User login
For MD-IQ use only
Diffuse Pruritic Keratotic Papules
Diffuse Pruritic Keratotic Papules
THE DIAGNOSIS: Reactive Perforating Collagenosis
Histopathology revealed invagination of the epidermis with hyperkeratosis; prominent epidermal hyperplasia; and a central basophilic plug of keratin, collagen, and inflammatory debris. Transepidermal elimination of bright eosinophilic altered collagen fibers was seen (Figure). The findings were consistent with a diagnosis of reactive perforating collagenosis (RPC).
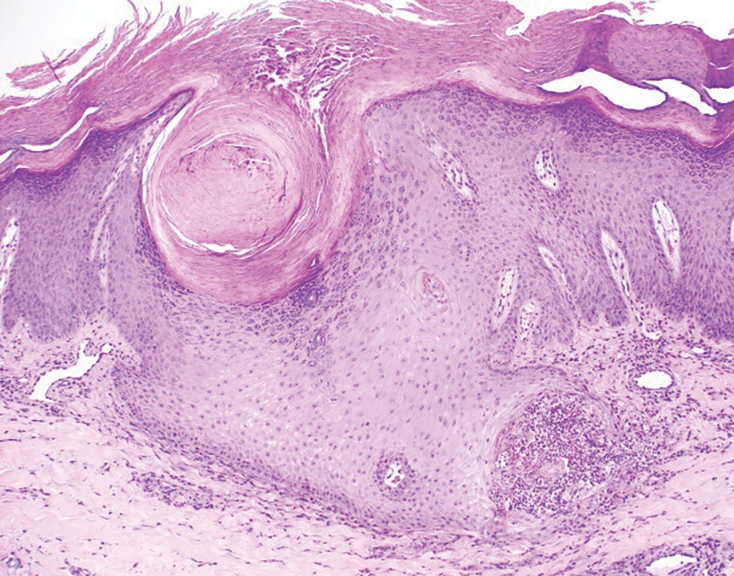
Reactive perforating collagenosis, a subtype of perforating dermatosis, is a rare skin condition in which altered collagen is eliminated through the epidermis.1 There are 2 forms of RPC: the inherited form, which is very rare and manifests in childhood, and the acquired form, which manifests in adulthood and is associated with systemic diseases, most notably diabetes and/or chronic renal failure, both of which our patient had been diagnosed with.1,2 The clinical presentation of RPC includes erythematous papules or nodules that evolve into umbilicated 4- to 10-mm craterlike ulcerations with a central keratotic plug. The lesions favor a linear distribution along the extensor surfaces of the arms and legs, trunk, and gluteal area. Involvement of the head, neck, and scalp has been reported less commonly, which makes our case particularly unique.3 Histopathologically, RPC is characterized by a cup-shaped depression of the epidermis with an overlying keratin plug containing inflammatory cells, keratinous debris, and collagen fibers. Vertically oriented collagen fibers are seen extruded through the epidermis.4,5
While the pathogenesis of RPC remains unknown, it is believed that superficial trauma due to chronic scratching results in transepithelial elimination of collagen. Due to the association of acquired RPC (ARPC) with diabetes, it also has been proposed that scratching can cause microtrauma and necrosis of the dermal structures, potentially due to diabetic microangiopathy.3 Additionally, RPC is associated with overexpression of transforming growth factor beta 3 in lesional skin, suggesting that transforming growth factor beta 3 is involved with tissue repair and extracellular remodeling in this condition.6
Treatment of ARPC should include the management of underlying disease. While no definitive treatment has been reported to date, topical corticosteroids, retinoids, keratolytics, emollients, antihistamines, narrow-band UVB phototherapy, and psoralen plus UVA phototherapy have been used with varying degrees of improvement. Typically, the lesions self-resolve within 6 to 8 weeks; however, they often recur and usually leave scarring with or without hyperpigmentation.2,7-10
Acquired RPC can be misdiagnosed initially, as it mimics several other conditions and commonly is associated with systemic diseases. While biopsy is necessary for diagnosis, if it cannot be performed or the results are indeterminate, dermoscopy can serve as a helpful diagnostic tool. The most common dermoscopic patterns seen in RPC include a yellow-brown structureless area in the center of the lesion with a peripheral surface crust and surrounding white rim—thought to represent epidermal invagination or keratinous debris. Additionally, inflammation with visible vessels both centrally and peripherally is represented by an outer pink circle on dermoscopy.5,11
The differential diagnoses for RPC include perforating folliculitis (PF), elastosis perforans serpiginosa (EPS), prurigo nodularis, and keratoacanthomas. The primary perforating dermatoses (PF, EPS, and RPC) are similarly characterized by elimination of altered dermal material through the epidermis. As these conditions manifest with similar features on clinical examination, differentiation is made by the type of epidermal damage and the features of elimination material, making histopathologic examination paramount for definitive diagnosis.
Perforating folliculitis manifests as erythematous, follicular papules with a small central keratotic core or a central hair. Histopathologically, PF reveals a widely dilated follicle containing keratin, necrotic debris, and degenerated inflammatory cells. Elastosis perforans serpiginosa manifests clinically as hyperkeratotic papules in serpiginous patterns rather than the linear pattern commonly seen with ARPC. Histopathologically, EPS reveals thickened elastic fibers, rather than collagen fibers as seen in ARPC, extruded through the epidermis. Prurigo nodularis manifests clinically as dome-shaped papules with possible excoriation and crusting. Histopathologic examination reveals epidermal hyperplasia and hyperkeratosis; however, the characteristic features of transepithelial elimination of collagen and invaginations of epidermis differentiate ARPC from prurigo nodularis.12,13 Keratoacanthomas manifest clinically as an eruption of small, round, pink papules that rapidly grow and evolve into 1- to 2-cm dome-shaped nodules with central keratinaceous plugs, mimicking a crateriform appearance. Histopathologic examination reveals a circumscribed proliferation of well-differentiated keratinocytes. Multilobular exophytic or endophytic cystlike invaginations of the epidermis also are noted. The expulsion of collagen from the epidermis is more consistent with ARPC.14
- Cohen RW, Auerbach R. Acquired reactive perforating collagenosis. J Am Acad Dermatol. 1989;20(2 pt 1):287-289. doi:10.1016/s0190 -9622(89)80059-3
- Bejjanki H, Siroy AE, Koratala A. Reactive perforating collagenosis in end-stage renal disease: not all that itches is uremic pruritis! Am J Med. 2019;132:E658-E660. doi:10.1016/j.amjmed.2019.03.015
- Gontijo JRV, Júnior FF, Pereira LB, et al. Trauma-induced acquired reactive perforating collagenosis. An Bras Dermatol. 2021;96:392-393. doi:10.1016/j.abd.2020.06.022
- Ambalathinkal JJ, Phiske MM, Someshwar SJ. Acquired reactive perforating collagenosis, a rare entity at uncommon site. Indian J Pathol Microbiol. 2022;65:895-897. doi:10.4103/ijpm.ijpm_333_21
- Ormerod E, Atwan A, Intzedy L, et al. Dermoscopy features of acquired reactive perforating collagenosis: a case series. Dermatol Pract Concept. 2018;8:303-305. doi:10.5826/dpc.0804a11
- Fei C, Wang Y, Gong Y, et al. Acquired reactive perforating collagenosis: a report of a typical case. Medicine (Baltimore). 2016;95:E4305. doi:10.1097/md.0000000000004305
- Bartling SJ, Naff JL, Canevari MM, et al. Pruritic rash in an elderly patient with uncontrolled diabetes mellitus. AACE Clin Case Rep. 2018;5:E146-E149. doi:10.4158/ACCR-2018-0388
- Kollipara H, Satya RS, Rao GR, et al. Acquired reactive perforating collagenosis: case series. Indian Dermatol Online J. 2023;14:72-76. doi:10.4103/idoj.idoj_373_22
- Wang C, Liu YH, Wang YX, et al. Acquired reactive perforating collagenosis. Chin Med J (Engl). 2020;133:2119-2120. doi:10.1097 /cm9.0000000000000906
- Harbaoui S, Litaiem N. Acquired perforating dermatosis. StatPearls [Internet]. Updated February 13, 2023. Accessed August 13, 2025. https://www.ncbi.nlm.nih.gov/books/NBK539715/
- Elmas ÖF, Kilitci A, Uyar B. Dermoscopic patterns of acquired reactive perforating collagenosis. Dermatol Pract Concept. 2021;11:E2020085. doi:10.5826/dpc.1101a85
- Patterson JW. The perforating disorders. J Am Acad Dermatol. 1984;10:561-581. doi:10.1016/s0190-9622(84)80259-5
- Huang AH, Williams KA, Kwatra SG. Prurigo nodularis: epidemiology and clinical features. J Am Acad Dermatol. 2020;83:1559-1565. doi:10.1016/j.jaad.2020.04.183
- Zito PM, Scharf R. Keratoacanthoma. StatPearls [Internet]. Updated August 8, 2023. Accessed August 13, 2025. https://www.ncbi.nlm.nih.gov/books/NBK499931/
THE DIAGNOSIS: Reactive Perforating Collagenosis
Histopathology revealed invagination of the epidermis with hyperkeratosis; prominent epidermal hyperplasia; and a central basophilic plug of keratin, collagen, and inflammatory debris. Transepidermal elimination of bright eosinophilic altered collagen fibers was seen (Figure). The findings were consistent with a diagnosis of reactive perforating collagenosis (RPC).
Reactive perforating collagenosis, a subtype of perforating dermatosis, is a rare skin condition in which altered collagen is eliminated through the epidermis.1 There are 2 forms of RPC: the inherited form, which is very rare and manifests in childhood, and the acquired form, which manifests in adulthood and is associated with systemic diseases, most notably diabetes and/or chronic renal failure, both of which our patient had been diagnosed with.1,2 The clinical presentation of RPC includes erythematous papules or nodules that evolve into umbilicated 4- to 10-mm craterlike ulcerations with a central keratotic plug. The lesions favor a linear distribution along the extensor surfaces of the arms and legs, trunk, and gluteal area. Involvement of the head, neck, and scalp has been reported less commonly, which makes our case particularly unique.3 Histopathologically, RPC is characterized by a cup-shaped depression of the epidermis with an overlying keratin plug containing inflammatory cells, keratinous debris, and collagen fibers. Vertically oriented collagen fibers are seen extruded through the epidermis.4,5
While the pathogenesis of RPC remains unknown, it is believed that superficial trauma due to chronic scratching results in transepithelial elimination of collagen. Due to the association of acquired RPC (ARPC) with diabetes, it also has been proposed that scratching can cause microtrauma and necrosis of the dermal structures, potentially due to diabetic microangiopathy.3 Additionally, RPC is associated with overexpression of transforming growth factor beta 3 in lesional skin, suggesting that transforming growth factor beta 3 is involved with tissue repair and extracellular remodeling in this condition.6
Treatment of ARPC should include the management of underlying disease. While no definitive treatment has been reported to date, topical corticosteroids, retinoids, keratolytics, emollients, antihistamines, narrow-band UVB phototherapy, and psoralen plus UVA phototherapy have been used with varying degrees of improvement. Typically, the lesions self-resolve within 6 to 8 weeks; however, they often recur and usually leave scarring with or without hyperpigmentation.2,7-10
Acquired RPC can be misdiagnosed initially, as it mimics several other conditions and commonly is associated with systemic diseases. While biopsy is necessary for diagnosis, if it cannot be performed or the results are indeterminate, dermoscopy can serve as a helpful diagnostic tool. The most common dermoscopic patterns seen in RPC include a yellow-brown structureless area in the center of the lesion with a peripheral surface crust and surrounding white rim—thought to represent epidermal invagination or keratinous debris. Additionally, inflammation with visible vessels both centrally and peripherally is represented by an outer pink circle on dermoscopy.5,11
The differential diagnoses for RPC include perforating folliculitis (PF), elastosis perforans serpiginosa (EPS), prurigo nodularis, and keratoacanthomas. The primary perforating dermatoses (PF, EPS, and RPC) are similarly characterized by elimination of altered dermal material through the epidermis. As these conditions manifest with similar features on clinical examination, differentiation is made by the type of epidermal damage and the features of elimination material, making histopathologic examination paramount for definitive diagnosis.
Perforating folliculitis manifests as erythematous, follicular papules with a small central keratotic core or a central hair. Histopathologically, PF reveals a widely dilated follicle containing keratin, necrotic debris, and degenerated inflammatory cells. Elastosis perforans serpiginosa manifests clinically as hyperkeratotic papules in serpiginous patterns rather than the linear pattern commonly seen with ARPC. Histopathologically, EPS reveals thickened elastic fibers, rather than collagen fibers as seen in ARPC, extruded through the epidermis. Prurigo nodularis manifests clinically as dome-shaped papules with possible excoriation and crusting. Histopathologic examination reveals epidermal hyperplasia and hyperkeratosis; however, the characteristic features of transepithelial elimination of collagen and invaginations of epidermis differentiate ARPC from prurigo nodularis.12,13 Keratoacanthomas manifest clinically as an eruption of small, round, pink papules that rapidly grow and evolve into 1- to 2-cm dome-shaped nodules with central keratinaceous plugs, mimicking a crateriform appearance. Histopathologic examination reveals a circumscribed proliferation of well-differentiated keratinocytes. Multilobular exophytic or endophytic cystlike invaginations of the epidermis also are noted. The expulsion of collagen from the epidermis is more consistent with ARPC.14
THE DIAGNOSIS: Reactive Perforating Collagenosis
Histopathology revealed invagination of the epidermis with hyperkeratosis; prominent epidermal hyperplasia; and a central basophilic plug of keratin, collagen, and inflammatory debris. Transepidermal elimination of bright eosinophilic altered collagen fibers was seen (Figure). The findings were consistent with a diagnosis of reactive perforating collagenosis (RPC).
Reactive perforating collagenosis, a subtype of perforating dermatosis, is a rare skin condition in which altered collagen is eliminated through the epidermis.1 There are 2 forms of RPC: the inherited form, which is very rare and manifests in childhood, and the acquired form, which manifests in adulthood and is associated with systemic diseases, most notably diabetes and/or chronic renal failure, both of which our patient had been diagnosed with.1,2 The clinical presentation of RPC includes erythematous papules or nodules that evolve into umbilicated 4- to 10-mm craterlike ulcerations with a central keratotic plug. The lesions favor a linear distribution along the extensor surfaces of the arms and legs, trunk, and gluteal area. Involvement of the head, neck, and scalp has been reported less commonly, which makes our case particularly unique.3 Histopathologically, RPC is characterized by a cup-shaped depression of the epidermis with an overlying keratin plug containing inflammatory cells, keratinous debris, and collagen fibers. Vertically oriented collagen fibers are seen extruded through the epidermis.4,5
While the pathogenesis of RPC remains unknown, it is believed that superficial trauma due to chronic scratching results in transepithelial elimination of collagen. Due to the association of acquired RPC (ARPC) with diabetes, it also has been proposed that scratching can cause microtrauma and necrosis of the dermal structures, potentially due to diabetic microangiopathy.3 Additionally, RPC is associated with overexpression of transforming growth factor beta 3 in lesional skin, suggesting that transforming growth factor beta 3 is involved with tissue repair and extracellular remodeling in this condition.6
Treatment of ARPC should include the management of underlying disease. While no definitive treatment has been reported to date, topical corticosteroids, retinoids, keratolytics, emollients, antihistamines, narrow-band UVB phototherapy, and psoralen plus UVA phototherapy have been used with varying degrees of improvement. Typically, the lesions self-resolve within 6 to 8 weeks; however, they often recur and usually leave scarring with or without hyperpigmentation.2,7-10
Acquired RPC can be misdiagnosed initially, as it mimics several other conditions and commonly is associated with systemic diseases. While biopsy is necessary for diagnosis, if it cannot be performed or the results are indeterminate, dermoscopy can serve as a helpful diagnostic tool. The most common dermoscopic patterns seen in RPC include a yellow-brown structureless area in the center of the lesion with a peripheral surface crust and surrounding white rim—thought to represent epidermal invagination or keratinous debris. Additionally, inflammation with visible vessels both centrally and peripherally is represented by an outer pink circle on dermoscopy.5,11
The differential diagnoses for RPC include perforating folliculitis (PF), elastosis perforans serpiginosa (EPS), prurigo nodularis, and keratoacanthomas. The primary perforating dermatoses (PF, EPS, and RPC) are similarly characterized by elimination of altered dermal material through the epidermis. As these conditions manifest with similar features on clinical examination, differentiation is made by the type of epidermal damage and the features of elimination material, making histopathologic examination paramount for definitive diagnosis.
Perforating folliculitis manifests as erythematous, follicular papules with a small central keratotic core or a central hair. Histopathologically, PF reveals a widely dilated follicle containing keratin, necrotic debris, and degenerated inflammatory cells. Elastosis perforans serpiginosa manifests clinically as hyperkeratotic papules in serpiginous patterns rather than the linear pattern commonly seen with ARPC. Histopathologically, EPS reveals thickened elastic fibers, rather than collagen fibers as seen in ARPC, extruded through the epidermis. Prurigo nodularis manifests clinically as dome-shaped papules with possible excoriation and crusting. Histopathologic examination reveals epidermal hyperplasia and hyperkeratosis; however, the characteristic features of transepithelial elimination of collagen and invaginations of epidermis differentiate ARPC from prurigo nodularis.12,13 Keratoacanthomas manifest clinically as an eruption of small, round, pink papules that rapidly grow and evolve into 1- to 2-cm dome-shaped nodules with central keratinaceous plugs, mimicking a crateriform appearance. Histopathologic examination reveals a circumscribed proliferation of well-differentiated keratinocytes. Multilobular exophytic or endophytic cystlike invaginations of the epidermis also are noted. The expulsion of collagen from the epidermis is more consistent with ARPC.14
- Cohen RW, Auerbach R. Acquired reactive perforating collagenosis. J Am Acad Dermatol. 1989;20(2 pt 1):287-289. doi:10.1016/s0190 -9622(89)80059-3
- Bejjanki H, Siroy AE, Koratala A. Reactive perforating collagenosis in end-stage renal disease: not all that itches is uremic pruritis! Am J Med. 2019;132:E658-E660. doi:10.1016/j.amjmed.2019.03.015
- Gontijo JRV, Júnior FF, Pereira LB, et al. Trauma-induced acquired reactive perforating collagenosis. An Bras Dermatol. 2021;96:392-393. doi:10.1016/j.abd.2020.06.022
- Ambalathinkal JJ, Phiske MM, Someshwar SJ. Acquired reactive perforating collagenosis, a rare entity at uncommon site. Indian J Pathol Microbiol. 2022;65:895-897. doi:10.4103/ijpm.ijpm_333_21
- Ormerod E, Atwan A, Intzedy L, et al. Dermoscopy features of acquired reactive perforating collagenosis: a case series. Dermatol Pract Concept. 2018;8:303-305. doi:10.5826/dpc.0804a11
- Fei C, Wang Y, Gong Y, et al. Acquired reactive perforating collagenosis: a report of a typical case. Medicine (Baltimore). 2016;95:E4305. doi:10.1097/md.0000000000004305
- Bartling SJ, Naff JL, Canevari MM, et al. Pruritic rash in an elderly patient with uncontrolled diabetes mellitus. AACE Clin Case Rep. 2018;5:E146-E149. doi:10.4158/ACCR-2018-0388
- Kollipara H, Satya RS, Rao GR, et al. Acquired reactive perforating collagenosis: case series. Indian Dermatol Online J. 2023;14:72-76. doi:10.4103/idoj.idoj_373_22
- Wang C, Liu YH, Wang YX, et al. Acquired reactive perforating collagenosis. Chin Med J (Engl). 2020;133:2119-2120. doi:10.1097 /cm9.0000000000000906
- Harbaoui S, Litaiem N. Acquired perforating dermatosis. StatPearls [Internet]. Updated February 13, 2023. Accessed August 13, 2025. https://www.ncbi.nlm.nih.gov/books/NBK539715/
- Elmas ÖF, Kilitci A, Uyar B. Dermoscopic patterns of acquired reactive perforating collagenosis. Dermatol Pract Concept. 2021;11:E2020085. doi:10.5826/dpc.1101a85
- Patterson JW. The perforating disorders. J Am Acad Dermatol. 1984;10:561-581. doi:10.1016/s0190-9622(84)80259-5
- Huang AH, Williams KA, Kwatra SG. Prurigo nodularis: epidemiology and clinical features. J Am Acad Dermatol. 2020;83:1559-1565. doi:10.1016/j.jaad.2020.04.183
- Zito PM, Scharf R. Keratoacanthoma. StatPearls [Internet]. Updated August 8, 2023. Accessed August 13, 2025. https://www.ncbi.nlm.nih.gov/books/NBK499931/
- Cohen RW, Auerbach R. Acquired reactive perforating collagenosis. J Am Acad Dermatol. 1989;20(2 pt 1):287-289. doi:10.1016/s0190 -9622(89)80059-3
- Bejjanki H, Siroy AE, Koratala A. Reactive perforating collagenosis in end-stage renal disease: not all that itches is uremic pruritis! Am J Med. 2019;132:E658-E660. doi:10.1016/j.amjmed.2019.03.015
- Gontijo JRV, Júnior FF, Pereira LB, et al. Trauma-induced acquired reactive perforating collagenosis. An Bras Dermatol. 2021;96:392-393. doi:10.1016/j.abd.2020.06.022
- Ambalathinkal JJ, Phiske MM, Someshwar SJ. Acquired reactive perforating collagenosis, a rare entity at uncommon site. Indian J Pathol Microbiol. 2022;65:895-897. doi:10.4103/ijpm.ijpm_333_21
- Ormerod E, Atwan A, Intzedy L, et al. Dermoscopy features of acquired reactive perforating collagenosis: a case series. Dermatol Pract Concept. 2018;8:303-305. doi:10.5826/dpc.0804a11
- Fei C, Wang Y, Gong Y, et al. Acquired reactive perforating collagenosis: a report of a typical case. Medicine (Baltimore). 2016;95:E4305. doi:10.1097/md.0000000000004305
- Bartling SJ, Naff JL, Canevari MM, et al. Pruritic rash in an elderly patient with uncontrolled diabetes mellitus. AACE Clin Case Rep. 2018;5:E146-E149. doi:10.4158/ACCR-2018-0388
- Kollipara H, Satya RS, Rao GR, et al. Acquired reactive perforating collagenosis: case series. Indian Dermatol Online J. 2023;14:72-76. doi:10.4103/idoj.idoj_373_22
- Wang C, Liu YH, Wang YX, et al. Acquired reactive perforating collagenosis. Chin Med J (Engl). 2020;133:2119-2120. doi:10.1097 /cm9.0000000000000906
- Harbaoui S, Litaiem N. Acquired perforating dermatosis. StatPearls [Internet]. Updated February 13, 2023. Accessed August 13, 2025. https://www.ncbi.nlm.nih.gov/books/NBK539715/
- Elmas ÖF, Kilitci A, Uyar B. Dermoscopic patterns of acquired reactive perforating collagenosis. Dermatol Pract Concept. 2021;11:E2020085. doi:10.5826/dpc.1101a85
- Patterson JW. The perforating disorders. J Am Acad Dermatol. 1984;10:561-581. doi:10.1016/s0190-9622(84)80259-5
- Huang AH, Williams KA, Kwatra SG. Prurigo nodularis: epidemiology and clinical features. J Am Acad Dermatol. 2020;83:1559-1565. doi:10.1016/j.jaad.2020.04.183
- Zito PM, Scharf R. Keratoacanthoma. StatPearls [Internet]. Updated August 8, 2023. Accessed August 13, 2025. https://www.ncbi.nlm.nih.gov/books/NBK499931/
Diffuse Pruritic Keratotic Papules
Diffuse Pruritic Keratotic Papules
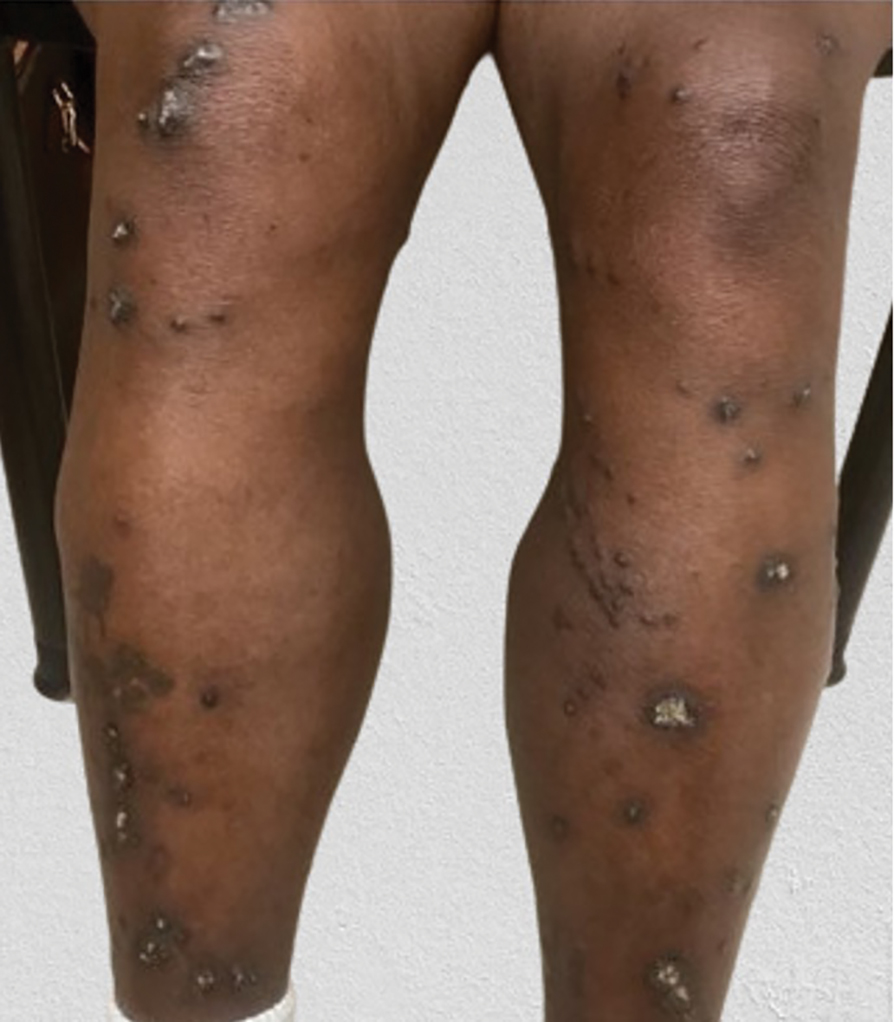
A 65-year-old woman presented to dermatology with an intensely pruritic rash on the arms, legs, neck, and face of several months’ duration. The patient reported scratching the lesions but denied any recent trauma to the affected areas. She previously had been evaluated by her primary care provider, who prescribed cephalexin with no improvement. Her medical history was remarkable for chronic renal failure on dialysis, diabetes, hypertension, and congestive heart failure. Physical examination of the skin revealed hard white cutaneous nodules distributed on the proximal posterior upper arms, bilateral proximal pretibial regions, right elbow, and left knee. Two shave biopsies from the right elbow and left knee were obtained for histopathology.

Bridging the Funding Gap
Federal grants have supported cutting-edge research in scientific and biomedical fields since the mid-20th century, fueling public health breakthroughs and health innovations. This crucial support has been greatly diminished in recent months with deep cuts to federal research dollars.

As these acute policy changes continue to disrupt academic institutions and their investigators, introducing financial strain and operational uncertainty, the importance of research support from professional societies and foundations has become increasingly evident. Their targeted funding plays a critical role in sustaining biomedical research, which directly impacts clinical innovation and patient care. As one example, the AGA Research Foundation provides over $2 million annually to spur discoveries in gastroenterology and hepatology. This vital research support, awarded to 74 unique recipients (including 7 early-career Research Scholar Award recipients) in 2025, represents one of the most important investments that AGA makes in the future of gastroenterology and the patients we treat.
While foundation awards such as these cannot completely close the federal funding gap, they serve as an important lifeline both in supporting the core work of early career and established investigators in an uncertain funding environment and in funding high-risk, high-reward research that more conservative funders are often hesitant to invest in. – now more than ever, the funding it provides has tremendous impact.
In this issue of GI & Hepatology News, we update you on the FDA’s recent approval of semaglutide as a treatment for MASH with fibrosis and highlight a recent target trial emulation study that casts doubt on our traditional understanding regarding the link between common medications such as PPIs and NSAIDs and microscopic colitis in older adults. We also summarize newly-released, global guidelines for pregnancy and IBD, which deserve a careful read. In this month’s Member Spotlight, we feature Pascale White, MD, MBA, MS (Mount Sinai), a recent recipient of the AGA-Pfizer Beacon of Hope Award for Gender and Health Equity, who shares her inspirational work to improve colorectal cancer screening among underserved, high-risk patients in East Harlem. We hope you enjoy this and all the exciting content in our October issue.
Megan A. Adams, MD, JD, MSc
Editor in Chief
Federal grants have supported cutting-edge research in scientific and biomedical fields since the mid-20th century, fueling public health breakthroughs and health innovations. This crucial support has been greatly diminished in recent months with deep cuts to federal research dollars.
As these acute policy changes continue to disrupt academic institutions and their investigators, introducing financial strain and operational uncertainty, the importance of research support from professional societies and foundations has become increasingly evident. Their targeted funding plays a critical role in sustaining biomedical research, which directly impacts clinical innovation and patient care. As one example, the AGA Research Foundation provides over $2 million annually to spur discoveries in gastroenterology and hepatology. This vital research support, awarded to 74 unique recipients (including 7 early-career Research Scholar Award recipients) in 2025, represents one of the most important investments that AGA makes in the future of gastroenterology and the patients we treat.
While foundation awards such as these cannot completely close the federal funding gap, they serve as an important lifeline both in supporting the core work of early career and established investigators in an uncertain funding environment and in funding high-risk, high-reward research that more conservative funders are often hesitant to invest in. – now more than ever, the funding it provides has tremendous impact.
In this issue of GI & Hepatology News, we update you on the FDA’s recent approval of semaglutide as a treatment for MASH with fibrosis and highlight a recent target trial emulation study that casts doubt on our traditional understanding regarding the link between common medications such as PPIs and NSAIDs and microscopic colitis in older adults. We also summarize newly-released, global guidelines for pregnancy and IBD, which deserve a careful read. In this month’s Member Spotlight, we feature Pascale White, MD, MBA, MS (Mount Sinai), a recent recipient of the AGA-Pfizer Beacon of Hope Award for Gender and Health Equity, who shares her inspirational work to improve colorectal cancer screening among underserved, high-risk patients in East Harlem. We hope you enjoy this and all the exciting content in our October issue.
Megan A. Adams, MD, JD, MSc
Editor in Chief
Federal grants have supported cutting-edge research in scientific and biomedical fields since the mid-20th century, fueling public health breakthroughs and health innovations. This crucial support has been greatly diminished in recent months with deep cuts to federal research dollars.
As these acute policy changes continue to disrupt academic institutions and their investigators, introducing financial strain and operational uncertainty, the importance of research support from professional societies and foundations has become increasingly evident. Their targeted funding plays a critical role in sustaining biomedical research, which directly impacts clinical innovation and patient care. As one example, the AGA Research Foundation provides over $2 million annually to spur discoveries in gastroenterology and hepatology. This vital research support, awarded to 74 unique recipients (including 7 early-career Research Scholar Award recipients) in 2025, represents one of the most important investments that AGA makes in the future of gastroenterology and the patients we treat.
While foundation awards such as these cannot completely close the federal funding gap, they serve as an important lifeline both in supporting the core work of early career and established investigators in an uncertain funding environment and in funding high-risk, high-reward research that more conservative funders are often hesitant to invest in. – now more than ever, the funding it provides has tremendous impact.
In this issue of GI & Hepatology News, we update you on the FDA’s recent approval of semaglutide as a treatment for MASH with fibrosis and highlight a recent target trial emulation study that casts doubt on our traditional understanding regarding the link between common medications such as PPIs and NSAIDs and microscopic colitis in older adults. We also summarize newly-released, global guidelines for pregnancy and IBD, which deserve a careful read. In this month’s Member Spotlight, we feature Pascale White, MD, MBA, MS (Mount Sinai), a recent recipient of the AGA-Pfizer Beacon of Hope Award for Gender and Health Equity, who shares her inspirational work to improve colorectal cancer screening among underserved, high-risk patients in East Harlem. We hope you enjoy this and all the exciting content in our October issue.
Megan A. Adams, MD, JD, MSc
Editor in Chief
Hyperpigmented Macules Caused by Burrowing Bugs (Cydnidae) May Mimic More Serious Conditions
Hyperpigmented Macules Caused by Burrowing Bugs (Cydnidae) May Mimic More Serious Conditions
Cydnidae is a family of small to medium-sized shield bugs with spiny legs that commonly are known as burrowing bugs (or burrower bugs). The family Cydnidae includes more than 100 genera and approximately 600 species worldwide.1 These insects are arthropods of the order Hemiptera (suborder: Heteroptera; superfamily: Pentatomoidae) and largely are concentrated in tropical and temperate regions. Approximately 145 species have been recorded in the Neotropical Region and have been included in the subfamilies Amnestinae, Cephalocteinae, and Sehirinae, in addition to Cydnidae.2 Burrowing bugs are ovoid in shape and 2 to 20 mm in length and morphologically are well adapted for burrowing. Their life span is 100 to 300 days. Being phytophagous, they burrow to feed on plants and roots. Adult burrowing bugs have wings and can fly. They have specialized glands located in either the abdomen (nymph) or thorax (adult) that secrete odorous chemicals for self-protection.3 The secretions contain hydrocarbonates that function as repellents and danger signals, can cause paralysis in prey, and act as a chemoattractant for mates.4-6 They also cause hyperpigmentation upon contact with the skin.
In this article, we present a series of cases from the same community to demonstrate the characteristic features of hyperpigmented macules caused by exposure to burrowing bugs. Dermatologists should be aware of this entity to prevent misdiagnosis and unnecessary investigations and treatment.
Case Series
A 36-year-old woman and 6 children (age range, 6-12 years) presented with a widespread, acute, brown-pigmented, macular eruption with lesions that increased in number over a 1-week period. All 7 patients resided in the same locality and were otherwise systemically healthy. Initially, the index case, a 7-year-old girl, was referred to our tertiary care center by a dermatologist with a provisional diagnosis of idiopathic macular eruptive pigmentation. The patient’s mother recalled noticing a tiny black insect on the child's scalp that left pigment on the skin when she crushed it between her fingers. The rest of the patients presented over the next few days: 3 of the children belonged to the same household as the index case, and there was history of all 6 children playing in the neighborhood park during late evening hours. The adult patient was the parent of one of the affected children. The lesions were associated with mild itching and tingling in 3 children but were asymptomatic in the other patients.
Clinical examination of the patients revealed multiple dark- to light-brown, discrete, irregularly shaped macules over the trunk, arms, and soles (eFigure 1). Dermoscopic examination of a pigmented macule showed an irregularly shaped, brownish, structureless area with accentuation of the pigment at skin creases and perieccrine pigmentation (eFigure 2). The pigmentation was unaffected by rubbing with alcohol or water. Clinicoepidemiologic parameters of the patients are summarized in the eTable.
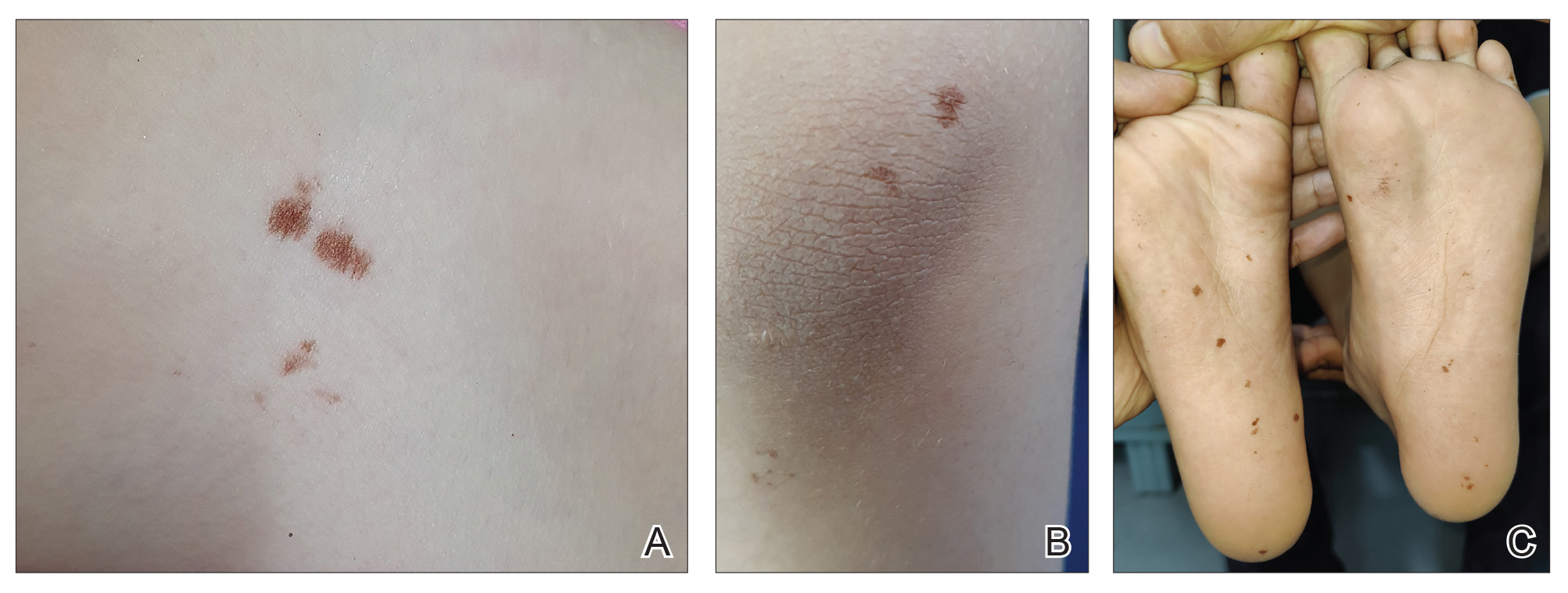
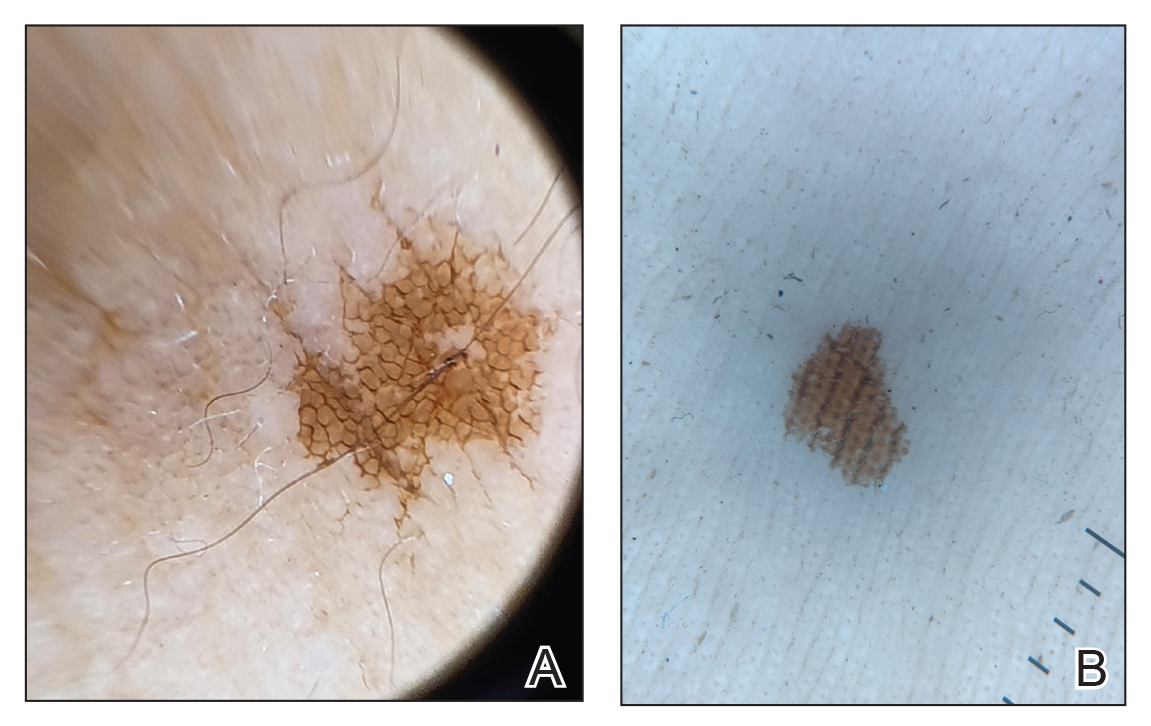
One of the children’s parents conducted a geological examination of the ground in the neighborhood park during evening hours and found tiny burrowing bugs (eFigure 3). When crushed between the fingers, these insects left a similar brownish hyperpigmentation on the skin. The parents were counseled on the nature of the eruption, and the patients were kept under observation for 2 weeks. On follow-up after 5 days, the lesions showed markedly decreased intensity of hyperpigmentation, and no new lesions were observed in any of the 7 patients.

