User login
Benign Lesion on the Posterior Aspect of the Neck
Nuchal-Type Fibroma
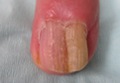
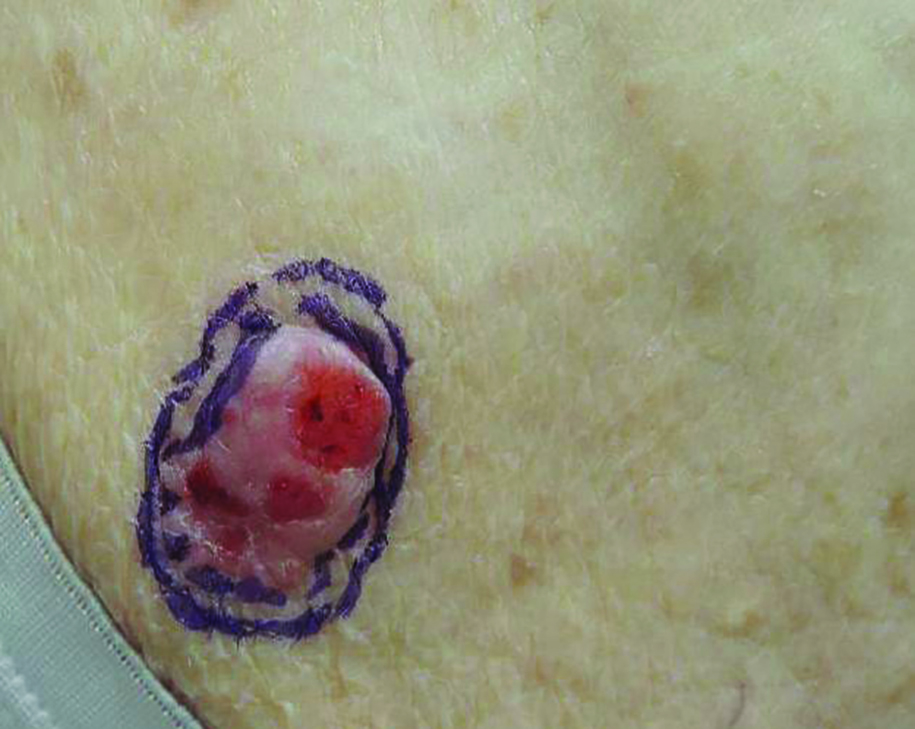
Nuchal-type fibroma (NTF) is a rare benign proliferation of the dermis and subcutis associated with diabetes mellitus and Gardner syndrome.1,2 Forty-four percent of patients with NTF have diabetes mellitus.2 The posterior aspect of the neck is the most frequently affected site, but lesions also may present on the upper back, lumbosacral area, buttocks, and face. Physical examination generally reveals an indurated, asymptomatic, ill-defined, 3-cm or smaller nodule that is hard and white, unencapsulated, and poorly circumscribed.
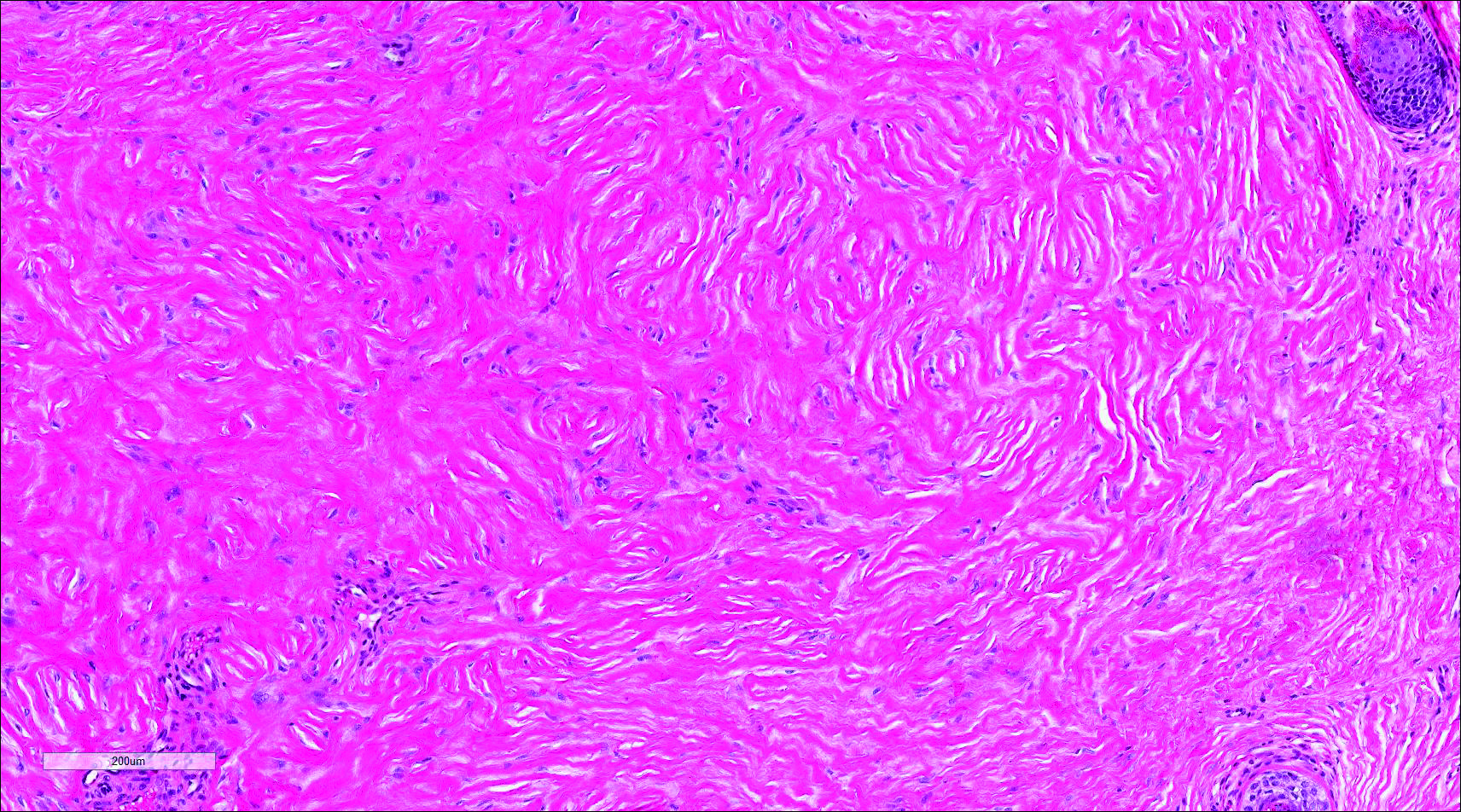
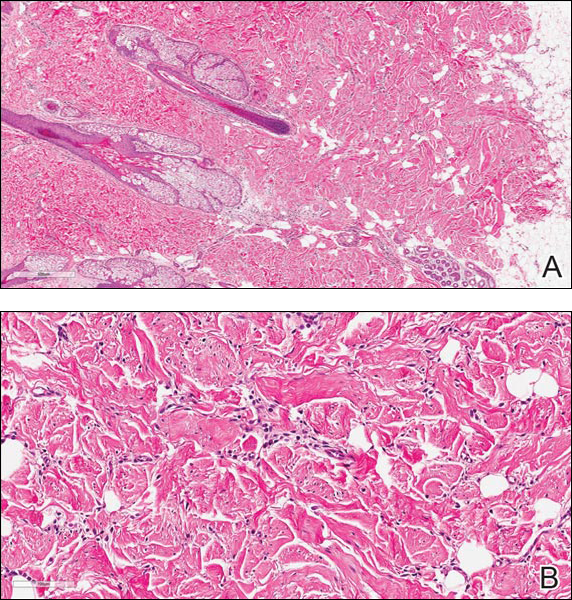
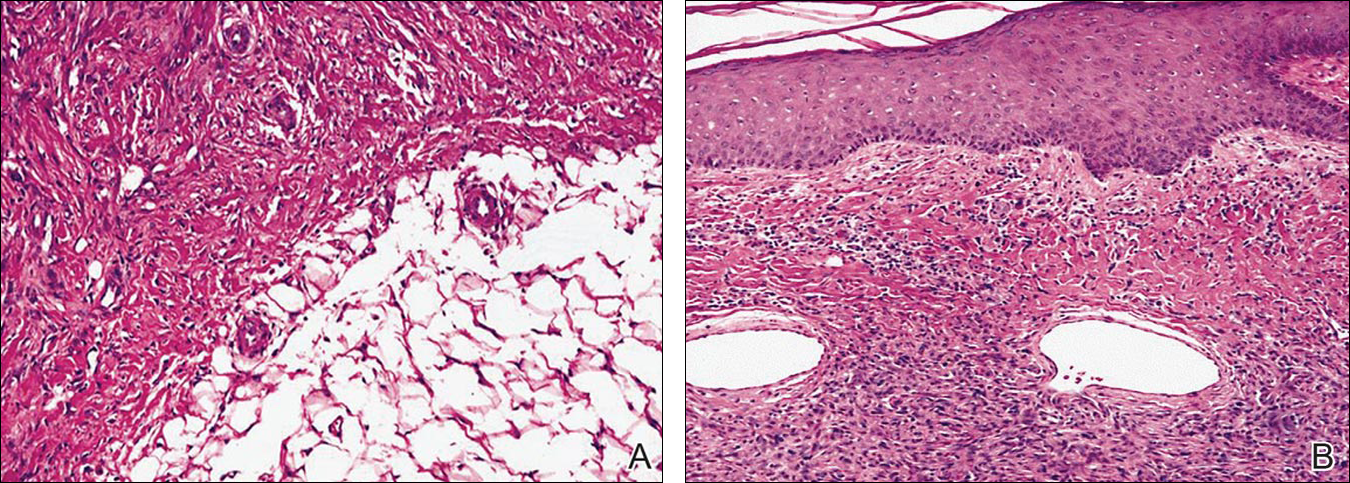
Histopathologic examination of NTF typically reveals a nodular paucicellular proliferation of thick collagen bundles with inconspicuous fibroblasts, radiation of collagenous septa into the subcutaneous fat, and entrapment of mature adipose tissue and small nerves (quiz image A). Collagen bundles are thickened with entrapment of adipose tissue without increased cellularity (quiz image B). S-100 staining can show the entrapped nerves.
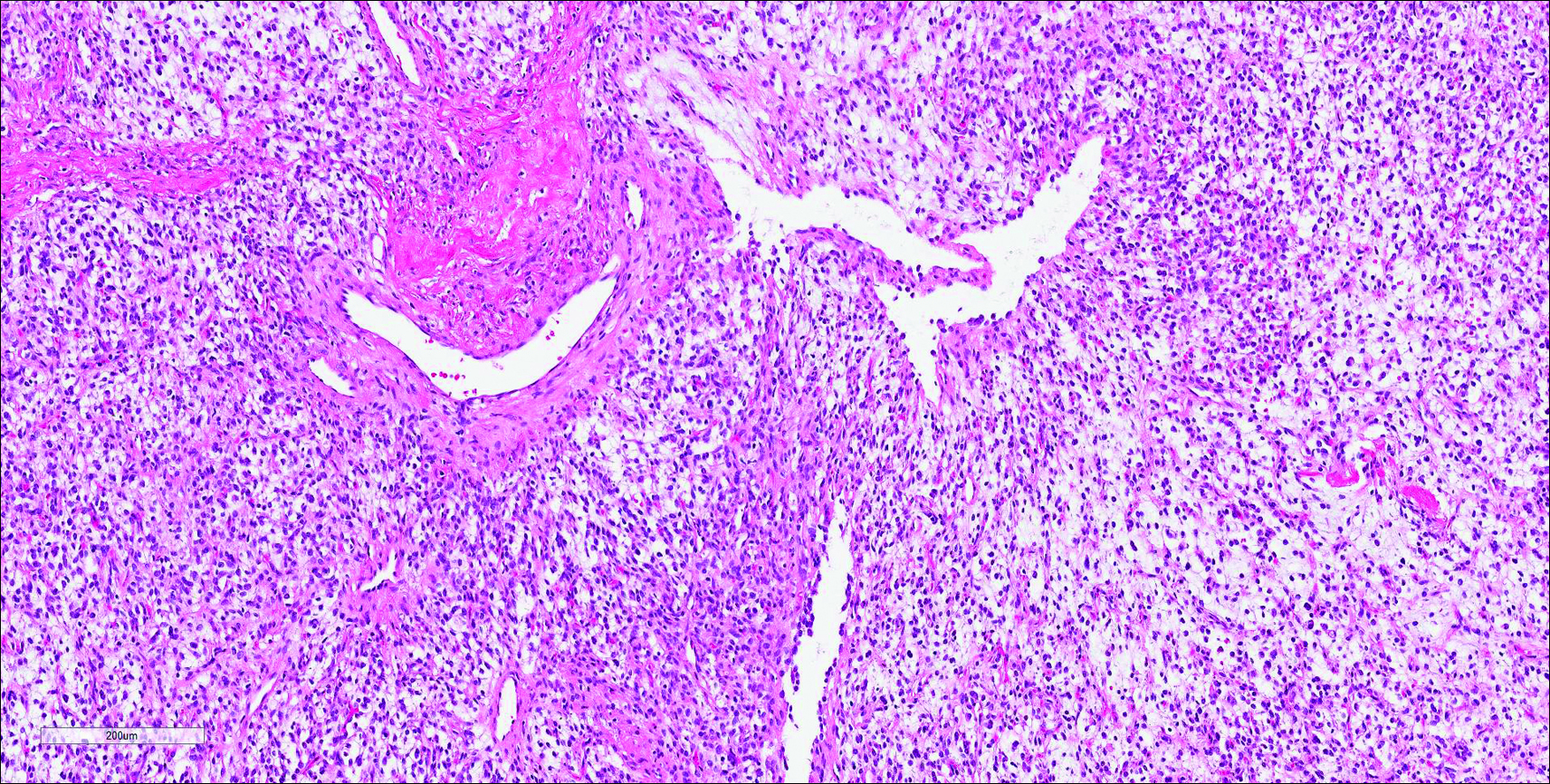
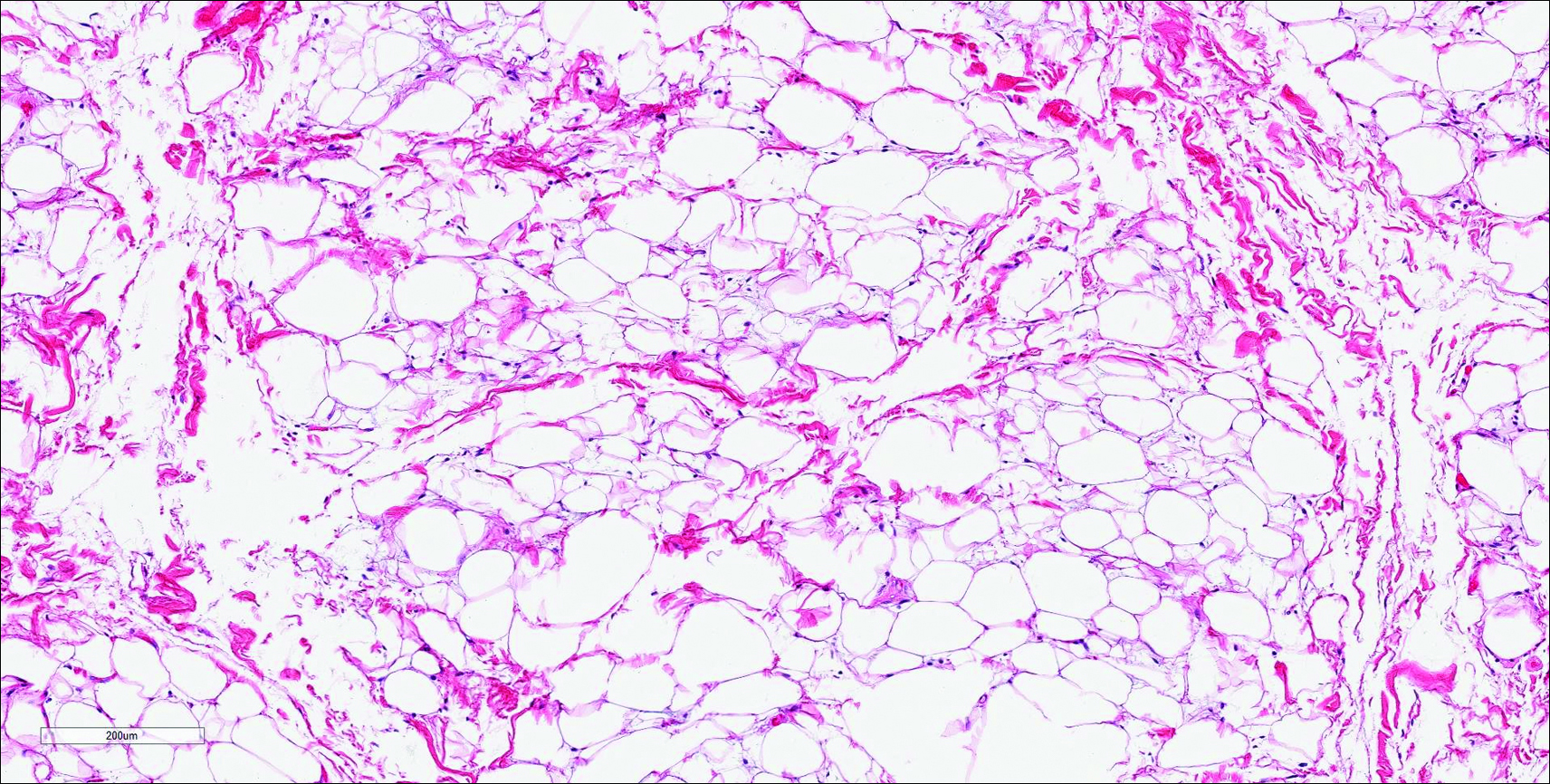
Similar to NTF, sclerotic fibroma is a firm dermal nodule with histologic examination usually demonstrating a paucicellular collagenous tumor. In sclerotic fibromas, the collagen pattern resembles Vincent van Gogh’s painting “The Starry Night” and may be a marker for Cowden disease (Figure 1).3 Solitary fibrous tumors are distinguished by more hypercellular areas, patternless pattern, and staghorn-shaped blood vessels (Figure 2).4 Spindle cell lipoma classically demonstrates a mixture of mature adipocytes and bland spindle cells in a mucinous or fibrous background with thick collagen bundles with no storiform pattern (Figure 3). Some variants of spindle cell lipoma have minimal or no fat.5 All of these conditions have positive immunohistochemical staining for CD34.
However, dermatofibroma is CD34‒. Dermatofibroma is characterized by an interstitial spindle cell proliferation with a loose storiform pattern, collagen trapping at the outer edges of the tumor, overlying platelike acanthosis, and sometimes follicular induction (Figure 4).

Nuchal-type fibroma also can resemble scleredema. Both lesions can show increased and thickened collagen bundles without notable fibroblast proliferation; the difference is the occurrence of mucin in scleredema. However, incases of late-stage scleredema, mucin is not always demonstrated. Therefore, one can conclude that histologically NTF is closely associated with late-stage scleredema.6
- Dawes LC, La Hei ER, Tobias V, et al. Nuchal fibroma should be recognized as a new extracolonic manifestation of Gardner-variant familial adenomatous polyposis. Aust N Z J Surg. 2000;70:824-826.
- Michal M, Fetsch JF, Hes O, et al. Nuchal-type fibroma: a clinicopathologic study of 52 cases. Cancer. 1999;85:156-163.
- Pernet C, Durand L, Bessis D, et al. Solitary sclerotic fibroma of the skin: a possible clue for Cowden syndrome. Eur J Dermatol. 2012;22:278-279.
- Omori Y, Saeki H, Ito K, et al. Solitary fibrous tumour of the scalp. Clin Exp Dermatol. 2014;39:539-541.
- Billings SD, Folpe AL. Diagnostically challenging spindle cell lipomas: a report of 34 “low-fat” and “fat-free” variants. Am J Dermatopathol. 2007;29:437-442.
- Banney LA, Weedon D, Muir JB. Nuchal fibroma associated with scleredema, diabetes mellitus and organic solvent exposure. Australas J Dermatol. 2000;41:39-41.
Nuchal-Type Fibroma
Nuchal-type fibroma (NTF) is a rare benign proliferation of the dermis and subcutis associated with diabetes mellitus and Gardner syndrome.1,2 Forty-four percent of patients with NTF have diabetes mellitus.2 The posterior aspect of the neck is the most frequently affected site, but lesions also may present on the upper back, lumbosacral area, buttocks, and face. Physical examination generally reveals an indurated, asymptomatic, ill-defined, 3-cm or smaller nodule that is hard and white, unencapsulated, and poorly circumscribed.
Histopathologic examination of NTF typically reveals a nodular paucicellular proliferation of thick collagen bundles with inconspicuous fibroblasts, radiation of collagenous septa into the subcutaneous fat, and entrapment of mature adipose tissue and small nerves (quiz image A). Collagen bundles are thickened with entrapment of adipose tissue without increased cellularity (quiz image B). S-100 staining can show the entrapped nerves.
Similar to NTF, sclerotic fibroma is a firm dermal nodule with histologic examination usually demonstrating a paucicellular collagenous tumor. In sclerotic fibromas, the collagen pattern resembles Vincent van Gogh’s painting “The Starry Night” and may be a marker for Cowden disease (Figure 1).3 Solitary fibrous tumors are distinguished by more hypercellular areas, patternless pattern, and staghorn-shaped blood vessels (Figure 2).4 Spindle cell lipoma classically demonstrates a mixture of mature adipocytes and bland spindle cells in a mucinous or fibrous background with thick collagen bundles with no storiform pattern (Figure 3). Some variants of spindle cell lipoma have minimal or no fat.5 All of these conditions have positive immunohistochemical staining for CD34.
However, dermatofibroma is CD34‒. Dermatofibroma is characterized by an interstitial spindle cell proliferation with a loose storiform pattern, collagen trapping at the outer edges of the tumor, overlying platelike acanthosis, and sometimes follicular induction (Figure 4).
Nuchal-type fibroma also can resemble scleredema. Both lesions can show increased and thickened collagen bundles without notable fibroblast proliferation; the difference is the occurrence of mucin in scleredema. However, incases of late-stage scleredema, mucin is not always demonstrated. Therefore, one can conclude that histologically NTF is closely associated with late-stage scleredema.6
Nuchal-Type Fibroma
Nuchal-type fibroma (NTF) is a rare benign proliferation of the dermis and subcutis associated with diabetes mellitus and Gardner syndrome.1,2 Forty-four percent of patients with NTF have diabetes mellitus.2 The posterior aspect of the neck is the most frequently affected site, but lesions also may present on the upper back, lumbosacral area, buttocks, and face. Physical examination generally reveals an indurated, asymptomatic, ill-defined, 3-cm or smaller nodule that is hard and white, unencapsulated, and poorly circumscribed.
Histopathologic examination of NTF typically reveals a nodular paucicellular proliferation of thick collagen bundles with inconspicuous fibroblasts, radiation of collagenous septa into the subcutaneous fat, and entrapment of mature adipose tissue and small nerves (quiz image A). Collagen bundles are thickened with entrapment of adipose tissue without increased cellularity (quiz image B). S-100 staining can show the entrapped nerves.
Similar to NTF, sclerotic fibroma is a firm dermal nodule with histologic examination usually demonstrating a paucicellular collagenous tumor. In sclerotic fibromas, the collagen pattern resembles Vincent van Gogh’s painting “The Starry Night” and may be a marker for Cowden disease (Figure 1).3 Solitary fibrous tumors are distinguished by more hypercellular areas, patternless pattern, and staghorn-shaped blood vessels (Figure 2).4 Spindle cell lipoma classically demonstrates a mixture of mature adipocytes and bland spindle cells in a mucinous or fibrous background with thick collagen bundles with no storiform pattern (Figure 3). Some variants of spindle cell lipoma have minimal or no fat.5 All of these conditions have positive immunohistochemical staining for CD34.
However, dermatofibroma is CD34‒. Dermatofibroma is characterized by an interstitial spindle cell proliferation with a loose storiform pattern, collagen trapping at the outer edges of the tumor, overlying platelike acanthosis, and sometimes follicular induction (Figure 4).
Nuchal-type fibroma also can resemble scleredema. Both lesions can show increased and thickened collagen bundles without notable fibroblast proliferation; the difference is the occurrence of mucin in scleredema. However, incases of late-stage scleredema, mucin is not always demonstrated. Therefore, one can conclude that histologically NTF is closely associated with late-stage scleredema.6
- Dawes LC, La Hei ER, Tobias V, et al. Nuchal fibroma should be recognized as a new extracolonic manifestation of Gardner-variant familial adenomatous polyposis. Aust N Z J Surg. 2000;70:824-826.
- Michal M, Fetsch JF, Hes O, et al. Nuchal-type fibroma: a clinicopathologic study of 52 cases. Cancer. 1999;85:156-163.
- Pernet C, Durand L, Bessis D, et al. Solitary sclerotic fibroma of the skin: a possible clue for Cowden syndrome. Eur J Dermatol. 2012;22:278-279.
- Omori Y, Saeki H, Ito K, et al. Solitary fibrous tumour of the scalp. Clin Exp Dermatol. 2014;39:539-541.
- Billings SD, Folpe AL. Diagnostically challenging spindle cell lipomas: a report of 34 “low-fat” and “fat-free” variants. Am J Dermatopathol. 2007;29:437-442.
- Banney LA, Weedon D, Muir JB. Nuchal fibroma associated with scleredema, diabetes mellitus and organic solvent exposure. Australas J Dermatol. 2000;41:39-41.
- Dawes LC, La Hei ER, Tobias V, et al. Nuchal fibroma should be recognized as a new extracolonic manifestation of Gardner-variant familial adenomatous polyposis. Aust N Z J Surg. 2000;70:824-826.
- Michal M, Fetsch JF, Hes O, et al. Nuchal-type fibroma: a clinicopathologic study of 52 cases. Cancer. 1999;85:156-163.
- Pernet C, Durand L, Bessis D, et al. Solitary sclerotic fibroma of the skin: a possible clue for Cowden syndrome. Eur J Dermatol. 2012;22:278-279.
- Omori Y, Saeki H, Ito K, et al. Solitary fibrous tumour of the scalp. Clin Exp Dermatol. 2014;39:539-541.
- Billings SD, Folpe AL. Diagnostically challenging spindle cell lipomas: a report of 34 “low-fat” and “fat-free” variants. Am J Dermatopathol. 2007;29:437-442.
- Banney LA, Weedon D, Muir JB. Nuchal fibroma associated with scleredema, diabetes mellitus and organic solvent exposure. Australas J Dermatol. 2000;41:39-41.
The best diagnosis is:
a. dermatofibroma
b. nuchal-type fibroma
c. sclerotic fibroma
d. solitary fibrous tumor
e. spindle cell lipoma
Continue to the next page for the diagnosis >>
Irregular, Smooth, Pink Plaque on the Back
The Diagnosis: Fibroepithelioma of Pinkus
Fibroepithelioma of Pinkus (FeP) was first described in 19531 and was thought to be premalignant as evidenced by the proposed name premalignant fibroepithelial tumor of the skin. This neoplasm now is largely believed to represent a rare form of basal cell carcinoma (BCC). Typical presentation is a smooth, flesh-colored or pink plaque or nodule.2 Fibroepithelioma of Pinkus has a predilection for the lumbosacral back, though the groin also has been reported as a common site of incidence.1,3 Similar to other BCCs, it is seen in older individuals, typically those older than 50 years.3,4
Clinical diagnosis of FeP can be difficult. The differential diagnosis of FeP can include acrochordon, amelanotic melanoma, compound nevus, hemangioma, neurofibroma, nevus sebaceous, pyogenic granuloma, and seborrheic keratosis.5 Dermoscopic evaluation can aid in the diagnosis. A vascular network composed of fine arborizing vessels with or without dotted vessels and white streaks are characteristic findings of FeP. Patients with pigment also demonstrate structureless gray-brown areas and gray-blue dots.6
Biopsy with subsequent histopathologic evaluation confirms the diagnosis of FeP. The characteristic microscopic findings of thin eosinophilic epithelial strands with eccrine ducts anastomosing in an abundant fibromyxoid stroma with collections of basophilic cells located at the ends of the epithelial strands were demonstrated in our patient’s histopathologic specimen (Figure). The histologic appearance is similar to syringofibroadenoma of Mascaro. Recognition of basaloid nests, which often demonstrate retraction, and mitotic activity can differentiate FeP from syringofibroadenoma of Mascaro.7
Treatment of FeP is largely the same as other BCCs including destruction by electrodesiccation and curettage or complete removal by surgical excision. Several studies have demonstrated effective treatment of nonaggressive BCCs with curettage alone and subjectively reported improved cosmesis compared to electrodesiccation and curettage.8-10 Although methyl aminolevulinate photodynamic therapy has demonstrated some therapeutic efficacy for superficial and nodular BCCs,11 a case report utilizing the same modality for FeP did not provide adequate response.12 However, adequate data are not available to assess potential use of this less invasive therapy.
- Pinkus H. Premalignant fibroepithelial tumors of skin. AMA Arch Derm Syphilol. 1953;67:598-615.
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012.
- Barr RJ, Herten RJ, Stone OJ. Multiple premalignant fibroepitheliomas of Pinkus: a case report and review of the literature. Cutis. 1978;21:335-337.
- Betti R, Inselvini E, Carducci M, et al. Age and site prevalence of histologic subtypes of basal cell carcinomas. Int J Dermatol. 1995;34:174-176.
- Cohen PR, Tschen JA. Fibroepithelioma of Pinkus presenting as a sessile thigh nodule. Skinmed. 2003;2:385-387.
- Zalaudek I, Ferrara G, Broganelli P, et al. Dermoscopy patterns of fibroepithelioma of Pinkus. Arch Dermatol. 2006;142:1318-1322.
- Schadt CR, Boyd AS. Eccrine syringofibroadenoma with co-existent squamous cell carcinoma. J Cutan Pathol. 2007;34(suppl 1):71-74.
- Barlow JO, Zalla MJ, Kyle A, et al. Treatment of basal cell carcinoma with curettage alone. J Am Acad Dermatol. 2006;54:1039-1045.
- McDaniel WE. Therapy for basal cell epitheliomas by curettage only. further study. Arch Dermatol. 1983;119:901-903.
- Reymann F. 15 Years’ experience with treatment of basal cell carcinomas of the skin with curettage. Acta Derm Venereol Suppl (Stockh). 1985;120:56-59.
- Fai D, Arpaia N, Romano I, et al. Methyl-aminolevulinate photodynamic therapy for the treatment of actinic keratoses and non-melanoma skin cancers: a retrospective analysis of response in 462 patients. G Ital Dermatol Venereol. 2009;144:281-285.
- Park MY, Kim YC. Fibroepithelioma of Pinkus: poor response to topical photodynamic therapy. Eur J Dermatol. 2010;20:133-134.
The Diagnosis: Fibroepithelioma of Pinkus
Fibroepithelioma of Pinkus (FeP) was first described in 19531 and was thought to be premalignant as evidenced by the proposed name premalignant fibroepithelial tumor of the skin. This neoplasm now is largely believed to represent a rare form of basal cell carcinoma (BCC). Typical presentation is a smooth, flesh-colored or pink plaque or nodule.2 Fibroepithelioma of Pinkus has a predilection for the lumbosacral back, though the groin also has been reported as a common site of incidence.1,3 Similar to other BCCs, it is seen in older individuals, typically those older than 50 years.3,4
Clinical diagnosis of FeP can be difficult. The differential diagnosis of FeP can include acrochordon, amelanotic melanoma, compound nevus, hemangioma, neurofibroma, nevus sebaceous, pyogenic granuloma, and seborrheic keratosis.5 Dermoscopic evaluation can aid in the diagnosis. A vascular network composed of fine arborizing vessels with or without dotted vessels and white streaks are characteristic findings of FeP. Patients with pigment also demonstrate structureless gray-brown areas and gray-blue dots.6
Biopsy with subsequent histopathologic evaluation confirms the diagnosis of FeP. The characteristic microscopic findings of thin eosinophilic epithelial strands with eccrine ducts anastomosing in an abundant fibromyxoid stroma with collections of basophilic cells located at the ends of the epithelial strands were demonstrated in our patient’s histopathologic specimen (Figure). The histologic appearance is similar to syringofibroadenoma of Mascaro. Recognition of basaloid nests, which often demonstrate retraction, and mitotic activity can differentiate FeP from syringofibroadenoma of Mascaro.7
Treatment of FeP is largely the same as other BCCs including destruction by electrodesiccation and curettage or complete removal by surgical excision. Several studies have demonstrated effective treatment of nonaggressive BCCs with curettage alone and subjectively reported improved cosmesis compared to electrodesiccation and curettage.8-10 Although methyl aminolevulinate photodynamic therapy has demonstrated some therapeutic efficacy for superficial and nodular BCCs,11 a case report utilizing the same modality for FeP did not provide adequate response.12 However, adequate data are not available to assess potential use of this less invasive therapy.
