduction in IgE-associated AD.6 Kalliomäki et al7 demonstrated that L rhamnosus GG alone reduced AD by half in at-risk infants in a double-blind, placebo-controlled trial. On the other hand, Taylor et al8 performed a study of probiotic supplementation in which patients at high risk for AD developed higher rates of allergen sensitization. The most successful recent trial involved the randomization of 415 pregnant women to receive interventions from 36 weeks’ gestation until 3 months postpartum.9 The intervention was a randomized comparison of milk without probiotics versus a blend of probiotic milk containing L rhamnosus GG, Lactobacillus acidophilus La-5, and Bifidobacterium animalis subsp lactis Bb-12. At 6 years of age, 81 babies who consumed probiotic milk and 82 babies who consumed milk without probiotics were available for testing. The strategy caused a statistically significant reduction in AD in the complete case analysis (odds ratio, 0.48; 95% confidence interval, 0.25-0.92; P=.027; number needed to treat, 6). Sadly, other allergic diseases were not prevented in this study.9
MANAGEMENT OF AD
There currently is no cure or perfected prevention technique for AD. As a result, therapy focuses on avoiding triggers and alleviating symptoms.10 Recent guidelines from the AAD state that“[t]he ultimate judgment regarding the propriety of any specific therapy must be made by the physician and the patient in light of all the circumstances presented by the individual patient, and the known variability and biologic behavior of the disease.”11 Skin-directed therapies are the first line of treatment including emollients, gentle skin care, and topical medicaments. In AD, therapies are needed to reduce disease activity and flare severity, clear flares, and provide relief.
Parental education and written eczema action plans are recommended to help patients and parents/guardians follow recommended regimens12; Tollefson and Bruckner13 for the American Academy of Pediatrics provide an action plan to guide the care of children with atopic dermatitis that is simple, but many others exist online. The eczema action plan usually provides information on how to bathe and what to do when the skin is actively inflamed.
In 2014, a 4-part series of guidelines of care for the management of AD was published by the AAD, replacing prior guidelines.4,11,14,15 The following sections review some of the important parameters of care highlighted in these management guidelines.
Psychological Support
Appropriate psychological support for AD patients can be sought through counselors, therapists, psychiatrists, and support groups such as the National Eczema Association (https://nationaleczema.org/).
Education
Education is the leading form of medical therapy in patients with AD. Eczema schools are popular in Europe and are just beginning to form in the United States (http://tuh.templehealth.org/content/eczema_school.htm), which can be helpful to educate caregivers and patients with AD. Patient resources online and through support groups with an online presence, in-person meetings, and patient/family conventions can be helpful to AD patients. Often, an initial office visit with a dermatologist involves a review of avoidance of triggers, usage of gentle skin care including bland emollients, and therapeutic regimens for disease activity. This form of verbal education is to be paired with an eczema action plan, a written document that allows individuals to reference recommendations and share information with other caregivers.12,13,16
Emollients and Gentle Skin Care
Gentle skin care regimens, which includes the usage of synthetic cleansers with a low pH to help maintain the acidity (acid mantle) of the skin, seek to reduce irritation and have been rated as level IA (highest level) in recent AAD guidelines.14 Although bathing frequency has been emphasized in the guidelines, AD severity as reflected by SCORAD (SCORing Atopic Dermatitis) was not different for daily bathing versus twice weekly.17 The American Academy of Pediatrics recommended a skin care regimen of bathing every 2 to 3 days in lukewarm water for 10 to 15 minutes, followed by application of emollients that are fragrance free and have few preservatives.13 Topical emollients with additives such as colloidal oatmeal, avenanthramides, or ceramides can be used to enhance the skin barrier and are well tolerated in all age groups.18,19 Despite enhanced emollients, the therapy of AD still requires usage of prescription or over-the-counter TCs and/or topical calcineurin inhibitors (TCIs) in many cases.20
Topical Medication
Children have a relatively higher body surface area–to-weight ratio, allowing for greater potential absorption of topical medicaments and potential side effects from absorption. Types of vehicle, cost, site of application, and availability may impact patient and physician preference in choice of therapeutic topical agent.14
Topical Corticosteroids
Topical corticosteroids (TCs) are the mainstay of treatment for AD and have been used for more than 60 years.14,20 Topical corticosteroids provide anti-inflammatory effects on T cells, monocytes, and macrophages, producing altered cytokine activity locally. Topical corticosteroids inhibit collagen synthesis, potentially causing skin atrophy. They also inhibit IL-1, IL-2, IL-6, IFN-α, and tumor necrosis factor α.21 Topical corticosteroids are classified as class I (ultra-high potency) to class VII (low potency). In children, low-potency TCs generally are applied to the face, intertriginous areas, groin, and genitalia, and mid-potency corticosteroids are applied to the body, arms, and legs. An even higher-strength agent can be prescribed as a rescue medication in severe cases. After clearance with once- or twice-daily therapy, twice-weekly usage can benefit disease activity.22 Topical corticosteroids reduce inflammation as well as Staphylococcus aureus load through inhibition of cytokines that inhibit antimicrobial peptides. Topical corticosteroids have been endorsed as level IA evidence therapy by the AAD guidelines.14
Topical corticosteroids, particularly prolonged usage of mid- to high-potency products, have been associated with side effects such as skin atrophy, striae, telangiectases, hypopigmentation, rosacea, acneiform eruptions, focal hypertrichosis, perioral dermatitis, and acne23; potential systemic side effects include hypothalamic-pituitary-adrenal axis suppression, cataracts, glaucoma (with periocular application), Cushing syndrome, hyperglycemia, hypertension,23 and growth retardation.14 Long-term corticosteroid therapy is associated with tachyphylaxis and potential rebound of disease with discontinuation.24 Based on the potential risk of side effects with TCs, the least potent product for the shortest time needed is recommended, with special care for thin skin. Discontinuation when clearance occurs is advised. Allergy to TCs and/or vehicle ingredients such as propylene glycol should be suspected in severe unremitting cases.14 A recent registry review of children screened for contact dermatitis demonstrated that children with AD had higher sensitization to the steroid tixocortol pivalate.25
Topical Calcineurin Inhibitors
Topical calcineurin inhibitors include pimecrolimus cream 1%, which is approved for mild to moderate AD in adults and children 2 years and older, and tacrolimus ointment 0.03% and 0.1%, which are approved for moderate to severe AD in adults and children aged 2 to 15 years (0.03% formulation only). Topical calcineurin inhibitors can be used as second-line agents in AD in patients who have inadequate response to TCs or who may not be able to use TCs due to the disease site.10,13,14 Guidelines from the AAD also have endorsed TCIs as level IA evidence for steroid-sparing agents.
Concerns about the reporting of cancers and lymphomas prompted the US Food and Drug Administration to issue a black box warning on TCIs more than 10 years ago. Pimecrolimus, which has little absorption and no notable immunosuppressive effects, has been used without detrimental effect on vaccination and delayed-type hypersensitivities, but many decades of data are lacking.10,13,14,17,26-29 Topical calcineurin inhibitors can be used as steroid-sparing agents in lieu of corticosteroids in specific locations such as the face and eyelids and for long-term suppressive therapy twice weekly.30 Intermittent usage and cycling with corticosteroids is advisable,28 but usage intermittently beyond 1 year has not been evaluated.
Topical calcineurin inhibitors are recommended as effective for acute and chronic AD. Their use as maintenance therapy in adults and children, for AD recalcitrant to steroids, for AD in sensitive areas, for steroid-induced atrophy, and for long-term uninterrupted topical steroid usage carries a level IA evidence recommendation. Furthermore, the AAD guidelines have recommended TCIs as steroid-sparing agents with level IA evidence and off-label use of TCIs in children younger than 2 years with level IA evidence. Pretreatment with TCs to reduce stinging has level IIB evidence. Usage for flare prevention is level IA evidence. Routine blood monitoring of TCI-treated patients was not recommended; in fact, the AAD guidelines provided this recommendation as level IA evidence against routine laboratory monitoring of TCI-treated patients.14
Topical Antibiotics
Topical antibiotics are indicated for the therapy of impetigo and can be used in the setting of impetiginized AD in conjunction with TCs. Recent AAD guidelines suggested against routine usage of topical antistaphylococcal agents as level IA evidence.14 There is one study supporting usage of topical mupirocin in addition to TCs to heal children with eczema area and severity index scores more than 7 more rapidly in the first week of AD therapy, but in the same study, additive benefit was not demonstrated in AD beyond the first week.31 There also are data supporting usage of intranasal mupirocin adjunctively with bleach baths in patients with moderate to severe AD, which was rated as level IIB evidence in the AAD guidelines.14,32 There are limited data on the long-term utility of topical anti-infectives in AD. The risks of long-term usage could include resistance formation to agents such as mupirocin, contact dermatitis, and lack of efficacy.
Additional Therapeutics
Wet Wraps
Penetration through the stratum corneum is needed for drug activity in AD. Penetration can be enhanced using wet wrap therapy or using ointments, which produce higher relative potency.13 Wet wraps overlying a dilute topical medicament have been described as effective in AD and are recommended in AAD guidelines as level IIB evidence.14 Different wet wrap techniques can be used, including wet pajamas covered by dry pajamas or saline-soaked gauze wrapped around the affected areas and then dry gauze applied over the wet gauze. The methodology used should be tailored to the patient as well as to whether the individual is an inpatient or outpatient.
Bleach Baths
Dilute sodium hypochlorite solution 0.005% (one-quarter cup bleach in 20 gallons of water) has been demonstrated to be beneficial in reduction of disease activity in AD patients with recurrent bacterial infections.32 This simple technique in addition to intranasal mupirocin can reduce AD severity and improve quality of life and is the only ongoing S aureus therapeutic management endorsed by the AAD guidelines for the management of AD.14,32
Topical and Oral Delivery
Antihistamines
Topical antihistamines are ineffective in AD. Oral antihistamines can be used to reduce pruritus and are most effective when given as sedating agents for sleep enhancement but may be given as nonsedating agents for patients with concomitant allergic disorders such as allergic rhinoconjunctivitis. Paradoxical hyperreactivity with sedating antihistamines is not uncommon in small children, and sedating antihistamine usage should be discontinued in these instances.13 Parents of children with AD have reported giving the child antihistamines to sleep was helpful, as well as putting on creams, using special clothes (eg, all cotton), and keeping the room cool.33 There is level IIIC evidence against use of systemic antihistamines and level IIA evidence for sedating and nonsedating, according to the AAD guidelines.14
Systemic Therapeutics
Oral therapeutics range from oral antihistamines to oral antibiotics and immunosuppressive medications. Oral antibiotics (level IIB evidence) are reserved for superinfected AD, which is not easily defined for the following reasons: there is no consensus definition of superinfected AD; the majority of active AD lesions when cultured will demonstrate S aureus growth; and most AD lesions ooze, thereby creating the appearance of superinfection. In real-world practice, superinfection can be diagnosed based on the presence of pustules; furuncles; or signs of infection such as tracking erythema, tenderness, severe erosions, or maceration. Clinical judgment is always required.
The immunosuppressive medications used in AD include leukotriene inhibitors, which rarely are effective for AD.34 More effective systemic agents for AD include cyclosporine (level I to IIB evidence), azathioprine (level IIB evidence), mycophenolate mofetil (level IIIC evidence), and methotrexate (level IIB evidence). These agents are indicated for pediatric or adult patients when topical agents and/or phototherapy have failed.15 Monitoring these agents for side effects includes ongoing evaluation for renal and liver toxicity. Short courses (ie, 6 months) are preferred to minimize side effects.35
Dupilumab, an injectable AD therapy, is approved in the United States. This agent is injected every 2 weeks and binds to the IL-4Rα shared by IL-4 and IL-13. In 4 weeks of monotherapy, 85% of adult patients treated had 50% or greater clearance.36 Recently published consensus opinion from the International Eczema Council recommends assessment of a variety of factors before initiating systemic therapy including comorbid illnesses such as contact allergy, trigger avoidance, superinfection, and impact on quality of life.37
Oral Corticosteroids
Systemic corticosteroids clear patients quickly but provide no sustained improvement; in fact, many patients rebound or have tachyphylaxis. Although short-term corticosteroid usage can break the itch-scratch cycle, long-term usage is associated with osteoporosis, Cushing syndrome, and aseptic necrosis of the femoral head. Decreased linear growth will occur during therapy in children; therefore, systemic steroids are not recommended in children with AD, except for additional or comorbid conditions (eg, asthma or contact dermatitis).4
Phototherapy
Phototherapy has been recommended in the AAD guidelines as a second-line treatment after failure of first-line agents (ie, TCIs and TCs) for clearance and or maintenance and should be tailored to the patient’s skin tone by an experienced physician. Narrowband UVB phototherapy may act through the suppression of T-cell activity in the skin and possibly via suppression of staphylococcal superantigens; however, many phototherapy types have been described for AD.38,39 Usage can be effective in school-aged children and teenagers but may be limited due to school attendance. Phototherapy was graded as level IIB evidence in the AAD guidelines.15 Side effects include aggravation of AD by exposure to heat and UV light, actinic damage, tenderness, erythema, pruritus, burning, and stinging. Lentigines; skin cancers (melanoma and nonmelanoma); folliculitis; and ocular toxicity, especially cataracts, can occur.15 Children younger than 6 years will find it difficult to stand in a phototherapy booth and may be poor candidates.15,38,39
Complementary and Alternative Medicine
Complementary and alternative medicine (CAM) also has been used for AD in the United States. In a review of the 2007 National Health Interview Survey of 9417 children aged 0 to 17 years, CAM was used for AD by 0.99% of children. Some CAM techniques were associated with worsening severity of AD, including herbal therapy, vitamins, homeopathic agents, diet, and movement techniques.40 Usage of Chinese herbal medications for AD can be associated with liver toxicity.41 Only one CAM therapy—massage therapy—has some mild supportive data.42
Allergen Avoidance and Diet
Bronsnick et al43 discussed the possible benefit of prenatal and postnatal probiotics for prevention of AD, which were not supported in the AAD guidelines for management of AD4; postnatal prebiotic supplementation; and exclusive breastfeeding and/or supplementation with hydrolyzed formula in at-risk children. Elimination diets for children and mothers were not recommended. The authors found no beneficial role of supplements including vitamin D, selenium, fish oil, borage oil, and zinc sulfate.43
A National Institute of Allergy and Infectious Diseases consensus group recommended avoidance of proven but not random elimination of food allergens in AD, asthma, and/or eosinophilic esophagitis.44 Restricted maternal diet was not recommended, and breastfeeding exclusively for the first 4 to 6 months was recommended. Hydrolyzed formulas were suggested as a possible preventive strategy in at-risk infants as a breastfeeding alternative, with cost of these formulas being a problem.44
In children younger than 5 years, food allergy screening for the most common allergens (eg, milk, eggs, peanuts, wheat, soy) should be considered in children with persistent unremitting dermatitis and/or known food challenge–induced reactions.4 Conservative measures to avoid house dust mite exposure in known sensitized individuals including dust covers for pillows and mattresses may be beneficial.4,45
Emerging Therapies
Recently approved therapies include better-targeted agents that appear to have a reasonable safety profile and may fulfill unmet needs in AD care. Of these agents, crisaborole, a topical boron-based phosphodiesterase 4 inhibitor, was approved in December 2016 for mild to moderate AD in patients 2 years and older.Topically, this agent seems to be efficacious in the absence of notable carcinogenicity.46
The systemic (injectable) biologic agent dupilumab was approved in March 2017 for moderate to severe AD. Phase 3 studies in adults with AD showed excellent success in adults with moderate to severe AD.37 This agent is a monoclonal antibody targeted at blockade of the crucial atopic inflammatory triggering pathway via blockade of the IL-4A receptor site, targeting IL-4 and IL-13 activity.36,47 There are many medications in the pipeline, which Renert-Yuval and Guttman-Yassky48 review. However, an overview of the landscape demonstrates that Janus kinase (JAK) inhibitors49 and biologic medications in addition to dupilumab affecting targeted inflammatory cascades in AD are in development. In particular, the JAK inhibitors appear promising due to availability both as oral and topical agents.49
Need for Ongoing Care and Monitoring
Atopic dermatitis is a chronic inflammatory skin disorder with a genetic basis. Once initiated, the process of AD may persist throughout the patient’s life and become a systemic disorder with comorbidities including sleep disturbance, reduced quality of life, and cardiovascular disease.50 Ongoing management of AD includes topical reduction in irritants and triggers, topical medicaments, and management of pruritus and infections. At this time, emollients and irritant avoidance paired with judicious topical medicaments including TCs and second-line or site-specific (eg, eyelids) usage of TCIs or phosphodiesterase 4 inhibitors remain the backbone of therapy. Ongoing review of therapeutics for associated morbidities is underway, which may guide future therapeutic interventions into AD. The future of prevention and therapy look bright, but time will tell.
- Kelleher M, Dunn-Galvin A, Hourihane JO, et al. Skin barrier dysfunction measured by transepidermal water loss at 2 days and 2 months predates and predicts atopic dermatitis at 1 year. J Allergy Clin Immunol. 2015;135:930-935.
- Simpson EL, Chalmers JR, Hanifin JM, et al. Emollient enhancement of the skin barrier from birth offers effective atopic dermatitis prevention. J Allergy Clin Immunol. 2014;134:818-823.
- Tsitoura S, Nestoridou K, Botis P, et al. Randomized trial to prevent sensitization to mite allergens in toddlers and preschoolers by allergen reduction and education: one-year results. Arch Pediatr Adolesc Med. 2002;156:1021-1027.
- Sidbury R, Tom WL, Bergman JN, et al. Guidelines of care for the management of atopic dermatitis: section 4. prevention of disease flares and use of adjunctive therapies and approaches. J Am Acad Dermatol. 2014;71:1218-1233.
- Foolad N, Brezinski EA, Chase EP, et al. Effect of nutrient supplementation on atopic dermatitis in children: a systematic review of probiotics, prebiotics, formula, and fatty acids. JAMA Dermatol. 2013;149:350-355.
- Abrahamsson TR, Jakobsson T, Böttcher MF, et al. Probiotics in prevention of IgE-associated eczema: a double-blind, randomized, placebo-controlled trial. J Allergy Clin Immunol. 2007;119:1174-1180.
- Kalliomäki M, Salminen S, Arvilommi H, et al. Probiotics in primary prevention of atopic disease: a randomised placebo-controlled trial. Lancet. 2001;357:1076-1079.
- Taylor AL, Dunstan JA, Prescott SL. Probiotic supplementation for the first 6 months of life fails to reduce the risk of atopic dermatitis and increases the risk of allergen sensitization in high-risk children: a randomized controlled trial. J Allergy Clin Immunol. 2007;119:184-191.
- Simpson MR, Dotterud CK, Storrø O, et al. Perinatal probiotic supplementation in the prevention of allergy related disease: 6 year follow up of a randomised controlled trial. BMC Dermatol. 2015;15:13. doi:10.1186/s12895-015-0030-1.
- Carr WW. Topical calcineurin inhibitors for atopic dermatitis: review and treatment recommendations. Paediatr Drugs. 2013;15:303-310.
- Eichenfield LF, Tom WL, Chamlin SL, et al. Guidelines of care for the management of atopic dermatitis: section 1. diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. 2014;70:338-351.
- Silverberg NB. Creating an action plan for eczema patients. Cutis. 2015;96:362-363.
- Tollefson MM, Bruckner AL; Section on Dermatology. Atopic dermatitis: skin-directed management. Pediatrics. 2014;134:E1735-E1744.
- Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 2. management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71:116-132.
- Sidbury R, Davis DM, Cohen DE, et al; American Academy of Dermatology. Guidelines of care for the management of atopic dermatitis: section 3. management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71:327-349.
- Shi VY, Nanda S, Lee K, et al. Improving patient education with an eczema action plan: a randomized controlled trial. JAMA Dermatol. 2013;149:481-483.
- Koutroulis I, Petrova K, Kratimenos P, et al. Frequency of bathing in the management of atopic dermatitis: to bathe or not to bathe? Clin Pediatr (Phila). 2014;53:677-681.
- Fowler JF, Nebus J, Wallo W, et al. Colloidal oatmeal formulations as adjunct treatments in atopic dermatitis. J Drugs Dermatol. 2012;11:804-807.
- Fowler J Jr, Silverberg N. Active naturals have a key role in atopic dermatitis. Semin Cutan Med Surg. 2008;27:8-10.
- Eichenfield LF. Consensus guidelines in diagnosis and treatment of atopic dermatitis. Allergy. 2004;59:86-92.
- Nghiem P, Pearson G, Langley RG. Tacrolimus and pimecrolimus: from clever prokaryotes to inhibiting calcineurin and treating atopic dermatitis. J Am Acad Dermatol. 2002;46:228-241.
- Schmitt J. Commentary: eczema and cancer risk. Br J Dermatol. 2011;165:463-464.
- Abramovits W, Hung P, Tong KB. Efficacy and economics of topical calcineurin inhibitors for the treatment of atopic dermatitis. Am J Clin Dermatol. 2006;7:213-222.
- Takahashi-Ando N, Jones MA, Fujisawa S, et al. Patient-reported outcomes after discontinuation of long-term topical corticosteroid treatment for atopic dermatitis: a targeted cross-sectional survey. Drug Healthc Patient Saf. 2015;7:57-62.
- Jacob SE, McGowan M, Silverberg NB, et al. Pediatric contact dermatitis registry data on contact allergy in children with atopic dermatitis. JAMA Dermatol. 2017;153:765-770.
- Werfel T. Topical use of pimecrolimus in atopic dermatitis: update on the safety and efficacy. J Dtsch Dermatol Ges. 2009;7:739-742.
- Wahn U, Bos JD, Goodfield M, et al. Efficacy and safety of pimecrolimus cream in the long-term management of atopic dermatitis in children. Pediatrics. 2002;110(1, pt 1):E2.
- Berger TG, Duvic M, Van Voorhees AS, et al; American Academy of Dermatology Association Task Force. The use of topical calcineurin inhibitors in dermatology: safety concerns. report of the American Academy of Dermatology Association Task Force. J Am Acad Dermatol. 2006;54:818-823.
- Paller AS. Latest approaches to treating atopic dermatitis. Chem Immunol Allergy. 2012;96:132-140.
- Thaçi D, Reitamo S, Gonzalez Ensenat MA, et al. Proactive disease management with 0.03% tacrolimus ointment for children with atopic dermatitis: results of a randomized, multicentre, comparative study. Br J Dermatol. 2008;159:1348-1356.
- Gong JQ, Lin L, Lin T, et al. Skin colonization by Staphylococcus aureus in patients with eczema and atopic dermatitis and relevant combined topical therapy: a double-blind multicentre randomized controlled trial. Br J Dermatol. 2006;155:680-687.
- Huang JT, Abrams M, Tlougan B, et al. Treatment of Staphylococcus aureus colonization in atopic dermatitis decreases disease severity. Pediatrics. 2009;123:E808-E814.
- Reid P, Lewis-Jones MS. Sleep difficulties and their management in preschoolers with atopic eczema. Clin Exp Dermatol. 1995;20:38-41.
- Silverberg NB, Paller AS. Leukotriene receptor antagonists are ineffective for severe atopic dermatitis. J Am Acad Dermatol. 2004;50:485-486.
- Wolverton SE. Comprehensive Dermatologic Drug Therapy. 3rd ed. New York, NY: Elsevier Saunders; 2013.
- Beck LA, Thaçi D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
- Simpson EL, Bruin-Weller M, Flohr C, et al. When does atopic dermatitis warrant systemic therapy? recommendations from an expert panel of the International Eczema Council [published online August 10, 2017]. J Am Acad Dermatol. doi:10.1016/j.jaad.2017.06.042.
- Veith W, DeLeo V, Silverberg N. Medical phototherapy in childhood skin diseases. Minerva Pediatr. 2011;63:327-333.
- Song E, Reja D, Silverberg N, et al. Phototherapy: kids are not just little people. Clin Dermatol. 2015;33:672-680.
- Silverberg JI, Lee-Wong M, Silverberg NB. Complementary and alternative medicines and childhood eczema: a US population-based study. Dermatitis. 2014;25:246-254.
- Stickel F, Shouval D. Hepatotoxicity of herbal and dietary supplements: an update. Arch Toxicol. 2015;89:851-865.
- Schachner L, Field T, Hernandez-Reif M, et al. Atopic dermatitis symptoms decreased in children following massage therapy. Pediatr Dermatol. 1998;15:390-395.
- Bronsnick T, Murzaku EC, Rao BK. Diet in dermatology: part I. atopic dermatitis, acne, and nonmelanoma skin cancer. J Am Acad Dermatol. 2014;71:1039.e1-1039.e12.
- Boyce JA, Assa’ad A, Burks AW, et al. Guidelines for the diagnosis and management of food allergy in the United States: summary of the NIAID-sponsored expert panel report. Nutr Res. 2011;31:61-75.
- Silverberg NB, Lee-Wong M, Yosipovitch G. Diet and atopic dermatitis. Cutis. 2016;97:227-232.
- Hanifin JM, Chan SC, Cheng JB, et al. Type phosphodiesterase inhibitors have clinical and in vitro anti-inflammatory effects in atopic dermatitis. J Invest Dermatol. 1996;107:51-56.
- Boguniewicz M, Leung DY. Targeted therapy for allergic diseases: at the intersection of cutting-edge science and clinical practice. J Allergy Clin Immunol. 2015;135:354-356.
- Renert-Yuval Y, Guttman-Yassky E. Systemic therapies in atopic dermatitis: the pipeline. Clin Dermatol. 2017;35:387-397.
- Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76:736-744.
- Brunner PM, Silverberg JI, Guttman-Yassky E, et al. Increasing comorbidities suggest that atopic dermatitis is a systemic disorder. J Invest Dermatol. 2017;137:18-25.
Atopic dermatitis (AD) is a disease that finally is coming of age in dermatology research. New topical agents and systemic biologic agents offer patients with AD other options for medical management. This article provides a practical review of prevention strategies and treatment guidelines for AD.
PREVENTION
Prevention strategies for AD have been largely unsuccessful in the past, which may relate to factors such as prenatal triggers.1 However, some newer interventional studies have shown some promise in AD prevention in specific settings. For example, a randomized trial of infants in the United States and United Kingdom at high risk for AD (ie, family history of atopy) reported that the AD risk was reduced by 50% when patients were treated with at least once-daily application of full-body emollients for 6 months (beginning by 3 weeks of life).2 The strategy of daily application of emollients for avoidance of AD in infants with a family history of AD is reasonable but may not offer lifetime prevention, and the benefit in children not from AD families is unknown.
