User login
Three Conditions for Which Cannabis Appears to Help
The utility of cannabinoids to treat most medical conditions remains uncertain at best, but for at least three indications the data lean in favor of effectiveness, Ellie Grossman, MD, MPH, told attendees recently at the 2024 American College of Physicians Internal Medicine meeting.
Those are neuropathic pain, chemotherapy-induced nausea or vomiting, and spasticity in people with multiple sclerosis, said Dr. Grossman, an instructor at Harvard Medical School in Boston and medical director for primary care/behavioral health integration at Cambridge Health Alliance in Somerville, Massachusetts.
Dearth of Research Persists
Research is sorely lacking and of low quality in the field for many reasons, Dr. Grossman said. Most of the products tested come from outside the United States and often are synthetic and taken orally — which does not match the real-world use when patients go to dispensaries for cannabis derived directly from plants (or the plant product itself). And studies often rely on self-report.
Chronic pain is by far the top reason patients say they use medical cannabis, Dr. Grossman said. A Cochrane review of 16 studies found only that the potential benefits of cannabis may outweigh the potential harms for chronic neuropathic pain.
No Evidence in OUD
Dr. Grossman said she is frequently asked if cannabis can help people quit taking opioids. The answer seems to be no. A study published earlier this year in states with legalized medical or recreational cannabis found no difference between rates of opioid overdose compared with states with no such laws. “It seems like it doesn’t do anything to help us with our opioid problem,” she said.
Nor does high-quality evidence exist showing use of cannabis can improve sleep, she said. A 2022 systematic review found fewer than half of studies showed the substance useful for sleep outcomes. “Where studies were positives, it was in people who had chronic pain,” Dr. Grossman noted. Research indicates cannabis may have substantial benefit for chronic pain compared with placebo.
Potential Harms
If the medical benefits of cannabis are murky, the evidence for its potential harms, at least in the short term, are clearer, according to Dr. Grossman. A simplified guideline for prescribing medical cannabinoids in primary care includes sedation, feeling high, dizziness, speech disorders, muscle twitching, hypotension, and several other conditions among the potential hazards of the drug.
But the potential for long-term harm is uncertain. “All the evidence comes from people who have been using it for recreational reasons,” where there may be co-use of tobacco, self-reported outcomes, and recall bias, she said. The characteristics of people using cannabis recreationally often differ from those using it medicinally.
Use With Other Controlled Substances
Dr. Grossman said clinicians should consider whether the co-use of cannabis and other controlled substances, such as benzodiazepines, opioids, or Adderall, raises the potential risks associated with those drugs. “Ultimately it comes down to talking to your patients,” she said. If a toxicity screen shows the presence of controlled substances, ask about their experience with the drugs they are using and let them know your main concern is their safety.
Dr. Grossman reported no relevant financial conflicts of interest.
A version of this article appeared on Medscape.com.
The utility of cannabinoids to treat most medical conditions remains uncertain at best, but for at least three indications the data lean in favor of effectiveness, Ellie Grossman, MD, MPH, told attendees recently at the 2024 American College of Physicians Internal Medicine meeting.
Those are neuropathic pain, chemotherapy-induced nausea or vomiting, and spasticity in people with multiple sclerosis, said Dr. Grossman, an instructor at Harvard Medical School in Boston and medical director for primary care/behavioral health integration at Cambridge Health Alliance in Somerville, Massachusetts.
Dearth of Research Persists
Research is sorely lacking and of low quality in the field for many reasons, Dr. Grossman said. Most of the products tested come from outside the United States and often are synthetic and taken orally — which does not match the real-world use when patients go to dispensaries for cannabis derived directly from plants (or the plant product itself). And studies often rely on self-report.
Chronic pain is by far the top reason patients say they use medical cannabis, Dr. Grossman said. A Cochrane review of 16 studies found only that the potential benefits of cannabis may outweigh the potential harms for chronic neuropathic pain.
No Evidence in OUD
Dr. Grossman said she is frequently asked if cannabis can help people quit taking opioids. The answer seems to be no. A study published earlier this year in states with legalized medical or recreational cannabis found no difference between rates of opioid overdose compared with states with no such laws. “It seems like it doesn’t do anything to help us with our opioid problem,” she said.
Nor does high-quality evidence exist showing use of cannabis can improve sleep, she said. A 2022 systematic review found fewer than half of studies showed the substance useful for sleep outcomes. “Where studies were positives, it was in people who had chronic pain,” Dr. Grossman noted. Research indicates cannabis may have substantial benefit for chronic pain compared with placebo.
Potential Harms
If the medical benefits of cannabis are murky, the evidence for its potential harms, at least in the short term, are clearer, according to Dr. Grossman. A simplified guideline for prescribing medical cannabinoids in primary care includes sedation, feeling high, dizziness, speech disorders, muscle twitching, hypotension, and several other conditions among the potential hazards of the drug.
But the potential for long-term harm is uncertain. “All the evidence comes from people who have been using it for recreational reasons,” where there may be co-use of tobacco, self-reported outcomes, and recall bias, she said. The characteristics of people using cannabis recreationally often differ from those using it medicinally.
Use With Other Controlled Substances
Dr. Grossman said clinicians should consider whether the co-use of cannabis and other controlled substances, such as benzodiazepines, opioids, or Adderall, raises the potential risks associated with those drugs. “Ultimately it comes down to talking to your patients,” she said. If a toxicity screen shows the presence of controlled substances, ask about their experience with the drugs they are using and let them know your main concern is their safety.
Dr. Grossman reported no relevant financial conflicts of interest.
A version of this article appeared on Medscape.com.
The utility of cannabinoids to treat most medical conditions remains uncertain at best, but for at least three indications the data lean in favor of effectiveness, Ellie Grossman, MD, MPH, told attendees recently at the 2024 American College of Physicians Internal Medicine meeting.
Those are neuropathic pain, chemotherapy-induced nausea or vomiting, and spasticity in people with multiple sclerosis, said Dr. Grossman, an instructor at Harvard Medical School in Boston and medical director for primary care/behavioral health integration at Cambridge Health Alliance in Somerville, Massachusetts.
Dearth of Research Persists
Research is sorely lacking and of low quality in the field for many reasons, Dr. Grossman said. Most of the products tested come from outside the United States and often are synthetic and taken orally — which does not match the real-world use when patients go to dispensaries for cannabis derived directly from plants (or the plant product itself). And studies often rely on self-report.
Chronic pain is by far the top reason patients say they use medical cannabis, Dr. Grossman said. A Cochrane review of 16 studies found only that the potential benefits of cannabis may outweigh the potential harms for chronic neuropathic pain.
No Evidence in OUD
Dr. Grossman said she is frequently asked if cannabis can help people quit taking opioids. The answer seems to be no. A study published earlier this year in states with legalized medical or recreational cannabis found no difference between rates of opioid overdose compared with states with no such laws. “It seems like it doesn’t do anything to help us with our opioid problem,” she said.
Nor does high-quality evidence exist showing use of cannabis can improve sleep, she said. A 2022 systematic review found fewer than half of studies showed the substance useful for sleep outcomes. “Where studies were positives, it was in people who had chronic pain,” Dr. Grossman noted. Research indicates cannabis may have substantial benefit for chronic pain compared with placebo.
Potential Harms
If the medical benefits of cannabis are murky, the evidence for its potential harms, at least in the short term, are clearer, according to Dr. Grossman. A simplified guideline for prescribing medical cannabinoids in primary care includes sedation, feeling high, dizziness, speech disorders, muscle twitching, hypotension, and several other conditions among the potential hazards of the drug.
But the potential for long-term harm is uncertain. “All the evidence comes from people who have been using it for recreational reasons,” where there may be co-use of tobacco, self-reported outcomes, and recall bias, she said. The characteristics of people using cannabis recreationally often differ from those using it medicinally.
Use With Other Controlled Substances
Dr. Grossman said clinicians should consider whether the co-use of cannabis and other controlled substances, such as benzodiazepines, opioids, or Adderall, raises the potential risks associated with those drugs. “Ultimately it comes down to talking to your patients,” she said. If a toxicity screen shows the presence of controlled substances, ask about their experience with the drugs they are using and let them know your main concern is their safety.
Dr. Grossman reported no relevant financial conflicts of interest.
A version of this article appeared on Medscape.com.
Girls Catching Up With Boys in Substance Use
, warned the authors of a new report detailing trends across several regions between 2018 and 2022. The latest 4-yearly Health Behaviour in School-Aged Children study, in collaboration with the World Health Organization (WHO) Regional Office for Europe, concluded that substance use remains “a crucial public health problem among adolescents” despite overall declines in smoking, alcohol, and cannabis use.
The new report: A focus on adolescent substance use in Europe, central Asia, and Canada, detailed substance use among adolescents aged 11, 13, and 15 years across 44 countries and regions in Europe, Central Asia, and Canada in the 2021-2022 school-based survey.
Principal findings included:
- Cigarette smoking: Lifetime smoking declined between 2018 and 2022, particularly among 13-year-old boys and 15-year-old boys and girls. There was also a small but significant decrease in current smoking among 15-year-old boys.
- Alcohol use: Lifetime use decreased overall in boys between 2018 and 2022, particularly among 15-year-olds. An increase was observed among 11- and 13-year-old girls but not 15-year-old girls. There was a small but significant decrease in the proportion of current drinkers among 15-year-old boys, with no change among 11- and 13-year-old boys. Current alcohol use increased among girls in all age groups.
- Cannabis use: Lifetime use among 15-year-olds decreased slightly from 14% to 12% between 2018 and 2022, while 6% of 15-year-olds reported having used cannabis in the previous 30 days.
- Vaping: In 2022 vapes (e-cigarettes) were more popular among adolescents than conventional tobacco cigarettes.
Traditional Gender Gap Narrowing or Reversing
Report coauthor Judith Brown from the University of Glasgow, Glasgow, Scotland, and a project manager for the Scottish survey, said that “there was an overall increase in current alcohol use and drunkenness among older girls” despite the overall decrease in boys’ alcohol use.
She explained: “Substance use has traditionally been more prevalent among boys, and the survey findings confirm a well-established gender difference, with higher prevalence in boys than in girls among 11-year-olds. By the age of 13, however, gender differences diminish or even disappear in many countries and regions.”
“Among 15-year-olds, girls often reported more frequent substance use than boys. While this pattern has been known for cigarette smoking in many countries and regions for about two decades, especially among 15-year-olds, it is a new phenomenon for behaviors related to other substances (such as alcohol consumption and drunkenness) in most countries and regions. Historically, prevalence for these behaviors has been higher among boys than girls.”
The new survey results highlight this gender reversal for several substances, she said. “Cannabis is the only substance for which both lifetime and current use is consistently higher in boys.”
Vaping Is an Emerging Public Health Concern
Dr. Brown added that the 2022 survey was the first time that vaping data had been collected from all countries. Although this is against the background of continuing decreases in smoking rates, “researchers suggest the transition to e-cigarettes, as a more popular choice than conventional cigarettes, highlights an urgent need for more targeted interventions to address this emerging public health concern.”
The report authors commented that because young people’s brains are still developing, they are “very sensitive to substances such as nicotine,” making it “easier for them to get hooked.”
Margreet de Looze, PhD, assistant professor of interdisciplinary social science at Utrecht University in Utrecht, the Netherlands, agreed with the authors’ concerns. “Vaping is extremely attractive for young people,” she said, “because the taste is more attractive than that of traditional cigarettes.” Until recently, many people were not aware of health hazards attached to vaping. “While more research is needed, vaping may function as a first step toward tobacco use and is hazardous for young people’s health. Therefore, it should be strongly discouraged.”
Substance Use Trends May Be Stabilizing or Rising Again
Increased awareness of the harmful effects of alcohol for adolescent development is also one postulated reason for declining adolescent alcohol consumption in both Europe and North America over the past two decades, which Dr. de Looze’s research has explored. Her work has also noted the “growing trend” of young people abstaining from alcohol altogether and some evidence of reductions in adolescent risk behaviors more generally, including early sexual initiation and juvenile crime.
“It may be good to realize that, in fact, the current generation of youth in many respects is healthier and reports less risky health behaviors as compared to previous generations,” she said.
However, “The declining trend in adolescent substance use that took place in many countries since the beginning of the 21st century seems to have stabilized, and moreover, in some countries and subgroups of adolescents, substance use appears to be on the rise again.” She cited particularly an overall increase in current alcohol use and drunkenness among older girls between 2018 and 2022. “It appears that, especially for girls, recent trends over time are less favorable as compared with boys.”
Multiple Influences on Adolescent Substance Abuse
Peer group influences are known to come to the fore during adolescence, and Dr. de Looze added that the early 21st century saw marked reductions in adolescent face-to-face contacts with their peers due to the rise in digital communications. “Adolescents typically use substances in the presence of peers (and in the absence of adults/parents), as it increases their status in their peer group.” Reduced in person interactions with friends may therefore have contributed to the earlier decline in substance use.
However, her team had found that adolescents who spend much time online with friends often also spend much time with friends offline. “They are what you could call the ‘social’ youth, who just spend much time with peers, be it offline or online,” she said. “More research is needed to disentangle exactly how, what kind, for whom the digital environment may be related to young people’s substance use,” she said.
“We also see that young people actively select their friends. So, if you are curious and a bit of a sensation-seeker yourself, you are more likely to become friends with youth who are just like you, and together, you may be more likely to try out substances.”
Factors underlying adolescent substance use and differences between countries are influenced by a complex interplay of factors, said Carina Ferreira-Borges, PhD, regional adviser for alcohol, illicit drugs, and prison health at the WHO Regional Office for Europe.
“Prevention measures definitely play a critical role in reducing substance use,” she said, “but other factors, such as cultural norms and socioeconomic conditions, also significantly impact these patterns.”
“Variations in substance use among countries can be attributed to different levels of implemented polices, public health initiatives, and the extent to which substance use is normalized or stigmatized within each society.”
Policy Efforts Must Be Targeted
“To address these disparities effectively, interventions and population-level policies need to be culturally adapted and target the specific environments where substance use is normalized among adolescents. By understanding and modifying the broader context in which young people make choices about substance use, we can better influence their behavior and health outcomes.”
Dr. de Looze cautioned, “In the past two decades, public health efforts in many countries have focused on reducing young people’s engagement in substance use. It is important that these efforts continue, as every year a new generation of youth is born. If public health efforts do not continue to focus on supporting a healthy lifestyle among young people, it should not come as a surprise that rates start or continue to rise again.”
A version of this article appeared on Medscape.com.
, warned the authors of a new report detailing trends across several regions between 2018 and 2022. The latest 4-yearly Health Behaviour in School-Aged Children study, in collaboration with the World Health Organization (WHO) Regional Office for Europe, concluded that substance use remains “a crucial public health problem among adolescents” despite overall declines in smoking, alcohol, and cannabis use.
The new report: A focus on adolescent substance use in Europe, central Asia, and Canada, detailed substance use among adolescents aged 11, 13, and 15 years across 44 countries and regions in Europe, Central Asia, and Canada in the 2021-2022 school-based survey.
Principal findings included:
- Cigarette smoking: Lifetime smoking declined between 2018 and 2022, particularly among 13-year-old boys and 15-year-old boys and girls. There was also a small but significant decrease in current smoking among 15-year-old boys.
- Alcohol use: Lifetime use decreased overall in boys between 2018 and 2022, particularly among 15-year-olds. An increase was observed among 11- and 13-year-old girls but not 15-year-old girls. There was a small but significant decrease in the proportion of current drinkers among 15-year-old boys, with no change among 11- and 13-year-old boys. Current alcohol use increased among girls in all age groups.
- Cannabis use: Lifetime use among 15-year-olds decreased slightly from 14% to 12% between 2018 and 2022, while 6% of 15-year-olds reported having used cannabis in the previous 30 days.
- Vaping: In 2022 vapes (e-cigarettes) were more popular among adolescents than conventional tobacco cigarettes.
Traditional Gender Gap Narrowing or Reversing
Report coauthor Judith Brown from the University of Glasgow, Glasgow, Scotland, and a project manager for the Scottish survey, said that “there was an overall increase in current alcohol use and drunkenness among older girls” despite the overall decrease in boys’ alcohol use.
She explained: “Substance use has traditionally been more prevalent among boys, and the survey findings confirm a well-established gender difference, with higher prevalence in boys than in girls among 11-year-olds. By the age of 13, however, gender differences diminish or even disappear in many countries and regions.”
“Among 15-year-olds, girls often reported more frequent substance use than boys. While this pattern has been known for cigarette smoking in many countries and regions for about two decades, especially among 15-year-olds, it is a new phenomenon for behaviors related to other substances (such as alcohol consumption and drunkenness) in most countries and regions. Historically, prevalence for these behaviors has been higher among boys than girls.”
The new survey results highlight this gender reversal for several substances, she said. “Cannabis is the only substance for which both lifetime and current use is consistently higher in boys.”
Vaping Is an Emerging Public Health Concern
Dr. Brown added that the 2022 survey was the first time that vaping data had been collected from all countries. Although this is against the background of continuing decreases in smoking rates, “researchers suggest the transition to e-cigarettes, as a more popular choice than conventional cigarettes, highlights an urgent need for more targeted interventions to address this emerging public health concern.”
The report authors commented that because young people’s brains are still developing, they are “very sensitive to substances such as nicotine,” making it “easier for them to get hooked.”
Margreet de Looze, PhD, assistant professor of interdisciplinary social science at Utrecht University in Utrecht, the Netherlands, agreed with the authors’ concerns. “Vaping is extremely attractive for young people,” she said, “because the taste is more attractive than that of traditional cigarettes.” Until recently, many people were not aware of health hazards attached to vaping. “While more research is needed, vaping may function as a first step toward tobacco use and is hazardous for young people’s health. Therefore, it should be strongly discouraged.”
Substance Use Trends May Be Stabilizing or Rising Again
Increased awareness of the harmful effects of alcohol for adolescent development is also one postulated reason for declining adolescent alcohol consumption in both Europe and North America over the past two decades, which Dr. de Looze’s research has explored. Her work has also noted the “growing trend” of young people abstaining from alcohol altogether and some evidence of reductions in adolescent risk behaviors more generally, including early sexual initiation and juvenile crime.
“It may be good to realize that, in fact, the current generation of youth in many respects is healthier and reports less risky health behaviors as compared to previous generations,” she said.
However, “The declining trend in adolescent substance use that took place in many countries since the beginning of the 21st century seems to have stabilized, and moreover, in some countries and subgroups of adolescents, substance use appears to be on the rise again.” She cited particularly an overall increase in current alcohol use and drunkenness among older girls between 2018 and 2022. “It appears that, especially for girls, recent trends over time are less favorable as compared with boys.”
Multiple Influences on Adolescent Substance Abuse
Peer group influences are known to come to the fore during adolescence, and Dr. de Looze added that the early 21st century saw marked reductions in adolescent face-to-face contacts with their peers due to the rise in digital communications. “Adolescents typically use substances in the presence of peers (and in the absence of adults/parents), as it increases their status in their peer group.” Reduced in person interactions with friends may therefore have contributed to the earlier decline in substance use.
However, her team had found that adolescents who spend much time online with friends often also spend much time with friends offline. “They are what you could call the ‘social’ youth, who just spend much time with peers, be it offline or online,” she said. “More research is needed to disentangle exactly how, what kind, for whom the digital environment may be related to young people’s substance use,” she said.
