User login
Insurance status linked to survival in FL patients

Having health insurance can mean the difference between life and death for US patients with follicular lymphoma (FL), according to research published in Blood.
The study showed that patients with private health insurance had nearly 2-fold better survival outcomes than patients without insurance or those who were covered by Medicare or Medicaid.
A review of records on more than 43,000 FL patients showed that, compared with patients under age 65 with private insurance, the hazard ratios (HR) for death among patients in the same age bracket were 1.96 for those with no insurance, 1.83 for those with Medicaid, and 1.96 for those with Medicare (P<0.0001 for each comparison).
“Our study finds that insurance status contributes to survival disparities in FL,” Christopher R. Flowers, MD, of Emory University in Atlanta, Georgia, and his colleagues wrote in Blood.
“Future studies on outcomes in FL should include insurance status as an important predictor. Further research on prognosis for FL should examine the impact of public policy, such as the passage of the [Affordable Care Act], on FL outcomes, as well as examine other factors that influence access to care, such as individual-level socioeconomic status, regular primary care visits, access to prescription medications, and care affordability.”
Earlier research showed that patients with Medicaid or no insurance were more likely than privately insured patients to be diagnosed with cancers at advanced stages, and some patients with aggressive non-Hodgkin lymphomas have been shown to have insurance-related disparities in treatments and outcomes.
To see whether the same could be true for patients with indolent-histology lymphomas such as FL, Dr Flowers and his colleagues extracted data from the National Cancer Database, a nationwide hospital-based cancer registry sponsored jointly by the American College of Surgeons and the American Cancer Society.
The investigators identified 43,648 patients, age 18 and older, who were diagnosed with FL from 2004 through 2014. The team looked at patients ages 18 to 64 as well as patients age 65 and older to account for changes in insurance with Medicare eligibility.
Overall survival among patients younger than 65 was significantly worse for patients with public insurance (Medicaid or Medicare) or no insurance in Cox proportional hazard models controlling for available data on sociodemographic factors and prognostic indicators.
However, compared with patients age 65 and older with private insurance, only patients with Medicare as their sole source of insurance had significantly worse overall survival (HR, 1.28; P<0.0001).
Patients who were uninsured or had Medicaid were more likely than others to have lower socioeconomic status, present with advanced-stage disease, have systemic symptoms, and have multiple comorbidities that persisted after controlling for known sociodemographic and prognostic factors.
The investigators found that, among patients under age 65, those with a comorbidity score of 1 had an HR for death of 1.71, compared with patients with no comorbidities, and patients with a score of 2 or greater had an HR of 3.1 (P<0.0001 for each comparison).
“The findings of the study indicate that improving access to affordable, quality healthcare may reduce disparities in survival for those currently lacking coverage,” the investigators wrote.
The study was supported by Emory University, the National Institutes of Health, and the National Center for Advancing Translational Sciences. Dr Flowers reported financial relationships with AbbVie, Spectrum, Celgene, and several other companies. The other authors reported having nothing to disclose.
Having health insurance can mean the difference between life and death for US patients with follicular lymphoma (FL), according to research published in Blood.
The study showed that patients with private health insurance had nearly 2-fold better survival outcomes than patients without insurance or those who were covered by Medicare or Medicaid.
A review of records on more than 43,000 FL patients showed that, compared with patients under age 65 with private insurance, the hazard ratios (HR) for death among patients in the same age bracket were 1.96 for those with no insurance, 1.83 for those with Medicaid, and 1.96 for those with Medicare (P<0.0001 for each comparison).
“Our study finds that insurance status contributes to survival disparities in FL,” Christopher R. Flowers, MD, of Emory University in Atlanta, Georgia, and his colleagues wrote in Blood.
“Future studies on outcomes in FL should include insurance status as an important predictor. Further research on prognosis for FL should examine the impact of public policy, such as the passage of the [Affordable Care Act], on FL outcomes, as well as examine other factors that influence access to care, such as individual-level socioeconomic status, regular primary care visits, access to prescription medications, and care affordability.”
Earlier research showed that patients with Medicaid or no insurance were more likely than privately insured patients to be diagnosed with cancers at advanced stages, and some patients with aggressive non-Hodgkin lymphomas have been shown to have insurance-related disparities in treatments and outcomes.
To see whether the same could be true for patients with indolent-histology lymphomas such as FL, Dr Flowers and his colleagues extracted data from the National Cancer Database, a nationwide hospital-based cancer registry sponsored jointly by the American College of Surgeons and the American Cancer Society.
The investigators identified 43,648 patients, age 18 and older, who were diagnosed with FL from 2004 through 2014. The team looked at patients ages 18 to 64 as well as patients age 65 and older to account for changes in insurance with Medicare eligibility.
Overall survival among patients younger than 65 was significantly worse for patients with public insurance (Medicaid or Medicare) or no insurance in Cox proportional hazard models controlling for available data on sociodemographic factors and prognostic indicators.
However, compared with patients age 65 and older with private insurance, only patients with Medicare as their sole source of insurance had significantly worse overall survival (HR, 1.28; P<0.0001).
Patients who were uninsured or had Medicaid were more likely than others to have lower socioeconomic status, present with advanced-stage disease, have systemic symptoms, and have multiple comorbidities that persisted after controlling for known sociodemographic and prognostic factors.
The investigators found that, among patients under age 65, those with a comorbidity score of 1 had an HR for death of 1.71, compared with patients with no comorbidities, and patients with a score of 2 or greater had an HR of 3.1 (P<0.0001 for each comparison).
“The findings of the study indicate that improving access to affordable, quality healthcare may reduce disparities in survival for those currently lacking coverage,” the investigators wrote.
The study was supported by Emory University, the National Institutes of Health, and the National Center for Advancing Translational Sciences. Dr Flowers reported financial relationships with AbbVie, Spectrum, Celgene, and several other companies. The other authors reported having nothing to disclose.
Having health insurance can mean the difference between life and death for US patients with follicular lymphoma (FL), according to research published in Blood.
The study showed that patients with private health insurance had nearly 2-fold better survival outcomes than patients without insurance or those who were covered by Medicare or Medicaid.
A review of records on more than 43,000 FL patients showed that, compared with patients under age 65 with private insurance, the hazard ratios (HR) for death among patients in the same age bracket were 1.96 for those with no insurance, 1.83 for those with Medicaid, and 1.96 for those with Medicare (P<0.0001 for each comparison).
“Our study finds that insurance status contributes to survival disparities in FL,” Christopher R. Flowers, MD, of Emory University in Atlanta, Georgia, and his colleagues wrote in Blood.
“Future studies on outcomes in FL should include insurance status as an important predictor. Further research on prognosis for FL should examine the impact of public policy, such as the passage of the [Affordable Care Act], on FL outcomes, as well as examine other factors that influence access to care, such as individual-level socioeconomic status, regular primary care visits, access to prescription medications, and care affordability.”
Earlier research showed that patients with Medicaid or no insurance were more likely than privately insured patients to be diagnosed with cancers at advanced stages, and some patients with aggressive non-Hodgkin lymphomas have been shown to have insurance-related disparities in treatments and outcomes.
To see whether the same could be true for patients with indolent-histology lymphomas such as FL, Dr Flowers and his colleagues extracted data from the National Cancer Database, a nationwide hospital-based cancer registry sponsored jointly by the American College of Surgeons and the American Cancer Society.
The investigators identified 43,648 patients, age 18 and older, who were diagnosed with FL from 2004 through 2014. The team looked at patients ages 18 to 64 as well as patients age 65 and older to account for changes in insurance with Medicare eligibility.
Overall survival among patients younger than 65 was significantly worse for patients with public insurance (Medicaid or Medicare) or no insurance in Cox proportional hazard models controlling for available data on sociodemographic factors and prognostic indicators.
However, compared with patients age 65 and older with private insurance, only patients with Medicare as their sole source of insurance had significantly worse overall survival (HR, 1.28; P<0.0001).
Patients who were uninsured or had Medicaid were more likely than others to have lower socioeconomic status, present with advanced-stage disease, have systemic symptoms, and have multiple comorbidities that persisted after controlling for known sociodemographic and prognostic factors.
The investigators found that, among patients under age 65, those with a comorbidity score of 1 had an HR for death of 1.71, compared with patients with no comorbidities, and patients with a score of 2 or greater had an HR of 3.1 (P<0.0001 for each comparison).
“The findings of the study indicate that improving access to affordable, quality healthcare may reduce disparities in survival for those currently lacking coverage,” the investigators wrote.
The study was supported by Emory University, the National Institutes of Health, and the National Center for Advancing Translational Sciences. Dr Flowers reported financial relationships with AbbVie, Spectrum, Celgene, and several other companies. The other authors reported having nothing to disclose.
Clozapine-induced GI hypomotility: From constipation to bowel obstruction
Patients who are treated with clozapine—a second-generation antipsychotic approved for treatment-resistant schizophrenia—require monitoring for serious adverse effects. Many of these adverse effects, such as agranulocytosis or seizures, are familiar to clinicians; however, gastrointestinal (GI) hypomotility is not always recognized as a potentially serious adverse effect, even though it is one of the most common causes for hospital admission.1 Its manifestations range from being relatively benign (nausea, vomiting, constipation) to potentially severe (fecal impaction) or even life-threatening (bowel obstruction, ileus, toxic megacolon).2
GI hypomotility is caused by clozapine’s strong anticholinergic properties, which lead to slowed smooth muscle contractions and delayed bowel transit time. It is further compounded by clozapine’s 5-HT3 antagonism, which is also known to slow bowel transit time. To avoid the potentially serious risks associated with GI hypomotility, we offer simple approaches for clinicians to follow when treating patients with clozapine.
Before starting a patient on clozapine, and at all subsequent visits, ask him or her about bowel habits and GI symptoms. Because the onset of GI hypomotility can be subtle, ask patients to pay close attention to their bowel habits and keep a diary to document GI symptoms and bowel movements. Signs of bowel obstruction can include an inability to pass stool, nausea, vomiting, abdominal pain, and a bloated abdomen. Staff who care for patients taking clozapine who live in a supervised setting should be educated about the relevance of a patient’s changing bowel habits or GI complaints. Also, teach patients about simple lifestyle modifications they can make to counteract constipation, including increased physical activity, adequate hydration, and consuming a fiber-rich diet.

If possible, avoid prescribing anticholinergic medications to a patient receiving clozapine because such agents may add to clozapine’s anticholinergic load. Some patients may require medical management of chronic constipation with stool softeners and stimulant laxatives. The Table outlines a typical bowel regimen we use for patients at our clinic. Some clinicians may prefer earlier and more regular use of senna glycoside. Also, patients might need a referral to their primary care physician if prevention has failed and fecal impaction requires enemas or mechanical disimpaction. An urgent referral to the emergency department is needed if a patient has a suspected bowel obstruction.
Acknowledgment
The authors thank Travis Baggett, MD, Department of Medicine, Massachusetts General Hospital, Boston, Massachusetts, for reviewing the medical management of constipation.
1. Stroup TS, Gerhard T, Crystal S, et al. Comparative effectiveness of clozapine and standard antipsychotic treatment in adults with schizophrenia. Am J Psychiatry. 2016;173(2):166-173.
2. Every-Palmer S, Ellis PM. Clozapine-induced gastrointestinal hypomotility: a 22-year bi-national pharmacovigilance study of serious or fatal ‘slow gut’ reactions, and comparison with international drug safety advice. CNS Drugs. 2017;31(8):699-709.
Patients who are treated with clozapine—a second-generation antipsychotic approved for treatment-resistant schizophrenia—require monitoring for serious adverse effects. Many of these adverse effects, such as agranulocytosis or seizures, are familiar to clinicians; however, gastrointestinal (GI) hypomotility is not always recognized as a potentially serious adverse effect, even though it is one of the most common causes for hospital admission.1 Its manifestations range from being relatively benign (nausea, vomiting, constipation) to potentially severe (fecal impaction) or even life-threatening (bowel obstruction, ileus, toxic megacolon).2
GI hypomotility is caused by clozapine’s strong anticholinergic properties, which lead to slowed smooth muscle contractions and delayed bowel transit time. It is further compounded by clozapine’s 5-HT3 antagonism, which is also known to slow bowel transit time. To avoid the potentially serious risks associated with GI hypomotility, we offer simple approaches for clinicians to follow when treating patients with clozapine.
