User login
Bullous Pemphigoid Associated With a Lymphoepithelial Cyst of the Pancreas
Bullous pemphigoid (BP) is an acquired, autoimmune, subepidermal blistering disease that is more common in elderly patients.1 An association with internal neoplasms and BP has been established; however, there is debate regarding the precise nature of the relationship.2 Several gastrointestinal tract tumors have been associated with BP, including adenocarcinoma of the colon, adenosquamous cell carcinoma and adenocarcinoma of the stomach, adenocarcinoma of the rectum, and liver and bile duct malignancies.3-5 Association with pancreatic neoplasms (eg, carcinoma of the pancreas) rarely has been reported.5-7 We present an unusual case of a lymphoepithelial cyst of the pancreas in a patient with BP.
Case Report
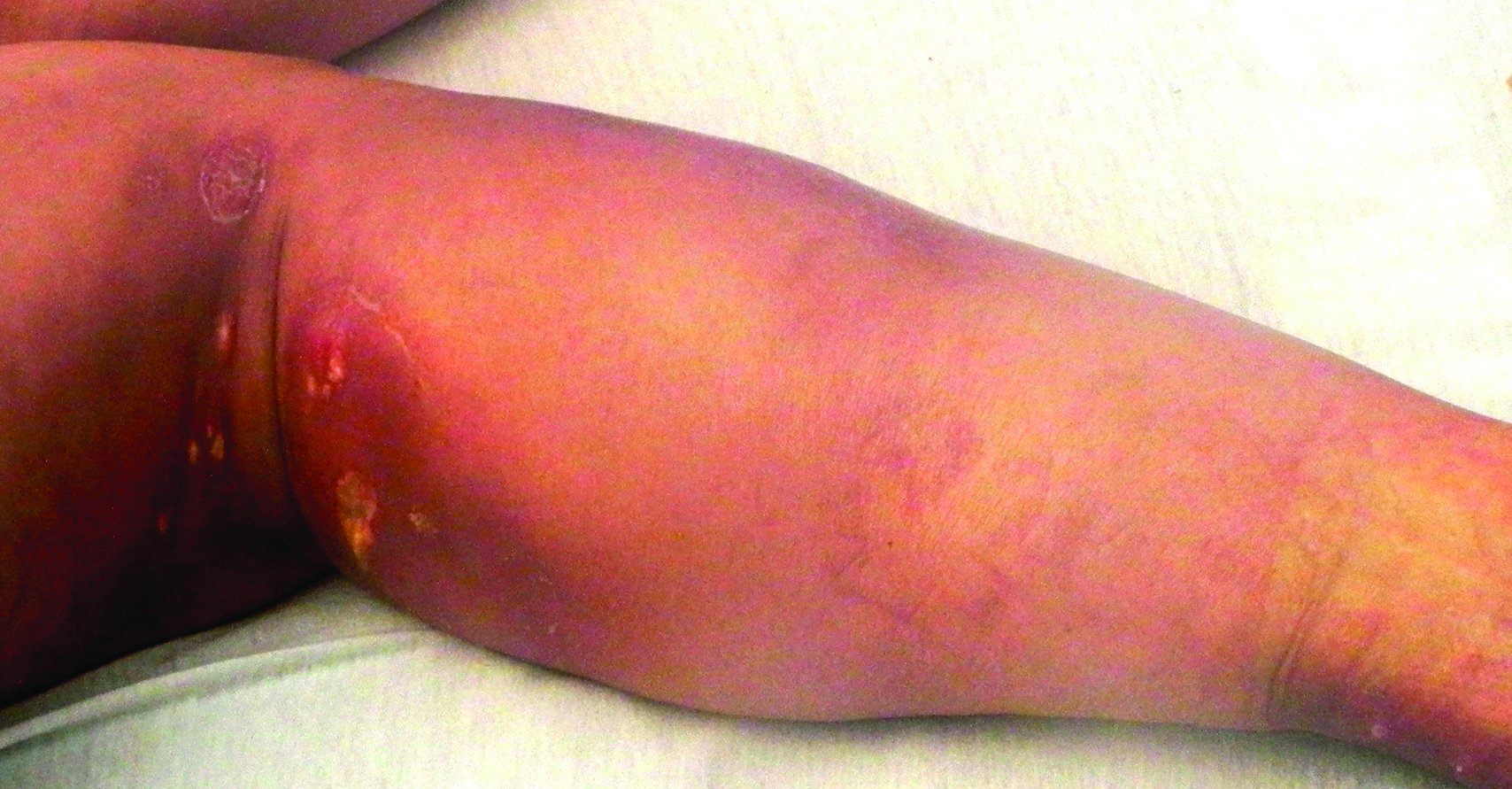
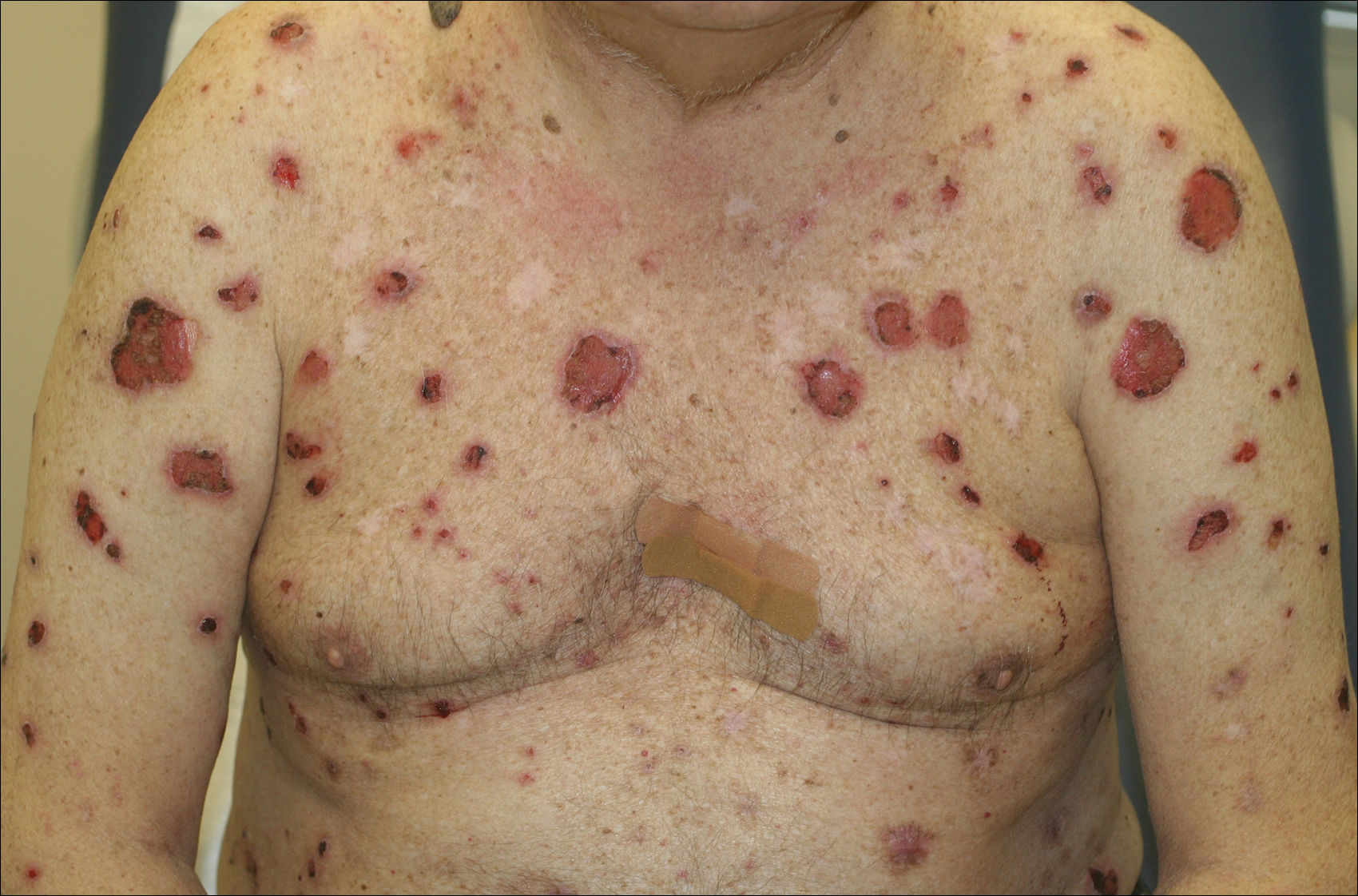
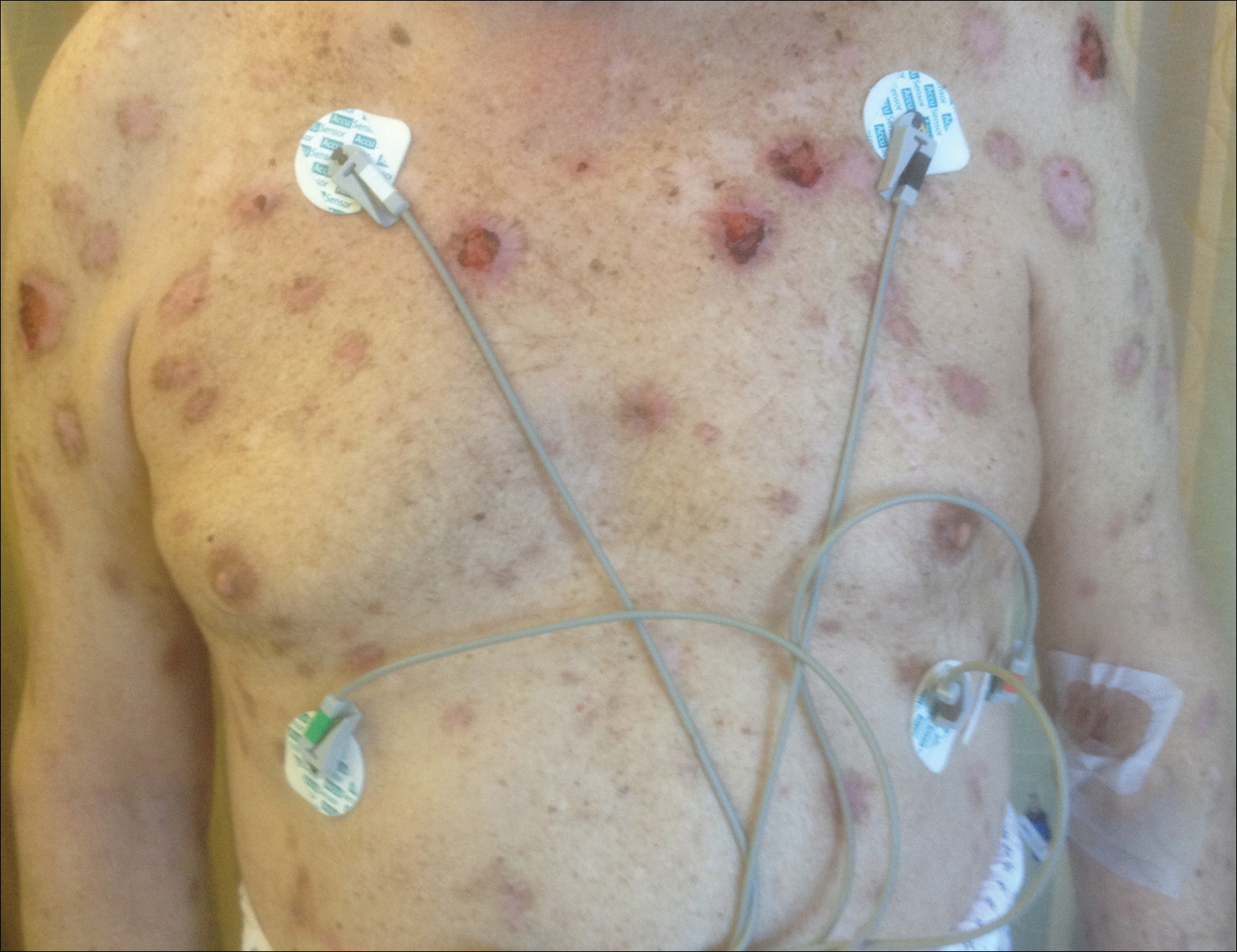
A 67-year-old man presented with erythematous crusted plaques and pink scars over the chest, back, arms, and legs (Figure 1). A 1.5-cm tense bullous lesion was observed on the right knee. The patient’s medical history was notable for biopsy-proven BP of 8 months’ duration as well as diabetes mellitus and hypothyroidism. The patient was being followed by his surgeon for a 1.9-cm soft-tissue lesion in the pancreatic tail and was awaiting surgical excision at the time of the current presentation. The pancreatic lesion was discovered incidentally on magnetic resonance imaging performed following urologic concerns. At the current presentation, the patient’s medications included nifedipine, hydralazine, metoprolol, torsemide, aspirin, levothyroxine, atorvastatin, insulin lispro, and insulin glargine. He had previously been treated for BP with prednisone at a maximum dosage of 60 mg daily, clobetasol propionate cream 0.05%, and mupirocin ointment 2% without improvement. Because of substantial weight gain and poorly controlled diabetes, prednisone was discontinued.
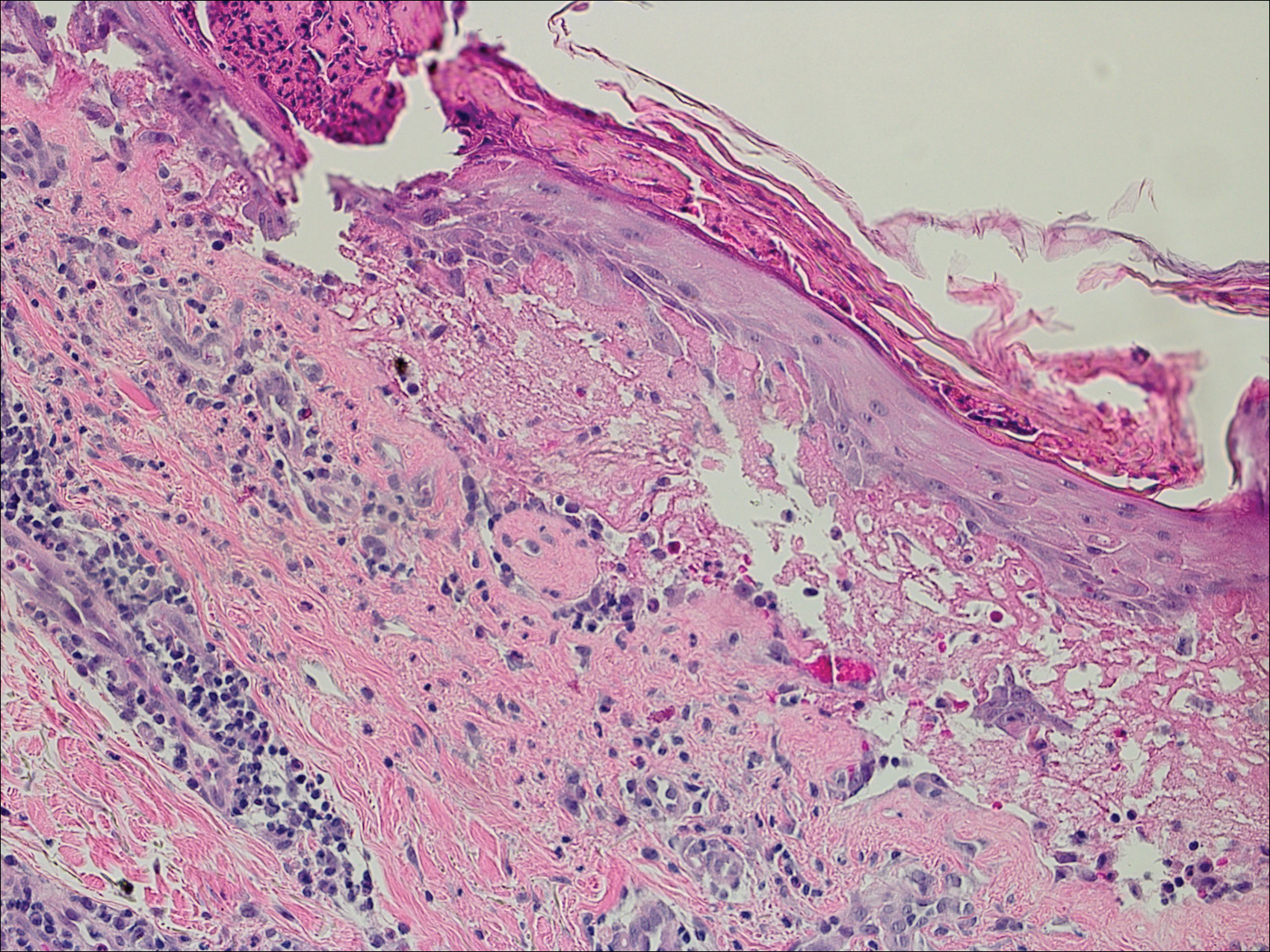
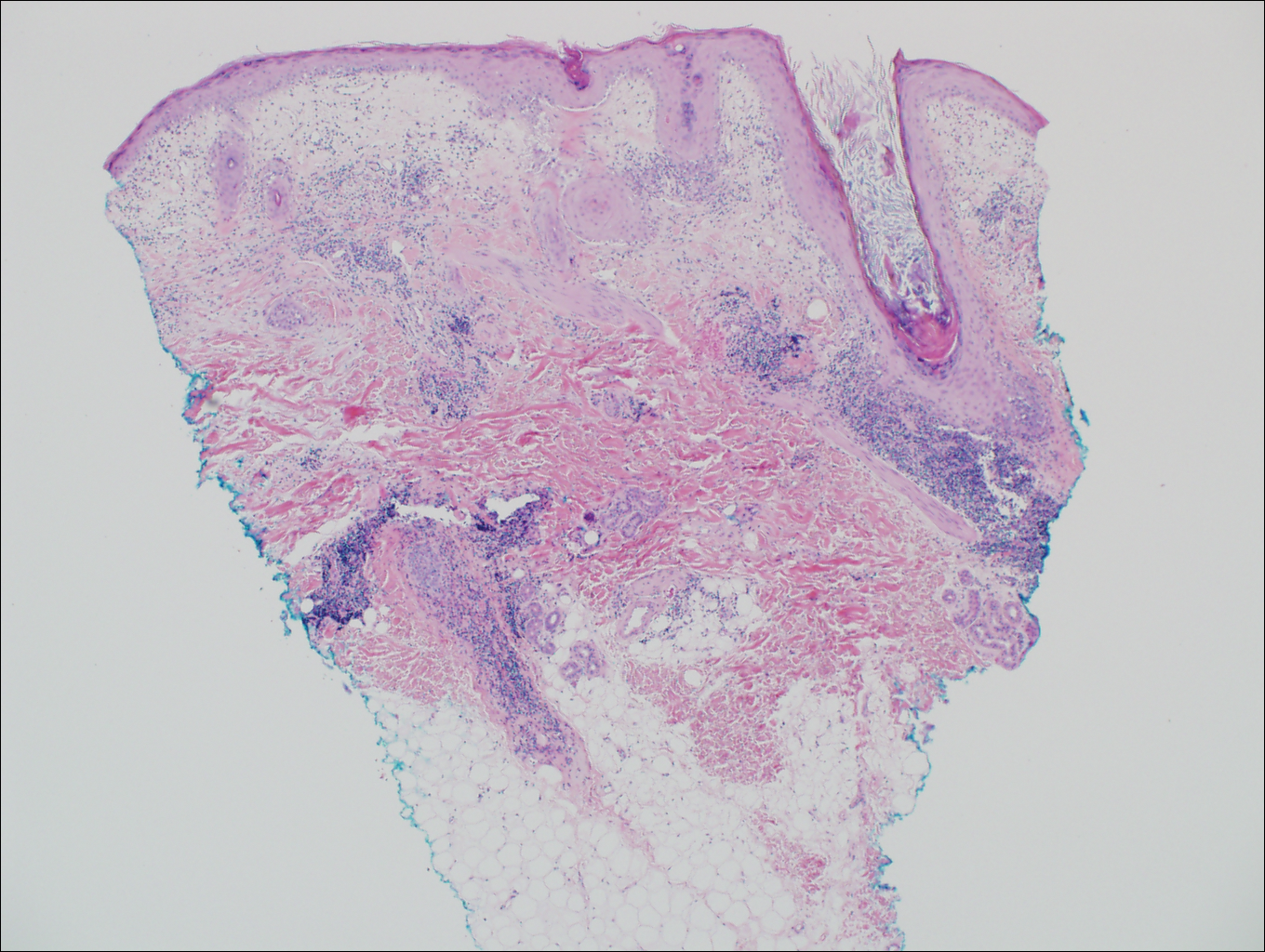
Bullous pemphigoid had been diagnosed histopathologically by a prior dermatologist. Hematoxylin and eosin staining demonstrated a subepidermal separation with eosinophils within the perivascular infiltrate (Figure 2). Direct immunofluorescence was noted in a linear pattern at the dermoepidermal junction with IgG and C3. Bullous pemphigoid antigen antibodies 1 and 2 were obtained via enzyme-linked immunosorbent assay with a positive BP-1 antigen antibody of 19 U/mL (positive, >15 U/mL) and a normal BP-2 antigen antibody of less than 9 U/mL (reference range, <9 U/mL). The glucagon level was elevated at 245 pg/mL (reference range, ≤134 pg/mL).
The patient was prescribed minocycline 100 mg twice daily and niacinamide 500 mg 3 times daily. Topical treatment with clobetasol and mupirocin was continued. One month later, the patient returned with an increase in disease activity. Changes to his therapeutic regimen were deferred until after excision of the pancreatic lesion based on the decision not to start immunosuppressive therapy until the precise nature of the pancreatic lesion was determined.
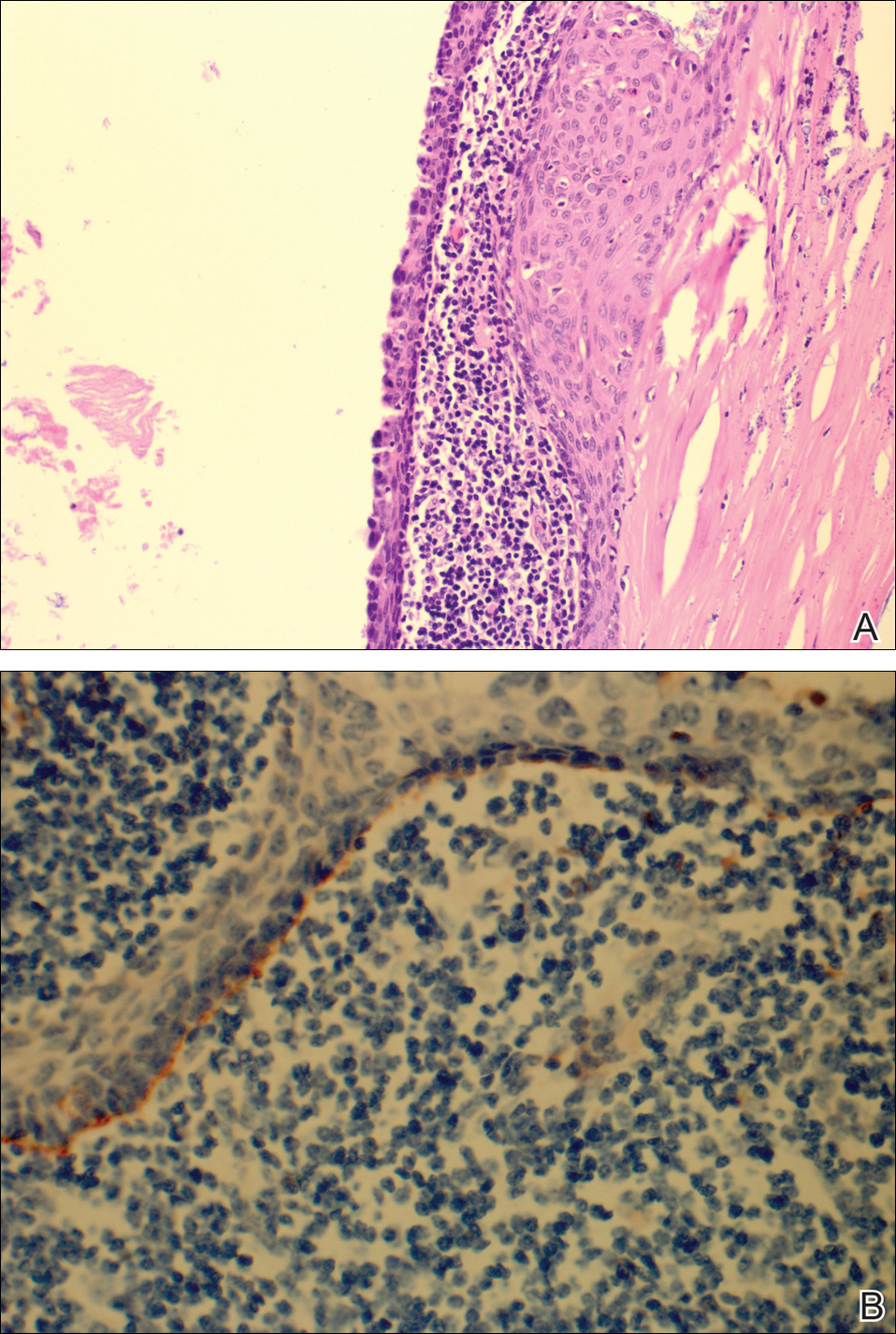
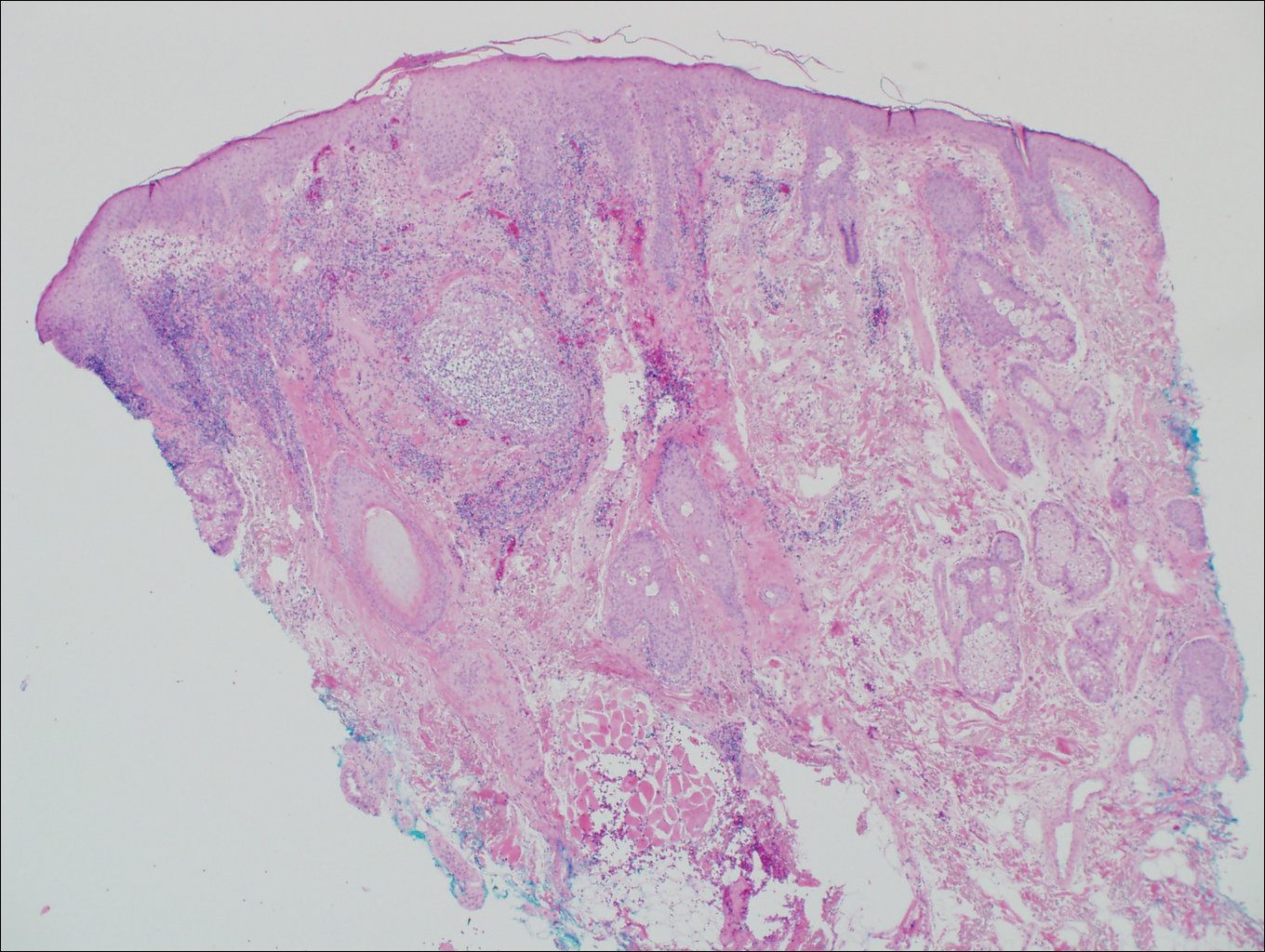
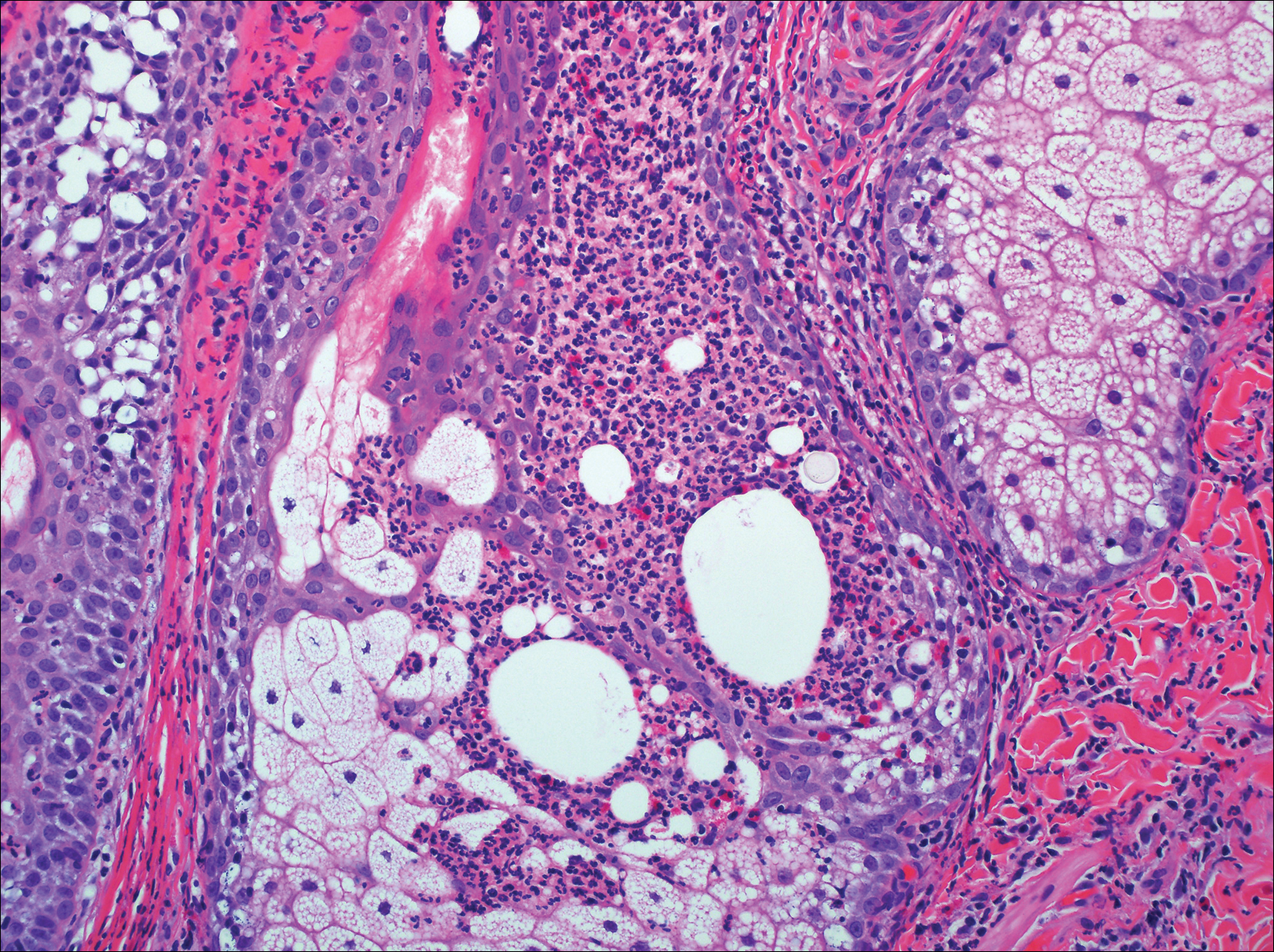
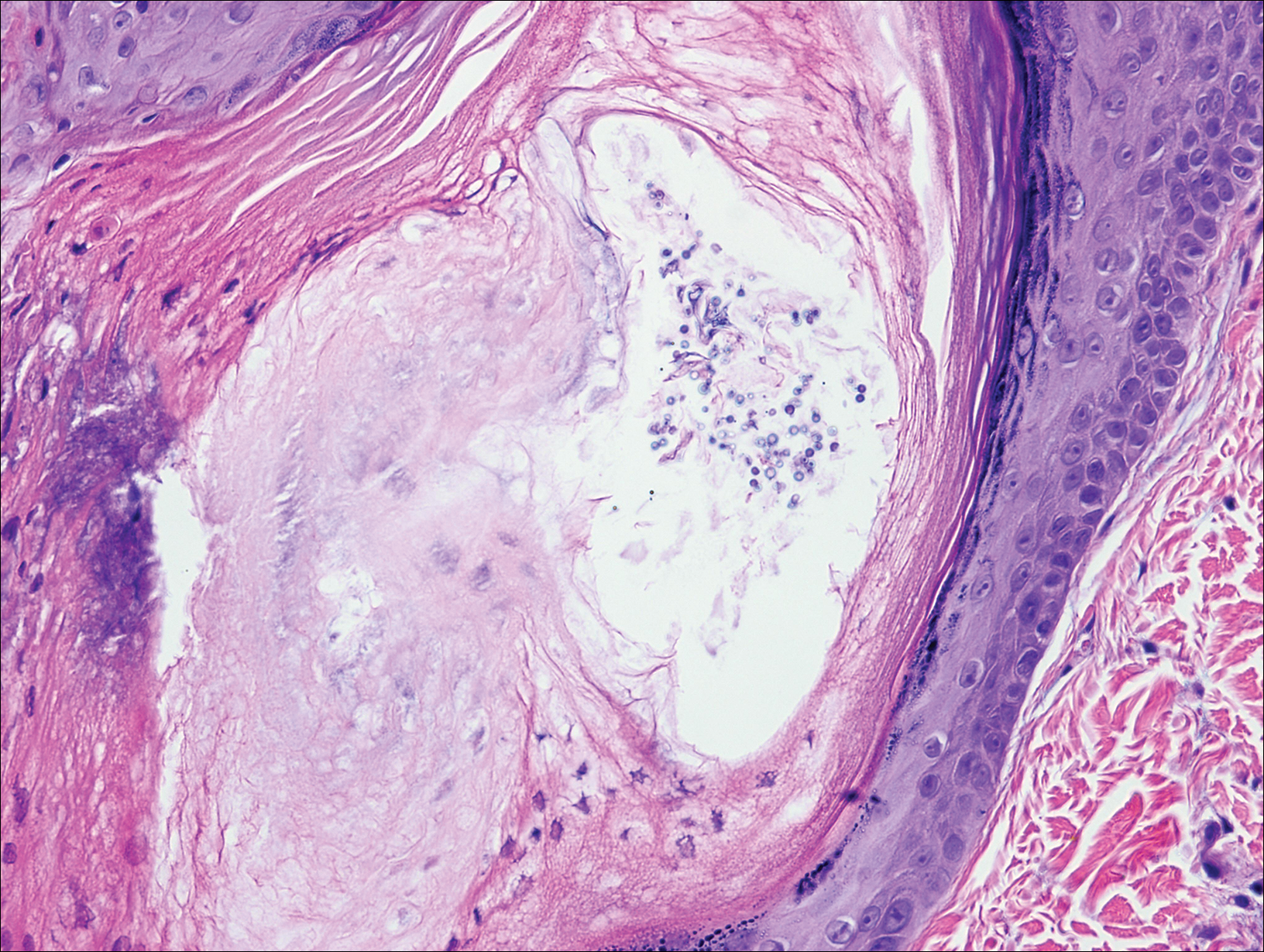
The patient underwent excision of the pancreatic lesion approximately 3 months later, which proved to be a benign lymphoepithelial cyst of the pancreas. Histology of the cyst consisted of dense fibrous tissue with a squamous epithelial lining focally infiltrated by lymphocytes (Figure 3A). Immunoperoxidase staining of the cyst revealed focal linear areas of C3d staining along the basement membrane of the stratified squamous epithelium (Figure 3B).
The patient stated that his skin started to improve virtually immediately following the excision without systemic treatment for BP. On follow-up examination 3 weeks postoperatively, no bullae were observed and there was a notable decrease in erythematous crusted plaques (Figure 4).
Comment
Paraneoplastic BP has been documented; however, lymphoepithelial cysts of the pancreas in association with BP are rare. We propose that the lymphoepithelial cyst of the pancreas provided the immunologic stimulus for the development of cutaneous BP based on the observation that our patient’s condition remarkably improved with resection of the tumor.
There are fewer than 100 cases of lymphoepithelial cysts of the pancreas reported in the literature.8 The histologic appearance is consistent with a true cyst exhibiting a well-differentiated stratified squamous epithelium, often with keratinization, surrounded by lymphoid tissue. These tumors are most commonly seen in middle-aged men and are frequently found incidentally,8-10 as was the case with our patient. Although histologically similar, lymphoepithelial cysts of the pancreas are considered distinct from lymphoepithelial cysts of the parotid gland or head and neck region.10 Lymphoepithelial cysts of the pancreas are unrelated to elevated glucagon levels; it is likely that our patient’s glucagon levels were associated with his history of diabetes.11
The diagnosis of BP is characteristically confirmed by direct immunofluorescence. Although it was performed for our patient’s cutaneous lesions, it was not obtained for the lymphoepithelial cyst of the pancreas. Once the diagnosis of the lymphoepithelial cyst of the pancreas was established, as direct immunofluorescence could not be performed in formalin-fixed tissue, immunoperoxidase staining with C3d was obtained. C3 has a well-established role in activation of complement and as a marker in BP. Deposition of C3d is a result of deactivation of C3b, a cleavage product of C3. In a study of 6 autoimmune blistering disorders that included 32 patients with BP, Pfaltz et al12 found positive immunoperoxidase staining for C3d in 31 of 32 patients, which translated to a sensitivity of 97%, a positive predictive value of 100%, and a negative predictive value of 98% among the blistering diseases being studied. Similarly, Magro and Dyrsen13 had positive staining of C3d in 17 of 17 (100%) patients with BP.