The Diagnosis: Fibroepithelioma of Pinkus
Fibroepithelioma of Pinkus (FeP) was first described in 19531 and was thought to be premalignant as evidenced by the proposed name premalignant fibroepithelial tumor of the skin. This neoplasm now is largely believed to represent a rare form of basal cell carcinoma (BCC). Typical presentation is a smooth, flesh-colored or pink plaque or nodule.2 Fibroepithelioma of Pinkus has a predilection for the lumbosacral back, though the groin also has been reported as a common site of incidence.1,3 Similar to other BCCs, it is seen in older individuals, typically those older than 50 years.3,4
Clinical diagnosis of FeP can be difficult. The differential diagnosis of FeP can include acrochordon, amelanotic melanoma, compound nevus, hemangioma, neurofibroma, nevus sebaceous, pyogenic granuloma, and seborrheic keratosis.5 Dermoscopic evaluation can aid in the diagnosis. A vascular network composed of fine arborizing vessels with or without dotted vessels and white streaks are characteristic findings of FeP. Patients with pigment also demonstrate structureless gray-brown areas and gray-blue dots.6
Biopsy with subsequent histopathologic evaluation confirms the diagnosis of FeP. The characteristic microscopic findings of thin eosinophilic epithelial strands with eccrine ducts anastomosing in an abundant fibromyxoid stroma with collections of basophilic cells located at the ends of the epithelial strands were demonstrated in our patient’s histopathologic specimen (Figure). The histologic appearance is similar to syringofibroadenoma of Mascaro. Recognition of basaloid nests, which often demonstrate retraction, and mitotic activity can differentiate FeP from syringofibroadenoma of Mascaro.7
Treatment of FeP is largely the same as other BCCs including destruction by electrodesiccation and curettage or complete removal by surgical excision. Several studies have demonstrated effective treatment of nonaggressive BCCs with curettage alone and subjectively reported improved cosmesis compared to electrodesiccation and curettage.8-10 Although methyl aminolevulinate photodynamic therapy has demonstrated some therapeutic efficacy for superficial and nodular BCCs,11 a case report utilizing the same modality for FeP did not provide adequate response.12 However, adequate data are not available to assess potential use of this less invasive therapy.
- Pinkus H. Premalignant fibroepithelial tumors of skin. AMA Arch Derm Syphilol. 1953;67:598-615.
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012.
- Barr RJ, Herten RJ, Stone OJ. Multiple premalignant fibroepitheliomas of Pinkus: a case report and review of the literature. Cutis. 1978;21:335-337.
- Betti R, Inselvini E, Carducci M, et al. Age and site prevalence of histologic subtypes of basal cell carcinomas. Int J Dermatol. 1995;34:174-176.
- Cohen PR, Tschen JA. Fibroepithelioma of Pinkus presenting as a sessile thigh nodule. Skinmed. 2003;2:385-387.
- Zalaudek I, Ferrara G, Broganelli P, et al. Dermoscopy patterns of fibroepithelioma of Pinkus. Arch Dermatol. 2006;142:1318-1322.
- Schadt CR, Boyd AS. Eccrine syringofibroadenoma with co-existent squamous cell carcinoma. J Cutan Pathol. 2007;34(suppl 1):71-74.
- Barlow JO, Zalla MJ, Kyle A, et al. Treatment of basal cell carcinoma with curettage alone. J Am Acad Dermatol. 2006;54:1039-1045.
- McDaniel WE. Therapy for basal cell epitheliomas by curettage only. further study. Arch Dermatol. 1983;119:901-903.
- Reymann F. 15 Years’ experience with treatment of basal cell carcinomas of the skin with curettage. Acta Derm Venereol Suppl (Stockh). 1985;120:56-59.
- Fai D, Arpaia N, Romano I, et al. Methyl-aminolevulinate photodynamic therapy for the treatment of actinic keratoses and non-melanoma skin cancers: a retrospective analysis of response in 462 patients. G Ital Dermatol Venereol. 2009;144:281-285.
- Park MY, Kim YC. Fibroepithelioma of Pinkus: poor response to topical photodynamic therapy. Eur J Dermatol. 2010;20:133-134.
- Pinkus H. Premalignant fibroepithelial tumors of skin. AMA Arch Derm Syphilol. 1953;67:598-615.
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012.
- Barr RJ, Herten RJ, Stone OJ. Multiple premalignant fibroepitheliomas of Pinkus: a case report and review of the literature. Cutis. 1978;21:335-337.
- Betti R, Inselvini E, Carducci M, et al. Age and site prevalence of histologic subtypes of basal cell carcinomas. Int J Dermatol. 1995;34:174-176.
- Cohen PR, Tschen JA. Fibroepithelioma of Pinkus presenting as a sessile thigh nodule. Skinmed. 2003;2:385-387.
- Zalaudek I, Ferrara G, Broganelli P, et al. Dermoscopy patterns of fibroepithelioma of Pinkus. Arch Dermatol. 2006;142:1318-1322.
- Schadt CR, Boyd AS. Eccrine syringofibroadenoma with co-existent squamous cell carcinoma. J Cutan Pathol. 2007;34(suppl 1):71-74.
- Barlow JO, Zalla MJ, Kyle A, et al. Treatment of basal cell carcinoma with curettage alone. J Am Acad Dermatol. 2006;54:1039-1045.
- McDaniel WE. Therapy for basal cell epitheliomas by curettage only. further study. Arch Dermatol. 1983;119:901-903.
- Reymann F. 15 Years’ experience with treatment of basal cell carcinomas of the skin with curettage. Acta Derm Venereol Suppl (Stockh). 1985;120:56-59.
- Fai D, Arpaia N, Romano I, et al. Methyl-aminolevulinate photodynamic therapy for the treatment of actinic keratoses and non-melanoma skin cancers: a retrospective analysis of response in 462 patients. G Ital Dermatol Venereol. 2009;144:281-285.
- Park MY, Kim YC. Fibroepithelioma of Pinkus: poor response to topical photodynamic therapy. Eur J Dermatol. 2010;20:133-134.
Regional Lymphomatoid Papulosis of the Breast Restricted to an Area of Prior Radiotherapy
Lymphomatoid papulosis (LyP) is a clinicopathologic variant of CD30+ primary cutaneous T-cell lymphoproliferative disorder characterized by a chronic, recurrent, self-healing eruption of papules and small nodules. From a clinical point of view, LyP is not considered a malignant disorder despite demonstration of clonality in most cases.1 From a histopathologic point of view, there are 5 types of LyP: (1) type A, the most common type, which is characterized by a wedge-shaped infiltrate composed of clustered large atypical cells admixed with neutrophils, eosinophils, histiocytes, and small lymphocytes; (2) type B, a rare variant characterized by a bandlike infiltrate of small- to medium-sized pleomorphic and hyperchromatic lymphocytes involving the superficial dermis with epidermotropism; (3) type C, which consists of a nodular infiltrate of large atypical cells with a cohesive arrangement closely similar to anaplastic large-cell lymphoma; (4) type D, a variant with histopathologic features that resemble primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma, but neoplastic cells express CD30 and a T-cell cytotoxic phenotype (βF1+, CD3+, CD4‒, CD8+), and follow-up usually does not reveal development of systemic involvement or signs of other cutaneous lymphomas2; and (5) type E, which is characterized by oligolesional papules that rapidly ulcerate and evolve into large, necrotic, escharlike lesions with a diameter of 1 to 4 cm and an angiocentric and angiodestructive infiltrate of small- to medium-sized atypical lymphocytes expressing CD30 and frequently CD8.3
The clinical appearance of LyP usually is polymorphic, with lesions in different stages of evolution scattered all over the skin; however, the lesions are occasionally localized only to one area of the skin, the so-called regional or agminated LyP.4-14 We report a case of regional LyP that exclusively involved the skin of the left breast, which had previously received radiotherapy for treatment of breast carcinoma. Lymphomatoid papulosis with cutaneous lesions involving only an area of irradiated skin is rare.
Case Report
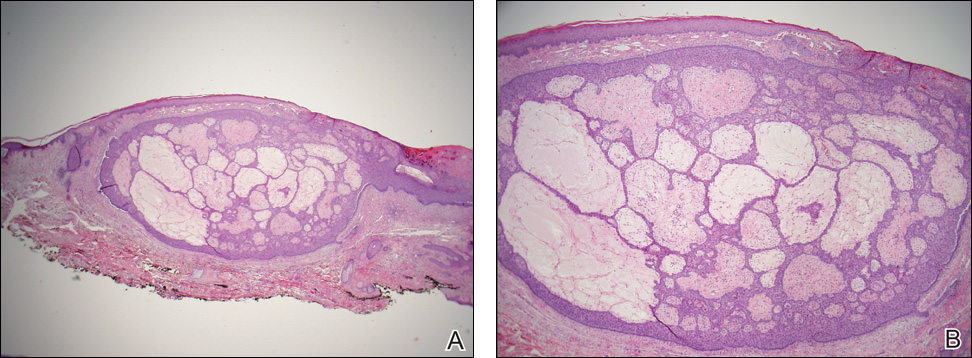
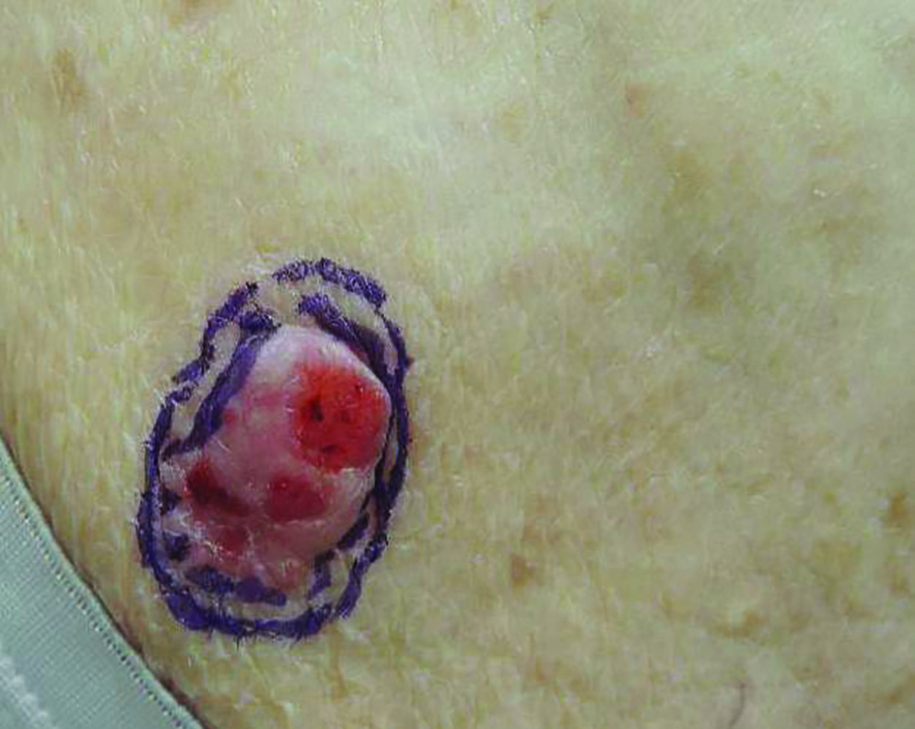
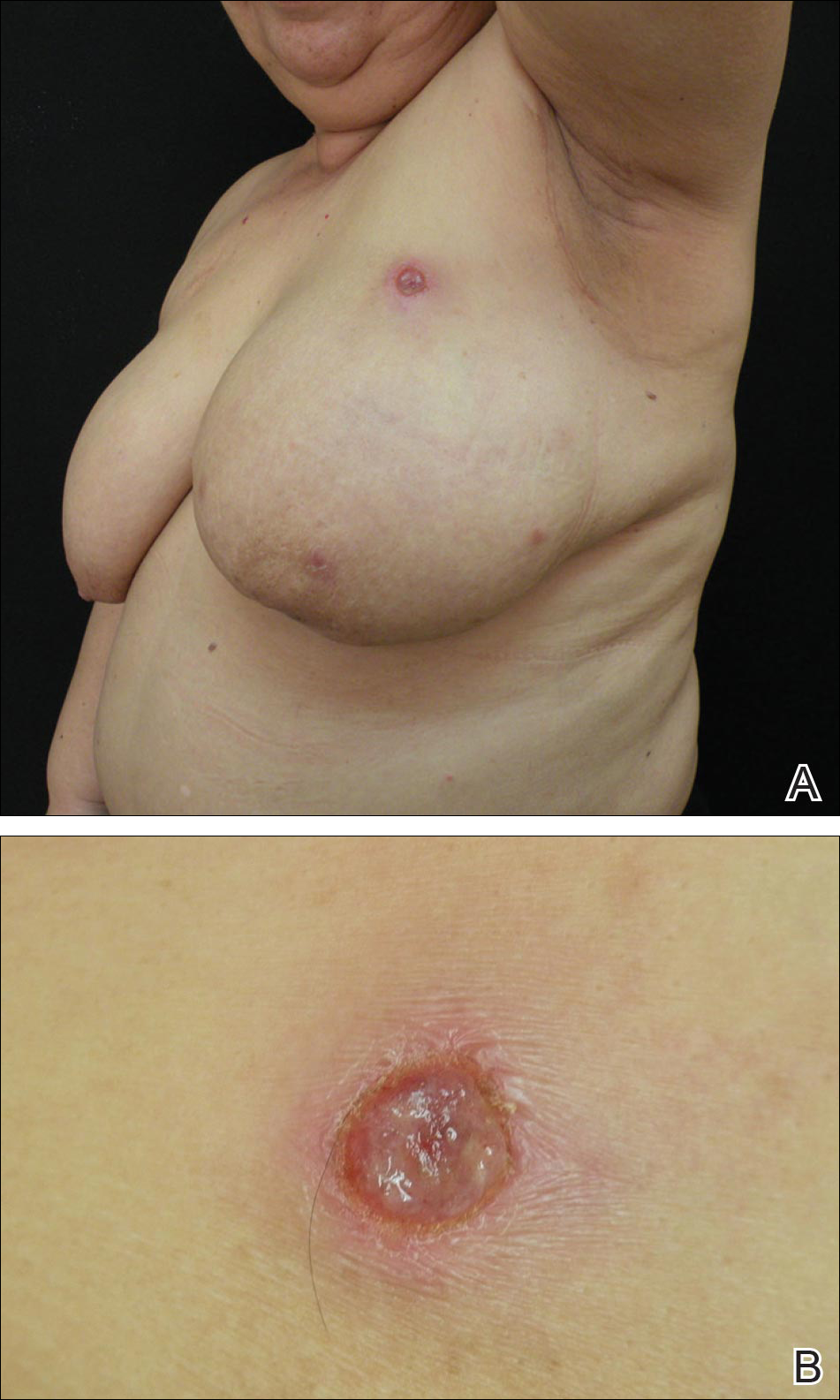
A 59-year-old woman presented with new-onset cutaneous lesions on the left breast. The patient had a history of invasive ductal carcinoma of the left breast, which had been treated 5 years prior with a partial mastectomy and radiotherapy (10 Gy per week for 5 consecutive weeks [50 Gy total]). Physical examination revealed a large nodular lesion with a necrotic surface on the upper half of the left breast as well as 3 small papular lesions with eroded surfaces on the lower half of the breast (Figure 1). A clinical diagnosis of cutaneous metastases from breast carcinoma was suspected.
Biopsies from one small papule and the large nodular lesion showed similar findings consisting of a necrotic epidermis covered by crusts and a wedge-shaped infiltrate involving the superficial dermis (Figure 2A). The infiltrate was mostly composed of large atypical mononuclear cells with oval to kidney-shaped nuclei, prominent nucleoli, and ample basophilic cytoplasm. Many mitotic figures were seen within the infiltrate (Figure 2B). The infiltrate of atypical cells was admixed with small lymphocytes, histiocytes, and some eosinophils. Immunohistochemically, the large atypical cells expressed CD2, CD3, CD4, CD45, CD30, and epithelial membrane antigen (Figures 2C and 2D). A few atypical cells also expressed CD8 and T-cell intracellular antigen 1. Approximately 60% of the nuclei of the atypical cells showed MIB-1 positivity, while CD20, CD56, AE1/AE3, S-100 protein, CD34, and CD31 were negative. The anaplastic lymphoma kinase was not expressed in atypical cells. Monoclonal rearrangement of the γ T-cell receptor was demonstrated on polymerase chain reaction. Physical examination showed no lymphadenopathy in any lymph node chains. Computed tomography of the chest and abdomen failed to demonstrate systemic involvement. On the basis of these clinical, histologic, immunohistochemical, and molecular results, a diagnosis of type A regional LyP was established.

The patient was treated with 2 daily applications of clobetasol propionate cream 0.5 mg/g and 10 mg of oral methotrexate per week for 4 weeks. After 4 weeks of treatment, the lesions on the left breast had resolved leaving slightly atrophic scars. Six months later, an episode of recurrent papular lesions occurred in the same area and responded to the same treatment, but no systemic involvement had been found.
Comment
Regional LyP is a rare variant, with only a few reported cases in the literature.4-18 Scarisbrick et al4 originally reported 4 patients with LyP limited to specific regions. Interestingly, one of the patients had mycosis fungoides and the LyP lesions were confined to the same region where the mycosis fungoides lesions were observed.4 In a review of LyP in patients from the Netherlands (n=118), lesions limited to a specific region of the body were observed in 13% of cases.5 Cases of LyP limited to acral skin also have been reported.6-8 Heald et al9 described 7 patients who had continuing eruptions of papulonodules with histopathologic features of LyP within well-circumscribed areas of the skin. The investigators interpreted this localized variant of LyP as an equivalent of the limited plaque stage of mycosis fungoides. Interestingly, one of the patients with LyP eventually developed plaques of mycosis fungoides in other areas of the skin not involved by LyP.9 Sharma et al10 described an additional example of regional LyP, and Nakahigashi et al11 described a patient with tumor-stage mycosis fungoides who subsequently developed regional LyP involving the right side of the chest. Kim et al12 described a patient with recurrent episodes of regional LyP exclusively involving the periorbital skin, and Torrelo et al13 reported a 12-year-old boy with persistent lesions of LyP involving the skin of the right side of the abdomen. Coelho et al14 reported a 13-year-old adolescent girl who presented with recurrent papules of LyP exclusively involving the left upper arm. Buder et al15 reported a case of LyP limited to Becker melanosis. Shang et al16 described an additional caseof regional LyP that was successfully controlled by interferon alfa-2b and nitrogen mustard solution. Haus et al17 reported type A LyP confined to the cutaneous area within a red tattoo. Finally, Wang et al18 reported a case of regional LyP in association with pseudoepitheliomatous hyperplasia
Several dermatoses may appear as specific isomorphic responses to various external stimuli, and it is possible that radiotherapy induces some damage that favors the location of the lesions because the irradiated skin behaves as a locus minoris resistentiae. Pemphigus vulgaris,19,20 Sweet syndrome,21 cutaneous angiosarcoma,22-32 and cutaneous metastases from malignant melanoma also have been reported to be confined to irradiated skin.33 However, in our PubMed search of articles indexed for MEDLINE using the terms lymphomatoid papules and regional, none of the previously reported cases of regional LyP had a history of radiotherapy, and in no instance did the lesions develop on a previously irradiated area of the skin.4-18 The localization of the lesions in our patient could have been the result of the so-called radiation recall phenomenon. Recall dermatitis is defined as a skin reaction in a previously irradiated field, usually subsequent to the administration of cytotoxic drugs or antibiotics.34 It may appear days to years after exposure to ionizing radiation and has mostly been associated with chemotherapy drugs, but recall dermatitis is neither exclusive of chemotherapy medications nor strictly radiotherapy induced. The concept of recall dermatitis has been expanded beyond radiation recall dermatitis to include dermatitis induced by other stimuli, including other drugs, contact irritants, and UV radiation, as well as residual herpes zoster. Nevertheless, in recall dermatitis the triggering drug or agent recalls a prior dermatitis in the involved area, such as sunburn or radiodermatitis. In our patient, there was no history of LyP prior to irradiation of the left breast; therefore, the most plausible interpretation of the peculiar localization of the lesions in our patient seems to be that the eruption resulted as expression of a locus minoris resistentiae.
Distinction between primary cutaneous anaplastic large-cell lymphoma and LyP may be difficult because the histopathologic and immunophenotypic features may overlap. In our case, the presence of several papular lesions and one large nodule are more consistent, from a clinical point of view, with a diagnosis of LyP rather than primary cutaneous anaplastic large-cell lymphoma, which usually presents with a solitary and often large, ulcerated, reddish brown tumor. In our patient, the absence of lymphadenopathy, negative results of the computed tomography of the chest and abdomen, and lack of expression for anaplastic lymphoma kinase in atypical cells of the infiltrate militate against a diagnosis of secondary cutaneous involvement from nodal disease.
The histopathologic differential diagnosis of the current case also included cutaneous CD30+ epithelioid angiosarcoma of the breast. Weed and Folpe35 reported the case of an 85-year-old woman who developed a CD30+ epithelioid angiosarcoma on the breast after undergoing breast-conserving surgery and adjuvant radiotherapy for treatment of an infiltrating ductal carcinoma of the breast. Histopathology showed a diffuse replacement of the dermis by a highly malignant-appearing epithelioid neoplasm growing in a solid sheet. Neoplastic cells expressed strong CD30 immunoreactivity with absence of immunoexpression for cytokeratins, S-100 protein, and CD45. Additional immunostaining demonstrated that neoplastic cells also expressed strong immunoreactivity for CD31 and the friend leukemia virus integration 1 gene, FLI-1, and focal positivity for von Willebrand factor, supporting a diagnosis of epithelioid angiosarcoma.35 In our patient, CD34 and CD31 were negative, which ruled out the endothelial nature of neoplastic cells.
Conclusion
In summary, we report an example of regional LyP limited to the left breast of a woman with a history of partial mastectomy and adjuvant radiotherapy for treatment of invasive ductal breast carcinoma. It is a rare case of regional LyP exclusively involving an irradiated area of the skin.
- Ralfkiaer E, Willemze R, Paulli M, et al. Primary cutaneous CD30-positive T-cell lymphoproliferative disorders. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphomatoid Tissues. Lyon, France: IARC Press, 2008:300-301.
- Saggini A, Gulia A, Argenyi Z, et al. A variant of lymphomatoid papulosis simulating primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma. description of 9 cases. Am J Surg Pathol. 2010;34:1168-1175.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Scarisbrick JJ, Evans AV, Woolford AJ, et al. Regional lymphomatoid papulosis: a report of four cases. Br J Dermatol. 1999;141:1125-1128.
- Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30+ lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000;95:3653-3661.
- Thomas GJ, Conejo-Mir JS, Ruiz AP, et al. Lymphomatoid papulosis in childhood with exclusive acral involvement. Pediatr Dermatol. 1998;15:146-147.
- Deroo-Berger MC, Skowson F, Roner S, et al. Lymphomatoid papulosis: a localized form with acral pustular involvement. Dermatology. 2002;205:60-62.
- Kagaya M, Kondo S, Kamada A, et al. Localized lymphomatoid papulosis. Dermatology. 2002;204:72-74.
- Heald P, Subtil A, Breneman D, et al. Persistent agmination of lymphomatoid papulosis: an equivalent of limited plaque mycosis fungoides type of cutaneous T-cell lymphoma. J Am Acad Dermatol. 2007;57:1005-1011.
- Sharma V, Xu G, Petronic-Rosic V, et al. Clinicopathologic challenge. regional lymphomatoid papulosis, type A. Int J Dermatol. 2007;46:905-909.
- Nakahigashi K, Ishida Y, Matsumura Y, et al. Large cell transformation mimicking regional lymphomatoid papulosis in a patient with mycosis fungoides. J Dermatol. 2008;35:283-288.
- Kim YJ, Rho YK, Yoo KH, et al. Case of regional lymphomatoid papulosis confined to the periorbital areas. J Dermatol. 2009;36:163-165.
- Torrelo A, Colmenero I, Hernández A, et al. Persistent agmination of lymphomatoid papulosis. Pediatr Dermatol. 2009;26:762-764.
- Coelho JD, Afonso A, Feio AB. Regional lymphomatoid papulosis in a child—treatment with a UVB phototherapy handpiece. J Cosmet Laser Ther. 2010;12:155-156.
- Buder K, Wendel AM, Cerroni L, et al. A case of lymphomatoid papulosis limited to Becker’s melanosis. Dermatology. 2013;226:124-127.
- Shang SX, Chen H, Sun JF, et al. Regional lymphomatoid papulosis successfully controlled by interferon α-2b and nitrogen mustard solution. Chin Med J (Engl). 2013;126:3194-3195.
- Haus G, Utikal J, Geraud C, et al. CD30-positive lymphoproliferative disorder in a red tattoo: regional lymphomatoid papulosis type C or pseudolymphoma? Br J Dermatol. 2014;171:668-670.
- Wang T, Guo CL, Xu CC, et al. Regional lymphomatoid papulosis in association with pseudoepitheliomatous hyperplasia: 13 years follow-up. J Eur Acad Dermatol Venereol. 2015;29:1853-1854.
- Davis M, Feverman EJ. Induction of pemphigus by X-ray irradiation. Clin Exp Dermatol. 1987;12:197-199.
- Crovato F, Descrello G, Nazzari G, et al. Liner pemphigus vulgaris after X-ray irradiation. Dermatologica. 1989;179:135-136.
- Vergara G, Vargas-Machuca I, Pastor MA, et al. Localized Sweet’s syndrome in radiation-induced locus minoris resistentae. J Am Acad Dermatol. 2003;49:907-909.
- Caldwell JB, Ryan MT, Benson PM, et al. Cutaneous angiosarcoma arising in the radiation site of a congenital hemangioma. J Am Acad Dermatol. 1995;33:865-870.
- Stone NM, Holden CA. Postirradiation angiosarcoma. Clin Exp Dermatol. 1997;22:46-47.
- Goette EK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12:922-926.
- Chen TK, Goffman KD, Hendricks EJ. Angiosarcoma following therapeutic irradiation. Cancer. 1979;44:2044-2048.
- Rubin E, Maddox WA, Mazur MT. Cutaneous angiosarcoma of the breast 7 years after lumpectomy and radiation therapy. Radiology. 1990;174:258-260.
- Stokkel MPM, Peterse HL. Angiosarcoma of the breast after lumpectomy and radiation therapy for adenocarcinoma. Cancer. 1992;69:2965-2968.
- Moskaluk CA, Merino MJ, Danforth DN, et al. Low-grade angiosarcoma of the skin of the breast: a complication of lumpectomy and radiation therapy for breast carcinoma. Hum Pathol. 1992;23:710-714.
- Parham DM, Fisher C. Angiosarcomas of the breast developing post radiotherapy. Histopathology. 1997;31:189-195.
- Rao J, DeKoven JG, Beatty JD, et al. Cutaneous angiosarcoma as a delayed complication of radiation therapy for carcinoma of the breast. J Am Acad Dermatol. 2003;49:532-538.
- Billings SD, McKenney JK, Folpe Al, et al. Cutaneous angiosarcoma following breast-conserving surgery and radiation. an analysis of 27 cases. Am J Surg Pathol. 2004;28:781-788.
- Fodor J, Orosz Z, Szabo E, et al. Angiosarcoma after conservation treatment for breast carcinoma: our experience and a review of the literature. J Am Acad Dermatol. 2006;54:499-504.
- Roses DP, Harris MN, Rigel D, et al. Local and in-transit metastases following definitive excision from primary cutaneous malignant melanoma. Ann Surg. 1983;198:65-69.
- Burris HA 3rd, Hurtig J. Radiation recall with anticancer agents. Oncologist. 2010;15:1227-1237.
- Weed BR, Folpe AL. Cutaneous CD30-positive epithelioid angiosarcoma following breast-conserving therapy and irradiation. a potential diagnostic pitfall. Am J Dermatopathol. 2008;30:370-372.
Lymphomatoid papulosis (LyP) is a clinicopathologic variant of CD30+ primary cutaneous T-cell lymphoproliferative disorder characterized by a chronic, recurrent, self-healing eruption of papules and small nodules. From a clinical point of view, LyP is not considered a malignant disorder despite demonstration of clonality in most cases.1 From a histopathologic point of view, there are 5 types of LyP: (1) type A, the most common type, which is characterized by a wedge-shaped infiltrate composed of clustered large atypical cells admixed with neutrophils, eosinophils, histiocytes, and small lymphocytes; (2) type B, a rare variant characterized by a bandlike infiltrate of small- to medium-sized pleomorphic and hyperchromatic lymphocytes involving the superficial dermis with epidermotropism; (3) type C, which consists of a nodular infiltrate of large atypical cells with a cohesive arrangement closely similar to anaplastic large-cell lymphoma; (4) type D, a variant with histopathologic features that resemble primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma, but neoplastic cells express CD30 and a T-cell cytotoxic phenotype (βF1+, CD3+, CD4‒, CD8+), and follow-up usually does not reveal development of systemic involvement or signs of other cutaneous lymphomas2; and (5) type E, which is characterized by oligolesional papules that rapidly ulcerate and evolve into large, necrotic, escharlike lesions with a diameter of 1 to 4 cm and an angiocentric and angiodestructive infiltrate of small- to medium-sized atypical lymphocytes expressing CD30 and frequently CD8.3
The clinical appearance of LyP usually is polymorphic, with lesions in different stages of evolution scattered all over the skin; however, the lesions are occasionally localized only to one area of the skin, the so-called regional or agminated LyP.4-14 We report a case of regional LyP that exclusively involved the skin of the left breast, which had previously received radiotherapy for treatment of breast carcinoma. Lymphomatoid papulosis with cutaneous lesions involving only an area of irradiated skin is rare.
Case Report
A 59-year-old woman presented with new-onset cutaneous lesions on the left breast. The patient had a history of invasive ductal carcinoma of the left breast, which had been treated 5 years prior with a partial mastectomy and radiotherapy (10 Gy per week for 5 consecutive weeks [50 Gy total]). Physical examination revealed a large nodular lesion with a necrotic surface on the upper half of the left breast as well as 3 small papular lesions with eroded surfaces on the lower half of the breast (Figure 1). A clinical diagnosis of cutaneous metastases from breast carcinoma was suspected.
Biopsies from one small papule and the large nodular lesion showed similar findings consisting of a necrotic epidermis covered by crusts and a wedge-shaped infiltrate involving the superficial dermis (Figure 2A). The infiltrate was mostly composed of large atypical mononuclear cells with oval to kidney-shaped nuclei, prominent nucleoli, and ample basophilic cytoplasm. Many mitotic figures were seen within the infiltrate (Figure 2B). The infiltrate of atypical cells was admixed with small lymphocytes, histiocytes, and some eosinophils. Immunohistochemically, the large atypical cells expressed CD2, CD3, CD4, CD45, CD30, and epithelial membrane antigen (Figures 2C and 2D). A few atypical cells also expressed CD8 and T-cell intracellular antigen 1. Approximately 60% of the nuclei of the atypical cells showed MIB-1 positivity, while CD20, CD56, AE1/AE3, S-100 protein, CD34, and CD31 were negative. The anaplastic lymphoma kinase was not expressed in atypical cells. Monoclonal rearrangement of the γ T-cell receptor was demonstrated on polymerase chain reaction. Physical examination showed no lymphadenopathy in any lymph node chains. Computed tomography of the chest and abdomen failed to demonstrate systemic involvement. On the basis of these clinical, histologic, immunohistochemical, and molecular results, a diagnosis of type A regional LyP was established.
