User login
FDA approves Cassipa for opioid dependence

Cassipa sublingual film, made by Teva Pharmaceuticals, is a 16 mg/4 mg dosage of buprenorphine and naloxone for the maintenance treatment of opioid dependence. Buprenorphine and naloxone sublingual film also is approved in both brand name and generic versions and in various strengths, the FDA said in a press release.
Cassipa should be used as part of a complete treatment plan that includes counseling and psychosocial support and should be used only after patient induction and stabilization up to a dose of 16 mg of buprenorphine using another marketed product. These products may only be prescribed by Drug Addiction Treatment Act (DATA)–certified prescribers.
“There’s an urgent need to ensure access to, and wider use and understanding of, medication-assisted treatment for opioid use disorder ... the FDA recently described a streamlined approach to drug development for certain medication-assisted treatments that are based on buprenorphine. This streamlined approach can reduce drug development costs, so products may be offered at a lower price to patients and we can broaden access to treatment,” FDA Commissioner Scott Gottlieb, MD, said in the statement.
He added that “individuals who successfully transition onto medication-assisted treatment are not swapping one addiction for another. Opioid replacement therapy can be an important part of effective treatment. Opioid use disorder should be viewed similarly to any other chronic condition that is treated with medication.”
Medication-assisted treatment (MAT) is a comprehensive approach that combines FDA-approved medications (currently methadone, buprenorphine, or naltrexone) with counseling and other behavioral therapies to treat patients with opioid use disorder. Regular adherence to MAT with buprenorphine reduces opioid withdrawal symptoms and the desire to use opioids. According to the Substance Abuse and Mental Health Services Administration, patients receiving MAT for treatment of their opioid use disorder cut their risk of death from all causes in half.
Improving access to prevention, treatment, and recovery services, including the full range of MAT, is part of the Department of Health and Human Services’ Five-Point Strategy to Combat the Opioid Crisis. Last month, the FDA issued draft guidance outlining new ways for drug developers to consider measuring and demonstrating the effectiveness and benefits of new or existing MAT products, building on another draft guidance issued in April outlining the agency’s current thinking about drug development and trial design issues relevant to the study of depot buprenorphine products (i.e., modified-release products for injection or implantation). In June, the agency also approved the first generic versions of Suboxone (buprenorphine and naloxone) sublingual film in multiple strengths, the statement said.
Cassipa was approved through the abbreviated 505(b)(2) approval pathway and its application relied, in part, on the FDA’s finding of safety and effectiveness for Suboxone sublingual film to support approval. The applicant demonstrated that reliance on the FDA’s finding of safety and effectiveness for Suboxone was scientifically justified and provided Cassipa-specific pharmacokinetic data to establish the drug’s safety and efficacy for its approved uses, according to the FDA.
Adverse events commonly observed with the buprenorphine and naloxone sublingual film are oral hypoesthesia, glossodynia, oral mucosal erythema, headache, nausea, vomiting, hyperhidrosis, constipation, signs and symptoms of withdrawal, insomnia, pain, and peripheral edema.
Cassipa sublingual film, made by Teva Pharmaceuticals, is a 16 mg/4 mg dosage of buprenorphine and naloxone for the maintenance treatment of opioid dependence. Buprenorphine and naloxone sublingual film also is approved in both brand name and generic versions and in various strengths, the FDA said in a press release.
Cassipa should be used as part of a complete treatment plan that includes counseling and psychosocial support and should be used only after patient induction and stabilization up to a dose of 16 mg of buprenorphine using another marketed product. These products may only be prescribed by Drug Addiction Treatment Act (DATA)–certified prescribers.
“There’s an urgent need to ensure access to, and wider use and understanding of, medication-assisted treatment for opioid use disorder ... the FDA recently described a streamlined approach to drug development for certain medication-assisted treatments that are based on buprenorphine. This streamlined approach can reduce drug development costs, so products may be offered at a lower price to patients and we can broaden access to treatment,” FDA Commissioner Scott Gottlieb, MD, said in the statement.
He added that “individuals who successfully transition onto medication-assisted treatment are not swapping one addiction for another. Opioid replacement therapy can be an important part of effective treatment. Opioid use disorder should be viewed similarly to any other chronic condition that is treated with medication.”
Medication-assisted treatment (MAT) is a comprehensive approach that combines FDA-approved medications (currently methadone, buprenorphine, or naltrexone) with counseling and other behavioral therapies to treat patients with opioid use disorder. Regular adherence to MAT with buprenorphine reduces opioid withdrawal symptoms and the desire to use opioids. According to the Substance Abuse and Mental Health Services Administration, patients receiving MAT for treatment of their opioid use disorder cut their risk of death from all causes in half.
Improving access to prevention, treatment, and recovery services, including the full range of MAT, is part of the Department of Health and Human Services’ Five-Point Strategy to Combat the Opioid Crisis. Last month, the FDA issued draft guidance outlining new ways for drug developers to consider measuring and demonstrating the effectiveness and benefits of new or existing MAT products, building on another draft guidance issued in April outlining the agency’s current thinking about drug development and trial design issues relevant to the study of depot buprenorphine products (i.e., modified-release products for injection or implantation). In June, the agency also approved the first generic versions of Suboxone (buprenorphine and naloxone) sublingual film in multiple strengths, the statement said.
Cassipa was approved through the abbreviated 505(b)(2) approval pathway and its application relied, in part, on the FDA’s finding of safety and effectiveness for Suboxone sublingual film to support approval. The applicant demonstrated that reliance on the FDA’s finding of safety and effectiveness for Suboxone was scientifically justified and provided Cassipa-specific pharmacokinetic data to establish the drug’s safety and efficacy for its approved uses, according to the FDA.
Adverse events commonly observed with the buprenorphine and naloxone sublingual film are oral hypoesthesia, glossodynia, oral mucosal erythema, headache, nausea, vomiting, hyperhidrosis, constipation, signs and symptoms of withdrawal, insomnia, pain, and peripheral edema.
Cassipa sublingual film, made by Teva Pharmaceuticals, is a 16 mg/4 mg dosage of buprenorphine and naloxone for the maintenance treatment of opioid dependence. Buprenorphine and naloxone sublingual film also is approved in both brand name and generic versions and in various strengths, the FDA said in a press release.
Cassipa should be used as part of a complete treatment plan that includes counseling and psychosocial support and should be used only after patient induction and stabilization up to a dose of 16 mg of buprenorphine using another marketed product. These products may only be prescribed by Drug Addiction Treatment Act (DATA)–certified prescribers.
“There’s an urgent need to ensure access to, and wider use and understanding of, medication-assisted treatment for opioid use disorder ... the FDA recently described a streamlined approach to drug development for certain medication-assisted treatments that are based on buprenorphine. This streamlined approach can reduce drug development costs, so products may be offered at a lower price to patients and we can broaden access to treatment,” FDA Commissioner Scott Gottlieb, MD, said in the statement.
He added that “individuals who successfully transition onto medication-assisted treatment are not swapping one addiction for another. Opioid replacement therapy can be an important part of effective treatment. Opioid use disorder should be viewed similarly to any other chronic condition that is treated with medication.”
Medication-assisted treatment (MAT) is a comprehensive approach that combines FDA-approved medications (currently methadone, buprenorphine, or naltrexone) with counseling and other behavioral therapies to treat patients with opioid use disorder. Regular adherence to MAT with buprenorphine reduces opioid withdrawal symptoms and the desire to use opioids. According to the Substance Abuse and Mental Health Services Administration, patients receiving MAT for treatment of their opioid use disorder cut their risk of death from all causes in half.
Improving access to prevention, treatment, and recovery services, including the full range of MAT, is part of the Department of Health and Human Services’ Five-Point Strategy to Combat the Opioid Crisis. Last month, the FDA issued draft guidance outlining new ways for drug developers to consider measuring and demonstrating the effectiveness and benefits of new or existing MAT products, building on another draft guidance issued in April outlining the agency’s current thinking about drug development and trial design issues relevant to the study of depot buprenorphine products (i.e., modified-release products for injection or implantation). In June, the agency also approved the first generic versions of Suboxone (buprenorphine and naloxone) sublingual film in multiple strengths, the statement said.
Cassipa was approved through the abbreviated 505(b)(2) approval pathway and its application relied, in part, on the FDA’s finding of safety and effectiveness for Suboxone sublingual film to support approval. The applicant demonstrated that reliance on the FDA’s finding of safety and effectiveness for Suboxone was scientifically justified and provided Cassipa-specific pharmacokinetic data to establish the drug’s safety and efficacy for its approved uses, according to the FDA.
Adverse events commonly observed with the buprenorphine and naloxone sublingual film are oral hypoesthesia, glossodynia, oral mucosal erythema, headache, nausea, vomiting, hyperhidrosis, constipation, signs and symptoms of withdrawal, insomnia, pain, and peripheral edema.
Risk Factors for Epilepsy after Traumatic Brain Injury
Among patients who have experienced a traumatic brain injury (TBI), the likelihood of developing post-traumatic epilepsy (PTE) increases with age, the early onset of seizures, and the severity of the brain injury, according to an analysis of insurance claims from 2004 to 2014.
- There were approximately 2.8 million emergency room visits, hospitalizations and deaths from traumatic brain injuries in the US in 2013.
- Early seizures occurred in 0.5% of patients with TBI.
- Over a 9 year period, the incidence of post-traumatic epilepsy (PTE) increased from 1 to 4% in this population.
- Early onset of seizures, older age, and TBI severity increased the likelihood of PTE.
- Prophylactic acetazolamide seemed to reduce the risk of PTE, when compared to patients who had received no anti-epilepsy drugs.
DeGrauw X, Thurman D, Xu L, et al. Epidemiology of traumatic brain injury-associated epilepsy and early use of anti-epilepsy drugs: An analysis of insurance claims data, 2004-2014. [Published online ahead of print July 23, 2018] Epilepsy Res. doi: 10.1016/j.eplepsyres.2018.07.012
Among patients who have experienced a traumatic brain injury (TBI), the likelihood of developing post-traumatic epilepsy (PTE) increases with age, the early onset of seizures, and the severity of the brain injury, according to an analysis of insurance claims from 2004 to 2014.
- There were approximately 2.8 million emergency room visits, hospitalizations and deaths from traumatic brain injuries in the US in 2013.
- Early seizures occurred in 0.5% of patients with TBI.
- Over a 9 year period, the incidence of post-traumatic epilepsy (PTE) increased from 1 to 4% in this population.
- Early onset of seizures, older age, and TBI severity increased the likelihood of PTE.
- Prophylactic acetazolamide seemed to reduce the risk of PTE, when compared to patients who had received no anti-epilepsy drugs.
DeGrauw X, Thurman D, Xu L, et al. Epidemiology of traumatic brain injury-associated epilepsy and early use of anti-epilepsy drugs: An analysis of insurance claims data, 2004-2014. [Published online ahead of print July 23, 2018] Epilepsy Res. doi: 10.1016/j.eplepsyres.2018.07.012
Among patients who have experienced a traumatic brain injury (TBI), the likelihood of developing post-traumatic epilepsy (PTE) increases with age, the early onset of seizures, and the severity of the brain injury, according to an analysis of insurance claims from 2004 to 2014.
- There were approximately 2.8 million emergency room visits, hospitalizations and deaths from traumatic brain injuries in the US in 2013.
- Early seizures occurred in 0.5% of patients with TBI.
- Over a 9 year period, the incidence of post-traumatic epilepsy (PTE) increased from 1 to 4% in this population.
- Early onset of seizures, older age, and TBI severity increased the likelihood of PTE.
- Prophylactic acetazolamide seemed to reduce the risk of PTE, when compared to patients who had received no anti-epilepsy drugs.
DeGrauw X, Thurman D, Xu L, et al. Epidemiology of traumatic brain injury-associated epilepsy and early use of anti-epilepsy drugs: An analysis of insurance claims data, 2004-2014. [Published online ahead of print July 23, 2018] Epilepsy Res. doi: 10.1016/j.eplepsyres.2018.07.012
Is It Possible to Predict Seizures?
Conventional medical wisdom insists that it is not possible to predict the onset of seizures with any certainty, but recent research suggests otherwise, according to a review published in Nature Reviews Neurology.
- An influential 2007 review of the medical literature concluded that there was insufficient evidence to foresee the onset of seizures.
- However, an international team of experts summarized several recent advances that suggest seizure prediction may be possible.
- The new review discusses a small experiment in which intracranial EEGs were used prospectively to predict seizures.
- Additional advances include EEG databases, seizure prediction competitions, and a better understanding of the mechanism of action behind seizures.
Kuhlmann L, Lehnertz K, Richardson MP, et al. Seizure prediction - ready for a new era. [Published online ahead of print August 21, 2018] Nat Rev Neurol. doi: 10.1038/s41582-018-0055-2
Conventional medical wisdom insists that it is not possible to predict the onset of seizures with any certainty, but recent research suggests otherwise, according to a review published in Nature Reviews Neurology.
- An influential 2007 review of the medical literature concluded that there was insufficient evidence to foresee the onset of seizures.
- However, an international team of experts summarized several recent advances that suggest seizure prediction may be possible.
- The new review discusses a small experiment in which intracranial EEGs were used prospectively to predict seizures.
- Additional advances include EEG databases, seizure prediction competitions, and a better understanding of the mechanism of action behind seizures.
Kuhlmann L, Lehnertz K, Richardson MP, et al. Seizure prediction - ready for a new era. [Published online ahead of print August 21, 2018] Nat Rev Neurol. doi: 10.1038/s41582-018-0055-2
Conventional medical wisdom insists that it is not possible to predict the onset of seizures with any certainty, but recent research suggests otherwise, according to a review published in Nature Reviews Neurology.
- An influential 2007 review of the medical literature concluded that there was insufficient evidence to foresee the onset of seizures.
- However, an international team of experts summarized several recent advances that suggest seizure prediction may be possible.
- The new review discusses a small experiment in which intracranial EEGs were used prospectively to predict seizures.
- Additional advances include EEG databases, seizure prediction competitions, and a better understanding of the mechanism of action behind seizures.
Kuhlmann L, Lehnertz K, Richardson MP, et al. Seizure prediction - ready for a new era. [Published online ahead of print August 21, 2018] Nat Rev Neurol. doi: 10.1038/s41582-018-0055-2
Managing Sleep Disorders in Epilepsy
Understanding the relationship between epilepsy and sleep disorders can help clinicians improve the management of both disorders according to a recent review in Epilepsy Research.
- Sleep apnea, insomnia, restless legs syndrome and parasomnias often occur in patients with epilepsy.
- Researchers from Brigham and Women’s Hospital in Boston and Mayo Clinic reviewed the symptoms and diagnosis of these sleep disorders to help clinicians screen patients with epilepsy at risk for these problems.
