User login
Flesh-Colored Papular Eruption
Papular Mucinosis/Scleromyxedema
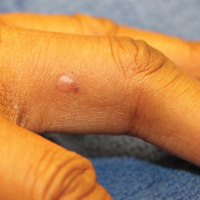
Papular mucinosis/scleromyxedema, also known as generalized lichen myxedematosus, is a rare dermal mucinosis characterized by a papular eruption that can have an associated IgG λ paraproteinemia. The clinical presentation is gradual with the development of firm, flesh-colored, 2- to 3-mm papules often involving the hands, face, and neck that can progress to plaques that cover the entire body. Skin stiffening also can be seen.1 Extracutaneous symptoms are common and include dysphagia, arthralgia, myopathy, and cardiac dysfunction.2 Occasionally, central nervous system involvement can lead to the often fatal dermato-neuro syndrome.3,4
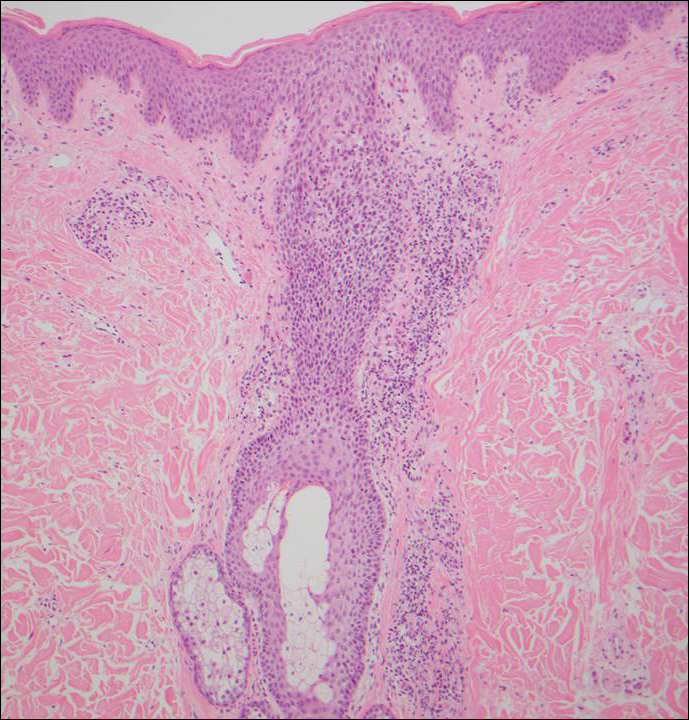
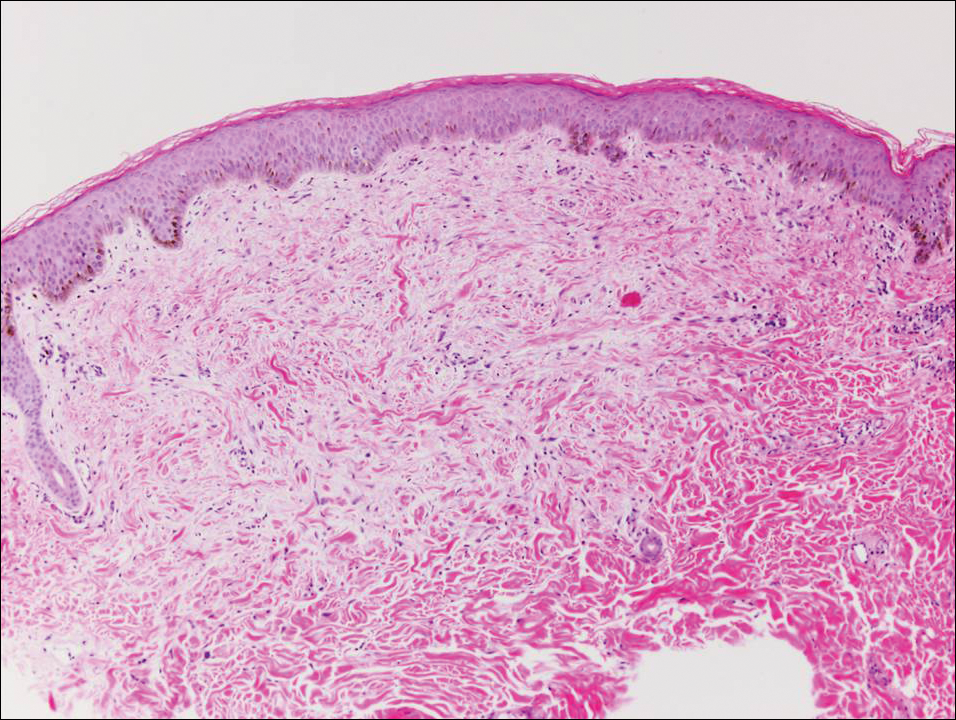
Histologically, papular mucinosis/scleromyxedema demonstrates increased, irregularly arranged fibroblasts in the reticular dermis with increased dermal mucin deposition (quiz image and Figure 1). The epidermis is normal or slightly thinned due to pressure from dermal changes. There may be a mild superficial perivascular lymphocytic infiltrate and atrophy of hair follicles.5 In this case, the clinical and histologic findings best supported a diagnosis of papular mucinosis/scleromyxedema.

Infundibulofolliculitis is a pruritic follicular papular eruption typically involving the neck, trunk, and proximal upper arms and shoulders. It is most common in black men who reside in hot and humid climates. Although infundibulofolliculitis would be included in the clinical differential diagnosis for the current patient, the histopathologic findings were quite distinct for the correct diagnosis of papular mucinosis/scleromyxedema. Infundibulofolliculitis shows widening of the upper part of the hair follicle (infundibulum) and infundibular inflammatory infiltrate with follicular spongiosis (Figure 2). Neither mucin deposition nor fibroblast proliferation is appreciated in infundibulofolliculitis.6,7
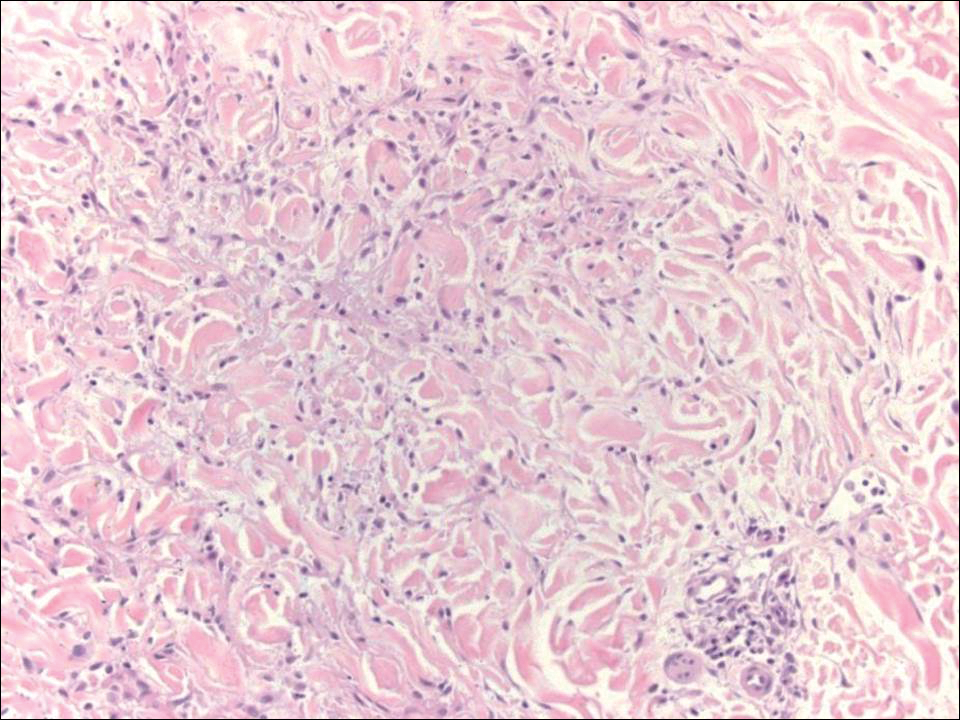
Granuloma annulare (GA) often can be distinguished clinically from papular mucinosis/scleromyxedema due to the annular appearance of papules and plaques in GA and the lack of stiffness of underlying skin. Interstitial granuloma annulare is a histologic variant of GA that can be included in the histologic differential diagnosis of papular mucinosis/scleromyxedema. Histologically, there is an interstitial infiltrate of cytologically bland histiocytes dissecting between collagen bundles in interstitial GA (Figure 3). Necrobiosis and collections of mucin often are inconspicuous. Occasionally, the presence of eosinophils can be a helpful clue.8 A fibroblast proliferation is not a feature of GA.
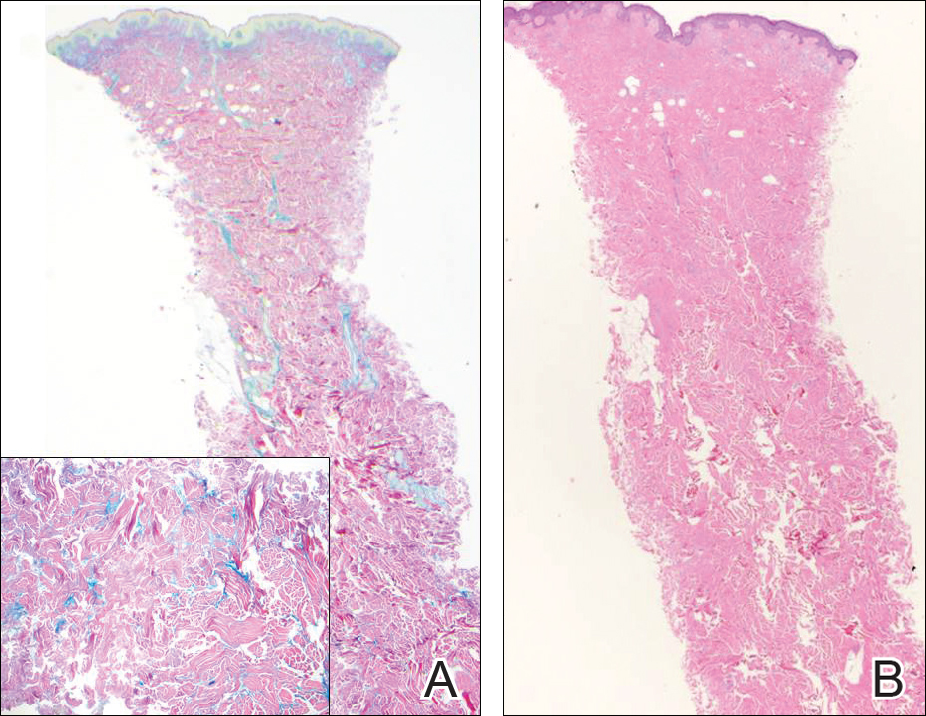
Reticular erythematous mucinosis also is a type of cutaneous mucinosis but with a classic clinical appearance of a reticulated erythematous plaque on the chest or back, making it clinically distinct from papular mucinosis/scleromyxedema and the presentation described in the current patient. Reticular erythematous mucinosis can be histologically distinguished from papular mucinosis/scleromyxedema by the presence of a superficial and deep perivascular lymphocytic infiltrate with increased dermal mucin deposition (Figure 4). It often shows a positive IgM deposition on the basement membrane on direct immunofluorescence.9

Similar to papular mucinosis/scleromyxedema, scleredema shows thickening of the skin with decreased movement of involved areas. Scleredema often involves the upper back, shoulders, and neck where affected areas often have a peau d'orange appearance. Scleredema is classified into 3 clinical forms based on clinical associations. Type 1 often is preceded by an infection, classically Streptococcus pyogenes. Type 2 is associated with a hematologic dyscrasia such as multiple myeloma, or it can have an associated paraproteinemia that is typically of the IgA κ type, which is distinct from papular mucinosis/scleromyxedema where IgG λ paraproteinemia typically is seen. Type 3 is associated with diabetes mellitus. Histologically, scleredema also is distinct from papular mucinosis/scleromyxedema. Although increased mucin is seen in the dermis, the mucin is classically more prominent in the deep reticular dermis as compared with papular mucinosis/scleromyxedema (Figure 5). Additionally, collagen bundles are thickened with clear separation between them. Hyperplasia of fibroblasts in the dermis that is a characteristic feature of papular mucinosis/scleromyxedema is not observed in scleredema.10
- Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Fleming KE, Virmani D, Sutton E, et al. Scleromyxedema and the dermato-neuro syndrome: case report and review of the literature. J Cutan Pathol. 2012;39:508-517.
- Hummers LK. Scleromyxedema. Curr Opin Rheumatol. 2014;26:658-662.
- Rongioleti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Owen WR, Wood C. Disseminate and recurrent infundibulofolliculitis. Arch Dermatol. 1979;5:174-175.
- Soyinka F. Recurrent disseminated infundibulofolliculitis. Int J Dermatol. 1973;12:314-317.
- Keimig EL. Granuloma annulare. Dermatol Clin. 2015;33:315-329.
- Thareja S, Paghdal K, Lein MH, et al. Reticular erythematous mucinosis--a review. Int J Dermatol. 2012;51:903-909.
- Beers WH, Ince AI, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
Papular Mucinosis/Scleromyxedema
Papular mucinosis/scleromyxedema, also known as generalized lichen myxedematosus, is a rare dermal mucinosis characterized by a papular eruption that can have an associated IgG λ paraproteinemia. The clinical presentation is gradual with the development of firm, flesh-colored, 2- to 3-mm papules often involving the hands, face, and neck that can progress to plaques that cover the entire body. Skin stiffening also can be seen.1 Extracutaneous symptoms are common and include dysphagia, arthralgia, myopathy, and cardiac dysfunction.2 Occasionally, central nervous system involvement can lead to the often fatal dermato-neuro syndrome.3,4
Histologically, papular mucinosis/scleromyxedema demonstrates increased, irregularly arranged fibroblasts in the reticular dermis with increased dermal mucin deposition (quiz image and Figure 1). The epidermis is normal or slightly thinned due to pressure from dermal changes. There may be a mild superficial perivascular lymphocytic infiltrate and atrophy of hair follicles.5 In this case, the clinical and histologic findings best supported a diagnosis of papular mucinosis/scleromyxedema.
Infundibulofolliculitis is a pruritic follicular papular eruption typically involving the neck, trunk, and proximal upper arms and shoulders. It is most common in black men who reside in hot and humid climates. Although infundibulofolliculitis would be included in the clinical differential diagnosis for the current patient, the histopathologic findings were quite distinct for the correct diagnosis of papular mucinosis/scleromyxedema. Infundibulofolliculitis shows widening of the upper part of the hair follicle (infundibulum) and infundibular inflammatory infiltrate with follicular spongiosis (Figure 2). Neither mucin deposition nor fibroblast proliferation is appreciated in infundibulofolliculitis.6,7
Granuloma annulare (GA) often can be distinguished clinically from papular mucinosis/scleromyxedema due to the annular appearance of papules and plaques in GA and the lack of stiffness of underlying skin. Interstitial granuloma annulare is a histologic variant of GA that can be included in the histologic differential diagnosis of papular mucinosis/scleromyxedema. Histologically, there is an interstitial infiltrate of cytologically bland histiocytes dissecting between collagen bundles in interstitial GA (Figure 3). Necrobiosis and collections of mucin often are inconspicuous. Occasionally, the presence of eosinophils can be a helpful clue.8 A fibroblast proliferation is not a feature of GA.
Reticular erythematous mucinosis also is a type of cutaneous mucinosis but with a classic clinical appearance of a reticulated erythematous plaque on the chest or back, making it clinically distinct from papular mucinosis/scleromyxedema and the presentation described in the current patient. Reticular erythematous mucinosis can be histologically distinguished from papular mucinosis/scleromyxedema by the presence of a superficial and deep perivascular lymphocytic infiltrate with increased dermal mucin deposition (Figure 4). It often shows a positive IgM deposition on the basement membrane on direct immunofluorescence.9
Similar to papular mucinosis/scleromyxedema, scleredema shows thickening of the skin with decreased movement of involved areas. Scleredema often involves the upper back, shoulders, and neck where affected areas often have a peau d'orange appearance. Scleredema is classified into 3 clinical forms based on clinical associations. Type 1 often is preceded by an infection, classically Streptococcus pyogenes. Type 2 is associated with a hematologic dyscrasia such as multiple myeloma, or it can have an associated paraproteinemia that is typically of the IgA κ type, which is distinct from papular mucinosis/scleromyxedema where IgG λ paraproteinemia typically is seen. Type 3 is associated with diabetes mellitus. Histologically, scleredema also is distinct from papular mucinosis/scleromyxedema. Although increased mucin is seen in the dermis, the mucin is classically more prominent in the deep reticular dermis as compared with papular mucinosis/scleromyxedema (Figure 5). Additionally, collagen bundles are thickened with clear separation between them. Hyperplasia of fibroblasts in the dermis that is a characteristic feature of papular mucinosis/scleromyxedema is not observed in scleredema.10
Papular Mucinosis/Scleromyxedema
Papular mucinosis/scleromyxedema, also known as generalized lichen myxedematosus, is a rare dermal mucinosis characterized by a papular eruption that can have an associated IgG λ paraproteinemia. The clinical presentation is gradual with the development of firm, flesh-colored, 2- to 3-mm papules often involving the hands, face, and neck that can progress to plaques that cover the entire body. Skin stiffening also can be seen.1 Extracutaneous symptoms are common and include dysphagia, arthralgia, myopathy, and cardiac dysfunction.2 Occasionally, central nervous system involvement can lead to the often fatal dermato-neuro syndrome.3,4
Histologically, papular mucinosis/scleromyxedema demonstrates increased, irregularly arranged fibroblasts in the reticular dermis with increased dermal mucin deposition (quiz image and Figure 1). The epidermis is normal or slightly thinned due to pressure from dermal changes. There may be a mild superficial perivascular lymphocytic infiltrate and atrophy of hair follicles.5 In this case, the clinical and histologic findings best supported a diagnosis of papular mucinosis/scleromyxedema.
Infundibulofolliculitis is a pruritic follicular papular eruption typically involving the neck, trunk, and proximal upper arms and shoulders. It is most common in black men who reside in hot and humid climates. Although infundibulofolliculitis would be included in the clinical differential diagnosis for the current patient, the histopathologic findings were quite distinct for the correct diagnosis of papular mucinosis/scleromyxedema. Infundibulofolliculitis shows widening of the upper part of the hair follicle (infundibulum) and infundibular inflammatory infiltrate with follicular spongiosis (Figure 2). Neither mucin deposition nor fibroblast proliferation is appreciated in infundibulofolliculitis.6,7
Granuloma annulare (GA) often can be distinguished clinically from papular mucinosis/scleromyxedema due to the annular appearance of papules and plaques in GA and the lack of stiffness of underlying skin. Interstitial granuloma annulare is a histologic variant of GA that can be included in the histologic differential diagnosis of papular mucinosis/scleromyxedema. Histologically, there is an interstitial infiltrate of cytologically bland histiocytes dissecting between collagen bundles in interstitial GA (Figure 3). Necrobiosis and collections of mucin often are inconspicuous. Occasionally, the presence of eosinophils can be a helpful clue.8 A fibroblast proliferation is not a feature of GA.
Reticular erythematous mucinosis also is a type of cutaneous mucinosis but with a classic clinical appearance of a reticulated erythematous plaque on the chest or back, making it clinically distinct from papular mucinosis/scleromyxedema and the presentation described in the current patient. Reticular erythematous mucinosis can be histologically distinguished from papular mucinosis/scleromyxedema by the presence of a superficial and deep perivascular lymphocytic infiltrate with increased dermal mucin deposition (Figure 4). It often shows a positive IgM deposition on the basement membrane on direct immunofluorescence.9
Similar to papular mucinosis/scleromyxedema, scleredema shows thickening of the skin with decreased movement of involved areas. Scleredema often involves the upper back, shoulders, and neck where affected areas often have a peau d'orange appearance. Scleredema is classified into 3 clinical forms based on clinical associations. Type 1 often is preceded by an infection, classically Streptococcus pyogenes. Type 2 is associated with a hematologic dyscrasia such as multiple myeloma, or it can have an associated paraproteinemia that is typically of the IgA κ type, which is distinct from papular mucinosis/scleromyxedema where IgG λ paraproteinemia typically is seen. Type 3 is associated with diabetes mellitus. Histologically, scleredema also is distinct from papular mucinosis/scleromyxedema. Although increased mucin is seen in the dermis, the mucin is classically more prominent in the deep reticular dermis as compared with papular mucinosis/scleromyxedema (Figure 5). Additionally, collagen bundles are thickened with clear separation between them. Hyperplasia of fibroblasts in the dermis that is a characteristic feature of papular mucinosis/scleromyxedema is not observed in scleredema.10
- Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Fleming KE, Virmani D, Sutton E, et al. Scleromyxedema and the dermato-neuro syndrome: case report and review of the literature. J Cutan Pathol. 2012;39:508-517.
- Hummers LK. Scleromyxedema. Curr Opin Rheumatol. 2014;26:658-662.
- Rongioleti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Owen WR, Wood C. Disseminate and recurrent infundibulofolliculitis. Arch Dermatol. 1979;5:174-175.
- Soyinka F. Recurrent disseminated infundibulofolliculitis. Int J Dermatol. 1973;12:314-317.
- Keimig EL. Granuloma annulare. Dermatol Clin. 2015;33:315-329.
- Thareja S, Paghdal K, Lein MH, et al. Reticular erythematous mucinosis--a review. Int J Dermatol. 2012;51:903-909.
- Beers WH, Ince AI, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
- Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Fleming KE, Virmani D, Sutton E, et al. Scleromyxedema and the dermato-neuro syndrome: case report and review of the literature. J Cutan Pathol. 2012;39:508-517.
- Hummers LK. Scleromyxedema. Curr Opin Rheumatol. 2014;26:658-662.
- Rongioleti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Owen WR, Wood C. Disseminate and recurrent infundibulofolliculitis. Arch Dermatol. 1979;5:174-175.
- Soyinka F. Recurrent disseminated infundibulofolliculitis. Int J Dermatol. 1973;12:314-317.
- Keimig EL. Granuloma annulare. Dermatol Clin. 2015;33:315-329.
- Thareja S, Paghdal K, Lein MH, et al. Reticular erythematous mucinosis--a review. Int J Dermatol. 2012;51:903-909.
- Beers WH, Ince AI, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
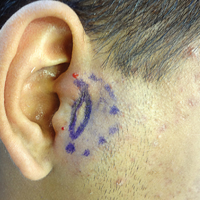
A 48-year-old black man presented with a rash of 7 months' duration that started on the face and spread to the body. He had extreme pruritus, increased stiffness in the hands and joints, and paresthesia. Physical examination revealed an eruption of 2- to 4-mm, flesh-colored papules with follicular accentuation on the face, neck, bilateral upper extremities, back, and thighs.
Sarcoidosis and Squamous Cell Carcinoma: A Connection Documented in a Case Series of 3 Patients
Sarcoidosis is a multisystem granulomatous disease of unknown etiology that most commonly affects the lungs, eyes, and skin. Cutaneous involvement is reported in 25% to 35% of patients with sarcoidosis and may occur in a variety of forms including macules, papules, plaques, and lupus pernio.1,2 Dermatologists commonly are confronted with the diagnosis and management of sarcoidosis because of its high incidence of cutaneous involvement. Due to the protean nature of the disease, skin biopsy plays a key role in confirming the diagnosis. Histological evidence of noncaseating granulomas in combination with an appropriate clinical and radiographic picture is necessary for the diagnosis of sarcoidosis.1,2 Brincker and Wilbek
We describe 3 patients with sarcoidosis who developed squamous cell carcinoma (SCC) of the skin, including 2 black patients, which highlights the potential for SCC development.
Case Reports
Patient 1
A black woman in her 60s with a history of sarcoidosis affecting the lungs and skin that was well controlled with biweekly adalimumab 40 mg subcutaneous injections presented with a new dark painful lesion on the right third finger. She reported the lesion had been present for 1 to 2 years prior to the current presentation and was increasing in size. She had no history of prior skin cancers.
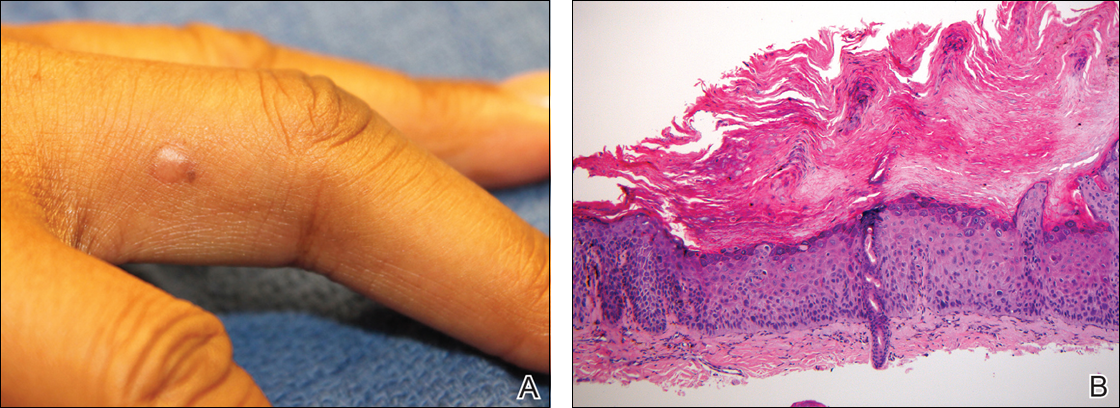
Physical examination revealed a waxy, brown-pigmented papule with overlying scale on the ulnar aspect of the right third digit near the web space (Figure 1A). A shave biopsy revealed atypical keratinocytes involving all layers of the epidermis along with associated parakeratotic scale consistent with a diagnosis of SCC in situ (Figure 1B). Human papillomavirus staining was negative. Due to the location of the lesion, the patient underwent Mohs micrographic surgery and the lesion was completely excised.
Patient 2
A black woman in her 60s with a history of cutaneous sarcoidosis that was maintained on minocycline 100 mg twice daily, chloroquine 250 mg daily, tacrolimus ointment 0.1%, tretinoin cream 0.025%, and intermittent intralesional triamcinolone acetonide injections to the nose, as well as quiescent pulmonary sarcoidosis, developed a new, growing, asymptomatic, hyperpigmented lesion on the left side of the submandibular neck over a period of a few months. A biopsy was performed and the lesion was found to be an SCC, which subsequently was completely excised.
Patient 3
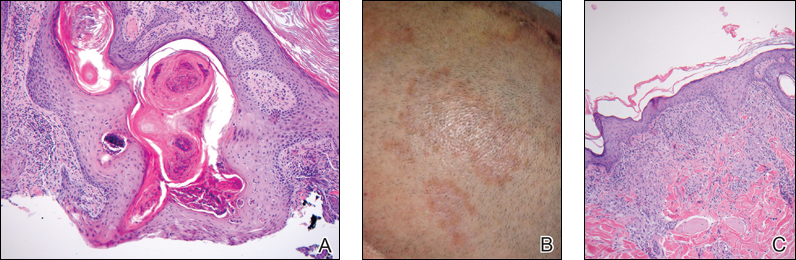
A white man in his 60s with a history of prior quiescent pulmonary sarcoidosis, remote melanoma, and multiple nonmelanoma skin cancers developed scaly papules on the scalp for months, one that was interpreted by an outside pathologist as an invasive SCC (Figure 2A). He was referred to our institution for Mohs micrographic surgery. On presentation when his scalp was shaved for surgery, he was noted to have several violaceous, annular, thin plaques on the scalp (Figure 2B). A biopsy of an annular plaque demonstrated several areas of granulomatous dermatitis consistent with a diagnosis of cutaneous sarcoidosis (Figure 2C). The patient had clinical lymphadenopathy of the neck and supraclavicular region. Given the patient’s history, the differential diagnosis for these lesions included metastatic SCC, lymphoma, and sarcoidosis. The patient underwent a positron emission tomography scan, which demonstrated fluorodeoxyglucose-positive regions in both lungs and the right side of the neck. After evaluation by the pulmonary and otorhinolaryngology departments, including a lymph node biopsy, the positron emission tomography–enhancing lesions were ultimately determined to be consistent with sarcoidosis.
The patient underwent Mohs micrographic surgery for treatment of the scalp SCC and was started on triamcinolone cream 0.1% for the body, clobetasol propionate foam 0.05% for the scalp, and hydroxychloroquine sulfate 400 mg daily for the cutaneous sarcoidosis. His annular scalp lesions resolved, but over the following 12 months the patient had numerous clinically suspicious skin lesions that were biopsied and were consistent with multiple basal cell carcinomas, actinic keratoses, and SCC in situ. They were treated with surgery, cryosurgical destruction with liquid nitrogen, and 5-fluorouracil cream.
Over the 3 years subsequent to initial presentation, the patient developed ocular inflammation attributed to his sarcoidosis and atrial fibrillation, which was determined to be unrelated. He also developed 5 scaly hyperkeratotic plaques on the vertex aspect of the scalp. Biopsy of 2 lesions revealed mild keratinocyte atypia and epidermal hyperplasia, favored to represent SCC over pseudoepitheliomatous hyperplasia overlying associated granulomatous inflammation. These lesions ultimately were believed to represent new SCCs, while biopsies of 2 other lesions revealed isolated granulomatous inflammation that was believed to represent hyperkeratotic cutaneous sarcoidosis clinically resembling his SCCs. The patient was again referred for Mohs micrographic surgery and the malignancies were completely removed, while the cutaneous sarcoidosis was again treated with topical corticosteroids with complete resolution.