Other trials to prevent AD have included usage of dust avoidance and dust covers for mattresses. This strategy showed modest benefit in reducing the incidence of atopic diatheses in the first year3 but did not gain endorsement by the most recent guidelines of the American Academy of Dermatology (AAD).4
Prenatal and postnatal (maternal and child) supplementation of Lactobacillus rhamnosus has shown promise in prevention.5 The exact regimen likely makes an impact on efficacy. An early study showed the usage of probiotics (eg, Lactobacillus reuteri) prenatally in pregnant women and postnatally in infants resulted in no reduction in occurrence of AD and possible reduction in IgE-associated AD.6 Kalliomäki et al7 demonstrated that L rhamnosus GG alone reduced AD by half in at-risk infants in a double-blind, placebo-controlled trial. On the other hand, Taylor et al8 performed a study of probiotic supplementation in which patients at high risk for AD developed higher rates of allergen sensitization. The most successful recent trial involved the randomization of 415 pregnant women to receive interventions from 36 weeks’ gestation until 3 months postpartum.9 The intervention was a randomized comparison of milk without probiotics versus a blend of probiotic milk containing L rhamnosus GG, Lactobacillus acidophilus La-5, and Bifidobacterium animalis subsp lactis Bb-12. At 6 years of age, 81 babies who consumed probiotic milk and 82 babies who consumed milk without probiotics were available for testing. The strategy caused a statistically significant reduction in AD in the complete case analysis (odds ratio, 0.48; 95% confidence interval, 0.25-0.92; P=.027; number needed to treat, 6). Sadly, other allergic diseases were not prevented in this study.9
MANAGEMENT OF AD
There currently is no cure or perfected prevention technique for AD. As a result, therapy focuses on avoiding triggers and alleviating symptoms.10 Recent guidelines from the AAD state that“[t]he ultimate judgment regarding the propriety of any specific therapy must be made by the physician and the patient in light of all the circumstances presented by the individual patient, and the known variability and biologic behavior of the disease.”11 Skin-directed therapies are the first line of treatment including emollients, gentle skin care, and topical medicaments. In AD, therapies are needed to reduce disease activity and flare severity, clear flares, and provide relief.
Parental education and written eczema action plans are recommended to help patients and parents/guardians follow recommended regimens12; Tollefson and Bruckner13 for the American Academy of Pediatrics provide an action plan to guide the care of children with atopic dermatitis that is simple, but many others exist online. The eczema action plan usually provides information on how to bathe and what to do when the skin is actively inflamed.
In 2014, a 4-part series of guidelines of care for the management of AD was published by the AAD, replacing prior guidelines.4,11,14,15 The following sections review some of the important parameters of care highlighted in these management guidelines.
Psychological Support
Appropriate psychological support for AD patients can be sought through counselors, therapists, psychiatrists, and support groups such as the National Eczema Association (https://nationaleczema.org/).
Education
Education is the leading form of medical therapy in patients with AD. Eczema schools are popular in Europe and are just beginning to form in the United States (http://tuh.templehealth.org/content/eczema_school.htm), which can be helpful to educate caregivers and patients with AD. Patient resources online and through support groups with an online presence, in-person meetings, and patient/family conventions can be helpful to AD patients. Often, an initial office visit with a dermatologist involves a review of avoidance of triggers, usage of gentle skin care including bland emollients, and therapeutic regimens for disease activity. This form of verbal education is to be paired with an eczema action plan, a written document that allows individuals to reference recommendations and share information with other caregivers.12,13,16
Emollients and Gentle Skin Care
Gentle skin care regimens, which includes the usage of synthetic cleansers with a low pH to help maintain the acidity (acid mantle) of the skin, seek to reduce irritation and have been rated as level IA (highest level) in recent AAD guidelines.14 Although bathing frequency has been emphasized in the guidelines, AD severity as reflected by SCORAD (SCORing Atopic Dermatitis) was not different for daily bathing versus twice weekly.17 The American Academy of Pediatrics recommended a skin care regimen of bathing every 2 to 3 days in lukewarm water for 10 to 15 minutes, followed by application of emollients that are fragrance free and have few preservatives.13 Topical emollients with additives such as colloidal oatmeal, avenanthramides, or ceramides can be used to enhance the skin barrier and are well tolerated in all age groups.18,19 Despite enhanced emollients, the therapy of AD still requires usage of prescription or over-the-counter TCs and/or topical calcineurin inhibitors (TCIs) in many cases.20
Topical Medication
Children have a relatively higher body surface area–to-weight ratio, allowing for greater potential absorption of topical medicaments and potential side effects from absorption. Types of vehicle, cost, site of application, and availability may impact patient and physician preference in choice of therapeutic topical agent.14
Topical Corticosteroids
Topical corticosteroids (TCs) are the mainstay of treatment for AD and have been used for more than 60 years.14,20 Topical corticosteroids provide anti-inflammatory effects on T cells, monocytes, and macrophages, producing altered cytokine activity locally. Topical corticosteroids inhibit collagen synthesis, potentially causing skin atrophy. They also inhibit IL-1, IL-2, IL-6, IFN-α, and tumor necrosis factor α.21 Topical corticosteroids are classified as class I (ultra-high potency) to class VII (low potency). In children, low-potency TCs generally are applied to the face, intertriginous areas, groin, and genitalia, and mid-potency corticosteroids are applied to the body, arms, and legs. An even higher-strength agent can be prescribed as a rescue medication in severe cases. After clearance with once- or twice-daily therapy, twice-weekly usage can benefit disease activity.22 Topical corticosteroids reduce inflammation as well as Staphylococcus aureus load through inhibition of cytokines that inhibit antimicrobial peptides. Topical corticosteroids have been endorsed as level IA evidence therapy by the AAD guidelines.14
Topical corticosteroids, particularly prolonged usage of mid- to high-potency products, have been associated with side effects such as skin atrophy, striae, telangiectases, hypopigmentation, rosacea, acneiform eruptions, focal hypertrichosis, perioral dermatitis, and acne23; potential systemic side effects include hypothalamic-pituitary-adrenal axis suppression, cataracts, glaucoma (with periocular application), Cushing syndrome, hyperglycemia, hypertension,23 and growth retardation.14 Long-term corticosteroid therapy is associated with tachyphylaxis and potential rebound of disease with discontinuation.24 Based on the potential risk of side effects with TCs, the least potent product for the shortest time needed is recommended, with special care for thin skin. Discontinuation when clearance occurs is advised. Allergy to TCs and/or vehicle ingredients such as propylene glycol should be suspected in severe unremitting cases.14 A recent registry review of children screened for contact dermatitis demonstrated that children with AD had higher sensitization to the steroid tixocortol pivalate.25
Topical Calcineurin Inhibitors
Topical calcineurin inhibitors include pimecrolimus cream 1%, which is approved for mild to moderate AD in adults and children 2 years and older, and tacrolimus ointment 0.03% and 0.1%, which are approved for moderate to severe AD in adults and children aged 2 to 15 years (0.03% formulation only). Topical calcineurin inhibitors can be used as second-line agents in AD in patients who have inadequate response to TCs or who may not be able to use TCs due to the disease site.10,13,14 Guidelines from the AAD also have endorsed TCIs as level IA evidence for steroid-sparing agents.
Concerns about the reporting of cancers and lymphomas prompted the US Food and Drug Administration to issue a black box warning on TCIs more than 10 years ago. Pimecrolimus, which has little absorption and no notable immunosuppressive effects, has been used without detrimental effect on vaccination and delayed-type hypersensitivities, but many decades of data are lacking.10,13,14,17,26-29 Topical calcineurin inhibitors can be used as steroid-sparing agents in lieu of corticosteroids in specific locations such as the face and eyelids and for long-term suppressive therapy twice weekly.30 Intermittent usage and cycling with corticosteroids is advisable,28 but usage intermittently beyond 1 year has not been evaluated.
Topical calcineurin inhibitors are recommended as effective for acute and chronic AD. Their use as maintenance therapy in adults and children, for AD recalcitrant to steroids, for AD in sensitive areas, for steroid-induced atrophy, and for long-term uninterrupted topical steroid usage carries a level IA evidence recommendation. Furthermore, the AAD guidelines have recommended TCIs as steroid-sparing agents with level IA evidence and off-label use of TCIs in children younger than 2 years with level IA evidence. Pretreatment with TCs to reduce stinging has level IIB evidence. Usage for flare prevention is level IA evidence. Routine blood monitoring of TCI-treated patients was not recommended; in fact, the AAD guidelines provided this recommendation as level IA evidence against routine laboratory monitoring of TCI-treated patients.14
Topical Antibiotics
Topical antibiotics are indicated for the therapy of impetigo and can be used in the setting of impetiginized AD in conjunction with TCs. Recent AAD guidelines suggested against routine usage of topical antistaphylococcal agents as level IA evidence.14 There is one study supporting usage of topical mupirocin in addition to TCs to heal children with eczema area and severity index scores more than 7 more rapidly in the first week of AD therapy, but in the same study, additive benefit was not demonstrated in AD beyond the first week.31 There also are data supporting usage of intranasal mupirocin adjunctively with bleach baths in patients with moderate to severe AD, which was rated as level IIB evidence in the AAD guidelines.14,32 There are limited data on the long-term utility of topical anti-infectives in AD. The risks of long-term usage could include resistance formation to agents such as mupirocin, contact dermatitis, and lack of efficacy.
Additional Therapeutics
Wet Wraps
Penetration through the stratum corneum is needed for drug activity in AD. Penetration can be enhanced using wet wrap therapy or using ointments, which produce higher relative potency.13 Wet wraps overlying a dilute topical medicament have been described as effective in AD and are recommended in AAD guidelines as level IIB evidence.14 Different wet wrap techniques can be used, including wet pajamas covered by dry pajamas or saline-soaked gauze wrapped around the affected areas and then dry gauze applied over the wet gauze. The methodology used should be tailored to the patient as well as to whether the individual is an inpatient or outpatient.
Bleach Baths
Dilute sodium hypochlorite solution 0.005% (one-quarter cup bleach in 20 gallons of water) has been demonstrated to be beneficial in reduction of disease activity in AD patients with recurrent bacterial infections.32 This simple technique in addition to intranasal mupirocin can reduce AD severity and improve quality of life and is the only ongoing S aureus therapeutic management endorsed by the AAD guidelines for the management of AD.14,32
Topical and Oral Delivery
Antihistamines
Topical antihistamines are ineffective in AD. Oral antihistamines can be used to reduce pruritus and are most effective when given as sedating agents for sleep enhancement but may be given as nonsedating agents for patients with concomitant allergic disorders such as allergic rhinoconjunctivitis. Paradoxical hyperreactivity with sedating antihistamines is not uncommon in small children, and sedating antihistamine usage should be discontinued in these instances.13 Parents of children with AD have reported giving the child antihistamines to sleep was helpful, as well as putting on creams, using special clothes (eg, all cotton), and keeping the room cool.33 There is level IIIC evidence against use of systemic antihistamines and level IIA evidence for sedating and nonsedating, according to the AAD guidelines.14
Systemic Therapeutics
Oral therapeutics range from oral antihistamines to oral antibiotics and immunosuppressive medications. Oral antibiotics (level IIB evidence) are reserved for superinfected AD, which is not easily defined for the following reasons: there is no consensus definition of superinfected AD; the majority of active AD lesions when cultured will demonstrate S aureus growth; and most AD lesions ooze, thereby creating the appearance of superinfection. In real-world practice, superinfection can be diagnosed based on the presence of pustules; furuncles; or signs of infection such as tracking erythema, tenderness, severe erosions, or maceration. Clinical judgment is always required.
The immunosuppressive medications used in AD include leukotriene inhibitors, which rarely are effective for AD.34 More effective systemic agents for AD include cyclosporine (level I to IIB evidence), azathioprine (level IIB evidence), mycophenolate mofetil (level IIIC evidence), and methotrexate (level IIB evidence). These agents are indicated for pediatric or adult patients when topical agents and/or phototherapy have failed.15 Monitoring these agents for side effects includes ongoing evaluation for renal and liver toxicity. Short courses (ie, 6 months) are preferred to minimize side effects.35
Dupilumab, an injectable AD therapy, is approved in the United States. This agent is injected every 2 weeks and binds to the IL-4Rα shared by IL-4 and IL-13. In 4 weeks of monotherapy, 85% of adult patients treated had 50% or greater clearance.36 Recently published consensus opinion from the International Eczema Council recommends assessment of a variety of factors before initiating systemic therapy including comorbid illnesses such as contact allergy, trigger avoidance, superinfection, and impact on quality of life.37
Oral Corticosteroids
Systemic corticosteroids clear patients quickly but provide no sustained improvement; in fact, many patients rebound or have tachyphylaxis. Although short-term corticosteroid usage can break the itch-scratch cycle, long-term usage is associated with osteoporosis, Cushing syndrome, and aseptic necrosis of the femoral head. Decreased linear growth will occur during therapy in children; therefore, systemic steroids are not recommended in children with AD, except for additional or comorbid conditions (eg, asthma or contact dermatitis).4
Phototherapy
Phototherapy has been recommended in the AAD guidelines as a second-line treatment after failure of first-line agents (ie, TCIs and TCs) for clearance and or maintenance and should be tailored to the patient’s skin tone by an experienced physician. Narrowband UVB phototherapy may act through the suppression of T-cell activity in the skin and possibly via suppression of staphylococcal superantigens; however, many phototherapy types have been described for AD.38,39 Usage can be effective in school-aged children and teenagers but may be limited due to school attendance. Phototherapy was graded as level IIB evidence in the AAD guidelines.15 Side effects include aggravation of AD by exposure to heat and UV light, actinic damage, tenderness, erythema, pruritus, burning, and stinging. Lentigines; skin cancers (melanoma and nonmelanoma); folliculitis; and ocular toxicity, especially cataracts, can occur.15 Children younger than 6 years will find it difficult to stand in a phototherapy booth and may be poor candidates.15,38,39
Complementary and Alternative Medicine
Complementary and alternative medicine (CAM) also has been used for AD in the United States. In a review of the 2007 National Health Interview Survey of 9417 children aged 0 to 17 years, CAM was used for AD by 0.99% of children. Some CAM techniques were associated with worsening severity of AD, including herbal therapy, vitamins, homeopathic agents, diet, and movement techniques.40 Usage of Chinese herbal medications for AD can be associated with liver toxicity.41 Only one CAM therapy—massage therapy—has some mild supportive data.42
Allergen Avoidance and Diet
Bronsnick et al43 discussed the possible benefit of prenatal and postnatal probiotics for prevention of AD, which were not supported in the AAD guidelines for management of AD4; postnatal prebiotic supplementation; and exclusive breastfeeding and/or supplementation with hydrolyzed formula in at-risk children. Elimination diets for children and mothers were not recommended. The authors found no beneficial role of supplements including vitamin D, selenium, fish oil, borage oil, and zinc sulfate.43
A National Institute of Allergy and Infectious Diseases consensus group recommended avoidance of proven but not random elimination of food allergens in AD, asthma, and/or eosinophilic esophagitis.44 Restricted maternal diet was not recommended, and breastfeeding exclusively for the first 4 to 6 months was recommended. Hydrolyzed formulas were suggested as a possible preventive strategy in at-risk infants as a breastfeeding alternative, with cost of these formulas being a problem.44
In children younger than 5 years, food allergy screening for the most common allergens (eg, milk, eggs, peanuts, wheat, soy) should be considered in children with persistent unremitting dermatitis and/or known food challenge–induced reactions.4 Conservative measures to avoid house dust mite exposure in known sensitized individuals including dust covers for pillows and mattresses may be beneficial.4,45
Emerging Therapies
Recently approved therapies include better-targeted agents that appear to have a reasonable safety profile and may fulfill unmet needs in AD care. Of these agents, crisaborole, a topical boron-based phosphodiesterase 4 inhibitor, was approved in December 2016 for mild to moderate AD in patients 2 years and older.Topically, this agent seems to be efficacious in the absence of notable carcinogenicity.46
The systemic (injectable) biologic agent dupilumab was approved in March 2017 for moderate to severe AD. Phase 3 studies in adults with AD showed excellent success in adults with moderate to severe AD.37 This agent is a monoclonal antibody targeted at blockade of the crucial atopic inflammatory triggering pathway via blockade of the IL-4A receptor site, targeting IL-4 and IL-13 activity.36,47 There are many medications in the pipeline, which Renert-Yuval and Guttman-Yassky48 review. However, an overview of the landscape demonstrates that Janus kinase (JAK) inhibitors49 and biologic medications in addition to dupilumab affecting targeted inflammatory cascades in AD are in development. In particular, the JAK inhibitors appear promising due to availability both as oral and topical agents.49
Need for Ongoing Care and Monitoring
Atopic dermatitis is a chronic inflammatory skin disorder with a genetic basis. Once initiated, the process of AD may persist throughout the patient’s life and become a systemic disorder with comorbidities including sleep disturbance, reduced quality of life, and cardiovascular disease.50 Ongoing management of AD includes topical reduction in irritants and triggers, topical medicaments, and management of pruritus and infections. At this time, emollients and irritant avoidance paired with judicious topical medicaments including TCs and second-line or site-specific (eg, eyelids) usage of TCIs or phosphodiesterase 4 inhibitors remain the backbone of therapy. Ongoing review of therapeutics for associated morbidities is underway, which may guide future therapeutic interventions into AD. The future of prevention and therapy look bright, but time will tell.
Atopic dermatitis (AD) is a disease that finally is coming of age in dermatology research. New topical agents and systemic biologic agents offer patients with AD other options for medical management. This article provides a practical review of prevention strategies and treatment guidelines for AD.
PREVENTION
Prevention strategies for AD have been largely unsuccessful in the past, which may relate to factors such as prenatal triggers.1 However, some newer interventional studies have shown some promise in AD prevention in specific settings. For example, a randomized trial of infants in the United States and United Kingdom at high risk for AD (ie, family history of atopy) reported that the AD risk was reduced by 50% when patients were treated with at least once-daily application of full-body emollients for 6 months (beginning by 3 weeks of life).2 The strategy of daily application of emollients for avoidance of AD in infants with a family history of AD is reasonable but may not offer lifetime prevention, and the benefit in children not from AD families is unknown.
Other trials to prevent AD have included usage of dust avoidance and dust covers for mattresses. This strategy showed modest benefit in reducing the incidence of atopic diatheses in the first year3 but did not gain endorsement by the most recent guidelines of the American Academy of Dermatology (AAD).4
Prenatal and postnatal (maternal and child) supplementation of Lactobacillus rhamnosus has shown promise in prevention.5 The exact regimen likely makes an impact on efficacy. An early study showed the usage of probiotics (eg, Lactobacillus reuteri) prenatally in pregnant women and postnatally in infants resulted in no reduction in occurrence of AD and possible reduction in IgE-associated AD.6 Kalliomäki et al7 demonstrated that L rhamnosus GG alone reduced AD by half in at-risk infants in a double-blind, placebo-controlled trial. On the other hand, Taylor et al8 performed a study of probiotic supplementation in which patients at high risk for AD developed higher rates of allergen sensitization. The most successful recent trial involved the randomization of 415 pregnant women to receive interventions from 36 weeks’ gestation until 3 months postpartum.9 The intervention was a randomized comparison of milk without probiotics versus a blend of probiotic milk containing L rhamnosus GG, Lactobacillus acidophilus La-5, and Bifidobacterium animalis subsp lactis Bb-12. At 6 years of age, 81 babies who consumed probiotic milk and 82 babies who consumed milk without probiotics were available for testing. The strategy caused a statistically significant reduction in AD in the complete case analysis (odds ratio, 0.48; 95% confidence interval, 0.25-0.92; P=.027; number needed to treat, 6). Sadly, other allergic diseases were not prevented in this study.9
MANAGEMENT OF AD
There currently is no cure or perfected prevention technique for AD. As a result, therapy focuses on avoiding triggers and alleviating symptoms.10 Recent guidelines from the AAD state that“[t]he ultimate judgment regarding the propriety of any specific therapy must be made by the physician and the patient in light of all the circumstances presented by the individual patient, and the known variability and biologic behavior of the disease.”11 Skin-directed therapies are the first line of treatment including emollients, gentle skin care, and topical medicaments. In AD, therapies are needed to reduce disease activity and flare severity, clear flares, and provide relief.
Parental education and written eczema action plans are recommended to help patients and parents/guardians follow recommended regimens12; Tollefson and Bruckner13 for the American Academy of Pediatrics provide an action plan to guide the care of children with atopic dermatitis that is simple, but many others exist online. The eczema action plan usually provides information on how to bathe and what to do when the skin is actively inflamed.
In 2014, a 4-part series of guidelines of care for the management of AD was published by the AAD, replacing prior guidelines.4,11,14,15 The following sections review some of the important parameters of care highlighted in these management guidelines.
Psychological Support
Appropriate psychological support for AD patients can be sought through counselors, therapists, psychiatrists, and support groups such as the National Eczema Association (https://nationaleczema.org/).
Education
Education is the leading form of medical therapy in patients with AD. Eczema schools are popular in Europe and are just beginning to form in the United States (http://tuh.templehealth.org/content/eczema_school.htm), which can be helpful to educate caregivers and patients with AD. Patient resources online and through support groups with an online presence, in-person meetings, and patient/family conventions can be helpful to AD patients. Often, an initial office visit with a dermatologist involves a review of avoidance of triggers, usage of gentle skin care including bland emollients, and therapeutic regimens for disease activity. This form of verbal education is to be paired with an eczema action plan, a written document that allows individuals to reference recommendations and share information with other caregivers.12,13,16
Emollients and Gentle Skin Care
Gentle skin care regimens, which includes the usage of synthetic cleansers with a low pH to help maintain the acidity (acid mantle) of the skin, seek to reduce irritation and have been rated as level IA (highest level) in recent AAD guidelines.14 Although bathing frequency has been emphasized in the guidelines, AD severity as reflected by SCORAD (SCORing Atopic Dermatitis) was not different for daily bathing versus twice weekly.17 The American Academy of Pediatrics recommended a skin care regimen of bathing every 2 to 3 days in lukewarm water for 10 to 15 minutes, followed by application of emollients that are fragrance free and have few preservatives.13 Topical emollients with additives such as colloidal oatmeal, avenanthramides, or ceramides can be used to enhance the skin barrier and are well tolerated in all age groups.18,19 Despite enhanced emollients, the therapy of AD still requires usage of prescription or over-the-counter TCs and/or topical calcineurin inhibitors (TCIs) in many cases.20
Topical Medication
Children have a relatively higher body surface area–to-weight ratio, allowing for greater potential absorption of topical medicaments and potential side effects from absorption. Types of vehicle, cost, site of application, and availability may impact patient and physician preference in choice of therapeutic topical agent.14
Topical Corticosteroids
Topical corticosteroids (TCs) are the mainstay of treatment for AD and have been used for more than 60 years.14,20 Topical corticosteroids provide anti-inflammatory effects on T cells, monocytes, and macrophages, producing altered cytokine activity locally. Topical corticosteroids inhibit collagen synthesis, potentially causing skin atrophy. They also inhibit IL-1, IL-2, IL-6, IFN-α, and tumor necrosis factor α.21 Topical corticosteroids are classified as class I (ultra-high potency) to class VII (low potency). In children, low-potency TCs generally are applied to the face, intertriginous areas, groin, and genitalia, and mid-potency corticosteroids are applied to the body, arms, and legs. An even higher-strength agent can be prescribed as a rescue medication in severe cases. After clearance with once- or twice-daily therapy, twice-weekly usage can benefit disease activity.22 Topical corticosteroids reduce inflammation as well as Staphylococcus aureus load through inhibition of cytokines that inhibit antimicrobial peptides. Topical corticosteroids have been endorsed as level IA evidence therapy by the AAD guidelines.14
Topical corticosteroids, particularly prolonged usage of mid- to high-potency products, have been associated with side effects such as skin atrophy, striae, telangiectases, hypopigmentation, rosacea, acneiform eruptions, focal hypertrichosis, perioral dermatitis, and acne23; potential systemic side effects include hypothalamic-pituitary-adrenal axis suppression, cataracts, glaucoma (with periocular application), Cushing syndrome, hyperglycemia, hypertension,23 and growth retardation.14 Long-term corticosteroid therapy is associated with tachyphylaxis and potential rebound of disease with discontinuation.24 Based on the potential risk of side effects with TCs, the least potent product for the shortest time needed is recommended, with special care for thin skin. Discontinuation when clearance occurs is advised. Allergy to TCs and/or vehicle ingredients such as propylene glycol should be suspected in severe unremitting cases.14 A recent registry review of children screened for contact dermatitis demonstrated that children with AD had higher sensitization to the steroid tixocortol pivalate.25
Topical Calcineurin Inhibitors
Topical calcineurin inhibitors include pimecrolimus cream 1%, which is approved for mild to moderate AD in adults and children 2 years and older, and tacrolimus ointment 0.03% and 0.1%, which are approved for moderate to severe AD in adults and children aged 2 to 15 years (0.03% formulation only). Topical calcineurin inhibitors can be used as second-line agents in AD in patients who have inadequate response to TCs or who may not be able to use TCs due to the disease site.10,13,14 Guidelines from the AAD also have endorsed TCIs as level IA evidence for steroid-sparing agents.
Concerns about the reporting of cancers and lymphomas prompted the US Food and Drug Administration to issue a black box warning on TCIs more than 10 years ago. Pimecrolimus, which has little absorption and no notable immunosuppressive effects, has been used without detrimental effect on vaccination and delayed-type hypersensitivities, but many decades of data are lacking.10,13,14,17,26-29 Topical calcineurin inhibitors can be used as steroid-sparing agents in lieu of corticosteroids in specific locations such as the face and eyelids and for long-term suppressive therapy twice weekly.30 Intermittent usage and cycling with corticosteroids is advisable,28 but usage intermittently beyond 1 year has not been evaluated.
Topical calcineurin inhibitors are recommended as effective for acute and chronic AD. Their use as maintenance therapy in adults and children, for AD recalcitrant to steroids, for AD in sensitive areas, for steroid-induced atrophy, and for long-term uninterrupted topical steroid usage carries a level IA evidence recommendation. Furthermore, the AAD guidelines have recommended TCIs as steroid-sparing agents with level IA evidence and off-label use of TCIs in children younger than 2 years with level IA evidence. Pretreatment with TCs to reduce stinging has level IIB evidence. Usage for flare prevention is level IA evidence. Routine blood monitoring of TCI-treated patients was not recommended; in fact, the AAD guidelines provided this recommendation as level IA evidence against routine laboratory monitoring of TCI-treated patients.14
Topical Antibiotics
Topical antibiotics are indicated for the therapy of impetigo and can be used in the setting of impetiginized AD in conjunction with TCs. Recent AAD guidelines suggested against routine usage of topical antistaphylococcal agents as level IA evidence.14 There is one study supporting usage of topical mupirocin in addition to TCs to heal children with eczema area and severity index scores more than 7 more rapidly in the first week of AD therapy, but in the same study, additive benefit was not demonstrated in AD beyond the first week.31 There also are data supporting usage of intranasal mupirocin adjunctively with bleach baths in patients with moderate to severe AD, which was rated as level IIB evidence in the AAD guidelines.14,32 There are limited data on the long-term utility of topical anti-infectives in AD. The risks of long-term usage could include resistance formation to agents such as mupirocin, contact dermatitis, and lack of efficacy.
Additional Therapeutics
Wet Wraps
Penetration through the stratum corneum is needed for drug activity in AD. Penetration can be enhanced using wet wrap therapy or using ointments, which produce higher relative potency.13 Wet wraps overlying a dilute topical medicament have been described as effective in AD and are recommended in AAD guidelines as level IIB evidence.14 Different wet wrap techniques can be used, including wet pajamas covered by dry pajamas or saline-soaked gauze wrapped around the affected areas and then dry gauze applied over the wet gauze. The methodology used should be tailored to the patient as well as to whether the individual is an inpatient or outpatient.