- The review also explores treatment options and several case reports that illustrate the consequences of said sleep disorders in those with epilepsy.
- Latreille et al believe that employing the latest diagnostic and treatment approaches to co-morbid sleep disorders may improve patients’ functional status, alertness, quality of life and seizure burden.
Latreille V, St. Louis EK, Pavlova M. Co-morbid sleep disorders and epilepsy: A narrative review and case examples. Epilepsy Res. 2018; 145:185-197. https://doi.org/10.1016/j.eplepsyres.2018.07.005
Understanding the relationship between epilepsy and sleep disorders can help clinicians improve the management of both disorders according to a recent review in Epilepsy Research.
- Sleep apnea, insomnia, restless legs syndrome and parasomnias often occur in patients with epilepsy.
- Researchers from Brigham and Women’s Hospital in Boston and Mayo Clinic reviewed the symptoms and diagnosis of these sleep disorders to help clinicians screen patients with epilepsy at risk for these problems.
- The review also explores treatment options and several case reports that illustrate the consequences of said sleep disorders in those with epilepsy.
- Latreille et al believe that employing the latest diagnostic and treatment approaches to co-morbid sleep disorders may improve patients’ functional status, alertness, quality of life and seizure burden.
Latreille V, St. Louis EK, Pavlova M. Co-morbid sleep disorders and epilepsy: A narrative review and case examples. Epilepsy Res. 2018; 145:185-197. https://doi.org/10.1016/j.eplepsyres.2018.07.005
Understanding the relationship between epilepsy and sleep disorders can help clinicians improve the management of both disorders according to a recent review in Epilepsy Research.
- Sleep apnea, insomnia, restless legs syndrome and parasomnias often occur in patients with epilepsy.
- Researchers from Brigham and Women’s Hospital in Boston and Mayo Clinic reviewed the symptoms and diagnosis of these sleep disorders to help clinicians screen patients with epilepsy at risk for these problems.
- The review also explores treatment options and several case reports that illustrate the consequences of said sleep disorders in those with epilepsy.
- Latreille et al believe that employing the latest diagnostic and treatment approaches to co-morbid sleep disorders may improve patients’ functional status, alertness, quality of life and seizure burden.
Latreille V, St. Louis EK, Pavlova M. Co-morbid sleep disorders and epilepsy: A narrative review and case examples. Epilepsy Res. 2018; 145:185-197. https://doi.org/10.1016/j.eplepsyres.2018.07.005
ASCO updates guidance on prophylaxis for adults with cancer-related immunosuppression
Fluoroquinolones are recommended for adults with cancer-related immunosuppression if they are at high risk of infection, according to an updated clinical practice guideline on antimicrobial prophylaxis.
By contrast, patients with solid tumors are not routinely recommended to receive antibiotic prophylaxis, according to the guideline, developed by the American Society of Clinical Oncology (ASCO) with the Infectious Diseases Society of America (IDSA).
The guideline includes antibacterial, antifungal, and antiviral prophylaxis recommendations, along with additional precautions such as hand hygiene that may reduce infection risk.
Released in the Journal of Clinical Oncology, the updated guidelines were developed by an expert panel cochaired by Christopher R. Flowers, MD of Emory University, Atlanta, and Randy A. Taplitz, MD of the University of California, San Diego, Health.
For the most part, the panel endorsed the previous ASCO recommendations, published in 2013. However, the panel considered six new high-quality studies and six new or updated meta-analyses to make modifications and add some new recommendations.
Fluoroquinolones, in the 2013 guideline, were recommended over trimethoprim-sulfamethoxazole because of fewer adverse events leading to treatment discontinuation. Panelists for the new guidelines said they continued to support that recommendation, based on an updated literature review.
That review showed significant reductions in both febrile neutropenia incidence and all-cause mortality, not only for patients at high risk of febrile neutropenia or profound, protracted neutropenia but also for lower-risk patients with solid tumors, they said.
However, the benefits did not sufficiently outweigh the harms to justify recommending fluoroquinolone prophylaxis for all patients with solid tumors or lymphoma, according to the report from the expert panel.
Those harms could include antibiotic-associated adverse effects, emergence of resistance, and Clostridium difficile infections, they said.
Accordingly, they recommended fluoroquinolone prophylaxis for the high-risk patients, including most patients with acute myeloid leukemia/myelodysplastic syndromes (AML/MDS) or those undergoing hematopoietic stem-cell transplantation (HSCT).
Similarly, the panel recommended that high-risk patients should receive antifungal prophylaxis with an oral triazole or parenteral echinocandin, while prophylaxis would not be routinely recommended for solid tumor patients.
By contrast, all patients undergoing chemotherapy for malignancy should receive yearly influenza vaccination with an inactivated quadrivalent vaccine, the panel said in its antiviral prophylaxis recommendations.
Family members, household contacts, and health care providers also should receive influenza vaccinations, said the panel, endorsing recommendations from the Centers for Disease Control and Prevention that were also cited in the 2013 ASCO guidelines.
Health care workers should follow hand hygiene and respiratory hygiene/cough etiquette to reduce risk of pathogen transmission, the panel said, endorsing CDC recommendations cited in the previous guideline.
However, the panel said they recommend against interventions such as neutropenic diet, footwear exchange, nutritional supplements, and surgical masks.
“Evidence of clinical benefit is lacking” for those interventions, they said.
Participants in the expert panel disclosed potential conflicts of interest related to Merck, Chimerix, GlyPharma Therapeutic, Pfizer, Cidara Therapeutics, Celgene, Astellas Pharma, Gilead Sciences, and Allergan, among other entities.
SOURCE: Taplitz RA et al. J Clin Oncol. 2018 Sept 4. doi: 10.1200/JCO.18.00374.
Fluoroquinolones are recommended for adults with cancer-related immunosuppression if they are at high risk of infection, according to an updated clinical practice guideline on antimicrobial prophylaxis.
By contrast, patients with solid tumors are not routinely recommended to receive antibiotic prophylaxis, according to the guideline, developed by the American Society of Clinical Oncology (ASCO) with the Infectious Diseases Society of America (IDSA).
The guideline includes antibacterial, antifungal, and antiviral prophylaxis recommendations, along with additional precautions such as hand hygiene that may reduce infection risk.
Released in the Journal of Clinical Oncology, the updated guidelines were developed by an expert panel cochaired by Christopher R. Flowers, MD of Emory University, Atlanta, and Randy A. Taplitz, MD of the University of California, San Diego, Health.
For the most part, the panel endorsed the previous ASCO recommendations, published in 2013. However, the panel considered six new high-quality studies and six new or updated meta-analyses to make modifications and add some new recommendations.
Fluoroquinolones, in the 2013 guideline, were recommended over trimethoprim-sulfamethoxazole because of fewer adverse events leading to treatment discontinuation. Panelists for the new guidelines said they continued to support that recommendation, based on an updated literature review.
That review showed significant reductions in both febrile neutropenia incidence and all-cause mortality, not only for patients at high risk of febrile neutropenia or profound, protracted neutropenia but also for lower-risk patients with solid tumors, they said.
However, the benefits did not sufficiently outweigh the harms to justify recommending fluoroquinolone prophylaxis for all patients with solid tumors or lymphoma, according to the report from the expert panel.
Those harms could include antibiotic-associated adverse effects, emergence of resistance, and Clostridium difficile infections, they said.
Accordingly, they recommended fluoroquinolone prophylaxis for the high-risk patients, including most patients with acute myeloid leukemia/myelodysplastic syndromes (AML/MDS) or those undergoing hematopoietic stem-cell transplantation (HSCT).
Similarly, the panel recommended that high-risk patients should receive antifungal prophylaxis with an oral triazole or parenteral echinocandin, while prophylaxis would not be routinely recommended for solid tumor patients.
By contrast, all patients undergoing chemotherapy for malignancy should receive yearly influenza vaccination with an inactivated quadrivalent vaccine, the panel said in its antiviral prophylaxis recommendations.
Family members, household contacts, and health care providers also should receive influenza vaccinations, said the panel, endorsing recommendations from the Centers for Disease Control and Prevention that were also cited in the 2013 ASCO guidelines.
Health care workers should follow hand hygiene and respiratory hygiene/cough etiquette to reduce risk of pathogen transmission, the panel said, endorsing CDC recommendations cited in the previous guideline.
However, the panel said they recommend against interventions such as neutropenic diet, footwear exchange, nutritional supplements, and surgical masks.
“Evidence of clinical benefit is lacking” for those interventions, they said.
Participants in the expert panel disclosed potential conflicts of interest related to Merck, Chimerix, GlyPharma Therapeutic, Pfizer, Cidara Therapeutics, Celgene, Astellas Pharma, Gilead Sciences, and Allergan, among other entities.
SOURCE: Taplitz RA et al. J Clin Oncol. 2018 Sept 4. doi: 10.1200/JCO.18.00374.
Fluoroquinolones are recommended for adults with cancer-related immunosuppression if they are at high risk of infection, according to an updated clinical practice guideline on antimicrobial prophylaxis.
By contrast, patients with solid tumors are not routinely recommended to receive antibiotic prophylaxis, according to the guideline, developed by the American Society of Clinical Oncology (ASCO) with the Infectious Diseases Society of America (IDSA).
The guideline includes antibacterial, antifungal, and antiviral prophylaxis recommendations, along with additional precautions such as hand hygiene that may reduce infection risk.
Released in the Journal of Clinical Oncology, the updated guidelines were developed by an expert panel cochaired by Christopher R. Flowers, MD of Emory University, Atlanta, and Randy A. Taplitz, MD of the University of California, San Diego, Health.
For the most part, the panel endorsed the previous ASCO recommendations, published in 2013. However, the panel considered six new high-quality studies and six new or updated meta-analyses to make modifications and add some new recommendations.
Fluoroquinolones, in the 2013 guideline, were recommended over trimethoprim-sulfamethoxazole because of fewer adverse events leading to treatment discontinuation. Panelists for the new guidelines said they continued to support that recommendation, based on an updated literature review.
That review showed significant reductions in both febrile neutropenia incidence and all-cause mortality, not only for patients at high risk of febrile neutropenia or profound, protracted neutropenia but also for lower-risk patients with solid tumors, they said.
However, the benefits did not sufficiently outweigh the harms to justify recommending fluoroquinolone prophylaxis for all patients with solid tumors or lymphoma, according to the report from the expert panel.
Those harms could include antibiotic-associated adverse effects, emergence of resistance, and Clostridium difficile infections, they said.
Accordingly, they recommended fluoroquinolone prophylaxis for the high-risk patients, including most patients with acute myeloid leukemia/myelodysplastic syndromes (AML/MDS) or those undergoing hematopoietic stem-cell transplantation (HSCT).
Similarly, the panel recommended that high-risk patients should receive antifungal prophylaxis with an oral triazole or parenteral echinocandin, while prophylaxis would not be routinely recommended for solid tumor patients.
By contrast, all patients undergoing chemotherapy for malignancy should receive yearly influenza vaccination with an inactivated quadrivalent vaccine, the panel said in its antiviral prophylaxis recommendations.
Family members, household contacts, and health care providers also should receive influenza vaccinations, said the panel, endorsing recommendations from the Centers for Disease Control and Prevention that were also cited in the 2013 ASCO guidelines.
Health care workers should follow hand hygiene and respiratory hygiene/cough etiquette to reduce risk of pathogen transmission, the panel said, endorsing CDC recommendations cited in the previous guideline.
However, the panel said they recommend against interventions such as neutropenic diet, footwear exchange, nutritional supplements, and surgical masks.
“Evidence of clinical benefit is lacking” for those interventions, they said.
Participants in the expert panel disclosed potential conflicts of interest related to Merck, Chimerix, GlyPharma Therapeutic, Pfizer, Cidara Therapeutics, Celgene, Astellas Pharma, Gilead Sciences, and Allergan, among other entities.
SOURCE: Taplitz RA et al. J Clin Oncol. 2018 Sept 4. doi: 10.1200/JCO.18.00374.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
A Three-View Radiographic Approach to Femoroacetabular Impingement
ABSTRACT
Femoroacetabular impingement (FAI) is an abnormality of the hip joint that is increasingly being recognized as a cause of athletic disability and early degenerative hip disease. Despite significant advances in the knowledge of FAI, it remains a frequently unrecognized cause of hip pain in adolescents and young adults among orthopedic providers. The purpose of this article is to present a simple 3-view radiographic approach to young adults with hip pain. The radiographs include a standing anteroposterior view of the pelvis, a cross-table lateral view, and a false profile view. Good quality radiographs showing the common sites of potential impingement combined with a basic understanding of certain radiographic parameters may allow faster diagnosis, eliminate unnecessary studies, and allow earlier referral and management.
Continue to: The prevalence of femoroacetabular impingement...
The prevalence of femoroacetabular impingement (FAI) in the general population is estimated at 23.1%.1 While FAI is often bilateral,2 patients usually present with unilateral symptoms.3 Young, highly active individuals are most commonly affected.3 Despite significant improvement in our understanding of FAI in recent years, it remains a poorly recognized cause of hip pain among orthopedic providers. Clohisy and colleagues3 found that the average time to diagnosis was 3.1 years (range, 3-15 years) and the average number of providers seen before correct diagnosis was 4.2 (range, 1-16) with nearly half those providers being orthopedic specialists. This is likely attributed to limited training and lack of appropriate imaging. Multiple comprehensive radiographic approaches have been described, including plain films, computed tomography, and magnetic resonance imaging.2,4 The objective of this article is to present a simple 3-view plain film approach for young adults with hip pain. While history and physical examination remain key to FAI diagnosis, a basic knowledge of the common sites of impingement with appropriate radiographic views to visualize these sites may help eliminate unnecessary imaging and delayed diagnosis.
