User login
Spurring innovation in digital health
Owing to digital advances, we’re experiencing a reimagination of health care delivery. Consumers are now empowered to take more control of their own health information to make better informed decisions about their medical care and healthy living. These advances enable better health outcomes for patients.

This opportunity is supported by a new technological paradigm of digital health tools, like apps, that enable consumers to have more active engagement and access to real-time information about their health and activities. These tools allow consumers and providers to supersede the traditional, physical constraints of health care delivery and make the most of the opportunities offered by mobile technology.
With these advances has come a new swath of companies that are investing in these new opportunities. These firms may be new to health care products and may not be accustomed to navigating the regulatory landscape that has traditionally surrounded these areas. A great example is the announcement of two mobile medical apps designed by Apple to work on the Apple Watch. One app creates an electrocardiogram, similar to traditional electrocardiograms, to detect the presence of atrial fibrillation and regular heart rhythm, while the other app analyzes pulse rate data to identify irregular heart rhythms suggestive of atrial fibrillation and notify the user. The FDA [Food and Drug Administration] worked closely with the company as they developed and tested these software products, which may help millions of users identify health concerns more quickly. Health care products on ubiquitous devices, like smartwatches, may help users seek treatment earlier and will truly empower them with more information about their health.
In the last few years, the FDA has been taking steps to encourage more development and greater innovation in the digital health space. With the launch of our Digital Health Innovation Action Plan last summer, we committed to implementing policies, adding expertise, and exploring a software precertification pilot program to bring clarity and efficiency to how we regulate digital health products.
This commitment is not only reflected in actions like approving or clearing new apps and launching our Digital Health Innovation Action Plan but also in what we hope to do in the future. That’s why in the FDA’s Fiscal Year 2019 Budget, we proposed to create a Center of Excellence for Digital Health that would advance modernizing our regulatory approach to help this industry grow and reach its full potential, while protecting patients.
Dr. Gottlieb is commissioner of the FDA and Dr. Shuren in director of the FDA Center for Devices and Radiological Health. Their comments are excerpted from an FDA statement released Sept. 12, 2018.
Owing to digital advances, we’re experiencing a reimagination of health care delivery. Consumers are now empowered to take more control of their own health information to make better informed decisions about their medical care and healthy living. These advances enable better health outcomes for patients.
This opportunity is supported by a new technological paradigm of digital health tools, like apps, that enable consumers to have more active engagement and access to real-time information about their health and activities. These tools allow consumers and providers to supersede the traditional, physical constraints of health care delivery and make the most of the opportunities offered by mobile technology.
With these advances has come a new swath of companies that are investing in these new opportunities. These firms may be new to health care products and may not be accustomed to navigating the regulatory landscape that has traditionally surrounded these areas. A great example is the announcement of two mobile medical apps designed by Apple to work on the Apple Watch. One app creates an electrocardiogram, similar to traditional electrocardiograms, to detect the presence of atrial fibrillation and regular heart rhythm, while the other app analyzes pulse rate data to identify irregular heart rhythms suggestive of atrial fibrillation and notify the user. The FDA [Food and Drug Administration] worked closely with the company as they developed and tested these software products, which may help millions of users identify health concerns more quickly. Health care products on ubiquitous devices, like smartwatches, may help users seek treatment earlier and will truly empower them with more information about their health.
In the last few years, the FDA has been taking steps to encourage more development and greater innovation in the digital health space. With the launch of our Digital Health Innovation Action Plan last summer, we committed to implementing policies, adding expertise, and exploring a software precertification pilot program to bring clarity and efficiency to how we regulate digital health products.
This commitment is not only reflected in actions like approving or clearing new apps and launching our Digital Health Innovation Action Plan but also in what we hope to do in the future. That’s why in the FDA’s Fiscal Year 2019 Budget, we proposed to create a Center of Excellence for Digital Health that would advance modernizing our regulatory approach to help this industry grow and reach its full potential, while protecting patients.
Dr. Gottlieb is commissioner of the FDA and Dr. Shuren in director of the FDA Center for Devices and Radiological Health. Their comments are excerpted from an FDA statement released Sept. 12, 2018.
Owing to digital advances, we’re experiencing a reimagination of health care delivery. Consumers are now empowered to take more control of their own health information to make better informed decisions about their medical care and healthy living. These advances enable better health outcomes for patients.
This opportunity is supported by a new technological paradigm of digital health tools, like apps, that enable consumers to have more active engagement and access to real-time information about their health and activities. These tools allow consumers and providers to supersede the traditional, physical constraints of health care delivery and make the most of the opportunities offered by mobile technology.
With these advances has come a new swath of companies that are investing in these new opportunities. These firms may be new to health care products and may not be accustomed to navigating the regulatory landscape that has traditionally surrounded these areas. A great example is the announcement of two mobile medical apps designed by Apple to work on the Apple Watch. One app creates an electrocardiogram, similar to traditional electrocardiograms, to detect the presence of atrial fibrillation and regular heart rhythm, while the other app analyzes pulse rate data to identify irregular heart rhythms suggestive of atrial fibrillation and notify the user. The FDA [Food and Drug Administration] worked closely with the company as they developed and tested these software products, which may help millions of users identify health concerns more quickly. Health care products on ubiquitous devices, like smartwatches, may help users seek treatment earlier and will truly empower them with more information about their health.
In the last few years, the FDA has been taking steps to encourage more development and greater innovation in the digital health space. With the launch of our Digital Health Innovation Action Plan last summer, we committed to implementing policies, adding expertise, and exploring a software precertification pilot program to bring clarity and efficiency to how we regulate digital health products.
This commitment is not only reflected in actions like approving or clearing new apps and launching our Digital Health Innovation Action Plan but also in what we hope to do in the future. That’s why in the FDA’s Fiscal Year 2019 Budget, we proposed to create a Center of Excellence for Digital Health that would advance modernizing our regulatory approach to help this industry grow and reach its full potential, while protecting patients.
Dr. Gottlieb is commissioner of the FDA and Dr. Shuren in director of the FDA Center for Devices and Radiological Health. Their comments are excerpted from an FDA statement released Sept. 12, 2018.
E/M comments may fall on deaf ears at CMS
Doctors’ dismay at the proposed flattening of evaluation and management (E/M) payments seems to be falling on deaf ears.
More than 170 medical societies and organizations expressed their concern about the new payment structure for E/M codes proposed as part of the 2019 Medicare Physician Fee Schedule (PFS) in comments on the draft rule.
Yet, in the final days of the comment period, Seema Verma, administrator of the Centers for Medicare & Medicaid Services, took to Twitter to defend her agency’s plan.

The controversial proposal would set the payment rate for a level 1 E/M office visit for a new patient at $44, down from the current $45. Payment for levels 2-5 would be $135. Currently, payments for level 2 new patient visits are set at $76, level 3 at $110, level 4 at $167, and level 5 at $211.
For E/M office visits with established patients, the proposed rate would be $24 for level 1, up from the current $22. Payment for levels 2-5 would be $93. Under the current methodology, payments for established patient level 2 visits are set at $45, level 3 at $74, level 4 at $109, and level 5 at $148.
Offsetting the changes in payment are several new proposed add-on codes, according to CMS.
Despite the lower payment for more complex patient care, Ms. Verma touted the scheme’s budget neutrality.

Ms. Verma’s tweets come as medical societies filed their formal complaints on the proposal, mirroring concerns expressed in two letters sent to the agency ahead of the comment deadline. The letters, sent at the end of August and between the two of them signed by more than 170 medical associations, aimed to preempt the comment process. They called for the E/M proposal to be rescinded, claiming that the cuts would reduce access to Medicare services by patients and hurt physicians that treat the sickest patients and those who provide comprehensive primary care because the expected lower reimbursement. One suggested that the changes exacerbate workforce shortages.
In its formal comments, the American Medical Association said that given “the groundswell of opposition from individual physicians and nearly every physician and health professional organization in the country, including the AMA, we ask that CMS set aside its proposal to restructure payment and coding for E/M office and other outpatient visits while an expert physician work group, with input from a broad spectrum of physicians and other health professionals, develops an alternative that could be implemented in 2020.”
The proposed E/M changes “are not an improvement over the current documentation requirements and payment structure. The structure is flawed, and the proposal to reduce payments when E/M services are reported with procedures fails to account for fee schedule reductions that have already been taken on these codes,” according to comments submitted by the American Academy of Dermatology Association.
The American Society of Clinical Oncology said it “supports the Agency’s proposal to reduce documentation burdens for E&M services but pairing it with reductions in payment will negatively impact patient access and should be avoided.”
ASCO also called on the agency to withdraw its proposal to consolidate E/M payments, noting that offsetting payments from add-on codes do “not appear to fully offset the direct and indirect cuts to oncology reimbursement, is ambiguous, and lacks assurances of long-term durability.”
Surgeons “cannot support the collapse of work RVU [relative value unit] values into one single rate under the [physician fee schedule] that would be paid for services using the current CPT codes for level 2 through 5 E/M visits because this single rate is a calculation of several values that were resourced-based, but in and of itself is not a resource-based value. There is no assurance that the underlying math used to derive this single value correctly reflects the resources used to deliver care across a wide spectrum of providers in America,” according to comments submitted by the American College of Surgeons.
ACS also argued that it is “not possible to fully analyze the repercussions and potential distortions to the PFS from these policies individually or taken as a whole during the 60-day comment period.” The comments noted that ACS favors documentation reduction efforts included in the proposal, but urged CMS to delay finalizing any E/M changes until more work can be done in tandem with stakeholders to craft a better solution.
The American College of Cardiology voiced support for the documentation reduction aspects of the E/M proposal but urged CMS to “not finalize any E/M payment changes for 2019. The Agency makes it clear it believes documentation proposals are intrinsically linked to the payment proposals. It is not clear to the ACC exactly why that must be the case.”
ACC also voiced concern over a provision that would halve the least-expensive procedure or the E/M visit code when a physician bills for both simultaneously. “No data are described to indicate that 50% is a correct reduction. Instead, it appears that CMS chose 50% because the reduction is equivalent to the 6.7 million RVUs needed to offset other proposed changes for compressing E/M payment into single levels and allowing use of the new add-on codes.”
A key concern for the American Academy of Family Physicians was collapsing the levels 2 through 5 E/M visits into a single payment level.
Instead, AAFP recommended that CMS work with it and other medical societies to develop new codes and values to ensure proper payment for services. Instead of a primary care add-on code, CMS should increase E/M payments by 15% for services provided “by physicians who list their primary practice designation as family medicine, internal medicine, or geriatrics,” according to the comments.
AAFP is “concerned that the changes included in the proposed rule may harm the quality and cost of care for Medicare beneficiaries,” the comment letter states, adding that it is “possible that beneficiary out-of-pocket costs would increase due to more frequent physician or clinician visits.”
The American College of Rheumatology voiced its support for the focus “on reducing physician burden by simplifying documentation requirements,” but said it had “serious concerns about the changes to evaluation and management (E/M) codes that result in cuts in reimbursement to cognitive specialists for the complex services they provide.” It added that while there is support for the documentation reduction efforts, “we are skeptical that this proposal will simplify the reporting burden on providers in the Quality Payment Program. As proposed, the new plan proposes several ‘add-on’ codes that would likely prevent reduction in audits or documentation.”
The estimated 51 hours per doctor per year of time saved “are insufficient to offset the proposed cuts to reimbursement. For example, if a physician sees around 100 patients a week, this translates to under 40 seconds per patient, which is not a benefit that outweighs the proposed reduction in reimbursement.”
The American College of Physicians voiced its opposition to the E/M proposal, noting that it “strongly believes that cognitive care of more complex patients must be appropriately recognized with higher allowed payment rates than less complex care patients.” ACP said the even with the proposed add-on codes, the proposed changes undervalue cognitive care for the most complex patients.
Doctors’ dismay at the proposed flattening of evaluation and management (E/M) payments seems to be falling on deaf ears.
More than 170 medical societies and organizations expressed their concern about the new payment structure for E/M codes proposed as part of the 2019 Medicare Physician Fee Schedule (PFS) in comments on the draft rule.
Yet, in the final days of the comment period, Seema Verma, administrator of the Centers for Medicare & Medicaid Services, took to Twitter to defend her agency’s plan.
The controversial proposal would set the payment rate for a level 1 E/M office visit for a new patient at $44, down from the current $45. Payment for levels 2-5 would be $135. Currently, payments for level 2 new patient visits are set at $76, level 3 at $110, level 4 at $167, and level 5 at $211.
For E/M office visits with established patients, the proposed rate would be $24 for level 1, up from the current $22. Payment for levels 2-5 would be $93. Under the current methodology, payments for established patient level 2 visits are set at $45, level 3 at $74, level 4 at $109, and level 5 at $148.
Offsetting the changes in payment are several new proposed add-on codes, according to CMS.
Despite the lower payment for more complex patient care, Ms. Verma touted the scheme’s budget neutrality.
Ms. Verma’s tweets come as medical societies filed their formal complaints on the proposal, mirroring concerns expressed in two letters sent to the agency ahead of the comment deadline. The letters, sent at the end of August and between the two of them signed by more than 170 medical associations, aimed to preempt the comment process. They called for the E/M proposal to be rescinded, claiming that the cuts would reduce access to Medicare services by patients and hurt physicians that treat the sickest patients and those who provide comprehensive primary care because the expected lower reimbursement. One suggested that the changes exacerbate workforce shortages.
In its formal comments, the American Medical Association said that given “the groundswell of opposition from individual physicians and nearly every physician and health professional organization in the country, including the AMA, we ask that CMS set aside its proposal to restructure payment and coding for E/M office and other outpatient visits while an expert physician work group, with input from a broad spectrum of physicians and other health professionals, develops an alternative that could be implemented in 2020.”
The proposed E/M changes “are not an improvement over the current documentation requirements and payment structure. The structure is flawed, and the proposal to reduce payments when E/M services are reported with procedures fails to account for fee schedule reductions that have already been taken on these codes,” according to comments submitted by the American Academy of Dermatology Association.
The American Society of Clinical Oncology said it “supports the Agency’s proposal to reduce documentation burdens for E&M services but pairing it with reductions in payment will negatively impact patient access and should be avoided.”
ASCO also called on the agency to withdraw its proposal to consolidate E/M payments, noting that offsetting payments from add-on codes do “not appear to fully offset the direct and indirect cuts to oncology reimbursement, is ambiguous, and lacks assurances of long-term durability.”
Surgeons “cannot support the collapse of work RVU [relative value unit] values into one single rate under the [physician fee schedule] that would be paid for services using the current CPT codes for level 2 through 5 E/M visits because this single rate is a calculation of several values that were resourced-based, but in and of itself is not a resource-based value. There is no assurance that the underlying math used to derive this single value correctly reflects the resources used to deliver care across a wide spectrum of providers in America,” according to comments submitted by the American College of Surgeons.
ACS also argued that it is “not possible to fully analyze the repercussions and potential distortions to the PFS from these policies individually or taken as a whole during the 60-day comment period.” The comments noted that ACS favors documentation reduction efforts included in the proposal, but urged CMS to delay finalizing any E/M changes until more work can be done in tandem with stakeholders to craft a better solution.
The American College of Cardiology voiced support for the documentation reduction aspects of the E/M proposal but urged CMS to “not finalize any E/M payment changes for 2019. The Agency makes it clear it believes documentation proposals are intrinsically linked to the payment proposals. It is not clear to the ACC exactly why that must be the case.”
ACC also voiced concern over a provision that would halve the least-expensive procedure or the E/M visit code when a physician bills for both simultaneously. “No data are described to indicate that 50% is a correct reduction. Instead, it appears that CMS chose 50% because the reduction is equivalent to the 6.7 million RVUs needed to offset other proposed changes for compressing E/M payment into single levels and allowing use of the new add-on codes.”
A key concern for the American Academy of Family Physicians was collapsing the levels 2 through 5 E/M visits into a single payment level.
Instead, AAFP recommended that CMS work with it and other medical societies to develop new codes and values to ensure proper payment for services. Instead of a primary care add-on code, CMS should increase E/M payments by 15% for services provided “by physicians who list their primary practice designation as family medicine, internal medicine, or geriatrics,” according to the comments.
AAFP is “concerned that the changes included in the proposed rule may harm the quality and cost of care for Medicare beneficiaries,” the comment letter states, adding that it is “possible that beneficiary out-of-pocket costs would increase due to more frequent physician or clinician visits.”
The American College of Rheumatology voiced its support for the focus “on reducing physician burden by simplifying documentation requirements,” but said it had “serious concerns about the changes to evaluation and management (E/M) codes that result in cuts in reimbursement to cognitive specialists for the complex services they provide.” It added that while there is support for the documentation reduction efforts, “we are skeptical that this proposal will simplify the reporting burden on providers in the Quality Payment Program. As proposed, the new plan proposes several ‘add-on’ codes that would likely prevent reduction in audits or documentation.”
The estimated 51 hours per doctor per year of time saved “are insufficient to offset the proposed cuts to reimbursement. For example, if a physician sees around 100 patients a week, this translates to under 40 seconds per patient, which is not a benefit that outweighs the proposed reduction in reimbursement.”
The American College of Physicians voiced its opposition to the E/M proposal, noting that it “strongly believes that cognitive care of more complex patients must be appropriately recognized with higher allowed payment rates than less complex care patients.” ACP said the even with the proposed add-on codes, the proposed changes undervalue cognitive care for the most complex patients.
Doctors’ dismay at the proposed flattening of evaluation and management (E/M) payments seems to be falling on deaf ears.
More than 170 medical societies and organizations expressed their concern about the new payment structure for E/M codes proposed as part of the 2019 Medicare Physician Fee Schedule (PFS) in comments on the draft rule.
Yet, in the final days of the comment period, Seema Verma, administrator of the Centers for Medicare & Medicaid Services, took to Twitter to defend her agency’s plan.
The controversial proposal would set the payment rate for a level 1 E/M office visit for a new patient at $44, down from the current $45. Payment for levels 2-5 would be $135. Currently, payments for level 2 new patient visits are set at $76, level 3 at $110, level 4 at $167, and level 5 at $211.
For E/M office visits with established patients, the proposed rate would be $24 for level 1, up from the current $22. Payment for levels 2-5 would be $93. Under the current methodology, payments for established patient level 2 visits are set at $45, level 3 at $74, level 4 at $109, and level 5 at $148.
Offsetting the changes in payment are several new proposed add-on codes, according to CMS.
Despite the lower payment for more complex patient care, Ms. Verma touted the scheme’s budget neutrality.
Ms. Verma’s tweets come as medical societies filed their formal complaints on the proposal, mirroring concerns expressed in two letters sent to the agency ahead of the comment deadline. The letters, sent at the end of August and between the two of them signed by more than 170 medical associations, aimed to preempt the comment process. They called for the E/M proposal to be rescinded, claiming that the cuts would reduce access to Medicare services by patients and hurt physicians that treat the sickest patients and those who provide comprehensive primary care because the expected lower reimbursement. One suggested that the changes exacerbate workforce shortages.
In its formal comments, the American Medical Association said that given “the groundswell of opposition from individual physicians and nearly every physician and health professional organization in the country, including the AMA, we ask that CMS set aside its proposal to restructure payment and coding for E/M office and other outpatient visits while an expert physician work group, with input from a broad spectrum of physicians and other health professionals, develops an alternative that could be implemented in 2020.”
The proposed E/M changes “are not an improvement over the current documentation requirements and payment structure. The structure is flawed, and the proposal to reduce payments when E/M services are reported with procedures fails to account for fee schedule reductions that have already been taken on these codes,” according to comments submitted by the American Academy of Dermatology Association.
The American Society of Clinical Oncology said it “supports the Agency’s proposal to reduce documentation burdens for E&M services but pairing it with reductions in payment will negatively impact patient access and should be avoided.”
ASCO also called on the agency to withdraw its proposal to consolidate E/M payments, noting that offsetting payments from add-on codes do “not appear to fully offset the direct and indirect cuts to oncology reimbursement, is ambiguous, and lacks assurances of long-term durability.”
Surgeons “cannot support the collapse of work RVU [relative value unit] values into one single rate under the [physician fee schedule] that would be paid for services using the current CPT codes for level 2 through 5 E/M visits because this single rate is a calculation of several values that were resourced-based, but in and of itself is not a resource-based value. There is no assurance that the underlying math used to derive this single value correctly reflects the resources used to deliver care across a wide spectrum of providers in America,” according to comments submitted by the American College of Surgeons.
ACS also argued that it is “not possible to fully analyze the repercussions and potential distortions to the PFS from these policies individually or taken as a whole during the 60-day comment period.” The comments noted that ACS favors documentation reduction efforts included in the proposal, but urged CMS to delay finalizing any E/M changes until more work can be done in tandem with stakeholders to craft a better solution.
The American College of Cardiology voiced support for the documentation reduction aspects of the E/M proposal but urged CMS to “not finalize any E/M payment changes for 2019. The Agency makes it clear it believes documentation proposals are intrinsically linked to the payment proposals. It is not clear to the ACC exactly why that must be the case.”
ACC also voiced concern over a provision that would halve the least-expensive procedure or the E/M visit code when a physician bills for both simultaneously. “No data are described to indicate that 50% is a correct reduction. Instead, it appears that CMS chose 50% because the reduction is equivalent to the 6.7 million RVUs needed to offset other proposed changes for compressing E/M payment into single levels and allowing use of the new add-on codes.”
A key concern for the American Academy of Family Physicians was collapsing the levels 2 through 5 E/M visits into a single payment level.