Comment
Pentatomoidae insects generally are benign and harmless to humans. There have been isolated reports of erythematous plaques caused by Antiteuchus mixtus and Edessa maculate.7 Malhotra et al8 reported the first known series of cases with Cydnidae insect–induced hyperpigmented macules. The reported patients presented with asymptomatic, brown, hyperpigmented macules over exposed sites such as the feet, neck, and chest. All the cases occurred during the monsoon season in tropical and temperate regions of the world, and the patients were characteristically clustered in similar geographic areas. The causative insect was identified as Chilocoris assmuthi Breddin, 1904, belonging to the family Cydnidae. When it was crushed between the fingers, the skin became hyperpigmented, confirming the role of the secretions from the insect in the etiology.8
A second case was described by Sonthalia,9 who also described the dermoscopic features of hyperpigmented macules caused by burrowing bugs. The lesions showed a stuck-on, clustered appearance of ovoid and bizarre pigmented clods, globules, and granules.9 Although the lesions occur mainly over exposed sites, pigmented macules occurring over unusual sites such as the abdomen and back also have been reported in association with burrowing bugs.10 Characteristically, the lesions initially are faint and darken with time and usually fade within a week. They can be rubbed off with acetone but persist when washed with soap and water. The fleeting nature of the pigmentation also has led to the term transient pseudo-lentigines sign to describe hyperpigmentation caused by burrowing bugs.11
Soil and plants are burrowing bugs’ natural habitats, and the insects typically are seen in vegetation-rich, moist areas adjoining human dwellings (eg, parks, gardens), where clusters of cases can occur. These insects proliferate during the monsoon season in tropical and temperate areas, leading to more cases occurring during these months.
Compared to prior reports,8,9 a few of our patients had predominant trunk and neck involvement with an occasional tingling sensation or pruritus while the rest were asymptomatic. Dermoscopic features from our patients shared similar reported features of Cydnidae pigmentation.4,5 The accentuation of pigment over skin creases seen on dermoscopy was due to accumulation of Cydnidae secretion at these sites.
The differential diagnosis commonly includes idiopathic macular eruptive pigmentation, which is characterized by an asymptomatic progressive eruption of hyperpigmented macules over the trunk that persists from a few months up to 3 years. Other conditions in the differential include benign conditions such as acral benign melanocytic nevi, lentigines, pigmented purpuric dermatosis, and postinflammatory hyperpigmentation, as well as malignant conditions such as acral melanoma. Dermoscopy is a helpful, easy-to-use tool in differentiating these pigmentation disorders, obviating the need for an invasive investigation such as histopathologic analysis. Simultaneous involvement in a group of people living together or visiting the same place, abrupt onset, predominant involvement of the exposed sites, characteristic clinical and dermoscopic features, self-limiting course, and timing with the monsoon season should suggest a possibility of Cydnidae dermatitis/pigmentation, which can be confirmed by finding the causative bug in the affected locality.
Management
No specific treatment is required for the pigmentation caused by Cydnidae, as it is self-resolving. The macules can, however, be removed with acetone. Patients must be counseled regarding the benign and fleeting nature of this condition, as the abrupt onset may alarm them of a systemic disease. Affected patients should be advised against walking barefoot in areas where the insects can be found. Spraying insecticides in the affected locality also helps to reduce the presence of burrowing bugs.
- Hosokawa T, Kikuchi Y, Nikoh N, et al. Polyphyly of gut symbionts in stinkbugs of the family Cydnidae. Appl Environ Microbiol. 2012; 78:4758-4761.
- Schwertner CF, Nardi C. Burrower bugs (Cydnidae). In: Panizzi A, Grazia J, eds. True Bugs (Heteroptera) of the Neotropics. Entomology in Focus, vol 2. Springer; 2015.
- Lis JA. Burrower bugs of the Old World: a catalogue (Hemiptera: Heteroptera: Cydnidae). Genus (Wroclaw). 1999;10:165-249.
- Hayashi N, Yamamura Y, Ôhama S, et al. Defensive substances from stink bugs of Cydnidae. Experientia. 1976;32:418-419.
- Smith RM. The defensive secretion of the bugs Lampropharadifasciata, Adrisanumeensis, and Tectocorisdiophthalmus from Fiji. NZ J Zool. 1978;5:821-822.
- Krall BS, Zilkowski BW, Kight SL, et al. Chemistry and defensive efficacy of secretion of burrowing bugs. J Chem Ecol. 1997;23:1951-1962.
- Haddad V Jr, Cardoso J, Moraes R. Skin lesions caused by stink bugs (Insecta: Heteroptera: Pentatomidae): first report of dermatological injuries in humans. Wilderness Environ Med. 2002;13:48-50.
- Malhotra AK, Lis JA, Ramam M. Cydnidae (burrowing bug) pigmentation: a novel arthropod dermatosis. JAMA Dermatol. 2015;151:232-233.
- Sonthalia S. Dermoscopy of Cydnidae pigmentation: a novel disorder of pigmentation. Dermatol Pract Concept. 2019;9:228-229.
- Poojary S, Baddireddy K. Demystifying the stinking reddish brown stains through the dermoscope: Cydnidae pigmentation. Indian Dermatol Online J. 2019;10:757-758.
- Amrani A, Das A. Cydnidae pigmentation: unusual location on the abdomen and back. Br J Dermatol. 2021;184:E125.
Cydnidae is a family of small to medium-sized shield bugs with spiny legs that commonly are known as burrowing bugs (or burrower bugs). The family Cydnidae includes more than 100 genera and approximately 600 species worldwide.1 These insects are arthropods of the order Hemiptera (suborder: Heteroptera; superfamily: Pentatomoidae) and largely are concentrated in tropical and temperate regions. Approximately 145 species have been recorded in the Neotropical Region and have been included in the subfamilies Amnestinae, Cephalocteinae, and Sehirinae, in addition to Cydnidae.2 Burrowing bugs are ovoid in shape and 2 to 20 mm in length and morphologically are well adapted for burrowing. Their life span is 100 to 300 days. Being phytophagous, they burrow to feed on plants and roots. Adult burrowing bugs have wings and can fly. They have specialized glands located in either the abdomen (nymph) or thorax (adult) that secrete odorous chemicals for self-protection.3 The secretions contain hydrocarbonates that function as repellents and danger signals, can cause paralysis in prey, and act as a chemoattractant for mates.4-6 They also cause hyperpigmentation upon contact with the skin.
In this article, we present a series of cases from the same community to demonstrate the characteristic features of hyperpigmented macules caused by exposure to burrowing bugs. Dermatologists should be aware of this entity to prevent misdiagnosis and unnecessary investigations and treatment.
Case Series
A 36-year-old woman and 6 children (age range, 6-12 years) presented with a widespread, acute, brown-pigmented, macular eruption with lesions that increased in number over a 1-week period. All 7 patients resided in the same locality and were otherwise systemically healthy. Initially, the index case, a 7-year-old girl, was referred to our tertiary care center by a dermatologist with a provisional diagnosis of idiopathic macular eruptive pigmentation. The patient’s mother recalled noticing a tiny black insect on the child's scalp that left pigment on the skin when she crushed it between her fingers. The rest of the patients presented over the next few days: 3 of the children belonged to the same household as the index case, and there was history of all 6 children playing in the neighborhood park during late evening hours. The adult patient was the parent of one of the affected children. The lesions were associated with mild itching and tingling in 3 children but were asymptomatic in the other patients.
Clinical examination of the patients revealed multiple dark- to light-brown, discrete, irregularly shaped macules over the trunk, arms, and soles (eFigure 1). Dermoscopic examination of a pigmented macule showed an irregularly shaped, brownish, structureless area with accentuation of the pigment at skin creases and perieccrine pigmentation (eFigure 2). The pigmentation was unaffected by rubbing with alcohol or water. Clinicoepidemiologic parameters of the patients are summarized in the eTable.
One of the children’s parents conducted a geological examination of the ground in the neighborhood park during evening hours and found tiny burrowing bugs (eFigure 3). When crushed between the fingers, these insects left a similar brownish hyperpigmentation on the skin. The parents were counseled on the nature of the eruption, and the patients were kept under observation for 2 weeks. On follow-up after 5 days, the lesions showed markedly decreased intensity of hyperpigmentation, and no new lesions were observed in any of the 7 patients.
Comment
Pentatomoidae insects generally are benign and harmless to humans. There have been isolated reports of erythematous plaques caused by Antiteuchus mixtus and Edessa maculate.7 Malhotra et al8 reported the first known series of cases with Cydnidae insect–induced hyperpigmented macules. The reported patients presented with asymptomatic, brown, hyperpigmented macules over exposed sites such as the feet, neck, and chest. All the cases occurred during the monsoon season in tropical and temperate regions of the world, and the patients were characteristically clustered in similar geographic areas. The causative insect was identified as Chilocoris assmuthi Breddin, 1904, belonging to the family Cydnidae. When it was crushed between the fingers, the skin became hyperpigmented, confirming the role of the secretions from the insect in the etiology.8
A second case was described by Sonthalia,9 who also described the dermoscopic features of hyperpigmented macules caused by burrowing bugs. The lesions showed a stuck-on, clustered appearance of ovoid and bizarre pigmented clods, globules, and granules.9 Although the lesions occur mainly over exposed sites, pigmented macules occurring over unusual sites such as the abdomen and back also have been reported in association with burrowing bugs.10 Characteristically, the lesions initially are faint and darken with time and usually fade within a week. They can be rubbed off with acetone but persist when washed with soap and water. The fleeting nature of the pigmentation also has led to the term transient pseudo-lentigines sign to describe hyperpigmentation caused by burrowing bugs.11
Soil and plants are burrowing bugs’ natural habitats, and the insects typically are seen in vegetation-rich, moist areas adjoining human dwellings (eg, parks, gardens), where clusters of cases can occur. These insects proliferate during the monsoon season in tropical and temperate areas, leading to more cases occurring during these months.
Compared to prior reports,8,9 a few of our patients had predominant trunk and neck involvement with an occasional tingling sensation or pruritus while the rest were asymptomatic. Dermoscopic features from our patients shared similar reported features of Cydnidae pigmentation.4,5 The accentuation of pigment over skin creases seen on dermoscopy was due to accumulation of Cydnidae secretion at these sites.
The differential diagnosis commonly includes idiopathic macular eruptive pigmentation, which is characterized by an asymptomatic progressive eruption of hyperpigmented macules over the trunk that persists from a few months up to 3 years. Other conditions in the differential include benign conditions such as acral benign melanocytic nevi, lentigines, pigmented purpuric dermatosis, and postinflammatory hyperpigmentation, as well as malignant conditions such as acral melanoma. Dermoscopy is a helpful, easy-to-use tool in differentiating these pigmentation disorders, obviating the need for an invasive investigation such as histopathologic analysis. Simultaneous involvement in a group of people living together or visiting the same place, abrupt onset, predominant involvement of the exposed sites, characteristic clinical and dermoscopic features, self-limiting course, and timing with the monsoon season should suggest a possibility of Cydnidae dermatitis/pigmentation, which can be confirmed by finding the causative bug in the affected locality.
Management
No specific treatment is required for the pigmentation caused by Cydnidae, as it is self-resolving. The macules can, however, be removed with acetone. Patients must be counseled regarding the benign and fleeting nature of this condition, as the abrupt onset may alarm them of a systemic disease. Affected patients should be advised against walking barefoot in areas where the insects can be found. Spraying insecticides in the affected locality also helps to reduce the presence of burrowing bugs.
Cydnidae is a family of small to medium-sized shield bugs with spiny legs that commonly are known as burrowing bugs (or burrower bugs). The family Cydnidae includes more than 100 genera and approximately 600 species worldwide.1 These insects are arthropods of the order Hemiptera (suborder: Heteroptera; superfamily: Pentatomoidae) and largely are concentrated in tropical and temperate regions. Approximately 145 species have been recorded in the Neotropical Region and have been included in the subfamilies Amnestinae, Cephalocteinae, and Sehirinae, in addition to Cydnidae.2 Burrowing bugs are ovoid in shape and 2 to 20 mm in length and morphologically are well adapted for burrowing. Their life span is 100 to 300 days. Being phytophagous, they burrow to feed on plants and roots. Adult burrowing bugs have wings and can fly. They have specialized glands located in either the abdomen (nymph) or thorax (adult) that secrete odorous chemicals for self-protection.3 The secretions contain hydrocarbonates that function as repellents and danger signals, can cause paralysis in prey, and act as a chemoattractant for mates.4-6 They also cause hyperpigmentation upon contact with the skin.
In this article, we present a series of cases from the same community to demonstrate the characteristic features of hyperpigmented macules caused by exposure to burrowing bugs. Dermatologists should be aware of this entity to prevent misdiagnosis and unnecessary investigations and treatment.
Case Series
A 36-year-old woman and 6 children (age range, 6-12 years) presented with a widespread, acute, brown-pigmented, macular eruption with lesions that increased in number over a 1-week period. All 7 patients resided in the same locality and were otherwise systemically healthy. Initially, the index case, a 7-year-old girl, was referred to our tertiary care center by a dermatologist with a provisional diagnosis of idiopathic macular eruptive pigmentation. The patient’s mother recalled noticing a tiny black insect on the child's scalp that left pigment on the skin when she crushed it between her fingers. The rest of the patients presented over the next few days: 3 of the children belonged to the same household as the index case, and there was history of all 6 children playing in the neighborhood park during late evening hours. The adult patient was the parent of one of the affected children. The lesions were associated with mild itching and tingling in 3 children but were asymptomatic in the other patients.
Clinical examination of the patients revealed multiple dark- to light-brown, discrete, irregularly shaped macules over the trunk, arms, and soles (eFigure 1). Dermoscopic examination of a pigmented macule showed an irregularly shaped, brownish, structureless area with accentuation of the pigment at skin creases and perieccrine pigmentation (eFigure 2). The pigmentation was unaffected by rubbing with alcohol or water. Clinicoepidemiologic parameters of the patients are summarized in the eTable.
One of the children’s parents conducted a geological examination of the ground in the neighborhood park during evening hours and found tiny burrowing bugs (eFigure 3). When crushed between the fingers, these insects left a similar brownish hyperpigmentation on the skin. The parents were counseled on the nature of the eruption, and the patients were kept under observation for 2 weeks. On follow-up after 5 days, the lesions showed markedly decreased intensity of hyperpigmentation, and no new lesions were observed in any of the 7 patients.
Comment
Pentatomoidae insects generally are benign and harmless to humans. There have been isolated reports of erythematous plaques caused by Antiteuchus mixtus and Edessa maculate.7 Malhotra et al8 reported the first known series of cases with Cydnidae insect–induced hyperpigmented macules. The reported patients presented with asymptomatic, brown, hyperpigmented macules over exposed sites such as the feet, neck, and chest. All the cases occurred during the monsoon season in tropical and temperate regions of the world, and the patients were characteristically clustered in similar geographic areas. The causative insect was identified as Chilocoris assmuthi Breddin, 1904, belonging to the family Cydnidae. When it was crushed between the fingers, the skin became hyperpigmented, confirming the role of the secretions from the insect in the etiology.8
A second case was described by Sonthalia,9 who also described the dermoscopic features of hyperpigmented macules caused by burrowing bugs. The lesions showed a stuck-on, clustered appearance of ovoid and bizarre pigmented clods, globules, and granules.9 Although the lesions occur mainly over exposed sites, pigmented macules occurring over unusual sites such as the abdomen and back also have been reported in association with burrowing bugs.10 Characteristically, the lesions initially are faint and darken with time and usually fade within a week. They can be rubbed off with acetone but persist when washed with soap and water. The fleeting nature of the pigmentation also has led to the term transient pseudo-lentigines sign to describe hyperpigmentation caused by burrowing bugs.11
Soil and plants are burrowing bugs’ natural habitats, and the insects typically are seen in vegetation-rich, moist areas adjoining human dwellings (eg, parks, gardens), where clusters of cases can occur. These insects proliferate during the monsoon season in tropical and temperate areas, leading to more cases occurring during these months.
Compared to prior reports,8,9 a few of our patients had predominant trunk and neck involvement with an occasional tingling sensation or pruritus while the rest were asymptomatic. Dermoscopic features from our patients shared similar reported features of Cydnidae pigmentation.4,5 The accentuation of pigment over skin creases seen on dermoscopy was due to accumulation of Cydnidae secretion at these sites.
The differential diagnosis commonly includes idiopathic macular eruptive pigmentation, which is characterized by an asymptomatic progressive eruption of hyperpigmented macules over the trunk that persists from a few months up to 3 years. Other conditions in the differential include benign conditions such as acral benign melanocytic nevi, lentigines, pigmented purpuric dermatosis, and postinflammatory hyperpigmentation, as well as malignant conditions such as acral melanoma. Dermoscopy is a helpful, easy-to-use tool in differentiating these pigmentation disorders, obviating the need for an invasive investigation such as histopathologic analysis. Simultaneous involvement in a group of people living together or visiting the same place, abrupt onset, predominant involvement of the exposed sites, characteristic clinical and dermoscopic features, self-limiting course, and timing with the monsoon season should suggest a possibility of Cydnidae dermatitis/pigmentation, which can be confirmed by finding the causative bug in the affected locality.
Management
No specific treatment is required for the pigmentation caused by Cydnidae, as it is self-resolving. The macules can, however, be removed with acetone. Patients must be counseled regarding the benign and fleeting nature of this condition, as the abrupt onset may alarm them of a systemic disease. Affected patients should be advised against walking barefoot in areas where the insects can be found. Spraying insecticides in the affected locality also helps to reduce the presence of burrowing bugs.
- Hosokawa T, Kikuchi Y, Nikoh N, et al. Polyphyly of gut symbionts in stinkbugs of the family Cydnidae. Appl Environ Microbiol. 2012; 78:4758-4761.
- Schwertner CF, Nardi C. Burrower bugs (Cydnidae). In: Panizzi A, Grazia J, eds. True Bugs (Heteroptera) of the Neotropics. Entomology in Focus, vol 2. Springer; 2015.
- Lis JA. Burrower bugs of the Old World: a catalogue (Hemiptera: Heteroptera: Cydnidae). Genus (Wroclaw). 1999;10:165-249.
- Hayashi N, Yamamura Y, Ôhama S, et al. Defensive substances from stink bugs of Cydnidae. Experientia. 1976;32:418-419.
- Smith RM. The defensive secretion of the bugs Lampropharadifasciata, Adrisanumeensis, and Tectocorisdiophthalmus from Fiji. NZ J Zool. 1978;5:821-822.
- Krall BS, Zilkowski BW, Kight SL, et al. Chemistry and defensive efficacy of secretion of burrowing bugs. J Chem Ecol. 1997;23:1951-1962.
- Haddad V Jr, Cardoso J, Moraes R. Skin lesions caused by stink bugs (Insecta: Heteroptera: Pentatomidae): first report of dermatological injuries in humans. Wilderness Environ Med. 2002;13:48-50.
- Malhotra AK, Lis JA, Ramam M. Cydnidae (burrowing bug) pigmentation: a novel arthropod dermatosis. JAMA Dermatol. 2015;151:232-233.
- Sonthalia S. Dermoscopy of Cydnidae pigmentation: a novel disorder of pigmentation. Dermatol Pract Concept. 2019;9:228-229.
- Poojary S, Baddireddy K. Demystifying the stinking reddish brown stains through the dermoscope: Cydnidae pigmentation. Indian Dermatol Online J. 2019;10:757-758.
- Amrani A, Das A. Cydnidae pigmentation: unusual location on the abdomen and back. Br J Dermatol. 2021;184:E125.
- Hosokawa T, Kikuchi Y, Nikoh N, et al. Polyphyly of gut symbionts in stinkbugs of the family Cydnidae. Appl Environ Microbiol. 2012; 78:4758-4761.
- Schwertner CF, Nardi C. Burrower bugs (Cydnidae). In: Panizzi A, Grazia J, eds. True Bugs (Heteroptera) of the Neotropics. Entomology in Focus, vol 2. Springer; 2015.
- Lis JA. Burrower bugs of the Old World: a catalogue (Hemiptera: Heteroptera: Cydnidae). Genus (Wroclaw). 1999;10:165-249.
- Hayashi N, Yamamura Y, Ôhama S, et al. Defensive substances from stink bugs of Cydnidae. Experientia. 1976;32:418-419.
- Smith RM. The defensive secretion of the bugs Lampropharadifasciata, Adrisanumeensis, and Tectocorisdiophthalmus from Fiji. NZ J Zool. 1978;5:821-822.
- Krall BS, Zilkowski BW, Kight SL, et al. Chemistry and defensive efficacy of secretion of burrowing bugs. J Chem Ecol. 1997;23:1951-1962.
- Haddad V Jr, Cardoso J, Moraes R. Skin lesions caused by stink bugs (Insecta: Heteroptera: Pentatomidae): first report of dermatological injuries in humans. Wilderness Environ Med. 2002;13:48-50.
- Malhotra AK, Lis JA, Ramam M. Cydnidae (burrowing bug) pigmentation: a novel arthropod dermatosis. JAMA Dermatol. 2015;151:232-233.
- Sonthalia S. Dermoscopy of Cydnidae pigmentation: a novel disorder of pigmentation. Dermatol Pract Concept. 2019;9:228-229.
- Poojary S, Baddireddy K. Demystifying the stinking reddish brown stains through the dermoscope: Cydnidae pigmentation. Indian Dermatol Online J. 2019;10:757-758.
- Amrani A, Das A. Cydnidae pigmentation: unusual location on the abdomen and back. Br J Dermatol. 2021;184:E125.
Hyperpigmented Macules Caused by Burrowing Bugs (Cydnidae) May Mimic More Serious Conditions
Hyperpigmented Macules Caused by Burrowing Bugs (Cydnidae) May Mimic More Serious Conditions
Practice Points
- Burrowing bugs (Cydnidae) are phytophagous and burrow to feed on plants and roots. They are more numerous during the monsoon season in tropical and temperate regions.
- Secretions from burrowing bugs cause asymptomatic, hyperpigmented, irregularly shaped macules suggestive of an exogenous cause that commonly affect clusters of patients from the same geographic locality.
- The lesions are self-limiting and must be differentiated from close mimickers to ensure adequate and appropriate patient counseling.

Tapping Into Relief: A Distraction Technique to Reduce Pain During Dermatologic Procedures
Tapping Into Relief: A Distraction Technique to Reduce Pain During Dermatologic Procedures
Practice Gap
Pain during minimally invasive dermatologic procedures such as lidocaine injections, cryotherapy, nail unit injections, and cosmetic procedures including neurotoxin injections can cause patient discomfort leading to procedural anxiety, poor compliance with treatment regimens, and avoidance of necessary care. Current solutions to manage pain during dermatologic procedures present several limitations; for example, topical anesthetics seldom alleviate procedural pain,1 particularly in sensitive areas (eg, nail unit, face) or for patients with a needle phobia. Additionally, topical anesthetics often require up to 2 hours to take effect, making them impractical for quick outpatient procedures. Other pain reduction strategies including vibration devices or cold sprays2,3 can be effective but are an added expense to the physician or clinic, which may preclude their use in resource-limited settings. Psychological distraction techniques such as deep breathing require active patient participation and might reinforce pain expectations and increase patient anxiety.4 Given these challenges, there is a need for effective, affordable, nonpharmacologic pain reduction strategies that can be integrated seamlessly into clinical practice to enhance the patient experience.
The Technique
Tapping is a simple noninvasive distraction technique that may alleviate procedural pain by exploiting the gate control theory of pain.5 According to this theory, tactile stimuli activate mechanoreceptors that send inhibitory signals to the spinal cord, effectively closing the gate to pain transmission. Unlike the Helfer skin tap technique,6 which involves 15 preinjection taps and 3 postinjection taps directly on the injection site, our approach targets distant bony prominences. This modification allows for immediate needle insertion without interfering with the sterile field or increasing the risk for needlestick injuries from tapping near the injection site. Bony sites such as the shoulder or knee are ideal for this technique due to their high density and rigidity that efficiently transmit tactile stimuli––similar to how sound travels faster through solids than through liquids or gases.7
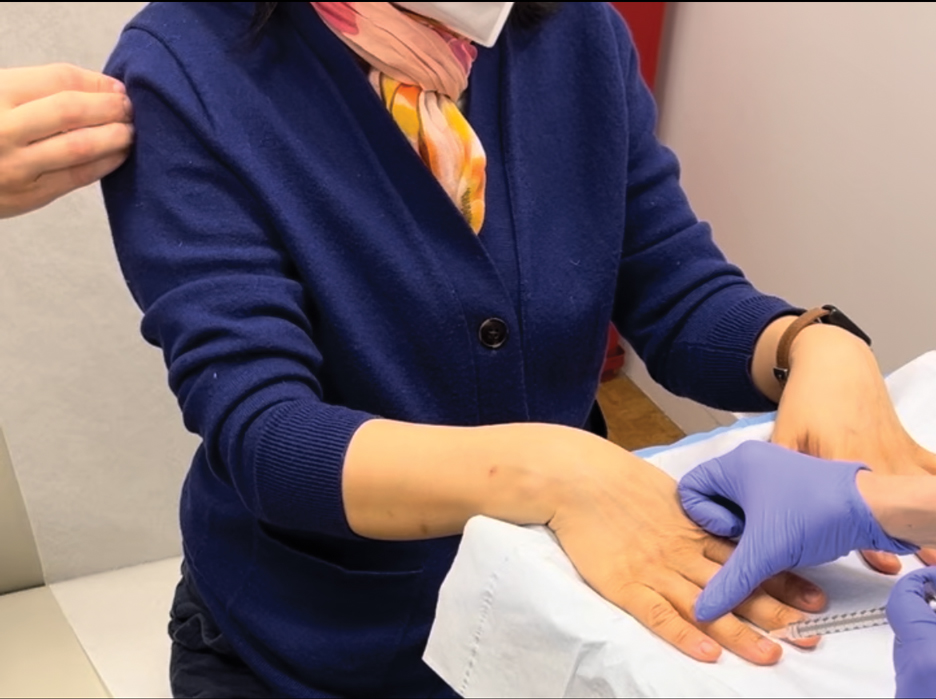
To implement this technique in practice, we first stabilize the injection site to reduce movement from tapping. This can be done by stabilizing the injection site (eg, resting the hand on an instrument stand during a nail unit injection). A second person—such as a medical assistant, medical student, resident, or even the patient’s family member—taps at a distant site at least an arm’s length away from the injection site (Figure). The tapping pressure should be firm enough for the patient to feel the vibration but not forceful enough that it becomes unpleasant or disrupts the injection area. Tapping starts just before needle insertion and continues through the injection. No warning is given to the patient, as the surprise element may help distract them from pain. Varying the rhythm, intensity, or location of the tapping can enhance its distracting effect.
This tapping technique can be effectively combined with other pain reduction strategies in a multimodal approach; for example, when used concurrently with topical anesthetics, both the central (tapping) and peripheral (anesthetic) pain pathways are addressed, potentially yielding additive effects. For patients with a needle phobia, pairing tapping with cognitive distraction (eg, talkesthesia) may further reduce anxiety. In our nail specialty clinic at Weill Cornell Medicine (New York, New York), we often combine tapping with cold sprays and talkesthesia, which improves patient comfort without prolonging the visit. Importantly, the technique enables seamless integration with most pharmacologic and nonpharmacologic methods, eliminating the need for additional patient education or procedure time.
Practice Implications
The tapping technique described here is free, easy to implement, and requires no additional resources aside from another person to tap the patient during the procedure. It can be used for a wide range of dermatologic procedures, including biopsies, intralesional injections, and cosmetic treatments, including neurotoxin injections. The minimal learning curve and ease of integration into procedural workflows make this technique a valuable tool for dermatologists aiming to improve patient comfort without disrupting workflow. In our practice, we have observed that tapping reduces self-reported pain and helps ease anxiety, particularly in patients with a needle phobia. Its simplicity and accessibility make it a valuable addition to a wide range of dermatologic procedures. Prospective studies investigating patient-reported outcomes could help establish this technique’s role in clinical practice.
- Navarro-Rodriguez JM, Suarez-Serrano C, Martin-Valero R, et al. Effectiveness of topical anesthetics in pain management for dermal injuries: a systematic review. J Clin Med. 2021;10:2522. doi:10.3390/jcm10112522
- Lipner SR. Pain-minimizing strategies for nail surgery. Cutis. 2018;101:76-77.
- Ricardo JW, Lipner SR. Air cooling for improved analgesia during local anesthetic infiltration for nail surgery. J Am Acad Dermatol. 2021;84:e231-e232. doi:10.1016/j.jaad.2019.11.032
- Hill RC, Chernoff KA, Lipner SR. A breath of fresh air: use of deep breathing technique to minimize pain with nail injections. J Am Acad Dermatol. 2024;90:e163. doi:10.1016/j.jaad.2023.10.043
- Mendell LM. Constructing and deconstructing the gate theory of pain. Pain. 2014;155:210-216. doi:10.1016/j.pain.2013.12.010
- Jyoti G, Arora S, Sharma B. Helfer Skin Tap Tech Technique for the IM injection pain among adult patients. Nursing & Midwifery Research Journal. 2018;14:18-30. doi:10.1177/0974150X20180304
- Iowa State University. Nondestructive Evaluation Physics: Sound. Published 2021. Accessed July 31, 2025. https://www.nde-ed.org/Physics/Sound/speedinmaterials.xhtml
Practice Gap
Pain during minimally invasive dermatologic procedures such as lidocaine injections, cryotherapy, nail unit injections, and cosmetic procedures including neurotoxin injections can cause patient discomfort leading to procedural anxiety, poor compliance with treatment regimens, and avoidance of necessary care. Current solutions to manage pain during dermatologic procedures present several limitations; for example, topical anesthetics seldom alleviate procedural pain,1 particularly in sensitive areas (eg, nail unit, face) or for patients with a needle phobia. Additionally, topical anesthetics often require up to 2 hours to take effect, making them impractical for quick outpatient procedures. Other pain reduction strategies including vibration devices or cold sprays2,3 can be effective but are an added expense to the physician or clinic, which may preclude their use in resource-limited settings. Psychological distraction techniques such as deep breathing require active patient participation and might reinforce pain expectations and increase patient anxiety.4 Given these challenges, there is a need for effective, affordable, nonpharmacologic pain reduction strategies that can be integrated seamlessly into clinical practice to enhance the patient experience.
The Technique
Tapping is a simple noninvasive distraction technique that may alleviate procedural pain by exploiting the gate control theory of pain.5 According to this theory, tactile stimuli activate mechanoreceptors that send inhibitory signals to the spinal cord, effectively closing the gate to pain transmission. Unlike the Helfer skin tap technique,6 which involves 15 preinjection taps and 3 postinjection taps directly on the injection site, our approach targets distant bony prominences. This modification allows for immediate needle insertion without interfering with the sterile field or increasing the risk for needlestick injuries from tapping near the injection site. Bony sites such as the shoulder or knee are ideal for this technique due to their high density and rigidity that efficiently transmit tactile stimuli––similar to how sound travels faster through solids than through liquids or gases.7
To implement this technique in practice, we first stabilize the injection site to reduce movement from tapping. This can be done by stabilizing the injection site (eg, resting the hand on an instrument stand during a nail unit injection). A second person—such as a medical assistant, medical student, resident, or even the patient’s family member—taps at a distant site at least an arm’s length away from the injection site (Figure). The tapping pressure should be firm enough for the patient to feel the vibration but not forceful enough that it becomes unpleasant or disrupts the injection area. Tapping starts just before needle insertion and continues through the injection. No warning is given to the patient, as the surprise element may help distract them from pain. Varying the rhythm, intensity, or location of the tapping can enhance its distracting effect.
This tapping technique can be effectively combined with other pain reduction strategies in a multimodal approach; for example, when used concurrently with topical anesthetics, both the central (tapping) and peripheral (anesthetic) pain pathways are addressed, potentially yielding additive effects. For patients with a needle phobia, pairing tapping with cognitive distraction (eg, talkesthesia) may further reduce anxiety. In our nail specialty clinic at Weill Cornell Medicine (New York, New York), we often combine tapping with cold sprays and talkesthesia, which improves patient comfort without prolonging the visit. Importantly, the technique enables seamless integration with most pharmacologic and nonpharmacologic methods, eliminating the need for additional patient education or procedure time.
Practice Implications
The tapping technique described here is free, easy to implement, and requires no additional resources aside from another person to tap the patient during the procedure. It can be used for a wide range of dermatologic procedures, including biopsies, intralesional injections, and cosmetic treatments, including neurotoxin injections. The minimal learning curve and ease of integration into procedural workflows make this technique a valuable tool for dermatologists aiming to improve patient comfort without disrupting workflow. In our practice, we have observed that tapping reduces self-reported pain and helps ease anxiety, particularly in patients with a needle phobia. Its simplicity and accessibility make it a valuable addition to a wide range of dermatologic procedures. Prospective studies investigating patient-reported outcomes could help establish this technique’s role in clinical practice.
Practice Gap
Pain during minimally invasive dermatologic procedures such as lidocaine injections, cryotherapy, nail unit injections, and cosmetic procedures including neurotoxin injections can cause patient discomfort leading to procedural anxiety, poor compliance with treatment regimens, and avoidance of necessary care. Current solutions to manage pain during dermatologic procedures present several limitations; for example, topical anesthetics seldom alleviate procedural pain,1 particularly in sensitive areas (eg, nail unit, face) or for patients with a needle phobia. Additionally, topical anesthetics often require up to 2 hours to take effect, making them impractical for quick outpatient procedures. Other pain reduction strategies including vibration devices or cold sprays2,3 can be effective but are an added expense to the physician or clinic, which may preclude their use in resource-limited settings. Psychological distraction techniques such as deep breathing require active patient participation and might reinforce pain expectations and increase patient anxiety.4 Given these challenges, there is a need for effective, affordable, nonpharmacologic pain reduction strategies that can be integrated seamlessly into clinical practice to enhance the patient experience.
The Technique
Tapping is a simple noninvasive distraction technique that may alleviate procedural pain by exploiting the gate control theory of pain.5 According to this theory, tactile stimuli activate mechanoreceptors that send inhibitory signals to the spinal cord, effectively closing the gate to pain transmission. Unlike the Helfer skin tap technique,6 which involves 15 preinjection taps and 3 postinjection taps directly on the injection site, our approach targets distant bony prominences. This modification allows for immediate needle insertion without interfering with the sterile field or increasing the risk for needlestick injuries from tapping near the injection site. Bony sites such as the shoulder or knee are ideal for this technique due to their high density and rigidity that efficiently transmit tactile stimuli––similar to how sound travels faster through solids than through liquids or gases.7
To implement this technique in practice, we first stabilize the injection site to reduce movement from tapping. This can be done by stabilizing the injection site (eg, resting the hand on an instrument stand during a nail unit injection). A second person—such as a medical assistant, medical student, resident, or even the patient’s family member—taps at a distant site at least an arm’s length away from the injection site (Figure). The tapping pressure should be firm enough for the patient to feel the vibration but not forceful enough that it becomes unpleasant or disrupts the injection area. Tapping starts just before needle insertion and continues through the injection. No warning is given to the patient, as the surprise element may help distract them from pain. Varying the rhythm, intensity, or location of the tapping can enhance its distracting effect.
This tapping technique can be effectively combined with other pain reduction strategies in a multimodal approach; for example, when used concurrently with topical anesthetics, both the central (tapping) and peripheral (anesthetic) pain pathways are addressed, potentially yielding additive effects. For patients with a needle phobia, pairing tapping with cognitive distraction (eg, talkesthesia) may further reduce anxiety. In our nail specialty clinic at Weill Cornell Medicine (New York, New York), we often combine tapping with cold sprays and talkesthesia, which improves patient comfort without prolonging the visit. Importantly, the technique enables seamless integration with most pharmacologic and nonpharmacologic methods, eliminating the need for additional patient education or procedure time.
Practice Implications
The tapping technique described here is free, easy to implement, and requires no additional resources aside from another person to tap the patient during the procedure. It can be used for a wide range of dermatologic procedures, including biopsies, intralesional injections, and cosmetic treatments, including neurotoxin injections. The minimal learning curve and ease of integration into procedural workflows make this technique a valuable tool for dermatologists aiming to improve patient comfort without disrupting workflow. In our practice, we have observed that tapping reduces self-reported pain and helps ease anxiety, particularly in patients with a needle phobia. Its simplicity and accessibility make it a valuable addition to a wide range of dermatologic procedures. Prospective studies investigating patient-reported outcomes could help establish this technique’s role in clinical practice.
- Navarro-Rodriguez JM, Suarez-Serrano C, Martin-Valero R, et al. Effectiveness of topical anesthetics in pain management for dermal injuries: a systematic review. J Clin Med. 2021;10:2522. doi:10.3390/jcm10112522
- Lipner SR. Pain-minimizing strategies for nail surgery. Cutis. 2018;101:76-77.
- Ricardo JW, Lipner SR. Air cooling for improved analgesia during local anesthetic infiltration for nail surgery. J Am Acad Dermatol. 2021;84:e231-e232. doi:10.1016/j.jaad.2019.11.032
- Hill RC, Chernoff KA, Lipner SR. A breath of fresh air: use of deep breathing technique to minimize pain with nail injections. J Am Acad Dermatol. 2024;90:e163. doi:10.1016/j.jaad.2023.10.043
- Mendell LM. Constructing and deconstructing the gate theory of pain. Pain. 2014;155:210-216. doi:10.1016/j.pain.2013.12.010
- Jyoti G, Arora S, Sharma B. Helfer Skin Tap Tech Technique for the IM injection pain among adult patients. Nursing & Midwifery Research Journal. 2018;14:18-30. doi:10.1177/0974150X20180304
- Iowa State University. Nondestructive Evaluation Physics: Sound. Published 2021. Accessed July 31, 2025. https://www.nde-ed.org/Physics/Sound/speedinmaterials.xhtml
- Navarro-Rodriguez JM, Suarez-Serrano C, Martin-Valero R, et al. Effectiveness of topical anesthetics in pain management for dermal injuries: a systematic review. J Clin Med. 2021;10:2522. doi:10.3390/jcm10112522
- Lipner SR. Pain-minimizing strategies for nail surgery. Cutis. 2018;101:76-77.
- Ricardo JW, Lipner SR. Air cooling for improved analgesia during local anesthetic infiltration for nail surgery. J Am Acad Dermatol. 2021;84:e231-e232. doi:10.1016/j.jaad.2019.11.032
- Hill RC, Chernoff KA, Lipner SR. A breath of fresh air: use of deep breathing technique to minimize pain with nail injections. J Am Acad Dermatol. 2024;90:e163. doi:10.1016/j.jaad.2023.10.043
- Mendell LM. Constructing and deconstructing the gate theory of pain. Pain. 2014;155:210-216. doi:10.1016/j.pain.2013.12.010
- Jyoti G, Arora S, Sharma B. Helfer Skin Tap Tech Technique for the IM injection pain among adult patients. Nursing & Midwifery Research Journal. 2018;14:18-30. doi:10.1177/0974150X20180304
- Iowa State University. Nondestructive Evaluation Physics: Sound. Published 2021. Accessed July 31, 2025. https://www.nde-ed.org/Physics/Sound/speedinmaterials.xhtml
Tapping Into Relief: A Distraction Technique to Reduce Pain During Dermatologic Procedures
Tapping Into Relief: A Distraction Technique to Reduce Pain During Dermatologic Procedures

A Cross-Sectional Analysis of TikTok Skin Care Routines and the Associated Environmental Impact
A Cross-Sectional Analysis of TikTok Skin Care Routines and the Associated Environmental Impact
To the Editor:
The popularity of the social media platform TikTok, which is known for its short-form videos, has surged in recent years. Viral videos demonstrating skin care routines reach millions of viewers,1 showcasing specific products, detailing beauty regimens, and setting fads that many users eagerly follow. These trends often influence consumer behavior—in 2023, viral videos using the tag #TikTokMadeMeBuy lead to a 14% growth in the sale of skin care products.2 However, they also encourage purchasing decisions that may escalate environmental waste through plastic packaging and single-use products. In this study, we analyzed videos on TikTok to assess the environmental impact of trending skin care routines. By examining the types of products promoted, their packaging, and the frequency with which they appear in viral content, we aimed to investigate how these trends, which may be imitated by users, impact the environment.
A search of TikTok videos using #skincareroutine was conducted on June 21, 2024. Sponsored content, non–English language videos, videos without demonstrated skin care routines, and videos showing makeup routines were excluded from our analysis. Data collected from each video included username, date posted, number of likes, total number of skin care products used, number of single-use skin care products used, average amount of product used, number of skin care applicators used, and number of single-use applicators used. Single-use items, defined as those intended for one-time use and subsequent disposal, were identified visually by packaging, manufacturer intent, and common consumer usage patterns. The amount of product used per application was graded on a scale of 1 to 3 (1=pea-sized amount or less; 2=single full pump/spray; 3=multiple pumps/sprays). Videos were categorized as personal (ie, skin care routine walk-throughs by the creator) or autonomous sensory meridian response (ASMR)(focused on product sounds and aesthetics).3 A Mann-Whitney U test was utilized to statistically compare the 2 groups. Statistical analysis was performed using Microsoft Excel (α=0.05).