STANDING ANTEROPOSTERIOR VIEW OF THE PELVIS
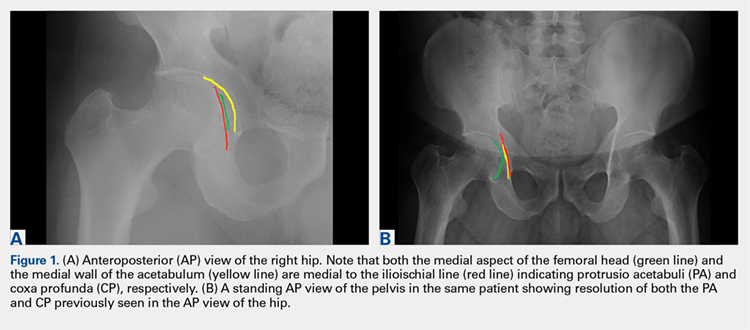
An anteroposterior (AP) view of the pelvis, as opposed to an AP view of the hip, is an important first radiograph in the evaluation of young patients presenting with hip pain. Not only does it permit visualization of the contralateral hip for comparison, but it also allows more accurate measurements of several radiographic parameters (Table). An AP view of the hip often gives the false impression of global over coverage, such as coxa profunda2 and protrusio acetabuli (Figures 1A, 1B), and may overestimate the amount of acetabular anteversion.2
Table. Summary of Common Radiographic Parameters When Assessing Young Adults with Hip Pain2,4
Sign | Best Radiographic View | Measurement | Quoted Normal Valuesa | Clinical Relevance of Abnormal Values |
Acetabular depth | AP pelvis | Medial wall of the acetabulum (MWA) relative to the ilioischial line (IIL) | MWA is lateral to IIL | Global overcoverage (ie, coxa profunda) |
Femoral depth | AP pelvis | Medial surface of the femoral head (MFH) relative to the IIL | MFH is lateral and within 10 mm of the IIL | >10 mm may indicate undercoverage (ie, dysplasia)
MFH medial to IIL may indicate overcoverage (ie, protrusio acetabuli) |
Tonnis angle | AP pelvis | Angle between the weight-bearing surface of the acetabulum and a line parallel to the horizontal axis of the pelvis (eg, inter-teardrop line) | 0°-10° | >10° may indicate undercoverage (ie, dysplasia)
<0° may indicate overcoverage (ie, pincer-type FAI) |
Lateral center edge angle | AP pelvis | Angle between a line perpendicular to the horizontal axis of the pelvis through the center of the femoral head and a line connecting the center of the femoral head to the lateral most edge of the acetabular weight-bearing surface | 25°-40° | >40° may indicate overcoverage (ie, pincer-type FAI)
<25° may indicate undercoverage (ie, dysplasia) |
Crossover sign | AP pelvis | Intersection between the anterior and posterior rims of the acetabulum | Crossover occurs at the lateral most aspect of the acetabular weight-bearing surface | Crossover occurring distal to the lateral most aspect of the acetabular weight-bearing surface may indicate acetabular retroversion |
Femoral neck-shaft angle | AP pelvis | Angle between the femoral shaft and the longitudinal axis of the neck | 135° ± 5° | >140° may indicate coxa valga
<130° may indicate coxa vara |
Alpha angle | Cross-table lateral | Angle between a line connecting the center of the femoral neck to the center of the femoral head and a line connecting the center of the head to a point on the anterolateral aspect of the head-neck junction where the head sphericity ends | >55° | Decreased head-neck offset (ie, cam-type impingement) |
Anterior head-neck offset | Cross-table lateral | Distance between 2 lines parallel to the longitudinal axis of the femoral neck: 1 line tangent to the anterior most aspect of the neck and 1 line tangent to the anterior surface of the femoral head | >10 mm | Decreased head-neck offset (ie, cam-type impingement) |
Anterior head-neck offset ratio | Cross-table lateral | Anterior head-neck offset divided by the diameter of the femoral head | >0.14 | Decreased head-neck offset (ie, cam-type impingement) |
Femoral version | Cross-table lateral | Angle between the longitudinal axis of the femoral neck and the longitudinal axis of the femoral shaft | 15° ± 5° | Developmental disorders (eg, dysplasia, slipped capital femoral epiphysis) |
Anterior center edge angle | False profile view | Angle between a vertical line through the center of the femoral head and a line connecting the center of the femoral head to the anterior most edge of the acetabular weight-bearing surface | >20° | Undercoverage (ie, dysplasia) |
aNormal values are provided for reference only and should not be solely relied on for diagnosis.
Abbreviations: AP, anteroposterior; FAI, femoroacetabular impingement.
A good quality radiograph is important for accurate assessment. The X-ray beam should be perpendicular to the coronal plane of the pelvis. Neutral rotation of the pelvis is a prerequisite and can be confirmed by the presence of symmetric obturator foramina, iliac wings, and coccyx vertically in line with the pubic symphysis. Deviations from this configuration can significantly affect the ability to accurately assess the acetabular version. This is because the rotational profile of the acetabulum is sensitive to pelvic rotation.5,6
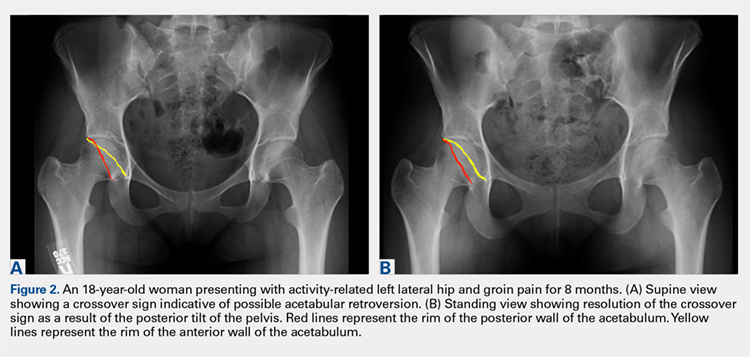
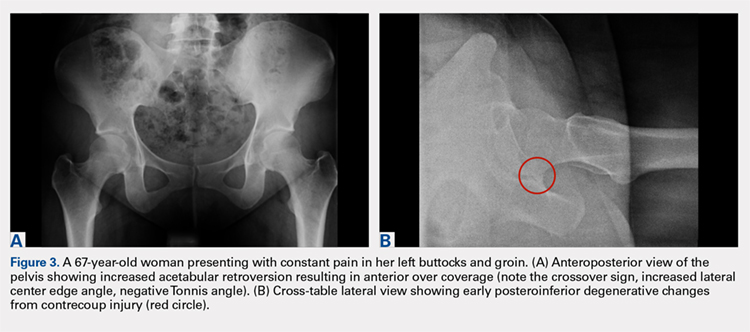
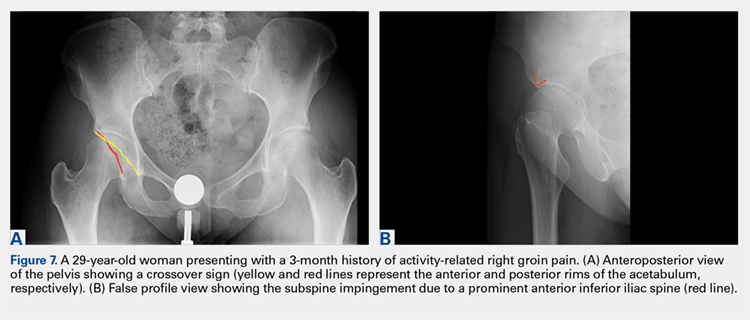
While the AP view of the pelvis can be obtained in either supine or standing positions, the standing position is recommended. A supine view tends to increase the likelihood of finding a crossover sign that often disappears in the standing position (Figures 2A, 2B). This is attributed to the posterior tilt of the pelvis in the sagittal plane with standing, which functionally increases acetabular anteversion, eliminating the crossover sign.5,6 In contrast, a crossover sign that persists in the standing position combined with other abnormal radiographic parameters, such as a negative Tonnis angle and/or increased lateral center edge angle, are concerning for pincer-type FAI (Figures 3A, 3B). An isolated crossover sign may be a normal variant in young asymptomatic patients7 and is not a reliable indicator of acetabular retroversion.5
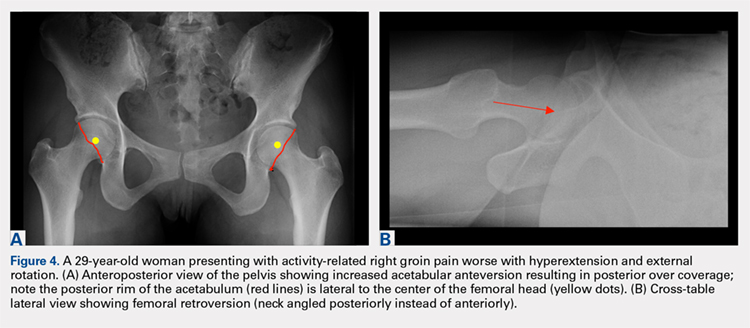
In addition to assessing the acetabular coverage and version (Figures 1A, 1B, 3A, 3B, and 4A, 4B), the AP view of the pelvis can provide valuable information regarding the proximal femur. One should pay attention to the sphericity of the head (pistol grip cam lesions are most obvious on this view), congruency between the femoral head and the acetabulum, femoral offset, and neck-shaft angle. While we tend to traditionally classify FAI into cam and pincer osseous bumps, alterations in hip dynamics (i.e., coxa vara and coxa breva) can result in functional impingement even in the absence of the osseous bumps.
Continue to: CROSS-TABLE LATERAL...
CROSS-TABLE LATERAL
A cross-table lateral of the affected hip is another important radiographic adjunct in the evaluation of hip pain in young patients. This view provides AP axial visualization of the hip joint identifying potential pathologies such as anterior cam lesions that may not be apparent on frog-leg lateral radiographs (Figures 5A, 5B and 6A, 6B). The cross-table lateral view can also show posterior impingement and/or joint space narrowing from countercoup lesions associated with pincer-type FAI (Figures 3A, 3B). In addition, the rotational profile of the proximal femur is best assessed in this view (Figure 4B). The challenge with a cross-table lateral, however, is that it is operator-dependent. In circumstances where a good quality cross-table lateral cannot be obtained, we default to a frog-leg lateral to avoid excess radiation exposure.

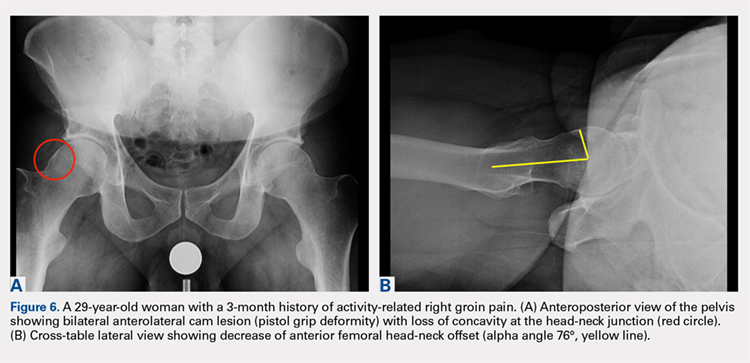
FALSE PROFILE VIEW
A false profile view provides a good visualization of the anterosuperior aspect of the acetabulum. It can show anterior acetabular over or under coverage. It may also show sub-spine impingement (Figures 7A, 7B). Sub-spine impingement is characterized by a prominent anterior inferior iliac spine (AIIS) that extends to the level of the anterosuperior acetabular rim. The prominent AIIS can impinge on the femoral head-neck junction during hip flexion. A prominent AIIS has also been shown to give the false impression of a crossover sign.8
CONCLUSION
Even to the trained eye, radiographic findings of FAI can be quite subtle and easily missed. A systematic approach when interpreting plain radiographs is important. Radiographic assessment starts with good quality X-rays with the pelvis in neutral rotation. Because of the young age of most patients, radiation exposure should be minimized. An understanding of the potential sites of impingement and the specific radiographs to visualize these sites minimizes radiation exposure and other unnecessary imaging. In our experience, the 3-view radiographic approach presented combined with supportive history and physical examination findings are highly sensitive to identify cases of FAI. Advanced imaging is reserved for patients who have failed conservative management or considering surgical intervention.
1. Fernquest S, Arnold C, Palmer A, et al. Osseous impingement occurs early in flexion in cam-type femoroacetabular impingement: a 4D CT model. Bone Joint J. 2017;99-B(4 Supple B):41-48. doi:10.1302/0301-620X.99B4.BJJ-2016-1274.R1.
2. Tannast M, Siebenrock KA, Anderson SE. Femoroacetabular impingement: radiographic diagnosis--what the radiologist should know. AJR Am J Roentgenol. 2007;188(6):1540-1552. doi:10.2214/AJR.06.0921.
3. Clohisy JC, Knaus ER, Hunt DM, Lesher JM, Harris-Hayes M, Prather H. Clinical presentation of patients with symptomatic anterior hip impingement. Clin Orthop Relat Res. 2009;467(3):638-644. doi:10.1007/s11999-008-0680-y.
4. Clohisy JC, Carlisle JC, Beaule PE, et al. A systematic approach to the plain radiographic evaluation of the young adult hip. J Bone Joint Surg Am. 2008;90 Suppl 4:47-66. doi:10.2106/JBJS.H.00756.
5. Dandachli W, Islam SU, Liu M, Richards R, Hall-Craggs M, Witt J. Three-dimensional CT analysis to determine acetabular retroversion and the implications for the management of femoro-acetabular impingement. J Bone Joint Surg Br. 2009;91(8):1031-1036. doi:10.1302/0301-620X.91B8.22389.
6. Dandachli W, Kannan V, Richards R, Shah Z, Hall-Craggs M, Witt J. Analysis of cover of the femoral head in normal and dysplastic hips: new CT-based technique. J Bone Joint Surg Br. 2008;90(11):1428-1434. doi:10.1302/0301-620X.90B11.20073.
7. Larson CM, Moreau-Gaudry A, Kelly BT, et al. Are normal hips being labeled as pathologic? A CT-based method for defining normal acetabular coverage. Clin Orthop Relat Res. 2015;473(4):1247-1254. doi:10.1007/s11999-014-4055-2.
8. Zaltz I, Kelly BT, Hetsroni I, Bedi A. The crossover sign overestimates acetabular retroversion. Clin Orthop Relat Res. 2013;471(8):2463-2470. doi:10.1007/s11999-012-2689-5.
ABSTRACT
Femoroacetabular impingement (FAI) is an abnormality of the hip joint that is increasingly being recognized as a cause of athletic disability and early degenerative hip disease. Despite significant advances in the knowledge of FAI, it remains a frequently unrecognized cause of hip pain in adolescents and young adults among orthopedic providers. The purpose of this article is to present a simple 3-view radiographic approach to young adults with hip pain. The radiographs include a standing anteroposterior view of the pelvis, a cross-table lateral view, and a false profile view. Good quality radiographs showing the common sites of potential impingement combined with a basic understanding of certain radiographic parameters may allow faster diagnosis, eliminate unnecessary studies, and allow earlier referral and management.
Continue to: The prevalence of femoroacetabular impingement...
The prevalence of femoroacetabular impingement (FAI) in the general population is estimated at 23.1%.1 While FAI is often bilateral,2 patients usually present with unilateral symptoms.3 Young, highly active individuals are most commonly affected.3 Despite significant improvement in our understanding of FAI in recent years, it remains a poorly recognized cause of hip pain among orthopedic providers. Clohisy and colleagues3 found that the average time to diagnosis was 3.1 years (range, 3-15 years) and the average number of providers seen before correct diagnosis was 4.2 (range, 1-16) with nearly half those providers being orthopedic specialists. This is likely attributed to limited training and lack of appropriate imaging. Multiple comprehensive radiographic approaches have been described, including plain films, computed tomography, and magnetic resonance imaging.2,4 The objective of this article is to present a simple 3-view plain film approach for young adults with hip pain. While history and physical examination remain key to FAI diagnosis, a basic knowledge of the common sites of impingement with appropriate radiographic views to visualize these sites may help eliminate unnecessary imaging and delayed diagnosis.
