User login
Oozing puncture wound on foot
A 49-year-old, unkempt-looking Indian man came into our emergency department in Singapore complaining of increasing right foot swelling and worsening pain that he’d had for a month after having stepped on a fish bone while walking on the beach. Our patient pulled the bone out himself and did not seek immediate medical attention.
Our patient said that he had occasional fever with chills and rigors. His medical history was unremarkable, and there was no history of previous injury or surgery to the right foot. He worked as an “oiler” refueling ships, and said that he did not drink alcohol or smoke.
He was dehydrated, and there were no other sources of infection or sores on his body. Our patient was febrile (38.3°C [100.9°F]), his blood pressure was 112/74 mm Hg, and he was tachycardic at 117 beats per minute.
His right foot was boggy, swollen, and tender with significant crepitus. Brownish discharge was oozing from the sole of his foot (FIGURE 1). There were large infected subcutaneous bullae on the dorsum of his foot (FIGURE 2). We could feel a popliteal pulse in both legs, but could not feel a right dorsalis pedis pulse.
What is your diagnosis?
FIGURE 1
Swollen, dusky foot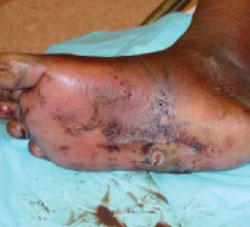
FIGURE 2
Dark bullae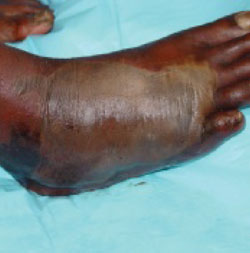
Diagnosis: Necrotizing fasciitis
Necrotizing fasciitis infections are characterized by fulminant destruction of tissue, systemic signs of toxicity, and high rates of mortality. The incidence of necrotizing fasciitis in adults is 0.40 cases per 100,000 population, while the incidence in children is 0.08 cases per 100,000 population. Mortality rates as high as 73% have been reported.1 Early clinical suspicion, early surgical intervention (surgical debridement), and systemic antibiotics are of the utmost importance.
Necrotizing fasciitis is caused by gram positive, gram negative, and/or anaerobic bacteria. Local tissue hypoxia from trauma, surgery, or a medically compromised state creates an ideal opportunistic environment for bacterial proliferation.
Necrotizing fasciitis can be broadly classified into 2 main types:
Type 1 necrotizing fasciitis is a polymicrobial infection caused by facultative bacteria along with anaerobes. Polymicrobial infections with anaerobes are common (up to 74%) in infected puncture wounds in patients with diabetes.2
Type 2 necrotizing fasciitis is caused by group A streptococci alone, but sometimes in association with Staphylococcus aureus. Vibrio vulnificus infection has also been reported in fish bone piercing injuries leading to necrotizing fasciitis.3
Typical early signs and symptoms include severe pain, rapidly progressing erythema, dusky or purplish skin discoloration, and systemic signs of septic toxicity such as fever, tachycardia, a generalized unwell feeling, and even hypotension. (A lack of classic tissue inflammatory signs may mask an ongoing necrotizing fasciitis beneath the skin.) The involved region may become numb due to the necrosis of the innervating nerve fibers. Discharge or crepitus may also be noted.
Late clinical signs of necrotizing fasciitis include cellulitis, skin discoloration, discharge of “dishwater” fluid, blistering, and hemorrhagic bullae. Findings of crepitus and soft tissue air on plain radiographs are seen in 37% and 57% of patients, respectively.4 Our patient’s X-ray findings revealed extensive gas pockets within soft tissue and osteomyelitis changes of the 5th metatarsal head.
The differential diagnosis for necrotizing fasciitis includes:
- pyogenic soft tissue cellulitis
- clostridial cellulitis (which may also present with soft tissue crepitus)
- nonclostridial anaerobic cellulitis
- acute febrile neutrophilic dermatosis
- acute hemorrhagic edema of infancy
- erythema induratum (nodular vasculitis).
Diagnosis is often made on clinical evaluation
The definitive diagnosis of necrotizing fasciitis is made by histological examination of the debrided specimen or deep skin tissue biopsy. Fascial necrosis with thrombosed blood vessels and a dense infiltrate of inflammatory cells is seen on histological evaluation.
However, diagnosis is often reached on clinical evaluation. Rapidly deteriorating local signs and symptoms together with systemic toxicity should prompt a working diagnosis of necrotizing fasciitis.
Laboratory tests (white blood cell count, blood urea nitrogen level, sodium levels, creatinine levels, erythrocyte sedimentation rates, C-reactive protein levels) and radiographic evaluation (X-rays, computed tomography [CT], and magnetic resonance imaging [MRI]) are useful adjuncts in reaching the diagnosis.
Prompt treatment is the name of the game
Antibiotic therapy is guided by gram stain and bacterial culture results. (When the clinical suspicion of necrotizing fasciitis is reached, empirical antibiotics should be started right away.) Broad-spectrum antibiotics covering gram positive, gram negative, and anaerobic bacteria should be used. The patient’s age, weight, and liver and renal function should also be considered before starting antibiotics.
Choices of antibiotics include penicillin for gram positive cover and an aminoglycoside or third-generation cephalosporin for gram negative counteraction. Metronidazole (Flagyl) may be considered for anaerobic superimposed infections. Adjunctive clindamycin is also recommended—especially in group A streptococcal infections—because it suppresses toxin production, inhibits M-protein synthesis, and facilitates phagocytosis.5 In addition, hyperbaric oxygen therapy and intravenous immunoglobulin are increasingly being utilized in the management of necrotizing fasciitis.6 Their efficacy has yet to be conclusively established.
Surgical debridement. The importance of prompt and thorough surgical debridement cannot be stressed enough. Large soft-tissue defects created by surgery can be treated with vacuum-assisted closure dressings, local or free soft-tissue flaps, and/or skin grafts. In extreme cases, limb amputation may be necessary.
Aggressive steps were needed for our patient
In the emergency department, we treated our patient with intravenous cloxacillin, gentamicin, and metronidazole. He later underwent surgical debridement and had pus drained from deep abscesses in his foot. Intraoperative findings indicated that there was necrotic tissue involving 80% of the dorsum of the foot and the necrosis extended proximally to the ankle and distally to the toes.
Wound cultures grew group B Streptococcus (Peptostreptococcus species) and Bacteroides. He was treated with intravenous amoxicillin and clavulanic acid (IV Augmentin, available in Singapore) and oral metronidazole. An endocrinologist evaluated him and determined that he had diabetes. He was started on insulin.
Postoperatively, our patient remained septic. Five days later, he underwent a below-the-knee amputation; the surgeons noted that his foot was gangrenous.
Our patient stayed in the hospital for 7 more days and was discharged about 2 weeks after his arrival at the ER.
Correspondence
Ramesh Subramaniam, MBBS, National University Hospital, 5 Lower Kent Ridge Road, Singapore 119074
1. Trent JT, Kirsner RS. Necrotizing fasciitis. Wounds. 2002;14:284-292.
2. Lavery LA, Harkless LB, Felder-Johnson K, et al. Bacterial pathogens in infected puncture wounds in adults with diabetes. J Foot Ankle Surg. 1994;33:91-97.
3. Oliver JD. Wound infections caused by Vibrio vulnificus and other marine bacteria. Epidemiol Infect. 2005;133:383-391.
4. Elliott DC, Kufera JA, Myers RA. Necrotizing soft tissue infections. Risk factors for mortality and strategies for management. Ann Surg. 1996;224:672-683.
5. Stevens DL. Streptococcal toxic shock syndrome: spectrum of disease, pathogenesis and new concepts in treatment. Emerg Infect Dis. 1995;1:69.
6. Young MH, Aronoff DM, Engleberg NC. Necrotizing fasciitis: pathogenesis and treatment. Expert Rev Anti Infect Ther. 2005;3:279-294.
A 49-year-old, unkempt-looking Indian man came into our emergency department in Singapore complaining of increasing right foot swelling and worsening pain that he’d had for a month after having stepped on a fish bone while walking on the beach. Our patient pulled the bone out himself and did not seek immediate medical attention.
Our patient said that he had occasional fever with chills and rigors. His medical history was unremarkable, and there was no history of previous injury or surgery to the right foot. He worked as an “oiler” refueling ships, and said that he did not drink alcohol or smoke.
He was dehydrated, and there were no other sources of infection or sores on his body. Our patient was febrile (38.3°C [100.9°F]), his blood pressure was 112/74 mm Hg, and he was tachycardic at 117 beats per minute.
His right foot was boggy, swollen, and tender with significant crepitus. Brownish discharge was oozing from the sole of his foot (FIGURE 1). There were large infected subcutaneous bullae on the dorsum of his foot (FIGURE 2). We could feel a popliteal pulse in both legs, but could not feel a right dorsalis pedis pulse.
What is your diagnosis?
FIGURE 1
Swollen, dusky foot
FIGURE 2
Dark bullae
Diagnosis: Necrotizing fasciitis
Necrotizing fasciitis infections are characterized by fulminant destruction of tissue, systemic signs of toxicity, and high rates of mortality. The incidence of necrotizing fasciitis in adults is 0.40 cases per 100,000 population, while the incidence in children is 0.08 cases per 100,000 population. Mortality rates as high as 73% have been reported.1 Early clinical suspicion, early surgical intervention (surgical debridement), and systemic antibiotics are of the utmost importance.
Necrotizing fasciitis is caused by gram positive, gram negative, and/or anaerobic bacteria. Local tissue hypoxia from trauma, surgery, or a medically compromised state creates an ideal opportunistic environment for bacterial proliferation.
Necrotizing fasciitis can be broadly classified into 2 main types:
Type 1 necrotizing fasciitis is a polymicrobial infection caused by facultative bacteria along with anaerobes. Polymicrobial infections with anaerobes are common (up to 74%) in infected puncture wounds in patients with diabetes.2
Type 2 necrotizing fasciitis is caused by group A streptococci alone, but sometimes in association with Staphylococcus aureus. Vibrio vulnificus infection has also been reported in fish bone piercing injuries leading to necrotizing fasciitis.3
Typical early signs and symptoms include severe pain, rapidly progressing erythema, dusky or purplish skin discoloration, and systemic signs of septic toxicity such as fever, tachycardia, a generalized unwell feeling, and even hypotension. (A lack of classic tissue inflammatory signs may mask an ongoing necrotizing fasciitis beneath the skin.) The involved region may become numb due to the necrosis of the innervating nerve fibers. Discharge or crepitus may also be noted.
Late clinical signs of necrotizing fasciitis include cellulitis, skin discoloration, discharge of “dishwater” fluid, blistering, and hemorrhagic bullae. Findings of crepitus and soft tissue air on plain radiographs are seen in 37% and 57% of patients, respectively.4 Our patient’s X-ray findings revealed extensive gas pockets within soft tissue and osteomyelitis changes of the 5th metatarsal head.
The differential diagnosis for necrotizing fasciitis includes:
- pyogenic soft tissue cellulitis
- clostridial cellulitis (which may also present with soft tissue crepitus)
- nonclostridial anaerobic cellulitis
- acute febrile neutrophilic dermatosis
- acute hemorrhagic edema of infancy
- erythema induratum (nodular vasculitis).
Diagnosis is often made on clinical evaluation
The definitive diagnosis of necrotizing fasciitis is made by histological examination of the debrided specimen or deep skin tissue biopsy. Fascial necrosis with thrombosed blood vessels and a dense infiltrate of inflammatory cells is seen on histological evaluation.
However, diagnosis is often reached on clinical evaluation. Rapidly deteriorating local signs and symptoms together with systemic toxicity should prompt a working diagnosis of necrotizing fasciitis.
Laboratory tests (white blood cell count, blood urea nitrogen level, sodium levels, creatinine levels, erythrocyte sedimentation rates, C-reactive protein levels) and radiographic evaluation (X-rays, computed tomography [CT], and magnetic resonance imaging [MRI]) are useful adjuncts in reaching the diagnosis.
Prompt treatment is the name of the game
Antibiotic therapy is guided by gram stain and bacterial culture results. (When the clinical suspicion of necrotizing fasciitis is reached, empirical antibiotics should be started right away.) Broad-spectrum antibiotics covering gram positive, gram negative, and anaerobic bacteria should be used. The patient’s age, weight, and liver and renal function should also be considered before starting antibiotics.
Choices of antibiotics include penicillin for gram positive cover and an aminoglycoside or third-generation cephalosporin for gram negative counteraction. Metronidazole (Flagyl) may be considered for anaerobic superimposed infections. Adjunctive clindamycin is also recommended—especially in group A streptococcal infections—because it suppresses toxin production, inhibits M-protein synthesis, and facilitates phagocytosis.5 In addition, hyperbaric oxygen therapy and intravenous immunoglobulin are increasingly being utilized in the management of necrotizing fasciitis.6 Their efficacy has yet to be conclusively established.
Surgical debridement. The importance of prompt and thorough surgical debridement cannot be stressed enough. Large soft-tissue defects created by surgery can be treated with vacuum-assisted closure dressings, local or free soft-tissue flaps, and/or skin grafts. In extreme cases, limb amputation may be necessary.
Aggressive steps were needed for our patient
In the emergency department, we treated our patient with intravenous cloxacillin, gentamicin, and metronidazole. He later underwent surgical debridement and had pus drained from deep abscesses in his foot. Intraoperative findings indicated that there was necrotic tissue involving 80% of the dorsum of the foot and the necrosis extended proximally to the ankle and distally to the toes.
Wound cultures grew group B Streptococcus (Peptostreptococcus species) and Bacteroides. He was treated with intravenous amoxicillin and clavulanic acid (IV Augmentin, available in Singapore) and oral metronidazole. An endocrinologist evaluated him and determined that he had diabetes. He was started on insulin.
Postoperatively, our patient remained septic. Five days later, he underwent a below-the-knee amputation; the surgeons noted that his foot was gangrenous.
Our patient stayed in the hospital for 7 more days and was discharged about 2 weeks after his arrival at the ER.
Correspondence
Ramesh Subramaniam, MBBS, National University Hospital, 5 Lower Kent Ridge Road, Singapore 119074
A 49-year-old, unkempt-looking Indian man came into our emergency department in Singapore complaining of increasing right foot swelling and worsening pain that he’d had for a month after having stepped on a fish bone while walking on the beach. Our patient pulled the bone out himself and did not seek immediate medical attention.
Our patient said that he had occasional fever with chills and rigors. His medical history was unremarkable, and there was no history of previous injury or surgery to the right foot. He worked as an “oiler” refueling ships, and said that he did not drink alcohol or smoke.
He was dehydrated, and there were no other sources of infection or sores on his body. Our patient was febrile (38.3°C [100.9°F]), his blood pressure was 112/74 mm Hg, and he was tachycardic at 117 beats per minute.
His right foot was boggy, swollen, and tender with significant crepitus. Brownish discharge was oozing from the sole of his foot (FIGURE 1). There were large infected subcutaneous bullae on the dorsum of his foot (FIGURE 2). We could feel a popliteal pulse in both legs, but could not feel a right dorsalis pedis pulse.
What is your diagnosis?
FIGURE 1
Swollen, dusky foot
FIGURE 2
Dark bullae
Diagnosis: Necrotizing fasciitis
Necrotizing fasciitis infections are characterized by fulminant destruction of tissue, systemic signs of toxicity, and high rates of mortality. The incidence of necrotizing fasciitis in adults is 0.40 cases per 100,000 population, while the incidence in children is 0.08 cases per 100,000 population. Mortality rates as high as 73% have been reported.1 Early clinical suspicion, early surgical intervention (surgical debridement), and systemic antibiotics are of the utmost importance.
Necrotizing fasciitis is caused by gram positive, gram negative, and/or anaerobic bacteria. Local tissue hypoxia from trauma, surgery, or a medically compromised state creates an ideal opportunistic environment for bacterial proliferation.
Necrotizing fasciitis can be broadly classified into 2 main types:
Type 1 necrotizing fasciitis is a polymicrobial infection caused by facultative bacteria along with anaerobes. Polymicrobial infections with anaerobes are common (up to 74%) in infected puncture wounds in patients with diabetes.2
Type 2 necrotizing fasciitis is caused by group A streptococci alone, but sometimes in association with Staphylococcus aureus. Vibrio vulnificus infection has also been reported in fish bone piercing injuries leading to necrotizing fasciitis.3
Typical early signs and symptoms include severe pain, rapidly progressing erythema, dusky or purplish skin discoloration, and systemic signs of septic toxicity such as fever, tachycardia, a generalized unwell feeling, and even hypotension. (A lack of classic tissue inflammatory signs may mask an ongoing necrotizing fasciitis beneath the skin.) The involved region may become numb due to the necrosis of the innervating nerve fibers. Discharge or crepitus may also be noted.
Late clinical signs of necrotizing fasciitis include cellulitis, skin discoloration, discharge of “dishwater” fluid, blistering, and hemorrhagic bullae. Findings of crepitus and soft tissue air on plain radiographs are seen in 37% and 57% of patients, respectively.4 Our patient’s X-ray findings revealed extensive gas pockets within soft tissue and osteomyelitis changes of the 5th metatarsal head.
The differential diagnosis for necrotizing fasciitis includes:
- pyogenic soft tissue cellulitis
- clostridial cellulitis (which may also present with soft tissue crepitus)
- nonclostridial anaerobic cellulitis
- acute febrile neutrophilic dermatosis
- acute hemorrhagic edema of infancy
- erythema induratum (nodular vasculitis).
Diagnosis is often made on clinical evaluation
The definitive diagnosis of necrotizing fasciitis is made by histological examination of the debrided specimen or deep skin tissue biopsy. Fascial necrosis with thrombosed blood vessels and a dense infiltrate of inflammatory cells is seen on histological evaluation.
However, diagnosis is often reached on clinical evaluation. Rapidly deteriorating local signs and symptoms together with systemic toxicity should prompt a working diagnosis of necrotizing fasciitis.
Laboratory tests (white blood cell count, blood urea nitrogen level, sodium levels, creatinine levels, erythrocyte sedimentation rates, C-reactive protein levels) and radiographic evaluation (X-rays, computed tomography [CT], and magnetic resonance imaging [MRI]) are useful adjuncts in reaching the diagnosis.
Prompt treatment is the name of the game
Antibiotic therapy is guided by gram stain and bacterial culture results. (When the clinical suspicion of necrotizing fasciitis is reached, empirical antibiotics should be started right away.) Broad-spectrum antibiotics covering gram positive, gram negative, and anaerobic bacteria should be used. The patient’s age, weight, and liver and renal function should also be considered before starting antibiotics.
Choices of antibiotics include penicillin for gram positive cover and an aminoglycoside or third-generation cephalosporin for gram negative counteraction. Metronidazole (Flagyl) may be considered for anaerobic superimposed infections. Adjunctive clindamycin is also recommended—especially in group A streptococcal infections—because it suppresses toxin production, inhibits M-protein synthesis, and facilitates phagocytosis.5 In addition, hyperbaric oxygen therapy and intravenous immunoglobulin are increasingly being utilized in the management of necrotizing fasciitis.6 Their efficacy has yet to be conclusively established.
Surgical debridement. The importance of prompt and thorough surgical debridement cannot be stressed enough. Large soft-tissue defects created by surgery can be treated with vacuum-assisted closure dressings, local or free soft-tissue flaps, and/or skin grafts. In extreme cases, limb amputation may be necessary.
Aggressive steps were needed for our patient
In the emergency department, we treated our patient with intravenous cloxacillin, gentamicin, and metronidazole. He later underwent surgical debridement and had pus drained from deep abscesses in his foot. Intraoperative findings indicated that there was necrotic tissue involving 80% of the dorsum of the foot and the necrosis extended proximally to the ankle and distally to the toes.
Wound cultures grew group B Streptococcus (Peptostreptococcus species) and Bacteroides. He was treated with intravenous amoxicillin and clavulanic acid (IV Augmentin, available in Singapore) and oral metronidazole. An endocrinologist evaluated him and determined that he had diabetes. He was started on insulin.
Postoperatively, our patient remained septic. Five days later, he underwent a below-the-knee amputation; the surgeons noted that his foot was gangrenous.
Our patient stayed in the hospital for 7 more days and was discharged about 2 weeks after his arrival at the ER.
Correspondence
Ramesh Subramaniam, MBBS, National University Hospital, 5 Lower Kent Ridge Road, Singapore 119074
1. Trent JT, Kirsner RS. Necrotizing fasciitis. Wounds. 2002;14:284-292.
2. Lavery LA, Harkless LB, Felder-Johnson K, et al. Bacterial pathogens in infected puncture wounds in adults with diabetes. J Foot Ankle Surg. 1994;33:91-97.
3. Oliver JD. Wound infections caused by Vibrio vulnificus and other marine bacteria. Epidemiol Infect. 2005;133:383-391.
4. Elliott DC, Kufera JA, Myers RA. Necrotizing soft tissue infections. Risk factors for mortality and strategies for management. Ann Surg. 1996;224:672-683.
5. Stevens DL. Streptococcal toxic shock syndrome: spectrum of disease, pathogenesis and new concepts in treatment. Emerg Infect Dis. 1995;1:69.
6. Young MH, Aronoff DM, Engleberg NC. Necrotizing fasciitis: pathogenesis and treatment. Expert Rev Anti Infect Ther. 2005;3:279-294.
1. Trent JT, Kirsner RS. Necrotizing fasciitis. Wounds. 2002;14:284-292.
2. Lavery LA, Harkless LB, Felder-Johnson K, et al. Bacterial pathogens in infected puncture wounds in adults with diabetes. J Foot Ankle Surg. 1994;33:91-97.
3. Oliver JD. Wound infections caused by Vibrio vulnificus and other marine bacteria. Epidemiol Infect. 2005;133:383-391.
4. Elliott DC, Kufera JA, Myers RA. Necrotizing soft tissue infections. Risk factors for mortality and strategies for management. Ann Surg. 1996;224:672-683.
5. Stevens DL. Streptococcal toxic shock syndrome: spectrum of disease, pathogenesis and new concepts in treatment. Emerg Infect Dis. 1995;1:69.
