User login
Subungual Onycholemmal Cyst of the Toenail Mimicking Subungual Melanoma
Case Report
A 23-year-old woman presented with a horizontal split along the midline of the right great toenail associated with some tenderness of 2 to 3 months’ duration. Approximately 5 years prior, she noticed a bluish-colored area under the nail that had been steadily increasing in size. She denied a history of trauma, drainage, or bleeding. There was no history of other nail abnormalities. Her medications and personal, family, and social history were noncontributory.
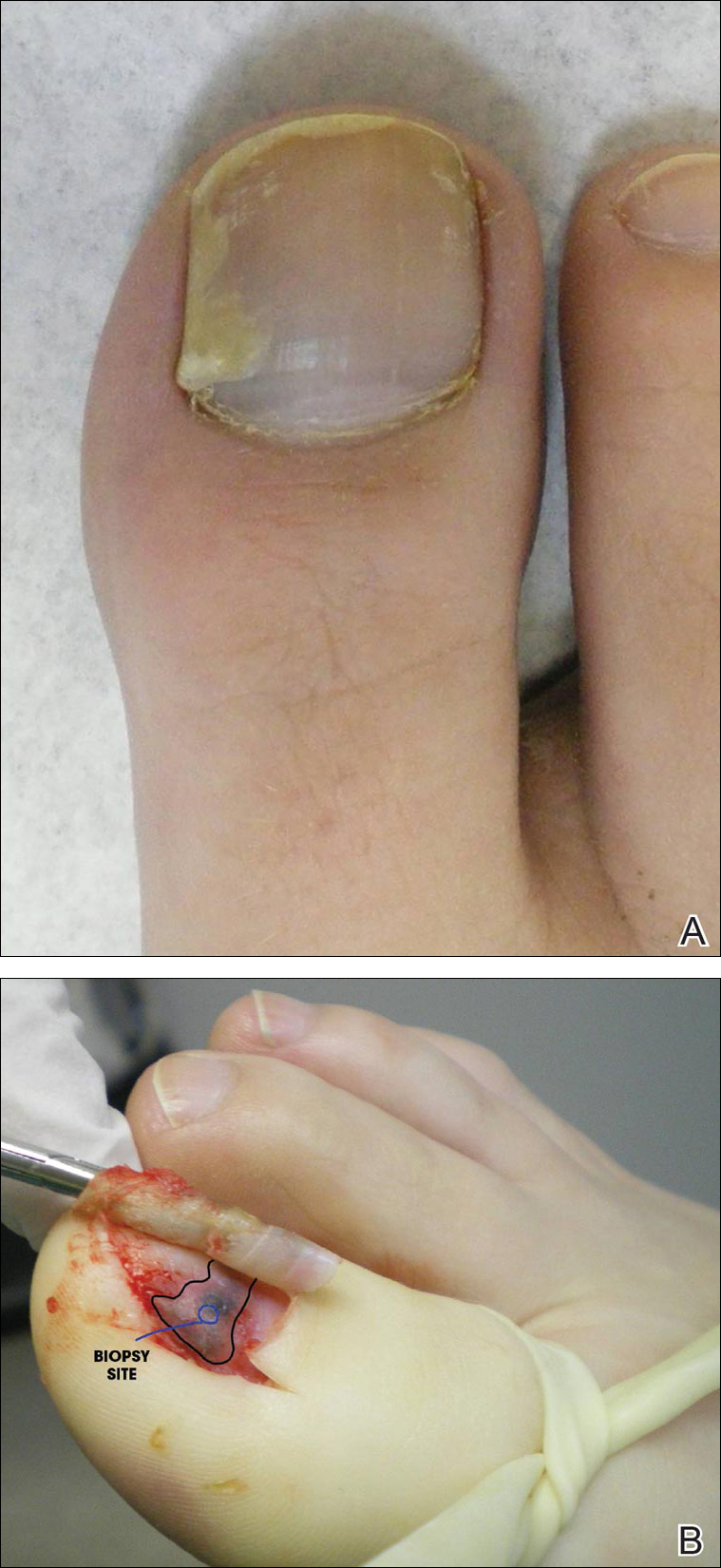
Physical examination of the right great toenail revealed a horizontal split of the nail plate with a bluish hue visible under the nail plate (Figure 1A). The remaining toenails and fingernails were normal. A punch biopsy of the nail bed was performed with a presumptive clinical diagnosis of subungual melanoma versus melanocytic nevus versus cyst (Figure 1B). Nail plate avulsion revealed a blackened nail bed dotted with areas of bluish color and a red friable nodule present focally. Upon further inspection, extension was apparent into the distal matrix.
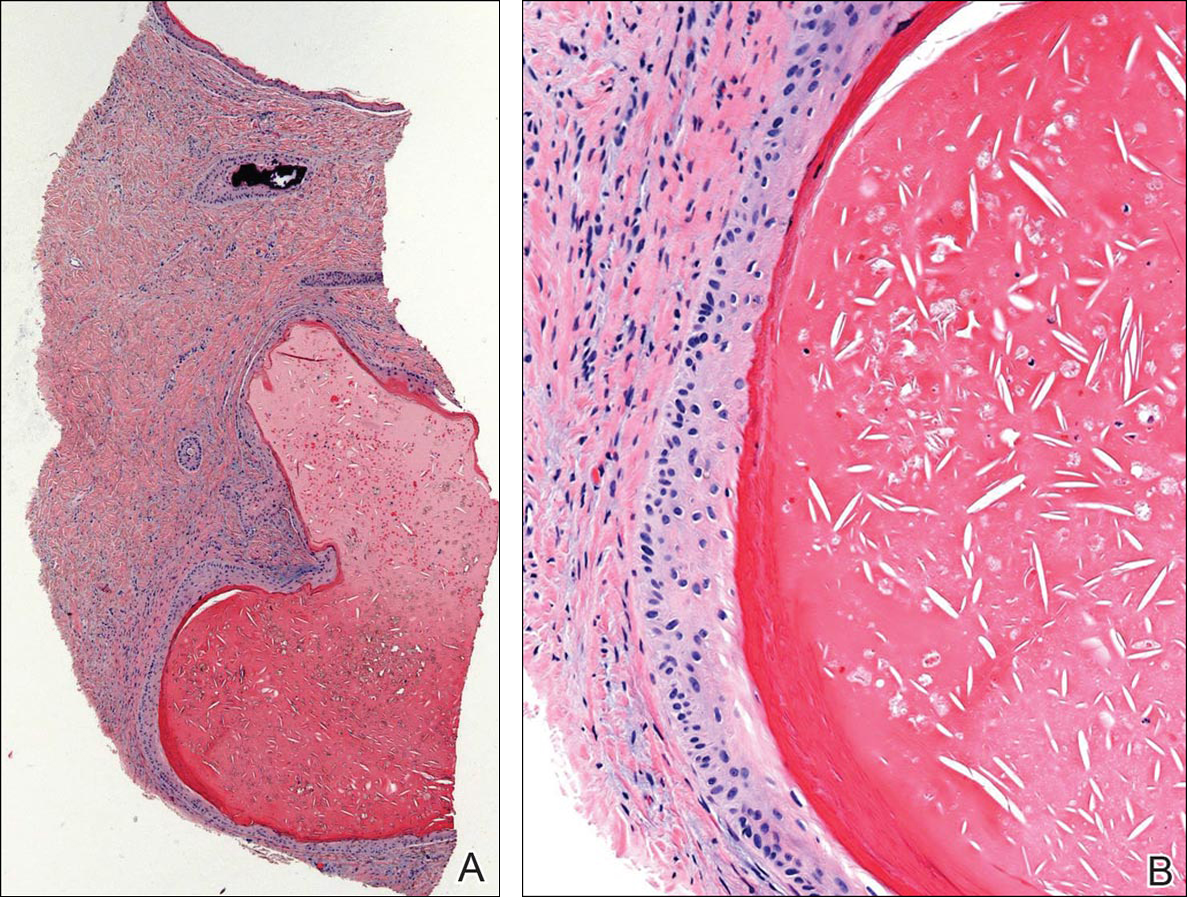
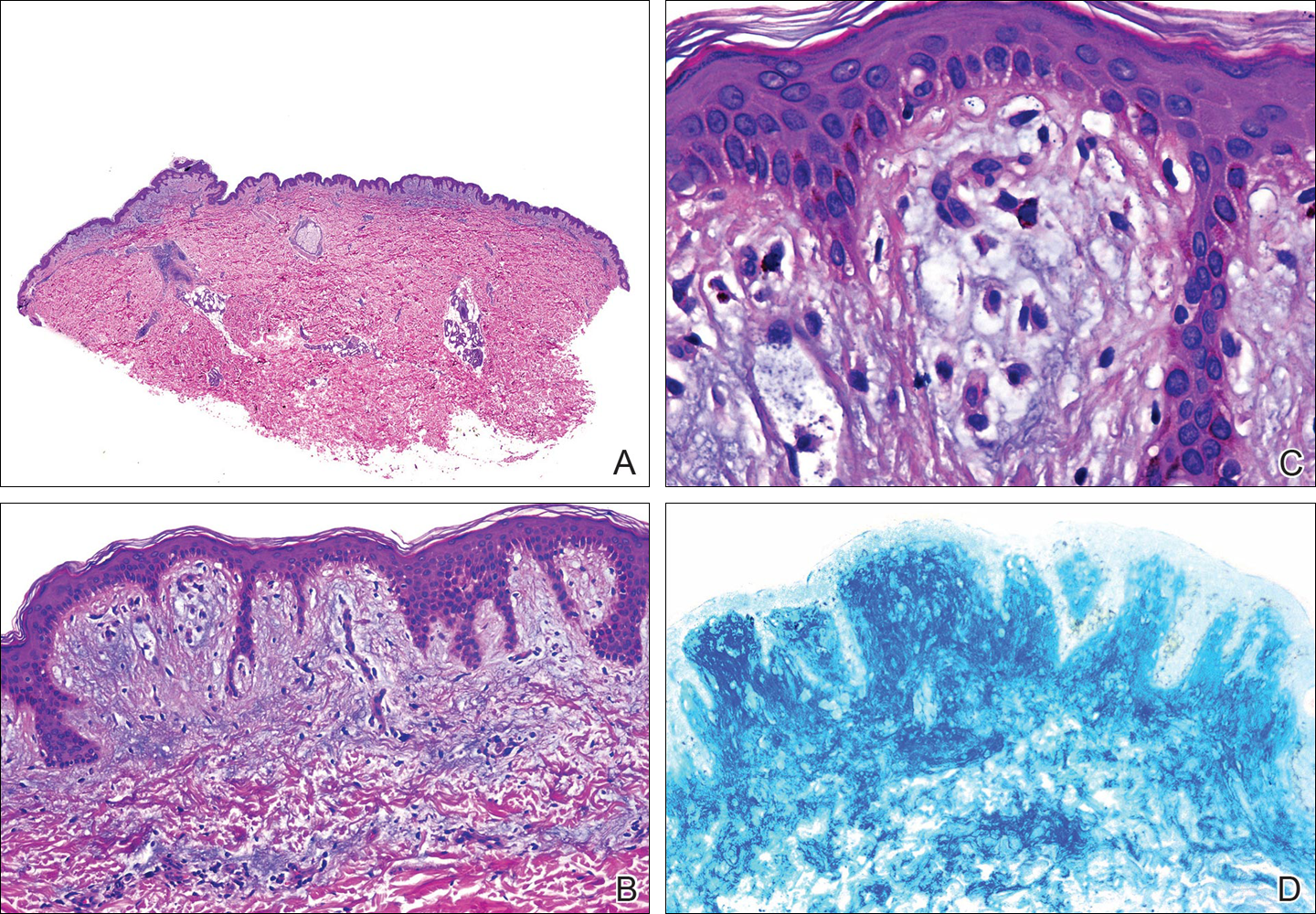
Histopathologic examination revealed a cystic structure with an epithelial lining mostly reminiscent of an isthmus catagen cyst admixed with the presence of both an intermittent focal granular layer and an eosinophilic cuticle surrounding pink laminated keratin, most consistent with a diagnosis of subungual onycholemmal cyst (SOC)(Figure 2). A reexcision was performed with removal of half of the nail bed, including a portion of the distal matrix extending inferiorly to the bone. Variably sized, epithelium-lined, keratin-filled cystic structures emanated from the nail bed epithelium. There were foci of hemorrhage and granulation tissue secondary to cyst rupture (Figure 3). The defect healed by secondary intention. No clinical evidence of recurrence was seen at 6-month follow-up.

Subungual onycholemmal cysts, also known as subungual epidermoid cysts or subungual epidermoid inclusions, are rare and distinctive nail abnormalities occurring in the dermis of the nail bed. We present a case of an SOC in a toenail mimicking subungual malignant melanoma.
Originally described by Samman1 in 1959, SOCs were attributed to trauma to the nail with resultant implantation of the epidermis into the deeper tissue. Lewin2,3 examined 90 postmortem fingernail and nail bed samples and found 8 subungual epidermoid cysts associated with clubbing of the fingernails. He postulated that the early pathogenesis of clubbing involved dermal fibroblast proliferation in the nail bed, leading to sequestration of nail bed epithelium into the dermis with resultant cyst formation. Microscopic subungual cysts also were identified in normal-appearing nails without evidence of trauma, thought to have arisen from the tips of the nail bed rete ridges by a process of bulbous proliferation rather than sequestration. These findings in normal nails suggest that SOCs may represent a more common entity than previously recognized.
It is imperative to recognize the presence of nail unit tumors early because of the risk for permanent nail plate dystrophy and the possibility of a malignant tumor.4,5 Subungual onycholemmal cysts may present with a wide spectrum of clinical findings including marked subungual hyperkeratosis, onychodystrophy, ridging, nail bed pigmentation, clubbing, thickening, or less often a normal-appearing nail. Based on reported cases, several trends are evident. Although nail dystrophy is most often asymptomatic, pain is not uncommon.5,6 It most commonly involves single digits, predominantly thumbs and great toenails.7,8 This predilection suggests that trauma or other local factors may be involved in its pathogenesis. Of note, trauma to the nail may occur years before the development of the lesions or it may not be recalled at all.
Diagnosis requires a degree of clinical suspicion and a nail bed biopsy with partial or total nail plate avulsion to visualize the pathologic portion of the nail bed. Because surgical intervention may lead to the implantation of epithelium, recurrences after nail biopsy or excision may occur.
In contrast to epidermal inclusion cysts arising in the skin, most SOCs do not have a granular layer.9 Hair and nails represent analogous differentiation products of the ectoderm. The nail matrix is homologous to portions of the hair matrix, while the nail bed epithelium is comparable to the outer root sheath of the hair follicle.7 Subungual onycholemmal cysts originate from the nail bed epithelium, which keratinizes in the absence of a granular layer, similar to the follicular isthmus outer root sheath. Thus, SOCs are comparable to the outer root sheath–derived isthmus-catagen cysts because of their abrupt central keratinization.8
Subungual onycholemmal cysts also must be distinguished from slowly growing malignant tumors of the nail bed epithelium, referred to as onycholemmal carcinomas by Alessi et al.10 This entity characteristically presents in elderly patients as a slowly growing, circumscribed, subungual discoloration that may ulcerate, destroying the nail apparatus and penetrating the phalangeal bone. On histopathology, it is characterized by small cysts filled with eosinophilic keratin devoid of a granular layer and lined by atypical squamous epithelium accompanied by solid nests and strands of atypical keratinocytes within the dermis.11 When a cystic component and clear cells predominate, the designation of malignant proliferating onycholemmal cyst has been applied. Its infiltrative growth pattern with destruction of the underlying bone makes it an important entity to exclude when considering the differential diagnosis of tumors of the nail bed.
Subungual melanomas comprise only 1% to 3% of malignant melanomas and 85% are initially misdiagnosed due to their rarity and nonspecific variable presentation. Aside from clinical evidence of Hutchinson sign in the early stages in almost all cases, accurate diagnosis of subungual melanoma and differentiation from SOCs relies on histopathology. A biopsy is necessary to make the diagnosis, but even microscopic findings may be nonspecific during the early stages.
Conclusion
We report a case of a 23-year-old woman with horizontal ridging and tenderness of the right great toenail associated with pigmentation of 5 years’ duration due to an SOC. The etiology of these subungual cysts, with or without nail abnormalities, still remains unclear. Its predilection for the thumbs and great toenails suggests that trauma or other local factors may be involved in its pathogenesis. Because of the rarity of this entity, there are no guidelines for surgical treatment. Subungual onycholemmal cysts may be an underrecognized and more common entity that must be considered when discussing tumors of the nail unit.
- Samman PD. The human toe nail. its genesis and blood supply. Br J Dermatol. 1959;71:296-302.
- Lewin K. The normal fingernail. Br J Dermatol. 1965;77:421-430.
- Lewin K. Subungual epidermoid inclusions. Br J Dermatol. 1969;81:671-675.
- Dominguez-Cherit J, Chanussot-Deprez C, Maria-Sarti H, et al. Nail unit tumors: a study of 234 patients in the dermatology department of the “Dr. Manuel Gea González” General Hospital in Mexico City. Dermatol Surg. 2008;34:1363-1371.
- Sáez-de-Ocariz MM, Domínguez-Cherit J, García-Corona C. Subungual epidermoid cysts. Int J Dermatol. 2001;40:524-526.
- Molly DO, Herbert K. Subungual epidermoid cyst. J Hand Surg Br. 2006;31:345.
- Telang GH, Jellinek N. Multiple calcified subungual epidermoid inclusions. J Am Acad Dermatol. 2007;56:336-339.
- Fanti PA, Tosti A. Subungual epidermoid inclusions: report of 8 cases. Dermatologica. 1989;178:209-212.
- Takiyoshi N, Nakano H, Matsuzaki T, et al. An eclipse in the subungual space: a diagnostic sign for a subungual epidermal cyst? Br J Dermatol. 2009;161:962-963.
- Alessi E, Coggi A, Gianotti R, et al. Onycholemmal carcinoma. Am J Dermatopathol. 2004;26:397-402.
- Inaoki M, Makino E, Adachi M, et al. Onycholemmal carcinoma. J Cutan Pathol. 2006;33:577-580.
Case Report
A 23-year-old woman presented with a horizontal split along the midline of the right great toenail associated with some tenderness of 2 to 3 months’ duration. Approximately 5 years prior, she noticed a bluish-colored area under the nail that had been steadily increasing in size. She denied a history of trauma, drainage, or bleeding. There was no history of other nail abnormalities. Her medications and personal, family, and social history were noncontributory.
Physical examination of the right great toenail revealed a horizontal split of the nail plate with a bluish hue visible under the nail plate (Figure 1A). The remaining toenails and fingernails were normal. A punch biopsy of the nail bed was performed with a presumptive clinical diagnosis of subungual melanoma versus melanocytic nevus versus cyst (Figure 1B). Nail plate avulsion revealed a blackened nail bed dotted with areas of bluish color and a red friable nodule present focally. Upon further inspection, extension was apparent into the distal matrix.
Histopathologic examination revealed a cystic structure with an epithelial lining mostly reminiscent of an isthmus catagen cyst admixed with the presence of both an intermittent focal granular layer and an eosinophilic cuticle surrounding pink laminated keratin, most consistent with a diagnosis of subungual onycholemmal cyst (SOC)(Figure 2). A reexcision was performed with removal of half of the nail bed, including a portion of the distal matrix extending inferiorly to the bone. Variably sized, epithelium-lined, keratin-filled cystic structures emanated from the nail bed epithelium. There were foci of hemorrhage and granulation tissue secondary to cyst rupture (Figure 3). The defect healed by secondary intention. No clinical evidence of recurrence was seen at 6-month follow-up.
Subungual onycholemmal cysts, also known as subungual epidermoid cysts or subungual epidermoid inclusions, are rare and distinctive nail abnormalities occurring in the dermis of the nail bed. We present a case of an SOC in a toenail mimicking subungual malignant melanoma.
Originally described by Samman1 in 1959, SOCs were attributed to trauma to the nail with resultant implantation of the epidermis into the deeper tissue. Lewin2,3 examined 90 postmortem fingernail and nail bed samples and found 8 subungual epidermoid cysts associated with clubbing of the fingernails. He postulated that the early pathogenesis of clubbing involved dermal fibroblast proliferation in the nail bed, leading to sequestration of nail bed epithelium into the dermis with resultant cyst formation. Microscopic subungual cysts also were identified in normal-appearing nails without evidence of trauma, thought to have arisen from the tips of the nail bed rete ridges by a process of bulbous proliferation rather than sequestration. These findings in normal nails suggest that SOCs may represent a more common entity than previously recognized.
It is imperative to recognize the presence of nail unit tumors early because of the risk for permanent nail plate dystrophy and the possibility of a malignant tumor.4,5 Subungual onycholemmal cysts may present with a wide spectrum of clinical findings including marked subungual hyperkeratosis, onychodystrophy, ridging, nail bed pigmentation, clubbing, thickening, or less often a normal-appearing nail. Based on reported cases, several trends are evident. Although nail dystrophy is most often asymptomatic, pain is not uncommon.5,6 It most commonly involves single digits, predominantly thumbs and great toenails.7,8 This predilection suggests that trauma or other local factors may be involved in its pathogenesis. Of note, trauma to the nail may occur years before the development of the lesions or it may not be recalled at all.
Diagnosis requires a degree of clinical suspicion and a nail bed biopsy with partial or total nail plate avulsion to visualize the pathologic portion of the nail bed. Because surgical intervention may lead to the implantation of epithelium, recurrences after nail biopsy or excision may occur.
In contrast to epidermal inclusion cysts arising in the skin, most SOCs do not have a granular layer.9 Hair and nails represent analogous differentiation products of the ectoderm. The nail matrix is homologous to portions of the hair matrix, while the nail bed epithelium is comparable to the outer root sheath of the hair follicle.7 Subungual onycholemmal cysts originate from the nail bed epithelium, which keratinizes in the absence of a granular layer, similar to the follicular isthmus outer root sheath. Thus, SOCs are comparable to the outer root sheath–derived isthmus-catagen cysts because of their abrupt central keratinization.8
Subungual onycholemmal cysts also must be distinguished from slowly growing malignant tumors of the nail bed epithelium, referred to as onycholemmal carcinomas by Alessi et al.10 This entity characteristically presents in elderly patients as a slowly growing, circumscribed, subungual discoloration that may ulcerate, destroying the nail apparatus and penetrating the phalangeal bone. On histopathology, it is characterized by small cysts filled with eosinophilic keratin devoid of a granular layer and lined by atypical squamous epithelium accompanied by solid nests and strands of atypical keratinocytes within the dermis.11 When a cystic component and clear cells predominate, the designation of malignant proliferating onycholemmal cyst has been applied. Its infiltrative growth pattern with destruction of the underlying bone makes it an important entity to exclude when considering the differential diagnosis of tumors of the nail bed.
Subungual melanomas comprise only 1% to 3% of malignant melanomas and 85% are initially misdiagnosed due to their rarity and nonspecific variable presentation. Aside from clinical evidence of Hutchinson sign in the early stages in almost all cases, accurate diagnosis of subungual melanoma and differentiation from SOCs relies on histopathology. A biopsy is necessary to make the diagnosis, but even microscopic findings may be nonspecific during the early stages.
Conclusion
We report a case of a 23-year-old woman with horizontal ridging and tenderness of the right great toenail associated with pigmentation of 5 years’ duration due to an SOC. The etiology of these subungual cysts, with or without nail abnormalities, still remains unclear. Its predilection for the thumbs and great toenails suggests that trauma or other local factors may be involved in its pathogenesis. Because of the rarity of this entity, there are no guidelines for surgical treatment. Subungual onycholemmal cysts may be an underrecognized and more common entity that must be considered when discussing tumors of the nail unit.
Case Report
A 23-year-old woman presented with a horizontal split along the midline of the right great toenail associated with some tenderness of 2 to 3 months’ duration. Approximately 5 years prior, she noticed a bluish-colored area under the nail that had been steadily increasing in size. She denied a history of trauma, drainage, or bleeding. There was no history of other nail abnormalities. Her medications and personal, family, and social history were noncontributory.
Physical examination of the right great toenail revealed a horizontal split of the nail plate with a bluish hue visible under the nail plate (Figure 1A). The remaining toenails and fingernails were normal. A punch biopsy of the nail bed was performed with a presumptive clinical diagnosis of subungual melanoma versus melanocytic nevus versus cyst (Figure 1B). Nail plate avulsion revealed a blackened nail bed dotted with areas of bluish color and a red friable nodule present focally. Upon further inspection, extension was apparent into the distal matrix.
Histopathologic examination revealed a cystic structure with an epithelial lining mostly reminiscent of an isthmus catagen cyst admixed with the presence of both an intermittent focal granular layer and an eosinophilic cuticle surrounding pink laminated keratin, most consistent with a diagnosis of subungual onycholemmal cyst (SOC)(Figure 2). A reexcision was performed with removal of half of the nail bed, including a portion of the distal matrix extending inferiorly to the bone. Variably sized, epithelium-lined, keratin-filled cystic structures emanated from the nail bed epithelium. There were foci of hemorrhage and granulation tissue secondary to cyst rupture (Figure 3). The defect healed by secondary intention. No clinical evidence of recurrence was seen at 6-month follow-up.
Subungual onycholemmal cysts, also known as subungual epidermoid cysts or subungual epidermoid inclusions, are rare and distinctive nail abnormalities occurring in the dermis of the nail bed. We present a case of an SOC in a toenail mimicking subungual malignant melanoma.
Originally described by Samman1 in 1959, SOCs were attributed to trauma to the nail with resultant implantation of the epidermis into the deeper tissue. Lewin2,3 examined 90 postmortem fingernail and nail bed samples and found 8 subungual epidermoid cysts associated with clubbing of the fingernails. He postulated that the early pathogenesis of clubbing involved dermal fibroblast proliferation in the nail bed, leading to sequestration of nail bed epithelium into the dermis with resultant cyst formation. Microscopic subungual cysts also were identified in normal-appearing nails without evidence of trauma, thought to have arisen from the tips of the nail bed rete ridges by a process of bulbous proliferation rather than sequestration. These findings in normal nails suggest that SOCs may represent a more common entity than previously recognized.
It is imperative to recognize the presence of nail unit tumors early because of the risk for permanent nail plate dystrophy and the possibility of a malignant tumor.4,5 Subungual onycholemmal cysts may present with a wide spectrum of clinical findings including marked subungual hyperkeratosis, onychodystrophy, ridging, nail bed pigmentation, clubbing, thickening, or less often a normal-appearing nail. Based on reported cases, several trends are evident. Although nail dystrophy is most often asymptomatic, pain is not uncommon.5,6 It most commonly involves single digits, predominantly thumbs and great toenails.7,8 This predilection suggests that trauma or other local factors may be involved in its pathogenesis. Of note, trauma to the nail may occur years before the development of the lesions or it may not be recalled at all.
Diagnosis requires a degree of clinical suspicion and a nail bed biopsy with partial or total nail plate avulsion to visualize the pathologic portion of the nail bed. Because surgical intervention may lead to the implantation of epithelium, recurrences after nail biopsy or excision may occur.
In contrast to epidermal inclusion cysts arising in the skin, most SOCs do not have a granular layer.9 Hair and nails represent analogous differentiation products of the ectoderm. The nail matrix is homologous to portions of the hair matrix, while the nail bed epithelium is comparable to the outer root sheath of the hair follicle.7 Subungual onycholemmal cysts originate from the nail bed epithelium, which keratinizes in the absence of a granular layer, similar to the follicular isthmus outer root sheath. Thus, SOCs are comparable to the outer root sheath–derived isthmus-catagen cysts because of their abrupt central keratinization.8
Subungual onycholemmal cysts also must be distinguished from slowly growing malignant tumors of the nail bed epithelium, referred to as onycholemmal carcinomas by Alessi et al.10 This entity characteristically presents in elderly patients as a slowly growing, circumscribed, subungual discoloration that may ulcerate, destroying the nail apparatus and penetrating the phalangeal bone. On histopathology, it is characterized by small cysts filled with eosinophilic keratin devoid of a granular layer and lined by atypical squamous epithelium accompanied by solid nests and strands of atypical keratinocytes within the dermis.11 When a cystic component and clear cells predominate, the designation of malignant proliferating onycholemmal cyst has been applied. Its infiltrative growth pattern with destruction of the underlying bone makes it an important entity to exclude when considering the differential diagnosis of tumors of the nail bed.
Subungual melanomas comprise only 1% to 3% of malignant melanomas and 85% are initially misdiagnosed due to their rarity and nonspecific variable presentation. Aside from clinical evidence of Hutchinson sign in the early stages in almost all cases, accurate diagnosis of subungual melanoma and differentiation from SOCs relies on histopathology. A biopsy is necessary to make the diagnosis, but even microscopic findings may be nonspecific during the early stages.
Conclusion
We report a case of a 23-year-old woman with horizontal ridging and tenderness of the right great toenail associated with pigmentation of 5 years’ duration due to an SOC. The etiology of these subungual cysts, with or without nail abnormalities, still remains unclear. Its predilection for the thumbs and great toenails suggests that trauma or other local factors may be involved in its pathogenesis. Because of the rarity of this entity, there are no guidelines for surgical treatment. Subungual onycholemmal cysts may be an underrecognized and more common entity that must be considered when discussing tumors of the nail unit.
- Samman PD. The human toe nail. its genesis and blood supply. Br J Dermatol. 1959;71:296-302.
- Lewin K. The normal fingernail. Br J Dermatol. 1965;77:421-430.
- Lewin K. Subungual epidermoid inclusions. Br J Dermatol. 1969;81:671-675.
- Dominguez-Cherit J, Chanussot-Deprez C, Maria-Sarti H, et al. Nail unit tumors: a study of 234 patients in the dermatology department of the “Dr. Manuel Gea González” General Hospital in Mexico City. Dermatol Surg. 2008;34:1363-1371.
- Sáez-de-Ocariz MM, Domínguez-Cherit J, García-Corona C. Subungual epidermoid cysts. Int J Dermatol. 2001;40:524-526.
- Molly DO, Herbert K. Subungual epidermoid cyst. J Hand Surg Br. 2006;31:345.
- Telang GH, Jellinek N. Multiple calcified subungual epidermoid inclusions. J Am Acad Dermatol. 2007;56:336-339.
- Fanti PA, Tosti A. Subungual epidermoid inclusions: report of 8 cases. Dermatologica. 1989;178:209-212.
- Takiyoshi N, Nakano H, Matsuzaki T, et al. An eclipse in the subungual space: a diagnostic sign for a subungual epidermal cyst? Br J Dermatol. 2009;161:962-963.
- Alessi E, Coggi A, Gianotti R, et al. Onycholemmal carcinoma. Am J Dermatopathol. 2004;26:397-402.
- Inaoki M, Makino E, Adachi M, et al. Onycholemmal carcinoma. J Cutan Pathol. 2006;33:577-580.
- Samman PD. The human toe nail. its genesis and blood supply. Br J Dermatol. 1959;71:296-302.
- Lewin K. The normal fingernail. Br J Dermatol. 1965;77:421-430.
- Lewin K. Subungual epidermoid inclusions. Br J Dermatol. 1969;81:671-675.
- Dominguez-Cherit J, Chanussot-Deprez C, Maria-Sarti H, et al. Nail unit tumors: a study of 234 patients in the dermatology department of the “Dr. Manuel Gea González” General Hospital in Mexico City. Dermatol Surg. 2008;34:1363-1371.
- Sáez-de-Ocariz MM, Domínguez-Cherit J, García-Corona C. Subungual epidermoid cysts. Int J Dermatol. 2001;40:524-526.
- Molly DO, Herbert K. Subungual epidermoid cyst. J Hand Surg Br. 2006;31:345.
- Telang GH, Jellinek N. Multiple calcified subungual epidermoid inclusions. J Am Acad Dermatol. 2007;56:336-339.
- Fanti PA, Tosti A. Subungual epidermoid inclusions: report of 8 cases. Dermatologica. 1989;178:209-212.
- Takiyoshi N, Nakano H, Matsuzaki T, et al. An eclipse in the subungual space: a diagnostic sign for a subungual epidermal cyst? Br J Dermatol. 2009;161:962-963.
- Alessi E, Coggi A, Gianotti R, et al. Onycholemmal carcinoma. Am J Dermatopathol. 2004;26:397-402.
- Inaoki M, Makino E, Adachi M, et al. Onycholemmal carcinoma. J Cutan Pathol. 2006;33:577-580.
Practice Points
- Trauma to the nail may occur years before the development of subungual onycholemmal cysts or it may not be recalled at all.
- Diagnosis requires a degree of clinical suspicion and a nail bed biopsy.
- Subungual onycholemmal cysts must be distinguished from slowly growing malignant tumors of the nail bed epithelium.
Circumscribed Nodule in a Renal Transplant Patient
The Diagnosis: Subcutaneous Phaeohyphomycosis
Subcutaneous phaeohyphomycosis (SP), also called mycotic cyst, is characterized by a painless, nodular lesion that develops in response to traumatic implantation of dematiaceous, pigment-forming fungi.1 Similar to other fungal infections, SP can arise opportunistically in immunocompromised patients.2,3 More than 60 genera (and more than 100 species) are known etiologic agents of phaeohyphomycosis; the 2 main causes of infection are Bipolaris spicifera and Exophiala jeanselmei.4,5 Given this variety, phaeohyphomycosis can present superficially as black piedra or tinea nigra, cutaneously as scytalidiosis, subcutaneously as SP, or disseminated as sinusitis or systemic phaeohyphomycosis.
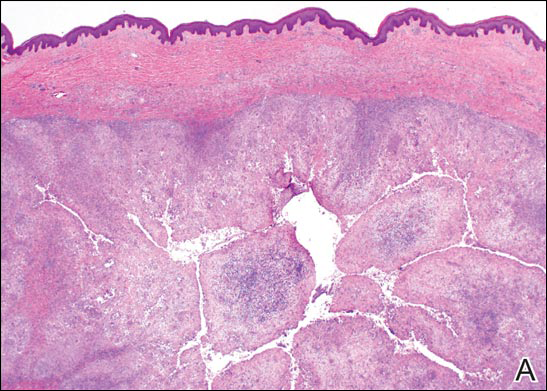
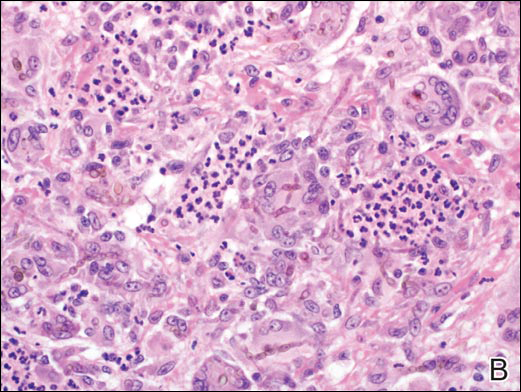
Coined in 1974 by Ajello et al,6 the term phaeohyphomycosis translates to “condition of dark hyphal fungus,” a term used to designate mycoses caused by fungi with melanized hyphae. Histologically, SP demonstrates a circumscribed chronic cyst or abscess with a dense fibrous wall (quiz image A). At high power, the wall is composed of chronic granulomatous inflammation with foamy macrophages, and the cystic cavity contains necrotic debris admixed with neutrophils. Pigmented filamentous hyphae and yeastlike entities can be seen in the cyst wall, in multinucleated giant cells, in the necrotic debris, or directly attached to the implanted foreign material (quiz image B).7 The first-line treatment of SP is wide local excision and oral itraconazole. It often requires adjustments to dosage or change to antifungal due to recurrence and etiologic variation.8 Furthermore, if SP is not definitively treated, immunocompromised patients are at an increased risk for developing potentially fatal systemic phaeohyphomycosis.3
Chromoblastomycosis (CBM), also caused by dematiaceous fungi, is characterized by an initially indolent clinical presentation. Typically found on the legs and lower thighs of agricultural workers, the lesion begins as a slow-growing, nodular papule with subsequent transformation into an edematous verrucous plaque with peripheral erythema.9 Lesions can be annular with central clearing, and lymphedema with elephantiasis may be present.10 Histologically, CBM shows pseudoepitheliomatous hyperplasia and intraepidermal pustules as the host rids the infection via transepithelial elimination. Dematiaceous fungi often are seen in the dermis, either freestanding or attached to foreign plant material. Medlar bodies, also called copper penny spores or sclerotic bodies, are the most defining histologic finding and are characterized by groups of brown, thick-walled cells found in giant cells or neutrophil abscesses (Figure 1). Hyphae are not typically found in this type of infection.11

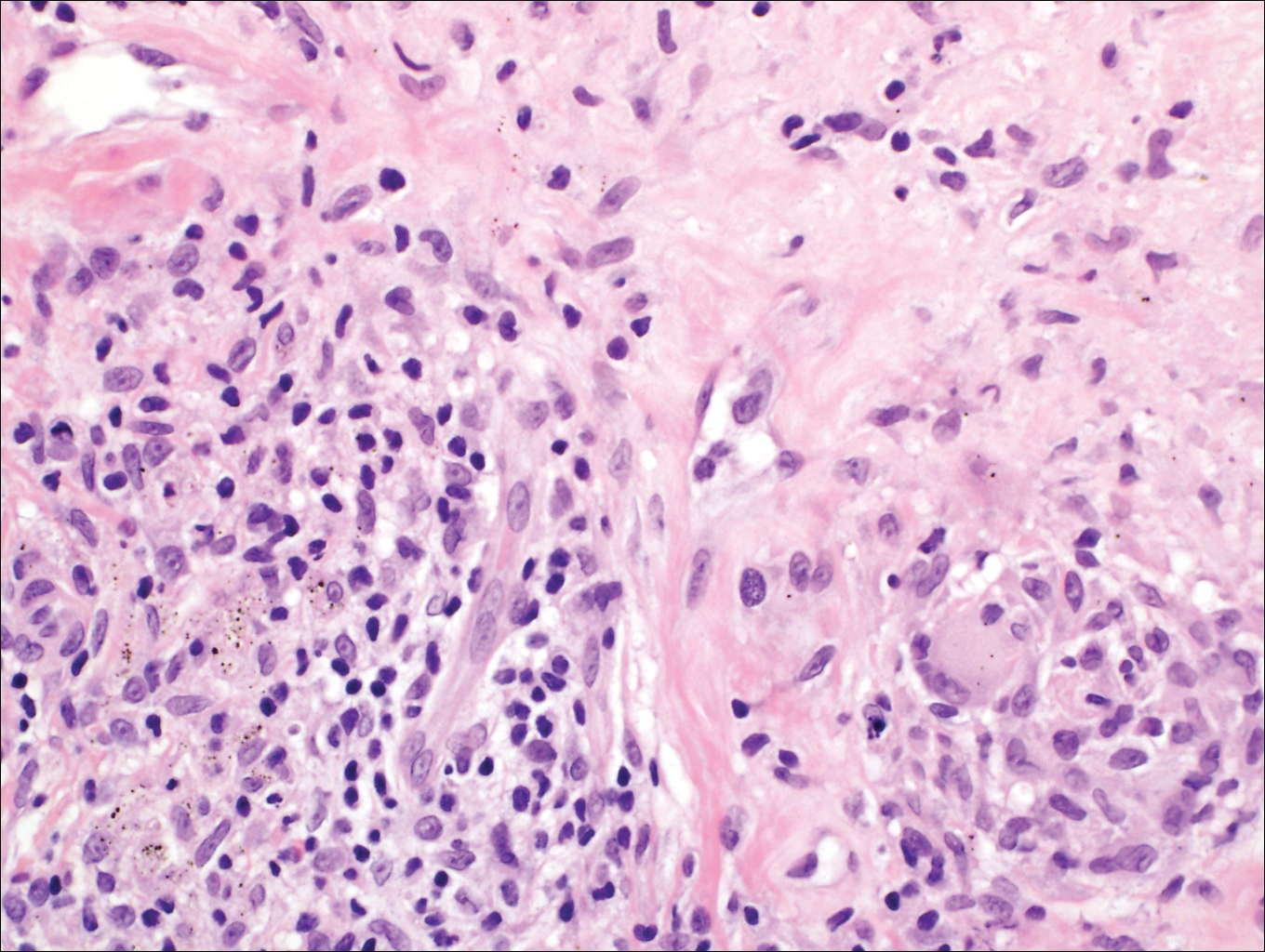
Granulomatous foreign body reactions occur in response to the inoculation of nonhuman material and are characterized by dermal or subcutaneous nodules. Tissue macrophages phagocytize material not removed shortly after implantation, which initiates an inflammatory response that attempts to isolate the material from the uninvolved surrounding tissue. Vegetative foreign bodies will cause the most severe inflammatory reactions.12 Histologically, foreign body granulomas are noncaseating with epithelioid histiocytes surrounding a central foreign body (Figure 2). Occasionally, foreign bodies may be difficult to detect; some are birefringent to polarized light.13 Additionally, inoculation injuries can predispose patients to SP, CBM, and other fungal infections.