The patient was treated with 2 daily applications of clobetasol propionate cream 0.5 mg/g and 10 mg of oral methotrexate per week for 4 weeks. After 4 weeks of treatment, the lesions on the left breast had resolved leaving slightly atrophic scars. Six months later, an episode of recurrent papular lesions occurred in the same area and responded to the same treatment, but no systemic involvement had been found.
Comment
Regional LyP is a rare variant, with only a few reported cases in the literature.4-18 Scarisbrick et al4 originally reported 4 patients with LyP limited to specific regions. Interestingly, one of the patients had mycosis fungoides and the LyP lesions were confined to the same region where the mycosis fungoides lesions were observed.4 In a review of LyP in patients from the Netherlands (n=118), lesions limited to a specific region of the body were observed in 13% of cases.5 Cases of LyP limited to acral skin also have been reported.6-8 Heald et al9 described 7 patients who had continuing eruptions of papulonodules with histopathologic features of LyP within well-circumscribed areas of the skin. The investigators interpreted this localized variant of LyP as an equivalent of the limited plaque stage of mycosis fungoides. Interestingly, one of the patients with LyP eventually developed plaques of mycosis fungoides in other areas of the skin not involved by LyP.9 Sharma et al10 described an additional example of regional LyP, and Nakahigashi et al11 described a patient with tumor-stage mycosis fungoides who subsequently developed regional LyP involving the right side of the chest. Kim et al12 described a patient with recurrent episodes of regional LyP exclusively involving the periorbital skin, and Torrelo et al13 reported a 12-year-old boy with persistent lesions of LyP involving the skin of the right side of the abdomen. Coelho et al14 reported a 13-year-old adolescent girl who presented with recurrent papules of LyP exclusively involving the left upper arm. Buder et al15 reported a case of LyP limited to Becker melanosis. Shang et al16 described an additional caseof regional LyP that was successfully controlled by interferon alfa-2b and nitrogen mustard solution. Haus et al17 reported type A LyP confined to the cutaneous area within a red tattoo. Finally, Wang et al18 reported a case of regional LyP in association with pseudoepitheliomatous hyperplasia
Several dermatoses may appear as specific isomorphic responses to various external stimuli, and it is possible that radiotherapy induces some damage that favors the location of the lesions because the irradiated skin behaves as a locus minoris resistentiae. Pemphigus vulgaris,19,20 Sweet syndrome,21 cutaneous angiosarcoma,22-32 and cutaneous metastases from malignant melanoma also have been reported to be confined to irradiated skin.33 However, in our PubMed search of articles indexed for MEDLINE using the terms lymphomatoid papules and regional, none of the previously reported cases of regional LyP had a history of radiotherapy, and in no instance did the lesions develop on a previously irradiated area of the skin.4-18 The localization of the lesions in our patient could have been the result of the so-called radiation recall phenomenon. Recall dermatitis is defined as a skin reaction in a previously irradiated field, usually subsequent to the administration of cytotoxic drugs or antibiotics.34 It may appear days to years after exposure to ionizing radiation and has mostly been associated with chemotherapy drugs, but recall dermatitis is neither exclusive of chemotherapy medications nor strictly radiotherapy induced. The concept of recall dermatitis has been expanded beyond radiation recall dermatitis to include dermatitis induced by other stimuli, including other drugs, contact irritants, and UV radiation, as well as residual herpes zoster. Nevertheless, in recall dermatitis the triggering drug or agent recalls a prior dermatitis in the involved area, such as sunburn or radiodermatitis. In our patient, there was no history of LyP prior to irradiation of the left breast; therefore, the most plausible interpretation of the peculiar localization of the lesions in our patient seems to be that the eruption resulted as expression of a locus minoris resistentiae.
Distinction between primary cutaneous anaplastic large-cell lymphoma and LyP may be difficult because the histopathologic and immunophenotypic features may overlap. In our case, the presence of several papular lesions and one large nodule are more consistent, from a clinical point of view, with a diagnosis of LyP rather than primary cutaneous anaplastic large-cell lymphoma, which usually presents with a solitary and often large, ulcerated, reddish brown tumor. In our patient, the absence of lymphadenopathy, negative results of the computed tomography of the chest and abdomen, and lack of expression for anaplastic lymphoma kinase in atypical cells of the infiltrate militate against a diagnosis of secondary cutaneous involvement from nodal disease.
The histopathologic differential diagnosis of the current case also included cutaneous CD30+ epithelioid angiosarcoma of the breast. Weed and Folpe35 reported the case of an 85-year-old woman who developed a CD30+ epithelioid angiosarcoma on the breast after undergoing breast-conserving surgery and adjuvant radiotherapy for treatment of an infiltrating ductal carcinoma of the breast. Histopathology showed a diffuse replacement of the dermis by a highly malignant-appearing epithelioid neoplasm growing in a solid sheet. Neoplastic cells expressed strong CD30 immunoreactivity with absence of immunoexpression for cytokeratins, S-100 protein, and CD45. Additional immunostaining demonstrated that neoplastic cells also expressed strong immunoreactivity for CD31 and the friend leukemia virus integration 1 gene, FLI-1, and focal positivity for von Willebrand factor, supporting a diagnosis of epithelioid angiosarcoma.35 In our patient, CD34 and CD31 were negative, which ruled out the endothelial nature of neoplastic cells.
Conclusion
In summary, we report an example of regional LyP limited to the left breast of a woman with a history of partial mastectomy and adjuvant radiotherapy for treatment of invasive ductal breast carcinoma. It is a rare case of regional LyP exclusively involving an irradiated area of the skin.
Lymphomatoid papulosis (LyP) is a clinicopathologic variant of CD30+ primary cutaneous T-cell lymphoproliferative disorder characterized by a chronic, recurrent, self-healing eruption of papules and small nodules. From a clinical point of view, LyP is not considered a malignant disorder despite demonstration of clonality in most cases.1 From a histopathologic point of view, there are 5 types of LyP: (1) type A, the most common type, which is characterized by a wedge-shaped infiltrate composed of clustered large atypical cells admixed with neutrophils, eosinophils, histiocytes, and small lymphocytes; (2) type B, a rare variant characterized by a bandlike infiltrate of small- to medium-sized pleomorphic and hyperchromatic lymphocytes involving the superficial dermis with epidermotropism; (3) type C, which consists of a nodular infiltrate of large atypical cells with a cohesive arrangement closely similar to anaplastic large-cell lymphoma; (4) type D, a variant with histopathologic features that resemble primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma, but neoplastic cells express CD30 and a T-cell cytotoxic phenotype (βF1+, CD3+, CD4‒, CD8+), and follow-up usually does not reveal development of systemic involvement or signs of other cutaneous lymphomas2; and (5) type E, which is characterized by oligolesional papules that rapidly ulcerate and evolve into large, necrotic, escharlike lesions with a diameter of 1 to 4 cm and an angiocentric and angiodestructive infiltrate of small- to medium-sized atypical lymphocytes expressing CD30 and frequently CD8.3
The clinical appearance of LyP usually is polymorphic, with lesions in different stages of evolution scattered all over the skin; however, the lesions are occasionally localized only to one area of the skin, the so-called regional or agminated LyP.4-14 We report a case of regional LyP that exclusively involved the skin of the left breast, which had previously received radiotherapy for treatment of breast carcinoma. Lymphomatoid papulosis with cutaneous lesions involving only an area of irradiated skin is rare.
Case Report
A 59-year-old woman presented with new-onset cutaneous lesions on the left breast. The patient had a history of invasive ductal carcinoma of the left breast, which had been treated 5 years prior with a partial mastectomy and radiotherapy (10 Gy per week for 5 consecutive weeks [50 Gy total]). Physical examination revealed a large nodular lesion with a necrotic surface on the upper half of the left breast as well as 3 small papular lesions with eroded surfaces on the lower half of the breast (Figure 1). A clinical diagnosis of cutaneous metastases from breast carcinoma was suspected.
Biopsies from one small papule and the large nodular lesion showed similar findings consisting of a necrotic epidermis covered by crusts and a wedge-shaped infiltrate involving the superficial dermis (Figure 2A). The infiltrate was mostly composed of large atypical mononuclear cells with oval to kidney-shaped nuclei, prominent nucleoli, and ample basophilic cytoplasm. Many mitotic figures were seen within the infiltrate (Figure 2B). The infiltrate of atypical cells was admixed with small lymphocytes, histiocytes, and some eosinophils. Immunohistochemically, the large atypical cells expressed CD2, CD3, CD4, CD45, CD30, and epithelial membrane antigen (Figures 2C and 2D). A few atypical cells also expressed CD8 and T-cell intracellular antigen 1. Approximately 60% of the nuclei of the atypical cells showed MIB-1 positivity, while CD20, CD56, AE1/AE3, S-100 protein, CD34, and CD31 were negative. The anaplastic lymphoma kinase was not expressed in atypical cells. Monoclonal rearrangement of the γ T-cell receptor was demonstrated on polymerase chain reaction. Physical examination showed no lymphadenopathy in any lymph node chains. Computed tomography of the chest and abdomen failed to demonstrate systemic involvement. On the basis of these clinical, histologic, immunohistochemical, and molecular results, a diagnosis of type A regional LyP was established.
The patient was treated with 2 daily applications of clobetasol propionate cream 0.5 mg/g and 10 mg of oral methotrexate per week for 4 weeks. After 4 weeks of treatment, the lesions on the left breast had resolved leaving slightly atrophic scars. Six months later, an episode of recurrent papular lesions occurred in the same area and responded to the same treatment, but no systemic involvement had been found.
Comment
Regional LyP is a rare variant, with only a few reported cases in the literature.4-18 Scarisbrick et al4 originally reported 4 patients with LyP limited to specific regions. Interestingly, one of the patients had mycosis fungoides and the LyP lesions were confined to the same region where the mycosis fungoides lesions were observed.4 In a review of LyP in patients from the Netherlands (n=118), lesions limited to a specific region of the body were observed in 13% of cases.5 Cases of LyP limited to acral skin also have been reported.6-8 Heald et al9 described 7 patients who had continuing eruptions of papulonodules with histopathologic features of LyP within well-circumscribed areas of the skin. The investigators interpreted this localized variant of LyP as an equivalent of the limited plaque stage of mycosis fungoides. Interestingly, one of the patients with LyP eventually developed plaques of mycosis fungoides in other areas of the skin not involved by LyP.9 Sharma et al10 described an additional example of regional LyP, and Nakahigashi et al11 described a patient with tumor-stage mycosis fungoides who subsequently developed regional LyP involving the right side of the chest. Kim et al12 described a patient with recurrent episodes of regional LyP exclusively involving the periorbital skin, and Torrelo et al13 reported a 12-year-old boy with persistent lesions of LyP involving the skin of the right side of the abdomen. Coelho et al14 reported a 13-year-old adolescent girl who presented with recurrent papules of LyP exclusively involving the left upper arm. Buder et al15 reported a case of LyP limited to Becker melanosis. Shang et al16 described an additional caseof regional LyP that was successfully controlled by interferon alfa-2b and nitrogen mustard solution. Haus et al17 reported type A LyP confined to the cutaneous area within a red tattoo. Finally, Wang et al18 reported a case of regional LyP in association with pseudoepitheliomatous hyperplasia
Several dermatoses may appear as specific isomorphic responses to various external stimuli, and it is possible that radiotherapy induces some damage that favors the location of the lesions because the irradiated skin behaves as a locus minoris resistentiae. Pemphigus vulgaris,19,20 Sweet syndrome,21 cutaneous angiosarcoma,22-32 and cutaneous metastases from malignant melanoma also have been reported to be confined to irradiated skin.33 However, in our PubMed search of articles indexed for MEDLINE using the terms lymphomatoid papules and regional, none of the previously reported cases of regional LyP had a history of radiotherapy, and in no instance did the lesions develop on a previously irradiated area of the skin.4-18 The localization of the lesions in our patient could have been the result of the so-called radiation recall phenomenon. Recall dermatitis is defined as a skin reaction in a previously irradiated field, usually subsequent to the administration of cytotoxic drugs or antibiotics.34 It may appear days to years after exposure to ionizing radiation and has mostly been associated with chemotherapy drugs, but recall dermatitis is neither exclusive of chemotherapy medications nor strictly radiotherapy induced. The concept of recall dermatitis has been expanded beyond radiation recall dermatitis to include dermatitis induced by other stimuli, including other drugs, contact irritants, and UV radiation, as well as residual herpes zoster. Nevertheless, in recall dermatitis the triggering drug or agent recalls a prior dermatitis in the involved area, such as sunburn or radiodermatitis. In our patient, there was no history of LyP prior to irradiation of the left breast; therefore, the most plausible interpretation of the peculiar localization of the lesions in our patient seems to be that the eruption resulted as expression of a locus minoris resistentiae.
Distinction between primary cutaneous anaplastic large-cell lymphoma and LyP may be difficult because the histopathologic and immunophenotypic features may overlap. In our case, the presence of several papular lesions and one large nodule are more consistent, from a clinical point of view, with a diagnosis of LyP rather than primary cutaneous anaplastic large-cell lymphoma, which usually presents with a solitary and often large, ulcerated, reddish brown tumor. In our patient, the absence of lymphadenopathy, negative results of the computed tomography of the chest and abdomen, and lack of expression for anaplastic lymphoma kinase in atypical cells of the infiltrate militate against a diagnosis of secondary cutaneous involvement from nodal disease.
The histopathologic differential diagnosis of the current case also included cutaneous CD30+ epithelioid angiosarcoma of the breast. Weed and Folpe35 reported the case of an 85-year-old woman who developed a CD30+ epithelioid angiosarcoma on the breast after undergoing breast-conserving surgery and adjuvant radiotherapy for treatment of an infiltrating ductal carcinoma of the breast. Histopathology showed a diffuse replacement of the dermis by a highly malignant-appearing epithelioid neoplasm growing in a solid sheet. Neoplastic cells expressed strong CD30 immunoreactivity with absence of immunoexpression for cytokeratins, S-100 protein, and CD45. Additional immunostaining demonstrated that neoplastic cells also expressed strong immunoreactivity for CD31 and the friend leukemia virus integration 1 gene, FLI-1, and focal positivity for von Willebrand factor, supporting a diagnosis of epithelioid angiosarcoma.35 In our patient, CD34 and CD31 were negative, which ruled out the endothelial nature of neoplastic cells.
Conclusion
In summary, we report an example of regional LyP limited to the left breast of a woman with a history of partial mastectomy and adjuvant radiotherapy for treatment of invasive ductal breast carcinoma. It is a rare case of regional LyP exclusively involving an irradiated area of the skin.
- Ralfkiaer E, Willemze R, Paulli M, et al. Primary cutaneous CD30-positive T-cell lymphoproliferative disorders. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphomatoid Tissues. Lyon, France: IARC Press, 2008:300-301.
- Saggini A, Gulia A, Argenyi Z, et al. A variant of lymphomatoid papulosis simulating primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma. description of 9 cases. Am J Surg Pathol. 2010;34:1168-1175.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Scarisbrick JJ, Evans AV, Woolford AJ, et al. Regional lymphomatoid papulosis: a report of four cases. Br J Dermatol. 1999;141:1125-1128.
- Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30+ lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000;95:3653-3661.
- Thomas GJ, Conejo-Mir JS, Ruiz AP, et al. Lymphomatoid papulosis in childhood with exclusive acral involvement. Pediatr Dermatol. 1998;15:146-147.
- Deroo-Berger MC, Skowson F, Roner S, et al. Lymphomatoid papulosis: a localized form with acral pustular involvement. Dermatology. 2002;205:60-62.
- Kagaya M, Kondo S, Kamada A, et al. Localized lymphomatoid papulosis. Dermatology. 2002;204:72-74.
- Heald P, Subtil A, Breneman D, et al. Persistent agmination of lymphomatoid papulosis: an equivalent of limited plaque mycosis fungoides type of cutaneous T-cell lymphoma. J Am Acad Dermatol. 2007;57:1005-1011.
- Sharma V, Xu G, Petronic-Rosic V, et al. Clinicopathologic challenge. regional lymphomatoid papulosis, type A. Int J Dermatol. 2007;46:905-909.
- Nakahigashi K, Ishida Y, Matsumura Y, et al. Large cell transformation mimicking regional lymphomatoid papulosis in a patient with mycosis fungoides. J Dermatol. 2008;35:283-288.
- Kim YJ, Rho YK, Yoo KH, et al. Case of regional lymphomatoid papulosis confined to the periorbital areas. J Dermatol. 2009;36:163-165.
- Torrelo A, Colmenero I, Hernández A, et al. Persistent agmination of lymphomatoid papulosis. Pediatr Dermatol. 2009;26:762-764.
- Coelho JD, Afonso A, Feio AB. Regional lymphomatoid papulosis in a child—treatment with a UVB phototherapy handpiece. J Cosmet Laser Ther. 2010;12:155-156.
- Buder K, Wendel AM, Cerroni L, et al. A case of lymphomatoid papulosis limited to Becker’s melanosis. Dermatology. 2013;226:124-127.
- Shang SX, Chen H, Sun JF, et al. Regional lymphomatoid papulosis successfully controlled by interferon α-2b and nitrogen mustard solution. Chin Med J (Engl). 2013;126:3194-3195.
- Haus G, Utikal J, Geraud C, et al. CD30-positive lymphoproliferative disorder in a red tattoo: regional lymphomatoid papulosis type C or pseudolymphoma? Br J Dermatol. 2014;171:668-670.
- Wang T, Guo CL, Xu CC, et al. Regional lymphomatoid papulosis in association with pseudoepitheliomatous hyperplasia: 13 years follow-up. J Eur Acad Dermatol Venereol. 2015;29:1853-1854.
- Davis M, Feverman EJ. Induction of pemphigus by X-ray irradiation. Clin Exp Dermatol. 1987;12:197-199.
- Crovato F, Descrello G, Nazzari G, et al. Liner pemphigus vulgaris after X-ray irradiation. Dermatologica. 1989;179:135-136.
- Vergara G, Vargas-Machuca I, Pastor MA, et al. Localized Sweet’s syndrome in radiation-induced locus minoris resistentae. J Am Acad Dermatol. 2003;49:907-909.
- Caldwell JB, Ryan MT, Benson PM, et al. Cutaneous angiosarcoma arising in the radiation site of a congenital hemangioma. J Am Acad Dermatol. 1995;33:865-870.
- Stone NM, Holden CA. Postirradiation angiosarcoma. Clin Exp Dermatol. 1997;22:46-47.
- Goette EK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12:922-926.
- Chen TK, Goffman KD, Hendricks EJ. Angiosarcoma following therapeutic irradiation. Cancer. 1979;44:2044-2048.
- Rubin E, Maddox WA, Mazur MT. Cutaneous angiosarcoma of the breast 7 years after lumpectomy and radiation therapy. Radiology. 1990;174:258-260.
- Stokkel MPM, Peterse HL. Angiosarcoma of the breast after lumpectomy and radiation therapy for adenocarcinoma. Cancer. 1992;69:2965-2968.
- Moskaluk CA, Merino MJ, Danforth DN, et al. Low-grade angiosarcoma of the skin of the breast: a complication of lumpectomy and radiation therapy for breast carcinoma. Hum Pathol. 1992;23:710-714.
- Parham DM, Fisher C. Angiosarcomas of the breast developing post radiotherapy. Histopathology. 1997;31:189-195.
- Rao J, DeKoven JG, Beatty JD, et al. Cutaneous angiosarcoma as a delayed complication of radiation therapy for carcinoma of the breast. J Am Acad Dermatol. 2003;49:532-538.
- Billings SD, McKenney JK, Folpe Al, et al. Cutaneous angiosarcoma following breast-conserving surgery and radiation. an analysis of 27 cases. Am J Surg Pathol. 2004;28:781-788.
- Fodor J, Orosz Z, Szabo E, et al. Angiosarcoma after conservation treatment for breast carcinoma: our experience and a review of the literature. J Am Acad Dermatol. 2006;54:499-504.
- Roses DP, Harris MN, Rigel D, et al. Local and in-transit metastases following definitive excision from primary cutaneous malignant melanoma. Ann Surg. 1983;198:65-69.
- Burris HA 3rd, Hurtig J. Radiation recall with anticancer agents. Oncologist. 2010;15:1227-1237.
- Weed BR, Folpe AL. Cutaneous CD30-positive epithelioid angiosarcoma following breast-conserving therapy and irradiation. a potential diagnostic pitfall. Am J Dermatopathol. 2008;30:370-372.
- Ralfkiaer E, Willemze R, Paulli M, et al. Primary cutaneous CD30-positive T-cell lymphoproliferative disorders. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphomatoid Tissues. Lyon, France: IARC Press, 2008:300-301.
- Saggini A, Gulia A, Argenyi Z, et al. A variant of lymphomatoid papulosis simulating primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma. description of 9 cases. Am J Surg Pathol. 2010;34:1168-1175.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Scarisbrick JJ, Evans AV, Woolford AJ, et al. Regional lymphomatoid papulosis: a report of four cases. Br J Dermatol. 1999;141:1125-1128.
- Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30+ lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000;95:3653-3661.
- Thomas GJ, Conejo-Mir JS, Ruiz AP, et al. Lymphomatoid papulosis in childhood with exclusive acral involvement. Pediatr Dermatol. 1998;15:146-147.
- Deroo-Berger MC, Skowson F, Roner S, et al. Lymphomatoid papulosis: a localized form with acral pustular involvement. Dermatology. 2002;205:60-62.
- Kagaya M, Kondo S, Kamada A, et al. Localized lymphomatoid papulosis. Dermatology. 2002;204:72-74.
- Heald P, Subtil A, Breneman D, et al. Persistent agmination of lymphomatoid papulosis: an equivalent of limited plaque mycosis fungoides type of cutaneous T-cell lymphoma. J Am Acad Dermatol. 2007;57:1005-1011.
- Sharma V, Xu G, Petronic-Rosic V, et al. Clinicopathologic challenge. regional lymphomatoid papulosis, type A. Int J Dermatol. 2007;46:905-909.
- Nakahigashi K, Ishida Y, Matsumura Y, et al. Large cell transformation mimicking regional lymphomatoid papulosis in a patient with mycosis fungoides. J Dermatol. 2008;35:283-288.
- Kim YJ, Rho YK, Yoo KH, et al. Case of regional lymphomatoid papulosis confined to the periorbital areas. J Dermatol. 2009;36:163-165.
- Torrelo A, Colmenero I, Hernández A, et al. Persistent agmination of lymphomatoid papulosis. Pediatr Dermatol. 2009;26:762-764.
- Coelho JD, Afonso A, Feio AB. Regional lymphomatoid papulosis in a child—treatment with a UVB phototherapy handpiece. J Cosmet Laser Ther. 2010;12:155-156.
- Buder K, Wendel AM, Cerroni L, et al. A case of lymphomatoid papulosis limited to Becker’s melanosis. Dermatology. 2013;226:124-127.
- Shang SX, Chen H, Sun JF, et al. Regional lymphomatoid papulosis successfully controlled by interferon α-2b and nitrogen mustard solution. Chin Med J (Engl). 2013;126:3194-3195.
- Haus G, Utikal J, Geraud C, et al. CD30-positive lymphoproliferative disorder in a red tattoo: regional lymphomatoid papulosis type C or pseudolymphoma? Br J Dermatol. 2014;171:668-670.
- Wang T, Guo CL, Xu CC, et al. Regional lymphomatoid papulosis in association with pseudoepitheliomatous hyperplasia: 13 years follow-up. J Eur Acad Dermatol Venereol. 2015;29:1853-1854.
- Davis M, Feverman EJ. Induction of pemphigus by X-ray irradiation. Clin Exp Dermatol. 1987;12:197-199.
- Crovato F, Descrello G, Nazzari G, et al. Liner pemphigus vulgaris after X-ray irradiation. Dermatologica. 1989;179:135-136.
- Vergara G, Vargas-Machuca I, Pastor MA, et al. Localized Sweet’s syndrome in radiation-induced locus minoris resistentae. J Am Acad Dermatol. 2003;49:907-909.
- Caldwell JB, Ryan MT, Benson PM, et al. Cutaneous angiosarcoma arising in the radiation site of a congenital hemangioma. J Am Acad Dermatol. 1995;33:865-870.
- Stone NM, Holden CA. Postirradiation angiosarcoma. Clin Exp Dermatol. 1997;22:46-47.
- Goette EK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12:922-926.
- Chen TK, Goffman KD, Hendricks EJ. Angiosarcoma following therapeutic irradiation. Cancer. 1979;44:2044-2048.
- Rubin E, Maddox WA, Mazur MT. Cutaneous angiosarcoma of the breast 7 years after lumpectomy and radiation therapy. Radiology. 1990;174:258-260.
- Stokkel MPM, Peterse HL. Angiosarcoma of the breast after lumpectomy and radiation therapy for adenocarcinoma. Cancer. 1992;69:2965-2968.
- Moskaluk CA, Merino MJ, Danforth DN, et al. Low-grade angiosarcoma of the skin of the breast: a complication of lumpectomy and radiation therapy for breast carcinoma. Hum Pathol. 1992;23:710-714.
- Parham DM, Fisher C. Angiosarcomas of the breast developing post radiotherapy. Histopathology. 1997;31:189-195.
- Rao J, DeKoven JG, Beatty JD, et al. Cutaneous angiosarcoma as a delayed complication of radiation therapy for carcinoma of the breast. J Am Acad Dermatol. 2003;49:532-538.
- Billings SD, McKenney JK, Folpe Al, et al. Cutaneous angiosarcoma following breast-conserving surgery and radiation. an analysis of 27 cases. Am J Surg Pathol. 2004;28:781-788.
- Fodor J, Orosz Z, Szabo E, et al. Angiosarcoma after conservation treatment for breast carcinoma: our experience and a review of the literature. J Am Acad Dermatol. 2006;54:499-504.
- Roses DP, Harris MN, Rigel D, et al. Local and in-transit metastases following definitive excision from primary cutaneous malignant melanoma. Ann Surg. 1983;198:65-69.
- Burris HA 3rd, Hurtig J. Radiation recall with anticancer agents. Oncologist. 2010;15:1227-1237.
- Weed BR, Folpe AL. Cutaneous CD30-positive epithelioid angiosarcoma following breast-conserving therapy and irradiation. a potential diagnostic pitfall. Am J Dermatopathol. 2008;30:370-372.
Practice Points
- Cutaneous lesions of lymphomatoid papulosis (LyP) sometimes are confined to only one area of the skin, which is known as regional LyP.
- Patients with regional LyP have the same prognosis as those with widespread LyP, and no specific association has been reported with this clinical variant.
- Lesions of regional LyP respond to the same treatments as widespread LyP.

The Elongated Dermatofibroma: A New Dermoscopic Variant?
To the Editor:
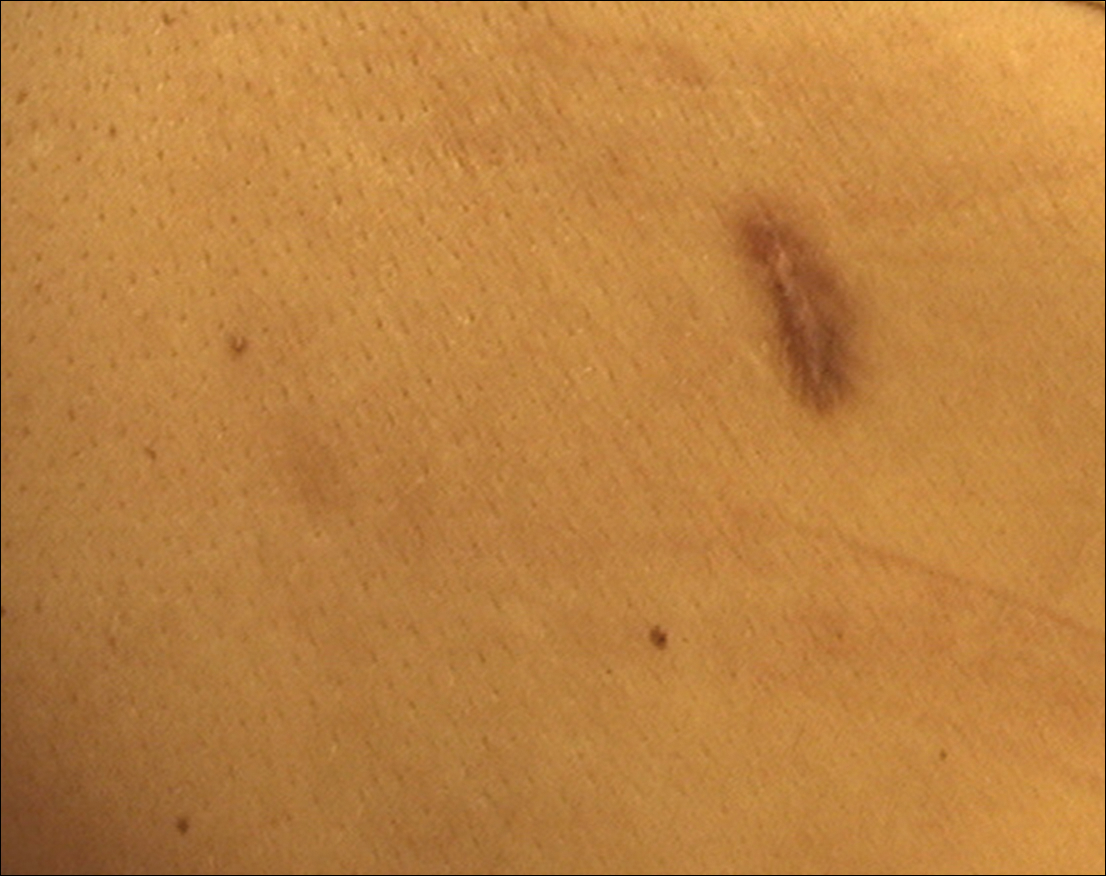
Dermatofibroma is a common cutaneous lesion that most frequently affects young or middle-aged adults, especially women.1 Clinically, it appears as a firm, pink or brown nodule. It may be painful or show a tendency for scarring. The pathognomonic feature of dermatofibroma, regarded as a fibrohistiocytic tumor, is the so-called button sign caused by skin depression following pressure. We present a unique case of elongated dermatofibroma with a linear, white, scarlike patch with a brownish pigmented network and globules.
A 40-year-old woman presented with a linear elongated lesion localized to the right side of the infrascapular region of 10 years’ duration. The lesion initially was a small brownish plaque. There was no history of trauma or scratching. Over the next 10 years, the lesion slowly progressed, finally becoming a linear, atrophic, brownish plaque that was 2.5-cm long (Figure 1). The button sign was positive. On dermoscopy the central, elongated, white patch was visualized not as a typical round patch but as a scarlike white line (Figure 2A) surrounded by a brownish network that was especially pronounced in the distal parts of the lesion. In the upper part of the lesion, multiple marginally disseminated, dark brown dots were present. Brownish globules within the linear white patch also were observed in the lower central part. Figure 2B presents a dermoscopic picture of the linear variant of dermatofibroma. For cosmetic reasons, the patient underwent total surgical excision of the lesion. Histopathology revealed distinct characteristics of dermatofibroma (Figures 3A and 3B).