“We also see that young people actively select their friends. So, if you are curious and a bit of a sensation-seeker yourself, you are more likely to become friends with youth who are just like you, and together, you may be more likely to try out substances.”
Factors underlying adolescent substance use and differences between countries are influenced by a complex interplay of factors, said Carina Ferreira-Borges, PhD, regional adviser for alcohol, illicit drugs, and prison health at the WHO Regional Office for Europe.
“Prevention measures definitely play a critical role in reducing substance use,” she said, “but other factors, such as cultural norms and socioeconomic conditions, also significantly impact these patterns.”
“Variations in substance use among countries can be attributed to different levels of implemented polices, public health initiatives, and the extent to which substance use is normalized or stigmatized within each society.”
Policy Efforts Must Be Targeted
“To address these disparities effectively, interventions and population-level policies need to be culturally adapted and target the specific environments where substance use is normalized among adolescents. By understanding and modifying the broader context in which young people make choices about substance use, we can better influence their behavior and health outcomes.”
Dr. de Looze cautioned, “In the past two decades, public health efforts in many countries have focused on reducing young people’s engagement in substance use. It is important that these efforts continue, as every year a new generation of youth is born. If public health efforts do not continue to focus on supporting a healthy lifestyle among young people, it should not come as a surprise that rates start or continue to rise again.”
A version of this article appeared on Medscape.com.
, warned the authors of a new report detailing trends across several regions between 2018 and 2022. The latest 4-yearly Health Behaviour in School-Aged Children study, in collaboration with the World Health Organization (WHO) Regional Office for Europe, concluded that substance use remains “a crucial public health problem among adolescents” despite overall declines in smoking, alcohol, and cannabis use.
The new report: A focus on adolescent substance use in Europe, central Asia, and Canada, detailed substance use among adolescents aged 11, 13, and 15 years across 44 countries and regions in Europe, Central Asia, and Canada in the 2021-2022 school-based survey.
Principal findings included:
- Cigarette smoking: Lifetime smoking declined between 2018 and 2022, particularly among 13-year-old boys and 15-year-old boys and girls. There was also a small but significant decrease in current smoking among 15-year-old boys.
- Alcohol use: Lifetime use decreased overall in boys between 2018 and 2022, particularly among 15-year-olds. An increase was observed among 11- and 13-year-old girls but not 15-year-old girls. There was a small but significant decrease in the proportion of current drinkers among 15-year-old boys, with no change among 11- and 13-year-old boys. Current alcohol use increased among girls in all age groups.
- Cannabis use: Lifetime use among 15-year-olds decreased slightly from 14% to 12% between 2018 and 2022, while 6% of 15-year-olds reported having used cannabis in the previous 30 days.
- Vaping: In 2022 vapes (e-cigarettes) were more popular among adolescents than conventional tobacco cigarettes.
Traditional Gender Gap Narrowing or Reversing
Report coauthor Judith Brown from the University of Glasgow, Glasgow, Scotland, and a project manager for the Scottish survey, said that “there was an overall increase in current alcohol use and drunkenness among older girls” despite the overall decrease in boys’ alcohol use.
She explained: “Substance use has traditionally been more prevalent among boys, and the survey findings confirm a well-established gender difference, with higher prevalence in boys than in girls among 11-year-olds. By the age of 13, however, gender differences diminish or even disappear in many countries and regions.”
“Among 15-year-olds, girls often reported more frequent substance use than boys. While this pattern has been known for cigarette smoking in many countries and regions for about two decades, especially among 15-year-olds, it is a new phenomenon for behaviors related to other substances (such as alcohol consumption and drunkenness) in most countries and regions. Historically, prevalence for these behaviors has been higher among boys than girls.”
The new survey results highlight this gender reversal for several substances, she said. “Cannabis is the only substance for which both lifetime and current use is consistently higher in boys.”
Vaping Is an Emerging Public Health Concern
Dr. Brown added that the 2022 survey was the first time that vaping data had been collected from all countries. Although this is against the background of continuing decreases in smoking rates, “researchers suggest the transition to e-cigarettes, as a more popular choice than conventional cigarettes, highlights an urgent need for more targeted interventions to address this emerging public health concern.”
The report authors commented that because young people’s brains are still developing, they are “very sensitive to substances such as nicotine,” making it “easier for them to get hooked.”
Margreet de Looze, PhD, assistant professor of interdisciplinary social science at Utrecht University in Utrecht, the Netherlands, agreed with the authors’ concerns. “Vaping is extremely attractive for young people,” she said, “because the taste is more attractive than that of traditional cigarettes.” Until recently, many people were not aware of health hazards attached to vaping. “While more research is needed, vaping may function as a first step toward tobacco use and is hazardous for young people’s health. Therefore, it should be strongly discouraged.”
Substance Use Trends May Be Stabilizing or Rising Again
Increased awareness of the harmful effects of alcohol for adolescent development is also one postulated reason for declining adolescent alcohol consumption in both Europe and North America over the past two decades, which Dr. de Looze’s research has explored. Her work has also noted the “growing trend” of young people abstaining from alcohol altogether and some evidence of reductions in adolescent risk behaviors more generally, including early sexual initiation and juvenile crime.
“It may be good to realize that, in fact, the current generation of youth in many respects is healthier and reports less risky health behaviors as compared to previous generations,” she said.
However, “The declining trend in adolescent substance use that took place in many countries since the beginning of the 21st century seems to have stabilized, and moreover, in some countries and subgroups of adolescents, substance use appears to be on the rise again.” She cited particularly an overall increase in current alcohol use and drunkenness among older girls between 2018 and 2022. “It appears that, especially for girls, recent trends over time are less favorable as compared with boys.”
Multiple Influences on Adolescent Substance Abuse
Peer group influences are known to come to the fore during adolescence, and Dr. de Looze added that the early 21st century saw marked reductions in adolescent face-to-face contacts with their peers due to the rise in digital communications. “Adolescents typically use substances in the presence of peers (and in the absence of adults/parents), as it increases their status in their peer group.” Reduced in person interactions with friends may therefore have contributed to the earlier decline in substance use.
However, her team had found that adolescents who spend much time online with friends often also spend much time with friends offline. “They are what you could call the ‘social’ youth, who just spend much time with peers, be it offline or online,” she said. “More research is needed to disentangle exactly how, what kind, for whom the digital environment may be related to young people’s substance use,” she said.
“We also see that young people actively select their friends. So, if you are curious and a bit of a sensation-seeker yourself, you are more likely to become friends with youth who are just like you, and together, you may be more likely to try out substances.”
Factors underlying adolescent substance use and differences between countries are influenced by a complex interplay of factors, said Carina Ferreira-Borges, PhD, regional adviser for alcohol, illicit drugs, and prison health at the WHO Regional Office for Europe.
“Prevention measures definitely play a critical role in reducing substance use,” she said, “but other factors, such as cultural norms and socioeconomic conditions, also significantly impact these patterns.”
“Variations in substance use among countries can be attributed to different levels of implemented polices, public health initiatives, and the extent to which substance use is normalized or stigmatized within each society.”
Policy Efforts Must Be Targeted
“To address these disparities effectively, interventions and population-level policies need to be culturally adapted and target the specific environments where substance use is normalized among adolescents. By understanding and modifying the broader context in which young people make choices about substance use, we can better influence their behavior and health outcomes.”
Dr. de Looze cautioned, “In the past two decades, public health efforts in many countries have focused on reducing young people’s engagement in substance use. It is important that these efforts continue, as every year a new generation of youth is born. If public health efforts do not continue to focus on supporting a healthy lifestyle among young people, it should not come as a surprise that rates start or continue to rise again.”
A version of this article appeared on Medscape.com.
FDA Requests More Information for RDEB Rx Under Review
The Food and Drug Administration (RDEB), requesting more information from the manufacturer.
Pz-cel, which comprises autologous, COL7A1 gene–corrected epidermal sheets, is being evaluated for its ability to enable normal type VII collagen expression in a patient’s skin cells and to facilitate wound healing and pain reduction in wounds in patients with RDEB after a one-time application procedure. The cause of RDEB is a defect in the COL7A1 gene that “results in the inability to produce type VII collagen,” a press release from the manufacturer noted.
On April 22, 2024, the manufacturer Abeona Therapeutics announced that following a meeting with the FDA in March and in a subsequent request for information, the agency requires additional information to satisfy certain Chemistry Manufacturing and Controls requirements before the BLA for pz-cel can be approved. According to a press release from the company, the information pertains to validation requirements for certain manufacturing and release testing methods, including some that were observed during the FDA’s pre-licensing inspection.
The complete response letter did not identify any issues related to the clinical efficacy or safety data in the BLA, and the FDA did not request any new clinical trials or clinical data to support approval, according to the company.
The company anticipates completing the BLA resubmission in the third quarter of 2024. The application is supported by clinical efficacy and safety data from the pivotal phase 3 VIITAL study and a phase 1/2a study in patients with RDEB.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration (RDEB), requesting more information from the manufacturer.
Pz-cel, which comprises autologous, COL7A1 gene–corrected epidermal sheets, is being evaluated for its ability to enable normal type VII collagen expression in a patient’s skin cells and to facilitate wound healing and pain reduction in wounds in patients with RDEB after a one-time application procedure. The cause of RDEB is a defect in the COL7A1 gene that “results in the inability to produce type VII collagen,” a press release from the manufacturer noted.
On April 22, 2024, the manufacturer Abeona Therapeutics announced that following a meeting with the FDA in March and in a subsequent request for information, the agency requires additional information to satisfy certain Chemistry Manufacturing and Controls requirements before the BLA for pz-cel can be approved. According to a press release from the company, the information pertains to validation requirements for certain manufacturing and release testing methods, including some that were observed during the FDA’s pre-licensing inspection.
The complete response letter did not identify any issues related to the clinical efficacy or safety data in the BLA, and the FDA did not request any new clinical trials or clinical data to support approval, according to the company.
The company anticipates completing the BLA resubmission in the third quarter of 2024. The application is supported by clinical efficacy and safety data from the pivotal phase 3 VIITAL study and a phase 1/2a study in patients with RDEB.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration (RDEB), requesting more information from the manufacturer.
Pz-cel, which comprises autologous, COL7A1 gene–corrected epidermal sheets, is being evaluated for its ability to enable normal type VII collagen expression in a patient’s skin cells and to facilitate wound healing and pain reduction in wounds in patients with RDEB after a one-time application procedure. The cause of RDEB is a defect in the COL7A1 gene that “results in the inability to produce type VII collagen,” a press release from the manufacturer noted.
On April 22, 2024, the manufacturer Abeona Therapeutics announced that following a meeting with the FDA in March and in a subsequent request for information, the agency requires additional information to satisfy certain Chemistry Manufacturing and Controls requirements before the BLA for pz-cel can be approved. According to a press release from the company, the information pertains to validation requirements for certain manufacturing and release testing methods, including some that were observed during the FDA’s pre-licensing inspection.
The complete response letter did not identify any issues related to the clinical efficacy or safety data in the BLA, and the FDA did not request any new clinical trials or clinical data to support approval, according to the company.
The company anticipates completing the BLA resubmission in the third quarter of 2024. The application is supported by clinical efficacy and safety data from the pivotal phase 3 VIITAL study and a phase 1/2a study in patients with RDEB.
A version of this article first appeared on Medscape.com.
Antidepressants and Dementia Risk: Reassuring Data
TOPLINE:
, new research suggests.
METHODOLOGY:
- Investigators studied 5511 individuals (58% women; mean age, 71 years) from the Rotterdam study, an ongoing prospective population-based cohort study.
- Participants were free from dementia at baseline, and incident dementia was monitored from baseline until 2018 with repeated cognitive assessments using the Mini-Mental Status Examination (MMSE) and the Geriatric Mental Schedule, as well as MRIs.
- Information on participants’ antidepressant use was extracted from pharmacy records from 1992 until baseline (2002-2008).
- During a mean follow-up of 10 years, 12% of participants developed dementia.
TAKEAWAY:
- Overall, 17% of participants had used antidepressants during the roughly 10-year period prior to baseline, and 4.1% were still using antidepressants at baseline.
- Medication use at baseline was more common in women than in men (21% vs 18%), and use increased with age: From 2.1% in participants aged between 45 and 50 years to 4.5% in those older than 80 years.
- After adjustment for confounders, there was no association between antidepressant use and dementia risk (hazard ratio [HR], 1.14; 95% CI, 0.92-1.41), accelerated cognitive decline, or atrophy of white and gray matter.
- However, tricyclic antidepressant use was associated with increased dementia risk (HR, 1.36; 95% CI, 1.01-1.83) compared with the use of selective serotonin reuptake inhibitors (HR, 1.12; 95% CI, 0.81-1.54).
IN PRACTICE:
“Although prescription of antidepressant medication in older individuals, in particular those with some cognitive impairment, may have acute symptomatic anticholinergic effects that warrant consideration in clinical practice, our results show that long-term antidepressant use does not have lasting effects on cognition or brain health in older adults without indication of cognitive impairment,” the authors wrote.
SOURCE:
Frank J. Wolters, MD, of the Department of Epidemiology and the Department of Radiology and Nuclear Medicine and Alzheimer Center, Erasmus University Medical Center, Rotterdam, the Netherlands, was the senior author on this study that was published online in Alzheimer’s and Dementia.
LIMITATIONS:
Limitations included the concern that although exclusion of participants with MMSE < 26 at baseline prevented reversed causation (ie, antidepressant use in response to depression during the prodromal phase of dementia), it may have introduced selection bias by disregarding the effects of antidepressant use prior to baseline and excluding participants with lower education.
DISCLOSURES:
This study was conducted as part of the Netherlands Consortium of Dementia Cohorts, which receives funding in the context of Deltaplan Dementie from ZonMW Memorabel and Alzheimer Nederland. Further funding was also obtained from the Stichting Erasmus Trustfonds. This study was further supported by a 2020 NARSAD Young Investigator Grant from the Brain & Behavior Research Foundation. The authors reported no conflicts of interest or relevant financial relationships.
A version of this article appeared on Medscape.com.
TOPLINE:
, new research suggests.
METHODOLOGY:
- Investigators studied 5511 individuals (58% women; mean age, 71 years) from the Rotterdam study, an ongoing prospective population-based cohort study.
- Participants were free from dementia at baseline, and incident dementia was monitored from baseline until 2018 with repeated cognitive assessments using the Mini-Mental Status Examination (MMSE) and the Geriatric Mental Schedule, as well as MRIs.
- Information on participants’ antidepressant use was extracted from pharmacy records from 1992 until baseline (2002-2008).
- During a mean follow-up of 10 years, 12% of participants developed dementia.
TAKEAWAY:
- Overall, 17% of participants had used antidepressants during the roughly 10-year period prior to baseline, and 4.1% were still using antidepressants at baseline.
- Medication use at baseline was more common in women than in men (21% vs 18%), and use increased with age: From 2.1% in participants aged between 45 and 50 years to 4.5% in those older than 80 years.
- After adjustment for confounders, there was no association between antidepressant use and dementia risk (hazard ratio [HR], 1.14; 95% CI, 0.92-1.41), accelerated cognitive decline, or atrophy of white and gray matter.
- However, tricyclic antidepressant use was associated with increased dementia risk (HR, 1.36; 95% CI, 1.01-1.83) compared with the use of selective serotonin reuptake inhibitors (HR, 1.12; 95% CI, 0.81-1.54).
IN PRACTICE:
“Although prescription of antidepressant medication in older individuals, in particular those with some cognitive impairment, may have acute symptomatic anticholinergic effects that warrant consideration in clinical practice, our results show that long-term antidepressant use does not have lasting effects on cognition or brain health in older adults without indication of cognitive impairment,” the authors wrote.
SOURCE:
Frank J. Wolters, MD, of the Department of Epidemiology and the Department of Radiology and Nuclear Medicine and Alzheimer Center, Erasmus University Medical Center, Rotterdam, the Netherlands, was the senior author on this study that was published online in Alzheimer’s and Dementia.
LIMITATIONS:
Limitations included the concern that although exclusion of participants with MMSE < 26 at baseline prevented reversed causation (ie, antidepressant use in response to depression during the prodromal phase of dementia), it may have introduced selection bias by disregarding the effects of antidepressant use prior to baseline and excluding participants with lower education.
DISCLOSURES:
This study was conducted as part of the Netherlands Consortium of Dementia Cohorts, which receives funding in the context of Deltaplan Dementie from ZonMW Memorabel and Alzheimer Nederland. Further funding was also obtained from the Stichting Erasmus Trustfonds. This study was further supported by a 2020 NARSAD Young Investigator Grant from the Brain & Behavior Research Foundation. The authors reported no conflicts of interest or relevant financial relationships.
A version of this article appeared on Medscape.com.
TOPLINE:
, new research suggests.
METHODOLOGY:
- Investigators studied 5511 individuals (58% women; mean age, 71 years) from the Rotterdam study, an ongoing prospective population-based cohort study.
- Participants were free from dementia at baseline, and incident dementia was monitored from baseline until 2018 with repeated cognitive assessments using the Mini-Mental Status Examination (MMSE) and the Geriatric Mental Schedule, as well as MRIs.
- Information on participants’ antidepressant use was extracted from pharmacy records from 1992 until baseline (2002-2008).
- During a mean follow-up of 10 years, 12% of participants developed dementia.
TAKEAWAY:
- Overall, 17% of participants had used antidepressants during the roughly 10-year period prior to baseline, and 4.1% were still using antidepressants at baseline.
- Medication use at baseline was more common in women than in men (21% vs 18%), and use increased with age: From 2.1% in participants aged between 45 and 50 years to 4.5% in those older than 80 years.
- After adjustment for confounders, there was no association between antidepressant use and dementia risk (hazard ratio [HR], 1.14; 95% CI, 0.92-1.41), accelerated cognitive decline, or atrophy of white and gray matter.
- However, tricyclic antidepressant use was associated with increased dementia risk (HR, 1.36; 95% CI, 1.01-1.83) compared with the use of selective serotonin reuptake inhibitors (HR, 1.12; 95% CI, 0.81-1.54).
IN PRACTICE:
“Although prescription of antidepressant medication in older individuals, in particular those with some cognitive impairment, may have acute symptomatic anticholinergic effects that warrant consideration in clinical practice, our results show that long-term antidepressant use does not have lasting effects on cognition or brain health in older adults without indication of cognitive impairment,” the authors wrote.
SOURCE:
Frank J. Wolters, MD, of the Department of Epidemiology and the Department of Radiology and Nuclear Medicine and Alzheimer Center, Erasmus University Medical Center, Rotterdam, the Netherlands, was the senior author on this study that was published online in Alzheimer’s and Dementia.
LIMITATIONS:
Limitations included the concern that although exclusion of participants with MMSE < 26 at baseline prevented reversed causation (ie, antidepressant use in response to depression during the prodromal phase of dementia), it may have introduced selection bias by disregarding the effects of antidepressant use prior to baseline and excluding participants with lower education.
DISCLOSURES:
This study was conducted as part of the Netherlands Consortium of Dementia Cohorts, which receives funding in the context of Deltaplan Dementie from ZonMW Memorabel and Alzheimer Nederland. Further funding was also obtained from the Stichting Erasmus Trustfonds. This study was further supported by a 2020 NARSAD Young Investigator Grant from the Brain & Behavior Research Foundation. The authors reported no conflicts of interest or relevant financial relationships.
