User login
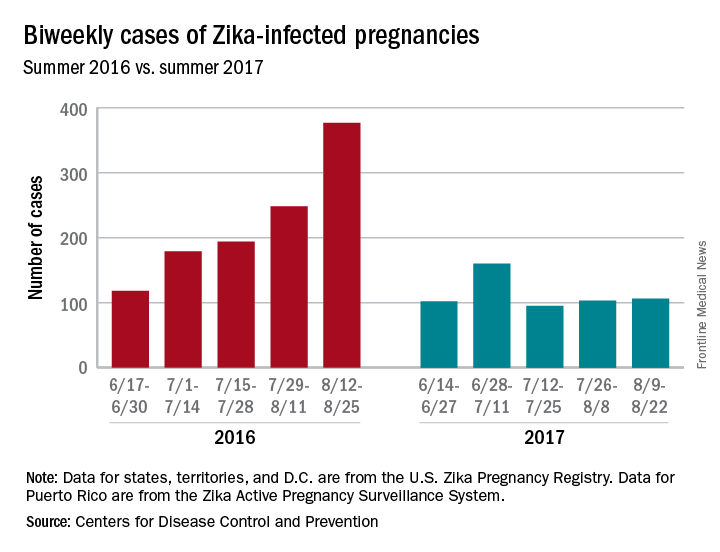
Zika’s 2017 summer less active than 2016
Zika may not have gone away this summer, but it didn’t make a comeback, either.
New cases in pregnant women are still being reported, but the numbers are much lower than a year ago, when the infection was kicking into high gear. For the 2 weeks ending Aug. 22, 106 pregnant women with laboratory evidence of Zika virus infection were reported: 43 in the U.S. states and the District of Columbia, and 63 in the U.S. territories, according to the Centers for Disease Control and Prevention.
The total cases reported for the previous 2-week periods, going back to mid-June, look like this: 102 (June 14-27), 160 (June 28–July 11), 95 (July 12-25), and 103 (July 26–Aug. 8). In the summer of 2016, the 2-week period of Aug. 12-25 produced 375 new reports of Zika-infected pregnant women, the CDC data show.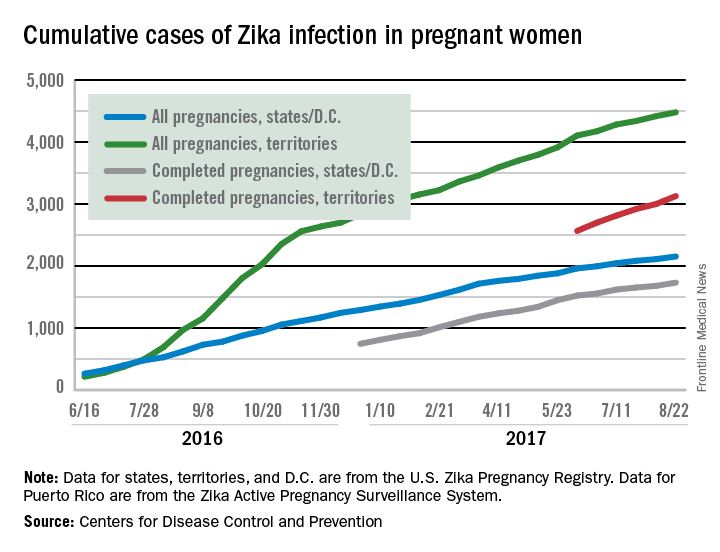
Zika may not have gone away this summer, but it didn’t make a comeback, either.
New cases in pregnant women are still being reported, but the numbers are much lower than a year ago, when the infection was kicking into high gear. For the 2 weeks ending Aug. 22, 106 pregnant women with laboratory evidence of Zika virus infection were reported: 43 in the U.S. states and the District of Columbia, and 63 in the U.S. territories, according to the Centers for Disease Control and Prevention.
The total cases reported for the previous 2-week periods, going back to mid-June, look like this: 102 (June 14-27), 160 (June 28–July 11), 95 (July 12-25), and 103 (July 26–Aug. 8). In the summer of 2016, the 2-week period of Aug. 12-25 produced 375 new reports of Zika-infected pregnant women, the CDC data show.
Zika may not have gone away this summer, but it didn’t make a comeback, either.
New cases in pregnant women are still being reported, but the numbers are much lower than a year ago, when the infection was kicking into high gear. For the 2 weeks ending Aug. 22, 106 pregnant women with laboratory evidence of Zika virus infection were reported: 43 in the U.S. states and the District of Columbia, and 63 in the U.S. territories, according to the Centers for Disease Control and Prevention.
The total cases reported for the previous 2-week periods, going back to mid-June, look like this: 102 (June 14-27), 160 (June 28–July 11), 95 (July 12-25), and 103 (July 26–Aug. 8). In the summer of 2016, the 2-week period of Aug. 12-25 produced 375 new reports of Zika-infected pregnant women, the CDC data show.
No obvious choice for treating pruritus in PBC patients
While multiple options exist for the treatment of pruritus in patients with primary biliary cholangitis (PBC), none have compelling evidence regarding their long-term efficacy and safety, according to a narrative review of literature by Hirsh D. Trivedi, MD, and associates.
There are four treatments commonly used for treating pruritus in PBC patients: bile acid–binding resins, rifampicin, opioid antagonists, and sertraline. In the cases of bile acid–binding resins, rifampicin, and opioid antagonists, significant side effects and a lack of proof of long-term efficacy prevent the treatments from standing out. Sertraline seems to have no significant side effects, but research is lacking, and further investigation is required.
Several experimental treatments for refractory pruritus also exist: These include phototherapy, plasmapheresis, albumin dialysis, nasobiliary drainage, ileal bile acid transporter–inhibitors, methotrexate and colchicine, and fibrates. In extreme cases, liver transplant can also be utilized to reduce pruritus symptoms.
“Our ongoing learning [about] this multifaceted symptom will hopefully lead to the development of more effective therapies and improve the quality of life for patients with primary biliary cholangitis,” the investigators concluded.
Find the full narrative review in the American Journal of Medicine (doi: 10.1016/j.amjmed.2017.01.037).
While multiple options exist for the treatment of pruritus in patients with primary biliary cholangitis (PBC), none have compelling evidence regarding their long-term efficacy and safety, according to a narrative review of literature by Hirsh D. Trivedi, MD, and associates.
There are four treatments commonly used for treating pruritus in PBC patients: bile acid–binding resins, rifampicin, opioid antagonists, and sertraline. In the cases of bile acid–binding resins, rifampicin, and opioid antagonists, significant side effects and a lack of proof of long-term efficacy prevent the treatments from standing out. Sertraline seems to have no significant side effects, but research is lacking, and further investigation is required.
Several experimental treatments for refractory pruritus also exist: These include phototherapy, plasmapheresis, albumin dialysis, nasobiliary drainage, ileal bile acid transporter–inhibitors, methotrexate and colchicine, and fibrates. In extreme cases, liver transplant can also be utilized to reduce pruritus symptoms.
“Our ongoing learning [about] this multifaceted symptom will hopefully lead to the development of more effective therapies and improve the quality of life for patients with primary biliary cholangitis,” the investigators concluded.
Find the full narrative review in the American Journal of Medicine (doi: 10.1016/j.amjmed.2017.01.037).
While multiple options exist for the treatment of pruritus in patients with primary biliary cholangitis (PBC), none have compelling evidence regarding their long-term efficacy and safety, according to a narrative review of literature by Hirsh D. Trivedi, MD, and associates.
There are four treatments commonly used for treating pruritus in PBC patients: bile acid–binding resins, rifampicin, opioid antagonists, and sertraline. In the cases of bile acid–binding resins, rifampicin, and opioid antagonists, significant side effects and a lack of proof of long-term efficacy prevent the treatments from standing out. Sertraline seems to have no significant side effects, but research is lacking, and further investigation is required.
Several experimental treatments for refractory pruritus also exist: These include phototherapy, plasmapheresis, albumin dialysis, nasobiliary drainage, ileal bile acid transporter–inhibitors, methotrexate and colchicine, and fibrates. In extreme cases, liver transplant can also be utilized to reduce pruritus symptoms.
“Our ongoing learning [about] this multifaceted symptom will hopefully lead to the development of more effective therapies and improve the quality of life for patients with primary biliary cholangitis,” the investigators concluded.
Find the full narrative review in the American Journal of Medicine (doi: 10.1016/j.amjmed.2017.01.037).
FROM THE AMERICAN JOURNAL OF MEDICINE
Vaccinate and consider tofacitinib monotherapy to prevent herpes zoster in RA
The results of two studies of tofacitinib treatment for rheumatoid arthritis offer evidence to support the use of the drug without concomitant conventional synthetic disease-modifying antirheumatic drugs in order to reduce the risk of risk of herpes zoster infection and the safety of starting the drug 2-3 weeks after administering live zoster vaccine.
The risk of herpes zoster infection was elevated among rheumatoid arthritis (RA) patients receiving tofacitinib (Xeljanz) with glucocorticoids, compared with those receiving tofacitinib monotherapy, according to an analysis of data from 19 phase 2, phase 3, and long-term extension studies of tofacitinib.

“Further, physicians should continue to consider shingles vaccination prior to starting tofacitinib or biologic therapy,” they wrote.
Dr. Winthrop is the first author of a separate phase 2 study that also appears in Arthritis & Rheumatology, which suggests that live zoster vaccine (LZV) is safe in RA patients who start tofacitinib 2-3 weeks after vaccination. The study also showed that varicella-zoster virus (VZV)-specific humoral and cell-mediated immune responses to LZV are similar in tofacitinib- and placebo-treated patients.
“Our study provides the first data with this vaccine in the RA setting and suggests that these patients, even while using nonbiologic DMARDs at the time of vaccination, are capable of mounting adequate immune responses to this vaccine. Further, our data suggest that the use of tofacitinib following VZV vaccination in the RA setting did not impact negatively the vaccine immunogenicity or the time course of the immune response to the vaccine,” he and his colleagues wrote (Arthritis Rheumatol. 2017 Aug 28. doi: 10.1002/art.40187).
Increased herpes zoster risk in combination therapy
In the first study, herpes zoster (HZ) was reported in 636 of 6,192 patients over a median follow-up of 3 years of tofacitinib exposure, for an incident rate (IR) of 4.0 per 100 patient-years. However, IRs varied by region, ranging from 2.4 in Eastern Europe to 8.0 and 8.4 in Japan and Korea, respectively.
Further, in phase 3 studies, the IRs varied by tofacitinib dose, background use of conventional csDMARDs, and baseline glucocorticoid use; the rates were lowest among patients on tofacitinib monotherapy at a dose of 5 mg twice daily (IR, 0.6), and highest in those on tofacitinib at 10 mg twice daily with csDMARDs and glucocorticoids (IR, 5.4), the investigators found.
Independent risk factors for HZ included age, glucocorticoid use, tofacitinib dose, and enrollment within Asia, they said.
“Shingles, or reactivation of varicella virus, is a common and potentially debilitating illness. Around one-third of the general population will develop HZ in their lifetime, and approximately 10% of these patients develop postherpetic neuralgia which can last months to years and cause significant pain and morbidity,” the investigators wrote, adding that RA patients are at 1.5- to 2-fold greater risk vs. similarly aged individuals in the general population.
RA itself and treatment with glucocorticoids are known to increase HZ risk, but recent data have suggested that Janus kinase inhibitors, such as tofacitinib, and tumor necrosis factor antagonists are also associated with a higher rate of HZ. Additionally, a theoretical risk exists with various csDMARDs, they said.
“Given the increased risk of HZ observed among patients with RA versus the general population and the risk associated with RA therapies, it is possible that risk of HZ may be further increased when such therapies are combined,” they wrote.
Indeed, the findings of the study demonstrate an increased risk of HZ with tofacitinib in combination with glucocorticoids vs. tofacitinib monotherapy.
Further research is necessary to understand why Japanese and Korean patients are at elevated risk, and to understand the mechanism for the effects of combination therapy on VZV reactivation, they concluded.
LZV immunogenicity holds up during tofacitinib treatment
In the second study, 112 patients aged 50 years and older with active RA on background methotrexate received LZV and were then randomized to receive 5 mg tofacitinib twice daily or placebo 2-3 weeks after vaccination.
At 6 weeks after vaccination, VZV-specific IgG geometric mean fold rise (GMFR) was 2.11 and 1.74 in the tofacitinib and placebo patients, respectively; at all postvaccination time points at which VZV-specific IgG levels were evaluated, there was a trend toward numerically higher GMFR in tofacitinib patients, but the differences were not statistically significant. Also, the proportion of patients developing a 1.5-fold or greater postvaccination rise in IgG levels at 6 weeks trended higher for tofacitinib (57.4% vs. 43.4% with placebo).
VZV-specific T-cell GMFR at 6 weeks increased similarly in the groups (1.50 with tofacitinib and 1.29 with placebo).
Serious adverse events occurred in three patients in the tofacitinib group (5.5%) and in none of the placebo patients.
“The three SAEs included one case each of cholangitis and bronchitis, and once case of disseminated primary varicella,” the investigators said, noting that the onset of the latter was 16 days postvaccination, 2 days after starting tofacitinib. The rash resolved with discontinuation of tofacitinib and treatment with valacyclovir for 7 days.
The findings suggest that patients with active RA develop robust immune responses to HZ vaccine, and that starting tofacitinib 2-3 weeks after vaccination has no negative impact on the established immune response.