In theory, any process that involves deposition of C3 should be positive for C3d on immunoperoxidase staining. Other dermatologic inflammatory conditions stain positively with C3d, such as systemic lupus erythematosus, discoid lupus erythematosus, subacute cutaneous lupus erythematosus, and dermatomysositis.13 The staining for these diseases correlates with the site of the associated inflammatory component seen on hematoxylin and eosin staining. The staining of C3d along the basement membrane of stratified squamous epithelium in the lymphoepithelial cyst of the pancreas seen in our patient closely resembles the staining seen in cutaneous BP.
A proposed mechanism for BP in our patient would be exposure of BP-1 antigen in the pancreatic cyst leading to antibody recognition and C3 deposition along the basement membrane in the cyst, as evidenced by C3d immunoperoxidase staining. The IgG and C3 deposition along the cutaneous basement membrane would then represent a systemic response to the antigen exposure in the cyst. Thus, the lymphoepithelial cyst provided the immunologic stimulus for the development of the cutaneous BP. This theory is based on the observation of our patient’s rapid improvement without a change in his treatment regimen immediately after surgical excision of the cyst.
Despite the plausibility of our hypothesis, several questions remain regarding the validity of our assumptions. Although sensitive for C3 deposition, C3d immunoperoxidase staining is not specific for BP. If the proposed mechanism for causation is true, one might have expected that a subepithelial cleft along the basement membrane of the pancreatic cyst would be observed, which was not seen. A repeat BP antigen antibody was not obtained, which would have been helpful in determining if there was clearance of the antibody that would have correlated with the clinical resolution of the BP lesions.
Conclusion
Our case suggests that paraneoplastic BP is a genuine entity. Indeed, the primary tumor itself may be the immunologic stimulus in the development of BP. Recalcitrant BP should raise the question of a neoplastic process that is exposing the BP antigen. If a thorough review of systems accompanied by corroborating laboratory studies suggests a neoplastic process, the suspect lesion should be further evaluated and surgically excised if clinically indicated. Further evaluation of neoplasms with advanced staining methods may aid in establishing the causative nature of tumors in the development of BP.
Acknowledgments
We are grateful to John Stanley, MD, and Aimee Payne, MD (both from Philadelphia, Pennsylvania), for theirinsights into this case.
- Charneux J, Lorin J, Vitry F, et al. Usefulness of BP230 and BP180-NC16a enzyme-linked immunosorbent assays in the initial diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:286-291.
- Patel M, Sniha AA, Gilbert E. Bullous pemphigoid associated with renal cell carcinoma and invasive squamous cell carcinoma. J Drugs Dermatol. 2012;11:234-238.
- Song HJ, Han SH, Hong WK, et al. Paraneoplastic bullous pemphigoid: clinical disease activity correlated with enzyme-linked immunosorbent assay index for NC16A domain of BP180. J Dermatol. 2009;36:66-68.
- Muramatsu T, Iida T, Tada H, et al. Bullous pemphigoid associated with internal malignancies: identification of 180-kDa antigen by Western immunoblotting. Br J Dermatol. 1996;135:782-784.
- Ogawa H, Sakuma M, Morioka S, et al. The incidence of internal malignancies in pemphigus and bullous pemphigoid in Japan. J Dermatol Sci. 1995;9:136-141.
- Boyd RV. Pemphigoid and carcinoma of the pancreas. Br Med J. 1964;1:1092.
- Eustace S, Morrow G, O’Loughlin S, et al. The role of computed tomography and sonography in acute bullous pemphigoid. Ir J Med Sci. 1993;162:401-404.
- Clemente G, Sarno G, De Rose AM, et al. Lymphoepithelial cyst of the pancreas: case report and review of the literature. Acta Gastroenterol Belg. 2011;74:343-346.
- Frezza E, Wachtel MS. Lymphoepithelial cyst of the pancreas tail. case report and review of the literature. JOP. 2008;9:46-49.
- Basturk O, Coban I, Adsay NV. Pancreatic cysts: pathologic classification, differential diagnosis and clinical implications. Arch Pathol Lab Med. 2009;133:423-438.
- Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest. 2012;122:4-12.
- Pfaltz K, Mertz K, Rose C, et al. C3d immunohistochemistry on formalin-fixed tissue is a valuable tool in the diagnosis of bullous pemphigoid of the skin. J Cutan Pathol. 2010;37:654-658.
- Magro CM, Dyrsen ME. The use of C3d and C4d immunohistochemistry on formalin-fixed tissue as a diagnostic adjunct in the assessment of inflammatory skin disease. J Am Acad Dermatol. 2008;59:822-833.
Bullous pemphigoid (BP) is an acquired, autoimmune, subepidermal blistering disease that is more common in elderly patients.1 An association with internal neoplasms and BP has been established; however, there is debate regarding the precise nature of the relationship.2 Several gastrointestinal tract tumors have been associated with BP, including adenocarcinoma of the colon, adenosquamous cell carcinoma and adenocarcinoma of the stomach, adenocarcinoma of the rectum, and liver and bile duct malignancies.3-5 Association with pancreatic neoplasms (eg, carcinoma of the pancreas) rarely has been reported.5-7 We present an unusual case of a lymphoepithelial cyst of the pancreas in a patient with BP.
Case Report
A 67-year-old man presented with erythematous crusted plaques and pink scars over the chest, back, arms, and legs (Figure 1). A 1.5-cm tense bullous lesion was observed on the right knee. The patient’s medical history was notable for biopsy-proven BP of 8 months’ duration as well as diabetes mellitus and hypothyroidism. The patient was being followed by his surgeon for a 1.9-cm soft-tissue lesion in the pancreatic tail and was awaiting surgical excision at the time of the current presentation. The pancreatic lesion was discovered incidentally on magnetic resonance imaging performed following urologic concerns. At the current presentation, the patient’s medications included nifedipine, hydralazine, metoprolol, torsemide, aspirin, levothyroxine, atorvastatin, insulin lispro, and insulin glargine. He had previously been treated for BP with prednisone at a maximum dosage of 60 mg daily, clobetasol propionate cream 0.05%, and mupirocin ointment 2% without improvement. Because of substantial weight gain and poorly controlled diabetes, prednisone was discontinued.
Bullous pemphigoid had been diagnosed histopathologically by a prior dermatologist. Hematoxylin and eosin staining demonstrated a subepidermal separation with eosinophils within the perivascular infiltrate (Figure 2). Direct immunofluorescence was noted in a linear pattern at the dermoepidermal junction with IgG and C3. Bullous pemphigoid antigen antibodies 1 and 2 were obtained via enzyme-linked immunosorbent assay with a positive BP-1 antigen antibody of 19 U/mL (positive, >15 U/mL) and a normal BP-2 antigen antibody of less than 9 U/mL (reference range, <9 U/mL). The glucagon level was elevated at 245 pg/mL (reference range, ≤134 pg/mL).
The patient was prescribed minocycline 100 mg twice daily and niacinamide 500 mg 3 times daily. Topical treatment with clobetasol and mupirocin was continued. One month later, the patient returned with an increase in disease activity. Changes to his therapeutic regimen were deferred until after excision of the pancreatic lesion based on the decision not to start immunosuppressive therapy until the precise nature of the pancreatic lesion was determined.
The patient underwent excision of the pancreatic lesion approximately 3 months later, which proved to be a benign lymphoepithelial cyst of the pancreas. Histology of the cyst consisted of dense fibrous tissue with a squamous epithelial lining focally infiltrated by lymphocytes (Figure 3A). Immunoperoxidase staining of the cyst revealed focal linear areas of C3d staining along the basement membrane of the stratified squamous epithelium (Figure 3B).
The patient stated that his skin started to improve virtually immediately following the excision without systemic treatment for BP. On follow-up examination 3 weeks postoperatively, no bullae were observed and there was a notable decrease in erythematous crusted plaques (Figure 4).
Comment
Paraneoplastic BP has been documented; however, lymphoepithelial cysts of the pancreas in association with BP are rare. We propose that the lymphoepithelial cyst of the pancreas provided the immunologic stimulus for the development of cutaneous BP based on the observation that our patient’s condition remarkably improved with resection of the tumor.
There are fewer than 100 cases of lymphoepithelial cysts of the pancreas reported in the literature.8 The histologic appearance is consistent with a true cyst exhibiting a well-differentiated stratified squamous epithelium, often with keratinization, surrounded by lymphoid tissue. These tumors are most commonly seen in middle-aged men and are frequently found incidentally,8-10 as was the case with our patient. Although histologically similar, lymphoepithelial cysts of the pancreas are considered distinct from lymphoepithelial cysts of the parotid gland or head and neck region.10 Lymphoepithelial cysts of the pancreas are unrelated to elevated glucagon levels; it is likely that our patient’s glucagon levels were associated with his history of diabetes.11
The diagnosis of BP is characteristically confirmed by direct immunofluorescence. Although it was performed for our patient’s cutaneous lesions, it was not obtained for the lymphoepithelial cyst of the pancreas. Once the diagnosis of the lymphoepithelial cyst of the pancreas was established, as direct immunofluorescence could not be performed in formalin-fixed tissue, immunoperoxidase staining with C3d was obtained. C3 has a well-established role in activation of complement and as a marker in BP. Deposition of C3d is a result of deactivation of C3b, a cleavage product of C3. In a study of 6 autoimmune blistering disorders that included 32 patients with BP, Pfaltz et al12 found positive immunoperoxidase staining for C3d in 31 of 32 patients, which translated to a sensitivity of 97%, a positive predictive value of 100%, and a negative predictive value of 98% among the blistering diseases being studied. Similarly, Magro and Dyrsen13 had positive staining of C3d in 17 of 17 (100%) patients with BP.
In theory, any process that involves deposition of C3 should be positive for C3d on immunoperoxidase staining. Other dermatologic inflammatory conditions stain positively with C3d, such as systemic lupus erythematosus, discoid lupus erythematosus, subacute cutaneous lupus erythematosus, and dermatomysositis.13 The staining for these diseases correlates with the site of the associated inflammatory component seen on hematoxylin and eosin staining. The staining of C3d along the basement membrane of stratified squamous epithelium in the lymphoepithelial cyst of the pancreas seen in our patient closely resembles the staining seen in cutaneous BP.
A proposed mechanism for BP in our patient would be exposure of BP-1 antigen in the pancreatic cyst leading to antibody recognition and C3 deposition along the basement membrane in the cyst, as evidenced by C3d immunoperoxidase staining. The IgG and C3 deposition along the cutaneous basement membrane would then represent a systemic response to the antigen exposure in the cyst. Thus, the lymphoepithelial cyst provided the immunologic stimulus for the development of the cutaneous BP. This theory is based on the observation of our patient’s rapid improvement without a change in his treatment regimen immediately after surgical excision of the cyst.
Despite the plausibility of our hypothesis, several questions remain regarding the validity of our assumptions. Although sensitive for C3 deposition, C3d immunoperoxidase staining is not specific for BP. If the proposed mechanism for causation is true, one might have expected that a subepithelial cleft along the basement membrane of the pancreatic cyst would be observed, which was not seen. A repeat BP antigen antibody was not obtained, which would have been helpful in determining if there was clearance of the antibody that would have correlated with the clinical resolution of the BP lesions.
Conclusion
Our case suggests that paraneoplastic BP is a genuine entity. Indeed, the primary tumor itself may be the immunologic stimulus in the development of BP. Recalcitrant BP should raise the question of a neoplastic process that is exposing the BP antigen. If a thorough review of systems accompanied by corroborating laboratory studies suggests a neoplastic process, the suspect lesion should be further evaluated and surgically excised if clinically indicated. Further evaluation of neoplasms with advanced staining methods may aid in establishing the causative nature of tumors in the development of BP.
Acknowledgments
We are grateful to John Stanley, MD, and Aimee Payne, MD (both from Philadelphia, Pennsylvania), for theirinsights into this case.
Bullous pemphigoid (BP) is an acquired, autoimmune, subepidermal blistering disease that is more common in elderly patients.1 An association with internal neoplasms and BP has been established; however, there is debate regarding the precise nature of the relationship.2 Several gastrointestinal tract tumors have been associated with BP, including adenocarcinoma of the colon, adenosquamous cell carcinoma and adenocarcinoma of the stomach, adenocarcinoma of the rectum, and liver and bile duct malignancies.3-5 Association with pancreatic neoplasms (eg, carcinoma of the pancreas) rarely has been reported.5-7 We present an unusual case of a lymphoepithelial cyst of the pancreas in a patient with BP.
Case Report
A 67-year-old man presented with erythematous crusted plaques and pink scars over the chest, back, arms, and legs (Figure 1). A 1.5-cm tense bullous lesion was observed on the right knee. The patient’s medical history was notable for biopsy-proven BP of 8 months’ duration as well as diabetes mellitus and hypothyroidism. The patient was being followed by his surgeon for a 1.9-cm soft-tissue lesion in the pancreatic tail and was awaiting surgical excision at the time of the current presentation. The pancreatic lesion was discovered incidentally on magnetic resonance imaging performed following urologic concerns. At the current presentation, the patient’s medications included nifedipine, hydralazine, metoprolol, torsemide, aspirin, levothyroxine, atorvastatin, insulin lispro, and insulin glargine. He had previously been treated for BP with prednisone at a maximum dosage of 60 mg daily, clobetasol propionate cream 0.05%, and mupirocin ointment 2% without improvement. Because of substantial weight gain and poorly controlled diabetes, prednisone was discontinued.
Bullous pemphigoid had been diagnosed histopathologically by a prior dermatologist. Hematoxylin and eosin staining demonstrated a subepidermal separation with eosinophils within the perivascular infiltrate (Figure 2). Direct immunofluorescence was noted in a linear pattern at the dermoepidermal junction with IgG and C3. Bullous pemphigoid antigen antibodies 1 and 2 were obtained via enzyme-linked immunosorbent assay with a positive BP-1 antigen antibody of 19 U/mL (positive, >15 U/mL) and a normal BP-2 antigen antibody of less than 9 U/mL (reference range, <9 U/mL). The glucagon level was elevated at 245 pg/mL (reference range, ≤134 pg/mL).
The patient was prescribed minocycline 100 mg twice daily and niacinamide 500 mg 3 times daily. Topical treatment with clobetasol and mupirocin was continued. One month later, the patient returned with an increase in disease activity. Changes to his therapeutic regimen were deferred until after excision of the pancreatic lesion based on the decision not to start immunosuppressive therapy until the precise nature of the pancreatic lesion was determined.
The patient underwent excision of the pancreatic lesion approximately 3 months later, which proved to be a benign lymphoepithelial cyst of the pancreas. Histology of the cyst consisted of dense fibrous tissue with a squamous epithelial lining focally infiltrated by lymphocytes (Figure 3A). Immunoperoxidase staining of the cyst revealed focal linear areas of C3d staining along the basement membrane of the stratified squamous epithelium (Figure 3B).
The patient stated that his skin started to improve virtually immediately following the excision without systemic treatment for BP. On follow-up examination 3 weeks postoperatively, no bullae were observed and there was a notable decrease in erythematous crusted plaques (Figure 4).
Comment
Paraneoplastic BP has been documented; however, lymphoepithelial cysts of the pancreas in association with BP are rare. We propose that the lymphoepithelial cyst of the pancreas provided the immunologic stimulus for the development of cutaneous BP based on the observation that our patient’s condition remarkably improved with resection of the tumor.
There are fewer than 100 cases of lymphoepithelial cysts of the pancreas reported in the literature.8 The histologic appearance is consistent with a true cyst exhibiting a well-differentiated stratified squamous epithelium, often with keratinization, surrounded by lymphoid tissue. These tumors are most commonly seen in middle-aged men and are frequently found incidentally,8-10 as was the case with our patient. Although histologically similar, lymphoepithelial cysts of the pancreas are considered distinct from lymphoepithelial cysts of the parotid gland or head and neck region.10 Lymphoepithelial cysts of the pancreas are unrelated to elevated glucagon levels; it is likely that our patient’s glucagon levels were associated with his history of diabetes.11
The diagnosis of BP is characteristically confirmed by direct immunofluorescence. Although it was performed for our patient’s cutaneous lesions, it was not obtained for the lymphoepithelial cyst of the pancreas. Once the diagnosis of the lymphoepithelial cyst of the pancreas was established, as direct immunofluorescence could not be performed in formalin-fixed tissue, immunoperoxidase staining with C3d was obtained. C3 has a well-established role in activation of complement and as a marker in BP. Deposition of C3d is a result of deactivation of C3b, a cleavage product of C3. In a study of 6 autoimmune blistering disorders that included 32 patients with BP, Pfaltz et al12 found positive immunoperoxidase staining for C3d in 31 of 32 patients, which translated to a sensitivity of 97%, a positive predictive value of 100%, and a negative predictive value of 98% among the blistering diseases being studied. Similarly, Magro and Dyrsen13 had positive staining of C3d in 17 of 17 (100%) patients with BP.
In theory, any process that involves deposition of C3 should be positive for C3d on immunoperoxidase staining. Other dermatologic inflammatory conditions stain positively with C3d, such as systemic lupus erythematosus, discoid lupus erythematosus, subacute cutaneous lupus erythematosus, and dermatomysositis.13 The staining for these diseases correlates with the site of the associated inflammatory component seen on hematoxylin and eosin staining. The staining of C3d along the basement membrane of stratified squamous epithelium in the lymphoepithelial cyst of the pancreas seen in our patient closely resembles the staining seen in cutaneous BP.
A proposed mechanism for BP in our patient would be exposure of BP-1 antigen in the pancreatic cyst leading to antibody recognition and C3 deposition along the basement membrane in the cyst, as evidenced by C3d immunoperoxidase staining. The IgG and C3 deposition along the cutaneous basement membrane would then represent a systemic response to the antigen exposure in the cyst. Thus, the lymphoepithelial cyst provided the immunologic stimulus for the development of the cutaneous BP. This theory is based on the observation of our patient’s rapid improvement without a change in his treatment regimen immediately after surgical excision of the cyst.
Despite the plausibility of our hypothesis, several questions remain regarding the validity of our assumptions. Although sensitive for C3 deposition, C3d immunoperoxidase staining is not specific for BP. If the proposed mechanism for causation is true, one might have expected that a subepithelial cleft along the basement membrane of the pancreatic cyst would be observed, which was not seen. A repeat BP antigen antibody was not obtained, which would have been helpful in determining if there was clearance of the antibody that would have correlated with the clinical resolution of the BP lesions.
Conclusion
Our case suggests that paraneoplastic BP is a genuine entity. Indeed, the primary tumor itself may be the immunologic stimulus in the development of BP. Recalcitrant BP should raise the question of a neoplastic process that is exposing the BP antigen. If a thorough review of systems accompanied by corroborating laboratory studies suggests a neoplastic process, the suspect lesion should be further evaluated and surgically excised if clinically indicated. Further evaluation of neoplasms with advanced staining methods may aid in establishing the causative nature of tumors in the development of BP.
Acknowledgments
We are grateful to John Stanley, MD, and Aimee Payne, MD (both from Philadelphia, Pennsylvania), for theirinsights into this case.
- Charneux J, Lorin J, Vitry F, et al. Usefulness of BP230 and BP180-NC16a enzyme-linked immunosorbent assays in the initial diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:286-291.
- Patel M, Sniha AA, Gilbert E. Bullous pemphigoid associated with renal cell carcinoma and invasive squamous cell carcinoma. J Drugs Dermatol. 2012;11:234-238.
- Song HJ, Han SH, Hong WK, et al. Paraneoplastic bullous pemphigoid: clinical disease activity correlated with enzyme-linked immunosorbent assay index for NC16A domain of BP180. J Dermatol. 2009;36:66-68.
- Muramatsu T, Iida T, Tada H, et al. Bullous pemphigoid associated with internal malignancies: identification of 180-kDa antigen by Western immunoblotting. Br J Dermatol. 1996;135:782-784.
- Ogawa H, Sakuma M, Morioka S, et al. The incidence of internal malignancies in pemphigus and bullous pemphigoid in Japan. J Dermatol Sci. 1995;9:136-141.
- Boyd RV. Pemphigoid and carcinoma of the pancreas. Br Med J. 1964;1:1092.
- Eustace S, Morrow G, O’Loughlin S, et al. The role of computed tomography and sonography in acute bullous pemphigoid. Ir J Med Sci. 1993;162:401-404.
- Clemente G, Sarno G, De Rose AM, et al. Lymphoepithelial cyst of the pancreas: case report and review of the literature. Acta Gastroenterol Belg. 2011;74:343-346.
- Frezza E, Wachtel MS. Lymphoepithelial cyst of the pancreas tail. case report and review of the literature. JOP. 2008;9:46-49.
- Basturk O, Coban I, Adsay NV. Pancreatic cysts: pathologic classification, differential diagnosis and clinical implications. Arch Pathol Lab Med. 2009;133:423-438.
- Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest. 2012;122:4-12.
- Pfaltz K, Mertz K, Rose C, et al. C3d immunohistochemistry on formalin-fixed tissue is a valuable tool in the diagnosis of bullous pemphigoid of the skin. J Cutan Pathol. 2010;37:654-658.
- Magro CM, Dyrsen ME. The use of C3d and C4d immunohistochemistry on formalin-fixed tissue as a diagnostic adjunct in the assessment of inflammatory skin disease. J Am Acad Dermatol. 2008;59:822-833.
- Charneux J, Lorin J, Vitry F, et al. Usefulness of BP230 and BP180-NC16a enzyme-linked immunosorbent assays in the initial diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:286-291.
- Patel M, Sniha AA, Gilbert E. Bullous pemphigoid associated with renal cell carcinoma and invasive squamous cell carcinoma. J Drugs Dermatol. 2012;11:234-238.
- Song HJ, Han SH, Hong WK, et al. Paraneoplastic bullous pemphigoid: clinical disease activity correlated with enzyme-linked immunosorbent assay index for NC16A domain of BP180. J Dermatol. 2009;36:66-68.
- Muramatsu T, Iida T, Tada H, et al. Bullous pemphigoid associated with internal malignancies: identification of 180-kDa antigen by Western immunoblotting. Br J Dermatol. 1996;135:782-784.
- Ogawa H, Sakuma M, Morioka S, et al. The incidence of internal malignancies in pemphigus and bullous pemphigoid in Japan. J Dermatol Sci. 1995;9:136-141.
- Boyd RV. Pemphigoid and carcinoma of the pancreas. Br Med J. 1964;1:1092.
- Eustace S, Morrow G, O’Loughlin S, et al. The role of computed tomography and sonography in acute bullous pemphigoid. Ir J Med Sci. 1993;162:401-404.
- Clemente G, Sarno G, De Rose AM, et al. Lymphoepithelial cyst of the pancreas: case report and review of the literature. Acta Gastroenterol Belg. 2011;74:343-346.
- Frezza E, Wachtel MS. Lymphoepithelial cyst of the pancreas tail. case report and review of the literature. JOP. 2008;9:46-49.
- Basturk O, Coban I, Adsay NV. Pancreatic cysts: pathologic classification, differential diagnosis and clinical implications. Arch Pathol Lab Med. 2009;133:423-438.
- Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest. 2012;122:4-12.
- Pfaltz K, Mertz K, Rose C, et al. C3d immunohistochemistry on formalin-fixed tissue is a valuable tool in the diagnosis of bullous pemphigoid of the skin. J Cutan Pathol. 2010;37:654-658.
- Magro CM, Dyrsen ME. The use of C3d and C4d immunohistochemistry on formalin-fixed tissue as a diagnostic adjunct in the assessment of inflammatory skin disease. J Am Acad Dermatol. 2008;59:822-833.
Pruritic Papules on the Scalp and Arms
Folliculotropic Mycosis Fungoides
Folliculotropic mycosis fungoides (FMF) is a variant of mycosis fungoides (MF) that occurs mostly in adults with a male predilection. The disease clinically favors the head and neck. Patients commonly present with pruritic papules that often are grouped, alopecia, and frequent secondary bacterial infections. Less commonly patients present with acneiform lesions and mucinorrhea. Patients often experience more pruritus in FMF than in classic MF, which can provide a good means of assessing disease activity. Disease-specific 5-year survival is approximately 70% to 80%, which is worse than classic plaque-stage MF and similar to tumor-stage MF.1
Treatment of FMF differs from classic MF in that the lesions are less responsive to skin-targeted therapies due to the perifollicular nature of dermal infiltrates. Superficial skin lesions can be treated with psoralen plus UVA (PUVA) therapy. Other options include PUVA in combination with interferon alfa or retinoids and local radiotherapy for solitary thick tumors; however, in patients who have more infiltrative skin lesions or had PUVA therapy that failed, total skin electron beam therapy may be required.2
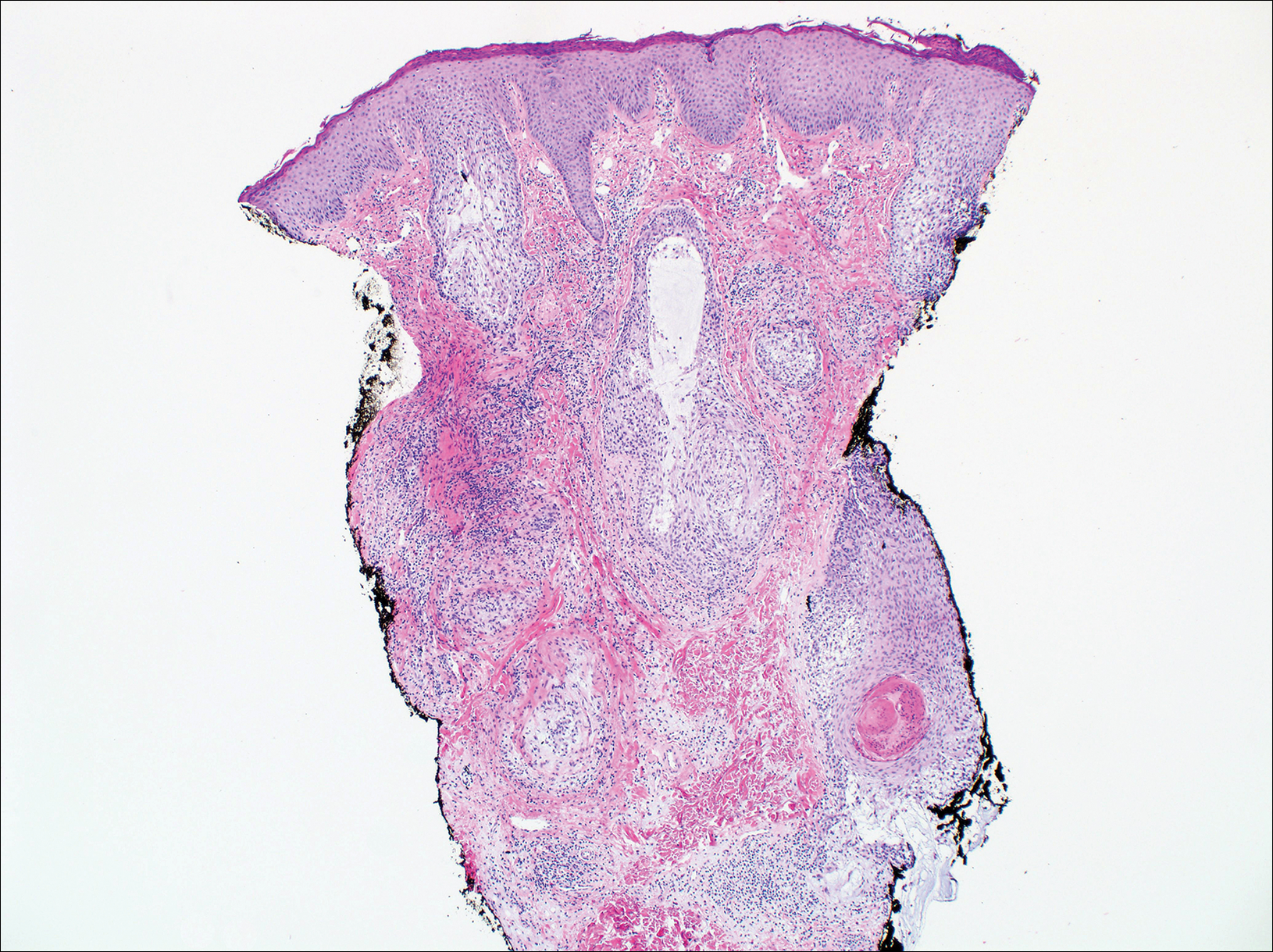
On histologic examination, there typically is perivascular and periadnexal localization of dermal infiltrates with varied involvement of the follicular epithelium and damage to hair follicles by atypical small, medium, and large hyperchromatic lymphocytes with cerebriform nuclei. Mucinous degeneration of the follicular epithelium can be seen, as highlighted on Alcian blue staining, and a mixed infiltrate of eosinophils and plasma cells often is present (quiz image and Figure 1). Frequent sparing of the epidermis is noteworthy.2-4 In most cases, the neoplastic T lymphocytes are characterized by a CD3+CD4+CD8-immunophenotype as is seen in classic MF. Sometimes an admixture of CD30+ blast cells is seen.1
Histologic differential considerations for FMF include eosinophilic pustular folliculitis (EPF), primary follicular mucinosis, lupus erythematosus, and pityrosporum folliculitis.
Eosinophilic pustular folliculitis has several clinical subtypes, such as classic Ofuji disease and immunosuppression-associated EPF secondary to human immunodeficiency virus. Histologically, EPF is characterized by spongiosis of the hair follicle epithelium with exocytosis of a mixed infiltrate of lymphocytes and eosinophils extending from the sebaceous gland and its duct to the infundibulum with formation of hallmark eosinophilic pustules (Figure 2). Infiltration of neutrophils in inflamed lesions generally is seen. Eosinophilic pustular folliculitis is an important differential for FMF, as follicular mucinosis has been observed in lesions of EPF.5 Both EPF and FMF can exhibit eosinophils and lymphocytes in the upper dermis, spongiosis of the hair follicle epithelium, and mucinous degeneration of follicles,6 though lymphocytic atypia and relatively fewer eosinophils are suggestive of the latter.
Primary follicular mucinosis (PFM) tends to occur as a solitary lesion in younger female patients in contrast to the multiple lesions that typically appear in older male patients with FMF. Histologically, PFM usually manifests as large, cystic, mucin-filled spaces and polyclonal perivascular and periadnexal lymphocytic infiltrate without notable cellular atypia or epidermotropism (Figure 3). Because follicular mucinosis is a common feature of FMF, its distinction from PFM can be challenging and often is aided by the absence of cellular atypia and relatively mild lymphocytic infiltrate in the latter.7
Cutaneous lupus erythematosus with its characteristic folliculocentric lymphocytic infiltration and associated dermal mucin also qualifies as a potential differential possibility for FMF; however, the perivascular and periadnexal pattern of lymphocytic infiltration as well as the localization of mucin to the reticular dermal interstitium8,9 are key histopathologic distinctions (Figure 4). Furthermore, although the histologic presentation of lupus erythematosus can be variable, it also classically shows interface dermatitis, basement membrane thickening, and follicular plugging.
Pityrosporum folliculitis is the most common cause of fungal folliculitis and is caused by the Malassezia species. On histology, there typically is an unremarkable epithelium with plugged follicles and suppurative folliculitis. Serial sections of the biopsy specimen often are required to identify dilated, follicle-containing, budding yeast cells (Figure 5). Organisms are located predominantly within the infundibulum and orifice of follicular lumen, are positive for periodic acid-Schiff, and are diastase resistant.10
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- van Doorn R, Scheffer E, Willemze R. Follicular mycosis fungoides, a distinct disease entity with or without associated follicular mucinosis. a clinicopathologic and follow-up study of 51 patients. Arch Dermatol. 2002;138:191-198.
- DeBloom J 2nd, Severson J, Gaspari A, et al. Follicular mycosis fungoides: a case report and review of the literature. J Cutan Pathol. 2001;28:318-324.
- Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides: a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525-530.
- Fujiyama T, Tokura Y. Clinical and histopathological differential diagnosis of eosinophilic pustular folliculitis. J Dermatol. 2013;40:419-423.
- Lee JY, Tsai YM, Sheu HM. Ofuji's disease with follicular mucinosis and its differential diagnosis from alopecia mucinosa. J Cutan Pathol. 2003;30:307-313.
- Rongioletti F, De Lucchi S, Meyes D, et al. Follicular mucinosis: a clinicopathologic, histochemical, immunohistochemical and molecular study comparing the primary benign form and the mycosis fungoides-associated follicular mucinosis. J Cutan Pathol. 2010;37:15-19.
- Vincent JG, Chan MP. Specificity of dermal mucin in the diagnosis of lupus erythematosus: comparison with other dermatitides and normal skin. J Cutan Pathol. 2015;42:722-729.
- Yell JA, Mbuagbaw J, Burge SM. Cutaneous manifestations of systemic lupus erythematosus. Br J Dermatol. 1996;135:355-362.
- Durdu M, Ilkit M. First step in the differential diagnosis of folliculitis: cytology. Crit Rev Microbiol. 2013;39:9-25.
Folliculotropic Mycosis Fungoides
Folliculotropic mycosis fungoides (FMF) is a variant of mycosis fungoides (MF) that occurs mostly in adults with a male predilection. The disease clinically favors the head and neck. Patients commonly present with pruritic papules that often are grouped, alopecia, and frequent secondary bacterial infections. Less commonly patients present with acneiform lesions and mucinorrhea. Patients often experience more pruritus in FMF than in classic MF, which can provide a good means of assessing disease activity. Disease-specific 5-year survival is approximately 70% to 80%, which is worse than classic plaque-stage MF and similar to tumor-stage MF.1
Treatment of FMF differs from classic MF in that the lesions are less responsive to skin-targeted therapies due to the perifollicular nature of dermal infiltrates. Superficial skin lesions can be treated with psoralen plus UVA (PUVA) therapy. Other options include PUVA in combination with interferon alfa or retinoids and local radiotherapy for solitary thick tumors; however, in patients who have more infiltrative skin lesions or had PUVA therapy that failed, total skin electron beam therapy may be required.2
On histologic examination, there typically is perivascular and periadnexal localization of dermal infiltrates with varied involvement of the follicular epithelium and damage to hair follicles by atypical small, medium, and large hyperchromatic lymphocytes with cerebriform nuclei. Mucinous degeneration of the follicular epithelium can be seen, as highlighted on Alcian blue staining, and a mixed infiltrate of eosinophils and plasma cells often is present (quiz image and Figure 1). Frequent sparing of the epidermis is noteworthy.2-4 In most cases, the neoplastic T lymphocytes are characterized by a CD3+CD4+CD8-immunophenotype as is seen in classic MF. Sometimes an admixture of CD30+ blast cells is seen.1
Histologic differential considerations for FMF include eosinophilic pustular folliculitis (EPF), primary follicular mucinosis, lupus erythematosus, and pityrosporum folliculitis.
Eosinophilic pustular folliculitis has several clinical subtypes, such as classic Ofuji disease and immunosuppression-associated EPF secondary to human immunodeficiency virus. Histologically, EPF is characterized by spongiosis of the hair follicle epithelium with exocytosis of a mixed infiltrate of lymphocytes and eosinophils extending from the sebaceous gland and its duct to the infundibulum with formation of hallmark eosinophilic pustules (Figure 2). Infiltration of neutrophils in inflamed lesions generally is seen. Eosinophilic pustular folliculitis is an important differential for FMF, as follicular mucinosis has been observed in lesions of EPF.5 Both EPF and FMF can exhibit eosinophils and lymphocytes in the upper dermis, spongiosis of the hair follicle epithelium, and mucinous degeneration of follicles,6 though lymphocytic atypia and relatively fewer eosinophils are suggestive of the latter.
Primary follicular mucinosis (PFM) tends to occur as a solitary lesion in younger female patients in contrast to the multiple lesions that typically appear in older male patients with FMF. Histologically, PFM usually manifests as large, cystic, mucin-filled spaces and polyclonal perivascular and periadnexal lymphocytic infiltrate without notable cellular atypia or epidermotropism (Figure 3). Because follicular mucinosis is a common feature of FMF, its distinction from PFM can be challenging and often is aided by the absence of cellular atypia and relatively mild lymphocytic infiltrate in the latter.7
Cutaneous lupus erythematosus with its characteristic folliculocentric lymphocytic infiltration and associated dermal mucin also qualifies as a potential differential possibility for FMF; however, the perivascular and periadnexal pattern of lymphocytic infiltration as well as the localization of mucin to the reticular dermal interstitium8,9 are key histopathologic distinctions (Figure 4). Furthermore, although the histologic presentation of lupus erythematosus can be variable, it also classically shows interface dermatitis, basement membrane thickening, and follicular plugging.
Pityrosporum folliculitis is the most common cause of fungal folliculitis and is caused by the Malassezia species. On histology, there typically is an unremarkable epithelium with plugged follicles and suppurative folliculitis. Serial sections of the biopsy specimen often are required to identify dilated, follicle-containing, budding yeast cells (Figure 5). Organisms are located predominantly within the infundibulum and orifice of follicular lumen, are positive for periodic acid-Schiff, and are diastase resistant.10
Folliculotropic Mycosis Fungoides
Folliculotropic mycosis fungoides (FMF) is a variant of mycosis fungoides (MF) that occurs mostly in adults with a male predilection. The disease clinically favors the head and neck. Patients commonly present with pruritic papules that often are grouped, alopecia, and frequent secondary bacterial infections. Less commonly patients present with acneiform lesions and mucinorrhea. Patients often experience more pruritus in FMF than in classic MF, which can provide a good means of assessing disease activity. Disease-specific 5-year survival is approximately 70% to 80%, which is worse than classic plaque-stage MF and similar to tumor-stage MF.1
Treatment of FMF differs from classic MF in that the lesions are less responsive to skin-targeted therapies due to the perifollicular nature of dermal infiltrates. Superficial skin lesions can be treated with psoralen plus UVA (PUVA) therapy. Other options include PUVA in combination with interferon alfa or retinoids and local radiotherapy for solitary thick tumors; however, in patients who have more infiltrative skin lesions or had PUVA therapy that failed, total skin electron beam therapy may be required.2
On histologic examination, there typically is perivascular and periadnexal localization of dermal infiltrates with varied involvement of the follicular epithelium and damage to hair follicles by atypical small, medium, and large hyperchromatic lymphocytes with cerebriform nuclei. Mucinous degeneration of the follicular epithelium can be seen, as highlighted on Alcian blue staining, and a mixed infiltrate of eosinophils and plasma cells often is present (quiz image and Figure 1). Frequent sparing of the epidermis is noteworthy.2-4 In most cases, the neoplastic T lymphocytes are characterized by a CD3+CD4+CD8-immunophenotype as is seen in classic MF. Sometimes an admixture of CD30+ blast cells is seen.1
Histologic differential considerations for FMF include eosinophilic pustular folliculitis (EPF), primary follicular mucinosis, lupus erythematosus, and pityrosporum folliculitis.
