User login
Exophytic Papule on the Chin of a Child
Exophytic Papule on the Chin of a Child
THE DIAGNOSIS: Rhabdomyomatous Mesenchymal Hamartoma
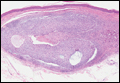
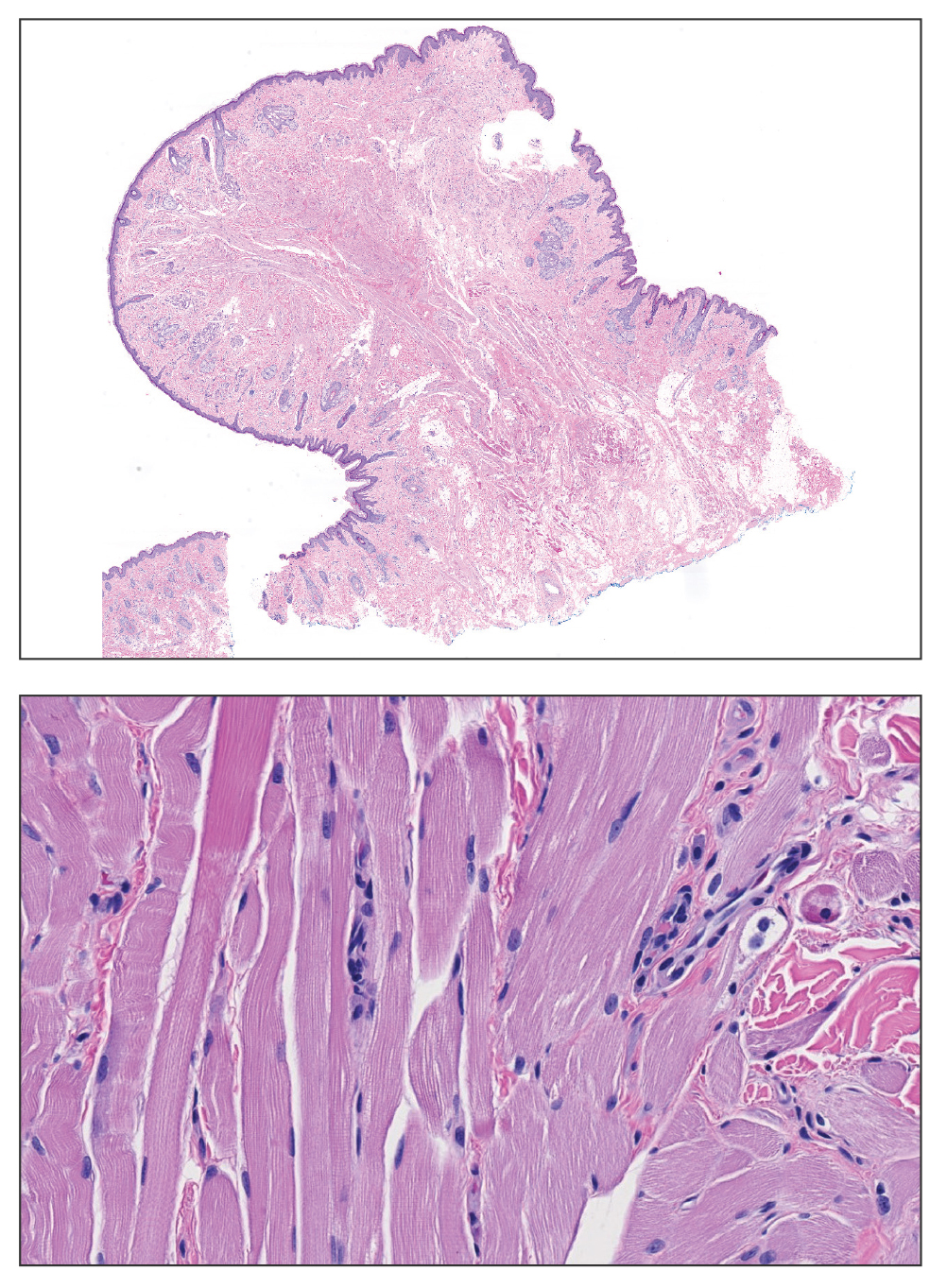
Histopathologic examination of the excised tissue revealed haphazardly arranged bundles of mature striated muscle within the dermis and subcutaneous tissue admixed with adipose tissue, adnexal structures, blood vessels, and nerves. The presence of the lesion since birth, midline clinical presentation, and histologic findings were consistent with a diagnosis of rhabdomyomatous mesenchymal hamartoma (RMH).
Also referred to as striated muscle hamartoma, RMH is a rare benign lesion thought to have embryonic origin due to its midline presentation.1 The etiology of RMH is unknown but is hypothesized to be due to abnormal migration or growth of embryonic mesenchymal tissue. Rhabdomyomatous mesenchymal hamartoma typically manifests in infancy or early childhood as a solitary midline papule on the head or neck, although there have been rare reports of development in adulthood.2-4 Lesions often are polypoid or exophytic but may manifest as smooth papules or subcutaneous nodules.2 Although benign, RMH may be associated with other congenital abnormalities and conditions, such as Delleman syndrome, which is caused by a sporadic genetic abnormality and results in defects of the eye, central nervous system, and skin.5 Treatment for RMH is not needed, but surgical excision for cosmetic purposes can be performed with low risk for recurrence. Histologically, RMH demonstrates a normal epidermis overlying disorganized bundles of skeletal muscle accompanied by varying amounts of other mature dermal and subcutaneous tissues including nerves, blood vessels, adipose tissue, and other adnexal structures.2,6 Myoglobin and desmin are positive within the skeletal muscle bundles.7
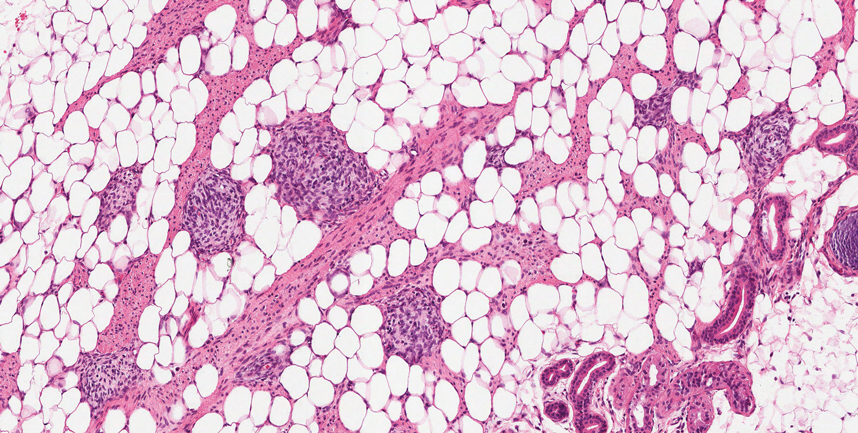
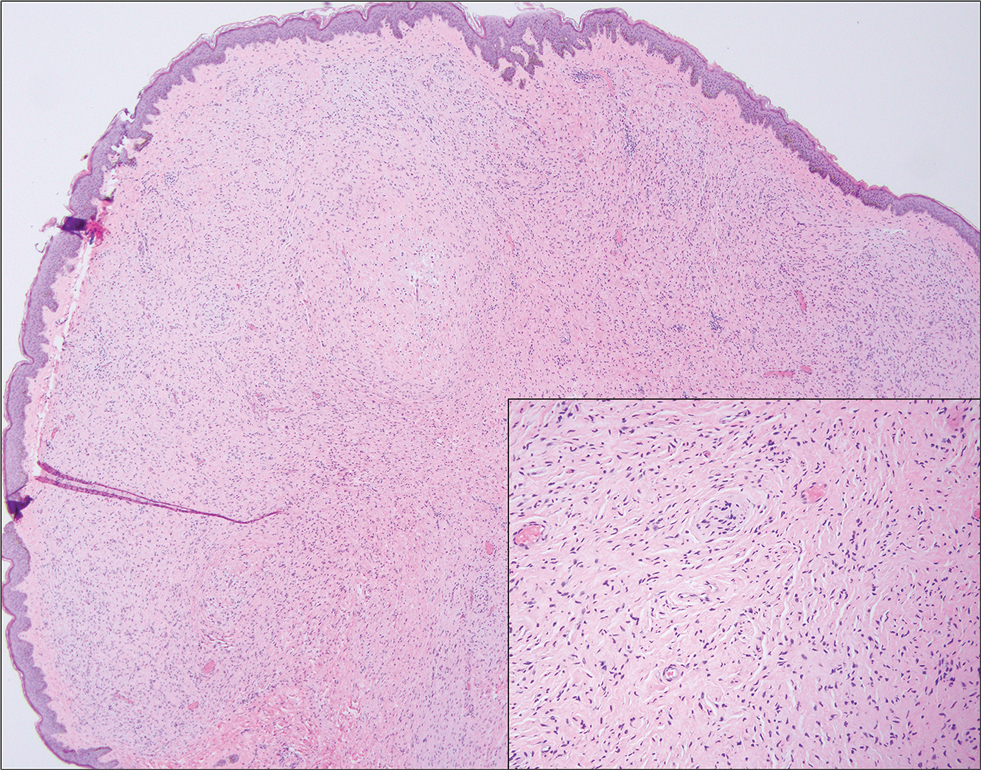
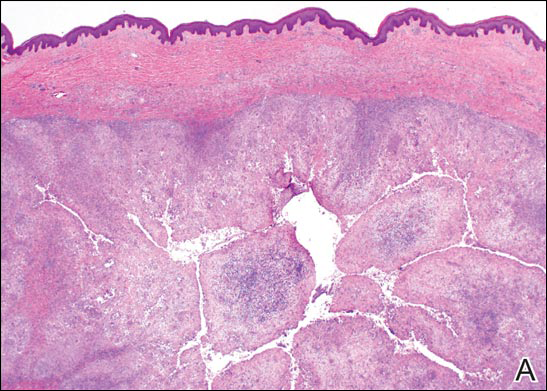
Fibrous hamartoma of infancy (FHI) often manifests as a movable, ill-defined nodule within the subcutaneous tissue. While also occurring in young children—typically within the first 2 years of life—FHI primarily is found on the upper arms, back, and axillae, in contrast to FHI.8 The classic histopathologic presentation of FHI consists of a triphasic morphology consisting of undifferentiated mesenchymal cells and dense fibroblastic/myofibroblastic tissue with mature adipose tissue woven throughout in islands (Figure 1).9 Skeletal muscle is not a component of this tumor.
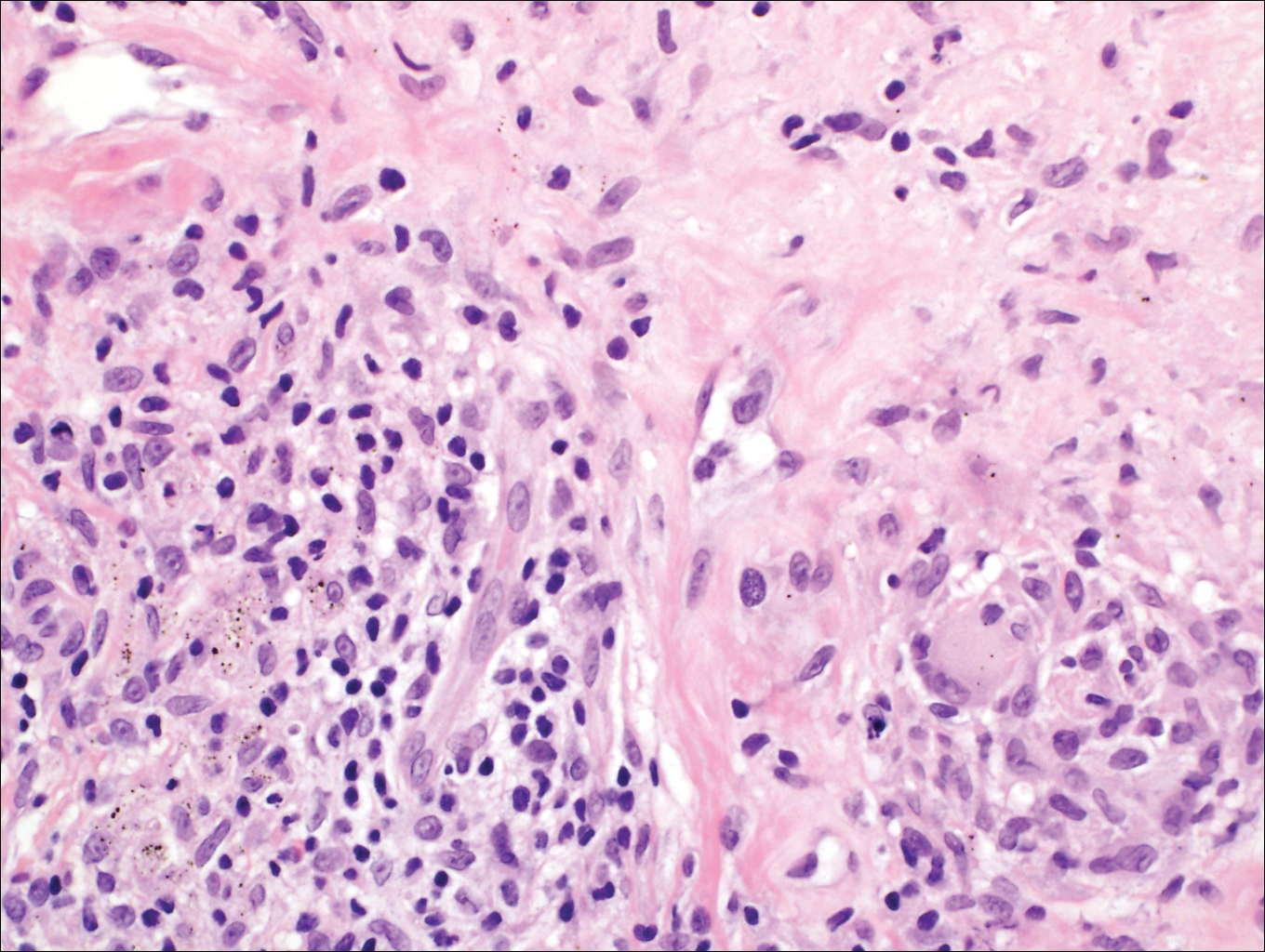
Neurofibromas also may manifest clinically as papules or nodules and arise from the peripheral nerve sheath. There are 3 major subtypes of neurofibromas—localized, diffuse, and plexiform—with the last being strongly associated with neurofibromatosis type 1.10 The plexiform type has a rare risk for malignant transformation. The typical histopathologic finding of a localized cutaneous neurofibroma is a dermal proliferation of spindle cells with wavy nuclei within a variably myxoid stroma (Figure 2).11 Interspersed mast cells also can be seen. A plexiform neurofibroma typically involves multiple nerve fascicles and comprises multinodular or tortuous bundles of cytologically bland spindle cells. Compared to RMH, skeletal muscle is not a component of this tumor.