A version of this article appeared on Medscape.com.
Children With Chronic Skin Disorders Face Substantial Stigma
TOPLINE:
METHODOLOGY:
- Stigmatization has been addressed for several chronic medical conditions, such as HIV/AIDS, obesity, and mental illness; however, it has received limited attention in children living with chronic skin disorders.
- This cross-sectional, single-visit study examined the prevalence of stigma, its dependence on disease visibility and severity, and its association with mental health and QoL in children with chronic skin disorders.
- A total of 1671 children aged 8-17 years (57.9% girls; mean age, 13.7 years) were recruited from 32 pediatric dermatology centers in the United States and Canada from November 2018 to November 2021. The most common conditions were acne, atopic dermatitis/eczematous disorders, alopecia, and psoriasis, but rare genetic disorders were also represented.
- The primary outcome was the extent of stigmatization in relation to disease visibility, assessed using the Patient-Reported Outcomes Measurement Instrumentation System Pediatric Stigma-Skin.
- Secondary outcomes were the extent of stigmatization in relation to disease severity, along with QoL, depression, anxiety, and poor peer relationships.
TAKEAWAY:
- Approximately half (56.4%) of the children self-reported their skin condition as highly visible; 50.5% reported their disease severity as moderate, while 21.3% reported it as severe.
- Stigma was experienced by 73% of children and adolescents with chronic skin disease, with 43.8% reporting moderate stigma.
- Stigma scores correlated strongly with impaired QOL (Spearman’s rank correlation coefficient = 0.73) and child-reported scores for depression (Spearman’s rank correlation coefficient = 0.61) and moderately with anxiety (Spearman’s rank correlation coefficient = 0.54) and peer relationships (Spearman’s rank correlation coefficient = −0.49; all P < .001).
- Although stigma is increased for children with higher disease visibility and severity, the relatively weak correlation between child-assessed disease visibility and stigma (Spearman’s rank correlation coefficient = 0.22) showed that stigma is common in children even when diseases are not highly visible.
IN PRACTICE:
“Better treatment approaches for chronic skin diseases in children remain an unmet need. Increased awareness and instituting medical and psychological interventions to identify and reduce stigma and disease severity are important directions for improving QOL,” the authors concluded.
SOURCE:
Amy S. Paller, MD, professor of pediatrics and dermatology, Northwestern University, Chicago, led the study, which was published online in JAMA Dermatology.
LIMITATIONS:
Stigmatization needs to be assessed in children from low- and middle-income countries. Investigators enrolled children who had physician-assessed moderate to severe disease severity and/or at least some visibility of skin disease while wearing clothing, which resulted in exclusion of children with mild chronic disease, and the pandemic limited enrollment.
DISCLOSURES:
This study was funded through a grant from the Pediatric Dermatology Research Alliance (PeDRA). The authors declared receiving grants, personal fees, and honorarium and having other ties with various sources.
A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- Stigmatization has been addressed for several chronic medical conditions, such as HIV/AIDS, obesity, and mental illness; however, it has received limited attention in children living with chronic skin disorders.
- This cross-sectional, single-visit study examined the prevalence of stigma, its dependence on disease visibility and severity, and its association with mental health and QoL in children with chronic skin disorders.
- A total of 1671 children aged 8-17 years (57.9% girls; mean age, 13.7 years) were recruited from 32 pediatric dermatology centers in the United States and Canada from November 2018 to November 2021. The most common conditions were acne, atopic dermatitis/eczematous disorders, alopecia, and psoriasis, but rare genetic disorders were also represented.
- The primary outcome was the extent of stigmatization in relation to disease visibility, assessed using the Patient-Reported Outcomes Measurement Instrumentation System Pediatric Stigma-Skin.
- Secondary outcomes were the extent of stigmatization in relation to disease severity, along with QoL, depression, anxiety, and poor peer relationships.
TAKEAWAY:
- Approximately half (56.4%) of the children self-reported their skin condition as highly visible; 50.5% reported their disease severity as moderate, while 21.3% reported it as severe.
- Stigma was experienced by 73% of children and adolescents with chronic skin disease, with 43.8% reporting moderate stigma.
- Stigma scores correlated strongly with impaired QOL (Spearman’s rank correlation coefficient = 0.73) and child-reported scores for depression (Spearman’s rank correlation coefficient = 0.61) and moderately with anxiety (Spearman’s rank correlation coefficient = 0.54) and peer relationships (Spearman’s rank correlation coefficient = −0.49; all P < .001).
- Although stigma is increased for children with higher disease visibility and severity, the relatively weak correlation between child-assessed disease visibility and stigma (Spearman’s rank correlation coefficient = 0.22) showed that stigma is common in children even when diseases are not highly visible.
IN PRACTICE:
“Better treatment approaches for chronic skin diseases in children remain an unmet need. Increased awareness and instituting medical and psychological interventions to identify and reduce stigma and disease severity are important directions for improving QOL,” the authors concluded.
SOURCE:
Amy S. Paller, MD, professor of pediatrics and dermatology, Northwestern University, Chicago, led the study, which was published online in JAMA Dermatology.
LIMITATIONS:
Stigmatization needs to be assessed in children from low- and middle-income countries. Investigators enrolled children who had physician-assessed moderate to severe disease severity and/or at least some visibility of skin disease while wearing clothing, which resulted in exclusion of children with mild chronic disease, and the pandemic limited enrollment.
DISCLOSURES:
This study was funded through a grant from the Pediatric Dermatology Research Alliance (PeDRA). The authors declared receiving grants, personal fees, and honorarium and having other ties with various sources.
A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- Stigmatization has been addressed for several chronic medical conditions, such as HIV/AIDS, obesity, and mental illness; however, it has received limited attention in children living with chronic skin disorders.
- This cross-sectional, single-visit study examined the prevalence of stigma, its dependence on disease visibility and severity, and its association with mental health and QoL in children with chronic skin disorders.
- A total of 1671 children aged 8-17 years (57.9% girls; mean age, 13.7 years) were recruited from 32 pediatric dermatology centers in the United States and Canada from November 2018 to November 2021. The most common conditions were acne, atopic dermatitis/eczematous disorders, alopecia, and psoriasis, but rare genetic disorders were also represented.
- The primary outcome was the extent of stigmatization in relation to disease visibility, assessed using the Patient-Reported Outcomes Measurement Instrumentation System Pediatric Stigma-Skin.
- Secondary outcomes were the extent of stigmatization in relation to disease severity, along with QoL, depression, anxiety, and poor peer relationships.
TAKEAWAY:
- Approximately half (56.4%) of the children self-reported their skin condition as highly visible; 50.5% reported their disease severity as moderate, while 21.3% reported it as severe.
- Stigma was experienced by 73% of children and adolescents with chronic skin disease, with 43.8% reporting moderate stigma.
- Stigma scores correlated strongly with impaired QOL (Spearman’s rank correlation coefficient = 0.73) and child-reported scores for depression (Spearman’s rank correlation coefficient = 0.61) and moderately with anxiety (Spearman’s rank correlation coefficient = 0.54) and peer relationships (Spearman’s rank correlation coefficient = −0.49; all P < .001).
- Although stigma is increased for children with higher disease visibility and severity, the relatively weak correlation between child-assessed disease visibility and stigma (Spearman’s rank correlation coefficient = 0.22) showed that stigma is common in children even when diseases are not highly visible.
IN PRACTICE:
“Better treatment approaches for chronic skin diseases in children remain an unmet need. Increased awareness and instituting medical and psychological interventions to identify and reduce stigma and disease severity are important directions for improving QOL,” the authors concluded.
SOURCE:
Amy S. Paller, MD, professor of pediatrics and dermatology, Northwestern University, Chicago, led the study, which was published online in JAMA Dermatology.
LIMITATIONS:
Stigmatization needs to be assessed in children from low- and middle-income countries. Investigators enrolled children who had physician-assessed moderate to severe disease severity and/or at least some visibility of skin disease while wearing clothing, which resulted in exclusion of children with mild chronic disease, and the pandemic limited enrollment.
DISCLOSURES:
This study was funded through a grant from the Pediatric Dermatology Research Alliance (PeDRA). The authors declared receiving grants, personal fees, and honorarium and having other ties with various sources.
A version of this article appeared on Medscape.com.
A Simplified Approach to Pelvic Floor Dysfunction
Pelvic floor dysfunction (PFD) represents a spectrum of symptoms involving sensory and emptying abnormalities of the bowel and bladder and pelvic organ prolapse. The pelvic floor refers to a group of muscles that spans the pelvic outlet, providing support to the pelvic organs and coordinating constrictor mechanisms to control urination and defecation. Symptoms reported by patients experiencing PFD include involuntary loss of stool or urine, incomplete emptying of the bowel and bladder, a sensation of fullness, bulging in the vagina, and sexual dysfunction.1
As such, symptoms related to PFD are very common concerns raised by patients to their gastroenterologists. Data from the National Health and Nutrition Examination Survey show that 23.7% of women over the age of 20 had at least one symptom of PFD.2 Unfortunately, patients experiencing pelvic floor dysfunction often are hesitant to seek care because of embarrassment or perception that limited treatment options exist for their symptoms.
Pelvic Floor Anatomy
Regions of the pelvis are often referred to by anatomic compartment: anterior (bladder and urethra), middle (vagina and uterus or prostate), and posterior (colon, rectum, and anal canal). Supporting these compartments is the levator ani, a muscle group that is used synonymously with the term “pelvic diaphragm.”

Continence of stool is provided by the anal sphincter muscles and the puborectalis muscle, which wraps around the posterior aspect of the anorectal canal. Damage to the musculature or sensory perception to this area may result in fecal incontinence. Defecation is a coordinated process during which the abdominal and rectal muscles contract, while the anal sphincter muscles and puborectalis simultaneously relax. A disturbance in neuromuscular coordination (dyssynergic defecation) or structural pathology such as pelvic organ prolapse may lead to obstructed defecation.
PFD is thought to be a result of one or more insults to the pelvic floor such as chronic straining, childbirth, iatrogenic injury, or systemic disease such as diabetes.3
Evaluation of PFD Symptoms
Patients presenting with suspected PFD necessitate a comprehensive interdisciplinary assessment. In addition to obtaining a medical, surgical, and obstetric history, details about symptoms and lifestyle should include toileting habits, diet, and physical activity. The Pelvic Floor Distress Inventory (PFDI-20) is a commonly used tool that can be employed in the clinical setting.4
A pelvic exam can reveal pelvic organ prolapse and other mucosal pathology. The Pelvic Organ Prolapse Quantification System (POP-Q) is a widely used classification system for describing pelvic organ prolapse.5 Protrusion of the rectal wall into the vagina is referred to as a rectocele, while prolapse of small bowel into the upper posterior wall of the vagina is called an enterocele. While the finding of a rectocele on exam is common in parous women and may not cause any symptoms, a larger rectocele may cause a sensation of incomplete evacuation of stool.

A digital rectal exam (DRE) should be performed to assess pelvic floor function and help identify structural abnormalities.
Initial Management
A stepwise approach to the management of PFD can allow many patients to be effectively treated without the need for surgical intervention. For patients reporting liquid stool consistency, the evaluation should pivot toward the workup and management of diarrhea, which can easily overwhelm continence mechanisms and cause fecal incontinence. Fiber supplementation to normalize stool consistency is considered first-line therapy for patients presenting with both fecal incontinence and obstructed defecation. Other tools for fecal incontinence include avoiding foods that trigger diarrhea and use of loperamide.6 For patients with obstructed defecation, a trial of laxatives can be followed by a prescription agent if needed, such as a secretagogue or prokinetic.7
Vaginal splinting is a technique that can be used in patients with rectocele, whereby a finger is inserted into the vagina and pressure is applied on the posterior vaginal wall toward the rectum. Reducing the rectocele can facilitate emptying stool from the rectum and prevent leakage of retained stool.8 Similarly, use of rectal irrigation enemas can also help clear retained stool.
Pelvic floor physical therapists examine the strength, coordination, and tone of the pelvic floor muscles. When hypertonic musculature is present, manual interventions may be performed including trigger point release, myofascial release, and dry needling.9 When hypotonic musculature or dyssynergia is present, strengthening and neuromuscular re-education are recommended. Biofeedback can be administered via surface electromyography and/or balloon training to improve rectal sensitivity. Proper defecation techniques, including positioning, breathing, and behavioral modifications, improve clinical outcomes.

Diagnostic Testing
For patients who do not improve with conservative management, further testing is recommended to characterize the underlying pathology. Typically, anorectal manometry (ARM) is performed in conjunction with the balloon expulsion test and imaging. Each modality has its strengths and limitations (see Table 1).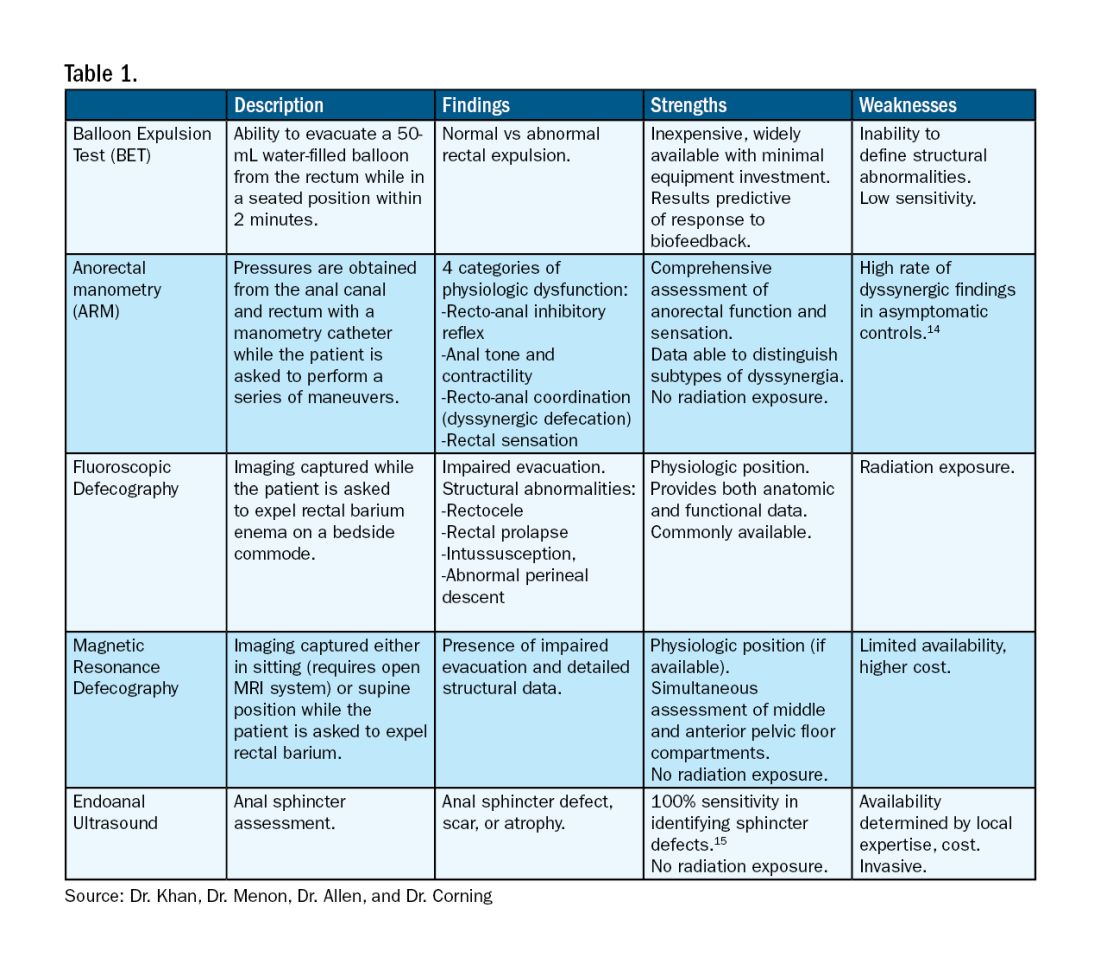
ARM allows for the assessment of rectal sensation and recto-anal pressures and coordination.10
Dynamic imaging, by barium defecography under fluoroscopy or MRI, captures anatomy at rest and with simulated defecation to identify pelvic organ prolapse, compartmental defects, and organ mobility.11 Endoanal ultrasonography is considered in patients experiencing fecal incontinence to evaluate the integrity of the anal sphincter muscles.
Minimally Invasive Procedures and Surgical Options for PFD
Functional abnormalities such as dyssynergia often coexist with structural abnormalities. Because structural abnormalities are commonly found in asymptomatic patients, noninvasive functional therapy, such as pelvic floor physical therapy and anorectal biofeedback, are preferred prior to surgical repair of a structural finding. For patients with fecal incontinence, sacral nerve stimulation (SNS) has emerged as a preferred therapy due to demonstrated efficacy in symptom improvement.12 Sphincteroplasty is reserved for those with acute sphincter injury or failure of SNS.

In patients with findings of intussusception, prolapse, or rectocele that have not responded to conservative therapy, referral for surgical repair may be considered. While the specific surgical approach will depend on many factors, the goal is typically excision and/or suspension of rectal tissue and reinforcement of the rectovaginal septum.
It is critical that we are equipped with the available knowledge and tools to provide these patients with optimal care.
Dr. Khan, Dr. Menon, Dr. Allen, and Dr. Corning are based at the University of Texas Medical Branch in Galveston, Texas. They report no conflicts of interest.
References
1. Grimes WR and Stratton M. Pelvic floor dysfunction. 2023 Jun 26. In: StatPearls [Internet]. Treasure Island (Fla.): StatPearls Publishing; 2024 Jan. PMID: 32644672.
2. Nygaard I et al. Prevalence of symptomatic pelvic floor disorders in US women. JAMA. 2008 Sep 17. doi: 10.1001/jama.300.11.1311.
3. Lawrence JM et al. Pelvic floor disorders, diabetes, and obesity in women: Findings from the Kaiser Permanente Continence Associated Risk Epidemiology Study. Diabetes Care. 2007 Oct. doi: 10.2337/dc07-0262.
4. Barber MD et al. Short forms of two condition-specific quality-of-life questionnaires for women with pelvic floor disorders (PFDI-20 and PFIQ-7). Am J Obstet Gynecol. 2005 Jul. doi: 10.1016/j.ajog.2004.12.025.
5. Persu C et al. Pelvic Organ Prolapse Quantification System (POP-Q) — A new era in pelvic prolapse staging. J Med Life. 2011 Jan-Mar. PMID: 21505577.
6. Wald A et al. ACG Clinical Guidelines: Management of benign anorectal disorders. Am J Gastroenterol. 2021 Oct 1. doi: 10.14309/ajg.0000000000001507.
7. Bharucha AE and Lacy BE. Mechanisms, evaluation, and management of chronic constipation. Gastroenterology. 2020 Apr. doi: 10.1053/j.gastro.2019.12.034.
8. Menees S and Chey WD. Fecal incontinence: Pathogenesis, diagnosis, and updated treatment strategies. Gastroenterol Clin North Am. 2022 Mar. doi: 10.1016/j.gtc.2021.10.005.