Before starting a patient on clozapine, and at all subsequent visits, ask him or her about bowel habits and GI symptoms. Because the onset of GI hypomotility can be subtle, ask patients to pay close attention to their bowel habits and keep a diary to document GI symptoms and bowel movements. Signs of bowel obstruction can include an inability to pass stool, nausea, vomiting, abdominal pain, and a bloated abdomen. Staff who care for patients taking clozapine who live in a supervised setting should be educated about the relevance of a patient’s changing bowel habits or GI complaints. Also, teach patients about simple lifestyle modifications they can make to counteract constipation, including increased physical activity, adequate hydration, and consuming a fiber-rich diet.
If possible, avoid prescribing anticholinergic medications to a patient receiving clozapine because such agents may add to clozapine’s anticholinergic load. Some patients may require medical management of chronic constipation with stool softeners and stimulant laxatives. The Table outlines a typical bowel regimen we use for patients at our clinic. Some clinicians may prefer earlier and more regular use of senna glycoside. Also, patients might need a referral to their primary care physician if prevention has failed and fecal impaction requires enemas or mechanical disimpaction. An urgent referral to the emergency department is needed if a patient has a suspected bowel obstruction.
Acknowledgment
The authors thank Travis Baggett, MD, Department of Medicine, Massachusetts General Hospital, Boston, Massachusetts, for reviewing the medical management of constipation.
Patients who are treated with clozapine—a second-generation antipsychotic approved for treatment-resistant schizophrenia—require monitoring for serious adverse effects. Many of these adverse effects, such as agranulocytosis or seizures, are familiar to clinicians; however, gastrointestinal (GI) hypomotility is not always recognized as a potentially serious adverse effect, even though it is one of the most common causes for hospital admission.1 Its manifestations range from being relatively benign (nausea, vomiting, constipation) to potentially severe (fecal impaction) or even life-threatening (bowel obstruction, ileus, toxic megacolon).2
GI hypomotility is caused by clozapine’s strong anticholinergic properties, which lead to slowed smooth muscle contractions and delayed bowel transit time. It is further compounded by clozapine’s 5-HT3 antagonism, which is also known to slow bowel transit time. To avoid the potentially serious risks associated with GI hypomotility, we offer simple approaches for clinicians to follow when treating patients with clozapine.
Before starting a patient on clozapine, and at all subsequent visits, ask him or her about bowel habits and GI symptoms. Because the onset of GI hypomotility can be subtle, ask patients to pay close attention to their bowel habits and keep a diary to document GI symptoms and bowel movements. Signs of bowel obstruction can include an inability to pass stool, nausea, vomiting, abdominal pain, and a bloated abdomen. Staff who care for patients taking clozapine who live in a supervised setting should be educated about the relevance of a patient’s changing bowel habits or GI complaints. Also, teach patients about simple lifestyle modifications they can make to counteract constipation, including increased physical activity, adequate hydration, and consuming a fiber-rich diet.
If possible, avoid prescribing anticholinergic medications to a patient receiving clozapine because such agents may add to clozapine’s anticholinergic load. Some patients may require medical management of chronic constipation with stool softeners and stimulant laxatives. The Table outlines a typical bowel regimen we use for patients at our clinic. Some clinicians may prefer earlier and more regular use of senna glycoside. Also, patients might need a referral to their primary care physician if prevention has failed and fecal impaction requires enemas or mechanical disimpaction. An urgent referral to the emergency department is needed if a patient has a suspected bowel obstruction.
Acknowledgment
The authors thank Travis Baggett, MD, Department of Medicine, Massachusetts General Hospital, Boston, Massachusetts, for reviewing the medical management of constipation.
1. Stroup TS, Gerhard T, Crystal S, et al. Comparative effectiveness of clozapine and standard antipsychotic treatment in adults with schizophrenia. Am J Psychiatry. 2016;173(2):166-173.
2. Every-Palmer S, Ellis PM. Clozapine-induced gastrointestinal hypomotility: a 22-year bi-national pharmacovigilance study of serious or fatal ‘slow gut’ reactions, and comparison with international drug safety advice. CNS Drugs. 2017;31(8):699-709.
1. Stroup TS, Gerhard T, Crystal S, et al. Comparative effectiveness of clozapine and standard antipsychotic treatment in adults with schizophrenia. Am J Psychiatry. 2016;173(2):166-173.
2. Every-Palmer S, Ellis PM. Clozapine-induced gastrointestinal hypomotility: a 22-year bi-national pharmacovigilance study of serious or fatal ‘slow gut’ reactions, and comparison with international drug safety advice. CNS Drugs. 2017;31(8):699-709.
FDA approves vaginal ring contraceptive Annovera
The first vaginal ring contraceptive that can be used for 1 year has been approved by the Food and Drug Administration.
The FDA granted approval of Annovera (segesterone acetate and ethinyl estradiol vaginal system), a reusable donut-shaped ring, to the Population Council. The nonbiodegradable, flexible vaginal system is placed in the vagina for 3 weeks followed by 1 week out of the vagina, at which time women may experience a menstrual period. This schedule is repeated every 4 weeks for 13 28-day menstrual cycles.
The contraceptive ring is washed and stored in a compact case for the 7 days when it is not in use. Annovera does not require refrigeration prior to dispensing and can withstand storage temperatures up to 30° C (86° F).
“Today’s approval builds on available birth control options,” said Victor Crentsil, MD, acting deputy director of the Office of Drug Evaluation III in FDA’s Center for Drug Evaluation and Research.
The efficacy and safety of Annovera were studied in three, open-label clinical trials with healthy women ranging from 18 to 40 years of age. Based on the results, about 2%-4% of women may get pregnant during the first year they use Annovera.
Annovera carries a boxed warning regarding cigarette smoking and serious cardiovascular events. Women over age 35 who smoke should not use Annovera. Cigarette smoking increases the risk of serious cardiovascular events from combination hormonal contraceptive use.
Annovera is contraindicated for women with a high risk of arterial or venous thrombotic diseases; a history of breast cancer or another estrogen- or progestin-sensitive cancer; liver tumors, acute hepatitis, or severe (decompensated) cirrhosis; undiagnosed abnormal uterine bleeding; hypersensitivity to any of the components of Annovera; and use of hepatitis C drug combinations containing ombitasvir/paritaprevir/ritonavir, with or without dasabuvir, according to an FDA press release.
The most common side effects of Annovera are similar to those of other combined hormonal contraceptive products and include headache, nausea and vomiting, yeast infections, abdominal pain, dysmenorrhea, breast tenderness, irregular bleeding, diarrhea, and genital itching.
The FDA is requiring postmarketing studies to further evaluate the risks of venous thromboembolism and the effects of CYP3A-modulating drugs and tampon use on the pharmacokinetics of Annovera.
It is very exciting to see ongoing research and development for new contraceptives and approval of these new methods! We know that each person’s contraceptive needs are unique and having more options from which to choose will help us as providers connect our patients to methods that meet their goals and individual needs. These two methods fill important gaps in our current contraceptive portfolio: a patient-controlled, long-acting method and a facilitated non-hormonal, non-prescription method (the Natural Cycles app). I am looking forward to hearing feedback from patients about their thoughts and experiences with these new methods.

Melissa Kottke, MD, MPH, MBA, is an associate professor of obstetrics and gynecology and director of the Jane Fonda Center for Adolescent Reproductive Health, Emory University, Atlanta.
It is very exciting to see ongoing research and development for new contraceptives and approval of these new methods! We know that each person’s contraceptive needs are unique and having more options from which to choose will help us as providers connect our patients to methods that meet their goals and individual needs. These two methods fill important gaps in our current contraceptive portfolio: a patient-controlled, long-acting method and a facilitated non-hormonal, non-prescription method (the Natural Cycles app). I am looking forward to hearing feedback from patients about their thoughts and experiences with these new methods.
Melissa Kottke, MD, MPH, MBA, is an associate professor of obstetrics and gynecology and director of the Jane Fonda Center for Adolescent Reproductive Health, Emory University, Atlanta.
It is very exciting to see ongoing research and development for new contraceptives and approval of these new methods! We know that each person’s contraceptive needs are unique and having more options from which to choose will help us as providers connect our patients to methods that meet their goals and individual needs. These two methods fill important gaps in our current contraceptive portfolio: a patient-controlled, long-acting method and a facilitated non-hormonal, non-prescription method (the Natural Cycles app). I am looking forward to hearing feedback from patients about their thoughts and experiences with these new methods.
Melissa Kottke, MD, MPH, MBA, is an associate professor of obstetrics and gynecology and director of the Jane Fonda Center for Adolescent Reproductive Health, Emory University, Atlanta.
The first vaginal ring contraceptive that can be used for 1 year has been approved by the Food and Drug Administration.
The FDA granted approval of Annovera (segesterone acetate and ethinyl estradiol vaginal system), a reusable donut-shaped ring, to the Population Council. The nonbiodegradable, flexible vaginal system is placed in the vagina for 3 weeks followed by 1 week out of the vagina, at which time women may experience a menstrual period. This schedule is repeated every 4 weeks for 13 28-day menstrual cycles.
The contraceptive ring is washed and stored in a compact case for the 7 days when it is not in use. Annovera does not require refrigeration prior to dispensing and can withstand storage temperatures up to 30° C (86° F).
“Today’s approval builds on available birth control options,” said Victor Crentsil, MD, acting deputy director of the Office of Drug Evaluation III in FDA’s Center for Drug Evaluation and Research.
The efficacy and safety of Annovera were studied in three, open-label clinical trials with healthy women ranging from 18 to 40 years of age. Based on the results, about 2%-4% of women may get pregnant during the first year they use Annovera.
Annovera carries a boxed warning regarding cigarette smoking and serious cardiovascular events. Women over age 35 who smoke should not use Annovera. Cigarette smoking increases the risk of serious cardiovascular events from combination hormonal contraceptive use.
Annovera is contraindicated for women with a high risk of arterial or venous thrombotic diseases; a history of breast cancer or another estrogen- or progestin-sensitive cancer; liver tumors, acute hepatitis, or severe (decompensated) cirrhosis; undiagnosed abnormal uterine bleeding; hypersensitivity to any of the components of Annovera; and use of hepatitis C drug combinations containing ombitasvir/paritaprevir/ritonavir, with or without dasabuvir, according to an FDA press release.
The most common side effects of Annovera are similar to those of other combined hormonal contraceptive products and include headache, nausea and vomiting, yeast infections, abdominal pain, dysmenorrhea, breast tenderness, irregular bleeding, diarrhea, and genital itching.
The FDA is requiring postmarketing studies to further evaluate the risks of venous thromboembolism and the effects of CYP3A-modulating drugs and tampon use on the pharmacokinetics of Annovera.
The first vaginal ring contraceptive that can be used for 1 year has been approved by the Food and Drug Administration.
The FDA granted approval of Annovera (segesterone acetate and ethinyl estradiol vaginal system), a reusable donut-shaped ring, to the Population Council. The nonbiodegradable, flexible vaginal system is placed in the vagina for 3 weeks followed by 1 week out of the vagina, at which time women may experience a menstrual period. This schedule is repeated every 4 weeks for 13 28-day menstrual cycles.
The contraceptive ring is washed and stored in a compact case for the 7 days when it is not in use. Annovera does not require refrigeration prior to dispensing and can withstand storage temperatures up to 30° C (86° F).
“Today’s approval builds on available birth control options,” said Victor Crentsil, MD, acting deputy director of the Office of Drug Evaluation III in FDA’s Center for Drug Evaluation and Research.
The efficacy and safety of Annovera were studied in three, open-label clinical trials with healthy women ranging from 18 to 40 years of age. Based on the results, about 2%-4% of women may get pregnant during the first year they use Annovera.
Annovera carries a boxed warning regarding cigarette smoking and serious cardiovascular events. Women over age 35 who smoke should not use Annovera. Cigarette smoking increases the risk of serious cardiovascular events from combination hormonal contraceptive use.