STANDING ANTEROPOSTERIOR VIEW OF THE PELVIS
An anteroposterior (AP) view of the pelvis, as opposed to an AP view of the hip, is an important first radiograph in the evaluation of young patients presenting with hip pain. Not only does it permit visualization of the contralateral hip for comparison, but it also allows more accurate measurements of several radiographic parameters (Table). An AP view of the hip often gives the false impression of global over coverage, such as coxa profunda2 and protrusio acetabuli (Figures 1A, 1B), and may overestimate the amount of acetabular anteversion.2
Table. Summary of Common Radiographic Parameters When Assessing Young Adults with Hip Pain2,4
Sign | Best Radiographic View | Measurement | Quoted Normal Valuesa | Clinical Relevance of Abnormal Values |
Acetabular depth | AP pelvis | Medial wall of the acetabulum (MWA) relative to the ilioischial line (IIL) | MWA is lateral to IIL | Global overcoverage (ie, coxa profunda) |
Femoral depth | AP pelvis | Medial surface of the femoral head (MFH) relative to the IIL | MFH is lateral and within 10 mm of the IIL | >10 mm may indicate undercoverage (ie, dysplasia)
MFH medial to IIL may indicate overcoverage (ie, protrusio acetabuli) |
Tonnis angle | AP pelvis | Angle between the weight-bearing surface of the acetabulum and a line parallel to the horizontal axis of the pelvis (eg, inter-teardrop line) | 0°-10° | >10° may indicate undercoverage (ie, dysplasia)
<0° may indicate overcoverage (ie, pincer-type FAI) |
Lateral center edge angle | AP pelvis | Angle between a line perpendicular to the horizontal axis of the pelvis through the center of the femoral head and a line connecting the center of the femoral head to the lateral most edge of the acetabular weight-bearing surface | 25°-40° | >40° may indicate overcoverage (ie, pincer-type FAI)
<25° may indicate undercoverage (ie, dysplasia) |
Crossover sign | AP pelvis | Intersection between the anterior and posterior rims of the acetabulum | Crossover occurs at the lateral most aspect of the acetabular weight-bearing surface | Crossover occurring distal to the lateral most aspect of the acetabular weight-bearing surface may indicate acetabular retroversion |
Femoral neck-shaft angle | AP pelvis | Angle between the femoral shaft and the longitudinal axis of the neck | 135° ± 5° | >140° may indicate coxa valga
<130° may indicate coxa vara |
Alpha angle | Cross-table lateral | Angle between a line connecting the center of the femoral neck to the center of the femoral head and a line connecting the center of the head to a point on the anterolateral aspect of the head-neck junction where the head sphericity ends | >55° | Decreased head-neck offset (ie, cam-type impingement) |
Anterior head-neck offset | Cross-table lateral | Distance between 2 lines parallel to the longitudinal axis of the femoral neck: 1 line tangent to the anterior most aspect of the neck and 1 line tangent to the anterior surface of the femoral head | >10 mm | Decreased head-neck offset (ie, cam-type impingement) |
Anterior head-neck offset ratio | Cross-table lateral | Anterior head-neck offset divided by the diameter of the femoral head | >0.14 | Decreased head-neck offset (ie, cam-type impingement) |
Femoral version | Cross-table lateral | Angle between the longitudinal axis of the femoral neck and the longitudinal axis of the femoral shaft | 15° ± 5° | Developmental disorders (eg, dysplasia, slipped capital femoral epiphysis) |
Anterior center edge angle | False profile view | Angle between a vertical line through the center of the femoral head and a line connecting the center of the femoral head to the anterior most edge of the acetabular weight-bearing surface | >20° | Undercoverage (ie, dysplasia) |
aNormal values are provided for reference only and should not be solely relied on for diagnosis.
Abbreviations: AP, anteroposterior; FAI, femoroacetabular impingement.
A good quality radiograph is important for accurate assessment. The X-ray beam should be perpendicular to the coronal plane of the pelvis. Neutral rotation of the pelvis is a prerequisite and can be confirmed by the presence of symmetric obturator foramina, iliac wings, and coccyx vertically in line with the pubic symphysis. Deviations from this configuration can significantly affect the ability to accurately assess the acetabular version. This is because the rotational profile of the acetabulum is sensitive to pelvic rotation.5,6
While the AP view of the pelvis can be obtained in either supine or standing positions, the standing position is recommended. A supine view tends to increase the likelihood of finding a crossover sign that often disappears in the standing position (Figures 2A, 2B). This is attributed to the posterior tilt of the pelvis in the sagittal plane with standing, which functionally increases acetabular anteversion, eliminating the crossover sign.5,6 In contrast, a crossover sign that persists in the standing position combined with other abnormal radiographic parameters, such as a negative Tonnis angle and/or increased lateral center edge angle, are concerning for pincer-type FAI (Figures 3A, 3B). An isolated crossover sign may be a normal variant in young asymptomatic patients7 and is not a reliable indicator of acetabular retroversion.5
In addition to assessing the acetabular coverage and version (Figures 1A, 1B, 3A, 3B, and 4A, 4B), the AP view of the pelvis can provide valuable information regarding the proximal femur. One should pay attention to the sphericity of the head (pistol grip cam lesions are most obvious on this view), congruency between the femoral head and the acetabulum, femoral offset, and neck-shaft angle. While we tend to traditionally classify FAI into cam and pincer osseous bumps, alterations in hip dynamics (i.e., coxa vara and coxa breva) can result in functional impingement even in the absence of the osseous bumps.
Continue to: CROSS-TABLE LATERAL...
CROSS-TABLE LATERAL
A cross-table lateral of the affected hip is another important radiographic adjunct in the evaluation of hip pain in young patients. This view provides AP axial visualization of the hip joint identifying potential pathologies such as anterior cam lesions that may not be apparent on frog-leg lateral radiographs (Figures 5A, 5B and 6A, 6B). The cross-table lateral view can also show posterior impingement and/or joint space narrowing from countercoup lesions associated with pincer-type FAI (Figures 3A, 3B). In addition, the rotational profile of the proximal femur is best assessed in this view (Figure 4B). The challenge with a cross-table lateral, however, is that it is operator-dependent. In circumstances where a good quality cross-table lateral cannot be obtained, we default to a frog-leg lateral to avoid excess radiation exposure.
FALSE PROFILE VIEW
A false profile view provides a good visualization of the anterosuperior aspect of the acetabulum. It can show anterior acetabular over or under coverage. It may also show sub-spine impingement (Figures 7A, 7B). Sub-spine impingement is characterized by a prominent anterior inferior iliac spine (AIIS) that extends to the level of the anterosuperior acetabular rim. The prominent AIIS can impinge on the femoral head-neck junction during hip flexion. A prominent AIIS has also been shown to give the false impression of a crossover sign.8
CONCLUSION
Even to the trained eye, radiographic findings of FAI can be quite subtle and easily missed. A systematic approach when interpreting plain radiographs is important. Radiographic assessment starts with good quality X-rays with the pelvis in neutral rotation. Because of the young age of most patients, radiation exposure should be minimized. An understanding of the potential sites of impingement and the specific radiographs to visualize these sites minimizes radiation exposure and other unnecessary imaging. In our experience, the 3-view radiographic approach presented combined with supportive history and physical examination findings are highly sensitive to identify cases of FAI. Advanced imaging is reserved for patients who have failed conservative management or considering surgical intervention.
ABSTRACT
Femoroacetabular impingement (FAI) is an abnormality of the hip joint that is increasingly being recognized as a cause of athletic disability and early degenerative hip disease. Despite significant advances in the knowledge of FAI, it remains a frequently unrecognized cause of hip pain in adolescents and young adults among orthopedic providers. The purpose of this article is to present a simple 3-view radiographic approach to young adults with hip pain. The radiographs include a standing anteroposterior view of the pelvis, a cross-table lateral view, and a false profile view. Good quality radiographs showing the common sites of potential impingement combined with a basic understanding of certain radiographic parameters may allow faster diagnosis, eliminate unnecessary studies, and allow earlier referral and management.
Continue to: The prevalence of femoroacetabular impingement...
The prevalence of femoroacetabular impingement (FAI) in the general population is estimated at 23.1%.1 While FAI is often bilateral,2 patients usually present with unilateral symptoms.3 Young, highly active individuals are most commonly affected.3 Despite significant improvement in our understanding of FAI in recent years, it remains a poorly recognized cause of hip pain among orthopedic providers. Clohisy and colleagues3 found that the average time to diagnosis was 3.1 years (range, 3-15 years) and the average number of providers seen before correct diagnosis was 4.2 (range, 1-16) with nearly half those providers being orthopedic specialists. This is likely attributed to limited training and lack of appropriate imaging. Multiple comprehensive radiographic approaches have been described, including plain films, computed tomography, and magnetic resonance imaging.2,4 The objective of this article is to present a simple 3-view plain film approach for young adults with hip pain. While history and physical examination remain key to FAI diagnosis, a basic knowledge of the common sites of impingement with appropriate radiographic views to visualize these sites may help eliminate unnecessary imaging and delayed diagnosis.
STANDING ANTEROPOSTERIOR VIEW OF THE PELVIS
An anteroposterior (AP) view of the pelvis, as opposed to an AP view of the hip, is an important first radiograph in the evaluation of young patients presenting with hip pain. Not only does it permit visualization of the contralateral hip for comparison, but it also allows more accurate measurements of several radiographic parameters (Table). An AP view of the hip often gives the false impression of global over coverage, such as coxa profunda2 and protrusio acetabuli (Figures 1A, 1B), and may overestimate the amount of acetabular anteversion.2
Table. Summary of Common Radiographic Parameters When Assessing Young Adults with Hip Pain2,4
Sign | Best Radiographic View | Measurement | Quoted Normal Valuesa | Clinical Relevance of Abnormal Values |
Acetabular depth | AP pelvis | Medial wall of the acetabulum (MWA) relative to the ilioischial line (IIL) | MWA is lateral to IIL | Global overcoverage (ie, coxa profunda) |
Femoral depth | AP pelvis | Medial surface of the femoral head (MFH) relative to the IIL | MFH is lateral and within 10 mm of the IIL | >10 mm may indicate undercoverage (ie, dysplasia)
MFH medial to IIL may indicate overcoverage (ie, protrusio acetabuli) |
Tonnis angle | AP pelvis | Angle between the weight-bearing surface of the acetabulum and a line parallel to the horizontal axis of the pelvis (eg, inter-teardrop line) | 0°-10° | >10° may indicate undercoverage (ie, dysplasia)
<0° may indicate overcoverage (ie, pincer-type FAI) |
Lateral center edge angle | AP pelvis | Angle between a line perpendicular to the horizontal axis of the pelvis through the center of the femoral head and a line connecting the center of the femoral head to the lateral most edge of the acetabular weight-bearing surface | 25°-40° | >40° may indicate overcoverage (ie, pincer-type FAI)
<25° may indicate undercoverage (ie, dysplasia) |
Crossover sign | AP pelvis | Intersection between the anterior and posterior rims of the acetabulum | Crossover occurs at the lateral most aspect of the acetabular weight-bearing surface | Crossover occurring distal to the lateral most aspect of the acetabular weight-bearing surface may indicate acetabular retroversion |
Femoral neck-shaft angle | AP pelvis | Angle between the femoral shaft and the longitudinal axis of the neck | 135° ± 5° | >140° may indicate coxa valga
<130° may indicate coxa vara |
Alpha angle | Cross-table lateral | Angle between a line connecting the center of the femoral neck to the center of the femoral head and a line connecting the center of the head to a point on the anterolateral aspect of the head-neck junction where the head sphericity ends | >55° | Decreased head-neck offset (ie, cam-type impingement) |
Anterior head-neck offset | Cross-table lateral | Distance between 2 lines parallel to the longitudinal axis of the femoral neck: 1 line tangent to the anterior most aspect of the neck and 1 line tangent to the anterior surface of the femoral head | >10 mm | Decreased head-neck offset (ie, cam-type impingement) |
Anterior head-neck offset ratio | Cross-table lateral | Anterior head-neck offset divided by the diameter of the femoral head | >0.14 | Decreased head-neck offset (ie, cam-type impingement) |
Femoral version | Cross-table lateral | Angle between the longitudinal axis of the femoral neck and the longitudinal axis of the femoral shaft | 15° ± 5° | Developmental disorders (eg, dysplasia, slipped capital femoral epiphysis) |
Anterior center edge angle | False profile view | Angle between a vertical line through the center of the femoral head and a line connecting the center of the femoral head to the anterior most edge of the acetabular weight-bearing surface | >20° | Undercoverage (ie, dysplasia) |
aNormal values are provided for reference only and should not be solely relied on for diagnosis.
Abbreviations: AP, anteroposterior; FAI, femoroacetabular impingement.
A good quality radiograph is important for accurate assessment. The X-ray beam should be perpendicular to the coronal plane of the pelvis. Neutral rotation of the pelvis is a prerequisite and can be confirmed by the presence of symmetric obturator foramina, iliac wings, and coccyx vertically in line with the pubic symphysis. Deviations from this configuration can significantly affect the ability to accurately assess the acetabular version. This is because the rotational profile of the acetabulum is sensitive to pelvic rotation.5,6
While the AP view of the pelvis can be obtained in either supine or standing positions, the standing position is recommended. A supine view tends to increase the likelihood of finding a crossover sign that often disappears in the standing position (Figures 2A, 2B). This is attributed to the posterior tilt of the pelvis in the sagittal plane with standing, which functionally increases acetabular anteversion, eliminating the crossover sign.5,6 In contrast, a crossover sign that persists in the standing position combined with other abnormal radiographic parameters, such as a negative Tonnis angle and/or increased lateral center edge angle, are concerning for pincer-type FAI (Figures 3A, 3B). An isolated crossover sign may be a normal variant in young asymptomatic patients7 and is not a reliable indicator of acetabular retroversion.5
In addition to assessing the acetabular coverage and version (Figures 1A, 1B, 3A, 3B, and 4A, 4B), the AP view of the pelvis can provide valuable information regarding the proximal femur. One should pay attention to the sphericity of the head (pistol grip cam lesions are most obvious on this view), congruency between the femoral head and the acetabulum, femoral offset, and neck-shaft angle. While we tend to traditionally classify FAI into cam and pincer osseous bumps, alterations in hip dynamics (i.e., coxa vara and coxa breva) can result in functional impingement even in the absence of the osseous bumps.
Continue to: CROSS-TABLE LATERAL...