Tattoos are characterized by exogenous pigment deposition into the dermis.14 Histologically, tattoos display exogenous pigment deposited throughout the reticular dermis, attached to collagen bundles, within macrophages, or adjacent to adnexal structures (eg, pilosebaceous units or eccrine glands). Although all tattoo pigments can cause adverse reactions, hypersensitivity reactions occur most commonly in response to red pigment, resulting in discrete areas of spongiosis and granulomatous or lichenoid inflammation. Occasionally, hypersensitivity reactions can induce necrobiotic granulomatous reactions characterized by collagen alteration surrounded by palisaded histiocytes and lymphocytes (Figure 3).15,16 There also may be focally dense areas of superficial and deep perivascular lymphohistiocytic infiltrate. Clinical context is important, as brown tattoo pigment (Figure 3) can be easily confused with the pigmented hyphae of phaeohyphomycosis, melanin, or hemosiderin.
Subcutaneous hyalohyphomycosis is a nondemat-iaceous (nonpigmented) infection that is caused by hyaline septate hyphal cells.17 Hyalohyphomycosis skin lesions can present as painful erythematous nodules that evolve into excoriated pustules.18 Hyalohyphomycosis most often arises in immunocompromised patients. Causative organisms are ubiquitous soil saprophytes and plant pathogens, most often Aspergillus and Fusarium species, with a predilection for affecting severely immunocompromised hosts, particularly children.19 These species tend to be vasculotropic, which can result in tissue necrosis and systemic dissemination. Histologically, fungi are dispersed within tissue. They have a bright, bubbly, mildly basophilic cytoplasm and are nonpigmented, branching, and septate (Figure 4).11

- Isa-Isa R, García C, Isa M, et al. Subcutaneous phaeohyphomycosis (mycotic cyst). Clin Dermatol. 2012;30:425-431.
- Rubin RH. Infectious disease complications of renal transplantation. Kidney Int. 1993;44:221-236.
- Ogawa MM, Galante NZ, Godoy P, et al. Treatment of subcutaneous phaeohyphomycosis and prospective follow-up of 17 kidney transplant recipients. J Am Acad Dermatol. 2009;61:977-985.
- Matsumoto T, Ajello L, Matsuda T, et al. Developments in hyalohyphomycosis and phaeohyphomycosis. J Med Vet Mycol. 1994;32(suppl 1):329-349.
- Rinaldi MG. Phaeohyphomycosis. Dermatol Clin. 1996;14:147-153.
- Ajello L, Georg LK, Steigbigel RT, et al. A case of phaeohyphomycosis caused by a new species of Phialophora. Mycologia. 1974;66:490-498.
- Patterson J. Weedon’s Skin Pathology. 4th ed. London, England: Churchill Livingstone Elsevier; 2014.
- Patel U, Chu J, Patel R, et al. Subcutaneous dematiaceous fungal infection. Dermatol Online J. 2011;17:19.
- Bonifaz A, Carrasco-Gerard E, Saúl A. Chromoblastomycosis: clinical and mycologic experience of 51 cases. Mycoses. 2001;44:1-7.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Elston D, Ferringer T, Peckham S, et al, eds. Dermatopathology. 2nd ed. St. Louis, MO: Elsevier Saunders; 2014.
- Lammers RL. Soft tissue foreign bodies. In: Tintinalli J, Stapczynski S, Ma O, et al, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw Hill Professional; 2011.
- Murphy GF, Saavedra AP, Mihm MC. Nodular/interstitial dermatitis. In: Murphy GF, Saavedra AP, Mihm MC, eds. Atlas of Nontumor Pathology: Inflammatory Disorders of the Skin. Vol 10. Washington, DC: American Registry of Pathology; 2012:337-395.
- Laumann A. Body art. In: Goldsmith L, Katz S, Gilchrest B, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012. http://access medicine.mhmedical.com.proxy.lib.uiowa.edu/content.aspx?bookid=392&Sectionid=41138811. Accessed July 17,2016.
- Wood A, Hamilton SA, Wallace WA, et al. Necrobiotic granulomatous tattoo reaction: report of an unusual case showing features of both necrobiosis lipoidica and granuloma annulare patterns. Am J Dermatopathol. 2014;36:e152-e155.
- Mortimer N, Chave T, Johnston G. Red tattoo reactions. Clin Exp Dermatol. 2003;28:508-510.
- Ajello L. Hyalohyphomycosis and phaeohyphomycosis: two global disease entities of public health importance. Eur J Epidemiol. 1986;2:243-251.
- Safdar A. Progressive cutaneous hyalohyphomycosis due to Paecilomyces lilacinus: rapid response to treatment with caspofungin and itraconazole. Clin Infect Dis. 2002;34:1415-1417.
- Marcoux D, Jafarian F, Joncas V, et al. Deep cutaneous fungal infections in immunocompromised children. J Am Acad Dermatol. 2009;61:857-864.
The Diagnosis: Subcutaneous Phaeohyphomycosis
Subcutaneous phaeohyphomycosis (SP), also called mycotic cyst, is characterized by a painless, nodular lesion that develops in response to traumatic implantation of dematiaceous, pigment-forming fungi.1 Similar to other fungal infections, SP can arise opportunistically in immunocompromised patients.2,3 More than 60 genera (and more than 100 species) are known etiologic agents of phaeohyphomycosis; the 2 main causes of infection are Bipolaris spicifera and Exophiala jeanselmei.4,5 Given this variety, phaeohyphomycosis can present superficially as black piedra or tinea nigra, cutaneously as scytalidiosis, subcutaneously as SP, or disseminated as sinusitis or systemic phaeohyphomycosis.
Coined in 1974 by Ajello et al,6 the term phaeohyphomycosis translates to “condition of dark hyphal fungus,” a term used to designate mycoses caused by fungi with melanized hyphae. Histologically, SP demonstrates a circumscribed chronic cyst or abscess with a dense fibrous wall (quiz image A). At high power, the wall is composed of chronic granulomatous inflammation with foamy macrophages, and the cystic cavity contains necrotic debris admixed with neutrophils. Pigmented filamentous hyphae and yeastlike entities can be seen in the cyst wall, in multinucleated giant cells, in the necrotic debris, or directly attached to the implanted foreign material (quiz image B).7 The first-line treatment of SP is wide local excision and oral itraconazole. It often requires adjustments to dosage or change to antifungal due to recurrence and etiologic variation.8 Furthermore, if SP is not definitively treated, immunocompromised patients are at an increased risk for developing potentially fatal systemic phaeohyphomycosis.3
Chromoblastomycosis (CBM), also caused by dematiaceous fungi, is characterized by an initially indolent clinical presentation. Typically found on the legs and lower thighs of agricultural workers, the lesion begins as a slow-growing, nodular papule with subsequent transformation into an edematous verrucous plaque with peripheral erythema.9 Lesions can be annular with central clearing, and lymphedema with elephantiasis may be present.10 Histologically, CBM shows pseudoepitheliomatous hyperplasia and intraepidermal pustules as the host rids the infection via transepithelial elimination. Dematiaceous fungi often are seen in the dermis, either freestanding or attached to foreign plant material. Medlar bodies, also called copper penny spores or sclerotic bodies, are the most defining histologic finding and are characterized by groups of brown, thick-walled cells found in giant cells or neutrophil abscesses (Figure 1). Hyphae are not typically found in this type of infection.11
Granulomatous foreign body reactions occur in response to the inoculation of nonhuman material and are characterized by dermal or subcutaneous nodules. Tissue macrophages phagocytize material not removed shortly after implantation, which initiates an inflammatory response that attempts to isolate the material from the uninvolved surrounding tissue. Vegetative foreign bodies will cause the most severe inflammatory reactions.12 Histologically, foreign body granulomas are noncaseating with epithelioid histiocytes surrounding a central foreign body (Figure 2). Occasionally, foreign bodies may be difficult to detect; some are birefringent to polarized light.13 Additionally, inoculation injuries can predispose patients to SP, CBM, and other fungal infections.
Tattoos are characterized by exogenous pigment deposition into the dermis.14 Histologically, tattoos display exogenous pigment deposited throughout the reticular dermis, attached to collagen bundles, within macrophages, or adjacent to adnexal structures (eg, pilosebaceous units or eccrine glands). Although all tattoo pigments can cause adverse reactions, hypersensitivity reactions occur most commonly in response to red pigment, resulting in discrete areas of spongiosis and granulomatous or lichenoid inflammation. Occasionally, hypersensitivity reactions can induce necrobiotic granulomatous reactions characterized by collagen alteration surrounded by palisaded histiocytes and lymphocytes (Figure 3).15,16 There also may be focally dense areas of superficial and deep perivascular lymphohistiocytic infiltrate. Clinical context is important, as brown tattoo pigment (Figure 3) can be easily confused with the pigmented hyphae of phaeohyphomycosis, melanin, or hemosiderin.
Subcutaneous hyalohyphomycosis is a nondemat-iaceous (nonpigmented) infection that is caused by hyaline septate hyphal cells.17 Hyalohyphomycosis skin lesions can present as painful erythematous nodules that evolve into excoriated pustules.18 Hyalohyphomycosis most often arises in immunocompromised patients. Causative organisms are ubiquitous soil saprophytes and plant pathogens, most often Aspergillus and Fusarium species, with a predilection for affecting severely immunocompromised hosts, particularly children.19 These species tend to be vasculotropic, which can result in tissue necrosis and systemic dissemination. Histologically, fungi are dispersed within tissue. They have a bright, bubbly, mildly basophilic cytoplasm and are nonpigmented, branching, and septate (Figure 4).11
The Diagnosis: Subcutaneous Phaeohyphomycosis
Subcutaneous phaeohyphomycosis (SP), also called mycotic cyst, is characterized by a painless, nodular lesion that develops in response to traumatic implantation of dematiaceous, pigment-forming fungi.1 Similar to other fungal infections, SP can arise opportunistically in immunocompromised patients.2,3 More than 60 genera (and more than 100 species) are known etiologic agents of phaeohyphomycosis; the 2 main causes of infection are Bipolaris spicifera and Exophiala jeanselmei.4,5 Given this variety, phaeohyphomycosis can present superficially as black piedra or tinea nigra, cutaneously as scytalidiosis, subcutaneously as SP, or disseminated as sinusitis or systemic phaeohyphomycosis.
Coined in 1974 by Ajello et al,6 the term phaeohyphomycosis translates to “condition of dark hyphal fungus,” a term used to designate mycoses caused by fungi with melanized hyphae. Histologically, SP demonstrates a circumscribed chronic cyst or abscess with a dense fibrous wall (quiz image A). At high power, the wall is composed of chronic granulomatous inflammation with foamy macrophages, and the cystic cavity contains necrotic debris admixed with neutrophils. Pigmented filamentous hyphae and yeastlike entities can be seen in the cyst wall, in multinucleated giant cells, in the necrotic debris, or directly attached to the implanted foreign material (quiz image B).7 The first-line treatment of SP is wide local excision and oral itraconazole. It often requires adjustments to dosage or change to antifungal due to recurrence and etiologic variation.8 Furthermore, if SP is not definitively treated, immunocompromised patients are at an increased risk for developing potentially fatal systemic phaeohyphomycosis.3
Chromoblastomycosis (CBM), also caused by dematiaceous fungi, is characterized by an initially indolent clinical presentation. Typically found on the legs and lower thighs of agricultural workers, the lesion begins as a slow-growing, nodular papule with subsequent transformation into an edematous verrucous plaque with peripheral erythema.9 Lesions can be annular with central clearing, and lymphedema with elephantiasis may be present.10 Histologically, CBM shows pseudoepitheliomatous hyperplasia and intraepidermal pustules as the host rids the infection via transepithelial elimination. Dematiaceous fungi often are seen in the dermis, either freestanding or attached to foreign plant material. Medlar bodies, also called copper penny spores or sclerotic bodies, are the most defining histologic finding and are characterized by groups of brown, thick-walled cells found in giant cells or neutrophil abscesses (Figure 1). Hyphae are not typically found in this type of infection.11
Granulomatous foreign body reactions occur in response to the inoculation of nonhuman material and are characterized by dermal or subcutaneous nodules. Tissue macrophages phagocytize material not removed shortly after implantation, which initiates an inflammatory response that attempts to isolate the material from the uninvolved surrounding tissue. Vegetative foreign bodies will cause the most severe inflammatory reactions.12 Histologically, foreign body granulomas are noncaseating with epithelioid histiocytes surrounding a central foreign body (Figure 2). Occasionally, foreign bodies may be difficult to detect; some are birefringent to polarized light.13 Additionally, inoculation injuries can predispose patients to SP, CBM, and other fungal infections.
Tattoos are characterized by exogenous pigment deposition into the dermis.14 Histologically, tattoos display exogenous pigment deposited throughout the reticular dermis, attached to collagen bundles, within macrophages, or adjacent to adnexal structures (eg, pilosebaceous units or eccrine glands). Although all tattoo pigments can cause adverse reactions, hypersensitivity reactions occur most commonly in response to red pigment, resulting in discrete areas of spongiosis and granulomatous or lichenoid inflammation. Occasionally, hypersensitivity reactions can induce necrobiotic granulomatous reactions characterized by collagen alteration surrounded by palisaded histiocytes and lymphocytes (Figure 3).15,16 There also may be focally dense areas of superficial and deep perivascular lymphohistiocytic infiltrate. Clinical context is important, as brown tattoo pigment (Figure 3) can be easily confused with the pigmented hyphae of phaeohyphomycosis, melanin, or hemosiderin.
Subcutaneous hyalohyphomycosis is a nondemat-iaceous (nonpigmented) infection that is caused by hyaline septate hyphal cells.17 Hyalohyphomycosis skin lesions can present as painful erythematous nodules that evolve into excoriated pustules.18 Hyalohyphomycosis most often arises in immunocompromised patients. Causative organisms are ubiquitous soil saprophytes and plant pathogens, most often Aspergillus and Fusarium species, with a predilection for affecting severely immunocompromised hosts, particularly children.19 These species tend to be vasculotropic, which can result in tissue necrosis and systemic dissemination. Histologically, fungi are dispersed within tissue. They have a bright, bubbly, mildly basophilic cytoplasm and are nonpigmented, branching, and septate (Figure 4).11
- Isa-Isa R, García C, Isa M, et al. Subcutaneous phaeohyphomycosis (mycotic cyst). Clin Dermatol. 2012;30:425-431.
- Rubin RH. Infectious disease complications of renal transplantation. Kidney Int. 1993;44:221-236.
- Ogawa MM, Galante NZ, Godoy P, et al. Treatment of subcutaneous phaeohyphomycosis and prospective follow-up of 17 kidney transplant recipients. J Am Acad Dermatol. 2009;61:977-985.
- Matsumoto T, Ajello L, Matsuda T, et al. Developments in hyalohyphomycosis and phaeohyphomycosis. J Med Vet Mycol. 1994;32(suppl 1):329-349.
- Rinaldi MG. Phaeohyphomycosis. Dermatol Clin. 1996;14:147-153.
- Ajello L, Georg LK, Steigbigel RT, et al. A case of phaeohyphomycosis caused by a new species of Phialophora. Mycologia. 1974;66:490-498.
- Patterson J. Weedon’s Skin Pathology. 4th ed. London, England: Churchill Livingstone Elsevier; 2014.
- Patel U, Chu J, Patel R, et al. Subcutaneous dematiaceous fungal infection. Dermatol Online J. 2011;17:19.
- Bonifaz A, Carrasco-Gerard E, Saúl A. Chromoblastomycosis: clinical and mycologic experience of 51 cases. Mycoses. 2001;44:1-7.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Elston D, Ferringer T, Peckham S, et al, eds. Dermatopathology. 2nd ed. St. Louis, MO: Elsevier Saunders; 2014.
- Lammers RL. Soft tissue foreign bodies. In: Tintinalli J, Stapczynski S, Ma O, et al, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw Hill Professional; 2011.
- Murphy GF, Saavedra AP, Mihm MC. Nodular/interstitial dermatitis. In: Murphy GF, Saavedra AP, Mihm MC, eds. Atlas of Nontumor Pathology: Inflammatory Disorders of the Skin. Vol 10. Washington, DC: American Registry of Pathology; 2012:337-395.
- Laumann A. Body art. In: Goldsmith L, Katz S, Gilchrest B, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012. http://access medicine.mhmedical.com.proxy.lib.uiowa.edu/content.aspx?bookid=392&Sectionid=41138811. Accessed July 17,2016.
- Wood A, Hamilton SA, Wallace WA, et al. Necrobiotic granulomatous tattoo reaction: report of an unusual case showing features of both necrobiosis lipoidica and granuloma annulare patterns. Am J Dermatopathol. 2014;36:e152-e155.
- Mortimer N, Chave T, Johnston G. Red tattoo reactions. Clin Exp Dermatol. 2003;28:508-510.
- Ajello L. Hyalohyphomycosis and phaeohyphomycosis: two global disease entities of public health importance. Eur J Epidemiol. 1986;2:243-251.
- Safdar A. Progressive cutaneous hyalohyphomycosis due to Paecilomyces lilacinus: rapid response to treatment with caspofungin and itraconazole. Clin Infect Dis. 2002;34:1415-1417.
- Marcoux D, Jafarian F, Joncas V, et al. Deep cutaneous fungal infections in immunocompromised children. J Am Acad Dermatol. 2009;61:857-864.
- Isa-Isa R, García C, Isa M, et al. Subcutaneous phaeohyphomycosis (mycotic cyst). Clin Dermatol. 2012;30:425-431.
- Rubin RH. Infectious disease complications of renal transplantation. Kidney Int. 1993;44:221-236.
- Ogawa MM, Galante NZ, Godoy P, et al. Treatment of subcutaneous phaeohyphomycosis and prospective follow-up of 17 kidney transplant recipients. J Am Acad Dermatol. 2009;61:977-985.
- Matsumoto T, Ajello L, Matsuda T, et al. Developments in hyalohyphomycosis and phaeohyphomycosis. J Med Vet Mycol. 1994;32(suppl 1):329-349.
- Rinaldi MG. Phaeohyphomycosis. Dermatol Clin. 1996;14:147-153.
- Ajello L, Georg LK, Steigbigel RT, et al. A case of phaeohyphomycosis caused by a new species of Phialophora. Mycologia. 1974;66:490-498.
- Patterson J. Weedon’s Skin Pathology. 4th ed. London, England: Churchill Livingstone Elsevier; 2014.
- Patel U, Chu J, Patel R, et al. Subcutaneous dematiaceous fungal infection. Dermatol Online J. 2011;17:19.
- Bonifaz A, Carrasco-Gerard E, Saúl A. Chromoblastomycosis: clinical and mycologic experience of 51 cases. Mycoses. 2001;44:1-7.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Elston D, Ferringer T, Peckham S, et al, eds. Dermatopathology. 2nd ed. St. Louis, MO: Elsevier Saunders; 2014.
- Lammers RL. Soft tissue foreign bodies. In: Tintinalli J, Stapczynski S, Ma O, et al, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw Hill Professional; 2011.
- Murphy GF, Saavedra AP, Mihm MC. Nodular/interstitial dermatitis. In: Murphy GF, Saavedra AP, Mihm MC, eds. Atlas of Nontumor Pathology: Inflammatory Disorders of the Skin. Vol 10. Washington, DC: American Registry of Pathology; 2012:337-395.
- Laumann A. Body art. In: Goldsmith L, Katz S, Gilchrest B, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012. http://access medicine.mhmedical.com.proxy.lib.uiowa.edu/content.aspx?bookid=392&Sectionid=41138811. Accessed July 17,2016.
- Wood A, Hamilton SA, Wallace WA, et al. Necrobiotic granulomatous tattoo reaction: report of an unusual case showing features of both necrobiosis lipoidica and granuloma annulare patterns. Am J Dermatopathol. 2014;36:e152-e155.
- Mortimer N, Chave T, Johnston G. Red tattoo reactions. Clin Exp Dermatol. 2003;28:508-510.
- Ajello L. Hyalohyphomycosis and phaeohyphomycosis: two global disease entities of public health importance. Eur J Epidemiol. 1986;2:243-251.
- Safdar A. Progressive cutaneous hyalohyphomycosis due to Paecilomyces lilacinus: rapid response to treatment with caspofungin and itraconazole. Clin Infect Dis. 2002;34:1415-1417.
- Marcoux D, Jafarian F, Joncas V, et al. Deep cutaneous fungal infections in immunocompromised children. J Am Acad Dermatol. 2009;61:857-864.
A 63-year-old man on immunosuppressive therapy following renal transplantation 5 years prior presented with a nontender circumscribed nodule above the left knee of 6 months’ duration. The patient denied any trauma or injury to the site.
Primary Cutaneous Dermal Mucinosis on Herpes Zoster Scars
Mucin is an amorphous gelatinous substance that is found in a large variety of tissues. There are 2 types of cutaneous mucin: dermal and epithelial. Both types appear as basophilic shreds and granules with hematoxylin and eosin stain.1 Epithelial mucin (sialomucin) is found mainly in the gastrointestinal tract and lungs. In the skin, it is present in the cytoplasm of the dark cells of the eccrine glands and in the apocrine secretory cells. Epithelial mucin contains both neutral and acid glycosaminoglycans, stains positive with Alcian blue (pH 2.5) and periodic acid–Schiff, is resistant to hyaluronidase, and does not stain metachromatically with toluidine blue. Dermal mucin is composed of acid glycosaminoglycans (eg, dermatan sulfate, chondroitin 6-sulfate, chondroitin 4-sulfate, hyaluronic acid) and normally is produced by dermal fibroblasts. Dermal mucin stains positive with Alcian blue (pH 2.5); is periodic acid–Schiff negative and sensitive to hyaluronidase; and shows metachromasia with toluidine blue, methylene blue, and thionine.
Cutaneous mucinosis comprises a heterogeneous group of skin disorders characterized by the deposition of mucin in the interstices of the dermis. These diseases may be classified as primary mucinosis with the mucin deposition as the main histologic feature resulting in clinically distinctive lesions and secondary mucinosis with the mucin deposition as an additional histologic finding within the context of an independent skin disease or lesion (eg, basal cell carcinoma) with deposits of mucin in the stroma. Primary cutaneous mucinosis may be subclassified into 2 groups: degenerative-inflammatory mucinoses and neoplastic-hamartomatous mucinoses. According to the histologic features, the degenerative-inflammatory mucinoses are better divided into dermal and follicular mucinoses.2 We describe a case of primary cutaneous dermal mucinosis on herpes zoster (HZ) scars as an isotopic response.
Case Report
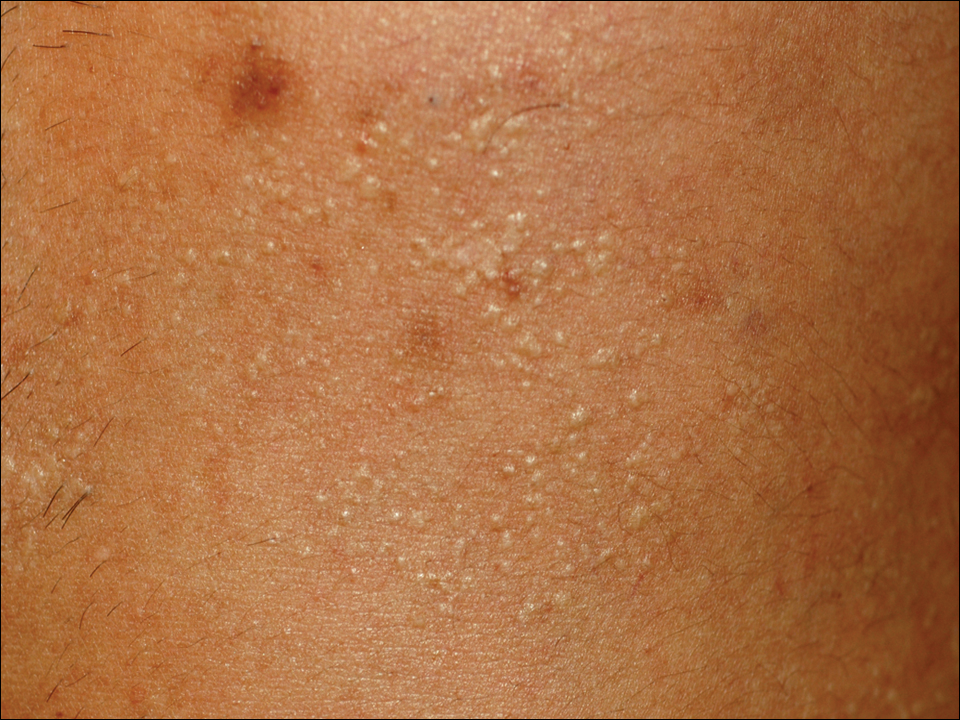
A 33-year-old man presented to the dermatology department with slightly pruritic lesions on the left side of the chest and back that had appeared progressively at the site of HZ scars that had healed without treatment 9 months prior. Dermatologic examination revealed sharply defined whitish papules (Figure 1) measuring 2 to 4 mm in diameter with a smooth surface and linear distribution over the area of the left T8 and T9 dermatomes. The patient reported no postherpetic neuralgia and was otherwise healthy. Laboratory tests including a complete blood cell count, biochemistry, urinalysis, and determination of free thyroid hormones were within reference range. Serologic tests for human immunodeficiency virus, hepatitis B and C viruses, and syphilis were negative. Antinuclear antibodies also were negative.
Histopathology demonstrated abundant bluish granular material between collagen bundles of the papillary dermis (Figure 2). No cytopathologic signs of active herpetic infection were seen. The Alcian blue stain at pH 2.5 was strongly positive for mucin, which confirmed the diagnosis of primary cutaneous dermal mucinosis.
Topical corticosteroids were applied for 2 months with no notable improvement. The lesions gradually improved without any other therapy during the subsequent 6 months.
Comment
The occurrence of a new skin disease at the exact site of a prior unrelated cutaneous disorder that had already resolved was first reported by Wyburn-Mason3 in 1955. Forty years later, the term isotopic response was coined by Wolf et al4 to describe this phenomenon. Diverse types of skin diseases such as herpes simplex virus,5 varicella-zoster infections,4 and thrombophlebitis4 have been implicated in cases of isotopic response, but the most frequently associated primary disorder by far is cutaneous HZ.
Several benign and malignant disorders may occur at sites of resolved HZ lesions, including granulomatous dermatitis,6 granuloma annulare,7 fungal granuloma,8 fungal folliculitis,9 psoriasis,10 morphea,11 lichen sclerosus,12 Kaposi sarcoma,13 the lichenoid variant of chronic graft-versus-host disease,14 cutaneous sarcoidosis,15 granulomatous folliculitis,16 comedones,17 furuncles,18 erythema annulare centrifugum,19 eosinophilic dermatosis,20 cutaneous pseudolymphoma,21 granulomatous vasculitis,22 Rosai-Dorfman disease,12 xanthomatous changes,23 tuberculoid granulomas,24 acneform eruption,25 lichen planus,26 acquired reactive perforating collagenosis,27 lymphoma,28 leukemia,29 angiosarcoma,30 basal cell carcinoma,31 squamous cell carcinoma, and cutaneous metastasis from internal carcinoma.32 The interval between the acute HZ episode and presentation of the second disease is quite variable, ranging from days to several months. Postzoster isotopic response has been described in individuals with varying degrees of immune response, affecting both immunocompetent12 and immunocompromised patients.14 There is no predilection for age, sex, or race. It also seems that antiviral treatment during the active episode does not prevent the development of secondary reactions.Kim et al33 reported a 59-year-old woman who developed flesh-colored or erythematous papules on HZ scars over the area of the left T1 and T2 dermatomes 1 week after the active viral process. Histopathologic study demonstrated deposition of mucin between collagen bundles in the dermis. The authors established the diagnosis of secondary cutaneous mucinosis as an isotopic response.33 Nevertheless, we believe that based on the aforementioned classification of cutaneous mucinosis,2 both this case and our case are better considered as primary cutaneous dermal mucinosis, as the mucin deposition in the dermis was the main histologic finding resulting in a distinctive cutaneous disorder. In the case reported by Kim et al,33 a possible relationship between cutaneous mucinosis and postherpetic neuralgia was suggested based on the slow regression of skin lesions in accordance with the improvement of the neuralgic pain; however, our patient did not have postherpetic neuralgia and the lesions persisted unchanged several months after the acute HZ episode. In the literature, there are reports of primary cutaneous dermal mucinosis associated with altered thyroid function34; autoimmune connective tissue diseases, mostly lupus erythematosus35; monoclonal gammopathy36; and human immunodeficiency virus infection,37 but these possibilities were ruled out in our patient by pertinent laboratory studies.
The pathogenesis of the postherpetic isotopic response remains unknown, but several mechanisms have been proposed. Some authors have suggested that postzoster dermatoses may represent isomorphic response of Köbner phenomenon.13,15 Although isomorphic and isotopic responses share some similarities, these terms describe 2 different phenomena: the first refers to the appearance of the same cutaneous disorder at a different site favored by trauma, while the second manifests a new and unrelated disease at the same location.38 Local anatomic changes such as altered microcirculation, collagen rearrangement, and an imperfect skin barrier may promote a prolonged local inflammatory response. Moreover, the destruction of nerve fibers by the varicella-zoster virus may indirectly influence the local immune system through the release of specific neuropeptides in the skin.39 It has been speculated that some secondary reactions may be the result of type III and type IV hypersensitivity reactions40 to viral antigens or to tissue antigens modified by the virus, inducing either immune hypersensitivity or local immune suppression.41 Some authors have documented the presence of varicella-zoster DNA within early postzoster lesions6,7 by using polymerase chain reaction in early lesions but not in late-stage and residual lesions.12,22 Nikkels et al42 studied early granulomatous lesions by immunohistochemistry and in situ hybridization techniques and concluded that major viral envelope glycoproteins (glycoproteins I and II) rather than complete viral particles could be responsible for delayed-type hypersensitivity reactions. All these findings suggest that secondary reactions presenting on HZ scars are mainly the result of atypical immune reactions to local antigenic stimuli.
The pathogenesis of our case is unknown. From a theoretical point of view, it is possible that varicella-zoster virus may induce fibroblastic proliferation and mucin production on HZ scars; however, if HZ is a frequent process and the virus may induce mucin production, then focal dermal mucinosis in an HZ scar should be a common finding. In our patient, there was no associated disease favoring the development of the cutaneous mucinosis. These localized variants of primary cutaneous mucinosis usually do not require therapy, and a wait-and-see approach is recommended. Topical applications of corticosteroids, pimecrolimus, or tacrolimus, as well as oral isotretinoin, may have some benefit,43 but spontaneous resolution may occur.44 In our patient, topical corticosteroids were applied 2 months following initial presentation without any benefit and the cutaneous lesions gradually improved without any therapy during the subsequent 6 months. Focal dermal mucinosis should be added to the list of cutaneous reactions that may develop in HZ scars.
- Truhan AP, Roenigk HH Jr. The cutaneous mucinoses. J Am Acad Dermatol. 1986;14:1-18.
- Rongioletti F, Rebora A. Cutaneous mucinoses: microscopic criteria for diagnosis. Am J Dermatopathol. 2001;23:257-267.
- Wyburn-Mason R. Malignant change arising in tissues affected by herpes. BMJ. 1955;2:1106-1109.
- Wolf R, Brenner S, Ruocco V, et al. Isotopic response. Int J Dermatol. 1995;34:341-348.
- Ruocco E. Genital warts at the site of healed herpes progenitalis: the isotopic response. Int J Dermatol. 2000;39:705-706.
- Serfling U, Penneys NS, Zhu WY, et al. Varicella-zoster virus DNA in granulomatous skin lesions following herpes zoster. a study by the polymerase chain reaction. J Cutan Pathol. 1993;20:28-33.
- Gibney MD, Nahass GT, Leonardi CL. Cutaneous reactions following herpes zoster infections: report of three cases and a review of the literature. Br J Dermatol. 1996;134:504-509.
- Huang CW, Tu ME, Wu YH, et al. Isotopic response of fungal granuloma following facial herpes zoster infections-report of three cases. Int J Dermatol. 2007;46:1141-1145.
- Tüzün Y, Işçimen A, Göksügür N, et al. Wolf’s isotopic response: Trichophyton rubrum folliculitis appearing on a herpes zoster scar. Int J Dermatol. 2000;39:766-768.
- Allegue F, Fachal C, Romo M, et al. Psoriasis at the site of healed herpes zoster: Wolf’s isotopic response. Actas Dermosifiliogr. 2007;98:576-578.
- Forschner A, Metzler G, Rassner G, et al. Morphea with features of lichen sclerosus et atrophicus at the site of a herpes zoster scar: another case of an isotopic response. Int J Dermatol. 2005;44:524-525.
- Requena L, Kutzner H, Escalonilla P, et al. Cutaneous reactions at sites of herpes zoster scars: an expanded spectrum. Br J Dermatol. 1998;138:161-168.
- Niedt GW, Prioleau PG. Kaposi’s sarcoma occurring in a dermatome previously involved by herpes zoster. J Am Acad Dermatol. 1988;18:448-451.
- Sanli H, Anadolu R, Arat M, et al. Dermatomal lichenoid graft-versus-host disease within herpes zoster scars. Int J Dermatol. 2003;42:562-564.
- Cecchi R, Giomi A. Scar sarcoidosis following herpes zoster. J Eur Acad Dermatol Venereol. 1999;12:280-282.
- Fernández-Redondo V, Amrouni B, Varela E, et al. Granulomatous folliculitis at sites of herpes zoster scars: Wolf’s isotopic response. J Eur Acad Dermatol Venereol. 2002;16:628-630.
- Sanchez-Salas MP. Appearance of comedones at the site of healed herpes zoster: Wolf’s isotopic response. Int J Dermatol. 2011;50:633-634.
- Ghorpade A. Wolf’s isotopic response—furuncles at the site of healed herpes zoster in an Indian male. Int J Dermatol. 2010;49:105-107.
- Lee HW, Lee DK, Rhee DY, et al. Erythema annulare centrifugum following herpes zoster infection: Wolf’s isotopic response? Br J Dermatol. 2005;153:1241-1243.
- Mitsuhashi Y, Kondo S. Post-zoster eosinophilic dermatosis. Br J Dermatol. 1997;136:465-466.
- Roo E, Villegas C, Lopez-Bran E, et al. Postzoster cutaneous pseudolymphoma. Arch Dermatol. 1994;130:661-663.
- Langenberg A, Yen TS, LeBoit PE. Granulomatous vasculitis occurring after cutaneous herpes zoster despite absence of viral genome. J Am Acad Dermatol. 1991;24:429-433.
- Weidman F, Boston LN. Generalized xanthoma tuberosum with xantomathous changes in fresh scars of intercurrent zoster. Arch Intern Med. 1937;59:793-822.
- Olalquiaga J, Minaño R, Barrio J. Granuloma tuberculoide post-herpético en un paciente con leucemia linfocítica crónica. Med Cutan ILA. 1995;23:113-115.
- Stubbings JM, Goodfield MJ. An unusual distribution of an acneiform rash due to herpes zoster infection. Clin Exp Dermatol. 1993;18:92-93.
- Shemer A, Weiss G, Trau H. Wolf’s isotopic response: a case of zosteriform lichen planus on the site of healed herpes zoster. J Eur Acad Dermatol Venereol. 2001;15:445-447.
- Bang SW, Kim YK, Whang KU. Acquired reactive perforating collagenosis: unilateral umbilicated papules along the lesions of herpes zoster. J Am Acad Dermatol. 1997;36:778-779.
- Paydaş S, Sahin B, Yavuz S, et al. Lymphomatous skin infiltration at the site of previous varicella zoster virus infection in a patient with T cell lymphoma. Leuk Lymphoma. 2000;37:229-232.
- Cerroni L, Kerl H. Cutaneous localization of B-cell chronic lymphocytic leukemia at the site of varicella/herpes virus eruptions. J Am Acad Dermatol. 1997;37:1022.
- Hudson CP, Hanno R, Callen JP. Cutaneous angiosarcoma in a site of healed herpes zoster. Int J Dermatol. 1984;23:404-407.
- Wyburn-Mason R. Visceral lesions in herpes zoster. Br Med J. 1957;1:678-681.
- Caroti A. Metastasi cutanee di a adenocarcinoma papillifero ovarico in sede di herpes zoster. Chron Dermatol. 1987;18:769-773.
- Kim MB, Jwa SW, Ko HC, et al. A case of secondary cutaneous mucinosis following herpes zoster: Wolf’s isotopic response. Int J Dermatol. 2009;48:212-214.
- Burman KD, McKinley-Grant L. Dermatologic aspects of thyroid disease. Clin Dermatol. 2006;24:247-255.
- Shekari AM, Ghiasi M, Ghasemi E, et al. Papulonodular mucinosis indicating systemic lupus erythematosus. Clin Exp Dermatol. 2009;34:558-560.
- Dinneen AM, Dicken CH. Scleromyxedema. J Am Acad Dermatol. 1995;33:37-43.
- Rongioletti F, Ghigliotti G, De Marchi R, et al. Cutaneous mucinoses and HIV infection. Br J Dermatol. 1998;139:1077-1080.
- Krahl D, Hartschuh W, Tilgen W. Granuloma annulare perforans in herpes zoster scars. J Am Acad Dermatol. 1993;29:859-862.
- Wolf R, Lotti T, Ruocco V. Isomorphic versus isotopic response: data and hypotheses. J Eur Acad Dermatol Venereol. 2003;17:123-125.
- Fisher G, Jaworski R. Granuloma formation in herpes zoster scars. J Am Acad Dermatol. 1987;16:1261-1263.
- Ruocco V, Grimaldi Filioli F. La risposta isotopica post-erpetica: possibile sequela di un locus minoris resistentiae acquisito. G Ital Dermatol Venereol. 1999;134:547-552.