Eosinophilic pustular folliculitis has several clinical subtypes, such as classic Ofuji disease and immunosuppression-associated EPF secondary to human immunodeficiency virus. Histologically, EPF is characterized by spongiosis of the hair follicle epithelium with exocytosis of a mixed infiltrate of lymphocytes and eosinophils extending from the sebaceous gland and its duct to the infundibulum with formation of hallmark eosinophilic pustules (Figure 2). Infiltration of neutrophils in inflamed lesions generally is seen. Eosinophilic pustular folliculitis is an important differential for FMF, as follicular mucinosis has been observed in lesions of EPF.5 Both EPF and FMF can exhibit eosinophils and lymphocytes in the upper dermis, spongiosis of the hair follicle epithelium, and mucinous degeneration of follicles,6 though lymphocytic atypia and relatively fewer eosinophils are suggestive of the latter.
Primary follicular mucinosis (PFM) tends to occur as a solitary lesion in younger female patients in contrast to the multiple lesions that typically appear in older male patients with FMF. Histologically, PFM usually manifests as large, cystic, mucin-filled spaces and polyclonal perivascular and periadnexal lymphocytic infiltrate without notable cellular atypia or epidermotropism (Figure 3). Because follicular mucinosis is a common feature of FMF, its distinction from PFM can be challenging and often is aided by the absence of cellular atypia and relatively mild lymphocytic infiltrate in the latter.7
Cutaneous lupus erythematosus with its characteristic folliculocentric lymphocytic infiltration and associated dermal mucin also qualifies as a potential differential possibility for FMF; however, the perivascular and periadnexal pattern of lymphocytic infiltration as well as the localization of mucin to the reticular dermal interstitium8,9 are key histopathologic distinctions (Figure 4). Furthermore, although the histologic presentation of lupus erythematosus can be variable, it also classically shows interface dermatitis, basement membrane thickening, and follicular plugging.
Pityrosporum folliculitis is the most common cause of fungal folliculitis and is caused by the Malassezia species. On histology, there typically is an unremarkable epithelium with plugged follicles and suppurative folliculitis. Serial sections of the biopsy specimen often are required to identify dilated, follicle-containing, budding yeast cells (Figure 5). Organisms are located predominantly within the infundibulum and orifice of follicular lumen, are positive for periodic acid-Schiff, and are diastase resistant.10
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- van Doorn R, Scheffer E, Willemze R. Follicular mycosis fungoides, a distinct disease entity with or without associated follicular mucinosis. a clinicopathologic and follow-up study of 51 patients. Arch Dermatol. 2002;138:191-198.
- DeBloom J 2nd, Severson J, Gaspari A, et al. Follicular mycosis fungoides: a case report and review of the literature. J Cutan Pathol. 2001;28:318-324.
- Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides: a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525-530.
- Fujiyama T, Tokura Y. Clinical and histopathological differential diagnosis of eosinophilic pustular folliculitis. J Dermatol. 2013;40:419-423.
- Lee JY, Tsai YM, Sheu HM. Ofuji's disease with follicular mucinosis and its differential diagnosis from alopecia mucinosa. J Cutan Pathol. 2003;30:307-313.
- Rongioletti F, De Lucchi S, Meyes D, et al. Follicular mucinosis: a clinicopathologic, histochemical, immunohistochemical and molecular study comparing the primary benign form and the mycosis fungoides-associated follicular mucinosis. J Cutan Pathol. 2010;37:15-19.
- Vincent JG, Chan MP. Specificity of dermal mucin in the diagnosis of lupus erythematosus: comparison with other dermatitides and normal skin. J Cutan Pathol. 2015;42:722-729.
- Yell JA, Mbuagbaw J, Burge SM. Cutaneous manifestations of systemic lupus erythematosus. Br J Dermatol. 1996;135:355-362.
- Durdu M, Ilkit M. First step in the differential diagnosis of folliculitis: cytology. Crit Rev Microbiol. 2013;39:9-25.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- van Doorn R, Scheffer E, Willemze R. Follicular mycosis fungoides, a distinct disease entity with or without associated follicular mucinosis. a clinicopathologic and follow-up study of 51 patients. Arch Dermatol. 2002;138:191-198.
- DeBloom J 2nd, Severson J, Gaspari A, et al. Follicular mycosis fungoides: a case report and review of the literature. J Cutan Pathol. 2001;28:318-324.
- Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides: a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525-530.
- Fujiyama T, Tokura Y. Clinical and histopathological differential diagnosis of eosinophilic pustular folliculitis. J Dermatol. 2013;40:419-423.
- Lee JY, Tsai YM, Sheu HM. Ofuji's disease with follicular mucinosis and its differential diagnosis from alopecia mucinosa. J Cutan Pathol. 2003;30:307-313.
- Rongioletti F, De Lucchi S, Meyes D, et al. Follicular mucinosis: a clinicopathologic, histochemical, immunohistochemical and molecular study comparing the primary benign form and the mycosis fungoides-associated follicular mucinosis. J Cutan Pathol. 2010;37:15-19.
- Vincent JG, Chan MP. Specificity of dermal mucin in the diagnosis of lupus erythematosus: comparison with other dermatitides and normal skin. J Cutan Pathol. 2015;42:722-729.
- Yell JA, Mbuagbaw J, Burge SM. Cutaneous manifestations of systemic lupus erythematosus. Br J Dermatol. 1996;135:355-362.
- Durdu M, Ilkit M. First step in the differential diagnosis of folliculitis: cytology. Crit Rev Microbiol. 2013;39:9-25.
A 60-year-old man presented with a 3-month history of itchy bumps on the scalp and arms. He also noticed some patches of hair loss in these areas. He had no history of other skin conditions and was otherwise healthy with no other medical comorbidities.
The Proposed Rule and Payments for 2017: The Good, the Bad, and the Ugly
Just as Charlie Brown looks forward to the coming of the Great Pumpkin each Halloween, those of us who dance in the minefields of payment policy await the publication of the Proposed Rule, more formally known as the “Revisions to Payment Policies under the Physician Fee Schedule and Other Revisions to Part B for CY 2017.”1,2 You could read the entire tome—a mere 316 pages (excluding the hundreds of pages of granular supplement data discussed in the last few columns)—or simply read what I have outlined as the good, the bad, and the ugly for the Proposed Rule for 2017.
The Good
In 2017, dermatology will increase its share of the pie by 1% to $3.505 billion of a total $89.467 billion expected to be expended for physician services.1 The effect on individual providers will vary by geographic location and practice mix. Half is from the 0.5% increase that has come to all physicians across the board as mandated by the Medicare Access and CHIP Reauthorization Act (MACRA).3
Current Procedural Terminology (CPT) codes for reflectance confocal microscopy (96931–96936) will have Centers for Medicare & Medicaid Services valuations beginning in 2017, and individuals performing this service should be able to report it and be paid for their efforts.1 The values are below the American Medical Association/Specialty Society Relative Value Scale Update Committee (RUC) recommendations.
The Bad
Payment rates for 2017 will be based on a conversion factor of 35.7751,1 a drop from the 2016 conversion factor of 35.8043. Cuts will be made for some specialties. Gastroenterology, nephrology, neurosurgery, radiology, urology, and radiation therapy centers will take a 1% hit; ophthalmology, pathology, and vascular surgery will take 2% cuts; and interventional radiology will lose 7%.1 A special case within dermatology and pathology is a 15% cut to the technical component of slide preparation for CPT code 883054 due to a redefinition of the valuation of eosin stains.2 While the accuracy and precision of the value of these practice expense inputs can be debated, the government by definition makes the rules and involved specialties had an opportunity to appeal this change through the comment process that ended on September 6, 2016. The government can take comments into account, but substantial changes usually are not made from the Proposed Rule to the Final Rule, which usually arrives around the beginning of November; however, in an election year, the Final Rule can be a few weeks late.
The Ugly
The government will increase its unfunded mandates with the creation of new Medicare G codes (global services codes) that will allow the government to track the provision of postoperative care for all 010 and 090 global service periods (Table 1). The codes look mostly at time and do not clearly take into account the severity or complexity of the conditions being cared for and will be reported on claim forms as an unfunded mandate with more confusion and cost.1 Because not all claim-paying intermediaries are likely to have these G codes smoothly set up in their systems, there will still be a cost to filing the claim. Unless changes occur in the Final Rule, which is unlikely, there will be no payment for the time and effort of submitting these claims. The goal of the US Government is to hone in on postoperative services and parse them down so they can cut payments wherever possible beginning in 2019.1 Everyone wants to save money, from the consumer5 to the payer, and the ultimate payer is playing hardball. Additional validation efforts likely will lower physician fee-for-service payments further.

The US Government also is taking a shot at what they call “misvalued services” that have not had recent refinement within the RUC process.1 The work list for 2017 includes a number of 000 global period codes where additional evaluation and management services are reported using modifier -25, which implies a substantial, separately identifiable cognitive service performed by the same physician on the day of a procedure above and beyond other services provided or beyond the usual preservice and postservice care associated with the procedure that was performed. Although codes such as biopsies (11100 and 11101) and premalignant destructions (17000–17004) have an adjustment built in and dermatologists who provide services on the same day are actually penalized for the multiple built-in reductions that are already additive, the government is concerned that 19% of the 000 global services were billed more than 50% of the time with an evaluation and management code with modifier -25. Eighty-three codes met the criteria for which the government believes it may be overpaying1; the codes of interest to dermatology are shown in Table 2.1

The refinement of global periods will be an ongoing exercise through 2017, and beyond, with results likely to play an important role in the 2019 fee schedule. These global period reviews combined with some Stark law refinement relating the leasing of space at market rates while disallowing the landlord physician from receiving patient referrals from the tenant may also affect practitioner income.1,6 I never cease to be amazed that former Congressman Fortney Hillman “Pete” Stark (D), who has an antikickback scheme that keeps expanding, never went after the banking and brokerage industries. The founder of the $1.1 billion Security National Bank, a small bank in Walnut Creek, California,7 never focused on regulating banks. In his 40-year congressional career, he decided physicians make better targets. His efforts have not helped physicians but have helped lawyers, as he is quick to acknowledge.8
Final Thoughts
I end this column with an appeal to the dermatologists of America. Go to the American Academy of Dermatology Association Political Action Committee website (https://skinpac.org/), the home page for the only political action committee that represents the dermatology specialty, and consider making a donation. Emergency medicine physicians created the “Giving a Shift” campaign, which is a donation to their national political action committee of one shift’s earnings, and most of us could easily donate a half day’s income, as the only way to potentially change the increasingly onerous burdens on practitioners is through political action. As we say at RUC meetings, you can eat lunch or be lunch. The choice is yours.
- Medicare Program; Revisions to Payment Policies Under the Physician Fee Schedule and Other Revisions to Part B for CY 2017; Medicare Advantage Pricing Data Release; Medicare Advantage and Part D Medical Low Ratio Data Release; Medicare Advantage Provider Network Requirements; Expansion of Medicare Diabetes Prevention Program Model. Fed Regist. 2016;81(136):46162-46476. To be codified at 42 CFR §405, 410, 411, et al. https://www.gpo.gov/fdsys/pkg/FR-2016-07-15/pdf/2016-16097.pdf. Accessed September 7, 2016.
- Revisions to Payment Policies under the Physician Fee Schedule and Other Revisions to Part B for CY 2017. Centers for Medicare & Medicaid Services website. https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/PhysicianFeeSched/PFS-Federal-Regulation-Notices-Items/CMS-1654-P.html. Accessed September 7, 2016.
- Text of the Medicare Access and CHIP Reauthorization Act of 2015. GovTrack website. https://www.govtrack.us/congress/bills/114/hr2/text. Accessed September 9, 2016.
- Kaplan KJ. Proposed Medicare 2017 reimbursement schedule whacks biopsy payments; digital pathology payments up. Digital Pathology Blog website. http://tissuepathology.com/2016/07/20/proposed-medicare-2017-reimbursement-schedule-whacks-biopsy-payments-digital-pathology-payments-up/#ixzz4HEqBLgzu. Published July 20, 2016. Accessed September 7, 2016.
- Abelson R. Cost, not choice, is top concern of health insurance customers. The New York Times. http://www.nytimes.com/2016/08/13/business/cost-not-choice-is-top-concern-of-health-insurance-customers.html?_r=0. Published August 12, 2016. Accessed September 7, 2016.
- Stark Law website. http://starklaw.org/. Accessed September 7, 2016.
- Pete Stark. Freedom From Religion website. https://ffrf.org/news/day/dayitems/item/14800-pete-stark. Accessed September 19, 2016.
- Adamy J. Pete Stark: Law regulating doctors mostly helped lawyers. The Wall Street Journal. October 22, 2014. http://blogs.wsj.com/washwire/2014/10/22/pete-stark-law-regulating-doctors-mostly-helped-lawyers/. Accessed September 19, 2016.
Just as Charlie Brown looks forward to the coming of the Great Pumpkin each Halloween, those of us who dance in the minefields of payment policy await the publication of the Proposed Rule, more formally known as the “Revisions to Payment Policies under the Physician Fee Schedule and Other Revisions to Part B for CY 2017.”1,2 You could read the entire tome—a mere 316 pages (excluding the hundreds of pages of granular supplement data discussed in the last few columns)—or simply read what I have outlined as the good, the bad, and the ugly for the Proposed Rule for 2017.
The Good
In 2017, dermatology will increase its share of the pie by 1% to $3.505 billion of a total $89.467 billion expected to be expended for physician services.1 The effect on individual providers will vary by geographic location and practice mix. Half is from the 0.5% increase that has come to all physicians across the board as mandated by the Medicare Access and CHIP Reauthorization Act (MACRA).3
Current Procedural Terminology (CPT) codes for reflectance confocal microscopy (96931–96936) will have Centers for Medicare & Medicaid Services valuations beginning in 2017, and individuals performing this service should be able to report it and be paid for their efforts.1 The values are below the American Medical Association/Specialty Society Relative Value Scale Update Committee (RUC) recommendations.
The Bad
Payment rates for 2017 will be based on a conversion factor of 35.7751,1 a drop from the 2016 conversion factor of 35.8043. Cuts will be made for some specialties. Gastroenterology, nephrology, neurosurgery, radiology, urology, and radiation therapy centers will take a 1% hit; ophthalmology, pathology, and vascular surgery will take 2% cuts; and interventional radiology will lose 7%.1 A special case within dermatology and pathology is a 15% cut to the technical component of slide preparation for CPT code 883054 due to a redefinition of the valuation of eosin stains.2 While the accuracy and precision of the value of these practice expense inputs can be debated, the government by definition makes the rules and involved specialties had an opportunity to appeal this change through the comment process that ended on September 6, 2016. The government can take comments into account, but substantial changes usually are not made from the Proposed Rule to the Final Rule, which usually arrives around the beginning of November; however, in an election year, the Final Rule can be a few weeks late.
The Ugly
The government will increase its unfunded mandates with the creation of new Medicare G codes (global services codes) that will allow the government to track the provision of postoperative care for all 010 and 090 global service periods (Table 1). The codes look mostly at time and do not clearly take into account the severity or complexity of the conditions being cared for and will be reported on claim forms as an unfunded mandate with more confusion and cost.1 Because not all claim-paying intermediaries are likely to have these G codes smoothly set up in their systems, there will still be a cost to filing the claim. Unless changes occur in the Final Rule, which is unlikely, there will be no payment for the time and effort of submitting these claims. The goal of the US Government is to hone in on postoperative services and parse them down so they can cut payments wherever possible beginning in 2019.1 Everyone wants to save money, from the consumer5 to the payer, and the ultimate payer is playing hardball. Additional validation efforts likely will lower physician fee-for-service payments further.
The US Government also is taking a shot at what they call “misvalued services” that have not had recent refinement within the RUC process.1 The work list for 2017 includes a number of 000 global period codes where additional evaluation and management services are reported using modifier -25, which implies a substantial, separately identifiable cognitive service performed by the same physician on the day of a procedure above and beyond other services provided or beyond the usual preservice and postservice care associated with the procedure that was performed. Although codes such as biopsies (11100 and 11101) and premalignant destructions (17000–17004) have an adjustment built in and dermatologists who provide services on the same day are actually penalized for the multiple built-in reductions that are already additive, the government is concerned that 19% of the 000 global services were billed more than 50% of the time with an evaluation and management code with modifier -25. Eighty-three codes met the criteria for which the government believes it may be overpaying1; the codes of interest to dermatology are shown in Table 2.1
The refinement of global periods will be an ongoing exercise through 2017, and beyond, with results likely to play an important role in the 2019 fee schedule. These global period reviews combined with some Stark law refinement relating the leasing of space at market rates while disallowing the landlord physician from receiving patient referrals from the tenant may also affect practitioner income.1,6 I never cease to be amazed that former Congressman Fortney Hillman “Pete” Stark (D), who has an antikickback scheme that keeps expanding, never went after the banking and brokerage industries. The founder of the $1.1 billion Security National Bank, a small bank in Walnut Creek, California,7 never focused on regulating banks. In his 40-year congressional career, he decided physicians make better targets. His efforts have not helped physicians but have helped lawyers, as he is quick to acknowledge.8
Final Thoughts
I end this column with an appeal to the dermatologists of America. Go to the American Academy of Dermatology Association Political Action Committee website (https://skinpac.org/), the home page for the only political action committee that represents the dermatology specialty, and consider making a donation. Emergency medicine physicians created the “Giving a Shift” campaign, which is a donation to their national political action committee of one shift’s earnings, and most of us could easily donate a half day’s income, as the only way to potentially change the increasingly onerous burdens on practitioners is through political action. As we say at RUC meetings, you can eat lunch or be lunch. The choice is yours.
Just as Charlie Brown looks forward to the coming of the Great Pumpkin each Halloween, those of us who dance in the minefields of payment policy await the publication of the Proposed Rule, more formally known as the “Revisions to Payment Policies under the Physician Fee Schedule and Other Revisions to Part B for CY 2017.”1,2 You could read the entire tome—a mere 316 pages (excluding the hundreds of pages of granular supplement data discussed in the last few columns)—or simply read what I have outlined as the good, the bad, and the ugly for the Proposed Rule for 2017.
The Good
In 2017, dermatology will increase its share of the pie by 1% to $3.505 billion of a total $89.467 billion expected to be expended for physician services.1 The effect on individual providers will vary by geographic location and practice mix. Half is from the 0.5% increase that has come to all physicians across the board as mandated by the Medicare Access and CHIP Reauthorization Act (MACRA).3
Current Procedural Terminology (CPT) codes for reflectance confocal microscopy (96931–96936) will have Centers for Medicare & Medicaid Services valuations beginning in 2017, and individuals performing this service should be able to report it and be paid for their efforts.1 The values are below the American Medical Association/Specialty Society Relative Value Scale Update Committee (RUC) recommendations.
The Bad
Payment rates for 2017 will be based on a conversion factor of 35.7751,1 a drop from the 2016 conversion factor of 35.8043. Cuts will be made for some specialties. Gastroenterology, nephrology, neurosurgery, radiology, urology, and radiation therapy centers will take a 1% hit; ophthalmology, pathology, and vascular surgery will take 2% cuts; and interventional radiology will lose 7%.1 A special case within dermatology and pathology is a 15% cut to the technical component of slide preparation for CPT code 883054 due to a redefinition of the valuation of eosin stains.2 While the accuracy and precision of the value of these practice expense inputs can be debated, the government by definition makes the rules and involved specialties had an opportunity to appeal this change through the comment process that ended on September 6, 2016. The government can take comments into account, but substantial changes usually are not made from the Proposed Rule to the Final Rule, which usually arrives around the beginning of November; however, in an election year, the Final Rule can be a few weeks late.
The Ugly
The government will increase its unfunded mandates with the creation of new Medicare G codes (global services codes) that will allow the government to track the provision of postoperative care for all 010 and 090 global service periods (Table 1). The codes look mostly at time and do not clearly take into account the severity or complexity of the conditions being cared for and will be reported on claim forms as an unfunded mandate with more confusion and cost.1 Because not all claim-paying intermediaries are likely to have these G codes smoothly set up in their systems, there will still be a cost to filing the claim. Unless changes occur in the Final Rule, which is unlikely, there will be no payment for the time and effort of submitting these claims. The goal of the US Government is to hone in on postoperative services and parse them down so they can cut payments wherever possible beginning in 2019.1 Everyone wants to save money, from the consumer5 to the payer, and the ultimate payer is playing hardball. Additional validation efforts likely will lower physician fee-for-service payments further.
The US Government also is taking a shot at what they call “misvalued services” that have not had recent refinement within the RUC process.1 The work list for 2017 includes a number of 000 global period codes where additional evaluation and management services are reported using modifier -25, which implies a substantial, separately identifiable cognitive service performed by the same physician on the day of a procedure above and beyond other services provided or beyond the usual preservice and postservice care associated with the procedure that was performed. Although codes such as biopsies (11100 and 11101) and premalignant destructions (17000–17004) have an adjustment built in and dermatologists who provide services on the same day are actually penalized for the multiple built-in reductions that are already additive, the government is concerned that 19% of the 000 global services were billed more than 50% of the time with an evaluation and management code with modifier -25. Eighty-three codes met the criteria for which the government believes it may be overpaying1; the codes of interest to dermatology are shown in Table 2.1
The refinement of global periods will be an ongoing exercise through 2017, and beyond, with results likely to play an important role in the 2019 fee schedule. These global period reviews combined with some Stark law refinement relating the leasing of space at market rates while disallowing the landlord physician from receiving patient referrals from the tenant may also affect practitioner income.1,6 I never cease to be amazed that former Congressman Fortney Hillman “Pete” Stark (D), who has an antikickback scheme that keeps expanding, never went after the banking and brokerage industries. The founder of the $1.1 billion Security National Bank, a small bank in Walnut Creek, California,7 never focused on regulating banks. In his 40-year congressional career, he decided physicians make better targets. His efforts have not helped physicians but have helped lawyers, as he is quick to acknowledge.8
Final Thoughts
I end this column with an appeal to the dermatologists of America. Go to the American Academy of Dermatology Association Political Action Committee website (https://skinpac.org/), the home page for the only political action committee that represents the dermatology specialty, and consider making a donation. Emergency medicine physicians created the “Giving a Shift” campaign, which is a donation to their national political action committee of one shift’s earnings, and most of us could easily donate a half day’s income, as the only way to potentially change the increasingly onerous burdens on practitioners is through political action. As we say at RUC meetings, you can eat lunch or be lunch. The choice is yours.
- Medicare Program; Revisions to Payment Policies Under the Physician Fee Schedule and Other Revisions to Part B for CY 2017; Medicare Advantage Pricing Data Release; Medicare Advantage and Part D Medical Low Ratio Data Release; Medicare Advantage Provider Network Requirements; Expansion of Medicare Diabetes Prevention Program Model. Fed Regist. 2016;81(136):46162-46476. To be codified at 42 CFR §405, 410, 411, et al. https://www.gpo.gov/fdsys/pkg/FR-2016-07-15/pdf/2016-16097.pdf. Accessed September 7, 2016.
- Revisions to Payment Policies under the Physician Fee Schedule and Other Revisions to Part B for CY 2017. Centers for Medicare & Medicaid Services website. https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/PhysicianFeeSched/PFS-Federal-Regulation-Notices-Items/CMS-1654-P.html. Accessed September 7, 2016.
- Text of the Medicare Access and CHIP Reauthorization Act of 2015. GovTrack website. https://www.govtrack.us/congress/bills/114/hr2/text. Accessed September 9, 2016.
- Kaplan KJ. Proposed Medicare 2017 reimbursement schedule whacks biopsy payments; digital pathology payments up. Digital Pathology Blog website. http://tissuepathology.com/2016/07/20/proposed-medicare-2017-reimbursement-schedule-whacks-biopsy-payments-digital-pathology-payments-up/#ixzz4HEqBLgzu. Published July 20, 2016. Accessed September 7, 2016.
- Abelson R. Cost, not choice, is top concern of health insurance customers. The New York Times. http://www.nytimes.com/2016/08/13/business/cost-not-choice-is-top-concern-of-health-insurance-customers.html?_r=0. Published August 12, 2016. Accessed September 7, 2016.
- Stark Law website. http://starklaw.org/. Accessed September 7, 2016.
- Pete Stark. Freedom From Religion website. https://ffrf.org/news/day/dayitems/item/14800-pete-stark. Accessed September 19, 2016.
- Adamy J. Pete Stark: Law regulating doctors mostly helped lawyers. The Wall Street Journal. October 22, 2014. http://blogs.wsj.com/washwire/2014/10/22/pete-stark-law-regulating-doctors-mostly-helped-lawyers/. Accessed September 19, 2016.
- Medicare Program; Revisions to Payment Policies Under the Physician Fee Schedule and Other Revisions to Part B for CY 2017; Medicare Advantage Pricing Data Release; Medicare Advantage and Part D Medical Low Ratio Data Release; Medicare Advantage Provider Network Requirements; Expansion of Medicare Diabetes Prevention Program Model. Fed Regist. 2016;81(136):46162-46476. To be codified at 42 CFR §405, 410, 411, et al. https://www.gpo.gov/fdsys/pkg/FR-2016-07-15/pdf/2016-16097.pdf. Accessed September 7, 2016.
- Revisions to Payment Policies under the Physician Fee Schedule and Other Revisions to Part B for CY 2017. Centers for Medicare & Medicaid Services website. https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/PhysicianFeeSched/PFS-Federal-Regulation-Notices-Items/CMS-1654-P.html. Accessed September 7, 2016.
- Text of the Medicare Access and CHIP Reauthorization Act of 2015. GovTrack website. https://www.govtrack.us/congress/bills/114/hr2/text. Accessed September 9, 2016.
- Kaplan KJ. Proposed Medicare 2017 reimbursement schedule whacks biopsy payments; digital pathology payments up. Digital Pathology Blog website. http://tissuepathology.com/2016/07/20/proposed-medicare-2017-reimbursement-schedule-whacks-biopsy-payments-digital-pathology-payments-up/#ixzz4HEqBLgzu. Published July 20, 2016. Accessed September 7, 2016.
- Abelson R. Cost, not choice, is top concern of health insurance customers. The New York Times. http://www.nytimes.com/2016/08/13/business/cost-not-choice-is-top-concern-of-health-insurance-customers.html?_r=0. Published August 12, 2016. Accessed September 7, 2016.
- Stark Law website. http://starklaw.org/. Accessed September 7, 2016.
- Pete Stark. Freedom From Religion website. https://ffrf.org/news/day/dayitems/item/14800-pete-stark. Accessed September 19, 2016.
- Adamy J. Pete Stark: Law regulating doctors mostly helped lawyers. The Wall Street Journal. October 22, 2014. http://blogs.wsj.com/washwire/2014/10/22/pete-stark-law-regulating-doctors-mostly-helped-lawyers/. Accessed September 19, 2016.
Practice Points
- The Proposed Rule outlines the probable payment levels for calendar year 2017.
- The rule also announces how the Medicare Access and CHIP Reauthorization Act (MACRA) may be implemented.

Development of Bullous Pemphigoid in a Patient With Psoriasis and Metabolic Syndrome
Bullous pemphigoid (BP) is an autoimmune subepidermal blistering disease.1 The majority of BP cases are idiopathic and occur in patients older than 60 years. The disease is characterized by the development of circulating IgG autoantibodies reacting with the BP180 antigen of the basement membrane zone.1 Psoriasis vulgaris (PV) is a common, chronic, immune-mediated disease affecting approximately 2% of the world’s population including children and adults.2 Both entities may coexist with internal disorders such as hypertension, diabetes mellitus, coronary heart disease, congestive heart failure, hyperlipidemia, and cerebrovascular accident. It has been postulated that BP more often coexists with neurological disorders, such as stroke and Parkinson disease,3 whereas PV usually is associated with cardiovascular disorders and diabetes mellitus.2 We report the case of a 35-year-old man with chronic PV and metabolic syndrome who developed BP that was successfully treated with methotrexate (MTX).
Case Report
A 35-year-old man with a 15-year history of PV, class 3 obesity (body mass index, 69.2), and thrombosis of the left leg was referred to the dermatology department due to a sudden extensive erythematous and bullous eruption located on the trunk, arms, and legs with involvement of the oral mucosa that had started 4 weeks prior. The skin lesions were accompanied by severe pruritus. On admission to the hospital, the patient presented with stable psoriatic plaques located on the trunk, arms, and proximal part of the lower legs with a psoriasis area severity index score of 11.8 (Figure 1A). He also had disseminated tense blisters and erosions partially arranged in an annular pattern located on the border of the psoriatic plaques as well as on an erythematous base or within unaffected skin (Figure 1B). Additionally, a few small erosions were present on the oral mucosa.
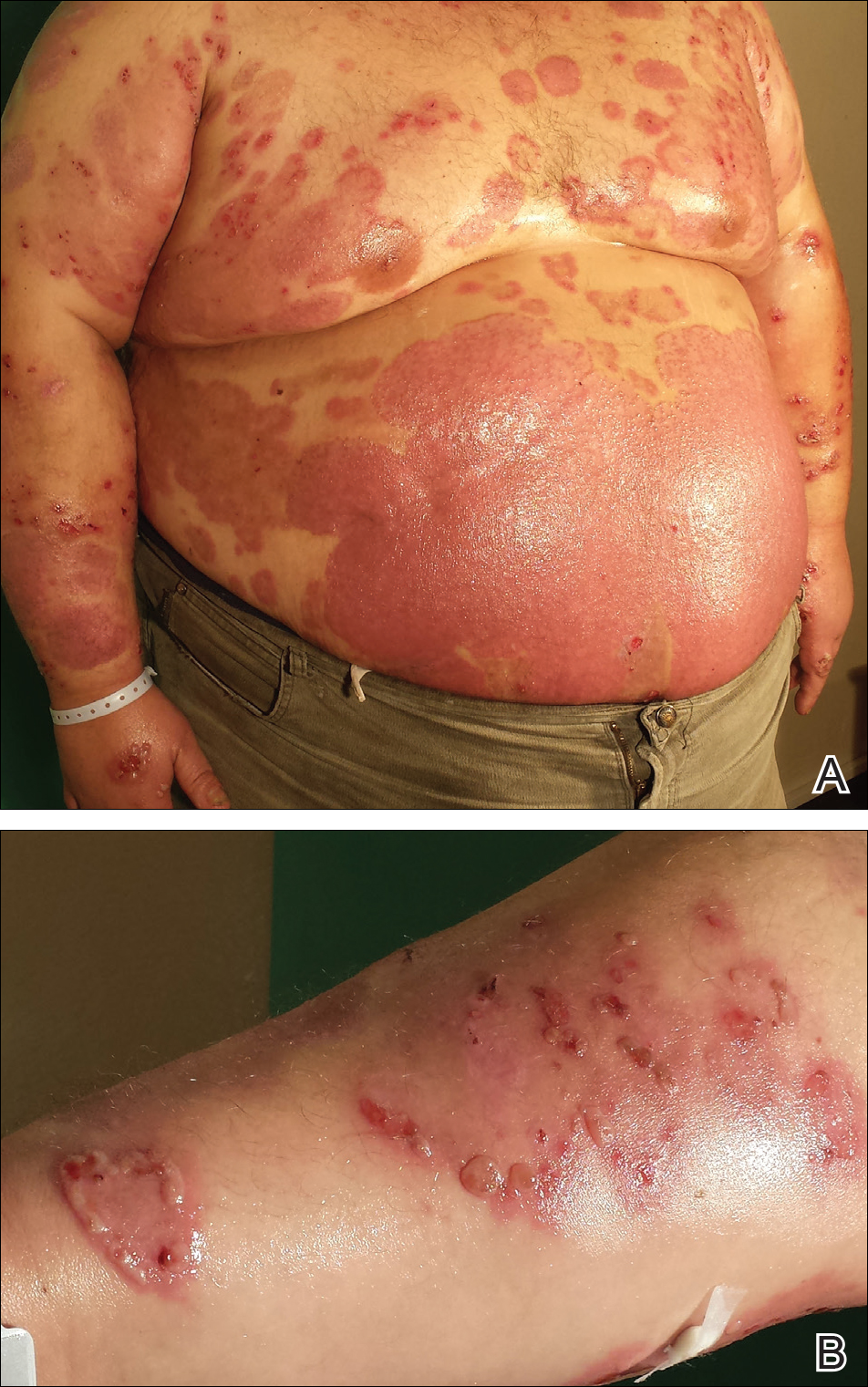
The patient’s father had a history of PV, but there was no family history of obesity or autoimmune blistering disorders. On physical examination, central obesity was noted with a waist circumference of 180 cm and a body mass index of 69.2; his blood pressure was 220/150 mm Hg. Laboratory tests revealed leukocytosis (20.06×109/L [reference range, 4.5–11.0×109/L]) with neutrophilia (16.2×109/L [reference range, 1.6–7.6×109/L]; 80.9% [reference range, 40.0%–70.0%]), eosinophilia (1.01×109/L [reference range, 0–0.5×109/L]), elevated C-reactive protein levels (49.4 mg/L [reference range, 0.0–9.0 mg/L]), elevated erythrocyte sedimentation rate (35 mm/h [reference range, 0–12 mm/h]), elevated γ-glutamyltransferase (66 U/L [reference range, 0–55 U/L]), decreased high-density lipoprotein levels (38 mg/dL [reference range, ≥40 mg/dL]), elevated fasting plasma glucose (116 mg/dL or 6.4 mmol/L [reference range, 70–99 mg/dL or 3.9–5.5 mmol/L]), elevated total IgE (1540 µg/L [reference range, 0–1000 µg/L]), elevated D-dimer (3.21 µg/mL [reference range, <0.5 µg/mL]), and low free triiodothyronine levels (130 pg/dL [reference range, 171–371 pg/dL]). The total protein level was 6.5 g/dL (reference range, 6.0–8.0 g/dL) and albumin level was 3.2 g/dL (reference range, 4.02–4.76 g/dL). A chest radiograph showed no abnormalities.
Based on the physical examination and laboratory testing, it was determined that the patient fulfilled 4 of 5 criteria for metabolic syndrome described by the International Diabetes Federation in 2006 (Table).4 Direct immunofluorescence performed on normal-appearing perilesional skin demonstrated linear IgG and C3 deposits along the basement membrane zone. Indirect immunofluorescence detected circulating IgG autoantibodies at a titer of 1:80. Serum studies using biochip mosaics5 revealed the reactivity of circulating IgG antibodies to the epidermal side of salt-split skin and with antigen dots of tetrameric BP180-NC16a, which prompted the diagnosis of BP (Figure 2).

Oral treatment with MTX 12.5 mg once weekly with clobetasol propionate cream applied to affected skin was initiated for 4 weeks. The PV resolved completely and blister formation stopped. A few weeks later BP reappeared, even though the patient was still taking MTX. The treatment failure may have been related to the patient’s class 3 obesity; therefore, the dose was increased to 20 mg once weekly for 8 weeks, which led to rapid healing of BP erosions. The patient was monitored for 2 months with no symptoms of recurrence.
Comment
Psoriasis Comorbidities
The correlation between PV and cardiovascular disorders such as myocardial infarction, cerebrovascular accident, and pulmonary embolism has been well established and is widely accepted.2 It also has been documented that the risk for metabolic syndrome with components such as diabetes mellitus, hypertension, lipid abnormalities, obesity, and arteriosclerosis is notably increased in PV patients.6 Moreover, associated internal disorders are responsible for a 3- to 4-year reduction in life expectancy in patients with moderate to severe PV.7
Correlation of PV and BP
Psoriasis also may coexist with autoimmune disorders such as rheumatoid arthritis, lupus erythematosus, and blistering disorders.8 There are more than 60 known cases reporting PV in association with various types of subepidermal blistering diseases, including pemphigus vulgaris, epidermolysis bullosa acquisita, anti-p200 pemphigoid, and BP.8,9 The pathogenetic relationship between BP and PV remains obscure. In most published cases, PV preceded BP by 5 to 30 years, possibly ascribable to patients being diagnosed with PV at a younger age.9 In general, patients with BP and PV are younger than patients with BP only, with a mean age of 62 years.9 Because our patient was in his mid-30s when he developed BP, in such cases physicians should take under consideration any triggering factors (eg, drugs). Physical examination and detailed laboratory findings allowed us to make the patient aware of the potential for development of metabolic syndrome. This condition in combination with PV could be a predisposing factor for BP development. According to more recent research, PV is considered a generalized inflammatory process rather than a disorder limited to the skin and joints.10 The chronic inflammatory process in psoriatic skin results in exposure of autoantigens, leading to an immune response and the production of BP antibodies. The neutrophil elastase enzyme present in psoriatic lesions also may take part in dermoepidermal junction degradation and blister formation of BP.11 According to other observations, some antipsoriatic therapies (eg, psoralen plus UVA, UVB, dithranol, coal tar) could be associated with development of BP.12 Moreover, it was shown that psoralen plus UVA therapy, which is widely used in PV treatment, alters the cytokine profile from helper T cells TH1 to TH2.12 TH2-dependent cytokines predominate the sera and erosions in BP patients and seem to be notably relevant to the pathophysiology of the disease.13 The history of our patient’s psoriatic treatment included only topical corticosteroids, keratolytic agents, and occasionally dithranol and coal tar; however, UV phototherapy or any other systemic therapies had never been utilized. Three previously reported cases of patients with PV and BP also revealed no history of UV phototherapy,8,9 which suggests that mechanisms responsible for coexistence of PV and BP are more complex. It has been proven that proinflammatory cytokines secreted by TH1 and TH17 cells, in particular tumor necrosis factor α, IL-17, IL-22, and IL-23, play an important role in the development of psoriatic lesions.10 On the other hand, these cytokines are known to contribute to vascular inflammation, leading to development of arteriosclerosis, as well as to regulate adipogenesis and obesity.14,15 Arakawa et al16 reported increased expression of IL-17 in lesional skin in BP. They concluded that IL-17 may contribute to the recruitment of eosinophils and neutrophils and tissue damage in BP. Therefore, it is highly likely that IL-17 might be a common factor underlying the coexistence of BP with PV and metabolic syndrome. More such reports are required for better understanding this association.