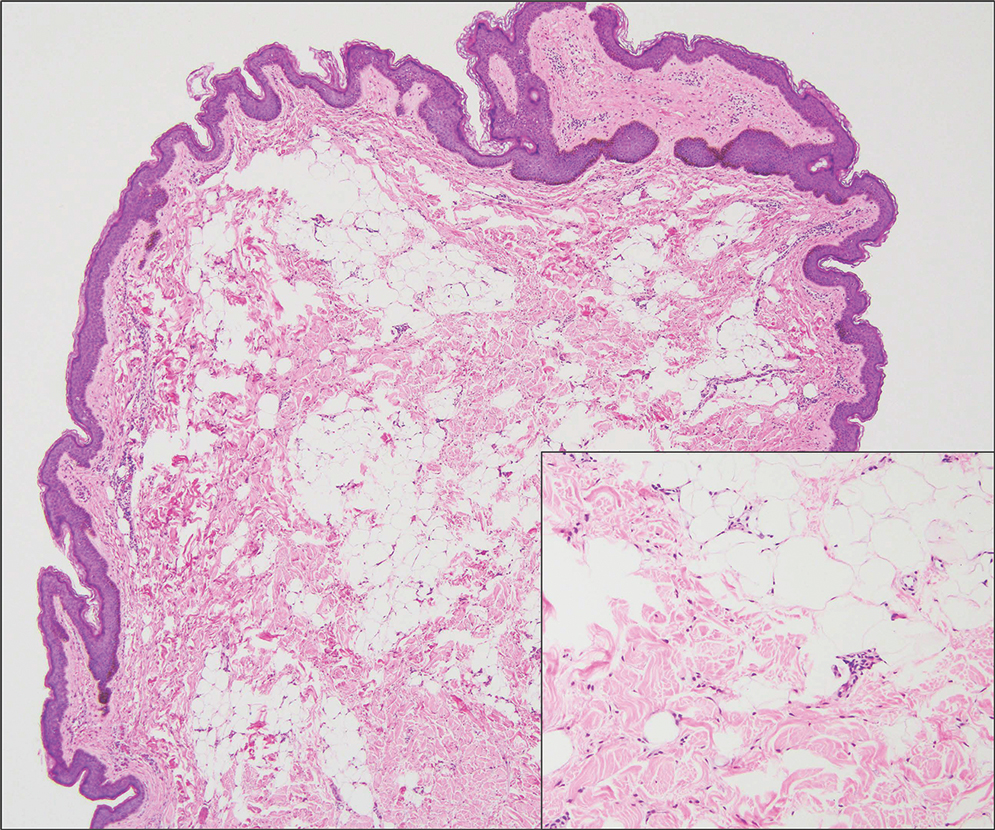
Nevus lipomatosus superficialis is a benign hamartoma that can manifest as a pedunculated or exophytic papule. The lesions may be solitary or multiple and, unlike RMH, are most common on the buttocks, upper thighs, and trunk.12 The histopathologic features of nevus lipomatosus superficialis include clusters of mature adipose tissue in the superficial dermis admixed with collagen fibers and variably increased vasculature (Figure 3).13 Nevus lipomatosus superficialis does not contain skeletal muscle within the tumor in comparison to RMH.
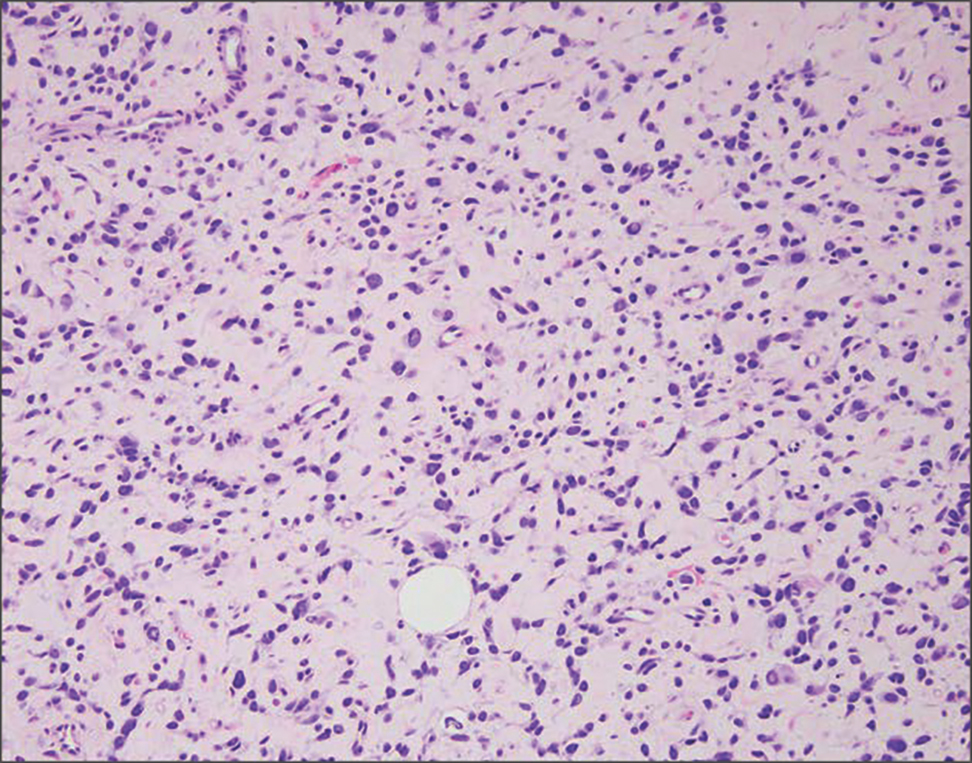
It is important to distinguish rhabdomyosarcoma (RMS) from RMH, as it is associated with increased mortality and morbidity. Rhabdomyosarcoma is the most common soft-tissue sarcoma in children and is derived from mesenchyme with variable degrees of skeletal muscle differentiation.14 Due to its mesenchymal origin, these tumors can manifest in a variety of places but most commonly on the head and neck and in the genital region.15 The most common subtype is embryonal rhabdomyosarcoma. Histologically, embryonal RMS shows a moderately cellular tumor composed of sheets of spindle-shaped or round cells with scant or eosinophilic cytoplasm (Figure 4). The absence of genetic translocation in the paired box-forkhead box protein 01 (PAX-FOXO1) gene helps distinguish it from solid alveolar RMS, the second most common and more aggressive subtype.12 Positive immunohistochemical staining for desmin, myoblast determination protein 1 (MyoD1), and myogenin supports myogenic differentiation.14
- Bernal-Mañas CM, Isaac-Montero MA, Vargas-Uribe MC, et al. Hamartoma mesenquimal rabdomiomatoso [rhabdomyomatous mesenchymal hamartoma]. An Pediatr (Barc). 2013;78:260-262. doi:10.1016/j.anpedi.2012.08.005
- Al Amri R, De Stefano DV, Wang Q, et al. Morphologic spectrum of rhabdomyomatous mesenchymal hamartomas (striated muscle hamartomas) in pediatric dermatopathology. Am J Dermatopathol. 2022;44:170-173. doi:10.1097/DAD.0000000000002062
- Carboni A, Fomin D. A rare adult presentation of a congenital tumor discovered incidentally after trauma. JAAD Case Rep. 2022;31:121-123. doi:10.1016/j.jdcr.2022.10.024
- Chang CP, Chen GS. Rhabdomyomatous mesenchymal hamartoma: a plaque-type variant in an adult. Kaohsiung J Med Sci. 2005; 21(4):185-188. doi:10.1016/S1607-551X(09)70299-2
- Bahmani M, Naseri R, Iraniparast A, et al. Oculocerebrocutaneous syndrome (Delleman syndrome): a case with a novel presentation of orbital involvement. Case Rep Pediatr. 2021;2021:5524131. doi:10.1155/2021/5524131
- Kim H, Chung JH, Sung HM, et al. Rhabdomyomatous mesenchymal hamartoma presenting as a midline mass on a chin. Arch Craniofac Surg. 2017;18:292-295. doi:10.7181/acfs.2017.18.4.292.
- Lin CP, Nguyen JM, Aboutalebi S, et al. Incidental rhabdomyomatous mesenchymal hamartoma. Proc (Bayl Univ Med Cent). 2020;34:161-162. doi:10.1080/08998280.2020.1801087
- Ji Y, Hu P, Zhang C, et al. Fibrous hamartoma of infancy: radiologic features and literature review. BMC Musculoskelet Disord. 2019;20:356. doi:10.1186/s12891-019-2743-5
- Yu G, Wang Y, Wang G, et al. Fibrous hamartoma of infancy: a clinical pathological analysis of seventeen cases. Int J Clin Exp Pathol. 2015;8:3374-3377.
- Ferner RE, O’Doherty MJ. Neurofibroma and schwannoma. Curr Opin Neurol. 2002;15:679-684. doi:10.1097/01.wco.0000044763.39452.aa
- Miettinen MM, Antonescu CR, Fletcher CDM, et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum Pathol. 2017;67:1-10. doi:10.1016/j.humpath.2017.05.010
- Kim RH, Stevenson ML, Hale CS, et al. Nevus lipomatosus superficialis. Dermatol Online J. 2014;20:13030/qt2cb3c5t3.
- Singh P, Anandani GM. Nevus lipomatosus superficialis, an unusual case report. J Family Med Prim Care. 2022;11:4045-4047. doi:10.4103/jfmpc.jfmpc_2352_21
- Shern JF, Yohe ME, Khan J. Pediatric rhabdomyosarcoma. Crit Rev Oncog. 2015;20:227-243. doi:10.1615/critrevoncog.2015013800
- Rogers TN, Dasgupta R. Management of rhabdomyosarcoma in pediatric patients. Surg Oncol Clin N Am. 2021;30:339-353. doi:10.1016/j.soc.2020.11.003
- Machado I, Mayordomo-Aranda E, Giner F, et al. The role of immunohistochemistry in rhabdomyosarcoma diagnosis using tissue microarray technology and a xenograft model. Fetal Pediatr Pathol. 2015;34:271-281. doi:10.3109/15513815.2015.1042604
THE DIAGNOSIS: Rhabdomyomatous Mesenchymal Hamartoma
Histopathologic examination of the excised tissue revealed haphazardly arranged bundles of mature striated muscle within the dermis and subcutaneous tissue admixed with adipose tissue, adnexal structures, blood vessels, and nerves. The presence of the lesion since birth, midline clinical presentation, and histologic findings were consistent with a diagnosis of rhabdomyomatous mesenchymal hamartoma (RMH).
Also referred to as striated muscle hamartoma, RMH is a rare benign lesion thought to have embryonic origin due to its midline presentation.1 The etiology of RMH is unknown but is hypothesized to be due to abnormal migration or growth of embryonic mesenchymal tissue. Rhabdomyomatous mesenchymal hamartoma typically manifests in infancy or early childhood as a solitary midline papule on the head or neck, although there have been rare reports of development in adulthood.2-4 Lesions often are polypoid or exophytic but may manifest as smooth papules or subcutaneous nodules.2 Although benign, RMH may be associated with other congenital abnormalities and conditions, such as Delleman syndrome, which is caused by a sporadic genetic abnormality and results in defects of the eye, central nervous system, and skin.5 Treatment for RMH is not needed, but surgical excision for cosmetic purposes can be performed with low risk for recurrence. Histologically, RMH demonstrates a normal epidermis overlying disorganized bundles of skeletal muscle accompanied by varying amounts of other mature dermal and subcutaneous tissues including nerves, blood vessels, adipose tissue, and other adnexal structures.2,6 Myoglobin and desmin are positive within the skeletal muscle bundles.7
Fibrous hamartoma of infancy (FHI) often manifests as a movable, ill-defined nodule within the subcutaneous tissue. While also occurring in young children—typically within the first 2 years of life—FHI primarily is found on the upper arms, back, and axillae, in contrast to FHI.8 The classic histopathologic presentation of FHI consists of a triphasic morphology consisting of undifferentiated mesenchymal cells and dense fibroblastic/myofibroblastic tissue with mature adipose tissue woven throughout in islands (Figure 1).9 Skeletal muscle is not a component of this tumor.
Neurofibromas also may manifest clinically as papules or nodules and arise from the peripheral nerve sheath. There are 3 major subtypes of neurofibromas—localized, diffuse, and plexiform—with the last being strongly associated with neurofibromatosis type 1.10 The plexiform type has a rare risk for malignant transformation. The typical histopathologic finding of a localized cutaneous neurofibroma is a dermal proliferation of spindle cells with wavy nuclei within a variably myxoid stroma (Figure 2).11 Interspersed mast cells also can be seen. A plexiform neurofibroma typically involves multiple nerve fascicles and comprises multinodular or tortuous bundles of cytologically bland spindle cells. Compared to RMH, skeletal muscle is not a component of this tumor.
Nevus lipomatosus superficialis is a benign hamartoma that can manifest as a pedunculated or exophytic papule. The lesions may be solitary or multiple and, unlike RMH, are most common on the buttocks, upper thighs, and trunk.12 The histopathologic features of nevus lipomatosus superficialis include clusters of mature adipose tissue in the superficial dermis admixed with collagen fibers and variably increased vasculature (Figure 3).13 Nevus lipomatosus superficialis does not contain skeletal muscle within the tumor in comparison to RMH.
It is important to distinguish rhabdomyosarcoma (RMS) from RMH, as it is associated with increased mortality and morbidity. Rhabdomyosarcoma is the most common soft-tissue sarcoma in children and is derived from mesenchyme with variable degrees of skeletal muscle differentiation.14 Due to its mesenchymal origin, these tumors can manifest in a variety of places but most commonly on the head and neck and in the genital region.15 The most common subtype is embryonal rhabdomyosarcoma. Histologically, embryonal RMS shows a moderately cellular tumor composed of sheets of spindle-shaped or round cells with scant or eosinophilic cytoplasm (Figure 4). The absence of genetic translocation in the paired box-forkhead box protein 01 (PAX-FOXO1) gene helps distinguish it from solid alveolar RMS, the second most common and more aggressive subtype.12 Positive immunohistochemical staining for desmin, myoblast determination protein 1 (MyoD1), and myogenin supports myogenic differentiation.14
THE DIAGNOSIS: Rhabdomyomatous Mesenchymal Hamartoma
Histopathologic examination of the excised tissue revealed haphazardly arranged bundles of mature striated muscle within the dermis and subcutaneous tissue admixed with adipose tissue, adnexal structures, blood vessels, and nerves. The presence of the lesion since birth, midline clinical presentation, and histologic findings were consistent with a diagnosis of rhabdomyomatous mesenchymal hamartoma (RMH).