Bleach Baths
Dilute sodium hypochlorite solution 0.005% (one-quarter cup bleach in 20 gallons of water) has been demonstrated to be beneficial in reduction of disease activity in AD patients with recurrent bacterial infections.32 This simple technique in addition to intranasal mupirocin can reduce AD severity and improve quality of life and is the only ongoing S aureus therapeutic management endorsed by the AAD guidelines for the management of AD.14,32
Topical and Oral Delivery
Antihistamines
Topical antihistamines are ineffective in AD. Oral antihistamines can be used to reduce pruritus and are most effective when given as sedating agents for sleep enhancement but may be given as nonsedating agents for patients with concomitant allergic disorders such as allergic rhinoconjunctivitis. Paradoxical hyperreactivity with sedating antihistamines is not uncommon in small children, and sedating antihistamine usage should be discontinued in these instances.13 Parents of children with AD have reported giving the child antihistamines to sleep was helpful, as well as putting on creams, using special clothes (eg, all cotton), and keeping the room cool.33 There is level IIIC evidence against use of systemic antihistamines and level IIA evidence for sedating and nonsedating, according to the AAD guidelines.14
Systemic Therapeutics
Oral therapeutics range from oral antihistamines to oral antibiotics and immunosuppressive medications. Oral antibiotics (level IIB evidence) are reserved for superinfected AD, which is not easily defined for the following reasons: there is no consensus definition of superinfected AD; the majority of active AD lesions when cultured will demonstrate S aureus growth; and most AD lesions ooze, thereby creating the appearance of superinfection. In real-world practice, superinfection can be diagnosed based on the presence of pustules; furuncles; or signs of infection such as tracking erythema, tenderness, severe erosions, or maceration. Clinical judgment is always required.
The immunosuppressive medications used in AD include leukotriene inhibitors, which rarely are effective for AD.34 More effective systemic agents for AD include cyclosporine (level I to IIB evidence), azathioprine (level IIB evidence), mycophenolate mofetil (level IIIC evidence), and methotrexate (level IIB evidence). These agents are indicated for pediatric or adult patients when topical agents and/or phototherapy have failed.15 Monitoring these agents for side effects includes ongoing evaluation for renal and liver toxicity. Short courses (ie, 6 months) are preferred to minimize side effects.35
Dupilumab, an injectable AD therapy, is approved in the United States. This agent is injected every 2 weeks and binds to the IL-4Rα shared by IL-4 and IL-13. In 4 weeks of monotherapy, 85% of adult patients treated had 50% or greater clearance.36 Recently published consensus opinion from the International Eczema Council recommends assessment of a variety of factors before initiating systemic therapy including comorbid illnesses such as contact allergy, trigger avoidance, superinfection, and impact on quality of life.37
Oral Corticosteroids
Systemic corticosteroids clear patients quickly but provide no sustained improvement; in fact, many patients rebound or have tachyphylaxis. Although short-term corticosteroid usage can break the itch-scratch cycle, long-term usage is associated with osteoporosis, Cushing syndrome, and aseptic necrosis of the femoral head. Decreased linear growth will occur during therapy in children; therefore, systemic steroids are not recommended in children with AD, except for additional or comorbid conditions (eg, asthma or contact dermatitis).4
Phototherapy
Phototherapy has been recommended in the AAD guidelines as a second-line treatment after failure of first-line agents (ie, TCIs and TCs) for clearance and or maintenance and should be tailored to the patient’s skin tone by an experienced physician. Narrowband UVB phototherapy may act through the suppression of T-cell activity in the skin and possibly via suppression of staphylococcal superantigens; however, many phototherapy types have been described for AD.38,39 Usage can be effective in school-aged children and teenagers but may be limited due to school attendance. Phototherapy was graded as level IIB evidence in the AAD guidelines.15 Side effects include aggravation of AD by exposure to heat and UV light, actinic damage, tenderness, erythema, pruritus, burning, and stinging. Lentigines; skin cancers (melanoma and nonmelanoma); folliculitis; and ocular toxicity, especially cataracts, can occur.15 Children younger than 6 years will find it difficult to stand in a phototherapy booth and may be poor candidates.15,38,39
Complementary and Alternative Medicine
Complementary and alternative medicine (CAM) also has been used for AD in the United States. In a review of the 2007 National Health Interview Survey of 9417 children aged 0 to 17 years, CAM was used for AD by 0.99% of children. Some CAM techniques were associated with worsening severity of AD, including herbal therapy, vitamins, homeopathic agents, diet, and movement techniques.40 Usage of Chinese herbal medications for AD can be associated with liver toxicity.41 Only one CAM therapy—massage therapy—has some mild supportive data.42
Allergen Avoidance and Diet
Bronsnick et al43 discussed the possible benefit of prenatal and postnatal probiotics for prevention of AD, which were not supported in the AAD guidelines for management of AD4; postnatal prebiotic supplementation; and exclusive breastfeeding and/or supplementation with hydrolyzed formula in at-risk children. Elimination diets for children and mothers were not recommended. The authors found no beneficial role of supplements including vitamin D, selenium, fish oil, borage oil, and zinc sulfate.43
A National Institute of Allergy and Infectious Diseases consensus group recommended avoidance of proven but not random elimination of food allergens in AD, asthma, and/or eosinophilic esophagitis.44 Restricted maternal diet was not recommended, and breastfeeding exclusively for the first 4 to 6 months was recommended. Hydrolyzed formulas were suggested as a possible preventive strategy in at-risk infants as a breastfeeding alternative, with cost of these formulas being a problem.44
In children younger than 5 years, food allergy screening for the most common allergens (eg, milk, eggs, peanuts, wheat, soy) should be considered in children with persistent unremitting dermatitis and/or known food challenge–induced reactions.4 Conservative measures to avoid house dust mite exposure in known sensitized individuals including dust covers for pillows and mattresses may be beneficial.4,45
Emerging Therapies
Recently approved therapies include better-targeted agents that appear to have a reasonable safety profile and may fulfill unmet needs in AD care. Of these agents, crisaborole, a topical boron-based phosphodiesterase 4 inhibitor, was approved in December 2016 for mild to moderate AD in patients 2 years and older.Topically, this agent seems to be efficacious in the absence of notable carcinogenicity.46
The systemic (injectable) biologic agent dupilumab was approved in March 2017 for moderate to severe AD. Phase 3 studies in adults with AD showed excellent success in adults with moderate to severe AD.37 This agent is a monoclonal antibody targeted at blockade of the crucial atopic inflammatory triggering pathway via blockade of the IL-4A receptor site, targeting IL-4 and IL-13 activity.36,47 There are many medications in the pipeline, which Renert-Yuval and Guttman-Yassky48 review. However, an overview of the landscape demonstrates that Janus kinase (JAK) inhibitors49 and biologic medications in addition to dupilumab affecting targeted inflammatory cascades in AD are in development. In particular, the JAK inhibitors appear promising due to availability both as oral and topical agents.49
Need for Ongoing Care and Monitoring
Atopic dermatitis is a chronic inflammatory skin disorder with a genetic basis. Once initiated, the process of AD may persist throughout the patient’s life and become a systemic disorder with comorbidities including sleep disturbance, reduced quality of life, and cardiovascular disease.50 Ongoing management of AD includes topical reduction in irritants and triggers, topical medicaments, and management of pruritus and infections. At this time, emollients and irritant avoidance paired with judicious topical medicaments including TCs and second-line or site-specific (eg, eyelids) usage of TCIs or phosphodiesterase 4 inhibitors remain the backbone of therapy. Ongoing review of therapeutics for associated morbidities is underway, which may guide future therapeutic interventions into AD. The future of prevention and therapy look bright, but time will tell.
- Kelleher M, Dunn-Galvin A, Hourihane JO, et al. Skin barrier dysfunction measured by transepidermal water loss at 2 days and 2 months predates and predicts atopic dermatitis at 1 year. J Allergy Clin Immunol. 2015;135:930-935.
- Simpson EL, Chalmers JR, Hanifin JM, et al. Emollient enhancement of the skin barrier from birth offers effective atopic dermatitis prevention. J Allergy Clin Immunol. 2014;134:818-823.
- Tsitoura S, Nestoridou K, Botis P, et al. Randomized trial to prevent sensitization to mite allergens in toddlers and preschoolers by allergen reduction and education: one-year results. Arch Pediatr Adolesc Med. 2002;156:1021-1027.
- Sidbury R, Tom WL, Bergman JN, et al. Guidelines of care for the management of atopic dermatitis: section 4. prevention of disease flares and use of adjunctive therapies and approaches. J Am Acad Dermatol. 2014;71:1218-1233.
- Foolad N, Brezinski EA, Chase EP, et al. Effect of nutrient supplementation on atopic dermatitis in children: a systematic review of probiotics, prebiotics, formula, and fatty acids. JAMA Dermatol. 2013;149:350-355.
- Abrahamsson TR, Jakobsson T, Böttcher MF, et al. Probiotics in prevention of IgE-associated eczema: a double-blind, randomized, placebo-controlled trial. J Allergy Clin Immunol. 2007;119:1174-1180.
- Kalliomäki M, Salminen S, Arvilommi H, et al. Probiotics in primary prevention of atopic disease: a randomised placebo-controlled trial. Lancet. 2001;357:1076-1079.
- Taylor AL, Dunstan JA, Prescott SL. Probiotic supplementation for the first 6 months of life fails to reduce the risk of atopic dermatitis and increases the risk of allergen sensitization in high-risk children: a randomized controlled trial. J Allergy Clin Immunol. 2007;119:184-191.
- Simpson MR, Dotterud CK, Storrø O, et al. Perinatal probiotic supplementation in the prevention of allergy related disease: 6 year follow up of a randomised controlled trial. BMC Dermatol. 2015;15:13. doi:10.1186/s12895-015-0030-1.
- Carr WW. Topical calcineurin inhibitors for atopic dermatitis: review and treatment recommendations. Paediatr Drugs. 2013;15:303-310.
- Eichenfield LF, Tom WL, Chamlin SL, et al. Guidelines of care for the management of atopic dermatitis: section 1. diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. 2014;70:338-351.
- Silverberg NB. Creating an action plan for eczema patients. Cutis. 2015;96:362-363.
- Tollefson MM, Bruckner AL; Section on Dermatology. Atopic dermatitis: skin-directed management. Pediatrics. 2014;134:E1735-E1744.
- Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 2. management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71:116-132.
- Sidbury R, Davis DM, Cohen DE, et al; American Academy of Dermatology. Guidelines of care for the management of atopic dermatitis: section 3. management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71:327-349.
- Shi VY, Nanda S, Lee K, et al. Improving patient education with an eczema action plan: a randomized controlled trial. JAMA Dermatol. 2013;149:481-483.
- Koutroulis I, Petrova K, Kratimenos P, et al. Frequency of bathing in the management of atopic dermatitis: to bathe or not to bathe? Clin Pediatr (Phila). 2014;53:677-681.
- Fowler JF, Nebus J, Wallo W, et al. Colloidal oatmeal formulations as adjunct treatments in atopic dermatitis. J Drugs Dermatol. 2012;11:804-807.
- Fowler J Jr, Silverberg N. Active naturals have a key role in atopic dermatitis. Semin Cutan Med Surg. 2008;27:8-10.
- Eichenfield LF. Consensus guidelines in diagnosis and treatment of atopic dermatitis. Allergy. 2004;59:86-92.
- Nghiem P, Pearson G, Langley RG. Tacrolimus and pimecrolimus: from clever prokaryotes to inhibiting calcineurin and treating atopic dermatitis. J Am Acad Dermatol. 2002;46:228-241.
- Schmitt J. Commentary: eczema and cancer risk. Br J Dermatol. 2011;165:463-464.
- Abramovits W, Hung P, Tong KB. Efficacy and economics of topical calcineurin inhibitors for the treatment of atopic dermatitis. Am J Clin Dermatol. 2006;7:213-222.
- Takahashi-Ando N, Jones MA, Fujisawa S, et al. Patient-reported outcomes after discontinuation of long-term topical corticosteroid treatment for atopic dermatitis: a targeted cross-sectional survey. Drug Healthc Patient Saf. 2015;7:57-62.
- Jacob SE, McGowan M, Silverberg NB, et al. Pediatric contact dermatitis registry data on contact allergy in children with atopic dermatitis. JAMA Dermatol. 2017;153:765-770.
- Werfel T. Topical use of pimecrolimus in atopic dermatitis: update on the safety and efficacy. J Dtsch Dermatol Ges. 2009;7:739-742.
- Wahn U, Bos JD, Goodfield M, et al. Efficacy and safety of pimecrolimus cream in the long-term management of atopic dermatitis in children. Pediatrics. 2002;110(1, pt 1):E2.
- Berger TG, Duvic M, Van Voorhees AS, et al; American Academy of Dermatology Association Task Force. The use of topical calcineurin inhibitors in dermatology: safety concerns. report of the American Academy of Dermatology Association Task Force. J Am Acad Dermatol. 2006;54:818-823.
- Paller AS. Latest approaches to treating atopic dermatitis. Chem Immunol Allergy. 2012;96:132-140.
- Thaçi D, Reitamo S, Gonzalez Ensenat MA, et al. Proactive disease management with 0.03% tacrolimus ointment for children with atopic dermatitis: results of a randomized, multicentre, comparative study. Br J Dermatol. 2008;159:1348-1356.
- Gong JQ, Lin L, Lin T, et al. Skin colonization by Staphylococcus aureus in patients with eczema and atopic dermatitis and relevant combined topical therapy: a double-blind multicentre randomized controlled trial. Br J Dermatol. 2006;155:680-687.
- Huang JT, Abrams M, Tlougan B, et al. Treatment of Staphylococcus aureus colonization in atopic dermatitis decreases disease severity. Pediatrics. 2009;123:E808-E814.
- Reid P, Lewis-Jones MS. Sleep difficulties and their management in preschoolers with atopic eczema. Clin Exp Dermatol. 1995;20:38-41.
- Silverberg NB, Paller AS. Leukotriene receptor antagonists are ineffective for severe atopic dermatitis. J Am Acad Dermatol. 2004;50:485-486.
- Wolverton SE. Comprehensive Dermatologic Drug Therapy. 3rd ed. New York, NY: Elsevier Saunders; 2013.
- Beck LA, Thaçi D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
- Simpson EL, Bruin-Weller M, Flohr C, et al. When does atopic dermatitis warrant systemic therapy? recommendations from an expert panel of the International Eczema Council [published online August 10, 2017]. J Am Acad Dermatol. doi:10.1016/j.jaad.2017.06.042.
- Veith W, DeLeo V, Silverberg N. Medical phototherapy in childhood skin diseases. Minerva Pediatr. 2011;63:327-333.
- Song E, Reja D, Silverberg N, et al. Phototherapy: kids are not just little people. Clin Dermatol. 2015;33:672-680.
- Silverberg JI, Lee-Wong M, Silverberg NB. Complementary and alternative medicines and childhood eczema: a US population-based study. Dermatitis. 2014;25:246-254.
- Stickel F, Shouval D. Hepatotoxicity of herbal and dietary supplements: an update. Arch Toxicol. 2015;89:851-865.
- Schachner L, Field T, Hernandez-Reif M, et al. Atopic dermatitis symptoms decreased in children following massage therapy. Pediatr Dermatol. 1998;15:390-395.
- Bronsnick T, Murzaku EC, Rao BK. Diet in dermatology: part I. atopic dermatitis, acne, and nonmelanoma skin cancer. J Am Acad Dermatol. 2014;71:1039.e1-1039.e12.
- Boyce JA, Assa’ad A, Burks AW, et al. Guidelines for the diagnosis and management of food allergy in the United States: summary of the NIAID-sponsored expert panel report. Nutr Res. 2011;31:61-75.
- Silverberg NB, Lee-Wong M, Yosipovitch G. Diet and atopic dermatitis. Cutis. 2016;97:227-232.
- Hanifin JM, Chan SC, Cheng JB, et al. Type phosphodiesterase inhibitors have clinical and in vitro anti-inflammatory effects in atopic dermatitis. J Invest Dermatol. 1996;107:51-56.
- Boguniewicz M, Leung DY. Targeted therapy for allergic diseases: at the intersection of cutting-edge science and clinical practice. J Allergy Clin Immunol. 2015;135:354-356.
- Renert-Yuval Y, Guttman-Yassky E. Systemic therapies in atopic dermatitis: the pipeline. Clin Dermatol. 2017;35:387-397.
- Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76:736-744.
- Brunner PM, Silverberg JI, Guttman-Yassky E, et al. Increasing comorbidities suggest that atopic dermatitis is a systemic disorder. J Invest Dermatol. 2017;137:18-25.
- Kelleher M, Dunn-Galvin A, Hourihane JO, et al. Skin barrier dysfunction measured by transepidermal water loss at 2 days and 2 months predates and predicts atopic dermatitis at 1 year. J Allergy Clin Immunol. 2015;135:930-935.
- Simpson EL, Chalmers JR, Hanifin JM, et al. Emollient enhancement of the skin barrier from birth offers effective atopic dermatitis prevention. J Allergy Clin Immunol. 2014;134:818-823.
- Tsitoura S, Nestoridou K, Botis P, et al. Randomized trial to prevent sensitization to mite allergens in toddlers and preschoolers by allergen reduction and education: one-year results. Arch Pediatr Adolesc Med. 2002;156:1021-1027.
- Sidbury R, Tom WL, Bergman JN, et al. Guidelines of care for the management of atopic dermatitis: section 4. prevention of disease flares and use of adjunctive therapies and approaches. J Am Acad Dermatol. 2014;71:1218-1233.
- Foolad N, Brezinski EA, Chase EP, et al. Effect of nutrient supplementation on atopic dermatitis in children: a systematic review of probiotics, prebiotics, formula, and fatty acids. JAMA Dermatol. 2013;149:350-355.
- Abrahamsson TR, Jakobsson T, Böttcher MF, et al. Probiotics in prevention of IgE-associated eczema: a double-blind, randomized, placebo-controlled trial. J Allergy Clin Immunol. 2007;119:1174-1180.
- Kalliomäki M, Salminen S, Arvilommi H, et al. Probiotics in primary prevention of atopic disease: a randomised placebo-controlled trial. Lancet. 2001;357:1076-1079.
- Taylor AL, Dunstan JA, Prescott SL. Probiotic supplementation for the first 6 months of life fails to reduce the risk of atopic dermatitis and increases the risk of allergen sensitization in high-risk children: a randomized controlled trial. J Allergy Clin Immunol. 2007;119:184-191.
- Simpson MR, Dotterud CK, Storrø O, et al. Perinatal probiotic supplementation in the prevention of allergy related disease: 6 year follow up of a randomised controlled trial. BMC Dermatol. 2015;15:13. doi:10.1186/s12895-015-0030-1.
- Carr WW. Topical calcineurin inhibitors for atopic dermatitis: review and treatment recommendations. Paediatr Drugs. 2013;15:303-310.
- Eichenfield LF, Tom WL, Chamlin SL, et al. Guidelines of care for the management of atopic dermatitis: section 1. diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. 2014;70:338-351.
- Silverberg NB. Creating an action plan for eczema patients. Cutis. 2015;96:362-363.
- Tollefson MM, Bruckner AL; Section on Dermatology. Atopic dermatitis: skin-directed management. Pediatrics. 2014;134:E1735-E1744.
- Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 2. management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71:116-132.
- Sidbury R, Davis DM, Cohen DE, et al; American Academy of Dermatology. Guidelines of care for the management of atopic dermatitis: section 3. management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71:327-349.
- Shi VY, Nanda S, Lee K, et al. Improving patient education with an eczema action plan: a randomized controlled trial. JAMA Dermatol. 2013;149:481-483.
- Koutroulis I, Petrova K, Kratimenos P, et al. Frequency of bathing in the management of atopic dermatitis: to bathe or not to bathe? Clin Pediatr (Phila). 2014;53:677-681.
- Fowler JF, Nebus J, Wallo W, et al. Colloidal oatmeal formulations as adjunct treatments in atopic dermatitis. J Drugs Dermatol. 2012;11:804-807.
- Fowler J Jr, Silverberg N. Active naturals have a key role in atopic dermatitis. Semin Cutan Med Surg. 2008;27:8-10.
- Eichenfield LF. Consensus guidelines in diagnosis and treatment of atopic dermatitis. Allergy. 2004;59:86-92.
- Nghiem P, Pearson G, Langley RG. Tacrolimus and pimecrolimus: from clever prokaryotes to inhibiting calcineurin and treating atopic dermatitis. J Am Acad Dermatol. 2002;46:228-241.
- Schmitt J. Commentary: eczema and cancer risk. Br J Dermatol. 2011;165:463-464.
- Abramovits W, Hung P, Tong KB. Efficacy and economics of topical calcineurin inhibitors for the treatment of atopic dermatitis. Am J Clin Dermatol. 2006;7:213-222.
- Takahashi-Ando N, Jones MA, Fujisawa S, et al. Patient-reported outcomes after discontinuation of long-term topical corticosteroid treatment for atopic dermatitis: a targeted cross-sectional survey. Drug Healthc Patient Saf. 2015;7:57-62.
- Jacob SE, McGowan M, Silverberg NB, et al. Pediatric contact dermatitis registry data on contact allergy in children with atopic dermatitis. JAMA Dermatol. 2017;153:765-770.
- Werfel T. Topical use of pimecrolimus in atopic dermatitis: update on the safety and efficacy. J Dtsch Dermatol Ges. 2009;7:739-742.
- Wahn U, Bos JD, Goodfield M, et al. Efficacy and safety of pimecrolimus cream in the long-term management of atopic dermatitis in children. Pediatrics. 2002;110(1, pt 1):E2.
- Berger TG, Duvic M, Van Voorhees AS, et al; American Academy of Dermatology Association Task Force. The use of topical calcineurin inhibitors in dermatology: safety concerns. report of the American Academy of Dermatology Association Task Force. J Am Acad Dermatol. 2006;54:818-823.
- Paller AS. Latest approaches to treating atopic dermatitis. Chem Immunol Allergy. 2012;96:132-140.
- Thaçi D, Reitamo S, Gonzalez Ensenat MA, et al. Proactive disease management with 0.03% tacrolimus ointment for children with atopic dermatitis: results of a randomized, multicentre, comparative study. Br J Dermatol. 2008;159:1348-1356.
- Gong JQ, Lin L, Lin T, et al. Skin colonization by Staphylococcus aureus in patients with eczema and atopic dermatitis and relevant combined topical therapy: a double-blind multicentre randomized controlled trial. Br J Dermatol. 2006;155:680-687.
- Huang JT, Abrams M, Tlougan B, et al. Treatment of Staphylococcus aureus colonization in atopic dermatitis decreases disease severity. Pediatrics. 2009;123:E808-E814.
- Reid P, Lewis-Jones MS. Sleep difficulties and their management in preschoolers with atopic eczema. Clin Exp Dermatol. 1995;20:38-41.
- Silverberg NB, Paller AS. Leukotriene receptor antagonists are ineffective for severe atopic dermatitis. J Am Acad Dermatol. 2004;50:485-486.
- Wolverton SE. Comprehensive Dermatologic Drug Therapy. 3rd ed. New York, NY: Elsevier Saunders; 2013.
- Beck LA, Thaçi D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
- Simpson EL, Bruin-Weller M, Flohr C, et al. When does atopic dermatitis warrant systemic therapy? recommendations from an expert panel of the International Eczema Council [published online August 10, 2017]. J Am Acad Dermatol. doi:10.1016/j.jaad.2017.06.042.
- Veith W, DeLeo V, Silverberg N. Medical phototherapy in childhood skin diseases. Minerva Pediatr. 2011;63:327-333.
- Song E, Reja D, Silverberg N, et al. Phototherapy: kids are not just little people. Clin Dermatol. 2015;33:672-680.
- Silverberg JI, Lee-Wong M, Silverberg NB. Complementary and alternative medicines and childhood eczema: a US population-based study. Dermatitis. 2014;25:246-254.
- Stickel F, Shouval D. Hepatotoxicity of herbal and dietary supplements: an update. Arch Toxicol. 2015;89:851-865.
- Schachner L, Field T, Hernandez-Reif M, et al. Atopic dermatitis symptoms decreased in children following massage therapy. Pediatr Dermatol. 1998;15:390-395.
- Bronsnick T, Murzaku EC, Rao BK. Diet in dermatology: part I. atopic dermatitis, acne, and nonmelanoma skin cancer. J Am Acad Dermatol. 2014;71:1039.e1-1039.e12.
- Boyce JA, Assa’ad A, Burks AW, et al. Guidelines for the diagnosis and management of food allergy in the United States: summary of the NIAID-sponsored expert panel report. Nutr Res. 2011;31:61-75.
- Silverberg NB, Lee-Wong M, Yosipovitch G. Diet and atopic dermatitis. Cutis. 2016;97:227-232.
- Hanifin JM, Chan SC, Cheng JB, et al. Type phosphodiesterase inhibitors have clinical and in vitro anti-inflammatory effects in atopic dermatitis. J Invest Dermatol. 1996;107:51-56.
- Boguniewicz M, Leung DY. Targeted therapy for allergic diseases: at the intersection of cutting-edge science and clinical practice. J Allergy Clin Immunol. 2015;135:354-356.
- Renert-Yuval Y, Guttman-Yassky E. Systemic therapies in atopic dermatitis: the pipeline. Clin Dermatol. 2017;35:387-397.
- Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76:736-744.
- Brunner PM, Silverberg JI, Guttman-Yassky E, et al. Increasing comorbidities suggest that atopic dermatitis is a systemic disorder. J Invest Dermatol. 2017;137:18-25.
Practice Points
- Prevention of atopic dermatitis is desired in high-risk settings (ie, 1 or more relatives with atopy).
- Emollient therapy from early infancy has been described as one method.
- Other forms of disease prevention have not yet been adequately developed.
Clinical Pearl: Mastering the Flexible Scalpel Blade With the Banana Practice Model
The flexible scalpel blade (FSB) is a 2-sided handheld razor blade that serves as a pivotal instrument in certain dermatologic procedures. Its unrivaled sharpness1 permits pinpoint precision for shave biopsies, excisions of superficial lesions,2 scar contouring, and harvesting of split-thickness skin grafts.3 Given its flexibility and long edge, considerable manual dexterity and skill are required to maximize its full potential.
Practice Gap
Prior to practicing on live patients, students on clinical rotation would benefit from in vitro skin simulators to practice correct hand position, FSB control for concave and convex surface cutting, and safety. Prior practice models have included mannequins, tomatoes, and eggplants.4,5 Here, the authors recommend the use of a banana (genus Musa). In addition to its year-round availability, economic feasibility, simplicity, and portability, the banana has colored skin that well represents the epidermis, dermis, and subcutaneous tissue, allowing for visual feedback. Furthermore, its contour irregularities simulate convexities and concavities for various anatomic locations. Although the firmness of a yellow-green banana provides immediate tissue feedback, the softness and pliability of a ripe banana simulates the consistency of older skin and the use of appropriate traction.