Comment
The potential increased risk for malignancy in patients with sarcoidosis has been well documented.3-6 Brincker and Wilbek3 first reported this association after studying 2544 patients with pulmonary sarcoidosis from 1962 to 1971. In particular, they noted a difference between the expected and observed number of cases of malignancy, particularly lung cancer and lymphoma, in the sarcoidosis population.3 In a study of 10,037 hospitalized sarcoidosis patients from 1964 to 2004, Ji et al5 noted a 40% overall increase in the incidence of cancer and found that the risk for malignancy was highest in the year following hospitalization. Interestingly, they found that the risk for developing cutaneous SCC was elevated in sarcoidosis patients even after the first year following hospitalization.5 In a retrospective cohort study examining more than 9000 patients, Askling et al4 also confirmed the increased incidence of malignancy in sarcoidosis patients. Specifically, the authors found a higher than expected occurrence of skin cancer, both melanoma (standardized incidence ratio, 1.6; 95% confidence interval, 1.1-2.3) and nonmelanoma skin cancer (standardized incidence ratio, 2.8; 95% confidence interval, 2.0-3.8) in patients with sarcoidosis.4 Reich et al7 cross-matched 30,000 cases from the Kaiser Permanente Northwest Region Tumor Registry against a sarcoidosis registry of 243 cases to evaluate for evidence of linkage between sarcoidosis and malignancy. They concluded that there may be an etiologic relationship between sarcoidosis and malignancy in at least one-quarter of cases in which both are present and hypothesized that granulomas may be the result of a cell-mediated reaction to tumor antigens.7
Few published studies specifically address the incidence of malignancy in patients with primarily cutaneous sarcoidosis. Cutaneous sarcoidosis includes nonspecific lesions, such as erythema nodosum, as well as specific lesions, such as papules, plaques, nodules, and lupus pernio.8 Alexandrescu et al6 evaluated 110 patients with a diagnosis of both sarcoidosis (cutaneous and noncutaneous) and malignancy. Through their analysis, they found that cutaneous sarcoidosis is seen more commonly in patients presenting with sarcoidosis and malignancy (56.4%) than in the total sarcoidosis population (20%–25%). From these findings, the authors concluded that cutaneous sarcoidosis appears to be a subtype of sarcoidosis associated with cancer.6
We report 3 cases that specifically illustrate a link between cutaneous sarcoidosis and an increased risk for cutaneous SCC. Because sarcoidosis commonly affects the skin, patients often present to dermatologists for care. Once the initial diagnosis of cutaneous sarcoidosis is made via biopsy, it is natural to be tempted to attribute any new skin lesions to worsening or active disease; however, as cutaneous sarcoidosis may take on a variety of nonspecific forms, it is important to biopsy any unusual lesions. In our case series, patient 3 presented at several different points with scaly scalp lesions. Upon biopsy, several of these lesions were found to be SCCs, while others demonstrated regions of granulomatous inflammation consistent with a diagnosis of cutaneous sarcoidosis. On further review of pathology during the preparation of this manuscript after the initial diagnoses were made, it was further noted that it is challenging to distinguish granulomatous inflammation with reactive pseudoepitheliomatous hyperplasia from SCC. The fact that these lesions were clinically indistinguishable illustrates the critical importance of appropriate-depth biopsy in this situation, and the histopathologic challenges highlighted herein are important for pathologists to remember.
Patients 1 and 2 were both black women, and the fact that these patients both presented with cutaneous SCCs—one of whom was immunosuppressed due to treatment with adalimumab, the other without systemic immunosuppression—exemplifies the need for comprehensive skin examinations in sarcoidosis patients as well as for biopsies of new or unusual lesions.
The mechanism for the development of malignancy in patients with sarcoidosis is unknown and likely is multifactorial. Multiple theories have been proposed.1,2,5,6,8 Sarcoidosis is marked by the development of granulomas secondary to the interaction between CD4+ T cells and antigen-presenting cells, which is mediated by various cytokines and chemokines, including IL-2 and IFN-γ. Patients with sarcoidosis have been found to have oligoclonal T-cell lineages with a limited receptor repertoire, suggestive of selective immune system activation, as well as a deficiency of certain types of regulatory cells, namely natural killer cells.1,2 This immune dysregulation has been postulated to play an etiologic role in the development of malignancy in sarcoidosis patients.1,2,5 Furthermore, the chronic inflammation found in the organs commonly affected by both sarcoidosis and malignancy is another possible mechanism.6,8 Finally, immunosuppression and mutagenesis secondary to the treatment modalities used in sarcoidosis may be another contributing factor.6
Conclusion
An association between sarcoidosis and malignancy has been suggested for several decades. We specifically report 3 cases of patients with cutaneous sarcoidosis who presented with concurrent cutaneous SCCs. Given the varied and often nonspecific nature of cutaneous sarcoidosis, these cases highlight the importance of biopsy when sarcoidosis patients present with new and unusual skin lesions. Additionally, they illustrate the importance of thorough skin examinations in sarcoidosis patients as well as some of the challenges these patients pose for dermatologists.
- Iannuzzi MC, Rybicki BA, Teirsten AS. Sarcoidosis. N Engl J Med. 2007;357:2153-2165.
- Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis and therapeutics. JAMA. 2011;305:391-399.
- Brincker H, Wilbek E. The incidence of malignant tumours in patients with respiratory sarcoidosis. Br J Cancer. 1974;29:247-251.
- Askling J, Grunewald J, Eklund A, et al. Increased risk for cancer following sarcoidosis. Am J Respir Crit Care Med. 1999;160(5, pt 1):1668-1672.
- Ji J, Shu X, Li X, et al. Cancer risk in hospitalized sarcoidosis patients: a follow-up study in Sweden. Ann Oncol. 2009;20:1121-1126.
- Alexandrescu DT, Kauffman CL, Ichim TE, et al. Cutaneous sarcoidosis and malignancy: an association between sarcoidosis with skin manifestations and systemic neoplasia. Dermatol Online J. 2011;17:2.
- Reich JM, Mullooly JP, Johnson RE. Linkage analysis of malignancy-associated sarcoidosis. Chest. 1995;107:605-613.
- Cohen PR, Kurzrock R. Sarcoidosis and malignancy. Clin Dermatol. 2007;25:326-333.
Sarcoidosis is a multisystem granulomatous disease of unknown etiology that most commonly affects the lungs, eyes, and skin. Cutaneous involvement is reported in 25% to 35% of patients with sarcoidosis and may occur in a variety of forms including macules, papules, plaques, and lupus pernio.1,2 Dermatologists commonly are confronted with the diagnosis and management of sarcoidosis because of its high incidence of cutaneous involvement. Due to the protean nature of the disease, skin biopsy plays a key role in confirming the diagnosis. Histological evidence of noncaseating granulomas in combination with an appropriate clinical and radiographic picture is necessary for the diagnosis of sarcoidosis.1,2 Brincker and Wilbek
We describe 3 patients with sarcoidosis who developed squamous cell carcinoma (SCC) of the skin, including 2 black patients, which highlights the potential for SCC development.
Case Reports
Patient 1
A black woman in her 60s with a history of sarcoidosis affecting the lungs and skin that was well controlled with biweekly adalimumab 40 mg subcutaneous injections presented with a new dark painful lesion on the right third finger. She reported the lesion had been present for 1 to 2 years prior to the current presentation and was increasing in size. She had no history of prior skin cancers.
Physical examination revealed a waxy, brown-pigmented papule with overlying scale on the ulnar aspect of the right third digit near the web space (Figure 1A). A shave biopsy revealed atypical keratinocytes involving all layers of the epidermis along with associated parakeratotic scale consistent with a diagnosis of SCC in situ (Figure 1B). Human papillomavirus staining was negative. Due to the location of the lesion, the patient underwent Mohs micrographic surgery and the lesion was completely excised.
Patient 2
A black woman in her 60s with a history of cutaneous sarcoidosis that was maintained on minocycline 100 mg twice daily, chloroquine 250 mg daily, tacrolimus ointment 0.1%, tretinoin cream 0.025%, and intermittent intralesional triamcinolone acetonide injections to the nose, as well as quiescent pulmonary sarcoidosis, developed a new, growing, asymptomatic, hyperpigmented lesion on the left side of the submandibular neck over a period of a few months. A biopsy was performed and the lesion was found to be an SCC, which subsequently was completely excised.
Patient 3
A white man in his 60s with a history of prior quiescent pulmonary sarcoidosis, remote melanoma, and multiple nonmelanoma skin cancers developed scaly papules on the scalp for months, one that was interpreted by an outside pathologist as an invasive SCC (Figure 2A). He was referred to our institution for Mohs micrographic surgery. On presentation when his scalp was shaved for surgery, he was noted to have several violaceous, annular, thin plaques on the scalp (Figure 2B). A biopsy of an annular plaque demonstrated several areas of granulomatous dermatitis consistent with a diagnosis of cutaneous sarcoidosis (Figure 2C). The patient had clinical lymphadenopathy of the neck and supraclavicular region. Given the patient’s history, the differential diagnosis for these lesions included metastatic SCC, lymphoma, and sarcoidosis. The patient underwent a positron emission tomography scan, which demonstrated fluorodeoxyglucose-positive regions in both lungs and the right side of the neck. After evaluation by the pulmonary and otorhinolaryngology departments, including a lymph node biopsy, the positron emission tomography–enhancing lesions were ultimately determined to be consistent with sarcoidosis.
The patient underwent Mohs micrographic surgery for treatment of the scalp SCC and was started on triamcinolone cream 0.1% for the body, clobetasol propionate foam 0.05% for the scalp, and hydroxychloroquine sulfate 400 mg daily for the cutaneous sarcoidosis. His annular scalp lesions resolved, but over the following 12 months the patient had numerous clinically suspicious skin lesions that were biopsied and were consistent with multiple basal cell carcinomas, actinic keratoses, and SCC in situ. They were treated with surgery, cryosurgical destruction with liquid nitrogen, and 5-fluorouracil cream.
Over the 3 years subsequent to initial presentation, the patient developed ocular inflammation attributed to his sarcoidosis and atrial fibrillation, which was determined to be unrelated. He also developed 5 scaly hyperkeratotic plaques on the vertex aspect of the scalp. Biopsy of 2 lesions revealed mild keratinocyte atypia and epidermal hyperplasia, favored to represent SCC over pseudoepitheliomatous hyperplasia overlying associated granulomatous inflammation. These lesions ultimately were believed to represent new SCCs, while biopsies of 2 other lesions revealed isolated granulomatous inflammation that was believed to represent hyperkeratotic cutaneous sarcoidosis clinically resembling his SCCs. The patient was again referred for Mohs micrographic surgery and the malignancies were completely removed, while the cutaneous sarcoidosis was again treated with topical corticosteroids with complete resolution.
Comment
The potential increased risk for malignancy in patients with sarcoidosis has been well documented.3-6 Brincker and Wilbek3 first reported this association after studying 2544 patients with pulmonary sarcoidosis from 1962 to 1971. In particular, they noted a difference between the expected and observed number of cases of malignancy, particularly lung cancer and lymphoma, in the sarcoidosis population.3 In a study of 10,037 hospitalized sarcoidosis patients from 1964 to 2004, Ji et al5 noted a 40% overall increase in the incidence of cancer and found that the risk for malignancy was highest in the year following hospitalization. Interestingly, they found that the risk for developing cutaneous SCC was elevated in sarcoidosis patients even after the first year following hospitalization.5 In a retrospective cohort study examining more than 9000 patients, Askling et al4 also confirmed the increased incidence of malignancy in sarcoidosis patients. Specifically, the authors found a higher than expected occurrence of skin cancer, both melanoma (standardized incidence ratio, 1.6; 95% confidence interval, 1.1-2.3) and nonmelanoma skin cancer (standardized incidence ratio, 2.8; 95% confidence interval, 2.0-3.8) in patients with sarcoidosis.4 Reich et al7 cross-matched 30,000 cases from the Kaiser Permanente Northwest Region Tumor Registry against a sarcoidosis registry of 243 cases to evaluate for evidence of linkage between sarcoidosis and malignancy. They concluded that there may be an etiologic relationship between sarcoidosis and malignancy in at least one-quarter of cases in which both are present and hypothesized that granulomas may be the result of a cell-mediated reaction to tumor antigens.7
Few published studies specifically address the incidence of malignancy in patients with primarily cutaneous sarcoidosis. Cutaneous sarcoidosis includes nonspecific lesions, such as erythema nodosum, as well as specific lesions, such as papules, plaques, nodules, and lupus pernio.8 Alexandrescu et al6 evaluated 110 patients with a diagnosis of both sarcoidosis (cutaneous and noncutaneous) and malignancy. Through their analysis, they found that cutaneous sarcoidosis is seen more commonly in patients presenting with sarcoidosis and malignancy (56.4%) than in the total sarcoidosis population (20%–25%). From these findings, the authors concluded that cutaneous sarcoidosis appears to be a subtype of sarcoidosis associated with cancer.6
We report 3 cases that specifically illustrate a link between cutaneous sarcoidosis and an increased risk for cutaneous SCC. Because sarcoidosis commonly affects the skin, patients often present to dermatologists for care. Once the initial diagnosis of cutaneous sarcoidosis is made via biopsy, it is natural to be tempted to attribute any new skin lesions to worsening or active disease; however, as cutaneous sarcoidosis may take on a variety of nonspecific forms, it is important to biopsy any unusual lesions. In our case series, patient 3 presented at several different points with scaly scalp lesions. Upon biopsy, several of these lesions were found to be SCCs, while others demonstrated regions of granulomatous inflammation consistent with a diagnosis of cutaneous sarcoidosis. On further review of pathology during the preparation of this manuscript after the initial diagnoses were made, it was further noted that it is challenging to distinguish granulomatous inflammation with reactive pseudoepitheliomatous hyperplasia from SCC. The fact that these lesions were clinically indistinguishable illustrates the critical importance of appropriate-depth biopsy in this situation, and the histopathologic challenges highlighted herein are important for pathologists to remember.
Patients 1 and 2 were both black women, and the fact that these patients both presented with cutaneous SCCs—one of whom was immunosuppressed due to treatment with adalimumab, the other without systemic immunosuppression—exemplifies the need for comprehensive skin examinations in sarcoidosis patients as well as for biopsies of new or unusual lesions.
The mechanism for the development of malignancy in patients with sarcoidosis is unknown and likely is multifactorial. Multiple theories have been proposed.1,2,5,6,8 Sarcoidosis is marked by the development of granulomas secondary to the interaction between CD4+ T cells and antigen-presenting cells, which is mediated by various cytokines and chemokines, including IL-2 and IFN-γ. Patients with sarcoidosis have been found to have oligoclonal T-cell lineages with a limited receptor repertoire, suggestive of selective immune system activation, as well as a deficiency of certain types of regulatory cells, namely natural killer cells.1,2 This immune dysregulation has been postulated to play an etiologic role in the development of malignancy in sarcoidosis patients.1,2,5 Furthermore, the chronic inflammation found in the organs commonly affected by both sarcoidosis and malignancy is another possible mechanism.6,8 Finally, immunosuppression and mutagenesis secondary to the treatment modalities used in sarcoidosis may be another contributing factor.6
Conclusion
An association between sarcoidosis and malignancy has been suggested for several decades. We specifically report 3 cases of patients with cutaneous sarcoidosis who presented with concurrent cutaneous SCCs. Given the varied and often nonspecific nature of cutaneous sarcoidosis, these cases highlight the importance of biopsy when sarcoidosis patients present with new and unusual skin lesions. Additionally, they illustrate the importance of thorough skin examinations in sarcoidosis patients as well as some of the challenges these patients pose for dermatologists.
Sarcoidosis is a multisystem granulomatous disease of unknown etiology that most commonly affects the lungs, eyes, and skin. Cutaneous involvement is reported in 25% to 35% of patients with sarcoidosis and may occur in a variety of forms including macules, papules, plaques, and lupus pernio.1,2 Dermatologists commonly are confronted with the diagnosis and management of sarcoidosis because of its high incidence of cutaneous involvement. Due to the protean nature of the disease, skin biopsy plays a key role in confirming the diagnosis. Histological evidence of noncaseating granulomas in combination with an appropriate clinical and radiographic picture is necessary for the diagnosis of sarcoidosis.1,2 Brincker and Wilbek
We describe 3 patients with sarcoidosis who developed squamous cell carcinoma (SCC) of the skin, including 2 black patients, which highlights the potential for SCC development.
Case Reports
Patient 1
A black woman in her 60s with a history of sarcoidosis affecting the lungs and skin that was well controlled with biweekly adalimumab 40 mg subcutaneous injections presented with a new dark painful lesion on the right third finger. She reported the lesion had been present for 1 to 2 years prior to the current presentation and was increasing in size. She had no history of prior skin cancers.
Physical examination revealed a waxy, brown-pigmented papule with overlying scale on the ulnar aspect of the right third digit near the web space (Figure 1A). A shave biopsy revealed atypical keratinocytes involving all layers of the epidermis along with associated parakeratotic scale consistent with a diagnosis of SCC in situ (Figure 1B). Human papillomavirus staining was negative. Due to the location of the lesion, the patient underwent Mohs micrographic surgery and the lesion was completely excised.
Patient 2
A black woman in her 60s with a history of cutaneous sarcoidosis that was maintained on minocycline 100 mg twice daily, chloroquine 250 mg daily, tacrolimus ointment 0.1%, tretinoin cream 0.025%, and intermittent intralesional triamcinolone acetonide injections to the nose, as well as quiescent pulmonary sarcoidosis, developed a new, growing, asymptomatic, hyperpigmented lesion on the left side of the submandibular neck over a period of a few months. A biopsy was performed and the lesion was found to be an SCC, which subsequently was completely excised.
Patient 3
A white man in his 60s with a history of prior quiescent pulmonary sarcoidosis, remote melanoma, and multiple nonmelanoma skin cancers developed scaly papules on the scalp for months, one that was interpreted by an outside pathologist as an invasive SCC (Figure 2A). He was referred to our institution for Mohs micrographic surgery. On presentation when his scalp was shaved for surgery, he was noted to have several violaceous, annular, thin plaques on the scalp (Figure 2B). A biopsy of an annular plaque demonstrated several areas of granulomatous dermatitis consistent with a diagnosis of cutaneous sarcoidosis (Figure 2C). The patient had clinical lymphadenopathy of the neck and supraclavicular region. Given the patient’s history, the differential diagnosis for these lesions included metastatic SCC, lymphoma, and sarcoidosis. The patient underwent a positron emission tomography scan, which demonstrated fluorodeoxyglucose-positive regions in both lungs and the right side of the neck. After evaluation by the pulmonary and otorhinolaryngology departments, including a lymph node biopsy, the positron emission tomography–enhancing lesions were ultimately determined to be consistent with sarcoidosis.
The patient underwent Mohs micrographic surgery for treatment of the scalp SCC and was started on triamcinolone cream 0.1% for the body, clobetasol propionate foam 0.05% for the scalp, and hydroxychloroquine sulfate 400 mg daily for the cutaneous sarcoidosis. His annular scalp lesions resolved, but over the following 12 months the patient had numerous clinically suspicious skin lesions that were biopsied and were consistent with multiple basal cell carcinomas, actinic keratoses, and SCC in situ. They were treated with surgery, cryosurgical destruction with liquid nitrogen, and 5-fluorouracil cream.
Over the 3 years subsequent to initial presentation, the patient developed ocular inflammation attributed to his sarcoidosis and atrial fibrillation, which was determined to be unrelated. He also developed 5 scaly hyperkeratotic plaques on the vertex aspect of the scalp. Biopsy of 2 lesions revealed mild keratinocyte atypia and epidermal hyperplasia, favored to represent SCC over pseudoepitheliomatous hyperplasia overlying associated granulomatous inflammation. These lesions ultimately were believed to represent new SCCs, while biopsies of 2 other lesions revealed isolated granulomatous inflammation that was believed to represent hyperkeratotic cutaneous sarcoidosis clinically resembling his SCCs. The patient was again referred for Mohs micrographic surgery and the malignancies were completely removed, while the cutaneous sarcoidosis was again treated with topical corticosteroids with complete resolution.
Comment
The potential increased risk for malignancy in patients with sarcoidosis has been well documented.3-6 Brincker and Wilbek3 first reported this association after studying 2544 patients with pulmonary sarcoidosis from 1962 to 1971. In particular, they noted a difference between the expected and observed number of cases of malignancy, particularly lung cancer and lymphoma, in the sarcoidosis population.3 In a study of 10,037 hospitalized sarcoidosis patients from 1964 to 2004, Ji et al5 noted a 40% overall increase in the incidence of cancer and found that the risk for malignancy was highest in the year following hospitalization. Interestingly, they found that the risk for developing cutaneous SCC was elevated in sarcoidosis patients even after the first year following hospitalization.5 In a retrospective cohort study examining more than 9000 patients, Askling et al4 also confirmed the increased incidence of malignancy in sarcoidosis patients. Specifically, the authors found a higher than expected occurrence of skin cancer, both melanoma (standardized incidence ratio, 1.6; 95% confidence interval, 1.1-2.3) and nonmelanoma skin cancer (standardized incidence ratio, 2.8; 95% confidence interval, 2.0-3.8) in patients with sarcoidosis.4 Reich et al7 cross-matched 30,000 cases from the Kaiser Permanente Northwest Region Tumor Registry against a sarcoidosis registry of 243 cases to evaluate for evidence of linkage between sarcoidosis and malignancy. They concluded that there may be an etiologic relationship between sarcoidosis and malignancy in at least one-quarter of cases in which both are present and hypothesized that granulomas may be the result of a cell-mediated reaction to tumor antigens.7
Few published studies specifically address the incidence of malignancy in patients with primarily cutaneous sarcoidosis. Cutaneous sarcoidosis includes nonspecific lesions, such as erythema nodosum, as well as specific lesions, such as papules, plaques, nodules, and lupus pernio.8 Alexandrescu et al6 evaluated 110 patients with a diagnosis of both sarcoidosis (cutaneous and noncutaneous) and malignancy. Through their analysis, they found that cutaneous sarcoidosis is seen more commonly in patients presenting with sarcoidosis and malignancy (56.4%) than in the total sarcoidosis population (20%–25%). From these findings, the authors concluded that cutaneous sarcoidosis appears to be a subtype of sarcoidosis associated with cancer.6
We report 3 cases that specifically illustrate a link between cutaneous sarcoidosis and an increased risk for cutaneous SCC. Because sarcoidosis commonly affects the skin, patients often present to dermatologists for care. Once the initial diagnosis of cutaneous sarcoidosis is made via biopsy, it is natural to be tempted to attribute any new skin lesions to worsening or active disease; however, as cutaneous sarcoidosis may take on a variety of nonspecific forms, it is important to biopsy any unusual lesions. In our case series, patient 3 presented at several different points with scaly scalp lesions. Upon biopsy, several of these lesions were found to be SCCs, while others demonstrated regions of granulomatous inflammation consistent with a diagnosis of cutaneous sarcoidosis. On further review of pathology during the preparation of this manuscript after the initial diagnoses were made, it was further noted that it is challenging to distinguish granulomatous inflammation with reactive pseudoepitheliomatous hyperplasia from SCC. The fact that these lesions were clinically indistinguishable illustrates the critical importance of appropriate-depth biopsy in this situation, and the histopathologic challenges highlighted herein are important for pathologists to remember.
Patients 1 and 2 were both black women, and the fact that these patients both presented with cutaneous SCCs—one of whom was immunosuppressed due to treatment with adalimumab, the other without systemic immunosuppression—exemplifies the need for comprehensive skin examinations in sarcoidosis patients as well as for biopsies of new or unusual lesions.
The mechanism for the development of malignancy in patients with sarcoidosis is unknown and likely is multifactorial. Multiple theories have been proposed.1,2,5,6,8 Sarcoidosis is marked by the development of granulomas secondary to the interaction between CD4+ T cells and antigen-presenting cells, which is mediated by various cytokines and chemokines, including IL-2 and IFN-γ. Patients with sarcoidosis have been found to have oligoclonal T-cell lineages with a limited receptor repertoire, suggestive of selective immune system activation, as well as a deficiency of certain types of regulatory cells, namely natural killer cells.1,2 This immune dysregulation has been postulated to play an etiologic role in the development of malignancy in sarcoidosis patients.1,2,5 Furthermore, the chronic inflammation found in the organs commonly affected by both sarcoidosis and malignancy is another possible mechanism.6,8 Finally, immunosuppression and mutagenesis secondary to the treatment modalities used in sarcoidosis may be another contributing factor.6
Conclusion
An association between sarcoidosis and malignancy has been suggested for several decades. We specifically report 3 cases of patients with cutaneous sarcoidosis who presented with concurrent cutaneous SCCs. Given the varied and often nonspecific nature of cutaneous sarcoidosis, these cases highlight the importance of biopsy when sarcoidosis patients present with new and unusual skin lesions. Additionally, they illustrate the importance of thorough skin examinations in sarcoidosis patients as well as some of the challenges these patients pose for dermatologists.
- Iannuzzi MC, Rybicki BA, Teirsten AS. Sarcoidosis. N Engl J Med. 2007;357:2153-2165.
- Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis and therapeutics. JAMA. 2011;305:391-399.
- Brincker H, Wilbek E. The incidence of malignant tumours in patients with respiratory sarcoidosis. Br J Cancer. 1974;29:247-251.
- Askling J, Grunewald J, Eklund A, et al. Increased risk for cancer following sarcoidosis. Am J Respir Crit Care Med. 1999;160(5, pt 1):1668-1672.
- Ji J, Shu X, Li X, et al. Cancer risk in hospitalized sarcoidosis patients: a follow-up study in Sweden. Ann Oncol. 2009;20:1121-1126.
- Alexandrescu DT, Kauffman CL, Ichim TE, et al. Cutaneous sarcoidosis and malignancy: an association between sarcoidosis with skin manifestations and systemic neoplasia. Dermatol Online J. 2011;17:2.
- Reich JM, Mullooly JP, Johnson RE. Linkage analysis of malignancy-associated sarcoidosis. Chest. 1995;107:605-613.
- Cohen PR, Kurzrock R. Sarcoidosis and malignancy. Clin Dermatol. 2007;25:326-333.
- Iannuzzi MC, Rybicki BA, Teirsten AS. Sarcoidosis. N Engl J Med. 2007;357:2153-2165.
- Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis and therapeutics. JAMA. 2011;305:391-399.
- Brincker H, Wilbek E. The incidence of malignant tumours in patients with respiratory sarcoidosis. Br J Cancer. 1974;29:247-251.
- Askling J, Grunewald J, Eklund A, et al. Increased risk for cancer following sarcoidosis. Am J Respir Crit Care Med. 1999;160(5, pt 1):1668-1672.
- Ji J, Shu X, Li X, et al. Cancer risk in hospitalized sarcoidosis patients: a follow-up study in Sweden. Ann Oncol. 2009;20:1121-1126.
- Alexandrescu DT, Kauffman CL, Ichim TE, et al. Cutaneous sarcoidosis and malignancy: an association between sarcoidosis with skin manifestations and systemic neoplasia. Dermatol Online J. 2011;17:2.
- Reich JM, Mullooly JP, Johnson RE. Linkage analysis of malignancy-associated sarcoidosis. Chest. 1995;107:605-613.
- Cohen PR, Kurzrock R. Sarcoidosis and malignancy. Clin Dermatol. 2007;25:326-333.
Practice Points
- There may be an increased risk of skin cancer in patients with sarcoidosis.
- Sarcoidosis may present with multiple morphologies, including verrucous or hyperkeratotic lesions; superficial biopsy of this type of lesion may be mistaken for a squamous cell carcinoma.
- A biopsy diagnosis of squamous cell carcinoma in a black patient with sarcoidosis should be carefully reviewed for evidence of deeper granulomatous inflammation.
Low-dose IL-2 shows promise for refractory lupus
WASHINGTON – A novel biologic treatment strategy involving subcutaneous low-dose interleukin-2 therapy for refractory systemic lupus erythematosus (SLE) showed promise in a single-center, combined phase I/IIa trial.
In 12 patients with active and refractory SLE – that is, patients with SLE disease activity index (SLEDAI) score of at least 6 who were on at least two different immunosuppressive therapies – low-dose IL-2 treatment led to an effective and cycle-dependent increase in the percentage of CD25hi cells among regulatory T cells (Treg). The increase was statistically significant (P less than .001), Jens Humrich, MD, and his colleagues reported in a late-breaking poster at the annual meeting of the American College of Rheumatology.
However, a reduction in levels of anti-dsDNA antibodies was not observed, said Dr. Humrich of University Hospital Schleswig-Holstein in Lubeck, Germany.
Treatment was safe; treatment-related adverse events were generally mild and transient, Dr. Humrich noted.
Study subjects received four treatment cycles each, with daily subcutaneous injections of recombinant human IL-2 (aldesleukin) at single doses of 0.75, 1.5, or 3.0 million IU on 5 consecutive days. Cycles were separated by a washout period of 9-16 days. Subjects were then followed for 9 weeks.
IL-2 is crucial for the growth and survival of Treg (and thus for the control of autoimmunity). Prior studies demonstrated the significance of acquired IL-2 deficiency and related Treg defects in the pathogenesis of SLE – and that compensation for IL-2 deficiency with low-dose IL-2 can correct these defects, he explained.
In the current study, the primary aim was to show at least a twofold increase in the percentage of CD25hi cells among CD3+CD4+Foxp3+CD127lo Treg cells after the fourth treatment cycle vs. baseline, and secondary aims included clinical responses assessed by SLEDAI and changes in serologic and other immunologic parameters, he said.