Also referred to as striated muscle hamartoma, RMH is a rare benign lesion thought to have embryonic origin due to its midline presentation.1 The etiology of RMH is unknown but is hypothesized to be due to abnormal migration or growth of embryonic mesenchymal tissue. Rhabdomyomatous mesenchymal hamartoma typically manifests in infancy or early childhood as a solitary midline papule on the head or neck, although there have been rare reports of development in adulthood.2-4 Lesions often are polypoid or exophytic but may manifest as smooth papules or subcutaneous nodules.2 Although benign, RMH may be associated with other congenital abnormalities and conditions, such as Delleman syndrome, which is caused by a sporadic genetic abnormality and results in defects of the eye, central nervous system, and skin.5 Treatment for RMH is not needed, but surgical excision for cosmetic purposes can be performed with low risk for recurrence. Histologically, RMH demonstrates a normal epidermis overlying disorganized bundles of skeletal muscle accompanied by varying amounts of other mature dermal and subcutaneous tissues including nerves, blood vessels, adipose tissue, and other adnexal structures.2,6 Myoglobin and desmin are positive within the skeletal muscle bundles.7
Fibrous hamartoma of infancy (FHI) often manifests as a movable, ill-defined nodule within the subcutaneous tissue. While also occurring in young children—typically within the first 2 years of life—FHI primarily is found on the upper arms, back, and axillae, in contrast to FHI.8 The classic histopathologic presentation of FHI consists of a triphasic morphology consisting of undifferentiated mesenchymal cells and dense fibroblastic/myofibroblastic tissue with mature adipose tissue woven throughout in islands (Figure 1).9 Skeletal muscle is not a component of this tumor.
Neurofibromas also may manifest clinically as papules or nodules and arise from the peripheral nerve sheath. There are 3 major subtypes of neurofibromas—localized, diffuse, and plexiform—with the last being strongly associated with neurofibromatosis type 1.10 The plexiform type has a rare risk for malignant transformation. The typical histopathologic finding of a localized cutaneous neurofibroma is a dermal proliferation of spindle cells with wavy nuclei within a variably myxoid stroma (Figure 2).11 Interspersed mast cells also can be seen. A plexiform neurofibroma typically involves multiple nerve fascicles and comprises multinodular or tortuous bundles of cytologically bland spindle cells. Compared to RMH, skeletal muscle is not a component of this tumor.
Nevus lipomatosus superficialis is a benign hamartoma that can manifest as a pedunculated or exophytic papule. The lesions may be solitary or multiple and, unlike RMH, are most common on the buttocks, upper thighs, and trunk.12 The histopathologic features of nevus lipomatosus superficialis include clusters of mature adipose tissue in the superficial dermis admixed with collagen fibers and variably increased vasculature (Figure 3).13 Nevus lipomatosus superficialis does not contain skeletal muscle within the tumor in comparison to RMH.
It is important to distinguish rhabdomyosarcoma (RMS) from RMH, as it is associated with increased mortality and morbidity. Rhabdomyosarcoma is the most common soft-tissue sarcoma in children and is derived from mesenchyme with variable degrees of skeletal muscle differentiation.14 Due to its mesenchymal origin, these tumors can manifest in a variety of places but most commonly on the head and neck and in the genital region.15 The most common subtype is embryonal rhabdomyosarcoma. Histologically, embryonal RMS shows a moderately cellular tumor composed of sheets of spindle-shaped or round cells with scant or eosinophilic cytoplasm (Figure 4). The absence of genetic translocation in the paired box-forkhead box protein 01 (PAX-FOXO1) gene helps distinguish it from solid alveolar RMS, the second most common and more aggressive subtype.12 Positive immunohistochemical staining for desmin, myoblast determination protein 1 (MyoD1), and myogenin supports myogenic differentiation.14
- Bernal-Mañas CM, Isaac-Montero MA, Vargas-Uribe MC, et al. Hamartoma mesenquimal rabdomiomatoso [rhabdomyomatous mesenchymal hamartoma]. An Pediatr (Barc). 2013;78:260-262. doi:10.1016/j.anpedi.2012.08.005
- Al Amri R, De Stefano DV, Wang Q, et al. Morphologic spectrum of rhabdomyomatous mesenchymal hamartomas (striated muscle hamartomas) in pediatric dermatopathology. Am J Dermatopathol. 2022;44:170-173. doi:10.1097/DAD.0000000000002062
- Carboni A, Fomin D. A rare adult presentation of a congenital tumor discovered incidentally after trauma. JAAD Case Rep. 2022;31:121-123. doi:10.1016/j.jdcr.2022.10.024
- Chang CP, Chen GS. Rhabdomyomatous mesenchymal hamartoma: a plaque-type variant in an adult. Kaohsiung J Med Sci. 2005; 21(4):185-188. doi:10.1016/S1607-551X(09)70299-2
- Bahmani M, Naseri R, Iraniparast A, et al. Oculocerebrocutaneous syndrome (Delleman syndrome): a case with a novel presentation of orbital involvement. Case Rep Pediatr. 2021;2021:5524131. doi:10.1155/2021/5524131
- Kim H, Chung JH, Sung HM, et al. Rhabdomyomatous mesenchymal hamartoma presenting as a midline mass on a chin. Arch Craniofac Surg. 2017;18:292-295. doi:10.7181/acfs.2017.18.4.292.
- Lin CP, Nguyen JM, Aboutalebi S, et al. Incidental rhabdomyomatous mesenchymal hamartoma. Proc (Bayl Univ Med Cent). 2020;34:161-162. doi:10.1080/08998280.2020.1801087
- Ji Y, Hu P, Zhang C, et al. Fibrous hamartoma of infancy: radiologic features and literature review. BMC Musculoskelet Disord. 2019;20:356. doi:10.1186/s12891-019-2743-5
- Yu G, Wang Y, Wang G, et al. Fibrous hamartoma of infancy: a clinical pathological analysis of seventeen cases. Int J Clin Exp Pathol. 2015;8:3374-3377.
- Ferner RE, O’Doherty MJ. Neurofibroma and schwannoma. Curr Opin Neurol. 2002;15:679-684. doi:10.1097/01.wco.0000044763.39452.aa
- Miettinen MM, Antonescu CR, Fletcher CDM, et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum Pathol. 2017;67:1-10. doi:10.1016/j.humpath.2017.05.010
- Kim RH, Stevenson ML, Hale CS, et al. Nevus lipomatosus superficialis. Dermatol Online J. 2014;20:13030/qt2cb3c5t3.
- Singh P, Anandani GM. Nevus lipomatosus superficialis, an unusual case report. J Family Med Prim Care. 2022;11:4045-4047. doi:10.4103/jfmpc.jfmpc_2352_21
- Shern JF, Yohe ME, Khan J. Pediatric rhabdomyosarcoma. Crit Rev Oncog. 2015;20:227-243. doi:10.1615/critrevoncog.2015013800
- Rogers TN, Dasgupta R. Management of rhabdomyosarcoma in pediatric patients. Surg Oncol Clin N Am. 2021;30:339-353. doi:10.1016/j.soc.2020.11.003
- Machado I, Mayordomo-Aranda E, Giner F, et al. The role of immunohistochemistry in rhabdomyosarcoma diagnosis using tissue microarray technology and a xenograft model. Fetal Pediatr Pathol. 2015;34:271-281. doi:10.3109/15513815.2015.1042604
- Bernal-Mañas CM, Isaac-Montero MA, Vargas-Uribe MC, et al. Hamartoma mesenquimal rabdomiomatoso [rhabdomyomatous mesenchymal hamartoma]. An Pediatr (Barc). 2013;78:260-262. doi:10.1016/j.anpedi.2012.08.005
- Al Amri R, De Stefano DV, Wang Q, et al. Morphologic spectrum of rhabdomyomatous mesenchymal hamartomas (striated muscle hamartomas) in pediatric dermatopathology. Am J Dermatopathol. 2022;44:170-173. doi:10.1097/DAD.0000000000002062
- Carboni A, Fomin D. A rare adult presentation of a congenital tumor discovered incidentally after trauma. JAAD Case Rep. 2022;31:121-123. doi:10.1016/j.jdcr.2022.10.024
- Chang CP, Chen GS. Rhabdomyomatous mesenchymal hamartoma: a plaque-type variant in an adult. Kaohsiung J Med Sci. 2005; 21(4):185-188. doi:10.1016/S1607-551X(09)70299-2
- Bahmani M, Naseri R, Iraniparast A, et al. Oculocerebrocutaneous syndrome (Delleman syndrome): a case with a novel presentation of orbital involvement. Case Rep Pediatr. 2021;2021:5524131. doi:10.1155/2021/5524131
- Kim H, Chung JH, Sung HM, et al. Rhabdomyomatous mesenchymal hamartoma presenting as a midline mass on a chin. Arch Craniofac Surg. 2017;18:292-295. doi:10.7181/acfs.2017.18.4.292.
- Lin CP, Nguyen JM, Aboutalebi S, et al. Incidental rhabdomyomatous mesenchymal hamartoma. Proc (Bayl Univ Med Cent). 2020;34:161-162. doi:10.1080/08998280.2020.1801087
- Ji Y, Hu P, Zhang C, et al. Fibrous hamartoma of infancy: radiologic features and literature review. BMC Musculoskelet Disord. 2019;20:356. doi:10.1186/s12891-019-2743-5
- Yu G, Wang Y, Wang G, et al. Fibrous hamartoma of infancy: a clinical pathological analysis of seventeen cases. Int J Clin Exp Pathol. 2015;8:3374-3377.
- Ferner RE, O’Doherty MJ. Neurofibroma and schwannoma. Curr Opin Neurol. 2002;15:679-684. doi:10.1097/01.wco.0000044763.39452.aa
- Miettinen MM, Antonescu CR, Fletcher CDM, et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum Pathol. 2017;67:1-10. doi:10.1016/j.humpath.2017.05.010
- Kim RH, Stevenson ML, Hale CS, et al. Nevus lipomatosus superficialis. Dermatol Online J. 2014;20:13030/qt2cb3c5t3.
- Singh P, Anandani GM. Nevus lipomatosus superficialis, an unusual case report. J Family Med Prim Care. 2022;11:4045-4047. doi:10.4103/jfmpc.jfmpc_2352_21
- Shern JF, Yohe ME, Khan J. Pediatric rhabdomyosarcoma. Crit Rev Oncog. 2015;20:227-243. doi:10.1615/critrevoncog.2015013800
- Rogers TN, Dasgupta R. Management of rhabdomyosarcoma in pediatric patients. Surg Oncol Clin N Am. 2021;30:339-353. doi:10.1016/j.soc.2020.11.003
- Machado I, Mayordomo-Aranda E, Giner F, et al. The role of immunohistochemistry in rhabdomyosarcoma diagnosis using tissue microarray technology and a xenograft model. Fetal Pediatr Pathol. 2015;34:271-281. doi:10.3109/15513815.2015.1042604
Exophytic Papule on the Chin of a Child
Exophytic Papule on the Chin of a Child
A 3-year-old boy presented to the dermatology department for evaluation of an asymptomatic papule on the chin that had been present since birth. His medical history was otherwise unremarkable. Physical examination revealed a 4×2-mm, flesh-colored, exophytic papule on the midline chin. An excisional biopsy was performed.

Healthy infant with a blistering rash
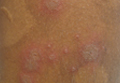
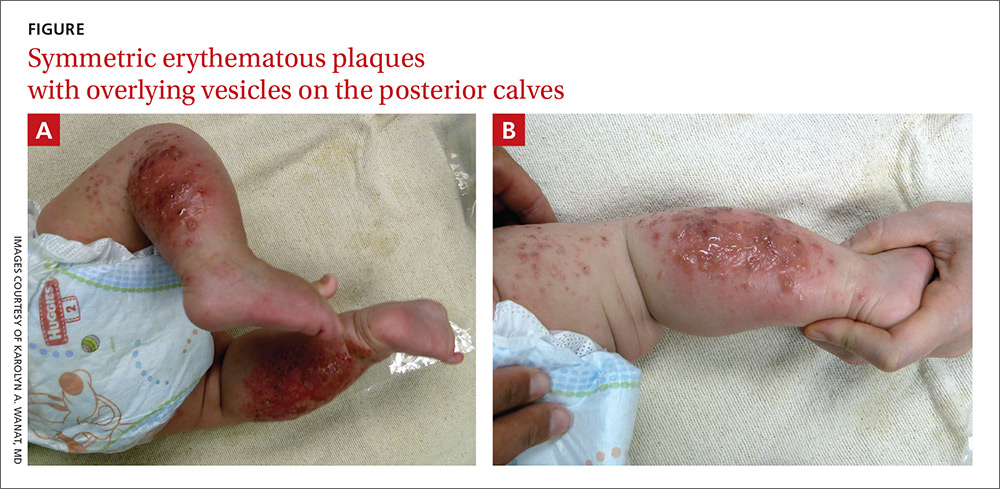
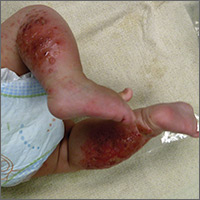
A 4-month-old girl was brought to our clinic with a 4-week history of blisters on her arms and legs. The eruption started on her right posterior and lateral calf and then appeared on her left calf and bilateral elbows. Other than the blisters, the girl appeared well and was eating and growing normally. Her parents said she had not been in contact with anyone with a similar rash or itching. They also denied recent outdoor activities, camping trips, or environmental exposures.
The child had been previously treated with topical and oral steroids and oral antibiotics by a pediatrician, but the rash barely improved. On physical examination, she was afebrile with well-demarcated erythematous papules and plaques with bullae, and erosions with honey-colored crusts. The rash was distributed symmetrically on the bilateral posterior and lateral lower legs and lateral upper arms (FIGURE).
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Allergic contact dermatitis from a car seat
The appearance and distribution of the rash on the infant’s posterior and lateral lower legs and lateral upper arms prompted us to conclude that this was a case of allergic contact dermatitis from a car seat, along with secondary impetiginization.
The incidence of car seat contact dermatitis is unknown, although it is suspected to be both under-recognized and under-reported. In fact, the number of cases may be on the rise,1 given the increasing number of synthetic liners now being used in car seats, high chairs, and other infant support products.