- Nikkels AF, Debrus S, Delvenne P, et al. Viral glycoproteins in herpesviridae granulomas. Am J Dermatopathol. 1994;16:588-592.
- Rongioletti F, Zaccaria E, Cozzani E, et al. Treatment of localized lichen myxedematosus of discrete type with tacrolimus ointment. J Am Acad Dermatol. 2008;5:530-532.
- Kwon OS, Moon SE, Kim JA, et al. Lichen myxodematosus with rapid spontaneous regression. Br J Dermatol. 1997;136:295-296.
Mucin is an amorphous gelatinous substance that is found in a large variety of tissues. There are 2 types of cutaneous mucin: dermal and epithelial. Both types appear as basophilic shreds and granules with hematoxylin and eosin stain.1 Epithelial mucin (sialomucin) is found mainly in the gastrointestinal tract and lungs. In the skin, it is present in the cytoplasm of the dark cells of the eccrine glands and in the apocrine secretory cells. Epithelial mucin contains both neutral and acid glycosaminoglycans, stains positive with Alcian blue (pH 2.5) and periodic acid–Schiff, is resistant to hyaluronidase, and does not stain metachromatically with toluidine blue. Dermal mucin is composed of acid glycosaminoglycans (eg, dermatan sulfate, chondroitin 6-sulfate, chondroitin 4-sulfate, hyaluronic acid) and normally is produced by dermal fibroblasts. Dermal mucin stains positive with Alcian blue (pH 2.5); is periodic acid–Schiff negative and sensitive to hyaluronidase; and shows metachromasia with toluidine blue, methylene blue, and thionine.
Cutaneous mucinosis comprises a heterogeneous group of skin disorders characterized by the deposition of mucin in the interstices of the dermis. These diseases may be classified as primary mucinosis with the mucin deposition as the main histologic feature resulting in clinically distinctive lesions and secondary mucinosis with the mucin deposition as an additional histologic finding within the context of an independent skin disease or lesion (eg, basal cell carcinoma) with deposits of mucin in the stroma. Primary cutaneous mucinosis may be subclassified into 2 groups: degenerative-inflammatory mucinoses and neoplastic-hamartomatous mucinoses. According to the histologic features, the degenerative-inflammatory mucinoses are better divided into dermal and follicular mucinoses.2 We describe a case of primary cutaneous dermal mucinosis on herpes zoster (HZ) scars as an isotopic response.
Case Report
A 33-year-old man presented to the dermatology department with slightly pruritic lesions on the left side of the chest and back that had appeared progressively at the site of HZ scars that had healed without treatment 9 months prior. Dermatologic examination revealed sharply defined whitish papules (Figure 1) measuring 2 to 4 mm in diameter with a smooth surface and linear distribution over the area of the left T8 and T9 dermatomes. The patient reported no postherpetic neuralgia and was otherwise healthy. Laboratory tests including a complete blood cell count, biochemistry, urinalysis, and determination of free thyroid hormones were within reference range. Serologic tests for human immunodeficiency virus, hepatitis B and C viruses, and syphilis were negative. Antinuclear antibodies also were negative.
Histopathology demonstrated abundant bluish granular material between collagen bundles of the papillary dermis (Figure 2). No cytopathologic signs of active herpetic infection were seen. The Alcian blue stain at pH 2.5 was strongly positive for mucin, which confirmed the diagnosis of primary cutaneous dermal mucinosis.
Topical corticosteroids were applied for 2 months with no notable improvement. The lesions gradually improved without any other therapy during the subsequent 6 months.
Comment
The occurrence of a new skin disease at the exact site of a prior unrelated cutaneous disorder that had already resolved was first reported by Wyburn-Mason3 in 1955. Forty years later, the term isotopic response was coined by Wolf et al4 to describe this phenomenon. Diverse types of skin diseases such as herpes simplex virus,5 varicella-zoster infections,4 and thrombophlebitis4 have been implicated in cases of isotopic response, but the most frequently associated primary disorder by far is cutaneous HZ.
Several benign and malignant disorders may occur at sites of resolved HZ lesions, including granulomatous dermatitis,6 granuloma annulare,7 fungal granuloma,8 fungal folliculitis,9 psoriasis,10 morphea,11 lichen sclerosus,12 Kaposi sarcoma,13 the lichenoid variant of chronic graft-versus-host disease,14 cutaneous sarcoidosis,15 granulomatous folliculitis,16 comedones,17 furuncles,18 erythema annulare centrifugum,19 eosinophilic dermatosis,20 cutaneous pseudolymphoma,21 granulomatous vasculitis,22 Rosai-Dorfman disease,12 xanthomatous changes,23 tuberculoid granulomas,24 acneform eruption,25 lichen planus,26 acquired reactive perforating collagenosis,27 lymphoma,28 leukemia,29 angiosarcoma,30 basal cell carcinoma,31 squamous cell carcinoma, and cutaneous metastasis from internal carcinoma.32 The interval between the acute HZ episode and presentation of the second disease is quite variable, ranging from days to several months. Postzoster isotopic response has been described in individuals with varying degrees of immune response, affecting both immunocompetent12 and immunocompromised patients.14 There is no predilection for age, sex, or race. It also seems that antiviral treatment during the active episode does not prevent the development of secondary reactions.Kim et al33 reported a 59-year-old woman who developed flesh-colored or erythematous papules on HZ scars over the area of the left T1 and T2 dermatomes 1 week after the active viral process. Histopathologic study demonstrated deposition of mucin between collagen bundles in the dermis. The authors established the diagnosis of secondary cutaneous mucinosis as an isotopic response.33 Nevertheless, we believe that based on the aforementioned classification of cutaneous mucinosis,2 both this case and our case are better considered as primary cutaneous dermal mucinosis, as the mucin deposition in the dermis was the main histologic finding resulting in a distinctive cutaneous disorder. In the case reported by Kim et al,33 a possible relationship between cutaneous mucinosis and postherpetic neuralgia was suggested based on the slow regression of skin lesions in accordance with the improvement of the neuralgic pain; however, our patient did not have postherpetic neuralgia and the lesions persisted unchanged several months after the acute HZ episode. In the literature, there are reports of primary cutaneous dermal mucinosis associated with altered thyroid function34; autoimmune connective tissue diseases, mostly lupus erythematosus35; monoclonal gammopathy36; and human immunodeficiency virus infection,37 but these possibilities were ruled out in our patient by pertinent laboratory studies.
The pathogenesis of the postherpetic isotopic response remains unknown, but several mechanisms have been proposed. Some authors have suggested that postzoster dermatoses may represent isomorphic response of Köbner phenomenon.13,15 Although isomorphic and isotopic responses share some similarities, these terms describe 2 different phenomena: the first refers to the appearance of the same cutaneous disorder at a different site favored by trauma, while the second manifests a new and unrelated disease at the same location.38 Local anatomic changes such as altered microcirculation, collagen rearrangement, and an imperfect skin barrier may promote a prolonged local inflammatory response. Moreover, the destruction of nerve fibers by the varicella-zoster virus may indirectly influence the local immune system through the release of specific neuropeptides in the skin.39 It has been speculated that some secondary reactions may be the result of type III and type IV hypersensitivity reactions40 to viral antigens or to tissue antigens modified by the virus, inducing either immune hypersensitivity or local immune suppression.41 Some authors have documented the presence of varicella-zoster DNA within early postzoster lesions6,7 by using polymerase chain reaction in early lesions but not in late-stage and residual lesions.12,22 Nikkels et al42 studied early granulomatous lesions by immunohistochemistry and in situ hybridization techniques and concluded that major viral envelope glycoproteins (glycoproteins I and II) rather than complete viral particles could be responsible for delayed-type hypersensitivity reactions. All these findings suggest that secondary reactions presenting on HZ scars are mainly the result of atypical immune reactions to local antigenic stimuli.
The pathogenesis of our case is unknown. From a theoretical point of view, it is possible that varicella-zoster virus may induce fibroblastic proliferation and mucin production on HZ scars; however, if HZ is a frequent process and the virus may induce mucin production, then focal dermal mucinosis in an HZ scar should be a common finding. In our patient, there was no associated disease favoring the development of the cutaneous mucinosis. These localized variants of primary cutaneous mucinosis usually do not require therapy, and a wait-and-see approach is recommended. Topical applications of corticosteroids, pimecrolimus, or tacrolimus, as well as oral isotretinoin, may have some benefit,43 but spontaneous resolution may occur.44 In our patient, topical corticosteroids were applied 2 months following initial presentation without any benefit and the cutaneous lesions gradually improved without any therapy during the subsequent 6 months. Focal dermal mucinosis should be added to the list of cutaneous reactions that may develop in HZ scars.
Mucin is an amorphous gelatinous substance that is found in a large variety of tissues. There are 2 types of cutaneous mucin: dermal and epithelial. Both types appear as basophilic shreds and granules with hematoxylin and eosin stain.1 Epithelial mucin (sialomucin) is found mainly in the gastrointestinal tract and lungs. In the skin, it is present in the cytoplasm of the dark cells of the eccrine glands and in the apocrine secretory cells. Epithelial mucin contains both neutral and acid glycosaminoglycans, stains positive with Alcian blue (pH 2.5) and periodic acid–Schiff, is resistant to hyaluronidase, and does not stain metachromatically with toluidine blue. Dermal mucin is composed of acid glycosaminoglycans (eg, dermatan sulfate, chondroitin 6-sulfate, chondroitin 4-sulfate, hyaluronic acid) and normally is produced by dermal fibroblasts. Dermal mucin stains positive with Alcian blue (pH 2.5); is periodic acid–Schiff negative and sensitive to hyaluronidase; and shows metachromasia with toluidine blue, methylene blue, and thionine.
Cutaneous mucinosis comprises a heterogeneous group of skin disorders characterized by the deposition of mucin in the interstices of the dermis. These diseases may be classified as primary mucinosis with the mucin deposition as the main histologic feature resulting in clinically distinctive lesions and secondary mucinosis with the mucin deposition as an additional histologic finding within the context of an independent skin disease or lesion (eg, basal cell carcinoma) with deposits of mucin in the stroma. Primary cutaneous mucinosis may be subclassified into 2 groups: degenerative-inflammatory mucinoses and neoplastic-hamartomatous mucinoses. According to the histologic features, the degenerative-inflammatory mucinoses are better divided into dermal and follicular mucinoses.2 We describe a case of primary cutaneous dermal mucinosis on herpes zoster (HZ) scars as an isotopic response.
Case Report
A 33-year-old man presented to the dermatology department with slightly pruritic lesions on the left side of the chest and back that had appeared progressively at the site of HZ scars that had healed without treatment 9 months prior. Dermatologic examination revealed sharply defined whitish papules (Figure 1) measuring 2 to 4 mm in diameter with a smooth surface and linear distribution over the area of the left T8 and T9 dermatomes. The patient reported no postherpetic neuralgia and was otherwise healthy. Laboratory tests including a complete blood cell count, biochemistry, urinalysis, and determination of free thyroid hormones were within reference range. Serologic tests for human immunodeficiency virus, hepatitis B and C viruses, and syphilis were negative. Antinuclear antibodies also were negative.
Histopathology demonstrated abundant bluish granular material between collagen bundles of the papillary dermis (Figure 2). No cytopathologic signs of active herpetic infection were seen. The Alcian blue stain at pH 2.5 was strongly positive for mucin, which confirmed the diagnosis of primary cutaneous dermal mucinosis.
Topical corticosteroids were applied for 2 months with no notable improvement. The lesions gradually improved without any other therapy during the subsequent 6 months.
Comment
The occurrence of a new skin disease at the exact site of a prior unrelated cutaneous disorder that had already resolved was first reported by Wyburn-Mason3 in 1955. Forty years later, the term isotopic response was coined by Wolf et al4 to describe this phenomenon. Diverse types of skin diseases such as herpes simplex virus,5 varicella-zoster infections,4 and thrombophlebitis4 have been implicated in cases of isotopic response, but the most frequently associated primary disorder by far is cutaneous HZ.
Several benign and malignant disorders may occur at sites of resolved HZ lesions, including granulomatous dermatitis,6 granuloma annulare,7 fungal granuloma,8 fungal folliculitis,9 psoriasis,10 morphea,11 lichen sclerosus,12 Kaposi sarcoma,13 the lichenoid variant of chronic graft-versus-host disease,14 cutaneous sarcoidosis,15 granulomatous folliculitis,16 comedones,17 furuncles,18 erythema annulare centrifugum,19 eosinophilic dermatosis,20 cutaneous pseudolymphoma,21 granulomatous vasculitis,22 Rosai-Dorfman disease,12 xanthomatous changes,23 tuberculoid granulomas,24 acneform eruption,25 lichen planus,26 acquired reactive perforating collagenosis,27 lymphoma,28 leukemia,29 angiosarcoma,30 basal cell carcinoma,31 squamous cell carcinoma, and cutaneous metastasis from internal carcinoma.32 The interval between the acute HZ episode and presentation of the second disease is quite variable, ranging from days to several months. Postzoster isotopic response has been described in individuals with varying degrees of immune response, affecting both immunocompetent12 and immunocompromised patients.14 There is no predilection for age, sex, or race. It also seems that antiviral treatment during the active episode does not prevent the development of secondary reactions.Kim et al33 reported a 59-year-old woman who developed flesh-colored or erythematous papules on HZ scars over the area of the left T1 and T2 dermatomes 1 week after the active viral process. Histopathologic study demonstrated deposition of mucin between collagen bundles in the dermis. The authors established the diagnosis of secondary cutaneous mucinosis as an isotopic response.33 Nevertheless, we believe that based on the aforementioned classification of cutaneous mucinosis,2 both this case and our case are better considered as primary cutaneous dermal mucinosis, as the mucin deposition in the dermis was the main histologic finding resulting in a distinctive cutaneous disorder. In the case reported by Kim et al,33 a possible relationship between cutaneous mucinosis and postherpetic neuralgia was suggested based on the slow regression of skin lesions in accordance with the improvement of the neuralgic pain; however, our patient did not have postherpetic neuralgia and the lesions persisted unchanged several months after the acute HZ episode. In the literature, there are reports of primary cutaneous dermal mucinosis associated with altered thyroid function34; autoimmune connective tissue diseases, mostly lupus erythematosus35; monoclonal gammopathy36; and human immunodeficiency virus infection,37 but these possibilities were ruled out in our patient by pertinent laboratory studies.
The pathogenesis of the postherpetic isotopic response remains unknown, but several mechanisms have been proposed. Some authors have suggested that postzoster dermatoses may represent isomorphic response of Köbner phenomenon.13,15 Although isomorphic and isotopic responses share some similarities, these terms describe 2 different phenomena: the first refers to the appearance of the same cutaneous disorder at a different site favored by trauma, while the second manifests a new and unrelated disease at the same location.38 Local anatomic changes such as altered microcirculation, collagen rearrangement, and an imperfect skin barrier may promote a prolonged local inflammatory response. Moreover, the destruction of nerve fibers by the varicella-zoster virus may indirectly influence the local immune system through the release of specific neuropeptides in the skin.39 It has been speculated that some secondary reactions may be the result of type III and type IV hypersensitivity reactions40 to viral antigens or to tissue antigens modified by the virus, inducing either immune hypersensitivity or local immune suppression.41 Some authors have documented the presence of varicella-zoster DNA within early postzoster lesions6,7 by using polymerase chain reaction in early lesions but not in late-stage and residual lesions.12,22 Nikkels et al42 studied early granulomatous lesions by immunohistochemistry and in situ hybridization techniques and concluded that major viral envelope glycoproteins (glycoproteins I and II) rather than complete viral particles could be responsible for delayed-type hypersensitivity reactions. All these findings suggest that secondary reactions presenting on HZ scars are mainly the result of atypical immune reactions to local antigenic stimuli.
The pathogenesis of our case is unknown. From a theoretical point of view, it is possible that varicella-zoster virus may induce fibroblastic proliferation and mucin production on HZ scars; however, if HZ is a frequent process and the virus may induce mucin production, then focal dermal mucinosis in an HZ scar should be a common finding. In our patient, there was no associated disease favoring the development of the cutaneous mucinosis. These localized variants of primary cutaneous mucinosis usually do not require therapy, and a wait-and-see approach is recommended. Topical applications of corticosteroids, pimecrolimus, or tacrolimus, as well as oral isotretinoin, may have some benefit,43 but spontaneous resolution may occur.44 In our patient, topical corticosteroids were applied 2 months following initial presentation without any benefit and the cutaneous lesions gradually improved without any therapy during the subsequent 6 months. Focal dermal mucinosis should be added to the list of cutaneous reactions that may develop in HZ scars.
- Truhan AP, Roenigk HH Jr. The cutaneous mucinoses. J Am Acad Dermatol. 1986;14:1-18.
- Rongioletti F, Rebora A. Cutaneous mucinoses: microscopic criteria for diagnosis. Am J Dermatopathol. 2001;23:257-267.
- Wyburn-Mason R. Malignant change arising in tissues affected by herpes. BMJ. 1955;2:1106-1109.
- Wolf R, Brenner S, Ruocco V, et al. Isotopic response. Int J Dermatol. 1995;34:341-348.
- Ruocco E. Genital warts at the site of healed herpes progenitalis: the isotopic response. Int J Dermatol. 2000;39:705-706.
- Serfling U, Penneys NS, Zhu WY, et al. Varicella-zoster virus DNA in granulomatous skin lesions following herpes zoster. a study by the polymerase chain reaction. J Cutan Pathol. 1993;20:28-33.
- Gibney MD, Nahass GT, Leonardi CL. Cutaneous reactions following herpes zoster infections: report of three cases and a review of the literature. Br J Dermatol. 1996;134:504-509.
- Huang CW, Tu ME, Wu YH, et al. Isotopic response of fungal granuloma following facial herpes zoster infections-report of three cases. Int J Dermatol. 2007;46:1141-1145.
- Tüzün Y, Işçimen A, Göksügür N, et al. Wolf’s isotopic response: Trichophyton rubrum folliculitis appearing on a herpes zoster scar. Int J Dermatol. 2000;39:766-768.
- Allegue F, Fachal C, Romo M, et al. Psoriasis at the site of healed herpes zoster: Wolf’s isotopic response. Actas Dermosifiliogr. 2007;98:576-578.
- Forschner A, Metzler G, Rassner G, et al. Morphea with features of lichen sclerosus et atrophicus at the site of a herpes zoster scar: another case of an isotopic response. Int J Dermatol. 2005;44:524-525.
- Requena L, Kutzner H, Escalonilla P, et al. Cutaneous reactions at sites of herpes zoster scars: an expanded spectrum. Br J Dermatol. 1998;138:161-168.
- Niedt GW, Prioleau PG. Kaposi’s sarcoma occurring in a dermatome previously involved by herpes zoster. J Am Acad Dermatol. 1988;18:448-451.
- Sanli H, Anadolu R, Arat M, et al. Dermatomal lichenoid graft-versus-host disease within herpes zoster scars. Int J Dermatol. 2003;42:562-564.
- Cecchi R, Giomi A. Scar sarcoidosis following herpes zoster. J Eur Acad Dermatol Venereol. 1999;12:280-282.
- Fernández-Redondo V, Amrouni B, Varela E, et al. Granulomatous folliculitis at sites of herpes zoster scars: Wolf’s isotopic response. J Eur Acad Dermatol Venereol. 2002;16:628-630.
- Sanchez-Salas MP. Appearance of comedones at the site of healed herpes zoster: Wolf’s isotopic response. Int J Dermatol. 2011;50:633-634.
- Ghorpade A. Wolf’s isotopic response—furuncles at the site of healed herpes zoster in an Indian male. Int J Dermatol. 2010;49:105-107.
- Lee HW, Lee DK, Rhee DY, et al. Erythema annulare centrifugum following herpes zoster infection: Wolf’s isotopic response? Br J Dermatol. 2005;153:1241-1243.
- Mitsuhashi Y, Kondo S. Post-zoster eosinophilic dermatosis. Br J Dermatol. 1997;136:465-466.
- Roo E, Villegas C, Lopez-Bran E, et al. Postzoster cutaneous pseudolymphoma. Arch Dermatol. 1994;130:661-663.
- Langenberg A, Yen TS, LeBoit PE. Granulomatous vasculitis occurring after cutaneous herpes zoster despite absence of viral genome. J Am Acad Dermatol. 1991;24:429-433.
- Weidman F, Boston LN. Generalized xanthoma tuberosum with xantomathous changes in fresh scars of intercurrent zoster. Arch Intern Med. 1937;59:793-822.
- Olalquiaga J, Minaño R, Barrio J. Granuloma tuberculoide post-herpético en un paciente con leucemia linfocítica crónica. Med Cutan ILA. 1995;23:113-115.
- Stubbings JM, Goodfield MJ. An unusual distribution of an acneiform rash due to herpes zoster infection. Clin Exp Dermatol. 1993;18:92-93.
- Shemer A, Weiss G, Trau H. Wolf’s isotopic response: a case of zosteriform lichen planus on the site of healed herpes zoster. J Eur Acad Dermatol Venereol. 2001;15:445-447.
- Bang SW, Kim YK, Whang KU. Acquired reactive perforating collagenosis: unilateral umbilicated papules along the lesions of herpes zoster. J Am Acad Dermatol. 1997;36:778-779.
- Paydaş S, Sahin B, Yavuz S, et al. Lymphomatous skin infiltration at the site of previous varicella zoster virus infection in a patient with T cell lymphoma. Leuk Lymphoma. 2000;37:229-232.
- Cerroni L, Kerl H. Cutaneous localization of B-cell chronic lymphocytic leukemia at the site of varicella/herpes virus eruptions. J Am Acad Dermatol. 1997;37:1022.
- Hudson CP, Hanno R, Callen JP. Cutaneous angiosarcoma in a site of healed herpes zoster. Int J Dermatol. 1984;23:404-407.
- Wyburn-Mason R. Visceral lesions in herpes zoster. Br Med J. 1957;1:678-681.
- Caroti A. Metastasi cutanee di a adenocarcinoma papillifero ovarico in sede di herpes zoster. Chron Dermatol. 1987;18:769-773.
- Kim MB, Jwa SW, Ko HC, et al. A case of secondary cutaneous mucinosis following herpes zoster: Wolf’s isotopic response. Int J Dermatol. 2009;48:212-214.
- Burman KD, McKinley-Grant L. Dermatologic aspects of thyroid disease. Clin Dermatol. 2006;24:247-255.
- Shekari AM, Ghiasi M, Ghasemi E, et al. Papulonodular mucinosis indicating systemic lupus erythematosus. Clin Exp Dermatol. 2009;34:558-560.
- Dinneen AM, Dicken CH. Scleromyxedema. J Am Acad Dermatol. 1995;33:37-43.
- Rongioletti F, Ghigliotti G, De Marchi R, et al. Cutaneous mucinoses and HIV infection. Br J Dermatol. 1998;139:1077-1080.
- Krahl D, Hartschuh W, Tilgen W. Granuloma annulare perforans in herpes zoster scars. J Am Acad Dermatol. 1993;29:859-862.
- Wolf R, Lotti T, Ruocco V. Isomorphic versus isotopic response: data and hypotheses. J Eur Acad Dermatol Venereol. 2003;17:123-125.
- Fisher G, Jaworski R. Granuloma formation in herpes zoster scars. J Am Acad Dermatol. 1987;16:1261-1263.
- Ruocco V, Grimaldi Filioli F. La risposta isotopica post-erpetica: possibile sequela di un locus minoris resistentiae acquisito. G Ital Dermatol Venereol. 1999;134:547-552.
- Nikkels AF, Debrus S, Delvenne P, et al. Viral glycoproteins in herpesviridae granulomas. Am J Dermatopathol. 1994;16:588-592.
- Rongioletti F, Zaccaria E, Cozzani E, et al. Treatment of localized lichen myxedematosus of discrete type with tacrolimus ointment. J Am Acad Dermatol. 2008;5:530-532.
- Kwon OS, Moon SE, Kim JA, et al. Lichen myxodematosus with rapid spontaneous regression. Br J Dermatol. 1997;136:295-296.
- Truhan AP, Roenigk HH Jr. The cutaneous mucinoses. J Am Acad Dermatol. 1986;14:1-18.
- Rongioletti F, Rebora A. Cutaneous mucinoses: microscopic criteria for diagnosis. Am J Dermatopathol. 2001;23:257-267.
- Wyburn-Mason R. Malignant change arising in tissues affected by herpes. BMJ. 1955;2:1106-1109.
- Wolf R, Brenner S, Ruocco V, et al. Isotopic response. Int J Dermatol. 1995;34:341-348.
- Ruocco E. Genital warts at the site of healed herpes progenitalis: the isotopic response. Int J Dermatol. 2000;39:705-706.
- Serfling U, Penneys NS, Zhu WY, et al. Varicella-zoster virus DNA in granulomatous skin lesions following herpes zoster. a study by the polymerase chain reaction. J Cutan Pathol. 1993;20:28-33.
- Gibney MD, Nahass GT, Leonardi CL. Cutaneous reactions following herpes zoster infections: report of three cases and a review of the literature. Br J Dermatol. 1996;134:504-509.
- Huang CW, Tu ME, Wu YH, et al. Isotopic response of fungal granuloma following facial herpes zoster infections-report of three cases. Int J Dermatol. 2007;46:1141-1145.
- Tüzün Y, Işçimen A, Göksügür N, et al. Wolf’s isotopic response: Trichophyton rubrum folliculitis appearing on a herpes zoster scar. Int J Dermatol. 2000;39:766-768.
- Allegue F, Fachal C, Romo M, et al. Psoriasis at the site of healed herpes zoster: Wolf’s isotopic response. Actas Dermosifiliogr. 2007;98:576-578.
- Forschner A, Metzler G, Rassner G, et al. Morphea with features of lichen sclerosus et atrophicus at the site of a herpes zoster scar: another case of an isotopic response. Int J Dermatol. 2005;44:524-525.
- Requena L, Kutzner H, Escalonilla P, et al. Cutaneous reactions at sites of herpes zoster scars: an expanded spectrum. Br J Dermatol. 1998;138:161-168.
- Niedt GW, Prioleau PG. Kaposi’s sarcoma occurring in a dermatome previously involved by herpes zoster. J Am Acad Dermatol. 1988;18:448-451.
- Sanli H, Anadolu R, Arat M, et al. Dermatomal lichenoid graft-versus-host disease within herpes zoster scars. Int J Dermatol. 2003;42:562-564.
- Cecchi R, Giomi A. Scar sarcoidosis following herpes zoster. J Eur Acad Dermatol Venereol. 1999;12:280-282.
- Fernández-Redondo V, Amrouni B, Varela E, et al. Granulomatous folliculitis at sites of herpes zoster scars: Wolf’s isotopic response. J Eur Acad Dermatol Venereol. 2002;16:628-630.
- Sanchez-Salas MP. Appearance of comedones at the site of healed herpes zoster: Wolf’s isotopic response. Int J Dermatol. 2011;50:633-634.
- Ghorpade A. Wolf’s isotopic response—furuncles at the site of healed herpes zoster in an Indian male. Int J Dermatol. 2010;49:105-107.
- Lee HW, Lee DK, Rhee DY, et al. Erythema annulare centrifugum following herpes zoster infection: Wolf’s isotopic response? Br J Dermatol. 2005;153:1241-1243.
- Mitsuhashi Y, Kondo S. Post-zoster eosinophilic dermatosis. Br J Dermatol. 1997;136:465-466.
- Roo E, Villegas C, Lopez-Bran E, et al. Postzoster cutaneous pseudolymphoma. Arch Dermatol. 1994;130:661-663.
- Langenberg A, Yen TS, LeBoit PE. Granulomatous vasculitis occurring after cutaneous herpes zoster despite absence of viral genome. J Am Acad Dermatol. 1991;24:429-433.
- Weidman F, Boston LN. Generalized xanthoma tuberosum with xantomathous changes in fresh scars of intercurrent zoster. Arch Intern Med. 1937;59:793-822.
- Olalquiaga J, Minaño R, Barrio J. Granuloma tuberculoide post-herpético en un paciente con leucemia linfocítica crónica. Med Cutan ILA. 1995;23:113-115.
- Stubbings JM, Goodfield MJ. An unusual distribution of an acneiform rash due to herpes zoster infection. Clin Exp Dermatol. 1993;18:92-93.
- Shemer A, Weiss G, Trau H. Wolf’s isotopic response: a case of zosteriform lichen planus on the site of healed herpes zoster. J Eur Acad Dermatol Venereol. 2001;15:445-447.
- Bang SW, Kim YK, Whang KU. Acquired reactive perforating collagenosis: unilateral umbilicated papules along the lesions of herpes zoster. J Am Acad Dermatol. 1997;36:778-779.
- Paydaş S, Sahin B, Yavuz S, et al. Lymphomatous skin infiltration at the site of previous varicella zoster virus infection in a patient with T cell lymphoma. Leuk Lymphoma. 2000;37:229-232.
- Cerroni L, Kerl H. Cutaneous localization of B-cell chronic lymphocytic leukemia at the site of varicella/herpes virus eruptions. J Am Acad Dermatol. 1997;37:1022.
- Hudson CP, Hanno R, Callen JP. Cutaneous angiosarcoma in a site of healed herpes zoster. Int J Dermatol. 1984;23:404-407.
- Wyburn-Mason R. Visceral lesions in herpes zoster. Br Med J. 1957;1:678-681.
- Caroti A. Metastasi cutanee di a adenocarcinoma papillifero ovarico in sede di herpes zoster. Chron Dermatol. 1987;18:769-773.
- Kim MB, Jwa SW, Ko HC, et al. A case of secondary cutaneous mucinosis following herpes zoster: Wolf’s isotopic response. Int J Dermatol. 2009;48:212-214.
- Burman KD, McKinley-Grant L. Dermatologic aspects of thyroid disease. Clin Dermatol. 2006;24:247-255.
- Shekari AM, Ghiasi M, Ghasemi E, et al. Papulonodular mucinosis indicating systemic lupus erythematosus. Clin Exp Dermatol. 2009;34:558-560.
- Dinneen AM, Dicken CH. Scleromyxedema. J Am Acad Dermatol. 1995;33:37-43.
- Rongioletti F, Ghigliotti G, De Marchi R, et al. Cutaneous mucinoses and HIV infection. Br J Dermatol. 1998;139:1077-1080.
- Krahl D, Hartschuh W, Tilgen W. Granuloma annulare perforans in herpes zoster scars. J Am Acad Dermatol. 1993;29:859-862.
- Wolf R, Lotti T, Ruocco V. Isomorphic versus isotopic response: data and hypotheses. J Eur Acad Dermatol Venereol. 2003;17:123-125.
- Fisher G, Jaworski R. Granuloma formation in herpes zoster scars. J Am Acad Dermatol. 1987;16:1261-1263.
- Ruocco V, Grimaldi Filioli F. La risposta isotopica post-erpetica: possibile sequela di un locus minoris resistentiae acquisito. G Ital Dermatol Venereol. 1999;134:547-552.
- Nikkels AF, Debrus S, Delvenne P, et al. Viral glycoproteins in herpesviridae granulomas. Am J Dermatopathol. 1994;16:588-592.
- Rongioletti F, Zaccaria E, Cozzani E, et al. Treatment of localized lichen myxedematosus of discrete type with tacrolimus ointment. J Am Acad Dermatol. 2008;5:530-532.
- Kwon OS, Moon SE, Kim JA, et al. Lichen myxodematosus with rapid spontaneous regression. Br J Dermatol. 1997;136:295-296.
Practice Points
- Focal mucinosis is a histopathologic finding that may be seen in different cutaneous disorders. It is an exceptional histopathologic finding that has rarely been described in herpes zoster scars.
- In most cases, focal mucinosis is just a histopathologic finding with no therapeutic consequences.
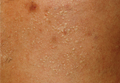
Brown Papules and a Plaque on the Calf
The Diagnosis: Irritated Seborrheic Keratosis
Biopsies of one of the protruding papules and the underlying plaque were performed. The specimen from the papule showed hyperkeratosis, acanthosis, papillomatosis, and a flattened dermoepidermal junction with demarcated horizontal margin, which demonstrated apparent upward growth of the epidermis. Moderate lymphocytic infiltration in the upper dermis also was observed (Figure, A). The histologic findings of the plaque showed acanthosis, several pseudohorn cysts, hyperpigmentation of the basal layer, and a horizontal demarcation of the dermoepidermal junction (Figure, B).
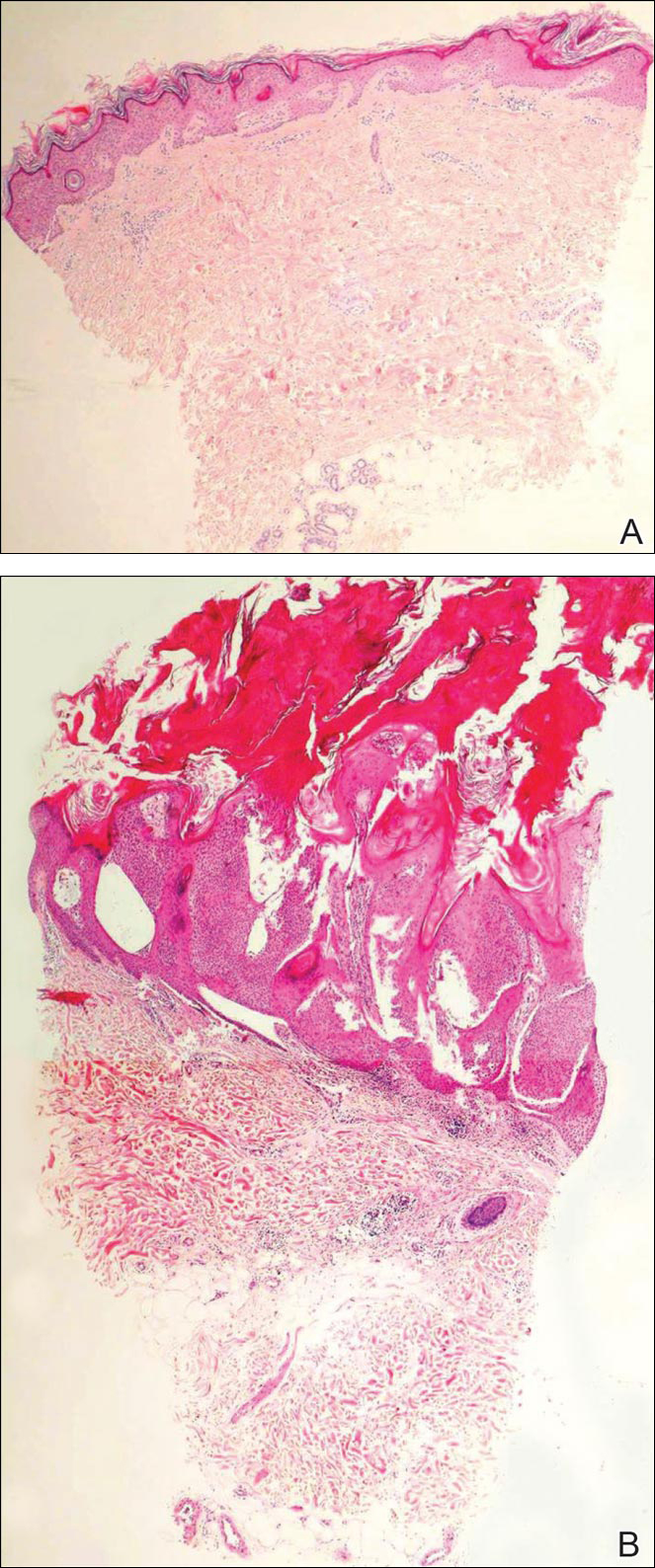
Seborrheic keratosis is the most common benign epidermal tumor of the skin with variable appearance.1 It usually begins with well-circumscribed, dull, flat, tan or brown patches that then grow into waxy verrucous papules.1 There are many clinicopathologic variants of SK such as common SK, stucco keratosis, and dermatosis papulosa nigra in clinical variation, as well as acanthotic, hyperkeratotic, clonal, reticulated, irritated, and melanoacanthoma subtypes based on histological variation.2,3
Seborrheic keratosis is a tumor of keratinocytic origin. Although genetics, sun exposure,4 and human papillomavirus infection5 are thought to be causative factors, the precise etiology of SK is unknown.1
The histology of SK shows monotonous basaloid tumor cells without atypia. It generally is comprised of focal acanthosis and papillomatosis with a sharp flat base. Intraepithelial horn pseudocysts are notable features of SK and increased melanin often is seen.2,6
Irritated SK is a histologic variant of SK that has been mechanically or chemically irritated or is involved in immunologic responses. Histologically, the dermis underlying an SK lesion filled with a dense lymphocytic infiltration is characteristic.1,2
For symptomatic or cosmetically undesirable lesions, complete removal of the lesion is the preferred treatment. Cryotherapy, electrodesiccation followed by curettage, curettage followed by desiccation, laser ablation, and surgical excision are effective treatments.1
- Valencia DT, Nicholas RS, Ken KL, et al. Benign epithelial tumors, hamartomas, and hyperplasias. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Professional; 2012:1319-1336.
- Kirkharn N. Tumors and cysts of the epidermis. In: Elder DE, Elenitsas R, Johnson BL Jr, eds. Lever’s Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:791-850.
- Rajesh G, Thappa DM, Jaisankar TJ, et al. Spectrum of seborrheic keratoses in South Indians: a clinical and dermoscopic study. Indian J Dermatol Venereol Leprol. 2011;77:483-488.
- Yeatman JM, Kilkenny M, Marks R. The prevalence of seborrhoeic keratoses in an Australian population: does exposure to sunlight play a part in their frequency? Br J Dermatol. 1997;137:411-414.
- Li YH, Chen G, Dong XP, et al. Detection of epidermodysplasia verruciformis-associated human papillomavirus DNA in nongenital seborrhoeic keratosis. Br J Dermatol. 2004;151:1060-1065.
- Brinster NK, Liu V, Diwan AH, et al. Dermatopathology. Philadelphia, PA: Saunders/Elsevier; 2011.
The Diagnosis: Irritated Seborrheic Keratosis
Biopsies of one of the protruding papules and the underlying plaque were performed. The specimen from the papule showed hyperkeratosis, acanthosis, papillomatosis, and a flattened dermoepidermal junction with demarcated horizontal margin, which demonstrated apparent upward growth of the epidermis. Moderate lymphocytic infiltration in the upper dermis also was observed (Figure, A). The histologic findings of the plaque showed acanthosis, several pseudohorn cysts, hyperpigmentation of the basal layer, and a horizontal demarcation of the dermoepidermal junction (Figure, B).