Tools
To begin, one simply requires a marking pen, banana, and razor blade. Various shapes, including a circle, ellipse, rectangle, trapezoid, triangle, and multilobed lesion are demarcated by students or attendings (Figure 1).

The Technique
To handle the FSB, one can hold the lateral edges of the blade between the thumb and index finger or between the thumb and middle finger. The thumb and index finger position allows for additional flexible working space and visualization, increased traction by the remaining 3 fingers, and greater ease of removal of lesions with considerable height. The thumb and middle finger hold allows for versatile use of the index finger of the same hand for stabilizing the center of the blade, fixing the tissue on the FSB while it is removed, and sliding the specimen off the FSB. It is important to maintain a fixed distance from the blade to the metacarpals at all times to ensure smooth advancement of the blade and visualization. Beginners can lift the pinky finger of the hand holding the FSB and move the finger up and down to control the angle of the blade.
Practice Implications
Generally, we utilize various techniques of shaving using the FSB. We approach the target lesion 2 to 3 mm from the marked location and slide parallel to the skin surface and perpendicular to the lesion until the epidermis is penetrated. Second, we advance the blade toward the lesion with careful attention paid to the perimeter of the lesion and the points of contact of the FSB. For lesions with hardier consistencies, a sawing motion of the blade is employed, which also requires controlled tilting of the wrist to maintain an even depth and smooth bevel. To cut deeper, flexing the FSB with lateral pressure is helpful. More shallow lesions require the instrument to be flatter and less bowed. When finishing the shave, it is important to start angling the blade upward early, either at the center of the targeted lesion or 2 to 3 mm before the demarcated edge of the skin graft, while applying traction away from the lesion and slight downward pressure with the nondominant hand.
For larger lesions, the perimeter may be more difficult to remove precisely and can be achieved by rotating the blade around the lesion with focus on one point of contact of the FSB to cut and glide through the tissue’s perimeter. To achieve a more exact wound edge and to preclude jagged borders, a No. 15 blade can be used to score the perimeter very superficially to the papillary dermis prior to shave removal. The main disadvantage, however, is that the beveled edge is removed.
In summary, the FSB is an exceptional tool for biopsies, tumor removal, scar contouring, and split-thickness skin grafts. Through the banana practice model, one can attain fine control and reap the benefits of the FSB after meticulous and dedicated training.
- Awadalla B, Hexsel C, Goldberg LH. The sharpness of blades used in dermatologic surgery. Dermatol Surg. 2016;42:105-107.
- Vergilis-Kalner IJ, Goldberg LH, Firoz B, et al. Horizontal excision of in situ epidermal tumors using a flexible blade. Dermatol Surg. 2011;37:234-236.
- Hexsel CL, Loosemore M, Goldberg LH, et al. Postauricular skin: an excellent donor site for split-thickness skin grafts for the head, neck, and upper chest. Dermatol Surg. 2015;41:48-52.
- Chen TM, Mellette JR. Surgical pearl: tomato—an alternative model for shave biopsy training. J Am Acad Dermatol. 2006;54:517-518.
- Wang X, Albahrani Y, Pan M, et al. Skin simulators for dermatological procedures. Dermatol Online J. 2015;21. pii:13030/qt33j6x4nx.
The flexible scalpel blade (FSB) is a 2-sided handheld razor blade that serves as a pivotal instrument in certain dermatologic procedures. Its unrivaled sharpness1 permits pinpoint precision for shave biopsies, excisions of superficial lesions,2 scar contouring, and harvesting of split-thickness skin grafts.3 Given its flexibility and long edge, considerable manual dexterity and skill are required to maximize its full potential.
Practice Gap
Prior to practicing on live patients, students on clinical rotation would benefit from in vitro skin simulators to practice correct hand position, FSB control for concave and convex surface cutting, and safety. Prior practice models have included mannequins, tomatoes, and eggplants.4,5 Here, the authors recommend the use of a banana (genus Musa). In addition to its year-round availability, economic feasibility, simplicity, and portability, the banana has colored skin that well represents the epidermis, dermis, and subcutaneous tissue, allowing for visual feedback. Furthermore, its contour irregularities simulate convexities and concavities for various anatomic locations. Although the firmness of a yellow-green banana provides immediate tissue feedback, the softness and pliability of a ripe banana simulates the consistency of older skin and the use of appropriate traction.
Tools
To begin, one simply requires a marking pen, banana, and razor blade. Various shapes, including a circle, ellipse, rectangle, trapezoid, triangle, and multilobed lesion are demarcated by students or attendings (Figure 1).
The Technique
To handle the FSB, one can hold the lateral edges of the blade between the thumb and index finger or between the thumb and middle finger. The thumb and index finger position allows for additional flexible working space and visualization, increased traction by the remaining 3 fingers, and greater ease of removal of lesions with considerable height. The thumb and middle finger hold allows for versatile use of the index finger of the same hand for stabilizing the center of the blade, fixing the tissue on the FSB while it is removed, and sliding the specimen off the FSB. It is important to maintain a fixed distance from the blade to the metacarpals at all times to ensure smooth advancement of the blade and visualization. Beginners can lift the pinky finger of the hand holding the FSB and move the finger up and down to control the angle of the blade.
Practice Implications
Generally, we utilize various techniques of shaving using the FSB. We approach the target lesion 2 to 3 mm from the marked location and slide parallel to the skin surface and perpendicular to the lesion until the epidermis is penetrated. Second, we advance the blade toward the lesion with careful attention paid to the perimeter of the lesion and the points of contact of the FSB. For lesions with hardier consistencies, a sawing motion of the blade is employed, which also requires controlled tilting of the wrist to maintain an even depth and smooth bevel. To cut deeper, flexing the FSB with lateral pressure is helpful. More shallow lesions require the instrument to be flatter and less bowed. When finishing the shave, it is important to start angling the blade upward early, either at the center of the targeted lesion or 2 to 3 mm before the demarcated edge of the skin graft, while applying traction away from the lesion and slight downward pressure with the nondominant hand.
For larger lesions, the perimeter may be more difficult to remove precisely and can be achieved by rotating the blade around the lesion with focus on one point of contact of the FSB to cut and glide through the tissue’s perimeter. To achieve a more exact wound edge and to preclude jagged borders, a No. 15 blade can be used to score the perimeter very superficially to the papillary dermis prior to shave removal. The main disadvantage, however, is that the beveled edge is removed.
In summary, the FSB is an exceptional tool for biopsies, tumor removal, scar contouring, and split-thickness skin grafts. Through the banana practice model, one can attain fine control and reap the benefits of the FSB after meticulous and dedicated training.
The flexible scalpel blade (FSB) is a 2-sided handheld razor blade that serves as a pivotal instrument in certain dermatologic procedures. Its unrivaled sharpness1 permits pinpoint precision for shave biopsies, excisions of superficial lesions,2 scar contouring, and harvesting of split-thickness skin grafts.3 Given its flexibility and long edge, considerable manual dexterity and skill are required to maximize its full potential.
Practice Gap
Prior to practicing on live patients, students on clinical rotation would benefit from in vitro skin simulators to practice correct hand position, FSB control for concave and convex surface cutting, and safety. Prior practice models have included mannequins, tomatoes, and eggplants.4,5 Here, the authors recommend the use of a banana (genus Musa). In addition to its year-round availability, economic feasibility, simplicity, and portability, the banana has colored skin that well represents the epidermis, dermis, and subcutaneous tissue, allowing for visual feedback. Furthermore, its contour irregularities simulate convexities and concavities for various anatomic locations. Although the firmness of a yellow-green banana provides immediate tissue feedback, the softness and pliability of a ripe banana simulates the consistency of older skin and the use of appropriate traction.
Tools
To begin, one simply requires a marking pen, banana, and razor blade. Various shapes, including a circle, ellipse, rectangle, trapezoid, triangle, and multilobed lesion are demarcated by students or attendings (Figure 1).
The Technique
To handle the FSB, one can hold the lateral edges of the blade between the thumb and index finger or between the thumb and middle finger. The thumb and index finger position allows for additional flexible working space and visualization, increased traction by the remaining 3 fingers, and greater ease of removal of lesions with considerable height. The thumb and middle finger hold allows for versatile use of the index finger of the same hand for stabilizing the center of the blade, fixing the tissue on the FSB while it is removed, and sliding the specimen off the FSB. It is important to maintain a fixed distance from the blade to the metacarpals at all times to ensure smooth advancement of the blade and visualization. Beginners can lift the pinky finger of the hand holding the FSB and move the finger up and down to control the angle of the blade.
Practice Implications
Generally, we utilize various techniques of shaving using the FSB. We approach the target lesion 2 to 3 mm from the marked location and slide parallel to the skin surface and perpendicular to the lesion until the epidermis is penetrated. Second, we advance the blade toward the lesion with careful attention paid to the perimeter of the lesion and the points of contact of the FSB. For lesions with hardier consistencies, a sawing motion of the blade is employed, which also requires controlled tilting of the wrist to maintain an even depth and smooth bevel. To cut deeper, flexing the FSB with lateral pressure is helpful. More shallow lesions require the instrument to be flatter and less bowed. When finishing the shave, it is important to start angling the blade upward early, either at the center of the targeted lesion or 2 to 3 mm before the demarcated edge of the skin graft, while applying traction away from the lesion and slight downward pressure with the nondominant hand.
For larger lesions, the perimeter may be more difficult to remove precisely and can be achieved by rotating the blade around the lesion with focus on one point of contact of the FSB to cut and glide through the tissue’s perimeter. To achieve a more exact wound edge and to preclude jagged borders, a No. 15 blade can be used to score the perimeter very superficially to the papillary dermis prior to shave removal. The main disadvantage, however, is that the beveled edge is removed.
In summary, the FSB is an exceptional tool for biopsies, tumor removal, scar contouring, and split-thickness skin grafts. Through the banana practice model, one can attain fine control and reap the benefits of the FSB after meticulous and dedicated training.
- Awadalla B, Hexsel C, Goldberg LH. The sharpness of blades used in dermatologic surgery. Dermatol Surg. 2016;42:105-107.
- Vergilis-Kalner IJ, Goldberg LH, Firoz B, et al. Horizontal excision of in situ epidermal tumors using a flexible blade. Dermatol Surg. 2011;37:234-236.
- Hexsel CL, Loosemore M, Goldberg LH, et al. Postauricular skin: an excellent donor site for split-thickness skin grafts for the head, neck, and upper chest. Dermatol Surg. 2015;41:48-52.
- Chen TM, Mellette JR. Surgical pearl: tomato—an alternative model for shave biopsy training. J Am Acad Dermatol. 2006;54:517-518.
- Wang X, Albahrani Y, Pan M, et al. Skin simulators for dermatological procedures. Dermatol Online J. 2015;21. pii:13030/qt33j6x4nx.
- Awadalla B, Hexsel C, Goldberg LH. The sharpness of blades used in dermatologic surgery. Dermatol Surg. 2016;42:105-107.
- Vergilis-Kalner IJ, Goldberg LH, Firoz B, et al. Horizontal excision of in situ epidermal tumors using a flexible blade. Dermatol Surg. 2011;37:234-236.
- Hexsel CL, Loosemore M, Goldberg LH, et al. Postauricular skin: an excellent donor site for split-thickness skin grafts for the head, neck, and upper chest. Dermatol Surg. 2015;41:48-52.
- Chen TM, Mellette JR. Surgical pearl: tomato—an alternative model for shave biopsy training. J Am Acad Dermatol. 2006;54:517-518.
- Wang X, Albahrani Y, Pan M, et al. Skin simulators for dermatological procedures. Dermatol Online J. 2015;21. pii:13030/qt33j6x4nx.
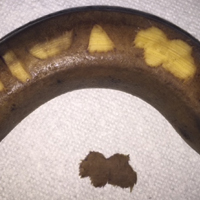
Photosensitive Atopic Dermatitis Exacerbated by UVB Exposure
Atopic dermatitis (AD) is the most common inflammatory skin condition, affecting approximately 15% to 20% of the global population.1,2 Atopic dermatitis is characterized by a chronic relapsing dermatitis with pruritus, often beginning in infancy or childhood. Atopic dermatitis is caused by a defect in epidermal barrier function, which results in increased transepidermal water loss.1 The criteria for AD include a pruritic skin condition plus 3 or more of the following: history of involvement of the skin creases, history of asthma or hay fever, history of AD in a first-degree relative (in children), 1-year history of generally dry skin, visible flexural eczema, and an age of onset of less than 2 years. Adults with AD frequently present with hand or facial dermatitis.1
UV light therapies including narrowband UVB (NB-UVB), UVA1, and psoralen plus UVA (PUVA) have all been used as effective treatments of AD.3,4 UV light is beneficial for AD patients due to its immunomodulatory effects, thickening of the stratum corneum, and the reduction of Staphylococcus aureus in the skin.2 Most patients with AD improve with light therapy; however, it is estimated that 1% to 3% of patients with AD will experience a paradoxical worsening of their AD after exposure to UV light.2,5 This condition is referred to as photosensitive AD and is characterized by a photodistributed rash in patients who fulfill the criteria of AD. Photosensitive AD has a female predominance and generally affects patients with late-onset disease with development of AD after puberty.2,5 The pathogenesis for the development of photosensitivity in patients with AD who previously tolerated exposure to sunlight is unknown.5 We describe a case of photosensitive AD exacerbated by UVB exposure.
Case Report
On physical examination the patient had thin, well-demarcated, erythematous papules and plaques with scaling, primarily on sun-exposed skin on the forehead (Figure 1A), cheeks (Figure 1B), eyelids, upper lip, neck (Figures 1B and 1C), upper chest (Figure 1C), and dorsal aspect of the hands, with excoriated pink papules on the forearms, shoulders, and back. A punch biopsy of the right neck showed spongiotic dermatitis with a perivascular lymphohistiocytic infiltrate (Figure 2). Further workup was pursued including complete blood cell count, comprehensive metabolic profile, liver function panel, Sjögren syndrome antigen A/Sjögren syndrome antigen B test, antinuclear antibody test, human immunodeficiency virus 1/2 antigen/antibody test, hepatitis panel, and mycobacterium tuberculosis test, which were all within reference range. Photodermatosis was suspected and she underwent phototesting including UVA, NB-UVB, and visible light. Phototesting confirmed she had a UVB photosensitivity with a markedly decreased minimal erythema dose (MED) to NB-UVB. The MED to NB-UVB was positive at 24 hours to all tested sites, the lowest of which was 0.135 J/cm2. Eczematous changes began to develop at day 6 at doses of 0.945 and 1.080 J/cm2. The patient also underwent visible light testing, which was negative. The patient was patch tested for multiple standardized agents as well as personal products, all of which were negative. Subsequent photopatch testing revealed a slightly positive reaction to benzophenone 4, a common ingredient in sunscreens.
The patient was then started on mycophenolate mofetil and prednisone. Repeat MED testing to NB-UVB was performed. Her repeat MED to NB-UVB was determined to be 0.405 J/cm2, and hardening commenced at 3 times per week at 70% of the MED (0.2835 J/cm2). She began to flare and develop an eczematous reaction, thus the dose was decreased to 50% of the MED (0.2025 J/cm2), which she tolerated.
Comment
Classification and Clinical Presentation
The literature on photosensitive AD is scant, and this disease entity is rare. Alternative names include photoaggravated AD, photosensitive eczema, and light-exacerbated eczema.5 Two main studies have been conducted in recent years that were intended to characterize photosensitive AD. ten Berge et al5 conducted a retrospective study of 145 patients with AD that were phototested in 2009. They found that 3% of their total AD patient population had photosensitive AD.5 In 2016, Ellenbogen et al2 performed a similar single-center retrospective analysis of 17 patients with long-standing AD who suddenly developed photosensitivity.
Patients with photosensitive AD typically present with lesions on sun-exposed skin with coexisting eczematous lesions in sites with a predilection for AD.2 In the study conducted by ten Berge et al,5 2 main reaction patterns were observed: erythematous papules with pruritus and an eczematous reaction.
Histopathology
The histopathologic findings of photosensitive AD are nonspecific but are characterized by spongiotic dermatitis with a perivascular lymphohistiocytic infiltrate.2
Diagnosis With Phototesting
Phototesting of patients with AD should be considered if there is a suspicion for photosensitivity based on persistent disease despite use of photoprotection and local treatment.5-7 Patients may not notice a correlation of skin exacerbations with UV exposure, especially if they are only sensitive to UVA, as it is still present on cloudy days and can penetrate glass windows.8 Phototesting evaluates the degree of sensitivity to UV light and the specific wavelength eliciting the cutaneous response. Phototesting consists of determining the MED to UVA and UVB, the minimal phototoxic dose for PUVA, and visible light exposure. Further evaluation may include photoprovocation testing or photopatch testing, as these patients can have coexisting photocontact allergies.
The MED is defined as the minimal dose of UV light needed to induce perceptible erythema in exposed skin.5 It is dependent on the light source and patient’s skin type, and individual units may vary. To determine the MED to UVA or UVB, 2×2-cm skin fields are irradiated with increasing cumulative UVA/UVB. The dose varies by skin type and it is then read at 24 hours. The majority of patients with photosensitive AD are reported to have a normal MED; however, some studies have reported the MED to be decreased.5,7-9 ten Berge et al5 found 7% of their study participants exhibited a lower MED, as seen in our patient.
The minimal phototoxic dose for PUVA is defined as the least exposure dose of UVA 1 hour after ingestion of 0.4 mg/kg of methoxsalen that produces pink erythema with 4 distinct borders at 48, 72, or 96 hours after ingestion.10 Visible light exposure is tested using a slide projector as the light source to an approximately 10×5-cm area of skin for 45 minutes. Any immediate or delayed reaction is abnormal and considered positive.10
Photoprovocation testing has been performed in several studies.2,5 It consists of exposing an 8-cm area of skin to 80 J/cm2 UVA and 10 mJ/cm2 UVB, which is read at 24, 48, or 72 hours. A papular or eczematous reaction is considered positive.2,11
The results of phototesting have varied between studies. ten Berge et al5 phototested 107 patients with AD and photosensitivity and 17% were found to be solely sensitive to UVA whereas 67% were found to be sensitive to UVA and UVB. In contrast, Ellenbogen et al2 only tested 17 patients with AD and photosensitivity and they found that 56% (9/16) were sensitive to UVA alone while only 44% (7/16) were sensitive to UVA and UVB.
Photopatch testing can help to rule out photosensitivity due to a substance in the presence of UV light. In studies of patients with photosensitive AD (N=125), photocontact reactions occurred in 23% and were predominantly associated with sunscreens, skin care products, and fragrances.5,12 Photopatch testing is done by placing duplicate sets of patches on nonlesional skin using the Finn Chamber technique. A published list of allergens, which were agreed upon by the European Society of Contact Dermatitis and the European Society for Photodermatology in 2000 are seen in Table 1.13 The list contains mainly UV filters and drugs. The patients’ own products also should be tested in addition to the published list of allergens, but a maximum of 30 patches should be placed at one time. The patches are removed at either 24 or 48 hours; some researchers have found greater sensitivity with the 48-hour time period, while others have not found a significant difference.10 One set of skin fields then is covered with an impermeable occlusive dressing as a control while the other is irradiated with 5 J/cm2 of a broad-spectrum UVA light source. UVA fluorescent lamps are the light source of choice because of their widespread availability, reproducible broad spectrum, and beam uniformity.10 In the study conducted by ten Berge et al,5 photopatch testing was performed on 125 patients, and 29 patients were found to be positive to one or more substances. Ellenbogen et al2 photopatch tested 5 patients with photosensitive AD and a clinical suspicion of photoallergy; however, all 5 were negative. Our patient underwent traditional patch testing due to clinical suspicion of a coexisting contact allergy, which was negative.
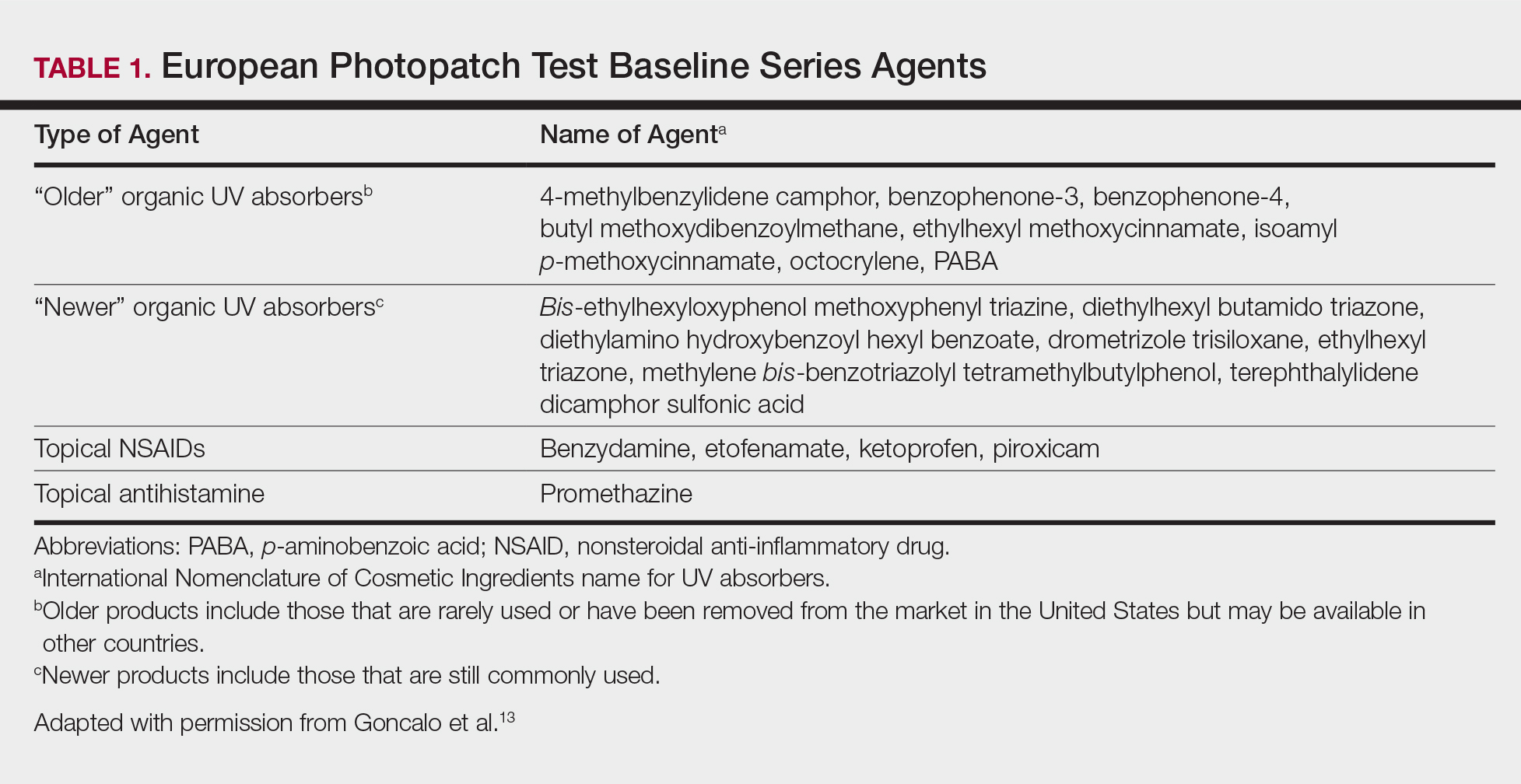
Differential Diagnosis
The differential diagnosis for photosensitive AD includes PMLE with coexisting AD, chronic AD, and photoallergic contact dermatitis. Photosensitive AD worsens with increasing exposure to uncontrolled sunlight, in contrast to patients with PMLE who experience UV radiation (UVR) hardening with increasing UV exposure during the summer months, resulting in improvement of skin lesions. Patients with chronic AD generally report a history of chronic ambient sun exposure and exhibit well-demarcated eczematous lesions in a photodistributed pattern with sparing of sun-protected skin.2 In contrast, photosensitive AD involves both sun-exposed and covered areas of the body. Chronic AD will have a positive photoprovocation test with a decreased MED (Table 2). Photoallergic contact dermatitis also will have photodistributed eczematous lesions with relative sparing of non–sun-exposed skin; however, these patients generally have negative photoprovocation testing with a normal MED.2 These patients may or may not have a history of reaction to a known allergen, but they likely will have a positive photopatch test.
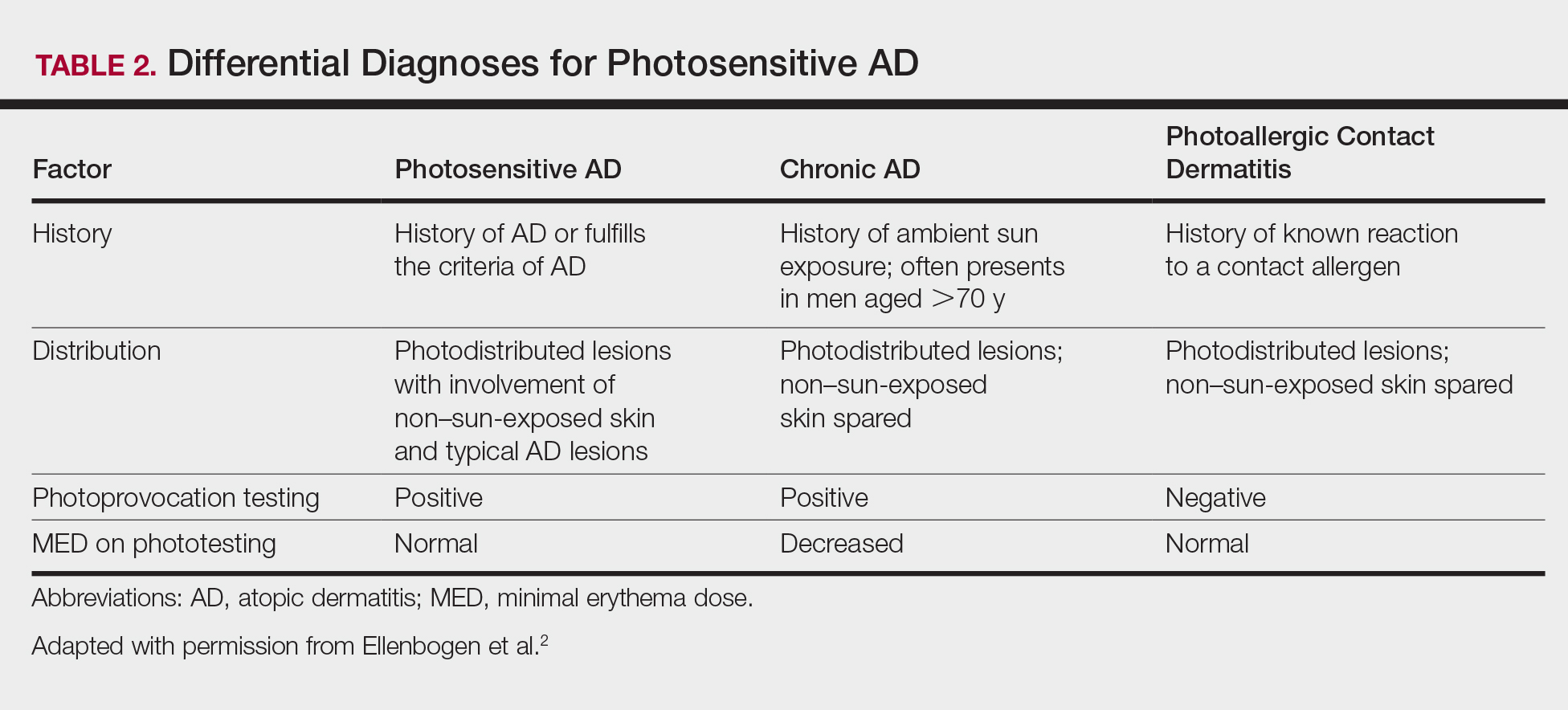
Treatment
The treatment of photosensitive AD is based on the severity of the photosensitivity. Treatment for mild disease is limited to sun protection in addition to topical corticosteroids or topical calcineurin inhibitors. For moderate disease and unsatisfactory relief with proper sun protection, UVR hardening is recommended. If severe disease is present, immunosuppression with medications such as corticosteroids, cyclosporine, and mycophenolate mofetil is suggested to prevent flaring of disease during UVR hardening.2,5,8,14
Conclusion
Photosensitive AD is a rare entity characterized by a photodistributed rash and involvement of non–sun-exposed skin. Patients will either have a history of AD or fulfill the criteria of AD. They have positive photoprovocation testing and generally have a normal MED. They may have positive photopatch testing with coexisting photoallergies. Histopathology is nonspecific but shows spongiotic dermatitis with perivascular lymphohistiocytic infiltrate. Diagnosis is essential, as this disease can be life altering and affect quality of life. Effective treatment options are available, and the therapeutic ladder is based on severity of disease.2,5
- Bieber T, Bussmann C. Atopic dermatitis. In: Bolognia JL, Jorizzo J, Rapini R, eds. Dermatology. 3rd ed. New York, NY: Elsevier; 2012:203-230.