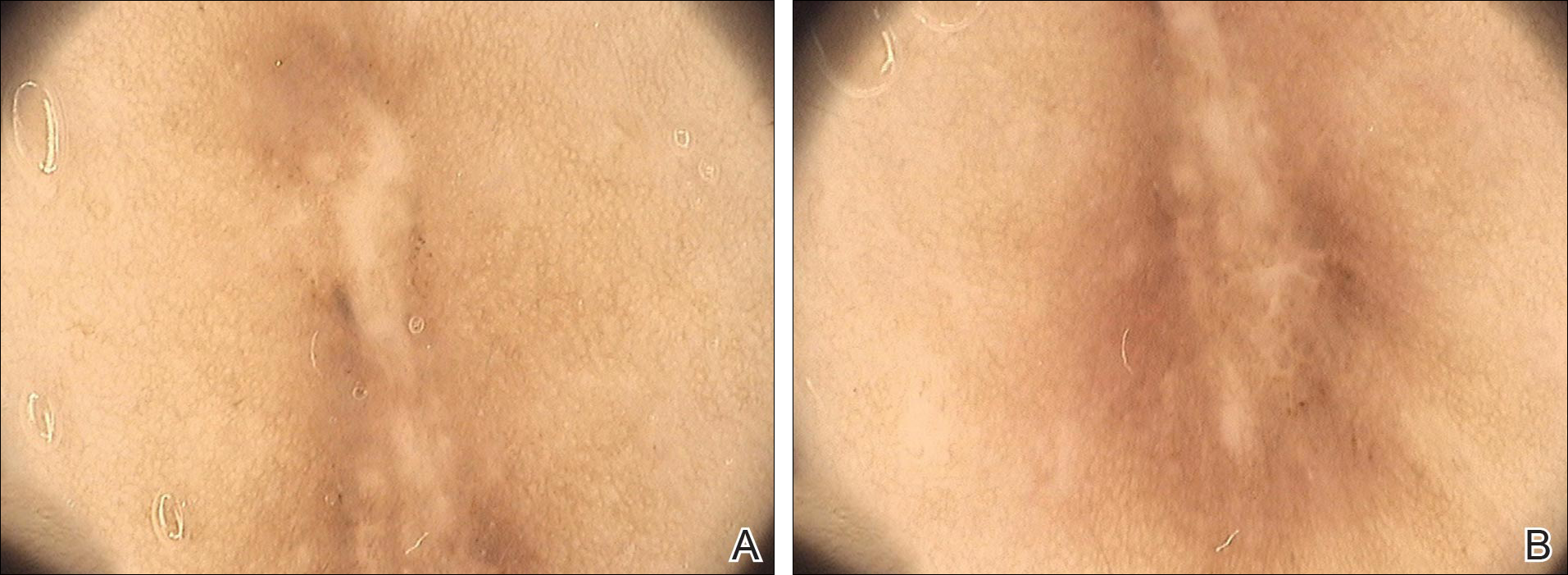
The most common features of dermatofibromas seen in polarized and nonpolarized dermoscopy are central white scarlike patches, brown globulelike structures, vascular structures, and a peripheral fine pigmented network.2 Kilinc Karaarslan et al3 described atypical dermatofibromas with linear irregular crypts, which were seen in 26.9% of all studied cases. These irregular crypts were mainly medium in size (10 lesions), with only 2 lesions being tiny and regularly distributed. Only one lesion had atypical clinical and dermoscopic features occurring as an atrophic plaque with multiple small scarlike areas and peripherally distributed pigment network.3 Based on this typology, we believe our patient represents a case of elongated dermatofibroma that could be an atrophic variant of dermatofibroma. This form would not appear as a small scarlike area with pigment network in a somewhat patchy distribution3 but as a scarlike linear chord with a bipolar pigment network. Zaballos et al1 described 10 dermoscopic patterns of dermatofibroma (N=412); the most common was a central white patch and peripheral pigment network in approximately 35% of cases. A white scarlike patch was observed in 57.0% of dermat-ofibromas in 4 variants: (1) a solitary structure located in the center; (2) multiple white scarlike patches; (3) white scarlike patch extending throughout the lesion or irregularly distributed; and (4) white network (central, total, or irregular).1 Agero et al2 first described the new feature as a central white patch characterized by shiny white streaks. The most frequent dermoscopic pattern associated with dermatofibromas is the central white scarlike patch and peripheral delicate pigment network.1,4 Arpaia et al4 observed that dermoscopic patterns may correspond to distinct sequential stages of the formation of dermatofibroma. The linear character we described may be related to a variant of scarring keloid dermatofibroma.5
- Zaballos P, Puig S, Llambrich A, et al. Dermoscopy of dermatofibromas: a prospective morphological study of 412 cases. Arch Dermatol. 2008;144:75-83.
- Agero AL, Taliercio S, Dusza SW, et al. Conventional and polarized dermoscopy features of dermatofibroma. Arch Dermatol. 2006;142:1431-1437.
- Kilinc Karaarslan I, Gencoglan G, Akalin T, et al. Different dermoscopic faces of dermatofibromas. J Am Acad Dermatol. 2007;57:401-406.
- Arpaia N, Cassano N, Vena GA. Dermoscopic patterns of dermatofibroma. Dermatol Surg. 2005;31:1336-1339.
- Kuo TT, Hu S, Chan HL. Keloidal dermatofibroma: report of 10 cases of a new variant. Am J Surg Pathol. 1998;22:564-568.
To the Editor:
Dermatofibroma is a common cutaneous lesion that most frequently affects young or middle-aged adults, especially women.1 Clinically, it appears as a firm, pink or brown nodule. It may be painful or show a tendency for scarring. The pathognomonic feature of dermatofibroma, regarded as a fibrohistiocytic tumor, is the so-called button sign caused by skin depression following pressure. We present a unique case of elongated dermatofibroma with a linear, white, scarlike patch with a brownish pigmented network and globules.
A 40-year-old woman presented with a linear elongated lesion localized to the right side of the infrascapular region of 10 years’ duration. The lesion initially was a small brownish plaque. There was no history of trauma or scratching. Over the next 10 years, the lesion slowly progressed, finally becoming a linear, atrophic, brownish plaque that was 2.5-cm long (Figure 1). The button sign was positive. On dermoscopy the central, elongated, white patch was visualized not as a typical round patch but as a scarlike white line (Figure 2A) surrounded by a brownish network that was especially pronounced in the distal parts of the lesion. In the upper part of the lesion, multiple marginally disseminated, dark brown dots were present. Brownish globules within the linear white patch also were observed in the lower central part. Figure 2B presents a dermoscopic picture of the linear variant of dermatofibroma. For cosmetic reasons, the patient underwent total surgical excision of the lesion. Histopathology revealed distinct characteristics of dermatofibroma (Figures 3A and 3B).
The most common features of dermatofibromas seen in polarized and nonpolarized dermoscopy are central white scarlike patches, brown globulelike structures, vascular structures, and a peripheral fine pigmented network.2 Kilinc Karaarslan et al3 described atypical dermatofibromas with linear irregular crypts, which were seen in 26.9% of all studied cases. These irregular crypts were mainly medium in size (10 lesions), with only 2 lesions being tiny and regularly distributed. Only one lesion had atypical clinical and dermoscopic features occurring as an atrophic plaque with multiple small scarlike areas and peripherally distributed pigment network.3 Based on this typology, we believe our patient represents a case of elongated dermatofibroma that could be an atrophic variant of dermatofibroma. This form would not appear as a small scarlike area with pigment network in a somewhat patchy distribution3 but as a scarlike linear chord with a bipolar pigment network. Zaballos et al1 described 10 dermoscopic patterns of dermatofibroma (N=412); the most common was a central white patch and peripheral pigment network in approximately 35% of cases. A white scarlike patch was observed in 57.0% of dermat-ofibromas in 4 variants: (1) a solitary structure located in the center; (2) multiple white scarlike patches; (3) white scarlike patch extending throughout the lesion or irregularly distributed; and (4) white network (central, total, or irregular).1 Agero et al2 first described the new feature as a central white patch characterized by shiny white streaks. The most frequent dermoscopic pattern associated with dermatofibromas is the central white scarlike patch and peripheral delicate pigment network.1,4 Arpaia et al4 observed that dermoscopic patterns may correspond to distinct sequential stages of the formation of dermatofibroma. The linear character we described may be related to a variant of scarring keloid dermatofibroma.5
To the Editor:
Dermatofibroma is a common cutaneous lesion that most frequently affects young or middle-aged adults, especially women.1 Clinically, it appears as a firm, pink or brown nodule. It may be painful or show a tendency for scarring. The pathognomonic feature of dermatofibroma, regarded as a fibrohistiocytic tumor, is the so-called button sign caused by skin depression following pressure. We present a unique case of elongated dermatofibroma with a linear, white, scarlike patch with a brownish pigmented network and globules.
A 40-year-old woman presented with a linear elongated lesion localized to the right side of the infrascapular region of 10 years’ duration. The lesion initially was a small brownish plaque. There was no history of trauma or scratching. Over the next 10 years, the lesion slowly progressed, finally becoming a linear, atrophic, brownish plaque that was 2.5-cm long (Figure 1). The button sign was positive. On dermoscopy the central, elongated, white patch was visualized not as a typical round patch but as a scarlike white line (Figure 2A) surrounded by a brownish network that was especially pronounced in the distal parts of the lesion. In the upper part of the lesion, multiple marginally disseminated, dark brown dots were present. Brownish globules within the linear white patch also were observed in the lower central part. Figure 2B presents a dermoscopic picture of the linear variant of dermatofibroma. For cosmetic reasons, the patient underwent total surgical excision of the lesion. Histopathology revealed distinct characteristics of dermatofibroma (Figures 3A and 3B).
The most common features of dermatofibromas seen in polarized and nonpolarized dermoscopy are central white scarlike patches, brown globulelike structures, vascular structures, and a peripheral fine pigmented network.2 Kilinc Karaarslan et al3 described atypical dermatofibromas with linear irregular crypts, which were seen in 26.9% of all studied cases. These irregular crypts were mainly medium in size (10 lesions), with only 2 lesions being tiny and regularly distributed. Only one lesion had atypical clinical and dermoscopic features occurring as an atrophic plaque with multiple small scarlike areas and peripherally distributed pigment network.3 Based on this typology, we believe our patient represents a case of elongated dermatofibroma that could be an atrophic variant of dermatofibroma. This form would not appear as a small scarlike area with pigment network in a somewhat patchy distribution3 but as a scarlike linear chord with a bipolar pigment network. Zaballos et al1 described 10 dermoscopic patterns of dermatofibroma (N=412); the most common was a central white patch and peripheral pigment network in approximately 35% of cases. A white scarlike patch was observed in 57.0% of dermat-ofibromas in 4 variants: (1) a solitary structure located in the center; (2) multiple white scarlike patches; (3) white scarlike patch extending throughout the lesion or irregularly distributed; and (4) white network (central, total, or irregular).1 Agero et al2 first described the new feature as a central white patch characterized by shiny white streaks. The most frequent dermoscopic pattern associated with dermatofibromas is the central white scarlike patch and peripheral delicate pigment network.1,4 Arpaia et al4 observed that dermoscopic patterns may correspond to distinct sequential stages of the formation of dermatofibroma. The linear character we described may be related to a variant of scarring keloid dermatofibroma.5
- Zaballos P, Puig S, Llambrich A, et al. Dermoscopy of dermatofibromas: a prospective morphological study of 412 cases. Arch Dermatol. 2008;144:75-83.
- Agero AL, Taliercio S, Dusza SW, et al. Conventional and polarized dermoscopy features of dermatofibroma. Arch Dermatol. 2006;142:1431-1437.
- Kilinc Karaarslan I, Gencoglan G, Akalin T, et al. Different dermoscopic faces of dermatofibromas. J Am Acad Dermatol. 2007;57:401-406.
- Arpaia N, Cassano N, Vena GA. Dermoscopic patterns of dermatofibroma. Dermatol Surg. 2005;31:1336-1339.
- Kuo TT, Hu S, Chan HL. Keloidal dermatofibroma: report of 10 cases of a new variant. Am J Surg Pathol. 1998;22:564-568.
- Zaballos P, Puig S, Llambrich A, et al. Dermoscopy of dermatofibromas: a prospective morphological study of 412 cases. Arch Dermatol. 2008;144:75-83.
- Agero AL, Taliercio S, Dusza SW, et al. Conventional and polarized dermoscopy features of dermatofibroma. Arch Dermatol. 2006;142:1431-1437.
- Kilinc Karaarslan I, Gencoglan G, Akalin T, et al. Different dermoscopic faces of dermatofibromas. J Am Acad Dermatol. 2007;57:401-406.
- Arpaia N, Cassano N, Vena GA. Dermoscopic patterns of dermatofibroma. Dermatol Surg. 2005;31:1336-1339.
- Kuo TT, Hu S, Chan HL. Keloidal dermatofibroma: report of 10 cases of a new variant. Am J Surg Pathol. 1998;22:564-568.
Practice Points
- The most common features of dermatofibromas are white scarlike patches, brown globulelike structures, vascular structures, and a peripheral fine pigmented network.
- Dermoscopy may be used in the diagnostic workup of pigmented nonmelanocytic lesions.

Palmoplantar Pustular Eruption Due to Dabigatran
To the Editor:
A 71-year-old woman with hypertension and atrial fibrillation due to thyrotoxicosis was prescribed dabigatran for stroke prevention by her cardiologist. She also was taking pantoprazole, methimazole, and amiodarone at the time of presentation, all managed by her endocrinologist. She had no known drug allergies but reported a remote history of a palmar rash after eating shellfish. She otherwise had never had any problems with her skin and had no family history of psoriasis. She had a history of smoking 50 packs per year but had quit 6 months prior to presentation. After two 150-mg doses of dabigatran, she noticed numerous mildly tender and itchy eruptions on the palmar and plantar surfaces with no associated respiratory, oropharyngeal, or constitutional symptoms. She denied any recent shellfish ingestion. On dermatologic examination, numerous discreet pustules were present on the bilateral palmar and plantar surfaces with minimal erythema of the underlying skin (Figure).
A punch biopsy was taken from a newly forming lesion on the right palm. Histopathology revealed mild hyperkeratosis, spongiosis with lymphocyte exocytosis, intraepidermal vesiculation, and a sparse upper dermal and perivascular lymphohistiocytic infiltration. No neutrophils or microabscesses were seen. Staining with periodic acid–Schiff revealed no fungi, and S-100 staining revealed numerous Langerhans cells in the epidermis. Although the skin lesions clinically appeared pustular, the results were consistent with an eczematous drug reaction. Laboratory values, including a complete blood cell count, iron studies, chemistry panels, liver function, thyroid function, and coagulation studies, were remarkable only for mild anemia. The patient declined any topical or systemic skin treatment. Dabigatran was discontinued, and the lesions began to clear immediately thereafter. Dabigatran was not reintroduced. Enoxaparin subsequently was prescribed for anticoagulation. The diagnosis of a drug reaction due to dabigatran was made, which was supported with a score of 7 on the Naranjo scale (0=doubtful; 1–4=possible; 5–8=probable; ≥9=definite) for determining probability of drug-induced adverse reactions.1 The differential diagnosis for the skin eruption included palmoplantar pustular psoriasis, dyshidrotic eczema, and allergic contact dermatitis, but the clinical history did not support these diagnoses.
Dabigatran is a direct thrombin inhibitor used to reduce the risk for stroke and systemic embolism in patients with nonvalvular atrial fibrillation. Based on results of the RE-LY (Randomization Evaluation of Long-term Anticoagulation Therapy) trial published in 2009, dabigatran 150 mg twice daily significantly reduced the risk for stroke and systemic emboli in patients with atrial fibrillation compared to warfarin (annual risk, 1.11% vs 1.69%; relative risk, 0.66; 95% CI, 0.53-0.82; P<.001) with the advantage of not requiring frequent monitoring of the international normalized ratio.2 The most common adverse effect of dabigatran in this trial was dyspepsia (11.3% vs 5.8%). Drug hypersensitivity, allergic edema, and anaphylaxis were reported in less than 0.1% of patients taking dabigatran.2
According to a PubMed search of articles indexed for MEDLINE using the search terms dabigatran cutaneous reaction and dabigatran rash, 4 case reports of cutaneous eruption due to dabigatran were identified. In one report, a 20-year-old man with atrial fibrillation developed an eruption similar to our patient on the thigh and forearm after 2 weeks of taking oral dabigatran 150 mg twice daily. It resolved without complication after topical corticosteroid use and discontinuation of dabigatran.3 In another report, a 78-year-old man presented to the emergency department after taking two 150-mg doses of dabigatran with a diffuse, full-body, pruritic rash that resolved with oral diphenhydramine and discontinuation of dabigatran.4 A third case described a 59-year-old man who was taking 150 mg dabigatran twice daily for 5 days before developing a rash.5 The fourth case involved a 74-year-old woman who developed leukocytoclastic vasculitis 1 week after taking dabigatran 150 mg twice daily.6
It is important to monitor for and report hypersensitivity reactions in patients taking dabigatran. Drug exanthems may cause discomfort or even herald more serious hypersensitivity reactions. Patients experiencing these reactions may discontinue therapy without notifying a physician and consequently place themselves at risk for embolism or stroke.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation [published online August 30, 2009]. N Engl J Med. 2009;361:1139-1151.
- Whitehead H, Boyd J, Blais D, et al. Drug induced exanthem following dabigatran. Ann Pharmacother. 2011;45:e53.
- Eid TJ, Shah SA. Dabigatran-induced rash. Am J Health Syst Pharm. 2011;68:1489-1490.
- To K, Reynolds C, Spinler SA. Rash associated with dabigatran etrexilate. Pharmacotherapy. 2013;33:e23-e27.
- Cakmak MA, Sahin S, Cinar N, et al. Adverse skin reaction caused by dabigatran. Eur Rev Med Pharmacol Sci. 2014;18:2595.
To the Editor:
A 71-year-old woman with hypertension and atrial fibrillation due to thyrotoxicosis was prescribed dabigatran for stroke prevention by her cardiologist. She also was taking pantoprazole, methimazole, and amiodarone at the time of presentation, all managed by her endocrinologist. She had no known drug allergies but reported a remote history of a palmar rash after eating shellfish. She otherwise had never had any problems with her skin and had no family history of psoriasis. She had a history of smoking 50 packs per year but had quit 6 months prior to presentation. After two 150-mg doses of dabigatran, she noticed numerous mildly tender and itchy eruptions on the palmar and plantar surfaces with no associated respiratory, oropharyngeal, or constitutional symptoms. She denied any recent shellfish ingestion. On dermatologic examination, numerous discreet pustules were present on the bilateral palmar and plantar surfaces with minimal erythema of the underlying skin (Figure).
A punch biopsy was taken from a newly forming lesion on the right palm. Histopathology revealed mild hyperkeratosis, spongiosis with lymphocyte exocytosis, intraepidermal vesiculation, and a sparse upper dermal and perivascular lymphohistiocytic infiltration. No neutrophils or microabscesses were seen. Staining with periodic acid–Schiff revealed no fungi, and S-100 staining revealed numerous Langerhans cells in the epidermis. Although the skin lesions clinically appeared pustular, the results were consistent with an eczematous drug reaction. Laboratory values, including a complete blood cell count, iron studies, chemistry panels, liver function, thyroid function, and coagulation studies, were remarkable only for mild anemia. The patient declined any topical or systemic skin treatment. Dabigatran was discontinued, and the lesions began to clear immediately thereafter. Dabigatran was not reintroduced. Enoxaparin subsequently was prescribed for anticoagulation. The diagnosis of a drug reaction due to dabigatran was made, which was supported with a score of 7 on the Naranjo scale (0=doubtful; 1–4=possible; 5–8=probable; ≥9=definite) for determining probability of drug-induced adverse reactions.1 The differential diagnosis for the skin eruption included palmoplantar pustular psoriasis, dyshidrotic eczema, and allergic contact dermatitis, but the clinical history did not support these diagnoses.
Dabigatran is a direct thrombin inhibitor used to reduce the risk for stroke and systemic embolism in patients with nonvalvular atrial fibrillation. Based on results of the RE-LY (Randomization Evaluation of Long-term Anticoagulation Therapy) trial published in 2009, dabigatran 150 mg twice daily significantly reduced the risk for stroke and systemic emboli in patients with atrial fibrillation compared to warfarin (annual risk, 1.11% vs 1.69%; relative risk, 0.66; 95% CI, 0.53-0.82; P<.001) with the advantage of not requiring frequent monitoring of the international normalized ratio.2 The most common adverse effect of dabigatran in this trial was dyspepsia (11.3% vs 5.8%). Drug hypersensitivity, allergic edema, and anaphylaxis were reported in less than 0.1% of patients taking dabigatran.2
According to a PubMed search of articles indexed for MEDLINE using the search terms dabigatran cutaneous reaction and dabigatran rash, 4 case reports of cutaneous eruption due to dabigatran were identified. In one report, a 20-year-old man with atrial fibrillation developed an eruption similar to our patient on the thigh and forearm after 2 weeks of taking oral dabigatran 150 mg twice daily. It resolved without complication after topical corticosteroid use and discontinuation of dabigatran.3 In another report, a 78-year-old man presented to the emergency department after taking two 150-mg doses of dabigatran with a diffuse, full-body, pruritic rash that resolved with oral diphenhydramine and discontinuation of dabigatran.4 A third case described a 59-year-old man who was taking 150 mg dabigatran twice daily for 5 days before developing a rash.5 The fourth case involved a 74-year-old woman who developed leukocytoclastic vasculitis 1 week after taking dabigatran 150 mg twice daily.6
It is important to monitor for and report hypersensitivity reactions in patients taking dabigatran. Drug exanthems may cause discomfort or even herald more serious hypersensitivity reactions. Patients experiencing these reactions may discontinue therapy without notifying a physician and consequently place themselves at risk for embolism or stroke.
To the Editor:
A 71-year-old woman with hypertension and atrial fibrillation due to thyrotoxicosis was prescribed dabigatran for stroke prevention by her cardiologist. She also was taking pantoprazole, methimazole, and amiodarone at the time of presentation, all managed by her endocrinologist. She had no known drug allergies but reported a remote history of a palmar rash after eating shellfish. She otherwise had never had any problems with her skin and had no family history of psoriasis. She had a history of smoking 50 packs per year but had quit 6 months prior to presentation. After two 150-mg doses of dabigatran, she noticed numerous mildly tender and itchy eruptions on the palmar and plantar surfaces with no associated respiratory, oropharyngeal, or constitutional symptoms. She denied any recent shellfish ingestion. On dermatologic examination, numerous discreet pustules were present on the bilateral palmar and plantar surfaces with minimal erythema of the underlying skin (Figure).
A punch biopsy was taken from a newly forming lesion on the right palm. Histopathology revealed mild hyperkeratosis, spongiosis with lymphocyte exocytosis, intraepidermal vesiculation, and a sparse upper dermal and perivascular lymphohistiocytic infiltration. No neutrophils or microabscesses were seen. Staining with periodic acid–Schiff revealed no fungi, and S-100 staining revealed numerous Langerhans cells in the epidermis. Although the skin lesions clinically appeared pustular, the results were consistent with an eczematous drug reaction. Laboratory values, including a complete blood cell count, iron studies, chemistry panels, liver function, thyroid function, and coagulation studies, were remarkable only for mild anemia. The patient declined any topical or systemic skin treatment. Dabigatran was discontinued, and the lesions began to clear immediately thereafter. Dabigatran was not reintroduced. Enoxaparin subsequently was prescribed for anticoagulation. The diagnosis of a drug reaction due to dabigatran was made, which was supported with a score of 7 on the Naranjo scale (0=doubtful; 1–4=possible; 5–8=probable; ≥9=definite) for determining probability of drug-induced adverse reactions.1 The differential diagnosis for the skin eruption included palmoplantar pustular psoriasis, dyshidrotic eczema, and allergic contact dermatitis, but the clinical history did not support these diagnoses.
Dabigatran is a direct thrombin inhibitor used to reduce the risk for stroke and systemic embolism in patients with nonvalvular atrial fibrillation. Based on results of the RE-LY (Randomization Evaluation of Long-term Anticoagulation Therapy) trial published in 2009, dabigatran 150 mg twice daily significantly reduced the risk for stroke and systemic emboli in patients with atrial fibrillation compared to warfarin (annual risk, 1.11% vs 1.69%; relative risk, 0.66; 95% CI, 0.53-0.82; P<.001) with the advantage of not requiring frequent monitoring of the international normalized ratio.2 The most common adverse effect of dabigatran in this trial was dyspepsia (11.3% vs 5.8%). Drug hypersensitivity, allergic edema, and anaphylaxis were reported in less than 0.1% of patients taking dabigatran.2
According to a PubMed search of articles indexed for MEDLINE using the search terms dabigatran cutaneous reaction and dabigatran rash, 4 case reports of cutaneous eruption due to dabigatran were identified. In one report, a 20-year-old man with atrial fibrillation developed an eruption similar to our patient on the thigh and forearm after 2 weeks of taking oral dabigatran 150 mg twice daily. It resolved without complication after topical corticosteroid use and discontinuation of dabigatran.3 In another report, a 78-year-old man presented to the emergency department after taking two 150-mg doses of dabigatran with a diffuse, full-body, pruritic rash that resolved with oral diphenhydramine and discontinuation of dabigatran.4 A third case described a 59-year-old man who was taking 150 mg dabigatran twice daily for 5 days before developing a rash.5 The fourth case involved a 74-year-old woman who developed leukocytoclastic vasculitis 1 week after taking dabigatran 150 mg twice daily.6
It is important to monitor for and report hypersensitivity reactions in patients taking dabigatran. Drug exanthems may cause discomfort or even herald more serious hypersensitivity reactions. Patients experiencing these reactions may discontinue therapy without notifying a physician and consequently place themselves at risk for embolism or stroke.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation [published online August 30, 2009]. N Engl J Med. 2009;361:1139-1151.
- Whitehead H, Boyd J, Blais D, et al. Drug induced exanthem following dabigatran. Ann Pharmacother. 2011;45:e53.
- Eid TJ, Shah SA. Dabigatran-induced rash. Am J Health Syst Pharm. 2011;68:1489-1490.
- To K, Reynolds C, Spinler SA. Rash associated with dabigatran etrexilate. Pharmacotherapy. 2013;33:e23-e27.
- Cakmak MA, Sahin S, Cinar N, et al. Adverse skin reaction caused by dabigatran. Eur Rev Med Pharmacol Sci. 2014;18:2595.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation [published online August 30, 2009]. N Engl J Med. 2009;361:1139-1151.
- Whitehead H, Boyd J, Blais D, et al. Drug induced exanthem following dabigatran. Ann Pharmacother. 2011;45:e53.
- Eid TJ, Shah SA. Dabigatran-induced rash. Am J Health Syst Pharm. 2011;68:1489-1490.
- To K, Reynolds C, Spinler SA. Rash associated with dabigatran etrexilate. Pharmacotherapy. 2013;33:e23-e27.
- Cakmak MA, Sahin S, Cinar N, et al. Adverse skin reaction caused by dabigatran. Eur Rev Med Pharmacol Sci. 2014;18:2595.
Practice Points
- Dabigatran is a direct thrombin inhibitor used in patients with atrial fibrillation to prevent thromboembolic events.
- Although the most common adverse effects of dabigatran are bleeding and dyspepsia, clinicians also should be aware of the potential for cutaneous hypersensitivity reactions to this drug.
Exophytic Scalp Tumor
The Diagnosis: Primary Cutaneous Carcinosarcoma
A generous shave biopsy and debulking performed on the initial visit revealed an infiltrating tumor consisting of malignant epithelial and stromal components (Figure). The basaloid and squamoid epithelial cells were keratin positive. The stromal cells demonstrated positivity for CD10 but were keratin negative. The epithelial portion of the tumor was composed mostly of basaloid islands of cells with nuclear pleomorphism, scattered mitoses, and focal sebaceous differentiation. The mesenchymal portion of the tumor displayed florid pleomorphism and polymorphism, with many large atypical cells and proliferation. A diagnosis of primary cutaneous carcinosarcoma (PCC) was rendered. Head and neck computed tomography showed tumor penetration of less than 1 cm into scalp soft tissues with no involvement of the underlying bone. There was some evidence of swelling of the supragaleal soft tissues without indication of perineural spread. An 11-mm hyperlucent lower cervical lymph node on the left side that likely represented an incidental finding was noted. Surgical excision with margin evaluation was recommended, but the patient declined. He instead received radiation therapy to the left side of the posterior scalp with a total dose of 30 Gy at 6 Gy per fraction and 1 fraction daily. The patient was found to have a well-healed scar with no evidence of recurrence at 4-week follow-up and again at 5 months after radiation therapy.
|
|
|
| A generous shave biopsy and debulking performed on the initial visit revealed an inflitrating tumor consisting on malignant epithelial and stromal components (A-C)(H&E; original magnifications ×10, ×20, and ×40, respectively). |
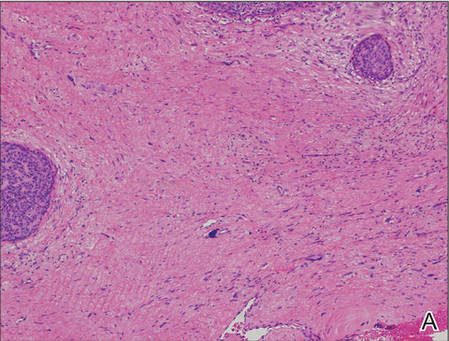
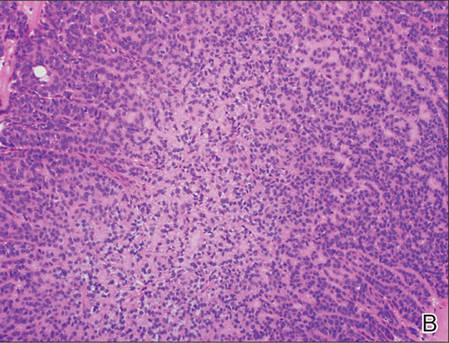
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm of unknown etiology that is characterized by the presence of both malignant epithelial and mesenchymal components.1 Carcinosarcomas have been reported in both the male and female reproductive tracts, urinary tract, gastrointestinal tract, lungs, breasts, larynx, thymus, and thyroid but is uncommon as a primary neoplasm of the skin.2 Epidermal PCC occurs with greater frequency in males than in females and typically presents in the eighth or ninth decades of life.3 These tumors tend to arise in sun-exposed regions, most commonly on the face and scalp.2
Morphologically, PCCs typically are exophytic growths that often feature surface ulceration and may or may not bleed upon palpation.4 Primary cutaneous carcinosarcomas may present as long-standing lesions that have undergone rapid transformation in the weeks preceding presentation.4 It is not uncommon for PCC lesions to carry the clinical diagnosis of squamous cell carcinoma, which suggests notable morphologic overlap between these entities. Histopathologically, PCC shows a basal cell carcinoma and/or a squamous cell carcinoma epithelial component intimately admixed with a sarcomatous component.5 The mesenchymal component of PCC typically resembles a superficial malignant fibrous histiocytoma characterized by pleomorphic nuclei and cytoplasm, necrosis, and an increased number of mitotic figures.2 Immunohistochemistry can be beneficial in the diagnosis of PCC. A combination of p63 and AE1/AE3 stains can be used to confirm cells of epithelial origin. Staining with vimentin, CD10, or caldesmon can help to delineate the mesenchymal component of PCC.
Epidermal PCC most commonly affects elderly individuals with a history of extensive sun exposure. It has been suggested that p53 mutations due to UV damage are key in tumor formation for both epithelial and mesenchymal elements.5 Literature supports a monoclonal origin for the epithelial and mesenchymal components of this tumor; however, there is insufficient evidence.6 Surgical excision is the primary treatment modality for epidermal PCC, but adjuvant or substitutive radiotherapy has been used in some cases.4 The prognosis of PCC is notably better than its visceral counterpart due to early diagnosis and treatment of easily visible lesions. Epidermal PCC has a 70% 5-year disease-free survival rate, while adnexal PCC tends to occur in younger patients and has a 25% 5-year disease-free survival rate.3 Due to the rarity of reported cases and limited follow-up, the long-term prognosis for PCC remains unclear.
We report an unusual case of PCC on the scalp that was successfully treated with radiation therapy alone. This modality should be considered in patients with large tumors who refuse surgery or are not good surgical candidates.
