User login
LVAD use soars in elderly Americans
DALLAS – The percentage of left ventricular assist devices placed in U.S. heart failure patients at least 75 years of age jumped sharply during 2003-2014, and concurrently the short-term survival of these patients improved dramatically, according to data collected by the National Inpatient Sample.
During the 12-year period examined, the percentage of left-ventricular assist devices (LVADs) placed in U.S. heart failure patients aged 75 years and older rose from 3% of all LVADs in 2003 to 11% in 2014, Aniket S. Rali, MD, said at the annual scientific meeting of the Heart Failure Society of America.

The U.S. national numbers also showed that throughout the period studied, elderly U.S. patients who received an LVAD were increasingly sicker, with steadily increasing numbers of patients with a Charlson Comorbidity Index score of four or greater. Despite this, in-hospital mortality rates of elderly patients receiving an LVAD plummeted, dropping from 61% of elderly LVAD recipients in 2003 to 18% in 2014. During the same time, the percentage of elderly patients with a Charlson Comorbidity Index score greater than four doubled from 33% in 2003 to 66% in 2014, said Dr. Rali, a cardiologist at the University of Kansas Medical Center in Kansas City.
“If the Charlson Comorbidity Index score is increasing but in-hospital mortality is decreasing, then increased LVAD use is not a bad trend,” Dr. Rali said in an interview. He hopes that future analysis of longitudinal data from patients could identify clinical factors that link with better patient survival and help target LVAD placement to the patients who stand to gain the most benefit.
“We may be able to give these elderly patients not just longer life but improved quality of life” by a more informed targeting of LVADs, he suggested. “I think these numbers will help convince people that all is not lost,” he noted, for elderly heart failure patients who receive an LVAD as destination therapy. Patients at least 75 years old are not eligible for heart transplantation, so when these patients receive an LVAD it is, by definition, destination therapy.
The data also showed a marked sex disparity in LVAD use, with LVAD placement in men at least 75 years old rising from 1.4/1,000 patients in 2003 to 2.78/1,000 patients in 2014. In contrast, among women these rates rose from 0.8/1,000 patients in 2003 to 1.36/1,000 patients in 2014.
The average age for elderly U.S. LVAD recipients for the entire 12-year period studied was 77.6 years among a total of 2,090 recipients. For all 21,323 U.S. LVAD recipients during 2003-2014 the average age was 51.5 years old.
[email protected]
On Twitter @mitchelzoler
DALLAS – The percentage of left ventricular assist devices placed in U.S. heart failure patients at least 75 years of age jumped sharply during 2003-2014, and concurrently the short-term survival of these patients improved dramatically, according to data collected by the National Inpatient Sample.
During the 12-year period examined, the percentage of left-ventricular assist devices (LVADs) placed in U.S. heart failure patients aged 75 years and older rose from 3% of all LVADs in 2003 to 11% in 2014, Aniket S. Rali, MD, said at the annual scientific meeting of the Heart Failure Society of America.
The U.S. national numbers also showed that throughout the period studied, elderly U.S. patients who received an LVAD were increasingly sicker, with steadily increasing numbers of patients with a Charlson Comorbidity Index score of four or greater. Despite this, in-hospital mortality rates of elderly patients receiving an LVAD plummeted, dropping from 61% of elderly LVAD recipients in 2003 to 18% in 2014. During the same time, the percentage of elderly patients with a Charlson Comorbidity Index score greater than four doubled from 33% in 2003 to 66% in 2014, said Dr. Rali, a cardiologist at the University of Kansas Medical Center in Kansas City.
“If the Charlson Comorbidity Index score is increasing but in-hospital mortality is decreasing, then increased LVAD use is not a bad trend,” Dr. Rali said in an interview. He hopes that future analysis of longitudinal data from patients could identify clinical factors that link with better patient survival and help target LVAD placement to the patients who stand to gain the most benefit.
“We may be able to give these elderly patients not just longer life but improved quality of life” by a more informed targeting of LVADs, he suggested. “I think these numbers will help convince people that all is not lost,” he noted, for elderly heart failure patients who receive an LVAD as destination therapy. Patients at least 75 years old are not eligible for heart transplantation, so when these patients receive an LVAD it is, by definition, destination therapy.
The data also showed a marked sex disparity in LVAD use, with LVAD placement in men at least 75 years old rising from 1.4/1,000 patients in 2003 to 2.78/1,000 patients in 2014. In contrast, among women these rates rose from 0.8/1,000 patients in 2003 to 1.36/1,000 patients in 2014.
The average age for elderly U.S. LVAD recipients for the entire 12-year period studied was 77.6 years among a total of 2,090 recipients. For all 21,323 U.S. LVAD recipients during 2003-2014 the average age was 51.5 years old.
[email protected]
On Twitter @mitchelzoler
DALLAS – The percentage of left ventricular assist devices placed in U.S. heart failure patients at least 75 years of age jumped sharply during 2003-2014, and concurrently the short-term survival of these patients improved dramatically, according to data collected by the National Inpatient Sample.
During the 12-year period examined, the percentage of left-ventricular assist devices (LVADs) placed in U.S. heart failure patients aged 75 years and older rose from 3% of all LVADs in 2003 to 11% in 2014, Aniket S. Rali, MD, said at the annual scientific meeting of the Heart Failure Society of America.
The U.S. national numbers also showed that throughout the period studied, elderly U.S. patients who received an LVAD were increasingly sicker, with steadily increasing numbers of patients with a Charlson Comorbidity Index score of four or greater. Despite this, in-hospital mortality rates of elderly patients receiving an LVAD plummeted, dropping from 61% of elderly LVAD recipients in 2003 to 18% in 2014. During the same time, the percentage of elderly patients with a Charlson Comorbidity Index score greater than four doubled from 33% in 2003 to 66% in 2014, said Dr. Rali, a cardiologist at the University of Kansas Medical Center in Kansas City.
“If the Charlson Comorbidity Index score is increasing but in-hospital mortality is decreasing, then increased LVAD use is not a bad trend,” Dr. Rali said in an interview. He hopes that future analysis of longitudinal data from patients could identify clinical factors that link with better patient survival and help target LVAD placement to the patients who stand to gain the most benefit.
“We may be able to give these elderly patients not just longer life but improved quality of life” by a more informed targeting of LVADs, he suggested. “I think these numbers will help convince people that all is not lost,” he noted, for elderly heart failure patients who receive an LVAD as destination therapy. Patients at least 75 years old are not eligible for heart transplantation, so when these patients receive an LVAD it is, by definition, destination therapy.
The data also showed a marked sex disparity in LVAD use, with LVAD placement in men at least 75 years old rising from 1.4/1,000 patients in 2003 to 2.78/1,000 patients in 2014. In contrast, among women these rates rose from 0.8/1,000 patients in 2003 to 1.36/1,000 patients in 2014.
The average age for elderly U.S. LVAD recipients for the entire 12-year period studied was 77.6 years among a total of 2,090 recipients. For all 21,323 U.S. LVAD recipients during 2003-2014 the average age was 51.5 years old.
[email protected]
On Twitter @mitchelzoler
AT THE HFSA ANNUAL SCIENTIFIC MEETING
Key clinical point:
Major finding: Elderly U.S. patients receiving an LVAD rose from 3% of all LVADs placed in 2003 to 11% in 2014.
Data source: The U.S. National Inpatient Survey during 2003-2014.
Disclosures: Dr. Rali had no disclosures.
Tips and Tricks: Using a ‘Roman sandal’ after compartment syndrome treatment
Compartment syndrome is a common complication after revascularization for acute lower extremity ischemia. Treatment with four compartment fasciotomy can result in significant morbidity, with wounds that are challenging to manage for both the patient and practitioner.
An alternative to wound VAC or simple wet-to-dry dressings, the “Roman sandal” technique makes bedside closure of these wounds possible, especially in patients with minimal postdecompression muscle edema.
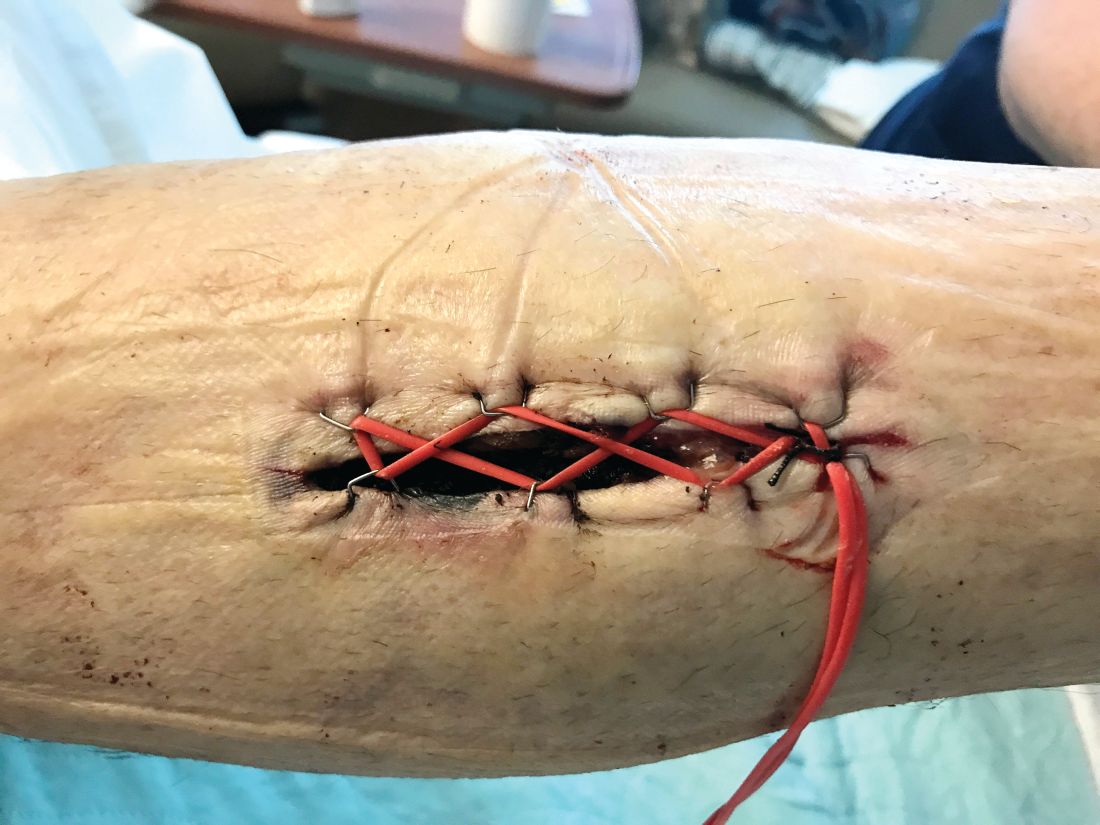

Compartment syndrome is a common complication after revascularization for acute lower extremity ischemia. Treatment with four compartment fasciotomy can result in significant morbidity, with wounds that are challenging to manage for both the patient and practitioner.
An alternative to wound VAC or simple wet-to-dry dressings, the “Roman sandal” technique makes bedside closure of these wounds possible, especially in patients with minimal postdecompression muscle edema.
Compartment syndrome is a common complication after revascularization for acute lower extremity ischemia. Treatment with four compartment fasciotomy can result in significant morbidity, with wounds that are challenging to manage for both the patient and practitioner.
An alternative to wound VAC or simple wet-to-dry dressings, the “Roman sandal” technique makes bedside closure of these wounds possible, especially in patients with minimal postdecompression muscle edema.
The Role of Synovial Cytokines in the Diagnosis of Periprosthetic Joint Infections: Current Concepts
Take-Home Points
- In cases of failed TJA, it is important to differentiate between septic and aseptic etiologies.
- Chronic and low-grade infections are challenging for orthopedic surgeons, as the symptoms often overlap with aseptic etiologies.
- Verification of infection eradication before beginning the second-stage reimplantation surgery is extremely important, but pre- and intraoperative findings can be unreliable.
- Synovial fluid cytokines have been shown to accurately diagnose PJIs.
- Synovial fluid cytokines may help surgeons differentiate between septic and aseptic cases of failed TJA.
Total joint arthroplasty (TJA) is an effective procedure that has been extensively used to relieve pain and improve quality of life in patients with various forms of joint disease. Although advances in technology and surgical technique have improved the success of TJA, periprosthetic joint infection (PJI) remains a serious complication. In the United States, it is estimated that PJI is the most common reason for total knee arthroplasty failure and the third most common reason for total hip arthroplasty revision.1 Although the incidence of PJI is 1% to 2%, the dramatic increase in TJA volume is expected to be accompanied by a similar rise in the number of infected TJAs; that number is expected to exceed 60,000 in the United States by 2020.2 Moreover, management of PJI is expensive and imposes a heavy burden on the healthcare system, with costs expected to hit $20 billion by 2020 in the US.2 Therefore, treating asepsis cases as infections imposes a heavy burden on the healthcare system and may result in excessive morbidity.3 At the same time, inadequate management of a PJI may result in recurrences that require infection treatment with morbid procedures, such as arthrodesis or amputation. Accurate diagnosis of PJI is of paramount importance in preventing potential implications of a misdiagnosed case. Unfortunately, the PJI diagnosis is extremely challenging, and the available diagnostic tests are often unreliable.4 Thus, research has recently focused on use of several synovial fluid cytokines in the detection of PJI.5-7 In this article, we provide an overview of the synovial biomarkers being used to diagnose PJI.
Diagnosis of Periprosthetic Joint Infection
Differentiating between septic and aseptic failed TJA is important, as the treatment options differ considerably. PJI can be broadly classified as acute or early postoperative (<6 weeks), late chronic (indolent onset), and acute-on-chronic (acute onset in well-functioning prosthesis, secondary to hematogenous spread).8 The acute and acute-on-chronic presentations are often associated with obvious signs of infection.9 However, chronic and low-grade infections pose a challenge to modern orthopedic practice, as the symptoms often overlap with that of aseptic causes of TJA failure.10 As a result, the International Consensus Group on Periprosthetic Joint Infection developed complex criteria using the Musculoskeletal Infection Society definition of PJI and involving a battery of tests for PJI diagnosis.11 According to these criteria, PJI is diagnosed when 1 of the 2 major criteria or 3 of the 5 minor criteria are met (Table 1).
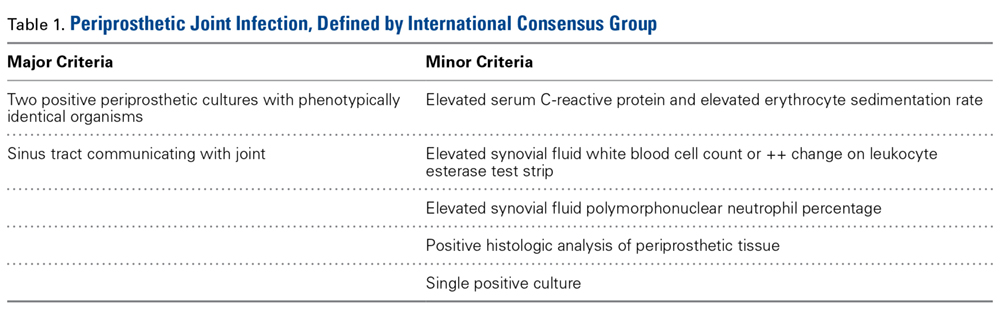
Although these criteria constitute the most agreed on and widely used standard for PJI diagnosis, the definition is complex and often incomplete until surgical intervention. An ideal diagnostic test would aid in managing a PJI and provide results before a treatment decision is made. Many revision surgeries are being performed with insufficient information about the true diagnosis, and the diagnosis might change during or after surgery. About 10% of the revisions presumed to be aseptic may unexpectedly grow cultures during surgery and thereby satisfy the criteria for PJI after surgery.12 Moreover, with the use of novel methods such as polymerase chain reaction, microorganisms were identified in more than three-fourths of the presumed aseptic revisions.13 The optimal management of such cases is controversial, and it is unclear whether positive cultures should be treated as possible contaminants or true infection.12,14
Verification of Infection Eradication
A 2-stage revision procedure, widely accepted as the standard treatment for PJI, has success rates approaching 94%.15 In this procedure, it is important to verify infection eradication before beginning the second-stage reimplantation. Verification is crucial in avoiding reimplantation of an infected joint.16 After the first stage, patients are usually administered intravenous antibiotics for at least 6 weeks; these antibiotics are then withheld, and systemic inflammatory markers are evaluated for infection eradication. Although reliable criteria have been established for PJI diagnosis, guidelines for detecting eradication of infection are rudimentary. Most surgeons monitor the decrease in serologic markers, such as erythrocyte sedimentation rate and C-reactive protein (CRP) level, to assess the response to treatment. However, noninfectious etiologies may result in continued elevation of these markers.17 Even though aspirations are often performed to diagnose persistent infection before the second-stage procedure, their diagnostic utility may be limited.18 Use of cultures is also limited, as presence of antibiotic-loaded spacers can decrease the sensitivity of culture.19 Inadequate diagnosis often leads to unnecessary continuation of antimicrobial therapy or additional surgical débridement. Nuclear scans often remain positive because of aseptic inflammation related to surgery and are not useful in documenting sepsis arrest.20 Given the limitations of available tests, novel strategies for identifying the presence of infection at the second stage are being tested.
Synovial Fluid Cytokines
PJI pathogenesis begins with colonization of the implant surfaces with microorganisms and subsequent formation of biofilms.21 The human immune system is activated by the microbial products, cell wall components, and various biofilm proteins. Immune cells are recruited to the site, where they secrete a myriad of inflammatory biomarkers, such as cytokines, which promote further recruitment of inflammatory cells and aid in the eradication of pathogens.9 These inflammatory cytokines and cells are involved in aseptic inflammatory joint conditions, such as rheumatoid arthritis22,23; however, some are specifically involved in immune pathways combating pathogens.24 This action is the basis for increasing interest in using various synovial fluid cytokines and other biomarkers in the diagnosis of PJI. Here we describe some of the commonly studied cytokines.
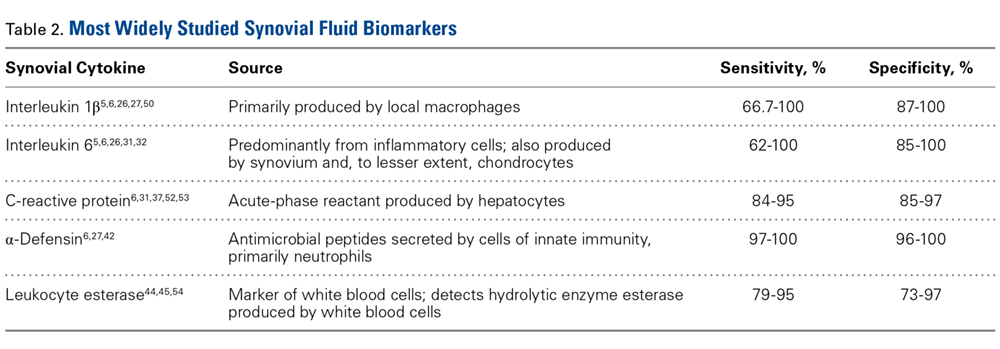
Interleukin 1β
Interleukin 1β (IL-1β) is a major proinflammatory cytokine that is synthesized by multiple cells, including macrophages and monocytes.25 IL-1β is produced in response to microorganisms, other cytokines, antigen-presenting cells, and immune complexes; stimulates production of acute-phase proteins by the liver; and is an important pyrogen.25 Deirmengian and colleagues5 found that synovial IL-1β increased 258-fold in patients with a PJI. Studies have found that synovial IL-1β has sensitivity ranging from 66.7% to 100% and specificity ranging from 87% to 100%, with 1 study reporting an accuracy of 100%.5,6,26,27
Interleukin 6
Also produced by macrophages and monocytes, interleukin 6 (IL-6) is a potent stimulator of acute-phase proteins.28,29 IL-6 has a role as a chemoattractant and helps with cell differentiation when changing from innate to acquired immunity.30 It is also used as an aid in diagnosing PJI; it has sensitivity ranging from 62% to 100% and specificity ranging from 85% to 100%.5,6,26,31,32 Synovial IL-6 measurements were more accurate than serum IL-6 measurements.26 Furthermore, synovial IL-6 can be increased up to 27-fold in PJI cases.5 In one study, synovial IL-6 levels >2100 pg/mL had sensitivity of 62.5% and specificity of 85.7% in PJI diagnosis26; in another study, an IL-6 threshold of 4270 pg/mL had sensitivity of 87.1%, specificity of 100%, and accuracy of 94.6%.31
C-Reactive Protein
CRP is an acute-phase reactant. Blood levels increase in response to aseptic inflammatory processes and systemic infection.33 CRP plays an important role in host defense by activating complement and helping mediate phagocytosis.33,34 Although serum CRP levels have been used in diagnosing PJIs,6 they can yield false-negative results.35,36 Therefore, attention turned to synovial CRP levels, which were found to be increased 13-fold in PJI cases.5 It has been shown that synovial CRP levels are significantly higher in infected vs noninfected prosthetic joints34 and had diagnostic accuracy better than that of serum CRP levels in diagnosing PJI.37 One study found that CRP at a threshold of 3.7 mg/L had sensitivity of 84%, specificity of 97.1%, and accuracy of 91.5%,37 whereas another study found that CRP at a threshold of 3.61 mg/L had sensitivity of 87.1%, specificity of 97.7%, and accuracy of 93.3%.31
α-Defensin
α-Defensin, a natural peptide produced and secreted by neutrophils in response to pathogens, has antimicrobial and cytotoxic properties,38-40 signals for the secretion of various cytokines, and acts as a chemoattractant for various immune cells.41 Deirmengian and colleagues6 found that α-defensin was consistently elevated in patients with PJI. α-Defensin is extremely accurate in diagnosing PJI; it has sensitivity ranging from 97% to 100% and specificity ranging from 96% to 100%.6,27,42 Moreover, α-defensin was effective in diagnosing PJI caused by a wide spectrum of organisms, including various low-virulence bacteria and fungi.43
Leukocyte Esterase
Leukocyte esterase is an enzyme produced and secreted by neutrophils at sites of active infection.7,44 Testing for this enzyme is performed with a colorimetric strip and was originally performed for the diagnosis of urinary tract infections.44,45 In a study conducted by Parvizi and colleagues,7 this strip was used to test for leukocyte esterase in synovial fluid samples; a ++ reading was found to have sensitivity of 80.6% and specificity of 100% in diagnosing knee PJI. Similarly, De Vecchi and colleagues45 found sensitivity of 92.6% and specificity of 97%.