Seborrheic keratosis is the most common benign epidermal tumor of the skin with variable appearance.1 It usually begins with well-circumscribed, dull, flat, tan or brown patches that then grow into waxy verrucous papules.1 There are many clinicopathologic variants of SK such as common SK, stucco keratosis, and dermatosis papulosa nigra in clinical variation, as well as acanthotic, hyperkeratotic, clonal, reticulated, irritated, and melanoacanthoma subtypes based on histological variation.2,3
Seborrheic keratosis is a tumor of keratinocytic origin. Although genetics, sun exposure,4 and human papillomavirus infection5 are thought to be causative factors, the precise etiology of SK is unknown.1
The histology of SK shows monotonous basaloid tumor cells without atypia. It generally is comprised of focal acanthosis and papillomatosis with a sharp flat base. Intraepithelial horn pseudocysts are notable features of SK and increased melanin often is seen.2,6
Irritated SK is a histologic variant of SK that has been mechanically or chemically irritated or is involved in immunologic responses. Histologically, the dermis underlying an SK lesion filled with a dense lymphocytic infiltration is characteristic.1,2
For symptomatic or cosmetically undesirable lesions, complete removal of the lesion is the preferred treatment. Cryotherapy, electrodesiccation followed by curettage, curettage followed by desiccation, laser ablation, and surgical excision are effective treatments.1
The Diagnosis: Irritated Seborrheic Keratosis
Biopsies of one of the protruding papules and the underlying plaque were performed. The specimen from the papule showed hyperkeratosis, acanthosis, papillomatosis, and a flattened dermoepidermal junction with demarcated horizontal margin, which demonstrated apparent upward growth of the epidermis. Moderate lymphocytic infiltration in the upper dermis also was observed (Figure, A). The histologic findings of the plaque showed acanthosis, several pseudohorn cysts, hyperpigmentation of the basal layer, and a horizontal demarcation of the dermoepidermal junction (Figure, B).
Seborrheic keratosis is the most common benign epidermal tumor of the skin with variable appearance.1 It usually begins with well-circumscribed, dull, flat, tan or brown patches that then grow into waxy verrucous papules.1 There are many clinicopathologic variants of SK such as common SK, stucco keratosis, and dermatosis papulosa nigra in clinical variation, as well as acanthotic, hyperkeratotic, clonal, reticulated, irritated, and melanoacanthoma subtypes based on histological variation.2,3
Seborrheic keratosis is a tumor of keratinocytic origin. Although genetics, sun exposure,4 and human papillomavirus infection5 are thought to be causative factors, the precise etiology of SK is unknown.1
The histology of SK shows monotonous basaloid tumor cells without atypia. It generally is comprised of focal acanthosis and papillomatosis with a sharp flat base. Intraepithelial horn pseudocysts are notable features of SK and increased melanin often is seen.2,6
Irritated SK is a histologic variant of SK that has been mechanically or chemically irritated or is involved in immunologic responses. Histologically, the dermis underlying an SK lesion filled with a dense lymphocytic infiltration is characteristic.1,2
For symptomatic or cosmetically undesirable lesions, complete removal of the lesion is the preferred treatment. Cryotherapy, electrodesiccation followed by curettage, curettage followed by desiccation, laser ablation, and surgical excision are effective treatments.1
- Valencia DT, Nicholas RS, Ken KL, et al. Benign epithelial tumors, hamartomas, and hyperplasias. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Professional; 2012:1319-1336.
- Kirkharn N. Tumors and cysts of the epidermis. In: Elder DE, Elenitsas R, Johnson BL Jr, eds. Lever’s Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:791-850.
- Rajesh G, Thappa DM, Jaisankar TJ, et al. Spectrum of seborrheic keratoses in South Indians: a clinical and dermoscopic study. Indian J Dermatol Venereol Leprol. 2011;77:483-488.
- Yeatman JM, Kilkenny M, Marks R. The prevalence of seborrhoeic keratoses in an Australian population: does exposure to sunlight play a part in their frequency? Br J Dermatol. 1997;137:411-414.
- Li YH, Chen G, Dong XP, et al. Detection of epidermodysplasia verruciformis-associated human papillomavirus DNA in nongenital seborrhoeic keratosis. Br J Dermatol. 2004;151:1060-1065.
- Brinster NK, Liu V, Diwan AH, et al. Dermatopathology. Philadelphia, PA: Saunders/Elsevier; 2011.
- Valencia DT, Nicholas RS, Ken KL, et al. Benign epithelial tumors, hamartomas, and hyperplasias. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Professional; 2012:1319-1336.
- Kirkharn N. Tumors and cysts of the epidermis. In: Elder DE, Elenitsas R, Johnson BL Jr, eds. Lever’s Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:791-850.
- Rajesh G, Thappa DM, Jaisankar TJ, et al. Spectrum of seborrheic keratoses in South Indians: a clinical and dermoscopic study. Indian J Dermatol Venereol Leprol. 2011;77:483-488.
- Yeatman JM, Kilkenny M, Marks R. The prevalence of seborrhoeic keratoses in an Australian population: does exposure to sunlight play a part in their frequency? Br J Dermatol. 1997;137:411-414.
- Li YH, Chen G, Dong XP, et al. Detection of epidermodysplasia verruciformis-associated human papillomavirus DNA in nongenital seborrhoeic keratosis. Br J Dermatol. 2004;151:1060-1065.
- Brinster NK, Liu V, Diwan AH, et al. Dermatopathology. Philadelphia, PA: Saunders/Elsevier; 2011.
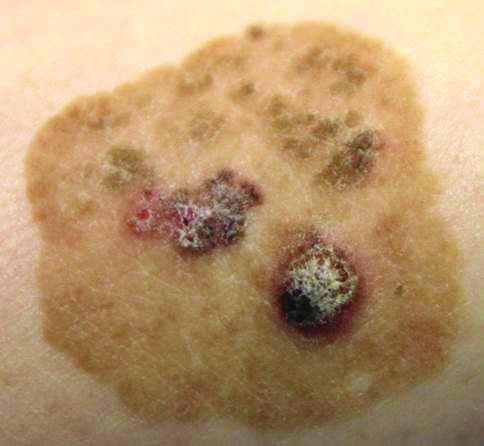
Lichen Planus Pemphigoides Associated With Pregnancy Mimicking Pemphigoid Gestationis
Case Report
A 25-year-old woman with a 5-month history of severe lichen planus (LP) on the arms, legs, and trunk presented to the emergency department with generalized blisters and erythema over the entire body, including the face and soles, of 2 days’ duration. She was evaluated for the LP 1 week prior in a referral dermatology clinic, and in addition to topical corticosteroids, she received 1 injection of 40 mg intramuscular triamcinolone acetonide. Hours following the injection she developed nausea, vomiting, and fever. The patient reported that her last menstrual period was 3 weeks prior to the current presentation.
Physical examination revealed numerous lichenified, flat-topped, pink-violaceous, hyperpigmented, scaly papules and plaques (Figure 1), as well as tense, yellow, fluid-filled vesicles and bullae of various sizes on the neck, arms (Figure 2), legs, trunk, and dorsal aspect of the feet. The vesicles occurred on both normal skin and the lichenified plaques with a negative Nikolsky sign. There also were urticarial erythematous papules and plaques on the arms, trunk, neck, and face, some of which had vesicles or a violaceous dusky central hue (Figure 3). Vesicles were noted within both nostrils (nasal mucosa), and there were extremely tender erythematous patches and thick sheets of scales on the soles.
An elevated β human chorionic gonadotropin level and transvaginal ultrasonography confirmed an intrauterine pregnancy of 12 weeks’ gestation despite the patient’s report of the last menstrual period.
Histologic examination of a vesicle on the right arm revealed hyperkeratosis with hypergranulosis, vacuolar alteration of the basal layer with a paucicellular subepidermal vesicle, and melanophages in the superficial dermis consistent with vesicular LP (Figure 4). Histologic examination of an erythematous edematous plaque on the right upper leg revealed edema in the upper dermis with a perivascular and interstitial lymphocytic infiltrate with eosinophils. A third biopsy of a lichenoid flat-topped papule on the left arm revealed a mild bandlike infiltrate of lymphocytes and scattered eosinophils, eosinophilic colloid bodies and edema in the papillary dermis, and subepidermal vesicles and vacuolar alteration of the basal layer consistent with a vesicular lichenoid dermatitis (Figure 5). Direct immunofluorescence (DIF) of perilesional skin showed linear deposition of C3 and IgM along the basement membrane zone (BMZ) in addition to a shaggy pattern with cytoid bodies (Figure 6). There also was a faint linear deposit of IgA along the BMZ with cytoid bodies but negative for IgG. These results were interpreted as consistent with LP pemphigoides (LPP). Neither an enzyme-linked immunosorbent assay nor an immunoblot analysis was performed.
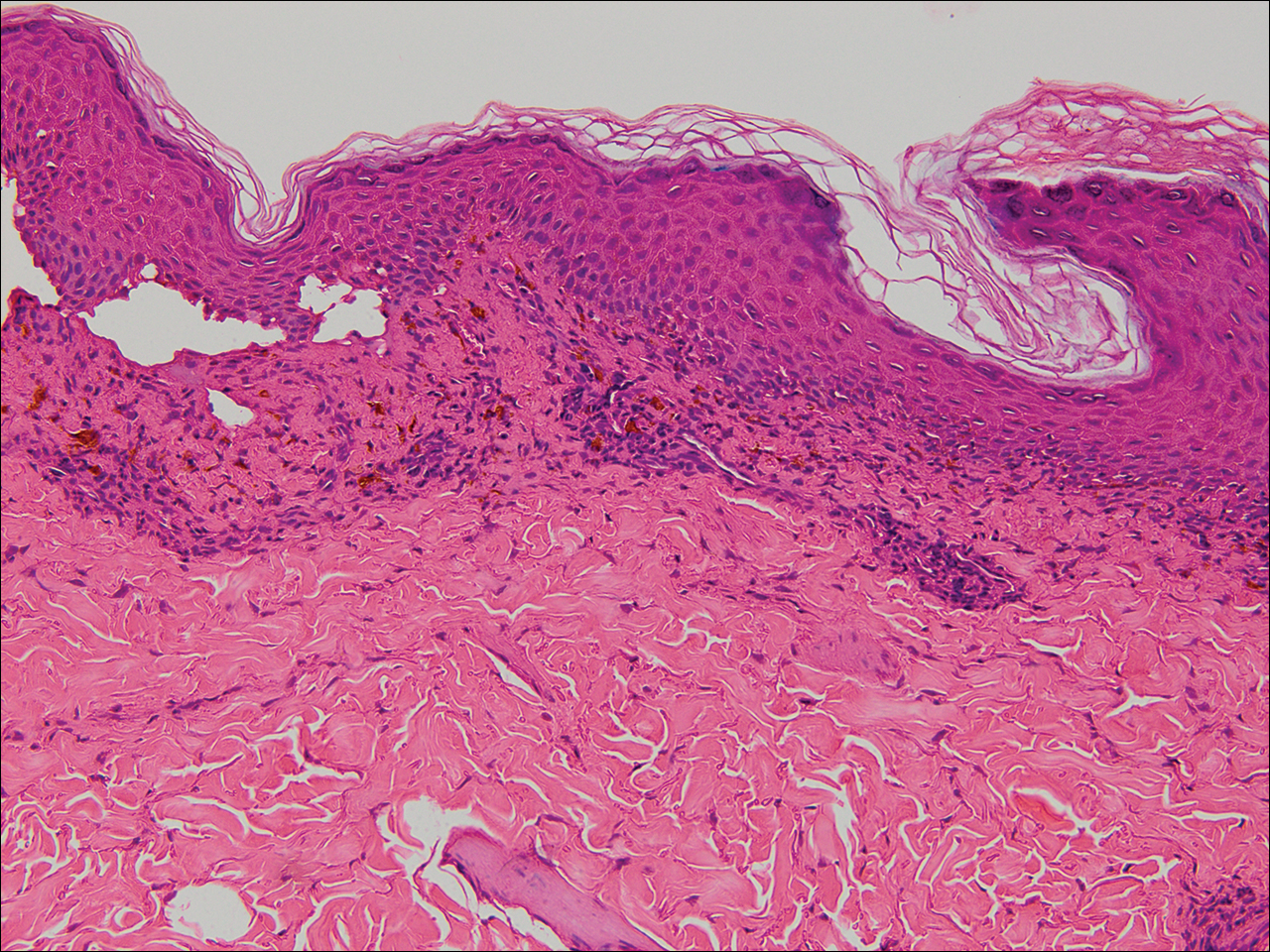
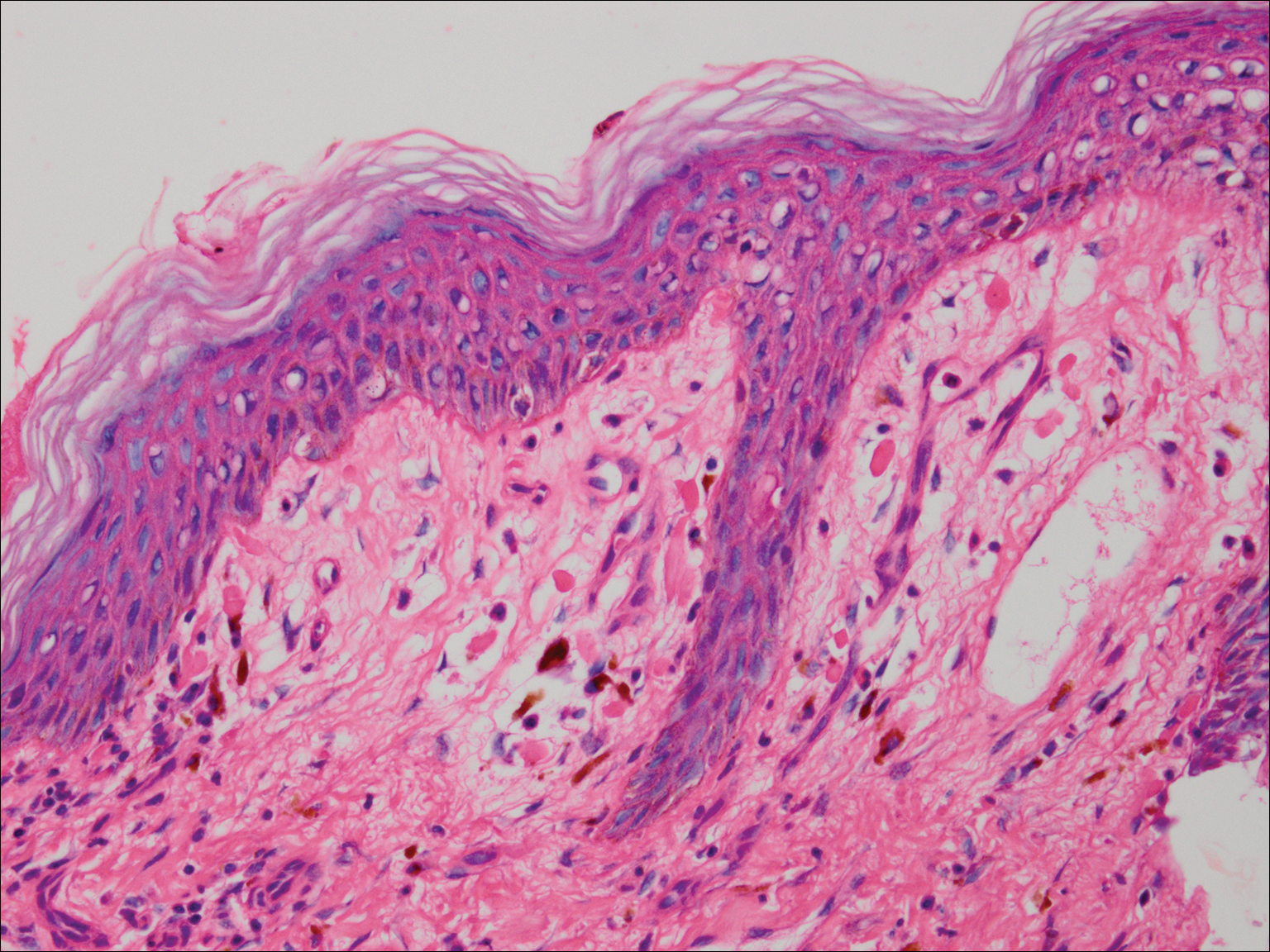
Because the patient was pregnant and had failed to respond to topical and intramuscular corticosteroids, she was started on intravenous methylprednisolone in the emergency department until new lesions stopped appearing. She was then discharged home on oral prednisone 50 mg (0.5 mg/kg/d), with close observation by her obstetrician. She also used clobetasol propionate ointment 0.05% for more severe lesions and triamcinolone acetonide cream 0.1% for less severe lesions until lesions resolved.
During treatment, the patient developed cellulitis on the leg that presented as pustules and erythema at a site of an eroded bulla, inframammary and axillary cutaneous candidiasis, and hyperglycemia at 19 weeks’ gestation. The cutaneous infections resolved with oral clindamycin 300 mg 3 times daily for 10 days. Topical mupirocin was used to treat the cellulitis and a mixture of zinc oxide, econazole cream, and desonide cream twice daily treated the candidiasis. Her obstetrician managed the hyperglycemia.
The bullous lesions and LP completely resolved after 2 months of treatment with oral prednisone 50 mg daily. The patient tolerated a corticosteroid taper (dose decreased by 5 mg every 2 weeks) until arriving at 10 mg, which was then decreased to 7.5 mg until delivery. A cesarean delivery was performed due to a large-for-gestational-age fetus, and an internist was consulted for the necessary precautions to increase the steroid dose during delivery due to the stress of the surgery and the risk for a hypothalamic crisis. There were no peripartum complications, and the baby was born without cutaneous lesions and remains healthy 1 year later. The patient remained disease free over 2 months postpartum, until new LP lesions developed without vesicles or bullae, which were then controlled with topical therapy. She was subsequently lost to follow-up.
Comment
Kaposi first described LPP in 1892 and used the term lichen ruber pemphigoides to describe a case of typical LP together with a widespread bullous eruption. Lichen planus pemphigoides is characterized by tense blisters that arise on lesions of LP as well as on skin unaffected by LP. In contrast, bullous LP blisters are confined to LP lesions only and occur from intense lichenoid inflammation and extensive liquefactive degeneration of basal keratinocytes. The vesicle formation in LPP is a result of autoantibodies to the bullous pemphigoid (BP) antigen BPAg2, which can be explained by the epitope spreading epiphenomenon whereby epidermotropic cytotoxic T cells damage the basal keratinocytes in LP by targeting unknown epidermal antigens, resulting in the exposure of BP180 and therefore instigating the autoimmune response.1 The process of epitope spreading takes months to develop; the mean duration of LP before LPP is 8 weeks in children and 12 weeks in adults,2 which is comparable to the current case.
Pathogenesis
Lichen planus pemphigoides usually is idiopathic; however, there have been cases reported in association with various medications including calcium channel blockers such as diltiazem, Chinese herbs,3 simvastatin,4 ramipril,5,6 captopril,7 psoralen plus UVA phototherapy,8 and cinnarizine.9 In addition, in a case-controlled study, the use of neuroleptics or diuretics was found to be a risk factor for LPP development.10
This case is unique because it shows an association of LPP with an intrauterine pregnancy. Despite the fact that we did not perform the required studies to determine the exact cause, there probably exists an association between LPP and the pregnancy, as the patient presented with a 5-month history of severe LP prior to vesicle formation. The patient only developed the vesicular lesions during pregnancy, which were later controlled with systemic steroids and then recurred postpartum only as LP lesions, suggesting that the patient’s pregnancy may have contributed in the pathogenesis as an inducing factor. We suspect that the LP was aggravated by the pregnancy and continued to worsen, so much as to cause epitope spreading and lead to the bullous eruption at the end of the first trimester.2
Differential Diagnosis
Initially, we suspected a diagnosis of pemphigoid gestationis (PG), previously known as herpes gestationis. The classic presentation of PG starts with an intense pruritus followed by the emergence of pruritic urticarial papules and plaques in the umbilical or periumbilical areas. The lesions may become targetlike or polycyclic and may spread to other areas of the trunk, arms, and legs, often including the palms and soles.11-15 Just as in our case, vesicles and bullous lesions appear at both the site of the urticarial plaques as well as on normal skin.16 The clinical features noted in our patient that were not typical of PG included the multiple lesions on the face and inside the nostrils. Only 20% of PG cases are associated with mucosal involvement,11,12,15 and there are no documented reports of PG occurring in a patient with LP, according to a PubMed search of articles indexed for MEDLINE using the search terms pemphigoid gestationis, herpes gestationis, and lichen planus.
Lichen planus pemphigoides can be easily differentiated from BP. Lichen planus pemphigoides occurs in younger patients, with a mean age of 35 years, unlike BP, which commonly affects elderly men.17 Lichen planus pemphigoides also is less severe and has a better response to treatment than BP. It also affects the palms and soles, which are rarely affected in BP. There are no reports in the literature of BP developing during pregnancy, according to a PubMed search using the terms bullous pemphigoid and pregnancy. However, LPP and BP share a common antibody, the BP180 antigen, and differences exist in the epitope where the antibody binds in each condition.18,19
Diagnosing LPP
In LPP, DIF typically shows linear deposits of IgG, IgM, IgA, fibrinogen, and C3 along the BMZ, of which IgG and C3 are most commonly seen.3 Our patient had linear deposition of C3, IgM, and IgA along the BMZ, which excluded bullous LP from the differential diagnosis. Bullous LP is not an autoimmune condition but rather is on the severe spectrum of LP where Max Joseph spaces become so large so as to lead to vesicle and bullae formation. In addition to the linear deposit at the BMZ, LPP typically reveals immunoglobulin (mainly IgM but also IgA), C3, and fibrinogen staining of colloid bodies in the papillary dermis on DIF; however, some cases of LPP only present with a linear deposition of C3 along the BMZ, which is why, similar to PG, these diagnoses by DIF are similar. Direct immunofluorescence of PG reveals linear IgG1 and IgG3 along the BMZ. IgG1 and IgG3 immunoglobulins are known to fix complement better than other immunoglobulins, thus linear C3 along the BMZ is the most consistently positive immunoreactant. Less common positive immunoreactivity with the same pattern has been seen with IgA, IgM, C1, and C4 (Table).14,15,18 The lack of linear IgG and the presence of IgM is more suggestive of LPP.
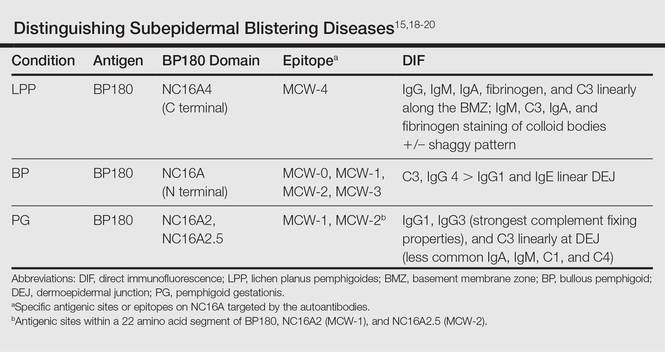
The differential diagnosis of the subepidermal autoimmune blistering diseases associated with antibodies against BP180, including BP, LPP, and PG, often is challenging.15 However, LPP can now be distinguished by immunological studies including immunoblot analysis of the immunodominant region of NC16A of the BP180 antigen and the immunoglobulin subclass that reacts to 180-, 200-,20 and 230-kDa antigens within the BMZ (Table).15,18-20 The Table summarizes the different autoantibodies, antigens, and epitopes to distinguish subepidermal autoimmune blistering diseases.
Despite not performing these studies in our patient, we concluded that the clinical, histological, and DIF findings of this case are more consistent with LPP than with the other subepidermal blistering diseases. However, we cannot exclude the possibility of the patient having a new entity with a unique antibody from epitope spreading.
Conclusion
We present a case of lichenoid papules and plaques consistent with LP, with the development of vesicles and bullae after the first trimester of pregnancy. The clinical, pathologic, and DIF findings were highly suggestive of LPP. Although the exact pathogenic mechanism is not fully known, we suspect that pregnancy may have contributed to the origin of the disease. Further evaluation of pregnant patients with lichenoid lesions who develop blisters are needed for the elucidation of the mechanism, which may be secondary to epitope spreading that led to new autoantibody formation.
- Stingl G, Holubar K. Coexistence of lichen planus and bullous pemphigoid. an immunopathological study. Br J Dermatol. 1975;93:313-320.
- Paige DG, Bhogal BS, Black MM, et al. Lichen planus pemphigoides in a child—immunopathological findings. Clin Exp Dermatol. 1993;18:552-554.
- Xu HH, Xiao T, He CD, et al. Lichen planus pemphigoides associated with Chinese herbs. Clin Exp Dermatol. 2009;34:329-332.
- Stoebner PE, Michot C, Ligeron C, et al. Simvastatin induced lichen planus pemphigoides. Ann Dermatol Venereol. 2003;130:187-190.
- Zhu YI, Fitzpatrick JE, Kornfeld BW. Lichen planus pemphigoides associated with Ramipril. Int J Dermatol. 2006;45:1453-1455.
- Ogg GS, Bhogal BS, Hashimoto T, et al. Ramipril-associated lichen planus pemphigoides. Br J Dermatol. 1997;136:412-414.
- Flageul B, Foldes C, Wallach D, et al. Captopril-induced lichen planus pemphigoides with pemphigus-like features. a case report. Dermatologica. 1986;173:248-255.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Miyagawa S, Ohi H, Muramatsu T, et al. Lichen planus pemphigoides-like lesions induced by Cinnarizine. Br J Dermatol. 1985;112:607-613.
- Bastuji-Garin S, Joly P, Picard-Dahan C, et al. Drugs associated with bullous pemphigoid. a case-control study. Arch Dermatol. 1996;132:272-276.
- Ambros-Rudolph CM. Dermatoses of pregnancy-clues to diagnosis, fetal risk and therapy. Ann Dermatol. 2011;23:265-275.
- DiZenzo G, Calabresi V, Grosso F, et al. The intracellular and extracellular domains of BP180 antigen comprise novel epitopes targeted by pemphigoid gestationis autoantibodies. J Invest Dermatol. 2006;127:864-873.
- Jenkis RE, Hern S, Black MM. Clinical features and management of 87 patients with pemphigus gestationis. Clin Exp Dermatol. 1999;24:255-259.
- Kasperkiewicz M, Zillikens D, Schmidt E. Pemphigoid diseases: pathogenesis, diagnosis, and treatment. Autoimmunity. 2012;45:55-70.
- Cobo MF, Santi CG, Maruta CW, et al. Pemphigoid gestationis: clinical and laboratory evaluation. Clinics. 2009;64:1042-1047.
- Hsu S, Ghohestani RF, Uitto J. Lichen planus pemphigoides with IgG autoantibodies to the 180 kd bullous pemphigoid antigen (type XVII collagen). J Am Acad Dermatol. 2000;42:136-141.
- Harjai B, Mendiratta V, Kakkar S, et al. Childhood lichen planus pemphigoides—a rare entity. J Eur Acad Dermatol Venereol. 2006;20:117-118.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Zillikens D. BP180 as the common autoantigen in blistering diseases with different clinical phenotypes. Keio J Med. 2002;51:21-28.
- Davis AL, Bhogal BS, Whitehead P, et al. Lichen planus pemphigoides: its relationship to bullous pemphigoid. Br J Dermatol. 1991;125:263-271.
Case Report
A 25-year-old woman with a 5-month history of severe lichen planus (LP) on the arms, legs, and trunk presented to the emergency department with generalized blisters and erythema over the entire body, including the face and soles, of 2 days’ duration. She was evaluated for the LP 1 week prior in a referral dermatology clinic, and in addition to topical corticosteroids, she received 1 injection of 40 mg intramuscular triamcinolone acetonide. Hours following the injection she developed nausea, vomiting, and fever. The patient reported that her last menstrual period was 3 weeks prior to the current presentation.
Physical examination revealed numerous lichenified, flat-topped, pink-violaceous, hyperpigmented, scaly papules and plaques (Figure 1), as well as tense, yellow, fluid-filled vesicles and bullae of various sizes on the neck, arms (Figure 2), legs, trunk, and dorsal aspect of the feet. The vesicles occurred on both normal skin and the lichenified plaques with a negative Nikolsky sign. There also were urticarial erythematous papules and plaques on the arms, trunk, neck, and face, some of which had vesicles or a violaceous dusky central hue (Figure 3). Vesicles were noted within both nostrils (nasal mucosa), and there were extremely tender erythematous patches and thick sheets of scales on the soles.
An elevated β human chorionic gonadotropin level and transvaginal ultrasonography confirmed an intrauterine pregnancy of 12 weeks’ gestation despite the patient’s report of the last menstrual period.
Histologic examination of a vesicle on the right arm revealed hyperkeratosis with hypergranulosis, vacuolar alteration of the basal layer with a paucicellular subepidermal vesicle, and melanophages in the superficial dermis consistent with vesicular LP (Figure 4). Histologic examination of an erythematous edematous plaque on the right upper leg revealed edema in the upper dermis with a perivascular and interstitial lymphocytic infiltrate with eosinophils. A third biopsy of a lichenoid flat-topped papule on the left arm revealed a mild bandlike infiltrate of lymphocytes and scattered eosinophils, eosinophilic colloid bodies and edema in the papillary dermis, and subepidermal vesicles and vacuolar alteration of the basal layer consistent with a vesicular lichenoid dermatitis (Figure 5). Direct immunofluorescence (DIF) of perilesional skin showed linear deposition of C3 and IgM along the basement membrane zone (BMZ) in addition to a shaggy pattern with cytoid bodies (Figure 6). There also was a faint linear deposit of IgA along the BMZ with cytoid bodies but negative for IgG. These results were interpreted as consistent with LP pemphigoides (LPP). Neither an enzyme-linked immunosorbent assay nor an immunoblot analysis was performed.
Because the patient was pregnant and had failed to respond to topical and intramuscular corticosteroids, she was started on intravenous methylprednisolone in the emergency department until new lesions stopped appearing. She was then discharged home on oral prednisone 50 mg (0.5 mg/kg/d), with close observation by her obstetrician. She also used clobetasol propionate ointment 0.05% for more severe lesions and triamcinolone acetonide cream 0.1% for less severe lesions until lesions resolved.
During treatment, the patient developed cellulitis on the leg that presented as pustules and erythema at a site of an eroded bulla, inframammary and axillary cutaneous candidiasis, and hyperglycemia at 19 weeks’ gestation. The cutaneous infections resolved with oral clindamycin 300 mg 3 times daily for 10 days. Topical mupirocin was used to treat the cellulitis and a mixture of zinc oxide, econazole cream, and desonide cream twice daily treated the candidiasis. Her obstetrician managed the hyperglycemia.
The bullous lesions and LP completely resolved after 2 months of treatment with oral prednisone 50 mg daily. The patient tolerated a corticosteroid taper (dose decreased by 5 mg every 2 weeks) until arriving at 10 mg, which was then decreased to 7.5 mg until delivery. A cesarean delivery was performed due to a large-for-gestational-age fetus, and an internist was consulted for the necessary precautions to increase the steroid dose during delivery due to the stress of the surgery and the risk for a hypothalamic crisis. There were no peripartum complications, and the baby was born without cutaneous lesions and remains healthy 1 year later. The patient remained disease free over 2 months postpartum, until new LP lesions developed without vesicles or bullae, which were then controlled with topical therapy. She was subsequently lost to follow-up.
Comment
Kaposi first described LPP in 1892 and used the term lichen ruber pemphigoides to describe a case of typical LP together with a widespread bullous eruption. Lichen planus pemphigoides is characterized by tense blisters that arise on lesions of LP as well as on skin unaffected by LP. In contrast, bullous LP blisters are confined to LP lesions only and occur from intense lichenoid inflammation and extensive liquefactive degeneration of basal keratinocytes. The vesicle formation in LPP is a result of autoantibodies to the bullous pemphigoid (BP) antigen BPAg2, which can be explained by the epitope spreading epiphenomenon whereby epidermotropic cytotoxic T cells damage the basal keratinocytes in LP by targeting unknown epidermal antigens, resulting in the exposure of BP180 and therefore instigating the autoimmune response.1 The process of epitope spreading takes months to develop; the mean duration of LP before LPP is 8 weeks in children and 12 weeks in adults,2 which is comparable to the current case.
Pathogenesis
Lichen planus pemphigoides usually is idiopathic; however, there have been cases reported in association with various medications including calcium channel blockers such as diltiazem, Chinese herbs,3 simvastatin,4 ramipril,5,6 captopril,7 psoralen plus UVA phototherapy,8 and cinnarizine.9 In addition, in a case-controlled study, the use of neuroleptics or diuretics was found to be a risk factor for LPP development.10
This case is unique because it shows an association of LPP with an intrauterine pregnancy. Despite the fact that we did not perform the required studies to determine the exact cause, there probably exists an association between LPP and the pregnancy, as the patient presented with a 5-month history of severe LP prior to vesicle formation. The patient only developed the vesicular lesions during pregnancy, which were later controlled with systemic steroids and then recurred postpartum only as LP lesions, suggesting that the patient’s pregnancy may have contributed in the pathogenesis as an inducing factor. We suspect that the LP was aggravated by the pregnancy and continued to worsen, so much as to cause epitope spreading and lead to the bullous eruption at the end of the first trimester.2
Differential Diagnosis
Initially, we suspected a diagnosis of pemphigoid gestationis (PG), previously known as herpes gestationis. The classic presentation of PG starts with an intense pruritus followed by the emergence of pruritic urticarial papules and plaques in the umbilical or periumbilical areas. The lesions may become targetlike or polycyclic and may spread to other areas of the trunk, arms, and legs, often including the palms and soles.11-15 Just as in our case, vesicles and bullous lesions appear at both the site of the urticarial plaques as well as on normal skin.16 The clinical features noted in our patient that were not typical of PG included the multiple lesions on the face and inside the nostrils. Only 20% of PG cases are associated with mucosal involvement,11,12,15 and there are no documented reports of PG occurring in a patient with LP, according to a PubMed search of articles indexed for MEDLINE using the search terms pemphigoid gestationis, herpes gestationis, and lichen planus.
Lichen planus pemphigoides can be easily differentiated from BP. Lichen planus pemphigoides occurs in younger patients, with a mean age of 35 years, unlike BP, which commonly affects elderly men.17 Lichen planus pemphigoides also is less severe and has a better response to treatment than BP. It also affects the palms and soles, which are rarely affected in BP. There are no reports in the literature of BP developing during pregnancy, according to a PubMed search using the terms bullous pemphigoid and pregnancy. However, LPP and BP share a common antibody, the BP180 antigen, and differences exist in the epitope where the antibody binds in each condition.18,19
Diagnosing LPP
In LPP, DIF typically shows linear deposits of IgG, IgM, IgA, fibrinogen, and C3 along the BMZ, of which IgG and C3 are most commonly seen.3 Our patient had linear deposition of C3, IgM, and IgA along the BMZ, which excluded bullous LP from the differential diagnosis. Bullous LP is not an autoimmune condition but rather is on the severe spectrum of LP where Max Joseph spaces become so large so as to lead to vesicle and bullae formation. In addition to the linear deposit at the BMZ, LPP typically reveals immunoglobulin (mainly IgM but also IgA), C3, and fibrinogen staining of colloid bodies in the papillary dermis on DIF; however, some cases of LPP only present with a linear deposition of C3 along the BMZ, which is why, similar to PG, these diagnoses by DIF are similar. Direct immunofluorescence of PG reveals linear IgG1 and IgG3 along the BMZ. IgG1 and IgG3 immunoglobulins are known to fix complement better than other immunoglobulins, thus linear C3 along the BMZ is the most consistently positive immunoreactant. Less common positive immunoreactivity with the same pattern has been seen with IgA, IgM, C1, and C4 (Table).14,15,18 The lack of linear IgG and the presence of IgM is more suggestive of LPP.
The differential diagnosis of the subepidermal autoimmune blistering diseases associated with antibodies against BP180, including BP, LPP, and PG, often is challenging.15 However, LPP can now be distinguished by immunological studies including immunoblot analysis of the immunodominant region of NC16A of the BP180 antigen and the immunoglobulin subclass that reacts to 180-, 200-,20 and 230-kDa antigens within the BMZ (Table).15,18-20 The Table summarizes the different autoantibodies, antigens, and epitopes to distinguish subepidermal autoimmune blistering diseases.
Despite not performing these studies in our patient, we concluded that the clinical, histological, and DIF findings of this case are more consistent with LPP than with the other subepidermal blistering diseases. However, we cannot exclude the possibility of the patient having a new entity with a unique antibody from epitope spreading.
Conclusion
We present a case of lichenoid papules and plaques consistent with LP, with the development of vesicles and bullae after the first trimester of pregnancy. The clinical, pathologic, and DIF findings were highly suggestive of LPP. Although the exact pathogenic mechanism is not fully known, we suspect that pregnancy may have contributed to the origin of the disease. Further evaluation of pregnant patients with lichenoid lesions who develop blisters are needed for the elucidation of the mechanism, which may be secondary to epitope spreading that led to new autoantibody formation.
Case Report
A 25-year-old woman with a 5-month history of severe lichen planus (LP) on the arms, legs, and trunk presented to the emergency department with generalized blisters and erythema over the entire body, including the face and soles, of 2 days’ duration. She was evaluated for the LP 1 week prior in a referral dermatology clinic, and in addition to topical corticosteroids, she received 1 injection of 40 mg intramuscular triamcinolone acetonide. Hours following the injection she developed nausea, vomiting, and fever. The patient reported that her last menstrual period was 3 weeks prior to the current presentation.
Physical examination revealed numerous lichenified, flat-topped, pink-violaceous, hyperpigmented, scaly papules and plaques (Figure 1), as well as tense, yellow, fluid-filled vesicles and bullae of various sizes on the neck, arms (Figure 2), legs, trunk, and dorsal aspect of the feet. The vesicles occurred on both normal skin and the lichenified plaques with a negative Nikolsky sign. There also were urticarial erythematous papules and plaques on the arms, trunk, neck, and face, some of which had vesicles or a violaceous dusky central hue (Figure 3). Vesicles were noted within both nostrils (nasal mucosa), and there were extremely tender erythematous patches and thick sheets of scales on the soles.
An elevated β human chorionic gonadotropin level and transvaginal ultrasonography confirmed an intrauterine pregnancy of 12 weeks’ gestation despite the patient’s report of the last menstrual period.
Histologic examination of a vesicle on the right arm revealed hyperkeratosis with hypergranulosis, vacuolar alteration of the basal layer with a paucicellular subepidermal vesicle, and melanophages in the superficial dermis consistent with vesicular LP (Figure 4). Histologic examination of an erythematous edematous plaque on the right upper leg revealed edema in the upper dermis with a perivascular and interstitial lymphocytic infiltrate with eosinophils. A third biopsy of a lichenoid flat-topped papule on the left arm revealed a mild bandlike infiltrate of lymphocytes and scattered eosinophils, eosinophilic colloid bodies and edema in the papillary dermis, and subepidermal vesicles and vacuolar alteration of the basal layer consistent with a vesicular lichenoid dermatitis (Figure 5). Direct immunofluorescence (DIF) of perilesional skin showed linear deposition of C3 and IgM along the basement membrane zone (BMZ) in addition to a shaggy pattern with cytoid bodies (Figure 6). There also was a faint linear deposit of IgA along the BMZ with cytoid bodies but negative for IgG. These results were interpreted as consistent with LP pemphigoides (LPP). Neither an enzyme-linked immunosorbent assay nor an immunoblot analysis was performed.
Because the patient was pregnant and had failed to respond to topical and intramuscular corticosteroids, she was started on intravenous methylprednisolone in the emergency department until new lesions stopped appearing. She was then discharged home on oral prednisone 50 mg (0.5 mg/kg/d), with close observation by her obstetrician. She also used clobetasol propionate ointment 0.05% for more severe lesions and triamcinolone acetonide cream 0.1% for less severe lesions until lesions resolved.
