User login
Bilateral Ankle Ulcerations and Gangrene of the Toes
Bilateral Ankle Ulcerations and Gangrene of the Toes
THE DIAGNOSIS: Rheumatoid Vasculitis
A diagnosis of rheumatoid vasculitis (RV) was made based on the clinical features, histopathology, and laboratory results in the setting of rheumatoid arthritis (RA). The distal gangrene was surgically managed with bilateral transmetatarsal amputation followed by ankle collagen graft placement. The patient was started on a prednisone taper for 1 month (40 mg/d for 3 days, then 30 mg/d for 3 days, then 20 mg/d for 24 days) before transitioning to rituximab (375 mg/m2 once weekly for 4 weeks), which improved the size and depth of the ulcers.
Rheumatoid vasculitis is an inflammatory disease that affects small- to medium-sized blood vessels in patients with RA. The pathogenesis involves immune complex deposition and complement system activation, leading to vessel wall destruction.1 Rheumatoid vasculitis is an extra-articular complication of RA that primarily is observed in seropositive patients with long-standing severe disease.1,2 The mean duration between RA diagnosis and RV onset is 10 to 14 years.2 Rheumatoid vasculitis manifests heterogeneously and can affect many organs; however, it most frequently affects the skin. Cutaneous manifestations vary in severity. Palpable purpura, pyoderma gangrenosum, and distal ulcers can be seen in addition to extensive digital ischemia with necrosis, as was present in our patient.1
When RA patients present with skin changes that are concerning for vasculitis, RV should be suspected. Currently, there are no validated diagnostic criteria for RV. Diagnosis is made based on clinical presentation and tissue biopsy. Histopathology shows small- and medium-sized vessel wall destruction with neutrophilic, granulomatous, or lymphocytic infiltration, which may be observed only in the lower dermis sparing superficial vessels.3 Direct immunofluorescence shows IgM, IgA, and C3 deposition within and around vessels.3,4 Laboratory findings including elevated inflammatory markers, positive rheumatoid factor, positive anti–cyclic citrullinated peptide, and hypocomplementemia support a diagnosis of RV.1,2
Mortality rates for RV remain high, necessitating aggressive treatment. High-dose corticosteroids typically are combined with immunosuppressant or biologic agents, frequently cyclophosphamide or rituximab.1 Consistent with other reported cases, our patient’s ulcers improved with rituximab and oral steroids.
The differential diagnosis for our patient included type I cryoglobulinemia, cutaneous polyarteritis nodosa (CPAN), peripheral vascular disease (PVD), and nonuremic calciphylaxis. Type I cryoglobulinemia manifests due to direct occlusion of vessels by precipitation of monoclonal immunoglobulin.5 It commonly is associated with lymphoproliferative diseases such as Waldenström macroglobulinemia and multiple myeloma. While our patient’s history of RA was a risk factor for mixed cryoglobulinemia as opposed to type I cryoglobulinemia, the clinical presentation aligned more closely with type I cryoglobulinemia. The clinical manifestations of type I cryoglobulinemia are related to intravascular obstruction, including Raynaud phenomenon, retiform purpura, ischemic ulcers, distal gangrene, and cold-induced urticaria.6-8 Type I cryoglobulinemia also frequently has neurologic and renal manifestations. Histopathology, along with the detection of serum cryoglobulins, is the gold standard for diagnosing cryoglobulinemia.6 On histopathology, type I cryoglobulinemia typically shows a thrombotic vasculopathy with amorphous eosinophilic periodic acid–Schiff–positive thrombi.7 False-negative results are particularly common with serum cryoglobulins, so repeat testing often is needed. While many clinical features overlap, RV is the most likely diagnosis in a patient with long-standing RA who is negative for cryoglobulins and has no history of lymphoproliferative disorders.
Cutaneous polyarteritis nodosa is a necrotizing vasculitis that similarly affects small- and medium-sized vessels. The exact etiology is unknown, but the high prevalence of anti–phosphatidylserine/prothrombin complex antibodies among patients with CPAN suggests that prothrombin bound to apoptotic endothelial cells may initiate the immune response.9 Underlying infection and inflammatory and autoimmune diseases (including group A beta-hemolytic streptococcus, hepatitis B, inflammatory bowel disease, myasthenia gravis, and RA) also may trigger CPAN.9,10,11 The most common clinical manifestations of CPAN are tender subcutaneous nodules, livedo reticularis, leg ulcers, and cutaneous necrosis. Extracutaneous symptoms such as myalgias and arthralgias also can be associated with CPAN. There is no specific serologic test to diagnose CPAN; the diagnosis is made based on clinicopathologic correlation, with characteristic histopathology showing leukocytoclastic vasculitis in the small- and medium-sized arteries of the deep dermis or hypodermis.9
Peripheral vascular disease is a manifestation of atherosclerosis that affects the legs. Risk factors for atherosclerosis, especially smoking and diabetes mellitus, similarly increase the risk for PVD.12 The most common clinical manifestation of PVD is intermittent claudication, but rarely PVD can progress to critical limb ischemia, which is characterized by pain at rest, nonhealing ulcers, or gangrene of the legs.12 Common findings on physical examination include diminished or absent pedal pulses, abnormal skin color, and skin that is cool to the touch.12 The standard diagnostic test for PVD affecting the legs is evaluation via the ankle-brachial index, with a score of 0.90 or lower being diagnostic of PVD, a score of 0.91 to 1.00 being borderline, and a score of 1.01 to 1.40 being normal.13
Calciphylaxis most frequently is seen in patients with end-stage kidney disease; however, it also has been less commonly reported in patients with normal kidney function, known as nonuremic calciphylaxis. It is characterized by calcification of arteries, arterioles, and soft tissues, which can lead to thrombosis and eventually ischemia and necrosis of the skin.14 Calciphylaxis initially causes tender, indurated, erythematous to purpuric plaques that quickly progress to retiform and stellate ulcers with overlying necrotic eschars.15 Disease typically occurs on the legs and areas that are rich in adipose tissue, such as the abdomen and thighs.16 Skin biopsy is needed for diagnosis of calciphylaxis. Characteristic histopathologic findings include calcification, microvascular thrombosis, and fibrointimal hyperplasia of small dermal and subcutaneous arteries and arterioles.16
We present a rare case of RV in a patient with well-controlled RA. While the incidence of RV is decreasing in the United States and United Kingdom due to the initiation of earlier and more aggressive RA therapies, mortality remains high.1 Thus, it is important to include RV in the differential diagnosis when there are skin changes concerning vasculitis in patients with seropositive, longstanding RA, even if the RA is well controlled.
- Kishore S, Maher L, Majithia V. Rheumatoid vasculitis: a diminishing yet devastating menace. Curr Rheumatol Rep. 2017;19:39. doi:10.1007/s11926-017-0667-3
- Makol A, Matteson EL, Warrington KJ. Rheumatoid vasculitis: an update. Curr Opin Rheumatol. 2015;27:63-70. doi:10.1097 /BOR.0000000000000126
- Patterson J. The vasculopathic reaction pattern. In: Patterson J, ed. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:241-301.
- Lora V, Cerroni L, Cota C. Skin manifestations of rheumatoid arthritis. G Ital Dermatol Venereol. 2018;153:243-255. doi:10.23736 /S0392-0488.18.05872-8
- Kolopp-Sarda MN, Miossec P. Cryoglobulinemic vasculitis: pathophysiological mechanisms and diagnosis. Curr Opin Rheumatol. 2021;33:1-7. doi:10.1097/BOR.0000000000000757
- Silva F, Pinto C, Barbosa A, et al. New insights in cryoglobulinemic vasculitis. J Autoimmun. 2019;105:102313. doi:10.1016 /j.jaut.2019.102313
- Harel S, Mohr M, Jahn I, et al. Clinico-biological characteristics and treatment of type I monoclonal cryoglobulinaemia: a study of 64 cases. Br J Haematol. 2015;168:671-678. doi:10.1111/bjh.13196
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j.jbspin.2019.01.016
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756. doi:10.1111/j.1365-4632.2010.04522.