Other Synovial Markers
Research has identified numerous molecular biomarkers that may be associated with the pathogenesis of PJI. Although several (eg, cytokines) have demonstrated higher levels in synovial fluid in patients with PJI than in normal controls, only a few have had clinically relevant diagnostic utility.6 Deirmengian and colleagues6 screened 43 synovial fluid biomarkers that potentially could be used in the diagnosis of PJI. Besides the cytokine α-defensin, 4 other biomarkers—lactoferrin, neutrophil gelatinase-associated lipocalcin, neutrophil elastase 2, and bactericidal/permeability-increasing protein—had accuracy of 100%. In addition, 8 cytokines and biomarkers (IL-8, CRP, resistin, thrombospondin, IL-1β, IL-6, IL-10, IL-1α) had area under the curve values higher than 0.9. Studies have also evaluated the diagnostic utility of metabolic products such as lactate, lactate dehydrogenase, and glucose; their accuracy was comparable to that of serum CRP.32
Serum Markers
In addition to the synovial fluid cytokines, several serum inflammatory cytokines have been studied as potential targets in diagnosing infection. Serum IL-6 has had excellent diagnostic accuracy46 and, when combined with CRP, could increase sensitivity in diagnosing PJI; such a combination (vs either test alone) could be useful in screening patients.47,48 Biomarkers such as tumor necrosis factor α and procalcitonin are considered very specific for PJI and may be useful in confirmatory testing.48 Evidence also suggests that toll-like receptor 2 proteins are elevated in the serum of patients with PJI and therefore are a potential diagnostic tool.49
Limitations of Synovial Cytokines
The literature suggests that some synovial fluid cytokines have promise.6 However, the best biomarker or combination of biomarkers is yet to be determined. Results have been consistent with α-defensin and other cytokines but mixed with IL-6 and still others32,42,50 (Table 2).
Information on the utility of synovial biomarkers in detecting persistent infection is limited. Frangiamore and colleagues50 found that IL-1 and IL-6 levels decreased between the stages of 2-stage revision. Unfortunately, none of the synovial fluid cytokines investigated (IL-1, IL-2, IL-6, IL-8, Il-10, interferon γ, granulocyte macrophage-colony stimulating factor, tumor necrosis factor α, IL-12p70) satisfactorily detected resolution of infection in the setting of prior treatment for PJI. Although cytokines are expected to be elevated in the presence of infection, the internal milieu at the time of stage 2 of the revision makes diagnosis of infection difficult. In addition, presence of spacer particles and recent surgery may activate immune pathways and yield false-positive results. Furthermore, antibiotic cement spacers may suppress the microorganisms to very low levels and yield false-negative results even if these organisms remain virulent.19
Even though the synovial molecular markers can detect the presence of infection, they are unable to identify pathogens. As identifying the pathogen is important in the treatment of PJI, there has been interest in using polymerase chain reaction (PCR) techniques.51 These tests may also provide specific information about the pathogen, such as its antibiotic sensitivity. A recently developed technology, the Ibis T5000 Universal Biosensor (Ibis Biosciences), uses novel pan-domain primers in a series of PCRs. This biosensor is useful in diagnosing infections when cultures are negative and appears to be more accurate than conventional PCR.13 As reported by Jacovides and colleagues,13 this novel PCR technique identified an organism in about 88% of presumed cases of aseptic revision.
Conclusion
PJI poses an extreme challenge to the healthcare system. Given the morbidity associated with improper management of PJI, accurate diagnosis is of paramount importance. Given the limitations of current tests, synovial fluid cytokines hold promise in the diagnosis of PJIs. However, these cytokines are expensive, and their clinical utility in PJI management is not well established. More research is needed before guidelines for synovial fluid cytokines and biomarkers can replace or be incorporated into guidelines for the treatment of PJIs.
1 Parvizi J, Adeli B, Zmistowski B, Restrepo C, Greenwald AS. Management of periprosthetic joint infection: the current knowledge: AAOS exhibit selection. J Bone Joint Surg Am. 2012;94(14):e104.
2. Kurtz SM, Lau E, Watson H, Schmier JK, Parvizi J. Economic burden of periprosthetic joint infection in the United States. J Arthroplasty. 2012;27(8 suppl):61-65.e1.
3. Sierra RJ, Trousdale RT, Pagnano MW. Above-the-knee amputation after a total knee replacement: prevalence, etiology, and functional outcome. J Bone Joint Surg Am. 2003;85(6):1000-1004.
4. Bauer TW, Parvizi J, Kobayashi N, Krebs V. Diagnosis of periprosthetic infection. J Bone Joint Surg Am. 2006;88(4):869-882.
5. Deirmengian C, Hallab N, Tarabishy A, et al. Synovial fluid biomarkers for periprosthetic infection. Clin Orthop Relat Res. 2010;468(8):2017-2023.
6. Deirmengian C, Kardos K, Kilmartin P, Cameron A, Schiller K, Parvizi J. Diagnosing periprosthetic joint infection: has the era of the biomarker arrived? Clin Orthop Relat Res. 2014;472(11):3254-3262.
7. Parvizi J, Jacovides C, Antoci V, Ghanem E. Diagnosis of periprosthetic joint infection: the utility of a simple yet unappreciated enzyme. J Bone Joint Surg Am. 2011;93(24):2242-2248.
8. Kuiper JW, Willink RT, Moojen DJF, van den Bekerom MP, Colen S. Treatment of acute periprosthetic infections with prosthesis retention: review of current concepts. World J Orthop. 2014;5(5):667-676.
9. Zimmerli W, Trampuz A, Ochsner PE. Prosthetic-joint infections. N Engl J Med. 2004;351(16):1645-1654.
10. Osmon DR, Berbari EF, Berendt AR, et al. Diagnosis and management of prosthetic joint infection: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2013;56(1):e1-e25.
11. Parvizi J, Gehrke T; International Consensus Group on Periprosthetic Joint Infection. Definition of periprosthetic joint infection. J Arthroplasty. 2014;29(7):1331.
12. Saleh A, Guirguis A, Klika AK, Johnson L, Higuera CA, Barsoum WK. Unexpected positive intraoperative cultures in aseptic revision arthroplasty. J Arthroplasty. 2014;29(11):2181-2186.
13. Jacovides CL, Kreft R, Adeli B, Hozack B, Ehrlich GD, Parvizi J. Successful identification of pathogens by polymerase chain reaction (PCR)–based electron spray ionization time-of-flight mass spectrometry (ESI-TOF-MS) in culture-negative periprosthetic joint infection. J Bone Joint Surg Am. 2012;94(24):2247-2254.
14. Barrack RL, Aggarwal A, Burnett RS, et al. The fate of the unexpected positive intraoperative cultures after revision total knee arthroplasty. J Arthroplasty. 2007;22(6 suppl 2):94-99.
15. Macheras GA, Koutsostathis SD, Kateros K, Papadakis S, Anastasopoulos P. A two stage re-implantation protocol for the treatment of deep periprosthetic hip infection. Mid to long-term results. Hip Int. 2012;22(suppl 8):S54-S61.
16. George J, Kwiecien G, Klika AK, et al. Are frozen sections and MSIS criteria reliable at the time of reimplantation of two-stage revision arthroplasty? Clin Orthop Relat Res. 2016;474(7):1619-1626.
17. Kusuma SK, Ward J, Jacofsky M, Sporer SM, Della Valle CJ. What is the role of serological testing between stages of two-stage reconstruction of the infected prosthetic knee? Clin Orthop Relat Res. 2011;469(4):1002-1008.
18. Lonner JH, Siliski JM, Della Valle C, DiCesare P, Lotke PA. Role of knee aspiration after resection of the infected total knee arthroplasty. Am J Orthop. 2001;30(4):305-309.
19. Mont MA, Waldman BJ, Hungerford DS. Evaluation of preoperative cultures before second-stage reimplantation of a total knee prosthesis complicated by infection. A comparison-group study. J Bone Joint Surg Am. 2000;82(11):1552-1557.
20. Love C, Marwin SE, Palestro CJ. Nuclear medicine and the infected joint replacement. Semin Nucl Med. 2009;39(1):66-78.
21. Zimmerli W, Moser C. Pathogenesis and treatment concepts of orthopaedic biofilm infections. FEMS Immunol Med Microbiol. 2012;65(2):158-168.
22. Fontana A, Hengartner H, Weber E, Fehr K, Grob PJ, Cohen G. Interleukin 1 activity in the synovial fluid of patients with rheumatoid arthritis. Rheumatol Int. 1982;2(2):49-53.
23. Guerne PA, Zuraw BL, Vaughan JH, Carson DA, Lotz M. Synovium as a source of interleukin 6 in vitro. Contribution to local and systemic manifestations of arthritis. J Clin Invest. 1989;83(2):585-592.
24. Wang G. Human antimicrobial peptides and proteins. Pharmaceuticals (Basel). 2014;7(5):545-594.
25. Stylianou E, Saklatvala J. Interleukin-1. Int J Biochem Cell Biol. 1998;30(10):1075-1079.
26. Gollwitzer H, Dombrowski Y, Prodinger PM, et al. Antimicrobial peptides and proinflammatory cytokines in periprosthetic joint infection. J Bone Joint Surg Am. 2013;95(7):644-651.
27. Deirmengian C, Kardos K, Kilmartin P, Cameron A, Schiller K, Parvizi J. Combined measurement of synovial fluid α-defensin and C-reactive protein levels: highly accurate for diagnosing periprosthetic joint infection. J Bone Joint Surg Am. 2014;96(17):1439-1445.
28. Randau TM, Friedrich MJ, Wimmer MD, et al. Interleukin-6 in serum and in synovial fluid enhances the differentiation between periprosthetic joint infection and aseptic loosening. PLoS One. 2014;9(2):e89045.
29. Heinrich PC, Castell JV, Andus T. Interleukin-6 and the acute phase response. Biochem J. 1990;265(3):621-636.
30. Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011;1813(5):878-888.
31. Jacovides CL, Parvizi J, Adeli B, Jung KA. Molecular markers for diagnosis of periprosthetic joint infection. J Arthroplasty. 2011;26(6 suppl):99-103.e1.
32. Lenski M, Scherer MA. Synovial IL-6 as inflammatory marker in periprosthetic joint infections. J Arthroplasty. 2014;29(6):1105-1109.
33. Mortensen RF. C-reactive protein, inflammation, and innate immunity. Immunol Res. 2001;24(2):163-176.
34. Parvizi J, McKenzie JC, Cashman JP. Diagnosis of periprosthetic joint infection using synovial C-reactive protein. J Arthroplasty. 2012;27(8 suppl):12-16.
35. Ghanem E, Antoci V, Pulido L, Joshi A, Hozack W, Parvizi J. The use of receiver operating characteristics analysis in determining erythrocyte sedimentation rate and C-reactive protein levels in diagnosing periprosthetic infection prior to revision total hip arthroplasty. Int J Infect Dis. 2009;13(6):e444-e449.
36. Johnson AJ, Zywiel MG, Stroh A, Marker DR, Mont MA. Serological markers can lead to false negative diagnoses of periprosthetic infections following total knee arthroplasty. Int Orthop. 2011;35(11):1621-1626.
37. Parvizi J, Jacovides C, Adeli B, Jung KA, Hozack WJ. Mark B. Coventry award: synovial C-reactive protein: a prospective evaluation of a molecular marker for periprosthetic knee joint infection. Clin Orthop Relat Res. 2012;470(1):54-60.
38. Lehrer RI, Lichtenstein AK, Ganz T. Defensins: antimicrobial and cytotoxic peptides of mammalian cells. Annu Rev Immunol. 1993;11:105-128.
39. Ganz T, Selsted ME, Szklarek D, et al. Defensins. Natural peptide antibiotics of human neutrophils. J Clin Invest. 1985;76(4):1427-1435.
40. Chalifour A, Jeannin P, Gauchat JF, et al. Direct bacterial protein PAMP recognition by human NK cells involves TLRs and triggers alpha-defensin production. Blood. 2004;104(6):1778-1783.
41. Ulm H, Wilmes M, Shai Y, Sahl HG. Antimicrobial host defensins—specific antibiotic activities and innate defense modulation. Front Immunol. 2012;3:249.
42. Bingham J, Clarke H, Spangehl M, Schwartz A, Beauchamp C, Goldberg B. The alpha defensin-1 biomarker assay can be used to evaluate the potentially infected total joint arthroplasty. Clin Orthop Relat Res. 2014;472(12):4006-4009.
43. Deirmengian C, Kardos K, Kilmartin P, Gulati S, Citrano P, Booth RE. The alpha-defensin test for periprosthetic joint infection responds to a wide spectrum of organisms. Clin Orthop Relat Res. 2015;473(7):2229-2235.
44. Tischler EH, Cavanaugh PK, Parvizi J. Leukocyte esterase strip test: matched for Musculoskeletal Infection Society criteria. J Bone Joint Surg Am. 2014;96(22):1917-1920.
45. De Vecchi E, Villa F, Bortolin M, et al. Leucocyte esterase, glucose and C-reactive protein in the diagnosis of prosthetic joint infections: a prospective study. Clin Microbiol Infect. 2016;22(6):555-560.
46. Di Cesare PE, Chang E, Preston CF, Liu C. Serum interleukin-6 as a marker of periprosthetic infection following total hip and knee arthroplasty. J Bone Joint Surg Am. 2005;87(9):1921-1927.
47. Ettinger M, Calliess T, Kielstein JT, et al. Circulating biomarkers for discrimination between aseptic joint failure, low-grade infection, and high-grade septic failure. Clin Infect Dis. 2015;61(3):332-341.
48. Bottner F, Wegner A, Winkelmann W, Becker K, Erren M, Götze C. Interleukin-6, procalcitonin and TNF-alpha: markers of peri-prosthetic infection following total joint replacement. J Bone Joint Surg Br. 2007;89(1):94-99.
49. Galliera E, Drago L, Vassena C, et al. Toll-like receptor 2 in serum: a potential diagnostic marker of prosthetic joint infection? J Clin Microbiol. 2014;52(2):620-623.
50. Frangiamore SJ, Siqueira MB, Saleh A, Daly T, Higuera CA, Barsoum WK. Synovial cytokines and the MSIS criteria are not useful for determining infection resolution after periprosthetic joint infection explantation. Clin Orthop Relat Res. 2016;474(7):1630-1639.
51. Rasouli MR, Harandi AA, Adeli B, Purtill JJ, Parvizi J. Revision total knee arthroplasty: infection should be ruled out in all cases. J Arthroplasty. 2012;27(6):1239-1243.e1-e2.
52. Omar M, Ettinger M, Reichling M, et al. Synovial C-reactive protein as a marker for chronic periprosthetic infection in total hip arthroplasty. Bone Joint J. 2015;97(2):173-176.
53. Tetreault MW, Wetters NG, Moric M, Gross CE, Della Valle CJ. Is synovial C-reactive protein a useful marker for periprosthetic joint infection? Clin Orthop Relat Res. 2014;472(12):3997-4003.
54. Omar M, Ettinger M, Reichling M, et al. Preliminary results of a new test for rapid diagnosis of septic arthritis with use of leukocyte esterase and glucose reagent strips. J Bone Joint Surg Am. 2014;96(24):2032-2037.
Take-Home Points
- In cases of failed TJA, it is important to differentiate between septic and aseptic etiologies.
- Chronic and low-grade infections are challenging for orthopedic surgeons, as the symptoms often overlap with aseptic etiologies.
- Verification of infection eradication before beginning the second-stage reimplantation surgery is extremely important, but pre- and intraoperative findings can be unreliable.
- Synovial fluid cytokines have been shown to accurately diagnose PJIs.
- Synovial fluid cytokines may help surgeons differentiate between septic and aseptic cases of failed TJA.
Total joint arthroplasty (TJA) is an effective procedure that has been extensively used to relieve pain and improve quality of life in patients with various forms of joint disease. Although advances in technology and surgical technique have improved the success of TJA, periprosthetic joint infection (PJI) remains a serious complication. In the United States, it is estimated that PJI is the most common reason for total knee arthroplasty failure and the third most common reason for total hip arthroplasty revision.1 Although the incidence of PJI is 1% to 2%, the dramatic increase in TJA volume is expected to be accompanied by a similar rise in the number of infected TJAs; that number is expected to exceed 60,000 in the United States by 2020.2 Moreover, management of PJI is expensive and imposes a heavy burden on the healthcare system, with costs expected to hit $20 billion by 2020 in the US.2 Therefore, treating asepsis cases as infections imposes a heavy burden on the healthcare system and may result in excessive morbidity.3 At the same time, inadequate management of a PJI may result in recurrences that require infection treatment with morbid procedures, such as arthrodesis or amputation. Accurate diagnosis of PJI is of paramount importance in preventing potential implications of a misdiagnosed case. Unfortunately, the PJI diagnosis is extremely challenging, and the available diagnostic tests are often unreliable.4 Thus, research has recently focused on use of several synovial fluid cytokines in the detection of PJI.5-7 In this article, we provide an overview of the synovial biomarkers being used to diagnose PJI.
Diagnosis of Periprosthetic Joint Infection
Differentiating between septic and aseptic failed TJA is important, as the treatment options differ considerably. PJI can be broadly classified as acute or early postoperative (<6 weeks), late chronic (indolent onset), and acute-on-chronic (acute onset in well-functioning prosthesis, secondary to hematogenous spread).8 The acute and acute-on-chronic presentations are often associated with obvious signs of infection.9 However, chronic and low-grade infections pose a challenge to modern orthopedic practice, as the symptoms often overlap with that of aseptic causes of TJA failure.10 As a result, the International Consensus Group on Periprosthetic Joint Infection developed complex criteria using the Musculoskeletal Infection Society definition of PJI and involving a battery of tests for PJI diagnosis.11 According to these criteria, PJI is diagnosed when 1 of the 2 major criteria or 3 of the 5 minor criteria are met (Table 1).
Although these criteria constitute the most agreed on and widely used standard for PJI diagnosis, the definition is complex and often incomplete until surgical intervention. An ideal diagnostic test would aid in managing a PJI and provide results before a treatment decision is made. Many revision surgeries are being performed with insufficient information about the true diagnosis, and the diagnosis might change during or after surgery. About 10% of the revisions presumed to be aseptic may unexpectedly grow cultures during surgery and thereby satisfy the criteria for PJI after surgery.12 Moreover, with the use of novel methods such as polymerase chain reaction, microorganisms were identified in more than three-fourths of the presumed aseptic revisions.13 The optimal management of such cases is controversial, and it is unclear whether positive cultures should be treated as possible contaminants or true infection.12,14
Verification of Infection Eradication
A 2-stage revision procedure, widely accepted as the standard treatment for PJI, has success rates approaching 94%.15 In this procedure, it is important to verify infection eradication before beginning the second-stage reimplantation. Verification is crucial in avoiding reimplantation of an infected joint.16 After the first stage, patients are usually administered intravenous antibiotics for at least 6 weeks; these antibiotics are then withheld, and systemic inflammatory markers are evaluated for infection eradication. Although reliable criteria have been established for PJI diagnosis, guidelines for detecting eradication of infection are rudimentary. Most surgeons monitor the decrease in serologic markers, such as erythrocyte sedimentation rate and C-reactive protein (CRP) level, to assess the response to treatment. However, noninfectious etiologies may result in continued elevation of these markers.17 Even though aspirations are often performed to diagnose persistent infection before the second-stage procedure, their diagnostic utility may be limited.18 Use of cultures is also limited, as presence of antibiotic-loaded spacers can decrease the sensitivity of culture.19 Inadequate diagnosis often leads to unnecessary continuation of antimicrobial therapy or additional surgical débridement. Nuclear scans often remain positive because of aseptic inflammation related to surgery and are not useful in documenting sepsis arrest.20 Given the limitations of available tests, novel strategies for identifying the presence of infection at the second stage are being tested.
Synovial Fluid Cytokines
PJI pathogenesis begins with colonization of the implant surfaces with microorganisms and subsequent formation of biofilms.21 The human immune system is activated by the microbial products, cell wall components, and various biofilm proteins. Immune cells are recruited to the site, where they secrete a myriad of inflammatory biomarkers, such as cytokines, which promote further recruitment of inflammatory cells and aid in the eradication of pathogens.9 These inflammatory cytokines and cells are involved in aseptic inflammatory joint conditions, such as rheumatoid arthritis22,23; however, some are specifically involved in immune pathways combating pathogens.24 This action is the basis for increasing interest in using various synovial fluid cytokines and other biomarkers in the diagnosis of PJI. Here we describe some of the commonly studied cytokines.
Interleukin 1β
Interleukin 1β (IL-1β) is a major proinflammatory cytokine that is synthesized by multiple cells, including macrophages and monocytes.25 IL-1β is produced in response to microorganisms, other cytokines, antigen-presenting cells, and immune complexes; stimulates production of acute-phase proteins by the liver; and is an important pyrogen.25 Deirmengian and colleagues5 found that synovial IL-1β increased 258-fold in patients with a PJI. Studies have found that synovial IL-1β has sensitivity ranging from 66.7% to 100% and specificity ranging from 87% to 100%, with 1 study reporting an accuracy of 100%.5,6,26,27
Interleukin 6
Also produced by macrophages and monocytes, interleukin 6 (IL-6) is a potent stimulator of acute-phase proteins.28,29 IL-6 has a role as a chemoattractant and helps with cell differentiation when changing from innate to acquired immunity.30 It is also used as an aid in diagnosing PJI; it has sensitivity ranging from 62% to 100% and specificity ranging from 85% to 100%.5,6,26,31,32 Synovial IL-6 measurements were more accurate than serum IL-6 measurements.26 Furthermore, synovial IL-6 can be increased up to 27-fold in PJI cases.5 In one study, synovial IL-6 levels >2100 pg/mL had sensitivity of 62.5% and specificity of 85.7% in PJI diagnosis26; in another study, an IL-6 threshold of 4270 pg/mL had sensitivity of 87.1%, specificity of 100%, and accuracy of 94.6%.31
C-Reactive Protein
CRP is an acute-phase reactant. Blood levels increase in response to aseptic inflammatory processes and systemic infection.33 CRP plays an important role in host defense by activating complement and helping mediate phagocytosis.33,34 Although serum CRP levels have been used in diagnosing PJIs,6 they can yield false-negative results.35,36 Therefore, attention turned to synovial CRP levels, which were found to be increased 13-fold in PJI cases.5 It has been shown that synovial CRP levels are significantly higher in infected vs noninfected prosthetic joints34 and had diagnostic accuracy better than that of serum CRP levels in diagnosing PJI.37 One study found that CRP at a threshold of 3.7 mg/L had sensitivity of 84%, specificity of 97.1%, and accuracy of 91.5%,37 whereas another study found that CRP at a threshold of 3.61 mg/L had sensitivity of 87.1%, specificity of 97.7%, and accuracy of 93.3%.31
α-Defensin
α-Defensin, a natural peptide produced and secreted by neutrophils in response to pathogens, has antimicrobial and cytotoxic properties,38-40 signals for the secretion of various cytokines, and acts as a chemoattractant for various immune cells.41 Deirmengian and colleagues6 found that α-defensin was consistently elevated in patients with PJI. α-Defensin is extremely accurate in diagnosing PJI; it has sensitivity ranging from 97% to 100% and specificity ranging from 96% to 100%.6,27,42 Moreover, α-defensin was effective in diagnosing PJI caused by a wide spectrum of organisms, including various low-virulence bacteria and fungi.43
Leukocyte Esterase
Leukocyte esterase is an enzyme produced and secreted by neutrophils at sites of active infection.7,44 Testing for this enzyme is performed with a colorimetric strip and was originally performed for the diagnosis of urinary tract infections.44,45 In a study conducted by Parvizi and colleagues,7 this strip was used to test for leukocyte esterase in synovial fluid samples; a ++ reading was found to have sensitivity of 80.6% and specificity of 100% in diagnosing knee PJI. Similarly, De Vecchi and colleagues45 found sensitivity of 92.6% and specificity of 97%.