9. Wallace SL et al. Pelvic floor physical therapy in the treatment of pelvic floor dysfunction in women. Curr Opin Obstet Gynecol. 2019 Dec. doi: 10.1097/GCO.0000000000000584.
10. Carrington EV et al. The international anorectal physiology working group (IAPWG) recommendations: Standardized testing protocol and the London classification for disorders of anorectal function. Neurogastroenterol Motil. 2020 Jan. doi: 10.1111/nmo.13679.
11. El Sayed RF et al. Magnetic resonance imaging of pelvic floor dysfunction — Joint recommendations of the ESUR and ESGAR Pelvic Floor Working Group. Eur Radiol. 2017 May. doi: 10.1007/s00330-016-4471-7.
12. Thaha MA et al. Sacral nerve stimulation for faecal incontinence and constipation in adults. Cochrane Database Syst Rev. 2015 Aug 24. doi: 10.1002/14651858.CD004464.pub3.
13. Chiarioni G et al. Biofeedback benefits only patients with outlet dysfunction, not patients with isolated slow transit constipation. Gastroenterology. 2005 Jul. doi: 10.1053/j.gastro.2005.05.015.
14. Grossi U et al. Diagnostic accuracy study of anorectal manometry for diagnosis of dyssynergic defecation. Gut. 2016 Mar. doi: 10.1136/gutjnl-2014-308835.
15. Albuquerque A. Endoanal ultrasonography in fecal incontinence: Current and future perspectives. World J Gastrointest Endosc. 2015 Jun 10. doi: 10.4253/wjge.v7.i6.575.
Pelvic floor dysfunction (PFD) represents a spectrum of symptoms involving sensory and emptying abnormalities of the bowel and bladder and pelvic organ prolapse. The pelvic floor refers to a group of muscles that spans the pelvic outlet, providing support to the pelvic organs and coordinating constrictor mechanisms to control urination and defecation. Symptoms reported by patients experiencing PFD include involuntary loss of stool or urine, incomplete emptying of the bowel and bladder, a sensation of fullness, bulging in the vagina, and sexual dysfunction.1
As such, symptoms related to PFD are very common concerns raised by patients to their gastroenterologists. Data from the National Health and Nutrition Examination Survey show that 23.7% of women over the age of 20 had at least one symptom of PFD.2 Unfortunately, patients experiencing pelvic floor dysfunction often are hesitant to seek care because of embarrassment or perception that limited treatment options exist for their symptoms.
Pelvic Floor Anatomy
Regions of the pelvis are often referred to by anatomic compartment: anterior (bladder and urethra), middle (vagina and uterus or prostate), and posterior (colon, rectum, and anal canal). Supporting these compartments is the levator ani, a muscle group that is used synonymously with the term “pelvic diaphragm.”
Continence of stool is provided by the anal sphincter muscles and the puborectalis muscle, which wraps around the posterior aspect of the anorectal canal. Damage to the musculature or sensory perception to this area may result in fecal incontinence. Defecation is a coordinated process during which the abdominal and rectal muscles contract, while the anal sphincter muscles and puborectalis simultaneously relax. A disturbance in neuromuscular coordination (dyssynergic defecation) or structural pathology such as pelvic organ prolapse may lead to obstructed defecation.
PFD is thought to be a result of one or more insults to the pelvic floor such as chronic straining, childbirth, iatrogenic injury, or systemic disease such as diabetes.3
Evaluation of PFD Symptoms
Patients presenting with suspected PFD necessitate a comprehensive interdisciplinary assessment. In addition to obtaining a medical, surgical, and obstetric history, details about symptoms and lifestyle should include toileting habits, diet, and physical activity. The Pelvic Floor Distress Inventory (PFDI-20) is a commonly used tool that can be employed in the clinical setting.4
A pelvic exam can reveal pelvic organ prolapse and other mucosal pathology. The Pelvic Organ Prolapse Quantification System (POP-Q) is a widely used classification system for describing pelvic organ prolapse.5 Protrusion of the rectal wall into the vagina is referred to as a rectocele, while prolapse of small bowel into the upper posterior wall of the vagina is called an enterocele. While the finding of a rectocele on exam is common in parous women and may not cause any symptoms, a larger rectocele may cause a sensation of incomplete evacuation of stool.
A digital rectal exam (DRE) should be performed to assess pelvic floor function and help identify structural abnormalities.
Initial Management
A stepwise approach to the management of PFD can allow many patients to be effectively treated without the need for surgical intervention. For patients reporting liquid stool consistency, the evaluation should pivot toward the workup and management of diarrhea, which can easily overwhelm continence mechanisms and cause fecal incontinence. Fiber supplementation to normalize stool consistency is considered first-line therapy for patients presenting with both fecal incontinence and obstructed defecation. Other tools for fecal incontinence include avoiding foods that trigger diarrhea and use of loperamide.6 For patients with obstructed defecation, a trial of laxatives can be followed by a prescription agent if needed, such as a secretagogue or prokinetic.7
Vaginal splinting is a technique that can be used in patients with rectocele, whereby a finger is inserted into the vagina and pressure is applied on the posterior vaginal wall toward the rectum. Reducing the rectocele can facilitate emptying stool from the rectum and prevent leakage of retained stool.8 Similarly, use of rectal irrigation enemas can also help clear retained stool.
Pelvic floor physical therapists examine the strength, coordination, and tone of the pelvic floor muscles. When hypertonic musculature is present, manual interventions may be performed including trigger point release, myofascial release, and dry needling.9 When hypotonic musculature or dyssynergia is present, strengthening and neuromuscular re-education are recommended. Biofeedback can be administered via surface electromyography and/or balloon training to improve rectal sensitivity. Proper defecation techniques, including positioning, breathing, and behavioral modifications, improve clinical outcomes.
Diagnostic Testing
For patients who do not improve with conservative management, further testing is recommended to characterize the underlying pathology. Typically, anorectal manometry (ARM) is performed in conjunction with the balloon expulsion test and imaging. Each modality has its strengths and limitations (see Table 1).
ARM allows for the assessment of rectal sensation and recto-anal pressures and coordination.10
Dynamic imaging, by barium defecography under fluoroscopy or MRI, captures anatomy at rest and with simulated defecation to identify pelvic organ prolapse, compartmental defects, and organ mobility.11 Endoanal ultrasonography is considered in patients experiencing fecal incontinence to evaluate the integrity of the anal sphincter muscles.
Minimally Invasive Procedures and Surgical Options for PFD
Functional abnormalities such as dyssynergia often coexist with structural abnormalities. Because structural abnormalities are commonly found in asymptomatic patients, noninvasive functional therapy, such as pelvic floor physical therapy and anorectal biofeedback, are preferred prior to surgical repair of a structural finding. For patients with fecal incontinence, sacral nerve stimulation (SNS) has emerged as a preferred therapy due to demonstrated efficacy in symptom improvement.12 Sphincteroplasty is reserved for those with acute sphincter injury or failure of SNS.
In patients with findings of intussusception, prolapse, or rectocele that have not responded to conservative therapy, referral for surgical repair may be considered. While the specific surgical approach will depend on many factors, the goal is typically excision and/or suspension of rectal tissue and reinforcement of the rectovaginal septum.
It is critical that we are equipped with the available knowledge and tools to provide these patients with optimal care.
Dr. Khan, Dr. Menon, Dr. Allen, and Dr. Corning are based at the University of Texas Medical Branch in Galveston, Texas. They report no conflicts of interest.
References
1. Grimes WR and Stratton M. Pelvic floor dysfunction. 2023 Jun 26. In: StatPearls [Internet]. Treasure Island (Fla.): StatPearls Publishing; 2024 Jan. PMID: 32644672.
2. Nygaard I et al. Prevalence of symptomatic pelvic floor disorders in US women. JAMA. 2008 Sep 17. doi: 10.1001/jama.300.11.1311.
3. Lawrence JM et al. Pelvic floor disorders, diabetes, and obesity in women: Findings from the Kaiser Permanente Continence Associated Risk Epidemiology Study. Diabetes Care. 2007 Oct. doi: 10.2337/dc07-0262.
4. Barber MD et al. Short forms of two condition-specific quality-of-life questionnaires for women with pelvic floor disorders (PFDI-20 and PFIQ-7). Am J Obstet Gynecol. 2005 Jul. doi: 10.1016/j.ajog.2004.12.025.
5. Persu C et al. Pelvic Organ Prolapse Quantification System (POP-Q) — A new era in pelvic prolapse staging. J Med Life. 2011 Jan-Mar. PMID: 21505577.
6. Wald A et al. ACG Clinical Guidelines: Management of benign anorectal disorders. Am J Gastroenterol. 2021 Oct 1. doi: 10.14309/ajg.0000000000001507.
7. Bharucha AE and Lacy BE. Mechanisms, evaluation, and management of chronic constipation. Gastroenterology. 2020 Apr. doi: 10.1053/j.gastro.2019.12.034.
8. Menees S and Chey WD. Fecal incontinence: Pathogenesis, diagnosis, and updated treatment strategies. Gastroenterol Clin North Am. 2022 Mar. doi: 10.1016/j.gtc.2021.10.005.
9. Wallace SL et al. Pelvic floor physical therapy in the treatment of pelvic floor dysfunction in women. Curr Opin Obstet Gynecol. 2019 Dec. doi: 10.1097/GCO.0000000000000584.
10. Carrington EV et al. The international anorectal physiology working group (IAPWG) recommendations: Standardized testing protocol and the London classification for disorders of anorectal function. Neurogastroenterol Motil. 2020 Jan. doi: 10.1111/nmo.13679.
11. El Sayed RF et al. Magnetic resonance imaging of pelvic floor dysfunction — Joint recommendations of the ESUR and ESGAR Pelvic Floor Working Group. Eur Radiol. 2017 May. doi: 10.1007/s00330-016-4471-7.
12. Thaha MA et al. Sacral nerve stimulation for faecal incontinence and constipation in adults. Cochrane Database Syst Rev. 2015 Aug 24. doi: 10.1002/14651858.CD004464.pub3.
13. Chiarioni G et al. Biofeedback benefits only patients with outlet dysfunction, not patients with isolated slow transit constipation. Gastroenterology. 2005 Jul. doi: 10.1053/j.gastro.2005.05.015.
14. Grossi U et al. Diagnostic accuracy study of anorectal manometry for diagnosis of dyssynergic defecation. Gut. 2016 Mar. doi: 10.1136/gutjnl-2014-308835.
15. Albuquerque A. Endoanal ultrasonography in fecal incontinence: Current and future perspectives. World J Gastrointest Endosc. 2015 Jun 10. doi: 10.4253/wjge.v7.i6.575.
Pelvic floor dysfunction (PFD) represents a spectrum of symptoms involving sensory and emptying abnormalities of the bowel and bladder and pelvic organ prolapse. The pelvic floor refers to a group of muscles that spans the pelvic outlet, providing support to the pelvic organs and coordinating constrictor mechanisms to control urination and defecation. Symptoms reported by patients experiencing PFD include involuntary loss of stool or urine, incomplete emptying of the bowel and bladder, a sensation of fullness, bulging in the vagina, and sexual dysfunction.1
As such, symptoms related to PFD are very common concerns raised by patients to their gastroenterologists. Data from the National Health and Nutrition Examination Survey show that 23.7% of women over the age of 20 had at least one symptom of PFD.2 Unfortunately, patients experiencing pelvic floor dysfunction often are hesitant to seek care because of embarrassment or perception that limited treatment options exist for their symptoms.
Pelvic Floor Anatomy
Regions of the pelvis are often referred to by anatomic compartment: anterior (bladder and urethra), middle (vagina and uterus or prostate), and posterior (colon, rectum, and anal canal). Supporting these compartments is the levator ani, a muscle group that is used synonymously with the term “pelvic diaphragm.”
Continence of stool is provided by the anal sphincter muscles and the puborectalis muscle, which wraps around the posterior aspect of the anorectal canal. Damage to the musculature or sensory perception to this area may result in fecal incontinence. Defecation is a coordinated process during which the abdominal and rectal muscles contract, while the anal sphincter muscles and puborectalis simultaneously relax. A disturbance in neuromuscular coordination (dyssynergic defecation) or structural pathology such as pelvic organ prolapse may lead to obstructed defecation.
PFD is thought to be a result of one or more insults to the pelvic floor such as chronic straining, childbirth, iatrogenic injury, or systemic disease such as diabetes.3
Evaluation of PFD Symptoms
Patients presenting with suspected PFD necessitate a comprehensive interdisciplinary assessment. In addition to obtaining a medical, surgical, and obstetric history, details about symptoms and lifestyle should include toileting habits, diet, and physical activity. The Pelvic Floor Distress Inventory (PFDI-20) is a commonly used tool that can be employed in the clinical setting.4
A pelvic exam can reveal pelvic organ prolapse and other mucosal pathology. The Pelvic Organ Prolapse Quantification System (POP-Q) is a widely used classification system for describing pelvic organ prolapse.5 Protrusion of the rectal wall into the vagina is referred to as a rectocele, while prolapse of small bowel into the upper posterior wall of the vagina is called an enterocele. While the finding of a rectocele on exam is common in parous women and may not cause any symptoms, a larger rectocele may cause a sensation of incomplete evacuation of stool.
A digital rectal exam (DRE) should be performed to assess pelvic floor function and help identify structural abnormalities.
Initial Management
A stepwise approach to the management of PFD can allow many patients to be effectively treated without the need for surgical intervention. For patients reporting liquid stool consistency, the evaluation should pivot toward the workup and management of diarrhea, which can easily overwhelm continence mechanisms and cause fecal incontinence. Fiber supplementation to normalize stool consistency is considered first-line therapy for patients presenting with both fecal incontinence and obstructed defecation. Other tools for fecal incontinence include avoiding foods that trigger diarrhea and use of loperamide.6 For patients with obstructed defecation, a trial of laxatives can be followed by a prescription agent if needed, such as a secretagogue or prokinetic.7
Vaginal splinting is a technique that can be used in patients with rectocele, whereby a finger is inserted into the vagina and pressure is applied on the posterior vaginal wall toward the rectum. Reducing the rectocele can facilitate emptying stool from the rectum and prevent leakage of retained stool.8 Similarly, use of rectal irrigation enemas can also help clear retained stool.
Pelvic floor physical therapists examine the strength, coordination, and tone of the pelvic floor muscles. When hypertonic musculature is present, manual interventions may be performed including trigger point release, myofascial release, and dry needling.9 When hypotonic musculature or dyssynergia is present, strengthening and neuromuscular re-education are recommended. Biofeedback can be administered via surface electromyography and/or balloon training to improve rectal sensitivity. Proper defecation techniques, including positioning, breathing, and behavioral modifications, improve clinical outcomes.
Diagnostic Testing
For patients who do not improve with conservative management, further testing is recommended to characterize the underlying pathology. Typically, anorectal manometry (ARM) is performed in conjunction with the balloon expulsion test and imaging. Each modality has its strengths and limitations (see Table 1).
ARM allows for the assessment of rectal sensation and recto-anal pressures and coordination.10
Dynamic imaging, by barium defecography under fluoroscopy or MRI, captures anatomy at rest and with simulated defecation to identify pelvic organ prolapse, compartmental defects, and organ mobility.11 Endoanal ultrasonography is considered in patients experiencing fecal incontinence to evaluate the integrity of the anal sphincter muscles.
Minimally Invasive Procedures and Surgical Options for PFD
Functional abnormalities such as dyssynergia often coexist with structural abnormalities. Because structural abnormalities are commonly found in asymptomatic patients, noninvasive functional therapy, such as pelvic floor physical therapy and anorectal biofeedback, are preferred prior to surgical repair of a structural finding. For patients with fecal incontinence, sacral nerve stimulation (SNS) has emerged as a preferred therapy due to demonstrated efficacy in symptom improvement.12 Sphincteroplasty is reserved for those with acute sphincter injury or failure of SNS.
In patients with findings of intussusception, prolapse, or rectocele that have not responded to conservative therapy, referral for surgical repair may be considered. While the specific surgical approach will depend on many factors, the goal is typically excision and/or suspension of rectal tissue and reinforcement of the rectovaginal septum.
It is critical that we are equipped with the available knowledge and tools to provide these patients with optimal care.
Dr. Khan, Dr. Menon, Dr. Allen, and Dr. Corning are based at the University of Texas Medical Branch in Galveston, Texas. They report no conflicts of interest.
References
1. Grimes WR and Stratton M. Pelvic floor dysfunction. 2023 Jun 26. In: StatPearls [Internet]. Treasure Island (Fla.): StatPearls Publishing; 2024 Jan. PMID: 32644672.
2. Nygaard I et al. Prevalence of symptomatic pelvic floor disorders in US women. JAMA. 2008 Sep 17. doi: 10.1001/jama.300.11.1311.
3. Lawrence JM et al. Pelvic floor disorders, diabetes, and obesity in women: Findings from the Kaiser Permanente Continence Associated Risk Epidemiology Study. Diabetes Care. 2007 Oct. doi: 10.2337/dc07-0262.
4. Barber MD et al. Short forms of two condition-specific quality-of-life questionnaires for women with pelvic floor disorders (PFDI-20 and PFIQ-7). Am J Obstet Gynecol. 2005 Jul. doi: 10.1016/j.ajog.2004.12.025.
5. Persu C et al. Pelvic Organ Prolapse Quantification System (POP-Q) — A new era in pelvic prolapse staging. J Med Life. 2011 Jan-Mar. PMID: 21505577.
6. Wald A et al. ACG Clinical Guidelines: Management of benign anorectal disorders. Am J Gastroenterol. 2021 Oct 1. doi: 10.14309/ajg.0000000000001507.
7. Bharucha AE and Lacy BE. Mechanisms, evaluation, and management of chronic constipation. Gastroenterology. 2020 Apr. doi: 10.1053/j.gastro.2019.12.034.
8. Menees S and Chey WD. Fecal incontinence: Pathogenesis, diagnosis, and updated treatment strategies. Gastroenterol Clin North Am. 2022 Mar. doi: 10.1016/j.gtc.2021.10.005.
9. Wallace SL et al. Pelvic floor physical therapy in the treatment of pelvic floor dysfunction in women. Curr Opin Obstet Gynecol. 2019 Dec. doi: 10.1097/GCO.0000000000000584.
10. Carrington EV et al. The international anorectal physiology working group (IAPWG) recommendations: Standardized testing protocol and the London classification for disorders of anorectal function. Neurogastroenterol Motil. 2020 Jan. doi: 10.1111/nmo.13679.
11. El Sayed RF et al. Magnetic resonance imaging of pelvic floor dysfunction — Joint recommendations of the ESUR and ESGAR Pelvic Floor Working Group. Eur Radiol. 2017 May. doi: 10.1007/s00330-016-4471-7.
12. Thaha MA et al. Sacral nerve stimulation for faecal incontinence and constipation in adults. Cochrane Database Syst Rev. 2015 Aug 24. doi: 10.1002/14651858.CD004464.pub3.
13. Chiarioni G et al. Biofeedback benefits only patients with outlet dysfunction, not patients with isolated slow transit constipation. Gastroenterology. 2005 Jul. doi: 10.1053/j.gastro.2005.05.015.
14. Grossi U et al. Diagnostic accuracy study of anorectal manometry for diagnosis of dyssynergic defecation. Gut. 2016 Mar. doi: 10.1136/gutjnl-2014-308835.