- Criado PR, Marques GF, Morita TC, et al. Epidemiological, clinical and laboratory profiles of cutaneous polyarteritis nodosa patients: report of 22 cases and literature review. Autoimmun Rev. 2016;15:558-563. doi:10.1016/j.autrev.2016.02.010
- Daoud MS, Hutton KP, Gibson LE. Cutaneous periarteritis nodosa: a clinicopathological study of 79 cases. Br J Dermatol. 1997;136:706-713.
- Campia U, Gerhard-Herman M, Piazza G, et al. Peripheral artery disease: past, present, and future. Am J Med. 2019;132:1133-1141. doi:10.1016/j.amjmed.2019.04.043
- Aboyans V, Criqui MH, Abraham P, et al. Measurement and interpretation of the ankle-brachial index: a scientific statement from the American Heart Association [published correction appears in Circulation. 2013 Jan 1;127:e264]. Circulation. 2012;126:2890-2909. doi:10.1161/CIR.0b013e318276fbcb
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Nigwekar SU, Thadhani R, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714. doi:10.1056/NEJMra1505292
- Gomes F, La Feria P, Costa C, et al. Non-uremic calciphylaxis: a rare diagnosis with limited therapeutic strategies. Eur J Case Rep Intern Med.
THE DIAGNOSIS: Rheumatoid Vasculitis
A diagnosis of rheumatoid vasculitis (RV) was made based on the clinical features, histopathology, and laboratory results in the setting of rheumatoid arthritis (RA). The distal gangrene was surgically managed with bilateral transmetatarsal amputation followed by ankle collagen graft placement. The patient was started on a prednisone taper for 1 month (40 mg/d for 3 days, then 30 mg/d for 3 days, then 20 mg/d for 24 days) before transitioning to rituximab (375 mg/m2 once weekly for 4 weeks), which improved the size and depth of the ulcers.
Rheumatoid vasculitis is an inflammatory disease that affects small- to medium-sized blood vessels in patients with RA. The pathogenesis involves immune complex deposition and complement system activation, leading to vessel wall destruction.1 Rheumatoid vasculitis is an extra-articular complication of RA that primarily is observed in seropositive patients with long-standing severe disease.1,2 The mean duration between RA diagnosis and RV onset is 10 to 14 years.2 Rheumatoid vasculitis manifests heterogeneously and can affect many organs; however, it most frequently affects the skin. Cutaneous manifestations vary in severity. Palpable purpura, pyoderma gangrenosum, and distal ulcers can be seen in addition to extensive digital ischemia with necrosis, as was present in our patient.1
When RA patients present with skin changes that are concerning for vasculitis, RV should be suspected. Currently, there are no validated diagnostic criteria for RV. Diagnosis is made based on clinical presentation and tissue biopsy. Histopathology shows small- and medium-sized vessel wall destruction with neutrophilic, granulomatous, or lymphocytic infiltration, which may be observed only in the lower dermis sparing superficial vessels.3 Direct immunofluorescence shows IgM, IgA, and C3 deposition within and around vessels.3,4 Laboratory findings including elevated inflammatory markers, positive rheumatoid factor, positive anti–cyclic citrullinated peptide, and hypocomplementemia support a diagnosis of RV.1,2
Mortality rates for RV remain high, necessitating aggressive treatment. High-dose corticosteroids typically are combined with immunosuppressant or biologic agents, frequently cyclophosphamide or rituximab.1 Consistent with other reported cases, our patient’s ulcers improved with rituximab and oral steroids.
The differential diagnosis for our patient included type I cryoglobulinemia, cutaneous polyarteritis nodosa (CPAN), peripheral vascular disease (PVD), and nonuremic calciphylaxis. Type I cryoglobulinemia manifests due to direct occlusion of vessels by precipitation of monoclonal immunoglobulin.5 It commonly is associated with lymphoproliferative diseases such as Waldenström macroglobulinemia and multiple myeloma. While our patient’s history of RA was a risk factor for mixed cryoglobulinemia as opposed to type I cryoglobulinemia, the clinical presentation aligned more closely with type I cryoglobulinemia. The clinical manifestations of type I cryoglobulinemia are related to intravascular obstruction, including Raynaud phenomenon, retiform purpura, ischemic ulcers, distal gangrene, and cold-induced urticaria.6-8 Type I cryoglobulinemia also frequently has neurologic and renal manifestations. Histopathology, along with the detection of serum cryoglobulins, is the gold standard for diagnosing cryoglobulinemia.6 On histopathology, type I cryoglobulinemia typically shows a thrombotic vasculopathy with amorphous eosinophilic periodic acid–Schiff–positive thrombi.7 False-negative results are particularly common with serum cryoglobulins, so repeat testing often is needed. While many clinical features overlap, RV is the most likely diagnosis in a patient with long-standing RA who is negative for cryoglobulins and has no history of lymphoproliferative disorders.
Cutaneous polyarteritis nodosa is a necrotizing vasculitis that similarly affects small- and medium-sized vessels. The exact etiology is unknown, but the high prevalence of anti–phosphatidylserine/prothrombin complex antibodies among patients with CPAN suggests that prothrombin bound to apoptotic endothelial cells may initiate the immune response.9 Underlying infection and inflammatory and autoimmune diseases (including group A beta-hemolytic streptococcus, hepatitis B, inflammatory bowel disease, myasthenia gravis, and RA) also may trigger CPAN.9,10,11 The most common clinical manifestations of CPAN are tender subcutaneous nodules, livedo reticularis, leg ulcers, and cutaneous necrosis. Extracutaneous symptoms such as myalgias and arthralgias also can be associated with CPAN. There is no specific serologic test to diagnose CPAN; the diagnosis is made based on clinicopathologic correlation, with characteristic histopathology showing leukocytoclastic vasculitis in the small- and medium-sized arteries of the deep dermis or hypodermis.9
Peripheral vascular disease is a manifestation of atherosclerosis that affects the legs. Risk factors for atherosclerosis, especially smoking and diabetes mellitus, similarly increase the risk for PVD.12 The most common clinical manifestation of PVD is intermittent claudication, but rarely PVD can progress to critical limb ischemia, which is characterized by pain at rest, nonhealing ulcers, or gangrene of the legs.12 Common findings on physical examination include diminished or absent pedal pulses, abnormal skin color, and skin that is cool to the touch.12 The standard diagnostic test for PVD affecting the legs is evaluation via the ankle-brachial index, with a score of 0.90 or lower being diagnostic of PVD, a score of 0.91 to 1.00 being borderline, and a score of 1.01 to 1.40 being normal.13
Calciphylaxis most frequently is seen in patients with end-stage kidney disease; however, it also has been less commonly reported in patients with normal kidney function, known as nonuremic calciphylaxis. It is characterized by calcification of arteries, arterioles, and soft tissues, which can lead to thrombosis and eventually ischemia and necrosis of the skin.14 Calciphylaxis initially causes tender, indurated, erythematous to purpuric plaques that quickly progress to retiform and stellate ulcers with overlying necrotic eschars.15 Disease typically occurs on the legs and areas that are rich in adipose tissue, such as the abdomen and thighs.16 Skin biopsy is needed for diagnosis of calciphylaxis. Characteristic histopathologic findings include calcification, microvascular thrombosis, and fibrointimal hyperplasia of small dermal and subcutaneous arteries and arterioles.16
We present a rare case of RV in a patient with well-controlled RA. While the incidence of RV is decreasing in the United States and United Kingdom due to the initiation of earlier and more aggressive RA therapies, mortality remains high.1 Thus, it is important to include RV in the differential diagnosis when there are skin changes concerning vasculitis in patients with seropositive, longstanding RA, even if the RA is well controlled.
THE DIAGNOSIS: Rheumatoid Vasculitis
A diagnosis of rheumatoid vasculitis (RV) was made based on the clinical features, histopathology, and laboratory results in the setting of rheumatoid arthritis (RA). The distal gangrene was surgically managed with bilateral transmetatarsal amputation followed by ankle collagen graft placement. The patient was started on a prednisone taper for 1 month (40 mg/d for 3 days, then 30 mg/d for 3 days, then 20 mg/d for 24 days) before transitioning to rituximab (375 mg/m2 once weekly for 4 weeks), which improved the size and depth of the ulcers.