The findings suggest that low-dose IL-2 therapy can safely and selectively expand the Treg population and decrease disease activity in patients with active and refractory SLE.
“This study provides the basis for larger and placebo-controlled clinical studies aiming to prove the efficacy of this novel biologic treatment strategy,” the investigators concluded.
Dr. Humrich reported having no disclosures.
WASHINGTON – A novel biologic treatment strategy involving subcutaneous low-dose interleukin-2 therapy for refractory systemic lupus erythematosus (SLE) showed promise in a single-center, combined phase I/IIa trial.
In 12 patients with active and refractory SLE – that is, patients with SLE disease activity index (SLEDAI) score of at least 6 who were on at least two different immunosuppressive therapies – low-dose IL-2 treatment led to an effective and cycle-dependent increase in the percentage of CD25hi cells among regulatory T cells (Treg). The increase was statistically significant (P less than .001), Jens Humrich, MD, and his colleagues reported in a late-breaking poster at the annual meeting of the American College of Rheumatology.
However, a reduction in levels of anti-dsDNA antibodies was not observed, said Dr. Humrich of University Hospital Schleswig-Holstein in Lubeck, Germany.
Treatment was safe; treatment-related adverse events were generally mild and transient, Dr. Humrich noted.
Study subjects received four treatment cycles each, with daily subcutaneous injections of recombinant human IL-2 (aldesleukin) at single doses of 0.75, 1.5, or 3.0 million IU on 5 consecutive days. Cycles were separated by a washout period of 9-16 days. Subjects were then followed for 9 weeks.
IL-2 is crucial for the growth and survival of Treg (and thus for the control of autoimmunity). Prior studies demonstrated the significance of acquired IL-2 deficiency and related Treg defects in the pathogenesis of SLE – and that compensation for IL-2 deficiency with low-dose IL-2 can correct these defects, he explained.
In the current study, the primary aim was to show at least a twofold increase in the percentage of CD25hi cells among CD3+CD4+Foxp3+CD127lo Treg cells after the fourth treatment cycle vs. baseline, and secondary aims included clinical responses assessed by SLEDAI and changes in serologic and other immunologic parameters, he said.
The findings suggest that low-dose IL-2 therapy can safely and selectively expand the Treg population and decrease disease activity in patients with active and refractory SLE.
“This study provides the basis for larger and placebo-controlled clinical studies aiming to prove the efficacy of this novel biologic treatment strategy,” the investigators concluded.
Dr. Humrich reported having no disclosures.
WASHINGTON – A novel biologic treatment strategy involving subcutaneous low-dose interleukin-2 therapy for refractory systemic lupus erythematosus (SLE) showed promise in a single-center, combined phase I/IIa trial.
In 12 patients with active and refractory SLE – that is, patients with SLE disease activity index (SLEDAI) score of at least 6 who were on at least two different immunosuppressive therapies – low-dose IL-2 treatment led to an effective and cycle-dependent increase in the percentage of CD25hi cells among regulatory T cells (Treg). The increase was statistically significant (P less than .001), Jens Humrich, MD, and his colleagues reported in a late-breaking poster at the annual meeting of the American College of Rheumatology.
However, a reduction in levels of anti-dsDNA antibodies was not observed, said Dr. Humrich of University Hospital Schleswig-Holstein in Lubeck, Germany.
Treatment was safe; treatment-related adverse events were generally mild and transient, Dr. Humrich noted.
Study subjects received four treatment cycles each, with daily subcutaneous injections of recombinant human IL-2 (aldesleukin) at single doses of 0.75, 1.5, or 3.0 million IU on 5 consecutive days. Cycles were separated by a washout period of 9-16 days. Subjects were then followed for 9 weeks.
IL-2 is crucial for the growth and survival of Treg (and thus for the control of autoimmunity). Prior studies demonstrated the significance of acquired IL-2 deficiency and related Treg defects in the pathogenesis of SLE – and that compensation for IL-2 deficiency with low-dose IL-2 can correct these defects, he explained.
In the current study, the primary aim was to show at least a twofold increase in the percentage of CD25hi cells among CD3+CD4+Foxp3+CD127lo Treg cells after the fourth treatment cycle vs. baseline, and secondary aims included clinical responses assessed by SLEDAI and changes in serologic and other immunologic parameters, he said.
The findings suggest that low-dose IL-2 therapy can safely and selectively expand the Treg population and decrease disease activity in patients with active and refractory SLE.
“This study provides the basis for larger and placebo-controlled clinical studies aiming to prove the efficacy of this novel biologic treatment strategy,” the investigators concluded.
Dr. Humrich reported having no disclosures.
Key clinical point:
Major finding: A reduction in SLEDAI was seen in 10 patients (83.3%), and a clinical response occurred in 8 (66.7%).
Data source: A combined phase I/IIa trial involving 12 patients.
Disclosures: Dr. Humrich reported having no disclosures.
Verrucous Plaque on the Leg
Blastomycosis
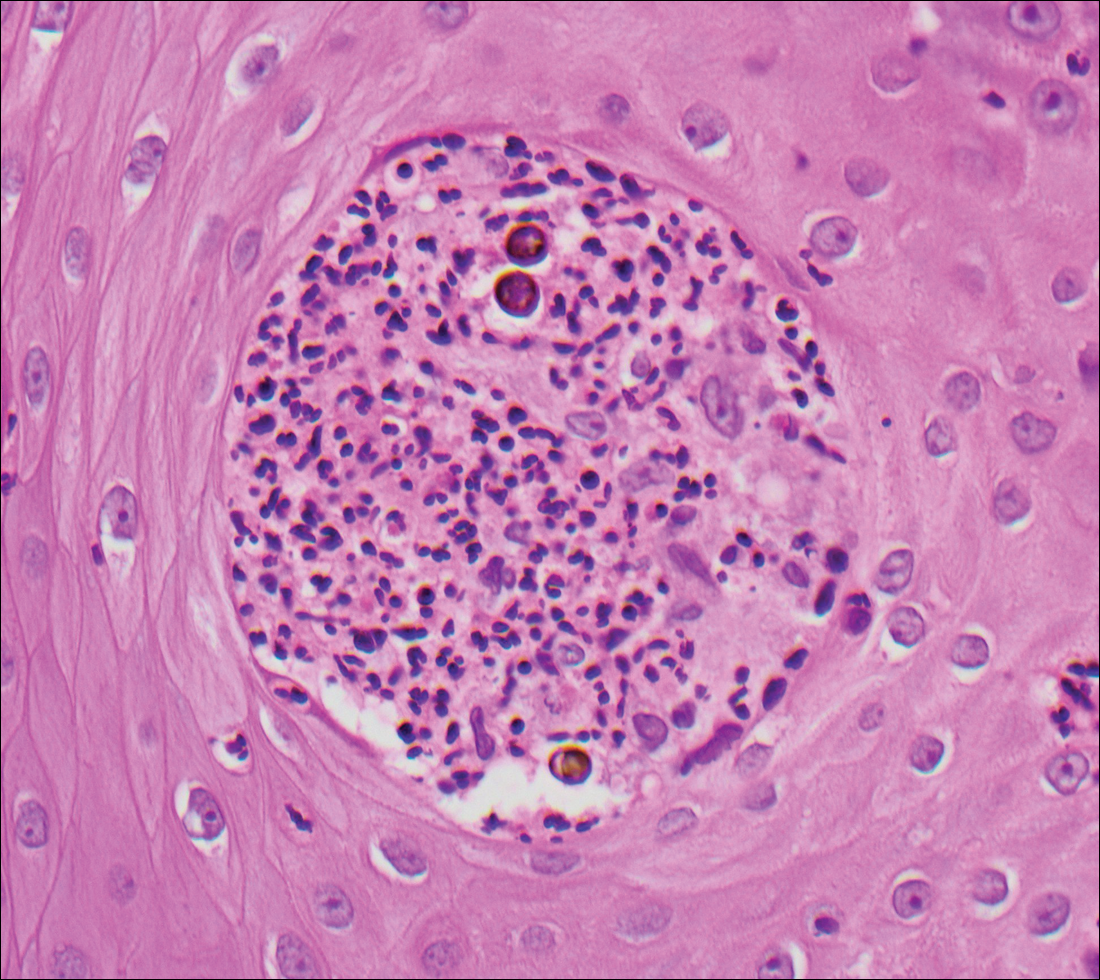
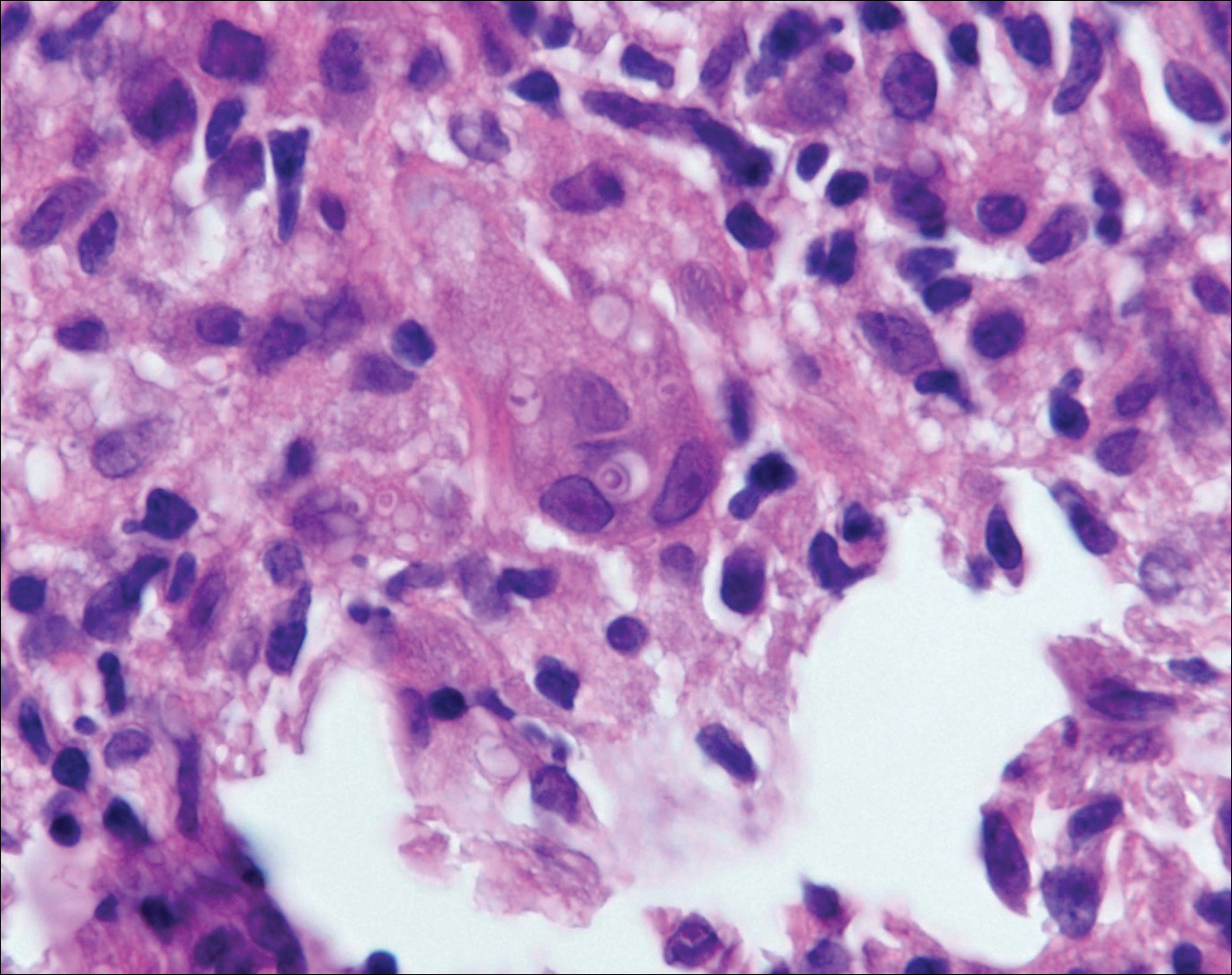
Blastomycosis is caused by Blastomyces dermatitidis, which is endemic in the Midwestern and southeastern United States where it occurs environmentally in wood and soil. Unlike many fungal infections, blastomycosis most often develops in immunocompetent hosts. Infection is usually acquired via inhalation,1 and cutaneous disease typically is secondary to pulmonary infection. Although not common, traumatic inoculation also can cause cutaneous blastomycosis. Skin lesions include crusted verrucous nodules and plaques with elevated borders.1,2 Histologic features include pseudoepitheliomatous hyperplasia with intraepidermal neutrophilic microabscesses (Figure 1), and a neutrophilic and granulomatous dermal infiltrate. Organisms often are found within histiocytes (quiz image) or small abscesses. The yeasts usually are 8 to 15 µm in diameter with a thick cell wall and occasionally display broad-based budding.
Chromoblastomycosis is caused by dematiaceous (pigmented) fungi, including Fonsecaea, Phialophora, Cladophialophora, and Rhinocladiella species,3 which are present in soil and vegetable debris in tropical and subtropical regions. Infection typically occurs in the foot or lower leg from traumatic inoculation, such as a thorn or splinter injury.2 Histologically, chromoblastomycosis is characterized by pseudoepitheliomatous hyperplasia; suppurative and granulomatous dermatitis; and sclerotic (Medlar) bodies, which are 5 to 12 µm in diameter, round, brown, sometimes septate cells resembling copper pennies (Figure 2).2
Coccidioidomycosis is caused by Coccidioides immitis, which is found in soil in the southwestern United States. Infection most often occurs via inhalation of airborne arthrospores.2 Cutaneous lesions occasionally are observed following dissemination or rarely following primary inoculation injury. They may present as papules, nodules, pustules, plaques, and ulcers, with the face being the most commonly affected site.1 Histologically, coccidioidomycosis is characterized by pseudoepitheliomatous hyperplasia, suppurative and granulomatous dermatitis, and large spherules (up to 100 µm in diameter) containing numerous small endospores (Figure 3).
Cryptococcosis is caused by Cryptococcus neoformans, a fungus found in soil, fruit, and pigeon droppings throughout the world.2,3 The most common route of infection is via the respiratory tract. Systemic spread and central nervous system involvement may occur in immunocompromised hosts.2 Skin involvement is uncommon and may present on the head and neck with umbilicated papules, pustules, nodules, plaques, or ulcers. Histologically, Cryptococcus is a spherical yeast, often 4 to 20 µm in diameter. Replication is by narrow-based budding. A characteristic feature is a mucoid capsule, which retracts during processing, leaving a clear space around the yeast (Figure 4). When present, the mucoid capsule can be highlighted on mucicarmine or Alcian blue staining. Histologic variants of cryptococcosis include granulomatous (high host immune response), gelatinous (low host immune response), and suppurative types.3
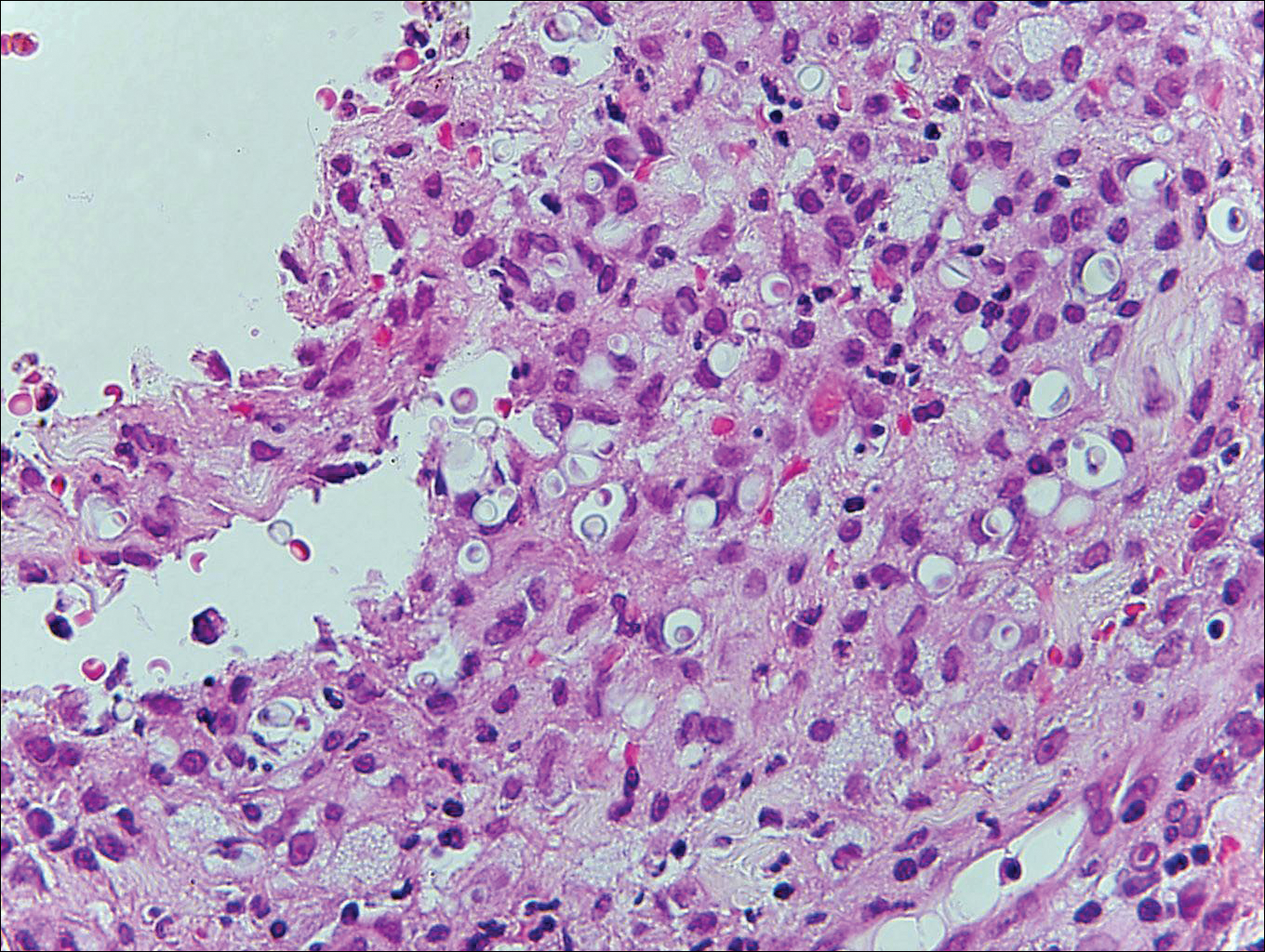
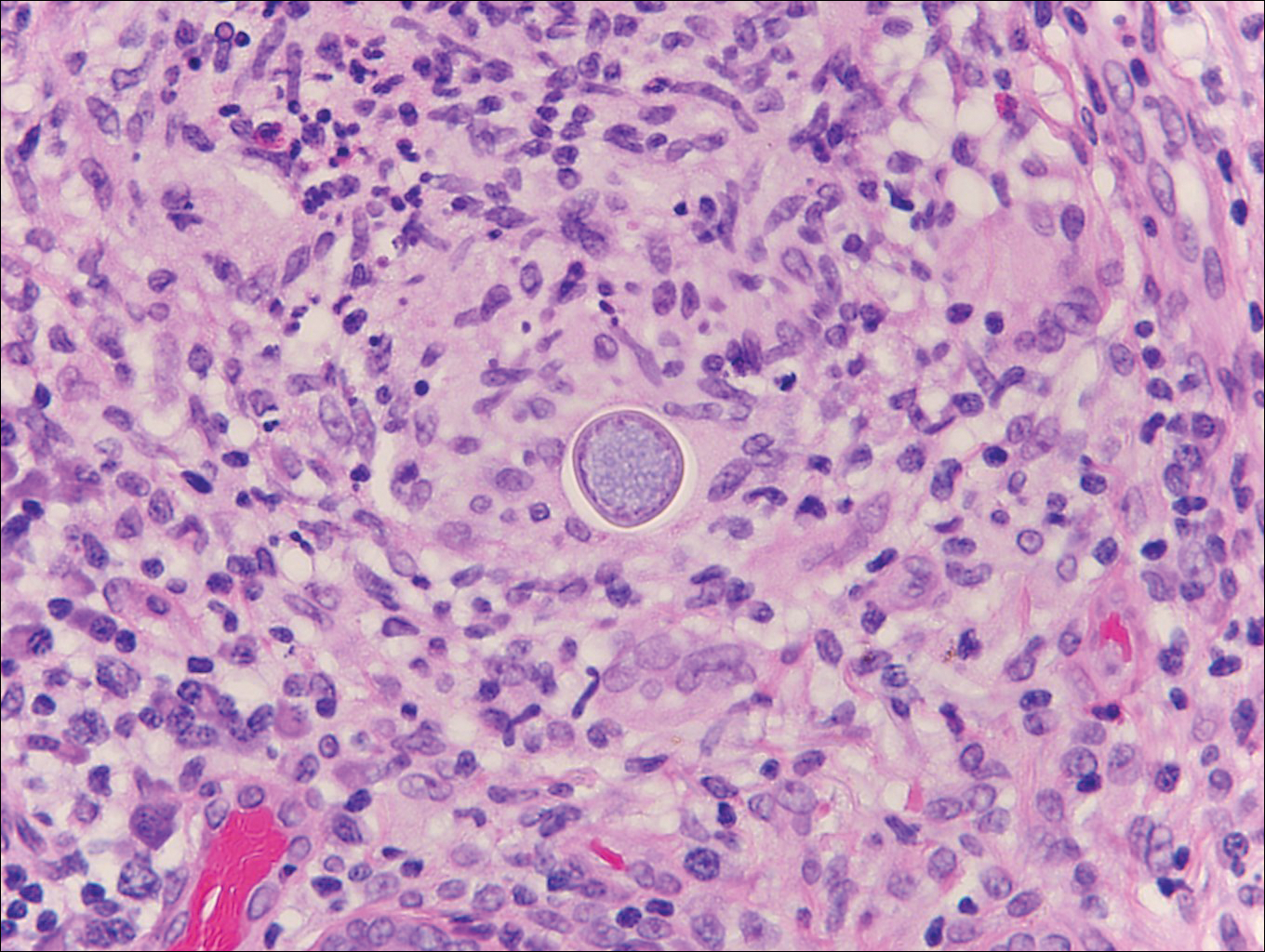
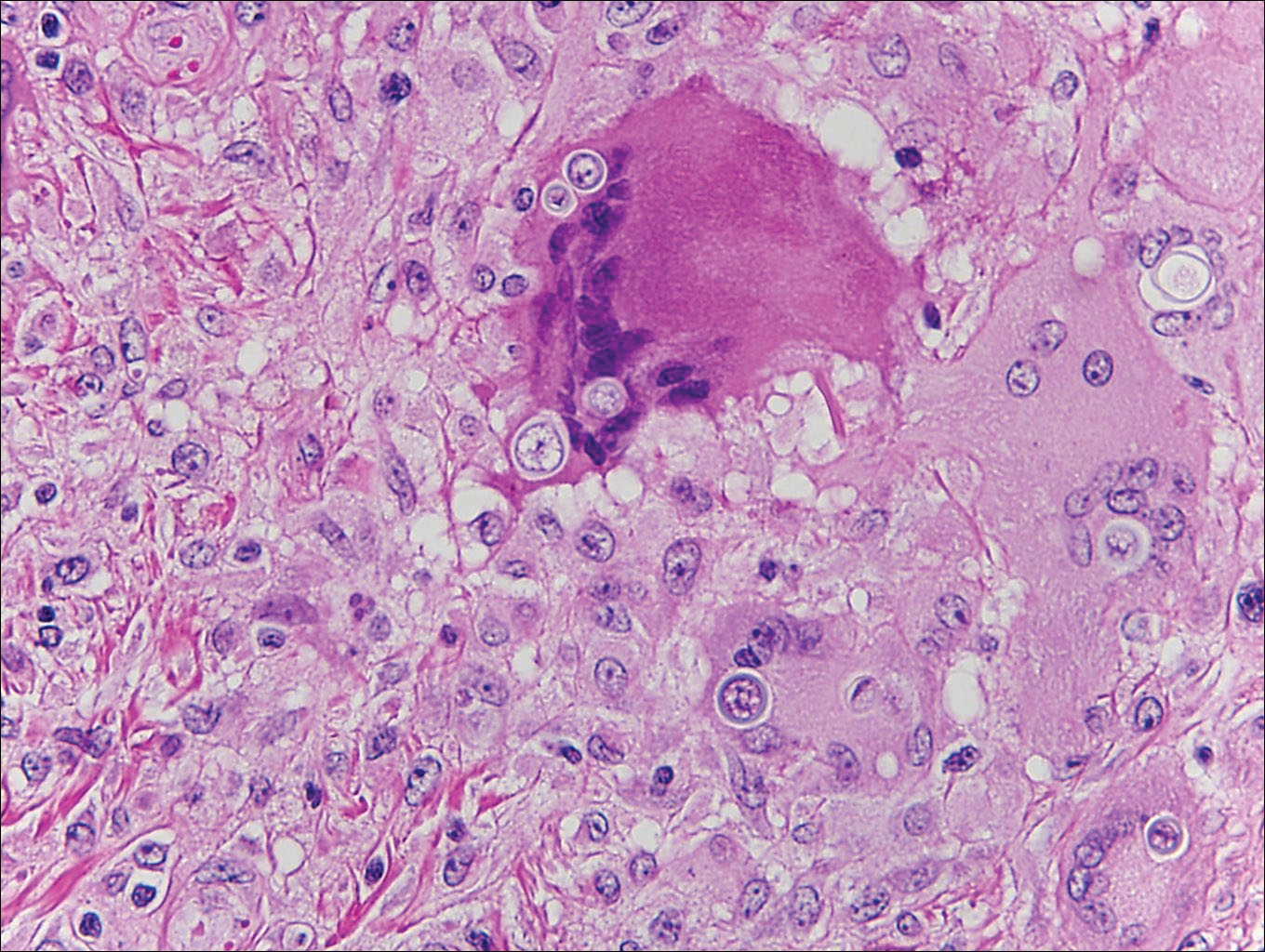
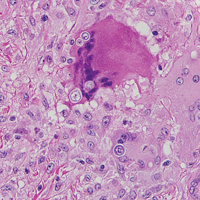
Histoplasmosis is caused by Histoplasma capsulatum, which occurs in soil and bird and bat droppings, with exposure primarily via inhalation. Cutaneous histoplasmosis is almost always a feature of disseminated disease, which occurs most commonly in immunosuppressed individuals.1 Skin lesions may present as macules, papules, indurated plaques, ulcers, purpura, panniculitis, and subcutaneous nodules.2 Histologically, there is a granulomatous and neutrophilic infiltrate within the dermis and subcutis. Yeasts are small (2-4 µm in diameter) and are observed within the cytoplasm of macrophages (Figure 5) where they appear as basophilic dots, sometimes surrounded by an artifactual clear space (pseudocapsule).2
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Vol 2. Philadelphia, PA: Elsevier/Saunders; 2012.
- Calonje JE, Brenn T, Lazar AJ, et al. McKee's Pathology of the Skin. 4th ed. St. Louis, MO: Elsevier/Saunders; 2012.
- Schwarzenberger K, Werchniak A, Ko C. Requisites in Dermatology: General Dermatology. Philadelphia, PA: Elsevier/Saunders; 2009.
Blastomycosis
Blastomycosis is caused by Blastomyces dermatitidis, which is endemic in the Midwestern and southeastern United States where it occurs environmentally in wood and soil. Unlike many fungal infections, blastomycosis most often develops in immunocompetent hosts. Infection is usually acquired via inhalation,1 and cutaneous disease typically is secondary to pulmonary infection. Although not common, traumatic inoculation also can cause cutaneous blastomycosis. Skin lesions include crusted verrucous nodules and plaques with elevated borders.1,2 Histologic features include pseudoepitheliomatous hyperplasia with intraepidermal neutrophilic microabscesses (Figure 1), and a neutrophilic and granulomatous dermal infiltrate. Organisms often are found within histiocytes (quiz image) or small abscesses. The yeasts usually are 8 to 15 µm in diameter with a thick cell wall and occasionally display broad-based budding.
Chromoblastomycosis is caused by dematiaceous (pigmented) fungi, including Fonsecaea, Phialophora, Cladophialophora, and Rhinocladiella species,3 which are present in soil and vegetable debris in tropical and subtropical regions. Infection typically occurs in the foot or lower leg from traumatic inoculation, such as a thorn or splinter injury.2 Histologically, chromoblastomycosis is characterized by pseudoepitheliomatous hyperplasia; suppurative and granulomatous dermatitis; and sclerotic (Medlar) bodies, which are 5 to 12 µm in diameter, round, brown, sometimes septate cells resembling copper pennies (Figure 2).2
Coccidioidomycosis is caused by Coccidioides immitis, which is found in soil in the southwestern United States. Infection most often occurs via inhalation of airborne arthrospores.2 Cutaneous lesions occasionally are observed following dissemination or rarely following primary inoculation injury. They may present as papules, nodules, pustules, plaques, and ulcers, with the face being the most commonly affected site.1 Histologically, coccidioidomycosis is characterized by pseudoepitheliomatous hyperplasia, suppurative and granulomatous dermatitis, and large spherules (up to 100 µm in diameter) containing numerous small endospores (Figure 3).
