User login
FDA approves first tocilizumab biosimilar
The Food and Drug Administration has approved the biosimilar tocilizumab-bavi (Tofidence), Biogen, the drug’s manufacturer, announced on Sept. 29.
It is the first tocilizumab biosimilar approved by the FDA. The reference product, Actemra (Genentech), was first approved by the agency in 2010.
“The approval of Tofidence in the U.S. marks another positive step toward helping more people with chronic autoimmune conditions gain access to leading therapies,” Ian Henshaw, global head of biosimilars at Biogen, said in a statement. “With the increasing numbers of approved biosimilars, we expect increased savings and sustainability for health care systems and an increase in physician choice and patient access to biologics.”
Biogen’s pricing for tocilizumab-bavi will be available closer to the product’s launch date, which has yet to be determined, a company spokesman said. The U.S. average monthly cost of Actemra for rheumatoid arthritis, administered intravenously, is $2,134-$4,268 depending on dosage, according to a Genentech spokesperson.
Tocilizumab-bavi is an intravenous formulation (20 mg/mL) indicated for treatment of moderately to severely active RA, polyarticular juvenile idiopathic arthritis (PJIA), and systemic juvenile idiopathic arthritis (SJIA). The medication is administered every 4 weeks in RA and PJIA and every 8 weeks in SJIA as a single intravenous drip infusion over 1 hour.
The European Commission approved its first tocilizumab biosimilar, Tyenne (Fresenius Kabi), earlier in 2023 in both subcutaneous and intravenous formulations. Biogen did not comment on whether the company is working on a subcutaneous formulation for tocilizumab-bavi.
A version of this article appeared on Medscape.com.
The Food and Drug Administration has approved the biosimilar tocilizumab-bavi (Tofidence), Biogen, the drug’s manufacturer, announced on Sept. 29.
It is the first tocilizumab biosimilar approved by the FDA. The reference product, Actemra (Genentech), was first approved by the agency in 2010.
“The approval of Tofidence in the U.S. marks another positive step toward helping more people with chronic autoimmune conditions gain access to leading therapies,” Ian Henshaw, global head of biosimilars at Biogen, said in a statement. “With the increasing numbers of approved biosimilars, we expect increased savings and sustainability for health care systems and an increase in physician choice and patient access to biologics.”
Biogen’s pricing for tocilizumab-bavi will be available closer to the product’s launch date, which has yet to be determined, a company spokesman said. The U.S. average monthly cost of Actemra for rheumatoid arthritis, administered intravenously, is $2,134-$4,268 depending on dosage, according to a Genentech spokesperson.
Tocilizumab-bavi is an intravenous formulation (20 mg/mL) indicated for treatment of moderately to severely active RA, polyarticular juvenile idiopathic arthritis (PJIA), and systemic juvenile idiopathic arthritis (SJIA). The medication is administered every 4 weeks in RA and PJIA and every 8 weeks in SJIA as a single intravenous drip infusion over 1 hour.
The European Commission approved its first tocilizumab biosimilar, Tyenne (Fresenius Kabi), earlier in 2023 in both subcutaneous and intravenous formulations. Biogen did not comment on whether the company is working on a subcutaneous formulation for tocilizumab-bavi.
A version of this article appeared on Medscape.com.
The Food and Drug Administration has approved the biosimilar tocilizumab-bavi (Tofidence), Biogen, the drug’s manufacturer, announced on Sept. 29.
It is the first tocilizumab biosimilar approved by the FDA. The reference product, Actemra (Genentech), was first approved by the agency in 2010.
“The approval of Tofidence in the U.S. marks another positive step toward helping more people with chronic autoimmune conditions gain access to leading therapies,” Ian Henshaw, global head of biosimilars at Biogen, said in a statement. “With the increasing numbers of approved biosimilars, we expect increased savings and sustainability for health care systems and an increase in physician choice and patient access to biologics.”
Biogen’s pricing for tocilizumab-bavi will be available closer to the product’s launch date, which has yet to be determined, a company spokesman said. The U.S. average monthly cost of Actemra for rheumatoid arthritis, administered intravenously, is $2,134-$4,268 depending on dosage, according to a Genentech spokesperson.
Tocilizumab-bavi is an intravenous formulation (20 mg/mL) indicated for treatment of moderately to severely active RA, polyarticular juvenile idiopathic arthritis (PJIA), and systemic juvenile idiopathic arthritis (SJIA). The medication is administered every 4 weeks in RA and PJIA and every 8 weeks in SJIA as a single intravenous drip infusion over 1 hour.
The European Commission approved its first tocilizumab biosimilar, Tyenne (Fresenius Kabi), earlier in 2023 in both subcutaneous and intravenous formulations. Biogen did not comment on whether the company is working on a subcutaneous formulation for tocilizumab-bavi.
A version of this article appeared on Medscape.com.
Triple therapy boosts anaplastic thyroid cancer survival
WASHINGTON – - particularly when administered in a neoadjuvant fashion, prior to surgery. Overall survival rates in the study exceeded 5 years.
“The very long median overall survival in the study’s neoadjuvant group is quite remarkable for a group of patients who used to have a very poor prognosis,” senior author Maria E. Cabanillas, MD, associate professor in the department of endocrine neoplasia and hormonal disorders at the University of Texas MD Anderson Cancer Center in Houston, said in an interview.
“This median overall survival definitely exceeds any other treatments thus far in BRAF-mutated anaplastic thyroid cancer.”
The research was presented at the annual meeting of the American Thyroid Association.
Anaplastic thyroid cancer, though rare, is the most aggressive form of thyroid cancer. It accounts for just 1% of the cancers but causes about 50% of thyroid cancer mortality.
The historical median overall survival is 5-6 months.
With research in recent years showing that as many as 40% of anaplastic thyroid cancers harbor BRAF V600E mutations, the door has opened for potential benefits with the combination of the BRAF inhibitor dabrafenib with the MEK-inhibitor drug trametinib.
The treatment combination was shown in research that included the phase 2 ROAR trial to yield important responses. It was approved by the Food and Drug Administration in 2018 for locally advanced or metastatic BRAF V600E-mutant anaplastic thyroid cancer, as well as other cancers.
However, a key caveat of DT is that patients eventually develop resistance mutations, leading to disease progression.
To overcome the problem, Dr. Cabanillas and her team found two key strategies that show promise – the addition of immunotherapy, such as pembrolizumab to DT, and the use of a neoadjuvant approach, with surgery performed after an initial response to the triplet therapy.
Triple therapy showed highly favorable results
In a study presented at the 2022 ATA annual meeting, researchers reported on the triple therapy of BRAF/MEK inhibitors vemurafenib and cobimetinib plus immunotherapy with atezolizumab. Results were highly favorable, with an overall response rate of 72% and an impressive 2-year survival of 67%.
However, a major limitation was that the study lacked a control arm. In the current study, the addition of pembrolizumab to DT was compared with DT alone. The investigators also sought to determine the survival benefits of a neoadjuvant strategy.
For the study, first author Sarah Hamidi, MD, also of the MD Anderson Cancer Center, and her colleagues identified 94 patients with BRAF-mutated anaplastic thyroid cancer who were treated either with first‐line DT or DT plus pembrolizumab between 2014 and 2023, either outside of a trial or in a reported clinical trial.
The study compared three treatment regimens – DT alone (n = 23), DT with pembrolizumab added before or after disease progression (n = 48), and DT with neoadjuvant pembrolizumab added prior to or after surgery (n = 23).
There were no significant differences in baseline characteristics between the groups. Metastatic disease was present at the start of treatment among 87.0% of the DT group, 79.2% of the pembrolizumab group prior to or after disease progression, and 65.2% of the neoadjuvant pembrolizumab group.
The median follow-up of the three groups was 102 months, 28 months, and 42 months, respectively. The median overall survival was 9 months with DT alone, vs. 17 months with DT plus pembrolizumab before or after progression and 63 months with neoadjuvant pembrolizumab plus DT (P < .001).
The 12- and 24-month survival rates with DT alone were 33.7% and 28.9%, respectively; for DT plus pembrolizumab before or after progression, the rates were 60.2% and 36.5%; and for neoadjuvant pembrolizumab plus DT, the rates were 80.7% and 74.5%.
In an analysis that did not include the neoadjuvant group, median progression-free survival was significantly longer with DT plus pembrolizumab as an initial treatment (11.0 months) compared with DT alone (4.0 months; P = .049).
A subanalysis that evaluated the timing of the addition of pembrolizumab to DT before or after disease progression showed no significant differences between the two in median overall survival (17 months vs. 16 months; P = .554).
“This is valuable information, especially for centers where pembrolizumab cannot be easily obtained as a first-line therapy for anaplastic thyroid cancer,” Dr. Hamidi said in presenting the findings.
She noted, however, that the results should be interpreted with caution, given the small number of patients who received pembrolizumab before progression (n = 34) and especially after progression (n = 14).
In terms of safety, there were no grade 5 adverse events (AEs); 32.4% of patients experienced immune‐related AEs, most frequently, colitis and hepatitis.
Therapies “improve survival”
Overall, the results are important, Dr. Cabanillas said.
The results are “very exciting when you think about the fact that 10 years ago, patients with anaplastic thyroid cancer had a median overall survival measured in months, and now we see that those with a BRAF mutation have a real chance at survival when managed appropriately from the start,” she told this news organization.
She noted that a key caveat is the study’s retrospective nature. Other important considerations are that pembrolizumab adds toxicity as well as cost, and it is largely used off label in anaplastic thyroid cancer.
Nevertheless, “it does feel like there needs to be a call to action in the guidelines for this disease so that it includes neoadjuvant DT or DT plus pembrolizumab as the primary treatment of patients with BRAF-mutated anaplastic thyroid cancer because the initial treatment is critical here,” Dr. Cabanillas said.
She added that a phase 2 trial with neoadjuvant DT plus pembrolizumab is ongoing. Enrollment is expected to be completed soon.
Commenting on the findings, Sarimar Agosto Salgado, MD, of the department of head and neck – endocrine oncology, H. Lee Moffitt Cancer Center and Research Institute, Tampa, Fla., who was a comoderator of the session, said the results are encouraging.
“These findings are promising because they open the landscape of options of therapies that we can provide these patients,” she said in an interview.
“Anaplastic thyroid cancer has been a disease with a very short survival despite aggressive therapies, but we are seeing that not only have these therapies been able to improve survival but also patients’ quality of life.”
Particularly encouraging is how quickly the therapies can work, Dr. Salgado added.
“Many times when patients present to the clinic, the rapid response to these systemic therapies can even [allow them to avoid] having a tracheostomy, and we’re also seeing that some of these patients are able to go from unresectable disease to resectable disease, and then by having the main tumor out, their survival improves.
“So, this is definitely a big ray of hope for these patients.”
Dr. Cabanillas has received research funding from Merck. Dr. Hamidi has disclosed no relevant financial relationships. Dr. Salgado has relationships with Lilly and Exelixis.
A version of this article appeared on Medscape.com.
WASHINGTON – - particularly when administered in a neoadjuvant fashion, prior to surgery. Overall survival rates in the study exceeded 5 years.
“The very long median overall survival in the study’s neoadjuvant group is quite remarkable for a group of patients who used to have a very poor prognosis,” senior author Maria E. Cabanillas, MD, associate professor in the department of endocrine neoplasia and hormonal disorders at the University of Texas MD Anderson Cancer Center in Houston, said in an interview.
“This median overall survival definitely exceeds any other treatments thus far in BRAF-mutated anaplastic thyroid cancer.”
The research was presented at the annual meeting of the American Thyroid Association.
Anaplastic thyroid cancer, though rare, is the most aggressive form of thyroid cancer. It accounts for just 1% of the cancers but causes about 50% of thyroid cancer mortality.
The historical median overall survival is 5-6 months.
With research in recent years showing that as many as 40% of anaplastic thyroid cancers harbor BRAF V600E mutations, the door has opened for potential benefits with the combination of the BRAF inhibitor dabrafenib with the MEK-inhibitor drug trametinib.
The treatment combination was shown in research that included the phase 2 ROAR trial to yield important responses. It was approved by the Food and Drug Administration in 2018 for locally advanced or metastatic BRAF V600E-mutant anaplastic thyroid cancer, as well as other cancers.
However, a key caveat of DT is that patients eventually develop resistance mutations, leading to disease progression.
To overcome the problem, Dr. Cabanillas and her team found two key strategies that show promise – the addition of immunotherapy, such as pembrolizumab to DT, and the use of a neoadjuvant approach, with surgery performed after an initial response to the triplet therapy.
Triple therapy showed highly favorable results
In a study presented at the 2022 ATA annual meeting, researchers reported on the triple therapy of BRAF/MEK inhibitors vemurafenib and cobimetinib plus immunotherapy with atezolizumab. Results were highly favorable, with an overall response rate of 72% and an impressive 2-year survival of 67%.
However, a major limitation was that the study lacked a control arm. In the current study, the addition of pembrolizumab to DT was compared with DT alone. The investigators also sought to determine the survival benefits of a neoadjuvant strategy.
For the study, first author Sarah Hamidi, MD, also of the MD Anderson Cancer Center, and her colleagues identified 94 patients with BRAF-mutated anaplastic thyroid cancer who were treated either with first‐line DT or DT plus pembrolizumab between 2014 and 2023, either outside of a trial or in a reported clinical trial.
The study compared three treatment regimens – DT alone (n = 23), DT with pembrolizumab added before or after disease progression (n = 48), and DT with neoadjuvant pembrolizumab added prior to or after surgery (n = 23).
There were no significant differences in baseline characteristics between the groups. Metastatic disease was present at the start of treatment among 87.0% of the DT group, 79.2% of the pembrolizumab group prior to or after disease progression, and 65.2% of the neoadjuvant pembrolizumab group.
The median follow-up of the three groups was 102 months, 28 months, and 42 months, respectively. The median overall survival was 9 months with DT alone, vs. 17 months with DT plus pembrolizumab before or after progression and 63 months with neoadjuvant pembrolizumab plus DT (P < .001).
The 12- and 24-month survival rates with DT alone were 33.7% and 28.9%, respectively; for DT plus pembrolizumab before or after progression, the rates were 60.2% and 36.5%; and for neoadjuvant pembrolizumab plus DT, the rates were 80.7% and 74.5%.
In an analysis that did not include the neoadjuvant group, median progression-free survival was significantly longer with DT plus pembrolizumab as an initial treatment (11.0 months) compared with DT alone (4.0 months; P = .049).
A subanalysis that evaluated the timing of the addition of pembrolizumab to DT before or after disease progression showed no significant differences between the two in median overall survival (17 months vs. 16 months; P = .554).
“This is valuable information, especially for centers where pembrolizumab cannot be easily obtained as a first-line therapy for anaplastic thyroid cancer,” Dr. Hamidi said in presenting the findings.
She noted, however, that the results should be interpreted with caution, given the small number of patients who received pembrolizumab before progression (n = 34) and especially after progression (n = 14).
In terms of safety, there were no grade 5 adverse events (AEs); 32.4% of patients experienced immune‐related AEs, most frequently, colitis and hepatitis.
Therapies “improve survival”
Overall, the results are important, Dr. Cabanillas said.
The results are “very exciting when you think about the fact that 10 years ago, patients with anaplastic thyroid cancer had a median overall survival measured in months, and now we see that those with a BRAF mutation have a real chance at survival when managed appropriately from the start,” she told this news organization.
She noted that a key caveat is the study’s retrospective nature. Other important considerations are that pembrolizumab adds toxicity as well as cost, and it is largely used off label in anaplastic thyroid cancer.
Nevertheless, “it does feel like there needs to be a call to action in the guidelines for this disease so that it includes neoadjuvant DT or DT plus pembrolizumab as the primary treatment of patients with BRAF-mutated anaplastic thyroid cancer because the initial treatment is critical here,” Dr. Cabanillas said.
She added that a phase 2 trial with neoadjuvant DT plus pembrolizumab is ongoing. Enrollment is expected to be completed soon.
Commenting on the findings, Sarimar Agosto Salgado, MD, of the department of head and neck – endocrine oncology, H. Lee Moffitt Cancer Center and Research Institute, Tampa, Fla., who was a comoderator of the session, said the results are encouraging.
“These findings are promising because they open the landscape of options of therapies that we can provide these patients,” she said in an interview.
“Anaplastic thyroid cancer has been a disease with a very short survival despite aggressive therapies, but we are seeing that not only have these therapies been able to improve survival but also patients’ quality of life.”
Particularly encouraging is how quickly the therapies can work, Dr. Salgado added.
“Many times when patients present to the clinic, the rapid response to these systemic therapies can even [allow them to avoid] having a tracheostomy, and we’re also seeing that some of these patients are able to go from unresectable disease to resectable disease, and then by having the main tumor out, their survival improves.
“So, this is definitely a big ray of hope for these patients.”
Dr. Cabanillas has received research funding from Merck. Dr. Hamidi has disclosed no relevant financial relationships. Dr. Salgado has relationships with Lilly and Exelixis.
A version of this article appeared on Medscape.com.
WASHINGTON – - particularly when administered in a neoadjuvant fashion, prior to surgery. Overall survival rates in the study exceeded 5 years.
“The very long median overall survival in the study’s neoadjuvant group is quite remarkable for a group of patients who used to have a very poor prognosis,” senior author Maria E. Cabanillas, MD, associate professor in the department of endocrine neoplasia and hormonal disorders at the University of Texas MD Anderson Cancer Center in Houston, said in an interview.
“This median overall survival definitely exceeds any other treatments thus far in BRAF-mutated anaplastic thyroid cancer.”
The research was presented at the annual meeting of the American Thyroid Association.
Anaplastic thyroid cancer, though rare, is the most aggressive form of thyroid cancer. It accounts for just 1% of the cancers but causes about 50% of thyroid cancer mortality.
The historical median overall survival is 5-6 months.
With research in recent years showing that as many as 40% of anaplastic thyroid cancers harbor BRAF V600E mutations, the door has opened for potential benefits with the combination of the BRAF inhibitor dabrafenib with the MEK-inhibitor drug trametinib.