Rheumatoid vasculitis is an inflammatory disease that affects small- to medium-sized blood vessels in patients with RA. The pathogenesis involves immune complex deposition and complement system activation, leading to vessel wall destruction.1 Rheumatoid vasculitis is an extra-articular complication of RA that primarily is observed in seropositive patients with long-standing severe disease.1,2 The mean duration between RA diagnosis and RV onset is 10 to 14 years.2 Rheumatoid vasculitis manifests heterogeneously and can affect many organs; however, it most frequently affects the skin. Cutaneous manifestations vary in severity. Palpable purpura, pyoderma gangrenosum, and distal ulcers can be seen in addition to extensive digital ischemia with necrosis, as was present in our patient.1
When RA patients present with skin changes that are concerning for vasculitis, RV should be suspected. Currently, there are no validated diagnostic criteria for RV. Diagnosis is made based on clinical presentation and tissue biopsy. Histopathology shows small- and medium-sized vessel wall destruction with neutrophilic, granulomatous, or lymphocytic infiltration, which may be observed only in the lower dermis sparing superficial vessels.3 Direct immunofluorescence shows IgM, IgA, and C3 deposition within and around vessels.3,4 Laboratory findings including elevated inflammatory markers, positive rheumatoid factor, positive anti–cyclic citrullinated peptide, and hypocomplementemia support a diagnosis of RV.1,2
Mortality rates for RV remain high, necessitating aggressive treatment. High-dose corticosteroids typically are combined with immunosuppressant or biologic agents, frequently cyclophosphamide or rituximab.1 Consistent with other reported cases, our patient’s ulcers improved with rituximab and oral steroids.
The differential diagnosis for our patient included type I cryoglobulinemia, cutaneous polyarteritis nodosa (CPAN), peripheral vascular disease (PVD), and nonuremic calciphylaxis. Type I cryoglobulinemia manifests due to direct occlusion of vessels by precipitation of monoclonal immunoglobulin.5 It commonly is associated with lymphoproliferative diseases such as Waldenström macroglobulinemia and multiple myeloma. While our patient’s history of RA was a risk factor for mixed cryoglobulinemia as opposed to type I cryoglobulinemia, the clinical presentation aligned more closely with type I cryoglobulinemia. The clinical manifestations of type I cryoglobulinemia are related to intravascular obstruction, including Raynaud phenomenon, retiform purpura, ischemic ulcers, distal gangrene, and cold-induced urticaria.6-8 Type I cryoglobulinemia also frequently has neurologic and renal manifestations. Histopathology, along with the detection of serum cryoglobulins, is the gold standard for diagnosing cryoglobulinemia.6 On histopathology, type I cryoglobulinemia typically shows a thrombotic vasculopathy with amorphous eosinophilic periodic acid–Schiff–positive thrombi.7 False-negative results are particularly common with serum cryoglobulins, so repeat testing often is needed. While many clinical features overlap, RV is the most likely diagnosis in a patient with long-standing RA who is negative for cryoglobulins and has no history of lymphoproliferative disorders.
Cutaneous polyarteritis nodosa is a necrotizing vasculitis that similarly affects small- and medium-sized vessels. The exact etiology is unknown, but the high prevalence of anti–phosphatidylserine/prothrombin complex antibodies among patients with CPAN suggests that prothrombin bound to apoptotic endothelial cells may initiate the immune response.9 Underlying infection and inflammatory and autoimmune diseases (including group A beta-hemolytic streptococcus, hepatitis B, inflammatory bowel disease, myasthenia gravis, and RA) also may trigger CPAN.9,10,11 The most common clinical manifestations of CPAN are tender subcutaneous nodules, livedo reticularis, leg ulcers, and cutaneous necrosis. Extracutaneous symptoms such as myalgias and arthralgias also can be associated with CPAN. There is no specific serologic test to diagnose CPAN; the diagnosis is made based on clinicopathologic correlation, with characteristic histopathology showing leukocytoclastic vasculitis in the small- and medium-sized arteries of the deep dermis or hypodermis.9
Peripheral vascular disease is a manifestation of atherosclerosis that affects the legs. Risk factors for atherosclerosis, especially smoking and diabetes mellitus, similarly increase the risk for PVD.12 The most common clinical manifestation of PVD is intermittent claudication, but rarely PVD can progress to critical limb ischemia, which is characterized by pain at rest, nonhealing ulcers, or gangrene of the legs.12 Common findings on physical examination include diminished or absent pedal pulses, abnormal skin color, and skin that is cool to the touch.12 The standard diagnostic test for PVD affecting the legs is evaluation via the ankle-brachial index, with a score of 0.90 or lower being diagnostic of PVD, a score of 0.91 to 1.00 being borderline, and a score of 1.01 to 1.40 being normal.13
Calciphylaxis most frequently is seen in patients with end-stage kidney disease; however, it also has been less commonly reported in patients with normal kidney function, known as nonuremic calciphylaxis. It is characterized by calcification of arteries, arterioles, and soft tissues, which can lead to thrombosis and eventually ischemia and necrosis of the skin.14 Calciphylaxis initially causes tender, indurated, erythematous to purpuric plaques that quickly progress to retiform and stellate ulcers with overlying necrotic eschars.15 Disease typically occurs on the legs and areas that are rich in adipose tissue, such as the abdomen and thighs.16 Skin biopsy is needed for diagnosis of calciphylaxis. Characteristic histopathologic findings include calcification, microvascular thrombosis, and fibrointimal hyperplasia of small dermal and subcutaneous arteries and arterioles.16
We present a rare case of RV in a patient with well-controlled RA. While the incidence of RV is decreasing in the United States and United Kingdom due to the initiation of earlier and more aggressive RA therapies, mortality remains high.1 Thus, it is important to include RV in the differential diagnosis when there are skin changes concerning vasculitis in patients with seropositive, longstanding RA, even if the RA is well controlled.
- Kishore S, Maher L, Majithia V. Rheumatoid vasculitis: a diminishing yet devastating menace. Curr Rheumatol Rep. 2017;19:39. doi:10.1007/s11926-017-0667-3
- Makol A, Matteson EL, Warrington KJ. Rheumatoid vasculitis: an update. Curr Opin Rheumatol. 2015;27:63-70. doi:10.1097 /BOR.0000000000000126
- Patterson J. The vasculopathic reaction pattern. In: Patterson J, ed. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:241-301.
- Lora V, Cerroni L, Cota C. Skin manifestations of rheumatoid arthritis. G Ital Dermatol Venereol. 2018;153:243-255. doi:10.23736 /S0392-0488.18.05872-8
- Kolopp-Sarda MN, Miossec P. Cryoglobulinemic vasculitis: pathophysiological mechanisms and diagnosis. Curr Opin Rheumatol. 2021;33:1-7. doi:10.1097/BOR.0000000000000757
- Silva F, Pinto C, Barbosa A, et al. New insights in cryoglobulinemic vasculitis. J Autoimmun. 2019;105:102313. doi:10.1016 /j.jaut.2019.102313
- Harel S, Mohr M, Jahn I, et al. Clinico-biological characteristics and treatment of type I monoclonal cryoglobulinaemia: a study of 64 cases. Br J Haematol. 2015;168:671-678. doi:10.1111/bjh.13196
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j.jbspin.2019.01.016
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756. doi:10.1111/j.1365-4632.2010.04522.
- Criado PR, Marques GF, Morita TC, et al. Epidemiological, clinical and laboratory profiles of cutaneous polyarteritis nodosa patients: report of 22 cases and literature review. Autoimmun Rev. 2016;15:558-563. doi:10.1016/j.autrev.2016.02.010
- Daoud MS, Hutton KP, Gibson LE. Cutaneous periarteritis nodosa: a clinicopathological study of 79 cases. Br J Dermatol. 1997;136:706-713.
- Campia U, Gerhard-Herman M, Piazza G, et al. Peripheral artery disease: past, present, and future. Am J Med. 2019;132:1133-1141. doi:10.1016/j.amjmed.2019.04.043
- Aboyans V, Criqui MH, Abraham P, et al. Measurement and interpretation of the ankle-brachial index: a scientific statement from the American Heart Association [published correction appears in Circulation. 2013 Jan 1;127:e264]. Circulation. 2012;126:2890-2909. doi:10.1161/CIR.0b013e318276fbcb
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Nigwekar SU, Thadhani R, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714. doi:10.1056/NEJMra1505292
- Gomes F, La Feria P, Costa C, et al. Non-uremic calciphylaxis: a rare diagnosis with limited therapeutic strategies. Eur J Case Rep Intern Med.
