User login
Ulipristal acetate reduced bleeding for women with fibroids
SAN ANTONIO – About half of women with uterine fibroids became amenorrheic when taking the selective progesterone receptor modulator ulipristal acetate (UPA) during a 12-week study cycle, and women taking UPA experienced significant improvement of quality of life, compared with those taking placebo.
Of those women taking 5 mg of UPA, 40.5%-42% became amenorrheic; of those taking 10 mg, 54.8%-57.3% became amenorrheic, James Liu, MD, reported at the annual meeting of the American Society for Reproductive Medicine. These results compared to amenorrhea rates of 0%-8% for women on placebo (P less than .0001 for all values).
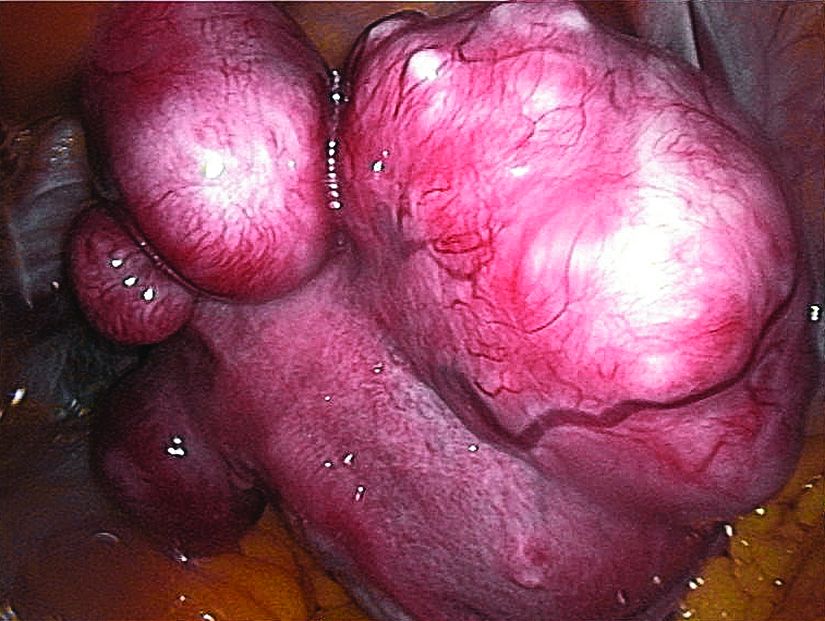
The primary aim of VENUS II was to evaluate UPA’s efficacy and safety as intermittent treatment of abnormal uterine bleeding associated with uterine fibroids. Patients received UPA at either 5 mg or 10 mg orally. Secondary efficacy measures included the maintenance effect of UPA at both doses when compared to placebo, by assessing the rate of amenorrhea and the time to amenorrhea. Another secondary measure assessed uterine fibroid–related quality of life.
Safety was assessed by tracking adverse events through both courses of treatment. The study was not powered to compare the two doses against each other, but rather compared each against placebo.
Altogether, 432 patients were randomized to one of the treatment arms, which was begun after an initial screening period of about 10-12 weeks. The first treatment course lasted 12 weeks, after which patients went off treatment for two menstrual cycles. They then began another treatment course for 12 weeks and were followed for an additional 12 weeks after treatment was stopped.
Patients were included if they were premenopausal, aged 18-50 years old, had prolonged bleeding in at least 4 of the last 6 menstrual periods, had menstrual blood loss of at least 80 mL by cycle by the alkaline hematin method, and had at least one discrete leiomyoma without a uterine size greater than 20 weeks. About two-thirds of patients were black, reflecting the higher prevalence of uterine fibroids in this population, said Dr. Liu, professor of medicine and reproductive biology at Case Western Reserve University, Cleveland.
Most patients (60%-80%) had their bleeding controlled on either dose of UPA, compared with fewer than 10% of women taking placebo.
Quality of life data, presented separately at ASRM by Lee Shulman, MD, examined the impact of treatment on patients’ physical and social activities, and also on the severity of symptoms and health-related quality of life.
“The majority of patients on UPA versus a minority of patients on placebo described their menstrual/vaginal bleeding at the end of treatment course 1 as ‘much better’ or ‘very much better,’ ” said Dr. Shulman, chief of clinical genetics in the department of ob.gyn. at Northwestern University in Evanston, Ill.
Of patients taking 5 mg UPA, 75% reported that degree of improvement, as did 87% of those on 10 mg UPA, compared with 17.9% of those taking placebo. .
Adverse events were rare, with hot flashes occurring in about 10% of women taking UPA, compared with less than 2% of those taking placebo. Headaches, fatigue, and nausea were also reported, but rates were not significantly different from rates for those taking placebo. One serious adverse event that was deemed treatment related was a uterine hemorrhage experienced by a woman taking UPA.
“Even in treatment course 1 we already had profound and statistically significant effects in symptoms across the board,” Dr. Shulman said. “How long will they last? We obviously need more data. But the study suggests that the benefits last significantly longer than that associated with leuprolide acetate.”
Ulipristal acetate is already approved by the Food and Drug Administration at a different dosage as emergency contraception. The VENUS II data support its use for women with uterine fibroids, the researchers said. “Our results provide reassurance,” Dr. Liu said. “We can conclude that UPA is effective and safe in the management of uterine fibroids in the U.S. population.”
Dr. Liu reported that he had no relevant disclosures. Dr. Shulman reported relationships with multiple pharmaceutical companies, including Allergan, which funded the VENUS II study.
[email protected]
On Twitter @karioakes
SAN ANTONIO – About half of women with uterine fibroids became amenorrheic when taking the selective progesterone receptor modulator ulipristal acetate (UPA) during a 12-week study cycle, and women taking UPA experienced significant improvement of quality of life, compared with those taking placebo.
Of those women taking 5 mg of UPA, 40.5%-42% became amenorrheic; of those taking 10 mg, 54.8%-57.3% became amenorrheic, James Liu, MD, reported at the annual meeting of the American Society for Reproductive Medicine. These results compared to amenorrhea rates of 0%-8% for women on placebo (P less than .0001 for all values).
The primary aim of VENUS II was to evaluate UPA’s efficacy and safety as intermittent treatment of abnormal uterine bleeding associated with uterine fibroids. Patients received UPA at either 5 mg or 10 mg orally. Secondary efficacy measures included the maintenance effect of UPA at both doses when compared to placebo, by assessing the rate of amenorrhea and the time to amenorrhea. Another secondary measure assessed uterine fibroid–related quality of life.
Safety was assessed by tracking adverse events through both courses of treatment. The study was not powered to compare the two doses against each other, but rather compared each against placebo.
Altogether, 432 patients were randomized to one of the treatment arms, which was begun after an initial screening period of about 10-12 weeks. The first treatment course lasted 12 weeks, after which patients went off treatment for two menstrual cycles. They then began another treatment course for 12 weeks and were followed for an additional 12 weeks after treatment was stopped.
Patients were included if they were premenopausal, aged 18-50 years old, had prolonged bleeding in at least 4 of the last 6 menstrual periods, had menstrual blood loss of at least 80 mL by cycle by the alkaline hematin method, and had at least one discrete leiomyoma without a uterine size greater than 20 weeks. About two-thirds of patients were black, reflecting the higher prevalence of uterine fibroids in this population, said Dr. Liu, professor of medicine and reproductive biology at Case Western Reserve University, Cleveland.
Most patients (60%-80%) had their bleeding controlled on either dose of UPA, compared with fewer than 10% of women taking placebo.
Quality of life data, presented separately at ASRM by Lee Shulman, MD, examined the impact of treatment on patients’ physical and social activities, and also on the severity of symptoms and health-related quality of life.
“The majority of patients on UPA versus a minority of patients on placebo described their menstrual/vaginal bleeding at the end of treatment course 1 as ‘much better’ or ‘very much better,’ ” said Dr. Shulman, chief of clinical genetics in the department of ob.gyn. at Northwestern University in Evanston, Ill.