15. Albuquerque A. Endoanal ultrasonography in fecal incontinence: Current and future perspectives. World J Gastrointest Endosc. 2015 Jun 10. doi: 10.4253/wjge.v7.i6.575.

Excess Thrombotic Risk in RA Has No Clear Driving Factor
LIVERPOOL, ENGLAND — People with rheumatoid arthritis (RA) have a consistently higher risk for venous thromboembolism (VTE) than the general population, but the reasons for this remain unclear, research presented at the annual meeting of the British Society for Rheumatology (BSR) reaffirmed.
Regardless of age, sex, body mass index (BMI), duration of disease, use of estrogen-based oral contraceptives, or hormone replacement therapy (HRT), people with RA are more likely to experience a pulmonary embolism or deep vein thrombosis than those without RA.
However, “these are rare events,” James Galloway, MBChB, PhD, professor of rheumatology and deputy head of the Centre for Rheumatic Diseases at King’s College London in England, said at the meeting.
In one analysis of data from 117,050 individuals living in England and Wales that are held within a large primary care practice database, Dr. Galloway and colleagues found that the unadjusted incidence of VTE in people diagnosed with RA (n = 23,410) was 0.44% vs 0.26% for matched controls within the general population (n = 93,640).
RA and VTE Risk
The overall risk for VTE was 46% higher among people with RA than among those without, although the absolute difference was small, Dr. Galloway reported.
“RA is associated with an increased risk of VTE; that’s been well described over the years,” Dr. Galloway told this news organization. Past research into why there is an elevated risk for VTE in patients with RA has often focused on the role of disease activity and inflammation.
“In the last few years, a new class of drugs, the JAK [Janus kinase] inhibitors, have emerged in which we have seen a signal of increased VTE risk from a number of studies. And I think that puts a spotlight on our understanding of VTE risk,” Dr. Galloway said.
He added “JAK inhibitors are very powerful at controlling inflammation, but if you take away inflammation, there is still an excess risk. What else could be driving that?”
To examine the excess risk for VTE seen in people with RA, Dr. Galloway and colleagues performed three separate analyses using data collected between January 1999 and December 2018 by the Royal College of General Practitioners Research and Surveillance Center.
One analysis looked at VTE risk according to age, sex, and BMI; another looked at the effect of the duration of RA; and a third analysis focused on the use of estrogen-based oral contraceptives or HRT.
For all three analyses, those with RA were matched in a 4:1 ratio to people from the general population without RA on the basis of current age, sex, calendar time, and years since registration at the primary care practice.
Observational Data Challenged
“These are observational data, so it’s important to weigh up the strengths and limitations,” Dr. Galloway acknowledged. Strengths are the large sample size and long follow-up provided by the database, which assesses and monitors more than 2000 primary care practices in England and Wales.
Confounding is still possible, despite adjusting for multiple factors that included sociodemographic factors; clinical features; and VTE risk factors such as smoking status, alcohol use, thrombophilia, reduced mobility, lower limb fracture, and a family history of VTE if data had been available. There wasn’t information on disease activity, for example, and disease duration was used as a surrogate marker for this.
Sitting in the audience, Marwan Bukhari, MBBS, PhD, challenged the population-matching process.
“Do you think maybe it was the matching that was the problem?” asked Dr. Bukhari, who is consultant rheumatologist at University Hospitals of Morecambe Bay NHS Foundation Trust and an honorary senior lecturer at the University of Manchester, both in England.
“They’re not entirely matched completely, correctly. Even if it is 4:1, there’s a difference between the populations,” he said.
Age, Sex, and Bodyweight
Over an average of 8.2 years’ follow-up, the adjusted hazard ratios (aHRs) comparing VTE risk in women and men with and without RA were a respective 1.62 and 1.52. The corresponding aHRs for VTE according to different age groups were 2.13 for age 18-49 years, 1.57 for age 50-69 years, and 1.34 for age 70 years and older.
“The highest excess risk was in the youngest age group,” Dr. Galloway pointed out, “but all age groups showing a significant increased risk of venous thromboembolism.”
Similar findings were seen across different BMI categories, with the highest risk occurring in those in the lowest BMI group. The aHRs were 1.66, 1.60, and 1.41 for the BMI categories of less than 25 kg/m2, 25-30 kg/m2, and more than 30 kg/m2, respectively.
Duration of RA
As for disease duration, nearly two thirds (63.9%) of the 23,410 adults with RA included in this analysis were included at or within 2 years of a diagnosis of RA, 7.8% within 2-5 years of diagnosis, 9.8% within 5-10 years of diagnosis, and 18.5% at 10 or more years after diagnosis.
The aHR for an increased relative risk for VTE in people with RA vs the control group ranged from 1.49 for 0-2 years of diagnosis up to 1.63 for more than 10 years since diagnosis.
“We could see no evidence that the VTE excess risk in rheumatoid arthritis was with a specific time since diagnosis,” Dr. Galloway said in the interview. “It appears that the risk is increased in people with established RA, whether you’ve had the disease for 2 years or 10 years.”
Similar findings were also seen when they looked at aHRs for pulmonary embolism (1.46-2.02) and deep vein thrombosis (1.43-1.89) separately.
Oral Contraceptives and HRT
Data on the use of estrogen-based oral contraceptives or HRT were detailed in a virtual poster presentation. In this analysis, there were 16,664 women with and 65,448 without RA, and the average follow-up was 8.3 years.
“The number of people available for this analysis was small, and bigger studies are needed,” Dr. Galloway said in the interview. Indeed, in the RA group, just 3.3% had used an estrogen-based oral contraceptive and 4.5% had used HRT compared with 3.9% and 3.8% in the control group, respectively.
The overall VTE risk was 52% higher in women with RA than in those without RA.
Risk for VTE was higher among women with RA regardless of the use of estrogen-based oral contraceptives or not (aHRs, 1.43 and 1.52, respectively) and regardless of the use of HRT or not (aHRs, 2.32 and 1.51).
Assess and Monitor
Together these data increase understanding of how age, gender, obesity, duration of disease, and estrogen-based contraception and HRT may make a difference to someone’s VTE risk.
“In all people with RA, we observe an increased risk of venous thromboembolism, and that is both relevant in a contemporary era when we think about prescribing and the different risks of drugs we use for therapeutic strategies,” Dr. Galloway said.
The overall take-home message, he said, is that VTE risk should be considered in everyone with RA and assessed and monitored accordingly. This includes those who may have traditionally been thought of as having a lower risk than others, such as men vs women, younger vs older individuals, and those who may have had RA for a few years.
The research was funded by Pfizer. Dr. Galloway reported receiving honoraria from Pfizer, AbbVie, Biovitrum, Bristol Myers Squibb, Celgene, Chugai, Galapagos, Janssen, Lilly, Novartis, Roche, Sanofi, Sobi, and UCB. Two coauthors of the work were employees of Pfizer. Dr. Bukhari had no conflicts of interest and was not involved in the research.
A version of this article appeared on Medscape.com.
LIVERPOOL, ENGLAND — People with rheumatoid arthritis (RA) have a consistently higher risk for venous thromboembolism (VTE) than the general population, but the reasons for this remain unclear, research presented at the annual meeting of the British Society for Rheumatology (BSR) reaffirmed.
Regardless of age, sex, body mass index (BMI), duration of disease, use of estrogen-based oral contraceptives, or hormone replacement therapy (HRT), people with RA are more likely to experience a pulmonary embolism or deep vein thrombosis than those without RA.
However, “these are rare events,” James Galloway, MBChB, PhD, professor of rheumatology and deputy head of the Centre for Rheumatic Diseases at King’s College London in England, said at the meeting.
In one analysis of data from 117,050 individuals living in England and Wales that are held within a large primary care practice database, Dr. Galloway and colleagues found that the unadjusted incidence of VTE in people diagnosed with RA (n = 23,410) was 0.44% vs 0.26% for matched controls within the general population (n = 93,640).
RA and VTE Risk
The overall risk for VTE was 46% higher among people with RA than among those without, although the absolute difference was small, Dr. Galloway reported.
“RA is associated with an increased risk of VTE; that’s been well described over the years,” Dr. Galloway told this news organization. Past research into why there is an elevated risk for VTE in patients with RA has often focused on the role of disease activity and inflammation.
“In the last few years, a new class of drugs, the JAK [Janus kinase] inhibitors, have emerged in which we have seen a signal of increased VTE risk from a number of studies. And I think that puts a spotlight on our understanding of VTE risk,” Dr. Galloway said.
He added “JAK inhibitors are very powerful at controlling inflammation, but if you take away inflammation, there is still an excess risk. What else could be driving that?”
To examine the excess risk for VTE seen in people with RA, Dr. Galloway and colleagues performed three separate analyses using data collected between January 1999 and December 2018 by the Royal College of General Practitioners Research and Surveillance Center.
One analysis looked at VTE risk according to age, sex, and BMI; another looked at the effect of the duration of RA; and a third analysis focused on the use of estrogen-based oral contraceptives or HRT.
For all three analyses, those with RA were matched in a 4:1 ratio to people from the general population without RA on the basis of current age, sex, calendar time, and years since registration at the primary care practice.
Observational Data Challenged
“These are observational data, so it’s important to weigh up the strengths and limitations,” Dr. Galloway acknowledged. Strengths are the large sample size and long follow-up provided by the database, which assesses and monitors more than 2000 primary care practices in England and Wales.
Confounding is still possible, despite adjusting for multiple factors that included sociodemographic factors; clinical features; and VTE risk factors such as smoking status, alcohol use, thrombophilia, reduced mobility, lower limb fracture, and a family history of VTE if data had been available. There wasn’t information on disease activity, for example, and disease duration was used as a surrogate marker for this.
Sitting in the audience, Marwan Bukhari, MBBS, PhD, challenged the population-matching process.
“Do you think maybe it was the matching that was the problem?” asked Dr. Bukhari, who is consultant rheumatologist at University Hospitals of Morecambe Bay NHS Foundation Trust and an honorary senior lecturer at the University of Manchester, both in England.
“They’re not entirely matched completely, correctly. Even if it is 4:1, there’s a difference between the populations,” he said.
Age, Sex, and Bodyweight
Over an average of 8.2 years’ follow-up, the adjusted hazard ratios (aHRs) comparing VTE risk in women and men with and without RA were a respective 1.62 and 1.52. The corresponding aHRs for VTE according to different age groups were 2.13 for age 18-49 years, 1.57 for age 50-69 years, and 1.34 for age 70 years and older.
“The highest excess risk was in the youngest age group,” Dr. Galloway pointed out, “but all age groups showing a significant increased risk of venous thromboembolism.”
Similar findings were seen across different BMI categories, with the highest risk occurring in those in the lowest BMI group. The aHRs were 1.66, 1.60, and 1.41 for the BMI categories of less than 25 kg/m2, 25-30 kg/m2, and more than 30 kg/m2, respectively.
Duration of RA
As for disease duration, nearly two thirds (63.9%) of the 23,410 adults with RA included in this analysis were included at or within 2 years of a diagnosis of RA, 7.8% within 2-5 years of diagnosis, 9.8% within 5-10 years of diagnosis, and 18.5% at 10 or more years after diagnosis.
The aHR for an increased relative risk for VTE in people with RA vs the control group ranged from 1.49 for 0-2 years of diagnosis up to 1.63 for more than 10 years since diagnosis.
“We could see no evidence that the VTE excess risk in rheumatoid arthritis was with a specific time since diagnosis,” Dr. Galloway said in the interview. “It appears that the risk is increased in people with established RA, whether you’ve had the disease for 2 years or 10 years.”
Similar findings were also seen when they looked at aHRs for pulmonary embolism (1.46-2.02) and deep vein thrombosis (1.43-1.89) separately.
Oral Contraceptives and HRT
Data on the use of estrogen-based oral contraceptives or HRT were detailed in a virtual poster presentation. In this analysis, there were 16,664 women with and 65,448 without RA, and the average follow-up was 8.3 years.
“The number of people available for this analysis was small, and bigger studies are needed,” Dr. Galloway said in the interview. Indeed, in the RA group, just 3.3% had used an estrogen-based oral contraceptive and 4.5% had used HRT compared with 3.9% and 3.8% in the control group, respectively.
The overall VTE risk was 52% higher in women with RA than in those without RA.
Risk for VTE was higher among women with RA regardless of the use of estrogen-based oral contraceptives or not (aHRs, 1.43 and 1.52, respectively) and regardless of the use of HRT or not (aHRs, 2.32 and 1.51).
Assess and Monitor
Together these data increase understanding of how age, gender, obesity, duration of disease, and estrogen-based contraception and HRT may make a difference to someone’s VTE risk.
“In all people with RA, we observe an increased risk of venous thromboembolism, and that is both relevant in a contemporary era when we think about prescribing and the different risks of drugs we use for therapeutic strategies,” Dr. Galloway said.
The overall take-home message, he said, is that VTE risk should be considered in everyone with RA and assessed and monitored accordingly. This includes those who may have traditionally been thought of as having a lower risk than others, such as men vs women, younger vs older individuals, and those who may have had RA for a few years.
The research was funded by Pfizer. Dr. Galloway reported receiving honoraria from Pfizer, AbbVie, Biovitrum, Bristol Myers Squibb, Celgene, Chugai, Galapagos, Janssen, Lilly, Novartis, Roche, Sanofi, Sobi, and UCB. Two coauthors of the work were employees of Pfizer. Dr. Bukhari had no conflicts of interest and was not involved in the research.
A version of this article appeared on Medscape.com.
LIVERPOOL, ENGLAND — People with rheumatoid arthritis (RA) have a consistently higher risk for venous thromboembolism (VTE) than the general population, but the reasons for this remain unclear, research presented at the annual meeting of the British Society for Rheumatology (BSR) reaffirmed.
Regardless of age, sex, body mass index (BMI), duration of disease, use of estrogen-based oral contraceptives, or hormone replacement therapy (HRT), people with RA are more likely to experience a pulmonary embolism or deep vein thrombosis than those without RA.
However, “these are rare events,” James Galloway, MBChB, PhD, professor of rheumatology and deputy head of the Centre for Rheumatic Diseases at King’s College London in England, said at the meeting.
In one analysis of data from 117,050 individuals living in England and Wales that are held within a large primary care practice database, Dr. Galloway and colleagues found that the unadjusted incidence of VTE in people diagnosed with RA (n = 23,410) was 0.44% vs 0.26% for matched controls within the general population (n = 93,640).
RA and VTE Risk
The overall risk for VTE was 46% higher among people with RA than among those without, although the absolute difference was small, Dr. Galloway reported.
“RA is associated with an increased risk of VTE; that’s been well described over the years,” Dr. Galloway told this news organization. Past research into why there is an elevated risk for VTE in patients with RA has often focused on the role of disease activity and inflammation.
“In the last few years, a new class of drugs, the JAK [Janus kinase] inhibitors, have emerged in which we have seen a signal of increased VTE risk from a number of studies. And I think that puts a spotlight on our understanding of VTE risk,” Dr. Galloway said.
He added “JAK inhibitors are very powerful at controlling inflammation, but if you take away inflammation, there is still an excess risk. What else could be driving that?”
To examine the excess risk for VTE seen in people with RA, Dr. Galloway and colleagues performed three separate analyses using data collected between January 1999 and December 2018 by the Royal College of General Practitioners Research and Surveillance Center.
One analysis looked at VTE risk according to age, sex, and BMI; another looked at the effect of the duration of RA; and a third analysis focused on the use of estrogen-based oral contraceptives or HRT.
For all three analyses, those with RA were matched in a 4:1 ratio to people from the general population without RA on the basis of current age, sex, calendar time, and years since registration at the primary care practice.
Observational Data Challenged
“These are observational data, so it’s important to weigh up the strengths and limitations,” Dr. Galloway acknowledged. Strengths are the large sample size and long follow-up provided by the database, which assesses and monitors more than 2000 primary care practices in England and Wales.
Confounding is still possible, despite adjusting for multiple factors that included sociodemographic factors; clinical features; and VTE risk factors such as smoking status, alcohol use, thrombophilia, reduced mobility, lower limb fracture, and a family history of VTE if data had been available. There wasn’t information on disease activity, for example, and disease duration was used as a surrogate marker for this.
Sitting in the audience, Marwan Bukhari, MBBS, PhD, challenged the population-matching process.
“Do you think maybe it was the matching that was the problem?” asked Dr. Bukhari, who is consultant rheumatologist at University Hospitals of Morecambe Bay NHS Foundation Trust and an honorary senior lecturer at the University of Manchester, both in England.
“They’re not entirely matched completely, correctly. Even if it is 4:1, there’s a difference between the populations,” he said.
Age, Sex, and Bodyweight
Over an average of 8.2 years’ follow-up, the adjusted hazard ratios (aHRs) comparing VTE risk in women and men with and without RA were a respective 1.62 and 1.52. The corresponding aHRs for VTE according to different age groups were 2.13 for age 18-49 years, 1.57 for age 50-69 years, and 1.34 for age 70 years and older.
“The highest excess risk was in the youngest age group,” Dr. Galloway pointed out, “but all age groups showing a significant increased risk of venous thromboembolism.”
Similar findings were seen across different BMI categories, with the highest risk occurring in those in the lowest BMI group. The aHRs were 1.66, 1.60, and 1.41 for the BMI categories of less than 25 kg/m2, 25-30 kg/m2, and more than 30 kg/m2, respectively.
Duration of RA
As for disease duration, nearly two thirds (63.9%) of the 23,410 adults with RA included in this analysis were included at or within 2 years of a diagnosis of RA, 7.8% within 2-5 years of diagnosis, 9.8% within 5-10 years of diagnosis, and 18.5% at 10 or more years after diagnosis.
The aHR for an increased relative risk for VTE in people with RA vs the control group ranged from 1.49 for 0-2 years of diagnosis up to 1.63 for more than 10 years since diagnosis.
“We could see no evidence that the VTE excess risk in rheumatoid arthritis was with a specific time since diagnosis,” Dr. Galloway said in the interview. “It appears that the risk is increased in people with established RA, whether you’ve had the disease for 2 years or 10 years.”
Similar findings were also seen when they looked at aHRs for pulmonary embolism (1.46-2.02) and deep vein thrombosis (1.43-1.89) separately.
Oral Contraceptives and HRT
Data on the use of estrogen-based oral contraceptives or HRT were detailed in a virtual poster presentation. In this analysis, there were 16,664 women with and 65,448 without RA, and the average follow-up was 8.3 years.
“The number of people available for this analysis was small, and bigger studies are needed,” Dr. Galloway said in the interview. Indeed, in the RA group, just 3.3% had used an estrogen-based oral contraceptive and 4.5% had used HRT compared with 3.9% and 3.8% in the control group, respectively.
The overall VTE risk was 52% higher in women with RA than in those without RA.
Risk for VTE was higher among women with RA regardless of the use of estrogen-based oral contraceptives or not (aHRs, 1.43 and 1.52, respectively) and regardless of the use of HRT or not (aHRs, 2.32 and 1.51).
Assess and Monitor
Together these data increase understanding of how age, gender, obesity, duration of disease, and estrogen-based contraception and HRT may make a difference to someone’s VTE risk.
“In all people with RA, we observe an increased risk of venous thromboembolism, and that is both relevant in a contemporary era when we think about prescribing and the different risks of drugs we use for therapeutic strategies,” Dr. Galloway said.
