User login
Methylphenidate shows enduring sleep benefits in pediatric ADHD
PARIS – Methylphenidate therapy for attention-deficit/hyperactivity disorder in medication-naive boys significantly improved their sleep quality, timing, and duration in a double-blind randomized trial, Michelle M. Solleveld, MD, reported at the annual congress of the European College of Neuropsychopharmacology.
Moreover, these salutary effects on sleep persisted for at least 1 week after methylphenidate was stopped at the end of the 16-week study, added Dr. Solleveld of the University of Amsterdam.

Indeed, while parents embrace the improvement in behavioral symptoms of ADHD provided by methylphenidate, they often express concern about the possible adverse effects of stimulant medication on their child’s sleep. The new study findings are reassuring on that score.
Sleep difficulties are a major problem in patients with ADHD: They tend to fall asleep later and have more frequent awakenings during the night, which results in decreased total sleep time and sleep efficiency, Dr. Solleveld noted.
Prior studies of methylphenidate’s effects on sleep in pediatric ADHD have yielded mixed results. The negative studies were too brief to provide meaningful results, according to Dr. Solleveld, who said at least 8 weeks of treatment are required in order to evaluate the drug’s effect on sleep problems properly.
She presented a randomized, double-blind, 16-week, placebo-controlled clinical trial involving 50 medication-naive boys with ADHD who were 10-12 years old. Their sleep was assessed via actigraphy measurements taken over 5 consecutive nights, keeping a sleep diary, and answering questionnaires, including the Epworth Sleepiness Scale, at three time points: prior to randomization, 8 weeks into the trial, and finally 1 week after the study ended.
Sleep efficiency – the primary study outcome – showed a strong 5% improvement in the methylphenidate group but was unchanged from baseline in placebo-treated controls. The boys who received methylphenidate fell asleep earlier, had a shorter latency of sleep onset, and slept for longer, compared with their baseline measures or with the sleep results in controls.
The finding that the methylphenidate-induced improvements in sleep persisted for a week after drug clearance is consistent with brain imaging studies carried out by Dr. Solleveld and her coinvestigators. They believe that the effects of stimulant therapy may be age dependent. The investigators previously have shown that adults with ADHD who began treatment with stimulants before age 16 years – when brain development is still ongoing – had lower levels of basal gamma-aminobutyric acid (GABA) and higher GABA response to an oral methylphenidate than did those who began treatment with stimulants after age 23 years. This is thought to be attributable to prolonged reductions in dopamine turnover induced by methylphenidate in the developing brain (Neuroimage Clin. 2017 Jun 2;15:812-8).
The Dutch investigators also have reported that methylphenidate therapy in children with ADHD – but not in affected adults – increased the cerebral blood flow response within the thalamus to a dopamine challenge (JAMA Psychiatry. 2016 Sep 1;73[9]:955-62).
The clinical ramifications of these apparently long-lasting, drug-related alterations in GABA neurotransmission are the subject of ongoing research.
Dr. Solleveld reported having no financial conflicts of interest.
PARIS – Methylphenidate therapy for attention-deficit/hyperactivity disorder in medication-naive boys significantly improved their sleep quality, timing, and duration in a double-blind randomized trial, Michelle M. Solleveld, MD, reported at the annual congress of the European College of Neuropsychopharmacology.
Moreover, these salutary effects on sleep persisted for at least 1 week after methylphenidate was stopped at the end of the 16-week study, added Dr. Solleveld of the University of Amsterdam.
Indeed, while parents embrace the improvement in behavioral symptoms of ADHD provided by methylphenidate, they often express concern about the possible adverse effects of stimulant medication on their child’s sleep. The new study findings are reassuring on that score.
Sleep difficulties are a major problem in patients with ADHD: They tend to fall asleep later and have more frequent awakenings during the night, which results in decreased total sleep time and sleep efficiency, Dr. Solleveld noted.
Prior studies of methylphenidate’s effects on sleep in pediatric ADHD have yielded mixed results. The negative studies were too brief to provide meaningful results, according to Dr. Solleveld, who said at least 8 weeks of treatment are required in order to evaluate the drug’s effect on sleep problems properly.
She presented a randomized, double-blind, 16-week, placebo-controlled clinical trial involving 50 medication-naive boys with ADHD who were 10-12 years old. Their sleep was assessed via actigraphy measurements taken over 5 consecutive nights, keeping a sleep diary, and answering questionnaires, including the Epworth Sleepiness Scale, at three time points: prior to randomization, 8 weeks into the trial, and finally 1 week after the study ended.
Sleep efficiency – the primary study outcome – showed a strong 5% improvement in the methylphenidate group but was unchanged from baseline in placebo-treated controls. The boys who received methylphenidate fell asleep earlier, had a shorter latency of sleep onset, and slept for longer, compared with their baseline measures or with the sleep results in controls.
The finding that the methylphenidate-induced improvements in sleep persisted for a week after drug clearance is consistent with brain imaging studies carried out by Dr. Solleveld and her coinvestigators. They believe that the effects of stimulant therapy may be age dependent. The investigators previously have shown that adults with ADHD who began treatment with stimulants before age 16 years – when brain development is still ongoing – had lower levels of basal gamma-aminobutyric acid (GABA) and higher GABA response to an oral methylphenidate than did those who began treatment with stimulants after age 23 years. This is thought to be attributable to prolonged reductions in dopamine turnover induced by methylphenidate in the developing brain (Neuroimage Clin. 2017 Jun 2;15:812-8).
The Dutch investigators also have reported that methylphenidate therapy in children with ADHD – but not in affected adults – increased the cerebral blood flow response within the thalamus to a dopamine challenge (JAMA Psychiatry. 2016 Sep 1;73[9]:955-62).
The clinical ramifications of these apparently long-lasting, drug-related alterations in GABA neurotransmission are the subject of ongoing research.
Dr. Solleveld reported having no financial conflicts of interest.
PARIS – Methylphenidate therapy for attention-deficit/hyperactivity disorder in medication-naive boys significantly improved their sleep quality, timing, and duration in a double-blind randomized trial, Michelle M. Solleveld, MD, reported at the annual congress of the European College of Neuropsychopharmacology.
Moreover, these salutary effects on sleep persisted for at least 1 week after methylphenidate was stopped at the end of the 16-week study, added Dr. Solleveld of the University of Amsterdam.
Indeed, while parents embrace the improvement in behavioral symptoms of ADHD provided by methylphenidate, they often express concern about the possible adverse effects of stimulant medication on their child’s sleep. The new study findings are reassuring on that score.
Sleep difficulties are a major problem in patients with ADHD: They tend to fall asleep later and have more frequent awakenings during the night, which results in decreased total sleep time and sleep efficiency, Dr. Solleveld noted.
Prior studies of methylphenidate’s effects on sleep in pediatric ADHD have yielded mixed results. The negative studies were too brief to provide meaningful results, according to Dr. Solleveld, who said at least 8 weeks of treatment are required in order to evaluate the drug’s effect on sleep problems properly.
She presented a randomized, double-blind, 16-week, placebo-controlled clinical trial involving 50 medication-naive boys with ADHD who were 10-12 years old. Their sleep was assessed via actigraphy measurements taken over 5 consecutive nights, keeping a sleep diary, and answering questionnaires, including the Epworth Sleepiness Scale, at three time points: prior to randomization, 8 weeks into the trial, and finally 1 week after the study ended.
Sleep efficiency – the primary study outcome – showed a strong 5% improvement in the methylphenidate group but was unchanged from baseline in placebo-treated controls. The boys who received methylphenidate fell asleep earlier, had a shorter latency of sleep onset, and slept for longer, compared with their baseline measures or with the sleep results in controls.
The finding that the methylphenidate-induced improvements in sleep persisted for a week after drug clearance is consistent with brain imaging studies carried out by Dr. Solleveld and her coinvestigators. They believe that the effects of stimulant therapy may be age dependent. The investigators previously have shown that adults with ADHD who began treatment with stimulants before age 16 years – when brain development is still ongoing – had lower levels of basal gamma-aminobutyric acid (GABA) and higher GABA response to an oral methylphenidate than did those who began treatment with stimulants after age 23 years. This is thought to be attributable to prolonged reductions in dopamine turnover induced by methylphenidate in the developing brain (Neuroimage Clin. 2017 Jun 2;15:812-8).
The Dutch investigators also have reported that methylphenidate therapy in children with ADHD – but not in affected adults – increased the cerebral blood flow response within the thalamus to a dopamine challenge (JAMA Psychiatry. 2016 Sep 1;73[9]:955-62).
The clinical ramifications of these apparently long-lasting, drug-related alterations in GABA neurotransmission are the subject of ongoing research.
Dr. Solleveld reported having no financial conflicts of interest.
AT THE ECNP CONGRESS
Key clinical point:
Major finding: Sleep efficiency in boys with ADHD improved significantly by 5% in response to methylphenidate therapy.
Data source: This randomized, double-blind, placebo-controlled clinical trial included 50 medication-naive boys aged 10-12 years with ADHD.
Disclosures: The study presenter reported having no financial conflicts of interest.
Worse survival with cetuximab plus chemo for CRC liver mets
MADRID – Adding cetuximab (Erbitux) to chemotherapy before or after surgery for resectable colorectal liver metastases resulted in significantly worse survival compared with perioperative chemotherapy alone, mature results of the randomized New EPOC trial confirmed.
Among 257 patients with KRAS exon 2 wild-type resectable colorectal liver metastases (CRLM) or suboptimally resectable CRLM, median overall survival (OS) was 81 months for patients assigned to neoadjuvant and adjuvant chemotherapy alone, compared with 55.4 months for patients who received perioperative chemotherapy plus cetuximab, reported John Bridgewater, MD, of University College London Cancer Institute.

Median progression-free survival (PFS) was 22.2 months without cetuximab, vs. 15.5 months with chemotherapy plus cetuximab, a difference that was not statistically significant. However, when the primary analysis of the trial was reported in 2014 in The Lancet: Oncology, cetuximab was associated with significantly shorter PFS. The trial was halted in 2012 on the recommendation of the data monitoring committee.
“In the context of perioperative therapy for resectable CRLM, chemotherapy plus cetuximab confers a shorter OS and PPS,” Dr. Bridgewater and his associates wrote.”This is primarily in those with conventionally favorable prognostic features suggesting that cetuximab induces adverse biology in some patients, whose biomarker profile is ongoing.”
They also found that there were no differences in overall survival among patients with responses to chemotherapy according to Response Evaluation Criteria in Solid Tumors (RECIST) and those who did not have responses, “suggesting that in this predominantly operable population, any conferred benefit of systemic treatment is through elimination of micro-metastatic disease rather than by downsizing of radiologically evaluable disease.
The randomized phase 3 Medical Research Council COIN trial, results of which were reported at the 2010 annual meeting of the American Society of Clinical Oncology and later published in The Lancet, failed to show a benefit for the addition of cetuximab to front-line oxaliplatin-based chemotherapy for metastatic CRC. Median OS in KRAS wild-type patients was 17 months when cetuximab was added to chemotherapy with oxaliplatin and an intravenous or oral fluoropyrimidine, compared with 17.9 months when chemotherapy was delivered without cetuximab. Median PFS held at 8.6 months in both arms of the trial.
The New EPOC investigators described their study as a “natural extension” of the COIN study as well as earlier trials and phase 2 studies of neoadjuvant and adjuvant therapy in this patient population.
From 2007 through 2012, 257 patients were randomly assigned to receive chemotherapy alone, or chemotherapy with rituximab. Chemotherapy consisted of one of two regimens: oxaliplatin 85 mg/m2 intravenously over 2 hours and fluorouracil bolus 400 mg/m2 intravenously over 5 minutes followed by a 46-hour infusion of fluorouracil 2,400 mg/m2 repeated every 2 weeks; or oxaliplatin 130 mg/m2 intravenously over 2 hours and oral capecitabine (Xeloda) 1,000 mg/m2 twice daily on days 1-14 repeated every 3 weeks.
After a median follow-up of 69 months, there were 130 deaths from any cause. As noted before, median OS was significantly shorter with cetuximab. The hazard ratio for death with cetuximab was 1.45 (P = .035). Median PFS did not differ in this most recent analysis, however.
Median PPS was 35.4 months for chemotherapy alone, compared with 23.5 months for chemotherapy plus cetuximab (P = .014).
The poor overall survival and PFS results indicate that “cetuximab cannot be recommended for perioperative treatment in patients with resectable disease,” said Thomas Gruenberger, MD, an oncologic surgeon at Rudolf Hospital in Vienna, who was invited to review the results in a poster discussion session.
Cancer Research UK supported the study. Dr. Bridgewater disclosed honoraria and speakers fees from Merck, Celgene, and Servier, and travel support from Amgen, Merck Sharpe Dohme, and Servier.
MADRID – Adding cetuximab (Erbitux) to chemotherapy before or after surgery for resectable colorectal liver metastases resulted in significantly worse survival compared with perioperative chemotherapy alone, mature results of the randomized New EPOC trial confirmed.
Among 257 patients with KRAS exon 2 wild-type resectable colorectal liver metastases (CRLM) or suboptimally resectable CRLM, median overall survival (OS) was 81 months for patients assigned to neoadjuvant and adjuvant chemotherapy alone, compared with 55.4 months for patients who received perioperative chemotherapy plus cetuximab, reported John Bridgewater, MD, of University College London Cancer Institute.
Median progression-free survival (PFS) was 22.2 months without cetuximab, vs. 15.5 months with chemotherapy plus cetuximab, a difference that was not statistically significant. However, when the primary analysis of the trial was reported in 2014 in The Lancet: Oncology, cetuximab was associated with significantly shorter PFS. The trial was halted in 2012 on the recommendation of the data monitoring committee.
“In the context of perioperative therapy for resectable CRLM, chemotherapy plus cetuximab confers a shorter OS and PPS,” Dr. Bridgewater and his associates wrote.”This is primarily in those with conventionally favorable prognostic features suggesting that cetuximab induces adverse biology in some patients, whose biomarker profile is ongoing.”
They also found that there were no differences in overall survival among patients with responses to chemotherapy according to Response Evaluation Criteria in Solid Tumors (RECIST) and those who did not have responses, “suggesting that in this predominantly operable population, any conferred benefit of systemic treatment is through elimination of micro-metastatic disease rather than by downsizing of radiologically evaluable disease.
The randomized phase 3 Medical Research Council COIN trial, results of which were reported at the 2010 annual meeting of the American Society of Clinical Oncology and later published in The Lancet, failed to show a benefit for the addition of cetuximab to front-line oxaliplatin-based chemotherapy for metastatic CRC. Median OS in KRAS wild-type patients was 17 months when cetuximab was added to chemotherapy with oxaliplatin and an intravenous or oral fluoropyrimidine, compared with 17.9 months when chemotherapy was delivered without cetuximab. Median PFS held at 8.6 months in both arms of the trial.
The New EPOC investigators described their study as a “natural extension” of the COIN study as well as earlier trials and phase 2 studies of neoadjuvant and adjuvant therapy in this patient population.
From 2007 through 2012, 257 patients were randomly assigned to receive chemotherapy alone, or chemotherapy with rituximab. Chemotherapy consisted of one of two regimens: oxaliplatin 85 mg/m2 intravenously over 2 hours and fluorouracil bolus 400 mg/m2 intravenously over 5 minutes followed by a 46-hour infusion of fluorouracil 2,400 mg/m2 repeated every 2 weeks; or oxaliplatin 130 mg/m2 intravenously over 2 hours and oral capecitabine (Xeloda) 1,000 mg/m2 twice daily on days 1-14 repeated every 3 weeks.