Of patients taking 5 mg UPA, 75% reported that degree of improvement, as did 87% of those on 10 mg UPA, compared with 17.9% of those taking placebo. .
Adverse events were rare, with hot flashes occurring in about 10% of women taking UPA, compared with less than 2% of those taking placebo. Headaches, fatigue, and nausea were also reported, but rates were not significantly different from rates for those taking placebo. One serious adverse event that was deemed treatment related was a uterine hemorrhage experienced by a woman taking UPA.
“Even in treatment course 1 we already had profound and statistically significant effects in symptoms across the board,” Dr. Shulman said. “How long will they last? We obviously need more data. But the study suggests that the benefits last significantly longer than that associated with leuprolide acetate.”
Ulipristal acetate is already approved by the Food and Drug Administration at a different dosage as emergency contraception. The VENUS II data support its use for women with uterine fibroids, the researchers said. “Our results provide reassurance,” Dr. Liu said. “We can conclude that UPA is effective and safe in the management of uterine fibroids in the U.S. population.”
Dr. Liu reported that he had no relevant disclosures. Dr. Shulman reported relationships with multiple pharmaceutical companies, including Allergan, which funded the VENUS II study.
[email protected]
On Twitter @karioakes
SAN ANTONIO – About half of women with uterine fibroids became amenorrheic when taking the selective progesterone receptor modulator ulipristal acetate (UPA) during a 12-week study cycle, and women taking UPA experienced significant improvement of quality of life, compared with those taking placebo.
Of those women taking 5 mg of UPA, 40.5%-42% became amenorrheic; of those taking 10 mg, 54.8%-57.3% became amenorrheic, James Liu, MD, reported at the annual meeting of the American Society for Reproductive Medicine. These results compared to amenorrhea rates of 0%-8% for women on placebo (P less than .0001 for all values).
The primary aim of VENUS II was to evaluate UPA’s efficacy and safety as intermittent treatment of abnormal uterine bleeding associated with uterine fibroids. Patients received UPA at either 5 mg or 10 mg orally. Secondary efficacy measures included the maintenance effect of UPA at both doses when compared to placebo, by assessing the rate of amenorrhea and the time to amenorrhea. Another secondary measure assessed uterine fibroid–related quality of life.
Safety was assessed by tracking adverse events through both courses of treatment. The study was not powered to compare the two doses against each other, but rather compared each against placebo.
Altogether, 432 patients were randomized to one of the treatment arms, which was begun after an initial screening period of about 10-12 weeks. The first treatment course lasted 12 weeks, after which patients went off treatment for two menstrual cycles. They then began another treatment course for 12 weeks and were followed for an additional 12 weeks after treatment was stopped.
Patients were included if they were premenopausal, aged 18-50 years old, had prolonged bleeding in at least 4 of the last 6 menstrual periods, had menstrual blood loss of at least 80 mL by cycle by the alkaline hematin method, and had at least one discrete leiomyoma without a uterine size greater than 20 weeks. About two-thirds of patients were black, reflecting the higher prevalence of uterine fibroids in this population, said Dr. Liu, professor of medicine and reproductive biology at Case Western Reserve University, Cleveland.
Most patients (60%-80%) had their bleeding controlled on either dose of UPA, compared with fewer than 10% of women taking placebo.
Quality of life data, presented separately at ASRM by Lee Shulman, MD, examined the impact of treatment on patients’ physical and social activities, and also on the severity of symptoms and health-related quality of life.
“The majority of patients on UPA versus a minority of patients on placebo described their menstrual/vaginal bleeding at the end of treatment course 1 as ‘much better’ or ‘very much better,’ ” said Dr. Shulman, chief of clinical genetics in the department of ob.gyn. at Northwestern University in Evanston, Ill.
Of patients taking 5 mg UPA, 75% reported that degree of improvement, as did 87% of those on 10 mg UPA, compared with 17.9% of those taking placebo. .
Adverse events were rare, with hot flashes occurring in about 10% of women taking UPA, compared with less than 2% of those taking placebo. Headaches, fatigue, and nausea were also reported, but rates were not significantly different from rates for those taking placebo. One serious adverse event that was deemed treatment related was a uterine hemorrhage experienced by a woman taking UPA.
“Even in treatment course 1 we already had profound and statistically significant effects in symptoms across the board,” Dr. Shulman said. “How long will they last? We obviously need more data. But the study suggests that the benefits last significantly longer than that associated with leuprolide acetate.”
Ulipristal acetate is already approved by the Food and Drug Administration at a different dosage as emergency contraception. The VENUS II data support its use for women with uterine fibroids, the researchers said. “Our results provide reassurance,” Dr. Liu said. “We can conclude that UPA is effective and safe in the management of uterine fibroids in the U.S. population.”
Dr. Liu reported that he had no relevant disclosures. Dr. Shulman reported relationships with multiple pharmaceutical companies, including Allergan, which funded the VENUS II study.
[email protected]
On Twitter @karioakes
AT ASRM 2017
Key clinical point:
Major finding: Between 40.5% and 57.3% of women taking ulipristal acetate (UPA) achieved amenorrhea, compared with 0%-8% of women on placebo (P less than .0001).
Data source: Venus II, a phase 3 prospective, randomized, double-blind, double-dummy, placebo-controlled study that was partially parallel and partially crossover, with 432 patients.
Disclosures: Dr. Liu reported no relevant disclosures; Dr. Shulman reported financial relationships with multiple pharmaceutical companies, including Allergan, which funded the trial.
Cosmetic Corner: Dermatologists Weigh in on Men’s Moisturizers
To improve patient care and outcomes, leading dermatologists offered their recommendations on men’s moisturizers. Consideration must be given to:
- Clinique For Men Oil Control Mattifying Moisturizer
Clinique Laboratories, LLC
“I recommend this product for men with oily or combination skin. It’s very lightweight and provides good hydration benefits without leaving the skin shiny.”—Jeannette Graf, MD, Great Neck, New York
- Facial Fuel Energizing Moisture Treatment for Men
Kiehl’s
“I commonly recommend this moisturizer. The Facial Fuel line is great for most skin types and the products are moderately priced.”—Gary Goldenberg, MD, New York, New York
- Neutrogena Men Triple Protect Face Lotion With Sunscreen
Johnson & Johnson Consumer Inc
“This is a light, daily moisturizer with broad-spectrum UV protection.”—Shari Lipner, MD, PhD, New York, New York
- Triple Lipid Restore 2:4:2
SkinCeuticals
“This moisturizer has the precise lipid content needed by the skin.”— Jerome Potozkin, MD, Danville, California
Cutis invites readers to send us their recommendations. Wet skin moisturizer, lip plumper, and pigment corrector will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on men’s moisturizers. Consideration must be given to:
- Clinique For Men Oil Control Mattifying Moisturizer
Clinique Laboratories, LLC
“I recommend this product for men with oily or combination skin. It’s very lightweight and provides good hydration benefits without leaving the skin shiny.”—Jeannette Graf, MD, Great Neck, New York
- Facial Fuel Energizing Moisture Treatment for Men
Kiehl’s
“I commonly recommend this moisturizer. The Facial Fuel line is great for most skin types and the products are moderately priced.”—Gary Goldenberg, MD, New York, New York
- Neutrogena Men Triple Protect Face Lotion With Sunscreen
Johnson & Johnson Consumer Inc
“This is a light, daily moisturizer with broad-spectrum UV protection.”—Shari Lipner, MD, PhD, New York, New York
- Triple Lipid Restore 2:4:2
SkinCeuticals
“This moisturizer has the precise lipid content needed by the skin.”— Jerome Potozkin, MD, Danville, California
Cutis invites readers to send us their recommendations. Wet skin moisturizer, lip plumper, and pigment corrector will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on men’s moisturizers. Consideration must be given to:
- Clinique For Men Oil Control Mattifying Moisturizer
Clinique Laboratories, LLC
“I recommend this product for men with oily or combination skin. It’s very lightweight and provides good hydration benefits without leaving the skin shiny.”—Jeannette Graf, MD, Great Neck, New York
- Facial Fuel Energizing Moisture Treatment for Men
Kiehl’s
“I commonly recommend this moisturizer. The Facial Fuel line is great for most skin types and the products are moderately priced.”—Gary Goldenberg, MD, New York, New York
- Neutrogena Men Triple Protect Face Lotion With Sunscreen
Johnson & Johnson Consumer Inc
“This is a light, daily moisturizer with broad-spectrum UV protection.”—Shari Lipner, MD, PhD, New York, New York
- Triple Lipid Restore 2:4:2
SkinCeuticals
“This moisturizer has the precise lipid content needed by the skin.”— Jerome Potozkin, MD, Danville, California
Cutis invites readers to send us their recommendations. Wet skin moisturizer, lip plumper, and pigment corrector will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.