A total of 50 videos met the inclusion criteria and were included in the analysis. The average number of likes per video was 499,696.15, with skin care routines featuring an average of 6.4 unique products (Table). There was a weak positive correlation (r=0.1809) between the number of skin care products used and the number of likes. A total of 320 products were used across the videos, 23 of which were single-use (7.2%).On average, single-use skin care items were used 0.46 times per routine, comprising a mean 7.99% of total products per video. The average score for the amount of product used per application was 2.18. There was no difference in personal vs ASMR videos with regard to the total number of skin care products used or the average amount of product used per application (P>.05). Thirty-three (70.2%) of the 47 applicators used across all videos were single-use. An average of 0.94 applicators per routine were utilized, with a mean 68.83% being single-use applicators. Common single-use products were toner wipes and eye patches, and single-use applicators included cotton pads and plastic spatulas.
Our findings indicated a prevalence of multiple products and large amount of product used in trending skin care routines, suggesting a shift toward multistep skin care. This implies a high rate of product consumption that may accelerate the carbon footprint associated with skin care products,3 which could contribute to climate change and environmental degradation. Consumers also may feel compelled to purchase and discard numerous partially used products in order to keep up with the latest trends, exacerbating the environmental impact. Furthermore, the utilization of single-use products and applicators contributes to increased plastic waste, pollution, and resource depletion. Single-use items often are difficult to recycle due to their mixed materials and small size,4,5 and therefore they can accumulate in landfills and oceans. This impact can be mitigated by switching to reusable applicators, refillable packaging, and biodegradable materials.
The substantial average number of likes per video indicates high engagement with skin care content among TikTok users. The continued popularity of complex multistep skin care routines, despite a weak correlation between the number of skin care products used and the number of likes per video, likely stems from factors such as aesthetic appeal, ASMR effects, and creators’ established followings, which may drive user engagement to contribute to unsustainable consumption patterns. Factors such as presentation style, aesthetics, or creators’ pre-existing online following may have a major impact on how well a video performs on TikTok. The similarity between personal and ASMR videos, particularly in the number of products used and the amount applied, suggests that both formats employ common approaches to meet audience expectations and align with promotional trends, relying more on sensory and aesthetic strategies than substantive differences in skin care routines.
Our use of only one tag in our search as well as the subjective quantity scale limits the generalizability of these findings to broader TikTok skin care content.
Overall, our study underscores the role of brands and social media influencers in skin care education and promotion of sustainable practices. The extensive number of products used and generous application of each product in skin care routines demonstrated in TikTok videos may mislead viewers into believing that using more product improves outcomes, when often, less is more. We recommend that dermatologists counsel patients about informed skin care regimens that prioritize individual needs over social media fads.
- Pagani K, Lukac D, Martinez R, et al. Slugging: TikTokTM as a source of a viral “harmless” beauty trend. Clin Dermatol. 2022;40:810-812. doi:10.1016/j.clindermatol.2022.08.005
- Stern C. TikTok drives $31.7B in beauty sales: how viral trends are shaping the future of cosmetics. CosmeticsDesign. August 20, 2024. Accessed June 24, 2025. https://www.cosmeticsdesign.com/Article/2024/08/20/tiktok-drives-31.7b-in-beauty-sales-how-viral-trends-are-shaping-the-future-of-cosmetics/
- Fountain C. ASMR content saw huge growth on YouTube, but now creators are flocking to TikTok instead. Business Insider. July 4, 2022. Accessed June 24, 2025. https://www.businessinsider.com/asmr-tiktok-instead-of-youtube-growth-subscribers-2022-7
- Rathore S, Schuler B, Park J. Life cycle assessment of multiple dispensing systems used for cosmetic product packaging. Packaging Technol Sci. 2023;36:533-547. doi:10.1002/pts.2729
- Shaw S. How to actually recycle your empty beauty products. CNN Underscored. Updated April 17, 2024. Accessed June 24, 2025. https://www.cnn.com/cnn-underscored/beauty/how-to-recycle-beauty-products
To the Editor:
The popularity of the social media platform TikTok, which is known for its short-form videos, has surged in recent years. Viral videos demonstrating skin care routines reach millions of viewers,1 showcasing specific products, detailing beauty regimens, and setting fads that many users eagerly follow. These trends often influence consumer behavior—in 2023, viral videos using the tag #TikTokMadeMeBuy lead to a 14% growth in the sale of skin care products.2 However, they also encourage purchasing decisions that may escalate environmental waste through plastic packaging and single-use products. In this study, we analyzed videos on TikTok to assess the environmental impact of trending skin care routines. By examining the types of products promoted, their packaging, and the frequency with which they appear in viral content, we aimed to investigate how these trends, which may be imitated by users, impact the environment.
A search of TikTok videos using #skincareroutine was conducted on June 21, 2024. Sponsored content, non–English language videos, videos without demonstrated skin care routines, and videos showing makeup routines were excluded from our analysis. Data collected from each video included username, date posted, number of likes, total number of skin care products used, number of single-use skin care products used, average amount of product used, number of skin care applicators used, and number of single-use applicators used. Single-use items, defined as those intended for one-time use and subsequent disposal, were identified visually by packaging, manufacturer intent, and common consumer usage patterns. The amount of product used per application was graded on a scale of 1 to 3 (1=pea-sized amount or less; 2=single full pump/spray; 3=multiple pumps/sprays). Videos were categorized as personal (ie, skin care routine walk-throughs by the creator) or autonomous sensory meridian response (ASMR)(focused on product sounds and aesthetics).3 A Mann-Whitney U test was utilized to statistically compare the 2 groups. Statistical analysis was performed using Microsoft Excel (α=0.05).
A total of 50 videos met the inclusion criteria and were included in the analysis. The average number of likes per video was 499,696.15, with skin care routines featuring an average of 6.4 unique products (Table). There was a weak positive correlation (r=0.1809) between the number of skin care products used and the number of likes. A total of 320 products were used across the videos, 23 of which were single-use (7.2%).On average, single-use skin care items were used 0.46 times per routine, comprising a mean 7.99% of total products per video. The average score for the amount of product used per application was 2.18. There was no difference in personal vs ASMR videos with regard to the total number of skin care products used or the average amount of product used per application (P>.05). Thirty-three (70.2%) of the 47 applicators used across all videos were single-use. An average of 0.94 applicators per routine were utilized, with a mean 68.83% being single-use applicators. Common single-use products were toner wipes and eye patches, and single-use applicators included cotton pads and plastic spatulas.
Our findings indicated a prevalence of multiple products and large amount of product used in trending skin care routines, suggesting a shift toward multistep skin care. This implies a high rate of product consumption that may accelerate the carbon footprint associated with skin care products,3 which could contribute to climate change and environmental degradation. Consumers also may feel compelled to purchase and discard numerous partially used products in order to keep up with the latest trends, exacerbating the environmental impact. Furthermore, the utilization of single-use products and applicators contributes to increased plastic waste, pollution, and resource depletion. Single-use items often are difficult to recycle due to their mixed materials and small size,4,5 and therefore they can accumulate in landfills and oceans. This impact can be mitigated by switching to reusable applicators, refillable packaging, and biodegradable materials.
The substantial average number of likes per video indicates high engagement with skin care content among TikTok users. The continued popularity of complex multistep skin care routines, despite a weak correlation between the number of skin care products used and the number of likes per video, likely stems from factors such as aesthetic appeal, ASMR effects, and creators’ established followings, which may drive user engagement to contribute to unsustainable consumption patterns. Factors such as presentation style, aesthetics, or creators’ pre-existing online following may have a major impact on how well a video performs on TikTok. The similarity between personal and ASMR videos, particularly in the number of products used and the amount applied, suggests that both formats employ common approaches to meet audience expectations and align with promotional trends, relying more on sensory and aesthetic strategies than substantive differences in skin care routines.
Our use of only one tag in our search as well as the subjective quantity scale limits the generalizability of these findings to broader TikTok skin care content.
Overall, our study underscores the role of brands and social media influencers in skin care education and promotion of sustainable practices. The extensive number of products used and generous application of each product in skin care routines demonstrated in TikTok videos may mislead viewers into believing that using more product improves outcomes, when often, less is more. We recommend that dermatologists counsel patients about informed skin care regimens that prioritize individual needs over social media fads.
To the Editor:
The popularity of the social media platform TikTok, which is known for its short-form videos, has surged in recent years. Viral videos demonstrating skin care routines reach millions of viewers,1 showcasing specific products, detailing beauty regimens, and setting fads that many users eagerly follow. These trends often influence consumer behavior—in 2023, viral videos using the tag #TikTokMadeMeBuy lead to a 14% growth in the sale of skin care products.2 However, they also encourage purchasing decisions that may escalate environmental waste through plastic packaging and single-use products. In this study, we analyzed videos on TikTok to assess the environmental impact of trending skin care routines. By examining the types of products promoted, their packaging, and the frequency with which they appear in viral content, we aimed to investigate how these trends, which may be imitated by users, impact the environment.
A search of TikTok videos using #skincareroutine was conducted on June 21, 2024. Sponsored content, non–English language videos, videos without demonstrated skin care routines, and videos showing makeup routines were excluded from our analysis. Data collected from each video included username, date posted, number of likes, total number of skin care products used, number of single-use skin care products used, average amount of product used, number of skin care applicators used, and number of single-use applicators used. Single-use items, defined as those intended for one-time use and subsequent disposal, were identified visually by packaging, manufacturer intent, and common consumer usage patterns. The amount of product used per application was graded on a scale of 1 to 3 (1=pea-sized amount or less; 2=single full pump/spray; 3=multiple pumps/sprays). Videos were categorized as personal (ie, skin care routine walk-throughs by the creator) or autonomous sensory meridian response (ASMR)(focused on product sounds and aesthetics).3 A Mann-Whitney U test was utilized to statistically compare the 2 groups. Statistical analysis was performed using Microsoft Excel (α=0.05).
A total of 50 videos met the inclusion criteria and were included in the analysis. The average number of likes per video was 499,696.15, with skin care routines featuring an average of 6.4 unique products (Table). There was a weak positive correlation (r=0.1809) between the number of skin care products used and the number of likes. A total of 320 products were used across the videos, 23 of which were single-use (7.2%).On average, single-use skin care items were used 0.46 times per routine, comprising a mean 7.99% of total products per video. The average score for the amount of product used per application was 2.18. There was no difference in personal vs ASMR videos with regard to the total number of skin care products used or the average amount of product used per application (P>.05). Thirty-three (70.2%) of the 47 applicators used across all videos were single-use. An average of 0.94 applicators per routine were utilized, with a mean 68.83% being single-use applicators. Common single-use products were toner wipes and eye patches, and single-use applicators included cotton pads and plastic spatulas.
Our findings indicated a prevalence of multiple products and large amount of product used in trending skin care routines, suggesting a shift toward multistep skin care. This implies a high rate of product consumption that may accelerate the carbon footprint associated with skin care products,3 which could contribute to climate change and environmental degradation. Consumers also may feel compelled to purchase and discard numerous partially used products in order to keep up with the latest trends, exacerbating the environmental impact. Furthermore, the utilization of single-use products and applicators contributes to increased plastic waste, pollution, and resource depletion. Single-use items often are difficult to recycle due to their mixed materials and small size,4,5 and therefore they can accumulate in landfills and oceans. This impact can be mitigated by switching to reusable applicators, refillable packaging, and biodegradable materials.
The substantial average number of likes per video indicates high engagement with skin care content among TikTok users. The continued popularity of complex multistep skin care routines, despite a weak correlation between the number of skin care products used and the number of likes per video, likely stems from factors such as aesthetic appeal, ASMR effects, and creators’ established followings, which may drive user engagement to contribute to unsustainable consumption patterns. Factors such as presentation style, aesthetics, or creators’ pre-existing online following may have a major impact on how well a video performs on TikTok. The similarity between personal and ASMR videos, particularly in the number of products used and the amount applied, suggests that both formats employ common approaches to meet audience expectations and align with promotional trends, relying more on sensory and aesthetic strategies than substantive differences in skin care routines.
Our use of only one tag in our search as well as the subjective quantity scale limits the generalizability of these findings to broader TikTok skin care content.
Overall, our study underscores the role of brands and social media influencers in skin care education and promotion of sustainable practices. The extensive number of products used and generous application of each product in skin care routines demonstrated in TikTok videos may mislead viewers into believing that using more product improves outcomes, when often, less is more. We recommend that dermatologists counsel patients about informed skin care regimens that prioritize individual needs over social media fads.
- Pagani K, Lukac D, Martinez R, et al. Slugging: TikTokTM as a source of a viral “harmless” beauty trend. Clin Dermatol. 2022;40:810-812. doi:10.1016/j.clindermatol.2022.08.005
- Stern C. TikTok drives $31.7B in beauty sales: how viral trends are shaping the future of cosmetics. CosmeticsDesign. August 20, 2024. Accessed June 24, 2025. https://www.cosmeticsdesign.com/Article/2024/08/20/tiktok-drives-31.7b-in-beauty-sales-how-viral-trends-are-shaping-the-future-of-cosmetics/
- Fountain C. ASMR content saw huge growth on YouTube, but now creators are flocking to TikTok instead. Business Insider. July 4, 2022. Accessed June 24, 2025. https://www.businessinsider.com/asmr-tiktok-instead-of-youtube-growth-subscribers-2022-7
- Rathore S, Schuler B, Park J. Life cycle assessment of multiple dispensing systems used for cosmetic product packaging. Packaging Technol Sci. 2023;36:533-547. doi:10.1002/pts.2729
- Shaw S. How to actually recycle your empty beauty products. CNN Underscored. Updated April 17, 2024. Accessed June 24, 2025. https://www.cnn.com/cnn-underscored/beauty/how-to-recycle-beauty-products
- Pagani K, Lukac D, Martinez R, et al. Slugging: TikTokTM as a source of a viral “harmless” beauty trend. Clin Dermatol. 2022;40:810-812. doi:10.1016/j.clindermatol.2022.08.005
- Stern C. TikTok drives $31.7B in beauty sales: how viral trends are shaping the future of cosmetics. CosmeticsDesign. August 20, 2024. Accessed June 24, 2025. https://www.cosmeticsdesign.com/Article/2024/08/20/tiktok-drives-31.7b-in-beauty-sales-how-viral-trends-are-shaping-the-future-of-cosmetics/
- Fountain C. ASMR content saw huge growth on YouTube, but now creators are flocking to TikTok instead. Business Insider. July 4, 2022. Accessed June 24, 2025. https://www.businessinsider.com/asmr-tiktok-instead-of-youtube-growth-subscribers-2022-7
- Rathore S, Schuler B, Park J. Life cycle assessment of multiple dispensing systems used for cosmetic product packaging. Packaging Technol Sci. 2023;36:533-547. doi:10.1002/pts.2729
- Shaw S. How to actually recycle your empty beauty products. CNN Underscored. Updated April 17, 2024. Accessed June 24, 2025. https://www.cnn.com/cnn-underscored/beauty/how-to-recycle-beauty-products
A Cross-Sectional Analysis of TikTok Skin Care Routines and the Associated Environmental Impact
A Cross-Sectional Analysis of TikTok Skin Care Routines and the Associated Environmental Impact
PRACTICE POINTS
- Social media platforms are increasingly influential in shaping consumer skin care habits, particularly among younger demographics.
- Dermatologists should be aware of the aesthetic-driven nature of online skin care trends when advising patients on product use.
- Viral skin care routines often feature multiple products and applicators, potentially encouraging excessive product use and waste.

From Refractory to Responsive: The Expanding Therapeutic Landscape of Prurigo Nodularis
From Refractory to Responsive: The Expanding Therapeutic Landscape of Prurigo Nodularis
Prurigo nodularis (PN) is a chronic, severely pruritic neuroimmunologic skin disorder characterized by multiple firm hyperkeratotic nodules and intense pruritus, often leading to considerable impairment in quality of life and increased rates of depression and anxiety.1 It is considered difficult to treat due to its complex pathogenesis, the severity and chronicity of pruritus, and the limited efficacy of conventional therapies.2,3 The disease is driven by a self-perpetuating itch-scratch cycle, underpinned by dysregulation of both immune and neural pathways including type 2 (interleukin [IL] 4, IL-13, IL-31), Th17, and Th22 cytokines as well as neuropeptides and altered cutaneous nerve architecture.1,3 This results in persistent severe pruritus and nodular lesions that are highly refractory to standard treatments.1 Conventional therapies (eg, locally acting agents, phototherapy, and systemic immunomodulators and neuromodulators) have varied efficacy and notable adverse effect profiles.3 While the approval of targeted biologics has transformed the therapeutic landscape, several other treatment options also are being explored in clinical trials. Herein, we review all recently approved therapies as well as emerging treatments currently under investigation.
Dupilumab
Dupilumab, the first therapy for PN approved by the US Food and Drug Administration (FDA) in 2022—is a monoclonal antibody that inhibits signaling of IL-4 and IL-13, key drivers of type 2 inflammation implicated in PN pathogenesis.4,5 In 2 pivotal phase 3 randomized controlled trials (LIBERTY-PN PRIME and PRIME2),5 dupilumab demonstrated notable efficacy in adults with moderate to severe PN. A reduction of 4 points or more on the Worst Itch Numeric Rating Scale (WI-NRS) was achieved by 60.0% (45/75) of patients treated with dupilumab at week 24 compared with 18.4% (14/76) receiving placebo in the PRIME trial. In PRIME2, the same outcome was achieved by 37.2% (29/78) of patients receiving dupilumab at week 12 compared with 22.0% (18/82) of patients receiving placebo.5 Dupilumab also led to a greater proportion of patients achieving a substantial reduction in nodule count (≤5 nodules) and improved quality of life compared with placebo.5,6 The safety profile of dupilumab for treatment of PN was favorable and consistent with prior experience in atopic dermatitis; conjunctivitis was the most common adverse event.5,6
Nemolizumab
Nemolizumab, an IL-31 receptor A antagonist, is the most recent agent approved by the FDA for PN in 2024.7 In the OLYMPIA 1 and OLYMPIA 2 phase 3 trials,8 nemolizumab produced a clinically meaningful reduction in itch (defined as a ≥4-point improvement in the Peak Pruritus Numerical Rating Scale score) in 56.3% (103/183) of patients at week 16 compared with 20.9% (19/91) receiving placebo. Additionally, 37.7% (69/183) of patients receiving nemolizumab achieved clear or almost clear skin (Investigator’s Global Assessment score of 0 or 1 with a ≥2-point reduction) vs 11.0% with placebo (both P<.001). Benefits were observed as early as week 4, including rapid improvements in itch, sleep disturbance, and nodule count.8 Nemolizumab also improved quality of life and reduced symptoms of anxiety and depression. The safety profile was favorable, with headache and atopic dermatitis the most common adverse events; serious adverse events were infrequent and similar between groups.8
Abrocitinib
Abrocitinib, an oral selective Janus kinase 1 inhibitor, is an investigational therapy for PN and currently has not been approved by the FDA for this indication. In a phase 2 open-label trial, abrocitinib 200 mg daily for 12 weeks led to a 78.3% reduction in weekly Peak Pruritus Numerical Rating Scale scores in PN, with 80.0% (8/10) of patients achieving a clinically meaningful improvement of 4 points or higher. Nodule counts and quality of life also improved, with an onset of itch relief as early as week 2. The safety profile was favorable, with acneform eruptions the most common adverse event and no serious adverse events reported9; however, these results are based on small, nonrandomized studies and require confirmation in larger randomized controlled trials before abrocitinib can be considered a standard therapy for PN.
Cryosim-1
Transient receptor potential melastatin 8 (TRPM8) is a cold-sensing ion channel found in unmyelinated sensory neurons within the dorsal root and trigeminal ganglia.10 It is activated by cool temperatures (15-28 °C) and compounds such as menthol, leading to calcium influx and a cooling sensation. In a randomized, double-blind, vehicle-controlled trial, researchers investigated the efficacy of cryosim-1 (a synthetic TRPM8 agonist) in treating PN.10 Thirty patients were enrolled, with 18 (60.0%) receiving cryosim-1 and 12 (40.0%) receiving placebo over 8 weeks. By week 8, cryosim-1 significantly reduced itch severity (mean numerical rating scale score postapplication, 2.8 vs 4.3; P=.031) and improved sleep disturbances (2.2 vs 4.2; P=.031) compared to placebo. Patients reported higher satisfaction with itch relief, and no adverse effects were observed. The study concluded that cryosim-1 is a safe, effective topical therapy for PN, likely working by interrupting the itch-scratch cycle and potentially modulating inflammatory pathways involved in chronic itch.10
Nalbuphine
Nalbuphine is a κ opioid receptor agonist and μ opioid receptor antagonist that has been investigated for the treatment of PN.11 In a phase 2 randomized controlled trial, oral nalbuphine extended release (NAL-ER) 162 mg twice daily provided measurable antipruritic efficacy, with 44.4% (8/18) of patients achieving at least a 30% reduction in 7-day WI-NRS at week 10 compared with 36.4% (8/22) in the placebo group. Among those who completed the study, 66.7% (8/12) of patients receiving NAL-ER 162 mg achieved significant itch reduction vs 40% (8/20) receiving placebo (P=.03). At least a 50% reduction in WI-NRS was achieved by 33.3% (6/18) of patients receiving NAL-ER 162 mg twice daily. Extended open-label treatment was associated with further improvements in itch and lesion activity. Adverse events were mostly mild to moderate (eg, nausea, dizziness, headache, and fatigue) and occurred during dose titration. Physiologic opioid withdrawal symptoms were limited and resolved within a few days of discontinuing the medication.11
Final Thoughts
In conclusion, PN remains one of the most challenging chronic dermatologic conditions to manage and is driven by a complex interplay of neuroimmune mechanisms and resistance to many conventional therapies. The approval of dupilumab and nemolizumab has marked a pivotal shift in the therapeutic landscape, offering hope to patients who previously had limited options5,8; however, the burden of PN remains substantial, and many patients continue to experience relentless itch, poor sleep, and reduced quality of life.1 Emerging therapies such as TRPM8 agonists, Janus kinase inhibitors, and opioid modulators represent promising additions to the treatment options, targeting novel pathways beyond traditional immunosuppression.9-11
- Williams KA, Huang AH, Belzberg M, et al. Prurigo nodularis: pathogenesis and management. J Am Acad Dermatol. 2020;83:1567-1575. doi:10.1016/j.jaad.2020.04.182
- Gründel S, Pereira MP, Storck M, et al. Analysis of 325 patients with chronic nodular prurigo: clinics, burden of disease and course of treatment. Acta Derm Venereol. 2020;100:adv00269. doi:10.2340/00015555-3571
- Liao V, Cornman HL, Ma E, et al. Prurigo nodularis: new insights into pathogenesis and novel therapeutics. Br J Dermatol. 2024;190:798-810. doi:10.1093/bjd/ljae052
- Elmariah SB, Tao L, Valdes-Rodriguez R, et al. Individual article: management of prurigo nodularis. J Drugs Dermatol. 2023;22:SF365502s15-SF365502s22. doi:10.36849/JDD.SF365502
- Yosipovitch G, Mollanazar N, Ständer S, et al. Dupilumab in patients with prurigo nodularis: two randomized, double-blind, placebo-controlled phase 3 trials. Nat Med. 2023;29:1180-1190. doi:10.1038/s41591-023-02320-9
- Cao P, Xu W, Jiang S, et al. Dupilumab for the treatment of prurigo nodularis: a systematic review. Front Immunol. 2023;14:1092685. doi:10.3389/fimmu.2023.1092685
- Dagenet CB, Saadi C, Phillips MA, et al. Landscape of prurigo nodularis clinical trials. JAAD Rev. 2024;2:127-136. doi:10.1016/j.jdrv.2024.09.006
- Kwatra SG, Yosipovitch G, Legat FJ, et al. Phase 3 trial of nemolizumab in patients with prurigo nodularis. N Engl J Med. 2023;389:1579-1589. doi:10.1056/NEJMoa2301333
- Kwatra SG, Bordeaux ZA, Parthasarathy V, et al. Efficacy and safety of abrocitinib in prurigo nodularis and chronic pruritus of unknown origin: a nonrandomized controlled trial. JAMA Dermatol. 2024;160:717-724. doi:10.1001/jamadermatol.2024.1464
- Choi ME, Lee JH, Jung CJ, et al. A randomized, double-blinded, vehicle-controlled clinical trial of topical cryosim-1, a synthetic TRPM8 agonist, in prurigo nodularis. J Cosmet Dermatol. 2024;23:931-937. doi:10.1111/jocd.16079
- Weisshaar E, Szepietowski JC, Bernhard JD, et al. Efficacy and safety of oral nalbuphine extended release in prurigo nodularis: results of a phase 2 randomized controlled trial with an open-label extension phase. J Eur Acad Dermatol Venereol. 2022;36:453-461. doi:10.1111/jdv.17816
Prurigo nodularis (PN) is a chronic, severely pruritic neuroimmunologic skin disorder characterized by multiple firm hyperkeratotic nodules and intense pruritus, often leading to considerable impairment in quality of life and increased rates of depression and anxiety.1 It is considered difficult to treat due to its complex pathogenesis, the severity and chronicity of pruritus, and the limited efficacy of conventional therapies.2,3 The disease is driven by a self-perpetuating itch-scratch cycle, underpinned by dysregulation of both immune and neural pathways including type 2 (interleukin [IL] 4, IL-13, IL-31), Th17, and Th22 cytokines as well as neuropeptides and altered cutaneous nerve architecture.1,3 This results in persistent severe pruritus and nodular lesions that are highly refractory to standard treatments.1 Conventional therapies (eg, locally acting agents, phototherapy, and systemic immunomodulators and neuromodulators) have varied efficacy and notable adverse effect profiles.3 While the approval of targeted biologics has transformed the therapeutic landscape, several other treatment options also are being explored in clinical trials. Herein, we review all recently approved therapies as well as emerging treatments currently under investigation.
Dupilumab
Dupilumab, the first therapy for PN approved by the US Food and Drug Administration (FDA) in 2022—is a monoclonal antibody that inhibits signaling of IL-4 and IL-13, key drivers of type 2 inflammation implicated in PN pathogenesis.4,5 In 2 pivotal phase 3 randomized controlled trials (LIBERTY-PN PRIME and PRIME2),5 dupilumab demonstrated notable efficacy in adults with moderate to severe PN. A reduction of 4 points or more on the Worst Itch Numeric Rating Scale (WI-NRS) was achieved by 60.0% (45/75) of patients treated with dupilumab at week 24 compared with 18.4% (14/76) receiving placebo in the PRIME trial. In PRIME2, the same outcome was achieved by 37.2% (29/78) of patients receiving dupilumab at week 12 compared with 22.0% (18/82) of patients receiving placebo.5 Dupilumab also led to a greater proportion of patients achieving a substantial reduction in nodule count (≤5 nodules) and improved quality of life compared with placebo.5,6 The safety profile of dupilumab for treatment of PN was favorable and consistent with prior experience in atopic dermatitis; conjunctivitis was the most common adverse event.5,6
Nemolizumab
Nemolizumab, an IL-31 receptor A antagonist, is the most recent agent approved by the FDA for PN in 2024.7 In the OLYMPIA 1 and OLYMPIA 2 phase 3 trials,8 nemolizumab produced a clinically meaningful reduction in itch (defined as a ≥4-point improvement in the Peak Pruritus Numerical Rating Scale score) in 56.3% (103/183) of patients at week 16 compared with 20.9% (19/91) receiving placebo. Additionally, 37.7% (69/183) of patients receiving nemolizumab achieved clear or almost clear skin (Investigator’s Global Assessment score of 0 or 1 with a ≥2-point reduction) vs 11.0% with placebo (both P<.001). Benefits were observed as early as week 4, including rapid improvements in itch, sleep disturbance, and nodule count.8 Nemolizumab also improved quality of life and reduced symptoms of anxiety and depression. The safety profile was favorable, with headache and atopic dermatitis the most common adverse events; serious adverse events were infrequent and similar between groups.8
Abrocitinib
Abrocitinib, an oral selective Janus kinase 1 inhibitor, is an investigational therapy for PN and currently has not been approved by the FDA for this indication. In a phase 2 open-label trial, abrocitinib 200 mg daily for 12 weeks led to a 78.3% reduction in weekly Peak Pruritus Numerical Rating Scale scores in PN, with 80.0% (8/10) of patients achieving a clinically meaningful improvement of 4 points or higher. Nodule counts and quality of life also improved, with an onset of itch relief as early as week 2. The safety profile was favorable, with acneform eruptions the most common adverse event and no serious adverse events reported9; however, these results are based on small, nonrandomized studies and require confirmation in larger randomized controlled trials before abrocitinib can be considered a standard therapy for PN.
Cryosim-1
Transient receptor potential melastatin 8 (TRPM8) is a cold-sensing ion channel found in unmyelinated sensory neurons within the dorsal root and trigeminal ganglia.10 It is activated by cool temperatures (15-28 °C) and compounds such as menthol, leading to calcium influx and a cooling sensation. In a randomized, double-blind, vehicle-controlled trial, researchers investigated the efficacy of cryosim-1 (a synthetic TRPM8 agonist) in treating PN.10 Thirty patients were enrolled, with 18 (60.0%) receiving cryosim-1 and 12 (40.0%) receiving placebo over 8 weeks. By week 8, cryosim-1 significantly reduced itch severity (mean numerical rating scale score postapplication, 2.8 vs 4.3; P=.031) and improved sleep disturbances (2.2 vs 4.2; P=.031) compared to placebo. Patients reported higher satisfaction with itch relief, and no adverse effects were observed. The study concluded that cryosim-1 is a safe, effective topical therapy for PN, likely working by interrupting the itch-scratch cycle and potentially modulating inflammatory pathways involved in chronic itch.10
Nalbuphine
Nalbuphine is a κ opioid receptor agonist and μ opioid receptor antagonist that has been investigated for the treatment of PN.11 In a phase 2 randomized controlled trial, oral nalbuphine extended release (NAL-ER) 162 mg twice daily provided measurable antipruritic efficacy, with 44.4% (8/18) of patients achieving at least a 30% reduction in 7-day WI-NRS at week 10 compared with 36.4% (8/22) in the placebo group. Among those who completed the study, 66.7% (8/12) of patients receiving NAL-ER 162 mg achieved significant itch reduction vs 40% (8/20) receiving placebo (P=.03). At least a 50% reduction in WI-NRS was achieved by 33.3% (6/18) of patients receiving NAL-ER 162 mg twice daily. Extended open-label treatment was associated with further improvements in itch and lesion activity. Adverse events were mostly mild to moderate (eg, nausea, dizziness, headache, and fatigue) and occurred during dose titration. Physiologic opioid withdrawal symptoms were limited and resolved within a few days of discontinuing the medication.11
Final Thoughts
In conclusion, PN remains one of the most challenging chronic dermatologic conditions to manage and is driven by a complex interplay of neuroimmune mechanisms and resistance to many conventional therapies. The approval of dupilumab and nemolizumab has marked a pivotal shift in the therapeutic landscape, offering hope to patients who previously had limited options5,8; however, the burden of PN remains substantial, and many patients continue to experience relentless itch, poor sleep, and reduced quality of life.1 Emerging therapies such as TRPM8 agonists, Janus kinase inhibitors, and opioid modulators represent promising additions to the treatment options, targeting novel pathways beyond traditional immunosuppression.9-11
Prurigo nodularis (PN) is a chronic, severely pruritic neuroimmunologic skin disorder characterized by multiple firm hyperkeratotic nodules and intense pruritus, often leading to considerable impairment in quality of life and increased rates of depression and anxiety.1 It is considered difficult to treat due to its complex pathogenesis, the severity and chronicity of pruritus, and the limited efficacy of conventional therapies.2,3 The disease is driven by a self-perpetuating itch-scratch cycle, underpinned by dysregulation of both immune and neural pathways including type 2 (interleukin [IL] 4, IL-13, IL-31), Th17, and Th22 cytokines as well as neuropeptides and altered cutaneous nerve architecture.1,3 This results in persistent severe pruritus and nodular lesions that are highly refractory to standard treatments.1 Conventional therapies (eg, locally acting agents, phototherapy, and systemic immunomodulators and neuromodulators) have varied efficacy and notable adverse effect profiles.3 While the approval of targeted biologics has transformed the therapeutic landscape, several other treatment options also are being explored in clinical trials. Herein, we review all recently approved therapies as well as emerging treatments currently under investigation.
Dupilumab
Dupilumab, the first therapy for PN approved by the US Food and Drug Administration (FDA) in 2022—is a monoclonal antibody that inhibits signaling of IL-4 and IL-13, key drivers of type 2 inflammation implicated in PN pathogenesis.4,5 In 2 pivotal phase 3 randomized controlled trials (LIBERTY-PN PRIME and PRIME2),5 dupilumab demonstrated notable efficacy in adults with moderate to severe PN. A reduction of 4 points or more on the Worst Itch Numeric Rating Scale (WI-NRS) was achieved by 60.0% (45/75) of patients treated with dupilumab at week 24 compared with 18.4% (14/76) receiving placebo in the PRIME trial. In PRIME2, the same outcome was achieved by 37.2% (29/78) of patients receiving dupilumab at week 12 compared with 22.0% (18/82) of patients receiving placebo.5 Dupilumab also led to a greater proportion of patients achieving a substantial reduction in nodule count (≤5 nodules) and improved quality of life compared with placebo.5,6 The safety profile of dupilumab for treatment of PN was favorable and consistent with prior experience in atopic dermatitis; conjunctivitis was the most common adverse event.5,6
Nemolizumab
Nemolizumab, an IL-31 receptor A antagonist, is the most recent agent approved by the FDA for PN in 2024.7 In the OLYMPIA 1 and OLYMPIA 2 phase 3 trials,8 nemolizumab produced a clinically meaningful reduction in itch (defined as a ≥4-point improvement in the Peak Pruritus Numerical Rating Scale score) in 56.3% (103/183) of patients at week 16 compared with 20.9% (19/91) receiving placebo. Additionally, 37.7% (69/183) of patients receiving nemolizumab achieved clear or almost clear skin (Investigator’s Global Assessment score of 0 or 1 with a ≥2-point reduction) vs 11.0% with placebo (both P<.001). Benefits were observed as early as week 4, including rapid improvements in itch, sleep disturbance, and nodule count.8 Nemolizumab also improved quality of life and reduced symptoms of anxiety and depression. The safety profile was favorable, with headache and atopic dermatitis the most common adverse events; serious adverse events were infrequent and similar between groups.8
Abrocitinib
Abrocitinib, an oral selective Janus kinase 1 inhibitor, is an investigational therapy for PN and currently has not been approved by the FDA for this indication. In a phase 2 open-label trial, abrocitinib 200 mg daily for 12 weeks led to a 78.3% reduction in weekly Peak Pruritus Numerical Rating Scale scores in PN, with 80.0% (8/10) of patients achieving a clinically meaningful improvement of 4 points or higher. Nodule counts and quality of life also improved, with an onset of itch relief as early as week 2. The safety profile was favorable, with acneform eruptions the most common adverse event and no serious adverse events reported9; however, these results are based on small, nonrandomized studies and require confirmation in larger randomized controlled trials before abrocitinib can be considered a standard therapy for PN.
Cryosim-1
Transient receptor potential melastatin 8 (TRPM8) is a cold-sensing ion channel found in unmyelinated sensory neurons within the dorsal root and trigeminal ganglia.10 It is activated by cool temperatures (15-28 °C) and compounds such as menthol, leading to calcium influx and a cooling sensation. In a randomized, double-blind, vehicle-controlled trial, researchers investigated the efficacy of cryosim-1 (a synthetic TRPM8 agonist) in treating PN.10 Thirty patients were enrolled, with 18 (60.0%) receiving cryosim-1 and 12 (40.0%) receiving placebo over 8 weeks. By week 8, cryosim-1 significantly reduced itch severity (mean numerical rating scale score postapplication, 2.8 vs 4.3; P=.031) and improved sleep disturbances (2.2 vs 4.2; P=.031) compared to placebo. Patients reported higher satisfaction with itch relief, and no adverse effects were observed. The study concluded that cryosim-1 is a safe, effective topical therapy for PN, likely working by interrupting the itch-scratch cycle and potentially modulating inflammatory pathways involved in chronic itch.10
Nalbuphine
Nalbuphine is a κ opioid receptor agonist and μ opioid receptor antagonist that has been investigated for the treatment of PN.11 In a phase 2 randomized controlled trial, oral nalbuphine extended release (NAL-ER) 162 mg twice daily provided measurable antipruritic efficacy, with 44.4% (8/18) of patients achieving at least a 30% reduction in 7-day WI-NRS at week 10 compared with 36.4% (8/22) in the placebo group. Among those who completed the study, 66.7% (8/12) of patients receiving NAL-ER 162 mg achieved significant itch reduction vs 40% (8/20) receiving placebo (P=.03). At least a 50% reduction in WI-NRS was achieved by 33.3% (6/18) of patients receiving NAL-ER 162 mg twice daily. Extended open-label treatment was associated with further improvements in itch and lesion activity. Adverse events were mostly mild to moderate (eg, nausea, dizziness, headache, and fatigue) and occurred during dose titration. Physiologic opioid withdrawal symptoms were limited and resolved within a few days of discontinuing the medication.11
Final Thoughts
In conclusion, PN remains one of the most challenging chronic dermatologic conditions to manage and is driven by a complex interplay of neuroimmune mechanisms and resistance to many conventional therapies. The approval of dupilumab and nemolizumab has marked a pivotal shift in the therapeutic landscape, offering hope to patients who previously had limited options5,8; however, the burden of PN remains substantial, and many patients continue to experience relentless itch, poor sleep, and reduced quality of life.1 Emerging therapies such as TRPM8 agonists, Janus kinase inhibitors, and opioid modulators represent promising additions to the treatment options, targeting novel pathways beyond traditional immunosuppression.9-11
- Williams KA, Huang AH, Belzberg M, et al. Prurigo nodularis: pathogenesis and management. J Am Acad Dermatol. 2020;83:1567-1575. doi:10.1016/j.jaad.2020.04.182
- Gründel S, Pereira MP, Storck M, et al. Analysis of 325 patients with chronic nodular prurigo: clinics, burden of disease and course of treatment. Acta Derm Venereol. 2020;100:adv00269. doi:10.2340/00015555-3571
- Liao V, Cornman HL, Ma E, et al. Prurigo nodularis: new insights into pathogenesis and novel therapeutics. Br J Dermatol. 2024;190:798-810. doi:10.1093/bjd/ljae052
- Elmariah SB, Tao L, Valdes-Rodriguez R, et al. Individual article: management of prurigo nodularis. J Drugs Dermatol. 2023;22:SF365502s15-SF365502s22. doi:10.36849/JDD.SF365502
- Yosipovitch G, Mollanazar N, Ständer S, et al. Dupilumab in patients with prurigo nodularis: two randomized, double-blind, placebo-controlled phase 3 trials. Nat Med. 2023;29:1180-1190. doi:10.1038/s41591-023-02320-9
- Cao P, Xu W, Jiang S, et al. Dupilumab for the treatment of prurigo nodularis: a systematic review. Front Immunol. 2023;14:1092685. doi:10.3389/fimmu.2023.1092685
- Dagenet CB, Saadi C, Phillips MA, et al. Landscape of prurigo nodularis clinical trials. JAAD Rev. 2024;2:127-136. doi:10.1016/j.jdrv.2024.09.006
- Kwatra SG, Yosipovitch G, Legat FJ, et al. Phase 3 trial of nemolizumab in patients with prurigo nodularis. N Engl J Med. 2023;389:1579-1589. doi:10.1056/NEJMoa2301333
- Kwatra SG, Bordeaux ZA, Parthasarathy V, et al. Efficacy and safety of abrocitinib in prurigo nodularis and chronic pruritus of unknown origin: a nonrandomized controlled trial. JAMA Dermatol. 2024;160:717-724. doi:10.1001/jamadermatol.2024.1464
- Choi ME, Lee JH, Jung CJ, et al. A randomized, double-blinded, vehicle-controlled clinical trial of topical cryosim-1, a synthetic TRPM8 agonist, in prurigo nodularis. J Cosmet Dermatol. 2024;23:931-937. doi:10.1111/jocd.16079
- Weisshaar E, Szepietowski JC, Bernhard JD, et al. Efficacy and safety of oral nalbuphine extended release in prurigo nodularis: results of a phase 2 randomized controlled trial with an open-label extension phase. J Eur Acad Dermatol Venereol. 2022;36:453-461. doi:10.1111/jdv.17816
- Williams KA, Huang AH, Belzberg M, et al. Prurigo nodularis: pathogenesis and management. J Am Acad Dermatol. 2020;83:1567-1575. doi:10.1016/j.jaad.2020.04.182
- Gründel S, Pereira MP, Storck M, et al. Analysis of 325 patients with chronic nodular prurigo: clinics, burden of disease and course of treatment. Acta Derm Venereol. 2020;100:adv00269. doi:10.2340/00015555-3571
- Liao V, Cornman HL, Ma E, et al. Prurigo nodularis: new insights into pathogenesis and novel therapeutics. Br J Dermatol. 2024;190:798-810. doi:10.1093/bjd/ljae052
- Elmariah SB, Tao L, Valdes-Rodriguez R, et al. Individual article: management of prurigo nodularis. J Drugs Dermatol. 2023;22:SF365502s15-SF365502s22. doi:10.36849/JDD.SF365502
- Yosipovitch G, Mollanazar N, Ständer S, et al. Dupilumab in patients with prurigo nodularis: two randomized, double-blind, placebo-controlled phase 3 trials. Nat Med. 2023;29:1180-1190. doi:10.1038/s41591-023-02320-9
- Cao P, Xu W, Jiang S, et al. Dupilumab for the treatment of prurigo nodularis: a systematic review. Front Immunol. 2023;14:1092685. doi:10.3389/fimmu.2023.1092685
- Dagenet CB, Saadi C, Phillips MA, et al. Landscape of prurigo nodularis clinical trials. JAAD Rev. 2024;2:127-136. doi:10.1016/j.jdrv.2024.09.006
- Kwatra SG, Yosipovitch G, Legat FJ, et al. Phase 3 trial of nemolizumab in patients with prurigo nodularis. N Engl J Med. 2023;389:1579-1589. doi:10.1056/NEJMoa2301333
- Kwatra SG, Bordeaux ZA, Parthasarathy V, et al. Efficacy and safety of abrocitinib in prurigo nodularis and chronic pruritus of unknown origin: a nonrandomized controlled trial. JAMA Dermatol. 2024;160:717-724. doi:10.1001/jamadermatol.2024.1464
- Choi ME, Lee JH, Jung CJ, et al. A randomized, double-blinded, vehicle-controlled clinical trial of topical cryosim-1, a synthetic TRPM8 agonist, in prurigo nodularis. J Cosmet Dermatol. 2024;23:931-937. doi:10.1111/jocd.16079
- Weisshaar E, Szepietowski JC, Bernhard JD, et al. Efficacy and safety of oral nalbuphine extended release in prurigo nodularis: results of a phase 2 randomized controlled trial with an open-label extension phase. J Eur Acad Dermatol Venereol. 2022;36:453-461. doi:10.1111/jdv.17816
From Refractory to Responsive: The Expanding Therapeutic Landscape of Prurigo Nodularis
From Refractory to Responsive: The Expanding Therapeutic Landscape of Prurigo Nodularis
Type VII Collagen Disorders Simplified
Type VII Collagen Disorders Simplified
There are 3 uncommon types of mechanobullous skin diseases caused by relative reduction or complete loss of functional type VII collagen, which is the main component of anchoring fibrils in the lamina densa of the basement membrane zone (BMZ) of the skin and mucous membrane epithelium.1 The function of the anchoring fibrils is to maintain adherence of the basement membrane of the epithelium to the connective tissue of the papillary dermis and submucosa.1 The mechanism of action of the loss of type VII collagen function is via autoimmunity in epidermolysis bullosa acquisita (EBA)2 and
Epidermolysis Bullosa
Epidermolysis bullosa consists of a heterogeneous family of 4 major genetic mechanobullous diseases that affect the skin and mucous membranes with more than 30 subtypes.1 Dystrophic EB is caused by mutations in the COL7A1 gene, which encodes for the α-1 chain of collagen type VII. Classically, EB is divided into 4 main variants based on the location of the cleavage plane or split occurring in the epithelium, which in turn helps to predict the severity of the illness.