The treatment combination was shown in research that included the phase 2 ROAR trial to yield important responses. It was approved by the Food and Drug Administration in 2018 for locally advanced or metastatic BRAF V600E-mutant anaplastic thyroid cancer, as well as other cancers.
However, a key caveat of DT is that patients eventually develop resistance mutations, leading to disease progression.
To overcome the problem, Dr. Cabanillas and her team found two key strategies that show promise – the addition of immunotherapy, such as pembrolizumab to DT, and the use of a neoadjuvant approach, with surgery performed after an initial response to the triplet therapy.
Triple therapy showed highly favorable results
In a study presented at the 2022 ATA annual meeting, researchers reported on the triple therapy of BRAF/MEK inhibitors vemurafenib and cobimetinib plus immunotherapy with atezolizumab. Results were highly favorable, with an overall response rate of 72% and an impressive 2-year survival of 67%.
However, a major limitation was that the study lacked a control arm. In the current study, the addition of pembrolizumab to DT was compared with DT alone. The investigators also sought to determine the survival benefits of a neoadjuvant strategy.
For the study, first author Sarah Hamidi, MD, also of the MD Anderson Cancer Center, and her colleagues identified 94 patients with BRAF-mutated anaplastic thyroid cancer who were treated either with first‐line DT or DT plus pembrolizumab between 2014 and 2023, either outside of a trial or in a reported clinical trial.
The study compared three treatment regimens – DT alone (n = 23), DT with pembrolizumab added before or after disease progression (n = 48), and DT with neoadjuvant pembrolizumab added prior to or after surgery (n = 23).
There were no significant differences in baseline characteristics between the groups. Metastatic disease was present at the start of treatment among 87.0% of the DT group, 79.2% of the pembrolizumab group prior to or after disease progression, and 65.2% of the neoadjuvant pembrolizumab group.
The median follow-up of the three groups was 102 months, 28 months, and 42 months, respectively. The median overall survival was 9 months with DT alone, vs. 17 months with DT plus pembrolizumab before or after progression and 63 months with neoadjuvant pembrolizumab plus DT (P < .001).
The 12- and 24-month survival rates with DT alone were 33.7% and 28.9%, respectively; for DT plus pembrolizumab before or after progression, the rates were 60.2% and 36.5%; and for neoadjuvant pembrolizumab plus DT, the rates were 80.7% and 74.5%.
In an analysis that did not include the neoadjuvant group, median progression-free survival was significantly longer with DT plus pembrolizumab as an initial treatment (11.0 months) compared with DT alone (4.0 months; P = .049).
A subanalysis that evaluated the timing of the addition of pembrolizumab to DT before or after disease progression showed no significant differences between the two in median overall survival (17 months vs. 16 months; P = .554).
“This is valuable information, especially for centers where pembrolizumab cannot be easily obtained as a first-line therapy for anaplastic thyroid cancer,” Dr. Hamidi said in presenting the findings.
She noted, however, that the results should be interpreted with caution, given the small number of patients who received pembrolizumab before progression (n = 34) and especially after progression (n = 14).
In terms of safety, there were no grade 5 adverse events (AEs); 32.4% of patients experienced immune‐related AEs, most frequently, colitis and hepatitis.
Therapies “improve survival”
Overall, the results are important, Dr. Cabanillas said.
The results are “very exciting when you think about the fact that 10 years ago, patients with anaplastic thyroid cancer had a median overall survival measured in months, and now we see that those with a BRAF mutation have a real chance at survival when managed appropriately from the start,” she told this news organization.
She noted that a key caveat is the study’s retrospective nature. Other important considerations are that pembrolizumab adds toxicity as well as cost, and it is largely used off label in anaplastic thyroid cancer.
Nevertheless, “it does feel like there needs to be a call to action in the guidelines for this disease so that it includes neoadjuvant DT or DT plus pembrolizumab as the primary treatment of patients with BRAF-mutated anaplastic thyroid cancer because the initial treatment is critical here,” Dr. Cabanillas said.
She added that a phase 2 trial with neoadjuvant DT plus pembrolizumab is ongoing. Enrollment is expected to be completed soon.
Commenting on the findings, Sarimar Agosto Salgado, MD, of the department of head and neck – endocrine oncology, H. Lee Moffitt Cancer Center and Research Institute, Tampa, Fla., who was a comoderator of the session, said the results are encouraging.
“These findings are promising because they open the landscape of options of therapies that we can provide these patients,” she said in an interview.
“Anaplastic thyroid cancer has been a disease with a very short survival despite aggressive therapies, but we are seeing that not only have these therapies been able to improve survival but also patients’ quality of life.”
Particularly encouraging is how quickly the therapies can work, Dr. Salgado added.
“Many times when patients present to the clinic, the rapid response to these systemic therapies can even [allow them to avoid] having a tracheostomy, and we’re also seeing that some of these patients are able to go from unresectable disease to resectable disease, and then by having the main tumor out, their survival improves.
“So, this is definitely a big ray of hope for these patients.”
Dr. Cabanillas has received research funding from Merck. Dr. Hamidi has disclosed no relevant financial relationships. Dr. Salgado has relationships with Lilly and Exelixis.
A version of this article appeared on Medscape.com.
AT ATA 2023
Idiopathic Granulomatous Lobular Mastitis: A Mimicker of Inflammatory Breast Cancer
Idiopathic granulomatous lobular mastitis (IGLM) is a rare, chronic inflammatory breast disease first described in 1972.1 IGLM usually affects women during reproductive years and has similar clinical features to breast cancer.2 Ultrasonography and mammography yield nonspecific results and cannot adequately differentiate between malignancy and inflammation.3 Magnetic resonance imaging (MRI) is known to be more sensitive in detecting lesions in dense breasts; however, it does not differentiate between granulomatous lesions and other disorders.4,5 Histopathology is the gold standard for diagnosis.1-12
Infectious and autoimmune causes of granulomatous mastitis must be excluded before establishing an IGLM diagnosis. The clinical quandary that remains is how to adequately manage the disease. Although there are no defined treatment guidelines, current literature has proposed a multimodal strategy.6,9 In this report, we describe a case of IGLM successfully treated with surgical excision after failed medical therapy.
Case Presentation
A 43-year-old gravida 5, para 4 White woman presented with a 2-week history of right breast tenderness, heaviness, warmth, and redness that was refractory to cephalexin and dicloxacillin. She had no personal or family history of breast cancer; never had breast surgery and breastfed all 4 children.

An examination of the right breast demonstrated erythema and an 8-cm tender mass in the right lower outer quadrant but no skin retraction or dimpling (Figure 1). The mammography, concerning for inflammatory breast cancer, was category BI-RADS 4 and demonstrated a suspicious right axillary lymph node (Figure 2).

A core needle breast biopsy revealed granulomatous mastitis (Figure 3A), without evidence of malignancy. Rheumatology and endocrinology excluded secondary causes of granulomatous mastitis (ie, sarcoidosis, tuberculosis, granulomatosis with polyangiitis, and other autoimmune conditions). A pituitary MRI to assess an elevated serum prolactin level showed no evidence of microadenoma.
After a prolonged course of 8 months of unsuccessful therapy with prednisone and methotrexate, the patient was referred for surgical excision. Culture and special stains (Gram stain, periodic acid-Schiff stain, acid-fast Bacillus culture, Fite stain, and Brown and Benn stain) of the breast tissue were negative for organisms (Figure 3B). Seven months after excision the patient was doing well and had no evidence of recurrence.
Discussion
IGLM is a rare, chronic benign inflammatory breast disease of unknown etiology and more commonly reported in individuals of Mediterranean descent.13 It is believed that hyperprolactinemia causing extravasation of fat and protein during milk letdown leads to lymphocyte and macrophage migration, resulting in a localized autoimmune response in the breast ducts.10,14
There are 2 types of granulomatous mastitis: idiopathic and specific. Infectious, autoimmune, and malignant causes of granulomatous mastitis (ie, tuberculosis, sarcoidosis, Corynebacterium spp, granulomatosis with polyangiitis, systemic lupus erythematosus, Behçet disease, ductal ectasia, or granulomatous reaction in a carcinoma) must be excluded prior to establishing an IGLM diagnosis, as these can be fatal if left untreated.15 The most frequent findings on ultrasound and mammography are hypoechoic masses and focal asymmetric densities, respectively.3,5 MRI has been proposed more for surveillance in patients with chronic IGLM.4,5 Histopathology—featuring lobular noncaseating granulomas with epithelioid histiocytes; and multinucleated giant cells in a background of neutrophils, lymphocytes, plasma cells, and eosinophils—is the gold standard for diagnosing IGLM.1-12
There are currently no universal treatment guidelines and management usually consists of observation, systemic and topical steroids, or surgery.3,13 Topical and injectable steroids have been effective in treating both initial and recurrent IGLM in patients who are unable to be treated with systemic steroids.16-18 Due to reported high recurrence rates with steroid tapers, adjunctive therapy with methotrexate, azathioprine, colchicine, and hydroxychloroquine have been proposed.1,3-6,10-12
Additionally, antibiotics are recommended only in the management of IGLM when microbial co-infection is concerning, such as with Corynebacterium spp.9,11,19-22 Histologically, this bacterium is distinct from IGLM and demonstrates granulomatous, neutrophilic inflammation within cystic spaces.19-21 Wide surgical excision with negative margins is the only definitive treatment to reduce recurrence and expedite recovery time.2,3,7-10 Notably, surgical excision has been associated with poor wound healing and occasional recurrence compared with medication alone.5,11
Although IGLM is normally a benign process, chronic disease has been related (without causality) to infiltrating breast carcinoma.4 A proposed theory for the development of malignancy suggests that chronic inflammation leading to free radical formation can result in cellular dysplasia and cancer.23
Conclusions
Fifty years after its first description, IGLM is still a poorly understood disease. There remains no consensus behind its etiology or management. In our case, we demonstrated a stepwise treatment progression, beginning with medical therapy before proceeding to surgical cure. Given concerns for poor wound healing and postsurgical infections, monitoring the response and recurrence to an initial trial of conservative medical treatment is not unreasonable. Because of possible risk for malignancy with chronic IGLM, patients should not delay surgical excision if their condition remains refractory to medical therapy alone.
1. Garcia-Rodiguez JA, Pattullo A. Idiopathic granulomatous mastitis: a mimicking disease in a pregnant woman: a case report. BMC Res Notes. 2013;6:95. doi.10.1186/1756-0500-6-95
2. Gurleyik G, Aktekin A, Aker F, Karagulle H, Saglamc A. Medical and surgical treatment of idiopathic granulomatous lobular mastitis: a benign inflammatory disease mimicking invasive carcinoma. J Breast Cancer. 2012;15(1):119-123. doi:10.4048/jbc.2012.15.1.119
3. Hovanessian Larsen LJ, Peyvandi B, Klipfel N, Grant E, Iyengar G. Granulomatous lobular mastitis: imaging, diagnosis, and treatment. AJR Am J Roentgenol. 2009;193(2):574-581. doi:10.2214/AJR.08.1528
4. Mazlan L, Suhaimi SN, Jasmin SJ, Latar NH, Adzman S, Muhammad R. Breast carcinoma occurring from chronic granulomatous mastitis. Malays J Med Sci. 2012;19(2):82-85.
5. Patel RA, Strickland P, Sankara IR, Pinkston G, Many W Jr, Rodriguez M. Idiopathic granulomatous mastitis: case reports and review of literature. J Gen Intern Med. 2010;25(3):270-273. doi:10.1007/s11606-009-1207-2
6. Akbulut S, Yilmaz D, Bakir S. Methotrexate in the management of idiopathic granulomatous mastitis: review of 108 published cases and report of four cases. Breast J. 2011;17(6):661-668. doi:10.1111/j.1524-4741.2011.01162.x
7. Ergin AB, Cristofanilli M, Daw H, Tahan G, Gong Y. Recurrent granulomatous mastitis mimicking inflammatory breast cancer. BMJ Case Rep. 2011;2011:bcr0720103156. doi:10.1136/bcr.07.2010.3156
8. Hladik M, Schoeller T, Ensat F, Wechselberger G. Idiopathic granulomatous mastitis: successful treatment by mastectomy and immediate breast reconstruction. J Plast Reconstr Aesthet Surg. 2011;64(12):1604-1607. doi:10.1016/j.bjps.2011.07.01
9. Hur SM, Cho DH, Lee SK, et al. Experience of treatment of patients with granulomatous lobular mastitis. J Korean Surg Soc. 2013;85(1):1-6. doi:10.4174/jkss.2013.85.1.
10. Kayahan M, Kadioglu H, Muslumanoglu M. Management of patients with granulomatous mastitis: analysis of 31 cases. Breast Care (Basel). 2012;7(3):226-230. doi:10.1159/000337758
11. Neel A, Hello M, Cottereau A, et al. Long-term outcome in idiopathic granulomatous mastitis: a western multicentre study. QJM. 2013;106(5):433-441. doi:10.1093/qjmed/hct040
12. Seo HR, Na KY, Yim HE, et al. Differential diagnosis in idiopathic granulomatous mastitis and tuberculous mastitis. J Breast Cancer. 2012;15(1):111-118. doi:10.4048/jbc.2012.15.1.111
13. Martinez-Ramos D, Simon-Monterde L, Suelves-Piqueres C, et al. Idiopathic granulomatous mastitis: a systematic review of 3060 patients. Breast J. 2019;25(6):1245-1250. doi:10.1111/tbj.13446
14. Lin CH, Hsu CW, Tsao TY, Chou J. Idiopathic granulomatous mastitis associated with risperidone-induced hyperprolactinemia. Diagn Pathol. 2012;7:2. doi:10.1186/1746-1596-7-2
15. Goulabchand R, Hafidi A, Van de Perre P, et al. Mastitis in autoimmune diseases: review of the literature, diagnostic pathway, and pathophysiological key players. J Clin Med. 2020;9(4):958. doi:10.3390/jcm9040958
16. Altintoprak F. Topical steroids to treat granulomatous mastitis: a case report. Korean J Intern Med. 2011;26(3):356-359. doi:10.3904/kjim.2011.26.3.356
17. Tang A, Dominguez DA, Edquilang JK, et al. Granulomatous mastitis: comparison of novel treatment of steroid injection and current management. J Surg Res. 2020;254:300-305. doi:10.1016/j.jss.2020.04.018
18. Toktas O, Toprak N. Treatment results of intralesional steroid injection and topical steroid administration in pregnant women with idiopathic granulomatous mastitis. Eur J Breast Health. 2021;17(3):283-287. doi:10.4274/ejbh.galenos.2021.2021-2-4
19. Bercot B, Kannengiesser C, Oudin C, et al. First description of NOD2 variant associated with defective neutrophil responses in a woman with granulomatous mastitis related to corynebacteria. J Clin Microbiol. 2009;47(9):3034-3037. doi:10.1128/JCM.00561-09
20. Renshaw AA, Derhagopian RP, Gould EW. Cystic neutrophilic granulomatous mastitis: an underappreciated pattern strongly associated with gram-positive bacilli. Am J Clin Pathol. 2011;136(3):424-427. doi:10.1309/AJCP1W9JBRYOQSNZ
21. Stary CM, Lee YS, Balfour J. Idiopathic granulomatous mastitis associated with corynebacterium sp. Infection. Hawaii Med J. 2011;70(5):99-101.
22. Taylor GB, Paviour SD, Musaad S, Jones WO, Holland DJ. A clinicopathological review of 34 cases of inflammatory breast disease showing an association between corynebacteria infection and granulomatous mastitis. Pathology. 2003;35(2):109-119.
23. Rakoff-Nahoum S. Why cancer and inflammation? Yale J Biol Med. 2006;79(3-4):123-130.
Idiopathic granulomatous lobular mastitis (IGLM) is a rare, chronic inflammatory breast disease first described in 1972.1 IGLM usually affects women during reproductive years and has similar clinical features to breast cancer.2 Ultrasonography and mammography yield nonspecific results and cannot adequately differentiate between malignancy and inflammation.3 Magnetic resonance imaging (MRI) is known to be more sensitive in detecting lesions in dense breasts; however, it does not differentiate between granulomatous lesions and other disorders.4,5 Histopathology is the gold standard for diagnosis.1-12
Infectious and autoimmune causes of granulomatous mastitis must be excluded before establishing an IGLM diagnosis. The clinical quandary that remains is how to adequately manage the disease. Although there are no defined treatment guidelines, current literature has proposed a multimodal strategy.6,9 In this report, we describe a case of IGLM successfully treated with surgical excision after failed medical therapy.
Case Presentation
A 43-year-old gravida 5, para 4 White woman presented with a 2-week history of right breast tenderness, heaviness, warmth, and redness that was refractory to cephalexin and dicloxacillin. She had no personal or family history of breast cancer; never had breast surgery and breastfed all 4 children.
An examination of the right breast demonstrated erythema and an 8-cm tender mass in the right lower outer quadrant but no skin retraction or dimpling (Figure 1). The mammography, concerning for inflammatory breast cancer, was category BI-RADS 4 and demonstrated a suspicious right axillary lymph node (Figure 2).
A core needle breast biopsy revealed granulomatous mastitis (Figure 3A), without evidence of malignancy. Rheumatology and endocrinology excluded secondary causes of granulomatous mastitis (ie, sarcoidosis, tuberculosis, granulomatosis with polyangiitis, and other autoimmune conditions). A pituitary MRI to assess an elevated serum prolactin level showed no evidence of microadenoma.
After a prolonged course of 8 months of unsuccessful therapy with prednisone and methotrexate, the patient was referred for surgical excision. Culture and special stains (Gram stain, periodic acid-Schiff stain, acid-fast Bacillus culture, Fite stain, and Brown and Benn stain) of the breast tissue were negative for organisms (Figure 3B). Seven months after excision the patient was doing well and had no evidence of recurrence.