Other Synovial Markers
Research has identified numerous molecular biomarkers that may be associated with the pathogenesis of PJI. Although several (eg, cytokines) have demonstrated higher levels in synovial fluid in patients with PJI than in normal controls, only a few have had clinically relevant diagnostic utility.6 Deirmengian and colleagues6 screened 43 synovial fluid biomarkers that potentially could be used in the diagnosis of PJI. Besides the cytokine α-defensin, 4 other biomarkers—lactoferrin, neutrophil gelatinase-associated lipocalcin, neutrophil elastase 2, and bactericidal/permeability-increasing protein—had accuracy of 100%. In addition, 8 cytokines and biomarkers (IL-8, CRP, resistin, thrombospondin, IL-1β, IL-6, IL-10, IL-1α) had area under the curve values higher than 0.9. Studies have also evaluated the diagnostic utility of metabolic products such as lactate, lactate dehydrogenase, and glucose; their accuracy was comparable to that of serum CRP.32
Serum Markers
In addition to the synovial fluid cytokines, several serum inflammatory cytokines have been studied as potential targets in diagnosing infection. Serum IL-6 has had excellent diagnostic accuracy46 and, when combined with CRP, could increase sensitivity in diagnosing PJI; such a combination (vs either test alone) could be useful in screening patients.47,48 Biomarkers such as tumor necrosis factor α and procalcitonin are considered very specific for PJI and may be useful in confirmatory testing.48 Evidence also suggests that toll-like receptor 2 proteins are elevated in the serum of patients with PJI and therefore are a potential diagnostic tool.49
Limitations of Synovial Cytokines
The literature suggests that some synovial fluid cytokines have promise.6 However, the best biomarker or combination of biomarkers is yet to be determined. Results have been consistent with α-defensin and other cytokines but mixed with IL-6 and still others32,42,50 (Table 2).
Information on the utility of synovial biomarkers in detecting persistent infection is limited. Frangiamore and colleagues50 found that IL-1 and IL-6 levels decreased between the stages of 2-stage revision. Unfortunately, none of the synovial fluid cytokines investigated (IL-1, IL-2, IL-6, IL-8, Il-10, interferon γ, granulocyte macrophage-colony stimulating factor, tumor necrosis factor α, IL-12p70) satisfactorily detected resolution of infection in the setting of prior treatment for PJI. Although cytokines are expected to be elevated in the presence of infection, the internal milieu at the time of stage 2 of the revision makes diagnosis of infection difficult. In addition, presence of spacer particles and recent surgery may activate immune pathways and yield false-positive results. Furthermore, antibiotic cement spacers may suppress the microorganisms to very low levels and yield false-negative results even if these organisms remain virulent.19
Even though the synovial molecular markers can detect the presence of infection, they are unable to identify pathogens. As identifying the pathogen is important in the treatment of PJI, there has been interest in using polymerase chain reaction (PCR) techniques.51 These tests may also provide specific information about the pathogen, such as its antibiotic sensitivity. A recently developed technology, the Ibis T5000 Universal Biosensor (Ibis Biosciences), uses novel pan-domain primers in a series of PCRs. This biosensor is useful in diagnosing infections when cultures are negative and appears to be more accurate than conventional PCR.13 As reported by Jacovides and colleagues,13 this novel PCR technique identified an organism in about 88% of presumed cases of aseptic revision.
Conclusion
PJI poses an extreme challenge to the healthcare system. Given the morbidity associated with improper management of PJI, accurate diagnosis is of paramount importance. Given the limitations of current tests, synovial fluid cytokines hold promise in the diagnosis of PJIs. However, these cytokines are expensive, and their clinical utility in PJI management is not well established. More research is needed before guidelines for synovial fluid cytokines and biomarkers can replace or be incorporated into guidelines for the treatment of PJIs.
Take-Home Points
- In cases of failed TJA, it is important to differentiate between septic and aseptic etiologies.
- Chronic and low-grade infections are challenging for orthopedic surgeons, as the symptoms often overlap with aseptic etiologies.
- Verification of infection eradication before beginning the second-stage reimplantation surgery is extremely important, but pre- and intraoperative findings can be unreliable.
- Synovial fluid cytokines have been shown to accurately diagnose PJIs.
- Synovial fluid cytokines may help surgeons differentiate between septic and aseptic cases of failed TJA.
Total joint arthroplasty (TJA) is an effective procedure that has been extensively used to relieve pain and improve quality of life in patients with various forms of joint disease. Although advances in technology and surgical technique have improved the success of TJA, periprosthetic joint infection (PJI) remains a serious complication. In the United States, it is estimated that PJI is the most common reason for total knee arthroplasty failure and the third most common reason for total hip arthroplasty revision.1 Although the incidence of PJI is 1% to 2%, the dramatic increase in TJA volume is expected to be accompanied by a similar rise in the number of infected TJAs; that number is expected to exceed 60,000 in the United States by 2020.2 Moreover, management of PJI is expensive and imposes a heavy burden on the healthcare system, with costs expected to hit $20 billion by 2020 in the US.2 Therefore, treating asepsis cases as infections imposes a heavy burden on the healthcare system and may result in excessive morbidity.3 At the same time, inadequate management of a PJI may result in recurrences that require infection treatment with morbid procedures, such as arthrodesis or amputation. Accurate diagnosis of PJI is of paramount importance in preventing potential implications of a misdiagnosed case. Unfortunately, the PJI diagnosis is extremely challenging, and the available diagnostic tests are often unreliable.4 Thus, research has recently focused on use of several synovial fluid cytokines in the detection of PJI.5-7 In this article, we provide an overview of the synovial biomarkers being used to diagnose PJI.
Diagnosis of Periprosthetic Joint Infection
Differentiating between septic and aseptic failed TJA is important, as the treatment options differ considerably. PJI can be broadly classified as acute or early postoperative (<6 weeks), late chronic (indolent onset), and acute-on-chronic (acute onset in well-functioning prosthesis, secondary to hematogenous spread).8 The acute and acute-on-chronic presentations are often associated with obvious signs of infection.9 However, chronic and low-grade infections pose a challenge to modern orthopedic practice, as the symptoms often overlap with that of aseptic causes of TJA failure.10 As a result, the International Consensus Group on Periprosthetic Joint Infection developed complex criteria using the Musculoskeletal Infection Society definition of PJI and involving a battery of tests for PJI diagnosis.11 According to these criteria, PJI is diagnosed when 1 of the 2 major criteria or 3 of the 5 minor criteria are met (Table 1).
Although these criteria constitute the most agreed on and widely used standard for PJI diagnosis, the definition is complex and often incomplete until surgical intervention. An ideal diagnostic test would aid in managing a PJI and provide results before a treatment decision is made. Many revision surgeries are being performed with insufficient information about the true diagnosis, and the diagnosis might change during or after surgery. About 10% of the revisions presumed to be aseptic may unexpectedly grow cultures during surgery and thereby satisfy the criteria for PJI after surgery.12 Moreover, with the use of novel methods such as polymerase chain reaction, microorganisms were identified in more than three-fourths of the presumed aseptic revisions.13 The optimal management of such cases is controversial, and it is unclear whether positive cultures should be treated as possible contaminants or true infection.12,14
Verification of Infection Eradication
A 2-stage revision procedure, widely accepted as the standard treatment for PJI, has success rates approaching 94%.15 In this procedure, it is important to verify infection eradication before beginning the second-stage reimplantation. Verification is crucial in avoiding reimplantation of an infected joint.16 After the first stage, patients are usually administered intravenous antibiotics for at least 6 weeks; these antibiotics are then withheld, and systemic inflammatory markers are evaluated for infection eradication. Although reliable criteria have been established for PJI diagnosis, guidelines for detecting eradication of infection are rudimentary. Most surgeons monitor the decrease in serologic markers, such as erythrocyte sedimentation rate and C-reactive protein (CRP) level, to assess the response to treatment. However, noninfectious etiologies may result in continued elevation of these markers.17 Even though aspirations are often performed to diagnose persistent infection before the second-stage procedure, their diagnostic utility may be limited.18 Use of cultures is also limited, as presence of antibiotic-loaded spacers can decrease the sensitivity of culture.19 Inadequate diagnosis often leads to unnecessary continuation of antimicrobial therapy or additional surgical débridement. Nuclear scans often remain positive because of aseptic inflammation related to surgery and are not useful in documenting sepsis arrest.20 Given the limitations of available tests, novel strategies for identifying the presence of infection at the second stage are being tested.
Synovial Fluid Cytokines
PJI pathogenesis begins with colonization of the implant surfaces with microorganisms and subsequent formation of biofilms.21 The human immune system is activated by the microbial products, cell wall components, and various biofilm proteins. Immune cells are recruited to the site, where they secrete a myriad of inflammatory biomarkers, such as cytokines, which promote further recruitment of inflammatory cells and aid in the eradication of pathogens.9 These inflammatory cytokines and cells are involved in aseptic inflammatory joint conditions, such as rheumatoid arthritis22,23; however, some are specifically involved in immune pathways combating pathogens.24 This action is the basis for increasing interest in using various synovial fluid cytokines and other biomarkers in the diagnosis of PJI. Here we describe some of the commonly studied cytokines.
Interleukin 1β
Interleukin 1β (IL-1β) is a major proinflammatory cytokine that is synthesized by multiple cells, including macrophages and monocytes.25 IL-1β is produced in response to microorganisms, other cytokines, antigen-presenting cells, and immune complexes; stimulates production of acute-phase proteins by the liver; and is an important pyrogen.25 Deirmengian and colleagues5 found that synovial IL-1β increased 258-fold in patients with a PJI. Studies have found that synovial IL-1β has sensitivity ranging from 66.7% to 100% and specificity ranging from 87% to 100%, with 1 study reporting an accuracy of 100%.5,6,26,27
Interleukin 6
Also produced by macrophages and monocytes, interleukin 6 (IL-6) is a potent stimulator of acute-phase proteins.28,29 IL-6 has a role as a chemoattractant and helps with cell differentiation when changing from innate to acquired immunity.30 It is also used as an aid in diagnosing PJI; it has sensitivity ranging from 62% to 100% and specificity ranging from 85% to 100%.5,6,26,31,32 Synovial IL-6 measurements were more accurate than serum IL-6 measurements.26 Furthermore, synovial IL-6 can be increased up to 27-fold in PJI cases.5 In one study, synovial IL-6 levels >2100 pg/mL had sensitivity of 62.5% and specificity of 85.7% in PJI diagnosis26; in another study, an IL-6 threshold of 4270 pg/mL had sensitivity of 87.1%, specificity of 100%, and accuracy of 94.6%.31
C-Reactive Protein
CRP is an acute-phase reactant. Blood levels increase in response to aseptic inflammatory processes and systemic infection.33 CRP plays an important role in host defense by activating complement and helping mediate phagocytosis.33,34 Although serum CRP levels have been used in diagnosing PJIs,6 they can yield false-negative results.35,36 Therefore, attention turned to synovial CRP levels, which were found to be increased 13-fold in PJI cases.5 It has been shown that synovial CRP levels are significantly higher in infected vs noninfected prosthetic joints34 and had diagnostic accuracy better than that of serum CRP levels in diagnosing PJI.37 One study found that CRP at a threshold of 3.7 mg/L had sensitivity of 84%, specificity of 97.1%, and accuracy of 91.5%,37 whereas another study found that CRP at a threshold of 3.61 mg/L had sensitivity of 87.1%, specificity of 97.7%, and accuracy of 93.3%.31
α-Defensin
α-Defensin, a natural peptide produced and secreted by neutrophils in response to pathogens, has antimicrobial and cytotoxic properties,38-40 signals for the secretion of various cytokines, and acts as a chemoattractant for various immune cells.41 Deirmengian and colleagues6 found that α-defensin was consistently elevated in patients with PJI. α-Defensin is extremely accurate in diagnosing PJI; it has sensitivity ranging from 97% to 100% and specificity ranging from 96% to 100%.6,27,42 Moreover, α-defensin was effective in diagnosing PJI caused by a wide spectrum of organisms, including various low-virulence bacteria and fungi.43
Leukocyte Esterase
Leukocyte esterase is an enzyme produced and secreted by neutrophils at sites of active infection.7,44 Testing for this enzyme is performed with a colorimetric strip and was originally performed for the diagnosis of urinary tract infections.44,45 In a study conducted by Parvizi and colleagues,7 this strip was used to test for leukocyte esterase in synovial fluid samples; a ++ reading was found to have sensitivity of 80.6% and specificity of 100% in diagnosing knee PJI. Similarly, De Vecchi and colleagues45 found sensitivity of 92.6% and specificity of 97%.
Other Synovial Markers
Research has identified numerous molecular biomarkers that may be associated with the pathogenesis of PJI. Although several (eg, cytokines) have demonstrated higher levels in synovial fluid in patients with PJI than in normal controls, only a few have had clinically relevant diagnostic utility.6 Deirmengian and colleagues6 screened 43 synovial fluid biomarkers that potentially could be used in the diagnosis of PJI. Besides the cytokine α-defensin, 4 other biomarkers—lactoferrin, neutrophil gelatinase-associated lipocalcin, neutrophil elastase 2, and bactericidal/permeability-increasing protein—had accuracy of 100%. In addition, 8 cytokines and biomarkers (IL-8, CRP, resistin, thrombospondin, IL-1β, IL-6, IL-10, IL-1α) had area under the curve values higher than 0.9. Studies have also evaluated the diagnostic utility of metabolic products such as lactate, lactate dehydrogenase, and glucose; their accuracy was comparable to that of serum CRP.32
Serum Markers
In addition to the synovial fluid cytokines, several serum inflammatory cytokines have been studied as potential targets in diagnosing infection. Serum IL-6 has had excellent diagnostic accuracy46 and, when combined with CRP, could increase sensitivity in diagnosing PJI; such a combination (vs either test alone) could be useful in screening patients.47,48 Biomarkers such as tumor necrosis factor α and procalcitonin are considered very specific for PJI and may be useful in confirmatory testing.48 Evidence also suggests that toll-like receptor 2 proteins are elevated in the serum of patients with PJI and therefore are a potential diagnostic tool.49
Limitations of Synovial Cytokines
The literature suggests that some synovial fluid cytokines have promise.6 However, the best biomarker or combination of biomarkers is yet to be determined. Results have been consistent with α-defensin and other cytokines but mixed with IL-6 and still others32,42,50 (Table 2).
Information on the utility of synovial biomarkers in detecting persistent infection is limited. Frangiamore and colleagues50 found that IL-1 and IL-6 levels decreased between the stages of 2-stage revision. Unfortunately, none of the synovial fluid cytokines investigated (IL-1, IL-2, IL-6, IL-8, Il-10, interferon γ, granulocyte macrophage-colony stimulating factor, tumor necrosis factor α, IL-12p70) satisfactorily detected resolution of infection in the setting of prior treatment for PJI. Although cytokines are expected to be elevated in the presence of infection, the internal milieu at the time of stage 2 of the revision makes diagnosis of infection difficult. In addition, presence of spacer particles and recent surgery may activate immune pathways and yield false-positive results. Furthermore, antibiotic cement spacers may suppress the microorganisms to very low levels and yield false-negative results even if these organisms remain virulent.19
Even though the synovial molecular markers can detect the presence of infection, they are unable to identify pathogens. As identifying the pathogen is important in the treatment of PJI, there has been interest in using polymerase chain reaction (PCR) techniques.51 These tests may also provide specific information about the pathogen, such as its antibiotic sensitivity. A recently developed technology, the Ibis T5000 Universal Biosensor (Ibis Biosciences), uses novel pan-domain primers in a series of PCRs. This biosensor is useful in diagnosing infections when cultures are negative and appears to be more accurate than conventional PCR.13 As reported by Jacovides and colleagues,13 this novel PCR technique identified an organism in about 88% of presumed cases of aseptic revision.
Conclusion
PJI poses an extreme challenge to the healthcare system. Given the morbidity associated with improper management of PJI, accurate diagnosis is of paramount importance. Given the limitations of current tests, synovial fluid cytokines hold promise in the diagnosis of PJIs. However, these cytokines are expensive, and their clinical utility in PJI management is not well established. More research is needed before guidelines for synovial fluid cytokines and biomarkers can replace or be incorporated into guidelines for the treatment of PJIs.
1 Parvizi J, Adeli B, Zmistowski B, Restrepo C, Greenwald AS. Management of periprosthetic joint infection: the current knowledge: AAOS exhibit selection. J Bone Joint Surg Am. 2012;94(14):e104.
2. Kurtz SM, Lau E, Watson H, Schmier JK, Parvizi J. Economic burden of periprosthetic joint infection in the United States. J Arthroplasty. 2012;27(8 suppl):61-65.e1.
3. Sierra RJ, Trousdale RT, Pagnano MW. Above-the-knee amputation after a total knee replacement: prevalence, etiology, and functional outcome. J Bone Joint Surg Am. 2003;85(6):1000-1004.
4. Bauer TW, Parvizi J, Kobayashi N, Krebs V. Diagnosis of periprosthetic infection. J Bone Joint Surg Am. 2006;88(4):869-882.
5. Deirmengian C, Hallab N, Tarabishy A, et al. Synovial fluid biomarkers for periprosthetic infection. Clin Orthop Relat Res. 2010;468(8):2017-2023.
6. Deirmengian C, Kardos K, Kilmartin P, Cameron A, Schiller K, Parvizi J. Diagnosing periprosthetic joint infection: has the era of the biomarker arrived? Clin Orthop Relat Res. 2014;472(11):3254-3262.
7. Parvizi J, Jacovides C, Antoci V, Ghanem E. Diagnosis of periprosthetic joint infection: the utility of a simple yet unappreciated enzyme. J Bone Joint Surg Am. 2011;93(24):2242-2248.
8. Kuiper JW, Willink RT, Moojen DJF, van den Bekerom MP, Colen S. Treatment of acute periprosthetic infections with prosthesis retention: review of current concepts. World J Orthop. 2014;5(5):667-676.
9. Zimmerli W, Trampuz A, Ochsner PE. Prosthetic-joint infections. N Engl J Med. 2004;351(16):1645-1654.
10. Osmon DR, Berbari EF, Berendt AR, et al. Diagnosis and management of prosthetic joint infection: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2013;56(1):e1-e25.
11. Parvizi J, Gehrke T; International Consensus Group on Periprosthetic Joint Infection. Definition of periprosthetic joint infection. J Arthroplasty. 2014;29(7):1331.
12. Saleh A, Guirguis A, Klika AK, Johnson L, Higuera CA, Barsoum WK. Unexpected positive intraoperative cultures in aseptic revision arthroplasty. J Arthroplasty. 2014;29(11):2181-2186.
13. Jacovides CL, Kreft R, Adeli B, Hozack B, Ehrlich GD, Parvizi J. Successful identification of pathogens by polymerase chain reaction (PCR)–based electron spray ionization time-of-flight mass spectrometry (ESI-TOF-MS) in culture-negative periprosthetic joint infection. J Bone Joint Surg Am. 2012;94(24):2247-2254.
14. Barrack RL, Aggarwal A, Burnett RS, et al. The fate of the unexpected positive intraoperative cultures after revision total knee arthroplasty. J Arthroplasty. 2007;22(6 suppl 2):94-99.
15. Macheras GA, Koutsostathis SD, Kateros K, Papadakis S, Anastasopoulos P. A two stage re-implantation protocol for the treatment of deep periprosthetic hip infection. Mid to long-term results. Hip Int. 2012;22(suppl 8):S54-S61.
16. George J, Kwiecien G, Klika AK, et al. Are frozen sections and MSIS criteria reliable at the time of reimplantation of two-stage revision arthroplasty? Clin Orthop Relat Res. 2016;474(7):1619-1626.
17. Kusuma SK, Ward J, Jacofsky M, Sporer SM, Della Valle CJ. What is the role of serological testing between stages of two-stage reconstruction of the infected prosthetic knee? Clin Orthop Relat Res. 2011;469(4):1002-1008.
18. Lonner JH, Siliski JM, Della Valle C, DiCesare P, Lotke PA. Role of knee aspiration after resection of the infected total knee arthroplasty. Am J Orthop. 2001;30(4):305-309.
19. Mont MA, Waldman BJ, Hungerford DS. Evaluation of preoperative cultures before second-stage reimplantation of a total knee prosthesis complicated by infection. A comparison-group study. J Bone Joint Surg Am. 2000;82(11):1552-1557.
20. Love C, Marwin SE, Palestro CJ. Nuclear medicine and the infected joint replacement. Semin Nucl Med. 2009;39(1):66-78.
21. Zimmerli W, Moser C. Pathogenesis and treatment concepts of orthopaedic biofilm infections. FEMS Immunol Med Microbiol. 2012;65(2):158-168.
22. Fontana A, Hengartner H, Weber E, Fehr K, Grob PJ, Cohen G. Interleukin 1 activity in the synovial fluid of patients with rheumatoid arthritis. Rheumatol Int. 1982;2(2):49-53.
23. Guerne PA, Zuraw BL, Vaughan JH, Carson DA, Lotz M. Synovium as a source of interleukin 6 in vitro. Contribution to local and systemic manifestations of arthritis. J Clin Invest. 1989;83(2):585-592.
24. Wang G. Human antimicrobial peptides and proteins. Pharmaceuticals (Basel). 2014;7(5):545-594.
25. Stylianou E, Saklatvala J. Interleukin-1. Int J Biochem Cell Biol. 1998;30(10):1075-1079.
26. Gollwitzer H, Dombrowski Y, Prodinger PM, et al. Antimicrobial peptides and proinflammatory cytokines in periprosthetic joint infection. J Bone Joint Surg Am. 2013;95(7):644-651.
27. Deirmengian C, Kardos K, Kilmartin P, Cameron A, Schiller K, Parvizi J. Combined measurement of synovial fluid α-defensin and C-reactive protein levels: highly accurate for diagnosing periprosthetic joint infection. J Bone Joint Surg Am. 2014;96(17):1439-1445.
28. Randau TM, Friedrich MJ, Wimmer MD, et al. Interleukin-6 in serum and in synovial fluid enhances the differentiation between periprosthetic joint infection and aseptic loosening. PLoS One. 2014;9(2):e89045.
29. Heinrich PC, Castell JV, Andus T. Interleukin-6 and the acute phase response. Biochem J. 1990;265(3):621-636.
30. Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011;1813(5):878-888.
31. Jacovides CL, Parvizi J, Adeli B, Jung KA. Molecular markers for diagnosis of periprosthetic joint infection. J Arthroplasty. 2011;26(6 suppl):99-103.e1.
32. Lenski M, Scherer MA. Synovial IL-6 as inflammatory marker in periprosthetic joint infections. J Arthroplasty. 2014;29(6):1105-1109.
33. Mortensen RF. C-reactive protein, inflammation, and innate immunity. Immunol Res. 2001;24(2):163-176.
34. Parvizi J, McKenzie JC, Cashman JP. Diagnosis of periprosthetic joint infection using synovial C-reactive protein. J Arthroplasty. 2012;27(8 suppl):12-16.
35. Ghanem E, Antoci V, Pulido L, Joshi A, Hozack W, Parvizi J. The use of receiver operating characteristics analysis in determining erythrocyte sedimentation rate and C-reactive protein levels in diagnosing periprosthetic infection prior to revision total hip arthroplasty. Int J Infect Dis. 2009;13(6):e444-e449.