Instead, AAFP recommended that CMS work with it and other medical societies to develop new codes and values to ensure proper payment for services. Instead of a primary care add-on code, CMS should increase E/M payments by 15% for services provided “by physicians who list their primary practice designation as family medicine, internal medicine, or geriatrics,” according to the comments.
AAFP is “concerned that the changes included in the proposed rule may harm the quality and cost of care for Medicare beneficiaries,” the comment letter states, adding that it is “possible that beneficiary out-of-pocket costs would increase due to more frequent physician or clinician visits.”
The American College of Rheumatology voiced its support for the focus “on reducing physician burden by simplifying documentation requirements,” but said it had “serious concerns about the changes to evaluation and management (E/M) codes that result in cuts in reimbursement to cognitive specialists for the complex services they provide.” It added that while there is support for the documentation reduction efforts, “we are skeptical that this proposal will simplify the reporting burden on providers in the Quality Payment Program. As proposed, the new plan proposes several ‘add-on’ codes that would likely prevent reduction in audits or documentation.”
The estimated 51 hours per doctor per year of time saved “are insufficient to offset the proposed cuts to reimbursement. For example, if a physician sees around 100 patients a week, this translates to under 40 seconds per patient, which is not a benefit that outweighs the proposed reduction in reimbursement.”
The American College of Physicians voiced its opposition to the E/M proposal, noting that it “strongly believes that cognitive care of more complex patients must be appropriately recognized with higher allowed payment rates than less complex care patients.” ACP said the even with the proposed add-on codes, the proposed changes undervalue cognitive care for the most complex patients.
The Flint Lock: A Novel Technique in Total Knee Arthroplasty Closure
ABSTRACT
Conventional interrupted sutures are traditionally used in extensor mechanism closure during total knee arthroplasty (TKA). In recent years, barbed suture has been introduced with the proposed benefits of decreased closure time and a watertight seal that is superior to interrupted sutures. Complication rates using barbed sutures and conventional interrupted sutures are similar. We propose a novel closure technique known as the Flint Lock, which is a double continuous interlocking stitch. The Flint Lock provides a quick and efficient closure to the extensor mechanism in TKA. In addition, similar to barbed suture, the Flint Lock should provide a superior watertight seal. It utilizes relatively inexpensive and readily available materials.
Continue to: In 2003, more than 400,000 total knee replacements...
In 2003, more than 400,000 total knee replacements were performed in the United States. This number is expected to increase in the coming decades to 3 million by the year 2030.1 The surgical approach to knee arthroplasty always involves a capsular incision that needs to be repaired after implantation of the components. The capsular incision repair should be strong enough to allow for immediate range of motion.
Traditionally, repair of the arthrotomy is performed using interrupted sutures. Recently, a running technique using barbed suture has been demonstrated to enable faster closure times.2-6 In addition, a running suture technique using barbed suture provides a superior watertight closure compared with an interrupted suture.7 It has been reported that the barbed suture has the same safety profile as that of interrupted sutures,2,3,4 although extensor mechanism repair failure8 and wound complications9,10 have been reported.
This study proposes a novel technique for arthrotomy closure in total knee arthroplasty (TKA). It is a double continuous interlocking stitch, termed the “Flint Lock.” Based on our clinical experience using this method, this technique has been found to be safe and effective.
TECHNIQUE
The Flint Lock was developed for closure in TKA, which was performed through a standard medial parapatellar approach. Before creating the arthrotomy, a horizontal line is drawn along the medial side of the patella to ensure anatomic alignment of the extensor mechanism during closure of the capsule.
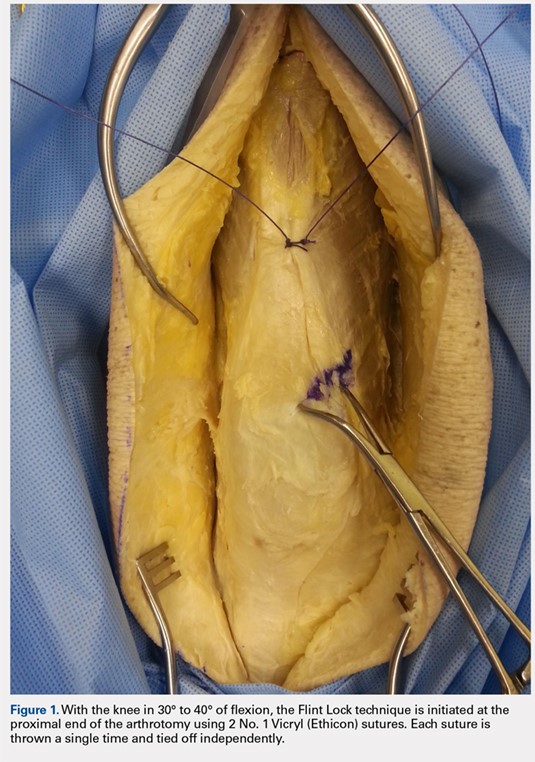
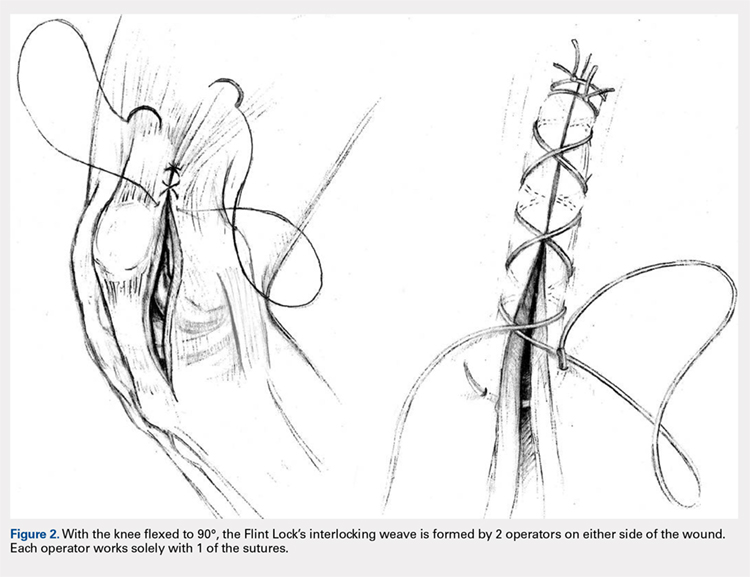
The Flint Lock is performed by 2 people working simultaneously. Closure begins at the proximal end of the arthrotomy using 2 No. 1 Vicryl (Ethicon) sutures. Each suture is thrown a single time at the most proximal extent of the arthrotomy with the knee in 30° to 40° of flexion. These sutures are tied off independently from each other (Figure 1). At this point, the knee is flexed to 90° and the sutures are thrown alternately, with the first operator passing medial to lateral through the capsule and the second operator passing lateral to medial. While 1 operator is passing a suture, the other operator holds the other suture tight to maintain tension on the closure. The alternating throws create an interlocking weave as the pattern is repeated and progressively moves distally (Figure 2). This technique results in 2 continuous sutures running in opposing directions. Each No. 1 Vicryl suture is specific to each operator. Therefore, each operator uses the same suture for the entirety of the closure.
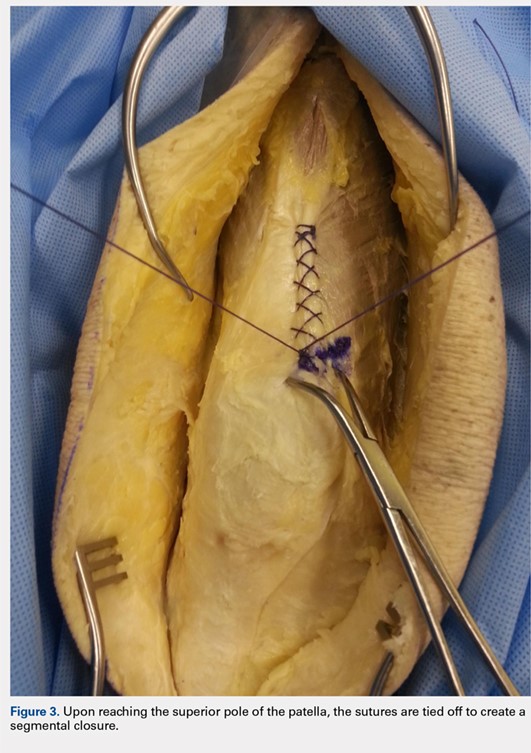
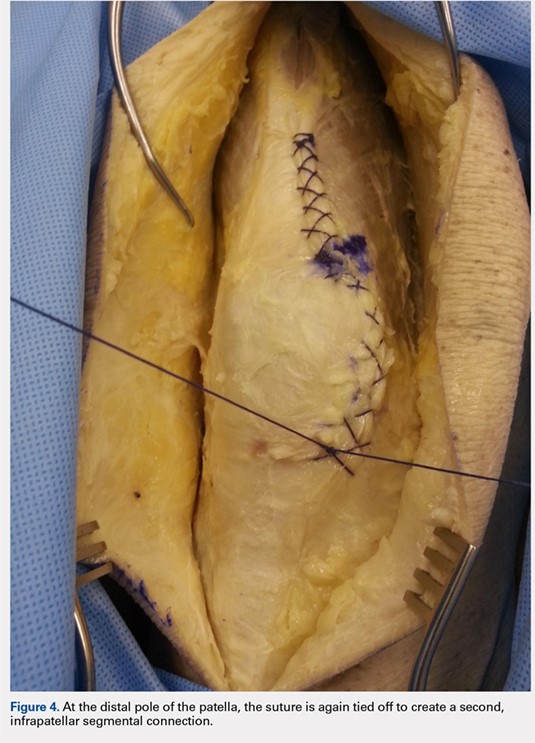
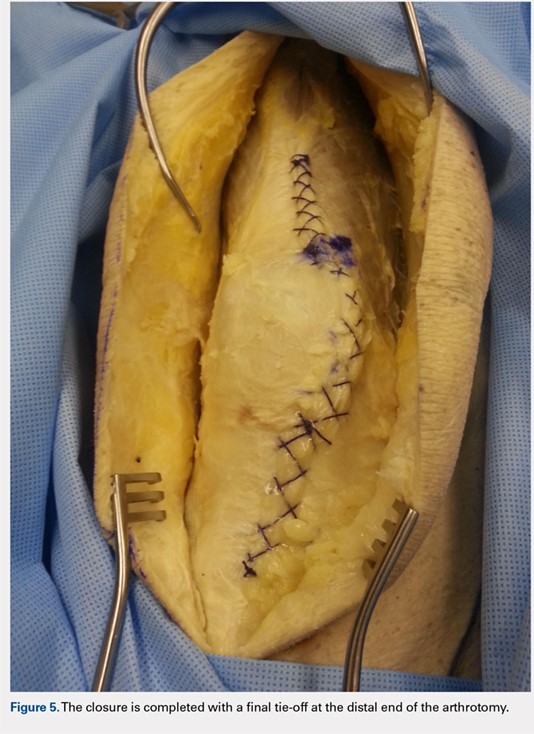
When the superior pole of the patella is reached, the 2 sutures are tied together, thus creating a segmental closure (Figure 3). Following this tie off, the closure is continued in a similar manner until the inferior pole of the patella is reached. The sutures are then tied off to each other again, creating another segmental closure (Figure 4). The remainder of the arthrotomy is closed continuing the Flint Lock technique, and the 2 sutures are tied off to each other at the distal end of the arthrotomy and cut (Figure 5).
Continue to: The superficial layers are closed at the surgeon’s discretion...
The superficial layers are closed at the surgeon’s discretion. The authors prefer interrupted 2-0 Vicryl sutures followed by a running 3-0 Monocryl (Ethicon) suture in the subcutaneous layer. Dermabond (Ethicon) skin glue and an Aquacel Ag (ConvaTec) dressing are applied, followed by a compressive bandage.
DISCUSSION
The importance of a strong, tight closure of the arthrotomy in TKA is critical to the success of the procedure. Nevertheless, there are multiple methods to achieve closure. The Flint Lock technique is a novel method that employs basic concepts of surgical technique in an original manner. The continuous nature of the closure should provide a tighter seal, leading to less wound drainage. Persistent wound drainage has been associated with deep wound infections following total joint arthroplasty.11,12 In addition, the double suture provides a safeguard to a single suture rupture, while the segmental quality protects against complete arthrotomy failure.
A potential downside of this technique is that it requires 2 individuals operating 2 needles simultaneously. This presents a potential for a sharp injury to the operators; however, this has not occurred in our experience. A comparable risk with interrupted sutures is probably present because there are often multiple sutures utilized during closure via the interrupted technique.
In 2015, the cost of a single No. 1 barbed suture was $13.14 at our institution, whereas the cost of 2 No. 1 Vicryl sutures was $3.66. Although pricing differs across hospitals, the Vicryl sutures are probably less costly compared with the barbed sutures.
Our experience with the Flint Lock technique has been favorable thus far, with no incidences of postoperative drainage, infection, or extensor mechanism failure. Our current use has been in closure of the knee, but it could be considered in closure of long incisions about the hip as well. A more in-depth analysis of relevant factors, such as time for closure, mechanical strength, cost savings, and clinical outcomes, is needed to further evaluate this method of closure. In addition, biomechanical analysis of the technique would aid in its evaluation. Future studies are needed to analyze these factors to verify the benefits and viability of the Flint Lock technique.
1. Kurtz S, Ong K, Lau E, Mowat F, Halpern M. Projections of primary and revision hip and knee arthroplasty in the United States from 2005 to 2030. J Bone Joint Surg Am. 2007;89(4):780-785. doi:10.2106/JBJS.F.00222.
2. Eickmann T, Quane E. Total knee arthroplasty closure with barbed sutures. J Knee Surg. 2010;23(3):163-167. doi:10.1055/s-0030-1268692.
3. Gililland JM, Anderson LA, Sun G, Erickson JA, Peters CL. Perioperative closure-related complication rates and cost analysis of barbed suture for closure in TKA. Clin Orthop Relat Res. 2012;470(1):125-129. doi:10.1007/s11999-011-2104-7.
4. Ting NT, Moric MM, Della Valle CJ, Levine BR. Use of knotless suture for closure of total hip and knee arthroplasties: a prospective, randomized clinical trial. J Arthroplasty. 2012;27(10):1783-1788. doi:10.1016/j.arth.2012.05.022.
5. Stephens S, Politi J, Taylor BC. Evaluation of primary total knee arthroplasty incision closure with use of continuous bidirectional barbed suture. Surg Technol Int. 2011;21:199-203.
6. Levine BR, Ting N, Della Valle CJ. Use of a barbed suture in the closure of hip and knee arthroplasty wounds. Orthopedics. 2011;34(9):e473-e475. doi:10.3928/01477447-20110714-35.
7. Nett M, Avelar R, Sheehan M, Cushner F. Water-tight knee arthrotomy closure: comparison of a novel single bidirectional barbed self-retaining running suture versus conventional interrupted sutures. J Knee Surg. 2011;24(1):55-59. doi:10.1055/s-0031-1275400.
8. Wright RC, Gillis CT, Yacoubian SV, Raven RB 3rd, Falkinstein Y, Yacoubian SV. Extensor mechanism repair failure with use of birectional barbed suture in total knee arthroplasty. J Arthroplasty. 2012;27(7):1413.e1-e4. doi:10.1016/j.arth.2011.08.013.
9. Campbell AL, Patrick DA Jr, Liabaud B, Geller JA. Superficial wound closure complications with barbed sutures following knee arthroplasty. J Arthroplasty. 2014;29(5):966-969. doi:10.1016/j.arth.2013.09.045.
10. Smith EL, DiSegna ST, Shukla PY, Matzkin EG. Barbed versus traditional sutures: closure time, cost, and wound related outcomes in total joint arthroplasty. J Arthroplasty. 2014;29(2):283-287. doi:10.1016/j.arth.2013.05.031.
11. Saleh K, Olson M, Resig S, et al. Predictors of wound infection in hip and knee joint replacement: results from a 20 year surveillance program. J Orthop Res. 2002;20(3):506-515. doi:10.1016/S0736-0266(01)00153-X.
12. Weiss AP, Krackow KA. Persistent wound drainage after primary total knee arthroplasty. J Arthroplasty. 1993;8(3):285-289. doi:10.1016/S0883-5403(06)80091-4.
ABSTRACT
Conventional interrupted sutures are traditionally used in extensor mechanism closure during total knee arthroplasty (TKA). In recent years, barbed suture has been introduced with the proposed benefits of decreased closure time and a watertight seal that is superior to interrupted sutures. Complication rates using barbed sutures and conventional interrupted sutures are similar. We propose a novel closure technique known as the Flint Lock, which is a double continuous interlocking stitch. The Flint Lock provides a quick and efficient closure to the extensor mechanism in TKA. In addition, similar to barbed suture, the Flint Lock should provide a superior watertight seal. It utilizes relatively inexpensive and readily available materials.
Continue to: In 2003, more than 400,000 total knee replacements...
In 2003, more than 400,000 total knee replacements were performed in the United States. This number is expected to increase in the coming decades to 3 million by the year 2030.1 The surgical approach to knee arthroplasty always involves a capsular incision that needs to be repaired after implantation of the components. The capsular incision repair should be strong enough to allow for immediate range of motion.
Traditionally, repair of the arthrotomy is performed using interrupted sutures. Recently, a running technique using barbed suture has been demonstrated to enable faster closure times.2-6 In addition, a running suture technique using barbed suture provides a superior watertight closure compared with an interrupted suture.7 It has been reported that the barbed suture has the same safety profile as that of interrupted sutures,2,3,4 although extensor mechanism repair failure8 and wound complications9,10 have been reported.
This study proposes a novel technique for arthrotomy closure in total knee arthroplasty (TKA). It is a double continuous interlocking stitch, termed the “Flint Lock.” Based on our clinical experience using this method, this technique has been found to be safe and effective.
TECHNIQUE
The Flint Lock was developed for closure in TKA, which was performed through a standard medial parapatellar approach. Before creating the arthrotomy, a horizontal line is drawn along the medial side of the patella to ensure anatomic alignment of the extensor mechanism during closure of the capsule.
The Flint Lock is performed by 2 people working simultaneously. Closure begins at the proximal end of the arthrotomy using 2 No. 1 Vicryl (Ethicon) sutures. Each suture is thrown a single time at the most proximal extent of the arthrotomy with the knee in 30° to 40° of flexion. These sutures are tied off independently from each other (Figure 1). At this point, the knee is flexed to 90° and the sutures are thrown alternately, with the first operator passing medial to lateral through the capsule and the second operator passing lateral to medial. While 1 operator is passing a suture, the other operator holds the other suture tight to maintain tension on the closure. The alternating throws create an interlocking weave as the pattern is repeated and progressively moves distally (Figure 2). This technique results in 2 continuous sutures running in opposing directions. Each No. 1 Vicryl suture is specific to each operator. Therefore, each operator uses the same suture for the entirety of the closure.
When the superior pole of the patella is reached, the 2 sutures are tied together, thus creating a segmental closure (Figure 3). Following this tie off, the closure is continued in a similar manner until the inferior pole of the patella is reached. The sutures are then tied off to each other again, creating another segmental closure (Figure 4). The remainder of the arthrotomy is closed continuing the Flint Lock technique, and the 2 sutures are tied off to each other at the distal end of the arthrotomy and cut (Figure 5).
Continue to: The superficial layers are closed at the surgeon’s discretion...
The superficial layers are closed at the surgeon’s discretion. The authors prefer interrupted 2-0 Vicryl sutures followed by a running 3-0 Monocryl (Ethicon) suture in the subcutaneous layer. Dermabond (Ethicon) skin glue and an Aquacel Ag (ConvaTec) dressing are applied, followed by a compressive bandage.
DISCUSSION
The importance of a strong, tight closure of the arthrotomy in TKA is critical to the success of the procedure. Nevertheless, there are multiple methods to achieve closure. The Flint Lock technique is a novel method that employs basic concepts of surgical technique in an original manner. The continuous nature of the closure should provide a tighter seal, leading to less wound drainage. Persistent wound drainage has been associated with deep wound infections following total joint arthroplasty.11,12 In addition, the double suture provides a safeguard to a single suture rupture, while the segmental quality protects against complete arthrotomy failure.
A potential downside of this technique is that it requires 2 individuals operating 2 needles simultaneously. This presents a potential for a sharp injury to the operators; however, this has not occurred in our experience. A comparable risk with interrupted sutures is probably present because there are often multiple sutures utilized during closure via the interrupted technique.
In 2015, the cost of a single No. 1 barbed suture was $13.14 at our institution, whereas the cost of 2 No. 1 Vicryl sutures was $3.66. Although pricing differs across hospitals, the Vicryl sutures are probably less costly compared with the barbed sutures.
Our experience with the Flint Lock technique has been favorable thus far, with no incidences of postoperative drainage, infection, or extensor mechanism failure. Our current use has been in closure of the knee, but it could be considered in closure of long incisions about the hip as well. A more in-depth analysis of relevant factors, such as time for closure, mechanical strength, cost savings, and clinical outcomes, is needed to further evaluate this method of closure. In addition, biomechanical analysis of the technique would aid in its evaluation. Future studies are needed to analyze these factors to verify the benefits and viability of the Flint Lock technique.