Discussion
IGLM is a rare, chronic benign inflammatory breast disease of unknown etiology and more commonly reported in individuals of Mediterranean descent.13 It is believed that hyperprolactinemia causing extravasation of fat and protein during milk letdown leads to lymphocyte and macrophage migration, resulting in a localized autoimmune response in the breast ducts.10,14
There are 2 types of granulomatous mastitis: idiopathic and specific. Infectious, autoimmune, and malignant causes of granulomatous mastitis (ie, tuberculosis, sarcoidosis, Corynebacterium spp, granulomatosis with polyangiitis, systemic lupus erythematosus, Behçet disease, ductal ectasia, or granulomatous reaction in a carcinoma) must be excluded prior to establishing an IGLM diagnosis, as these can be fatal if left untreated.15 The most frequent findings on ultrasound and mammography are hypoechoic masses and focal asymmetric densities, respectively.3,5 MRI has been proposed more for surveillance in patients with chronic IGLM.4,5 Histopathology—featuring lobular noncaseating granulomas with epithelioid histiocytes; and multinucleated giant cells in a background of neutrophils, lymphocytes, plasma cells, and eosinophils—is the gold standard for diagnosing IGLM.1-12
There are currently no universal treatment guidelines and management usually consists of observation, systemic and topical steroids, or surgery.3,13 Topical and injectable steroids have been effective in treating both initial and recurrent IGLM in patients who are unable to be treated with systemic steroids.16-18 Due to reported high recurrence rates with steroid tapers, adjunctive therapy with methotrexate, azathioprine, colchicine, and hydroxychloroquine have been proposed.1,3-6,10-12
Additionally, antibiotics are recommended only in the management of IGLM when microbial co-infection is concerning, such as with Corynebacterium spp.9,11,19-22 Histologically, this bacterium is distinct from IGLM and demonstrates granulomatous, neutrophilic inflammation within cystic spaces.19-21 Wide surgical excision with negative margins is the only definitive treatment to reduce recurrence and expedite recovery time.2,3,7-10 Notably, surgical excision has been associated with poor wound healing and occasional recurrence compared with medication alone.5,11
Although IGLM is normally a benign process, chronic disease has been related (without causality) to infiltrating breast carcinoma.4 A proposed theory for the development of malignancy suggests that chronic inflammation leading to free radical formation can result in cellular dysplasia and cancer.23
Conclusions
Fifty years after its first description, IGLM is still a poorly understood disease. There remains no consensus behind its etiology or management. In our case, we demonstrated a stepwise treatment progression, beginning with medical therapy before proceeding to surgical cure. Given concerns for poor wound healing and postsurgical infections, monitoring the response and recurrence to an initial trial of conservative medical treatment is not unreasonable. Because of possible risk for malignancy with chronic IGLM, patients should not delay surgical excision if their condition remains refractory to medical therapy alone.
Idiopathic granulomatous lobular mastitis (IGLM) is a rare, chronic inflammatory breast disease first described in 1972.1 IGLM usually affects women during reproductive years and has similar clinical features to breast cancer.2 Ultrasonography and mammography yield nonspecific results and cannot adequately differentiate between malignancy and inflammation.3 Magnetic resonance imaging (MRI) is known to be more sensitive in detecting lesions in dense breasts; however, it does not differentiate between granulomatous lesions and other disorders.4,5 Histopathology is the gold standard for diagnosis.1-12
Infectious and autoimmune causes of granulomatous mastitis must be excluded before establishing an IGLM diagnosis. The clinical quandary that remains is how to adequately manage the disease. Although there are no defined treatment guidelines, current literature has proposed a multimodal strategy.6,9 In this report, we describe a case of IGLM successfully treated with surgical excision after failed medical therapy.
Case Presentation
A 43-year-old gravida 5, para 4 White woman presented with a 2-week history of right breast tenderness, heaviness, warmth, and redness that was refractory to cephalexin and dicloxacillin. She had no personal or family history of breast cancer; never had breast surgery and breastfed all 4 children.
An examination of the right breast demonstrated erythema and an 8-cm tender mass in the right lower outer quadrant but no skin retraction or dimpling (Figure 1). The mammography, concerning for inflammatory breast cancer, was category BI-RADS 4 and demonstrated a suspicious right axillary lymph node (Figure 2).
A core needle breast biopsy revealed granulomatous mastitis (Figure 3A), without evidence of malignancy. Rheumatology and endocrinology excluded secondary causes of granulomatous mastitis (ie, sarcoidosis, tuberculosis, granulomatosis with polyangiitis, and other autoimmune conditions). A pituitary MRI to assess an elevated serum prolactin level showed no evidence of microadenoma.
After a prolonged course of 8 months of unsuccessful therapy with prednisone and methotrexate, the patient was referred for surgical excision. Culture and special stains (Gram stain, periodic acid-Schiff stain, acid-fast Bacillus culture, Fite stain, and Brown and Benn stain) of the breast tissue were negative for organisms (Figure 3B). Seven months after excision the patient was doing well and had no evidence of recurrence.
Discussion
IGLM is a rare, chronic benign inflammatory breast disease of unknown etiology and more commonly reported in individuals of Mediterranean descent.13 It is believed that hyperprolactinemia causing extravasation of fat and protein during milk letdown leads to lymphocyte and macrophage migration, resulting in a localized autoimmune response in the breast ducts.10,14
There are 2 types of granulomatous mastitis: idiopathic and specific. Infectious, autoimmune, and malignant causes of granulomatous mastitis (ie, tuberculosis, sarcoidosis, Corynebacterium spp, granulomatosis with polyangiitis, systemic lupus erythematosus, Behçet disease, ductal ectasia, or granulomatous reaction in a carcinoma) must be excluded prior to establishing an IGLM diagnosis, as these can be fatal if left untreated.15 The most frequent findings on ultrasound and mammography are hypoechoic masses and focal asymmetric densities, respectively.3,5 MRI has been proposed more for surveillance in patients with chronic IGLM.4,5 Histopathology—featuring lobular noncaseating granulomas with epithelioid histiocytes; and multinucleated giant cells in a background of neutrophils, lymphocytes, plasma cells, and eosinophils—is the gold standard for diagnosing IGLM.1-12
There are currently no universal treatment guidelines and management usually consists of observation, systemic and topical steroids, or surgery.3,13 Topical and injectable steroids have been effective in treating both initial and recurrent IGLM in patients who are unable to be treated with systemic steroids.16-18 Due to reported high recurrence rates with steroid tapers, adjunctive therapy with methotrexate, azathioprine, colchicine, and hydroxychloroquine have been proposed.1,3-6,10-12
Additionally, antibiotics are recommended only in the management of IGLM when microbial co-infection is concerning, such as with Corynebacterium spp.9,11,19-22 Histologically, this bacterium is distinct from IGLM and demonstrates granulomatous, neutrophilic inflammation within cystic spaces.19-21 Wide surgical excision with negative margins is the only definitive treatment to reduce recurrence and expedite recovery time.2,3,7-10 Notably, surgical excision has been associated with poor wound healing and occasional recurrence compared with medication alone.5,11
Although IGLM is normally a benign process, chronic disease has been related (without causality) to infiltrating breast carcinoma.4 A proposed theory for the development of malignancy suggests that chronic inflammation leading to free radical formation can result in cellular dysplasia and cancer.23
Conclusions
Fifty years after its first description, IGLM is still a poorly understood disease. There remains no consensus behind its etiology or management. In our case, we demonstrated a stepwise treatment progression, beginning with medical therapy before proceeding to surgical cure. Given concerns for poor wound healing and postsurgical infections, monitoring the response and recurrence to an initial trial of conservative medical treatment is not unreasonable. Because of possible risk for malignancy with chronic IGLM, patients should not delay surgical excision if their condition remains refractory to medical therapy alone.
1. Garcia-Rodiguez JA, Pattullo A. Idiopathic granulomatous mastitis: a mimicking disease in a pregnant woman: a case report. BMC Res Notes. 2013;6:95. doi.10.1186/1756-0500-6-95
2. Gurleyik G, Aktekin A, Aker F, Karagulle H, Saglamc A. Medical and surgical treatment of idiopathic granulomatous lobular mastitis: a benign inflammatory disease mimicking invasive carcinoma. J Breast Cancer. 2012;15(1):119-123. doi:10.4048/jbc.2012.15.1.119
3. Hovanessian Larsen LJ, Peyvandi B, Klipfel N, Grant E, Iyengar G. Granulomatous lobular mastitis: imaging, diagnosis, and treatment. AJR Am J Roentgenol. 2009;193(2):574-581. doi:10.2214/AJR.08.1528
4. Mazlan L, Suhaimi SN, Jasmin SJ, Latar NH, Adzman S, Muhammad R. Breast carcinoma occurring from chronic granulomatous mastitis. Malays J Med Sci. 2012;19(2):82-85.
5. Patel RA, Strickland P, Sankara IR, Pinkston G, Many W Jr, Rodriguez M. Idiopathic granulomatous mastitis: case reports and review of literature. J Gen Intern Med. 2010;25(3):270-273. doi:10.1007/s11606-009-1207-2
6. Akbulut S, Yilmaz D, Bakir S. Methotrexate in the management of idiopathic granulomatous mastitis: review of 108 published cases and report of four cases. Breast J. 2011;17(6):661-668. doi:10.1111/j.1524-4741.2011.01162.x
7. Ergin AB, Cristofanilli M, Daw H, Tahan G, Gong Y. Recurrent granulomatous mastitis mimicking inflammatory breast cancer. BMJ Case Rep. 2011;2011:bcr0720103156. doi:10.1136/bcr.07.2010.3156
8. Hladik M, Schoeller T, Ensat F, Wechselberger G. Idiopathic granulomatous mastitis: successful treatment by mastectomy and immediate breast reconstruction. J Plast Reconstr Aesthet Surg. 2011;64(12):1604-1607. doi:10.1016/j.bjps.2011.07.01
9. Hur SM, Cho DH, Lee SK, et al. Experience of treatment of patients with granulomatous lobular mastitis. J Korean Surg Soc. 2013;85(1):1-6. doi:10.4174/jkss.2013.85.1.
10. Kayahan M, Kadioglu H, Muslumanoglu M. Management of patients with granulomatous mastitis: analysis of 31 cases. Breast Care (Basel). 2012;7(3):226-230. doi:10.1159/000337758
11. Neel A, Hello M, Cottereau A, et al. Long-term outcome in idiopathic granulomatous mastitis: a western multicentre study. QJM. 2013;106(5):433-441. doi:10.1093/qjmed/hct040
12. Seo HR, Na KY, Yim HE, et al. Differential diagnosis in idiopathic granulomatous mastitis and tuberculous mastitis. J Breast Cancer. 2012;15(1):111-118. doi:10.4048/jbc.2012.15.1.111
13. Martinez-Ramos D, Simon-Monterde L, Suelves-Piqueres C, et al. Idiopathic granulomatous mastitis: a systematic review of 3060 patients. Breast J. 2019;25(6):1245-1250. doi:10.1111/tbj.13446
14. Lin CH, Hsu CW, Tsao TY, Chou J. Idiopathic granulomatous mastitis associated with risperidone-induced hyperprolactinemia. Diagn Pathol. 2012;7:2. doi:10.1186/1746-1596-7-2
15. Goulabchand R, Hafidi A, Van de Perre P, et al. Mastitis in autoimmune diseases: review of the literature, diagnostic pathway, and pathophysiological key players. J Clin Med. 2020;9(4):958. doi:10.3390/jcm9040958
16. Altintoprak F. Topical steroids to treat granulomatous mastitis: a case report. Korean J Intern Med. 2011;26(3):356-359. doi:10.3904/kjim.2011.26.3.356
17. Tang A, Dominguez DA, Edquilang JK, et al. Granulomatous mastitis: comparison of novel treatment of steroid injection and current management. J Surg Res. 2020;254:300-305. doi:10.1016/j.jss.2020.04.018
18. Toktas O, Toprak N. Treatment results of intralesional steroid injection and topical steroid administration in pregnant women with idiopathic granulomatous mastitis. Eur J Breast Health. 2021;17(3):283-287. doi:10.4274/ejbh.galenos.2021.2021-2-4
19. Bercot B, Kannengiesser C, Oudin C, et al. First description of NOD2 variant associated with defective neutrophil responses in a woman with granulomatous mastitis related to corynebacteria. J Clin Microbiol. 2009;47(9):3034-3037. doi:10.1128/JCM.00561-09
20. Renshaw AA, Derhagopian RP, Gould EW. Cystic neutrophilic granulomatous mastitis: an underappreciated pattern strongly associated with gram-positive bacilli. Am J Clin Pathol. 2011;136(3):424-427. doi:10.1309/AJCP1W9JBRYOQSNZ
21. Stary CM, Lee YS, Balfour J. Idiopathic granulomatous mastitis associated with corynebacterium sp. Infection. Hawaii Med J. 2011;70(5):99-101.
22. Taylor GB, Paviour SD, Musaad S, Jones WO, Holland DJ. A clinicopathological review of 34 cases of inflammatory breast disease showing an association between corynebacteria infection and granulomatous mastitis. Pathology. 2003;35(2):109-119.
23. Rakoff-Nahoum S. Why cancer and inflammation? Yale J Biol Med. 2006;79(3-4):123-130.
1. Garcia-Rodiguez JA, Pattullo A. Idiopathic granulomatous mastitis: a mimicking disease in a pregnant woman: a case report. BMC Res Notes. 2013;6:95. doi.10.1186/1756-0500-6-95
2. Gurleyik G, Aktekin A, Aker F, Karagulle H, Saglamc A. Medical and surgical treatment of idiopathic granulomatous lobular mastitis: a benign inflammatory disease mimicking invasive carcinoma. J Breast Cancer. 2012;15(1):119-123. doi:10.4048/jbc.2012.15.1.119
3. Hovanessian Larsen LJ, Peyvandi B, Klipfel N, Grant E, Iyengar G. Granulomatous lobular mastitis: imaging, diagnosis, and treatment. AJR Am J Roentgenol. 2009;193(2):574-581. doi:10.2214/AJR.08.1528
4. Mazlan L, Suhaimi SN, Jasmin SJ, Latar NH, Adzman S, Muhammad R. Breast carcinoma occurring from chronic granulomatous mastitis. Malays J Med Sci. 2012;19(2):82-85.
5. Patel RA, Strickland P, Sankara IR, Pinkston G, Many W Jr, Rodriguez M. Idiopathic granulomatous mastitis: case reports and review of literature. J Gen Intern Med. 2010;25(3):270-273. doi:10.1007/s11606-009-1207-2
6. Akbulut S, Yilmaz D, Bakir S. Methotrexate in the management of idiopathic granulomatous mastitis: review of 108 published cases and report of four cases. Breast J. 2011;17(6):661-668. doi:10.1111/j.1524-4741.2011.01162.x
7. Ergin AB, Cristofanilli M, Daw H, Tahan G, Gong Y. Recurrent granulomatous mastitis mimicking inflammatory breast cancer. BMJ Case Rep. 2011;2011:bcr0720103156. doi:10.1136/bcr.07.2010.3156
8. Hladik M, Schoeller T, Ensat F, Wechselberger G. Idiopathic granulomatous mastitis: successful treatment by mastectomy and immediate breast reconstruction. J Plast Reconstr Aesthet Surg. 2011;64(12):1604-1607. doi:10.1016/j.bjps.2011.07.01
9. Hur SM, Cho DH, Lee SK, et al. Experience of treatment of patients with granulomatous lobular mastitis. J Korean Surg Soc. 2013;85(1):1-6. doi:10.4174/jkss.2013.85.1.
10. Kayahan M, Kadioglu H, Muslumanoglu M. Management of patients with granulomatous mastitis: analysis of 31 cases. Breast Care (Basel). 2012;7(3):226-230. doi:10.1159/000337758
11. Neel A, Hello M, Cottereau A, et al. Long-term outcome in idiopathic granulomatous mastitis: a western multicentre study. QJM. 2013;106(5):433-441. doi:10.1093/qjmed/hct040
12. Seo HR, Na KY, Yim HE, et al. Differential diagnosis in idiopathic granulomatous mastitis and tuberculous mastitis. J Breast Cancer. 2012;15(1):111-118. doi:10.4048/jbc.2012.15.1.111
13. Martinez-Ramos D, Simon-Monterde L, Suelves-Piqueres C, et al. Idiopathic granulomatous mastitis: a systematic review of 3060 patients. Breast J. 2019;25(6):1245-1250. doi:10.1111/tbj.13446
14. Lin CH, Hsu CW, Tsao TY, Chou J. Idiopathic granulomatous mastitis associated with risperidone-induced hyperprolactinemia. Diagn Pathol. 2012;7:2. doi:10.1186/1746-1596-7-2
15. Goulabchand R, Hafidi A, Van de Perre P, et al. Mastitis in autoimmune diseases: review of the literature, diagnostic pathway, and pathophysiological key players. J Clin Med. 2020;9(4):958. doi:10.3390/jcm9040958
16. Altintoprak F. Topical steroids to treat granulomatous mastitis: a case report. Korean J Intern Med. 2011;26(3):356-359. doi:10.3904/kjim.2011.26.3.356
17. Tang A, Dominguez DA, Edquilang JK, et al. Granulomatous mastitis: comparison of novel treatment of steroid injection and current management. J Surg Res. 2020;254:300-305. doi:10.1016/j.jss.2020.04.018
18. Toktas O, Toprak N. Treatment results of intralesional steroid injection and topical steroid administration in pregnant women with idiopathic granulomatous mastitis. Eur J Breast Health. 2021;17(3):283-287. doi:10.4274/ejbh.galenos.2021.2021-2-4
19. Bercot B, Kannengiesser C, Oudin C, et al. First description of NOD2 variant associated with defective neutrophil responses in a woman with granulomatous mastitis related to corynebacteria. J Clin Microbiol. 2009;47(9):3034-3037. doi:10.1128/JCM.00561-09
20. Renshaw AA, Derhagopian RP, Gould EW. Cystic neutrophilic granulomatous mastitis: an underappreciated pattern strongly associated with gram-positive bacilli. Am J Clin Pathol. 2011;136(3):424-427. doi:10.1309/AJCP1W9JBRYOQSNZ
21. Stary CM, Lee YS, Balfour J. Idiopathic granulomatous mastitis associated with corynebacterium sp. Infection. Hawaii Med J. 2011;70(5):99-101.