1. El Harroudi T, Ech-Charif S, Amrani M, et al. Primary carcinosarcoma of the skin. J Hand Microsurg. 2010;2:79-81.
2. Patel NK, McKee PH, Smith NP. Primary metaplastic carcinoma (carcinosarcoma) of the skin: a clinicopathologic study of four cases and review of the literature. Am J Dermatopathol. 1997;19:363-372.
3. Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
4. Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
5. Tran TA, Muller S, Chaudahri PJ, et al. Cutaneous carcinosarcoma: adnexal vs. epidermal types define high- and low-risk tumors. results of a meta-analysis. J Cutan Pathol. 2005;32:2-11.
6. Paniz Mondolfi AE, Jour G, Johnson M, et al. Primary cutaneous carcinosarcoma: insights into its clonal origin and mutational pattern expression analysis through next-generation sequencing. Hum Pathol. 2013;44:2853-2860.
The Diagnosis: Primary Cutaneous Carcinosarcoma
A generous shave biopsy and debulking performed on the initial visit revealed an infiltrating tumor consisting of malignant epithelial and stromal components (Figure). The basaloid and squamoid epithelial cells were keratin positive. The stromal cells demonstrated positivity for CD10 but were keratin negative. The epithelial portion of the tumor was composed mostly of basaloid islands of cells with nuclear pleomorphism, scattered mitoses, and focal sebaceous differentiation. The mesenchymal portion of the tumor displayed florid pleomorphism and polymorphism, with many large atypical cells and proliferation. A diagnosis of primary cutaneous carcinosarcoma (PCC) was rendered. Head and neck computed tomography showed tumor penetration of less than 1 cm into scalp soft tissues with no involvement of the underlying bone. There was some evidence of swelling of the supragaleal soft tissues without indication of perineural spread. An 11-mm hyperlucent lower cervical lymph node on the left side that likely represented an incidental finding was noted. Surgical excision with margin evaluation was recommended, but the patient declined. He instead received radiation therapy to the left side of the posterior scalp with a total dose of 30 Gy at 6 Gy per fraction and 1 fraction daily. The patient was found to have a well-healed scar with no evidence of recurrence at 4-week follow-up and again at 5 months after radiation therapy.
|
|
|
|
| A generous shave biopsy and debulking performed on the initial visit revealed an inflitrating tumor consisting on malignant epithelial and stromal components (A-C)(H&E; original magnifications ×10, ×20, and ×40, respectively). |
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm of unknown etiology that is characterized by the presence of both malignant epithelial and mesenchymal components.1 Carcinosarcomas have been reported in both the male and female reproductive tracts, urinary tract, gastrointestinal tract, lungs, breasts, larynx, thymus, and thyroid but is uncommon as a primary neoplasm of the skin.2 Epidermal PCC occurs with greater frequency in males than in females and typically presents in the eighth or ninth decades of life.3 These tumors tend to arise in sun-exposed regions, most commonly on the face and scalp.2
Morphologically, PCCs typically are exophytic growths that often feature surface ulceration and may or may not bleed upon palpation.4 Primary cutaneous carcinosarcomas may present as long-standing lesions that have undergone rapid transformation in the weeks preceding presentation.4 It is not uncommon for PCC lesions to carry the clinical diagnosis of squamous cell carcinoma, which suggests notable morphologic overlap between these entities. Histopathologically, PCC shows a basal cell carcinoma and/or a squamous cell carcinoma epithelial component intimately admixed with a sarcomatous component.5 The mesenchymal component of PCC typically resembles a superficial malignant fibrous histiocytoma characterized by pleomorphic nuclei and cytoplasm, necrosis, and an increased number of mitotic figures.2 Immunohistochemistry can be beneficial in the diagnosis of PCC. A combination of p63 and AE1/AE3 stains can be used to confirm cells of epithelial origin. Staining with vimentin, CD10, or caldesmon can help to delineate the mesenchymal component of PCC.
Epidermal PCC most commonly affects elderly individuals with a history of extensive sun exposure. It has been suggested that p53 mutations due to UV damage are key in tumor formation for both epithelial and mesenchymal elements.5 Literature supports a monoclonal origin for the epithelial and mesenchymal components of this tumor; however, there is insufficient evidence.6 Surgical excision is the primary treatment modality for epidermal PCC, but adjuvant or substitutive radiotherapy has been used in some cases.4 The prognosis of PCC is notably better than its visceral counterpart due to early diagnosis and treatment of easily visible lesions. Epidermal PCC has a 70% 5-year disease-free survival rate, while adnexal PCC tends to occur in younger patients and has a 25% 5-year disease-free survival rate.3 Due to the rarity of reported cases and limited follow-up, the long-term prognosis for PCC remains unclear.
We report an unusual case of PCC on the scalp that was successfully treated with radiation therapy alone. This modality should be considered in patients with large tumors who refuse surgery or are not good surgical candidates.
The Diagnosis: Primary Cutaneous Carcinosarcoma
A generous shave biopsy and debulking performed on the initial visit revealed an infiltrating tumor consisting of malignant epithelial and stromal components (Figure). The basaloid and squamoid epithelial cells were keratin positive. The stromal cells demonstrated positivity for CD10 but were keratin negative. The epithelial portion of the tumor was composed mostly of basaloid islands of cells with nuclear pleomorphism, scattered mitoses, and focal sebaceous differentiation. The mesenchymal portion of the tumor displayed florid pleomorphism and polymorphism, with many large atypical cells and proliferation. A diagnosis of primary cutaneous carcinosarcoma (PCC) was rendered. Head and neck computed tomography showed tumor penetration of less than 1 cm into scalp soft tissues with no involvement of the underlying bone. There was some evidence of swelling of the supragaleal soft tissues without indication of perineural spread. An 11-mm hyperlucent lower cervical lymph node on the left side that likely represented an incidental finding was noted. Surgical excision with margin evaluation was recommended, but the patient declined. He instead received radiation therapy to the left side of the posterior scalp with a total dose of 30 Gy at 6 Gy per fraction and 1 fraction daily. The patient was found to have a well-healed scar with no evidence of recurrence at 4-week follow-up and again at 5 months after radiation therapy.
|
|
|
|
| A generous shave biopsy and debulking performed on the initial visit revealed an inflitrating tumor consisting on malignant epithelial and stromal components (A-C)(H&E; original magnifications ×10, ×20, and ×40, respectively). |
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm of unknown etiology that is characterized by the presence of both malignant epithelial and mesenchymal components.1 Carcinosarcomas have been reported in both the male and female reproductive tracts, urinary tract, gastrointestinal tract, lungs, breasts, larynx, thymus, and thyroid but is uncommon as a primary neoplasm of the skin.2 Epidermal PCC occurs with greater frequency in males than in females and typically presents in the eighth or ninth decades of life.3 These tumors tend to arise in sun-exposed regions, most commonly on the face and scalp.2
Morphologically, PCCs typically are exophytic growths that often feature surface ulceration and may or may not bleed upon palpation.4 Primary cutaneous carcinosarcomas may present as long-standing lesions that have undergone rapid transformation in the weeks preceding presentation.4 It is not uncommon for PCC lesions to carry the clinical diagnosis of squamous cell carcinoma, which suggests notable morphologic overlap between these entities. Histopathologically, PCC shows a basal cell carcinoma and/or a squamous cell carcinoma epithelial component intimately admixed with a sarcomatous component.5 The mesenchymal component of PCC typically resembles a superficial malignant fibrous histiocytoma characterized by pleomorphic nuclei and cytoplasm, necrosis, and an increased number of mitotic figures.2 Immunohistochemistry can be beneficial in the diagnosis of PCC. A combination of p63 and AE1/AE3 stains can be used to confirm cells of epithelial origin. Staining with vimentin, CD10, or caldesmon can help to delineate the mesenchymal component of PCC.
Epidermal PCC most commonly affects elderly individuals with a history of extensive sun exposure. It has been suggested that p53 mutations due to UV damage are key in tumor formation for both epithelial and mesenchymal elements.5 Literature supports a monoclonal origin for the epithelial and mesenchymal components of this tumor; however, there is insufficient evidence.6 Surgical excision is the primary treatment modality for epidermal PCC, but adjuvant or substitutive radiotherapy has been used in some cases.4 The prognosis of PCC is notably better than its visceral counterpart due to early diagnosis and treatment of easily visible lesions. Epidermal PCC has a 70% 5-year disease-free survival rate, while adnexal PCC tends to occur in younger patients and has a 25% 5-year disease-free survival rate.3 Due to the rarity of reported cases and limited follow-up, the long-term prognosis for PCC remains unclear.
We report an unusual case of PCC on the scalp that was successfully treated with radiation therapy alone. This modality should be considered in patients with large tumors who refuse surgery or are not good surgical candidates.
1. El Harroudi T, Ech-Charif S, Amrani M, et al. Primary carcinosarcoma of the skin. J Hand Microsurg. 2010;2:79-81.
2. Patel NK, McKee PH, Smith NP. Primary metaplastic carcinoma (carcinosarcoma) of the skin: a clinicopathologic study of four cases and review of the literature. Am J Dermatopathol. 1997;19:363-372.
3. Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
4. Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
5. Tran TA, Muller S, Chaudahri PJ, et al. Cutaneous carcinosarcoma: adnexal vs. epidermal types define high- and low-risk tumors. results of a meta-analysis. J Cutan Pathol. 2005;32:2-11.
6. Paniz Mondolfi AE, Jour G, Johnson M, et al. Primary cutaneous carcinosarcoma: insights into its clonal origin and mutational pattern expression analysis through next-generation sequencing. Hum Pathol. 2013;44:2853-2860.
1. El Harroudi T, Ech-Charif S, Amrani M, et al. Primary carcinosarcoma of the skin. J Hand Microsurg. 2010;2:79-81.
2. Patel NK, McKee PH, Smith NP. Primary metaplastic carcinoma (carcinosarcoma) of the skin: a clinicopathologic study of four cases and review of the literature. Am J Dermatopathol. 1997;19:363-372.
3. Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
4. Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
5. Tran TA, Muller S, Chaudahri PJ, et al. Cutaneous carcinosarcoma: adnexal vs. epidermal types define high- and low-risk tumors. results of a meta-analysis. J Cutan Pathol. 2005;32:2-11.
6. Paniz Mondolfi AE, Jour G, Johnson M, et al. Primary cutaneous carcinosarcoma: insights into its clonal origin and mutational pattern expression analysis through next-generation sequencing. Hum Pathol. 2013;44:2853-2860.
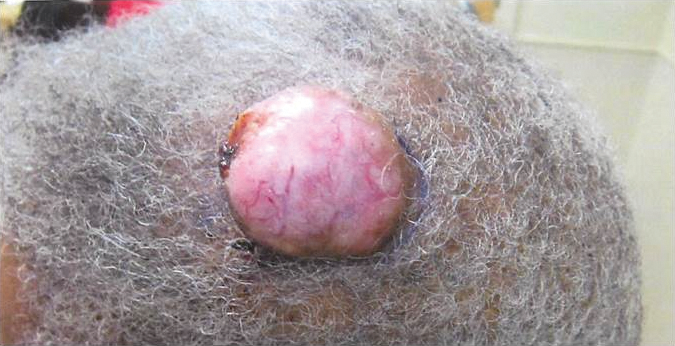
An 81-year-old man presented with a 3.5×3.0-cm pink exophytic tumor with an eroded surface and prominent vascularity on the left side of the parietal scalp. The patient reported that the tumor had been present for more than 30 years but recently had grown larger in size. He denied pain or pruritus in association with the lesion and did not report any systemic symptoms. He had received no prior treatments for the tumor.

Boards Review Resources
Books
There are a number of classic textbooks that serve as primary resources for dermatology training1-4; however, there also are other options if memorizing these books seems a little daunting. The “first aid” books of dermatology are the Derm In-Review binder and Jain’s5 Dermatology: Illustrated Study Guide and Comprehensive Board Review. Mariwalla and Leffell’s6Primer in Dermatologic Surgery: A Study Companion is helpful for surgical review and is available at a discounted price for members of the American Society for Dermatologic Surgery (https://www.asds.net/store/product.aspx?id=3914&terms=primer%20in%20Dermatologic%20surgery). The American Academy of Dermatology (AAD) provides a list of additional textbooks that dermatology residents may find useful for board review.7
Guided Study
The AAD offers board review courses for dermatology residents (cost varies).7 The Florida Dermatology and Dermatopathology Board Review Course (http://dermatology.med.ufl.edu/education/florida-dermatology-and-dermatopathology-board-review-course/) is an annual review course held in Tampa (early registration fee, $800 [does not include travel costs]). The Oakstone Institute also offers its Dermatology Board Review Course, which is a self-study program that can be completed online for approximately $1195 (http://www.oakstone.com/dermatology-board-review-course). Be sure to take advantage of free didactics lectures, society meetings with board review courses, and study groups, as these resources can be just as helpful and more budget friendly.
Digital Resources
The Derm In-Review question bank (http://dermatologyinreview.com/Merz) is probably one of the most popular board review resources and is free to US dermatology residents; however, be cautious when using this resource, as a fair number of the answers to questions may actually be outdated or based on older studies. A group study session can help tease out why certain answers are erroneous and provide a forum for discussing what would be a more correct answer. Take advantage of the opportunity to provide feedback on this website, as your comments will improve this resource for future dermatology residents.
Beyond traditional dermatopathology textbooks, there also are some excellent mobile applications (apps) available. The Clearpath app is a user-friendly dermatopathology study tool that is free for download in the iTunes store (https://itunes.apple.com/us/app/clearpath/id540260769?mt=8). However, the app is only compatible with iPads. The Clearpath website also offers virtual study slide sets that are easier to access (http://dermpathlab.com/slide-study-set-program). Your institution’s glass slide sets also are useful for building pattern recognition skills and practicing for the actual board examination. The DermOID website (http://www.derm-oid.com), which is powered by the David Geffen School of Medicine at the University of California, Los Angeles, is another online dermatopathology study database with free registration for access to the site. Another fun way to test your dermatopathology skills is in the exhibition hall at the AAD annual meeting where some vendors may offer daily dermatopathology quizzes and prizes for the residents with the most correct answers. Also, it is worth reviewing the Cutis® Fast Facts for Board Review (http://www.cutis.com/articles/fast-facts-for-board-review/), as this section offers many outstanding fact sheets that are an easy read and an efficient way to gain board knowledge. Some recent topics include fillers, paraneoplastic skin conditions, and medications in dermatology.
Many residents enjoy using the Anki flashcard app (http://ankisrs.net) for reviewing kodachromes. The AAD website also includes a Boards’ Fodder archive that is worth reviewing (https://www.aad.org/members/residents-fellows/boards-study-tools/boards-fodder/boards-fodder). New board review resources are constantly being posted on the AAD website, so definitely check this out. You may be able to access this resource through your residency program; it is also available for purchase ($425 for AAD members; $850 for nonmembers).
Journals
All the major dermatology journals are helpful in preparing for the board examination. Your resident journal club will likely review many of the most clinically relevant dermatology articles published over the course of your residency. Some other helpful journal resources that are recommended for board review include the Journal of the American Medical Association’s Clinical Challenge, which has many dermatologic cases (http://jama.jamanetwork.com/public/QuizzesAndPolls.aspx), and the New England Journal of Medicine’s Journal Watch (http://www.jwatch.org) and Image Challenge (http://www.nejm.org/image-challenge).
Practice Examinations
The American Board of Dermatology’s In-Training Examination is the most well-known practice examination among dermatology residents.8 A link to an additional practice examination usually is provided a few weeks prior to the examination. The Derm In-Review website also offers diagnostic practice examinations with some ability to custom select questions for your studying needs.
Conclusion
There are many board review resources out there. Find the ones that work for you, and be encouraged that your studying and hard work will pay off!
1. Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012.
2. James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 11th ed. Philadelphia, PA: Elsevier Saunders; 2011.
3. Spitz JL. Genodermatoses: A Clinical Guide to Genetic Skin Diseases. 2nd ed. Baltimore, MD: Lippincott Williams & Wilkins; 2004.
4. Weedon D. Weedon’s Skin Pathology. 3rd ed. London, England: Churchill Livingstone; 2010.
5. Jain S. Dermatology: Illustrated Study Guide and Comprehensive Board Review. New York, NY; Springer: 2012.
6. Mariwalla K, Leffell DJ. Primer in Dermatologic Surgery: A Study Companion. 2nd ed. Rolling Meadows, IL: American Society for Dermatologic Surgery; 2011.
7. Additional boards resources. American Academy of Dermatology website. https://www.aad.org/members/residents-fellows/boards-study-tools/more-boards-resources. Accessed March 31, 2016.
8. In-training examination (ITE). American Board of Dermatology website. https://www.abderm.org/residents-and-fellows/in-training-and-primary-certification-examinations/in-training-examination-ite.aspx. Accessed March 22, 2016.
Books
There are a number of classic textbooks that serve as primary resources for dermatology training1-4; however, there also are other options if memorizing these books seems a little daunting. The “first aid” books of dermatology are the Derm In-Review binder and Jain’s5 Dermatology: Illustrated Study Guide and Comprehensive Board Review. Mariwalla and Leffell’s6Primer in Dermatologic Surgery: A Study Companion is helpful for surgical review and is available at a discounted price for members of the American Society for Dermatologic Surgery (https://www.asds.net/store/product.aspx?id=3914&terms=primer%20in%20Dermatologic%20surgery). The American Academy of Dermatology (AAD) provides a list of additional textbooks that dermatology residents may find useful for board review.7
Guided Study
The AAD offers board review courses for dermatology residents (cost varies).7 The Florida Dermatology and Dermatopathology Board Review Course (http://dermatology.med.ufl.edu/education/florida-dermatology-and-dermatopathology-board-review-course/) is an annual review course held in Tampa (early registration fee, $800 [does not include travel costs]). The Oakstone Institute also offers its Dermatology Board Review Course, which is a self-study program that can be completed online for approximately $1195 (http://www.oakstone.com/dermatology-board-review-course). Be sure to take advantage of free didactics lectures, society meetings with board review courses, and study groups, as these resources can be just as helpful and more budget friendly.
Digital Resources
The Derm In-Review question bank (http://dermatologyinreview.com/Merz) is probably one of the most popular board review resources and is free to US dermatology residents; however, be cautious when using this resource, as a fair number of the answers to questions may actually be outdated or based on older studies. A group study session can help tease out why certain answers are erroneous and provide a forum for discussing what would be a more correct answer. Take advantage of the opportunity to provide feedback on this website, as your comments will improve this resource for future dermatology residents.
Beyond traditional dermatopathology textbooks, there also are some excellent mobile applications (apps) available. The Clearpath app is a user-friendly dermatopathology study tool that is free for download in the iTunes store (https://itunes.apple.com/us/app/clearpath/id540260769?mt=8). However, the app is only compatible with iPads. The Clearpath website also offers virtual study slide sets that are easier to access (http://dermpathlab.com/slide-study-set-program). Your institution’s glass slide sets also are useful for building pattern recognition skills and practicing for the actual board examination. The DermOID website (http://www.derm-oid.com), which is powered by the David Geffen School of Medicine at the University of California, Los Angeles, is another online dermatopathology study database with free registration for access to the site. Another fun way to test your dermatopathology skills is in the exhibition hall at the AAD annual meeting where some vendors may offer daily dermatopathology quizzes and prizes for the residents with the most correct answers. Also, it is worth reviewing the Cutis® Fast Facts for Board Review (http://www.cutis.com/articles/fast-facts-for-board-review/), as this section offers many outstanding fact sheets that are an easy read and an efficient way to gain board knowledge. Some recent topics include fillers, paraneoplastic skin conditions, and medications in dermatology.
Many residents enjoy using the Anki flashcard app (http://ankisrs.net) for reviewing kodachromes. The AAD website also includes a Boards’ Fodder archive that is worth reviewing (https://www.aad.org/members/residents-fellows/boards-study-tools/boards-fodder/boards-fodder). New board review resources are constantly being posted on the AAD website, so definitely check this out. You may be able to access this resource through your residency program; it is also available for purchase ($425 for AAD members; $850 for nonmembers).
Journals
All the major dermatology journals are helpful in preparing for the board examination. Your resident journal club will likely review many of the most clinically relevant dermatology articles published over the course of your residency. Some other helpful journal resources that are recommended for board review include the Journal of the American Medical Association’s Clinical Challenge, which has many dermatologic cases (http://jama.jamanetwork.com/public/QuizzesAndPolls.aspx), and the New England Journal of Medicine’s Journal Watch (http://www.jwatch.org) and Image Challenge (http://www.nejm.org/image-challenge).
Practice Examinations
The American Board of Dermatology’s In-Training Examination is the most well-known practice examination among dermatology residents.8 A link to an additional practice examination usually is provided a few weeks prior to the examination. The Derm In-Review website also offers diagnostic practice examinations with some ability to custom select questions for your studying needs.
Conclusion
There are many board review resources out there. Find the ones that work for you, and be encouraged that your studying and hard work will pay off!
Books
There are a number of classic textbooks that serve as primary resources for dermatology training1-4; however, there also are other options if memorizing these books seems a little daunting. The “first aid” books of dermatology are the Derm In-Review binder and Jain’s5 Dermatology: Illustrated Study Guide and Comprehensive Board Review. Mariwalla and Leffell’s6Primer in Dermatologic Surgery: A Study Companion is helpful for surgical review and is available at a discounted price for members of the American Society for Dermatologic Surgery (https://www.asds.net/store/product.aspx?id=3914&terms=primer%20in%20Dermatologic%20surgery). The American Academy of Dermatology (AAD) provides a list of additional textbooks that dermatology residents may find useful for board review.7
Guided Study
The AAD offers board review courses for dermatology residents (cost varies).7 The Florida Dermatology and Dermatopathology Board Review Course (http://dermatology.med.ufl.edu/education/florida-dermatology-and-dermatopathology-board-review-course/) is an annual review course held in Tampa (early registration fee, $800 [does not include travel costs]). The Oakstone Institute also offers its Dermatology Board Review Course, which is a self-study program that can be completed online for approximately $1195 (http://www.oakstone.com/dermatology-board-review-course). Be sure to take advantage of free didactics lectures, society meetings with board review courses, and study groups, as these resources can be just as helpful and more budget friendly.
Digital Resources
The Derm In-Review question bank (http://dermatologyinreview.com/Merz) is probably one of the most popular board review resources and is free to US dermatology residents; however, be cautious when using this resource, as a fair number of the answers to questions may actually be outdated or based on older studies. A group study session can help tease out why certain answers are erroneous and provide a forum for discussing what would be a more correct answer. Take advantage of the opportunity to provide feedback on this website, as your comments will improve this resource for future dermatology residents.
Beyond traditional dermatopathology textbooks, there also are some excellent mobile applications (apps) available. The Clearpath app is a user-friendly dermatopathology study tool that is free for download in the iTunes store (https://itunes.apple.com/us/app/clearpath/id540260769?mt=8). However, the app is only compatible with iPads. The Clearpath website also offers virtual study slide sets that are easier to access (http://dermpathlab.com/slide-study-set-program). Your institution’s glass slide sets also are useful for building pattern recognition skills and practicing for the actual board examination. The DermOID website (http://www.derm-oid.com), which is powered by the David Geffen School of Medicine at the University of California, Los Angeles, is another online dermatopathology study database with free registration for access to the site. Another fun way to test your dermatopathology skills is in the exhibition hall at the AAD annual meeting where some vendors may offer daily dermatopathology quizzes and prizes for the residents with the most correct answers. Also, it is worth reviewing the Cutis® Fast Facts for Board Review (http://www.cutis.com/articles/fast-facts-for-board-review/), as this section offers many outstanding fact sheets that are an easy read and an efficient way to gain board knowledge. Some recent topics include fillers, paraneoplastic skin conditions, and medications in dermatology.
Many residents enjoy using the Anki flashcard app (http://ankisrs.net) for reviewing kodachromes. The AAD website also includes a Boards’ Fodder archive that is worth reviewing (https://www.aad.org/members/residents-fellows/boards-study-tools/boards-fodder/boards-fodder). New board review resources are constantly being posted on the AAD website, so definitely check this out. You may be able to access this resource through your residency program; it is also available for purchase ($425 for AAD members; $850 for nonmembers).
Journals
All the major dermatology journals are helpful in preparing for the board examination. Your resident journal club will likely review many of the most clinically relevant dermatology articles published over the course of your residency. Some other helpful journal resources that are recommended for board review include the Journal of the American Medical Association’s Clinical Challenge, which has many dermatologic cases (http://jama.jamanetwork.com/public/QuizzesAndPolls.aspx), and the New England Journal of Medicine’s Journal Watch (http://www.jwatch.org) and Image Challenge (http://www.nejm.org/image-challenge).
Practice Examinations
The American Board of Dermatology’s In-Training Examination is the most well-known practice examination among dermatology residents.8 A link to an additional practice examination usually is provided a few weeks prior to the examination. The Derm In-Review website also offers diagnostic practice examinations with some ability to custom select questions for your studying needs.
Conclusion
There are many board review resources out there. Find the ones that work for you, and be encouraged that your studying and hard work will pay off!
1. Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012.
2. James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 11th ed. Philadelphia, PA: Elsevier Saunders; 2011.
3. Spitz JL. Genodermatoses: A Clinical Guide to Genetic Skin Diseases. 2nd ed. Baltimore, MD: Lippincott Williams & Wilkins; 2004.
4. Weedon D. Weedon’s Skin Pathology. 3rd ed. London, England: Churchill Livingstone; 2010.
5. Jain S. Dermatology: Illustrated Study Guide and Comprehensive Board Review. New York, NY; Springer: 2012.
6. Mariwalla K, Leffell DJ. Primer in Dermatologic Surgery: A Study Companion. 2nd ed. Rolling Meadows, IL: American Society for Dermatologic Surgery; 2011.
7. Additional boards resources. American Academy of Dermatology website. https://www.aad.org/members/residents-fellows/boards-study-tools/more-boards-resources. Accessed March 31, 2016.
8. In-training examination (ITE). American Board of Dermatology website. https://www.abderm.org/residents-and-fellows/in-training-and-primary-certification-examinations/in-training-examination-ite.aspx. Accessed March 22, 2016.
1. Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012.
2. James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 11th ed. Philadelphia, PA: Elsevier Saunders; 2011.
3. Spitz JL. Genodermatoses: A Clinical Guide to Genetic Skin Diseases. 2nd ed. Baltimore, MD: Lippincott Williams & Wilkins; 2004.
4. Weedon D. Weedon’s Skin Pathology. 3rd ed. London, England: Churchill Livingstone; 2010.
5. Jain S. Dermatology: Illustrated Study Guide and Comprehensive Board Review. New York, NY; Springer: 2012.
6. Mariwalla K, Leffell DJ. Primer in Dermatologic Surgery: A Study Companion. 2nd ed. Rolling Meadows, IL: American Society for Dermatologic Surgery; 2011.
7. Additional boards resources. American Academy of Dermatology website. https://www.aad.org/members/residents-fellows/boards-study-tools/more-boards-resources. Accessed March 31, 2016.
8. In-training examination (ITE). American Board of Dermatology website. https://www.abderm.org/residents-and-fellows/in-training-and-primary-certification-examinations/in-training-examination-ite.aspx. Accessed March 22, 2016.

Merkel Cell Carcinoma: A Review
Merkel cells originally were described by German histopathologist Friedrich Sigmund Merkel in 1875. These unique tactile cells were described as epidermal, nondendritic, and nonkeratinizing. Merkel cells are thought to arise from the neural crest and are believed to be primary neural cells found within the basal layer of the epidermis.1,2 They likely function primarily as slowly adapting type I mechanoreceptors. Origin from the neural crest is controversial, as other investigators have suggested derivation from epidermal keratinocytes.1,2 Tumor cells have been linked to the amine precursor uptake and decarboxylation system.3 In 1972, Toker4 described several cases of trabecular or sweat gland carcinomas of the skin. Upon further investigation, the cells that comprised these tumors were found to have dense core granules on electron microscopy, typical of Merkel cells.1,2 Other terms such as neuroendocrine carcinoma of the skin, small cell carcinoma of the skin, and anaplastic carcinoma of the skin have been used to describe Merkel cell carcinoma (MCC),1 which was suggested by De Wolf-Peeters et al5 in 1980.
Despite being a rare malignancy, MCC follows an aggressive clinical course. Upon presentation, approximately 66% of patients have local disease, 27% have nodal involvement, and 7% have distant metastasis.1 Future treatments will likely center around the novel Merkel cell polyomavirus (MCPyV) and modification of immune responses toward tumor cells. Standardization continues to be lacking in both staging and treatment of this aggressive tumor.