Epidermolysis bullosa may be inherited in an autosomal-dominant or autosomal-recessive fashion, or it may occur as a spontaneous mutation. All sexes and races are affected equally. Patients present at birth or in early childhood with fragile skin and mucous membranes that may develop blisters, erosions, and ulcerations after minor trauma.7 These lesions are marked by slow healing and scar formation and often are associated with itching and pain.
Dystrophic Epidermolysis Bullosa
Dystrophic EB accounts for approximately 25%6 of all EB cases in the United States and may be inherited as either a dominant or recessive trait. Hundreds of different pathogenic mutations have been discovered in the COL7A1 gene in the subtypes of DEB.4,8 Dominant DEB tends to cause milder disease because the patients retain one normal COL7A1 allele and produce some type VII collagen (Figure 1), whereas patients with recessive DEB lack type VII collagen completely.9 The cleavage plane is between the lamina densa and the superficial dermis or submucosa. Severity is variable and ranges from localization to the hands and feet to severe generalized blistering and painful ulcerations depending on which of the many possible gene mutations have been inherited. Sequelae include mitten deformities, malalignment and tooth decay, and the development of early aggressive squamous cell carcinomas, which may be fatal. The most severe cases of recessive DEB also may have internal organ involvement.
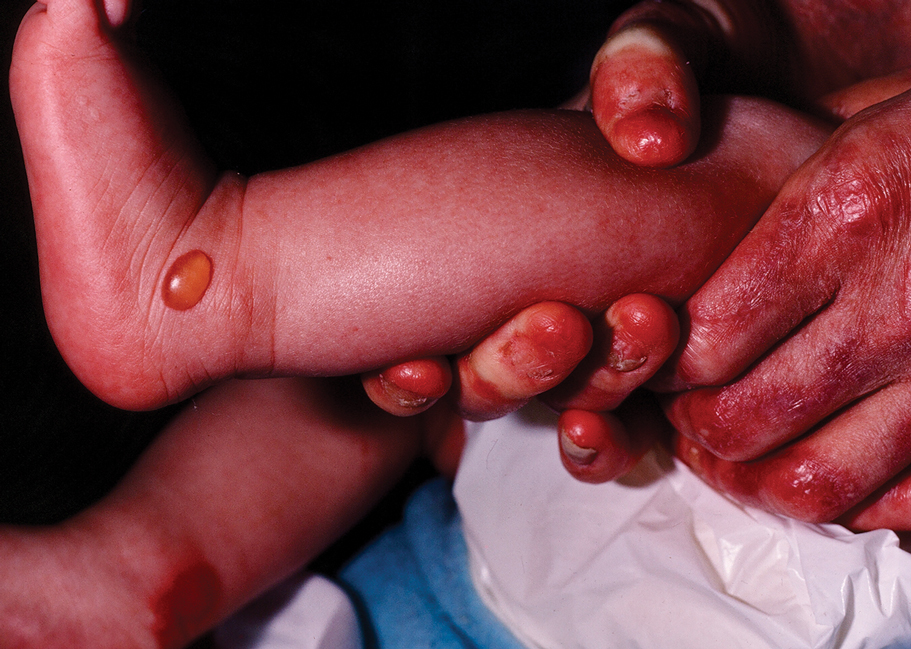
Epidermolysis Bullosa Simplex
Epidermolysis bullosa simplex is the most common variant, comprising approximately 70%of EB cases in the United States.6 Epidermolysis bullosa simplex usually is inherited as autosomal-dominant mutations in the keratin 5 or keratin 14 genes,10 not COL7A1. Skin blistering results from cleavage within the basal cell layer where the keratin genes are primarily expressed. Blisters tend to occur in acral areas such as hands and feet and may heal without scarring in the localized form of epidermolysis bullosa simplex (Figure 2).
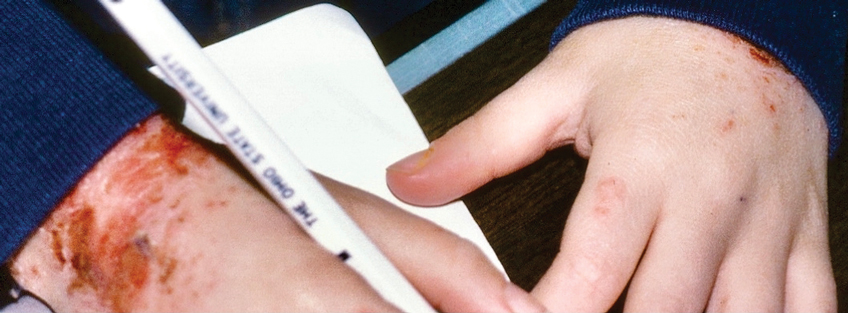
Junctional Epidermolysis Bullosa and Kindler Syndrome
Junctional epidermolysis bullosa (JEB) and Kindler syndrome11 are the rarest of the autosomal-recessive EB variants.6 The plane of cleavage in JEB is through the lamina lucida of the BMZ. Junctional epidermolysis bullosa is caused by mutations of the genes that encode for the 3 chains of laminin 332 protein and type XVII collagen,5,12 not to be confused with type VII collagen. As with DEB, there is a wide range of severity in JEB, from localized effects on the eyes, oral cavity, and tooth enamel to widespread blistering and skin cancers. In JEB cases involving newborns, nonhealing wounds on the face, buttocks, fingers, and toes may be seen, with devastating complications in the oral cavity, esophagus, and larynx. Life expectancy is limited to 2 years or less.6 There have only been approximately 40013 cases of Kindler syndrome reported worldwide6 and there is clinical overlap with DEB. Patients also may demonstrate poikiloderma and photosensitivity. Kindler syndrome is caused by mutations in the FERMT1 gene which encodes for kindlin-1. This protein mediates anchorage between the actin cytoskeleton and the extracellular matrix.5,11 Loss of function produces variable cleavage planes around the dermoepidermal junction.
Clinical management of all EB variants, especially the severe recessive types, traditionally has been limited to the prevention of trauma to the skin and mucous membranes and supportive care, including dressing changes to erosions and ulcerations, antibiotic ointments as needed, and amelioration of pain and pruritus. Bone marrow and pluripotential stem cell transplants have been attempted.12 Complications of EB, such as deformities of the hands and feet caused by excessive scarring, esophageal strictures, poor dentition, and squamous cell carcinomas, must be addressed by a multidisciplinary team of specialists, including plastic surgery, gastroenterology, dentistry/oral surgery, ophthalmology, and dermatology/Mohs surgery.
Until recently, there were no medications approved by the US Food and Drug Administration (FDA) specifically indicated for EB. In 2023, topical gene therapy was approved by the FDA for both recessive and dominant forms of DEB. Normal COL7A1 sequences are delivered by an attenuated herpes simplex virus 1 vector (beremagene geperpavec) in a gel applied directly to the wounds of patients with DEB. In a clinical trial, matching wounds on 31 patients (62 wounds total) were treated with the active agent or placebo gel. After 6 months, complete wound closure was observed in 67% (21/31) of those treated with the active agent and 22% (7/31) of those treated with placebo (P=.002).14 In a single case report, a patient with recessive DEB and cicatrizing conjunctivitis (Figure 3) was given ophthalmic beremagene geperpavec after surgery and had improved visual acuity.15 A topical gel consisting of birch triterpenes to promote healing of partial-thickness wounds also was approved for patients with DEB and JEB by the FDA and the European Commission. In a study of 223 patients, 41% of those using active gel and 29% of those using placebo gel achieved the primary end point of percentage of target wounds that had first complete closure at 45 days.16
The most recent FDA approval for DEB involves transferring the functional COL7A1 gene to the patient’s skin cells, then expanding the gene-corrected cells into sheets of keratinocytes that can be surgically applied to the chronic wound sites. In a phase 3 trial of prademagene zamikeracel (pz-cel), 11 patients with 86 matched wounds were randomized to receive pz-cel (50%) or standard wound care (50%). After 24 weeks, 35 wounds treated with pz-cel were at least 50% healed compared to 7 control wounds.17 The results for healing and reduction of pain were statistically significant (P<.0001 and P<.0002, respectively).17 Recombinant collagen VII as replacement therapy also is under study to be given by intravenous infusion to increase tissue collagen VII where it is lacking. This treatment has shown early biologic and therapeutic effects.9,18 Larger long-term follow-up studies are necessary to confirm persistence of the gene-corrected skin cells, the functionality of the replacement collagen VII, and the potential risk for the development of autoantibodies to type VII collagen.
Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita is a rare autoimmune subepithelial bullous disease that primarily affects middle-aged adults but also has been reported in children.19 Epidermolysis bullosa acquisita is caused by circulating pathogenic IgG autoantibodies that target and bind to type VII collagen in the anchoring fibrils,20-22 thereby disrupting the attachment of the epithelium to its underlying connective tissue.
The 2 major clinical manifestations of EBA include a mechanobullous disease resembling inherited forms of DEB (Figure 4) and an inflammatory bullous pemphigoid (BP)–like disease,23 as well as a combination of both types of skin lesions (Figure 5). The skin and mucous membranes of the oral cavity, esophagus, eyes, and urogenital areas are affected in both types; scarring may cause functional disabilities. In the mechanobullous type of EBA, it is common for blisters and erosions to develop in trauma-prone areas such as the hands, feet, elbows, and knees. The blisters tend to heal with scarring and milia formation as might be seen in porphyria cutanea tarda or cicatricial pemphigoid, which are in the differential diagnosis. Dystrophy of the fingernails or complete nail loss may be observed, resembling DEB. In the BP-like presentation, tense blisters arise upon inflamed or urticarial skin and mucous membranes, which may then become generalized.
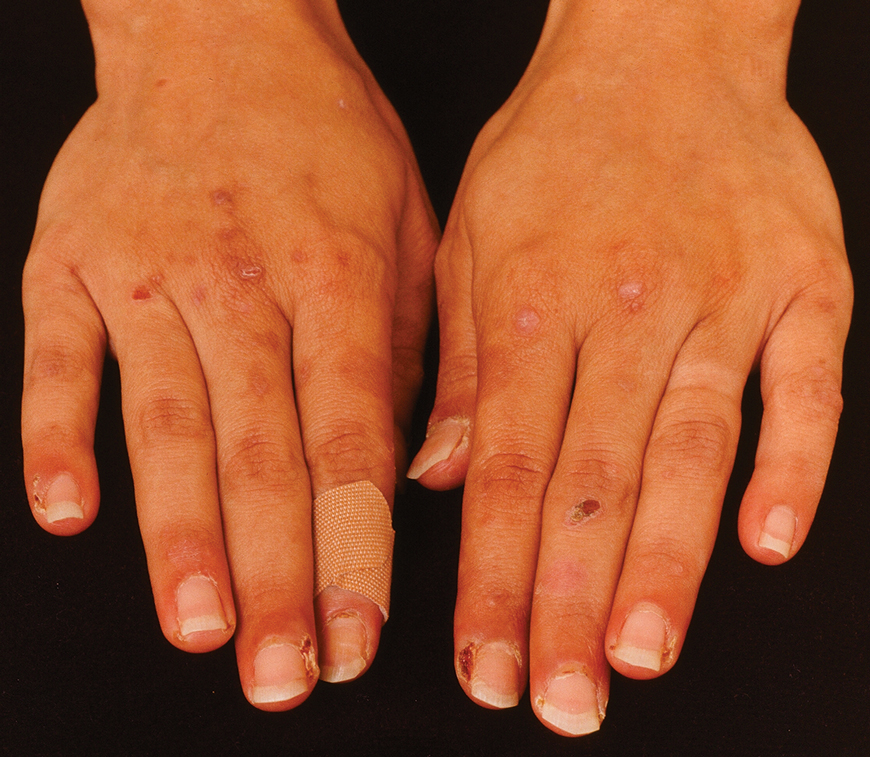
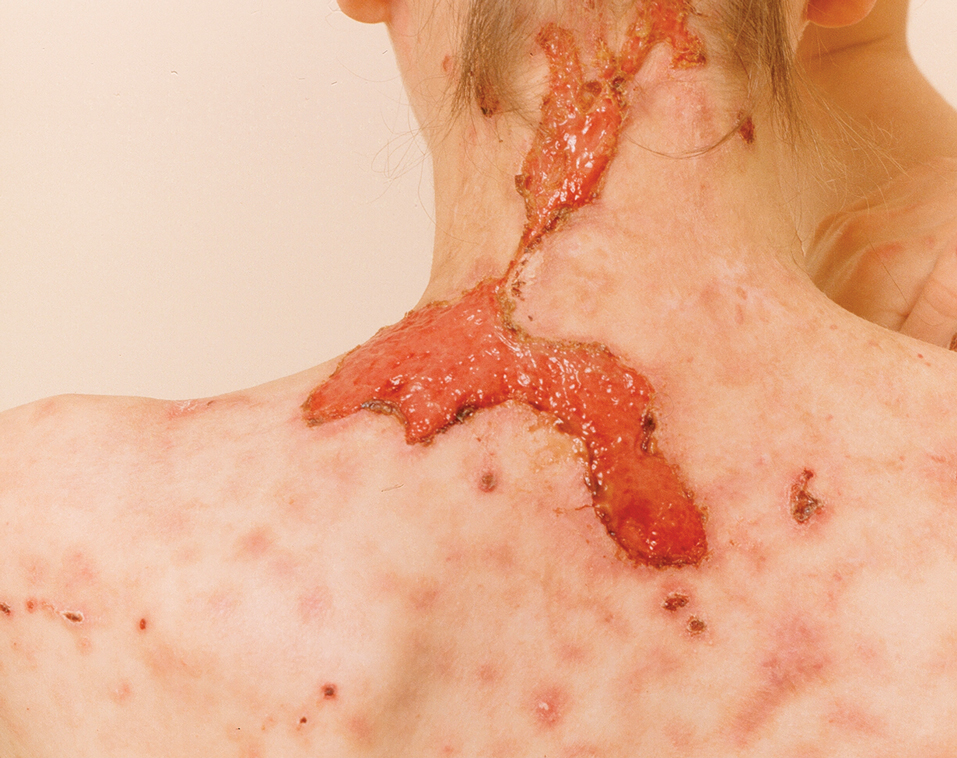
Histopathology in both forms of EBA demonstrates subepithelial separation as clefts or blisters. The mechanobullous type shows a sparse inflammatory infiltrate compared to large collections of neutrophils and eosinophils in the blister cavity and in the superficial dermis in the BP-like cases. The final diagnosis rests on the results of immunopathology testing.24 Direct immunofluorescence of perilesional skin and mucosa shows a linear-granular band of IgG and C3 and other conjugates along the BMZ. Deposits of IgA alone in EBA occur in only about 2.4% of cases and are observed more often when there is mucous membrane involvement.2 Indirect immunofluorescence of sera against salt-split skin substrates detects immunoreactants in the floor of the blister rather than in the roof, as would be seen in BP. Highly specific and sensitive enzyme-linked immunosorbent assay (ELISA) kits now are commercially available and can detect autoantibodies against the N-terminal domain of type VII collagen in more than 90% of cases of EBA.25
Inflammatory bowel disease (IBD), particularly Crohn disease (CD), precedes the onset of EBA in approximately 25% of cases.26,27 Ulcerative colitis is much less common. Type VII collagen is normally present in the basement membrane of intestinal epithelium. In a survey of patients with IBD, 68% of those with CD and 13% of those with ulcerative colitis had circulating anti–type VII collagen antibodies detected by ELISA without having symptoms of EBA.28 A case report of a patient with both well-proven EBA and CD highlighted the clinical difficulty of controlling EBA: treatment with prednisolone and sulfasalazine improved the CD but had little effect on the skin blisters.29 A variety of malignancies have been reported in association with EBA, including cancers of the uterine cervix,30 thyroid, and pancreas,31 lymphoma, and chronic lymphatic leukemia. Some of these cases have met the criteria for classification as paraneoplastic, whereas others may have been coincidental.
Treatment for chronic EBA generally has been limited.2,24 Putative antineutrophil drugs such as dapsone and colchicine combined with systemic corticosteroids may be useful in milder or juvenile cases, which tend to have a better prognosis than adult cases.19 In more severe EBA, systemic corticosteroids and/or immunosuppressive drugs such as azathioprine,23 cyclophosphamide,23 mycophenolate mofetil,31 methotrexate,23 cyclosporine,33 and infliximab23 have been used. More recently, rituximab infusion monotherapy33 and rituximab combined with intravenous immunoglobulin or
Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus is a rare and specific autoimmune skin complication that mostly is seen in patients with an established diagnosis of systemic lupus erythematosus (SLE) who are experiencing a disease flare. Although more common in women, it has been reported in all sexes and races as well as in children. Vesicles and bullae may arise on sun-exposed (Figure 6) and sun-protected areas of skin.
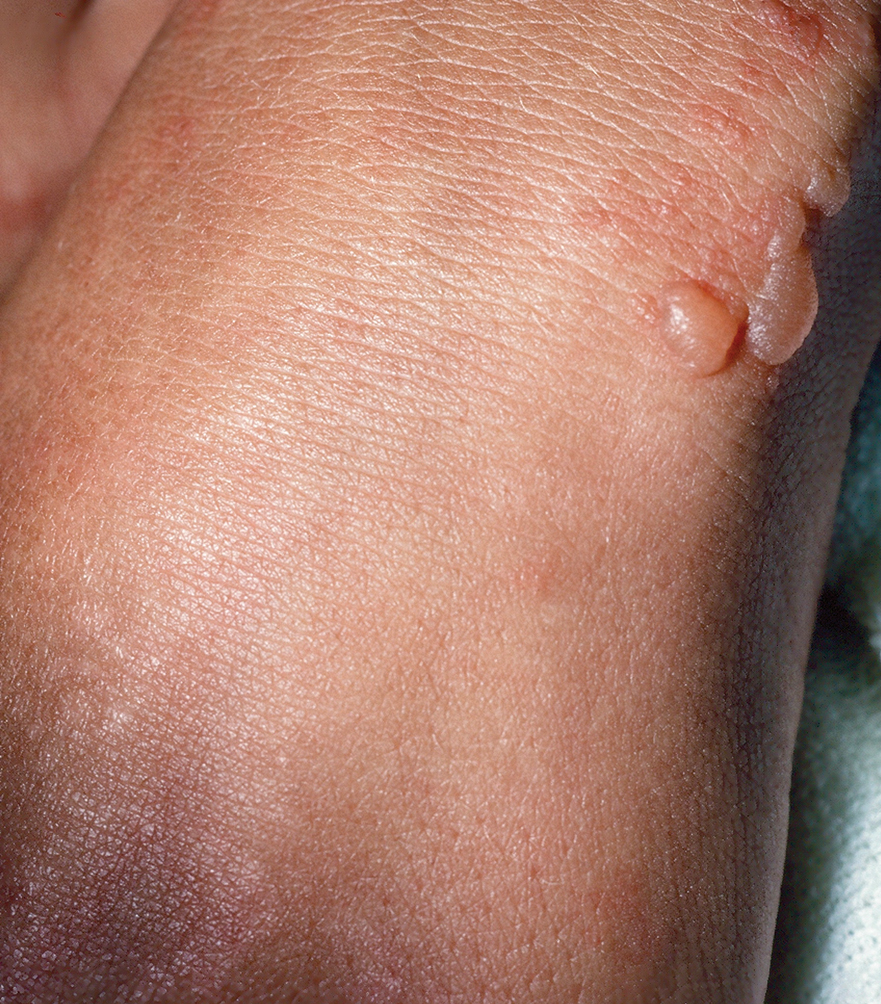
Histopathology shows subepidermal separation with collections of neutrophils and nuclear fragments in the blister cavity. The differential diagnosis of BSLE includes EBA, BP, dermatitis herpetiformis, and linear IgA bullous dermatosis. Direct immunofluorescence testing shows linear-granular deposits of IgG and/or IgM and IgA along the BMZ.34 When utilizing the indirect immunofluorescence split-skin assay, the autoantibody to type VII collagen would be detected in the floor of the blister if the serum titer was sufficiently high.3 Proposed criteria for the diagnosis of BSLE have been published: 1) diagnosis of SLE now based on the 2019 European League Against Rheumatism/American College of Rheumatology classification35; 2) vesicles and bullae arising upon but not limited to sun-exposed skin; 3) histopathology featuring neutrophil-rich subepithelial bullae; 4) positive indirect immunofluorescence for circulating BMZ antibodies using separated human skin as substrate; 5) and direct immunofluorescence showing IgG and/or IgM and often IgA at the BMZ.36 Using ELISA to detect circulating antibodies against type VII collagen24 should now be added to the criteria. The new criteria for SLE34 do not include BSLE, perhaps because it occurs in less than 1% of patients with SLE.37
Further investigation by Gammon et al3 confirmed that the autoantibodies in BSLE are identical to those found in EBA (ie, directed against type VII collagen in the lamina densa). Bullous systemic lupus erythematosus is not considered to be the coexistence of EBA with SLE but rather a specific entity wherein type VII collagen autoantibodies are expressed in the autoimmune spectrum of SLE. It is especially important to make the diagnosis of BSLE because it is predictive of more serious systemic complications of SLE (eg, hematologic and renal disease is found in up to 90% of cases).38
The natural course of BSLE is variable. Treatments include systemic corticosteroids, dapsone, and immunosuppressive drugs such as azathioprine, methotrexate, mycophenolate mofetil, and cyclophosphamide, especially in cases with nephritis.37 There may be spontaneous resolution of the rash as the inflammatory activity of SLE subsides. Rituximab has been used effectively in several refractory cases of BSLE that failed to respond to all other conventional treatments.39
Conclusion
Anchoring fibrils are composed primarily of type VII collagen. Their role is to maintain the attachment of epithelium to the upper dermis and submucosa. The reduction or complete loss of type VII collagen caused by mutations of the COL7A1 gene results in dominant DEB or recessive DEB, respectively. Two distinct non-heritable immunobullous diseases, EBA and BSLE, are caused by autoantibodies that target type VII collagen. A comparison of the 4 type VII collagen disorders can be found in the eTable.
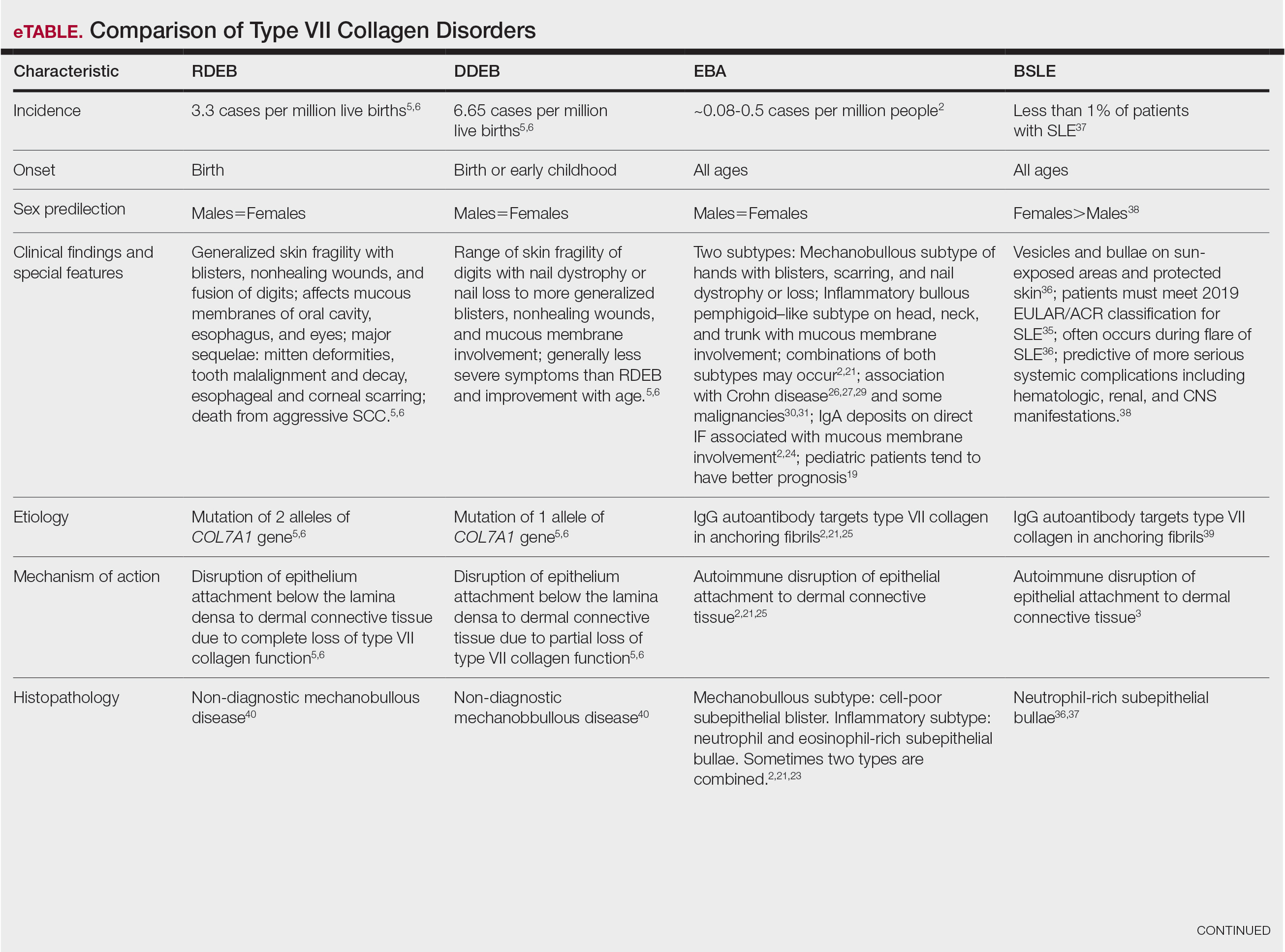
- Bardhan A, Bruckner-Tuderman L, Chapple ILC, et al. Epidermolysis bullosa. Nat Rev Dis Primers. 2020;6:78. doi:10.1038/s41572-020-0210-0
- Miyamoto D, Gordilho JO, Santi CG, et al. Epidermolysis bullosa acquisita. An Bras Dermatol. 2022;97:409-423. doi:10.1016/j.abd.2021.09.010.
- Gammon WR, Woodley DT, Dole KC, et al. Evidence that anti-basement membrane zone antibodies in bullous eruption of systemic lupus erythematosus recognize epidermolysis bullosa acquisita autoantigen. J Invest Dermatol. 1985;84:472-476. doi:10.1111/1523-1747.ep12272402.
- Yadav RS, Jaswal A, Shrestha S, et al. Dystrophic epidermolysis bullosa. J Nepal Med Assoc. 2018;56:879-882. doi:10.31729/jnma.3791
- Mariath LM, Santin JT, Schuler-Faccini L, et al. Inherited epidermolysis bullosa: update on the clinical and genetic aspects. An Bras Dermatol. 2020;95:551-569. doi:10.1016/j.abd.2020.05.001
- Understanding epidermolysis bullosa (EB). DEBRA website. Accessed August 17, 2025. https://www.debra.org/about-eb/understanding-epidermolysis-bullosa-eb
- Hon KL, Chu S, Leung AKC. Epidermolysis bullosa: pediatric perspectives. Curr Pediatr Rev. 2022;18:182-190. doi:10.2174/1573396317666210525161252
- Dang N, Klingberg S, Marr P, et al. Review of collagen VII sequence variants found in Australasian patients with dystrophic epidermolysis bullosa reveals nine COL7A1 variants. J Dermatol Sci. 2007;46:169-178. doi:10.1016/j.jdermsci.2007.02.006
- Payne AS. Topical gene therapy for epidermolysis bullosa. N Engl J Med. 2022;387:2281-2284. doi:10.1056/NEJMe2213203
- Khani P, Ghazi F, Zekri A, et al. Keratins and epidermolysis bullosa simplex. J Cell Physiol. 2018;234:289-297. doi:10.1002/jcp.26898
- Lai-Cheong JE, Tanaka A, Hawche G, et al. Kindler syndrome: a focal adhesion genodermatosis. Br J Dermatol. 2009;160:233-242. doi:10.1111/j.1365-2133.2008.08976.x
- Hou P-C, Wang H-T, Abhee S, et al. Investigational treatments for epidermolysis bullosa. Am J Clin Dermatol. 2021;22:801-817. doi:10.1007/s40257-021-00626-3
- Youseffian L, Vahidnezhad H, Uitto J. Kindler Syndrome. GeneReviews [Internet]. Updated January 6, 2022. Accessed August 21, 2025.
- Guide SV, Gonzalez ME, Bagci S, et al. Trial of beremagene geperpavec (B-VEC) for dystrophic epidermolysis bullosa. N Engl J Med. 2022;387:2211-2219. doi:10.1056/NEJMoa2206663
- Vetencourt AT, Sayed-Ahmed I, Gomez J, et al. Ocular gene therapy in a patient with dystrophic epidermolysis bullosa. N Engl J Med. 2024;390:530-535. doi:10.1056/NEJMoa2301244
- Kern JS, Sprecher E, Fernandez MF, et al. Efficacy and safety of Oleogel-S10 (birch triterpenes for epidermolysis bullosa: results from the phase III randomized double-blind phase of the EASE study. Br J Dermatol. 2023;188:12-21. doi:10.1093/bjd/ljac001
- Tang JY, Marinkovich MP, Wiss K, et al. Prademagene zamikeracel for recessive dystrophic epidermolysis bullosa wounds (VIITAL): a two-centre, randomized, open-label, intrapatient-controlled phase 3 trial. Lancet. 2025;406:163-173. doi:10.1016/S0140-6736(25)00778-0
- Gretzmeier C, Pin D, Kern JS, et al. Systemic collagen VII replacement therapy for advanced recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2022;142:1094-1102. doi:10.1016/j.jid.2021.09.008
- Hignett E, Sami N. Pediatric epidermolysis bullosa acquisita. A review. Pediatr Dermatol. 2021;38:1047-1050. doi:10.1111/pde.14722
- Chen M, Kim GH, Prakash L, et al. Autoimmunity to anchoring fibril collagen. Autoimmunity. 2012;45:91-101. doi:10.1007/s12016-007-0027-6.
- Kridin K, Kneiber D, Kowalski EH, et al. Epidermolysis bullosa acquisita: a comprehensive review. Autoimmun Rev. 2019;18:786-795. doi:10.1016/j.autrev.2019.06.007
- Hofmann SC, Weidinger A. Epidermolysis bullosa acquisita. Hautarzt. 2019;70:265-270. doi:10.1007/s00105-019-4387-7
- Ishi N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230. doi:10.1111/j.1346-8138.2009.00799.x
- Iwata H, Vorobyev A, Koga H, et al. Meta-analysis of the clinical and immunopathological characteristics and treatment outcomes in epidermolysis bullosa acquisita patients. Orphanet J Rare Dis. 2018;13:153. doi:10.1186/s13023-018-0896-1
- Komorowski L, Muller R, Vorobyev A, et al. Sensitive and specific assays for routine serological diagnosis of epidermolysis bullosa acquisita. J Am Acad Dermatol. 2013;68:e89-95. doi:10.1016/j.jaad.2011.12.032
- Antonelli E, Bassotti G, Tramontana M, et al. Dermatological manifestations in inflammatory bowel diseases. J Clin Med. 2021;10:364-390. doi:10.3390/jcm10020364
- Bezzio C, Della Corte C, Vernero M, et al. Inflammatory bowel disease and immune-mediated inflammatory diseases: looking at less frequent associations. Therap Adv Gastroenterol. 2022;15:17562848221115312. doi:10.1177/17562848221115312
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have antibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064. doi:10.1046/j.1523-1747.2002.01772.x
- Labeille B, Gineston JL, Denoeux JP, et al. Epidermolysis bullosa acquisita and Crohn’s disease. A case report with immunological and electron microscopic studies. Arch Intern Med. 1988;148:1457-1459.
- Etienne A, Ruffieux P, Didierjean L, et al. Epidermolysis bullosa acquisita and metastatic cancer of the uterine cervix. Ann Dermatol Venereol. 1998;125:321-323.
- Busch J-O, Sticherling M. Epidermolysis bullosa acquisita and neuroendocrine pancreatic cancer-Coincidence or patho-genetic relationship? J Dtsch Dermatol Ges. 2007;5:916-918. doi:10.111/j.1610-0387.2007.06338.x
- Bevans SL, Sami N. The use of rituximab in treatment of epidermolysis bullosa acquisita: three new cases and a review of the literature. Dermatol Ther. 2018;31:e12726. doi:10.1111/j.1610-0387.2007.06338.x
- Yang A, Kim M, Craig P, et al. A case report of the use of rituximab and the epidermolysis bullosa disease activity scoring index (EBDASI) in a patient with epidermolysis bullosa acquisita with extensive esophageal involvement. Arch Dermatovenerol Croat. 2018;26:325-328.
- Burrows NP, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338. doi:10.1111/j.1365-2133.1993.tb00180.x
- Aringer M, Leuchten N, Johnson SR. New criteria for lupus. Curr Rheum Rep. 2020;22:18. doi:10.1007/s11926-020-00896-6
- Camisa C. Vesiculobullous systemic lupus erythematosus. A report of four cases. J Am Acad Dermatol. 1988;18:93-100. doi:10.1016/s0190-9622(88)70014-6
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:167064. doi:10.1155/2015/167064
- Sprow G, Afarideh M, Dan J, et al. Bullous systemic lupus erythematosus in females. Int J Womens Dermatol. 2022;8:e034. doi:10.1097/JW9.0000000000000034
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524. doi:10.1007/s40257-014-0098-0
- Fine JD, Mellerio JE. Epidermolysis bullosa. In: Bolognia JL, Jorizzo JL, Schaffer JV (eds), Dermatology (ed 3), Elsevier Saunders; 2012: 501-513.
There are 3 uncommon types of mechanobullous skin diseases caused by relative reduction or complete loss of functional type VII collagen, which is the main component of anchoring fibrils in the lamina densa of the basement membrane zone (BMZ) of the skin and mucous membrane epithelium.1 The function of the anchoring fibrils is to maintain adherence of the basement membrane of the epithelium to the connective tissue of the papillary dermis and submucosa.1 The mechanism of action of the loss of type VII collagen function is via autoimmunity in epidermolysis bullosa acquisita (EBA)2 and
Epidermolysis Bullosa
Epidermolysis bullosa consists of a heterogeneous family of 4 major genetic mechanobullous diseases that affect the skin and mucous membranes with more than 30 subtypes.1 Dystrophic EB is caused by mutations in the COL7A1 gene, which encodes for the α-1 chain of collagen type VII. Classically, EB is divided into 4 main variants based on the location of the cleavage plane or split occurring in the epithelium, which in turn helps to predict the severity of the illness.
Epidermolysis bullosa may be inherited in an autosomal-dominant or autosomal-recessive fashion, or it may occur as a spontaneous mutation. All sexes and races are affected equally. Patients present at birth or in early childhood with fragile skin and mucous membranes that may develop blisters, erosions, and ulcerations after minor trauma.7 These lesions are marked by slow healing and scar formation and often are associated with itching and pain.
Dystrophic Epidermolysis Bullosa
Dystrophic EB accounts for approximately 25%6 of all EB cases in the United States and may be inherited as either a dominant or recessive trait. Hundreds of different pathogenic mutations have been discovered in the COL7A1 gene in the subtypes of DEB.4,8 Dominant DEB tends to cause milder disease because the patients retain one normal COL7A1 allele and produce some type VII collagen (Figure 1), whereas patients with recessive DEB lack type VII collagen completely.9 The cleavage plane is between the lamina densa and the superficial dermis or submucosa. Severity is variable and ranges from localization to the hands and feet to severe generalized blistering and painful ulcerations depending on which of the many possible gene mutations have been inherited. Sequelae include mitten deformities, malalignment and tooth decay, and the development of early aggressive squamous cell carcinomas, which may be fatal. The most severe cases of recessive DEB also may have internal organ involvement.
Epidermolysis Bullosa Simplex
Epidermolysis bullosa simplex is the most common variant, comprising approximately 70%of EB cases in the United States.6 Epidermolysis bullosa simplex usually is inherited as autosomal-dominant mutations in the keratin 5 or keratin 14 genes,10 not COL7A1. Skin blistering results from cleavage within the basal cell layer where the keratin genes are primarily expressed. Blisters tend to occur in acral areas such as hands and feet and may heal without scarring in the localized form of epidermolysis bullosa simplex (Figure 2).
Junctional Epidermolysis Bullosa and Kindler Syndrome
Junctional epidermolysis bullosa (JEB) and Kindler syndrome11 are the rarest of the autosomal-recessive EB variants.6 The plane of cleavage in JEB is through the lamina lucida of the BMZ. Junctional epidermolysis bullosa is caused by mutations of the genes that encode for the 3 chains of laminin 332 protein and type XVII collagen,5,12 not to be confused with type VII collagen. As with DEB, there is a wide range of severity in JEB, from localized effects on the eyes, oral cavity, and tooth enamel to widespread blistering and skin cancers. In JEB cases involving newborns, nonhealing wounds on the face, buttocks, fingers, and toes may be seen, with devastating complications in the oral cavity, esophagus, and larynx. Life expectancy is limited to 2 years or less.6 There have only been approximately 40013 cases of Kindler syndrome reported worldwide6 and there is clinical overlap with DEB. Patients also may demonstrate poikiloderma and photosensitivity. Kindler syndrome is caused by mutations in the FERMT1 gene which encodes for kindlin-1. This protein mediates anchorage between the actin cytoskeleton and the extracellular matrix.5,11 Loss of function produces variable cleavage planes around the dermoepidermal junction.