ABSTRACT
Conventional interrupted sutures are traditionally used in extensor mechanism closure during total knee arthroplasty (TKA). In recent years, barbed suture has been introduced with the proposed benefits of decreased closure time and a watertight seal that is superior to interrupted sutures. Complication rates using barbed sutures and conventional interrupted sutures are similar. We propose a novel closure technique known as the Flint Lock, which is a double continuous interlocking stitch. The Flint Lock provides a quick and efficient closure to the extensor mechanism in TKA. In addition, similar to barbed suture, the Flint Lock should provide a superior watertight seal. It utilizes relatively inexpensive and readily available materials.
Continue to: In 2003, more than 400,000 total knee replacements...
In 2003, more than 400,000 total knee replacements were performed in the United States. This number is expected to increase in the coming decades to 3 million by the year 2030.1 The surgical approach to knee arthroplasty always involves a capsular incision that needs to be repaired after implantation of the components. The capsular incision repair should be strong enough to allow for immediate range of motion.
Traditionally, repair of the arthrotomy is performed using interrupted sutures. Recently, a running technique using barbed suture has been demonstrated to enable faster closure times.2-6 In addition, a running suture technique using barbed suture provides a superior watertight closure compared with an interrupted suture.7 It has been reported that the barbed suture has the same safety profile as that of interrupted sutures,2,3,4 although extensor mechanism repair failure8 and wound complications9,10 have been reported.
This study proposes a novel technique for arthrotomy closure in total knee arthroplasty (TKA). It is a double continuous interlocking stitch, termed the “Flint Lock.” Based on our clinical experience using this method, this technique has been found to be safe and effective.
TECHNIQUE
The Flint Lock was developed for closure in TKA, which was performed through a standard medial parapatellar approach. Before creating the arthrotomy, a horizontal line is drawn along the medial side of the patella to ensure anatomic alignment of the extensor mechanism during closure of the capsule.
The Flint Lock is performed by 2 people working simultaneously. Closure begins at the proximal end of the arthrotomy using 2 No. 1 Vicryl (Ethicon) sutures. Each suture is thrown a single time at the most proximal extent of the arthrotomy with the knee in 30° to 40° of flexion. These sutures are tied off independently from each other (Figure 1). At this point, the knee is flexed to 90° and the sutures are thrown alternately, with the first operator passing medial to lateral through the capsule and the second operator passing lateral to medial. While 1 operator is passing a suture, the other operator holds the other suture tight to maintain tension on the closure. The alternating throws create an interlocking weave as the pattern is repeated and progressively moves distally (Figure 2). This technique results in 2 continuous sutures running in opposing directions. Each No. 1 Vicryl suture is specific to each operator. Therefore, each operator uses the same suture for the entirety of the closure.
When the superior pole of the patella is reached, the 2 sutures are tied together, thus creating a segmental closure (Figure 3). Following this tie off, the closure is continued in a similar manner until the inferior pole of the patella is reached. The sutures are then tied off to each other again, creating another segmental closure (Figure 4). The remainder of the arthrotomy is closed continuing the Flint Lock technique, and the 2 sutures are tied off to each other at the distal end of the arthrotomy and cut (Figure 5).
Continue to: The superficial layers are closed at the surgeon’s discretion...
The superficial layers are closed at the surgeon’s discretion. The authors prefer interrupted 2-0 Vicryl sutures followed by a running 3-0 Monocryl (Ethicon) suture in the subcutaneous layer. Dermabond (Ethicon) skin glue and an Aquacel Ag (ConvaTec) dressing are applied, followed by a compressive bandage.
DISCUSSION
The importance of a strong, tight closure of the arthrotomy in TKA is critical to the success of the procedure. Nevertheless, there are multiple methods to achieve closure. The Flint Lock technique is a novel method that employs basic concepts of surgical technique in an original manner. The continuous nature of the closure should provide a tighter seal, leading to less wound drainage. Persistent wound drainage has been associated with deep wound infections following total joint arthroplasty.11,12 In addition, the double suture provides a safeguard to a single suture rupture, while the segmental quality protects against complete arthrotomy failure.
A potential downside of this technique is that it requires 2 individuals operating 2 needles simultaneously. This presents a potential for a sharp injury to the operators; however, this has not occurred in our experience. A comparable risk with interrupted sutures is probably present because there are often multiple sutures utilized during closure via the interrupted technique.
In 2015, the cost of a single No. 1 barbed suture was $13.14 at our institution, whereas the cost of 2 No. 1 Vicryl sutures was $3.66. Although pricing differs across hospitals, the Vicryl sutures are probably less costly compared with the barbed sutures.
Our experience with the Flint Lock technique has been favorable thus far, with no incidences of postoperative drainage, infection, or extensor mechanism failure. Our current use has been in closure of the knee, but it could be considered in closure of long incisions about the hip as well. A more in-depth analysis of relevant factors, such as time for closure, mechanical strength, cost savings, and clinical outcomes, is needed to further evaluate this method of closure. In addition, biomechanical analysis of the technique would aid in its evaluation. Future studies are needed to analyze these factors to verify the benefits and viability of the Flint Lock technique.
1. Kurtz S, Ong K, Lau E, Mowat F, Halpern M. Projections of primary and revision hip and knee arthroplasty in the United States from 2005 to 2030. J Bone Joint Surg Am. 2007;89(4):780-785. doi:10.2106/JBJS.F.00222.
2. Eickmann T, Quane E. Total knee arthroplasty closure with barbed sutures. J Knee Surg. 2010;23(3):163-167. doi:10.1055/s-0030-1268692.
3. Gililland JM, Anderson LA, Sun G, Erickson JA, Peters CL. Perioperative closure-related complication rates and cost analysis of barbed suture for closure in TKA. Clin Orthop Relat Res. 2012;470(1):125-129. doi:10.1007/s11999-011-2104-7.
4. Ting NT, Moric MM, Della Valle CJ, Levine BR. Use of knotless suture for closure of total hip and knee arthroplasties: a prospective, randomized clinical trial. J Arthroplasty. 2012;27(10):1783-1788. doi:10.1016/j.arth.2012.05.022.
5. Stephens S, Politi J, Taylor BC. Evaluation of primary total knee arthroplasty incision closure with use of continuous bidirectional barbed suture. Surg Technol Int. 2011;21:199-203.
6. Levine BR, Ting N, Della Valle CJ. Use of a barbed suture in the closure of hip and knee arthroplasty wounds. Orthopedics. 2011;34(9):e473-e475. doi:10.3928/01477447-20110714-35.
7. Nett M, Avelar R, Sheehan M, Cushner F. Water-tight knee arthrotomy closure: comparison of a novel single bidirectional barbed self-retaining running suture versus conventional interrupted sutures. J Knee Surg. 2011;24(1):55-59. doi:10.1055/s-0031-1275400.
8. Wright RC, Gillis CT, Yacoubian SV, Raven RB 3rd, Falkinstein Y, Yacoubian SV. Extensor mechanism repair failure with use of birectional barbed suture in total knee arthroplasty. J Arthroplasty. 2012;27(7):1413.e1-e4. doi:10.1016/j.arth.2011.08.013.
9. Campbell AL, Patrick DA Jr, Liabaud B, Geller JA. Superficial wound closure complications with barbed sutures following knee arthroplasty. J Arthroplasty. 2014;29(5):966-969. doi:10.1016/j.arth.2013.09.045.
10. Smith EL, DiSegna ST, Shukla PY, Matzkin EG. Barbed versus traditional sutures: closure time, cost, and wound related outcomes in total joint arthroplasty. J Arthroplasty. 2014;29(2):283-287. doi:10.1016/j.arth.2013.05.031.
11. Saleh K, Olson M, Resig S, et al. Predictors of wound infection in hip and knee joint replacement: results from a 20 year surveillance program. J Orthop Res. 2002;20(3):506-515. doi:10.1016/S0736-0266(01)00153-X.
12. Weiss AP, Krackow KA. Persistent wound drainage after primary total knee arthroplasty. J Arthroplasty. 1993;8(3):285-289. doi:10.1016/S0883-5403(06)80091-4.
1. Kurtz S, Ong K, Lau E, Mowat F, Halpern M. Projections of primary and revision hip and knee arthroplasty in the United States from 2005 to 2030. J Bone Joint Surg Am. 2007;89(4):780-785. doi:10.2106/JBJS.F.00222.
2. Eickmann T, Quane E. Total knee arthroplasty closure with barbed sutures. J Knee Surg. 2010;23(3):163-167. doi:10.1055/s-0030-1268692.
3. Gililland JM, Anderson LA, Sun G, Erickson JA, Peters CL. Perioperative closure-related complication rates and cost analysis of barbed suture for closure in TKA. Clin Orthop Relat Res. 2012;470(1):125-129. doi:10.1007/s11999-011-2104-7.
4. Ting NT, Moric MM, Della Valle CJ, Levine BR. Use of knotless suture for closure of total hip and knee arthroplasties: a prospective, randomized clinical trial. J Arthroplasty. 2012;27(10):1783-1788. doi:10.1016/j.arth.2012.05.022.
5. Stephens S, Politi J, Taylor BC. Evaluation of primary total knee arthroplasty incision closure with use of continuous bidirectional barbed suture. Surg Technol Int. 2011;21:199-203.
6. Levine BR, Ting N, Della Valle CJ. Use of a barbed suture in the closure of hip and knee arthroplasty wounds. Orthopedics. 2011;34(9):e473-e475. doi:10.3928/01477447-20110714-35.
7. Nett M, Avelar R, Sheehan M, Cushner F. Water-tight knee arthrotomy closure: comparison of a novel single bidirectional barbed self-retaining running suture versus conventional interrupted sutures. J Knee Surg. 2011;24(1):55-59. doi:10.1055/s-0031-1275400.
8. Wright RC, Gillis CT, Yacoubian SV, Raven RB 3rd, Falkinstein Y, Yacoubian SV. Extensor mechanism repair failure with use of birectional barbed suture in total knee arthroplasty. J Arthroplasty. 2012;27(7):1413.e1-e4. doi:10.1016/j.arth.2011.08.013.
9. Campbell AL, Patrick DA Jr, Liabaud B, Geller JA. Superficial wound closure complications with barbed sutures following knee arthroplasty. J Arthroplasty. 2014;29(5):966-969. doi:10.1016/j.arth.2013.09.045.
10. Smith EL, DiSegna ST, Shukla PY, Matzkin EG. Barbed versus traditional sutures: closure time, cost, and wound related outcomes in total joint arthroplasty. J Arthroplasty. 2014;29(2):283-287. doi:10.1016/j.arth.2013.05.031.
11. Saleh K, Olson M, Resig S, et al. Predictors of wound infection in hip and knee joint replacement: results from a 20 year surveillance program. J Orthop Res. 2002;20(3):506-515. doi:10.1016/S0736-0266(01)00153-X.
12. Weiss AP, Krackow KA. Persistent wound drainage after primary total knee arthroplasty. J Arthroplasty. 1993;8(3):285-289. doi:10.1016/S0883-5403(06)80091-4.
TAKE-HOME POINTS
- The Flint Lock is a novel technique in TKA closure.
- Its continuous nature provides a tight seal with extensor mechanism closure.
- The utilization of a segmental closure with double suture provides a safeguard for suture failure.
- The suture used in the technique is less expensive than barbed suture.
- Future investigation is warranted to further validate the use of the Flint Lock.
The Effect of Insurance Type on Patient Access to Ankle Fracture Care Under the Affordable Care Act
ABSTRACT
The purpose of this study is to assess the effect of insurance type (Medicaid, Medicare, private insurance) on the ability for patients with operative ankle fractures to access orthopedic traumatologists. The research team called 245 board-certified orthopedic surgeons specializing in orthopedic trauma within 8 representative states. The caller requested an appointment for their fictitious mother in order to be evaluated for an ankle fracture which was previously evaluated by her primary care physician and believed to require surgery. Each office was called 3 times to assess the response for each insurance type. For each call, information was documented regarding whether the patient was able to receive an appointment and the barriers the patient confronted to receive an appointment. Overall, 35.7% of offices scheduled an appointment for a patient with Medicaid, in comparison to 81.4%and 88.6% for Medicare and BlueCross, respectively (P < .0001). Medicaid patients confronted more barriers for receiving appointments. There was no statistically significant difference in access for Medicaid patients in states that had expanded Medicaid eligibility vs states that had not expanded Medicaid. Medicaid reimbursement for open reduction and internal fixation of an ankle fracture did not significantly correlate with appointment success rates or wait times. Despite the passage of the Affordable Care Act, patients with Medicaid have reduced access to orthopedic surgeons and more complex barriers to receiving appointments. A more robust strategy for increasing care-access for patients with Medicaid would be more equitable.
Continue to: In 2010, the Patient Protection and Affordable Care Act...
In 2010, the Patient Protection and Affordable Care Act (PPACA) expanded the eligibility criteria for Medicaid to all individuals with an income up to 138% of the poverty level.1 A Supreme Court ruling stated that the decision to expand Medicaid was to be decided by individual states.2 Currently, 31 states have chosen to expand Medicaid eligibility to their residents.2 This expansion has allowed an additional 11.7 million people to enroll in Medicaid and the Children’s Health Insurance Program by May 2015.3-5
Even with the passage of the PPACA, Medicaid patients seeking specialty orthopedic care have experienced more barriers to accessing care than Medicare or commercially-insured patients.2,6-10 One major cited reason is Medicaid’s low reimbursement, which may discourage physicians from open panel participation in Medicaid.11,12
A common fundamental teaching for orthopedic traumatologists is the notion that they should be available to treat all injuries regardless of the patient’s ability to pay.13 This has resulted in both trauma centers and trauma surgeons becoming financially challenged due to the higher proportion of Medicaid and uninsured trauma patients and lower Medicaid reimbursement levels.14,15
This study focuses on the effect of different types of insurance (Medicaid, Medicare, or commercial insurance) on the ability of patients to obtain care for operative ankle fractures. The purpose of this study is to evaluate, in the context of the PPACA, patient access to orthopedic surgeons for operative ankle fractures based on insurance-type. We hypothesized that patients with Medicaid would face a greater volume of obstacles when seeking appointments for an ankle fracture, even after the PPACA.
Continue to: MATERIALS AND METHODS...
MATERIALS AND METHODS
The study population included board-certified orthopedic surgeons who belonged to the Orthopaedic Trauma Association (OTA) from 8 representative states; 4 states with expanded Medicaid eligibility (California, Massachusetts, New York, Ohio) and 4 states without expanded Medicaid eligibility (Florida, North Carolina, Georgia, Texas). These states were selected due to their ability to represent diverse healthcare marketplaces throughout the country. Using the OTA website’s “Find a Surgeon” search tool,16 we created a list of surgeons for each state and matched each surgeon with a random number. The list of surgeons was ordered according to the value of the surgeon’s associated random number, and surgeons were called in ascending order. We excluded disconnected or inaccurate numbers from the calling list. Surgeons who did not manage ankle fractures were removed from the dataset. Approximately 30 orthopedic trauma surgeons per state were contacted.
Each office was called to make an appointment for the caller’s mother. Every surgeon’s office was specifically asked if the surgeon would accept the patient to be evaluated for an ankle fracture that occurred out-of-state. The caller had a standardized protocol to limit intra- and inter-office variations (Appendix). The scenario involved a request to be evaluated for an unstable ankle fracture, with the patient having Medicaid, Medicare, or BlueCross insurance. The scenario required 3 separate calls to the same surgeon in order to obtain data regarding each insurance-type. The calls were separated by at least 1 week to avoid caller recognition by the surgeon’s office.
Appendix
Scenario
1. Date of Birth: Medicaid–2/07/55; BlueCross PPO–2/09/55; Medicare–7/31/45.
2. Ankle fracture evaluated by primary care physician 1 or 2 days ago
3. Not seen previously by your clinic or hospital, she would be a new patient
4. Asked how early she could be scheduled for an appointment
5. Script:
“I’m calling for my mother who injured her ankle a few days ago. Her family doctor took an X-ray and believes she has a fracture and needs surgery. Is Dr. X accepting new patients for evaluation and treatment of ankle fractures?” If YES →
“I was wondering if you take Medicaid/Medicare/BlueCross plan?” If YES →
“When is your soonest available appointment?”
The date of each phone call and date of appointment, if provided, were recorded. If the office did not give an appointment, we asked for reasons why. If an appointment was denied for a patient with Medicaid, we asked for a referral to another office that accepted Medicaid. We considered barriers to obtaining an initial appointment, such as requiring a referral from a primary care physician (PCP), as an unsuccessful attempt at making an appointment. We determined the waiting period for an appointment by calculating the time between the date of the call and the date of the appointment. Appointments were not scheduled to ensure that actual patients were not disadvantaged. For both appointment success rates and waiting periods, we stratified the data into 2 groups: states with expanded Medicaid eligibility (California, Massachusetts, New York, Ohio) and states without expanded Medicaid eligibility (Florida, North Carolina, Georgia, Texas).
We obtained Medicaid reimbursement rates for open reduction and internal fixation of an ankle fracture by querying each state’s reimbursement rate using Current Procedural Terminology code 27822.
Chi-square test or Fisher’s exact test was used to analyze acceptance rate differences based on the patient’s type of insurance. To compare the waiting periods for an appointment, we used an independent samples t-test after applying natural log-transformation, as the data was not normally distributed. We performed logistic regression analysis to detect whether reimbursement was a significant predictor of successfully making an appointment for patients, and a linear regression analysis was used to evaluate whether reimbursement predicted waiting periods. Unless otherwise stated, all statistical testing was performed two-tailed at an alpha-level of 0.05.
This study was approved by the Institutional Review Board of Yale University School of Medicine (HIC No. 1363).
Continue to: RESULTS...
RESULTS
In total, 350 offices were contacted across 8 states (4 states with and 4 states without expanded Medicaid eligibility) of which we identified 245 orthopedic surgeons who would surgically treat ankle fractures. The 245 surgeons’ offices were called 3 times for each separate insurance-type.
Table 1. Appointment Success Rate
| Medicaid | Medicare | Private |
All states |
|
| |
Yes (%) | 100 (35.7) | 228 (81.4) | 248 (88.6) |
No (%) | 180 (64.3) | 52 (18.60 | 32 (11.4) |
P-valuea |
| 0.0001 | 0.0001 |
States with expanded Medicaid eligibility |
|
|
|
Yes (%) | 55 (39.6) | 116 (83.5) | 124 (89.2) |
No (%) | 84 (60.4) | 23 (16.5) | 15 (10.8) |
P-valuea |
| 0.0001 | 0.0001 |
States without expanded Medicaid eligibility |
|
|
|
Yes (%) | 45 (31.9) | 112 (79.4) | 124 (87.9) |
No (%) | 96 (68.1) | 29 (20.6) | 17 (12.1) |
P-valuea |
| 0.0001 | 0.0001 |
aComparison to Medicaid.
The overall rate of successfully being offered an appointment with Medicaid was 35.7%, 81.4% for Medicare, and 88.6% for BlueCross (Table 1). For states with expanded Medicaid eligibility, the success rate for obtaining an appointment was 39.6%, 83.5%, and 89.2% for Medicaid, Medicare, and BlueCross, respectively. For states without expanded Medicaid eligibility, the success rate for obtaining an appointment was 31.9% for Medicaid, 79.4% for Medicare, and 87.9% for BlueCross. In all cases, the success rate for obtaining an appointment was significantly lower for Medicaid, compared to Medicare (P < .0001) or BlueCross (P < .0001). Medicaid appointment success rate was 39.6% in expanded states vs 31.9% in non-expanded states, however, the difference was not statistically significant (Table 2).
Table 2. Medicaid Appointment Success Rate in Expanded Vs Non-Expanded States
| Expanded states | Non-expanded states | P-value |
Yes (%) | 55 (39.6) | 45 (31.9) | .181 |
No (%) | 84 (60.4) | 96 (68.1) |
|
In 43.7% of occasions, patients with Medicaid did not have their insurance accepted, compared to 7.3% for Medicare and 0% for BlueCross. The majority of offices which did not accept Medicaid were not able to refer patients to another surgeon who would accept Medicaid. The requirement to have a primary care referral was the second most common reason for Medicaid patients not obtaining an appointment. No Medicare (10.4% vs 0.0%, P < .0001) or BlueCross (10.4% vs 0.0%, P < .0001) patients experienced this requirement (Table 3). There was no difference found between the percent of Medicaid patients who were required to have referrals in states with and without expanded Medicaid eligibility (Table 4).
Table 3. Referral Rate
| Medicaid | Medicare | Private |
All states |
|
|
|
Yes (%) | 29 (10.4) | 0 (0) | 0 (0) |
No (%) | 251 (89.6) | 280 (100) | 280 (100) |
P-valuea |
| 0.0001 | 0.0001 |
States with expanded Medicaid eligibility |
|
|
|
Yes (%) | 12 (8.6) | 0 (0) | 0 (0) |
No (%) | 127 (91.4) | 139 (100) | 139 (100) |
P-valuea |
| 0.0001 | 0.0001 |
States without expanded Medicaid eligibility |
|
|
|
Yes (%) | 17 (12.1) | 0 (0) | 0 (0) |
No (%) | 124 (87.9) | 141 (100) | 141 (100) |
P-valuea |
| 0.0001 | 0.0001 |
aComparison to Medicaid.
Table 4. Medicaid Referral Rates in Expanded Vs Non-Expanded States
| Expanded states | Non-expanded states | P-value |
Yes (%) | 12 (9.7) | 17 (14.0) | .35 |
No (%) | 127 (91.4) | 124 (87.9) |
|
Reimbursements for ankle fracture varied across states (Table 5). For Medicaid, Georgia paid the highest reimbursement ($1049.95) and Florida paid the lowest ($469.44). Logistic and linear regression analysis did not demonstrate a significant relationship between reimbursement and appointment success rate or waiting periods.