The overall take-home message, he said, is that VTE risk should be considered in everyone with RA and assessed and monitored accordingly. This includes those who may have traditionally been thought of as having a lower risk than others, such as men vs women, younger vs older individuals, and those who may have had RA for a few years.
The research was funded by Pfizer. Dr. Galloway reported receiving honoraria from Pfizer, AbbVie, Biovitrum, Bristol Myers Squibb, Celgene, Chugai, Galapagos, Janssen, Lilly, Novartis, Roche, Sanofi, Sobi, and UCB. Two coauthors of the work were employees of Pfizer. Dr. Bukhari had no conflicts of interest and was not involved in the research.
A version of this article appeared on Medscape.com.
FROM BSR 2024

Clinical Manifestation of Degos Disease: Painful Penile Ulcers
To the Editor:
A 56-year-old man was referred to our Grand Rounds by another dermatologist in our health system for evaluation of a red scaly rash on the trunk that had been present for more than a year. More recently, over the course of approximately 9 months he experienced recurrent painful penile ulcers that lasted for approximately 4 weeks and then self-resolved. He had a medical history of central retinal vein occlusion, primary hyperparathyroidism, and nonspecific colitis. A family history was notable for lung cancer in the patient’s father and myelodysplastic syndrome and breast cancer in his mother; however, there was no family history of a similar rash. A bacterial culture of the penile ulcer was negative. Testing for antibodies against HIV and herpes simplex virus (HSV) types 1 and 2 was negative. Results of a serum VDRL test were nonreactive, which ruled out syphilis. The patient was treated by the referring dermatologist with azithromycin for possible chancroid without relief.
The patient was being followed by the referring dermatologist who initially was concerned for Degos disease based on clinical examination findings, prompting biopsy of a lesion on the back, which revealed vacuolar interface dermatitis, a sparse superficial perivascular lymphocytic infiltrate, and increased mucin—all highly suspicious for connective tissue disease (Figure 1). An antinuclear antibody test was positive, with a titer of 1:640. The patient was started on prednisone and referred to rheumatology; however, further evaluation by rheumatology for an autoimmune process—including anticardiolipin antibodies—was unremarkable. A few months prior to the current presentation, he also had mildly elevated liver function test results. A colonoscopy was performed, and a biopsy revealed nonspecific colitis. A biopsy of the penile ulcer also was nonspecific, showing only ulceration and acute and chronic inflammation. No epidermal interface change was seen. Results from a Grocott-Gomori methenamine-silver stain, Treponema pallidum immunostain, and HSV polymerase chain reaction were negative for fungal organisms, spirochetes, and HSV, respectively. The differential diagnosis included trauma, aphthous ulceration, and Behçet disease. Behçet disease was suspected by the referring dermatologist, and the patient was treated with colchicine, prednisone, pimecrolimus cream, and topical lidocaine; however, the lesions persisted, and he was subsequently referred to our Grand Rounds for further evaluation.

At the current presentation, physical examination revealed several small papules with white atrophic centers and erythematous rims on the trunk and extremities (Figure 2A). An ulceration was noted on the penile shaft (Figure 2B). Further evaluation for Behçet disease, including testing for pathergy and HLA-B51, was negative. Degos disease was strongly suspected clinically, and a repeat biopsy was performed of a lesion on the abdomen, which revealed central epidermal necrosis, atrophy, and parakeratosis with an underlying wedge-shaped dermal infarct surrounded by multiple small occluded dermal vessels, perivascular inflammation, and dermal edema (Figure 3). Direct immunofluorescence was performed using antibodies against IgG, IgA, IgM, fibrinogen, albumin, and C3, which was negative. These findings from direct immunofluorescence and histopathology as well as the clinical presentation were considered compatible with Degos disease. The patient was started on aspirin and pentoxifylline. Pentoxifylline 400 mg twice daily appeared to lessen some of the pain. Pain management specialists started the patient on gabapentin.

Approximately 4 months after the Grand Rounds evaluation, during which time he continued treatment with pentoxifylline, he was admitted to the hospital for intractable nausea and vomiting. His condition acutely declined due to bowel perforation, and he was started on eculizumab 1200 mg every 14 days. Because of an increased risk for meningococcal meningitis while on this medication, he also was given erythromycin 500 mg twice daily prophylactically. He was being followed by hematology for the vasculopathy, and they were planning to monitor for any disease changes with computed tomography of the chest, abdomen, and pelvis every 3 months, as well as echocardiogram every 6 months for any development of pericardial or pleural fibrosis. Approximately 1 month later, the patient was admitted to the hospital again but died after 1 week from gastrointestinal complications (approximately 22 months after the onset of the rash).

Degos disease (atrophic papulosis) is a rare small vessel vasculopathy of unknown etiology, but complement-mediated endothelial injury plays a role.1,2 It typically occurs in the fourth decade of life, with a slight female predominance.3,4 The skin lesions are characteristic and described as 5- to 10-mm papules with atrophic white centers and erythematous telangiectatic rims, most commonly on the upper body and typically sparing the head, palms, and soles.1 Penile ulceration is an uncommon cutaneous feature, with only a few cases reported in the literature.5,6 Approximately one-third of patients will have only skin lesions, but two-thirds will develop systemic involvement 1 to 2 years after onset, with the gastrointestinal tract and central nervous system most commonly involved. For those with systemic involvement, the 5-year survival rate is approximately 55%, and the most common causes of death are bowel perforation, peritonitis, and stroke.3,4 Because some patients appear to never develop systemic complications, Theodoridis et al4 proposed that the disease be classified as either malignant atrophic papulosis or benign atrophic papulosis to indicate the malignant systemic form and the benign cutaneous form, respectively.
The histopathology of Degos disease changes as the lesions evolve.7 Early lesions show a superficial and deep perivascular and periadnexal lymphocytic infiltrate, possible interface dermatitis, and dermal mucin resembling lupus. The more fully developed lesions show a greater degree of inflammation and interface change as well as lymphocytic vasculitis. This stage also may have epidermal atrophy and early papillary dermal sclerosis resembling lichen sclerosus. The late-stage lesions, clinically observed as papules with atrophic white centers and surrounding erythema, show the classic pathology of wedge-shaped dermal sclerosis and central epidermal atrophy with surrounding hyperkeratosis. Interface dermatitis and dermal mucin can be seen in all stages, though mucin is diminished in the later stage.
Effective treatment options are limited; however, antithrombotics or compounds that facilitate blood perfusion, such as aspirin or pentoxifylline, initially can be used.1 Eculizumab, a humanized monoclonal antibody that prevents the cleavage of C5, has been used for salvage therapy,8 as in our case. Treprostinil, a prostacyclin analog that causes arterial vasodilation and inhibition of platelet aggregation, has been reported to improve bowel and cutaneous lesions, functional status, and neurologic symptoms.9
Our case highlights important features of Degos disease. First, it is important for both the clinician and the pathologist to recognize that the histopathology of Degos disease changes as the lesions evolve. In our case, although the lesions were characteristic of Degos disease clinically, the initial biopsy was suspicious for connective tissue disease, which led to an autoimmune evaluation that ultimately was unremarkable. Recognizing that early lesions of Degos disease can resemble connective tissue disease histologically could have prevented this delay in diagnosis. However, Degos disease has been reported in association with autoimmune diseases.10 Second, although penile ulceration is uncommon, it can be a prominent cutaneous manifestation of the disease. Finally, eculizumab and treprostinil are therapeutic options that have shown some efficacy in improving symptoms and cutaneous lesions.8,9
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease)—a review. Orphanet J Rare Dis. 2013;8:10. doi:10.1186/1750-1172-8-10
- Magro CM, Poe JC, Kim C, et al. Degos disease: a C5b-9/interferon-α-mediated endotheliopathy syndrome. Am J Clin Pathol. 2011;135:599-610. doi:10.1309/AJCP66QIMFARLZKI
- Hu P, Mao Z, Liu C, et al. Malignant atrophic papulosis with motor aphasia and intestinal perforation: a case report and review of published works. J Dermatol. 2018;45:723-726. doi:10.1111/1346-8138.14280
- Theodoridis A, Konstantinidou A, Makrantonaki E, et al. Malignant and benign forms of atrophic papulosis (Köhlmeier-Degos disease): systemic involvement determines the prognosis. Br J Dermatol. 2014;170:110-115. doi:10.1111/bjd.12642
- Thomson KF, Highet AS. Penile ulceration in fatal malignant atrophic papulosis (Degos’ disease). Br J Dermatol. 2000;143:1320-1322. doi:10.1046/j.1365-2133.2000.03911.x
- Aydogan K, Alkan G, Karadogan Koran S, et al. Painful penile ulceration in a patient with malignant atrophic papulosis. J Eur Acad Dermatol Venereol. 2005;19:612-616. doi:10.1111/j.1468-3083.2005.01227.x
- Harvell JD, Williford PL, White WL. Benign cutaneous Degos’ disease: a case report with emphasis on histopathology as papules chronologically evolve. Am J Dermatopathol. 2001;23:116-123. doi:10.1097/00000372-200104000-00006
- Oliver B, Boehm M, Rosing DR, et al. Diffuse atrophic papules and plaques, intermittent abdominal pain, paresthesias, and cardiac abnormalities in a 55-year-old woman. J Am Acad Dermatol. 2016;75:1274-1277. doi:10.1016/j.jaad.2016.09.015
- Shapiro LS, Toledo-Garcia AE, Farrell JF. Effective treatment of malignant atrophic papulosis (Köhlmeier-Degos disease) with treprostinil—early experience. Orphanet J Rare Dis. 2013;8:52. doi:10.1186/1750-1172-8-52
- Burgin S, Stone JH, Shenoy-Bhangle AS, et al. Case records of the Massachusetts General Hospital. Case 18-2014. A 32-year-old man with a rash, myalgia, and weakness. N Engl J Med. 2014;370:2327-2337. doi:10.1056/NEJMcpc1304161
To the Editor:
A 56-year-old man was referred to our Grand Rounds by another dermatologist in our health system for evaluation of a red scaly rash on the trunk that had been present for more than a year. More recently, over the course of approximately 9 months he experienced recurrent painful penile ulcers that lasted for approximately 4 weeks and then self-resolved. He had a medical history of central retinal vein occlusion, primary hyperparathyroidism, and nonspecific colitis. A family history was notable for lung cancer in the patient’s father and myelodysplastic syndrome and breast cancer in his mother; however, there was no family history of a similar rash. A bacterial culture of the penile ulcer was negative. Testing for antibodies against HIV and herpes simplex virus (HSV) types 1 and 2 was negative. Results of a serum VDRL test were nonreactive, which ruled out syphilis. The patient was treated by the referring dermatologist with azithromycin for possible chancroid without relief.
The patient was being followed by the referring dermatologist who initially was concerned for Degos disease based on clinical examination findings, prompting biopsy of a lesion on the back, which revealed vacuolar interface dermatitis, a sparse superficial perivascular lymphocytic infiltrate, and increased mucin—all highly suspicious for connective tissue disease (Figure 1). An antinuclear antibody test was positive, with a titer of 1:640. The patient was started on prednisone and referred to rheumatology; however, further evaluation by rheumatology for an autoimmune process—including anticardiolipin antibodies—was unremarkable. A few months prior to the current presentation, he also had mildly elevated liver function test results. A colonoscopy was performed, and a biopsy revealed nonspecific colitis. A biopsy of the penile ulcer also was nonspecific, showing only ulceration and acute and chronic inflammation. No epidermal interface change was seen. Results from a Grocott-Gomori methenamine-silver stain, Treponema pallidum immunostain, and HSV polymerase chain reaction were negative for fungal organisms, spirochetes, and HSV, respectively. The differential diagnosis included trauma, aphthous ulceration, and Behçet disease. Behçet disease was suspected by the referring dermatologist, and the patient was treated with colchicine, prednisone, pimecrolimus cream, and topical lidocaine; however, the lesions persisted, and he was subsequently referred to our Grand Rounds for further evaluation.
At the current presentation, physical examination revealed several small papules with white atrophic centers and erythematous rims on the trunk and extremities (Figure 2A). An ulceration was noted on the penile shaft (Figure 2B). Further evaluation for Behçet disease, including testing for pathergy and HLA-B51, was negative. Degos disease was strongly suspected clinically, and a repeat biopsy was performed of a lesion on the abdomen, which revealed central epidermal necrosis, atrophy, and parakeratosis with an underlying wedge-shaped dermal infarct surrounded by multiple small occluded dermal vessels, perivascular inflammation, and dermal edema (Figure 3). Direct immunofluorescence was performed using antibodies against IgG, IgA, IgM, fibrinogen, albumin, and C3, which was negative. These findings from direct immunofluorescence and histopathology as well as the clinical presentation were considered compatible with Degos disease. The patient was started on aspirin and pentoxifylline. Pentoxifylline 400 mg twice daily appeared to lessen some of the pain. Pain management specialists started the patient on gabapentin.
Approximately 4 months after the Grand Rounds evaluation, during which time he continued treatment with pentoxifylline, he was admitted to the hospital for intractable nausea and vomiting. His condition acutely declined due to bowel perforation, and he was started on eculizumab 1200 mg every 14 days. Because of an increased risk for meningococcal meningitis while on this medication, he also was given erythromycin 500 mg twice daily prophylactically. He was being followed by hematology for the vasculopathy, and they were planning to monitor for any disease changes with computed tomography of the chest, abdomen, and pelvis every 3 months, as well as echocardiogram every 6 months for any development of pericardial or pleural fibrosis. Approximately 1 month later, the patient was admitted to the hospital again but died after 1 week from gastrointestinal complications (approximately 22 months after the onset of the rash).
Degos disease (atrophic papulosis) is a rare small vessel vasculopathy of unknown etiology, but complement-mediated endothelial injury plays a role.1,2 It typically occurs in the fourth decade of life, with a slight female predominance.3,4 The skin lesions are characteristic and described as 5- to 10-mm papules with atrophic white centers and erythematous telangiectatic rims, most commonly on the upper body and typically sparing the head, palms, and soles.1 Penile ulceration is an uncommon cutaneous feature, with only a few cases reported in the literature.5,6 Approximately one-third of patients will have only skin lesions, but two-thirds will develop systemic involvement 1 to 2 years after onset, with the gastrointestinal tract and central nervous system most commonly involved. For those with systemic involvement, the 5-year survival rate is approximately 55%, and the most common causes of death are bowel perforation, peritonitis, and stroke.3,4 Because some patients appear to never develop systemic complications, Theodoridis et al4 proposed that the disease be classified as either malignant atrophic papulosis or benign atrophic papulosis to indicate the malignant systemic form and the benign cutaneous form, respectively.
The histopathology of Degos disease changes as the lesions evolve.7 Early lesions show a superficial and deep perivascular and periadnexal lymphocytic infiltrate, possible interface dermatitis, and dermal mucin resembling lupus. The more fully developed lesions show a greater degree of inflammation and interface change as well as lymphocytic vasculitis. This stage also may have epidermal atrophy and early papillary dermal sclerosis resembling lichen sclerosus. The late-stage lesions, clinically observed as papules with atrophic white centers and surrounding erythema, show the classic pathology of wedge-shaped dermal sclerosis and central epidermal atrophy with surrounding hyperkeratosis. Interface dermatitis and dermal mucin can be seen in all stages, though mucin is diminished in the later stage.
Effective treatment options are limited; however, antithrombotics or compounds that facilitate blood perfusion, such as aspirin or pentoxifylline, initially can be used.1 Eculizumab, a humanized monoclonal antibody that prevents the cleavage of C5, has been used for salvage therapy,8 as in our case. Treprostinil, a prostacyclin analog that causes arterial vasodilation and inhibition of platelet aggregation, has been reported to improve bowel and cutaneous lesions, functional status, and neurologic symptoms.9
Our case highlights important features of Degos disease. First, it is important for both the clinician and the pathologist to recognize that the histopathology of Degos disease changes as the lesions evolve. In our case, although the lesions were characteristic of Degos disease clinically, the initial biopsy was suspicious for connective tissue disease, which led to an autoimmune evaluation that ultimately was unremarkable. Recognizing that early lesions of Degos disease can resemble connective tissue disease histologically could have prevented this delay in diagnosis. However, Degos disease has been reported in association with autoimmune diseases.10 Second, although penile ulceration is uncommon, it can be a prominent cutaneous manifestation of the disease. Finally, eculizumab and treprostinil are therapeutic options that have shown some efficacy in improving symptoms and cutaneous lesions.8,9
To the Editor:
A 56-year-old man was referred to our Grand Rounds by another dermatologist in our health system for evaluation of a red scaly rash on the trunk that had been present for more than a year. More recently, over the course of approximately 9 months he experienced recurrent painful penile ulcers that lasted for approximately 4 weeks and then self-resolved. He had a medical history of central retinal vein occlusion, primary hyperparathyroidism, and nonspecific colitis. A family history was notable for lung cancer in the patient’s father and myelodysplastic syndrome and breast cancer in his mother; however, there was no family history of a similar rash. A bacterial culture of the penile ulcer was negative. Testing for antibodies against HIV and herpes simplex virus (HSV) types 1 and 2 was negative. Results of a serum VDRL test were nonreactive, which ruled out syphilis. The patient was treated by the referring dermatologist with azithromycin for possible chancroid without relief.
The patient was being followed by the referring dermatologist who initially was concerned for Degos disease based on clinical examination findings, prompting biopsy of a lesion on the back, which revealed vacuolar interface dermatitis, a sparse superficial perivascular lymphocytic infiltrate, and increased mucin—all highly suspicious for connective tissue disease (Figure 1). An antinuclear antibody test was positive, with a titer of 1:640. The patient was started on prednisone and referred to rheumatology; however, further evaluation by rheumatology for an autoimmune process—including anticardiolipin antibodies—was unremarkable. A few months prior to the current presentation, he also had mildly elevated liver function test results. A colonoscopy was performed, and a biopsy revealed nonspecific colitis. A biopsy of the penile ulcer also was nonspecific, showing only ulceration and acute and chronic inflammation. No epidermal interface change was seen. Results from a Grocott-Gomori methenamine-silver stain, Treponema pallidum immunostain, and HSV polymerase chain reaction were negative for fungal organisms, spirochetes, and HSV, respectively. The differential diagnosis included trauma, aphthous ulceration, and Behçet disease. Behçet disease was suspected by the referring dermatologist, and the patient was treated with colchicine, prednisone, pimecrolimus cream, and topical lidocaine; however, the lesions persisted, and he was subsequently referred to our Grand Rounds for further evaluation.
At the current presentation, physical examination revealed several small papules with white atrophic centers and erythematous rims on the trunk and extremities (Figure 2A). An ulceration was noted on the penile shaft (Figure 2B). Further evaluation for Behçet disease, including testing for pathergy and HLA-B51, was negative. Degos disease was strongly suspected clinically, and a repeat biopsy was performed of a lesion on the abdomen, which revealed central epidermal necrosis, atrophy, and parakeratosis with an underlying wedge-shaped dermal infarct surrounded by multiple small occluded dermal vessels, perivascular inflammation, and dermal edema (Figure 3). Direct immunofluorescence was performed using antibodies against IgG, IgA, IgM, fibrinogen, albumin, and C3, which was negative. These findings from direct immunofluorescence and histopathology as well as the clinical presentation were considered compatible with Degos disease. The patient was started on aspirin and pentoxifylline. Pentoxifylline 400 mg twice daily appeared to lessen some of the pain. Pain management specialists started the patient on gabapentin.