Clinical management of all EB variants, especially the severe recessive types, traditionally has been limited to the prevention of trauma to the skin and mucous membranes and supportive care, including dressing changes to erosions and ulcerations, antibiotic ointments as needed, and amelioration of pain and pruritus. Bone marrow and pluripotential stem cell transplants have been attempted.12 Complications of EB, such as deformities of the hands and feet caused by excessive scarring, esophageal strictures, poor dentition, and squamous cell carcinomas, must be addressed by a multidisciplinary team of specialists, including plastic surgery, gastroenterology, dentistry/oral surgery, ophthalmology, and dermatology/Mohs surgery.
Until recently, there were no medications approved by the US Food and Drug Administration (FDA) specifically indicated for EB. In 2023, topical gene therapy was approved by the FDA for both recessive and dominant forms of DEB. Normal COL7A1 sequences are delivered by an attenuated herpes simplex virus 1 vector (beremagene geperpavec) in a gel applied directly to the wounds of patients with DEB. In a clinical trial, matching wounds on 31 patients (62 wounds total) were treated with the active agent or placebo gel. After 6 months, complete wound closure was observed in 67% (21/31) of those treated with the active agent and 22% (7/31) of those treated with placebo (P=.002).14 In a single case report, a patient with recessive DEB and cicatrizing conjunctivitis (Figure 3) was given ophthalmic beremagene geperpavec after surgery and had improved visual acuity.15 A topical gel consisting of birch triterpenes to promote healing of partial-thickness wounds also was approved for patients with DEB and JEB by the FDA and the European Commission. In a study of 223 patients, 41% of those using active gel and 29% of those using placebo gel achieved the primary end point of percentage of target wounds that had first complete closure at 45 days.16
The most recent FDA approval for DEB involves transferring the functional COL7A1 gene to the patient’s skin cells, then expanding the gene-corrected cells into sheets of keratinocytes that can be surgically applied to the chronic wound sites. In a phase 3 trial of prademagene zamikeracel (pz-cel), 11 patients with 86 matched wounds were randomized to receive pz-cel (50%) or standard wound care (50%). After 24 weeks, 35 wounds treated with pz-cel were at least 50% healed compared to 7 control wounds.17 The results for healing and reduction of pain were statistically significant (P<.0001 and P<.0002, respectively).17 Recombinant collagen VII as replacement therapy also is under study to be given by intravenous infusion to increase tissue collagen VII where it is lacking. This treatment has shown early biologic and therapeutic effects.9,18 Larger long-term follow-up studies are necessary to confirm persistence of the gene-corrected skin cells, the functionality of the replacement collagen VII, and the potential risk for the development of autoantibodies to type VII collagen.
Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita is a rare autoimmune subepithelial bullous disease that primarily affects middle-aged adults but also has been reported in children.19 Epidermolysis bullosa acquisita is caused by circulating pathogenic IgG autoantibodies that target and bind to type VII collagen in the anchoring fibrils,20-22 thereby disrupting the attachment of the epithelium to its underlying connective tissue.
The 2 major clinical manifestations of EBA include a mechanobullous disease resembling inherited forms of DEB (Figure 4) and an inflammatory bullous pemphigoid (BP)–like disease,23 as well as a combination of both types of skin lesions (Figure 5). The skin and mucous membranes of the oral cavity, esophagus, eyes, and urogenital areas are affected in both types; scarring may cause functional disabilities. In the mechanobullous type of EBA, it is common for blisters and erosions to develop in trauma-prone areas such as the hands, feet, elbows, and knees. The blisters tend to heal with scarring and milia formation as might be seen in porphyria cutanea tarda or cicatricial pemphigoid, which are in the differential diagnosis. Dystrophy of the fingernails or complete nail loss may be observed, resembling DEB. In the BP-like presentation, tense blisters arise upon inflamed or urticarial skin and mucous membranes, which may then become generalized.
Histopathology in both forms of EBA demonstrates subepithelial separation as clefts or blisters. The mechanobullous type shows a sparse inflammatory infiltrate compared to large collections of neutrophils and eosinophils in the blister cavity and in the superficial dermis in the BP-like cases. The final diagnosis rests on the results of immunopathology testing.24 Direct immunofluorescence of perilesional skin and mucosa shows a linear-granular band of IgG and C3 and other conjugates along the BMZ. Deposits of IgA alone in EBA occur in only about 2.4% of cases and are observed more often when there is mucous membrane involvement.2 Indirect immunofluorescence of sera against salt-split skin substrates detects immunoreactants in the floor of the blister rather than in the roof, as would be seen in BP. Highly specific and sensitive enzyme-linked immunosorbent assay (ELISA) kits now are commercially available and can detect autoantibodies against the N-terminal domain of type VII collagen in more than 90% of cases of EBA.25
Inflammatory bowel disease (IBD), particularly Crohn disease (CD), precedes the onset of EBA in approximately 25% of cases.26,27 Ulcerative colitis is much less common. Type VII collagen is normally present in the basement membrane of intestinal epithelium. In a survey of patients with IBD, 68% of those with CD and 13% of those with ulcerative colitis had circulating anti–type VII collagen antibodies detected by ELISA without having symptoms of EBA.28 A case report of a patient with both well-proven EBA and CD highlighted the clinical difficulty of controlling EBA: treatment with prednisolone and sulfasalazine improved the CD but had little effect on the skin blisters.29 A variety of malignancies have been reported in association with EBA, including cancers of the uterine cervix,30 thyroid, and pancreas,31 lymphoma, and chronic lymphatic leukemia. Some of these cases have met the criteria for classification as paraneoplastic, whereas others may have been coincidental.
Treatment for chronic EBA generally has been limited.2,24 Putative antineutrophil drugs such as dapsone and colchicine combined with systemic corticosteroids may be useful in milder or juvenile cases, which tend to have a better prognosis than adult cases.19 In more severe EBA, systemic corticosteroids and/or immunosuppressive drugs such as azathioprine,23 cyclophosphamide,23 mycophenolate mofetil,31 methotrexate,23 cyclosporine,33 and infliximab23 have been used. More recently, rituximab infusion monotherapy33 and rituximab combined with intravenous immunoglobulin or
Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus is a rare and specific autoimmune skin complication that mostly is seen in patients with an established diagnosis of systemic lupus erythematosus (SLE) who are experiencing a disease flare. Although more common in women, it has been reported in all sexes and races as well as in children. Vesicles and bullae may arise on sun-exposed (Figure 6) and sun-protected areas of skin.
Histopathology shows subepidermal separation with collections of neutrophils and nuclear fragments in the blister cavity. The differential diagnosis of BSLE includes EBA, BP, dermatitis herpetiformis, and linear IgA bullous dermatosis. Direct immunofluorescence testing shows linear-granular deposits of IgG and/or IgM and IgA along the BMZ.34 When utilizing the indirect immunofluorescence split-skin assay, the autoantibody to type VII collagen would be detected in the floor of the blister if the serum titer was sufficiently high.3 Proposed criteria for the diagnosis of BSLE have been published: 1) diagnosis of SLE now based on the 2019 European League Against Rheumatism/American College of Rheumatology classification35; 2) vesicles and bullae arising upon but not limited to sun-exposed skin; 3) histopathology featuring neutrophil-rich subepithelial bullae; 4) positive indirect immunofluorescence for circulating BMZ antibodies using separated human skin as substrate; 5) and direct immunofluorescence showing IgG and/or IgM and often IgA at the BMZ.36 Using ELISA to detect circulating antibodies against type VII collagen24 should now be added to the criteria. The new criteria for SLE34 do not include BSLE, perhaps because it occurs in less than 1% of patients with SLE.37
Further investigation by Gammon et al3 confirmed that the autoantibodies in BSLE are identical to those found in EBA (ie, directed against type VII collagen in the lamina densa). Bullous systemic lupus erythematosus is not considered to be the coexistence of EBA with SLE but rather a specific entity wherein type VII collagen autoantibodies are expressed in the autoimmune spectrum of SLE. It is especially important to make the diagnosis of BSLE because it is predictive of more serious systemic complications of SLE (eg, hematologic and renal disease is found in up to 90% of cases).38
The natural course of BSLE is variable. Treatments include systemic corticosteroids, dapsone, and immunosuppressive drugs such as azathioprine, methotrexate, mycophenolate mofetil, and cyclophosphamide, especially in cases with nephritis.37 There may be spontaneous resolution of the rash as the inflammatory activity of SLE subsides. Rituximab has been used effectively in several refractory cases of BSLE that failed to respond to all other conventional treatments.39
Conclusion
Anchoring fibrils are composed primarily of type VII collagen. Their role is to maintain the attachment of epithelium to the upper dermis and submucosa. The reduction or complete loss of type VII collagen caused by mutations of the COL7A1 gene results in dominant DEB or recessive DEB, respectively. Two distinct non-heritable immunobullous diseases, EBA and BSLE, are caused by autoantibodies that target type VII collagen. A comparison of the 4 type VII collagen disorders can be found in the eTable.
There are 3 uncommon types of mechanobullous skin diseases caused by relative reduction or complete loss of functional type VII collagen, which is the main component of anchoring fibrils in the lamina densa of the basement membrane zone (BMZ) of the skin and mucous membrane epithelium.1 The function of the anchoring fibrils is to maintain adherence of the basement membrane of the epithelium to the connective tissue of the papillary dermis and submucosa.1 The mechanism of action of the loss of type VII collagen function is via autoimmunity in epidermolysis bullosa acquisita (EBA)2 and
Epidermolysis Bullosa
Epidermolysis bullosa consists of a heterogeneous family of 4 major genetic mechanobullous diseases that affect the skin and mucous membranes with more than 30 subtypes.1 Dystrophic EB is caused by mutations in the COL7A1 gene, which encodes for the α-1 chain of collagen type VII. Classically, EB is divided into 4 main variants based on the location of the cleavage plane or split occurring in the epithelium, which in turn helps to predict the severity of the illness.
Epidermolysis bullosa may be inherited in an autosomal-dominant or autosomal-recessive fashion, or it may occur as a spontaneous mutation. All sexes and races are affected equally. Patients present at birth or in early childhood with fragile skin and mucous membranes that may develop blisters, erosions, and ulcerations after minor trauma.7 These lesions are marked by slow healing and scar formation and often are associated with itching and pain.
Dystrophic Epidermolysis Bullosa
Dystrophic EB accounts for approximately 25%6 of all EB cases in the United States and may be inherited as either a dominant or recessive trait. Hundreds of different pathogenic mutations have been discovered in the COL7A1 gene in the subtypes of DEB.4,8 Dominant DEB tends to cause milder disease because the patients retain one normal COL7A1 allele and produce some type VII collagen (Figure 1), whereas patients with recessive DEB lack type VII collagen completely.9 The cleavage plane is between the lamina densa and the superficial dermis or submucosa. Severity is variable and ranges from localization to the hands and feet to severe generalized blistering and painful ulcerations depending on which of the many possible gene mutations have been inherited. Sequelae include mitten deformities, malalignment and tooth decay, and the development of early aggressive squamous cell carcinomas, which may be fatal. The most severe cases of recessive DEB also may have internal organ involvement.
Epidermolysis Bullosa Simplex
Epidermolysis bullosa simplex is the most common variant, comprising approximately 70%of EB cases in the United States.6 Epidermolysis bullosa simplex usually is inherited as autosomal-dominant mutations in the keratin 5 or keratin 14 genes,10 not COL7A1. Skin blistering results from cleavage within the basal cell layer where the keratin genes are primarily expressed. Blisters tend to occur in acral areas such as hands and feet and may heal without scarring in the localized form of epidermolysis bullosa simplex (Figure 2).
Junctional Epidermolysis Bullosa and Kindler Syndrome
Junctional epidermolysis bullosa (JEB) and Kindler syndrome11 are the rarest of the autosomal-recessive EB variants.6 The plane of cleavage in JEB is through the lamina lucida of the BMZ. Junctional epidermolysis bullosa is caused by mutations of the genes that encode for the 3 chains of laminin 332 protein and type XVII collagen,5,12 not to be confused with type VII collagen. As with DEB, there is a wide range of severity in JEB, from localized effects on the eyes, oral cavity, and tooth enamel to widespread blistering and skin cancers. In JEB cases involving newborns, nonhealing wounds on the face, buttocks, fingers, and toes may be seen, with devastating complications in the oral cavity, esophagus, and larynx. Life expectancy is limited to 2 years or less.6 There have only been approximately 40013 cases of Kindler syndrome reported worldwide6 and there is clinical overlap with DEB. Patients also may demonstrate poikiloderma and photosensitivity. Kindler syndrome is caused by mutations in the FERMT1 gene which encodes for kindlin-1. This protein mediates anchorage between the actin cytoskeleton and the extracellular matrix.5,11 Loss of function produces variable cleavage planes around the dermoepidermal junction.
Clinical management of all EB variants, especially the severe recessive types, traditionally has been limited to the prevention of trauma to the skin and mucous membranes and supportive care, including dressing changes to erosions and ulcerations, antibiotic ointments as needed, and amelioration of pain and pruritus. Bone marrow and pluripotential stem cell transplants have been attempted.12 Complications of EB, such as deformities of the hands and feet caused by excessive scarring, esophageal strictures, poor dentition, and squamous cell carcinomas, must be addressed by a multidisciplinary team of specialists, including plastic surgery, gastroenterology, dentistry/oral surgery, ophthalmology, and dermatology/Mohs surgery.
Until recently, there were no medications approved by the US Food and Drug Administration (FDA) specifically indicated for EB. In 2023, topical gene therapy was approved by the FDA for both recessive and dominant forms of DEB. Normal COL7A1 sequences are delivered by an attenuated herpes simplex virus 1 vector (beremagene geperpavec) in a gel applied directly to the wounds of patients with DEB. In a clinical trial, matching wounds on 31 patients (62 wounds total) were treated with the active agent or placebo gel. After 6 months, complete wound closure was observed in 67% (21/31) of those treated with the active agent and 22% (7/31) of those treated with placebo (P=.002).14 In a single case report, a patient with recessive DEB and cicatrizing conjunctivitis (Figure 3) was given ophthalmic beremagene geperpavec after surgery and had improved visual acuity.15 A topical gel consisting of birch triterpenes to promote healing of partial-thickness wounds also was approved for patients with DEB and JEB by the FDA and the European Commission. In a study of 223 patients, 41% of those using active gel and 29% of those using placebo gel achieved the primary end point of percentage of target wounds that had first complete closure at 45 days.16
The most recent FDA approval for DEB involves transferring the functional COL7A1 gene to the patient’s skin cells, then expanding the gene-corrected cells into sheets of keratinocytes that can be surgically applied to the chronic wound sites. In a phase 3 trial of prademagene zamikeracel (pz-cel), 11 patients with 86 matched wounds were randomized to receive pz-cel (50%) or standard wound care (50%). After 24 weeks, 35 wounds treated with pz-cel were at least 50% healed compared to 7 control wounds.17 The results for healing and reduction of pain were statistically significant (P<.0001 and P<.0002, respectively).17 Recombinant collagen VII as replacement therapy also is under study to be given by intravenous infusion to increase tissue collagen VII where it is lacking. This treatment has shown early biologic and therapeutic effects.9,18 Larger long-term follow-up studies are necessary to confirm persistence of the gene-corrected skin cells, the functionality of the replacement collagen VII, and the potential risk for the development of autoantibodies to type VII collagen.
Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita is a rare autoimmune subepithelial bullous disease that primarily affects middle-aged adults but also has been reported in children.19 Epidermolysis bullosa acquisita is caused by circulating pathogenic IgG autoantibodies that target and bind to type VII collagen in the anchoring fibrils,20-22 thereby disrupting the attachment of the epithelium to its underlying connective tissue.
The 2 major clinical manifestations of EBA include a mechanobullous disease resembling inherited forms of DEB (Figure 4) and an inflammatory bullous pemphigoid (BP)–like disease,23 as well as a combination of both types of skin lesions (Figure 5). The skin and mucous membranes of the oral cavity, esophagus, eyes, and urogenital areas are affected in both types; scarring may cause functional disabilities. In the mechanobullous type of EBA, it is common for blisters and erosions to develop in trauma-prone areas such as the hands, feet, elbows, and knees. The blisters tend to heal with scarring and milia formation as might be seen in porphyria cutanea tarda or cicatricial pemphigoid, which are in the differential diagnosis. Dystrophy of the fingernails or complete nail loss may be observed, resembling DEB. In the BP-like presentation, tense blisters arise upon inflamed or urticarial skin and mucous membranes, which may then become generalized.
Histopathology in both forms of EBA demonstrates subepithelial separation as clefts or blisters. The mechanobullous type shows a sparse inflammatory infiltrate compared to large collections of neutrophils and eosinophils in the blister cavity and in the superficial dermis in the BP-like cases. The final diagnosis rests on the results of immunopathology testing.24 Direct immunofluorescence of perilesional skin and mucosa shows a linear-granular band of IgG and C3 and other conjugates along the BMZ. Deposits of IgA alone in EBA occur in only about 2.4% of cases and are observed more often when there is mucous membrane involvement.2 Indirect immunofluorescence of sera against salt-split skin substrates detects immunoreactants in the floor of the blister rather than in the roof, as would be seen in BP. Highly specific and sensitive enzyme-linked immunosorbent assay (ELISA) kits now are commercially available and can detect autoantibodies against the N-terminal domain of type VII collagen in more than 90% of cases of EBA.25
Inflammatory bowel disease (IBD), particularly Crohn disease (CD), precedes the onset of EBA in approximately 25% of cases.26,27 Ulcerative colitis is much less common. Type VII collagen is normally present in the basement membrane of intestinal epithelium. In a survey of patients with IBD, 68% of those with CD and 13% of those with ulcerative colitis had circulating anti–type VII collagen antibodies detected by ELISA without having symptoms of EBA.28 A case report of a patient with both well-proven EBA and CD highlighted the clinical difficulty of controlling EBA: treatment with prednisolone and sulfasalazine improved the CD but had little effect on the skin blisters.29 A variety of malignancies have been reported in association with EBA, including cancers of the uterine cervix,30 thyroid, and pancreas,31 lymphoma, and chronic lymphatic leukemia. Some of these cases have met the criteria for classification as paraneoplastic, whereas others may have been coincidental.
Treatment for chronic EBA generally has been limited.2,24 Putative antineutrophil drugs such as dapsone and colchicine combined with systemic corticosteroids may be useful in milder or juvenile cases, which tend to have a better prognosis than adult cases.19 In more severe EBA, systemic corticosteroids and/or immunosuppressive drugs such as azathioprine,23 cyclophosphamide,23 mycophenolate mofetil,31 methotrexate,23 cyclosporine,33 and infliximab23 have been used. More recently, rituximab infusion monotherapy33 and rituximab combined with intravenous immunoglobulin or
Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus is a rare and specific autoimmune skin complication that mostly is seen in patients with an established diagnosis of systemic lupus erythematosus (SLE) who are experiencing a disease flare. Although more common in women, it has been reported in all sexes and races as well as in children. Vesicles and bullae may arise on sun-exposed (Figure 6) and sun-protected areas of skin.
Histopathology shows subepidermal separation with collections of neutrophils and nuclear fragments in the blister cavity. The differential diagnosis of BSLE includes EBA, BP, dermatitis herpetiformis, and linear IgA bullous dermatosis. Direct immunofluorescence testing shows linear-granular deposits of IgG and/or IgM and IgA along the BMZ.34 When utilizing the indirect immunofluorescence split-skin assay, the autoantibody to type VII collagen would be detected in the floor of the blister if the serum titer was sufficiently high.3 Proposed criteria for the diagnosis of BSLE have been published: 1) diagnosis of SLE now based on the 2019 European League Against Rheumatism/American College of Rheumatology classification35; 2) vesicles and bullae arising upon but not limited to sun-exposed skin; 3) histopathology featuring neutrophil-rich subepithelial bullae; 4) positive indirect immunofluorescence for circulating BMZ antibodies using separated human skin as substrate; 5) and direct immunofluorescence showing IgG and/or IgM and often IgA at the BMZ.36 Using ELISA to detect circulating antibodies against type VII collagen24 should now be added to the criteria. The new criteria for SLE34 do not include BSLE, perhaps because it occurs in less than 1% of patients with SLE.37
Further investigation by Gammon et al3 confirmed that the autoantibodies in BSLE are identical to those found in EBA (ie, directed against type VII collagen in the lamina densa). Bullous systemic lupus erythematosus is not considered to be the coexistence of EBA with SLE but rather a specific entity wherein type VII collagen autoantibodies are expressed in the autoimmune spectrum of SLE. It is especially important to make the diagnosis of BSLE because it is predictive of more serious systemic complications of SLE (eg, hematologic and renal disease is found in up to 90% of cases).38
The natural course of BSLE is variable. Treatments include systemic corticosteroids, dapsone, and immunosuppressive drugs such as azathioprine, methotrexate, mycophenolate mofetil, and cyclophosphamide, especially in cases with nephritis.37 There may be spontaneous resolution of the rash as the inflammatory activity of SLE subsides. Rituximab has been used effectively in several refractory cases of BSLE that failed to respond to all other conventional treatments.39
Conclusion
Anchoring fibrils are composed primarily of type VII collagen. Their role is to maintain the attachment of epithelium to the upper dermis and submucosa. The reduction or complete loss of type VII collagen caused by mutations of the COL7A1 gene results in dominant DEB or recessive DEB, respectively. Two distinct non-heritable immunobullous diseases, EBA and BSLE, are caused by autoantibodies that target type VII collagen. A comparison of the 4 type VII collagen disorders can be found in the eTable.
- Bardhan A, Bruckner-Tuderman L, Chapple ILC, et al. Epidermolysis bullosa. Nat Rev Dis Primers. 2020;6:78. doi:10.1038/s41572-020-0210-0
- Miyamoto D, Gordilho JO, Santi CG, et al. Epidermolysis bullosa acquisita. An Bras Dermatol. 2022;97:409-423. doi:10.1016/j.abd.2021.09.010.
- Gammon WR, Woodley DT, Dole KC, et al. Evidence that anti-basement membrane zone antibodies in bullous eruption of systemic lupus erythematosus recognize epidermolysis bullosa acquisita autoantigen. J Invest Dermatol. 1985;84:472-476. doi:10.1111/1523-1747.ep12272402.
- Yadav RS, Jaswal A, Shrestha S, et al. Dystrophic epidermolysis bullosa. J Nepal Med Assoc. 2018;56:879-882. doi:10.31729/jnma.3791
- Mariath LM, Santin JT, Schuler-Faccini L, et al. Inherited epidermolysis bullosa: update on the clinical and genetic aspects. An Bras Dermatol. 2020;95:551-569. doi:10.1016/j.abd.2020.05.001
- Understanding epidermolysis bullosa (EB). DEBRA website. Accessed August 17, 2025. https://www.debra.org/about-eb/understanding-epidermolysis-bullosa-eb
- Hon KL, Chu S, Leung AKC. Epidermolysis bullosa: pediatric perspectives. Curr Pediatr Rev. 2022;18:182-190. doi:10.2174/1573396317666210525161252
- Dang N, Klingberg S, Marr P, et al. Review of collagen VII sequence variants found in Australasian patients with dystrophic epidermolysis bullosa reveals nine COL7A1 variants. J Dermatol Sci. 2007;46:169-178. doi:10.1016/j.jdermsci.2007.02.006
- Payne AS. Topical gene therapy for epidermolysis bullosa. N Engl J Med. 2022;387:2281-2284. doi:10.1056/NEJMe2213203
- Khani P, Ghazi F, Zekri A, et al. Keratins and epidermolysis bullosa simplex. J Cell Physiol. 2018;234:289-297. doi:10.1002/jcp.26898
- Lai-Cheong JE, Tanaka A, Hawche G, et al. Kindler syndrome: a focal adhesion genodermatosis. Br J Dermatol. 2009;160:233-242. doi:10.1111/j.1365-2133.2008.08976.x
- Hou P-C, Wang H-T, Abhee S, et al. Investigational treatments for epidermolysis bullosa. Am J Clin Dermatol. 2021;22:801-817. doi:10.1007/s40257-021-00626-3
- Youseffian L, Vahidnezhad H, Uitto J. Kindler Syndrome. GeneReviews [Internet]. Updated January 6, 2022. Accessed August 21, 2025.
- Guide SV, Gonzalez ME, Bagci S, et al. Trial of beremagene geperpavec (B-VEC) for dystrophic epidermolysis bullosa. N Engl J Med. 2022;387:2211-2219. doi:10.1056/NEJMoa2206663
- Vetencourt AT, Sayed-Ahmed I, Gomez J, et al. Ocular gene therapy in a patient with dystrophic epidermolysis bullosa. N Engl J Med. 2024;390:530-535. doi:10.1056/NEJMoa2301244
- Kern JS, Sprecher E, Fernandez MF, et al. Efficacy and safety of Oleogel-S10 (birch triterpenes for epidermolysis bullosa: results from the phase III randomized double-blind phase of the EASE study. Br J Dermatol. 2023;188:12-21. doi:10.1093/bjd/ljac001
- Tang JY, Marinkovich MP, Wiss K, et al. Prademagene zamikeracel for recessive dystrophic epidermolysis bullosa wounds (VIITAL): a two-centre, randomized, open-label, intrapatient-controlled phase 3 trial. Lancet. 2025;406:163-173. doi:10.1016/S0140-6736(25)00778-0
- Gretzmeier C, Pin D, Kern JS, et al. Systemic collagen VII replacement therapy for advanced recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2022;142:1094-1102. doi:10.1016/j.jid.2021.09.008
- Hignett E, Sami N. Pediatric epidermolysis bullosa acquisita. A review. Pediatr Dermatol. 2021;38:1047-1050. doi:10.1111/pde.14722
- Chen M, Kim GH, Prakash L, et al. Autoimmunity to anchoring fibril collagen. Autoimmunity. 2012;45:91-101. doi:10.1007/s12016-007-0027-6.
- Kridin K, Kneiber D, Kowalski EH, et al. Epidermolysis bullosa acquisita: a comprehensive review. Autoimmun Rev. 2019;18:786-795. doi:10.1016/j.autrev.2019.06.007
- Hofmann SC, Weidinger A. Epidermolysis bullosa acquisita. Hautarzt. 2019;70:265-270. doi:10.1007/s00105-019-4387-7
- Ishi N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230. doi:10.1111/j.1346-8138.2009.00799.x
- Iwata H, Vorobyev A, Koga H, et al. Meta-analysis of the clinical and immunopathological characteristics and treatment outcomes in epidermolysis bullosa acquisita patients. Orphanet J Rare Dis. 2018;13:153. doi:10.1186/s13023-018-0896-1
- Komorowski L, Muller R, Vorobyev A, et al. Sensitive and specific assays for routine serological diagnosis of epidermolysis bullosa acquisita. J Am Acad Dermatol. 2013;68:e89-95. doi:10.1016/j.jaad.2011.12.032
- Antonelli E, Bassotti G, Tramontana M, et al. Dermatological manifestations in inflammatory bowel diseases. J Clin Med. 2021;10:364-390. doi:10.3390/jcm10020364
- Bezzio C, Della Corte C, Vernero M, et al. Inflammatory bowel disease and immune-mediated inflammatory diseases: looking at less frequent associations. Therap Adv Gastroenterol. 2022;15:17562848221115312. doi:10.1177/17562848221115312
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have antibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064. doi:10.1046/j.1523-1747.2002.01772.x
- Labeille B, Gineston JL, Denoeux JP, et al. Epidermolysis bullosa acquisita and Crohn’s disease. A case report with immunological and electron microscopic studies. Arch Intern Med. 1988;148:1457-1459.
- Etienne A, Ruffieux P, Didierjean L, et al. Epidermolysis bullosa acquisita and metastatic cancer of the uterine cervix. Ann Dermatol Venereol. 1998;125:321-323.
- Busch J-O, Sticherling M. Epidermolysis bullosa acquisita and neuroendocrine pancreatic cancer-Coincidence or patho-genetic relationship? J Dtsch Dermatol Ges. 2007;5:916-918. doi:10.111/j.1610-0387.2007.06338.x
- Bevans SL, Sami N. The use of rituximab in treatment of epidermolysis bullosa acquisita: three new cases and a review of the literature. Dermatol Ther. 2018;31:e12726. doi:10.1111/j.1610-0387.2007.06338.x
- Yang A, Kim M, Craig P, et al. A case report of the use of rituximab and the epidermolysis bullosa disease activity scoring index (EBDASI) in a patient with epidermolysis bullosa acquisita with extensive esophageal involvement. Arch Dermatovenerol Croat. 2018;26:325-328.
- Burrows NP, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338. doi:10.1111/j.1365-2133.1993.tb00180.x
- Aringer M, Leuchten N, Johnson SR. New criteria for lupus. Curr Rheum Rep. 2020;22:18. doi:10.1007/s11926-020-00896-6
- Camisa C. Vesiculobullous systemic lupus erythematosus. A report of four cases. J Am Acad Dermatol. 1988;18:93-100. doi:10.1016/s0190-9622(88)70014-6
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:167064. doi:10.1155/2015/167064
- Sprow G, Afarideh M, Dan J, et al. Bullous systemic lupus erythematosus in females. Int J Womens Dermatol. 2022;8:e034. doi:10.1097/JW9.0000000000000034
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524. doi:10.1007/s40257-014-0098-0
- Fine JD, Mellerio JE. Epidermolysis bullosa. In: Bolognia JL, Jorizzo JL, Schaffer JV (eds), Dermatology (ed 3), Elsevier Saunders; 2012: 501-513.
- Bardhan A, Bruckner-Tuderman L, Chapple ILC, et al. Epidermolysis bullosa. Nat Rev Dis Primers. 2020;6:78. doi:10.1038/s41572-020-0210-0
- Miyamoto D, Gordilho JO, Santi CG, et al. Epidermolysis bullosa acquisita. An Bras Dermatol. 2022;97:409-423. doi:10.1016/j.abd.2021.09.010.
- Gammon WR, Woodley DT, Dole KC, et al. Evidence that anti-basement membrane zone antibodies in bullous eruption of systemic lupus erythematosus recognize epidermolysis bullosa acquisita autoantigen. J Invest Dermatol. 1985;84:472-476. doi:10.1111/1523-1747.ep12272402.
- Yadav RS, Jaswal A, Shrestha S, et al. Dystrophic epidermolysis bullosa. J Nepal Med Assoc. 2018;56:879-882. doi:10.31729/jnma.3791
- Mariath LM, Santin JT, Schuler-Faccini L, et al. Inherited epidermolysis bullosa: update on the clinical and genetic aspects. An Bras Dermatol. 2020;95:551-569. doi:10.1016/j.abd.2020.05.001
- Understanding epidermolysis bullosa (EB). DEBRA website. Accessed August 17, 2025. https://www.debra.org/about-eb/understanding-epidermolysis-bullosa-eb
- Hon KL, Chu S, Leung AKC. Epidermolysis bullosa: pediatric perspectives. Curr Pediatr Rev. 2022;18:182-190. doi:10.2174/1573396317666210525161252
- Dang N, Klingberg S, Marr P, et al. Review of collagen VII sequence variants found in Australasian patients with dystrophic epidermolysis bullosa reveals nine COL7A1 variants. J Dermatol Sci. 2007;46:169-178. doi:10.1016/j.jdermsci.2007.02.006
- Payne AS. Topical gene therapy for epidermolysis bullosa. N Engl J Med. 2022;387:2281-2284. doi:10.1056/NEJMe2213203
- Khani P, Ghazi F, Zekri A, et al. Keratins and epidermolysis bullosa simplex. J Cell Physiol. 2018;234:289-297. doi:10.1002/jcp.26898
- Lai-Cheong JE, Tanaka A, Hawche G, et al. Kindler syndrome: a focal adhesion genodermatosis. Br J Dermatol. 2009;160:233-242. doi:10.1111/j.1365-2133.2008.08976.x
- Hou P-C, Wang H-T, Abhee S, et al. Investigational treatments for epidermolysis bullosa. Am J Clin Dermatol. 2021;22:801-817. doi:10.1007/s40257-021-00626-3
- Youseffian L, Vahidnezhad H, Uitto J. Kindler Syndrome. GeneReviews [Internet]. Updated January 6, 2022. Accessed August 21, 2025.
- Guide SV, Gonzalez ME, Bagci S, et al. Trial of beremagene geperpavec (B-VEC) for dystrophic epidermolysis bullosa. N Engl J Med. 2022;387:2211-2219. doi:10.1056/NEJMoa2206663
- Vetencourt AT, Sayed-Ahmed I, Gomez J, et al. Ocular gene therapy in a patient with dystrophic epidermolysis bullosa. N Engl J Med. 2024;390:530-535. doi:10.1056/NEJMoa2301244
- Kern JS, Sprecher E, Fernandez MF, et al. Efficacy and safety of Oleogel-S10 (birch triterpenes for epidermolysis bullosa: results from the phase III randomized double-blind phase of the EASE study. Br J Dermatol. 2023;188:12-21. doi:10.1093/bjd/ljac001
- Tang JY, Marinkovich MP, Wiss K, et al. Prademagene zamikeracel for recessive dystrophic epidermolysis bullosa wounds (VIITAL): a two-centre, randomized, open-label, intrapatient-controlled phase 3 trial. Lancet. 2025;406:163-173. doi:10.1016/S0140-6736(25)00778-0
- Gretzmeier C, Pin D, Kern JS, et al. Systemic collagen VII replacement therapy for advanced recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2022;142:1094-1102. doi:10.1016/j.jid.2021.09.008
- Hignett E, Sami N. Pediatric epidermolysis bullosa acquisita. A review. Pediatr Dermatol. 2021;38:1047-1050. doi:10.1111/pde.14722
- Chen M, Kim GH, Prakash L, et al. Autoimmunity to anchoring fibril collagen. Autoimmunity. 2012;45:91-101. doi:10.1007/s12016-007-0027-6.
- Kridin K, Kneiber D, Kowalski EH, et al. Epidermolysis bullosa acquisita: a comprehensive review. Autoimmun Rev. 2019;18:786-795. doi:10.1016/j.autrev.2019.06.007
- Hofmann SC, Weidinger A. Epidermolysis bullosa acquisita. Hautarzt. 2019;70:265-270. doi:10.1007/s00105-019-4387-7
- Ishi N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230. doi:10.1111/j.1346-8138.2009.00799.x
- Iwata H, Vorobyev A, Koga H, et al. Meta-analysis of the clinical and immunopathological characteristics and treatment outcomes in epidermolysis bullosa acquisita patients. Orphanet J Rare Dis. 2018;13:153. doi:10.1186/s13023-018-0896-1
- Komorowski L, Muller R, Vorobyev A, et al. Sensitive and specific assays for routine serological diagnosis of epidermolysis bullosa acquisita. J Am Acad Dermatol. 2013;68:e89-95. doi:10.1016/j.jaad.2011.12.032
- Antonelli E, Bassotti G, Tramontana M, et al. Dermatological manifestations in inflammatory bowel diseases. J Clin Med. 2021;10:364-390. doi:10.3390/jcm10020364
- Bezzio C, Della Corte C, Vernero M, et al. Inflammatory bowel disease and immune-mediated inflammatory diseases: looking at less frequent associations. Therap Adv Gastroenterol. 2022;15:17562848221115312. doi:10.1177/17562848221115312
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have antibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064. doi:10.1046/j.1523-1747.2002.01772.x
- Labeille B, Gineston JL, Denoeux JP, et al. Epidermolysis bullosa acquisita and Crohn’s disease. A case report with immunological and electron microscopic studies. Arch Intern Med. 1988;148:1457-1459.
- Etienne A, Ruffieux P, Didierjean L, et al. Epidermolysis bullosa acquisita and metastatic cancer of the uterine cervix. Ann Dermatol Venereol. 1998;125:321-323.
- Busch J-O, Sticherling M. Epidermolysis bullosa acquisita and neuroendocrine pancreatic cancer-Coincidence or patho-genetic relationship? J Dtsch Dermatol Ges. 2007;5:916-918. doi:10.111/j.1610-0387.2007.06338.x
- Bevans SL, Sami N. The use of rituximab in treatment of epidermolysis bullosa acquisita: three new cases and a review of the literature. Dermatol Ther. 2018;31:e12726. doi:10.1111/j.1610-0387.2007.06338.x
- Yang A, Kim M, Craig P, et al. A case report of the use of rituximab and the epidermolysis bullosa disease activity scoring index (EBDASI) in a patient with epidermolysis bullosa acquisita with extensive esophageal involvement. Arch Dermatovenerol Croat. 2018;26:325-328.
- Burrows NP, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338. doi:10.1111/j.1365-2133.1993.tb00180.x
- Aringer M, Leuchten N, Johnson SR. New criteria for lupus. Curr Rheum Rep. 2020;22:18. doi:10.1007/s11926-020-00896-6
- Camisa C. Vesiculobullous systemic lupus erythematosus. A report of four cases. J Am Acad Dermatol. 1988;18:93-100. doi:10.1016/s0190-9622(88)70014-6
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:167064. doi:10.1155/2015/167064
- Sprow G, Afarideh M, Dan J, et al. Bullous systemic lupus erythematosus in females. Int J Womens Dermatol. 2022;8:e034. doi:10.1097/JW9.0000000000000034
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524. doi:10.1007/s40257-014-0098-0
- Fine JD, Mellerio JE. Epidermolysis bullosa. In: Bolognia JL, Jorizzo JL, Schaffer JV (eds), Dermatology (ed 3), Elsevier Saunders; 2012: 501-513.
Type VII Collagen Disorders Simplified
Type VII Collagen Disorders Simplified
PRACTICE POINTS
- The full complement of type VII collagen is required for the normal assembly of anchoring fibrils, whose function is to adhere the basement membrane to the underlying connective tissue of skin and mucous membranes.
- In the heritable epidermolysis bullosa (EB) family of diseases, only dominant and recessive dystrophic epidermolysis bullosa are caused by partial or total loss of type VII collagen function.
- New treatments that have been approved for EB include topical gene therapy with COL7A1, topical birch triterpene gel, and skin cells from patients that are genetically corrected with a functional COL7A1 gene.
- Epidermolysis bullosa acquisita and bullous systemic lupus erythematosus are rare distinct autoimmune subepithelial bullous diseases caused by IgG antibodies that target type VII collagen in the anchoring fibrils.

Generalized Erythematous Plaques and Pustules in a Pregnant Patient
Generalized Erythematous Plaques and Pustules in a Pregnant Patient
THE DIAGNOSIS: Impetigo Herpetiformis
Histopathology revealed epidermal acanthosis and spongiosis with overlying parakeratosis associated with subcorneal and intracorneal neutrophils, papillary dermal edema, and dermal mixed inflammation with neutrophils and eosinophils. Both direct immunofluorescence and periodic acid–Schiff studies were negative. Blood and pustule cultures were sterile and the skin flora were normal. Based on these findings, a diagnosis of impetigo herpetiformis (IH) was made. The condition improved with systemic and topical steroids, supportive care, and an intravenous infusion of infliximab 5 mg/kg. At 3 weeks’ follow-up, the patient demonstrated near-complete resolution and later delivered successfully at 40 weeks’ gestation without complications.
Impetigo herpetiformis, also known as pustular psoriasis of pregnancy, is an exceedingly rare gestational dermatosis that typically manifests in the third trimester and can be life-threatening for both the mother and fetus. The term was first used in 1872 to describe 5 pregnant women with extensive acute pustular eruptions, all in unstable condition; 4 (80%)of the cases resulted in maternal death, and all resulted in fetal death.1 Impetigo herpetiformis is characterized by pruritic and painful erythematous patches studded at the periphery with subcorneal pustules. Eruptions usually occur in the flexural areas and spread centrifugally, with extension of the lesions peripherally as the center erodes and crusts. Sparing of the face, palms, and soles is expected, and mucosal involvement is rare. Generalized involvement and exfoliation may occur in extreme cases.2 While IH typically manifests during the third trimester, it may occur any time throughout pregnancy or immediately postpartum.3 A few cases have been reported in the puerperium.2 Common symptoms include fever, chills, malaise, anorexia, nausea, vomiting, diarrhea, and arthralgias. Less common complications include hypoalbuminemia and severe hypocalcemia leading to tetany, seizures, and delirium.2,3 While maternal mortality is uncommon, fetal mortality often is a more pressing risk and is attributed to placental insufficiency.3,4 For this reason, early delivery commonly is considered in severe cases.