6. Young MH, Aronoff DM, Engleberg NC. Necrotizing fasciitis: pathogenesis and treatment. Expert Rev Anti Infect Ther. 2005;3:279-294.
Bilateral thumbnail deformity
During my recent deployment to Afghanistan, a 31-year-old man came into our aid station with a bilateral thumbnail deformity that he’d had for several weeks. He was concerned about a thumbnail infection and wanted to discuss antifungal treatment. His history was unremarkable, and he said he had no nail fold infections or other skin conditions. He was taking doxycycline for malaria prophylaxis. He also mentioned that he had recently started working as a military intelligence officer, putting in 18-hour days.
His exam revealed sharp, closely spaced horizontal grooves on both of his thumbnails (FIGURE). His nails had no hyperkeratotic debris or pitting. His nail folds appeared hypopigmented with scaling, but they were nontender to palpation. His other fingernails were normal in appearance. The remainder of the skin exam was unremarkable.
FIGURE
Horizontal grooves on both thumbnails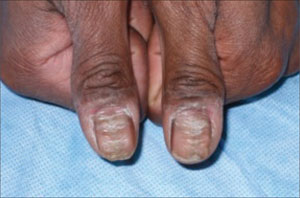
What is your diagnosis?
How would you manage this condition?
Diagnosis: Habit-tic deformity
Habit-tic deformity is a common nail condition caused by a conscious or unconscious rubbing or picking of the proximal nail folds. Horizontal grooves are formed proximally due to nail matrix damage and subsequently move distally with fingernail growth. The dominant thumbnail is most often affected by frequent rubbing with the ipsilateral index fingernail, although all nails can be involved.
This deformity is more common during periods of stress—which my patient was experiencing in his new role as an intelligence officer—and is believed to result from a compulsive or impulse control disorder.
Differential diagnosis includes onychomycosis
The differential diagnosis of external nail deformation includes median nail dystrophy, chronic proximal nail fold inflammation, onychomycosis, psoriasis, Beau’s lines, and habit-tic-like dystrophy.
Median nail dystrophy. This nail deformity has a distinctive longitudinal split in the center of the nail plate with several cracks projecting laterally, resembling a fir tree. The etiology of this disorder is unknown, although some have suggested that habit-tic deformity and median nail dystrophy are different manifestations of the same disorder.1
Chronic proximal nail fold inflammation. Chronic paronychia is an inflammation of the proximal nail fold, and is one of 2 forms of inflammation included in the differential. It is characterized by tenderness and swelling around the proximal and lateral nail folds, most often due to contact irritant exposure. The cuticle separates from the nail plate, which creates a space for infection. The nail plate surface subsequently becomes brown and develops ripples that can mimic the habit-tic deformity. Chronic eczematous inflammation is the second form of inflammation in the differential and can produce results that are similar to chronic paronychia. Both forms of proximal nail fold inflammation tend to resemble rounded waves, as opposed to the narrow, closely spaced grooves seen in habit-tic deformity.
Onychomycosis. Habit-tic deformity is often confused with onychomycosis, though the 2 can be easily distinguished upon close inspection. Unlike habit-tic deformity, onychomycosis classically has nail thickening and hyperkeratotic debris. Diagnosis can be solidified by a potassium hydroxide examination and culture.
Psoriasis. Psoriatic nail lesions can occur in the absence of classic skin findings. Nail pitting is the hallmark nail finding in psoriasis. Other findings such as onycholysis (separation of the nail from the nail bed), nail fragmentation and crumbling, and splinter hemorrhages can distinguish psoriatic nails from habit-tic deformity.
Beau’s lines. This disorder is characterized by horizontal depressions in most or all of the nails. These depressions occur at the proximal portion of the nail weeks after trauma, medication use, or illness interrupts nail formation. Chemotherapeutic agents, systemic illnesses such as syphilis, myocarditis, peripheral vascular disease, and uncontrolled diabetes, as well as illnesses associated with high fevers, such as scarlet fever, hand-foot-mouth disease, and pneumonia, have all been linked to Beau’s lines.2
The lines (usually one per nail) eventually move distally to the nail’s free edge. The nail on either side of the depression is normal in appearance.
Habit-tic-like dystrophy. Nail deformities that closely resemble habit-tic deformity on exam have been reported in patients who explicitly deny trauma. One report in the literature indicates that 2 patients who denied trauma—one with systemic lupus erythematous and another with no significant past medical history—responded to treatment with a multivitamin rich in biotin, vitamins B6, C, and E, and riboflavin.3 Another report describes habit-tic-like deformity that occurred in patients who were taking aromatic retinoids. The disorder resolved when the patients stopped taking the medication.1
Low-tech approach to treatment
The simplest approach to cosmetic improvement is to have the patient tape the proximal portion of the affected nails during the day when he or she picks at and rubs them. The tape acts as a barrier to the repetitive trauma and also serves as a reminder to the patient. The patient can expect improvement in several weeks if the tape is applied consistently. In addition, selective serotonin reuptake inhibitors (SSRIs) may play a role in treatment, because habit-tic deformity is believed to be a compulsive disorder.4
My patient tried the taping method for 2 months and had moderate improvement of his nail deformity. He was not, however, completely satisfied with the results, so we discussed vitamin supplementation as an additional therapy. His tour ended shortly thereafter, and he was lost to follow-up.
Disclosure
The author reported no potential conflict of interest relevant to this article. The opinions or assertions contained herein are the private views of the author and are not to be construed as official or as reflecting the views of the Department of Defense.
Correspondence
Edwin A. Farnell IV, MD, Urgent Care Clinic, Moncrief Army Community Hospital, 4500 Stuart Street, Fort Jackson, SC 29207-5720; [email protected]
1. Griego RD, Orengo IF, Scher RK. Median nail dystrophy and habit tic deformity: are they different forms of the same disorder? Int J Dermatol. 1995;34:799-800.
2. Habif TB. Nail diseases. In: Clinical Dermatology. 4th ed. Philadelphia: Mosby–Elsevier; 2004:869-884.
3. Gloster H. Habit-tic-like and median nail-like dystrophies treated with multivitamins. J Am Acad Dermatol. 2005;53:543-544.
4. Vittorio C, Phillips K. Treatment of habit-tic deformity with fluoxetine. Arch Dermatol. 1997;133:1203-1204.
During my recent deployment to Afghanistan, a 31-year-old man came into our aid station with a bilateral thumbnail deformity that he’d had for several weeks. He was concerned about a thumbnail infection and wanted to discuss antifungal treatment. His history was unremarkable, and he said he had no nail fold infections or other skin conditions. He was taking doxycycline for malaria prophylaxis. He also mentioned that he had recently started working as a military intelligence officer, putting in 18-hour days.
His exam revealed sharp, closely spaced horizontal grooves on both of his thumbnails (FIGURE). His nails had no hyperkeratotic debris or pitting. His nail folds appeared hypopigmented with scaling, but they were nontender to palpation. His other fingernails were normal in appearance. The remainder of the skin exam was unremarkable.
FIGURE
Horizontal grooves on both thumbnails
What is your diagnosis?
How would you manage this condition?
Diagnosis: Habit-tic deformity
Habit-tic deformity is a common nail condition caused by a conscious or unconscious rubbing or picking of the proximal nail folds. Horizontal grooves are formed proximally due to nail matrix damage and subsequently move distally with fingernail growth. The dominant thumbnail is most often affected by frequent rubbing with the ipsilateral index fingernail, although all nails can be involved.
This deformity is more common during periods of stress—which my patient was experiencing in his new role as an intelligence officer—and is believed to result from a compulsive or impulse control disorder.
Differential diagnosis includes onychomycosis
The differential diagnosis of external nail deformation includes median nail dystrophy, chronic proximal nail fold inflammation, onychomycosis, psoriasis, Beau’s lines, and habit-tic-like dystrophy.
Median nail dystrophy. This nail deformity has a distinctive longitudinal split in the center of the nail plate with several cracks projecting laterally, resembling a fir tree. The etiology of this disorder is unknown, although some have suggested that habit-tic deformity and median nail dystrophy are different manifestations of the same disorder.1
Chronic proximal nail fold inflammation. Chronic paronychia is an inflammation of the proximal nail fold, and is one of 2 forms of inflammation included in the differential. It is characterized by tenderness and swelling around the proximal and lateral nail folds, most often due to contact irritant exposure. The cuticle separates from the nail plate, which creates a space for infection. The nail plate surface subsequently becomes brown and develops ripples that can mimic the habit-tic deformity. Chronic eczematous inflammation is the second form of inflammation in the differential and can produce results that are similar to chronic paronychia. Both forms of proximal nail fold inflammation tend to resemble rounded waves, as opposed to the narrow, closely spaced grooves seen in habit-tic deformity.
Onychomycosis. Habit-tic deformity is often confused with onychomycosis, though the 2 can be easily distinguished upon close inspection. Unlike habit-tic deformity, onychomycosis classically has nail thickening and hyperkeratotic debris. Diagnosis can be solidified by a potassium hydroxide examination and culture.
Psoriasis. Psoriatic nail lesions can occur in the absence of classic skin findings. Nail pitting is the hallmark nail finding in psoriasis. Other findings such as onycholysis (separation of the nail from the nail bed), nail fragmentation and crumbling, and splinter hemorrhages can distinguish psoriatic nails from habit-tic deformity.
Beau’s lines. This disorder is characterized by horizontal depressions in most or all of the nails. These depressions occur at the proximal portion of the nail weeks after trauma, medication use, or illness interrupts nail formation. Chemotherapeutic agents, systemic illnesses such as syphilis, myocarditis, peripheral vascular disease, and uncontrolled diabetes, as well as illnesses associated with high fevers, such as scarlet fever, hand-foot-mouth disease, and pneumonia, have all been linked to Beau’s lines.2
The lines (usually one per nail) eventually move distally to the nail’s free edge. The nail on either side of the depression is normal in appearance.
Habit-tic-like dystrophy. Nail deformities that closely resemble habit-tic deformity on exam have been reported in patients who explicitly deny trauma. One report in the literature indicates that 2 patients who denied trauma—one with systemic lupus erythematous and another with no significant past medical history—responded to treatment with a multivitamin rich in biotin, vitamins B6, C, and E, and riboflavin.3 Another report describes habit-tic-like deformity that occurred in patients who were taking aromatic retinoids. The disorder resolved when the patients stopped taking the medication.1
Low-tech approach to treatment
The simplest approach to cosmetic improvement is to have the patient tape the proximal portion of the affected nails during the day when he or she picks at and rubs them. The tape acts as a barrier to the repetitive trauma and also serves as a reminder to the patient. The patient can expect improvement in several weeks if the tape is applied consistently. In addition, selective serotonin reuptake inhibitors (SSRIs) may play a role in treatment, because habit-tic deformity is believed to be a compulsive disorder.4
My patient tried the taping method for 2 months and had moderate improvement of his nail deformity. He was not, however, completely satisfied with the results, so we discussed vitamin supplementation as an additional therapy. His tour ended shortly thereafter, and he was lost to follow-up.
Disclosure
The author reported no potential conflict of interest relevant to this article. The opinions or assertions contained herein are the private views of the author and are not to be construed as official or as reflecting the views of the Department of Defense.
Correspondence
Edwin A. Farnell IV, MD, Urgent Care Clinic, Moncrief Army Community Hospital, 4500 Stuart Street, Fort Jackson, SC 29207-5720; [email protected]
During my recent deployment to Afghanistan, a 31-year-old man came into our aid station with a bilateral thumbnail deformity that he’d had for several weeks. He was concerned about a thumbnail infection and wanted to discuss antifungal treatment. His history was unremarkable, and he said he had no nail fold infections or other skin conditions. He was taking doxycycline for malaria prophylaxis. He also mentioned that he had recently started working as a military intelligence officer, putting in 18-hour days.
His exam revealed sharp, closely spaced horizontal grooves on both of his thumbnails (FIGURE). His nails had no hyperkeratotic debris or pitting. His nail folds appeared hypopigmented with scaling, but they were nontender to palpation. His other fingernails were normal in appearance. The remainder of the skin exam was unremarkable.
FIGURE
Horizontal grooves on both thumbnails
What is your diagnosis?
How would you manage this condition?
Diagnosis: Habit-tic deformity
Habit-tic deformity is a common nail condition caused by a conscious or unconscious rubbing or picking of the proximal nail folds. Horizontal grooves are formed proximally due to nail matrix damage and subsequently move distally with fingernail growth. The dominant thumbnail is most often affected by frequent rubbing with the ipsilateral index fingernail, although all nails can be involved.
This deformity is more common during periods of stress—which my patient was experiencing in his new role as an intelligence officer—and is believed to result from a compulsive or impulse control disorder.
Differential diagnosis includes onychomycosis
The differential diagnosis of external nail deformation includes median nail dystrophy, chronic proximal nail fold inflammation, onychomycosis, psoriasis, Beau’s lines, and habit-tic-like dystrophy.
Median nail dystrophy. This nail deformity has a distinctive longitudinal split in the center of the nail plate with several cracks projecting laterally, resembling a fir tree. The etiology of this disorder is unknown, although some have suggested that habit-tic deformity and median nail dystrophy are different manifestations of the same disorder.1
Chronic proximal nail fold inflammation. Chronic paronychia is an inflammation of the proximal nail fold, and is one of 2 forms of inflammation included in the differential. It is characterized by tenderness and swelling around the proximal and lateral nail folds, most often due to contact irritant exposure. The cuticle separates from the nail plate, which creates a space for infection. The nail plate surface subsequently becomes brown and develops ripples that can mimic the habit-tic deformity. Chronic eczematous inflammation is the second form of inflammation in the differential and can produce results that are similar to chronic paronychia. Both forms of proximal nail fold inflammation tend to resemble rounded waves, as opposed to the narrow, closely spaced grooves seen in habit-tic deformity.
Onychomycosis. Habit-tic deformity is often confused with onychomycosis, though the 2 can be easily distinguished upon close inspection. Unlike habit-tic deformity, onychomycosis classically has nail thickening and hyperkeratotic debris. Diagnosis can be solidified by a potassium hydroxide examination and culture.
Psoriasis. Psoriatic nail lesions can occur in the absence of classic skin findings. Nail pitting is the hallmark nail finding in psoriasis. Other findings such as onycholysis (separation of the nail from the nail bed), nail fragmentation and crumbling, and splinter hemorrhages can distinguish psoriatic nails from habit-tic deformity.
Beau’s lines. This disorder is characterized by horizontal depressions in most or all of the nails. These depressions occur at the proximal portion of the nail weeks after trauma, medication use, or illness interrupts nail formation. Chemotherapeutic agents, systemic illnesses such as syphilis, myocarditis, peripheral vascular disease, and uncontrolled diabetes, as well as illnesses associated with high fevers, such as scarlet fever, hand-foot-mouth disease, and pneumonia, have all been linked to Beau’s lines.2
The lines (usually one per nail) eventually move distally to the nail’s free edge. The nail on either side of the depression is normal in appearance.
Habit-tic-like dystrophy. Nail deformities that closely resemble habit-tic deformity on exam have been reported in patients who explicitly deny trauma. One report in the literature indicates that 2 patients who denied trauma—one with systemic lupus erythematous and another with no significant past medical history—responded to treatment with a multivitamin rich in biotin, vitamins B6, C, and E, and riboflavin.3 Another report describes habit-tic-like deformity that occurred in patients who were taking aromatic retinoids. The disorder resolved when the patients stopped taking the medication.1
Low-tech approach to treatment
The simplest approach to cosmetic improvement is to have the patient tape the proximal portion of the affected nails during the day when he or she picks at and rubs them. The tape acts as a barrier to the repetitive trauma and also serves as a reminder to the patient. The patient can expect improvement in several weeks if the tape is applied consistently. In addition, selective serotonin reuptake inhibitors (SSRIs) may play a role in treatment, because habit-tic deformity is believed to be a compulsive disorder.4
My patient tried the taping method for 2 months and had moderate improvement of his nail deformity. He was not, however, completely satisfied with the results, so we discussed vitamin supplementation as an additional therapy. His tour ended shortly thereafter, and he was lost to follow-up.
Disclosure
The author reported no potential conflict of interest relevant to this article. The opinions or assertions contained herein are the private views of the author and are not to be construed as official or as reflecting the views of the Department of Defense.
Correspondence
Edwin A. Farnell IV, MD, Urgent Care Clinic, Moncrief Army Community Hospital, 4500 Stuart Street, Fort Jackson, SC 29207-5720; [email protected]
1. Griego RD, Orengo IF, Scher RK. Median nail dystrophy and habit tic deformity: are they different forms of the same disorder? Int J Dermatol. 1995;34:799-800.
2. Habif TB. Nail diseases. In: Clinical Dermatology. 4th ed. Philadelphia: Mosby–Elsevier; 2004:869-884.
3. Gloster H. Habit-tic-like and median nail-like dystrophies treated with multivitamins. J Am Acad Dermatol. 2005;53:543-544.
4. Vittorio C, Phillips K. Treatment of habit-tic deformity with fluoxetine. Arch Dermatol. 1997;133:1203-1204.
1. Griego RD, Orengo IF, Scher RK. Median nail dystrophy and habit tic deformity: are they different forms of the same disorder? Int J Dermatol. 1995;34:799-800.
2. Habif TB. Nail diseases. In: Clinical Dermatology. 4th ed. Philadelphia: Mosby–Elsevier; 2004:869-884.
3. Gloster H. Habit-tic-like and median nail-like dystrophies treated with multivitamins. J Am Acad Dermatol. 2005;53:543-544.
4. Vittorio C, Phillips K. Treatment of habit-tic deformity with fluoxetine. Arch Dermatol. 1997;133:1203-1204.
Tumor with central crusting
A 63-year-old man came into our dermatologic surgery clinic with a growth on his left cheek just anterior to his sideburn (FIGURE). Our patient indicated that the lesion, which appeared 6 weeks earlier, started as a small, hard papule with a central depression and rapidly grew to reach its current size and shape.
The patient’s face was sun damaged, and he had a prominent 2.1×1.8 cm well-circumscribed, skin-colored tumor on his cheek. The tumor had a central depression covered by a crust that appeared to conceal a deep keratinous plug. The tumor also had a volcano-like shape and was firm in texture, but tender to palpation and pressure. No lymphadenopathy was present.
The patient had a history of extensive sun exposure, and he’d had previous nonmelanoma skin cancers treated with various medical and surgical techniques. The rest of his history and exam were within normal limits. We performed a biopsy to confirm our clinical diagnosis.
FIGURE
Rapidly growing skin-colored tumor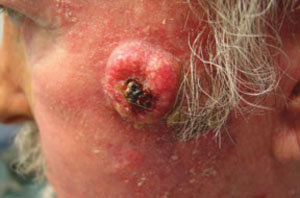
What is your diagnosis?
How would you manage this condition?
Diagnosis: Solitary keratoacanthoma
Our patient had a solitary keratoacanthoma, a unique epidermal tumor that’s characterized by rapid, abundant growth and spontaneous resolution. This tumor goes by many names—molluscum sebaceum, molluscum pseudocarcinomatosum, cutaneous sebaceous neoplasm, and self-healing squamous epithelioma—but keratoacanthoma is the preferred term.1,2
There are several types of keratoacanthoma, but solitary keratoacanthoma remains the most common. It is typically found in light-skinned people in hair-bearing, sun-exposed areas. Peak incidence occurs between the ages of 50 and 69, although the tumors have been reported in patients of all ages. Both sexes are about equally affected, although there is a slight predilection for males. Keratoacanthomas mainly develop on the face (lower lip, cheek, nose, and eyelid), neck, and hands.
The tumors are often considered benign, but they become aggressive in 20% of cases—showing signs of perineural, perivascular, and intravascular invasion and metastases to regional lymph nodes. As a result, some clinicians consider keratoacanthoma to be a pseudomalignancy with self-regressing potential, while others view it as a pseudo-benign tumor progressing into an invasive squamous cell carcinoma (SCC).1,2
The exact etiology of keratoacanthoma is unknown. However, several precipitating factors have been implicated. Primary among them is exposure to ultraviolet light. Other etiological factors include:3-6
- chemical carcinogens (tar, pitch, mineral oil, and cigarette smoking)
- trauma (body peel, carbon dioxide laser resurfacing, megavoltage radiation therapy, and cryosurgery)
- immunosuppression
- surgical scar
- human papilloma virus (HPV).
A tumor with 3 stages
Keratoacanthoma undergoes a proliferative, mature, and involution stage.
In the proliferative stage, there is a rapid increase in tumor size; the tumor can get as big as 10 to 25 mm in diameter in 6 to 8 weeks.1,7
In the mature stage, the tumor stops growing and maintains a typical volcano-like form with a central keratin-filled crater.
In the involution stage, up to 50% of keratoacanthomas undergo spontaneous resolution with expulsion of the keratin plug and resorption of the tumoral mass. The process lasts 4 to 6 weeks, on average, but may take up to 1 year. What’s left behind is a residual atrophic and hypopigmented scar.8
Some lesions persist for a year or more, although the entire process from start to spontaneous resolution usually takes about 4 to 9 months.2,7,8
Is it an SCC or keratoacanthoma?
Histology is the gold standard in diagnosing a keratoacanthoma. A deep biopsy specimen that preferably includes part or full subcutaneous fat with excision of the entire lesion should allow for good histologic interpretation and diagnosis. Keratoacanthoma presents as a downgrowth of well-differentiated squamous epithelium. However, even with a well-performed biopsy, the diagnosis of keratoacanthoma remains challenging due to the lack of sufficient sensitive or specific histological features that can distinguish between keratoacanthomas and SCCs.
As a rule, a normal surface epithelium surrounding the keratin plug with sharp demarcation between tumor and stroma favors keratoacanthoma, whereas ulceration, numerous mitoses, and marked pleomorphism/anaplasia favor SCC. Because of the lack of a universal diagnostic criterion, several experts recommend that all keratoacanthomas be considered potential SCCs and thus treated as such.1,2
When in doubt, cut it out
Although the natural course of a keratoacanthoma is spontaneous regression, the lack of reliable criteria to differentiate it from an SCC with confidence renders therapeutic intervention the safest approach. Solitary keratoacanthomas respond well to surgical excision and may require aggressive procedures if they become too large or invade other structures. Since Mohs’ micrographic surgery is tissue sparing, consider it the treatment of choice if the keratoacanthoma is located in a sensitive area, such as the face.