- Ellenbogen E, Wesselmann U, Hofmann SC, et al. Photosensitive atopic dermatitis—a neglected subset: clinical, laboratory, histological and photobiological workup. J Eur Acad Dermatol Venereol. 2016;30:270-275.
- Yule S, Dawe RS, Cameron H, et al. Does narrow-band ultraviolet B phototherapy work in atopic dermatitis through a local or a systemic effect? Photodermatol Photoimmunol Photomed. 2005;21:333-335.
- Sidbury R, Davis DM, Cohen DE, et al. Guidelines of care for the management of atopic dermatitis. section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71:327-349.
- ten Berge O, van Weelden H, Bruijnzeel-Koomen CA, et al. Throwing a light on photosensitivity in atopic dermatitis: a retrospective study. Am J Clin Dermatol. 2009;10:119-123.
- O’Gorman SM, Murphy GM. Photoaggravated disorders. Dermatol Clin. 2014;32:385-398.
- Crouch RB, Foley PA, Baker CS. Analysis of patients with suspected photosensitivity referred for investigation to an Australian photodermatology clinic. J Am Acad Dermatol. 2003;48:714-720.
- Russell SC, Dawes RS, Collins P, et al. The photosensitivity dermatitis and actinic reticuloid syndrome (chronic actinic dermatitis) occurring in seven young atopic dermatitis patients. Br J Dermatol. 1998;138:496-501.
- Tajima T, Ibe M, Matsushita T, et al. A variety of skin responses to ultraviolet irradiation in patients with atopic dermatitis. J Dermatol Sci. 1998;17:101-107.
- Faurschou A, Wulf HC. European Dermatology Guideline for the photodermatoses: phototesting. European Dermatology Forum website. http://www.euroderm.org/edf/index.php/edf-guidelines/category/3-guidelines-on-photodermatoses. Accessed August 21, 2017.
- Keong CH, Kurumaji Y, Miyamoto C, et al. Photosensitivity in atopic dermatitis: demonstration of abnormal response to UVB. J Dermatol. 1992;19:342-347.
- Lee PA, Freeman S. Photosensitivity: the 9-year experience at a Sydney contact dermatitis clinic. Australas J Dermatol. 2002;43:289-292.
- Goncalo M, Ferguson J, Bonevalle A, et al. Photopatch testing: recommendations for a European photopatch test baseline series. Contact Dermatitis. 2013;68:239-243.
- Amon U, Mangalo S, Roth A. Clinical relevance of increased UV-sensitivity in patients with atopic dermatitis. J Allergy Clin Immunol. 2011;127:AB39.
Atopic dermatitis (AD) is the most common inflammatory skin condition, affecting approximately 15% to 20% of the global population.1,2 Atopic dermatitis is characterized by a chronic relapsing dermatitis with pruritus, often beginning in infancy or childhood. Atopic dermatitis is caused by a defect in epidermal barrier function, which results in increased transepidermal water loss.1 The criteria for AD include a pruritic skin condition plus 3 or more of the following: history of involvement of the skin creases, history of asthma or hay fever, history of AD in a first-degree relative (in children), 1-year history of generally dry skin, visible flexural eczema, and an age of onset of less than 2 years. Adults with AD frequently present with hand or facial dermatitis.1
UV light therapies including narrowband UVB (NB-UVB), UVA1, and psoralen plus UVA (PUVA) have all been used as effective treatments of AD.3,4 UV light is beneficial for AD patients due to its immunomodulatory effects, thickening of the stratum corneum, and the reduction of Staphylococcus aureus in the skin.2 Most patients with AD improve with light therapy; however, it is estimated that 1% to 3% of patients with AD will experience a paradoxical worsening of their AD after exposure to UV light.2,5 This condition is referred to as photosensitive AD and is characterized by a photodistributed rash in patients who fulfill the criteria of AD. Photosensitive AD has a female predominance and generally affects patients with late-onset disease with development of AD after puberty.2,5 The pathogenesis for the development of photosensitivity in patients with AD who previously tolerated exposure to sunlight is unknown.5 We describe a case of photosensitive AD exacerbated by UVB exposure.
Case Report
On physical examination the patient had thin, well-demarcated, erythematous papules and plaques with scaling, primarily on sun-exposed skin on the forehead (Figure 1A), cheeks (Figure 1B), eyelids, upper lip, neck (Figures 1B and 1C), upper chest (Figure 1C), and dorsal aspect of the hands, with excoriated pink papules on the forearms, shoulders, and back. A punch biopsy of the right neck showed spongiotic dermatitis with a perivascular lymphohistiocytic infiltrate (Figure 2). Further workup was pursued including complete blood cell count, comprehensive metabolic profile, liver function panel, Sjögren syndrome antigen A/Sjögren syndrome antigen B test, antinuclear antibody test, human immunodeficiency virus 1/2 antigen/antibody test, hepatitis panel, and mycobacterium tuberculosis test, which were all within reference range. Photodermatosis was suspected and she underwent phototesting including UVA, NB-UVB, and visible light. Phototesting confirmed she had a UVB photosensitivity with a markedly decreased minimal erythema dose (MED) to NB-UVB. The MED to NB-UVB was positive at 24 hours to all tested sites, the lowest of which was 0.135 J/cm2. Eczematous changes began to develop at day 6 at doses of 0.945 and 1.080 J/cm2. The patient also underwent visible light testing, which was negative. The patient was patch tested for multiple standardized agents as well as personal products, all of which were negative. Subsequent photopatch testing revealed a slightly positive reaction to benzophenone 4, a common ingredient in sunscreens.
The patient was then started on mycophenolate mofetil and prednisone. Repeat MED testing to NB-UVB was performed. Her repeat MED to NB-UVB was determined to be 0.405 J/cm2, and hardening commenced at 3 times per week at 70% of the MED (0.2835 J/cm2). She began to flare and develop an eczematous reaction, thus the dose was decreased to 50% of the MED (0.2025 J/cm2), which she tolerated.
Comment
Classification and Clinical Presentation
The literature on photosensitive AD is scant, and this disease entity is rare. Alternative names include photoaggravated AD, photosensitive eczema, and light-exacerbated eczema.5 Two main studies have been conducted in recent years that were intended to characterize photosensitive AD. ten Berge et al5 conducted a retrospective study of 145 patients with AD that were phototested in 2009. They found that 3% of their total AD patient population had photosensitive AD.5 In 2016, Ellenbogen et al2 performed a similar single-center retrospective analysis of 17 patients with long-standing AD who suddenly developed photosensitivity.
Patients with photosensitive AD typically present with lesions on sun-exposed skin with coexisting eczematous lesions in sites with a predilection for AD.2 In the study conducted by ten Berge et al,5 2 main reaction patterns were observed: erythematous papules with pruritus and an eczematous reaction.
Histopathology
The histopathologic findings of photosensitive AD are nonspecific but are characterized by spongiotic dermatitis with a perivascular lymphohistiocytic infiltrate.2
Diagnosis With Phototesting
Phototesting of patients with AD should be considered if there is a suspicion for photosensitivity based on persistent disease despite use of photoprotection and local treatment.5-7 Patients may not notice a correlation of skin exacerbations with UV exposure, especially if they are only sensitive to UVA, as it is still present on cloudy days and can penetrate glass windows.8 Phototesting evaluates the degree of sensitivity to UV light and the specific wavelength eliciting the cutaneous response. Phototesting consists of determining the MED to UVA and UVB, the minimal phototoxic dose for PUVA, and visible light exposure. Further evaluation may include photoprovocation testing or photopatch testing, as these patients can have coexisting photocontact allergies.
The MED is defined as the minimal dose of UV light needed to induce perceptible erythema in exposed skin.5 It is dependent on the light source and patient’s skin type, and individual units may vary. To determine the MED to UVA or UVB, 2×2-cm skin fields are irradiated with increasing cumulative UVA/UVB. The dose varies by skin type and it is then read at 24 hours. The majority of patients with photosensitive AD are reported to have a normal MED; however, some studies have reported the MED to be decreased.5,7-9 ten Berge et al5 found 7% of their study participants exhibited a lower MED, as seen in our patient.
The minimal phototoxic dose for PUVA is defined as the least exposure dose of UVA 1 hour after ingestion of 0.4 mg/kg of methoxsalen that produces pink erythema with 4 distinct borders at 48, 72, or 96 hours after ingestion.10 Visible light exposure is tested using a slide projector as the light source to an approximately 10×5-cm area of skin for 45 minutes. Any immediate or delayed reaction is abnormal and considered positive.10
Photoprovocation testing has been performed in several studies.2,5 It consists of exposing an 8-cm area of skin to 80 J/cm2 UVA and 10 mJ/cm2 UVB, which is read at 24, 48, or 72 hours. A papular or eczematous reaction is considered positive.2,11
The results of phototesting have varied between studies. ten Berge et al5 phototested 107 patients with AD and photosensitivity and 17% were found to be solely sensitive to UVA whereas 67% were found to be sensitive to UVA and UVB. In contrast, Ellenbogen et al2 only tested 17 patients with AD and photosensitivity and they found that 56% (9/16) were sensitive to UVA alone while only 44% (7/16) were sensitive to UVA and UVB.
Photopatch testing can help to rule out photosensitivity due to a substance in the presence of UV light. In studies of patients with photosensitive AD (N=125), photocontact reactions occurred in 23% and were predominantly associated with sunscreens, skin care products, and fragrances.5,12 Photopatch testing is done by placing duplicate sets of patches on nonlesional skin using the Finn Chamber technique. A published list of allergens, which were agreed upon by the European Society of Contact Dermatitis and the European Society for Photodermatology in 2000 are seen in Table 1.13 The list contains mainly UV filters and drugs. The patients’ own products also should be tested in addition to the published list of allergens, but a maximum of 30 patches should be placed at one time. The patches are removed at either 24 or 48 hours; some researchers have found greater sensitivity with the 48-hour time period, while others have not found a significant difference.10 One set of skin fields then is covered with an impermeable occlusive dressing as a control while the other is irradiated with 5 J/cm2 of a broad-spectrum UVA light source. UVA fluorescent lamps are the light source of choice because of their widespread availability, reproducible broad spectrum, and beam uniformity.10 In the study conducted by ten Berge et al,5 photopatch testing was performed on 125 patients, and 29 patients were found to be positive to one or more substances. Ellenbogen et al2 photopatch tested 5 patients with photosensitive AD and a clinical suspicion of photoallergy; however, all 5 were negative. Our patient underwent traditional patch testing due to clinical suspicion of a coexisting contact allergy, which was negative.
Differential Diagnosis
The differential diagnosis for photosensitive AD includes PMLE with coexisting AD, chronic AD, and photoallergic contact dermatitis. Photosensitive AD worsens with increasing exposure to uncontrolled sunlight, in contrast to patients with PMLE who experience UV radiation (UVR) hardening with increasing UV exposure during the summer months, resulting in improvement of skin lesions. Patients with chronic AD generally report a history of chronic ambient sun exposure and exhibit well-demarcated eczematous lesions in a photodistributed pattern with sparing of sun-protected skin.2 In contrast, photosensitive AD involves both sun-exposed and covered areas of the body. Chronic AD will have a positive photoprovocation test with a decreased MED (Table 2). Photoallergic contact dermatitis also will have photodistributed eczematous lesions with relative sparing of non–sun-exposed skin; however, these patients generally have negative photoprovocation testing with a normal MED.2 These patients may or may not have a history of reaction to a known allergen, but they likely will have a positive photopatch test.
Treatment
The treatment of photosensitive AD is based on the severity of the photosensitivity. Treatment for mild disease is limited to sun protection in addition to topical corticosteroids or topical calcineurin inhibitors. For moderate disease and unsatisfactory relief with proper sun protection, UVR hardening is recommended. If severe disease is present, immunosuppression with medications such as corticosteroids, cyclosporine, and mycophenolate mofetil is suggested to prevent flaring of disease during UVR hardening.2,5,8,14
Conclusion
Photosensitive AD is a rare entity characterized by a photodistributed rash and involvement of non–sun-exposed skin. Patients will either have a history of AD or fulfill the criteria of AD. They have positive photoprovocation testing and generally have a normal MED. They may have positive photopatch testing with coexisting photoallergies. Histopathology is nonspecific but shows spongiotic dermatitis with perivascular lymphohistiocytic infiltrate. Diagnosis is essential, as this disease can be life altering and affect quality of life. Effective treatment options are available, and the therapeutic ladder is based on severity of disease.2,5
Atopic dermatitis (AD) is the most common inflammatory skin condition, affecting approximately 15% to 20% of the global population.1,2 Atopic dermatitis is characterized by a chronic relapsing dermatitis with pruritus, often beginning in infancy or childhood. Atopic dermatitis is caused by a defect in epidermal barrier function, which results in increased transepidermal water loss.1 The criteria for AD include a pruritic skin condition plus 3 or more of the following: history of involvement of the skin creases, history of asthma or hay fever, history of AD in a first-degree relative (in children), 1-year history of generally dry skin, visible flexural eczema, and an age of onset of less than 2 years. Adults with AD frequently present with hand or facial dermatitis.1
UV light therapies including narrowband UVB (NB-UVB), UVA1, and psoralen plus UVA (PUVA) have all been used as effective treatments of AD.3,4 UV light is beneficial for AD patients due to its immunomodulatory effects, thickening of the stratum corneum, and the reduction of Staphylococcus aureus in the skin.2 Most patients with AD improve with light therapy; however, it is estimated that 1% to 3% of patients with AD will experience a paradoxical worsening of their AD after exposure to UV light.2,5 This condition is referred to as photosensitive AD and is characterized by a photodistributed rash in patients who fulfill the criteria of AD. Photosensitive AD has a female predominance and generally affects patients with late-onset disease with development of AD after puberty.2,5 The pathogenesis for the development of photosensitivity in patients with AD who previously tolerated exposure to sunlight is unknown.5 We describe a case of photosensitive AD exacerbated by UVB exposure.
Case Report
On physical examination the patient had thin, well-demarcated, erythematous papules and plaques with scaling, primarily on sun-exposed skin on the forehead (Figure 1A), cheeks (Figure 1B), eyelids, upper lip, neck (Figures 1B and 1C), upper chest (Figure 1C), and dorsal aspect of the hands, with excoriated pink papules on the forearms, shoulders, and back. A punch biopsy of the right neck showed spongiotic dermatitis with a perivascular lymphohistiocytic infiltrate (Figure 2). Further workup was pursued including complete blood cell count, comprehensive metabolic profile, liver function panel, Sjögren syndrome antigen A/Sjögren syndrome antigen B test, antinuclear antibody test, human immunodeficiency virus 1/2 antigen/antibody test, hepatitis panel, and mycobacterium tuberculosis test, which were all within reference range. Photodermatosis was suspected and she underwent phototesting including UVA, NB-UVB, and visible light. Phototesting confirmed she had a UVB photosensitivity with a markedly decreased minimal erythema dose (MED) to NB-UVB. The MED to NB-UVB was positive at 24 hours to all tested sites, the lowest of which was 0.135 J/cm2. Eczematous changes began to develop at day 6 at doses of 0.945 and 1.080 J/cm2. The patient also underwent visible light testing, which was negative. The patient was patch tested for multiple standardized agents as well as personal products, all of which were negative. Subsequent photopatch testing revealed a slightly positive reaction to benzophenone 4, a common ingredient in sunscreens.
The patient was then started on mycophenolate mofetil and prednisone. Repeat MED testing to NB-UVB was performed. Her repeat MED to NB-UVB was determined to be 0.405 J/cm2, and hardening commenced at 3 times per week at 70% of the MED (0.2835 J/cm2). She began to flare and develop an eczematous reaction, thus the dose was decreased to 50% of the MED (0.2025 J/cm2), which she tolerated.
Comment
Classification and Clinical Presentation
The literature on photosensitive AD is scant, and this disease entity is rare. Alternative names include photoaggravated AD, photosensitive eczema, and light-exacerbated eczema.5 Two main studies have been conducted in recent years that were intended to characterize photosensitive AD. ten Berge et al5 conducted a retrospective study of 145 patients with AD that were phototested in 2009. They found that 3% of their total AD patient population had photosensitive AD.5 In 2016, Ellenbogen et al2 performed a similar single-center retrospective analysis of 17 patients with long-standing AD who suddenly developed photosensitivity.
Patients with photosensitive AD typically present with lesions on sun-exposed skin with coexisting eczematous lesions in sites with a predilection for AD.2 In the study conducted by ten Berge et al,5 2 main reaction patterns were observed: erythematous papules with pruritus and an eczematous reaction.
Histopathology
The histopathologic findings of photosensitive AD are nonspecific but are characterized by spongiotic dermatitis with a perivascular lymphohistiocytic infiltrate.2
Diagnosis With Phototesting
Phototesting of patients with AD should be considered if there is a suspicion for photosensitivity based on persistent disease despite use of photoprotection and local treatment.5-7 Patients may not notice a correlation of skin exacerbations with UV exposure, especially if they are only sensitive to UVA, as it is still present on cloudy days and can penetrate glass windows.8 Phototesting evaluates the degree of sensitivity to UV light and the specific wavelength eliciting the cutaneous response. Phototesting consists of determining the MED to UVA and UVB, the minimal phototoxic dose for PUVA, and visible light exposure. Further evaluation may include photoprovocation testing or photopatch testing, as these patients can have coexisting photocontact allergies.
The MED is defined as the minimal dose of UV light needed to induce perceptible erythema in exposed skin.5 It is dependent on the light source and patient’s skin type, and individual units may vary. To determine the MED to UVA or UVB, 2×2-cm skin fields are irradiated with increasing cumulative UVA/UVB. The dose varies by skin type and it is then read at 24 hours. The majority of patients with photosensitive AD are reported to have a normal MED; however, some studies have reported the MED to be decreased.5,7-9 ten Berge et al5 found 7% of their study participants exhibited a lower MED, as seen in our patient.
The minimal phototoxic dose for PUVA is defined as the least exposure dose of UVA 1 hour after ingestion of 0.4 mg/kg of methoxsalen that produces pink erythema with 4 distinct borders at 48, 72, or 96 hours after ingestion.10 Visible light exposure is tested using a slide projector as the light source to an approximately 10×5-cm area of skin for 45 minutes. Any immediate or delayed reaction is abnormal and considered positive.10
Photoprovocation testing has been performed in several studies.2,5 It consists of exposing an 8-cm area of skin to 80 J/cm2 UVA and 10 mJ/cm2 UVB, which is read at 24, 48, or 72 hours. A papular or eczematous reaction is considered positive.2,11
The results of phototesting have varied between studies. ten Berge et al5 phototested 107 patients with AD and photosensitivity and 17% were found to be solely sensitive to UVA whereas 67% were found to be sensitive to UVA and UVB. In contrast, Ellenbogen et al2 only tested 17 patients with AD and photosensitivity and they found that 56% (9/16) were sensitive to UVA alone while only 44% (7/16) were sensitive to UVA and UVB.
Photopatch testing can help to rule out photosensitivity due to a substance in the presence of UV light. In studies of patients with photosensitive AD (N=125), photocontact reactions occurred in 23% and were predominantly associated with sunscreens, skin care products, and fragrances.5,12 Photopatch testing is done by placing duplicate sets of patches on nonlesional skin using the Finn Chamber technique. A published list of allergens, which were agreed upon by the European Society of Contact Dermatitis and the European Society for Photodermatology in 2000 are seen in Table 1.13 The list contains mainly UV filters and drugs. The patients’ own products also should be tested in addition to the published list of allergens, but a maximum of 30 patches should be placed at one time. The patches are removed at either 24 or 48 hours; some researchers have found greater sensitivity with the 48-hour time period, while others have not found a significant difference.10 One set of skin fields then is covered with an impermeable occlusive dressing as a control while the other is irradiated with 5 J/cm2 of a broad-spectrum UVA light source. UVA fluorescent lamps are the light source of choice because of their widespread availability, reproducible broad spectrum, and beam uniformity.10 In the study conducted by ten Berge et al,5 photopatch testing was performed on 125 patients, and 29 patients were found to be positive to one or more substances. Ellenbogen et al2 photopatch tested 5 patients with photosensitive AD and a clinical suspicion of photoallergy; however, all 5 were negative. Our patient underwent traditional patch testing due to clinical suspicion of a coexisting contact allergy, which was negative.
Differential Diagnosis
The differential diagnosis for photosensitive AD includes PMLE with coexisting AD, chronic AD, and photoallergic contact dermatitis. Photosensitive AD worsens with increasing exposure to uncontrolled sunlight, in contrast to patients with PMLE who experience UV radiation (UVR) hardening with increasing UV exposure during the summer months, resulting in improvement of skin lesions. Patients with chronic AD generally report a history of chronic ambient sun exposure and exhibit well-demarcated eczematous lesions in a photodistributed pattern with sparing of sun-protected skin.2 In contrast, photosensitive AD involves both sun-exposed and covered areas of the body. Chronic AD will have a positive photoprovocation test with a decreased MED (Table 2). Photoallergic contact dermatitis also will have photodistributed eczematous lesions with relative sparing of non–sun-exposed skin; however, these patients generally have negative photoprovocation testing with a normal MED.2 These patients may or may not have a history of reaction to a known allergen, but they likely will have a positive photopatch test.
Treatment
The treatment of photosensitive AD is based on the severity of the photosensitivity. Treatment for mild disease is limited to sun protection in addition to topical corticosteroids or topical calcineurin inhibitors. For moderate disease and unsatisfactory relief with proper sun protection, UVR hardening is recommended. If severe disease is present, immunosuppression with medications such as corticosteroids, cyclosporine, and mycophenolate mofetil is suggested to prevent flaring of disease during UVR hardening.2,5,8,14
Conclusion
Photosensitive AD is a rare entity characterized by a photodistributed rash and involvement of non–sun-exposed skin. Patients will either have a history of AD or fulfill the criteria of AD. They have positive photoprovocation testing and generally have a normal MED. They may have positive photopatch testing with coexisting photoallergies. Histopathology is nonspecific but shows spongiotic dermatitis with perivascular lymphohistiocytic infiltrate. Diagnosis is essential, as this disease can be life altering and affect quality of life. Effective treatment options are available, and the therapeutic ladder is based on severity of disease.2,5
- Bieber T, Bussmann C. Atopic dermatitis. In: Bolognia JL, Jorizzo J, Rapini R, eds. Dermatology. 3rd ed. New York, NY: Elsevier; 2012:203-230.
- Ellenbogen E, Wesselmann U, Hofmann SC, et al. Photosensitive atopic dermatitis—a neglected subset: clinical, laboratory, histological and photobiological workup. J Eur Acad Dermatol Venereol. 2016;30:270-275.
- Yule S, Dawe RS, Cameron H, et al. Does narrow-band ultraviolet B phototherapy work in atopic dermatitis through a local or a systemic effect? Photodermatol Photoimmunol Photomed. 2005;21:333-335.
- Sidbury R, Davis DM, Cohen DE, et al. Guidelines of care for the management of atopic dermatitis. section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71:327-349.
- ten Berge O, van Weelden H, Bruijnzeel-Koomen CA, et al. Throwing a light on photosensitivity in atopic dermatitis: a retrospective study. Am J Clin Dermatol. 2009;10:119-123.
- O’Gorman SM, Murphy GM. Photoaggravated disorders. Dermatol Clin. 2014;32:385-398.
- Crouch RB, Foley PA, Baker CS. Analysis of patients with suspected photosensitivity referred for investigation to an Australian photodermatology clinic. J Am Acad Dermatol. 2003;48:714-720.
- Russell SC, Dawes RS, Collins P, et al. The photosensitivity dermatitis and actinic reticuloid syndrome (chronic actinic dermatitis) occurring in seven young atopic dermatitis patients. Br J Dermatol. 1998;138:496-501.
- Tajima T, Ibe M, Matsushita T, et al. A variety of skin responses to ultraviolet irradiation in patients with atopic dermatitis. J Dermatol Sci. 1998;17:101-107.
- Faurschou A, Wulf HC. European Dermatology Guideline for the photodermatoses: phototesting. European Dermatology Forum website. http://www.euroderm.org/edf/index.php/edf-guidelines/category/3-guidelines-on-photodermatoses. Accessed August 21, 2017.
- Keong CH, Kurumaji Y, Miyamoto C, et al. Photosensitivity in atopic dermatitis: demonstration of abnormal response to UVB. J Dermatol. 1992;19:342-347.
- Lee PA, Freeman S. Photosensitivity: the 9-year experience at a Sydney contact dermatitis clinic. Australas J Dermatol. 2002;43:289-292.
- Goncalo M, Ferguson J, Bonevalle A, et al. Photopatch testing: recommendations for a European photopatch test baseline series. Contact Dermatitis. 2013;68:239-243.
- Amon U, Mangalo S, Roth A. Clinical relevance of increased UV-sensitivity in patients with atopic dermatitis. J Allergy Clin Immunol. 2011;127:AB39.
- Bieber T, Bussmann C. Atopic dermatitis. In: Bolognia JL, Jorizzo J, Rapini R, eds. Dermatology. 3rd ed. New York, NY: Elsevier; 2012:203-230.
- Ellenbogen E, Wesselmann U, Hofmann SC, et al. Photosensitive atopic dermatitis—a neglected subset: clinical, laboratory, histological and photobiological workup. J Eur Acad Dermatol Venereol. 2016;30:270-275.