Epidemiology of MCC

| ||
Figure 1. A 2.3×1.5×1.2-cm, hemorrhagic, crusted, | ||
| ||
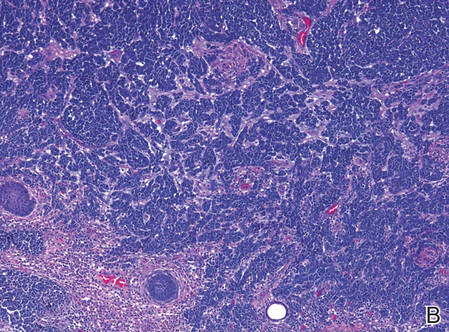
| ||
| Figure 2. Merkel cells are small- to medium-sized cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen (A)(H&E, original magnification ×40). Merkel cell carcinoma with trabecular pattern (B) (H&E, original magnification ×10). | ||
Between 1986 and 2006, the incidence of MCC grew substantially.1,2 Figures have been reported at 0.15 cases per 100,000 individuals to 0.6 cases per 100,000 individuals worldwide. In the United States, the age-adjusted incidence of MCC is estimated at 0.24 per 100,000 person-years, which is higher than the estimated 0.13 per 100,000 person-years found in Europe.3 The highest incidence worldwide has been noted in Western Australia, likely due to high levels of UV exposure.1 The incidence of MCC in psoriasis patients who are treated with oral methoxsalen (psoralen) and UVA photochemotherapy is 100 times greater than in the general population, further supporting the role of UV light in the development of MCC.1 White individuals have the highest incidence of MCC worldwide, with men being affected more frequently than women.1,3 The majority of patients with MCC are diagnosed at 70 years or older.1 Approximately 5% of reported MCC patients are diagnosed before 50 years of age.2 Immunosuppression and immunodeficiency likely play a role in the pathogenesis of MCC, and the incidence is increased in solid organ transplant recipients, most commonly renal transplant recipients,1 as well as individuals with chronic lymphocytic leukemia, human immunodeficiency virus infection, and AIDS.1,3 Patients with autoimmune diseases such as rheumatoid arthritis also are at increased risk for MCC.3 Individuals who are diagnosed with MCC are at an increased risk for development of other malignancies including nonmelanoma skin cancers, chronic lymphocytic leukemia, Hodgkin lymphoma, and non-Hodgkin lymphoma.3
Clinical Presentation of MCC
The clinical presentation of MCC can be variable. Most tumors present as firm, red to purple, nontender papules or nodules (Figure 1).1 Tumor size may range from 2 to 200 mm but is most commonly less than 20 mm.2 Growth can be rapid, and tumors are most commonly located on sun-exposed skin. The head and neck areas account for 48% of all MCC cases,1 with the eyelids being frequently involved.2 Merkel cell carcinoma also has been reported on the arms, legs, trunk, back, and buttocks.1 Non–sun-exposed areas are less commonly affected. Mucosal sites (eg, larynx, nasal cavity, pharynx, mouth) account for 5% of primary MCCs.1 Merkel cell carcinoma also has been reported to affect the vulva and penis. Subcutaneous primary MCC has presented without overlying epidermal changes.1 In a case series by Heath et al,6 14% (27/195) of MCC patients presented with nodal disease without any identifiable primary tumor, with the inguinal nodal chain being the most common for this presentation. It currently is not known whether these nodal tumors are primary tumors or metastatic disease with a regression of the primary tumor.1
|
Histopathology of MCC
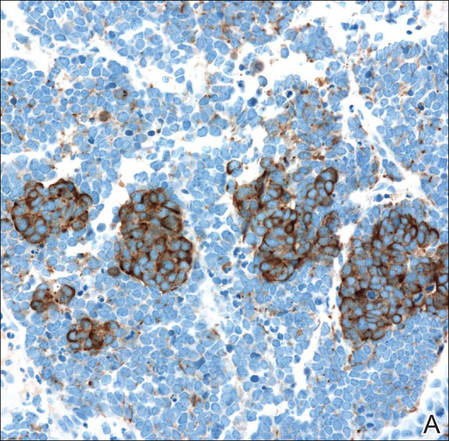
| ||
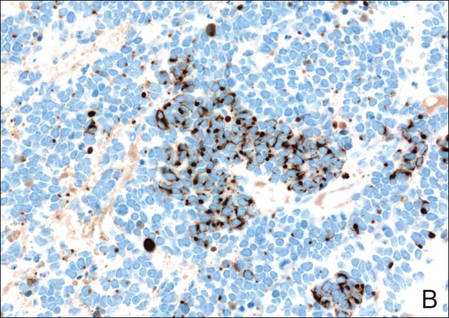
| ||
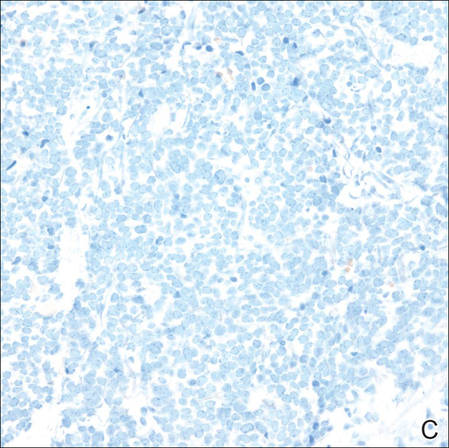
| ||
Figure 3. Positive chromogranin staining (A)(original | ||
Merkel cells are small- to medium-sized basophilic cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen on histopathology (Figure 2A).1 Some tumor cells have more vesicular chromatin, multiple small nucleoli, irregular contours, and more abundant cytoplasm. In some reports, irregular contours and abundant cytoplasm were associated with no detectible MCPyV infection.1,3 Merkel cell carcinomas have a primarily nodular architecture, and classification is based on growth pattern and cell size. Three histopathologic growth patterns have been described: nodular, infiltrative, and trabecular. The trabecular pattern is composed of interconnecting strands ofcells (Figure 2B). Tumors with solely intraepidermal involvement (MCC in situ) have been described but are exceedingly rare.1 Cell types are classified according to size, with the intermediate cell type being the most common. The small cell variant may be mistaken for a lymphocytic infiltrate due to the similar size and appearance of both types of cells.1,3
Merkel cell carcinomas can have histopathological overlap with lymphomas, small cell lung cancers, carcinoid tumors, primitive neuroectodermal tumors, neuroblastomas, small cell osteosarcomas, rhabdomyosarcomas, or Ewing sarcomas.1,3 Specifically, differentiation from small cell carcinoma of the lung is of utmost importance. Merkel cell carcinoma stains positively for cytokeratins 8, 18, 19, and 20. The neuroendocrine markers chromogranin (Figure 3A), synaptophysin, and neuron-specific enolase also may show positive staining. Cytokeratin 20, low-molecular-weight cytokeratins (CAM 5.2), and neurofilament immunostains have a high sensitivity for MCC and are the most frequently used.1 Cytokeratin 20 stains in the characteristic paranuclear dot–like pattern, which is a hallmark of MCC (Figure 3B). Cytokeratin 20 positivity in conjunction with negative staining for thyroid transcription factor 1 (Figure 3C) and cytokeratin 7 can aid in differentiation from small cell carcinoma of the lung.1,3
Pathogenesis of MCC
In 2008, Feng et al7 discovered a novel polyomavirus associated with the development of MCC. This novel polyomavirus, MCPyV, is found in approximately 80% of all cases of MCC. Seventeen members of the polyomavirus family have been identified, 9 of which have been found to infect humans, including BK virus, JC virus, WU, MCPyV, human polyomavirus 6, human polyomavirus 7, trichodysplasia spinulosa–associated polyomavirus, human polyomavirus 9, and Simian virus 40.1 Merkel cell polyomavirus infection is found in approximately 60% of the general population and exposure likely occurs early in life. The virus likely is transmitted through skin shedding and nasal secretions, though it also has been found in urine specimens.3 Currently, there is no evidence to suggest vertical viral transmission from mother to fetus.
Merkel cell polyomavirus is composed of early and late gene regions. The early gene region contains both large T antigen (LT) and small T antigen reading frames, which are necessary for viral replication.8 The late region is responsible for encoding viral proteins necessary for viral capsid assembly. Mutations found in viral protein 1 prevent formation of viral particles.9 Large T antigen is substantially overexpressed in MCC and is responsible for tumor suppression through retinoblastoma tumor suppressor protein. It also serves as a binding domain for both heat shock proteins and helicases.8,10 These domains allow the polyomaviruses to use host-cell machinery for viral genome replication while targeting tumor suppressor proteins.8 Upon viral integration into host DNA, viral replication ceases while oncogenic function persists.
The exact mechanism by which the MCPyV contributes to the development of MCC still has yet to be identified. Hypotheses suggest a combination of viral infection with external mutagens (eg, UV radiation). Experimental observations suggest viral contribution is likely due to the large percentage of MCCs that are positive for MCPyV, the identification of LT antigen expression and the role it plays in preserving cell cycle progression, and the role persistent LT antigen expression plays in continued growth of MCC cell lines in vitro.8 Two important cell line preservation mechanisms ensure continued tumor growth, including prevention of apoptosis triggered by DNA damage response mechanisms following UV damage and interaction with the retinoblastoma tumor suppressor protein allowing continued growth.8,11 Other important factors in tumor growth and survival may be the inhibition of apoptosis through the BCL2 (B-cell chronic lymphocytic leukemia/lymphoma 2) proto-oncogene and survivin (baculoviral inhibitor of apoptosis repeat-containing 5 [BIRC5]).12 Survivin has been found to play an important role in MCPyV-positive MCCs.12,13 It has been suggested that lymphangiogenesis in MCC likely is driven by vascular endothelial growth factor-C+CD68+CD163+ M2 macrophages.14 Another survival mechanism specific to polyomaviruses is their ability to interfere with the p53 tumor suppressor pathway.8 Loss of p53 expression by tumor cell nuclei has been associated with poor prognosis.15
Immune Response
Immune response as a role in tumor progression can be primarily centered on the concept of persistent antigen expression as a means of immune downregulation. Dunn et al16 suggested that cancer cells must interact through 3 consecutive phases with the host immune system (immunoediting hypothesis). In the elimination phase, the host immune system is able to recognize and destroy newly transformed cells through both the innate and adaptive immune systems. The second equilibrium phase allows the tumor to remain dormant and growth remains stagnant. Lastly, the tumor is allowed to evade the immune system through the escape phase.8
Host immune responses play an important role in both the progression and prognosis of MCC. High anti-MCPyV capsid antibody titers have been associated with better progression-free survival in some patients.8 Patients with high antibody titers (>10,000) likely have better progression-free survival than those with low antibody titers (<10,000).17 Antibody titers to the LT antigen may serve as a biomarker of MCC disease burden in the future. Rising LT antigen titers have been shown to correlate with disease progression and falling titers correlate with successful treatment.8 Tumoral infiltration of CD8+ T lymphocytes has been shown to be a predictor of survival compared to no intratumoral infiltration.6 Sihto et al18 suggested that this better prognosis from high intratumoral infiltration is not specific to MCPyV-positive MCC; however, it does highlight an important aspect of tumor evasion through the downregulation of cell surface expression of class I major histocompatibility complex antigens, which allows presentation of tumor intracellular peptides to CD8+ T lymphocytes.8 Upregulation of this specific immune response may play a role in the future treatment of MCC.
Staging and Prognosis
Due to the extremely aggressive nature of MCC, patients with local disease and tumors 2 cm or smaller in diameter have a 66% survival at 5 years.1,3 The 5-year survival rate for patients with local metastasis to regional lymph nodes ranges from 26% to 42%. Patients with distant metastasis have an 18% survival rate at 5 years.1,3 Data suggest that sentinel lymph node biopsy should be performed on all patients with MCC regardless of tumor size.1 There are no consensus guidelines to date regarding imaging for the staging of MCC patients. It is suggested that (18F)fluorodeoxyglucose positron emission tomography alone or in combination with computed tomography (CT) may be of value as a single whole-body diagnostic tool for accurate staging.10 It also has been suggested that (18F)fluorodeoxyglucose positron emission tomography and CT may offer more accurate staging than other screening modalities such as CT alone or magnetic resonance imaging.14,19
Treatment of MCC
Surgery remains the mainstay of treatment of MCC. Current National Comprehensive Cancer Network guidelines20 recommend 1- to 2-cm margins for wide local excision or treatment with Mohs micrographic surgery. Sentinel lymph node biopsy should be performed intraoperatively in patients undergoing wide local excision and preoperatively for patients undergoing Mohs micrographic surgery due to potential alterations in lymphatic drainage that may affect lymphoscintigraphy.1
Radiation may be used as primary or adjuvant therapy in patients with MCC. Radiation as primary therapy generally is reserved for patients who are not surgical candidates. It has been suggested that there was no difference in outcome in a small group of patients treated with radiation alone compared to patients who underwent surgery and radiation to the tumor bed.1 Current guidelines suggest a small group of patients may not require adjuvant therapy following adequate resection of some small tumors, and clinical observation may be appropriate.1,3 Chemotherapy may play a palliative role in patients with metastatic MCC. Merkel cell carcinoma has been shown to be chemosensitive but with a high recurrence rate.1 Because the immune system plays an important role in disease prognosis, having an intact immune system likely is paramount in the prevention of further disease progression.
Future Treatments of MCC
Future treatment of MCC may be focused on the viral etiology of most tumors and upregulation of the immune response, which may lead to the possibility of specifically interfering with virus-specific oncoproteins and stimulation of immune responses to virally infected tumor cells.8 The MCPyV large T antigen has been found to be overexpressed in some tumors and may serve as a specific target of therapy.10,21 Survivin, a key cell cycle protein encoded by LT antigen, may be an interesting target given its implication in other cancers.13 Other potential nonviral molecular target antigens include the oncoprotein H1P1 that interacts with c-KIT.8 Specific immunostimulatory cytokines that may be used to upregulate the immune response to tumoral cells may include IL-2, IL-12, IL-15, or IL-21. Therapeutic agents that may be studied in the future to target the immune exhaustion phenomenon associated with tumorigenesis include ipilimumab (cytotoxic T lymphocyte antigen 4 receptor-blocking agent) as well as programmed cell death 1 and programmed cell death 1 ligand 1 (PD-1/PD-L1).8 Neuroendocrine tumors including MCC tend to be highly vascular and express vascular endothelial growth factors and platelet-derived growth factors, which may be other potential therapeutic targets. It has been reported that approximately 95% of MCC patients have CD56+ tumors, and current clinical trials suggest a promising therapeutic response with the immunogen anti-CD56 monoclonal antibody.3
Conclusion
Merkel cell carcinoma is a rare aggressive neuroendocrine tumor that has been associated with a novel polyomavirus. Merkel cell carcinoma tends to affect elderly and immunocompromised patients as well as white individuals. Tumors are most often found in areas of high UV exposure and clinically on sun-exposed skin. Merkel cell polyomavirus is associated with approximately 80% of tumors, and tumorigenesis likely is caused by a number of sequential steps from viral integration into host DNA, mutagenic events, and specific immune responses. Currently there are no consensus guidelines for using imaging for staging of MCCs, but sentinel lymph node biopsy is recommended for all cases due to the aggressive nature of even smaller tumors. Surgery remains the mainstay of treatment, and radiation therapy may be used as a primary or adjuvant treatment. Chemotherapy usually is reserved for patients with metastatic disease purely for palliation. Future treatments of MCC likely will center on the viral etiology of MCC and upregulation of immune responses to virally infected tumor cells.
1. Han S, North J, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
2. Goessling W, McKee P, Mayer R. Merkel cell carcinoma. J Clin Oncol. 2002;20:588-598.
3. Donepudi S, DeConti R, Samlowski W. Recent advances in the understanding of the genetics, etiology, and treatment of merkel cell carcinoma. Semin Oncol. 2012;39:163-172.
4. Toker C. Trabecular carcinoma of the skin. Arch Dermatol. 1972;105:107-110.
5. De Wolff-Peeters C, Marien K, Mebis J, et al. A cutaneous APUDoma or Merkel cell tumor? a morphologically recognizable tumor with a biological and histological malignant aspect in contrast with its clinical behavior. Cancer. 1980;46:810-816.
6. Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375-381.
7. Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
8. Bhatia S, Afanasiev O, Nghiem P. Immunobiology of Merkel cell carcinoma: implications for immunotherapy of a polyomavirus-associated cancer. Curr Oncol Rep. 2011;13:488-497.
9. Amber K, McLeod M, Nouri K. The Merkel cell polyomavirus and its involvement in Merkel cell carcinoma. Dermatol Surg. 2013;39:232-238.
10. Erovic B, Al Habeeb A, Harris L, et al. Significant overexpression of the Merkel cell polyomavirus (MCPyV) large T antigen in Merkel cell carcinoma. Head Neck. 2013;35:184-189.
11. Demetriou S, Ona-Vu K, Sullivan E, et al. Defective DNA repair and cell cycle arrest in cells expressing Merkel cell polyomavirus T antigen. Int J Cancer. 2012;131:1818-1827.
12. Sahi H, Koljonen V, Kavola H, et al. Bcl-2 expression indicates better prognosis of Merkel cell carcinoma regardless of the presence of Merkel cell polyomavirus. Virchows Arch. 2012;461:553-559.
13. Arora R, Shuda M, Guastafierro A, et al. Survivin is a therapeutic target in Merkel cell carcinoma. Sci Transl Med. 2012;4:1-11.
14. Hawryluk E, O’Regan K, Sheehy N, et al. Positron emission tomography/computed tomography imaging in Merkel cell carcinoma: a study of 270 scans in 97 patients at the Dana-Farber/Brigham and Women’s Cancer Center. J Am Acad Dermatol. 2013;68:592-599.
15. Hall B, Pincus L, Yu S, et al. Immunohistochemical prognostication of Merkel cell carcinoma: p63 expression but not polyomavirus status correlates with outcome. J Cutan Pathol. 2012;39:911-917.
16. Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991-998.
17. Touze A, Le Bidre E, Laude H, et al. High levels of antibodies against Merkel cell polyomavirus identify a subset of patients with Merkel cell carcinoma with better clinical outcome. J Clin Oncol. 2011;29:1612-1619.
18. Sihto H, Bohling T, Kavola H, et al. Tumor infiltrating immune cells and outcome of Merkel cell carcinoma: a population-based study. Clin Cancer Res. 2012;18:2872-2881.
19. Colgan M, Tarantola T, Weaver A, et al. The predictive value of imaging studies in evaluating regional lymph node involvement in Merkel cell carcinoma. J Am Acad Dermatol. 2012;67:1250-1256.
20. NCCN Clinical Practice Guidelines in Oncology. National Comprehensive Cancer Network website. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#site. Accessed March 22, 2016.
21. Angermeyer S, Hesbacher S, Becker J, et al. Merkel cell polyomavirus-positive Merkel cell carcinoma cells do not require expression of the viral small T antigen. J Invest Dermatol. 2013;133:1-6.
Merkel cells originally were described by German histopathologist Friedrich Sigmund Merkel in 1875. These unique tactile cells were described as epidermal, nondendritic, and nonkeratinizing. Merkel cells are thought to arise from the neural crest and are believed to be primary neural cells found within the basal layer of the epidermis.1,2 They likely function primarily as slowly adapting type I mechanoreceptors. Origin from the neural crest is controversial, as other investigators have suggested derivation from epidermal keratinocytes.1,2 Tumor cells have been linked to the amine precursor uptake and decarboxylation system.3 In 1972, Toker4 described several cases of trabecular or sweat gland carcinomas of the skin. Upon further investigation, the cells that comprised these tumors were found to have dense core granules on electron microscopy, typical of Merkel cells.1,2 Other terms such as neuroendocrine carcinoma of the skin, small cell carcinoma of the skin, and anaplastic carcinoma of the skin have been used to describe Merkel cell carcinoma (MCC),1 which was suggested by De Wolf-Peeters et al5 in 1980.
Despite being a rare malignancy, MCC follows an aggressive clinical course. Upon presentation, approximately 66% of patients have local disease, 27% have nodal involvement, and 7% have distant metastasis.1 Future treatments will likely center around the novel Merkel cell polyomavirus (MCPyV) and modification of immune responses toward tumor cells. Standardization continues to be lacking in both staging and treatment of this aggressive tumor.
Epidemiology of MCC
|
| ||
Figure 1. A 2.3×1.5×1.2-cm, hemorrhagic, crusted, | ||
|
| ||
|
| ||
| Figure 2. Merkel cells are small- to medium-sized cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen (A)(H&E, original magnification ×40). Merkel cell carcinoma with trabecular pattern (B) (H&E, original magnification ×10). | ||
Between 1986 and 2006, the incidence of MCC grew substantially.1,2 Figures have been reported at 0.15 cases per 100,000 individuals to 0.6 cases per 100,000 individuals worldwide. In the United States, the age-adjusted incidence of MCC is estimated at 0.24 per 100,000 person-years, which is higher than the estimated 0.13 per 100,000 person-years found in Europe.3 The highest incidence worldwide has been noted in Western Australia, likely due to high levels of UV exposure.1 The incidence of MCC in psoriasis patients who are treated with oral methoxsalen (psoralen) and UVA photochemotherapy is 100 times greater than in the general population, further supporting the role of UV light in the development of MCC.1 White individuals have the highest incidence of MCC worldwide, with men being affected more frequently than women.1,3 The majority of patients with MCC are diagnosed at 70 years or older.1 Approximately 5% of reported MCC patients are diagnosed before 50 years of age.2 Immunosuppression and immunodeficiency likely play a role in the pathogenesis of MCC, and the incidence is increased in solid organ transplant recipients, most commonly renal transplant recipients,1 as well as individuals with chronic lymphocytic leukemia, human immunodeficiency virus infection, and AIDS.1,3 Patients with autoimmune diseases such as rheumatoid arthritis also are at increased risk for MCC.3 Individuals who are diagnosed with MCC are at an increased risk for development of other malignancies including nonmelanoma skin cancers, chronic lymphocytic leukemia, Hodgkin lymphoma, and non-Hodgkin lymphoma.3
Clinical Presentation of MCC
The clinical presentation of MCC can be variable. Most tumors present as firm, red to purple, nontender papules or nodules (Figure 1).1 Tumor size may range from 2 to 200 mm but is most commonly less than 20 mm.2 Growth can be rapid, and tumors are most commonly located on sun-exposed skin. The head and neck areas account for 48% of all MCC cases,1 with the eyelids being frequently involved.2 Merkel cell carcinoma also has been reported on the arms, legs, trunk, back, and buttocks.1 Non–sun-exposed areas are less commonly affected. Mucosal sites (eg, larynx, nasal cavity, pharynx, mouth) account for 5% of primary MCCs.1 Merkel cell carcinoma also has been reported to affect the vulva and penis. Subcutaneous primary MCC has presented without overlying epidermal changes.1 In a case series by Heath et al,6 14% (27/195) of MCC patients presented with nodal disease without any identifiable primary tumor, with the inguinal nodal chain being the most common for this presentation. It currently is not known whether these nodal tumors are primary tumors or metastatic disease with a regression of the primary tumor.1
|
Histopathology of MCC
|
| ||
|
| ||
|
| ||
Figure 3. Positive chromogranin staining (A)(original | ||
Merkel cells are small- to medium-sized basophilic cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen on histopathology (Figure 2A).1 Some tumor cells have more vesicular chromatin, multiple small nucleoli, irregular contours, and more abundant cytoplasm. In some reports, irregular contours and abundant cytoplasm were associated with no detectible MCPyV infection.1,3 Merkel cell carcinomas have a primarily nodular architecture, and classification is based on growth pattern and cell size. Three histopathologic growth patterns have been described: nodular, infiltrative, and trabecular. The trabecular pattern is composed of interconnecting strands ofcells (Figure 2B). Tumors with solely intraepidermal involvement (MCC in situ) have been described but are exceedingly rare.1 Cell types are classified according to size, with the intermediate cell type being the most common. The small cell variant may be mistaken for a lymphocytic infiltrate due to the similar size and appearance of both types of cells.1,3
Merkel cell carcinomas can have histopathological overlap with lymphomas, small cell lung cancers, carcinoid tumors, primitive neuroectodermal tumors, neuroblastomas, small cell osteosarcomas, rhabdomyosarcomas, or Ewing sarcomas.1,3 Specifically, differentiation from small cell carcinoma of the lung is of utmost importance. Merkel cell carcinoma stains positively for cytokeratins 8, 18, 19, and 20. The neuroendocrine markers chromogranin (Figure 3A), synaptophysin, and neuron-specific enolase also may show positive staining. Cytokeratin 20, low-molecular-weight cytokeratins (CAM 5.2), and neurofilament immunostains have a high sensitivity for MCC and are the most frequently used.1 Cytokeratin 20 stains in the characteristic paranuclear dot–like pattern, which is a hallmark of MCC (Figure 3B). Cytokeratin 20 positivity in conjunction with negative staining for thyroid transcription factor 1 (Figure 3C) and cytokeratin 7 can aid in differentiation from small cell carcinoma of the lung.1,3
Pathogenesis of MCC
In 2008, Feng et al7 discovered a novel polyomavirus associated with the development of MCC. This novel polyomavirus, MCPyV, is found in approximately 80% of all cases of MCC. Seventeen members of the polyomavirus family have been identified, 9 of which have been found to infect humans, including BK virus, JC virus, WU, MCPyV, human polyomavirus 6, human polyomavirus 7, trichodysplasia spinulosa–associated polyomavirus, human polyomavirus 9, and Simian virus 40.1 Merkel cell polyomavirus infection is found in approximately 60% of the general population and exposure likely occurs early in life. The virus likely is transmitted through skin shedding and nasal secretions, though it also has been found in urine specimens.3 Currently, there is no evidence to suggest vertical viral transmission from mother to fetus.
Merkel cell polyomavirus is composed of early and late gene regions. The early gene region contains both large T antigen (LT) and small T antigen reading frames, which are necessary for viral replication.8 The late region is responsible for encoding viral proteins necessary for viral capsid assembly. Mutations found in viral protein 1 prevent formation of viral particles.9 Large T antigen is substantially overexpressed in MCC and is responsible for tumor suppression through retinoblastoma tumor suppressor protein. It also serves as a binding domain for both heat shock proteins and helicases.8,10 These domains allow the polyomaviruses to use host-cell machinery for viral genome replication while targeting tumor suppressor proteins.8 Upon viral integration into host DNA, viral replication ceases while oncogenic function persists.
The exact mechanism by which the MCPyV contributes to the development of MCC still has yet to be identified. Hypotheses suggest a combination of viral infection with external mutagens (eg, UV radiation). Experimental observations suggest viral contribution is likely due to the large percentage of MCCs that are positive for MCPyV, the identification of LT antigen expression and the role it plays in preserving cell cycle progression, and the role persistent LT antigen expression plays in continued growth of MCC cell lines in vitro.8 Two important cell line preservation mechanisms ensure continued tumor growth, including prevention of apoptosis triggered by DNA damage response mechanisms following UV damage and interaction with the retinoblastoma tumor suppressor protein allowing continued growth.8,11 Other important factors in tumor growth and survival may be the inhibition of apoptosis through the BCL2 (B-cell chronic lymphocytic leukemia/lymphoma 2) proto-oncogene and survivin (baculoviral inhibitor of apoptosis repeat-containing 5 [BIRC5]).12 Survivin has been found to play an important role in MCPyV-positive MCCs.12,13 It has been suggested that lymphangiogenesis in MCC likely is driven by vascular endothelial growth factor-C+CD68+CD163+ M2 macrophages.14 Another survival mechanism specific to polyomaviruses is their ability to interfere with the p53 tumor suppressor pathway.8 Loss of p53 expression by tumor cell nuclei has been associated with poor prognosis.15
Immune Response
Immune response as a role in tumor progression can be primarily centered on the concept of persistent antigen expression as a means of immune downregulation. Dunn et al16 suggested that cancer cells must interact through 3 consecutive phases with the host immune system (immunoediting hypothesis). In the elimination phase, the host immune system is able to recognize and destroy newly transformed cells through both the innate and adaptive immune systems. The second equilibrium phase allows the tumor to remain dormant and growth remains stagnant. Lastly, the tumor is allowed to evade the immune system through the escape phase.8
Host immune responses play an important role in both the progression and prognosis of MCC. High anti-MCPyV capsid antibody titers have been associated with better progression-free survival in some patients.8 Patients with high antibody titers (>10,000) likely have better progression-free survival than those with low antibody titers (<10,000).17 Antibody titers to the LT antigen may serve as a biomarker of MCC disease burden in the future. Rising LT antigen titers have been shown to correlate with disease progression and falling titers correlate with successful treatment.8 Tumoral infiltration of CD8+ T lymphocytes has been shown to be a predictor of survival compared to no intratumoral infiltration.6 Sihto et al18 suggested that this better prognosis from high intratumoral infiltration is not specific to MCPyV-positive MCC; however, it does highlight an important aspect of tumor evasion through the downregulation of cell surface expression of class I major histocompatibility complex antigens, which allows presentation of tumor intracellular peptides to CD8+ T lymphocytes.8 Upregulation of this specific immune response may play a role in the future treatment of MCC.
Staging and Prognosis
Due to the extremely aggressive nature of MCC, patients with local disease and tumors 2 cm or smaller in diameter have a 66% survival at 5 years.1,3 The 5-year survival rate for patients with local metastasis to regional lymph nodes ranges from 26% to 42%. Patients with distant metastasis have an 18% survival rate at 5 years.1,3 Data suggest that sentinel lymph node biopsy should be performed on all patients with MCC regardless of tumor size.1 There are no consensus guidelines to date regarding imaging for the staging of MCC patients. It is suggested that (18F)fluorodeoxyglucose positron emission tomography alone or in combination with computed tomography (CT) may be of value as a single whole-body diagnostic tool for accurate staging.10 It also has been suggested that (18F)fluorodeoxyglucose positron emission tomography and CT may offer more accurate staging than other screening modalities such as CT alone or magnetic resonance imaging.14,19
Treatment of MCC
Surgery remains the mainstay of treatment of MCC. Current National Comprehensive Cancer Network guidelines20 recommend 1- to 2-cm margins for wide local excision or treatment with Mohs micrographic surgery. Sentinel lymph node biopsy should be performed intraoperatively in patients undergoing wide local excision and preoperatively for patients undergoing Mohs micrographic surgery due to potential alterations in lymphatic drainage that may affect lymphoscintigraphy.1
Radiation may be used as primary or adjuvant therapy in patients with MCC. Radiation as primary therapy generally is reserved for patients who are not surgical candidates. It has been suggested that there was no difference in outcome in a small group of patients treated with radiation alone compared to patients who underwent surgery and radiation to the tumor bed.1 Current guidelines suggest a small group of patients may not require adjuvant therapy following adequate resection of some small tumors, and clinical observation may be appropriate.1,3 Chemotherapy may play a palliative role in patients with metastatic MCC. Merkel cell carcinoma has been shown to be chemosensitive but with a high recurrence rate.1 Because the immune system plays an important role in disease prognosis, having an intact immune system likely is paramount in the prevention of further disease progression.
Future Treatments of MCC
Future treatment of MCC may be focused on the viral etiology of most tumors and upregulation of the immune response, which may lead to the possibility of specifically interfering with virus-specific oncoproteins and stimulation of immune responses to virally infected tumor cells.8 The MCPyV large T antigen has been found to be overexpressed in some tumors and may serve as a specific target of therapy.10,21 Survivin, a key cell cycle protein encoded by LT antigen, may be an interesting target given its implication in other cancers.13 Other potential nonviral molecular target antigens include the oncoprotein H1P1 that interacts with c-KIT.8 Specific immunostimulatory cytokines that may be used to upregulate the immune response to tumoral cells may include IL-2, IL-12, IL-15, or IL-21. Therapeutic agents that may be studied in the future to target the immune exhaustion phenomenon associated with tumorigenesis include ipilimumab (cytotoxic T lymphocyte antigen 4 receptor-blocking agent) as well as programmed cell death 1 and programmed cell death 1 ligand 1 (PD-1/PD-L1).8 Neuroendocrine tumors including MCC tend to be highly vascular and express vascular endothelial growth factors and platelet-derived growth factors, which may be other potential therapeutic targets. It has been reported that approximately 95% of MCC patients have CD56+ tumors, and current clinical trials suggest a promising therapeutic response with the immunogen anti-CD56 monoclonal antibody.3
Conclusion
Merkel cell carcinoma is a rare aggressive neuroendocrine tumor that has been associated with a novel polyomavirus. Merkel cell carcinoma tends to affect elderly and immunocompromised patients as well as white individuals. Tumors are most often found in areas of high UV exposure and clinically on sun-exposed skin. Merkel cell polyomavirus is associated with approximately 80% of tumors, and tumorigenesis likely is caused by a number of sequential steps from viral integration into host DNA, mutagenic events, and specific immune responses. Currently there are no consensus guidelines for using imaging for staging of MCCs, but sentinel lymph node biopsy is recommended for all cases due to the aggressive nature of even smaller tumors. Surgery remains the mainstay of treatment, and radiation therapy may be used as a primary or adjuvant treatment. Chemotherapy usually is reserved for patients with metastatic disease purely for palliation. Future treatments of MCC likely will center on the viral etiology of MCC and upregulation of immune responses to virally infected tumor cells.
Merkel cells originally were described by German histopathologist Friedrich Sigmund Merkel in 1875. These unique tactile cells were described as epidermal, nondendritic, and nonkeratinizing. Merkel cells are thought to arise from the neural crest and are believed to be primary neural cells found within the basal layer of the epidermis.1,2 They likely function primarily as slowly adapting type I mechanoreceptors. Origin from the neural crest is controversial, as other investigators have suggested derivation from epidermal keratinocytes.1,2 Tumor cells have been linked to the amine precursor uptake and decarboxylation system.3 In 1972, Toker4 described several cases of trabecular or sweat gland carcinomas of the skin. Upon further investigation, the cells that comprised these tumors were found to have dense core granules on electron microscopy, typical of Merkel cells.1,2 Other terms such as neuroendocrine carcinoma of the skin, small cell carcinoma of the skin, and anaplastic carcinoma of the skin have been used to describe Merkel cell carcinoma (MCC),1 which was suggested by De Wolf-Peeters et al5 in 1980.
Despite being a rare malignancy, MCC follows an aggressive clinical course. Upon presentation, approximately 66% of patients have local disease, 27% have nodal involvement, and 7% have distant metastasis.1 Future treatments will likely center around the novel Merkel cell polyomavirus (MCPyV) and modification of immune responses toward tumor cells. Standardization continues to be lacking in both staging and treatment of this aggressive tumor.