Cryotherapy with liquid nitrogen, electrodessication and curettage, radiation therapy, and CO2 laser surgery have all been used in small solitary keratoacanthomas with good success.9,10 Other treatment options include intralesional and/or topical treatment using several compounds, such as 5-fluorouracil, corticosteroids, bleomycin, imiquimod, interferon alpha 2b, and methotrexate.7,9-13
Keratoacanthoma patients are often UV light sensitive, so they must avoid excessive sun exposure and use sunscreen with high SPF at all times to prevent recurrence and minimize scarring.
We opted for Mohs’ surgery for our patient
Given the cosmetically sensitive location of our patient’s keratoacanthoma, the size of it, and the patient’s history of skin cancers, we decided to use Mohs’ micrographic surgery for the management of this tumor, with good clinical outcome. There were no new lesions or recurrence on follow-up visit 6 months later.
Correspondence
Amor Khachemoune, MD, CWS, Assistant Professor, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016; [email protected].
1. Beham A, Regauer S, Soyer HP, Beham-Schmid C. Keratoacanthoma: a clinically distinct variant of well differentiated squamous cell carcinoma. Ad Anat Pathol. 1998;5:269-280.
2. Schwartz RA. Keratoacanthoma: a clinico-pathologic enigma. Dermatol Surg. 2004;30(2 Pt 2):326-333; discussion 333.
3. Miot HA, Miot LD, da Costa AL, Matsuo CY, Stolf HO, Marques ME. Association between solitary keratoacanthoma and cigarette smoking: a case-control study. Dermatol Online J. 2006;12(2):2.-
4. Pattee SF, Silvis NG. Keratoacanthoma developing in sites of previous trauma: a report of two cases and review of the literature. J Am Acad Dermatol. 2003;48(2 suppl):S35-S38.
5. Goldberg LH, Silapunt S, Beyrau KK, Peterson SR, Friedman PM, Alam M. Keratoacanthoma as a postoperative complication of skin cancer excision. J Am Acad Dermatol. 2004;50:753-758.
6. Stockfleth E, Meinke B, Arndt R, Christophers E, Meyer T. Identification of DNA sequences of both genital and cutaneous HPV types in a small number of keratoacanthomas of nonimmunosuppressed patients. Dermatology. 1999;198:122-125.
7. Oh CK, Son HS, Lee JB, Jang HS, Kwon KS. Intralesional interferon alfa-2b treatment of keratoacanthomas. J Am Acad Dermatol. 2004;51(5 suppl):S177-S180.
8. Griffiths RW. Keratoacanthoma observed. Br J Plast Surg. 2004;57:485-501.
9. Caccialanza M, Sopelana N. Radiation therapy of keratoacanthomas: results in 55 patients. Int J Radiat Oncol Biol Phys. 1989;16:475-477.
10. Gray RJ, Meland NB. Topical 5-fluorouracil as primary therapy for keratoacanthoma. Ann Plast Surg. 2000;44:82-85.
11. Sanders S, Busam KJ, Halpern AC, Nehal KS. Intralesional corticosteroid treatment of multiple eruptive keratoacanthomas: case report and review of a controversial therapy. Dermatol Surg. 2002;28:954-958.
12. Di Lernia V, Ricci C, Albertini G. Spontaneous regression of keratoacanthoma can be promoted by topical treatment with imiquimod cream. J Eur Acad Dermatol Venereol. 2004;18:626-629.
13. Cohen PR, Schulze KE, Teller CF, Nelson BR. Intralesional methotrexate for keratoacanthoma of the nose. Skinmed. 2005;4:393-395.
A 63-year-old man came into our dermatologic surgery clinic with a growth on his left cheek just anterior to his sideburn (FIGURE). Our patient indicated that the lesion, which appeared 6 weeks earlier, started as a small, hard papule with a central depression and rapidly grew to reach its current size and shape.
The patient’s face was sun damaged, and he had a prominent 2.1×1.8 cm well-circumscribed, skin-colored tumor on his cheek. The tumor had a central depression covered by a crust that appeared to conceal a deep keratinous plug. The tumor also had a volcano-like shape and was firm in texture, but tender to palpation and pressure. No lymphadenopathy was present.
The patient had a history of extensive sun exposure, and he’d had previous nonmelanoma skin cancers treated with various medical and surgical techniques. The rest of his history and exam were within normal limits. We performed a biopsy to confirm our clinical diagnosis.
FIGURE
Rapidly growing skin-colored tumor
What is your diagnosis?
How would you manage this condition?
Diagnosis: Solitary keratoacanthoma
Our patient had a solitary keratoacanthoma, a unique epidermal tumor that’s characterized by rapid, abundant growth and spontaneous resolution. This tumor goes by many names—molluscum sebaceum, molluscum pseudocarcinomatosum, cutaneous sebaceous neoplasm, and self-healing squamous epithelioma—but keratoacanthoma is the preferred term.1,2
There are several types of keratoacanthoma, but solitary keratoacanthoma remains the most common. It is typically found in light-skinned people in hair-bearing, sun-exposed areas. Peak incidence occurs between the ages of 50 and 69, although the tumors have been reported in patients of all ages. Both sexes are about equally affected, although there is a slight predilection for males. Keratoacanthomas mainly develop on the face (lower lip, cheek, nose, and eyelid), neck, and hands.
The tumors are often considered benign, but they become aggressive in 20% of cases—showing signs of perineural, perivascular, and intravascular invasion and metastases to regional lymph nodes. As a result, some clinicians consider keratoacanthoma to be a pseudomalignancy with self-regressing potential, while others view it as a pseudo-benign tumor progressing into an invasive squamous cell carcinoma (SCC).1,2
The exact etiology of keratoacanthoma is unknown. However, several precipitating factors have been implicated. Primary among them is exposure to ultraviolet light. Other etiological factors include:3-6
- chemical carcinogens (tar, pitch, mineral oil, and cigarette smoking)
- trauma (body peel, carbon dioxide laser resurfacing, megavoltage radiation therapy, and cryosurgery)
- immunosuppression
- surgical scar
- human papilloma virus (HPV).
A tumor with 3 stages
Keratoacanthoma undergoes a proliferative, mature, and involution stage.
In the proliferative stage, there is a rapid increase in tumor size; the tumor can get as big as 10 to 25 mm in diameter in 6 to 8 weeks.1,7
In the mature stage, the tumor stops growing and maintains a typical volcano-like form with a central keratin-filled crater.
In the involution stage, up to 50% of keratoacanthomas undergo spontaneous resolution with expulsion of the keratin plug and resorption of the tumoral mass. The process lasts 4 to 6 weeks, on average, but may take up to 1 year. What’s left behind is a residual atrophic and hypopigmented scar.8
Some lesions persist for a year or more, although the entire process from start to spontaneous resolution usually takes about 4 to 9 months.2,7,8
Is it an SCC or keratoacanthoma?
Histology is the gold standard in diagnosing a keratoacanthoma. A deep biopsy specimen that preferably includes part or full subcutaneous fat with excision of the entire lesion should allow for good histologic interpretation and diagnosis. Keratoacanthoma presents as a downgrowth of well-differentiated squamous epithelium. However, even with a well-performed biopsy, the diagnosis of keratoacanthoma remains challenging due to the lack of sufficient sensitive or specific histological features that can distinguish between keratoacanthomas and SCCs.
As a rule, a normal surface epithelium surrounding the keratin plug with sharp demarcation between tumor and stroma favors keratoacanthoma, whereas ulceration, numerous mitoses, and marked pleomorphism/anaplasia favor SCC. Because of the lack of a universal diagnostic criterion, several experts recommend that all keratoacanthomas be considered potential SCCs and thus treated as such.1,2
When in doubt, cut it out
Although the natural course of a keratoacanthoma is spontaneous regression, the lack of reliable criteria to differentiate it from an SCC with confidence renders therapeutic intervention the safest approach. Solitary keratoacanthomas respond well to surgical excision and may require aggressive procedures if they become too large or invade other structures. Since Mohs’ micrographic surgery is tissue sparing, consider it the treatment of choice if the keratoacanthoma is located in a sensitive area, such as the face.
Cryotherapy with liquid nitrogen, electrodessication and curettage, radiation therapy, and CO2 laser surgery have all been used in small solitary keratoacanthomas with good success.9,10 Other treatment options include intralesional and/or topical treatment using several compounds, such as 5-fluorouracil, corticosteroids, bleomycin, imiquimod, interferon alpha 2b, and methotrexate.7,9-13
Keratoacanthoma patients are often UV light sensitive, so they must avoid excessive sun exposure and use sunscreen with high SPF at all times to prevent recurrence and minimize scarring.
We opted for Mohs’ surgery for our patient
Given the cosmetically sensitive location of our patient’s keratoacanthoma, the size of it, and the patient’s history of skin cancers, we decided to use Mohs’ micrographic surgery for the management of this tumor, with good clinical outcome. There were no new lesions or recurrence on follow-up visit 6 months later.
Correspondence
Amor Khachemoune, MD, CWS, Assistant Professor, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016; [email protected].
A 63-year-old man came into our dermatologic surgery clinic with a growth on his left cheek just anterior to his sideburn (FIGURE). Our patient indicated that the lesion, which appeared 6 weeks earlier, started as a small, hard papule with a central depression and rapidly grew to reach its current size and shape.
The patient’s face was sun damaged, and he had a prominent 2.1×1.8 cm well-circumscribed, skin-colored tumor on his cheek. The tumor had a central depression covered by a crust that appeared to conceal a deep keratinous plug. The tumor also had a volcano-like shape and was firm in texture, but tender to palpation and pressure. No lymphadenopathy was present.
The patient had a history of extensive sun exposure, and he’d had previous nonmelanoma skin cancers treated with various medical and surgical techniques. The rest of his history and exam were within normal limits. We performed a biopsy to confirm our clinical diagnosis.
FIGURE
Rapidly growing skin-colored tumor
What is your diagnosis?
How would you manage this condition?
Diagnosis: Solitary keratoacanthoma
Our patient had a solitary keratoacanthoma, a unique epidermal tumor that’s characterized by rapid, abundant growth and spontaneous resolution. This tumor goes by many names—molluscum sebaceum, molluscum pseudocarcinomatosum, cutaneous sebaceous neoplasm, and self-healing squamous epithelioma—but keratoacanthoma is the preferred term.1,2
There are several types of keratoacanthoma, but solitary keratoacanthoma remains the most common. It is typically found in light-skinned people in hair-bearing, sun-exposed areas. Peak incidence occurs between the ages of 50 and 69, although the tumors have been reported in patients of all ages. Both sexes are about equally affected, although there is a slight predilection for males. Keratoacanthomas mainly develop on the face (lower lip, cheek, nose, and eyelid), neck, and hands.
The tumors are often considered benign, but they become aggressive in 20% of cases—showing signs of perineural, perivascular, and intravascular invasion and metastases to regional lymph nodes. As a result, some clinicians consider keratoacanthoma to be a pseudomalignancy with self-regressing potential, while others view it as a pseudo-benign tumor progressing into an invasive squamous cell carcinoma (SCC).1,2
The exact etiology of keratoacanthoma is unknown. However, several precipitating factors have been implicated. Primary among them is exposure to ultraviolet light. Other etiological factors include:3-6
- chemical carcinogens (tar, pitch, mineral oil, and cigarette smoking)
- trauma (body peel, carbon dioxide laser resurfacing, megavoltage radiation therapy, and cryosurgery)
- immunosuppression
- surgical scar
- human papilloma virus (HPV).
A tumor with 3 stages
Keratoacanthoma undergoes a proliferative, mature, and involution stage.
In the proliferative stage, there is a rapid increase in tumor size; the tumor can get as big as 10 to 25 mm in diameter in 6 to 8 weeks.1,7
In the mature stage, the tumor stops growing and maintains a typical volcano-like form with a central keratin-filled crater.
In the involution stage, up to 50% of keratoacanthomas undergo spontaneous resolution with expulsion of the keratin plug and resorption of the tumoral mass. The process lasts 4 to 6 weeks, on average, but may take up to 1 year. What’s left behind is a residual atrophic and hypopigmented scar.8
Some lesions persist for a year or more, although the entire process from start to spontaneous resolution usually takes about 4 to 9 months.2,7,8
Is it an SCC or keratoacanthoma?
Histology is the gold standard in diagnosing a keratoacanthoma. A deep biopsy specimen that preferably includes part or full subcutaneous fat with excision of the entire lesion should allow for good histologic interpretation and diagnosis. Keratoacanthoma presents as a downgrowth of well-differentiated squamous epithelium. However, even with a well-performed biopsy, the diagnosis of keratoacanthoma remains challenging due to the lack of sufficient sensitive or specific histological features that can distinguish between keratoacanthomas and SCCs.
As a rule, a normal surface epithelium surrounding the keratin plug with sharp demarcation between tumor and stroma favors keratoacanthoma, whereas ulceration, numerous mitoses, and marked pleomorphism/anaplasia favor SCC. Because of the lack of a universal diagnostic criterion, several experts recommend that all keratoacanthomas be considered potential SCCs and thus treated as such.1,2
When in doubt, cut it out
Although the natural course of a keratoacanthoma is spontaneous regression, the lack of reliable criteria to differentiate it from an SCC with confidence renders therapeutic intervention the safest approach. Solitary keratoacanthomas respond well to surgical excision and may require aggressive procedures if they become too large or invade other structures. Since Mohs’ micrographic surgery is tissue sparing, consider it the treatment of choice if the keratoacanthoma is located in a sensitive area, such as the face.
Cryotherapy with liquid nitrogen, electrodessication and curettage, radiation therapy, and CO2 laser surgery have all been used in small solitary keratoacanthomas with good success.9,10 Other treatment options include intralesional and/or topical treatment using several compounds, such as 5-fluorouracil, corticosteroids, bleomycin, imiquimod, interferon alpha 2b, and methotrexate.7,9-13
Keratoacanthoma patients are often UV light sensitive, so they must avoid excessive sun exposure and use sunscreen with high SPF at all times to prevent recurrence and minimize scarring.
We opted for Mohs’ surgery for our patient
Given the cosmetically sensitive location of our patient’s keratoacanthoma, the size of it, and the patient’s history of skin cancers, we decided to use Mohs’ micrographic surgery for the management of this tumor, with good clinical outcome. There were no new lesions or recurrence on follow-up visit 6 months later.
Correspondence
Amor Khachemoune, MD, CWS, Assistant Professor, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016; [email protected].
1. Beham A, Regauer S, Soyer HP, Beham-Schmid C. Keratoacanthoma: a clinically distinct variant of well differentiated squamous cell carcinoma. Ad Anat Pathol. 1998;5:269-280.
2. Schwartz RA. Keratoacanthoma: a clinico-pathologic enigma. Dermatol Surg. 2004;30(2 Pt 2):326-333; discussion 333.
3. Miot HA, Miot LD, da Costa AL, Matsuo CY, Stolf HO, Marques ME. Association between solitary keratoacanthoma and cigarette smoking: a case-control study. Dermatol Online J. 2006;12(2):2.-
4. Pattee SF, Silvis NG. Keratoacanthoma developing in sites of previous trauma: a report of two cases and review of the literature. J Am Acad Dermatol. 2003;48(2 suppl):S35-S38.
5. Goldberg LH, Silapunt S, Beyrau KK, Peterson SR, Friedman PM, Alam M. Keratoacanthoma as a postoperative complication of skin cancer excision. J Am Acad Dermatol. 2004;50:753-758.
6. Stockfleth E, Meinke B, Arndt R, Christophers E, Meyer T. Identification of DNA sequences of both genital and cutaneous HPV types in a small number of keratoacanthomas of nonimmunosuppressed patients. Dermatology. 1999;198:122-125.
7. Oh CK, Son HS, Lee JB, Jang HS, Kwon KS. Intralesional interferon alfa-2b treatment of keratoacanthomas. J Am Acad Dermatol. 2004;51(5 suppl):S177-S180.
8. Griffiths RW. Keratoacanthoma observed. Br J Plast Surg. 2004;57:485-501.
9. Caccialanza M, Sopelana N. Radiation therapy of keratoacanthomas: results in 55 patients. Int J Radiat Oncol Biol Phys. 1989;16:475-477.
10. Gray RJ, Meland NB. Topical 5-fluorouracil as primary therapy for keratoacanthoma. Ann Plast Surg. 2000;44:82-85.
11. Sanders S, Busam KJ, Halpern AC, Nehal KS. Intralesional corticosteroid treatment of multiple eruptive keratoacanthomas: case report and review of a controversial therapy. Dermatol Surg. 2002;28:954-958.
12. Di Lernia V, Ricci C, Albertini G. Spontaneous regression of keratoacanthoma can be promoted by topical treatment with imiquimod cream. J Eur Acad Dermatol Venereol. 2004;18:626-629.
13. Cohen PR, Schulze KE, Teller CF, Nelson BR. Intralesional methotrexate for keratoacanthoma of the nose. Skinmed. 2005;4:393-395.
1. Beham A, Regauer S, Soyer HP, Beham-Schmid C. Keratoacanthoma: a clinically distinct variant of well differentiated squamous cell carcinoma. Ad Anat Pathol. 1998;5:269-280.
2. Schwartz RA. Keratoacanthoma: a clinico-pathologic enigma. Dermatol Surg. 2004;30(2 Pt 2):326-333; discussion 333.
3. Miot HA, Miot LD, da Costa AL, Matsuo CY, Stolf HO, Marques ME. Association between solitary keratoacanthoma and cigarette smoking: a case-control study. Dermatol Online J. 2006;12(2):2.-
4. Pattee SF, Silvis NG. Keratoacanthoma developing in sites of previous trauma: a report of two cases and review of the literature. J Am Acad Dermatol. 2003;48(2 suppl):S35-S38.
5. Goldberg LH, Silapunt S, Beyrau KK, Peterson SR, Friedman PM, Alam M. Keratoacanthoma as a postoperative complication of skin cancer excision. J Am Acad Dermatol. 2004;50:753-758.
6. Stockfleth E, Meinke B, Arndt R, Christophers E, Meyer T. Identification of DNA sequences of both genital and cutaneous HPV types in a small number of keratoacanthomas of nonimmunosuppressed patients. Dermatology. 1999;198:122-125.
7. Oh CK, Son HS, Lee JB, Jang HS, Kwon KS. Intralesional interferon alfa-2b treatment of keratoacanthomas. J Am Acad Dermatol. 2004;51(5 suppl):S177-S180.
8. Griffiths RW. Keratoacanthoma observed. Br J Plast Surg. 2004;57:485-501.
9. Caccialanza M, Sopelana N. Radiation therapy of keratoacanthomas: results in 55 patients. Int J Radiat Oncol Biol Phys. 1989;16:475-477.
10. Gray RJ, Meland NB. Topical 5-fluorouracil as primary therapy for keratoacanthoma. Ann Plast Surg. 2000;44:82-85.
11. Sanders S, Busam KJ, Halpern AC, Nehal KS. Intralesional corticosteroid treatment of multiple eruptive keratoacanthomas: case report and review of a controversial therapy. Dermatol Surg. 2002;28:954-958.
12. Di Lernia V, Ricci C, Albertini G. Spontaneous regression of keratoacanthoma can be promoted by topical treatment with imiquimod cream. J Eur Acad Dermatol Venereol. 2004;18:626-629.
13. Cohen PR, Schulze KE, Teller CF, Nelson BR. Intralesional methotrexate for keratoacanthoma of the nose. Skinmed. 2005;4:393-395.
Growing plaque on foot
An 82-year-old African American woman with a history of pancreatic cancer came into the clinic for evaluation of a growing, asymptomatic lesion on her right dorsal foot. She first noticed the lesion a year ago, when it was pinpoint size. It was now a 2.5 cm × 1 cm hyperpigmented plaque.
The lesion was dark brown and black and had irregular borders. It also had a central hyperkeratotic area (FIGURE 1). There was no inguinal lymphadenopathy. We performed an incisional biopsy.
FIGURE 1
Growing lesion with irregular borders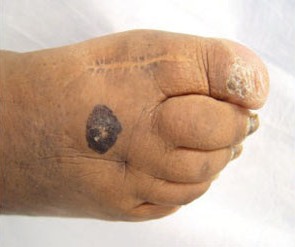
What is your diagnosis?
How would you manage this condition?
Diagnosis: Pigmented Bowen’s disease
Histopathological evaluation of the lesion revealed hyperkeratosis, large atypical keratinocytes, increased mitotic figures, and an intact basement membrane (FIGURE 2), leading us to diagnose pigmented Bowen’s disease.
Bowen’s disease, an intraepidermal squamous cell carcinoma (carcinoma in situ), is a common type of nonmelanoma skin cancer. However, the form our patient had—pigmented Bowen’s disease—is a rare form of squamous cell carcinoma (SCC) in situ (2% of cases).1 The pigmented form of Bowen’s disease is more common in individuals with darker skin tones, while the nonpigmented is more common in fair-skinned individuals.
Bowen’s disease typically presents as a slow-growing, sharply demarcated, scaly erythematous plaque ranging in size from a few millimeters to several centimeters. Crusting, fissuring, hyperkeratosis, and pigmentation, as seen in our case, are also associated findings.2 Bowen’s disease often presents as a solitary lesion, with most cases (approximately 75%) associated with sun damage.3
The most common sites for Bowen’s disease include the head, neck, and hands. Rarely, the nail bed, oral mucosa, or anogenital region may be affected.