“Importantly, while our results suggest the vaccine is safe for patients with RA with prior VZV exposure, they also indicate the potential need to either screen for prior exposure before giving this vaccine or waiting longer than 2-3 weeks before starting immunosuppression with tofacitinib,” they said, noting that the current data suggest 4 weeks might be preferable.
Alternatively, testing patients who don’t recollect a history of chickenpox to ensure prior VZV exposure prior to vaccination could also mitigate the risk, they said.
“Further research is necessary to understand the risk of this complication, as well as the long-term effectiveness of this vaccine to prevent HZ in this high-risk population,” they concluded.
Both studies were sponsored by Pfizer. Dr. Winthrop has received research grants from and served as a scientific consultant to Pfizer. Other authors also reported financial relationships with Pfizer.
At a symposium during the 2015 annual meeting of the American College of Rheumatology, William Schaffner, MD, highlighted the connection between the seriousness of an infection, and the respect one has for the solution.
Dr. Schaffner said that “if you don’t fear the infection, you won’t value the solution.” Herpes zoster (HZ) should be feared, and the solution – the live zoster vaccine – valued.

Live zoster vaccine (LZV) was approved in 2006 on the basis of a trial involving more than 38,500 adults over age 60 years, which showed a 51% HZ prevention rate (64% protection in the 60-69 year age group) and a two-thirds reduction in postherpetic neuralgia. Complications and disseminated infection were rare.
HZ vaccination should be offered regardless of a history of varicella infection or prior shingles, as HZ may recur.
There is an imperative need to know who is at risk, when and how they should be vaccinated, and what other risk reduction measures should be considered.
John J. Cush, MD, is director of clinical rheumatology at Baylor Scott & White Research Institute and professor of medicine and rheumatology at Baylor University Medical Center, both in Dallas. His comments are taken from his editorial accompanying the two studies by Winthrop et al. (Arthritis Rheumatol. 2017 Aug 28. doi: 10.1002/art.40188).
At a symposium during the 2015 annual meeting of the American College of Rheumatology, William Schaffner, MD, highlighted the connection between the seriousness of an infection, and the respect one has for the solution.
Dr. Schaffner said that “if you don’t fear the infection, you won’t value the solution.” Herpes zoster (HZ) should be feared, and the solution – the live zoster vaccine – valued.
Live zoster vaccine (LZV) was approved in 2006 on the basis of a trial involving more than 38,500 adults over age 60 years, which showed a 51% HZ prevention rate (64% protection in the 60-69 year age group) and a two-thirds reduction in postherpetic neuralgia. Complications and disseminated infection were rare.
HZ vaccination should be offered regardless of a history of varicella infection or prior shingles, as HZ may recur.
There is an imperative need to know who is at risk, when and how they should be vaccinated, and what other risk reduction measures should be considered.
John J. Cush, MD, is director of clinical rheumatology at Baylor Scott & White Research Institute and professor of medicine and rheumatology at Baylor University Medical Center, both in Dallas. His comments are taken from his editorial accompanying the two studies by Winthrop et al. (Arthritis Rheumatol. 2017 Aug 28. doi: 10.1002/art.40188).
At a symposium during the 2015 annual meeting of the American College of Rheumatology, William Schaffner, MD, highlighted the connection between the seriousness of an infection, and the respect one has for the solution.
Dr. Schaffner said that “if you don’t fear the infection, you won’t value the solution.” Herpes zoster (HZ) should be feared, and the solution – the live zoster vaccine – valued.
Live zoster vaccine (LZV) was approved in 2006 on the basis of a trial involving more than 38,500 adults over age 60 years, which showed a 51% HZ prevention rate (64% protection in the 60-69 year age group) and a two-thirds reduction in postherpetic neuralgia. Complications and disseminated infection were rare.
HZ vaccination should be offered regardless of a history of varicella infection or prior shingles, as HZ may recur.
There is an imperative need to know who is at risk, when and how they should be vaccinated, and what other risk reduction measures should be considered.
John J. Cush, MD, is director of clinical rheumatology at Baylor Scott & White Research Institute and professor of medicine and rheumatology at Baylor University Medical Center, both in Dallas. His comments are taken from his editorial accompanying the two studies by Winthrop et al. (Arthritis Rheumatol. 2017 Aug 28. doi: 10.1002/art.40188).
The results of two studies of tofacitinib treatment for rheumatoid arthritis offer evidence to support the use of the drug without concomitant conventional synthetic disease-modifying antirheumatic drugs in order to reduce the risk of risk of herpes zoster infection and the safety of starting the drug 2-3 weeks after administering live zoster vaccine.
The risk of herpes zoster infection was elevated among rheumatoid arthritis (RA) patients receiving tofacitinib (Xeljanz) with glucocorticoids, compared with those receiving tofacitinib monotherapy, according to an analysis of data from 19 phase 2, phase 3, and long-term extension studies of tofacitinib.
“Further, physicians should continue to consider shingles vaccination prior to starting tofacitinib or biologic therapy,” they wrote.
Dr. Winthrop is the first author of a separate phase 2 study that also appears in Arthritis & Rheumatology, which suggests that live zoster vaccine (LZV) is safe in RA patients who start tofacitinib 2-3 weeks after vaccination. The study also showed that varicella-zoster virus (VZV)-specific humoral and cell-mediated immune responses to LZV are similar in tofacitinib- and placebo-treated patients.
“Our study provides the first data with this vaccine in the RA setting and suggests that these patients, even while using nonbiologic DMARDs at the time of vaccination, are capable of mounting adequate immune responses to this vaccine. Further, our data suggest that the use of tofacitinib following VZV vaccination in the RA setting did not impact negatively the vaccine immunogenicity or the time course of the immune response to the vaccine,” he and his colleagues wrote (Arthritis Rheumatol. 2017 Aug 28. doi: 10.1002/art.40187).
Increased herpes zoster risk in combination therapy
In the first study, herpes zoster (HZ) was reported in 636 of 6,192 patients over a median follow-up of 3 years of tofacitinib exposure, for an incident rate (IR) of 4.0 per 100 patient-years. However, IRs varied by region, ranging from 2.4 in Eastern Europe to 8.0 and 8.4 in Japan and Korea, respectively.
Further, in phase 3 studies, the IRs varied by tofacitinib dose, background use of conventional csDMARDs, and baseline glucocorticoid use; the rates were lowest among patients on tofacitinib monotherapy at a dose of 5 mg twice daily (IR, 0.6), and highest in those on tofacitinib at 10 mg twice daily with csDMARDs and glucocorticoids (IR, 5.4), the investigators found.
Independent risk factors for HZ included age, glucocorticoid use, tofacitinib dose, and enrollment within Asia, they said.
“Shingles, or reactivation of varicella virus, is a common and potentially debilitating illness. Around one-third of the general population will develop HZ in their lifetime, and approximately 10% of these patients develop postherpetic neuralgia which can last months to years and cause significant pain and morbidity,” the investigators wrote, adding that RA patients are at 1.5- to 2-fold greater risk vs. similarly aged individuals in the general population.
RA itself and treatment with glucocorticoids are known to increase HZ risk, but recent data have suggested that Janus kinase inhibitors, such as tofacitinib, and tumor necrosis factor antagonists are also associated with a higher rate of HZ. Additionally, a theoretical risk exists with various csDMARDs, they said.
“Given the increased risk of HZ observed among patients with RA versus the general population and the risk associated with RA therapies, it is possible that risk of HZ may be further increased when such therapies are combined,” they wrote.
Indeed, the findings of the study demonstrate an increased risk of HZ with tofacitinib in combination with glucocorticoids vs. tofacitinib monotherapy.
Further research is necessary to understand why Japanese and Korean patients are at elevated risk, and to understand the mechanism for the effects of combination therapy on VZV reactivation, they concluded.
LZV immunogenicity holds up during tofacitinib treatment
In the second study, 112 patients aged 50 years and older with active RA on background methotrexate received LZV and were then randomized to receive 5 mg tofacitinib twice daily or placebo 2-3 weeks after vaccination.
At 6 weeks after vaccination, VZV-specific IgG geometric mean fold rise (GMFR) was 2.11 and 1.74 in the tofacitinib and placebo patients, respectively; at all postvaccination time points at which VZV-specific IgG levels were evaluated, there was a trend toward numerically higher GMFR in tofacitinib patients, but the differences were not statistically significant. Also, the proportion of patients developing a 1.5-fold or greater postvaccination rise in IgG levels at 6 weeks trended higher for tofacitinib (57.4% vs. 43.4% with placebo).
VZV-specific T-cell GMFR at 6 weeks increased similarly in the groups (1.50 with tofacitinib and 1.29 with placebo).
Serious adverse events occurred in three patients in the tofacitinib group (5.5%) and in none of the placebo patients.
“The three SAEs included one case each of cholangitis and bronchitis, and once case of disseminated primary varicella,” the investigators said, noting that the onset of the latter was 16 days postvaccination, 2 days after starting tofacitinib. The rash resolved with discontinuation of tofacitinib and treatment with valacyclovir for 7 days.
The findings suggest that patients with active RA develop robust immune responses to HZ vaccine, and that starting tofacitinib 2-3 weeks after vaccination has no negative impact on the established immune response.
“Importantly, while our results suggest the vaccine is safe for patients with RA with prior VZV exposure, they also indicate the potential need to either screen for prior exposure before giving this vaccine or waiting longer than 2-3 weeks before starting immunosuppression with tofacitinib,” they said, noting that the current data suggest 4 weeks might be preferable.
Alternatively, testing patients who don’t recollect a history of chickenpox to ensure prior VZV exposure prior to vaccination could also mitigate the risk, they said.
“Further research is necessary to understand the risk of this complication, as well as the long-term effectiveness of this vaccine to prevent HZ in this high-risk population,” they concluded.
Both studies were sponsored by Pfizer. Dr. Winthrop has received research grants from and served as a scientific consultant to Pfizer. Other authors also reported financial relationships with Pfizer.
The results of two studies of tofacitinib treatment for rheumatoid arthritis offer evidence to support the use of the drug without concomitant conventional synthetic disease-modifying antirheumatic drugs in order to reduce the risk of risk of herpes zoster infection and the safety of starting the drug 2-3 weeks after administering live zoster vaccine.
The risk of herpes zoster infection was elevated among rheumatoid arthritis (RA) patients receiving tofacitinib (Xeljanz) with glucocorticoids, compared with those receiving tofacitinib monotherapy, according to an analysis of data from 19 phase 2, phase 3, and long-term extension studies of tofacitinib.
“Further, physicians should continue to consider shingles vaccination prior to starting tofacitinib or biologic therapy,” they wrote.
Dr. Winthrop is the first author of a separate phase 2 study that also appears in Arthritis & Rheumatology, which suggests that live zoster vaccine (LZV) is safe in RA patients who start tofacitinib 2-3 weeks after vaccination. The study also showed that varicella-zoster virus (VZV)-specific humoral and cell-mediated immune responses to LZV are similar in tofacitinib- and placebo-treated patients.
“Our study provides the first data with this vaccine in the RA setting and suggests that these patients, even while using nonbiologic DMARDs at the time of vaccination, are capable of mounting adequate immune responses to this vaccine. Further, our data suggest that the use of tofacitinib following VZV vaccination in the RA setting did not impact negatively the vaccine immunogenicity or the time course of the immune response to the vaccine,” he and his colleagues wrote (Arthritis Rheumatol. 2017 Aug 28. doi: 10.1002/art.40187).
Increased herpes zoster risk in combination therapy
In the first study, herpes zoster (HZ) was reported in 636 of 6,192 patients over a median follow-up of 3 years of tofacitinib exposure, for an incident rate (IR) of 4.0 per 100 patient-years. However, IRs varied by region, ranging from 2.4 in Eastern Europe to 8.0 and 8.4 in Japan and Korea, respectively.
Further, in phase 3 studies, the IRs varied by tofacitinib dose, background use of conventional csDMARDs, and baseline glucocorticoid use; the rates were lowest among patients on tofacitinib monotherapy at a dose of 5 mg twice daily (IR, 0.6), and highest in those on tofacitinib at 10 mg twice daily with csDMARDs and glucocorticoids (IR, 5.4), the investigators found.
Independent risk factors for HZ included age, glucocorticoid use, tofacitinib dose, and enrollment within Asia, they said.
“Shingles, or reactivation of varicella virus, is a common and potentially debilitating illness. Around one-third of the general population will develop HZ in their lifetime, and approximately 10% of these patients develop postherpetic neuralgia which can last months to years and cause significant pain and morbidity,” the investigators wrote, adding that RA patients are at 1.5- to 2-fold greater risk vs. similarly aged individuals in the general population.
RA itself and treatment with glucocorticoids are known to increase HZ risk, but recent data have suggested that Janus kinase inhibitors, such as tofacitinib, and tumor necrosis factor antagonists are also associated with a higher rate of HZ. Additionally, a theoretical risk exists with various csDMARDs, they said.
“Given the increased risk of HZ observed among patients with RA versus the general population and the risk associated with RA therapies, it is possible that risk of HZ may be further increased when such therapies are combined,” they wrote.
Indeed, the findings of the study demonstrate an increased risk of HZ with tofacitinib in combination with glucocorticoids vs. tofacitinib monotherapy.
Further research is necessary to understand why Japanese and Korean patients are at elevated risk, and to understand the mechanism for the effects of combination therapy on VZV reactivation, they concluded.