Epidemiology of MCC
|
| ||
Figure 1. A 2.3×1.5×1.2-cm, hemorrhagic, crusted, | ||
|
| ||
|
| ||
| Figure 2. Merkel cells are small- to medium-sized cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen (A)(H&E, original magnification ×40). Merkel cell carcinoma with trabecular pattern (B) (H&E, original magnification ×10). | ||
Between 1986 and 2006, the incidence of MCC grew substantially.1,2 Figures have been reported at 0.15 cases per 100,000 individuals to 0.6 cases per 100,000 individuals worldwide. In the United States, the age-adjusted incidence of MCC is estimated at 0.24 per 100,000 person-years, which is higher than the estimated 0.13 per 100,000 person-years found in Europe.3 The highest incidence worldwide has been noted in Western Australia, likely due to high levels of UV exposure.1 The incidence of MCC in psoriasis patients who are treated with oral methoxsalen (psoralen) and UVA photochemotherapy is 100 times greater than in the general population, further supporting the role of UV light in the development of MCC.1 White individuals have the highest incidence of MCC worldwide, with men being affected more frequently than women.1,3 The majority of patients with MCC are diagnosed at 70 years or older.1 Approximately 5% of reported MCC patients are diagnosed before 50 years of age.2 Immunosuppression and immunodeficiency likely play a role in the pathogenesis of MCC, and the incidence is increased in solid organ transplant recipients, most commonly renal transplant recipients,1 as well as individuals with chronic lymphocytic leukemia, human immunodeficiency virus infection, and AIDS.1,3 Patients with autoimmune diseases such as rheumatoid arthritis also are at increased risk for MCC.3 Individuals who are diagnosed with MCC are at an increased risk for development of other malignancies including nonmelanoma skin cancers, chronic lymphocytic leukemia, Hodgkin lymphoma, and non-Hodgkin lymphoma.3
Clinical Presentation of MCC
The clinical presentation of MCC can be variable. Most tumors present as firm, red to purple, nontender papules or nodules (Figure 1).1 Tumor size may range from 2 to 200 mm but is most commonly less than 20 mm.2 Growth can be rapid, and tumors are most commonly located on sun-exposed skin. The head and neck areas account for 48% of all MCC cases,1 with the eyelids being frequently involved.2 Merkel cell carcinoma also has been reported on the arms, legs, trunk, back, and buttocks.1 Non–sun-exposed areas are less commonly affected. Mucosal sites (eg, larynx, nasal cavity, pharynx, mouth) account for 5% of primary MCCs.1 Merkel cell carcinoma also has been reported to affect the vulva and penis. Subcutaneous primary MCC has presented without overlying epidermal changes.1 In a case series by Heath et al,6 14% (27/195) of MCC patients presented with nodal disease without any identifiable primary tumor, with the inguinal nodal chain being the most common for this presentation. It currently is not known whether these nodal tumors are primary tumors or metastatic disease with a regression of the primary tumor.1
|
Histopathology of MCC
|
| ||
|
| ||
|
| ||
Figure 3. Positive chromogranin staining (A)(original | ||
Merkel cells are small- to medium-sized basophilic cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen on histopathology (Figure 2A).1 Some tumor cells have more vesicular chromatin, multiple small nucleoli, irregular contours, and more abundant cytoplasm. In some reports, irregular contours and abundant cytoplasm were associated with no detectible MCPyV infection.1,3 Merkel cell carcinomas have a primarily nodular architecture, and classification is based on growth pattern and cell size. Three histopathologic growth patterns have been described: nodular, infiltrative, and trabecular. The trabecular pattern is composed of interconnecting strands ofcells (Figure 2B). Tumors with solely intraepidermal involvement (MCC in situ) have been described but are exceedingly rare.1 Cell types are classified according to size, with the intermediate cell type being the most common. The small cell variant may be mistaken for a lymphocytic infiltrate due to the similar size and appearance of both types of cells.1,3
Merkel cell carcinomas can have histopathological overlap with lymphomas, small cell lung cancers, carcinoid tumors, primitive neuroectodermal tumors, neuroblastomas, small cell osteosarcomas, rhabdomyosarcomas, or Ewing sarcomas.1,3 Specifically, differentiation from small cell carcinoma of the lung is of utmost importance. Merkel cell carcinoma stains positively for cytokeratins 8, 18, 19, and 20. The neuroendocrine markers chromogranin (Figure 3A), synaptophysin, and neuron-specific enolase also may show positive staining. Cytokeratin 20, low-molecular-weight cytokeratins (CAM 5.2), and neurofilament immunostains have a high sensitivity for MCC and are the most frequently used.1 Cytokeratin 20 stains in the characteristic paranuclear dot–like pattern, which is a hallmark of MCC (Figure 3B). Cytokeratin 20 positivity in conjunction with negative staining for thyroid transcription factor 1 (Figure 3C) and cytokeratin 7 can aid in differentiation from small cell carcinoma of the lung.1,3
Pathogenesis of MCC
In 2008, Feng et al7 discovered a novel polyomavirus associated with the development of MCC. This novel polyomavirus, MCPyV, is found in approximately 80% of all cases of MCC. Seventeen members of the polyomavirus family have been identified, 9 of which have been found to infect humans, including BK virus, JC virus, WU, MCPyV, human polyomavirus 6, human polyomavirus 7, trichodysplasia spinulosa–associated polyomavirus, human polyomavirus 9, and Simian virus 40.1 Merkel cell polyomavirus infection is found in approximately 60% of the general population and exposure likely occurs early in life. The virus likely is transmitted through skin shedding and nasal secretions, though it also has been found in urine specimens.3 Currently, there is no evidence to suggest vertical viral transmission from mother to fetus.
Merkel cell polyomavirus is composed of early and late gene regions. The early gene region contains both large T antigen (LT) and small T antigen reading frames, which are necessary for viral replication.8 The late region is responsible for encoding viral proteins necessary for viral capsid assembly. Mutations found in viral protein 1 prevent formation of viral particles.9 Large T antigen is substantially overexpressed in MCC and is responsible for tumor suppression through retinoblastoma tumor suppressor protein. It also serves as a binding domain for both heat shock proteins and helicases.8,10 These domains allow the polyomaviruses to use host-cell machinery for viral genome replication while targeting tumor suppressor proteins.8 Upon viral integration into host DNA, viral replication ceases while oncogenic function persists.
The exact mechanism by which the MCPyV contributes to the development of MCC still has yet to be identified. Hypotheses suggest a combination of viral infection with external mutagens (eg, UV radiation). Experimental observations suggest viral contribution is likely due to the large percentage of MCCs that are positive for MCPyV, the identification of LT antigen expression and the role it plays in preserving cell cycle progression, and the role persistent LT antigen expression plays in continued growth of MCC cell lines in vitro.8 Two important cell line preservation mechanisms ensure continued tumor growth, including prevention of apoptosis triggered by DNA damage response mechanisms following UV damage and interaction with the retinoblastoma tumor suppressor protein allowing continued growth.8,11 Other important factors in tumor growth and survival may be the inhibition of apoptosis through the BCL2 (B-cell chronic lymphocytic leukemia/lymphoma 2) proto-oncogene and survivin (baculoviral inhibitor of apoptosis repeat-containing 5 [BIRC5]).12 Survivin has been found to play an important role in MCPyV-positive MCCs.12,13 It has been suggested that lymphangiogenesis in MCC likely is driven by vascular endothelial growth factor-C+CD68+CD163+ M2 macrophages.14 Another survival mechanism specific to polyomaviruses is their ability to interfere with the p53 tumor suppressor pathway.8 Loss of p53 expression by tumor cell nuclei has been associated with poor prognosis.15
Immune Response
Immune response as a role in tumor progression can be primarily centered on the concept of persistent antigen expression as a means of immune downregulation. Dunn et al16 suggested that cancer cells must interact through 3 consecutive phases with the host immune system (immunoediting hypothesis). In the elimination phase, the host immune system is able to recognize and destroy newly transformed cells through both the innate and adaptive immune systems. The second equilibrium phase allows the tumor to remain dormant and growth remains stagnant. Lastly, the tumor is allowed to evade the immune system through the escape phase.8
Host immune responses play an important role in both the progression and prognosis of MCC. High anti-MCPyV capsid antibody titers have been associated with better progression-free survival in some patients.8 Patients with high antibody titers (>10,000) likely have better progression-free survival than those with low antibody titers (<10,000).17 Antibody titers to the LT antigen may serve as a biomarker of MCC disease burden in the future. Rising LT antigen titers have been shown to correlate with disease progression and falling titers correlate with successful treatment.8 Tumoral infiltration of CD8+ T lymphocytes has been shown to be a predictor of survival compared to no intratumoral infiltration.6 Sihto et al18 suggested that this better prognosis from high intratumoral infiltration is not specific to MCPyV-positive MCC; however, it does highlight an important aspect of tumor evasion through the downregulation of cell surface expression of class I major histocompatibility complex antigens, which allows presentation of tumor intracellular peptides to CD8+ T lymphocytes.8 Upregulation of this specific immune response may play a role in the future treatment of MCC.
Staging and Prognosis
Due to the extremely aggressive nature of MCC, patients with local disease and tumors 2 cm or smaller in diameter have a 66% survival at 5 years.1,3 The 5-year survival rate for patients with local metastasis to regional lymph nodes ranges from 26% to 42%. Patients with distant metastasis have an 18% survival rate at 5 years.1,3 Data suggest that sentinel lymph node biopsy should be performed on all patients with MCC regardless of tumor size.1 There are no consensus guidelines to date regarding imaging for the staging of MCC patients. It is suggested that (18F)fluorodeoxyglucose positron emission tomography alone or in combination with computed tomography (CT) may be of value as a single whole-body diagnostic tool for accurate staging.10 It also has been suggested that (18F)fluorodeoxyglucose positron emission tomography and CT may offer more accurate staging than other screening modalities such as CT alone or magnetic resonance imaging.14,19
Treatment of MCC
Surgery remains the mainstay of treatment of MCC. Current National Comprehensive Cancer Network guidelines20 recommend 1- to 2-cm margins for wide local excision or treatment with Mohs micrographic surgery. Sentinel lymph node biopsy should be performed intraoperatively in patients undergoing wide local excision and preoperatively for patients undergoing Mohs micrographic surgery due to potential alterations in lymphatic drainage that may affect lymphoscintigraphy.1
Radiation may be used as primary or adjuvant therapy in patients with MCC. Radiation as primary therapy generally is reserved for patients who are not surgical candidates. It has been suggested that there was no difference in outcome in a small group of patients treated with radiation alone compared to patients who underwent surgery and radiation to the tumor bed.1 Current guidelines suggest a small group of patients may not require adjuvant therapy following adequate resection of some small tumors, and clinical observation may be appropriate.1,3 Chemotherapy may play a palliative role in patients with metastatic MCC. Merkel cell carcinoma has been shown to be chemosensitive but with a high recurrence rate.1 Because the immune system plays an important role in disease prognosis, having an intact immune system likely is paramount in the prevention of further disease progression.
Future Treatments of MCC
Future treatment of MCC may be focused on the viral etiology of most tumors and upregulation of the immune response, which may lead to the possibility of specifically interfering with virus-specific oncoproteins and stimulation of immune responses to virally infected tumor cells.8 The MCPyV large T antigen has been found to be overexpressed in some tumors and may serve as a specific target of therapy.10,21 Survivin, a key cell cycle protein encoded by LT antigen, may be an interesting target given its implication in other cancers.13 Other potential nonviral molecular target antigens include the oncoprotein H1P1 that interacts with c-KIT.8 Specific immunostimulatory cytokines that may be used to upregulate the immune response to tumoral cells may include IL-2, IL-12, IL-15, or IL-21. Therapeutic agents that may be studied in the future to target the immune exhaustion phenomenon associated with tumorigenesis include ipilimumab (cytotoxic T lymphocyte antigen 4 receptor-blocking agent) as well as programmed cell death 1 and programmed cell death 1 ligand 1 (PD-1/PD-L1).8 Neuroendocrine tumors including MCC tend to be highly vascular and express vascular endothelial growth factors and platelet-derived growth factors, which may be other potential therapeutic targets. It has been reported that approximately 95% of MCC patients have CD56+ tumors, and current clinical trials suggest a promising therapeutic response with the immunogen anti-CD56 monoclonal antibody.3
Conclusion
Merkel cell carcinoma is a rare aggressive neuroendocrine tumor that has been associated with a novel polyomavirus. Merkel cell carcinoma tends to affect elderly and immunocompromised patients as well as white individuals. Tumors are most often found in areas of high UV exposure and clinically on sun-exposed skin. Merkel cell polyomavirus is associated with approximately 80% of tumors, and tumorigenesis likely is caused by a number of sequential steps from viral integration into host DNA, mutagenic events, and specific immune responses. Currently there are no consensus guidelines for using imaging for staging of MCCs, but sentinel lymph node biopsy is recommended for all cases due to the aggressive nature of even smaller tumors. Surgery remains the mainstay of treatment, and radiation therapy may be used as a primary or adjuvant treatment. Chemotherapy usually is reserved for patients with metastatic disease purely for palliation. Future treatments of MCC likely will center on the viral etiology of MCC and upregulation of immune responses to virally infected tumor cells.
1. Han S, North J, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
2. Goessling W, McKee P, Mayer R. Merkel cell carcinoma. J Clin Oncol. 2002;20:588-598.
3. Donepudi S, DeConti R, Samlowski W. Recent advances in the understanding of the genetics, etiology, and treatment of merkel cell carcinoma. Semin Oncol. 2012;39:163-172.
4. Toker C. Trabecular carcinoma of the skin. Arch Dermatol. 1972;105:107-110.
5. De Wolff-Peeters C, Marien K, Mebis J, et al. A cutaneous APUDoma or Merkel cell tumor? a morphologically recognizable tumor with a biological and histological malignant aspect in contrast with its clinical behavior. Cancer. 1980;46:810-816.
6. Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375-381.
7. Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
8. Bhatia S, Afanasiev O, Nghiem P. Immunobiology of Merkel cell carcinoma: implications for immunotherapy of a polyomavirus-associated cancer. Curr Oncol Rep. 2011;13:488-497.
9. Amber K, McLeod M, Nouri K. The Merkel cell polyomavirus and its involvement in Merkel cell carcinoma. Dermatol Surg. 2013;39:232-238.
10. Erovic B, Al Habeeb A, Harris L, et al. Significant overexpression of the Merkel cell polyomavirus (MCPyV) large T antigen in Merkel cell carcinoma. Head Neck. 2013;35:184-189.
11. Demetriou S, Ona-Vu K, Sullivan E, et al. Defective DNA repair and cell cycle arrest in cells expressing Merkel cell polyomavirus T antigen. Int J Cancer. 2012;131:1818-1827.
12. Sahi H, Koljonen V, Kavola H, et al. Bcl-2 expression indicates better prognosis of Merkel cell carcinoma regardless of the presence of Merkel cell polyomavirus. Virchows Arch. 2012;461:553-559.
13. Arora R, Shuda M, Guastafierro A, et al. Survivin is a therapeutic target in Merkel cell carcinoma. Sci Transl Med. 2012;4:1-11.
14. Hawryluk E, O’Regan K, Sheehy N, et al. Positron emission tomography/computed tomography imaging in Merkel cell carcinoma: a study of 270 scans in 97 patients at the Dana-Farber/Brigham and Women’s Cancer Center. J Am Acad Dermatol. 2013;68:592-599.
15. Hall B, Pincus L, Yu S, et al. Immunohistochemical prognostication of Merkel cell carcinoma: p63 expression but not polyomavirus status correlates with outcome. J Cutan Pathol. 2012;39:911-917.
16. Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991-998.
17. Touze A, Le Bidre E, Laude H, et al. High levels of antibodies against Merkel cell polyomavirus identify a subset of patients with Merkel cell carcinoma with better clinical outcome. J Clin Oncol. 2011;29:1612-1619.
18. Sihto H, Bohling T, Kavola H, et al. Tumor infiltrating immune cells and outcome of Merkel cell carcinoma: a population-based study. Clin Cancer Res. 2012;18:2872-2881.
19. Colgan M, Tarantola T, Weaver A, et al. The predictive value of imaging studies in evaluating regional lymph node involvement in Merkel cell carcinoma. J Am Acad Dermatol. 2012;67:1250-1256.
20. NCCN Clinical Practice Guidelines in Oncology. National Comprehensive Cancer Network website. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#site. Accessed March 22, 2016.
21. Angermeyer S, Hesbacher S, Becker J, et al. Merkel cell polyomavirus-positive Merkel cell carcinoma cells do not require expression of the viral small T antigen. J Invest Dermatol. 2013;133:1-6.
1. Han S, North J, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
2. Goessling W, McKee P, Mayer R. Merkel cell carcinoma. J Clin Oncol. 2002;20:588-598.
3. Donepudi S, DeConti R, Samlowski W. Recent advances in the understanding of the genetics, etiology, and treatment of merkel cell carcinoma. Semin Oncol. 2012;39:163-172.
4. Toker C. Trabecular carcinoma of the skin. Arch Dermatol. 1972;105:107-110.
5. De Wolff-Peeters C, Marien K, Mebis J, et al. A cutaneous APUDoma or Merkel cell tumor? a morphologically recognizable tumor with a biological and histological malignant aspect in contrast with its clinical behavior. Cancer. 1980;46:810-816.
6. Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375-381.
7. Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
8. Bhatia S, Afanasiev O, Nghiem P. Immunobiology of Merkel cell carcinoma: implications for immunotherapy of a polyomavirus-associated cancer. Curr Oncol Rep. 2011;13:488-497.
9. Amber K, McLeod M, Nouri K. The Merkel cell polyomavirus and its involvement in Merkel cell carcinoma. Dermatol Surg. 2013;39:232-238.
10. Erovic B, Al Habeeb A, Harris L, et al. Significant overexpression of the Merkel cell polyomavirus (MCPyV) large T antigen in Merkel cell carcinoma. Head Neck. 2013;35:184-189.
11. Demetriou S, Ona-Vu K, Sullivan E, et al. Defective DNA repair and cell cycle arrest in cells expressing Merkel cell polyomavirus T antigen. Int J Cancer. 2012;131:1818-1827.
12. Sahi H, Koljonen V, Kavola H, et al. Bcl-2 expression indicates better prognosis of Merkel cell carcinoma regardless of the presence of Merkel cell polyomavirus. Virchows Arch. 2012;461:553-559.
13. Arora R, Shuda M, Guastafierro A, et al. Survivin is a therapeutic target in Merkel cell carcinoma. Sci Transl Med. 2012;4:1-11.
14. Hawryluk E, O’Regan K, Sheehy N, et al. Positron emission tomography/computed tomography imaging in Merkel cell carcinoma: a study of 270 scans in 97 patients at the Dana-Farber/Brigham and Women’s Cancer Center. J Am Acad Dermatol. 2013;68:592-599.
15. Hall B, Pincus L, Yu S, et al. Immunohistochemical prognostication of Merkel cell carcinoma: p63 expression but not polyomavirus status correlates with outcome. J Cutan Pathol. 2012;39:911-917.
16. Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991-998.
17. Touze A, Le Bidre E, Laude H, et al. High levels of antibodies against Merkel cell polyomavirus identify a subset of patients with Merkel cell carcinoma with better clinical outcome. J Clin Oncol. 2011;29:1612-1619.
18. Sihto H, Bohling T, Kavola H, et al. Tumor infiltrating immune cells and outcome of Merkel cell carcinoma: a population-based study. Clin Cancer Res. 2012;18:2872-2881.
19. Colgan M, Tarantola T, Weaver A, et al. The predictive value of imaging studies in evaluating regional lymph node involvement in Merkel cell carcinoma. J Am Acad Dermatol. 2012;67:1250-1256.
20. NCCN Clinical Practice Guidelines in Oncology. National Comprehensive Cancer Network website. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#site. Accessed March 22, 2016.
21. Angermeyer S, Hesbacher S, Becker J, et al. Merkel cell polyomavirus-positive Merkel cell carcinoma cells do not require expression of the viral small T antigen. J Invest Dermatol. 2013;133:1-6.
Practice Points
- Merkel cell carcinoma has been associated with a novel polyomavirus.
- Merkel cell carcinoma follows a very aggressive course and is most likely metastatic at diagnosis.
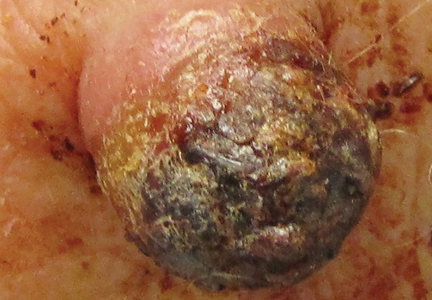
Cyst on the Eyebrow
The best diagnosis is:
a. bronchogenic cyst
b. dermoid cyst
c. epidermal inclusion cyst
d. hidrocystoma
e. steatocystoma
|
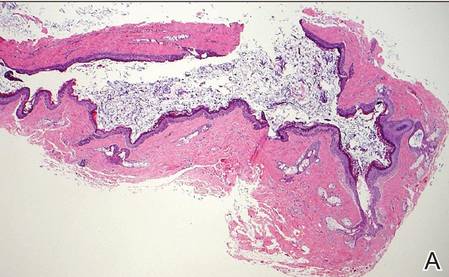
|
| H&E, original magnification ×40. |
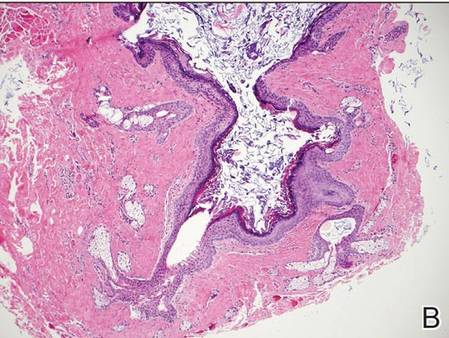
|
| H&E, original magnification ×100. |
Continue to the next page for the diagnosis >>
Dermoid Cyst
Dermoid cysts often present clinically as firm subcutaneous nodules on the head or neck in young children. They tend to arise along the lateral aspect of the eyebrow but also can occur on the nose, forehead, neck, chest, or scalp.1 Dermoid cysts are thought to arise from the sequestration of ectodermal tissues along the embryonic fusion planes during development.2 As such, they represent congenital defects and often are identified at birth; however, some are not noticed until much later when they enlarge or become inflamed or infected. Midline dermoid cysts may be associated with underlying dysraphism or intracranial extension.3,4 Thus, any midline lesion warrants evaluation that incorporates imaging with computed tomography or magnetic resonance imaging.4,5 Histologically, dermoid cysts are lined by a keratinizing stratified squamous epithelium (quiz image A), but the lining may be brightly eosinophilic and wavy resembling shark teeth.1,3 The wall of a dermoid cyst commonly contains mature adnexal structures such as terminal hair follicles, sebaceous glands, apocrine glands, and/or eccrine glands (quiz image B).1 Smooth muscle also may be seen within the lining; however, bone and cartilage are not commonly reported in dermoid cysts.2 Lamellar keratin is typical of the cyst contents, and terminal hair shafts also are sometimes noted within the cystic space (quiz image B).1,2 Treatment options include excision at the time of diagnosis or close clinical monitoring with subsequent excision if the lesion grows or becomes symptomatic.4,5 Many practitioners opt to excise these cysts at diagnosis, as untreated lesions are at risk for infection and/or inflammation or may be cosmetically deforming.6,7 Surgical resection, including removal of the wall of the cyst, is curative and reoccurrence is rare.5
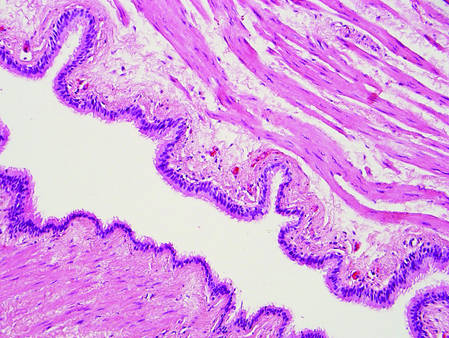
| |
Figure 1. Bronchogenic cyst demonstrating a ciliated pseudostratified epithelial lining encircled by smooth muscle (H&E, original magnification ×200). | |
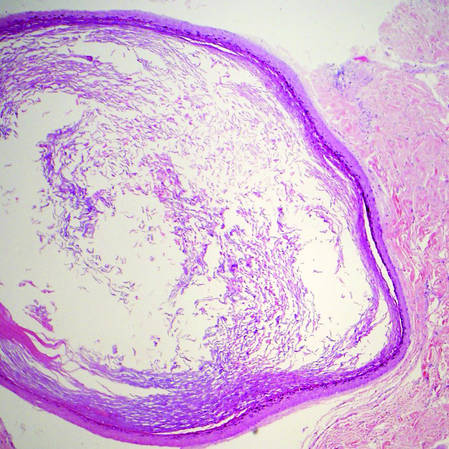
| |
| Figure 2. Epidermal inclusion cyst containing loose lamellar keratin and a lining that closely resembles the surface epidermis (H&E, original magnification ×40). |
|
Bronchogenic cysts demonstrate an epithelial lining that often is pseudostratified cuboidal or columnar as well as ciliated (Figure 1). Goblet cells are present in the lining in approximately 50% of cases. Smooth muscle may be seen circumferentially surrounding the cyst lining, and rare cases also contain cartilage.1 In contrast to dermoid cysts, other types of adnexal structures are not found within the lining. Bronchogenic cysts that arise in the skin are extremely rare.2 These cysts are thought to arise from respiratory epithelium that has been sequestered during embryologic formation of the tracheobronchial tree. They often are seen overlying the suprasternal notch and occasionally are found on the anterior aspect of the neck or chin. These cysts also are present at birth, similar to dermoid cysts.3
Epidermal inclusion cysts have a lining that histologically bears close resemblance to the surface epidermis. These cysts contain loose lamellar keratin, similar to a dermoid cyst. In contrast, the lining of an epidermal inclusion cyst will lack adnexal structures (Figure 2).1 Clinically, epidermal inclusion cysts often present as smooth, dome-shaped papules and nodules with a central punctum. They are classically found on the face, neck, and trunk. These cysts are thought to arise after a traumatic insult to the pilosebaceous unit.2
Hidrocystomas can be apocrine or eccrine.3 Eccrine hidrocystomas are unilocular cysts that are lined by 2 layers of flattened to cuboidal epithelial cells (Figure 3). The cysts are filled with clear fluid and often are found adjacent to normal eccrine glands.1 Apocrine hidrocystomas are unilocular or multilocular cysts that are lined by 1 to several layers of epithelial cells. The lining of an apocrine hidrocystoma will often exhibit luminal decapitation secretion.3 Apocrine and eccrine hidrocystomas are clinically identical and appear as blue translucent papules on the cheeks or eyelids of adults.1-3 They usually occur periorbitally but also can be seen on the trunk, popliteal fossa, external ears, or vulva. Eccrine hidrocystomas can wax and wane in accordance with the amount of sweat produced; thus, they often expand in size during the summer months.2
Steatocystomas, or simple sebaceous duct cysts, histologically demonstrate a characteristically wavy and eosinophilic cuticle resembling shark teeth (Figure 4) similar to the lining of the sebaceous duct where it enters the follicle.1 Sebaceous glands are an almost invariable feature, either present within the lining of the cyst (Figure 4) or in the adjacent tissue.2 In comparison, dermoid cysts may have a red wavy cuticle but also will usually have terminal hair follicles or eccrine or apocrine glands within the wall of the cyst. Steatocystomas typically are collapsed and empty or only contain sebaceous debris (Figure 4) rather than the lamellar keratin seen in dermoid and epidermoid inclusion cysts. Steatocystomas can occur as solitary (steatocystoma simplex) or multiple (steatocystoma multiplex) lesions.1,3 They are clinically comprised of small dome-shaped papules that often are translucent and yellow. These cysts are commonly found on the sternum of males and the axillae or groin of females.2
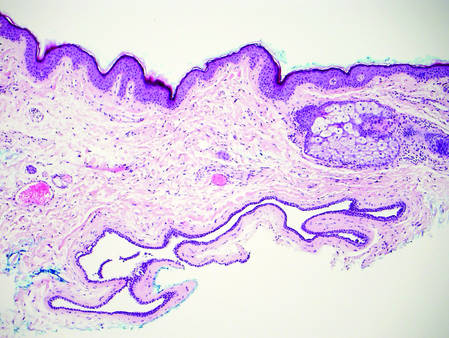
| 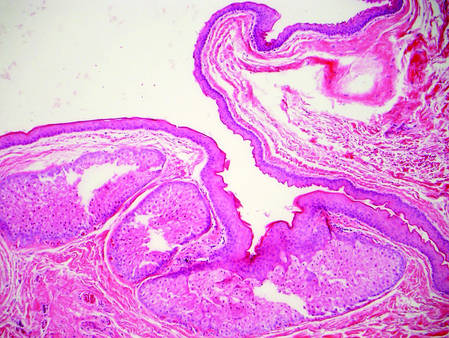
| |
Figure 3. Eccrine hidrocystoma with clear contents and lined by 2 layers of cuboidal epithelial cells (H&E, original magnification ×100). | Figure 4. Steatocystoma with a red wavy cuticle, sparse sebaceous contents, and sebaceous glands within the lining (H&E, original magnification ×100). |
|
1. Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
2. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin. 4th ed. St Louis, MO: Elsevier/Saunders; 2012.
3. Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012.
4. Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
5. Sorenson EP, Powel JE, Rozzelle CJ, et al. Scalp dermoids: a review of their anatomy, diagnosis, and treatment. Childs Nerv Syst. 2013;29:375-380.
6. Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolarynol Head Neck Surg. 2005;132:938-942.
7. Abou-Rayyah Y, Rose GE, Konrad H, et al. Clinical, radiological and pathological examination of periocular dermoid cysts: evidence of inflammation from an early age. Eye (Lond). 2002;16:507-512.