The mean age of diagnosis occurs in the sixth decade4 and there is an equal incidence in men and women. Bowen’s disease in men usually occurs on the scalp and ears, while in women, the lower legs are the most common site.5 Three to eight percent of Bowen’s disease cases progress to invasive carcinoma if left untreated.6
FIGURE 2
Histopathology of pigmented Bowen’s disease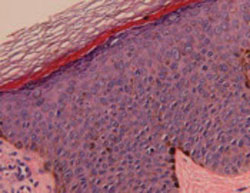
A disease that’s linked to the sun—but also, HPV
The development of Bowen’s disease has been linked to sunlight exposure, human papilloma virus (HPV), and chronic arsenic intoxication.7
Sunlight exposure. Cumulative ultraviolet sunlight exposure is one of the most important etiologic factors. There is a doubling in the incidence of SCC for every 8- to 10-degree decrease in latitude.8
HPV. Human papillomavirus is common in patients who have SCCs in their genital areas.9 There is a poor correlation between nonanogenital Bowen’s disease and HPV infection. However, HPV types 2, 16, 18, 34, and 35 are occasionally identified in these lesions.10
Patients with penile Bowen’s disease (referred to as erythroplasia of Queyrat) are typically uncircumcised men with red, velvety plaques on the glans penis. Occasional itching and bleeding may be associated symptoms.
Arsenic intoxication. Chronic arsenic poisoning from drinking water is a documented cause of cancers occurring in the lung, bladder, kidney, liver, and skin. The US Geological Survey found the highest arsenic contamination levels of ground-water in the West, Midwest, and North-east United States.11
Unlike non-arsenical Bowen’s disease, arsenic-induced Bowen’s disease (As-BD) can occur on non-sun-exposed skin. As-BD typically appears 10 years after initial arsenic exposure, with pulmonary carcinoma appearing 30 years after exposure. As a result, it’s advisable to screen all patients with As-BD for cancer of the lung and bladder.12
Is it Bowen’s, or something more serious?
The differential diagnosis of this lesion includes superficial spreading melanoma, pigmented basal cell carcinoma, atypical melanocytic nevus, and seborrheic keratosis. These different skin conditions may be difficult to distinguish on clinical examination and ultimately may require a biopsy.
Although pigmented Bowen’s disease can occur in anyone, Caucasian patients (as noted earlier) tend to have the more typical nonpigmented, erythematous scaly plaques in sun-exposed sites (FIGURE 3). Darker pigmented individuals are more likely to present with pigmented cutaneous lesions, which may mimic malignant melanoma,13 as was the case with our patient.
FIGURE 3
Nonpigmented Bowen’s disease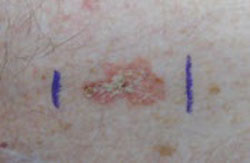
Surgical excision is extremely effective
Bowen’s disease can be treated with cryotherapy; curettage and electrodesiccation; surgical excision, including Mohs micrographic surgery; laser surgery; photodynamic therapy; radiation therapy; topical 5-fluorouracil; and topical imiquimod. Invasive or higher risk lesions require surgical excision or Mohs surgery. Surgical excision of SCCs is extremely effective, with 5-year cure rates of 92%.14
A delay in treatment for our patient
Our patient was scheduled to undergo surgical excision with graft repair of the site. However, she was receiving chemotherapy for mucinous adenocarcinoma of the pancreas and declined excision due to concerns about possible infection.
She later underwent curettage and electrodesiccation, followed by topical imiquimod therapy for 10 weeks. She remains free of any Bowen’s disease recurrences 2 years after her diagnosis.
Correspondence
Claudia Hernandez, MD, Department of Dermatology, University of Illinois at Chicago, 808 S Wood St, MC 624, Chicago, IL 60612-7300.
1. Ragi G, Turner MS, Klein LE, Stoll HL. Pigmented Bowen’s disease and review of 420 Bowen’s disease lesions. J Dermatol Surg Oncol. 1988;14:756-759.
2. Rinker MH, Fenske NA, Scalf LA, Glass LF. Histologic variants of squamous cell carcinoma of the skin. Can Control. 2001;8:354-363.
3. Lee MM, Wick MM. Bowen’s disease. CA Cancer J Clin. 1990;40:237-242.
4. Kossard S, Rosen R. Cutaneous Bowen’s disease. J Am Acad Dermatol. 1992;27:406-410.
5. VanderSpek LAL, Pond GR, Wells W, Tsang RW. Radiation therapy for Bowen’s disease of the skin. Int J Radiat Oncol Biol Phys. 2005;63:505-510.
6. Leibovitch I, Huilgol SC, Selva D, Richards S, Paver R. Cutaneous squamous carcinoma in situ (Bowen’s disease: treatment with Mohs micrographic surgery. J Am Acad Dermatol. 2005;52:997-1002.
7. Stante M, De Giorgi V, Massi D, Chiarugi A, Carli P. Pigmented Bowen’s disease mimicking cutaneous melanoma: clinical and dermoscopic aspects. Dermatol Surg. 2004;30:541-544.
8. Rundel RD. Promotional effects of ultraviolet radiation on human basal and squamous cell carcinoma. Photochem Photobiol. 1983;38:569-575.
9. Crum CP, Ikenberg H, Richart RM, et al. Human papillomavirus type 16 and early cervical neoplasia. N Engl J Med. 1984;310:880-883.
10. Amagi N, Feldman B, Lobel S, Williams C. Irregularly pigmented hyperkeratotic plaque on the thumb. Arch Dermatol. 1993;129:1045-1048.
11. United States Geologic Survey. Arsenic in Ground-water Resources of the United States. Available at: http://water.usgs.gov/nawqa/trace/pubs/fs-063-00/fs-063-00.pdf. Accessed July 29, 2008.
12. Yu HS, Liao WT, Chai CY. Arsenic carcinogenesis in the skin. J Biomed Sci. 2006;13:657-66.
13. Krishnan R, Lewis A, Orengo IF, Rosen T. Pigmented Bowen’s disease (squamous cell carcinoma in situ): a mimic of malignant melanoma. Dermatol Surg. 2001;27:673-674.
14. Miller SJ, Moresi JM. Actinic keratosis, basal cell carcinoma, and squamous cell carcinoma. In: Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. Vol. 2. New York, NY: Mosby; 2003:1677-1696.
An 82-year-old African American woman with a history of pancreatic cancer came into the clinic for evaluation of a growing, asymptomatic lesion on her right dorsal foot. She first noticed the lesion a year ago, when it was pinpoint size. It was now a 2.5 cm × 1 cm hyperpigmented plaque.
The lesion was dark brown and black and had irregular borders. It also had a central hyperkeratotic area (FIGURE 1). There was no inguinal lymphadenopathy. We performed an incisional biopsy.
FIGURE 1
Growing lesion with irregular borders
What is your diagnosis?
How would you manage this condition?
Diagnosis: Pigmented Bowen’s disease
Histopathological evaluation of the lesion revealed hyperkeratosis, large atypical keratinocytes, increased mitotic figures, and an intact basement membrane (FIGURE 2), leading us to diagnose pigmented Bowen’s disease.
Bowen’s disease, an intraepidermal squamous cell carcinoma (carcinoma in situ), is a common type of nonmelanoma skin cancer. However, the form our patient had—pigmented Bowen’s disease—is a rare form of squamous cell carcinoma (SCC) in situ (2% of cases).1 The pigmented form of Bowen’s disease is more common in individuals with darker skin tones, while the nonpigmented is more common in fair-skinned individuals.
Bowen’s disease typically presents as a slow-growing, sharply demarcated, scaly erythematous plaque ranging in size from a few millimeters to several centimeters. Crusting, fissuring, hyperkeratosis, and pigmentation, as seen in our case, are also associated findings.2 Bowen’s disease often presents as a solitary lesion, with most cases (approximately 75%) associated with sun damage.3
The most common sites for Bowen’s disease include the head, neck, and hands. Rarely, the nail bed, oral mucosa, or anogenital region may be affected.
The mean age of diagnosis occurs in the sixth decade4 and there is an equal incidence in men and women. Bowen’s disease in men usually occurs on the scalp and ears, while in women, the lower legs are the most common site.5 Three to eight percent of Bowen’s disease cases progress to invasive carcinoma if left untreated.6
FIGURE 2
Histopathology of pigmented Bowen’s disease
A disease that’s linked to the sun—but also, HPV
The development of Bowen’s disease has been linked to sunlight exposure, human papilloma virus (HPV), and chronic arsenic intoxication.7
Sunlight exposure. Cumulative ultraviolet sunlight exposure is one of the most important etiologic factors. There is a doubling in the incidence of SCC for every 8- to 10-degree decrease in latitude.8
HPV. Human papillomavirus is common in patients who have SCCs in their genital areas.9 There is a poor correlation between nonanogenital Bowen’s disease and HPV infection. However, HPV types 2, 16, 18, 34, and 35 are occasionally identified in these lesions.10
Patients with penile Bowen’s disease (referred to as erythroplasia of Queyrat) are typically uncircumcised men with red, velvety plaques on the glans penis. Occasional itching and bleeding may be associated symptoms.
Arsenic intoxication. Chronic arsenic poisoning from drinking water is a documented cause of cancers occurring in the lung, bladder, kidney, liver, and skin. The US Geological Survey found the highest arsenic contamination levels of ground-water in the West, Midwest, and North-east United States.11
Unlike non-arsenical Bowen’s disease, arsenic-induced Bowen’s disease (As-BD) can occur on non-sun-exposed skin. As-BD typically appears 10 years after initial arsenic exposure, with pulmonary carcinoma appearing 30 years after exposure. As a result, it’s advisable to screen all patients with As-BD for cancer of the lung and bladder.12
Is it Bowen’s, or something more serious?
The differential diagnosis of this lesion includes superficial spreading melanoma, pigmented basal cell carcinoma, atypical melanocytic nevus, and seborrheic keratosis. These different skin conditions may be difficult to distinguish on clinical examination and ultimately may require a biopsy.
Although pigmented Bowen’s disease can occur in anyone, Caucasian patients (as noted earlier) tend to have the more typical nonpigmented, erythematous scaly plaques in sun-exposed sites (FIGURE 3). Darker pigmented individuals are more likely to present with pigmented cutaneous lesions, which may mimic malignant melanoma,13 as was the case with our patient.
FIGURE 3
Nonpigmented Bowen’s disease
Surgical excision is extremely effective
Bowen’s disease can be treated with cryotherapy; curettage and electrodesiccation; surgical excision, including Mohs micrographic surgery; laser surgery; photodynamic therapy; radiation therapy; topical 5-fluorouracil; and topical imiquimod. Invasive or higher risk lesions require surgical excision or Mohs surgery. Surgical excision of SCCs is extremely effective, with 5-year cure rates of 92%.14
A delay in treatment for our patient
Our patient was scheduled to undergo surgical excision with graft repair of the site. However, she was receiving chemotherapy for mucinous adenocarcinoma of the pancreas and declined excision due to concerns about possible infection.
She later underwent curettage and electrodesiccation, followed by topical imiquimod therapy for 10 weeks. She remains free of any Bowen’s disease recurrences 2 years after her diagnosis.
Correspondence
Claudia Hernandez, MD, Department of Dermatology, University of Illinois at Chicago, 808 S Wood St, MC 624, Chicago, IL 60612-7300.
An 82-year-old African American woman with a history of pancreatic cancer came into the clinic for evaluation of a growing, asymptomatic lesion on her right dorsal foot. She first noticed the lesion a year ago, when it was pinpoint size. It was now a 2.5 cm × 1 cm hyperpigmented plaque.
The lesion was dark brown and black and had irregular borders. It also had a central hyperkeratotic area (FIGURE 1). There was no inguinal lymphadenopathy. We performed an incisional biopsy.
FIGURE 1
Growing lesion with irregular borders
What is your diagnosis?
How would you manage this condition?
Diagnosis: Pigmented Bowen’s disease
Histopathological evaluation of the lesion revealed hyperkeratosis, large atypical keratinocytes, increased mitotic figures, and an intact basement membrane (FIGURE 2), leading us to diagnose pigmented Bowen’s disease.
Bowen’s disease, an intraepidermal squamous cell carcinoma (carcinoma in situ), is a common type of nonmelanoma skin cancer. However, the form our patient had—pigmented Bowen’s disease—is a rare form of squamous cell carcinoma (SCC) in situ (2% of cases).1 The pigmented form of Bowen’s disease is more common in individuals with darker skin tones, while the nonpigmented is more common in fair-skinned individuals.
Bowen’s disease typically presents as a slow-growing, sharply demarcated, scaly erythematous plaque ranging in size from a few millimeters to several centimeters. Crusting, fissuring, hyperkeratosis, and pigmentation, as seen in our case, are also associated findings.2 Bowen’s disease often presents as a solitary lesion, with most cases (approximately 75%) associated with sun damage.3
The most common sites for Bowen’s disease include the head, neck, and hands. Rarely, the nail bed, oral mucosa, or anogenital region may be affected.
The mean age of diagnosis occurs in the sixth decade4 and there is an equal incidence in men and women. Bowen’s disease in men usually occurs on the scalp and ears, while in women, the lower legs are the most common site.5 Three to eight percent of Bowen’s disease cases progress to invasive carcinoma if left untreated.6
FIGURE 2
Histopathology of pigmented Bowen’s disease
A disease that’s linked to the sun—but also, HPV
The development of Bowen’s disease has been linked to sunlight exposure, human papilloma virus (HPV), and chronic arsenic intoxication.7
Sunlight exposure. Cumulative ultraviolet sunlight exposure is one of the most important etiologic factors. There is a doubling in the incidence of SCC for every 8- to 10-degree decrease in latitude.8
HPV. Human papillomavirus is common in patients who have SCCs in their genital areas.9 There is a poor correlation between nonanogenital Bowen’s disease and HPV infection. However, HPV types 2, 16, 18, 34, and 35 are occasionally identified in these lesions.10
Patients with penile Bowen’s disease (referred to as erythroplasia of Queyrat) are typically uncircumcised men with red, velvety plaques on the glans penis. Occasional itching and bleeding may be associated symptoms.
Arsenic intoxication. Chronic arsenic poisoning from drinking water is a documented cause of cancers occurring in the lung, bladder, kidney, liver, and skin. The US Geological Survey found the highest arsenic contamination levels of ground-water in the West, Midwest, and North-east United States.11
Unlike non-arsenical Bowen’s disease, arsenic-induced Bowen’s disease (As-BD) can occur on non-sun-exposed skin. As-BD typically appears 10 years after initial arsenic exposure, with pulmonary carcinoma appearing 30 years after exposure. As a result, it’s advisable to screen all patients with As-BD for cancer of the lung and bladder.12
Is it Bowen’s, or something more serious?
The differential diagnosis of this lesion includes superficial spreading melanoma, pigmented basal cell carcinoma, atypical melanocytic nevus, and seborrheic keratosis. These different skin conditions may be difficult to distinguish on clinical examination and ultimately may require a biopsy.
Although pigmented Bowen’s disease can occur in anyone, Caucasian patients (as noted earlier) tend to have the more typical nonpigmented, erythematous scaly plaques in sun-exposed sites (FIGURE 3). Darker pigmented individuals are more likely to present with pigmented cutaneous lesions, which may mimic malignant melanoma,13 as was the case with our patient.
FIGURE 3
Nonpigmented Bowen’s disease
Surgical excision is extremely effective
Bowen’s disease can be treated with cryotherapy; curettage and electrodesiccation; surgical excision, including Mohs micrographic surgery; laser surgery; photodynamic therapy; radiation therapy; topical 5-fluorouracil; and topical imiquimod. Invasive or higher risk lesions require surgical excision or Mohs surgery. Surgical excision of SCCs is extremely effective, with 5-year cure rates of 92%.14
A delay in treatment for our patient
Our patient was scheduled to undergo surgical excision with graft repair of the site. However, she was receiving chemotherapy for mucinous adenocarcinoma of the pancreas and declined excision due to concerns about possible infection.
She later underwent curettage and electrodesiccation, followed by topical imiquimod therapy for 10 weeks. She remains free of any Bowen’s disease recurrences 2 years after her diagnosis.
Correspondence
Claudia Hernandez, MD, Department of Dermatology, University of Illinois at Chicago, 808 S Wood St, MC 624, Chicago, IL 60612-7300.
1. Ragi G, Turner MS, Klein LE, Stoll HL. Pigmented Bowen’s disease and review of 420 Bowen’s disease lesions. J Dermatol Surg Oncol. 1988;14:756-759.
2. Rinker MH, Fenske NA, Scalf LA, Glass LF. Histologic variants of squamous cell carcinoma of the skin. Can Control. 2001;8:354-363.
3. Lee MM, Wick MM. Bowen’s disease. CA Cancer J Clin. 1990;40:237-242.
4. Kossard S, Rosen R. Cutaneous Bowen’s disease. J Am Acad Dermatol. 1992;27:406-410.
5. VanderSpek LAL, Pond GR, Wells W, Tsang RW. Radiation therapy for Bowen’s disease of the skin. Int J Radiat Oncol Biol Phys. 2005;63:505-510.
6. Leibovitch I, Huilgol SC, Selva D, Richards S, Paver R. Cutaneous squamous carcinoma in situ (Bowen’s disease: treatment with Mohs micrographic surgery. J Am Acad Dermatol. 2005;52:997-1002.
7. Stante M, De Giorgi V, Massi D, Chiarugi A, Carli P. Pigmented Bowen’s disease mimicking cutaneous melanoma: clinical and dermoscopic aspects. Dermatol Surg. 2004;30:541-544.
8. Rundel RD. Promotional effects of ultraviolet radiation on human basal and squamous cell carcinoma. Photochem Photobiol. 1983;38:569-575.
9. Crum CP, Ikenberg H, Richart RM, et al. Human papillomavirus type 16 and early cervical neoplasia. N Engl J Med. 1984;310:880-883.
10. Amagi N, Feldman B, Lobel S, Williams C. Irregularly pigmented hyperkeratotic plaque on the thumb. Arch Dermatol. 1993;129:1045-1048.
11. United States Geologic Survey. Arsenic in Ground-water Resources of the United States. Available at: http://water.usgs.gov/nawqa/trace/pubs/fs-063-00/fs-063-00.pdf. Accessed July 29, 2008.
12. Yu HS, Liao WT, Chai CY. Arsenic carcinogenesis in the skin. J Biomed Sci. 2006;13:657-66.
13. Krishnan R, Lewis A, Orengo IF, Rosen T. Pigmented Bowen’s disease (squamous cell carcinoma in situ): a mimic of malignant melanoma. Dermatol Surg. 2001;27:673-674.
14. Miller SJ, Moresi JM. Actinic keratosis, basal cell carcinoma, and squamous cell carcinoma. In: Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. Vol. 2. New York, NY: Mosby; 2003:1677-1696.
1. Ragi G, Turner MS, Klein LE, Stoll HL. Pigmented Bowen’s disease and review of 420 Bowen’s disease lesions. J Dermatol Surg Oncol. 1988;14:756-759.
2. Rinker MH, Fenske NA, Scalf LA, Glass LF. Histologic variants of squamous cell carcinoma of the skin. Can Control. 2001;8:354-363.
3. Lee MM, Wick MM. Bowen’s disease. CA Cancer J Clin. 1990;40:237-242.
4. Kossard S, Rosen R. Cutaneous Bowen’s disease. J Am Acad Dermatol. 1992;27:406-410.
5. VanderSpek LAL, Pond GR, Wells W, Tsang RW. Radiation therapy for Bowen’s disease of the skin. Int J Radiat Oncol Biol Phys. 2005;63:505-510.
6. Leibovitch I, Huilgol SC, Selva D, Richards S, Paver R. Cutaneous squamous carcinoma in situ (Bowen’s disease: treatment with Mohs micrographic surgery. J Am Acad Dermatol. 2005;52:997-1002.
7. Stante M, De Giorgi V, Massi D, Chiarugi A, Carli P. Pigmented Bowen’s disease mimicking cutaneous melanoma: clinical and dermoscopic aspects. Dermatol Surg. 2004;30:541-544.
8. Rundel RD. Promotional effects of ultraviolet radiation on human basal and squamous cell carcinoma. Photochem Photobiol. 1983;38:569-575.
9. Crum CP, Ikenberg H, Richart RM, et al. Human papillomavirus type 16 and early cervical neoplasia. N Engl J Med. 1984;310:880-883.
10. Amagi N, Feldman B, Lobel S, Williams C. Irregularly pigmented hyperkeratotic plaque on the thumb. Arch Dermatol. 1993;129:1045-1048.
11. United States Geologic Survey. Arsenic in Ground-water Resources of the United States. Available at: http://water.usgs.gov/nawqa/trace/pubs/fs-063-00/fs-063-00.pdf. Accessed July 29, 2008.
12. Yu HS, Liao WT, Chai CY. Arsenic carcinogenesis in the skin. J Biomed Sci. 2006;13:657-66.
13. Krishnan R, Lewis A, Orengo IF, Rosen T. Pigmented Bowen’s disease (squamous cell carcinoma in situ): a mimic of malignant melanoma. Dermatol Surg. 2001;27:673-674.
14. Miller SJ, Moresi JM. Actinic keratosis, basal cell carcinoma, and squamous cell carcinoma. In: Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. Vol. 2. New York, NY: Mosby; 2003:1677-1696.
An unusual skin nodule
A 53-year-old African American woman with progressive shortness of breath for 1 month and a productive cough with intermittent blood-tinged sputum sought treatment at our emergency room. Over the past 2 months, she had lost 20 pounds—despite a normal appetite—and had noticed multiple “bumps” on her body.
The patient had no changes in her bowel habits, nor had she experienced nausea, vomiting, hematochezia, hemoptysis, odynophagia, dysphagia, chills, fever, or night sweats. Her past medical and surgical histories were unremarkable.
She had a 40-year history of smoking 1 to 3 packs of cigarettes per day, and a 30-year history of drinking a 1-liter bottle of hard liquor a day. She also occasionally used marijuana.
While in the ER, the patient was comfortable breathing on room air and afebrile with stable vital signs. We noticed decreased breath sounds bilaterally, as well as rhonchi. The patient had diffuse lymphadenopathy, as well as multiple, fixed, painless subcutaneous nodules on the neck, arms, flanks, back, and proximal right thigh. There was a 4 × 3.5 cm exophytic beefy-red tumor on the left inner arm (FIGURE 1). Some smaller nodules had surface hyperpigmentation, while others were subcutaneous, with no epidermal surface changes. All of the lesions were asymptomatic.