Debunking Acne Myths: Does Back Acne Need to Be Treated?
Myth: Back acne will clear on its own
Patients with back acne may not seek treatment options because they assume it will clear on its own; however, deep painful lesions typically require treatment by a dermatologist. Mild to moderate cases may respond to less aggressive over-the-counter treatments but also may benefit from a combination of topical and systemic antibiotic therapies. According to James Q. Del Rosso, MD, a dermatologist in Las Vegas, Nevada, “Many dermatologists believe that truncal acne vulgaris warrants use of systemic antibiotic therapy, which may not necessarily be the case, especially in patients presenting with mild to moderate acne severity.” Cases of deep inflammatory acne on the back often warrant using systemic therapies such as oral isotretinoin. These severe cases may be less responsive to standard therapies and may require repeated treatment.
Although back acne is not as cosmetically visible as facial acne, it has been associated with sexual and bodily self-consciousness in both males and females. Preliminary data from one study showed that 78% of patients with truncal acne (n=141) on the back and/or chest indicated they were definitely interested in treatment, but truncal acne was not mentioned by these patients without direct inquiry from a physician. As a result, it may be beneficial for dermatologists to ask acne patients about lesions presenting on the back and inform them that treatment options are available.
Treatment application also is a consideration for back acne. Benzoyl peroxide cleanser/wash formulations are convenient, and the foam formulation of clindamycin phosphate allows for easy use due to its spreadability, rapid penetration, and lack of residue and fabric bleaching.
It is important to inform patients that back acne can flare even during active treatment. Patients should be instructed to wear loose-fitting clothes made of cotton or other sweat-wicking fabrics when working out and to shower and change clothes immediately after. Sheets and pillowcases should be changed regularly to avoid exposure to dead skin cells and bacteria, which can exacerbate acne on the back. Backpacks and handbags also can rub against the skin on the back, causing acne to flare. As an alternative, patients should be encouraged to carry handheld bags or bags with shoulder straps to avoid irritation of the skin on the back.
Back acne: how to see clearer skin. American Academy of Dermatology website. https://www.aad.org/public/diseases/acne-and-rosacea/back-acne-how-to-see-clearer-skin. Accessed October 30, 2017.
Del Rosso JQ. Management of truncal acne vulgaris: current perspectives on treatment. Cutis. 2006;77:285-289.
Dunn LK, O’Neill JL, Feldman SR. Acne in adolescents: quality of life, self-esteem, mood, and psychological disorders. Dermatol Online J. 2011;17:1.
Hassan J, Grogan S, Clark-Carter D, et al. The individual health burden of acne: appearance-related distress in male and female adolescents and adults with back, chest and facial acne. J Health Psychol. 2009;14:1105-11118.
Myth: Back acne will clear on its own
Patients with back acne may not seek treatment options because they assume it will clear on its own; however, deep painful lesions typically require treatment by a dermatologist. Mild to moderate cases may respond to less aggressive over-the-counter treatments but also may benefit from a combination of topical and systemic antibiotic therapies. According to James Q. Del Rosso, MD, a dermatologist in Las Vegas, Nevada, “Many dermatologists believe that truncal acne vulgaris warrants use of systemic antibiotic therapy, which may not necessarily be the case, especially in patients presenting with mild to moderate acne severity.” Cases of deep inflammatory acne on the back often warrant using systemic therapies such as oral isotretinoin. These severe cases may be less responsive to standard therapies and may require repeated treatment.
Although back acne is not as cosmetically visible as facial acne, it has been associated with sexual and bodily self-consciousness in both males and females. Preliminary data from one study showed that 78% of patients with truncal acne (n=141) on the back and/or chest indicated they were definitely interested in treatment, but truncal acne was not mentioned by these patients without direct inquiry from a physician. As a result, it may be beneficial for dermatologists to ask acne patients about lesions presenting on the back and inform them that treatment options are available.
Treatment application also is a consideration for back acne. Benzoyl peroxide cleanser/wash formulations are convenient, and the foam formulation of clindamycin phosphate allows for easy use due to its spreadability, rapid penetration, and lack of residue and fabric bleaching.
It is important to inform patients that back acne can flare even during active treatment. Patients should be instructed to wear loose-fitting clothes made of cotton or other sweat-wicking fabrics when working out and to shower and change clothes immediately after. Sheets and pillowcases should be changed regularly to avoid exposure to dead skin cells and bacteria, which can exacerbate acne on the back. Backpacks and handbags also can rub against the skin on the back, causing acne to flare. As an alternative, patients should be encouraged to carry handheld bags or bags with shoulder straps to avoid irritation of the skin on the back.
Myth: Back acne will clear on its own
Patients with back acne may not seek treatment options because they assume it will clear on its own; however, deep painful lesions typically require treatment by a dermatologist. Mild to moderate cases may respond to less aggressive over-the-counter treatments but also may benefit from a combination of topical and systemic antibiotic therapies. According to James Q. Del Rosso, MD, a dermatologist in Las Vegas, Nevada, “Many dermatologists believe that truncal acne vulgaris warrants use of systemic antibiotic therapy, which may not necessarily be the case, especially in patients presenting with mild to moderate acne severity.” Cases of deep inflammatory acne on the back often warrant using systemic therapies such as oral isotretinoin. These severe cases may be less responsive to standard therapies and may require repeated treatment.
Although back acne is not as cosmetically visible as facial acne, it has been associated with sexual and bodily self-consciousness in both males and females. Preliminary data from one study showed that 78% of patients with truncal acne (n=141) on the back and/or chest indicated they were definitely interested in treatment, but truncal acne was not mentioned by these patients without direct inquiry from a physician. As a result, it may be beneficial for dermatologists to ask acne patients about lesions presenting on the back and inform them that treatment options are available.
Treatment application also is a consideration for back acne. Benzoyl peroxide cleanser/wash formulations are convenient, and the foam formulation of clindamycin phosphate allows for easy use due to its spreadability, rapid penetration, and lack of residue and fabric bleaching.
It is important to inform patients that back acne can flare even during active treatment. Patients should be instructed to wear loose-fitting clothes made of cotton or other sweat-wicking fabrics when working out and to shower and change clothes immediately after. Sheets and pillowcases should be changed regularly to avoid exposure to dead skin cells and bacteria, which can exacerbate acne on the back. Backpacks and handbags also can rub against the skin on the back, causing acne to flare. As an alternative, patients should be encouraged to carry handheld bags or bags with shoulder straps to avoid irritation of the skin on the back.
Back acne: how to see clearer skin. American Academy of Dermatology website. https://www.aad.org/public/diseases/acne-and-rosacea/back-acne-how-to-see-clearer-skin. Accessed October 30, 2017.
Del Rosso JQ. Management of truncal acne vulgaris: current perspectives on treatment. Cutis. 2006;77:285-289.
Dunn LK, O’Neill JL, Feldman SR. Acne in adolescents: quality of life, self-esteem, mood, and psychological disorders. Dermatol Online J. 2011;17:1.
Hassan J, Grogan S, Clark-Carter D, et al. The individual health burden of acne: appearance-related distress in male and female adolescents and adults with back, chest and facial acne. J Health Psychol. 2009;14:1105-11118.