Table 5. Medicaid Reimbursements for Ankle Fracture Repair (CPT and HCPCS 27822) in 2014
State | Medicaid reimbursement |
Californiaa | $785.55 |
Texas | $678.95 |
Florida | $469.44 |
Ohioa | $617.08 |
New Yorka | $500.02 |
North Carolina | $621.63 |
Massachusettsa | $627.94 |
Georgia | $1,049.95 |
Average | $668.82 |
aStates with expanded Medicaid eligibility.
Abbreviations: CPT, Current Procedural Terminology; HCPCS, Healthcare Common Procedure Coding System.
Waiting periods (Table 6) varied significantly by the type of insurance (7.3 days for Medicaid, 6.0 days for Medicare, and 6.0 days for BlueCross; P = .002). For states with expanded Medicaid eligibility, waiting periods varied significantly by insurance (7.7 days for Medicaid, 6.2 days for Medicare, P = .003; and 6.1 days for BlueCross, P = .01). Waiting periods did not vary significantly for states without expanded Medicaid. Additionally, waiting periods did not differ significantly when comparing between states with and without Medicaid expansion.
Table 6. Waiting Period (Days) by Insurance Type.
| Medicaid | Medicare | Private |
Comparison by Insurance Type |
|
|
|
All states |
|
|
|
Waiting period | 7.3 | 6.0 | 6.0 |
P-value |
| 0.002 | 0.002 |
States with expanded Medicaid eligibility |
|
|
|
Waiting period | 7.7 | 6.2 | 6.1 |
P-value |
| 0.003 | 0.01 |
States without expanded Medicaid eligibility |
|
|
|
Waiting period | 6.9 | 5.9 | 5.9 |
P-value |
| 0.15 | 0.15 |
Comparison by Medicaid Expansion |
|
|
|
States with expanded Medicaid eligibility | 7.7 | 6.2 | 6.1 |
States without expanded Medicaid eligibility | 6.9 | 5.9 | 5.9 |
P-value | 0.17 | 0.13 | 0.07 |
Continue to: DISCUSSION...
DISCUSSION
This study assessed how insurance type (Medicaid, Medicare, and BlueCross) affects patient access to orthopedic trauma surgeons in 8 geographically representative states. We selected unstable ankle fractures as they are basic fractures treated by nearly all trauma surgeons and should often be surgically treated to prevent serious long-term consequences. Our hypothesis stated that despite the passage of the PPACA, patients with Medicaid would have reduced access to care. As the PPACA has changed the healthcare marketplace by increasing the number of Medicaid enrollees, it is important to ensure that patient access to care improves.
This nationwide survey of orthopedic trauma surgeons demonstrates that Medicaid patients experience added barriers to care that ultimately results in lower rates of successfully obtaining care. This is consistent with other investigations which have assessed Medicaid patient healthcare access.6,8,10,17-19 This study did not demonstrate a statistically significant difference between Medicaid patients’ ability to obtain appointments in states with expanded Medicaid eligibility vs in states without expanded Medicaid eligibility (39.6% vs 31.9%, P < .18); this has been demonstrated in the literature.6
A barrier that was unique to Medicaid patients was the requirement to have a PCP referral (Table 3). A PCP referral was not a barrier to receiving an appointment for patients with Medicare or BlueCross. One reason to explain why Medicaid patients may be required to have PCP referrals is due to their increased medical complexity, extra documentation requirements, and low reimbursement.4 Patients who have obtained a PCP referral may be characterized as being more medically compliant.
It is important to note that the Medicaid policies for 4 states included in this study (Massachusetts, North Carolina, Texas, and New York) required a PCP referral in order to see a specialist. However, we found that many orthopedic trauma practices in these states scheduled appointments for Medicaid patients without a PCP referral, suggesting that the decision depended on individual policy. In addition, the majority of offices within these states cited that they simply did not accept Medicaid as an insurance policy, and not that they required a referral.
Our regression analysis did not find a significant relationship between being able to successfully obtain an appointment to be evaluated for an ankle fracture and reimbursement rates for Medicaid. Although studies have stressed the importance of Medicaid reimbursements on physician participation, this result is consistent with previous studies regarding carpal tunnel release and total ankle replacements.17,19 Long20 suggested that although reimbursements may help, additional strategies for promoting Medicaid acceptance may be needed, including: lowering the costs of participating in Medicaid by simplifying administrative processes, speeding up reimbursement, and reducing the costs associated with caring for those patients.
Continue to: Previous studies have demonstrated...
Previous studies have demonstrated that more physicians may accept Medicaid if reimbursements increased.4,12 Given the high percentage of trauma patients with Medicaid as their primary insurance or whom are emergently enrolled in Medicaid by hospital systems, it is concerning that the PPACA is reducing payments under the Medicare and Medicaid Disproportionate Share Hospital programs which provide hospitals for uncompensated care given to low-income and uninsured patients.21 Trauma centers generally operate at a deficit due to the higher proportion of Medicaid and uninsured patients.14 This is currently worsened by additional federal funding cuts for supporting trauma service’s humane mission.21
This study has several limitations. While the study evaluated access to care in 8 representative states, a thorough nationwide survey would be more representative. Some results may have become statistically significant if we had performed the study with a larger sample size. In addition, we were unable to control for many factors which could impact appointment wait times, such as physician call schedules and vacations. Socioeconomic factors can influence a patient’s ability to attend an appointment, such as transportation costs, time off from work, and childcare availability. In addition, this study did not assess access for the uninsured, who are predominantly the working poor who cannot afford health insurance, even with federal and state subsidies.
The authors apologize for inconveniencing these offices, however, data collection could not be achieved in a better manner. We hope that the value of this study compensates any inconvenience.
CONCLUSION
Overall, our results demonstrate that despite the ratification of the PPACA, Medicaid patients are confronted with more barriers to accessing care by comparison to patients with Medicare and BlueCross insurance. Medicaid patients have worse baseline health22 and are at an increased risk of complications. These disparities are thought to be due to decreased healthcare access,23,24 as well as socioeconomic challenges. Interventions, such as increasing Medicaid’s reimbursement levels, reducing burdensome administrative responsibilities, and establishing partnerships between trauma centers and trauma surgeons, may enable underinsured patients to be appropriately cared for.
This paper will be judged for the Resident Writer’s Award.
1. Blumenthal D, Collins SR. Health care coverage under the affordable care act--a progress report. N Engl J Med. 2014;371(3):275-281. doi:10.1056/NEJMhpr1405667.
2. Sommers BD. Health care reform's unfinished work--remaining barriers to coverage and access. N Engl J Med. 2015;373(25):2395-2397. doi:10.1056/NEJMp1509462.
3. US Department of Health and Human Services. Centers for Medicare & Medicaid Services. Medicaid & CHIP: February 2015 monthly applications, eligibility determinations and enrollment report. https://www.medicaid.gov/medicaid/program-information/downloads/medicaid-and-chip-february-2015-application-eligibility-and-enrollment-data.pdf. Published May 1, 2015. Accessed May 2015.
4. Iglehart JK, Sommers BD. Medicaid at 50--from welfare program to nation's largest health insurer. N Engl J Med. 2015;372(22):2152-2159. doi:10.1056/NEJMhpr1500791.
5. Kaiser Family Foundation. Medicaid moving forward. http://kff.org/medicaid/fact-sheet/the-medicaid-program-at-a-glance-update/. Updated 2014. Accessed October 10, 2014.
6. Kim CY, Wiznia DH, Hsiang WR, Pelker RR. The effect of insurance type on patient access to knee arthroplasty and revision under the affordable care act. J Arthroplasty. 2015;30(9):1498-1501. doi:10.1016/j.arth.2015.03.015.
7. Draeger RW, Patterson BM, Olsson EC, Schaffer A, Patterson JM. The influence of patient insurance status on access to outpatient orthopedic care for flexor tendon lacerations. J Hand Surg Am. 2014;39(3):527-533. doi:10.1016/j.jhsa.2013.10.031.
8. Patterson BM, Spang JT, Draeger RW, Olsson EC, Creighton RA, Kamath GV. Access to outpatient care for adult rotator cuff patients with private insurance versus Medicaid in North Carolina. J Shoulder Elbow Surg. 2013;22(12):1623-1627. doi:10.1016/j.jse.2013.07.051.
9. Patterson BM, Draeger RW, Olsson EC, Spang JT, Lin FC, Kamath GV. A regional assessment of medicaid access to outpatient orthopaedic care: the influence of population density and proximity to academic medical centers on patient access. J Bone Joint Surg Am. 2014;96(18):e156. doi:10.2106/JBJS.M.01188.
10. Schwarzkopf R, Phan D, Hoang M, Ross S, Mukamel D. Do patients with income-based insurance have access to total joint arthroplasty? J Arthroplasty. 2014;29(6):1083-1086. doi:10.1016/j.arth.2013.11.022.
11. Decker SL. In 2011 nearly one-third of physicians said they would not accept new Medicaid patients, but rising fees may help. Health Aff (Millwood). 2012;31(8):1673-1679 doi:10.1377/hlthaff.2012.0294.
12. Perloff JD, Kletke P, Fossett JW. Which physicians limit their Medicaid participation, and why. Health Serv Res. 1995;30(1):7-26.
13. Althausen PL. Building a successful trauma practice in a community setting. J Orthop Trauma. 2011;25 Suppl 3:S113-S117. doi:10.1097/BOT.0b013e318237bcce.
14. Greenberg S, Mir HR, Jahangir AA, Mehta S, Sethi MK. Impacting policy change for orthopaedic trauma. J Orthop Trauma. 2014;28 Suppl 10:S14-S16. doi:10.1097/BOT.0000000000000216.
15. Wiznia DH, Averbukh L, Kim CY, Goel A, Leslie MP. Motorcycle helmets: The economic burden of an incomplete helmet law to medical care in the state of Connecticut. Conn Med. 2015;79(8):453-459.
16. Orthopaedic Trauma Association. Find a surgeon. https://online.ota.org/otassa/otacenssafindasurgeon.query_page. Updated 2015. Accessed July, 2015.
17. Kim CY, Wiznia DH, Roth AS, Walls RJ, Pelker RR. Survey of patient insurance status on access to specialty foot and ankle care under the affordable care act. Foot Ankle Int. 2016;37(7):776-781. doi:1071100716642015.
18. Patterson BM, Draeger RW, Olsson EC, Spang JT, Lin FC, Kamath GV. A regional assessment of Medicaid access to outpatient orthopaedic care: the influence of population density and proximity to academic medical centers on patient access. J Bone Joint Surg Am. 2014;96(18):e156. doi:10.2106/JBJS.M.01188.
19. Kim CY, Wiznia DH, Wang Y, et al. The effect of insurance type on patient access to carpal tunnel release under the affordable care act. J Hand Surg Am. 2016;41(4):503-509.e1. doi:S0363-5023(16)00104-0.
20. Long SK. Physicians may need more than higher reimbursements to expand Medicaid participation: findings from Washington state. Health Aff (Millwood). 2013;32(9):1560-1567. doi:10.1377/hlthaff.2012.1010.
21. Issar NM, Jahangir AA. The affordable care act and orthopaedic trauma. J Orthop Trauma. 2014;28 Suppl 10:S5-S7. doi:10.1097/BOT.0000000000000211.
22. Hahn B, Flood AB. No insurance, public insurance, and private insurance: do these options contribute to differences in general health? J Health Care Poor Underserved. 1995;6(1):41-59.
23. Hinman A, Bozic KJ. Impact of payer type on resource utilization, outcomes and access to care in total hip arthroplasty. J Arthroplasty. 2008;23(6 Suppl 1):9-14. doi:10.1016/j.arth.2008.05.010.
24. Schoenfeld AJ, Tipirneni R, Nelson JH, Carpenter JE, Iwashyna TJ. The influence of race and ethnicity on complications and mortality after orthopedic surgery: A systematic review of the literature. Med Care. 2014;52(9):842-851. doi:10.1097/MLR.0000000000000177.
ABSTRACT
The purpose of this study is to assess the effect of insurance type (Medicaid, Medicare, private insurance) on the ability for patients with operative ankle fractures to access orthopedic traumatologists. The research team called 245 board-certified orthopedic surgeons specializing in orthopedic trauma within 8 representative states. The caller requested an appointment for their fictitious mother in order to be evaluated for an ankle fracture which was previously evaluated by her primary care physician and believed to require surgery. Each office was called 3 times to assess the response for each insurance type. For each call, information was documented regarding whether the patient was able to receive an appointment and the barriers the patient confronted to receive an appointment. Overall, 35.7% of offices scheduled an appointment for a patient with Medicaid, in comparison to 81.4%and 88.6% for Medicare and BlueCross, respectively (P < .0001). Medicaid patients confronted more barriers for receiving appointments. There was no statistically significant difference in access for Medicaid patients in states that had expanded Medicaid eligibility vs states that had not expanded Medicaid. Medicaid reimbursement for open reduction and internal fixation of an ankle fracture did not significantly correlate with appointment success rates or wait times. Despite the passage of the Affordable Care Act, patients with Medicaid have reduced access to orthopedic surgeons and more complex barriers to receiving appointments. A more robust strategy for increasing care-access for patients with Medicaid would be more equitable.
Continue to: In 2010, the Patient Protection and Affordable Care Act...
In 2010, the Patient Protection and Affordable Care Act (PPACA) expanded the eligibility criteria for Medicaid to all individuals with an income up to 138% of the poverty level.1 A Supreme Court ruling stated that the decision to expand Medicaid was to be decided by individual states.2 Currently, 31 states have chosen to expand Medicaid eligibility to their residents.2 This expansion has allowed an additional 11.7 million people to enroll in Medicaid and the Children’s Health Insurance Program by May 2015.3-5
Even with the passage of the PPACA, Medicaid patients seeking specialty orthopedic care have experienced more barriers to accessing care than Medicare or commercially-insured patients.2,6-10 One major cited reason is Medicaid’s low reimbursement, which may discourage physicians from open panel participation in Medicaid.11,12
A common fundamental teaching for orthopedic traumatologists is the notion that they should be available to treat all injuries regardless of the patient’s ability to pay.13 This has resulted in both trauma centers and trauma surgeons becoming financially challenged due to the higher proportion of Medicaid and uninsured trauma patients and lower Medicaid reimbursement levels.14,15
This study focuses on the effect of different types of insurance (Medicaid, Medicare, or commercial insurance) on the ability of patients to obtain care for operative ankle fractures. The purpose of this study is to evaluate, in the context of the PPACA, patient access to orthopedic surgeons for operative ankle fractures based on insurance-type. We hypothesized that patients with Medicaid would face a greater volume of obstacles when seeking appointments for an ankle fracture, even after the PPACA.
Continue to: MATERIALS AND METHODS...
MATERIALS AND METHODS
The study population included board-certified orthopedic surgeons who belonged to the Orthopaedic Trauma Association (OTA) from 8 representative states; 4 states with expanded Medicaid eligibility (California, Massachusetts, New York, Ohio) and 4 states without expanded Medicaid eligibility (Florida, North Carolina, Georgia, Texas). These states were selected due to their ability to represent diverse healthcare marketplaces throughout the country. Using the OTA website’s “Find a Surgeon” search tool,16 we created a list of surgeons for each state and matched each surgeon with a random number. The list of surgeons was ordered according to the value of the surgeon’s associated random number, and surgeons were called in ascending order. We excluded disconnected or inaccurate numbers from the calling list. Surgeons who did not manage ankle fractures were removed from the dataset. Approximately 30 orthopedic trauma surgeons per state were contacted.
Each office was called to make an appointment for the caller’s mother. Every surgeon’s office was specifically asked if the surgeon would accept the patient to be evaluated for an ankle fracture that occurred out-of-state. The caller had a standardized protocol to limit intra- and inter-office variations (Appendix). The scenario involved a request to be evaluated for an unstable ankle fracture, with the patient having Medicaid, Medicare, or BlueCross insurance. The scenario required 3 separate calls to the same surgeon in order to obtain data regarding each insurance-type. The calls were separated by at least 1 week to avoid caller recognition by the surgeon’s office.
Appendix
Scenario
1. Date of Birth: Medicaid–2/07/55; BlueCross PPO–2/09/55; Medicare–7/31/45.
2. Ankle fracture evaluated by primary care physician 1 or 2 days ago
3. Not seen previously by your clinic or hospital, she would be a new patient
4. Asked how early she could be scheduled for an appointment
5. Script:
“I’m calling for my mother who injured her ankle a few days ago. Her family doctor took an X-ray and believes she has a fracture and needs surgery. Is Dr. X accepting new patients for evaluation and treatment of ankle fractures?” If YES →
“I was wondering if you take Medicaid/Medicare/BlueCross plan?” If YES →
“When is your soonest available appointment?”
The date of each phone call and date of appointment, if provided, were recorded. If the office did not give an appointment, we asked for reasons why. If an appointment was denied for a patient with Medicaid, we asked for a referral to another office that accepted Medicaid. We considered barriers to obtaining an initial appointment, such as requiring a referral from a primary care physician (PCP), as an unsuccessful attempt at making an appointment. We determined the waiting period for an appointment by calculating the time between the date of the call and the date of the appointment. Appointments were not scheduled to ensure that actual patients were not disadvantaged. For both appointment success rates and waiting periods, we stratified the data into 2 groups: states with expanded Medicaid eligibility (California, Massachusetts, New York, Ohio) and states without expanded Medicaid eligibility (Florida, North Carolina, Georgia, Texas).
We obtained Medicaid reimbursement rates for open reduction and internal fixation of an ankle fracture by querying each state’s reimbursement rate using Current Procedural Terminology code 27822.
Chi-square test or Fisher’s exact test was used to analyze acceptance rate differences based on the patient’s type of insurance. To compare the waiting periods for an appointment, we used an independent samples t-test after applying natural log-transformation, as the data was not normally distributed. We performed logistic regression analysis to detect whether reimbursement was a significant predictor of successfully making an appointment for patients, and a linear regression analysis was used to evaluate whether reimbursement predicted waiting periods. Unless otherwise stated, all statistical testing was performed two-tailed at an alpha-level of 0.05.
This study was approved by the Institutional Review Board of Yale University School of Medicine (HIC No. 1363).
Continue to: RESULTS...
RESULTS
In total, 350 offices were contacted across 8 states (4 states with and 4 states without expanded Medicaid eligibility) of which we identified 245 orthopedic surgeons who would surgically treat ankle fractures. The 245 surgeons’ offices were called 3 times for each separate insurance-type.
Table 1. Appointment Success Rate
| Medicaid | Medicare | Private |
All states |
|
| |
Yes (%) | 100 (35.7) | 228 (81.4) | 248 (88.6) |
No (%) | 180 (64.3) | 52 (18.60 | 32 (11.4) |
P-valuea |
| 0.0001 | 0.0001 |
States with expanded Medicaid eligibility |
|
|
|
Yes (%) | 55 (39.6) | 116 (83.5) | 124 (89.2) |
No (%) | 84 (60.4) | 23 (16.5) | 15 (10.8) |
P-valuea |
| 0.0001 | 0.0001 |
States without expanded Medicaid eligibility |
|
|
|
Yes (%) | 45 (31.9) | 112 (79.4) | 124 (87.9) |
No (%) | 96 (68.1) | 29 (20.6) | 17 (12.1) |
P-valuea |
| 0.0001 | 0.0001 |
aComparison to Medicaid.
The overall rate of successfully being offered an appointment with Medicaid was 35.7%, 81.4% for Medicare, and 88.6% for BlueCross (Table 1). For states with expanded Medicaid eligibility, the success rate for obtaining an appointment was 39.6%, 83.5%, and 89.2% for Medicaid, Medicare, and BlueCross, respectively. For states without expanded Medicaid eligibility, the success rate for obtaining an appointment was 31.9% for Medicaid, 79.4% for Medicare, and 87.9% for BlueCross. In all cases, the success rate for obtaining an appointment was significantly lower for Medicaid, compared to Medicare (P < .0001) or BlueCross (P < .0001). Medicaid appointment success rate was 39.6% in expanded states vs 31.9% in non-expanded states, however, the difference was not statistically significant (Table 2).
Table 2. Medicaid Appointment Success Rate in Expanded Vs Non-Expanded States
| Expanded states | Non-expanded states | P-value |
Yes (%) | 55 (39.6) | 45 (31.9) | .181 |
No (%) | 84 (60.4) | 96 (68.1) |
|
In 43.7% of occasions, patients with Medicaid did not have their insurance accepted, compared to 7.3% for Medicare and 0% for BlueCross. The majority of offices which did not accept Medicaid were not able to refer patients to another surgeon who would accept Medicaid. The requirement to have a primary care referral was the second most common reason for Medicaid patients not obtaining an appointment. No Medicare (10.4% vs 0.0%, P < .0001) or BlueCross (10.4% vs 0.0%, P < .0001) patients experienced this requirement (Table 3). There was no difference found between the percent of Medicaid patients who were required to have referrals in states with and without expanded Medicaid eligibility (Table 4).