Annovera is contraindicated for women with a high risk of arterial or venous thrombotic diseases; a history of breast cancer or another estrogen- or progestin-sensitive cancer; liver tumors, acute hepatitis, or severe (decompensated) cirrhosis; undiagnosed abnormal uterine bleeding; hypersensitivity to any of the components of Annovera; and use of hepatitis C drug combinations containing ombitasvir/paritaprevir/ritonavir, with or without dasabuvir, according to an FDA press release.
The most common side effects of Annovera are similar to those of other combined hormonal contraceptive products and include headache, nausea and vomiting, yeast infections, abdominal pain, dysmenorrhea, breast tenderness, irregular bleeding, diarrhea, and genital itching.
The FDA is requiring postmarketing studies to further evaluate the risks of venous thromboembolism and the effects of CYP3A-modulating drugs and tampon use on the pharmacokinetics of Annovera.
SHM aids national infection prevention and control effort
Multidisciplinary teams celebrate achievements in getting to zero
The Society of Hospital Medicine is pleased to share successes and resources from a 3-year national quality improvement program called STRIVE (States Targeting Reduction in Infections Via Engagement). This program targeted opportunities to streamline and enhance infection prevention and control efforts in participating hospitals.
SHM was a key partner in the STRIVE program, which was managed by the Health Research & Educational Trust, the not-for-profit research and education affiliate of the American Hospital Association. Other partners included the American Society for Healthcare Engineering, Association for Professionals in Infection Control and Epidemiology, University of Michigan, Ann Arbor, and experts from academic institutions and professional societies such as Cornell University, Ithaca, N.Y.; Rush University, Chicago; and the Association for the Healthcare Environment. SHM provided specific knowledge and experience on HAI prevention and helped develop the STRIVE curriculum and resources. Faculty coaches from SHM also supported STRIVE hospitals by presenting on webinars, attending in-person meetings, and consulting on calls.
Following the U.S. experience with Ebola, the Centers for Disease Control and Infection identified the critical importance of enhancing infection control for all infectious threats to protect both patients and health care personnel. The CDC also recognized that many state and regional organizations and agencies work with the same health care facilities in order to coordinate efforts to address infectious threats. The STRIVE program provided tools and resources to help communities strengthen the relationships among diverse organizations to maximize improvement and coordination.
Closely aligned with SHM’s mission to promote exceptional care for hospitalized patients, the CDC’s STRIVE program goals were as follows:
- To expand the CDC’s Targeting Assessment for Prevention (TAP) strategy of using surveillance data to identify hospitals with a disproportionately high burden of health care–associated infections (HAIs),
- To build and strengthen relationships between state and regional organizations that help hospitals with infection control and prevention, and
- To provide technical assistance to hospitals to improve implementation of infection control practices in existing and newly constructed health care facilities.
The participants in this program included 449 hospitals from 28 states and the District of Columbia. Short-stay and long-term acute care hospitals that had a high burden of Clostridium difficile infection, and a high burden of one or more of the following HAIs – central line–associated bloodstream infection, catheter associated urinary tract infection, and health care–associated methicillin-resistant Staphylococcus aureus (MRSA) bacteremia – were targeted. Each participant had access to specific education modules, webinars, and learning networks designed to enhance collaboration, performance improvement, and understanding of the successes and barriers to coordinating hospital- and community-based services. Hospitals joined the program in cohorts and engaged in a year-long effort to reduce infection burden. During the program implementation period, many hospitals showed measurable improvement by achieving an HAI-specific relative rate reduction or maintenance of a rate of zero between baseline and intervention periods.
Key successes of the program centered around development of multidisciplinary teams that engaged not only the infection preventionists but also such areas as environmental services and other departments that may not have traditionally been included in infection prevention efforts. These teams focused on establishing competency-based trainings and processes for auditing competencies. One series of STRIVE resources helped hospitals learn new ways to implement best practices and communicate with diverse departments so every team member could participate in removing barriers to infection prevention in the hospital.
SHM was especially pleased to be a part of a program that brought together state health departments, state hospital associations, quality innovation network–quality improvement organizations, and other agencies and health systems committed to infection prevention. The collaboration and partnerships among the STRIVE program participants helped minimize duplication of work and improve efficiency and effectiveness of infection prevention efforts lead by hospitals.
To learn more about the STRIVE resources, visit www.hret.org/quality/projects/strive.shtml.
Multidisciplinary teams celebrate achievements in getting to zero
Multidisciplinary teams celebrate achievements in getting to zero
The Society of Hospital Medicine is pleased to share successes and resources from a 3-year national quality improvement program called STRIVE (States Targeting Reduction in Infections Via Engagement). This program targeted opportunities to streamline and enhance infection prevention and control efforts in participating hospitals.
SHM was a key partner in the STRIVE program, which was managed by the Health Research & Educational Trust, the not-for-profit research and education affiliate of the American Hospital Association. Other partners included the American Society for Healthcare Engineering, Association for Professionals in Infection Control and Epidemiology, University of Michigan, Ann Arbor, and experts from academic institutions and professional societies such as Cornell University, Ithaca, N.Y.; Rush University, Chicago; and the Association for the Healthcare Environment. SHM provided specific knowledge and experience on HAI prevention and helped develop the STRIVE curriculum and resources. Faculty coaches from SHM also supported STRIVE hospitals by presenting on webinars, attending in-person meetings, and consulting on calls.
Following the U.S. experience with Ebola, the Centers for Disease Control and Infection identified the critical importance of enhancing infection control for all infectious threats to protect both patients and health care personnel. The CDC also recognized that many state and regional organizations and agencies work with the same health care facilities in order to coordinate efforts to address infectious threats. The STRIVE program provided tools and resources to help communities strengthen the relationships among diverse organizations to maximize improvement and coordination.
Closely aligned with SHM’s mission to promote exceptional care for hospitalized patients, the CDC’s STRIVE program goals were as follows:
- To expand the CDC’s Targeting Assessment for Prevention (TAP) strategy of using surveillance data to identify hospitals with a disproportionately high burden of health care–associated infections (HAIs),
- To build and strengthen relationships between state and regional organizations that help hospitals with infection control and prevention, and
- To provide technical assistance to hospitals to improve implementation of infection control practices in existing and newly constructed health care facilities.
The participants in this program included 449 hospitals from 28 states and the District of Columbia. Short-stay and long-term acute care hospitals that had a high burden of Clostridium difficile infection, and a high burden of one or more of the following HAIs – central line–associated bloodstream infection, catheter associated urinary tract infection, and health care–associated methicillin-resistant Staphylococcus aureus (MRSA) bacteremia – were targeted. Each participant had access to specific education modules, webinars, and learning networks designed to enhance collaboration, performance improvement, and understanding of the successes and barriers to coordinating hospital- and community-based services. Hospitals joined the program in cohorts and engaged in a year-long effort to reduce infection burden. During the program implementation period, many hospitals showed measurable improvement by achieving an HAI-specific relative rate reduction or maintenance of a rate of zero between baseline and intervention periods.
Key successes of the program centered around development of multidisciplinary teams that engaged not only the infection preventionists but also such areas as environmental services and other departments that may not have traditionally been included in infection prevention efforts. These teams focused on establishing competency-based trainings and processes for auditing competencies. One series of STRIVE resources helped hospitals learn new ways to implement best practices and communicate with diverse departments so every team member could participate in removing barriers to infection prevention in the hospital.
SHM was especially pleased to be a part of a program that brought together state health departments, state hospital associations, quality innovation network–quality improvement organizations, and other agencies and health systems committed to infection prevention. The collaboration and partnerships among the STRIVE program participants helped minimize duplication of work and improve efficiency and effectiveness of infection prevention efforts lead by hospitals.
To learn more about the STRIVE resources, visit www.hret.org/quality/projects/strive.shtml.
The Society of Hospital Medicine is pleased to share successes and resources from a 3-year national quality improvement program called STRIVE (States Targeting Reduction in Infections Via Engagement). This program targeted opportunities to streamline and enhance infection prevention and control efforts in participating hospitals.
SHM was a key partner in the STRIVE program, which was managed by the Health Research & Educational Trust, the not-for-profit research and education affiliate of the American Hospital Association. Other partners included the American Society for Healthcare Engineering, Association for Professionals in Infection Control and Epidemiology, University of Michigan, Ann Arbor, and experts from academic institutions and professional societies such as Cornell University, Ithaca, N.Y.; Rush University, Chicago; and the Association for the Healthcare Environment. SHM provided specific knowledge and experience on HAI prevention and helped develop the STRIVE curriculum and resources. Faculty coaches from SHM also supported STRIVE hospitals by presenting on webinars, attending in-person meetings, and consulting on calls.
Following the U.S. experience with Ebola, the Centers for Disease Control and Infection identified the critical importance of enhancing infection control for all infectious threats to protect both patients and health care personnel. The CDC also recognized that many state and regional organizations and agencies work with the same health care facilities in order to coordinate efforts to address infectious threats. The STRIVE program provided tools and resources to help communities strengthen the relationships among diverse organizations to maximize improvement and coordination.
Closely aligned with SHM’s mission to promote exceptional care for hospitalized patients, the CDC’s STRIVE program goals were as follows:
- To expand the CDC’s Targeting Assessment for Prevention (TAP) strategy of using surveillance data to identify hospitals with a disproportionately high burden of health care–associated infections (HAIs),
- To build and strengthen relationships between state and regional organizations that help hospitals with infection control and prevention, and
- To provide technical assistance to hospitals to improve implementation of infection control practices in existing and newly constructed health care facilities.
The participants in this program included 449 hospitals from 28 states and the District of Columbia. Short-stay and long-term acute care hospitals that had a high burden of Clostridium difficile infection, and a high burden of one or more of the following HAIs – central line–associated bloodstream infection, catheter associated urinary tract infection, and health care–associated methicillin-resistant Staphylococcus aureus (MRSA) bacteremia – were targeted. Each participant had access to specific education modules, webinars, and learning networks designed to enhance collaboration, performance improvement, and understanding of the successes and barriers to coordinating hospital- and community-based services. Hospitals joined the program in cohorts and engaged in a year-long effort to reduce infection burden. During the program implementation period, many hospitals showed measurable improvement by achieving an HAI-specific relative rate reduction or maintenance of a rate of zero between baseline and intervention periods.
Key successes of the program centered around development of multidisciplinary teams that engaged not only the infection preventionists but also such areas as environmental services and other departments that may not have traditionally been included in infection prevention efforts. These teams focused on establishing competency-based trainings and processes for auditing competencies. One series of STRIVE resources helped hospitals learn new ways to implement best practices and communicate with diverse departments so every team member could participate in removing barriers to infection prevention in the hospital.
SHM was especially pleased to be a part of a program that brought together state health departments, state hospital associations, quality innovation network–quality improvement organizations, and other agencies and health systems committed to infection prevention. The collaboration and partnerships among the STRIVE program participants helped minimize duplication of work and improve efficiency and effectiveness of infection prevention efforts lead by hospitals.
To learn more about the STRIVE resources, visit www.hret.org/quality/projects/strive.shtml.
Continuation, complication rates similar for implants, IUDs
A new study of Medicaid-covered women who sought long-acting reversible contraception via subdermal etonogestrel implants or IUDs found that levels of continuation at 1 year were high, and complication rates were low.