Approximately 4 months after the Grand Rounds evaluation, during which time he continued treatment with pentoxifylline, he was admitted to the hospital for intractable nausea and vomiting. His condition acutely declined due to bowel perforation, and he was started on eculizumab 1200 mg every 14 days. Because of an increased risk for meningococcal meningitis while on this medication, he also was given erythromycin 500 mg twice daily prophylactically. He was being followed by hematology for the vasculopathy, and they were planning to monitor for any disease changes with computed tomography of the chest, abdomen, and pelvis every 3 months, as well as echocardiogram every 6 months for any development of pericardial or pleural fibrosis. Approximately 1 month later, the patient was admitted to the hospital again but died after 1 week from gastrointestinal complications (approximately 22 months after the onset of the rash).
Degos disease (atrophic papulosis) is a rare small vessel vasculopathy of unknown etiology, but complement-mediated endothelial injury plays a role.1,2 It typically occurs in the fourth decade of life, with a slight female predominance.3,4 The skin lesions are characteristic and described as 5- to 10-mm papules with atrophic white centers and erythematous telangiectatic rims, most commonly on the upper body and typically sparing the head, palms, and soles.1 Penile ulceration is an uncommon cutaneous feature, with only a few cases reported in the literature.5,6 Approximately one-third of patients will have only skin lesions, but two-thirds will develop systemic involvement 1 to 2 years after onset, with the gastrointestinal tract and central nervous system most commonly involved. For those with systemic involvement, the 5-year survival rate is approximately 55%, and the most common causes of death are bowel perforation, peritonitis, and stroke.3,4 Because some patients appear to never develop systemic complications, Theodoridis et al4 proposed that the disease be classified as either malignant atrophic papulosis or benign atrophic papulosis to indicate the malignant systemic form and the benign cutaneous form, respectively.
The histopathology of Degos disease changes as the lesions evolve.7 Early lesions show a superficial and deep perivascular and periadnexal lymphocytic infiltrate, possible interface dermatitis, and dermal mucin resembling lupus. The more fully developed lesions show a greater degree of inflammation and interface change as well as lymphocytic vasculitis. This stage also may have epidermal atrophy and early papillary dermal sclerosis resembling lichen sclerosus. The late-stage lesions, clinically observed as papules with atrophic white centers and surrounding erythema, show the classic pathology of wedge-shaped dermal sclerosis and central epidermal atrophy with surrounding hyperkeratosis. Interface dermatitis and dermal mucin can be seen in all stages, though mucin is diminished in the later stage.
Effective treatment options are limited; however, antithrombotics or compounds that facilitate blood perfusion, such as aspirin or pentoxifylline, initially can be used.1 Eculizumab, a humanized monoclonal antibody that prevents the cleavage of C5, has been used for salvage therapy,8 as in our case. Treprostinil, a prostacyclin analog that causes arterial vasodilation and inhibition of platelet aggregation, has been reported to improve bowel and cutaneous lesions, functional status, and neurologic symptoms.9
Our case highlights important features of Degos disease. First, it is important for both the clinician and the pathologist to recognize that the histopathology of Degos disease changes as the lesions evolve. In our case, although the lesions were characteristic of Degos disease clinically, the initial biopsy was suspicious for connective tissue disease, which led to an autoimmune evaluation that ultimately was unremarkable. Recognizing that early lesions of Degos disease can resemble connective tissue disease histologically could have prevented this delay in diagnosis. However, Degos disease has been reported in association with autoimmune diseases.10 Second, although penile ulceration is uncommon, it can be a prominent cutaneous manifestation of the disease. Finally, eculizumab and treprostinil are therapeutic options that have shown some efficacy in improving symptoms and cutaneous lesions.8,9
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease)—a review. Orphanet J Rare Dis. 2013;8:10. doi:10.1186/1750-1172-8-10
- Magro CM, Poe JC, Kim C, et al. Degos disease: a C5b-9/interferon-α-mediated endotheliopathy syndrome. Am J Clin Pathol. 2011;135:599-610. doi:10.1309/AJCP66QIMFARLZKI
- Hu P, Mao Z, Liu C, et al. Malignant atrophic papulosis with motor aphasia and intestinal perforation: a case report and review of published works. J Dermatol. 2018;45:723-726. doi:10.1111/1346-8138.14280
- Theodoridis A, Konstantinidou A, Makrantonaki E, et al. Malignant and benign forms of atrophic papulosis (Köhlmeier-Degos disease): systemic involvement determines the prognosis. Br J Dermatol. 2014;170:110-115. doi:10.1111/bjd.12642
- Thomson KF, Highet AS. Penile ulceration in fatal malignant atrophic papulosis (Degos’ disease). Br J Dermatol. 2000;143:1320-1322. doi:10.1046/j.1365-2133.2000.03911.x
- Aydogan K, Alkan G, Karadogan Koran S, et al. Painful penile ulceration in a patient with malignant atrophic papulosis. J Eur Acad Dermatol Venereol. 2005;19:612-616. doi:10.1111/j.1468-3083.2005.01227.x
- Harvell JD, Williford PL, White WL. Benign cutaneous Degos’ disease: a case report with emphasis on histopathology as papules chronologically evolve. Am J Dermatopathol. 2001;23:116-123. doi:10.1097/00000372-200104000-00006
- Oliver B, Boehm M, Rosing DR, et al. Diffuse atrophic papules and plaques, intermittent abdominal pain, paresthesias, and cardiac abnormalities in a 55-year-old woman. J Am Acad Dermatol. 2016;75:1274-1277. doi:10.1016/j.jaad.2016.09.015
- Shapiro LS, Toledo-Garcia AE, Farrell JF. Effective treatment of malignant atrophic papulosis (Köhlmeier-Degos disease) with treprostinil—early experience. Orphanet J Rare Dis. 2013;8:52. doi:10.1186/1750-1172-8-52
- Burgin S, Stone JH, Shenoy-Bhangle AS, et al. Case records of the Massachusetts General Hospital. Case 18-2014. A 32-year-old man with a rash, myalgia, and weakness. N Engl J Med. 2014;370:2327-2337. doi:10.1056/NEJMcpc1304161
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease)—a review. Orphanet J Rare Dis. 2013;8:10. doi:10.1186/1750-1172-8-10
- Magro CM, Poe JC, Kim C, et al. Degos disease: a C5b-9/interferon-α-mediated endotheliopathy syndrome. Am J Clin Pathol. 2011;135:599-610. doi:10.1309/AJCP66QIMFARLZKI
- Hu P, Mao Z, Liu C, et al. Malignant atrophic papulosis with motor aphasia and intestinal perforation: a case report and review of published works. J Dermatol. 2018;45:723-726. doi:10.1111/1346-8138.14280
- Theodoridis A, Konstantinidou A, Makrantonaki E, et al. Malignant and benign forms of atrophic papulosis (Köhlmeier-Degos disease): systemic involvement determines the prognosis. Br J Dermatol. 2014;170:110-115. doi:10.1111/bjd.12642
- Thomson KF, Highet AS. Penile ulceration in fatal malignant atrophic papulosis (Degos’ disease). Br J Dermatol. 2000;143:1320-1322. doi:10.1046/j.1365-2133.2000.03911.x
- Aydogan K, Alkan G, Karadogan Koran S, et al. Painful penile ulceration in a patient with malignant atrophic papulosis. J Eur Acad Dermatol Venereol. 2005;19:612-616. doi:10.1111/j.1468-3083.2005.01227.x
- Harvell JD, Williford PL, White WL. Benign cutaneous Degos’ disease: a case report with emphasis on histopathology as papules chronologically evolve. Am J Dermatopathol. 2001;23:116-123. doi:10.1097/00000372-200104000-00006
- Oliver B, Boehm M, Rosing DR, et al. Diffuse atrophic papules and plaques, intermittent abdominal pain, paresthesias, and cardiac abnormalities in a 55-year-old woman. J Am Acad Dermatol. 2016;75:1274-1277. doi:10.1016/j.jaad.2016.09.015
- Shapiro LS, Toledo-Garcia AE, Farrell JF. Effective treatment of malignant atrophic papulosis (Köhlmeier-Degos disease) with treprostinil—early experience. Orphanet J Rare Dis. 2013;8:52. doi:10.1186/1750-1172-8-52
- Burgin S, Stone JH, Shenoy-Bhangle AS, et al. Case records of the Massachusetts General Hospital. Case 18-2014. A 32-year-old man with a rash, myalgia, and weakness. N Engl J Med. 2014;370:2327-2337. doi:10.1056/NEJMcpc1304161
PRACTICE POINTS
- Papules with atrophic white centers and erythematous telangiectatic rims are the characteristic skin lesions found in Degos disease.
- A painful penile ulceration also may occur in Degos disease, though it is uncommon.
- The histopathology of skin lesions changes as the lesions evolve. Early lesions may resemble connective tissue disease. Late lesions show the classic pathology of wedge-shaped dermal sclerosis.

Mandatory DMV Reporting Tied to Dementia Underdiagnosis
, new research suggests.
Investigators found that primary care physicians (PCPs) in states with clinician reporting mandates had a 59% higher probability of underdiagnosing dementia compared with their counterparts in states that require patients to self-report or that have no reporting mandates.
“Our findings in this cross-sectional study raise concerns about potential adverse effects of mandatory clinician reporting for dementia diagnosis and underscore the need for careful consideration of the effect of such policies,” wrote the investigators, led by Soeren Mattke, MD, DSc, director of the USC Brain Health Observatory and research professor of economics at the University of Southern California, Los Angeles.
The study was published online in JAMA Network Open.
Lack of Guidance
As the US population ages, the number of older drivers is increasing, with 55.8 million drivers 65 years old or older. Approximately 7 million people in this age group have dementia — an estimate that is expected to increase to nearly 12 million by 2040.
The aging population raises a “critical policy question” about how to ensure road safety. Although the American Medical Association’s Code of Ethics outlines a physician’s obligation to identify drivers with medical impairments that impede safe driving, guidance restricting cognitively impaired drivers from driving is lacking.
In addition, evidence as to whether cognitive impairment indeed poses a threat to driving safety is mixed and has led to a lack of uniform policies with respect to reporting dementia.
Four states explicitly require clinicians to report dementia diagnoses to the DMV, which will then determine the patient’s fitness to drive, whereas 14 states require people with dementia to self-report. The remaining states have no explicit reporting requirements.
The issue of mandatory reporting is controversial, the researchers noted. On the one hand, physicians could protect patients and others by reporting potentially unsafe drivers.
On the other hand, evidence of an association with lower accident risks in patients with dementia is sparse and mandatory reporting may adversely affect physician-patient relationships. Empirical evidence for unintended consequences of reporting laws is lacking.
To examine the potential link between dementia underdiagnosis and mandatory reporting policies, the investigators analyzed the 100% data from the Medicare fee-for-service program and Medicare Advantage plans from 2017 to 2019, which included 223,036 PCPs with a panel of 25 or more Medicare patients.
The researchers examined dementia diagnosis rates in the patient panel of PCPs, rather than neurologists or gerontologists, regardless of who documented the diagnosis. Dr. Mattke said that it is possible that the diagnosis was established after referral to a specialist.
Each physician’s expected number of dementia cases was estimated using a predictive model based on patient characteristics. The researchers then compared the estimate with observed dementia diagnoses, thereby identifying clinicians who underdiagnosed dementia after sampling errors were accounted for.
‘Heavy-Handed Interference’
The researchers adjusted for several covariates potentially associated with a clinician’s probability of underdiagnosing dementia. These included sex, office location, practice specialty, racial/ethnic composition of the patient panel, and percentage of patients dually eligible for Medicare and Medicaid. The table shows PCP characteristics.
Adjusted results showed that PCPs practicing in states with clinician reporting mandates had a 12.4% (95% confidence interval [CI], 10.5%-14.2%) probability of underdiagnosing dementia versus 7.8% (95% CI, 6.9%-8.7%) in states with self-reporting and 7.7% (95% CI, 6.9%-8.4%) in states with no mandates, translating into a 4–percentage point difference (P < .001).
“Our study is the first to provide empirical evidence for the potential adverse effects of reporting policies,” the researchers noted. “Although we found that some clinicians underdiagnosed dementia regardless of state mandates, the key finding of this study reveals that primary care clinicians who practice in states with clinician reporting mandates were 59% more likely to do so…compared with those states with no reporting requirements…or driver self-reporting requirements.”
The investigators suggested that one potential explanation for underdiagnosis is patient resistance to cognitive testing. If patients were aware that the clinician was obligated by law to report their dementia diagnosis to the DMV, “they might be more inclined to conceal their symptoms or refuse further assessments, in addition to the general stigma and resistance to a formal assessment after a positive dementia screening result.”
“The findings suggest that policymakers might want to rethink those physician reporting mandates, since we also could not find conclusive evidence that they improve road safety,” Dr. Mattke said. “Maybe patients and their physicians can arrive at a sensible approach to determine driving fitness without such heavy-handed interference.”
However, he cautioned that the findings are not definitive and further study is needed before firm recommendations either for or against mandatory reporting.
In addition, the researchers noted several study limitations. One is that dementia underdiagnosis may also be associated with factors not captured in their model, including physician-patient relationships, health literacy, or language barriers.
However, Dr. Mattke noted, “ my sense is that those unobservable factors are not systematically related to state reporting policies and having omitted them would therefore not bias our results.”
Experts Weigh In
Commenting on the research, Morgan Daven, MA, the Alzheimer’s Association vice president of health systems, said that dementia is widely and significantly underdiagnosed, and not only in the states with dementia reporting mandates. Many factors may contribute to underdiagnosis, and although the study shows an association between reporting mandates and underdiagnosis, it does not demonstrate causation.
That said, Mr. Daven added, “fear and stigma related to dementia may inhibit the clinician, the patient, and their family from pursuing detection and diagnosis for dementia. As a society, we need to address dementia fear and stigma for all parties.”
He noted that useful tools include healthcare policies, workforce training, public awareness and education, and public policies to mitigate fear and stigma and their negative effects on diagnosis, care, support, and communication.
A potential study limitation is that it relied only on diagnoses by PCPs. Mr. Daven noted that the diagnosis of Alzheimer’ disease — the most common cause of dementia — is confirmation of amyloid buildup via a biomarker test, using PET or cerebrospinal fluid analysis.
“Both of these tests are extremely limited in their use and accessibility in a primary care setting. Inclusion of diagnoses by dementia specialists would provide a more complete picture,” he said.
Mr. Daven added that the Alzheimer’s Association encourages families to proactively discuss driving and other disease-related safety concerns as soon as possible. The Alzheimer’s Association Dementia and Driving webpage offers tips and strategies to discuss driving concerns with a family member.
In an accompanying editorial, Donald Redelmeier, MD, MS(HSR), and Vidhi Bhatt, BSc, both of the Department of Medicine, University of Toronto, differentiate the mandate for physicians to warn patients with dementia about traffic safety from the mandate for reporting child maltreatment, gunshot victims, or communicable diseases. They noted that mandated warnings “are not easy, can engender patient dissatisfaction, and need to be handled with tact.”
Yet, they pointed out, “breaking bad news is what practicing medicine entails.” They emphasized that, regardless of government mandates, “counseling patients for more road safety is an essential skill for clinicians in diverse states who hope to help their patients avoid becoming more traffic statistics.”
Research reported in this publication was supported by Genentech, a member of the Roche Group, and a grant from the National Institute on Aging of the National Institutes of Health. Dr. Mattke reported receiving grants from Genentech for a research contract with USC during the conduct of the study; personal fees from Eisai, Biogen, C2N, Novo Nordisk, Novartis, and Roche Genentech; and serving on the Senscio Systems board of directors, ALZpath scientific advisory board, AiCure scientific advisory board, and Boston Millennia Partners scientific advisory board outside the submitted work. The other authors’ disclosures are listed on the original paper. The editorial was supported by the Canada Research Chair in Medical Decision Sciences, the Canadian Institutes of Health Research, Kimel-Schatzky Traumatic Brain Injury Research Fund, and the Graduate Diploma Program in Health Research at the University of Toronto. The editorial authors report no other relevant financial relationships.
A version of this article appeared on Medscape.com.
, new research suggests.
Investigators found that primary care physicians (PCPs) in states with clinician reporting mandates had a 59% higher probability of underdiagnosing dementia compared with their counterparts in states that require patients to self-report or that have no reporting mandates.
“Our findings in this cross-sectional study raise concerns about potential adverse effects of mandatory clinician reporting for dementia diagnosis and underscore the need for careful consideration of the effect of such policies,” wrote the investigators, led by Soeren Mattke, MD, DSc, director of the USC Brain Health Observatory and research professor of economics at the University of Southern California, Los Angeles.
The study was published online in JAMA Network Open.
Lack of Guidance
As the US population ages, the number of older drivers is increasing, with 55.8 million drivers 65 years old or older. Approximately 7 million people in this age group have dementia — an estimate that is expected to increase to nearly 12 million by 2040.
The aging population raises a “critical policy question” about how to ensure road safety. Although the American Medical Association’s Code of Ethics outlines a physician’s obligation to identify drivers with medical impairments that impede safe driving, guidance restricting cognitively impaired drivers from driving is lacking.
In addition, evidence as to whether cognitive impairment indeed poses a threat to driving safety is mixed and has led to a lack of uniform policies with respect to reporting dementia.
Four states explicitly require clinicians to report dementia diagnoses to the DMV, which will then determine the patient’s fitness to drive, whereas 14 states require people with dementia to self-report. The remaining states have no explicit reporting requirements.
The issue of mandatory reporting is controversial, the researchers noted. On the one hand, physicians could protect patients and others by reporting potentially unsafe drivers.
On the other hand, evidence of an association with lower accident risks in patients with dementia is sparse and mandatory reporting may adversely affect physician-patient relationships. Empirical evidence for unintended consequences of reporting laws is lacking.
To examine the potential link between dementia underdiagnosis and mandatory reporting policies, the investigators analyzed the 100% data from the Medicare fee-for-service program and Medicare Advantage plans from 2017 to 2019, which included 223,036 PCPs with a panel of 25 or more Medicare patients.
The researchers examined dementia diagnosis rates in the patient panel of PCPs, rather than neurologists or gerontologists, regardless of who documented the diagnosis. Dr. Mattke said that it is possible that the diagnosis was established after referral to a specialist.
Each physician’s expected number of dementia cases was estimated using a predictive model based on patient characteristics. The researchers then compared the estimate with observed dementia diagnoses, thereby identifying clinicians who underdiagnosed dementia after sampling errors were accounted for.
‘Heavy-Handed Interference’
The researchers adjusted for several covariates potentially associated with a clinician’s probability of underdiagnosing dementia. These included sex, office location, practice specialty, racial/ethnic composition of the patient panel, and percentage of patients dually eligible for Medicare and Medicaid. The table shows PCP characteristics.
Adjusted results showed that PCPs practicing in states with clinician reporting mandates had a 12.4% (95% confidence interval [CI], 10.5%-14.2%) probability of underdiagnosing dementia versus 7.8% (95% CI, 6.9%-8.7%) in states with self-reporting and 7.7% (95% CI, 6.9%-8.4%) in states with no mandates, translating into a 4–percentage point difference (P < .001).