More common in summer months. Car seat dermatitis is commonly reported in warmer months, when an infant’s skin is more likely to be in direct contact with the car seat and sweating is increased.1 In the acute setting, clinical morphology usually takes the form of inflamed papules or vesicles, while in chronic presentations, lichenified eczematous plaques may be seen. Distribution is typically symmetric and involves areas in direct contact with the car seat, such as the elbows, upper lateral or posterior thighs, lower lateral legs, and sometimes, the occipital scalp.1 The presence of a secondary infection or autoeczematization can complicate the clinical presentation.
Which car seat materials are to blame? Previous reports have described the shiny, nylon-like material overlying the car seat cushion as the cause of the contact allergy, but no specific allergens have yet been identified.1 Attempts at identifying specific allergens in car seat liners have been thwarted by the proprietary nature of manufacturers’ formulas and the unwillingness of companies to divulge the chemicals used in the manufacture of their car seats. Potential allergens include bromine, chlorine, and flame-retardants.1 These allergens differ from the usual contact allergens in children and adolescents, which include nickel sulfate, cobalt chloride, potassium dichromate, fragrance mix, thimerosal, neomycin sulfate, and para-tertiary-butylphenol formaldehyde resin.2
Differential includes other conditions with blisters, plaques
The differential diagnosis includes eczema herpeticum, bullous impetigo, and psoriasis.
Infants with eczema herpeticum usually have eczematous plaques in locations such as the cheeks, neck, antecubital fossa, popliteal fossa, and ankles, with numerous “punched-out” shallow erosions. Children with extensive eczema herpeticum can be systemically ill.
Bullous impetigo is seen as flaccid bullae in infants, which can easily rupture and leave behind superficial erosions. These blisters tend to appear on normal skin. (This is quite different from the thick, erythematous plaques seen in contact dermatitis.) In patients with superficial erosions, a polymerase chain reaction test for the herpes virus and a bacterial culture should be obtained.
Psoriasis often presents with well-demarcated erythematous plaques with overlying silver scale. Although it can be symmetric on extensor surfaces, the weeping vesicles with acute onset that were seen in this case would be unusual.
Look for a pattern. The well-demarcated symmetric plaques corresponding directly to areas in contact with the car seat should be a strong clue for contact dermatitis. While patch testing for relevant chemicals is often indicated in patients for whom there is a clinical suspicion of a contact allergy,3,4 we did not perform such testing because the specific chemicals involved in car seat manufacturing are unknown.
Topical steroids and avoidance of the allergen help resolve the rash
The mainstay of treatment for allergic contact dermatitis is avoiding the contact allergen. In car seat contact dermatitis, parents should be counseled to avoid contact between the child’s bare skin and the car seat liner. Given that the precise allergen is unknown, it is impossible to know if a new car seat would contain the same material. Instead, we recommend covering the car seat with a cotton blanket to avoid irritation/allergens.
Depending on the extent of the rash, the patient should be treated with a mid- or high-potency topical steroid until the erythema and blistering resolve.5-8 A 3-week prednisone taper can also be considered for severe cases. For patients who have >25% of their body surface involved, oral steroids are recommended.6 Any secondary infection should be treated with topical and oral antibiotics, as appropriate.
Our patient. Due to the extent and severity of the eruption, we put the patient on a 3-week oral prednisone taper and advised the parents to apply clobetasol 0.05% ointment to the affected areas 2 times a day. We also prescribed a 7-day course of cephalexin 50 mg/kg divided in 3 doses a day and topical mupirocin ointment (to be applied 2 times a day) for the secondary impetiginization.
We advised the parents to use a cotton blanket over the baby’s car seat to prevent further outbreaks. The eruption resolved within 2 months.
CORRESPONDENCE
Karolyn A. Wanat, MD, Department of Dermatology, University of Iowa Hospitals and Clinics, 200 Hawkins Drive, 40000 PFP, Iowa City, IA 52242; [email protected].
1. Ghali FE. “Car seat dermatitis”: a newly described form of contact dermatitis. Pediatr Dermatol. 2011;28:321-326.
2. Mortz CG, Andersen KE. Allergic contact dermatitis in children and adolescents. Contact Dermatitis. 1999;41:121-130.
3. van der Valk PG, Devos SA, Coenraads PJ. Evidence-based diagnosis in patch testing. Contact Dermatitis. 2003;48:121-125.
4. Krob HA, Fleischer AB Jr, D’Agostino R Jr, et al. Prevalence and relevance of contact dermatitis allergens: a meta-analysis of 15 years of published T.R.U.E. test data. J Am Acad Dermatol. 2004;51:349-353.
5. Cohen DE, Heidary N. Treatment of irritant and allergic contact dermatitis. Dermatol Ther. 2004;17:334-340.
6. Belsito DV. The diagnostic evaluation, treatment, and prevention of allergic contact dermatitis in the new millennium. J Allergy Clin Immunol. 2000;105:409-420.
7. Hachem JP, De Paepe K, Vanpée E, et al. Efficacy of topical corticosteroids in nickel-induced contact allergy. Clin Exp Dermatol. 2002;27:47-50.
8. Saary J, Qureshi R, Palda V, et al. A systematic review of contact dermatitis treatment and prevention. J Am Acad Dermatol. 2005;53:845.
A 4-month-old girl was brought to our clinic with a 4-week history of blisters on her arms and legs. The eruption started on her right posterior and lateral calf and then appeared on her left calf and bilateral elbows. Other than the blisters, the girl appeared well and was eating and growing normally. Her parents said she had not been in contact with anyone with a similar rash or itching. They also denied recent outdoor activities, camping trips, or environmental exposures.
The child had been previously treated with topical and oral steroids and oral antibiotics by a pediatrician, but the rash barely improved. On physical examination, she was afebrile with well-demarcated erythematous papules and plaques with bullae, and erosions with honey-colored crusts. The rash was distributed symmetrically on the bilateral posterior and lateral lower legs and lateral upper arms (FIGURE).
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Allergic contact dermatitis from a car seat
The appearance and distribution of the rash on the infant’s posterior and lateral lower legs and lateral upper arms prompted us to conclude that this was a case of allergic contact dermatitis from a car seat, along with secondary impetiginization.
The incidence of car seat contact dermatitis is unknown, although it is suspected to be both under-recognized and under-reported. In fact, the number of cases may be on the rise,1 given the increasing number of synthetic liners now being used in car seats, high chairs, and other infant support products.
More common in summer months. Car seat dermatitis is commonly reported in warmer months, when an infant’s skin is more likely to be in direct contact with the car seat and sweating is increased.1 In the acute setting, clinical morphology usually takes the form of inflamed papules or vesicles, while in chronic presentations, lichenified eczematous plaques may be seen. Distribution is typically symmetric and involves areas in direct contact with the car seat, such as the elbows, upper lateral or posterior thighs, lower lateral legs, and sometimes, the occipital scalp.1 The presence of a secondary infection or autoeczematization can complicate the clinical presentation.
Which car seat materials are to blame? Previous reports have described the shiny, nylon-like material overlying the car seat cushion as the cause of the contact allergy, but no specific allergens have yet been identified.1 Attempts at identifying specific allergens in car seat liners have been thwarted by the proprietary nature of manufacturers’ formulas and the unwillingness of companies to divulge the chemicals used in the manufacture of their car seats. Potential allergens include bromine, chlorine, and flame-retardants.1 These allergens differ from the usual contact allergens in children and adolescents, which include nickel sulfate, cobalt chloride, potassium dichromate, fragrance mix, thimerosal, neomycin sulfate, and para-tertiary-butylphenol formaldehyde resin.2
Differential includes other conditions with blisters, plaques
The differential diagnosis includes eczema herpeticum, bullous impetigo, and psoriasis.
Infants with eczema herpeticum usually have eczematous plaques in locations such as the cheeks, neck, antecubital fossa, popliteal fossa, and ankles, with numerous “punched-out” shallow erosions. Children with extensive eczema herpeticum can be systemically ill.
Bullous impetigo is seen as flaccid bullae in infants, which can easily rupture and leave behind superficial erosions. These blisters tend to appear on normal skin. (This is quite different from the thick, erythematous plaques seen in contact dermatitis.) In patients with superficial erosions, a polymerase chain reaction test for the herpes virus and a bacterial culture should be obtained.
Psoriasis often presents with well-demarcated erythematous plaques with overlying silver scale. Although it can be symmetric on extensor surfaces, the weeping vesicles with acute onset that were seen in this case would be unusual.
Look for a pattern. The well-demarcated symmetric plaques corresponding directly to areas in contact with the car seat should be a strong clue for contact dermatitis. While patch testing for relevant chemicals is often indicated in patients for whom there is a clinical suspicion of a contact allergy,3,4 we did not perform such testing because the specific chemicals involved in car seat manufacturing are unknown.
Topical steroids and avoidance of the allergen help resolve the rash
The mainstay of treatment for allergic contact dermatitis is avoiding the contact allergen. In car seat contact dermatitis, parents should be counseled to avoid contact between the child’s bare skin and the car seat liner. Given that the precise allergen is unknown, it is impossible to know if a new car seat would contain the same material. Instead, we recommend covering the car seat with a cotton blanket to avoid irritation/allergens.
Depending on the extent of the rash, the patient should be treated with a mid- or high-potency topical steroid until the erythema and blistering resolve.5-8 A 3-week prednisone taper can also be considered for severe cases. For patients who have >25% of their body surface involved, oral steroids are recommended.6 Any secondary infection should be treated with topical and oral antibiotics, as appropriate.
Our patient. Due to the extent and severity of the eruption, we put the patient on a 3-week oral prednisone taper and advised the parents to apply clobetasol 0.05% ointment to the affected areas 2 times a day. We also prescribed a 7-day course of cephalexin 50 mg/kg divided in 3 doses a day and topical mupirocin ointment (to be applied 2 times a day) for the secondary impetiginization.
We advised the parents to use a cotton blanket over the baby’s car seat to prevent further outbreaks. The eruption resolved within 2 months.
CORRESPONDENCE
Karolyn A. Wanat, MD, Department of Dermatology, University of Iowa Hospitals and Clinics, 200 Hawkins Drive, 40000 PFP, Iowa City, IA 52242; [email protected].
A 4-month-old girl was brought to our clinic with a 4-week history of blisters on her arms and legs. The eruption started on her right posterior and lateral calf and then appeared on her left calf and bilateral elbows. Other than the blisters, the girl appeared well and was eating and growing normally. Her parents said she had not been in contact with anyone with a similar rash or itching. They also denied recent outdoor activities, camping trips, or environmental exposures.
The child had been previously treated with topical and oral steroids and oral antibiotics by a pediatrician, but the rash barely improved. On physical examination, she was afebrile with well-demarcated erythematous papules and plaques with bullae, and erosions with honey-colored crusts. The rash was distributed symmetrically on the bilateral posterior and lateral lower legs and lateral upper arms (FIGURE).
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Allergic contact dermatitis from a car seat
The appearance and distribution of the rash on the infant’s posterior and lateral lower legs and lateral upper arms prompted us to conclude that this was a case of allergic contact dermatitis from a car seat, along with secondary impetiginization.
The incidence of car seat contact dermatitis is unknown, although it is suspected to be both under-recognized and under-reported. In fact, the number of cases may be on the rise,1 given the increasing number of synthetic liners now being used in car seats, high chairs, and other infant support products.
More common in summer months. Car seat dermatitis is commonly reported in warmer months, when an infant’s skin is more likely to be in direct contact with the car seat and sweating is increased.1 In the acute setting, clinical morphology usually takes the form of inflamed papules or vesicles, while in chronic presentations, lichenified eczematous plaques may be seen. Distribution is typically symmetric and involves areas in direct contact with the car seat, such as the elbows, upper lateral or posterior thighs, lower lateral legs, and sometimes, the occipital scalp.1 The presence of a secondary infection or autoeczematization can complicate the clinical presentation.
Which car seat materials are to blame? Previous reports have described the shiny, nylon-like material overlying the car seat cushion as the cause of the contact allergy, but no specific allergens have yet been identified.1 Attempts at identifying specific allergens in car seat liners have been thwarted by the proprietary nature of manufacturers’ formulas and the unwillingness of companies to divulge the chemicals used in the manufacture of their car seats. Potential allergens include bromine, chlorine, and flame-retardants.1 These allergens differ from the usual contact allergens in children and adolescents, which include nickel sulfate, cobalt chloride, potassium dichromate, fragrance mix, thimerosal, neomycin sulfate, and para-tertiary-butylphenol formaldehyde resin.2
Differential includes other conditions with blisters, plaques
The differential diagnosis includes eczema herpeticum, bullous impetigo, and psoriasis.
Infants with eczema herpeticum usually have eczematous plaques in locations such as the cheeks, neck, antecubital fossa, popliteal fossa, and ankles, with numerous “punched-out” shallow erosions. Children with extensive eczema herpeticum can be systemically ill.
Bullous impetigo is seen as flaccid bullae in infants, which can easily rupture and leave behind superficial erosions. These blisters tend to appear on normal skin. (This is quite different from the thick, erythematous plaques seen in contact dermatitis.) In patients with superficial erosions, a polymerase chain reaction test for the herpes virus and a bacterial culture should be obtained.
Psoriasis often presents with well-demarcated erythematous plaques with overlying silver scale. Although it can be symmetric on extensor surfaces, the weeping vesicles with acute onset that were seen in this case would be unusual.
Look for a pattern. The well-demarcated symmetric plaques corresponding directly to areas in contact with the car seat should be a strong clue for contact dermatitis. While patch testing for relevant chemicals is often indicated in patients for whom there is a clinical suspicion of a contact allergy,3,4 we did not perform such testing because the specific chemicals involved in car seat manufacturing are unknown.
Topical steroids and avoidance of the allergen help resolve the rash
The mainstay of treatment for allergic contact dermatitis is avoiding the contact allergen. In car seat contact dermatitis, parents should be counseled to avoid contact between the child’s bare skin and the car seat liner. Given that the precise allergen is unknown, it is impossible to know if a new car seat would contain the same material. Instead, we recommend covering the car seat with a cotton blanket to avoid irritation/allergens.
Depending on the extent of the rash, the patient should be treated with a mid- or high-potency topical steroid until the erythema and blistering resolve.5-8 A 3-week prednisone taper can also be considered for severe cases. For patients who have >25% of their body surface involved, oral steroids are recommended.6 Any secondary infection should be treated with topical and oral antibiotics, as appropriate.
Our patient. Due to the extent and severity of the eruption, we put the patient on a 3-week oral prednisone taper and advised the parents to apply clobetasol 0.05% ointment to the affected areas 2 times a day. We also prescribed a 7-day course of cephalexin 50 mg/kg divided in 3 doses a day and topical mupirocin ointment (to be applied 2 times a day) for the secondary impetiginization.