LZV immunogenicity holds up during tofacitinib treatment
In the second study, 112 patients aged 50 years and older with active RA on background methotrexate received LZV and were then randomized to receive 5 mg tofacitinib twice daily or placebo 2-3 weeks after vaccination.
At 6 weeks after vaccination, VZV-specific IgG geometric mean fold rise (GMFR) was 2.11 and 1.74 in the tofacitinib and placebo patients, respectively; at all postvaccination time points at which VZV-specific IgG levels were evaluated, there was a trend toward numerically higher GMFR in tofacitinib patients, but the differences were not statistically significant. Also, the proportion of patients developing a 1.5-fold or greater postvaccination rise in IgG levels at 6 weeks trended higher for tofacitinib (57.4% vs. 43.4% with placebo).
VZV-specific T-cell GMFR at 6 weeks increased similarly in the groups (1.50 with tofacitinib and 1.29 with placebo).
Serious adverse events occurred in three patients in the tofacitinib group (5.5%) and in none of the placebo patients.
“The three SAEs included one case each of cholangitis and bronchitis, and once case of disseminated primary varicella,” the investigators said, noting that the onset of the latter was 16 days postvaccination, 2 days after starting tofacitinib. The rash resolved with discontinuation of tofacitinib and treatment with valacyclovir for 7 days.
The findings suggest that patients with active RA develop robust immune responses to HZ vaccine, and that starting tofacitinib 2-3 weeks after vaccination has no negative impact on the established immune response.
“Importantly, while our results suggest the vaccine is safe for patients with RA with prior VZV exposure, they also indicate the potential need to either screen for prior exposure before giving this vaccine or waiting longer than 2-3 weeks before starting immunosuppression with tofacitinib,” they said, noting that the current data suggest 4 weeks might be preferable.
Alternatively, testing patients who don’t recollect a history of chickenpox to ensure prior VZV exposure prior to vaccination could also mitigate the risk, they said.
“Further research is necessary to understand the risk of this complication, as well as the long-term effectiveness of this vaccine to prevent HZ in this high-risk population,” they concluded.
Both studies were sponsored by Pfizer. Dr. Winthrop has received research grants from and served as a scientific consultant to Pfizer. Other authors also reported financial relationships with Pfizer.
FROM ARTHRITIS & RHEUMATOLOGY
Key clinical point:
Major finding: HZ rates were lowest among patients on tofacitinib monotherapy at a dose of 5 mg twice daily (IR, 0.6).
Data source: A phase 2 trial of 112 patients, and a review of 19 studies involving 6,192 patients.
Disclosures: Both studies were sponsored by Pfizer. Dr. Winthrop has received research grants from and served as a scientific consultant to Pfizer. Other authors also reported financial relationships with Pfizer.
Heart failure guidelines updated
Clinical Question: What new evidence is available to guide heart failure (HF) management?
Background: New data has become available since the 2013 HF guidelines.
Study Design: A focused update.
Setting: Ongoing review of HF literature.

Synopsis: Beta-natriuretic peptide (BNP) is recommended to screen at risk patients (IIaB), on admission (IA), and prior to discharge (IIaB). The combination of ARB and neprilysin inhibitor (ARB-NI) is recommended in symptomatic patients with HF with reduced ejection fraction (HFrEF) who are tolerant of ACE inhibition (IB). For these patients, transitioning from ACE-inhibitor to the ARB-NI combination, valsartan-sacubitril significantly reduced hospitalization and mortality. Optimal dose and titration strategies remain unclear. ARB-NIs should not be used in patients with a history of angioedema (IIIC) or within 36 hours of receiving ACE-inhibitors (IIIB). Ivabradine, a selective inhibitor of the If current in the sinoatrial node, is recommended to reduce hospitalizations for patients with HFrEF with stable symptoms with resting sinus heart rate greater than or equal to 70 despite maximally-tolerated beta-blockade (IIaB). Intravenous iron replacement is recommended to improve function and quality of life for patients with symptomatic HF and iron deficiency (IIbB).
Bottom Line: Updates support use of BNP, ARB-NIs, ivabradine, and IV iron for HFrEF.
Citation: Yancy CW, et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: A report of the American college of cardiology/American heart association task force on clinical practice guidelines and the heart failure society of America. Published online, 2017 Apr 28. Circulation. doi: 10.1161/CIR.0000000000000509.
Dr. Sweigart is an assistant professor in the University of Kentucky division of hospital medicine and Lexington VA Medical Center.
Clinical Question: What new evidence is available to guide heart failure (HF) management?
Background: New data has become available since the 2013 HF guidelines.
Study Design: A focused update.
Setting: Ongoing review of HF literature.
Synopsis: Beta-natriuretic peptide (BNP) is recommended to screen at risk patients (IIaB), on admission (IA), and prior to discharge (IIaB). The combination of ARB and neprilysin inhibitor (ARB-NI) is recommended in symptomatic patients with HF with reduced ejection fraction (HFrEF) who are tolerant of ACE inhibition (IB). For these patients, transitioning from ACE-inhibitor to the ARB-NI combination, valsartan-sacubitril significantly reduced hospitalization and mortality. Optimal dose and titration strategies remain unclear. ARB-NIs should not be used in patients with a history of angioedema (IIIC) or within 36 hours of receiving ACE-inhibitors (IIIB). Ivabradine, a selective inhibitor of the If current in the sinoatrial node, is recommended to reduce hospitalizations for patients with HFrEF with stable symptoms with resting sinus heart rate greater than or equal to 70 despite maximally-tolerated beta-blockade (IIaB). Intravenous iron replacement is recommended to improve function and quality of life for patients with symptomatic HF and iron deficiency (IIbB).
Bottom Line: Updates support use of BNP, ARB-NIs, ivabradine, and IV iron for HFrEF.
Citation: Yancy CW, et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: A report of the American college of cardiology/American heart association task force on clinical practice guidelines and the heart failure society of America. Published online, 2017 Apr 28. Circulation. doi: 10.1161/CIR.0000000000000509.
Dr. Sweigart is an assistant professor in the University of Kentucky division of hospital medicine and Lexington VA Medical Center.
Clinical Question: What new evidence is available to guide heart failure (HF) management?
Background: New data has become available since the 2013 HF guidelines.
Study Design: A focused update.
Setting: Ongoing review of HF literature.
Synopsis: Beta-natriuretic peptide (BNP) is recommended to screen at risk patients (IIaB), on admission (IA), and prior to discharge (IIaB). The combination of ARB and neprilysin inhibitor (ARB-NI) is recommended in symptomatic patients with HF with reduced ejection fraction (HFrEF) who are tolerant of ACE inhibition (IB). For these patients, transitioning from ACE-inhibitor to the ARB-NI combination, valsartan-sacubitril significantly reduced hospitalization and mortality. Optimal dose and titration strategies remain unclear. ARB-NIs should not be used in patients with a history of angioedema (IIIC) or within 36 hours of receiving ACE-inhibitors (IIIB). Ivabradine, a selective inhibitor of the If current in the sinoatrial node, is recommended to reduce hospitalizations for patients with HFrEF with stable symptoms with resting sinus heart rate greater than or equal to 70 despite maximally-tolerated beta-blockade (IIaB). Intravenous iron replacement is recommended to improve function and quality of life for patients with symptomatic HF and iron deficiency (IIbB).
Bottom Line: Updates support use of BNP, ARB-NIs, ivabradine, and IV iron for HFrEF.
Citation: Yancy CW, et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: A report of the American college of cardiology/American heart association task force on clinical practice guidelines and the heart failure society of America. Published online, 2017 Apr 28. Circulation. doi: 10.1161/CIR.0000000000000509.
Dr. Sweigart is an assistant professor in the University of Kentucky division of hospital medicine and Lexington VA Medical Center.
Study linked H2 receptor antagonists, but not PPIs, to dementia
A large prospective study of middle-aged and older women found no convincing evidence that using proton pump inhibitors increased their risk of dementia, investigators reported.
However, using H2 receptor antagonists for at least 9 years was associated with a slight decrease in scores of learning and working memory (mean decrease, –0.2; 95% confidence interval, –0.3 to –0.08; P less than .001), Paul Lochhead, MBChB, PhD, and his associates wrote in the October issue of Gastroenterology (doi: 10.1053/j.gastro.2017.06.061). “Since our primary hypothesis related to PPI [proton pump inhibitor] use, our findings for [H2 receptor antagonists] should be interpreted with caution,” they said.
Source: American Gastroenterological Association
In a recent German study of a medical claims database, use of PPIs was associated with a 44% increase in the likelihood of incident dementia (JAMA Neurol. 2016;73:410-6). “The existence of a causal mechanism linking PPI use to dementia is suggested by observations from cellular and animal models of Alzheimer’s disease, where PPI exposure appears to influence amyloid-beta metabolism,” Dr. Lochhead and his associates wrote. “However, other preclinical data on PPIs and Alzheimer’s disease are conflicting.” Noting that cognitive function predicts dementia later in life, they analyzed prospective data on medications and other potential risk factors from 13,864 participants in the Nurses’ Health Study II who had completed Cogstate, a computerized, self-administered neuropsychological battery.
Study participants averaged 61 years old when they underwent cognitive testing, ranging in age from 50 to 70 years. Users of PPIs tended to be older, had more comorbidities, were less physically active, had higher body mass indexes, had less education, and ate a lower-quality diet than women who did not use PPIs. After adjusting for such confounders, using PPIs for 9-14 years was associated with a modest decrease in scores for psychomotor speed and attention (mean score difference, compared with never users, –0.06; 95% CI, –0.11 to 0.00; P = .03). “For comparison, in multivariable models, a 1-year increase in age was associated with mean score decreases of 0.03 for psychomotor speed and attention, 0.02 for learning and working memory, and 0.03 for overall cognition,” the researchers wrote.
Next, they examined links between use of H2 receptor antagonists and cognitive scores among 10,778 study participants who had used PPIs for 2 years or less. Use of H2 receptor antagonists for 9-14 years predicted poorer scores on learning, working memory, and overall cognition, even after controlling for potential confounders (P less than or equal to .002). “The magnitudes of mean score differences were larger than those observed in the analysis of PPI use, particularly for learning and working memory,” the researchers noted. Additionally, PPI use did not predict lower cognitive scores among individuals who had never used H2 receptor antagonists.
On the other hand, using PPIs for 9-14 years was associated with the equivalent of about 2 years of age-related cognitive decline, and controlling for exposure to H2 receptor antagonists weakened even this modest effect, the investigators said. Users and nonusers of PPIs tend to differ on many measures, and analyses of claims data, such as the German study above, are less able to account for these potential confounders, they noted. “Nonjudicious PPI prescribing is especially frequent among the elderly and those with cognitive impairment,” they added. “Therefore, elderly individuals who have frequent contact with health providers are at increased risk of both PPI prescription and dementia diagnosis. This bias may not be completely mitigated by adjustment for comorbidities or polypharmacy.”
The findings regarding H2 receptor antagonists reflect those of three smaller cohort studies, and these medications are known to cause central nervous system effects in the elderly, including delirium, the researchers said. Ranitidine and cimetidine have anticholinergic effects that also could “pose a risk for adverse cognitive effects with long-term use.”
Dr. Lochhead reported having no conflicts. Two coinvestigators disclosed ties to Bayer Healthcare, Pfizer, Aralez Pharmaceuticals, AbbVie, Samsung Bioepis, and Takeda.
Numerous possible PPI-related adverse events have been reported within the past few years; some resultant media attention has caused anxiety for patients. Dementia is a dreaded diagnosis. Therefore, initial reports that PPI treatment might be associated with an increased risk of dementia attracted considerable media attention, much of which was unbalanced and uninformed. There is no obvious biological rationale for such an association, and the risks reported initially were of small magnitude (for example, hazard ratios of approximately 1.4). However, patients cannot reliably assess levels of risk from media coverage that often veers toward sensationalism.

The study by Lochhead et al. is a welcome contribution to the topic of PPI safety. Using the Nurses’ Health Study II database, the investigators measured cognitive function in a large group of female PPI users and nonusers. Unsurprisingly, PPI users were older and sicker than nonusers. There were quantitatively small changes in some measures of cognitive function among PPI users. However, learning and working memory scores, which are more predictive of Alzheimer’s-type cognitive decline, were unaffected by PPI use.
For those prescribers with residual concerns about any association between PPIs and dementia, these prospectively collected data on cognitive function should provide further reassurance. It is appropriate that this study should have been highlighted in GI & Hepatology News, but since it lacks the potential sensationalism of studies that report a putative association, we should not expect it to be discussed on the TV evening news anytime soon!
Colin W. Howden, MD, AGAF, is chief of gastroenterology at University of Tennessee Health Science Center, Memphis. He has been a consultant, investigator, and/or speaker for all PPI manufacturers at some time. He is currently a consultant for Takeda, Aralez, and Pfizer Consumer Health.
Numerous possible PPI-related adverse events have been reported within the past few years; some resultant media attention has caused anxiety for patients. Dementia is a dreaded diagnosis. Therefore, initial reports that PPI treatment might be associated with an increased risk of dementia attracted considerable media attention, much of which was unbalanced and uninformed. There is no obvious biological rationale for such an association, and the risks reported initially were of small magnitude (for example, hazard ratios of approximately 1.4). However, patients cannot reliably assess levels of risk from media coverage that often veers toward sensationalism.
The study by Lochhead et al. is a welcome contribution to the topic of PPI safety. Using the Nurses’ Health Study II database, the investigators measured cognitive function in a large group of female PPI users and nonusers. Unsurprisingly, PPI users were older and sicker than nonusers. There were quantitatively small changes in some measures of cognitive function among PPI users. However, learning and working memory scores, which are more predictive of Alzheimer’s-type cognitive decline, were unaffected by PPI use.