22. Taylor GB, Paviour SD, Musaad S, Jones WO, Holland DJ. A clinicopathological review of 34 cases of inflammatory breast disease showing an association between corynebacteria infection and granulomatous mastitis. Pathology. 2003;35(2):109-119.
23. Rakoff-Nahoum S. Why cancer and inflammation? Yale J Biol Med. 2006;79(3-4):123-130.
Disseminated Papules and Nodules on the Skin and Oral Mucosa in an Infant
The Diagnosis: Congenital Cutaneous Langerhans Cell Histiocytosis
Although the infectious workup was positive for herpes simplex virus type 1 and cytomegalovirus antibodies, serologies for the rest of the TORCH (toxoplasmosis, other agents [syphilis, hepatitis B virus], rubella, cytomegalovirus) group of infections, as well as other bacterial, fungal, and viral infections, were negative. A skin biopsy from the right fifth toe showed a dense infiltrate of CD1a+ histiocytic cells with folded or kidney-shaped nuclei mixed with eosinophils, which was consistent with Langerhans cell histiocytosis (LCH) (Figure 1). Skin lesions were treated with hydrocortisone cream 2.5% and progressively faded over a few weeks.
.")
Langerhans cell histiocytosis is a rare disorder with a variable clinical presentation depending on the sites affected and the extent of involvement. It can involve multiple organ systems, most commonly the skeletal system and the skin. Organ involvement is characterized by histiocyte infiltration. Acute disseminated multisystem disease most commonly is seen in children younger than 3 years.1
Congenital cutaneous LCH presents with variable skin lesions ranging from papules to vesicles, pustules, and ulcers, with onset at birth or in the neonatal period. Various morphologic traits of skin lesions have been described; the most common presentation is multiple red to yellow-brown, crusted papules with accompanying hemorrhage or erosion.1 Other cases have described an eczematous, seborrheic, diffuse eruption or erosive intertrigo. One case of a child with a solitary necrotic nodule on the scalp has been reported.2
Our patient presented with disseminated, nonblanching, purple to dark red papules and nodules of the skin and oral mucosa, as well as nail dystrophy (Figure 2). However, LCH in a neonate can mimic other causes of congenital papulonodular eruptions. Red-brown papules and nodules with or without crusting in a newborn can be mistaken for erythema toxicum neonatorum, transient neonatal pustular melanosis, congenital leukemia cutis, neonatal erythropoiesis, disseminated neonatal hemangiomatosis, infantile acropustulosis, or congenital TORCH infections such as rubella or syphilis. When LCH presents as vesicles or eroded papules or nodules in a newborn, the differential diagnosis includes incontinentia pigmenti and hereditary epidermolysis bullosa.

Langerhans cell histiocytosis may even present with a classic blueberry muffin rash that can lead clinicians to consider cutaneous metastasis from various hematologic malignancies or the more common TORCH infections. Several diagnostic tests can be performed to clarify the diagnosis, including bacterial and viral cultures and stains, serology, immunohistochemistry, flow cytometry, bone marrow aspiration, or skin biopsy.3 Langerhans cell histiocytosis is diagnosed with a combination of histology, immunohistochemistry, and clinical presentation; however, a skin biopsy is crucial. Tissue should be taken from the most easily accessible yet representative lesion. The characteristic appearance of LCH lesions is described as a dense infiltrate of histiocytic cells mixed with numerous eosinophils in the dermis.1 Histiocytes usually have folded nuclei and eosinophilic cytoplasm or kidney-shaped nuclei with prominent nucleoli. Positive CD1a and/or CD207 (Langerin) staining of the cells is required for definitive diagnosis.4 After diagnosis, it is important to obtain baseline laboratory and radiographic studies to determine the extent of systemic involvement.
Treatment of congenital LCH is tailored to the extent of organ involvement. The dermatologic manifestations resolve without medications in many cases. However, true self-resolving LCH can only be diagnosed retrospectively after a full evaluation for other sites of disease. Disseminated disease can be life-threatening and requires more active management. In cases of skin-limited disease, therapies include topical steroids, nitrogen mustard, or imiquimod; surgical resection of isolated lesions; phototherapy; or systemic therapies such as methotrexate, 6-mercaptopurine, vinblastine/vincristine, cladribine, and/or cytarabine. Symptomatic patients initially are treated with methotrexate and 6-mercaptopurine.5 Asymptomatic infants with skin-limited involvement can be managed with topical treatments.
Our patient had skin-limited disease. Abdominal ultrasonography, skeletal survey, and magnetic resonance imaging of the brain revealed no abnormalities. The patient’s family was advised to monitor him for reoccurrence of the skin lesions and to continue close follow-up with hematology and dermatology. Although congenital LCH often is self-resolving, extensive skin involvement increases the risk for internal organ involvement for several years.6 These patients require long-term follow-up for potential musculoskeletal, ophthalmologic, endocrine, hepatic, and/or pulmonary disease.
- Pan Y, Zeng X, Ge J, et al. Congenital self-healing Langerhans cell histiocytosis: clinical and pathological characteristics. Int J Clin Exp Pathol. 2019;12:2275-2278.
- Morren MA, Vanden Broecke K, Vangeebergen L, et al. Diverse cutaneous presentations of Langerhans cell histiocytosis in children: a retrospective cohort study. Pediatr Blood Cancer. 2016;63:486-492. doi:10.1002/pbc.25834
- Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: diagnosis, differential diagnosis, treatment, sequelae, and standardized follow-up. J Am Acad Dermatol. 2018;78:1047-1056. doi:10.1016/j.jaad.2017.05.060
- Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
- Allen CE, Ladisch S, McClain KL. How I treat Langerhans cell histiocytosis. Blood. 2015;126:26-35. doi:10.1182/blood-2014-12-569301
- Jezierska M, Stefanowicz J, Romanowicz G, et al. Langerhans cell histiocytosis in children—a disease with many faces. recent advances in pathogenesis, diagnostic examinations and treatment. Postepy Dermatol Alergol. 2018;35:6-17. doi:10.5114/pdia.2017.67095
The Diagnosis: Congenital Cutaneous Langerhans Cell Histiocytosis
Although the infectious workup was positive for herpes simplex virus type 1 and cytomegalovirus antibodies, serologies for the rest of the TORCH (toxoplasmosis, other agents [syphilis, hepatitis B virus], rubella, cytomegalovirus) group of infections, as well as other bacterial, fungal, and viral infections, were negative. A skin biopsy from the right fifth toe showed a dense infiltrate of CD1a+ histiocytic cells with folded or kidney-shaped nuclei mixed with eosinophils, which was consistent with Langerhans cell histiocytosis (LCH) (Figure 1). Skin lesions were treated with hydrocortisone cream 2.5% and progressively faded over a few weeks.
Langerhans cell histiocytosis is a rare disorder with a variable clinical presentation depending on the sites affected and the extent of involvement. It can involve multiple organ systems, most commonly the skeletal system and the skin. Organ involvement is characterized by histiocyte infiltration. Acute disseminated multisystem disease most commonly is seen in children younger than 3 years.1
Congenital cutaneous LCH presents with variable skin lesions ranging from papules to vesicles, pustules, and ulcers, with onset at birth or in the neonatal period. Various morphologic traits of skin lesions have been described; the most common presentation is multiple red to yellow-brown, crusted papules with accompanying hemorrhage or erosion.1 Other cases have described an eczematous, seborrheic, diffuse eruption or erosive intertrigo. One case of a child with a solitary necrotic nodule on the scalp has been reported.2
Our patient presented with disseminated, nonblanching, purple to dark red papules and nodules of the skin and oral mucosa, as well as nail dystrophy (Figure 2). However, LCH in a neonate can mimic other causes of congenital papulonodular eruptions. Red-brown papules and nodules with or without crusting in a newborn can be mistaken for erythema toxicum neonatorum, transient neonatal pustular melanosis, congenital leukemia cutis, neonatal erythropoiesis, disseminated neonatal hemangiomatosis, infantile acropustulosis, or congenital TORCH infections such as rubella or syphilis. When LCH presents as vesicles or eroded papules or nodules in a newborn, the differential diagnosis includes incontinentia pigmenti and hereditary epidermolysis bullosa.
Langerhans cell histiocytosis may even present with a classic blueberry muffin rash that can lead clinicians to consider cutaneous metastasis from various hematologic malignancies or the more common TORCH infections. Several diagnostic tests can be performed to clarify the diagnosis, including bacterial and viral cultures and stains, serology, immunohistochemistry, flow cytometry, bone marrow aspiration, or skin biopsy.3 Langerhans cell histiocytosis is diagnosed with a combination of histology, immunohistochemistry, and clinical presentation; however, a skin biopsy is crucial. Tissue should be taken from the most easily accessible yet representative lesion. The characteristic appearance of LCH lesions is described as a dense infiltrate of histiocytic cells mixed with numerous eosinophils in the dermis.1 Histiocytes usually have folded nuclei and eosinophilic cytoplasm or kidney-shaped nuclei with prominent nucleoli. Positive CD1a and/or CD207 (Langerin) staining of the cells is required for definitive diagnosis.4 After diagnosis, it is important to obtain baseline laboratory and radiographic studies to determine the extent of systemic involvement.
Treatment of congenital LCH is tailored to the extent of organ involvement. The dermatologic manifestations resolve without medications in many cases. However, true self-resolving LCH can only be diagnosed retrospectively after a full evaluation for other sites of disease. Disseminated disease can be life-threatening and requires more active management. In cases of skin-limited disease, therapies include topical steroids, nitrogen mustard, or imiquimod; surgical resection of isolated lesions; phototherapy; or systemic therapies such as methotrexate, 6-mercaptopurine, vinblastine/vincristine, cladribine, and/or cytarabine. Symptomatic patients initially are treated with methotrexate and 6-mercaptopurine.5 Asymptomatic infants with skin-limited involvement can be managed with topical treatments.
Our patient had skin-limited disease. Abdominal ultrasonography, skeletal survey, and magnetic resonance imaging of the brain revealed no abnormalities. The patient’s family was advised to monitor him for reoccurrence of the skin lesions and to continue close follow-up with hematology and dermatology. Although congenital LCH often is self-resolving, extensive skin involvement increases the risk for internal organ involvement for several years.6 These patients require long-term follow-up for potential musculoskeletal, ophthalmologic, endocrine, hepatic, and/or pulmonary disease.
The Diagnosis: Congenital Cutaneous Langerhans Cell Histiocytosis
Although the infectious workup was positive for herpes simplex virus type 1 and cytomegalovirus antibodies, serologies for the rest of the TORCH (toxoplasmosis, other agents [syphilis, hepatitis B virus], rubella, cytomegalovirus) group of infections, as well as other bacterial, fungal, and viral infections, were negative. A skin biopsy from the right fifth toe showed a dense infiltrate of CD1a+ histiocytic cells with folded or kidney-shaped nuclei mixed with eosinophils, which was consistent with Langerhans cell histiocytosis (LCH) (Figure 1). Skin lesions were treated with hydrocortisone cream 2.5% and progressively faded over a few weeks.
Langerhans cell histiocytosis is a rare disorder with a variable clinical presentation depending on the sites affected and the extent of involvement. It can involve multiple organ systems, most commonly the skeletal system and the skin. Organ involvement is characterized by histiocyte infiltration. Acute disseminated multisystem disease most commonly is seen in children younger than 3 years.1
Congenital cutaneous LCH presents with variable skin lesions ranging from papules to vesicles, pustules, and ulcers, with onset at birth or in the neonatal period. Various morphologic traits of skin lesions have been described; the most common presentation is multiple red to yellow-brown, crusted papules with accompanying hemorrhage or erosion.1 Other cases have described an eczematous, seborrheic, diffuse eruption or erosive intertrigo. One case of a child with a solitary necrotic nodule on the scalp has been reported.2
Our patient presented with disseminated, nonblanching, purple to dark red papules and nodules of the skin and oral mucosa, as well as nail dystrophy (Figure 2). However, LCH in a neonate can mimic other causes of congenital papulonodular eruptions. Red-brown papules and nodules with or without crusting in a newborn can be mistaken for erythema toxicum neonatorum, transient neonatal pustular melanosis, congenital leukemia cutis, neonatal erythropoiesis, disseminated neonatal hemangiomatosis, infantile acropustulosis, or congenital TORCH infections such as rubella or syphilis. When LCH presents as vesicles or eroded papules or nodules in a newborn, the differential diagnosis includes incontinentia pigmenti and hereditary epidermolysis bullosa.
Langerhans cell histiocytosis may even present with a classic blueberry muffin rash that can lead clinicians to consider cutaneous metastasis from various hematologic malignancies or the more common TORCH infections. Several diagnostic tests can be performed to clarify the diagnosis, including bacterial and viral cultures and stains, serology, immunohistochemistry, flow cytometry, bone marrow aspiration, or skin biopsy.3 Langerhans cell histiocytosis is diagnosed with a combination of histology, immunohistochemistry, and clinical presentation; however, a skin biopsy is crucial. Tissue should be taken from the most easily accessible yet representative lesion. The characteristic appearance of LCH lesions is described as a dense infiltrate of histiocytic cells mixed with numerous eosinophils in the dermis.1 Histiocytes usually have folded nuclei and eosinophilic cytoplasm or kidney-shaped nuclei with prominent nucleoli. Positive CD1a and/or CD207 (Langerin) staining of the cells is required for definitive diagnosis.4 After diagnosis, it is important to obtain baseline laboratory and radiographic studies to determine the extent of systemic involvement.
Treatment of congenital LCH is tailored to the extent of organ involvement. The dermatologic manifestations resolve without medications in many cases. However, true self-resolving LCH can only be diagnosed retrospectively after a full evaluation for other sites of disease. Disseminated disease can be life-threatening and requires more active management. In cases of skin-limited disease, therapies include topical steroids, nitrogen mustard, or imiquimod; surgical resection of isolated lesions; phototherapy; or systemic therapies such as methotrexate, 6-mercaptopurine, vinblastine/vincristine, cladribine, and/or cytarabine. Symptomatic patients initially are treated with methotrexate and 6-mercaptopurine.5 Asymptomatic infants with skin-limited involvement can be managed with topical treatments.
Our patient had skin-limited disease. Abdominal ultrasonography, skeletal survey, and magnetic resonance imaging of the brain revealed no abnormalities. The patient’s family was advised to monitor him for reoccurrence of the skin lesions and to continue close follow-up with hematology and dermatology. Although congenital LCH often is self-resolving, extensive skin involvement increases the risk for internal organ involvement for several years.6 These patients require long-term follow-up for potential musculoskeletal, ophthalmologic, endocrine, hepatic, and/or pulmonary disease.
- Pan Y, Zeng X, Ge J, et al. Congenital self-healing Langerhans cell histiocytosis: clinical and pathological characteristics. Int J Clin Exp Pathol. 2019;12:2275-2278.
- Morren MA, Vanden Broecke K, Vangeebergen L, et al. Diverse cutaneous presentations of Langerhans cell histiocytosis in children: a retrospective cohort study. Pediatr Blood Cancer. 2016;63:486-492. doi:10.1002/pbc.25834
- Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: diagnosis, differential diagnosis, treatment, sequelae, and standardized follow-up. J Am Acad Dermatol. 2018;78:1047-1056. doi:10.1016/j.jaad.2017.05.060
- Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
- Allen CE, Ladisch S, McClain KL. How I treat Langerhans cell histiocytosis. Blood. 2015;126:26-35. doi:10.1182/blood-2014-12-569301
- Jezierska M, Stefanowicz J, Romanowicz G, et al. Langerhans cell histiocytosis in children—a disease with many faces. recent advances in pathogenesis, diagnostic examinations and treatment. Postepy Dermatol Alergol. 2018;35:6-17. doi:10.5114/pdia.2017.67095
- Pan Y, Zeng X, Ge J, et al. Congenital self-healing Langerhans cell histiocytosis: clinical and pathological characteristics. Int J Clin Exp Pathol. 2019;12:2275-2278.
- Morren MA, Vanden Broecke K, Vangeebergen L, et al. Diverse cutaneous presentations of Langerhans cell histiocytosis in children: a retrospective cohort study. Pediatr Blood Cancer. 2016;63:486-492. doi:10.1002/pbc.25834
- Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: diagnosis, differential diagnosis, treatment, sequelae, and standardized follow-up. J Am Acad Dermatol. 2018;78:1047-1056. doi:10.1016/j.jaad.2017.05.060
- Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
- Allen CE, Ladisch S, McClain KL. How I treat Langerhans cell histiocytosis. Blood. 2015;126:26-35. doi:10.1182/blood-2014-12-569301
- Jezierska M, Stefanowicz J, Romanowicz G, et al. Langerhans cell histiocytosis in children—a disease with many faces. recent advances in pathogenesis, diagnostic examinations and treatment. Postepy Dermatol Alergol. 2018;35:6-17. doi:10.5114/pdia.2017.67095
A 38-week-old infant boy presented at birth with disseminated, nonblanching, purple to dark red papules and nodules on the skin and oral mucosa. He was born spontaneously after an uncomplicated pregnancy. The mother experienced an episode of oral herpes simplex virus during pregnancy. The infant was otherwise healthy. Laboratory tests including a complete blood cell count and routine serum biochemical analyses were within reference range; however, an infectious workup was positive for herpes simplex virus type 1 and cytomegalovirus antibodies. Ophthalmologic and auditory screenings were normal.


Interstitial lung disease plus pulmonary hypertension equals poor outcomes in systemic sclerosis
, based on data from more than 3,000 individuals.
Pulmonary complications are now the most common causes of death in adults with systemic sclerosis (SSc), but the impact of patient characteristics and risk factors such as interstitial lung disease (ILD) and pulmonary hypertension (PH) on SSc outcomes remains unclear, wrote Pia Moinzadeh, MD, of University Hospital Cologne (Germany) and colleagues.
Although the role of ILD and PH in different SSc subtypes has been studied, larger studies of the effects of ILD and combining ILD and PH on outcomes are needed, since survival rates can change over time with new classification criteria, diagnostic tools, and improved therapies, they said.