- Kishore S, Maher L, Majithia V. Rheumatoid vasculitis: a diminishing yet devastating menace. Curr Rheumatol Rep. 2017;19:39. doi:10.1007/s11926-017-0667-3
- Makol A, Matteson EL, Warrington KJ. Rheumatoid vasculitis: an update. Curr Opin Rheumatol. 2015;27:63-70. doi:10.1097 /BOR.0000000000000126
- Patterson J. The vasculopathic reaction pattern. In: Patterson J, ed. Weedon’s Skin Pathology. 5th ed. Elsevier; 2021:241-301.
- Lora V, Cerroni L, Cota C. Skin manifestations of rheumatoid arthritis. G Ital Dermatol Venereol. 2018;153:243-255. doi:10.23736 /S0392-0488.18.05872-8
- Kolopp-Sarda MN, Miossec P. Cryoglobulinemic vasculitis: pathophysiological mechanisms and diagnosis. Curr Opin Rheumatol. 2021;33:1-7. doi:10.1097/BOR.0000000000000757
- Silva F, Pinto C, Barbosa A, et al. New insights in cryoglobulinemic vasculitis. J Autoimmun. 2019;105:102313. doi:10.1016 /j.jaut.2019.102313
- Harel S, Mohr M, Jahn I, et al. Clinico-biological characteristics and treatment of type I monoclonal cryoglobulinaemia: a study of 64 cases. Br J Haematol. 2015;168:671-678. doi:10.1111/bjh.13196
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j.jbspin.2019.01.016
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756. doi:10.1111/j.1365-4632.2010.04522.
- Criado PR, Marques GF, Morita TC, et al. Epidemiological, clinical and laboratory profiles of cutaneous polyarteritis nodosa patients: report of 22 cases and literature review. Autoimmun Rev. 2016;15:558-563. doi:10.1016/j.autrev.2016.02.010
- Daoud MS, Hutton KP, Gibson LE. Cutaneous periarteritis nodosa: a clinicopathological study of 79 cases. Br J Dermatol. 1997;136:706-713.
- Campia U, Gerhard-Herman M, Piazza G, et al. Peripheral artery disease: past, present, and future. Am J Med. 2019;132:1133-1141. doi:10.1016/j.amjmed.2019.04.043
- Aboyans V, Criqui MH, Abraham P, et al. Measurement and interpretation of the ankle-brachial index: a scientific statement from the American Heart Association [published correction appears in Circulation. 2013 Jan 1;127:e264]. Circulation. 2012;126:2890-2909. doi:10.1161/CIR.0b013e318276fbcb
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Nigwekar SU, Thadhani R, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714. doi:10.1056/NEJMra1505292
- Gomes F, La Feria P, Costa C, et al. Non-uremic calciphylaxis: a rare diagnosis with limited therapeutic strategies. Eur J Case Rep Intern Med.
Bilateral Ankle Ulcerations and Gangrene of the Toes
Bilateral Ankle Ulcerations and Gangrene of the Toes
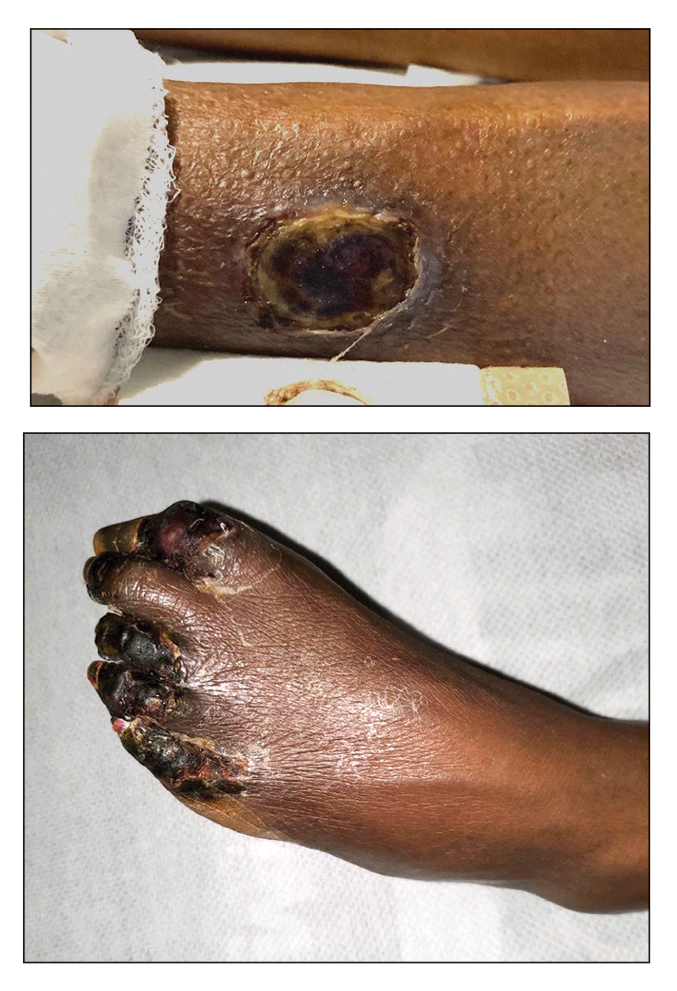
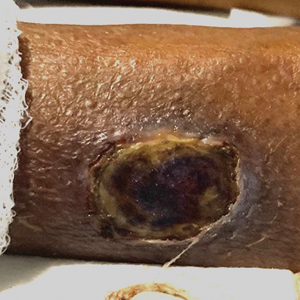
A 74-year-old woman presented to the hospital with large tender ulcerations on both ankles as well as gangrene of the toes of 6 to 8 weeks’ duration. The patient had a history of hypertension as well as seropositive nonerosive rheumatoid arthritis that had been diagnosed 8 years prior and was well controlled with leflunomide and prednisone as needed for flares. She denied any history of similar ulcers as well as any recent illnesses, medication changes, or joint pain or swelling. She was evaluated by vascular surgery 1 week prior to the current presentation, at which time her ankle-brachial index score was normal. Skin examination revealed noninflammatory retiform purpura surrounding ulcerations on both ankles (top) and necrosis of all toes (bottom) with peripheral retiform purpura. Joint examination revealed swan neck deformities of multiple fingers with normal range of motion, and there was no effusion or tenderness of the joints of the fingers on palpation. No rheumatoid nodules were present. Laboratory testing revealed elevated rheumatoid factor, anti–cyclic citrullinated peptide, C-reactive protein, and anti–Sjögren syndrome–related antigen A levels and low C4 levels. Cryoglobulins, antineutrophil cytoplasmic antibodies, and serum protein electrophoresis were negative. Biopsy of an ulcer on the right ankle showed medium-sized vessel vasculitis with fibrinoid necrosis, including endothelium necrosis and a perivascular lymphocytic infiltrate. Direct immunofluorescence demonstrated dense, granular, intraperivascular deposition of IgM and IgG with slightly weaker deposition of IgA, C3, and C5b-9 in the dermis and subcutis with a greater effect on medium-sized vessels.
Sudden-Onset Blistering Rash
The Diagnosis: Generalized Bullous Fixed Drug Eruption
A punch biopsy from the left thigh revealed a vacuolar interface dermatitis with full-thickness necrosis of the epidermis and a patchy lichenoid inflammatory cell infiltrate in the superficial dermis consistent with a generalized bullous fixed drug eruption (GBFDE). The patient received supportive care and methylprednisolone with improvement of symptoms.