We ordered a chest radiograph (FIGURE 2) and blood work. Abnormal hematologic test results were: alkaline phosphatase (236 IU/L), gamma-glutamyl transferase (435 IU/L), and serum Ca++ (8.2 mg/dL).
FIGURE 1
Tumor on left inner arm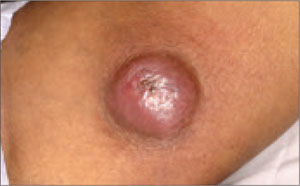
FIGURE 2
Chest x-ray brings Dx into focus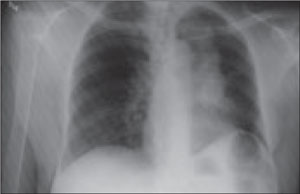
What is your diagnosis?
Dx: SCC of the lungs with skin metastases
Our patient’s chest radiograph—which revealed a left pulmonary mass—combined with her blood work and lesional biopsies, led us to diagnose primary squamous cell carcinoma (SCC) of the lung with skin metastases.
Approximately 30% of all lung cancers are classified as SCC.1 Pulmonary SCC metastatic to the skin tends to remain localized within the skin in the areas of the abdomen, anterior chest, back, scalp, and neck.2-6 Though cutaneous metastases of internal malignancies occur in 0.7% to 9.0% of all cancer patients, the incidence of skin metastases from lung cancer ranges from 1% to 12%.6-8
Lesions are painless and moveable
Most cutaneous metastases consist of single or multiple lesions that are painless, movable, and nonspecific. These lesions may also be firm to palpation, ulcerated, or exudative and able to penetrate the dermis or subcutaneous layer of skin with intact overlying epidermis.7-9
Skin eruptions may be the first sign of trouble
Prompt identification and diagnosis of these lesions are tantamount in tumor staging and therapy.
Schwartz reported that “in a series of patients with skin metastases seen in dermatologic consultation, the underlying cancer had been undiagnosed in 60% with lung cancer, in 53% with renal cancer, and in 40% with ovarian cancer.”7 Similar findings have been reported elsewhere.4,8 Thus, metastatic carcinoma to the skin should always be included in the differential diagnosis for newly eruptive skin lesions, regardless of whether the patient has ever been diagnosed with any form of cancer.10
A grim prognosis
Current therapies for skin metastases from primary lung carcinomas include surgical excision, symptomatic therapy, and chemotherapy—with and without radiation. After a patient’s course of treatment, skin metastases may be an important diagnostic marker for tumor recurrence.
Kamiyoshihara et al8 found that when skin metastases are detected, only 23% of the underlying primary cancers are operable, noting that most “portent a fatal outcome.”
Our patient declined further treatment
A CT of the patient’s chest showed a left hilar mass. A radionucleotide bone scan revealed metastases to the right elbow. We estimated that our patient had 1 year to live. In light of her very dismal prognosis, our patient declined any further work-up or treatment.
Our patient died 5 months later.
Correspondence
David A. Kasper DO, MBA, 2064 West Market St., Potts-ville, PA 17901; [email protected]
1. Parkin DM, Pisani P, Ferlay J. Global cancer statistics. CA Cancer J Clin. 1999;49:33.-
2. Ambrogi V, Nofroni I, Tonini G, Mineo TC. Skin metastases in lung cancer: analysis of a 10-year experience. Oncol Rep. 2001;8:57-61.
3. Altundag K, Yalcin S, Ozkaya O, Guler N. Synchronous squamous cell carcinoma of the stomach, the lung and the skin. Onkologie. 2004;27:291-293.
4. Brownstein MH, Helwig EB. Patterns of cutaneous metastasis. Arch Dermatol. 1972;105:862-868.
5. Reingold IM. Cutaneous metastases from internal carcinoma. Cancer. 1966;19:162-168.
6. Terashima T, Kanazawa M. Lung cancer with skin metastasis. Chest. 1994;106:1448-1450.
7. Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33:161-182.
8. Kamiyoshihara M, Sakata K, Otani Y, Kawashima O, Takahashi T, Morishita Y. Solitary skin metastasis after lung cancer resection. Jpn J Thorac Cardiovasc Surg. 2002;50:343-346.
9. Kaplan RP. Specific cutaneous manifestations of internal malignancy. Adv Dermatol. 1986;1:3-42.
10. Nakamura H, Shimizu T, Kodama K, Shimizu H. Metastasis of lung cancer to the finger: a report of two cases. Int J Dermatol. 2005;44:47-49.
A 53-year-old African American woman with progressive shortness of breath for 1 month and a productive cough with intermittent blood-tinged sputum sought treatment at our emergency room. Over the past 2 months, she had lost 20 pounds—despite a normal appetite—and had noticed multiple “bumps” on her body.
The patient had no changes in her bowel habits, nor had she experienced nausea, vomiting, hematochezia, hemoptysis, odynophagia, dysphagia, chills, fever, or night sweats. Her past medical and surgical histories were unremarkable.
She had a 40-year history of smoking 1 to 3 packs of cigarettes per day, and a 30-year history of drinking a 1-liter bottle of hard liquor a day. She also occasionally used marijuana.
While in the ER, the patient was comfortable breathing on room air and afebrile with stable vital signs. We noticed decreased breath sounds bilaterally, as well as rhonchi. The patient had diffuse lymphadenopathy, as well as multiple, fixed, painless subcutaneous nodules on the neck, arms, flanks, back, and proximal right thigh. There was a 4 × 3.5 cm exophytic beefy-red tumor on the left inner arm (FIGURE 1). Some smaller nodules had surface hyperpigmentation, while others were subcutaneous, with no epidermal surface changes. All of the lesions were asymptomatic.
We ordered a chest radiograph (FIGURE 2) and blood work. Abnormal hematologic test results were: alkaline phosphatase (236 IU/L), gamma-glutamyl transferase (435 IU/L), and serum Ca++ (8.2 mg/dL).
FIGURE 1
Tumor on left inner arm
FIGURE 2
Chest x-ray brings Dx into focus
What is your diagnosis?
Dx: SCC of the lungs with skin metastases
Our patient’s chest radiograph—which revealed a left pulmonary mass—combined with her blood work and lesional biopsies, led us to diagnose primary squamous cell carcinoma (SCC) of the lung with skin metastases.
Approximately 30% of all lung cancers are classified as SCC.1 Pulmonary SCC metastatic to the skin tends to remain localized within the skin in the areas of the abdomen, anterior chest, back, scalp, and neck.2-6 Though cutaneous metastases of internal malignancies occur in 0.7% to 9.0% of all cancer patients, the incidence of skin metastases from lung cancer ranges from 1% to 12%.6-8
Lesions are painless and moveable
Most cutaneous metastases consist of single or multiple lesions that are painless, movable, and nonspecific. These lesions may also be firm to palpation, ulcerated, or exudative and able to penetrate the dermis or subcutaneous layer of skin with intact overlying epidermis.7-9
Skin eruptions may be the first sign of trouble
Prompt identification and diagnosis of these lesions are tantamount in tumor staging and therapy.
Schwartz reported that “in a series of patients with skin metastases seen in dermatologic consultation, the underlying cancer had been undiagnosed in 60% with lung cancer, in 53% with renal cancer, and in 40% with ovarian cancer.”7 Similar findings have been reported elsewhere.4,8 Thus, metastatic carcinoma to the skin should always be included in the differential diagnosis for newly eruptive skin lesions, regardless of whether the patient has ever been diagnosed with any form of cancer.10
A grim prognosis
Current therapies for skin metastases from primary lung carcinomas include surgical excision, symptomatic therapy, and chemotherapy—with and without radiation. After a patient’s course of treatment, skin metastases may be an important diagnostic marker for tumor recurrence.
Kamiyoshihara et al8 found that when skin metastases are detected, only 23% of the underlying primary cancers are operable, noting that most “portent a fatal outcome.”
Our patient declined further treatment
A CT of the patient’s chest showed a left hilar mass. A radionucleotide bone scan revealed metastases to the right elbow. We estimated that our patient had 1 year to live. In light of her very dismal prognosis, our patient declined any further work-up or treatment.
Our patient died 5 months later.
Correspondence
David A. Kasper DO, MBA, 2064 West Market St., Potts-ville, PA 17901; [email protected]
A 53-year-old African American woman with progressive shortness of breath for 1 month and a productive cough with intermittent blood-tinged sputum sought treatment at our emergency room. Over the past 2 months, she had lost 20 pounds—despite a normal appetite—and had noticed multiple “bumps” on her body.
The patient had no changes in her bowel habits, nor had she experienced nausea, vomiting, hematochezia, hemoptysis, odynophagia, dysphagia, chills, fever, or night sweats. Her past medical and surgical histories were unremarkable.
She had a 40-year history of smoking 1 to 3 packs of cigarettes per day, and a 30-year history of drinking a 1-liter bottle of hard liquor a day. She also occasionally used marijuana.
While in the ER, the patient was comfortable breathing on room air and afebrile with stable vital signs. We noticed decreased breath sounds bilaterally, as well as rhonchi. The patient had diffuse lymphadenopathy, as well as multiple, fixed, painless subcutaneous nodules on the neck, arms, flanks, back, and proximal right thigh. There was a 4 × 3.5 cm exophytic beefy-red tumor on the left inner arm (FIGURE 1). Some smaller nodules had surface hyperpigmentation, while others were subcutaneous, with no epidermal surface changes. All of the lesions were asymptomatic.
We ordered a chest radiograph (FIGURE 2) and blood work. Abnormal hematologic test results were: alkaline phosphatase (236 IU/L), gamma-glutamyl transferase (435 IU/L), and serum Ca++ (8.2 mg/dL).
FIGURE 1
Tumor on left inner arm
FIGURE 2
Chest x-ray brings Dx into focus
What is your diagnosis?
Dx: SCC of the lungs with skin metastases
Our patient’s chest radiograph—which revealed a left pulmonary mass—combined with her blood work and lesional biopsies, led us to diagnose primary squamous cell carcinoma (SCC) of the lung with skin metastases.
Approximately 30% of all lung cancers are classified as SCC.1 Pulmonary SCC metastatic to the skin tends to remain localized within the skin in the areas of the abdomen, anterior chest, back, scalp, and neck.2-6 Though cutaneous metastases of internal malignancies occur in 0.7% to 9.0% of all cancer patients, the incidence of skin metastases from lung cancer ranges from 1% to 12%.6-8
Lesions are painless and moveable
Most cutaneous metastases consist of single or multiple lesions that are painless, movable, and nonspecific. These lesions may also be firm to palpation, ulcerated, or exudative and able to penetrate the dermis or subcutaneous layer of skin with intact overlying epidermis.7-9
Skin eruptions may be the first sign of trouble
Prompt identification and diagnosis of these lesions are tantamount in tumor staging and therapy.
Schwartz reported that “in a series of patients with skin metastases seen in dermatologic consultation, the underlying cancer had been undiagnosed in 60% with lung cancer, in 53% with renal cancer, and in 40% with ovarian cancer.”7 Similar findings have been reported elsewhere.4,8 Thus, metastatic carcinoma to the skin should always be included in the differential diagnosis for newly eruptive skin lesions, regardless of whether the patient has ever been diagnosed with any form of cancer.10
A grim prognosis
Current therapies for skin metastases from primary lung carcinomas include surgical excision, symptomatic therapy, and chemotherapy—with and without radiation. After a patient’s course of treatment, skin metastases may be an important diagnostic marker for tumor recurrence.
Kamiyoshihara et al8 found that when skin metastases are detected, only 23% of the underlying primary cancers are operable, noting that most “portent a fatal outcome.”
Our patient declined further treatment
A CT of the patient’s chest showed a left hilar mass. A radionucleotide bone scan revealed metastases to the right elbow. We estimated that our patient had 1 year to live. In light of her very dismal prognosis, our patient declined any further work-up or treatment.
Our patient died 5 months later.
Correspondence
David A. Kasper DO, MBA, 2064 West Market St., Potts-ville, PA 17901; [email protected]
1. Parkin DM, Pisani P, Ferlay J. Global cancer statistics. CA Cancer J Clin. 1999;49:33.-
2. Ambrogi V, Nofroni I, Tonini G, Mineo TC. Skin metastases in lung cancer: analysis of a 10-year experience. Oncol Rep. 2001;8:57-61.
3. Altundag K, Yalcin S, Ozkaya O, Guler N. Synchronous squamous cell carcinoma of the stomach, the lung and the skin. Onkologie. 2004;27:291-293.
4. Brownstein MH, Helwig EB. Patterns of cutaneous metastasis. Arch Dermatol. 1972;105:862-868.
5. Reingold IM. Cutaneous metastases from internal carcinoma. Cancer. 1966;19:162-168.
6. Terashima T, Kanazawa M. Lung cancer with skin metastasis. Chest. 1994;106:1448-1450.
7. Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33:161-182.
8. Kamiyoshihara M, Sakata K, Otani Y, Kawashima O, Takahashi T, Morishita Y. Solitary skin metastasis after lung cancer resection. Jpn J Thorac Cardiovasc Surg. 2002;50:343-346.
9. Kaplan RP. Specific cutaneous manifestations of internal malignancy. Adv Dermatol. 1986;1:3-42.
10. Nakamura H, Shimizu T, Kodama K, Shimizu H. Metastasis of lung cancer to the finger: a report of two cases. Int J Dermatol. 2005;44:47-49.
1. Parkin DM, Pisani P, Ferlay J. Global cancer statistics. CA Cancer J Clin. 1999;49:33.-
2. Ambrogi V, Nofroni I, Tonini G, Mineo TC. Skin metastases in lung cancer: analysis of a 10-year experience. Oncol Rep. 2001;8:57-61.
3. Altundag K, Yalcin S, Ozkaya O, Guler N. Synchronous squamous cell carcinoma of the stomach, the lung and the skin. Onkologie. 2004;27:291-293.
4. Brownstein MH, Helwig EB. Patterns of cutaneous metastasis. Arch Dermatol. 1972;105:862-868.
5. Reingold IM. Cutaneous metastases from internal carcinoma. Cancer. 1966;19:162-168.
6. Terashima T, Kanazawa M. Lung cancer with skin metastasis. Chest. 1994;106:1448-1450.
7. Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33:161-182.
8. Kamiyoshihara M, Sakata K, Otani Y, Kawashima O, Takahashi T, Morishita Y. Solitary skin metastasis after lung cancer resection. Jpn J Thorac Cardiovasc Surg. 2002;50:343-346.
9. Kaplan RP. Specific cutaneous manifestations of internal malignancy. Adv Dermatol. 1986;1:3-42.
10. Nakamura H, Shimizu T, Kodama K, Shimizu H. Metastasis of lung cancer to the finger: a report of two cases. Int J Dermatol. 2005;44:47-49.
Tender growth on toe
A 50-year-old Caucasian man came to the dermatology clinic with a tender growth on his great toe. He first noticed it a year earlier after sustaining trauma to the area. The growth had been getting bigger and had intermittently bled. It had also oozed, so his primary care physician treated it as an infection with topical antibiotics, followed by several courses of oral antibiotics.
Despite therapy, the lesion continued to grow and the patient was referred to us. The patient had a 3.4 cm × 1.8 cm exophytic erythematous nodule on the left lateral great toe (FIGURE).
What is your diagnosis?
How would you manage this condition?
FIGURE
Erythematous nodule on left great toe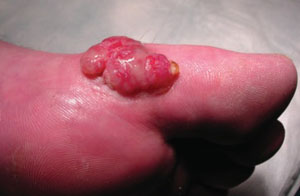
Diagnosis: Acral lentiginous melanoma
A wedge biopsy of the lesion showed that the patient had acral lentiginous melanoma. ALM is a less common subtype of melanoma. (The other subtypes are superficial spreading malignant melanoma, nodular melanoma, and lentigo maligna melanoma.) Acral refers to its typical location, occurring in the distal extremities. The term lentiginous refers to its characteristic histopathologic finding.1,2 Unlike the other subtypes of melanoma, ALM typically affects all ethnic groups. In non-white populations it constitutes 29% to 72% of all melanomas.3 In Caucasian patients it accounts for 2% to 8% of all melanomas.
In many cases, ALM presents with the typical “ABCD” features of melanoma. These include asymmetry, border irregularity, color variegation, and a diameter greater than 6 mm. Lesions on the palmo-plantar surface may be macular, papular, or nodular, and may ulcerate. ALM may involve the nail as well.
As in our case, however, not all cases are typical. Amelanotic lesions may mimic a variety of skin conditions and pose a diagnostic challenge.
ALM is not linked to sun exposure
The pathogenesis of ALM is unclear. Unlike the other types of melanoma, in which sun exposure is clearly identified as an etiologic factor, ALM has not been definitively linked to exposure. In fact, ALM occurs in patients with more darkly pigmented skin, a group not typically considered at risk for melanoma. In African American patients, Hispanic patients, and Asian patients, ALM is the most common subtype of melanoma.4
BRAF/NRAS mutations seem to play an important role in all the major subtypes of cutaneous melanoma.5 Recent studies indicate that nevoid precursor lesions, high total body nevus count, and plantar nevi act as predisposing lesions to ALM.2,6
Injury to the hands and feet. Another common association with ALM is penetrative injury of the hands and feet. There is no clinical or research-based evidence to support this association and therefore the role of trauma in the pathogenesis of ALM is controversial. This association may be seen in some cases because acral sites are more prone to trauma than the rest of the body.7
Trauma leading to hemorrhage mimics ALM
The differential diagnosis includes both melanocytic lesions and non-melanocytic lesions. Benign nevi or dysplastic nevi are included in the first category. Among non-melanocytic lesions, trauma leading to hemorrhage may mimic ALM.2 ALM has also been misdiagnosed as pyogenic granuloma, ischemic ulceration, wart, a foreign body, callus, a nonhealing wound, or other benign skin lesions such as seborrheic keratosis.2,8,9
Amelanotic lesions are particularly misleading on the foot, as they may be frequently misdiagnosed as hyperkeratotic benign lesions.8 Bleeding, itching, satellite lesions, and sudden growth, however, should raise your suspicion of melanoma.10
Excision is best, plus sentinel node mapping
The most effective treatment for ALM is excision—either conservatively or with wide margins—along with sentinel lymph node mapping.11 Systemic chemotherapy is more palliative than curative, as it has not been shown to improve the number of recurrences.2 Patients require lifelong regular full body skin exams with early biopsy of any suspicious skin lesions.9
The prognosis of ALM is poorer than other subtypes of melanoma, mainly because of delayed diagnosis.2,9 The single most important and accurate prognostic factor is the depth of invasion of the tumor as measured by Breslow thickness.10
Other factors that are predictive of a poorer prognosis in ALM patients are male sex and amelanosis.12 It’s important to recognize that the palms, soles, and nails may be sites for melanoma—despite the lack of direct sun exposure.
Our patient required amputation
Our patient’s toe was amputated and the melanoma had a Breslow thickness of 7 mm. Sentinel lymph node biopsy was negative. Our patient received adjuvant chemotherapy with interferon alfa-2B and was followed by oncology.
If you said the face or the arms, you’d be wrong. Given that increased sun exposure and a history of multiple sunburns are recognized as important risk factors for melanoma, it’s easy to assume that the most common sites for melanoma would be exposed areas of the body, such as the face and arms.
Interestingly, though, superficial spreading malignant melanoma—the most common type of malignant melanoma13—is most often found on the legs in women and on the back in men.3,10 One theory is that repeated intense exposure to sunlight may be a more important risk factor than continuous sun exposure.14,15
Correspondence
Rajani Katta, MD, 6620 Main Street, Suite 1425, Houston, TX 77030.
1. Freedberg I. Acral lentiginous melanoma. In: Fitzpatrick’s Dermatology in General Medicine. 5th ed. New York: McGraw-Hill; 1999;767:1081-1083.
2. Stalkup JR, Orengo IF, Katta R. Controversies in acral lentiginous melanoma. Dermatol Surg. 2002;28:1051-1059.
3. Swetter SM. Malignant melanoma. Available at: http://www.emedicine.com/DERM/topic257.htm. Accessed June 6, 2008.
4. Cress RD, Holly EA. Incidence of cutaneous melanoma among non-Hispanic whites, Hispanics, Asians, and blacks: an analysis of California cancer registry data, 1988-93. Cancer Causes Control. 1997;8:246-252.
5. Saldanha G, Potter L, Daforno P, Pringle JH. Cutaneous melanoma subtypes show different BRAF and NRAS mutation frequencies. Clin Cancer Res. 2006;12:4499-4505.
6. Green A, McCredie M, MacKie R, et al. A casecontrol study of melanomas of the soles and palms (Australia and Scotland). Cancer Causes Control. 1999;10:21-25.
7. Kaskel P, Kind P, Sander S, Peter RU, Krähn G. Trauma and melanoma formation: a true association? Br J Dermatol. 2000;143:749-753.
8. Soon SL, Solomon AR, Jr, Papadopoulos D, Murray DR, McAlpine B, Washington CV. Acral lentiginous melanoma mimicking benign disease: the Emory experience. J Am Acad Dermatol. 2003;48:183-188.
9. Rosen T. Acral lentiginous melanoma misdiagnosed as verruca plantaris: a case report. Dermatology Online Journal. 2006;12(4):3.-Available at: http://dermatology.cdlib.org/124/case_reports/melanoma/rosen.html. Accessed June 6, 2008.
10. Bruce AJ, Brodland DG. Overview of skin cancer detection and prevention for the primary care physician. Mayo Clin Proc. 2000;75:491-500.
11. Lang PG. Current concepts in the management of patients with melanoma. Am J Clin Dermatol. 2002;3:401-426.
12. Phan A, Touzet S, Dalle S, Ronger-Savle S, Balme B, Thomas L. Acral lentiginous melanoma: a clinicoprognostic study of 126 cases. Br J Dermatol. 2006;155:561-569.
13. Kang S, Barnhill RL, Mihm MC, Jr, et al. Multiple primary cutaneous melanomas. Cancer. 1992;70:1911-1916.
14. Elwood JM. Melanoma and sun exposure: contrasts between intermittent and chronic exposure. World J Surg. 1992;16:157-165.