BP Treatment
Selecting a therapy for BP with coexistent PV is challenging, especially in patients with extreme obesity and metabolic syndrome. It is well established that obesity correlates with a higher incidence of PV and more severe disease. On the other hand, obesity also influences response to therapy. Systemic corticosteroids are contraindicated in psoriasis patients because of severe side effects, such as rebound phenomenon of psoriatic lesions and risk for development of generalized pustular PV. Although systemic corticosteroids are effective in BP, high-dose therapy may potentially be life-threatening, particularly in these obese patients with conditions such as hypertension and diabetes mellitus, among others,1 as was observed in our case. Taking into consideration the above mentioned conditions and our experience on such cases, the current patient had received MTX (12.5 mg once weekly) and clobetasol propionate cream, which led to the rapid healing of the psoriatic plaques, whereas BP was more resistant to this therapy. This response may be explained by our patient’s class 3 obesity (body mass index, 69.2). Therefore, the dose of MTX was increased to 20 mg once weekly and was successful. The decision to use MTX was supported by evidence that this medicine may reduce the risk for arteriosclerosis and cardiovascular disorders.17
There are some alternative therapeutic options for patients with coexisting BP and PV, such as cyclosporine,18 combination low-dose cyclosporine and low-dose systemic corticosteroids,19 dapsone,20 azathioprine,21 mycophenolate mofetil,22 and acitretin.23 It also has been shown that biologics (eg, ustekinumab) may be a successful solution in patients with PV and antilaminin-γ1 pemphigoid.24 However, these alternative therapeutic regimens could not be considered in our patient because of serious coexisting internal disorders.
Conclusion
We present a case of concomitant BP and PV in a patient with metabolic syndrome. Although the pathogenic role of this unique coexistence is not fully understood, MTX proved suitable and effective in this single case. Further studies should be performed to elucidate the pathogenic relationship and therapeutic solutions for cases with coexisting PV, BP, and metabolic syndrome.
- Rzany B, Partscht K, Jung M, et al. Risk factors for lethal outcome in patients with bullous pemphigoid: low serum albumin level, high dosage of gluco-corticosteroids, and old age. Arch Dermatol. 2002;138:903-908.
- Pietrzak A, Bartosinska J, Chodorowska G, et al. Cardiovascular aspects of psoriasis vulgaris. Int J Dermatol. 2013;52:153-162.
- Stinco G, Codutti R, Scarbolo M, et al. A retrospective epidemiological study on the association of bullous pemphigoid and neurological diseases. Acta Derm Venereol. 2005;85:136-139.
- International Diabetes Federation. The IDF Consensus Worldwide Definition of the Metabolic Syndrome. Brussels, Belgium: International Diabetes Foundation; 2006. http://www.idf.org/webdata/docs/IDF_Meta_def_final.pdf. Accessed September 14, 2016.
- Van Beek N, Rentzsch K, Probst C, et al. Serological diagnosis of autoimmune bullous skin diseases: prospective comparison of the BIOCHIP mosaic-based indirect immunofluorescence technique with the conventional multi-step single test strategy. Orphanet J Rare Dis. 2012;7:49.
- Sommer DM, Jenisch S, Suchan M, et al. Increased prevalence of the metabolic syndrome in patients with moderate to severe psoriasis. Arch Dermatol Res. 2006;298:321-328.
- Gelfand JM, Troxel AB, Lewis JD, et al. The risk of mortality in patients with psoriasis: results from a population-based study. Arch Dermatol. 2007;143:1493-1499.
- Lazarczyk M, Wozniak K, Ishii N, et al. Coexistence of psoriasis and pemphigoid—only a coincidence? Int J Mol Med. 2006;18:619-623.
- Yasuda H, Tomita Y, Shibaki A, et al. Two cases of subepidermal blistering disease with anti-p200 or 180-kD bullous pemphigoid antigen associated with psoriasis. Dermatology. 2004;209:149-155.
- Malakouti M, Brown GE, Wang E, et al. The role of IL-17 in psoriasis [published online February 20, 2014]. J Dermatolog Treat. 2015;26:41-44.
- Glinski W, Jarzabek-Chorzelska M, Pierozynska-Dubowska M, et al. Basement membrane zone as a target for human neutrophil elastase in psoriasis. Arch Dermatol Res. 1990;282:506-511.
- Klosner G, Trautinger F, Knobler R, et al. Treatment of peripheral blood mononuclear cells with 8-methoxypsoralen plus ultraviolet A radiation induces a shift in cytokine expression from a Th1 to a Th2 response. J Invest Dermatol. 2001;116:459-462.
- Gounni AS, Wellemans V, Agouli M, et al. Increased expression of Th2-associated chemokines in bullous pemphigoid disease. role of eosinophils in the production and release of these chemokines. Clin Immunol. 2006;120:220-231.
- Gao Q, Jiang Y, Ma T, et al. A critical function of Th17 proinflammatory cells in the development of atherosclerotic plaque in mice. J Immunol. 2010;185:5820-5827.
- Zúñiga LA, Shen WJ, Joyce-Shaikh B, et al. IL-17 regulates adipogenesis, glucose homeostasis, and obesity. J Immunol. 2010;185:6947-6959.
- Arakawa M, Dainichi T, Ishii N, et al. Lesional Th17 cells and regulatory T cells in bullous pemphigoid. Exp Dermatol. 2011;20:1022-1024.
- Everett BM, Pradhan AD, Solomon DH, et al. Rationale and design of the Cardiovascular Inflammation Reduction Trial: a test of the inflammatory hypothesis of atherothrombosis. Am Heart J. 2013;166:199-207.
- Boixeda JP, Soria C, Medina S, et al. Bullous pemphigoid and psoriasis: treatment with cyclosporine. J Am Acad Dermatol. 1991;24:152.
- Bianchi L, Gatti S, Nini G. Bullous pemphigoid and severe erythrodermic psoriasis: combined low-dose treatment with cyclosporine and systemic steroids. J Am Acad Dermatol. 1992;27(2, pt 1):278.
- Hisler BM, Blumenthal NC, Aronson PJ, et al. Bullous pemphigoid in psoriatic lesions. J Am Acad Dermatol. 1989;20:683-684.
- Primka EJ III, Camisa C. Psoriasis and bullous pemphigoid treated with azathioprine. J Am Acad Dermatol. 1998;39:121-123.
- Nousari HC, Sragovich A, Kimyai-Asadi A, et al. Mycophenolate mofetil in autoimmune and inflammatory skin disorders. J Am Acad Dermatol. 1999;40:265-268.
- Kobayashi TT, Elston DM, Libow LF, et al. A case of bullous pemphigoid limited to psoriatic plaques. Cutis. 2002;70:283-287.
- Maijima Y, Yagi H, Tateishi C, et al. A successful treatment with ustekinumab in case of antilaminin-γ1 pemphigoid associated with psoriasis. Br J Dermatol. 2013;168:1367-1369.
Bullous pemphigoid (BP) is an autoimmune subepidermal blistering disease.1 The majority of BP cases are idiopathic and occur in patients older than 60 years. The disease is characterized by the development of circulating IgG autoantibodies reacting with the BP180 antigen of the basement membrane zone.1 Psoriasis vulgaris (PV) is a common, chronic, immune-mediated disease affecting approximately 2% of the world’s population including children and adults.2 Both entities may coexist with internal disorders such as hypertension, diabetes mellitus, coronary heart disease, congestive heart failure, hyperlipidemia, and cerebrovascular accident. It has been postulated that BP more often coexists with neurological disorders, such as stroke and Parkinson disease,3 whereas PV usually is associated with cardiovascular disorders and diabetes mellitus.2 We report the case of a 35-year-old man with chronic PV and metabolic syndrome who developed BP that was successfully treated with methotrexate (MTX).
Case Report
A 35-year-old man with a 15-year history of PV, class 3 obesity (body mass index, 69.2), and thrombosis of the left leg was referred to the dermatology department due to a sudden extensive erythematous and bullous eruption located on the trunk, arms, and legs with involvement of the oral mucosa that had started 4 weeks prior. The skin lesions were accompanied by severe pruritus. On admission to the hospital, the patient presented with stable psoriatic plaques located on the trunk, arms, and proximal part of the lower legs with a psoriasis area severity index score of 11.8 (Figure 1A). He also had disseminated tense blisters and erosions partially arranged in an annular pattern located on the border of the psoriatic plaques as well as on an erythematous base or within unaffected skin (Figure 1B). Additionally, a few small erosions were present on the oral mucosa.
The patient’s father had a history of PV, but there was no family history of obesity or autoimmune blistering disorders. On physical examination, central obesity was noted with a waist circumference of 180 cm and a body mass index of 69.2; his blood pressure was 220/150 mm Hg. Laboratory tests revealed leukocytosis (20.06×109/L [reference range, 4.5–11.0×109/L]) with neutrophilia (16.2×109/L [reference range, 1.6–7.6×109/L]; 80.9% [reference range, 40.0%–70.0%]), eosinophilia (1.01×109/L [reference range, 0–0.5×109/L]), elevated C-reactive protein levels (49.4 mg/L [reference range, 0.0–9.0 mg/L]), elevated erythrocyte sedimentation rate (35 mm/h [reference range, 0–12 mm/h]), elevated γ-glutamyltransferase (66 U/L [reference range, 0–55 U/L]), decreased high-density lipoprotein levels (38 mg/dL [reference range, ≥40 mg/dL]), elevated fasting plasma glucose (116 mg/dL or 6.4 mmol/L [reference range, 70–99 mg/dL or 3.9–5.5 mmol/L]), elevated total IgE (1540 µg/L [reference range, 0–1000 µg/L]), elevated D-dimer (3.21 µg/mL [reference range, <0.5 µg/mL]), and low free triiodothyronine levels (130 pg/dL [reference range, 171–371 pg/dL]). The total protein level was 6.5 g/dL (reference range, 6.0–8.0 g/dL) and albumin level was 3.2 g/dL (reference range, 4.02–4.76 g/dL). A chest radiograph showed no abnormalities.
Based on the physical examination and laboratory testing, it was determined that the patient fulfilled 4 of 5 criteria for metabolic syndrome described by the International Diabetes Federation in 2006 (Table).4 Direct immunofluorescence performed on normal-appearing perilesional skin demonstrated linear IgG and C3 deposits along the basement membrane zone. Indirect immunofluorescence detected circulating IgG autoantibodies at a titer of 1:80. Serum studies using biochip mosaics5 revealed the reactivity of circulating IgG antibodies to the epidermal side of salt-split skin and with antigen dots of tetrameric BP180-NC16a, which prompted the diagnosis of BP (Figure 2).
Oral treatment with MTX 12.5 mg once weekly with clobetasol propionate cream applied to affected skin was initiated for 4 weeks. The PV resolved completely and blister formation stopped. A few weeks later BP reappeared, even though the patient was still taking MTX. The treatment failure may have been related to the patient’s class 3 obesity; therefore, the dose was increased to 20 mg once weekly for 8 weeks, which led to rapid healing of BP erosions. The patient was monitored for 2 months with no symptoms of recurrence.
Comment
Psoriasis Comorbidities
The correlation between PV and cardiovascular disorders such as myocardial infarction, cerebrovascular accident, and pulmonary embolism has been well established and is widely accepted.2 It also has been documented that the risk for metabolic syndrome with components such as diabetes mellitus, hypertension, lipid abnormalities, obesity, and arteriosclerosis is notably increased in PV patients.6 Moreover, associated internal disorders are responsible for a 3- to 4-year reduction in life expectancy in patients with moderate to severe PV.7
Correlation of PV and BP
Psoriasis also may coexist with autoimmune disorders such as rheumatoid arthritis, lupus erythematosus, and blistering disorders.8 There are more than 60 known cases reporting PV in association with various types of subepidermal blistering diseases, including pemphigus vulgaris, epidermolysis bullosa acquisita, anti-p200 pemphigoid, and BP.8,9 The pathogenetic relationship between BP and PV remains obscure. In most published cases, PV preceded BP by 5 to 30 years, possibly ascribable to patients being diagnosed with PV at a younger age.9 In general, patients with BP and PV are younger than patients with BP only, with a mean age of 62 years.9 Because our patient was in his mid-30s when he developed BP, in such cases physicians should take under consideration any triggering factors (eg, drugs). Physical examination and detailed laboratory findings allowed us to make the patient aware of the potential for development of metabolic syndrome. This condition in combination with PV could be a predisposing factor for BP development. According to more recent research, PV is considered a generalized inflammatory process rather than a disorder limited to the skin and joints.10 The chronic inflammatory process in psoriatic skin results in exposure of autoantigens, leading to an immune response and the production of BP antibodies. The neutrophil elastase enzyme present in psoriatic lesions also may take part in dermoepidermal junction degradation and blister formation of BP.11 According to other observations, some antipsoriatic therapies (eg, psoralen plus UVA, UVB, dithranol, coal tar) could be associated with development of BP.12 Moreover, it was shown that psoralen plus UVA therapy, which is widely used in PV treatment, alters the cytokine profile from helper T cells TH1 to TH2.12 TH2-dependent cytokines predominate the sera and erosions in BP patients and seem to be notably relevant to the pathophysiology of the disease.13 The history of our patient’s psoriatic treatment included only topical corticosteroids, keratolytic agents, and occasionally dithranol and coal tar; however, UV phototherapy or any other systemic therapies had never been utilized. Three previously reported cases of patients with PV and BP also revealed no history of UV phototherapy,8,9 which suggests that mechanisms responsible for coexistence of PV and BP are more complex. It has been proven that proinflammatory cytokines secreted by TH1 and TH17 cells, in particular tumor necrosis factor α, IL-17, IL-22, and IL-23, play an important role in the development of psoriatic lesions.10 On the other hand, these cytokines are known to contribute to vascular inflammation, leading to development of arteriosclerosis, as well as to regulate adipogenesis and obesity.14,15 Arakawa et al16 reported increased expression of IL-17 in lesional skin in BP. They concluded that IL-17 may contribute to the recruitment of eosinophils and neutrophils and tissue damage in BP. Therefore, it is highly likely that IL-17 might be a common factor underlying the coexistence of BP with PV and metabolic syndrome. More such reports are required for better understanding this association.
BP Treatment
Selecting a therapy for BP with coexistent PV is challenging, especially in patients with extreme obesity and metabolic syndrome. It is well established that obesity correlates with a higher incidence of PV and more severe disease. On the other hand, obesity also influences response to therapy. Systemic corticosteroids are contraindicated in psoriasis patients because of severe side effects, such as rebound phenomenon of psoriatic lesions and risk for development of generalized pustular PV. Although systemic corticosteroids are effective in BP, high-dose therapy may potentially be life-threatening, particularly in these obese patients with conditions such as hypertension and diabetes mellitus, among others,1 as was observed in our case. Taking into consideration the above mentioned conditions and our experience on such cases, the current patient had received MTX (12.5 mg once weekly) and clobetasol propionate cream, which led to the rapid healing of the psoriatic plaques, whereas BP was more resistant to this therapy. This response may be explained by our patient’s class 3 obesity (body mass index, 69.2). Therefore, the dose of MTX was increased to 20 mg once weekly and was successful. The decision to use MTX was supported by evidence that this medicine may reduce the risk for arteriosclerosis and cardiovascular disorders.17
There are some alternative therapeutic options for patients with coexisting BP and PV, such as cyclosporine,18 combination low-dose cyclosporine and low-dose systemic corticosteroids,19 dapsone,20 azathioprine,21 mycophenolate mofetil,22 and acitretin.23 It also has been shown that biologics (eg, ustekinumab) may be a successful solution in patients with PV and antilaminin-γ1 pemphigoid.24 However, these alternative therapeutic regimens could not be considered in our patient because of serious coexisting internal disorders.
Conclusion
We present a case of concomitant BP and PV in a patient with metabolic syndrome. Although the pathogenic role of this unique coexistence is not fully understood, MTX proved suitable and effective in this single case. Further studies should be performed to elucidate the pathogenic relationship and therapeutic solutions for cases with coexisting PV, BP, and metabolic syndrome.
Bullous pemphigoid (BP) is an autoimmune subepidermal blistering disease.1 The majority of BP cases are idiopathic and occur in patients older than 60 years. The disease is characterized by the development of circulating IgG autoantibodies reacting with the BP180 antigen of the basement membrane zone.1 Psoriasis vulgaris (PV) is a common, chronic, immune-mediated disease affecting approximately 2% of the world’s population including children and adults.2 Both entities may coexist with internal disorders such as hypertension, diabetes mellitus, coronary heart disease, congestive heart failure, hyperlipidemia, and cerebrovascular accident. It has been postulated that BP more often coexists with neurological disorders, such as stroke and Parkinson disease,3 whereas PV usually is associated with cardiovascular disorders and diabetes mellitus.2 We report the case of a 35-year-old man with chronic PV and metabolic syndrome who developed BP that was successfully treated with methotrexate (MTX).
Case Report
A 35-year-old man with a 15-year history of PV, class 3 obesity (body mass index, 69.2), and thrombosis of the left leg was referred to the dermatology department due to a sudden extensive erythematous and bullous eruption located on the trunk, arms, and legs with involvement of the oral mucosa that had started 4 weeks prior. The skin lesions were accompanied by severe pruritus. On admission to the hospital, the patient presented with stable psoriatic plaques located on the trunk, arms, and proximal part of the lower legs with a psoriasis area severity index score of 11.8 (Figure 1A). He also had disseminated tense blisters and erosions partially arranged in an annular pattern located on the border of the psoriatic plaques as well as on an erythematous base or within unaffected skin (Figure 1B). Additionally, a few small erosions were present on the oral mucosa.
The patient’s father had a history of PV, but there was no family history of obesity or autoimmune blistering disorders. On physical examination, central obesity was noted with a waist circumference of 180 cm and a body mass index of 69.2; his blood pressure was 220/150 mm Hg. Laboratory tests revealed leukocytosis (20.06×109/L [reference range, 4.5–11.0×109/L]) with neutrophilia (16.2×109/L [reference range, 1.6–7.6×109/L]; 80.9% [reference range, 40.0%–70.0%]), eosinophilia (1.01×109/L [reference range, 0–0.5×109/L]), elevated C-reactive protein levels (49.4 mg/L [reference range, 0.0–9.0 mg/L]), elevated erythrocyte sedimentation rate (35 mm/h [reference range, 0–12 mm/h]), elevated γ-glutamyltransferase (66 U/L [reference range, 0–55 U/L]), decreased high-density lipoprotein levels (38 mg/dL [reference range, ≥40 mg/dL]), elevated fasting plasma glucose (116 mg/dL or 6.4 mmol/L [reference range, 70–99 mg/dL or 3.9–5.5 mmol/L]), elevated total IgE (1540 µg/L [reference range, 0–1000 µg/L]), elevated D-dimer (3.21 µg/mL [reference range, <0.5 µg/mL]), and low free triiodothyronine levels (130 pg/dL [reference range, 171–371 pg/dL]). The total protein level was 6.5 g/dL (reference range, 6.0–8.0 g/dL) and albumin level was 3.2 g/dL (reference range, 4.02–4.76 g/dL). A chest radiograph showed no abnormalities.
Based on the physical examination and laboratory testing, it was determined that the patient fulfilled 4 of 5 criteria for metabolic syndrome described by the International Diabetes Federation in 2006 (Table).4 Direct immunofluorescence performed on normal-appearing perilesional skin demonstrated linear IgG and C3 deposits along the basement membrane zone. Indirect immunofluorescence detected circulating IgG autoantibodies at a titer of 1:80. Serum studies using biochip mosaics5 revealed the reactivity of circulating IgG antibodies to the epidermal side of salt-split skin and with antigen dots of tetrameric BP180-NC16a, which prompted the diagnosis of BP (Figure 2).
Oral treatment with MTX 12.5 mg once weekly with clobetasol propionate cream applied to affected skin was initiated for 4 weeks. The PV resolved completely and blister formation stopped. A few weeks later BP reappeared, even though the patient was still taking MTX. The treatment failure may have been related to the patient’s class 3 obesity; therefore, the dose was increased to 20 mg once weekly for 8 weeks, which led to rapid healing of BP erosions. The patient was monitored for 2 months with no symptoms of recurrence.
Comment
Psoriasis Comorbidities
The correlation between PV and cardiovascular disorders such as myocardial infarction, cerebrovascular accident, and pulmonary embolism has been well established and is widely accepted.2 It also has been documented that the risk for metabolic syndrome with components such as diabetes mellitus, hypertension, lipid abnormalities, obesity, and arteriosclerosis is notably increased in PV patients.6 Moreover, associated internal disorders are responsible for a 3- to 4-year reduction in life expectancy in patients with moderate to severe PV.7
Correlation of PV and BP
Psoriasis also may coexist with autoimmune disorders such as rheumatoid arthritis, lupus erythematosus, and blistering disorders.8 There are more than 60 known cases reporting PV in association with various types of subepidermal blistering diseases, including pemphigus vulgaris, epidermolysis bullosa acquisita, anti-p200 pemphigoid, and BP.8,9 The pathogenetic relationship between BP and PV remains obscure. In most published cases, PV preceded BP by 5 to 30 years, possibly ascribable to patients being diagnosed with PV at a younger age.9 In general, patients with BP and PV are younger than patients with BP only, with a mean age of 62 years.9 Because our patient was in his mid-30s when he developed BP, in such cases physicians should take under consideration any triggering factors (eg, drugs). Physical examination and detailed laboratory findings allowed us to make the patient aware of the potential for development of metabolic syndrome. This condition in combination with PV could be a predisposing factor for BP development. According to more recent research, PV is considered a generalized inflammatory process rather than a disorder limited to the skin and joints.10 The chronic inflammatory process in psoriatic skin results in exposure of autoantigens, leading to an immune response and the production of BP antibodies. The neutrophil elastase enzyme present in psoriatic lesions also may take part in dermoepidermal junction degradation and blister formation of BP.11 According to other observations, some antipsoriatic therapies (eg, psoralen plus UVA, UVB, dithranol, coal tar) could be associated with development of BP.12 Moreover, it was shown that psoralen plus UVA therapy, which is widely used in PV treatment, alters the cytokine profile from helper T cells TH1 to TH2.12 TH2-dependent cytokines predominate the sera and erosions in BP patients and seem to be notably relevant to the pathophysiology of the disease.13 The history of our patient’s psoriatic treatment included only topical corticosteroids, keratolytic agents, and occasionally dithranol and coal tar; however, UV phototherapy or any other systemic therapies had never been utilized. Three previously reported cases of patients with PV and BP also revealed no history of UV phototherapy,8,9 which suggests that mechanisms responsible for coexistence of PV and BP are more complex. It has been proven that proinflammatory cytokines secreted by TH1 and TH17 cells, in particular tumor necrosis factor α, IL-17, IL-22, and IL-23, play an important role in the development of psoriatic lesions.10 On the other hand, these cytokines are known to contribute to vascular inflammation, leading to development of arteriosclerosis, as well as to regulate adipogenesis and obesity.14,15 Arakawa et al16 reported increased expression of IL-17 in lesional skin in BP. They concluded that IL-17 may contribute to the recruitment of eosinophils and neutrophils and tissue damage in BP. Therefore, it is highly likely that IL-17 might be a common factor underlying the coexistence of BP with PV and metabolic syndrome. More such reports are required for better understanding this association.
BP Treatment
Selecting a therapy for BP with coexistent PV is challenging, especially in patients with extreme obesity and metabolic syndrome. It is well established that obesity correlates with a higher incidence of PV and more severe disease. On the other hand, obesity also influences response to therapy. Systemic corticosteroids are contraindicated in psoriasis patients because of severe side effects, such as rebound phenomenon of psoriatic lesions and risk for development of generalized pustular PV. Although systemic corticosteroids are effective in BP, high-dose therapy may potentially be life-threatening, particularly in these obese patients with conditions such as hypertension and diabetes mellitus, among others,1 as was observed in our case. Taking into consideration the above mentioned conditions and our experience on such cases, the current patient had received MTX (12.5 mg once weekly) and clobetasol propionate cream, which led to the rapid healing of the psoriatic plaques, whereas BP was more resistant to this therapy. This response may be explained by our patient’s class 3 obesity (body mass index, 69.2). Therefore, the dose of MTX was increased to 20 mg once weekly and was successful. The decision to use MTX was supported by evidence that this medicine may reduce the risk for arteriosclerosis and cardiovascular disorders.17
There are some alternative therapeutic options for patients with coexisting BP and PV, such as cyclosporine,18 combination low-dose cyclosporine and low-dose systemic corticosteroids,19 dapsone,20 azathioprine,21 mycophenolate mofetil,22 and acitretin.23 It also has been shown that biologics (eg, ustekinumab) may be a successful solution in patients with PV and antilaminin-γ1 pemphigoid.24 However, these alternative therapeutic regimens could not be considered in our patient because of serious coexisting internal disorders.
Conclusion
We present a case of concomitant BP and PV in a patient with metabolic syndrome. Although the pathogenic role of this unique coexistence is not fully understood, MTX proved suitable and effective in this single case. Further studies should be performed to elucidate the pathogenic relationship and therapeutic solutions for cases with coexisting PV, BP, and metabolic syndrome.
- Rzany B, Partscht K, Jung M, et al. Risk factors for lethal outcome in patients with bullous pemphigoid: low serum albumin level, high dosage of gluco-corticosteroids, and old age. Arch Dermatol. 2002;138:903-908.
- Pietrzak A, Bartosinska J, Chodorowska G, et al. Cardiovascular aspects of psoriasis vulgaris. Int J Dermatol. 2013;52:153-162.
- Stinco G, Codutti R, Scarbolo M, et al. A retrospective epidemiological study on the association of bullous pemphigoid and neurological diseases. Acta Derm Venereol. 2005;85:136-139.
- International Diabetes Federation. The IDF Consensus Worldwide Definition of the Metabolic Syndrome. Brussels, Belgium: International Diabetes Foundation; 2006. http://www.idf.org/webdata/docs/IDF_Meta_def_final.pdf. Accessed September 14, 2016.
- Van Beek N, Rentzsch K, Probst C, et al. Serological diagnosis of autoimmune bullous skin diseases: prospective comparison of the BIOCHIP mosaic-based indirect immunofluorescence technique with the conventional multi-step single test strategy. Orphanet J Rare Dis. 2012;7:49.
- Sommer DM, Jenisch S, Suchan M, et al. Increased prevalence of the metabolic syndrome in patients with moderate to severe psoriasis. Arch Dermatol Res. 2006;298:321-328.
- Gelfand JM, Troxel AB, Lewis JD, et al. The risk of mortality in patients with psoriasis: results from a population-based study. Arch Dermatol. 2007;143:1493-1499.
- Lazarczyk M, Wozniak K, Ishii N, et al. Coexistence of psoriasis and pemphigoid—only a coincidence? Int J Mol Med. 2006;18:619-623.
- Yasuda H, Tomita Y, Shibaki A, et al. Two cases of subepidermal blistering disease with anti-p200 or 180-kD bullous pemphigoid antigen associated with psoriasis. Dermatology. 2004;209:149-155.
- Malakouti M, Brown GE, Wang E, et al. The role of IL-17 in psoriasis [published online February 20, 2014]. J Dermatolog Treat. 2015;26:41-44.
- Glinski W, Jarzabek-Chorzelska M, Pierozynska-Dubowska M, et al. Basement membrane zone as a target for human neutrophil elastase in psoriasis. Arch Dermatol Res. 1990;282:506-511.
- Klosner G, Trautinger F, Knobler R, et al. Treatment of peripheral blood mononuclear cells with 8-methoxypsoralen plus ultraviolet A radiation induces a shift in cytokine expression from a Th1 to a Th2 response. J Invest Dermatol. 2001;116:459-462.
- Gounni AS, Wellemans V, Agouli M, et al. Increased expression of Th2-associated chemokines in bullous pemphigoid disease. role of eosinophils in the production and release of these chemokines. Clin Immunol. 2006;120:220-231.
- Gao Q, Jiang Y, Ma T, et al. A critical function of Th17 proinflammatory cells in the development of atherosclerotic plaque in mice. J Immunol. 2010;185:5820-5827.
- Zúñiga LA, Shen WJ, Joyce-Shaikh B, et al. IL-17 regulates adipogenesis, glucose homeostasis, and obesity. J Immunol. 2010;185:6947-6959.
- Arakawa M, Dainichi T, Ishii N, et al. Lesional Th17 cells and regulatory T cells in bullous pemphigoid. Exp Dermatol. 2011;20:1022-1024.
- Everett BM, Pradhan AD, Solomon DH, et al. Rationale and design of the Cardiovascular Inflammation Reduction Trial: a test of the inflammatory hypothesis of atherothrombosis. Am Heart J. 2013;166:199-207.
- Boixeda JP, Soria C, Medina S, et al. Bullous pemphigoid and psoriasis: treatment with cyclosporine. J Am Acad Dermatol. 1991;24:152.
- Bianchi L, Gatti S, Nini G. Bullous pemphigoid and severe erythrodermic psoriasis: combined low-dose treatment with cyclosporine and systemic steroids. J Am Acad Dermatol. 1992;27(2, pt 1):278.
- Hisler BM, Blumenthal NC, Aronson PJ, et al. Bullous pemphigoid in psoriatic lesions. J Am Acad Dermatol. 1989;20:683-684.
- Primka EJ III, Camisa C. Psoriasis and bullous pemphigoid treated with azathioprine. J Am Acad Dermatol. 1998;39:121-123.
- Nousari HC, Sragovich A, Kimyai-Asadi A, et al. Mycophenolate mofetil in autoimmune and inflammatory skin disorders. J Am Acad Dermatol. 1999;40:265-268.
- Kobayashi TT, Elston DM, Libow LF, et al. A case of bullous pemphigoid limited to psoriatic plaques. Cutis. 2002;70:283-287.
- Maijima Y, Yagi H, Tateishi C, et al. A successful treatment with ustekinumab in case of antilaminin-γ1 pemphigoid associated with psoriasis. Br J Dermatol. 2013;168:1367-1369.
- Rzany B, Partscht K, Jung M, et al. Risk factors for lethal outcome in patients with bullous pemphigoid: low serum albumin level, high dosage of gluco-corticosteroids, and old age. Arch Dermatol. 2002;138:903-908.
- Pietrzak A, Bartosinska J, Chodorowska G, et al. Cardiovascular aspects of psoriasis vulgaris. Int J Dermatol. 2013;52:153-162.
- Stinco G, Codutti R, Scarbolo M, et al. A retrospective epidemiological study on the association of bullous pemphigoid and neurological diseases. Acta Derm Venereol. 2005;85:136-139.
- International Diabetes Federation. The IDF Consensus Worldwide Definition of the Metabolic Syndrome. Brussels, Belgium: International Diabetes Foundation; 2006. http://www.idf.org/webdata/docs/IDF_Meta_def_final.pdf. Accessed September 14, 2016.
- Van Beek N, Rentzsch K, Probst C, et al. Serological diagnosis of autoimmune bullous skin diseases: prospective comparison of the BIOCHIP mosaic-based indirect immunofluorescence technique with the conventional multi-step single test strategy. Orphanet J Rare Dis. 2012;7:49.
- Sommer DM, Jenisch S, Suchan M, et al. Increased prevalence of the metabolic syndrome in patients with moderate to severe psoriasis. Arch Dermatol Res. 2006;298:321-328.
- Gelfand JM, Troxel AB, Lewis JD, et al. The risk of mortality in patients with psoriasis: results from a population-based study. Arch Dermatol. 2007;143:1493-1499.
- Lazarczyk M, Wozniak K, Ishii N, et al. Coexistence of psoriasis and pemphigoid—only a coincidence? Int J Mol Med. 2006;18:619-623.
- Yasuda H, Tomita Y, Shibaki A, et al. Two cases of subepidermal blistering disease with anti-p200 or 180-kD bullous pemphigoid antigen associated with psoriasis. Dermatology. 2004;209:149-155.
- Malakouti M, Brown GE, Wang E, et al. The role of IL-17 in psoriasis [published online February 20, 2014]. J Dermatolog Treat. 2015;26:41-44.
- Glinski W, Jarzabek-Chorzelska M, Pierozynska-Dubowska M, et al. Basement membrane zone as a target for human neutrophil elastase in psoriasis. Arch Dermatol Res. 1990;282:506-511.
- Klosner G, Trautinger F, Knobler R, et al. Treatment of peripheral blood mononuclear cells with 8-methoxypsoralen plus ultraviolet A radiation induces a shift in cytokine expression from a Th1 to a Th2 response. J Invest Dermatol. 2001;116:459-462.
- Gounni AS, Wellemans V, Agouli M, et al. Increased expression of Th2-associated chemokines in bullous pemphigoid disease. role of eosinophils in the production and release of these chemokines. Clin Immunol. 2006;120:220-231.
- Gao Q, Jiang Y, Ma T, et al. A critical function of Th17 proinflammatory cells in the development of atherosclerotic plaque in mice. J Immunol. 2010;185:5820-5827.
- Zúñiga LA, Shen WJ, Joyce-Shaikh B, et al. IL-17 regulates adipogenesis, glucose homeostasis, and obesity. J Immunol. 2010;185:6947-6959.
- Arakawa M, Dainichi T, Ishii N, et al. Lesional Th17 cells and regulatory T cells in bullous pemphigoid. Exp Dermatol. 2011;20:1022-1024.
- Everett BM, Pradhan AD, Solomon DH, et al. Rationale and design of the Cardiovascular Inflammation Reduction Trial: a test of the inflammatory hypothesis of atherothrombosis. Am Heart J. 2013;166:199-207.
- Boixeda JP, Soria C, Medina S, et al. Bullous pemphigoid and psoriasis: treatment with cyclosporine. J Am Acad Dermatol. 1991;24:152.
- Bianchi L, Gatti S, Nini G. Bullous pemphigoid and severe erythrodermic psoriasis: combined low-dose treatment with cyclosporine and systemic steroids. J Am Acad Dermatol. 1992;27(2, pt 1):278.
- Hisler BM, Blumenthal NC, Aronson PJ, et al. Bullous pemphigoid in psoriatic lesions. J Am Acad Dermatol. 1989;20:683-684.
- Primka EJ III, Camisa C. Psoriasis and bullous pemphigoid treated with azathioprine. J Am Acad Dermatol. 1998;39:121-123.
- Nousari HC, Sragovich A, Kimyai-Asadi A, et al. Mycophenolate mofetil in autoimmune and inflammatory skin disorders. J Am Acad Dermatol. 1999;40:265-268.
- Kobayashi TT, Elston DM, Libow LF, et al. A case of bullous pemphigoid limited to psoriatic plaques. Cutis. 2002;70:283-287.
- Maijima Y, Yagi H, Tateishi C, et al. A successful treatment with ustekinumab in case of antilaminin-γ1 pemphigoid associated with psoriasis. Br J Dermatol. 2013;168:1367-1369.
Practice Points
- Metabolic syndrome and psoriasis vulgaris (PV) may promote development of bullous pemphigoid (BP) in patients younger than 60 years.
- Methotrexate may be a therapeutic solution for BP coexisting with PV and metabolic syndrome.
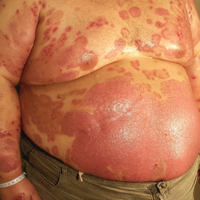
Metastatic Crohn Disease Clinically Reminiscent of Erythema Nodosum on the Right Leg
Metastatic Crohn disease (MCD) is defined by the presence of cutaneous noncaseating granulomatous lesions that are noncontiguous with the gastrointestinal (GI) tract or fistulae.1 The clinical presentation of MCD is so variable that its diagnosis requires a high index of suspicion.1,2 In particular, the presence of erythematous tender nodules on the legs is easily mistaken for erythema nodosum (EN). Skin biopsy has an important role in confirming the diagnosis, as histopathological examination would reveal a noncaseating granuloma similar to those in the involved GI tract.2 Herein, we report a case of MCD on the right leg that was clinically reminiscent of unilateral EN.
Case Report
A 21-year-old woman presented to the dermatology department with 2 painful erythematous nodules on the lower right leg of 2 weeks’ duration. She also reported abdominal pain, diarrhea, and bloody stool. She had been diagnosed with Crohn disease (CD) 6 years prior that had been well controlled with systemic low-dose steroids (5–15 mg/d), metronidazole (750 mg/d), and intermittent mesalamine and antidiarrheal drugs. However, she had not taken her medication for several weeks on her own authority. Subsequently, the patient developed skin lesions, which were characterized by ill-defined erythematous nodules with tenderness on the right lower leg along with GI symptoms (Figure 1). Laboratory studies revealed anemia (hemoglobin, 9.9 g/dL [reference range, 12.0–16.0 g/dL]) and an elevated C-reactive protein level (4.3 mg/dL [reference range, 0–0.3 mg/dL]). Other routine laboratory findings were normal.
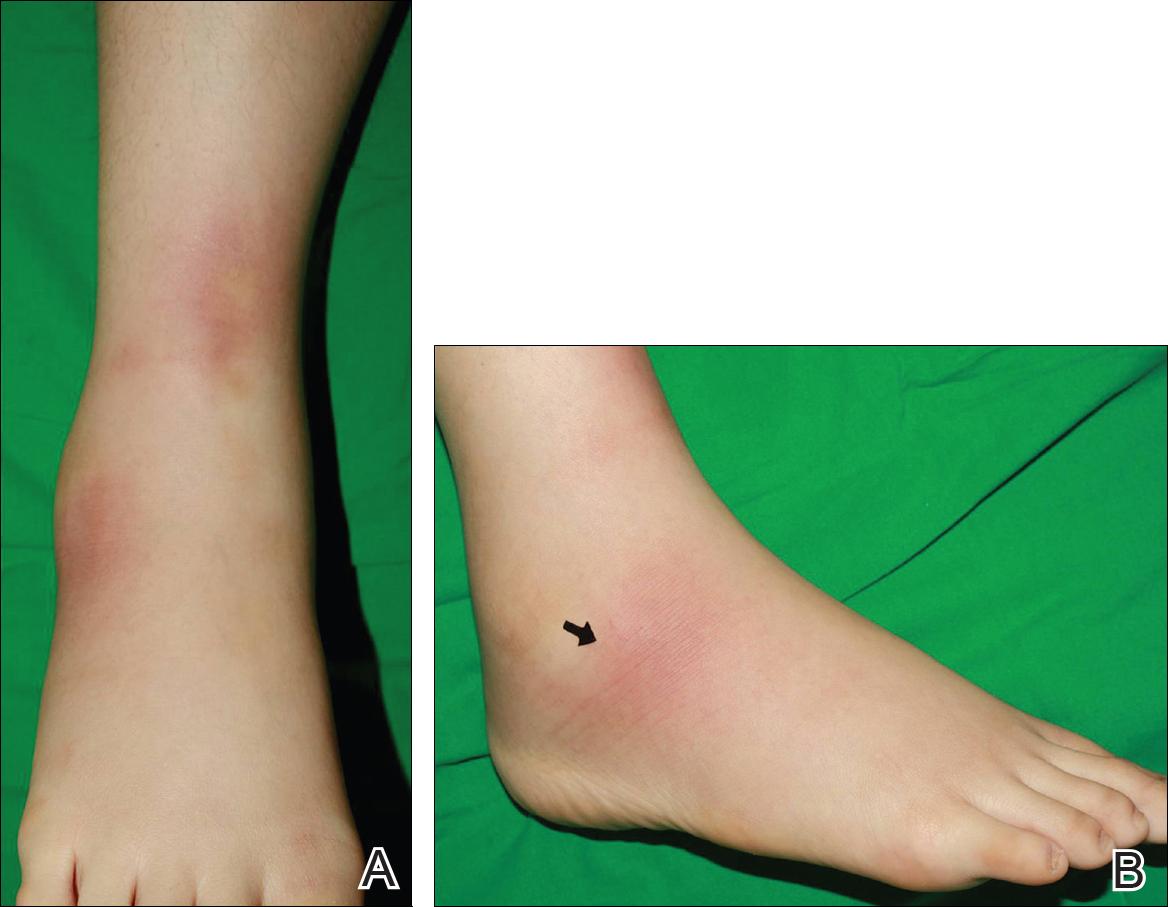
Histopathologically, a skin biopsy from the right ankle showed vague, ill-defined, noncaseating granulomas scattered in the deep dermis and lobules of the subcutis (Figure 2). The granulomas were composed of epithelioid cells and Langerhans-type giant cells. Lymphocytes and neutrophils also were present, but eosinophils were absent. Immunohistochemical staining revealed that the infiltrating cells were mostly CD4+ helper/inducer T cells intermixed with CD8+ suppressor/cytotoxic T cells. The CD4:CD8 ratio was approximately 2:1. Counts of CD20+ B cells were low. Epithelioid cells and giant cells were positive for CD68.