After a median follow-up of 69 months, there were 130 deaths from any cause. As noted before, median OS was significantly shorter with cetuximab. The hazard ratio for death with cetuximab was 1.45 (P = .035). Median PFS did not differ in this most recent analysis, however.
Median PPS was 35.4 months for chemotherapy alone, compared with 23.5 months for chemotherapy plus cetuximab (P = .014).
The poor overall survival and PFS results indicate that “cetuximab cannot be recommended for perioperative treatment in patients with resectable disease,” said Thomas Gruenberger, MD, an oncologic surgeon at Rudolf Hospital in Vienna, who was invited to review the results in a poster discussion session.
Cancer Research UK supported the study. Dr. Bridgewater disclosed honoraria and speakers fees from Merck, Celgene, and Servier, and travel support from Amgen, Merck Sharpe Dohme, and Servier.
MADRID – Adding cetuximab (Erbitux) to chemotherapy before or after surgery for resectable colorectal liver metastases resulted in significantly worse survival compared with perioperative chemotherapy alone, mature results of the randomized New EPOC trial confirmed.
Among 257 patients with KRAS exon 2 wild-type resectable colorectal liver metastases (CRLM) or suboptimally resectable CRLM, median overall survival (OS) was 81 months for patients assigned to neoadjuvant and adjuvant chemotherapy alone, compared with 55.4 months for patients who received perioperative chemotherapy plus cetuximab, reported John Bridgewater, MD, of University College London Cancer Institute.
Median progression-free survival (PFS) was 22.2 months without cetuximab, vs. 15.5 months with chemotherapy plus cetuximab, a difference that was not statistically significant. However, when the primary analysis of the trial was reported in 2014 in The Lancet: Oncology, cetuximab was associated with significantly shorter PFS. The trial was halted in 2012 on the recommendation of the data monitoring committee.
“In the context of perioperative therapy for resectable CRLM, chemotherapy plus cetuximab confers a shorter OS and PPS,” Dr. Bridgewater and his associates wrote.”This is primarily in those with conventionally favorable prognostic features suggesting that cetuximab induces adverse biology in some patients, whose biomarker profile is ongoing.”
They also found that there were no differences in overall survival among patients with responses to chemotherapy according to Response Evaluation Criteria in Solid Tumors (RECIST) and those who did not have responses, “suggesting that in this predominantly operable population, any conferred benefit of systemic treatment is through elimination of micro-metastatic disease rather than by downsizing of radiologically evaluable disease.
The randomized phase 3 Medical Research Council COIN trial, results of which were reported at the 2010 annual meeting of the American Society of Clinical Oncology and later published in The Lancet, failed to show a benefit for the addition of cetuximab to front-line oxaliplatin-based chemotherapy for metastatic CRC. Median OS in KRAS wild-type patients was 17 months when cetuximab was added to chemotherapy with oxaliplatin and an intravenous or oral fluoropyrimidine, compared with 17.9 months when chemotherapy was delivered without cetuximab. Median PFS held at 8.6 months in both arms of the trial.
The New EPOC investigators described their study as a “natural extension” of the COIN study as well as earlier trials and phase 2 studies of neoadjuvant and adjuvant therapy in this patient population.
From 2007 through 2012, 257 patients were randomly assigned to receive chemotherapy alone, or chemotherapy with rituximab. Chemotherapy consisted of one of two regimens: oxaliplatin 85 mg/m2 intravenously over 2 hours and fluorouracil bolus 400 mg/m2 intravenously over 5 minutes followed by a 46-hour infusion of fluorouracil 2,400 mg/m2 repeated every 2 weeks; or oxaliplatin 130 mg/m2 intravenously over 2 hours and oral capecitabine (Xeloda) 1,000 mg/m2 twice daily on days 1-14 repeated every 3 weeks.
After a median follow-up of 69 months, there were 130 deaths from any cause. As noted before, median OS was significantly shorter with cetuximab. The hazard ratio for death with cetuximab was 1.45 (P = .035). Median PFS did not differ in this most recent analysis, however.
Median PPS was 35.4 months for chemotherapy alone, compared with 23.5 months for chemotherapy plus cetuximab (P = .014).
The poor overall survival and PFS results indicate that “cetuximab cannot be recommended for perioperative treatment in patients with resectable disease,” said Thomas Gruenberger, MD, an oncologic surgeon at Rudolf Hospital in Vienna, who was invited to review the results in a poster discussion session.
Cancer Research UK supported the study. Dr. Bridgewater disclosed honoraria and speakers fees from Merck, Celgene, and Servier, and travel support from Amgen, Merck Sharpe Dohme, and Servier.
AT ESMO 2017
Key clinical point: Patients with resectable colorectal liver metastases (CRLM) fared worse when cetuximab was added to neoadjuvant and adjuvant chemotherapy.
Major finding: Median overall survival was 81 months for patients assigned to chemotherapy alone, vs. 55.4 months for patients assigned to chemotherapy plus cetuximab.
Data source: Mature analysis of a randomized phase 3 trial in 257 patients with resectable CRLM. The trial was halted for futility in 2012.
Disclosures: Cancer Research UK supported the study. Dr. Bridgewater disclosed honoraria and speakers fees from Merck, Celgene, and Servier, and travel support from Amgen, Merck Sharpe Dohme, and Servier.
Just over half of pregnant women got flu vaccine in 2016-2017
Influenza vaccination among pregnant women during the 2016-2017 flu season was slightly higher than during the 2015-2016 season, according to the Centers for Disease Control and Prevention.
Overall coverage for 2016-2017 was 53.6% among pregnant women, compared with 49.9% in 2015-2016, continuing the overall rise seen over the last several flu seasons. Among pregnant women who received a recommendation from a health care provider and were offered vaccination, coverage was 70.5% in 2016-2017, while coverage was 43.7% among women who received a recommendation but no offer and 14.8% among those who did not receive a recommendation, the CDC reported (MMWR. 2017 Sep 29;66[38]:1016-22).
Among other subgroups, coverage by age for the 2016-2017 flu season was 41.7% for those aged 18-24 years, 58.4% for those aged 25-34 years, and 58.5% for those 35-49 years old. There also was considerable variation by race/ethnicity, with coverage at 61.2% for Hispanics, 55.4% for whites, 42.3% for blacks, and 51.7% for others. Coverage for the subgroups corresponded with the rates at which vaccination was recommended: Younger women were less likely than older women to receive a recommendation, and Hispanic and white women more likely to receive recommendations than did blacks and other races/ethnicities, the CDC said.
The 2017 data include 1,893 responses to an Internet panel survey conducted from March 28 to April 7, 2017. The analysis of the 2016 panel survey, which was conducted from March 29 to April 7, 2016, included responses from 1,692 women.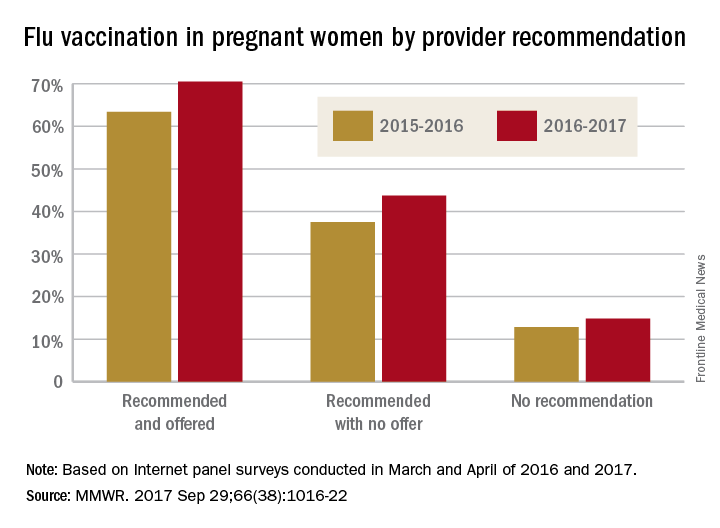
Influenza vaccination among pregnant women during the 2016-2017 flu season was slightly higher than during the 2015-2016 season, according to the Centers for Disease Control and Prevention.
Overall coverage for 2016-2017 was 53.6% among pregnant women, compared with 49.9% in 2015-2016, continuing the overall rise seen over the last several flu seasons. Among pregnant women who received a recommendation from a health care provider and were offered vaccination, coverage was 70.5% in 2016-2017, while coverage was 43.7% among women who received a recommendation but no offer and 14.8% among those who did not receive a recommendation, the CDC reported (MMWR. 2017 Sep 29;66[38]:1016-22).
Among other subgroups, coverage by age for the 2016-2017 flu season was 41.7% for those aged 18-24 years, 58.4% for those aged 25-34 years, and 58.5% for those 35-49 years old. There also was considerable variation by race/ethnicity, with coverage at 61.2% for Hispanics, 55.4% for whites, 42.3% for blacks, and 51.7% for others. Coverage for the subgroups corresponded with the rates at which vaccination was recommended: Younger women were less likely than older women to receive a recommendation, and Hispanic and white women more likely to receive recommendations than did blacks and other races/ethnicities, the CDC said.
The 2017 data include 1,893 responses to an Internet panel survey conducted from March 28 to April 7, 2017. The analysis of the 2016 panel survey, which was conducted from March 29 to April 7, 2016, included responses from 1,692 women.
Influenza vaccination among pregnant women during the 2016-2017 flu season was slightly higher than during the 2015-2016 season, according to the Centers for Disease Control and Prevention.
Overall coverage for 2016-2017 was 53.6% among pregnant women, compared with 49.9% in 2015-2016, continuing the overall rise seen over the last several flu seasons. Among pregnant women who received a recommendation from a health care provider and were offered vaccination, coverage was 70.5% in 2016-2017, while coverage was 43.7% among women who received a recommendation but no offer and 14.8% among those who did not receive a recommendation, the CDC reported (MMWR. 2017 Sep 29;66[38]:1016-22).
Among other subgroups, coverage by age for the 2016-2017 flu season was 41.7% for those aged 18-24 years, 58.4% for those aged 25-34 years, and 58.5% for those 35-49 years old. There also was considerable variation by race/ethnicity, with coverage at 61.2% for Hispanics, 55.4% for whites, 42.3% for blacks, and 51.7% for others. Coverage for the subgroups corresponded with the rates at which vaccination was recommended: Younger women were less likely than older women to receive a recommendation, and Hispanic and white women more likely to receive recommendations than did blacks and other races/ethnicities, the CDC said.
The 2017 data include 1,893 responses to an Internet panel survey conducted from March 28 to April 7, 2017. The analysis of the 2016 panel survey, which was conducted from March 29 to April 7, 2016, included responses from 1,692 women.
FROM MMWR
Talking with vaccine-hesitant parents takes training and finesse
CHICAGO – Addressing vaccine-hesitant parents can cause physicians considerable stress. However, they can feel more confident by adopting one of two communication strategies after gauging the strength of antivaccine beliefs, results of a pilot study suggest.
“We found that physicians frequently feel anxious and uncomfortable when confronted with parents who are strongly vaccine hesitant. They frequently lack confidence in dispelling the various safety concerns raised by parents and find themselves frequently combating an internal desire to just avoid the conflict,” said Paul J. Carson, MD, an expert in infectious diseases in the department of public health at North Dakota State University in Fargo.
The AAP suggests pediatricians adopt the “CASE method,” which stands for Corroborate parents’ concern, talk About me, describe the Science, and Explain/advise why they should vaccinate. The academy also recommends motivational interviewing as an additional tool to achieve vaccine acceptance. Ms. Dybsand, Dr. Carson, and their colleagues examined these two different approaches after training five pediatric providers. They also assessed physician perceptions about confidence and satisfaction regarding each method.
The pediatric providers were trained during a 7-hour retreat and 10 subsequent 1-hour training/debriefing sessions over 9 months. Explanations of vaccine safety and efficacy, vaccine licensure, how to refute common vaccine myths, and the two differing communication strategies were included in the training. Participants implemented the presumptive/CASE approach for 4 months then crossed over and used motivational interviewing for an additional 4 months.
“Some intensive training and education on the vaccine safety process and scientific evidence dispelling the common myths about vaccine safety were very helpful in boosting provider confidence,” Dr. Carson said.
“We want to be able to give them the tools to approach these conversations in an educated manner. We want them to feel like they have some ammunition behind the conversation,” said Ms. Dybsand, a graduate research assistant at the university.
The study revealed that the CASE approach was easier to learn and used more readily when pediatricians encountered a moderately hesitant parent. However, the investigators found the pediatricians perceived motivational interviewing as useful for the more strongly resistant parent. “For those really resistant parents who have looked at all the websites and are very concerned about vaccines, maybe motivational interviewing is the way to go,” Ms. Dybsand said. The goal of motivational interviewing is to build a trusting relationship over time. “You may not be giving that vaccine today, but you may be able to convince them in the future to vaccinate.”
The frequency and duration of training may be essential to success. “We didn’t really set out to find this, but it really takes more than 1 day of training to get providers to make a meaningful change in their communication strategies,” Ms. Dybsand said. When asked how long it might take the average pediatrician to become proficient in both techniques, she said that likely is a focus of future study.
The investigators plan to build on the success of the pilot study by expanding the research to multiple sites. In addition, they want to go beyond assessing provider perceptions of the communication techniques. Dr. Carson said, “These strategies need to be tested in formal clinical trials to see what is successful in actually increasing vaccine acceptance.”
Ms. Dybsand and Dr. Carson had no relevant financial disclosures.
CHICAGO – Addressing vaccine-hesitant parents can cause physicians considerable stress. However, they can feel more confident by adopting one of two communication strategies after gauging the strength of antivaccine beliefs, results of a pilot study suggest.
“We found that physicians frequently feel anxious and uncomfortable when confronted with parents who are strongly vaccine hesitant. They frequently lack confidence in dispelling the various safety concerns raised by parents and find themselves frequently combating an internal desire to just avoid the conflict,” said Paul J. Carson, MD, an expert in infectious diseases in the department of public health at North Dakota State University in Fargo.
The AAP suggests pediatricians adopt the “CASE method,” which stands for Corroborate parents’ concern, talk About me, describe the Science, and Explain/advise why they should vaccinate. The academy also recommends motivational interviewing as an additional tool to achieve vaccine acceptance. Ms. Dybsand, Dr. Carson, and their colleagues examined these two different approaches after training five pediatric providers. They also assessed physician perceptions about confidence and satisfaction regarding each method.
The pediatric providers were trained during a 7-hour retreat and 10 subsequent 1-hour training/debriefing sessions over 9 months. Explanations of vaccine safety and efficacy, vaccine licensure, how to refute common vaccine myths, and the two differing communication strategies were included in the training. Participants implemented the presumptive/CASE approach for 4 months then crossed over and used motivational interviewing for an additional 4 months.
“Some intensive training and education on the vaccine safety process and scientific evidence dispelling the common myths about vaccine safety were very helpful in boosting provider confidence,” Dr. Carson said.
“We want to be able to give them the tools to approach these conversations in an educated manner. We want them to feel like they have some ammunition behind the conversation,” said Ms. Dybsand, a graduate research assistant at the university.
The study revealed that the CASE approach was easier to learn and used more readily when pediatricians encountered a moderately hesitant parent. However, the investigators found the pediatricians perceived motivational interviewing as useful for the more strongly resistant parent. “For those really resistant parents who have looked at all the websites and are very concerned about vaccines, maybe motivational interviewing is the way to go,” Ms. Dybsand said. The goal of motivational interviewing is to build a trusting relationship over time. “You may not be giving that vaccine today, but you may be able to convince them in the future to vaccinate.”