Generalized bullous fixed drug eruption is a rare, potentially life-threatening form of a fixed drug eruption (FDE), a cutaneous drug reaction that occurs in response to a causative medication. It typically presents with welldemarcated, dusky, erythematous patches or plaques that recur in the same sites with repeat exposure.1 The pathogenesis of FDE has been hypothesized to involve epidermal CD8+ T cells, which are activated by drug exposure and release cytotoxic molecules including Fas, Fas ligand, perforin, and granzyme B, resulting in lysis of the surrounding keratinocytes.1-3 Common eliciting drugs include nonsteroidal anti-inflammatory drugs, antibacterial agents (particularly trimethoprim-sulfamethoxazole), barbiturates, acetaminophen, and antimalarials.1 In addition to the findings seen in FDE, GBFDE is characterized by widespread bullous skin lesions.1-4 Typical histologic patterns seen in GBFDE are dispersed epidermal apoptotic keratinocytes, prominent dermal eosinophilic and lymphocytic infiltrates, and dermal melanophages.3 Discontinuing the causative agent and diligent prevention of re-exposure are the most important steps in management, as additional exposures can increase the number of lesions and overall severity. Symptoms typically resolve 7 to 14 days after drug discontinuation, often with postinflammatory hyperpigmentation.3
Generalized bullous fixed drug eruption presents a diagnostic challenge, as it sometimes involves the oral mucosa and can exhibit the Nikolsky sign. Thus, it often is confused with Stevens-Johnson syndrome (SJS) or toxic epidermal necrolysis (TEN).1,4 Stevens-Johnson syndrome and TEN are severe cutaneous drug eruptions that also can present with diffuse bullous skin lesions. Stevens-Johnson syndrome and TEN are thought to be a spectrum of the same disease that initially presents with dusky red macules that can coalesce, develop central blistering, and lead to skin detachment.5 Stevens-Johnson syndrome is defined as skin detachment of less than 10% body surface area (BSA); TEN is defined as skin detachment of more than 30% BSA. Stevens-Johnson syndrome/TEN overlap syndrome includes skin detachment of 10% to 30% BSA.5
Causative medications overlap substantially with GBFDE and include anticonvulsants, sulfa-containing drugs, antibiotics, nonsteroidal anti-inflammatory drugs, and uric acid–lowering agents. The histology of SJS/TEN also is quite similar to GBFDE, and these entities may be indistinguishable without clinical information.5 Lee et al1 found that absence of grouped necrotic keratinocytes (fire flag sign), deep inflammatory infiltrates, notable pigment incontinence, and higher eosinophil counts appear to be more common in GBFDE than SJS/TEN. Constitutional symptoms and mucosal involvement also were more frequent in SJS/TEN.
The timing of clinical presentation and medical history can be useful in differentiating between SJS/TEN and GBFDE. In SJS/TEN, drug exposure typically occurs 1 to 3 weeks before onset of symptoms vs 30 minutes to 24 hours in GBFDE.3 Additionally, a history of similar eruption in the same location is pathognomonic for GBFDE. Although GBFDE has been thought to have a better prognosis than SJS/TEN, more recent data suggest mortality rates may be similar.3 A case-control study found a mortality rate of 22% (13/58) in patients with GBFDE compared to 28% (n=170) in SJS/TEN patients.4
Erythema multiforme (EM) is an uncommon immunemediated disorder that typically presents as targetoid lesions with central epidermal necrosis in an acral distribution. Erythema multiforme can arise from a variety of factors, but up to 90% of cases are due to infection, most commonly herpes simplex virus; medications account for less than 10% of cases.6 Previously, EM has been thought to be on the same disease spectrum as SJS and TEN. It is now clear that EM is a separate entity with similar mucosal erosions but different cutaneous findings,6 mainly typical target lesions that differ from the atypical targets seen in SJS.
Staphylococcal scalded skin syndrome is a blistering skin disorder associated with local Staphylococcus aureus infection. It most commonly is seen in children and rarely occurs in adults who are not on dialysis. Some Staphylococcus strains produce exfoliative toxins A and B, which are serine proteases that target and cleave desmoglein 1, a mediator of keratinocyte adhesion. Staphylococcal scalded skin syndrome initially presents with erythema accentuated in the skin folds that becomes generalized. The disruption of keratinocyte adhesion leads to bullae formation in areas of erythema and diffuse sheetlike desquamation. Pathology reveals subcorneal rather than subepidermal blistering, which is seen in GBFDE and SJS/TEN. Treatment involves antistaphylococcal antibiotics and supportive care. With proper treatment, most cases resolve within 2 to 3 weeks.7
Mycoplasma pneumoniae–induced rash and mucositis presents with prominent mucositis and can have cutaneous findings of sparse vesiculobullous or targetoid eruption.8 Mycoplasma pneumoniae typically infects the lungs and is a leading cause of community-acquired pneumonia. However, a subset of patients can have extrapulmonary disease presenting as mucocutaneous eruptions, which is preceded by an approximately weeklong prodrome of fever, cough, and malaise.7 Mycoplasma pneumoniae–induced rash and mucositis also affect children and young patients and is more common in males.8
- Lee CH, Chen YC, Cho YT, et al. Fixed-drug eruption: a retrospective study in a single referral center in northern Taiwan. Dermatologica Sinica. 2012;30:11-15. doi:10.1016/j.dsi.2012.02.002
- Cho Y-T, Lin J-W, Chen Y-C, et al. Generalized bullous fixed drug eruption is distinct from Stevens-Johnson syndrome/toxic epidermal necrolysis by immunohistopathological features. J Am Acad Dermatol. 2014;70:539-548. doi:10.1016/j.jaad.2013.11.015
- Mitre V, Applebaum DS, Albahrani Y, et al. Generalized bullous fixed drug eruption imitating toxic epidermal necrolysis: a case report and literature review. Dermatol Online J. 2017;23: 13030/qt25v009gs.
- Lipowicz S, Sekula P, Ingen-Housz-Oro S, et al. Prognosis of generalized bullous fixed drug eruption: comparison with StevensJohnson syndrome and toxic epidermal necrolysis. Br J Dermatol. 2013;168:726-732. doi:10.1111/bjd.12133
- Cho Y-T, Chu C-Y. Treatments for severe cutaneous adverse reactions [published online December 27, 2017]. J Immunol Res. doi:10.1155/2017/1503709
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902. doi:10.1111/j.1365-4632.2011.05348.x
- Leung AKC, Barankin B, Leong KF. Staphylococcal-scalded skin syndrome: evaluation, diagnosis, and management. World J Pediatr. 2018;14:116-120.
- Canavan TN, Mathes EF, Frieden I, et al. Mycoplasma pneumoniae–induced rash and mucositis as a syndrome distinct from Stevens-Johnson syndrome and erythema multiforme: a systematic review. J Am Acad Dermatol. 2015;72:239-245. doi:10.1016/j .jaad.2014.06.026
The Diagnosis: Generalized Bullous Fixed Drug Eruption
A punch biopsy from the left thigh revealed a vacuolar interface dermatitis with full-thickness necrosis of the epidermis and a patchy lichenoid inflammatory cell infiltrate in the superficial dermis consistent with a generalized bullous fixed drug eruption (GBFDE). The patient received supportive care and methylprednisolone with improvement of symptoms.
Generalized bullous fixed drug eruption is a rare, potentially life-threatening form of a fixed drug eruption (FDE), a cutaneous drug reaction that occurs in response to a causative medication. It typically presents with welldemarcated, dusky, erythematous patches or plaques that recur in the same sites with repeat exposure.1 The pathogenesis of FDE has been hypothesized to involve epidermal CD8+ T cells, which are activated by drug exposure and release cytotoxic molecules including Fas, Fas ligand, perforin, and granzyme B, resulting in lysis of the surrounding keratinocytes.1-3 Common eliciting drugs include nonsteroidal anti-inflammatory drugs, antibacterial agents (particularly trimethoprim-sulfamethoxazole), barbiturates, acetaminophen, and antimalarials.1 In addition to the findings seen in FDE, GBFDE is characterized by widespread bullous skin lesions.1-4 Typical histologic patterns seen in GBFDE are dispersed epidermal apoptotic keratinocytes, prominent dermal eosinophilic and lymphocytic infiltrates, and dermal melanophages.3 Discontinuing the causative agent and diligent prevention of re-exposure are the most important steps in management, as additional exposures can increase the number of lesions and overall severity. Symptoms typically resolve 7 to 14 days after drug discontinuation, often with postinflammatory hyperpigmentation.3
Generalized bullous fixed drug eruption presents a diagnostic challenge, as it sometimes involves the oral mucosa and can exhibit the Nikolsky sign. Thus, it often is confused with Stevens-Johnson syndrome (SJS) or toxic epidermal necrolysis (TEN).1,4 Stevens-Johnson syndrome and TEN are severe cutaneous drug eruptions that also can present with diffuse bullous skin lesions. Stevens-Johnson syndrome and TEN are thought to be a spectrum of the same disease that initially presents with dusky red macules that can coalesce, develop central blistering, and lead to skin detachment.5 Stevens-Johnson syndrome is defined as skin detachment of less than 10% body surface area (BSA); TEN is defined as skin detachment of more than 30% BSA. Stevens-Johnson syndrome/TEN overlap syndrome includes skin detachment of 10% to 30% BSA.5
Causative medications overlap substantially with GBFDE and include anticonvulsants, sulfa-containing drugs, antibiotics, nonsteroidal anti-inflammatory drugs, and uric acid–lowering agents. The histology of SJS/TEN also is quite similar to GBFDE, and these entities may be indistinguishable without clinical information.5 Lee et al1 found that absence of grouped necrotic keratinocytes (fire flag sign), deep inflammatory infiltrates, notable pigment incontinence, and higher eosinophil counts appear to be more common in GBFDE than SJS/TEN. Constitutional symptoms and mucosal involvement also were more frequent in SJS/TEN.