CROSS-TABLE LATERAL
A cross-table lateral of the affected hip is another important radiographic adjunct in the evaluation of hip pain in young patients. This view provides AP axial visualization of the hip joint identifying potential pathologies such as anterior cam lesions that may not be apparent on frog-leg lateral radiographs (Figures 5A, 5B and 6A, 6B). The cross-table lateral view can also show posterior impingement and/or joint space narrowing from countercoup lesions associated with pincer-type FAI (Figures 3A, 3B). In addition, the rotational profile of the proximal femur is best assessed in this view (Figure 4B). The challenge with a cross-table lateral, however, is that it is operator-dependent. In circumstances where a good quality cross-table lateral cannot be obtained, we default to a frog-leg lateral to avoid excess radiation exposure.
FALSE PROFILE VIEW
A false profile view provides a good visualization of the anterosuperior aspect of the acetabulum. It can show anterior acetabular over or under coverage. It may also show sub-spine impingement (Figures 7A, 7B). Sub-spine impingement is characterized by a prominent anterior inferior iliac spine (AIIS) that extends to the level of the anterosuperior acetabular rim. The prominent AIIS can impinge on the femoral head-neck junction during hip flexion. A prominent AIIS has also been shown to give the false impression of a crossover sign.8
CONCLUSION
Even to the trained eye, radiographic findings of FAI can be quite subtle and easily missed. A systematic approach when interpreting plain radiographs is important. Radiographic assessment starts with good quality X-rays with the pelvis in neutral rotation. Because of the young age of most patients, radiation exposure should be minimized. An understanding of the potential sites of impingement and the specific radiographs to visualize these sites minimizes radiation exposure and other unnecessary imaging. In our experience, the 3-view radiographic approach presented combined with supportive history and physical examination findings are highly sensitive to identify cases of FAI. Advanced imaging is reserved for patients who have failed conservative management or considering surgical intervention.
1. Fernquest S, Arnold C, Palmer A, et al. Osseous impingement occurs early in flexion in cam-type femoroacetabular impingement: a 4D CT model. Bone Joint J. 2017;99-B(4 Supple B):41-48. doi:10.1302/0301-620X.99B4.BJJ-2016-1274.R1.
2. Tannast M, Siebenrock KA, Anderson SE. Femoroacetabular impingement: radiographic diagnosis--what the radiologist should know. AJR Am J Roentgenol. 2007;188(6):1540-1552. doi:10.2214/AJR.06.0921.
3. Clohisy JC, Knaus ER, Hunt DM, Lesher JM, Harris-Hayes M, Prather H. Clinical presentation of patients with symptomatic anterior hip impingement. Clin Orthop Relat Res. 2009;467(3):638-644. doi:10.1007/s11999-008-0680-y.
4. Clohisy JC, Carlisle JC, Beaule PE, et al. A systematic approach to the plain radiographic evaluation of the young adult hip. J Bone Joint Surg Am. 2008;90 Suppl 4:47-66. doi:10.2106/JBJS.H.00756.
5. Dandachli W, Islam SU, Liu M, Richards R, Hall-Craggs M, Witt J. Three-dimensional CT analysis to determine acetabular retroversion and the implications for the management of femoro-acetabular impingement. J Bone Joint Surg Br. 2009;91(8):1031-1036. doi:10.1302/0301-620X.91B8.22389.
6. Dandachli W, Kannan V, Richards R, Shah Z, Hall-Craggs M, Witt J. Analysis of cover of the femoral head in normal and dysplastic hips: new CT-based technique. J Bone Joint Surg Br. 2008;90(11):1428-1434. doi:10.1302/0301-620X.90B11.20073.
7. Larson CM, Moreau-Gaudry A, Kelly BT, et al. Are normal hips being labeled as pathologic? A CT-based method for defining normal acetabular coverage. Clin Orthop Relat Res. 2015;473(4):1247-1254. doi:10.1007/s11999-014-4055-2.
8. Zaltz I, Kelly BT, Hetsroni I, Bedi A. The crossover sign overestimates acetabular retroversion. Clin Orthop Relat Res. 2013;471(8):2463-2470. doi:10.1007/s11999-012-2689-5.
1. Fernquest S, Arnold C, Palmer A, et al. Osseous impingement occurs early in flexion in cam-type femoroacetabular impingement: a 4D CT model. Bone Joint J. 2017;99-B(4 Supple B):41-48. doi:10.1302/0301-620X.99B4.BJJ-2016-1274.R1.
2. Tannast M, Siebenrock KA, Anderson SE. Femoroacetabular impingement: radiographic diagnosis--what the radiologist should know. AJR Am J Roentgenol. 2007;188(6):1540-1552. doi:10.2214/AJR.06.0921.
3. Clohisy JC, Knaus ER, Hunt DM, Lesher JM, Harris-Hayes M, Prather H. Clinical presentation of patients with symptomatic anterior hip impingement. Clin Orthop Relat Res. 2009;467(3):638-644. doi:10.1007/s11999-008-0680-y.
4. Clohisy JC, Carlisle JC, Beaule PE, et al. A systematic approach to the plain radiographic evaluation of the young adult hip. J Bone Joint Surg Am. 2008;90 Suppl 4:47-66. doi:10.2106/JBJS.H.00756.
5. Dandachli W, Islam SU, Liu M, Richards R, Hall-Craggs M, Witt J. Three-dimensional CT analysis to determine acetabular retroversion and the implications for the management of femoro-acetabular impingement. J Bone Joint Surg Br. 2009;91(8):1031-1036. doi:10.1302/0301-620X.91B8.22389.
6. Dandachli W, Kannan V, Richards R, Shah Z, Hall-Craggs M, Witt J. Analysis of cover of the femoral head in normal and dysplastic hips: new CT-based technique. J Bone Joint Surg Br. 2008;90(11):1428-1434. doi:10.1302/0301-620X.90B11.20073.
7. Larson CM, Moreau-Gaudry A, Kelly BT, et al. Are normal hips being labeled as pathologic? A CT-based method for defining normal acetabular coverage. Clin Orthop Relat Res. 2015;473(4):1247-1254. doi:10.1007/s11999-014-4055-2.
8. Zaltz I, Kelly BT, Hetsroni I, Bedi A. The crossover sign overestimates acetabular retroversion. Clin Orthop Relat Res. 2013;471(8):2463-2470. doi:10.1007/s11999-012-2689-5.
TAKE-HOME POINTS
- FAI is a frequently unrecognized cause of hip pain in adolescents and young adults.
- Understanding the potential sites of impingement and the specific radiographs to visualize these sites can help avoid unnecessary imaging and delayed diagnosis.
- A simple radiographic approach consisting of a standing AP view of the pelvis, a cross-table lateral view, and a false profile view is often a sufficient screening tool.
- While we tend to classify FAI into cam and pincer osseous bumps, alterations in hip dynamics can result in functional impingement even in the absence of the osseous bumps.
- Advanced imaging is reserved for patients who have failed conservative management or are considering surgical intervention.
Novartis nabs first CAR T approval in Canada
the first chimeric antigen receptor (CAR) T-cell therapy to receive regulatory approval in Canada.
Tisagenlecleucel is approved to treat patients aged 3-25 years who have B-cell acute lymphoblastic leukemia (ALL) and relapsed after allogenic stem cell transplant (SCT) or are otherwise ineligible for SCT, have experienced second or later relapse, or have refractory disease.
Tisagenlecleucel is also approved in Canada to treat adults who have received two or more lines of systemic therapy and have relapsed or refractory diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high-grade B-cell lymphoma, or DLBCL arising from follicular lymphoma.
Novartis, the company marketing tisagenlecleucel, said it is working with qualified treatment centers in Canada to prepare for the delivery of tisagenlecleucel. Certification and training are underway at these centers and Novartis is enhancing manufacturing capacity to meet patient needs.
Tisagenlecleucel has been studied in a pair of phase 2 trials – JULIET and ELIANA.
JULIET enrolled 165 adults with relapsed/refractory DLBCL, 111 of whom received a single infusion of tisagenlecleucel.
The overall response rate was 52% and the complete response (CR) rate was 40%. The median duration of response was not reached with a median follow-up of 13.9 months. At last follow-up, none of the responders had gone on to SCT.
The 12-month overall survival (OS) rate was 49%; the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome.
These results were presented at the 2018 annual congress of the European Hematology Association in June.
The ELIANA trial included 75 children and young adults with relapsed/refractory ALL. All patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The overall remission rate was 81%, with 60% of patients achieving a CR and 21% achieving CR with incomplete hematologic recovery. All patients whose best response was CR with incomplete hematologic recovery were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to SCT while in remission. At last follow-up, four were still in remission, and four had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
About 95% of patients had adverse events thought to be related to tisagenlecleucel. The incidence of treatment-related grade 3/4 adverse eventss was 73% (N Engl J Med 2018; 378:439-48).
the first chimeric antigen receptor (CAR) T-cell therapy to receive regulatory approval in Canada.
Tisagenlecleucel is approved to treat patients aged 3-25 years who have B-cell acute lymphoblastic leukemia (ALL) and relapsed after allogenic stem cell transplant (SCT) or are otherwise ineligible for SCT, have experienced second or later relapse, or have refractory disease.
Tisagenlecleucel is also approved in Canada to treat adults who have received two or more lines of systemic therapy and have relapsed or refractory diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high-grade B-cell lymphoma, or DLBCL arising from follicular lymphoma.
Novartis, the company marketing tisagenlecleucel, said it is working with qualified treatment centers in Canada to prepare for the delivery of tisagenlecleucel. Certification and training are underway at these centers and Novartis is enhancing manufacturing capacity to meet patient needs.
Tisagenlecleucel has been studied in a pair of phase 2 trials – JULIET and ELIANA.
JULIET enrolled 165 adults with relapsed/refractory DLBCL, 111 of whom received a single infusion of tisagenlecleucel.
The overall response rate was 52% and the complete response (CR) rate was 40%. The median duration of response was not reached with a median follow-up of 13.9 months. At last follow-up, none of the responders had gone on to SCT.
The 12-month overall survival (OS) rate was 49%; the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome.
These results were presented at the 2018 annual congress of the European Hematology Association in June.
The ELIANA trial included 75 children and young adults with relapsed/refractory ALL. All patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The overall remission rate was 81%, with 60% of patients achieving a CR and 21% achieving CR with incomplete hematologic recovery. All patients whose best response was CR with incomplete hematologic recovery were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to SCT while in remission. At last follow-up, four were still in remission, and four had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
About 95% of patients had adverse events thought to be related to tisagenlecleucel. The incidence of treatment-related grade 3/4 adverse eventss was 73% (N Engl J Med 2018; 378:439-48).
the first chimeric antigen receptor (CAR) T-cell therapy to receive regulatory approval in Canada.
Tisagenlecleucel is approved to treat patients aged 3-25 years who have B-cell acute lymphoblastic leukemia (ALL) and relapsed after allogenic stem cell transplant (SCT) or are otherwise ineligible for SCT, have experienced second or later relapse, or have refractory disease.
Tisagenlecleucel is also approved in Canada to treat adults who have received two or more lines of systemic therapy and have relapsed or refractory diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high-grade B-cell lymphoma, or DLBCL arising from follicular lymphoma.
Novartis, the company marketing tisagenlecleucel, said it is working with qualified treatment centers in Canada to prepare for the delivery of tisagenlecleucel. Certification and training are underway at these centers and Novartis is enhancing manufacturing capacity to meet patient needs.
Tisagenlecleucel has been studied in a pair of phase 2 trials – JULIET and ELIANA.
JULIET enrolled 165 adults with relapsed/refractory DLBCL, 111 of whom received a single infusion of tisagenlecleucel.
The overall response rate was 52% and the complete response (CR) rate was 40%. The median duration of response was not reached with a median follow-up of 13.9 months. At last follow-up, none of the responders had gone on to SCT.
The 12-month overall survival (OS) rate was 49%; the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome.
These results were presented at the 2018 annual congress of the European Hematology Association in June.
The ELIANA trial included 75 children and young adults with relapsed/refractory ALL. All patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The overall remission rate was 81%, with 60% of patients achieving a CR and 21% achieving CR with incomplete hematologic recovery. All patients whose best response was CR with incomplete hematologic recovery were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to SCT while in remission. At last follow-up, four were still in remission, and four had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
About 95% of patients had adverse events thought to be related to tisagenlecleucel. The incidence of treatment-related grade 3/4 adverse eventss was 73% (N Engl J Med 2018; 378:439-48).
CDC releases guidelines for pediatric mTBI
and should base management and prognostication on clinical decision-making tools and symptom rating scales, according to new practice guidelines issued by a working group of the Centers for Disease Control and Prevention (JAMA Pediatrics. 2018 Sep 4. doi: 10.1001/jamapediatrics.2018.2853.
The guidelines were released simultaneously with a systematic review, conducted by the same authors, of the existing literature regarding pediatric mTBI (JAMA Pediatrics 2018 Sep 4. doi: 10.1001/jamapediatrics.2018.2847). As the evaluators sorted through the literature to find high-quality studies for this population, the funnel rapidly narrowed: From an initial pool of over 15,000 studies conducted between 1990 and 2015, findings from just 75 studies were eventually included in the systematic review.
The review’s findings formed the basis for the guidelines and allowed Angela Lumba-Brown, MD, a pediatric emergency medicine physician at Stanford (Calif.) University, and her coauthors to ascribe a level of confidence in the inference from study data for a given recommendation. Recommendations also are categorized by strength and accordingly indicate that clinicians “should” or “may” follow them. Exceptions are carved out for practices, such as the use of hypertonic 3% saline solution for acute headache in the ED, that should not be used outside research settings.
In the end, the guidelines cover 19 main topics, sorted into guidance regarding the diagnosis, prognosis, and management and treatment of mTBI in children.
Diagnosis
The recommendations regarding mTBI diagnosis center around determining which children are at risk for significant intracranial injury (ICI). The guidelines recommend, with moderate confidence, that clinicians usually should not obtain a head CT for children with mTBI. Validated clinical decision rules should be used for risk stratification to determine which children can safely avoid imaging and which children should be considered for head CT, wrote Dr. Lumba-Brown and her coauthors. Magnetic resonance imaging is not recommended for initial evaluation of mTBI, nor should skull radiographs be ordered in the absence of clinical suspicion for skull fracture.
From the systematic review, Dr. Lumba-Brown and her colleagues found that several risk factors taken together may mean that significant ICI is more likely. These include patient age younger than 2 years; any vomiting, loss of consciousness, or amnesia; a severe mechanism of injury, severe or worsening headache, or nonfrontal scalp hematoma; a Glasgow Coma Scale (GCS) score of less than 15; and clinical suspicion for skull fracture. Clinicians should give consideration to the risks of ionizing radiation to the head, and balance this against their assessment of risk for severe – and perhaps actionable – injury.