Cryptococcosis is caused by Cryptococcus neoformans, a fungus found in soil, fruit, and pigeon droppings throughout the world.2,3 The most common route of infection is via the respiratory tract. Systemic spread and central nervous system involvement may occur in immunocompromised hosts.2 Skin involvement is uncommon and may present on the head and neck with umbilicated papules, pustules, nodules, plaques, or ulcers. Histologically, Cryptococcus is a spherical yeast, often 4 to 20 µm in diameter. Replication is by narrow-based budding. A characteristic feature is a mucoid capsule, which retracts during processing, leaving a clear space around the yeast (Figure 4). When present, the mucoid capsule can be highlighted on mucicarmine or Alcian blue staining. Histologic variants of cryptococcosis include granulomatous (high host immune response), gelatinous (low host immune response), and suppurative types.3
Histoplasmosis is caused by Histoplasma capsulatum, which occurs in soil and bird and bat droppings, with exposure primarily via inhalation. Cutaneous histoplasmosis is almost always a feature of disseminated disease, which occurs most commonly in immunosuppressed individuals.1 Skin lesions may present as macules, papules, indurated plaques, ulcers, purpura, panniculitis, and subcutaneous nodules.2 Histologically, there is a granulomatous and neutrophilic infiltrate within the dermis and subcutis. Yeasts are small (2-4 µm in diameter) and are observed within the cytoplasm of macrophages (Figure 5) where they appear as basophilic dots, sometimes surrounded by an artifactual clear space (pseudocapsule).2
Blastomycosis
Blastomycosis is caused by Blastomyces dermatitidis, which is endemic in the Midwestern and southeastern United States where it occurs environmentally in wood and soil. Unlike many fungal infections, blastomycosis most often develops in immunocompetent hosts. Infection is usually acquired via inhalation,1 and cutaneous disease typically is secondary to pulmonary infection. Although not common, traumatic inoculation also can cause cutaneous blastomycosis. Skin lesions include crusted verrucous nodules and plaques with elevated borders.1,2 Histologic features include pseudoepitheliomatous hyperplasia with intraepidermal neutrophilic microabscesses (Figure 1), and a neutrophilic and granulomatous dermal infiltrate. Organisms often are found within histiocytes (quiz image) or small abscesses. The yeasts usually are 8 to 15 µm in diameter with a thick cell wall and occasionally display broad-based budding.
Chromoblastomycosis is caused by dematiaceous (pigmented) fungi, including Fonsecaea, Phialophora, Cladophialophora, and Rhinocladiella species,3 which are present in soil and vegetable debris in tropical and subtropical regions. Infection typically occurs in the foot or lower leg from traumatic inoculation, such as a thorn or splinter injury.2 Histologically, chromoblastomycosis is characterized by pseudoepitheliomatous hyperplasia; suppurative and granulomatous dermatitis; and sclerotic (Medlar) bodies, which are 5 to 12 µm in diameter, round, brown, sometimes septate cells resembling copper pennies (Figure 2).2
Coccidioidomycosis is caused by Coccidioides immitis, which is found in soil in the southwestern United States. Infection most often occurs via inhalation of airborne arthrospores.2 Cutaneous lesions occasionally are observed following dissemination or rarely following primary inoculation injury. They may present as papules, nodules, pustules, plaques, and ulcers, with the face being the most commonly affected site.1 Histologically, coccidioidomycosis is characterized by pseudoepitheliomatous hyperplasia, suppurative and granulomatous dermatitis, and large spherules (up to 100 µm in diameter) containing numerous small endospores (Figure 3).
Cryptococcosis is caused by Cryptococcus neoformans, a fungus found in soil, fruit, and pigeon droppings throughout the world.2,3 The most common route of infection is via the respiratory tract. Systemic spread and central nervous system involvement may occur in immunocompromised hosts.2 Skin involvement is uncommon and may present on the head and neck with umbilicated papules, pustules, nodules, plaques, or ulcers. Histologically, Cryptococcus is a spherical yeast, often 4 to 20 µm in diameter. Replication is by narrow-based budding. A characteristic feature is a mucoid capsule, which retracts during processing, leaving a clear space around the yeast (Figure 4). When present, the mucoid capsule can be highlighted on mucicarmine or Alcian blue staining. Histologic variants of cryptococcosis include granulomatous (high host immune response), gelatinous (low host immune response), and suppurative types.3
Histoplasmosis is caused by Histoplasma capsulatum, which occurs in soil and bird and bat droppings, with exposure primarily via inhalation. Cutaneous histoplasmosis is almost always a feature of disseminated disease, which occurs most commonly in immunosuppressed individuals.1 Skin lesions may present as macules, papules, indurated plaques, ulcers, purpura, panniculitis, and subcutaneous nodules.2 Histologically, there is a granulomatous and neutrophilic infiltrate within the dermis and subcutis. Yeasts are small (2-4 µm in diameter) and are observed within the cytoplasm of macrophages (Figure 5) where they appear as basophilic dots, sometimes surrounded by an artifactual clear space (pseudocapsule).2
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Vol 2. Philadelphia, PA: Elsevier/Saunders; 2012.
- Calonje JE, Brenn T, Lazar AJ, et al. McKee's Pathology of the Skin. 4th ed. St. Louis, MO: Elsevier/Saunders; 2012.
- Schwarzenberger K, Werchniak A, Ko C. Requisites in Dermatology: General Dermatology. Philadelphia, PA: Elsevier/Saunders; 2009.
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Vol 2. Philadelphia, PA: Elsevier/Saunders; 2012.
- Calonje JE, Brenn T, Lazar AJ, et al. McKee's Pathology of the Skin. 4th ed. St. Louis, MO: Elsevier/Saunders; 2012.
- Schwarzenberger K, Werchniak A, Ko C. Requisites in Dermatology: General Dermatology. Philadelphia, PA: Elsevier/Saunders; 2009.
Aquatic Antagonists: Cutaneous Sea Urchin Spine Injury
Sea urchin injuries are commonly seen in coastal regions near both warm and cold salt water with frequent recreational water activities or fishing. Sea urchins belong to the class Echinoidea with approximately 600 species, of which roughly 80 are poisonous to humans.1,2 When a human comes in contact with a sea urchin, the spines of the sea urchin (made of calcium carbonate) can penetrate the skin and break off from the sea urchin, becoming embedded in the skin. Injuries from sea urchin spines are most commonly seen on the hands and feet, as the likelihood of contact with a sea urchin is greater on these sites. The severity of sea urchin spine injuries can vary widely, from minimal local trauma and pain to arthritis, synovitis, and occasionally systemic illness.1,3 It is important to recognize the wide variety of responses to sea urchin spine injuries and the impact of prompt treatment. Many published reports on injuries from sea urchin spines describe arthritis and synovitis from spines in the joints.1,2,4-6 Fewer reports discuss nonjoint injuries and the dermatologic aspects of sea urchin spine injuries.3,7,8 We pre-sent a case of a patient with a puncture injury from sea urchin spines that resulted in painful granulomas.
Case Report
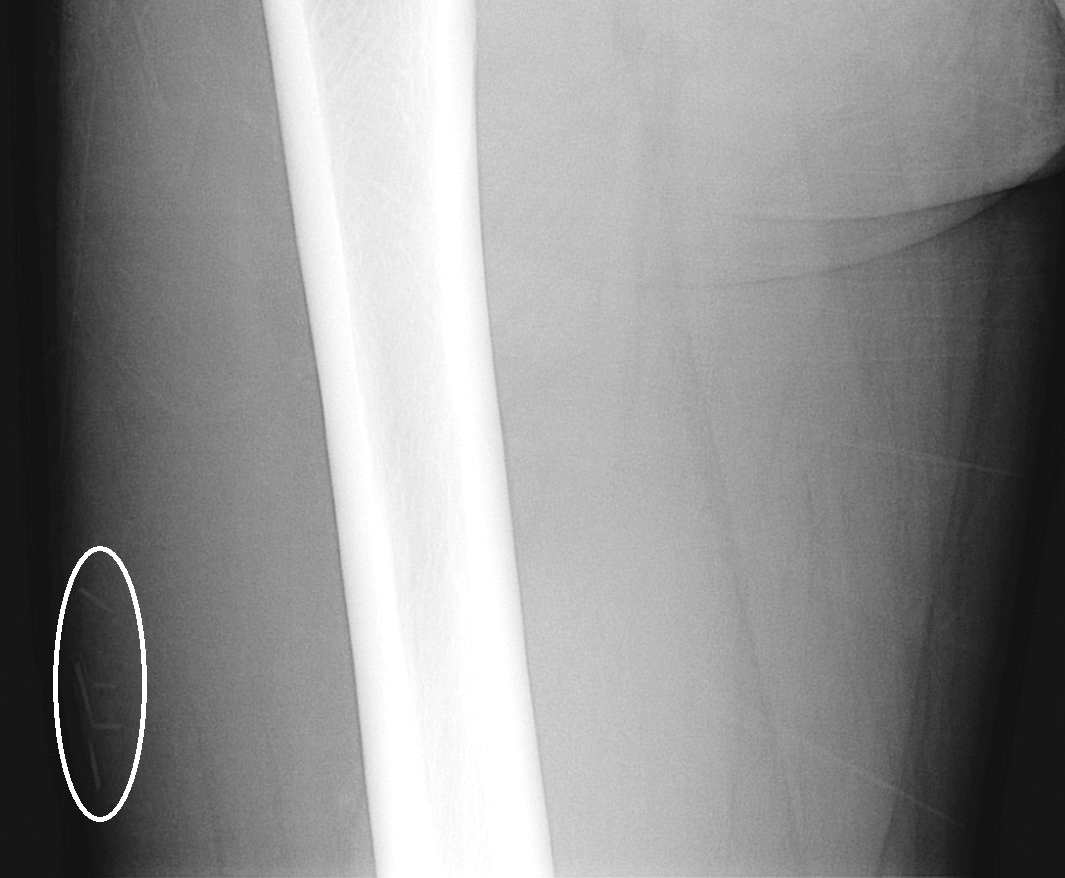
A 29-year-old otherwise healthy man was referred to our dermatology clinic by the university student health center due to continued pain in the right thigh. Five weeks prior to presentation to the student health center, the patient had fallen on a sea urchin while snorkeling in Hawaii. Sea urchin spines became lodged in the right thigh, some of which were removed in a local medical clinic in Hawaii. He was given oral antibiotics prior to his return home. A plain film radiograph of the affected area ordered by the student health center showed several punctate and linear densities in the lateral aspect of the right mid thigh (Figure 1). These findings were consistent with sea urchin spines within the superficial soft tissues of the lateral thigh.
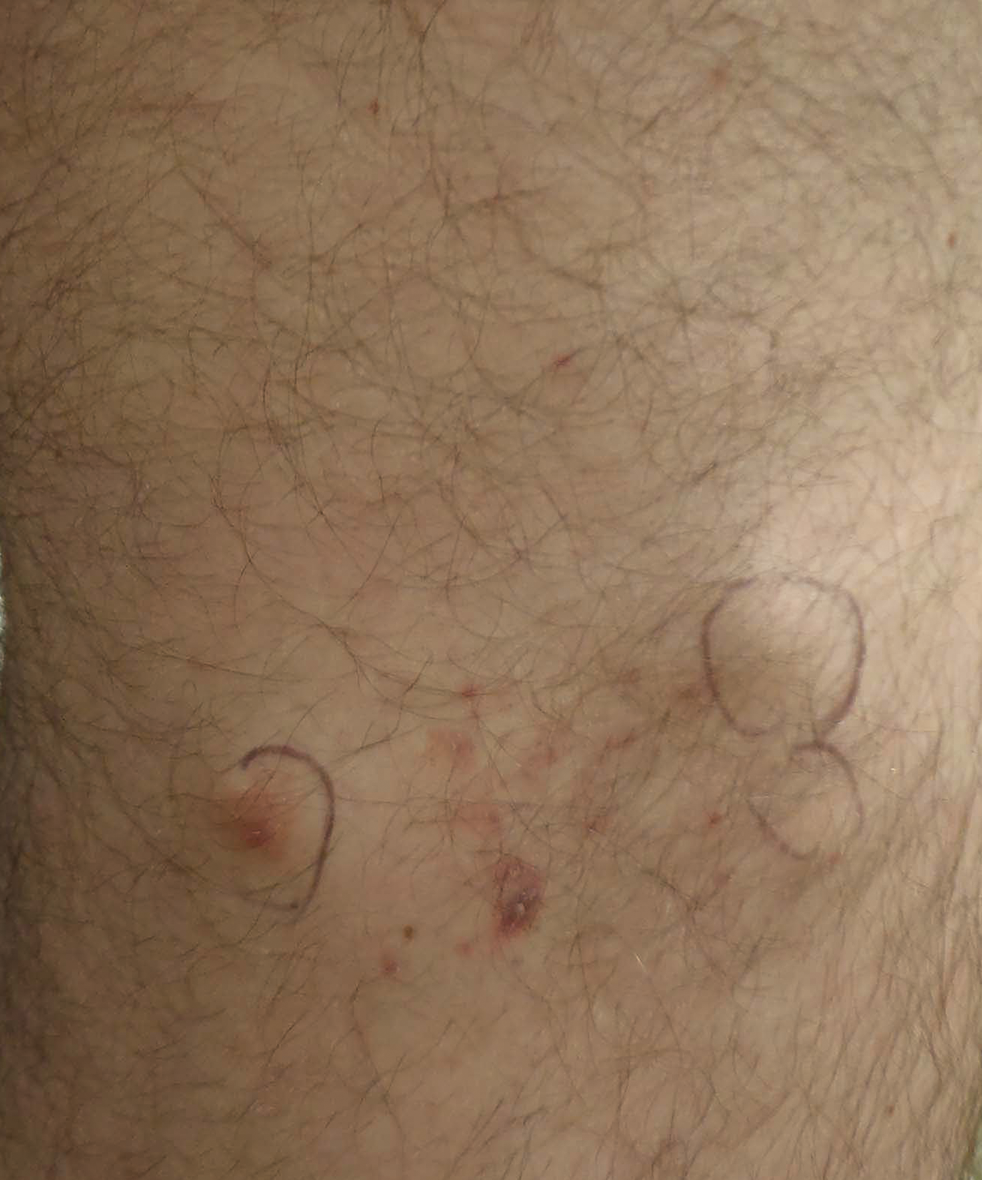
At the time of presentation to our dermatology clinic, the patient reported sharp intermittent pain localized to the right thigh. The patient denied any fever, chills, or pain in the joints. On physical examination, there were several firm nodules on the right thigh, ranging from 4 to 20 mm in diameter (Figure 2). The nodules were tender to palpation with some surrounding edema. Drainage was not noted. Several scars were visible at sites of the original puncture injuries and removal of the spines.
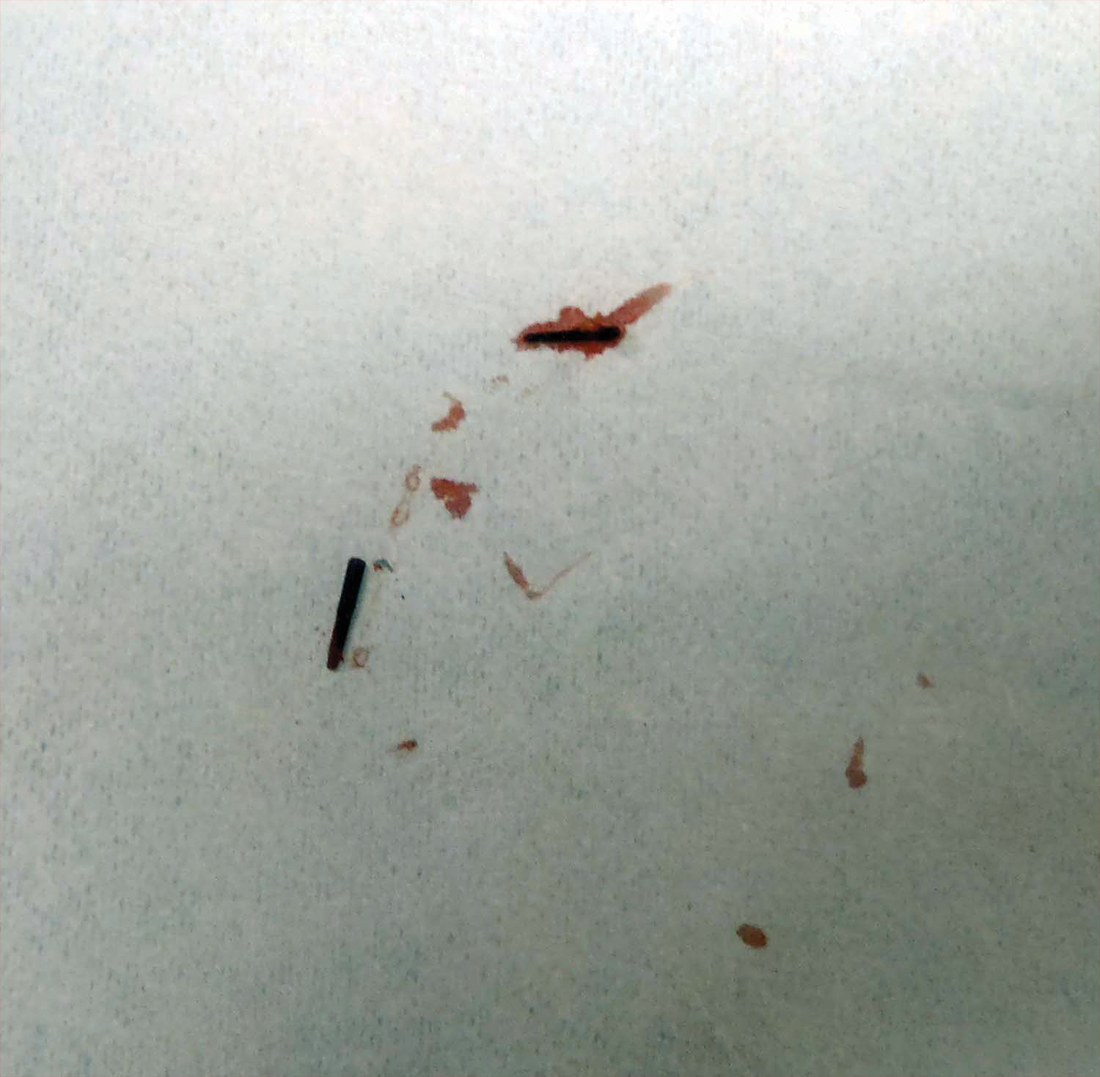
Two 6-mm punch biopsies were performed on representative nodules on the right thigh for histopathologic examination. Along with the biopsy tissue, firm, brown-black, linear foreign bodies consistent with sea urchin spines were extracted with forceps (Figure 3). Histopathologic examination revealed a dense, diffuse, mixed inflammatory cell infiltrate in the dermis predominantly composed of lymphocytes, histiocytes, and numerous eosinophils. Proliferation of small vessels was noted. In one of the biopsies, small fragments of necrotic tissue were present. These findings were consistent with granulomatous inflammation and granulation tissue due to a foreign body.
At the time of suture removal 2 weeks later, the biopsied areas were well healed with minimal erythema. The patient reported decreased pain in the involved areas. He was not seen in clinic again due to resolution of the nodules and associated pain.
Comment
Sea urchin spine injuries are commonly seen in coastal regions with frequent participation in recreational and occupational water activities. A wide variety of responses can be seen in sea urchin spine injuries. There generally are 2 types of cutaneous reaction patterns to sea urchin spines: a primary initial reaction and a secondary delayed/granulomatous reaction. When the spines initially penetrate the skin, the primary initial reaction consists of sharp localized pain that worsens with applied pressure. In addition to pain, bleeding, erythema, edema, and myalgia can occur.3 These symptoms typically subside a few hours after complete removal of the spines from the skin.6 If some spines remain in the skin, a secondary delayed/granulomatous reaction can occur, which can lead to the formation of granulomas that can manifest as nodules or papules and can be diffuse.
Many patients may think their painful encounter with a sea urchin was just an unfortunate event, but depending on the location of the injury, more serious extracutaneous reactions and chronic symptoms may occur. Some cases have described the development of arthritis and synovitis from the implantation of spines into joints.1,2,4-6 Other extracutaneous complications include neuropathy and paresthesia, local bone destruction, radiating pain, muscular weakness, and hypotension.3
The severity of the injury also can depend on the sea urchin species and the number of spines implanted. There are approximately 80 poisonous sea urchin species possessing toxins in venomous spines, resulting in edema and change in the leukocyte-endothelial interaction.9 Substances identified in the spines include proteins, steroids, serotonin, histamine, and glycosides.3,9 The number of spines implanted, particularly the number of venomous spines, can lead to more severe complications. Penetration of 15 or more venomous spines can commonly lead to extracutaneous symptoms.3 Another concern, irrespective of species type, is the potential for secondary infection associated with the spine penetration or implantation into the skin. Mycobacterium marinum infections have been reported in some sea urchin granulomas,10 as well as fungal infection, bacterial infection, and tetanus.3
The diagnosis of sea urchin spine injuries starts with a thorough history and physical examination. A positive history of sea urchin contact suggests the diagnosis, and radiographs can be useful to find the location of the spine(s), especially if there are no visible nodules on the skin. However, small fragments of spine may not be completely observed on plain radiographs. Any signs or symptoms of infection should prompt a culture for confirmation and guidance for management. Cutaneous biopsies can be helpful for both diagnosis confirmation and symptomatic relief. Reported cases have described granulomatous reactions in the vast majority of the histologic specimens, with necrosis an additional common finding.7,8 Sea urchin granulomas can be of varying types, the majority being foreign-body and sarcoid types.3,6,7
Treatment of sea urchin spine injuries primarily involves removal of the spines by a physician. Patients may soak the affected areas in warm water prior to the removal of the spines to aid in pain relief. Surgical removal with local anesthesia and cutaneous extraction is a common treatment method, and more extensive surgical removal of the spines is another option, especially in areas around the joints.2 The use of liquid nitrogen or skin punch biopsy also have been described as possible methods to remove the spines.11,12
Conclusion
Sea urchin spine injuries can result in a wide range of cutaneous and systemic complications. Prompt diagnosis and treatment to remove the sea urchin spines can lessen the associated pain and is important in the prevention of more serious complications.
- Liram N, Gomori M, Perouansky M. Sea urchin puncture resulting in PIP joint synovial arthritis: case report and MRI study. J Travel Med. 2000;7:43-45.
- Dahl WJ, Jebson P, Louis DS. Sea urchin injuries to the hand: a case report and review of the literature. Iowa Orthop J. 2010;30:153-156.
- Rossetto AL, de Macedo Mora J, Haddad Junior V. Sea urchin granuloma. Rev Inst Med Trop Sao Paulo. 2006;48:303-306.
- Ahmad R, McCann PA, Barakat M, et al. Sea urchin spine injuries of the hand. J Hand Surg Eur Vol. 2008;33:670-671.
- Schefflein J, Umans H, Ellenbogen D, et al. Sea urchin spine arthritis in the foot. Skeletal Radiol. 2012;41:1327-1331.
- Wada T, Soma T, Gaman K, et al. Sea urchin spine arthritis of the hand. J Hand Surg. 2008;33:398-401.
- Suárez-Peñaranda JM, Vieites B, Del Río E, et al. Histopathologic and immunohistochemical features of sea urchin granulomas. J Cutan Pathol. 2013;40:550-556.
- De La Torre C, Toribio J. Sea-urchin granuloma: histologic profile. a pathologic study of 50 biopsies. J Cutan Pathol. 2001;28:223-228.
- Sciani JM, Zychar BC, Gonçalves LR, et al. Pro-inflammatory effects of the aqueous extract of Echinometra lucunter sea urchin spines. Exp Biol Med (Maywood). 2011;236:277-280.
- De la Torre C, Vega A, Carracedo A, et al. Identification of Mycobacterium marinum in sea-urchin granulomas. Br J Dermatol. 2001;145:114-116.
- Gargus MD, Morohashi DK. A sea-urchin spine chilling remedy. N Engl J Med. 2012;367:1867-1868.
- Sjøberg T, de Weerd L. The usefulness of a skin biopsy punch to remove sea urchin spines. ANZ J Surg. 2010;80:383.
Sea urchin injuries are commonly seen in coastal regions near both warm and cold salt water with frequent recreational water activities or fishing. Sea urchins belong to the class Echinoidea with approximately 600 species, of which roughly 80 are poisonous to humans.1,2 When a human comes in contact with a sea urchin, the spines of the sea urchin (made of calcium carbonate) can penetrate the skin and break off from the sea urchin, becoming embedded in the skin. Injuries from sea urchin spines are most commonly seen on the hands and feet, as the likelihood of contact with a sea urchin is greater on these sites. The severity of sea urchin spine injuries can vary widely, from minimal local trauma and pain to arthritis, synovitis, and occasionally systemic illness.1,3 It is important to recognize the wide variety of responses to sea urchin spine injuries and the impact of prompt treatment. Many published reports on injuries from sea urchin spines describe arthritis and synovitis from spines in the joints.1,2,4-6 Fewer reports discuss nonjoint injuries and the dermatologic aspects of sea urchin spine injuries.3,7,8 We pre-sent a case of a patient with a puncture injury from sea urchin spines that resulted in painful granulomas.
Case Report
A 29-year-old otherwise healthy man was referred to our dermatology clinic by the university student health center due to continued pain in the right thigh. Five weeks prior to presentation to the student health center, the patient had fallen on a sea urchin while snorkeling in Hawaii. Sea urchin spines became lodged in the right thigh, some of which were removed in a local medical clinic in Hawaii. He was given oral antibiotics prior to his return home. A plain film radiograph of the affected area ordered by the student health center showed several punctate and linear densities in the lateral aspect of the right mid thigh (Figure 1). These findings were consistent with sea urchin spines within the superficial soft tissues of the lateral thigh.
At the time of presentation to our dermatology clinic, the patient reported sharp intermittent pain localized to the right thigh. The patient denied any fever, chills, or pain in the joints. On physical examination, there were several firm nodules on the right thigh, ranging from 4 to 20 mm in diameter (Figure 2). The nodules were tender to palpation with some surrounding edema. Drainage was not noted. Several scars were visible at sites of the original puncture injuries and removal of the spines.
Two 6-mm punch biopsies were performed on representative nodules on the right thigh for histopathologic examination. Along with the biopsy tissue, firm, brown-black, linear foreign bodies consistent with sea urchin spines were extracted with forceps (Figure 3). Histopathologic examination revealed a dense, diffuse, mixed inflammatory cell infiltrate in the dermis predominantly composed of lymphocytes, histiocytes, and numerous eosinophils. Proliferation of small vessels was noted. In one of the biopsies, small fragments of necrotic tissue were present. These findings were consistent with granulomatous inflammation and granulation tissue due to a foreign body.
At the time of suture removal 2 weeks later, the biopsied areas were well healed with minimal erythema. The patient reported decreased pain in the involved areas. He was not seen in clinic again due to resolution of the nodules and associated pain.
Comment
Sea urchin spine injuries are commonly seen in coastal regions with frequent participation in recreational and occupational water activities. A wide variety of responses can be seen in sea urchin spine injuries. There generally are 2 types of cutaneous reaction patterns to sea urchin spines: a primary initial reaction and a secondary delayed/granulomatous reaction. When the spines initially penetrate the skin, the primary initial reaction consists of sharp localized pain that worsens with applied pressure. In addition to pain, bleeding, erythema, edema, and myalgia can occur.3 These symptoms typically subside a few hours after complete removal of the spines from the skin.6 If some spines remain in the skin, a secondary delayed/granulomatous reaction can occur, which can lead to the formation of granulomas that can manifest as nodules or papules and can be diffuse.
Many patients may think their painful encounter with a sea urchin was just an unfortunate event, but depending on the location of the injury, more serious extracutaneous reactions and chronic symptoms may occur. Some cases have described the development of arthritis and synovitis from the implantation of spines into joints.1,2,4-6 Other extracutaneous complications include neuropathy and paresthesia, local bone destruction, radiating pain, muscular weakness, and hypotension.3
The severity of the injury also can depend on the sea urchin species and the number of spines implanted. There are approximately 80 poisonous sea urchin species possessing toxins in venomous spines, resulting in edema and change in the leukocyte-endothelial interaction.9 Substances identified in the spines include proteins, steroids, serotonin, histamine, and glycosides.3,9 The number of spines implanted, particularly the number of venomous spines, can lead to more severe complications. Penetration of 15 or more venomous spines can commonly lead to extracutaneous symptoms.3 Another concern, irrespective of species type, is the potential for secondary infection associated with the spine penetration or implantation into the skin. Mycobacterium marinum infections have been reported in some sea urchin granulomas,10 as well as fungal infection, bacterial infection, and tetanus.3
The diagnosis of sea urchin spine injuries starts with a thorough history and physical examination. A positive history of sea urchin contact suggests the diagnosis, and radiographs can be useful to find the location of the spine(s), especially if there are no visible nodules on the skin. However, small fragments of spine may not be completely observed on plain radiographs. Any signs or symptoms of infection should prompt a culture for confirmation and guidance for management. Cutaneous biopsies can be helpful for both diagnosis confirmation and symptomatic relief. Reported cases have described granulomatous reactions in the vast majority of the histologic specimens, with necrosis an additional common finding.7,8 Sea urchin granulomas can be of varying types, the majority being foreign-body and sarcoid types.3,6,7
Treatment of sea urchin spine injuries primarily involves removal of the spines by a physician. Patients may soak the affected areas in warm water prior to the removal of the spines to aid in pain relief. Surgical removal with local anesthesia and cutaneous extraction is a common treatment method, and more extensive surgical removal of the spines is another option, especially in areas around the joints.2 The use of liquid nitrogen or skin punch biopsy also have been described as possible methods to remove the spines.11,12
Conclusion
Sea urchin spine injuries can result in a wide range of cutaneous and systemic complications. Prompt diagnosis and treatment to remove the sea urchin spines can lessen the associated pain and is important in the prevention of more serious complications.