- Yule S, Dawe RS, Cameron H, et al. Does narrow-band ultraviolet B phototherapy work in atopic dermatitis through a local or a systemic effect? Photodermatol Photoimmunol Photomed. 2005;21:333-335.
- Sidbury R, Davis DM, Cohen DE, et al. Guidelines of care for the management of atopic dermatitis. section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71:327-349.
- ten Berge O, van Weelden H, Bruijnzeel-Koomen CA, et al. Throwing a light on photosensitivity in atopic dermatitis: a retrospective study. Am J Clin Dermatol. 2009;10:119-123.
- O’Gorman SM, Murphy GM. Photoaggravated disorders. Dermatol Clin. 2014;32:385-398.
- Crouch RB, Foley PA, Baker CS. Analysis of patients with suspected photosensitivity referred for investigation to an Australian photodermatology clinic. J Am Acad Dermatol. 2003;48:714-720.
- Russell SC, Dawes RS, Collins P, et al. The photosensitivity dermatitis and actinic reticuloid syndrome (chronic actinic dermatitis) occurring in seven young atopic dermatitis patients. Br J Dermatol. 1998;138:496-501.
- Tajima T, Ibe M, Matsushita T, et al. A variety of skin responses to ultraviolet irradiation in patients with atopic dermatitis. J Dermatol Sci. 1998;17:101-107.
- Faurschou A, Wulf HC. European Dermatology Guideline for the photodermatoses: phototesting. European Dermatology Forum website. http://www.euroderm.org/edf/index.php/edf-guidelines/category/3-guidelines-on-photodermatoses. Accessed August 21, 2017.
- Keong CH, Kurumaji Y, Miyamoto C, et al. Photosensitivity in atopic dermatitis: demonstration of abnormal response to UVB. J Dermatol. 1992;19:342-347.
- Lee PA, Freeman S. Photosensitivity: the 9-year experience at a Sydney contact dermatitis clinic. Australas J Dermatol. 2002;43:289-292.
- Goncalo M, Ferguson J, Bonevalle A, et al. Photopatch testing: recommendations for a European photopatch test baseline series. Contact Dermatitis. 2013;68:239-243.
- Amon U, Mangalo S, Roth A. Clinical relevance of increased UV-sensitivity in patients with atopic dermatitis. J Allergy Clin Immunol. 2011;127:AB39.
Practice Points
- Photosensitive atopic dermatitis (AD) is rare but should be considered in patients with uncontrolled AD with a rash on sun-exposed skin.
- A thorough history and physical examination of these patients can provide the necessary clues for further workup.
- Phototesting should be performed to confirm the diagnosis and evaluate the degree of sensitivity to UV light and the specific wavelength eliciting the cutaneous response.
- Photoprovocation and photopatch testing also can be useful to confirm the diagnosis.
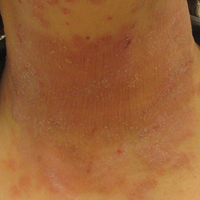
Midyear formulary changes wreak havoc on diabetes care
Instability of formulary coverage for drug and insulin can make it tough to treat patients with diabetes, and the problem is getting worse.
Just one example: CVS Caremark removed 22 insulin products and drugs to treat diabetes in its July 2017 formulary update.
“This is a real problem,” Philip Levy, MD, an endocrinologist at Banner University Medical Group in Phoenix, said in an interview. “We have people that are doing well on one medication and then all of a sudden, their insurance carrier says you have to use a different medication and they don’t tolerate it. It’s a real problem.”
Claresa Levetan, MD, with Chestnut Hill Endocrinology, Diabetes & Metabolic Associates of Philadelphia, agreed. “It’s a very big problem. ... In my practice, it’s a nightmare because I cannot choose to put them on the insulin or the medications that would be best for them.”

The American Diabetes Association also is concerned. The association is “deeply concerned with recent trends in prescription drug formulary designs that result in frequent changes in drug coverage for individuals with diabetes,” according to a statement.
The situation is made more challenging by the sheer volume of money spent on insulins and diabetes drugs.
Diabetes prescription ranked first among therapeutic classes in terms of total spend (13%), ahead of autoimmune (11.9%) and multiple sclerosis and HIV treatments (both tied at 4.7% each) for its commercial clients, according to pharmacy benefit manager (PBM) Prime Therapeutics. Three of the top 10 drugs by drug spend are diabetes medication (No. 4: Lantus; No. 7: NovoLog; and No. 8: Victoza). For its Medicare Part D clients, diabetes drugs also topped the list in terms of total spend per therapeutic class (14.2%), ahead of oral cancer therapies (11.3%) and respiratory (6.5%). Three of the top 10 drugs by drug spend among Medicare Part D clients are diabetes drugs (No. 4: Lantus; No. 6: Januvia; and No. 10: Humalog KwikPen).
Express Scripts reports similar trends. Diabetes prescriptions were the second highest therapeutic class in terms of per member per year spending in 2016 at $108.80, behind inflammatory conditions ($118.21) and ahead of oncology ($60.70).
Utilization of diabetes drugs increased 5.3%, according to the PBM, while unit costs for these treatments rose 14.1%. Express Scripts identified the top five most costly medications as Lantus, Humalog KwikPen, metformin, Januvia, and Invokana.
“Formulary changes made during the year are particularly troubling because most individuals do not have the option of changing to a different insurance plan that would cover his/her prescription medication,” the ADA said in its statement. “Therefore, the association is strongly opposed to formulary changes such as removing medications from formularies or moving medications to a higher tier during the plan year.”
Beyond cost, Dr. Levetan suggested that another issue is the lack of clinician input at the PBM level.
“I think the problem is not only just companies switching to a cheaper bid mid-year between [manufacturers], but there is really no input from physicians, especially the endocrinologists and the specialists,” she said. “This is potentially life threatening.”
Finding alternatives can also have adverse effects on outcomes and can also be time consuming.
“If somebody is doing well, there is no real reason to change that or to stop it,” Dr. Levy said. “There are some medications that are pretty interchangeable, but others, they all have slightly different side effects and we try to use what we think is the best medication for that patient based on the patient’s characteristics.”
ADA is recommending that PBMs, insurance plans, and employers “provide an expedited and standard process for gaining access to medications not included in the plan’s formulary. Responses to exception requests must be timely to ensure no delay in obtaining a needed medication, thereby preventing a gap in treatment. In addition, the association recommends PBMs, plans, and employers temporarily cover nonformulary drugs as if they are on formulary during the first 30 days after a formulary list is changed.”
Instability of formulary coverage for drug and insulin can make it tough to treat patients with diabetes, and the problem is getting worse.
Just one example: CVS Caremark removed 22 insulin products and drugs to treat diabetes in its July 2017 formulary update.
“This is a real problem,” Philip Levy, MD, an endocrinologist at Banner University Medical Group in Phoenix, said in an interview. “We have people that are doing well on one medication and then all of a sudden, their insurance carrier says you have to use a different medication and they don’t tolerate it. It’s a real problem.”
Claresa Levetan, MD, with Chestnut Hill Endocrinology, Diabetes & Metabolic Associates of Philadelphia, agreed. “It’s a very big problem. ... In my practice, it’s a nightmare because I cannot choose to put them on the insulin or the medications that would be best for them.”
The American Diabetes Association also is concerned. The association is “deeply concerned with recent trends in prescription drug formulary designs that result in frequent changes in drug coverage for individuals with diabetes,” according to a statement.
The situation is made more challenging by the sheer volume of money spent on insulins and diabetes drugs.
Diabetes prescription ranked first among therapeutic classes in terms of total spend (13%), ahead of autoimmune (11.9%) and multiple sclerosis and HIV treatments (both tied at 4.7% each) for its commercial clients, according to pharmacy benefit manager (PBM) Prime Therapeutics. Three of the top 10 drugs by drug spend are diabetes medication (No. 4: Lantus; No. 7: NovoLog; and No. 8: Victoza). For its Medicare Part D clients, diabetes drugs also topped the list in terms of total spend per therapeutic class (14.2%), ahead of oral cancer therapies (11.3%) and respiratory (6.5%). Three of the top 10 drugs by drug spend among Medicare Part D clients are diabetes drugs (No. 4: Lantus; No. 6: Januvia; and No. 10: Humalog KwikPen).
Express Scripts reports similar trends. Diabetes prescriptions were the second highest therapeutic class in terms of per member per year spending in 2016 at $108.80, behind inflammatory conditions ($118.21) and ahead of oncology ($60.70).
Utilization of diabetes drugs increased 5.3%, according to the PBM, while unit costs for these treatments rose 14.1%. Express Scripts identified the top five most costly medications as Lantus, Humalog KwikPen, metformin, Januvia, and Invokana.
“Formulary changes made during the year are particularly troubling because most individuals do not have the option of changing to a different insurance plan that would cover his/her prescription medication,” the ADA said in its statement. “Therefore, the association is strongly opposed to formulary changes such as removing medications from formularies or moving medications to a higher tier during the plan year.”
Beyond cost, Dr. Levetan suggested that another issue is the lack of clinician input at the PBM level.
“I think the problem is not only just companies switching to a cheaper bid mid-year between [manufacturers], but there is really no input from physicians, especially the endocrinologists and the specialists,” she said. “This is potentially life threatening.”
Finding alternatives can also have adverse effects on outcomes and can also be time consuming.
“If somebody is doing well, there is no real reason to change that or to stop it,” Dr. Levy said. “There are some medications that are pretty interchangeable, but others, they all have slightly different side effects and we try to use what we think is the best medication for that patient based on the patient’s characteristics.”
ADA is recommending that PBMs, insurance plans, and employers “provide an expedited and standard process for gaining access to medications not included in the plan’s formulary. Responses to exception requests must be timely to ensure no delay in obtaining a needed medication, thereby preventing a gap in treatment. In addition, the association recommends PBMs, plans, and employers temporarily cover nonformulary drugs as if they are on formulary during the first 30 days after a formulary list is changed.”
Instability of formulary coverage for drug and insulin can make it tough to treat patients with diabetes, and the problem is getting worse.
Just one example: CVS Caremark removed 22 insulin products and drugs to treat diabetes in its July 2017 formulary update.
“This is a real problem,” Philip Levy, MD, an endocrinologist at Banner University Medical Group in Phoenix, said in an interview. “We have people that are doing well on one medication and then all of a sudden, their insurance carrier says you have to use a different medication and they don’t tolerate it. It’s a real problem.”
Claresa Levetan, MD, with Chestnut Hill Endocrinology, Diabetes & Metabolic Associates of Philadelphia, agreed. “It’s a very big problem. ... In my practice, it’s a nightmare because I cannot choose to put them on the insulin or the medications that would be best for them.”
The American Diabetes Association also is concerned. The association is “deeply concerned with recent trends in prescription drug formulary designs that result in frequent changes in drug coverage for individuals with diabetes,” according to a statement.
The situation is made more challenging by the sheer volume of money spent on insulins and diabetes drugs.
Diabetes prescription ranked first among therapeutic classes in terms of total spend (13%), ahead of autoimmune (11.9%) and multiple sclerosis and HIV treatments (both tied at 4.7% each) for its commercial clients, according to pharmacy benefit manager (PBM) Prime Therapeutics. Three of the top 10 drugs by drug spend are diabetes medication (No. 4: Lantus; No. 7: NovoLog; and No. 8: Victoza). For its Medicare Part D clients, diabetes drugs also topped the list in terms of total spend per therapeutic class (14.2%), ahead of oral cancer therapies (11.3%) and respiratory (6.5%). Three of the top 10 drugs by drug spend among Medicare Part D clients are diabetes drugs (No. 4: Lantus; No. 6: Januvia; and No. 10: Humalog KwikPen).
Express Scripts reports similar trends. Diabetes prescriptions were the second highest therapeutic class in terms of per member per year spending in 2016 at $108.80, behind inflammatory conditions ($118.21) and ahead of oncology ($60.70).
Utilization of diabetes drugs increased 5.3%, according to the PBM, while unit costs for these treatments rose 14.1%. Express Scripts identified the top five most costly medications as Lantus, Humalog KwikPen, metformin, Januvia, and Invokana.
“Formulary changes made during the year are particularly troubling because most individuals do not have the option of changing to a different insurance plan that would cover his/her prescription medication,” the ADA said in its statement. “Therefore, the association is strongly opposed to formulary changes such as removing medications from formularies or moving medications to a higher tier during the plan year.”
Beyond cost, Dr. Levetan suggested that another issue is the lack of clinician input at the PBM level.
“I think the problem is not only just companies switching to a cheaper bid mid-year between [manufacturers], but there is really no input from physicians, especially the endocrinologists and the specialists,” she said. “This is potentially life threatening.”
Finding alternatives can also have adverse effects on outcomes and can also be time consuming.
“If somebody is doing well, there is no real reason to change that or to stop it,” Dr. Levy said. “There are some medications that are pretty interchangeable, but others, they all have slightly different side effects and we try to use what we think is the best medication for that patient based on the patient’s characteristics.”
ADA is recommending that PBMs, insurance plans, and employers “provide an expedited and standard process for gaining access to medications not included in the plan’s formulary. Responses to exception requests must be timely to ensure no delay in obtaining a needed medication, thereby preventing a gap in treatment. In addition, the association recommends PBMs, plans, and employers temporarily cover nonformulary drugs as if they are on formulary during the first 30 days after a formulary list is changed.”
Experimental Drug Slows Niemann-Pick Disease
“Encouraging” results from a study with an experimental drug offer hope to patients with Niemann-Pick disease type C1 (NPC1), a fatal neurologic disease that affects children and adolescents.
Symptoms of NPC1 result from cholesterol building in brain cells. The drug VTS-270 showed signs of improving cholesterol metabolism in neurons. After treatment, a molecule derived from cholesterol metabolism in neurons had increased, and 2 proteins indicative of brain injury had decreased.
In a phase 1/2a clinical trial, 14 participants received the experimental drug once a month for 12 to 18 months. Another 3 participants received the drug every 2 weeks for 18 months. After observing that the drug was safe and well tolerated, the researchers increased dosing for all participants. Their progress was compared with that of 21 participants in a previous study of NPC1.
The researchers also evaluated the drug’s effectiveness using a neurologic severity score (higher scores indicated more severe disease effects). Participants treated with VTS-270 had lower scores in cognition, speech, and mobility, indicating that the drug can stabilize or slow disease progression.
No one was observed to have serious adverse outcomes, but earlier studies had shown that the treatment carries the risk for hearing loss. After treatment in this study, participants, most of whom had already lost some hearing due to the disease, had further loss, for which they compensated with hearing aids.
“Encouraging” results from a study with an experimental drug offer hope to patients with Niemann-Pick disease type C1 (NPC1), a fatal neurologic disease that affects children and adolescents.
Symptoms of NPC1 result from cholesterol building in brain cells. The drug VTS-270 showed signs of improving cholesterol metabolism in neurons. After treatment, a molecule derived from cholesterol metabolism in neurons had increased, and 2 proteins indicative of brain injury had decreased.
In a phase 1/2a clinical trial, 14 participants received the experimental drug once a month for 12 to 18 months. Another 3 participants received the drug every 2 weeks for 18 months. After observing that the drug was safe and well tolerated, the researchers increased dosing for all participants. Their progress was compared with that of 21 participants in a previous study of NPC1.
The researchers also evaluated the drug’s effectiveness using a neurologic severity score (higher scores indicated more severe disease effects). Participants treated with VTS-270 had lower scores in cognition, speech, and mobility, indicating that the drug can stabilize or slow disease progression.
No one was observed to have serious adverse outcomes, but earlier studies had shown that the treatment carries the risk for hearing loss. After treatment in this study, participants, most of whom had already lost some hearing due to the disease, had further loss, for which they compensated with hearing aids.
“Encouraging” results from a study with an experimental drug offer hope to patients with Niemann-Pick disease type C1 (NPC1), a fatal neurologic disease that affects children and adolescents.
Symptoms of NPC1 result from cholesterol building in brain cells. The drug VTS-270 showed signs of improving cholesterol metabolism in neurons. After treatment, a molecule derived from cholesterol metabolism in neurons had increased, and 2 proteins indicative of brain injury had decreased.
In a phase 1/2a clinical trial, 14 participants received the experimental drug once a month for 12 to 18 months. Another 3 participants received the drug every 2 weeks for 18 months. After observing that the drug was safe and well tolerated, the researchers increased dosing for all participants. Their progress was compared with that of 21 participants in a previous study of NPC1.
The researchers also evaluated the drug’s effectiveness using a neurologic severity score (higher scores indicated more severe disease effects). Participants treated with VTS-270 had lower scores in cognition, speech, and mobility, indicating that the drug can stabilize or slow disease progression.
No one was observed to have serious adverse outcomes, but earlier studies had shown that the treatment carries the risk for hearing loss. After treatment in this study, participants, most of whom had already lost some hearing due to the disease, had further loss, for which they compensated with hearing aids.
Study confirms zoonotic transmission of malaria

Molecular analysis has confirmed zoonotic transmission of malaria in southern Brazil, according to an article published in The Lancet Global Health.
Researchers identified 28 humans infected with Plasmodium simium, a malaria parasite usually only found in monkeys.
The researchers said screening of local monkeys and mosquitoes will be required to evaluate the extent of the emerging zoonotic threat to public health.
Malaria was thought to have been eliminated from southern Brazil over 50 years ago. However, between 2006 and 2014, 43 cases of malaria were reported in the Atlantic Forest area in southern Brazil. An additional 49 cases were reported from 2015 through 2016.
This prompted researchers to investigate the possibility of zoonotic transmission. The team looked at the 49 cases reported in 2015 and 2016, and they were able to sequence DNA samples from 28 of the 49 patients.
In all 28 cases, the parasite was confirmed to be P simium—not the human parasite Plasmodium vivax, as previously thought.
P simium is transmitted via the Anopheles mosquito and is known to infect some species of howler and capuchin monkeys in the Atlantic Forest region.
All 28 humans infected with P simium had entered the forest or visited the surrounding area. The patients’ main symptom was fever, none of them were admitted to the hospital, and all made full recoveries following treatment.
“There is no evidence that zoonotic malaria can be transmitted from human to human via mosquitoes,” said study author Patrícia Brasil, MD, of Instituto Nacional de Infectologia Evandro Chagas in Rio de Janeiro, Brazil.
“In addition, there is no current threat to people in the city of Rio de Janeiro or in other non-forest areas of the Rio de Janeiro state where transmission of the disease does not exist. However, its unique mode of transmission via monkeys and the fact that it occurs in areas of high forest coverage mean that zoonotic malaria poses a unique problem for malaria control efforts and may complicate the drive towards eventual elimination of the disease. Although [this type of malaria is] benign and treatable, visitors should follow measures to avoid insect bites when going into the forest.”
Dr Brasil and her colleagues noted that samples from previous malaria cases reported in the Atlantic Forest area have not yet been tested. Therefore, it is not possible to establish whether P simium has only recently acquired the ability to infect human beings or if zoonotic malaria has previously infected humans in this region.
“In the 1960s, there was a probable case of zoonotic malaria described in a forest guard in the Atlantic Forest of São Paulo, but, until now, there has been no molecular evidence of the parasite being present in humans,” said study author Cláudio Tadeu Daniel-Ribeiro, MD, of Instituto Oswaldo Cruz in Rio de Janeiro, Brazil.
“This is the first demonstration of P simium naturally infecting human beings in forest locations in a region considered to have eliminated transmission of malaria at least 50 years ago.” ![]()
Molecular analysis has confirmed zoonotic transmission of malaria in southern Brazil, according to an article published in The Lancet Global Health.
Researchers identified 28 humans infected with Plasmodium simium, a malaria parasite usually only found in monkeys.
The researchers said screening of local monkeys and mosquitoes will be required to evaluate the extent of the emerging zoonotic threat to public health.
Malaria was thought to have been eliminated from southern Brazil over 50 years ago. However, between 2006 and 2014, 43 cases of malaria were reported in the Atlantic Forest area in southern Brazil. An additional 49 cases were reported from 2015 through 2016.
This prompted researchers to investigate the possibility of zoonotic transmission. The team looked at the 49 cases reported in 2015 and 2016, and they were able to sequence DNA samples from 28 of the 49 patients.
In all 28 cases, the parasite was confirmed to be P simium—not the human parasite Plasmodium vivax, as previously thought.
P simium is transmitted via the Anopheles mosquito and is known to infect some species of howler and capuchin monkeys in the Atlantic Forest region.
All 28 humans infected with P simium had entered the forest or visited the surrounding area. The patients’ main symptom was fever, none of them were admitted to the hospital, and all made full recoveries following treatment.
“There is no evidence that zoonotic malaria can be transmitted from human to human via mosquitoes,” said study author Patrícia Brasil, MD, of Instituto Nacional de Infectologia Evandro Chagas in Rio de Janeiro, Brazil.
“In addition, there is no current threat to people in the city of Rio de Janeiro or in other non-forest areas of the Rio de Janeiro state where transmission of the disease does not exist. However, its unique mode of transmission via monkeys and the fact that it occurs in areas of high forest coverage mean that zoonotic malaria poses a unique problem for malaria control efforts and may complicate the drive towards eventual elimination of the disease. Although [this type of malaria is] benign and treatable, visitors should follow measures to avoid insect bites when going into the forest.”
Dr Brasil and her colleagues noted that samples from previous malaria cases reported in the Atlantic Forest area have not yet been tested. Therefore, it is not possible to establish whether P simium has only recently acquired the ability to infect human beings or if zoonotic malaria has previously infected humans in this region.
“In the 1960s, there was a probable case of zoonotic malaria described in a forest guard in the Atlantic Forest of São Paulo, but, until now, there has been no molecular evidence of the parasite being present in humans,” said study author Cláudio Tadeu Daniel-Ribeiro, MD, of Instituto Oswaldo Cruz in Rio de Janeiro, Brazil.
“This is the first demonstration of P simium naturally infecting human beings in forest locations in a region considered to have eliminated transmission of malaria at least 50 years ago.” ![]()
Molecular analysis has confirmed zoonotic transmission of malaria in southern Brazil, according to an article published in The Lancet Global Health.
Researchers identified 28 humans infected with Plasmodium simium, a malaria parasite usually only found in monkeys.
The researchers said screening of local monkeys and mosquitoes will be required to evaluate the extent of the emerging zoonotic threat to public health.
Malaria was thought to have been eliminated from southern Brazil over 50 years ago. However, between 2006 and 2014, 43 cases of malaria were reported in the Atlantic Forest area in southern Brazil. An additional 49 cases were reported from 2015 through 2016.
This prompted researchers to investigate the possibility of zoonotic transmission. The team looked at the 49 cases reported in 2015 and 2016, and they were able to sequence DNA samples from 28 of the 49 patients.
In all 28 cases, the parasite was confirmed to be P simium—not the human parasite Plasmodium vivax, as previously thought.
P simium is transmitted via the Anopheles mosquito and is known to infect some species of howler and capuchin monkeys in the Atlantic Forest region.
All 28 humans infected with P simium had entered the forest or visited the surrounding area. The patients’ main symptom was fever, none of them were admitted to the hospital, and all made full recoveries following treatment.
“There is no evidence that zoonotic malaria can be transmitted from human to human via mosquitoes,” said study author Patrícia Brasil, MD, of Instituto Nacional de Infectologia Evandro Chagas in Rio de Janeiro, Brazil.
“In addition, there is no current threat to people in the city of Rio de Janeiro or in other non-forest areas of the Rio de Janeiro state where transmission of the disease does not exist. However, its unique mode of transmission via monkeys and the fact that it occurs in areas of high forest coverage mean that zoonotic malaria poses a unique problem for malaria control efforts and may complicate the drive towards eventual elimination of the disease. Although [this type of malaria is] benign and treatable, visitors should follow measures to avoid insect bites when going into the forest.”
Dr Brasil and her colleagues noted that samples from previous malaria cases reported in the Atlantic Forest area have not yet been tested. Therefore, it is not possible to establish whether P simium has only recently acquired the ability to infect human beings or if zoonotic malaria has previously infected humans in this region.
“In the 1960s, there was a probable case of zoonotic malaria described in a forest guard in the Atlantic Forest of São Paulo, but, until now, there has been no molecular evidence of the parasite being present in humans,” said study author Cláudio Tadeu Daniel-Ribeiro, MD, of Instituto Oswaldo Cruz in Rio de Janeiro, Brazil.
“This is the first demonstration of P simium naturally infecting human beings in forest locations in a region considered to have eliminated transmission of malaria at least 50 years ago.” ![]()
Man, 32, With Severe Scrotal Pain and Swelling
IN THIS ARTICLE
- Lab values for case patient
- Differential diagnoses
- Case outcome
A 32-year-old man presents to the urgent care center at a community hospital with severe scrotal pain and swelling of five days’ duration. What began as mild left scrotal discomfort is now causing increasing pain, swelling, hematuria, dysuria, low-grade fever, and nausea, prompting him to seek medical attention.
The patient, who is a pipefitter in a hospital, was at work when his symptoms began. He denies any history of scrotal trauma, and his review of systems is otherwise unremarkable. His medical history is significant for mild hypertension and morbid obesity, but he is not immunocompromised. Two months ago, he had an excision and repair of a left ureterocele, for which he was treated prophylactically with ciprofloxacin for one week. He has a 3–pack-year history of smoking and consumes three alcoholic beverages per week. He denies illicit drug use and has no report of sexually transmitted infection.
Upon arrival to urgent care, the patient appears to be in moderate distress, with a blood pressure (BP) of 111/79 mm Hg; pulse, 104 beats/min; respiratory rate, 18 breaths/min-1; temperature, 100.1°F; and SpO2, 94%. Physical exam reveals left scrotal erythema, severe tenderness upon palpation, marked scrotal edema, and a slight amount of foul-smelling discharge seeping from a pinpoint opening in the left perineum (see Figure 1a). Given his scrotal presentation, he is quickly transferred to a regional emergency department (ED) for a urology consult.

In the ED, lab testing yields significant findings (see Table 1). His ECG demonstrates sinus tachycardia at 126 beats/min without rhythm or ST changes. His urinalysis reveals a cloudy appearance, a protein level of 100 mg/dL, and trace leukocyte esterase.