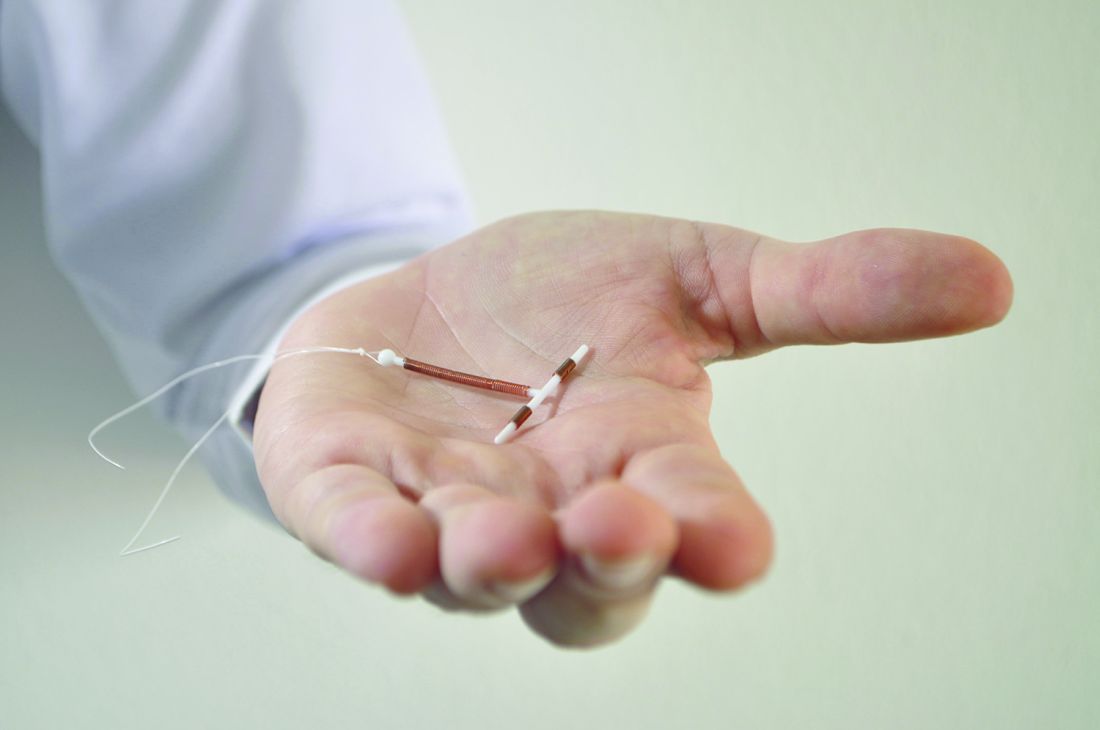
There was little difference between the groups on both fronts.
“This study demonstrates high rates of continued use of subdermal and intrauterine contraceptives by Medicaid clients, particularly among adolescents, and therefore, interventions that increase adolescent access to these contraceptives are likely to be cost effective from the payer perspective,” the study authors wrote in Contraception.
The study, led by Max J. Romano, MD, MPH, of MedStar Franklin Square Medical Center in Baltimore, retrospectively examined the medical claims of 3,305 women treated via MedStar Family Choice, a Medicaid managed-choice insurer.
The women, who lived in Maryland and the District of Columbia, were aged 15-44 years when they underwent contraceptive treatment during 2012-2015. Of the women, 1,335 had subdermal etonogestrel implants inserted, and 1,970 had IUDs inserted. Researchers followed the women for a mean 344 days.
The implant users were younger than the IUD group (mean age, 25 years vs. 28 years; P = less than .001), and women under age 20 years were more than more than twice as likely to get implants than IUDs (71% vs. 29%).
Women older than 20 years were more likely to get IUDs than implants, with those older than 25 preferring them by a ratio of 70% to 30%.
After researchers controlled for such factors as age group and year of insertion, they found that implant recipients were slightly more likely to still have the contraception tool in place at 1 year: 81% (implants) vs. IUDs (77%; P = .01). It’s not clear why women had their implants or IUDs removed.
Claims for complications were similar between the groups (8% for implants, 7% for IUDs), and researchers didn’t find a statistically significant difference after they controlled for various factors. Dysfunctional uterine bleeding was the most common complication, followed by excessive or frequent menstruation.
Few women reported pregnancy among the implant (0.82%) and IUD (0.86%) groups, and there was no statistically significant difference between the groups.
The researchers reported that their study has various limitations: The insurance claims information may provide misleading information about minor complications, and it doesn’t include details about abortions. The claims also may not report IUD expulsions and self-removals.
The authors also noted that they didn’t control for factors like socioeconomic status, psychological conditions, and multimorbidity.
MedStar Family Choice provided study funding. Dr. Romano and Loral Patchen, PhD, disclosed receiving free CME trainings and insertion certification regarding the long-acting contraceptive devices discussed in the study from Merck, Bayer, and Teva. Dr. Patchen disclosed serving on the Diclegis speakers bureau.
A new study of Medicaid-covered women who sought long-acting reversible contraception via subdermal etonogestrel implants or IUDs found that levels of continuation at 1 year were high, and complication rates were low.
There was little difference between the groups on both fronts.
“This study demonstrates high rates of continued use of subdermal and intrauterine contraceptives by Medicaid clients, particularly among adolescents, and therefore, interventions that increase adolescent access to these contraceptives are likely to be cost effective from the payer perspective,” the study authors wrote in Contraception.
The study, led by Max J. Romano, MD, MPH, of MedStar Franklin Square Medical Center in Baltimore, retrospectively examined the medical claims of 3,305 women treated via MedStar Family Choice, a Medicaid managed-choice insurer.
The women, who lived in Maryland and the District of Columbia, were aged 15-44 years when they underwent contraceptive treatment during 2012-2015. Of the women, 1,335 had subdermal etonogestrel implants inserted, and 1,970 had IUDs inserted. Researchers followed the women for a mean 344 days.
The implant users were younger than the IUD group (mean age, 25 years vs. 28 years; P = less than .001), and women under age 20 years were more than more than twice as likely to get implants than IUDs (71% vs. 29%).
Women older than 20 years were more likely to get IUDs than implants, with those older than 25 preferring them by a ratio of 70% to 30%.
After researchers controlled for such factors as age group and year of insertion, they found that implant recipients were slightly more likely to still have the contraception tool in place at 1 year: 81% (implants) vs. IUDs (77%; P = .01). It’s not clear why women had their implants or IUDs removed.
Claims for complications were similar between the groups (8% for implants, 7% for IUDs), and researchers didn’t find a statistically significant difference after they controlled for various factors. Dysfunctional uterine bleeding was the most common complication, followed by excessive or frequent menstruation.
Few women reported pregnancy among the implant (0.82%) and IUD (0.86%) groups, and there was no statistically significant difference between the groups.
The researchers reported that their study has various limitations: The insurance claims information may provide misleading information about minor complications, and it doesn’t include details about abortions. The claims also may not report IUD expulsions and self-removals.
The authors also noted that they didn’t control for factors like socioeconomic status, psychological conditions, and multimorbidity.
MedStar Family Choice provided study funding. Dr. Romano and Loral Patchen, PhD, disclosed receiving free CME trainings and insertion certification regarding the long-acting contraceptive devices discussed in the study from Merck, Bayer, and Teva. Dr. Patchen disclosed serving on the Diclegis speakers bureau.
A new study of Medicaid-covered women who sought long-acting reversible contraception via subdermal etonogestrel implants or IUDs found that levels of continuation at 1 year were high, and complication rates were low.
There was little difference between the groups on both fronts.
“This study demonstrates high rates of continued use of subdermal and intrauterine contraceptives by Medicaid clients, particularly among adolescents, and therefore, interventions that increase adolescent access to these contraceptives are likely to be cost effective from the payer perspective,” the study authors wrote in Contraception.
The study, led by Max J. Romano, MD, MPH, of MedStar Franklin Square Medical Center in Baltimore, retrospectively examined the medical claims of 3,305 women treated via MedStar Family Choice, a Medicaid managed-choice insurer.
The women, who lived in Maryland and the District of Columbia, were aged 15-44 years when they underwent contraceptive treatment during 2012-2015. Of the women, 1,335 had subdermal etonogestrel implants inserted, and 1,970 had IUDs inserted. Researchers followed the women for a mean 344 days.
The implant users were younger than the IUD group (mean age, 25 years vs. 28 years; P = less than .001), and women under age 20 years were more than more than twice as likely to get implants than IUDs (71% vs. 29%).
Women older than 20 years were more likely to get IUDs than implants, with those older than 25 preferring them by a ratio of 70% to 30%.
After researchers controlled for such factors as age group and year of insertion, they found that implant recipients were slightly more likely to still have the contraception tool in place at 1 year: 81% (implants) vs. IUDs (77%; P = .01). It’s not clear why women had their implants or IUDs removed.
Claims for complications were similar between the groups (8% for implants, 7% for IUDs), and researchers didn’t find a statistically significant difference after they controlled for various factors. Dysfunctional uterine bleeding was the most common complication, followed by excessive or frequent menstruation.
Few women reported pregnancy among the implant (0.82%) and IUD (0.86%) groups, and there was no statistically significant difference between the groups.
The researchers reported that their study has various limitations: The insurance claims information may provide misleading information about minor complications, and it doesn’t include details about abortions. The claims also may not report IUD expulsions and self-removals.
The authors also noted that they didn’t control for factors like socioeconomic status, psychological conditions, and multimorbidity.
MedStar Family Choice provided study funding. Dr. Romano and Loral Patchen, PhD, disclosed receiving free CME trainings and insertion certification regarding the long-acting contraceptive devices discussed in the study from Merck, Bayer, and Teva. Dr. Patchen disclosed serving on the Diclegis speakers bureau.
FROM CONTRACEPTION
Key clinical point:
Major finding: For implants and IUDS, respectively, 1-year continuation rates were similar (adjusted 81% vs. 77%, P = .01), as were complication rates (unadjusted 8% for implants, 7% for IUDs).
Study details: Retrospective analysis of 3,305 Medicaid-covered women treated with the two types of contraceptives during 2012-2015 in the District of Columbia and Maryland.
Disclosures: MedStar Family Choice provided study funding. Dr. Romano and Loral Patchen, PhD, disclosed receiving free CME trainings and insertion certification regarding the long-acting contraceptive devices discussed in the study from Merck, Bayer, and Teva. Dr. Patchen discloses serving on the Diclegis speakers bureau.
Source: Romano MJ et al. Contraception. 2018 Aug;98:125-9.
New IUD expelled less often after C-section than older device
that’s inserted the same way, according to results of a Turkish study.
The study authors, led by Ceren Unal, MD, of Zeynep Kamil Women’s and Children’s Disease Training and Research Hospital, Istanbul, wrote that the new device, the frameless copper-releasing Gyn-CS IUD, “could be a major advance, potentially suitable for general use due to the ease and safety of the insertion procedure,” which requires limited training.
According to the study authors, most IUDs are retained in the uterus, and most have a T-shape configuration. Some are inserted immediately following expulsion of the placenta at birth, although techniques “are far from being optimal,” and devices are frequently displaced and sometimes expelled.
The newly developed Gyn-CS IUD, a redesigned version of a previous model, has an alternative “anchor” design.
The investigators tracked 140 pregnant women – 106 who underwent elective cesarean section and 34 who underwent emergency cesarean section – who had the Gyn-CS or the TCu380A IUD inserted shortly after they gave birth. Their median age was around 30 years , all were white, and all were married.
The TCu380A IUD, a decades-old model that’s been recommended by the World Health Organization, and the Gyn-CS IUD were each inserted in 70 women. The researchers then followed them for 3 months. Three women (one in the Gyn-CS IUD group and two in the TCu380A IUD group) were lost to follow-up.
In total, 61 of 70 IUDs (88%) remained in place in the Gyn-CS group, and 54 of 70 (79%) in the TCu380A group (P = .30). One (1%) Gyn-CS IUD was expelled , possibly because it was incorrectly anchored, and eight (11%) TCu380A IUDs were expelled (P = .04). There were equal numbers of medical removals (four) and nonmedical removals (two) in the two groups, and one “other medical removal” in the Gyn-CS IUD group.
“The very low expulsion rate of Gyn-CS, compared with TCu380A, is a very strong argument in favor of the anchored IUD, preventing expulsion and displacement,” Dr. Unal and colleagues reported in Contraception.
They noted that the study has a limited 3-month tracking period, but they also pointed out that most expulsions inserted post partum occurred in the initial 6 weeks.
The low expulsion rate of the Gyn-CS device “will prevent more women becoming pregnant too soon which constitutes an important safety issue during future pregnancy,” the authors wrote. “The device, preferably its high-load 10-year version, could also interest many women as a reversible alternative for tubal sterilization. Additional clinical experience with the Gyn-CS IUD is, therefore, urgently warranted.”
No study funding is reported, although Contrel Research provided the Gyn-CS devices at no charge. The study authors reported no relevant disclosures.
that’s inserted the same way, according to results of a Turkish study.
The study authors, led by Ceren Unal, MD, of Zeynep Kamil Women’s and Children’s Disease Training and Research Hospital, Istanbul, wrote that the new device, the frameless copper-releasing Gyn-CS IUD, “could be a major advance, potentially suitable for general use due to the ease and safety of the insertion procedure,” which requires limited training.