15. Elwood JM, Gallagher RP. Body site distribution of cutaneous malignant melanoma in relationship to patterns of sun exposure. Int J Cancer. 1998;78:276-280.
A 50-year-old Caucasian man came to the dermatology clinic with a tender growth on his great toe. He first noticed it a year earlier after sustaining trauma to the area. The growth had been getting bigger and had intermittently bled. It had also oozed, so his primary care physician treated it as an infection with topical antibiotics, followed by several courses of oral antibiotics.
Despite therapy, the lesion continued to grow and the patient was referred to us. The patient had a 3.4 cm × 1.8 cm exophytic erythematous nodule on the left lateral great toe (FIGURE).
What is your diagnosis?
How would you manage this condition?
FIGURE
Erythematous nodule on left great toe
Diagnosis: Acral lentiginous melanoma
A wedge biopsy of the lesion showed that the patient had acral lentiginous melanoma. ALM is a less common subtype of melanoma. (The other subtypes are superficial spreading malignant melanoma, nodular melanoma, and lentigo maligna melanoma.) Acral refers to its typical location, occurring in the distal extremities. The term lentiginous refers to its characteristic histopathologic finding.1,2 Unlike the other subtypes of melanoma, ALM typically affects all ethnic groups. In non-white populations it constitutes 29% to 72% of all melanomas.3 In Caucasian patients it accounts for 2% to 8% of all melanomas.
In many cases, ALM presents with the typical “ABCD” features of melanoma. These include asymmetry, border irregularity, color variegation, and a diameter greater than 6 mm. Lesions on the palmo-plantar surface may be macular, papular, or nodular, and may ulcerate. ALM may involve the nail as well.
As in our case, however, not all cases are typical. Amelanotic lesions may mimic a variety of skin conditions and pose a diagnostic challenge.
ALM is not linked to sun exposure
The pathogenesis of ALM is unclear. Unlike the other types of melanoma, in which sun exposure is clearly identified as an etiologic factor, ALM has not been definitively linked to exposure. In fact, ALM occurs in patients with more darkly pigmented skin, a group not typically considered at risk for melanoma. In African American patients, Hispanic patients, and Asian patients, ALM is the most common subtype of melanoma.4
BRAF/NRAS mutations seem to play an important role in all the major subtypes of cutaneous melanoma.5 Recent studies indicate that nevoid precursor lesions, high total body nevus count, and plantar nevi act as predisposing lesions to ALM.2,6
Injury to the hands and feet. Another common association with ALM is penetrative injury of the hands and feet. There is no clinical or research-based evidence to support this association and therefore the role of trauma in the pathogenesis of ALM is controversial. This association may be seen in some cases because acral sites are more prone to trauma than the rest of the body.7
Trauma leading to hemorrhage mimics ALM
The differential diagnosis includes both melanocytic lesions and non-melanocytic lesions. Benign nevi or dysplastic nevi are included in the first category. Among non-melanocytic lesions, trauma leading to hemorrhage may mimic ALM.2 ALM has also been misdiagnosed as pyogenic granuloma, ischemic ulceration, wart, a foreign body, callus, a nonhealing wound, or other benign skin lesions such as seborrheic keratosis.2,8,9
Amelanotic lesions are particularly misleading on the foot, as they may be frequently misdiagnosed as hyperkeratotic benign lesions.8 Bleeding, itching, satellite lesions, and sudden growth, however, should raise your suspicion of melanoma.10
Excision is best, plus sentinel node mapping
The most effective treatment for ALM is excision—either conservatively or with wide margins—along with sentinel lymph node mapping.11 Systemic chemotherapy is more palliative than curative, as it has not been shown to improve the number of recurrences.2 Patients require lifelong regular full body skin exams with early biopsy of any suspicious skin lesions.9
The prognosis of ALM is poorer than other subtypes of melanoma, mainly because of delayed diagnosis.2,9 The single most important and accurate prognostic factor is the depth of invasion of the tumor as measured by Breslow thickness.10
Other factors that are predictive of a poorer prognosis in ALM patients are male sex and amelanosis.12 It’s important to recognize that the palms, soles, and nails may be sites for melanoma—despite the lack of direct sun exposure.
Our patient required amputation
Our patient’s toe was amputated and the melanoma had a Breslow thickness of 7 mm. Sentinel lymph node biopsy was negative. Our patient received adjuvant chemotherapy with interferon alfa-2B and was followed by oncology.
If you said the face or the arms, you’d be wrong. Given that increased sun exposure and a history of multiple sunburns are recognized as important risk factors for melanoma, it’s easy to assume that the most common sites for melanoma would be exposed areas of the body, such as the face and arms.
Interestingly, though, superficial spreading malignant melanoma—the most common type of malignant melanoma13—is most often found on the legs in women and on the back in men.3,10 One theory is that repeated intense exposure to sunlight may be a more important risk factor than continuous sun exposure.14,15
Correspondence
Rajani Katta, MD, 6620 Main Street, Suite 1425, Houston, TX 77030.
A 50-year-old Caucasian man came to the dermatology clinic with a tender growth on his great toe. He first noticed it a year earlier after sustaining trauma to the area. The growth had been getting bigger and had intermittently bled. It had also oozed, so his primary care physician treated it as an infection with topical antibiotics, followed by several courses of oral antibiotics.
Despite therapy, the lesion continued to grow and the patient was referred to us. The patient had a 3.4 cm × 1.8 cm exophytic erythematous nodule on the left lateral great toe (FIGURE).
What is your diagnosis?
How would you manage this condition?
FIGURE
Erythematous nodule on left great toe
Diagnosis: Acral lentiginous melanoma
A wedge biopsy of the lesion showed that the patient had acral lentiginous melanoma. ALM is a less common subtype of melanoma. (The other subtypes are superficial spreading malignant melanoma, nodular melanoma, and lentigo maligna melanoma.) Acral refers to its typical location, occurring in the distal extremities. The term lentiginous refers to its characteristic histopathologic finding.1,2 Unlike the other subtypes of melanoma, ALM typically affects all ethnic groups. In non-white populations it constitutes 29% to 72% of all melanomas.3 In Caucasian patients it accounts for 2% to 8% of all melanomas.
In many cases, ALM presents with the typical “ABCD” features of melanoma. These include asymmetry, border irregularity, color variegation, and a diameter greater than 6 mm. Lesions on the palmo-plantar surface may be macular, papular, or nodular, and may ulcerate. ALM may involve the nail as well.
As in our case, however, not all cases are typical. Amelanotic lesions may mimic a variety of skin conditions and pose a diagnostic challenge.
ALM is not linked to sun exposure
The pathogenesis of ALM is unclear. Unlike the other types of melanoma, in which sun exposure is clearly identified as an etiologic factor, ALM has not been definitively linked to exposure. In fact, ALM occurs in patients with more darkly pigmented skin, a group not typically considered at risk for melanoma. In African American patients, Hispanic patients, and Asian patients, ALM is the most common subtype of melanoma.4
BRAF/NRAS mutations seem to play an important role in all the major subtypes of cutaneous melanoma.5 Recent studies indicate that nevoid precursor lesions, high total body nevus count, and plantar nevi act as predisposing lesions to ALM.2,6
Injury to the hands and feet. Another common association with ALM is penetrative injury of the hands and feet. There is no clinical or research-based evidence to support this association and therefore the role of trauma in the pathogenesis of ALM is controversial. This association may be seen in some cases because acral sites are more prone to trauma than the rest of the body.7
Trauma leading to hemorrhage mimics ALM
The differential diagnosis includes both melanocytic lesions and non-melanocytic lesions. Benign nevi or dysplastic nevi are included in the first category. Among non-melanocytic lesions, trauma leading to hemorrhage may mimic ALM.2 ALM has also been misdiagnosed as pyogenic granuloma, ischemic ulceration, wart, a foreign body, callus, a nonhealing wound, or other benign skin lesions such as seborrheic keratosis.2,8,9
Amelanotic lesions are particularly misleading on the foot, as they may be frequently misdiagnosed as hyperkeratotic benign lesions.8 Bleeding, itching, satellite lesions, and sudden growth, however, should raise your suspicion of melanoma.10
Excision is best, plus sentinel node mapping
The most effective treatment for ALM is excision—either conservatively or with wide margins—along with sentinel lymph node mapping.11 Systemic chemotherapy is more palliative than curative, as it has not been shown to improve the number of recurrences.2 Patients require lifelong regular full body skin exams with early biopsy of any suspicious skin lesions.9
The prognosis of ALM is poorer than other subtypes of melanoma, mainly because of delayed diagnosis.2,9 The single most important and accurate prognostic factor is the depth of invasion of the tumor as measured by Breslow thickness.10
Other factors that are predictive of a poorer prognosis in ALM patients are male sex and amelanosis.12 It’s important to recognize that the palms, soles, and nails may be sites for melanoma—despite the lack of direct sun exposure.
Our patient required amputation
Our patient’s toe was amputated and the melanoma had a Breslow thickness of 7 mm. Sentinel lymph node biopsy was negative. Our patient received adjuvant chemotherapy with interferon alfa-2B and was followed by oncology.
If you said the face or the arms, you’d be wrong. Given that increased sun exposure and a history of multiple sunburns are recognized as important risk factors for melanoma, it’s easy to assume that the most common sites for melanoma would be exposed areas of the body, such as the face and arms.
Interestingly, though, superficial spreading malignant melanoma—the most common type of malignant melanoma13—is most often found on the legs in women and on the back in men.3,10 One theory is that repeated intense exposure to sunlight may be a more important risk factor than continuous sun exposure.14,15
Correspondence
Rajani Katta, MD, 6620 Main Street, Suite 1425, Houston, TX 77030.
1. Freedberg I. Acral lentiginous melanoma. In: Fitzpatrick’s Dermatology in General Medicine. 5th ed. New York: McGraw-Hill; 1999;767:1081-1083.
2. Stalkup JR, Orengo IF, Katta R. Controversies in acral lentiginous melanoma. Dermatol Surg. 2002;28:1051-1059.
3. Swetter SM. Malignant melanoma. Available at: http://www.emedicine.com/DERM/topic257.htm. Accessed June 6, 2008.
4. Cress RD, Holly EA. Incidence of cutaneous melanoma among non-Hispanic whites, Hispanics, Asians, and blacks: an analysis of California cancer registry data, 1988-93. Cancer Causes Control. 1997;8:246-252.
5. Saldanha G, Potter L, Daforno P, Pringle JH. Cutaneous melanoma subtypes show different BRAF and NRAS mutation frequencies. Clin Cancer Res. 2006;12:4499-4505.
6. Green A, McCredie M, MacKie R, et al. A casecontrol study of melanomas of the soles and palms (Australia and Scotland). Cancer Causes Control. 1999;10:21-25.
7. Kaskel P, Kind P, Sander S, Peter RU, Krähn G. Trauma and melanoma formation: a true association? Br J Dermatol. 2000;143:749-753.
8. Soon SL, Solomon AR, Jr, Papadopoulos D, Murray DR, McAlpine B, Washington CV. Acral lentiginous melanoma mimicking benign disease: the Emory experience. J Am Acad Dermatol. 2003;48:183-188.
9. Rosen T. Acral lentiginous melanoma misdiagnosed as verruca plantaris: a case report. Dermatology Online Journal. 2006;12(4):3.-Available at: http://dermatology.cdlib.org/124/case_reports/melanoma/rosen.html. Accessed June 6, 2008.
10. Bruce AJ, Brodland DG. Overview of skin cancer detection and prevention for the primary care physician. Mayo Clin Proc. 2000;75:491-500.
11. Lang PG. Current concepts in the management of patients with melanoma. Am J Clin Dermatol. 2002;3:401-426.
12. Phan A, Touzet S, Dalle S, Ronger-Savle S, Balme B, Thomas L. Acral lentiginous melanoma: a clinicoprognostic study of 126 cases. Br J Dermatol. 2006;155:561-569.
13. Kang S, Barnhill RL, Mihm MC, Jr, et al. Multiple primary cutaneous melanomas. Cancer. 1992;70:1911-1916.
14. Elwood JM. Melanoma and sun exposure: contrasts between intermittent and chronic exposure. World J Surg. 1992;16:157-165.
15. Elwood JM, Gallagher RP. Body site distribution of cutaneous malignant melanoma in relationship to patterns of sun exposure. Int J Cancer. 1998;78:276-280.
1. Freedberg I. Acral lentiginous melanoma. In: Fitzpatrick’s Dermatology in General Medicine. 5th ed. New York: McGraw-Hill; 1999;767:1081-1083.
2. Stalkup JR, Orengo IF, Katta R. Controversies in acral lentiginous melanoma. Dermatol Surg. 2002;28:1051-1059.
3. Swetter SM. Malignant melanoma. Available at: http://www.emedicine.com/DERM/topic257.htm. Accessed June 6, 2008.
4. Cress RD, Holly EA. Incidence of cutaneous melanoma among non-Hispanic whites, Hispanics, Asians, and blacks: an analysis of California cancer registry data, 1988-93. Cancer Causes Control. 1997;8:246-252.
5. Saldanha G, Potter L, Daforno P, Pringle JH. Cutaneous melanoma subtypes show different BRAF and NRAS mutation frequencies. Clin Cancer Res. 2006;12:4499-4505.
6. Green A, McCredie M, MacKie R, et al. A casecontrol study of melanomas of the soles and palms (Australia and Scotland). Cancer Causes Control. 1999;10:21-25.
7. Kaskel P, Kind P, Sander S, Peter RU, Krähn G. Trauma and melanoma formation: a true association? Br J Dermatol. 2000;143:749-753.
8. Soon SL, Solomon AR, Jr, Papadopoulos D, Murray DR, McAlpine B, Washington CV. Acral lentiginous melanoma mimicking benign disease: the Emory experience. J Am Acad Dermatol. 2003;48:183-188.
9. Rosen T. Acral lentiginous melanoma misdiagnosed as verruca plantaris: a case report. Dermatology Online Journal. 2006;12(4):3.-Available at: http://dermatology.cdlib.org/124/case_reports/melanoma/rosen.html. Accessed June 6, 2008.
10. Bruce AJ, Brodland DG. Overview of skin cancer detection and prevention for the primary care physician. Mayo Clin Proc. 2000;75:491-500.
11. Lang PG. Current concepts in the management of patients with melanoma. Am J Clin Dermatol. 2002;3:401-426.
12. Phan A, Touzet S, Dalle S, Ronger-Savle S, Balme B, Thomas L. Acral lentiginous melanoma: a clinicoprognostic study of 126 cases. Br J Dermatol. 2006;155:561-569.
13. Kang S, Barnhill RL, Mihm MC, Jr, et al. Multiple primary cutaneous melanomas. Cancer. 1992;70:1911-1916.
14. Elwood JM. Melanoma and sun exposure: contrasts between intermittent and chronic exposure. World J Surg. 1992;16:157-165.
15. Elwood JM, Gallagher RP. Body site distribution of cutaneous malignant melanoma in relationship to patterns of sun exposure. Int J Cancer. 1998;78:276-280.
Purple-red papules on foot
An 88-year-old Caucasian man of Italian ancestry came into our clinic with multiple, painful purple-red “growths” on his left foot that he’d had for several years (FIGURE 1).
The patient had no systemic complaints (no fever, chills, weight loss, night sweats). He had a history of hypertension, a cardiac valve replacement, and chronic back pain (secondary to a motor vehicle accident). He was taking warfarin and nadolol.
The patient had multiple, 0.1– to 0.5-cm purple-red papules and nodules on the dorsal and plantar surfaces of the left foot, with associated moderate lower extremity edema and mottled dyspigmentation.
We did a punch biopsy, which showed a nodular neoplasm composed of moderately plump, spindle-shaped cells in short interweaving fascicles and numerous extravasated erythrocytes in the spaces (“vascular slits”) between the spindle-shaped cells (FIGURE 2).
FIGURE 1
Painful papules and nodules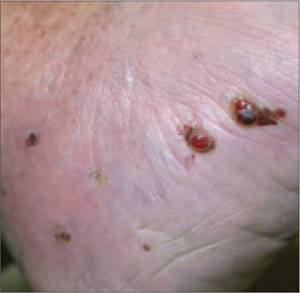
FIGURE 2
Hematoxylin/eosin stain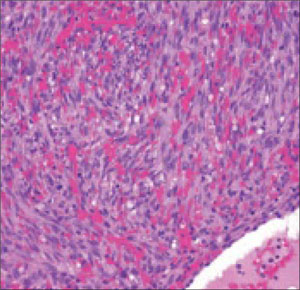
What is your diagnosis?
How would you manage this condition?
Diagnosis: Kaposi’s sarcoma
Classic Kaposi’s sarcoma is a rare mesenchymal tumor most often seen in elderly men of Mediterranean or Ashkenazi Jewish origin with an annual incidence in the United States of between 0.02% and 0.06%, with a peak occurring in the 5th to 8th decade of life.1 (Two-thirds of cases develop after the age of 50.) Population-based studies in the United States have shown a male-to-female ratio of 4:1.1
First described by the Hungarian dermatologist Moritz Kaposi in 1872, Kaposi’s sarcoma assumed prominence during the emerging HIV epidemic and is now the most common tumor in patients with acquired immune deficiency syndrome (AIDS).2
Recent research has implicated the human herpes virus–8 (HHV–8) as an inductive agent (necessary though not sufficient) in all epidemiologic subsets of the disease.2
There are 4 principal clinical variants of Kaposi’s sarcoma:
- classic (or chronic),
- African endemic (includes childhood lymphadenopathic),
- transplant-associated, and
- AIDS-related.
What you’ll see
Clinically, classic Kaposi’s sarcoma often first manifests as blue-red, well-demarcated, painless macules confined to the distal lower extremities.3 These slow-growing lesions may enlarge to forms papules and plaques, or progress to nodules and tumors. Unilateral involvement is often observed at the outset of the disease, with potential centripetal spread occurring late-in-course.3
Early lesions are generally soft, spongy, and “angiomatous,” while in the advanced state, lesional skin becomes hard, solid, and brown in color.3 Edema of the surrounding tissue is common. In addition to the skin, classic Kaposi’s sarcoma also involves mucosal sites (especially the oral and gastrointestinal mucosae).
Differential includes melanocytic nevus
A differential diagnosis for classic Kaposi’s sarcoma includes stasis dermatitis (“acroangiodermatitis”), melanocytic nevus, pyogenic granuloma, hemangioma, granuloma annulare, arthropod assault, and dermatofibroma/dermatofibrosarcoma protuberans (DF/DFSP).
Melanocytic nevi, pyogenic granuloma, hemangioma, granuloma annulare, and DF/DFSP ordinarily feature single lesions, while Kaposi’s sarcoma has multiple lesions. An arthropod assault is pruritic, and stasis dermatitis typically has dilated/varicose veins.
Histology will confirm your suspicions
While epidemiological and clinical factors may suggest classic Kaposi’s sarcoma, a final diagnosis ultimately rests on confirmatory histology. The pathology of classic Kaposi’s sarcoma (like all of the variant subtypes) is based solely on stage of the lesion.
Early patch-stage lesions exhibit papillary dermal proliferation of small, angulated vessels lined by bland endothelial cells with an accompanying sparse infiltrate of lymphocytes and plasma cells.
As the disease progresses to the plaque stage, the vascular proliferation expands into the reticular dermis and subcutis. The transition to nodular Kaposi’s sarcoma develops when a population of spindle cells expressing endothelial markers occurs between the “vascular slits” (FIGURE 2).
Chemotherapy for rapidly progressive disease
There is minimal evidence-based data for the treatment of Kaposi’s sarcoma. Treatment options for limited disease include surgical excision, cryotherapy, laser ablation, topical retinoids (alitretinoin), interferon-alpha, and radiation.1
If rapidly progressive disease (>10 new lesions per month) exists, the most effective treatment remains systemic chemotherapy (vincristine, doxorubicin, vinblastine,4 bleomycin,4 or paclitaxel5). The benefits of chemotherapy can last for months—and even years.
Liquid nitrogen cryotherapy does the trick
We treated our patient with liquid nitrogen cryotherapy that was applied at regular 4- to 6-week intervals over several months. After 3 months, our patient’s lesions were nearly resolved. We followed him monthly thereafter.
Correspondence
John Patrick Welsh, MD, Associates in Dermatology, 4727 Friendship Avenue, Suite 300, Pittsburgh, PA 15224-1778; [email protected].
1. Iscovich J, Boffetta P, Franceschi S, Azizi E, Sarid R. Classic Kaposi sarcoma: epidemiology and risk factors. Cancer. 2000;88:500-517.
2. Pellet C, Kerob D, Dupuy A, et al. Kaposi’s sarcoma-associated herpesvirus viremia is associated with the progression of classic and endemic Kaposi’s sarcoma. J Invest Dermatol. 2006;126:621-627.
3. Schwartz R. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151.
4. Brambilla L, Miedico A, Ferrucci S, et al. Combination of vinblastine and bleomycin as first line therapy in advanced classic Kaposi’s sarcoma. J Eur Acad Dermatol Venereol. 2006;20:1090-1094.
5. Baskan EB, Tunali S, Adim SB, et al. Treatment of advanced classic Kaposi’s sarcoma with weekly low-dose paclitaxel therapy. Int J Dermatol. 2006;45:1441-1443.
An 88-year-old Caucasian man of Italian ancestry came into our clinic with multiple, painful purple-red “growths” on his left foot that he’d had for several years (FIGURE 1).
The patient had no systemic complaints (no fever, chills, weight loss, night sweats). He had a history of hypertension, a cardiac valve replacement, and chronic back pain (secondary to a motor vehicle accident). He was taking warfarin and nadolol.
The patient had multiple, 0.1– to 0.5-cm purple-red papules and nodules on the dorsal and plantar surfaces of the left foot, with associated moderate lower extremity edema and mottled dyspigmentation.