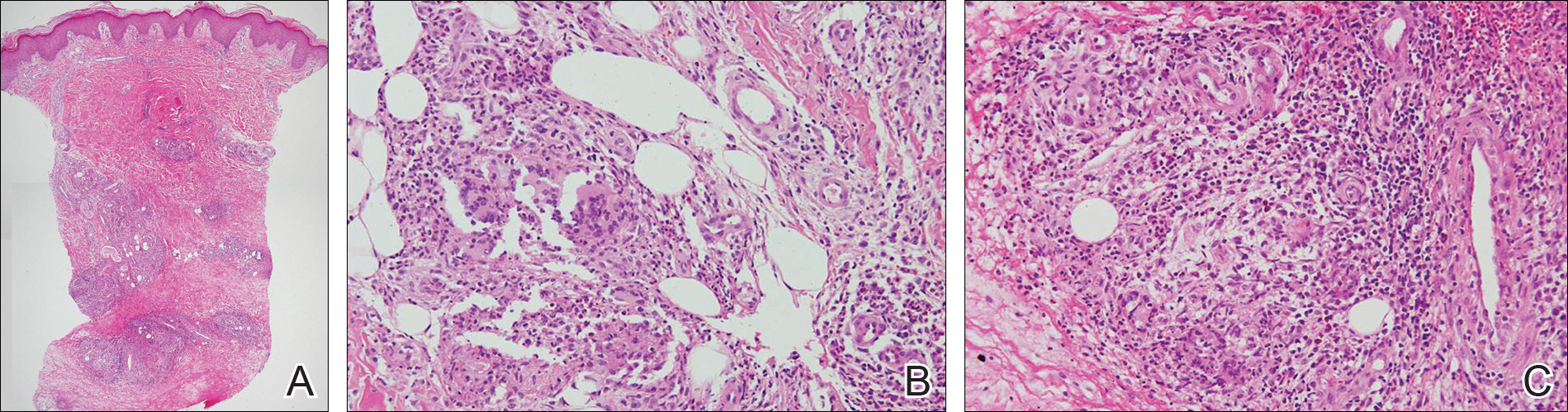
×20). The skin biopsy showed granulomas composed of epithelioid cells and multinucleated giant cells in the deep dermis and in the lobules of the subcutis (B)(H&E, original magnification ×200). Histopathologic features such as small vessel vasculitis characterized by a fibrin deposit in the small blood vessels and swelling of the endothelial cells as well as granulomatous perivasculitis with perivascular infiltration of the epithelioid cells were present (C)(H&E, original magnification ×200).
A colonoscopy was performed to evaluate the aggravation of CD. Multiple longitudinal ulcers were observed in the ileocecal valve area and from the transverse colon to the sigmoid colon (Figure 3A). Histopathologic findings from the colon showed mucosal ulceration and noncaseating granulomas with heavy infiltration of lymphocytes and plasma cells (Figure 3B). Staining for infectious microorganisms (eg, Ziehl-Neelsen, periodic acid–Schiff, Gram) was negative. A polymerase chain reaction performed on sections cut from the paraffin block of the skin biopsy was negative for Mycobacterium tuberculosis DNA.
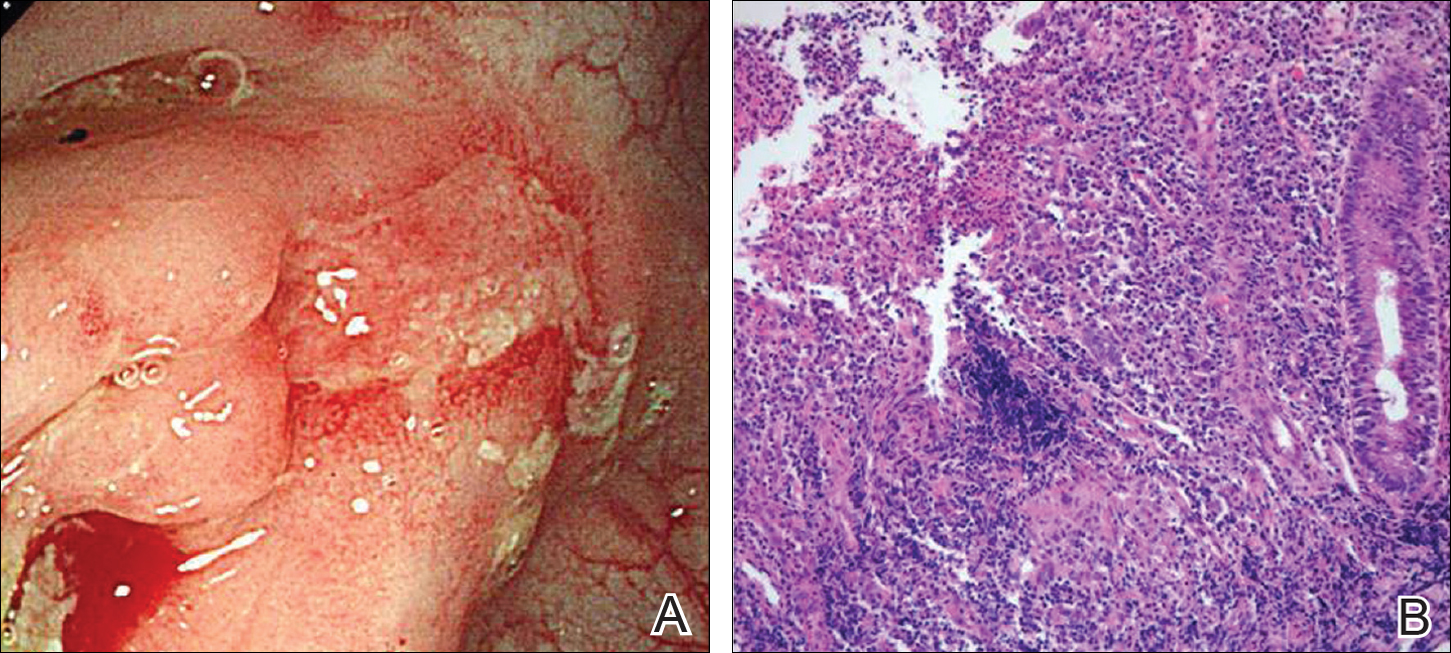
Based on the clinical and histopathologic findings, the patient was diagnosed with MCD that was clinically reminiscent of unilateral EN. Four weeks after the initiation of therapy with systemic corticosteroids (25 mg/d), oral metronidazole (750 mg/d), and mesalamine (1200 mg/d) for CD, the skin lesions were completely resolved and the patient’s GI symptoms improved simultaneously.
Comment
Crohn disease is a chronic inflammatory granulomatous disease of the GI tract that often is associated with reactive cutaneous lesions including EN, pyoderma gangrenosum, necrotizing vasculitis, and epidermolysis bullosa acquisita. Of these, EN is the most common to appear in CD patients and has been reported to occur in 1% to 15% of patients.3-5 In particular, skin lesions on the leg presenting as tender erythematous nodules and patches are often diagnosed as EN, which is relatively common. In our case, we initially suspected EN due to the rare presentation of MCD and lack of specific clinical features; however, the skin biopsy revealed noncaseating granulomas in the mid to deep dermis and subcutis consistent with MCD.
Metastatic Crohn disease is a rare disease entity and is characterized by the presence of noncaseating granulomas of the skin at sites separated from the GI tract by normal tissue.1 Although its pathogenesis is unclear, it has been suggested that immune complexes deposited in the skin could be responsible for the granulomatous reactions.4 A T lymphocyte–mediated type IV hypersensitivity reaction also could be responsible.6,7 Because antimicrobial therapy can be curative for infection-related MCD, special histologic stains and/or tissue cultures can help to exclude an infectious etiology.8
Clinical presentations of MCD vary greatly, with observations such as single or multiple erythematous swellings, papules, plaques, nodules, abscesses, and ulcers.1,2 The relationship between these clinical presentations and the intestinal activity of CD still is unknown; in some cases, however, the metastatic granulomatous lesions and the bowel disease show comparable severity.2,9,10 In a review of the literature, MCD was generally reported to present in the genital area in children. In adults, lesions most frequently present in the genital area, followed by ulcers on the arms and legs.1,2 These variations in clinical features and location resemble benign or infectious disease and can lead to delays in diagnosis.
Histopathologically, MCD lesions usually are ill-defined noncaseating granulomas with numerous multinucleated giant cells and lymphomononuclear cells located mostly in the dermis and occasionally extending into the subcutis. The cutaneous granulomata are similar to those present in the affected GI tract. Lymphocytes and plasma cells also are commonly present and eosinophils can be prominent.1,2,11 In some cases of MCD, granulomatous vasculitis of small- to medium-sized vessels can be found and is associated with dermal and subcutaneous granulomatous inflammation.8,11,12 Misago and Narisawa13 suggested that granulomatous vasculitis and panniculitis associated with CD is considered to be a rare subtype of MCD. Few cases of MCD presenting as granulomatous panniculitis have been described in the literature.14-16 Our patient presented with lesions that clinically resembled EN; however, the biopsy was more consistent with MCD. The Table summarizes the distinguishing clinical and histopathological features of MCD in our case and classic EN.
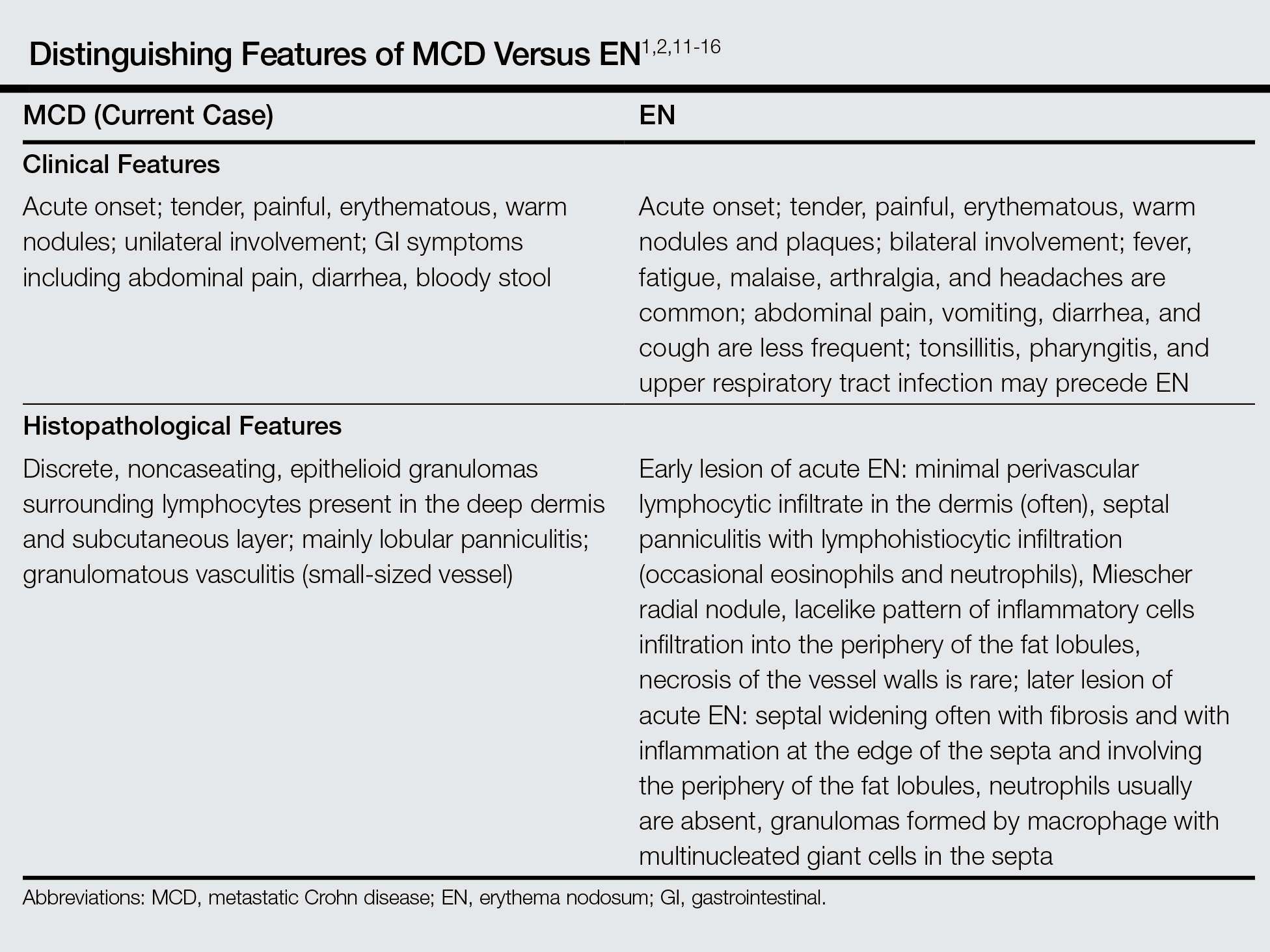
Although some authors believe that MCD is not related to CD activity, others assert that MCD lesions may parallel GI activity.1,2 Our patient was treated with systemic corticosteroids, oral metronidazole, and mesalamine to control the GI symptoms associated with CD. Four weeks after treatment, the GI symptoms and skin lesions improved simultaneously without any additional dermatologic treatment. We believe that MCD has the potential to serve as an early marker of the recurrence of CD and can help with the early diagnosis of CD aggravation, though an association between MCD and CD activity has not been confirmed.
Conclusion
We reported a case of MCD that was clinically reminiscent of unilateral EN and associated with GI disease activity. Physicians should be aware of the possibility of skin manifestations in CD, especially when erythematous nodular lesions are present on the leg.
- Calonje E, Brenn T, Lazar AJ, et al. Mckee’s Pathology of the Skin: With Clinical Correlations. 4th ed. Philadelphia, PA: Saunders Elsevier; 2012.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn’s disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Sonia F, Richard SB. Inflammatory bowel disease. In: Kasper DL, Braunwald E, Fauci AS, et al, eds. Harrison’s Principles of Internal Medicine. 16th ed. New York, NY: McGraw-Hill; 2005:1776-1789.
- Burgdorf W. Cutaneous manifestations of Crohn’s disease. J Am Acad Dermatol. 1981;5:689-695.
- Crowson AN, Nuovo GJ, Mihm MC Jr, et al. Cutaneous manifestations of Crohn’s disease, its spectrum, and its pathogenesis: intracellular consensus bacterial 16S rRNA is associated with the gastrointestinal but not the cutaneous manifestations of Crohn’s disease. Hum Pathol. 2003;34:1185-1192.
- Tatnall FM, Dodd HJ, Sarkany I. Crohn’s disease with metastatic cutaneous involvement and granulomatous cheilitis. J R Soc Med. 1987;80:49-51.
- Shum DT, Guenther L. Metastatic Crohn’s disease. case report and review of the literature. Arch Dermatol. 1990;126:645-648.
- Emanuel PO, Phelps RG. Metastatic Crohn’s disease: a histopathologic study of 12 cases. J Cutan Pathol. 2008;35:457-461.
- Chalvardjian A, Nethercott JR. Cutaneous granulomatous vasculitis associated with Crohn’s disease. Cutis. 1982;30:645-655.
- Lebwohl M, Fleischmajer R, Janowitz H, et al. Metastatic Crohn’s disease. J Am Acad Dermatol. 1984;10:33-38.
- Sabat M, Leulmo J, Saez A. Cutaneous granulomatous vasculitis in metastatic Crohn’s disease. J Eur Acad Dermatol Venereol. 2005;19:652-653.
- Burns AM, Walsh N, Green PJ. Granulomatous vasculitis in Crohn’s disease: a clinicopathologic correlate of two unusual cases. J Cutan Pathol. 2010;37:1077-1083.
- Misago N, Narisawa Y. Erythema induratum (nodular vasculitis) associated with Crohn’s disease: a rare type of metastatic Crohn’s disease. Am J Dermatopathol. 2012;34:325-329.
- Liebermann TR, Greene JF Jr. Transient subcutaneous granulomatosis of the upper extremities in Crohn’s disease. Am J Gastroenterol. 1979;72:89-91.
- Levine N, Bangert J. Cutaneous granulomatosis in Crohn’s disease. Arch Dermatol. 1982;118:1006-1009.
- Hackzell-Bradley M, Hedblad MA, Stephansson EA. Metastatic Crohn’s disease. report of 3 cases with special reference to histopathologic findings. Arch Dermatol. 1996;132:928-932.
Metastatic Crohn disease (MCD) is defined by the presence of cutaneous noncaseating granulomatous lesions that are noncontiguous with the gastrointestinal (GI) tract or fistulae.1 The clinical presentation of MCD is so variable that its diagnosis requires a high index of suspicion.1,2 In particular, the presence of erythematous tender nodules on the legs is easily mistaken for erythema nodosum (EN). Skin biopsy has an important role in confirming the diagnosis, as histopathological examination would reveal a noncaseating granuloma similar to those in the involved GI tract.2 Herein, we report a case of MCD on the right leg that was clinically reminiscent of unilateral EN.
Case Report
A 21-year-old woman presented to the dermatology department with 2 painful erythematous nodules on the lower right leg of 2 weeks’ duration. She also reported abdominal pain, diarrhea, and bloody stool. She had been diagnosed with Crohn disease (CD) 6 years prior that had been well controlled with systemic low-dose steroids (5–15 mg/d), metronidazole (750 mg/d), and intermittent mesalamine and antidiarrheal drugs. However, she had not taken her medication for several weeks on her own authority. Subsequently, the patient developed skin lesions, which were characterized by ill-defined erythematous nodules with tenderness on the right lower leg along with GI symptoms (Figure 1). Laboratory studies revealed anemia (hemoglobin, 9.9 g/dL [reference range, 12.0–16.0 g/dL]) and an elevated C-reactive protein level (4.3 mg/dL [reference range, 0–0.3 mg/dL]). Other routine laboratory findings were normal.
Histopathologically, a skin biopsy from the right ankle showed vague, ill-defined, noncaseating granulomas scattered in the deep dermis and lobules of the subcutis (Figure 2). The granulomas were composed of epithelioid cells and Langerhans-type giant cells. Lymphocytes and neutrophils also were present, but eosinophils were absent. Immunohistochemical staining revealed that the infiltrating cells were mostly CD4+ helper/inducer T cells intermixed with CD8+ suppressor/cytotoxic T cells. The CD4:CD8 ratio was approximately 2:1. Counts of CD20+ B cells were low. Epithelioid cells and giant cells were positive for CD68.
×20). The skin biopsy showed granulomas composed of epithelioid cells and multinucleated giant cells in the deep dermis and in the lobules of the subcutis (B)(H&E, original magnification ×200). Histopathologic features such as small vessel vasculitis characterized by a fibrin deposit in the small blood vessels and swelling of the endothelial cells as well as granulomatous perivasculitis with perivascular infiltration of the epithelioid cells were present (C)(H&E, original magnification ×200).
A colonoscopy was performed to evaluate the aggravation of CD. Multiple longitudinal ulcers were observed in the ileocecal valve area and from the transverse colon to the sigmoid colon (Figure 3A). Histopathologic findings from the colon showed mucosal ulceration and noncaseating granulomas with heavy infiltration of lymphocytes and plasma cells (Figure 3B). Staining for infectious microorganisms (eg, Ziehl-Neelsen, periodic acid–Schiff, Gram) was negative. A polymerase chain reaction performed on sections cut from the paraffin block of the skin biopsy was negative for Mycobacterium tuberculosis DNA.
Based on the clinical and histopathologic findings, the patient was diagnosed with MCD that was clinically reminiscent of unilateral EN. Four weeks after the initiation of therapy with systemic corticosteroids (25 mg/d), oral metronidazole (750 mg/d), and mesalamine (1200 mg/d) for CD, the skin lesions were completely resolved and the patient’s GI symptoms improved simultaneously.
Comment
Crohn disease is a chronic inflammatory granulomatous disease of the GI tract that often is associated with reactive cutaneous lesions including EN, pyoderma gangrenosum, necrotizing vasculitis, and epidermolysis bullosa acquisita. Of these, EN is the most common to appear in CD patients and has been reported to occur in 1% to 15% of patients.3-5 In particular, skin lesions on the leg presenting as tender erythematous nodules and patches are often diagnosed as EN, which is relatively common. In our case, we initially suspected EN due to the rare presentation of MCD and lack of specific clinical features; however, the skin biopsy revealed noncaseating granulomas in the mid to deep dermis and subcutis consistent with MCD.
Metastatic Crohn disease is a rare disease entity and is characterized by the presence of noncaseating granulomas of the skin at sites separated from the GI tract by normal tissue.1 Although its pathogenesis is unclear, it has been suggested that immune complexes deposited in the skin could be responsible for the granulomatous reactions.4 A T lymphocyte–mediated type IV hypersensitivity reaction also could be responsible.6,7 Because antimicrobial therapy can be curative for infection-related MCD, special histologic stains and/or tissue cultures can help to exclude an infectious etiology.8
Clinical presentations of MCD vary greatly, with observations such as single or multiple erythematous swellings, papules, plaques, nodules, abscesses, and ulcers.1,2 The relationship between these clinical presentations and the intestinal activity of CD still is unknown; in some cases, however, the metastatic granulomatous lesions and the bowel disease show comparable severity.2,9,10 In a review of the literature, MCD was generally reported to present in the genital area in children. In adults, lesions most frequently present in the genital area, followed by ulcers on the arms and legs.1,2 These variations in clinical features and location resemble benign or infectious disease and can lead to delays in diagnosis.
Histopathologically, MCD lesions usually are ill-defined noncaseating granulomas with numerous multinucleated giant cells and lymphomononuclear cells located mostly in the dermis and occasionally extending into the subcutis. The cutaneous granulomata are similar to those present in the affected GI tract. Lymphocytes and plasma cells also are commonly present and eosinophils can be prominent.1,2,11 In some cases of MCD, granulomatous vasculitis of small- to medium-sized vessels can be found and is associated with dermal and subcutaneous granulomatous inflammation.8,11,12 Misago and Narisawa13 suggested that granulomatous vasculitis and panniculitis associated with CD is considered to be a rare subtype of MCD. Few cases of MCD presenting as granulomatous panniculitis have been described in the literature.14-16 Our patient presented with lesions that clinically resembled EN; however, the biopsy was more consistent with MCD. The Table summarizes the distinguishing clinical and histopathological features of MCD in our case and classic EN.
Although some authors believe that MCD is not related to CD activity, others assert that MCD lesions may parallel GI activity.1,2 Our patient was treated with systemic corticosteroids, oral metronidazole, and mesalamine to control the GI symptoms associated with CD. Four weeks after treatment, the GI symptoms and skin lesions improved simultaneously without any additional dermatologic treatment. We believe that MCD has the potential to serve as an early marker of the recurrence of CD and can help with the early diagnosis of CD aggravation, though an association between MCD and CD activity has not been confirmed.
Conclusion
We reported a case of MCD that was clinically reminiscent of unilateral EN and associated with GI disease activity. Physicians should be aware of the possibility of skin manifestations in CD, especially when erythematous nodular lesions are present on the leg.
Metastatic Crohn disease (MCD) is defined by the presence of cutaneous noncaseating granulomatous lesions that are noncontiguous with the gastrointestinal (GI) tract or fistulae.1 The clinical presentation of MCD is so variable that its diagnosis requires a high index of suspicion.1,2 In particular, the presence of erythematous tender nodules on the legs is easily mistaken for erythema nodosum (EN). Skin biopsy has an important role in confirming the diagnosis, as histopathological examination would reveal a noncaseating granuloma similar to those in the involved GI tract.2 Herein, we report a case of MCD on the right leg that was clinically reminiscent of unilateral EN.
Case Report
A 21-year-old woman presented to the dermatology department with 2 painful erythematous nodules on the lower right leg of 2 weeks’ duration. She also reported abdominal pain, diarrhea, and bloody stool. She had been diagnosed with Crohn disease (CD) 6 years prior that had been well controlled with systemic low-dose steroids (5–15 mg/d), metronidazole (750 mg/d), and intermittent mesalamine and antidiarrheal drugs. However, she had not taken her medication for several weeks on her own authority. Subsequently, the patient developed skin lesions, which were characterized by ill-defined erythematous nodules with tenderness on the right lower leg along with GI symptoms (Figure 1). Laboratory studies revealed anemia (hemoglobin, 9.9 g/dL [reference range, 12.0–16.0 g/dL]) and an elevated C-reactive protein level (4.3 mg/dL [reference range, 0–0.3 mg/dL]). Other routine laboratory findings were normal.
Histopathologically, a skin biopsy from the right ankle showed vague, ill-defined, noncaseating granulomas scattered in the deep dermis and lobules of the subcutis (Figure 2). The granulomas were composed of epithelioid cells and Langerhans-type giant cells. Lymphocytes and neutrophils also were present, but eosinophils were absent. Immunohistochemical staining revealed that the infiltrating cells were mostly CD4+ helper/inducer T cells intermixed with CD8+ suppressor/cytotoxic T cells. The CD4:CD8 ratio was approximately 2:1. Counts of CD20+ B cells were low. Epithelioid cells and giant cells were positive for CD68.
×20). The skin biopsy showed granulomas composed of epithelioid cells and multinucleated giant cells in the deep dermis and in the lobules of the subcutis (B)(H&E, original magnification ×200). Histopathologic features such as small vessel vasculitis characterized by a fibrin deposit in the small blood vessels and swelling of the endothelial cells as well as granulomatous perivasculitis with perivascular infiltration of the epithelioid cells were present (C)(H&E, original magnification ×200).
A colonoscopy was performed to evaluate the aggravation of CD. Multiple longitudinal ulcers were observed in the ileocecal valve area and from the transverse colon to the sigmoid colon (Figure 3A). Histopathologic findings from the colon showed mucosal ulceration and noncaseating granulomas with heavy infiltration of lymphocytes and plasma cells (Figure 3B). Staining for infectious microorganisms (eg, Ziehl-Neelsen, periodic acid–Schiff, Gram) was negative. A polymerase chain reaction performed on sections cut from the paraffin block of the skin biopsy was negative for Mycobacterium tuberculosis DNA.
Based on the clinical and histopathologic findings, the patient was diagnosed with MCD that was clinically reminiscent of unilateral EN. Four weeks after the initiation of therapy with systemic corticosteroids (25 mg/d), oral metronidazole (750 mg/d), and mesalamine (1200 mg/d) for CD, the skin lesions were completely resolved and the patient’s GI symptoms improved simultaneously.
Comment
Crohn disease is a chronic inflammatory granulomatous disease of the GI tract that often is associated with reactive cutaneous lesions including EN, pyoderma gangrenosum, necrotizing vasculitis, and epidermolysis bullosa acquisita. Of these, EN is the most common to appear in CD patients and has been reported to occur in 1% to 15% of patients.3-5 In particular, skin lesions on the leg presenting as tender erythematous nodules and patches are often diagnosed as EN, which is relatively common. In our case, we initially suspected EN due to the rare presentation of MCD and lack of specific clinical features; however, the skin biopsy revealed noncaseating granulomas in the mid to deep dermis and subcutis consistent with MCD.
Metastatic Crohn disease is a rare disease entity and is characterized by the presence of noncaseating granulomas of the skin at sites separated from the GI tract by normal tissue.1 Although its pathogenesis is unclear, it has been suggested that immune complexes deposited in the skin could be responsible for the granulomatous reactions.4 A T lymphocyte–mediated type IV hypersensitivity reaction also could be responsible.6,7 Because antimicrobial therapy can be curative for infection-related MCD, special histologic stains and/or tissue cultures can help to exclude an infectious etiology.8
Clinical presentations of MCD vary greatly, with observations such as single or multiple erythematous swellings, papules, plaques, nodules, abscesses, and ulcers.1,2 The relationship between these clinical presentations and the intestinal activity of CD still is unknown; in some cases, however, the metastatic granulomatous lesions and the bowel disease show comparable severity.2,9,10 In a review of the literature, MCD was generally reported to present in the genital area in children. In adults, lesions most frequently present in the genital area, followed by ulcers on the arms and legs.1,2 These variations in clinical features and location resemble benign or infectious disease and can lead to delays in diagnosis.
Histopathologically, MCD lesions usually are ill-defined noncaseating granulomas with numerous multinucleated giant cells and lymphomononuclear cells located mostly in the dermis and occasionally extending into the subcutis. The cutaneous granulomata are similar to those present in the affected GI tract. Lymphocytes and plasma cells also are commonly present and eosinophils can be prominent.1,2,11 In some cases of MCD, granulomatous vasculitis of small- to medium-sized vessels can be found and is associated with dermal and subcutaneous granulomatous inflammation.8,11,12 Misago and Narisawa13 suggested that granulomatous vasculitis and panniculitis associated with CD is considered to be a rare subtype of MCD. Few cases of MCD presenting as granulomatous panniculitis have been described in the literature.14-16 Our patient presented with lesions that clinically resembled EN; however, the biopsy was more consistent with MCD. The Table summarizes the distinguishing clinical and histopathological features of MCD in our case and classic EN.
Although some authors believe that MCD is not related to CD activity, others assert that MCD lesions may parallel GI activity.1,2 Our patient was treated with systemic corticosteroids, oral metronidazole, and mesalamine to control the GI symptoms associated with CD. Four weeks after treatment, the GI symptoms and skin lesions improved simultaneously without any additional dermatologic treatment. We believe that MCD has the potential to serve as an early marker of the recurrence of CD and can help with the early diagnosis of CD aggravation, though an association between MCD and CD activity has not been confirmed.
Conclusion
We reported a case of MCD that was clinically reminiscent of unilateral EN and associated with GI disease activity. Physicians should be aware of the possibility of skin manifestations in CD, especially when erythematous nodular lesions are present on the leg.
- Calonje E, Brenn T, Lazar AJ, et al. Mckee’s Pathology of the Skin: With Clinical Correlations. 4th ed. Philadelphia, PA: Saunders Elsevier; 2012.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn’s disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Sonia F, Richard SB. Inflammatory bowel disease. In: Kasper DL, Braunwald E, Fauci AS, et al, eds. Harrison’s Principles of Internal Medicine. 16th ed. New York, NY: McGraw-Hill; 2005:1776-1789.
- Burgdorf W. Cutaneous manifestations of Crohn’s disease. J Am Acad Dermatol. 1981;5:689-695.
- Crowson AN, Nuovo GJ, Mihm MC Jr, et al. Cutaneous manifestations of Crohn’s disease, its spectrum, and its pathogenesis: intracellular consensus bacterial 16S rRNA is associated with the gastrointestinal but not the cutaneous manifestations of Crohn’s disease. Hum Pathol. 2003;34:1185-1192.
- Tatnall FM, Dodd HJ, Sarkany I. Crohn’s disease with metastatic cutaneous involvement and granulomatous cheilitis. J R Soc Med. 1987;80:49-51.
- Shum DT, Guenther L. Metastatic Crohn’s disease. case report and review of the literature. Arch Dermatol. 1990;126:645-648.
- Emanuel PO, Phelps RG. Metastatic Crohn’s disease: a histopathologic study of 12 cases. J Cutan Pathol. 2008;35:457-461.
- Chalvardjian A, Nethercott JR. Cutaneous granulomatous vasculitis associated with Crohn’s disease. Cutis. 1982;30:645-655.
- Lebwohl M, Fleischmajer R, Janowitz H, et al. Metastatic Crohn’s disease. J Am Acad Dermatol. 1984;10:33-38.
- Sabat M, Leulmo J, Saez A. Cutaneous granulomatous vasculitis in metastatic Crohn’s disease. J Eur Acad Dermatol Venereol. 2005;19:652-653.
- Burns AM, Walsh N, Green PJ. Granulomatous vasculitis in Crohn’s disease: a clinicopathologic correlate of two unusual cases. J Cutan Pathol. 2010;37:1077-1083.
- Misago N, Narisawa Y. Erythema induratum (nodular vasculitis) associated with Crohn’s disease: a rare type of metastatic Crohn’s disease. Am J Dermatopathol. 2012;34:325-329.
- Liebermann TR, Greene JF Jr. Transient subcutaneous granulomatosis of the upper extremities in Crohn’s disease. Am J Gastroenterol. 1979;72:89-91.
- Levine N, Bangert J. Cutaneous granulomatosis in Crohn’s disease. Arch Dermatol. 1982;118:1006-1009.
- Hackzell-Bradley M, Hedblad MA, Stephansson EA. Metastatic Crohn’s disease. report of 3 cases with special reference to histopathologic findings. Arch Dermatol. 1996;132:928-932.
- Calonje E, Brenn T, Lazar AJ, et al. Mckee’s Pathology of the Skin: With Clinical Correlations. 4th ed. Philadelphia, PA: Saunders Elsevier; 2012.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn’s disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Sonia F, Richard SB. Inflammatory bowel disease. In: Kasper DL, Braunwald E, Fauci AS, et al, eds. Harrison’s Principles of Internal Medicine. 16th ed. New York, NY: McGraw-Hill; 2005:1776-1789.
- Burgdorf W. Cutaneous manifestations of Crohn’s disease. J Am Acad Dermatol. 1981;5:689-695.
- Crowson AN, Nuovo GJ, Mihm MC Jr, et al. Cutaneous manifestations of Crohn’s disease, its spectrum, and its pathogenesis: intracellular consensus bacterial 16S rRNA is associated with the gastrointestinal but not the cutaneous manifestations of Crohn’s disease. Hum Pathol. 2003;34:1185-1192.
- Tatnall FM, Dodd HJ, Sarkany I. Crohn’s disease with metastatic cutaneous involvement and granulomatous cheilitis. J R Soc Med. 1987;80:49-51.
- Shum DT, Guenther L. Metastatic Crohn’s disease. case report and review of the literature. Arch Dermatol. 1990;126:645-648.
- Emanuel PO, Phelps RG. Metastatic Crohn’s disease: a histopathologic study of 12 cases. J Cutan Pathol. 2008;35:457-461.
- Chalvardjian A, Nethercott JR. Cutaneous granulomatous vasculitis associated with Crohn’s disease. Cutis. 1982;30:645-655.
- Lebwohl M, Fleischmajer R, Janowitz H, et al. Metastatic Crohn’s disease. J Am Acad Dermatol. 1984;10:33-38.
- Sabat M, Leulmo J, Saez A. Cutaneous granulomatous vasculitis in metastatic Crohn’s disease. J Eur Acad Dermatol Venereol. 2005;19:652-653.
- Burns AM, Walsh N, Green PJ. Granulomatous vasculitis in Crohn’s disease: a clinicopathologic correlate of two unusual cases. J Cutan Pathol. 2010;37:1077-1083.
- Misago N, Narisawa Y. Erythema induratum (nodular vasculitis) associated with Crohn’s disease: a rare type of metastatic Crohn’s disease. Am J Dermatopathol. 2012;34:325-329.
- Liebermann TR, Greene JF Jr. Transient subcutaneous granulomatosis of the upper extremities in Crohn’s disease. Am J Gastroenterol. 1979;72:89-91.
- Levine N, Bangert J. Cutaneous granulomatosis in Crohn’s disease. Arch Dermatol. 1982;118:1006-1009.
- Hackzell-Bradley M, Hedblad MA, Stephansson EA. Metastatic Crohn’s disease. report of 3 cases with special reference to histopathologic findings. Arch Dermatol. 1996;132:928-932.
Practice Points
- Metastatic Crohn disease (MCD) may be an initial sign indicating the aggravation of intestinal Crohn disease (CD).
- Metastatic Crohn disease on the legs could be clinically reminiscent of erythema nodosum (EN).
- Physicians should be aware of the possibility of MCD when encountering EN-like lesions on the legs in a CD patient.
Desmoplastic Hairless Hypopigmented Nevus
To the Editor:
We report 2 cases of desmoplastic hairless hypopigmented nevi (DHHN), which are giant congenital melanocytic nevi (GCMN) that show sclerosis with progressive loss of pigment and hair. These changes in GCMN could be considered signs of regression.
A 6-year-old boy presented in the dermatology department with an asymptomatic skin lesion on the right buttock since birth. The parents claimed that the lesion was darkly pigmented at birth and gradually increased in size, with progressive reduction in color in the last 2 years. Physical examination revealed a 10×6-cm, well-defined, raised plaque on the upper medial side of the right buttock (Figure 1). The plaque was firm with a shiny smooth surface and was devoid of hair. The surface was flesh colored with scattered pigmented spots. A punch biopsy of the lesion showed increased melanin content in the basal cell layer. The upper dermis showed small nests of epithelioid nevus cells, most of them containing melanin pigment (Figure 2). In the lower two-thirds of the dermis, nevus cells were both epithelioid and spindle shaped and were arranged in between thick sclerotic collagen bundles with an increased number of fibroblasts. There was a marked reduction in the number of hair follicles. Immunohistochemical staining results were S-100 positive and CD34 negative.