Sea urchin injuries are commonly seen in coastal regions near both warm and cold salt water with frequent recreational water activities or fishing. Sea urchins belong to the class Echinoidea with approximately 600 species, of which roughly 80 are poisonous to humans.1,2 When a human comes in contact with a sea urchin, the spines of the sea urchin (made of calcium carbonate) can penetrate the skin and break off from the sea urchin, becoming embedded in the skin. Injuries from sea urchin spines are most commonly seen on the hands and feet, as the likelihood of contact with a sea urchin is greater on these sites. The severity of sea urchin spine injuries can vary widely, from minimal local trauma and pain to arthritis, synovitis, and occasionally systemic illness.1,3 It is important to recognize the wide variety of responses to sea urchin spine injuries and the impact of prompt treatment. Many published reports on injuries from sea urchin spines describe arthritis and synovitis from spines in the joints.1,2,4-6 Fewer reports discuss nonjoint injuries and the dermatologic aspects of sea urchin spine injuries.3,7,8 We pre-sent a case of a patient with a puncture injury from sea urchin spines that resulted in painful granulomas.
Case Report
A 29-year-old otherwise healthy man was referred to our dermatology clinic by the university student health center due to continued pain in the right thigh. Five weeks prior to presentation to the student health center, the patient had fallen on a sea urchin while snorkeling in Hawaii. Sea urchin spines became lodged in the right thigh, some of which were removed in a local medical clinic in Hawaii. He was given oral antibiotics prior to his return home. A plain film radiograph of the affected area ordered by the student health center showed several punctate and linear densities in the lateral aspect of the right mid thigh (Figure 1). These findings were consistent with sea urchin spines within the superficial soft tissues of the lateral thigh.
At the time of presentation to our dermatology clinic, the patient reported sharp intermittent pain localized to the right thigh. The patient denied any fever, chills, or pain in the joints. On physical examination, there were several firm nodules on the right thigh, ranging from 4 to 20 mm in diameter (Figure 2). The nodules were tender to palpation with some surrounding edema. Drainage was not noted. Several scars were visible at sites of the original puncture injuries and removal of the spines.
Two 6-mm punch biopsies were performed on representative nodules on the right thigh for histopathologic examination. Along with the biopsy tissue, firm, brown-black, linear foreign bodies consistent with sea urchin spines were extracted with forceps (Figure 3). Histopathologic examination revealed a dense, diffuse, mixed inflammatory cell infiltrate in the dermis predominantly composed of lymphocytes, histiocytes, and numerous eosinophils. Proliferation of small vessels was noted. In one of the biopsies, small fragments of necrotic tissue were present. These findings were consistent with granulomatous inflammation and granulation tissue due to a foreign body.
At the time of suture removal 2 weeks later, the biopsied areas were well healed with minimal erythema. The patient reported decreased pain in the involved areas. He was not seen in clinic again due to resolution of the nodules and associated pain.
Comment
Sea urchin spine injuries are commonly seen in coastal regions with frequent participation in recreational and occupational water activities. A wide variety of responses can be seen in sea urchin spine injuries. There generally are 2 types of cutaneous reaction patterns to sea urchin spines: a primary initial reaction and a secondary delayed/granulomatous reaction. When the spines initially penetrate the skin, the primary initial reaction consists of sharp localized pain that worsens with applied pressure. In addition to pain, bleeding, erythema, edema, and myalgia can occur.3 These symptoms typically subside a few hours after complete removal of the spines from the skin.6 If some spines remain in the skin, a secondary delayed/granulomatous reaction can occur, which can lead to the formation of granulomas that can manifest as nodules or papules and can be diffuse.
Many patients may think their painful encounter with a sea urchin was just an unfortunate event, but depending on the location of the injury, more serious extracutaneous reactions and chronic symptoms may occur. Some cases have described the development of arthritis and synovitis from the implantation of spines into joints.1,2,4-6 Other extracutaneous complications include neuropathy and paresthesia, local bone destruction, radiating pain, muscular weakness, and hypotension.3
The severity of the injury also can depend on the sea urchin species and the number of spines implanted. There are approximately 80 poisonous sea urchin species possessing toxins in venomous spines, resulting in edema and change in the leukocyte-endothelial interaction.9 Substances identified in the spines include proteins, steroids, serotonin, histamine, and glycosides.3,9 The number of spines implanted, particularly the number of venomous spines, can lead to more severe complications. Penetration of 15 or more venomous spines can commonly lead to extracutaneous symptoms.3 Another concern, irrespective of species type, is the potential for secondary infection associated with the spine penetration or implantation into the skin. Mycobacterium marinum infections have been reported in some sea urchin granulomas,10 as well as fungal infection, bacterial infection, and tetanus.3
The diagnosis of sea urchin spine injuries starts with a thorough history and physical examination. A positive history of sea urchin contact suggests the diagnosis, and radiographs can be useful to find the location of the spine(s), especially if there are no visible nodules on the skin. However, small fragments of spine may not be completely observed on plain radiographs. Any signs or symptoms of infection should prompt a culture for confirmation and guidance for management. Cutaneous biopsies can be helpful for both diagnosis confirmation and symptomatic relief. Reported cases have described granulomatous reactions in the vast majority of the histologic specimens, with necrosis an additional common finding.7,8 Sea urchin granulomas can be of varying types, the majority being foreign-body and sarcoid types.3,6,7
Treatment of sea urchin spine injuries primarily involves removal of the spines by a physician. Patients may soak the affected areas in warm water prior to the removal of the spines to aid in pain relief. Surgical removal with local anesthesia and cutaneous extraction is a common treatment method, and more extensive surgical removal of the spines is another option, especially in areas around the joints.2 The use of liquid nitrogen or skin punch biopsy also have been described as possible methods to remove the spines.11,12
Conclusion
Sea urchin spine injuries can result in a wide range of cutaneous and systemic complications. Prompt diagnosis and treatment to remove the sea urchin spines can lessen the associated pain and is important in the prevention of more serious complications.
- Liram N, Gomori M, Perouansky M. Sea urchin puncture resulting in PIP joint synovial arthritis: case report and MRI study. J Travel Med. 2000;7:43-45.
- Dahl WJ, Jebson P, Louis DS. Sea urchin injuries to the hand: a case report and review of the literature. Iowa Orthop J. 2010;30:153-156.
- Rossetto AL, de Macedo Mora J, Haddad Junior V. Sea urchin granuloma. Rev Inst Med Trop Sao Paulo. 2006;48:303-306.
- Ahmad R, McCann PA, Barakat M, et al. Sea urchin spine injuries of the hand. J Hand Surg Eur Vol. 2008;33:670-671.
- Schefflein J, Umans H, Ellenbogen D, et al. Sea urchin spine arthritis in the foot. Skeletal Radiol. 2012;41:1327-1331.
- Wada T, Soma T, Gaman K, et al. Sea urchin spine arthritis of the hand. J Hand Surg. 2008;33:398-401.
- Suárez-Peñaranda JM, Vieites B, Del Río E, et al. Histopathologic and immunohistochemical features of sea urchin granulomas. J Cutan Pathol. 2013;40:550-556.
- De La Torre C, Toribio J. Sea-urchin granuloma: histologic profile. a pathologic study of 50 biopsies. J Cutan Pathol. 2001;28:223-228.
- Sciani JM, Zychar BC, Gonçalves LR, et al. Pro-inflammatory effects of the aqueous extract of Echinometra lucunter sea urchin spines. Exp Biol Med (Maywood). 2011;236:277-280.
- De la Torre C, Vega A, Carracedo A, et al. Identification of Mycobacterium marinum in sea-urchin granulomas. Br J Dermatol. 2001;145:114-116.
- Gargus MD, Morohashi DK. A sea-urchin spine chilling remedy. N Engl J Med. 2012;367:1867-1868.
- Sjøberg T, de Weerd L. The usefulness of a skin biopsy punch to remove sea urchin spines. ANZ J Surg. 2010;80:383.
- Liram N, Gomori M, Perouansky M. Sea urchin puncture resulting in PIP joint synovial arthritis: case report and MRI study. J Travel Med. 2000;7:43-45.
- Dahl WJ, Jebson P, Louis DS. Sea urchin injuries to the hand: a case report and review of the literature. Iowa Orthop J. 2010;30:153-156.
- Rossetto AL, de Macedo Mora J, Haddad Junior V. Sea urchin granuloma. Rev Inst Med Trop Sao Paulo. 2006;48:303-306.
- Ahmad R, McCann PA, Barakat M, et al. Sea urchin spine injuries of the hand. J Hand Surg Eur Vol. 2008;33:670-671.
- Schefflein J, Umans H, Ellenbogen D, et al. Sea urchin spine arthritis in the foot. Skeletal Radiol. 2012;41:1327-1331.
- Wada T, Soma T, Gaman K, et al. Sea urchin spine arthritis of the hand. J Hand Surg. 2008;33:398-401.
- Suárez-Peñaranda JM, Vieites B, Del Río E, et al. Histopathologic and immunohistochemical features of sea urchin granulomas. J Cutan Pathol. 2013;40:550-556.
- De La Torre C, Toribio J. Sea-urchin granuloma: histologic profile. a pathologic study of 50 biopsies. J Cutan Pathol. 2001;28:223-228.
- Sciani JM, Zychar BC, Gonçalves LR, et al. Pro-inflammatory effects of the aqueous extract of Echinometra lucunter sea urchin spines. Exp Biol Med (Maywood). 2011;236:277-280.
- De la Torre C, Vega A, Carracedo A, et al. Identification of Mycobacterium marinum in sea-urchin granulomas. Br J Dermatol. 2001;145:114-116.
- Gargus MD, Morohashi DK. A sea-urchin spine chilling remedy. N Engl J Med. 2012;367:1867-1868.
- Sjøberg T, de Weerd L. The usefulness of a skin biopsy punch to remove sea urchin spines. ANZ J Surg. 2010;80:383.
Practice Points
- Radiographic imaging may aid in the identification of sea urchin spines, especially if there are no visible or palpable skin nodules.
- Treatment of sea urchin spine injuries typically involves surgical removal of the spines with local anesthesia and cutaneous extraction.
- Prompt extraction of sea urchin spines can improve pain symptoms and decrease the likelihood of granuloma formation, infection, and extracutaneous complications.
Necrotic Lesion of the Ear
The Diagnosis: Chondrodermatitis Nodularis Chronica Helicis
Histopathologic examination revealed focal epidermal erosion and ulceration directly overlying the hyaline cartilage with degenerative changes (Figure). The dermis was relatively noninflamed with fibroplasia of the vasculature. The blood vessels indirectly beneath the ulceration were found to be unremarkable with no indications of fibrinoid necrosis, vasculitis, or the presence of thrombi. The patient was informed of the diagnosis, at which point she reported that she slept on the right side. The excisional biopsy site healed well without recurrence of chondrodermatitis nodularis chronica helicis (CNH).
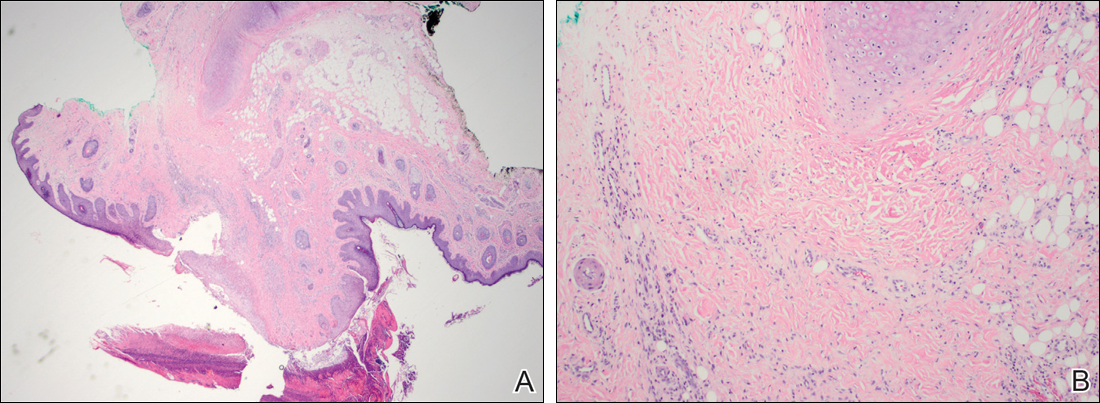
Chondrodermatitis nodularis chronica helicis, also known as clavus helicis, is a benign, usually solitary, painful lesion. Historically, it was first described in 1915 by Winkler1 and in the 1960s the most common documented cases were attributed to the headpieces of telephone operators and nuns.2 In the early 2000s, cell phones were determined to be a growing cause.3 Chondrodermatitis nodularis chronica helicis is most commonly found on the helix with the antihelix being affected less often.4 The condition is more common in men, with a male to female ratio being reported as high as 10:1. Possible causes of this disorder stem from damage to cartilage associated with pressure, sun exposure, cold temperatures, and microvascular disease. Additionally, some researchers have hypothesized that the cartilaginous damage resulting from solar elastosis and minor trauma leaves a susceptibility to CNH. This disorder usually presents as a small, exquisitely tender nodule that may ulcerate and crust.4 Chondrodermatitis nodularis chronica helicis may be mistaken for basal cell carcinoma, squamous cell carcinoma, actinic keratosis, and weathering nodules, though CNH tends to be more painful.
The diagnosis of CNH often is clinical but may require a skin biopsy. Histopathology of CNH shows a benign inflammatory lesion with an acanthotic hyperkeratotic epidermis that may be ulcerated. A primarily lymphocytic infiltrate usually is observed with variable presence of histiocytes and neutrophils. Cartilaginous changes range from simple perichondral thickening to notable areas of degeneration with calcification and ossification.4
Although the diagnosis of CNH often is straightforward, the remarkable necrosis present in our case made for an interesting differential diagnosis. Pernio, cryoglobulinemia, and levamisole-induced vasculopathy were all considered. Pernio, caused by cold-induced vasoconstriction and hypoxemia, classically presents as erythematous lesions with a symmetrical distribution on acral sites.5 Cryoglobulinemia involves proteins that precipitate at cold temperatures causing damage via an occlusive vasculopathy or an immune complex-mediated vasculitis. The presence of cryoglobulinemia is strongly associated with concomitant hepatitis C virus infection.6 Ulcerated and purpuric lesions of cryoglobulinemia may become necrotic. Levamisole is a veterinary antihelminthic drug and common cocaine contaminant, often added to cocaine as a cutting agent. Levamisole-induced vasculopathy favors acral sites and often is noted on the ears as purpuric patches, sometimes with necrosis.7
Several therapies for CNH have been reported with variable effectiveness.8 First-line treatments are the use of pressure-relieving devices including a doughnut-shaped pillow during sleep and intralesional corticosteroids.9 Surgical treatments including cryotherapy, simple excision, electrodesiccation and curettage, wedge resection with helical rim advancement flap, punch and graft technique, and CO2 laser have been tried.8 Photodynamic therapy and topical nitroglycerine also have shown to be of benefit.8,9
Our case of CNH is unique because of the remarkable degree of necrosis present on clinical examination. Chondrodermatitis nodularis chronica helicis with such an impressive necrotic presentation is rare. We speculate that the patient's underlying hypercoagulable state may have contributed to the dramatic presentation. It is important to keep CNH in mind when evaluating any necrotic lesion on the ear.
- Winkler M. Knötcehnformige Erkrankung am helix. chondrodermatitis nodularis chronic helicis. Arch für Dermatologie und Syphilis. 1915;121:278-285.
- Barker L, Young AW, Sachs W. Chondrodermatitis of the ears: a differential study of nodules of the helix and antihelix. Arch Dermatol. 1960;81:15-25.
- Elgart M. Cell phone chondrodermatitis. Arch Dermatol. 2000;136:1568.
- Cribier B, Scrivener Y, Peltre B. Neural hyperplasia in chondrodermatitis nodularis chronica helicis. J Am Acad Dermatol. 2006;55:844-848.
- King JM, Plotner AN, Adams BB. Perniosis induced by a cold-therapy system. Arch Dermatol. 2012;148:1101-1102.
- Berk DR, Mallory SB, Keeffe EB, et al. Dermatologic disorders associated with chronic hepatitis C: effect of interferon therapy. Clin Gastroenterol Hepatol. 2007;5:142-151.
- Hennings C, Miller J. Illicit drugs: what dermatologists need to know. J Am Acad Dermatol. 2013;69:135-142.
- Flynn V, Chisholm C, Grimwood R. Topical nitroglycerin: a promising treatment option for chondrodermatitis nodularis helicis. J Am Acad Dermatol. 2011;64:531-536.
- Gilaberte Y, Frias M, Pérez-Lorenz J. Chondrodermatitis nodularis helicis successfully treated with photodynamic therapy. Arch Dermatol. 2010;146:1080-1082.
The Diagnosis: Chondrodermatitis Nodularis Chronica Helicis
Histopathologic examination revealed focal epidermal erosion and ulceration directly overlying the hyaline cartilage with degenerative changes (Figure). The dermis was relatively noninflamed with fibroplasia of the vasculature. The blood vessels indirectly beneath the ulceration were found to be unremarkable with no indications of fibrinoid necrosis, vasculitis, or the presence of thrombi. The patient was informed of the diagnosis, at which point she reported that she slept on the right side. The excisional biopsy site healed well without recurrence of chondrodermatitis nodularis chronica helicis (CNH).
Chondrodermatitis nodularis chronica helicis, also known as clavus helicis, is a benign, usually solitary, painful lesion. Historically, it was first described in 1915 by Winkler1 and in the 1960s the most common documented cases were attributed to the headpieces of telephone operators and nuns.2 In the early 2000s, cell phones were determined to be a growing cause.3 Chondrodermatitis nodularis chronica helicis is most commonly found on the helix with the antihelix being affected less often.4 The condition is more common in men, with a male to female ratio being reported as high as 10:1. Possible causes of this disorder stem from damage to cartilage associated with pressure, sun exposure, cold temperatures, and microvascular disease. Additionally, some researchers have hypothesized that the cartilaginous damage resulting from solar elastosis and minor trauma leaves a susceptibility to CNH. This disorder usually presents as a small, exquisitely tender nodule that may ulcerate and crust.4 Chondrodermatitis nodularis chronica helicis may be mistaken for basal cell carcinoma, squamous cell carcinoma, actinic keratosis, and weathering nodules, though CNH tends to be more painful.
The diagnosis of CNH often is clinical but may require a skin biopsy. Histopathology of CNH shows a benign inflammatory lesion with an acanthotic hyperkeratotic epidermis that may be ulcerated. A primarily lymphocytic infiltrate usually is observed with variable presence of histiocytes and neutrophils. Cartilaginous changes range from simple perichondral thickening to notable areas of degeneration with calcification and ossification.4
Although the diagnosis of CNH often is straightforward, the remarkable necrosis present in our case made for an interesting differential diagnosis. Pernio, cryoglobulinemia, and levamisole-induced vasculopathy were all considered. Pernio, caused by cold-induced vasoconstriction and hypoxemia, classically presents as erythematous lesions with a symmetrical distribution on acral sites.5 Cryoglobulinemia involves proteins that precipitate at cold temperatures causing damage via an occlusive vasculopathy or an immune complex-mediated vasculitis. The presence of cryoglobulinemia is strongly associated with concomitant hepatitis C virus infection.6 Ulcerated and purpuric lesions of cryoglobulinemia may become necrotic. Levamisole is a veterinary antihelminthic drug and common cocaine contaminant, often added to cocaine as a cutting agent. Levamisole-induced vasculopathy favors acral sites and often is noted on the ears as purpuric patches, sometimes with necrosis.7
Several therapies for CNH have been reported with variable effectiveness.8 First-line treatments are the use of pressure-relieving devices including a doughnut-shaped pillow during sleep and intralesional corticosteroids.9 Surgical treatments including cryotherapy, simple excision, electrodesiccation and curettage, wedge resection with helical rim advancement flap, punch and graft technique, and CO2 laser have been tried.8 Photodynamic therapy and topical nitroglycerine also have shown to be of benefit.8,9
Our case of CNH is unique because of the remarkable degree of necrosis present on clinical examination. Chondrodermatitis nodularis chronica helicis with such an impressive necrotic presentation is rare. We speculate that the patient's underlying hypercoagulable state may have contributed to the dramatic presentation. It is important to keep CNH in mind when evaluating any necrotic lesion on the ear.
The Diagnosis: Chondrodermatitis Nodularis Chronica Helicis
Histopathologic examination revealed focal epidermal erosion and ulceration directly overlying the hyaline cartilage with degenerative changes (Figure). The dermis was relatively noninflamed with fibroplasia of the vasculature. The blood vessels indirectly beneath the ulceration were found to be unremarkable with no indications of fibrinoid necrosis, vasculitis, or the presence of thrombi. The patient was informed of the diagnosis, at which point she reported that she slept on the right side. The excisional biopsy site healed well without recurrence of chondrodermatitis nodularis chronica helicis (CNH).
Chondrodermatitis nodularis chronica helicis, also known as clavus helicis, is a benign, usually solitary, painful lesion. Historically, it was first described in 1915 by Winkler1 and in the 1960s the most common documented cases were attributed to the headpieces of telephone operators and nuns.2 In the early 2000s, cell phones were determined to be a growing cause.3 Chondrodermatitis nodularis chronica helicis is most commonly found on the helix with the antihelix being affected less often.4 The condition is more common in men, with a male to female ratio being reported as high as 10:1. Possible causes of this disorder stem from damage to cartilage associated with pressure, sun exposure, cold temperatures, and microvascular disease. Additionally, some researchers have hypothesized that the cartilaginous damage resulting from solar elastosis and minor trauma leaves a susceptibility to CNH. This disorder usually presents as a small, exquisitely tender nodule that may ulcerate and crust.4 Chondrodermatitis nodularis chronica helicis may be mistaken for basal cell carcinoma, squamous cell carcinoma, actinic keratosis, and weathering nodules, though CNH tends to be more painful.
The diagnosis of CNH often is clinical but may require a skin biopsy. Histopathology of CNH shows a benign inflammatory lesion with an acanthotic hyperkeratotic epidermis that may be ulcerated. A primarily lymphocytic infiltrate usually is observed with variable presence of histiocytes and neutrophils. Cartilaginous changes range from simple perichondral thickening to notable areas of degeneration with calcification and ossification.4
Although the diagnosis of CNH often is straightforward, the remarkable necrosis present in our case made for an interesting differential diagnosis. Pernio, cryoglobulinemia, and levamisole-induced vasculopathy were all considered. Pernio, caused by cold-induced vasoconstriction and hypoxemia, classically presents as erythematous lesions with a symmetrical distribution on acral sites.5 Cryoglobulinemia involves proteins that precipitate at cold temperatures causing damage via an occlusive vasculopathy or an immune complex-mediated vasculitis. The presence of cryoglobulinemia is strongly associated with concomitant hepatitis C virus infection.6 Ulcerated and purpuric lesions of cryoglobulinemia may become necrotic. Levamisole is a veterinary antihelminthic drug and common cocaine contaminant, often added to cocaine as a cutting agent. Levamisole-induced vasculopathy favors acral sites and often is noted on the ears as purpuric patches, sometimes with necrosis.7
Several therapies for CNH have been reported with variable effectiveness.8 First-line treatments are the use of pressure-relieving devices including a doughnut-shaped pillow during sleep and intralesional corticosteroids.9 Surgical treatments including cryotherapy, simple excision, electrodesiccation and curettage, wedge resection with helical rim advancement flap, punch and graft technique, and CO2 laser have been tried.8 Photodynamic therapy and topical nitroglycerine also have shown to be of benefit.8,9
Our case of CNH is unique because of the remarkable degree of necrosis present on clinical examination. Chondrodermatitis nodularis chronica helicis with such an impressive necrotic presentation is rare. We speculate that the patient's underlying hypercoagulable state may have contributed to the dramatic presentation. It is important to keep CNH in mind when evaluating any necrotic lesion on the ear.
- Winkler M. Knötcehnformige Erkrankung am helix. chondrodermatitis nodularis chronic helicis. Arch für Dermatologie und Syphilis. 1915;121:278-285.
- Barker L, Young AW, Sachs W. Chondrodermatitis of the ears: a differential study of nodules of the helix and antihelix. Arch Dermatol. 1960;81:15-25.
- Elgart M. Cell phone chondrodermatitis. Arch Dermatol. 2000;136:1568.
- Cribier B, Scrivener Y, Peltre B. Neural hyperplasia in chondrodermatitis nodularis chronica helicis. J Am Acad Dermatol. 2006;55:844-848.
- King JM, Plotner AN, Adams BB. Perniosis induced by a cold-therapy system. Arch Dermatol. 2012;148:1101-1102.
- Berk DR, Mallory SB, Keeffe EB, et al. Dermatologic disorders associated with chronic hepatitis C: effect of interferon therapy. Clin Gastroenterol Hepatol. 2007;5:142-151.
- Hennings C, Miller J. Illicit drugs: what dermatologists need to know. J Am Acad Dermatol. 2013;69:135-142.
- Flynn V, Chisholm C, Grimwood R. Topical nitroglycerin: a promising treatment option for chondrodermatitis nodularis helicis. J Am Acad Dermatol. 2011;64:531-536.
- Gilaberte Y, Frias M, Pérez-Lorenz J. Chondrodermatitis nodularis helicis successfully treated with photodynamic therapy. Arch Dermatol. 2010;146:1080-1082.
- Winkler M. Knötcehnformige Erkrankung am helix. chondrodermatitis nodularis chronic helicis. Arch für Dermatologie und Syphilis. 1915;121:278-285.
- Barker L, Young AW, Sachs W. Chondrodermatitis of the ears: a differential study of nodules of the helix and antihelix. Arch Dermatol. 1960;81:15-25.
- Elgart M. Cell phone chondrodermatitis. Arch Dermatol. 2000;136:1568.
- Cribier B, Scrivener Y, Peltre B. Neural hyperplasia in chondrodermatitis nodularis chronica helicis. J Am Acad Dermatol. 2006;55:844-848.
- King JM, Plotner AN, Adams BB. Perniosis induced by a cold-therapy system. Arch Dermatol. 2012;148:1101-1102.
- Berk DR, Mallory SB, Keeffe EB, et al. Dermatologic disorders associated with chronic hepatitis C: effect of interferon therapy. Clin Gastroenterol Hepatol. 2007;5:142-151.
- Hennings C, Miller J. Illicit drugs: what dermatologists need to know. J Am Acad Dermatol. 2013;69:135-142.
- Flynn V, Chisholm C, Grimwood R. Topical nitroglycerin: a promising treatment option for chondrodermatitis nodularis helicis. J Am Acad Dermatol. 2011;64:531-536.
- Gilaberte Y, Frias M, Pérez-Lorenz J. Chondrodermatitis nodularis helicis successfully treated with photodynamic therapy. Arch Dermatol. 2010;146:1080-1082.
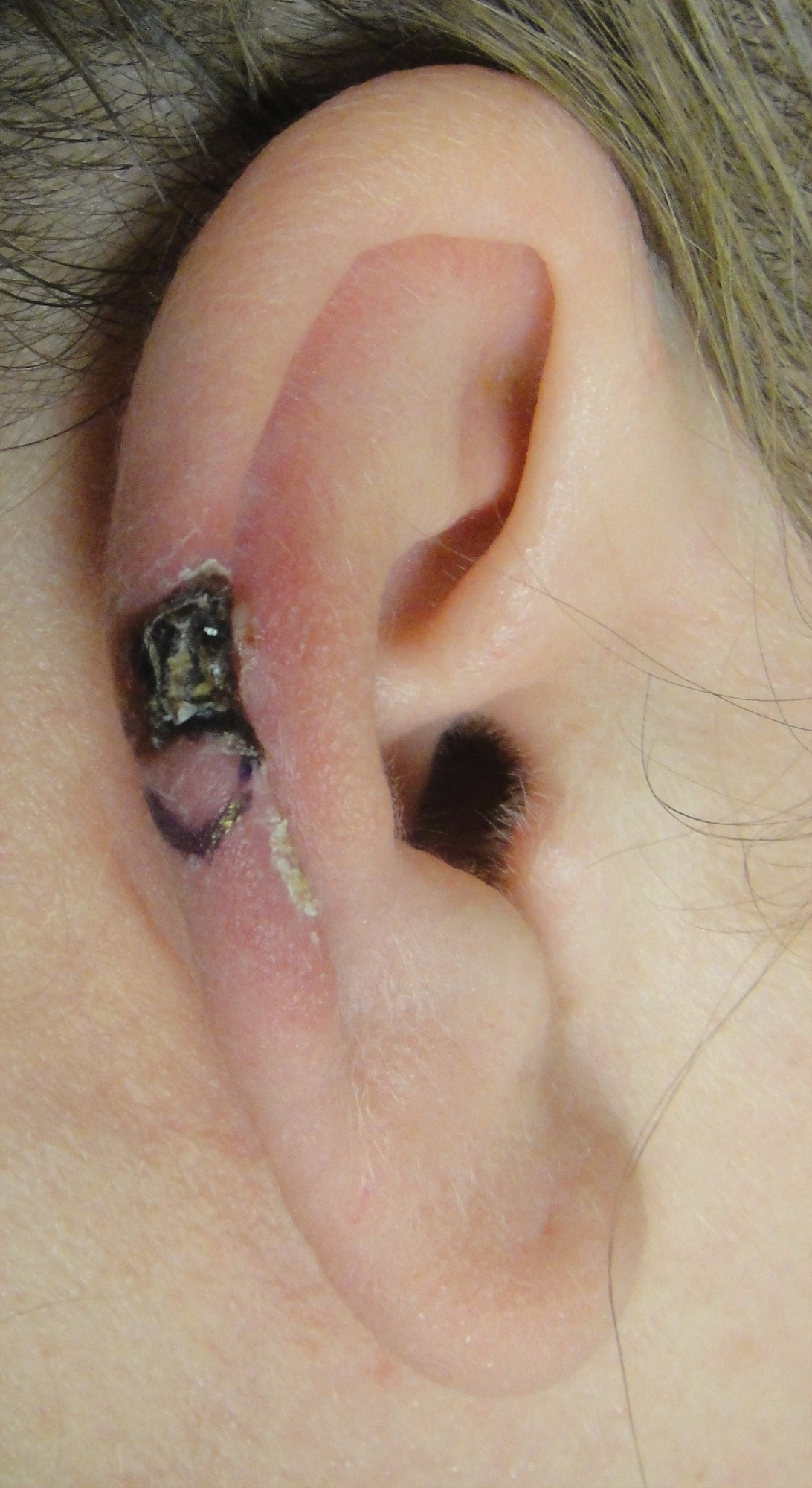
A 43-year-old woman presented with a painful necrotic lesion on the right ear of 1 month's duration. She denied trauma to the ear and had no other skin lesions elsewhere on the body. A course of doxycycline prior to presentation did not result in improvement. Her medical history was remarkable for diabetes mellitus, deep vein thrombosis, depression, and gastroesophageal reflux disease. She had been taking warfarin regularly for years. She denied using recreational drugs. On physical examination, the right ear demonstrated a 6-mm necrotic area with surrounding tender erythema. Examinations of the left ear, face, and legs were normal. An excisional biopsy was performed.