A validated symptom rating scale, used in an age-appropriate way, should be used as part of the evaluation of children with mTBI. For children aged 6 and older, the Graded Symptom Checklist is an appropriate tool within 2 days after injury, while the Post Concussion Symptom Scale as part of computerized neurocognitive testing can differentiate which high school athletes have mTBI when used within 4 days of injury, according to the guidelines, which also identify other validated symptom rating scales.
The guidelines authors recommend, with high confidence, that serum biomarkers should not be used outside of research settings in the diagnosis of mTBI in children at present.
Prognosis
Families should be counseled that symptoms mostly resolve within 1-3 months for up to 80% of children with mTBI, but families also should know that “each child’s recovery from mTBI is unique and will follow its own trajectory,” wrote Dr. Lumba-Brown and her coauthors, in a moderate-strength recommendation.
Some factors have been associated with slower recovery from mTBI, and either upon evaluation for mTBI or in routine sports examinations, families should be told about this potential if risk factors are present, said the guidelines, although the evidence supporting the associations is of “varying strength,” wrote Dr. Lumba-Brown and her coauthors. Children with previous mTBIs and those with a history of premorbid neurologic and psychiatric problems, learning problems, or family and social stress all may have delayed recovery. For children with ICI, lower cognitive ability also is associated with delayed recovery.
Demographic factors such as lower socioeconomic status and being of Hispanic ethnicity also may increase the risk for delayed mTBI recovery. Older children and adolescents may recover more slowly. Those with more severe initial presentation and more symptoms in the immediate post-mTBI phase also may have a slower recovery course, said Dr. Lumba-Brown and her coauthors.

A validated prediction rule can be used in the ED to gather information about these discrete risk factors to guide family counseling, according to the guidelines, which note that research has found that “an empirically derived set of risk factors predicted the risk of persistent post-concussion symptoms at 28 days” for children seen in the ED with mTBI.
During the recovery phase, a combination of tools should be used to track recovery from mTBI; these can include validated symptom scales, validated cognitive testing, reaction time measures, and, in adolescent athletes, balance testing. Using a combination of tools is a valuable strategy, the researchers wrote. “No single assessment tool is strongly predictive of outcome in children with mTBI,” they noted.
When prognosis is poor, or recovery is not proceeding as expected, clinicians should have a low threshold for initiating other interventions and referrals.
Management and treatment
Although the guideline authors acknowledged significant knowledge gaps in all areas of pediatric mTBI diagnosis and management, evidence is especially scant for best practices for treatment, rest, and return to play and school after a child sustains mTBI, said Dr. Lumba-Brown and her coauthors.
However, families should be given information about warning signs for serious head injury and how to monitor symptoms, as well as information about mTBI and the expected recovery course. Other forward-looking instructions should cover the importance of preventing new head injuries, managing the gradual return to normal cognitive and physical activities, and clear instructions regarding return to school and recreational activities. The guideline authors made a strong recommendation to provide this information, with high confidence in the data.
However, little strong evidence points the way to a clear set of criteria for when children are ready for school, play, and athletic participation. These decisions must be customized to the individual child, and decision making, particularly about return to school and academic activities, should be a collaborative affair, with schools, clinicians, and families all communicating to make sure the pace of return to normal life is keeping pace with the child’s recovery. “Because postconcussive symptoms resolve at different rates in different children after mTBI, individualization of return-to-school programming is necessary,” wrote Dr. Lumba-Brown and her coauthors.
The guideline authors cite evidence that “suggests that early rest (within the first 3 days) may be beneficial but that inactivity beyond this period for most children may worsen their self-reported symptoms.”
Psychosocial support may be beneficial for certain children, wrote the researchers, drawing on evidence showing that such support is beneficial in frank TBI, and is probably beneficial in mTBI.
Active rehabilitation as tolerated is recommended after an initial period of rest, with exertion kept to a level that does not exacerbate symptoms. Children should not participate in contact activities until symptoms are fully resolved.
A posttraumatic headache that is severe or worsens in the ED should prompt consideration of emergent neuroimaging, according to the guidelines. In the postacute phase, however, children can have nonopioid analgesia, although parents should know about such risks as rebound headache. When chronic headache follows a mTBI, the guidelines recommend that clinicians refer patients for a multidisciplinary evaluation that can assess the many factors – including analgesic overuse – that can be contributors.
Drawing on the larger body of adult TBI research, the authors recommend that insufficient or disordered sleep be addressed, because “the maintenance of appropriate sleep and the management of disrupted sleep may be a critical target of treatment for the child with mTBI.”
Children who suffer a mTBI may experience cognitive dysfunction as a direct result of injury to the brain or secondary to the effects of other symptoms such as sleep disruptions, headache pain, fatigue, or low tolerance of frustration. Clinicians may want to perform or refer their patients for a neuropsychological evaluation to determine what is causing the cognitive dysfunction, the authors said.
Dr. Lumba-Brown and her coauthors, who formed the CDC’s Pediatric Mild Traumatic Brain Injury Guideline Workgroup, also recommended that clinicians use the term “mild traumatic brain injury” to describe head injuries that cause confusion or disorientation, without loss of consciousness, or loss of consciousness of up to 30 minutes or less, or posttraumatic amnesia of less than 24 hours duration, and that are associated with a GCS of 13-15 by 30 minutes after injury or at the time of initial medical assessment. This practice, they said, may reduce the risk of misinterpretation by medical professionals and the public that can occur when the terms “mTBI,” “concussion,” and “minor head injury” all may refer to the same injury.
The CDC has developed a suite of materials to assist both health care providers and the public in guideline implementation. The agency also is using its HEADS UP campaign to publicize the guidelines and related materials, and plans ongoing evaluation of the guidelines and implementation materials.
Many study authors, including Dr. Lumba-Brown, had relationships with medical device or pharmaceutical companies. The systematic review and guideline development were funded by the CDC.
A growing realization that mTBI can have persistent and significant deleterious effects has informed medical and public attitudes toward concussion in children, which now results in almost 1 million annual ED visits.
Progress at the laboratory bench has elucidated much of the neurometabolic cascade that occurs with the insult of mTBI, and has allowed researchers to document the path of brain healing after injury. Neuroimaging now can go beyond static images to trace neural networks and detect previously unseen and subtle functional deficits engendered by mTBI.
In particular, 21st century magnetic resonance imaging (MRI) has shown increased sensitivity over CT alone. In the TRACK-TBI study, over one in four patients whose CTs were read as normal had MRI findings consistent with trauma-induced pathology. Both multimodal MRI and serum biomarkers show promise, although more research regarding their utility is needed, particularly in the case of proteomic biomarkers.
Still, high-quality studies of pediatric mTBI are scant, and translation of burgeoning research into clinical practice is severely impeded by the numerous knowledge gaps that exist in the field.
Dr. Lumba-Brown and her colleagues have synthesized research that supports a neurobiopsychosocial model of mTBI in children that comes into play most prominently in the postacute phase, when non–injury-related factors such as demographics, socioeconomic status, and premorbid psychological conditions are strong mediators of the recovery trajectory.
With children as with adults, scant research guides the path forward for treatment and recovery from mTBI. For children, clinicians are still grappling with issues surrounding return to full participation in the academic and recreational activities of the school environment.
Data from two currently active studies should help light the way forward, however. The TRACK-TBI study, funded by the National Institutes of Health, will include almost 200 children among its 2,700 enrollees who have sustained all levels of TBI.
The Concussion Assessment, Research, and Education (CARE) Consortium is funded jointly by the National College Athletic Association and the Department of Defense. Between student athletes and military cadets, over 40,000 individuals are now part of the study.
The two studies’ testing modalities and methodologies align, offering the opportunity for a powerful pooled analysis that includes civilians, athletes, and those in the military.
Until then, these guidelines provide a way forward to an individualized approach to the best care for a child with mTBI.
Michael McCrea, PhD, is professor of neurology and neurosurgery, and director of brain injury research at the Medical College of Wisconsin, Milwaukee. Geoff Manley, MD, PhD, is professor of neurologic surgery at the University of California, San Francisco. Neither author reported conflicts of interest. These remarks were drawn from an editorial accompanying the guidelines and systematic review (JAMA Pediatrics. 2018 Sep 4. doi: 10.1001/jamapediatrics.2018.2846).
A growing realization that mTBI can have persistent and significant deleterious effects has informed medical and public attitudes toward concussion in children, which now results in almost 1 million annual ED visits.
Progress at the laboratory bench has elucidated much of the neurometabolic cascade that occurs with the insult of mTBI, and has allowed researchers to document the path of brain healing after injury. Neuroimaging now can go beyond static images to trace neural networks and detect previously unseen and subtle functional deficits engendered by mTBI.
In particular, 21st century magnetic resonance imaging (MRI) has shown increased sensitivity over CT alone. In the TRACK-TBI study, over one in four patients whose CTs were read as normal had MRI findings consistent with trauma-induced pathology. Both multimodal MRI and serum biomarkers show promise, although more research regarding their utility is needed, particularly in the case of proteomic biomarkers.
Still, high-quality studies of pediatric mTBI are scant, and translation of burgeoning research into clinical practice is severely impeded by the numerous knowledge gaps that exist in the field.
Dr. Lumba-Brown and her colleagues have synthesized research that supports a neurobiopsychosocial model of mTBI in children that comes into play most prominently in the postacute phase, when non–injury-related factors such as demographics, socioeconomic status, and premorbid psychological conditions are strong mediators of the recovery trajectory.
With children as with adults, scant research guides the path forward for treatment and recovery from mTBI. For children, clinicians are still grappling with issues surrounding return to full participation in the academic and recreational activities of the school environment.
Data from two currently active studies should help light the way forward, however. The TRACK-TBI study, funded by the National Institutes of Health, will include almost 200 children among its 2,700 enrollees who have sustained all levels of TBI.
The Concussion Assessment, Research, and Education (CARE) Consortium is funded jointly by the National College Athletic Association and the Department of Defense. Between student athletes and military cadets, over 40,000 individuals are now part of the study.
The two studies’ testing modalities and methodologies align, offering the opportunity for a powerful pooled analysis that includes civilians, athletes, and those in the military.
Until then, these guidelines provide a way forward to an individualized approach to the best care for a child with mTBI.
Michael McCrea, PhD, is professor of neurology and neurosurgery, and director of brain injury research at the Medical College of Wisconsin, Milwaukee. Geoff Manley, MD, PhD, is professor of neurologic surgery at the University of California, San Francisco. Neither author reported conflicts of interest. These remarks were drawn from an editorial accompanying the guidelines and systematic review (JAMA Pediatrics. 2018 Sep 4. doi: 10.1001/jamapediatrics.2018.2846).
A growing realization that mTBI can have persistent and significant deleterious effects has informed medical and public attitudes toward concussion in children, which now results in almost 1 million annual ED visits.
Progress at the laboratory bench has elucidated much of the neurometabolic cascade that occurs with the insult of mTBI, and has allowed researchers to document the path of brain healing after injury. Neuroimaging now can go beyond static images to trace neural networks and detect previously unseen and subtle functional deficits engendered by mTBI.
In particular, 21st century magnetic resonance imaging (MRI) has shown increased sensitivity over CT alone. In the TRACK-TBI study, over one in four patients whose CTs were read as normal had MRI findings consistent with trauma-induced pathology. Both multimodal MRI and serum biomarkers show promise, although more research regarding their utility is needed, particularly in the case of proteomic biomarkers.
Still, high-quality studies of pediatric mTBI are scant, and translation of burgeoning research into clinical practice is severely impeded by the numerous knowledge gaps that exist in the field.
Dr. Lumba-Brown and her colleagues have synthesized research that supports a neurobiopsychosocial model of mTBI in children that comes into play most prominently in the postacute phase, when non–injury-related factors such as demographics, socioeconomic status, and premorbid psychological conditions are strong mediators of the recovery trajectory.
With children as with adults, scant research guides the path forward for treatment and recovery from mTBI. For children, clinicians are still grappling with issues surrounding return to full participation in the academic and recreational activities of the school environment.
Data from two currently active studies should help light the way forward, however. The TRACK-TBI study, funded by the National Institutes of Health, will include almost 200 children among its 2,700 enrollees who have sustained all levels of TBI.
The Concussion Assessment, Research, and Education (CARE) Consortium is funded jointly by the National College Athletic Association and the Department of Defense. Between student athletes and military cadets, over 40,000 individuals are now part of the study.
The two studies’ testing modalities and methodologies align, offering the opportunity for a powerful pooled analysis that includes civilians, athletes, and those in the military.
Until then, these guidelines provide a way forward to an individualized approach to the best care for a child with mTBI.
Michael McCrea, PhD, is professor of neurology and neurosurgery, and director of brain injury research at the Medical College of Wisconsin, Milwaukee. Geoff Manley, MD, PhD, is professor of neurologic surgery at the University of California, San Francisco. Neither author reported conflicts of interest. These remarks were drawn from an editorial accompanying the guidelines and systematic review (JAMA Pediatrics. 2018 Sep 4. doi: 10.1001/jamapediatrics.2018.2846).
and should base management and prognostication on clinical decision-making tools and symptom rating scales, according to new practice guidelines issued by a working group of the Centers for Disease Control and Prevention (JAMA Pediatrics. 2018 Sep 4. doi: 10.1001/jamapediatrics.2018.2853.
The guidelines were released simultaneously with a systematic review, conducted by the same authors, of the existing literature regarding pediatric mTBI (JAMA Pediatrics 2018 Sep 4. doi: 10.1001/jamapediatrics.2018.2847). As the evaluators sorted through the literature to find high-quality studies for this population, the funnel rapidly narrowed: From an initial pool of over 15,000 studies conducted between 1990 and 2015, findings from just 75 studies were eventually included in the systematic review.
The review’s findings formed the basis for the guidelines and allowed Angela Lumba-Brown, MD, a pediatric emergency medicine physician at Stanford (Calif.) University, and her coauthors to ascribe a level of confidence in the inference from study data for a given recommendation. Recommendations also are categorized by strength and accordingly indicate that clinicians “should” or “may” follow them. Exceptions are carved out for practices, such as the use of hypertonic 3% saline solution for acute headache in the ED, that should not be used outside research settings.
In the end, the guidelines cover 19 main topics, sorted into guidance regarding the diagnosis, prognosis, and management and treatment of mTBI in children.
Diagnosis
The recommendations regarding mTBI diagnosis center around determining which children are at risk for significant intracranial injury (ICI). The guidelines recommend, with moderate confidence, that clinicians usually should not obtain a head CT for children with mTBI. Validated clinical decision rules should be used for risk stratification to determine which children can safely avoid imaging and which children should be considered for head CT, wrote Dr. Lumba-Brown and her coauthors. Magnetic resonance imaging is not recommended for initial evaluation of mTBI, nor should skull radiographs be ordered in the absence of clinical suspicion for skull fracture.