Whether IH is a separate entity or a variant of pustular psoriasis remains heavily debated. Although the histopathology of IH is identical to pustular psoriasis, the lack of a personal and family history of psoriasis, symptom resolution with delivery, and possible recurrence during successive pregnancies help differentiate IH from generalized pustular psoriasis.2,5 Earlier onset, diffuse involvement, faster progression, and recurrence in subsequent pregnancies all have been linked to a worse prognosis.4
The differential diagnosis for IH includes acute generalized exanthematous pustulosis, pemphigoid gestationis, dermatitis herpetiformis, and subcorneal pustular dermatosis. Acute generalized exanthematous pustulosis is an uncommon severe cutaneous drug reaction characterized by the sudden onset of numerous sterile pustules on erythematous skin within 48 hours of exposure. The most common offending medications include pristinamycin and beta-lactam antibiotics. A high fever, neutrophilic leukocytosis, and hypocalcemia often accompany acute generalized exanthematous pustulosis.6 Prompt diagnosis and withdrawal of the offending drug as well as supportive care and symptomatic treatment are crucial for disease management, as systemic symptoms and even organ involvement may occur.6
Pemphigoid gestationis, also known as gestational pemphigoid or herpes gestationis, is a rare autoimmune blistering disorder that primarily affects pregnant women. It typically manifests in the second or third trimester or shortly after delivery. Clinically, it manifests as an intensely pruritic polymorphic eruption of urticarial papules and plaques accompanied by vesicles and bullae and often is distributed on the abdomen and extends to other body regions. Although the exact etiology is unknown, pemphigoid gestationis is caused by autoantibodies targeting the BP180 and BP230 hemidesmosomal proteins.7 Treatment usually involves systemic corticosteroids and may require additional immunosuppressive therapy. In most cases, patients see resolution within 6 months of delivery.7
Dermatitis herpetiformis is a chronic autoimmune blistering skin disorder characterized by intensely pruritic, grouped vesicles and papules, often distributed symmetrically on extensor surfaces such as the elbows, knees, buttocks, and back. It is closely associated with celiac disease and is triggered by gluten ingestion in genetically predisposed individuals with human leukocyte antigen DQ2 and DQ8 haplotypes. Dermatitis herpetiformis is caused by deposition of IgA antibodies that target tissue transglutaminase 3 at the dermal papillae, leading to inflammation and blister formation. 8 Treatment typically involves a gluten-free diet and medications such as dapsone to alleviate symptoms and prevent recurrence.
Subcorneal pustular dermatosis, also known as Sneddon-Wilkinson disease, is a rare chronic relapsing pustular dermatosis characterized by sterile superficial pustules arranged in annular or circinate patterns on erythematous plaques. It predominantly affects middleaged women and often is associated with underlying conditions such as IgA gammopathy or monoclonal gammopathy of undetermined significance. The pathogenesis remains unclear, but immune dysregulation is thought to play a role. Some authors still question whether subcorneal pustular dermatosis is a distinct entity from pustular psoriasis.4,5,12 Dapsone is the preferred first-line treatment, with adjunct therapies including topical or systemic corticosteroids, other immunosuppressive agents, tumor necrosis factor inhibitors, and UV light therapy.9
Definitive management of IH is achieved through early delivery; however, systemic corticosteroids often are used in varying doses to control the disease and to extend the pregnancy period closer to term or until delivery is considered viable. Additional therapies that can be considered include infliximab, cyclosporine, and topical corticosteroids, in conjunction with fluid and electrolyte maintenance.2,4,10 If symptoms persist despite supportive care and pharmacologic intervention, induction of labor or termination of pregnancy may be indicated. In nonbreastfeeding postpartum mothers with persistent disease, therapies commonly used in generalized pustular psoriasis may be given.11
- Hebra F. Ueber einzelne wahrend Schwangerschaft, des wacherbette unde bei uterinal. Krankheiten der Frauen zu beobachtende Hautkrankheiten. Wien Med Wochenschr. 1872;48:1197-1202.
- Fouda UM, Fouda RM, Ammar HM, et al. Impetigo herpetiformis during the puerperium triggered by secondary hypoparathyroidism: a case report. Cases J. 2009;2:9338. doi:10.1186/1757-1626-2-9338
- Kroumpouzos G, Cohen LM. Dermatoses of pregnancy. J Am Acad Dermatol. 2001;45:1-22. doi:10.1067/mjd.2001.114595
- Liu J, Ali K, Lou H, et al. First-trimester impetigo herpetiformis leads to stillbirth: a case report. Dermatol Ther (Heidelb). 2022;12:1271-1279. doi:10.1007/s13555-022-00735-9
- Lotem M, Katzenelson V, Rotem A, et al. Impetigo herpetiformis: a variant of pustular psoriasis or a separate entity? J Am Acad Dermatol. 1989;20:338-41. doi:10.1016/s0190-9622(89)70042-6
- Stadler PC, Oschmann A, Kerl-French K, et al. Acute generalized exanthematous pustulosis: clinical characteristics, pathogenesis, and management. Dermatology. 2023;239:328-333. doi:10.1159/000529218
- Abdelhafez MMA, Ahmed KAM, Daud MNBM, et al. Pemphigoid gestationis and adverse pregnancy outcomes: a literature review. J Gynecol Obstet Hum Reprod. 2022;51:102370. doi:10.1016 /j.jogoh.2022.102370
- Reunala T, Hervonen K, Salmi T. Dermatitis herpetiformis: an update on diagnosis and management. Am J Clin Dermatol. 2021;22:329-338. doi:10.1007/s40257-020-00584-2
- Watts PJ, Khachemoune A. Subcorneal pustular dermatosis: a review of 30 years of progress. Am J Clin Dermatol. 2016;17:653-671. doi:10.1007 /s40257-016-0202-8
- Robinson A, Van Voorhees AS, Hsu S, et al. Treatment of pustular psoriasis: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol. 2012;67:279-288. doi:10.1016/j.jaad.2011.01.032
- Bukhari IA. Impetigo herpetiformis in a primigravida: successful treatment with etanercept. J Drugs Dermatol. 2004;3:449-451.
- Chang SE, Kim HH, Choi JH, et al. Impetigo herpetiformis followed by generalized pustular psoriasis: more evidence of same disease entity. Int J Dermatol. 2003;42(9):754-755.
THE DIAGNOSIS: Impetigo Herpetiformis
Histopathology revealed epidermal acanthosis and spongiosis with overlying parakeratosis associated with subcorneal and intracorneal neutrophils, papillary dermal edema, and dermal mixed inflammation with neutrophils and eosinophils. Both direct immunofluorescence and periodic acid–Schiff studies were negative. Blood and pustule cultures were sterile and the skin flora were normal. Based on these findings, a diagnosis of impetigo herpetiformis (IH) was made. The condition improved with systemic and topical steroids, supportive care, and an intravenous infusion of infliximab 5 mg/kg. At 3 weeks’ follow-up, the patient demonstrated near-complete resolution and later delivered successfully at 40 weeks’ gestation without complications.
Impetigo herpetiformis, also known as pustular psoriasis of pregnancy, is an exceedingly rare gestational dermatosis that typically manifests in the third trimester and can be life-threatening for both the mother and fetus. The term was first used in 1872 to describe 5 pregnant women with extensive acute pustular eruptions, all in unstable condition; 4 (80%)of the cases resulted in maternal death, and all resulted in fetal death.1 Impetigo herpetiformis is characterized by pruritic and painful erythematous patches studded at the periphery with subcorneal pustules. Eruptions usually occur in the flexural areas and spread centrifugally, with extension of the lesions peripherally as the center erodes and crusts. Sparing of the face, palms, and soles is expected, and mucosal involvement is rare. Generalized involvement and exfoliation may occur in extreme cases.2 While IH typically manifests during the third trimester, it may occur any time throughout pregnancy or immediately postpartum.3 A few cases have been reported in the puerperium.2 Common symptoms include fever, chills, malaise, anorexia, nausea, vomiting, diarrhea, and arthralgias. Less common complications include hypoalbuminemia and severe hypocalcemia leading to tetany, seizures, and delirium.2,3 While maternal mortality is uncommon, fetal mortality often is a more pressing risk and is attributed to placental insufficiency.3,4 For this reason, early delivery commonly is considered in severe cases.
Whether IH is a separate entity or a variant of pustular psoriasis remains heavily debated. Although the histopathology of IH is identical to pustular psoriasis, the lack of a personal and family history of psoriasis, symptom resolution with delivery, and possible recurrence during successive pregnancies help differentiate IH from generalized pustular psoriasis.2,5 Earlier onset, diffuse involvement, faster progression, and recurrence in subsequent pregnancies all have been linked to a worse prognosis.4
The differential diagnosis for IH includes acute generalized exanthematous pustulosis, pemphigoid gestationis, dermatitis herpetiformis, and subcorneal pustular dermatosis. Acute generalized exanthematous pustulosis is an uncommon severe cutaneous drug reaction characterized by the sudden onset of numerous sterile pustules on erythematous skin within 48 hours of exposure. The most common offending medications include pristinamycin and beta-lactam antibiotics. A high fever, neutrophilic leukocytosis, and hypocalcemia often accompany acute generalized exanthematous pustulosis.6 Prompt diagnosis and withdrawal of the offending drug as well as supportive care and symptomatic treatment are crucial for disease management, as systemic symptoms and even organ involvement may occur.6
Pemphigoid gestationis, also known as gestational pemphigoid or herpes gestationis, is a rare autoimmune blistering disorder that primarily affects pregnant women. It typically manifests in the second or third trimester or shortly after delivery. Clinically, it manifests as an intensely pruritic polymorphic eruption of urticarial papules and plaques accompanied by vesicles and bullae and often is distributed on the abdomen and extends to other body regions. Although the exact etiology is unknown, pemphigoid gestationis is caused by autoantibodies targeting the BP180 and BP230 hemidesmosomal proteins.7 Treatment usually involves systemic corticosteroids and may require additional immunosuppressive therapy. In most cases, patients see resolution within 6 months of delivery.7
Dermatitis herpetiformis is a chronic autoimmune blistering skin disorder characterized by intensely pruritic, grouped vesicles and papules, often distributed symmetrically on extensor surfaces such as the elbows, knees, buttocks, and back. It is closely associated with celiac disease and is triggered by gluten ingestion in genetically predisposed individuals with human leukocyte antigen DQ2 and DQ8 haplotypes. Dermatitis herpetiformis is caused by deposition of IgA antibodies that target tissue transglutaminase 3 at the dermal papillae, leading to inflammation and blister formation. 8 Treatment typically involves a gluten-free diet and medications such as dapsone to alleviate symptoms and prevent recurrence.
Subcorneal pustular dermatosis, also known as Sneddon-Wilkinson disease, is a rare chronic relapsing pustular dermatosis characterized by sterile superficial pustules arranged in annular or circinate patterns on erythematous plaques. It predominantly affects middleaged women and often is associated with underlying conditions such as IgA gammopathy or monoclonal gammopathy of undetermined significance. The pathogenesis remains unclear, but immune dysregulation is thought to play a role. Some authors still question whether subcorneal pustular dermatosis is a distinct entity from pustular psoriasis.4,5,12 Dapsone is the preferred first-line treatment, with adjunct therapies including topical or systemic corticosteroids, other immunosuppressive agents, tumor necrosis factor inhibitors, and UV light therapy.9
Definitive management of IH is achieved through early delivery; however, systemic corticosteroids often are used in varying doses to control the disease and to extend the pregnancy period closer to term or until delivery is considered viable. Additional therapies that can be considered include infliximab, cyclosporine, and topical corticosteroids, in conjunction with fluid and electrolyte maintenance.2,4,10 If symptoms persist despite supportive care and pharmacologic intervention, induction of labor or termination of pregnancy may be indicated. In nonbreastfeeding postpartum mothers with persistent disease, therapies commonly used in generalized pustular psoriasis may be given.11
THE DIAGNOSIS: Impetigo Herpetiformis
Histopathology revealed epidermal acanthosis and spongiosis with overlying parakeratosis associated with subcorneal and intracorneal neutrophils, papillary dermal edema, and dermal mixed inflammation with neutrophils and eosinophils. Both direct immunofluorescence and periodic acid–Schiff studies were negative. Blood and pustule cultures were sterile and the skin flora were normal. Based on these findings, a diagnosis of impetigo herpetiformis (IH) was made. The condition improved with systemic and topical steroids, supportive care, and an intravenous infusion of infliximab 5 mg/kg. At 3 weeks’ follow-up, the patient demonstrated near-complete resolution and later delivered successfully at 40 weeks’ gestation without complications.
Impetigo herpetiformis, also known as pustular psoriasis of pregnancy, is an exceedingly rare gestational dermatosis that typically manifests in the third trimester and can be life-threatening for both the mother and fetus. The term was first used in 1872 to describe 5 pregnant women with extensive acute pustular eruptions, all in unstable condition; 4 (80%)of the cases resulted in maternal death, and all resulted in fetal death.1 Impetigo herpetiformis is characterized by pruritic and painful erythematous patches studded at the periphery with subcorneal pustules. Eruptions usually occur in the flexural areas and spread centrifugally, with extension of the lesions peripherally as the center erodes and crusts. Sparing of the face, palms, and soles is expected, and mucosal involvement is rare. Generalized involvement and exfoliation may occur in extreme cases.2 While IH typically manifests during the third trimester, it may occur any time throughout pregnancy or immediately postpartum.3 A few cases have been reported in the puerperium.2 Common symptoms include fever, chills, malaise, anorexia, nausea, vomiting, diarrhea, and arthralgias. Less common complications include hypoalbuminemia and severe hypocalcemia leading to tetany, seizures, and delirium.2,3 While maternal mortality is uncommon, fetal mortality often is a more pressing risk and is attributed to placental insufficiency.3,4 For this reason, early delivery commonly is considered in severe cases.
Whether IH is a separate entity or a variant of pustular psoriasis remains heavily debated. Although the histopathology of IH is identical to pustular psoriasis, the lack of a personal and family history of psoriasis, symptom resolution with delivery, and possible recurrence during successive pregnancies help differentiate IH from generalized pustular psoriasis.2,5 Earlier onset, diffuse involvement, faster progression, and recurrence in subsequent pregnancies all have been linked to a worse prognosis.4
The differential diagnosis for IH includes acute generalized exanthematous pustulosis, pemphigoid gestationis, dermatitis herpetiformis, and subcorneal pustular dermatosis. Acute generalized exanthematous pustulosis is an uncommon severe cutaneous drug reaction characterized by the sudden onset of numerous sterile pustules on erythematous skin within 48 hours of exposure. The most common offending medications include pristinamycin and beta-lactam antibiotics. A high fever, neutrophilic leukocytosis, and hypocalcemia often accompany acute generalized exanthematous pustulosis.6 Prompt diagnosis and withdrawal of the offending drug as well as supportive care and symptomatic treatment are crucial for disease management, as systemic symptoms and even organ involvement may occur.6
Pemphigoid gestationis, also known as gestational pemphigoid or herpes gestationis, is a rare autoimmune blistering disorder that primarily affects pregnant women. It typically manifests in the second or third trimester or shortly after delivery. Clinically, it manifests as an intensely pruritic polymorphic eruption of urticarial papules and plaques accompanied by vesicles and bullae and often is distributed on the abdomen and extends to other body regions. Although the exact etiology is unknown, pemphigoid gestationis is caused by autoantibodies targeting the BP180 and BP230 hemidesmosomal proteins.7 Treatment usually involves systemic corticosteroids and may require additional immunosuppressive therapy. In most cases, patients see resolution within 6 months of delivery.7
Dermatitis herpetiformis is a chronic autoimmune blistering skin disorder characterized by intensely pruritic, grouped vesicles and papules, often distributed symmetrically on extensor surfaces such as the elbows, knees, buttocks, and back. It is closely associated with celiac disease and is triggered by gluten ingestion in genetically predisposed individuals with human leukocyte antigen DQ2 and DQ8 haplotypes. Dermatitis herpetiformis is caused by deposition of IgA antibodies that target tissue transglutaminase 3 at the dermal papillae, leading to inflammation and blister formation. 8 Treatment typically involves a gluten-free diet and medications such as dapsone to alleviate symptoms and prevent recurrence.
Subcorneal pustular dermatosis, also known as Sneddon-Wilkinson disease, is a rare chronic relapsing pustular dermatosis characterized by sterile superficial pustules arranged in annular or circinate patterns on erythematous plaques. It predominantly affects middleaged women and often is associated with underlying conditions such as IgA gammopathy or monoclonal gammopathy of undetermined significance. The pathogenesis remains unclear, but immune dysregulation is thought to play a role. Some authors still question whether subcorneal pustular dermatosis is a distinct entity from pustular psoriasis.4,5,12 Dapsone is the preferred first-line treatment, with adjunct therapies including topical or systemic corticosteroids, other immunosuppressive agents, tumor necrosis factor inhibitors, and UV light therapy.9
Definitive management of IH is achieved through early delivery; however, systemic corticosteroids often are used in varying doses to control the disease and to extend the pregnancy period closer to term or until delivery is considered viable. Additional therapies that can be considered include infliximab, cyclosporine, and topical corticosteroids, in conjunction with fluid and electrolyte maintenance.2,4,10 If symptoms persist despite supportive care and pharmacologic intervention, induction of labor or termination of pregnancy may be indicated. In nonbreastfeeding postpartum mothers with persistent disease, therapies commonly used in generalized pustular psoriasis may be given.11
- Hebra F. Ueber einzelne wahrend Schwangerschaft, des wacherbette unde bei uterinal. Krankheiten der Frauen zu beobachtende Hautkrankheiten. Wien Med Wochenschr. 1872;48:1197-1202.
- Fouda UM, Fouda RM, Ammar HM, et al. Impetigo herpetiformis during the puerperium triggered by secondary hypoparathyroidism: a case report. Cases J. 2009;2:9338. doi:10.1186/1757-1626-2-9338
- Kroumpouzos G, Cohen LM. Dermatoses of pregnancy. J Am Acad Dermatol. 2001;45:1-22. doi:10.1067/mjd.2001.114595
- Liu J, Ali K, Lou H, et al. First-trimester impetigo herpetiformis leads to stillbirth: a case report. Dermatol Ther (Heidelb). 2022;12:1271-1279. doi:10.1007/s13555-022-00735-9
- Lotem M, Katzenelson V, Rotem A, et al. Impetigo herpetiformis: a variant of pustular psoriasis or a separate entity? J Am Acad Dermatol. 1989;20:338-41. doi:10.1016/s0190-9622(89)70042-6
- Stadler PC, Oschmann A, Kerl-French K, et al. Acute generalized exanthematous pustulosis: clinical characteristics, pathogenesis, and management. Dermatology. 2023;239:328-333. doi:10.1159/000529218
- Abdelhafez MMA, Ahmed KAM, Daud MNBM, et al. Pemphigoid gestationis and adverse pregnancy outcomes: a literature review. J Gynecol Obstet Hum Reprod. 2022;51:102370. doi:10.1016 /j.jogoh.2022.102370
- Reunala T, Hervonen K, Salmi T. Dermatitis herpetiformis: an update on diagnosis and management. Am J Clin Dermatol. 2021;22:329-338. doi:10.1007/s40257-020-00584-2
- Watts PJ, Khachemoune A. Subcorneal pustular dermatosis: a review of 30 years of progress. Am J Clin Dermatol. 2016;17:653-671. doi:10.1007 /s40257-016-0202-8
- Robinson A, Van Voorhees AS, Hsu S, et al. Treatment of pustular psoriasis: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol. 2012;67:279-288. doi:10.1016/j.jaad.2011.01.032
- Bukhari IA. Impetigo herpetiformis in a primigravida: successful treatment with etanercept. J Drugs Dermatol. 2004;3:449-451.
- Chang SE, Kim HH, Choi JH, et al. Impetigo herpetiformis followed by generalized pustular psoriasis: more evidence of same disease entity. Int J Dermatol. 2003;42(9):754-755.
- Hebra F. Ueber einzelne wahrend Schwangerschaft, des wacherbette unde bei uterinal. Krankheiten der Frauen zu beobachtende Hautkrankheiten. Wien Med Wochenschr. 1872;48:1197-1202.
- Fouda UM, Fouda RM, Ammar HM, et al. Impetigo herpetiformis during the puerperium triggered by secondary hypoparathyroidism: a case report. Cases J. 2009;2:9338. doi:10.1186/1757-1626-2-9338
- Kroumpouzos G, Cohen LM. Dermatoses of pregnancy. J Am Acad Dermatol. 2001;45:1-22. doi:10.1067/mjd.2001.114595
- Liu J, Ali K, Lou H, et al. First-trimester impetigo herpetiformis leads to stillbirth: a case report. Dermatol Ther (Heidelb). 2022;12:1271-1279. doi:10.1007/s13555-022-00735-9
- Lotem M, Katzenelson V, Rotem A, et al. Impetigo herpetiformis: a variant of pustular psoriasis or a separate entity? J Am Acad Dermatol. 1989;20:338-41. doi:10.1016/s0190-9622(89)70042-6
- Stadler PC, Oschmann A, Kerl-French K, et al. Acute generalized exanthematous pustulosis: clinical characteristics, pathogenesis, and management. Dermatology. 2023;239:328-333. doi:10.1159/000529218
- Abdelhafez MMA, Ahmed KAM, Daud MNBM, et al. Pemphigoid gestationis and adverse pregnancy outcomes: a literature review. J Gynecol Obstet Hum Reprod. 2022;51:102370. doi:10.1016 /j.jogoh.2022.102370
- Reunala T, Hervonen K, Salmi T. Dermatitis herpetiformis: an update on diagnosis and management. Am J Clin Dermatol. 2021;22:329-338. doi:10.1007/s40257-020-00584-2
- Watts PJ, Khachemoune A. Subcorneal pustular dermatosis: a review of 30 years of progress. Am J Clin Dermatol. 2016;17:653-671. doi:10.1007 /s40257-016-0202-8
- Robinson A, Van Voorhees AS, Hsu S, et al. Treatment of pustular psoriasis: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol. 2012;67:279-288. doi:10.1016/j.jaad.2011.01.032
- Bukhari IA. Impetigo herpetiformis in a primigravida: successful treatment with etanercept. J Drugs Dermatol. 2004;3:449-451.
- Chang SE, Kim HH, Choi JH, et al. Impetigo herpetiformis followed by generalized pustular psoriasis: more evidence of same disease entity. Int J Dermatol. 2003;42(9):754-755.
Generalized Erythematous Plaques and Pustules in a Pregnant Patient
Generalized Erythematous Plaques and Pustules in a Pregnant Patient
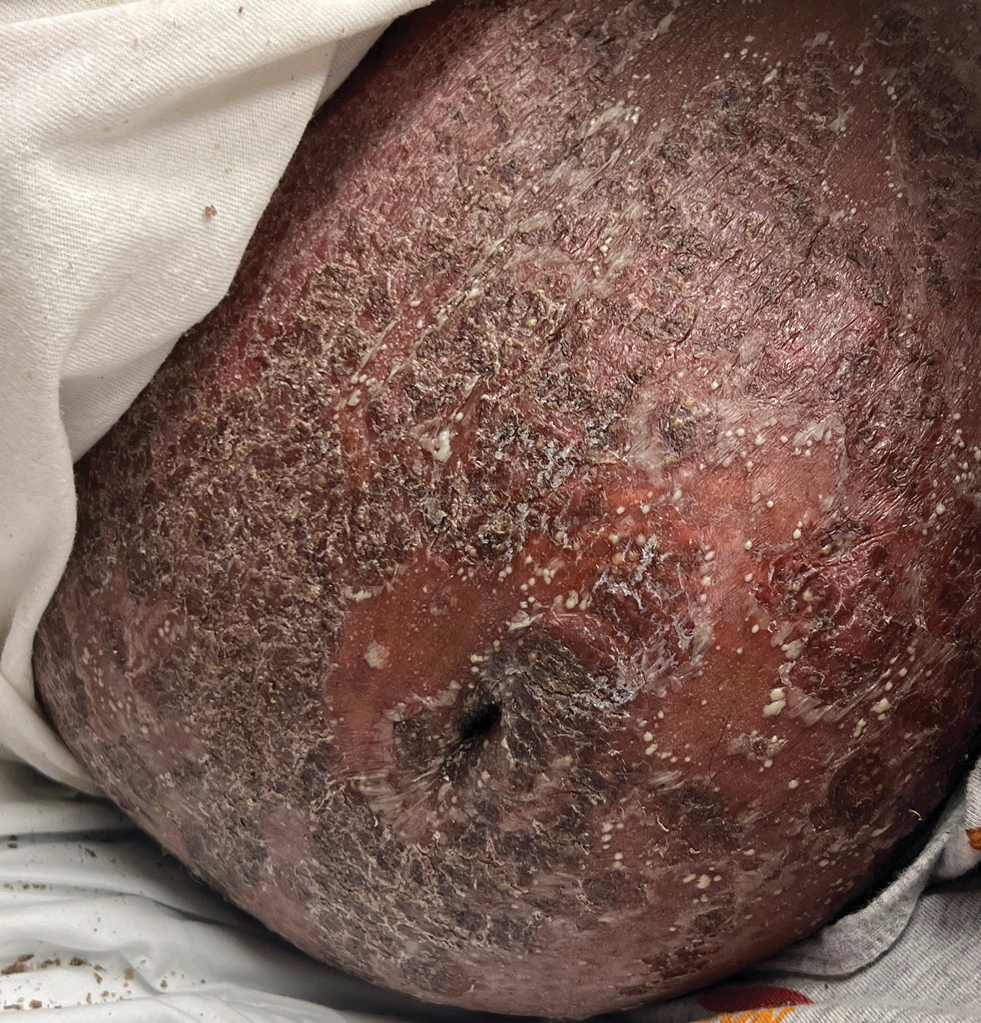
A 17-year-old girl was admitted to the hospital at 19 weeks' gestation for a widespread eruption of erythematous plaques with pustules covering more than 60% of the body and signs of sepsis. The rash initially appeared as a few small spots on the upper chest and under the breasts 5 weeks prior to hospital admission with subsequent spread to the abdomen and groin. At admission, the patient had a mild fever and tachycardia. She reported a history of eczema, herpes simplex virus, and intertrigo. Physical examination performed by dermatology revealed generalized erythematous plaques with pustule-studded margins and overlying scale involving the neck, torso, arms, and legs favoring the flexural areas. There was no involvement of the face, eyes, oral mucosa, palms, soles, or nails. Laboratory testing revealed hypoalbuminemia (2.4 g/dL [reference range, 3.5-5.5 g/dL]) and elevated inflammatory markers, including leukocytosis (15.83×103μL [reference range, 4.50- 11.00×103/μL]), absolute neutrophil count (12.87×103/μL [reference range, 1.50-8.00×103/μL]), and erythrocyte sedimentation rate (124 mm/h [reference range, 0-20 mm/h]). A culture from an abdominal pustule grew 1 colony of taphylococcus epidermidis, a suspected contaminant. A biopsy from a lesion on the right chest was performed.

Fluoroscopy-Induced Chronic Radiation Dermatitis: A Comprehensive Review and Reappraisal
Fluoroscopy-Induced Chronic Radiation Dermatitis: A Comprehensive Review and Reappraisal
Fluoroscopy is an imaging technique that allows for real-time visualization of internal structures in the body using continuous radiography beams. More than 1 million fluoroscopy-guided procedures are performed annually in the United States.1 Utilization of these procedures continues to increase, and so does the probability of related complications, as prolonged exposure to ionizing radiation can cause skin injuries.2 Fortunately, the incidence of radiation-induced skin injuries compared with the total number of fluoroscopic procedures performed remains small,2 although one study suggested the incidence may be as high as 8.9% in at-risk populations.3
Radiation dermatitis is well recognized in dermatology as a complication of oncologic management; however, radiation dermatitis as a complication of fluoroscopic procedures is underrecognized.4 Fluoroscopy-induced radiation dermatitis can be categorized as acute, subacute, or chronic.5 Common fluoroscopic procedures that have been associated with fluoroscopy-induced radiation dermatitis include interventional cardiac procedures, neurovascular procedures, transjugular intrahepatic portosystemic shunt procedures, and endovascular abdominal aortic aneurysm repairs.6,7
Patients with fluoroscopy-induced radiation dermatitis, particularly fluoroscopy-induced chronic radiation dermatitis (FICRD), can present to dermatology up to several years after the initial fluoroscopy procedure with no awareness of the association between the procedure and their skin findings. This presents a diagnostic challenge, and FICRD often is overlooked.5,8-10
We conducted a literature search of PubMed articles indexed for MEDLINE using the search terms fluoroscopy and dermatitis. In this reappraisal, we will provide a comprehensive overview of fluoroscopy-induced radiation dermatitis with an emphasis on FICRD, covering its clinical manifestations, pathophysiology, risk factors, differential diagnosis, histology, and management. The aim of this review is to highlight the salient features and mimickers of FICRD and inform readers how to approach suspected cases, leading to accurate diagnosis and effective management.
Pathophysiology
Fluoroscopy-induced radiation dermatitis is the result of dose-dependent radiation-induced tissue damage. As the peak skin dosage (PSD) of radiation increases over the course of a procedure or multiple procedures, the severity of skin injury predictably increases. During fluoroscopic procedures, the standard irradiation dosage ranges from 0.02 Gy/min to 0.05 Gy/min.11 Transient skin changes may start to be seen around 2 Gy of cumulative exposure. Fluoroscopic procedures typically range in duration from 60 to 120 minutes; however, complex cases may exceed that. Additionally, multiple procedures performed within shorter intervals can result in greater PSD accumulation. Shorter intervals between procedures do not allow enough time for damage repair from the previous procedure and can result in further severe damage when the skin is re-exposed to radiation.2 The American College of Radiology recommends medical follow-up after 10 Gy of cumulative exposure, while cumulative exposure above 15 Gy within a 6- to 12-month period is defined as a sentinel event, according to The Joint Commission.12-14
Depending on the patient’s total radiation dosage during one or more procedures, the result of the tissue damage manifests differently at varying times: early skin changes are categorized as fluoroscopy-induced acute radiation dermatitis, and late skin changes are categorized as FICRD (Table 1).

Clinical Manifestations
Acute radiation dermatitis from fluoroscopic procedures manifests within hours to days up to 90 days following radiation exposure and can be characterized by erythema with blistering, desquamation, epilation, pigmentation changes, and even necrosis if the accumulated dosage exceeds 15 Gy.15 Chronic radiation dermatitis (which as related to fluoroscopic procedures is termed FICRD) has a longer onset of weeks to years and is clinically characterized by telangiectasias, permanent erythema, dermal atrophy, or ulcerations. Clinically, subacute radiation dermatitis shares features of both acute and chronic radiation dermatitis; therefore, it is differentiated based on its histologic features.5,16
Although fluoroscopy-induced acute radiation dermatitis (Table 1) may precede FICRD, acute manifestations of fluoroscopy-related dermatitis can be subtle and often manifest in areas not easily visualized. Because referrals to dermatologists for full-skin examinations after fluoroscopy procedures are not standard, patients may not be aware of the association between these procedures and the development of skin lesions. Nonetheless, some patients may report a history of skin changes such as redness days or weeks after a fluoroscopic procedure with accompanying pain and pruritus limited to the fluoroscopy-exposed region, which tend to self-resolve.17 The risk for FICRD is thought to increase if a history of fluoroscopy-induced acute radiation dermatitis is present.18
The location of the skin findings correlates to the area exposed to prolonged radiation during the procedure(s). The most common areas include the scapular and subscapular regions, the right lateral trunk inferior to the axilla, the mid back, and the right anterolateral chest.16,19,20 These regions are associated with more complex (eg, cardiac) procedures that have been reported to lead to prolonged radiation exposure. The skin findings in FICRD are described as geometric, corresponding to the squarish or rectangular radiography beam that is directed at the patient. Additionally, radiography beams spread outward as they travel in space; therefore, skin injuries are common at the region more distal to the path of origination of the beam.21-23 Subsequently, a geometric, dyspigmented, indurated or atrophic plaque with telangiectasias and erosions or ulcerations with progressive worsening is a common manifestation of FICRD.5,16,23 Patients also commonly present with pruritus or severe pain associated with the lesion.24,25
Dermatologic Manifestations of FICRD
Skin responses seen weeks to years after a fluoroscopic procedure and typically after cumulative radiation exposure of 10 Gy or greater are categorized as FICRD (Table 2). These changes also can be clinically graded based on the Radiation Therapy Oncology Group classification of radiation dermatitis (Tables 3 and 4).26 Chronic changes in the skin largely result from remodeling of the vasculature and the subcutaneous tissue over time. Unlike acute changes, chronic changes typically persist and continue to worsen.27

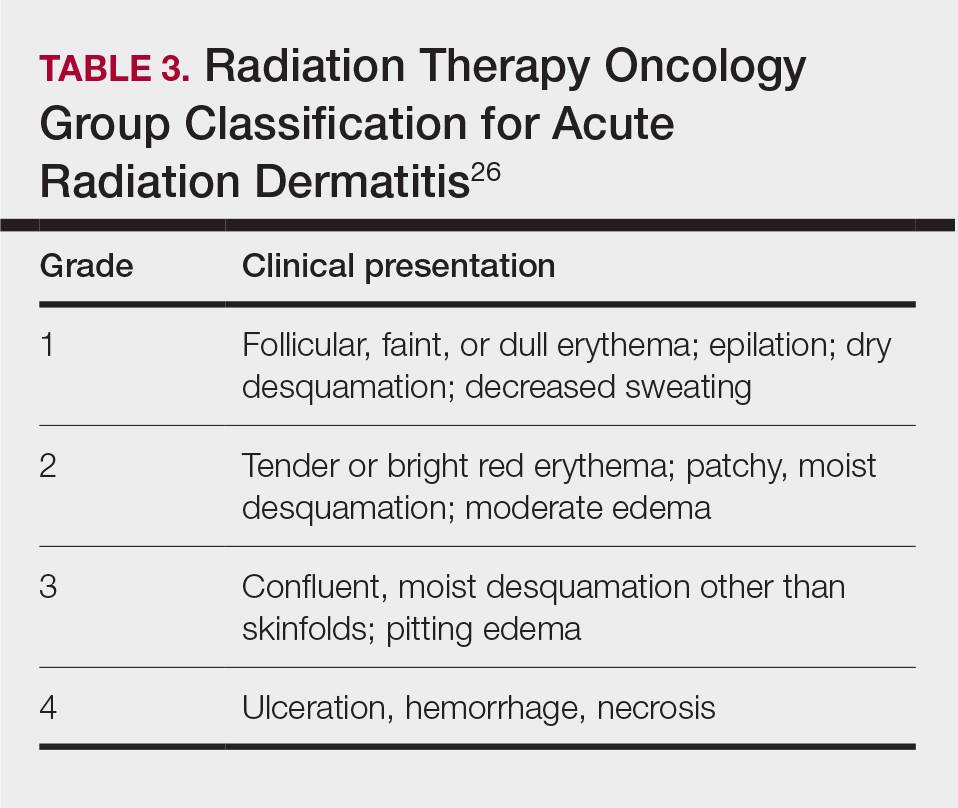
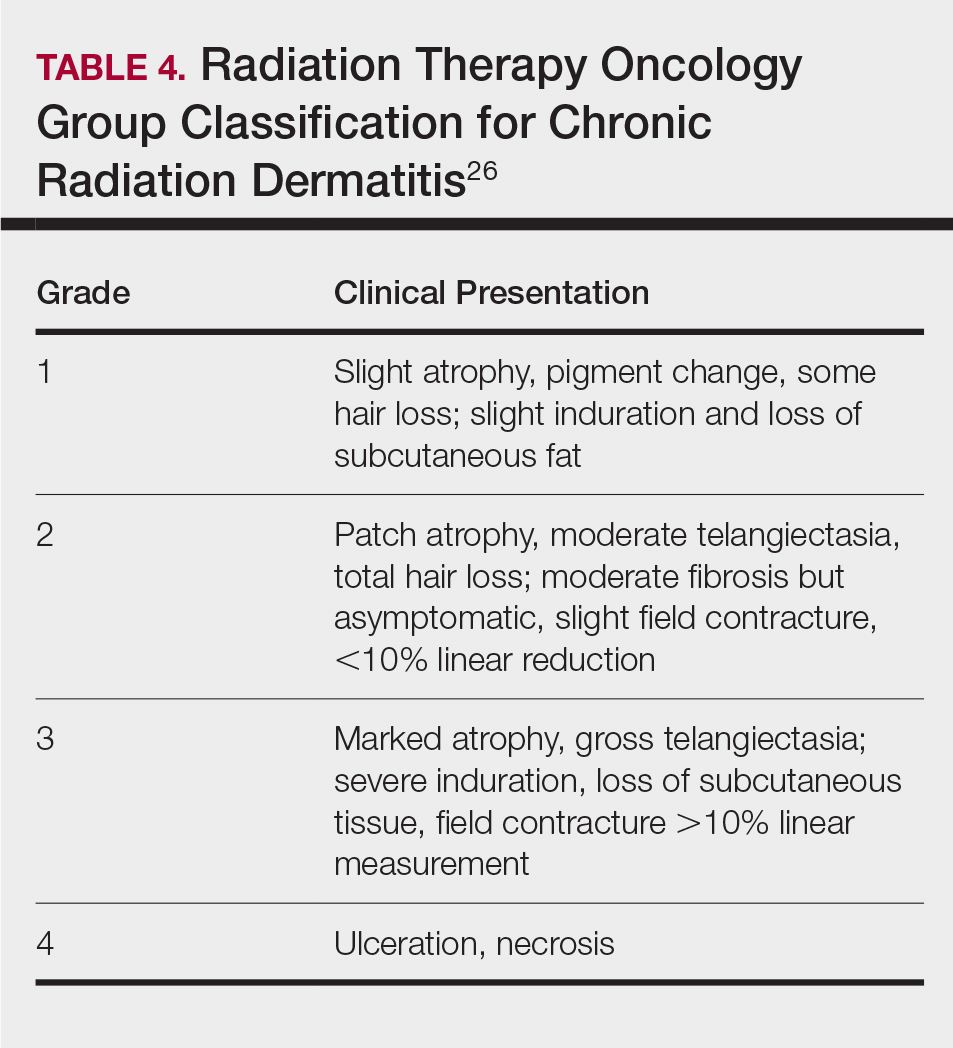
Telangiectasias—Anywhere from months to 1 year after exposure to 10 Gy of radiation, proliferation of atypical superficial vessels in the dermis can be seen, typically manifesting as telangiectasias on physical examination. Telangiectasias can increase with time and can even exhibit a dose-dependent relationship to the radiation exposure.28
Atrophy—Atrophic-appearing skin after radiation exposure is the result of direct injury to both the epidermis and fibroblasts in the dermis. The destruction of keratinocytes leads to a thin epidermis, and destruction of dermal fibroblasts causes insufficient collagen production.29 Clinically, this process manifests as an atrophic plaque that can be seen 12 weeks to 1 year after the procedure.
Fibrosis—Approximately 1 year after the exposure, the initial damage can lead to disruption of molecular pathways, causing fibrosis. Transforming growth factor (TGF) β1 is the main factor involved.29 Damage to the endothelial cells results in increased TGF-β1 levels, which causes increased stimulation of remaining atypical fibroblasts and thus increased irregular collagen deposition.30 Further adding to this knowledge, Wei et al31 recently proposed that damage to the epidermal keratinocytes leads to disruption of yes-associated protein 1, which is a protective factor released from keratinocytes that regulates the dermal fibroblasts. However, extensive damage to the keratinocytes can lead to lower yes-associated protein 1 levels and its downstream activity, leading to increased levels of TGF-β1 and fibroblast activity.31 Clinically, this fibrotic stage is seen as indurated plaques in patients.
Necrosis—There are 2 forms of necrosis that can be seen. Ischemic dermal necrosis typically occurs in the acute phase after 10 weeks and approximately 18 Gy of cumulative exposure. It results from substantial skin damage, including microvascular damage and reduction in dermal capillaries, leading to ischemia of the tissue.2 Late dermal necrosis is the process seen in the chronic stage of FICRD and radiation dermatitis not related to fluoroscopy. It results from the inability of the fibrotic dermis to vascularly support the epidermis above it.2 It can be seen anywhere from 1 to 4 years after the procedure. This stage clinically manifests as worsening ulcerations with major pain and increased risk for secondary infections.16
Dyspigmentation—Dyspigmentation at the site of the radiation exposure can be seen acutely and chronically. Dosage above 15 to 18 Gy can lead to destruction of melanocytes, which can cause hypopigmentation in exposed areas. However, melanocytes are relatively resistant to radiation; therefore, dosages below the threshold of destruction of 15 to 18 Gy can cause melanocytic hyperactivity leading to hyperpigmentation.32 Hence, pigmentary changes can vary greatly. Classically, a central area of hypopigmentation with surrounding hyperpigmentation is seen.