The timing of clinical presentation and medical history can be useful in differentiating between SJS/TEN and GBFDE. In SJS/TEN, drug exposure typically occurs 1 to 3 weeks before onset of symptoms vs 30 minutes to 24 hours in GBFDE.3 Additionally, a history of similar eruption in the same location is pathognomonic for GBFDE. Although GBFDE has been thought to have a better prognosis than SJS/TEN, more recent data suggest mortality rates may be similar.3 A case-control study found a mortality rate of 22% (13/58) in patients with GBFDE compared to 28% (n=170) in SJS/TEN patients.4
Erythema multiforme (EM) is an uncommon immunemediated disorder that typically presents as targetoid lesions with central epidermal necrosis in an acral distribution. Erythema multiforme can arise from a variety of factors, but up to 90% of cases are due to infection, most commonly herpes simplex virus; medications account for less than 10% of cases.6 Previously, EM has been thought to be on the same disease spectrum as SJS and TEN. It is now clear that EM is a separate entity with similar mucosal erosions but different cutaneous findings,6 mainly typical target lesions that differ from the atypical targets seen in SJS.
Staphylococcal scalded skin syndrome is a blistering skin disorder associated with local Staphylococcus aureus infection. It most commonly is seen in children and rarely occurs in adults who are not on dialysis. Some Staphylococcus strains produce exfoliative toxins A and B, which are serine proteases that target and cleave desmoglein 1, a mediator of keratinocyte adhesion. Staphylococcal scalded skin syndrome initially presents with erythema accentuated in the skin folds that becomes generalized. The disruption of keratinocyte adhesion leads to bullae formation in areas of erythema and diffuse sheetlike desquamation. Pathology reveals subcorneal rather than subepidermal blistering, which is seen in GBFDE and SJS/TEN. Treatment involves antistaphylococcal antibiotics and supportive care. With proper treatment, most cases resolve within 2 to 3 weeks.7
Mycoplasma pneumoniae–induced rash and mucositis presents with prominent mucositis and can have cutaneous findings of sparse vesiculobullous or targetoid eruption.8 Mycoplasma pneumoniae typically infects the lungs and is a leading cause of community-acquired pneumonia. However, a subset of patients can have extrapulmonary disease presenting as mucocutaneous eruptions, which is preceded by an approximately weeklong prodrome of fever, cough, and malaise.7 Mycoplasma pneumoniae–induced rash and mucositis also affect children and young patients and is more common in males.8
The Diagnosis: Generalized Bullous Fixed Drug Eruption
A punch biopsy from the left thigh revealed a vacuolar interface dermatitis with full-thickness necrosis of the epidermis and a patchy lichenoid inflammatory cell infiltrate in the superficial dermis consistent with a generalized bullous fixed drug eruption (GBFDE). The patient received supportive care and methylprednisolone with improvement of symptoms.
Generalized bullous fixed drug eruption is a rare, potentially life-threatening form of a fixed drug eruption (FDE), a cutaneous drug reaction that occurs in response to a causative medication. It typically presents with welldemarcated, dusky, erythematous patches or plaques that recur in the same sites with repeat exposure.1 The pathogenesis of FDE has been hypothesized to involve epidermal CD8+ T cells, which are activated by drug exposure and release cytotoxic molecules including Fas, Fas ligand, perforin, and granzyme B, resulting in lysis of the surrounding keratinocytes.1-3 Common eliciting drugs include nonsteroidal anti-inflammatory drugs, antibacterial agents (particularly trimethoprim-sulfamethoxazole), barbiturates, acetaminophen, and antimalarials.1 In addition to the findings seen in FDE, GBFDE is characterized by widespread bullous skin lesions.1-4 Typical histologic patterns seen in GBFDE are dispersed epidermal apoptotic keratinocytes, prominent dermal eosinophilic and lymphocytic infiltrates, and dermal melanophages.3 Discontinuing the causative agent and diligent prevention of re-exposure are the most important steps in management, as additional exposures can increase the number of lesions and overall severity. Symptoms typically resolve 7 to 14 days after drug discontinuation, often with postinflammatory hyperpigmentation.3
Generalized bullous fixed drug eruption presents a diagnostic challenge, as it sometimes involves the oral mucosa and can exhibit the Nikolsky sign. Thus, it often is confused with Stevens-Johnson syndrome (SJS) or toxic epidermal necrolysis (TEN).1,4 Stevens-Johnson syndrome and TEN are severe cutaneous drug eruptions that also can present with diffuse bullous skin lesions. Stevens-Johnson syndrome and TEN are thought to be a spectrum of the same disease that initially presents with dusky red macules that can coalesce, develop central blistering, and lead to skin detachment.5 Stevens-Johnson syndrome is defined as skin detachment of less than 10% body surface area (BSA); TEN is defined as skin detachment of more than 30% BSA. Stevens-Johnson syndrome/TEN overlap syndrome includes skin detachment of 10% to 30% BSA.5
Causative medications overlap substantially with GBFDE and include anticonvulsants, sulfa-containing drugs, antibiotics, nonsteroidal anti-inflammatory drugs, and uric acid–lowering agents. The histology of SJS/TEN also is quite similar to GBFDE, and these entities may be indistinguishable without clinical information.5 Lee et al1 found that absence of grouped necrotic keratinocytes (fire flag sign), deep inflammatory infiltrates, notable pigment incontinence, and higher eosinophil counts appear to be more common in GBFDE than SJS/TEN. Constitutional symptoms and mucosal involvement also were more frequent in SJS/TEN.
The timing of clinical presentation and medical history can be useful in differentiating between SJS/TEN and GBFDE. In SJS/TEN, drug exposure typically occurs 1 to 3 weeks before onset of symptoms vs 30 minutes to 24 hours in GBFDE.3 Additionally, a history of similar eruption in the same location is pathognomonic for GBFDE. Although GBFDE has been thought to have a better prognosis than SJS/TEN, more recent data suggest mortality rates may be similar.3 A case-control study found a mortality rate of 22% (13/58) in patients with GBFDE compared to 28% (n=170) in SJS/TEN patients.4
Erythema multiforme (EM) is an uncommon immunemediated disorder that typically presents as targetoid lesions with central epidermal necrosis in an acral distribution. Erythema multiforme can arise from a variety of factors, but up to 90% of cases are due to infection, most commonly herpes simplex virus; medications account for less than 10% of cases.6 Previously, EM has been thought to be on the same disease spectrum as SJS and TEN. It is now clear that EM is a separate entity with similar mucosal erosions but different cutaneous findings,6 mainly typical target lesions that differ from the atypical targets seen in SJS.