We advised the parents to use a cotton blanket over the baby’s car seat to prevent further outbreaks. The eruption resolved within 2 months.
CORRESPONDENCE
Karolyn A. Wanat, MD, Department of Dermatology, University of Iowa Hospitals and Clinics, 200 Hawkins Drive, 40000 PFP, Iowa City, IA 52242; [email protected].
1. Ghali FE. “Car seat dermatitis”: a newly described form of contact dermatitis. Pediatr Dermatol. 2011;28:321-326.
2. Mortz CG, Andersen KE. Allergic contact dermatitis in children and adolescents. Contact Dermatitis. 1999;41:121-130.
3. van der Valk PG, Devos SA, Coenraads PJ. Evidence-based diagnosis in patch testing. Contact Dermatitis. 2003;48:121-125.
4. Krob HA, Fleischer AB Jr, D’Agostino R Jr, et al. Prevalence and relevance of contact dermatitis allergens: a meta-analysis of 15 years of published T.R.U.E. test data. J Am Acad Dermatol. 2004;51:349-353.
5. Cohen DE, Heidary N. Treatment of irritant and allergic contact dermatitis. Dermatol Ther. 2004;17:334-340.
6. Belsito DV. The diagnostic evaluation, treatment, and prevention of allergic contact dermatitis in the new millennium. J Allergy Clin Immunol. 2000;105:409-420.
7. Hachem JP, De Paepe K, Vanpée E, et al. Efficacy of topical corticosteroids in nickel-induced contact allergy. Clin Exp Dermatol. 2002;27:47-50.
8. Saary J, Qureshi R, Palda V, et al. A systematic review of contact dermatitis treatment and prevention. J Am Acad Dermatol. 2005;53:845.
1. Ghali FE. “Car seat dermatitis”: a newly described form of contact dermatitis. Pediatr Dermatol. 2011;28:321-326.
2. Mortz CG, Andersen KE. Allergic contact dermatitis in children and adolescents. Contact Dermatitis. 1999;41:121-130.
3. van der Valk PG, Devos SA, Coenraads PJ. Evidence-based diagnosis in patch testing. Contact Dermatitis. 2003;48:121-125.
4. Krob HA, Fleischer AB Jr, D’Agostino R Jr, et al. Prevalence and relevance of contact dermatitis allergens: a meta-analysis of 15 years of published T.R.U.E. test data. J Am Acad Dermatol. 2004;51:349-353.
5. Cohen DE, Heidary N. Treatment of irritant and allergic contact dermatitis. Dermatol Ther. 2004;17:334-340.
6. Belsito DV. The diagnostic evaluation, treatment, and prevention of allergic contact dermatitis in the new millennium. J Allergy Clin Immunol. 2000;105:409-420.
7. Hachem JP, De Paepe K, Vanpée E, et al. Efficacy of topical corticosteroids in nickel-induced contact allergy. Clin Exp Dermatol. 2002;27:47-50.
8. Saary J, Qureshi R, Palda V, et al. A systematic review of contact dermatitis treatment and prevention. J Am Acad Dermatol. 2005;53:845.
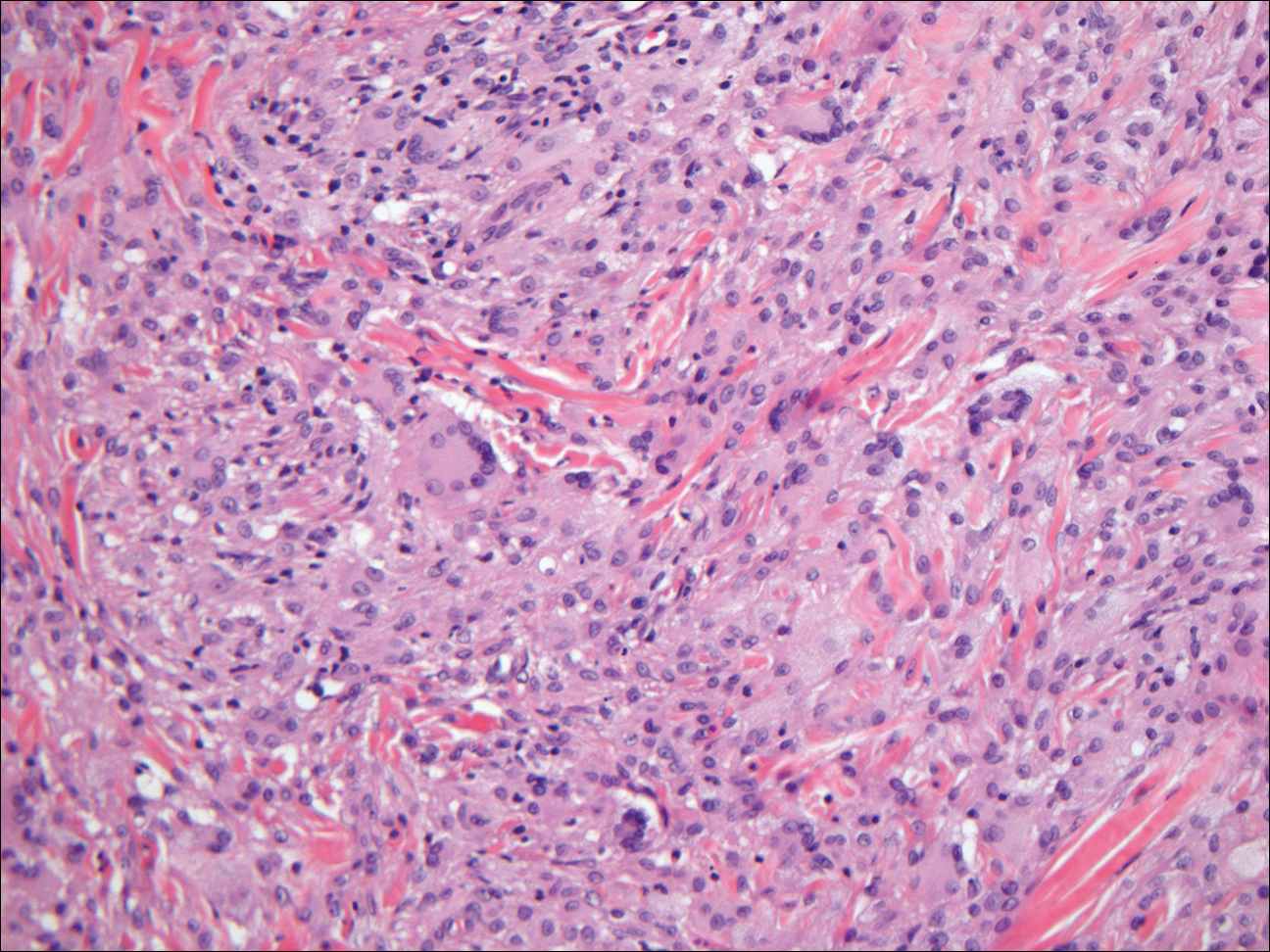
Circumscribed Nodule in a Renal Transplant Patient
The Diagnosis: Subcutaneous Phaeohyphomycosis
Subcutaneous phaeohyphomycosis (SP), also called mycotic cyst, is characterized by a painless, nodular lesion that develops in response to traumatic implantation of dematiaceous, pigment-forming fungi.1 Similar to other fungal infections, SP can arise opportunistically in immunocompromised patients.2,3 More than 60 genera (and more than 100 species) are known etiologic agents of phaeohyphomycosis; the 2 main causes of infection are Bipolaris spicifera and Exophiala jeanselmei.4,5 Given this variety, phaeohyphomycosis can present superficially as black piedra or tinea nigra, cutaneously as scytalidiosis, subcutaneously as SP, or disseminated as sinusitis or systemic phaeohyphomycosis.
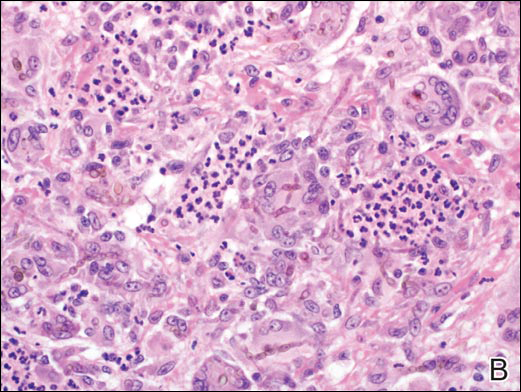
Coined in 1974 by Ajello et al,6 the term phaeohyphomycosis translates to “condition of dark hyphal fungus,” a term used to designate mycoses caused by fungi with melanized hyphae. Histologically, SP demonstrates a circumscribed chronic cyst or abscess with a dense fibrous wall (quiz image A). At high power, the wall is composed of chronic granulomatous inflammation with foamy macrophages, and the cystic cavity contains necrotic debris admixed with neutrophils. Pigmented filamentous hyphae and yeastlike entities can be seen in the cyst wall, in multinucleated giant cells, in the necrotic debris, or directly attached to the implanted foreign material (quiz image B).7 The first-line treatment of SP is wide local excision and oral itraconazole. It often requires adjustments to dosage or change to antifungal due to recurrence and etiologic variation.8 Furthermore, if SP is not definitively treated, immunocompromised patients are at an increased risk for developing potentially fatal systemic phaeohyphomycosis.3
Chromoblastomycosis (CBM), also caused by dematiaceous fungi, is characterized by an initially indolent clinical presentation. Typically found on the legs and lower thighs of agricultural workers, the lesion begins as a slow-growing, nodular papule with subsequent transformation into an edematous verrucous plaque with peripheral erythema.9 Lesions can be annular with central clearing, and lymphedema with elephantiasis may be present.10 Histologically, CBM shows pseudoepitheliomatous hyperplasia and intraepidermal pustules as the host rids the infection via transepithelial elimination. Dematiaceous fungi often are seen in the dermis, either freestanding or attached to foreign plant material. Medlar bodies, also called copper penny spores or sclerotic bodies, are the most defining histologic finding and are characterized by groups of brown, thick-walled cells found in giant cells or neutrophil abscesses (Figure 1). Hyphae are not typically found in this type of infection.11

Granulomatous foreign body reactions occur in response to the inoculation of nonhuman material and are characterized by dermal or subcutaneous nodules. Tissue macrophages phagocytize material not removed shortly after implantation, which initiates an inflammatory response that attempts to isolate the material from the uninvolved surrounding tissue. Vegetative foreign bodies will cause the most severe inflammatory reactions.12 Histologically, foreign body granulomas are noncaseating with epithelioid histiocytes surrounding a central foreign body (Figure 2). Occasionally, foreign bodies may be difficult to detect; some are birefringent to polarized light.13 Additionally, inoculation injuries can predispose patients to SP, CBM, and other fungal infections.

Tattoos are characterized by exogenous pigment deposition into the dermis.14 Histologically, tattoos display exogenous pigment deposited throughout the reticular dermis, attached to collagen bundles, within macrophages, or adjacent to adnexal structures (eg, pilosebaceous units or eccrine glands). Although all tattoo pigments can cause adverse reactions, hypersensitivity reactions occur most commonly in response to red pigment, resulting in discrete areas of spongiosis and granulomatous or lichenoid inflammation. Occasionally, hypersensitivity reactions can induce necrobiotic granulomatous reactions characterized by collagen alteration surrounded by palisaded histiocytes and lymphocytes (Figure 3).15,16 There also may be focally dense areas of superficial and deep perivascular lymphohistiocytic infiltrate. Clinical context is important, as brown tattoo pigment (Figure 3) can be easily confused with the pigmented hyphae of phaeohyphomycosis, melanin, or hemosiderin.
Subcutaneous hyalohyphomycosis is a nondemat-iaceous (nonpigmented) infection that is caused by hyaline septate hyphal cells.17 Hyalohyphomycosis skin lesions can present as painful erythematous nodules that evolve into excoriated pustules.18 Hyalohyphomycosis most often arises in immunocompromised patients. Causative organisms are ubiquitous soil saprophytes and plant pathogens, most often Aspergillus and Fusarium species, with a predilection for affecting severely immunocompromised hosts, particularly children.19 These species tend to be vasculotropic, which can result in tissue necrosis and systemic dissemination. Histologically, fungi are dispersed within tissue. They have a bright, bubbly, mildly basophilic cytoplasm and are nonpigmented, branching, and septate (Figure 4).11

- Isa-Isa R, García C, Isa M, et al. Subcutaneous phaeohyphomycosis (mycotic cyst). Clin Dermatol. 2012;30:425-431.
- Rubin RH. Infectious disease complications of renal transplantation. Kidney Int. 1993;44:221-236.
- Ogawa MM, Galante NZ, Godoy P, et al. Treatment of subcutaneous phaeohyphomycosis and prospective follow-up of 17 kidney transplant recipients. J Am Acad Dermatol. 2009;61:977-985.
- Matsumoto T, Ajello L, Matsuda T, et al. Developments in hyalohyphomycosis and phaeohyphomycosis. J Med Vet Mycol. 1994;32(suppl 1):329-349.
- Rinaldi MG. Phaeohyphomycosis. Dermatol Clin. 1996;14:147-153.
- Ajello L, Georg LK, Steigbigel RT, et al. A case of phaeohyphomycosis caused by a new species of Phialophora. Mycologia. 1974;66:490-498.
- Patterson J. Weedon’s Skin Pathology. 4th ed. London, England: Churchill Livingstone Elsevier; 2014.
- Patel U, Chu J, Patel R, et al. Subcutaneous dematiaceous fungal infection. Dermatol Online J. 2011;17:19.
- Bonifaz A, Carrasco-Gerard E, Saúl A. Chromoblastomycosis: clinical and mycologic experience of 51 cases. Mycoses. 2001;44:1-7.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Elston D, Ferringer T, Peckham S, et al, eds. Dermatopathology. 2nd ed. St. Louis, MO: Elsevier Saunders; 2014.
- Lammers RL. Soft tissue foreign bodies. In: Tintinalli J, Stapczynski S, Ma O, et al, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw Hill Professional; 2011.
- Murphy GF, Saavedra AP, Mihm MC. Nodular/interstitial dermatitis. In: Murphy GF, Saavedra AP, Mihm MC, eds. Atlas of Nontumor Pathology: Inflammatory Disorders of the Skin. Vol 10. Washington, DC: American Registry of Pathology; 2012:337-395.
- Laumann A. Body art. In: Goldsmith L, Katz S, Gilchrest B, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012. http://access medicine.mhmedical.com.proxy.lib.uiowa.edu/content.aspx?bookid=392&Sectionid=41138811. Accessed July 17,2016.