For those prescribers with residual concerns about any association between PPIs and dementia, these prospectively collected data on cognitive function should provide further reassurance. It is appropriate that this study should have been highlighted in GI & Hepatology News, but since it lacks the potential sensationalism of studies that report a putative association, we should not expect it to be discussed on the TV evening news anytime soon!
Colin W. Howden, MD, AGAF, is chief of gastroenterology at University of Tennessee Health Science Center, Memphis. He has been a consultant, investigator, and/or speaker for all PPI manufacturers at some time. He is currently a consultant for Takeda, Aralez, and Pfizer Consumer Health.
Numerous possible PPI-related adverse events have been reported within the past few years; some resultant media attention has caused anxiety for patients. Dementia is a dreaded diagnosis. Therefore, initial reports that PPI treatment might be associated with an increased risk of dementia attracted considerable media attention, much of which was unbalanced and uninformed. There is no obvious biological rationale for such an association, and the risks reported initially were of small magnitude (for example, hazard ratios of approximately 1.4). However, patients cannot reliably assess levels of risk from media coverage that often veers toward sensationalism.
The study by Lochhead et al. is a welcome contribution to the topic of PPI safety. Using the Nurses’ Health Study II database, the investigators measured cognitive function in a large group of female PPI users and nonusers. Unsurprisingly, PPI users were older and sicker than nonusers. There were quantitatively small changes in some measures of cognitive function among PPI users. However, learning and working memory scores, which are more predictive of Alzheimer’s-type cognitive decline, were unaffected by PPI use.
For those prescribers with residual concerns about any association between PPIs and dementia, these prospectively collected data on cognitive function should provide further reassurance. It is appropriate that this study should have been highlighted in GI & Hepatology News, but since it lacks the potential sensationalism of studies that report a putative association, we should not expect it to be discussed on the TV evening news anytime soon!
Colin W. Howden, MD, AGAF, is chief of gastroenterology at University of Tennessee Health Science Center, Memphis. He has been a consultant, investigator, and/or speaker for all PPI manufacturers at some time. He is currently a consultant for Takeda, Aralez, and Pfizer Consumer Health.
A large prospective study of middle-aged and older women found no convincing evidence that using proton pump inhibitors increased their risk of dementia, investigators reported.
However, using H2 receptor antagonists for at least 9 years was associated with a slight decrease in scores of learning and working memory (mean decrease, –0.2; 95% confidence interval, –0.3 to –0.08; P less than .001), Paul Lochhead, MBChB, PhD, and his associates wrote in the October issue of Gastroenterology (doi: 10.1053/j.gastro.2017.06.061). “Since our primary hypothesis related to PPI [proton pump inhibitor] use, our findings for [H2 receptor antagonists] should be interpreted with caution,” they said.
Source: American Gastroenterological Association
In a recent German study of a medical claims database, use of PPIs was associated with a 44% increase in the likelihood of incident dementia (JAMA Neurol. 2016;73:410-6). “The existence of a causal mechanism linking PPI use to dementia is suggested by observations from cellular and animal models of Alzheimer’s disease, where PPI exposure appears to influence amyloid-beta metabolism,” Dr. Lochhead and his associates wrote. “However, other preclinical data on PPIs and Alzheimer’s disease are conflicting.” Noting that cognitive function predicts dementia later in life, they analyzed prospective data on medications and other potential risk factors from 13,864 participants in the Nurses’ Health Study II who had completed Cogstate, a computerized, self-administered neuropsychological battery.
Study participants averaged 61 years old when they underwent cognitive testing, ranging in age from 50 to 70 years. Users of PPIs tended to be older, had more comorbidities, were less physically active, had higher body mass indexes, had less education, and ate a lower-quality diet than women who did not use PPIs. After adjusting for such confounders, using PPIs for 9-14 years was associated with a modest decrease in scores for psychomotor speed and attention (mean score difference, compared with never users, –0.06; 95% CI, –0.11 to 0.00; P = .03). “For comparison, in multivariable models, a 1-year increase in age was associated with mean score decreases of 0.03 for psychomotor speed and attention, 0.02 for learning and working memory, and 0.03 for overall cognition,” the researchers wrote.
Next, they examined links between use of H2 receptor antagonists and cognitive scores among 10,778 study participants who had used PPIs for 2 years or less. Use of H2 receptor antagonists for 9-14 years predicted poorer scores on learning, working memory, and overall cognition, even after controlling for potential confounders (P less than or equal to .002). “The magnitudes of mean score differences were larger than those observed in the analysis of PPI use, particularly for learning and working memory,” the researchers noted. Additionally, PPI use did not predict lower cognitive scores among individuals who had never used H2 receptor antagonists.
On the other hand, using PPIs for 9-14 years was associated with the equivalent of about 2 years of age-related cognitive decline, and controlling for exposure to H2 receptor antagonists weakened even this modest effect, the investigators said. Users and nonusers of PPIs tend to differ on many measures, and analyses of claims data, such as the German study above, are less able to account for these potential confounders, they noted. “Nonjudicious PPI prescribing is especially frequent among the elderly and those with cognitive impairment,” they added. “Therefore, elderly individuals who have frequent contact with health providers are at increased risk of both PPI prescription and dementia diagnosis. This bias may not be completely mitigated by adjustment for comorbidities or polypharmacy.”
The findings regarding H2 receptor antagonists reflect those of three smaller cohort studies, and these medications are known to cause central nervous system effects in the elderly, including delirium, the researchers said. Ranitidine and cimetidine have anticholinergic effects that also could “pose a risk for adverse cognitive effects with long-term use.”
Dr. Lochhead reported having no conflicts. Two coinvestigators disclosed ties to Bayer Healthcare, Pfizer, Aralez Pharmaceuticals, AbbVie, Samsung Bioepis, and Takeda.
A large prospective study of middle-aged and older women found no convincing evidence that using proton pump inhibitors increased their risk of dementia, investigators reported.
However, using H2 receptor antagonists for at least 9 years was associated with a slight decrease in scores of learning and working memory (mean decrease, –0.2; 95% confidence interval, –0.3 to –0.08; P less than .001), Paul Lochhead, MBChB, PhD, and his associates wrote in the October issue of Gastroenterology (doi: 10.1053/j.gastro.2017.06.061). “Since our primary hypothesis related to PPI [proton pump inhibitor] use, our findings for [H2 receptor antagonists] should be interpreted with caution,” they said.
Source: American Gastroenterological Association
In a recent German study of a medical claims database, use of PPIs was associated with a 44% increase in the likelihood of incident dementia (JAMA Neurol. 2016;73:410-6). “The existence of a causal mechanism linking PPI use to dementia is suggested by observations from cellular and animal models of Alzheimer’s disease, where PPI exposure appears to influence amyloid-beta metabolism,” Dr. Lochhead and his associates wrote. “However, other preclinical data on PPIs and Alzheimer’s disease are conflicting.” Noting that cognitive function predicts dementia later in life, they analyzed prospective data on medications and other potential risk factors from 13,864 participants in the Nurses’ Health Study II who had completed Cogstate, a computerized, self-administered neuropsychological battery.
Study participants averaged 61 years old when they underwent cognitive testing, ranging in age from 50 to 70 years. Users of PPIs tended to be older, had more comorbidities, were less physically active, had higher body mass indexes, had less education, and ate a lower-quality diet than women who did not use PPIs. After adjusting for such confounders, using PPIs for 9-14 years was associated with a modest decrease in scores for psychomotor speed and attention (mean score difference, compared with never users, –0.06; 95% CI, –0.11 to 0.00; P = .03). “For comparison, in multivariable models, a 1-year increase in age was associated with mean score decreases of 0.03 for psychomotor speed and attention, 0.02 for learning and working memory, and 0.03 for overall cognition,” the researchers wrote.
Next, they examined links between use of H2 receptor antagonists and cognitive scores among 10,778 study participants who had used PPIs for 2 years or less. Use of H2 receptor antagonists for 9-14 years predicted poorer scores on learning, working memory, and overall cognition, even after controlling for potential confounders (P less than or equal to .002). “The magnitudes of mean score differences were larger than those observed in the analysis of PPI use, particularly for learning and working memory,” the researchers noted. Additionally, PPI use did not predict lower cognitive scores among individuals who had never used H2 receptor antagonists.
On the other hand, using PPIs for 9-14 years was associated with the equivalent of about 2 years of age-related cognitive decline, and controlling for exposure to H2 receptor antagonists weakened even this modest effect, the investigators said. Users and nonusers of PPIs tend to differ on many measures, and analyses of claims data, such as the German study above, are less able to account for these potential confounders, they noted. “Nonjudicious PPI prescribing is especially frequent among the elderly and those with cognitive impairment,” they added. “Therefore, elderly individuals who have frequent contact with health providers are at increased risk of both PPI prescription and dementia diagnosis. This bias may not be completely mitigated by adjustment for comorbidities or polypharmacy.”
The findings regarding H2 receptor antagonists reflect those of three smaller cohort studies, and these medications are known to cause central nervous system effects in the elderly, including delirium, the researchers said. Ranitidine and cimetidine have anticholinergic effects that also could “pose a risk for adverse cognitive effects with long-term use.”
Dr. Lochhead reported having no conflicts. Two coinvestigators disclosed ties to Bayer Healthcare, Pfizer, Aralez Pharmaceuticals, AbbVie, Samsung Bioepis, and Takeda.
FROM GASTROENTEROLOGY
Key clinical point: A large prospective cohort study linked long-term use of H2 receptor antagonists, but not PPIs, to dementia.
Major finding: Use of PPIs did not significantly predict incident dementia in the adjusted analysis. However, using H2 receptor antagonists for at least 9 years was associated with a slight decrease in scores of learning and working memory (mean decrease, –0.2; P less than .001).
Data source: A population-based cohort study of 13,864 middle-aged and older women.
Disclosures: Dr. Lochhead reported having no conflicts. Two coinvestigators disclosed ties to Bayer Healthcare, Pfizer, Aralez Pharmaceuticals, AbbVie, Samsung Bioepis, and Takeda.
Burden of HCV-induced cirrhosis expected to shift from men to women
Prevalence of hepatitis C virus (HCV) complications among women grew at a rate similar to that of men, while mortality among men was nearly double that of women, according to a study conducted through the Veterans Affairs office.
While men still have a higher prevalence of conditions such as cirrhosis, investigators expect to see a shift in the burden of care as women with HCV complications outlive men with similar diagnoses.
“The current and near-term burden in HCV-related cirrhosis was disproportionately attributed to men,” according to Jennifer Kramer, PhD, investigator at the Center for Innovations in Quality, Effectiveness and Safety, Michael E. DeBakey Veterans Affairs Medical Center, Houston. “However, the trends are expected to change after 2020.”
The retrospective cohort study analyzed 264,409 HCV-infected veterans, 7,162 of whom were women, between January 2000 and December 2013.
Investigators found annual average prevalence change (AAPC) among men and women was 13.1% and 15.2%, respectively, for cirrhosis, while overall mortality was 28.7% for men, compared with 15.5% for women (J Viral Hepat. 2017 Aug 16. doi: 10.1111/jvh.12728).
Dr. Kramer and her fellow investigators also found similar rates among decompensated cirrhosis between 15.6% and 16.9% for women and men, respectively, and hepatocellular cancer, 21% and 25.3%, respectively.
Women included in the cohort were, on average, younger (48 years vs. 53 years), were less likely to use alcohol (33% vs. 45%), and were less likely to have contracted diabetes (30% vs. 39%).
While men’s prevalence growth was equal to women’s, male patients are 1.7 times more likely to be infected with HCV (J Hepatol. 2012 Jun 2 doi: 10.1016/j.jhep.2012.05.018), which is reflected in overall incidence rates of complications.
As expected, overall incidence of cirrhosis was higher in men than in women, with incidence rates for men at 28.2% compared with 20.1% of women.
Similar differences were found in rates of decompensated cirrhosis, 18.6% in men compared with 12.4% in women, and hepatocellular cancer, 5.3% in men compared with 1.5% in women.
Shifting trends in burden of care toward women have investigators worried about current HCV treatment practices for female patients.
“The increasing burden of HCV complications in women is concerning,” the researchers wrote. “Studies show that women are less likely to receive antiviral treatment than men.”
Contrary to this claim, antiviral treatment rates among men and women in this study were almost identical: 23.6% of women and 23.3% of men.
While the difference in treatment is not evident, the low rate of treatment for both men and women is another concern for Dr. Kramer and her colleagues.
“In the U.S., HCV infection remains undiagnosed in over 50% of all persons with HCV disease,” the investigators wrote. “Access to highly affective yet expensive direct acting antiviral treatment remains a challenge.”
Findings from this study may not be a true representation of the U.S. HCV-infected population because patients were veterans, with differences such as a higher rate of alcohol use among women.
The researchers reported no relevant financial disclosures.
AGA Resource
Through the HCV Clinical Service Line, AGA offers tools to help you become more efficient, understand quality standards and improve the process of care for patients. Learn more at http://www.gastro.org/patient-care/conditions-diseases/hepatitis-c
[email protected]
On Twitter @eaztweets
Prevalence of hepatitis C virus (HCV) complications among women grew at a rate similar to that of men, while mortality among men was nearly double that of women, according to a study conducted through the Veterans Affairs office.