36. Johnson AJ, Zywiel MG, Stroh A, Marker DR, Mont MA. Serological markers can lead to false negative diagnoses of periprosthetic infections following total knee arthroplasty. Int Orthop. 2011;35(11):1621-1626.
37. Parvizi J, Jacovides C, Adeli B, Jung KA, Hozack WJ. Mark B. Coventry award: synovial C-reactive protein: a prospective evaluation of a molecular marker for periprosthetic knee joint infection. Clin Orthop Relat Res. 2012;470(1):54-60.
38. Lehrer RI, Lichtenstein AK, Ganz T. Defensins: antimicrobial and cytotoxic peptides of mammalian cells. Annu Rev Immunol. 1993;11:105-128.
39. Ganz T, Selsted ME, Szklarek D, et al. Defensins. Natural peptide antibiotics of human neutrophils. J Clin Invest. 1985;76(4):1427-1435.
40. Chalifour A, Jeannin P, Gauchat JF, et al. Direct bacterial protein PAMP recognition by human NK cells involves TLRs and triggers alpha-defensin production. Blood. 2004;104(6):1778-1783.
41. Ulm H, Wilmes M, Shai Y, Sahl HG. Antimicrobial host defensins—specific antibiotic activities and innate defense modulation. Front Immunol. 2012;3:249.
42. Bingham J, Clarke H, Spangehl M, Schwartz A, Beauchamp C, Goldberg B. The alpha defensin-1 biomarker assay can be used to evaluate the potentially infected total joint arthroplasty. Clin Orthop Relat Res. 2014;472(12):4006-4009.
43. Deirmengian C, Kardos K, Kilmartin P, Gulati S, Citrano P, Booth RE. The alpha-defensin test for periprosthetic joint infection responds to a wide spectrum of organisms. Clin Orthop Relat Res. 2015;473(7):2229-2235.
44. Tischler EH, Cavanaugh PK, Parvizi J. Leukocyte esterase strip test: matched for Musculoskeletal Infection Society criteria. J Bone Joint Surg Am. 2014;96(22):1917-1920.
45. De Vecchi E, Villa F, Bortolin M, et al. Leucocyte esterase, glucose and C-reactive protein in the diagnosis of prosthetic joint infections: a prospective study. Clin Microbiol Infect. 2016;22(6):555-560.
46. Di Cesare PE, Chang E, Preston CF, Liu C. Serum interleukin-6 as a marker of periprosthetic infection following total hip and knee arthroplasty. J Bone Joint Surg Am. 2005;87(9):1921-1927.
47. Ettinger M, Calliess T, Kielstein JT, et al. Circulating biomarkers for discrimination between aseptic joint failure, low-grade infection, and high-grade septic failure. Clin Infect Dis. 2015;61(3):332-341.
48. Bottner F, Wegner A, Winkelmann W, Becker K, Erren M, Götze C. Interleukin-6, procalcitonin and TNF-alpha: markers of peri-prosthetic infection following total joint replacement. J Bone Joint Surg Br. 2007;89(1):94-99.
49. Galliera E, Drago L, Vassena C, et al. Toll-like receptor 2 in serum: a potential diagnostic marker of prosthetic joint infection? J Clin Microbiol. 2014;52(2):620-623.
50. Frangiamore SJ, Siqueira MB, Saleh A, Daly T, Higuera CA, Barsoum WK. Synovial cytokines and the MSIS criteria are not useful for determining infection resolution after periprosthetic joint infection explantation. Clin Orthop Relat Res. 2016;474(7):1630-1639.
51. Rasouli MR, Harandi AA, Adeli B, Purtill JJ, Parvizi J. Revision total knee arthroplasty: infection should be ruled out in all cases. J Arthroplasty. 2012;27(6):1239-1243.e1-e2.
52. Omar M, Ettinger M, Reichling M, et al. Synovial C-reactive protein as a marker for chronic periprosthetic infection in total hip arthroplasty. Bone Joint J. 2015;97(2):173-176.
53. Tetreault MW, Wetters NG, Moric M, Gross CE, Della Valle CJ. Is synovial C-reactive protein a useful marker for periprosthetic joint infection? Clin Orthop Relat Res. 2014;472(12):3997-4003.
54. Omar M, Ettinger M, Reichling M, et al. Preliminary results of a new test for rapid diagnosis of septic arthritis with use of leukocyte esterase and glucose reagent strips. J Bone Joint Surg Am. 2014;96(24):2032-2037.
1 Parvizi J, Adeli B, Zmistowski B, Restrepo C, Greenwald AS. Management of periprosthetic joint infection: the current knowledge: AAOS exhibit selection. J Bone Joint Surg Am. 2012;94(14):e104.
2. Kurtz SM, Lau E, Watson H, Schmier JK, Parvizi J. Economic burden of periprosthetic joint infection in the United States. J Arthroplasty. 2012;27(8 suppl):61-65.e1.
3. Sierra RJ, Trousdale RT, Pagnano MW. Above-the-knee amputation after a total knee replacement: prevalence, etiology, and functional outcome. J Bone Joint Surg Am. 2003;85(6):1000-1004.
4. Bauer TW, Parvizi J, Kobayashi N, Krebs V. Diagnosis of periprosthetic infection. J Bone Joint Surg Am. 2006;88(4):869-882.
5. Deirmengian C, Hallab N, Tarabishy A, et al. Synovial fluid biomarkers for periprosthetic infection. Clin Orthop Relat Res. 2010;468(8):2017-2023.
6. Deirmengian C, Kardos K, Kilmartin P, Cameron A, Schiller K, Parvizi J. Diagnosing periprosthetic joint infection: has the era of the biomarker arrived? Clin Orthop Relat Res. 2014;472(11):3254-3262.
7. Parvizi J, Jacovides C, Antoci V, Ghanem E. Diagnosis of periprosthetic joint infection: the utility of a simple yet unappreciated enzyme. J Bone Joint Surg Am. 2011;93(24):2242-2248.
8. Kuiper JW, Willink RT, Moojen DJF, van den Bekerom MP, Colen S. Treatment of acute periprosthetic infections with prosthesis retention: review of current concepts. World J Orthop. 2014;5(5):667-676.
9. Zimmerli W, Trampuz A, Ochsner PE. Prosthetic-joint infections. N Engl J Med. 2004;351(16):1645-1654.
10. Osmon DR, Berbari EF, Berendt AR, et al. Diagnosis and management of prosthetic joint infection: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2013;56(1):e1-e25.
11. Parvizi J, Gehrke T; International Consensus Group on Periprosthetic Joint Infection. Definition of periprosthetic joint infection. J Arthroplasty. 2014;29(7):1331.
12. Saleh A, Guirguis A, Klika AK, Johnson L, Higuera CA, Barsoum WK. Unexpected positive intraoperative cultures in aseptic revision arthroplasty. J Arthroplasty. 2014;29(11):2181-2186.
13. Jacovides CL, Kreft R, Adeli B, Hozack B, Ehrlich GD, Parvizi J. Successful identification of pathogens by polymerase chain reaction (PCR)–based electron spray ionization time-of-flight mass spectrometry (ESI-TOF-MS) in culture-negative periprosthetic joint infection. J Bone Joint Surg Am. 2012;94(24):2247-2254.
14. Barrack RL, Aggarwal A, Burnett RS, et al. The fate of the unexpected positive intraoperative cultures after revision total knee arthroplasty. J Arthroplasty. 2007;22(6 suppl 2):94-99.
15. Macheras GA, Koutsostathis SD, Kateros K, Papadakis S, Anastasopoulos P. A two stage re-implantation protocol for the treatment of deep periprosthetic hip infection. Mid to long-term results. Hip Int. 2012;22(suppl 8):S54-S61.
16. George J, Kwiecien G, Klika AK, et al. Are frozen sections and MSIS criteria reliable at the time of reimplantation of two-stage revision arthroplasty? Clin Orthop Relat Res. 2016;474(7):1619-1626.
17. Kusuma SK, Ward J, Jacofsky M, Sporer SM, Della Valle CJ. What is the role of serological testing between stages of two-stage reconstruction of the infected prosthetic knee? Clin Orthop Relat Res. 2011;469(4):1002-1008.
18. Lonner JH, Siliski JM, Della Valle C, DiCesare P, Lotke PA. Role of knee aspiration after resection of the infected total knee arthroplasty. Am J Orthop. 2001;30(4):305-309.
19. Mont MA, Waldman BJ, Hungerford DS. Evaluation of preoperative cultures before second-stage reimplantation of a total knee prosthesis complicated by infection. A comparison-group study. J Bone Joint Surg Am. 2000;82(11):1552-1557.
20. Love C, Marwin SE, Palestro CJ. Nuclear medicine and the infected joint replacement. Semin Nucl Med. 2009;39(1):66-78.
21. Zimmerli W, Moser C. Pathogenesis and treatment concepts of orthopaedic biofilm infections. FEMS Immunol Med Microbiol. 2012;65(2):158-168.
22. Fontana A, Hengartner H, Weber E, Fehr K, Grob PJ, Cohen G. Interleukin 1 activity in the synovial fluid of patients with rheumatoid arthritis. Rheumatol Int. 1982;2(2):49-53.
23. Guerne PA, Zuraw BL, Vaughan JH, Carson DA, Lotz M. Synovium as a source of interleukin 6 in vitro. Contribution to local and systemic manifestations of arthritis. J Clin Invest. 1989;83(2):585-592.
24. Wang G. Human antimicrobial peptides and proteins. Pharmaceuticals (Basel). 2014;7(5):545-594.
25. Stylianou E, Saklatvala J. Interleukin-1. Int J Biochem Cell Biol. 1998;30(10):1075-1079.
26. Gollwitzer H, Dombrowski Y, Prodinger PM, et al. Antimicrobial peptides and proinflammatory cytokines in periprosthetic joint infection. J Bone Joint Surg Am. 2013;95(7):644-651.
27. Deirmengian C, Kardos K, Kilmartin P, Cameron A, Schiller K, Parvizi J. Combined measurement of synovial fluid α-defensin and C-reactive protein levels: highly accurate for diagnosing periprosthetic joint infection. J Bone Joint Surg Am. 2014;96(17):1439-1445.
28. Randau TM, Friedrich MJ, Wimmer MD, et al. Interleukin-6 in serum and in synovial fluid enhances the differentiation between periprosthetic joint infection and aseptic loosening. PLoS One. 2014;9(2):e89045.
29. Heinrich PC, Castell JV, Andus T. Interleukin-6 and the acute phase response. Biochem J. 1990;265(3):621-636.
30. Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011;1813(5):878-888.
31. Jacovides CL, Parvizi J, Adeli B, Jung KA. Molecular markers for diagnosis of periprosthetic joint infection. J Arthroplasty. 2011;26(6 suppl):99-103.e1.
32. Lenski M, Scherer MA. Synovial IL-6 as inflammatory marker in periprosthetic joint infections. J Arthroplasty. 2014;29(6):1105-1109.
33. Mortensen RF. C-reactive protein, inflammation, and innate immunity. Immunol Res. 2001;24(2):163-176.
34. Parvizi J, McKenzie JC, Cashman JP. Diagnosis of periprosthetic joint infection using synovial C-reactive protein. J Arthroplasty. 2012;27(8 suppl):12-16.
35. Ghanem E, Antoci V, Pulido L, Joshi A, Hozack W, Parvizi J. The use of receiver operating characteristics analysis in determining erythrocyte sedimentation rate and C-reactive protein levels in diagnosing periprosthetic infection prior to revision total hip arthroplasty. Int J Infect Dis. 2009;13(6):e444-e449.
36. Johnson AJ, Zywiel MG, Stroh A, Marker DR, Mont MA. Serological markers can lead to false negative diagnoses of periprosthetic infections following total knee arthroplasty. Int Orthop. 2011;35(11):1621-1626.
37. Parvizi J, Jacovides C, Adeli B, Jung KA, Hozack WJ. Mark B. Coventry award: synovial C-reactive protein: a prospective evaluation of a molecular marker for periprosthetic knee joint infection. Clin Orthop Relat Res. 2012;470(1):54-60.
38. Lehrer RI, Lichtenstein AK, Ganz T. Defensins: antimicrobial and cytotoxic peptides of mammalian cells. Annu Rev Immunol. 1993;11:105-128.
39. Ganz T, Selsted ME, Szklarek D, et al. Defensins. Natural peptide antibiotics of human neutrophils. J Clin Invest. 1985;76(4):1427-1435.
40. Chalifour A, Jeannin P, Gauchat JF, et al. Direct bacterial protein PAMP recognition by human NK cells involves TLRs and triggers alpha-defensin production. Blood. 2004;104(6):1778-1783.
41. Ulm H, Wilmes M, Shai Y, Sahl HG. Antimicrobial host defensins—specific antibiotic activities and innate defense modulation. Front Immunol. 2012;3:249.
42. Bingham J, Clarke H, Spangehl M, Schwartz A, Beauchamp C, Goldberg B. The alpha defensin-1 biomarker assay can be used to evaluate the potentially infected total joint arthroplasty. Clin Orthop Relat Res. 2014;472(12):4006-4009.
43. Deirmengian C, Kardos K, Kilmartin P, Gulati S, Citrano P, Booth RE. The alpha-defensin test for periprosthetic joint infection responds to a wide spectrum of organisms. Clin Orthop Relat Res. 2015;473(7):2229-2235.
44. Tischler EH, Cavanaugh PK, Parvizi J. Leukocyte esterase strip test: matched for Musculoskeletal Infection Society criteria. J Bone Joint Surg Am. 2014;96(22):1917-1920.
45. De Vecchi E, Villa F, Bortolin M, et al. Leucocyte esterase, glucose and C-reactive protein in the diagnosis of prosthetic joint infections: a prospective study. Clin Microbiol Infect. 2016;22(6):555-560.
46. Di Cesare PE, Chang E, Preston CF, Liu C. Serum interleukin-6 as a marker of periprosthetic infection following total hip and knee arthroplasty. J Bone Joint Surg Am. 2005;87(9):1921-1927.
47. Ettinger M, Calliess T, Kielstein JT, et al. Circulating biomarkers for discrimination between aseptic joint failure, low-grade infection, and high-grade septic failure. Clin Infect Dis. 2015;61(3):332-341.
48. Bottner F, Wegner A, Winkelmann W, Becker K, Erren M, Götze C. Interleukin-6, procalcitonin and TNF-alpha: markers of peri-prosthetic infection following total joint replacement. J Bone Joint Surg Br. 2007;89(1):94-99.
49. Galliera E, Drago L, Vassena C, et al. Toll-like receptor 2 in serum: a potential diagnostic marker of prosthetic joint infection? J Clin Microbiol. 2014;52(2):620-623.
50. Frangiamore SJ, Siqueira MB, Saleh A, Daly T, Higuera CA, Barsoum WK. Synovial cytokines and the MSIS criteria are not useful for determining infection resolution after periprosthetic joint infection explantation. Clin Orthop Relat Res. 2016;474(7):1630-1639.
51. Rasouli MR, Harandi AA, Adeli B, Purtill JJ, Parvizi J. Revision total knee arthroplasty: infection should be ruled out in all cases. J Arthroplasty. 2012;27(6):1239-1243.e1-e2.
52. Omar M, Ettinger M, Reichling M, et al. Synovial C-reactive protein as a marker for chronic periprosthetic infection in total hip arthroplasty. Bone Joint J. 2015;97(2):173-176.
53. Tetreault MW, Wetters NG, Moric M, Gross CE, Della Valle CJ. Is synovial C-reactive protein a useful marker for periprosthetic joint infection? Clin Orthop Relat Res. 2014;472(12):3997-4003.
54. Omar M, Ettinger M, Reichling M, et al. Preliminary results of a new test for rapid diagnosis of septic arthritis with use of leukocyte esterase and glucose reagent strips. J Bone Joint Surg Am. 2014;96(24):2032-2037.

Can Anti-Tau Therapies Treat Neurodegenerative Disorders?
LONDON—Several experimental therapies targeting tau are currently under investigation in phase I and II clinical trials. Researchers at the 2017 Alzheimer’s Association International Conference described the design of, and early results from, studies of two monoclonal antibodies and an antisense oligonucleotide.

RO7105705 for Alzheimer’s Disease
Dr. Kerchner presented data from a phase I trial of RO7105705, a humanized anti-tau monoclonal antibody. RO7105705 binds specifically to tau and is intended to intercept tau in the extracellular space of the brain, blocking its cell-to-cell spread.
The primary objective of the study was to evaluate the safety of single and multiple doses of the drug, compared with placebo. The secondary objective was to look at the pharmacokinetic profile following IV and subcutaneous doses. Study participants included healthy volunteers ages 18 to 80 and patients with probable Alzheimer’s disease. Patients with Alzheimer’s disease were between ages 50 and 80 and had a Mini-Mental State Examination score of 16 to 28; a Clinical Dementia Rating global score of 0.5, 1.0, or 2.0; and 18F-florbetapir PET scan evidence of cerebral amyloid pathology.
In the single-dose escalation phase, six cohorts of eight healthy volunteers each received IV doses that ranged from 225 mg to 16,800 mg. Another cohort received 1,200 mg of the drug subcutaneously. In the multiple-dose phase, a cohort of healthy volunteers and a cohort of patients with mild-to-moderate Alzheimer’s disease received four weekly doses of 8.4 g.
“The drug was well tolerated, even at these high doses,” Dr. Kerchner said. “So far, there have been no dose-limiting adverse events, no serious adverse events, no deaths, and no one who stopped the drug due to adverse events.” In the single-dose cohorts, adverse events that occurred in more than one participant included headache, infusion/injection site reaction, upper respiratory tract infection, nausea, vomiting, and gastrointestinal viral infection. In the multiple-dose cohorts, adverse events that occurred in more than one participant included vessel puncture site complications and postural dizziness.
ABBV-8E12 for PSP and Alzheimer’s Disease
Kumar Budur, MD, of AbbVie, Chicago, presented the results of a phase I study of ABBV-8E12, a humanized anti-tau monoclonal antibody, in patients with progressive supranuclear palsy (PSP). He also gave an overview of two ongoing phase II studies of ABBV-8E12 for early Alzheimer’s disease and PSP.
“PSP is a chronic progressive neurodegenerative disorder that affects movement, control of gait and balance, speech, swallowing, vision, mood and behavior, and thinking,” Dr. Budur explained. “The time from the onset of symptoms to death is only seven years. There currently are no approved treatments for this condition.” PSP affects approximately 20,000 people in the United States, and symptoms typically begin after age 60.
The phase I trial in patients with PSP was a double-blind, placebo-controlled, single ascending dose study assessing the drug’s safety, tolerability, pharmacokinetics, and immunogenicity. Investigators randomized 30 participants to a single IV infusion of ABBV-8E12 (2.5, 7.5, 15, 25, or 50 mg/kg) or placebo in blocks of four subjects, with one subject in each block randomized to placebo and three to an active ABBV-8E12 dose. Researchers monitored safety and pharmacokinetics for 84 days post dosing.
Twenty-three patients received ABBV-8E12, and seven patients received placebo. At baseline, subjects’ mean age was 68.8; 16 (53%) were men. Patients had to be able to walk with minimal assistance to be included in the study. Twenty-seven subjects completed the 84-day follow-up and one subject (3.3%) withdrew from the study due to an adverse event.
Twenty-one subjects experienced an adverse event, including dermatitis (n = 5) and fall (n = 5). Three participants experienced serious adverse events. One patient had a subdural hematoma following two falls, one had agitation/anxiety/perseverative behavior, and one had hypertension. The serious adverse events occurred in the 15, 25, and 50 mg/kg cohorts, respectively. ABBV-8E12 exhibited an acceptable safety and tolerability profile to support repeat-dose testing in subjects with tauopathies, Dr. Budur said.
Dr. Budur and colleagues are recruiting subjects for a multinational phase II study evaluating ABBV-8E12 to delay the progression of Alzheimer’s disease. Eligible subjects (n = 400) will meet criteria for early Alzheimer’s disease and have a positive amyloid PET scan. “Subjects will be randomized to three doses of ABBV-8E12 versus placebo, 100 subjects per arm,” he said. In addition, investigators are recruiting patients with PSP for a phase II study evaluating the 52-week efficacy and safety of ABBV-8E12. The trial will assess whether the therapy can slow disease progression in 180 patients with PSP.
Antisense Oligonucleotide to Reduce Total Tau Expression in Mild Alzheimer’s Disease
Roger M. Lane, MD, MPH, of lonis Pharmaceuticals, Carlsbad, California, described a study that is designed to the test safety and pharmacokinetics of a tau-lowering antisense oligonucleotide (ASO) in patients with mild Alzheimer’s disease. The ASO, lonis-MAPTRX, targets microtubule-associated protein tau (MAPT) messenger RNA to reduce the synthesis of tau in the CNS, Dr. Lane said. “In contrast, current investigational biologic or small molecule anti-tau drugs target the tau protein,” he said.

In a recent study, DeVos and colleagues identified ASOs that selectively decreased human tau mRNA and protein in mice expressing mutant P301S human tau. After a reduction of human tau in this mouse model of tauopathy, fewer tau inclusions developed, and pre-existing tau pathology was reversed. The resolution of tau pathology was accompanied by the prevention of hippocampal volume loss, neuronal death, and nesting deficits. In nonhuman primates, ASOs distributed throughout the brain and spinal cord, and reduced tau mRNA and protein in the brain and spinal cord, and tau protein in the CSF.
Based on these findings, researchers are studying lonis-MAPTRX, a second generation 2’-O-methoxyethyl (2’-MOE) chimeric ASO identified as suitable for clinical trials. It is designed to bind and degrade the pre-mRNA transcribed from the MAPT gene and thereby reduce synthesis of tau protein.
“The first clinical trial is set up in 12 centers in Canada and five European countries,” Dr. Lane said. “Patients aged 50 to 75 with mild Alzheimer’s disease are being randomized to multiple ascending doses of lonis-MAPTRX administered intrathecally.” The study drug will be given four times at monthly intervals, and there will be six months of follow-up. End points include CSF biomarkers, neuroimaging, and clinical outcomes. “We are expecting a 50–85% reduction in tau production in the cerebral cortex,” Dr. Lane said.
—Adriene Marshall
Suggested Reading
DeVos SL, Miller RL, Schoch KM, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. 2017;9(374).
LONDON—Several experimental therapies targeting tau are currently under investigation in phase I and II clinical trials. Researchers at the 2017 Alzheimer’s Association International Conference described the design of, and early results from, studies of two monoclonal antibodies and an antisense oligonucleotide.
RO7105705 for Alzheimer’s Disease
Dr. Kerchner presented data from a phase I trial of RO7105705, a humanized anti-tau monoclonal antibody. RO7105705 binds specifically to tau and is intended to intercept tau in the extracellular space of the brain, blocking its cell-to-cell spread.
The primary objective of the study was to evaluate the safety of single and multiple doses of the drug, compared with placebo. The secondary objective was to look at the pharmacokinetic profile following IV and subcutaneous doses. Study participants included healthy volunteers ages 18 to 80 and patients with probable Alzheimer’s disease. Patients with Alzheimer’s disease were between ages 50 and 80 and had a Mini-Mental State Examination score of 16 to 28; a Clinical Dementia Rating global score of 0.5, 1.0, or 2.0; and 18F-florbetapir PET scan evidence of cerebral amyloid pathology.