The frequency and duration of training may be essential to success. “We didn’t really set out to find this, but it really takes more than 1 day of training to get providers to make a meaningful change in their communication strategies,” Ms. Dybsand said. When asked how long it might take the average pediatrician to become proficient in both techniques, she said that likely is a focus of future study.
The investigators plan to build on the success of the pilot study by expanding the research to multiple sites. In addition, they want to go beyond assessing provider perceptions of the communication techniques. Dr. Carson said, “These strategies need to be tested in formal clinical trials to see what is successful in actually increasing vaccine acceptance.”
Ms. Dybsand and Dr. Carson had no relevant financial disclosures.
CHICAGO – Addressing vaccine-hesitant parents can cause physicians considerable stress. However, they can feel more confident by adopting one of two communication strategies after gauging the strength of antivaccine beliefs, results of a pilot study suggest.
“We found that physicians frequently feel anxious and uncomfortable when confronted with parents who are strongly vaccine hesitant. They frequently lack confidence in dispelling the various safety concerns raised by parents and find themselves frequently combating an internal desire to just avoid the conflict,” said Paul J. Carson, MD, an expert in infectious diseases in the department of public health at North Dakota State University in Fargo.
The AAP suggests pediatricians adopt the “CASE method,” which stands for Corroborate parents’ concern, talk About me, describe the Science, and Explain/advise why they should vaccinate. The academy also recommends motivational interviewing as an additional tool to achieve vaccine acceptance. Ms. Dybsand, Dr. Carson, and their colleagues examined these two different approaches after training five pediatric providers. They also assessed physician perceptions about confidence and satisfaction regarding each method.
The pediatric providers were trained during a 7-hour retreat and 10 subsequent 1-hour training/debriefing sessions over 9 months. Explanations of vaccine safety and efficacy, vaccine licensure, how to refute common vaccine myths, and the two differing communication strategies were included in the training. Participants implemented the presumptive/CASE approach for 4 months then crossed over and used motivational interviewing for an additional 4 months.
“Some intensive training and education on the vaccine safety process and scientific evidence dispelling the common myths about vaccine safety were very helpful in boosting provider confidence,” Dr. Carson said.
“We want to be able to give them the tools to approach these conversations in an educated manner. We want them to feel like they have some ammunition behind the conversation,” said Ms. Dybsand, a graduate research assistant at the university.
The study revealed that the CASE approach was easier to learn and used more readily when pediatricians encountered a moderately hesitant parent. However, the investigators found the pediatricians perceived motivational interviewing as useful for the more strongly resistant parent. “For those really resistant parents who have looked at all the websites and are very concerned about vaccines, maybe motivational interviewing is the way to go,” Ms. Dybsand said. The goal of motivational interviewing is to build a trusting relationship over time. “You may not be giving that vaccine today, but you may be able to convince them in the future to vaccinate.”
The frequency and duration of training may be essential to success. “We didn’t really set out to find this, but it really takes more than 1 day of training to get providers to make a meaningful change in their communication strategies,” Ms. Dybsand said. When asked how long it might take the average pediatrician to become proficient in both techniques, she said that likely is a focus of future study.
The investigators plan to build on the success of the pilot study by expanding the research to multiple sites. In addition, they want to go beyond assessing provider perceptions of the communication techniques. Dr. Carson said, “These strategies need to be tested in formal clinical trials to see what is successful in actually increasing vaccine acceptance.”
Ms. Dybsand and Dr. Carson had no relevant financial disclosures.
AT AAP 2017
Key clinical point: Pediatricians can use tactics to help reduce the anxiety of discussing the importance of immunization with vaccine-hesitant parents.
Major finding: The CASE approach was easier to learn and used more readily when pediatricians encountered a moderately hesitant parent, but pediatricians perceived motivational interviewing as useful for the more strongly resistant parent.
Data source: Pilot study of five pediatric providers who received comprehensive training and ongoing support using different communication techniques.
Disclosures: Ms. Dybsand and Dr. Carson had no relevant financial disclosures.
Upfront preparation key to QI projects
Editor’s note: The Society of Hospital Medicine’s (SHM’s) Physician in Training Committee launched a scholarship program in 2015 for medical students to help transform healthcare and revolutionize patient care. The program has been expanded for the 2017-2018 year, offering two options for students to receive funding and engage in scholarly work during their first, second, and third years of medical school. As a part of the longitudinal (18-month) program, recipients are required to write about their experience on a monthly basis.
I am currently working with my mentor, Ian Jenkins, MD, an attending in the Division of Hospital Medicine at the University of California, San Diego, to begin preliminary data collection for our project to cut catheter-associated urinary tract infections (CAUTI). The project time line is on track, and we hope to have things up and running in the next month.
Up to this point, we have been working to identify the most relevant data to collect to best explore our outcome variable. A key goal for our project is to show that increased education measures can ultimately lead to reductions in patient harm. Rather than directly measuring harm reduction, we have settled on tracking the closely identified process measure of the number of inappropriate Foley catheters removed. This measure is potentially more accessible for health care providers than measuring CAUTI rates would be because individual CAUTI events are rare.
In addition to starting data collection, I am quickly learning that conducting a quality improvement project requires a large amount of upfront preparation. Namely, it requires not only identifying the outcome measures you would like to track but also prospectively strategizing about how to track this measure to facilitate future data presentation and publication. Dr. Jenkins has been instrumental as a resource for bouncing off various ideas regarding how to streamline data collection and presentation. He has also been valuable in helping me to identify appropriate units for data collection and teaching me to be forward thinking regarding the best way to collect data for my project. This has truly saved me a significant amount of time and increased the project’s efficiency.
Outside of data collection, we have continued to engage as many stakeholders as we can to ensure the success of the project. Because our project was deemed high priority because of the high CAUTI rates at UCSD, we engaged higher-level hospital administrators who could be onboard with the project, as well as provide their own input to improve project’s effects. Separately, we have continued to collaborate directly with nursing and physician staff to not only share our ongoing project with them but also directly engage them in the project so we can better ensure that the project is not only theoretically palatable but will be realistically implemented as well.
A quality improvement project certainly presents its own unique set of challenges, but I am truly enjoying collaborating and troubleshooting in hopes of ultimately improving patient care.
Victor Ekuta is a third-year medical student at UC San Diego.
Editor’s note: The Society of Hospital Medicine’s (SHM’s) Physician in Training Committee launched a scholarship program in 2015 for medical students to help transform healthcare and revolutionize patient care. The program has been expanded for the 2017-2018 year, offering two options for students to receive funding and engage in scholarly work during their first, second, and third years of medical school. As a part of the longitudinal (18-month) program, recipients are required to write about their experience on a monthly basis.
I am currently working with my mentor, Ian Jenkins, MD, an attending in the Division of Hospital Medicine at the University of California, San Diego, to begin preliminary data collection for our project to cut catheter-associated urinary tract infections (CAUTI). The project time line is on track, and we hope to have things up and running in the next month.
Up to this point, we have been working to identify the most relevant data to collect to best explore our outcome variable. A key goal for our project is to show that increased education measures can ultimately lead to reductions in patient harm. Rather than directly measuring harm reduction, we have settled on tracking the closely identified process measure of the number of inappropriate Foley catheters removed. This measure is potentially more accessible for health care providers than measuring CAUTI rates would be because individual CAUTI events are rare.
In addition to starting data collection, I am quickly learning that conducting a quality improvement project requires a large amount of upfront preparation. Namely, it requires not only identifying the outcome measures you would like to track but also prospectively strategizing about how to track this measure to facilitate future data presentation and publication. Dr. Jenkins has been instrumental as a resource for bouncing off various ideas regarding how to streamline data collection and presentation. He has also been valuable in helping me to identify appropriate units for data collection and teaching me to be forward thinking regarding the best way to collect data for my project. This has truly saved me a significant amount of time and increased the project’s efficiency.
Outside of data collection, we have continued to engage as many stakeholders as we can to ensure the success of the project. Because our project was deemed high priority because of the high CAUTI rates at UCSD, we engaged higher-level hospital administrators who could be onboard with the project, as well as provide their own input to improve project’s effects. Separately, we have continued to collaborate directly with nursing and physician staff to not only share our ongoing project with them but also directly engage them in the project so we can better ensure that the project is not only theoretically palatable but will be realistically implemented as well.
A quality improvement project certainly presents its own unique set of challenges, but I am truly enjoying collaborating and troubleshooting in hopes of ultimately improving patient care.
Victor Ekuta is a third-year medical student at UC San Diego.
Editor’s note: The Society of Hospital Medicine’s (SHM’s) Physician in Training Committee launched a scholarship program in 2015 for medical students to help transform healthcare and revolutionize patient care. The program has been expanded for the 2017-2018 year, offering two options for students to receive funding and engage in scholarly work during their first, second, and third years of medical school. As a part of the longitudinal (18-month) program, recipients are required to write about their experience on a monthly basis.
I am currently working with my mentor, Ian Jenkins, MD, an attending in the Division of Hospital Medicine at the University of California, San Diego, to begin preliminary data collection for our project to cut catheter-associated urinary tract infections (CAUTI). The project time line is on track, and we hope to have things up and running in the next month.
Up to this point, we have been working to identify the most relevant data to collect to best explore our outcome variable. A key goal for our project is to show that increased education measures can ultimately lead to reductions in patient harm. Rather than directly measuring harm reduction, we have settled on tracking the closely identified process measure of the number of inappropriate Foley catheters removed. This measure is potentially more accessible for health care providers than measuring CAUTI rates would be because individual CAUTI events are rare.
In addition to starting data collection, I am quickly learning that conducting a quality improvement project requires a large amount of upfront preparation. Namely, it requires not only identifying the outcome measures you would like to track but also prospectively strategizing about how to track this measure to facilitate future data presentation and publication. Dr. Jenkins has been instrumental as a resource for bouncing off various ideas regarding how to streamline data collection and presentation. He has also been valuable in helping me to identify appropriate units for data collection and teaching me to be forward thinking regarding the best way to collect data for my project. This has truly saved me a significant amount of time and increased the project’s efficiency.
Outside of data collection, we have continued to engage as many stakeholders as we can to ensure the success of the project. Because our project was deemed high priority because of the high CAUTI rates at UCSD, we engaged higher-level hospital administrators who could be onboard with the project, as well as provide their own input to improve project’s effects. Separately, we have continued to collaborate directly with nursing and physician staff to not only share our ongoing project with them but also directly engage them in the project so we can better ensure that the project is not only theoretically palatable but will be realistically implemented as well.
A quality improvement project certainly presents its own unique set of challenges, but I am truly enjoying collaborating and troubleshooting in hopes of ultimately improving patient care.
Victor Ekuta is a third-year medical student at UC San Diego.
JFP PCEC Injectable Insulin Supplement
Click Here to Read the Supplement.

Topics include:
- Medications for Type 2 Diabetes Mellitus: A Work in Progress
- Role of Injectable Medications in Type 2 Diabetes Treatment
- Basal Insulins
- Glucagon-Like Peptide-1 Receptor Agonists
- Using Combinations of a Basal Insulin and a Glucagon-Like Peptide-1 Receptor Agonist
Click Here to Read the Supplement.
Topics include:
- Medications for Type 2 Diabetes Mellitus: A Work in Progress
- Role of Injectable Medications in Type 2 Diabetes Treatment
- Basal Insulins
- Glucagon-Like Peptide-1 Receptor Agonists
- Using Combinations of a Basal Insulin and a Glucagon-Like Peptide-1 Receptor Agonist
Click Here to Read the Supplement.
Topics include:
- Medications for Type 2 Diabetes Mellitus: A Work in Progress
- Role of Injectable Medications in Type 2 Diabetes Treatment
- Basal Insulins
- Glucagon-Like Peptide-1 Receptor Agonists
- Using Combinations of a Basal Insulin and a Glucagon-Like Peptide-1 Receptor Agonist

Dabigatran, rivaroxaban linked to slight increase in GI bleeding risk
Compared with conventional anticoagulants, both dabigatran and rivaroxaban conferred small but statistically significant increases in the risk of major gastrointestinal bleeding in a systematic review and meta-analysis of randomized trials reported in the November issue of Clinical Gastroenterology and Hepatology. (doi: 10.1016/j.cgh.2017.04.031)
But other novel oral anticoagulants (NOACs) showed no such effect compared with warfarin, aspirin, or placebo, reported Corey S. Miller, MD, of McGill University, Montreal, and his associates. “The potentially increased risk of GI bleeding associated with dabigatran and rivaroxaban observed in some of our subgroup analyses merits further consideration,” they wrote.
The NOACs (also known as non–vitamin K antagonist oral anticoagulants) help prevent stroke in patients with atrial fibrillation and prevent and treat venous thromboembolism. However, large AF trials have linked all except apixaban to an increased risk of major GI bleeding compared with warfarin. Dabigatran currently is the only NOAC with an approved reversal agent, “making the question of GI bleeding risk even more consequential,” the authors wrote.
They searched the MEDLINE, EMBASE, Cochrane, and ISI Web of Knowledge databases for reports of randomized trials of NOACs for approved indications published between 1980 and January 2016, which identified 43 trials of 166,289 patients. Most used warfarin as the comparator, but one study compared apixaban with aspirin and six studies compared apixaban, rivaroxaban, or dabigatran with placebo. Fifteen trials failed to specify bleeding sources and therefore could not be evaluated for the primary endpoint, the reviewers noted.
In the remaining 28 trials, 1.5% of NOAC recipients developed major GI bleeding, compared with 1.3% of recipients of conventional anticoagulants (odds ratio, 0.98; 95% confidence interval, 0.80-1.21). Five trials of dabigatran showed a 2% risk of major GI bleeding, compared with 1.4% with conventional anticoagulation, a slight but significant increase (OR, 1.27; 95% CI, 1.04-1.55). Eight trials of rivaroxaban showed a similar trend (bleeding risk, 1.7% vs. 1.3%; OR, 1.40; 95% CI, 1.15-1.70). In contrast, subgroup analyses of apixaban and edoxaban found no difference in risk of major GI bleeding versus conventional treatment.
Subgroup analyses by region found no differences except in Asia, where NOACs were associated with a significantly lower odds of major GI bleeding (0.5% and 1.2%, respectively; OR, 0.45; 95% CI, 0.22-0.91).
Most studies did not report minor or nonsevere bleeds or specify bleeding location within the GI tract, the reviewers noted. Given those caveats, NOACs and conventional anticoagulants conferred similar risks of clinically relevant nonmajor bleeding (0.6% and 0.6%, respectively), upper GI bleeding (1.5% and 1.6%), and lower GI bleeding (1.0% and 1.0%).
A post hoc analysis using a random-effects model found no significant difference in risk of major GI bleeding between either rivaroxaban or dabigatran and conventional therapy, the reviewers said. In addition, the increased risk of bleeding with dabigatran was confined to the RELY and ROCKET trials of AF, both of which exposed patients to longer treatment periods. Dabigatran is coated with tartaric acid, which might have a “direct caustic effect on the intestinal lumen,” they wrote. Also, NOACs are incompletely absorbed across the GI mucosa and therefore have some anticoagulant activity in the GI lumen, unlike warfarin or parenteral anticoagulants.
The reviewers disclosed no funding sources. Dr. Miller and another author reported having no conflicts of interest. One author received research grants and speaker honoraria from Boehringer Ingelheim Canada, Bayer Canada, Daiichi Sankyo, Bristol Myers Squibb, and Pfizer Canada; another author disclosed serving as a consultant to Pendopharm, Boston Scientific, and Cook.