In a study published in the journal Chest, the researchers reviewed data from 3,257 adults aged 18 years and older with SSc over a mean follow-up of 3.45 years. Participants were part of the German Network for Systemic Sclerosis (DNSS) that included 25 clinical centers in Germany. The participants were divided into SSc subsets: 54.2% with limited cutaneous SSc (lcSSc), 31.4% with diffuse cutaneous SSc (dcSSc), and 14.4% SSc overlapping syndromes.
The baseline prevalence of ILD was 34.5%, including 200 patients with ILD-PH and 923 with ILD but without PH. The baseline prevalence of PH without ILD was 4.5%. ILD was defined as SSc associated when other causes were excluded. PH was defined as an increase in mean arterial pressure of at least 25 mm Hg at rest, and also was defined by an estimated right ventricular systolic pressure greater than 35 mm Hg based on echocardiography.
By the end of the study period, 47.6% of SSc patients had ILD, 15.2% had ILD-PH, and 6.5% had pulmonary arterial hypertension (PAH). Of the SSc patients with ILD, 57.3% had dcSSc; the prevalence of PAH was not significantly different between the SSc subtypes. Patients with dcSSc were more likely to develop ILD-PH (52.2%) and ILD without PH (52.1%); patients with lcSSc were more likely to have PAH (64.9%) or no pulmonary involvement (64.1%).
“For all subsets, a significant increase in the frequency of SSc-ILD was observed during follow-ups,” the researchers noted.
Overall survival at 5 years was worst for patients with both ILD and PH (79.1%). Five-year OS for patients with PAH was 85.0%. OS at 5 years was significantly better for patients with ILD without PH (92.8%) and those with no pulmonary involvement (96.4%), compared with the ILD and PH patients (P < 0.001).
In a multivariate analysis, the risk of death was more than five times higher for patients with ILD-PH, compared with the reference group of patients without pulmonary involvement (hazard ratio, 5.3). Factors associated with reduced risk of death included female sex (HR, 0.3), higher body mass index (HR, 0.9), and higher diffusing capacity of the lung for carbon monoxide (HR, 0.98).
The findings were limited by several factors including the incomplete data for patients enrolled early in the registry, lack of complete radiology data, and the inability to determine whether the association between pulmonary involvement and survival was related to ILD or to pulmonary vascular disease, the researchers noted.
However, the results suggest that a combination of ILD and PH is the main predictor of death in patients with SSc and ILD, although the overall survival for SSc patients with and without pulmonary involvement has improved in recent decades thanks to improved therapies, multidisciplinary care, and greater attention to the disease worldwide, they concluded.
The study received no outside funding. Dr. Moinzadeh disclosed lecture fees from Boehringer Ingelheim.
, based on data from more than 3,000 individuals.
Pulmonary complications are now the most common causes of death in adults with systemic sclerosis (SSc), but the impact of patient characteristics and risk factors such as interstitial lung disease (ILD) and pulmonary hypertension (PH) on SSc outcomes remains unclear, wrote Pia Moinzadeh, MD, of University Hospital Cologne (Germany) and colleagues.
Although the role of ILD and PH in different SSc subtypes has been studied, larger studies of the effects of ILD and combining ILD and PH on outcomes are needed, since survival rates can change over time with new classification criteria, diagnostic tools, and improved therapies, they said.
In a study published in the journal Chest, the researchers reviewed data from 3,257 adults aged 18 years and older with SSc over a mean follow-up of 3.45 years. Participants were part of the German Network for Systemic Sclerosis (DNSS) that included 25 clinical centers in Germany. The participants were divided into SSc subsets: 54.2% with limited cutaneous SSc (lcSSc), 31.4% with diffuse cutaneous SSc (dcSSc), and 14.4% SSc overlapping syndromes.
The baseline prevalence of ILD was 34.5%, including 200 patients with ILD-PH and 923 with ILD but without PH. The baseline prevalence of PH without ILD was 4.5%. ILD was defined as SSc associated when other causes were excluded. PH was defined as an increase in mean arterial pressure of at least 25 mm Hg at rest, and also was defined by an estimated right ventricular systolic pressure greater than 35 mm Hg based on echocardiography.
By the end of the study period, 47.6% of SSc patients had ILD, 15.2% had ILD-PH, and 6.5% had pulmonary arterial hypertension (PAH). Of the SSc patients with ILD, 57.3% had dcSSc; the prevalence of PAH was not significantly different between the SSc subtypes. Patients with dcSSc were more likely to develop ILD-PH (52.2%) and ILD without PH (52.1%); patients with lcSSc were more likely to have PAH (64.9%) or no pulmonary involvement (64.1%).
“For all subsets, a significant increase in the frequency of SSc-ILD was observed during follow-ups,” the researchers noted.
Overall survival at 5 years was worst for patients with both ILD and PH (79.1%). Five-year OS for patients with PAH was 85.0%. OS at 5 years was significantly better for patients with ILD without PH (92.8%) and those with no pulmonary involvement (96.4%), compared with the ILD and PH patients (P < 0.001).
In a multivariate analysis, the risk of death was more than five times higher for patients with ILD-PH, compared with the reference group of patients without pulmonary involvement (hazard ratio, 5.3). Factors associated with reduced risk of death included female sex (HR, 0.3), higher body mass index (HR, 0.9), and higher diffusing capacity of the lung for carbon monoxide (HR, 0.98).
The findings were limited by several factors including the incomplete data for patients enrolled early in the registry, lack of complete radiology data, and the inability to determine whether the association between pulmonary involvement and survival was related to ILD or to pulmonary vascular disease, the researchers noted.
However, the results suggest that a combination of ILD and PH is the main predictor of death in patients with SSc and ILD, although the overall survival for SSc patients with and without pulmonary involvement has improved in recent decades thanks to improved therapies, multidisciplinary care, and greater attention to the disease worldwide, they concluded.
The study received no outside funding. Dr. Moinzadeh disclosed lecture fees from Boehringer Ingelheim.
, based on data from more than 3,000 individuals.
Pulmonary complications are now the most common causes of death in adults with systemic sclerosis (SSc), but the impact of patient characteristics and risk factors such as interstitial lung disease (ILD) and pulmonary hypertension (PH) on SSc outcomes remains unclear, wrote Pia Moinzadeh, MD, of University Hospital Cologne (Germany) and colleagues.
Although the role of ILD and PH in different SSc subtypes has been studied, larger studies of the effects of ILD and combining ILD and PH on outcomes are needed, since survival rates can change over time with new classification criteria, diagnostic tools, and improved therapies, they said.
In a study published in the journal Chest, the researchers reviewed data from 3,257 adults aged 18 years and older with SSc over a mean follow-up of 3.45 years. Participants were part of the German Network for Systemic Sclerosis (DNSS) that included 25 clinical centers in Germany. The participants were divided into SSc subsets: 54.2% with limited cutaneous SSc (lcSSc), 31.4% with diffuse cutaneous SSc (dcSSc), and 14.4% SSc overlapping syndromes.
The baseline prevalence of ILD was 34.5%, including 200 patients with ILD-PH and 923 with ILD but without PH. The baseline prevalence of PH without ILD was 4.5%. ILD was defined as SSc associated when other causes were excluded. PH was defined as an increase in mean arterial pressure of at least 25 mm Hg at rest, and also was defined by an estimated right ventricular systolic pressure greater than 35 mm Hg based on echocardiography.
By the end of the study period, 47.6% of SSc patients had ILD, 15.2% had ILD-PH, and 6.5% had pulmonary arterial hypertension (PAH). Of the SSc patients with ILD, 57.3% had dcSSc; the prevalence of PAH was not significantly different between the SSc subtypes. Patients with dcSSc were more likely to develop ILD-PH (52.2%) and ILD without PH (52.1%); patients with lcSSc were more likely to have PAH (64.9%) or no pulmonary involvement (64.1%).
“For all subsets, a significant increase in the frequency of SSc-ILD was observed during follow-ups,” the researchers noted.
Overall survival at 5 years was worst for patients with both ILD and PH (79.1%). Five-year OS for patients with PAH was 85.0%. OS at 5 years was significantly better for patients with ILD without PH (92.8%) and those with no pulmonary involvement (96.4%), compared with the ILD and PH patients (P < 0.001).
In a multivariate analysis, the risk of death was more than five times higher for patients with ILD-PH, compared with the reference group of patients without pulmonary involvement (hazard ratio, 5.3). Factors associated with reduced risk of death included female sex (HR, 0.3), higher body mass index (HR, 0.9), and higher diffusing capacity of the lung for carbon monoxide (HR, 0.98).
The findings were limited by several factors including the incomplete data for patients enrolled early in the registry, lack of complete radiology data, and the inability to determine whether the association between pulmonary involvement and survival was related to ILD or to pulmonary vascular disease, the researchers noted.
However, the results suggest that a combination of ILD and PH is the main predictor of death in patients with SSc and ILD, although the overall survival for SSc patients with and without pulmonary involvement has improved in recent decades thanks to improved therapies, multidisciplinary care, and greater attention to the disease worldwide, they concluded.
The study received no outside funding. Dr. Moinzadeh disclosed lecture fees from Boehringer Ingelheim.
FROM THE JOURNAL CHEST
Mohs found to confer survival benefit in localized Merkel cell carcinoma
results from a national retrospective cohort study suggest.
The study found that, in patients with pathologically confirmed, localized T1/T2 MCC, “treatment with MMS was associated with an approximately 40% reduction in hazard of death compared with WLE,” reported John A. Carucci, MD, PhD, and colleagues in the department of dermatology at NYU Langone Health, New York. The results provide “preliminary data suggesting that treatment of localized, early-stage MCC with MMS may result in the most optimal patient survival outcomes for this aggressive form of skin cancer,” they added. The study was published online in JAMA Dermatology.
“Although data for keratinocytic nonmelanoma skin cancers have been definitive in demonstrating the advantage of peripheral and deep en face margin assessment over conventional WLE or NME [narrow-margin excision], the data for MCC, likely because of the disease’s rarity and limitations of available data sets, have been mixed,” they wrote.
Results from national studies published in the Journal of the National Cancer Institute and the Journal of the American Academy of Dermatology found no difference in survival among patients with localized MCC treated with WLE versus MMS. “However, these studies did not have confirmed pathologic node status, a substantial limitation considering that clinically node-negative cases of localized MCC have sentinel lymph node positivity rates ranging from 25% to 40%,” the authors noted.
To evaluate the association of the surgical excision modality and patient survival for pathologically confirmed localized T1/T2 MCC, Dr. Carucci and coauthors examined a cohort of 2,313 patients from the National Cancer Database with T1/T2 MCC diagnosed between Jan. 1, 2004, and Dec. 31, 2018, with pathologically confirmed, negative regional lymph nodes and treated with surgery. Their mean age was 71 years and 57.9% were male. Of the 2,313 patients, 1,452 underwent WLE, 104 underwent MMS, and 757 underwent NME.
The unadjusted analysis revealed that, compared with WLE, excision with MMS had the best unadjusted mean survival rates: 87.4% versus 86.1%, respectively, at 3 years, 84.5% versus 76.9% at 5 years, and 81.8% versus 60.9% at 10 years. Patients treated with NME had similar mean survival rates as those treated with WLE: 84.8% at 3 years, 78.3% at 5 years, and 60.8% at 10 years.
Multivariable survival analysis demonstrated that treatment with MMS was associated with significantly improved survival, compared with WLE (hazard ratio, 0.59; 95% CI, 0.36-0.97; P = .04).
“These data suggest that MMS may provide a survival benefit in the treatment of localized MCC, although further prospective work studying this issue is required,” the authors concluded. “Future directions may also focus on elucidating the benefit of adjuvant radiotherapy in localized cases treated with MMS.”
They acknowledged certain limitations of the study, including the fewer numbers of patients receiving MMS surgery, lack of randomization, and potential for selection bias.
In an interview, Travis W. Blalock, MD, director of dermatologic surgery, Mohs micrographic surgery, and cutaneous oncology at Emory University, Atlanta, who was asked to comment on the study, said that the field of MCC “has undergone rapid and robust transformation over the past 20 years. These changes encompass advancements in diagnosing the condition, identifying linked viruses, and developing systemic treatments.”
The study findings “imply that comprehensive assessment of histologic margins might offer advantages beyond minimizing scars, minimizing functional impact, and reducing the likelihood of local recurrence,” he said.
“It’s beyond doubt,” he added, that the study “furnishes us with yet another set of real-world insights that will undoubtedly influence patient outcomes. These insights serve to bring clarity to the ways in which we can deliver precisely targeted surgical treatment with durable outcomes for localized MCC.”
Patricia M. Richey, MD, director of Mohs surgery at Boston University, who was also asked to comment on the study, added that, because of the nature of the National Cancer Database, “the authors of this study were unfortunately unable to report disease-specific survival or immunosuppression status. That being said, the preliminary data presented are convincing and should result in us further exploring this topic, as well as readdressing and questioning related issues such as whether or not adjuvant radiotherapy is truly beneficial in cases with histologic clearance via Mohs.”
Dr. Carucci reported receiving grant funding from Regeneron for investigator-initiated basic research. No other author disclosures were reported. Neither Dr. Blalock nor Dr. Richey had relevant disclosures.
results from a national retrospective cohort study suggest.
The study found that, in patients with pathologically confirmed, localized T1/T2 MCC, “treatment with MMS was associated with an approximately 40% reduction in hazard of death compared with WLE,” reported John A. Carucci, MD, PhD, and colleagues in the department of dermatology at NYU Langone Health, New York. The results provide “preliminary data suggesting that treatment of localized, early-stage MCC with MMS may result in the most optimal patient survival outcomes for this aggressive form of skin cancer,” they added. The study was published online in JAMA Dermatology.
“Although data for keratinocytic nonmelanoma skin cancers have been definitive in demonstrating the advantage of peripheral and deep en face margin assessment over conventional WLE or NME [narrow-margin excision], the data for MCC, likely because of the disease’s rarity and limitations of available data sets, have been mixed,” they wrote.
Results from national studies published in the Journal of the National Cancer Institute and the Journal of the American Academy of Dermatology found no difference in survival among patients with localized MCC treated with WLE versus MMS. “However, these studies did not have confirmed pathologic node status, a substantial limitation considering that clinically node-negative cases of localized MCC have sentinel lymph node positivity rates ranging from 25% to 40%,” the authors noted.
To evaluate the association of the surgical excision modality and patient survival for pathologically confirmed localized T1/T2 MCC, Dr. Carucci and coauthors examined a cohort of 2,313 patients from the National Cancer Database with T1/T2 MCC diagnosed between Jan. 1, 2004, and Dec. 31, 2018, with pathologically confirmed, negative regional lymph nodes and treated with surgery. Their mean age was 71 years and 57.9% were male. Of the 2,313 patients, 1,452 underwent WLE, 104 underwent MMS, and 757 underwent NME.
The unadjusted analysis revealed that, compared with WLE, excision with MMS had the best unadjusted mean survival rates: 87.4% versus 86.1%, respectively, at 3 years, 84.5% versus 76.9% at 5 years, and 81.8% versus 60.9% at 10 years. Patients treated with NME had similar mean survival rates as those treated with WLE: 84.8% at 3 years, 78.3% at 5 years, and 60.8% at 10 years.
Multivariable survival analysis demonstrated that treatment with MMS was associated with significantly improved survival, compared with WLE (hazard ratio, 0.59; 95% CI, 0.36-0.97; P = .04).
“These data suggest that MMS may provide a survival benefit in the treatment of localized MCC, although further prospective work studying this issue is required,” the authors concluded. “Future directions may also focus on elucidating the benefit of adjuvant radiotherapy in localized cases treated with MMS.”
They acknowledged certain limitations of the study, including the fewer numbers of patients receiving MMS surgery, lack of randomization, and potential for selection bias.
In an interview, Travis W. Blalock, MD, director of dermatologic surgery, Mohs micrographic surgery, and cutaneous oncology at Emory University, Atlanta, who was asked to comment on the study, said that the field of MCC “has undergone rapid and robust transformation over the past 20 years. These changes encompass advancements in diagnosing the condition, identifying linked viruses, and developing systemic treatments.”
The study findings “imply that comprehensive assessment of histologic margins might offer advantages beyond minimizing scars, minimizing functional impact, and reducing the likelihood of local recurrence,” he said.
“It’s beyond doubt,” he added, that the study “furnishes us with yet another set of real-world insights that will undoubtedly influence patient outcomes. These insights serve to bring clarity to the ways in which we can deliver precisely targeted surgical treatment with durable outcomes for localized MCC.”
Patricia M. Richey, MD, director of Mohs surgery at Boston University, who was also asked to comment on the study, added that, because of the nature of the National Cancer Database, “the authors of this study were unfortunately unable to report disease-specific survival or immunosuppression status. That being said, the preliminary data presented are convincing and should result in us further exploring this topic, as well as readdressing and questioning related issues such as whether or not adjuvant radiotherapy is truly beneficial in cases with histologic clearance via Mohs.”
Dr. Carucci reported receiving grant funding from Regeneron for investigator-initiated basic research. No other author disclosures were reported. Neither Dr. Blalock nor Dr. Richey had relevant disclosures.
results from a national retrospective cohort study suggest.
The study found that, in patients with pathologically confirmed, localized T1/T2 MCC, “treatment with MMS was associated with an approximately 40% reduction in hazard of death compared with WLE,” reported John A. Carucci, MD, PhD, and colleagues in the department of dermatology at NYU Langone Health, New York. The results provide “preliminary data suggesting that treatment of localized, early-stage MCC with MMS may result in the most optimal patient survival outcomes for this aggressive form of skin cancer,” they added. The study was published online in JAMA Dermatology.
“Although data for keratinocytic nonmelanoma skin cancers have been definitive in demonstrating the advantage of peripheral and deep en face margin assessment over conventional WLE or NME [narrow-margin excision], the data for MCC, likely because of the disease’s rarity and limitations of available data sets, have been mixed,” they wrote.
Results from national studies published in the Journal of the National Cancer Institute and the Journal of the American Academy of Dermatology found no difference in survival among patients with localized MCC treated with WLE versus MMS. “However, these studies did not have confirmed pathologic node status, a substantial limitation considering that clinically node-negative cases of localized MCC have sentinel lymph node positivity rates ranging from 25% to 40%,” the authors noted.