Table 3. Referral Rate
| Medicaid | Medicare | Private |
All states |
|
|
|
Yes (%) | 29 (10.4) | 0 (0) | 0 (0) |
No (%) | 251 (89.6) | 280 (100) | 280 (100) |
P-valuea |
| 0.0001 | 0.0001 |
States with expanded Medicaid eligibility |
|
|
|
Yes (%) | 12 (8.6) | 0 (0) | 0 (0) |
No (%) | 127 (91.4) | 139 (100) | 139 (100) |
P-valuea |
| 0.0001 | 0.0001 |
States without expanded Medicaid eligibility |
|
|
|
Yes (%) | 17 (12.1) | 0 (0) | 0 (0) |
No (%) | 124 (87.9) | 141 (100) | 141 (100) |
P-valuea |
| 0.0001 | 0.0001 |
aComparison to Medicaid.
Table 4. Medicaid Referral Rates in Expanded Vs Non-Expanded States
| Expanded states | Non-expanded states | P-value |
Yes (%) | 12 (9.7) | 17 (14.0) | .35 |
No (%) | 127 (91.4) | 124 (87.9) |
|
Reimbursements for ankle fracture varied across states (Table 5). For Medicaid, Georgia paid the highest reimbursement ($1049.95) and Florida paid the lowest ($469.44). Logistic and linear regression analysis did not demonstrate a significant relationship between reimbursement and appointment success rate or waiting periods.
Table 5. Medicaid Reimbursements for Ankle Fracture Repair (CPT and HCPCS 27822) in 2014
State | Medicaid reimbursement |
Californiaa | $785.55 |
Texas | $678.95 |
Florida | $469.44 |
Ohioa | $617.08 |
New Yorka | $500.02 |
North Carolina | $621.63 |
Massachusettsa | $627.94 |
Georgia | $1,049.95 |
Average | $668.82 |
aStates with expanded Medicaid eligibility.
Abbreviations: CPT, Current Procedural Terminology; HCPCS, Healthcare Common Procedure Coding System.
Waiting periods (Table 6) varied significantly by the type of insurance (7.3 days for Medicaid, 6.0 days for Medicare, and 6.0 days for BlueCross; P = .002). For states with expanded Medicaid eligibility, waiting periods varied significantly by insurance (7.7 days for Medicaid, 6.2 days for Medicare, P = .003; and 6.1 days for BlueCross, P = .01). Waiting periods did not vary significantly for states without expanded Medicaid. Additionally, waiting periods did not differ significantly when comparing between states with and without Medicaid expansion.
Table 6. Waiting Period (Days) by Insurance Type.
| Medicaid | Medicare | Private |
Comparison by Insurance Type |
|
|
|
All states |
|
|
|
Waiting period | 7.3 | 6.0 | 6.0 |
P-value |
| 0.002 | 0.002 |
States with expanded Medicaid eligibility |
|
|
|
Waiting period | 7.7 | 6.2 | 6.1 |
P-value |
| 0.003 | 0.01 |
States without expanded Medicaid eligibility |
|
|
|
Waiting period | 6.9 | 5.9 | 5.9 |
P-value |
| 0.15 | 0.15 |
Comparison by Medicaid Expansion |
|
|
|
States with expanded Medicaid eligibility | 7.7 | 6.2 | 6.1 |
States without expanded Medicaid eligibility | 6.9 | 5.9 | 5.9 |
P-value | 0.17 | 0.13 | 0.07 |
Continue to: DISCUSSION...
DISCUSSION
This study assessed how insurance type (Medicaid, Medicare, and BlueCross) affects patient access to orthopedic trauma surgeons in 8 geographically representative states. We selected unstable ankle fractures as they are basic fractures treated by nearly all trauma surgeons and should often be surgically treated to prevent serious long-term consequences. Our hypothesis stated that despite the passage of the PPACA, patients with Medicaid would have reduced access to care. As the PPACA has changed the healthcare marketplace by increasing the number of Medicaid enrollees, it is important to ensure that patient access to care improves.
This nationwide survey of orthopedic trauma surgeons demonstrates that Medicaid patients experience added barriers to care that ultimately results in lower rates of successfully obtaining care. This is consistent with other investigations which have assessed Medicaid patient healthcare access.6,8,10,17-19 This study did not demonstrate a statistically significant difference between Medicaid patients’ ability to obtain appointments in states with expanded Medicaid eligibility vs in states without expanded Medicaid eligibility (39.6% vs 31.9%, P < .18); this has been demonstrated in the literature.6
A barrier that was unique to Medicaid patients was the requirement to have a PCP referral (Table 3). A PCP referral was not a barrier to receiving an appointment for patients with Medicare or BlueCross. One reason to explain why Medicaid patients may be required to have PCP referrals is due to their increased medical complexity, extra documentation requirements, and low reimbursement.4 Patients who have obtained a PCP referral may be characterized as being more medically compliant.
It is important to note that the Medicaid policies for 4 states included in this study (Massachusetts, North Carolina, Texas, and New York) required a PCP referral in order to see a specialist. However, we found that many orthopedic trauma practices in these states scheduled appointments for Medicaid patients without a PCP referral, suggesting that the decision depended on individual policy. In addition, the majority of offices within these states cited that they simply did not accept Medicaid as an insurance policy, and not that they required a referral.
Our regression analysis did not find a significant relationship between being able to successfully obtain an appointment to be evaluated for an ankle fracture and reimbursement rates for Medicaid. Although studies have stressed the importance of Medicaid reimbursements on physician participation, this result is consistent with previous studies regarding carpal tunnel release and total ankle replacements.17,19 Long20 suggested that although reimbursements may help, additional strategies for promoting Medicaid acceptance may be needed, including: lowering the costs of participating in Medicaid by simplifying administrative processes, speeding up reimbursement, and reducing the costs associated with caring for those patients.
Continue to: Previous studies have demonstrated...
Previous studies have demonstrated that more physicians may accept Medicaid if reimbursements increased.4,12 Given the high percentage of trauma patients with Medicaid as their primary insurance or whom are emergently enrolled in Medicaid by hospital systems, it is concerning that the PPACA is reducing payments under the Medicare and Medicaid Disproportionate Share Hospital programs which provide hospitals for uncompensated care given to low-income and uninsured patients.21 Trauma centers generally operate at a deficit due to the higher proportion of Medicaid and uninsured patients.14 This is currently worsened by additional federal funding cuts for supporting trauma service’s humane mission.21
This study has several limitations. While the study evaluated access to care in 8 representative states, a thorough nationwide survey would be more representative. Some results may have become statistically significant if we had performed the study with a larger sample size. In addition, we were unable to control for many factors which could impact appointment wait times, such as physician call schedules and vacations. Socioeconomic factors can influence a patient’s ability to attend an appointment, such as transportation costs, time off from work, and childcare availability. In addition, this study did not assess access for the uninsured, who are predominantly the working poor who cannot afford health insurance, even with federal and state subsidies.
The authors apologize for inconveniencing these offices, however, data collection could not be achieved in a better manner. We hope that the value of this study compensates any inconvenience.
CONCLUSION
Overall, our results demonstrate that despite the ratification of the PPACA, Medicaid patients are confronted with more barriers to accessing care by comparison to patients with Medicare and BlueCross insurance. Medicaid patients have worse baseline health22 and are at an increased risk of complications. These disparities are thought to be due to decreased healthcare access,23,24 as well as socioeconomic challenges. Interventions, such as increasing Medicaid’s reimbursement levels, reducing burdensome administrative responsibilities, and establishing partnerships between trauma centers and trauma surgeons, may enable underinsured patients to be appropriately cared for.
This paper will be judged for the Resident Writer’s Award.
ABSTRACT
The purpose of this study is to assess the effect of insurance type (Medicaid, Medicare, private insurance) on the ability for patients with operative ankle fractures to access orthopedic traumatologists. The research team called 245 board-certified orthopedic surgeons specializing in orthopedic trauma within 8 representative states. The caller requested an appointment for their fictitious mother in order to be evaluated for an ankle fracture which was previously evaluated by her primary care physician and believed to require surgery. Each office was called 3 times to assess the response for each insurance type. For each call, information was documented regarding whether the patient was able to receive an appointment and the barriers the patient confronted to receive an appointment. Overall, 35.7% of offices scheduled an appointment for a patient with Medicaid, in comparison to 81.4%and 88.6% for Medicare and BlueCross, respectively (P < .0001). Medicaid patients confronted more barriers for receiving appointments. There was no statistically significant difference in access for Medicaid patients in states that had expanded Medicaid eligibility vs states that had not expanded Medicaid. Medicaid reimbursement for open reduction and internal fixation of an ankle fracture did not significantly correlate with appointment success rates or wait times. Despite the passage of the Affordable Care Act, patients with Medicaid have reduced access to orthopedic surgeons and more complex barriers to receiving appointments. A more robust strategy for increasing care-access for patients with Medicaid would be more equitable.
Continue to: In 2010, the Patient Protection and Affordable Care Act...
In 2010, the Patient Protection and Affordable Care Act (PPACA) expanded the eligibility criteria for Medicaid to all individuals with an income up to 138% of the poverty level.1 A Supreme Court ruling stated that the decision to expand Medicaid was to be decided by individual states.2 Currently, 31 states have chosen to expand Medicaid eligibility to their residents.2 This expansion has allowed an additional 11.7 million people to enroll in Medicaid and the Children’s Health Insurance Program by May 2015.3-5
Even with the passage of the PPACA, Medicaid patients seeking specialty orthopedic care have experienced more barriers to accessing care than Medicare or commercially-insured patients.2,6-10 One major cited reason is Medicaid’s low reimbursement, which may discourage physicians from open panel participation in Medicaid.11,12
A common fundamental teaching for orthopedic traumatologists is the notion that they should be available to treat all injuries regardless of the patient’s ability to pay.13 This has resulted in both trauma centers and trauma surgeons becoming financially challenged due to the higher proportion of Medicaid and uninsured trauma patients and lower Medicaid reimbursement levels.14,15
This study focuses on the effect of different types of insurance (Medicaid, Medicare, or commercial insurance) on the ability of patients to obtain care for operative ankle fractures. The purpose of this study is to evaluate, in the context of the PPACA, patient access to orthopedic surgeons for operative ankle fractures based on insurance-type. We hypothesized that patients with Medicaid would face a greater volume of obstacles when seeking appointments for an ankle fracture, even after the PPACA.
Continue to: MATERIALS AND METHODS...
MATERIALS AND METHODS
The study population included board-certified orthopedic surgeons who belonged to the Orthopaedic Trauma Association (OTA) from 8 representative states; 4 states with expanded Medicaid eligibility (California, Massachusetts, New York, Ohio) and 4 states without expanded Medicaid eligibility (Florida, North Carolina, Georgia, Texas). These states were selected due to their ability to represent diverse healthcare marketplaces throughout the country. Using the OTA website’s “Find a Surgeon” search tool,16 we created a list of surgeons for each state and matched each surgeon with a random number. The list of surgeons was ordered according to the value of the surgeon’s associated random number, and surgeons were called in ascending order. We excluded disconnected or inaccurate numbers from the calling list. Surgeons who did not manage ankle fractures were removed from the dataset. Approximately 30 orthopedic trauma surgeons per state were contacted.
Each office was called to make an appointment for the caller’s mother. Every surgeon’s office was specifically asked if the surgeon would accept the patient to be evaluated for an ankle fracture that occurred out-of-state. The caller had a standardized protocol to limit intra- and inter-office variations (Appendix). The scenario involved a request to be evaluated for an unstable ankle fracture, with the patient having Medicaid, Medicare, or BlueCross insurance. The scenario required 3 separate calls to the same surgeon in order to obtain data regarding each insurance-type. The calls were separated by at least 1 week to avoid caller recognition by the surgeon’s office.
Appendix
Scenario
1. Date of Birth: Medicaid–2/07/55; BlueCross PPO–2/09/55; Medicare–7/31/45.
2. Ankle fracture evaluated by primary care physician 1 or 2 days ago
3. Not seen previously by your clinic or hospital, she would be a new patient
4. Asked how early she could be scheduled for an appointment
5. Script:
“I’m calling for my mother who injured her ankle a few days ago. Her family doctor took an X-ray and believes she has a fracture and needs surgery. Is Dr. X accepting new patients for evaluation and treatment of ankle fractures?” If YES →
“I was wondering if you take Medicaid/Medicare/BlueCross plan?” If YES →
“When is your soonest available appointment?”
The date of each phone call and date of appointment, if provided, were recorded. If the office did not give an appointment, we asked for reasons why. If an appointment was denied for a patient with Medicaid, we asked for a referral to another office that accepted Medicaid. We considered barriers to obtaining an initial appointment, such as requiring a referral from a primary care physician (PCP), as an unsuccessful attempt at making an appointment. We determined the waiting period for an appointment by calculating the time between the date of the call and the date of the appointment. Appointments were not scheduled to ensure that actual patients were not disadvantaged. For both appointment success rates and waiting periods, we stratified the data into 2 groups: states with expanded Medicaid eligibility (California, Massachusetts, New York, Ohio) and states without expanded Medicaid eligibility (Florida, North Carolina, Georgia, Texas).
We obtained Medicaid reimbursement rates for open reduction and internal fixation of an ankle fracture by querying each state’s reimbursement rate using Current Procedural Terminology code 27822.
Chi-square test or Fisher’s exact test was used to analyze acceptance rate differences based on the patient’s type of insurance. To compare the waiting periods for an appointment, we used an independent samples t-test after applying natural log-transformation, as the data was not normally distributed. We performed logistic regression analysis to detect whether reimbursement was a significant predictor of successfully making an appointment for patients, and a linear regression analysis was used to evaluate whether reimbursement predicted waiting periods. Unless otherwise stated, all statistical testing was performed two-tailed at an alpha-level of 0.05.
This study was approved by the Institutional Review Board of Yale University School of Medicine (HIC No. 1363).
Continue to: RESULTS...
RESULTS
In total, 350 offices were contacted across 8 states (4 states with and 4 states without expanded Medicaid eligibility) of which we identified 245 orthopedic surgeons who would surgically treat ankle fractures. The 245 surgeons’ offices were called 3 times for each separate insurance-type.
Table 1. Appointment Success Rate
| Medicaid | Medicare | Private |
All states |
|
| |
Yes (%) | 100 (35.7) | 228 (81.4) | 248 (88.6) |
No (%) | 180 (64.3) | 52 (18.60 | 32 (11.4) |
P-valuea |
| 0.0001 | 0.0001 |
States with expanded Medicaid eligibility |
|
|
|
Yes (%) | 55 (39.6) | 116 (83.5) | 124 (89.2) |
No (%) | 84 (60.4) | 23 (16.5) | 15 (10.8) |
P-valuea |
| 0.0001 | 0.0001 |
States without expanded Medicaid eligibility |
|
|
|
Yes (%) | 45 (31.9) | 112 (79.4) | 124 (87.9) |
No (%) | 96 (68.1) | 29 (20.6) | 17 (12.1) |
P-valuea |
| 0.0001 | 0.0001 |
aComparison to Medicaid.
The overall rate of successfully being offered an appointment with Medicaid was 35.7%, 81.4% for Medicare, and 88.6% for BlueCross (Table 1). For states with expanded Medicaid eligibility, the success rate for obtaining an appointment was 39.6%, 83.5%, and 89.2% for Medicaid, Medicare, and BlueCross, respectively. For states without expanded Medicaid eligibility, the success rate for obtaining an appointment was 31.9% for Medicaid, 79.4% for Medicare, and 87.9% for BlueCross. In all cases, the success rate for obtaining an appointment was significantly lower for Medicaid, compared to Medicare (P < .0001) or BlueCross (P < .0001). Medicaid appointment success rate was 39.6% in expanded states vs 31.9% in non-expanded states, however, the difference was not statistically significant (Table 2).
Table 2. Medicaid Appointment Success Rate in Expanded Vs Non-Expanded States
| Expanded states | Non-expanded states | P-value |
Yes (%) | 55 (39.6) | 45 (31.9) | .181 |
No (%) | 84 (60.4) | 96 (68.1) |
|
In 43.7% of occasions, patients with Medicaid did not have their insurance accepted, compared to 7.3% for Medicare and 0% for BlueCross. The majority of offices which did not accept Medicaid were not able to refer patients to another surgeon who would accept Medicaid. The requirement to have a primary care referral was the second most common reason for Medicaid patients not obtaining an appointment. No Medicare (10.4% vs 0.0%, P < .0001) or BlueCross (10.4% vs 0.0%, P < .0001) patients experienced this requirement (Table 3). There was no difference found between the percent of Medicaid patients who were required to have referrals in states with and without expanded Medicaid eligibility (Table 4).
Table 3. Referral Rate
| Medicaid | Medicare | Private |
All states |
|
|
|
Yes (%) | 29 (10.4) | 0 (0) | 0 (0) |
No (%) | 251 (89.6) | 280 (100) | 280 (100) |
P-valuea |
| 0.0001 | 0.0001 |
States with expanded Medicaid eligibility |
|
|
|
Yes (%) | 12 (8.6) | 0 (0) | 0 (0) |
No (%) | 127 (91.4) | 139 (100) | 139 (100) |
P-valuea |
| 0.0001 | 0.0001 |
States without expanded Medicaid eligibility |
|
|
|
Yes (%) | 17 (12.1) | 0 (0) | 0 (0) |
No (%) | 124 (87.9) | 141 (100) | 141 (100) |
P-valuea |
| 0.0001 | 0.0001 |
aComparison to Medicaid.
Table 4. Medicaid Referral Rates in Expanded Vs Non-Expanded States
| Expanded states | Non-expanded states | P-value |
Yes (%) | 12 (9.7) | 17 (14.0) | .35 |
No (%) | 127 (91.4) | 124 (87.9) |
|
Reimbursements for ankle fracture varied across states (Table 5). For Medicaid, Georgia paid the highest reimbursement ($1049.95) and Florida paid the lowest ($469.44). Logistic and linear regression analysis did not demonstrate a significant relationship between reimbursement and appointment success rate or waiting periods.
Table 5. Medicaid Reimbursements for Ankle Fracture Repair (CPT and HCPCS 27822) in 2014
State | Medicaid reimbursement |
Californiaa | $785.55 |
Texas | $678.95 |
Florida | $469.44 |
Ohioa | $617.08 |
New Yorka | $500.02 |
North Carolina | $621.63 |
Massachusettsa | $627.94 |
Georgia | $1,049.95 |
Average | $668.82 |
aStates with expanded Medicaid eligibility.
Abbreviations: CPT, Current Procedural Terminology; HCPCS, Healthcare Common Procedure Coding System.
Waiting periods (Table 6) varied significantly by the type of insurance (7.3 days for Medicaid, 6.0 days for Medicare, and 6.0 days for BlueCross; P = .002). For states with expanded Medicaid eligibility, waiting periods varied significantly by insurance (7.7 days for Medicaid, 6.2 days for Medicare, P = .003; and 6.1 days for BlueCross, P = .01). Waiting periods did not vary significantly for states without expanded Medicaid. Additionally, waiting periods did not differ significantly when comparing between states with and without Medicaid expansion.
Table 6. Waiting Period (Days) by Insurance Type.
| Medicaid | Medicare | Private |
Comparison by Insurance Type |
|
|
|
All states |
|
|
|
Waiting period | 7.3 | 6.0 | 6.0 |
P-value |
| 0.002 | 0.002 |
States with expanded Medicaid eligibility |
|
|
|
Waiting period | 7.7 | 6.2 | 6.1 |
P-value |
| 0.003 | 0.01 |
States without expanded Medicaid eligibility |
|
|
|
Waiting period | 6.9 | 5.9 | 5.9 |
P-value |
| 0.15 | 0.15 |
Comparison by Medicaid Expansion |
|
|
|
States with expanded Medicaid eligibility | 7.7 | 6.2 | 6.1 |
States without expanded Medicaid eligibility | 6.9 | 5.9 | 5.9 |
P-value | 0.17 | 0.13 | 0.07 |
Continue to: DISCUSSION...
DISCUSSION
This study assessed how insurance type (Medicaid, Medicare, and BlueCross) affects patient access to orthopedic trauma surgeons in 8 geographically representative states. We selected unstable ankle fractures as they are basic fractures treated by nearly all trauma surgeons and should often be surgically treated to prevent serious long-term consequences. Our hypothesis stated that despite the passage of the PPACA, patients with Medicaid would have reduced access to care. As the PPACA has changed the healthcare marketplace by increasing the number of Medicaid enrollees, it is important to ensure that patient access to care improves.
This nationwide survey of orthopedic trauma surgeons demonstrates that Medicaid patients experience added barriers to care that ultimately results in lower rates of successfully obtaining care. This is consistent with other investigations which have assessed Medicaid patient healthcare access.6,8,10,17-19 This study did not demonstrate a statistically significant difference between Medicaid patients’ ability to obtain appointments in states with expanded Medicaid eligibility vs in states without expanded Medicaid eligibility (39.6% vs 31.9%, P < .18); this has been demonstrated in the literature.6
A barrier that was unique to Medicaid patients was the requirement to have a PCP referral (Table 3). A PCP referral was not a barrier to receiving an appointment for patients with Medicare or BlueCross. One reason to explain why Medicaid patients may be required to have PCP referrals is due to their increased medical complexity, extra documentation requirements, and low reimbursement.4 Patients who have obtained a PCP referral may be characterized as being more medically compliant.
It is important to note that the Medicaid policies for 4 states included in this study (Massachusetts, North Carolina, Texas, and New York) required a PCP referral in order to see a specialist. However, we found that many orthopedic trauma practices in these states scheduled appointments for Medicaid patients without a PCP referral, suggesting that the decision depended on individual policy. In addition, the majority of offices within these states cited that they simply did not accept Medicaid as an insurance policy, and not that they required a referral.