Novel oral anticoagulants (NOACs) receive a lot of press now. In randomized controlled trials (RCTs) comparing NOACs to warfarin for prevention of strokes and thromboembolism in atrial fibrillation (AF) and venous thromboembolism (VTE), fewer thromboembolisms are reported, but risks of gastrointestinal bleeding vary. To expand analyses for gastrointestinal bleeding, several systematic reviews and meta-analyses are reported, including this one by CS Miller et al. Their goals were to delineate risks of gastrointestinal bleeding for different NOACs compared with warfarin. What can GI clinicians now recommend about gastrointestinal bleeding for patients requiring anticoagulants? While we lack RCTs to give the highest quality of evidence about GIB as a primary outcome, conclusions now depend on the weight of evidence from recent secondary data analyses and I have some recommendations. First, although there may be differences among NOACs in risks of bleeding, all are likely to increase the risk of GI bleeding, comparable with warfarin. Some report that dabigatran and rivaroxaban have a higher risk of GI bleeding than other NOACs or warfarin, but differences are small. Second, some patients who need NOACs/warfarin have increased risks of ulcer bleeds including elderly patients and those with a history of upper GI bleeding, renal or hepatic impairment, low body weight, and concomitant antiplatelet agents. Such high-risk patients warrant treatment with a proton pump inhibitor or histamine2-receptor agonists for primary prevention while on anticoagulants. Finally, for patients with severe ulcer bleeding who require anticoagulation, warfarin or NOACs should be restarted after successful endoscopic hemostasis and proton pump inhibitors, usually within 3-5 days.
Dr. Jensen is professor of medicine at the University of California, Los Angeles; associate director of the CURE: DDRC, where he directs the Human Studies Core; a full-time staff physician in the UCLA division of digestive diseases; and a part-time staff physician in the GI section of the VA Greater Los Angeles Healthcare Center.
Novel oral anticoagulants (NOACs) receive a lot of press now. In randomized controlled trials (RCTs) comparing NOACs to warfarin for prevention of strokes and thromboembolism in atrial fibrillation (AF) and venous thromboembolism (VTE), fewer thromboembolisms are reported, but risks of gastrointestinal bleeding vary. To expand analyses for gastrointestinal bleeding, several systematic reviews and meta-analyses are reported, including this one by CS Miller et al. Their goals were to delineate risks of gastrointestinal bleeding for different NOACs compared with warfarin. What can GI clinicians now recommend about gastrointestinal bleeding for patients requiring anticoagulants? While we lack RCTs to give the highest quality of evidence about GIB as a primary outcome, conclusions now depend on the weight of evidence from recent secondary data analyses and I have some recommendations. First, although there may be differences among NOACs in risks of bleeding, all are likely to increase the risk of GI bleeding, comparable with warfarin. Some report that dabigatran and rivaroxaban have a higher risk of GI bleeding than other NOACs or warfarin, but differences are small. Second, some patients who need NOACs/warfarin have increased risks of ulcer bleeds including elderly patients and those with a history of upper GI bleeding, renal or hepatic impairment, low body weight, and concomitant antiplatelet agents. Such high-risk patients warrant treatment with a proton pump inhibitor or histamine2-receptor agonists for primary prevention while on anticoagulants. Finally, for patients with severe ulcer bleeding who require anticoagulation, warfarin or NOACs should be restarted after successful endoscopic hemostasis and proton pump inhibitors, usually within 3-5 days.
Dr. Jensen is professor of medicine at the University of California, Los Angeles; associate director of the CURE: DDRC, where he directs the Human Studies Core; a full-time staff physician in the UCLA division of digestive diseases; and a part-time staff physician in the GI section of the VA Greater Los Angeles Healthcare Center.
Novel oral anticoagulants (NOACs) receive a lot of press now. In randomized controlled trials (RCTs) comparing NOACs to warfarin for prevention of strokes and thromboembolism in atrial fibrillation (AF) and venous thromboembolism (VTE), fewer thromboembolisms are reported, but risks of gastrointestinal bleeding vary. To expand analyses for gastrointestinal bleeding, several systematic reviews and meta-analyses are reported, including this one by CS Miller et al. Their goals were to delineate risks of gastrointestinal bleeding for different NOACs compared with warfarin. What can GI clinicians now recommend about gastrointestinal bleeding for patients requiring anticoagulants? While we lack RCTs to give the highest quality of evidence about GIB as a primary outcome, conclusions now depend on the weight of evidence from recent secondary data analyses and I have some recommendations. First, although there may be differences among NOACs in risks of bleeding, all are likely to increase the risk of GI bleeding, comparable with warfarin. Some report that dabigatran and rivaroxaban have a higher risk of GI bleeding than other NOACs or warfarin, but differences are small. Second, some patients who need NOACs/warfarin have increased risks of ulcer bleeds including elderly patients and those with a history of upper GI bleeding, renal or hepatic impairment, low body weight, and concomitant antiplatelet agents. Such high-risk patients warrant treatment with a proton pump inhibitor or histamine2-receptor agonists for primary prevention while on anticoagulants. Finally, for patients with severe ulcer bleeding who require anticoagulation, warfarin or NOACs should be restarted after successful endoscopic hemostasis and proton pump inhibitors, usually within 3-5 days.
Dr. Jensen is professor of medicine at the University of California, Los Angeles; associate director of the CURE: DDRC, where he directs the Human Studies Core; a full-time staff physician in the UCLA division of digestive diseases; and a part-time staff physician in the GI section of the VA Greater Los Angeles Healthcare Center.
Compared with conventional anticoagulants, both dabigatran and rivaroxaban conferred small but statistically significant increases in the risk of major gastrointestinal bleeding in a systematic review and meta-analysis of randomized trials reported in the November issue of Clinical Gastroenterology and Hepatology. (doi: 10.1016/j.cgh.2017.04.031)
But other novel oral anticoagulants (NOACs) showed no such effect compared with warfarin, aspirin, or placebo, reported Corey S. Miller, MD, of McGill University, Montreal, and his associates. “The potentially increased risk of GI bleeding associated with dabigatran and rivaroxaban observed in some of our subgroup analyses merits further consideration,” they wrote.
The NOACs (also known as non–vitamin K antagonist oral anticoagulants) help prevent stroke in patients with atrial fibrillation and prevent and treat venous thromboembolism. However, large AF trials have linked all except apixaban to an increased risk of major GI bleeding compared with warfarin. Dabigatran currently is the only NOAC with an approved reversal agent, “making the question of GI bleeding risk even more consequential,” the authors wrote.
They searched the MEDLINE, EMBASE, Cochrane, and ISI Web of Knowledge databases for reports of randomized trials of NOACs for approved indications published between 1980 and January 2016, which identified 43 trials of 166,289 patients. Most used warfarin as the comparator, but one study compared apixaban with aspirin and six studies compared apixaban, rivaroxaban, or dabigatran with placebo. Fifteen trials failed to specify bleeding sources and therefore could not be evaluated for the primary endpoint, the reviewers noted.
In the remaining 28 trials, 1.5% of NOAC recipients developed major GI bleeding, compared with 1.3% of recipients of conventional anticoagulants (odds ratio, 0.98; 95% confidence interval, 0.80-1.21). Five trials of dabigatran showed a 2% risk of major GI bleeding, compared with 1.4% with conventional anticoagulation, a slight but significant increase (OR, 1.27; 95% CI, 1.04-1.55). Eight trials of rivaroxaban showed a similar trend (bleeding risk, 1.7% vs. 1.3%; OR, 1.40; 95% CI, 1.15-1.70). In contrast, subgroup analyses of apixaban and edoxaban found no difference in risk of major GI bleeding versus conventional treatment.
Subgroup analyses by region found no differences except in Asia, where NOACs were associated with a significantly lower odds of major GI bleeding (0.5% and 1.2%, respectively; OR, 0.45; 95% CI, 0.22-0.91).
Most studies did not report minor or nonsevere bleeds or specify bleeding location within the GI tract, the reviewers noted. Given those caveats, NOACs and conventional anticoagulants conferred similar risks of clinically relevant nonmajor bleeding (0.6% and 0.6%, respectively), upper GI bleeding (1.5% and 1.6%), and lower GI bleeding (1.0% and 1.0%).
A post hoc analysis using a random-effects model found no significant difference in risk of major GI bleeding between either rivaroxaban or dabigatran and conventional therapy, the reviewers said. In addition, the increased risk of bleeding with dabigatran was confined to the RELY and ROCKET trials of AF, both of which exposed patients to longer treatment periods. Dabigatran is coated with tartaric acid, which might have a “direct caustic effect on the intestinal lumen,” they wrote. Also, NOACs are incompletely absorbed across the GI mucosa and therefore have some anticoagulant activity in the GI lumen, unlike warfarin or parenteral anticoagulants.
The reviewers disclosed no funding sources. Dr. Miller and another author reported having no conflicts of interest. One author received research grants and speaker honoraria from Boehringer Ingelheim Canada, Bayer Canada, Daiichi Sankyo, Bristol Myers Squibb, and Pfizer Canada; another author disclosed serving as a consultant to Pendopharm, Boston Scientific, and Cook.
Compared with conventional anticoagulants, both dabigatran and rivaroxaban conferred small but statistically significant increases in the risk of major gastrointestinal bleeding in a systematic review and meta-analysis of randomized trials reported in the November issue of Clinical Gastroenterology and Hepatology. (doi: 10.1016/j.cgh.2017.04.031)
But other novel oral anticoagulants (NOACs) showed no such effect compared with warfarin, aspirin, or placebo, reported Corey S. Miller, MD, of McGill University, Montreal, and his associates. “The potentially increased risk of GI bleeding associated with dabigatran and rivaroxaban observed in some of our subgroup analyses merits further consideration,” they wrote.
The NOACs (also known as non–vitamin K antagonist oral anticoagulants) help prevent stroke in patients with atrial fibrillation and prevent and treat venous thromboembolism. However, large AF trials have linked all except apixaban to an increased risk of major GI bleeding compared with warfarin. Dabigatran currently is the only NOAC with an approved reversal agent, “making the question of GI bleeding risk even more consequential,” the authors wrote.
They searched the MEDLINE, EMBASE, Cochrane, and ISI Web of Knowledge databases for reports of randomized trials of NOACs for approved indications published between 1980 and January 2016, which identified 43 trials of 166,289 patients. Most used warfarin as the comparator, but one study compared apixaban with aspirin and six studies compared apixaban, rivaroxaban, or dabigatran with placebo. Fifteen trials failed to specify bleeding sources and therefore could not be evaluated for the primary endpoint, the reviewers noted.
In the remaining 28 trials, 1.5% of NOAC recipients developed major GI bleeding, compared with 1.3% of recipients of conventional anticoagulants (odds ratio, 0.98; 95% confidence interval, 0.80-1.21). Five trials of dabigatran showed a 2% risk of major GI bleeding, compared with 1.4% with conventional anticoagulation, a slight but significant increase (OR, 1.27; 95% CI, 1.04-1.55). Eight trials of rivaroxaban showed a similar trend (bleeding risk, 1.7% vs. 1.3%; OR, 1.40; 95% CI, 1.15-1.70). In contrast, subgroup analyses of apixaban and edoxaban found no difference in risk of major GI bleeding versus conventional treatment.
Subgroup analyses by region found no differences except in Asia, where NOACs were associated with a significantly lower odds of major GI bleeding (0.5% and 1.2%, respectively; OR, 0.45; 95% CI, 0.22-0.91).
Most studies did not report minor or nonsevere bleeds or specify bleeding location within the GI tract, the reviewers noted. Given those caveats, NOACs and conventional anticoagulants conferred similar risks of clinically relevant nonmajor bleeding (0.6% and 0.6%, respectively), upper GI bleeding (1.5% and 1.6%), and lower GI bleeding (1.0% and 1.0%).
A post hoc analysis using a random-effects model found no significant difference in risk of major GI bleeding between either rivaroxaban or dabigatran and conventional therapy, the reviewers said. In addition, the increased risk of bleeding with dabigatran was confined to the RELY and ROCKET trials of AF, both of which exposed patients to longer treatment periods. Dabigatran is coated with tartaric acid, which might have a “direct caustic effect on the intestinal lumen,” they wrote. Also, NOACs are incompletely absorbed across the GI mucosa and therefore have some anticoagulant activity in the GI lumen, unlike warfarin or parenteral anticoagulants.
The reviewers disclosed no funding sources. Dr. Miller and another author reported having no conflicts of interest. One author received research grants and speaker honoraria from Boehringer Ingelheim Canada, Bayer Canada, Daiichi Sankyo, Bristol Myers Squibb, and Pfizer Canada; another author disclosed serving as a consultant to Pendopharm, Boston Scientific, and Cook.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Key clinical point: Compared with conventional anticoagulants, novel oral anticoagulants (NOACs) were not associated with increased risk of major gastrointestinal bleeding, with the possible exception of dabigatran and rivaroxaban.
Major finding: In the overall analysis, risk of major GI bleeding was 1.5% with NOACs and 1.3% with conventional anticoagulants (OR, 0.98; 95% CI, 0.80-1.21). In subgroup analyses, dabigatran conferred a 2% risk of major GI bleeding (OR, 1.3; 95% CI, 1.04-1.55), rivaroxaban conferred a 1.7% risk (OR, 1.40; 95% CI, 1.15-1.70).
Data source: A systematic review and meta-analysis of 43 randomized trials, comprising 166,289 patients.
Disclosures: The reviewers disclosed no funding sources. Dr. Miller and another author reported having no conflicts of interest. One author received research grants and speaker honoraria from Boehringer Ingelheim Canada, Bayer Canada, Daiichi Sankyo, Bristol Myers Squibb, and Pfizer Canada; another author disclosed serving as a consultant to Pendopharm, Boston Scientific, and Cook.
Concurrent Sturge-Weber Syndrome, Facial Infantile Hemangioma, and Cutis Marmorata Telangiectatica Congenita
Sturge-Weber syndrome (SWS) is a disease of dermatologic, neurologic, and ocular significance.1 The most distinctive manifestation is facial capillary malformation, commonly referred to as a port-wine stain or nevus flammeus. The dysregulated angiogenesis, caused by somatic mutations of the G protein subunit alpha Q gene, GNAQ, also affects the central nervous system.2 Seizures, intellectual disability, and glaucoma are common consequences.1 Not all port-wine stains are associated with SWS.3 Distribution in the ophthalmic dermatome is associated with increased risk for SWS, with 8% of patients with port-wine stains also having SWS.4 The disease is more serious when bilateral lesions are present.5 Diagnosis is clinical based on dermatologic, nervous system, and ophthalmologic findings.6 The disease is nonheritable because the mutation is found only in the somatic cell lines.2 The possibility of epigenetic influence on disease development has to be investigated. The treatment is aimed at managing complications, as there is no cure.7
Infantile hemangioma (IH) likewise represents a disruption in the process of vascular development but without the widespread consequences of SWS. The pathogenesis of hemangioma development has not been fully elucidated, with presence of GLUT1 (glucose transporter 1) protein implicated in lesions.4 Facial infantile hemangiomas have an incidence of approximately 5 in every 100 births, and the prevalence decreases with age. Most hemangiomas undergo growth followed by an involution process, with most lesions vanishing by 5 years of age.4 They typically are seen at 2 to 3 weeks of age, growing rapidly for the first 6 months, which is a contrast to the static nature of nevus flammeus. Infantile hemangiomas are regarded as sporadic, though autosomal-dominant inheritance patterns have been observed.4 Our patient demonstrated facial IH at birth, which is a rare and interesting finding suggesting that some epigenetic factors influenced this modification of the disease course in this patient.