According to the study authors, most IUDs are retained in the uterus, and most have a T-shape configuration. Some are inserted immediately following expulsion of the placenta at birth, although techniques “are far from being optimal,” and devices are frequently displaced and sometimes expelled.
The newly developed Gyn-CS IUD, a redesigned version of a previous model, has an alternative “anchor” design.
The investigators tracked 140 pregnant women – 106 who underwent elective cesarean section and 34 who underwent emergency cesarean section – who had the Gyn-CS or the TCu380A IUD inserted shortly after they gave birth. Their median age was around 30 years , all were white, and all were married.
The TCu380A IUD, a decades-old model that’s been recommended by the World Health Organization, and the Gyn-CS IUD were each inserted in 70 women. The researchers then followed them for 3 months. Three women (one in the Gyn-CS IUD group and two in the TCu380A IUD group) were lost to follow-up.
In total, 61 of 70 IUDs (88%) remained in place in the Gyn-CS group, and 54 of 70 (79%) in the TCu380A group (P = .30). One (1%) Gyn-CS IUD was expelled , possibly because it was incorrectly anchored, and eight (11%) TCu380A IUDs were expelled (P = .04). There were equal numbers of medical removals (four) and nonmedical removals (two) in the two groups, and one “other medical removal” in the Gyn-CS IUD group.
“The very low expulsion rate of Gyn-CS, compared with TCu380A, is a very strong argument in favor of the anchored IUD, preventing expulsion and displacement,” Dr. Unal and colleagues reported in Contraception.
They noted that the study has a limited 3-month tracking period, but they also pointed out that most expulsions inserted post partum occurred in the initial 6 weeks.
The low expulsion rate of the Gyn-CS device “will prevent more women becoming pregnant too soon which constitutes an important safety issue during future pregnancy,” the authors wrote. “The device, preferably its high-load 10-year version, could also interest many women as a reversible alternative for tubal sterilization. Additional clinical experience with the Gyn-CS IUD is, therefore, urgently warranted.”
No study funding is reported, although Contrel Research provided the Gyn-CS devices at no charge. The study authors reported no relevant disclosures.
that’s inserted the same way, according to results of a Turkish study.
The study authors, led by Ceren Unal, MD, of Zeynep Kamil Women’s and Children’s Disease Training and Research Hospital, Istanbul, wrote that the new device, the frameless copper-releasing Gyn-CS IUD, “could be a major advance, potentially suitable for general use due to the ease and safety of the insertion procedure,” which requires limited training.
According to the study authors, most IUDs are retained in the uterus, and most have a T-shape configuration. Some are inserted immediately following expulsion of the placenta at birth, although techniques “are far from being optimal,” and devices are frequently displaced and sometimes expelled.
The newly developed Gyn-CS IUD, a redesigned version of a previous model, has an alternative “anchor” design.
The investigators tracked 140 pregnant women – 106 who underwent elective cesarean section and 34 who underwent emergency cesarean section – who had the Gyn-CS or the TCu380A IUD inserted shortly after they gave birth. Their median age was around 30 years , all were white, and all were married.
The TCu380A IUD, a decades-old model that’s been recommended by the World Health Organization, and the Gyn-CS IUD were each inserted in 70 women. The researchers then followed them for 3 months. Three women (one in the Gyn-CS IUD group and two in the TCu380A IUD group) were lost to follow-up.
In total, 61 of 70 IUDs (88%) remained in place in the Gyn-CS group, and 54 of 70 (79%) in the TCu380A group (P = .30). One (1%) Gyn-CS IUD was expelled , possibly because it was incorrectly anchored, and eight (11%) TCu380A IUDs were expelled (P = .04). There were equal numbers of medical removals (four) and nonmedical removals (two) in the two groups, and one “other medical removal” in the Gyn-CS IUD group.
“The very low expulsion rate of Gyn-CS, compared with TCu380A, is a very strong argument in favor of the anchored IUD, preventing expulsion and displacement,” Dr. Unal and colleagues reported in Contraception.
They noted that the study has a limited 3-month tracking period, but they also pointed out that most expulsions inserted post partum occurred in the initial 6 weeks.
The low expulsion rate of the Gyn-CS device “will prevent more women becoming pregnant too soon which constitutes an important safety issue during future pregnancy,” the authors wrote. “The device, preferably its high-load 10-year version, could also interest many women as a reversible alternative for tubal sterilization. Additional clinical experience with the Gyn-CS IUD is, therefore, urgently warranted.”
No study funding is reported, although Contrel Research provided the Gyn-CS devices at no charge. The study authors reported no relevant disclosures.
FROM CONTRACEPTION
Key clinical point: A newly developed IUD appears to be expelled less frequently than an older device after insertion following cesarean section births.
Major finding: 61 of 70 IUDs (88%) remained in place in women who received the frameless copper-releasing Gyn-CS device, while 54 of 70 (79%) did so in those who received the TCu380A device (P = .30).
Study details: Randomized trial of 140 women who underwent cesarean section followed by insertion of one of the two IUD types (n = 70 for each).
Disclosures: No study funding is reported, although Contrel Research provided the Gyn-CS devices at no charge. The study authors reported no relevant disclosures.
Source: Unal C et al. Contraception. 2018 Aug;98:135-40.
FDA approves first mobile app for contraceptive use
The Food and Drug Administration has approved marketing of the first medical mobile application that can be used as a contraceptive, the agency announced in a written statement.

The app, called Natural Cycles, contains an algorithm that calculates the days of the month women are likely to be fertile, based on daily body temperature readings and menstrual cycle information, and is intended for premenopausal women aged 18 years and older. App users are instructed to take their daily body temperatures with a basal body thermometer and enter the information into the app. The more-sensitive basal body thermometer can detect temperature elevations during ovulation, and the app will display a warning on days when users are most fertile. During these days, women should either abstain from sex or use protection.
In clinical studies comprising more than 15,000 women, Natural Cycles had a “perfect use” failure rate of 1.8% and a “normal use” failure rate of 6.5%. This compares favorably with other forms of contraception and birth control: Male condoms have a perfect use failure rate of 2.0% and a normal use failure rate of 18.0%, and combined oral contraceptives have a perfect use failure rate of 0.3% and a normal use failure rate of 9.0%, according to the Association of Reproductive Care Professionals.
“Consumers are increasingly using digital health technologies to inform their everyday health decisions, and this new app can provide an effective method of contraception if it’s used carefully and correctly,” Terri Cornelison, MD, PhD, assistant director for the health of women in the FDA’s Center for Devices and Radiological Health, said in the statement.
The app should not be used by women with a condition in which a pregnancy would present risk to the mother or fetus or by women who are using a birth control or hormonal treatment that inhibits ovulation. Natural Cycles does not protect against sexually transmitted infections.
The Food and Drug Administration has approved marketing of the first medical mobile application that can be used as a contraceptive, the agency announced in a written statement.
The app, called Natural Cycles, contains an algorithm that calculates the days of the month women are likely to be fertile, based on daily body temperature readings and menstrual cycle information, and is intended for premenopausal women aged 18 years and older. App users are instructed to take their daily body temperatures with a basal body thermometer and enter the information into the app. The more-sensitive basal body thermometer can detect temperature elevations during ovulation, and the app will display a warning on days when users are most fertile. During these days, women should either abstain from sex or use protection.
In clinical studies comprising more than 15,000 women, Natural Cycles had a “perfect use” failure rate of 1.8% and a “normal use” failure rate of 6.5%. This compares favorably with other forms of contraception and birth control: Male condoms have a perfect use failure rate of 2.0% and a normal use failure rate of 18.0%, and combined oral contraceptives have a perfect use failure rate of 0.3% and a normal use failure rate of 9.0%, according to the Association of Reproductive Care Professionals.
“Consumers are increasingly using digital health technologies to inform their everyday health decisions, and this new app can provide an effective method of contraception if it’s used carefully and correctly,” Terri Cornelison, MD, PhD, assistant director for the health of women in the FDA’s Center for Devices and Radiological Health, said in the statement.
The app should not be used by women with a condition in which a pregnancy would present risk to the mother or fetus or by women who are using a birth control or hormonal treatment that inhibits ovulation. Natural Cycles does not protect against sexually transmitted infections.
The Food and Drug Administration has approved marketing of the first medical mobile application that can be used as a contraceptive, the agency announced in a written statement.
The app, called Natural Cycles, contains an algorithm that calculates the days of the month women are likely to be fertile, based on daily body temperature readings and menstrual cycle information, and is intended for premenopausal women aged 18 years and older. App users are instructed to take their daily body temperatures with a basal body thermometer and enter the information into the app. The more-sensitive basal body thermometer can detect temperature elevations during ovulation, and the app will display a warning on days when users are most fertile. During these days, women should either abstain from sex or use protection.
In clinical studies comprising more than 15,000 women, Natural Cycles had a “perfect use” failure rate of 1.8% and a “normal use” failure rate of 6.5%. This compares favorably with other forms of contraception and birth control: Male condoms have a perfect use failure rate of 2.0% and a normal use failure rate of 18.0%, and combined oral contraceptives have a perfect use failure rate of 0.3% and a normal use failure rate of 9.0%, according to the Association of Reproductive Care Professionals.
“Consumers are increasingly using digital health technologies to inform their everyday health decisions, and this new app can provide an effective method of contraception if it’s used carefully and correctly,” Terri Cornelison, MD, PhD, assistant director for the health of women in the FDA’s Center for Devices and Radiological Health, said in the statement.
The app should not be used by women with a condition in which a pregnancy would present risk to the mother or fetus or by women who are using a birth control or hormonal treatment that inhibits ovulation. Natural Cycles does not protect against sexually transmitted infections.
Adjuvant chemotherapy benefits high-risk sarcoma patients
CHICAGO – Patients with high-risk soft-tissue sarcomas identified by patient data and a risk calculator had significantly better overall and disease-free survival when they were treated with adjuvant doxorubicin and ifosfamide, a retrospective analysis from a randomized clinical trial showed.
The findings suggest that future clinical trials for sarcoma therapies may need to focus on specific risk categories, investigators said.
Among 290 patients with soft tissues sarcomas (STS) of the trunk wall or extremities, adjuvant chemotherapy with doxorubicin, ifosfamide, mesna, and lenograstim more than halved the risk of death for patients determined by the nomogram to have a low probability of 10-year overall survival, reported Sandro Pasquali, MD, from the Fondazione IRCCS Istituto Nazionale dei Tumori in Milan, and his colleagues.
“These findings interpret conflicting results of randomized controlled trials on perioperative chemotherapy in STS showing that inclusion of low-risk tumors have diluted the effect of chemotherapy leading to negative results and small study effect in meta-analysis,” they wrote in a poster presented at the annual meeting of the American Society of Clinical Oncology.
The clinical trial in question, EORTC-STBSG 62931, results of which were published in 2012 in The Lancet Oncology, was technically a failure, because it did not show a significant benefit of adjuvant chemotherapy vs. observation in patients with STS.
To see whether adjuvant chemotherapy may have benefited select patients, the investigators calculated 10-year probabilities of overall survival (P-OS) for 290 patients with STS of the trunk wall or extremities out of the total trial cohort of 351 patients. The P-OS for each of three categories – low (51% or less), intermediate (52%-66%), and high (67% or greater) – was calculated using individual patient data and the freely available smartphone-based nomogram Sarculator (available in the Apple App Store and Google Play).
They calculated disease-free survival at the study median follow-up of 8 years.
The tumor histologies included malignant fibrous histiocytoma/undifferentiated pleomorphic sarcoma, liposarcoma, leiomyosarcoma, rhabdomyosarcoma, angiosarcoma, synovial sarcoma, malignant peripheral nerve sheath tumor, and others.
A total of 52 patients were in the low P-OS group, including 24 treated with observation, and 28 with adjuvant chemotherapy. Respective numbers for the intermediate and high P-OS categories were 34/34, and 90/80.
The investigators found that for patients in the low P-OS group, adjuvant chemotherapy cut the risk of death by slightly more than half, with a hazard ratio of 0.46 (P = .033). In contrast, there were no significant differences in the risk of death for patients at either intermediate or high probability of 10 year OS (HR, 1.00 and 1.08, respectively; P values not significant).