Histology
Histologic appearance of radiation dermatitis varies depending on its stage. Acute radiation dermatitis primarily demonstrates superficial dermal edema, damage to the basal cell layer, small vessel dilation with thrombi, and hemorrhage along with a sparse inflammatory cell infiltrate.33 Histology typically is the only way to characterize subacute radiation dermatitis.5 Lichenoid tissue reaction is its characteristic feature. Mononuclear cells are found adjected to necrotic keratinocytes along with prominent vacuolization of the basal cell layer.33
The key histologic features of chronic radiation dermatitis include epidermal atrophy, hyperkeratosis, telangiectasias, loss of adnexal structures, and dermal fibrosis along with sparse atypical stellate fibroblasts.34 However, clinical context of fluoroscopic exposure is required for the dermatopathologist to differentiate chronic radiation dermatitis from its histologic differential of morphea and lichen sclerosus. In a cross-sectional study, only 1 of 6 cases (16.7%) was correctly diagnosed as chronic radiation dermatitis in the absence of correlating clinical history.35
Risk Factors for FICRD
Since the diagnosis of FICRD can be a clinical challenge, understanding the risk factors can be helpful. The general likelihood of developing FICRD is related to the duration, frequency, interval, intensity, and area of radiation exposure. Procedures exceeding the normal duration of 60 to 120 minutes have been well documented as a substantial risk factor for radiation dermatitis and FICRD.36-38 The risk tends to be higher in longer procedures because they result in more radiation exposure and higher accumulated PSD. Obesity (ie, body mass index >26) is the major risk factor that has been associated with longer procedure times, as higher radiation dosages are necessary to penetrate the body of a larger patient and a larger skin surface area is exposed.37-39
Other risk factors associated with FICRD relate to how prone a patient is to radiation-induced DNA damage. Older patients are at higher risk due to lower intrinsic ability of the tissue to repair itself.11 Patients with a history of connective tissue diseases—particularly lupus, scleroderma, and mixed connective tissue disease—are at an increased risk.40 Furthermore, patients with genetic disorders that impair DNA repair are more susceptible to radiation-induced DNA damage; therefore, patients with ataxia-telangiectasia, xeroderma pigmentosum, Fanconi anemia, and hereditary nevoid basal cell carcinoma are at higher risk for FICRD.39 Similarly, medications that can affect DNA repair also have been shown to be risk factors. These medications include chemotherapeutic agents such as actinomycin D, cyclophosphamide, doxorubicin, methotrexate, and 5-fluorouracil.2,39 Diabetes, hyperthyroidism, and tobacco use also have been shown to increase a patient’s risk for FICRD.39 It also is reasonable to believe that patients with defects in fibroblasts or with elastin or collagen disorders (eg, Ehlers-Danlos syndrome) would be at higher risk, but there are no known studies highlighting the association in the literature.
Differential Diagnosis of FICRD
Acute allergic or irritant contact dermatitis manifests with a localized area of erythematous skin accompanied by pruritus.41 Patients with FICRD can present with a localized area of erythema and hyperpigmentation with minimal atrophy. The lesion may accompany substantial pruritus, which can favor the more common diagnosis of contact dermatitis.35,42,43
Fixed-drug eruption manifests as a well-defined, hyperpigmented plaque in a fixed location that occurs upon ingestion of a drug.44 Fluoroscopy-induced chronic radiation dermatitis lesions are well demarcated and geometrically shaped and therefore can mimic lesions seen in fixed-drug eruptions.45 Additionally, the patient population undergoing fluoroscopic procedures tends to have major comorbidities requiring multiple medications.4
Decubitus ulcers are a result of vascular compromise to an area of skin due to constant pressure and are most commonly seen in the sacral region of patients with obesity.46 Ulcerated FICRD lesions can manifest on the lower midback. These lesions can be seen after endovascular repair of abdominal aortic aneurysm or prostatic artery embolization.20,21 The location of these lesions can mimic decubitus ulcers if fluoroscopic history is unknown. As mentioned, obesity also increases the risk for FICRD.
Morphea can manifest as a localized area of induration and hyperpigmentation of the skin.47 When FICRD has progressed to dermal fibrosis, patients can present with indurated plaques without ulcerations, which can be hard to differentiate from morphea.16,48 However, the presence of ulcerations or hyperkeratosis can differentiate morphea from FICRD.16
Ultimately, it is the location of FICRD lesions that remains the biggest diagnostic clue. Any suspicious lesion present on the scapular or subscapular areas, anterolateral chest, and/or mid back should prompt an investigation into recent or remote history of fluoroscopic procedures.
Management of FICRD
Diagnosis of FICRD should be made clinically based on the history and physical examination whenever possible, since a biopsy is not recommended.35 Wound healing in FICRD is delayed, and biopsies can lead to ulcerations or secondary infections.17 Therefore, it is important to remain suspicious for FICRD. Management of FICRD should correspond to the clinical findings outlined by a recent Delphi consensus survey.49 Regardless, the core of FICRD management framework should always include good hygiene, maintenance of skin hydration to improve epithelialization, and sufficient photoprotection.49,50
Among the first signs of FICRD are telangiectasias. Although asymptomatic, their appearance can be distressing for patients. Pulsed dye laser therapy is a first-line option that has been studied and has shown clinical efficacy for treatment of telangiectasias and vascular changes in patients with FICRD.49,51
If patients develop fibrotic changes, treatment options are limited. Fibrosis is hard to reverse, and the management approach is limited to symptomatic relief. Mechanical and deep-friction massages have been shown to be effective at reducing skin induration in patients.52 Fractional ablative lasers also may be utilized for skin contractures, especially if range of motion is affected.53,54 Although it comes with its own challenges, autologous fat grafting has shown promise in reducing postradiation fibrosis and inducing angiogenesis in tissue.55 Oral pentoxifylline also has shown mild efficacy, as it may be able to suppress TGF-β1 levels.53 However, prevention of fibrotic changes may be the most important. Wei et al31 suggested that low-dose oral prednisolone at 5 mg twice daily for 3 weeks might be an option to prevent the progression of skin changes and even reverse fibrosis to an extent; however, further evidence regarding its efficacy still is necessary. Additionally, no evidence was identified to support the use of topical corticosteroids for fibrotic changes seen in FICRD.56
Patients with FICRD or even acute radiation dermatitis after fluoroscopy tend to develop superficial ulcerations from minor traumas. Good wound hygiene, antiseptic care, and absorbent dressings, such as hydrogel and hydrocolloid, may be sufficient for treating these wounds, as seen in the Figure.42,48 However, once patients develop refractory ulcerations or necrosis, treatment options are then limited to surgical removal with a flap or graft.5,33,42,45
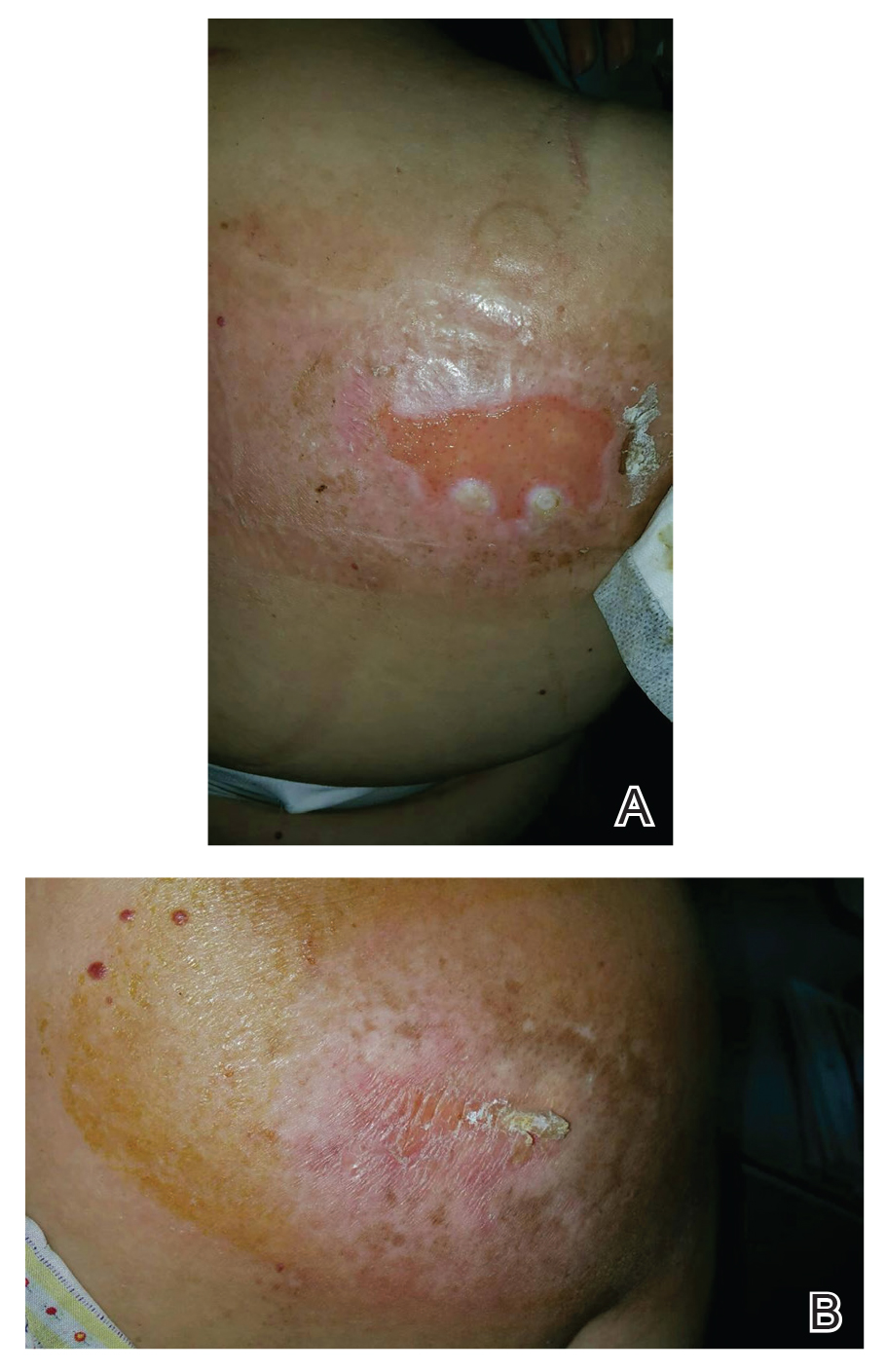
Risk for basal cell carcinomas and squamous cell carcinomas is higher in patients with radiation exposure; however, the exact risk from fluoroscopic procedures is unknown. One study demonstrated an increased risk of 6.9% in development of skin cancer after a median radiation exposure of 15.5 Gy and a mean latency period of 38.3 years,57 and in another retrospective study, the risk was higher in Fitzpatrick skin types I and II.58 Unlike the development of radiodermatitis itself, which shows a dose-dependent response, development of skin cancers follows a stochastic pattern (not dose dependent).59 Therefore, it is important to identify these high-risk patients and establish follow-up.
Conclusion
Fluoroscopy-induced chronic radiation dermatitis can be a diagnostic challenge, as skin changes may not be readily associated with the procedure by patients. Therefore, any lesion with a geometric shape and accompanying chronic radiation dermatitis features located on the scapular or subscapular areas, anterolateral chest, and midback should prompt an investigation into history of fluoroscopic procedures. Treatment of chronic skin changes in FICRD depends on the clinical manifestations. Good hygiene, skin hydration, and sufficient photoprotection are crucial. Finally, long-term monitoring with skin examinations is important to assess for the development of skin cancers in the treated area.
- Benjamin EJ, Muntner P, Alonso A, et al. Heart Disease and Stroke Statistics-2019 Update: a report from the American Heart Association. Circulation. 2019;139:E56-E528. doi:10.1161/CIR.0000000000000659. Published correction appears in Circulation. 2020;141:E33.
- Koenig TR, Wolff D, Mettler FA, et al. Skin injuries from fluoroscopically guided procedures: part 1, characteristics of radiation injury. AJR Am J Roentgenol. 2001;177:3-11. doi:10.2214/ajr.177.1.1770003
- Guesnier-Dopagne M, Boyer L, Pereira B, et al. Incidence of chronic radiodermatitis after fluoroscopically guided interventions: a retrospective study. J Vasc Interv Radiol. 2019;30:692-698.e13. doi:10.1016/j.jvir.2019.01.010
- Cunha N, Cardoso P, Cabete J. Subacute radiation dermatitis following an interventional cardiology procedure. Cutan Ocul Toxicol. 2017;36:297-299. doi:10.1080/15569527.2016.1254649
- Frazier TH, Richardson JB, Fabré VC, et al. Fluoroscopy-induced chronic radiation skin injury: a disease perhaps often overlooked. Arch Dermatol. 2007;143:637-640. doi:10.1001/archderm.143.5.637
- Koenig TR, Mettler FA, Wagner LK. Skin injuries from fluoroscopically guided procedures: part 2, review of 73 cases and recommendations for minimizing dose delivered to patient. AJR Am J Roentgenol. 2001;177:13-20. doi:10.2214/ajr.177.1.1770013
- Shope TB. Radiation-induced skin injuries from fluoroscopy. Radiographics. 1996;16:1195-1199. doi:10.1148/radiographics.16.5.8888398
- Tchanque-Fossuo CN, Isseroff RR, Silverstein MA. Fluoroscopy induced chronic radiation dermatitis should be included in the differential diagnosis of notalgia paresthetica. Dermatol Online J. 2016;22:13030/qt0kh726m9.
- Berlin L. Radiation-induced skin injuries and fluoroscopy. AJR Am J Roentgenol. 2001;177:21-25. doi:10.2214/ajr.177.1.1770021
- Tchanque-Fossuo CN, Kamangar F, Ho B, et al. Fluoroscopy-induced radionecrosis. Dermatol Online J. 2016;22:13030/qt68w910t2.
- Wagner LK, Eifel PJ, Geise RA. Potential biological effects following high X-ray dose interventional procedures. J Vasc Interv Radiol. 1994;5:71-84. doi:10.1016/s1051-0443(94)71456-1
- Balter S, Hopewell JW, Miller DL, et al. Fluoroscopically guided interventional procedures: a review of radiation effects on patients’ skin and hair. Radiology. 2010;254:326-341. doi:10.1148/radiol.2542082312
- Vance AZ, Weinberg BD, Arbique GM, et al. Fluoroscopic sentinel events in neuroendovascular procedures: how to screen, prevent, and address occurrence. AJNR Am J Neuroradiol. 2013;34:1513-1515. doi:10.3174/ajnr.A3185
- Aerts A, Decraene T, van den Oord JJ, et al. Chronic radiodermatitis following percutaneous coronary interventions: a report of two cases. J Eur Acad Dermatol Venereol. 2003;17:340-343. doi:10.1046/j.1468-3083.2003.00687.x
- Rosenthal A, Israilevich R, Moy R. Management of acute radiation dermatitis: a review of the literature and proposal for treatment algorithm. J Am Acad Dermatol. 2019;81:558-567. doi:10.1016/j.jaad.2019.02.047
- Boncher J, Bergfeld WF. Fluoroscopy-induced chronic radiation dermatitis: a report of two additional cases and a brief review of the literature. J Cutan Pathol. 2012;39:63-67. doi:10.1111/j.1600-0560.2011.01754.x
- Spiker A, Zinn Z, Carter WH, et al. Fluoroscopy-induced chronic radiation dermatitis. Am J Cardiol. 2012;110:1861-1863. doi:10.1016/j.amjcard.2012.08.023
- Batrani M, Kubba A, Sundharam J. Fluoroscopy-induced chronic radiation dermatitis masquerading as morphea: a diagnostic pitfall. Indian J Pathol Microbiol. 2018;61:393-396. doi:10.4103/IJPM.IJPM_566_17
- Jeskowiak A, Hubmer M, Prenner G, et al. Radiation induced cutaneous ulcer on the back in a patient with congenital anomaly of the upper cava system. Interact Cardiovasc Thorac Surg. 2011;12:290-292.
- Laborda A, De Assis AM, Ioakeim I, et al. Radiodermitis after prostatic artery embolization: case report and review of the literature. Cardiovasc Intervent Radiol. 2015;38:755-759. doi:10.1007/s00270-015-1083-6
- Lyons AB, Harvey VM, Gusev J. Fluoroscopy-induced chronic radiation dermatitis (FICRD) after endovascular abdominal aortic aneurysm endoleak repair. JAAD Case Rep. 2015;1:403-405. doi:10.1016/j.jdcr.2015.09.022
- Mossman KL. Analysis of risk in computerized tomography and other diagnostic radiology procedures. Comput Radiol. 1982;6:251-256. doi:10.1016/0730-4862(82)90109-3
- Henry MF, Maender JL, Shen Y, et al. Fluoroscopy-induced chronic radiation dermatitis: a report of three cases. Dermatol Online J. 2009;15:3.
- Balter S, Miller DL. Patient skin reactions from interventional fluoroscopy procedures. AJR Am J Roentgenol. 2014;202:W335-W342. doi:10.2214/AJR.13.12029
- Nishimoto S, Fukuda K, Kawai K, et al. Supplementation of bone marrow aspirate-derived platelet-rich plasma for treating radiation-induced ulcer after cardiac fluoroscopic procedures: a preliminary report. Indian J Plast Surg. 2012;45:109-114. doi:10.4103/0970-0358.96599
- Cox JD, Stetz J, Pajak TF. Toxicity criteria of the Radiation Therapy Oncology Group (RTOG) and the European Organization for Research and Treatment of Cancer (EORTC). Int J Radiat Oncol Biol Phys. 1995;31:1341-1346. doi:10.1016/0360-3016(95)00060-C
- Wong RK, Bensadoun RJ, Boers-Doets CB, et al. Clinical practice guidelines for the prevention and treatment of acute and late radiation reactions from the MASCC Skin Toxicity Study Group. Support Care Cancer. 2013;21:2933-2948. doi:10.1007/s00520-013-1896-2
- Turesson I, Notter G. The predictive value of skin telangiectasia for late radiation effects in different normal tissues. Int J Radiat Oncol Biol Phys. 1986;12:603-609. doi:10.1016/0360-3016(86)90069-6
- Hegedus F, Mathew LM, Schwartz RA. Radiation dermatitis: an overview. Int J Dermatol. 2017;56:909-914. doi:10.1111/ijd.13371
- Denham JW, Hauer-Jensen M. The radiotherapeutic injury—a complex ‘wound.’ Radiother Oncol. 2002;63:129-145. doi:10.1016/s0167-8140(02)00060-9
- Wei KC, Lai SF, Huang WL, et al. An innovative targeted therapy for fluoroscopy-induced chronic radiation dermatitis. J Mol Med (Berl). 2022;100:135-146. doi:10.1007/s00109-021-02146-3
- Sitton E. Early and late radiation-induced skin alterations. part I: mechanisms of skin changes. Oncol Nurs Forum. 1992;19:801-807.
- Pruitt LG, Rogers W, Byarlay JA, et al. Subacute radiation dermatitis after fluoroscopy. J Cutan Pathol. 2016;43:1091-1095. doi:10.1111/cup.12815
- Anderson EB, Draft KS, Lee RA, et al. Update in dermatopathology. Am J Clin Pathol. 2006;125(Suppl):S50-S70. doi:10.1309/GMUFNP6LFMPNR86R
- Wei KC, Yang KC, Mar GY, et al. STROBE—radiation ulcer: an overlooked complication of fluoroscopic intervention: a cross-sectional study. Medicine (Baltimore). 2015;94:e2178. doi:10.1097/MD.0000000000002178
- Otterburn D, Losken A. Iatrogenic fluoroscopy injury to the skin. Ann Plast Surg. 2010;65:462-465. doi:10.1097/SAP.0b013e3181d6e2d3
- Cha MJ, Jo SJ, Cho Y, et al. Patient characteristics and the incidence of radiation-induced dermatitis following radiofrequency catheter ablation. Korean Circ J. 2016;46:646-653. doi:10.4070/kcj.2016.46.5.646
- Dehen L, Vilmer C, Humilière C, et al. Chronic radiodermatitis following cardiac catheterisation: a report of two cases and a brief review of the literature. Heart. 1999;81:308-312. doi:10.1136/hrt.81.3.308
- Brown KR, Rzucidlo E. Acute and chronic radiation injury. J Vasc Surg. 2011;53(Suppl 1):15S-21S. doi:10.1016/j.jvs.2010.06.175. Published correction appears in J Vasc Surg. 2012;55:627.
- Hymes SR, Strom EA, Fife C. Radiation dermatitis: clinical presentation, pathophysiology, and treatment 2006. J Am Acad Dermatol. 2006;54:28-46. doi:10.1016/j.jaad.2005.08.054
- Scheinman PL, Vocanson M, Thyssen JP, et al. Contact dermatitis. Nat Rev Dis Primers. 2021;7:38. doi:10.1038/s41572-021-00271-4
- Cheng TT, Yang HJ. Chronic radiation dermatitis induced by cardiac catheterization: a case report and literature review. Acta Dermatovenerol Alp Pannonica Adriat. 2022;31:147-149.
- Minni JP, Nowak M, Usmani A, et al. A unique case of subacute radiodermatitis. Cutis. 2013;91:230-232.
- Flowers H, Brodell R, Brents M, et al. Fixed drug eruptions: presentation, diagnosis, and management. South Med J. 2014;107:724-727. doi:10.14423/SMJ.0000000000000195
- Hashimoto I, Sedo H, Inatsugi K, et al. Severe radiation-induced injury after cardiac catheter ablation: a case requiring free anterolateral thigh flap and vastus lateralis muscle flap reconstruction on the upper arm. J Plast Reconstr Aesthet Surg. 2008;61:704-708. doi:10.1016/j.bjps.2007.01.003
- Mervis JS, Phillips TJ. Pressure ulcers: pathophysiology, epidemiology, risk factors, and presentation. J Am Acad Dermatol. 2019;81:881-890. doi:10.1016/j.jaad.2018.12.069
- Careta MF, Romiti R. Localized scleroderma: clinical spectrum and therapeutic update. An Bras Dermatol. 2015;90:62-73. doi:10.1590/abd1806-4841.20152890
- Herz-Ruelas ME, Gómez-Flores M, Moxica-Del Angel J, et al. Ulcerated radiodermatitis induced after fluoroscopically guided stent implantation angioplasty. Case Rep Dermatol Med. 2014;2014:768624. doi:10.1155/2014/768624
- Wilson BN, Shah R, Menzer C, et al. Consensus on the clinical management of chronic radiation dermatitis and radiation fibrosis: a Delphi survey. Br J Dermatol. 2022;187:1054-1056. doi:10.1111/bjd.21852
- Khanna NR, Kumar DP, Laskar SG, et al. Radiation dermatitis: an overview. Indian J Burns. 2013;21:24-31. doi:10.4103/0971-653x.121877
- Spalek M. Chronic radiation-induced dermatitis: challenges and solutions. Clin Cosmet Investig Dermatol. 2016;9:473-482. doi:10.2147/CCID.S94320
- Bourgeois JF, Gourgou S, Kramar A, et al. A randomized, prospective study using the LPG technique in treating radiation-induced skin fibrosis: clinical and profilometric analysis. Skin Res Technol. 2008;14:71-76. doi:10.1111/j.1600-0846.2007.00263.x
- Borrelli MR, Shen AH, Lee GK, et al. Radiation-induced skinfibrosis: pathogenesis, current treatment options, and emerging therapeutics. Ann Plast Surg. 2019;83(4S Suppl 1):S59-S64. doi:10.1097/SAP.0000000000002098
- Wilson B, Shah R, Menzer C, et al. Laser therapy as a treatment for chronic radiation fibrosis. Lasers Surg Med. 2023;55:82-88. doi:10.1002/lsm.23617
- Rigotti G, Marchi A, Galiè M, et al. Clinical treatment of radiotherapy tissue damage by lipoaspirate transplant: a healing process mediated by adipose-derived adult stem cells. Plast Reconstr Surg. 2007;119:1409-1422. doi:10.1097/01.prs.0000256047.47909.71
- Leventhal J, Young MR. Radiation dermatitis: recognition, prevention, and management. Oncology (Williston Park). 2017;31:885-899.
- van Vloten WA, Hermans J, van Daal WA. Radiation-induced skin cancer and radiodermatitis of the head and neck. Cancer. 1987;59:411-414. doi:10.1002/1097-0142(19870201)59:3<411::aid-cncr2820590310>3.0.co;2-z
- Davis MM, Hanke CW, Zollinger TW, et al. Skin cancer in patients with chronic radiation dermatitis. J Am Acad Dermatol. 1989;20:608-616. doi:10.1016/s0190-9622(89)70072-4
- Miller DL, Balter S, Schueler BA, et al. Clinical radiation management for fluoroscopically guided interventional procedures. Radiology. 2010;257:321-332. doi:10.1148/radiol.10091269
Fluoroscopy is an imaging technique that allows for real-time visualization of internal structures in the body using continuous radiography beams. More than 1 million fluoroscopy-guided procedures are performed annually in the United States.1 Utilization of these procedures continues to increase, and so does the probability of related complications, as prolonged exposure to ionizing radiation can cause skin injuries.2 Fortunately, the incidence of radiation-induced skin injuries compared with the total number of fluoroscopic procedures performed remains small,2 although one study suggested the incidence may be as high as 8.9% in at-risk populations.3
Radiation dermatitis is well recognized in dermatology as a complication of oncologic management; however, radiation dermatitis as a complication of fluoroscopic procedures is underrecognized.4 Fluoroscopy-induced radiation dermatitis can be categorized as acute, subacute, or chronic.5 Common fluoroscopic procedures that have been associated with fluoroscopy-induced radiation dermatitis include interventional cardiac procedures, neurovascular procedures, transjugular intrahepatic portosystemic shunt procedures, and endovascular abdominal aortic aneurysm repairs.6,7
Patients with fluoroscopy-induced radiation dermatitis, particularly fluoroscopy-induced chronic radiation dermatitis (FICRD), can present to dermatology up to several years after the initial fluoroscopy procedure with no awareness of the association between the procedure and their skin findings. This presents a diagnostic challenge, and FICRD often is overlooked.5,8-10
We conducted a literature search of PubMed articles indexed for MEDLINE using the search terms fluoroscopy and dermatitis. In this reappraisal, we will provide a comprehensive overview of fluoroscopy-induced radiation dermatitis with an emphasis on FICRD, covering its clinical manifestations, pathophysiology, risk factors, differential diagnosis, histology, and management. The aim of this review is to highlight the salient features and mimickers of FICRD and inform readers how to approach suspected cases, leading to accurate diagnosis and effective management.
Pathophysiology
Fluoroscopy-induced radiation dermatitis is the result of dose-dependent radiation-induced tissue damage. As the peak skin dosage (PSD) of radiation increases over the course of a procedure or multiple procedures, the severity of skin injury predictably increases. During fluoroscopic procedures, the standard irradiation dosage ranges from 0.02 Gy/min to 0.05 Gy/min.11 Transient skin changes may start to be seen around 2 Gy of cumulative exposure. Fluoroscopic procedures typically range in duration from 60 to 120 minutes; however, complex cases may exceed that. Additionally, multiple procedures performed within shorter intervals can result in greater PSD accumulation. Shorter intervals between procedures do not allow enough time for damage repair from the previous procedure and can result in further severe damage when the skin is re-exposed to radiation.2 The American College of Radiology recommends medical follow-up after 10 Gy of cumulative exposure, while cumulative exposure above 15 Gy within a 6- to 12-month period is defined as a sentinel event, according to The Joint Commission.12-14
Depending on the patient’s total radiation dosage during one or more procedures, the result of the tissue damage manifests differently at varying times: early skin changes are categorized as fluoroscopy-induced acute radiation dermatitis, and late skin changes are categorized as FICRD (Table 1).
Clinical Manifestations
Acute radiation dermatitis from fluoroscopic procedures manifests within hours to days up to 90 days following radiation exposure and can be characterized by erythema with blistering, desquamation, epilation, pigmentation changes, and even necrosis if the accumulated dosage exceeds 15 Gy.15 Chronic radiation dermatitis (which as related to fluoroscopic procedures is termed FICRD) has a longer onset of weeks to years and is clinically characterized by telangiectasias, permanent erythema, dermal atrophy, or ulcerations. Clinically, subacute radiation dermatitis shares features of both acute and chronic radiation dermatitis; therefore, it is differentiated based on its histologic features.5,16
Although fluoroscopy-induced acute radiation dermatitis (Table 1) may precede FICRD, acute manifestations of fluoroscopy-related dermatitis can be subtle and often manifest in areas not easily visualized. Because referrals to dermatologists for full-skin examinations after fluoroscopy procedures are not standard, patients may not be aware of the association between these procedures and the development of skin lesions. Nonetheless, some patients may report a history of skin changes such as redness days or weeks after a fluoroscopic procedure with accompanying pain and pruritus limited to the fluoroscopy-exposed region, which tend to self-resolve.17 The risk for FICRD is thought to increase if a history of fluoroscopy-induced acute radiation dermatitis is present.18
The location of the skin findings correlates to the area exposed to prolonged radiation during the procedure(s). The most common areas include the scapular and subscapular regions, the right lateral trunk inferior to the axilla, the mid back, and the right anterolateral chest.16,19,20 These regions are associated with more complex (eg, cardiac) procedures that have been reported to lead to prolonged radiation exposure. The skin findings in FICRD are described as geometric, corresponding to the squarish or rectangular radiography beam that is directed at the patient. Additionally, radiography beams spread outward as they travel in space; therefore, skin injuries are common at the region more distal to the path of origination of the beam.21-23 Subsequently, a geometric, dyspigmented, indurated or atrophic plaque with telangiectasias and erosions or ulcerations with progressive worsening is a common manifestation of FICRD.5,16,23 Patients also commonly present with pruritus or severe pain associated with the lesion.24,25
Dermatologic Manifestations of FICRD
Skin responses seen weeks to years after a fluoroscopic procedure and typically after cumulative radiation exposure of 10 Gy or greater are categorized as FICRD (Table 2). These changes also can be clinically graded based on the Radiation Therapy Oncology Group classification of radiation dermatitis (Tables 3 and 4).26 Chronic changes in the skin largely result from remodeling of the vasculature and the subcutaneous tissue over time. Unlike acute changes, chronic changes typically persist and continue to worsen.27
Telangiectasias—Anywhere from months to 1 year after exposure to 10 Gy of radiation, proliferation of atypical superficial vessels in the dermis can be seen, typically manifesting as telangiectasias on physical examination. Telangiectasias can increase with time and can even exhibit a dose-dependent relationship to the radiation exposure.28
Atrophy—Atrophic-appearing skin after radiation exposure is the result of direct injury to both the epidermis and fibroblasts in the dermis. The destruction of keratinocytes leads to a thin epidermis, and destruction of dermal fibroblasts causes insufficient collagen production.29 Clinically, this process manifests as an atrophic plaque that can be seen 12 weeks to 1 year after the procedure.
Fibrosis—Approximately 1 year after the exposure, the initial damage can lead to disruption of molecular pathways, causing fibrosis. Transforming growth factor (TGF) β1 is the main factor involved.29 Damage to the endothelial cells results in increased TGF-β1 levels, which causes increased stimulation of remaining atypical fibroblasts and thus increased irregular collagen deposition.30 Further adding to this knowledge, Wei et al31 recently proposed that damage to the epidermal keratinocytes leads to disruption of yes-associated protein 1, which is a protective factor released from keratinocytes that regulates the dermal fibroblasts. However, extensive damage to the keratinocytes can lead to lower yes-associated protein 1 levels and its downstream activity, leading to increased levels of TGF-β1 and fibroblast activity.31 Clinically, this fibrotic stage is seen as indurated plaques in patients.
Necrosis—There are 2 forms of necrosis that can be seen. Ischemic dermal necrosis typically occurs in the acute phase after 10 weeks and approximately 18 Gy of cumulative exposure. It results from substantial skin damage, including microvascular damage and reduction in dermal capillaries, leading to ischemia of the tissue.2 Late dermal necrosis is the process seen in the chronic stage of FICRD and radiation dermatitis not related to fluoroscopy. It results from the inability of the fibrotic dermis to vascularly support the epidermis above it.2 It can be seen anywhere from 1 to 4 years after the procedure. This stage clinically manifests as worsening ulcerations with major pain and increased risk for secondary infections.16
Dyspigmentation—Dyspigmentation at the site of the radiation exposure can be seen acutely and chronically. Dosage above 15 to 18 Gy can lead to destruction of melanocytes, which can cause hypopigmentation in exposed areas. However, melanocytes are relatively resistant to radiation; therefore, dosages below the threshold of destruction of 15 to 18 Gy can cause melanocytic hyperactivity leading to hyperpigmentation.32 Hence, pigmentary changes can vary greatly. Classically, a central area of hypopigmentation with surrounding hyperpigmentation is seen.
Histology
Histologic appearance of radiation dermatitis varies depending on its stage. Acute radiation dermatitis primarily demonstrates superficial dermal edema, damage to the basal cell layer, small vessel dilation with thrombi, and hemorrhage along with a sparse inflammatory cell infiltrate.33 Histology typically is the only way to characterize subacute radiation dermatitis.5 Lichenoid tissue reaction is its characteristic feature. Mononuclear cells are found adjected to necrotic keratinocytes along with prominent vacuolization of the basal cell layer.33
The key histologic features of chronic radiation dermatitis include epidermal atrophy, hyperkeratosis, telangiectasias, loss of adnexal structures, and dermal fibrosis along with sparse atypical stellate fibroblasts.34 However, clinical context of fluoroscopic exposure is required for the dermatopathologist to differentiate chronic radiation dermatitis from its histologic differential of morphea and lichen sclerosus. In a cross-sectional study, only 1 of 6 cases (16.7%) was correctly diagnosed as chronic radiation dermatitis in the absence of correlating clinical history.35
Risk Factors for FICRD
Since the diagnosis of FICRD can be a clinical challenge, understanding the risk factors can be helpful. The general likelihood of developing FICRD is related to the duration, frequency, interval, intensity, and area of radiation exposure. Procedures exceeding the normal duration of 60 to 120 minutes have been well documented as a substantial risk factor for radiation dermatitis and FICRD.36-38 The risk tends to be higher in longer procedures because they result in more radiation exposure and higher accumulated PSD. Obesity (ie, body mass index >26) is the major risk factor that has been associated with longer procedure times, as higher radiation dosages are necessary to penetrate the body of a larger patient and a larger skin surface area is exposed.37-39
Other risk factors associated with FICRD relate to how prone a patient is to radiation-induced DNA damage. Older patients are at higher risk due to lower intrinsic ability of the tissue to repair itself.11 Patients with a history of connective tissue diseases—particularly lupus, scleroderma, and mixed connective tissue disease—are at an increased risk.40 Furthermore, patients with genetic disorders that impair DNA repair are more susceptible to radiation-induced DNA damage; therefore, patients with ataxia-telangiectasia, xeroderma pigmentosum, Fanconi anemia, and hereditary nevoid basal cell carcinoma are at higher risk for FICRD.39 Similarly, medications that can affect DNA repair also have been shown to be risk factors. These medications include chemotherapeutic agents such as actinomycin D, cyclophosphamide, doxorubicin, methotrexate, and 5-fluorouracil.2,39 Diabetes, hyperthyroidism, and tobacco use also have been shown to increase a patient’s risk for FICRD.39 It also is reasonable to believe that patients with defects in fibroblasts or with elastin or collagen disorders (eg, Ehlers-Danlos syndrome) would be at higher risk, but there are no known studies highlighting the association in the literature.
Differential Diagnosis of FICRD
Acute allergic or irritant contact dermatitis manifests with a localized area of erythematous skin accompanied by pruritus.41 Patients with FICRD can present with a localized area of erythema and hyperpigmentation with minimal atrophy. The lesion may accompany substantial pruritus, which can favor the more common diagnosis of contact dermatitis.35,42,43
Fixed-drug eruption manifests as a well-defined, hyperpigmented plaque in a fixed location that occurs upon ingestion of a drug.44 Fluoroscopy-induced chronic radiation dermatitis lesions are well demarcated and geometrically shaped and therefore can mimic lesions seen in fixed-drug eruptions.45 Additionally, the patient population undergoing fluoroscopic procedures tends to have major comorbidities requiring multiple medications.4
Decubitus ulcers are a result of vascular compromise to an area of skin due to constant pressure and are most commonly seen in the sacral region of patients with obesity.46 Ulcerated FICRD lesions can manifest on the lower midback. These lesions can be seen after endovascular repair of abdominal aortic aneurysm or prostatic artery embolization.20,21 The location of these lesions can mimic decubitus ulcers if fluoroscopic history is unknown. As mentioned, obesity also increases the risk for FICRD.
Morphea can manifest as a localized area of induration and hyperpigmentation of the skin.47 When FICRD has progressed to dermal fibrosis, patients can present with indurated plaques without ulcerations, which can be hard to differentiate from morphea.16,48 However, the presence of ulcerations or hyperkeratosis can differentiate morphea from FICRD.16
Ultimately, it is the location of FICRD lesions that remains the biggest diagnostic clue. Any suspicious lesion present on the scapular or subscapular areas, anterolateral chest, and/or mid back should prompt an investigation into recent or remote history of fluoroscopic procedures.
Management of FICRD
Diagnosis of FICRD should be made clinically based on the history and physical examination whenever possible, since a biopsy is not recommended.35 Wound healing in FICRD is delayed, and biopsies can lead to ulcerations or secondary infections.17 Therefore, it is important to remain suspicious for FICRD. Management of FICRD should correspond to the clinical findings outlined by a recent Delphi consensus survey.49 Regardless, the core of FICRD management framework should always include good hygiene, maintenance of skin hydration to improve epithelialization, and sufficient photoprotection.49,50
Among the first signs of FICRD are telangiectasias. Although asymptomatic, their appearance can be distressing for patients. Pulsed dye laser therapy is a first-line option that has been studied and has shown clinical efficacy for treatment of telangiectasias and vascular changes in patients with FICRD.49,51
If patients develop fibrotic changes, treatment options are limited. Fibrosis is hard to reverse, and the management approach is limited to symptomatic relief. Mechanical and deep-friction massages have been shown to be effective at reducing skin induration in patients.52 Fractional ablative lasers also may be utilized for skin contractures, especially if range of motion is affected.53,54 Although it comes with its own challenges, autologous fat grafting has shown promise in reducing postradiation fibrosis and inducing angiogenesis in tissue.55 Oral pentoxifylline also has shown mild efficacy, as it may be able to suppress TGF-β1 levels.53 However, prevention of fibrotic changes may be the most important. Wei et al31 suggested that low-dose oral prednisolone at 5 mg twice daily for 3 weeks might be an option to prevent the progression of skin changes and even reverse fibrosis to an extent; however, further evidence regarding its efficacy still is necessary. Additionally, no evidence was identified to support the use of topical corticosteroids for fibrotic changes seen in FICRD.56
Patients with FICRD or even acute radiation dermatitis after fluoroscopy tend to develop superficial ulcerations from minor traumas. Good wound hygiene, antiseptic care, and absorbent dressings, such as hydrogel and hydrocolloid, may be sufficient for treating these wounds, as seen in the Figure.42,48 However, once patients develop refractory ulcerations or necrosis, treatment options are then limited to surgical removal with a flap or graft.5,33,42,45
Risk for basal cell carcinomas and squamous cell carcinomas is higher in patients with radiation exposure; however, the exact risk from fluoroscopic procedures is unknown. One study demonstrated an increased risk of 6.9% in development of skin cancer after a median radiation exposure of 15.5 Gy and a mean latency period of 38.3 years,57 and in another retrospective study, the risk was higher in Fitzpatrick skin types I and II.58 Unlike the development of radiodermatitis itself, which shows a dose-dependent response, development of skin cancers follows a stochastic pattern (not dose dependent).59 Therefore, it is important to identify these high-risk patients and establish follow-up.
Conclusion
Fluoroscopy-induced chronic radiation dermatitis can be a diagnostic challenge, as skin changes may not be readily associated with the procedure by patients. Therefore, any lesion with a geometric shape and accompanying chronic radiation dermatitis features located on the scapular or subscapular areas, anterolateral chest, and midback should prompt an investigation into history of fluoroscopic procedures. Treatment of chronic skin changes in FICRD depends on the clinical manifestations. Good hygiene, skin hydration, and sufficient photoprotection are crucial. Finally, long-term monitoring with skin examinations is important to assess for the development of skin cancers in the treated area.
Fluoroscopy is an imaging technique that allows for real-time visualization of internal structures in the body using continuous radiography beams. More than 1 million fluoroscopy-guided procedures are performed annually in the United States.1 Utilization of these procedures continues to increase, and so does the probability of related complications, as prolonged exposure to ionizing radiation can cause skin injuries.2 Fortunately, the incidence of radiation-induced skin injuries compared with the total number of fluoroscopic procedures performed remains small,2 although one study suggested the incidence may be as high as 8.9% in at-risk populations.3
Radiation dermatitis is well recognized in dermatology as a complication of oncologic management; however, radiation dermatitis as a complication of fluoroscopic procedures is underrecognized.4 Fluoroscopy-induced radiation dermatitis can be categorized as acute, subacute, or chronic.5 Common fluoroscopic procedures that have been associated with fluoroscopy-induced radiation dermatitis include interventional cardiac procedures, neurovascular procedures, transjugular intrahepatic portosystemic shunt procedures, and endovascular abdominal aortic aneurysm repairs.6,7
Patients with fluoroscopy-induced radiation dermatitis, particularly fluoroscopy-induced chronic radiation dermatitis (FICRD), can present to dermatology up to several years after the initial fluoroscopy procedure with no awareness of the association between the procedure and their skin findings. This presents a diagnostic challenge, and FICRD often is overlooked.5,8-10
We conducted a literature search of PubMed articles indexed for MEDLINE using the search terms fluoroscopy and dermatitis. In this reappraisal, we will provide a comprehensive overview of fluoroscopy-induced radiation dermatitis with an emphasis on FICRD, covering its clinical manifestations, pathophysiology, risk factors, differential diagnosis, histology, and management. The aim of this review is to highlight the salient features and mimickers of FICRD and inform readers how to approach suspected cases, leading to accurate diagnosis and effective management.