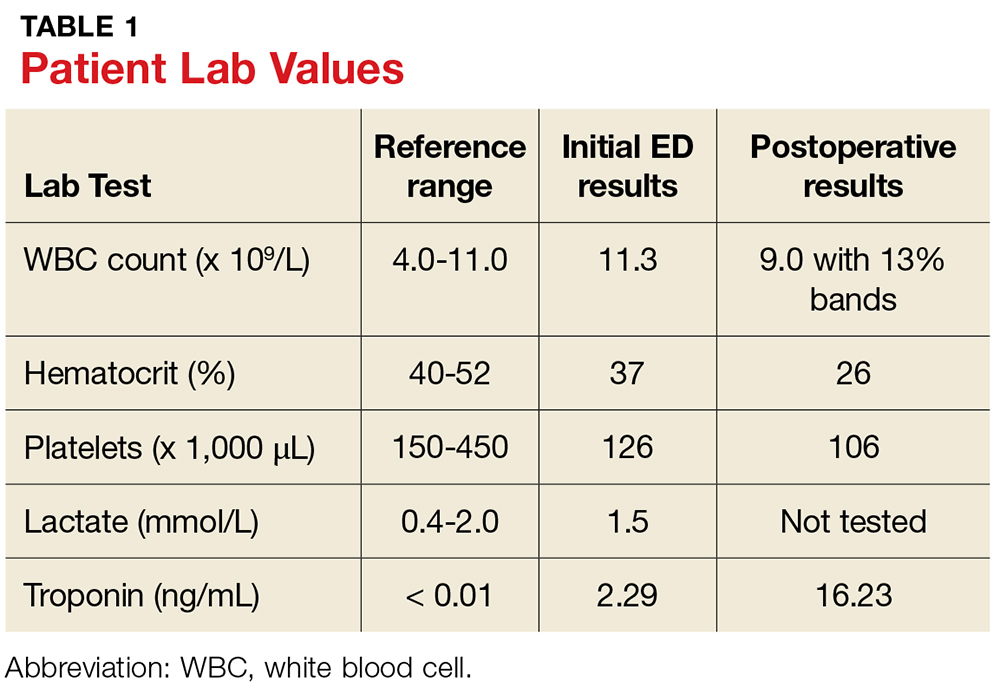
Urgent CT with contrast is obtained; it shows significant soft-tissue inflammatory changes in the left groin and scrotum that extend into the left thigh. In addition, a collection of fluid is seen in the inferior aspect of the left scrotal wall, indicating a probable abscess. There is no free air or lymphadenopathy.
Given the patient’s worsening condition and his apparent advancement to a systemic inflammatory response syndrome, surgical consult is obtained. He is diagnosed with a scrotal abscess and cellulitis; two blood and two scrotal cultures are obtained, and the patient is empirically started on IV ampicillin and gentamicin.
Two hours later, he has a BP of 122/74 mm Hg; pulse, 112 beats/min; respiratory rate, 20 breaths/min-1; and temperature, 103.1°F. His genital inflammation has advanced to the perineum and the left lower abdomen. The purulent, bloody, foul-smelling drainage from the opening in the left perineum is increasingly apparent. The patient is taken emergently to surgery for an incision and drainage, along with exploration of the scrotal abscess. During surgery, the patient is discovered to have Fournier’s gangrene.
DISCUSSION
Fournier’s gangrene (FG) is a necrotizing fasciitis of the perineal, perianal, and/or genital areas involving the superficial and deep fascial planes while sparing the deep muscular structures and overlying skin.1 A rare but potentially fatal disease, FG spreads at a rate of up to 3 cm/h.2,3
Mortality rates range from 7.5% to 88%, with the highest mortality occurring within the first 96 hours of hospitalization.1,4-7 Mortality is often related to the onset of sepsis.4,5 Survival requires early recognition; immediate, aggressive surgical debridement of all necrotic tissue; and concomitant, early administration of appropriate antibiotics.1,4,5,8 Mortality risk and prognosis are improved in patients younger than 60 with localized disease and no toxicity, along with sterile blood cultures.1
Risk Factors
FG is most commonly seen in males between the ages of 50 and 70, with a 10:1 male-to-female ratio.3,9 Impaired immunity typically increases a patient’s susceptibility to FG, with type 2 diabetes having the highest incidence (85% of patients).1,4,6,8,10 Other conditions that can increase the risk for FG include obesity, alcoholism, cirrhosis, cardiac disease, tobacco use, peripheral vascular disease, malignancy, chronic steroid use, renal insufficiency, IV drug abuse, and HIV.1,4,6,8,9,11
Trauma frequently initiates the infectious process,with urogenital trauma (eg, placement of urethral instrumentation, surgery, and urinary tract infection) being the main cause of bacterial introduction.1,3 Localized infection causes the development of an obliterative endarteritis, resulting in subcutaneous vascular ischemia, necrosis, and bacterial proliferation.3,7,9
Presentation and Diagnosis
Presenting symptoms of FG include intense, abrupt genital pain that is disproportionate to the physical exam findings.9 This rapidly escalates to include extreme swelling, erythema, bullae, discolored skin, and tissue crepitus with eventual necrosis.2,10 Lab results typically show leukocytosis > 18.0 × 109/L.4 The testicle and spermatic cord are generally unaffected (as in this patient), due to the anatomic relationship between the various layers of fascia within the scrotum and the anterior abdominal wall, as well as the independent blood supply of the compartmentalized testicular tissue.1-3
During an exam of the acute scrotum, the differential diagnosis includes cellulitis, scrotal abscess, acute epididymitis, and testicular torsion, with scrotal abscess being most frequently diagnosed (57% of patients).9,11,12 The distinguishing features of these diagnoses can be found in Table 2. Necrotizing fasciitis in the form of FG tends to be an unexpected, rare finding usually only diagnosed during the surgical draining of an abscess.12

CT is the test of choice to detect FG and determine the extent of its spread by identifying subcutaneous air/gas within the involved fascial planes.10,13 However, an incisional biopsy with culture is needed to confirm the diagnosis.3,9 Most patients with FG require an average of four surgeries (eg, reconstruction, skin grafting, and possibly colostomy if the infection has entered the peritoneal cavity) in order to eradicate the disease and achieve the best functional and cosmetic outcome.4
Etiology
About 83% of FG cases are polymicrobial infections comprised of enterobacter, enterococci, Escherichia coli, group A streptococci, pseudomonas, and clostridium, with symptoms evolving two to four days following the initial insult.4,7,11,14,15 Monomicrobial infections are much less common, but the symptoms progress even more rapidly.15 Methicillin-resistant Staphylococcus aureus (MRSA) necrotizing fasciitis infections occur in about 3% of monomicrobial cases.12 MRSA emerged in the early 2000s as an additional causative pathogen for polymicrobial necrotizing fasciitis infections.12,14,15 Prior to that time, S aureus strains were almost uniformly susceptible to penicillinase-resistant ß lactams.12
A distinction should be made between health care-associated (HA) MRSA and community-acquired (CA) MRSA due to treatment considerations. HA-MRSA infections are contracted through previous health care exposure (within the past year) and are less resistant to treatment.16,17 In contrast, CA-MRSA, which comprises 29% of MRSA cases, causes infections in previously healthy young patients without prior health care contact within the past year.16 CA-MRSA strains are more robust than HA-MRSA strains and can cause sepsis and other invasive, rapidly progressive, and possibly life-threatening infections due to the amount of tissue destruction and necrosis.16,18 Transmission of CA-MRSA is often associated with crowded environments, frequent skin-to-skin contact, compromised skin integrity, contaminated items or surfaces, and lack of cleanliness.16 Over the years, CA-MRSA has developed resistance to multiple antimicrobials; providers should therefore consider CA-MRSA on initial evaluation of necrotizing infections, to ensure appropriate initiation of treatment.12,16
CASE CONTINUED
Extensive debridement was completed down to healthy tissue in all affected areas (see Figure 1b). The necrotizing fasciitis had spared the left testicle and spermatic cord, and a colostomy was not required.
The patient’s initial postoperative vital signs were unremarkable, except for his BP (86/54 mm Hg). The patient was taken postoperatively to the surgical intensive care unit (SICU) with the diagnosis of FG. Aggressive IV fluids were administered for resuscitation, and he was closely monitored for increasing sepsis. Metronidazole was added for anaerobic and gram-positive coverage. His postoperative lab results can also be found in Table 1.
His ECG showed a normal sinus rhythm without ST changes, and he denied any cardiac symptoms. His physical exam was significant for mild pallor, dry mucus membranes, and a left scrotal and pelvic packed dressing. He was given two units of packed red blood cells for acute postoperative blood-loss anemia. The preliminary tissue culture results showed gram-positive cocci consistent with a staphylococcal infection; his antibiotics were then changed to IV ampicillin/sulbactam and clindamycin.
Approximately five hours postoperatively, an ECG suddenly showed acute ST elevation in leads II, II, and aVF, with reciprocal changes. The patient was diagnosed with an acute myocardial infarction (AMI). He denied any chest pain, shortness of breath, or diaphoresis. The SICU team initiated aspirin therapy and immediately contacted cardiology for an emergent coronary angiogram.
The angiogram and cardiac catheterization revealed an elevated left ventricular end diastolic (LVED) volume, inferior wall hypokinesis, a low-normal ejection fraction, and a 30% lesion in the first diagonal of his left anterior descending artery. A postprocedure echocardiogram demonstrated left ventricular (LV) ejection fraction of 50%, with LV hypokinesis in the inferior base and mild left atrial enlargement. The patient was started on metoprolol for myocardial protection and recovery.
Complications
Perioperative complications of FG, including AMI, must be considered due to the physiologic stress on the body.19 Most patients with perioperative AMI after noncardiac surgery do not experience ischemic symptoms.20
Growing evidence suggests the pivotal role of acute inflammation (postoperatively or from infection) as a precipitating event in AMI.20,21 Chemical mediators, such as inflammatory cytokines, endotoxins, and nitric oxide, may play a role in the development of an AMI.22
If cardiovascular disease and/or significant cardiovascular risk factors (ie, older age, male, cigarette smoking, cardiac family history, acute kidney injury) are present, the risk for AMI increases in the first two days following surgery.21,23 Acute infections and sepsis also initiate or increase systemic inflammatory activity via these same chemical mediators.21
Most suspected infectious agents also produce coronary artery sheer stress and destabilization of vulnerable plaques, leading to plaque rupture and thrombosis.19,24 Proinflammatory cytokines promote enhanced platelet activation and contribute to this thrombotic environment.21,23 Thrombus leads to obstructed coronary blood flow, myocardial ischemia, and finally, infarction.21
A reversible myocardial depression, cardiomyopathy, or myocardial ischemia may occur in patients with acute systemic infection or sepsis when the myocardium is functionally and structurally injured by these inflammatory chemical mediators.19,22-24 Characteristics of such a cardiomyopathy include left ventricle dilation with a low filling pressure, an abnormal increase in LVED volume, and a depressed ejection fraction.22
An acute infectious or septic process can raise troponin levels in 43% to 85% of patients.22,24 Troponin biomarkers can assist in predicting myocardial injury and events after surgery with nearly absolute myocardial tissue specificity.20 Cardiovascular involvement caused by myocardial injury–related sepsis is observed in up to 70% of patients in the ICU for these reasons.23 Therefore, providers should consider measuring troponin biomarkers during such infectious and septic processes, as this team did for the case patient. The providers were able to diagnose his AMI early and institute appropriate treatment measures to avoid extensive myocardial tissue damage.
Several studies have already demonstrated a correlation between pneumococcal pneumonia and an increased risk for AMI, and the same mechanisms are presumed responsible for any severe acute infectious state.21 More research is needed to understand the pathophysiology of AMI in sepsis and acute systemic infections.23
OUTCOME FOR THE CASE PATIENT
On postoperative day 2, the patient’s vital signs and lab results were normal. Additional lab results included an A1C of 5.2%. His ECG showed a resolving ST-elevation myocardial infarction (STEMI). The surgical wound had initiation of early granulation tissue without any further signs of necrosis.
A postoperative acute STEMI was unexpected in this patient, as his only risk factors included being male, mild hypertension, obesity, and tobacco use. At the time of his initial elevated troponin level, he had no cardiac symptoms or ECG changes. This initial high troponin level may have been stress-induced from the acute infectious process, and his acute inferior wall STEMI may have been secondary to a transient thrombotic event. The STEMI may then have resolved on its own during the cardiac catheterization with the administration of heparin, IV fluids, blood products, aspirin, or dye infiltration, thus enhancing reperfusion of the coronary artery system.
The final tissue culture showed MRSA. Given his job and his history of a genitourinary procedure, as well as the less fulminant form of disease and relatively quick recovery, it was likely HA-MRSA (rather than CA-MRSA). Only clindamycin was used for treatment.
The wound continued to have decreasing erythema, a reduction in tenderness, and evidence of viable, pink granulation tissue. HIV testing was not completed during his admission. The remainder of the patient’s hospital course was unremarkable, and he was discharged home with wound care, urology, and cardiology follow-up services.
CONCLUSION
Multiple factors contribute to a delayed or mistaken diagnosis of FG; it may be overlooked in the initial working diagnoses because of its low incidence and manifestations similar to those of other soft-tissue infections (eg, cellulitis, scrotal abscess). The cutaneous signs of FG often lag behind the disease manifestation, with minimal or no external presence while extensive internal tissue destruction is occurring. Constant review of symptoms is required when treating patients with soft-tissue infections, and early signs—such as pain out of proportion to physical findings—should prompt a clinician to include FG in the differential.
Early diagnosis with prompt debridement and antibiotic therapy are crucial to patient survival. Detecting FG within the first 24 hours is critical. Further differentiation between CA-MRSA and HA-MRSA can assist in patient recovery and survival by guiding appropriate antibiotic therapy. Perioperative risk assessment and serial troponin biomarkers may identify patients in need of intensive monitoring and management postoperatively to avoid an AMI, since patients may not experience ischemic symptoms.
1. Norton KS, Johnson LW, Perry T, et al. Management of Fournier’s gangrene: an eleven-year retrospective analysis of early recognition, diagnosis, and treatment. Am Surg. 2002;68(8):709-713.
2. Agostini T, Mori F, Perello R, et al. Successful combined approach to a severe Fournier’s gangrene. Indian J Plast Surg. 2014;47(1):132-136.
3. Cabrera G, March P. Fournier’s gangrene. Glendale, CA: Cinahl Information Systems; 2016.
4. Czymek R, Kujath P, Bruch HP, et al. Treatment, outcome and quality of life after Fournier’s gangrene: a multicentre study. Colorectal Dis. 2013;15(12):1529-1536.
5. Sugihara T, Yasunaga H, Horiguchi H, et al. Impact of surgical intervention timing on the case fatality rate for Fournier’s gangrene: an analysis of 379 cases. BJU Int. 2012;110(11c):E1096-1100.
6. Tuncel A, Keten T, Aslan Y, et al. Comparison of different scoring systems for outcome prediction in patients with Fournier’s gangrene: experience with 50 patients. Scand J Urol. 2014;48(4):393-399.
7. Taken K, Oncu MR, Ergun M, et al. Fournier’s gangrene: causes, presentation and survival of sixty-five patients. Pak J Med Sci. 2016;32(3):746-750.
8. Palvolgyi R, Kaji AH, Valeriano J, et al. Fournier’s gangrene: a model for early prediction. Am Surg. 2014;80(10):926-931.
9. Pais V, Santora T. Fournier gangrene. http://emedicine.medscape.com/article/2028899-overview. Accessed August 16, 2017.
10. Cottrill RR. A demonstration of clinical reasoning through a case of scrotal infection. Urol Nurs. 2013;33(1):33-37.
11. Summers A. Fournier’s gangrene. J Nurse Pract. 2014;10(8):582-587.
12. Miller LG, Perdreau-Remington F, Rieg G, et al. Necrotizing fasciitis caused by community-associated methicillin-resistant Staphylococcus aureus in Los Angeles. N Engl J Med. 2005;352(14):1445-1453.
13. Gupta N, Zinn K, Bansal I, Weinstein R. Fournier’s gangrene: ultrasound or computed tomography? A letter to the editor. Med Ultrason. 2014;16(4):389-390.
14. Bjurlin MA, O’Grady T, Kim DY, et al. Causative pathogens, antibiotic sensitivity, resistance patterns, and severity in a contemporary series of Fournier’s gangrene. Urol. 2013;81(4):752-758.
15. Goh T, Goh LG. Pitfalls in diagnosing necrotizing fasciitis. https://psnet.ahrq.gov/webmm/case/329/pitfalls-in-diagnos ing-necrotizing-fasciitis. Accessed August 16, 2017.
16. Kale P, Dhawan B. The changing face of community-acquired methicillin-resistant Staphylococcus aureus. Indian J Med Microbiol. 2016;34(3):275-285.
17. CDC. Necrotizing fasciitis. www.cdc.gov/Features/NecrotizingFasciitis/index.html. Accessed August 16, 2017.
18. Barnes BE, Sampson DA. A literature review on community-acquired methicillin-resistant Staphylococcus aureus in the United States: clinical information for primary care nurse practitioners. J Am Acad Nurse Pract. 2011;23(1):23-32.
19. Madjid M, Vela D, Khalili-Tabrizi H, et al. Systemic infections cause exaggerated local inflammation in atherosclerotic coronary arteries. Clues to the triggering effect of acute infections on acute coronary syndromes. Tex Heart Inst J. 2007;34(1):11-18.
20. Devereaux PJ, Chan MTV, Alonso-Coello PA, et al; VISION Study Investigators. Association between postoperative troponin levels and 30-day mortality among patients undergoing noncardiac surgery. JAMA. 2012;307(21):2295-2304.
21. Corrales-Medina VF, Fatemi O, Serpa J, et al. The association between Staphylococcus aureus bacteremia and acute myocardial infarction. Scand J Infect Dis. 2009;41(6-7):511-514.
22. Romero-Bermejo FJ, Ruiz-Bailen M, Gil-Cebrian J, Huertos-Ranchal MJ. Sepsis-induced cardiomyopathy. Curr Cardiol Rev. 2011;7(3):163-183.
23. Smilowitz NR, Gupta N, Guo Y, Bangalore S. Comparison of outcomes of patients with sepsis with versus without acute myocardial infarction and comparison of invasive versus noninvasive management of the patients with infarction. Am J Cardiol. 2016;117(7):1065-1071.
24. Mattson M. Sepsis and cardiac disease: improving outcomes through recognition and management. Prog Cardiovasc Nurs. 2009;24(4):199-201.
25. Papadakis MA, McPhee SJ. Current Medical Diagnosis & Treatment. 54th ed. New York, NY: McGraw Hill Education; 2015:137-138, 151-152, 937.
26. Eyre RC. Evaluation of the acute scrotum in adults. www.uptodate.com/contents/evaluation-of-the-acute-scrotum-in-adults. Accessed August 16, 2017.
IN THIS ARTICLE
- Lab values for case patient
- Differential diagnoses
- Case outcome
A 32-year-old man presents to the urgent care center at a community hospital with severe scrotal pain and swelling of five days’ duration. What began as mild left scrotal discomfort is now causing increasing pain, swelling, hematuria, dysuria, low-grade fever, and nausea, prompting him to seek medical attention.
The patient, who is a pipefitter in a hospital, was at work when his symptoms began. He denies any history of scrotal trauma, and his review of systems is otherwise unremarkable. His medical history is significant for mild hypertension and morbid obesity, but he is not immunocompromised. Two months ago, he had an excision and repair of a left ureterocele, for which he was treated prophylactically with ciprofloxacin for one week. He has a 3–pack-year history of smoking and consumes three alcoholic beverages per week. He denies illicit drug use and has no report of sexually transmitted infection.
Upon arrival to urgent care, the patient appears to be in moderate distress, with a blood pressure (BP) of 111/79 mm Hg; pulse, 104 beats/min; respiratory rate, 18 breaths/min-1; temperature, 100.1°F; and SpO2, 94%. Physical exam reveals left scrotal erythema, severe tenderness upon palpation, marked scrotal edema, and a slight amount of foul-smelling discharge seeping from a pinpoint opening in the left perineum (see Figure 1a). Given his scrotal presentation, he is quickly transferred to a regional emergency department (ED) for a urology consult.
In the ED, lab testing yields significant findings (see Table 1). His ECG demonstrates sinus tachycardia at 126 beats/min without rhythm or ST changes. His urinalysis reveals a cloudy appearance, a protein level of 100 mg/dL, and trace leukocyte esterase.
Urgent CT with contrast is obtained; it shows significant soft-tissue inflammatory changes in the left groin and scrotum that extend into the left thigh. In addition, a collection of fluid is seen in the inferior aspect of the left scrotal wall, indicating a probable abscess. There is no free air or lymphadenopathy.
Given the patient’s worsening condition and his apparent advancement to a systemic inflammatory response syndrome, surgical consult is obtained. He is diagnosed with a scrotal abscess and cellulitis; two blood and two scrotal cultures are obtained, and the patient is empirically started on IV ampicillin and gentamicin.
Two hours later, he has a BP of 122/74 mm Hg; pulse, 112 beats/min; respiratory rate, 20 breaths/min-1; and temperature, 103.1°F. His genital inflammation has advanced to the perineum and the left lower abdomen. The purulent, bloody, foul-smelling drainage from the opening in the left perineum is increasingly apparent. The patient is taken emergently to surgery for an incision and drainage, along with exploration of the scrotal abscess. During surgery, the patient is discovered to have Fournier’s gangrene.
DISCUSSION
Fournier’s gangrene (FG) is a necrotizing fasciitis of the perineal, perianal, and/or genital areas involving the superficial and deep fascial planes while sparing the deep muscular structures and overlying skin.1 A rare but potentially fatal disease, FG spreads at a rate of up to 3 cm/h.2,3
Mortality rates range from 7.5% to 88%, with the highest mortality occurring within the first 96 hours of hospitalization.1,4-7 Mortality is often related to the onset of sepsis.4,5 Survival requires early recognition; immediate, aggressive surgical debridement of all necrotic tissue; and concomitant, early administration of appropriate antibiotics.1,4,5,8 Mortality risk and prognosis are improved in patients younger than 60 with localized disease and no toxicity, along with sterile blood cultures.1
Risk Factors
FG is most commonly seen in males between the ages of 50 and 70, with a 10:1 male-to-female ratio.3,9 Impaired immunity typically increases a patient’s susceptibility to FG, with type 2 diabetes having the highest incidence (85% of patients).1,4,6,8,10 Other conditions that can increase the risk for FG include obesity, alcoholism, cirrhosis, cardiac disease, tobacco use, peripheral vascular disease, malignancy, chronic steroid use, renal insufficiency, IV drug abuse, and HIV.1,4,6,8,9,11
Trauma frequently initiates the infectious process,with urogenital trauma (eg, placement of urethral instrumentation, surgery, and urinary tract infection) being the main cause of bacterial introduction.1,3 Localized infection causes the development of an obliterative endarteritis, resulting in subcutaneous vascular ischemia, necrosis, and bacterial proliferation.3,7,9
Presentation and Diagnosis
Presenting symptoms of FG include intense, abrupt genital pain that is disproportionate to the physical exam findings.9 This rapidly escalates to include extreme swelling, erythema, bullae, discolored skin, and tissue crepitus with eventual necrosis.2,10 Lab results typically show leukocytosis > 18.0 × 109/L.4 The testicle and spermatic cord are generally unaffected (as in this patient), due to the anatomic relationship between the various layers of fascia within the scrotum and the anterior abdominal wall, as well as the independent blood supply of the compartmentalized testicular tissue.1-3
During an exam of the acute scrotum, the differential diagnosis includes cellulitis, scrotal abscess, acute epididymitis, and testicular torsion, with scrotal abscess being most frequently diagnosed (57% of patients).9,11,12 The distinguishing features of these diagnoses can be found in Table 2. Necrotizing fasciitis in the form of FG tends to be an unexpected, rare finding usually only diagnosed during the surgical draining of an abscess.12
CT is the test of choice to detect FG and determine the extent of its spread by identifying subcutaneous air/gas within the involved fascial planes.10,13 However, an incisional biopsy with culture is needed to confirm the diagnosis.3,9 Most patients with FG require an average of four surgeries (eg, reconstruction, skin grafting, and possibly colostomy if the infection has entered the peritoneal cavity) in order to eradicate the disease and achieve the best functional and cosmetic outcome.4
Etiology
About 83% of FG cases are polymicrobial infections comprised of enterobacter, enterococci, Escherichia coli, group A streptococci, pseudomonas, and clostridium, with symptoms evolving two to four days following the initial insult.4,7,11,14,15 Monomicrobial infections are much less common, but the symptoms progress even more rapidly.15 Methicillin-resistant Staphylococcus aureus (MRSA) necrotizing fasciitis infections occur in about 3% of monomicrobial cases.12 MRSA emerged in the early 2000s as an additional causative pathogen for polymicrobial necrotizing fasciitis infections.12,14,15 Prior to that time, S aureus strains were almost uniformly susceptible to penicillinase-resistant ß lactams.12
A distinction should be made between health care-associated (HA) MRSA and community-acquired (CA) MRSA due to treatment considerations. HA-MRSA infections are contracted through previous health care exposure (within the past year) and are less resistant to treatment.16,17 In contrast, CA-MRSA, which comprises 29% of MRSA cases, causes infections in previously healthy young patients without prior health care contact within the past year.16 CA-MRSA strains are more robust than HA-MRSA strains and can cause sepsis and other invasive, rapidly progressive, and possibly life-threatening infections due to the amount of tissue destruction and necrosis.16,18 Transmission of CA-MRSA is often associated with crowded environments, frequent skin-to-skin contact, compromised skin integrity, contaminated items or surfaces, and lack of cleanliness.16 Over the years, CA-MRSA has developed resistance to multiple antimicrobials; providers should therefore consider CA-MRSA on initial evaluation of necrotizing infections, to ensure appropriate initiation of treatment.12,16
CASE CONTINUED
Extensive debridement was completed down to healthy tissue in all affected areas (see Figure 1b). The necrotizing fasciitis had spared the left testicle and spermatic cord, and a colostomy was not required.
The patient’s initial postoperative vital signs were unremarkable, except for his BP (86/54 mm Hg). The patient was taken postoperatively to the surgical intensive care unit (SICU) with the diagnosis of FG. Aggressive IV fluids were administered for resuscitation, and he was closely monitored for increasing sepsis. Metronidazole was added for anaerobic and gram-positive coverage. His postoperative lab results can also be found in Table 1.
His ECG showed a normal sinus rhythm without ST changes, and he denied any cardiac symptoms. His physical exam was significant for mild pallor, dry mucus membranes, and a left scrotal and pelvic packed dressing. He was given two units of packed red blood cells for acute postoperative blood-loss anemia. The preliminary tissue culture results showed gram-positive cocci consistent with a staphylococcal infection; his antibiotics were then changed to IV ampicillin/sulbactam and clindamycin.
Approximately five hours postoperatively, an ECG suddenly showed acute ST elevation in leads II, II, and aVF, with reciprocal changes. The patient was diagnosed with an acute myocardial infarction (AMI). He denied any chest pain, shortness of breath, or diaphoresis. The SICU team initiated aspirin therapy and immediately contacted cardiology for an emergent coronary angiogram.
The angiogram and cardiac catheterization revealed an elevated left ventricular end diastolic (LVED) volume, inferior wall hypokinesis, a low-normal ejection fraction, and a 30% lesion in the first diagonal of his left anterior descending artery. A postprocedure echocardiogram demonstrated left ventricular (LV) ejection fraction of 50%, with LV hypokinesis in the inferior base and mild left atrial enlargement. The patient was started on metoprolol for myocardial protection and recovery.