During treatment, the patient developed cellulitis on the leg that presented as pustules and erythema at a site of an eroded bulla, inframammary and axillary cutaneous candidiasis, and hyperglycemia at 19 weeks’ gestation. The cutaneous infections resolved with oral clindamycin 300 mg 3 times daily for 10 days. Topical mupirocin was used to treat the cellulitis and a mixture of zinc oxide, econazole cream, and desonide cream twice daily treated the candidiasis. Her obstetrician managed the hyperglycemia.
The bullous lesions and LP completely resolved after 2 months of treatment with oral prednisone 50 mg daily. The patient tolerated a corticosteroid taper (dose decreased by 5 mg every 2 weeks) until arriving at 10 mg, which was then decreased to 7.5 mg until delivery. A cesarean delivery was performed due to a large-for-gestational-age fetus, and an internist was consulted for the necessary precautions to increase the steroid dose during delivery due to the stress of the surgery and the risk for a hypothalamic crisis. There were no peripartum complications, and the baby was born without cutaneous lesions and remains healthy 1 year later. The patient remained disease free over 2 months postpartum, until new LP lesions developed without vesicles or bullae, which were then controlled with topical therapy. She was subsequently lost to follow-up.
Comment
Kaposi first described LPP in 1892 and used the term lichen ruber pemphigoides to describe a case of typical LP together with a widespread bullous eruption. Lichen planus pemphigoides is characterized by tense blisters that arise on lesions of LP as well as on skin unaffected by LP. In contrast, bullous LP blisters are confined to LP lesions only and occur from intense lichenoid inflammation and extensive liquefactive degeneration of basal keratinocytes. The vesicle formation in LPP is a result of autoantibodies to the bullous pemphigoid (BP) antigen BPAg2, which can be explained by the epitope spreading epiphenomenon whereby epidermotropic cytotoxic T cells damage the basal keratinocytes in LP by targeting unknown epidermal antigens, resulting in the exposure of BP180 and therefore instigating the autoimmune response.1 The process of epitope spreading takes months to develop; the mean duration of LP before LPP is 8 weeks in children and 12 weeks in adults,2 which is comparable to the current case.
Pathogenesis
Lichen planus pemphigoides usually is idiopathic; however, there have been cases reported in association with various medications including calcium channel blockers such as diltiazem, Chinese herbs,3 simvastatin,4 ramipril,5,6 captopril,7 psoralen plus UVA phototherapy,8 and cinnarizine.9 In addition, in a case-controlled study, the use of neuroleptics or diuretics was found to be a risk factor for LPP development.10
This case is unique because it shows an association of LPP with an intrauterine pregnancy. Despite the fact that we did not perform the required studies to determine the exact cause, there probably exists an association between LPP and the pregnancy, as the patient presented with a 5-month history of severe LP prior to vesicle formation. The patient only developed the vesicular lesions during pregnancy, which were later controlled with systemic steroids and then recurred postpartum only as LP lesions, suggesting that the patient’s pregnancy may have contributed in the pathogenesis as an inducing factor. We suspect that the LP was aggravated by the pregnancy and continued to worsen, so much as to cause epitope spreading and lead to the bullous eruption at the end of the first trimester.2
Differential Diagnosis
Initially, we suspected a diagnosis of pemphigoid gestationis (PG), previously known as herpes gestationis. The classic presentation of PG starts with an intense pruritus followed by the emergence of pruritic urticarial papules and plaques in the umbilical or periumbilical areas. The lesions may become targetlike or polycyclic and may spread to other areas of the trunk, arms, and legs, often including the palms and soles.11-15 Just as in our case, vesicles and bullous lesions appear at both the site of the urticarial plaques as well as on normal skin.16 The clinical features noted in our patient that were not typical of PG included the multiple lesions on the face and inside the nostrils. Only 20% of PG cases are associated with mucosal involvement,11,12,15 and there are no documented reports of PG occurring in a patient with LP, according to a PubMed search of articles indexed for MEDLINE using the search terms pemphigoid gestationis, herpes gestationis, and lichen planus.
Lichen planus pemphigoides can be easily differentiated from BP. Lichen planus pemphigoides occurs in younger patients, with a mean age of 35 years, unlike BP, which commonly affects elderly men.17 Lichen planus pemphigoides also is less severe and has a better response to treatment than BP. It also affects the palms and soles, which are rarely affected in BP. There are no reports in the literature of BP developing during pregnancy, according to a PubMed search using the terms bullous pemphigoid and pregnancy. However, LPP and BP share a common antibody, the BP180 antigen, and differences exist in the epitope where the antibody binds in each condition.18,19
Diagnosing LPP
In LPP, DIF typically shows linear deposits of IgG, IgM, IgA, fibrinogen, and C3 along the BMZ, of which IgG and C3 are most commonly seen.3 Our patient had linear deposition of C3, IgM, and IgA along the BMZ, which excluded bullous LP from the differential diagnosis. Bullous LP is not an autoimmune condition but rather is on the severe spectrum of LP where Max Joseph spaces become so large so as to lead to vesicle and bullae formation. In addition to the linear deposit at the BMZ, LPP typically reveals immunoglobulin (mainly IgM but also IgA), C3, and fibrinogen staining of colloid bodies in the papillary dermis on DIF; however, some cases of LPP only present with a linear deposition of C3 along the BMZ, which is why, similar to PG, these diagnoses by DIF are similar. Direct immunofluorescence of PG reveals linear IgG1 and IgG3 along the BMZ. IgG1 and IgG3 immunoglobulins are known to fix complement better than other immunoglobulins, thus linear C3 along the BMZ is the most consistently positive immunoreactant. Less common positive immunoreactivity with the same pattern has been seen with IgA, IgM, C1, and C4 (Table).14,15,18 The lack of linear IgG and the presence of IgM is more suggestive of LPP.
The differential diagnosis of the subepidermal autoimmune blistering diseases associated with antibodies against BP180, including BP, LPP, and PG, often is challenging.15 However, LPP can now be distinguished by immunological studies including immunoblot analysis of the immunodominant region of NC16A of the BP180 antigen and the immunoglobulin subclass that reacts to 180-, 200-,20 and 230-kDa antigens within the BMZ (Table).15,18-20 The Table summarizes the different autoantibodies, antigens, and epitopes to distinguish subepidermal autoimmune blistering diseases.
Despite not performing these studies in our patient, we concluded that the clinical, histological, and DIF findings of this case are more consistent with LPP than with the other subepidermal blistering diseases. However, we cannot exclude the possibility of the patient having a new entity with a unique antibody from epitope spreading.
Conclusion
We present a case of lichenoid papules and plaques consistent with LP, with the development of vesicles and bullae after the first trimester of pregnancy. The clinical, pathologic, and DIF findings were highly suggestive of LPP. Although the exact pathogenic mechanism is not fully known, we suspect that pregnancy may have contributed to the origin of the disease. Further evaluation of pregnant patients with lichenoid lesions who develop blisters are needed for the elucidation of the mechanism, which may be secondary to epitope spreading that led to new autoantibody formation.
- Stingl G, Holubar K. Coexistence of lichen planus and bullous pemphigoid. an immunopathological study. Br J Dermatol. 1975;93:313-320.
- Paige DG, Bhogal BS, Black MM, et al. Lichen planus pemphigoides in a child—immunopathological findings. Clin Exp Dermatol. 1993;18:552-554.
- Xu HH, Xiao T, He CD, et al. Lichen planus pemphigoides associated with Chinese herbs. Clin Exp Dermatol. 2009;34:329-332.
- Stoebner PE, Michot C, Ligeron C, et al. Simvastatin induced lichen planus pemphigoides. Ann Dermatol Venereol. 2003;130:187-190.
- Zhu YI, Fitzpatrick JE, Kornfeld BW. Lichen planus pemphigoides associated with Ramipril. Int J Dermatol. 2006;45:1453-1455.
- Ogg GS, Bhogal BS, Hashimoto T, et al. Ramipril-associated lichen planus pemphigoides. Br J Dermatol. 1997;136:412-414.
- Flageul B, Foldes C, Wallach D, et al. Captopril-induced lichen planus pemphigoides with pemphigus-like features. a case report. Dermatologica. 1986;173:248-255.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Miyagawa S, Ohi H, Muramatsu T, et al. Lichen planus pemphigoides-like lesions induced by Cinnarizine. Br J Dermatol. 1985;112:607-613.
- Bastuji-Garin S, Joly P, Picard-Dahan C, et al. Drugs associated with bullous pemphigoid. a case-control study. Arch Dermatol. 1996;132:272-276.
- Ambros-Rudolph CM. Dermatoses of pregnancy-clues to diagnosis, fetal risk and therapy. Ann Dermatol. 2011;23:265-275.
- DiZenzo G, Calabresi V, Grosso F, et al. The intracellular and extracellular domains of BP180 antigen comprise novel epitopes targeted by pemphigoid gestationis autoantibodies. J Invest Dermatol. 2006;127:864-873.
- Jenkis RE, Hern S, Black MM. Clinical features and management of 87 patients with pemphigus gestationis. Clin Exp Dermatol. 1999;24:255-259.
- Kasperkiewicz M, Zillikens D, Schmidt E. Pemphigoid diseases: pathogenesis, diagnosis, and treatment. Autoimmunity. 2012;45:55-70.
- Cobo MF, Santi CG, Maruta CW, et al. Pemphigoid gestationis: clinical and laboratory evaluation. Clinics. 2009;64:1042-1047.
- Hsu S, Ghohestani RF, Uitto J. Lichen planus pemphigoides with IgG autoantibodies to the 180 kd bullous pemphigoid antigen (type XVII collagen). J Am Acad Dermatol. 2000;42:136-141.
- Harjai B, Mendiratta V, Kakkar S, et al. Childhood lichen planus pemphigoides—a rare entity. J Eur Acad Dermatol Venereol. 2006;20:117-118.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Zillikens D. BP180 as the common autoantigen in blistering diseases with different clinical phenotypes. Keio J Med. 2002;51:21-28.
- Davis AL, Bhogal BS, Whitehead P, et al. Lichen planus pemphigoides: its relationship to bullous pemphigoid. Br J Dermatol. 1991;125:263-271.
- Stingl G, Holubar K. Coexistence of lichen planus and bullous pemphigoid. an immunopathological study. Br J Dermatol. 1975;93:313-320.
- Paige DG, Bhogal BS, Black MM, et al. Lichen planus pemphigoides in a child—immunopathological findings. Clin Exp Dermatol. 1993;18:552-554.
- Xu HH, Xiao T, He CD, et al. Lichen planus pemphigoides associated with Chinese herbs. Clin Exp Dermatol. 2009;34:329-332.
- Stoebner PE, Michot C, Ligeron C, et al. Simvastatin induced lichen planus pemphigoides. Ann Dermatol Venereol. 2003;130:187-190.
- Zhu YI, Fitzpatrick JE, Kornfeld BW. Lichen planus pemphigoides associated with Ramipril. Int J Dermatol. 2006;45:1453-1455.
- Ogg GS, Bhogal BS, Hashimoto T, et al. Ramipril-associated lichen planus pemphigoides. Br J Dermatol. 1997;136:412-414.
- Flageul B, Foldes C, Wallach D, et al. Captopril-induced lichen planus pemphigoides with pemphigus-like features. a case report. Dermatologica. 1986;173:248-255.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Miyagawa S, Ohi H, Muramatsu T, et al. Lichen planus pemphigoides-like lesions induced by Cinnarizine. Br J Dermatol. 1985;112:607-613.
- Bastuji-Garin S, Joly P, Picard-Dahan C, et al. Drugs associated with bullous pemphigoid. a case-control study. Arch Dermatol. 1996;132:272-276.
- Ambros-Rudolph CM. Dermatoses of pregnancy-clues to diagnosis, fetal risk and therapy. Ann Dermatol. 2011;23:265-275.
- DiZenzo G, Calabresi V, Grosso F, et al. The intracellular and extracellular domains of BP180 antigen comprise novel epitopes targeted by pemphigoid gestationis autoantibodies. J Invest Dermatol. 2006;127:864-873.
- Jenkis RE, Hern S, Black MM. Clinical features and management of 87 patients with pemphigus gestationis. Clin Exp Dermatol. 1999;24:255-259.
- Kasperkiewicz M, Zillikens D, Schmidt E. Pemphigoid diseases: pathogenesis, diagnosis, and treatment. Autoimmunity. 2012;45:55-70.
- Cobo MF, Santi CG, Maruta CW, et al. Pemphigoid gestationis: clinical and laboratory evaluation. Clinics. 2009;64:1042-1047.
- Hsu S, Ghohestani RF, Uitto J. Lichen planus pemphigoides with IgG autoantibodies to the 180 kd bullous pemphigoid antigen (type XVII collagen). J Am Acad Dermatol. 2000;42:136-141.
- Harjai B, Mendiratta V, Kakkar S, et al. Childhood lichen planus pemphigoides—a rare entity. J Eur Acad Dermatol Venereol. 2006;20:117-118.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Zillikens D. BP180 as the common autoantigen in blistering diseases with different clinical phenotypes. Keio J Med. 2002;51:21-28.
- Davis AL, Bhogal BS, Whitehead P, et al. Lichen planus pemphigoides: its relationship to bullous pemphigoid. Br J Dermatol. 1991;125:263-271.
Practice Points
- Lichen planus pemphigoides (LPP) is characterized by tense blisters that arise not only on lichen planus lesions such as bullous lichen planus but also on skin unaffected by lichen planus.
- In LPP, the autoantibodies specifically target the MCW-4 epitope of the NC16A4 domain of the bullous pemphigoid antigen BPAg2, distinguishing it from other autoimmune blistering diseases against the NC16A domain.
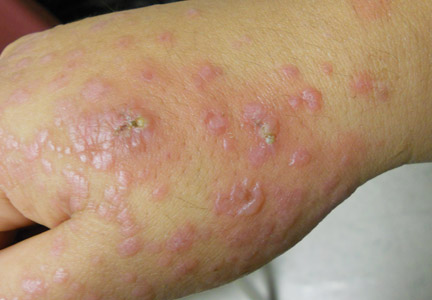
Pemphigus Vulgaris Successfully Treated With Doxycycline Monotherapy
To the Editor:
Pemphigus vulgaris (PV) is an acquired autoimmune bullous disease with notable morbidity and mortality if not treated appropriately due to loss of epidermal barrier function and subsequent infection and loss of body fluids. Although the use of systemic corticosteroids and immunosuppressive agents has improved the prognosis, these drugs also may have severe adverse effects, especially in elderly patients. Hence, alternative and safer therapies with anti-inflammatory and immunomodulatory agents such as tetracyclines and nicotinamide have been used with variable results. We report a case of PV that was successfully treated with doxycycline.
An 81-year-old man presented with well-demarcated erosions with overlying yellow crust as well as vesicles and pustules on the scalp (Figure 1A), forehead, bilateral cheeks, and upper back (Figure 1B) of 6 months’ duration. He used topical fluorouracil in the month prior to presentation for suspected actinic keratosis but had stopped its use after 2 weeks. At the first visit, a diagnosis of a reaction to topical fluorouracil with secondary bacterial infection was made and he was prescribed doxycycline hyclate 100 mg twice daily. The patient returned 4 weeks later for follow-up and reported initial notable improvement with subsequent worsening of lesions after he ran out of doxycycline. On physical examination the lesions had considerably improved from the last visit, but he still had a few erosions on the scalp and a few in the oral mucosa. A 1-cm shallow erosion with minimal surrounding erythema on the forehead was present, along with fewer scattered, edematous, erythematous plaques on the back and chest. Pemphigus vulgaris was suspected and 2 shave biopsies from the lesions on the back and cheek were obtained for confirmation. Histopathologic examination revealed epidermal hyperplasia and suprabasal acantholysis as well as moderate perivascular and perifollicular lymphocytic infiltrate with several eosinophils and plasma cells, characteristic of PV (Figure 2). Direct immunofluorescence showed moderate intercellular deposition of IgG within the basal layer and to a lesser extent within suprabasal layers as well as moderate intercellular deposition of C3 within the basal layer, characteristic of PV. IgA and IgM were not present. Indirect immunofluorescence using monkey esophagus revealed no antibodies against the intercellular space of the basement membrane zone. Due to the dramatic response, he continued on doxycycline 100 mg twice daily and remained in complete remission. Ten months after initiating treatment he discontinued doxycycline for 2 days and developed a 1-cm lesion on the left cheek. He resumed treatment with clearing of lesions and was slowly tapered to 50 mg of doxycycline once daily, remaining in complete remission (Figure 3). Doxycycline was discontinued 16 months after initiation; he has remained clear at 13 weeks.
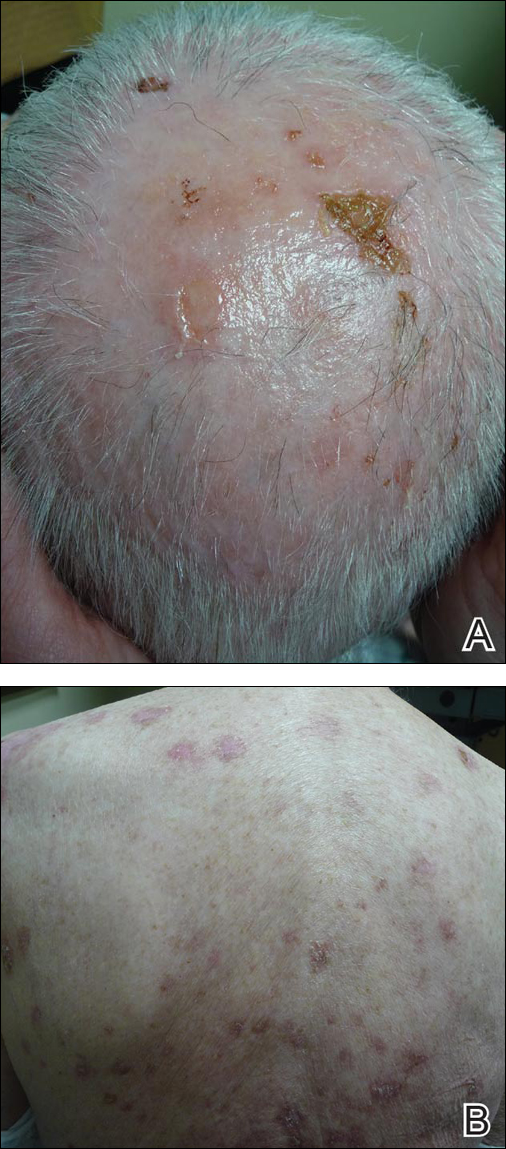
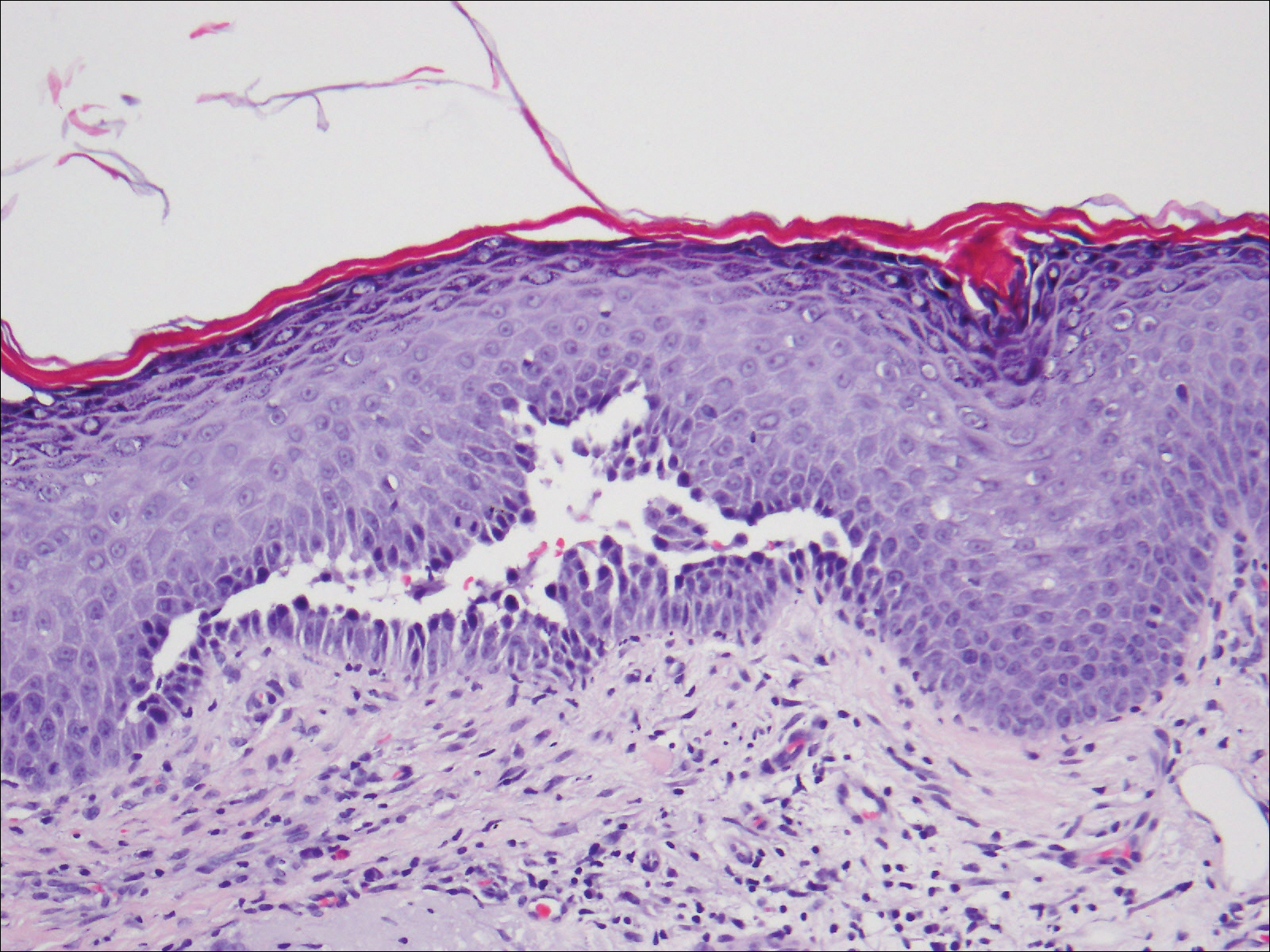
The treatment of PV is challenging given the multiple side effects of steroids, especially in elderly patients. Tetracyclines have an advantageous side-effect profile and they have been shown to be efficacious in treating PV when combined with nicotinamide or when used as adjuvant therapy to steroids.1-3 Our case shows a patient who was treated exclusively with doxycycline and achieved complete remission.
Tetracyclines have multiple biological activities in addition to their antimicrobial function that may provide a therapeutic benefit in PV. They possess immunomodulatory and anti-inflammatory effects by inhibiting leukocyte chemotaxis and activation4-8 and inhibiting cytokine release. They inhibit matrix metalloproteinases, which are the major enzymes responsible for breakdown of the extracellular matrix,9 and they indirectly inhibit neutrophil elastase by protecting α1-protease inhibitor from matrix metalloproteinase degradation.10 Additionally, tetracyclines increase the cohesion of the dermoepidermal junction11; whether they increase the adhesion between epidermal cells is unknown. It has been determined that CD4+ T cells play an essential role in the pathogenesis of PV by promoting anti-desmoglein 3 antibody production.12 Szeto et al13 reported that minocycline, a member of the tetracycline family, has suppressive effects on CD4+ T-cell activation by hindering the activation of nuclear factor of activated T cells (NFAT), a key regulatory factor in T-cell activation. We hypothesize that doxycycline exerted what appears to be immunomodulatory properties in our patient by suppressing CD4+ T-cell activity.
In conclusion, tetracyclines can be an effective and promising therapy for PV given their relatively few side effects and immunomodulating properties. However, further randomized controlled trials will be important to support our conclusion.
- Chaffins ML, Collison D, Fivenson DP. Treatment of pemphigus and linear IgA dermatosis with nicotinamide and tetracycline: a review of 13 cases. J Am Acad Dermatol. 1993;28:998-1000.
- Caelbotta A, Saenz AM, Gonzalez F, et al. Pemphigus vulgaris: benefits of tetracycline as adjuvant therapy in series of thirteen patients. Int J Dermatol. 1999;38:217-221.
- McCarty M, Fivenson D. Two decades of using the combination of tetracycline derivatives and niacinamide as steroid-sparing agents in the management of pemphigus defining a niche for these low toxicity agents. J Am Acad Dermatol. 2014;71:475-479.
- Majeski JA, McClellan MA, Alexander JW. Effect of antibiotics on the in vitro neutrophil chemotactic response. Am Surg. 1976;42:785-788.
- Esterly NB, Furley NL, Flanagan LE. The effect of antimicrobial agents on leukocyte chemotaxis. J Invest Dermatol. 1978;70:51-55.
- Gabler WL, Creamer HR. Suppression of human neutrophil functions by tetracyclines. J Periodontal Res. 1991;26:52-58.
- Esterly NB, Koransky JS, Furey NL, et al. Neutrophil chemotaxis in patients with acne receiving oral tetracycline therapy. Arch Dermatol. 1984;120:1308-1313.
- Sapadin AN, Fleischmajer R. Tetracyclines: nonantibiotic properties and their clinical implications. J Am Acad Dermatol. 2006;54:258-265.
- Monk E, Shalita A, Siegel DM. Clinical applications of non-antimicrobial tetracyclines in dermatology. Pharmacol Res. 2011;63:130-145.
- Golub LM, Evans RT, McNamara TF, et al. A nonantimicrobial tetracycline inhibits gingival matrix metalloproteinases and bone loss in Porphyromonas gingivalis–induced periodontitis in rats. Ann N Y Acad Sci. 1994;732:96-111.
- Humbert P, Treffel P, Chapius JF, et al. The tetracyclines in dermatology. J Am Acad Dermatol. 1991;25:691-697.
- Nishifuji K, Amagai M, Kuwana M, et al. Detection of antigen-specific B cells in patients with pemphigus vulgaris by enzyme-linked immunospot assay: requirement of T cell collaboration for autoantibody production. J Invest Dermatol. 2000;114:88-94.
- Szeto GL, Pomerantz JL, Graham DRM, et al. Minocycline suppresses activation of nuclear factor of activated T cells 1 (NFAT1) in human CD4 T Cells. J Biol Chem. 2011;286:11275-11282.
To the Editor:
Pemphigus vulgaris (PV) is an acquired autoimmune bullous disease with notable morbidity and mortality if not treated appropriately due to loss of epidermal barrier function and subsequent infection and loss of body fluids. Although the use of systemic corticosteroids and immunosuppressive agents has improved the prognosis, these drugs also may have severe adverse effects, especially in elderly patients. Hence, alternative and safer therapies with anti-inflammatory and immunomodulatory agents such as tetracyclines and nicotinamide have been used with variable results. We report a case of PV that was successfully treated with doxycycline.
An 81-year-old man presented with well-demarcated erosions with overlying yellow crust as well as vesicles and pustules on the scalp (Figure 1A), forehead, bilateral cheeks, and upper back (Figure 1B) of 6 months’ duration. He used topical fluorouracil in the month prior to presentation for suspected actinic keratosis but had stopped its use after 2 weeks. At the first visit, a diagnosis of a reaction to topical fluorouracil with secondary bacterial infection was made and he was prescribed doxycycline hyclate 100 mg twice daily. The patient returned 4 weeks later for follow-up and reported initial notable improvement with subsequent worsening of lesions after he ran out of doxycycline. On physical examination the lesions had considerably improved from the last visit, but he still had a few erosions on the scalp and a few in the oral mucosa. A 1-cm shallow erosion with minimal surrounding erythema on the forehead was present, along with fewer scattered, edematous, erythematous plaques on the back and chest. Pemphigus vulgaris was suspected and 2 shave biopsies from the lesions on the back and cheek were obtained for confirmation. Histopathologic examination revealed epidermal hyperplasia and suprabasal acantholysis as well as moderate perivascular and perifollicular lymphocytic infiltrate with several eosinophils and plasma cells, characteristic of PV (Figure 2). Direct immunofluorescence showed moderate intercellular deposition of IgG within the basal layer and to a lesser extent within suprabasal layers as well as moderate intercellular deposition of C3 within the basal layer, characteristic of PV. IgA and IgM were not present. Indirect immunofluorescence using monkey esophagus revealed no antibodies against the intercellular space of the basement membrane zone. Due to the dramatic response, he continued on doxycycline 100 mg twice daily and remained in complete remission. Ten months after initiating treatment he discontinued doxycycline for 2 days and developed a 1-cm lesion on the left cheek. He resumed treatment with clearing of lesions and was slowly tapered to 50 mg of doxycycline once daily, remaining in complete remission (Figure 3). Doxycycline was discontinued 16 months after initiation; he has remained clear at 13 weeks.
The treatment of PV is challenging given the multiple side effects of steroids, especially in elderly patients. Tetracyclines have an advantageous side-effect profile and they have been shown to be efficacious in treating PV when combined with nicotinamide or when used as adjuvant therapy to steroids.1-3 Our case shows a patient who was treated exclusively with doxycycline and achieved complete remission.
Tetracyclines have multiple biological activities in addition to their antimicrobial function that may provide a therapeutic benefit in PV. They possess immunomodulatory and anti-inflammatory effects by inhibiting leukocyte chemotaxis and activation4-8 and inhibiting cytokine release. They inhibit matrix metalloproteinases, which are the major enzymes responsible for breakdown of the extracellular matrix,9 and they indirectly inhibit neutrophil elastase by protecting α1-protease inhibitor from matrix metalloproteinase degradation.10 Additionally, tetracyclines increase the cohesion of the dermoepidermal junction11; whether they increase the adhesion between epidermal cells is unknown. It has been determined that CD4+ T cells play an essential role in the pathogenesis of PV by promoting anti-desmoglein 3 antibody production.12 Szeto et al13 reported that minocycline, a member of the tetracycline family, has suppressive effects on CD4+ T-cell activation by hindering the activation of nuclear factor of activated T cells (NFAT), a key regulatory factor in T-cell activation. We hypothesize that doxycycline exerted what appears to be immunomodulatory properties in our patient by suppressing CD4+ T-cell activity.
In conclusion, tetracyclines can be an effective and promising therapy for PV given their relatively few side effects and immunomodulating properties. However, further randomized controlled trials will be important to support our conclusion.
To the Editor:
Pemphigus vulgaris (PV) is an acquired autoimmune bullous disease with notable morbidity and mortality if not treated appropriately due to loss of epidermal barrier function and subsequent infection and loss of body fluids. Although the use of systemic corticosteroids and immunosuppressive agents has improved the prognosis, these drugs also may have severe adverse effects, especially in elderly patients. Hence, alternative and safer therapies with anti-inflammatory and immunomodulatory agents such as tetracyclines and nicotinamide have been used with variable results. We report a case of PV that was successfully treated with doxycycline.
An 81-year-old man presented with well-demarcated erosions with overlying yellow crust as well as vesicles and pustules on the scalp (Figure 1A), forehead, bilateral cheeks, and upper back (Figure 1B) of 6 months’ duration. He used topical fluorouracil in the month prior to presentation for suspected actinic keratosis but had stopped its use after 2 weeks. At the first visit, a diagnosis of a reaction to topical fluorouracil with secondary bacterial infection was made and he was prescribed doxycycline hyclate 100 mg twice daily. The patient returned 4 weeks later for follow-up and reported initial notable improvement with subsequent worsening of lesions after he ran out of doxycycline. On physical examination the lesions had considerably improved from the last visit, but he still had a few erosions on the scalp and a few in the oral mucosa. A 1-cm shallow erosion with minimal surrounding erythema on the forehead was present, along with fewer scattered, edematous, erythematous plaques on the back and chest. Pemphigus vulgaris was suspected and 2 shave biopsies from the lesions on the back and cheek were obtained for confirmation. Histopathologic examination revealed epidermal hyperplasia and suprabasal acantholysis as well as moderate perivascular and perifollicular lymphocytic infiltrate with several eosinophils and plasma cells, characteristic of PV (Figure 2). Direct immunofluorescence showed moderate intercellular deposition of IgG within the basal layer and to a lesser extent within suprabasal layers as well as moderate intercellular deposition of C3 within the basal layer, characteristic of PV. IgA and IgM were not present. Indirect immunofluorescence using monkey esophagus revealed no antibodies against the intercellular space of the basement membrane zone. Due to the dramatic response, he continued on doxycycline 100 mg twice daily and remained in complete remission. Ten months after initiating treatment he discontinued doxycycline for 2 days and developed a 1-cm lesion on the left cheek. He resumed treatment with clearing of lesions and was slowly tapered to 50 mg of doxycycline once daily, remaining in complete remission (Figure 3). Doxycycline was discontinued 16 months after initiation; he has remained clear at 13 weeks.
The treatment of PV is challenging given the multiple side effects of steroids, especially in elderly patients. Tetracyclines have an advantageous side-effect profile and they have been shown to be efficacious in treating PV when combined with nicotinamide or when used as adjuvant therapy to steroids.1-3 Our case shows a patient who was treated exclusively with doxycycline and achieved complete remission.
Tetracyclines have multiple biological activities in addition to their antimicrobial function that may provide a therapeutic benefit in PV. They possess immunomodulatory and anti-inflammatory effects by inhibiting leukocyte chemotaxis and activation4-8 and inhibiting cytokine release. They inhibit matrix metalloproteinases, which are the major enzymes responsible for breakdown of the extracellular matrix,9 and they indirectly inhibit neutrophil elastase by protecting α1-protease inhibitor from matrix metalloproteinase degradation.10 Additionally, tetracyclines increase the cohesion of the dermoepidermal junction11; whether they increase the adhesion between epidermal cells is unknown. It has been determined that CD4+ T cells play an essential role in the pathogenesis of PV by promoting anti-desmoglein 3 antibody production.12 Szeto et al13 reported that minocycline, a member of the tetracycline family, has suppressive effects on CD4+ T-cell activation by hindering the activation of nuclear factor of activated T cells (NFAT), a key regulatory factor in T-cell activation. We hypothesize that doxycycline exerted what appears to be immunomodulatory properties in our patient by suppressing CD4+ T-cell activity.
In conclusion, tetracyclines can be an effective and promising therapy for PV given their relatively few side effects and immunomodulating properties. However, further randomized controlled trials will be important to support our conclusion.
- Chaffins ML, Collison D, Fivenson DP. Treatment of pemphigus and linear IgA dermatosis with nicotinamide and tetracycline: a review of 13 cases. J Am Acad Dermatol. 1993;28:998-1000.
- Caelbotta A, Saenz AM, Gonzalez F, et al. Pemphigus vulgaris: benefits of tetracycline as adjuvant therapy in series of thirteen patients. Int J Dermatol. 1999;38:217-221.
- McCarty M, Fivenson D. Two decades of using the combination of tetracycline derivatives and niacinamide as steroid-sparing agents in the management of pemphigus defining a niche for these low toxicity agents. J Am Acad Dermatol. 2014;71:475-479.
- Majeski JA, McClellan MA, Alexander JW. Effect of antibiotics on the in vitro neutrophil chemotactic response. Am Surg. 1976;42:785-788.
- Esterly NB, Furley NL, Flanagan LE. The effect of antimicrobial agents on leukocyte chemotaxis. J Invest Dermatol. 1978;70:51-55.
- Gabler WL, Creamer HR. Suppression of human neutrophil functions by tetracyclines. J Periodontal Res. 1991;26:52-58.
- Esterly NB, Koransky JS, Furey NL, et al. Neutrophil chemotaxis in patients with acne receiving oral tetracycline therapy. Arch Dermatol. 1984;120:1308-1313.
- Sapadin AN, Fleischmajer R. Tetracyclines: nonantibiotic properties and their clinical implications. J Am Acad Dermatol. 2006;54:258-265.
- Monk E, Shalita A, Siegel DM. Clinical applications of non-antimicrobial tetracyclines in dermatology. Pharmacol Res. 2011;63:130-145.
- Golub LM, Evans RT, McNamara TF, et al. A nonantimicrobial tetracycline inhibits gingival matrix metalloproteinases and bone loss in Porphyromonas gingivalis–induced periodontitis in rats. Ann N Y Acad Sci. 1994;732:96-111.
- Humbert P, Treffel P, Chapius JF, et al. The tetracyclines in dermatology. J Am Acad Dermatol. 1991;25:691-697.
- Nishifuji K, Amagai M, Kuwana M, et al. Detection of antigen-specific B cells in patients with pemphigus vulgaris by enzyme-linked immunospot assay: requirement of T cell collaboration for autoantibody production. J Invest Dermatol. 2000;114:88-94.
- Szeto GL, Pomerantz JL, Graham DRM, et al. Minocycline suppresses activation of nuclear factor of activated T cells 1 (NFAT1) in human CD4 T Cells. J Biol Chem. 2011;286:11275-11282.
- Chaffins ML, Collison D, Fivenson DP. Treatment of pemphigus and linear IgA dermatosis with nicotinamide and tetracycline: a review of 13 cases. J Am Acad Dermatol. 1993;28:998-1000.
- Caelbotta A, Saenz AM, Gonzalez F, et al. Pemphigus vulgaris: benefits of tetracycline as adjuvant therapy in series of thirteen patients. Int J Dermatol. 1999;38:217-221.
- McCarty M, Fivenson D. Two decades of using the combination of tetracycline derivatives and niacinamide as steroid-sparing agents in the management of pemphigus defining a niche for these low toxicity agents. J Am Acad Dermatol. 2014;71:475-479.
- Majeski JA, McClellan MA, Alexander JW. Effect of antibiotics on the in vitro neutrophil chemotactic response. Am Surg. 1976;42:785-788.
- Esterly NB, Furley NL, Flanagan LE. The effect of antimicrobial agents on leukocyte chemotaxis. J Invest Dermatol. 1978;70:51-55.
- Gabler WL, Creamer HR. Suppression of human neutrophil functions by tetracyclines. J Periodontal Res. 1991;26:52-58.
- Esterly NB, Koransky JS, Furey NL, et al. Neutrophil chemotaxis in patients with acne receiving oral tetracycline therapy. Arch Dermatol. 1984;120:1308-1313.
- Sapadin AN, Fleischmajer R. Tetracyclines: nonantibiotic properties and their clinical implications. J Am Acad Dermatol. 2006;54:258-265.
- Monk E, Shalita A, Siegel DM. Clinical applications of non-antimicrobial tetracyclines in dermatology. Pharmacol Res. 2011;63:130-145.
- Golub LM, Evans RT, McNamara TF, et al. A nonantimicrobial tetracycline inhibits gingival matrix metalloproteinases and bone loss in Porphyromonas gingivalis–induced periodontitis in rats. Ann N Y Acad Sci. 1994;732:96-111.
- Humbert P, Treffel P, Chapius JF, et al. The tetracyclines in dermatology. J Am Acad Dermatol. 1991;25:691-697.
- Nishifuji K, Amagai M, Kuwana M, et al. Detection of antigen-specific B cells in patients with pemphigus vulgaris by enzyme-linked immunospot assay: requirement of T cell collaboration for autoantibody production. J Invest Dermatol. 2000;114:88-94.
- Szeto GL, Pomerantz JL, Graham DRM, et al. Minocycline suppresses activation of nuclear factor of activated T cells 1 (NFAT1) in human CD4 T Cells. J Biol Chem. 2011;286:11275-11282.
Practice Points
- The treatment of pemphigus vulgaris (PV) is challenging given the side-effect profile of commonly used systemic medications, including steroids, especially in elderly patients.