Cutis marmorata telangiectatica congenita (CMTC) is a rare cutaneous vascular condition found in newborns. Its extraordinary infrequency is reflected in the fact that only 300 cases have been reported worldwide.8 At birth, CMTC manifests as a pinkish reticulated pattern all over the body mimicking cutis marmorata; however, unlike cutis marmorata, the lesions do not improve with warming.9 The lesions of CMTC gradually lighten as the patient ages.8 Limb asymmetry is the most common extravascular complication of CMTC and, similar to SWS, glaucoma also can occur.10 Cutis marmorata telangiectatica congenita has been known to occur simultaneously with SWS or IH, but the combination of all 3 conditions in our patient is unique. Due to the scarcity of cases, the pathophysiology and treatment is poorly understood, with appropriate monitoring for sequelae recommended.9
Case Report
The patient was born at 39 weeks’ gestation following an uncomplicated pregnancy and delivery. She weighed 2950 g, her length was 19 in, and her head circumference was 13.25 in, correlating to the 10th, 50th, and 25th percentiles, respectively. Her Apgar score was 8/9 at 1 and 5 minutes. Her parents were nonconsanguineous and in good health. The patient’s family lived in poverty, which led us to conjecture about the role that toxins played in the epigenetics of the patient and her family. It was the mother’s third pregnancy; both prior pregnancies resulted in healthy children. The patient was breastfed. No family history of heritable vascular disorders was reported.
On the first day of life during the newborn examination, dark red pigment changes were noticed under the nose and erythematous pigmentation over the whole body was observed (Figure). On examination, 2-toned reticular lesions identified as extensive nevus flammeus were found bilaterally over the distribution of the ophthalmic division of the trigeminal nerve. A separate erythematous plaque over the maxilla also was recognized. The pediatrician suspected SWS and facial IH. The patient was discharged after 3 days with a referral to pediatric dermatology, and appropriate follow-up with a pediatrician was scheduled. The patient returned for these appointments and the significance of SWS was explained to her parents. Consultation with pediatric dermatology at 2 weeks of age confirmed the diagnosis of SWS as well as facial IH.
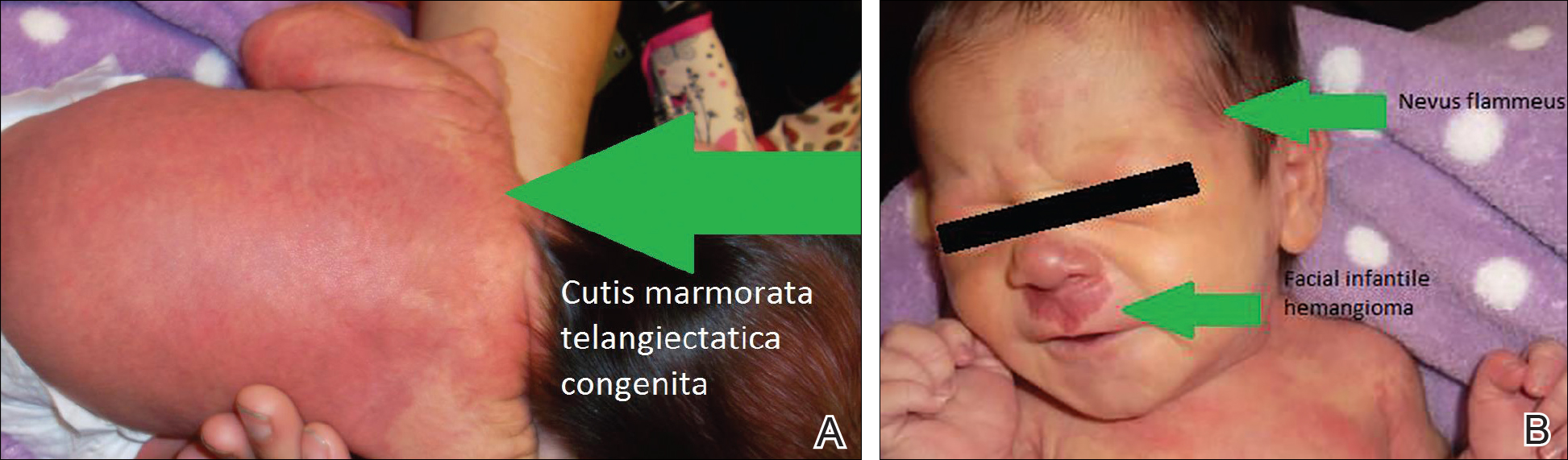
Upon further follow-up with pediatric dermatology at 2 months of age, the patient received an additional diagnosis of CMTC. These exceedingly rare lesions were located over the back, trunk, arms, and legs. The patient’s parents were counseled about the management of these conditions and appropriate follow-up.
Comment
This case describes 3 different vascular malformations in the same patient. Cutis marmorata telangiectatica congenita is rare and yet is described in this patient along with 2 other notable endothelial abnormalities. The clinical interest of this case is heightened by the presence of CMTC.
The causative factor of SWS is a well-documented mutation of the GNAQ gene, but there is considerable variability in how it affects the patient. Unlike in SWS, no single factor can be attributed to the development of IH. This case shows that these 3 diseases are not mutually exclusive and can present with unusually severe features when they occur concomitantly. The embryologic basis of SWS traces its roots back to the first trimester during vascular development, wher
The severity of the SWS in our patient was highlighted by the extensive nevus flammeus. These lesions occurred over the face, trunk, arms, and legs. The port-wine stain with dermatomal distribution of the ophthalmic nerve was the most concerning feature regarding the development of neurologic complications in this patient. Although the developmental delays associated with SWS can be serious, early intervention is important and can improve long-term outcomes. The facial IH arising at birth was contrary to the typical presentation. All of these factors will be kept in mind as the patient progresses and patient-centered care is provided. Because this patient’s presentation differed from other patients with IH, we will be more vigilant in providing close follow-up and monitoring for other medical problems involving other organs (eg, the brain); for instance, we will monitor for seizures and developmental delay.
Conclusion
In our patient, a unique pattern of SWS, facial IH, and CMTC are described in a pediatric patient. Many disciplines are involved in the treatment. In the patient’s first days of life, extensive collaboration between pediatrics and dermatologists was pivotal, with ophthalmology, pathology, and radiology consultations at hand. This case highlights that several vascular malformations of different origins can occur in the same patient. Epigenetic along with genetic factors likely contributed to this fascinating presentation. The importance of parental education and maintaining appropriate follow-up for this patient is crucial for a favorable outcome.
- Sinawat S, Auvichayapat N, Auvichayapat P, et al. 12-year retrospective study of Sturge-weber syndrome and literature review. J Med Assoc Thail. 2014;97:742-750.
- Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971-1979.
- Sudarsanam A, Ardern-Holmes SL. Sturge-Weber syndrome: from the past to the present [published online November 7, 2013]. Eur J Paediat Neurol. 2014;18:257-266.
- Paller AS, Mancini AJ. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. Philadelphia, PA: Elsevier Saunders; 2011.
- Sujansky E, Conradi S. Sturge-Weber syndrome: age of onset of seizures and glaucoma and the prognosis for affected children. J Child Neurol. 1995;10:49-58.
- Lo W, Marchuk DA, Ball KL, et al. Updates and future horizons on the understanding, diagnosis, and treatment of Sturge-Weber syndrome brain involvement. Dev Med Child Neurol. 2012;54:214-223.
- Comi AM. Update on Sturge-Weber syndrome: diagnosis, treatment, quantitative measures, and controversies. Lymphat Res Biol. 2007;5:257-264.
- Resende CI, Araujo C, Vieira AP, et al. Cutis marmorata telangiectatica congenital [published online October 17, 2013]. BMJ Case Rep. doi:10.1136/bcr-2013-200056.
- Levy R, Lam JM. Cutis marmorata telangiectatica congenita: a mimicker of a common disorder. CMAJ. 2011;183:E249-E251.
- Kienast AK, Hoeger PH. Cutis marmorata telangiectatica congenita: a prospective study of 27 cases and review of the literature with proposal of diagnostic criteria. Clin Exp Dermatol. 2009;34:319-323.
- Comi AM. Topical review: pathophysiology of Sturge-Weber syndrome. J Child Neurol. 2003;18:509-516.
Sturge-Weber syndrome (SWS) is a disease of dermatologic, neurologic, and ocular significance.1 The most distinctive manifestation is facial capillary malformation, commonly referred to as a port-wine stain or nevus flammeus. The dysregulated angiogenesis, caused by somatic mutations of the G protein subunit alpha Q gene, GNAQ, also affects the central nervous system.2 Seizures, intellectual disability, and glaucoma are common consequences.1 Not all port-wine stains are associated with SWS.3 Distribution in the ophthalmic dermatome is associated with increased risk for SWS, with 8% of patients with port-wine stains also having SWS.4 The disease is more serious when bilateral lesions are present.5 Diagnosis is clinical based on dermatologic, nervous system, and ophthalmologic findings.6 The disease is nonheritable because the mutation is found only in the somatic cell lines.2 The possibility of epigenetic influence on disease development has to be investigated. The treatment is aimed at managing complications, as there is no cure.7
Infantile hemangioma (IH) likewise represents a disruption in the process of vascular development but without the widespread consequences of SWS. The pathogenesis of hemangioma development has not been fully elucidated, with presence of GLUT1 (glucose transporter 1) protein implicated in lesions.4 Facial infantile hemangiomas have an incidence of approximately 5 in every 100 births, and the prevalence decreases with age. Most hemangiomas undergo growth followed by an involution process, with most lesions vanishing by 5 years of age.4 They typically are seen at 2 to 3 weeks of age, growing rapidly for the first 6 months, which is a contrast to the static nature of nevus flammeus. Infantile hemangiomas are regarded as sporadic, though autosomal-dominant inheritance patterns have been observed.4 Our patient demonstrated facial IH at birth, which is a rare and interesting finding suggesting that some epigenetic factors influenced this modification of the disease course in this patient.
Cutis marmorata telangiectatica congenita (CMTC) is a rare cutaneous vascular condition found in newborns. Its extraordinary infrequency is reflected in the fact that only 300 cases have been reported worldwide.8 At birth, CMTC manifests as a pinkish reticulated pattern all over the body mimicking cutis marmorata; however, unlike cutis marmorata, the lesions do not improve with warming.9 The lesions of CMTC gradually lighten as the patient ages.8 Limb asymmetry is the most common extravascular complication of CMTC and, similar to SWS, glaucoma also can occur.10 Cutis marmorata telangiectatica congenita has been known to occur simultaneously with SWS or IH, but the combination of all 3 conditions in our patient is unique. Due to the scarcity of cases, the pathophysiology and treatment is poorly understood, with appropriate monitoring for sequelae recommended.9
Case Report
The patient was born at 39 weeks’ gestation following an uncomplicated pregnancy and delivery. She weighed 2950 g, her length was 19 in, and her head circumference was 13.25 in, correlating to the 10th, 50th, and 25th percentiles, respectively. Her Apgar score was 8/9 at 1 and 5 minutes. Her parents were nonconsanguineous and in good health. The patient’s family lived in poverty, which led us to conjecture about the role that toxins played in the epigenetics of the patient and her family. It was the mother’s third pregnancy; both prior pregnancies resulted in healthy children. The patient was breastfed. No family history of heritable vascular disorders was reported.
On the first day of life during the newborn examination, dark red pigment changes were noticed under the nose and erythematous pigmentation over the whole body was observed (Figure). On examination, 2-toned reticular lesions identified as extensive nevus flammeus were found bilaterally over the distribution of the ophthalmic division of the trigeminal nerve. A separate erythematous plaque over the maxilla also was recognized. The pediatrician suspected SWS and facial IH. The patient was discharged after 3 days with a referral to pediatric dermatology, and appropriate follow-up with a pediatrician was scheduled. The patient returned for these appointments and the significance of SWS was explained to her parents. Consultation with pediatric dermatology at 2 weeks of age confirmed the diagnosis of SWS as well as facial IH.
Upon further follow-up with pediatric dermatology at 2 months of age, the patient received an additional diagnosis of CMTC. These exceedingly rare lesions were located over the back, trunk, arms, and legs. The patient’s parents were counseled about the management of these conditions and appropriate follow-up.
Comment
This case describes 3 different vascular malformations in the same patient. Cutis marmorata telangiectatica congenita is rare and yet is described in this patient along with 2 other notable endothelial abnormalities. The clinical interest of this case is heightened by the presence of CMTC.
The causative factor of SWS is a well-documented mutation of the GNAQ gene, but there is considerable variability in how it affects the patient. Unlike in SWS, no single factor can be attributed to the development of IH. This case shows that these 3 diseases are not mutually exclusive and can present with unusually severe features when they occur concomitantly. The embryologic basis of SWS traces its roots back to the first trimester during vascular development, wher
The severity of the SWS in our patient was highlighted by the extensive nevus flammeus. These lesions occurred over the face, trunk, arms, and legs. The port-wine stain with dermatomal distribution of the ophthalmic nerve was the most concerning feature regarding the development of neurologic complications in this patient. Although the developmental delays associated with SWS can be serious, early intervention is important and can improve long-term outcomes. The facial IH arising at birth was contrary to the typical presentation. All of these factors will be kept in mind as the patient progresses and patient-centered care is provided. Because this patient’s presentation differed from other patients with IH, we will be more vigilant in providing close follow-up and monitoring for other medical problems involving other organs (eg, the brain); for instance, we will monitor for seizures and developmental delay.
Conclusion
In our patient, a unique pattern of SWS, facial IH, and CMTC are described in a pediatric patient. Many disciplines are involved in the treatment. In the patient’s first days of life, extensive collaboration between pediatrics and dermatologists was pivotal, with ophthalmology, pathology, and radiology consultations at hand. This case highlights that several vascular malformations of different origins can occur in the same patient. Epigenetic along with genetic factors likely contributed to this fascinating presentation. The importance of parental education and maintaining appropriate follow-up for this patient is crucial for a favorable outcome.
Sturge-Weber syndrome (SWS) is a disease of dermatologic, neurologic, and ocular significance.1 The most distinctive manifestation is facial capillary malformation, commonly referred to as a port-wine stain or nevus flammeus. The dysregulated angiogenesis, caused by somatic mutations of the G protein subunit alpha Q gene, GNAQ, also affects the central nervous system.2 Seizures, intellectual disability, and glaucoma are common consequences.1 Not all port-wine stains are associated with SWS.3 Distribution in the ophthalmic dermatome is associated with increased risk for SWS, with 8% of patients with port-wine stains also having SWS.4 The disease is more serious when bilateral lesions are present.5 Diagnosis is clinical based on dermatologic, nervous system, and ophthalmologic findings.6 The disease is nonheritable because the mutation is found only in the somatic cell lines.2 The possibility of epigenetic influence on disease development has to be investigated. The treatment is aimed at managing complications, as there is no cure.7
Infantile hemangioma (IH) likewise represents a disruption in the process of vascular development but without the widespread consequences of SWS. The pathogenesis of hemangioma development has not been fully elucidated, with presence of GLUT1 (glucose transporter 1) protein implicated in lesions.4 Facial infantile hemangiomas have an incidence of approximately 5 in every 100 births, and the prevalence decreases with age. Most hemangiomas undergo growth followed by an involution process, with most lesions vanishing by 5 years of age.4 They typically are seen at 2 to 3 weeks of age, growing rapidly for the first 6 months, which is a contrast to the static nature of nevus flammeus. Infantile hemangiomas are regarded as sporadic, though autosomal-dominant inheritance patterns have been observed.4 Our patient demonstrated facial IH at birth, which is a rare and interesting finding suggesting that some epigenetic factors influenced this modification of the disease course in this patient.