While men still have a higher prevalence of conditions such as cirrhosis, investigators expect to see a shift in the burden of care as women with HCV complications outlive men with similar diagnoses.
“The current and near-term burden in HCV-related cirrhosis was disproportionately attributed to men,” according to Jennifer Kramer, PhD, investigator at the Center for Innovations in Quality, Effectiveness and Safety, Michael E. DeBakey Veterans Affairs Medical Center, Houston. “However, the trends are expected to change after 2020.”
The retrospective cohort study analyzed 264,409 HCV-infected veterans, 7,162 of whom were women, between January 2000 and December 2013.
Investigators found annual average prevalence change (AAPC) among men and women was 13.1% and 15.2%, respectively, for cirrhosis, while overall mortality was 28.7% for men, compared with 15.5% for women (J Viral Hepat. 2017 Aug 16. doi: 10.1111/jvh.12728).
Dr. Kramer and her fellow investigators also found similar rates among decompensated cirrhosis between 15.6% and 16.9% for women and men, respectively, and hepatocellular cancer, 21% and 25.3%, respectively.
Women included in the cohort were, on average, younger (48 years vs. 53 years), were less likely to use alcohol (33% vs. 45%), and were less likely to have contracted diabetes (30% vs. 39%).
While men’s prevalence growth was equal to women’s, male patients are 1.7 times more likely to be infected with HCV (J Hepatol. 2012 Jun 2 doi: 10.1016/j.jhep.2012.05.018), which is reflected in overall incidence rates of complications.
As expected, overall incidence of cirrhosis was higher in men than in women, with incidence rates for men at 28.2% compared with 20.1% of women.
Similar differences were found in rates of decompensated cirrhosis, 18.6% in men compared with 12.4% in women, and hepatocellular cancer, 5.3% in men compared with 1.5% in women.
Shifting trends in burden of care toward women have investigators worried about current HCV treatment practices for female patients.
“The increasing burden of HCV complications in women is concerning,” the researchers wrote. “Studies show that women are less likely to receive antiviral treatment than men.”
Contrary to this claim, antiviral treatment rates among men and women in this study were almost identical: 23.6% of women and 23.3% of men.
While the difference in treatment is not evident, the low rate of treatment for both men and women is another concern for Dr. Kramer and her colleagues.
“In the U.S., HCV infection remains undiagnosed in over 50% of all persons with HCV disease,” the investigators wrote. “Access to highly affective yet expensive direct acting antiviral treatment remains a challenge.”
Findings from this study may not be a true representation of the U.S. HCV-infected population because patients were veterans, with differences such as a higher rate of alcohol use among women.
The researchers reported no relevant financial disclosures.
AGA Resource
Through the HCV Clinical Service Line, AGA offers tools to help you become more efficient, understand quality standards and improve the process of care for patients. Learn more at http://www.gastro.org/patient-care/conditions-diseases/hepatitis-c
[email protected]
On Twitter @eaztweets
Prevalence of hepatitis C virus (HCV) complications among women grew at a rate similar to that of men, while mortality among men was nearly double that of women, according to a study conducted through the Veterans Affairs office.
While men still have a higher prevalence of conditions such as cirrhosis, investigators expect to see a shift in the burden of care as women with HCV complications outlive men with similar diagnoses.
“The current and near-term burden in HCV-related cirrhosis was disproportionately attributed to men,” according to Jennifer Kramer, PhD, investigator at the Center for Innovations in Quality, Effectiveness and Safety, Michael E. DeBakey Veterans Affairs Medical Center, Houston. “However, the trends are expected to change after 2020.”
The retrospective cohort study analyzed 264,409 HCV-infected veterans, 7,162 of whom were women, between January 2000 and December 2013.
Investigators found annual average prevalence change (AAPC) among men and women was 13.1% and 15.2%, respectively, for cirrhosis, while overall mortality was 28.7% for men, compared with 15.5% for women (J Viral Hepat. 2017 Aug 16. doi: 10.1111/jvh.12728).
Dr. Kramer and her fellow investigators also found similar rates among decompensated cirrhosis between 15.6% and 16.9% for women and men, respectively, and hepatocellular cancer, 21% and 25.3%, respectively.
Women included in the cohort were, on average, younger (48 years vs. 53 years), were less likely to use alcohol (33% vs. 45%), and were less likely to have contracted diabetes (30% vs. 39%).
While men’s prevalence growth was equal to women’s, male patients are 1.7 times more likely to be infected with HCV (J Hepatol. 2012 Jun 2 doi: 10.1016/j.jhep.2012.05.018), which is reflected in overall incidence rates of complications.
As expected, overall incidence of cirrhosis was higher in men than in women, with incidence rates for men at 28.2% compared with 20.1% of women.
Similar differences were found in rates of decompensated cirrhosis, 18.6% in men compared with 12.4% in women, and hepatocellular cancer, 5.3% in men compared with 1.5% in women.
Shifting trends in burden of care toward women have investigators worried about current HCV treatment practices for female patients.
“The increasing burden of HCV complications in women is concerning,” the researchers wrote. “Studies show that women are less likely to receive antiviral treatment than men.”
Contrary to this claim, antiviral treatment rates among men and women in this study were almost identical: 23.6% of women and 23.3% of men.
While the difference in treatment is not evident, the low rate of treatment for both men and women is another concern for Dr. Kramer and her colleagues.
“In the U.S., HCV infection remains undiagnosed in over 50% of all persons with HCV disease,” the investigators wrote. “Access to highly affective yet expensive direct acting antiviral treatment remains a challenge.”
Findings from this study may not be a true representation of the U.S. HCV-infected population because patients were veterans, with differences such as a higher rate of alcohol use among women.
The researchers reported no relevant financial disclosures.
AGA Resource
Through the HCV Clinical Service Line, AGA offers tools to help you become more efficient, understand quality standards and improve the process of care for patients. Learn more at http://www.gastro.org/patient-care/conditions-diseases/hepatitis-c
[email protected]
On Twitter @eaztweets
FROM JOURNAL OF VIRAL HEPATITIS
Key clinical point:
Major finding: Average annual change of cirrhosis prevalence in women was 15.2% for women and 13.1% for men, while total mortality was 15.5% compared with 28.7% for women and men, respectively.
Data source: Retrospective cohort study of 264,409 veterans diagnosed with HCV, from the Veterans Affairs corporate wellness data for January 2000 to December 2013.
Disclosures: The investigators reported no relevant financial disclosures.
Burden of HCV-induced cirrhosis expected to shift from men to women
Prevalence of hepatitis C virus (HCV) complications among women grew at a rate similar to that of men, while mortality among men was nearly double that of women, according to a study conducted through the Veterans Affairs office.
While men still have a higher prevalence of conditions such as cirrhosis, investigators expect to see a shift in the burden of care as women with HCV complications outlive men with similar diagnoses.
“The current and near-term burden in HCV-related cirrhosis was disproportionately attributed to men,” according to Jennifer Kramer, PhD, investigator at the Center for Innovations in Quality, Effectiveness and Safety, Michael E. DeBakey Veterans Affairs Medical Center, Houston. “However, the trends are expected to change after 2020.”
The retrospective cohort study analyzed 264,409 HCV-infected veterans, 7,162 of whom were women, between January 2000 and December 2013.
Investigators found annual average prevalence change (AAPC) among men and women was 13.1% and 15.2%, respectively, for cirrhosis, while overall mortality was 28.7% for men, compared with 15.5% for women (J Viral Hepat. 2017 Aug 16. doi: 10.1111/jvh.12728).
Dr. Kramer and her fellow investigators also found similar rates among decompensated cirrhosis between 15.6% and 16.9% for women and men, respectively, and hepatocellular cancer, 21% and 25.3%, respectively.
Women included in the cohort were, on average, younger (48 years vs. 53 years), were less likely to use alcohol (33% vs. 45%), and were less likely to have contracted diabetes (30% vs. 39%).
While men’s prevalence growth was equal to women’s, male patients are 1.7 times more likely to be infected with HCV (J Hepatol. 2012 Jun 2 doi: 10.1016/j.jhep.2012.05.018), which is reflected in overall incidence rates of complications.
As expected, overall incidence of cirrhosis was higher in men than in women, with incidence rates for men at 28.2% compared with 20.1% of women.
Similar differences were found in rates of decompensated cirrhosis, 18.6% in men compared with 12.4% in women, and hepatocellular cancer, 5.3% in men compared with 1.5% in women.
Shifting trends in burden of care toward women have investigators worried about current HCV treatment practices for female patients.
“The increasing burden of HCV complications in women is concerning,” the researchers wrote. “Studies show that women are less likely to receive antiviral treatment than men.”
Contrary to this claim, antiviral treatment rates among men and women in this study were almost identical: 23.6% of women and 23.3% of men.
While the difference in treatment is not evident, the low rate of treatment for both men and women is another concern for Dr. Kramer and her colleagues.
“In the U.S., HCV infection remains undiagnosed in over 50% of all persons with HCV disease,” the investigators wrote. “Access to highly affective yet expensive direct acting antiviral treatment remains a challenge.”
Findings from this study may not be a true representation of the U.S. HCV-infected population because patients were veterans, with differences such as a higher rate of alcohol use among women.
The researchers reported no relevant financial disclosures.
[email protected]
On Twitter @eaztweets
Prevalence of hepatitis C virus (HCV) complications among women grew at a rate similar to that of men, while mortality among men was nearly double that of women, according to a study conducted through the Veterans Affairs office.
While men still have a higher prevalence of conditions such as cirrhosis, investigators expect to see a shift in the burden of care as women with HCV complications outlive men with similar diagnoses.
“The current and near-term burden in HCV-related cirrhosis was disproportionately attributed to men,” according to Jennifer Kramer, PhD, investigator at the Center for Innovations in Quality, Effectiveness and Safety, Michael E. DeBakey Veterans Affairs Medical Center, Houston. “However, the trends are expected to change after 2020.”
The retrospective cohort study analyzed 264,409 HCV-infected veterans, 7,162 of whom were women, between January 2000 and December 2013.
Investigators found annual average prevalence change (AAPC) among men and women was 13.1% and 15.2%, respectively, for cirrhosis, while overall mortality was 28.7% for men, compared with 15.5% for women (J Viral Hepat. 2017 Aug 16. doi: 10.1111/jvh.12728).
Dr. Kramer and her fellow investigators also found similar rates among decompensated cirrhosis between 15.6% and 16.9% for women and men, respectively, and hepatocellular cancer, 21% and 25.3%, respectively.
Women included in the cohort were, on average, younger (48 years vs. 53 years), were less likely to use alcohol (33% vs. 45%), and were less likely to have contracted diabetes (30% vs. 39%).
While men’s prevalence growth was equal to women’s, male patients are 1.7 times more likely to be infected with HCV (J Hepatol. 2012 Jun 2 doi: 10.1016/j.jhep.2012.05.018), which is reflected in overall incidence rates of complications.
As expected, overall incidence of cirrhosis was higher in men than in women, with incidence rates for men at 28.2% compared with 20.1% of women.
Similar differences were found in rates of decompensated cirrhosis, 18.6% in men compared with 12.4% in women, and hepatocellular cancer, 5.3% in men compared with 1.5% in women.
Shifting trends in burden of care toward women have investigators worried about current HCV treatment practices for female patients.
“The increasing burden of HCV complications in women is concerning,” the researchers wrote. “Studies show that women are less likely to receive antiviral treatment than men.”
Contrary to this claim, antiviral treatment rates among men and women in this study were almost identical: 23.6% of women and 23.3% of men.
While the difference in treatment is not evident, the low rate of treatment for both men and women is another concern for Dr. Kramer and her colleagues.
“In the U.S., HCV infection remains undiagnosed in over 50% of all persons with HCV disease,” the investigators wrote. “Access to highly affective yet expensive direct acting antiviral treatment remains a challenge.”
Findings from this study may not be a true representation of the U.S. HCV-infected population because patients were veterans, with differences such as a higher rate of alcohol use among women.
The researchers reported no relevant financial disclosures.
[email protected]
On Twitter @eaztweets
Prevalence of hepatitis C virus (HCV) complications among women grew at a rate similar to that of men, while mortality among men was nearly double that of women, according to a study conducted through the Veterans Affairs office.
While men still have a higher prevalence of conditions such as cirrhosis, investigators expect to see a shift in the burden of care as women with HCV complications outlive men with similar diagnoses.
“The current and near-term burden in HCV-related cirrhosis was disproportionately attributed to men,” according to Jennifer Kramer, PhD, investigator at the Center for Innovations in Quality, Effectiveness and Safety, Michael E. DeBakey Veterans Affairs Medical Center, Houston. “However, the trends are expected to change after 2020.”
The retrospective cohort study analyzed 264,409 HCV-infected veterans, 7,162 of whom were women, between January 2000 and December 2013.
Investigators found annual average prevalence change (AAPC) among men and women was 13.1% and 15.2%, respectively, for cirrhosis, while overall mortality was 28.7% for men, compared with 15.5% for women (J Viral Hepat. 2017 Aug 16. doi: 10.1111/jvh.12728).
Dr. Kramer and her fellow investigators also found similar rates among decompensated cirrhosis between 15.6% and 16.9% for women and men, respectively, and hepatocellular cancer, 21% and 25.3%, respectively.
Women included in the cohort were, on average, younger (48 years vs. 53 years), were less likely to use alcohol (33% vs. 45%), and were less likely to have contracted diabetes (30% vs. 39%).