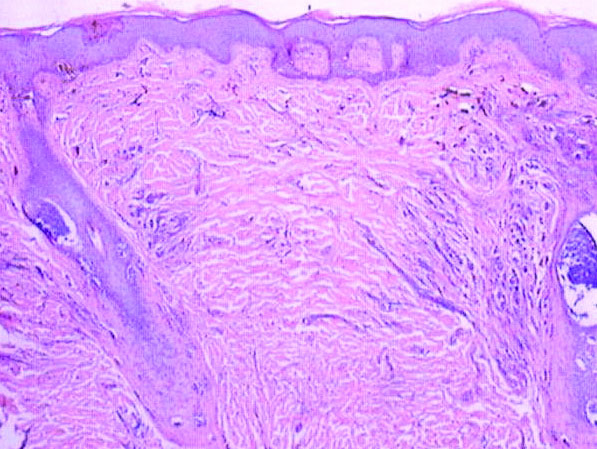
A 5-year-old boy presented in the dermatology department with a large hairy GCMN covering most of the trunk since birth. In the last 1.5 years the parents noted gradual fading of color, decreased hair density, and increased induration of the nevus. Physical examination revealed a large plaque covering the anterior aspect of the trunk (Figure 3) and the back extending down to the buttocks. The lesion formed large skin folds that were more pronounced on the back. The nevus was darkly pigmented with large areas of lighter color that were indurated, devoid of hair, and showed small spots of dark pigmentation. A punch biopsy from the lesion showed small nests of nevus cells in the upper part of the reticular dermis. In the lower part of the dermis, nevus cells were arranged in single units in between thick collagen bundles.
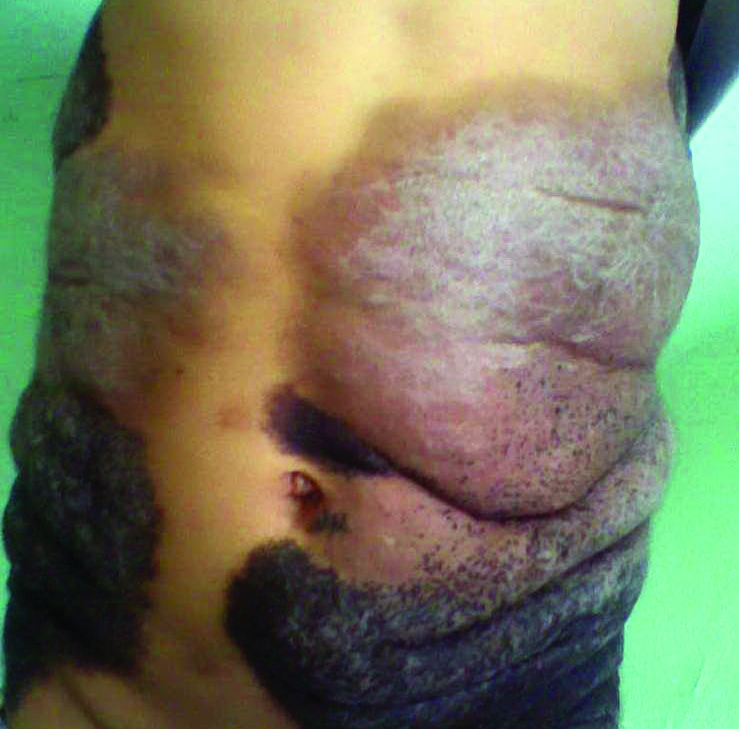
In 2003, Ruiz-Maldonado et al1 described 4 cases of GCMN that showed progressive loss of pigmentation, sclerosis, and hair loss. They proposed the term desmoplastic hairless hypopigmented nevus for their cases and considered it as a variant of GCMN.1 Prior to these reported cases, 2 similar cases were described. The first was a report by Hogan et al2 in 1988 of a 7-month-old girl with a GCMN involving the occipital area and the upper back that became indurated and ulcerated with progressive involution that led to complete disappearance of the nevus. The second was a report by Pattee et al3 in 2001 of a newborn with a GCMN located on the trunk with progressive sclerodermiform reaction. After surgical excision of the nevus, the sclerotic margin disappeared.3
Following the report by Ruiz-Maldonaldo et al,1 5 more cases of DHHN were described.4-8 All cases of DHHN share the same clinical and histopathological features. The clinical features include a GCMN present since birth with progressive sclerosis over time and loss of both pigmentation and hair. Histologically, DHHN shows the typical changes of a congenital melanocytic nevus with decreased numbers of nevus cells, thick sclerotic collagen bundles of the reticular dermis, increased number of fibroblasts, and decreased number of hair follicles. The progressive reduction in the number of nevus cells in melanocytic nevi is considered a sign of regression. Spontaneous regression was rarely described in GCMN, and all the reported cases of regression were associated with desmoplasia.4 Desmoplasia is thought to be induced by either melanocytes that function as adaptive fibroblasts or by fibroblasts themselves, as fibroblasts can show multifunctional differentiation capabilities.9 The direct correlation between the increased induration of DHHN and pigment depletion supports the former hypothesis. The absence of inflammatory cells within the sections of DHHN lesions is against the possibility of an immune-mediated reaction as a cause for the clinical and histological changes seen in this rare form of GCMN. The progressive hair loss in DHHN may be explained by the progressive fibrotic changes in the reticular dermis that affect the blood supply to follicles, leading to atrophy or even absence of the follicles. The progressive reduction in the number of nevus cells in DHHN reduces the potential for malignant transformation and hence following a watchful waiting strategy is a reasonable way to manage these nevi.
We present 2 patients with DHHN, which is a rare form of GCMN that shows signs of regression. The cause of these changes is still unclear.
- Ruiz-Maldonado R, Orozco-Covarrubias L, Ridaura-Sanz C, et al. Desmoplastic hairless hypopigmented naevus: a variant of giant congenital melanocytic naevus. Br J Dermatol. 2003;148:1253-1257.
- Hogan DJ, Murphy F, Bremner RM. Spontaneous resolution of a giant congenital melanocytic nevus. Pediatr Dermatol. 1988;5:170-172.
- Pattee SF, Hansen RC, Bangert JL, et al. Giant congenital nevus with progressive sclerodermoid reaction in a newborn. Pediatr Dermatol. 2001;18:321-324.
- Boente MC, Asial RA. Desmoplastic hairless hypopigmented nevus (DHHN). a distinct variant of giant melanocytic nevus. Eur J Dermatol. 2005;15:451-453.
- Bushby SA, Rajan NJ, Shehade SA. Spontaneous resolution of a giant melanocytic naevus involving a desmoplastic process. Br J Dermatol. 2005;153(suppl 1):13-19.
- Martin JM, Jorda E, Monteagudo C, et al. Desmoplastic giant congenital nevus with progressive depigmentation. J Am Acad Dermatol. 2007;56(suppl 2):S10-S14.
- Hermandez-Martin A, Torrelo A, Echevarria C, et al. Ulcerated sclerotic giant congenital melanocytic naevus: case report and review of the literature. Clin Exp Dermatol. 2007;32:529-532.
- Werner B, Carvalho VO, Nacif SB, et al. Desmoplastic hypopigmented hairless nevus: a variant with progressive depigmentation, induration and overgrowth [published online May 16, 2011]. Pediatr Dermatol. 2012;29:336-340.
- Fearns C, Dowdle EB. The desmoplastic response: induction of collagen synthesis by melanoma cells in vitro. Int J Cancer. 1992;50:621-627.
To the Editor:
We report 2 cases of desmoplastic hairless hypopigmented nevi (DHHN), which are giant congenital melanocytic nevi (GCMN) that show sclerosis with progressive loss of pigment and hair. These changes in GCMN could be considered signs of regression.
A 6-year-old boy presented in the dermatology department with an asymptomatic skin lesion on the right buttock since birth. The parents claimed that the lesion was darkly pigmented at birth and gradually increased in size, with progressive reduction in color in the last 2 years. Physical examination revealed a 10×6-cm, well-defined, raised plaque on the upper medial side of the right buttock (Figure 1). The plaque was firm with a shiny smooth surface and was devoid of hair. The surface was flesh colored with scattered pigmented spots. A punch biopsy of the lesion showed increased melanin content in the basal cell layer. The upper dermis showed small nests of epithelioid nevus cells, most of them containing melanin pigment (Figure 2). In the lower two-thirds of the dermis, nevus cells were both epithelioid and spindle shaped and were arranged in between thick sclerotic collagen bundles with an increased number of fibroblasts. There was a marked reduction in the number of hair follicles. Immunohistochemical staining results were S-100 positive and CD34 negative.
A 5-year-old boy presented in the dermatology department with a large hairy GCMN covering most of the trunk since birth. In the last 1.5 years the parents noted gradual fading of color, decreased hair density, and increased induration of the nevus. Physical examination revealed a large plaque covering the anterior aspect of the trunk (Figure 3) and the back extending down to the buttocks. The lesion formed large skin folds that were more pronounced on the back. The nevus was darkly pigmented with large areas of lighter color that were indurated, devoid of hair, and showed small spots of dark pigmentation. A punch biopsy from the lesion showed small nests of nevus cells in the upper part of the reticular dermis. In the lower part of the dermis, nevus cells were arranged in single units in between thick collagen bundles.
In 2003, Ruiz-Maldonado et al1 described 4 cases of GCMN that showed progressive loss of pigmentation, sclerosis, and hair loss. They proposed the term desmoplastic hairless hypopigmented nevus for their cases and considered it as a variant of GCMN.1 Prior to these reported cases, 2 similar cases were described. The first was a report by Hogan et al2 in 1988 of a 7-month-old girl with a GCMN involving the occipital area and the upper back that became indurated and ulcerated with progressive involution that led to complete disappearance of the nevus. The second was a report by Pattee et al3 in 2001 of a newborn with a GCMN located on the trunk with progressive sclerodermiform reaction. After surgical excision of the nevus, the sclerotic margin disappeared.3
Following the report by Ruiz-Maldonaldo et al,1 5 more cases of DHHN were described.4-8 All cases of DHHN share the same clinical and histopathological features. The clinical features include a GCMN present since birth with progressive sclerosis over time and loss of both pigmentation and hair. Histologically, DHHN shows the typical changes of a congenital melanocytic nevus with decreased numbers of nevus cells, thick sclerotic collagen bundles of the reticular dermis, increased number of fibroblasts, and decreased number of hair follicles. The progressive reduction in the number of nevus cells in melanocytic nevi is considered a sign of regression. Spontaneous regression was rarely described in GCMN, and all the reported cases of regression were associated with desmoplasia.4 Desmoplasia is thought to be induced by either melanocytes that function as adaptive fibroblasts or by fibroblasts themselves, as fibroblasts can show multifunctional differentiation capabilities.9 The direct correlation between the increased induration of DHHN and pigment depletion supports the former hypothesis. The absence of inflammatory cells within the sections of DHHN lesions is against the possibility of an immune-mediated reaction as a cause for the clinical and histological changes seen in this rare form of GCMN. The progressive hair loss in DHHN may be explained by the progressive fibrotic changes in the reticular dermis that affect the blood supply to follicles, leading to atrophy or even absence of the follicles. The progressive reduction in the number of nevus cells in DHHN reduces the potential for malignant transformation and hence following a watchful waiting strategy is a reasonable way to manage these nevi.
We present 2 patients with DHHN, which is a rare form of GCMN that shows signs of regression. The cause of these changes is still unclear.
To the Editor:
We report 2 cases of desmoplastic hairless hypopigmented nevi (DHHN), which are giant congenital melanocytic nevi (GCMN) that show sclerosis with progressive loss of pigment and hair. These changes in GCMN could be considered signs of regression.
A 6-year-old boy presented in the dermatology department with an asymptomatic skin lesion on the right buttock since birth. The parents claimed that the lesion was darkly pigmented at birth and gradually increased in size, with progressive reduction in color in the last 2 years. Physical examination revealed a 10×6-cm, well-defined, raised plaque on the upper medial side of the right buttock (Figure 1). The plaque was firm with a shiny smooth surface and was devoid of hair. The surface was flesh colored with scattered pigmented spots. A punch biopsy of the lesion showed increased melanin content in the basal cell layer. The upper dermis showed small nests of epithelioid nevus cells, most of them containing melanin pigment (Figure 2). In the lower two-thirds of the dermis, nevus cells were both epithelioid and spindle shaped and were arranged in between thick sclerotic collagen bundles with an increased number of fibroblasts. There was a marked reduction in the number of hair follicles. Immunohistochemical staining results were S-100 positive and CD34 negative.
A 5-year-old boy presented in the dermatology department with a large hairy GCMN covering most of the trunk since birth. In the last 1.5 years the parents noted gradual fading of color, decreased hair density, and increased induration of the nevus. Physical examination revealed a large plaque covering the anterior aspect of the trunk (Figure 3) and the back extending down to the buttocks. The lesion formed large skin folds that were more pronounced on the back. The nevus was darkly pigmented with large areas of lighter color that were indurated, devoid of hair, and showed small spots of dark pigmentation. A punch biopsy from the lesion showed small nests of nevus cells in the upper part of the reticular dermis. In the lower part of the dermis, nevus cells were arranged in single units in between thick collagen bundles.
In 2003, Ruiz-Maldonado et al1 described 4 cases of GCMN that showed progressive loss of pigmentation, sclerosis, and hair loss. They proposed the term desmoplastic hairless hypopigmented nevus for their cases and considered it as a variant of GCMN.1 Prior to these reported cases, 2 similar cases were described. The first was a report by Hogan et al2 in 1988 of a 7-month-old girl with a GCMN involving the occipital area and the upper back that became indurated and ulcerated with progressive involution that led to complete disappearance of the nevus. The second was a report by Pattee et al3 in 2001 of a newborn with a GCMN located on the trunk with progressive sclerodermiform reaction. After surgical excision of the nevus, the sclerotic margin disappeared.3
Following the report by Ruiz-Maldonaldo et al,1 5 more cases of DHHN were described.4-8 All cases of DHHN share the same clinical and histopathological features. The clinical features include a GCMN present since birth with progressive sclerosis over time and loss of both pigmentation and hair. Histologically, DHHN shows the typical changes of a congenital melanocytic nevus with decreased numbers of nevus cells, thick sclerotic collagen bundles of the reticular dermis, increased number of fibroblasts, and decreased number of hair follicles. The progressive reduction in the number of nevus cells in melanocytic nevi is considered a sign of regression. Spontaneous regression was rarely described in GCMN, and all the reported cases of regression were associated with desmoplasia.4 Desmoplasia is thought to be induced by either melanocytes that function as adaptive fibroblasts or by fibroblasts themselves, as fibroblasts can show multifunctional differentiation capabilities.9 The direct correlation between the increased induration of DHHN and pigment depletion supports the former hypothesis. The absence of inflammatory cells within the sections of DHHN lesions is against the possibility of an immune-mediated reaction as a cause for the clinical and histological changes seen in this rare form of GCMN. The progressive hair loss in DHHN may be explained by the progressive fibrotic changes in the reticular dermis that affect the blood supply to follicles, leading to atrophy or even absence of the follicles. The progressive reduction in the number of nevus cells in DHHN reduces the potential for malignant transformation and hence following a watchful waiting strategy is a reasonable way to manage these nevi.
We present 2 patients with DHHN, which is a rare form of GCMN that shows signs of regression. The cause of these changes is still unclear.
- Ruiz-Maldonado R, Orozco-Covarrubias L, Ridaura-Sanz C, et al. Desmoplastic hairless hypopigmented naevus: a variant of giant congenital melanocytic naevus. Br J Dermatol. 2003;148:1253-1257.
- Hogan DJ, Murphy F, Bremner RM. Spontaneous resolution of a giant congenital melanocytic nevus. Pediatr Dermatol. 1988;5:170-172.
- Pattee SF, Hansen RC, Bangert JL, et al. Giant congenital nevus with progressive sclerodermoid reaction in a newborn. Pediatr Dermatol. 2001;18:321-324.
- Boente MC, Asial RA. Desmoplastic hairless hypopigmented nevus (DHHN). a distinct variant of giant melanocytic nevus. Eur J Dermatol. 2005;15:451-453.
- Bushby SA, Rajan NJ, Shehade SA. Spontaneous resolution of a giant melanocytic naevus involving a desmoplastic process. Br J Dermatol. 2005;153(suppl 1):13-19.
- Martin JM, Jorda E, Monteagudo C, et al. Desmoplastic giant congenital nevus with progressive depigmentation. J Am Acad Dermatol. 2007;56(suppl 2):S10-S14.
- Hermandez-Martin A, Torrelo A, Echevarria C, et al. Ulcerated sclerotic giant congenital melanocytic naevus: case report and review of the literature. Clin Exp Dermatol. 2007;32:529-532.
- Werner B, Carvalho VO, Nacif SB, et al. Desmoplastic hypopigmented hairless nevus: a variant with progressive depigmentation, induration and overgrowth [published online May 16, 2011]. Pediatr Dermatol. 2012;29:336-340.
- Fearns C, Dowdle EB. The desmoplastic response: induction of collagen synthesis by melanoma cells in vitro. Int J Cancer. 1992;50:621-627.
- Ruiz-Maldonado R, Orozco-Covarrubias L, Ridaura-Sanz C, et al. Desmoplastic hairless hypopigmented naevus: a variant of giant congenital melanocytic naevus. Br J Dermatol. 2003;148:1253-1257.
- Hogan DJ, Murphy F, Bremner RM. Spontaneous resolution of a giant congenital melanocytic nevus. Pediatr Dermatol. 1988;5:170-172.
- Pattee SF, Hansen RC, Bangert JL, et al. Giant congenital nevus with progressive sclerodermoid reaction in a newborn. Pediatr Dermatol. 2001;18:321-324.
- Boente MC, Asial RA. Desmoplastic hairless hypopigmented nevus (DHHN). a distinct variant of giant melanocytic nevus. Eur J Dermatol. 2005;15:451-453.
- Bushby SA, Rajan NJ, Shehade SA. Spontaneous resolution of a giant melanocytic naevus involving a desmoplastic process. Br J Dermatol. 2005;153(suppl 1):13-19.
- Martin JM, Jorda E, Monteagudo C, et al. Desmoplastic giant congenital nevus with progressive depigmentation. J Am Acad Dermatol. 2007;56(suppl 2):S10-S14.
- Hermandez-Martin A, Torrelo A, Echevarria C, et al. Ulcerated sclerotic giant congenital melanocytic naevus: case report and review of the literature. Clin Exp Dermatol. 2007;32:529-532.
- Werner B, Carvalho VO, Nacif SB, et al. Desmoplastic hypopigmented hairless nevus: a variant with progressive depigmentation, induration and overgrowth [published online May 16, 2011]. Pediatr Dermatol. 2012;29:336-340.
- Fearns C, Dowdle EB. The desmoplastic response: induction of collagen synthesis by melanoma cells in vitro. Int J Cancer. 1992;50:621-627.
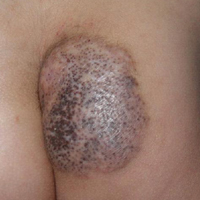
Painful Ulcerations Above the Malleoli
The Diagnosis: Livedoid Vasculopathy
Livedoid vasculopathy (LV) is a rare cutaneous disorder that most commonly affects the lower legs. It has an estimated incidence of 1 case per 100,000 per year and predominantly affects women.1 The disease pathogenesis is not fully understood but is thought to involve thrombosis and occlusion of dermal vessels resulting in tissue hypoxia.2 Both inherited and acquired thrombophilic conditions frequently are seen in patients with LV.3,4 Livedoid vasculopathy also has been described as idiopathic5 and is associated with immune complex deposition.6 However, the number of cases of idiopathic LV may be overestimated; as technological advancements to detect coagulation abnormalities improve, it is hypothesized that this entity will be identified less often.2,4
Livedoid vasculopathy has been described in the literature using the term PPURPLE (painful purpuric ulcers with reticular pattern of lower extremities).7 The triad of livedo racemosa, recurrent painful ulcerations, and residual healing with atrophie blanche characterizes the clinical manifestations of LV; however, all 3 characteristics do not need to appear simultaneously for a diagnosis to be made. The condition has a chronic course with spontaneous remissions and exacerbations. Episodic ulcerations occur, especially in the summertime, and heal slowly, leaving behind atrophic, porcelain white, stellate-shaped scars called atrophie blanche. Livedo racemosa also may be seen in Sneddon syndrome; however, these patients experience neurologic symptoms secondary to cerebrovascular occlusion. In contrast to livedo racemosa, acquired livedo reticularis represents a physiologic hypoperfusion pattern that occurs in response to cold exposure.8 A localized sharp pain, known as angina cutis, typically precedes the clinical symptom of painful ulcerations.9 Atrophie blanche once was thought to be specific to LV but has been seen in other diseases such as systemic lupus erythematosus and chronic venous insufficiency.2
The diagnosis of LV is based on identification of characteristic clinical features and skin biopsy. In almost all biopsy specimens, histopathology reveals fibrinoid occlusion of vessels in the superficial and mid dermis.4 Other findings may include epidermal necrosis and vessel wall hyalinization and infarction2 (Figure). Because LV is commonly misdiagnosed as vasculitis, the absence of hallmark features of vasculitis such as neutrophilic infiltrate of blood vessel walls and fibrinoid necrosis suggest the diagnosis. Extensive laboratory evaluation for inherited and acquired coagulation abnormalities should be performed.
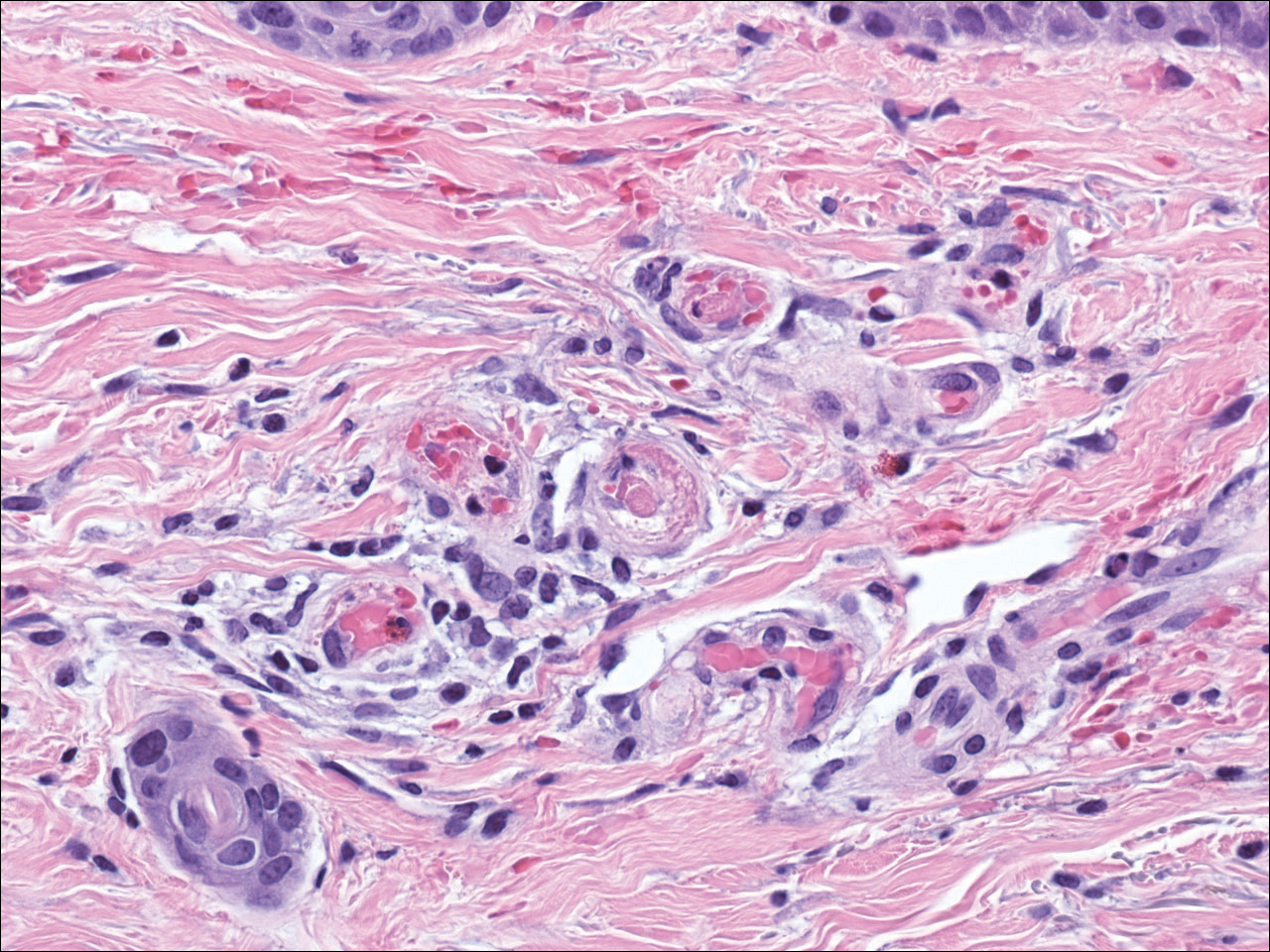
Treatment of LV is difficult, as there is currently no consensus on optimal therapy. The mainstay of therapy is to reduce pain, prevent infection, and reduce ulceration and development of atrophie blanche. Underlying causes should be identified and appropriately treated. Because the primary pathogenesis of LV is considered to be a hypercoagulable state, first-line treatment often includes therapies to enhance blood flow and prevent thrombosis such as smoking cessation, antiplatelet therapy, and pentoxifylline. Vasodilating agents, anti-inflammatory agents, anticoagulation, and fibrinolytic therapy also have been used with varying degrees of success.7
- Fritsch P, Zelger B. Livedo vasculitis [in German]. Hautarzt. 1995;46:215-224; quiz 222-223.
- Kerk N, Goerge T. Livedoid vasculopathy—a thrombotic disease. Vasa. 2013;42:317-322.
- Stevanovic DV. Atrophie blanche. a sign of dermal blood occlusion. Arch Dermatol. 1974;109:858-862.
- Hairston BR, Davis MD, Pittelkow MR, et al. Livedoid vasculopathy: further evidence for procoagulant pathogenesis. Arch Dermatol. 2006;142:1413-1418.
- Shornick JK, Nicholes BK, Bergstresser PR, et al. Idiopathic atrophie blanche. J Am Acad Dermatol. 1983;8:792-798.
- Feldaker M, Hines EA Jr, Kierland RR. Livedo reticularis with ulcerations. Circulation. 1956;13:196-216.
- Callen JP. Livedoid vasculopathy: what it is and how the patient should be evaluated and treated. Arch Dermatol. 2006;142:1481-1482.
- Copeman PW. Livedo reticularis. signs in the skin of disturbance of blood viscosity and of blood flow. Br J Dermatol. 1975;93:519-529.
- Goerge T. Livedoid vasculopathy. pathogenesis, diagnosis and treatment of cutaneous infarction [in German]. Hautarzt. 2011;62:627-634; quiz 635.
The Diagnosis: Livedoid Vasculopathy
Livedoid vasculopathy (LV) is a rare cutaneous disorder that most commonly affects the lower legs. It has an estimated incidence of 1 case per 100,000 per year and predominantly affects women.1 The disease pathogenesis is not fully understood but is thought to involve thrombosis and occlusion of dermal vessels resulting in tissue hypoxia.2 Both inherited and acquired thrombophilic conditions frequently are seen in patients with LV.3,4 Livedoid vasculopathy also has been described as idiopathic5 and is associated with immune complex deposition.6 However, the number of cases of idiopathic LV may be overestimated; as technological advancements to detect coagulation abnormalities improve, it is hypothesized that this entity will be identified less often.2,4
Livedoid vasculopathy has been described in the literature using the term PPURPLE (painful purpuric ulcers with reticular pattern of lower extremities).7 The triad of livedo racemosa, recurrent painful ulcerations, and residual healing with atrophie blanche characterizes the clinical manifestations of LV; however, all 3 characteristics do not need to appear simultaneously for a diagnosis to be made. The condition has a chronic course with spontaneous remissions and exacerbations. Episodic ulcerations occur, especially in the summertime, and heal slowly, leaving behind atrophic, porcelain white, stellate-shaped scars called atrophie blanche. Livedo racemosa also may be seen in Sneddon syndrome; however, these patients experience neurologic symptoms secondary to cerebrovascular occlusion. In contrast to livedo racemosa, acquired livedo reticularis represents a physiologic hypoperfusion pattern that occurs in response to cold exposure.8 A localized sharp pain, known as angina cutis, typically precedes the clinical symptom of painful ulcerations.9 Atrophie blanche once was thought to be specific to LV but has been seen in other diseases such as systemic lupus erythematosus and chronic venous insufficiency.2
The diagnosis of LV is based on identification of characteristic clinical features and skin biopsy. In almost all biopsy specimens, histopathology reveals fibrinoid occlusion of vessels in the superficial and mid dermis.4 Other findings may include epidermal necrosis and vessel wall hyalinization and infarction2 (Figure). Because LV is commonly misdiagnosed as vasculitis, the absence of hallmark features of vasculitis such as neutrophilic infiltrate of blood vessel walls and fibrinoid necrosis suggest the diagnosis. Extensive laboratory evaluation for inherited and acquired coagulation abnormalities should be performed.
Treatment of LV is difficult, as there is currently no consensus on optimal therapy. The mainstay of therapy is to reduce pain, prevent infection, and reduce ulceration and development of atrophie blanche. Underlying causes should be identified and appropriately treated. Because the primary pathogenesis of LV is considered to be a hypercoagulable state, first-line treatment often includes therapies to enhance blood flow and prevent thrombosis such as smoking cessation, antiplatelet therapy, and pentoxifylline. Vasodilating agents, anti-inflammatory agents, anticoagulation, and fibrinolytic therapy also have been used with varying degrees of success.7
The Diagnosis: Livedoid Vasculopathy
Livedoid vasculopathy (LV) is a rare cutaneous disorder that most commonly affects the lower legs. It has an estimated incidence of 1 case per 100,000 per year and predominantly affects women.1 The disease pathogenesis is not fully understood but is thought to involve thrombosis and occlusion of dermal vessels resulting in tissue hypoxia.2 Both inherited and acquired thrombophilic conditions frequently are seen in patients with LV.3,4 Livedoid vasculopathy also has been described as idiopathic5 and is associated with immune complex deposition.6 However, the number of cases of idiopathic LV may be overestimated; as technological advancements to detect coagulation abnormalities improve, it is hypothesized that this entity will be identified less often.2,4
Livedoid vasculopathy has been described in the literature using the term PPURPLE (painful purpuric ulcers with reticular pattern of lower extremities).7 The triad of livedo racemosa, recurrent painful ulcerations, and residual healing with atrophie blanche characterizes the clinical manifestations of LV; however, all 3 characteristics do not need to appear simultaneously for a diagnosis to be made. The condition has a chronic course with spontaneous remissions and exacerbations. Episodic ulcerations occur, especially in the summertime, and heal slowly, leaving behind atrophic, porcelain white, stellate-shaped scars called atrophie blanche. Livedo racemosa also may be seen in Sneddon syndrome; however, these patients experience neurologic symptoms secondary to cerebrovascular occlusion. In contrast to livedo racemosa, acquired livedo reticularis represents a physiologic hypoperfusion pattern that occurs in response to cold exposure.8 A localized sharp pain, known as angina cutis, typically precedes the clinical symptom of painful ulcerations.9 Atrophie blanche once was thought to be specific to LV but has been seen in other diseases such as systemic lupus erythematosus and chronic venous insufficiency.2
The diagnosis of LV is based on identification of characteristic clinical features and skin biopsy. In almost all biopsy specimens, histopathology reveals fibrinoid occlusion of vessels in the superficial and mid dermis.4 Other findings may include epidermal necrosis and vessel wall hyalinization and infarction2 (Figure). Because LV is commonly misdiagnosed as vasculitis, the absence of hallmark features of vasculitis such as neutrophilic infiltrate of blood vessel walls and fibrinoid necrosis suggest the diagnosis. Extensive laboratory evaluation for inherited and acquired coagulation abnormalities should be performed.
Treatment of LV is difficult, as there is currently no consensus on optimal therapy. The mainstay of therapy is to reduce pain, prevent infection, and reduce ulceration and development of atrophie blanche. Underlying causes should be identified and appropriately treated. Because the primary pathogenesis of LV is considered to be a hypercoagulable state, first-line treatment often includes therapies to enhance blood flow and prevent thrombosis such as smoking cessation, antiplatelet therapy, and pentoxifylline. Vasodilating agents, anti-inflammatory agents, anticoagulation, and fibrinolytic therapy also have been used with varying degrees of success.7
- Fritsch P, Zelger B. Livedo vasculitis [in German]. Hautarzt. 1995;46:215-224; quiz 222-223.
- Kerk N, Goerge T. Livedoid vasculopathy—a thrombotic disease. Vasa. 2013;42:317-322.
- Stevanovic DV. Atrophie blanche. a sign of dermal blood occlusion. Arch Dermatol. 1974;109:858-862.
- Hairston BR, Davis MD, Pittelkow MR, et al. Livedoid vasculopathy: further evidence for procoagulant pathogenesis. Arch Dermatol. 2006;142:1413-1418.
- Shornick JK, Nicholes BK, Bergstresser PR, et al. Idiopathic atrophie blanche. J Am Acad Dermatol. 1983;8:792-798.
- Feldaker M, Hines EA Jr, Kierland RR. Livedo reticularis with ulcerations. Circulation. 1956;13:196-216.
- Callen JP. Livedoid vasculopathy: what it is and how the patient should be evaluated and treated. Arch Dermatol. 2006;142:1481-1482.
- Copeman PW. Livedo reticularis. signs in the skin of disturbance of blood viscosity and of blood flow. Br J Dermatol. 1975;93:519-529.
- Goerge T. Livedoid vasculopathy. pathogenesis, diagnosis and treatment of cutaneous infarction [in German]. Hautarzt. 2011;62:627-634; quiz 635.
- Fritsch P, Zelger B. Livedo vasculitis [in German]. Hautarzt. 1995;46:215-224; quiz 222-223.
- Kerk N, Goerge T. Livedoid vasculopathy—a thrombotic disease. Vasa. 2013;42:317-322.
- Stevanovic DV. Atrophie blanche. a sign of dermal blood occlusion. Arch Dermatol. 1974;109:858-862.
- Hairston BR, Davis MD, Pittelkow MR, et al. Livedoid vasculopathy: further evidence for procoagulant pathogenesis. Arch Dermatol. 2006;142:1413-1418.
- Shornick JK, Nicholes BK, Bergstresser PR, et al. Idiopathic atrophie blanche. J Am Acad Dermatol. 1983;8:792-798.
- Feldaker M, Hines EA Jr, Kierland RR. Livedo reticularis with ulcerations. Circulation. 1956;13:196-216.
- Callen JP. Livedoid vasculopathy: what it is and how the patient should be evaluated and treated. Arch Dermatol. 2006;142:1481-1482.
- Copeman PW. Livedo reticularis. signs in the skin of disturbance of blood viscosity and of blood flow. Br J Dermatol. 1975;93:519-529.
- Goerge T. Livedoid vasculopathy. pathogenesis, diagnosis and treatment of cutaneous infarction [in German]. Hautarzt. 2011;62:627-634; quiz 635.
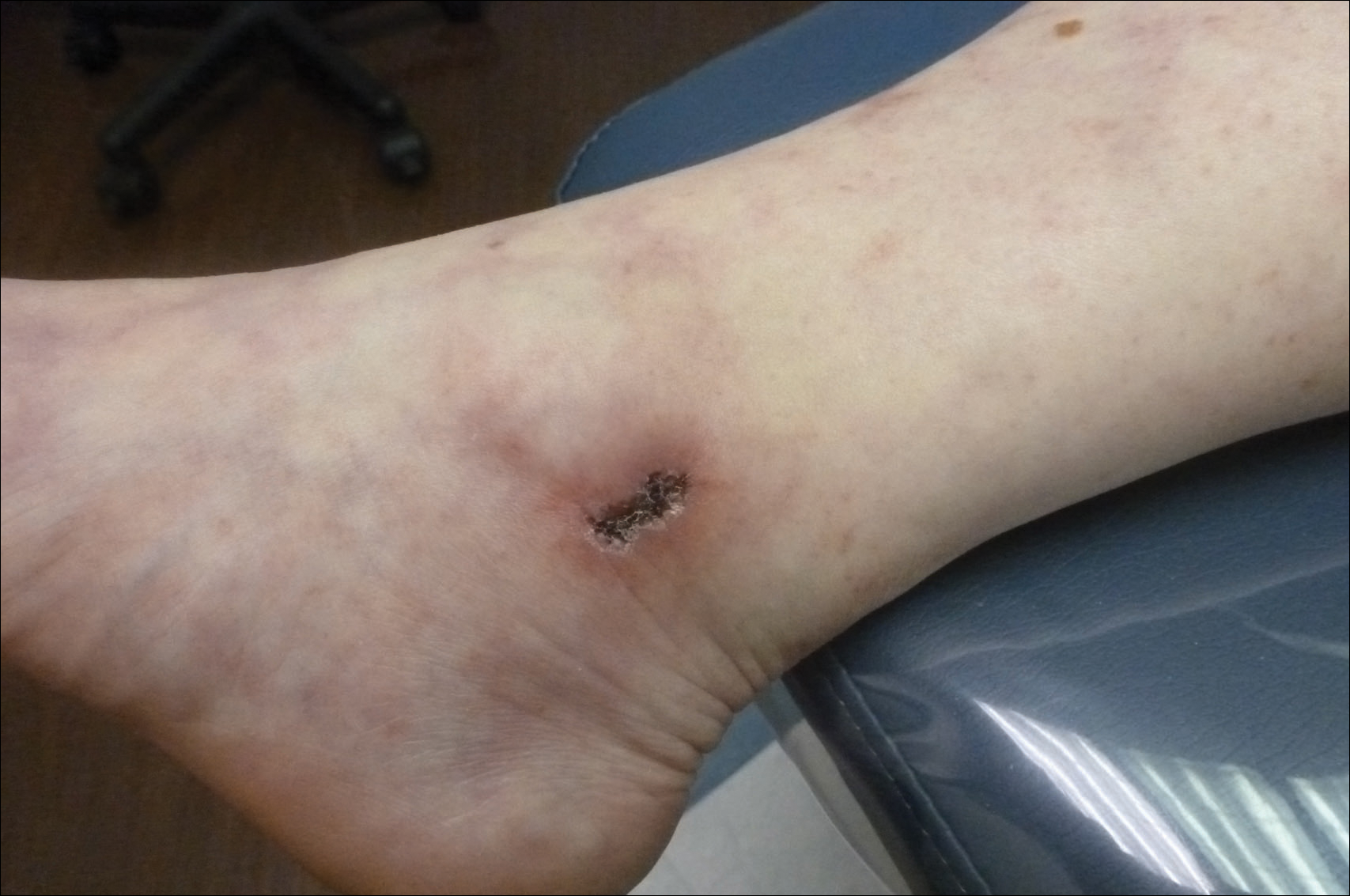
A 58-year-old woman presented in the summertime with skin discoloration of the bilateral lower legs and painful ulcerations above the medial and lateral malleoli of 15 years’ duration. She denied any recent trauma to the area or change in skin lesion appearance with cold exposure. Extensive laboratory evaluation for inherited and acquired coagulation abnormalities was negative. A punch biopsy specimen obtained from the left anterior lower leg revealed vascular thrombi with extravasated erythrocytes and a sparse perivascular inflammatory cell infiltrate.