Crusted Plaque in the Umbilicus
The Diagnosis: Sister Mary Joseph Nodule
The umbilical skin biopsy revealed a moderately differentiated adenocarcinoma (Figure) that was positive for cytokeratin 20 and CDX2 and negative for cytokeratin 7 and transcription termination factor 1. The patient subsequently underwent computed tomography of the abdomen and pelvis, which showed multiple soft-tissue nodules on the greater omentum, a soft-tissue density at the umbilicus, and thickening of the gastric mucosa. An upper endoscopy was then performed, which revealed a large fungating ulcerated mass in the stomach. Biopsy of this mass showed an invasive moderately differentiated adenocarcinoma, which was ERBB2 (formerly HER2) negative. Histopathologically, these pleomorphic glands looked similar to the glands seen in the original skin biopsy. With this diagnosis of metastatic gastric adenocarcinoma, our patient chose palliative chemotherapy but declined precipitously and died 2 months after the initial skin biopsy of the umbilical lesion.
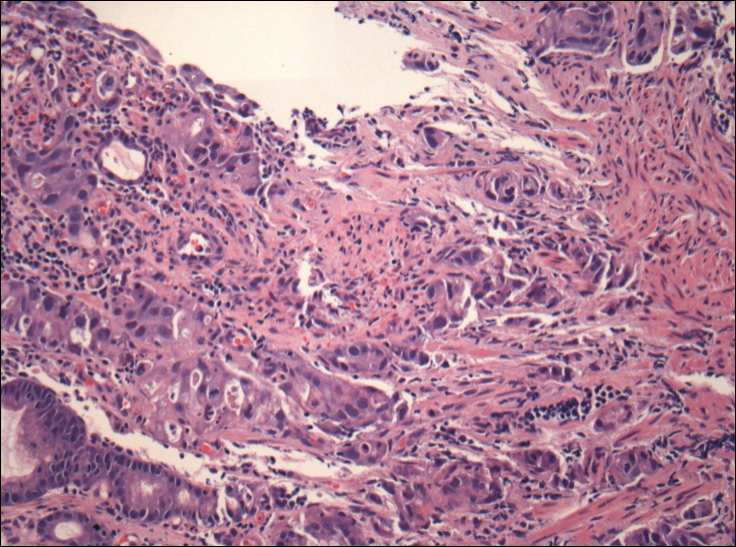
When encountering a patient with an umbilical lesion, it is important to consider benign and malignant lesions in the differential diagnosis. A benign lesion may include scar, cyst, pyogenic granuloma, hemangioma, umbilical hernia, endometriosis, polyp, abscess, or the presence of an omphalith.1 Inflammatory dermatoses such as psoriasis or eczema also should be considered. Malignant lesions could be either primary or secondary, with metastatic disease being the most common.2 Sister Mary Joseph nodule (SMJN) is the eponymgiven to an umbilical lesion representing metastatic disease. Sister Mary Joseph was a nurse and surgical assistant to Dr. William Mayo in Rochester, Minnesota, in what is now known as the Mayo Clinic. She is credited to be the first to observe and note the association between an umbilical nodule and intra-abdominal malignancy. Metastasis to the umbilicus is thought to occur by way of contiguous, hematogenous, lymphatic, or direct spread through embryologic remnants from primary cancers of nearby gastrointestinal or pelvic viscera. It is a rare cutaneous sign of internal malignancy, with an estimated prevalence of 1% to 3%.3 The most common primary cancer is gastric adenocarcinoma, though cases of metastasis from pancreatic, endometrial, and less commonly hematopoietic or supradiaphragmatic cancers have been reported.4 It is more common in women, likely due to the addition of gynecologic malignancies.1
The use of dermoscopy has been advocated as an adjuvant tool in delineating benign and malignant umbilical lesions when an atypical polymorphous vascular pattern indicating neovascularization has been observed with neoplastic growth.5 Once a suspicious umbilical lesion is identified, the first step should be to obtain a skin biopsy or to use fine needle aspiration for cytology.6 Biopsy is especially relevant in the background of cancer history because SMJN may present with cancer recurrence.3 Once one of these is obtained, histological and immunohistochemical analysis will guide further workup and diagnosis of the umbilical lesion.
The importance of reviewing such cases lies in the variable presentation of cutaneous metastases such as SMJN and the grim prognosis that accompanies this finding. It presents as a firm indurated plaque or nodule that may present with systemic symptoms suggestive of malignancy, though in 30% of cases it is the sole initial sign.7 The nodule may be painful if ulcerated or fissured. Bloody, serous, or purulent discharge may be present. After diagnosis of an SMJN, most patients succumb to the disease within 12 months. Thus, it is vital for dermatologists to investigate umbilical lesions with great caution and a high index of suspicion.
- Chalya PL, Mabula JB, Rambau PF, et al. Sister Mary Joseph's nodule at a University teaching hospital in northwestern Tanzania: a retrospective review of 34 cases. World J Surg Oncol. 2013;11:151.
- Papalas JA, Selim MA. Metastatic vs primary malignant neoplasms affecting the umbilicus: clinicopathologic features of 77 tumors. Ann Diagn Pathol. 2011;15:237-242.
- Palaniappan M, Jose WM, Mehta A, et al. Umbilical metastasis: a case series of four Sister Joseph nodules from four different visceral malignancies. Curr Oncol. 2010;17:78-81.
- Zhang YL, Selvaggi SM. Metastatic islet cell carcinoma to the umbilicus: diagnosis by fine-needle aspiration. Diagn Cytopathol. 2003;29:91-94.
- Mun JH, Kim JM, Ko HC, et al. Dermoscopy of a Sister Mary Joseph nodule. J Am Acad Dermatol. 2013;68:e190-e192.
- Handa U, Garg S, Mohan H. Fine-needle aspiration of Sister Mary Joseph's (paraumbilical) nodules. Diagn Cytopathol. 2008;36:348-350.
- Abu-Hilal M, Newman JS. Sister Mary Joseph and her nodule: historical and clinical perspective. Am J Med Sci. 2009;337:271-273.
The Diagnosis: Sister Mary Joseph Nodule
The umbilical skin biopsy revealed a moderately differentiated adenocarcinoma (Figure) that was positive for cytokeratin 20 and CDX2 and negative for cytokeratin 7 and transcription termination factor 1. The patient subsequently underwent computed tomography of the abdomen and pelvis, which showed multiple soft-tissue nodules on the greater omentum, a soft-tissue density at the umbilicus, and thickening of the gastric mucosa. An upper endoscopy was then performed, which revealed a large fungating ulcerated mass in the stomach. Biopsy of this mass showed an invasive moderately differentiated adenocarcinoma, which was ERBB2 (formerly HER2) negative. Histopathologically, these pleomorphic glands looked similar to the glands seen in the original skin biopsy. With this diagnosis of metastatic gastric adenocarcinoma, our patient chose palliative chemotherapy but declined precipitously and died 2 months after the initial skin biopsy of the umbilical lesion.
When encountering a patient with an umbilical lesion, it is important to consider benign and malignant lesions in the differential diagnosis. A benign lesion may include scar, cyst, pyogenic granuloma, hemangioma, umbilical hernia, endometriosis, polyp, abscess, or the presence of an omphalith.1 Inflammatory dermatoses such as psoriasis or eczema also should be considered. Malignant lesions could be either primary or secondary, with metastatic disease being the most common.2 Sister Mary Joseph nodule (SMJN) is the eponymgiven to an umbilical lesion representing metastatic disease. Sister Mary Joseph was a nurse and surgical assistant to Dr. William Mayo in Rochester, Minnesota, in what is now known as the Mayo Clinic. She is credited to be the first to observe and note the association between an umbilical nodule and intra-abdominal malignancy. Metastasis to the umbilicus is thought to occur by way of contiguous, hematogenous, lymphatic, or direct spread through embryologic remnants from primary cancers of nearby gastrointestinal or pelvic viscera. It is a rare cutaneous sign of internal malignancy, with an estimated prevalence of 1% to 3%.3 The most common primary cancer is gastric adenocarcinoma, though cases of metastasis from pancreatic, endometrial, and less commonly hematopoietic or supradiaphragmatic cancers have been reported.4 It is more common in women, likely due to the addition of gynecologic malignancies.1
The use of dermoscopy has been advocated as an adjuvant tool in delineating benign and malignant umbilical lesions when an atypical polymorphous vascular pattern indicating neovascularization has been observed with neoplastic growth.5 Once a suspicious umbilical lesion is identified, the first step should be to obtain a skin biopsy or to use fine needle aspiration for cytology.6 Biopsy is especially relevant in the background of cancer history because SMJN may present with cancer recurrence.3 Once one of these is obtained, histological and immunohistochemical analysis will guide further workup and diagnosis of the umbilical lesion.
The importance of reviewing such cases lies in the variable presentation of cutaneous metastases such as SMJN and the grim prognosis that accompanies this finding. It presents as a firm indurated plaque or nodule that may present with systemic symptoms suggestive of malignancy, though in 30% of cases it is the sole initial sign.7 The nodule may be painful if ulcerated or fissured. Bloody, serous, or purulent discharge may be present. After diagnosis of an SMJN, most patients succumb to the disease within 12 months. Thus, it is vital for dermatologists to investigate umbilical lesions with great caution and a high index of suspicion.
The Diagnosis: Sister Mary Joseph Nodule
The umbilical skin biopsy revealed a moderately differentiated adenocarcinoma (Figure) that was positive for cytokeratin 20 and CDX2 and negative for cytokeratin 7 and transcription termination factor 1. The patient subsequently underwent computed tomography of the abdomen and pelvis, which showed multiple soft-tissue nodules on the greater omentum, a soft-tissue density at the umbilicus, and thickening of the gastric mucosa. An upper endoscopy was then performed, which revealed a large fungating ulcerated mass in the stomach. Biopsy of this mass showed an invasive moderately differentiated adenocarcinoma, which was ERBB2 (formerly HER2) negative. Histopathologically, these pleomorphic glands looked similar to the glands seen in the original skin biopsy. With this diagnosis of metastatic gastric adenocarcinoma, our patient chose palliative chemotherapy but declined precipitously and died 2 months after the initial skin biopsy of the umbilical lesion.
When encountering a patient with an umbilical lesion, it is important to consider benign and malignant lesions in the differential diagnosis. A benign lesion may include scar, cyst, pyogenic granuloma, hemangioma, umbilical hernia, endometriosis, polyp, abscess, or the presence of an omphalith.1 Inflammatory dermatoses such as psoriasis or eczema also should be considered. Malignant lesions could be either primary or secondary, with metastatic disease being the most common.2 Sister Mary Joseph nodule (SMJN) is the eponymgiven to an umbilical lesion representing metastatic disease. Sister Mary Joseph was a nurse and surgical assistant to Dr. William Mayo in Rochester, Minnesota, in what is now known as the Mayo Clinic. She is credited to be the first to observe and note the association between an umbilical nodule and intra-abdominal malignancy. Metastasis to the umbilicus is thought to occur by way of contiguous, hematogenous, lymphatic, or direct spread through embryologic remnants from primary cancers of nearby gastrointestinal or pelvic viscera. It is a rare cutaneous sign of internal malignancy, with an estimated prevalence of 1% to 3%.3 The most common primary cancer is gastric adenocarcinoma, though cases of metastasis from pancreatic, endometrial, and less commonly hematopoietic or supradiaphragmatic cancers have been reported.4 It is more common in women, likely due to the addition of gynecologic malignancies.1
The use of dermoscopy has been advocated as an adjuvant tool in delineating benign and malignant umbilical lesions when an atypical polymorphous vascular pattern indicating neovascularization has been observed with neoplastic growth.5 Once a suspicious umbilical lesion is identified, the first step should be to obtain a skin biopsy or to use fine needle aspiration for cytology.6 Biopsy is especially relevant in the background of cancer history because SMJN may present with cancer recurrence.3 Once one of these is obtained, histological and immunohistochemical analysis will guide further workup and diagnosis of the umbilical lesion.
The importance of reviewing such cases lies in the variable presentation of cutaneous metastases such as SMJN and the grim prognosis that accompanies this finding. It presents as a firm indurated plaque or nodule that may present with systemic symptoms suggestive of malignancy, though in 30% of cases it is the sole initial sign.7 The nodule may be painful if ulcerated or fissured. Bloody, serous, or purulent discharge may be present. After diagnosis of an SMJN, most patients succumb to the disease within 12 months. Thus, it is vital for dermatologists to investigate umbilical lesions with great caution and a high index of suspicion.
- Chalya PL, Mabula JB, Rambau PF, et al. Sister Mary Joseph's nodule at a University teaching hospital in northwestern Tanzania: a retrospective review of 34 cases. World J Surg Oncol. 2013;11:151.
- Papalas JA, Selim MA. Metastatic vs primary malignant neoplasms affecting the umbilicus: clinicopathologic features of 77 tumors. Ann Diagn Pathol. 2011;15:237-242.
- Palaniappan M, Jose WM, Mehta A, et al. Umbilical metastasis: a case series of four Sister Joseph nodules from four different visceral malignancies. Curr Oncol. 2010;17:78-81.
- Zhang YL, Selvaggi SM. Metastatic islet cell carcinoma to the umbilicus: diagnosis by fine-needle aspiration. Diagn Cytopathol. 2003;29:91-94.
- Mun JH, Kim JM, Ko HC, et al. Dermoscopy of a Sister Mary Joseph nodule. J Am Acad Dermatol. 2013;68:e190-e192.
- Handa U, Garg S, Mohan H. Fine-needle aspiration of Sister Mary Joseph's (paraumbilical) nodules. Diagn Cytopathol. 2008;36:348-350.
- Abu-Hilal M, Newman JS. Sister Mary Joseph and her nodule: historical and clinical perspective. Am J Med Sci. 2009;337:271-273.
- Chalya PL, Mabula JB, Rambau PF, et al. Sister Mary Joseph's nodule at a University teaching hospital in northwestern Tanzania: a retrospective review of 34 cases. World J Surg Oncol. 2013;11:151.
- Papalas JA, Selim MA. Metastatic vs primary malignant neoplasms affecting the umbilicus: clinicopathologic features of 77 tumors. Ann Diagn Pathol. 2011;15:237-242.
- Palaniappan M, Jose WM, Mehta A, et al. Umbilical metastasis: a case series of four Sister Joseph nodules from four different visceral malignancies. Curr Oncol. 2010;17:78-81.
- Zhang YL, Selvaggi SM. Metastatic islet cell carcinoma to the umbilicus: diagnosis by fine-needle aspiration. Diagn Cytopathol. 2003;29:91-94.
- Mun JH, Kim JM, Ko HC, et al. Dermoscopy of a Sister Mary Joseph nodule. J Am Acad Dermatol. 2013;68:e190-e192.
- Handa U, Garg S, Mohan H. Fine-needle aspiration of Sister Mary Joseph's (paraumbilical) nodules. Diagn Cytopathol. 2008;36:348-350.
- Abu-Hilal M, Newman JS. Sister Mary Joseph and her nodule: historical and clinical perspective. Am J Med Sci. 2009;337:271-273.
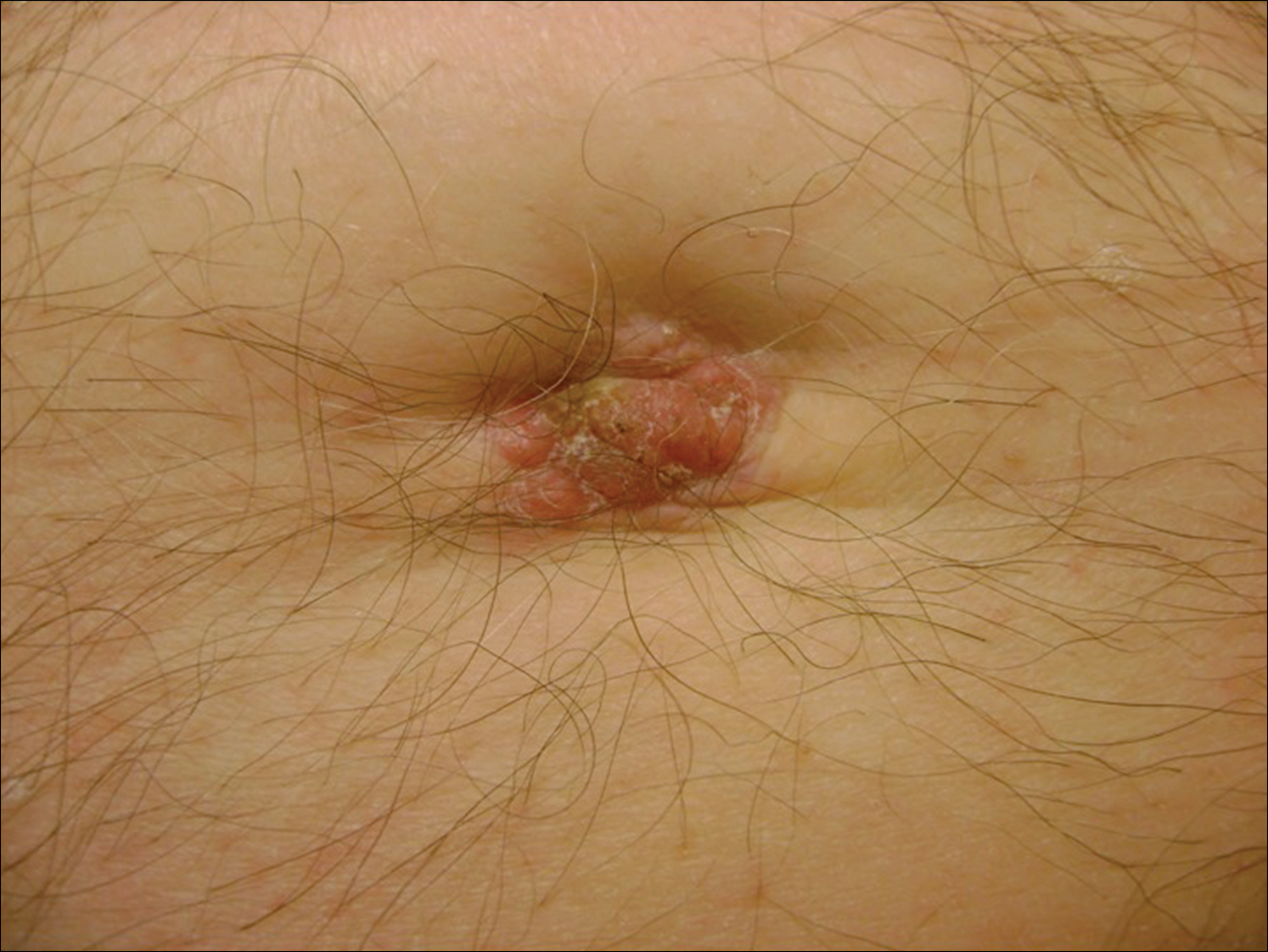
A 74-year-old man presented to our outpatient dermatology clinic with an asymptomatic umbilical lesion of unknown duration. The patient believed the lesion was a scar resulting from a prior laparoscopic repair of an umbilical hernia. However, the patient reported epigastric abdominal pain and diarrhea of 1 month's duration that he believed was due to the stomach flu. The patient denied fever, chills, loss of appetite, or weight loss. History was remarkable for hypertension, hyperlipidemia, coronary artery disease, chronic kidney disease, and emphysema. The patient had a surgical history of percutaneous transluminal coronary angioplasty in addition to the laparoscopic umbilical hernia repair. The patient's medications included pantoprazole, ondansetron, diphenoxylate-atropine as needed, amlodipine, lisinopril-hydrochlorothiazide, simvastatin, and aspirin. Physical examination revealed a 1×2-cm pink, nodular, firm plaque with crust at the umbilicus that was tender on palpation. A shave biopsy of the umbilicus was performed and sent for both pathological and immunohistochemical analysis.
Blaschkoid Unilateral Patch on the Chest
The Diagnosis: Lichen Striatus
Lichen striatus (LS) is an acquired and self-limited linear inflammatory dermatosis that most frequently occurs in children and less commonly in adults.1-3 Clinically, it is characterized by the sudden onset of an eruption consisting of slightly pigmented, erythematous, flat-topped papules with minimal scaling. These papules quickly coalesce to form a linear band that extends along a limb, the trunk, or the face, within Blaschko lines.1,4 In the adult form, patients tend to experience more diffuse lesions as well as severe pruritus with higher rates of relapse. It occasionally manifests in a dermatomal manner.1
The differential diagnosis includes other linear acquired inflammatory dermatoses such as blaschkitis, lichen planus, inflammatory linear verrucous epidermal nevus, and psoriasis. Blaschkitis has been described as a rare dermatosis that occurs along the Blaschko lines, affecting adults preferentially over children. Controversy exists whether blaschkitis and lichen striatus are the same disease or 2 separate entities.5 Clinically, both blaschkitis and lichen striatus can present with multiple linear papules and vesicles predominantly on the trunk. In blaschkitis, there is a predilection for males, with an older mean age at onset of 40 years.5 Lesions quickly resolve over months with frequent relapse compared to lichen striatus, which can persist for months to years.
Histopathologically, blaschkitis demonstrates spongiosis, usually without involvement of the adnexal structures. Lichenoid and spongiotic changes with adnexal extension are the hallmark features of lichen striatus. In our patient, biopsy showed several dense bandlike foci of lymphohistiocytic infiltrates along the dermoepidermal junction with spongiosis, basal cell liquefactive degeneration, and pigmentary incontinence (Figure 1). The focal areas were surfaced by parakeratotic and orthohyperkeratotic scale. Deep dermal perivascular and periadnexal extension was present (Figure 2). Periodic acid-Schiff stain was negative for fungi.
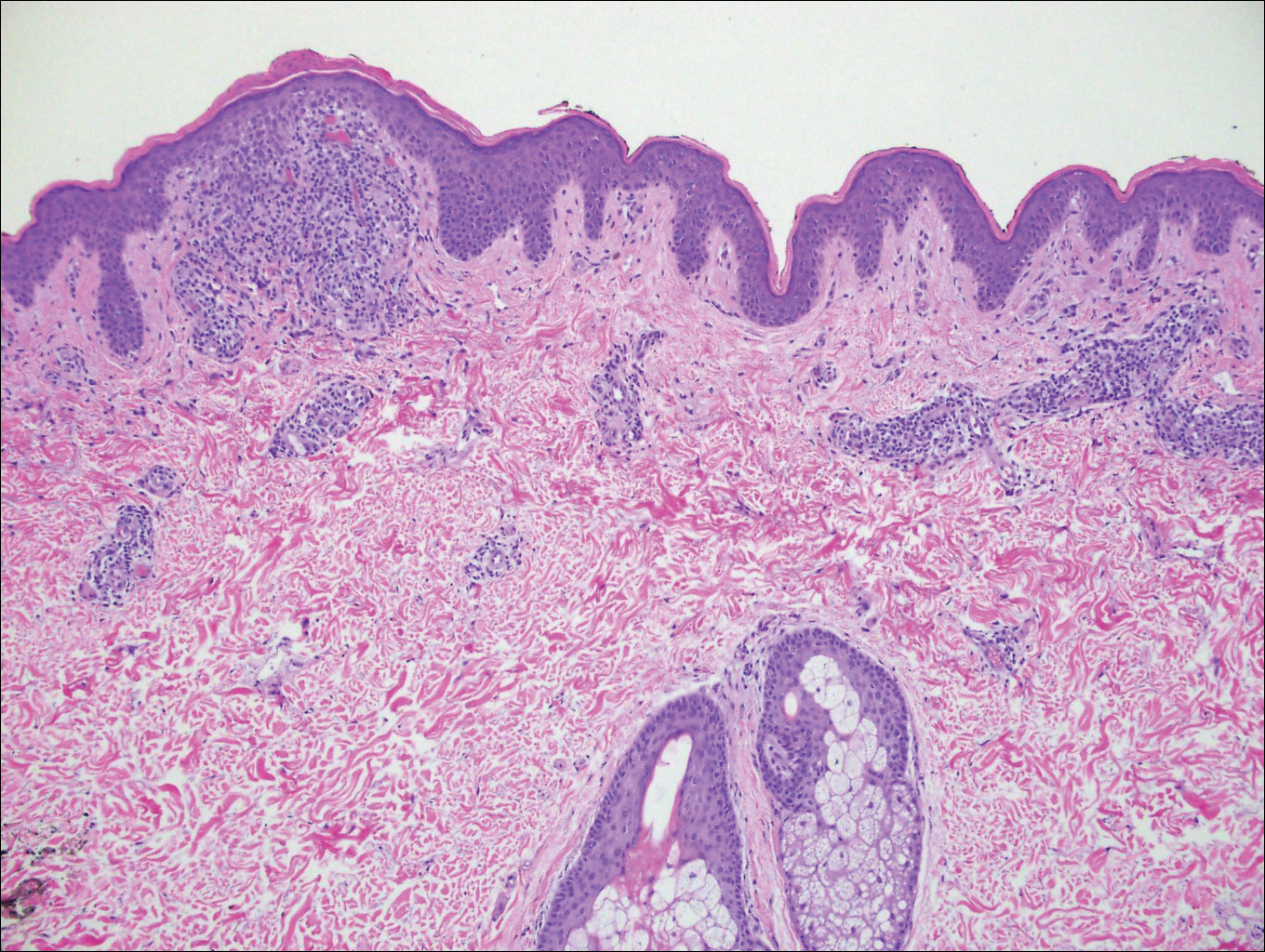
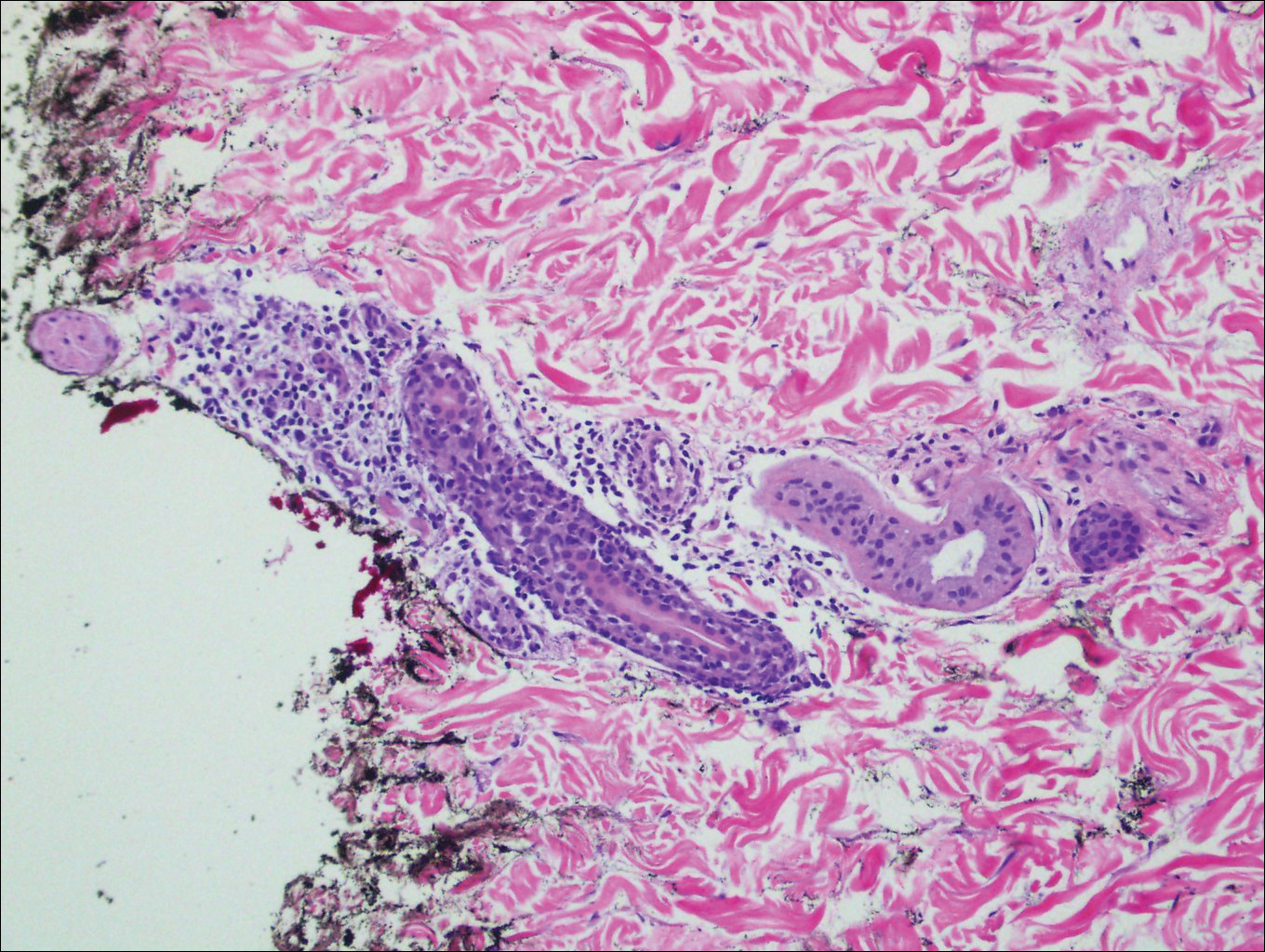
The pathogenesis of lichen striatus is not entirely understood, but it has been postulated that trauma, vaccinations, or viral infections may induce loss of immunologic tolerance to keratinocytes.1 This loss of tolerance can result in a T cell-mediated autoimmune reaction against malpighian cells, which show genetic mosaicism and are arranged along Blaschko lines.1,3 Familial cases also have been reported, suggesting that there may be an epigenetic mosaicism that contributes to this group of skin diseases.6,7
Lichen striatus tends to resolve on its own after approximately 6 to 9 months.8 Treatment typically consists of application of topical corticosteroids.1 Cases also have been successfully treated with tacrolimus and pimecrolimus.1,8 Our patient was treated with a midpotency topical steroid with improvement of the appearance but not complete resolution.