Staphylococcal scalded skin syndrome is a blistering skin disorder associated with local Staphylococcus aureus infection. It most commonly is seen in children and rarely occurs in adults who are not on dialysis. Some Staphylococcus strains produce exfoliative toxins A and B, which are serine proteases that target and cleave desmoglein 1, a mediator of keratinocyte adhesion. Staphylococcal scalded skin syndrome initially presents with erythema accentuated in the skin folds that becomes generalized. The disruption of keratinocyte adhesion leads to bullae formation in areas of erythema and diffuse sheetlike desquamation. Pathology reveals subcorneal rather than subepidermal blistering, which is seen in GBFDE and SJS/TEN. Treatment involves antistaphylococcal antibiotics and supportive care. With proper treatment, most cases resolve within 2 to 3 weeks.7
Mycoplasma pneumoniae–induced rash and mucositis presents with prominent mucositis and can have cutaneous findings of sparse vesiculobullous or targetoid eruption.8 Mycoplasma pneumoniae typically infects the lungs and is a leading cause of community-acquired pneumonia. However, a subset of patients can have extrapulmonary disease presenting as mucocutaneous eruptions, which is preceded by an approximately weeklong prodrome of fever, cough, and malaise.7 Mycoplasma pneumoniae–induced rash and mucositis also affect children and young patients and is more common in males.8
- Lee CH, Chen YC, Cho YT, et al. Fixed-drug eruption: a retrospective study in a single referral center in northern Taiwan. Dermatologica Sinica. 2012;30:11-15. doi:10.1016/j.dsi.2012.02.002
- Cho Y-T, Lin J-W, Chen Y-C, et al. Generalized bullous fixed drug eruption is distinct from Stevens-Johnson syndrome/toxic epidermal necrolysis by immunohistopathological features. J Am Acad Dermatol. 2014;70:539-548. doi:10.1016/j.jaad.2013.11.015
- Mitre V, Applebaum DS, Albahrani Y, et al. Generalized bullous fixed drug eruption imitating toxic epidermal necrolysis: a case report and literature review. Dermatol Online J. 2017;23: 13030/qt25v009gs.
- Lipowicz S, Sekula P, Ingen-Housz-Oro S, et al. Prognosis of generalized bullous fixed drug eruption: comparison with StevensJohnson syndrome and toxic epidermal necrolysis. Br J Dermatol. 2013;168:726-732. doi:10.1111/bjd.12133
- Cho Y-T, Chu C-Y. Treatments for severe cutaneous adverse reactions [published online December 27, 2017]. J Immunol Res. doi:10.1155/2017/1503709
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902. doi:10.1111/j.1365-4632.2011.05348.x
- Leung AKC, Barankin B, Leong KF. Staphylococcal-scalded skin syndrome: evaluation, diagnosis, and management. World J Pediatr. 2018;14:116-120.
- Canavan TN, Mathes EF, Frieden I, et al. Mycoplasma pneumoniae–induced rash and mucositis as a syndrome distinct from Stevens-Johnson syndrome and erythema multiforme: a systematic review. J Am Acad Dermatol. 2015;72:239-245. doi:10.1016/j .jaad.2014.06.026
- Lee CH, Chen YC, Cho YT, et al. Fixed-drug eruption: a retrospective study in a single referral center in northern Taiwan. Dermatologica Sinica. 2012;30:11-15. doi:10.1016/j.dsi.2012.02.002
- Cho Y-T, Lin J-W, Chen Y-C, et al. Generalized bullous fixed drug eruption is distinct from Stevens-Johnson syndrome/toxic epidermal necrolysis by immunohistopathological features. J Am Acad Dermatol. 2014;70:539-548. doi:10.1016/j.jaad.2013.11.015
- Mitre V, Applebaum DS, Albahrani Y, et al. Generalized bullous fixed drug eruption imitating toxic epidermal necrolysis: a case report and literature review. Dermatol Online J. 2017;23: 13030/qt25v009gs.
- Lipowicz S, Sekula P, Ingen-Housz-Oro S, et al. Prognosis of generalized bullous fixed drug eruption: comparison with StevensJohnson syndrome and toxic epidermal necrolysis. Br J Dermatol. 2013;168:726-732. doi:10.1111/bjd.12133
- Cho Y-T, Chu C-Y. Treatments for severe cutaneous adverse reactions [published online December 27, 2017]. J Immunol Res. doi:10.1155/2017/1503709
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902. doi:10.1111/j.1365-4632.2011.05348.x
- Leung AKC, Barankin B, Leong KF. Staphylococcal-scalded skin syndrome: evaluation, diagnosis, and management. World J Pediatr. 2018;14:116-120.
- Canavan TN, Mathes EF, Frieden I, et al. Mycoplasma pneumoniae–induced rash and mucositis as a syndrome distinct from Stevens-Johnson syndrome and erythema multiforme: a systematic review. J Am Acad Dermatol. 2015;72:239-245. doi:10.1016/j .jaad.2014.06.026
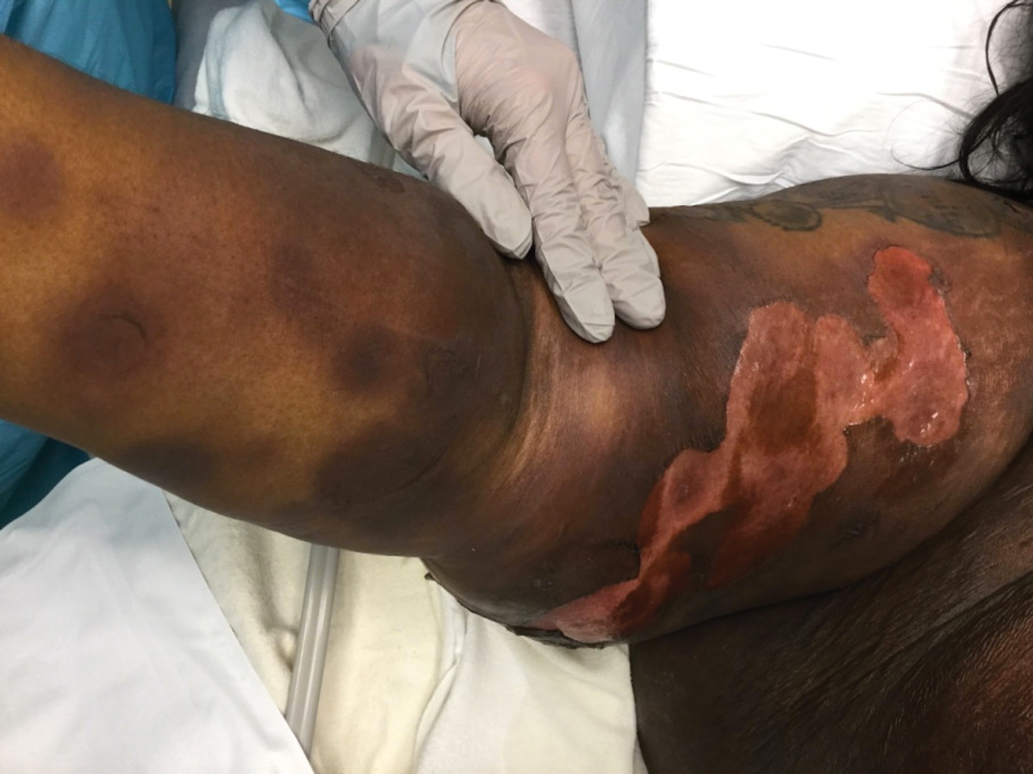
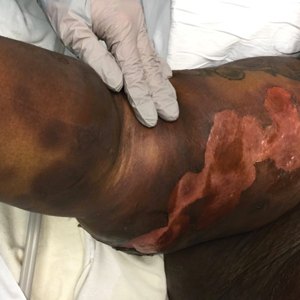
A 45-year-old woman presented with a diffuse rash 2 days after receiving ondansetron. She developed blisters on the arms, legs, trunk, and face 2 hours after exposure. There was no oral or vaginal involvement. She reported a history of leg blisters after prior exposure to ondansetron that were not as severe or numerous as the current episode. Physical examination revealed innumerable coalescing, ovoid and circular, dusky patches, some with central flaccid bullae, along with large areas of denuded skin on the trunk, arms, legs, and face. There were erosions on the lower eyelids without conjunctival or other mucosal involvement.
VEXAS syndrome: Implications for dermatologists
When I was a medical student, I always found it gratifying when there was a unifying mechanism that explained the symptoms of a disease. Part of the reason I chose dermatology as a specialty was how frequently we are able to “see” these mechanisms in the skin, both clinically and histologically. What’s even more interesting is that this condition is caused by a postzygotic somatic mutation, an apparently underrecognized cause of disease that we are just now beginning to understand. An example of a postzygotic somatic mutation causing a disease that we all learned about in medical school is paroxysmal nocturnal hemoglobinuria.