While men’s prevalence growth was equal to women’s, male patients are 1.7 times more likely to be infected with HCV (J Hepatol. 2012 Jun 2 doi: 10.1016/j.jhep.2012.05.018), which is reflected in overall incidence rates of complications.
As expected, overall incidence of cirrhosis was higher in men than in women, with incidence rates for men at 28.2% compared with 20.1% of women.
Similar differences were found in rates of decompensated cirrhosis, 18.6% in men compared with 12.4% in women, and hepatocellular cancer, 5.3% in men compared with 1.5% in women.
Shifting trends in burden of care toward women have investigators worried about current HCV treatment practices for female patients.
“The increasing burden of HCV complications in women is concerning,” the researchers wrote. “Studies show that women are less likely to receive antiviral treatment than men.”
Contrary to this claim, antiviral treatment rates among men and women in this study were almost identical: 23.6% of women and 23.3% of men.
While the difference in treatment is not evident, the low rate of treatment for both men and women is another concern for Dr. Kramer and her colleagues.
“In the U.S., HCV infection remains undiagnosed in over 50% of all persons with HCV disease,” the investigators wrote. “Access to highly affective yet expensive direct acting antiviral treatment remains a challenge.”
Findings from this study may not be a true representation of the U.S. HCV-infected population because patients were veterans, with differences such as a higher rate of alcohol use among women.
The researchers reported no relevant financial disclosures.
[email protected]
On Twitter @eaztweets
FROM THE JOURNAL OF VIRAL HEPATITIS
Key clinical point:
Major finding: Average annual change of cirrhosis prevalence in women was 15.2% for women and 13.1% for men, while total mortality was 15.5% compared with 28.7% for women and men, respectively.
Data source: Retrospective cohort study of 264,409 veterans diagnosed with HCV, from the Veterans Affairs corporate wellness data for January 2000 to December 2013.
Disclosures: The investigators reported no relevant financial disclosures.
Statin use cuts risks in compensated cirrhosis
For patients with compensated cirrhosis, statin therapy was associated with about a 46% decrease in the risk of hepatic decompensation and mortality and with a 27% drop in the risk of portal hypertension and variceal bleeding, according to moderate-quality evidence from a systematic review and meta-analysis of 13 studies.
Low-quality data also suggested that statins might help protect against the progression of noncirrhotic chronic liver disease, said Rebecca G. Kim of the University of California at San Diego and her associates. “Large, pragmatic randomized controlled trials in patients with compensated cirrhosis are required to confirm these observations,” they wrote in the October issue of Clinical Gastroenterology and Hepatology (doi: 10.1016/j.cgh.2017.04.039).
Prior studies have reported mixed findings on how statin therapy affects chronic liver disease. For their review, Ms. Kim and her associates searched MEDLINE, Embase, the Cochrane Central Register of Controlled Trials, Cochrane Database and Systematic Reviews, Scopus, Web of Science, and PubMed for randomized controlled trials or cohort studies published through March 25, 2017. They identified 10 cohort studies and three randomized controlled trials of adults with fibrosis without cirrhosis, compensated cirrhosis, or decompensated cirrhosis that evaluated statin exposure and reported associations between exposure and outcomes related to cirrhosis. They excluded case-control studies, cross-sectional studies, and studies that focused only on the relationship between statin use and the risk of hepatocellular carcinoma.
The resulting data set included 121,058 patients with chronic liver diseases, of whom 85% had chronic hepatitis C virus infection. A total of 46% of patients were exposed to statins, which appeared to reduce their risk of hepatic decompensation, variceal bleeding, and mortality. Among 87 such patients in five studies, statin use was associated with a 46% decrease in the risk of hepatic decompensation and death, with risk ratios of 0.54 (95% confidence intervals, 0.46-0.62 and 0.47-0.61, respectively). Statin use also was associated with a 27% lower risk of variceal bleeding or progression of portal hypertension, based on an analysis of 110 events in 236 patients from three trials (RR, 0.73; 95% CI, 0.59-0.91). Finally, statin use also was associated with a 58% lower risk of fibrosis progression or cirrhosis in patients with noncirrhotic chronic liver disease, but the 95% CIs for the risk estimate did not reach statistical significance (0.16-1.11).
Source: American Gastroenterological Association
Most studies lacked data on dose and duration of statin exposure, the researchers said. However, four cohort studies reported dose-dependent effects that were most pronounced after more than a year of treatment. “Similarly, several different types of statins were studied, and observed effects were assumed to be class-specific effects,” the reviewers wrote. “However, it is possible that lipophilic and lipophobic statins may have differential efficacy in decreasing fibrosis progression.”
Together, these findings support prior studies suggesting that statin therapy is safe and can potentially reduce the risk of hepatocellular carcinoma in this patient population, they concluded. Statins “may potentially improve patient-relevant outcomes in patients with chronic liver diseases and improve survival without significant additional costs.”
The reviewers acknowledged the American Gastroenterological Association Foundation, a T. Franklin Williams Scholarship Award, the National Institutes of Health, and the National Library of Medicine. They reported having no relevant conflicts of interest.
This story was updated on 9/13/2017.
The main mechanism in the development of cirrhosis in patients with chronic liver disease (CLD) is increased hepatic fibrogenesis. The initial consequence of cirrhosis is portal hypertension, which is the main driver of decompensation (defined as the presence of ascites, variceal hemorrhage, or encephalopathy).

Statins are widely used for reducing cholesterol levels and cardiovascular risk. However, statins ameliorate endothelial dysfunction and have additional antifibrotic, anti-inflammatory, and antithrombotic properties, all of them of potential benefit in preventing progression of CLD/cirrhosis. In fact, statins have been shown to reduce portal pressure in cirrhosis.
In a meta-analysis of 13 studies, Kim et al. demonstrated that statin use is associated with a 58% lower risk of developing cirrhosis/fibrosis progression in patients with CLD (not statistically significant), while in patients with compensated cirrhosis of any etiology, statin use was associated with a statistically significant 46% lower risk of developing decompensation and death.
Most studies in the meta-analysis were observational/retrospective. Although the authors jointly analyzed three randomized controlled trials, only one of the trials looked at clinical outcomes. This important double-blind, placebo-controlled study in patients with recent variceal hemorrhage showed a significantly lower mortality in patients randomized to simvastatin.
Therefore, although the evidence is not yet sufficient to recommend the widespread use of statins in patients with CLD/cirrhosis, providers should not avoid using statins in patients with CLD/cirrhosis who otherwise need them. In fact, they should actively look for indications that would justify their use.
Guadalupe Garcia-Tsao, MD, is professor of medicine at Yale University, chief of digestive diseases at the VA-CT Healthcare System, and director of the clinical core of the Yale Liver Center, New Haven, Conn. She had no conflicts of interest.
The main mechanism in the development of cirrhosis in patients with chronic liver disease (CLD) is increased hepatic fibrogenesis. The initial consequence of cirrhosis is portal hypertension, which is the main driver of decompensation (defined as the presence of ascites, variceal hemorrhage, or encephalopathy).
Statins are widely used for reducing cholesterol levels and cardiovascular risk. However, statins ameliorate endothelial dysfunction and have additional antifibrotic, anti-inflammatory, and antithrombotic properties, all of them of potential benefit in preventing progression of CLD/cirrhosis. In fact, statins have been shown to reduce portal pressure in cirrhosis.
In a meta-analysis of 13 studies, Kim et al. demonstrated that statin use is associated with a 58% lower risk of developing cirrhosis/fibrosis progression in patients with CLD (not statistically significant), while in patients with compensated cirrhosis of any etiology, statin use was associated with a statistically significant 46% lower risk of developing decompensation and death.
Most studies in the meta-analysis were observational/retrospective. Although the authors jointly analyzed three randomized controlled trials, only one of the trials looked at clinical outcomes. This important double-blind, placebo-controlled study in patients with recent variceal hemorrhage showed a significantly lower mortality in patients randomized to simvastatin.
Therefore, although the evidence is not yet sufficient to recommend the widespread use of statins in patients with CLD/cirrhosis, providers should not avoid using statins in patients with CLD/cirrhosis who otherwise need them. In fact, they should actively look for indications that would justify their use.
Guadalupe Garcia-Tsao, MD, is professor of medicine at Yale University, chief of digestive diseases at the VA-CT Healthcare System, and director of the clinical core of the Yale Liver Center, New Haven, Conn. She had no conflicts of interest.
The main mechanism in the development of cirrhosis in patients with chronic liver disease (CLD) is increased hepatic fibrogenesis. The initial consequence of cirrhosis is portal hypertension, which is the main driver of decompensation (defined as the presence of ascites, variceal hemorrhage, or encephalopathy).
Statins are widely used for reducing cholesterol levels and cardiovascular risk. However, statins ameliorate endothelial dysfunction and have additional antifibrotic, anti-inflammatory, and antithrombotic properties, all of them of potential benefit in preventing progression of CLD/cirrhosis. In fact, statins have been shown to reduce portal pressure in cirrhosis.
In a meta-analysis of 13 studies, Kim et al. demonstrated that statin use is associated with a 58% lower risk of developing cirrhosis/fibrosis progression in patients with CLD (not statistically significant), while in patients with compensated cirrhosis of any etiology, statin use was associated with a statistically significant 46% lower risk of developing decompensation and death.
Most studies in the meta-analysis were observational/retrospective. Although the authors jointly analyzed three randomized controlled trials, only one of the trials looked at clinical outcomes. This important double-blind, placebo-controlled study in patients with recent variceal hemorrhage showed a significantly lower mortality in patients randomized to simvastatin.
Therefore, although the evidence is not yet sufficient to recommend the widespread use of statins in patients with CLD/cirrhosis, providers should not avoid using statins in patients with CLD/cirrhosis who otherwise need them. In fact, they should actively look for indications that would justify their use.
Guadalupe Garcia-Tsao, MD, is professor of medicine at Yale University, chief of digestive diseases at the VA-CT Healthcare System, and director of the clinical core of the Yale Liver Center, New Haven, Conn. She had no conflicts of interest.
For patients with compensated cirrhosis, statin therapy was associated with about a 46% decrease in the risk of hepatic decompensation and mortality and with a 27% drop in the risk of portal hypertension and variceal bleeding, according to moderate-quality evidence from a systematic review and meta-analysis of 13 studies.
Low-quality data also suggested that statins might help protect against the progression of noncirrhotic chronic liver disease, said Rebecca G. Kim of the University of California at San Diego and her associates. “Large, pragmatic randomized controlled trials in patients with compensated cirrhosis are required to confirm these observations,” they wrote in the October issue of Clinical Gastroenterology and Hepatology (doi: 10.1016/j.cgh.2017.04.039).
Prior studies have reported mixed findings on how statin therapy affects chronic liver disease. For their review, Ms. Kim and her associates searched MEDLINE, Embase, the Cochrane Central Register of Controlled Trials, Cochrane Database and Systematic Reviews, Scopus, Web of Science, and PubMed for randomized controlled trials or cohort studies published through March 25, 2017. They identified 10 cohort studies and three randomized controlled trials of adults with fibrosis without cirrhosis, compensated cirrhosis, or decompensated cirrhosis that evaluated statin exposure and reported associations between exposure and outcomes related to cirrhosis. They excluded case-control studies, cross-sectional studies, and studies that focused only on the relationship between statin use and the risk of hepatocellular carcinoma.
The resulting data set included 121,058 patients with chronic liver diseases, of whom 85% had chronic hepatitis C virus infection. A total of 46% of patients were exposed to statins, which appeared to reduce their risk of hepatic decompensation, variceal bleeding, and mortality. Among 87 such patients in five studies, statin use was associated with a 46% decrease in the risk of hepatic decompensation and death, with risk ratios of 0.54 (95% confidence intervals, 0.46-0.62 and 0.47-0.61, respectively). Statin use also was associated with a 27% lower risk of variceal bleeding or progression of portal hypertension, based on an analysis of 110 events in 236 patients from three trials (RR, 0.73; 95% CI, 0.59-0.91). Finally, statin use also was associated with a 58% lower risk of fibrosis progression or cirrhosis in patients with noncirrhotic chronic liver disease, but the 95% CIs for the risk estimate did not reach statistical significance (0.16-1.11).
Source: American Gastroenterological Association
Most studies lacked data on dose and duration of statin exposure, the researchers said. However, four cohort studies reported dose-dependent effects that were most pronounced after more than a year of treatment. “Similarly, several different types of statins were studied, and observed effects were assumed to be class-specific effects,” the reviewers wrote. “However, it is possible that lipophilic and lipophobic statins may have differential efficacy in decreasing fibrosis progression.”
Together, these findings support prior studies suggesting that statin therapy is safe and can potentially reduce the risk of hepatocellular carcinoma in this patient population, they concluded. Statins “may potentially improve patient-relevant outcomes in patients with chronic liver diseases and improve survival without significant additional costs.”
The reviewers acknowledged the American Gastroenterological Association Foundation, a T. Franklin Williams Scholarship Award, the National Institutes of Health, and the National Library of Medicine. They reported having no relevant conflicts of interest.
This story was updated on 9/13/2017.
For patients with compensated cirrhosis, statin therapy was associated with about a 46% decrease in the risk of hepatic decompensation and mortality and with a 27% drop in the risk of portal hypertension and variceal bleeding, according to moderate-quality evidence from a systematic review and meta-analysis of 13 studies.