- Campanati A, Brandozzi G, Giangiacomi M, et al. Lichen striatus in adults and pimecrolimus: open, off-label clinical study. Int J Dermatol. 2008;47:732-736.
- Lee DY, Kim S, Kim CR, et al. Lichen striatus in an adult treated by a short course of low-dose systemic corticosteroid. J Dermatol. 2011;38:298-299.
- Hofer T. Lichen striatus in adults or "adult blaschkitis"? there is no need for a new naming. Dermatology. 2003;207:89-92.
- Shepherd V, Lun K, Strutton G. Lichen striatus in an adult following trauma. Australas J Dermatol. 2005;46:25-28.
- Müller CS, Schmaltz R, Vogt T, et al. Lichen striatus and blaschkitis reappraisal of the concept of blaschkolinear dermatoses. Br J Dermatol. 2011;164:257-262.
- Yaosaka M, Sawamura D, Iitoyo M, et al. Lichen striatus affecting a mother and her son. J Am Acad Dermatol. 2005;53:352-353.
- Jackson R. The lines of Blaschko: a review and reconsideration: observations of the cause of certain unusual linear conditions of the skin. Br J Dermatol. 1976;95:349-360.
- Sorgentini C, Allevato MA, Dahbar M, et al. Lichen striatus in an adult: successful treatment with tacrolimus. Br J Dermatol. 2004;150:776-777.
The Diagnosis: Lichen Striatus
Lichen striatus (LS) is an acquired and self-limited linear inflammatory dermatosis that most frequently occurs in children and less commonly in adults.1-3 Clinically, it is characterized by the sudden onset of an eruption consisting of slightly pigmented, erythematous, flat-topped papules with minimal scaling. These papules quickly coalesce to form a linear band that extends along a limb, the trunk, or the face, within Blaschko lines.1,4 In the adult form, patients tend to experience more diffuse lesions as well as severe pruritus with higher rates of relapse. It occasionally manifests in a dermatomal manner.1
The differential diagnosis includes other linear acquired inflammatory dermatoses such as blaschkitis, lichen planus, inflammatory linear verrucous epidermal nevus, and psoriasis. Blaschkitis has been described as a rare dermatosis that occurs along the Blaschko lines, affecting adults preferentially over children. Controversy exists whether blaschkitis and lichen striatus are the same disease or 2 separate entities.5 Clinically, both blaschkitis and lichen striatus can present with multiple linear papules and vesicles predominantly on the trunk. In blaschkitis, there is a predilection for males, with an older mean age at onset of 40 years.5 Lesions quickly resolve over months with frequent relapse compared to lichen striatus, which can persist for months to years.
Histopathologically, blaschkitis demonstrates spongiosis, usually without involvement of the adnexal structures. Lichenoid and spongiotic changes with adnexal extension are the hallmark features of lichen striatus. In our patient, biopsy showed several dense bandlike foci of lymphohistiocytic infiltrates along the dermoepidermal junction with spongiosis, basal cell liquefactive degeneration, and pigmentary incontinence (Figure 1). The focal areas were surfaced by parakeratotic and orthohyperkeratotic scale. Deep dermal perivascular and periadnexal extension was present (Figure 2). Periodic acid-Schiff stain was negative for fungi.
The pathogenesis of lichen striatus is not entirely understood, but it has been postulated that trauma, vaccinations, or viral infections may induce loss of immunologic tolerance to keratinocytes.1 This loss of tolerance can result in a T cell-mediated autoimmune reaction against malpighian cells, which show genetic mosaicism and are arranged along Blaschko lines.1,3 Familial cases also have been reported, suggesting that there may be an epigenetic mosaicism that contributes to this group of skin diseases.6,7
Lichen striatus tends to resolve on its own after approximately 6 to 9 months.8 Treatment typically consists of application of topical corticosteroids.1 Cases also have been successfully treated with tacrolimus and pimecrolimus.1,8 Our patient was treated with a midpotency topical steroid with improvement of the appearance but not complete resolution.
The Diagnosis: Lichen Striatus
Lichen striatus (LS) is an acquired and self-limited linear inflammatory dermatosis that most frequently occurs in children and less commonly in adults.1-3 Clinically, it is characterized by the sudden onset of an eruption consisting of slightly pigmented, erythematous, flat-topped papules with minimal scaling. These papules quickly coalesce to form a linear band that extends along a limb, the trunk, or the face, within Blaschko lines.1,4 In the adult form, patients tend to experience more diffuse lesions as well as severe pruritus with higher rates of relapse. It occasionally manifests in a dermatomal manner.1
The differential diagnosis includes other linear acquired inflammatory dermatoses such as blaschkitis, lichen planus, inflammatory linear verrucous epidermal nevus, and psoriasis. Blaschkitis has been described as a rare dermatosis that occurs along the Blaschko lines, affecting adults preferentially over children. Controversy exists whether blaschkitis and lichen striatus are the same disease or 2 separate entities.5 Clinically, both blaschkitis and lichen striatus can present with multiple linear papules and vesicles predominantly on the trunk. In blaschkitis, there is a predilection for males, with an older mean age at onset of 40 years.5 Lesions quickly resolve over months with frequent relapse compared to lichen striatus, which can persist for months to years.
Histopathologically, blaschkitis demonstrates spongiosis, usually without involvement of the adnexal structures. Lichenoid and spongiotic changes with adnexal extension are the hallmark features of lichen striatus. In our patient, biopsy showed several dense bandlike foci of lymphohistiocytic infiltrates along the dermoepidermal junction with spongiosis, basal cell liquefactive degeneration, and pigmentary incontinence (Figure 1). The focal areas were surfaced by parakeratotic and orthohyperkeratotic scale. Deep dermal perivascular and periadnexal extension was present (Figure 2). Periodic acid-Schiff stain was negative for fungi.
The pathogenesis of lichen striatus is not entirely understood, but it has been postulated that trauma, vaccinations, or viral infections may induce loss of immunologic tolerance to keratinocytes.1 This loss of tolerance can result in a T cell-mediated autoimmune reaction against malpighian cells, which show genetic mosaicism and are arranged along Blaschko lines.1,3 Familial cases also have been reported, suggesting that there may be an epigenetic mosaicism that contributes to this group of skin diseases.6,7
Lichen striatus tends to resolve on its own after approximately 6 to 9 months.8 Treatment typically consists of application of topical corticosteroids.1 Cases also have been successfully treated with tacrolimus and pimecrolimus.1,8 Our patient was treated with a midpotency topical steroid with improvement of the appearance but not complete resolution.
- Campanati A, Brandozzi G, Giangiacomi M, et al. Lichen striatus in adults and pimecrolimus: open, off-label clinical study. Int J Dermatol. 2008;47:732-736.
- Lee DY, Kim S, Kim CR, et al. Lichen striatus in an adult treated by a short course of low-dose systemic corticosteroid. J Dermatol. 2011;38:298-299.
- Hofer T. Lichen striatus in adults or "adult blaschkitis"? there is no need for a new naming. Dermatology. 2003;207:89-92.
- Shepherd V, Lun K, Strutton G. Lichen striatus in an adult following trauma. Australas J Dermatol. 2005;46:25-28.
- Müller CS, Schmaltz R, Vogt T, et al. Lichen striatus and blaschkitis reappraisal of the concept of blaschkolinear dermatoses. Br J Dermatol. 2011;164:257-262.
- Yaosaka M, Sawamura D, Iitoyo M, et al. Lichen striatus affecting a mother and her son. J Am Acad Dermatol. 2005;53:352-353.
- Jackson R. The lines of Blaschko: a review and reconsideration: observations of the cause of certain unusual linear conditions of the skin. Br J Dermatol. 1976;95:349-360.
- Sorgentini C, Allevato MA, Dahbar M, et al. Lichen striatus in an adult: successful treatment with tacrolimus. Br J Dermatol. 2004;150:776-777.
- Campanati A, Brandozzi G, Giangiacomi M, et al. Lichen striatus in adults and pimecrolimus: open, off-label clinical study. Int J Dermatol. 2008;47:732-736.
- Lee DY, Kim S, Kim CR, et al. Lichen striatus in an adult treated by a short course of low-dose systemic corticosteroid. J Dermatol. 2011;38:298-299.
- Hofer T. Lichen striatus in adults or "adult blaschkitis"? there is no need for a new naming. Dermatology. 2003;207:89-92.
- Shepherd V, Lun K, Strutton G. Lichen striatus in an adult following trauma. Australas J Dermatol. 2005;46:25-28.
- Müller CS, Schmaltz R, Vogt T, et al. Lichen striatus and blaschkitis reappraisal of the concept of blaschkolinear dermatoses. Br J Dermatol. 2011;164:257-262.
- Yaosaka M, Sawamura D, Iitoyo M, et al. Lichen striatus affecting a mother and her son. J Am Acad Dermatol. 2005;53:352-353.
- Jackson R. The lines of Blaschko: a review and reconsideration: observations of the cause of certain unusual linear conditions of the skin. Br J Dermatol. 1976;95:349-360.
- Sorgentini C, Allevato MA, Dahbar M, et al. Lichen striatus in an adult: successful treatment with tacrolimus. Br J Dermatol. 2004;150:776-777.
A 26-year-old man presented with erythematous, scaly, grouped papules along the right side of the chest of 3 weeks' duration, extending to the flank following a blaschkoid distribution on the right side of the chest and not crossing the midline. He reported occasional irritation but otherwise was asymptomatic. His medical history was unremarkable and he was not taking any medications. He also denied trauma to the area.
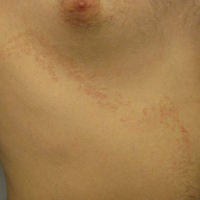
Epidermodysplasia Verruciformis and the Risk for Malignancy
To the Editor:
Epidermodysplasia verruciformis (EV) is a rare autosomal-recessive genodermatosis characterized by widespread infection with specific strains of human papillomavirus (HPV). Patients with EV have a unique susceptibility to acquire HPV due to defects in cellular immunity to the presenting antigens.1 These defects may be related to mutations of the EVER genes or due to acquisition of an immunosuppressive condition.2,3 Infections with HPV-3 and HPV-10 do not lead to the development of malignancies. However, infection with HPV-5, HPV-8, and HPV-14 can lead to the development of nonmelanoma skin cancers, usually squamous cell carcinomas (SCCs), in approximately 60% of patients.3,4 This viral condition lasts throughout the patient’s lifetime and presents as tinea versicolor–like macules and patches. These lesions may be confused with seborrheic keratosis or verruca plana.5 Lesions typically are hypopigmented but occasionally may be hyperpigmented or erythematous. They often are found on the trunk, but lesions on the face, arms, palms, legs, and soles have been reported.5 Mucous membranes are always spared. Epidermodysplasia verruciformis often presents in childhood, except in cases related to acquired immunosuppression. The condition has no sex or racial predilection and no geographical preference.5
A 7-year-old boy (Fitzpatrick skin type V) presented with an asymptomatic rash on the trunk (Figure 1), dorsal aspect of the hands, and forehead. The lesions first appeared 5 years prior on the upper back and upper chest and had recently spread to the forehead and frontal aspect of the scalp. The patient had a history of myelomeningocele, which was corrected at birth with surgical placement of a ventriculoperitoneal shunt. The patient was otherwise healthy and met all appropriate developmental milestones for his age group. Family history revealed consanguinity of the patient’s paternal grandparents who were first cousins. The patient’s mother denied any other family member having similar rashes or lesions.
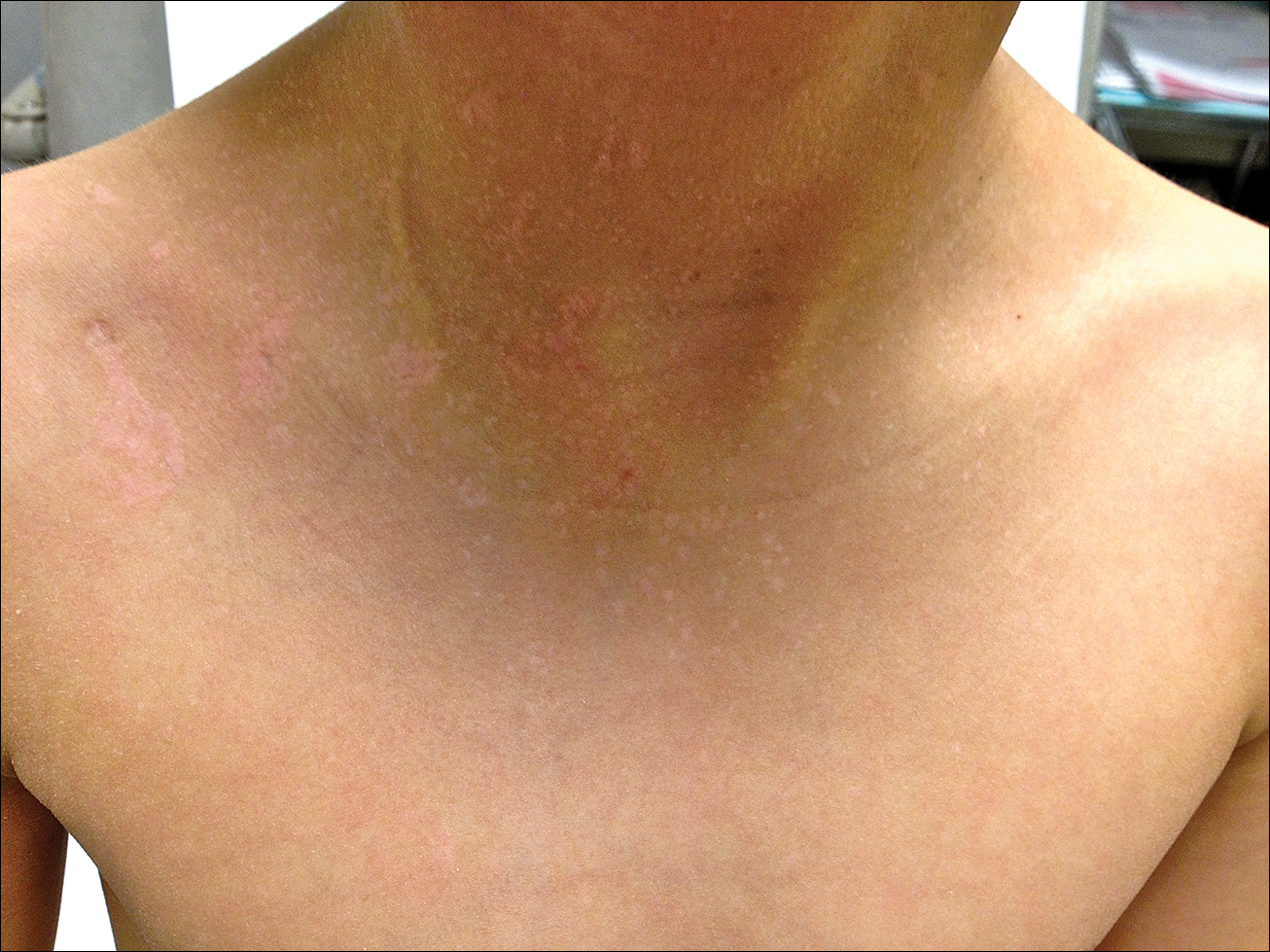
The patient had been treated for pityriasis versicolor on and off for 2 years by another dermatologist. His mother reported faithfully applying ketoconazole cream twice daily for several months with no improvement. She also reported using topical steroids, which did not provide any benefit. The patient and mother denied any associated pruritus, bleeding, burning, or physical discomfort.
Skin examination revealed diffuse, flat, polymorphous, hypopigmented and salmon-colored hyperkeratotic macules and patches with mild scaling on the upper region of the anterior aspect of the chest and upper back (Figure 2A). Additionally, the patient had an extensive number of lesions on the forehead and frontal aspect of the scalp (Figure 2B).

A shave biopsy demonstrated a thick basket weave stratum corneum, koilocytes, and large pale keratinocytes with characteristic blue cytoplasm. These findings were characteristic for EV.
At the patient’s 3-month follow-up visit, he again denied any symptoms associated with the lesions and reported that the appearance was diminishing in severity. On examination there was no evidence of SCC. The mother was advised to discontinue all topical treatments for the patient and return to the office every 3 to 6 months for regular skin surveillance. The mother was further advised to protect the patient from UV radiation with sunscreen and sun-protective clothing.
Epidermodysplasia verruciformis was first reported by Lewandowsky and Lutz6 in 1922. This rare condition often presents in childhood and is characterized by a persistent HPV infection and an autosomal-recessive inheritance pattern. Reports in the literature frequently involve kindreds. Often, patients with EV have a family history of first-degree or second-degree consanguinity.7
The clinical presentation of EV often resembles a pityriasis versicolor–like eruption. However, pityriasis versicolor is less commonly seen in childhood and is more prevalent in patients aged 21 to 30 years, likely due to increased sebum production and changing hormone levels. Furthermore, it is unusual to see pityriasis versicolor affect the face and scalp.8 Lesions of EV vary from hypopigmented and pinkish red macules to confluent patches and hyperkeratotic verrucalike lesions.3 Clinical characteristics also may include dyschromic patches; lesions that resemble flat warts on the trunk, face, and distal arms; and/or lesions that appear similar to seborrheic keratoses on the dorsal aspect of the hands.9,10
Mutations of the EVER gene downregulate a cell’s ability to adequately attack the HPV antigens.11 Although some patients with EV are found to have mutations of the EVER1 and EVER2 genes, a notable portion of patients with EV lack these mutations. Three other causes of EV include acquisition of immunosuppressive conditions including lymphoma, solid organ transplant, and human immunodeficiency virus. If one suspects autosomal-recessive inheritance of EV, genetic testing such as polymerase chain reaction DNA fragment analysis can be performed to determine if there are mutations on the EVER1 or EVER2 genes.12
The inability of patients with EV to mount an immune response to multiple types of HPV increases the risk for developing cutaneous malignancies.7 Additionally, it is known that UV radiation diminishes skin cell immunity, and the combination of EV and UV radiation further increases the risk for developing SCCs.11 The development of nonmelanoma skin cancers usually occurs on sun-exposed skin 20 to 30 years after the onset of lesions, with the highest occurrence of SCCs presenting in the fourth decade of life.1
Protection from UV light exposure is critical to reduce the risk for malignancy. Treatment options for EV lesions have included topical imiquimod 5%, 5-fluorouracil, oral isotretinoin, and intralesional interferon alfa, but patients are often refractory to these interventions. Curettage, surgical excision, electrosurgery, and laser ablation can be effective for individual lesions but carry a greater risk for scarring.1 Photodynamic therapy with aminolevulinic acid and blue light represents a promising option that deserves further study.
Epidermodysplasia verruciformis should be considered as a differential diagnosis in all patients presenting with disseminated lesions resembling pityriasis versicolor that are unresponsive to treatment. A biopsy will help to establish the diagnosis. Patients should minimize sun exposure and report any skin lesions that are changing in appearance.
- Hoffner MV, Camacho FM. Surgical treatment of epidermodysplasia verruciformis. Dermatol Surg. 2010;36:363-367.
- McDermott D, Gammon B, Snijders P. Autosomal dominant epidermodysplasia verruciformis lacking a known EVER1 or EVER2 mutation. Pediatr Dermatol. 2009;26:306-310.
- Patel T, Morrison K, Rady P, et al. Epidermodysplasia verruciformis and susceptibility to HPV. Dis Markers. 2010;29:199-206.
- Hultgren TL, Srinivasan SK, DiMaio DJ. Epidermodysplasia verruciformis occurring in a patient with human immunodeficiency virus: a case report. Cutis. 2007;79:308-311.
- Oliveira W, Netu C, Rady P, et al. Clinical aspects of epidermodysplasia verruciformis. J Eur Acad Dermatol Venereol. 2003;17:394-398.
- Lewandowsky F, Lutz W. Ein Fall einer bisher nicht beschriebenen Hauterkrankung (epidermodysplasia verruciformis). Arch Dermatol Syphilol. 1922;141:193-203.
- Prystowsky S, Herndon J, Freeman R, et al. Epidermodysplasia verruciformis. Am J Dis Child. 1976;130:437-440.
- Kyriakis KP, Terzoudi S, Palamaras I, et al. Pityriasis versicolor prevalence by age and gender. Mycoses. 2006;49:517-518.
- Nuovo G, Ishag M. The histologic spectrum of epidermodysplasia verruciformis. Am J Surg Pathol. 2000;24:1400-1406.
- Jacobelli S, Laude H, Carlotti A, et al. Epidermodysplasia verruciformis in human immunodeficiency virus-infected patients: a marker of human papillomavirus-related disorders not affected by antiretroviral therapy. Arch Dermatol. 2011;147:590-596.
- Rogers HD, MacGregor JL, Nord KM, et al. Acquired epidermodysplasia verruciformis. J Am Acad Dermatol. 2009;60:316-320.
- Gober MD, Rady PL, He Q, et al. Novel homozygous frameshift mutation of EVER1 gene in an epidermodysplasia verruciformis patient. J Invest Dermatol. 2007;127:817-820.
To the Editor:
Epidermodysplasia verruciformis (EV) is a rare autosomal-recessive genodermatosis characterized by widespread infection with specific strains of human papillomavirus (HPV). Patients with EV have a unique susceptibility to acquire HPV due to defects in cellular immunity to the presenting antigens.1 These defects may be related to mutations of the EVER genes or due to acquisition of an immunosuppressive condition.2,3 Infections with HPV-3 and HPV-10 do not lead to the development of malignancies. However, infection with HPV-5, HPV-8, and HPV-14 can lead to the development of nonmelanoma skin cancers, usually squamous cell carcinomas (SCCs), in approximately 60% of patients.3,4 This viral condition lasts throughout the patient’s lifetime and presents as tinea versicolor–like macules and patches. These lesions may be confused with seborrheic keratosis or verruca plana.5 Lesions typically are hypopigmented but occasionally may be hyperpigmented or erythematous. They often are found on the trunk, but lesions on the face, arms, palms, legs, and soles have been reported.5 Mucous membranes are always spared. Epidermodysplasia verruciformis often presents in childhood, except in cases related to acquired immunosuppression. The condition has no sex or racial predilection and no geographical preference.5
A 7-year-old boy (Fitzpatrick skin type V) presented with an asymptomatic rash on the trunk (Figure 1), dorsal aspect of the hands, and forehead. The lesions first appeared 5 years prior on the upper back and upper chest and had recently spread to the forehead and frontal aspect of the scalp. The patient had a history of myelomeningocele, which was corrected at birth with surgical placement of a ventriculoperitoneal shunt. The patient was otherwise healthy and met all appropriate developmental milestones for his age group. Family history revealed consanguinity of the patient’s paternal grandparents who were first cousins. The patient’s mother denied any other family member having similar rashes or lesions.
The patient had been treated for pityriasis versicolor on and off for 2 years by another dermatologist. His mother reported faithfully applying ketoconazole cream twice daily for several months with no improvement. She also reported using topical steroids, which did not provide any benefit. The patient and mother denied any associated pruritus, bleeding, burning, or physical discomfort.
Skin examination revealed diffuse, flat, polymorphous, hypopigmented and salmon-colored hyperkeratotic macules and patches with mild scaling on the upper region of the anterior aspect of the chest and upper back (Figure 2A). Additionally, the patient had an extensive number of lesions on the forehead and frontal aspect of the scalp (Figure 2B).
A shave biopsy demonstrated a thick basket weave stratum corneum, koilocytes, and large pale keratinocytes with characteristic blue cytoplasm. These findings were characteristic for EV.
At the patient’s 3-month follow-up visit, he again denied any symptoms associated with the lesions and reported that the appearance was diminishing in severity. On examination there was no evidence of SCC. The mother was advised to discontinue all topical treatments for the patient and return to the office every 3 to 6 months for regular skin surveillance. The mother was further advised to protect the patient from UV radiation with sunscreen and sun-protective clothing.
Epidermodysplasia verruciformis was first reported by Lewandowsky and Lutz6 in 1922. This rare condition often presents in childhood and is characterized by a persistent HPV infection and an autosomal-recessive inheritance pattern. Reports in the literature frequently involve kindreds. Often, patients with EV have a family history of first-degree or second-degree consanguinity.7
The clinical presentation of EV often resembles a pityriasis versicolor–like eruption. However, pityriasis versicolor is less commonly seen in childhood and is more prevalent in patients aged 21 to 30 years, likely due to increased sebum production and changing hormone levels. Furthermore, it is unusual to see pityriasis versicolor affect the face and scalp.8 Lesions of EV vary from hypopigmented and pinkish red macules to confluent patches and hyperkeratotic verrucalike lesions.3 Clinical characteristics also may include dyschromic patches; lesions that resemble flat warts on the trunk, face, and distal arms; and/or lesions that appear similar to seborrheic keratoses on the dorsal aspect of the hands.9,10
Mutations of the EVER gene downregulate a cell’s ability to adequately attack the HPV antigens.11 Although some patients with EV are found to have mutations of the EVER1 and EVER2 genes, a notable portion of patients with EV lack these mutations. Three other causes of EV include acquisition of immunosuppressive conditions including lymphoma, solid organ transplant, and human immunodeficiency virus. If one suspects autosomal-recessive inheritance of EV, genetic testing such as polymerase chain reaction DNA fragment analysis can be performed to determine if there are mutations on the EVER1 or EVER2 genes.12
The inability of patients with EV to mount an immune response to multiple types of HPV increases the risk for developing cutaneous malignancies.7 Additionally, it is known that UV radiation diminishes skin cell immunity, and the combination of EV and UV radiation further increases the risk for developing SCCs.11 The development of nonmelanoma skin cancers usually occurs on sun-exposed skin 20 to 30 years after the onset of lesions, with the highest occurrence of SCCs presenting in the fourth decade of life.1
Protection from UV light exposure is critical to reduce the risk for malignancy. Treatment options for EV lesions have included topical imiquimod 5%, 5-fluorouracil, oral isotretinoin, and intralesional interferon alfa, but patients are often refractory to these interventions. Curettage, surgical excision, electrosurgery, and laser ablation can be effective for individual lesions but carry a greater risk for scarring.1 Photodynamic therapy with aminolevulinic acid and blue light represents a promising option that deserves further study.
Epidermodysplasia verruciformis should be considered as a differential diagnosis in all patients presenting with disseminated lesions resembling pityriasis versicolor that are unresponsive to treatment. A biopsy will help to establish the diagnosis. Patients should minimize sun exposure and report any skin lesions that are changing in appearance.
To the Editor:
Epidermodysplasia verruciformis (EV) is a rare autosomal-recessive genodermatosis characterized by widespread infection with specific strains of human papillomavirus (HPV). Patients with EV have a unique susceptibility to acquire HPV due to defects in cellular immunity to the presenting antigens.1 These defects may be related to mutations of the EVER genes or due to acquisition of an immunosuppressive condition.2,3 Infections with HPV-3 and HPV-10 do not lead to the development of malignancies. However, infection with HPV-5, HPV-8, and HPV-14 can lead to the development of nonmelanoma skin cancers, usually squamous cell carcinomas (SCCs), in approximately 60% of patients.3,4 This viral condition lasts throughout the patient’s lifetime and presents as tinea versicolor–like macules and patches. These lesions may be confused with seborrheic keratosis or verruca plana.5 Lesions typically are hypopigmented but occasionally may be hyperpigmented or erythematous. They often are found on the trunk, but lesions on the face, arms, palms, legs, and soles have been reported.5 Mucous membranes are always spared. Epidermodysplasia verruciformis often presents in childhood, except in cases related to acquired immunosuppression. The condition has no sex or racial predilection and no geographical preference.5
A 7-year-old boy (Fitzpatrick skin type V) presented with an asymptomatic rash on the trunk (Figure 1), dorsal aspect of the hands, and forehead. The lesions first appeared 5 years prior on the upper back and upper chest and had recently spread to the forehead and frontal aspect of the scalp. The patient had a history of myelomeningocele, which was corrected at birth with surgical placement of a ventriculoperitoneal shunt. The patient was otherwise healthy and met all appropriate developmental milestones for his age group. Family history revealed consanguinity of the patient’s paternal grandparents who were first cousins. The patient’s mother denied any other family member having similar rashes or lesions.