To evaluate the association of the surgical excision modality and patient survival for pathologically confirmed localized T1/T2 MCC, Dr. Carucci and coauthors examined a cohort of 2,313 patients from the National Cancer Database with T1/T2 MCC diagnosed between Jan. 1, 2004, and Dec. 31, 2018, with pathologically confirmed, negative regional lymph nodes and treated with surgery. Their mean age was 71 years and 57.9% were male. Of the 2,313 patients, 1,452 underwent WLE, 104 underwent MMS, and 757 underwent NME.
The unadjusted analysis revealed that, compared with WLE, excision with MMS had the best unadjusted mean survival rates: 87.4% versus 86.1%, respectively, at 3 years, 84.5% versus 76.9% at 5 years, and 81.8% versus 60.9% at 10 years. Patients treated with NME had similar mean survival rates as those treated with WLE: 84.8% at 3 years, 78.3% at 5 years, and 60.8% at 10 years.
Multivariable survival analysis demonstrated that treatment with MMS was associated with significantly improved survival, compared with WLE (hazard ratio, 0.59; 95% CI, 0.36-0.97; P = .04).
“These data suggest that MMS may provide a survival benefit in the treatment of localized MCC, although further prospective work studying this issue is required,” the authors concluded. “Future directions may also focus on elucidating the benefit of adjuvant radiotherapy in localized cases treated with MMS.”
They acknowledged certain limitations of the study, including the fewer numbers of patients receiving MMS surgery, lack of randomization, and potential for selection bias.
In an interview, Travis W. Blalock, MD, director of dermatologic surgery, Mohs micrographic surgery, and cutaneous oncology at Emory University, Atlanta, who was asked to comment on the study, said that the field of MCC “has undergone rapid and robust transformation over the past 20 years. These changes encompass advancements in diagnosing the condition, identifying linked viruses, and developing systemic treatments.”
The study findings “imply that comprehensive assessment of histologic margins might offer advantages beyond minimizing scars, minimizing functional impact, and reducing the likelihood of local recurrence,” he said.
“It’s beyond doubt,” he added, that the study “furnishes us with yet another set of real-world insights that will undoubtedly influence patient outcomes. These insights serve to bring clarity to the ways in which we can deliver precisely targeted surgical treatment with durable outcomes for localized MCC.”
Patricia M. Richey, MD, director of Mohs surgery at Boston University, who was also asked to comment on the study, added that, because of the nature of the National Cancer Database, “the authors of this study were unfortunately unable to report disease-specific survival or immunosuppression status. That being said, the preliminary data presented are convincing and should result in us further exploring this topic, as well as readdressing and questioning related issues such as whether or not adjuvant radiotherapy is truly beneficial in cases with histologic clearance via Mohs.”
Dr. Carucci reported receiving grant funding from Regeneron for investigator-initiated basic research. No other author disclosures were reported. Neither Dr. Blalock nor Dr. Richey had relevant disclosures.
FROM JAMA DERMATOLOGY
FDA okays first-ever new drug for rare bone disorder
Affecting roughly 400 people in the United States and 900 worldwide, FOP is an autosomal dominant condition in which bone develops in soft connective tissue areas of the body where it isn’t normally present (heterotopic ossification), such as the ligaments, tendons, and skeletal muscles. This leads to severe restriction in mobility and function, to the point that people lose the ability to feed or care for themselves. Most are completely disabled by age 30 years and median life expectancy is 56 years, with death often caused by bone formation around the rib cage restricting respiration.
“As a clinician caring for patients with FOP, I personally see the daily challenges and stresses that our patients and their families must contend with ... since the accumulation of heterotopic ossification in FOP is progressive, irreversible, and life altering. This medication is an important treatment option for our FOP community,” said endocrinologist Edward Hsiao, MD, professor of medicine at the University of California, San Francisco, in a statement from Ipsen.
Taken orally, palovarotene selectively targets the gamma subtype of retinoic acid receptors that regulate skeletal development and ectopic bone in the retinoid signaling pathway. The drug mediates interactions between these receptors, growth factors, and proteins within that pathway to reduce new abnormal bone formation.
It is now FDA approved for the treatment of FOP in female patients aged 8 years or older and male patients aged 10 years or older. The recommended dosing is 5 mg daily or weight-based equivalent for pediatric patients under 14 years of age, which can be modified or increased for flare-up symptoms. It is contraindicated during pregnancy.
The FDA approval was based on 18-month data from the phase 3, multicenter, open-label MOVE trial that included 107 adult and pediatric patients, over 10% of the world’s population with FOP. All received oral palovarotene and were compared with untreated individuals from a prior natural history study of the condition. The drug reduced annualized heterotopic ossification volume by 54%.
Side effects were typical of those seen with other systemic retinoid drugs, including mucocutaneous events such as dryness of the skin and mucous membranes, alopecia, drug eruption, rash, and pruritus, and musculoskeletal events, such as arthralgia and premature growth plate closure in growing children.
According to Dr. Hsiao, who was a MOVE investigator, the study “showed that Sohonos can decrease new heterotopic ossification, and that palovarotene can be tolerated by many patients with FOP. Sohonos is not for everyone. As with all medicines there are risks in this case especially for young children who may develop early growth plate closure. In addition, Sohonos has the same side effects as other retinoids.”
The FDA approval of palovarotene follows its rejection for marketing authorization in the European Union in July 2023.
Reached for comment, an Ipsen spokesperson said in an interview: “We reached the end of the regulatory process in the European Union for Sohonos and are disappointed the European Commission decided not to approved palovarotene for people with FOP in Europe.”
The company is developing another drug, fidrisertib, for treating FOP. A pivotal phase 2 trial for that drug is now recruiting patients. Asked where Ipsen might try to market fidrisertib, the spokesperson replied:“At this point, our focus is on the completion of the pivotal trial.”
Meanwhile, in the United States, the FOP community is celebrating the palovarotene approval. In a statement, Michelle Davis, executive director of the International Fibrodysplasia Ossificans Progressiva Association, said: “FOP is life altering to the individuals diagnosed and their families. There’s not a day that goes by where those impacted don’t worry about the debilitating physical pain of muscle that is replaced by bone, another joint locking, or the relentless emotional toll of losing the ability to do an activity they love, or hold a loved one close. ... The first treatment for FOP has been proven to reduce the volume of new abnormal bone growth, which may result in better health outcomes for people living with FOP.”
Ipsen is offering a patient support program to assist with education, coverage, and reimbursement (1-866-435-5677).
A version of this article appeared on Medscape.com.
Affecting roughly 400 people in the United States and 900 worldwide, FOP is an autosomal dominant condition in which bone develops in soft connective tissue areas of the body where it isn’t normally present (heterotopic ossification), such as the ligaments, tendons, and skeletal muscles. This leads to severe restriction in mobility and function, to the point that people lose the ability to feed or care for themselves. Most are completely disabled by age 30 years and median life expectancy is 56 years, with death often caused by bone formation around the rib cage restricting respiration.
“As a clinician caring for patients with FOP, I personally see the daily challenges and stresses that our patients and their families must contend with ... since the accumulation of heterotopic ossification in FOP is progressive, irreversible, and life altering. This medication is an important treatment option for our FOP community,” said endocrinologist Edward Hsiao, MD, professor of medicine at the University of California, San Francisco, in a statement from Ipsen.
Taken orally, palovarotene selectively targets the gamma subtype of retinoic acid receptors that regulate skeletal development and ectopic bone in the retinoid signaling pathway. The drug mediates interactions between these receptors, growth factors, and proteins within that pathway to reduce new abnormal bone formation.
It is now FDA approved for the treatment of FOP in female patients aged 8 years or older and male patients aged 10 years or older. The recommended dosing is 5 mg daily or weight-based equivalent for pediatric patients under 14 years of age, which can be modified or increased for flare-up symptoms. It is contraindicated during pregnancy.
The FDA approval was based on 18-month data from the phase 3, multicenter, open-label MOVE trial that included 107 adult and pediatric patients, over 10% of the world’s population with FOP. All received oral palovarotene and were compared with untreated individuals from a prior natural history study of the condition. The drug reduced annualized heterotopic ossification volume by 54%.
Side effects were typical of those seen with other systemic retinoid drugs, including mucocutaneous events such as dryness of the skin and mucous membranes, alopecia, drug eruption, rash, and pruritus, and musculoskeletal events, such as arthralgia and premature growth plate closure in growing children.
According to Dr. Hsiao, who was a MOVE investigator, the study “showed that Sohonos can decrease new heterotopic ossification, and that palovarotene can be tolerated by many patients with FOP. Sohonos is not for everyone. As with all medicines there are risks in this case especially for young children who may develop early growth plate closure. In addition, Sohonos has the same side effects as other retinoids.”
The FDA approval of palovarotene follows its rejection for marketing authorization in the European Union in July 2023.
Reached for comment, an Ipsen spokesperson said in an interview: “We reached the end of the regulatory process in the European Union for Sohonos and are disappointed the European Commission decided not to approved palovarotene for people with FOP in Europe.”
The company is developing another drug, fidrisertib, for treating FOP. A pivotal phase 2 trial for that drug is now recruiting patients. Asked where Ipsen might try to market fidrisertib, the spokesperson replied:“At this point, our focus is on the completion of the pivotal trial.”
Meanwhile, in the United States, the FOP community is celebrating the palovarotene approval. In a statement, Michelle Davis, executive director of the International Fibrodysplasia Ossificans Progressiva Association, said: “FOP is life altering to the individuals diagnosed and their families. There’s not a day that goes by where those impacted don’t worry about the debilitating physical pain of muscle that is replaced by bone, another joint locking, or the relentless emotional toll of losing the ability to do an activity they love, or hold a loved one close. ... The first treatment for FOP has been proven to reduce the volume of new abnormal bone growth, which may result in better health outcomes for people living with FOP.”
Ipsen is offering a patient support program to assist with education, coverage, and reimbursement (1-866-435-5677).
A version of this article appeared on Medscape.com.
Affecting roughly 400 people in the United States and 900 worldwide, FOP is an autosomal dominant condition in which bone develops in soft connective tissue areas of the body where it isn’t normally present (heterotopic ossification), such as the ligaments, tendons, and skeletal muscles. This leads to severe restriction in mobility and function, to the point that people lose the ability to feed or care for themselves. Most are completely disabled by age 30 years and median life expectancy is 56 years, with death often caused by bone formation around the rib cage restricting respiration.
“As a clinician caring for patients with FOP, I personally see the daily challenges and stresses that our patients and their families must contend with ... since the accumulation of heterotopic ossification in FOP is progressive, irreversible, and life altering. This medication is an important treatment option for our FOP community,” said endocrinologist Edward Hsiao, MD, professor of medicine at the University of California, San Francisco, in a statement from Ipsen.
Taken orally, palovarotene selectively targets the gamma subtype of retinoic acid receptors that regulate skeletal development and ectopic bone in the retinoid signaling pathway. The drug mediates interactions between these receptors, growth factors, and proteins within that pathway to reduce new abnormal bone formation.
It is now FDA approved for the treatment of FOP in female patients aged 8 years or older and male patients aged 10 years or older. The recommended dosing is 5 mg daily or weight-based equivalent for pediatric patients under 14 years of age, which can be modified or increased for flare-up symptoms. It is contraindicated during pregnancy.
The FDA approval was based on 18-month data from the phase 3, multicenter, open-label MOVE trial that included 107 adult and pediatric patients, over 10% of the world’s population with FOP. All received oral palovarotene and were compared with untreated individuals from a prior natural history study of the condition. The drug reduced annualized heterotopic ossification volume by 54%.
Side effects were typical of those seen with other systemic retinoid drugs, including mucocutaneous events such as dryness of the skin and mucous membranes, alopecia, drug eruption, rash, and pruritus, and musculoskeletal events, such as arthralgia and premature growth plate closure in growing children.
According to Dr. Hsiao, who was a MOVE investigator, the study “showed that Sohonos can decrease new heterotopic ossification, and that palovarotene can be tolerated by many patients with FOP. Sohonos is not for everyone. As with all medicines there are risks in this case especially for young children who may develop early growth plate closure. In addition, Sohonos has the same side effects as other retinoids.”
The FDA approval of palovarotene follows its rejection for marketing authorization in the European Union in July 2023.
Reached for comment, an Ipsen spokesperson said in an interview: “We reached the end of the regulatory process in the European Union for Sohonos and are disappointed the European Commission decided not to approved palovarotene for people with FOP in Europe.”
The company is developing another drug, fidrisertib, for treating FOP. A pivotal phase 2 trial for that drug is now recruiting patients. Asked where Ipsen might try to market fidrisertib, the spokesperson replied:“At this point, our focus is on the completion of the pivotal trial.”
Meanwhile, in the United States, the FOP community is celebrating the palovarotene approval. In a statement, Michelle Davis, executive director of the International Fibrodysplasia Ossificans Progressiva Association, said: “FOP is life altering to the individuals diagnosed and their families. There’s not a day that goes by where those impacted don’t worry about the debilitating physical pain of muscle that is replaced by bone, another joint locking, or the relentless emotional toll of losing the ability to do an activity they love, or hold a loved one close. ... The first treatment for FOP has been proven to reduce the volume of new abnormal bone growth, which may result in better health outcomes for people living with FOP.”
Ipsen is offering a patient support program to assist with education, coverage, and reimbursement (1-866-435-5677).
A version of this article appeared on Medscape.com.
A 75-year-old White woman presented with diffuse erythema, scale, and pruritus on her scalp
The classical presentation includes symmetric proximal muscle weakness and underlying malignancy and is very common in adult patients. The etiology is unknown, however.
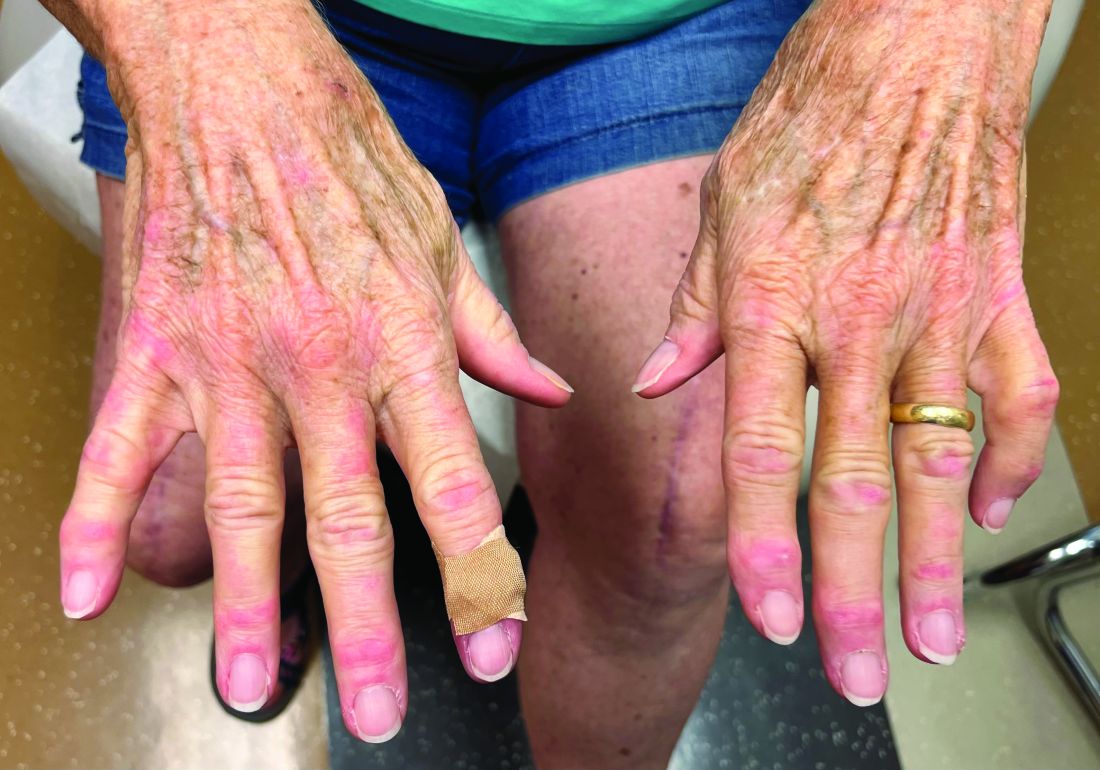
Some studies suggest people with certain HLA subtypes are at higher risk, and various infectious and pharmacological triggers are suspected to play a role in the pathogenesis of dermatomyositis. Infectious causes include Coxsackie B, enterovirus, and parvovirus. Drugs such as antineoplastic agents, antibiotics, and NSAIDs have been found to be triggers.
The pathogenesis of dermatomyositis involves immune-mediated damage to muscle capillaries and the endothelium of arterioles. In the typical humoral immune response, complement activation occurs. One mechanism of damage in dermatomyositis occurs when the membrane attack complex formed at the end of the complement process deposits in blood vessels, causing inflammation. B cells, autoantibodies, and interferon overexpression may also play a role in damaging the vasculature and muscle fibers. Hypoxia leads to muscular atrophy, resulting in degeneration and death of the fibers. On muscle biopsy, a perivascular and perimysial inflammatory infiltrate, perifascicular atrophy, and microangiopathy may be present. Skin histology reveals vacuolar changes in the basal layer, a lymphocytic infiltrate, and increased mucin production in the dermis.
On clinical examination, patients will have proximal muscle weakness and a skin rash that may include Gottron’s papules, heliotrope erythema, V-sign, shawl sign, holster sign, scalp erythema, midfacial erythema, and photosensitivity. Scalp erythema in dermatomyositis is highly linked to pruritus, alopecia, and telogen effluvium. Patients may experience small fiber neuropathy in dermatomyositis.
Serologies for this patient, who had previously been diagnosed and treated for dermatomyositis, were significant for a positive ANA 1:2560. Anti-Jo-1 antibody was negative. Her liver function tests, aldolase, creatinine kinase, sedimentation rate, C-reactive protein, and serum protein electrophoresis were normal. Imaging revealed mild chronic interstitial lung disease. A malignancy workup was negative.