Low-quality data also suggested that statins might help protect against the progression of noncirrhotic chronic liver disease, said Rebecca G. Kim of the University of California at San Diego and her associates. “Large, pragmatic randomized controlled trials in patients with compensated cirrhosis are required to confirm these observations,” they wrote in the October issue of Clinical Gastroenterology and Hepatology (doi: 10.1016/j.cgh.2017.04.039).
Prior studies have reported mixed findings on how statin therapy affects chronic liver disease. For their review, Ms. Kim and her associates searched MEDLINE, Embase, the Cochrane Central Register of Controlled Trials, Cochrane Database and Systematic Reviews, Scopus, Web of Science, and PubMed for randomized controlled trials or cohort studies published through March 25, 2017. They identified 10 cohort studies and three randomized controlled trials of adults with fibrosis without cirrhosis, compensated cirrhosis, or decompensated cirrhosis that evaluated statin exposure and reported associations between exposure and outcomes related to cirrhosis. They excluded case-control studies, cross-sectional studies, and studies that focused only on the relationship between statin use and the risk of hepatocellular carcinoma.
The resulting data set included 121,058 patients with chronic liver diseases, of whom 85% had chronic hepatitis C virus infection. A total of 46% of patients were exposed to statins, which appeared to reduce their risk of hepatic decompensation, variceal bleeding, and mortality. Among 87 such patients in five studies, statin use was associated with a 46% decrease in the risk of hepatic decompensation and death, with risk ratios of 0.54 (95% confidence intervals, 0.46-0.62 and 0.47-0.61, respectively). Statin use also was associated with a 27% lower risk of variceal bleeding or progression of portal hypertension, based on an analysis of 110 events in 236 patients from three trials (RR, 0.73; 95% CI, 0.59-0.91). Finally, statin use also was associated with a 58% lower risk of fibrosis progression or cirrhosis in patients with noncirrhotic chronic liver disease, but the 95% CIs for the risk estimate did not reach statistical significance (0.16-1.11).
Source: American Gastroenterological Association
Most studies lacked data on dose and duration of statin exposure, the researchers said. However, four cohort studies reported dose-dependent effects that were most pronounced after more than a year of treatment. “Similarly, several different types of statins were studied, and observed effects were assumed to be class-specific effects,” the reviewers wrote. “However, it is possible that lipophilic and lipophobic statins may have differential efficacy in decreasing fibrosis progression.”
Together, these findings support prior studies suggesting that statin therapy is safe and can potentially reduce the risk of hepatocellular carcinoma in this patient population, they concluded. Statins “may potentially improve patient-relevant outcomes in patients with chronic liver diseases and improve survival without significant additional costs.”
The reviewers acknowledged the American Gastroenterological Association Foundation, a T. Franklin Williams Scholarship Award, the National Institutes of Health, and the National Library of Medicine. They reported having no relevant conflicts of interest.
This story was updated on 9/13/2017.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Key clinical point: Statin therapy was associated with a significantly lower risk of hepatic decompensation and death in patients with compensated cirrhosis.
Major finding: Statin therapy was associated with a 46% decrease in the risk of both hepatic decompensation and mortality (risk ratios, 0.54) and with a 27% drop in the risk of portal hypertension and variceal bleeding (RR, 0.73).
Data source: A systematic review and meta-analysis of 10 cohort studies and three randomized controlled trials (121,058 patients).
Disclosures: The reviewers acknowledged the American Gastroenterological Association Foundation, a T. Franklin Williams Scholarship Award, the National Institutes of Health, and the National Library of Medicine. They reported having no relevant conflicts of interest.

Check children’s eyes early for best corrections
or its risk factors with the goal of improving visual acuity, publishing its final recommendation statement and evidence summary online Sept. 5 in JAMA.
A review of the latest evidence supports a B recommendation for vision screening at least once in children aged 3-5 years, but the evidence is insufficient to determine the balance of risks and benefits for vision screening in children younger than 3 years (meriting an I statement from the USPSTF). The recommendation updates the 2011 USPSTF recommendation, which also recommended vision screening for children aged 3-5 years with a B recommendation.
“The prevalence of amblyopia, strabismus, and anisometropia ranges from 1% to 6% among children younger than 6 years in the United States,” which can lead to permanent vision loss if left untreated, chair and corresponding author David C. Grossman, MD, of Kaiser Permanente Washington Health Research Institute, Seattle, and colleagues noted in the recommendation statement (JAMA. 2017;318:836-44).
The USPSTF found “inadequate evidence that treatment reduced the incidence of long-term amblyopia or improved school performance, functioning, or quality of life.” However, the USPSTF concluded that the harms of screening and treating preschool children for amblyopia and its risk factors were small, and that treatment improved visual acuity, “which is likely to result in permanent improvements throughout life.”
The benefits of early treatment were characterized as moderate because of the risk of permanent, uncorrectable vision loss associated with untreated amblyopia, “and the benefits of screening and treatment can be experienced over a child’s lifetime,” the researchers said.
The evidence report accompanying the recommendations contained data from 40 studies with 34,709 participants, and addressed issues including the benefits of screening, accuracy of vision screening tests, and the potential harms and benefits of treatments including eye patches and glasses (JAMA. 2017;318:845-58).
“Studies directly evaluating the effectiveness of screening were limited and do not establish whether vision screening in preschool children is better than no screening,” Daniel E. Jonas, MD, of RTI International–University of North Carolina at Chapel Hill Evidence-based Practice Center and his colleagues wrote in the evidence report.
Therefore, the Task Force called for additional research while recommending at least one screening.
The study was funded by the Agency for Healthcare Research and Quality. The researchers had no financial conflicts to disclose.
Read the complete recommendations online at http://www.uspreventiveservicestaskforce.org.
or its risk factors with the goal of improving visual acuity, publishing its final recommendation statement and evidence summary online Sept. 5 in JAMA.
A review of the latest evidence supports a B recommendation for vision screening at least once in children aged 3-5 years, but the evidence is insufficient to determine the balance of risks and benefits for vision screening in children younger than 3 years (meriting an I statement from the USPSTF). The recommendation updates the 2011 USPSTF recommendation, which also recommended vision screening for children aged 3-5 years with a B recommendation.
“The prevalence of amblyopia, strabismus, and anisometropia ranges from 1% to 6% among children younger than 6 years in the United States,” which can lead to permanent vision loss if left untreated, chair and corresponding author David C. Grossman, MD, of Kaiser Permanente Washington Health Research Institute, Seattle, and colleagues noted in the recommendation statement (JAMA. 2017;318:836-44).
The USPSTF found “inadequate evidence that treatment reduced the incidence of long-term amblyopia or improved school performance, functioning, or quality of life.” However, the USPSTF concluded that the harms of screening and treating preschool children for amblyopia and its risk factors were small, and that treatment improved visual acuity, “which is likely to result in permanent improvements throughout life.”
The benefits of early treatment were characterized as moderate because of the risk of permanent, uncorrectable vision loss associated with untreated amblyopia, “and the benefits of screening and treatment can be experienced over a child’s lifetime,” the researchers said.
The evidence report accompanying the recommendations contained data from 40 studies with 34,709 participants, and addressed issues including the benefits of screening, accuracy of vision screening tests, and the potential harms and benefits of treatments including eye patches and glasses (JAMA. 2017;318:845-58).
“Studies directly evaluating the effectiveness of screening were limited and do not establish whether vision screening in preschool children is better than no screening,” Daniel E. Jonas, MD, of RTI International–University of North Carolina at Chapel Hill Evidence-based Practice Center and his colleagues wrote in the evidence report.
Therefore, the Task Force called for additional research while recommending at least one screening.
The study was funded by the Agency for Healthcare Research and Quality. The researchers had no financial conflicts to disclose.
Read the complete recommendations online at http://www.uspreventiveservicestaskforce.org.
or its risk factors with the goal of improving visual acuity, publishing its final recommendation statement and evidence summary online Sept. 5 in JAMA.
A review of the latest evidence supports a B recommendation for vision screening at least once in children aged 3-5 years, but the evidence is insufficient to determine the balance of risks and benefits for vision screening in children younger than 3 years (meriting an I statement from the USPSTF). The recommendation updates the 2011 USPSTF recommendation, which also recommended vision screening for children aged 3-5 years with a B recommendation.
“The prevalence of amblyopia, strabismus, and anisometropia ranges from 1% to 6% among children younger than 6 years in the United States,” which can lead to permanent vision loss if left untreated, chair and corresponding author David C. Grossman, MD, of Kaiser Permanente Washington Health Research Institute, Seattle, and colleagues noted in the recommendation statement (JAMA. 2017;318:836-44).
The USPSTF found “inadequate evidence that treatment reduced the incidence of long-term amblyopia or improved school performance, functioning, or quality of life.” However, the USPSTF concluded that the harms of screening and treating preschool children for amblyopia and its risk factors were small, and that treatment improved visual acuity, “which is likely to result in permanent improvements throughout life.”
The benefits of early treatment were characterized as moderate because of the risk of permanent, uncorrectable vision loss associated with untreated amblyopia, “and the benefits of screening and treatment can be experienced over a child’s lifetime,” the researchers said.
The evidence report accompanying the recommendations contained data from 40 studies with 34,709 participants, and addressed issues including the benefits of screening, accuracy of vision screening tests, and the potential harms and benefits of treatments including eye patches and glasses (JAMA. 2017;318:845-58).
“Studies directly evaluating the effectiveness of screening were limited and do not establish whether vision screening in preschool children is better than no screening,” Daniel E. Jonas, MD, of RTI International–University of North Carolina at Chapel Hill Evidence-based Practice Center and his colleagues wrote in the evidence report.
Therefore, the Task Force called for additional research while recommending at least one screening.
The study was funded by the Agency for Healthcare Research and Quality. The researchers had no financial conflicts to disclose.
Read the complete recommendations online at http://www.uspreventiveservicestaskforce.org.
FROM JAMA
Q&A: CDC director Brenda Fitzgerald stresses ‘science and service’
Brenda Fitzgerald, MD, an ob.gyn. and most recently the public health commissioner for Georgia, was named the 17th director of the Centers for Disease Control and Prevention in July.
Dr. Fitzgerald comes to the CDC after 6 years as commissioner and state health officer for the Georgia Department of Public Health. She also has been a health care policy adviser to former House Speaker Newt Gingrich (R-Ga.) and ran for Congress herself in the 1990s. In addition to practicing medicine for 3 decades, she has served on the board and as president of the Georgia Obstetrical and Gynecological Society.

Question: What are you looking forward to as the new CDC director, and what are some of your top goals during your tenure?
Answer: The federal government holds responsibility to provide for the common defense. And CDC provides the common defense of the country against health threats. That’s why CDC’s mission of saving lives and protecting people means so much to me. As a doctor and a mother, I want to do everything I can to protect our future generations and keep them safe, whether it be from infectious or chronic diseases, natural or man-made disasters.
We are going to continue a legacy of protecting Americans from the things that we’ve been fighting for a long time, like heart disease and cancer, and from newer things like opioid overdose and the Zika virus. And we’ll continue to prepare for the next health threat, whatever it may be.
Q: How does your experience as an ob.gyn. shape your approach to this job?
A: There are two things that are important at CDC: science and service. CDC researchers are at the forefront of scientific research – and also provide a service by using scientific discoveries to improve health. That is the same approach I used as a practicing ob.gyn.: I brought the clinical lens that looked at the science, and then my work with my patients emphasized the service part.
As a practicing ob.gyn., it was my responsibility to address the questions and concerns of my patients. At CDC, I am just as committed to making science-based decisions and working with our experts to make sure doctors and patients have the state-of-the-art health information they need. I said when I went from being a clinician to being the state health official for Georgia that I went from treating one patient at a time to treating 10 million at a time. Now that’s changed to over 300 million.
Even though I am now CDC director and protecting the health of millions of people, my experiences as an ob.gyn. for 3 decades will forever shape how I work to improve the health of individual people and patients.
Q: What are the next steps for the agency in terms of its response to Zika virus?
A: Zika is the first infectious disease in 50 years that has been linked to birth defects. We know that the virus attacks neural tissue. We know that about 10% of babies are born with severe birth defects – and some babies born apparently normal later turned out to have neurologic problems, like blindness or deafness. And we suspect that there may be other problems we don’t know about yet.
I’m particularly concerned about developmental delays, as we know that early brain development is key to health and achievement for a child’s future. We are still learning more about Zika every day. It’s crucial that we – the health care and public health communities – work together and remain vigilant to ensure these babies receive the care they need. What Zika has taught us is that we need to establish a pregnancy and infant registry that can flag patterns of concerning health issues so that we can work together with health officials and clinicians to get ahead of emerging health threats.
Q: How can clinicians stay up to date on the rapidly changing guidance during an outbreak like Zika?
A: During outbreaks like Zika, public health officials are continually updating guidance so clinicians can get the latest information. You can check the latest CDC guidance about testing and follow-up care for pregnant women at www.cdc.gov/zika/hc-providers. Health care providers can also contact their state, local, or territorial health department to ensure the appropriate tests are ordered and interpreted correctly. CDC also maintains a 24/7 Zika consultation service for health officials and healthcare providers caring for pregnant women with possible Zika exposure.
Q: What steps will you take to decrease maternal mortality and to understand why this number is rising?
A: As an obstetrician, survival of mothers and babies is paramount to me. It’s so basic: mothers shouldn’t die. Sadly, about 700 women die each year in the U.S. as a result of pregnancy or delivery complications.
We know the leading causes of maternal mortality: cardiovascular disease, infections or sepsis, hemorrhage, and cardiomyopathy. We also know that an increasing number of pregnant women in the U.S. have chronic health conditions such as high blood pressure, diabetes, or heart disease that puts them at risk of pregnancy complications or death.