Pathophysiology
Fluoroscopy-induced radiation dermatitis is the result of dose-dependent radiation-induced tissue damage. As the peak skin dosage (PSD) of radiation increases over the course of a procedure or multiple procedures, the severity of skin injury predictably increases. During fluoroscopic procedures, the standard irradiation dosage ranges from 0.02 Gy/min to 0.05 Gy/min.11 Transient skin changes may start to be seen around 2 Gy of cumulative exposure. Fluoroscopic procedures typically range in duration from 60 to 120 minutes; however, complex cases may exceed that. Additionally, multiple procedures performed within shorter intervals can result in greater PSD accumulation. Shorter intervals between procedures do not allow enough time for damage repair from the previous procedure and can result in further severe damage when the skin is re-exposed to radiation.2 The American College of Radiology recommends medical follow-up after 10 Gy of cumulative exposure, while cumulative exposure above 15 Gy within a 6- to 12-month period is defined as a sentinel event, according to The Joint Commission.12-14
Depending on the patient’s total radiation dosage during one or more procedures, the result of the tissue damage manifests differently at varying times: early skin changes are categorized as fluoroscopy-induced acute radiation dermatitis, and late skin changes are categorized as FICRD (Table 1).
Clinical Manifestations
Acute radiation dermatitis from fluoroscopic procedures manifests within hours to days up to 90 days following radiation exposure and can be characterized by erythema with blistering, desquamation, epilation, pigmentation changes, and even necrosis if the accumulated dosage exceeds 15 Gy.15 Chronic radiation dermatitis (which as related to fluoroscopic procedures is termed FICRD) has a longer onset of weeks to years and is clinically characterized by telangiectasias, permanent erythema, dermal atrophy, or ulcerations. Clinically, subacute radiation dermatitis shares features of both acute and chronic radiation dermatitis; therefore, it is differentiated based on its histologic features.5,16
Although fluoroscopy-induced acute radiation dermatitis (Table 1) may precede FICRD, acute manifestations of fluoroscopy-related dermatitis can be subtle and often manifest in areas not easily visualized. Because referrals to dermatologists for full-skin examinations after fluoroscopy procedures are not standard, patients may not be aware of the association between these procedures and the development of skin lesions. Nonetheless, some patients may report a history of skin changes such as redness days or weeks after a fluoroscopic procedure with accompanying pain and pruritus limited to the fluoroscopy-exposed region, which tend to self-resolve.17 The risk for FICRD is thought to increase if a history of fluoroscopy-induced acute radiation dermatitis is present.18
The location of the skin findings correlates to the area exposed to prolonged radiation during the procedure(s). The most common areas include the scapular and subscapular regions, the right lateral trunk inferior to the axilla, the mid back, and the right anterolateral chest.16,19,20 These regions are associated with more complex (eg, cardiac) procedures that have been reported to lead to prolonged radiation exposure. The skin findings in FICRD are described as geometric, corresponding to the squarish or rectangular radiography beam that is directed at the patient. Additionally, radiography beams spread outward as they travel in space; therefore, skin injuries are common at the region more distal to the path of origination of the beam.21-23 Subsequently, a geometric, dyspigmented, indurated or atrophic plaque with telangiectasias and erosions or ulcerations with progressive worsening is a common manifestation of FICRD.5,16,23 Patients also commonly present with pruritus or severe pain associated with the lesion.24,25
Dermatologic Manifestations of FICRD
Skin responses seen weeks to years after a fluoroscopic procedure and typically after cumulative radiation exposure of 10 Gy or greater are categorized as FICRD (Table 2). These changes also can be clinically graded based on the Radiation Therapy Oncology Group classification of radiation dermatitis (Tables 3 and 4).26 Chronic changes in the skin largely result from remodeling of the vasculature and the subcutaneous tissue over time. Unlike acute changes, chronic changes typically persist and continue to worsen.27
Telangiectasias—Anywhere from months to 1 year after exposure to 10 Gy of radiation, proliferation of atypical superficial vessels in the dermis can be seen, typically manifesting as telangiectasias on physical examination. Telangiectasias can increase with time and can even exhibit a dose-dependent relationship to the radiation exposure.28
Atrophy—Atrophic-appearing skin after radiation exposure is the result of direct injury to both the epidermis and fibroblasts in the dermis. The destruction of keratinocytes leads to a thin epidermis, and destruction of dermal fibroblasts causes insufficient collagen production.29 Clinically, this process manifests as an atrophic plaque that can be seen 12 weeks to 1 year after the procedure.
Fibrosis—Approximately 1 year after the exposure, the initial damage can lead to disruption of molecular pathways, causing fibrosis. Transforming growth factor (TGF) β1 is the main factor involved.29 Damage to the endothelial cells results in increased TGF-β1 levels, which causes increased stimulation of remaining atypical fibroblasts and thus increased irregular collagen deposition.30 Further adding to this knowledge, Wei et al31 recently proposed that damage to the epidermal keratinocytes leads to disruption of yes-associated protein 1, which is a protective factor released from keratinocytes that regulates the dermal fibroblasts. However, extensive damage to the keratinocytes can lead to lower yes-associated protein 1 levels and its downstream activity, leading to increased levels of TGF-β1 and fibroblast activity.31 Clinically, this fibrotic stage is seen as indurated plaques in patients.
Necrosis—There are 2 forms of necrosis that can be seen. Ischemic dermal necrosis typically occurs in the acute phase after 10 weeks and approximately 18 Gy of cumulative exposure. It results from substantial skin damage, including microvascular damage and reduction in dermal capillaries, leading to ischemia of the tissue.2 Late dermal necrosis is the process seen in the chronic stage of FICRD and radiation dermatitis not related to fluoroscopy. It results from the inability of the fibrotic dermis to vascularly support the epidermis above it.2 It can be seen anywhere from 1 to 4 years after the procedure. This stage clinically manifests as worsening ulcerations with major pain and increased risk for secondary infections.16
Dyspigmentation—Dyspigmentation at the site of the radiation exposure can be seen acutely and chronically. Dosage above 15 to 18 Gy can lead to destruction of melanocytes, which can cause hypopigmentation in exposed areas. However, melanocytes are relatively resistant to radiation; therefore, dosages below the threshold of destruction of 15 to 18 Gy can cause melanocytic hyperactivity leading to hyperpigmentation.32 Hence, pigmentary changes can vary greatly. Classically, a central area of hypopigmentation with surrounding hyperpigmentation is seen.
Histology
Histologic appearance of radiation dermatitis varies depending on its stage. Acute radiation dermatitis primarily demonstrates superficial dermal edema, damage to the basal cell layer, small vessel dilation with thrombi, and hemorrhage along with a sparse inflammatory cell infiltrate.33 Histology typically is the only way to characterize subacute radiation dermatitis.5 Lichenoid tissue reaction is its characteristic feature. Mononuclear cells are found adjected to necrotic keratinocytes along with prominent vacuolization of the basal cell layer.33
The key histologic features of chronic radiation dermatitis include epidermal atrophy, hyperkeratosis, telangiectasias, loss of adnexal structures, and dermal fibrosis along with sparse atypical stellate fibroblasts.34 However, clinical context of fluoroscopic exposure is required for the dermatopathologist to differentiate chronic radiation dermatitis from its histologic differential of morphea and lichen sclerosus. In a cross-sectional study, only 1 of 6 cases (16.7%) was correctly diagnosed as chronic radiation dermatitis in the absence of correlating clinical history.35
Risk Factors for FICRD
Since the diagnosis of FICRD can be a clinical challenge, understanding the risk factors can be helpful. The general likelihood of developing FICRD is related to the duration, frequency, interval, intensity, and area of radiation exposure. Procedures exceeding the normal duration of 60 to 120 minutes have been well documented as a substantial risk factor for radiation dermatitis and FICRD.36-38 The risk tends to be higher in longer procedures because they result in more radiation exposure and higher accumulated PSD. Obesity (ie, body mass index >26) is the major risk factor that has been associated with longer procedure times, as higher radiation dosages are necessary to penetrate the body of a larger patient and a larger skin surface area is exposed.37-39
Other risk factors associated with FICRD relate to how prone a patient is to radiation-induced DNA damage. Older patients are at higher risk due to lower intrinsic ability of the tissue to repair itself.11 Patients with a history of connective tissue diseases—particularly lupus, scleroderma, and mixed connective tissue disease—are at an increased risk.40 Furthermore, patients with genetic disorders that impair DNA repair are more susceptible to radiation-induced DNA damage; therefore, patients with ataxia-telangiectasia, xeroderma pigmentosum, Fanconi anemia, and hereditary nevoid basal cell carcinoma are at higher risk for FICRD.39 Similarly, medications that can affect DNA repair also have been shown to be risk factors. These medications include chemotherapeutic agents such as actinomycin D, cyclophosphamide, doxorubicin, methotrexate, and 5-fluorouracil.2,39 Diabetes, hyperthyroidism, and tobacco use also have been shown to increase a patient’s risk for FICRD.39 It also is reasonable to believe that patients with defects in fibroblasts or with elastin or collagen disorders (eg, Ehlers-Danlos syndrome) would be at higher risk, but there are no known studies highlighting the association in the literature.
Differential Diagnosis of FICRD
Acute allergic or irritant contact dermatitis manifests with a localized area of erythematous skin accompanied by pruritus.41 Patients with FICRD can present with a localized area of erythema and hyperpigmentation with minimal atrophy. The lesion may accompany substantial pruritus, which can favor the more common diagnosis of contact dermatitis.35,42,43
Fixed-drug eruption manifests as a well-defined, hyperpigmented plaque in a fixed location that occurs upon ingestion of a drug.44 Fluoroscopy-induced chronic radiation dermatitis lesions are well demarcated and geometrically shaped and therefore can mimic lesions seen in fixed-drug eruptions.45 Additionally, the patient population undergoing fluoroscopic procedures tends to have major comorbidities requiring multiple medications.4
Decubitus ulcers are a result of vascular compromise to an area of skin due to constant pressure and are most commonly seen in the sacral region of patients with obesity.46 Ulcerated FICRD lesions can manifest on the lower midback. These lesions can be seen after endovascular repair of abdominal aortic aneurysm or prostatic artery embolization.20,21 The location of these lesions can mimic decubitus ulcers if fluoroscopic history is unknown. As mentioned, obesity also increases the risk for FICRD.
Morphea can manifest as a localized area of induration and hyperpigmentation of the skin.47 When FICRD has progressed to dermal fibrosis, patients can present with indurated plaques without ulcerations, which can be hard to differentiate from morphea.16,48 However, the presence of ulcerations or hyperkeratosis can differentiate morphea from FICRD.16
Ultimately, it is the location of FICRD lesions that remains the biggest diagnostic clue. Any suspicious lesion present on the scapular or subscapular areas, anterolateral chest, and/or mid back should prompt an investigation into recent or remote history of fluoroscopic procedures.
Management of FICRD
Diagnosis of FICRD should be made clinically based on the history and physical examination whenever possible, since a biopsy is not recommended.35 Wound healing in FICRD is delayed, and biopsies can lead to ulcerations or secondary infections.17 Therefore, it is important to remain suspicious for FICRD. Management of FICRD should correspond to the clinical findings outlined by a recent Delphi consensus survey.49 Regardless, the core of FICRD management framework should always include good hygiene, maintenance of skin hydration to improve epithelialization, and sufficient photoprotection.49,50
Among the first signs of FICRD are telangiectasias. Although asymptomatic, their appearance can be distressing for patients. Pulsed dye laser therapy is a first-line option that has been studied and has shown clinical efficacy for treatment of telangiectasias and vascular changes in patients with FICRD.49,51
If patients develop fibrotic changes, treatment options are limited. Fibrosis is hard to reverse, and the management approach is limited to symptomatic relief. Mechanical and deep-friction massages have been shown to be effective at reducing skin induration in patients.52 Fractional ablative lasers also may be utilized for skin contractures, especially if range of motion is affected.53,54 Although it comes with its own challenges, autologous fat grafting has shown promise in reducing postradiation fibrosis and inducing angiogenesis in tissue.55 Oral pentoxifylline also has shown mild efficacy, as it may be able to suppress TGF-β1 levels.53 However, prevention of fibrotic changes may be the most important. Wei et al31 suggested that low-dose oral prednisolone at 5 mg twice daily for 3 weeks might be an option to prevent the progression of skin changes and even reverse fibrosis to an extent; however, further evidence regarding its efficacy still is necessary. Additionally, no evidence was identified to support the use of topical corticosteroids for fibrotic changes seen in FICRD.56
Patients with FICRD or even acute radiation dermatitis after fluoroscopy tend to develop superficial ulcerations from minor traumas. Good wound hygiene, antiseptic care, and absorbent dressings, such as hydrogel and hydrocolloid, may be sufficient for treating these wounds, as seen in the Figure.42,48 However, once patients develop refractory ulcerations or necrosis, treatment options are then limited to surgical removal with a flap or graft.5,33,42,45
Risk for basal cell carcinomas and squamous cell carcinomas is higher in patients with radiation exposure; however, the exact risk from fluoroscopic procedures is unknown. One study demonstrated an increased risk of 6.9% in development of skin cancer after a median radiation exposure of 15.5 Gy and a mean latency period of 38.3 years,57 and in another retrospective study, the risk was higher in Fitzpatrick skin types I and II.58 Unlike the development of radiodermatitis itself, which shows a dose-dependent response, development of skin cancers follows a stochastic pattern (not dose dependent).59 Therefore, it is important to identify these high-risk patients and establish follow-up.
Conclusion
Fluoroscopy-induced chronic radiation dermatitis can be a diagnostic challenge, as skin changes may not be readily associated with the procedure by patients. Therefore, any lesion with a geometric shape and accompanying chronic radiation dermatitis features located on the scapular or subscapular areas, anterolateral chest, and midback should prompt an investigation into history of fluoroscopic procedures. Treatment of chronic skin changes in FICRD depends on the clinical manifestations. Good hygiene, skin hydration, and sufficient photoprotection are crucial. Finally, long-term monitoring with skin examinations is important to assess for the development of skin cancers in the treated area.
- Benjamin EJ, Muntner P, Alonso A, et al. Heart Disease and Stroke Statistics-2019 Update: a report from the American Heart Association. Circulation. 2019;139:E56-E528. doi:10.1161/CIR.0000000000000659. Published correction appears in Circulation. 2020;141:E33.
- Koenig TR, Wolff D, Mettler FA, et al. Skin injuries from fluoroscopically guided procedures: part 1, characteristics of radiation injury. AJR Am J Roentgenol. 2001;177:3-11. doi:10.2214/ajr.177.1.1770003
- Guesnier-Dopagne M, Boyer L, Pereira B, et al. Incidence of chronic radiodermatitis after fluoroscopically guided interventions: a retrospective study. J Vasc Interv Radiol. 2019;30:692-698.e13. doi:10.1016/j.jvir.2019.01.010
- Cunha N, Cardoso P, Cabete J. Subacute radiation dermatitis following an interventional cardiology procedure. Cutan Ocul Toxicol. 2017;36:297-299. doi:10.1080/15569527.2016.1254649
- Frazier TH, Richardson JB, Fabré VC, et al. Fluoroscopy-induced chronic radiation skin injury: a disease perhaps often overlooked. Arch Dermatol. 2007;143:637-640. doi:10.1001/archderm.143.5.637
- Koenig TR, Mettler FA, Wagner LK. Skin injuries from fluoroscopically guided procedures: part 2, review of 73 cases and recommendations for minimizing dose delivered to patient. AJR Am J Roentgenol. 2001;177:13-20. doi:10.2214/ajr.177.1.1770013
- Shope TB. Radiation-induced skin injuries from fluoroscopy. Radiographics. 1996;16:1195-1199. doi:10.1148/radiographics.16.5.8888398
- Tchanque-Fossuo CN, Isseroff RR, Silverstein MA. Fluoroscopy induced chronic radiation dermatitis should be included in the differential diagnosis of notalgia paresthetica. Dermatol Online J. 2016;22:13030/qt0kh726m9.
- Berlin L. Radiation-induced skin injuries and fluoroscopy. AJR Am J Roentgenol. 2001;177:21-25. doi:10.2214/ajr.177.1.1770021
- Tchanque-Fossuo CN, Kamangar F, Ho B, et al. Fluoroscopy-induced radionecrosis. Dermatol Online J. 2016;22:13030/qt68w910t2.
- Wagner LK, Eifel PJ, Geise RA. Potential biological effects following high X-ray dose interventional procedures. J Vasc Interv Radiol. 1994;5:71-84. doi:10.1016/s1051-0443(94)71456-1
- Balter S, Hopewell JW, Miller DL, et al. Fluoroscopically guided interventional procedures: a review of radiation effects on patients’ skin and hair. Radiology. 2010;254:326-341. doi:10.1148/radiol.2542082312
- Vance AZ, Weinberg BD, Arbique GM, et al. Fluoroscopic sentinel events in neuroendovascular procedures: how to screen, prevent, and address occurrence. AJNR Am J Neuroradiol. 2013;34:1513-1515. doi:10.3174/ajnr.A3185
- Aerts A, Decraene T, van den Oord JJ, et al. Chronic radiodermatitis following percutaneous coronary interventions: a report of two cases. J Eur Acad Dermatol Venereol. 2003;17:340-343. doi:10.1046/j.1468-3083.2003.00687.x
- Rosenthal A, Israilevich R, Moy R. Management of acute radiation dermatitis: a review of the literature and proposal for treatment algorithm. J Am Acad Dermatol. 2019;81:558-567. doi:10.1016/j.jaad.2019.02.047
- Boncher J, Bergfeld WF. Fluoroscopy-induced chronic radiation dermatitis: a report of two additional cases and a brief review of the literature. J Cutan Pathol. 2012;39:63-67. doi:10.1111/j.1600-0560.2011.01754.x
- Spiker A, Zinn Z, Carter WH, et al. Fluoroscopy-induced chronic radiation dermatitis. Am J Cardiol. 2012;110:1861-1863. doi:10.1016/j.amjcard.2012.08.023
- Batrani M, Kubba A, Sundharam J. Fluoroscopy-induced chronic radiation dermatitis masquerading as morphea: a diagnostic pitfall. Indian J Pathol Microbiol. 2018;61:393-396. doi:10.4103/IJPM.IJPM_566_17
- Jeskowiak A, Hubmer M, Prenner G, et al. Radiation induced cutaneous ulcer on the back in a patient with congenital anomaly of the upper cava system. Interact Cardiovasc Thorac Surg. 2011;12:290-292.
- Laborda A, De Assis AM, Ioakeim I, et al. Radiodermitis after prostatic artery embolization: case report and review of the literature. Cardiovasc Intervent Radiol. 2015;38:755-759. doi:10.1007/s00270-015-1083-6
- Lyons AB, Harvey VM, Gusev J. Fluoroscopy-induced chronic radiation dermatitis (FICRD) after endovascular abdominal aortic aneurysm endoleak repair. JAAD Case Rep. 2015;1:403-405. doi:10.1016/j.jdcr.2015.09.022
- Mossman KL. Analysis of risk in computerized tomography and other diagnostic radiology procedures. Comput Radiol. 1982;6:251-256. doi:10.1016/0730-4862(82)90109-3
- Henry MF, Maender JL, Shen Y, et al. Fluoroscopy-induced chronic radiation dermatitis: a report of three cases. Dermatol Online J. 2009;15:3.
- Balter S, Miller DL. Patient skin reactions from interventional fluoroscopy procedures. AJR Am J Roentgenol. 2014;202:W335-W342. doi:10.2214/AJR.13.12029
- Nishimoto S, Fukuda K, Kawai K, et al. Supplementation of bone marrow aspirate-derived platelet-rich plasma for treating radiation-induced ulcer after cardiac fluoroscopic procedures: a preliminary report. Indian J Plast Surg. 2012;45:109-114. doi:10.4103/0970-0358.96599
- Cox JD, Stetz J, Pajak TF. Toxicity criteria of the Radiation Therapy Oncology Group (RTOG) and the European Organization for Research and Treatment of Cancer (EORTC). Int J Radiat Oncol Biol Phys. 1995;31:1341-1346. doi:10.1016/0360-3016(95)00060-C
- Wong RK, Bensadoun RJ, Boers-Doets CB, et al. Clinical practice guidelines for the prevention and treatment of acute and late radiation reactions from the MASCC Skin Toxicity Study Group. Support Care Cancer. 2013;21:2933-2948. doi:10.1007/s00520-013-1896-2
- Turesson I, Notter G. The predictive value of skin telangiectasia for late radiation effects in different normal tissues. Int J Radiat Oncol Biol Phys. 1986;12:603-609. doi:10.1016/0360-3016(86)90069-6
- Hegedus F, Mathew LM, Schwartz RA. Radiation dermatitis: an overview. Int J Dermatol. 2017;56:909-914. doi:10.1111/ijd.13371
- Denham JW, Hauer-Jensen M. The radiotherapeutic injury—a complex ‘wound.’ Radiother Oncol. 2002;63:129-145. doi:10.1016/s0167-8140(02)00060-9
- Wei KC, Lai SF, Huang WL, et al. An innovative targeted therapy for fluoroscopy-induced chronic radiation dermatitis. J Mol Med (Berl). 2022;100:135-146. doi:10.1007/s00109-021-02146-3
- Sitton E. Early and late radiation-induced skin alterations. part I: mechanisms of skin changes. Oncol Nurs Forum. 1992;19:801-807.
- Pruitt LG, Rogers W, Byarlay JA, et al. Subacute radiation dermatitis after fluoroscopy. J Cutan Pathol. 2016;43:1091-1095. doi:10.1111/cup.12815
- Anderson EB, Draft KS, Lee RA, et al. Update in dermatopathology. Am J Clin Pathol. 2006;125(Suppl):S50-S70. doi:10.1309/GMUFNP6LFMPNR86R
- Wei KC, Yang KC, Mar GY, et al. STROBE—radiation ulcer: an overlooked complication of fluoroscopic intervention: a cross-sectional study. Medicine (Baltimore). 2015;94:e2178. doi:10.1097/MD.0000000000002178
- Otterburn D, Losken A. Iatrogenic fluoroscopy injury to the skin. Ann Plast Surg. 2010;65:462-465. doi:10.1097/SAP.0b013e3181d6e2d3
- Cha MJ, Jo SJ, Cho Y, et al. Patient characteristics and the incidence of radiation-induced dermatitis following radiofrequency catheter ablation. Korean Circ J. 2016;46:646-653. doi:10.4070/kcj.2016.46.5.646
- Dehen L, Vilmer C, Humilière C, et al. Chronic radiodermatitis following cardiac catheterisation: a report of two cases and a brief review of the literature. Heart. 1999;81:308-312. doi:10.1136/hrt.81.3.308
- Brown KR, Rzucidlo E. Acute and chronic radiation injury. J Vasc Surg. 2011;53(Suppl 1):15S-21S. doi:10.1016/j.jvs.2010.06.175. Published correction appears in J Vasc Surg. 2012;55:627.
- Hymes SR, Strom EA, Fife C. Radiation dermatitis: clinical presentation, pathophysiology, and treatment 2006. J Am Acad Dermatol. 2006;54:28-46. doi:10.1016/j.jaad.2005.08.054
- Scheinman PL, Vocanson M, Thyssen JP, et al. Contact dermatitis. Nat Rev Dis Primers. 2021;7:38. doi:10.1038/s41572-021-00271-4
- Cheng TT, Yang HJ. Chronic radiation dermatitis induced by cardiac catheterization: a case report and literature review. Acta Dermatovenerol Alp Pannonica Adriat. 2022;31:147-149.
- Minni JP, Nowak M, Usmani A, et al. A unique case of subacute radiodermatitis. Cutis. 2013;91:230-232.
- Flowers H, Brodell R, Brents M, et al. Fixed drug eruptions: presentation, diagnosis, and management. South Med J. 2014;107:724-727. doi:10.14423/SMJ.0000000000000195
- Hashimoto I, Sedo H, Inatsugi K, et al. Severe radiation-induced injury after cardiac catheter ablation: a case requiring free anterolateral thigh flap and vastus lateralis muscle flap reconstruction on the upper arm. J Plast Reconstr Aesthet Surg. 2008;61:704-708. doi:10.1016/j.bjps.2007.01.003
- Mervis JS, Phillips TJ. Pressure ulcers: pathophysiology, epidemiology, risk factors, and presentation. J Am Acad Dermatol. 2019;81:881-890. doi:10.1016/j.jaad.2018.12.069
- Careta MF, Romiti R. Localized scleroderma: clinical spectrum and therapeutic update. An Bras Dermatol. 2015;90:62-73. doi:10.1590/abd1806-4841.20152890
- Herz-Ruelas ME, Gómez-Flores M, Moxica-Del Angel J, et al. Ulcerated radiodermatitis induced after fluoroscopically guided stent implantation angioplasty. Case Rep Dermatol Med. 2014;2014:768624. doi:10.1155/2014/768624
- Wilson BN, Shah R, Menzer C, et al. Consensus on the clinical management of chronic radiation dermatitis and radiation fibrosis: a Delphi survey. Br J Dermatol. 2022;187:1054-1056. doi:10.1111/bjd.21852
- Khanna NR, Kumar DP, Laskar SG, et al. Radiation dermatitis: an overview. Indian J Burns. 2013;21:24-31. doi:10.4103/0971-653x.121877
- Spalek M. Chronic radiation-induced dermatitis: challenges and solutions. Clin Cosmet Investig Dermatol. 2016;9:473-482. doi:10.2147/CCID.S94320
- Bourgeois JF, Gourgou S, Kramar A, et al. A randomized, prospective study using the LPG technique in treating radiation-induced skin fibrosis: clinical and profilometric analysis. Skin Res Technol. 2008;14:71-76. doi:10.1111/j.1600-0846.2007.00263.x
- Borrelli MR, Shen AH, Lee GK, et al. Radiation-induced skinfibrosis: pathogenesis, current treatment options, and emerging therapeutics. Ann Plast Surg. 2019;83(4S Suppl 1):S59-S64. doi:10.1097/SAP.0000000000002098
- Wilson B, Shah R, Menzer C, et al. Laser therapy as a treatment for chronic radiation fibrosis. Lasers Surg Med. 2023;55:82-88. doi:10.1002/lsm.23617
- Rigotti G, Marchi A, Galiè M, et al. Clinical treatment of radiotherapy tissue damage by lipoaspirate transplant: a healing process mediated by adipose-derived adult stem cells. Plast Reconstr Surg. 2007;119:1409-1422. doi:10.1097/01.prs.0000256047.47909.71
- Leventhal J, Young MR. Radiation dermatitis: recognition, prevention, and management. Oncology (Williston Park). 2017;31:885-899.
- van Vloten WA, Hermans J, van Daal WA. Radiation-induced skin cancer and radiodermatitis of the head and neck. Cancer. 1987;59:411-414. doi:10.1002/1097-0142(19870201)59:3<411::aid-cncr2820590310>3.0.co;2-z
- Davis MM, Hanke CW, Zollinger TW, et al. Skin cancer in patients with chronic radiation dermatitis. J Am Acad Dermatol. 1989;20:608-616. doi:10.1016/s0190-9622(89)70072-4
- Miller DL, Balter S, Schueler BA, et al. Clinical radiation management for fluoroscopically guided interventional procedures. Radiology. 2010;257:321-332. doi:10.1148/radiol.10091269
- Benjamin EJ, Muntner P, Alonso A, et al. Heart Disease and Stroke Statistics-2019 Update: a report from the American Heart Association. Circulation. 2019;139:E56-E528. doi:10.1161/CIR.0000000000000659. Published correction appears in Circulation. 2020;141:E33.
- Koenig TR, Wolff D, Mettler FA, et al. Skin injuries from fluoroscopically guided procedures: part 1, characteristics of radiation injury. AJR Am J Roentgenol. 2001;177:3-11. doi:10.2214/ajr.177.1.1770003
- Guesnier-Dopagne M, Boyer L, Pereira B, et al. Incidence of chronic radiodermatitis after fluoroscopically guided interventions: a retrospective study. J Vasc Interv Radiol. 2019;30:692-698.e13. doi:10.1016/j.jvir.2019.01.010
- Cunha N, Cardoso P, Cabete J. Subacute radiation dermatitis following an interventional cardiology procedure. Cutan Ocul Toxicol. 2017;36:297-299. doi:10.1080/15569527.2016.1254649
- Frazier TH, Richardson JB, Fabré VC, et al. Fluoroscopy-induced chronic radiation skin injury: a disease perhaps often overlooked. Arch Dermatol. 2007;143:637-640. doi:10.1001/archderm.143.5.637
- Koenig TR, Mettler FA, Wagner LK. Skin injuries from fluoroscopically guided procedures: part 2, review of 73 cases and recommendations for minimizing dose delivered to patient. AJR Am J Roentgenol. 2001;177:13-20. doi:10.2214/ajr.177.1.1770013
- Shope TB. Radiation-induced skin injuries from fluoroscopy. Radiographics. 1996;16:1195-1199. doi:10.1148/radiographics.16.5.8888398
- Tchanque-Fossuo CN, Isseroff RR, Silverstein MA. Fluoroscopy induced chronic radiation dermatitis should be included in the differential diagnosis of notalgia paresthetica. Dermatol Online J. 2016;22:13030/qt0kh726m9.
- Berlin L. Radiation-induced skin injuries and fluoroscopy. AJR Am J Roentgenol. 2001;177:21-25. doi:10.2214/ajr.177.1.1770021
- Tchanque-Fossuo CN, Kamangar F, Ho B, et al. Fluoroscopy-induced radionecrosis. Dermatol Online J. 2016;22:13030/qt68w910t2.
- Wagner LK, Eifel PJ, Geise RA. Potential biological effects following high X-ray dose interventional procedures. J Vasc Interv Radiol. 1994;5:71-84. doi:10.1016/s1051-0443(94)71456-1
- Balter S, Hopewell JW, Miller DL, et al. Fluoroscopically guided interventional procedures: a review of radiation effects on patients’ skin and hair. Radiology. 2010;254:326-341. doi:10.1148/radiol.2542082312
- Vance AZ, Weinberg BD, Arbique GM, et al. Fluoroscopic sentinel events in neuroendovascular procedures: how to screen, prevent, and address occurrence. AJNR Am J Neuroradiol. 2013;34:1513-1515. doi:10.3174/ajnr.A3185
- Aerts A, Decraene T, van den Oord JJ, et al. Chronic radiodermatitis following percutaneous coronary interventions: a report of two cases. J Eur Acad Dermatol Venereol. 2003;17:340-343. doi:10.1046/j.1468-3083.2003.00687.x
- Rosenthal A, Israilevich R, Moy R. Management of acute radiation dermatitis: a review of the literature and proposal for treatment algorithm. J Am Acad Dermatol. 2019;81:558-567. doi:10.1016/j.jaad.2019.02.047
- Boncher J, Bergfeld WF. Fluoroscopy-induced chronic radiation dermatitis: a report of two additional cases and a brief review of the literature. J Cutan Pathol. 2012;39:63-67. doi:10.1111/j.1600-0560.2011.01754.x
- Spiker A, Zinn Z, Carter WH, et al. Fluoroscopy-induced chronic radiation dermatitis. Am J Cardiol. 2012;110:1861-1863. doi:10.1016/j.amjcard.2012.08.023
- Batrani M, Kubba A, Sundharam J. Fluoroscopy-induced chronic radiation dermatitis masquerading as morphea: a diagnostic pitfall. Indian J Pathol Microbiol. 2018;61:393-396. doi:10.4103/IJPM.IJPM_566_17
- Jeskowiak A, Hubmer M, Prenner G, et al. Radiation induced cutaneous ulcer on the back in a patient with congenital anomaly of the upper cava system. Interact Cardiovasc Thorac Surg. 2011;12:290-292.
- Laborda A, De Assis AM, Ioakeim I, et al. Radiodermitis after prostatic artery embolization: case report and review of the literature. Cardiovasc Intervent Radiol. 2015;38:755-759. doi:10.1007/s00270-015-1083-6
- Lyons AB, Harvey VM, Gusev J. Fluoroscopy-induced chronic radiation dermatitis (FICRD) after endovascular abdominal aortic aneurysm endoleak repair. JAAD Case Rep. 2015;1:403-405. doi:10.1016/j.jdcr.2015.09.022
- Mossman KL. Analysis of risk in computerized tomography and other diagnostic radiology procedures. Comput Radiol. 1982;6:251-256. doi:10.1016/0730-4862(82)90109-3
- Henry MF, Maender JL, Shen Y, et al. Fluoroscopy-induced chronic radiation dermatitis: a report of three cases. Dermatol Online J. 2009;15:3.
- Balter S, Miller DL. Patient skin reactions from interventional fluoroscopy procedures. AJR Am J Roentgenol. 2014;202:W335-W342. doi:10.2214/AJR.13.12029
- Nishimoto S, Fukuda K, Kawai K, et al. Supplementation of bone marrow aspirate-derived platelet-rich plasma for treating radiation-induced ulcer after cardiac fluoroscopic procedures: a preliminary report. Indian J Plast Surg. 2012;45:109-114. doi:10.4103/0970-0358.96599
- Cox JD, Stetz J, Pajak TF. Toxicity criteria of the Radiation Therapy Oncology Group (RTOG) and the European Organization for Research and Treatment of Cancer (EORTC). Int J Radiat Oncol Biol Phys. 1995;31:1341-1346. doi:10.1016/0360-3016(95)00060-C
- Wong RK, Bensadoun RJ, Boers-Doets CB, et al. Clinical practice guidelines for the prevention and treatment of acute and late radiation reactions from the MASCC Skin Toxicity Study Group. Support Care Cancer. 2013;21:2933-2948. doi:10.1007/s00520-013-1896-2
- Turesson I, Notter G. The predictive value of skin telangiectasia for late radiation effects in different normal tissues. Int J Radiat Oncol Biol Phys. 1986;12:603-609. doi:10.1016/0360-3016(86)90069-6
- Hegedus F, Mathew LM, Schwartz RA. Radiation dermatitis: an overview. Int J Dermatol. 2017;56:909-914. doi:10.1111/ijd.13371
- Denham JW, Hauer-Jensen M. The radiotherapeutic injury—a complex ‘wound.’ Radiother Oncol. 2002;63:129-145. doi:10.1016/s0167-8140(02)00060-9
- Wei KC, Lai SF, Huang WL, et al. An innovative targeted therapy for fluoroscopy-induced chronic radiation dermatitis. J Mol Med (Berl). 2022;100:135-146. doi:10.1007/s00109-021-02146-3
- Sitton E. Early and late radiation-induced skin alterations. part I: mechanisms of skin changes. Oncol Nurs Forum. 1992;19:801-807.
- Pruitt LG, Rogers W, Byarlay JA, et al. Subacute radiation dermatitis after fluoroscopy. J Cutan Pathol. 2016;43:1091-1095. doi:10.1111/cup.12815
- Anderson EB, Draft KS, Lee RA, et al. Update in dermatopathology. Am J Clin Pathol. 2006;125(Suppl):S50-S70. doi:10.1309/GMUFNP6LFMPNR86R
- Wei KC, Yang KC, Mar GY, et al. STROBE—radiation ulcer: an overlooked complication of fluoroscopic intervention: a cross-sectional study. Medicine (Baltimore). 2015;94:e2178. doi:10.1097/MD.0000000000002178
- Otterburn D, Losken A. Iatrogenic fluoroscopy injury to the skin. Ann Plast Surg. 2010;65:462-465. doi:10.1097/SAP.0b013e3181d6e2d3
- Cha MJ, Jo SJ, Cho Y, et al. Patient characteristics and the incidence of radiation-induced dermatitis following radiofrequency catheter ablation. Korean Circ J. 2016;46:646-653. doi:10.4070/kcj.2016.46.5.646
- Dehen L, Vilmer C, Humilière C, et al. Chronic radiodermatitis following cardiac catheterisation: a report of two cases and a brief review of the literature. Heart. 1999;81:308-312. doi:10.1136/hrt.81.3.308
- Brown KR, Rzucidlo E. Acute and chronic radiation injury. J Vasc Surg. 2011;53(Suppl 1):15S-21S. doi:10.1016/j.jvs.2010.06.175. Published correction appears in J Vasc Surg. 2012;55:627.
- Hymes SR, Strom EA, Fife C. Radiation dermatitis: clinical presentation, pathophysiology, and treatment 2006. J Am Acad Dermatol. 2006;54:28-46. doi:10.1016/j.jaad.2005.08.054
- Scheinman PL, Vocanson M, Thyssen JP, et al. Contact dermatitis. Nat Rev Dis Primers. 2021;7:38. doi:10.1038/s41572-021-00271-4
- Cheng TT, Yang HJ. Chronic radiation dermatitis induced by cardiac catheterization: a case report and literature review. Acta Dermatovenerol Alp Pannonica Adriat. 2022;31:147-149.
- Minni JP, Nowak M, Usmani A, et al. A unique case of subacute radiodermatitis. Cutis. 2013;91:230-232.
- Flowers H, Brodell R, Brents M, et al. Fixed drug eruptions: presentation, diagnosis, and management. South Med J. 2014;107:724-727. doi:10.14423/SMJ.0000000000000195
- Hashimoto I, Sedo H, Inatsugi K, et al. Severe radiation-induced injury after cardiac catheter ablation: a case requiring free anterolateral thigh flap and vastus lateralis muscle flap reconstruction on the upper arm. J Plast Reconstr Aesthet Surg. 2008;61:704-708. doi:10.1016/j.bjps.2007.01.003
- Mervis JS, Phillips TJ. Pressure ulcers: pathophysiology, epidemiology, risk factors, and presentation. J Am Acad Dermatol. 2019;81:881-890. doi:10.1016/j.jaad.2018.12.069
- Careta MF, Romiti R. Localized scleroderma: clinical spectrum and therapeutic update. An Bras Dermatol. 2015;90:62-73. doi:10.1590/abd1806-4841.20152890
- Herz-Ruelas ME, Gómez-Flores M, Moxica-Del Angel J, et al. Ulcerated radiodermatitis induced after fluoroscopically guided stent implantation angioplasty. Case Rep Dermatol Med. 2014;2014:768624. doi:10.1155/2014/768624
- Wilson BN, Shah R, Menzer C, et al. Consensus on the clinical management of chronic radiation dermatitis and radiation fibrosis: a Delphi survey. Br J Dermatol. 2022;187:1054-1056. doi:10.1111/bjd.21852
- Khanna NR, Kumar DP, Laskar SG, et al. Radiation dermatitis: an overview. Indian J Burns. 2013;21:24-31. doi:10.4103/0971-653x.121877
- Spalek M. Chronic radiation-induced dermatitis: challenges and solutions. Clin Cosmet Investig Dermatol. 2016;9:473-482. doi:10.2147/CCID.S94320
- Bourgeois JF, Gourgou S, Kramar A, et al. A randomized, prospective study using the LPG technique in treating radiation-induced skin fibrosis: clinical and profilometric analysis. Skin Res Technol. 2008;14:71-76. doi:10.1111/j.1600-0846.2007.00263.x
- Borrelli MR, Shen AH, Lee GK, et al. Radiation-induced skinfibrosis: pathogenesis, current treatment options, and emerging therapeutics. Ann Plast Surg. 2019;83(4S Suppl 1):S59-S64. doi:10.1097/SAP.0000000000002098
- Wilson B, Shah R, Menzer C, et al. Laser therapy as a treatment for chronic radiation fibrosis. Lasers Surg Med. 2023;55:82-88. doi:10.1002/lsm.23617
- Rigotti G, Marchi A, Galiè M, et al. Clinical treatment of radiotherapy tissue damage by lipoaspirate transplant: a healing process mediated by adipose-derived adult stem cells. Plast Reconstr Surg. 2007;119:1409-1422. doi:10.1097/01.prs.0000256047.47909.71
- Leventhal J, Young MR. Radiation dermatitis: recognition, prevention, and management. Oncology (Williston Park). 2017;31:885-899.
- van Vloten WA, Hermans J, van Daal WA. Radiation-induced skin cancer and radiodermatitis of the head and neck. Cancer. 1987;59:411-414. doi:10.1002/1097-0142(19870201)59:3<411::aid-cncr2820590310>3.0.co;2-z
- Davis MM, Hanke CW, Zollinger TW, et al. Skin cancer in patients with chronic radiation dermatitis. J Am Acad Dermatol. 1989;20:608-616. doi:10.1016/s0190-9622(89)70072-4
- Miller DL, Balter S, Schueler BA, et al. Clinical radiation management for fluoroscopically guided interventional procedures. Radiology. 2010;257:321-332. doi:10.1148/radiol.10091269
Fluoroscopy-Induced Chronic Radiation Dermatitis: A Comprehensive Review and Reappraisal
Fluoroscopy-Induced Chronic Radiation Dermatitis: A Comprehensive Review and Reappraisal
PRACTICE POINTS
- Fluoroscopy-induced chronic radiation dermatitis poses diagnostic challenges, as patients often are unable to associate a history of fluoroscopic procedures with the development of skin lesions.
- Scapular and subscapular lesions as well as those on the anterolateral chest and mid back should prompt clinicians to inquire about the patient’s history of fluoroscopic procedures.
- Because lesions can remain refractory to treatment, longterm monitoring is necessary if they are not excised.