Back acne: how to see clearer skin. American Academy of Dermatology website. https://www.aad.org/public/diseases/acne-and-rosacea/back-acne-how-to-see-clearer-skin. Accessed October 30, 2017.
Del Rosso JQ. Management of truncal acne vulgaris: current perspectives on treatment. Cutis. 2006;77:285-289.
Dunn LK, O’Neill JL, Feldman SR. Acne in adolescents: quality of life, self-esteem, mood, and psychological disorders. Dermatol Online J. 2011;17:1.
Hassan J, Grogan S, Clark-Carter D, et al. The individual health burden of acne: appearance-related distress in male and female adolescents and adults with back, chest and facial acne. J Health Psychol. 2009;14:1105-11118.

Improving transitions for elderly patients
Transitions are always a time of concern for hospitalists, and the transition from hospital to skilled nursing facilities (SNF) is no exception.
“During the transition and in the 30 days after discharge from the hospital to a SNF, patients are at high risk for death, rehospitalization, and high-cost health care,” said Amber Moore, MD, MPH, a hospitalist at Beth Israel Deaconess Medical Center, and instructor of medicine, Harvard Medical School. “Elderly adults are especially vulnerable because of impairments that may prevent them from participating in the discharge process and an increase in the risk that information is lost or incomplete during the care transition.”

To address this, she and several other physicians studied a novel video-conference program called Extension for Community Health Outcomes–Care Transitions (ECHO-CT) that connects an interdisciplinary hospital-based team with clinicians at SNFs to help reduce patient mortality, hospital readmission, skilled nursing facility length of stay, and 30-day health care costs.
The results of their study suggest that this intervention significantly decreased SNF length of stay, readmission rate, and costs of care, she says; the model they used is reproducible and has the potential to significantly improve care of these patients. “Our model was hospitalist run and is a mechanism to help hospitalists improve care to their patients during the transition time and beyond,” Dr. Moore said. “Furthermore, in participating in this model, hospitalists have the opportunity to better understand the challenges that face their patients after discharge and learn from postacute care providers.”
Ideally, she would like to see the model spread to other hospitals; she says hospitalists are well positioned to set up this program at their institution. “I also hope that our study highlights the incredible opportunity for improvement in the care of patients during transition from hospital to SNF and encourages hospitalists to look for innovative ways to improve care at this transition,” she said.
Reference
Moore AB, Krupp JE, Dufour AB, et al. Improving transitions to post-acute care for elderly patients using a novel video-conferencing program: ECHO-Care transitions. Am J Med. 2017 Oct;130(10):1199-204. Accessed June 6, 2017.
Transitions are always a time of concern for hospitalists, and the transition from hospital to skilled nursing facilities (SNF) is no exception.
“During the transition and in the 30 days after discharge from the hospital to a SNF, patients are at high risk for death, rehospitalization, and high-cost health care,” said Amber Moore, MD, MPH, a hospitalist at Beth Israel Deaconess Medical Center, and instructor of medicine, Harvard Medical School. “Elderly adults are especially vulnerable because of impairments that may prevent them from participating in the discharge process and an increase in the risk that information is lost or incomplete during the care transition.”
To address this, she and several other physicians studied a novel video-conference program called Extension for Community Health Outcomes–Care Transitions (ECHO-CT) that connects an interdisciplinary hospital-based team with clinicians at SNFs to help reduce patient mortality, hospital readmission, skilled nursing facility length of stay, and 30-day health care costs.
The results of their study suggest that this intervention significantly decreased SNF length of stay, readmission rate, and costs of care, she says; the model they used is reproducible and has the potential to significantly improve care of these patients. “Our model was hospitalist run and is a mechanism to help hospitalists improve care to their patients during the transition time and beyond,” Dr. Moore said. “Furthermore, in participating in this model, hospitalists have the opportunity to better understand the challenges that face their patients after discharge and learn from postacute care providers.”
Ideally, she would like to see the model spread to other hospitals; she says hospitalists are well positioned to set up this program at their institution. “I also hope that our study highlights the incredible opportunity for improvement in the care of patients during transition from hospital to SNF and encourages hospitalists to look for innovative ways to improve care at this transition,” she said.
Reference
Moore AB, Krupp JE, Dufour AB, et al. Improving transitions to post-acute care for elderly patients using a novel video-conferencing program: ECHO-Care transitions. Am J Med. 2017 Oct;130(10):1199-204. Accessed June 6, 2017.
Transitions are always a time of concern for hospitalists, and the transition from hospital to skilled nursing facilities (SNF) is no exception.
“During the transition and in the 30 days after discharge from the hospital to a SNF, patients are at high risk for death, rehospitalization, and high-cost health care,” said Amber Moore, MD, MPH, a hospitalist at Beth Israel Deaconess Medical Center, and instructor of medicine, Harvard Medical School. “Elderly adults are especially vulnerable because of impairments that may prevent them from participating in the discharge process and an increase in the risk that information is lost or incomplete during the care transition.”
To address this, she and several other physicians studied a novel video-conference program called Extension for Community Health Outcomes–Care Transitions (ECHO-CT) that connects an interdisciplinary hospital-based team with clinicians at SNFs to help reduce patient mortality, hospital readmission, skilled nursing facility length of stay, and 30-day health care costs.
The results of their study suggest that this intervention significantly decreased SNF length of stay, readmission rate, and costs of care, she says; the model they used is reproducible and has the potential to significantly improve care of these patients. “Our model was hospitalist run and is a mechanism to help hospitalists improve care to their patients during the transition time and beyond,” Dr. Moore said. “Furthermore, in participating in this model, hospitalists have the opportunity to better understand the challenges that face their patients after discharge and learn from postacute care providers.”
Ideally, she would like to see the model spread to other hospitals; she says hospitalists are well positioned to set up this program at their institution. “I also hope that our study highlights the incredible opportunity for improvement in the care of patients during transition from hospital to SNF and encourages hospitalists to look for innovative ways to improve care at this transition,” she said.
Reference
Moore AB, Krupp JE, Dufour AB, et al. Improving transitions to post-acute care for elderly patients using a novel video-conferencing program: ECHO-Care transitions. Am J Med. 2017 Oct;130(10):1199-204. Accessed June 6, 2017.
Some measures to control HAI sound better than they perform
SAN DIEGO – Some almost universally accepted measures against hospital-acquired infections are more costly, annoying, and time consuming than they’re worth, presenters agreed during a panel discussion at the annual clinical congress of the American College of Surgeons.
Intra-abdominal antibiotic irrigation, chlorhexidine bathing, and even postsurgical antibiotic infusions have not consistently been shown to reduce infections. These measures do, however, ratchet up costs and can contribute to antibiotic resistance.
Some of these and other measures to prevent nosocomial infections may indeed reduce the risk, but the gain is small, said Charles H. Cook, MD.
“Chlorhexidine bathing, for example, is touted by many as a panacea for all the infections we’re talking about,” said Dr. Cook, a critical care surgeon at Beth Israel Deaconess Medical Center, New York. “A recent meta-analysis in critical care units did find a reduced relative risk of 0.44 for central line bloodstream infections. But you needed to bathe 360 patients to prevent one infection. It’s what I call a long run for a short slide.”
Therese Duane, MD, FACS, agreed. A surgeon at the John Peter Smith Hospital, Ft. Worth, Tex., Dr. Duane reviewed three different guidelines for the prevention of surgical site infections: the ACS and Surgical Infection Society, the World Health Organization, and the Centers for Disease Control and Prevention. In looking for similarities between the documents, she said she found several well-accepted practices that just aren’t supported by good data.
Presurgical antimicrobial infusions got a strong thumbs-up from all the groups, but only under a very specific circumstance: The medication has to be administered well in advance of surgery for it to be effective.
“Your goal is to get the appropriate concentration into the tissues by the time of incision,” Dr. Duane said. “It takes time to get there – if you give it after the incision, you have bleeding and cellular death, and the antimicrobials cannot get to that incision and do their job. If I’m starting a case and they haven’t been given, I don’t ever start them after the incision, because then you have all of the risks and none of the benefits. In my opinion, we need to move to no further antimicrobials once the incision or case is over because it serves no purpose and is inconsistent with good antibiotic stewardship.”