Similarly, adjuvant chemotherapy cut the risk of disease progression by the same amount, with an HR of 0.46 (P = .021), whereas there was no additional benefit among patients at either intermediate or high probability of 10-year OS (HR 0.74 and 0.90; P values were not significant).
The absolute risk reduction for adjuvant chemotherapy was 21% (8-yr disease-free survival of 34% for adjuvant chemotherapy vs. 13% for observation), with a number needed to treated of 4.76.
The study was supported by the European Organization for Research and Treatment of Cancer. Dr. Pasquali reported having no conflicts of interest.
SOURCE: Pasquali S et al. ASCO 2018, Abstract 115118.
CHICAGO – Patients with high-risk soft-tissue sarcomas identified by patient data and a risk calculator had significantly better overall and disease-free survival when they were treated with adjuvant doxorubicin and ifosfamide, a retrospective analysis from a randomized clinical trial showed.
The findings suggest that future clinical trials for sarcoma therapies may need to focus on specific risk categories, investigators said.
Among 290 patients with soft tissues sarcomas (STS) of the trunk wall or extremities, adjuvant chemotherapy with doxorubicin, ifosfamide, mesna, and lenograstim more than halved the risk of death for patients determined by the nomogram to have a low probability of 10-year overall survival, reported Sandro Pasquali, MD, from the Fondazione IRCCS Istituto Nazionale dei Tumori in Milan, and his colleagues.
“These findings interpret conflicting results of randomized controlled trials on perioperative chemotherapy in STS showing that inclusion of low-risk tumors have diluted the effect of chemotherapy leading to negative results and small study effect in meta-analysis,” they wrote in a poster presented at the annual meeting of the American Society of Clinical Oncology.
The clinical trial in question, EORTC-STBSG 62931, results of which were published in 2012 in The Lancet Oncology, was technically a failure, because it did not show a significant benefit of adjuvant chemotherapy vs. observation in patients with STS.
To see whether adjuvant chemotherapy may have benefited select patients, the investigators calculated 10-year probabilities of overall survival (P-OS) for 290 patients with STS of the trunk wall or extremities out of the total trial cohort of 351 patients. The P-OS for each of three categories – low (51% or less), intermediate (52%-66%), and high (67% or greater) – was calculated using individual patient data and the freely available smartphone-based nomogram Sarculator (available in the Apple App Store and Google Play).
They calculated disease-free survival at the study median follow-up of 8 years.
The tumor histologies included malignant fibrous histiocytoma/undifferentiated pleomorphic sarcoma, liposarcoma, leiomyosarcoma, rhabdomyosarcoma, angiosarcoma, synovial sarcoma, malignant peripheral nerve sheath tumor, and others.
A total of 52 patients were in the low P-OS group, including 24 treated with observation, and 28 with adjuvant chemotherapy. Respective numbers for the intermediate and high P-OS categories were 34/34, and 90/80.
The investigators found that for patients in the low P-OS group, adjuvant chemotherapy cut the risk of death by slightly more than half, with a hazard ratio of 0.46 (P = .033). In contrast, there were no significant differences in the risk of death for patients at either intermediate or high probability of 10 year OS (HR, 1.00 and 1.08, respectively; P values not significant).
Similarly, adjuvant chemotherapy cut the risk of disease progression by the same amount, with an HR of 0.46 (P = .021), whereas there was no additional benefit among patients at either intermediate or high probability of 10-year OS (HR 0.74 and 0.90; P values were not significant).
The absolute risk reduction for adjuvant chemotherapy was 21% (8-yr disease-free survival of 34% for adjuvant chemotherapy vs. 13% for observation), with a number needed to treated of 4.76.
The study was supported by the European Organization for Research and Treatment of Cancer. Dr. Pasquali reported having no conflicts of interest.
SOURCE: Pasquali S et al. ASCO 2018, Abstract 115118.
CHICAGO – Patients with high-risk soft-tissue sarcomas identified by patient data and a risk calculator had significantly better overall and disease-free survival when they were treated with adjuvant doxorubicin and ifosfamide, a retrospective analysis from a randomized clinical trial showed.
The findings suggest that future clinical trials for sarcoma therapies may need to focus on specific risk categories, investigators said.
Among 290 patients with soft tissues sarcomas (STS) of the trunk wall or extremities, adjuvant chemotherapy with doxorubicin, ifosfamide, mesna, and lenograstim more than halved the risk of death for patients determined by the nomogram to have a low probability of 10-year overall survival, reported Sandro Pasquali, MD, from the Fondazione IRCCS Istituto Nazionale dei Tumori in Milan, and his colleagues.
“These findings interpret conflicting results of randomized controlled trials on perioperative chemotherapy in STS showing that inclusion of low-risk tumors have diluted the effect of chemotherapy leading to negative results and small study effect in meta-analysis,” they wrote in a poster presented at the annual meeting of the American Society of Clinical Oncology.
The clinical trial in question, EORTC-STBSG 62931, results of which were published in 2012 in The Lancet Oncology, was technically a failure, because it did not show a significant benefit of adjuvant chemotherapy vs. observation in patients with STS.
To see whether adjuvant chemotherapy may have benefited select patients, the investigators calculated 10-year probabilities of overall survival (P-OS) for 290 patients with STS of the trunk wall or extremities out of the total trial cohort of 351 patients. The P-OS for each of three categories – low (51% or less), intermediate (52%-66%), and high (67% or greater) – was calculated using individual patient data and the freely available smartphone-based nomogram Sarculator (available in the Apple App Store and Google Play).
They calculated disease-free survival at the study median follow-up of 8 years.
The tumor histologies included malignant fibrous histiocytoma/undifferentiated pleomorphic sarcoma, liposarcoma, leiomyosarcoma, rhabdomyosarcoma, angiosarcoma, synovial sarcoma, malignant peripheral nerve sheath tumor, and others.
A total of 52 patients were in the low P-OS group, including 24 treated with observation, and 28 with adjuvant chemotherapy. Respective numbers for the intermediate and high P-OS categories were 34/34, and 90/80.
The investigators found that for patients in the low P-OS group, adjuvant chemotherapy cut the risk of death by slightly more than half, with a hazard ratio of 0.46 (P = .033). In contrast, there were no significant differences in the risk of death for patients at either intermediate or high probability of 10 year OS (HR, 1.00 and 1.08, respectively; P values not significant).
Similarly, adjuvant chemotherapy cut the risk of disease progression by the same amount, with an HR of 0.46 (P = .021), whereas there was no additional benefit among patients at either intermediate or high probability of 10-year OS (HR 0.74 and 0.90; P values were not significant).
The absolute risk reduction for adjuvant chemotherapy was 21% (8-yr disease-free survival of 34% for adjuvant chemotherapy vs. 13% for observation), with a number needed to treated of 4.76.
The study was supported by the European Organization for Research and Treatment of Cancer. Dr. Pasquali reported having no conflicts of interest.
SOURCE: Pasquali S et al. ASCO 2018, Abstract 115118.
REPORTING FROM ASCO 2018
Key clinical point: Patients with high-risk soft-tissue sarcomas benefit from adjuvant chemotherapy.
Major finding: Hazard ratios for death and disease progression in patients with a low probability of 10-year overall survival were 0.46 for each.
Study details: Retrospective analysis of a randomized phase 3 trial comparing adjuvant chemotherapy with observation in 290 patients with soft tissue sarcomas of the trunk wall or extremities.
Disclosures: The study was supported by the European Organisation for Research and Treatment of Cancer. Dr. Pasquali reported having no conflicts of interest.
Source: Pasquali S et al. ASCO 2018, Abstract 115118.
Sonic hedgehog inhibitors have mixed efficacy for advanced BCC
For patients with locally advanced or metastatic basal cell carcinoma, Sonic hedgehog inhibitors (SSHi) are effective but are associated with primarily partial responses, and the two Food and Drug Administration–approved agents have significant toxicities, results of a systematic review and meta-analysis indicated.
Data on patients with metastatic or locally advanced basal cell carcinoma (BCC) treated with either vismodegib (Erivedge) or sonidegib (Odomzo) showed that the two agents had roughly similar overall response rates (ORR). Vismodegib, however, had a significantly higher rate of complete responses (CR) in patients with locally advanced disease, reported Pingxing Xie, MD, PhD, and Philippe Lefrançois, MD, PhD, from McGill University, Montreal.
“Common side effects of SHHi therapy tend to incapacitate patients, leading to high discontinuation rates. Over 25% of patients stopped treatment due to side effects. Most side effects are reversible after therapy cessation, except cases of persistent alopecia have been reported,” they wrote in the Journal of the American Academy of Dermatology.
The authors conducted the review to evaluate SHHi as a class and to get a better idea of the efficacy and safety of each agent in the class. They searched the literature to identify all studies using SHHi to treat BCC with vismodegib, sonidegib, itraconazole, or the investigational compound TAK-441, and identified 14 studies focused on vismodegib, 2 on sonidegib, and 1 each on itraconazole and TAK-441.
Of the 18 studies, data from 16 were pooled in fixed-effects linear models to analyze efficacy. The pooled ORR for all patients was 59.6%, “indicating that most patients receiving SHHi achieve at least a partial response.”
Combined ORR results showed a rate of 61.9% for vismodegib, 55.2% for sonidegib, 50% for itraconazole, and 20% for TAK-441, although data for the latter two agents were limited.
In studies looking at locally advanced and metastatic BCC separately, vismodegib was numerically better but statistically similar to sonidegib for locally advanced disease (ORR, 68.8% vs. 56.6%). However, vismodegib was significantly superior to sonidegib for patients with metastatic BCC (ORR, 39.7% vs. 14.7%; P = .007).
The pooled CR for all patients was 23.5%, and there were no CRs with either itraconazole or TAK-441. The combined CR rate for vismodegib was 28% (P = .012). The combined CR rate for sonidegib was just 8.9% and was not statistically significant, the investigators found.
In subgroup analyses for locally advanced BCC, the CR rate for vismodegib was 30.9% for vismodegib (P = .012), “meaning that many patients can expect cure.” In contrast, only 3% of patients treated for locally advanced disease had a CR with sonidegib. The difference between the drugs in this subpopulation was significant (P less than .0001).
Neither drug produced significant CRs in patients with metastatic melanoma, and the pooled clinical benefit rate (all patients with stable disease or better) was 94.9%, with rates similar among all four drugs.
Pooled prevalences of adverse events showed a 67.1% prevalence of muscle spasms, 54.1% prevalence of dysgeusia, and a 57.7% prevalence of alopecia. The proportions of side effects were similar between vismodegib and sonidegib, but sonidegib was associated with a higher prevalence of upper gastrointestinal tract distress than vismodegib.
The study was partially funded by the Canadian Dermatology Foundation. The authors reported having no conflicts of interest.
SOURCE: Xie P et al. J Am Acad Dermatol. 2018 Jul 9. doi: 10.1016/j.jaad.2018.07.004.
For patients with locally advanced or metastatic basal cell carcinoma, Sonic hedgehog inhibitors (SSHi) are effective but are associated with primarily partial responses, and the two Food and Drug Administration–approved agents have significant toxicities, results of a systematic review and meta-analysis indicated.
Data on patients with metastatic or locally advanced basal cell carcinoma (BCC) treated with either vismodegib (Erivedge) or sonidegib (Odomzo) showed that the two agents had roughly similar overall response rates (ORR). Vismodegib, however, had a significantly higher rate of complete responses (CR) in patients with locally advanced disease, reported Pingxing Xie, MD, PhD, and Philippe Lefrançois, MD, PhD, from McGill University, Montreal.
“Common side effects of SHHi therapy tend to incapacitate patients, leading to high discontinuation rates. Over 25% of patients stopped treatment due to side effects. Most side effects are reversible after therapy cessation, except cases of persistent alopecia have been reported,” they wrote in the Journal of the American Academy of Dermatology.
The authors conducted the review to evaluate SHHi as a class and to get a better idea of the efficacy and safety of each agent in the class. They searched the literature to identify all studies using SHHi to treat BCC with vismodegib, sonidegib, itraconazole, or the investigational compound TAK-441, and identified 14 studies focused on vismodegib, 2 on sonidegib, and 1 each on itraconazole and TAK-441.