Treatment of dermatomyositis involves lifestyle changes and pharmacologic therapy. Because of the intense photosensitivity, patients should be diligent with their sun protection. Methotrexate, azathioprine, and mycophenolate mofetil are considered first-line therapies for dermatomyositis. Therapies such as cyclophosphamide, rituximab, IVIg, and plasmapheresis may also be indicated in severe or refractory cases. Additionally, patients with pulmonary involvement should be given systemic steroids. The side effects of these drugs must be considered in the context of the patient’s demographics, comorbidities and lifestyle.
This case and the photos were submitted by Lucas Shapiro, BS, of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Natalie Y. Nasser, MD, of Kaiser Permanente Riverside Medical Center, Riverside, Calif. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Qudsiya Z and Waseem M. Dermatomyositis, in “StatPearls.” Treasure Island, Fla.: StatPearls Publishing, 2023 Jan.
2. Kamperman RG et al. Int J Mol Sci. 2022 Apr 13;23(8):4301.
3. Kassamali B et al. Int J WomensDermatol. 2021 Sep 24;7(5Part A):576-82.
4. Vázquez-Herrera NE et al. Skin Appendage Disord. 2018 Aug;4(3):187-99.
The classical presentation includes symmetric proximal muscle weakness and underlying malignancy and is very common in adult patients. The etiology is unknown, however.
Some studies suggest people with certain HLA subtypes are at higher risk, and various infectious and pharmacological triggers are suspected to play a role in the pathogenesis of dermatomyositis. Infectious causes include Coxsackie B, enterovirus, and parvovirus. Drugs such as antineoplastic agents, antibiotics, and NSAIDs have been found to be triggers.
The pathogenesis of dermatomyositis involves immune-mediated damage to muscle capillaries and the endothelium of arterioles. In the typical humoral immune response, complement activation occurs. One mechanism of damage in dermatomyositis occurs when the membrane attack complex formed at the end of the complement process deposits in blood vessels, causing inflammation. B cells, autoantibodies, and interferon overexpression may also play a role in damaging the vasculature and muscle fibers. Hypoxia leads to muscular atrophy, resulting in degeneration and death of the fibers. On muscle biopsy, a perivascular and perimysial inflammatory infiltrate, perifascicular atrophy, and microangiopathy may be present. Skin histology reveals vacuolar changes in the basal layer, a lymphocytic infiltrate, and increased mucin production in the dermis.
On clinical examination, patients will have proximal muscle weakness and a skin rash that may include Gottron’s papules, heliotrope erythema, V-sign, shawl sign, holster sign, scalp erythema, midfacial erythema, and photosensitivity. Scalp erythema in dermatomyositis is highly linked to pruritus, alopecia, and telogen effluvium. Patients may experience small fiber neuropathy in dermatomyositis.
Serologies for this patient, who had previously been diagnosed and treated for dermatomyositis, were significant for a positive ANA 1:2560. Anti-Jo-1 antibody was negative. Her liver function tests, aldolase, creatinine kinase, sedimentation rate, C-reactive protein, and serum protein electrophoresis were normal. Imaging revealed mild chronic interstitial lung disease. A malignancy workup was negative.
Treatment of dermatomyositis involves lifestyle changes and pharmacologic therapy. Because of the intense photosensitivity, patients should be diligent with their sun protection. Methotrexate, azathioprine, and mycophenolate mofetil are considered first-line therapies for dermatomyositis. Therapies such as cyclophosphamide, rituximab, IVIg, and plasmapheresis may also be indicated in severe or refractory cases. Additionally, patients with pulmonary involvement should be given systemic steroids. The side effects of these drugs must be considered in the context of the patient’s demographics, comorbidities and lifestyle.
This case and the photos were submitted by Lucas Shapiro, BS, of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Natalie Y. Nasser, MD, of Kaiser Permanente Riverside Medical Center, Riverside, Calif. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Qudsiya Z and Waseem M. Dermatomyositis, in “StatPearls.” Treasure Island, Fla.: StatPearls Publishing, 2023 Jan.
2. Kamperman RG et al. Int J Mol Sci. 2022 Apr 13;23(8):4301.
3. Kassamali B et al. Int J WomensDermatol. 2021 Sep 24;7(5Part A):576-82.
4. Vázquez-Herrera NE et al. Skin Appendage Disord. 2018 Aug;4(3):187-99.
The classical presentation includes symmetric proximal muscle weakness and underlying malignancy and is very common in adult patients. The etiology is unknown, however.
Some studies suggest people with certain HLA subtypes are at higher risk, and various infectious and pharmacological triggers are suspected to play a role in the pathogenesis of dermatomyositis. Infectious causes include Coxsackie B, enterovirus, and parvovirus. Drugs such as antineoplastic agents, antibiotics, and NSAIDs have been found to be triggers.
The pathogenesis of dermatomyositis involves immune-mediated damage to muscle capillaries and the endothelium of arterioles. In the typical humoral immune response, complement activation occurs. One mechanism of damage in dermatomyositis occurs when the membrane attack complex formed at the end of the complement process deposits in blood vessels, causing inflammation. B cells, autoantibodies, and interferon overexpression may also play a role in damaging the vasculature and muscle fibers. Hypoxia leads to muscular atrophy, resulting in degeneration and death of the fibers. On muscle biopsy, a perivascular and perimysial inflammatory infiltrate, perifascicular atrophy, and microangiopathy may be present. Skin histology reveals vacuolar changes in the basal layer, a lymphocytic infiltrate, and increased mucin production in the dermis.
On clinical examination, patients will have proximal muscle weakness and a skin rash that may include Gottron’s papules, heliotrope erythema, V-sign, shawl sign, holster sign, scalp erythema, midfacial erythema, and photosensitivity. Scalp erythema in dermatomyositis is highly linked to pruritus, alopecia, and telogen effluvium. Patients may experience small fiber neuropathy in dermatomyositis.
Serologies for this patient, who had previously been diagnosed and treated for dermatomyositis, were significant for a positive ANA 1:2560. Anti-Jo-1 antibody was negative. Her liver function tests, aldolase, creatinine kinase, sedimentation rate, C-reactive protein, and serum protein electrophoresis were normal. Imaging revealed mild chronic interstitial lung disease. A malignancy workup was negative.
Treatment of dermatomyositis involves lifestyle changes and pharmacologic therapy. Because of the intense photosensitivity, patients should be diligent with their sun protection. Methotrexate, azathioprine, and mycophenolate mofetil are considered first-line therapies for dermatomyositis. Therapies such as cyclophosphamide, rituximab, IVIg, and plasmapheresis may also be indicated in severe or refractory cases. Additionally, patients with pulmonary involvement should be given systemic steroids. The side effects of these drugs must be considered in the context of the patient’s demographics, comorbidities and lifestyle.
This case and the photos were submitted by Lucas Shapiro, BS, of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Natalie Y. Nasser, MD, of Kaiser Permanente Riverside Medical Center, Riverside, Calif. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Qudsiya Z and Waseem M. Dermatomyositis, in “StatPearls.” Treasure Island, Fla.: StatPearls Publishing, 2023 Jan.
2. Kamperman RG et al. Int J Mol Sci. 2022 Apr 13;23(8):4301.
3. Kassamali B et al. Int J WomensDermatol. 2021 Sep 24;7(5Part A):576-82.
4. Vázquez-Herrera NE et al. Skin Appendage Disord. 2018 Aug;4(3):187-99.
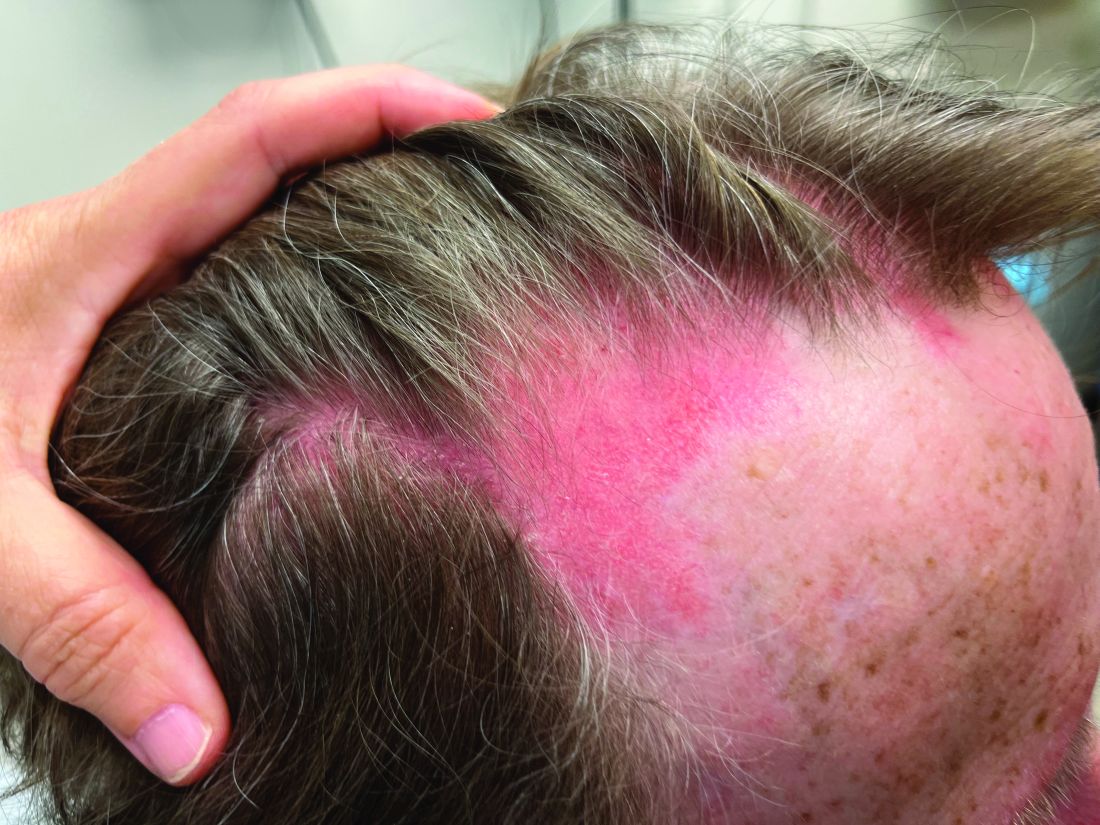
West Nile infections rising in the U.S.
Several signs are pointing to an impending surge in the number of human cases of West Nile virus in several regions of the United States.
West Nile virus is spread by infected mosquitoes and currently there is no cure or virus-specific treatment. In rare cases, it can be deadly. It can infect humans, birds, horses, and other mammals.
West Nile Virus is the leading cause of mosquito-borne disease in the continental United States. As of Aug. 8, 126 human cases had been identified across 22 states, according to the Centers for Disease Control and Prevention.
“Particularly here in California, it’s peak risk right now,” said Vicki Kramer, PhD, chief of vector-borne diseases in the California Department of Public Health. She said scientists there are seeing higher mosquito and infected mosquito numbers.
“Peak risk right now”
Dead birds are tested for the virus and by Aug. 4, 181 of the 913 birds tested in California have been positive, three times the total testing positive by this time in 2022.
“Last year at this time, we had 60 positive dead birds out of 817 tested,” Dr. Kramer said.
Severe flooding and high heat can contribute to the rise in mosquito populations and many parts of the country have seen plenty of both.
One of the ways scientists track infected mosquito patterns in California is by using flocks of strategically placed sentinel chickens.
“Chickens are a mosquito magnet,” Dr. Kramer said.
Chickens don’t get sick with the virus, but they do build antibodies to it. Surveillance teams check their blood every other week to track the virus.
Daniel Pastula, MD, MHS, chief of neuroinfectious diseases and global neurology at the University of Colorado School of Medicine and the Colorado School of Public Health, said the state is watching troubling signs as well.
“The concern this year,” Dr. Pastula said, “particularly along the Front Range in Colorado, is we’ve found many more mosquitoes [that are] positive for West Nile earlier in the season compared with other years.
“We’re bracing for higher-than-baseline human cases,” he said.
Asked about this year’s first human case, reported in Toronto, a region with a long winter and low incidence of the virus, he said that provides a further example that people need to be prepared even in climates not known to be mosquito-dense.
He added, however, that climate is only one factor in the severity of the season. Others include birds’ immunity and migratory patterns.
Dr. Pastula said that fluctuations in temperature and rainfall are rising with climate change and are disrupting normal baseline levels of West Nile.
“That shows we need to be prepared for West Nile virus and other mosquito-borne diseases in any place in North America or really the world. We recently saw malaria cases in the southern United States. It just shows you how dangerous mosquitoes can be.”
Avoid mosquito bites
Dr. Pastula and Dr. Kramer list the precautions people can take to protect themselves from West Nile virus:
- Limit outdoor exposure particularly at dusk and dawn.
- Wear protective clothing.
- Use .
- Repair window screens so mosquitoes cannot fly through.
- Dump and drain standing water on your property and maintain swimming pools.
Dr. Pastula noted that summer is the time human cases start to mount – typically from July and August to the first hard freeze.
“We have been warning people here up and down the Front Range of Colorado to take prevention very seriously,” Dr. Pastula said.
He pointed out that 80% who are infected with West Nile will have no symptoms.
About 20% will have flu-like illness – high fever, body and joint aches, rash, diarrhea, or headaches. Symptoms may last for weeks. About 1% of the time, he said, people can get neuroinvasive West Nile.
Dr. Pastula explained that the virus can infect the covering of the brain and spinal cord causing meningitis with very high fever, severe headaches, stiff neck, and sensitivity to light.
So far this year, there have been 89 neuroinvasive cases reported nationally, according to the CDC.
With West Nile encephalitis, the virus “can infect the brain itself causing altered mental status, movement disorders, or weakness,” Dr. Pastula said.
Sometimes it can infect the gray matter of the spinal cord causing a West Nile virus poliomyelitis, which brings polio-like symptoms.
“The West Nile encephalitis and poliomyelitis can cause permanent deficits or even death,” he said. “It’s uncommon but it’s not trivial.”
Several vaccine candidates are in development, Dr. Pastula said, but none has reached clinical trials. Part of the reason for that, he said, is that scientists must be able to predict the timing of an outbreak.
“We’re not really great at predicting outbreaks,” he said.
Although the risk for neuroinvasive disease is small, it can be higher in certain groups, he said – those who are over age 60 years or are immunocompromised; those who have diabetes, cancer, or kidney disease; or those who have undergone organ transplants.
Those infected should see a health care professional and may be able to get relief with the usual medications for flu-like illness.
Some with severe infection may need to go to the hospital, Dr. Pastula said.
A version of this article first appeared on Medscape.com.
Several signs are pointing to an impending surge in the number of human cases of West Nile virus in several regions of the United States.
West Nile virus is spread by infected mosquitoes and currently there is no cure or virus-specific treatment. In rare cases, it can be deadly. It can infect humans, birds, horses, and other mammals.
West Nile Virus is the leading cause of mosquito-borne disease in the continental United States. As of Aug. 8, 126 human cases had been identified across 22 states, according to the Centers for Disease Control and Prevention.
“Particularly here in California, it’s peak risk right now,” said Vicki Kramer, PhD, chief of vector-borne diseases in the California Department of Public Health. She said scientists there are seeing higher mosquito and infected mosquito numbers.
“Peak risk right now”
Dead birds are tested for the virus and by Aug. 4, 181 of the 913 birds tested in California have been positive, three times the total testing positive by this time in 2022.
“Last year at this time, we had 60 positive dead birds out of 817 tested,” Dr. Kramer said.
Severe flooding and high heat can contribute to the rise in mosquito populations and many parts of the country have seen plenty of both.
One of the ways scientists track infected mosquito patterns in California is by using flocks of strategically placed sentinel chickens.
“Chickens are a mosquito magnet,” Dr. Kramer said.
Chickens don’t get sick with the virus, but they do build antibodies to it. Surveillance teams check their blood every other week to track the virus.
Daniel Pastula, MD, MHS, chief of neuroinfectious diseases and global neurology at the University of Colorado School of Medicine and the Colorado School of Public Health, said the state is watching troubling signs as well.
“The concern this year,” Dr. Pastula said, “particularly along the Front Range in Colorado, is we’ve found many more mosquitoes [that are] positive for West Nile earlier in the season compared with other years.
“We’re bracing for higher-than-baseline human cases,” he said.
Asked about this year’s first human case, reported in Toronto, a region with a long winter and low incidence of the virus, he said that provides a further example that people need to be prepared even in climates not known to be mosquito-dense.
He added, however, that climate is only one factor in the severity of the season. Others include birds’ immunity and migratory patterns.
Dr. Pastula said that fluctuations in temperature and rainfall are rising with climate change and are disrupting normal baseline levels of West Nile.
“That shows we need to be prepared for West Nile virus and other mosquito-borne diseases in any place in North America or really the world. We recently saw malaria cases in the southern United States. It just shows you how dangerous mosquitoes can be.”
Avoid mosquito bites
Dr. Pastula and Dr. Kramer list the precautions people can take to protect themselves from West Nile virus:
- Limit outdoor exposure particularly at dusk and dawn.
- Wear protective clothing.
- Use .
- Repair window screens so mosquitoes cannot fly through.
- Dump and drain standing water on your property and maintain swimming pools.
Dr. Pastula noted that summer is the time human cases start to mount – typically from July and August to the first hard freeze.
“We have been warning people here up and down the Front Range of Colorado to take prevention very seriously,” Dr. Pastula said.
He pointed out that 80% who are infected with West Nile will have no symptoms.
About 20% will have flu-like illness – high fever, body and joint aches, rash, diarrhea, or headaches. Symptoms may last for weeks. About 1% of the time, he said, people can get neuroinvasive West Nile.
Dr. Pastula explained that the virus can infect the covering of the brain and spinal cord causing meningitis with very high fever, severe headaches, stiff neck, and sensitivity to light.
So far this year, there have been 89 neuroinvasive cases reported nationally, according to the CDC.
With West Nile encephalitis, the virus “can infect the brain itself causing altered mental status, movement disorders, or weakness,” Dr. Pastula said.
Sometimes it can infect the gray matter of the spinal cord causing a West Nile virus poliomyelitis, which brings polio-like symptoms.