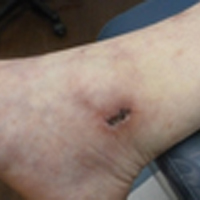
Enlarging Breast Lesion
The Diagnosis: Radiation-Associated Angiosarcoma
At the time of presentation, a 4-mm lesional punch biopsy was obtained (Figure), which revealed an epithelioid neoplasm within the dermis expressing CD31 and CD34, and staining negatively for S-100, CD45, and estrogen and progesterone receptors. The histologic and immunophenotypic findings were compatible with the diagnosis of angiosarcoma. Given the patient’s history of radiation for breast carcinoma several years ago, this tumor was consistent with radiation-associated angiosarcoma (RAAS).
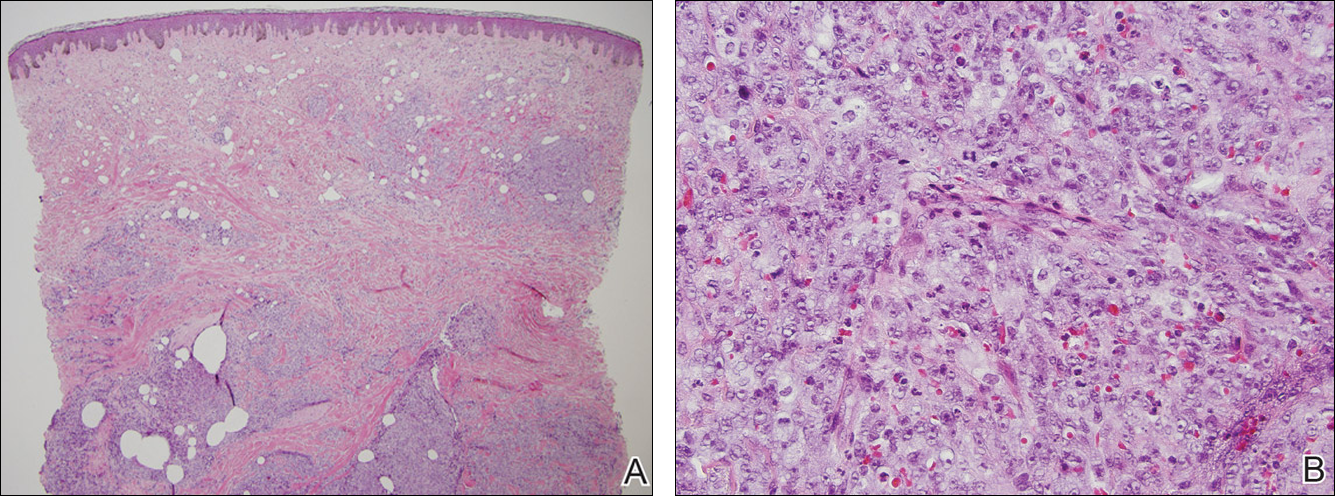
Development of secondary angiosarcoma has been linked to both prior radiation (RAAS) and chronic lymphedema (Stewart-Treves syndrome).1 Radiation-associated angiosarcoma is defined as a “pathologically confirmed breast or chest wall angiosarcoma arising within a previously irradiated field.”2 The incidence of RAAS is estimated to be 0.9 per 1000 individuals following radiation treatment of breast cancer over the subsequent 15 years and a mean time from radiation to development of 7 years.1 Incidence is expected to increase in the future due to improved likelihood of surviving early-stage breast carcinoma and the increased use of external beam radiation therapy for management of breast cancer.
Differentiating between primary and secondary angiosarcoma of the breast is important. Although primary breast angiosarcoma usually arises in women aged 30 to 40 years, RAAS tends to arise in older women (mean age, 68 years) and is seen only in those women with prior radiation.2 Additionally, high-level amplification of MYC, a known photo-oncogene, on chromosome 8 is a key genetic alteration of RAAS that helps to distinguish it from primary angiosarcoma, though this variance may be present in only half of RAAS cases.3 Immunohistochemical analysis of tumor cells for MYC expression correlates well with this amplification and also is helpful in distinguishing atypical vascular lesions from RAAS.4 Atypical vascular lesions, similar to RAAS, occur years after radiation exposure and may have a similar clinical presentation. Atypical vascular lesions do not progress to angiosarcoma in reported cases, but clinical and histologic overlap with RAAS make the diagnosis difficult.5 In these cases, analysis with fluorescence in situ hybridization or immunohistochemistry for the MYC amplification is important to differentiate these tumors.6
At the time of presentation, the majority of patients with RAAS of the breast have localized disease, often with a variable presentation. In all known cases, there have been skin changes present, emphasizing the importance of both patient and clinician vigilance on a regular basis in at-risk individuals. In one study, the most common presentation was breast ecchymosis, which was observed in 55% of patients.7 These lesions involve the dermis and are commonly mistaken for benign conditions such as infection or hemorrhage.2 In 2 other studies, RAAS most often manifested as a skin nodule or apparent tumor, closely followed by either a rash or bruiselike presentation.1,2
The overall recommendation for management of patients with ecchymotic skin lesions in previously irradiated regions is to obtain a biopsy specimen for tissue diagnosis. Although there is no standard of care for the management of RAAS, a multidisciplinary approach involving specialists from oncology, surgical oncology, and radiation oncology is recommended. Most often, radical surgery encompassing both the breast parenchyma and the at-risk radiated skin is performed. Extensive surgery has demonstrated the best survival benefits compared to mastectomy alone.7 Chemotherapeutics also may be used as adjuncts to surgery, which have been determined to decrease local recurrence rates but have no proven survival benefits.2 Adverse prognostic factors for survival are tumor size greater than 10 cm and development of local and/or distant metastases.2 Following the diagnosis of RAAS, our patient underwent radical mastectomy with adjuvant chemotherapy and remained disease free 6 months after surgery.
In summary, RAAS is a well-known, albeit relatively uncommon, consequence of radiation therapy. Dermatologists, oncologists, and primary care providers play an important role in recognizing this entity when evaluating patients with ecchymotic lesions as well as nodules or tumors within an irradiated field. Biopsy should be obtained promptly to prevent delay in diagnosis and to expedite referral to appropriate specialists for further evaluation and treatment.
- Seinen JM, Emelie S, Verstappen V, et al. Radiation-associated angiosarcoma after breast cancer: high recurrence rate and poor survival despite surgical treatment with R0 resection. Ann Surg Oncol. 2012;19:2700-2706.
- Torres KE, Ravi V, Kin K, et al. Long-term outcomes in patients with radiation-associated angiosarcomas of the breast following surgery and radiotherapy for breast cancer. Ann Surg Oncol. 2013;20:1267-1274.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Ginter PS, Mosquera JM, MacDonald TY, et al. Diagnostic utility of MYC amplification and anti-MYC immunohistochemistry in atypical vascular lesions, primary or radiation-induced mammary angiosarcomas, and primary angiosarcomas of other sites. Hum Pathol. 2014;45:709-716.
- Mentzel T, Schildhaus HU, Palmedo G, et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological immunohistochemical and molecular analysis of 66 cases. Mod Pathol. 2012;25:75-85.
- Fernandez AP, Sun Y, Tubbs RR, et al. FISH for MYC amplification and anti-MYC immunohistochemistry: useful diagnostic tools in the assessment of secondary angiosarcoma and atypical vascular proliferations. J Cutan Pathol. 2012;39:234-242.
- Morgan EA, Kozono DE, Wang Q, et al. Cutaneous radiation-associated angiosarcoma of the breast: poor prognosis in a rare secondary malignancy. Ann Surg Oncol. 2012;19:3801-3808.
The Diagnosis: Radiation-Associated Angiosarcoma
At the time of presentation, a 4-mm lesional punch biopsy was obtained (Figure), which revealed an epithelioid neoplasm within the dermis expressing CD31 and CD34, and staining negatively for S-100, CD45, and estrogen and progesterone receptors. The histologic and immunophenotypic findings were compatible with the diagnosis of angiosarcoma. Given the patient’s history of radiation for breast carcinoma several years ago, this tumor was consistent with radiation-associated angiosarcoma (RAAS).
Development of secondary angiosarcoma has been linked to both prior radiation (RAAS) and chronic lymphedema (Stewart-Treves syndrome).1 Radiation-associated angiosarcoma is defined as a “pathologically confirmed breast or chest wall angiosarcoma arising within a previously irradiated field.”2 The incidence of RAAS is estimated to be 0.9 per 1000 individuals following radiation treatment of breast cancer over the subsequent 15 years and a mean time from radiation to development of 7 years.1 Incidence is expected to increase in the future due to improved likelihood of surviving early-stage breast carcinoma and the increased use of external beam radiation therapy for management of breast cancer.
Differentiating between primary and secondary angiosarcoma of the breast is important. Although primary breast angiosarcoma usually arises in women aged 30 to 40 years, RAAS tends to arise in older women (mean age, 68 years) and is seen only in those women with prior radiation.2 Additionally, high-level amplification of MYC, a known photo-oncogene, on chromosome 8 is a key genetic alteration of RAAS that helps to distinguish it from primary angiosarcoma, though this variance may be present in only half of RAAS cases.3 Immunohistochemical analysis of tumor cells for MYC expression correlates well with this amplification and also is helpful in distinguishing atypical vascular lesions from RAAS.4 Atypical vascular lesions, similar to RAAS, occur years after radiation exposure and may have a similar clinical presentation. Atypical vascular lesions do not progress to angiosarcoma in reported cases, but clinical and histologic overlap with RAAS make the diagnosis difficult.5 In these cases, analysis with fluorescence in situ hybridization or immunohistochemistry for the MYC amplification is important to differentiate these tumors.6
At the time of presentation, the majority of patients with RAAS of the breast have localized disease, often with a variable presentation. In all known cases, there have been skin changes present, emphasizing the importance of both patient and clinician vigilance on a regular basis in at-risk individuals. In one study, the most common presentation was breast ecchymosis, which was observed in 55% of patients.7 These lesions involve the dermis and are commonly mistaken for benign conditions such as infection or hemorrhage.2 In 2 other studies, RAAS most often manifested as a skin nodule or apparent tumor, closely followed by either a rash or bruiselike presentation.1,2
The overall recommendation for management of patients with ecchymotic skin lesions in previously irradiated regions is to obtain a biopsy specimen for tissue diagnosis. Although there is no standard of care for the management of RAAS, a multidisciplinary approach involving specialists from oncology, surgical oncology, and radiation oncology is recommended. Most often, radical surgery encompassing both the breast parenchyma and the at-risk radiated skin is performed. Extensive surgery has demonstrated the best survival benefits compared to mastectomy alone.7 Chemotherapeutics also may be used as adjuncts to surgery, which have been determined to decrease local recurrence rates but have no proven survival benefits.2 Adverse prognostic factors for survival are tumor size greater than 10 cm and development of local and/or distant metastases.2 Following the diagnosis of RAAS, our patient underwent radical mastectomy with adjuvant chemotherapy and remained disease free 6 months after surgery.
In summary, RAAS is a well-known, albeit relatively uncommon, consequence of radiation therapy. Dermatologists, oncologists, and primary care providers play an important role in recognizing this entity when evaluating patients with ecchymotic lesions as well as nodules or tumors within an irradiated field. Biopsy should be obtained promptly to prevent delay in diagnosis and to expedite referral to appropriate specialists for further evaluation and treatment.
The Diagnosis: Radiation-Associated Angiosarcoma
At the time of presentation, a 4-mm lesional punch biopsy was obtained (Figure), which revealed an epithelioid neoplasm within the dermis expressing CD31 and CD34, and staining negatively for S-100, CD45, and estrogen and progesterone receptors. The histologic and immunophenotypic findings were compatible with the diagnosis of angiosarcoma. Given the patient’s history of radiation for breast carcinoma several years ago, this tumor was consistent with radiation-associated angiosarcoma (RAAS).
Development of secondary angiosarcoma has been linked to both prior radiation (RAAS) and chronic lymphedema (Stewart-Treves syndrome).1 Radiation-associated angiosarcoma is defined as a “pathologically confirmed breast or chest wall angiosarcoma arising within a previously irradiated field.”2 The incidence of RAAS is estimated to be 0.9 per 1000 individuals following radiation treatment of breast cancer over the subsequent 15 years and a mean time from radiation to development of 7 years.1 Incidence is expected to increase in the future due to improved likelihood of surviving early-stage breast carcinoma and the increased use of external beam radiation therapy for management of breast cancer.
Differentiating between primary and secondary angiosarcoma of the breast is important. Although primary breast angiosarcoma usually arises in women aged 30 to 40 years, RAAS tends to arise in older women (mean age, 68 years) and is seen only in those women with prior radiation.2 Additionally, high-level amplification of MYC, a known photo-oncogene, on chromosome 8 is a key genetic alteration of RAAS that helps to distinguish it from primary angiosarcoma, though this variance may be present in only half of RAAS cases.3 Immunohistochemical analysis of tumor cells for MYC expression correlates well with this amplification and also is helpful in distinguishing atypical vascular lesions from RAAS.4 Atypical vascular lesions, similar to RAAS, occur years after radiation exposure and may have a similar clinical presentation. Atypical vascular lesions do not progress to angiosarcoma in reported cases, but clinical and histologic overlap with RAAS make the diagnosis difficult.5 In these cases, analysis with fluorescence in situ hybridization or immunohistochemistry for the MYC amplification is important to differentiate these tumors.6
At the time of presentation, the majority of patients with RAAS of the breast have localized disease, often with a variable presentation. In all known cases, there have been skin changes present, emphasizing the importance of both patient and clinician vigilance on a regular basis in at-risk individuals. In one study, the most common presentation was breast ecchymosis, which was observed in 55% of patients.7 These lesions involve the dermis and are commonly mistaken for benign conditions such as infection or hemorrhage.2 In 2 other studies, RAAS most often manifested as a skin nodule or apparent tumor, closely followed by either a rash or bruiselike presentation.1,2
The overall recommendation for management of patients with ecchymotic skin lesions in previously irradiated regions is to obtain a biopsy specimen for tissue diagnosis. Although there is no standard of care for the management of RAAS, a multidisciplinary approach involving specialists from oncology, surgical oncology, and radiation oncology is recommended. Most often, radical surgery encompassing both the breast parenchyma and the at-risk radiated skin is performed. Extensive surgery has demonstrated the best survival benefits compared to mastectomy alone.7 Chemotherapeutics also may be used as adjuncts to surgery, which have been determined to decrease local recurrence rates but have no proven survival benefits.2 Adverse prognostic factors for survival are tumor size greater than 10 cm and development of local and/or distant metastases.2 Following the diagnosis of RAAS, our patient underwent radical mastectomy with adjuvant chemotherapy and remained disease free 6 months after surgery.
In summary, RAAS is a well-known, albeit relatively uncommon, consequence of radiation therapy. Dermatologists, oncologists, and primary care providers play an important role in recognizing this entity when evaluating patients with ecchymotic lesions as well as nodules or tumors within an irradiated field. Biopsy should be obtained promptly to prevent delay in diagnosis and to expedite referral to appropriate specialists for further evaluation and treatment.
- Seinen JM, Emelie S, Verstappen V, et al. Radiation-associated angiosarcoma after breast cancer: high recurrence rate and poor survival despite surgical treatment with R0 resection. Ann Surg Oncol. 2012;19:2700-2706.
- Torres KE, Ravi V, Kin K, et al. Long-term outcomes in patients with radiation-associated angiosarcomas of the breast following surgery and radiotherapy for breast cancer. Ann Surg Oncol. 2013;20:1267-1274.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Ginter PS, Mosquera JM, MacDonald TY, et al. Diagnostic utility of MYC amplification and anti-MYC immunohistochemistry in atypical vascular lesions, primary or radiation-induced mammary angiosarcomas, and primary angiosarcomas of other sites. Hum Pathol. 2014;45:709-716.
- Mentzel T, Schildhaus HU, Palmedo G, et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological immunohistochemical and molecular analysis of 66 cases. Mod Pathol. 2012;25:75-85.
- Fernandez AP, Sun Y, Tubbs RR, et al. FISH for MYC amplification and anti-MYC immunohistochemistry: useful diagnostic tools in the assessment of secondary angiosarcoma and atypical vascular proliferations. J Cutan Pathol. 2012;39:234-242.
- Morgan EA, Kozono DE, Wang Q, et al. Cutaneous radiation-associated angiosarcoma of the breast: poor prognosis in a rare secondary malignancy. Ann Surg Oncol. 2012;19:3801-3808.
- Seinen JM, Emelie S, Verstappen V, et al. Radiation-associated angiosarcoma after breast cancer: high recurrence rate and poor survival despite surgical treatment with R0 resection. Ann Surg Oncol. 2012;19:2700-2706.
- Torres KE, Ravi V, Kin K, et al. Long-term outcomes in patients with radiation-associated angiosarcomas of the breast following surgery and radiotherapy for breast cancer. Ann Surg Oncol. 2013;20:1267-1274.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Ginter PS, Mosquera JM, MacDonald TY, et al. Diagnostic utility of MYC amplification and anti-MYC immunohistochemistry in atypical vascular lesions, primary or radiation-induced mammary angiosarcomas, and primary angiosarcomas of other sites. Hum Pathol. 2014;45:709-716.
- Mentzel T, Schildhaus HU, Palmedo G, et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological immunohistochemical and molecular analysis of 66 cases. Mod Pathol. 2012;25:75-85.
- Fernandez AP, Sun Y, Tubbs RR, et al. FISH for MYC amplification and anti-MYC immunohistochemistry: useful diagnostic tools in the assessment of secondary angiosarcoma and atypical vascular proliferations. J Cutan Pathol. 2012;39:234-242.
- Morgan EA, Kozono DE, Wang Q, et al. Cutaneous radiation-associated angiosarcoma of the breast: poor prognosis in a rare secondary malignancy. Ann Surg Oncol. 2012;19:3801-3808.

A 75-year-old woman with a history of stage II invasive ductal carcinoma of the right breast presented to the dermatology clinic with an enlarging, indurated, ecchymotic plaque on the inferior aspect of the right breast of 2 months’ duration. The patient underwent a lumpectomy, radiation, and adjuvant chemotherapy 13 years prior to presentation. Review of systems was otherwise noncontributory.
Prednisone and Vardenafil Hydrochloride for Refractory Levamisole-Induced Vasculitis
Levamisole is an immunomodulatory drug that had been used to treat various medical conditions, including parasitic infections, nephrotic syndrome, and colorectal cancer,1 before being withdrawn from the US market in 2000. The most common reasons for levamisole discontinuation were leukopenia and rashes (1%–2%),1 many of which included leg ulcers and necrotizing purpura of the ears.1,2 The drug is currently available only as a deworming agent in veterinary medicine.
Since 2007, increasing amounts of levamisole have been used as an adulterant in cocaine. In 2007, less than 10% of cocaine was contaminated with levamisole, with an increase to 77% by 2010.3 In addition, 78% of 249 urine toxicology screens that were positive for cocaine in an inner city hospital also tested positive for levamisole.4 Levamisole-cut cocaine has become a concern because it is associated with a life-threatening syndrome involving a necrotizing purpuric rash, autoantibody production, and leukopenia.5
Levamisole-induced vasculitis is an independent entity from cocaine-induced vasculitis, which is associated with skin findings ranging from palpable purpura and chronic ulcers to digital infarction secondary to its vasospastic activity.6-8 Cocaine-induced vasculopathy has been related to cytoplasmic antineutrophil cytoplasmic antibody positivity and often resembles Wegener granulomatosis.6 Although both cocaine and levamisole have reportedly caused acrally distributed purpura and vasculopathy, levamisole is specifically associated with retiform purpura, ear involvement, and leukopenia.6,9 In addition, levamisole-induced skin reactions have been linked to specific antibodies, including antinuclear, antiphospholipid, and perinuclear antineutrophil cytoplasmic antibody (p-ANCA).2,5-7,9-14
We present a case of refractory levamisole-induced vasculitis and review its clinical presentation, diagnostic approach, laboratory findings, histology, and management. Furthermore, we discuss the possibility of a new treatment option for levamisole-induced vasculitis for patients with refractory disease or for patients who continue to use levamisole.
Case Report
A 49-year-old man with a history of polysubstance abuse presented with intermittent fevers and painful swollen ears as well as joint pain of 3 weeks’ duration. One week after the lesions developed on the ears, similar lesions were seen on the legs, arms, and trunk. He admitted to cocaine use 3 weeks prior to presentation when the symptoms began.
On physical examination, violaceous patches with necrotic bleeding edges and overlying black eschars were noted on the helices, antihelices, and ear lobules bilaterally (Figure 1). Retiform, purpuric to dark brown patches, some with signs of epidermal necrosis, were scattered on the arms, legs, and chest (Figure 2).
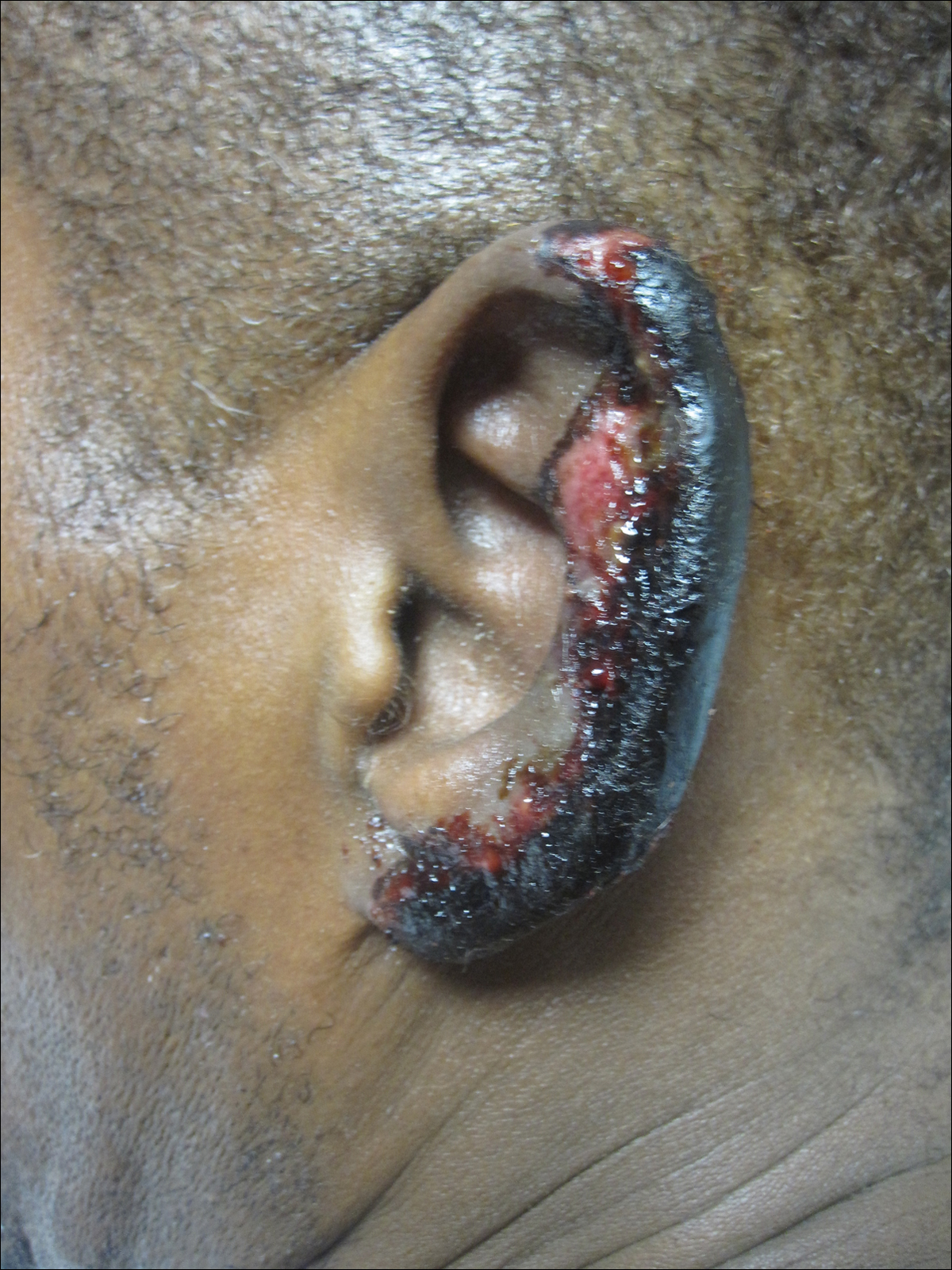
Laboratory examination revealed renal failure, anemia of chronic disease, and thrombocytosis (Table). The patient also screened positive for lupus anticoagulant and antinuclear antibodies and had elevated p-ANCA and anti–double-stranded DNA (Table). He also had an elevated sedimentation rate (109 mm/h [reference range, 0–20 mm/h]) and C-reactive protein level (11.3 mg/dL [reference range, 0–1.0 mg/dL])(Table). Urine toxicology was positive for cocaine.
A punch biopsy of the left thigh was performed on the edge of a retiform purpuric patch. Histopathologic examination revealed epidermal necrosis with subjacent intraluminal vascular thrombi along with extravasated red blood cells and neutrophilic debris (leukocytoclasis) and fibrin in and around vessel walls, consistent with vasculitis (Figure 3).
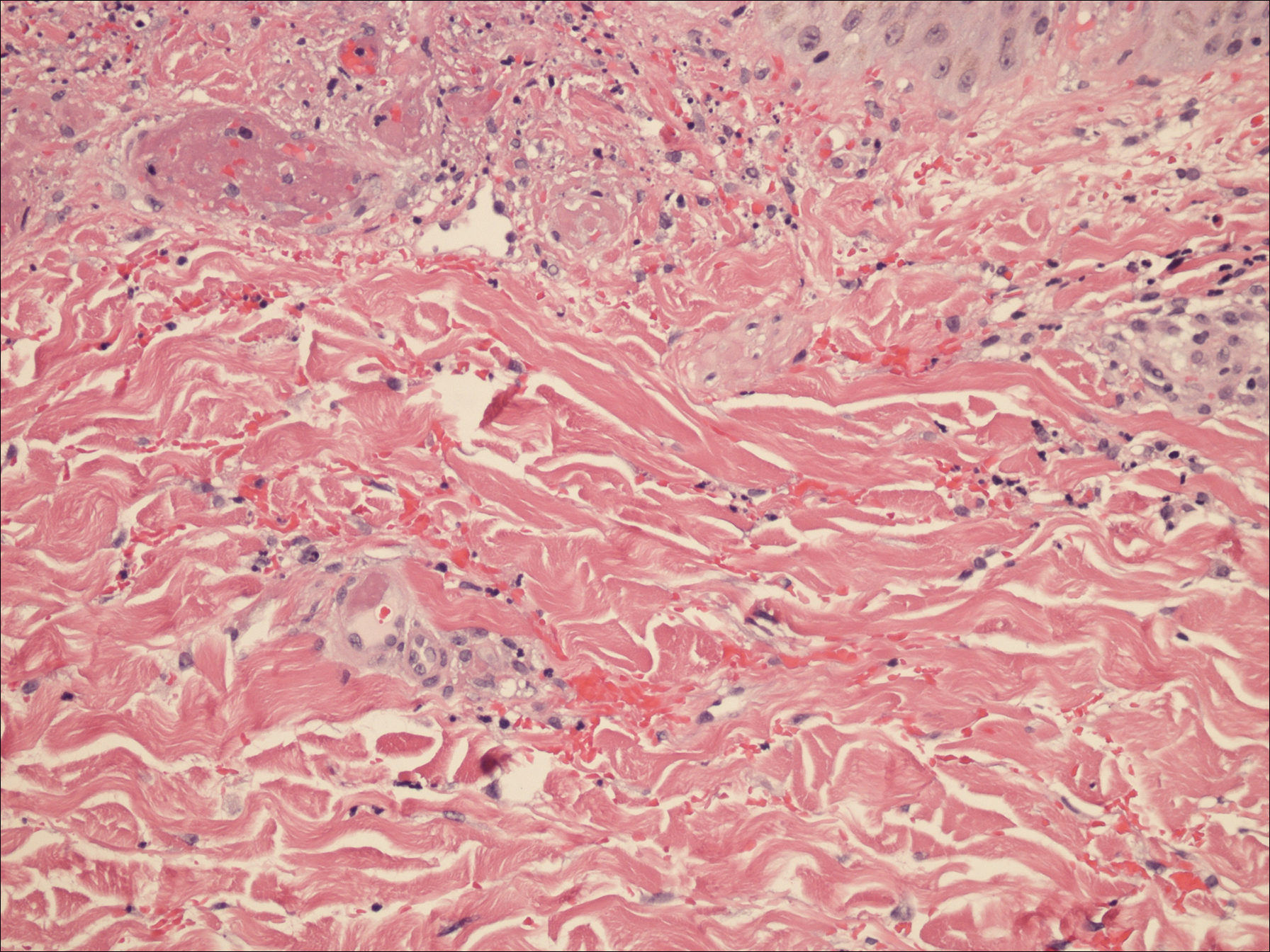
The patient was admitted to the hospital for pain management and wound care. Despite cocaine cessation and oral prednisone taper, the lesions on the legs worsened over the next several weeks. His condition was further complicated by wound infections, nonhealing ulcers, and subjective fevers and chills requiring frequent hospitalization. The patient was managed by the dermatology department as an outpatient and in clinic between hospital visits. He was treated with antibiotics, ulcer debridement, compression wraps, and aspirin (81 mg once daily) with moderate improvement.
Ten weeks after the first visit, the patient returned with worsening and recurrent leg and ear lesions. He denied any cocaine use since the initial hospital admission; however, a toxicology screen was never obtained. It was decided that the patient would need additional treatment along with traditional trigger (cocaine) avoidance and wound care. Combined treatment with aspirin (81 mg once daily), oral prednisone (40 mg once daily), and vardenafil hydrochloride (20 mg twice weekly) was initiated. At the end of week 1, the patient began to exhibit signs of improvement, which continued over the next 4 weeks. He was then lost to follow-up.
Comment
Our patient presented with severe necrotizing cutaneous vasculitis, likely secondary to levamisole exposure. Some of our patient’s cutaneous findings may be explained exclusively on the basis of cocaine exposure, but the characteristic lesion distribution and histopathologic findings along with the evidence of autoantibody positivity and concurrent arthralgias make the combination of levamisole and cocaine a more likely cause. Similar skin lesions were first described in children treated with levamisole for nephrotic syndrome.2 The most common site of clinical involvement in these children was the ears, as seen in our patient. Our patient tested positive for p-ANCA, which is the most commonly reported autoantibody associated with this patient population. Sixty-one percent (20/33) of patients with levamisole-induced vasculitis from 2 separate reviews showed p-ANCA positivity.7,10
On histopathology, our patient’s skin biopsy findings were consistent with those of prior reports of levamisole-induced vasculitis, which describe patterns of thrombotic vasculitis, leukocytoclasis, and fibrin deposition or occlusive disease.2,6,7,9-14 Mixed histologic findings of vasculitis and thrombosis, usually with varying ages of thrombi, are characteristic of levamisole-induced purpura. In addition, the disease can present nonspecifically with pure microvascular thrombosis without vasculitis, especially later in the course.9
The recommended management of levamisole-induced vasculitis currently involves the withdrawal of the culprit adulterated cocaine along with supportive treatment. Spontaneous and complete clinical resolution of lesions has been reported within 2 to 3 weeks and serology normalization within 2 to 14 months of levamisole cessation.2,6 A 2011 review of patients with levamisole-induced vasculitis reported 66% (19/29) of cases with either full cutaneous resolution after levamisole withdrawal or recurrence with resumed use, supporting a causal relationship.7 Walsh et al9 described 2 patients with recurrent and exacerbated retiform purpura following cocaine binges. Both of these patients had urine samples that tested positive for levamisole.9 In more severe cases, medications shown to be effective include colchicine, polidocanol, antibiotics, methotrexate, anticoagulants, and most commonly systemic corticosteroids.7,10,11,15 Nonsteroidal anti-inflammatory drugs were successful in treating lesions in 2 patients with concurrent arthralgia.7 Rarely, patients have required surgical debridement or skin grafting due to advanced disease at initial presentation.9,12-14 One of the most severe cases of levamisole-induced vasculitis reported in the literature involved 52% of the patient’s total body surface area with skin, soft tissue, and bony necrosis requiring nasal amputation, upper lip excision, skin grafting, and extremity amputation.14 Another severe case with widespread skin involvement was recently reported.16
For unclear reasons, our patient continued to develop cutaneous lesions despite self-reported cocaine cessation. Complete resolution required the combination of vardenafil, prednisone, and aspirin, along with debridement and wound care. Vardenafil, a selective phosphodiesterase 5 inhibitor, enhances the effect of nitrous oxide by increasing levels of cyclic guanosine monophosphate,17 which results in smooth muscle relaxation and vasodilatation. The primary indication for vardenafil is the treatment of erectile dysfunction, but it often is used off label in diseases that may benefit from vasodilatation. Because of its mechanism of action, it is understandable that a vasodilator such as vardenafil could be therapeutic in a condition associated with thrombosis. Moreover, the autoinflammatory nature of levamisole-induced vasculitis makes corticosteroid treatment effective. Given the 10-week delay in improvement, we suspect that it was the combination of treatment or an individual agent that led to our patient’s eventual recovery.
There are few reports in the literature focusing on optimal treatment of levamisole-induced vasculitis and none that mention alternative management for patients who continue to develop new lesions despite cocaine avoidance. Although the discontinuation of levamisole seems to be imperative for resolution of cutaneous lesions, it may not always be enough. It is possible that there is a subpopulation of patients that may not respond to the simple withdrawal of cocaine. It also should be mentioned that there was no urine toxicology screen obtained to support our patient’s reported cocaine cessation. Therefore, it is possible that his worsening condition was secondary to continued cocaine use. However, the patient successfully responded to the combination of vardenafil and prednisone, regardless of whether his condition persisted due to continued use of cocaine or not. This case suggests the possibility of a new treatment option for levamisole-induced vasculitis for patients who continue to use levamisole despite instruction for cessation or for patients with refractory disease.
Conclusion
A trial of prednisone and vardenafil should be considered for patients with refractory levamisole-induced vasculitis. Further studies and discussions of disease course are needed to identify the best treatment of this skin condition, especially for patients with refractory lesions.
- Scheinfeld N, Rosenberg JD, Weinberg JM. Levamisole in dermatology: a review. Am J Clin Dermatol. 2004;5:97-104.
- Rongioletti F, Ghio L, Ginevri F, et al. Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating long-term treatment with levamisole in children. Br J Dermatol. 1999;140:948-951.
- National Drug Threat Assessment 2011. US Department of Justice National Drug Intelligence Center website. https://www.justice.gov/archive/ndic/pubs44/44849/44849p.pdf. Published August 2011. Accessed August 7, 2016.
- Buchanan JA, Heard K, Burbach C, et al. Prevalence of levamisole in urine toxicology screens positive for cocaine in an inner-city hospital. JAMA. 2011;305:1657-1658.
- Gross RL, Brucker J, Bahce-Altuntas A, et al. A novel cutaneous vasculitis syndrome induced by levamisole-contaminated cocaine. Clin Rheumatol. 2011;30:1385-1392.
- Waller JM, Feramisco JD, Alberta-Wszolek L, et al. Cocaine-associated retiform purpura and neutropenia: is levamisole the culprit? J Am Acad Dermatol. 2010;63:530-535.
- Poon SH, Baliog CR, Sams RN, et al. Syndrome of cocaine-levamisole-induced cutaneous vasculitis and immune-mediated leukopenia. Semin Arthritis Rheum. 2011;41:434-444.
- Brewer JD, Meves A, Bostwick JM, et al. Cocaine abuse: dermatologic manifestations and therapeutic approaches. J Am Acad Dermatol. 2008;59:483-487.
- Walsh NMG, Green PJ, Burlingame RW, et al. Cocaine-related retiform purpura: evidence to incriminate the adulterant, levamisole. J Cutan Pathol. 2010;37:1212-1219.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia—a potential public health epidemic associated with levamisole adultered cocaine. J Am Acad Dermatol. 2011;65:722-725.
- Kahn TA, Cuchacovich R, Espinoza LR, et al. Vasculopathy, hematological, and immune abnormalities associated with levamisole-contaminated cocaine use. Semin Arthritis Rheum. 2011;41:445-454.
- Graf J, Lynch K, Yeh CL, et al. Purpura, cutaneous necrosis, and antineutrophil cytoplasmic antibodies associated with levamisole-adulterated cocaine. Arthritis Rheum. 2011;63:3998-4001.
- Farmer RW, Malhotra PS, Mays MP, et al. Necrotizing peripheral vasculitis/vasculopathy following the use of cocaine laced with levamisole. J Burn Care Res. 2012;33:e6-e11.
- Ching JA, Smith DJ Jr. Levamisole-induced skin necrosis of skin, soft tissue, and bone: case report and review of literature. J Burn Care Res. 2012;33:e1-e5.
- Buchanan JA, Vogel JA, Eberhardt AM. Levamisole-induced occlusive necrotizing vasculitis of the ears after use of cocaine contaminated with levamisole. J Med Toxicol. 2011;7:83-84.
- Graff N, Whitworth K, Trigger C. Purpuric skin eruption in an illicit drug user: levamisole-induced vasculitis. Am J Emer Med. 2016;34:1321.
- Schwartz BG, Kloner RA. Drug interactions with phosphodiesterase-5 inhibitors used for the treatment of erectile dysfunction or pulmonary hypertension. Circulation. 2010;122:88-95.
Levamisole is an immunomodulatory drug that had been used to treat various medical conditions, including parasitic infections, nephrotic syndrome, and colorectal cancer,1 before being withdrawn from the US market in 2000. The most common reasons for levamisole discontinuation were leukopenia and rashes (1%–2%),1 many of which included leg ulcers and necrotizing purpura of the ears.1,2 The drug is currently available only as a deworming agent in veterinary medicine.