- Tetracyclines have an advantageous side-effect profile and may be efficacious in treating PV.
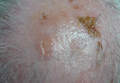
Firm Gray Nodule on the Scalp
The Diagnosis: Primary Cutaneous Mucinous Carcinoma
Primary cutaneous mucinous carcinoma is a rare tumor of the sweat glands that was first reported in 1952 by Lennox et al.1 These tumors are slow growing and have a predilection for the head and neck, with the eyelid being the most commonly reported location.2 In general, they present as erythematous asymptomatic nodules measuring less than 7 cm in diameter.2-4 Primary cutaneous mucinous carcinoma tends to have a good prognosis with complete resection, but cases of metastasis and recurrence have been reported.2 Although there is no standard of care, treatment typically consists of surgical management, as the tumors are nonresponsive to chemotherapy or radiation.4 Kamalpour et al2 compared outcomes for Mohs micrographic surgery versus standard excision, the former showing a lower percentage of poor outcomes. Of note, there were fewer cases treated with Mohs surgery in this study; only more recently reported cases have been treated with Mohs surgery.
Histologically, primary cutaneous mucinous carcinoma is composed of cords, tubules, and lobules of epithelial cells floating in large pools of basophilic mucin, separated by thin fibrovascular septa.5 It can be difficult to distinguish a primary tumor from a mucinous carcinoma metastasis with histology alone, especially on the breasts and in the gastrointestinal tract. Immunohistochemistry can be helpful in determining the origin of the tumor. A homologue of p53, p63 expressed in basal and myoepithelial cells of the skin can aid in the confirmation of a primary tumor when present.6,7 Negative staining for cytokeratin 20 and positive staining for cytokeratin 7 also are helpful in distinguishing a primary cutaneous mucinous carcinoma from a gastrointestinal tract metastasis.4,8
In our patient, no other symptoms were present that raised concern for an internal malignancy. Findings that supported a primary versus metastatic tumor included the clinicopathologic findings (Figure) as well as positive p63, cytokeratin 7, and negative cytokeratin 20 staining. The initial standard excision had tumor cells within 1 mm of the specimen margin; thus, a subsequent wider reexcision was performed. Reexcision was negative for tumor cells. Close follow-up with a primary care physician was recommended, with emphasis on colon and breast cancer screening. A follow-up mammogram was negative for breast cancer.
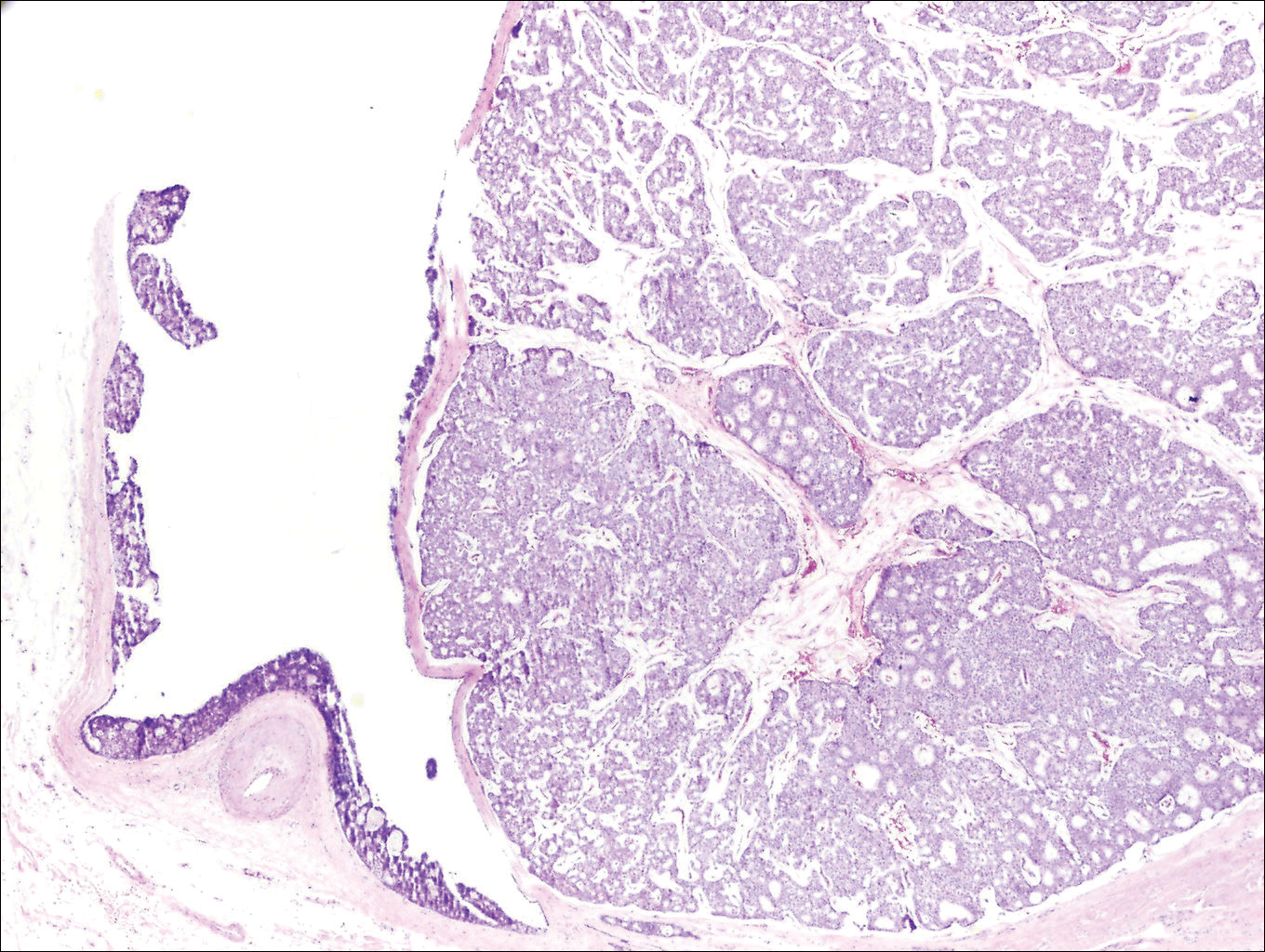
- Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin: with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-880.
- Kamalpour L, Brindise RT, Nodzenski M, et al. Primary cutaneous mucinous carcinoma a systematic review and meta-analysis of outcomes after surgery. JAMA Dermatol. 2014;150:380-384.
- Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathological and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
- Breiting L, Christensen L, Dahlstrom K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
- Walsh SN, Santa Cruz DJ. Adnexal carcinomas of the skin. In: Rigel DS, Robinson JK, Ross M, et al, eds. Cancer of the Skin. 2nd ed. Beijing, China: Elsevier Saunders; 2011:140-149.
- Jo VY, Fletcher CD. p63 Immunohistochemical staining is limited in soft tissue tumors. Am J Clin Pathol. 2011;136:762-766.
- Ivan D, Nash JW, Prieto VG, et al. Use of p63 expression in distinguishing primary and metastatic cutaneous adnexal neoplasms from metastatic adenocarcinoma to skin. J Cutan Pathol. 2006;34:478-489.
- Kazakov DV, Suster S, LeBoit PE, et al. Mucinous carcinoma of the skin, primary, and secondary: a clinicopathologic study of 63 cases with emphasis on the morphologic spectrum of primary cutaneous forms: homologies with mucinous lesions in the breast. Am J Surg Pathol. 2005;29:764-782.
The Diagnosis: Primary Cutaneous Mucinous Carcinoma
Primary cutaneous mucinous carcinoma is a rare tumor of the sweat glands that was first reported in 1952 by Lennox et al.1 These tumors are slow growing and have a predilection for the head and neck, with the eyelid being the most commonly reported location.2 In general, they present as erythematous asymptomatic nodules measuring less than 7 cm in diameter.2-4 Primary cutaneous mucinous carcinoma tends to have a good prognosis with complete resection, but cases of metastasis and recurrence have been reported.2 Although there is no standard of care, treatment typically consists of surgical management, as the tumors are nonresponsive to chemotherapy or radiation.4 Kamalpour et al2 compared outcomes for Mohs micrographic surgery versus standard excision, the former showing a lower percentage of poor outcomes. Of note, there were fewer cases treated with Mohs surgery in this study; only more recently reported cases have been treated with Mohs surgery.
Histologically, primary cutaneous mucinous carcinoma is composed of cords, tubules, and lobules of epithelial cells floating in large pools of basophilic mucin, separated by thin fibrovascular septa.5 It can be difficult to distinguish a primary tumor from a mucinous carcinoma metastasis with histology alone, especially on the breasts and in the gastrointestinal tract. Immunohistochemistry can be helpful in determining the origin of the tumor. A homologue of p53, p63 expressed in basal and myoepithelial cells of the skin can aid in the confirmation of a primary tumor when present.6,7 Negative staining for cytokeratin 20 and positive staining for cytokeratin 7 also are helpful in distinguishing a primary cutaneous mucinous carcinoma from a gastrointestinal tract metastasis.4,8
In our patient, no other symptoms were present that raised concern for an internal malignancy. Findings that supported a primary versus metastatic tumor included the clinicopathologic findings (Figure) as well as positive p63, cytokeratin 7, and negative cytokeratin 20 staining. The initial standard excision had tumor cells within 1 mm of the specimen margin; thus, a subsequent wider reexcision was performed. Reexcision was negative for tumor cells. Close follow-up with a primary care physician was recommended, with emphasis on colon and breast cancer screening. A follow-up mammogram was negative for breast cancer.
The Diagnosis: Primary Cutaneous Mucinous Carcinoma
Primary cutaneous mucinous carcinoma is a rare tumor of the sweat glands that was first reported in 1952 by Lennox et al.1 These tumors are slow growing and have a predilection for the head and neck, with the eyelid being the most commonly reported location.2 In general, they present as erythematous asymptomatic nodules measuring less than 7 cm in diameter.2-4 Primary cutaneous mucinous carcinoma tends to have a good prognosis with complete resection, but cases of metastasis and recurrence have been reported.2 Although there is no standard of care, treatment typically consists of surgical management, as the tumors are nonresponsive to chemotherapy or radiation.4 Kamalpour et al2 compared outcomes for Mohs micrographic surgery versus standard excision, the former showing a lower percentage of poor outcomes. Of note, there were fewer cases treated with Mohs surgery in this study; only more recently reported cases have been treated with Mohs surgery.
Histologically, primary cutaneous mucinous carcinoma is composed of cords, tubules, and lobules of epithelial cells floating in large pools of basophilic mucin, separated by thin fibrovascular septa.5 It can be difficult to distinguish a primary tumor from a mucinous carcinoma metastasis with histology alone, especially on the breasts and in the gastrointestinal tract. Immunohistochemistry can be helpful in determining the origin of the tumor. A homologue of p53, p63 expressed in basal and myoepithelial cells of the skin can aid in the confirmation of a primary tumor when present.6,7 Negative staining for cytokeratin 20 and positive staining for cytokeratin 7 also are helpful in distinguishing a primary cutaneous mucinous carcinoma from a gastrointestinal tract metastasis.4,8
In our patient, no other symptoms were present that raised concern for an internal malignancy. Findings that supported a primary versus metastatic tumor included the clinicopathologic findings (Figure) as well as positive p63, cytokeratin 7, and negative cytokeratin 20 staining. The initial standard excision had tumor cells within 1 mm of the specimen margin; thus, a subsequent wider reexcision was performed. Reexcision was negative for tumor cells. Close follow-up with a primary care physician was recommended, with emphasis on colon and breast cancer screening. A follow-up mammogram was negative for breast cancer.
- Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin: with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-880.
- Kamalpour L, Brindise RT, Nodzenski M, et al. Primary cutaneous mucinous carcinoma a systematic review and meta-analysis of outcomes after surgery. JAMA Dermatol. 2014;150:380-384.
- Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathological and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
- Breiting L, Christensen L, Dahlstrom K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
- Walsh SN, Santa Cruz DJ. Adnexal carcinomas of the skin. In: Rigel DS, Robinson JK, Ross M, et al, eds. Cancer of the Skin. 2nd ed. Beijing, China: Elsevier Saunders; 2011:140-149.
- Jo VY, Fletcher CD. p63 Immunohistochemical staining is limited in soft tissue tumors. Am J Clin Pathol. 2011;136:762-766.
- Ivan D, Nash JW, Prieto VG, et al. Use of p63 expression in distinguishing primary and metastatic cutaneous adnexal neoplasms from metastatic adenocarcinoma to skin. J Cutan Pathol. 2006;34:478-489.
- Kazakov DV, Suster S, LeBoit PE, et al. Mucinous carcinoma of the skin, primary, and secondary: a clinicopathologic study of 63 cases with emphasis on the morphologic spectrum of primary cutaneous forms: homologies with mucinous lesions in the breast. Am J Surg Pathol. 2005;29:764-782.
- Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin: with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-880.
- Kamalpour L, Brindise RT, Nodzenski M, et al. Primary cutaneous mucinous carcinoma a systematic review and meta-analysis of outcomes after surgery. JAMA Dermatol. 2014;150:380-384.
- Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathological and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
- Breiting L, Christensen L, Dahlstrom K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
- Walsh SN, Santa Cruz DJ. Adnexal carcinomas of the skin. In: Rigel DS, Robinson JK, Ross M, et al, eds. Cancer of the Skin. 2nd ed. Beijing, China: Elsevier Saunders; 2011:140-149.
- Jo VY, Fletcher CD. p63 Immunohistochemical staining is limited in soft tissue tumors. Am J Clin Pathol. 2011;136:762-766.
- Ivan D, Nash JW, Prieto VG, et al. Use of p63 expression in distinguishing primary and metastatic cutaneous adnexal neoplasms from metastatic adenocarcinoma to skin. J Cutan Pathol. 2006;34:478-489.
- Kazakov DV, Suster S, LeBoit PE, et al. Mucinous carcinoma of the skin, primary, and secondary: a clinicopathologic study of 63 cases with emphasis on the morphologic spectrum of primary cutaneous forms: homologies with mucinous lesions in the breast. Am J Surg Pathol. 2005;29:764-782.
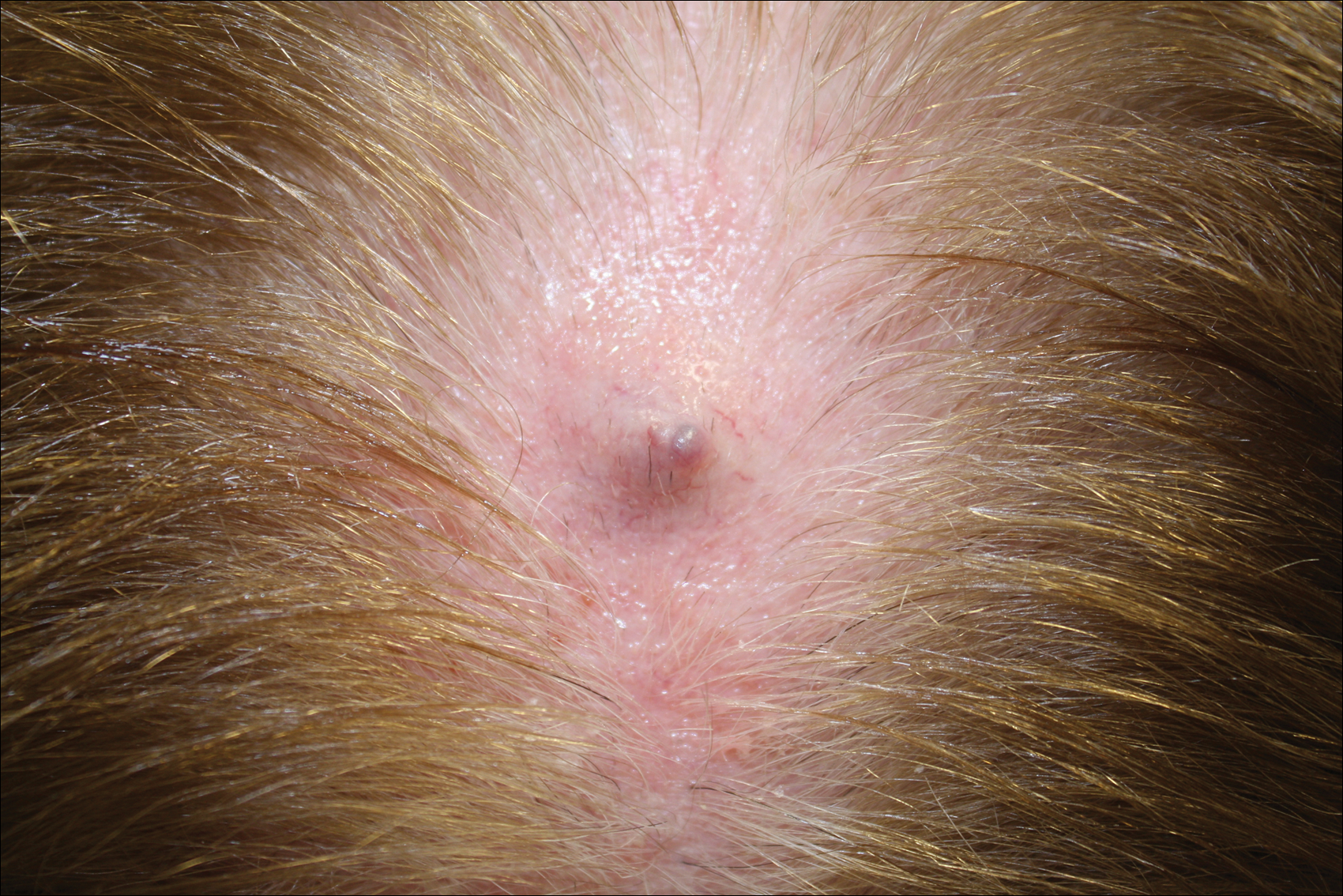
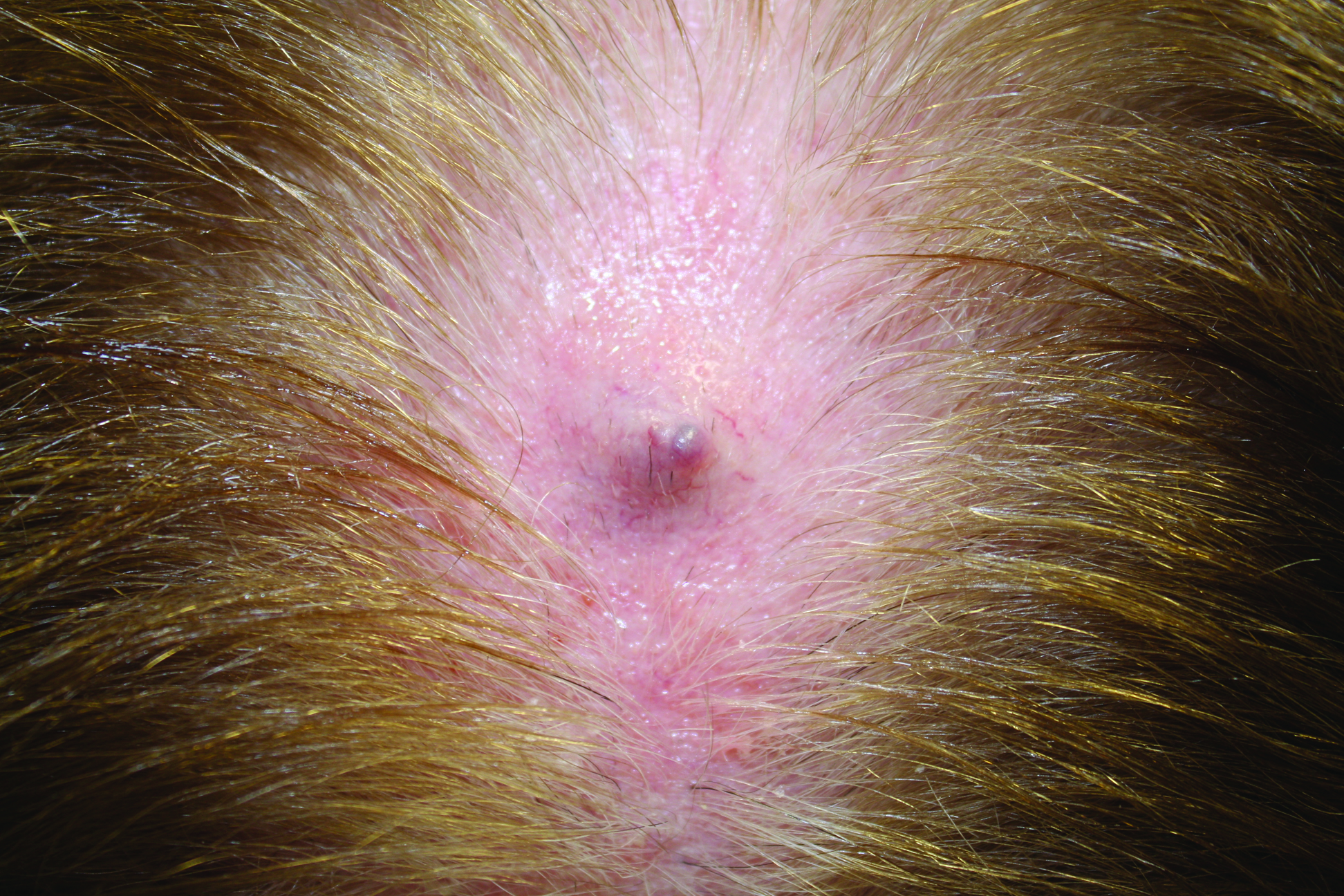
Growing Subcutaneous Mass on the Thigh
The Diagnosis: Eccrine Angiomatous Hamartoma
Given the progression of symptoms 3 months prior to presentation, an excisional biopsy was performed (Figure 1). Hematoxylin and eosin staining showed prominent eccrine sweat glands and vessels surrounded by superficially located adipose tissue in the mid and deep dermis (Figure 2).
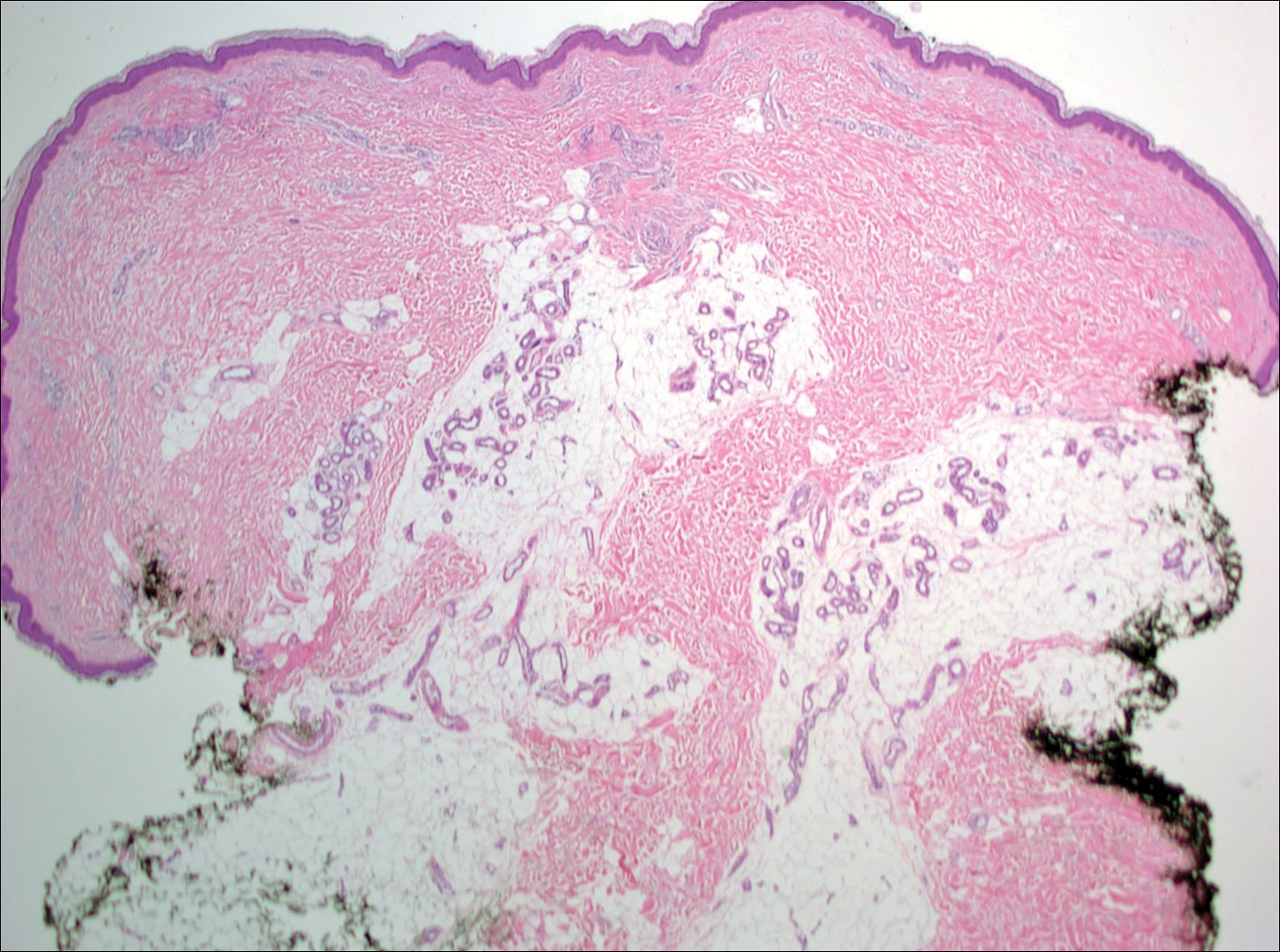
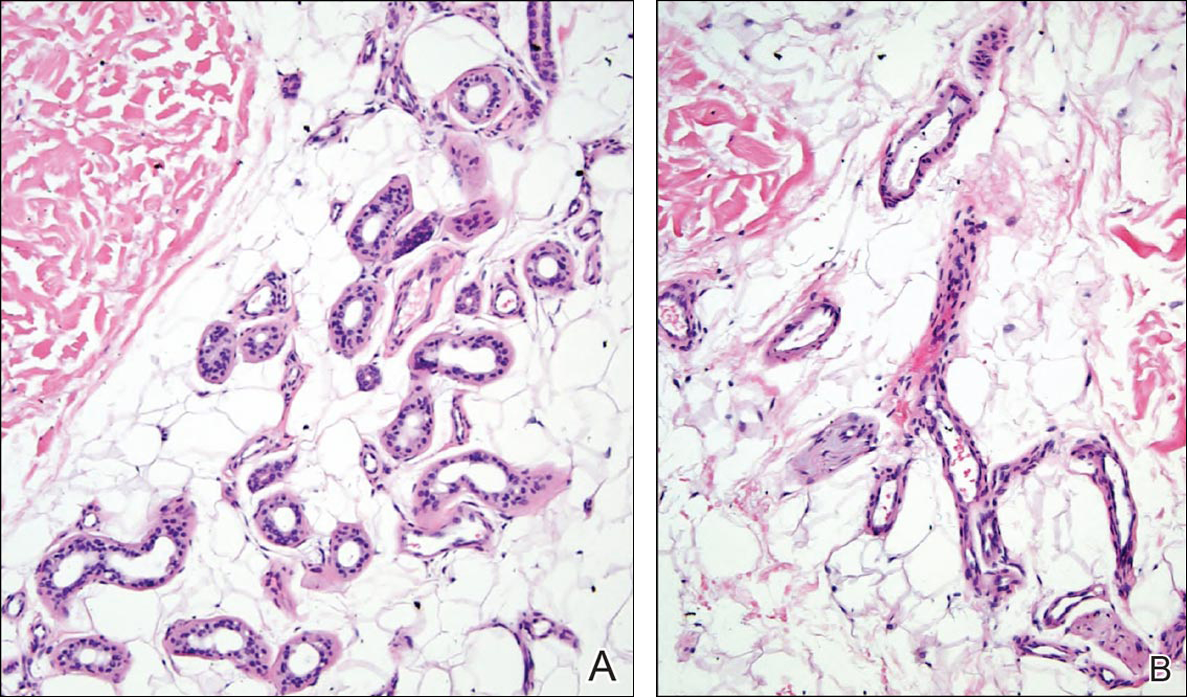
Eccrine angiomatous hamartoma (EAH) is an uncommon benign tumor typically located on the arms and legs or trunk. It is usually solitary, though cases with multiple lesions have been reported.1,2 Most cases are diagnosed in childhood as either congenital or acquired lesions. However, EAHs can develop in adulthood and have been described in patients up to 70 years of age.3 The median age of diagnosis is 10 years,2 indicating that EAH is primarily a pediatric tumor. There is no gender predilection.
Approximately 35% to 66% of patients report pain, pruritus, or hyperhidrosis associated with EAHs, though this incidence may be overrepresented because patients tend to present when the lesions become symptomatic.2-5 The pain is attributed to nerve fibers infiltrating the tumor. Hypertrichosis also has been described and is thought to be due to hair follicles within the hamartoma.
Histologically, EAHs are characterized by normal-appearing eccrine glands mingled with venules and capillaries. Additional variable pathologic findings include lipomatous, pilar, lymphatic, or mucinous features.2 Other vascular anomalies such as hemangiomas or arteriovenous malformations occasionally have been described in association with EAH. The vessels stain for ulex europaeus 1 and factor VIII. Eccrine glands stain for S-100 protein, carcinoembryonic antigen, epithelial membrane antigen, and cytokeratin CAM 5.2. In light of a publication proposing that EAH is a lymphatic proliferation,6 a D2-40 stain was performed on the specimen and was negative.
Eccrine angiomatous hamartoma has been reported to grow mainly during childhood, puberty, or pregnancy, presumably due to hormonal influences.7 There are few reports of EAH enlarging in middle-aged adults, and even fewer without pain during the growth phase. It is unclear what triggered the growth in our otherwise healthy postmenopausal patient.
Eccrine angiomatous hamartoma does not have malignant potential and thus treatment is optional and based on relief of symptoms. Simple excision of the EAH usually is curative, but recurrences can occur.4 Botulinum toxin also has been used to treat hyperhidrosis in tumors that are too large for resection. Treatment with lasers such as the pulsed dye laser and Nd:YAG laser has not been successful.8 A case of spontaneous regression has been reported.1
Liposuction was considered in our patient given the substantial adipose tissue on biopsy. The patient ultimately declined treatment. This case highlights that EAH can present in adulthood and should be considered in the differential diagnosis of an enlarging but otherwise asymptomatic vascular tumor.
- Tay YK, Sim CS. Eccrine angiomatous hamartoma associated with spontaneous regression. Pediatr Dermatol. 2006;23:516-517.
- Pelle MT, Pride HB, Tyler WB. Eccrine angiomatous hamartoma. J Am Acad Dermatol. 2002;47:429-435.
- Shin J, Jang YH, Kim SC, et al. Eccrine angiomatous hamartoma: a review of ten cases [published online May 10, 2013]. Ann Dermatol. 2013;25:208-212.
- Lin YT, Chen CM, Yang CH, et al. Eccrine angiomatous hamartoma: a retrospective study of 15 cases. Chang Gung Med J. 2012;35:167-177.
- Nakatsui TC, Schloss E, Krol A, et al. Eccrine angiomatous hamartoma: report of a case and literature review. J Am Acad Dermatol. 1999;41:109-111.
- Wang L, Wang S, Gao T, et al. Eccrine angiomatous hamartoma is a lymphatic proliferation. Eur J Dermatol. 2013;23:614-617.
- Kikusawa A, Oka M, Taguchi K, et al. Eccrine angiomatous hamartoma with sudden enlargement and pain in an adolescent girl after menarche [published online October 1, 2011]. Dermatoendocrinol. 2011;3:266-268.
- Barco D, Baselga E, Alegre M, et al. Successful treatment of eccrine angiomatous hamartoma with botulinum toxin. Arch Dermatol. 2009;145:241-243.
The Diagnosis: Eccrine Angiomatous Hamartoma
Given the progression of symptoms 3 months prior to presentation, an excisional biopsy was performed (Figure 1). Hematoxylin and eosin staining showed prominent eccrine sweat glands and vessels surrounded by superficially located adipose tissue in the mid and deep dermis (Figure 2).
Eccrine angiomatous hamartoma (EAH) is an uncommon benign tumor typically located on the arms and legs or trunk. It is usually solitary, though cases with multiple lesions have been reported.1,2 Most cases are diagnosed in childhood as either congenital or acquired lesions. However, EAHs can develop in adulthood and have been described in patients up to 70 years of age.3 The median age of diagnosis is 10 years,2 indicating that EAH is primarily a pediatric tumor. There is no gender predilection.
Approximately 35% to 66% of patients report pain, pruritus, or hyperhidrosis associated with EAHs, though this incidence may be overrepresented because patients tend to present when the lesions become symptomatic.2-5 The pain is attributed to nerve fibers infiltrating the tumor. Hypertrichosis also has been described and is thought to be due to hair follicles within the hamartoma.
Histologically, EAHs are characterized by normal-appearing eccrine glands mingled with venules and capillaries. Additional variable pathologic findings include lipomatous, pilar, lymphatic, or mucinous features.2 Other vascular anomalies such as hemangiomas or arteriovenous malformations occasionally have been described in association with EAH. The vessels stain for ulex europaeus 1 and factor VIII. Eccrine glands stain for S-100 protein, carcinoembryonic antigen, epithelial membrane antigen, and cytokeratin CAM 5.2. In light of a publication proposing that EAH is a lymphatic proliferation,6 a D2-40 stain was performed on the specimen and was negative.
Eccrine angiomatous hamartoma has been reported to grow mainly during childhood, puberty, or pregnancy, presumably due to hormonal influences.7 There are few reports of EAH enlarging in middle-aged adults, and even fewer without pain during the growth phase. It is unclear what triggered the growth in our otherwise healthy postmenopausal patient.
Eccrine angiomatous hamartoma does not have malignant potential and thus treatment is optional and based on relief of symptoms. Simple excision of the EAH usually is curative, but recurrences can occur.4 Botulinum toxin also has been used to treat hyperhidrosis in tumors that are too large for resection. Treatment with lasers such as the pulsed dye laser and Nd:YAG laser has not been successful.8 A case of spontaneous regression has been reported.1
Liposuction was considered in our patient given the substantial adipose tissue on biopsy. The patient ultimately declined treatment. This case highlights that EAH can present in adulthood and should be considered in the differential diagnosis of an enlarging but otherwise asymptomatic vascular tumor.
The Diagnosis: Eccrine Angiomatous Hamartoma
Given the progression of symptoms 3 months prior to presentation, an excisional biopsy was performed (Figure 1). Hematoxylin and eosin staining showed prominent eccrine sweat glands and vessels surrounded by superficially located adipose tissue in the mid and deep dermis (Figure 2).
Eccrine angiomatous hamartoma (EAH) is an uncommon benign tumor typically located on the arms and legs or trunk. It is usually solitary, though cases with multiple lesions have been reported.1,2 Most cases are diagnosed in childhood as either congenital or acquired lesions. However, EAHs can develop in adulthood and have been described in patients up to 70 years of age.3 The median age of diagnosis is 10 years,2 indicating that EAH is primarily a pediatric tumor. There is no gender predilection.
Approximately 35% to 66% of patients report pain, pruritus, or hyperhidrosis associated with EAHs, though this incidence may be overrepresented because patients tend to present when the lesions become symptomatic.2-5 The pain is attributed to nerve fibers infiltrating the tumor. Hypertrichosis also has been described and is thought to be due to hair follicles within the hamartoma.
Histologically, EAHs are characterized by normal-appearing eccrine glands mingled with venules and capillaries. Additional variable pathologic findings include lipomatous, pilar, lymphatic, or mucinous features.2 Other vascular anomalies such as hemangiomas or arteriovenous malformations occasionally have been described in association with EAH. The vessels stain for ulex europaeus 1 and factor VIII. Eccrine glands stain for S-100 protein, carcinoembryonic antigen, epithelial membrane antigen, and cytokeratin CAM 5.2. In light of a publication proposing that EAH is a lymphatic proliferation,6 a D2-40 stain was performed on the specimen and was negative.
Eccrine angiomatous hamartoma has been reported to grow mainly during childhood, puberty, or pregnancy, presumably due to hormonal influences.7 There are few reports of EAH enlarging in middle-aged adults, and even fewer without pain during the growth phase. It is unclear what triggered the growth in our otherwise healthy postmenopausal patient.
Eccrine angiomatous hamartoma does not have malignant potential and thus treatment is optional and based on relief of symptoms. Simple excision of the EAH usually is curative, but recurrences can occur.4 Botulinum toxin also has been used to treat hyperhidrosis in tumors that are too large for resection. Treatment with lasers such as the pulsed dye laser and Nd:YAG laser has not been successful.8 A case of spontaneous regression has been reported.1
Liposuction was considered in our patient given the substantial adipose tissue on biopsy. The patient ultimately declined treatment. This case highlights that EAH can present in adulthood and should be considered in the differential diagnosis of an enlarging but otherwise asymptomatic vascular tumor.
- Tay YK, Sim CS. Eccrine angiomatous hamartoma associated with spontaneous regression. Pediatr Dermatol. 2006;23:516-517.
- Pelle MT, Pride HB, Tyler WB. Eccrine angiomatous hamartoma. J Am Acad Dermatol. 2002;47:429-435.
- Shin J, Jang YH, Kim SC, et al. Eccrine angiomatous hamartoma: a review of ten cases [published online May 10, 2013]. Ann Dermatol. 2013;25:208-212.
- Lin YT, Chen CM, Yang CH, et al. Eccrine angiomatous hamartoma: a retrospective study of 15 cases. Chang Gung Med J. 2012;35:167-177.
- Nakatsui TC, Schloss E, Krol A, et al. Eccrine angiomatous hamartoma: report of a case and literature review. J Am Acad Dermatol. 1999;41:109-111.
- Wang L, Wang S, Gao T, et al. Eccrine angiomatous hamartoma is a lymphatic proliferation. Eur J Dermatol. 2013;23:614-617.
- Kikusawa A, Oka M, Taguchi K, et al. Eccrine angiomatous hamartoma with sudden enlargement and pain in an adolescent girl after menarche [published online October 1, 2011]. Dermatoendocrinol. 2011;3:266-268.
- Barco D, Baselga E, Alegre M, et al. Successful treatment of eccrine angiomatous hamartoma with botulinum toxin. Arch Dermatol. 2009;145:241-243.
- Tay YK, Sim CS. Eccrine angiomatous hamartoma associated with spontaneous regression. Pediatr Dermatol. 2006;23:516-517.
- Pelle MT, Pride HB, Tyler WB. Eccrine angiomatous hamartoma. J Am Acad Dermatol. 2002;47:429-435.
- Shin J, Jang YH, Kim SC, et al. Eccrine angiomatous hamartoma: a review of ten cases [published online May 10, 2013]. Ann Dermatol. 2013;25:208-212.
- Lin YT, Chen CM, Yang CH, et al. Eccrine angiomatous hamartoma: a retrospective study of 15 cases. Chang Gung Med J. 2012;35:167-177.
- Nakatsui TC, Schloss E, Krol A, et al. Eccrine angiomatous hamartoma: report of a case and literature review. J Am Acad Dermatol. 1999;41:109-111.