Complications
Perioperative complications of FG, including AMI, must be considered due to the physiologic stress on the body.19 Most patients with perioperative AMI after noncardiac surgery do not experience ischemic symptoms.20
Growing evidence suggests the pivotal role of acute inflammation (postoperatively or from infection) as a precipitating event in AMI.20,21 Chemical mediators, such as inflammatory cytokines, endotoxins, and nitric oxide, may play a role in the development of an AMI.22
If cardiovascular disease and/or significant cardiovascular risk factors (ie, older age, male, cigarette smoking, cardiac family history, acute kidney injury) are present, the risk for AMI increases in the first two days following surgery.21,23 Acute infections and sepsis also initiate or increase systemic inflammatory activity via these same chemical mediators.21
Most suspected infectious agents also produce coronary artery sheer stress and destabilization of vulnerable plaques, leading to plaque rupture and thrombosis.19,24 Proinflammatory cytokines promote enhanced platelet activation and contribute to this thrombotic environment.21,23 Thrombus leads to obstructed coronary blood flow, myocardial ischemia, and finally, infarction.21
A reversible myocardial depression, cardiomyopathy, or myocardial ischemia may occur in patients with acute systemic infection or sepsis when the myocardium is functionally and structurally injured by these inflammatory chemical mediators.19,22-24 Characteristics of such a cardiomyopathy include left ventricle dilation with a low filling pressure, an abnormal increase in LVED volume, and a depressed ejection fraction.22
An acute infectious or septic process can raise troponin levels in 43% to 85% of patients.22,24 Troponin biomarkers can assist in predicting myocardial injury and events after surgery with nearly absolute myocardial tissue specificity.20 Cardiovascular involvement caused by myocardial injury–related sepsis is observed in up to 70% of patients in the ICU for these reasons.23 Therefore, providers should consider measuring troponin biomarkers during such infectious and septic processes, as this team did for the case patient. The providers were able to diagnose his AMI early and institute appropriate treatment measures to avoid extensive myocardial tissue damage.
Several studies have already demonstrated a correlation between pneumococcal pneumonia and an increased risk for AMI, and the same mechanisms are presumed responsible for any severe acute infectious state.21 More research is needed to understand the pathophysiology of AMI in sepsis and acute systemic infections.23
OUTCOME FOR THE CASE PATIENT
On postoperative day 2, the patient’s vital signs and lab results were normal. Additional lab results included an A1C of 5.2%. His ECG showed a resolving ST-elevation myocardial infarction (STEMI). The surgical wound had initiation of early granulation tissue without any further signs of necrosis.
A postoperative acute STEMI was unexpected in this patient, as his only risk factors included being male, mild hypertension, obesity, and tobacco use. At the time of his initial elevated troponin level, he had no cardiac symptoms or ECG changes. This initial high troponin level may have been stress-induced from the acute infectious process, and his acute inferior wall STEMI may have been secondary to a transient thrombotic event. The STEMI may then have resolved on its own during the cardiac catheterization with the administration of heparin, IV fluids, blood products, aspirin, or dye infiltration, thus enhancing reperfusion of the coronary artery system.
The final tissue culture showed MRSA. Given his job and his history of a genitourinary procedure, as well as the less fulminant form of disease and relatively quick recovery, it was likely HA-MRSA (rather than CA-MRSA). Only clindamycin was used for treatment.
The wound continued to have decreasing erythema, a reduction in tenderness, and evidence of viable, pink granulation tissue. HIV testing was not completed during his admission. The remainder of the patient’s hospital course was unremarkable, and he was discharged home with wound care, urology, and cardiology follow-up services.
CONCLUSION
Multiple factors contribute to a delayed or mistaken diagnosis of FG; it may be overlooked in the initial working diagnoses because of its low incidence and manifestations similar to those of other soft-tissue infections (eg, cellulitis, scrotal abscess). The cutaneous signs of FG often lag behind the disease manifestation, with minimal or no external presence while extensive internal tissue destruction is occurring. Constant review of symptoms is required when treating patients with soft-tissue infections, and early signs—such as pain out of proportion to physical findings—should prompt a clinician to include FG in the differential.
Early diagnosis with prompt debridement and antibiotic therapy are crucial to patient survival. Detecting FG within the first 24 hours is critical. Further differentiation between CA-MRSA and HA-MRSA can assist in patient recovery and survival by guiding appropriate antibiotic therapy. Perioperative risk assessment and serial troponin biomarkers may identify patients in need of intensive monitoring and management postoperatively to avoid an AMI, since patients may not experience ischemic symptoms.
IN THIS ARTICLE
- Lab values for case patient
- Differential diagnoses
- Case outcome
A 32-year-old man presents to the urgent care center at a community hospital with severe scrotal pain and swelling of five days’ duration. What began as mild left scrotal discomfort is now causing increasing pain, swelling, hematuria, dysuria, low-grade fever, and nausea, prompting him to seek medical attention.
The patient, who is a pipefitter in a hospital, was at work when his symptoms began. He denies any history of scrotal trauma, and his review of systems is otherwise unremarkable. His medical history is significant for mild hypertension and morbid obesity, but he is not immunocompromised. Two months ago, he had an excision and repair of a left ureterocele, for which he was treated prophylactically with ciprofloxacin for one week. He has a 3–pack-year history of smoking and consumes three alcoholic beverages per week. He denies illicit drug use and has no report of sexually transmitted infection.
Upon arrival to urgent care, the patient appears to be in moderate distress, with a blood pressure (BP) of 111/79 mm Hg; pulse, 104 beats/min; respiratory rate, 18 breaths/min-1; temperature, 100.1°F; and SpO2, 94%. Physical exam reveals left scrotal erythema, severe tenderness upon palpation, marked scrotal edema, and a slight amount of foul-smelling discharge seeping from a pinpoint opening in the left perineum (see Figure 1a). Given his scrotal presentation, he is quickly transferred to a regional emergency department (ED) for a urology consult.
In the ED, lab testing yields significant findings (see Table 1). His ECG demonstrates sinus tachycardia at 126 beats/min without rhythm or ST changes. His urinalysis reveals a cloudy appearance, a protein level of 100 mg/dL, and trace leukocyte esterase.
Urgent CT with contrast is obtained; it shows significant soft-tissue inflammatory changes in the left groin and scrotum that extend into the left thigh. In addition, a collection of fluid is seen in the inferior aspect of the left scrotal wall, indicating a probable abscess. There is no free air or lymphadenopathy.
Given the patient’s worsening condition and his apparent advancement to a systemic inflammatory response syndrome, surgical consult is obtained. He is diagnosed with a scrotal abscess and cellulitis; two blood and two scrotal cultures are obtained, and the patient is empirically started on IV ampicillin and gentamicin.
Two hours later, he has a BP of 122/74 mm Hg; pulse, 112 beats/min; respiratory rate, 20 breaths/min-1; and temperature, 103.1°F. His genital inflammation has advanced to the perineum and the left lower abdomen. The purulent, bloody, foul-smelling drainage from the opening in the left perineum is increasingly apparent. The patient is taken emergently to surgery for an incision and drainage, along with exploration of the scrotal abscess. During surgery, the patient is discovered to have Fournier’s gangrene.
DISCUSSION
Fournier’s gangrene (FG) is a necrotizing fasciitis of the perineal, perianal, and/or genital areas involving the superficial and deep fascial planes while sparing the deep muscular structures and overlying skin.1 A rare but potentially fatal disease, FG spreads at a rate of up to 3 cm/h.2,3
Mortality rates range from 7.5% to 88%, with the highest mortality occurring within the first 96 hours of hospitalization.1,4-7 Mortality is often related to the onset of sepsis.4,5 Survival requires early recognition; immediate, aggressive surgical debridement of all necrotic tissue; and concomitant, early administration of appropriate antibiotics.1,4,5,8 Mortality risk and prognosis are improved in patients younger than 60 with localized disease and no toxicity, along with sterile blood cultures.1
Risk Factors
FG is most commonly seen in males between the ages of 50 and 70, with a 10:1 male-to-female ratio.3,9 Impaired immunity typically increases a patient’s susceptibility to FG, with type 2 diabetes having the highest incidence (85% of patients).1,4,6,8,10 Other conditions that can increase the risk for FG include obesity, alcoholism, cirrhosis, cardiac disease, tobacco use, peripheral vascular disease, malignancy, chronic steroid use, renal insufficiency, IV drug abuse, and HIV.1,4,6,8,9,11
Trauma frequently initiates the infectious process,with urogenital trauma (eg, placement of urethral instrumentation, surgery, and urinary tract infection) being the main cause of bacterial introduction.1,3 Localized infection causes the development of an obliterative endarteritis, resulting in subcutaneous vascular ischemia, necrosis, and bacterial proliferation.3,7,9
Presentation and Diagnosis
Presenting symptoms of FG include intense, abrupt genital pain that is disproportionate to the physical exam findings.9 This rapidly escalates to include extreme swelling, erythema, bullae, discolored skin, and tissue crepitus with eventual necrosis.2,10 Lab results typically show leukocytosis > 18.0 × 109/L.4 The testicle and spermatic cord are generally unaffected (as in this patient), due to the anatomic relationship between the various layers of fascia within the scrotum and the anterior abdominal wall, as well as the independent blood supply of the compartmentalized testicular tissue.1-3
During an exam of the acute scrotum, the differential diagnosis includes cellulitis, scrotal abscess, acute epididymitis, and testicular torsion, with scrotal abscess being most frequently diagnosed (57% of patients).9,11,12 The distinguishing features of these diagnoses can be found in Table 2. Necrotizing fasciitis in the form of FG tends to be an unexpected, rare finding usually only diagnosed during the surgical draining of an abscess.12
CT is the test of choice to detect FG and determine the extent of its spread by identifying subcutaneous air/gas within the involved fascial planes.10,13 However, an incisional biopsy with culture is needed to confirm the diagnosis.3,9 Most patients with FG require an average of four surgeries (eg, reconstruction, skin grafting, and possibly colostomy if the infection has entered the peritoneal cavity) in order to eradicate the disease and achieve the best functional and cosmetic outcome.4
Etiology
About 83% of FG cases are polymicrobial infections comprised of enterobacter, enterococci, Escherichia coli, group A streptococci, pseudomonas, and clostridium, with symptoms evolving two to four days following the initial insult.4,7,11,14,15 Monomicrobial infections are much less common, but the symptoms progress even more rapidly.15 Methicillin-resistant Staphylococcus aureus (MRSA) necrotizing fasciitis infections occur in about 3% of monomicrobial cases.12 MRSA emerged in the early 2000s as an additional causative pathogen for polymicrobial necrotizing fasciitis infections.12,14,15 Prior to that time, S aureus strains were almost uniformly susceptible to penicillinase-resistant ß lactams.12
A distinction should be made between health care-associated (HA) MRSA and community-acquired (CA) MRSA due to treatment considerations. HA-MRSA infections are contracted through previous health care exposure (within the past year) and are less resistant to treatment.16,17 In contrast, CA-MRSA, which comprises 29% of MRSA cases, causes infections in previously healthy young patients without prior health care contact within the past year.16 CA-MRSA strains are more robust than HA-MRSA strains and can cause sepsis and other invasive, rapidly progressive, and possibly life-threatening infections due to the amount of tissue destruction and necrosis.16,18 Transmission of CA-MRSA is often associated with crowded environments, frequent skin-to-skin contact, compromised skin integrity, contaminated items or surfaces, and lack of cleanliness.16 Over the years, CA-MRSA has developed resistance to multiple antimicrobials; providers should therefore consider CA-MRSA on initial evaluation of necrotizing infections, to ensure appropriate initiation of treatment.12,16
CASE CONTINUED
Extensive debridement was completed down to healthy tissue in all affected areas (see Figure 1b). The necrotizing fasciitis had spared the left testicle and spermatic cord, and a colostomy was not required.
The patient’s initial postoperative vital signs were unremarkable, except for his BP (86/54 mm Hg). The patient was taken postoperatively to the surgical intensive care unit (SICU) with the diagnosis of FG. Aggressive IV fluids were administered for resuscitation, and he was closely monitored for increasing sepsis. Metronidazole was added for anaerobic and gram-positive coverage. His postoperative lab results can also be found in Table 1.
His ECG showed a normal sinus rhythm without ST changes, and he denied any cardiac symptoms. His physical exam was significant for mild pallor, dry mucus membranes, and a left scrotal and pelvic packed dressing. He was given two units of packed red blood cells for acute postoperative blood-loss anemia. The preliminary tissue culture results showed gram-positive cocci consistent with a staphylococcal infection; his antibiotics were then changed to IV ampicillin/sulbactam and clindamycin.
Approximately five hours postoperatively, an ECG suddenly showed acute ST elevation in leads II, II, and aVF, with reciprocal changes. The patient was diagnosed with an acute myocardial infarction (AMI). He denied any chest pain, shortness of breath, or diaphoresis. The SICU team initiated aspirin therapy and immediately contacted cardiology for an emergent coronary angiogram.
The angiogram and cardiac catheterization revealed an elevated left ventricular end diastolic (LVED) volume, inferior wall hypokinesis, a low-normal ejection fraction, and a 30% lesion in the first diagonal of his left anterior descending artery. A postprocedure echocardiogram demonstrated left ventricular (LV) ejection fraction of 50%, with LV hypokinesis in the inferior base and mild left atrial enlargement. The patient was started on metoprolol for myocardial protection and recovery.
Complications
Perioperative complications of FG, including AMI, must be considered due to the physiologic stress on the body.19 Most patients with perioperative AMI after noncardiac surgery do not experience ischemic symptoms.20
Growing evidence suggests the pivotal role of acute inflammation (postoperatively or from infection) as a precipitating event in AMI.20,21 Chemical mediators, such as inflammatory cytokines, endotoxins, and nitric oxide, may play a role in the development of an AMI.22
If cardiovascular disease and/or significant cardiovascular risk factors (ie, older age, male, cigarette smoking, cardiac family history, acute kidney injury) are present, the risk for AMI increases in the first two days following surgery.21,23 Acute infections and sepsis also initiate or increase systemic inflammatory activity via these same chemical mediators.21
Most suspected infectious agents also produce coronary artery sheer stress and destabilization of vulnerable plaques, leading to plaque rupture and thrombosis.19,24 Proinflammatory cytokines promote enhanced platelet activation and contribute to this thrombotic environment.21,23 Thrombus leads to obstructed coronary blood flow, myocardial ischemia, and finally, infarction.21
A reversible myocardial depression, cardiomyopathy, or myocardial ischemia may occur in patients with acute systemic infection or sepsis when the myocardium is functionally and structurally injured by these inflammatory chemical mediators.19,22-24 Characteristics of such a cardiomyopathy include left ventricle dilation with a low filling pressure, an abnormal increase in LVED volume, and a depressed ejection fraction.22
An acute infectious or septic process can raise troponin levels in 43% to 85% of patients.22,24 Troponin biomarkers can assist in predicting myocardial injury and events after surgery with nearly absolute myocardial tissue specificity.20 Cardiovascular involvement caused by myocardial injury–related sepsis is observed in up to 70% of patients in the ICU for these reasons.23 Therefore, providers should consider measuring troponin biomarkers during such infectious and septic processes, as this team did for the case patient. The providers were able to diagnose his AMI early and institute appropriate treatment measures to avoid extensive myocardial tissue damage.
Several studies have already demonstrated a correlation between pneumococcal pneumonia and an increased risk for AMI, and the same mechanisms are presumed responsible for any severe acute infectious state.21 More research is needed to understand the pathophysiology of AMI in sepsis and acute systemic infections.23
OUTCOME FOR THE CASE PATIENT
On postoperative day 2, the patient’s vital signs and lab results were normal. Additional lab results included an A1C of 5.2%. His ECG showed a resolving ST-elevation myocardial infarction (STEMI). The surgical wound had initiation of early granulation tissue without any further signs of necrosis.
A postoperative acute STEMI was unexpected in this patient, as his only risk factors included being male, mild hypertension, obesity, and tobacco use. At the time of his initial elevated troponin level, he had no cardiac symptoms or ECG changes. This initial high troponin level may have been stress-induced from the acute infectious process, and his acute inferior wall STEMI may have been secondary to a transient thrombotic event. The STEMI may then have resolved on its own during the cardiac catheterization with the administration of heparin, IV fluids, blood products, aspirin, or dye infiltration, thus enhancing reperfusion of the coronary artery system.
The final tissue culture showed MRSA. Given his job and his history of a genitourinary procedure, as well as the less fulminant form of disease and relatively quick recovery, it was likely HA-MRSA (rather than CA-MRSA). Only clindamycin was used for treatment.
The wound continued to have decreasing erythema, a reduction in tenderness, and evidence of viable, pink granulation tissue. HIV testing was not completed during his admission. The remainder of the patient’s hospital course was unremarkable, and he was discharged home with wound care, urology, and cardiology follow-up services.
CONCLUSION
Multiple factors contribute to a delayed or mistaken diagnosis of FG; it may be overlooked in the initial working diagnoses because of its low incidence and manifestations similar to those of other soft-tissue infections (eg, cellulitis, scrotal abscess). The cutaneous signs of FG often lag behind the disease manifestation, with minimal or no external presence while extensive internal tissue destruction is occurring. Constant review of symptoms is required when treating patients with soft-tissue infections, and early signs—such as pain out of proportion to physical findings—should prompt a clinician to include FG in the differential.
Early diagnosis with prompt debridement and antibiotic therapy are crucial to patient survival. Detecting FG within the first 24 hours is critical. Further differentiation between CA-MRSA and HA-MRSA can assist in patient recovery and survival by guiding appropriate antibiotic therapy. Perioperative risk assessment and serial troponin biomarkers may identify patients in need of intensive monitoring and management postoperatively to avoid an AMI, since patients may not experience ischemic symptoms.
1. Norton KS, Johnson LW, Perry T, et al. Management of Fournier’s gangrene: an eleven-year retrospective analysis of early recognition, diagnosis, and treatment. Am Surg. 2002;68(8):709-713.
2. Agostini T, Mori F, Perello R, et al. Successful combined approach to a severe Fournier’s gangrene. Indian J Plast Surg. 2014;47(1):132-136.
3. Cabrera G, March P. Fournier’s gangrene. Glendale, CA: Cinahl Information Systems; 2016.
4. Czymek R, Kujath P, Bruch HP, et al. Treatment, outcome and quality of life after Fournier’s gangrene: a multicentre study. Colorectal Dis. 2013;15(12):1529-1536.
5. Sugihara T, Yasunaga H, Horiguchi H, et al. Impact of surgical intervention timing on the case fatality rate for Fournier’s gangrene: an analysis of 379 cases. BJU Int. 2012;110(11c):E1096-1100.
6. Tuncel A, Keten T, Aslan Y, et al. Comparison of different scoring systems for outcome prediction in patients with Fournier’s gangrene: experience with 50 patients. Scand J Urol. 2014;48(4):393-399.
7. Taken K, Oncu MR, Ergun M, et al. Fournier’s gangrene: causes, presentation and survival of sixty-five patients. Pak J Med Sci. 2016;32(3):746-750.
8. Palvolgyi R, Kaji AH, Valeriano J, et al. Fournier’s gangrene: a model for early prediction. Am Surg. 2014;80(10):926-931.
9. Pais V, Santora T. Fournier gangrene. http://emedicine.medscape.com/article/2028899-overview. Accessed August 16, 2017.
10. Cottrill RR. A demonstration of clinical reasoning through a case of scrotal infection. Urol Nurs. 2013;33(1):33-37.
11. Summers A. Fournier’s gangrene. J Nurse Pract. 2014;10(8):582-587.
12. Miller LG, Perdreau-Remington F, Rieg G, et al. Necrotizing fasciitis caused by community-associated methicillin-resistant Staphylococcus aureus in Los Angeles. N Engl J Med. 2005;352(14):1445-1453.
13. Gupta N, Zinn K, Bansal I, Weinstein R. Fournier’s gangrene: ultrasound or computed tomography? A letter to the editor. Med Ultrason. 2014;16(4):389-390.
14. Bjurlin MA, O’Grady T, Kim DY, et al. Causative pathogens, antibiotic sensitivity, resistance patterns, and severity in a contemporary series of Fournier’s gangrene. Urol. 2013;81(4):752-758.
15. Goh T, Goh LG. Pitfalls in diagnosing necrotizing fasciitis. https://psnet.ahrq.gov/webmm/case/329/pitfalls-in-diagnos ing-necrotizing-fasciitis. Accessed August 16, 2017.
16. Kale P, Dhawan B. The changing face of community-acquired methicillin-resistant Staphylococcus aureus. Indian J Med Microbiol. 2016;34(3):275-285.
17. CDC. Necrotizing fasciitis. www.cdc.gov/Features/NecrotizingFasciitis/index.html. Accessed August 16, 2017.
18. Barnes BE, Sampson DA. A literature review on community-acquired methicillin-resistant Staphylococcus aureus in the United States: clinical information for primary care nurse practitioners. J Am Acad Nurse Pract. 2011;23(1):23-32.
19. Madjid M, Vela D, Khalili-Tabrizi H, et al. Systemic infections cause exaggerated local inflammation in atherosclerotic coronary arteries. Clues to the triggering effect of acute infections on acute coronary syndromes. Tex Heart Inst J. 2007;34(1):11-18.
20. Devereaux PJ, Chan MTV, Alonso-Coello PA, et al; VISION Study Investigators. Association between postoperative troponin levels and 30-day mortality among patients undergoing noncardiac surgery. JAMA. 2012;307(21):2295-2304.
21. Corrales-Medina VF, Fatemi O, Serpa J, et al. The association between Staphylococcus aureus bacteremia and acute myocardial infarction. Scand J Infect Dis. 2009;41(6-7):511-514.
22. Romero-Bermejo FJ, Ruiz-Bailen M, Gil-Cebrian J, Huertos-Ranchal MJ. Sepsis-induced cardiomyopathy. Curr Cardiol Rev. 2011;7(3):163-183.
23. Smilowitz NR, Gupta N, Guo Y, Bangalore S. Comparison of outcomes of patients with sepsis with versus without acute myocardial infarction and comparison of invasive versus noninvasive management of the patients with infarction. Am J Cardiol. 2016;117(7):1065-1071.
24. Mattson M. Sepsis and cardiac disease: improving outcomes through recognition and management. Prog Cardiovasc Nurs. 2009;24(4):199-201.
25. Papadakis MA, McPhee SJ. Current Medical Diagnosis & Treatment. 54th ed. New York, NY: McGraw Hill Education; 2015:137-138, 151-152, 937.
26. Eyre RC. Evaluation of the acute scrotum in adults. www.uptodate.com/contents/evaluation-of-the-acute-scrotum-in-adults. Accessed August 16, 2017.
1. Norton KS, Johnson LW, Perry T, et al. Management of Fournier’s gangrene: an eleven-year retrospective analysis of early recognition, diagnosis, and treatment. Am Surg. 2002;68(8):709-713.
2. Agostini T, Mori F, Perello R, et al. Successful combined approach to a severe Fournier’s gangrene. Indian J Plast Surg. 2014;47(1):132-136.
3. Cabrera G, March P. Fournier’s gangrene. Glendale, CA: Cinahl Information Systems; 2016.
4. Czymek R, Kujath P, Bruch HP, et al. Treatment, outcome and quality of life after Fournier’s gangrene: a multicentre study. Colorectal Dis. 2013;15(12):1529-1536.
5. Sugihara T, Yasunaga H, Horiguchi H, et al. Impact of surgical intervention timing on the case fatality rate for Fournier’s gangrene: an analysis of 379 cases. BJU Int. 2012;110(11c):E1096-1100.
6. Tuncel A, Keten T, Aslan Y, et al. Comparison of different scoring systems for outcome prediction in patients with Fournier’s gangrene: experience with 50 patients. Scand J Urol. 2014;48(4):393-399.
7. Taken K, Oncu MR, Ergun M, et al. Fournier’s gangrene: causes, presentation and survival of sixty-five patients. Pak J Med Sci. 2016;32(3):746-750.
8. Palvolgyi R, Kaji AH, Valeriano J, et al. Fournier’s gangrene: a model for early prediction. Am Surg. 2014;80(10):926-931.
9. Pais V, Santora T. Fournier gangrene. http://emedicine.medscape.com/article/2028899-overview. Accessed August 16, 2017.
10. Cottrill RR. A demonstration of clinical reasoning through a case of scrotal infection. Urol Nurs. 2013;33(1):33-37.
11. Summers A. Fournier’s gangrene. J Nurse Pract. 2014;10(8):582-587.
12. Miller LG, Perdreau-Remington F, Rieg G, et al. Necrotizing fasciitis caused by community-associated methicillin-resistant Staphylococcus aureus in Los Angeles. N Engl J Med. 2005;352(14):1445-1453.
13. Gupta N, Zinn K, Bansal I, Weinstein R. Fournier’s gangrene: ultrasound or computed tomography? A letter to the editor. Med Ultrason. 2014;16(4):389-390.
14. Bjurlin MA, O’Grady T, Kim DY, et al. Causative pathogens, antibiotic sensitivity, resistance patterns, and severity in a contemporary series of Fournier’s gangrene. Urol. 2013;81(4):752-758.
15. Goh T, Goh LG. Pitfalls in diagnosing necrotizing fasciitis. https://psnet.ahrq.gov/webmm/case/329/pitfalls-in-diagnos ing-necrotizing-fasciitis. Accessed August 16, 2017.
16. Kale P, Dhawan B. The changing face of community-acquired methicillin-resistant Staphylococcus aureus. Indian J Med Microbiol. 2016;34(3):275-285.
17. CDC. Necrotizing fasciitis. www.cdc.gov/Features/NecrotizingFasciitis/index.html. Accessed August 16, 2017.
18. Barnes BE, Sampson DA. A literature review on community-acquired methicillin-resistant Staphylococcus aureus in the United States: clinical information for primary care nurse practitioners. J Am Acad Nurse Pract. 2011;23(1):23-32.
19. Madjid M, Vela D, Khalili-Tabrizi H, et al. Systemic infections cause exaggerated local inflammation in atherosclerotic coronary arteries. Clues to the triggering effect of acute infections on acute coronary syndromes. Tex Heart Inst J. 2007;34(1):11-18.
20. Devereaux PJ, Chan MTV, Alonso-Coello PA, et al; VISION Study Investigators. Association between postoperative troponin levels and 30-day mortality among patients undergoing noncardiac surgery. JAMA. 2012;307(21):2295-2304.
21. Corrales-Medina VF, Fatemi O, Serpa J, et al. The association between Staphylococcus aureus bacteremia and acute myocardial infarction. Scand J Infect Dis. 2009;41(6-7):511-514.
22. Romero-Bermejo FJ, Ruiz-Bailen M, Gil-Cebrian J, Huertos-Ranchal MJ. Sepsis-induced cardiomyopathy. Curr Cardiol Rev. 2011;7(3):163-183.
23. Smilowitz NR, Gupta N, Guo Y, Bangalore S. Comparison of outcomes of patients with sepsis with versus without acute myocardial infarction and comparison of invasive versus noninvasive management of the patients with infarction. Am J Cardiol. 2016;117(7):1065-1071.
24. Mattson M. Sepsis and cardiac disease: improving outcomes through recognition and management. Prog Cardiovasc Nurs. 2009;24(4):199-201.
25. Papadakis MA, McPhee SJ. Current Medical Diagnosis & Treatment. 54th ed. New York, NY: McGraw Hill Education; 2015:137-138, 151-152, 937.
26. Eyre RC. Evaluation of the acute scrotum in adults. www.uptodate.com/contents/evaluation-of-the-acute-scrotum-in-adults. Accessed August 16, 2017.