“The West Nile encephalitis and poliomyelitis can cause permanent deficits or even death,” he said. “It’s uncommon but it’s not trivial.”
Several vaccine candidates are in development, Dr. Pastula said, but none has reached clinical trials. Part of the reason for that, he said, is that scientists must be able to predict the timing of an outbreak.
“We’re not really great at predicting outbreaks,” he said.
Although the risk for neuroinvasive disease is small, it can be higher in certain groups, he said – those who are over age 60 years or are immunocompromised; those who have diabetes, cancer, or kidney disease; or those who have undergone organ transplants.
Those infected should see a health care professional and may be able to get relief with the usual medications for flu-like illness.
Some with severe infection may need to go to the hospital, Dr. Pastula said.
A version of this article first appeared on Medscape.com.
Several signs are pointing to an impending surge in the number of human cases of West Nile virus in several regions of the United States.
West Nile virus is spread by infected mosquitoes and currently there is no cure or virus-specific treatment. In rare cases, it can be deadly. It can infect humans, birds, horses, and other mammals.
West Nile Virus is the leading cause of mosquito-borne disease in the continental United States. As of Aug. 8, 126 human cases had been identified across 22 states, according to the Centers for Disease Control and Prevention.
“Particularly here in California, it’s peak risk right now,” said Vicki Kramer, PhD, chief of vector-borne diseases in the California Department of Public Health. She said scientists there are seeing higher mosquito and infected mosquito numbers.
“Peak risk right now”
Dead birds are tested for the virus and by Aug. 4, 181 of the 913 birds tested in California have been positive, three times the total testing positive by this time in 2022.
“Last year at this time, we had 60 positive dead birds out of 817 tested,” Dr. Kramer said.
Severe flooding and high heat can contribute to the rise in mosquito populations and many parts of the country have seen plenty of both.
One of the ways scientists track infected mosquito patterns in California is by using flocks of strategically placed sentinel chickens.
“Chickens are a mosquito magnet,” Dr. Kramer said.
Chickens don’t get sick with the virus, but they do build antibodies to it. Surveillance teams check their blood every other week to track the virus.
Daniel Pastula, MD, MHS, chief of neuroinfectious diseases and global neurology at the University of Colorado School of Medicine and the Colorado School of Public Health, said the state is watching troubling signs as well.
“The concern this year,” Dr. Pastula said, “particularly along the Front Range in Colorado, is we’ve found many more mosquitoes [that are] positive for West Nile earlier in the season compared with other years.
“We’re bracing for higher-than-baseline human cases,” he said.
Asked about this year’s first human case, reported in Toronto, a region with a long winter and low incidence of the virus, he said that provides a further example that people need to be prepared even in climates not known to be mosquito-dense.
He added, however, that climate is only one factor in the severity of the season. Others include birds’ immunity and migratory patterns.
Dr. Pastula said that fluctuations in temperature and rainfall are rising with climate change and are disrupting normal baseline levels of West Nile.
“That shows we need to be prepared for West Nile virus and other mosquito-borne diseases in any place in North America or really the world. We recently saw malaria cases in the southern United States. It just shows you how dangerous mosquitoes can be.”
Avoid mosquito bites
Dr. Pastula and Dr. Kramer list the precautions people can take to protect themselves from West Nile virus:
- Limit outdoor exposure particularly at dusk and dawn.
- Wear protective clothing.
- Use .
- Repair window screens so mosquitoes cannot fly through.
- Dump and drain standing water on your property and maintain swimming pools.
Dr. Pastula noted that summer is the time human cases start to mount – typically from July and August to the first hard freeze.
“We have been warning people here up and down the Front Range of Colorado to take prevention very seriously,” Dr. Pastula said.
He pointed out that 80% who are infected with West Nile will have no symptoms.
About 20% will have flu-like illness – high fever, body and joint aches, rash, diarrhea, or headaches. Symptoms may last for weeks. About 1% of the time, he said, people can get neuroinvasive West Nile.
Dr. Pastula explained that the virus can infect the covering of the brain and spinal cord causing meningitis with very high fever, severe headaches, stiff neck, and sensitivity to light.
So far this year, there have been 89 neuroinvasive cases reported nationally, according to the CDC.
With West Nile encephalitis, the virus “can infect the brain itself causing altered mental status, movement disorders, or weakness,” Dr. Pastula said.
Sometimes it can infect the gray matter of the spinal cord causing a West Nile virus poliomyelitis, which brings polio-like symptoms.
“The West Nile encephalitis and poliomyelitis can cause permanent deficits or even death,” he said. “It’s uncommon but it’s not trivial.”
Several vaccine candidates are in development, Dr. Pastula said, but none has reached clinical trials. Part of the reason for that, he said, is that scientists must be able to predict the timing of an outbreak.
“We’re not really great at predicting outbreaks,” he said.
Although the risk for neuroinvasive disease is small, it can be higher in certain groups, he said – those who are over age 60 years or are immunocompromised; those who have diabetes, cancer, or kidney disease; or those who have undergone organ transplants.
Those infected should see a health care professional and may be able to get relief with the usual medications for flu-like illness.
Some with severe infection may need to go to the hospital, Dr. Pastula said.
A version of this article first appeared on Medscape.com.
Study highlights diagnostic challenges of differentiating lichen sclerosus from vitiligo
of cases.
Researchers who tallied symptoms and physical exam findings observed fewer statistically significant differences between LS and vitiligo patients than expected, and LS and vitiligo were sometimes misdiagnosed as each other.
“LS must be treated aggressively to prevent long-term sequelae such as permanent scarring and vulvar squamous cell carcinoma, making an accurate diagnosis crucial,” the authors write in a poster they presented at the annual meeting of the Society for Pediatric Dermatology.

LS is symptomatic and has multiple exam findings, but once treated or quiescent, the discoloration can persist and create diagnostic uncertainty, lead study author Kaiane Habeshian, MD, a pediatric dermatologist at Children’s National Hospital, Washington, told this news organization following the SPD meeting.
The diagnostic uncertainty is especially true in patients with darker skin tones, who may have vitiligoid LS, an LS variant that has overlapping features of both LS and vitiligo.
Vitiligoid LS “presents clinically as a depigmented symmetric white vulvar and perianal white patch, often with minimal signs of inflammation, but is symptomatic and appears consistent with LS on histopathology,” Dr. Habeshian said.
“In our experience, in patients with medium to dark skin tones, there is a variable amount of repigmentation after treating LS,” she added. “After use of high potency topical corticosteroids, some patients almost completely repigment, while others have minimal repigmentation, and this can fluctuate over time, sometimes independent of other signs or symptoms of a flare up. This can lead to diagnostic confusion. For example, if an LS patient is examined after treatment, and their symptoms have resolved, they may subsequently be given a diagnosis of vitiligo.”
In the study, Dr. Habeshian and her coauthors aimed to characterize differences in LS and vitiligo based on history, physical exam, and demographic findings at the time of the initial clinic visit. She and her colleagues extracted and reviewed the medical records of 98 patients with a diagnosis of LS or vitiligo who were seen at a joint pediatric dermatology-gynecology vulvar clinic over 6.8 years. The median and mean age of the study population at diagnosis was about 6 years, with ages ranging from 2 to 20. The team used descriptive statistics and Z tests for data analysis.
The researchers found that pruritus, constipation, and dysuria were the most common symptoms experienced by both LS and vitiligo patients. All were experienced more frequently by LS patients, but only pruritus reached statistical significance (P = .040). Other symptoms experienced only by LS patients included vulvar pain, bleeding, and pain with defecation.
Meanwhile, apart from hypopigmentation and erythema, all physical exam findings were more frequent in LS patients, compared with vitiligo patients, including fissures and purpura/petechiae, but only epidermal atrophy and figure-of-8 distribution of hypopigmentation reached statistical significance (P values of .047 and .036, respectively).
In other findings, LS and vitiligo were misdiagnosed as each other 15 times. Nearly half of the misdiagnoses (46.7%) were made in Black patients, who composed 38.8% of all patients in the study.
“I suspect that some vitiligo cases that were previously ‘misdiagnosed’ as LS were actually LS that just didn’t repigment and then were labeled as vitiligo in the chart,” Dr. Habeshian said.
“And some of those LS cases that previously were misdiagnosed as vitiligo likely had other more subtle LS findings that were missed (shininess and wrinkling of the skin, small fissures, constipation) or that were attributed to comorbid irritant contact dermatitis or another condition,” she said. “It was interesting to see that even in a vulvar dermatology clinic there can be confusion between these diagnoses because the literature on pediatric LS in darker skin tones is so sparse.”
She emphasized that a close exam and detailed history are needed to properly diagnose patients with anogenital skin conditions.
“Don’t forget to ask about constipation and urinary symptoms as well as psychosocial and, in the appropriate patient, sexual and reproductive function,” Dr. Habeshian said. “Based on my experience, pediatric LS is much more common in our community than the literature would suggest. Its psychosocial impact is tremendous but not well documented, particularly in pediatric patients. In my experience, the longer LS is misdiagnosed or mistreated, the more challenging it becomes to manage. You don’t want to miss LS.”
She acknowledged certain limitations of the study, including the fact that photographs were not available for review for many of the earlier years of the clinic. “Therefore, we had to depend on the diagnosis given at the time of the visit,” she said. “This likely accounts in part for the smaller number than expected of significant exam and history findings between LS and vitiligo. We need further studies utilizing a standardized approach to accurate diagnosis.”
Her coauthors were Nikita Menta, Aneka Khilnani, MS, and Tazim Dowlut-McElroy, MD. The researchers reported having no financial disclosures.
of cases.
Researchers who tallied symptoms and physical exam findings observed fewer statistically significant differences between LS and vitiligo patients than expected, and LS and vitiligo were sometimes misdiagnosed as each other.
“LS must be treated aggressively to prevent long-term sequelae such as permanent scarring and vulvar squamous cell carcinoma, making an accurate diagnosis crucial,” the authors write in a poster they presented at the annual meeting of the Society for Pediatric Dermatology.
LS is symptomatic and has multiple exam findings, but once treated or quiescent, the discoloration can persist and create diagnostic uncertainty, lead study author Kaiane Habeshian, MD, a pediatric dermatologist at Children’s National Hospital, Washington, told this news organization following the SPD meeting.
The diagnostic uncertainty is especially true in patients with darker skin tones, who may have vitiligoid LS, an LS variant that has overlapping features of both LS and vitiligo.
Vitiligoid LS “presents clinically as a depigmented symmetric white vulvar and perianal white patch, often with minimal signs of inflammation, but is symptomatic and appears consistent with LS on histopathology,” Dr. Habeshian said.
“In our experience, in patients with medium to dark skin tones, there is a variable amount of repigmentation after treating LS,” she added. “After use of high potency topical corticosteroids, some patients almost completely repigment, while others have minimal repigmentation, and this can fluctuate over time, sometimes independent of other signs or symptoms of a flare up. This can lead to diagnostic confusion. For example, if an LS patient is examined after treatment, and their symptoms have resolved, they may subsequently be given a diagnosis of vitiligo.”
In the study, Dr. Habeshian and her coauthors aimed to characterize differences in LS and vitiligo based on history, physical exam, and demographic findings at the time of the initial clinic visit. She and her colleagues extracted and reviewed the medical records of 98 patients with a diagnosis of LS or vitiligo who were seen at a joint pediatric dermatology-gynecology vulvar clinic over 6.8 years. The median and mean age of the study population at diagnosis was about 6 years, with ages ranging from 2 to 20. The team used descriptive statistics and Z tests for data analysis.
The researchers found that pruritus, constipation, and dysuria were the most common symptoms experienced by both LS and vitiligo patients. All were experienced more frequently by LS patients, but only pruritus reached statistical significance (P = .040). Other symptoms experienced only by LS patients included vulvar pain, bleeding, and pain with defecation.
Meanwhile, apart from hypopigmentation and erythema, all physical exam findings were more frequent in LS patients, compared with vitiligo patients, including fissures and purpura/petechiae, but only epidermal atrophy and figure-of-8 distribution of hypopigmentation reached statistical significance (P values of .047 and .036, respectively).
In other findings, LS and vitiligo were misdiagnosed as each other 15 times. Nearly half of the misdiagnoses (46.7%) were made in Black patients, who composed 38.8% of all patients in the study.
“I suspect that some vitiligo cases that were previously ‘misdiagnosed’ as LS were actually LS that just didn’t repigment and then were labeled as vitiligo in the chart,” Dr. Habeshian said.
“And some of those LS cases that previously were misdiagnosed as vitiligo likely had other more subtle LS findings that were missed (shininess and wrinkling of the skin, small fissures, constipation) or that were attributed to comorbid irritant contact dermatitis or another condition,” she said. “It was interesting to see that even in a vulvar dermatology clinic there can be confusion between these diagnoses because the literature on pediatric LS in darker skin tones is so sparse.”
She emphasized that a close exam and detailed history are needed to properly diagnose patients with anogenital skin conditions.
“Don’t forget to ask about constipation and urinary symptoms as well as psychosocial and, in the appropriate patient, sexual and reproductive function,” Dr. Habeshian said. “Based on my experience, pediatric LS is much more common in our community than the literature would suggest. Its psychosocial impact is tremendous but not well documented, particularly in pediatric patients. In my experience, the longer LS is misdiagnosed or mistreated, the more challenging it becomes to manage. You don’t want to miss LS.”
She acknowledged certain limitations of the study, including the fact that photographs were not available for review for many of the earlier years of the clinic. “Therefore, we had to depend on the diagnosis given at the time of the visit,” she said. “This likely accounts in part for the smaller number than expected of significant exam and history findings between LS and vitiligo. We need further studies utilizing a standardized approach to accurate diagnosis.”
Her coauthors were Nikita Menta, Aneka Khilnani, MS, and Tazim Dowlut-McElroy, MD. The researchers reported having no financial disclosures.
of cases.
Researchers who tallied symptoms and physical exam findings observed fewer statistically significant differences between LS and vitiligo patients than expected, and LS and vitiligo were sometimes misdiagnosed as each other.
“LS must be treated aggressively to prevent long-term sequelae such as permanent scarring and vulvar squamous cell carcinoma, making an accurate diagnosis crucial,” the authors write in a poster they presented at the annual meeting of the Society for Pediatric Dermatology.
LS is symptomatic and has multiple exam findings, but once treated or quiescent, the discoloration can persist and create diagnostic uncertainty, lead study author Kaiane Habeshian, MD, a pediatric dermatologist at Children’s National Hospital, Washington, told this news organization following the SPD meeting.
The diagnostic uncertainty is especially true in patients with darker skin tones, who may have vitiligoid LS, an LS variant that has overlapping features of both LS and vitiligo.
Vitiligoid LS “presents clinically as a depigmented symmetric white vulvar and perianal white patch, often with minimal signs of inflammation, but is symptomatic and appears consistent with LS on histopathology,” Dr. Habeshian said.
“In our experience, in patients with medium to dark skin tones, there is a variable amount of repigmentation after treating LS,” she added. “After use of high potency topical corticosteroids, some patients almost completely repigment, while others have minimal repigmentation, and this can fluctuate over time, sometimes independent of other signs or symptoms of a flare up. This can lead to diagnostic confusion. For example, if an LS patient is examined after treatment, and their symptoms have resolved, they may subsequently be given a diagnosis of vitiligo.”
In the study, Dr. Habeshian and her coauthors aimed to characterize differences in LS and vitiligo based on history, physical exam, and demographic findings at the time of the initial clinic visit. She and her colleagues extracted and reviewed the medical records of 98 patients with a diagnosis of LS or vitiligo who were seen at a joint pediatric dermatology-gynecology vulvar clinic over 6.8 years. The median and mean age of the study population at diagnosis was about 6 years, with ages ranging from 2 to 20. The team used descriptive statistics and Z tests for data analysis.
The researchers found that pruritus, constipation, and dysuria were the most common symptoms experienced by both LS and vitiligo patients. All were experienced more frequently by LS patients, but only pruritus reached statistical significance (P = .040). Other symptoms experienced only by LS patients included vulvar pain, bleeding, and pain with defecation.
Meanwhile, apart from hypopigmentation and erythema, all physical exam findings were more frequent in LS patients, compared with vitiligo patients, including fissures and purpura/petechiae, but only epidermal atrophy and figure-of-8 distribution of hypopigmentation reached statistical significance (P values of .047 and .036, respectively).
In other findings, LS and vitiligo were misdiagnosed as each other 15 times. Nearly half of the misdiagnoses (46.7%) were made in Black patients, who composed 38.8% of all patients in the study.
“I suspect that some vitiligo cases that were previously ‘misdiagnosed’ as LS were actually LS that just didn’t repigment and then were labeled as vitiligo in the chart,” Dr. Habeshian said.
“And some of those LS cases that previously were misdiagnosed as vitiligo likely had other more subtle LS findings that were missed (shininess and wrinkling of the skin, small fissures, constipation) or that were attributed to comorbid irritant contact dermatitis or another condition,” she said. “It was interesting to see that even in a vulvar dermatology clinic there can be confusion between these diagnoses because the literature on pediatric LS in darker skin tones is so sparse.”
She emphasized that a close exam and detailed history are needed to properly diagnose patients with anogenital skin conditions.
“Don’t forget to ask about constipation and urinary symptoms as well as psychosocial and, in the appropriate patient, sexual and reproductive function,” Dr. Habeshian said. “Based on my experience, pediatric LS is much more common in our community than the literature would suggest. Its psychosocial impact is tremendous but not well documented, particularly in pediatric patients. In my experience, the longer LS is misdiagnosed or mistreated, the more challenging it becomes to manage. You don’t want to miss LS.”
She acknowledged certain limitations of the study, including the fact that photographs were not available for review for many of the earlier years of the clinic. “Therefore, we had to depend on the diagnosis given at the time of the visit,” she said. “This likely accounts in part for the smaller number than expected of significant exam and history findings between LS and vitiligo. We need further studies utilizing a standardized approach to accurate diagnosis.”
Her coauthors were Nikita Menta, Aneka Khilnani, MS, and Tazim Dowlut-McElroy, MD. The researchers reported having no financial disclosures.
FROM SPD 2023