CDC is committed to preventing pregnancy-related deaths and ensuring the best possible birth outcomes. We do this by conducting surveillance, supporting states in developing recommendations, and working with partners to promote evidence-based recommendations and best practices. State-based initiatives are important allies in bringing together OBs and the public health sector to show the importance of statewide efforts to reduce maternal mortality. There has been some exciting work done by the California Maternal Quality Care Collaborative, which has created a series of toolkits designed to help health care providers with pregnancy or childbirth complications. CMQCC’s work is inspiring other states to follow suit.
The best coalitions are between ob.gyns., public health, and hospital associations. For example, AWHONN’s [Association of Women’s Health, Obstetric and Neonatal Nurses] Postpartum Hemorrhage initiative in Georgia, New Jersey, and the District of Columbia aimed to improve the treatment of obstetric hemorrhage. Even though 54% to 93% of maternal hemorrhage-related deaths are preventable with improved clinical response, not one single hospital in Georgia or New Jersey had implemented all the recommended preparedness elements for postpartum hemorrhage events. We can save mothers’ lives – we know what to do. It just takes all of us – ob.gyns., public health and hospital associations – leading the way to make sure it happens.
Q: How can we increase HPV vaccination?
A: The latest news is that more girls and boys are getting the HPV vaccine. New CDC data showed that 60% of teens aged 13-17 had received one or more doses of HPV vaccine in 2016 – an increase of 4% from 2015. Plus, the latest statistics show that HPV vaccination has led to significant drops in HPV infections: HPV-related cancers and genital warts dropped by 71% among teen girls and 61% among young women. Still, too many children aren’t completing the HPV vaccine series, leaving them vulnerable to cancers caused by HPV infection.
Providers can help increase HPV vaccination rates by using every patient visit to review vaccination histories, providing strong clinical recommendations and education to parents for HPV and other recommended vaccines, and implementing systems to minimize missed opportunities.
[email protected]
On Twitter @legal_med
Brenda Fitzgerald, MD, an ob.gyn. and most recently the public health commissioner for Georgia, was named the 17th director of the Centers for Disease Control and Prevention in July.
Dr. Fitzgerald comes to the CDC after 6 years as commissioner and state health officer for the Georgia Department of Public Health. She also has been a health care policy adviser to former House Speaker Newt Gingrich (R-Ga.) and ran for Congress herself in the 1990s. In addition to practicing medicine for 3 decades, she has served on the board and as president of the Georgia Obstetrical and Gynecological Society.
Question: What are you looking forward to as the new CDC director, and what are some of your top goals during your tenure?
Answer: The federal government holds responsibility to provide for the common defense. And CDC provides the common defense of the country against health threats. That’s why CDC’s mission of saving lives and protecting people means so much to me. As a doctor and a mother, I want to do everything I can to protect our future generations and keep them safe, whether it be from infectious or chronic diseases, natural or man-made disasters.
We are going to continue a legacy of protecting Americans from the things that we’ve been fighting for a long time, like heart disease and cancer, and from newer things like opioid overdose and the Zika virus. And we’ll continue to prepare for the next health threat, whatever it may be.
Q: How does your experience as an ob.gyn. shape your approach to this job?
A: There are two things that are important at CDC: science and service. CDC researchers are at the forefront of scientific research – and also provide a service by using scientific discoveries to improve health. That is the same approach I used as a practicing ob.gyn.: I brought the clinical lens that looked at the science, and then my work with my patients emphasized the service part.
As a practicing ob.gyn., it was my responsibility to address the questions and concerns of my patients. At CDC, I am just as committed to making science-based decisions and working with our experts to make sure doctors and patients have the state-of-the-art health information they need. I said when I went from being a clinician to being the state health official for Georgia that I went from treating one patient at a time to treating 10 million at a time. Now that’s changed to over 300 million.
Even though I am now CDC director and protecting the health of millions of people, my experiences as an ob.gyn. for 3 decades will forever shape how I work to improve the health of individual people and patients.
Q: What are the next steps for the agency in terms of its response to Zika virus?
A: Zika is the first infectious disease in 50 years that has been linked to birth defects. We know that the virus attacks neural tissue. We know that about 10% of babies are born with severe birth defects – and some babies born apparently normal later turned out to have neurologic problems, like blindness or deafness. And we suspect that there may be other problems we don’t know about yet.
I’m particularly concerned about developmental delays, as we know that early brain development is key to health and achievement for a child’s future. We are still learning more about Zika every day. It’s crucial that we – the health care and public health communities – work together and remain vigilant to ensure these babies receive the care they need. What Zika has taught us is that we need to establish a pregnancy and infant registry that can flag patterns of concerning health issues so that we can work together with health officials and clinicians to get ahead of emerging health threats.
Q: How can clinicians stay up to date on the rapidly changing guidance during an outbreak like Zika?
A: During outbreaks like Zika, public health officials are continually updating guidance so clinicians can get the latest information. You can check the latest CDC guidance about testing and follow-up care for pregnant women at www.cdc.gov/zika/hc-providers. Health care providers can also contact their state, local, or territorial health department to ensure the appropriate tests are ordered and interpreted correctly. CDC also maintains a 24/7 Zika consultation service for health officials and healthcare providers caring for pregnant women with possible Zika exposure.
Q: What steps will you take to decrease maternal mortality and to understand why this number is rising?
A: As an obstetrician, survival of mothers and babies is paramount to me. It’s so basic: mothers shouldn’t die. Sadly, about 700 women die each year in the U.S. as a result of pregnancy or delivery complications.
We know the leading causes of maternal mortality: cardiovascular disease, infections or sepsis, hemorrhage, and cardiomyopathy. We also know that an increasing number of pregnant women in the U.S. have chronic health conditions such as high blood pressure, diabetes, or heart disease that puts them at risk of pregnancy complications or death.
CDC is committed to preventing pregnancy-related deaths and ensuring the best possible birth outcomes. We do this by conducting surveillance, supporting states in developing recommendations, and working with partners to promote evidence-based recommendations and best practices. State-based initiatives are important allies in bringing together OBs and the public health sector to show the importance of statewide efforts to reduce maternal mortality. There has been some exciting work done by the California Maternal Quality Care Collaborative, which has created a series of toolkits designed to help health care providers with pregnancy or childbirth complications. CMQCC’s work is inspiring other states to follow suit.
The best coalitions are between ob.gyns., public health, and hospital associations. For example, AWHONN’s [Association of Women’s Health, Obstetric and Neonatal Nurses] Postpartum Hemorrhage initiative in Georgia, New Jersey, and the District of Columbia aimed to improve the treatment of obstetric hemorrhage. Even though 54% to 93% of maternal hemorrhage-related deaths are preventable with improved clinical response, not one single hospital in Georgia or New Jersey had implemented all the recommended preparedness elements for postpartum hemorrhage events. We can save mothers’ lives – we know what to do. It just takes all of us – ob.gyns., public health and hospital associations – leading the way to make sure it happens.
Q: How can we increase HPV vaccination?
A: The latest news is that more girls and boys are getting the HPV vaccine. New CDC data showed that 60% of teens aged 13-17 had received one or more doses of HPV vaccine in 2016 – an increase of 4% from 2015. Plus, the latest statistics show that HPV vaccination has led to significant drops in HPV infections: HPV-related cancers and genital warts dropped by 71% among teen girls and 61% among young women. Still, too many children aren’t completing the HPV vaccine series, leaving them vulnerable to cancers caused by HPV infection.
Providers can help increase HPV vaccination rates by using every patient visit to review vaccination histories, providing strong clinical recommendations and education to parents for HPV and other recommended vaccines, and implementing systems to minimize missed opportunities.
[email protected]
On Twitter @legal_med
Brenda Fitzgerald, MD, an ob.gyn. and most recently the public health commissioner for Georgia, was named the 17th director of the Centers for Disease Control and Prevention in July.
Dr. Fitzgerald comes to the CDC after 6 years as commissioner and state health officer for the Georgia Department of Public Health. She also has been a health care policy adviser to former House Speaker Newt Gingrich (R-Ga.) and ran for Congress herself in the 1990s. In addition to practicing medicine for 3 decades, she has served on the board and as president of the Georgia Obstetrical and Gynecological Society.
Question: What are you looking forward to as the new CDC director, and what are some of your top goals during your tenure?
Answer: The federal government holds responsibility to provide for the common defense. And CDC provides the common defense of the country against health threats. That’s why CDC’s mission of saving lives and protecting people means so much to me. As a doctor and a mother, I want to do everything I can to protect our future generations and keep them safe, whether it be from infectious or chronic diseases, natural or man-made disasters.
We are going to continue a legacy of protecting Americans from the things that we’ve been fighting for a long time, like heart disease and cancer, and from newer things like opioid overdose and the Zika virus. And we’ll continue to prepare for the next health threat, whatever it may be.
Q: How does your experience as an ob.gyn. shape your approach to this job?
A: There are two things that are important at CDC: science and service. CDC researchers are at the forefront of scientific research – and also provide a service by using scientific discoveries to improve health. That is the same approach I used as a practicing ob.gyn.: I brought the clinical lens that looked at the science, and then my work with my patients emphasized the service part.
As a practicing ob.gyn., it was my responsibility to address the questions and concerns of my patients. At CDC, I am just as committed to making science-based decisions and working with our experts to make sure doctors and patients have the state-of-the-art health information they need. I said when I went from being a clinician to being the state health official for Georgia that I went from treating one patient at a time to treating 10 million at a time. Now that’s changed to over 300 million.
Even though I am now CDC director and protecting the health of millions of people, my experiences as an ob.gyn. for 3 decades will forever shape how I work to improve the health of individual people and patients.
Q: What are the next steps for the agency in terms of its response to Zika virus?
A: Zika is the first infectious disease in 50 years that has been linked to birth defects. We know that the virus attacks neural tissue. We know that about 10% of babies are born with severe birth defects – and some babies born apparently normal later turned out to have neurologic problems, like blindness or deafness. And we suspect that there may be other problems we don’t know about yet.
I’m particularly concerned about developmental delays, as we know that early brain development is key to health and achievement for a child’s future. We are still learning more about Zika every day. It’s crucial that we – the health care and public health communities – work together and remain vigilant to ensure these babies receive the care they need. What Zika has taught us is that we need to establish a pregnancy and infant registry that can flag patterns of concerning health issues so that we can work together with health officials and clinicians to get ahead of emerging health threats.
Q: How can clinicians stay up to date on the rapidly changing guidance during an outbreak like Zika?
A: During outbreaks like Zika, public health officials are continually updating guidance so clinicians can get the latest information. You can check the latest CDC guidance about testing and follow-up care for pregnant women at www.cdc.gov/zika/hc-providers. Health care providers can also contact their state, local, or territorial health department to ensure the appropriate tests are ordered and interpreted correctly. CDC also maintains a 24/7 Zika consultation service for health officials and healthcare providers caring for pregnant women with possible Zika exposure.
Q: What steps will you take to decrease maternal mortality and to understand why this number is rising?
A: As an obstetrician, survival of mothers and babies is paramount to me. It’s so basic: mothers shouldn’t die. Sadly, about 700 women die each year in the U.S. as a result of pregnancy or delivery complications.
We know the leading causes of maternal mortality: cardiovascular disease, infections or sepsis, hemorrhage, and cardiomyopathy. We also know that an increasing number of pregnant women in the U.S. have chronic health conditions such as high blood pressure, diabetes, or heart disease that puts them at risk of pregnancy complications or death.
CDC is committed to preventing pregnancy-related deaths and ensuring the best possible birth outcomes. We do this by conducting surveillance, supporting states in developing recommendations, and working with partners to promote evidence-based recommendations and best practices. State-based initiatives are important allies in bringing together OBs and the public health sector to show the importance of statewide efforts to reduce maternal mortality. There has been some exciting work done by the California Maternal Quality Care Collaborative, which has created a series of toolkits designed to help health care providers with pregnancy or childbirth complications. CMQCC’s work is inspiring other states to follow suit.
The best coalitions are between ob.gyns., public health, and hospital associations. For example, AWHONN’s [Association of Women’s Health, Obstetric and Neonatal Nurses] Postpartum Hemorrhage initiative in Georgia, New Jersey, and the District of Columbia aimed to improve the treatment of obstetric hemorrhage. Even though 54% to 93% of maternal hemorrhage-related deaths are preventable with improved clinical response, not one single hospital in Georgia or New Jersey had implemented all the recommended preparedness elements for postpartum hemorrhage events. We can save mothers’ lives – we know what to do. It just takes all of us – ob.gyns., public health and hospital associations – leading the way to make sure it happens.
Q: How can we increase HPV vaccination?
A: The latest news is that more girls and boys are getting the HPV vaccine. New CDC data showed that 60% of teens aged 13-17 had received one or more doses of HPV vaccine in 2016 – an increase of 4% from 2015. Plus, the latest statistics show that HPV vaccination has led to significant drops in HPV infections: HPV-related cancers and genital warts dropped by 71% among teen girls and 61% among young women. Still, too many children aren’t completing the HPV vaccine series, leaving them vulnerable to cancers caused by HPV infection.
Providers can help increase HPV vaccination rates by using every patient visit to review vaccination histories, providing strong clinical recommendations and education to parents for HPV and other recommended vaccines, and implementing systems to minimize missed opportunities.
[email protected]
On Twitter @legal_med