Adhesive drapes got a resounding “eh” from the guidelines, Dr. Duane said. “You really do not need them. They’re expensive and they’re not improving outcomes, so don’t waste your time or money. We need to think about minimizing what isn’t helpful and maximizing the things that are worthwhile. That’s the way to practice good socially responsible surgery without breaking the bank,” she said.
Antimicrobial sutures got weak recommendations, Dr. Duane said. The evidence supporting their use was not very strong, although she said she feels triclosan-coated sutures are helpful in all kinds of surgery. Preoperative showering with an antiseptic received strong support, with alcohol-containing preps superior to chlorhexidine, which is better than povidone-iodine–containing solutions.
Deep-space irrigation with aqueous iodophor also received a weak recommendation, but Dr. Duane said the evidence does not support the use of antibiotic-containing irrigation in either the abdomen or the incision. “And the guidelines came out strongly against using antimicrobial agents on the incision,” she said. None of the guidelines issued a recommendation for or against antimicrobial dressings.
Protocolized infection-control bundles are a very great help in reducing the incidence of surgical site infections, Dr. Duane added. “They increase attention to detail and decrease the rates of infection.”
Dr. Cook agreed. “Central line bundles are one of the things that work” for line-associated bloodstream infections, he said. Since their large-scale adoption, mortality from these infections has dropped significantly; it was hovering around 28,000 per year in the mid-2000s, he said. “That’s about how many men die from prostate cancer every year.”
Central line infections are very costly too, he added – around $46,000 per event. “That comes to around $2 billion in direct and indirect costs every year.”
A 2006 study demonstrated the efficacy of central line bundles in the fight against these potentially devastating infections.
The bundled intervention comprised hand washing, using full-barrier precautions during the insertion of central venous catheters, cleaning the skin with chlorhexidine, avoiding the femoral site if possible, and removing unnecessary catheters. The median rate of catheter-related bloodstream infections per 1,000 catheter-days decreased from 2.7 at baseline to 0 at 3 months after implementation of the study intervention.
Antibiotic-coated or impregnated catheters do not work as well. A 2016 Cochrane review of 57 studies determined that the devices didn’t improve sepsis, all-cause mortality, or catheter-related local infections.
The jury may still be out on coated dressings or securing devices, however. Another Cochrane review, of 22 studies, found a 40% decrease in central line–associated bloodstream infections with these items. “There was moderate evidence that tip colonization was reduced, but the authors said more research is needed.”
The evidence looks stronger for alcohol-impregnated port protectors, Dr. Cook said. Two studies in particular support their use. In an oncology unit, the rate of these infections dropped from 2.3 to 0.3 per 1,000 catheter days after the port protectors were instituted.
In the second study, infection rates declined from 1.43 to 0.69 per 1,000 line-days after the protectors came on board.
“The advantage was seen mostly in ICUs, so the recommendations are to use them there,” Dr. Cook said.
Neither Dr. Cook nor Dr. Duane had any relevant financial disclosures.
[email protected]
On Twitter @Alz Gal
SAN DIEGO – Some almost universally accepted measures against hospital-acquired infections are more costly, annoying, and time consuming than they’re worth, presenters agreed during a panel discussion at the annual clinical congress of the American College of Surgeons.
Intra-abdominal antibiotic irrigation, chlorhexidine bathing, and even postsurgical antibiotic infusions have not consistently been shown to reduce infections. These measures do, however, ratchet up costs and can contribute to antibiotic resistance.
Some of these and other measures to prevent nosocomial infections may indeed reduce the risk, but the gain is small, said Charles H. Cook, MD.
“Chlorhexidine bathing, for example, is touted by many as a panacea for all the infections we’re talking about,” said Dr. Cook, a critical care surgeon at Beth Israel Deaconess Medical Center, New York. “A recent meta-analysis in critical care units did find a reduced relative risk of 0.44 for central line bloodstream infections. But you needed to bathe 360 patients to prevent one infection. It’s what I call a long run for a short slide.”
Therese Duane, MD, FACS, agreed. A surgeon at the John Peter Smith Hospital, Ft. Worth, Tex., Dr. Duane reviewed three different guidelines for the prevention of surgical site infections: the ACS and Surgical Infection Society, the World Health Organization, and the Centers for Disease Control and Prevention. In looking for similarities between the documents, she said she found several well-accepted practices that just aren’t supported by good data.
Presurgical antimicrobial infusions got a strong thumbs-up from all the groups, but only under a very specific circumstance: The medication has to be administered well in advance of surgery for it to be effective.
“Your goal is to get the appropriate concentration into the tissues by the time of incision,” Dr. Duane said. “It takes time to get there – if you give it after the incision, you have bleeding and cellular death, and the antimicrobials cannot get to that incision and do their job. If I’m starting a case and they haven’t been given, I don’t ever start them after the incision, because then you have all of the risks and none of the benefits. In my opinion, we need to move to no further antimicrobials once the incision or case is over because it serves no purpose and is inconsistent with good antibiotic stewardship.”
Adhesive drapes got a resounding “eh” from the guidelines, Dr. Duane said. “You really do not need them. They’re expensive and they’re not improving outcomes, so don’t waste your time or money. We need to think about minimizing what isn’t helpful and maximizing the things that are worthwhile. That’s the way to practice good socially responsible surgery without breaking the bank,” she said.
Antimicrobial sutures got weak recommendations, Dr. Duane said. The evidence supporting their use was not very strong, although she said she feels triclosan-coated sutures are helpful in all kinds of surgery. Preoperative showering with an antiseptic received strong support, with alcohol-containing preps superior to chlorhexidine, which is better than povidone-iodine–containing solutions.
Deep-space irrigation with aqueous iodophor also received a weak recommendation, but Dr. Duane said the evidence does not support the use of antibiotic-containing irrigation in either the abdomen or the incision. “And the guidelines came out strongly against using antimicrobial agents on the incision,” she said. None of the guidelines issued a recommendation for or against antimicrobial dressings.
Protocolized infection-control bundles are a very great help in reducing the incidence of surgical site infections, Dr. Duane added. “They increase attention to detail and decrease the rates of infection.”
Dr. Cook agreed. “Central line bundles are one of the things that work” for line-associated bloodstream infections, he said. Since their large-scale adoption, mortality from these infections has dropped significantly; it was hovering around 28,000 per year in the mid-2000s, he said. “That’s about how many men die from prostate cancer every year.”
Central line infections are very costly too, he added – around $46,000 per event. “That comes to around $2 billion in direct and indirect costs every year.”
A 2006 study demonstrated the efficacy of central line bundles in the fight against these potentially devastating infections.
The bundled intervention comprised hand washing, using full-barrier precautions during the insertion of central venous catheters, cleaning the skin with chlorhexidine, avoiding the femoral site if possible, and removing unnecessary catheters. The median rate of catheter-related bloodstream infections per 1,000 catheter-days decreased from 2.7 at baseline to 0 at 3 months after implementation of the study intervention.
Antibiotic-coated or impregnated catheters do not work as well. A 2016 Cochrane review of 57 studies determined that the devices didn’t improve sepsis, all-cause mortality, or catheter-related local infections.
The jury may still be out on coated dressings or securing devices, however. Another Cochrane review, of 22 studies, found a 40% decrease in central line–associated bloodstream infections with these items. “There was moderate evidence that tip colonization was reduced, but the authors said more research is needed.”
The evidence looks stronger for alcohol-impregnated port protectors, Dr. Cook said. Two studies in particular support their use. In an oncology unit, the rate of these infections dropped from 2.3 to 0.3 per 1,000 catheter days after the port protectors were instituted.
In the second study, infection rates declined from 1.43 to 0.69 per 1,000 line-days after the protectors came on board.
“The advantage was seen mostly in ICUs, so the recommendations are to use them there,” Dr. Cook said.