Our regression analysis did not find a significant relationship between being able to successfully obtain an appointment to be evaluated for an ankle fracture and reimbursement rates for Medicaid. Although studies have stressed the importance of Medicaid reimbursements on physician participation, this result is consistent with previous studies regarding carpal tunnel release and total ankle replacements.17,19 Long20 suggested that although reimbursements may help, additional strategies for promoting Medicaid acceptance may be needed, including: lowering the costs of participating in Medicaid by simplifying administrative processes, speeding up reimbursement, and reducing the costs associated with caring for those patients.
Continue to: Previous studies have demonstrated...
Previous studies have demonstrated that more physicians may accept Medicaid if reimbursements increased.4,12 Given the high percentage of trauma patients with Medicaid as their primary insurance or whom are emergently enrolled in Medicaid by hospital systems, it is concerning that the PPACA is reducing payments under the Medicare and Medicaid Disproportionate Share Hospital programs which provide hospitals for uncompensated care given to low-income and uninsured patients.21 Trauma centers generally operate at a deficit due to the higher proportion of Medicaid and uninsured patients.14 This is currently worsened by additional federal funding cuts for supporting trauma service’s humane mission.21
This study has several limitations. While the study evaluated access to care in 8 representative states, a thorough nationwide survey would be more representative. Some results may have become statistically significant if we had performed the study with a larger sample size. In addition, we were unable to control for many factors which could impact appointment wait times, such as physician call schedules and vacations. Socioeconomic factors can influence a patient’s ability to attend an appointment, such as transportation costs, time off from work, and childcare availability. In addition, this study did not assess access for the uninsured, who are predominantly the working poor who cannot afford health insurance, even with federal and state subsidies.
The authors apologize for inconveniencing these offices, however, data collection could not be achieved in a better manner. We hope that the value of this study compensates any inconvenience.
CONCLUSION
Overall, our results demonstrate that despite the ratification of the PPACA, Medicaid patients are confronted with more barriers to accessing care by comparison to patients with Medicare and BlueCross insurance. Medicaid patients have worse baseline health22 and are at an increased risk of complications. These disparities are thought to be due to decreased healthcare access,23,24 as well as socioeconomic challenges. Interventions, such as increasing Medicaid’s reimbursement levels, reducing burdensome administrative responsibilities, and establishing partnerships between trauma centers and trauma surgeons, may enable underinsured patients to be appropriately cared for.
This paper will be judged for the Resident Writer’s Award.
1. Blumenthal D, Collins SR. Health care coverage under the affordable care act--a progress report. N Engl J Med. 2014;371(3):275-281. doi:10.1056/NEJMhpr1405667.
2. Sommers BD. Health care reform's unfinished work--remaining barriers to coverage and access. N Engl J Med. 2015;373(25):2395-2397. doi:10.1056/NEJMp1509462.
3. US Department of Health and Human Services. Centers for Medicare & Medicaid Services. Medicaid & CHIP: February 2015 monthly applications, eligibility determinations and enrollment report. https://www.medicaid.gov/medicaid/program-information/downloads/medicaid-and-chip-february-2015-application-eligibility-and-enrollment-data.pdf. Published May 1, 2015. Accessed May 2015.
4. Iglehart JK, Sommers BD. Medicaid at 50--from welfare program to nation's largest health insurer. N Engl J Med. 2015;372(22):2152-2159. doi:10.1056/NEJMhpr1500791.
5. Kaiser Family Foundation. Medicaid moving forward. http://kff.org/medicaid/fact-sheet/the-medicaid-program-at-a-glance-update/. Updated 2014. Accessed October 10, 2014.
6. Kim CY, Wiznia DH, Hsiang WR, Pelker RR. The effect of insurance type on patient access to knee arthroplasty and revision under the affordable care act. J Arthroplasty. 2015;30(9):1498-1501. doi:10.1016/j.arth.2015.03.015.
7. Draeger RW, Patterson BM, Olsson EC, Schaffer A, Patterson JM. The influence of patient insurance status on access to outpatient orthopedic care for flexor tendon lacerations. J Hand Surg Am. 2014;39(3):527-533. doi:10.1016/j.jhsa.2013.10.031.
8. Patterson BM, Spang JT, Draeger RW, Olsson EC, Creighton RA, Kamath GV. Access to outpatient care for adult rotator cuff patients with private insurance versus Medicaid in North Carolina. J Shoulder Elbow Surg. 2013;22(12):1623-1627. doi:10.1016/j.jse.2013.07.051.
9. Patterson BM, Draeger RW, Olsson EC, Spang JT, Lin FC, Kamath GV. A regional assessment of medicaid access to outpatient orthopaedic care: the influence of population density and proximity to academic medical centers on patient access. J Bone Joint Surg Am. 2014;96(18):e156. doi:10.2106/JBJS.M.01188.
10. Schwarzkopf R, Phan D, Hoang M, Ross S, Mukamel D. Do patients with income-based insurance have access to total joint arthroplasty? J Arthroplasty. 2014;29(6):1083-1086. doi:10.1016/j.arth.2013.11.022.
11. Decker SL. In 2011 nearly one-third of physicians said they would not accept new Medicaid patients, but rising fees may help. Health Aff (Millwood). 2012;31(8):1673-1679 doi:10.1377/hlthaff.2012.0294.
12. Perloff JD, Kletke P, Fossett JW. Which physicians limit their Medicaid participation, and why. Health Serv Res. 1995;30(1):7-26.
13. Althausen PL. Building a successful trauma practice in a community setting. J Orthop Trauma. 2011;25 Suppl 3:S113-S117. doi:10.1097/BOT.0b013e318237bcce.
14. Greenberg S, Mir HR, Jahangir AA, Mehta S, Sethi MK. Impacting policy change for orthopaedic trauma. J Orthop Trauma. 2014;28 Suppl 10:S14-S16. doi:10.1097/BOT.0000000000000216.
15. Wiznia DH, Averbukh L, Kim CY, Goel A, Leslie MP. Motorcycle helmets: The economic burden of an incomplete helmet law to medical care in the state of Connecticut. Conn Med. 2015;79(8):453-459.
16. Orthopaedic Trauma Association. Find a surgeon. https://online.ota.org/otassa/otacenssafindasurgeon.query_page. Updated 2015. Accessed July, 2015.
17. Kim CY, Wiznia DH, Roth AS, Walls RJ, Pelker RR. Survey of patient insurance status on access to specialty foot and ankle care under the affordable care act. Foot Ankle Int. 2016;37(7):776-781. doi:1071100716642015.
18. Patterson BM, Draeger RW, Olsson EC, Spang JT, Lin FC, Kamath GV. A regional assessment of Medicaid access to outpatient orthopaedic care: the influence of population density and proximity to academic medical centers on patient access. J Bone Joint Surg Am. 2014;96(18):e156. doi:10.2106/JBJS.M.01188.
19. Kim CY, Wiznia DH, Wang Y, et al. The effect of insurance type on patient access to carpal tunnel release under the affordable care act. J Hand Surg Am. 2016;41(4):503-509.e1. doi:S0363-5023(16)00104-0.
20. Long SK. Physicians may need more than higher reimbursements to expand Medicaid participation: findings from Washington state. Health Aff (Millwood). 2013;32(9):1560-1567. doi:10.1377/hlthaff.2012.1010.
21. Issar NM, Jahangir AA. The affordable care act and orthopaedic trauma. J Orthop Trauma. 2014;28 Suppl 10:S5-S7. doi:10.1097/BOT.0000000000000211.
22. Hahn B, Flood AB. No insurance, public insurance, and private insurance: do these options contribute to differences in general health? J Health Care Poor Underserved. 1995;6(1):41-59.
23. Hinman A, Bozic KJ. Impact of payer type on resource utilization, outcomes and access to care in total hip arthroplasty. J Arthroplasty. 2008;23(6 Suppl 1):9-14. doi:10.1016/j.arth.2008.05.010.
24. Schoenfeld AJ, Tipirneni R, Nelson JH, Carpenter JE, Iwashyna TJ. The influence of race and ethnicity on complications and mortality after orthopedic surgery: A systematic review of the literature. Med Care. 2014;52(9):842-851. doi:10.1097/MLR.0000000000000177.
1. Blumenthal D, Collins SR. Health care coverage under the affordable care act--a progress report. N Engl J Med. 2014;371(3):275-281. doi:10.1056/NEJMhpr1405667.
2. Sommers BD. Health care reform's unfinished work--remaining barriers to coverage and access. N Engl J Med. 2015;373(25):2395-2397. doi:10.1056/NEJMp1509462.
3. US Department of Health and Human Services. Centers for Medicare & Medicaid Services. Medicaid & CHIP: February 2015 monthly applications, eligibility determinations and enrollment report. https://www.medicaid.gov/medicaid/program-information/downloads/medicaid-and-chip-february-2015-application-eligibility-and-enrollment-data.pdf. Published May 1, 2015. Accessed May 2015.
4. Iglehart JK, Sommers BD. Medicaid at 50--from welfare program to nation's largest health insurer. N Engl J Med. 2015;372(22):2152-2159. doi:10.1056/NEJMhpr1500791.
5. Kaiser Family Foundation. Medicaid moving forward. http://kff.org/medicaid/fact-sheet/the-medicaid-program-at-a-glance-update/. Updated 2014. Accessed October 10, 2014.
6. Kim CY, Wiznia DH, Hsiang WR, Pelker RR. The effect of insurance type on patient access to knee arthroplasty and revision under the affordable care act. J Arthroplasty. 2015;30(9):1498-1501. doi:10.1016/j.arth.2015.03.015.
7. Draeger RW, Patterson BM, Olsson EC, Schaffer A, Patterson JM. The influence of patient insurance status on access to outpatient orthopedic care for flexor tendon lacerations. J Hand Surg Am. 2014;39(3):527-533. doi:10.1016/j.jhsa.2013.10.031.
8. Patterson BM, Spang JT, Draeger RW, Olsson EC, Creighton RA, Kamath GV. Access to outpatient care for adult rotator cuff patients with private insurance versus Medicaid in North Carolina. J Shoulder Elbow Surg. 2013;22(12):1623-1627. doi:10.1016/j.jse.2013.07.051.
9. Patterson BM, Draeger RW, Olsson EC, Spang JT, Lin FC, Kamath GV. A regional assessment of medicaid access to outpatient orthopaedic care: the influence of population density and proximity to academic medical centers on patient access. J Bone Joint Surg Am. 2014;96(18):e156. doi:10.2106/JBJS.M.01188.
10. Schwarzkopf R, Phan D, Hoang M, Ross S, Mukamel D. Do patients with income-based insurance have access to total joint arthroplasty? J Arthroplasty. 2014;29(6):1083-1086. doi:10.1016/j.arth.2013.11.022.
11. Decker SL. In 2011 nearly one-third of physicians said they would not accept new Medicaid patients, but rising fees may help. Health Aff (Millwood). 2012;31(8):1673-1679 doi:10.1377/hlthaff.2012.0294.
12. Perloff JD, Kletke P, Fossett JW. Which physicians limit their Medicaid participation, and why. Health Serv Res. 1995;30(1):7-26.
13. Althausen PL. Building a successful trauma practice in a community setting. J Orthop Trauma. 2011;25 Suppl 3:S113-S117. doi:10.1097/BOT.0b013e318237bcce.
14. Greenberg S, Mir HR, Jahangir AA, Mehta S, Sethi MK. Impacting policy change for orthopaedic trauma. J Orthop Trauma. 2014;28 Suppl 10:S14-S16. doi:10.1097/BOT.0000000000000216.
15. Wiznia DH, Averbukh L, Kim CY, Goel A, Leslie MP. Motorcycle helmets: The economic burden of an incomplete helmet law to medical care in the state of Connecticut. Conn Med. 2015;79(8):453-459.
16. Orthopaedic Trauma Association. Find a surgeon. https://online.ota.org/otassa/otacenssafindasurgeon.query_page. Updated 2015. Accessed July, 2015.
17. Kim CY, Wiznia DH, Roth AS, Walls RJ, Pelker RR. Survey of patient insurance status on access to specialty foot and ankle care under the affordable care act. Foot Ankle Int. 2016;37(7):776-781. doi:1071100716642015.
18. Patterson BM, Draeger RW, Olsson EC, Spang JT, Lin FC, Kamath GV. A regional assessment of Medicaid access to outpatient orthopaedic care: the influence of population density and proximity to academic medical centers on patient access. J Bone Joint Surg Am. 2014;96(18):e156. doi:10.2106/JBJS.M.01188.
19. Kim CY, Wiznia DH, Wang Y, et al. The effect of insurance type on patient access to carpal tunnel release under the affordable care act. J Hand Surg Am. 2016;41(4):503-509.e1. doi:S0363-5023(16)00104-0.
20. Long SK. Physicians may need more than higher reimbursements to expand Medicaid participation: findings from Washington state. Health Aff (Millwood). 2013;32(9):1560-1567. doi:10.1377/hlthaff.2012.1010.
21. Issar NM, Jahangir AA. The affordable care act and orthopaedic trauma. J Orthop Trauma. 2014;28 Suppl 10:S5-S7. doi:10.1097/BOT.0000000000000211.
22. Hahn B, Flood AB. No insurance, public insurance, and private insurance: do these options contribute to differences in general health? J Health Care Poor Underserved. 1995;6(1):41-59.
23. Hinman A, Bozic KJ. Impact of payer type on resource utilization, outcomes and access to care in total hip arthroplasty. J Arthroplasty. 2008;23(6 Suppl 1):9-14. doi:10.1016/j.arth.2008.05.010.
24. Schoenfeld AJ, Tipirneni R, Nelson JH, Carpenter JE, Iwashyna TJ. The influence of race and ethnicity on complications and mortality after orthopedic surgery: A systematic review of the literature. Med Care. 2014;52(9):842-851. doi:10.1097/MLR.0000000000000177.
TAKE-HOME POINTS
- One method in which the PPACA increased the number of individuals with health insurance coverage was by expanding Medicaid eligibility requirements.
- Despite this, Medicaid patients confronted more barriers to accessing care.
- The overall rate of successfully being offered an appointment with Medicaid was 35.7%, 81.4% for Medicare, and 88.6% for BlueCross. Patients with Medicaid also confronted longer appointment wait times.
- The disparity in access for this operative trauma scenario suggests that patients with Medicaid are likely to be excluded from the practice of their choice and may need to make considerably more effort to secure an appointment.
- Ultimately, Medicaid patients may have access to care through federally funded community health centers and public and non-profit safety net hospitals, which generally care for more uninsured and Medicaid patient populations.
When treating the lower face and neck, ‘don’t forget the platysma muscle’
SAN DIEGO – Of all the data points featured in a 2017 survey on cosmetic dermatologic procedures, one stands out to Jean Carruthers, MD: Among the 7,322 consumers surveyed, about 70% cited the lower face and submental contour as a significant cosmetic and social concern.

“That is quite a key statistic,” she said at the annual Masters of Aesthetics Symposium. The Consumer Survey on Cosmetic Dermatologic Procedures was conducted by the American Society for Dermatologic Surgery.
Dr. Carruthers, who, with her husband, Alastair Carruthers, MD, pioneered the cosmetic use of onabotulinumtoxinA (Botox), said that the world’s great sculptors “think that there is a difference between the classical faces of males and females, and we all know instinctually what those changes are, with a square face and larger jawline in males and a smooth oval, heart-shaped face in women,” she said.
“But what about the neck?” She cited an article by Greg J. Goodman, MD, and colleagues, which defined an ideal neck as the distinct inferior mandibular border from mentum to angle with no jowl overhang (Dermatol Surg 2016;42:S260-2). It includes subhyoid depression, which visually enhances the impression that the neck is thin and long; visible thyroid cartilage bulge; a visible anterior border of the sternocleidomastoid muscle, distinct in its entire course from the mastoid to the sternum; and a cervicomental angle between 105 and 120 degrees. An angle greater than 120 degrees appears as a double chin or heavy neck, according to the authors.
In the past, clinicians used to think of the lower face and neck as two separate cosmetic units, but now they are considered one cosmetic unit, said Dr. Carruthers, of the University of British Columbia, Vancouver. “Don’t forget the platysma muscle,” she added. “The platysma is a lower facial muscle of expression and it affects all the other muscles. It interdigitates with the depressor anguli oris, with orbicularis oris, and depressor labii inferioris muscles, and it goes backwards into the masseter muscle.”
She and two Brazilian investigators published a retrospective analysis of 161 patients treated by a Botox injection pattern encompassing the facial platysma components, aiming to block the lower face as a whole complex (Dermatol Surg. 2017;43[8]:1042-9). According to the article, results included “frontal and lateral enhancement of lower facial contour, relaxation of high horizontal lines located just below the lateral mandibular border, and lower deep vertical smile lines present lateral to the oral commissures and melomental folds.”
Fillers and tightening the face and neck envelope without surgery takes maintenance, Dr. Carruthers said. She also noted that deoxycholic acid works on jowls as well as submental fat. Noninvasive combinations for lower face and neck include neuromodulators and deep and superficial fillers, cryolipolysis and deoxycholic acid, energy-based devices, and microneedling. “I think that these are fantastic combinations, and they don’t have to all be done at the same time on the same day,” Dr. Carruthers said. “I’m very impressed with cryolipolysis as the unheralded skin tightener. I looked at our first 464 patients and saw tremendous skin tightening.”
Of all the injectable products on the market, she said that neuromodulators “have set a new gold standard for all aesthetic treatments. It’s the most powerful primary aesthetic modulator and enhances the result of everything else that you do.”
Dr. Carruthers disclosed that she is a consultant to and has received research support from Allergan, Alphaeon, Bonti, Merz, Revance, and Zeltiq.
SAN DIEGO – Of all the data points featured in a 2017 survey on cosmetic dermatologic procedures, one stands out to Jean Carruthers, MD: Among the 7,322 consumers surveyed, about 70% cited the lower face and submental contour as a significant cosmetic and social concern.
“That is quite a key statistic,” she said at the annual Masters of Aesthetics Symposium. The Consumer Survey on Cosmetic Dermatologic Procedures was conducted by the American Society for Dermatologic Surgery.
Dr. Carruthers, who, with her husband, Alastair Carruthers, MD, pioneered the cosmetic use of onabotulinumtoxinA (Botox), said that the world’s great sculptors “think that there is a difference between the classical faces of males and females, and we all know instinctually what those changes are, with a square face and larger jawline in males and a smooth oval, heart-shaped face in women,” she said.
“But what about the neck?” She cited an article by Greg J. Goodman, MD, and colleagues, which defined an ideal neck as the distinct inferior mandibular border from mentum to angle with no jowl overhang (Dermatol Surg 2016;42:S260-2). It includes subhyoid depression, which visually enhances the impression that the neck is thin and long; visible thyroid cartilage bulge; a visible anterior border of the sternocleidomastoid muscle, distinct in its entire course from the mastoid to the sternum; and a cervicomental angle between 105 and 120 degrees. An angle greater than 120 degrees appears as a double chin or heavy neck, according to the authors.
In the past, clinicians used to think of the lower face and neck as two separate cosmetic units, but now they are considered one cosmetic unit, said Dr. Carruthers, of the University of British Columbia, Vancouver. “Don’t forget the platysma muscle,” she added. “The platysma is a lower facial muscle of expression and it affects all the other muscles. It interdigitates with the depressor anguli oris, with orbicularis oris, and depressor labii inferioris muscles, and it goes backwards into the masseter muscle.”
She and two Brazilian investigators published a retrospective analysis of 161 patients treated by a Botox injection pattern encompassing the facial platysma components, aiming to block the lower face as a whole complex (Dermatol Surg. 2017;43[8]:1042-9). According to the article, results included “frontal and lateral enhancement of lower facial contour, relaxation of high horizontal lines located just below the lateral mandibular border, and lower deep vertical smile lines present lateral to the oral commissures and melomental folds.”
Fillers and tightening the face and neck envelope without surgery takes maintenance, Dr. Carruthers said. She also noted that deoxycholic acid works on jowls as well as submental fat. Noninvasive combinations for lower face and neck include neuromodulators and deep and superficial fillers, cryolipolysis and deoxycholic acid, energy-based devices, and microneedling. “I think that these are fantastic combinations, and they don’t have to all be done at the same time on the same day,” Dr. Carruthers said. “I’m very impressed with cryolipolysis as the unheralded skin tightener. I looked at our first 464 patients and saw tremendous skin tightening.”
Of all the injectable products on the market, she said that neuromodulators “have set a new gold standard for all aesthetic treatments. It’s the most powerful primary aesthetic modulator and enhances the result of everything else that you do.”
Dr. Carruthers disclosed that she is a consultant to and has received research support from Allergan, Alphaeon, Bonti, Merz, Revance, and Zeltiq.
SAN DIEGO – Of all the data points featured in a 2017 survey on cosmetic dermatologic procedures, one stands out to Jean Carruthers, MD: Among the 7,322 consumers surveyed, about 70% cited the lower face and submental contour as a significant cosmetic and social concern.
“That is quite a key statistic,” she said at the annual Masters of Aesthetics Symposium. The Consumer Survey on Cosmetic Dermatologic Procedures was conducted by the American Society for Dermatologic Surgery.
Dr. Carruthers, who, with her husband, Alastair Carruthers, MD, pioneered the cosmetic use of onabotulinumtoxinA (Botox), said that the world’s great sculptors “think that there is a difference between the classical faces of males and females, and we all know instinctually what those changes are, with a square face and larger jawline in males and a smooth oval, heart-shaped face in women,” she said.