Cutis marmorata telangiectatica congenita (CMTC) is a rare cutaneous vascular condition found in newborns. Its extraordinary infrequency is reflected in the fact that only 300 cases have been reported worldwide.8 At birth, CMTC manifests as a pinkish reticulated pattern all over the body mimicking cutis marmorata; however, unlike cutis marmorata, the lesions do not improve with warming.9 The lesions of CMTC gradually lighten as the patient ages.8 Limb asymmetry is the most common extravascular complication of CMTC and, similar to SWS, glaucoma also can occur.10 Cutis marmorata telangiectatica congenita has been known to occur simultaneously with SWS or IH, but the combination of all 3 conditions in our patient is unique. Due to the scarcity of cases, the pathophysiology and treatment is poorly understood, with appropriate monitoring for sequelae recommended.9
Case Report
The patient was born at 39 weeks’ gestation following an uncomplicated pregnancy and delivery. She weighed 2950 g, her length was 19 in, and her head circumference was 13.25 in, correlating to the 10th, 50th, and 25th percentiles, respectively. Her Apgar score was 8/9 at 1 and 5 minutes. Her parents were nonconsanguineous and in good health. The patient’s family lived in poverty, which led us to conjecture about the role that toxins played in the epigenetics of the patient and her family. It was the mother’s third pregnancy; both prior pregnancies resulted in healthy children. The patient was breastfed. No family history of heritable vascular disorders was reported.
On the first day of life during the newborn examination, dark red pigment changes were noticed under the nose and erythematous pigmentation over the whole body was observed (Figure). On examination, 2-toned reticular lesions identified as extensive nevus flammeus were found bilaterally over the distribution of the ophthalmic division of the trigeminal nerve. A separate erythematous plaque over the maxilla also was recognized. The pediatrician suspected SWS and facial IH. The patient was discharged after 3 days with a referral to pediatric dermatology, and appropriate follow-up with a pediatrician was scheduled. The patient returned for these appointments and the significance of SWS was explained to her parents. Consultation with pediatric dermatology at 2 weeks of age confirmed the diagnosis of SWS as well as facial IH.
Upon further follow-up with pediatric dermatology at 2 months of age, the patient received an additional diagnosis of CMTC. These exceedingly rare lesions were located over the back, trunk, arms, and legs. The patient’s parents were counseled about the management of these conditions and appropriate follow-up.
Comment
This case describes 3 different vascular malformations in the same patient. Cutis marmorata telangiectatica congenita is rare and yet is described in this patient along with 2 other notable endothelial abnormalities. The clinical interest of this case is heightened by the presence of CMTC.
The causative factor of SWS is a well-documented mutation of the GNAQ gene, but there is considerable variability in how it affects the patient. Unlike in SWS, no single factor can be attributed to the development of IH. This case shows that these 3 diseases are not mutually exclusive and can present with unusually severe features when they occur concomitantly. The embryologic basis of SWS traces its roots back to the first trimester during vascular development, wher
The severity of the SWS in our patient was highlighted by the extensive nevus flammeus. These lesions occurred over the face, trunk, arms, and legs. The port-wine stain with dermatomal distribution of the ophthalmic nerve was the most concerning feature regarding the development of neurologic complications in this patient. Although the developmental delays associated with SWS can be serious, early intervention is important and can improve long-term outcomes. The facial IH arising at birth was contrary to the typical presentation. All of these factors will be kept in mind as the patient progresses and patient-centered care is provided. Because this patient’s presentation differed from other patients with IH, we will be more vigilant in providing close follow-up and monitoring for other medical problems involving other organs (eg, the brain); for instance, we will monitor for seizures and developmental delay.
Conclusion
In our patient, a unique pattern of SWS, facial IH, and CMTC are described in a pediatric patient. Many disciplines are involved in the treatment. In the patient’s first days of life, extensive collaboration between pediatrics and dermatologists was pivotal, with ophthalmology, pathology, and radiology consultations at hand. This case highlights that several vascular malformations of different origins can occur in the same patient. Epigenetic along with genetic factors likely contributed to this fascinating presentation. The importance of parental education and maintaining appropriate follow-up for this patient is crucial for a favorable outcome.
- Sinawat S, Auvichayapat N, Auvichayapat P, et al. 12-year retrospective study of Sturge-weber syndrome and literature review. J Med Assoc Thail. 2014;97:742-750.
- Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971-1979.
- Sudarsanam A, Ardern-Holmes SL. Sturge-Weber syndrome: from the past to the present [published online November 7, 2013]. Eur J Paediat Neurol. 2014;18:257-266.
- Paller AS, Mancini AJ. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. Philadelphia, PA: Elsevier Saunders; 2011.
- Sujansky E, Conradi S. Sturge-Weber syndrome: age of onset of seizures and glaucoma and the prognosis for affected children. J Child Neurol. 1995;10:49-58.
- Lo W, Marchuk DA, Ball KL, et al. Updates and future horizons on the understanding, diagnosis, and treatment of Sturge-Weber syndrome brain involvement. Dev Med Child Neurol. 2012;54:214-223.
- Comi AM. Update on Sturge-Weber syndrome: diagnosis, treatment, quantitative measures, and controversies. Lymphat Res Biol. 2007;5:257-264.
- Resende CI, Araujo C, Vieira AP, et al. Cutis marmorata telangiectatica congenital [published online October 17, 2013]. BMJ Case Rep. doi:10.1136/bcr-2013-200056.
- Levy R, Lam JM. Cutis marmorata telangiectatica congenita: a mimicker of a common disorder. CMAJ. 2011;183:E249-E251.
- Kienast AK, Hoeger PH. Cutis marmorata telangiectatica congenita: a prospective study of 27 cases and review of the literature with proposal of diagnostic criteria. Clin Exp Dermatol. 2009;34:319-323.
- Comi AM. Topical review: pathophysiology of Sturge-Weber syndrome. J Child Neurol. 2003;18:509-516.
- Sinawat S, Auvichayapat N, Auvichayapat P, et al. 12-year retrospective study of Sturge-weber syndrome and literature review. J Med Assoc Thail. 2014;97:742-750.
- Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971-1979.
- Sudarsanam A, Ardern-Holmes SL. Sturge-Weber syndrome: from the past to the present [published online November 7, 2013]. Eur J Paediat Neurol. 2014;18:257-266.
- Paller AS, Mancini AJ. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. Philadelphia, PA: Elsevier Saunders; 2011.
- Sujansky E, Conradi S. Sturge-Weber syndrome: age of onset of seizures and glaucoma and the prognosis for affected children. J Child Neurol. 1995;10:49-58.
- Lo W, Marchuk DA, Ball KL, et al. Updates and future horizons on the understanding, diagnosis, and treatment of Sturge-Weber syndrome brain involvement. Dev Med Child Neurol. 2012;54:214-223.
- Comi AM. Update on Sturge-Weber syndrome: diagnosis, treatment, quantitative measures, and controversies. Lymphat Res Biol. 2007;5:257-264.
- Resende CI, Araujo C, Vieira AP, et al. Cutis marmorata telangiectatica congenital [published online October 17, 2013]. BMJ Case Rep. doi:10.1136/bcr-2013-200056.
- Levy R, Lam JM. Cutis marmorata telangiectatica congenita: a mimicker of a common disorder. CMAJ. 2011;183:E249-E251.
- Kienast AK, Hoeger PH. Cutis marmorata telangiectatica congenita: a prospective study of 27 cases and review of the literature with proposal of diagnostic criteria. Clin Exp Dermatol. 2009;34:319-323.
- Comi AM. Topical review: pathophysiology of Sturge-Weber syndrome. J Child Neurol. 2003;18:509-516.
Practice Points
- This case highlights that several vascular malformations of different origins can occur in the same patient.
- Epigenetic factors along with genetic factors can lead to development of complex vascular conditions.
- Close collaborations of different medical specialties is necessary to make an accurate diagnosis and to follow up to achieve optimal long-term outcomes for patients with complex medical conditions.
Harnessing vascular biology to rescue CVI sufferers
Despite affecting 25 million Americans, including two to six million with ulcer conditions, chronic venous insufficiency is relatively understudied compared to other vascular diseases. Yet for patients with venous leg ulcers, their condition is debilitating, painful and embarrassing.
Dr. Ulka Sachdev is studying the condition, hoping her “bench-side research” will develop “bedside” solutions. She received a five-year, National Institutes of Health K08 and SVS Foundation grants in 2012 and an SVS Foundation Clinical Research Seed Grant in 2017. “Their ulcers are very difficult to manage,” she said. “They are large, open, painful and wet. Patients’ daily routines are negatively affected by their wounds. It’s really sad.”
Since she regularly treats CVI patients, Dr. Sachdev wondered why some patients have benign venous insufficiency that ulcerates and why some ulcers recur. “If we could determine at an earlier stage how to mitigate the risk of new ulceration or recurrence, I think it would be worth it,” she said.
In her previous, K08-supported research she hypothesized that wound healing during ischemia is promoted by inflammatory proteins released by damaged tissue. She found that certain proteins known as danger signals can be released by damaged tissue and promote regenerative effects.
“My hypothesis is that specific danger signals can be manipulated, ideally with an oral drug,” Dr. Sachdev said. “If it works, this could be a mechanism that allows a dying muscle cell to say, ‘Hey, I need help.’ This might mean that someone who cannot get a bypass or a stent might not have to face amputation.’”
Her recent grant is for studying study patterns of inflammation in chronic venous insufficiency.
The goal of her studies is to determine whether patients with benign varicose veins and those with ulcerations express inflammatory mediators that predict their response to treatment. Later, perhaps, effective treatments will be found that change these patients’ lives.
Despite affecting 25 million Americans, including two to six million with ulcer conditions, chronic venous insufficiency is relatively understudied compared to other vascular diseases. Yet for patients with venous leg ulcers, their condition is debilitating, painful and embarrassing.
Dr. Ulka Sachdev is studying the condition, hoping her “bench-side research” will develop “bedside” solutions. She received a five-year, National Institutes of Health K08 and SVS Foundation grants in 2012 and an SVS Foundation Clinical Research Seed Grant in 2017. “Their ulcers are very difficult to manage,” she said. “They are large, open, painful and wet. Patients’ daily routines are negatively affected by their wounds. It’s really sad.”
Since she regularly treats CVI patients, Dr. Sachdev wondered why some patients have benign venous insufficiency that ulcerates and why some ulcers recur. “If we could determine at an earlier stage how to mitigate the risk of new ulceration or recurrence, I think it would be worth it,” she said.
In her previous, K08-supported research she hypothesized that wound healing during ischemia is promoted by inflammatory proteins released by damaged tissue. She found that certain proteins known as danger signals can be released by damaged tissue and promote regenerative effects.
“My hypothesis is that specific danger signals can be manipulated, ideally with an oral drug,” Dr. Sachdev said. “If it works, this could be a mechanism that allows a dying muscle cell to say, ‘Hey, I need help.’ This might mean that someone who cannot get a bypass or a stent might not have to face amputation.’”
Her recent grant is for studying study patterns of inflammation in chronic venous insufficiency.
The goal of her studies is to determine whether patients with benign varicose veins and those with ulcerations express inflammatory mediators that predict their response to treatment. Later, perhaps, effective treatments will be found that change these patients’ lives.
Despite affecting 25 million Americans, including two to six million with ulcer conditions, chronic venous insufficiency is relatively understudied compared to other vascular diseases. Yet for patients with venous leg ulcers, their condition is debilitating, painful and embarrassing.
Dr. Ulka Sachdev is studying the condition, hoping her “bench-side research” will develop “bedside” solutions. She received a five-year, National Institutes of Health K08 and SVS Foundation grants in 2012 and an SVS Foundation Clinical Research Seed Grant in 2017. “Their ulcers are very difficult to manage,” she said. “They are large, open, painful and wet. Patients’ daily routines are negatively affected by their wounds. It’s really sad.”
Since she regularly treats CVI patients, Dr. Sachdev wondered why some patients have benign venous insufficiency that ulcerates and why some ulcers recur. “If we could determine at an earlier stage how to mitigate the risk of new ulceration or recurrence, I think it would be worth it,” she said.
In her previous, K08-supported research she hypothesized that wound healing during ischemia is promoted by inflammatory proteins released by damaged tissue. She found that certain proteins known as danger signals can be released by damaged tissue and promote regenerative effects.
“My hypothesis is that specific danger signals can be manipulated, ideally with an oral drug,” Dr. Sachdev said. “If it works, this could be a mechanism that allows a dying muscle cell to say, ‘Hey, I need help.’ This might mean that someone who cannot get a bypass or a stent might not have to face amputation.’”
Her recent grant is for studying study patterns of inflammation in chronic venous insufficiency.
The goal of her studies is to determine whether patients with benign varicose veins and those with ulcerations express inflammatory mediators that predict their response to treatment. Later, perhaps, effective treatments will be found that change these patients’ lives.
Ideals of Facial Beauty
Several concepts of ideal aesthetic measurements can be traced back to ancient Greek and European Renaissance art. In examining canons of beauty, these classical ideals often are compared to modern-day standards, allowing clinicians to delineate the parameters of an attractive facial appearance and facilitate the planning of cosmetic procedures.
Given the growing number of available cosmetic interventions, dermatologists have a powerful ability to modify facial proportions; however, changes to individual structures should be made with a mindful approach to improving overall facial harmony. This article reviews the established parameters of facial beauty to assist the clinician in enhancing cosmetic outcomes.
Canons of Facial Aesthetics
Horizontal Thirds
In his writings on human anatomy, Leonardo da Vinci described dividing the face into equal thirds (Figure 1). The upper third measures from the trichion (the midline point of the normal hairline) to the glabella (the smooth prominence between the eyebrows). The middle third measures from the glabella to the subnasale (the midline point where the nasal septum meets the upper lip). The lower third measures from the subnasale to the menton (the most inferior point of the chin).1
Although the validity of the canon is intended to apply across race and gender, these proportions may vary by ethnicity (Table). In white individuals, the middle third of the face tends to be shorter than the upper and lower thirds.2 This same relationship has been observed in black males.3 In Chinese females, the upper third commonly is shorter than the middle and lower thirds, correlating with a less prominent forehead. In contrast, black females tend to have a relatively longer upper third.4

The relationship between modern perceptions of attractiveness and the neoclassical norm of equal thirds remains a topic of interest. Milutinovic et al1 examined facial thirds in white female celebrities from beauty and fashion magazines and compared them to a group of anonymous white females from the general population. The group of anonymous females showed statistically significant (P<.05) differences between the sizes of the 3 facial segments, whereas the group of celebrity faces demonstrated uniformity between the facial thirds.1
The lower face can itself be divided into thirds, with the upper third measured from the subnasale to the stomion (the midline point of the oral fissure when the lips are closed), and the lower two-thirds measured from the stomion to the menton (Figure 1). Mommaerts and Moerenhout5 examined photographs of 105 attractive celebrity faces and compared their proportions to those of classical sculptures of gods and goddesses (antique faces). The authors identified an upper one-third to lower two-thirds ratio of 69.8% in celebrity females and 69.1% in celebrity males; these ratios were not significantly different from the 72.4% seen in antique females and 73.1% in antique males. The authors concluded that a 30% upper lip to 70% lower lip-chin proportion may be the most appropriate to describe contemporary standards.5
Vertical Fifths
In the vertical dimension, the neoclassical canon of facial proportions divides the face into equal fifths (Figure 2).6 The 2 most lateral fifths are measured from the lateral helix of each ear to the exocanthus of each eye. The eye fissure lengths (measured between the endocanthion and exocanthion of each eye) represent one-fifth. The middle fifth is measured between the medial canthi of both eyes (endocanthion to endocanthion). This distance is equal to the width of the nose, as measured between both alae. Finally, the width of the mouth represents 1.5-times the width of the nose. These ratios of the vertical fifths apply to both males and females.6
Anthropometric studies have examined deviations from the neoclassical canon according to ethnicity. Wang et al7 compared the measurements of North American white and Han Chinese patients to these standards. White patients demonstrated a greater ratio of mouth width to nose width relative to the canon. In contrast, Han Chinese patients demonstrated a relatively wider nose and narrower mouth.7
In black individuals, it has been observed that the dimensions of most facial segments correspond to the neoclassical standards; however, nose width is relatively wider in black individuals relative to the canon as well as relative to white individuals.8
Milutinovic et al1 also compared vertical fifths between white celebrities and anonymous females. In the anonymous female group, statistically significant (P<.05) variations were found between the sizes of the different facial components. In contrast, the celebrity female group showed balance between the widths of vertical fifths.1
Lips
In the lower facial third, the lips represent a key element of attractiveness. Recently, lip augmentation, aimed at creating fuller and plumper lips, has dominated the popular culture and social media landscape.9 Although the aesthetic ideal of lips continues to evolve over time, recent studies have aimed at quantifying modern notions of attractive lip appearance.