Since 2007, increasing amounts of levamisole have been used as an adulterant in cocaine. In 2007, less than 10% of cocaine was contaminated with levamisole, with an increase to 77% by 2010.3 In addition, 78% of 249 urine toxicology screens that were positive for cocaine in an inner city hospital also tested positive for levamisole.4 Levamisole-cut cocaine has become a concern because it is associated with a life-threatening syndrome involving a necrotizing purpuric rash, autoantibody production, and leukopenia.5
Levamisole-induced vasculitis is an independent entity from cocaine-induced vasculitis, which is associated with skin findings ranging from palpable purpura and chronic ulcers to digital infarction secondary to its vasospastic activity.6-8 Cocaine-induced vasculopathy has been related to cytoplasmic antineutrophil cytoplasmic antibody positivity and often resembles Wegener granulomatosis.6 Although both cocaine and levamisole have reportedly caused acrally distributed purpura and vasculopathy, levamisole is specifically associated with retiform purpura, ear involvement, and leukopenia.6,9 In addition, levamisole-induced skin reactions have been linked to specific antibodies, including antinuclear, antiphospholipid, and perinuclear antineutrophil cytoplasmic antibody (p-ANCA).2,5-7,9-14
We present a case of refractory levamisole-induced vasculitis and review its clinical presentation, diagnostic approach, laboratory findings, histology, and management. Furthermore, we discuss the possibility of a new treatment option for levamisole-induced vasculitis for patients with refractory disease or for patients who continue to use levamisole.
Case Report
A 49-year-old man with a history of polysubstance abuse presented with intermittent fevers and painful swollen ears as well as joint pain of 3 weeks’ duration. One week after the lesions developed on the ears, similar lesions were seen on the legs, arms, and trunk. He admitted to cocaine use 3 weeks prior to presentation when the symptoms began.
On physical examination, violaceous patches with necrotic bleeding edges and overlying black eschars were noted on the helices, antihelices, and ear lobules bilaterally (Figure 1). Retiform, purpuric to dark brown patches, some with signs of epidermal necrosis, were scattered on the arms, legs, and chest (Figure 2).
Laboratory examination revealed renal failure, anemia of chronic disease, and thrombocytosis (Table). The patient also screened positive for lupus anticoagulant and antinuclear antibodies and had elevated p-ANCA and anti–double-stranded DNA (Table). He also had an elevated sedimentation rate (109 mm/h [reference range, 0–20 mm/h]) and C-reactive protein level (11.3 mg/dL [reference range, 0–1.0 mg/dL])(Table). Urine toxicology was positive for cocaine.
A punch biopsy of the left thigh was performed on the edge of a retiform purpuric patch. Histopathologic examination revealed epidermal necrosis with subjacent intraluminal vascular thrombi along with extravasated red blood cells and neutrophilic debris (leukocytoclasis) and fibrin in and around vessel walls, consistent with vasculitis (Figure 3).
The patient was admitted to the hospital for pain management and wound care. Despite cocaine cessation and oral prednisone taper, the lesions on the legs worsened over the next several weeks. His condition was further complicated by wound infections, nonhealing ulcers, and subjective fevers and chills requiring frequent hospitalization. The patient was managed by the dermatology department as an outpatient and in clinic between hospital visits. He was treated with antibiotics, ulcer debridement, compression wraps, and aspirin (81 mg once daily) with moderate improvement.
Ten weeks after the first visit, the patient returned with worsening and recurrent leg and ear lesions. He denied any cocaine use since the initial hospital admission; however, a toxicology screen was never obtained. It was decided that the patient would need additional treatment along with traditional trigger (cocaine) avoidance and wound care. Combined treatment with aspirin (81 mg once daily), oral prednisone (40 mg once daily), and vardenafil hydrochloride (20 mg twice weekly) was initiated. At the end of week 1, the patient began to exhibit signs of improvement, which continued over the next 4 weeks. He was then lost to follow-up.
Comment
Our patient presented with severe necrotizing cutaneous vasculitis, likely secondary to levamisole exposure. Some of our patient’s cutaneous findings may be explained exclusively on the basis of cocaine exposure, but the characteristic lesion distribution and histopathologic findings along with the evidence of autoantibody positivity and concurrent arthralgias make the combination of levamisole and cocaine a more likely cause. Similar skin lesions were first described in children treated with levamisole for nephrotic syndrome.2 The most common site of clinical involvement in these children was the ears, as seen in our patient. Our patient tested positive for p-ANCA, which is the most commonly reported autoantibody associated with this patient population. Sixty-one percent (20/33) of patients with levamisole-induced vasculitis from 2 separate reviews showed p-ANCA positivity.7,10
On histopathology, our patient’s skin biopsy findings were consistent with those of prior reports of levamisole-induced vasculitis, which describe patterns of thrombotic vasculitis, leukocytoclasis, and fibrin deposition or occlusive disease.2,6,7,9-14 Mixed histologic findings of vasculitis and thrombosis, usually with varying ages of thrombi, are characteristic of levamisole-induced purpura. In addition, the disease can present nonspecifically with pure microvascular thrombosis without vasculitis, especially later in the course.9
The recommended management of levamisole-induced vasculitis currently involves the withdrawal of the culprit adulterated cocaine along with supportive treatment. Spontaneous and complete clinical resolution of lesions has been reported within 2 to 3 weeks and serology normalization within 2 to 14 months of levamisole cessation.2,6 A 2011 review of patients with levamisole-induced vasculitis reported 66% (19/29) of cases with either full cutaneous resolution after levamisole withdrawal or recurrence with resumed use, supporting a causal relationship.7 Walsh et al9 described 2 patients with recurrent and exacerbated retiform purpura following cocaine binges. Both of these patients had urine samples that tested positive for levamisole.9 In more severe cases, medications shown to be effective include colchicine, polidocanol, antibiotics, methotrexate, anticoagulants, and most commonly systemic corticosteroids.7,10,11,15 Nonsteroidal anti-inflammatory drugs were successful in treating lesions in 2 patients with concurrent arthralgia.7 Rarely, patients have required surgical debridement or skin grafting due to advanced disease at initial presentation.9,12-14 One of the most severe cases of levamisole-induced vasculitis reported in the literature involved 52% of the patient’s total body surface area with skin, soft tissue, and bony necrosis requiring nasal amputation, upper lip excision, skin grafting, and extremity amputation.14 Another severe case with widespread skin involvement was recently reported.16
For unclear reasons, our patient continued to develop cutaneous lesions despite self-reported cocaine cessation. Complete resolution required the combination of vardenafil, prednisone, and aspirin, along with debridement and wound care. Vardenafil, a selective phosphodiesterase 5 inhibitor, enhances the effect of nitrous oxide by increasing levels of cyclic guanosine monophosphate,17 which results in smooth muscle relaxation and vasodilatation. The primary indication for vardenafil is the treatment of erectile dysfunction, but it often is used off label in diseases that may benefit from vasodilatation. Because of its mechanism of action, it is understandable that a vasodilator such as vardenafil could be therapeutic in a condition associated with thrombosis. Moreover, the autoinflammatory nature of levamisole-induced vasculitis makes corticosteroid treatment effective. Given the 10-week delay in improvement, we suspect that it was the combination of treatment or an individual agent that led to our patient’s eventual recovery.
There are few reports in the literature focusing on optimal treatment of levamisole-induced vasculitis and none that mention alternative management for patients who continue to develop new lesions despite cocaine avoidance. Although the discontinuation of levamisole seems to be imperative for resolution of cutaneous lesions, it may not always be enough. It is possible that there is a subpopulation of patients that may not respond to the simple withdrawal of cocaine. It also should be mentioned that there was no urine toxicology screen obtained to support our patient’s reported cocaine cessation. Therefore, it is possible that his worsening condition was secondary to continued cocaine use. However, the patient successfully responded to the combination of vardenafil and prednisone, regardless of whether his condition persisted due to continued use of cocaine or not. This case suggests the possibility of a new treatment option for levamisole-induced vasculitis for patients who continue to use levamisole despite instruction for cessation or for patients with refractory disease.
Conclusion
A trial of prednisone and vardenafil should be considered for patients with refractory levamisole-induced vasculitis. Further studies and discussions of disease course are needed to identify the best treatment of this skin condition, especially for patients with refractory lesions.
Levamisole is an immunomodulatory drug that had been used to treat various medical conditions, including parasitic infections, nephrotic syndrome, and colorectal cancer,1 before being withdrawn from the US market in 2000. The most common reasons for levamisole discontinuation were leukopenia and rashes (1%–2%),1 many of which included leg ulcers and necrotizing purpura of the ears.1,2 The drug is currently available only as a deworming agent in veterinary medicine.
Since 2007, increasing amounts of levamisole have been used as an adulterant in cocaine. In 2007, less than 10% of cocaine was contaminated with levamisole, with an increase to 77% by 2010.3 In addition, 78% of 249 urine toxicology screens that were positive for cocaine in an inner city hospital also tested positive for levamisole.4 Levamisole-cut cocaine has become a concern because it is associated with a life-threatening syndrome involving a necrotizing purpuric rash, autoantibody production, and leukopenia.5
Levamisole-induced vasculitis is an independent entity from cocaine-induced vasculitis, which is associated with skin findings ranging from palpable purpura and chronic ulcers to digital infarction secondary to its vasospastic activity.6-8 Cocaine-induced vasculopathy has been related to cytoplasmic antineutrophil cytoplasmic antibody positivity and often resembles Wegener granulomatosis.6 Although both cocaine and levamisole have reportedly caused acrally distributed purpura and vasculopathy, levamisole is specifically associated with retiform purpura, ear involvement, and leukopenia.6,9 In addition, levamisole-induced skin reactions have been linked to specific antibodies, including antinuclear, antiphospholipid, and perinuclear antineutrophil cytoplasmic antibody (p-ANCA).2,5-7,9-14
We present a case of refractory levamisole-induced vasculitis and review its clinical presentation, diagnostic approach, laboratory findings, histology, and management. Furthermore, we discuss the possibility of a new treatment option for levamisole-induced vasculitis for patients with refractory disease or for patients who continue to use levamisole.
Case Report
A 49-year-old man with a history of polysubstance abuse presented with intermittent fevers and painful swollen ears as well as joint pain of 3 weeks’ duration. One week after the lesions developed on the ears, similar lesions were seen on the legs, arms, and trunk. He admitted to cocaine use 3 weeks prior to presentation when the symptoms began.
On physical examination, violaceous patches with necrotic bleeding edges and overlying black eschars were noted on the helices, antihelices, and ear lobules bilaterally (Figure 1). Retiform, purpuric to dark brown patches, some with signs of epidermal necrosis, were scattered on the arms, legs, and chest (Figure 2).
Laboratory examination revealed renal failure, anemia of chronic disease, and thrombocytosis (Table). The patient also screened positive for lupus anticoagulant and antinuclear antibodies and had elevated p-ANCA and anti–double-stranded DNA (Table). He also had an elevated sedimentation rate (109 mm/h [reference range, 0–20 mm/h]) and C-reactive protein level (11.3 mg/dL [reference range, 0–1.0 mg/dL])(Table). Urine toxicology was positive for cocaine.
A punch biopsy of the left thigh was performed on the edge of a retiform purpuric patch. Histopathologic examination revealed epidermal necrosis with subjacent intraluminal vascular thrombi along with extravasated red blood cells and neutrophilic debris (leukocytoclasis) and fibrin in and around vessel walls, consistent with vasculitis (Figure 3).
The patient was admitted to the hospital for pain management and wound care. Despite cocaine cessation and oral prednisone taper, the lesions on the legs worsened over the next several weeks. His condition was further complicated by wound infections, nonhealing ulcers, and subjective fevers and chills requiring frequent hospitalization. The patient was managed by the dermatology department as an outpatient and in clinic between hospital visits. He was treated with antibiotics, ulcer debridement, compression wraps, and aspirin (81 mg once daily) with moderate improvement.
Ten weeks after the first visit, the patient returned with worsening and recurrent leg and ear lesions. He denied any cocaine use since the initial hospital admission; however, a toxicology screen was never obtained. It was decided that the patient would need additional treatment along with traditional trigger (cocaine) avoidance and wound care. Combined treatment with aspirin (81 mg once daily), oral prednisone (40 mg once daily), and vardenafil hydrochloride (20 mg twice weekly) was initiated. At the end of week 1, the patient began to exhibit signs of improvement, which continued over the next 4 weeks. He was then lost to follow-up.
Comment
Our patient presented with severe necrotizing cutaneous vasculitis, likely secondary to levamisole exposure. Some of our patient’s cutaneous findings may be explained exclusively on the basis of cocaine exposure, but the characteristic lesion distribution and histopathologic findings along with the evidence of autoantibody positivity and concurrent arthralgias make the combination of levamisole and cocaine a more likely cause. Similar skin lesions were first described in children treated with levamisole for nephrotic syndrome.2 The most common site of clinical involvement in these children was the ears, as seen in our patient. Our patient tested positive for p-ANCA, which is the most commonly reported autoantibody associated with this patient population. Sixty-one percent (20/33) of patients with levamisole-induced vasculitis from 2 separate reviews showed p-ANCA positivity.7,10
On histopathology, our patient’s skin biopsy findings were consistent with those of prior reports of levamisole-induced vasculitis, which describe patterns of thrombotic vasculitis, leukocytoclasis, and fibrin deposition or occlusive disease.2,6,7,9-14 Mixed histologic findings of vasculitis and thrombosis, usually with varying ages of thrombi, are characteristic of levamisole-induced purpura. In addition, the disease can present nonspecifically with pure microvascular thrombosis without vasculitis, especially later in the course.9
The recommended management of levamisole-induced vasculitis currently involves the withdrawal of the culprit adulterated cocaine along with supportive treatment. Spontaneous and complete clinical resolution of lesions has been reported within 2 to 3 weeks and serology normalization within 2 to 14 months of levamisole cessation.2,6 A 2011 review of patients with levamisole-induced vasculitis reported 66% (19/29) of cases with either full cutaneous resolution after levamisole withdrawal or recurrence with resumed use, supporting a causal relationship.7 Walsh et al9 described 2 patients with recurrent and exacerbated retiform purpura following cocaine binges. Both of these patients had urine samples that tested positive for levamisole.9 In more severe cases, medications shown to be effective include colchicine, polidocanol, antibiotics, methotrexate, anticoagulants, and most commonly systemic corticosteroids.7,10,11,15 Nonsteroidal anti-inflammatory drugs were successful in treating lesions in 2 patients with concurrent arthralgia.7 Rarely, patients have required surgical debridement or skin grafting due to advanced disease at initial presentation.9,12-14 One of the most severe cases of levamisole-induced vasculitis reported in the literature involved 52% of the patient’s total body surface area with skin, soft tissue, and bony necrosis requiring nasal amputation, upper lip excision, skin grafting, and extremity amputation.14 Another severe case with widespread skin involvement was recently reported.16
For unclear reasons, our patient continued to develop cutaneous lesions despite self-reported cocaine cessation. Complete resolution required the combination of vardenafil, prednisone, and aspirin, along with debridement and wound care. Vardenafil, a selective phosphodiesterase 5 inhibitor, enhances the effect of nitrous oxide by increasing levels of cyclic guanosine monophosphate,17 which results in smooth muscle relaxation and vasodilatation. The primary indication for vardenafil is the treatment of erectile dysfunction, but it often is used off label in diseases that may benefit from vasodilatation. Because of its mechanism of action, it is understandable that a vasodilator such as vardenafil could be therapeutic in a condition associated with thrombosis. Moreover, the autoinflammatory nature of levamisole-induced vasculitis makes corticosteroid treatment effective. Given the 10-week delay in improvement, we suspect that it was the combination of treatment or an individual agent that led to our patient’s eventual recovery.
There are few reports in the literature focusing on optimal treatment of levamisole-induced vasculitis and none that mention alternative management for patients who continue to develop new lesions despite cocaine avoidance. Although the discontinuation of levamisole seems to be imperative for resolution of cutaneous lesions, it may not always be enough. It is possible that there is a subpopulation of patients that may not respond to the simple withdrawal of cocaine. It also should be mentioned that there was no urine toxicology screen obtained to support our patient’s reported cocaine cessation. Therefore, it is possible that his worsening condition was secondary to continued cocaine use. However, the patient successfully responded to the combination of vardenafil and prednisone, regardless of whether his condition persisted due to continued use of cocaine or not. This case suggests the possibility of a new treatment option for levamisole-induced vasculitis for patients who continue to use levamisole despite instruction for cessation or for patients with refractory disease.
Conclusion
A trial of prednisone and vardenafil should be considered for patients with refractory levamisole-induced vasculitis. Further studies and discussions of disease course are needed to identify the best treatment of this skin condition, especially for patients with refractory lesions.
- Scheinfeld N, Rosenberg JD, Weinberg JM. Levamisole in dermatology: a review. Am J Clin Dermatol. 2004;5:97-104.
- Rongioletti F, Ghio L, Ginevri F, et al. Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating long-term treatment with levamisole in children. Br J Dermatol. 1999;140:948-951.
- National Drug Threat Assessment 2011. US Department of Justice National Drug Intelligence Center website. https://www.justice.gov/archive/ndic/pubs44/44849/44849p.pdf. Published August 2011. Accessed August 7, 2016.
- Buchanan JA, Heard K, Burbach C, et al. Prevalence of levamisole in urine toxicology screens positive for cocaine in an inner-city hospital. JAMA. 2011;305:1657-1658.
- Gross RL, Brucker J, Bahce-Altuntas A, et al. A novel cutaneous vasculitis syndrome induced by levamisole-contaminated cocaine. Clin Rheumatol. 2011;30:1385-1392.
- Waller JM, Feramisco JD, Alberta-Wszolek L, et al. Cocaine-associated retiform purpura and neutropenia: is levamisole the culprit? J Am Acad Dermatol. 2010;63:530-535.
- Poon SH, Baliog CR, Sams RN, et al. Syndrome of cocaine-levamisole-induced cutaneous vasculitis and immune-mediated leukopenia. Semin Arthritis Rheum. 2011;41:434-444.
- Brewer JD, Meves A, Bostwick JM, et al. Cocaine abuse: dermatologic manifestations and therapeutic approaches. J Am Acad Dermatol. 2008;59:483-487.
- Walsh NMG, Green PJ, Burlingame RW, et al. Cocaine-related retiform purpura: evidence to incriminate the adulterant, levamisole. J Cutan Pathol. 2010;37:1212-1219.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia—a potential public health epidemic associated with levamisole adultered cocaine. J Am Acad Dermatol. 2011;65:722-725.
- Kahn TA, Cuchacovich R, Espinoza LR, et al. Vasculopathy, hematological, and immune abnormalities associated with levamisole-contaminated cocaine use. Semin Arthritis Rheum. 2011;41:445-454.
- Graf J, Lynch K, Yeh CL, et al. Purpura, cutaneous necrosis, and antineutrophil cytoplasmic antibodies associated with levamisole-adulterated cocaine. Arthritis Rheum. 2011;63:3998-4001.
- Farmer RW, Malhotra PS, Mays MP, et al. Necrotizing peripheral vasculitis/vasculopathy following the use of cocaine laced with levamisole. J Burn Care Res. 2012;33:e6-e11.
- Ching JA, Smith DJ Jr. Levamisole-induced skin necrosis of skin, soft tissue, and bone: case report and review of literature. J Burn Care Res. 2012;33:e1-e5.
- Buchanan JA, Vogel JA, Eberhardt AM. Levamisole-induced occlusive necrotizing vasculitis of the ears after use of cocaine contaminated with levamisole. J Med Toxicol. 2011;7:83-84.
- Graff N, Whitworth K, Trigger C. Purpuric skin eruption in an illicit drug user: levamisole-induced vasculitis. Am J Emer Med. 2016;34:1321.
- Schwartz BG, Kloner RA. Drug interactions with phosphodiesterase-5 inhibitors used for the treatment of erectile dysfunction or pulmonary hypertension. Circulation. 2010;122:88-95.
- Scheinfeld N, Rosenberg JD, Weinberg JM. Levamisole in dermatology: a review. Am J Clin Dermatol. 2004;5:97-104.
- Rongioletti F, Ghio L, Ginevri F, et al. Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating long-term treatment with levamisole in children. Br J Dermatol. 1999;140:948-951.
- National Drug Threat Assessment 2011. US Department of Justice National Drug Intelligence Center website. https://www.justice.gov/archive/ndic/pubs44/44849/44849p.pdf. Published August 2011. Accessed August 7, 2016.
- Buchanan JA, Heard K, Burbach C, et al. Prevalence of levamisole in urine toxicology screens positive for cocaine in an inner-city hospital. JAMA. 2011;305:1657-1658.
- Gross RL, Brucker J, Bahce-Altuntas A, et al. A novel cutaneous vasculitis syndrome induced by levamisole-contaminated cocaine. Clin Rheumatol. 2011;30:1385-1392.
- Waller JM, Feramisco JD, Alberta-Wszolek L, et al. Cocaine-associated retiform purpura and neutropenia: is levamisole the culprit? J Am Acad Dermatol. 2010;63:530-535.
- Poon SH, Baliog CR, Sams RN, et al. Syndrome of cocaine-levamisole-induced cutaneous vasculitis and immune-mediated leukopenia. Semin Arthritis Rheum. 2011;41:434-444.
- Brewer JD, Meves A, Bostwick JM, et al. Cocaine abuse: dermatologic manifestations and therapeutic approaches. J Am Acad Dermatol. 2008;59:483-487.
- Walsh NMG, Green PJ, Burlingame RW, et al. Cocaine-related retiform purpura: evidence to incriminate the adulterant, levamisole. J Cutan Pathol. 2010;37:1212-1219.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia—a potential public health epidemic associated with levamisole adultered cocaine. J Am Acad Dermatol. 2011;65:722-725.
- Kahn TA, Cuchacovich R, Espinoza LR, et al. Vasculopathy, hematological, and immune abnormalities associated with levamisole-contaminated cocaine use. Semin Arthritis Rheum. 2011;41:445-454.
- Graf J, Lynch K, Yeh CL, et al. Purpura, cutaneous necrosis, and antineutrophil cytoplasmic antibodies associated with levamisole-adulterated cocaine. Arthritis Rheum. 2011;63:3998-4001.
- Farmer RW, Malhotra PS, Mays MP, et al. Necrotizing peripheral vasculitis/vasculopathy following the use of cocaine laced with levamisole. J Burn Care Res. 2012;33:e6-e11.
- Ching JA, Smith DJ Jr. Levamisole-induced skin necrosis of skin, soft tissue, and bone: case report and review of literature. J Burn Care Res. 2012;33:e1-e5.
- Buchanan JA, Vogel JA, Eberhardt AM. Levamisole-induced occlusive necrotizing vasculitis of the ears after use of cocaine contaminated with levamisole. J Med Toxicol. 2011;7:83-84.
- Graff N, Whitworth K, Trigger C. Purpuric skin eruption in an illicit drug user: levamisole-induced vasculitis. Am J Emer Med. 2016;34:1321.
- Schwartz BG, Kloner RA. Drug interactions with phosphodiesterase-5 inhibitors used for the treatment of erectile dysfunction or pulmonary hypertension. Circulation. 2010;122:88-95.
Practice Points
- Levamisole is an immunomodulatory drug that, before being withdrawn from the US market in 2000, was previously used to treat various medical conditions.
- A majority of the cocaine in the United States is contaminated with levamisole, which is added as an adulterant or bulking agent.
- Levamisole-cut cocaine is a concern because it is associated with a life-threatening syndrome involving a necrotizing purpuric rash, autoantibody production, and leukopenia.
- Although treatment of levamisole toxicity is primarily supportive and includes cessation of levamisole-cut cocaine, a trial of prednisone and vardenafil hydrochloride can be considered for refractory levamisole-induced vasculopathy or for patients who continue to use the drug.
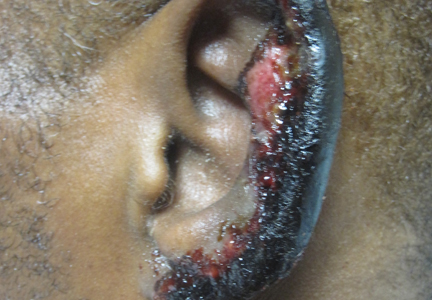
Sporotrichoid Fluctuant Nodules
The Diagnosis: Atypical Mycobacterial Infection
Punch biopsy specimens demonstrated necrotizing granulomatous inflammation in the dermis and subcutis (Figure). Special staining for microorganisms was negative. Tissue culture grew Mycobacterium avium-intracellulare (MAI). The patient began treatment with azithromycin, ethambutol, and rifabutin. Tissue susceptibilities later showed resistance to rifabutin and sensitivity to clarithromycin, moxifloxacin, and clofazimine. She subsequently was switched to azithromycin, clofazimine, and moxifloxacin with good response.
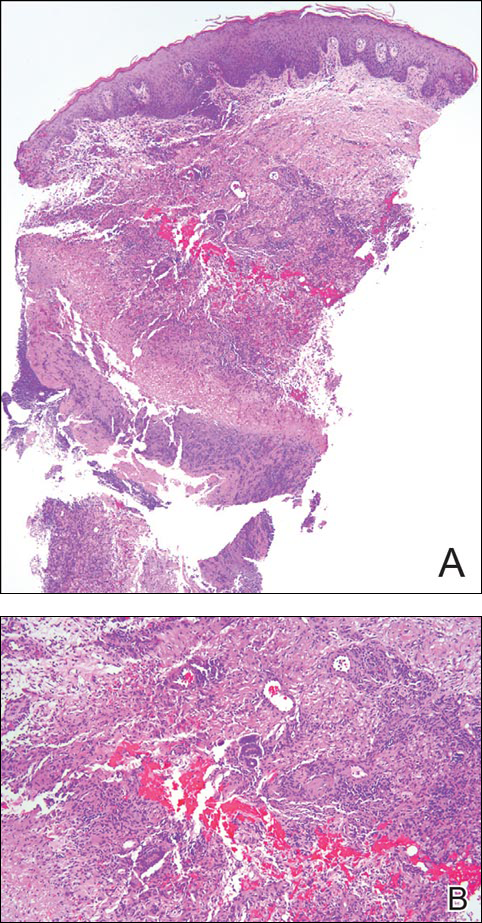
Mycobacterium avium-intracellulare is a slow-growing, nonchromogenic, atypical mycobacteria. Although ubiquitous, it tends to only cause serious infection in the setting of immunosuppression. Transmission usually is through the respiratory or gastrointestinal tract.1 Skin infections with MAI are uncommon and usually are secondary to seeding from disseminated infection or from direct inoculation.2
The clinical presentations of primary cutaneous MAI are myriad, including an isolated red nodule, multiple ulcers, abscesses, draining sinuses, facial nodules, granulomatous plaques, and panniculitis.2,3 Of 3 reported cases of primary cutaneous MAI in the form of sporotrichoid lesions, 2 involved patients with AIDS2 and 1 involved a cardiac transplant recipient.4
Cutaneous MAI is typically diagnosed with skin biopsy and tissue culture. Tissue culture is critical for determining the specific mycobacterial species and antibiotic susceptibilities. Polymerase chain reaction has been utilized to rapidly diagnose cutaneous MAI infection from an acid-fast bacilli–positive tissue sample in which the tissue culture was negative.5
Recommended treatment protocols for MAI involve multidrug regimens because of the intrinsic resistance of MAI and the concern for development of resistance with monotherapy.2 No definitive guidelines exist for treatment of primary cutaneous MAI infections. However, regimens for the treatment of pulmonary infection that also have been successfully utilized for cutaneous infection include a macrolide, ethambutol, and a rifamycin.6 Clinicians should be aware of MAI as a cause of primary cutaneous infections presenting as lymphocutaneous suppurative nodules and ulcerations.
- Hautmann G, Lotti T. Atypical mycobacterial infections of the skin. Dermatol Clin. 1994;12:657-668.
- Kayal JD, McCall CO. Sporotrichoid cutaneous Mycobacterium avium complex infection. J Am Acad Dermatol. 2002;47(5 suppl):S249-S250.
- Kullavanijaya P, Sirimachan S, Surarak S. Primary cutaneous infection with Mycobacterium avium-intracellulare complex resembling lupus vulgaris. Br J Dermatol. 1997;136:264-266.
- Wood C, Nickoloff BJ, Todes-Taylor NR. Pseudotumor resulting from atypical mycobacterial infection: a “histoid” variety of Mycobacterium avium-intracellulare complex infection. Am J Clin Pathol. 1985;83:524-527.
- Carlos CA, Tang YW, Adler DJ, et al. Mycobacterial infection identified with broad-range PCR amplification and suspension array identification. J Cutan Pathol. 2012;39:795-797.
- Griffith DE, Aksamit T, Brown-Elliot BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
The Diagnosis: Atypical Mycobacterial Infection
Punch biopsy specimens demonstrated necrotizing granulomatous inflammation in the dermis and subcutis (Figure). Special staining for microorganisms was negative. Tissue culture grew Mycobacterium avium-intracellulare (MAI). The patient began treatment with azithromycin, ethambutol, and rifabutin. Tissue susceptibilities later showed resistance to rifabutin and sensitivity to clarithromycin, moxifloxacin, and clofazimine. She subsequently was switched to azithromycin, clofazimine, and moxifloxacin with good response.
Mycobacterium avium-intracellulare is a slow-growing, nonchromogenic, atypical mycobacteria. Although ubiquitous, it tends to only cause serious infection in the setting of immunosuppression. Transmission usually is through the respiratory or gastrointestinal tract.1 Skin infections with MAI are uncommon and usually are secondary to seeding from disseminated infection or from direct inoculation.2
The clinical presentations of primary cutaneous MAI are myriad, including an isolated red nodule, multiple ulcers, abscesses, draining sinuses, facial nodules, granulomatous plaques, and panniculitis.2,3 Of 3 reported cases of primary cutaneous MAI in the form of sporotrichoid lesions, 2 involved patients with AIDS2 and 1 involved a cardiac transplant recipient.4
Cutaneous MAI is typically diagnosed with skin biopsy and tissue culture. Tissue culture is critical for determining the specific mycobacterial species and antibiotic susceptibilities. Polymerase chain reaction has been utilized to rapidly diagnose cutaneous MAI infection from an acid-fast bacilli–positive tissue sample in which the tissue culture was negative.5
Recommended treatment protocols for MAI involve multidrug regimens because of the intrinsic resistance of MAI and the concern for development of resistance with monotherapy.2 No definitive guidelines exist for treatment of primary cutaneous MAI infections. However, regimens for the treatment of pulmonary infection that also have been successfully utilized for cutaneous infection include a macrolide, ethambutol, and a rifamycin.6 Clinicians should be aware of MAI as a cause of primary cutaneous infections presenting as lymphocutaneous suppurative nodules and ulcerations.
The Diagnosis: Atypical Mycobacterial Infection
Punch biopsy specimens demonstrated necrotizing granulomatous inflammation in the dermis and subcutis (Figure). Special staining for microorganisms was negative. Tissue culture grew Mycobacterium avium-intracellulare (MAI). The patient began treatment with azithromycin, ethambutol, and rifabutin. Tissue susceptibilities later showed resistance to rifabutin and sensitivity to clarithromycin, moxifloxacin, and clofazimine. She subsequently was switched to azithromycin, clofazimine, and moxifloxacin with good response.
Mycobacterium avium-intracellulare is a slow-growing, nonchromogenic, atypical mycobacteria. Although ubiquitous, it tends to only cause serious infection in the setting of immunosuppression. Transmission usually is through the respiratory or gastrointestinal tract.1 Skin infections with MAI are uncommon and usually are secondary to seeding from disseminated infection or from direct inoculation.2
The clinical presentations of primary cutaneous MAI are myriad, including an isolated red nodule, multiple ulcers, abscesses, draining sinuses, facial nodules, granulomatous plaques, and panniculitis.2,3 Of 3 reported cases of primary cutaneous MAI in the form of sporotrichoid lesions, 2 involved patients with AIDS2 and 1 involved a cardiac transplant recipient.4
Cutaneous MAI is typically diagnosed with skin biopsy and tissue culture. Tissue culture is critical for determining the specific mycobacterial species and antibiotic susceptibilities. Polymerase chain reaction has been utilized to rapidly diagnose cutaneous MAI infection from an acid-fast bacilli–positive tissue sample in which the tissue culture was negative.5
Recommended treatment protocols for MAI involve multidrug regimens because of the intrinsic resistance of MAI and the concern for development of resistance with monotherapy.2 No definitive guidelines exist for treatment of primary cutaneous MAI infections. However, regimens for the treatment of pulmonary infection that also have been successfully utilized for cutaneous infection include a macrolide, ethambutol, and a rifamycin.6 Clinicians should be aware of MAI as a cause of primary cutaneous infections presenting as lymphocutaneous suppurative nodules and ulcerations.
- Hautmann G, Lotti T. Atypical mycobacterial infections of the skin. Dermatol Clin. 1994;12:657-668.
- Kayal JD, McCall CO. Sporotrichoid cutaneous Mycobacterium avium complex infection. J Am Acad Dermatol. 2002;47(5 suppl):S249-S250.
- Kullavanijaya P, Sirimachan S, Surarak S. Primary cutaneous infection with Mycobacterium avium-intracellulare complex resembling lupus vulgaris. Br J Dermatol. 1997;136:264-266.
- Wood C, Nickoloff BJ, Todes-Taylor NR. Pseudotumor resulting from atypical mycobacterial infection: a “histoid” variety of Mycobacterium avium-intracellulare complex infection. Am J Clin Pathol. 1985;83:524-527.
- Carlos CA, Tang YW, Adler DJ, et al. Mycobacterial infection identified with broad-range PCR amplification and suspension array identification. J Cutan Pathol. 2012;39:795-797.
- Griffith DE, Aksamit T, Brown-Elliot BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Hautmann G, Lotti T. Atypical mycobacterial infections of the skin. Dermatol Clin. 1994;12:657-668.
- Kayal JD, McCall CO. Sporotrichoid cutaneous Mycobacterium avium complex infection. J Am Acad Dermatol. 2002;47(5 suppl):S249-S250.
- Kullavanijaya P, Sirimachan S, Surarak S. Primary cutaneous infection with Mycobacterium avium-intracellulare complex resembling lupus vulgaris. Br J Dermatol. 1997;136:264-266.
- Wood C, Nickoloff BJ, Todes-Taylor NR. Pseudotumor resulting from atypical mycobacterial infection: a “histoid” variety of Mycobacterium avium-intracellulare complex infection. Am J Clin Pathol. 1985;83:524-527.
- Carlos CA, Tang YW, Adler DJ, et al. Mycobacterial infection identified with broad-range PCR amplification and suspension array identification. J Cutan Pathol. 2012;39:795-797.
- Griffith DE, Aksamit T, Brown-Elliot BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
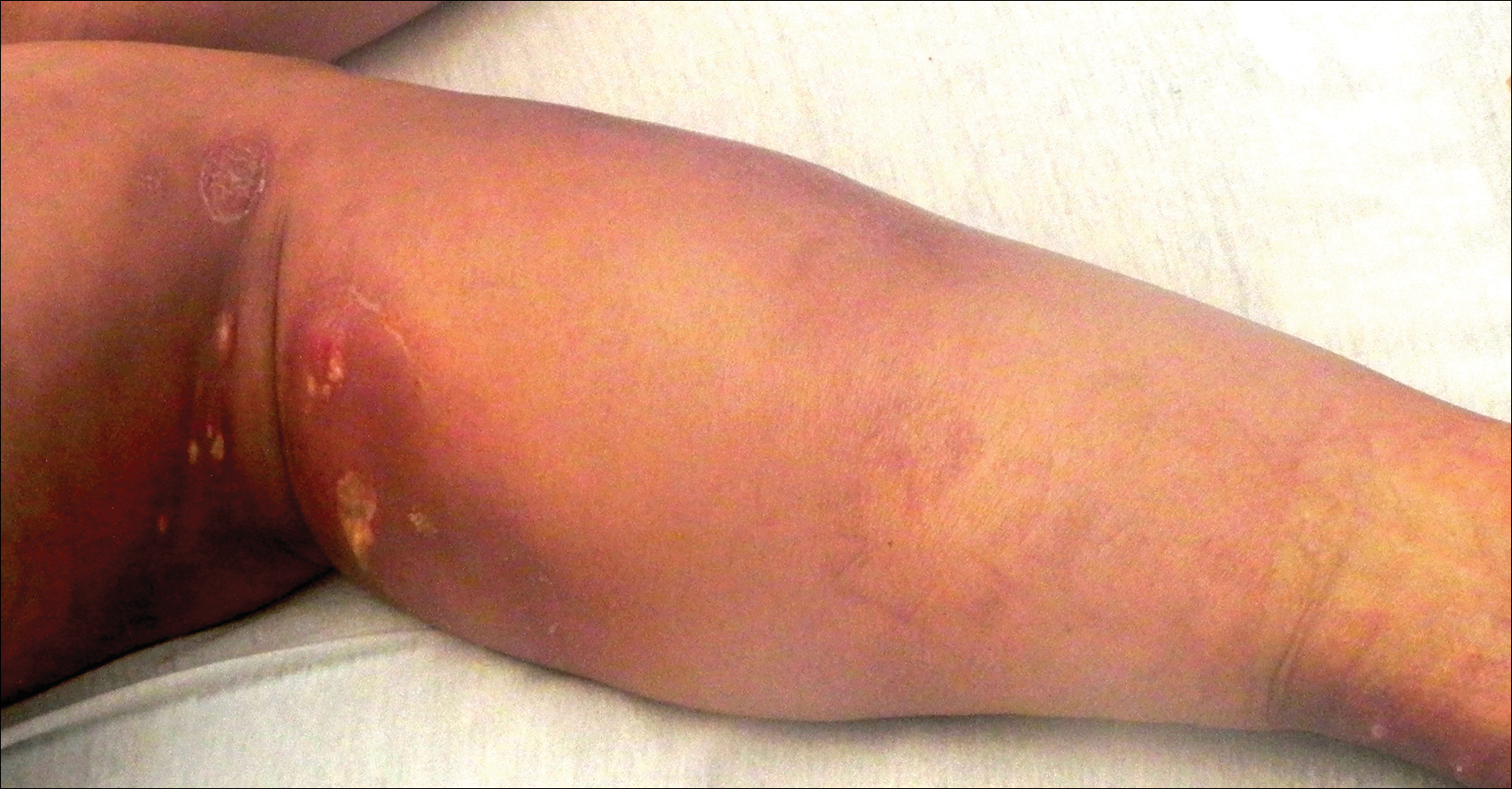
A woman in her 50s presented with low-grade subjective intermittent fevers and painful draining ulcerations on the legs of 7 months’ duration. Her medical history was remarkable for polymyositis and interstitial lung disease managed with prednisone and mycophenolate mofetil. While living in Taiwan, she developed lower extremity abscesses and persistent fevers. The patient denied any skin injuries or exposure to animals or brackish water. Mycophenolate mofetil was discontinued, and she was treated with multiple antibiotics alone and in combination without improvement, including amoxicillin–clavulanic acid, levofloxacin, azithromycin, moxifloxacin, rifampin, rifabutin, and ethambutol. She returned to the United States for evaluation. Physical examination revealed ulcerations with purulent drainage and interconnected sinus tracts with rare fluctuant nodules along the right leg. A single similar lesion was present on the right chest wall. There was no clinical evidence of disseminated disease.