- Wood A, Hamilton SA, Wallace WA, et al. Necrobiotic granulomatous tattoo reaction: report of an unusual case showing features of both necrobiosis lipoidica and granuloma annulare patterns. Am J Dermatopathol. 2014;36:e152-e155.
- Mortimer N, Chave T, Johnston G. Red tattoo reactions. Clin Exp Dermatol. 2003;28:508-510.
- Ajello L. Hyalohyphomycosis and phaeohyphomycosis: two global disease entities of public health importance. Eur J Epidemiol. 1986;2:243-251.
- Safdar A. Progressive cutaneous hyalohyphomycosis due to Paecilomyces lilacinus: rapid response to treatment with caspofungin and itraconazole. Clin Infect Dis. 2002;34:1415-1417.
- Marcoux D, Jafarian F, Joncas V, et al. Deep cutaneous fungal infections in immunocompromised children. J Am Acad Dermatol. 2009;61:857-864.
The Diagnosis: Subcutaneous Phaeohyphomycosis
Subcutaneous phaeohyphomycosis (SP), also called mycotic cyst, is characterized by a painless, nodular lesion that develops in response to traumatic implantation of dematiaceous, pigment-forming fungi.1 Similar to other fungal infections, SP can arise opportunistically in immunocompromised patients.2,3 More than 60 genera (and more than 100 species) are known etiologic agents of phaeohyphomycosis; the 2 main causes of infection are Bipolaris spicifera and Exophiala jeanselmei.4,5 Given this variety, phaeohyphomycosis can present superficially as black piedra or tinea nigra, cutaneously as scytalidiosis, subcutaneously as SP, or disseminated as sinusitis or systemic phaeohyphomycosis.
Coined in 1974 by Ajello et al,6 the term phaeohyphomycosis translates to “condition of dark hyphal fungus,” a term used to designate mycoses caused by fungi with melanized hyphae. Histologically, SP demonstrates a circumscribed chronic cyst or abscess with a dense fibrous wall (quiz image A). At high power, the wall is composed of chronic granulomatous inflammation with foamy macrophages, and the cystic cavity contains necrotic debris admixed with neutrophils. Pigmented filamentous hyphae and yeastlike entities can be seen in the cyst wall, in multinucleated giant cells, in the necrotic debris, or directly attached to the implanted foreign material (quiz image B).7 The first-line treatment of SP is wide local excision and oral itraconazole. It often requires adjustments to dosage or change to antifungal due to recurrence and etiologic variation.8 Furthermore, if SP is not definitively treated, immunocompromised patients are at an increased risk for developing potentially fatal systemic phaeohyphomycosis.3
Chromoblastomycosis (CBM), also caused by dematiaceous fungi, is characterized by an initially indolent clinical presentation. Typically found on the legs and lower thighs of agricultural workers, the lesion begins as a slow-growing, nodular papule with subsequent transformation into an edematous verrucous plaque with peripheral erythema.9 Lesions can be annular with central clearing, and lymphedema with elephantiasis may be present.10 Histologically, CBM shows pseudoepitheliomatous hyperplasia and intraepidermal pustules as the host rids the infection via transepithelial elimination. Dematiaceous fungi often are seen in the dermis, either freestanding or attached to foreign plant material. Medlar bodies, also called copper penny spores or sclerotic bodies, are the most defining histologic finding and are characterized by groups of brown, thick-walled cells found in giant cells or neutrophil abscesses (Figure 1). Hyphae are not typically found in this type of infection.11
Granulomatous foreign body reactions occur in response to the inoculation of nonhuman material and are characterized by dermal or subcutaneous nodules. Tissue macrophages phagocytize material not removed shortly after implantation, which initiates an inflammatory response that attempts to isolate the material from the uninvolved surrounding tissue. Vegetative foreign bodies will cause the most severe inflammatory reactions.12 Histologically, foreign body granulomas are noncaseating with epithelioid histiocytes surrounding a central foreign body (Figure 2). Occasionally, foreign bodies may be difficult to detect; some are birefringent to polarized light.13 Additionally, inoculation injuries can predispose patients to SP, CBM, and other fungal infections.
Tattoos are characterized by exogenous pigment deposition into the dermis.14 Histologically, tattoos display exogenous pigment deposited throughout the reticular dermis, attached to collagen bundles, within macrophages, or adjacent to adnexal structures (eg, pilosebaceous units or eccrine glands). Although all tattoo pigments can cause adverse reactions, hypersensitivity reactions occur most commonly in response to red pigment, resulting in discrete areas of spongiosis and granulomatous or lichenoid inflammation. Occasionally, hypersensitivity reactions can induce necrobiotic granulomatous reactions characterized by collagen alteration surrounded by palisaded histiocytes and lymphocytes (Figure 3).15,16 There also may be focally dense areas of superficial and deep perivascular lymphohistiocytic infiltrate. Clinical context is important, as brown tattoo pigment (Figure 3) can be easily confused with the pigmented hyphae of phaeohyphomycosis, melanin, or hemosiderin.
Subcutaneous hyalohyphomycosis is a nondemat-iaceous (nonpigmented) infection that is caused by hyaline septate hyphal cells.17 Hyalohyphomycosis skin lesions can present as painful erythematous nodules that evolve into excoriated pustules.18 Hyalohyphomycosis most often arises in immunocompromised patients. Causative organisms are ubiquitous soil saprophytes and plant pathogens, most often Aspergillus and Fusarium species, with a predilection for affecting severely immunocompromised hosts, particularly children.19 These species tend to be vasculotropic, which can result in tissue necrosis and systemic dissemination. Histologically, fungi are dispersed within tissue. They have a bright, bubbly, mildly basophilic cytoplasm and are nonpigmented, branching, and septate (Figure 4).11
The Diagnosis: Subcutaneous Phaeohyphomycosis
Subcutaneous phaeohyphomycosis (SP), also called mycotic cyst, is characterized by a painless, nodular lesion that develops in response to traumatic implantation of dematiaceous, pigment-forming fungi.1 Similar to other fungal infections, SP can arise opportunistically in immunocompromised patients.2,3 More than 60 genera (and more than 100 species) are known etiologic agents of phaeohyphomycosis; the 2 main causes of infection are Bipolaris spicifera and Exophiala jeanselmei.4,5 Given this variety, phaeohyphomycosis can present superficially as black piedra or tinea nigra, cutaneously as scytalidiosis, subcutaneously as SP, or disseminated as sinusitis or systemic phaeohyphomycosis.
Coined in 1974 by Ajello et al,6 the term phaeohyphomycosis translates to “condition of dark hyphal fungus,” a term used to designate mycoses caused by fungi with melanized hyphae. Histologically, SP demonstrates a circumscribed chronic cyst or abscess with a dense fibrous wall (quiz image A). At high power, the wall is composed of chronic granulomatous inflammation with foamy macrophages, and the cystic cavity contains necrotic debris admixed with neutrophils. Pigmented filamentous hyphae and yeastlike entities can be seen in the cyst wall, in multinucleated giant cells, in the necrotic debris, or directly attached to the implanted foreign material (quiz image B).7 The first-line treatment of SP is wide local excision and oral itraconazole. It often requires adjustments to dosage or change to antifungal due to recurrence and etiologic variation.8 Furthermore, if SP is not definitively treated, immunocompromised patients are at an increased risk for developing potentially fatal systemic phaeohyphomycosis.3
Chromoblastomycosis (CBM), also caused by dematiaceous fungi, is characterized by an initially indolent clinical presentation. Typically found on the legs and lower thighs of agricultural workers, the lesion begins as a slow-growing, nodular papule with subsequent transformation into an edematous verrucous plaque with peripheral erythema.9 Lesions can be annular with central clearing, and lymphedema with elephantiasis may be present.10 Histologically, CBM shows pseudoepitheliomatous hyperplasia and intraepidermal pustules as the host rids the infection via transepithelial elimination. Dematiaceous fungi often are seen in the dermis, either freestanding or attached to foreign plant material. Medlar bodies, also called copper penny spores or sclerotic bodies, are the most defining histologic finding and are characterized by groups of brown, thick-walled cells found in giant cells or neutrophil abscesses (Figure 1). Hyphae are not typically found in this type of infection.11
Granulomatous foreign body reactions occur in response to the inoculation of nonhuman material and are characterized by dermal or subcutaneous nodules. Tissue macrophages phagocytize material not removed shortly after implantation, which initiates an inflammatory response that attempts to isolate the material from the uninvolved surrounding tissue. Vegetative foreign bodies will cause the most severe inflammatory reactions.12 Histologically, foreign body granulomas are noncaseating with epithelioid histiocytes surrounding a central foreign body (Figure 2). Occasionally, foreign bodies may be difficult to detect; some are birefringent to polarized light.13 Additionally, inoculation injuries can predispose patients to SP, CBM, and other fungal infections.
Tattoos are characterized by exogenous pigment deposition into the dermis.14 Histologically, tattoos display exogenous pigment deposited throughout the reticular dermis, attached to collagen bundles, within macrophages, or adjacent to adnexal structures (eg, pilosebaceous units or eccrine glands). Although all tattoo pigments can cause adverse reactions, hypersensitivity reactions occur most commonly in response to red pigment, resulting in discrete areas of spongiosis and granulomatous or lichenoid inflammation. Occasionally, hypersensitivity reactions can induce necrobiotic granulomatous reactions characterized by collagen alteration surrounded by palisaded histiocytes and lymphocytes (Figure 3).15,16 There also may be focally dense areas of superficial and deep perivascular lymphohistiocytic infiltrate. Clinical context is important, as brown tattoo pigment (Figure 3) can be easily confused with the pigmented hyphae of phaeohyphomycosis, melanin, or hemosiderin.
Subcutaneous hyalohyphomycosis is a nondemat-iaceous (nonpigmented) infection that is caused by hyaline septate hyphal cells.17 Hyalohyphomycosis skin lesions can present as painful erythematous nodules that evolve into excoriated pustules.18 Hyalohyphomycosis most often arises in immunocompromised patients. Causative organisms are ubiquitous soil saprophytes and plant pathogens, most often Aspergillus and Fusarium species, with a predilection for affecting severely immunocompromised hosts, particularly children.19 These species tend to be vasculotropic, which can result in tissue necrosis and systemic dissemination. Histologically, fungi are dispersed within tissue. They have a bright, bubbly, mildly basophilic cytoplasm and are nonpigmented, branching, and septate (Figure 4).11
- Isa-Isa R, García C, Isa M, et al. Subcutaneous phaeohyphomycosis (mycotic cyst). Clin Dermatol. 2012;30:425-431.
- Rubin RH. Infectious disease complications of renal transplantation. Kidney Int. 1993;44:221-236.
- Ogawa MM, Galante NZ, Godoy P, et al. Treatment of subcutaneous phaeohyphomycosis and prospective follow-up of 17 kidney transplant recipients. J Am Acad Dermatol. 2009;61:977-985.
- Matsumoto T, Ajello L, Matsuda T, et al. Developments in hyalohyphomycosis and phaeohyphomycosis. J Med Vet Mycol. 1994;32(suppl 1):329-349.
- Rinaldi MG. Phaeohyphomycosis. Dermatol Clin. 1996;14:147-153.
- Ajello L, Georg LK, Steigbigel RT, et al. A case of phaeohyphomycosis caused by a new species of Phialophora. Mycologia. 1974;66:490-498.
- Patterson J. Weedon’s Skin Pathology. 4th ed. London, England: Churchill Livingstone Elsevier; 2014.
- Patel U, Chu J, Patel R, et al. Subcutaneous dematiaceous fungal infection. Dermatol Online J. 2011;17:19.
- Bonifaz A, Carrasco-Gerard E, Saúl A. Chromoblastomycosis: clinical and mycologic experience of 51 cases. Mycoses. 2001;44:1-7.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Elston D, Ferringer T, Peckham S, et al, eds. Dermatopathology. 2nd ed. St. Louis, MO: Elsevier Saunders; 2014.
- Lammers RL. Soft tissue foreign bodies. In: Tintinalli J, Stapczynski S, Ma O, et al, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw Hill Professional; 2011.
- Murphy GF, Saavedra AP, Mihm MC. Nodular/interstitial dermatitis. In: Murphy GF, Saavedra AP, Mihm MC, eds. Atlas of Nontumor Pathology: Inflammatory Disorders of the Skin. Vol 10. Washington, DC: American Registry of Pathology; 2012:337-395.
- Laumann A. Body art. In: Goldsmith L, Katz S, Gilchrest B, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012. http://access medicine.mhmedical.com.proxy.lib.uiowa.edu/content.aspx?bookid=392&Sectionid=41138811. Accessed July 17,2016.
- Wood A, Hamilton SA, Wallace WA, et al. Necrobiotic granulomatous tattoo reaction: report of an unusual case showing features of both necrobiosis lipoidica and granuloma annulare patterns. Am J Dermatopathol. 2014;36:e152-e155.
- Mortimer N, Chave T, Johnston G. Red tattoo reactions. Clin Exp Dermatol. 2003;28:508-510.
- Ajello L. Hyalohyphomycosis and phaeohyphomycosis: two global disease entities of public health importance. Eur J Epidemiol. 1986;2:243-251.
- Safdar A. Progressive cutaneous hyalohyphomycosis due to Paecilomyces lilacinus: rapid response to treatment with caspofungin and itraconazole. Clin Infect Dis. 2002;34:1415-1417.
- Marcoux D, Jafarian F, Joncas V, et al. Deep cutaneous fungal infections in immunocompromised children. J Am Acad Dermatol. 2009;61:857-864.
- Isa-Isa R, García C, Isa M, et al. Subcutaneous phaeohyphomycosis (mycotic cyst). Clin Dermatol. 2012;30:425-431.
- Rubin RH. Infectious disease complications of renal transplantation. Kidney Int. 1993;44:221-236.
- Ogawa MM, Galante NZ, Godoy P, et al. Treatment of subcutaneous phaeohyphomycosis and prospective follow-up of 17 kidney transplant recipients. J Am Acad Dermatol. 2009;61:977-985.
- Matsumoto T, Ajello L, Matsuda T, et al. Developments in hyalohyphomycosis and phaeohyphomycosis. J Med Vet Mycol. 1994;32(suppl 1):329-349.
- Rinaldi MG. Phaeohyphomycosis. Dermatol Clin. 1996;14:147-153.
- Ajello L, Georg LK, Steigbigel RT, et al. A case of phaeohyphomycosis caused by a new species of Phialophora. Mycologia. 1974;66:490-498.