In the single-dose escalation phase, six cohorts of eight healthy volunteers each received IV doses that ranged from 225 mg to 16,800 mg. Another cohort received 1,200 mg of the drug subcutaneously. In the multiple-dose phase, a cohort of healthy volunteers and a cohort of patients with mild-to-moderate Alzheimer’s disease received four weekly doses of 8.4 g.
“The drug was well tolerated, even at these high doses,” Dr. Kerchner said. “So far, there have been no dose-limiting adverse events, no serious adverse events, no deaths, and no one who stopped the drug due to adverse events.” In the single-dose cohorts, adverse events that occurred in more than one participant included headache, infusion/injection site reaction, upper respiratory tract infection, nausea, vomiting, and gastrointestinal viral infection. In the multiple-dose cohorts, adverse events that occurred in more than one participant included vessel puncture site complications and postural dizziness.
ABBV-8E12 for PSP and Alzheimer’s Disease
Kumar Budur, MD, of AbbVie, Chicago, presented the results of a phase I study of ABBV-8E12, a humanized anti-tau monoclonal antibody, in patients with progressive supranuclear palsy (PSP). He also gave an overview of two ongoing phase II studies of ABBV-8E12 for early Alzheimer’s disease and PSP.
“PSP is a chronic progressive neurodegenerative disorder that affects movement, control of gait and balance, speech, swallowing, vision, mood and behavior, and thinking,” Dr. Budur explained. “The time from the onset of symptoms to death is only seven years. There currently are no approved treatments for this condition.” PSP affects approximately 20,000 people in the United States, and symptoms typically begin after age 60.
The phase I trial in patients with PSP was a double-blind, placebo-controlled, single ascending dose study assessing the drug’s safety, tolerability, pharmacokinetics, and immunogenicity. Investigators randomized 30 participants to a single IV infusion of ABBV-8E12 (2.5, 7.5, 15, 25, or 50 mg/kg) or placebo in blocks of four subjects, with one subject in each block randomized to placebo and three to an active ABBV-8E12 dose. Researchers monitored safety and pharmacokinetics for 84 days post dosing.
Twenty-three patients received ABBV-8E12, and seven patients received placebo. At baseline, subjects’ mean age was 68.8; 16 (53%) were men. Patients had to be able to walk with minimal assistance to be included in the study. Twenty-seven subjects completed the 84-day follow-up and one subject (3.3%) withdrew from the study due to an adverse event.
Twenty-one subjects experienced an adverse event, including dermatitis (n = 5) and fall (n = 5). Three participants experienced serious adverse events. One patient had a subdural hematoma following two falls, one had agitation/anxiety/perseverative behavior, and one had hypertension. The serious adverse events occurred in the 15, 25, and 50 mg/kg cohorts, respectively. ABBV-8E12 exhibited an acceptable safety and tolerability profile to support repeat-dose testing in subjects with tauopathies, Dr. Budur said.
Dr. Budur and colleagues are recruiting subjects for a multinational phase II study evaluating ABBV-8E12 to delay the progression of Alzheimer’s disease. Eligible subjects (n = 400) will meet criteria for early Alzheimer’s disease and have a positive amyloid PET scan. “Subjects will be randomized to three doses of ABBV-8E12 versus placebo, 100 subjects per arm,” he said. In addition, investigators are recruiting patients with PSP for a phase II study evaluating the 52-week efficacy and safety of ABBV-8E12. The trial will assess whether the therapy can slow disease progression in 180 patients with PSP.
Antisense Oligonucleotide to Reduce Total Tau Expression in Mild Alzheimer’s Disease
Roger M. Lane, MD, MPH, of lonis Pharmaceuticals, Carlsbad, California, described a study that is designed to the test safety and pharmacokinetics of a tau-lowering antisense oligonucleotide (ASO) in patients with mild Alzheimer’s disease. The ASO, lonis-MAPTRX, targets microtubule-associated protein tau (MAPT) messenger RNA to reduce the synthesis of tau in the CNS, Dr. Lane said. “In contrast, current investigational biologic or small molecule anti-tau drugs target the tau protein,” he said.
In a recent study, DeVos and colleagues identified ASOs that selectively decreased human tau mRNA and protein in mice expressing mutant P301S human tau. After a reduction of human tau in this mouse model of tauopathy, fewer tau inclusions developed, and pre-existing tau pathology was reversed. The resolution of tau pathology was accompanied by the prevention of hippocampal volume loss, neuronal death, and nesting deficits. In nonhuman primates, ASOs distributed throughout the brain and spinal cord, and reduced tau mRNA and protein in the brain and spinal cord, and tau protein in the CSF.
Based on these findings, researchers are studying lonis-MAPTRX, a second generation 2’-O-methoxyethyl (2’-MOE) chimeric ASO identified as suitable for clinical trials. It is designed to bind and degrade the pre-mRNA transcribed from the MAPT gene and thereby reduce synthesis of tau protein.
“The first clinical trial is set up in 12 centers in Canada and five European countries,” Dr. Lane said. “Patients aged 50 to 75 with mild Alzheimer’s disease are being randomized to multiple ascending doses of lonis-MAPTRX administered intrathecally.” The study drug will be given four times at monthly intervals, and there will be six months of follow-up. End points include CSF biomarkers, neuroimaging, and clinical outcomes. “We are expecting a 50–85% reduction in tau production in the cerebral cortex,” Dr. Lane said.
—Adriene Marshall
Suggested Reading
DeVos SL, Miller RL, Schoch KM, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. 2017;9(374).
LONDON—Several experimental therapies targeting tau are currently under investigation in phase I and II clinical trials. Researchers at the 2017 Alzheimer’s Association International Conference described the design of, and early results from, studies of two monoclonal antibodies and an antisense oligonucleotide.
RO7105705 for Alzheimer’s Disease
Dr. Kerchner presented data from a phase I trial of RO7105705, a humanized anti-tau monoclonal antibody. RO7105705 binds specifically to tau and is intended to intercept tau in the extracellular space of the brain, blocking its cell-to-cell spread.
The primary objective of the study was to evaluate the safety of single and multiple doses of the drug, compared with placebo. The secondary objective was to look at the pharmacokinetic profile following IV and subcutaneous doses. Study participants included healthy volunteers ages 18 to 80 and patients with probable Alzheimer’s disease. Patients with Alzheimer’s disease were between ages 50 and 80 and had a Mini-Mental State Examination score of 16 to 28; a Clinical Dementia Rating global score of 0.5, 1.0, or 2.0; and 18F-florbetapir PET scan evidence of cerebral amyloid pathology.
In the single-dose escalation phase, six cohorts of eight healthy volunteers each received IV doses that ranged from 225 mg to 16,800 mg. Another cohort received 1,200 mg of the drug subcutaneously. In the multiple-dose phase, a cohort of healthy volunteers and a cohort of patients with mild-to-moderate Alzheimer’s disease received four weekly doses of 8.4 g.
“The drug was well tolerated, even at these high doses,” Dr. Kerchner said. “So far, there have been no dose-limiting adverse events, no serious adverse events, no deaths, and no one who stopped the drug due to adverse events.” In the single-dose cohorts, adverse events that occurred in more than one participant included headache, infusion/injection site reaction, upper respiratory tract infection, nausea, vomiting, and gastrointestinal viral infection. In the multiple-dose cohorts, adverse events that occurred in more than one participant included vessel puncture site complications and postural dizziness.
ABBV-8E12 for PSP and Alzheimer’s Disease
Kumar Budur, MD, of AbbVie, Chicago, presented the results of a phase I study of ABBV-8E12, a humanized anti-tau monoclonal antibody, in patients with progressive supranuclear palsy (PSP). He also gave an overview of two ongoing phase II studies of ABBV-8E12 for early Alzheimer’s disease and PSP.
“PSP is a chronic progressive neurodegenerative disorder that affects movement, control of gait and balance, speech, swallowing, vision, mood and behavior, and thinking,” Dr. Budur explained. “The time from the onset of symptoms to death is only seven years. There currently are no approved treatments for this condition.” PSP affects approximately 20,000 people in the United States, and symptoms typically begin after age 60.
The phase I trial in patients with PSP was a double-blind, placebo-controlled, single ascending dose study assessing the drug’s safety, tolerability, pharmacokinetics, and immunogenicity. Investigators randomized 30 participants to a single IV infusion of ABBV-8E12 (2.5, 7.5, 15, 25, or 50 mg/kg) or placebo in blocks of four subjects, with one subject in each block randomized to placebo and three to an active ABBV-8E12 dose. Researchers monitored safety and pharmacokinetics for 84 days post dosing.
Twenty-three patients received ABBV-8E12, and seven patients received placebo. At baseline, subjects’ mean age was 68.8; 16 (53%) were men. Patients had to be able to walk with minimal assistance to be included in the study. Twenty-seven subjects completed the 84-day follow-up and one subject (3.3%) withdrew from the study due to an adverse event.
Twenty-one subjects experienced an adverse event, including dermatitis (n = 5) and fall (n = 5). Three participants experienced serious adverse events. One patient had a subdural hematoma following two falls, one had agitation/anxiety/perseverative behavior, and one had hypertension. The serious adverse events occurred in the 15, 25, and 50 mg/kg cohorts, respectively. ABBV-8E12 exhibited an acceptable safety and tolerability profile to support repeat-dose testing in subjects with tauopathies, Dr. Budur said.
Dr. Budur and colleagues are recruiting subjects for a multinational phase II study evaluating ABBV-8E12 to delay the progression of Alzheimer’s disease. Eligible subjects (n = 400) will meet criteria for early Alzheimer’s disease and have a positive amyloid PET scan. “Subjects will be randomized to three doses of ABBV-8E12 versus placebo, 100 subjects per arm,” he said. In addition, investigators are recruiting patients with PSP for a phase II study evaluating the 52-week efficacy and safety of ABBV-8E12. The trial will assess whether the therapy can slow disease progression in 180 patients with PSP.
Antisense Oligonucleotide to Reduce Total Tau Expression in Mild Alzheimer’s Disease
Roger M. Lane, MD, MPH, of lonis Pharmaceuticals, Carlsbad, California, described a study that is designed to the test safety and pharmacokinetics of a tau-lowering antisense oligonucleotide (ASO) in patients with mild Alzheimer’s disease. The ASO, lonis-MAPTRX, targets microtubule-associated protein tau (MAPT) messenger RNA to reduce the synthesis of tau in the CNS, Dr. Lane said. “In contrast, current investigational biologic or small molecule anti-tau drugs target the tau protein,” he said.
In a recent study, DeVos and colleagues identified ASOs that selectively decreased human tau mRNA and protein in mice expressing mutant P301S human tau. After a reduction of human tau in this mouse model of tauopathy, fewer tau inclusions developed, and pre-existing tau pathology was reversed. The resolution of tau pathology was accompanied by the prevention of hippocampal volume loss, neuronal death, and nesting deficits. In nonhuman primates, ASOs distributed throughout the brain and spinal cord, and reduced tau mRNA and protein in the brain and spinal cord, and tau protein in the CSF.
Based on these findings, researchers are studying lonis-MAPTRX, a second generation 2’-O-methoxyethyl (2’-MOE) chimeric ASO identified as suitable for clinical trials. It is designed to bind and degrade the pre-mRNA transcribed from the MAPT gene and thereby reduce synthesis of tau protein.
“The first clinical trial is set up in 12 centers in Canada and five European countries,” Dr. Lane said. “Patients aged 50 to 75 with mild Alzheimer’s disease are being randomized to multiple ascending doses of lonis-MAPTRX administered intrathecally.” The study drug will be given four times at monthly intervals, and there will be six months of follow-up. End points include CSF biomarkers, neuroimaging, and clinical outcomes. “We are expecting a 50–85% reduction in tau production in the cerebral cortex,” Dr. Lane said.
—Adriene Marshall
Suggested Reading
DeVos SL, Miller RL, Schoch KM, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. 2017;9(374).
IDWeek 2017 opens in San Diego
facing infectious diseases clinicians and researchers in the 21st century.
Premeeting workshops and symposia occupy most of the first 2 days of the event, with highlights including a session on managing infections in opioid users and a “late breaker” symposium addressing the latest on the H7N9 outbreak in China, the current findings and recommendations regarding Candida auris, and the epidemiology of the recent Legionella outbreaks in the United States. Another late breaker session focuses on the recent spate of hepatitis A outbreaks, including one in conference host city San Diego, primarily among the homeless population.
IDWeek is the combined annual meeting of the Infectious Diseases Society of America (IDSA), the Society for Healthcare Epidemiology of America (SHEA), the HIV Medicine Association (HIVMA), and the Pediatric Infectious Diseases Society (PIDS). The first IDWeek was held in 2012.

One intriguing interactive session – aptly-titled “Nightmare Bugs” – will investigate the problems posed by multidrug-resistant organisms and the need for new antimicrobials to defeat them.
There are many sessions and posters addressing evergreen clinical topics for ID clinicians, such as antimicrobial resistance, antibiotic stewardship, surgical site infections, bacteremia and sepsis, Clostridium difficile, hepatitis care, and HIV care. But the education committee at IDWeek always manages to touch on topics in the news. For instance, one late breaker session will feature a discussion of the nexus between the opioid crisis and infectious diseases, the outbreak of cholera in Yemen, and the epidemiology of the yellow fever outbreak in Brazil.
Featured speakers at the event include James M. Hughes, MD, professor of medicine at Emory University, Atlanta, who will discuss the importance of a One Health approach to emerging microbial threats, and Connie Celum, MD, MPH, professor of global health and medicine, University of Washington, Seattle, who intends to describe the progress in effective HIV prevention interventions and lessons learned in implementation. Neil O. Fishman, MD, of the University of Pennsylvania Perelman School of Medicine, is delivering the annual SHEA lecture at IDWeek, and will explain how ID physicians and epidemiologists can promote interventions to achieve high reliability in health care. Renowned ID researcher Janet Englund, MD, of Seattle Children’s Hospital, will discuss the potential future therapies to prevent or treat respiratory viral infections in high-risk pediatric patients.
The 2017 conference will close with a three-part plenary – “21st Century Cures” – featuring ID luminaries Christopher Karp, MD, of the Bill & Melinda Gates Foundation, James E. Crowe Jr., MD, of Vanderbilt University Medical Center in Nashville, Tenn., and David Thomas, MD, MPH, of Johns Hopkins University in Baltimore.
[email protected]
On Twitter @richpizzi
facing infectious diseases clinicians and researchers in the 21st century.
Premeeting workshops and symposia occupy most of the first 2 days of the event, with highlights including a session on managing infections in opioid users and a “late breaker” symposium addressing the latest on the H7N9 outbreak in China, the current findings and recommendations regarding Candida auris, and the epidemiology of the recent Legionella outbreaks in the United States. Another late breaker session focuses on the recent spate of hepatitis A outbreaks, including one in conference host city San Diego, primarily among the homeless population.
IDWeek is the combined annual meeting of the Infectious Diseases Society of America (IDSA), the Society for Healthcare Epidemiology of America (SHEA), the HIV Medicine Association (HIVMA), and the Pediatric Infectious Diseases Society (PIDS). The first IDWeek was held in 2012.
One intriguing interactive session – aptly-titled “Nightmare Bugs” – will investigate the problems posed by multidrug-resistant organisms and the need for new antimicrobials to defeat them.
There are many sessions and posters addressing evergreen clinical topics for ID clinicians, such as antimicrobial resistance, antibiotic stewardship, surgical site infections, bacteremia and sepsis, Clostridium difficile, hepatitis care, and HIV care. But the education committee at IDWeek always manages to touch on topics in the news. For instance, one late breaker session will feature a discussion of the nexus between the opioid crisis and infectious diseases, the outbreak of cholera in Yemen, and the epidemiology of the yellow fever outbreak in Brazil.
Featured speakers at the event include James M. Hughes, MD, professor of medicine at Emory University, Atlanta, who will discuss the importance of a One Health approach to emerging microbial threats, and Connie Celum, MD, MPH, professor of global health and medicine, University of Washington, Seattle, who intends to describe the progress in effective HIV prevention interventions and lessons learned in implementation. Neil O. Fishman, MD, of the University of Pennsylvania Perelman School of Medicine, is delivering the annual SHEA lecture at IDWeek, and will explain how ID physicians and epidemiologists can promote interventions to achieve high reliability in health care. Renowned ID researcher Janet Englund, MD, of Seattle Children’s Hospital, will discuss the potential future therapies to prevent or treat respiratory viral infections in high-risk pediatric patients.
The 2017 conference will close with a three-part plenary – “21st Century Cures” – featuring ID luminaries Christopher Karp, MD, of the Bill & Melinda Gates Foundation, James E. Crowe Jr., MD, of Vanderbilt University Medical Center in Nashville, Tenn., and David Thomas, MD, MPH, of Johns Hopkins University in Baltimore.
[email protected]
On Twitter @richpizzi
facing infectious diseases clinicians and researchers in the 21st century.
Premeeting workshops and symposia occupy most of the first 2 days of the event, with highlights including a session on managing infections in opioid users and a “late breaker” symposium addressing the latest on the H7N9 outbreak in China, the current findings and recommendations regarding Candida auris, and the epidemiology of the recent Legionella outbreaks in the United States. Another late breaker session focuses on the recent spate of hepatitis A outbreaks, including one in conference host city San Diego, primarily among the homeless population.
IDWeek is the combined annual meeting of the Infectious Diseases Society of America (IDSA), the Society for Healthcare Epidemiology of America (SHEA), the HIV Medicine Association (HIVMA), and the Pediatric Infectious Diseases Society (PIDS). The first IDWeek was held in 2012.
One intriguing interactive session – aptly-titled “Nightmare Bugs” – will investigate the problems posed by multidrug-resistant organisms and the need for new antimicrobials to defeat them.
There are many sessions and posters addressing evergreen clinical topics for ID clinicians, such as antimicrobial resistance, antibiotic stewardship, surgical site infections, bacteremia and sepsis, Clostridium difficile, hepatitis care, and HIV care. But the education committee at IDWeek always manages to touch on topics in the news. For instance, one late breaker session will feature a discussion of the nexus between the opioid crisis and infectious diseases, the outbreak of cholera in Yemen, and the epidemiology of the yellow fever outbreak in Brazil.
Featured speakers at the event include James M. Hughes, MD, professor of medicine at Emory University, Atlanta, who will discuss the importance of a One Health approach to emerging microbial threats, and Connie Celum, MD, MPH, professor of global health and medicine, University of Washington, Seattle, who intends to describe the progress in effective HIV prevention interventions and lessons learned in implementation. Neil O. Fishman, MD, of the University of Pennsylvania Perelman School of Medicine, is delivering the annual SHEA lecture at IDWeek, and will explain how ID physicians and epidemiologists can promote interventions to achieve high reliability in health care. Renowned ID researcher Janet Englund, MD, of Seattle Children’s Hospital, will discuss the potential future therapies to prevent or treat respiratory viral infections in high-risk pediatric patients.
The 2017 conference will close with a three-part plenary – “21st Century Cures” – featuring ID luminaries Christopher Karp, MD, of the Bill & Melinda Gates Foundation, James E. Crowe Jr., MD, of Vanderbilt University Medical Center in Nashville, Tenn., and David Thomas, MD, MPH, of Johns Hopkins University in Baltimore.
[email protected]
On Twitter @richpizzi
Healthy youth sports participation excludes early specialization
CHICAGO – When a boy receives five college football scholarship offers and a girl commits to playing soccer for a university before either of them starts ninth grade, it’s time to take several steps back in youth sports.
, including the risk of potentially discouraging a lifetime of healthy athletic participation, according to Joel S. Brenner, MD, MPH, a sports medicine expert at the Children’s Hospital of The King’s Daughters and Eastern Virginia Medical School, both in Norfolk.
“This paradigm should be discouraged by society,” Dr. Brenner told attendees at the American Academy of Pediatrics annual meeting. “Sports specialization refers to focusing on one sport to the exclusion of all others, often playing that single sport year-round. Dr. Brenner authored the AAP’s 2016 clinical report on sports specialization and intensive training in young athletes.
Dr. Brenner emphasized the benefits of delaying sports specialization until after puberty, the risks of specializing sooner, and the importance of rest to prevent burnout and injuries.
This is not a new problem, he noted, showing the attendees two Time Magazine covers, from 1999 and 2017, that featured the concern of “Sports Crazed Kids.” But it is so far-reaching that it will requires more than just physicians to change.
“This is not just an athlete problem, a parent problem, a coach problem, or even a physician problem,” Dr. Brenner said. “It’s a societal problem, a youth sports culture problem, and one that all of us as stakeholders need to attack and try to change the culture.”
Youth sports offer a broad range of benefits, such as developing physical activity, and leadership skills, and promoting self-esteem, socialization, and teamwork, Dr. Brenner said.
“But one benefit that often gets forgotten by people, including the coaches, the parents, and the athletes, is that sports is supposed to be about having fun,” he said.
The old model of kids’ sports was loosely organized fun, with kids playing multiple sports throughout the year and less direct involvement from adults, such as street hockey games and pick-up basketball. But those bygone days, Dr. Brenner noted wistfully, have been replaced with a different paradigm today: Children specialize in a single sport very early, and parents and coaches are the driving forces behind their involvement.
Today’s culture of very early sports specialization and college recruitment increases pressure on parents and young athletes to play year-round on multiple teams to stay on the radar of scouts and colleges. And this specialization has expanded to younger and younger ages, with 7-year-olds participating in travel leagues and national rankings of children in their sport as early as sixth grade.
“We should not be ranking kids in middle school or even in early high school,” Dr. Brenner said to wide applause. “We should allow kids to develop in a low-pressure, healthy system before we do that.”
The effects of high pressure have potentially lifelong ramifications. By the time children are 13 years old, 70% have dropped out of organized sports, Dr. Brenner said, and injuries from overuse account for more than half of all sports-related injuries in youth.
Yet the alternative – early diversification and late specialization – can really benefit kids, he said. The early specialization paradigm of playing just one sport focuses on deliberate practice and performance from the start. By contrast, early diversification with multiple sports focuses on deliberate play, during which children develop foundational athletic skills. Children who play a variety of sports are more likely to participate for more years – and it meets youth’s more realistic, long-term needs for lifelong physical activity through “fun, variety, and play,” he said.
Dr. Brenner said that just 1% of high school athletes receive any athletic scholarships, and only 3%-11% of high school athletes compete at the college level. The numbers for high school athletes that go on to play at the professional level is, of course, even smaller: 0.03% to 0.5%, depending on the sport.
And the irony is that the goal of early specialization – producing such elite level athletes – is actually better accomplished through playing multiple sports, Dr. Brenner said. Most Division 1 National Collegiate Athletic Association (NCAA) athletes and 90% of National Football League (NFL) first-round picks played multiple sports in high school. So the benefits of waiting until late adolescence to specialize are twofold: a greater likelihood of athletic success, even at elite levels, and minimizing the risks of injury.