“But what about the neck?” She cited an article by Greg J. Goodman, MD, and colleagues, which defined an ideal neck as the distinct inferior mandibular border from mentum to angle with no jowl overhang (Dermatol Surg 2016;42:S260-2). It includes subhyoid depression, which visually enhances the impression that the neck is thin and long; visible thyroid cartilage bulge; a visible anterior border of the sternocleidomastoid muscle, distinct in its entire course from the mastoid to the sternum; and a cervicomental angle between 105 and 120 degrees. An angle greater than 120 degrees appears as a double chin or heavy neck, according to the authors.
In the past, clinicians used to think of the lower face and neck as two separate cosmetic units, but now they are considered one cosmetic unit, said Dr. Carruthers, of the University of British Columbia, Vancouver. “Don’t forget the platysma muscle,” she added. “The platysma is a lower facial muscle of expression and it affects all the other muscles. It interdigitates with the depressor anguli oris, with orbicularis oris, and depressor labii inferioris muscles, and it goes backwards into the masseter muscle.”
She and two Brazilian investigators published a retrospective analysis of 161 patients treated by a Botox injection pattern encompassing the facial platysma components, aiming to block the lower face as a whole complex (Dermatol Surg. 2017;43[8]:1042-9). According to the article, results included “frontal and lateral enhancement of lower facial contour, relaxation of high horizontal lines located just below the lateral mandibular border, and lower deep vertical smile lines present lateral to the oral commissures and melomental folds.”
Fillers and tightening the face and neck envelope without surgery takes maintenance, Dr. Carruthers said. She also noted that deoxycholic acid works on jowls as well as submental fat. Noninvasive combinations for lower face and neck include neuromodulators and deep and superficial fillers, cryolipolysis and deoxycholic acid, energy-based devices, and microneedling. “I think that these are fantastic combinations, and they don’t have to all be done at the same time on the same day,” Dr. Carruthers said. “I’m very impressed with cryolipolysis as the unheralded skin tightener. I looked at our first 464 patients and saw tremendous skin tightening.”
Of all the injectable products on the market, she said that neuromodulators “have set a new gold standard for all aesthetic treatments. It’s the most powerful primary aesthetic modulator and enhances the result of everything else that you do.”
Dr. Carruthers disclosed that she is a consultant to and has received research support from Allergan, Alphaeon, Bonti, Merz, Revance, and Zeltiq.
AT MOAS 2018
Multiday seizure cycles may be very common
Multiday epileptic seizure cycles may occur in a substantial number of individuals with epilepsy, results of a retrospective cohort study suggest.

While about 80% of patients in the study showed circadian modulation of their seizure rates, a substantial portion showed strong circaseptan (7 day) rhythms, according to the study’s senior author, Mark J. Cook, MD, of the department of medicine at St. Vincent’s Hospital, Melbourne.
Significant circaseptan cycles were seen in more than 20% of patients in one analysis from the study, which Dr. Cook and his colleagues reported in the Lancet Neurology.
The high prevalence of multiday seizure cycles could present an opportunity to improve treatment through development of patient-specific chronotherapy or the timing of medication to match when seizures would be most likely. “Even without fully understanding the mechanisms of seizure cycles, temporal patterns can be incorporated into patient management plans,” Dr. Cook said in a news release.
The study by Dr. Cook and his colleagues was based on two seizure datasets. One was a U.S. cohort of 1,118 patients who self-reported at least 100 episodes through the SeizureTracker website or mobile app. The other was an Australian cohort of 12 patients with focal epilepsy who had at least 30 recorded seizures in a study of an implanted electrocorticography device that tracked them between 6 months and 3 years.
In the SeizureTracker data, 86% of participants had at least one significant cycle in their seizure times, and 64% had more than one cycle. Most of the cycles (80%) were circadian, while 21% of people had significant 7-day cycles in one analysis using the Hodges-Ajne test, a statistical method used to assess circular data.
“Many patients also showed some evidence of cycles lasting up to a month,” wrote Dr. Cook and his coauthors.
A confirmatory analysis using Monte Carlo simulation found that 7% of people, or 77 individuals, had significant 7-day cycles. “The probability that 77 patients would randomly share a specific cycle [such as a 7-day cycle] is infinitesimal,” the authors wrote.
In the implantable device study, 11 out of 12 patients had strong rhythms at 24 hours, according to the investigators, while 1 had a significant cycle of exactly 1 week, and 2 others had cycles of approximately 1 week.
“Some people had stronger rhythms at time scales longer than 24 hours, which suggests that circadian regulation was not necessarily the strongest modulating factor of epileptic activity,” the investigators wrote.
The cause of longer seizure cycles remains unclear, according to the investigators, though peak seizure times might be linked to behavioral changes such as varying stress levels over the course of the week, seasonal changes in sleep quality, or biologic drivers such as menstruation.
“A better understanding of seizure cycles might provide new targets for treatment,” they wrote.
Funding for the study came from the Australian National Health and Medical Research Council. Dr. Cook declared no competing interests, while his coauthors reported support from sources outside of this study. One study author is a cofounder of SeizureTracker.com.
SOURCE: Cook MJ et al. Lancet Neurol. 2018 Sep 12. doi: 10.1016/S1474-4422(18)30274-6.
It is “remarkable” that there were such long-lasting cycling alterations in seizure propensity over the course of several weeks in this study, Andreas Schulze-Bonhage, PhD, wrote in a published commentary.
“Ultraslow oscillations in brain excitability are scarcely understood,” he wrote in The Lancet Neurology, adding that emerging evidence suggests slow cycles below well-known circadian rhythms influence both physiological functioning and disease states.
Of note, the study showed that longer cycle durations of several weeks were not limited to women, in whom such cycles might reflect monthly hormonal changes, Dr. Schulze-Bonhage wrote.
The authors of the study acknowledged the inherent limitations of analyzing patient-reported seizure information and long-term assessment of electrocorticography recordings. Some of those limitations might be overcome with technical advances, including seizure trackers that can be worn by large cohorts of patients, according to Dr. Schulze-Bonhage.
“The approach of generating hypotheses in small patient cohorts assessed with high-quality methods and testing them in much larger patient samples from whom only behavioral data can be obtained can be considered seminal for future investigations on big data,” he wrote.
Dr. Schulze-Bonhage is with the Epilepsy Center at University Medical Center, University of Freiburg (Germany). He reported no competing interests related to his editorial (Lancet Neurol. 2018 Sep 12. doi: 10.1016/S1474-4422(18)30337-5 ).
It is “remarkable” that there were such long-lasting cycling alterations in seizure propensity over the course of several weeks in this study, Andreas Schulze-Bonhage, PhD, wrote in a published commentary.
“Ultraslow oscillations in brain excitability are scarcely understood,” he wrote in The Lancet Neurology, adding that emerging evidence suggests slow cycles below well-known circadian rhythms influence both physiological functioning and disease states.
Of note, the study showed that longer cycle durations of several weeks were not limited to women, in whom such cycles might reflect monthly hormonal changes, Dr. Schulze-Bonhage wrote.
The authors of the study acknowledged the inherent limitations of analyzing patient-reported seizure information and long-term assessment of electrocorticography recordings. Some of those limitations might be overcome with technical advances, including seizure trackers that can be worn by large cohorts of patients, according to Dr. Schulze-Bonhage.
“The approach of generating hypotheses in small patient cohorts assessed with high-quality methods and testing them in much larger patient samples from whom only behavioral data can be obtained can be considered seminal for future investigations on big data,” he wrote.
Dr. Schulze-Bonhage is with the Epilepsy Center at University Medical Center, University of Freiburg (Germany). He reported no competing interests related to his editorial (Lancet Neurol. 2018 Sep 12. doi: 10.1016/S1474-4422(18)30337-5 ).
It is “remarkable” that there were such long-lasting cycling alterations in seizure propensity over the course of several weeks in this study, Andreas Schulze-Bonhage, PhD, wrote in a published commentary.
“Ultraslow oscillations in brain excitability are scarcely understood,” he wrote in The Lancet Neurology, adding that emerging evidence suggests slow cycles below well-known circadian rhythms influence both physiological functioning and disease states.
Of note, the study showed that longer cycle durations of several weeks were not limited to women, in whom such cycles might reflect monthly hormonal changes, Dr. Schulze-Bonhage wrote.
The authors of the study acknowledged the inherent limitations of analyzing patient-reported seizure information and long-term assessment of electrocorticography recordings. Some of those limitations might be overcome with technical advances, including seizure trackers that can be worn by large cohorts of patients, according to Dr. Schulze-Bonhage.
“The approach of generating hypotheses in small patient cohorts assessed with high-quality methods and testing them in much larger patient samples from whom only behavioral data can be obtained can be considered seminal for future investigations on big data,” he wrote.
Dr. Schulze-Bonhage is with the Epilepsy Center at University Medical Center, University of Freiburg (Germany). He reported no competing interests related to his editorial (Lancet Neurol. 2018 Sep 12. doi: 10.1016/S1474-4422(18)30337-5 ).
Multiday epileptic seizure cycles may occur in a substantial number of individuals with epilepsy, results of a retrospective cohort study suggest.
While about 80% of patients in the study showed circadian modulation of their seizure rates, a substantial portion showed strong circaseptan (7 day) rhythms, according to the study’s senior author, Mark J. Cook, MD, of the department of medicine at St. Vincent’s Hospital, Melbourne.
Significant circaseptan cycles were seen in more than 20% of patients in one analysis from the study, which Dr. Cook and his colleagues reported in the Lancet Neurology.
The high prevalence of multiday seizure cycles could present an opportunity to improve treatment through development of patient-specific chronotherapy or the timing of medication to match when seizures would be most likely. “Even without fully understanding the mechanisms of seizure cycles, temporal patterns can be incorporated into patient management plans,” Dr. Cook said in a news release.
The study by Dr. Cook and his colleagues was based on two seizure datasets. One was a U.S. cohort of 1,118 patients who self-reported at least 100 episodes through the SeizureTracker website or mobile app. The other was an Australian cohort of 12 patients with focal epilepsy who had at least 30 recorded seizures in a study of an implanted electrocorticography device that tracked them between 6 months and 3 years.
In the SeizureTracker data, 86% of participants had at least one significant cycle in their seizure times, and 64% had more than one cycle. Most of the cycles (80%) were circadian, while 21% of people had significant 7-day cycles in one analysis using the Hodges-Ajne test, a statistical method used to assess circular data.
“Many patients also showed some evidence of cycles lasting up to a month,” wrote Dr. Cook and his coauthors.
A confirmatory analysis using Monte Carlo simulation found that 7% of people, or 77 individuals, had significant 7-day cycles. “The probability that 77 patients would randomly share a specific cycle [such as a 7-day cycle] is infinitesimal,” the authors wrote.
In the implantable device study, 11 out of 12 patients had strong rhythms at 24 hours, according to the investigators, while 1 had a significant cycle of exactly 1 week, and 2 others had cycles of approximately 1 week.
“Some people had stronger rhythms at time scales longer than 24 hours, which suggests that circadian regulation was not necessarily the strongest modulating factor of epileptic activity,” the investigators wrote.
The cause of longer seizure cycles remains unclear, according to the investigators, though peak seizure times might be linked to behavioral changes such as varying stress levels over the course of the week, seasonal changes in sleep quality, or biologic drivers such as menstruation.
“A better understanding of seizure cycles might provide new targets for treatment,” they wrote.
Funding for the study came from the Australian National Health and Medical Research Council. Dr. Cook declared no competing interests, while his coauthors reported support from sources outside of this study. One study author is a cofounder of SeizureTracker.com.
SOURCE: Cook MJ et al. Lancet Neurol. 2018 Sep 12. doi: 10.1016/S1474-4422(18)30274-6.
Multiday epileptic seizure cycles may occur in a substantial number of individuals with epilepsy, results of a retrospective cohort study suggest.
While about 80% of patients in the study showed circadian modulation of their seizure rates, a substantial portion showed strong circaseptan (7 day) rhythms, according to the study’s senior author, Mark J. Cook, MD, of the department of medicine at St. Vincent’s Hospital, Melbourne.
Significant circaseptan cycles were seen in more than 20% of patients in one analysis from the study, which Dr. Cook and his colleagues reported in the Lancet Neurology.
The high prevalence of multiday seizure cycles could present an opportunity to improve treatment through development of patient-specific chronotherapy or the timing of medication to match when seizures would be most likely. “Even without fully understanding the mechanisms of seizure cycles, temporal patterns can be incorporated into patient management plans,” Dr. Cook said in a news release.
The study by Dr. Cook and his colleagues was based on two seizure datasets. One was a U.S. cohort of 1,118 patients who self-reported at least 100 episodes through the SeizureTracker website or mobile app. The other was an Australian cohort of 12 patients with focal epilepsy who had at least 30 recorded seizures in a study of an implanted electrocorticography device that tracked them between 6 months and 3 years.
In the SeizureTracker data, 86% of participants had at least one significant cycle in their seizure times, and 64% had more than one cycle. Most of the cycles (80%) were circadian, while 21% of people had significant 7-day cycles in one analysis using the Hodges-Ajne test, a statistical method used to assess circular data.
“Many patients also showed some evidence of cycles lasting up to a month,” wrote Dr. Cook and his coauthors.
A confirmatory analysis using Monte Carlo simulation found that 7% of people, or 77 individuals, had significant 7-day cycles. “The probability that 77 patients would randomly share a specific cycle [such as a 7-day cycle] is infinitesimal,” the authors wrote.
In the implantable device study, 11 out of 12 patients had strong rhythms at 24 hours, according to the investigators, while 1 had a significant cycle of exactly 1 week, and 2 others had cycles of approximately 1 week.
“Some people had stronger rhythms at time scales longer than 24 hours, which suggests that circadian regulation was not necessarily the strongest modulating factor of epileptic activity,” the investigators wrote.
The cause of longer seizure cycles remains unclear, according to the investigators, though peak seizure times might be linked to behavioral changes such as varying stress levels over the course of the week, seasonal changes in sleep quality, or biologic drivers such as menstruation.
“A better understanding of seizure cycles might provide new targets for treatment,” they wrote.
Funding for the study came from the Australian National Health and Medical Research Council. Dr. Cook declared no competing interests, while his coauthors reported support from sources outside of this study. One study author is a cofounder of SeizureTracker.com.
SOURCE: Cook MJ et al. Lancet Neurol. 2018 Sep 12. doi: 10.1016/S1474-4422(18)30274-6.
FROM THE LANCET NEUROLOGY
Key clinical point:
Major finding: Significant 7-day cycles were seen in more than 20% of patients in one analysis from this study.
Study details: A retrospective cohort analysis including 1,118 patients who self-reported episodes online and 12 patients who participated in an electrocorticography device study.
Disclosures: Funding for the study came from the Australian National Health and Medical Research Council. Dr. Cook declared no competing interests, while coauthors reported support from sources outside of this study. One study author is a cofounder of SeizureTracker.com.
Source: Cook MJ et al. Lancet Neurol. 2018 Sep 12. doi: 10.1016/S1474-4422(18)30274-6.
Terra Firma-Forme Dermatosis Mimicking Livedo Racemosa
To the Editor:
A 17-year-old adolescent boy presented with dark spots on the legs and back of 2 months’ duration. He was not taking any medications and the spots could not be washed away by scrubbing with soap and water. He denied symptoms, except occasional itching. Family history revealed a maternal uncle with protein C deficiency and a maternal grandmother with systemic lupus erythematosus. Review of systems was negative; the patient denied joint pain and contact with heating pads or laptop computers. Based on the initial presentation, an underlying systemic condition was suspected. Physical examination revealed reticulate, nonblanching, brown patches on the bilateral arms, legs, and back in an apparent livedoid pattern (Figure). The patient’s history and physical examination suggested terra firma-forme dermatosis, livedo racemosa, or another vasculopathic process. However, gentle rubbing of the skin with an alcohol swab removed the discoloration completely, leading to the diagnosis of terra firma-forme dermatosis.
Livedo racemosa appears as an irregular, focal, reticulated discoloration of the skin.1 The reticulated pattern of livedo racemosa has a branched or broken-up appearance.2 Livedo racemosa indicates a disruption in the vasculature due to inflammation or occlusion.1 The change is pathologic and does not blanch or resolve with warming.1,2 The condition can progress to pigmentation and ulceration.1 Livedo racemosa is a cutaneous manifestation of underlying vascular pathology. Due to a variety of causes, skin biopsy is nondiagnostic. Livedo racemosa can be caused by conditions such as systemic lupus erythematosus, syphilis, tuberculosis, polycythemia rubra vera, and Sneddon syndrome, among others.3-5
Terra firma-forme dermatosis was reported in 1987 by Duncan et al.6 The condition classically presents with an exasperated mother who is unable to clean the “dirt” off her child’s skin despite multiple vigorous scrubbing attempts. The condition most commonly occurs in the summer months on the neck, face, and ankles.7,8 Duncan et al6 reported that when the affected area was prepared for a biopsy, clean skin was revealed after wiping with an alcohol swab. No other cleansing agent has been reported to effectively remove the discoloration of terra firma-forme dermatosis. Hoping to elucidate a cause, Duncan et al6 performed both bacteriologic and fungal studies. The bacterial skin culture grew only normal flora, and fungal culture grew only normal contaminants consistent with the potassium hydroxide preparation of skin scraping. Histopathologic examination showed hyperkeratosis and orthokeratosis but not parakeratosis. Staining revealed melanin in the hyperkeratotic areas.6 Although the cause of this condition largely is unknown, it is thought that the epidermis in the affected areas could undergo altered maturation, resulting in trapping melanin that causes the skin to appear hyperkeratotic and hyperpigmented.1 In our case, wiping the skin revealed the unsuspected diagnosis of terra firma-forme dermatosis displaying an unusual pseudolivedoid pattern. With apparently hyperpigmented processes, rubbing the skin with alcohol may help avoid unnecessary aggressive workup.
- Parsi K, Partsch H, Rabe E, et al. Reticulate eruptions: part 2. historical perspectives, morphology, terminology and classification. Australas J Dermatol. 2011;52:237-244.
- Ehrmann S. A new vascular symptom in syphilis [in German]. Wien Med Wochenschr. 1907;57:777-782.
- Sneddon IB. Cerebrovascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Golden RL. Livedo reticularis in systemic lupus erythematosus. Arch Dermatol. 1963;87:299-301.
- Lyell A, Church R. The cutaneous manifestations of polyarteritis nodosa. Br J Dermatol. 1954;66:335-343.
- Duncan WC, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;23:297-300.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
To the Editor:
A 17-year-old adolescent boy presented with dark spots on the legs and back of 2 months’ duration. He was not taking any medications and the spots could not be washed away by scrubbing with soap and water. He denied symptoms, except occasional itching. Family history revealed a maternal uncle with protein C deficiency and a maternal grandmother with systemic lupus erythematosus. Review of systems was negative; the patient denied joint pain and contact with heating pads or laptop computers. Based on the initial presentation, an underlying systemic condition was suspected. Physical examination revealed reticulate, nonblanching, brown patches on the bilateral arms, legs, and back in an apparent livedoid pattern (Figure). The patient’s history and physical examination suggested terra firma-forme dermatosis, livedo racemosa, or another vasculopathic process. However, gentle rubbing of the skin with an alcohol swab removed the discoloration completely, leading to the diagnosis of terra firma-forme dermatosis.
Livedo racemosa appears as an irregular, focal, reticulated discoloration of the skin.1 The reticulated pattern of livedo racemosa has a branched or broken-up appearance.2 Livedo racemosa indicates a disruption in the vasculature due to inflammation or occlusion.1 The change is pathologic and does not blanch or resolve with warming.1,2 The condition can progress to pigmentation and ulceration.1 Livedo racemosa is a cutaneous manifestation of underlying vascular pathology. Due to a variety of causes, skin biopsy is nondiagnostic. Livedo racemosa can be caused by conditions such as systemic lupus erythematosus, syphilis, tuberculosis, polycythemia rubra vera, and Sneddon syndrome, among others.3-5
Terra firma-forme dermatosis was reported in 1987 by Duncan et al.6 The condition classically presents with an exasperated mother who is unable to clean the “dirt” off her child’s skin despite multiple vigorous scrubbing attempts. The condition most commonly occurs in the summer months on the neck, face, and ankles.7,8 Duncan et al6 reported that when the affected area was prepared for a biopsy, clean skin was revealed after wiping with an alcohol swab. No other cleansing agent has been reported to effectively remove the discoloration of terra firma-forme dermatosis. Hoping to elucidate a cause, Duncan et al6 performed both bacteriologic and fungal studies. The bacterial skin culture grew only normal flora, and fungal culture grew only normal contaminants consistent with the potassium hydroxide preparation of skin scraping. Histopathologic examination showed hyperkeratosis and orthokeratosis but not parakeratosis. Staining revealed melanin in the hyperkeratotic areas.6 Although the cause of this condition largely is unknown, it is thought that the epidermis in the affected areas could undergo altered maturation, resulting in trapping melanin that causes the skin to appear hyperkeratotic and hyperpigmented.1 In our case, wiping the skin revealed the unsuspected diagnosis of terra firma-forme dermatosis displaying an unusual pseudolivedoid pattern. With apparently hyperpigmented processes, rubbing the skin with alcohol may help avoid unnecessary aggressive workup.