“Our study is the first to provide empirical evidence for the potential adverse effects of reporting policies,” the researchers noted. “Although we found that some clinicians underdiagnosed dementia regardless of state mandates, the key finding of this study reveals that primary care clinicians who practice in states with clinician reporting mandates were 59% more likely to do so…compared with those states with no reporting requirements…or driver self-reporting requirements.”
The investigators suggested that one potential explanation for underdiagnosis is patient resistance to cognitive testing. If patients were aware that the clinician was obligated by law to report their dementia diagnosis to the DMV, “they might be more inclined to conceal their symptoms or refuse further assessments, in addition to the general stigma and resistance to a formal assessment after a positive dementia screening result.”
“The findings suggest that policymakers might want to rethink those physician reporting mandates, since we also could not find conclusive evidence that they improve road safety,” Dr. Mattke said. “Maybe patients and their physicians can arrive at a sensible approach to determine driving fitness without such heavy-handed interference.”
However, he cautioned that the findings are not definitive and further study is needed before firm recommendations either for or against mandatory reporting.
In addition, the researchers noted several study limitations. One is that dementia underdiagnosis may also be associated with factors not captured in their model, including physician-patient relationships, health literacy, or language barriers.
However, Dr. Mattke noted, “ my sense is that those unobservable factors are not systematically related to state reporting policies and having omitted them would therefore not bias our results.”
Experts Weigh In
Commenting on the research, Morgan Daven, MA, the Alzheimer’s Association vice president of health systems, said that dementia is widely and significantly underdiagnosed, and not only in the states with dementia reporting mandates. Many factors may contribute to underdiagnosis, and although the study shows an association between reporting mandates and underdiagnosis, it does not demonstrate causation.
That said, Mr. Daven added, “fear and stigma related to dementia may inhibit the clinician, the patient, and their family from pursuing detection and diagnosis for dementia. As a society, we need to address dementia fear and stigma for all parties.”
He noted that useful tools include healthcare policies, workforce training, public awareness and education, and public policies to mitigate fear and stigma and their negative effects on diagnosis, care, support, and communication.
A potential study limitation is that it relied only on diagnoses by PCPs. Mr. Daven noted that the diagnosis of Alzheimer’ disease — the most common cause of dementia — is confirmation of amyloid buildup via a biomarker test, using PET or cerebrospinal fluid analysis.
“Both of these tests are extremely limited in their use and accessibility in a primary care setting. Inclusion of diagnoses by dementia specialists would provide a more complete picture,” he said.
Mr. Daven added that the Alzheimer’s Association encourages families to proactively discuss driving and other disease-related safety concerns as soon as possible. The Alzheimer’s Association Dementia and Driving webpage offers tips and strategies to discuss driving concerns with a family member.
In an accompanying editorial, Donald Redelmeier, MD, MS(HSR), and Vidhi Bhatt, BSc, both of the Department of Medicine, University of Toronto, differentiate the mandate for physicians to warn patients with dementia about traffic safety from the mandate for reporting child maltreatment, gunshot victims, or communicable diseases. They noted that mandated warnings “are not easy, can engender patient dissatisfaction, and need to be handled with tact.”
Yet, they pointed out, “breaking bad news is what practicing medicine entails.” They emphasized that, regardless of government mandates, “counseling patients for more road safety is an essential skill for clinicians in diverse states who hope to help their patients avoid becoming more traffic statistics.”
Research reported in this publication was supported by Genentech, a member of the Roche Group, and a grant from the National Institute on Aging of the National Institutes of Health. Dr. Mattke reported receiving grants from Genentech for a research contract with USC during the conduct of the study; personal fees from Eisai, Biogen, C2N, Novo Nordisk, Novartis, and Roche Genentech; and serving on the Senscio Systems board of directors, ALZpath scientific advisory board, AiCure scientific advisory board, and Boston Millennia Partners scientific advisory board outside the submitted work. The other authors’ disclosures are listed on the original paper. The editorial was supported by the Canada Research Chair in Medical Decision Sciences, the Canadian Institutes of Health Research, Kimel-Schatzky Traumatic Brain Injury Research Fund, and the Graduate Diploma Program in Health Research at the University of Toronto. The editorial authors report no other relevant financial relationships.
A version of this article appeared on Medscape.com.
, new research suggests.
Investigators found that primary care physicians (PCPs) in states with clinician reporting mandates had a 59% higher probability of underdiagnosing dementia compared with their counterparts in states that require patients to self-report or that have no reporting mandates.
“Our findings in this cross-sectional study raise concerns about potential adverse effects of mandatory clinician reporting for dementia diagnosis and underscore the need for careful consideration of the effect of such policies,” wrote the investigators, led by Soeren Mattke, MD, DSc, director of the USC Brain Health Observatory and research professor of economics at the University of Southern California, Los Angeles.
The study was published online in JAMA Network Open.
Lack of Guidance
As the US population ages, the number of older drivers is increasing, with 55.8 million drivers 65 years old or older. Approximately 7 million people in this age group have dementia — an estimate that is expected to increase to nearly 12 million by 2040.
The aging population raises a “critical policy question” about how to ensure road safety. Although the American Medical Association’s Code of Ethics outlines a physician’s obligation to identify drivers with medical impairments that impede safe driving, guidance restricting cognitively impaired drivers from driving is lacking.
In addition, evidence as to whether cognitive impairment indeed poses a threat to driving safety is mixed and has led to a lack of uniform policies with respect to reporting dementia.
Four states explicitly require clinicians to report dementia diagnoses to the DMV, which will then determine the patient’s fitness to drive, whereas 14 states require people with dementia to self-report. The remaining states have no explicit reporting requirements.
The issue of mandatory reporting is controversial, the researchers noted. On the one hand, physicians could protect patients and others by reporting potentially unsafe drivers.
On the other hand, evidence of an association with lower accident risks in patients with dementia is sparse and mandatory reporting may adversely affect physician-patient relationships. Empirical evidence for unintended consequences of reporting laws is lacking.
To examine the potential link between dementia underdiagnosis and mandatory reporting policies, the investigators analyzed the 100% data from the Medicare fee-for-service program and Medicare Advantage plans from 2017 to 2019, which included 223,036 PCPs with a panel of 25 or more Medicare patients.
The researchers examined dementia diagnosis rates in the patient panel of PCPs, rather than neurologists or gerontologists, regardless of who documented the diagnosis. Dr. Mattke said that it is possible that the diagnosis was established after referral to a specialist.
Each physician’s expected number of dementia cases was estimated using a predictive model based on patient characteristics. The researchers then compared the estimate with observed dementia diagnoses, thereby identifying clinicians who underdiagnosed dementia after sampling errors were accounted for.
‘Heavy-Handed Interference’
The researchers adjusted for several covariates potentially associated with a clinician’s probability of underdiagnosing dementia. These included sex, office location, practice specialty, racial/ethnic composition of the patient panel, and percentage of patients dually eligible for Medicare and Medicaid. The table shows PCP characteristics.
Adjusted results showed that PCPs practicing in states with clinician reporting mandates had a 12.4% (95% confidence interval [CI], 10.5%-14.2%) probability of underdiagnosing dementia versus 7.8% (95% CI, 6.9%-8.7%) in states with self-reporting and 7.7% (95% CI, 6.9%-8.4%) in states with no mandates, translating into a 4–percentage point difference (P < .001).
“Our study is the first to provide empirical evidence for the potential adverse effects of reporting policies,” the researchers noted. “Although we found that some clinicians underdiagnosed dementia regardless of state mandates, the key finding of this study reveals that primary care clinicians who practice in states with clinician reporting mandates were 59% more likely to do so…compared with those states with no reporting requirements…or driver self-reporting requirements.”
The investigators suggested that one potential explanation for underdiagnosis is patient resistance to cognitive testing. If patients were aware that the clinician was obligated by law to report their dementia diagnosis to the DMV, “they might be more inclined to conceal their symptoms or refuse further assessments, in addition to the general stigma and resistance to a formal assessment after a positive dementia screening result.”
“The findings suggest that policymakers might want to rethink those physician reporting mandates, since we also could not find conclusive evidence that they improve road safety,” Dr. Mattke said. “Maybe patients and their physicians can arrive at a sensible approach to determine driving fitness without such heavy-handed interference.”
However, he cautioned that the findings are not definitive and further study is needed before firm recommendations either for or against mandatory reporting.
In addition, the researchers noted several study limitations. One is that dementia underdiagnosis may also be associated with factors not captured in their model, including physician-patient relationships, health literacy, or language barriers.
However, Dr. Mattke noted, “ my sense is that those unobservable factors are not systematically related to state reporting policies and having omitted them would therefore not bias our results.”
Experts Weigh In
Commenting on the research, Morgan Daven, MA, the Alzheimer’s Association vice president of health systems, said that dementia is widely and significantly underdiagnosed, and not only in the states with dementia reporting mandates. Many factors may contribute to underdiagnosis, and although the study shows an association between reporting mandates and underdiagnosis, it does not demonstrate causation.
That said, Mr. Daven added, “fear and stigma related to dementia may inhibit the clinician, the patient, and their family from pursuing detection and diagnosis for dementia. As a society, we need to address dementia fear and stigma for all parties.”
He noted that useful tools include healthcare policies, workforce training, public awareness and education, and public policies to mitigate fear and stigma and their negative effects on diagnosis, care, support, and communication.
A potential study limitation is that it relied only on diagnoses by PCPs. Mr. Daven noted that the diagnosis of Alzheimer’ disease — the most common cause of dementia — is confirmation of amyloid buildup via a biomarker test, using PET or cerebrospinal fluid analysis.
“Both of these tests are extremely limited in their use and accessibility in a primary care setting. Inclusion of diagnoses by dementia specialists would provide a more complete picture,” he said.
Mr. Daven added that the Alzheimer’s Association encourages families to proactively discuss driving and other disease-related safety concerns as soon as possible. The Alzheimer’s Association Dementia and Driving webpage offers tips and strategies to discuss driving concerns with a family member.
In an accompanying editorial, Donald Redelmeier, MD, MS(HSR), and Vidhi Bhatt, BSc, both of the Department of Medicine, University of Toronto, differentiate the mandate for physicians to warn patients with dementia about traffic safety from the mandate for reporting child maltreatment, gunshot victims, or communicable diseases. They noted that mandated warnings “are not easy, can engender patient dissatisfaction, and need to be handled with tact.”
Yet, they pointed out, “breaking bad news is what practicing medicine entails.” They emphasized that, regardless of government mandates, “counseling patients for more road safety is an essential skill for clinicians in diverse states who hope to help their patients avoid becoming more traffic statistics.”
Research reported in this publication was supported by Genentech, a member of the Roche Group, and a grant from the National Institute on Aging of the National Institutes of Health. Dr. Mattke reported receiving grants from Genentech for a research contract with USC during the conduct of the study; personal fees from Eisai, Biogen, C2N, Novo Nordisk, Novartis, and Roche Genentech; and serving on the Senscio Systems board of directors, ALZpath scientific advisory board, AiCure scientific advisory board, and Boston Millennia Partners scientific advisory board outside the submitted work. The other authors’ disclosures are listed on the original paper. The editorial was supported by the Canada Research Chair in Medical Decision Sciences, the Canadian Institutes of Health Research, Kimel-Schatzky Traumatic Brain Injury Research Fund, and the Graduate Diploma Program in Health Research at the University of Toronto. The editorial authors report no other relevant financial relationships.
A version of this article appeared on Medscape.com.
From JAMA Network Open
Defining Your ‘Success’
Dear Friends,
The prevailing theme of this issue is “Success.” I have learned that “success” is personal and personalized. What “success” looked like 10, or even 5, years ago to me is very different from how I perceive it now; and I know it may be different 5 years from now. My definition of success should not look like another’s — that was the best advice I have gotten over the years and it has kept me constantly redefining what is important to me and placing value on where I want to allocate my time and efforts, at work and at home.
This issue of The New Gastroenterologist highlights topics from successful GIs within their own realms of expertise, offering insights on advancing in academic medicine, navigating financial wellness with a financial adviser, and becoming a future leader in GI.

In this issue’s clinically-focused articles, we spotlight two very nuanced and challenging topics. Dr. Sachin Srinivasan and Dr. Prateek Sharma review Barrett’s esophagus management for our “In Focus” section, with a particular emphasis on Barrett’s endoscopic therapy modalities for dysplasia and early neoplasia. Dr. Brooke Corning and team simplify their approach to pelvic floor dysfunction (PFD) in our “Short Clinical Reviews.” They suggest validated ways to assess patient history, pros and cons of various diagnostic tests, and stepwise management of PFD.
Navigating academic promotion can be overwhelming and may not be at the forefront with our early career GIs’ priorities. In our “Early Career” section, Dr. Vineet Rolston interviews two highly accomplished professors in academic medicine, Dr. Sophie Balzora and Dr. Mark Schattner, for their insights into the promotion process and recommendations for junior faculty.
Dr. Anjuli K. Luthra, a therapeutic endoscopist and founder of The Scope of Finance, emphasizes financial wellness for physicians. She breaks down the search for a financial adviser, including the different types, what to ask when searching for the right fit, and what to expect.
Lastly, this issue highlights an AGA program that invests in the development of leaders for the field — the Future Leaders Program (FLP). Dr. Parakkal Deepak and Dr. Edward L. Barnes, along with their mentor, Dr. Aasma Shaukat, describe their experience as a mentee-mentor triad of FLP and how this program has impacted their careers.
If you are interested in contributing or have ideas for future TNG topics, please contact me ([email protected]), or Danielle Kiefer ([email protected]), managing editor of TNG.
Until next time, I leave you with a historical fun fact because we would not be where we are now without appreciating where we were: Dr. C.G. Stockton was the first AGA president in 1897, a Professor of the Principles and Practice of Medicine and Clinical Medicine at the University of Buffalo in New York, and published on the relationship between GI/Hepatology and gout in the Journal of the American Medical Association the same year of his presidency.
Yours truly,
Judy A. Trieu, MD, MPH
Editor-in-Chief
Interventional Endoscopy, Division of Gastroenterology
Washington University in St. Louis
Dear Friends,
The prevailing theme of this issue is “Success.” I have learned that “success” is personal and personalized. What “success” looked like 10, or even 5, years ago to me is very different from how I perceive it now; and I know it may be different 5 years from now. My definition of success should not look like another’s — that was the best advice I have gotten over the years and it has kept me constantly redefining what is important to me and placing value on where I want to allocate my time and efforts, at work and at home.
This issue of The New Gastroenterologist highlights topics from successful GIs within their own realms of expertise, offering insights on advancing in academic medicine, navigating financial wellness with a financial adviser, and becoming a future leader in GI.
In this issue’s clinically-focused articles, we spotlight two very nuanced and challenging topics. Dr. Sachin Srinivasan and Dr. Prateek Sharma review Barrett’s esophagus management for our “In Focus” section, with a particular emphasis on Barrett’s endoscopic therapy modalities for dysplasia and early neoplasia. Dr. Brooke Corning and team simplify their approach to pelvic floor dysfunction (PFD) in our “Short Clinical Reviews.” They suggest validated ways to assess patient history, pros and cons of various diagnostic tests, and stepwise management of PFD.
Navigating academic promotion can be overwhelming and may not be at the forefront with our early career GIs’ priorities. In our “Early Career” section, Dr. Vineet Rolston interviews two highly accomplished professors in academic medicine, Dr. Sophie Balzora and Dr. Mark Schattner, for their insights into the promotion process and recommendations for junior faculty.
Dr. Anjuli K. Luthra, a therapeutic endoscopist and founder of The Scope of Finance, emphasizes financial wellness for physicians. She breaks down the search for a financial adviser, including the different types, what to ask when searching for the right fit, and what to expect.
Lastly, this issue highlights an AGA program that invests in the development of leaders for the field — the Future Leaders Program (FLP). Dr. Parakkal Deepak and Dr. Edward L. Barnes, along with their mentor, Dr. Aasma Shaukat, describe their experience as a mentee-mentor triad of FLP and how this program has impacted their careers.
If you are interested in contributing or have ideas for future TNG topics, please contact me ([email protected]), or Danielle Kiefer ([email protected]), managing editor of TNG.
Until next time, I leave you with a historical fun fact because we would not be where we are now without appreciating where we were: Dr. C.G. Stockton was the first AGA president in 1897, a Professor of the Principles and Practice of Medicine and Clinical Medicine at the University of Buffalo in New York, and published on the relationship between GI/Hepatology and gout in the Journal of the American Medical Association the same year of his presidency.
Yours truly,
Judy A. Trieu, MD, MPH
Editor-in-Chief
Interventional Endoscopy, Division of Gastroenterology
Washington University in St. Louis
Dear Friends,
The prevailing theme of this issue is “Success.” I have learned that “success” is personal and personalized. What “success” looked like 10, or even 5, years ago to me is very different from how I perceive it now; and I know it may be different 5 years from now. My definition of success should not look like another’s — that was the best advice I have gotten over the years and it has kept me constantly redefining what is important to me and placing value on where I want to allocate my time and efforts, at work and at home.
This issue of The New Gastroenterologist highlights topics from successful GIs within their own realms of expertise, offering insights on advancing in academic medicine, navigating financial wellness with a financial adviser, and becoming a future leader in GI.
In this issue’s clinically-focused articles, we spotlight two very nuanced and challenging topics. Dr. Sachin Srinivasan and Dr. Prateek Sharma review Barrett’s esophagus management for our “In Focus” section, with a particular emphasis on Barrett’s endoscopic therapy modalities for dysplasia and early neoplasia. Dr. Brooke Corning and team simplify their approach to pelvic floor dysfunction (PFD) in our “Short Clinical Reviews.” They suggest validated ways to assess patient history, pros and cons of various diagnostic tests, and stepwise management of PFD.
Navigating academic promotion can be overwhelming and may not be at the forefront with our early career GIs’ priorities. In our “Early Career” section, Dr. Vineet Rolston interviews two highly accomplished professors in academic medicine, Dr. Sophie Balzora and Dr. Mark Schattner, for their insights into the promotion process and recommendations for junior faculty.
Dr. Anjuli K. Luthra, a therapeutic endoscopist and founder of The Scope of Finance, emphasizes financial wellness for physicians. She breaks down the search for a financial adviser, including the different types, what to ask when searching for the right fit, and what to expect.
Lastly, this issue highlights an AGA program that invests in the development of leaders for the field — the Future Leaders Program (FLP). Dr. Parakkal Deepak and Dr. Edward L. Barnes, along with their mentor, Dr. Aasma Shaukat, describe their experience as a mentee-mentor triad of FLP and how this program has impacted their careers.
If you are interested in contributing or have ideas for future TNG topics, please contact me ([email protected]), or Danielle Kiefer ([email protected]), managing editor of TNG.
Until next time, I leave you with a historical fun fact because we would not be where we are now without appreciating where we were: Dr. C.G. Stockton was the first AGA president in 1897, a Professor of the Principles and Practice of Medicine and Clinical Medicine at the University of Buffalo in New York, and published on the relationship between GI/Hepatology and gout in the Journal of the American Medical Association the same year of his presidency.
Yours truly,
Judy A. Trieu, MD, MPH
Editor-in-Chief
Interventional Endoscopy, Division of Gastroenterology
Washington University in St. Louis