Overuse injuries pose serious risks
More than half of sports injuries are from overuse, and a number of factors contribute to those injuries, such as muscle imbalance, playing surfaces, and training errors, Dr. Brenner said. But the biggest contributors are early specialization, playing year-round sports, and playing on multiple teams.
“This is a problem we see daily,” Dr. Brenner said. “We can see the young dancer, who’s dancing 6-7 days a week, who develops back pain and continues to dance, and develops a stress fracture in her lumbar spine known spondylosis.
“Or we see the young soccer player who plays on multiple teams and develops heel pain, who starts limping with activities of daily living, continues to play soccer despite limping, and develops calcaneal apophysitis, known as Sever’s disease. Or the young baseball pitcher, who pitches for two teams, who develops arm pain and weakness, who has a stress fracture through the proximal humeral epiphysis, known as Little League shoulder.” 
Two broad pieces of guidance can help reduce the risk of injuries, particularly from overuse. First, young athletes should take off at least 1 month from a specific sport at least three times a year to give them adequate time for physical and psychological recovery. Second, ensuring young athletes take at least 1 or 2 days off of practice each week further reduces the likelihood of injury.
In addition to the physical problems these young athletes may develop, they also risk anxiety, depression, burnout, early retirement, and social isolation from peers who don’t play their sport, Dr. Brenner said. Family members also may experience greater stress, he added. And then there’s the risk of missing out on learning other sports they may excel in that offer a lifetime of enjoyment, such as tennis or swimming.
It is not clear where the threshold of involvement is for reducing overuse injury, burnout, and attrition, but Dr. Brenner provided some guidelines as a starting place. High school athletes should not train more 16 hours a week, and organized sports should not exceed free play time by a greater ratio than 2:1. Another guideline is not to exceed more hours per week in organized sports than a child’s age in years.
The primary focus of sports should be learning lifelong physical activity skills and having fun, Dr. Brenner said. Pediatricians should encourage patients to play in a wide variety of sports at least until puberty, thereby decreasing the chance of injuries, stress, and burnout, he said. That can include sports that are not necessarily an official part of school or club competition. Waiting until later to specialize may lead to a higher likelihood of athletic success.
Dr. Brenner said he had no relevant financial disclosures.
CHICAGO – When a boy receives five college football scholarship offers and a girl commits to playing soccer for a university before either of them starts ninth grade, it’s time to take several steps back in youth sports.
, including the risk of potentially discouraging a lifetime of healthy athletic participation, according to Joel S. Brenner, MD, MPH, a sports medicine expert at the Children’s Hospital of The King’s Daughters and Eastern Virginia Medical School, both in Norfolk.
“This paradigm should be discouraged by society,” Dr. Brenner told attendees at the American Academy of Pediatrics annual meeting. “Sports specialization refers to focusing on one sport to the exclusion of all others, often playing that single sport year-round. Dr. Brenner authored the AAP’s 2016 clinical report on sports specialization and intensive training in young athletes.
Dr. Brenner emphasized the benefits of delaying sports specialization until after puberty, the risks of specializing sooner, and the importance of rest to prevent burnout and injuries.
This is not a new problem, he noted, showing the attendees two Time Magazine covers, from 1999 and 2017, that featured the concern of “Sports Crazed Kids.” But it is so far-reaching that it will requires more than just physicians to change.
“This is not just an athlete problem, a parent problem, a coach problem, or even a physician problem,” Dr. Brenner said. “It’s a societal problem, a youth sports culture problem, and one that all of us as stakeholders need to attack and try to change the culture.”
Youth sports offer a broad range of benefits, such as developing physical activity, and leadership skills, and promoting self-esteem, socialization, and teamwork, Dr. Brenner said.
“But one benefit that often gets forgotten by people, including the coaches, the parents, and the athletes, is that sports is supposed to be about having fun,” he said.
The old model of kids’ sports was loosely organized fun, with kids playing multiple sports throughout the year and less direct involvement from adults, such as street hockey games and pick-up basketball. But those bygone days, Dr. Brenner noted wistfully, have been replaced with a different paradigm today: Children specialize in a single sport very early, and parents and coaches are the driving forces behind their involvement.
Today’s culture of very early sports specialization and college recruitment increases pressure on parents and young athletes to play year-round on multiple teams to stay on the radar of scouts and colleges. And this specialization has expanded to younger and younger ages, with 7-year-olds participating in travel leagues and national rankings of children in their sport as early as sixth grade.
“We should not be ranking kids in middle school or even in early high school,” Dr. Brenner said to wide applause. “We should allow kids to develop in a low-pressure, healthy system before we do that.”
The effects of high pressure have potentially lifelong ramifications. By the time children are 13 years old, 70% have dropped out of organized sports, Dr. Brenner said, and injuries from overuse account for more than half of all sports-related injuries in youth.
Yet the alternative – early diversification and late specialization – can really benefit kids, he said. The early specialization paradigm of playing just one sport focuses on deliberate practice and performance from the start. By contrast, early diversification with multiple sports focuses on deliberate play, during which children develop foundational athletic skills. Children who play a variety of sports are more likely to participate for more years – and it meets youth’s more realistic, long-term needs for lifelong physical activity through “fun, variety, and play,” he said.
Dr. Brenner said that just 1% of high school athletes receive any athletic scholarships, and only 3%-11% of high school athletes compete at the college level. The numbers for high school athletes that go on to play at the professional level is, of course, even smaller: 0.03% to 0.5%, depending on the sport.
And the irony is that the goal of early specialization – producing such elite level athletes – is actually better accomplished through playing multiple sports, Dr. Brenner said. Most Division 1 National Collegiate Athletic Association (NCAA) athletes and 90% of National Football League (NFL) first-round picks played multiple sports in high school. So the benefits of waiting until late adolescence to specialize are twofold: a greater likelihood of athletic success, even at elite levels, and minimizing the risks of injury.
Overuse injuries pose serious risks
More than half of sports injuries are from overuse, and a number of factors contribute to those injuries, such as muscle imbalance, playing surfaces, and training errors, Dr. Brenner said. But the biggest contributors are early specialization, playing year-round sports, and playing on multiple teams.
“This is a problem we see daily,” Dr. Brenner said. “We can see the young dancer, who’s dancing 6-7 days a week, who develops back pain and continues to dance, and develops a stress fracture in her lumbar spine known spondylosis.
“Or we see the young soccer player who plays on multiple teams and develops heel pain, who starts limping with activities of daily living, continues to play soccer despite limping, and develops calcaneal apophysitis, known as Sever’s disease. Or the young baseball pitcher, who pitches for two teams, who develops arm pain and weakness, who has a stress fracture through the proximal humeral epiphysis, known as Little League shoulder.”
Two broad pieces of guidance can help reduce the risk of injuries, particularly from overuse. First, young athletes should take off at least 1 month from a specific sport at least three times a year to give them adequate time for physical and psychological recovery. Second, ensuring young athletes take at least 1 or 2 days off of practice each week further reduces the likelihood of injury.
In addition to the physical problems these young athletes may develop, they also risk anxiety, depression, burnout, early retirement, and social isolation from peers who don’t play their sport, Dr. Brenner said. Family members also may experience greater stress, he added. And then there’s the risk of missing out on learning other sports they may excel in that offer a lifetime of enjoyment, such as tennis or swimming.
It is not clear where the threshold of involvement is for reducing overuse injury, burnout, and attrition, but Dr. Brenner provided some guidelines as a starting place. High school athletes should not train more 16 hours a week, and organized sports should not exceed free play time by a greater ratio than 2:1. Another guideline is not to exceed more hours per week in organized sports than a child’s age in years.
The primary focus of sports should be learning lifelong physical activity skills and having fun, Dr. Brenner said. Pediatricians should encourage patients to play in a wide variety of sports at least until puberty, thereby decreasing the chance of injuries, stress, and burnout, he said. That can include sports that are not necessarily an official part of school or club competition. Waiting until later to specialize may lead to a higher likelihood of athletic success.
Dr. Brenner said he had no relevant financial disclosures.
CHICAGO – When a boy receives five college football scholarship offers and a girl commits to playing soccer for a university before either of them starts ninth grade, it’s time to take several steps back in youth sports.
, including the risk of potentially discouraging a lifetime of healthy athletic participation, according to Joel S. Brenner, MD, MPH, a sports medicine expert at the Children’s Hospital of The King’s Daughters and Eastern Virginia Medical School, both in Norfolk.
“This paradigm should be discouraged by society,” Dr. Brenner told attendees at the American Academy of Pediatrics annual meeting. “Sports specialization refers to focusing on one sport to the exclusion of all others, often playing that single sport year-round. Dr. Brenner authored the AAP’s 2016 clinical report on sports specialization and intensive training in young athletes.
Dr. Brenner emphasized the benefits of delaying sports specialization until after puberty, the risks of specializing sooner, and the importance of rest to prevent burnout and injuries.
This is not a new problem, he noted, showing the attendees two Time Magazine covers, from 1999 and 2017, that featured the concern of “Sports Crazed Kids.” But it is so far-reaching that it will requires more than just physicians to change.
“This is not just an athlete problem, a parent problem, a coach problem, or even a physician problem,” Dr. Brenner said. “It’s a societal problem, a youth sports culture problem, and one that all of us as stakeholders need to attack and try to change the culture.”
Youth sports offer a broad range of benefits, such as developing physical activity, and leadership skills, and promoting self-esteem, socialization, and teamwork, Dr. Brenner said.
“But one benefit that often gets forgotten by people, including the coaches, the parents, and the athletes, is that sports is supposed to be about having fun,” he said.
The old model of kids’ sports was loosely organized fun, with kids playing multiple sports throughout the year and less direct involvement from adults, such as street hockey games and pick-up basketball. But those bygone days, Dr. Brenner noted wistfully, have been replaced with a different paradigm today: Children specialize in a single sport very early, and parents and coaches are the driving forces behind their involvement.
Today’s culture of very early sports specialization and college recruitment increases pressure on parents and young athletes to play year-round on multiple teams to stay on the radar of scouts and colleges. And this specialization has expanded to younger and younger ages, with 7-year-olds participating in travel leagues and national rankings of children in their sport as early as sixth grade.
“We should not be ranking kids in middle school or even in early high school,” Dr. Brenner said to wide applause. “We should allow kids to develop in a low-pressure, healthy system before we do that.”
The effects of high pressure have potentially lifelong ramifications. By the time children are 13 years old, 70% have dropped out of organized sports, Dr. Brenner said, and injuries from overuse account for more than half of all sports-related injuries in youth.
Yet the alternative – early diversification and late specialization – can really benefit kids, he said. The early specialization paradigm of playing just one sport focuses on deliberate practice and performance from the start. By contrast, early diversification with multiple sports focuses on deliberate play, during which children develop foundational athletic skills. Children who play a variety of sports are more likely to participate for more years – and it meets youth’s more realistic, long-term needs for lifelong physical activity through “fun, variety, and play,” he said.
Dr. Brenner said that just 1% of high school athletes receive any athletic scholarships, and only 3%-11% of high school athletes compete at the college level. The numbers for high school athletes that go on to play at the professional level is, of course, even smaller: 0.03% to 0.5%, depending on the sport.
And the irony is that the goal of early specialization – producing such elite level athletes – is actually better accomplished through playing multiple sports, Dr. Brenner said. Most Division 1 National Collegiate Athletic Association (NCAA) athletes and 90% of National Football League (NFL) first-round picks played multiple sports in high school. So the benefits of waiting until late adolescence to specialize are twofold: a greater likelihood of athletic success, even at elite levels, and minimizing the risks of injury.
Overuse injuries pose serious risks
More than half of sports injuries are from overuse, and a number of factors contribute to those injuries, such as muscle imbalance, playing surfaces, and training errors, Dr. Brenner said. But the biggest contributors are early specialization, playing year-round sports, and playing on multiple teams.
“This is a problem we see daily,” Dr. Brenner said. “We can see the young dancer, who’s dancing 6-7 days a week, who develops back pain and continues to dance, and develops a stress fracture in her lumbar spine known spondylosis.
“Or we see the young soccer player who plays on multiple teams and develops heel pain, who starts limping with activities of daily living, continues to play soccer despite limping, and develops calcaneal apophysitis, known as Sever’s disease. Or the young baseball pitcher, who pitches for two teams, who develops arm pain and weakness, who has a stress fracture through the proximal humeral epiphysis, known as Little League shoulder.”
Two broad pieces of guidance can help reduce the risk of injuries, particularly from overuse. First, young athletes should take off at least 1 month from a specific sport at least three times a year to give them adequate time for physical and psychological recovery. Second, ensuring young athletes take at least 1 or 2 days off of practice each week further reduces the likelihood of injury.
In addition to the physical problems these young athletes may develop, they also risk anxiety, depression, burnout, early retirement, and social isolation from peers who don’t play their sport, Dr. Brenner said. Family members also may experience greater stress, he added. And then there’s the risk of missing out on learning other sports they may excel in that offer a lifetime of enjoyment, such as tennis or swimming.
It is not clear where the threshold of involvement is for reducing overuse injury, burnout, and attrition, but Dr. Brenner provided some guidelines as a starting place. High school athletes should not train more 16 hours a week, and organized sports should not exceed free play time by a greater ratio than 2:1. Another guideline is not to exceed more hours per week in organized sports than a child’s age in years.
The primary focus of sports should be learning lifelong physical activity skills and having fun, Dr. Brenner said. Pediatricians should encourage patients to play in a wide variety of sports at least until puberty, thereby decreasing the chance of injuries, stress, and burnout, he said. That can include sports that are not necessarily an official part of school or club competition. Waiting until later to specialize may lead to a higher likelihood of athletic success.
Dr. Brenner said he had no relevant financial disclosures.
EXPERT ANALYSIS FROM AAP 2017
Teratogenicity may not be a yes or no question
Since thalidomide, the medical community has sought to ensure that we do not miss any safety signal that a drug could cause malformations or developmental delays after in utero exposure.
At the time of a drug’s debut, there are relatively small numbers of exposures in pregnancy, and it’s difficult to decipher teratogenicity. As the number of exposures increases, we are generally able to answer “yes” or “no” to the question of teratogenicity. But in the last decade or so, data on drug exposure in pregnancy has become robust enough – thanks in part to large registries – to provide potentially more useful answers on safety. Specifically, .
Animal studies show us that there is a dose dependent effect in pregnancy, and not every dose causes harm to the fetus. However, that information is not easily translated into clinical practice because of the vast differences between animal and human pharmacokinetics and sensitivity to toxicity.
The first drug that came into focus as having dose dependent teratogenicity in pregnancy is the epilepsy drug valproic acid. In the late 1980s, studies showed that the drug was associated with an increased risk for spina bifida. Later, more congenital malformations were linked to valproic acid, including oral cleft, cardiac and limb defects, developmental delays, lower IQ, and even autism. But in the last few years, an increasing number of studies point to a lower dose that may represent an acceptable risk for some pregnant women.
Looking at data from EURAP, an international registry of antiepileptic drugs and pregnancy, researchers showed that the dose of valproic acid with the greatest risk for harm was 1,500 mg per day or greater, with a 24% frequency of major congenital malformations. But at less than 700 mg per day, the frequency of major malformations dropped to 5.9% (Neurology. 2015 Sep 8;85[10]:866-72).
Another analysis of the EURAP data showed the same dose-dependent relationship with other drugs. The researchers calculated rates of major congenital malformations in 1,402 pregnancies exposed to carbamazepine, 1,280 on lamotrigine, 1,010 on valproic acid, and 217 on phenobarbital, and all showed that the frequency of birth defects increased along with the dose of the drug.
The study identified the dose for each drug with the lowest rates of malformation. For lamotrigine, it was a dose of less than 300 mg per day, with a 2% frequency of malformations. Similarly, the dose was less than 400 mg per day for carbamazepine (3.4% rate of malformations). Overall, risks of malformation were significantly higher in valproic acid and phenobarbital at all tested doses and carbamazepine at doses greater than 400 mg per day, compared with lamotrigine monotherapy at less than 300 mg per day (Lancet Neurol. 2011 Jul;10[7]:609-17).
The study is important because it gives us a benchmark for these drugs, allowing us to see the risks at lower doses.
But not all the data are in agreement. In 2016, a Cochrane review of different antiepileptic drugs in pregnancy found that only with valproic acid could the risk of a malformation be clearly linked to the size of the dose (Cochrane Database Syst Rev. 2016 Nov 7;11:CD010224).
Most recently, a large U.S. database of Medicaid patients, which included more than 1.3 million pregnancies, showed the dose-dependent risk of malformations associated with lithium, still widely used in treating bipolar disorder. The researchers examined the risk of cardiac malformations after first-trimester lithium exposure.

With the publication of each of these studies, we are moving toward an era where the question of teratogenicity is no longer just “yes” or “no,” but dose dependent. Soon, I hope we will be able to expand our knowledge by evaluating doses in milligrams per kilogram, rather than just a per day dose, thus addressing body size in evaluating the risk.
As more than half of pregnancies are unplanned, there are often times when women have been exposed to teratogens during early pregnancy and knowing the size of the risk is an invaluable decision-making tool. We don’t have the full risk picture yet, but it is growing clearer.
Dr. Koren is professor of pediatrics at Western University in Ontario and Tel Aviv University in Israel, and is the founder of the Motherisk Program. He reported having no relevant financial disclosures.
Since thalidomide, the medical community has sought to ensure that we do not miss any safety signal that a drug could cause malformations or developmental delays after in utero exposure.
At the time of a drug’s debut, there are relatively small numbers of exposures in pregnancy, and it’s difficult to decipher teratogenicity. As the number of exposures increases, we are generally able to answer “yes” or “no” to the question of teratogenicity. But in the last decade or so, data on drug exposure in pregnancy has become robust enough – thanks in part to large registries – to provide potentially more useful answers on safety. Specifically, .
Animal studies show us that there is a dose dependent effect in pregnancy, and not every dose causes harm to the fetus. However, that information is not easily translated into clinical practice because of the vast differences between animal and human pharmacokinetics and sensitivity to toxicity.
The first drug that came into focus as having dose dependent teratogenicity in pregnancy is the epilepsy drug valproic acid. In the late 1980s, studies showed that the drug was associated with an increased risk for spina bifida. Later, more congenital malformations were linked to valproic acid, including oral cleft, cardiac and limb defects, developmental delays, lower IQ, and even autism. But in the last few years, an increasing number of studies point to a lower dose that may represent an acceptable risk for some pregnant women.
Looking at data from EURAP, an international registry of antiepileptic drugs and pregnancy, researchers showed that the dose of valproic acid with the greatest risk for harm was 1,500 mg per day or greater, with a 24% frequency of major congenital malformations. But at less than 700 mg per day, the frequency of major malformations dropped to 5.9% (Neurology. 2015 Sep 8;85[10]:866-72).
Another analysis of the EURAP data showed the same dose-dependent relationship with other drugs. The researchers calculated rates of major congenital malformations in 1,402 pregnancies exposed to carbamazepine, 1,280 on lamotrigine, 1,010 on valproic acid, and 217 on phenobarbital, and all showed that the frequency of birth defects increased along with the dose of the drug.
The study identified the dose for each drug with the lowest rates of malformation. For lamotrigine, it was a dose of less than 300 mg per day, with a 2% frequency of malformations. Similarly, the dose was less than 400 mg per day for carbamazepine (3.4% rate of malformations). Overall, risks of malformation were significantly higher in valproic acid and phenobarbital at all tested doses and carbamazepine at doses greater than 400 mg per day, compared with lamotrigine monotherapy at less than 300 mg per day (Lancet Neurol. 2011 Jul;10[7]:609-17).
The study is important because it gives us a benchmark for these drugs, allowing us to see the risks at lower doses.
But not all the data are in agreement. In 2016, a Cochrane review of different antiepileptic drugs in pregnancy found that only with valproic acid could the risk of a malformation be clearly linked to the size of the dose (Cochrane Database Syst Rev. 2016 Nov 7;11:CD010224).
Most recently, a large U.S. database of Medicaid patients, which included more than 1.3 million pregnancies, showed the dose-dependent risk of malformations associated with lithium, still widely used in treating bipolar disorder. The researchers examined the risk of cardiac malformations after first-trimester lithium exposure.
With the publication of each of these studies, we are moving toward an era where the question of teratogenicity is no longer just “yes” or “no,” but dose dependent. Soon, I hope we will be able to expand our knowledge by evaluating doses in milligrams per kilogram, rather than just a per day dose, thus addressing body size in evaluating the risk.
As more than half of pregnancies are unplanned, there are often times when women have been exposed to teratogens during early pregnancy and knowing the size of the risk is an invaluable decision-making tool. We don’t have the full risk picture yet, but it is growing clearer.
Dr. Koren is professor of pediatrics at Western University in Ontario and Tel Aviv University in Israel, and is the founder of the Motherisk Program. He reported having no relevant financial disclosures.
Since thalidomide, the medical community has sought to ensure that we do not miss any safety signal that a drug could cause malformations or developmental delays after in utero exposure.
At the time of a drug’s debut, there are relatively small numbers of exposures in pregnancy, and it’s difficult to decipher teratogenicity. As the number of exposures increases, we are generally able to answer “yes” or “no” to the question of teratogenicity. But in the last decade or so, data on drug exposure in pregnancy has become robust enough – thanks in part to large registries – to provide potentially more useful answers on safety. Specifically, .
Animal studies show us that there is a dose dependent effect in pregnancy, and not every dose causes harm to the fetus. However, that information is not easily translated into clinical practice because of the vast differences between animal and human pharmacokinetics and sensitivity to toxicity.
The first drug that came into focus as having dose dependent teratogenicity in pregnancy is the epilepsy drug valproic acid. In the late 1980s, studies showed that the drug was associated with an increased risk for spina bifida. Later, more congenital malformations were linked to valproic acid, including oral cleft, cardiac and limb defects, developmental delays, lower IQ, and even autism. But in the last few years, an increasing number of studies point to a lower dose that may represent an acceptable risk for some pregnant women.
Looking at data from EURAP, an international registry of antiepileptic drugs and pregnancy, researchers showed that the dose of valproic acid with the greatest risk for harm was 1,500 mg per day or greater, with a 24% frequency of major congenital malformations. But at less than 700 mg per day, the frequency of major malformations dropped to 5.9% (Neurology. 2015 Sep 8;85[10]:866-72).
Another analysis of the EURAP data showed the same dose-dependent relationship with other drugs. The researchers calculated rates of major congenital malformations in 1,402 pregnancies exposed to carbamazepine, 1,280 on lamotrigine, 1,010 on valproic acid, and 217 on phenobarbital, and all showed that the frequency of birth defects increased along with the dose of the drug.
The study identified the dose for each drug with the lowest rates of malformation. For lamotrigine, it was a dose of less than 300 mg per day, with a 2% frequency of malformations. Similarly, the dose was less than 400 mg per day for carbamazepine (3.4% rate of malformations). Overall, risks of malformation were significantly higher in valproic acid and phenobarbital at all tested doses and carbamazepine at doses greater than 400 mg per day, compared with lamotrigine monotherapy at less than 300 mg per day (Lancet Neurol. 2011 Jul;10[7]:609-17).
The study is important because it gives us a benchmark for these drugs, allowing us to see the risks at lower doses.
But not all the data are in agreement. In 2016, a Cochrane review of different antiepileptic drugs in pregnancy found that only with valproic acid could the risk of a malformation be clearly linked to the size of the dose (Cochrane Database Syst Rev. 2016 Nov 7;11:CD010224).