- Patterson J. Weedon’s Skin Pathology. 4th ed. London, England: Churchill Livingstone Elsevier; 2014.
- Patel U, Chu J, Patel R, et al. Subcutaneous dematiaceous fungal infection. Dermatol Online J. 2011;17:19.
- Bonifaz A, Carrasco-Gerard E, Saúl A. Chromoblastomycosis: clinical and mycologic experience of 51 cases. Mycoses. 2001;44:1-7.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Elston D, Ferringer T, Peckham S, et al, eds. Dermatopathology. 2nd ed. St. Louis, MO: Elsevier Saunders; 2014.
- Lammers RL. Soft tissue foreign bodies. In: Tintinalli J, Stapczynski S, Ma O, et al, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw Hill Professional; 2011.
- Murphy GF, Saavedra AP, Mihm MC. Nodular/interstitial dermatitis. In: Murphy GF, Saavedra AP, Mihm MC, eds. Atlas of Nontumor Pathology: Inflammatory Disorders of the Skin. Vol 10. Washington, DC: American Registry of Pathology; 2012:337-395.
- Laumann A. Body art. In: Goldsmith L, Katz S, Gilchrest B, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012. http://access medicine.mhmedical.com.proxy.lib.uiowa.edu/content.aspx?bookid=392&Sectionid=41138811. Accessed July 17,2016.
- Wood A, Hamilton SA, Wallace WA, et al. Necrobiotic granulomatous tattoo reaction: report of an unusual case showing features of both necrobiosis lipoidica and granuloma annulare patterns. Am J Dermatopathol. 2014;36:e152-e155.
- Mortimer N, Chave T, Johnston G. Red tattoo reactions. Clin Exp Dermatol. 2003;28:508-510.
- Ajello L. Hyalohyphomycosis and phaeohyphomycosis: two global disease entities of public health importance. Eur J Epidemiol. 1986;2:243-251.
- Safdar A. Progressive cutaneous hyalohyphomycosis due to Paecilomyces lilacinus: rapid response to treatment with caspofungin and itraconazole. Clin Infect Dis. 2002;34:1415-1417.
- Marcoux D, Jafarian F, Joncas V, et al. Deep cutaneous fungal infections in immunocompromised children. J Am Acad Dermatol. 2009;61:857-864.
A 63-year-old man on immunosuppressive therapy following renal transplantation 5 years prior presented with a nontender circumscribed nodule above the left knee of 6 months’ duration. The patient denied any trauma or injury to the site.
Growing Papule on the Right Shoulder of an Elderly Man
Granular Cell Basal Cell Carcinoma
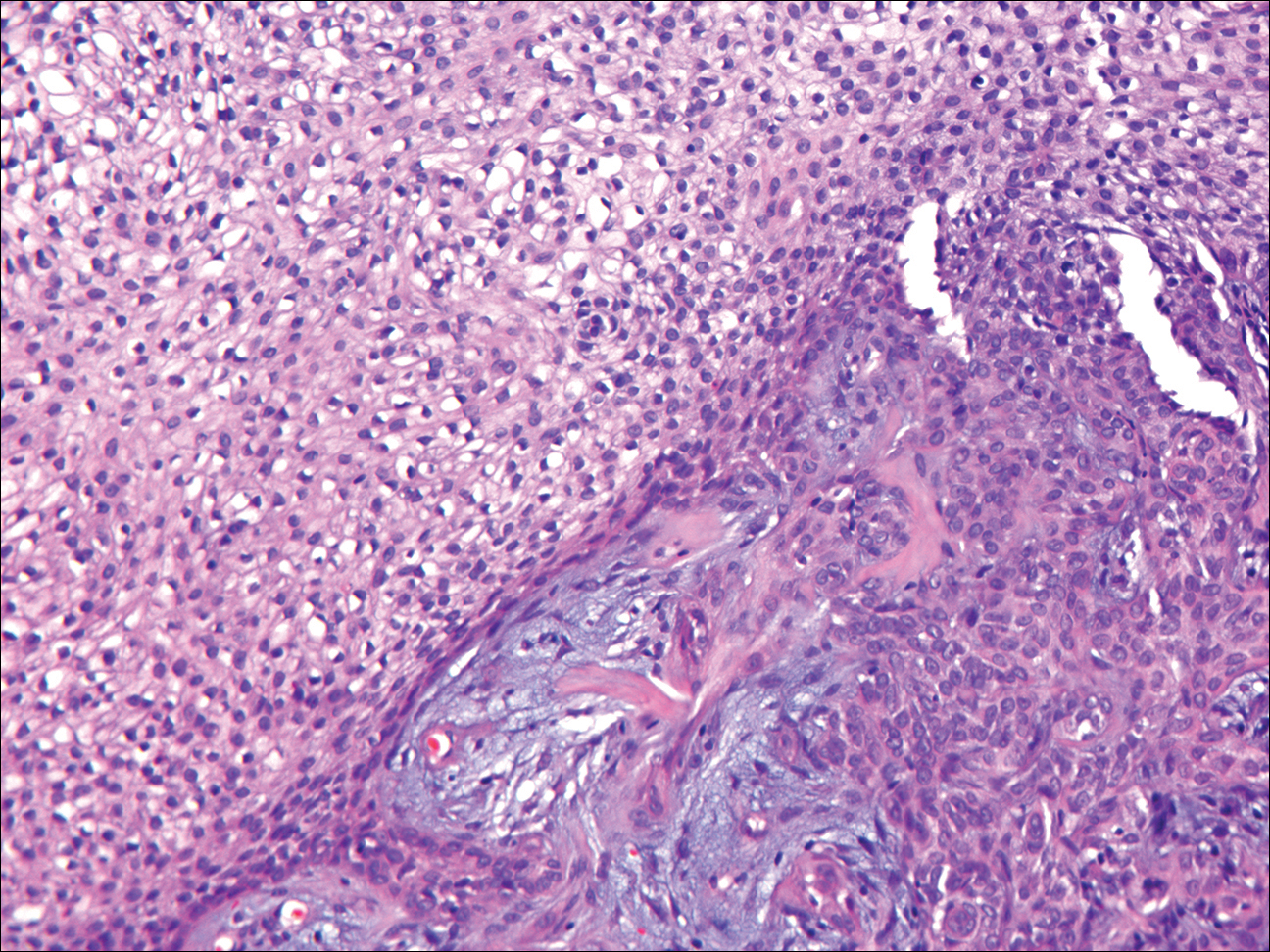
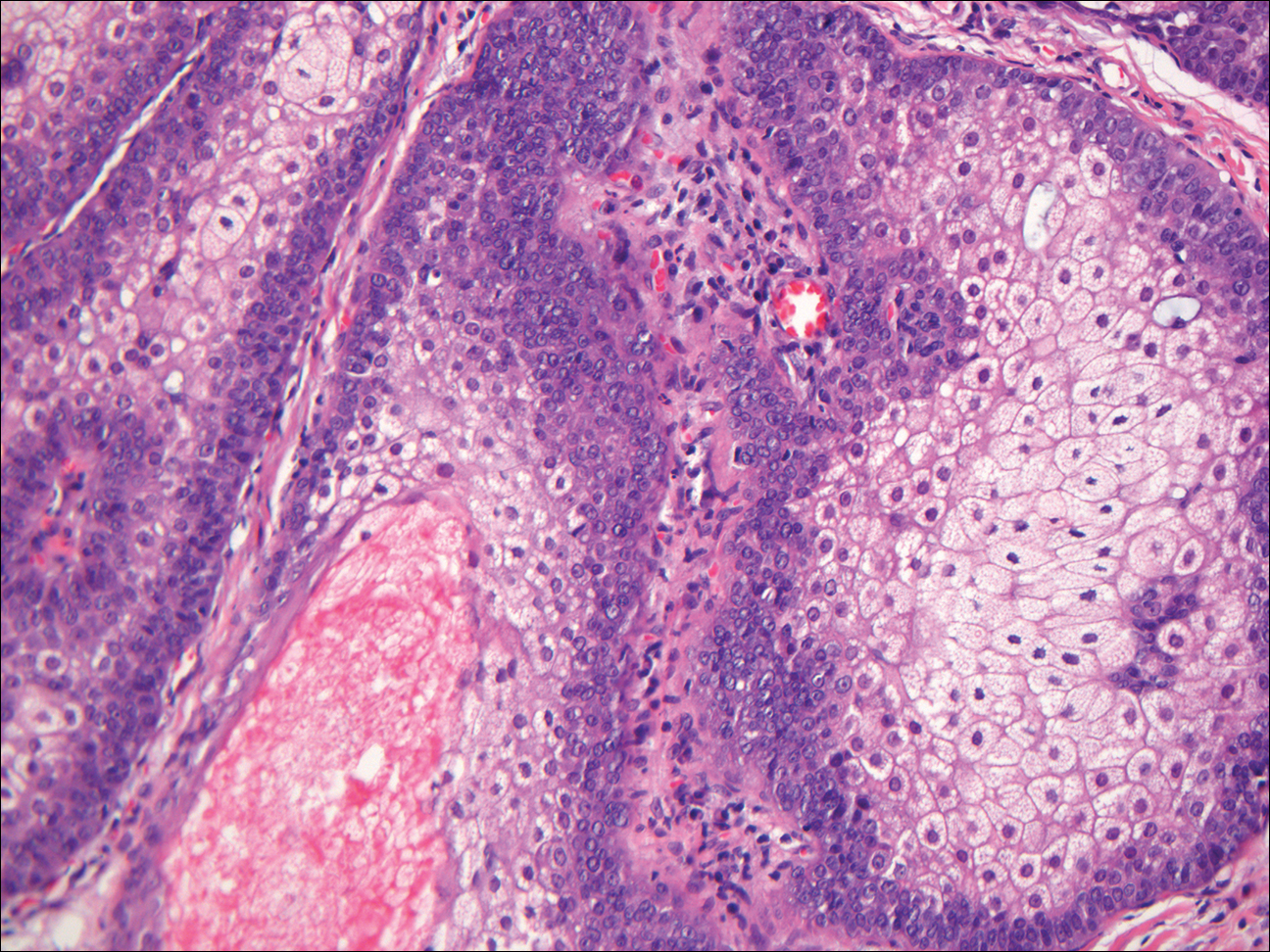
Basal cell carcinoma (BCC) is the most common human epithelial malignancy. There are several histologic variants, the rarest being granular cell BCC (GBCC).1 Granular cell BCC is reported most commonly in men with a mean age of 63 years. Sixty-four percent of cases develop on the face, with the remainder arising on the chest or trunk. Granular cell BCC has distinct histologic features but has no specific epidemiologic or clinical features that differentiate it from more common forms of BCC. Treatment of GBCC is identical to BCC and demonstrates similar outcomes. The presence of granular cells can make GBCC difficult to differentiate from other benign and malignant lesions that display similar granular histologic changes.1,2 Rarely, tumors that are histologically similar to human GBCC have been reported in animals.1
Histologically, GBCC commonly demonstrates the architecture of a nodular BCC or may extend from an existing nodular BCC (quiz images A and B). Granular cell BCC is comprised of large islands of basaloid cells extending from the epidermis with rare mitotic activity. Certain variants showing no epidermal attachments have been described,1,3 as in the current case. Classically, BCC and GBCC both demonstrate a peripheral palisade of blue basal cells; however, GBCC may lack this palisading feature in some cases. Therefore, GBCC may be comprised of granular cells only, which may be more easily confused with other tumors with granular cell differentiation. Even when GBCC retains the traditional peripheral palisade of blue basal cells, the central cells are filled with eosinophilic granules.1,2
Electron microscopy of GBCC usually reveals bundles of cytoplasmic tonofilaments and desmosomes in both granular cells and the peripherally palisaded cells. Electron microscopy imaging also demonstrates 0.1- to 0.5-µm membrane-bound lysosomelike structures. In certain reports, these structures show focal positivity for lysozymes.1,2 The etiology of the granules is unclear; however, they are thought to represent degenerative changes related to metabolic alteration and accumulation of lysosomelike structures. These lysosomelike structures have been highlighted with CD68 staining, which was negative in our case.1,2 The lesional cells in GBCC stain positively for cytokeratins, p63, and Ber-EP4, and negatively for S-100 protein, epithelial membrane antigen, and carcinoembryonic antigen. The granules in GBCC generally are positive on periodic acid–Schiff staining.1-4
The histologic differential diagnosis for GBCC includes granular cell tumor as well as other tumors that can present with granular cell changes such as ameloblastoma, leiomyoma, leiomyosarcoma, angiosarcoma, malignant peripheral nerve sheath tumor, and granular cell trichoblastoma. Granular cell ameloblastomas have histologic features and staining patterns that are identical to GBCC; however, ameloblastomas are distinguished by their location within the oral cavity. Granular cell tumors and malignant peripheral nerve sheath tumors stain positive for S-100 protein, and angiosarcomas stain positive for D2-40 and CD31. Leiomyomas and leiomyosarcomas can be differentiated by staining with smooth muscle actin or desmin.1 Granular cell trichoblastomas can be differentiated by the follicular stem cell marker protein PHLDA1 positivity.5
Desmoplastic trichilemmoma is difficult to distinguish from BCC. These tumors are comprised of superficial lobules of basaloid cells with a perilobular hyaline mantel surrounding a central desmoplastic stroma (Figure 1). The basaloid cells in desmoplastic trichoepithelioma demonstrate clear cell change; however, granular features are not seen. The cells within the desmoplastic areas are arranged haphazardly in cords and nests and can mimic an invasive carcinoma; however, nuclear atypia and mitotic activity generally are absent in desmoplastic trichilemmoma.6
Granular cell tumors generally are poorly circumscribed dermal nodules comprised of large polygonal cells with an eosinophilic granular cytoplasm (Figure 2). The nuclei are generally small and round, and cytological atypia, necrosis, and mitotic activity are uncommon. The cells are positive for S-100 protein and neuron-specific enolase but negative for CD68. The granules are positive for periodic acid–Schiff stain and are diastase resistant. Rarely, these tumors can be malignant.7

Sebaceous adenoma is a well-circumscribed tumor comprised of lobules of characteristic mature sebocytes with bubbly or multivacuolated cytoplasm and crenated nuclei (Figure 3). There is an expansion and increased prominence of the peripherally located basaloid cells; however, in contrast to sebaceous epithelioma, less than 50% of the tumor usually is comprised of these basaloid cells.8
Xanthogranuloma demonstrates a dense collection of histiocytes in the dermis, commonly with Touton giant cell formation (Figure 4). The cells often have a foamy cytoplasm and cytoplasmic vacuoles are observed. The histiocytes are positive for factor XIIIa and CD68, and generally negative for S-100 protein and CD1a, which allows for differentiation from Langerhans cells.9
- Kanitakis J, Chouvet B. Granular-cell basal cell carcinoma of the skin. Eur J Dermatol. 2005;15:301-303.