The best diagnosis is:
a. bronchogenic cyst
b. dermoid cyst
c. epidermal inclusion cyst
d. hidrocystoma
e. steatocystoma
|
|
|
| H&E, original magnification ×40. |
|
|
| H&E, original magnification ×100. |
Continue to the next page for the diagnosis >>
Dermoid Cyst
Dermoid cysts often present clinically as firm subcutaneous nodules on the head or neck in young children. They tend to arise along the lateral aspect of the eyebrow but also can occur on the nose, forehead, neck, chest, or scalp.1 Dermoid cysts are thought to arise from the sequestration of ectodermal tissues along the embryonic fusion planes during development.2 As such, they represent congenital defects and often are identified at birth; however, some are not noticed until much later when they enlarge or become inflamed or infected. Midline dermoid cysts may be associated with underlying dysraphism or intracranial extension.3,4 Thus, any midline lesion warrants evaluation that incorporates imaging with computed tomography or magnetic resonance imaging.4,5 Histologically, dermoid cysts are lined by a keratinizing stratified squamous epithelium (quiz image A), but the lining may be brightly eosinophilic and wavy resembling shark teeth.1,3 The wall of a dermoid cyst commonly contains mature adnexal structures such as terminal hair follicles, sebaceous glands, apocrine glands, and/or eccrine glands (quiz image B).1 Smooth muscle also may be seen within the lining; however, bone and cartilage are not commonly reported in dermoid cysts.2 Lamellar keratin is typical of the cyst contents, and terminal hair shafts also are sometimes noted within the cystic space (quiz image B).1,2 Treatment options include excision at the time of diagnosis or close clinical monitoring with subsequent excision if the lesion grows or becomes symptomatic.4,5 Many practitioners opt to excise these cysts at diagnosis, as untreated lesions are at risk for infection and/or inflammation or may be cosmetically deforming.6,7 Surgical resection, including removal of the wall of the cyst, is curative and reoccurrence is rare.5
|
| |
Figure 1. Bronchogenic cyst demonstrating a ciliated pseudostratified epithelial lining encircled by smooth muscle (H&E, original magnification ×200). | |
|
| |
| Figure 2. Epidermal inclusion cyst containing loose lamellar keratin and a lining that closely resembles the surface epidermis (H&E, original magnification ×40). |
|
Bronchogenic cysts demonstrate an epithelial lining that often is pseudostratified cuboidal or columnar as well as ciliated (Figure 1). Goblet cells are present in the lining in approximately 50% of cases. Smooth muscle may be seen circumferentially surrounding the cyst lining, and rare cases also contain cartilage.1 In contrast to dermoid cysts, other types of adnexal structures are not found within the lining. Bronchogenic cysts that arise in the skin are extremely rare.2 These cysts are thought to arise from respiratory epithelium that has been sequestered during embryologic formation of the tracheobronchial tree. They often are seen overlying the suprasternal notch and occasionally are found on the anterior aspect of the neck or chin. These cysts also are present at birth, similar to dermoid cysts.3
Epidermal inclusion cysts have a lining that histologically bears close resemblance to the surface epidermis. These cysts contain loose lamellar keratin, similar to a dermoid cyst. In contrast, the lining of an epidermal inclusion cyst will lack adnexal structures (Figure 2).1 Clinically, epidermal inclusion cysts often present as smooth, dome-shaped papules and nodules with a central punctum. They are classically found on the face, neck, and trunk. These cysts are thought to arise after a traumatic insult to the pilosebaceous unit.2
Hidrocystomas can be apocrine or eccrine.3 Eccrine hidrocystomas are unilocular cysts that are lined by 2 layers of flattened to cuboidal epithelial cells (Figure 3). The cysts are filled with clear fluid and often are found adjacent to normal eccrine glands.1 Apocrine hidrocystomas are unilocular or multilocular cysts that are lined by 1 to several layers of epithelial cells. The lining of an apocrine hidrocystoma will often exhibit luminal decapitation secretion.3 Apocrine and eccrine hidrocystomas are clinically identical and appear as blue translucent papules on the cheeks or eyelids of adults.1-3 They usually occur periorbitally but also can be seen on the trunk, popliteal fossa, external ears, or vulva. Eccrine hidrocystomas can wax and wane in accordance with the amount of sweat produced; thus, they often expand in size during the summer months.2
Steatocystomas, or simple sebaceous duct cysts, histologically demonstrate a characteristically wavy and eosinophilic cuticle resembling shark teeth (Figure 4) similar to the lining of the sebaceous duct where it enters the follicle.1 Sebaceous glands are an almost invariable feature, either present within the lining of the cyst (Figure 4) or in the adjacent tissue.2 In comparison, dermoid cysts may have a red wavy cuticle but also will usually have terminal hair follicles or eccrine or apocrine glands within the wall of the cyst. Steatocystomas typically are collapsed and empty or only contain sebaceous debris (Figure 4) rather than the lamellar keratin seen in dermoid and epidermoid inclusion cysts. Steatocystomas can occur as solitary (steatocystoma simplex) or multiple (steatocystoma multiplex) lesions.1,3 They are clinically comprised of small dome-shaped papules that often are translucent and yellow. These cysts are commonly found on the sternum of males and the axillae or groin of females.2
|
|
| |
Figure 3. Eccrine hidrocystoma with clear contents and lined by 2 layers of cuboidal epithelial cells (H&E, original magnification ×100). | Figure 4. Steatocystoma with a red wavy cuticle, sparse sebaceous contents, and sebaceous glands within the lining (H&E, original magnification ×100). |
|
The best diagnosis is:
a. bronchogenic cyst
b. dermoid cyst
c. epidermal inclusion cyst
d. hidrocystoma
e. steatocystoma
|
|
|
| H&E, original magnification ×40. |
|
|
| H&E, original magnification ×100. |
Continue to the next page for the diagnosis >>
Dermoid Cyst
Dermoid cysts often present clinically as firm subcutaneous nodules on the head or neck in young children. They tend to arise along the lateral aspect of the eyebrow but also can occur on the nose, forehead, neck, chest, or scalp.1 Dermoid cysts are thought to arise from the sequestration of ectodermal tissues along the embryonic fusion planes during development.2 As such, they represent congenital defects and often are identified at birth; however, some are not noticed until much later when they enlarge or become inflamed or infected. Midline dermoid cysts may be associated with underlying dysraphism or intracranial extension.3,4 Thus, any midline lesion warrants evaluation that incorporates imaging with computed tomography or magnetic resonance imaging.4,5 Histologically, dermoid cysts are lined by a keratinizing stratified squamous epithelium (quiz image A), but the lining may be brightly eosinophilic and wavy resembling shark teeth.1,3 The wall of a dermoid cyst commonly contains mature adnexal structures such as terminal hair follicles, sebaceous glands, apocrine glands, and/or eccrine glands (quiz image B).1 Smooth muscle also may be seen within the lining; however, bone and cartilage are not commonly reported in dermoid cysts.2 Lamellar keratin is typical of the cyst contents, and terminal hair shafts also are sometimes noted within the cystic space (quiz image B).1,2 Treatment options include excision at the time of diagnosis or close clinical monitoring with subsequent excision if the lesion grows or becomes symptomatic.4,5 Many practitioners opt to excise these cysts at diagnosis, as untreated lesions are at risk for infection and/or inflammation or may be cosmetically deforming.6,7 Surgical resection, including removal of the wall of the cyst, is curative and reoccurrence is rare.5
|
| |
Figure 1. Bronchogenic cyst demonstrating a ciliated pseudostratified epithelial lining encircled by smooth muscle (H&E, original magnification ×200). | |
|
| |
| Figure 2. Epidermal inclusion cyst containing loose lamellar keratin and a lining that closely resembles the surface epidermis (H&E, original magnification ×40). |
|
Bronchogenic cysts demonstrate an epithelial lining that often is pseudostratified cuboidal or columnar as well as ciliated (Figure 1). Goblet cells are present in the lining in approximately 50% of cases. Smooth muscle may be seen circumferentially surrounding the cyst lining, and rare cases also contain cartilage.1 In contrast to dermoid cysts, other types of adnexal structures are not found within the lining. Bronchogenic cysts that arise in the skin are extremely rare.2 These cysts are thought to arise from respiratory epithelium that has been sequestered during embryologic formation of the tracheobronchial tree. They often are seen overlying the suprasternal notch and occasionally are found on the anterior aspect of the neck or chin. These cysts also are present at birth, similar to dermoid cysts.3
Epidermal inclusion cysts have a lining that histologically bears close resemblance to the surface epidermis. These cysts contain loose lamellar keratin, similar to a dermoid cyst. In contrast, the lining of an epidermal inclusion cyst will lack adnexal structures (Figure 2).1 Clinically, epidermal inclusion cysts often present as smooth, dome-shaped papules and nodules with a central punctum. They are classically found on the face, neck, and trunk. These cysts are thought to arise after a traumatic insult to the pilosebaceous unit.2
Hidrocystomas can be apocrine or eccrine.3 Eccrine hidrocystomas are unilocular cysts that are lined by 2 layers of flattened to cuboidal epithelial cells (Figure 3). The cysts are filled with clear fluid and often are found adjacent to normal eccrine glands.1 Apocrine hidrocystomas are unilocular or multilocular cysts that are lined by 1 to several layers of epithelial cells. The lining of an apocrine hidrocystoma will often exhibit luminal decapitation secretion.3 Apocrine and eccrine hidrocystomas are clinically identical and appear as blue translucent papules on the cheeks or eyelids of adults.1-3 They usually occur periorbitally but also can be seen on the trunk, popliteal fossa, external ears, or vulva. Eccrine hidrocystomas can wax and wane in accordance with the amount of sweat produced; thus, they often expand in size during the summer months.2
Steatocystomas, or simple sebaceous duct cysts, histologically demonstrate a characteristically wavy and eosinophilic cuticle resembling shark teeth (Figure 4) similar to the lining of the sebaceous duct where it enters the follicle.1 Sebaceous glands are an almost invariable feature, either present within the lining of the cyst (Figure 4) or in the adjacent tissue.2 In comparison, dermoid cysts may have a red wavy cuticle but also will usually have terminal hair follicles or eccrine or apocrine glands within the wall of the cyst. Steatocystomas typically are collapsed and empty or only contain sebaceous debris (Figure 4) rather than the lamellar keratin seen in dermoid and epidermoid inclusion cysts. Steatocystomas can occur as solitary (steatocystoma simplex) or multiple (steatocystoma multiplex) lesions.1,3 They are clinically comprised of small dome-shaped papules that often are translucent and yellow. These cysts are commonly found on the sternum of males and the axillae or groin of females.2
|
|
| |
Figure 3. Eccrine hidrocystoma with clear contents and lined by 2 layers of cuboidal epithelial cells (H&E, original magnification ×100). | Figure 4. Steatocystoma with a red wavy cuticle, sparse sebaceous contents, and sebaceous glands within the lining (H&E, original magnification ×100). |
|
1. Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
2. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin. 4th ed. St Louis, MO: Elsevier/Saunders; 2012.
3. Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012.
4. Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
5. Sorenson EP, Powel JE, Rozzelle CJ, et al. Scalp dermoids: a review of their anatomy, diagnosis, and treatment. Childs Nerv Syst. 2013;29:375-380.
6. Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolarynol Head Neck Surg. 2005;132:938-942.
7. Abou-Rayyah Y, Rose GE, Konrad H, et al. Clinical, radiological and pathological examination of periocular dermoid cysts: evidence of inflammation from an early age. Eye (Lond). 2002;16:507-512.
1. Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
2. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin. 4th ed. St Louis, MO: Elsevier/Saunders; 2012.
3. Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012.
4. Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
5. Sorenson EP, Powel JE, Rozzelle CJ, et al. Scalp dermoids: a review of their anatomy, diagnosis, and treatment. Childs Nerv Syst. 2013;29:375-380.
6. Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolarynol Head Neck Surg. 2005;132:938-942.
7. Abou-Rayyah Y, Rose GE, Konrad H, et al. Clinical, radiological and pathological examination of periocular dermoid cysts: evidence of inflammation from an early age. Eye (Lond). 2002;16:507-512.
Onychomatricoma: A Rare Case of Unguioblastic Fibroma of the Fingernail Associated With Trauma
Onychomatricoma (OM) is a rare benign neoplasm of the nail matrix. Even less common is its possible association with both trauma to the nail apparatus and onychomycosis. This case illustrates both of these findings.
Case Report
A 72-year-old white man presented to the dermatology clinic with a 26-year history of a thickened nail plate on the right third finger that had developed soon after a baseball injury. The patient reported that the nail was completely normal prior to the trauma. According to the patient, the distal aspect of the finger was directly hit by a baseball and subsequently was wrapped by the patient for a few weeks. The nail then turned black and eventually fell off. When the nail grew back, it appeared abnormal and in its current state. The patient stated the lesion was asymptomatic at the time of presentation.
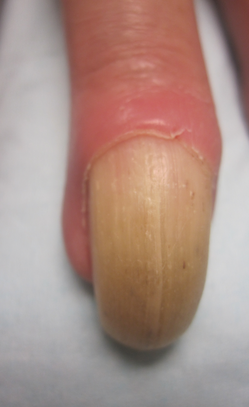
Physical examination revealed thickening, yellow discoloration, and transverse overcurvature of the nail plate on the right third finger with longitudinal ridging (Figure 1). A culture of the nail plate grew Chaetomium species. Application of topical clotrimazole for 3 months followed by a 6-week course of oral terbinafine produced no improvement. The patient then consented to a nail matrix incisional biopsy 6 months after initial presentation. After a digital nerve block was administered and a tourniquet of the proximal digit was applied, a nail avulsion was performed. Subsequently, a 3-mm punch biopsy was taken of the clinically apparent tumor in the nail matrix.
On microscopic examination of the removed tissue, a benign mixed epithelial and stromal proliferative lesion was noted. The basaloid epithelium, lacking a granular layer, arose from the surface epithelial layer and formed a reticulated pattern extending into the stromal component, which was moderately cellular with spindle to fusiform nuclei dissecting between collagen bundles arranged in parallel arrays (Figure 2). The stromal component predominated over the epithelial component in this neoplasm. The nail was preserved in formalin and underwent hematoxylin and eosin staining. It was thickened and grossly showed filiform fibrous projections extending into the nail plate. Histologically, the nail displayed prominent oval clear channels. Periodic acid–Schiff staining was negative for fungal organisms.
A diagnosis of unguioblastic fibroma–type OM was made. After receiving the diagnosis, expected course, and treatment options, the patient was offered conservative surgical excision but preferred clinical monitoring. At his last visit (6 months after the biopsy), the nail plate distal to the biopsy site had thinning and improvement, while the nail plate distal to the matrix that was not removed continued to show thickening, yellow discoloration, overcurvature, and longitudinal ridging (Figure 3).
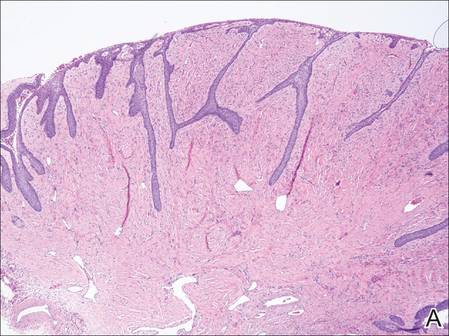
| 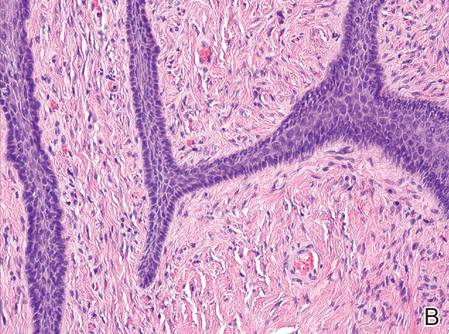
| |
Figure 2. The basaloid epithelium arose from the surface epithelial layer and formed a reticulated pattern extending into the stromal component (A)(H&E, original magnification ×2). At higher magnification, the stromal component was moderately cellular with spindle to fusiform nuclei dissecting between collagen bundles arranged in parallel arrays (B)(H&E, original magnification ×10). | ||
|
|
Comment
Onychomatricoma is a rare tumor originating from the nail matrix. The tumor was first described by Baran and Kint1 in 1992 using the term onychomatrixoma, but later the term onychomatricoma became more widely used.2 Onychomatricomas are more common in adults (mean age, 48 years) and white individuals with no gender predilection.3,4 Fingernail involvement is twice as common as toenail involvement.3 Onychomatricoma is the only tumor that actively produces a nail plate.4
Clinically, OM presents with yellow discoloration along the entire nail plate and proximal splinter hemorrhages. It has a tendency toward transverse overcurvature of the nail plate with prominent longitudinal ridging.4 Trauma has been associated in at least 3 cases reported in the literature, though the association was sometimes weak.3,4 Xanthonychia and onychodystrophy of the nail are common.3 Pterygium, melanonychia, nail bleeding, and cutaneous horns have been reported but are rare.3-5 The tumor typically is painless with no radiographic bone involvement.3 Onychomycosis can be present,3 which may either be a predisposing factor for the tumor or secondary due to the deformed nail plate.4
When the nail plate is avulsed and the proximal nail fold is turned back, the matrix tumor is exposed. This polypoid and filiform tumor has characteristic fingerlike fibrokeratogenous projections extending from the nail matrix into the nail plate.3
Histologically, the tumor is fibroepithelial or biphasic with stromal and epithelial components. It has a lobulated and papillary growth pattern with 2 distinct areas that correspond to 2 anatomic zones.3 The base of the tumor corresponds to the proximal anatomic zone, which begins at the root of the nail and extends to the cuticle. This area is composed of V-shaped keratinous zones similar to the normal matrix. If the nail is removed prior to excision, these areas can be avulsed, leaving clear clefts. The superficial aspect of the tumor corresponds to the distal anatomic zone, which is located in the region of the lunula. This area is composed of multiple digitate or fingerlike projections with a fibrous core and a thick matrical epithelial covering.3 These digitations extend into small cavities in the nail plate, which can be visualized as clear channels or woodwormlike holes in hematoxylin and eosin–stained specimens. A biphasic fibrous stroma also can be observed with the superficial dermis being cellular with fibrillary collagen and the deep dermis more hypocellular with thicker collagen bundles.3,4
An analysis of keratins in the nail matrix, bed, and isthmus showed that OM has the capacity to recapitulate the entire nail unit with differentiation toward the nail bed and isthmus.6 It appears that the mesenchymal component has an inductive effect that can lead to complete epithelial onychogenic differentiation.6
Due to the histological differences among the described cases of OM in the literature, a new classification based on the spectrum of epithelial to stromal ratio of stromal cellularity and the extent of nuclear pleomorphism was proposed in 2004.7 The prominent feature of the unguioblastoma type of OM is epithelial, while the cellular stroma is the prominent feature in the unguioblastic fibroma type. Atypical unguioblastic fibroma refers to a tumor with increased mitotic activity and nuclear pleomorphism among the stroma.7
Most OM tumors follow a benign clinical course; however, complete excision is advised to include the normal nail matrix proximal to the lesion, which may prevent recurrence and serves as a primary treatment.
Conclusion
Onychomatricoma is a benign neoplasm of the nail matrix that may be triggered by trauma; however, due to the weak association, further observations and studies should be conducted to substantiate this possibility. Patients with the classic clinical presentation possibly may be spared a nail avulsion and biopsy. Onychomycosis occurs in the setting of OM, and culture and treatment are unlikely to change the appearance or course of this nail condition.
1. Baran R, Kint A. Onychomatrixoma. filamentous tufted tumour in the matrix of a funnel-shaped nail: a new entity (report of three cases). Br J Dermatol. 1992;126:510-515.
2. Haneke E, Franken J. Onychomatricoma. Dermatol Surg. 1995;21:984-987.
3. Gaertner EM, Gordon M, Reed T. Onychomatricoma: case report of an unusual subungual tumor with literature review. J Cutan Pathol. 2009;36(suppl 1):66-69.
4. Cañueto J, Santos-Briz Á, García JL, et al. Onychomatricoma: genome-wide analyses of a rare nail matrix tumor. J Am Acad Dermatol. 2011;64:573-578.
5. Perrin C, Baran R. Onychomatricoma with dorsalpterygium: pathogenic mechanisms in 3 cases. J Am Acad Dermatol. 2008;59:990-994.
6. Perrin C, Langbein L, Schweizer J, et al. Onychomatricoma in the light of the microanatomy of the normal nail unit. Am J Dermatopathol. 2011;33:131-139.
7. Ko CJ, Shi L, Barr RJ, et al. Unguioblastoma and unguioblastic fibroma—an expanded spectrum of onychomatricoma. J Cutan Pathol. 2004;31:307-311.
Onychomatricoma (OM) is a rare benign neoplasm of the nail matrix. Even less common is its possible association with both trauma to the nail apparatus and onychomycosis. This case illustrates both of these findings.
Case Report
A 72-year-old white man presented to the dermatology clinic with a 26-year history of a thickened nail plate on the right third finger that had developed soon after a baseball injury. The patient reported that the nail was completely normal prior to the trauma. According to the patient, the distal aspect of the finger was directly hit by a baseball and subsequently was wrapped by the patient for a few weeks. The nail then turned black and eventually fell off. When the nail grew back, it appeared abnormal and in its current state. The patient stated the lesion was asymptomatic at the time of presentation.
Physical examination revealed thickening, yellow discoloration, and transverse overcurvature of the nail plate on the right third finger with longitudinal ridging (Figure 1). A culture of the nail plate grew Chaetomium species. Application of topical clotrimazole for 3 months followed by a 6-week course of oral terbinafine produced no improvement. The patient then consented to a nail matrix incisional biopsy 6 months after initial presentation. After a digital nerve block was administered and a tourniquet of the proximal digit was applied, a nail avulsion was performed. Subsequently, a 3-mm punch biopsy was taken of the clinically apparent tumor in the nail matrix.
On microscopic examination of the removed tissue, a benign mixed epithelial and stromal proliferative lesion was noted. The basaloid epithelium, lacking a granular layer, arose from the surface epithelial layer and formed a reticulated pattern extending into the stromal component, which was moderately cellular with spindle to fusiform nuclei dissecting between collagen bundles arranged in parallel arrays (Figure 2). The stromal component predominated over the epithelial component in this neoplasm. The nail was preserved in formalin and underwent hematoxylin and eosin staining. It was thickened and grossly showed filiform fibrous projections extending into the nail plate. Histologically, the nail displayed prominent oval clear channels. Periodic acid–Schiff staining was negative for fungal organisms.
A diagnosis of unguioblastic fibroma–type OM was made. After receiving the diagnosis, expected course, and treatment options, the patient was offered conservative surgical excision but preferred clinical monitoring. At his last visit (6 months after the biopsy), the nail plate distal to the biopsy site had thinning and improvement, while the nail plate distal to the matrix that was not removed continued to show thickening, yellow discoloration, overcurvature, and longitudinal ridging (Figure 3).
|
|
| |
Figure 2. The basaloid epithelium arose from the surface epithelial layer and formed a reticulated pattern extending into the stromal component (A)(H&E, original magnification ×2). At higher magnification, the stromal component was moderately cellular with spindle to fusiform nuclei dissecting between collagen bundles arranged in parallel arrays (B)(H&E, original magnification ×10). | ||
|
|
Comment
Onychomatricoma is a rare tumor originating from the nail matrix. The tumor was first described by Baran and Kint1 in 1992 using the term onychomatrixoma, but later the term onychomatricoma became more widely used.2 Onychomatricomas are more common in adults (mean age, 48 years) and white individuals with no gender predilection.3,4 Fingernail involvement is twice as common as toenail involvement.3 Onychomatricoma is the only tumor that actively produces a nail plate.4
Clinically, OM presents with yellow discoloration along the entire nail plate and proximal splinter hemorrhages. It has a tendency toward transverse overcurvature of the nail plate with prominent longitudinal ridging.4 Trauma has been associated in at least 3 cases reported in the literature, though the association was sometimes weak.3,4 Xanthonychia and onychodystrophy of the nail are common.3 Pterygium, melanonychia, nail bleeding, and cutaneous horns have been reported but are rare.3-5 The tumor typically is painless with no radiographic bone involvement.3 Onychomycosis can be present,3 which may either be a predisposing factor for the tumor or secondary due to the deformed nail plate.4
When the nail plate is avulsed and the proximal nail fold is turned back, the matrix tumor is exposed. This polypoid and filiform tumor has characteristic fingerlike fibrokeratogenous projections extending from the nail matrix into the nail plate.3
Histologically, the tumor is fibroepithelial or biphasic with stromal and epithelial components. It has a lobulated and papillary growth pattern with 2 distinct areas that correspond to 2 anatomic zones.3 The base of the tumor corresponds to the proximal anatomic zone, which begins at the root of the nail and extends to the cuticle. This area is composed of V-shaped keratinous zones similar to the normal matrix. If the nail is removed prior to excision, these areas can be avulsed, leaving clear clefts. The superficial aspect of the tumor corresponds to the distal anatomic zone, which is located in the region of the lunula. This area is composed of multiple digitate or fingerlike projections with a fibrous core and a thick matrical epithelial covering.3 These digitations extend into small cavities in the nail plate, which can be visualized as clear channels or woodwormlike holes in hematoxylin and eosin–stained specimens. A biphasic fibrous stroma also can be observed with the superficial dermis being cellular with fibrillary collagen and the deep dermis more hypocellular with thicker collagen bundles.3,4
An analysis of keratins in the nail matrix, bed, and isthmus showed that OM has the capacity to recapitulate the entire nail unit with differentiation toward the nail bed and isthmus.6 It appears that the mesenchymal component has an inductive effect that can lead to complete epithelial onychogenic differentiation.6
Due to the histological differences among the described cases of OM in the literature, a new classification based on the spectrum of epithelial to stromal ratio of stromal cellularity and the extent of nuclear pleomorphism was proposed in 2004.7 The prominent feature of the unguioblastoma type of OM is epithelial, while the cellular stroma is the prominent feature in the unguioblastic fibroma type. Atypical unguioblastic fibroma refers to a tumor with increased mitotic activity and nuclear pleomorphism among the stroma.7
Most OM tumors follow a benign clinical course; however, complete excision is advised to include the normal nail matrix proximal to the lesion, which may prevent recurrence and serves as a primary treatment.
Conclusion
Onychomatricoma is a benign neoplasm of the nail matrix that may be triggered by trauma; however, due to the weak association, further observations and studies should be conducted to substantiate this possibility. Patients with the classic clinical presentation possibly may be spared a nail avulsion and biopsy. Onychomycosis occurs in the setting of OM, and culture and treatment are unlikely to change the appearance or course of this nail condition.
Onychomatricoma (OM) is a rare benign neoplasm of the nail matrix. Even less common is its possible association with both trauma to the nail apparatus and onychomycosis. This case illustrates both of these findings.
Case Report
A 72-year-old white man presented to the dermatology clinic with a 26-year history of a thickened nail plate on the right third finger that had developed soon after a baseball injury. The patient reported that the nail was completely normal prior to the trauma. According to the patient, the distal aspect of the finger was directly hit by a baseball and subsequently was wrapped by the patient for a few weeks. The nail then turned black and eventually fell off. When the nail grew back, it appeared abnormal and in its current state. The patient stated the lesion was asymptomatic at the time of presentation.
Physical examination revealed thickening, yellow discoloration, and transverse overcurvature of the nail plate on the right third finger with longitudinal ridging (Figure 1). A culture of the nail plate grew Chaetomium species. Application of topical clotrimazole for 3 months followed by a 6-week course of oral terbinafine produced no improvement. The patient then consented to a nail matrix incisional biopsy 6 months after initial presentation. After a digital nerve block was administered and a tourniquet of the proximal digit was applied, a nail avulsion was performed. Subsequently, a 3-mm punch biopsy was taken of the clinically apparent tumor in the nail matrix.
On microscopic examination of the removed tissue, a benign mixed epithelial and stromal proliferative lesion was noted. The basaloid epithelium, lacking a granular layer, arose from the surface epithelial layer and formed a reticulated pattern extending into the stromal component, which was moderately cellular with spindle to fusiform nuclei dissecting between collagen bundles arranged in parallel arrays (Figure 2). The stromal component predominated over the epithelial component in this neoplasm. The nail was preserved in formalin and underwent hematoxylin and eosin staining. It was thickened and grossly showed filiform fibrous projections extending into the nail plate. Histologically, the nail displayed prominent oval clear channels. Periodic acid–Schiff staining was negative for fungal organisms.
A diagnosis of unguioblastic fibroma–type OM was made. After receiving the diagnosis, expected course, and treatment options, the patient was offered conservative surgical excision but preferred clinical monitoring. At his last visit (6 months after the biopsy), the nail plate distal to the biopsy site had thinning and improvement, while the nail plate distal to the matrix that was not removed continued to show thickening, yellow discoloration, overcurvature, and longitudinal ridging (Figure 3).
|
|
| |
Figure 2. The basaloid epithelium arose from the surface epithelial layer and formed a reticulated pattern extending into the stromal component (A)(H&E, original magnification ×2). At higher magnification, the stromal component was moderately cellular with spindle to fusiform nuclei dissecting between collagen bundles arranged in parallel arrays (B)(H&E, original magnification ×10). | ||
|
|
Comment
Onychomatricoma is a rare tumor originating from the nail matrix. The tumor was first described by Baran and Kint1 in 1992 using the term onychomatrixoma, but later the term onychomatricoma became more widely used.2 Onychomatricomas are more common in adults (mean age, 48 years) and white individuals with no gender predilection.3,4 Fingernail involvement is twice as common as toenail involvement.3 Onychomatricoma is the only tumor that actively produces a nail plate.4
Clinically, OM presents with yellow discoloration along the entire nail plate and proximal splinter hemorrhages. It has a tendency toward transverse overcurvature of the nail plate with prominent longitudinal ridging.4 Trauma has been associated in at least 3 cases reported in the literature, though the association was sometimes weak.3,4 Xanthonychia and onychodystrophy of the nail are common.3 Pterygium, melanonychia, nail bleeding, and cutaneous horns have been reported but are rare.3-5 The tumor typically is painless with no radiographic bone involvement.3 Onychomycosis can be present,3 which may either be a predisposing factor for the tumor or secondary due to the deformed nail plate.4
When the nail plate is avulsed and the proximal nail fold is turned back, the matrix tumor is exposed. This polypoid and filiform tumor has characteristic fingerlike fibrokeratogenous projections extending from the nail matrix into the nail plate.3
Histologically, the tumor is fibroepithelial or biphasic with stromal and epithelial components. It has a lobulated and papillary growth pattern with 2 distinct areas that correspond to 2 anatomic zones.3 The base of the tumor corresponds to the proximal anatomic zone, which begins at the root of the nail and extends to the cuticle. This area is composed of V-shaped keratinous zones similar to the normal matrix. If the nail is removed prior to excision, these areas can be avulsed, leaving clear clefts. The superficial aspect of the tumor corresponds to the distal anatomic zone, which is located in the region of the lunula. This area is composed of multiple digitate or fingerlike projections with a fibrous core and a thick matrical epithelial covering.3 These digitations extend into small cavities in the nail plate, which can be visualized as clear channels or woodwormlike holes in hematoxylin and eosin–stained specimens. A biphasic fibrous stroma also can be observed with the superficial dermis being cellular with fibrillary collagen and the deep dermis more hypocellular with thicker collagen bundles.3,4
An analysis of keratins in the nail matrix, bed, and isthmus showed that OM has the capacity to recapitulate the entire nail unit with differentiation toward the nail bed and isthmus.6 It appears that the mesenchymal component has an inductive effect that can lead to complete epithelial onychogenic differentiation.6
Due to the histological differences among the described cases of OM in the literature, a new classification based on the spectrum of epithelial to stromal ratio of stromal cellularity and the extent of nuclear pleomorphism was proposed in 2004.7 The prominent feature of the unguioblastoma type of OM is epithelial, while the cellular stroma is the prominent feature in the unguioblastic fibroma type. Atypical unguioblastic fibroma refers to a tumor with increased mitotic activity and nuclear pleomorphism among the stroma.7
Most OM tumors follow a benign clinical course; however, complete excision is advised to include the normal nail matrix proximal to the lesion, which may prevent recurrence and serves as a primary treatment.
Conclusion
Onychomatricoma is a benign neoplasm of the nail matrix that may be triggered by trauma; however, due to the weak association, further observations and studies should be conducted to substantiate this possibility. Patients with the classic clinical presentation possibly may be spared a nail avulsion and biopsy. Onychomycosis occurs in the setting of OM, and culture and treatment are unlikely to change the appearance or course of this nail condition.
1. Baran R, Kint A. Onychomatrixoma. filamentous tufted tumour in the matrix of a funnel-shaped nail: a new entity (report of three cases). Br J Dermatol. 1992;126:510-515.
2. Haneke E, Franken J. Onychomatricoma. Dermatol Surg. 1995;21:984-987.
3. Gaertner EM, Gordon M, Reed T. Onychomatricoma: case report of an unusual subungual tumor with literature review. J Cutan Pathol. 2009;36(suppl 1):66-69.
4. Cañueto J, Santos-Briz Á, García JL, et al. Onychomatricoma: genome-wide analyses of a rare nail matrix tumor. J Am Acad Dermatol. 2011;64:573-578.
5. Perrin C, Baran R. Onychomatricoma with dorsalpterygium: pathogenic mechanisms in 3 cases. J Am Acad Dermatol. 2008;59:990-994.
6. Perrin C, Langbein L, Schweizer J, et al. Onychomatricoma in the light of the microanatomy of the normal nail unit. Am J Dermatopathol. 2011;33:131-139.
7. Ko CJ, Shi L, Barr RJ, et al. Unguioblastoma and unguioblastic fibroma—an expanded spectrum of onychomatricoma. J Cutan Pathol. 2004;31:307-311.
1. Baran R, Kint A. Onychomatrixoma. filamentous tufted tumour in the matrix of a funnel-shaped nail: a new entity (report of three cases). Br J Dermatol. 1992;126:510-515.
2. Haneke E, Franken J. Onychomatricoma. Dermatol Surg. 1995;21:984-987.
3. Gaertner EM, Gordon M, Reed T. Onychomatricoma: case report of an unusual subungual tumor with literature review. J Cutan Pathol. 2009;36(suppl 1):66-69.
4. Cañueto J, Santos-Briz Á, García JL, et al. Onychomatricoma: genome-wide analyses of a rare nail matrix tumor. J Am Acad Dermatol. 2011;64:573-578.
5. Perrin C, Baran R. Onychomatricoma with dorsalpterygium: pathogenic mechanisms in 3 cases. J Am Acad Dermatol. 2008;59:990-994.
6. Perrin C, Langbein L, Schweizer J, et al. Onychomatricoma in the light of the microanatomy of the normal nail unit. Am J Dermatopathol. 2011;33:131-139.
7. Ko CJ, Shi L, Barr RJ, et al. Unguioblastoma and unguioblastic fibroma—an expanded spectrum of onychomatricoma. J Cutan Pathol. 2004;31:307-311.
Practice Points
- Onychomatricoma is a rare benign neoplasm of the nail matrix that actively produces a nail plate.
- Onychomatricoma should be in the differential diagnosis of a thickened discolored nail plate with transverse overcurvature.
- Onychomatricoma has been associated with onychomycosis and trauma to the nail apparatus.