Most recently, a large U.S. database of Medicaid patients, which included more than 1.3 million pregnancies, showed the dose-dependent risk of malformations associated with lithium, still widely used in treating bipolar disorder. The researchers examined the risk of cardiac malformations after first-trimester lithium exposure.
With the publication of each of these studies, we are moving toward an era where the question of teratogenicity is no longer just “yes” or “no,” but dose dependent. Soon, I hope we will be able to expand our knowledge by evaluating doses in milligrams per kilogram, rather than just a per day dose, thus addressing body size in evaluating the risk.
As more than half of pregnancies are unplanned, there are often times when women have been exposed to teratogens during early pregnancy and knowing the size of the risk is an invaluable decision-making tool. We don’t have the full risk picture yet, but it is growing clearer.
Dr. Koren is professor of pediatrics at Western University in Ontario and Tel Aviv University in Israel, and is the founder of the Motherisk Program. He reported having no relevant financial disclosures.
Too few RA patients get timely adjustment of DMARDs
Adjustment of disease-modifying antirheumatic drug (DMARD) therapy is not happening quickly enough for a substantial minority of rheumatoid arthritis patients with moderate to high disease activity, according to the findings of a registry study that is the first to evaluate this association.
Investigators led by Yomei Shaw, PhD, a research fellow with the National Data Bank for Rheumatic Diseases, retrospectively analyzed data from 538 patients in the university’s Rheumatoid Arthritis Comparative Effectiveness Research (RACER) registry who had moderate to high disease activity. Dr. Shaw conducted the study as a doctoral student in the department of health policy and management at the University of Pittsburgh.
In 40% of patients who had persistent disease activity of this severity, clinicians waited more than the 90 days recommended by treat-to-target (T2T) guidelines to adjust DMARDs, according to published results (Arthritis Care Res. 2017 Sep 21. doi: 10.1002/acr.23418). Such delay was more common for certain groups, including those on biologics and those who had rheumatoid arthritis (RA) for longer.
Compared with peers whose therapy was adjusted sooner, patients with such delayed adjustment were about one-quarter less likely to achieve low disease activity or remission during follow-up.
“The results of our survival analyses suggest that delays in DMARD adjustment of more than 3 months are common among RA patients with [moderate to high disease activity], highlighting an important potential gap in quality of care for these patients,” Dr. Shaw and her colleagues wrote.
They acknowledge that some of the delays in adjustment may have been unavoidable. For example, patients on biologic DMARDs may have needed insurance approval to start a new biologic, and patients with long-standing disease may have had fewer options remaining or symptoms that clinicians attributed to irreversible joint damage.
The study’s findings provide additional evidence of the importance of timely, guideline-adherent DMARD adjustment in reducing the amount of time patients spend in moderate to high disease activity, the investigators further contend.
“Promoting T2T in clinical practice requires coordinated change at the system, rheumatology practice, and individual levels,” such as consistently documenting the treatment target, assessing disease activity at every visit, and mandating a minimum visit frequency, they conclude.
Dr. Shaw and her colleagues studied patients having moderate to high disease activity, defined according to the 28-joint Disease Activity Score with C-reactive protein criteria (score of greater than 3.2).
The investigators reported a median time to adjustment of DMARD therapy of 154 days during 943.5 patient-years of follow-up. These adjustments could be adding, switching, or increasing the dose of a DMARD medication (excluding corticosteroids).
In multivariate analysis, patients had a longer time to DMARD adjustment if they were aged 75 years or older (subdistribution hazard ratio, 0.61; P = .02), had lower baseline disease activity (0.72; P less than .01), had longer duration of rheumatoid arthritis (0.98; P less than .01), or were receiving a biologic DMARD at baseline (0.71; P less than .01).
The median time to achieving low disease activity or remission was 301 days for the entire cohort. Patients had a longer time to achieve this goal if their DMARD therapy was not adjusted within 90 days (hazard ratio, 0.76; P = .01) or if they were African American (0.63; P = .01) or had higher baseline disease activity (0.75; P less than .01). They had a shorter time if they had better mental health (1.01; P = .03) or physical health (1.01; P = .02) as assessed with the 12-item Short Form Health Survey.
One of the study authors received research funding from Genentech and is currently employed by AbbVie. The registry used was funded by the National Institutes of Health and Genentech.
Randomized controlled trials demonstrate that statin lipid–lowering drugs reduce repeat cardiovascular events. Based on this evidence, clinicians agree that the vast majority of patients should receive statin lipid-lowering drugs after a CV event, and that providers not prescribing statins to these patients are providing suboptimal care.
By analogy, the data provided in this new analysis by Shaw and her colleagues suggests that many rheumatologists are providing suboptimal care. Many randomized, controlled trials testing a treat-to-target (T2T) paradigm demonstrate the clinical benefits of changing treatments for patients with rheumatoid arthritis who are in moderate or high disease activity. Yet, this analysis found many patients do not change treatments despite poor disease control. While the current study does not examine the reasons for suboptimal care, some correlates include:
• Patient preference to not change treatment;
• Provider desire to give treatments more time to work; and
• Health system issues such as drug payment complexities.

Continuing medical education may be necessary, but it is not a sufficient lever to push providers to implement T2T. Audit and feedback – providing individual providers with their performance metrics – alerts rheumatic disease providers when improvement is necessary. Yet, most providers need specific strategies within their practice to implement T2T. Collaborative learning between providers with a common purpose and coaches that understand the complexities of implementing T2T have been shown in the recently published TRACTION trial to produce improvements (Arthritis Rheumatol. 2017;69[7]:1374-80). The article by Shaw and her colleagues should sound the alarm to rheumatic disease providers: Care for RA needs improving if we want to produce optimal outcomes.
Daniel H. Solomon, MD, is a professor of medicine at Harvard University, and chief of the section of clinical sciences in the division of rheumatology at Brigham and Women’s Hospital, both in Boston. He has no relevant disclosures.
Randomized controlled trials demonstrate that statin lipid–lowering drugs reduce repeat cardiovascular events. Based on this evidence, clinicians agree that the vast majority of patients should receive statin lipid-lowering drugs after a CV event, and that providers not prescribing statins to these patients are providing suboptimal care.
By analogy, the data provided in this new analysis by Shaw and her colleagues suggests that many rheumatologists are providing suboptimal care. Many randomized, controlled trials testing a treat-to-target (T2T) paradigm demonstrate the clinical benefits of changing treatments for patients with rheumatoid arthritis who are in moderate or high disease activity. Yet, this analysis found many patients do not change treatments despite poor disease control. While the current study does not examine the reasons for suboptimal care, some correlates include:
• Patient preference to not change treatment;
• Provider desire to give treatments more time to work; and
• Health system issues such as drug payment complexities.
Continuing medical education may be necessary, but it is not a sufficient lever to push providers to implement T2T. Audit and feedback – providing individual providers with their performance metrics – alerts rheumatic disease providers when improvement is necessary. Yet, most providers need specific strategies within their practice to implement T2T. Collaborative learning between providers with a common purpose and coaches that understand the complexities of implementing T2T have been shown in the recently published TRACTION trial to produce improvements (Arthritis Rheumatol. 2017;69[7]:1374-80). The article by Shaw and her colleagues should sound the alarm to rheumatic disease providers: Care for RA needs improving if we want to produce optimal outcomes.
Daniel H. Solomon, MD, is a professor of medicine at Harvard University, and chief of the section of clinical sciences in the division of rheumatology at Brigham and Women’s Hospital, both in Boston. He has no relevant disclosures.
Randomized controlled trials demonstrate that statin lipid–lowering drugs reduce repeat cardiovascular events. Based on this evidence, clinicians agree that the vast majority of patients should receive statin lipid-lowering drugs after a CV event, and that providers not prescribing statins to these patients are providing suboptimal care.
By analogy, the data provided in this new analysis by Shaw and her colleagues suggests that many rheumatologists are providing suboptimal care. Many randomized, controlled trials testing a treat-to-target (T2T) paradigm demonstrate the clinical benefits of changing treatments for patients with rheumatoid arthritis who are in moderate or high disease activity. Yet, this analysis found many patients do not change treatments despite poor disease control. While the current study does not examine the reasons for suboptimal care, some correlates include:
• Patient preference to not change treatment;
• Provider desire to give treatments more time to work; and
• Health system issues such as drug payment complexities.
Continuing medical education may be necessary, but it is not a sufficient lever to push providers to implement T2T. Audit and feedback – providing individual providers with their performance metrics – alerts rheumatic disease providers when improvement is necessary. Yet, most providers need specific strategies within their practice to implement T2T. Collaborative learning between providers with a common purpose and coaches that understand the complexities of implementing T2T have been shown in the recently published TRACTION trial to produce improvements (Arthritis Rheumatol. 2017;69[7]:1374-80). The article by Shaw and her colleagues should sound the alarm to rheumatic disease providers: Care for RA needs improving if we want to produce optimal outcomes.
Daniel H. Solomon, MD, is a professor of medicine at Harvard University, and chief of the section of clinical sciences in the division of rheumatology at Brigham and Women’s Hospital, both in Boston. He has no relevant disclosures.
Adjustment of disease-modifying antirheumatic drug (DMARD) therapy is not happening quickly enough for a substantial minority of rheumatoid arthritis patients with moderate to high disease activity, according to the findings of a registry study that is the first to evaluate this association.
Investigators led by Yomei Shaw, PhD, a research fellow with the National Data Bank for Rheumatic Diseases, retrospectively analyzed data from 538 patients in the university’s Rheumatoid Arthritis Comparative Effectiveness Research (RACER) registry who had moderate to high disease activity. Dr. Shaw conducted the study as a doctoral student in the department of health policy and management at the University of Pittsburgh.
In 40% of patients who had persistent disease activity of this severity, clinicians waited more than the 90 days recommended by treat-to-target (T2T) guidelines to adjust DMARDs, according to published results (Arthritis Care Res. 2017 Sep 21. doi: 10.1002/acr.23418). Such delay was more common for certain groups, including those on biologics and those who had rheumatoid arthritis (RA) for longer.
Compared with peers whose therapy was adjusted sooner, patients with such delayed adjustment were about one-quarter less likely to achieve low disease activity or remission during follow-up.
“The results of our survival analyses suggest that delays in DMARD adjustment of more than 3 months are common among RA patients with [moderate to high disease activity], highlighting an important potential gap in quality of care for these patients,” Dr. Shaw and her colleagues wrote.
They acknowledge that some of the delays in adjustment may have been unavoidable. For example, patients on biologic DMARDs may have needed insurance approval to start a new biologic, and patients with long-standing disease may have had fewer options remaining or symptoms that clinicians attributed to irreversible joint damage.
The study’s findings provide additional evidence of the importance of timely, guideline-adherent DMARD adjustment in reducing the amount of time patients spend in moderate to high disease activity, the investigators further contend.
“Promoting T2T in clinical practice requires coordinated change at the system, rheumatology practice, and individual levels,” such as consistently documenting the treatment target, assessing disease activity at every visit, and mandating a minimum visit frequency, they conclude.
Dr. Shaw and her colleagues studied patients having moderate to high disease activity, defined according to the 28-joint Disease Activity Score with C-reactive protein criteria (score of greater than 3.2).
The investigators reported a median time to adjustment of DMARD therapy of 154 days during 943.5 patient-years of follow-up. These adjustments could be adding, switching, or increasing the dose of a DMARD medication (excluding corticosteroids).
In multivariate analysis, patients had a longer time to DMARD adjustment if they were aged 75 years or older (subdistribution hazard ratio, 0.61; P = .02), had lower baseline disease activity (0.72; P less than .01), had longer duration of rheumatoid arthritis (0.98; P less than .01), or were receiving a biologic DMARD at baseline (0.71; P less than .01).
The median time to achieving low disease activity or remission was 301 days for the entire cohort. Patients had a longer time to achieve this goal if their DMARD therapy was not adjusted within 90 days (hazard ratio, 0.76; P = .01) or if they were African American (0.63; P = .01) or had higher baseline disease activity (0.75; P less than .01). They had a shorter time if they had better mental health (1.01; P = .03) or physical health (1.01; P = .02) as assessed with the 12-item Short Form Health Survey.
One of the study authors received research funding from Genentech and is currently employed by AbbVie. The registry used was funded by the National Institutes of Health and Genentech.
Adjustment of disease-modifying antirheumatic drug (DMARD) therapy is not happening quickly enough for a substantial minority of rheumatoid arthritis patients with moderate to high disease activity, according to the findings of a registry study that is the first to evaluate this association.
Investigators led by Yomei Shaw, PhD, a research fellow with the National Data Bank for Rheumatic Diseases, retrospectively analyzed data from 538 patients in the university’s Rheumatoid Arthritis Comparative Effectiveness Research (RACER) registry who had moderate to high disease activity. Dr. Shaw conducted the study as a doctoral student in the department of health policy and management at the University of Pittsburgh.
In 40% of patients who had persistent disease activity of this severity, clinicians waited more than the 90 days recommended by treat-to-target (T2T) guidelines to adjust DMARDs, according to published results (Arthritis Care Res. 2017 Sep 21. doi: 10.1002/acr.23418). Such delay was more common for certain groups, including those on biologics and those who had rheumatoid arthritis (RA) for longer.
Compared with peers whose therapy was adjusted sooner, patients with such delayed adjustment were about one-quarter less likely to achieve low disease activity or remission during follow-up.
“The results of our survival analyses suggest that delays in DMARD adjustment of more than 3 months are common among RA patients with [moderate to high disease activity], highlighting an important potential gap in quality of care for these patients,” Dr. Shaw and her colleagues wrote.
They acknowledge that some of the delays in adjustment may have been unavoidable. For example, patients on biologic DMARDs may have needed insurance approval to start a new biologic, and patients with long-standing disease may have had fewer options remaining or symptoms that clinicians attributed to irreversible joint damage.
The study’s findings provide additional evidence of the importance of timely, guideline-adherent DMARD adjustment in reducing the amount of time patients spend in moderate to high disease activity, the investigators further contend.
“Promoting T2T in clinical practice requires coordinated change at the system, rheumatology practice, and individual levels,” such as consistently documenting the treatment target, assessing disease activity at every visit, and mandating a minimum visit frequency, they conclude.
Dr. Shaw and her colleagues studied patients having moderate to high disease activity, defined according to the 28-joint Disease Activity Score with C-reactive protein criteria (score of greater than 3.2).
The investigators reported a median time to adjustment of DMARD therapy of 154 days during 943.5 patient-years of follow-up. These adjustments could be adding, switching, or increasing the dose of a DMARD medication (excluding corticosteroids).
In multivariate analysis, patients had a longer time to DMARD adjustment if they were aged 75 years or older (subdistribution hazard ratio, 0.61; P = .02), had lower baseline disease activity (0.72; P less than .01), had longer duration of rheumatoid arthritis (0.98; P less than .01), or were receiving a biologic DMARD at baseline (0.71; P less than .01).
The median time to achieving low disease activity or remission was 301 days for the entire cohort. Patients had a longer time to achieve this goal if their DMARD therapy was not adjusted within 90 days (hazard ratio, 0.76; P = .01) or if they were African American (0.63; P = .01) or had higher baseline disease activity (0.75; P less than .01). They had a shorter time if they had better mental health (1.01; P = .03) or physical health (1.01; P = .02) as assessed with the 12-item Short Form Health Survey.
One of the study authors received research funding from Genentech and is currently employed by AbbVie. The registry used was funded by the National Institutes of Health and Genentech.
FROM ARTHRITIS CARE & RESEARCH
Key clinical point:
Major finding: In 40% of patients who had persistent disease activity of this severity, clinicians waited more than the 90 days recommended by treat-to-target guidelines to adjust DMARDs.
Data source: A retrospective cohort study of 538 patients with RA having moderate to high disease activity.
Disclosures: One of the study authors received research funding from Genentech and is currently employed by AbbVie. The study registry used was funded by the National Institutes of Health and Genentech.
Bearing the Standard
An esteemed ethics colleague forwarded me a Washington Post column that raises an important, neglected aspect of the Choice program and other VA forms of purchased care: potentially unequal and uneven standards of care.1 Congress authorized Choice to increase veterans’ ability to access needed clinical care in a timely and effective manner. The emphasis on access though may have inadvertently led to an equally serious gap in quality, especially for ethical standards of practice.
The Washington Post columnist Joe Davidson compares the often less demanding standard of opioid prescribing in the community with those of the VA and DoD. This difference in the monitoring of prescriptions the reporter suggests may be contributing to the epidemic of completed suicides—many by medication ingestion—and nonfatal but serious opioid overdoses. As Davidson writes, “The gap in coordination, adding to different clinical standards among VA and non-VA community providers, can be deadly. Health professionals outside the VA are not required to follow departmental guidelines.”1
The VA and DOD are required to follow rigorous, evidence-based practices, documented in the VA/DoD Practice Guideline for Opioid Therapy for Chronic Pain revised and reissued in February 2017. In addition, VA has a comprehensive, systematic, and standardized program of education and monitoring its Opioid Safety Initiative (OSI). The OSI was launched to improve and rationalize opioid prescribing especially when opioids are combined with benzodiazepines, which increases the risk of lethal outcomes from overdoses.
It is not just journalists who have expressed concerns about this disparity in prescribing rigor: The VA Office of Inspector General and several veterans service organizations also have called attention to what is in effect a double standard in care.2 All these entities have underscored another aspect of the Choice program that widens the quality and, hence, safety chasm—the fragmentation of clinical communication between community and VA providers. It is true that as of this writing, every state has passed prescription monitoring program (PMP) legislation. Prior to the change in federal regulation, VA was not permitted to release its controlled substance prescriptions to these pharmacy databases. But in the interest of patient safety, the privacy rules were modified to permit VA pharmacies to share records with the states. This has been a huge step forward in identifying patients who are receiving opioids, benzodiazepines, and stimulants, among other drugs, from a VAMC and 1 or more community prescribers.
Of course, it would be hubristic provincialism to think that there are not excellent clinicians and outstanding institutions in the community that equal or surpass the DoD/VA practice criteria. We are fortunate that because of Choice, veterans and service members now have available to them this level of expertise, which often is not present in smaller federal health care facilities. What is concerning, however, is those prescribers whose practice patterns are routinely and significantly below the bar and thereby place veterans in harms way.However, the efficacy of the PMPs to notify practitioners of prescribing patterns is dependent on the conscientiousness, given the death toll, even the conscience, of those who have prescribing privileges. I should emphasize that prescribing medications is a privilege and that states bestow this power only to those professionals who have met the stipulated education, training, credentialing, and licensing requirements. This professional preparation is crucial when there is not a shared medical record. Without the medical record, the practitioner, especiallyone who does not check the PMP or who does not have sufficient education and training in addiction and pain, is dependent on the history of the patient. The very substances being prescribed or sought may impair the ability of the patient to provide an accurate history due to ignorance, addiction, pain, or fear of losing pain relief.
There is a shortage of addiction and pain specialists in and outside the federal system.3,4 Therefore, we need Choice in order to meet the needs of service members and veterans. Congress has authorized bureaucratic mechanisms and payment sources to enable veterans to receive treatment from community providers. But a regulatory means to ensure that those providers adhere to the same high standards of care as that of VA and DoD practitioners must be established.
Critics of the VA have in many cases rightly made accountability the watchword of their campaigns. To its credit, the VA has embraced the cause in the I CARE value of integrity. “Act with high moral principle. Adhere to the highest professional standards. Maintain the trust and confidence of all with whom I engage.”
But that sense of responsibility must extend to all those who provide care to veterans—especially those who prescribe medications that have the immense double-edged potential to relieve pain and disability but at the same time also cause suffering and death.
While this inability to enforce VA/DoD responsible prescribing requirements in the community is likely more urgent and life threatening, there still are many other federal clinical and organizational policies and regulations to which the community is not required to adhere. I will discuss some of these and their potentially negative implications in future columns. For the promise of Choice to be realized, we all must work together to bear the highest possible standard of care for those who serve and have served.
1. Davidson J. Veterans’ health-care gap creates ‘greater risk’ for opioid use [news release]. Washington Post. August 7, 2017. https://www.washingtonpost.com/news/powerpost/wp/2017/08/07/veterans-health-care-gap-creates-greater-risk-for -opioid-abuse/?utm_term=.e4ec9596db6b. Accessed September 18, 2017.
2. Department of Veterans Affairs Office of Inspector General. Office of Healthcare Inspections. Report No. 17-01846-316. Opioid prescribing to high-risk veterans receiving VA purchased care. https://www.va.gov/oig/pubs/VAOIG-17-01846-316.pdf. Published July 31, 2017. Accessed September 18, 2017.
3. Vestal C. How severe is the shortage of substance abuse specialists? http://www.pewtrusts.org/en/research-and-analysis/blogs/stateline/2015/4/01/how-severe-is-the-shortage-of-substance-abuse -specialists. Published April 1, 2015. Accessed September 28, 2017.
4. Institute of Medicine (US) Committee on Advancing Pain Research, Care, and Education. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. Washington, DC: National Academies Press; 2011.
5. U.S. Department of Veterans Affairs. I care quick reference core values. https://www.va.gov/icare/docs/core_values_quick_reference.pdf. Accessed September 18, 2017.
An esteemed ethics colleague forwarded me a Washington Post column that raises an important, neglected aspect of the Choice program and other VA forms of purchased care: potentially unequal and uneven standards of care.1 Congress authorized Choice to increase veterans’ ability to access needed clinical care in a timely and effective manner. The emphasis on access though may have inadvertently led to an equally serious gap in quality, especially for ethical standards of practice.
The Washington Post columnist Joe Davidson compares the often less demanding standard of opioid prescribing in the community with those of the VA and DoD. This difference in the monitoring of prescriptions the reporter suggests may be contributing to the epidemic of completed suicides—many by medication ingestion—and nonfatal but serious opioid overdoses. As Davidson writes, “The gap in coordination, adding to different clinical standards among VA and non-VA community providers, can be deadly. Health professionals outside the VA are not required to follow departmental guidelines.”1
The VA and DOD are required to follow rigorous, evidence-based practices, documented in the VA/DoD Practice Guideline for Opioid Therapy for Chronic Pain revised and reissued in February 2017. In addition, VA has a comprehensive, systematic, and standardized program of education and monitoring its Opioid Safety Initiative (OSI). The OSI was launched to improve and rationalize opioid prescribing especially when opioids are combined with benzodiazepines, which increases the risk of lethal outcomes from overdoses.
It is not just journalists who have expressed concerns about this disparity in prescribing rigor: The VA Office of Inspector General and several veterans service organizations also have called attention to what is in effect a double standard in care.2 All these entities have underscored another aspect of the Choice program that widens the quality and, hence, safety chasm—the fragmentation of clinical communication between community and VA providers. It is true that as of this writing, every state has passed prescription monitoring program (PMP) legislation. Prior to the change in federal regulation, VA was not permitted to release its controlled substance prescriptions to these pharmacy databases. But in the interest of patient safety, the privacy rules were modified to permit VA pharmacies to share records with the states. This has been a huge step forward in identifying patients who are receiving opioids, benzodiazepines, and stimulants, among other drugs, from a VAMC and 1 or more community prescribers.