Neither Dr. Cook nor Dr. Duane had any relevant financial disclosures.
[email protected]
On Twitter @Alz Gal
SAN DIEGO – Some almost universally accepted measures against hospital-acquired infections are more costly, annoying, and time consuming than they’re worth, presenters agreed during a panel discussion at the annual clinical congress of the American College of Surgeons.
Intra-abdominal antibiotic irrigation, chlorhexidine bathing, and even postsurgical antibiotic infusions have not consistently been shown to reduce infections. These measures do, however, ratchet up costs and can contribute to antibiotic resistance.
Some of these and other measures to prevent nosocomial infections may indeed reduce the risk, but the gain is small, said Charles H. Cook, MD.
“Chlorhexidine bathing, for example, is touted by many as a panacea for all the infections we’re talking about,” said Dr. Cook, a critical care surgeon at Beth Israel Deaconess Medical Center, New York. “A recent meta-analysis in critical care units did find a reduced relative risk of 0.44 for central line bloodstream infections. But you needed to bathe 360 patients to prevent one infection. It’s what I call a long run for a short slide.”
Therese Duane, MD, FACS, agreed. A surgeon at the John Peter Smith Hospital, Ft. Worth, Tex., Dr. Duane reviewed three different guidelines for the prevention of surgical site infections: the ACS and Surgical Infection Society, the World Health Organization, and the Centers for Disease Control and Prevention. In looking for similarities between the documents, she said she found several well-accepted practices that just aren’t supported by good data.
Presurgical antimicrobial infusions got a strong thumbs-up from all the groups, but only under a very specific circumstance: The medication has to be administered well in advance of surgery for it to be effective.
“Your goal is to get the appropriate concentration into the tissues by the time of incision,” Dr. Duane said. “It takes time to get there – if you give it after the incision, you have bleeding and cellular death, and the antimicrobials cannot get to that incision and do their job. If I’m starting a case and they haven’t been given, I don’t ever start them after the incision, because then you have all of the risks and none of the benefits. In my opinion, we need to move to no further antimicrobials once the incision or case is over because it serves no purpose and is inconsistent with good antibiotic stewardship.”
Adhesive drapes got a resounding “eh” from the guidelines, Dr. Duane said. “You really do not need them. They’re expensive and they’re not improving outcomes, so don’t waste your time or money. We need to think about minimizing what isn’t helpful and maximizing the things that are worthwhile. That’s the way to practice good socially responsible surgery without breaking the bank,” she said.
Antimicrobial sutures got weak recommendations, Dr. Duane said. The evidence supporting their use was not very strong, although she said she feels triclosan-coated sutures are helpful in all kinds of surgery. Preoperative showering with an antiseptic received strong support, with alcohol-containing preps superior to chlorhexidine, which is better than povidone-iodine–containing solutions.
Deep-space irrigation with aqueous iodophor also received a weak recommendation, but Dr. Duane said the evidence does not support the use of antibiotic-containing irrigation in either the abdomen or the incision. “And the guidelines came out strongly against using antimicrobial agents on the incision,” she said. None of the guidelines issued a recommendation for or against antimicrobial dressings.
Protocolized infection-control bundles are a very great help in reducing the incidence of surgical site infections, Dr. Duane added. “They increase attention to detail and decrease the rates of infection.”
Dr. Cook agreed. “Central line bundles are one of the things that work” for line-associated bloodstream infections, he said. Since their large-scale adoption, mortality from these infections has dropped significantly; it was hovering around 28,000 per year in the mid-2000s, he said. “That’s about how many men die from prostate cancer every year.”
Central line infections are very costly too, he added – around $46,000 per event. “That comes to around $2 billion in direct and indirect costs every year.”
A 2006 study demonstrated the efficacy of central line bundles in the fight against these potentially devastating infections.
The bundled intervention comprised hand washing, using full-barrier precautions during the insertion of central venous catheters, cleaning the skin with chlorhexidine, avoiding the femoral site if possible, and removing unnecessary catheters. The median rate of catheter-related bloodstream infections per 1,000 catheter-days decreased from 2.7 at baseline to 0 at 3 months after implementation of the study intervention.
Antibiotic-coated or impregnated catheters do not work as well. A 2016 Cochrane review of 57 studies determined that the devices didn’t improve sepsis, all-cause mortality, or catheter-related local infections.
The jury may still be out on coated dressings or securing devices, however. Another Cochrane review, of 22 studies, found a 40% decrease in central line–associated bloodstream infections with these items. “There was moderate evidence that tip colonization was reduced, but the authors said more research is needed.”
The evidence looks stronger for alcohol-impregnated port protectors, Dr. Cook said. Two studies in particular support their use. In an oncology unit, the rate of these infections dropped from 2.3 to 0.3 per 1,000 catheter days after the port protectors were instituted.
In the second study, infection rates declined from 1.43 to 0.69 per 1,000 line-days after the protectors came on board.
“The advantage was seen mostly in ICUs, so the recommendations are to use them there,” Dr. Cook said.
Neither Dr. Cook nor Dr. Duane had any relevant financial disclosures.
[email protected]
On Twitter @Alz Gal
FROM THE ACS CLINICAL CONGRESS
Which Patients Respond Best to Repeat Epilepsy Surgery?
Repeat surgery is worth considering in patients with intractable epilepsy if they continue to have debilitating episodes after the first procedure, according to a meta-analysis and systematic review that looked at 782 patients from 36 studies. But the success of repeat resection is dependent on several positive and negative predictive factors.
- Krucoff et al conducted the first quantitative meta-analysis of theresearch literature from the last 30 years to determine the rate of successful repeat surgeries and to find predictors.
- Congruent electrophysiology data was a better predictor of seizure freedom when compared to noncongruent data, with an odds ratio (OR) of 3.6.
- Freedom from seizures after repeat surgery was better predicted for lesional than nonlesional epilepsy (OR, 3.2).
- Another predictor of seizure freedom was surgical limitations, when compared to disease-related factors that had been associated with failure of the first surgery. (OR, 2.6).
- 58% of patients were seizure free after repeat resection if they had at least one of these predictive factors.
Krucoff MO, Chan AY, Harward SC, et al. Rates and predictors of success and failure in repeat epilepsy surgery: a meta-analysis and systematic review. [Published online ahead of print October 10, 2017] Epilepsia. doi: 10.1111/epi.13920.
Repeat surgery is worth considering in patients with intractable epilepsy if they continue to have debilitating episodes after the first procedure, according to a meta-analysis and systematic review that looked at 782 patients from 36 studies. But the success of repeat resection is dependent on several positive and negative predictive factors.
- Krucoff et al conducted the first quantitative meta-analysis of theresearch literature from the last 30 years to determine the rate of successful repeat surgeries and to find predictors.
- Congruent electrophysiology data was a better predictor of seizure freedom when compared to noncongruent data, with an odds ratio (OR) of 3.6.
- Freedom from seizures after repeat surgery was better predicted for lesional than nonlesional epilepsy (OR, 3.2).
- Another predictor of seizure freedom was surgical limitations, when compared to disease-related factors that had been associated with failure of the first surgery. (OR, 2.6).
- 58% of patients were seizure free after repeat resection if they had at least one of these predictive factors.
Krucoff MO, Chan AY, Harward SC, et al. Rates and predictors of success and failure in repeat epilepsy surgery: a meta-analysis and systematic review. [Published online ahead of print October 10, 2017] Epilepsia. doi: 10.1111/epi.13920.
Repeat surgery is worth considering in patients with intractable epilepsy if they continue to have debilitating episodes after the first procedure, according to a meta-analysis and systematic review that looked at 782 patients from 36 studies. But the success of repeat resection is dependent on several positive and negative predictive factors.
- Krucoff et al conducted the first quantitative meta-analysis of theresearch literature from the last 30 years to determine the rate of successful repeat surgeries and to find predictors.
- Congruent electrophysiology data was a better predictor of seizure freedom when compared to noncongruent data, with an odds ratio (OR) of 3.6.