- Wang L, Wang S, Gao T, et al. Eccrine angiomatous hamartoma is a lymphatic proliferation. Eur J Dermatol. 2013;23:614-617.
- Kikusawa A, Oka M, Taguchi K, et al. Eccrine angiomatous hamartoma with sudden enlargement and pain in an adolescent girl after menarche [published online October 1, 2011]. Dermatoendocrinol. 2011;3:266-268.
- Barco D, Baselga E, Alegre M, et al. Successful treatment of eccrine angiomatous hamartoma with botulinum toxin. Arch Dermatol. 2009;145:241-243.
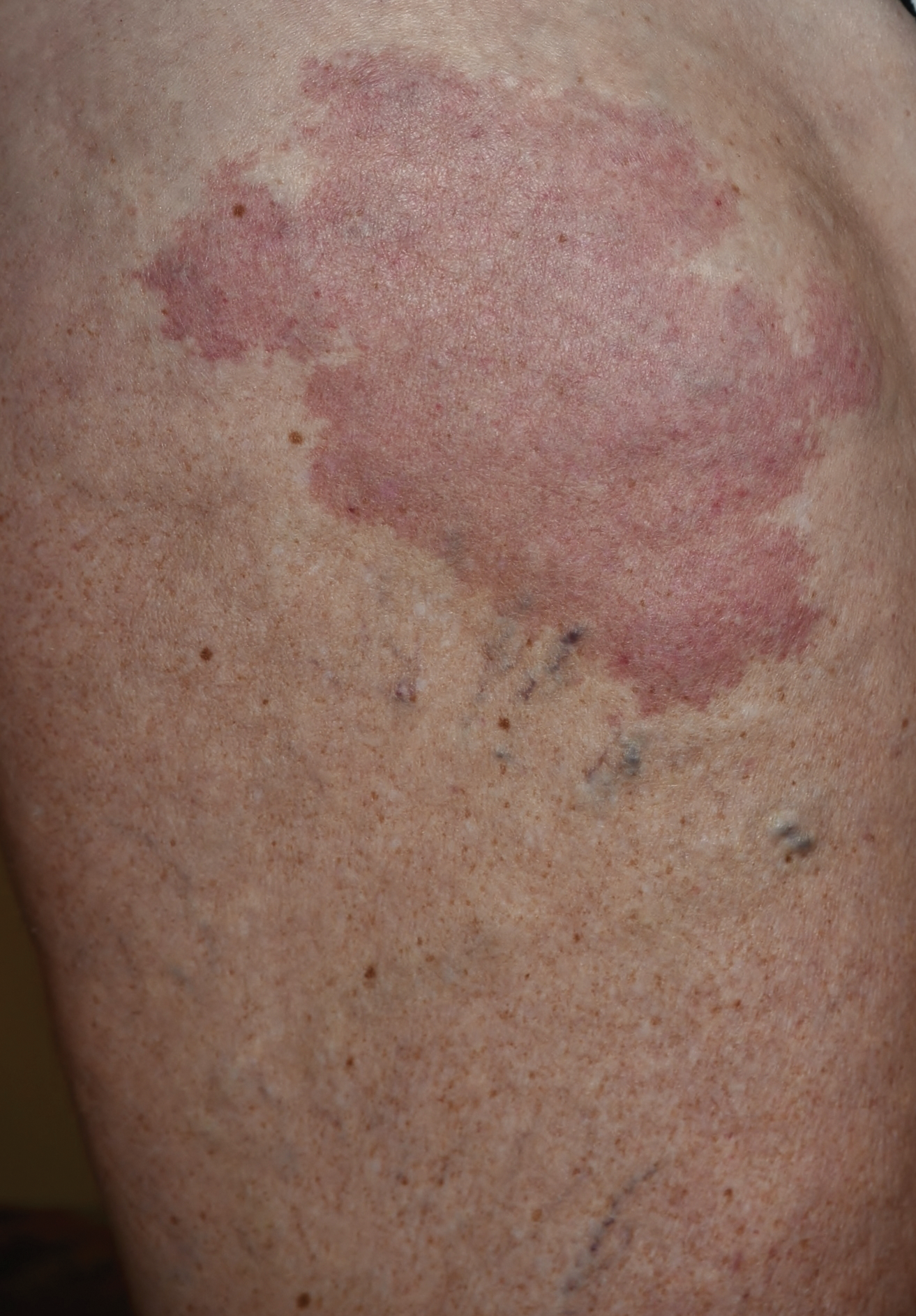
DRESS Syndrome With Autoimmune Hepatitis From Strontium Ranelate
Drug rash with eosinophilia and systemic symptoms (DRESS) syndrome refers to a severe, acute, potentially fatal, multisystem adverse drug reaction characterized by skin rash, fever, hematological abnormalities, and lymphadenopathy with involvement of several internal organs. The pathogenesis of DRESS syndrome is still unknown. Immunological factors such as a defect in detoxification of culprit drugs and infections seem to be involved. The most commonly associated drugs are anticonvulsants and sulfonamides, but dapsone, allopurinol, and minocycline also have been reported to be associated with DRESS syndrome.1
Although therapies for postmenopausal osteoporosis are considered to be safe from cutaneous side effects, there have been several reported cases of DRESS syndrome associated with strontium ranelate.2 Strontium ranelate is not used in the United States; nevertheless, some US patients may be taking this drug as an alternative to the current US Food and Drug Administration–approved drugs for osteoporosis. We report a case of DRESS syndrome in a woman who developed an extensive maculopapular rash, eosinophilia, dyspnea, bilateral cervical lymphadenopathy, and reactivation of Epstein-Barr virus (EBV) with liver damage 3 weeks after administration of strontium ranelate for postmenopausal osteoporosis. Approximately 6 months after total remission of skin conditions, the patient developed autoimmune hepatitis.
Case Report
A 64-year-old woman presented to the emergency department with dyspnea, fever (temperature, 38.5°C), and a generalized rash that had developed a few days prior. The patient reported that she was previously in good health and had no prior allergic episodes. She had been taking strontium ranelate for 3 weeks to treat postmenopausal osteoporosis and reported no other medication use. The patient was hospitalized because of worsening symptoms. Physical examination revealed a pruritic maculopapular rash involving the trunk, arms, and legs (Figure 1) with facial edema, mild inspiratory as well as expiratory dyspnea, and wheezing on all lung fields. An enlarged soft liver (6–7 cm from the right costal arch) and cervical bilateral lymphadenopathy were found.
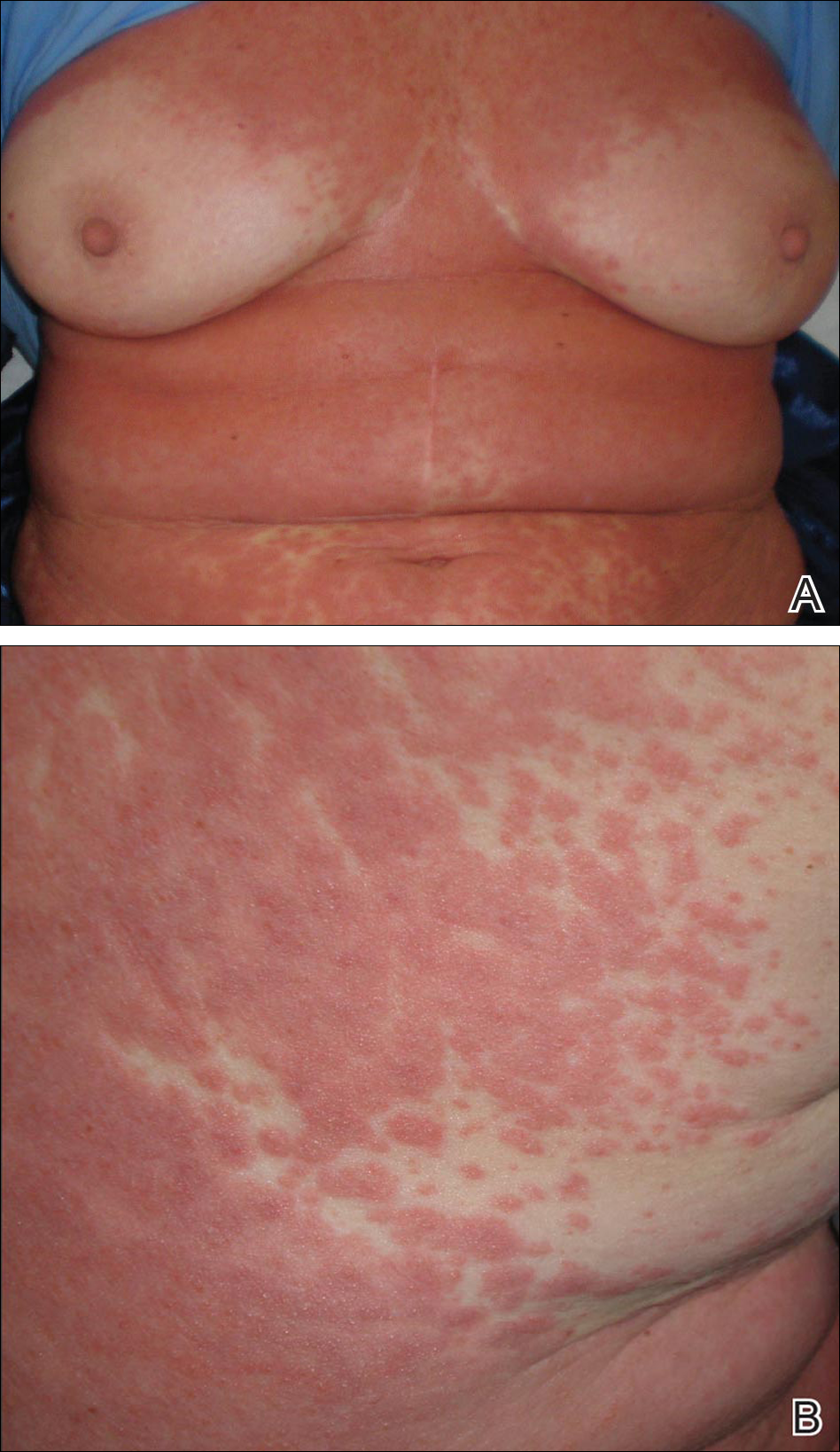
A chest radiograph detected a slight increase of the peribronchial thickening with interstitial involvement at the bilateral basal and perihilar levels, and an ultrasound of the chest confirmed the presence of many enlarged cervical bilateral lymph nodes between 2 and 4 cm in diameter.
Laboratory tests revealed the following values: leukocytosis (21,390/μL [reference range, 4500–11,000/μL]) with eosinophilia (27% [reference range, 2.7%]; 5780/μL [reference range, 0–450/μL]), elevated C-reactive protein (20 mg/L [reference range, 0.08–3.1 mg/L]), elevated erythrocyte sedimentation rate (35 mm/h [reference range, 0–20 mm/h]), a reactivation of EBV confirmed by simultaneous seropositivity to early antigen IgM and EBV nuclear antigen, liver damage with notable increases in liver function tests (aspartate aminotransferase, 51 U/L [reference range, 10–30 U/L]); alanine aminotransferase, 104 U/L [reference range 10–40 U/L]); γ-glutamyltransferase, 52 U/L [reference range, 2–30 U/L]), and no thyroid dysfunction.
Blood and urine cultures; antinuclear antibodies; and serology for hepatitis A, B, and C virus, as well as herpes simplex virus type 6 (HHV-6), chlamydia, Mycoplasma, and cytomegalovirus (CMV) were all negative. Histologic examination after skin biopsy showed keratinocytes with spongiosis, intraepidermal eosinophilic infiltration, suffusion of red blood cells with perivascular granulocytes, and lymphocyte inflammatory infiltrate (Figure 2).
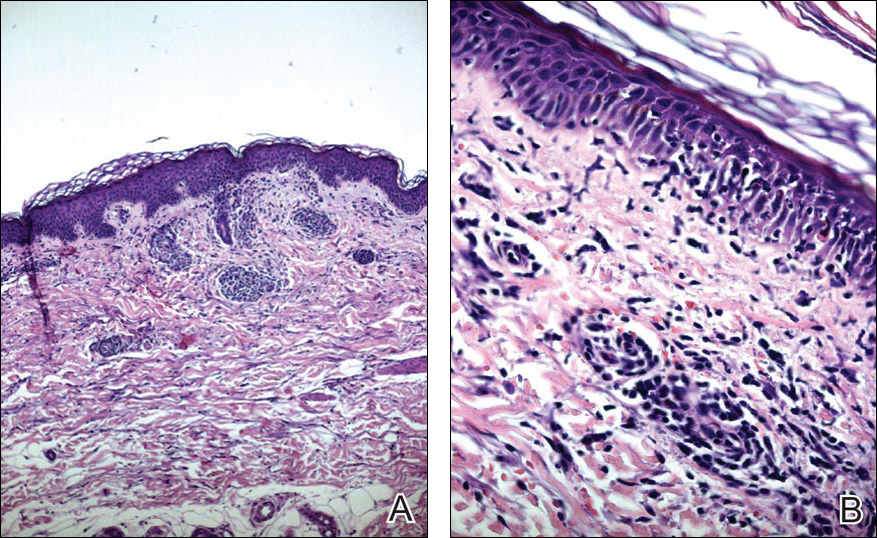
A diagnosis of DRESS syndrome was made on the basis of the following clinical data supported by laboratory findings: generalized maculopapular rash, eosinophilia, lung involvement with dyspnea, bilateral cervical lymphadenopathy, and liver damage, as well as an identified reactivation of EBV and onset of symptoms 3 weeks after treatment with strontium ranelate.
The patient was given intravenous methylprednisolone 120 mg once daily for 1 week in gradually decreasing doses. Three weeks of steroid therapy were necessary to obtain the first good results. Improvement of the patient’s clinical condition was considerably slow. Fever and rash gradually disappeared and the patient was discharged with oral corticosteroids. In the 2 months after starting systemic corticosteroid therapy, the lesions had not progressed and all other clinical symptoms improved. A slow but notable regression of the skin reaction was observed.
In a subsequent checkup approximately 8 months following initial presentation, the patient developed autoimmune hepatitis. There was a notable increase in liver enzymes and serum immunoglobulin content as well as positivity of antinuclear antibodies (1:160) and antimitochondrial antibodies (1:160). A liver biopsy was performed and confirmed the histologic pattern of autoimmune hepatitis. Thyroid function was reevaluated, but no other autoimmune disease was identified.
The patient was given another dose of steroids (prednisolone 25 mg daily). Liver function normalized within 1 month (aspartate aminotransferase levels went from 195 U/L to 21 U/L; alanine aminotransferase went from 324 U/L to 21 U/L; γ-glutamyltransferase went from 268 U/L to 63 U/L). The patient is currently taking a maintenance dose of prednisolone 5 mg and has normal liver function.
Comment
Uses of Strontium Ranelate
Strontium ranelate is recommended for reducing the risk for fracture in postmenopausal women 70 years and older with a bone mineral density T-score of –3.0 or lower (ie, primary prevention) as well as for the treatment of morphometric vertebral fracture in established postmenopausal osteoporosis (ie, secondary prevention). Strontium ranelate has a dual action that includes increasing bone formation and reducing bone resorption, leading to rebalancing of bone remodeling in favor of bone formation. Strontium ranelate was shown to increase the recruitment and activity of osteoblastic cells and to inhibit the recruitment and activity of osteoclasts.2 The recommended dose of oral strontium ranelate is 2 g once daily.
Side Effects of Strontium Ranelate
In a 3-year study of side effects associated with strontium ranelate, severe reactions were described in 23% of the reported adverse effects in 844 patients.3 In this study, cardiovascular effects, particularly thromboembolism, and DRESS syndrome were the most frequent side effects. Since its introduction in the market, at least 16 cases of DRESS syndrome related to strontium ranelate use have been reported in Europe, including 2 fatal cases.2 Two deaths have been reported to be associated with this drug,2 which was the basis of the warning document by the European Medicines Agency regarding the risk for strontium ranelate inducing DRESS syndrome.4
Development of DRESS Syndrome
The most common agents involved in DRESS syndrome are anticonvulsants, sulfonamides, dapsone, minocycline, allopurinol, and gold salts, as well as celecoxib, antituberculosis drugs, nonsteroidal anti-inflammatory drugs, antibiotics, calcium channel blockers, and antiretroviral drugs.5,6 The mortality rate of DRESS syndrome is 10%.6
The pathophysiology of DRESS syndrome is still unclear. Altered drug metabolism, genetic predisposition, and concomitant infection or reactivation of bacterial or viral infection (eg, HHV-6, EBV, CMV, human immunodeficiency virus, influenza, viral hepatitis) could be factors leading to development of DRESS. Autoimmune or connective-tissue diseases also have been suggested to increase the risk.7
Clinicians should suspect DRESS syndrome in any patient developing a rash 3 to 6 weeks after starting drug therapy. This disorder often starts with fever (temperature >38°C) and includes cutaneous symptoms such as generalized rash that may progress to exfoliative dermatitis. There usually is involvement of one or several internal organs with the development of hepatitis; interstitial pneumonia; interstitial nephritis; myopericarditis; myositis; pancreatitis; thyroiditis; and hematological abnormalities, primarily eosinophilia or atypical lymphocytosis. Facial edema and lymphadenopathy also may be present. A skin biopsy can confirm the clinical diagnosis of DRESS syndrome but is not specific because cutaneous histologic patterns often show a lymphocytic infiltrate that sometimes mimics cutaneous lymphoma. Other diseases that DRESS syndrome may mimic include Stevens-Johnson syndrome and toxic epidermal necrolysis as well as Kawasaki disease, Still disease, acute viral infections, idiopathic hypereosinophilic syndrome, and lymphoma, which should be excluded from the differential diagnosis.8
Diagnosis of DRESS Syndrome
There is no gold standard for the diagnosis of DRESS syndrome. In our case, the diagnosis of DRESS syndrome was based on the RegiSCAR (European Registry of Severe Cutaneous Adverse Reactions to Drugs) score as described by Kardaun et al,9 which grades DRESS syndrome cases as excluded (<2 points), possible (2–3 points), probable (4–5 points), or definite (>5 points) based on the following clinical criteria: fever (temperature >38.5°C; from a minimum of –1 point if absent to a maximum of 0 points if present); enlarged lymph nodes (from a minimum of 0 points if absent to a maximum of 1 point if present); eosinophilia (0 points if absent, 1 point if 10%–19% or 700–1500 μL, 2 points if ≥20% or >1500 μL); atypical lymphocytes (from a minimum of 0 points if absent to maximum of 1 point if present); skin involvement with rash (1 point if >50% of body surface area is involved, 1 point if there is a maculopapular rash, 1 point if skin biopsy suggests DRESS syndrome); organ involvement (1 point each for liver, kidneys, lungs, muscle/heart, pancreas, and other organs); resolution in at least 15 days (from a minimum of –1 point if absent to maximum of 0 points if present); and evaluation of other potential causes measuring antinuclear antibodies, blood culture, and serology for hepatitis virus (A–C), chlamydia, and Mycoplasma (1 point if 3 or more are negative and none positive). Virus reactivation also should be considered a main characteristic of DRESS syndrome. Therefore, its prevalence is not homogenous, so the absence of viral reactivation cannot be considered exclusion criteria. Several case reports and a few well-documented series have evidenced markers of virus reactivation in many cases of DRESS. Herpes simplex virus 6, CMV, and EBV are the most frequently reactivated.
The total RegiSCAR score of 8 in our case was taken as a definite indication of DRESS syndrome (temperature, 38.5°C [0 points]; enlarged lymph nodes [1 point]; eosinophilia, ≥20% or >1500 μL [2 points]; skin involvement with >50% body surface area involved [1 point] with a maculopapular rash [1 point] and histopathologic findings suggesting DRESS syndrome [1 point]; lung and liver involvement [2 points]). The causative drug was identified by carefully collecting the patient’s medication history and by evaluating clinical outcome characterized by improved skin and systemic symptoms after discontinuation of strontium ranelate.
Because of the high morbidity of DRESS syndrome, it needs to be diagnosed effectively and must be considered in the differential for any patient developing the triad of skin rash, hypereosinophilia, and systemic symptoms, as well as several other side effects when taking strontium ranelate.10
Therapies for DRESS Syndrome
Treatment of DRESS syndrome has not yet been standardized. Prompt withdrawal of the causative drug is the only mandatory activity in the treatment of DRESS syndrome. Systemic corticosteroids may be needed for organ or life-threatening disease, though the efficacy is controversial because it may result in activation of HHV-6, which in turn is probably involved in the pathogenesis of DRESS syndrome.
Conclusion
This case confirms that strontium ranelate should be considered a possible factor in the etiopathology of DRESS syndrome and in the development of autoimmune hepatitis as a part of DRESS syndrome. Case reports underline the importance of recognition of cutaneous adverse reactions in patients undergoing treatment of postmenopausal osteoporosis. The prognosis is good with immediate recognition followed by immediate and permanent withdrawal of the drug, along with hospitalization and systemic corticosteroids when necessary. The possibility of developing autoimmune hepatitis as a part of DRESS syndrome related to strontium ranelate has been reported,11 usually months after the acute episode.
- Tas S, Simonart T. Management of drug rash with eosinophilia and systemic symptoms (DRESS syndrome): an update. Dermatology. 2003;206:353-356.
- Le Merlouette M, Adamski H, Dinulescu M, et al. Strontium ranelate–induced DRESS syndrome. Ann Dermatol Venereol. 2011;138:124-128.
- Jonville-Bera AP, Autret-Leca E. Adverse drug reactions of strontium ranelate (Protelos®) in France. Presse Med. 2011;40:453-462.
- Assessment report for Protelos and Osseor. European Medicines Agency website. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Assessment_Report_-_Variation/human/000560/WC500131789.pdf). Published May 25, 2012. Accessed May 9, 2016.
- Breathnach S. Drug rash eosinophilia and systemic symptoms (DRESS) syndrome. types of clinical reaction: drug reaction. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. Vol 4. 8th ed. Hoboken, NJ: Oxford Wiley-Blackwell Publications; 2010:75.26.
- Lee JH, Park HK, Heo J, et al. Drug rash with eosinophilia and systemic symptoms (DRESS) syndrome induced by celecoxib and anti-tuberculosis drugs. J Korean Med Sci. 2008;23:521-525.
- Musette P, Brandi ML, Cacoub P, et al. Treatment of osteoporosis: recognizing and managing cutaneous adverse reactions and drug-induced hypersensitivity. Osteoporos Int. 2010;21:723-732.
- Telles Rudge de Aquino R, Vieitas Vergueiro CS, Ruffolo Magliari ME, et al. Sulfasalazine-induced DRESS syndrome (drug rash with eosinophilia and systemic symptoms). Sao Paulo Med J. 2008;126:225-226.
- Kardaun SH, Sidoroff A, Valeyrie-Allanore L, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2007;156:609-611.
- Pernicova I, Middleton ET, Aye M. Rash, strontium ranelate and DRESS syndrome put into perspective. European Medicine Agency on the alert [published online September 20, 2008]. Osteoporos Int. 2008;19:1811-1812.
- Kinyó A, Belsö N, Nagy N, et al. Strontium ranelate-induced DRESS syndrome with persistent autoimmune hepatitis. Acta Derm Venereol. 2011;91:205-206.
Drug rash with eosinophilia and systemic symptoms (DRESS) syndrome refers to a severe, acute, potentially fatal, multisystem adverse drug reaction characterized by skin rash, fever, hematological abnormalities, and lymphadenopathy with involvement of several internal organs. The pathogenesis of DRESS syndrome is still unknown. Immunological factors such as a defect in detoxification of culprit drugs and infections seem to be involved. The most commonly associated drugs are anticonvulsants and sulfonamides, but dapsone, allopurinol, and minocycline also have been reported to be associated with DRESS syndrome.1
Although therapies for postmenopausal osteoporosis are considered to be safe from cutaneous side effects, there have been several reported cases of DRESS syndrome associated with strontium ranelate.2 Strontium ranelate is not used in the United States; nevertheless, some US patients may be taking this drug as an alternative to the current US Food and Drug Administration–approved drugs for osteoporosis. We report a case of DRESS syndrome in a woman who developed an extensive maculopapular rash, eosinophilia, dyspnea, bilateral cervical lymphadenopathy, and reactivation of Epstein-Barr virus (EBV) with liver damage 3 weeks after administration of strontium ranelate for postmenopausal osteoporosis. Approximately 6 months after total remission of skin conditions, the patient developed autoimmune hepatitis.
Case Report
A 64-year-old woman presented to the emergency department with dyspnea, fever (temperature, 38.5°C), and a generalized rash that had developed a few days prior. The patient reported that she was previously in good health and had no prior allergic episodes. She had been taking strontium ranelate for 3 weeks to treat postmenopausal osteoporosis and reported no other medication use. The patient was hospitalized because of worsening symptoms. Physical examination revealed a pruritic maculopapular rash involving the trunk, arms, and legs (Figure 1) with facial edema, mild inspiratory as well as expiratory dyspnea, and wheezing on all lung fields. An enlarged soft liver (6–7 cm from the right costal arch) and cervical bilateral lymphadenopathy were found.
A chest radiograph detected a slight increase of the peribronchial thickening with interstitial involvement at the bilateral basal and perihilar levels, and an ultrasound of the chest confirmed the presence of many enlarged cervical bilateral lymph nodes between 2 and 4 cm in diameter.
Laboratory tests revealed the following values: leukocytosis (21,390/μL [reference range, 4500–11,000/μL]) with eosinophilia (27% [reference range, 2.7%]; 5780/μL [reference range, 0–450/μL]), elevated C-reactive protein (20 mg/L [reference range, 0.08–3.1 mg/L]), elevated erythrocyte sedimentation rate (35 mm/h [reference range, 0–20 mm/h]), a reactivation of EBV confirmed by simultaneous seropositivity to early antigen IgM and EBV nuclear antigen, liver damage with notable increases in liver function tests (aspartate aminotransferase, 51 U/L [reference range, 10–30 U/L]); alanine aminotransferase, 104 U/L [reference range 10–40 U/L]); γ-glutamyltransferase, 52 U/L [reference range, 2–30 U/L]), and no thyroid dysfunction.
Blood and urine cultures; antinuclear antibodies; and serology for hepatitis A, B, and C virus, as well as herpes simplex virus type 6 (HHV-6), chlamydia, Mycoplasma, and cytomegalovirus (CMV) were all negative. Histologic examination after skin biopsy showed keratinocytes with spongiosis, intraepidermal eosinophilic infiltration, suffusion of red blood cells with perivascular granulocytes, and lymphocyte inflammatory infiltrate (Figure 2).
A diagnosis of DRESS syndrome was made on the basis of the following clinical data supported by laboratory findings: generalized maculopapular rash, eosinophilia, lung involvement with dyspnea, bilateral cervical lymphadenopathy, and liver damage, as well as an identified reactivation of EBV and onset of symptoms 3 weeks after treatment with strontium ranelate.
The patient was given intravenous methylprednisolone 120 mg once daily for 1 week in gradually decreasing doses. Three weeks of steroid therapy were necessary to obtain the first good results. Improvement of the patient’s clinical condition was considerably slow. Fever and rash gradually disappeared and the patient was discharged with oral corticosteroids. In the 2 months after starting systemic corticosteroid therapy, the lesions had not progressed and all other clinical symptoms improved. A slow but notable regression of the skin reaction was observed.
In a subsequent checkup approximately 8 months following initial presentation, the patient developed autoimmune hepatitis. There was a notable increase in liver enzymes and serum immunoglobulin content as well as positivity of antinuclear antibodies (1:160) and antimitochondrial antibodies (1:160). A liver biopsy was performed and confirmed the histologic pattern of autoimmune hepatitis. Thyroid function was reevaluated, but no other autoimmune disease was identified.
The patient was given another dose of steroids (prednisolone 25 mg daily). Liver function normalized within 1 month (aspartate aminotransferase levels went from 195 U/L to 21 U/L; alanine aminotransferase went from 324 U/L to 21 U/L; γ-glutamyltransferase went from 268 U/L to 63 U/L). The patient is currently taking a maintenance dose of prednisolone 5 mg and has normal liver function.
Comment
Uses of Strontium Ranelate
Strontium ranelate is recommended for reducing the risk for fracture in postmenopausal women 70 years and older with a bone mineral density T-score of –3.0 or lower (ie, primary prevention) as well as for the treatment of morphometric vertebral fracture in established postmenopausal osteoporosis (ie, secondary prevention). Strontium ranelate has a dual action that includes increasing bone formation and reducing bone resorption, leading to rebalancing of bone remodeling in favor of bone formation. Strontium ranelate was shown to increase the recruitment and activity of osteoblastic cells and to inhibit the recruitment and activity of osteoclasts.2 The recommended dose of oral strontium ranelate is 2 g once daily.
Side Effects of Strontium Ranelate
In a 3-year study of side effects associated with strontium ranelate, severe reactions were described in 23% of the reported adverse effects in 844 patients.3 In this study, cardiovascular effects, particularly thromboembolism, and DRESS syndrome were the most frequent side effects. Since its introduction in the market, at least 16 cases of DRESS syndrome related to strontium ranelate use have been reported in Europe, including 2 fatal cases.2 Two deaths have been reported to be associated with this drug,2 which was the basis of the warning document by the European Medicines Agency regarding the risk for strontium ranelate inducing DRESS syndrome.4
Development of DRESS Syndrome
The most common agents involved in DRESS syndrome are anticonvulsants, sulfonamides, dapsone, minocycline, allopurinol, and gold salts, as well as celecoxib, antituberculosis drugs, nonsteroidal anti-inflammatory drugs, antibiotics, calcium channel blockers, and antiretroviral drugs.5,6 The mortality rate of DRESS syndrome is 10%.6
The pathophysiology of DRESS syndrome is still unclear. Altered drug metabolism, genetic predisposition, and concomitant infection or reactivation of bacterial or viral infection (eg, HHV-6, EBV, CMV, human immunodeficiency virus, influenza, viral hepatitis) could be factors leading to development of DRESS. Autoimmune or connective-tissue diseases also have been suggested to increase the risk.7
Clinicians should suspect DRESS syndrome in any patient developing a rash 3 to 6 weeks after starting drug therapy. This disorder often starts with fever (temperature >38°C) and includes cutaneous symptoms such as generalized rash that may progress to exfoliative dermatitis. There usually is involvement of one or several internal organs with the development of hepatitis; interstitial pneumonia; interstitial nephritis; myopericarditis; myositis; pancreatitis; thyroiditis; and hematological abnormalities, primarily eosinophilia or atypical lymphocytosis. Facial edema and lymphadenopathy also may be present. A skin biopsy can confirm the clinical diagnosis of DRESS syndrome but is not specific because cutaneous histologic patterns often show a lymphocytic infiltrate that sometimes mimics cutaneous lymphoma. Other diseases that DRESS syndrome may mimic include Stevens-Johnson syndrome and toxic epidermal necrolysis as well as Kawasaki disease, Still disease, acute viral infections, idiopathic hypereosinophilic syndrome, and lymphoma, which should be excluded from the differential diagnosis.8
Diagnosis of DRESS Syndrome
There is no gold standard for the diagnosis of DRESS syndrome. In our case, the diagnosis of DRESS syndrome was based on the RegiSCAR (European Registry of Severe Cutaneous Adverse Reactions to Drugs) score as described by Kardaun et al,9 which grades DRESS syndrome cases as excluded (<2 points), possible (2–3 points), probable (4–5 points), or definite (>5 points) based on the following clinical criteria: fever (temperature >38.5°C; from a minimum of –1 point if absent to a maximum of 0 points if present); enlarged lymph nodes (from a minimum of 0 points if absent to a maximum of 1 point if present); eosinophilia (0 points if absent, 1 point if 10%–19% or 700–1500 μL, 2 points if ≥20% or >1500 μL); atypical lymphocytes (from a minimum of 0 points if absent to maximum of 1 point if present); skin involvement with rash (1 point if >50% of body surface area is involved, 1 point if there is a maculopapular rash, 1 point if skin biopsy suggests DRESS syndrome); organ involvement (1 point each for liver, kidneys, lungs, muscle/heart, pancreas, and other organs); resolution in at least 15 days (from a minimum of –1 point if absent to maximum of 0 points if present); and evaluation of other potential causes measuring antinuclear antibodies, blood culture, and serology for hepatitis virus (A–C), chlamydia, and Mycoplasma (1 point if 3 or more are negative and none positive). Virus reactivation also should be considered a main characteristic of DRESS syndrome. Therefore, its prevalence is not homogenous, so the absence of viral reactivation cannot be considered exclusion criteria. Several case reports and a few well-documented series have evidenced markers of virus reactivation in many cases of DRESS. Herpes simplex virus 6, CMV, and EBV are the most frequently reactivated.
The total RegiSCAR score of 8 in our case was taken as a definite indication of DRESS syndrome (temperature, 38.5°C [0 points]; enlarged lymph nodes [1 point]; eosinophilia, ≥20% or >1500 μL [2 points]; skin involvement with >50% body surface area involved [1 point] with a maculopapular rash [1 point] and histopathologic findings suggesting DRESS syndrome [1 point]; lung and liver involvement [2 points]). The causative drug was identified by carefully collecting the patient’s medication history and by evaluating clinical outcome characterized by improved skin and systemic symptoms after discontinuation of strontium ranelate.
Because of the high morbidity of DRESS syndrome, it needs to be diagnosed effectively and must be considered in the differential for any patient developing the triad of skin rash, hypereosinophilia, and systemic symptoms, as well as several other side effects when taking strontium ranelate.10
Therapies for DRESS Syndrome
Treatment of DRESS syndrome has not yet been standardized. Prompt withdrawal of the causative drug is the only mandatory activity in the treatment of DRESS syndrome. Systemic corticosteroids may be needed for organ or life-threatening disease, though the efficacy is controversial because it may result in activation of HHV-6, which in turn is probably involved in the pathogenesis of DRESS syndrome.
Conclusion
This case confirms that strontium ranelate should be considered a possible factor in the etiopathology of DRESS syndrome and in the development of autoimmune hepatitis as a part of DRESS syndrome. Case reports underline the importance of recognition of cutaneous adverse reactions in patients undergoing treatment of postmenopausal osteoporosis. The prognosis is good with immediate recognition followed by immediate and permanent withdrawal of the drug, along with hospitalization and systemic corticosteroids when necessary. The possibility of developing autoimmune hepatitis as a part of DRESS syndrome related to strontium ranelate has been reported,11 usually months after the acute episode.
Drug rash with eosinophilia and systemic symptoms (DRESS) syndrome refers to a severe, acute, potentially fatal, multisystem adverse drug reaction characterized by skin rash, fever, hematological abnormalities, and lymphadenopathy with involvement of several internal organs. The pathogenesis of DRESS syndrome is still unknown. Immunological factors such as a defect in detoxification of culprit drugs and infections seem to be involved. The most commonly associated drugs are anticonvulsants and sulfonamides, but dapsone, allopurinol, and minocycline also have been reported to be associated with DRESS syndrome.1
Although therapies for postmenopausal osteoporosis are considered to be safe from cutaneous side effects, there have been several reported cases of DRESS syndrome associated with strontium ranelate.2 Strontium ranelate is not used in the United States; nevertheless, some US patients may be taking this drug as an alternative to the current US Food and Drug Administration–approved drugs for osteoporosis. We report a case of DRESS syndrome in a woman who developed an extensive maculopapular rash, eosinophilia, dyspnea, bilateral cervical lymphadenopathy, and reactivation of Epstein-Barr virus (EBV) with liver damage 3 weeks after administration of strontium ranelate for postmenopausal osteoporosis. Approximately 6 months after total remission of skin conditions, the patient developed autoimmune hepatitis.
Case Report
A 64-year-old woman presented to the emergency department with dyspnea, fever (temperature, 38.5°C), and a generalized rash that had developed a few days prior. The patient reported that she was previously in good health and had no prior allergic episodes. She had been taking strontium ranelate for 3 weeks to treat postmenopausal osteoporosis and reported no other medication use. The patient was hospitalized because of worsening symptoms. Physical examination revealed a pruritic maculopapular rash involving the trunk, arms, and legs (Figure 1) with facial edema, mild inspiratory as well as expiratory dyspnea, and wheezing on all lung fields. An enlarged soft liver (6–7 cm from the right costal arch) and cervical bilateral lymphadenopathy were found.
A chest radiograph detected a slight increase of the peribronchial thickening with interstitial involvement at the bilateral basal and perihilar levels, and an ultrasound of the chest confirmed the presence of many enlarged cervical bilateral lymph nodes between 2 and 4 cm in diameter.
Laboratory tests revealed the following values: leukocytosis (21,390/μL [reference range, 4500–11,000/μL]) with eosinophilia (27% [reference range, 2.7%]; 5780/μL [reference range, 0–450/μL]), elevated C-reactive protein (20 mg/L [reference range, 0.08–3.1 mg/L]), elevated erythrocyte sedimentation rate (35 mm/h [reference range, 0–20 mm/h]), a reactivation of EBV confirmed by simultaneous seropositivity to early antigen IgM and EBV nuclear antigen, liver damage with notable increases in liver function tests (aspartate aminotransferase, 51 U/L [reference range, 10–30 U/L]); alanine aminotransferase, 104 U/L [reference range 10–40 U/L]); γ-glutamyltransferase, 52 U/L [reference range, 2–30 U/L]), and no thyroid dysfunction.
Blood and urine cultures; antinuclear antibodies; and serology for hepatitis A, B, and C virus, as well as herpes simplex virus type 6 (HHV-6), chlamydia, Mycoplasma, and cytomegalovirus (CMV) were all negative. Histologic examination after skin biopsy showed keratinocytes with spongiosis, intraepidermal eosinophilic infiltration, suffusion of red blood cells with perivascular granulocytes, and lymphocyte inflammatory infiltrate (Figure 2).
A diagnosis of DRESS syndrome was made on the basis of the following clinical data supported by laboratory findings: generalized maculopapular rash, eosinophilia, lung involvement with dyspnea, bilateral cervical lymphadenopathy, and liver damage, as well as an identified reactivation of EBV and onset of symptoms 3 weeks after treatment with strontium ranelate.
The patient was given intravenous methylprednisolone 120 mg once daily for 1 week in gradually decreasing doses. Three weeks of steroid therapy were necessary to obtain the first good results. Improvement of the patient’s clinical condition was considerably slow. Fever and rash gradually disappeared and the patient was discharged with oral corticosteroids. In the 2 months after starting systemic corticosteroid therapy, the lesions had not progressed and all other clinical symptoms improved. A slow but notable regression of the skin reaction was observed.
In a subsequent checkup approximately 8 months following initial presentation, the patient developed autoimmune hepatitis. There was a notable increase in liver enzymes and serum immunoglobulin content as well as positivity of antinuclear antibodies (1:160) and antimitochondrial antibodies (1:160). A liver biopsy was performed and confirmed the histologic pattern of autoimmune hepatitis. Thyroid function was reevaluated, but no other autoimmune disease was identified.
The patient was given another dose of steroids (prednisolone 25 mg daily). Liver function normalized within 1 month (aspartate aminotransferase levels went from 195 U/L to 21 U/L; alanine aminotransferase went from 324 U/L to 21 U/L; γ-glutamyltransferase went from 268 U/L to 63 U/L). The patient is currently taking a maintenance dose of prednisolone 5 mg and has normal liver function.