Of course, it would be hubristic provincialism to think that there are not excellent clinicians and outstanding institutions in the community that equal or surpass the DoD/VA practice criteria. We are fortunate that because of Choice, veterans and service members now have available to them this level of expertise, which often is not present in smaller federal health care facilities. What is concerning, however, is those prescribers whose practice patterns are routinely and significantly below the bar and thereby place veterans in harms way.However, the efficacy of the PMPs to notify practitioners of prescribing patterns is dependent on the conscientiousness, given the death toll, even the conscience, of those who have prescribing privileges. I should emphasize that prescribing medications is a privilege and that states bestow this power only to those professionals who have met the stipulated education, training, credentialing, and licensing requirements. This professional preparation is crucial when there is not a shared medical record. Without the medical record, the practitioner, especiallyone who does not check the PMP or who does not have sufficient education and training in addiction and pain, is dependent on the history of the patient. The very substances being prescribed or sought may impair the ability of the patient to provide an accurate history due to ignorance, addiction, pain, or fear of losing pain relief.
There is a shortage of addiction and pain specialists in and outside the federal system.3,4 Therefore, we need Choice in order to meet the needs of service members and veterans. Congress has authorized bureaucratic mechanisms and payment sources to enable veterans to receive treatment from community providers. But a regulatory means to ensure that those providers adhere to the same high standards of care as that of VA and DoD practitioners must be established.
Critics of the VA have in many cases rightly made accountability the watchword of their campaigns. To its credit, the VA has embraced the cause in the I CARE value of integrity. “Act with high moral principle. Adhere to the highest professional standards. Maintain the trust and confidence of all with whom I engage.”
But that sense of responsibility must extend to all those who provide care to veterans—especially those who prescribe medications that have the immense double-edged potential to relieve pain and disability but at the same time also cause suffering and death.
While this inability to enforce VA/DoD responsible prescribing requirements in the community is likely more urgent and life threatening, there still are many other federal clinical and organizational policies and regulations to which the community is not required to adhere. I will discuss some of these and their potentially negative implications in future columns. For the promise of Choice to be realized, we all must work together to bear the highest possible standard of care for those who serve and have served.
An esteemed ethics colleague forwarded me a Washington Post column that raises an important, neglected aspect of the Choice program and other VA forms of purchased care: potentially unequal and uneven standards of care.1 Congress authorized Choice to increase veterans’ ability to access needed clinical care in a timely and effective manner. The emphasis on access though may have inadvertently led to an equally serious gap in quality, especially for ethical standards of practice.
The Washington Post columnist Joe Davidson compares the often less demanding standard of opioid prescribing in the community with those of the VA and DoD. This difference in the monitoring of prescriptions the reporter suggests may be contributing to the epidemic of completed suicides—many by medication ingestion—and nonfatal but serious opioid overdoses. As Davidson writes, “The gap in coordination, adding to different clinical standards among VA and non-VA community providers, can be deadly. Health professionals outside the VA are not required to follow departmental guidelines.”1
The VA and DOD are required to follow rigorous, evidence-based practices, documented in the VA/DoD Practice Guideline for Opioid Therapy for Chronic Pain revised and reissued in February 2017. In addition, VA has a comprehensive, systematic, and standardized program of education and monitoring its Opioid Safety Initiative (OSI). The OSI was launched to improve and rationalize opioid prescribing especially when opioids are combined with benzodiazepines, which increases the risk of lethal outcomes from overdoses.
It is not just journalists who have expressed concerns about this disparity in prescribing rigor: The VA Office of Inspector General and several veterans service organizations also have called attention to what is in effect a double standard in care.2 All these entities have underscored another aspect of the Choice program that widens the quality and, hence, safety chasm—the fragmentation of clinical communication between community and VA providers. It is true that as of this writing, every state has passed prescription monitoring program (PMP) legislation. Prior to the change in federal regulation, VA was not permitted to release its controlled substance prescriptions to these pharmacy databases. But in the interest of patient safety, the privacy rules were modified to permit VA pharmacies to share records with the states. This has been a huge step forward in identifying patients who are receiving opioids, benzodiazepines, and stimulants, among other drugs, from a VAMC and 1 or more community prescribers.
Of course, it would be hubristic provincialism to think that there are not excellent clinicians and outstanding institutions in the community that equal or surpass the DoD/VA practice criteria. We are fortunate that because of Choice, veterans and service members now have available to them this level of expertise, which often is not present in smaller federal health care facilities. What is concerning, however, is those prescribers whose practice patterns are routinely and significantly below the bar and thereby place veterans in harms way.However, the efficacy of the PMPs to notify practitioners of prescribing patterns is dependent on the conscientiousness, given the death toll, even the conscience, of those who have prescribing privileges. I should emphasize that prescribing medications is a privilege and that states bestow this power only to those professionals who have met the stipulated education, training, credentialing, and licensing requirements. This professional preparation is crucial when there is not a shared medical record. Without the medical record, the practitioner, especiallyone who does not check the PMP or who does not have sufficient education and training in addiction and pain, is dependent on the history of the patient. The very substances being prescribed or sought may impair the ability of the patient to provide an accurate history due to ignorance, addiction, pain, or fear of losing pain relief.
There is a shortage of addiction and pain specialists in and outside the federal system.3,4 Therefore, we need Choice in order to meet the needs of service members and veterans. Congress has authorized bureaucratic mechanisms and payment sources to enable veterans to receive treatment from community providers. But a regulatory means to ensure that those providers adhere to the same high standards of care as that of VA and DoD practitioners must be established.
Critics of the VA have in many cases rightly made accountability the watchword of their campaigns. To its credit, the VA has embraced the cause in the I CARE value of integrity. “Act with high moral principle. Adhere to the highest professional standards. Maintain the trust and confidence of all with whom I engage.”
But that sense of responsibility must extend to all those who provide care to veterans—especially those who prescribe medications that have the immense double-edged potential to relieve pain and disability but at the same time also cause suffering and death.
While this inability to enforce VA/DoD responsible prescribing requirements in the community is likely more urgent and life threatening, there still are many other federal clinical and organizational policies and regulations to which the community is not required to adhere. I will discuss some of these and their potentially negative implications in future columns. For the promise of Choice to be realized, we all must work together to bear the highest possible standard of care for those who serve and have served.
1. Davidson J. Veterans’ health-care gap creates ‘greater risk’ for opioid use [news release]. Washington Post. August 7, 2017. https://www.washingtonpost.com/news/powerpost/wp/2017/08/07/veterans-health-care-gap-creates-greater-risk-for -opioid-abuse/?utm_term=.e4ec9596db6b. Accessed September 18, 2017.
2. Department of Veterans Affairs Office of Inspector General. Office of Healthcare Inspections. Report No. 17-01846-316. Opioid prescribing to high-risk veterans receiving VA purchased care. https://www.va.gov/oig/pubs/VAOIG-17-01846-316.pdf. Published July 31, 2017. Accessed September 18, 2017.
3. Vestal C. How severe is the shortage of substance abuse specialists? http://www.pewtrusts.org/en/research-and-analysis/blogs/stateline/2015/4/01/how-severe-is-the-shortage-of-substance-abuse -specialists. Published April 1, 2015. Accessed September 28, 2017.
4. Institute of Medicine (US) Committee on Advancing Pain Research, Care, and Education. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. Washington, DC: National Academies Press; 2011.
5. U.S. Department of Veterans Affairs. I care quick reference core values. https://www.va.gov/icare/docs/core_values_quick_reference.pdf. Accessed September 18, 2017.
1. Davidson J. Veterans’ health-care gap creates ‘greater risk’ for opioid use [news release]. Washington Post. August 7, 2017. https://www.washingtonpost.com/news/powerpost/wp/2017/08/07/veterans-health-care-gap-creates-greater-risk-for -opioid-abuse/?utm_term=.e4ec9596db6b. Accessed September 18, 2017.
2. Department of Veterans Affairs Office of Inspector General. Office of Healthcare Inspections. Report No. 17-01846-316. Opioid prescribing to high-risk veterans receiving VA purchased care. https://www.va.gov/oig/pubs/VAOIG-17-01846-316.pdf. Published July 31, 2017. Accessed September 18, 2017.
3. Vestal C. How severe is the shortage of substance abuse specialists? http://www.pewtrusts.org/en/research-and-analysis/blogs/stateline/2015/4/01/how-severe-is-the-shortage-of-substance-abuse -specialists. Published April 1, 2015. Accessed September 28, 2017.
4. Institute of Medicine (US) Committee on Advancing Pain Research, Care, and Education. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. Washington, DC: National Academies Press; 2011.
5. U.S. Department of Veterans Affairs. I care quick reference core values. https://www.va.gov/icare/docs/core_values_quick_reference.pdf. Accessed September 18, 2017.

Caplacizumab may enhance treatment of aTTP
Caplacizumab can improve upon standard care for patients with acquired thrombotic thrombocytopenic purpura (aTTP), according to results reported by Ablynx, the company developing caplacizumab.
In the phase 3 HERCULES trial, researchers compared caplacizumab, an anti-von Willebrand factor nanobody, plus standard care to placebo plus standard care in patients with aTTP.
Patients who received caplacizumab had a significant reduction in time to platelet count response.
In addition, they were significantly less likely than patients who received placebo to achieve the combined endpoint of aTTP-related death, aTTP recurrence, and experiencing at least 1 major thromboembolic event during the treatment period.
The safety profile of caplacizumab in this trial was said to be consistent with results from the phase 2 TITAN trial.
“The results of this landmark trial constitute a complete game-changer for patients with aTTP,” said HERCULES investigator Marie Scully, MBBS, of the University College Hospital in London, UK.
“They will revolutionize how we manage the acute phase of the disease, which is when patients are at highest risk for organ damage, recurrence, and death.”
Treatment
The HERCULES trial included 145 patients with an acute episode of aTTP. They were randomized 1:1 to receive either caplacizumab or placebo in addition to daily plasma exchange and immunosuppression (standard of care).
Patients received a single intravenous bolus of 10 mg of caplacizumab or placebo followed by a daily subcutaneous dose of 10 mg of caplacizumab or placebo until 30 days after the last daily plasma exchange.
If, at the end of this treatment period, there was evidence of persistent underlying disease activity indicative of an imminent risk for recurrence, the treatment could be extended for additional 7-day periods up to a maximum of 28 days. Patients were followed for a further 28 days after discontinuation of treatment.
In all, 71 patients received caplacizumab, and 58 (80.6%) of them completed the treatment. Seventy-three patients received placebo, and 50 of these patients (68.5%) completed treatment.
Baseline characteristics
At baseline, the mean age was 44.9 in the caplacizumab arm and 47.3 in the placebo arm. A majority of patients in both arms were female—68.1% and 69.9%, respectively.
The proportion of patients with an initial aTTP episode was 66.7% in the caplacizumab arm and 46.6% in the placebo arm. The proportion with a recurrent episode was 33.3% and 53.4%, respectively.
Most patients in both arms had ADAMTS13 activity below 10% at baseline—81.7% in the caplacizumab arm and 90.3% in the placebo arm.
The mean platelet count at baseline was 32.0 x 109/L in the caplacizumab arm and 39.1 x 109/L in the placebo arm.
Efficacy
The study’s primary endpoint was the time to confirmed normalization of platelet count response. There was a significant reduction in time to platelet count response in the caplacizumab arm compared to the placebo arm. The platelet normalization rate ratio was 1.55 (P<0.01).
A key secondary endpoint was the combination of aTTP-related death, aTTP recurrence, and at least 1 major thromboembolic event during study treatment. The incidence of this combined endpoint was 12.7% (n=9) in the caplacizumab arm and 49.3% (n=36) in the placebo arm (P<0.0001).
The incidence of aTTP-related death was 0% (n=0) in the caplacizumab arm and 4.1% (n=3) in the placebo arm. The incidence of aTTP recurrence was 4.2% (n=3) and 38.4% (n=28), respectively. And the incidence of at least 1 major thromboembolic event was 8.5% (n=6) and 8.2% (n=6), respectively.
Another key secondary endpoint was the incidence of aTTP recurrence during the overall study period, which was 12.7% (n=9) in the caplacizumab arm and 38.4% (n=28) in the placebo arm (P<0.001).
The incidence of aTTP recurrence during the follow-up period alone was 9.1% (n=6) in the caplacizumab arm and 0% (n=0) in the placebo arm.
A third key secondary endpoint was the percentage of patients with refractory aTTP, which was 0% (n=0) in the caplacizumab arm and 4.2% (n=3) in the placebo arm (P=0.0572).
Safety
The number and nature of treatment-emergent adverse events (AEs) were similar between the treatment arms, according to Ablynx. The proportion of patients with at least 1 treatment-emergent AE was 97.2% in the caplacizumab arm and 97.3% in the placebo arm.
The proportion of patients with at least 1 study-drug-related AE was 57.7% in the caplacizumab arm and 43.8% in the placebo arm. The rate of discontinuation due to at least 1 AE was 7.0% and 12.3%, respectively.
The incidence of bleeding-related AEs was higher in the caplacizumab arm than the placebo arm—66.2% and 49.3%, respectively. However, most bleeding-related events were mild or moderate in severity.
The proportion of patients with at least 1 serious AE was 39.4% (n=28) in the caplacizumab arm and 53.4% (n=39) in the placebo arm. The proportion of patients with at least 1 study-drug-related serious AE was 14.1% (n=10) and 5.5% (n=4), respectively.
During the treatment period, there were no deaths in the caplacizumab arm and 3 deaths in the placebo arm. There was 1 death in the caplacizumab arm during the follow-up period, but it was considered unrelated to caplacizumab. ![]()
Caplacizumab can improve upon standard care for patients with acquired thrombotic thrombocytopenic purpura (aTTP), according to results reported by Ablynx, the company developing caplacizumab.
In the phase 3 HERCULES trial, researchers compared caplacizumab, an anti-von Willebrand factor nanobody, plus standard care to placebo plus standard care in patients with aTTP.
Patients who received caplacizumab had a significant reduction in time to platelet count response.
In addition, they were significantly less likely than patients who received placebo to achieve the combined endpoint of aTTP-related death, aTTP recurrence, and experiencing at least 1 major thromboembolic event during the treatment period.
The safety profile of caplacizumab in this trial was said to be consistent with results from the phase 2 TITAN trial.
“The results of this landmark trial constitute a complete game-changer for patients with aTTP,” said HERCULES investigator Marie Scully, MBBS, of the University College Hospital in London, UK.
“They will revolutionize how we manage the acute phase of the disease, which is when patients are at highest risk for organ damage, recurrence, and death.”
Treatment
The HERCULES trial included 145 patients with an acute episode of aTTP. They were randomized 1:1 to receive either caplacizumab or placebo in addition to daily plasma exchange and immunosuppression (standard of care).
Patients received a single intravenous bolus of 10 mg of caplacizumab or placebo followed by a daily subcutaneous dose of 10 mg of caplacizumab or placebo until 30 days after the last daily plasma exchange.
If, at the end of this treatment period, there was evidence of persistent underlying disease activity indicative of an imminent risk for recurrence, the treatment could be extended for additional 7-day periods up to a maximum of 28 days. Patients were followed for a further 28 days after discontinuation of treatment.
In all, 71 patients received caplacizumab, and 58 (80.6%) of them completed the treatment. Seventy-three patients received placebo, and 50 of these patients (68.5%) completed treatment.
Baseline characteristics
At baseline, the mean age was 44.9 in the caplacizumab arm and 47.3 in the placebo arm. A majority of patients in both arms were female—68.1% and 69.9%, respectively.
The proportion of patients with an initial aTTP episode was 66.7% in the caplacizumab arm and 46.6% in the placebo arm. The proportion with a recurrent episode was 33.3% and 53.4%, respectively.
Most patients in both arms had ADAMTS13 activity below 10% at baseline—81.7% in the caplacizumab arm and 90.3% in the placebo arm.
The mean platelet count at baseline was 32.0 x 109/L in the caplacizumab arm and 39.1 x 109/L in the placebo arm.
Efficacy
The study’s primary endpoint was the time to confirmed normalization of platelet count response. There was a significant reduction in time to platelet count response in the caplacizumab arm compared to the placebo arm. The platelet normalization rate ratio was 1.55 (P<0.01).
A key secondary endpoint was the combination of aTTP-related death, aTTP recurrence, and at least 1 major thromboembolic event during study treatment. The incidence of this combined endpoint was 12.7% (n=9) in the caplacizumab arm and 49.3% (n=36) in the placebo arm (P<0.0001).
The incidence of aTTP-related death was 0% (n=0) in the caplacizumab arm and 4.1% (n=3) in the placebo arm. The incidence of aTTP recurrence was 4.2% (n=3) and 38.4% (n=28), respectively. And the incidence of at least 1 major thromboembolic event was 8.5% (n=6) and 8.2% (n=6), respectively.
Another key secondary endpoint was the incidence of aTTP recurrence during the overall study period, which was 12.7% (n=9) in the caplacizumab arm and 38.4% (n=28) in the placebo arm (P<0.001).
The incidence of aTTP recurrence during the follow-up period alone was 9.1% (n=6) in the caplacizumab arm and 0% (n=0) in the placebo arm.
A third key secondary endpoint was the percentage of patients with refractory aTTP, which was 0% (n=0) in the caplacizumab arm and 4.2% (n=3) in the placebo arm (P=0.0572).
Safety
The number and nature of treatment-emergent adverse events (AEs) were similar between the treatment arms, according to Ablynx. The proportion of patients with at least 1 treatment-emergent AE was 97.2% in the caplacizumab arm and 97.3% in the placebo arm.
The proportion of patients with at least 1 study-drug-related AE was 57.7% in the caplacizumab arm and 43.8% in the placebo arm. The rate of discontinuation due to at least 1 AE was 7.0% and 12.3%, respectively.
The incidence of bleeding-related AEs was higher in the caplacizumab arm than the placebo arm—66.2% and 49.3%, respectively. However, most bleeding-related events were mild or moderate in severity.
The proportion of patients with at least 1 serious AE was 39.4% (n=28) in the caplacizumab arm and 53.4% (n=39) in the placebo arm. The proportion of patients with at least 1 study-drug-related serious AE was 14.1% (n=10) and 5.5% (n=4), respectively.
During the treatment period, there were no deaths in the caplacizumab arm and 3 deaths in the placebo arm. There was 1 death in the caplacizumab arm during the follow-up period, but it was considered unrelated to caplacizumab. ![]()
Caplacizumab can improve upon standard care for patients with acquired thrombotic thrombocytopenic purpura (aTTP), according to results reported by Ablynx, the company developing caplacizumab.
In the phase 3 HERCULES trial, researchers compared caplacizumab, an anti-von Willebrand factor nanobody, plus standard care to placebo plus standard care in patients with aTTP.
Patients who received caplacizumab had a significant reduction in time to platelet count response.
In addition, they were significantly less likely than patients who received placebo to achieve the combined endpoint of aTTP-related death, aTTP recurrence, and experiencing at least 1 major thromboembolic event during the treatment period.
The safety profile of caplacizumab in this trial was said to be consistent with results from the phase 2 TITAN trial.
“The results of this landmark trial constitute a complete game-changer for patients with aTTP,” said HERCULES investigator Marie Scully, MBBS, of the University College Hospital in London, UK.
“They will revolutionize how we manage the acute phase of the disease, which is when patients are at highest risk for organ damage, recurrence, and death.”
Treatment
The HERCULES trial included 145 patients with an acute episode of aTTP. They were randomized 1:1 to receive either caplacizumab or placebo in addition to daily plasma exchange and immunosuppression (standard of care).
Patients received a single intravenous bolus of 10 mg of caplacizumab or placebo followed by a daily subcutaneous dose of 10 mg of caplacizumab or placebo until 30 days after the last daily plasma exchange.
If, at the end of this treatment period, there was evidence of persistent underlying disease activity indicative of an imminent risk for recurrence, the treatment could be extended for additional 7-day periods up to a maximum of 28 days. Patients were followed for a further 28 days after discontinuation of treatment.
In all, 71 patients received caplacizumab, and 58 (80.6%) of them completed the treatment. Seventy-three patients received placebo, and 50 of these patients (68.5%) completed treatment.
Baseline characteristics
At baseline, the mean age was 44.9 in the caplacizumab arm and 47.3 in the placebo arm. A majority of patients in both arms were female—68.1% and 69.9%, respectively.
The proportion of patients with an initial aTTP episode was 66.7% in the caplacizumab arm and 46.6% in the placebo arm. The proportion with a recurrent episode was 33.3% and 53.4%, respectively.
Most patients in both arms had ADAMTS13 activity below 10% at baseline—81.7% in the caplacizumab arm and 90.3% in the placebo arm.
The mean platelet count at baseline was 32.0 x 109/L in the caplacizumab arm and 39.1 x 109/L in the placebo arm.
Efficacy
The study’s primary endpoint was the time to confirmed normalization of platelet count response. There was a significant reduction in time to platelet count response in the caplacizumab arm compared to the placebo arm. The platelet normalization rate ratio was 1.55 (P<0.01).
A key secondary endpoint was the combination of aTTP-related death, aTTP recurrence, and at least 1 major thromboembolic event during study treatment. The incidence of this combined endpoint was 12.7% (n=9) in the caplacizumab arm and 49.3% (n=36) in the placebo arm (P<0.0001).
The incidence of aTTP-related death was 0% (n=0) in the caplacizumab arm and 4.1% (n=3) in the placebo arm. The incidence of aTTP recurrence was 4.2% (n=3) and 38.4% (n=28), respectively. And the incidence of at least 1 major thromboembolic event was 8.5% (n=6) and 8.2% (n=6), respectively.
Another key secondary endpoint was the incidence of aTTP recurrence during the overall study period, which was 12.7% (n=9) in the caplacizumab arm and 38.4% (n=28) in the placebo arm (P<0.001).
The incidence of aTTP recurrence during the follow-up period alone was 9.1% (n=6) in the caplacizumab arm and 0% (n=0) in the placebo arm.
A third key secondary endpoint was the percentage of patients with refractory aTTP, which was 0% (n=0) in the caplacizumab arm and 4.2% (n=3) in the placebo arm (P=0.0572).
Safety
The number and nature of treatment-emergent adverse events (AEs) were similar between the treatment arms, according to Ablynx. The proportion of patients with at least 1 treatment-emergent AE was 97.2% in the caplacizumab arm and 97.3% in the placebo arm.
The proportion of patients with at least 1 study-drug-related AE was 57.7% in the caplacizumab arm and 43.8% in the placebo arm. The rate of discontinuation due to at least 1 AE was 7.0% and 12.3%, respectively.
The incidence of bleeding-related AEs was higher in the caplacizumab arm than the placebo arm—66.2% and 49.3%, respectively. However, most bleeding-related events were mild or moderate in severity.
The proportion of patients with at least 1 serious AE was 39.4% (n=28) in the caplacizumab arm and 53.4% (n=39) in the placebo arm. The proportion of patients with at least 1 study-drug-related serious AE was 14.1% (n=10) and 5.5% (n=4), respectively.
During the treatment period, there were no deaths in the caplacizumab arm and 3 deaths in the placebo arm. There was 1 death in the caplacizumab arm during the follow-up period, but it was considered unrelated to caplacizumab. ![]()