We did a punch biopsy, which showed a nodular neoplasm composed of moderately plump, spindle-shaped cells in short interweaving fascicles and numerous extravasated erythrocytes in the spaces (“vascular slits”) between the spindle-shaped cells (FIGURE 2).
FIGURE 1
Painful papules and nodules
FIGURE 2
Hematoxylin/eosin stain
What is your diagnosis?
How would you manage this condition?
Diagnosis: Kaposi’s sarcoma
Classic Kaposi’s sarcoma is a rare mesenchymal tumor most often seen in elderly men of Mediterranean or Ashkenazi Jewish origin with an annual incidence in the United States of between 0.02% and 0.06%, with a peak occurring in the 5th to 8th decade of life.1 (Two-thirds of cases develop after the age of 50.) Population-based studies in the United States have shown a male-to-female ratio of 4:1.1
First described by the Hungarian dermatologist Moritz Kaposi in 1872, Kaposi’s sarcoma assumed prominence during the emerging HIV epidemic and is now the most common tumor in patients with acquired immune deficiency syndrome (AIDS).2
Recent research has implicated the human herpes virus–8 (HHV–8) as an inductive agent (necessary though not sufficient) in all epidemiologic subsets of the disease.2
There are 4 principal clinical variants of Kaposi’s sarcoma:
- classic (or chronic),
- African endemic (includes childhood lymphadenopathic),
- transplant-associated, and
- AIDS-related.
What you’ll see
Clinically, classic Kaposi’s sarcoma often first manifests as blue-red, well-demarcated, painless macules confined to the distal lower extremities.3 These slow-growing lesions may enlarge to forms papules and plaques, or progress to nodules and tumors. Unilateral involvement is often observed at the outset of the disease, with potential centripetal spread occurring late-in-course.3
Early lesions are generally soft, spongy, and “angiomatous,” while in the advanced state, lesional skin becomes hard, solid, and brown in color.3 Edema of the surrounding tissue is common. In addition to the skin, classic Kaposi’s sarcoma also involves mucosal sites (especially the oral and gastrointestinal mucosae).
Differential includes melanocytic nevus
A differential diagnosis for classic Kaposi’s sarcoma includes stasis dermatitis (“acroangiodermatitis”), melanocytic nevus, pyogenic granuloma, hemangioma, granuloma annulare, arthropod assault, and dermatofibroma/dermatofibrosarcoma protuberans (DF/DFSP).
Melanocytic nevi, pyogenic granuloma, hemangioma, granuloma annulare, and DF/DFSP ordinarily feature single lesions, while Kaposi’s sarcoma has multiple lesions. An arthropod assault is pruritic, and stasis dermatitis typically has dilated/varicose veins.
Histology will confirm your suspicions
While epidemiological and clinical factors may suggest classic Kaposi’s sarcoma, a final diagnosis ultimately rests on confirmatory histology. The pathology of classic Kaposi’s sarcoma (like all of the variant subtypes) is based solely on stage of the lesion.
Early patch-stage lesions exhibit papillary dermal proliferation of small, angulated vessels lined by bland endothelial cells with an accompanying sparse infiltrate of lymphocytes and plasma cells.
As the disease progresses to the plaque stage, the vascular proliferation expands into the reticular dermis and subcutis. The transition to nodular Kaposi’s sarcoma develops when a population of spindle cells expressing endothelial markers occurs between the “vascular slits” (FIGURE 2).
Chemotherapy for rapidly progressive disease
There is minimal evidence-based data for the treatment of Kaposi’s sarcoma. Treatment options for limited disease include surgical excision, cryotherapy, laser ablation, topical retinoids (alitretinoin), interferon-alpha, and radiation.1
If rapidly progressive disease (>10 new lesions per month) exists, the most effective treatment remains systemic chemotherapy (vincristine, doxorubicin, vinblastine,4 bleomycin,4 or paclitaxel5). The benefits of chemotherapy can last for months—and even years.
Liquid nitrogen cryotherapy does the trick
We treated our patient with liquid nitrogen cryotherapy that was applied at regular 4- to 6-week intervals over several months. After 3 months, our patient’s lesions were nearly resolved. We followed him monthly thereafter.
Correspondence
John Patrick Welsh, MD, Associates in Dermatology, 4727 Friendship Avenue, Suite 300, Pittsburgh, PA 15224-1778; [email protected].
An 88-year-old Caucasian man of Italian ancestry came into our clinic with multiple, painful purple-red “growths” on his left foot that he’d had for several years (FIGURE 1).
The patient had no systemic complaints (no fever, chills, weight loss, night sweats). He had a history of hypertension, a cardiac valve replacement, and chronic back pain (secondary to a motor vehicle accident). He was taking warfarin and nadolol.
The patient had multiple, 0.1– to 0.5-cm purple-red papules and nodules on the dorsal and plantar surfaces of the left foot, with associated moderate lower extremity edema and mottled dyspigmentation.
We did a punch biopsy, which showed a nodular neoplasm composed of moderately plump, spindle-shaped cells in short interweaving fascicles and numerous extravasated erythrocytes in the spaces (“vascular slits”) between the spindle-shaped cells (FIGURE 2).
FIGURE 1
Painful papules and nodules
FIGURE 2
Hematoxylin/eosin stain
What is your diagnosis?
How would you manage this condition?
Diagnosis: Kaposi’s sarcoma
Classic Kaposi’s sarcoma is a rare mesenchymal tumor most often seen in elderly men of Mediterranean or Ashkenazi Jewish origin with an annual incidence in the United States of between 0.02% and 0.06%, with a peak occurring in the 5th to 8th decade of life.1 (Two-thirds of cases develop after the age of 50.) Population-based studies in the United States have shown a male-to-female ratio of 4:1.1
First described by the Hungarian dermatologist Moritz Kaposi in 1872, Kaposi’s sarcoma assumed prominence during the emerging HIV epidemic and is now the most common tumor in patients with acquired immune deficiency syndrome (AIDS).2
Recent research has implicated the human herpes virus–8 (HHV–8) as an inductive agent (necessary though not sufficient) in all epidemiologic subsets of the disease.2
There are 4 principal clinical variants of Kaposi’s sarcoma:
- classic (or chronic),
- African endemic (includes childhood lymphadenopathic),
- transplant-associated, and
- AIDS-related.
What you’ll see
Clinically, classic Kaposi’s sarcoma often first manifests as blue-red, well-demarcated, painless macules confined to the distal lower extremities.3 These slow-growing lesions may enlarge to forms papules and plaques, or progress to nodules and tumors. Unilateral involvement is often observed at the outset of the disease, with potential centripetal spread occurring late-in-course.3
Early lesions are generally soft, spongy, and “angiomatous,” while in the advanced state, lesional skin becomes hard, solid, and brown in color.3 Edema of the surrounding tissue is common. In addition to the skin, classic Kaposi’s sarcoma also involves mucosal sites (especially the oral and gastrointestinal mucosae).
Differential includes melanocytic nevus
A differential diagnosis for classic Kaposi’s sarcoma includes stasis dermatitis (“acroangiodermatitis”), melanocytic nevus, pyogenic granuloma, hemangioma, granuloma annulare, arthropod assault, and dermatofibroma/dermatofibrosarcoma protuberans (DF/DFSP).
Melanocytic nevi, pyogenic granuloma, hemangioma, granuloma annulare, and DF/DFSP ordinarily feature single lesions, while Kaposi’s sarcoma has multiple lesions. An arthropod assault is pruritic, and stasis dermatitis typically has dilated/varicose veins.
Histology will confirm your suspicions
While epidemiological and clinical factors may suggest classic Kaposi’s sarcoma, a final diagnosis ultimately rests on confirmatory histology. The pathology of classic Kaposi’s sarcoma (like all of the variant subtypes) is based solely on stage of the lesion.
Early patch-stage lesions exhibit papillary dermal proliferation of small, angulated vessels lined by bland endothelial cells with an accompanying sparse infiltrate of lymphocytes and plasma cells.
As the disease progresses to the plaque stage, the vascular proliferation expands into the reticular dermis and subcutis. The transition to nodular Kaposi’s sarcoma develops when a population of spindle cells expressing endothelial markers occurs between the “vascular slits” (FIGURE 2).
Chemotherapy for rapidly progressive disease
There is minimal evidence-based data for the treatment of Kaposi’s sarcoma. Treatment options for limited disease include surgical excision, cryotherapy, laser ablation, topical retinoids (alitretinoin), interferon-alpha, and radiation.1
If rapidly progressive disease (>10 new lesions per month) exists, the most effective treatment remains systemic chemotherapy (vincristine, doxorubicin, vinblastine,4 bleomycin,4 or paclitaxel5). The benefits of chemotherapy can last for months—and even years.
Liquid nitrogen cryotherapy does the trick
We treated our patient with liquid nitrogen cryotherapy that was applied at regular 4- to 6-week intervals over several months. After 3 months, our patient’s lesions were nearly resolved. We followed him monthly thereafter.
Correspondence
John Patrick Welsh, MD, Associates in Dermatology, 4727 Friendship Avenue, Suite 300, Pittsburgh, PA 15224-1778; [email protected].
1. Iscovich J, Boffetta P, Franceschi S, Azizi E, Sarid R. Classic Kaposi sarcoma: epidemiology and risk factors. Cancer. 2000;88:500-517.
2. Pellet C, Kerob D, Dupuy A, et al. Kaposi’s sarcoma-associated herpesvirus viremia is associated with the progression of classic and endemic Kaposi’s sarcoma. J Invest Dermatol. 2006;126:621-627.
3. Schwartz R. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151.
4. Brambilla L, Miedico A, Ferrucci S, et al. Combination of vinblastine and bleomycin as first line therapy in advanced classic Kaposi’s sarcoma. J Eur Acad Dermatol Venereol. 2006;20:1090-1094.
5. Baskan EB, Tunali S, Adim SB, et al. Treatment of advanced classic Kaposi’s sarcoma with weekly low-dose paclitaxel therapy. Int J Dermatol. 2006;45:1441-1443.
1. Iscovich J, Boffetta P, Franceschi S, Azizi E, Sarid R. Classic Kaposi sarcoma: epidemiology and risk factors. Cancer. 2000;88:500-517.
2. Pellet C, Kerob D, Dupuy A, et al. Kaposi’s sarcoma-associated herpesvirus viremia is associated with the progression of classic and endemic Kaposi’s sarcoma. J Invest Dermatol. 2006;126:621-627.
3. Schwartz R. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151.
4. Brambilla L, Miedico A, Ferrucci S, et al. Combination of vinblastine and bleomycin as first line therapy in advanced classic Kaposi’s sarcoma. J Eur Acad Dermatol Venereol. 2006;20:1090-1094.
5. Baskan EB, Tunali S, Adim SB, et al. Treatment of advanced classic Kaposi’s sarcoma with weekly low-dose paclitaxel therapy. Int J Dermatol. 2006;45:1441-1443.
A disfigured foot with ulcer
A 60-year-old woman came into our area hospital seeking relief for a foot wound that she’d had for several months (FIGURE 1A). The patient said that she had used several antibiotics (prescribed by her primary care physician over the past few months) and she changed her dressings daily, but the wound was not going away. Her podiatrist had evaluated her for osteomyelitis, and the results of the bone biopsy were pending.
The patient had a large wound on the plantar surface of her left foot. The skin over the surface had full-thickness breakdown of the epidermis and dermis, with partial necrosis of the subcutaneous tissue.
Following the wound edges did not reveal undermining, and there was no evidence of sinus tract formation. The ulcer did not extend through the fascia, and there was no gross damage to underlying muscle, bone, or tendon.
Her past medical history was significant for diabetes with neuropathy, nephropathy, and retinopathy. A radiograph was taken (FIGURE 1B).
FIGURE 1
Abnormally shaped foot and pressure ulcer

What is your diagnosis of her foot deformity?
What is the stage of her pressure ulcer?
Dx: “Rocker bottom foot,” Stage III pressure ulcer
The radiograph of the patient’s left foot (FIGURE 1B) revealed extensive collapse of the inner arch and a “rocker bottom foot”—the result of Charcot joint changes. Our patient also had an associated Stage III pressure ulcer.
Charcot joint, also known as neurogenic arthropathy, has been linked to tabes dorsalis, but is more commonly seen in diabetic neuropathy, syringomyelia, spinal cord injury, pernicious anemia, peripheral nerve injury, and from prolonged hydrocortisone injections into the joint.1 The prevalence of Charcot joint in diabetic patients with neuropathy is estimated to be 0.8% to 7.5%. Nine percent to 35% of affected patients have bilateral involvement.2
In patients like ours with diabetic neuropathy, secondary degenerative changes to the joints occur as normal muscle tone, proprioception, temperature perception, and pain perception are lost. The joints become loose, enlarged, and boggy. Though the joint is painless, there can be extensive cartilage erosion or osteophyte formation. The normal plantar and heel forces are increased, producing eccentric loading of the foot, leading to microfractures, ligament laxity, and progression to bony destruction.2
The area of the foot most likely to be involved is the midfoot (70%), with the forefoot and rearfoot comprising the other 30% (15% and 15%, respectively). Charcot joint can occur in a short amount of time in patients with diabetic neuropathy who have experienced even minor trauma.3 In our patient’s case, her trauma history was unknown, though she did comment that her feet initially would ache with walking and then became progressively more disfigured.
The ulcer connection
Approximately 50% of diabetic patients with Charcot joint will have an associated plantar ulcer, secondary to the pressure.3 Our patient had a Stage III pressure ulcer—an ulcer with full-thickness skin loss. With Stage III pressure ulcers, there is also damage or necrosis of the subcutaneous tissue, which may extend down to, but not through, the underlying fascia. (For more on staging pressure ulcers, see TABLE.4)
TABLE
Pressure ulcer staging4
| Suspected deep tissue injury | Localized area of discolored intact skin or blood-filled blister. Area may be surrounded by tissue that is painful, firm, mushy, boggy, or warmer/cooler than adjacent tissue |
| Stage I | Nonblanchable erythema of intact skin |
| Stage II | Partial-thickness loss of dermis that presents as a shallow open ulcer with a red/pink wound bed, without slough |
| Stage III | Full-thickness tissue loss; subcutaneous fat may be visible, though bone, tendon, or muscle are not |
| Stage IV | Full-thickness tissue loss with exposed bone, tendon, or muscle. Slough or eschar may be present |
| Unstageable | Full-thickness tissue loss in which the base of the ulcer is covered by slough and/or eschar |
A patient with her share of challenges
Our patient’s podiatrist had diagnosed her with Charcot joint several years prior, and more recently, her primary care physician had put her on antibiotics several times out of concern for cellulitis.
In fact, 2 weeks before we saw her, our patient had started taking antibiotics for a recurrence of infection. Wound cultures which had been sent on the patient were positive for Staphylococcus aureus and Enterococcus, so she began taking linezolid and clindamycin (she was allergic to penicillin and fluoroquinolones).
Worries that she might have a more serious infection prompted a trip to the podiatrist, who performed an MRI, which was questionable for osteomyelitis. The podiatrist then did a bone biopsy to determine if osteomyelitis was present, and provided mechanical debridement for the wound.
While awaiting the biopsy results, the patient developed increasing pain in her foot and developed new drainage from the ulcerated wound, so she sought treatment at our hospital over the weekend. Our pressing concern: Was this an acute process, such as osteomyelitis, or worsening Charcot joint?
The biopsy provided our answer: It was negative for osteomyelitis.
Simple test distinguishes Charcot from infection
A useful test to distinguish Charcot joint from infection in patients who have plantar ulcers is to elevate the affected extremity for 5 to 10 minutes. If swelling and redness persist when the foot is elevated, it is more likely an infectious process. If the swelling and redness resolve, a Charcot process is most likely.
In cases where it’s unclear whether the patient has Charcot joint or an infection, it may be necessary to obtain a synovial or bone biopsy to make the diagnosis.2
Treat the pressure ulcer before Charcot joint
The mainstays of treatment for pressure ulcers are relieving local pressure, keeping the area clean, using antibiotics for infection (if needed), debriding as necessary, and applying a dressing.
Relieving local pressure is important to reduce formation/progression of pressure ulcers. This can be achieved by careful evaluation of the patient’s skin and bony prominences. If the skin is red or irritated—suggesting that the tissue is under increased pressure—the patient can reduce pressure on the area. Custom orthotics can help toward that end. Frequent evaluation for skin breakdown is key to determining whether the intervention is working.
In our patient’s case, steps had been taken to relieve pressure. Her physician had instructed her to spend part of her days in a wheelchair (to reduce time spent with direct pressure on the soles of her feet) and the other part wearing custom orthotics. Unfortunately, she had not been wearing the orthotics. She felt that her regular shoes were more comfortable.
Keeping the area clean is of course essential, but there is no need to treat with systemic antibiotics unless there are signs of systemic cellulitis or osteomyelitis on MRI. Topical antibiotics may be used, but be sure to avoid any topical agents or disinfectants that can inadvertently damage new tissue growth.
Debriding necrotic tissue decreases bacterial load, increases effectiveness of topical antimicrobials, improves the body’s own antimicrobial function, and shortens the inflammatory phase of the wound. It can be accomplished in various ways, including: sharp debridement, mechanical debridement via gauze dressing changes or whirlpool, applied enzymatic agents, autolytic debridement via occlusive dressings, and biologic debridement using sterile maggots to remove necrotic tissue.
Applying an appropriate dressing is of course essential. Options include films, hydrogels, hydrocolloids, alginates, foams, and vacuum dressings. Topical dressings help maintain moisture and facilitate healing. Occlusive dressings facilitate autolytic debridement and act as a barrier against soiling. Hydrogels, hydrocolloids, and alginates help to absorb moisture. Vacuum dressings remove excess moisture and facilitate wound contracture and healing.
- Medline Plus, at www.nlm.nih.gov/medlineplus/pressuresores.html, provides access to basic information regarding pressure ulcers, as well as links to patient information pages from JAMA and the American Academy of Family Physicians.
- The National Pressure Ulcer Advisory Panel (NPUAP), at www.npuap.org, provides various resources, including staging information.
- The Braden Score, developed for neurosurgical patients to predict their risk of skin breakdown, may be a useful tool for evaluating other patients’ risk. See www.bradenscale.com/braden.PDF for the assessment form.
Next step: Apply a cast
A total contact cast is often utilized to relieve the pressure on the prone portion of the foot. As edema of the affected lower limb decreases over the first week, you’ll need to remove the cast and reapply another. Afterward, you can change the cast every 2 to 4 weeks. Recasting can continue for up to 4 months, with interval radiographs recommended every 4 to 6 weeks during this process.2
When the extremity is no longer swollen and erythematous, you can transition the patient to an ankle foot orthosis or patellar tendon-bearing brace. Surgical options include resecting bony prominences, osteotomies to re-approximate normal anatomy, and amputation.
Getting our patient back on her feet
Once we learned that our patient’s biopsy was negative for osteomyelitis, we began treating her with IV vancomycin for her resistant wound infection. We debrided the pressure ulcer for necrotic tissue.
We initially applied Accuzyme ointment, covered it with Telfa pads, and wrapped it in Kerlix. But the patient developed sensitivity to the Accuzyme, and after discussion with podiatry, we continued her wound care with wet to dry dressings alone for further debridement until cellulitis resolved. (Moist healing is usually recommended to allow new tissue growth.)
While we did not treat our patient’s pressure ulcers with casting, our plan was to do so when the infection resolved. The patient stayed in the hospital for 6 days and was discharged home with 4 weeks of IV vancomycin via a PICC.
Correspondence
Daniel L. Stulberg, MD, University of New Mexico, Department of Family and Community Medicine, MSC 095040, Albuquerque, NM 87131-0001; [email protected]
1. Tierney L, McPhee S, Papadakis M. Current Medical Diagnosis and Treatment. 4th ed. New York, New York: McGraw Hill Medical Publishing Division; 2004:830.
2. Sommer T, Lee T. Charcot Foot: The Diagnostic Dilemma. Am Fam Physician. 2001;64:1591-1598.
3. Armstrong DG, Todd WF, Lavery LA, Harkless LB, Bushman TR. The natural history of acute Charcot’s arthropathy in the diabetic foot specialty clinic. Diabet Med. 1997;14:357-363.
4. National Pressure Ulcer Advisory Panel. Pressure ulcer stages revised by NPUAP. Available at www.npuap.org. Accessed April 2, 2008.
A 60-year-old woman came into our area hospital seeking relief for a foot wound that she’d had for several months (FIGURE 1A). The patient said that she had used several antibiotics (prescribed by her primary care physician over the past few months) and she changed her dressings daily, but the wound was not going away. Her podiatrist had evaluated her for osteomyelitis, and the results of the bone biopsy were pending.
The patient had a large wound on the plantar surface of her left foot. The skin over the surface had full-thickness breakdown of the epidermis and dermis, with partial necrosis of the subcutaneous tissue.
Following the wound edges did not reveal undermining, and there was no evidence of sinus tract formation. The ulcer did not extend through the fascia, and there was no gross damage to underlying muscle, bone, or tendon.
Her past medical history was significant for diabetes with neuropathy, nephropathy, and retinopathy. A radiograph was taken (FIGURE 1B).
FIGURE 1
Abnormally shaped foot and pressure ulcer
What is your diagnosis of her foot deformity?
What is the stage of her pressure ulcer?
Dx: “Rocker bottom foot,” Stage III pressure ulcer
The radiograph of the patient’s left foot (FIGURE 1B) revealed extensive collapse of the inner arch and a “rocker bottom foot”—the result of Charcot joint changes. Our patient also had an associated Stage III pressure ulcer.
Charcot joint, also known as neurogenic arthropathy, has been linked to tabes dorsalis, but is more commonly seen in diabetic neuropathy, syringomyelia, spinal cord injury, pernicious anemia, peripheral nerve injury, and from prolonged hydrocortisone injections into the joint.1 The prevalence of Charcot joint in diabetic patients with neuropathy is estimated to be 0.8% to 7.5%. Nine percent to 35% of affected patients have bilateral involvement.2
In patients like ours with diabetic neuropathy, secondary degenerative changes to the joints occur as normal muscle tone, proprioception, temperature perception, and pain perception are lost. The joints become loose, enlarged, and boggy. Though the joint is painless, there can be extensive cartilage erosion or osteophyte formation. The normal plantar and heel forces are increased, producing eccentric loading of the foot, leading to microfractures, ligament laxity, and progression to bony destruction.2
The area of the foot most likely to be involved is the midfoot (70%), with the forefoot and rearfoot comprising the other 30% (15% and 15%, respectively). Charcot joint can occur in a short amount of time in patients with diabetic neuropathy who have experienced even minor trauma.3 In our patient’s case, her trauma history was unknown, though she did comment that her feet initially would ache with walking and then became progressively more disfigured.