- Dundr P, Stork J, Povysil C, et al. Granular cell basal cell carcinoma. Australas J Dermatol. 2004;45:70-72.
- Hayden AA, Shamma HN. Ber-EP4 and MNF-116 in a previously undescribed morphologic pattern of granular basal cell carcinoma. Am J Dermatopathol. 2001;23:530-532.
- Ansai S, Takayama R, Kimura T, et al. Ber-EP4 is a useful marker for follicular germinative cell differentiation of cutaneous epithelial neoplasms. J Dermatol. 2012;39:688-692.
- Battistella M, Peltre B, Cribier B. PHLDA1, a follicular stem cell marker, differentiates clear-cell/granular-cell trichoblastoma and clear-cell/granular cell basal cell carcinoma: a case-control study, with first description of granular-cell trichoblastoma. Am J Dermatopathol. 2014;36:643-650.
- Tellechea O, Reis JP, Baptista AP. Desmoplastic trichilemmoma. Am J Dermatopathol. 1992;14:107-114.
- Battistella M, Cribier B, Feugeas JP, et al. Vascular invasion and other invasive features in granular cell tumours of the skin: a multicentre study of 119 cases. J Clin Pathol. 2014;67:19-25.
- Shalin SC, Lyle S, Calonje E, et al. Sebaceous neoplasia and the Muir-Torre syndrome: important connections with clinical implications. Histopathology. 2010;56:133-147.
- Janssen D, Harms D. Juvenile xanthogranuloma in childhood and adolescence: a clinicopathologic study of 129 patients from the kiel pediatric tumor registry. Am J Surg Pathol. 2005;29:21-28.
Granular Cell Basal Cell Carcinoma
Basal cell carcinoma (BCC) is the most common human epithelial malignancy. There are several histologic variants, the rarest being granular cell BCC (GBCC).1 Granular cell BCC is reported most commonly in men with a mean age of 63 years. Sixty-four percent of cases develop on the face, with the remainder arising on the chest or trunk. Granular cell BCC has distinct histologic features but has no specific epidemiologic or clinical features that differentiate it from more common forms of BCC. Treatment of GBCC is identical to BCC and demonstrates similar outcomes. The presence of granular cells can make GBCC difficult to differentiate from other benign and malignant lesions that display similar granular histologic changes.1,2 Rarely, tumors that are histologically similar to human GBCC have been reported in animals.1
Histologically, GBCC commonly demonstrates the architecture of a nodular BCC or may extend from an existing nodular BCC (quiz images A and B). Granular cell BCC is comprised of large islands of basaloid cells extending from the epidermis with rare mitotic activity. Certain variants showing no epidermal attachments have been described,1,3 as in the current case. Classically, BCC and GBCC both demonstrate a peripheral palisade of blue basal cells; however, GBCC may lack this palisading feature in some cases. Therefore, GBCC may be comprised of granular cells only, which may be more easily confused with other tumors with granular cell differentiation. Even when GBCC retains the traditional peripheral palisade of blue basal cells, the central cells are filled with eosinophilic granules.1,2
Electron microscopy of GBCC usually reveals bundles of cytoplasmic tonofilaments and desmosomes in both granular cells and the peripherally palisaded cells. Electron microscopy imaging also demonstrates 0.1- to 0.5-µm membrane-bound lysosomelike structures. In certain reports, these structures show focal positivity for lysozymes.1,2 The etiology of the granules is unclear; however, they are thought to represent degenerative changes related to metabolic alteration and accumulation of lysosomelike structures. These lysosomelike structures have been highlighted with CD68 staining, which was negative in our case.1,2 The lesional cells in GBCC stain positively for cytokeratins, p63, and Ber-EP4, and negatively for S-100 protein, epithelial membrane antigen, and carcinoembryonic antigen. The granules in GBCC generally are positive on periodic acid–Schiff staining.1-4
The histologic differential diagnosis for GBCC includes granular cell tumor as well as other tumors that can present with granular cell changes such as ameloblastoma, leiomyoma, leiomyosarcoma, angiosarcoma, malignant peripheral nerve sheath tumor, and granular cell trichoblastoma. Granular cell ameloblastomas have histologic features and staining patterns that are identical to GBCC; however, ameloblastomas are distinguished by their location within the oral cavity. Granular cell tumors and malignant peripheral nerve sheath tumors stain positive for S-100 protein, and angiosarcomas stain positive for D2-40 and CD31. Leiomyomas and leiomyosarcomas can be differentiated by staining with smooth muscle actin or desmin.1 Granular cell trichoblastomas can be differentiated by the follicular stem cell marker protein PHLDA1 positivity.5
Desmoplastic trichilemmoma is difficult to distinguish from BCC. These tumors are comprised of superficial lobules of basaloid cells with a perilobular hyaline mantel surrounding a central desmoplastic stroma (Figure 1). The basaloid cells in desmoplastic trichoepithelioma demonstrate clear cell change; however, granular features are not seen. The cells within the desmoplastic areas are arranged haphazardly in cords and nests and can mimic an invasive carcinoma; however, nuclear atypia and mitotic activity generally are absent in desmoplastic trichilemmoma.6
Granular cell tumors generally are poorly circumscribed dermal nodules comprised of large polygonal cells with an eosinophilic granular cytoplasm (Figure 2). The nuclei are generally small and round, and cytological atypia, necrosis, and mitotic activity are uncommon. The cells are positive for S-100 protein and neuron-specific enolase but negative for CD68. The granules are positive for periodic acid–Schiff stain and are diastase resistant. Rarely, these tumors can be malignant.7
Sebaceous adenoma is a well-circumscribed tumor comprised of lobules of characteristic mature sebocytes with bubbly or multivacuolated cytoplasm and crenated nuclei (Figure 3). There is an expansion and increased prominence of the peripherally located basaloid cells; however, in contrast to sebaceous epithelioma, less than 50% of the tumor usually is comprised of these basaloid cells.8
Xanthogranuloma demonstrates a dense collection of histiocytes in the dermis, commonly with Touton giant cell formation (Figure 4). The cells often have a foamy cytoplasm and cytoplasmic vacuoles are observed. The histiocytes are positive for factor XIIIa and CD68, and generally negative for S-100 protein and CD1a, which allows for differentiation from Langerhans cells.9
Granular Cell Basal Cell Carcinoma
Basal cell carcinoma (BCC) is the most common human epithelial malignancy. There are several histologic variants, the rarest being granular cell BCC (GBCC).1 Granular cell BCC is reported most commonly in men with a mean age of 63 years. Sixty-four percent of cases develop on the face, with the remainder arising on the chest or trunk. Granular cell BCC has distinct histologic features but has no specific epidemiologic or clinical features that differentiate it from more common forms of BCC. Treatment of GBCC is identical to BCC and demonstrates similar outcomes. The presence of granular cells can make GBCC difficult to differentiate from other benign and malignant lesions that display similar granular histologic changes.1,2 Rarely, tumors that are histologically similar to human GBCC have been reported in animals.1
Histologically, GBCC commonly demonstrates the architecture of a nodular BCC or may extend from an existing nodular BCC (quiz images A and B). Granular cell BCC is comprised of large islands of basaloid cells extending from the epidermis with rare mitotic activity. Certain variants showing no epidermal attachments have been described,1,3 as in the current case. Classically, BCC and GBCC both demonstrate a peripheral palisade of blue basal cells; however, GBCC may lack this palisading feature in some cases. Therefore, GBCC may be comprised of granular cells only, which may be more easily confused with other tumors with granular cell differentiation. Even when GBCC retains the traditional peripheral palisade of blue basal cells, the central cells are filled with eosinophilic granules.1,2
Electron microscopy of GBCC usually reveals bundles of cytoplasmic tonofilaments and desmosomes in both granular cells and the peripherally palisaded cells. Electron microscopy imaging also demonstrates 0.1- to 0.5-µm membrane-bound lysosomelike structures. In certain reports, these structures show focal positivity for lysozymes.1,2 The etiology of the granules is unclear; however, they are thought to represent degenerative changes related to metabolic alteration and accumulation of lysosomelike structures. These lysosomelike structures have been highlighted with CD68 staining, which was negative in our case.1,2 The lesional cells in GBCC stain positively for cytokeratins, p63, and Ber-EP4, and negatively for S-100 protein, epithelial membrane antigen, and carcinoembryonic antigen. The granules in GBCC generally are positive on periodic acid–Schiff staining.1-4
The histologic differential diagnosis for GBCC includes granular cell tumor as well as other tumors that can present with granular cell changes such as ameloblastoma, leiomyoma, leiomyosarcoma, angiosarcoma, malignant peripheral nerve sheath tumor, and granular cell trichoblastoma. Granular cell ameloblastomas have histologic features and staining patterns that are identical to GBCC; however, ameloblastomas are distinguished by their location within the oral cavity. Granular cell tumors and malignant peripheral nerve sheath tumors stain positive for S-100 protein, and angiosarcomas stain positive for D2-40 and CD31. Leiomyomas and leiomyosarcomas can be differentiated by staining with smooth muscle actin or desmin.1 Granular cell trichoblastomas can be differentiated by the follicular stem cell marker protein PHLDA1 positivity.5
Desmoplastic trichilemmoma is difficult to distinguish from BCC. These tumors are comprised of superficial lobules of basaloid cells with a perilobular hyaline mantel surrounding a central desmoplastic stroma (Figure 1). The basaloid cells in desmoplastic trichoepithelioma demonstrate clear cell change; however, granular features are not seen. The cells within the desmoplastic areas are arranged haphazardly in cords and nests and can mimic an invasive carcinoma; however, nuclear atypia and mitotic activity generally are absent in desmoplastic trichilemmoma.6
Granular cell tumors generally are poorly circumscribed dermal nodules comprised of large polygonal cells with an eosinophilic granular cytoplasm (Figure 2). The nuclei are generally small and round, and cytological atypia, necrosis, and mitotic activity are uncommon. The cells are positive for S-100 protein and neuron-specific enolase but negative for CD68. The granules are positive for periodic acid–Schiff stain and are diastase resistant. Rarely, these tumors can be malignant.7
Sebaceous adenoma is a well-circumscribed tumor comprised of lobules of characteristic mature sebocytes with bubbly or multivacuolated cytoplasm and crenated nuclei (Figure 3). There is an expansion and increased prominence of the peripherally located basaloid cells; however, in contrast to sebaceous epithelioma, less than 50% of the tumor usually is comprised of these basaloid cells.8
Xanthogranuloma demonstrates a dense collection of histiocytes in the dermis, commonly with Touton giant cell formation (Figure 4). The cells often have a foamy cytoplasm and cytoplasmic vacuoles are observed. The histiocytes are positive for factor XIIIa and CD68, and generally negative for S-100 protein and CD1a, which allows for differentiation from Langerhans cells.9
- Kanitakis J, Chouvet B. Granular-cell basal cell carcinoma of the skin. Eur J Dermatol. 2005;15:301-303.
- Dundr P, Stork J, Povysil C, et al. Granular cell basal cell carcinoma. Australas J Dermatol. 2004;45:70-72.
- Hayden AA, Shamma HN. Ber-EP4 and MNF-116 in a previously undescribed morphologic pattern of granular basal cell carcinoma. Am J Dermatopathol. 2001;23:530-532.
- Ansai S, Takayama R, Kimura T, et al. Ber-EP4 is a useful marker for follicular germinative cell differentiation of cutaneous epithelial neoplasms. J Dermatol. 2012;39:688-692.
- Battistella M, Peltre B, Cribier B. PHLDA1, a follicular stem cell marker, differentiates clear-cell/granular-cell trichoblastoma and clear-cell/granular cell basal cell carcinoma: a case-control study, with first description of granular-cell trichoblastoma. Am J Dermatopathol. 2014;36:643-650.
- Tellechea O, Reis JP, Baptista AP. Desmoplastic trichilemmoma. Am J Dermatopathol. 1992;14:107-114.
- Battistella M, Cribier B, Feugeas JP, et al. Vascular invasion and other invasive features in granular cell tumours of the skin: a multicentre study of 119 cases. J Clin Pathol. 2014;67:19-25.
- Shalin SC, Lyle S, Calonje E, et al. Sebaceous neoplasia and the Muir-Torre syndrome: important connections with clinical implications. Histopathology. 2010;56:133-147.
- Janssen D, Harms D. Juvenile xanthogranuloma in childhood and adolescence: a clinicopathologic study of 129 patients from the kiel pediatric tumor registry. Am J Surg Pathol. 2005;29:21-28.
- Kanitakis J, Chouvet B. Granular-cell basal cell carcinoma of the skin. Eur J Dermatol. 2005;15:301-303.
- Dundr P, Stork J, Povysil C, et al. Granular cell basal cell carcinoma. Australas J Dermatol. 2004;45:70-72.
- Hayden AA, Shamma HN. Ber-EP4 and MNF-116 in a previously undescribed morphologic pattern of granular basal cell carcinoma. Am J Dermatopathol. 2001;23:530-532.
- Ansai S, Takayama R, Kimura T, et al. Ber-EP4 is a useful marker for follicular germinative cell differentiation of cutaneous epithelial neoplasms. J Dermatol. 2012;39:688-692.
- Battistella M, Peltre B, Cribier B. PHLDA1, a follicular stem cell marker, differentiates clear-cell/granular-cell trichoblastoma and clear-cell/granular cell basal cell carcinoma: a case-control study, with first description of granular-cell trichoblastoma. Am J Dermatopathol. 2014;36:643-650.
- Tellechea O, Reis JP, Baptista AP. Desmoplastic trichilemmoma. Am J Dermatopathol. 1992;14:107-114.
- Battistella M, Cribier B, Feugeas JP, et al. Vascular invasion and other invasive features in granular cell tumours of the skin: a multicentre study of 119 cases. J Clin Pathol. 2014;67:19-25.
- Shalin SC, Lyle S, Calonje E, et al. Sebaceous neoplasia and the Muir-Torre syndrome: important connections with clinical implications. Histopathology. 2010;56:133-147.
- Janssen D, Harms D. Juvenile xanthogranuloma in childhood and adolescence: a clinicopathologic study of 129 patients from the kiel pediatric tumor registry. Am J Surg Pathol. 2005;29:21-28.

The best diagnosis is:
a. desmoplastic trichilemmoma
b. granular cell basal cell carcinoma
c. granular cell tumor
d. sebaceous adenoma
e. xanthogranuloma
Continue to the next page for the diagnosis >>