To the Editor:
A 17-year-old adolescent boy presented with dark spots on the legs and back of 2 months’ duration. He was not taking any medications and the spots could not be washed away by scrubbing with soap and water. He denied symptoms, except occasional itching. Family history revealed a maternal uncle with protein C deficiency and a maternal grandmother with systemic lupus erythematosus. Review of systems was negative; the patient denied joint pain and contact with heating pads or laptop computers. Based on the initial presentation, an underlying systemic condition was suspected. Physical examination revealed reticulate, nonblanching, brown patches on the bilateral arms, legs, and back in an apparent livedoid pattern (Figure). The patient’s history and physical examination suggested terra firma-forme dermatosis, livedo racemosa, or another vasculopathic process. However, gentle rubbing of the skin with an alcohol swab removed the discoloration completely, leading to the diagnosis of terra firma-forme dermatosis.
Livedo racemosa appears as an irregular, focal, reticulated discoloration of the skin.1 The reticulated pattern of livedo racemosa has a branched or broken-up appearance.2 Livedo racemosa indicates a disruption in the vasculature due to inflammation or occlusion.1 The change is pathologic and does not blanch or resolve with warming.1,2 The condition can progress to pigmentation and ulceration.1 Livedo racemosa is a cutaneous manifestation of underlying vascular pathology. Due to a variety of causes, skin biopsy is nondiagnostic. Livedo racemosa can be caused by conditions such as systemic lupus erythematosus, syphilis, tuberculosis, polycythemia rubra vera, and Sneddon syndrome, among others.3-5
Terra firma-forme dermatosis was reported in 1987 by Duncan et al.6 The condition classically presents with an exasperated mother who is unable to clean the “dirt” off her child’s skin despite multiple vigorous scrubbing attempts. The condition most commonly occurs in the summer months on the neck, face, and ankles.7,8 Duncan et al6 reported that when the affected area was prepared for a biopsy, clean skin was revealed after wiping with an alcohol swab. No other cleansing agent has been reported to effectively remove the discoloration of terra firma-forme dermatosis. Hoping to elucidate a cause, Duncan et al6 performed both bacteriologic and fungal studies. The bacterial skin culture grew only normal flora, and fungal culture grew only normal contaminants consistent with the potassium hydroxide preparation of skin scraping. Histopathologic examination showed hyperkeratosis and orthokeratosis but not parakeratosis. Staining revealed melanin in the hyperkeratotic areas.6 Although the cause of this condition largely is unknown, it is thought that the epidermis in the affected areas could undergo altered maturation, resulting in trapping melanin that causes the skin to appear hyperkeratotic and hyperpigmented.1 In our case, wiping the skin revealed the unsuspected diagnosis of terra firma-forme dermatosis displaying an unusual pseudolivedoid pattern. With apparently hyperpigmented processes, rubbing the skin with alcohol may help avoid unnecessary aggressive workup.
- Parsi K, Partsch H, Rabe E, et al. Reticulate eruptions: part 2. historical perspectives, morphology, terminology and classification. Australas J Dermatol. 2011;52:237-244.
- Ehrmann S. A new vascular symptom in syphilis [in German]. Wien Med Wochenschr. 1907;57:777-782.
- Sneddon IB. Cerebrovascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Golden RL. Livedo reticularis in systemic lupus erythematosus. Arch Dermatol. 1963;87:299-301.
- Lyell A, Church R. The cutaneous manifestations of polyarteritis nodosa. Br J Dermatol. 1954;66:335-343.
- Duncan WC, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;23:297-300.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
- Parsi K, Partsch H, Rabe E, et al. Reticulate eruptions: part 2. historical perspectives, morphology, terminology and classification. Australas J Dermatol. 2011;52:237-244.
- Ehrmann S. A new vascular symptom in syphilis [in German]. Wien Med Wochenschr. 1907;57:777-782.
- Sneddon IB. Cerebrovascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Golden RL. Livedo reticularis in systemic lupus erythematosus. Arch Dermatol. 1963;87:299-301.
- Lyell A, Church R. The cutaneous manifestations of polyarteritis nodosa. Br J Dermatol. 1954;66:335-343.
- Duncan WC, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;23:297-300.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
Practice Points
- Clinicians should include terra firma-forme dermatosis in the differential diagnosis of any hyperpigmented condition, regardless of pattern of presentation.
- Clean the skin with an alcohol wipe to rule out a diagnosis of terra firma-forme dermatosis.
Brown spot on ear

The FP explained to the patient that this could be a skin cancer—specifically, a melanoma.
The FP performed a broad shave biopsy, being careful not to cut into the cartilage. (See the Watch & Learn video on “Shave biopsy.”) The FP did his best to include most of the pigmented area involved, but the convex surface made it difficult to biopsy the whole lesion. He was especially careful to include the darker area because it looked most atypical. The diagnosis came back as lentigo maligna.
The patient was referred for Mohs surgery for complete excision and repair. (Mohs surgery is recommended to spare tissue and maximize cure.) After complete excision, the patient learned that the melanoma was not invasive, but in situ. This suggested a very good prognosis.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Mayeaux, EJ, Usatine, R. Lentigo maligna. In: Usatine R, Smith M, Mayeaux EJ, et al. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:981-984.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/.
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com.
The FP explained to the patient that this could be a skin cancer—specifically, a melanoma.
The FP performed a broad shave biopsy, being careful not to cut into the cartilage. (See the Watch & Learn video on “Shave biopsy.”) The FP did his best to include most of the pigmented area involved, but the convex surface made it difficult to biopsy the whole lesion. He was especially careful to include the darker area because it looked most atypical. The diagnosis came back as lentigo maligna.
The patient was referred for Mohs surgery for complete excision and repair. (Mohs surgery is recommended to spare tissue and maximize cure.) After complete excision, the patient learned that the melanoma was not invasive, but in situ. This suggested a very good prognosis.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Mayeaux, EJ, Usatine, R. Lentigo maligna. In: Usatine R, Smith M, Mayeaux EJ, et al. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:981-984.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/.
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com.
The FP explained to the patient that this could be a skin cancer—specifically, a melanoma.
The FP performed a broad shave biopsy, being careful not to cut into the cartilage. (See the Watch & Learn video on “Shave biopsy.”) The FP did his best to include most of the pigmented area involved, but the convex surface made it difficult to biopsy the whole lesion. He was especially careful to include the darker area because it looked most atypical. The diagnosis came back as lentigo maligna.
The patient was referred for Mohs surgery for complete excision and repair. (Mohs surgery is recommended to spare tissue and maximize cure.) After complete excision, the patient learned that the melanoma was not invasive, but in situ. This suggested a very good prognosis.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Mayeaux, EJ, Usatine, R. Lentigo maligna. In: Usatine R, Smith M, Mayeaux EJ, et al. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:981-984.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/.
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com.

Insights could change treatment, classification of MPAL

An extensive analysis of mixed phenotype acute leukemia (MPAL) has led to new insights that may have implications for disease classification and treatment.
Researchers believe they have identified new subtypes of MPAL that should be included in the World Health Organization (WHO) classification for acute leukemia.
Each of these subtypes shares genomic characteristics with other acute leukemias, which suggests the new subtypes might respond to treatments that are already in use.
This research has also shed light on how MPAL evolves and appears to provide an explanation for why MPAL displays characteristics of both acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL).
“ALL and AML have very different treatments, but MPAL has features of both, so the question of how best to treat patients with MPAL has been challenging the leukemia community worldwide, and long-term survival of patients has been poor,” said Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
With these issues in mind, Dr. Mullighan and his colleagues conducted their study of MPAL and described their findings in Nature.
New classifications
The researchers used whole-genome, whole-exome, and RNA sequencing to analyze 115 samples from pediatric patients with MPAL.
The analysis revealed mutations that define the two most common subtypes of MPAL—B/myeloid and T/myeloid—and suggested these subtypes share similarities with other leukemia subtypes.
The researchers found that 48% of B/myeloid MPAL cases carried rearrangements in ZNF384, a characteristic that is also found in cases of B-cell ALL. In fact, the team said the gene expression profiles of ZNF384r B-ALL and ZNF384r MPAL were indistinguishable.
“That is biologically and clinically important,” Dr. Mullighan said. “The findings suggest the ZNF384 rearrangement defines a distinct leukemia subtype, and the alteration should be used to guide treatment.”
The researchers noted that patients with ZNF384r exhibited higher FLT3 expression than patients with other types of B/myeloid or T/myeloid MPAL, so patients with ZNF384r MPAL might respond well to treatment with a FLT3 inhibitor.
This study also showed that cases of B/myeloid MPAL without ZNF384r shared genomic features with other B-ALL subtypes, such as Ph-like B-ALL, which may have implications for treatment.
Another of the researchers’ discoveries was that T/myeloid MPAL and early T-cell precursor ALL have similar gene expression profiles.
The team identified several genes that were mutated at similar frequencies in T/myeloid MPAL and early T-cell precursor ALL, including WT1, ETV6, EZH2, and FLT3. WT1 was the most frequently mutated transcription factor gene in T/myeloid MPAL.
Based on these findings, the researchers said the WHO classification of acute leukemia should be updated to include:
- ZNF384r acute leukemia (either B-ALL or MPAL)
- WT1-mutant T/myeloid MPAL
- Ph-like B/myeloid MPAL.
Evolution of MPAL
The researchers’ analyses also revealed leukemia-initiating genetic alterations in early hematopoietic progenitors.
The team said this and other findings—including the common genomic features of ZNF384r MPAL and B-ALL—suggest the ambiguous phenotype of MPAL results from alterations in immature hematopoietic progenitors.
“These findings suggest that the founding mutation occurs early in blood cell development, in some cases in hematopoietic stem cells, and results in an acute leukemia with features of both myeloid and lymphoid cells,” said study author Thomas Alexander, MD, of the University of North Carolina at Chapel Hill.
“One previous theory was that the reason you have two different cancer types within the same patient is that they acquire different mutations that drive them to become AML or ALL, with genomically distinct tumors within the same patient. That doesn’t seem to be the case from our data. Our proposed model is that the mutations occur earlier in development in cells that retain the potential to acquire myeloid or lymphoid features.”
This research was supported by the National Cancer Institute, the National Institutes of Health, Cookies for Kids’ Cancer, and other organizations.
An extensive analysis of mixed phenotype acute leukemia (MPAL) has led to new insights that may have implications for disease classification and treatment.
Researchers believe they have identified new subtypes of MPAL that should be included in the World Health Organization (WHO) classification for acute leukemia.
Each of these subtypes shares genomic characteristics with other acute leukemias, which suggests the new subtypes might respond to treatments that are already in use.
This research has also shed light on how MPAL evolves and appears to provide an explanation for why MPAL displays characteristics of both acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL).
“ALL and AML have very different treatments, but MPAL has features of both, so the question of how best to treat patients with MPAL has been challenging the leukemia community worldwide, and long-term survival of patients has been poor,” said Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
With these issues in mind, Dr. Mullighan and his colleagues conducted their study of MPAL and described their findings in Nature.
New classifications
The researchers used whole-genome, whole-exome, and RNA sequencing to analyze 115 samples from pediatric patients with MPAL.
The analysis revealed mutations that define the two most common subtypes of MPAL—B/myeloid and T/myeloid—and suggested these subtypes share similarities with other leukemia subtypes.
The researchers found that 48% of B/myeloid MPAL cases carried rearrangements in ZNF384, a characteristic that is also found in cases of B-cell ALL. In fact, the team said the gene expression profiles of ZNF384r B-ALL and ZNF384r MPAL were indistinguishable.
“That is biologically and clinically important,” Dr. Mullighan said. “The findings suggest the ZNF384 rearrangement defines a distinct leukemia subtype, and the alteration should be used to guide treatment.”
The researchers noted that patients with ZNF384r exhibited higher FLT3 expression than patients with other types of B/myeloid or T/myeloid MPAL, so patients with ZNF384r MPAL might respond well to treatment with a FLT3 inhibitor.
This study also showed that cases of B/myeloid MPAL without ZNF384r shared genomic features with other B-ALL subtypes, such as Ph-like B-ALL, which may have implications for treatment.
Another of the researchers’ discoveries was that T/myeloid MPAL and early T-cell precursor ALL have similar gene expression profiles.
The team identified several genes that were mutated at similar frequencies in T/myeloid MPAL and early T-cell precursor ALL, including WT1, ETV6, EZH2, and FLT3. WT1 was the most frequently mutated transcription factor gene in T/myeloid MPAL.
Based on these findings, the researchers said the WHO classification of acute leukemia should be updated to include:
- ZNF384r acute leukemia (either B-ALL or MPAL)
- WT1-mutant T/myeloid MPAL
- Ph-like B/myeloid MPAL.
Evolution of MPAL
The researchers’ analyses also revealed leukemia-initiating genetic alterations in early hematopoietic progenitors.
The team said this and other findings—including the common genomic features of ZNF384r MPAL and B-ALL—suggest the ambiguous phenotype of MPAL results from alterations in immature hematopoietic progenitors.
“These findings suggest that the founding mutation occurs early in blood cell development, in some cases in hematopoietic stem cells, and results in an acute leukemia with features of both myeloid and lymphoid cells,” said study author Thomas Alexander, MD, of the University of North Carolina at Chapel Hill.
“One previous theory was that the reason you have two different cancer types within the same patient is that they acquire different mutations that drive them to become AML or ALL, with genomically distinct tumors within the same patient. That doesn’t seem to be the case from our data. Our proposed model is that the mutations occur earlier in development in cells that retain the potential to acquire myeloid or lymphoid features.”
This research was supported by the National Cancer Institute, the National Institutes of Health, Cookies for Kids’ Cancer, and other organizations.
An extensive analysis of mixed phenotype acute leukemia (MPAL) has led to new insights that may have implications for disease classification and treatment.
Researchers believe they have identified new subtypes of MPAL that should be included in the World Health Organization (WHO) classification for acute leukemia.
Each of these subtypes shares genomic characteristics with other acute leukemias, which suggests the new subtypes might respond to treatments that are already in use.
This research has also shed light on how MPAL evolves and appears to provide an explanation for why MPAL displays characteristics of both acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL).
“ALL and AML have very different treatments, but MPAL has features of both, so the question of how best to treat patients with MPAL has been challenging the leukemia community worldwide, and long-term survival of patients has been poor,” said Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
With these issues in mind, Dr. Mullighan and his colleagues conducted their study of MPAL and described their findings in Nature.
New classifications
The researchers used whole-genome, whole-exome, and RNA sequencing to analyze 115 samples from pediatric patients with MPAL.
The analysis revealed mutations that define the two most common subtypes of MPAL—B/myeloid and T/myeloid—and suggested these subtypes share similarities with other leukemia subtypes.
The researchers found that 48% of B/myeloid MPAL cases carried rearrangements in ZNF384, a characteristic that is also found in cases of B-cell ALL. In fact, the team said the gene expression profiles of ZNF384r B-ALL and ZNF384r MPAL were indistinguishable.
“That is biologically and clinically important,” Dr. Mullighan said. “The findings suggest the ZNF384 rearrangement defines a distinct leukemia subtype, and the alteration should be used to guide treatment.”
The researchers noted that patients with ZNF384r exhibited higher FLT3 expression than patients with other types of B/myeloid or T/myeloid MPAL, so patients with ZNF384r MPAL might respond well to treatment with a FLT3 inhibitor.
This study also showed that cases of B/myeloid MPAL without ZNF384r shared genomic features with other B-ALL subtypes, such as Ph-like B-ALL, which may have implications for treatment.
Another of the researchers’ discoveries was that T/myeloid MPAL and early T-cell precursor ALL have similar gene expression profiles.
The team identified several genes that were mutated at similar frequencies in T/myeloid MPAL and early T-cell precursor ALL, including WT1, ETV6, EZH2, and FLT3. WT1 was the most frequently mutated transcription factor gene in T/myeloid MPAL.
Based on these findings, the researchers said the WHO classification of acute leukemia should be updated to include:
- ZNF384r acute leukemia (either B-ALL or MPAL)
- WT1-mutant T/myeloid MPAL
- Ph-like B/myeloid MPAL.
Evolution of MPAL
The researchers’ analyses also revealed leukemia-initiating genetic alterations in early hematopoietic progenitors.
The team said this and other findings—including the common genomic features of ZNF384r MPAL and B-ALL—suggest the ambiguous phenotype of MPAL results from alterations in immature hematopoietic progenitors.
“These findings suggest that the founding mutation occurs early in blood cell development, in some cases in hematopoietic stem cells, and results in an acute leukemia with features of both myeloid and lymphoid cells,” said study author Thomas Alexander, MD, of the University of North Carolina at Chapel Hill.
“One previous theory was that the reason you have two different cancer types within the same patient is that they acquire different mutations that drive them to become AML or ALL, with genomically distinct tumors within the same patient. That doesn’t seem to be the case from our data. Our proposed model is that the mutations occur earlier in development in cells that retain the potential to acquire myeloid or lymphoid features.”
This research was supported by the National Cancer Institute, the National Institutes of Health, Cookies for Kids’ Cancer, and other organizations.
New U.S. cancer cases may exceed 2.3 million by 2035

The American Association for Cancer Research (AACR) has released its annual Cancer Progress Report, detailing recent advances in the fight against cancer and calling on elected officials to address the challenges that remain.
The AACR Cancer Progress Report 2018 lists the 22 new approvals for cancer treatments that have occurred during the last 12 months, including 12 therapies approved to treat hematologic malignancies.
However, the report also notes that cancer continues to pose immense public health challenges in the United States.
The estimated number of new cancer cases for 2018 is 1,735,350, and the estimated number of cancer deaths is 609,640.
The number of new cancer cases is predicted to increase to 2,387,304 in 2035. This is due, in large part, to the rising number of people age 65 and older, according to the report.
With this in mind, the AACR is calling on elected officials to:
Maintain “robust, sustained, and predictable growth” of the National Institutes of Health (NIH) budget, increasing it at least $2 billion in fiscal year (FY) 2019, for a total funding level of at least $39.1 billion.
Make sure the $711 million in funding provided through the 21st Century Cures Act for targeted initiatives—including the National Cancer Moonshot—“is fully appropriated in FY 2019 and is supplemental to the healthy increase for the NIH’s base budget.”
Raise the Food and Drug Administration’s base budget in FY 2019 to $3.1 billion—a $308 million increase above its FY 2018 level—to secure support for regulatory science and speed the development of medical products that are safe and effective.
Provide the Centers for Disease Control and Prevention’s Cancer Prevention and Control Programs with total funding of at least $517 million. This would include funding for “comprehensive cancer control, cancer registries, and screening and awareness programs for specific cancers.”
The American Association for Cancer Research (AACR) has released its annual Cancer Progress Report, detailing recent advances in the fight against cancer and calling on elected officials to address the challenges that remain.
The AACR Cancer Progress Report 2018 lists the 22 new approvals for cancer treatments that have occurred during the last 12 months, including 12 therapies approved to treat hematologic malignancies.
However, the report also notes that cancer continues to pose immense public health challenges in the United States.
The estimated number of new cancer cases for 2018 is 1,735,350, and the estimated number of cancer deaths is 609,640.
The number of new cancer cases is predicted to increase to 2,387,304 in 2035. This is due, in large part, to the rising number of people age 65 and older, according to the report.
With this in mind, the AACR is calling on elected officials to:
Maintain “robust, sustained, and predictable growth” of the National Institutes of Health (NIH) budget, increasing it at least $2 billion in fiscal year (FY) 2019, for a total funding level of at least $39.1 billion.
Make sure the $711 million in funding provided through the 21st Century Cures Act for targeted initiatives—including the National Cancer Moonshot—“is fully appropriated in FY 2019 and is supplemental to the healthy increase for the NIH’s base budget.”
Raise the Food and Drug Administration’s base budget in FY 2019 to $3.1 billion—a $308 million increase above its FY 2018 level—to secure support for regulatory science and speed the development of medical products that are safe and effective.
Provide the Centers for Disease Control and Prevention’s Cancer Prevention and Control Programs with total funding of at least $517 million. This would include funding for “comprehensive cancer control, cancer registries, and screening and awareness programs for specific cancers.”
The American Association for Cancer Research (AACR) has released its annual Cancer Progress Report, detailing recent advances in the fight against cancer and calling on elected officials to address the challenges that remain.
The AACR Cancer Progress Report 2018 lists the 22 new approvals for cancer treatments that have occurred during the last 12 months, including 12 therapies approved to treat hematologic malignancies.
However, the report also notes that cancer continues to pose immense public health challenges in the United States.
The estimated number of new cancer cases for 2018 is 1,735,350, and the estimated number of cancer deaths is 609,640.
The number of new cancer cases is predicted to increase to 2,387,304 in 2035. This is due, in large part, to the rising number of people age 65 and older, according to the report.
With this in mind, the AACR is calling on elected officials to:
Maintain “robust, sustained, and predictable growth” of the National Institutes of Health (NIH) budget, increasing it at least $2 billion in fiscal year (FY) 2019, for a total funding level of at least $39.1 billion.
Make sure the $711 million in funding provided through the 21st Century Cures Act for targeted initiatives—including the National Cancer Moonshot—“is fully appropriated in FY 2019 and is supplemental to the healthy increase for the NIH’s base budget.”
Raise the Food and Drug Administration’s base budget in FY 2019 to $3.1 billion—a $308 million increase above its FY 2018 level—to secure support for regulatory science and speed the development of medical products that are safe and effective.
Provide the Centers for Disease Control and Prevention’s Cancer Prevention and Control Programs with total funding of at least $517 million. This would include funding for “comprehensive cancer control, cancer registries, and screening and awareness programs for specific cancers.”