Popenko et al10 examined lip measurements using computer-generated images of white women with different variations of lip sizes and lower face proportions. Computer-generated faces were graded on attractiveness by more than 400 individuals from focus groups. An upper lip to lower lip ratio of 1:2 was judged to be the most attractive, while a ratio of 2:1 was judged to be the least attractive. Results also showed that the surface area of the most attractive lips comprised roughly 10% of the lower third of the face.10
Penna et al11 analyzed various parameters of the lips and lower facial third using photographs of 176 white males and females that were judged on attractiveness by 250 volunteer evaluators. Faces were graded on a scale from 1 (absolutely attractive) to 7 (absolutely unattractive). Attractive males and females (grades 1 and 2) both demonstrated an average ratio of upper vermilion height to nose-mouth distance (measured from the subnasalae to the lower edge of the upper vermilion border) of 0.28, which was significantly greater than the average ratio observed in less attractive individuals (grades 6 or 7)(P<.05). In addition, attractive males and females demonstrated a ratio of upper vermilion height to nose-chin distance (measured from the subnasalae to the menton) of 0.09, which again was larger than the average ratio seen in less attractive individuals. Figure 3 demonstrates an aesthetic ideal of the lips derived from these 2 studies, though consideration should be given to the fact that these studies were based in white populations.
Golden Ratio
The golden ratio, also known as Phi, can be observed in nature, art, and architecture. Approximately equal to 1.618, the golden ratio also has been identified as a possible marker of beauty in the human face and has garnered attention in the lay press. The ratio has been applied to several proportions and structures in the face, such as the ratio of mouth width to nose width or the ratio of tooth height to tooth width, with investigation providing varying levels of validation about whether these ratios truly correlate with perceptions of beauty.12 Swift and Remington13 advocated for application of the golden ratio toward a comprehensive set of facial proportions. Marquardt14 used the golden ratio to create a 3-dimensional representation of an idealized face, known as the golden decagon mask. Although the golden ratio and the golden decagon mask have been proposed as analytic tools, their utility in clinical practice may be limited. Firstly, due to its popularity in the lay press, the golden ratio has been inconsistently applied to a wide range of facial ratios, which may undermine confidence in its representation as truth rather than coincidence. Secondly, although some authors have found validity of the golden decagon mask in representing unified ratios of attractiveness, others have asserted that it characterizes a masculinized white female and fails to account for ethnic differences.15-19
Age-Related Changes
In addition to the facial proportions guided by genetics, several changes occur with increased age. Over the course of a lifetime, predictable patterns emerge in the dimensions of the skin, soft tissue, and bone. These alterations in structural proportions may ultimately lead to an unevenness in facial aesthetics.
In skeletal structure, gradual bone resorption and expansion causes a reduction in facial height as well as an increase in facial width and depth.20 Fat atrophy and hypertrophy affect soft tissue proportions, visualized as hollowing at the temples, cheeks, and around the eyes, along with fullness in the submental region and jowls.21 Finally, decreases in skin elasticity and collagen exacerbate the appearance of rhytides and sagging. In older patients who desire a more youthful appearance, various applications of dermal fillers, fat grafting, liposuction, and skin tightening techniques can help to mitigate these changes.
Conclusion
Improving facial aesthetics relies on an understanding of the norms of facial proportions. Although cosmetic interventions commonly are advertised or described based on a single anatomical unit, it is important to appreciate the relationships between facial structures. Most notably, clinicians should be mindful of facial ratios when considering the introduction of filler materials or implants. Augmentation procedures at the temples, zygomatic arch, jaw, chin, and lips all have the possibility to alter facial ratios. Changes should therefore be considered in the context of improving overall facial harmony, with the clinician remaining cognizant of the ideal vertical and horizontal divisions of the face. Understanding such concepts and communicating them to patients can help in appropriately addressing all target areas, thereby leading to greater patient satisfaction.
- Milutinovic J, Zelic K, Nedeljkovic N. Evaluation of facial beauty using anthropometric proportions. ScientificWorldJournal. 2014;2014:428250. doi:10.1155/2014/428250.
- Farkas LG, Hreczko TA, Kolar JC, et al. Vertical and horizontal proportions of the face in young-adult North-American Caucasians: revision of neoclassical canons. Plast Reconstr Surg. 1985;75:328-338.
- Porter JP. The average African American male face: an anthropometric analysis. Arch Facial Plast Surg. 2004;6:78-81.
- Porter JP, Olson KL. Anthropometric facial analysis of the African American woman. Arch Facial Plast Surg. 2001;3:191-197.
- Mommaerts MY, Moerenhout BA. Ideal proportions in full face front view, contemporary versus antique. J Craniomaxillofac Surg. 2011;39:107-110.
- Vegter F, Hage JJ. Clinical anthropometry and canons of the face in historical perspective. Plast Reconstr Surg. 2000;106:1090-1096.
- Wang D, Qian G, Zhang M, et al. Differences in horizontal, neoclassical facial canons in Chinese (Han) and North American Caucasian populations. Aesthetic Plast Surg. 1997;21:265-269.
- Farkas LG, Forrest CR, Litsas L. Revision of neoclassical facial canons in young adult Afro-Americans. Aesthetic Plast Surg. 2000;24:179-184.
- Coleman GG, Lindauer SJ, Tüfekçi E, et al. Influence of chin prominence on esthetic lip profile preferences. Am J Orthod Dentofacial Orthop. 2007;132:36-42.
- Popenko NA, Tripathi PB, Devcic Z, et al. A quantitative approach to determining the ideal female lip aesthetic and its effect on facial attractiveness. JAMA Facial Plast Surg. 2017;19:261-267.
- Penna V, Fricke A, Iblher N, et al. The attractive lip: a photomorphometric analysis. J Plast Reconstr Aesthet Surg. 2015;68:920-929.
- Prokopakis EP, Vlastos IM, Picavet VA, et al. The golden ratio in facial symmetry. Rhinology. 2013;51:18-21.
- Swift A, Remington K. BeautiPHIcationTM: a global approach to facial beauty. Clin Plast Surg. 2011;38:247-277.
- Marquardt SR. Dr. Stephen R. Marquardt on the Golden Decagon and human facial beauty. interview by Dr. Gottlieb. J Clin Orthod. 2002;36:339-347.
- Veerala G, Gandikota CS, Yadagiri PK, et al. Marquardt’s facial Golden Decagon mask and its fitness with South Indian facial traits. J Clin Diagn Res. 2016;10:ZC49-ZC52.
- Holland E. Marquardt’s Phi mask: pitfalls of relying on fashion models and the golden ratio to describe a beautiful face. Aesthetic Plast Surg. 2008;32:200-208.
- Alam MK, Mohd Noor NF, Basri R, et al. Multiracial facial golden ratio and evaluation of facial appearance. PLoS One. 2015;10:e0142914.
- Kim YH. Easy facial analysis using the facial golden mask. J Craniofac Surg. 2007;18:643-649.
- Bashour M. An objective system for measuring facial attractiveness. Plast Reconstr Surg. 2006;118:757-774; discussion 775-776.
- Bartlett SP, Grossman R, Whitaker LA. Age-related changes of the craniofacial skeleton: an anthropometric and histologic analysis. Plast Reconstr Surg. 1992;90:592-600.
- Donofrio LM. Fat distribution: a morphologic study of the aging face. Dermatol Surg. 2000;26:1107-1112.
Several concepts of ideal aesthetic measurements can be traced back to ancient Greek and European Renaissance art. In examining canons of beauty, these classical ideals often are compared to modern-day standards, allowing clinicians to delineate the parameters of an attractive facial appearance and facilitate the planning of cosmetic procedures.
Given the growing number of available cosmetic interventions, dermatologists have a powerful ability to modify facial proportions; however, changes to individual structures should be made with a mindful approach to improving overall facial harmony. This article reviews the established parameters of facial beauty to assist the clinician in enhancing cosmetic outcomes.
Canons of Facial Aesthetics
Horizontal Thirds
In his writings on human anatomy, Leonardo da Vinci described dividing the face into equal thirds (Figure 1). The upper third measures from the trichion (the midline point of the normal hairline) to the glabella (the smooth prominence between the eyebrows). The middle third measures from the glabella to the subnasale (the midline point where the nasal septum meets the upper lip). The lower third measures from the subnasale to the menton (the most inferior point of the chin).1
Although the validity of the canon is intended to apply across race and gender, these proportions may vary by ethnicity (Table). In white individuals, the middle third of the face tends to be shorter than the upper and lower thirds.2 This same relationship has been observed in black males.3 In Chinese females, the upper third commonly is shorter than the middle and lower thirds, correlating with a less prominent forehead. In contrast, black females tend to have a relatively longer upper third.4
The relationship between modern perceptions of attractiveness and the neoclassical norm of equal thirds remains a topic of interest. Milutinovic et al1 examined facial thirds in white female celebrities from beauty and fashion magazines and compared them to a group of anonymous white females from the general population. The group of anonymous females showed statistically significant (P<.05) differences between the sizes of the 3 facial segments, whereas the group of celebrity faces demonstrated uniformity between the facial thirds.1
The lower face can itself be divided into thirds, with the upper third measured from the subnasale to the stomion (the midline point of the oral fissure when the lips are closed), and the lower two-thirds measured from the stomion to the menton (Figure 1). Mommaerts and Moerenhout5 examined photographs of 105 attractive celebrity faces and compared their proportions to those of classical sculptures of gods and goddesses (antique faces). The authors identified an upper one-third to lower two-thirds ratio of 69.8% in celebrity females and 69.1% in celebrity males; these ratios were not significantly different from the 72.4% seen in antique females and 73.1% in antique males. The authors concluded that a 30% upper lip to 70% lower lip-chin proportion may be the most appropriate to describe contemporary standards.5
Vertical Fifths
In the vertical dimension, the neoclassical canon of facial proportions divides the face into equal fifths (Figure 2).6 The 2 most lateral fifths are measured from the lateral helix of each ear to the exocanthus of each eye. The eye fissure lengths (measured between the endocanthion and exocanthion of each eye) represent one-fifth. The middle fifth is measured between the medial canthi of both eyes (endocanthion to endocanthion). This distance is equal to the width of the nose, as measured between both alae. Finally, the width of the mouth represents 1.5-times the width of the nose. These ratios of the vertical fifths apply to both males and females.6
Anthropometric studies have examined deviations from the neoclassical canon according to ethnicity. Wang et al7 compared the measurements of North American white and Han Chinese patients to these standards. White patients demonstrated a greater ratio of mouth width to nose width relative to the canon. In contrast, Han Chinese patients demonstrated a relatively wider nose and narrower mouth.7
In black individuals, it has been observed that the dimensions of most facial segments correspond to the neoclassical standards; however, nose width is relatively wider in black individuals relative to the canon as well as relative to white individuals.8
Milutinovic et al1 also compared vertical fifths between white celebrities and anonymous females. In the anonymous female group, statistically significant (P<.05) variations were found between the sizes of the different facial components. In contrast, the celebrity female group showed balance between the widths of vertical fifths.1
Lips
In the lower facial third, the lips represent a key element of attractiveness. Recently, lip augmentation, aimed at creating fuller and plumper lips, has dominated the popular culture and social media landscape.9 Although the aesthetic ideal of lips continues to evolve over time, recent studies have aimed at quantifying modern notions of attractive lip appearance.
Popenko et al10 examined lip measurements using computer-generated images of white women with different variations of lip sizes and lower face proportions. Computer-generated faces were graded on attractiveness by more than 400 individuals from focus groups. An upper lip to lower lip ratio of 1:2 was judged to be the most attractive, while a ratio of 2:1 was judged to be the least attractive. Results also showed that the surface area of the most attractive lips comprised roughly 10% of the lower third of the face.10
Penna et al11 analyzed various parameters of the lips and lower facial third using photographs of 176 white males and females that were judged on attractiveness by 250 volunteer evaluators. Faces were graded on a scale from 1 (absolutely attractive) to 7 (absolutely unattractive). Attractive males and females (grades 1 and 2) both demonstrated an average ratio of upper vermilion height to nose-mouth distance (measured from the subnasalae to the lower edge of the upper vermilion border) of 0.28, which was significantly greater than the average ratio observed in less attractive individuals (grades 6 or 7)(P<.05). In addition, attractive males and females demonstrated a ratio of upper vermilion height to nose-chin distance (measured from the subnasalae to the menton) of 0.09, which again was larger than the average ratio seen in less attractive individuals. Figure 3 demonstrates an aesthetic ideal of the lips derived from these 2 studies, though consideration should be given to the fact that these studies were based in white populations.
Golden Ratio
The golden ratio, also known as Phi, can be observed in nature, art, and architecture. Approximately equal to 1.618, the golden ratio also has been identified as a possible marker of beauty in the human face and has garnered attention in the lay press. The ratio has been applied to several proportions and structures in the face, such as the ratio of mouth width to nose width or the ratio of tooth height to tooth width, with investigation providing varying levels of validation about whether these ratios truly correlate with perceptions of beauty.12 Swift and Remington13 advocated for application of the golden ratio toward a comprehensive set of facial proportions. Marquardt14 used the golden ratio to create a 3-dimensional representation of an idealized face, known as the golden decagon mask. Although the golden ratio and the golden decagon mask have been proposed as analytic tools, their utility in clinical practice may be limited. Firstly, due to its popularity in the lay press, the golden ratio has been inconsistently applied to a wide range of facial ratios, which may undermine confidence in its representation as truth rather than coincidence. Secondly, although some authors have found validity of the golden decagon mask in representing unified ratios of attractiveness, others have asserted that it characterizes a masculinized white female and fails to account for ethnic differences.15-19
Age-Related Changes
In addition to the facial proportions guided by genetics, several changes occur with increased age. Over the course of a lifetime, predictable patterns emerge in the dimensions of the skin, soft tissue, and bone. These alterations in structural proportions may ultimately lead to an unevenness in facial aesthetics.
In skeletal structure, gradual bone resorption and expansion causes a reduction in facial height as well as an increase in facial width and depth.20 Fat atrophy and hypertrophy affect soft tissue proportions, visualized as hollowing at the temples, cheeks, and around the eyes, along with fullness in the submental region and jowls.21 Finally, decreases in skin elasticity and collagen exacerbate the appearance of rhytides and sagging. In older patients who desire a more youthful appearance, various applications of dermal fillers, fat grafting, liposuction, and skin tightening techniques can help to mitigate these changes.