The patient had been treated for pityriasis versicolor on and off for 2 years by another dermatologist. His mother reported faithfully applying ketoconazole cream twice daily for several months with no improvement. She also reported using topical steroids, which did not provide any benefit. The patient and mother denied any associated pruritus, bleeding, burning, or physical discomfort.
Skin examination revealed diffuse, flat, polymorphous, hypopigmented and salmon-colored hyperkeratotic macules and patches with mild scaling on the upper region of the anterior aspect of the chest and upper back (Figure 2A). Additionally, the patient had an extensive number of lesions on the forehead and frontal aspect of the scalp (Figure 2B).
A shave biopsy demonstrated a thick basket weave stratum corneum, koilocytes, and large pale keratinocytes with characteristic blue cytoplasm. These findings were characteristic for EV.
At the patient’s 3-month follow-up visit, he again denied any symptoms associated with the lesions and reported that the appearance was diminishing in severity. On examination there was no evidence of SCC. The mother was advised to discontinue all topical treatments for the patient and return to the office every 3 to 6 months for regular skin surveillance. The mother was further advised to protect the patient from UV radiation with sunscreen and sun-protective clothing.
Epidermodysplasia verruciformis was first reported by Lewandowsky and Lutz6 in 1922. This rare condition often presents in childhood and is characterized by a persistent HPV infection and an autosomal-recessive inheritance pattern. Reports in the literature frequently involve kindreds. Often, patients with EV have a family history of first-degree or second-degree consanguinity.7
The clinical presentation of EV often resembles a pityriasis versicolor–like eruption. However, pityriasis versicolor is less commonly seen in childhood and is more prevalent in patients aged 21 to 30 years, likely due to increased sebum production and changing hormone levels. Furthermore, it is unusual to see pityriasis versicolor affect the face and scalp.8 Lesions of EV vary from hypopigmented and pinkish red macules to confluent patches and hyperkeratotic verrucalike lesions.3 Clinical characteristics also may include dyschromic patches; lesions that resemble flat warts on the trunk, face, and distal arms; and/or lesions that appear similar to seborrheic keratoses on the dorsal aspect of the hands.9,10
Mutations of the EVER gene downregulate a cell’s ability to adequately attack the HPV antigens.11 Although some patients with EV are found to have mutations of the EVER1 and EVER2 genes, a notable portion of patients with EV lack these mutations. Three other causes of EV include acquisition of immunosuppressive conditions including lymphoma, solid organ transplant, and human immunodeficiency virus. If one suspects autosomal-recessive inheritance of EV, genetic testing such as polymerase chain reaction DNA fragment analysis can be performed to determine if there are mutations on the EVER1 or EVER2 genes.12
The inability of patients with EV to mount an immune response to multiple types of HPV increases the risk for developing cutaneous malignancies.7 Additionally, it is known that UV radiation diminishes skin cell immunity, and the combination of EV and UV radiation further increases the risk for developing SCCs.11 The development of nonmelanoma skin cancers usually occurs on sun-exposed skin 20 to 30 years after the onset of lesions, with the highest occurrence of SCCs presenting in the fourth decade of life.1
Protection from UV light exposure is critical to reduce the risk for malignancy. Treatment options for EV lesions have included topical imiquimod 5%, 5-fluorouracil, oral isotretinoin, and intralesional interferon alfa, but patients are often refractory to these interventions. Curettage, surgical excision, electrosurgery, and laser ablation can be effective for individual lesions but carry a greater risk for scarring.1 Photodynamic therapy with aminolevulinic acid and blue light represents a promising option that deserves further study.
Epidermodysplasia verruciformis should be considered as a differential diagnosis in all patients presenting with disseminated lesions resembling pityriasis versicolor that are unresponsive to treatment. A biopsy will help to establish the diagnosis. Patients should minimize sun exposure and report any skin lesions that are changing in appearance.
- Hoffner MV, Camacho FM. Surgical treatment of epidermodysplasia verruciformis. Dermatol Surg. 2010;36:363-367.
- McDermott D, Gammon B, Snijders P. Autosomal dominant epidermodysplasia verruciformis lacking a known EVER1 or EVER2 mutation. Pediatr Dermatol. 2009;26:306-310.
- Patel T, Morrison K, Rady P, et al. Epidermodysplasia verruciformis and susceptibility to HPV. Dis Markers. 2010;29:199-206.
- Hultgren TL, Srinivasan SK, DiMaio DJ. Epidermodysplasia verruciformis occurring in a patient with human immunodeficiency virus: a case report. Cutis. 2007;79:308-311.
- Oliveira W, Netu C, Rady P, et al. Clinical aspects of epidermodysplasia verruciformis. J Eur Acad Dermatol Venereol. 2003;17:394-398.
- Lewandowsky F, Lutz W. Ein Fall einer bisher nicht beschriebenen Hauterkrankung (epidermodysplasia verruciformis). Arch Dermatol Syphilol. 1922;141:193-203.
- Prystowsky S, Herndon J, Freeman R, et al. Epidermodysplasia verruciformis. Am J Dis Child. 1976;130:437-440.
- Kyriakis KP, Terzoudi S, Palamaras I, et al. Pityriasis versicolor prevalence by age and gender. Mycoses. 2006;49:517-518.
- Nuovo G, Ishag M. The histologic spectrum of epidermodysplasia verruciformis. Am J Surg Pathol. 2000;24:1400-1406.
- Jacobelli S, Laude H, Carlotti A, et al. Epidermodysplasia verruciformis in human immunodeficiency virus-infected patients: a marker of human papillomavirus-related disorders not affected by antiretroviral therapy. Arch Dermatol. 2011;147:590-596.
- Rogers HD, MacGregor JL, Nord KM, et al. Acquired epidermodysplasia verruciformis. J Am Acad Dermatol. 2009;60:316-320.
- Gober MD, Rady PL, He Q, et al. Novel homozygous frameshift mutation of EVER1 gene in an epidermodysplasia verruciformis patient. J Invest Dermatol. 2007;127:817-820.
- Hoffner MV, Camacho FM. Surgical treatment of epidermodysplasia verruciformis. Dermatol Surg. 2010;36:363-367.
- McDermott D, Gammon B, Snijders P. Autosomal dominant epidermodysplasia verruciformis lacking a known EVER1 or EVER2 mutation. Pediatr Dermatol. 2009;26:306-310.
- Patel T, Morrison K, Rady P, et al. Epidermodysplasia verruciformis and susceptibility to HPV. Dis Markers. 2010;29:199-206.
- Hultgren TL, Srinivasan SK, DiMaio DJ. Epidermodysplasia verruciformis occurring in a patient with human immunodeficiency virus: a case report. Cutis. 2007;79:308-311.
- Oliveira W, Netu C, Rady P, et al. Clinical aspects of epidermodysplasia verruciformis. J Eur Acad Dermatol Venereol. 2003;17:394-398.
- Lewandowsky F, Lutz W. Ein Fall einer bisher nicht beschriebenen Hauterkrankung (epidermodysplasia verruciformis). Arch Dermatol Syphilol. 1922;141:193-203.
- Prystowsky S, Herndon J, Freeman R, et al. Epidermodysplasia verruciformis. Am J Dis Child. 1976;130:437-440.
- Kyriakis KP, Terzoudi S, Palamaras I, et al. Pityriasis versicolor prevalence by age and gender. Mycoses. 2006;49:517-518.
- Nuovo G, Ishag M. The histologic spectrum of epidermodysplasia verruciformis. Am J Surg Pathol. 2000;24:1400-1406.
- Jacobelli S, Laude H, Carlotti A, et al. Epidermodysplasia verruciformis in human immunodeficiency virus-infected patients: a marker of human papillomavirus-related disorders not affected by antiretroviral therapy. Arch Dermatol. 2011;147:590-596.
- Rogers HD, MacGregor JL, Nord KM, et al. Acquired epidermodysplasia verruciformis. J Am Acad Dermatol. 2009;60:316-320.
- Gober MD, Rady PL, He Q, et al. Novel homozygous frameshift mutation of EVER1 gene in an epidermodysplasia verruciformis patient. J Invest Dermatol. 2007;127:817-820.
Practice Points
- Epidermodysplasia verruciformis (EV) is a rare genodermatosis that usually presents in early childhood and presents as verrucous papules and plaques most commonly on the skin of the head, neck, and upper extremities. It often is misdiagnosed at pityriasis versicolor.
- Mutations of the EVER1 and EVER2 genes have been identified as a source for developing EV.
- Epidermodysplasia verruciformis produces wartlike lesions in individuals who have a unique susceptibility to acquiring the human papillomavirus and early onset of nonmelanoma skin cancers, most commonly squamous cell carcinomas related to viral oncogenesis.
- Avoidance and protection from UV exposure is a critical component of treatment plans for patients with EV.
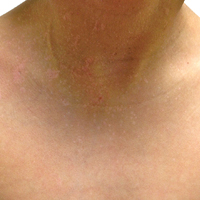
Diagnosis of a Rapidly Growing Preauricular Nodule: Chondroid Syringoma or Pleomorphic Adenoma?
To the Editor:
Chondroid syringoma is a rare benign mixed tumor that originates from the sweat glands, typically presenting with both epithelial and mesenchymal components.1 It differs from pleomorphic adenoma, which arises from salivary glands. The surgical approach for complete excision is different for the 2 tumors; therefore, definitive diagnosis is important. For chondroid syringoma, total excision is recommended,2 while for pleomorphic adenoma, extracapsular dissection or superficial parotidectomy is commonly indicated. We report a case of a preauricular nodule at presentation and highlight the importance of differentiating a chondroid syringoma from a pleomorphic adenoma. This case is unique because of the anatomic location of the nodule, making diagnosis difficult because the tumor was abutting the parotid gland and a biopsy included normal salivary gland cells.
A 19-year-old man with a history of moderate acne on the shoulders, back, and face presented with a rapidly growing, painless nodule on right preauricular region of 6 months’ duration. The nodule was originally thought to be acne related and monitored, as the patient was asymptomatic. On examination the patient was found to have a firm, fixed, nontender, subcutaneous nodule overlying the right temporomandibular joint just anterior to the right tragus (Figure 1). Laboratory results were unremarkable. Computed tomography showed a subcutaneous nonaggressive-appearing soft-tissue mass measuring 16×17×12 mm just anterior and inferior to the external auditory canal cartilaginous segment with no bony abnormalities. The patient was initially treated with incomplete excision of the area for a presumed sebaceous cyst; 2 months later, an abnormal biopsy prompted a complete excisional biopsy.
Histologically, the initial incomplete excision biopsy revealed myxoid and chondroid tissue with glandular elements and adjacent lymph node with strong positivity for S-100 protein and moderate positivity for glial fibrillary acid protein, consistent with chondroid syringoma (Figure 2). Histological findings on second excision biopsy performed 2 months later showed tumor cells surrounded by normal salivary gland cells; therefore, it was difficult to define the origin of this tumor. Subsequent magnetic resonance imaging showed no evidence of the tumor and normal parotid gland borders.
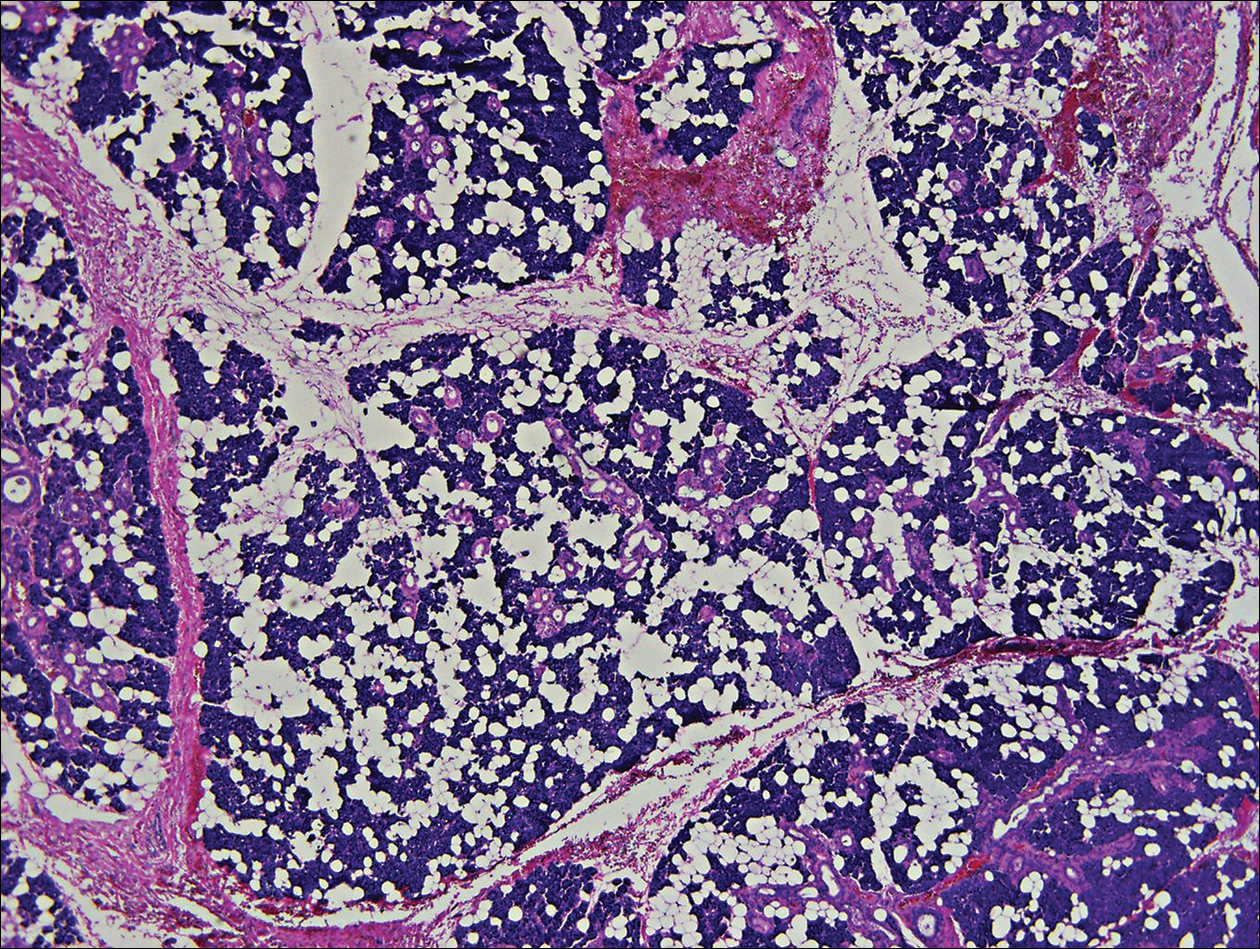
Chondroid syringoma is a rare nonmelanoma skin tumor of the head and neck, mostly benign in nature but with malignant potential. Predominantly, it presents in males as an asymptomatic, slow-growing, nontender nodule.2 Malignant chondroid syringomas are more rare, typically appear on the trunk and legs of females, and present as rapidly growing hard nodules. They can arise de novo or from a preexisting chondroid syringoma and can metastasize.3,4
Clinically and histologically, chondroid syringoma resembles a pleomorphic adenoma. Its diagnosis is dependent on the clinical location to exclude origin in a salivary gland.5 Folliculosebaceous and myoepithelial differentiation within the tumor has been reported.6 Immunocytochemistry is the same in both types and is used to identify 2 prominent components—epithelial and mesenchymal—found in both chondroid syringoma and pleomorphic adenoma. Immunocytochemistry differentiates the epithelial component, which expresses cytokeratin, epithelial membrane antigen, and carcinoembryonic antigen. In contrast, the mesenchymal component expresses S-100, vimentin, and neuron‐specific enolase, and less often glial fibrillary acidic protein, smooth muscle actin, calponin, or p63.5,7,8 Identification of both layers is a distinctive trait of both tumors, rendering it apart from other conditions in the differential diagnosis.5
Typical treatment options include excision, electrodesiccation, dermabrasion, and argon or CO2 laser. Total excision is recommended if there is a benign tumor and complete excision is a cure.2 One case of recurrent benign chondroid syringoma was treated by Mohs micrographic surgery on the eyebrow9; however, Mohs surgery was not recommended in our case due to concerns of spread if malignant as well as an unknown tumor depth, as these tumors have a tendency for deep infiltration.
Due to its anatomical location and presentation as an anterior preauricular mass, it was difficult to differentiate between chondroid syringoma from sweat gland origin and pleomorphic adenoma from the salivary gland. As seen in our case, it is important for physicians to be aware of the differential diagnosis for mixed tumors because it can have a notable effect on the type of surgical therapy and follow-up management.
- Hirsch P, Helwig EB. Chondroid syringoma. Arch Dermatol. 1961;84:835-847.
- Turhan-Haktanir N, Sahin O, Bukulmez A, et al. Chondroid syringoma in a child. Pediatr Dermatol. 2007;24:505-507.
- Mathiasen RA, Rasgon BM, Rumore G. Malignant chondroid syringoma of the face: a first reported case. Otolaryngol Head Neck Surg. 2005;133:305-307.
- Barnett MD, Wallack MK, Zuretti A, et al. Recurrent malignant chondroid syringoma of the foot: a case report and review of the literature. Am J Clin Oncol. 2000;23:227-232.
- Dubb M, Michelow P. Cytologic features of chondroid syringoma in fine needle aspiration biopsies a report of 3 cases. Acta Cytol. 2010;54:183-186.
- Rauso R, Santagata M, Tartaro G, et al. Chondroid syringoma: rare tumor of orofacial region. Minerva Stomatol. 2009;58:383-388.
- Metzler G, Schaumburg-Lever G, Hornstein O, et al. Malignant chondroid syringoma: immunohistopathology. Am J Dermatopathol. 1996;18:83-89.
- Argenyi ZB, Balogh K, Goeken JA. Immunohistochemical characterization of chondroid syringomas. Am J Clin Pathol. 1988;90:662-669.
- Walls AC, Deng A, Geist DE. Mohs micrographic surgery for recurrent chondroid syringoma of the eyebrow. Dermatol Surg. 2012;38:800-802.
To the Editor:
Chondroid syringoma is a rare benign mixed tumor that originates from the sweat glands, typically presenting with both epithelial and mesenchymal components.1 It differs from pleomorphic adenoma, which arises from salivary glands. The surgical approach for complete excision is different for the 2 tumors; therefore, definitive diagnosis is important. For chondroid syringoma, total excision is recommended,2 while for pleomorphic adenoma, extracapsular dissection or superficial parotidectomy is commonly indicated. We report a case of a preauricular nodule at presentation and highlight the importance of differentiating a chondroid syringoma from a pleomorphic adenoma. This case is unique because of the anatomic location of the nodule, making diagnosis difficult because the tumor was abutting the parotid gland and a biopsy included normal salivary gland cells.
A 19-year-old man with a history of moderate acne on the shoulders, back, and face presented with a rapidly growing, painless nodule on right preauricular region of 6 months’ duration. The nodule was originally thought to be acne related and monitored, as the patient was asymptomatic. On examination the patient was found to have a firm, fixed, nontender, subcutaneous nodule overlying the right temporomandibular joint just anterior to the right tragus (Figure 1). Laboratory results were unremarkable. Computed tomography showed a subcutaneous nonaggressive-appearing soft-tissue mass measuring 16×17×12 mm just anterior and inferior to the external auditory canal cartilaginous segment with no bony abnormalities. The patient was initially treated with incomplete excision of the area for a presumed sebaceous cyst; 2 months later, an abnormal biopsy prompted a complete excisional biopsy.
Histologically, the initial incomplete excision biopsy revealed myxoid and chondroid tissue with glandular elements and adjacent lymph node with strong positivity for S-100 protein and moderate positivity for glial fibrillary acid protein, consistent with chondroid syringoma (Figure 2). Histological findings on second excision biopsy performed 2 months later showed tumor cells surrounded by normal salivary gland cells; therefore, it was difficult to define the origin of this tumor. Subsequent magnetic resonance imaging showed no evidence of the tumor and normal parotid gland borders.
Chondroid syringoma is a rare nonmelanoma skin tumor of the head and neck, mostly benign in nature but with malignant potential. Predominantly, it presents in males as an asymptomatic, slow-growing, nontender nodule.2 Malignant chondroid syringomas are more rare, typically appear on the trunk and legs of females, and present as rapidly growing hard nodules. They can arise de novo or from a preexisting chondroid syringoma and can metastasize.3,4
Clinically and histologically, chondroid syringoma resembles a pleomorphic adenoma. Its diagnosis is dependent on the clinical location to exclude origin in a salivary gland.5 Folliculosebaceous and myoepithelial differentiation within the tumor has been reported.6 Immunocytochemistry is the same in both types and is used to identify 2 prominent components—epithelial and mesenchymal—found in both chondroid syringoma and pleomorphic adenoma. Immunocytochemistry differentiates the epithelial component, which expresses cytokeratin, epithelial membrane antigen, and carcinoembryonic antigen. In contrast, the mesenchymal component expresses S-100, vimentin, and neuron‐specific enolase, and less often glial fibrillary acidic protein, smooth muscle actin, calponin, or p63.5,7,8 Identification of both layers is a distinctive trait of both tumors, rendering it apart from other conditions in the differential diagnosis.5
Typical treatment options include excision, electrodesiccation, dermabrasion, and argon or CO2 laser. Total excision is recommended if there is a benign tumor and complete excision is a cure.2 One case of recurrent benign chondroid syringoma was treated by Mohs micrographic surgery on the eyebrow9; however, Mohs surgery was not recommended in our case due to concerns of spread if malignant as well as an unknown tumor depth, as these tumors have a tendency for deep infiltration.
Due to its anatomical location and presentation as an anterior preauricular mass, it was difficult to differentiate between chondroid syringoma from sweat gland origin and pleomorphic adenoma from the salivary gland. As seen in our case, it is important for physicians to be aware of the differential diagnosis for mixed tumors because it can have a notable effect on the type of surgical therapy and follow-up management.
To the Editor:
Chondroid syringoma is a rare benign mixed tumor that originates from the sweat glands, typically presenting with both epithelial and mesenchymal components.1 It differs from pleomorphic adenoma, which arises from salivary glands. The surgical approach for complete excision is different for the 2 tumors; therefore, definitive diagnosis is important. For chondroid syringoma, total excision is recommended,2 while for pleomorphic adenoma, extracapsular dissection or superficial parotidectomy is commonly indicated. We report a case of a preauricular nodule at presentation and highlight the importance of differentiating a chondroid syringoma from a pleomorphic adenoma. This case is unique because of the anatomic location of the nodule, making diagnosis difficult because the tumor was abutting the parotid gland and a biopsy included normal salivary gland cells.
A 19-year-old man with a history of moderate acne on the shoulders, back, and face presented with a rapidly growing, painless nodule on right preauricular region of 6 months’ duration. The nodule was originally thought to be acne related and monitored, as the patient was asymptomatic. On examination the patient was found to have a firm, fixed, nontender, subcutaneous nodule overlying the right temporomandibular joint just anterior to the right tragus (Figure 1). Laboratory results were unremarkable. Computed tomography showed a subcutaneous nonaggressive-appearing soft-tissue mass measuring 16×17×12 mm just anterior and inferior to the external auditory canal cartilaginous segment with no bony abnormalities. The patient was initially treated with incomplete excision of the area for a presumed sebaceous cyst; 2 months later, an abnormal biopsy prompted a complete excisional biopsy.
Histologically, the initial incomplete excision biopsy revealed myxoid and chondroid tissue with glandular elements and adjacent lymph node with strong positivity for S-100 protein and moderate positivity for glial fibrillary acid protein, consistent with chondroid syringoma (Figure 2). Histological findings on second excision biopsy performed 2 months later showed tumor cells surrounded by normal salivary gland cells; therefore, it was difficult to define the origin of this tumor. Subsequent magnetic resonance imaging showed no evidence of the tumor and normal parotid gland borders.
Chondroid syringoma is a rare nonmelanoma skin tumor of the head and neck, mostly benign in nature but with malignant potential. Predominantly, it presents in males as an asymptomatic, slow-growing, nontender nodule.2 Malignant chondroid syringomas are more rare, typically appear on the trunk and legs of females, and present as rapidly growing hard nodules. They can arise de novo or from a preexisting chondroid syringoma and can metastasize.3,4
Clinically and histologically, chondroid syringoma resembles a pleomorphic adenoma. Its diagnosis is dependent on the clinical location to exclude origin in a salivary gland.5 Folliculosebaceous and myoepithelial differentiation within the tumor has been reported.6 Immunocytochemistry is the same in both types and is used to identify 2 prominent components—epithelial and mesenchymal—found in both chondroid syringoma and pleomorphic adenoma. Immunocytochemistry differentiates the epithelial component, which expresses cytokeratin, epithelial membrane antigen, and carcinoembryonic antigen. In contrast, the mesenchymal component expresses S-100, vimentin, and neuron‐specific enolase, and less often glial fibrillary acidic protein, smooth muscle actin, calponin, or p63.5,7,8 Identification of both layers is a distinctive trait of both tumors, rendering it apart from other conditions in the differential diagnosis.5
Typical treatment options include excision, electrodesiccation, dermabrasion, and argon or CO2 laser. Total excision is recommended if there is a benign tumor and complete excision is a cure.2 One case of recurrent benign chondroid syringoma was treated by Mohs micrographic surgery on the eyebrow9; however, Mohs surgery was not recommended in our case due to concerns of spread if malignant as well as an unknown tumor depth, as these tumors have a tendency for deep infiltration.
Due to its anatomical location and presentation as an anterior preauricular mass, it was difficult to differentiate between chondroid syringoma from sweat gland origin and pleomorphic adenoma from the salivary gland. As seen in our case, it is important for physicians to be aware of the differential diagnosis for mixed tumors because it can have a notable effect on the type of surgical therapy and follow-up management.
- Hirsch P, Helwig EB. Chondroid syringoma. Arch Dermatol. 1961;84:835-847.
- Turhan-Haktanir N, Sahin O, Bukulmez A, et al. Chondroid syringoma in a child. Pediatr Dermatol. 2007;24:505-507.
- Mathiasen RA, Rasgon BM, Rumore G. Malignant chondroid syringoma of the face: a first reported case. Otolaryngol Head Neck Surg. 2005;133:305-307.
- Barnett MD, Wallack MK, Zuretti A, et al. Recurrent malignant chondroid syringoma of the foot: a case report and review of the literature. Am J Clin Oncol. 2000;23:227-232.
- Dubb M, Michelow P. Cytologic features of chondroid syringoma in fine needle aspiration biopsies a report of 3 cases. Acta Cytol. 2010;54:183-186.
- Rauso R, Santagata M, Tartaro G, et al. Chondroid syringoma: rare tumor of orofacial region. Minerva Stomatol. 2009;58:383-388.
- Metzler G, Schaumburg-Lever G, Hornstein O, et al. Malignant chondroid syringoma: immunohistopathology. Am J Dermatopathol. 1996;18:83-89.
- Argenyi ZB, Balogh K, Goeken JA. Immunohistochemical characterization of chondroid syringomas. Am J Clin Pathol. 1988;90:662-669.
- Walls AC, Deng A, Geist DE. Mohs micrographic surgery for recurrent chondroid syringoma of the eyebrow. Dermatol Surg. 2012;38:800-802.
- Hirsch P, Helwig EB. Chondroid syringoma. Arch Dermatol. 1961;84:835-847.
- Turhan-Haktanir N, Sahin O, Bukulmez A, et al. Chondroid syringoma in a child. Pediatr Dermatol. 2007;24:505-507.
- Mathiasen RA, Rasgon BM, Rumore G. Malignant chondroid syringoma of the face: a first reported case. Otolaryngol Head Neck Surg. 2005;133:305-307.
- Barnett MD, Wallack MK, Zuretti A, et al. Recurrent malignant chondroid syringoma of the foot: a case report and review of the literature. Am J Clin Oncol. 2000;23:227-232.
- Dubb M, Michelow P. Cytologic features of chondroid syringoma in fine needle aspiration biopsies a report of 3 cases. Acta Cytol. 2010;54:183-186.
- Rauso R, Santagata M, Tartaro G, et al. Chondroid syringoma: rare tumor of orofacial region. Minerva Stomatol. 2009;58:383-388.
- Metzler G, Schaumburg-Lever G, Hornstein O, et al. Malignant chondroid syringoma: immunohistopathology. Am J Dermatopathol. 1996;18:83-89.
- Argenyi ZB, Balogh K, Goeken JA. Immunohistochemical characterization of chondroid syringomas. Am J Clin Pathol. 1988;90:662-669.
- Walls AC, Deng A, Geist DE. Mohs micrographic surgery for recurrent chondroid syringoma of the eyebrow. Dermatol Surg. 2012;38:800-802.
Practice Points
- Clinically and histologically, pleomorphic adenomas and chondroid syringoma both have identical presentations. Differentiation can be determined by knowing where the mixed tumor originated.
- Both lesions warrant different surgical management techniques. Pleomorphic adenoma requires extracapsular dissection or superficial parotidectomy, while complete excision is recommended for chondroid syringoma.