Comment
Uses of Strontium Ranelate
Strontium ranelate is recommended for reducing the risk for fracture in postmenopausal women 70 years and older with a bone mineral density T-score of –3.0 or lower (ie, primary prevention) as well as for the treatment of morphometric vertebral fracture in established postmenopausal osteoporosis (ie, secondary prevention). Strontium ranelate has a dual action that includes increasing bone formation and reducing bone resorption, leading to rebalancing of bone remodeling in favor of bone formation. Strontium ranelate was shown to increase the recruitment and activity of osteoblastic cells and to inhibit the recruitment and activity of osteoclasts.2 The recommended dose of oral strontium ranelate is 2 g once daily.
Side Effects of Strontium Ranelate
In a 3-year study of side effects associated with strontium ranelate, severe reactions were described in 23% of the reported adverse effects in 844 patients.3 In this study, cardiovascular effects, particularly thromboembolism, and DRESS syndrome were the most frequent side effects. Since its introduction in the market, at least 16 cases of DRESS syndrome related to strontium ranelate use have been reported in Europe, including 2 fatal cases.2 Two deaths have been reported to be associated with this drug,2 which was the basis of the warning document by the European Medicines Agency regarding the risk for strontium ranelate inducing DRESS syndrome.4
Development of DRESS Syndrome
The most common agents involved in DRESS syndrome are anticonvulsants, sulfonamides, dapsone, minocycline, allopurinol, and gold salts, as well as celecoxib, antituberculosis drugs, nonsteroidal anti-inflammatory drugs, antibiotics, calcium channel blockers, and antiretroviral drugs.5,6 The mortality rate of DRESS syndrome is 10%.6
The pathophysiology of DRESS syndrome is still unclear. Altered drug metabolism, genetic predisposition, and concomitant infection or reactivation of bacterial or viral infection (eg, HHV-6, EBV, CMV, human immunodeficiency virus, influenza, viral hepatitis) could be factors leading to development of DRESS. Autoimmune or connective-tissue diseases also have been suggested to increase the risk.7
Clinicians should suspect DRESS syndrome in any patient developing a rash 3 to 6 weeks after starting drug therapy. This disorder often starts with fever (temperature >38°C) and includes cutaneous symptoms such as generalized rash that may progress to exfoliative dermatitis. There usually is involvement of one or several internal organs with the development of hepatitis; interstitial pneumonia; interstitial nephritis; myopericarditis; myositis; pancreatitis; thyroiditis; and hematological abnormalities, primarily eosinophilia or atypical lymphocytosis. Facial edema and lymphadenopathy also may be present. A skin biopsy can confirm the clinical diagnosis of DRESS syndrome but is not specific because cutaneous histologic patterns often show a lymphocytic infiltrate that sometimes mimics cutaneous lymphoma. Other diseases that DRESS syndrome may mimic include Stevens-Johnson syndrome and toxic epidermal necrolysis as well as Kawasaki disease, Still disease, acute viral infections, idiopathic hypereosinophilic syndrome, and lymphoma, which should be excluded from the differential diagnosis.8
Diagnosis of DRESS Syndrome
There is no gold standard for the diagnosis of DRESS syndrome. In our case, the diagnosis of DRESS syndrome was based on the RegiSCAR (European Registry of Severe Cutaneous Adverse Reactions to Drugs) score as described by Kardaun et al,9 which grades DRESS syndrome cases as excluded (<2 points), possible (2–3 points), probable (4–5 points), or definite (>5 points) based on the following clinical criteria: fever (temperature >38.5°C; from a minimum of –1 point if absent to a maximum of 0 points if present); enlarged lymph nodes (from a minimum of 0 points if absent to a maximum of 1 point if present); eosinophilia (0 points if absent, 1 point if 10%–19% or 700–1500 μL, 2 points if ≥20% or >1500 μL); atypical lymphocytes (from a minimum of 0 points if absent to maximum of 1 point if present); skin involvement with rash (1 point if >50% of body surface area is involved, 1 point if there is a maculopapular rash, 1 point if skin biopsy suggests DRESS syndrome); organ involvement (1 point each for liver, kidneys, lungs, muscle/heart, pancreas, and other organs); resolution in at least 15 days (from a minimum of –1 point if absent to maximum of 0 points if present); and evaluation of other potential causes measuring antinuclear antibodies, blood culture, and serology for hepatitis virus (A–C), chlamydia, and Mycoplasma (1 point if 3 or more are negative and none positive). Virus reactivation also should be considered a main characteristic of DRESS syndrome. Therefore, its prevalence is not homogenous, so the absence of viral reactivation cannot be considered exclusion criteria. Several case reports and a few well-documented series have evidenced markers of virus reactivation in many cases of DRESS. Herpes simplex virus 6, CMV, and EBV are the most frequently reactivated.
The total RegiSCAR score of 8 in our case was taken as a definite indication of DRESS syndrome (temperature, 38.5°C [0 points]; enlarged lymph nodes [1 point]; eosinophilia, ≥20% or >1500 μL [2 points]; skin involvement with >50% body surface area involved [1 point] with a maculopapular rash [1 point] and histopathologic findings suggesting DRESS syndrome [1 point]; lung and liver involvement [2 points]). The causative drug was identified by carefully collecting the patient’s medication history and by evaluating clinical outcome characterized by improved skin and systemic symptoms after discontinuation of strontium ranelate.
Because of the high morbidity of DRESS syndrome, it needs to be diagnosed effectively and must be considered in the differential for any patient developing the triad of skin rash, hypereosinophilia, and systemic symptoms, as well as several other side effects when taking strontium ranelate.10
Therapies for DRESS Syndrome
Treatment of DRESS syndrome has not yet been standardized. Prompt withdrawal of the causative drug is the only mandatory activity in the treatment of DRESS syndrome. Systemic corticosteroids may be needed for organ or life-threatening disease, though the efficacy is controversial because it may result in activation of HHV-6, which in turn is probably involved in the pathogenesis of DRESS syndrome.
Conclusion
This case confirms that strontium ranelate should be considered a possible factor in the etiopathology of DRESS syndrome and in the development of autoimmune hepatitis as a part of DRESS syndrome. Case reports underline the importance of recognition of cutaneous adverse reactions in patients undergoing treatment of postmenopausal osteoporosis. The prognosis is good with immediate recognition followed by immediate and permanent withdrawal of the drug, along with hospitalization and systemic corticosteroids when necessary. The possibility of developing autoimmune hepatitis as a part of DRESS syndrome related to strontium ranelate has been reported,11 usually months after the acute episode.
- Tas S, Simonart T. Management of drug rash with eosinophilia and systemic symptoms (DRESS syndrome): an update. Dermatology. 2003;206:353-356.
- Le Merlouette M, Adamski H, Dinulescu M, et al. Strontium ranelate–induced DRESS syndrome. Ann Dermatol Venereol. 2011;138:124-128.
- Jonville-Bera AP, Autret-Leca E. Adverse drug reactions of strontium ranelate (Protelos®) in France. Presse Med. 2011;40:453-462.
- Assessment report for Protelos and Osseor. European Medicines Agency website. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Assessment_Report_-_Variation/human/000560/WC500131789.pdf). Published May 25, 2012. Accessed May 9, 2016.
- Breathnach S. Drug rash eosinophilia and systemic symptoms (DRESS) syndrome. types of clinical reaction: drug reaction. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. Vol 4. 8th ed. Hoboken, NJ: Oxford Wiley-Blackwell Publications; 2010:75.26.
- Lee JH, Park HK, Heo J, et al. Drug rash with eosinophilia and systemic symptoms (DRESS) syndrome induced by celecoxib and anti-tuberculosis drugs. J Korean Med Sci. 2008;23:521-525.
- Musette P, Brandi ML, Cacoub P, et al. Treatment of osteoporosis: recognizing and managing cutaneous adverse reactions and drug-induced hypersensitivity. Osteoporos Int. 2010;21:723-732.
- Telles Rudge de Aquino R, Vieitas Vergueiro CS, Ruffolo Magliari ME, et al. Sulfasalazine-induced DRESS syndrome (drug rash with eosinophilia and systemic symptoms). Sao Paulo Med J. 2008;126:225-226.
- Kardaun SH, Sidoroff A, Valeyrie-Allanore L, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2007;156:609-611.
- Pernicova I, Middleton ET, Aye M. Rash, strontium ranelate and DRESS syndrome put into perspective. European Medicine Agency on the alert [published online September 20, 2008]. Osteoporos Int. 2008;19:1811-1812.
- Kinyó A, Belsö N, Nagy N, et al. Strontium ranelate-induced DRESS syndrome with persistent autoimmune hepatitis. Acta Derm Venereol. 2011;91:205-206.
- Tas S, Simonart T. Management of drug rash with eosinophilia and systemic symptoms (DRESS syndrome): an update. Dermatology. 2003;206:353-356.
- Le Merlouette M, Adamski H, Dinulescu M, et al. Strontium ranelate–induced DRESS syndrome. Ann Dermatol Venereol. 2011;138:124-128.
- Jonville-Bera AP, Autret-Leca E. Adverse drug reactions of strontium ranelate (Protelos®) in France. Presse Med. 2011;40:453-462.
- Assessment report for Protelos and Osseor. European Medicines Agency website. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Assessment_Report_-_Variation/human/000560/WC500131789.pdf). Published May 25, 2012. Accessed May 9, 2016.
- Breathnach S. Drug rash eosinophilia and systemic symptoms (DRESS) syndrome. types of clinical reaction: drug reaction. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. Vol 4. 8th ed. Hoboken, NJ: Oxford Wiley-Blackwell Publications; 2010:75.26.
- Lee JH, Park HK, Heo J, et al. Drug rash with eosinophilia and systemic symptoms (DRESS) syndrome induced by celecoxib and anti-tuberculosis drugs. J Korean Med Sci. 2008;23:521-525.
- Musette P, Brandi ML, Cacoub P, et al. Treatment of osteoporosis: recognizing and managing cutaneous adverse reactions and drug-induced hypersensitivity. Osteoporos Int. 2010;21:723-732.
- Telles Rudge de Aquino R, Vieitas Vergueiro CS, Ruffolo Magliari ME, et al. Sulfasalazine-induced DRESS syndrome (drug rash with eosinophilia and systemic symptoms). Sao Paulo Med J. 2008;126:225-226.
- Kardaun SH, Sidoroff A, Valeyrie-Allanore L, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2007;156:609-611.
- Pernicova I, Middleton ET, Aye M. Rash, strontium ranelate and DRESS syndrome put into perspective. European Medicine Agency on the alert [published online September 20, 2008]. Osteoporos Int. 2008;19:1811-1812.
- Kinyó A, Belsö N, Nagy N, et al. Strontium ranelate-induced DRESS syndrome with persistent autoimmune hepatitis. Acta Derm Venereol. 2011;91:205-206.
Practice Points
- Drug rash with eosinophilia and systemic symptoms (DRESS) syndrome refers to a severe, acute, potentially fatal, multisystem adverse drug reaction characterized by skin rash, fever, hematological abnormalities, and lymphadenopathy with involvement of several internal organs.
- Strontium ranelate should be considered as a possible factor in the etiopathology of DRESS syndrome and in the development of autoimmune hepatitis as a part of DRESS syndrome.

Lupus Erythematosus and Localized Scleroderma Coexistent at the Same Sites: A Rare Presentation of Overlap Syndrome of Connective-Tissue Diseases
Although lupus erythematosus (LE) and scleroderma are regarded as 2 distinct entities, there have been multiple cases described in the literature showing an overlap between these 2 disease processes. We report the case of a 60-year-old man with clinical and histopathologic findings consistent with the presence of localized scleroderma and discoid LE (DLE) within the same lesions. We also present a review of the literature and delineate the general patterns of coexistence of these 2 diseases based on our case and other reported cases.
Case Report
A 60-year-old man presented with a progressive pruritic rash on the face, neck, and upper back of approximately 20 to 30 years’ duration. On initial evaluation, the patient was found to have indurated hypopigmented plaques with follicular plugging bilaterally on the cheeks, temples, ears, and upper back (Figure 1). Punch biopsies were performed on the left cheek and upper back. Histopathology was notable for vacuolar interface dermatitis with dermal sclerosis at both sites. Specifically, interface changes, basement membrane thickening, and periadnexal inflammation were present on histopathologic examination from both biopsies supporting a diagnosis of DLE (Figure 2A). However, there also was sclerosis present in the reticular dermis, suggesting a diagnosis of localized scleroderma (Figure 2B). Direct immunofluorescence was negative for a lupus band. Laboratory workup was positive for antinuclear antibody (titer, 1:40; speckled pattern) and anti–Sjögren syndrome antigen A but negative for double-stranded DNA antibody, anti-Smith antibody, anti–Sjögren syndrome antigen B, and Scl-70.

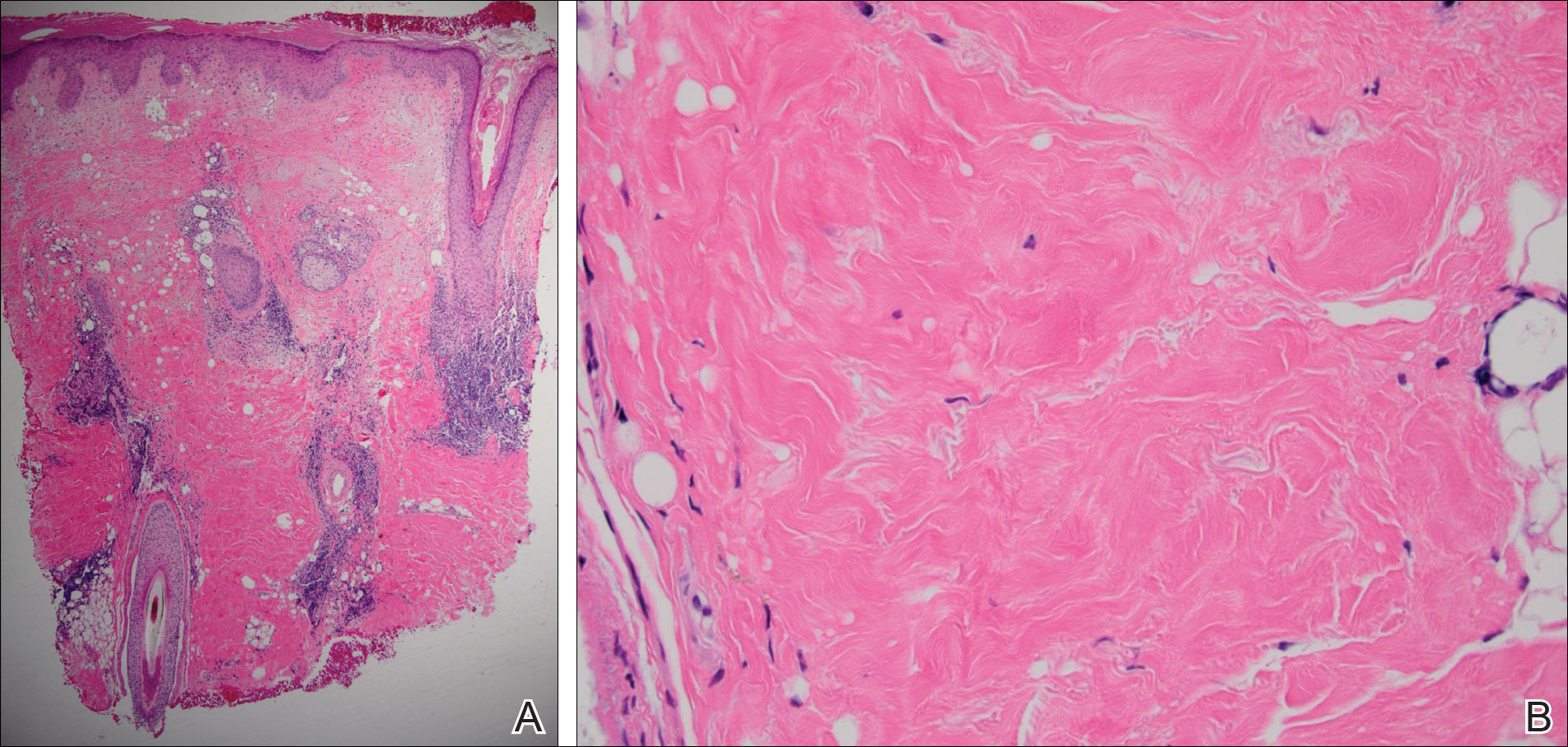
The patient was started on oral hydroxychloroquine 200 mg twice daily and clobetasol oint-ment 0.05% twice daily to affected areas. After 2 weeks of treatment, he developed urticaria on the trunk and the hydroxychloroquine was discontinued. He continued using only topical steroids following a regimen of applying clobetasol ointment 0.05% twice daily for 2 weeks, alternating with the use of triamcinolone ointment 0.1% twice daily for 2 weeks with improvement of the pruritus, but the induration and hypopigmentation remained unchanged. Alternative systemic medication was started with mycophenolate mofetil 1 g twice daily. The patient showed remarkable clinical improvement with a decrease in induration and partial resolution of follicular plugging after 4 months of treatment with mycophenolate mofetil in combination with the topical steroid regimen.
Comment
Autoimmune connective-tissue diseases (CTDs) often occur with a wide range of symptoms and signs. Most often patients affected by these diseases can be sorted into one of the named CTDs such as LE, rheumatoid arthritis, scleroderma, polymyositis/dermatomyositis, and Sjögren syndrome. On the other hand, it is widely recognized that patients with one classic autoimmune CTD are likely to possess multiple autoantibodies, and a small number of these patients develop symptoms and/or signs that satisfy the diagnostic criteria of a second autoimmune CTD; these latter patients are said to have an overlap syndrome.1 The development of a second identifiable CTD, hence indicating an overlap syndrome, may occur coincident to the initial CTD or may occur at a different time.1
Essentially all 5 of the CTDs mentioned above have been reported to occur in combination with one another. Most of the reports involving overlap among these 5 CTDs include patients with multiorgan systemic involvement without cutaneous involvement, leading to a fairly simple straightforward classification of overlap syndromes as viewed by rheumatologists.1
When the overlap occurs between the localized forms of scleroderma and purely cutaneous LE, the situation becomes even more complicated, as the skin lesions of the 2 diseases may occur at separate locations or coexistent disease may develop in the same location, as in our case.
More than 100 cases have been reported wherein LE and scleroderma coexist in the same patient.1 Most of these cases have been examples of type 1 overlap (Table 1), though a few have been type 2 overlap, with localized scleroderma coexisting with systemic LE or vice versa.1,2 There are rare reports of an overlap of the localized form of both of these entities (type 3 overlap), as demonstrated in our patient. According to a PubMed search of articles indexed for MEDLINE using the search terms localized scleroderma and morphea as well as discoid lupus erythematosus, we found 12 other cases describing type 3 overlap (Table 2).

The first case was described in 1976 as annular atrophic plaques on the face and neck of a 48-year-old man.3 As in our case, there were overlapping features of DLE and localized scleroderma. The investigators postulated that the entity was an atypical form of DLE.3 There were 4 more cases described in 1978, but the majority of these patients were young women with linear plaques. Instead of calling the disease a new form of DLE, the investigators considered it to be an overlap syndrome.4 Many years passed before another similar case was described in the literature in 1990.5 Interestingly, the investigators performed multiple biopsies on this patient over several years and observed that the pathology changed from subacute cutaneous LE to an overlap of subacute cutaneous LE and localized scleroderma to localized scleroderma, suggesting that localized scleroderma was the end result of persistent inflammation from the cutaneous LE lesions. The investigators compared the evolution of subacute cutaneous LE to localized scleroderma in the patient to the evolution of acute graft-versus-host disease (GVHD) to chronic GVHD. Acute GVHD has a lichenoid tissue reaction that develops into sclerosis in the chronic form.5
Additionally, there were 3 cases in the literature showing an overlap of lupus panniculitis with localized scleroderma.6,7 Stork and Vosmik6 described a case of a 22-year-old woman with lesions clinically suspicious for localized scleroderma, with lupus panniculitis demonstrated on histopathology. They discussed the difficulty in differentiating between lupus panniculitis and localized scleroderma but did not specify whether they believed the case represented a distinct entity or an overlap syndrome.6 Alternatively, Marzano et al7 reported 2 similar cases, which the investigators considered to be a specific new variant called sclerodermic linear lupus panniculitis.
In the last 10 years, there were 3 additional cases reported that described an overlap of DLE and localized scleroderma in the same anatomic location, similar to our patient.8-10 Although Julia et al8 considered their case to be an example of the distinct entity called sclerodermiform linear LE, the investigators in the other 2 cases described the possibility of an overlap syndrome.9,10
Based on reported cases, we found the following patterns in the overlap of cutaneous LE and localized scleroderma: predilection for young women, photodistributed lesions, DLE, linear morphology clinically, and positivity along the dermoepidermal junction on direct immunofluorescence. As in our case, the few affected men were older compared to affected women. Men ranged in age from 34 to 48 years compared to women who ranged in age from 7 to 29 years. We did not find a pattern in the laboratory findings in these patients. Most patients had a good response to antimalarials, topical steroids, or systemic steroids.
Conclusion
All 12 previously reported cases showed some form of overlap of cutaneous LE and localized scleroderma. As previously discussed, overlap syndromes are common in patients with CTDs. We postulate that our case represents a rare form of overlap syndrome, with the overlap occurring at the same clinical sites.
- Iaccarino L, Gatto M, Bettio S, et al. Overlap connective tissue disease syndromes [published online June 26, 2012]. Autoimmun Reviews. 2012;12:363-373.
- Balbir-Gurman A, Braun-Moscovici Y. Scleroderma overlap syndrome. Isr Med Assoc J. 2011;13:14-20.
- Chorzelski TP, Jablonska S, Blaszyczyk M, et al. Annular atrophic plaques of the face. Arch Dermatol. 1976;112:1143-1145.
- Umbert P, Winkelmann RK. Concurrent localized scleroderma and discoid lupus erythematosus. Arch Dermatol. 1978;114:1473-1478.
- Rao BK, Coldiron B, Freeman RG, et al. Subacute cutaneous lupus progressing to morphea erythematosus lesions. J Am Acad Dermatol. 1990;23(5, pt 2):1019-1022.
- Stork J, Vosmik F. Lupus erythematosus panniculitis with morphea-like lesions. Clin Exp Dermatol. 1994;19:79-82.
- Marzano AV, Tanzi C, Caputo R, et al. Sclerodermic linear lupus panniculitis: report of two cases. Dermatology. 2005;210:329-332.
- Julia M, Mascaro JM Jr, Guilaber A, et al. Sclerodermiform linear lupus erythematosus: a distinct entity or coexistence of two autoimmune diseases? J Am Acad Dermatol. 2008;58:665-667.
- Mir A, Tlougan B, O’Reilly K, et al. Morphea with discoid lupus erythematosus. Dermatol Online J. 2011;17:10.
- Khelifa E, Masouye I, Pham HC, et al. Linear sclerodermic lupus erythematosus, a distinct variant of linear morphea and chronic cutaneous lupus erythematosus. Int J Dermatol. 2011;50:1491-1495.
Although lupus erythematosus (LE) and scleroderma are regarded as 2 distinct entities, there have been multiple cases described in the literature showing an overlap between these 2 disease processes. We report the case of a 60-year-old man with clinical and histopathologic findings consistent with the presence of localized scleroderma and discoid LE (DLE) within the same lesions. We also present a review of the literature and delineate the general patterns of coexistence of these 2 diseases based on our case and other reported cases.
Case Report
A 60-year-old man presented with a progressive pruritic rash on the face, neck, and upper back of approximately 20 to 30 years’ duration. On initial evaluation, the patient was found to have indurated hypopigmented plaques with follicular plugging bilaterally on the cheeks, temples, ears, and upper back (Figure 1). Punch biopsies were performed on the left cheek and upper back. Histopathology was notable for vacuolar interface dermatitis with dermal sclerosis at both sites. Specifically, interface changes, basement membrane thickening, and periadnexal inflammation were present on histopathologic examination from both biopsies supporting a diagnosis of DLE (Figure 2A). However, there also was sclerosis present in the reticular dermis, suggesting a diagnosis of localized scleroderma (Figure 2B). Direct immunofluorescence was negative for a lupus band. Laboratory workup was positive for antinuclear antibody (titer, 1:40; speckled pattern) and anti–Sjögren syndrome antigen A but negative for double-stranded DNA antibody, anti-Smith antibody, anti–Sjögren syndrome antigen B, and Scl-70.
The patient was started on oral hydroxychloroquine 200 mg twice daily and clobetasol oint-ment 0.05% twice daily to affected areas. After 2 weeks of treatment, he developed urticaria on the trunk and the hydroxychloroquine was discontinued. He continued using only topical steroids following a regimen of applying clobetasol ointment 0.05% twice daily for 2 weeks, alternating with the use of triamcinolone ointment 0.1% twice daily for 2 weeks with improvement of the pruritus, but the induration and hypopigmentation remained unchanged. Alternative systemic medication was started with mycophenolate mofetil 1 g twice daily. The patient showed remarkable clinical improvement with a decrease in induration and partial resolution of follicular plugging after 4 months of treatment with mycophenolate mofetil in combination with the topical steroid regimen.
Comment
Autoimmune connective-tissue diseases (CTDs) often occur with a wide range of symptoms and signs. Most often patients affected by these diseases can be sorted into one of the named CTDs such as LE, rheumatoid arthritis, scleroderma, polymyositis/dermatomyositis, and Sjögren syndrome. On the other hand, it is widely recognized that patients with one classic autoimmune CTD are likely to possess multiple autoantibodies, and a small number of these patients develop symptoms and/or signs that satisfy the diagnostic criteria of a second autoimmune CTD; these latter patients are said to have an overlap syndrome.1 The development of a second identifiable CTD, hence indicating an overlap syndrome, may occur coincident to the initial CTD or may occur at a different time.1
Essentially all 5 of the CTDs mentioned above have been reported to occur in combination with one another. Most of the reports involving overlap among these 5 CTDs include patients with multiorgan systemic involvement without cutaneous involvement, leading to a fairly simple straightforward classification of overlap syndromes as viewed by rheumatologists.1
When the overlap occurs between the localized forms of scleroderma and purely cutaneous LE, the situation becomes even more complicated, as the skin lesions of the 2 diseases may occur at separate locations or coexistent disease may develop in the same location, as in our case.
More than 100 cases have been reported wherein LE and scleroderma coexist in the same patient.1 Most of these cases have been examples of type 1 overlap (Table 1), though a few have been type 2 overlap, with localized scleroderma coexisting with systemic LE or vice versa.1,2 There are rare reports of an overlap of the localized form of both of these entities (type 3 overlap), as demonstrated in our patient. According to a PubMed search of articles indexed for MEDLINE using the search terms localized scleroderma and morphea as well as discoid lupus erythematosus, we found 12 other cases describing type 3 overlap (Table 2).
The first case was described in 1976 as annular atrophic plaques on the face and neck of a 48-year-old man.3 As in our case, there were overlapping features of DLE and localized scleroderma. The investigators postulated that the entity was an atypical form of DLE.3 There were 4 more cases described in 1978, but the majority of these patients were young women with linear plaques. Instead of calling the disease a new form of DLE, the investigators considered it to be an overlap syndrome.4 Many years passed before another similar case was described in the literature in 1990.5 Interestingly, the investigators performed multiple biopsies on this patient over several years and observed that the pathology changed from subacute cutaneous LE to an overlap of subacute cutaneous LE and localized scleroderma to localized scleroderma, suggesting that localized scleroderma was the end result of persistent inflammation from the cutaneous LE lesions. The investigators compared the evolution of subacute cutaneous LE to localized scleroderma in the patient to the evolution of acute graft-versus-host disease (GVHD) to chronic GVHD. Acute GVHD has a lichenoid tissue reaction that develops into sclerosis in the chronic form.5
Additionally, there were 3 cases in the literature showing an overlap of lupus panniculitis with localized scleroderma.6,7 Stork and Vosmik6 described a case of a 22-year-old woman with lesions clinically suspicious for localized scleroderma, with lupus panniculitis demonstrated on histopathology. They discussed the difficulty in differentiating between lupus panniculitis and localized scleroderma but did not specify whether they believed the case represented a distinct entity or an overlap syndrome.6 Alternatively, Marzano et al7 reported 2 similar cases, which the investigators considered to be a specific new variant called sclerodermic linear lupus panniculitis.
In the last 10 years, there were 3 additional cases reported that described an overlap of DLE and localized scleroderma in the same anatomic location, similar to our patient.8-10 Although Julia et al8 considered their case to be an example of the distinct entity called sclerodermiform linear LE, the investigators in the other 2 cases described the possibility of an overlap syndrome.9,10
Based on reported cases, we found the following patterns in the overlap of cutaneous LE and localized scleroderma: predilection for young women, photodistributed lesions, DLE, linear morphology clinically, and positivity along the dermoepidermal junction on direct immunofluorescence. As in our case, the few affected men were older compared to affected women. Men ranged in age from 34 to 48 years compared to women who ranged in age from 7 to 29 years. We did not find a pattern in the laboratory findings in these patients. Most patients had a good response to antimalarials, topical steroids, or systemic steroids.
Conclusion
All 12 previously reported cases showed some form of overlap of cutaneous LE and localized scleroderma. As previously discussed, overlap syndromes are common in patients with CTDs. We postulate that our case represents a rare form of overlap syndrome, with the overlap occurring at the same clinical sites.
Although lupus erythematosus (LE) and scleroderma are regarded as 2 distinct entities, there have been multiple cases described in the literature showing an overlap between these 2 disease processes. We report the case of a 60-year-old man with clinical and histopathologic findings consistent with the presence of localized scleroderma and discoid LE (DLE) within the same lesions. We also present a review of the literature and delineate the general patterns of coexistence of these 2 diseases based on our case and other reported cases.
Case Report
A 60-year-old man presented with a progressive pruritic rash on the face, neck, and upper back of approximately 20 to 30 years’ duration. On initial evaluation, the patient was found to have indurated hypopigmented plaques with follicular plugging bilaterally on the cheeks, temples, ears, and upper back (Figure 1). Punch biopsies were performed on the left cheek and upper back. Histopathology was notable for vacuolar interface dermatitis with dermal sclerosis at both sites. Specifically, interface changes, basement membrane thickening, and periadnexal inflammation were present on histopathologic examination from both biopsies supporting a diagnosis of DLE (Figure 2A). However, there also was sclerosis present in the reticular dermis, suggesting a diagnosis of localized scleroderma (Figure 2B). Direct immunofluorescence was negative for a lupus band. Laboratory workup was positive for antinuclear antibody (titer, 1:40; speckled pattern) and anti–Sjögren syndrome antigen A but negative for double-stranded DNA antibody, anti-Smith antibody, anti–Sjögren syndrome antigen B, and Scl-70.
The patient was started on oral hydroxychloroquine 200 mg twice daily and clobetasol oint-ment 0.05% twice daily to affected areas. After 2 weeks of treatment, he developed urticaria on the trunk and the hydroxychloroquine was discontinued. He continued using only topical steroids following a regimen of applying clobetasol ointment 0.05% twice daily for 2 weeks, alternating with the use of triamcinolone ointment 0.1% twice daily for 2 weeks with improvement of the pruritus, but the induration and hypopigmentation remained unchanged. Alternative systemic medication was started with mycophenolate mofetil 1 g twice daily. The patient showed remarkable clinical improvement with a decrease in induration and partial resolution of follicular plugging after 4 months of treatment with mycophenolate mofetil in combination with the topical steroid regimen.
Comment
Autoimmune connective-tissue diseases (CTDs) often occur with a wide range of symptoms and signs. Most often patients affected by these diseases can be sorted into one of the named CTDs such as LE, rheumatoid arthritis, scleroderma, polymyositis/dermatomyositis, and Sjögren syndrome. On the other hand, it is widely recognized that patients with one classic autoimmune CTD are likely to possess multiple autoantibodies, and a small number of these patients develop symptoms and/or signs that satisfy the diagnostic criteria of a second autoimmune CTD; these latter patients are said to have an overlap syndrome.1 The development of a second identifiable CTD, hence indicating an overlap syndrome, may occur coincident to the initial CTD or may occur at a different time.1
Essentially all 5 of the CTDs mentioned above have been reported to occur in combination with one another. Most of the reports involving overlap among these 5 CTDs include patients with multiorgan systemic involvement without cutaneous involvement, leading to a fairly simple straightforward classification of overlap syndromes as viewed by rheumatologists.1
When the overlap occurs between the localized forms of scleroderma and purely cutaneous LE, the situation becomes even more complicated, as the skin lesions of the 2 diseases may occur at separate locations or coexistent disease may develop in the same location, as in our case.
More than 100 cases have been reported wherein LE and scleroderma coexist in the same patient.1 Most of these cases have been examples of type 1 overlap (Table 1), though a few have been type 2 overlap, with localized scleroderma coexisting with systemic LE or vice versa.1,2 There are rare reports of an overlap of the localized form of both of these entities (type 3 overlap), as demonstrated in our patient. According to a PubMed search of articles indexed for MEDLINE using the search terms localized scleroderma and morphea as well as discoid lupus erythematosus, we found 12 other cases describing type 3 overlap (Table 2).
The first case was described in 1976 as annular atrophic plaques on the face and neck of a 48-year-old man.3 As in our case, there were overlapping features of DLE and localized scleroderma. The investigators postulated that the entity was an atypical form of DLE.3 There were 4 more cases described in 1978, but the majority of these patients were young women with linear plaques. Instead of calling the disease a new form of DLE, the investigators considered it to be an overlap syndrome.4 Many years passed before another similar case was described in the literature in 1990.5 Interestingly, the investigators performed multiple biopsies on this patient over several years and observed that the pathology changed from subacute cutaneous LE to an overlap of subacute cutaneous LE and localized scleroderma to localized scleroderma, suggesting that localized scleroderma was the end result of persistent inflammation from the cutaneous LE lesions. The investigators compared the evolution of subacute cutaneous LE to localized scleroderma in the patient to the evolution of acute graft-versus-host disease (GVHD) to chronic GVHD. Acute GVHD has a lichenoid tissue reaction that develops into sclerosis in the chronic form.5
Additionally, there were 3 cases in the literature showing an overlap of lupus panniculitis with localized scleroderma.6,7 Stork and Vosmik6 described a case of a 22-year-old woman with lesions clinically suspicious for localized scleroderma, with lupus panniculitis demonstrated on histopathology. They discussed the difficulty in differentiating between lupus panniculitis and localized scleroderma but did not specify whether they believed the case represented a distinct entity or an overlap syndrome.6 Alternatively, Marzano et al7 reported 2 similar cases, which the investigators considered to be a specific new variant called sclerodermic linear lupus panniculitis.
In the last 10 years, there were 3 additional cases reported that described an overlap of DLE and localized scleroderma in the same anatomic location, similar to our patient.8-10 Although Julia et al8 considered their case to be an example of the distinct entity called sclerodermiform linear LE, the investigators in the other 2 cases described the possibility of an overlap syndrome.9,10
Based on reported cases, we found the following patterns in the overlap of cutaneous LE and localized scleroderma: predilection for young women, photodistributed lesions, DLE, linear morphology clinically, and positivity along the dermoepidermal junction on direct immunofluorescence. As in our case, the few affected men were older compared to affected women. Men ranged in age from 34 to 48 years compared to women who ranged in age from 7 to 29 years. We did not find a pattern in the laboratory findings in these patients. Most patients had a good response to antimalarials, topical steroids, or systemic steroids.
Conclusion
All 12 previously reported cases showed some form of overlap of cutaneous LE and localized scleroderma. As previously discussed, overlap syndromes are common in patients with CTDs. We postulate that our case represents a rare form of overlap syndrome, with the overlap occurring at the same clinical sites.
- Iaccarino L, Gatto M, Bettio S, et al. Overlap connective tissue disease syndromes [published online June 26, 2012]. Autoimmun Reviews. 2012;12:363-373.
- Balbir-Gurman A, Braun-Moscovici Y. Scleroderma overlap syndrome. Isr Med Assoc J. 2011;13:14-20.
- Chorzelski TP, Jablonska S, Blaszyczyk M, et al. Annular atrophic plaques of the face. Arch Dermatol. 1976;112:1143-1145.
- Umbert P, Winkelmann RK. Concurrent localized scleroderma and discoid lupus erythematosus. Arch Dermatol. 1978;114:1473-1478.
- Rao BK, Coldiron B, Freeman RG, et al. Subacute cutaneous lupus progressing to morphea erythematosus lesions. J Am Acad Dermatol. 1990;23(5, pt 2):1019-1022.
- Stork J, Vosmik F. Lupus erythematosus panniculitis with morphea-like lesions. Clin Exp Dermatol. 1994;19:79-82.
- Marzano AV, Tanzi C, Caputo R, et al. Sclerodermic linear lupus panniculitis: report of two cases. Dermatology. 2005;210:329-332.
- Julia M, Mascaro JM Jr, Guilaber A, et al. Sclerodermiform linear lupus erythematosus: a distinct entity or coexistence of two autoimmune diseases? J Am Acad Dermatol. 2008;58:665-667.
- Mir A, Tlougan B, O’Reilly K, et al. Morphea with discoid lupus erythematosus. Dermatol Online J. 2011;17:10.
- Khelifa E, Masouye I, Pham HC, et al. Linear sclerodermic lupus erythematosus, a distinct variant of linear morphea and chronic cutaneous lupus erythematosus. Int J Dermatol. 2011;50:1491-1495.
- Iaccarino L, Gatto M, Bettio S, et al. Overlap connective tissue disease syndromes [published online June 26, 2012]. Autoimmun Reviews. 2012;12:363-373.
- Balbir-Gurman A, Braun-Moscovici Y. Scleroderma overlap syndrome. Isr Med Assoc J. 2011;13:14-20.
- Chorzelski TP, Jablonska S, Blaszyczyk M, et al. Annular atrophic plaques of the face. Arch Dermatol. 1976;112:1143-1145.
- Umbert P, Winkelmann RK. Concurrent localized scleroderma and discoid lupus erythematosus. Arch Dermatol. 1978;114:1473-1478.
- Rao BK, Coldiron B, Freeman RG, et al. Subacute cutaneous lupus progressing to morphea erythematosus lesions. J Am Acad Dermatol. 1990;23(5, pt 2):1019-1022.
- Stork J, Vosmik F. Lupus erythematosus panniculitis with morphea-like lesions. Clin Exp Dermatol. 1994;19:79-82.
- Marzano AV, Tanzi C, Caputo R, et al. Sclerodermic linear lupus panniculitis: report of two cases. Dermatology. 2005;210:329-332.
- Julia M, Mascaro JM Jr, Guilaber A, et al. Sclerodermiform linear lupus erythematosus: a distinct entity or coexistence of two autoimmune diseases? J Am Acad Dermatol. 2008;58:665-667.
- Mir A, Tlougan B, O’Reilly K, et al. Morphea with discoid lupus erythematosus. Dermatol Online J. 2011;17:10.
- Khelifa E, Masouye I, Pham HC, et al. Linear sclerodermic lupus erythematosus, a distinct variant of linear morphea and chronic cutaneous lupus erythematosus. Int J Dermatol. 2011;50:1491-1495.
Practice Points
- Discoid lupus erythematosus and localized scleroderma may rarely overlap within the same lesions.
- Cutaneous overlap syndromes tend to respond well to antimalarials, topical steroids, and systemic steroids.