Conclusion
Improving facial aesthetics relies on an understanding of the norms of facial proportions. Although cosmetic interventions commonly are advertised or described based on a single anatomical unit, it is important to appreciate the relationships between facial structures. Most notably, clinicians should be mindful of facial ratios when considering the introduction of filler materials or implants. Augmentation procedures at the temples, zygomatic arch, jaw, chin, and lips all have the possibility to alter facial ratios. Changes should therefore be considered in the context of improving overall facial harmony, with the clinician remaining cognizant of the ideal vertical and horizontal divisions of the face. Understanding such concepts and communicating them to patients can help in appropriately addressing all target areas, thereby leading to greater patient satisfaction.
Several concepts of ideal aesthetic measurements can be traced back to ancient Greek and European Renaissance art. In examining canons of beauty, these classical ideals often are compared to modern-day standards, allowing clinicians to delineate the parameters of an attractive facial appearance and facilitate the planning of cosmetic procedures.
Given the growing number of available cosmetic interventions, dermatologists have a powerful ability to modify facial proportions; however, changes to individual structures should be made with a mindful approach to improving overall facial harmony. This article reviews the established parameters of facial beauty to assist the clinician in enhancing cosmetic outcomes.
Canons of Facial Aesthetics
Horizontal Thirds
In his writings on human anatomy, Leonardo da Vinci described dividing the face into equal thirds (Figure 1). The upper third measures from the trichion (the midline point of the normal hairline) to the glabella (the smooth prominence between the eyebrows). The middle third measures from the glabella to the subnasale (the midline point where the nasal septum meets the upper lip). The lower third measures from the subnasale to the menton (the most inferior point of the chin).1
Although the validity of the canon is intended to apply across race and gender, these proportions may vary by ethnicity (Table). In white individuals, the middle third of the face tends to be shorter than the upper and lower thirds.2 This same relationship has been observed in black males.3 In Chinese females, the upper third commonly is shorter than the middle and lower thirds, correlating with a less prominent forehead. In contrast, black females tend to have a relatively longer upper third.4
The relationship between modern perceptions of attractiveness and the neoclassical norm of equal thirds remains a topic of interest. Milutinovic et al1 examined facial thirds in white female celebrities from beauty and fashion magazines and compared them to a group of anonymous white females from the general population. The group of anonymous females showed statistically significant (P<.05) differences between the sizes of the 3 facial segments, whereas the group of celebrity faces demonstrated uniformity between the facial thirds.1
The lower face can itself be divided into thirds, with the upper third measured from the subnasale to the stomion (the midline point of the oral fissure when the lips are closed), and the lower two-thirds measured from the stomion to the menton (Figure 1). Mommaerts and Moerenhout5 examined photographs of 105 attractive celebrity faces and compared their proportions to those of classical sculptures of gods and goddesses (antique faces). The authors identified an upper one-third to lower two-thirds ratio of 69.8% in celebrity females and 69.1% in celebrity males; these ratios were not significantly different from the 72.4% seen in antique females and 73.1% in antique males. The authors concluded that a 30% upper lip to 70% lower lip-chin proportion may be the most appropriate to describe contemporary standards.5
Vertical Fifths
In the vertical dimension, the neoclassical canon of facial proportions divides the face into equal fifths (Figure 2).6 The 2 most lateral fifths are measured from the lateral helix of each ear to the exocanthus of each eye. The eye fissure lengths (measured between the endocanthion and exocanthion of each eye) represent one-fifth. The middle fifth is measured between the medial canthi of both eyes (endocanthion to endocanthion). This distance is equal to the width of the nose, as measured between both alae. Finally, the width of the mouth represents 1.5-times the width of the nose. These ratios of the vertical fifths apply to both males and females.6
Anthropometric studies have examined deviations from the neoclassical canon according to ethnicity. Wang et al7 compared the measurements of North American white and Han Chinese patients to these standards. White patients demonstrated a greater ratio of mouth width to nose width relative to the canon. In contrast, Han Chinese patients demonstrated a relatively wider nose and narrower mouth.7
In black individuals, it has been observed that the dimensions of most facial segments correspond to the neoclassical standards; however, nose width is relatively wider in black individuals relative to the canon as well as relative to white individuals.8
Milutinovic et al1 also compared vertical fifths between white celebrities and anonymous females. In the anonymous female group, statistically significant (P<.05) variations were found between the sizes of the different facial components. In contrast, the celebrity female group showed balance between the widths of vertical fifths.1
Lips
In the lower facial third, the lips represent a key element of attractiveness. Recently, lip augmentation, aimed at creating fuller and plumper lips, has dominated the popular culture and social media landscape.9 Although the aesthetic ideal of lips continues to evolve over time, recent studies have aimed at quantifying modern notions of attractive lip appearance.
Popenko et al10 examined lip measurements using computer-generated images of white women with different variations of lip sizes and lower face proportions. Computer-generated faces were graded on attractiveness by more than 400 individuals from focus groups. An upper lip to lower lip ratio of 1:2 was judged to be the most attractive, while a ratio of 2:1 was judged to be the least attractive. Results also showed that the surface area of the most attractive lips comprised roughly 10% of the lower third of the face.10
Penna et al11 analyzed various parameters of the lips and lower facial third using photographs of 176 white males and females that were judged on attractiveness by 250 volunteer evaluators. Faces were graded on a scale from 1 (absolutely attractive) to 7 (absolutely unattractive). Attractive males and females (grades 1 and 2) both demonstrated an average ratio of upper vermilion height to nose-mouth distance (measured from the subnasalae to the lower edge of the upper vermilion border) of 0.28, which was significantly greater than the average ratio observed in less attractive individuals (grades 6 or 7)(P<.05). In addition, attractive males and females demonstrated a ratio of upper vermilion height to nose-chin distance (measured from the subnasalae to the menton) of 0.09, which again was larger than the average ratio seen in less attractive individuals. Figure 3 demonstrates an aesthetic ideal of the lips derived from these 2 studies, though consideration should be given to the fact that these studies were based in white populations.
Golden Ratio
The golden ratio, also known as Phi, can be observed in nature, art, and architecture. Approximately equal to 1.618, the golden ratio also has been identified as a possible marker of beauty in the human face and has garnered attention in the lay press. The ratio has been applied to several proportions and structures in the face, such as the ratio of mouth width to nose width or the ratio of tooth height to tooth width, with investigation providing varying levels of validation about whether these ratios truly correlate with perceptions of beauty.12 Swift and Remington13 advocated for application of the golden ratio toward a comprehensive set of facial proportions. Marquardt14 used the golden ratio to create a 3-dimensional representation of an idealized face, known as the golden decagon mask. Although the golden ratio and the golden decagon mask have been proposed as analytic tools, their utility in clinical practice may be limited. Firstly, due to its popularity in the lay press, the golden ratio has been inconsistently applied to a wide range of facial ratios, which may undermine confidence in its representation as truth rather than coincidence. Secondly, although some authors have found validity of the golden decagon mask in representing unified ratios of attractiveness, others have asserted that it characterizes a masculinized white female and fails to account for ethnic differences.15-19
Age-Related Changes
In addition to the facial proportions guided by genetics, several changes occur with increased age. Over the course of a lifetime, predictable patterns emerge in the dimensions of the skin, soft tissue, and bone. These alterations in structural proportions may ultimately lead to an unevenness in facial aesthetics.
In skeletal structure, gradual bone resorption and expansion causes a reduction in facial height as well as an increase in facial width and depth.20 Fat atrophy and hypertrophy affect soft tissue proportions, visualized as hollowing at the temples, cheeks, and around the eyes, along with fullness in the submental region and jowls.21 Finally, decreases in skin elasticity and collagen exacerbate the appearance of rhytides and sagging. In older patients who desire a more youthful appearance, various applications of dermal fillers, fat grafting, liposuction, and skin tightening techniques can help to mitigate these changes.
Conclusion
Improving facial aesthetics relies on an understanding of the norms of facial proportions. Although cosmetic interventions commonly are advertised or described based on a single anatomical unit, it is important to appreciate the relationships between facial structures. Most notably, clinicians should be mindful of facial ratios when considering the introduction of filler materials or implants. Augmentation procedures at the temples, zygomatic arch, jaw, chin, and lips all have the possibility to alter facial ratios. Changes should therefore be considered in the context of improving overall facial harmony, with the clinician remaining cognizant of the ideal vertical and horizontal divisions of the face. Understanding such concepts and communicating them to patients can help in appropriately addressing all target areas, thereby leading to greater patient satisfaction.
- Milutinovic J, Zelic K, Nedeljkovic N. Evaluation of facial beauty using anthropometric proportions. ScientificWorldJournal. 2014;2014:428250. doi:10.1155/2014/428250.
- Farkas LG, Hreczko TA, Kolar JC, et al. Vertical and horizontal proportions of the face in young-adult North-American Caucasians: revision of neoclassical canons. Plast Reconstr Surg. 1985;75:328-338.
- Porter JP. The average African American male face: an anthropometric analysis. Arch Facial Plast Surg. 2004;6:78-81.
- Porter JP, Olson KL. Anthropometric facial analysis of the African American woman. Arch Facial Plast Surg. 2001;3:191-197.
- Mommaerts MY, Moerenhout BA. Ideal proportions in full face front view, contemporary versus antique. J Craniomaxillofac Surg. 2011;39:107-110.
- Vegter F, Hage JJ. Clinical anthropometry and canons of the face in historical perspective. Plast Reconstr Surg. 2000;106:1090-1096.
- Wang D, Qian G, Zhang M, et al. Differences in horizontal, neoclassical facial canons in Chinese (Han) and North American Caucasian populations. Aesthetic Plast Surg. 1997;21:265-269.
- Farkas LG, Forrest CR, Litsas L. Revision of neoclassical facial canons in young adult Afro-Americans. Aesthetic Plast Surg. 2000;24:179-184.
- Coleman GG, Lindauer SJ, Tüfekçi E, et al. Influence of chin prominence on esthetic lip profile preferences. Am J Orthod Dentofacial Orthop. 2007;132:36-42.
- Popenko NA, Tripathi PB, Devcic Z, et al. A quantitative approach to determining the ideal female lip aesthetic and its effect on facial attractiveness. JAMA Facial Plast Surg. 2017;19:261-267.
- Penna V, Fricke A, Iblher N, et al. The attractive lip: a photomorphometric analysis. J Plast Reconstr Aesthet Surg. 2015;68:920-929.
- Prokopakis EP, Vlastos IM, Picavet VA, et al. The golden ratio in facial symmetry. Rhinology. 2013;51:18-21.
- Swift A, Remington K. BeautiPHIcationTM: a global approach to facial beauty. Clin Plast Surg. 2011;38:247-277.
- Marquardt SR. Dr. Stephen R. Marquardt on the Golden Decagon and human facial beauty. interview by Dr. Gottlieb. J Clin Orthod. 2002;36:339-347.
- Veerala G, Gandikota CS, Yadagiri PK, et al. Marquardt’s facial Golden Decagon mask and its fitness with South Indian facial traits. J Clin Diagn Res. 2016;10:ZC49-ZC52.
- Holland E. Marquardt’s Phi mask: pitfalls of relying on fashion models and the golden ratio to describe a beautiful face. Aesthetic Plast Surg. 2008;32:200-208.
- Alam MK, Mohd Noor NF, Basri R, et al. Multiracial facial golden ratio and evaluation of facial appearance. PLoS One. 2015;10:e0142914.
- Kim YH. Easy facial analysis using the facial golden mask. J Craniofac Surg. 2007;18:643-649.
- Bashour M. An objective system for measuring facial attractiveness. Plast Reconstr Surg. 2006;118:757-774; discussion 775-776.
- Bartlett SP, Grossman R, Whitaker LA. Age-related changes of the craniofacial skeleton: an anthropometric and histologic analysis. Plast Reconstr Surg. 1992;90:592-600.
- Donofrio LM. Fat distribution: a morphologic study of the aging face. Dermatol Surg. 2000;26:1107-1112.
- Milutinovic J, Zelic K, Nedeljkovic N. Evaluation of facial beauty using anthropometric proportions. ScientificWorldJournal. 2014;2014:428250. doi:10.1155/2014/428250.
- Farkas LG, Hreczko TA, Kolar JC, et al. Vertical and horizontal proportions of the face in young-adult North-American Caucasians: revision of neoclassical canons. Plast Reconstr Surg. 1985;75:328-338.
- Porter JP. The average African American male face: an anthropometric analysis. Arch Facial Plast Surg. 2004;6:78-81.
- Porter JP, Olson KL. Anthropometric facial analysis of the African American woman. Arch Facial Plast Surg. 2001;3:191-197.
- Mommaerts MY, Moerenhout BA. Ideal proportions in full face front view, contemporary versus antique. J Craniomaxillofac Surg. 2011;39:107-110.
- Vegter F, Hage JJ. Clinical anthropometry and canons of the face in historical perspective. Plast Reconstr Surg. 2000;106:1090-1096.
- Wang D, Qian G, Zhang M, et al. Differences in horizontal, neoclassical facial canons in Chinese (Han) and North American Caucasian populations. Aesthetic Plast Surg. 1997;21:265-269.
- Farkas LG, Forrest CR, Litsas L. Revision of neoclassical facial canons in young adult Afro-Americans. Aesthetic Plast Surg. 2000;24:179-184.
- Coleman GG, Lindauer SJ, Tüfekçi E, et al. Influence of chin prominence on esthetic lip profile preferences. Am J Orthod Dentofacial Orthop. 2007;132:36-42.
- Popenko NA, Tripathi PB, Devcic Z, et al. A quantitative approach to determining the ideal female lip aesthetic and its effect on facial attractiveness. JAMA Facial Plast Surg. 2017;19:261-267.
- Penna V, Fricke A, Iblher N, et al. The attractive lip: a photomorphometric analysis. J Plast Reconstr Aesthet Surg. 2015;68:920-929.
- Prokopakis EP, Vlastos IM, Picavet VA, et al. The golden ratio in facial symmetry. Rhinology. 2013;51:18-21.
- Swift A, Remington K. BeautiPHIcationTM: a global approach to facial beauty. Clin Plast Surg. 2011;38:247-277.
- Marquardt SR. Dr. Stephen R. Marquardt on the Golden Decagon and human facial beauty. interview by Dr. Gottlieb. J Clin Orthod. 2002;36:339-347.
- Veerala G, Gandikota CS, Yadagiri PK, et al. Marquardt’s facial Golden Decagon mask and its fitness with South Indian facial traits. J Clin Diagn Res. 2016;10:ZC49-ZC52.
- Holland E. Marquardt’s Phi mask: pitfalls of relying on fashion models and the golden ratio to describe a beautiful face. Aesthetic Plast Surg. 2008;32:200-208.
- Alam MK, Mohd Noor NF, Basri R, et al. Multiracial facial golden ratio and evaluation of facial appearance. PLoS One. 2015;10:e0142914.
- Kim YH. Easy facial analysis using the facial golden mask. J Craniofac Surg. 2007;18:643-649.
- Bashour M. An objective system for measuring facial attractiveness. Plast Reconstr Surg. 2006;118:757-774; discussion 775-776.
- Bartlett SP, Grossman R, Whitaker LA. Age-related changes of the craniofacial skeleton: an anthropometric and histologic analysis. Plast Reconstr Surg. 1992;90:592-600.
- Donofrio LM. Fat distribution: a morphologic study of the aging face. Dermatol Surg. 2000;26:1107-1112.
Practice Points
- Canons of ideal facial dimensions have existed since antiquity and remain relevant in modern times.
- Horizontal and vertical anatomical ratios can provide a useful framework for cosmetic interventions.
- To maximize aesthetic results, alterations to individual cosmetic units should be made with thoughtful consideration of overall facial harmony.