Of the 18 studies, data from 16 were pooled in fixed-effects linear models to analyze efficacy. The pooled ORR for all patients was 59.6%, “indicating that most patients receiving SHHi achieve at least a partial response.”
Combined ORR results showed a rate of 61.9% for vismodegib, 55.2% for sonidegib, 50% for itraconazole, and 20% for TAK-441, although data for the latter two agents were limited.
In studies looking at locally advanced and metastatic BCC separately, vismodegib was numerically better but statistically similar to sonidegib for locally advanced disease (ORR, 68.8% vs. 56.6%). However, vismodegib was significantly superior to sonidegib for patients with metastatic BCC (ORR, 39.7% vs. 14.7%; P = .007).
The pooled CR for all patients was 23.5%, and there were no CRs with either itraconazole or TAK-441. The combined CR rate for vismodegib was 28% (P = .012). The combined CR rate for sonidegib was just 8.9% and was not statistically significant, the investigators found.
In subgroup analyses for locally advanced BCC, the CR rate for vismodegib was 30.9% for vismodegib (P = .012), “meaning that many patients can expect cure.” In contrast, only 3% of patients treated for locally advanced disease had a CR with sonidegib. The difference between the drugs in this subpopulation was significant (P less than .0001).
Neither drug produced significant CRs in patients with metastatic melanoma, and the pooled clinical benefit rate (all patients with stable disease or better) was 94.9%, with rates similar among all four drugs.
Pooled prevalences of adverse events showed a 67.1% prevalence of muscle spasms, 54.1% prevalence of dysgeusia, and a 57.7% prevalence of alopecia. The proportions of side effects were similar between vismodegib and sonidegib, but sonidegib was associated with a higher prevalence of upper gastrointestinal tract distress than vismodegib.
The study was partially funded by the Canadian Dermatology Foundation. The authors reported having no conflicts of interest.
SOURCE: Xie P et al. J Am Acad Dermatol. 2018 Jul 9. doi: 10.1016/j.jaad.2018.07.004.
For patients with locally advanced or metastatic basal cell carcinoma, Sonic hedgehog inhibitors (SSHi) are effective but are associated with primarily partial responses, and the two Food and Drug Administration–approved agents have significant toxicities, results of a systematic review and meta-analysis indicated.
Data on patients with metastatic or locally advanced basal cell carcinoma (BCC) treated with either vismodegib (Erivedge) or sonidegib (Odomzo) showed that the two agents had roughly similar overall response rates (ORR). Vismodegib, however, had a significantly higher rate of complete responses (CR) in patients with locally advanced disease, reported Pingxing Xie, MD, PhD, and Philippe Lefrançois, MD, PhD, from McGill University, Montreal.
“Common side effects of SHHi therapy tend to incapacitate patients, leading to high discontinuation rates. Over 25% of patients stopped treatment due to side effects. Most side effects are reversible after therapy cessation, except cases of persistent alopecia have been reported,” they wrote in the Journal of the American Academy of Dermatology.
The authors conducted the review to evaluate SHHi as a class and to get a better idea of the efficacy and safety of each agent in the class. They searched the literature to identify all studies using SHHi to treat BCC with vismodegib, sonidegib, itraconazole, or the investigational compound TAK-441, and identified 14 studies focused on vismodegib, 2 on sonidegib, and 1 each on itraconazole and TAK-441.
Of the 18 studies, data from 16 were pooled in fixed-effects linear models to analyze efficacy. The pooled ORR for all patients was 59.6%, “indicating that most patients receiving SHHi achieve at least a partial response.”
Combined ORR results showed a rate of 61.9% for vismodegib, 55.2% for sonidegib, 50% for itraconazole, and 20% for TAK-441, although data for the latter two agents were limited.
In studies looking at locally advanced and metastatic BCC separately, vismodegib was numerically better but statistically similar to sonidegib for locally advanced disease (ORR, 68.8% vs. 56.6%). However, vismodegib was significantly superior to sonidegib for patients with metastatic BCC (ORR, 39.7% vs. 14.7%; P = .007).
The pooled CR for all patients was 23.5%, and there were no CRs with either itraconazole or TAK-441. The combined CR rate for vismodegib was 28% (P = .012). The combined CR rate for sonidegib was just 8.9% and was not statistically significant, the investigators found.
In subgroup analyses for locally advanced BCC, the CR rate for vismodegib was 30.9% for vismodegib (P = .012), “meaning that many patients can expect cure.” In contrast, only 3% of patients treated for locally advanced disease had a CR with sonidegib. The difference between the drugs in this subpopulation was significant (P less than .0001).
Neither drug produced significant CRs in patients with metastatic melanoma, and the pooled clinical benefit rate (all patients with stable disease or better) was 94.9%, with rates similar among all four drugs.
Pooled prevalences of adverse events showed a 67.1% prevalence of muscle spasms, 54.1% prevalence of dysgeusia, and a 57.7% prevalence of alopecia. The proportions of side effects were similar between vismodegib and sonidegib, but sonidegib was associated with a higher prevalence of upper gastrointestinal tract distress than vismodegib.
The study was partially funded by the Canadian Dermatology Foundation. The authors reported having no conflicts of interest.
SOURCE: Xie P et al. J Am Acad Dermatol. 2018 Jul 9. doi: 10.1016/j.jaad.2018.07.004.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Key clinical point: Sonic hedgehog inhibitors (SHHi) have mixed efficacy against metastatic or locally advanced basal cell carcinoma (BCC).
Major finding: Combined overall response rate results showed a rate of 61.9% for vismodegib, 55.2% for sonidegib, 50% for itraconazole, and 20% for TAK-441.
Study details: A systematic review and meta-analysis of studies looking at SHHi in patients with BCC.
Disclosures: The study was partially funded by the Canadian Dermatology Foundation. The authors reported having no conflicts of interest.
Source: Xie P et al. J Am Acad Dermatol. 2018 Jul 9. doi:10.1016/j.jaad.2018.07.004.
Groups release guidelines for CAR T treatment in children
emphasize the need for a flexible approach to detect early signs of serious complications for younger patients treated with this emerging class of medicines.

Researchers at the University of Texas MD Anderson Cancer Center, Houston, and the Pediatric Acute Lung Injury and Sepsis Investigators Network (PALISI) developed the guidelines, which were published in Nature Reviews Clinical Oncology. The recommendations build on the guidelines for more general use of these medicines from MD Anderson’s CARTOX Program, which Nature Reviews Clinical Oncology published in 2017.
Among the chief concerns with this new class of medicines are cytokine-release syndrome (CRS) and CAR T cell-related encephalopathy syndrome (CRES), according to Kris Michael Mahadeo, MD MPH, of the MD Anderson Cancer Center and his coauthors of the new paper.
Some of the tools used for older patients in screening for complications with CAR T drugs don’t work as well with younger ones, Dr. Mahadeo said in an interview. For instance, at MD Anderson, a handwriting sample is used to monitor patients for CAR T cell-related encephalopathy syndrome, which has symptoms of confusion and delirium. Patients provide a baseline handwriting sample of a single sentence that’s scanned into the medical record, and then they are asked to write this again during their time in the hospital, he said. But this tool may not work for children too young to write well.
The new guidelines suggest using the Cornell Assessment of Pediatric Delirium (CAPD) or to evaluate a child’s mental state, asking questions about eye contact, and level of awareness and mood, Dr. Mahadeo said. An alternative for patients aged 12 years and older with greater cognitive ability is the CARTOX-10 grading system.
“The nurses who spent most of the day with these patients will observe them over their shift and kind of get an idea of what was normal and answer a series of questions” through the CAPD tool, which is already used in ICUs, Dr. Mahadeo said. “It takes into consideration both the nurses’ perception and the parents, or whoever is at the bedside with the child. So that if they have a concern, it gives them a point that actually escalates things upward.”
The newly published recommendations also remind physicians and others caring for young patients to pay attention to these reports.
“Parent and/or caregiver concerns should be addressed because early signs or symptoms of CRS can be subtle and best recognized by those who know the child best,” Dr. Mahadeo and his colleagues wrote in a summary of key recommendations in the paper.
The recommendations also noted a need for close monitoring for complications such as hypotension, hypocalcemia, and catheter-related pain in young patients who require a leukapheresis catheter for cell collection. Infant and younger children “might not verbalize these symptoms,” according to the researchers.
Other recommendations include:
- Obtaining the child’s assent when appropriate, with psychological services often aiding in this goal. Dr. Mahadeo and his colleagues recommend considering “age-appropriate advance directives.”
- Maintaining high vigilance for sinus tachycardia as an early sign of CRS, using age-specific normal range or baseline values.
- Giving pediatric dosing of tocilizumab, with patients weighing less than 30 kg receiving 12 mg/kg, and those weighing 30 kg or greater receiving 8 mg/kg.
- Considering participation with a prospective collaboration with intensive-care registries that could allow accurate data entry of cell-therapy variables into the Center for International Blood and Marrow Transplant Research registry by cell-therapy programs.
The Food and Drug Administration approved the first two CAR T-cell therapies in the United States in 2017: Novartis’ tisagenlecleucel (Kymriah) for children and young adults with B-cell precursor acute lymphoblastic leukemia and later for adults with large B-cell lymphoma; and axicabtagene ciloleucel (Yescarta), sold by Gilead, for adults with large B-cell lymphoma. The therapies involve reengineering a patient’s T cells such that they recognize the threat of cancer, and then introducing them back into the body. The European Medicines Agency’s Committee for Medicinal Products for Human Use in June recommended granting marketing authorization to these drugs.
In the new pediatric guidelines, Dr. Mahadeo and his colleagues noted the use of CAR T-cell therapies for treatment of solid tumors and other malignancies in children already “is being explored.” “Moreover, consideration of earlier or upfront use of CAR T-cell therapy might spare patients the acute and long-term toxicities associated with traditional chemotherapy and/or radiation regimens,” they wrote.

There’s been great interest in learning how to most safely use the CAR T cell therapies, said Helen Heslop, MD, of Baylor College of Medicine.
She pointed to a 2014 publication in the journal Blood from Daniel W. Lee and his colleagues as an earlier example of this research. By now, cancer centers will have worked out their own procedures for pediatric use of CAR T therapies, hewing to standards set by the Foundation for the Accreditation of Cellular Therapy (FACT), Dr. Heslop said.
Dr. Heslop also stressed the role of the FDA in requiring risk evaluation and management strategy programs for these drugs. All of this, including the new guidelines from Dr. Mahadeo and his colleagues, is part of a growing body of research into safe use of CAR T therapies, Dr. Heslop said.
“It’s an active area of research,” she said. “Most centers will look at all of it and then develop what works best in their own individual center for providing the best care for the patients.”
The newly published guidelines could prove an “important contribution” to managing the risk of CAR T therapies, Phyllis I. Warkentin, MD, chief medical officer for FACT, said in an interview, while stressing that they were not more or less important than other similar efforts. Physicians learning how to use the CAR T therapies may welcome new input, as most of what’s been published has been about adults, she said.
“You don’t have the luxury of a lot of time to be learning on the job, so to speak,” with CAR T therapies, she said. “Many of the toxicities are fairly severe and fairly sudden.”
Dr. Heslop has been on advisory board for Gilead and Novartis. Dr. Warkentin and Dr. Mahadeo each reported having no financial disclosures. Other authors of the guidelines paper reported a patent with applications in the field of gene-modified T cell therapy for cancer, as well as financial ties to Cellectis, NexImmune, Torque Pharma, Kite Pharma (a Gilead company), Poseida Therapeutics, Celgene, Novartis, and Unum Therapeutics.
SOURCE: Mahadeo KM et al. Nat Rev Clin Oncol. 2018 Aug 6. doi: 10.1038/s41571-018-0075-2.
emphasize the need for a flexible approach to detect early signs of serious complications for younger patients treated with this emerging class of medicines.
Researchers at the University of Texas MD Anderson Cancer Center, Houston, and the Pediatric Acute Lung Injury and Sepsis Investigators Network (PALISI) developed the guidelines, which were published in Nature Reviews Clinical Oncology. The recommendations build on the guidelines for more general use of these medicines from MD Anderson’s CARTOX Program, which Nature Reviews Clinical Oncology published in 2017.