Using a “bottom-up” approach, researchers at the National Institutes of Health and in the United Kingdom identified 25 patients with somatic UBA1 mutations and noticed that they had strikingly similar autoinflammatory syndromes. UBA1 encodes ubiquitin E1, which is part of the pathway the breaks down proteins as part of the normal cellular machine. It is localized to the X chromosome, so all 25 affected patients were males, and most were aged between 40 and 70 years. These patients had an autoinflammatory syndrome characterized by fever, chondritis (similar to relapsing polychondritis), vasculitis, and neutrophilic dermatoses. Many patients also had features of myelodysplastic syndrome and plasma cell dyscrasia. The inflammatory pattern in this condition seems to show elevations in tumor necrosis factor, interleukin-6, and interferon-gamma.
So why is this syndrome relevant to dermatology? We are often asked to evaluate patients for neutrophilic dermatosis and vasculitis, and many affected patients had clinical and histologic findings compatible with polyarteritis nodosa and Sweet syndrome. When confronted with a neutrophilic dermatosis, we’ve all been taught to evaluate for myelodysplastic syndrome, which many of these patients appeared to have, at least on the surface. When bone marrow biopsies were done, the myeloid cell precursors that give rise to neutrophils were noted to have prominent cytoplasmic vacuoles, hence the “V” in VEXAS.
In reading the article describing 25 patients with this syndrome, which was published in the New England Journal of Medicine, I was struck by how refractory they were to treatment. Most patients had been treated with systemic steroids, multiple biologics, and several nonbiologic medications that are mainstays of treatment for neutrophilic dermatosis like dapsone and colchicine. I was fortunate enough to speak to Amanda Ombrello, MD, of the National Human Genome Research Institute, one of the lead authors of the paper, who drew my attention to the supplementary appendix, which showed the marked injection-site reactions some patients had to anakinra – yet another reason why a patient might end up in a dermatology clinic. In my mind, all of these features could be a clue to a diagnosis of VEXAS syndrome.
Many patients seemed to fare poorly, with 40% of patients dying before the completion of the study. When it comes to extremely rare diseases, it seems that the more physicians who are aware of the existence of a particular syndrome, the more likely it is a patient will come under our care and be correctly diagnosed.
Dr. Saardi is a dermatologist and internist, and is director of the inpatient dermatology service at the George Washington University Hospital, Washington. He has no disclosures.
When I was a medical student, I always found it gratifying when there was a unifying mechanism that explained the symptoms of a disease. Part of the reason I chose dermatology as a specialty was how frequently we are able to “see” these mechanisms in the skin, both clinically and histologically. What’s even more interesting is that this condition is caused by a postzygotic somatic mutation, an apparently underrecognized cause of disease that we are just now beginning to understand. An example of a postzygotic somatic mutation causing a disease that we all learned about in medical school is paroxysmal nocturnal hemoglobinuria.
Using a “bottom-up” approach, researchers at the National Institutes of Health and in the United Kingdom identified 25 patients with somatic UBA1 mutations and noticed that they had strikingly similar autoinflammatory syndromes. UBA1 encodes ubiquitin E1, which is part of the pathway the breaks down proteins as part of the normal cellular machine. It is localized to the X chromosome, so all 25 affected patients were males, and most were aged between 40 and 70 years. These patients had an autoinflammatory syndrome characterized by fever, chondritis (similar to relapsing polychondritis), vasculitis, and neutrophilic dermatoses. Many patients also had features of myelodysplastic syndrome and plasma cell dyscrasia. The inflammatory pattern in this condition seems to show elevations in tumor necrosis factor, interleukin-6, and interferon-gamma.
So why is this syndrome relevant to dermatology? We are often asked to evaluate patients for neutrophilic dermatosis and vasculitis, and many affected patients had clinical and histologic findings compatible with polyarteritis nodosa and Sweet syndrome. When confronted with a neutrophilic dermatosis, we’ve all been taught to evaluate for myelodysplastic syndrome, which many of these patients appeared to have, at least on the surface. When bone marrow biopsies were done, the myeloid cell precursors that give rise to neutrophils were noted to have prominent cytoplasmic vacuoles, hence the “V” in VEXAS.
In reading the article describing 25 patients with this syndrome, which was published in the New England Journal of Medicine, I was struck by how refractory they were to treatment. Most patients had been treated with systemic steroids, multiple biologics, and several nonbiologic medications that are mainstays of treatment for neutrophilic dermatosis like dapsone and colchicine. I was fortunate enough to speak to Amanda Ombrello, MD, of the National Human Genome Research Institute, one of the lead authors of the paper, who drew my attention to the supplementary appendix, which showed the marked injection-site reactions some patients had to anakinra – yet another reason why a patient might end up in a dermatology clinic. In my mind, all of these features could be a clue to a diagnosis of VEXAS syndrome.
Many patients seemed to fare poorly, with 40% of patients dying before the completion of the study. When it comes to extremely rare diseases, it seems that the more physicians who are aware of the existence of a particular syndrome, the more likely it is a patient will come under our care and be correctly diagnosed.
Dr. Saardi is a dermatologist and internist, and is director of the inpatient dermatology service at the George Washington University Hospital, Washington. He has no disclosures.
When I was a medical student, I always found it gratifying when there was a unifying mechanism that explained the symptoms of a disease. Part of the reason I chose dermatology as a specialty was how frequently we are able to “see” these mechanisms in the skin, both clinically and histologically. What’s even more interesting is that this condition is caused by a postzygotic somatic mutation, an apparently underrecognized cause of disease that we are just now beginning to understand. An example of a postzygotic somatic mutation causing a disease that we all learned about in medical school is paroxysmal nocturnal hemoglobinuria.
Using a “bottom-up” approach, researchers at the National Institutes of Health and in the United Kingdom identified 25 patients with somatic UBA1 mutations and noticed that they had strikingly similar autoinflammatory syndromes. UBA1 encodes ubiquitin E1, which is part of the pathway the breaks down proteins as part of the normal cellular machine. It is localized to the X chromosome, so all 25 affected patients were males, and most were aged between 40 and 70 years. These patients had an autoinflammatory syndrome characterized by fever, chondritis (similar to relapsing polychondritis), vasculitis, and neutrophilic dermatoses. Many patients also had features of myelodysplastic syndrome and plasma cell dyscrasia. The inflammatory pattern in this condition seems to show elevations in tumor necrosis factor, interleukin-6, and interferon-gamma.
So why is this syndrome relevant to dermatology? We are often asked to evaluate patients for neutrophilic dermatosis and vasculitis, and many affected patients had clinical and histologic findings compatible with polyarteritis nodosa and Sweet syndrome. When confronted with a neutrophilic dermatosis, we’ve all been taught to evaluate for myelodysplastic syndrome, which many of these patients appeared to have, at least on the surface. When bone marrow biopsies were done, the myeloid cell precursors that give rise to neutrophils were noted to have prominent cytoplasmic vacuoles, hence the “V” in VEXAS.
In reading the article describing 25 patients with this syndrome, which was published in the New England Journal of Medicine, I was struck by how refractory they were to treatment. Most patients had been treated with systemic steroids, multiple biologics, and several nonbiologic medications that are mainstays of treatment for neutrophilic dermatosis like dapsone and colchicine. I was fortunate enough to speak to Amanda Ombrello, MD, of the National Human Genome Research Institute, one of the lead authors of the paper, who drew my attention to the supplementary appendix, which showed the marked injection-site reactions some patients had to anakinra – yet another reason why a patient might end up in a dermatology clinic. In my mind, all of these features could be a clue to a diagnosis of VEXAS syndrome.
Many patients seemed to fare poorly, with 40% of patients dying before the completion of the study. When it comes to extremely rare diseases, it seems that the more physicians who are aware of the existence of a particular syndrome, the more likely it is a patient will come under our care and be correctly diagnosed.
Dr. Saardi is a dermatologist and internist, and is director of the inpatient dermatology service at the George Washington University Hospital, Washington. He has no disclosures.