- Freedom from seizures after repeat surgery was better predicted for lesional than nonlesional epilepsy (OR, 3.2).
- Another predictor of seizure freedom was surgical limitations, when compared to disease-related factors that had been associated with failure of the first surgery. (OR, 2.6).
- 58% of patients were seizure free after repeat resection if they had at least one of these predictive factors.
Krucoff MO, Chan AY, Harward SC, et al. Rates and predictors of success and failure in repeat epilepsy surgery: a meta-analysis and systematic review. [Published online ahead of print October 10, 2017] Epilepsia. doi: 10.1111/epi.13920.
VIDEO: Smartphones could ‘democratize’ EEG
SAN DIEGO – Epilepsy is a common condition, but many people live in areas too poor or underserved to benefit from electroencephalogram (EEG) testing to confirm a diagnosis. Modern technology may be changing that. Smartphones wirelessly linked to headsets can collect EEG data and potentially serve as an alternate platform for diagnosis of epilepsy.
To test the efficacy of one such system, Farrah Mateen, MD, PhD, a neurologist at Massachusetts General Hospital, Boston, conducted a study in the remote, mountainous Kingdom of Bhutan, which lies between India and China. Her group compared the signals between a smartphone EEG and standard EEG.
The sensitivity was low, at around 40%, but the device’s specificity was 90%-95%. For now, the device performs better as a confirmatory test than as a screening device, but the researchers hope to adjust the lead location to improve sensitivity.
The headset used was designed for use by video gamers, and at a cost of less than $300, it represents a low-cost entry into portable EEG.
Dr. Mateen discussed the smartphone EEG study in a video interview at the annual meeting of the American Neurological Association.
SAN DIEGO – Epilepsy is a common condition, but many people live in areas too poor or underserved to benefit from electroencephalogram (EEG) testing to confirm a diagnosis. Modern technology may be changing that. Smartphones wirelessly linked to headsets can collect EEG data and potentially serve as an alternate platform for diagnosis of epilepsy.
To test the efficacy of one such system, Farrah Mateen, MD, PhD, a neurologist at Massachusetts General Hospital, Boston, conducted a study in the remote, mountainous Kingdom of Bhutan, which lies between India and China. Her group compared the signals between a smartphone EEG and standard EEG.
The sensitivity was low, at around 40%, but the device’s specificity was 90%-95%. For now, the device performs better as a confirmatory test than as a screening device, but the researchers hope to adjust the lead location to improve sensitivity.
The headset used was designed for use by video gamers, and at a cost of less than $300, it represents a low-cost entry into portable EEG.
Dr. Mateen discussed the smartphone EEG study in a video interview at the annual meeting of the American Neurological Association.
SAN DIEGO – Epilepsy is a common condition, but many people live in areas too poor or underserved to benefit from electroencephalogram (EEG) testing to confirm a diagnosis. Modern technology may be changing that. Smartphones wirelessly linked to headsets can collect EEG data and potentially serve as an alternate platform for diagnosis of epilepsy.
To test the efficacy of one such system, Farrah Mateen, MD, PhD, a neurologist at Massachusetts General Hospital, Boston, conducted a study in the remote, mountainous Kingdom of Bhutan, which lies between India and China. Her group compared the signals between a smartphone EEG and standard EEG.
The sensitivity was low, at around 40%, but the device’s specificity was 90%-95%. For now, the device performs better as a confirmatory test than as a screening device, but the researchers hope to adjust the lead location to improve sensitivity.
The headset used was designed for use by video gamers, and at a cost of less than $300, it represents a low-cost entry into portable EEG.
Dr. Mateen discussed the smartphone EEG study in a video interview at the annual meeting of the American Neurological Association.
AT ANA 2017

Laser Interstitial Thermal Therapy Versus Open Epilepsy Surgery
Magnetic resonance-guided laser interstitial therapy (MRgLITT) is as efficacious as open surgery for patients with medically intractable epilepsy, according to a recent review of the research literature.
- MRgLITT can effectively ablate seizure foci without disrupting neuropsychological functioning.
- The procedure allows clinicians to receive real-time feedback and lets them monitor tissue ablation.
- By contrast, open surgery can control seizures but risks complications, including cognitive problems and neurologic deficits.
- Patients who undergo MRgLITT will likely spend less time in the hospital and experience less postoperative pain, when compared to open surgical procedures.
- Although the research comparing open surgery to laser interstitial therapy suggests the latter offers advantages, the review points out that the research under consideration involved small sample sizes.
- Another limitation of the available studies is that they do not report the long-term outcomes of MRgLITT.
Shukla ND, Ho AL, Pendharkar AV, Sussman ES, Halpern CH. Laser interstitial thermal therapy for the treatment of epilepsy: evidence to date. Neuropsychiatr Dis Treat. 2017;13:2469-2475.
Magnetic resonance-guided laser interstitial therapy (MRgLITT) is as efficacious as open surgery for patients with medically intractable epilepsy, according to a recent review of the research literature.
- MRgLITT can effectively ablate seizure foci without disrupting neuropsychological functioning.
- The procedure allows clinicians to receive real-time feedback and lets them monitor tissue ablation.
- By contrast, open surgery can control seizures but risks complications, including cognitive problems and neurologic deficits.
- Patients who undergo MRgLITT will likely spend less time in the hospital and experience less postoperative pain, when compared to open surgical procedures.
- Although the research comparing open surgery to laser interstitial therapy suggests the latter offers advantages, the review points out that the research under consideration involved small sample sizes.
- Another limitation of the available studies is that they do not report the long-term outcomes of MRgLITT.
Shukla ND, Ho AL, Pendharkar AV, Sussman ES, Halpern CH. Laser interstitial thermal therapy for the treatment of epilepsy: evidence to date. Neuropsychiatr Dis Treat. 2017;13:2469-2475.
Magnetic resonance-guided laser interstitial therapy (MRgLITT) is as efficacious as open surgery for patients with medically intractable epilepsy, according to a recent review of the research literature.
- MRgLITT can effectively ablate seizure foci without disrupting neuropsychological functioning.
- The procedure allows clinicians to receive real-time feedback and lets them monitor tissue ablation.
- By contrast, open surgery can control seizures but risks complications, including cognitive problems and neurologic deficits.
- Patients who undergo MRgLITT will likely spend less time in the hospital and experience less postoperative pain, when compared to open surgical procedures.
- Although the research comparing open surgery to laser interstitial therapy suggests the latter offers advantages, the review points out that the research under consideration involved small sample sizes.
- Another limitation of the available studies is that they do not report the long-term outcomes of MRgLITT.
Shukla ND, Ho AL, Pendharkar AV, Sussman ES, Halpern CH. Laser interstitial thermal therapy for the treatment of epilepsy: evidence to date. Neuropsychiatr Dis Treat. 2017;13:2469-2475.
Should Adult AED Studies Determine Pediatric Usage?
Efficacy data from adult clinical trials of antiepileptic drugs (AEDs) can be used to determine the efficacy of these agents in children according to a recent analysis published in Epilepsia.
- The need for efficacious AEDs for children is urgent given the fact that epileptic seizures are the most common serious neurological problem in this population.
- The pathophysiology of focal epilepsy in children is similar to that found in adults, based on anatomical and neurophysiological evidence.
- The structural and physiological features that underlie seizures in adults and children aged 2 years and older are similar and justify extrapolation of efficacy data from adults to children.
- However, these similarities do not justify extrapolating pharmacokinetic, tolerability, and safety data from adults to children.
- Similarly, published data do not allow one to assume that long-term follow-up in children on AEDs will be the same as follow-up results in adults.
Pellock JM, Arzimanoglou A, D’Cruz O, Holmes GL, Nordli D, Shinnar S; Pediatric Epilepsy Academic Consortium for Extrapolation. Extrapolating evidence of antiepileptic drug efficacy in adults to children ≥2 years of age with focal seizures: The case for disease similarity. Epilepsia. 2017;58(10):1686-1696.
Efficacy data from adult clinical trials of antiepileptic drugs (AEDs) can be used to determine the efficacy of these agents in children according to a recent analysis published in Epilepsia.