Among the chief concerns with this new class of medicines are cytokine-release syndrome (CRS) and CAR T cell-related encephalopathy syndrome (CRES), according to Kris Michael Mahadeo, MD MPH, of the MD Anderson Cancer Center and his coauthors of the new paper.
Some of the tools used for older patients in screening for complications with CAR T drugs don’t work as well with younger ones, Dr. Mahadeo said in an interview. For instance, at MD Anderson, a handwriting sample is used to monitor patients for CAR T cell-related encephalopathy syndrome, which has symptoms of confusion and delirium. Patients provide a baseline handwriting sample of a single sentence that’s scanned into the medical record, and then they are asked to write this again during their time in the hospital, he said. But this tool may not work for children too young to write well.
The new guidelines suggest using the Cornell Assessment of Pediatric Delirium (CAPD) or to evaluate a child’s mental state, asking questions about eye contact, and level of awareness and mood, Dr. Mahadeo said. An alternative for patients aged 12 years and older with greater cognitive ability is the CARTOX-10 grading system.
“The nurses who spent most of the day with these patients will observe them over their shift and kind of get an idea of what was normal and answer a series of questions” through the CAPD tool, which is already used in ICUs, Dr. Mahadeo said. “It takes into consideration both the nurses’ perception and the parents, or whoever is at the bedside with the child. So that if they have a concern, it gives them a point that actually escalates things upward.”
The newly published recommendations also remind physicians and others caring for young patients to pay attention to these reports.
“Parent and/or caregiver concerns should be addressed because early signs or symptoms of CRS can be subtle and best recognized by those who know the child best,” Dr. Mahadeo and his colleagues wrote in a summary of key recommendations in the paper.
The recommendations also noted a need for close monitoring for complications such as hypotension, hypocalcemia, and catheter-related pain in young patients who require a leukapheresis catheter for cell collection. Infant and younger children “might not verbalize these symptoms,” according to the researchers.
Other recommendations include:
- Obtaining the child’s assent when appropriate, with psychological services often aiding in this goal. Dr. Mahadeo and his colleagues recommend considering “age-appropriate advance directives.”
- Maintaining high vigilance for sinus tachycardia as an early sign of CRS, using age-specific normal range or baseline values.
- Giving pediatric dosing of tocilizumab, with patients weighing less than 30 kg receiving 12 mg/kg, and those weighing 30 kg or greater receiving 8 mg/kg.
- Considering participation with a prospective collaboration with intensive-care registries that could allow accurate data entry of cell-therapy variables into the Center for International Blood and Marrow Transplant Research registry by cell-therapy programs.
The Food and Drug Administration approved the first two CAR T-cell therapies in the United States in 2017: Novartis’ tisagenlecleucel (Kymriah) for children and young adults with B-cell precursor acute lymphoblastic leukemia and later for adults with large B-cell lymphoma; and axicabtagene ciloleucel (Yescarta), sold by Gilead, for adults with large B-cell lymphoma. The therapies involve reengineering a patient’s T cells such that they recognize the threat of cancer, and then introducing them back into the body. The European Medicines Agency’s Committee for Medicinal Products for Human Use in June recommended granting marketing authorization to these drugs.
In the new pediatric guidelines, Dr. Mahadeo and his colleagues noted the use of CAR T-cell therapies for treatment of solid tumors and other malignancies in children already “is being explored.” “Moreover, consideration of earlier or upfront use of CAR T-cell therapy might spare patients the acute and long-term toxicities associated with traditional chemotherapy and/or radiation regimens,” they wrote.
There’s been great interest in learning how to most safely use the CAR T cell therapies, said Helen Heslop, MD, of Baylor College of Medicine.
She pointed to a 2014 publication in the journal Blood from Daniel W. Lee and his colleagues as an earlier example of this research. By now, cancer centers will have worked out their own procedures for pediatric use of CAR T therapies, hewing to standards set by the Foundation for the Accreditation of Cellular Therapy (FACT), Dr. Heslop said.
Dr. Heslop also stressed the role of the FDA in requiring risk evaluation and management strategy programs for these drugs. All of this, including the new guidelines from Dr. Mahadeo and his colleagues, is part of a growing body of research into safe use of CAR T therapies, Dr. Heslop said.
“It’s an active area of research,” she said. “Most centers will look at all of it and then develop what works best in their own individual center for providing the best care for the patients.”
The newly published guidelines could prove an “important contribution” to managing the risk of CAR T therapies, Phyllis I. Warkentin, MD, chief medical officer for FACT, said in an interview, while stressing that they were not more or less important than other similar efforts. Physicians learning how to use the CAR T therapies may welcome new input, as most of what’s been published has been about adults, she said.
“You don’t have the luxury of a lot of time to be learning on the job, so to speak,” with CAR T therapies, she said. “Many of the toxicities are fairly severe and fairly sudden.”
Dr. Heslop has been on advisory board for Gilead and Novartis. Dr. Warkentin and Dr. Mahadeo each reported having no financial disclosures. Other authors of the guidelines paper reported a patent with applications in the field of gene-modified T cell therapy for cancer, as well as financial ties to Cellectis, NexImmune, Torque Pharma, Kite Pharma (a Gilead company), Poseida Therapeutics, Celgene, Novartis, and Unum Therapeutics.
SOURCE: Mahadeo KM et al. Nat Rev Clin Oncol. 2018 Aug 6. doi: 10.1038/s41571-018-0075-2.
emphasize the need for a flexible approach to detect early signs of serious complications for younger patients treated with this emerging class of medicines.
Researchers at the University of Texas MD Anderson Cancer Center, Houston, and the Pediatric Acute Lung Injury and Sepsis Investigators Network (PALISI) developed the guidelines, which were published in Nature Reviews Clinical Oncology. The recommendations build on the guidelines for more general use of these medicines from MD Anderson’s CARTOX Program, which Nature Reviews Clinical Oncology published in 2017.
Among the chief concerns with this new class of medicines are cytokine-release syndrome (CRS) and CAR T cell-related encephalopathy syndrome (CRES), according to Kris Michael Mahadeo, MD MPH, of the MD Anderson Cancer Center and his coauthors of the new paper.
Some of the tools used for older patients in screening for complications with CAR T drugs don’t work as well with younger ones, Dr. Mahadeo said in an interview. For instance, at MD Anderson, a handwriting sample is used to monitor patients for CAR T cell-related encephalopathy syndrome, which has symptoms of confusion and delirium. Patients provide a baseline handwriting sample of a single sentence that’s scanned into the medical record, and then they are asked to write this again during their time in the hospital, he said. But this tool may not work for children too young to write well.
The new guidelines suggest using the Cornell Assessment of Pediatric Delirium (CAPD) or to evaluate a child’s mental state, asking questions about eye contact, and level of awareness and mood, Dr. Mahadeo said. An alternative for patients aged 12 years and older with greater cognitive ability is the CARTOX-10 grading system.
“The nurses who spent most of the day with these patients will observe them over their shift and kind of get an idea of what was normal and answer a series of questions” through the CAPD tool, which is already used in ICUs, Dr. Mahadeo said. “It takes into consideration both the nurses’ perception and the parents, or whoever is at the bedside with the child. So that if they have a concern, it gives them a point that actually escalates things upward.”
The newly published recommendations also remind physicians and others caring for young patients to pay attention to these reports.
“Parent and/or caregiver concerns should be addressed because early signs or symptoms of CRS can be subtle and best recognized by those who know the child best,” Dr. Mahadeo and his colleagues wrote in a summary of key recommendations in the paper.
The recommendations also noted a need for close monitoring for complications such as hypotension, hypocalcemia, and catheter-related pain in young patients who require a leukapheresis catheter for cell collection. Infant and younger children “might not verbalize these symptoms,” according to the researchers.
Other recommendations include:
- Obtaining the child’s assent when appropriate, with psychological services often aiding in this goal. Dr. Mahadeo and his colleagues recommend considering “age-appropriate advance directives.”
- Maintaining high vigilance for sinus tachycardia as an early sign of CRS, using age-specific normal range or baseline values.
- Giving pediatric dosing of tocilizumab, with patients weighing less than 30 kg receiving 12 mg/kg, and those weighing 30 kg or greater receiving 8 mg/kg.
- Considering participation with a prospective collaboration with intensive-care registries that could allow accurate data entry of cell-therapy variables into the Center for International Blood and Marrow Transplant Research registry by cell-therapy programs.
The Food and Drug Administration approved the first two CAR T-cell therapies in the United States in 2017: Novartis’ tisagenlecleucel (Kymriah) for children and young adults with B-cell precursor acute lymphoblastic leukemia and later for adults with large B-cell lymphoma; and axicabtagene ciloleucel (Yescarta), sold by Gilead, for adults with large B-cell lymphoma. The therapies involve reengineering a patient’s T cells such that they recognize the threat of cancer, and then introducing them back into the body. The European Medicines Agency’s Committee for Medicinal Products for Human Use in June recommended granting marketing authorization to these drugs.
In the new pediatric guidelines, Dr. Mahadeo and his colleagues noted the use of CAR T-cell therapies for treatment of solid tumors and other malignancies in children already “is being explored.” “Moreover, consideration of earlier or upfront use of CAR T-cell therapy might spare patients the acute and long-term toxicities associated with traditional chemotherapy and/or radiation regimens,” they wrote.
There’s been great interest in learning how to most safely use the CAR T cell therapies, said Helen Heslop, MD, of Baylor College of Medicine.
She pointed to a 2014 publication in the journal Blood from Daniel W. Lee and his colleagues as an earlier example of this research. By now, cancer centers will have worked out their own procedures for pediatric use of CAR T therapies, hewing to standards set by the Foundation for the Accreditation of Cellular Therapy (FACT), Dr. Heslop said.
Dr. Heslop also stressed the role of the FDA in requiring risk evaluation and management strategy programs for these drugs. All of this, including the new guidelines from Dr. Mahadeo and his colleagues, is part of a growing body of research into safe use of CAR T therapies, Dr. Heslop said.
“It’s an active area of research,” she said. “Most centers will look at all of it and then develop what works best in their own individual center for providing the best care for the patients.”
The newly published guidelines could prove an “important contribution” to managing the risk of CAR T therapies, Phyllis I. Warkentin, MD, chief medical officer for FACT, said in an interview, while stressing that they were not more or less important than other similar efforts. Physicians learning how to use the CAR T therapies may welcome new input, as most of what’s been published has been about adults, she said.
“You don’t have the luxury of a lot of time to be learning on the job, so to speak,” with CAR T therapies, she said. “Many of the toxicities are fairly severe and fairly sudden.”
Dr. Heslop has been on advisory board for Gilead and Novartis. Dr. Warkentin and Dr. Mahadeo each reported having no financial disclosures. Other authors of the guidelines paper reported a patent with applications in the field of gene-modified T cell therapy for cancer, as well as financial ties to Cellectis, NexImmune, Torque Pharma, Kite Pharma (a Gilead company), Poseida Therapeutics, Celgene, Novartis, and Unum Therapeutics.
SOURCE: Mahadeo KM et al. Nat Rev Clin Oncol. 2018 Aug 6. doi: 10.1038/s41571-018-0075-2.
FROM NATURE REVIEWS CLINICAL ONCOLOGY
Key clinical point: Multidisciplinary approach aids in managing CAR T-cell therapy’s severe potential toxicities in children.
Major finding: The guideline calls for pediatric dosing of tocilizumab, with patients weighing less than 30 kg receiving 12 mg/kg, and those weighing 30 kg or greater receiving 8 mg/kg.
Study details: Consensus guidelines on the care of children receiving CAR T-cell therapy from the Pediatric Acute Lung Injury and Sepsis Investigators and the MD Anderson Cancer Center CARTOX program.
Disclosures: Dr. Mahadeo reported having no financial disclosures. Other coauthors reported a patent with applications in the field of gene-modified T cell therapy for cancer, as well as financial ties to Cellectis, NexImmune, Torque Pharma, Kite Pharma (a Gilead company), Poseida Therapeutics, Celgene, Novartis, and Unum Therapeutics.
Source: Mahadeo KM et al. Nat Rev Clin Oncol. 2018 Aug 6. doi: 10.1038/s41571-018-0075-2.