From the systematic review, Dr. Lumba-Brown and her colleagues found that several risk factors taken together may mean that significant ICI is more likely. These include patient age younger than 2 years; any vomiting, loss of consciousness, or amnesia; a severe mechanism of injury, severe or worsening headache, or nonfrontal scalp hematoma; a Glasgow Coma Scale (GCS) score of less than 15; and clinical suspicion for skull fracture. Clinicians should give consideration to the risks of ionizing radiation to the head, and balance this against their assessment of risk for severe – and perhaps actionable – injury.
A validated symptom rating scale, used in an age-appropriate way, should be used as part of the evaluation of children with mTBI. For children aged 6 and older, the Graded Symptom Checklist is an appropriate tool within 2 days after injury, while the Post Concussion Symptom Scale as part of computerized neurocognitive testing can differentiate which high school athletes have mTBI when used within 4 days of injury, according to the guidelines, which also identify other validated symptom rating scales.
The guidelines authors recommend, with high confidence, that serum biomarkers should not be used outside of research settings in the diagnosis of mTBI in children at present.
Prognosis
Families should be counseled that symptoms mostly resolve within 1-3 months for up to 80% of children with mTBI, but families also should know that “each child’s recovery from mTBI is unique and will follow its own trajectory,” wrote Dr. Lumba-Brown and her coauthors, in a moderate-strength recommendation.
Some factors have been associated with slower recovery from mTBI, and either upon evaluation for mTBI or in routine sports examinations, families should be told about this potential if risk factors are present, said the guidelines, although the evidence supporting the associations is of “varying strength,” wrote Dr. Lumba-Brown and her coauthors. Children with previous mTBIs and those with a history of premorbid neurologic and psychiatric problems, learning problems, or family and social stress all may have delayed recovery. For children with ICI, lower cognitive ability also is associated with delayed recovery.
Demographic factors such as lower socioeconomic status and being of Hispanic ethnicity also may increase the risk for delayed mTBI recovery. Older children and adolescents may recover more slowly. Those with more severe initial presentation and more symptoms in the immediate post-mTBI phase also may have a slower recovery course, said Dr. Lumba-Brown and her coauthors.
A validated prediction rule can be used in the ED to gather information about these discrete risk factors to guide family counseling, according to the guidelines, which note that research has found that “an empirically derived set of risk factors predicted the risk of persistent post-concussion symptoms at 28 days” for children seen in the ED with mTBI.
During the recovery phase, a combination of tools should be used to track recovery from mTBI; these can include validated symptom scales, validated cognitive testing, reaction time measures, and, in adolescent athletes, balance testing. Using a combination of tools is a valuable strategy, the researchers wrote. “No single assessment tool is strongly predictive of outcome in children with mTBI,” they noted.
When prognosis is poor, or recovery is not proceeding as expected, clinicians should have a low threshold for initiating other interventions and referrals.
Management and treatment
Although the guideline authors acknowledged significant knowledge gaps in all areas of pediatric mTBI diagnosis and management, evidence is especially scant for best practices for treatment, rest, and return to play and school after a child sustains mTBI, said Dr. Lumba-Brown and her coauthors.
However, families should be given information about warning signs for serious head injury and how to monitor symptoms, as well as information about mTBI and the expected recovery course. Other forward-looking instructions should cover the importance of preventing new head injuries, managing the gradual return to normal cognitive and physical activities, and clear instructions regarding return to school and recreational activities. The guideline authors made a strong recommendation to provide this information, with high confidence in the data.
However, little strong evidence points the way to a clear set of criteria for when children are ready for school, play, and athletic participation. These decisions must be customized to the individual child, and decision making, particularly about return to school and academic activities, should be a collaborative affair, with schools, clinicians, and families all communicating to make sure the pace of return to normal life is keeping pace with the child’s recovery. “Because postconcussive symptoms resolve at different rates in different children after mTBI, individualization of return-to-school programming is necessary,” wrote Dr. Lumba-Brown and her coauthors.
The guideline authors cite evidence that “suggests that early rest (within the first 3 days) may be beneficial but that inactivity beyond this period for most children may worsen their self-reported symptoms.”
Psychosocial support may be beneficial for certain children, wrote the researchers, drawing on evidence showing that such support is beneficial in frank TBI, and is probably beneficial in mTBI.
Active rehabilitation as tolerated is recommended after an initial period of rest, with exertion kept to a level that does not exacerbate symptoms. Children should not participate in contact activities until symptoms are fully resolved.
A posttraumatic headache that is severe or worsens in the ED should prompt consideration of emergent neuroimaging, according to the guidelines. In the postacute phase, however, children can have nonopioid analgesia, although parents should know about such risks as rebound headache. When chronic headache follows a mTBI, the guidelines recommend that clinicians refer patients for a multidisciplinary evaluation that can assess the many factors – including analgesic overuse – that can be contributors.
Drawing on the larger body of adult TBI research, the authors recommend that insufficient or disordered sleep be addressed, because “the maintenance of appropriate sleep and the management of disrupted sleep may be a critical target of treatment for the child with mTBI.”
Children who suffer a mTBI may experience cognitive dysfunction as a direct result of injury to the brain or secondary to the effects of other symptoms such as sleep disruptions, headache pain, fatigue, or low tolerance of frustration. Clinicians may want to perform or refer their patients for a neuropsychological evaluation to determine what is causing the cognitive dysfunction, the authors said.
Dr. Lumba-Brown and her coauthors, who formed the CDC’s Pediatric Mild Traumatic Brain Injury Guideline Workgroup, also recommended that clinicians use the term “mild traumatic brain injury” to describe head injuries that cause confusion or disorientation, without loss of consciousness, or loss of consciousness of up to 30 minutes or less, or posttraumatic amnesia of less than 24 hours duration, and that are associated with a GCS of 13-15 by 30 minutes after injury or at the time of initial medical assessment. This practice, they said, may reduce the risk of misinterpretation by medical professionals and the public that can occur when the terms “mTBI,” “concussion,” and “minor head injury” all may refer to the same injury.
The CDC has developed a suite of materials to assist both health care providers and the public in guideline implementation. The agency also is using its HEADS UP campaign to publicize the guidelines and related materials, and plans ongoing evaluation of the guidelines and implementation materials.
Many study authors, including Dr. Lumba-Brown, had relationships with medical device or pharmaceutical companies. The systematic review and guideline development were funded by the CDC.
and should base management and prognostication on clinical decision-making tools and symptom rating scales, according to new practice guidelines issued by a working group of the Centers for Disease Control and Prevention (JAMA Pediatrics. 2018 Sep 4. doi: 10.1001/jamapediatrics.2018.2853.
The guidelines were released simultaneously with a systematic review, conducted by the same authors, of the existing literature regarding pediatric mTBI (JAMA Pediatrics 2018 Sep 4. doi: 10.1001/jamapediatrics.2018.2847). As the evaluators sorted through the literature to find high-quality studies for this population, the funnel rapidly narrowed: From an initial pool of over 15,000 studies conducted between 1990 and 2015, findings from just 75 studies were eventually included in the systematic review.
The review’s findings formed the basis for the guidelines and allowed Angela Lumba-Brown, MD, a pediatric emergency medicine physician at Stanford (Calif.) University, and her coauthors to ascribe a level of confidence in the inference from study data for a given recommendation. Recommendations also are categorized by strength and accordingly indicate that clinicians “should” or “may” follow them. Exceptions are carved out for practices, such as the use of hypertonic 3% saline solution for acute headache in the ED, that should not be used outside research settings.
In the end, the guidelines cover 19 main topics, sorted into guidance regarding the diagnosis, prognosis, and management and treatment of mTBI in children.
Diagnosis
The recommendations regarding mTBI diagnosis center around determining which children are at risk for significant intracranial injury (ICI). The guidelines recommend, with moderate confidence, that clinicians usually should not obtain a head CT for children with mTBI. Validated clinical decision rules should be used for risk stratification to determine which children can safely avoid imaging and which children should be considered for head CT, wrote Dr. Lumba-Brown and her coauthors. Magnetic resonance imaging is not recommended for initial evaluation of mTBI, nor should skull radiographs be ordered in the absence of clinical suspicion for skull fracture.
From the systematic review, Dr. Lumba-Brown and her colleagues found that several risk factors taken together may mean that significant ICI is more likely. These include patient age younger than 2 years; any vomiting, loss of consciousness, or amnesia; a severe mechanism of injury, severe or worsening headache, or nonfrontal scalp hematoma; a Glasgow Coma Scale (GCS) score of less than 15; and clinical suspicion for skull fracture. Clinicians should give consideration to the risks of ionizing radiation to the head, and balance this against their assessment of risk for severe – and perhaps actionable – injury.
A validated symptom rating scale, used in an age-appropriate way, should be used as part of the evaluation of children with mTBI. For children aged 6 and older, the Graded Symptom Checklist is an appropriate tool within 2 days after injury, while the Post Concussion Symptom Scale as part of computerized neurocognitive testing can differentiate which high school athletes have mTBI when used within 4 days of injury, according to the guidelines, which also identify other validated symptom rating scales.
The guidelines authors recommend, with high confidence, that serum biomarkers should not be used outside of research settings in the diagnosis of mTBI in children at present.
Prognosis
Families should be counseled that symptoms mostly resolve within 1-3 months for up to 80% of children with mTBI, but families also should know that “each child’s recovery from mTBI is unique and will follow its own trajectory,” wrote Dr. Lumba-Brown and her coauthors, in a moderate-strength recommendation.
Some factors have been associated with slower recovery from mTBI, and either upon evaluation for mTBI or in routine sports examinations, families should be told about this potential if risk factors are present, said the guidelines, although the evidence supporting the associations is of “varying strength,” wrote Dr. Lumba-Brown and her coauthors. Children with previous mTBIs and those with a history of premorbid neurologic and psychiatric problems, learning problems, or family and social stress all may have delayed recovery. For children with ICI, lower cognitive ability also is associated with delayed recovery.
Demographic factors such as lower socioeconomic status and being of Hispanic ethnicity also may increase the risk for delayed mTBI recovery. Older children and adolescents may recover more slowly. Those with more severe initial presentation and more symptoms in the immediate post-mTBI phase also may have a slower recovery course, said Dr. Lumba-Brown and her coauthors.
A validated prediction rule can be used in the ED to gather information about these discrete risk factors to guide family counseling, according to the guidelines, which note that research has found that “an empirically derived set of risk factors predicted the risk of persistent post-concussion symptoms at 28 days” for children seen in the ED with mTBI.
During the recovery phase, a combination of tools should be used to track recovery from mTBI; these can include validated symptom scales, validated cognitive testing, reaction time measures, and, in adolescent athletes, balance testing. Using a combination of tools is a valuable strategy, the researchers wrote. “No single assessment tool is strongly predictive of outcome in children with mTBI,” they noted.
When prognosis is poor, or recovery is not proceeding as expected, clinicians should have a low threshold for initiating other interventions and referrals.
Management and treatment
Although the guideline authors acknowledged significant knowledge gaps in all areas of pediatric mTBI diagnosis and management, evidence is especially scant for best practices for treatment, rest, and return to play and school after a child sustains mTBI, said Dr. Lumba-Brown and her coauthors.
However, families should be given information about warning signs for serious head injury and how to monitor symptoms, as well as information about mTBI and the expected recovery course. Other forward-looking instructions should cover the importance of preventing new head injuries, managing the gradual return to normal cognitive and physical activities, and clear instructions regarding return to school and recreational activities. The guideline authors made a strong recommendation to provide this information, with high confidence in the data.
However, little strong evidence points the way to a clear set of criteria for when children are ready for school, play, and athletic participation. These decisions must be customized to the individual child, and decision making, particularly about return to school and academic activities, should be a collaborative affair, with schools, clinicians, and families all communicating to make sure the pace of return to normal life is keeping pace with the child’s recovery. “Because postconcussive symptoms resolve at different rates in different children after mTBI, individualization of return-to-school programming is necessary,” wrote Dr. Lumba-Brown and her coauthors.
The guideline authors cite evidence that “suggests that early rest (within the first 3 days) may be beneficial but that inactivity beyond this period for most children may worsen their self-reported symptoms.”
Psychosocial support may be beneficial for certain children, wrote the researchers, drawing on evidence showing that such support is beneficial in frank TBI, and is probably beneficial in mTBI.
Active rehabilitation as tolerated is recommended after an initial period of rest, with exertion kept to a level that does not exacerbate symptoms. Children should not participate in contact activities until symptoms are fully resolved.
A posttraumatic headache that is severe or worsens in the ED should prompt consideration of emergent neuroimaging, according to the guidelines. In the postacute phase, however, children can have nonopioid analgesia, although parents should know about such risks as rebound headache. When chronic headache follows a mTBI, the guidelines recommend that clinicians refer patients for a multidisciplinary evaluation that can assess the many factors – including analgesic overuse – that can be contributors.
Drawing on the larger body of adult TBI research, the authors recommend that insufficient or disordered sleep be addressed, because “the maintenance of appropriate sleep and the management of disrupted sleep may be a critical target of treatment for the child with mTBI.”
Children who suffer a mTBI may experience cognitive dysfunction as a direct result of injury to the brain or secondary to the effects of other symptoms such as sleep disruptions, headache pain, fatigue, or low tolerance of frustration. Clinicians may want to perform or refer their patients for a neuropsychological evaluation to determine what is causing the cognitive dysfunction, the authors said.
Dr. Lumba-Brown and her coauthors, who formed the CDC’s Pediatric Mild Traumatic Brain Injury Guideline Workgroup, also recommended that clinicians use the term “mild traumatic brain injury” to describe head injuries that cause confusion or disorientation, without loss of consciousness, or loss of consciousness of up to 30 minutes or less, or posttraumatic amnesia of less than 24 hours duration, and that are associated with a GCS of 13-15 by 30 minutes after injury or at the time of initial medical assessment. This practice, they said, may reduce the risk of misinterpretation by medical professionals and the public that can occur when the terms “mTBI,” “concussion,” and “minor head injury” all may refer to the same injury.
The CDC has developed a suite of materials to assist both health care providers and the public in guideline implementation. The agency also is using its HEADS UP campaign to publicize the guidelines and related materials, and plans ongoing evaluation of the guidelines and implementation materials.
Many study authors, including Dr. Lumba-Brown, had relationships with medical device or pharmaceutical companies. The systematic review and guideline development were funded by the CDC.
FROM JAMA PEDIATRICS
ARRIVE: What are the perinatal and maternal consequences of labor induction at 39 weeks compared with expectant management?
WHAT DOES THIS MEAN FOR PRACTICE?
- Induction of labor at 39 weeks in low-risk nulliparas, irrespective of Bishop score, seems to be a reasonable option to be included in route of delivery discussions with patients as part of the principle of shared decision-making.
- The data in this trial would suggest that such an approach not only reduces adverse perinatal outcomes but also may reduce the need for subsequent cesarean delivery.