- The need for efficacious AEDs for children is urgent given the fact that epileptic seizures are the most common serious neurological problem in this population.
- The pathophysiology of focal epilepsy in children is similar to that found in adults, based on anatomical and neurophysiological evidence.
- The structural and physiological features that underlie seizures in adults and children aged 2 years and older are similar and justify extrapolation of efficacy data from adults to children.
- However, these similarities do not justify extrapolating pharmacokinetic, tolerability, and safety data from adults to children.
- Similarly, published data do not allow one to assume that long-term follow-up in children on AEDs will be the same as follow-up results in adults.
Pellock JM, Arzimanoglou A, D’Cruz O, Holmes GL, Nordli D, Shinnar S; Pediatric Epilepsy Academic Consortium for Extrapolation. Extrapolating evidence of antiepileptic drug efficacy in adults to children ≥2 years of age with focal seizures: The case for disease similarity. Epilepsia. 2017;58(10):1686-1696.
Efficacy data from adult clinical trials of antiepileptic drugs (AEDs) can be used to determine the efficacy of these agents in children according to a recent analysis published in Epilepsia.
- The need for efficacious AEDs for children is urgent given the fact that epileptic seizures are the most common serious neurological problem in this population.
- The pathophysiology of focal epilepsy in children is similar to that found in adults, based on anatomical and neurophysiological evidence.
- The structural and physiological features that underlie seizures in adults and children aged 2 years and older are similar and justify extrapolation of efficacy data from adults to children.
- However, these similarities do not justify extrapolating pharmacokinetic, tolerability, and safety data from adults to children.
- Similarly, published data do not allow one to assume that long-term follow-up in children on AEDs will be the same as follow-up results in adults.
Pellock JM, Arzimanoglou A, D’Cruz O, Holmes GL, Nordli D, Shinnar S; Pediatric Epilepsy Academic Consortium for Extrapolation. Extrapolating evidence of antiepileptic drug efficacy in adults to children ≥2 years of age with focal seizures: The case for disease similarity. Epilepsia. 2017;58(10):1686-1696.
Identifying the right database
Editor’s note: The Society of Hospital Medicine’s (SHM’s) Physician in Training Committee launched a scholarship program in 2015 for medical students to help transform health care and revolutionize patient care. The program has been expanded for the 2017-2018 year, offering two options for students to receive funding and engage in scholarly work during their first, second, and third years of medical school. As a part of the longitudinal (18-month) program, recipients are required to write about their experience on a monthly basis.
Vanderbilt University Medical Center will be converting to the most common electronic medical record (EMR) systems used today: Epic. Until that time, Vanderbilt used a homegrown system to keep track of patient data. The “system” was actual comprised of a few separate programs that integrated data, depending on the functions being accessed and who was accessing them.

For many research projects across the hospital, including my own, we are going to be limiting ourselves to data from the time period when our homegrown EMR was functioning. This is thinking a few steps ahead, but it would be interesting to see if our model, once validated, performed similarly in a new EMR environment. Unfortunately, this is thinking a few too many steps ahead for me, as I will have graduated (hopefully) by the time the new EMR is up and running reliably enough for EMR-based research like this project.
The first step in our study was identifying the right database to use, and now the next step will be extracting the data we need. Moving forward, I am continuing to work with my mentors, Dr. Eduard Vasilevskis and Dr. Jesse Ehrenfeld closely. We resubmitted our IRB application now that we have identified how we can pull the data we need, and we identified a few specialized patient populations for whom a separate scoring tool might be useful (e.g., stroke patients). I am looking forward to learning the particulars how our dataset will be built. The potential for finding the answers to many patient-care questions probably lies in the EMR data we already have, but you need to know how to get them to study them.
Monisha Bhatia, a native of Nashville, Tenn., is a fourth-year medical student at Vanderbilt University in Nashville. She is hoping to pursue either a residency in internal medicine or a combined internal medicine/emergency medicine program. Prior to medical school, she completed a JD/MPH program at Boston University, and she hopes to use her legal training in working with regulatory authorities to improve access to health care for all Americans.
Editor’s note: The Society of Hospital Medicine’s (SHM’s) Physician in Training Committee launched a scholarship program in 2015 for medical students to help transform health care and revolutionize patient care. The program has been expanded for the 2017-2018 year, offering two options for students to receive funding and engage in scholarly work during their first, second, and third years of medical school. As a part of the longitudinal (18-month) program, recipients are required to write about their experience on a monthly basis.
Vanderbilt University Medical Center will be converting to the most common electronic medical record (EMR) systems used today: Epic. Until that time, Vanderbilt used a homegrown system to keep track of patient data. The “system” was actual comprised of a few separate programs that integrated data, depending on the functions being accessed and who was accessing them.
For many research projects across the hospital, including my own, we are going to be limiting ourselves to data from the time period when our homegrown EMR was functioning. This is thinking a few steps ahead, but it would be interesting to see if our model, once validated, performed similarly in a new EMR environment. Unfortunately, this is thinking a few too many steps ahead for me, as I will have graduated (hopefully) by the time the new EMR is up and running reliably enough for EMR-based research like this project.
The first step in our study was identifying the right database to use, and now the next step will be extracting the data we need. Moving forward, I am continuing to work with my mentors, Dr. Eduard Vasilevskis and Dr. Jesse Ehrenfeld closely. We resubmitted our IRB application now that we have identified how we can pull the data we need, and we identified a few specialized patient populations for whom a separate scoring tool might be useful (e.g., stroke patients). I am looking forward to learning the particulars how our dataset will be built. The potential for finding the answers to many patient-care questions probably lies in the EMR data we already have, but you need to know how to get them to study them.
Monisha Bhatia, a native of Nashville, Tenn., is a fourth-year medical student at Vanderbilt University in Nashville. She is hoping to pursue either a residency in internal medicine or a combined internal medicine/emergency medicine program. Prior to medical school, she completed a JD/MPH program at Boston University, and she hopes to use her legal training in working with regulatory authorities to improve access to health care for all Americans.
Editor’s note: The Society of Hospital Medicine’s (SHM’s) Physician in Training Committee launched a scholarship program in 2015 for medical students to help transform health care and revolutionize patient care. The program has been expanded for the 2017-2018 year, offering two options for students to receive funding and engage in scholarly work during their first, second, and third years of medical school. As a part of the longitudinal (18-month) program, recipients are required to write about their experience on a monthly basis.
Vanderbilt University Medical Center will be converting to the most common electronic medical record (EMR) systems used today: Epic. Until that time, Vanderbilt used a homegrown system to keep track of patient data. The “system” was actual comprised of a few separate programs that integrated data, depending on the functions being accessed and who was accessing them.
For many research projects across the hospital, including my own, we are going to be limiting ourselves to data from the time period when our homegrown EMR was functioning. This is thinking a few steps ahead, but it would be interesting to see if our model, once validated, performed similarly in a new EMR environment. Unfortunately, this is thinking a few too many steps ahead for me, as I will have graduated (hopefully) by the time the new EMR is up and running reliably enough for EMR-based research like this project.
The first step in our study was identifying the right database to use, and now the next step will be extracting the data we need. Moving forward, I am continuing to work with my mentors, Dr. Eduard Vasilevskis and Dr. Jesse Ehrenfeld closely. We resubmitted our IRB application now that we have identified how we can pull the data we need, and we identified a few specialized patient populations for whom a separate scoring tool might be useful (e.g., stroke patients). I am looking forward to learning the particulars how our dataset will be built. The potential for finding the answers to many patient-care questions probably lies in the EMR data we already have, but you need to know how to get them to study them.
Monisha Bhatia, a native of Nashville, Tenn., is a fourth-year medical student at Vanderbilt University in Nashville. She is hoping to pursue either a residency in internal medicine or a combined internal medicine/emergency medicine program. Prior to medical school, she completed a JD/MPH program at Boston University, and she hopes to use her legal training in working with regulatory authorities to improve access to health care for all Americans.