Updates to EULAR hand OA management recommendations reflect current evidence
Updated EULAR recommendations on the management of hand osteoarthritis include five overarching principles as well as two new recommendations that reflect new research in the field.
The task force, led by Margreet Kloppenburg, MD, PhD, of the department of rheumatology at Leiden (the Netherlands) University Medical Center, noted that a decade had passed since the first recommendations were published in 2007.
“It was timely to update the recommendations, as many new studies had emerged during this period. In light of this new evidence, many of the 2007 recommendations were modified and new recommendations were added,” wrote Dr. Kloppenburg and her colleagues. The recommendations were published online in Annals of the Rheumatic Diseases.
They noted that the recommendations were targeted to all health professionals across primary and secondary care but also aimed to inform patients about their disease to “support shared decision making.”
In line with other EULAR sets of management recommendations, the update included five overarching principles that cover treatment goals, information and education for patients, individualization of treatment, shared decision making between clinicians and patients, and the need to take into consideration a multidisciplinary and multimodal (pharmacologic and nonpharmacologic) treatment approach.
The authors noted that for a long time hand OA was a “forgotten disease” and this was reflected by the paucity of clinical trials in the area. As a direct consequence, previous recommendations were based on expert opinion rather than evidence.
However, new data allowed the task force to recommend not to treat patients with hand OA with conventional synthetic or biologic disease-modifying antirheumatic drugs (DMARDs). The recommendation achieved the strongest level of evidence and a high level of agreement from the 19-member expert panel, which included 2 patient research partners. The authors said the recommendation was based on newer studies that demonstrated a lack of efficacy of csDMARDs and bDMARDs.
The authors also advised adapting the long-term follow-up of patients with hand OA to individual needs, although they noted this was based on expert opinion alone and that in the absence of a disease-modifying treatment, the goal of follow-up differs from that of many other rheumatic diseases. Individual needs will dictate the degree of follow-up required, based on the severity of symptoms, presence of erosive disease, reevaluation of the use of pharmacologic therapy, and a patient’s wishes and expectations. They also noted that “for most patients, standard radiographic follow-up is not useful at this moment” and that “follow-up does not necessarily have to be performed by a rheumatologist.
“Follow-up will likely increase adherence to nonpharmacological therapies like exercise or orthoses, and provides an opportunity for reevaluation of treatment,” they wrote.
The recommendations advise offering education and training in ergonomic principles and exercises to patients to improve function and muscle strength, as well as considering the use of orthoses in some patients.
Treatment recommendations suggested preferring topical treatments over systemic treatments and that oral analgesics, particularly NSAIDs, should be considered for a limited duration. The authors advised that chondroitin sulfate may be used in patients for pain relief and improvement in functioning and that intra-articular glucocorticoids should not generally be used but may be considered in patients with painful interphalangeal joints. Surgery should be considered for patients with structural abnormalities when other treatment modalities have not been sufficiently effective in relieving pain.
The recommendations were funded by EULAR. Several of the authors reported receiving consultancy fees and/or honoraria as well as research funding from industry.
SOURCE: Kloppenburg M et al. Ann Rheum Dis. 2018 Aug 28. doi: 10.1136/annrheumdis-2018-213826.
EULAR has updated its 2007 guidelines for the management of hand osteoarthritis. I find the recommendations helpful, and I have no disagreements.
The authors performed a systematic literature review that was more complete than the original guidelines. In addition, the methodology in developing the guidelines was updated utilizing the GRADE (Grading of Recommendations, Assessment, Development and Evaluations) system to guide the expert opinion. The manuscript presents recommendations that are carefully supported in the text. To understand guidelines, one really needs to read the text.

The update lists a set of research questions, similar to the 2007 recommendations.
The authors group their therapeutic recommendations according to nonpharmacologic, pharmacologic, and surgical approaches, as well as about the need for follow-up. The three nonpharmacologic recommendations include education and training, exercise and muscle strengthening, and the use of orthoses. The pharmacologic approach includes topical therapy as a first-line, oral NSAIDs and analgesics, chondroitin sulfate, and intra-articular injections. There is a negative recommendation for the use of biologics. The surgical recommendation is directed at the relief of pain. The last recommendation emphasizes the need for follow-up and individual care.
The differences between the recommendations include the removal of acetaminophen as a first-line therapy. Indeed, it seems to be barely recommended at all. In addition, there is an emphasis on topical therapy, particularly NSAIDs. The authors are equivocal on the recommendations for intra-articular therapy. Paraffin and local heat are no longer included. The recommendation against biologic therapy is new. They included agents used for rheumatoid arthritis, such as methotrexate, in this negative recommendation.
These new recommendations are an update of guidelines that are over 10 years old. They are practical and helpful. Unfortunately, more research is needed as the present day therapy is often inadequate.
Roy D. Altman, MD, is professor emeritus of medicine in the division of rheumatology and immunology at the University of California, Los Angeles. He is a consultant to Ferring, Flexion, GlaxoSmithKline, Novartis, Olatec, Pfizer, and Sorrento Therapeutics.
EULAR has updated its 2007 guidelines for the management of hand osteoarthritis. I find the recommendations helpful, and I have no disagreements.
The authors performed a systematic literature review that was more complete than the original guidelines. In addition, the methodology in developing the guidelines was updated utilizing the GRADE (Grading of Recommendations, Assessment, Development and Evaluations) system to guide the expert opinion. The manuscript presents recommendations that are carefully supported in the text. To understand guidelines, one really needs to read the text.
The update lists a set of research questions, similar to the 2007 recommendations.
The authors group their therapeutic recommendations according to nonpharmacologic, pharmacologic, and surgical approaches, as well as about the need for follow-up. The three nonpharmacologic recommendations include education and training, exercise and muscle strengthening, and the use of orthoses. The pharmacologic approach includes topical therapy as a first-line, oral NSAIDs and analgesics, chondroitin sulfate, and intra-articular injections. There is a negative recommendation for the use of biologics. The surgical recommendation is directed at the relief of pain. The last recommendation emphasizes the need for follow-up and individual care.
The differences between the recommendations include the removal of acetaminophen as a first-line therapy. Indeed, it seems to be barely recommended at all. In addition, there is an emphasis on topical therapy, particularly NSAIDs. The authors are equivocal on the recommendations for intra-articular therapy. Paraffin and local heat are no longer included. The recommendation against biologic therapy is new. They included agents used for rheumatoid arthritis, such as methotrexate, in this negative recommendation.
These new recommendations are an update of guidelines that are over 10 years old. They are practical and helpful. Unfortunately, more research is needed as the present day therapy is often inadequate.
Roy D. Altman, MD, is professor emeritus of medicine in the division of rheumatology and immunology at the University of California, Los Angeles. He is a consultant to Ferring, Flexion, GlaxoSmithKline, Novartis, Olatec, Pfizer, and Sorrento Therapeutics.
EULAR has updated its 2007 guidelines for the management of hand osteoarthritis. I find the recommendations helpful, and I have no disagreements.
The authors performed a systematic literature review that was more complete than the original guidelines. In addition, the methodology in developing the guidelines was updated utilizing the GRADE (Grading of Recommendations, Assessment, Development and Evaluations) system to guide the expert opinion. The manuscript presents recommendations that are carefully supported in the text. To understand guidelines, one really needs to read the text.
The update lists a set of research questions, similar to the 2007 recommendations.
The authors group their therapeutic recommendations according to nonpharmacologic, pharmacologic, and surgical approaches, as well as about the need for follow-up. The three nonpharmacologic recommendations include education and training, exercise and muscle strengthening, and the use of orthoses. The pharmacologic approach includes topical therapy as a first-line, oral NSAIDs and analgesics, chondroitin sulfate, and intra-articular injections. There is a negative recommendation for the use of biologics. The surgical recommendation is directed at the relief of pain. The last recommendation emphasizes the need for follow-up and individual care.
The differences between the recommendations include the removal of acetaminophen as a first-line therapy. Indeed, it seems to be barely recommended at all. In addition, there is an emphasis on topical therapy, particularly NSAIDs. The authors are equivocal on the recommendations for intra-articular therapy. Paraffin and local heat are no longer included. The recommendation against biologic therapy is new. They included agents used for rheumatoid arthritis, such as methotrexate, in this negative recommendation.
These new recommendations are an update of guidelines that are over 10 years old. They are practical and helpful. Unfortunately, more research is needed as the present day therapy is often inadequate.
Roy D. Altman, MD, is professor emeritus of medicine in the division of rheumatology and immunology at the University of California, Los Angeles. He is a consultant to Ferring, Flexion, GlaxoSmithKline, Novartis, Olatec, Pfizer, and Sorrento Therapeutics.
Updated EULAR recommendations on the management of hand osteoarthritis include five overarching principles as well as two new recommendations that reflect new research in the field.
The task force, led by Margreet Kloppenburg, MD, PhD, of the department of rheumatology at Leiden (the Netherlands) University Medical Center, noted that a decade had passed since the first recommendations were published in 2007.
“It was timely to update the recommendations, as many new studies had emerged during this period. In light of this new evidence, many of the 2007 recommendations were modified and new recommendations were added,” wrote Dr. Kloppenburg and her colleagues. The recommendations were published online in Annals of the Rheumatic Diseases.
They noted that the recommendations were targeted to all health professionals across primary and secondary care but also aimed to inform patients about their disease to “support shared decision making.”
In line with other EULAR sets of management recommendations, the update included five overarching principles that cover treatment goals, information and education for patients, individualization of treatment, shared decision making between clinicians and patients, and the need to take into consideration a multidisciplinary and multimodal (pharmacologic and nonpharmacologic) treatment approach.
The authors noted that for a long time hand OA was a “forgotten disease” and this was reflected by the paucity of clinical trials in the area. As a direct consequence, previous recommendations were based on expert opinion rather than evidence.
However, new data allowed the task force to recommend not to treat patients with hand OA with conventional synthetic or biologic disease-modifying antirheumatic drugs (DMARDs). The recommendation achieved the strongest level of evidence and a high level of agreement from the 19-member expert panel, which included 2 patient research partners. The authors said the recommendation was based on newer studies that demonstrated a lack of efficacy of csDMARDs and bDMARDs.
The authors also advised adapting the long-term follow-up of patients with hand OA to individual needs, although they noted this was based on expert opinion alone and that in the absence of a disease-modifying treatment, the goal of follow-up differs from that of many other rheumatic diseases. Individual needs will dictate the degree of follow-up required, based on the severity of symptoms, presence of erosive disease, reevaluation of the use of pharmacologic therapy, and a patient’s wishes and expectations. They also noted that “for most patients, standard radiographic follow-up is not useful at this moment” and that “follow-up does not necessarily have to be performed by a rheumatologist.
“Follow-up will likely increase adherence to nonpharmacological therapies like exercise or orthoses, and provides an opportunity for reevaluation of treatment,” they wrote.
The recommendations advise offering education and training in ergonomic principles and exercises to patients to improve function and muscle strength, as well as considering the use of orthoses in some patients.
Treatment recommendations suggested preferring topical treatments over systemic treatments and that oral analgesics, particularly NSAIDs, should be considered for a limited duration. The authors advised that chondroitin sulfate may be used in patients for pain relief and improvement in functioning and that intra-articular glucocorticoids should not generally be used but may be considered in patients with painful interphalangeal joints. Surgery should be considered for patients with structural abnormalities when other treatment modalities have not been sufficiently effective in relieving pain.
The recommendations were funded by EULAR. Several of the authors reported receiving consultancy fees and/or honoraria as well as research funding from industry.
SOURCE: Kloppenburg M et al. Ann Rheum Dis. 2018 Aug 28. doi: 10.1136/annrheumdis-2018-213826.
Updated EULAR recommendations on the management of hand osteoarthritis include five overarching principles as well as two new recommendations that reflect new research in the field.
The task force, led by Margreet Kloppenburg, MD, PhD, of the department of rheumatology at Leiden (the Netherlands) University Medical Center, noted that a decade had passed since the first recommendations were published in 2007.
“It was timely to update the recommendations, as many new studies had emerged during this period. In light of this new evidence, many of the 2007 recommendations were modified and new recommendations were added,” wrote Dr. Kloppenburg and her colleagues. The recommendations were published online in Annals of the Rheumatic Diseases.
They noted that the recommendations were targeted to all health professionals across primary and secondary care but also aimed to inform patients about their disease to “support shared decision making.”
In line with other EULAR sets of management recommendations, the update included five overarching principles that cover treatment goals, information and education for patients, individualization of treatment, shared decision making between clinicians and patients, and the need to take into consideration a multidisciplinary and multimodal (pharmacologic and nonpharmacologic) treatment approach.
The authors noted that for a long time hand OA was a “forgotten disease” and this was reflected by the paucity of clinical trials in the area. As a direct consequence, previous recommendations were based on expert opinion rather than evidence.
However, new data allowed the task force to recommend not to treat patients with hand OA with conventional synthetic or biologic disease-modifying antirheumatic drugs (DMARDs). The recommendation achieved the strongest level of evidence and a high level of agreement from the 19-member expert panel, which included 2 patient research partners. The authors said the recommendation was based on newer studies that demonstrated a lack of efficacy of csDMARDs and bDMARDs.
The authors also advised adapting the long-term follow-up of patients with hand OA to individual needs, although they noted this was based on expert opinion alone and that in the absence of a disease-modifying treatment, the goal of follow-up differs from that of many other rheumatic diseases. Individual needs will dictate the degree of follow-up required, based on the severity of symptoms, presence of erosive disease, reevaluation of the use of pharmacologic therapy, and a patient’s wishes and expectations. They also noted that “for most patients, standard radiographic follow-up is not useful at this moment” and that “follow-up does not necessarily have to be performed by a rheumatologist.
“Follow-up will likely increase adherence to nonpharmacological therapies like exercise or orthoses, and provides an opportunity for reevaluation of treatment,” they wrote.
The recommendations advise offering education and training in ergonomic principles and exercises to patients to improve function and muscle strength, as well as considering the use of orthoses in some patients.
Treatment recommendations suggested preferring topical treatments over systemic treatments and that oral analgesics, particularly NSAIDs, should be considered for a limited duration. The authors advised that chondroitin sulfate may be used in patients for pain relief and improvement in functioning and that intra-articular glucocorticoids should not generally be used but may be considered in patients with painful interphalangeal joints. Surgery should be considered for patients with structural abnormalities when other treatment modalities have not been sufficiently effective in relieving pain.
The recommendations were funded by EULAR. Several of the authors reported receiving consultancy fees and/or honoraria as well as research funding from industry.
SOURCE: Kloppenburg M et al. Ann Rheum Dis. 2018 Aug 28. doi: 10.1136/annrheumdis-2018-213826.
FROM ANNALS OF THE RHEUMATIC DISEASES