The ulcer connection
Approximately 50% of diabetic patients with Charcot joint will have an associated plantar ulcer, secondary to the pressure.3 Our patient had a Stage III pressure ulcer—an ulcer with full-thickness skin loss. With Stage III pressure ulcers, there is also damage or necrosis of the subcutaneous tissue, which may extend down to, but not through, the underlying fascia. (For more on staging pressure ulcers, see TABLE.4)
TABLE
Pressure ulcer staging4
| Suspected deep tissue injury | Localized area of discolored intact skin or blood-filled blister. Area may be surrounded by tissue that is painful, firm, mushy, boggy, or warmer/cooler than adjacent tissue |
| Stage I | Nonblanchable erythema of intact skin |
| Stage II | Partial-thickness loss of dermis that presents as a shallow open ulcer with a red/pink wound bed, without slough |
| Stage III | Full-thickness tissue loss; subcutaneous fat may be visible, though bone, tendon, or muscle are not |
| Stage IV | Full-thickness tissue loss with exposed bone, tendon, or muscle. Slough or eschar may be present |
| Unstageable | Full-thickness tissue loss in which the base of the ulcer is covered by slough and/or eschar |
A patient with her share of challenges
Our patient’s podiatrist had diagnosed her with Charcot joint several years prior, and more recently, her primary care physician had put her on antibiotics several times out of concern for cellulitis.
In fact, 2 weeks before we saw her, our patient had started taking antibiotics for a recurrence of infection. Wound cultures which had been sent on the patient were positive for Staphylococcus aureus and Enterococcus, so she began taking linezolid and clindamycin (she was allergic to penicillin and fluoroquinolones).
Worries that she might have a more serious infection prompted a trip to the podiatrist, who performed an MRI, which was questionable for osteomyelitis. The podiatrist then did a bone biopsy to determine if osteomyelitis was present, and provided mechanical debridement for the wound.
While awaiting the biopsy results, the patient developed increasing pain in her foot and developed new drainage from the ulcerated wound, so she sought treatment at our hospital over the weekend. Our pressing concern: Was this an acute process, such as osteomyelitis, or worsening Charcot joint?
The biopsy provided our answer: It was negative for osteomyelitis.
Simple test distinguishes Charcot from infection
A useful test to distinguish Charcot joint from infection in patients who have plantar ulcers is to elevate the affected extremity for 5 to 10 minutes. If swelling and redness persist when the foot is elevated, it is more likely an infectious process. If the swelling and redness resolve, a Charcot process is most likely.
In cases where it’s unclear whether the patient has Charcot joint or an infection, it may be necessary to obtain a synovial or bone biopsy to make the diagnosis.2
Treat the pressure ulcer before Charcot joint
The mainstays of treatment for pressure ulcers are relieving local pressure, keeping the area clean, using antibiotics for infection (if needed), debriding as necessary, and applying a dressing.
Relieving local pressure is important to reduce formation/progression of pressure ulcers. This can be achieved by careful evaluation of the patient’s skin and bony prominences. If the skin is red or irritated—suggesting that the tissue is under increased pressure—the patient can reduce pressure on the area. Custom orthotics can help toward that end. Frequent evaluation for skin breakdown is key to determining whether the intervention is working.
In our patient’s case, steps had been taken to relieve pressure. Her physician had instructed her to spend part of her days in a wheelchair (to reduce time spent with direct pressure on the soles of her feet) and the other part wearing custom orthotics. Unfortunately, she had not been wearing the orthotics. She felt that her regular shoes were more comfortable.
Keeping the area clean is of course essential, but there is no need to treat with systemic antibiotics unless there are signs of systemic cellulitis or osteomyelitis on MRI. Topical antibiotics may be used, but be sure to avoid any topical agents or disinfectants that can inadvertently damage new tissue growth.
Debriding necrotic tissue decreases bacterial load, increases effectiveness of topical antimicrobials, improves the body’s own antimicrobial function, and shortens the inflammatory phase of the wound. It can be accomplished in various ways, including: sharp debridement, mechanical debridement via gauze dressing changes or whirlpool, applied enzymatic agents, autolytic debridement via occlusive dressings, and biologic debridement using sterile maggots to remove necrotic tissue.
Applying an appropriate dressing is of course essential. Options include films, hydrogels, hydrocolloids, alginates, foams, and vacuum dressings. Topical dressings help maintain moisture and facilitate healing. Occlusive dressings facilitate autolytic debridement and act as a barrier against soiling. Hydrogels, hydrocolloids, and alginates help to absorb moisture. Vacuum dressings remove excess moisture and facilitate wound contracture and healing.
- Medline Plus, at www.nlm.nih.gov/medlineplus/pressuresores.html, provides access to basic information regarding pressure ulcers, as well as links to patient information pages from JAMA and the American Academy of Family Physicians.
- The National Pressure Ulcer Advisory Panel (NPUAP), at www.npuap.org, provides various resources, including staging information.
- The Braden Score, developed for neurosurgical patients to predict their risk of skin breakdown, may be a useful tool for evaluating other patients’ risk. See www.bradenscale.com/braden.PDF for the assessment form.
Next step: Apply a cast
A total contact cast is often utilized to relieve the pressure on the prone portion of the foot. As edema of the affected lower limb decreases over the first week, you’ll need to remove the cast and reapply another. Afterward, you can change the cast every 2 to 4 weeks. Recasting can continue for up to 4 months, with interval radiographs recommended every 4 to 6 weeks during this process.2
When the extremity is no longer swollen and erythematous, you can transition the patient to an ankle foot orthosis or patellar tendon-bearing brace. Surgical options include resecting bony prominences, osteotomies to re-approximate normal anatomy, and amputation.
Getting our patient back on her feet
Once we learned that our patient’s biopsy was negative for osteomyelitis, we began treating her with IV vancomycin for her resistant wound infection. We debrided the pressure ulcer for necrotic tissue.
We initially applied Accuzyme ointment, covered it with Telfa pads, and wrapped it in Kerlix. But the patient developed sensitivity to the Accuzyme, and after discussion with podiatry, we continued her wound care with wet to dry dressings alone for further debridement until cellulitis resolved. (Moist healing is usually recommended to allow new tissue growth.)
While we did not treat our patient’s pressure ulcers with casting, our plan was to do so when the infection resolved. The patient stayed in the hospital for 6 days and was discharged home with 4 weeks of IV vancomycin via a PICC.
Correspondence
Daniel L. Stulberg, MD, University of New Mexico, Department of Family and Community Medicine, MSC 095040, Albuquerque, NM 87131-0001; [email protected]
A 60-year-old woman came into our area hospital seeking relief for a foot wound that she’d had for several months (FIGURE 1A). The patient said that she had used several antibiotics (prescribed by her primary care physician over the past few months) and she changed her dressings daily, but the wound was not going away. Her podiatrist had evaluated her for osteomyelitis, and the results of the bone biopsy were pending.
The patient had a large wound on the plantar surface of her left foot. The skin over the surface had full-thickness breakdown of the epidermis and dermis, with partial necrosis of the subcutaneous tissue.
Following the wound edges did not reveal undermining, and there was no evidence of sinus tract formation. The ulcer did not extend through the fascia, and there was no gross damage to underlying muscle, bone, or tendon.
Her past medical history was significant for diabetes with neuropathy, nephropathy, and retinopathy. A radiograph was taken (FIGURE 1B).
FIGURE 1
Abnormally shaped foot and pressure ulcer
What is your diagnosis of her foot deformity?
What is the stage of her pressure ulcer?
Dx: “Rocker bottom foot,” Stage III pressure ulcer
The radiograph of the patient’s left foot (FIGURE 1B) revealed extensive collapse of the inner arch and a “rocker bottom foot”—the result of Charcot joint changes. Our patient also had an associated Stage III pressure ulcer.
Charcot joint, also known as neurogenic arthropathy, has been linked to tabes dorsalis, but is more commonly seen in diabetic neuropathy, syringomyelia, spinal cord injury, pernicious anemia, peripheral nerve injury, and from prolonged hydrocortisone injections into the joint.1 The prevalence of Charcot joint in diabetic patients with neuropathy is estimated to be 0.8% to 7.5%. Nine percent to 35% of affected patients have bilateral involvement.2
In patients like ours with diabetic neuropathy, secondary degenerative changes to the joints occur as normal muscle tone, proprioception, temperature perception, and pain perception are lost. The joints become loose, enlarged, and boggy. Though the joint is painless, there can be extensive cartilage erosion or osteophyte formation. The normal plantar and heel forces are increased, producing eccentric loading of the foot, leading to microfractures, ligament laxity, and progression to bony destruction.2
The area of the foot most likely to be involved is the midfoot (70%), with the forefoot and rearfoot comprising the other 30% (15% and 15%, respectively). Charcot joint can occur in a short amount of time in patients with diabetic neuropathy who have experienced even minor trauma.3 In our patient’s case, her trauma history was unknown, though she did comment that her feet initially would ache with walking and then became progressively more disfigured.
The ulcer connection
Approximately 50% of diabetic patients with Charcot joint will have an associated plantar ulcer, secondary to the pressure.3 Our patient had a Stage III pressure ulcer—an ulcer with full-thickness skin loss. With Stage III pressure ulcers, there is also damage or necrosis of the subcutaneous tissue, which may extend down to, but not through, the underlying fascia. (For more on staging pressure ulcers, see TABLE.4)
TABLE
Pressure ulcer staging4
| Suspected deep tissue injury | Localized area of discolored intact skin or blood-filled blister. Area may be surrounded by tissue that is painful, firm, mushy, boggy, or warmer/cooler than adjacent tissue |
| Stage I | Nonblanchable erythema of intact skin |
| Stage II | Partial-thickness loss of dermis that presents as a shallow open ulcer with a red/pink wound bed, without slough |
| Stage III | Full-thickness tissue loss; subcutaneous fat may be visible, though bone, tendon, or muscle are not |
| Stage IV | Full-thickness tissue loss with exposed bone, tendon, or muscle. Slough or eschar may be present |
| Unstageable | Full-thickness tissue loss in which the base of the ulcer is covered by slough and/or eschar |
A patient with her share of challenges
Our patient’s podiatrist had diagnosed her with Charcot joint several years prior, and more recently, her primary care physician had put her on antibiotics several times out of concern for cellulitis.
In fact, 2 weeks before we saw her, our patient had started taking antibiotics for a recurrence of infection. Wound cultures which had been sent on the patient were positive for Staphylococcus aureus and Enterococcus, so she began taking linezolid and clindamycin (she was allergic to penicillin and fluoroquinolones).
Worries that she might have a more serious infection prompted a trip to the podiatrist, who performed an MRI, which was questionable for osteomyelitis. The podiatrist then did a bone biopsy to determine if osteomyelitis was present, and provided mechanical debridement for the wound.
While awaiting the biopsy results, the patient developed increasing pain in her foot and developed new drainage from the ulcerated wound, so she sought treatment at our hospital over the weekend. Our pressing concern: Was this an acute process, such as osteomyelitis, or worsening Charcot joint?
The biopsy provided our answer: It was negative for osteomyelitis.
Simple test distinguishes Charcot from infection
A useful test to distinguish Charcot joint from infection in patients who have plantar ulcers is to elevate the affected extremity for 5 to 10 minutes. If swelling and redness persist when the foot is elevated, it is more likely an infectious process. If the swelling and redness resolve, a Charcot process is most likely.
In cases where it’s unclear whether the patient has Charcot joint or an infection, it may be necessary to obtain a synovial or bone biopsy to make the diagnosis.2
Treat the pressure ulcer before Charcot joint
The mainstays of treatment for pressure ulcers are relieving local pressure, keeping the area clean, using antibiotics for infection (if needed), debriding as necessary, and applying a dressing.
Relieving local pressure is important to reduce formation/progression of pressure ulcers. This can be achieved by careful evaluation of the patient’s skin and bony prominences. If the skin is red or irritated—suggesting that the tissue is under increased pressure—the patient can reduce pressure on the area. Custom orthotics can help toward that end. Frequent evaluation for skin breakdown is key to determining whether the intervention is working.
In our patient’s case, steps had been taken to relieve pressure. Her physician had instructed her to spend part of her days in a wheelchair (to reduce time spent with direct pressure on the soles of her feet) and the other part wearing custom orthotics. Unfortunately, she had not been wearing the orthotics. She felt that her regular shoes were more comfortable.
Keeping the area clean is of course essential, but there is no need to treat with systemic antibiotics unless there are signs of systemic cellulitis or osteomyelitis on MRI. Topical antibiotics may be used, but be sure to avoid any topical agents or disinfectants that can inadvertently damage new tissue growth.
Debriding necrotic tissue decreases bacterial load, increases effectiveness of topical antimicrobials, improves the body’s own antimicrobial function, and shortens the inflammatory phase of the wound. It can be accomplished in various ways, including: sharp debridement, mechanical debridement via gauze dressing changes or whirlpool, applied enzymatic agents, autolytic debridement via occlusive dressings, and biologic debridement using sterile maggots to remove necrotic tissue.
Applying an appropriate dressing is of course essential. Options include films, hydrogels, hydrocolloids, alginates, foams, and vacuum dressings. Topical dressings help maintain moisture and facilitate healing. Occlusive dressings facilitate autolytic debridement and act as a barrier against soiling. Hydrogels, hydrocolloids, and alginates help to absorb moisture. Vacuum dressings remove excess moisture and facilitate wound contracture and healing.
- Medline Plus, at www.nlm.nih.gov/medlineplus/pressuresores.html, provides access to basic information regarding pressure ulcers, as well as links to patient information pages from JAMA and the American Academy of Family Physicians.
- The National Pressure Ulcer Advisory Panel (NPUAP), at www.npuap.org, provides various resources, including staging information.
- The Braden Score, developed for neurosurgical patients to predict their risk of skin breakdown, may be a useful tool for evaluating other patients’ risk. See www.bradenscale.com/braden.PDF for the assessment form.
Next step: Apply a cast
A total contact cast is often utilized to relieve the pressure on the prone portion of the foot. As edema of the affected lower limb decreases over the first week, you’ll need to remove the cast and reapply another. Afterward, you can change the cast every 2 to 4 weeks. Recasting can continue for up to 4 months, with interval radiographs recommended every 4 to 6 weeks during this process.2
When the extremity is no longer swollen and erythematous, you can transition the patient to an ankle foot orthosis or patellar tendon-bearing brace. Surgical options include resecting bony prominences, osteotomies to re-approximate normal anatomy, and amputation.
Getting our patient back on her feet
Once we learned that our patient’s biopsy was negative for osteomyelitis, we began treating her with IV vancomycin for her resistant wound infection. We debrided the pressure ulcer for necrotic tissue.
We initially applied Accuzyme ointment, covered it with Telfa pads, and wrapped it in Kerlix. But the patient developed sensitivity to the Accuzyme, and after discussion with podiatry, we continued her wound care with wet to dry dressings alone for further debridement until cellulitis resolved. (Moist healing is usually recommended to allow new tissue growth.)
While we did not treat our patient’s pressure ulcers with casting, our plan was to do so when the infection resolved. The patient stayed in the hospital for 6 days and was discharged home with 4 weeks of IV vancomycin via a PICC.
Correspondence
Daniel L. Stulberg, MD, University of New Mexico, Department of Family and Community Medicine, MSC 095040, Albuquerque, NM 87131-0001; [email protected]
1. Tierney L, McPhee S, Papadakis M. Current Medical Diagnosis and Treatment. 4th ed. New York, New York: McGraw Hill Medical Publishing Division; 2004:830.
2. Sommer T, Lee T. Charcot Foot: The Diagnostic Dilemma. Am Fam Physician. 2001;64:1591-1598.
3. Armstrong DG, Todd WF, Lavery LA, Harkless LB, Bushman TR. The natural history of acute Charcot’s arthropathy in the diabetic foot specialty clinic. Diabet Med. 1997;14:357-363.
4. National Pressure Ulcer Advisory Panel. Pressure ulcer stages revised by NPUAP. Available at www.npuap.org. Accessed April 2, 2008.
1. Tierney L, McPhee S, Papadakis M. Current Medical Diagnosis and Treatment. 4th ed. New York, New York: McGraw Hill Medical Publishing Division; 2004:830.
2. Sommer T, Lee T. Charcot Foot: The Diagnostic Dilemma. Am Fam Physician. 2001;64:1591-1598.
3. Armstrong DG, Todd WF, Lavery LA, Harkless LB, Bushman TR. The natural history of acute Charcot’s arthropathy in the diabetic foot specialty clinic. Diabet Med. 1997;14:357-363.
4. National Pressure Ulcer Advisory Panel. Pressure ulcer stages revised by NPUAP. Available at www.npuap.org. Accessed April 2, 2008.
Bilateral axillary pustules
A healthy 29-year-old woman presented to the clinic with pustules and boil-like lesions in the axillary areas, on the left forearm, and on the right thigh (FIGURE). She indicated that she’d had the lesions for 4 weeks. She said that she had not had any recent infections, and there was no evidence of immune compromise in her history.
We incised a pustule in the right axilla and expressed the pus. We sent the sample out for bacterial culture and sensitivity testing.
We immediately considered bacterial folliculitis and hidradenitis suppurativa as part of the differential. The acute nature of the process led us to favor a diagnosis of bacterial folliculitis.
FIGURE
Papules and papulopustules on right axilla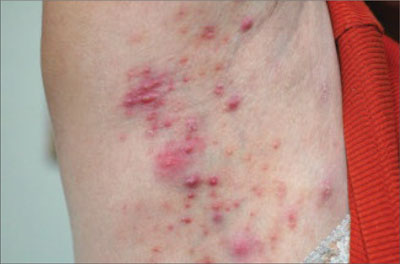
Would you favor this diagnosis?
What would you choose for initial empiric antibiotic treatment?
Diagnosis: MRSA folliculitis
We began empiric systemic antibiotic therapy with doxycycline 100 mg daily and topical treatment with clindamycin 1% lotion twice a day. The bacterial culture came back positive for methicillin-resistant Staphylococcus aureus (MRSA) and the organism proved to be tetracycline sensitive.
Staphylococci species have long been the most common causes of skin and soft tissue infections.1 A recent study in 11 major US cities identified MRSA as the most common cause of acute purulent skin and soft tissue infections in patients presenting to the emergency department.2 This rate, however, varied from 15% to 74% in various cities.2 Today, clinicians must consider MRSA as a potential causative agent whenever they treat a patient with a skin and soft tissue infection.
Community-acquired MRSA: Don’t expect typical risk factors
Community-acquired MRSA (CA-MRSA) cases differ from hospital-acquired MRSA (HA-MRSA) cases demographically, microbiologically, and clinically. CA-MRSA infections typically occur in young, healthy individuals without traditional MRSA risk factors such as recent hospitalization, residence in a long-term care facility, or prior antibiotic use.3,4
CA-MRSA outbreaks have been associated with participation in team sports, living in prison, dormitory, or group home settings, IV drug use, sharing personal items, and men who have sex with men.4,5 However, the prevalence of MRSA infections in patients without any recognized risk factors, like our patient, is increasing.6
CA-MRSA is genetically distinct from HA-MRSA in that it contains the Panton-Valentine leukocidin, an important virulence factor, and carries the type IV or type V SCCmec cassette, which resides at a different methicillin-resistant locus.7,8 CA-MRSA also does not demonstrate the multi-drug resistance typical of HA-MRSA, and these infections tend to be susceptible to most non-beta-lactam antibiotics.6
Is it MRSA or another pathogen?
When considering the differential diagnosis of MRSA folliculitis, consider methicillin-susceptible staphylococci species as equally likely pathogens. Gram-negative bacteria, including Pseudomonas aeruginosa, Malassezia furfur, and Candida species are less common, but notable causes of folliculitis. Other causes include eosinophilic folliculitis, non-bacterial/irritant folliculitis, pseudofolliculitis (chronic in-grown hairs), hidradenitis suppurativa, and acne vulgaris.
Use a low threshold for obtaining cultures CA-MRSA sometimes presents as an edematous, tender abscess, an expanding cellulitis, or both.9,10 These presentations, however, are neither sensitive nor specific. Cutaneous infection may also present as erythematous papules, nodules, pustules, crusted plaques, and infrequently suppurative folliculitis.3,10 Therefore, the clinician should have a low threshold to perform bacterial culture and sensitivity studies whenever a skin and soft tissue infection is suspected.
The potential complications of CA-MRSA infection, including pneumonia, sepsis, and endocarditis, can be avoided or minimized with early diagnosis and initiation of appropriate treatment.4,11,12
Consider community factors
In the past, folliculitis and other localized skin infections were traditionally caused by methicillin-susceptible Staphylococcus aureus and treated with beta-lactam antibiotics. In communities with high prevalence of CA-MRSA skin and soft tissue infections, beta-lactam antibiotics like cephalexin or dicloxacillin may no longer be appropriate.2,5 Therefore, let the prevalence of CA-MRSA in your community help guide your initial antibiotic choice while recognizing that empiric antibiotic therapy may be adjusted after cultures are available.
Good initial choices for CA-MRSA skin and soft tissue infections are trim-ethoprim/sulfamethoxazole with or without rifampin, clindamycin, gentamicin, and tetracycline,2,4,5,7 but inducible clindamycin resistance has been reported.13 if your community has a high prevalence of CA-MRSA, you may decide to begin empiric therapy that provides MRSA coverage.2 In addition, incision and drainage is necessary for adequate treatment of furuncles, carbuncles, and abscesses.5 Healthy, afebrile, immunocompetent patients without cellulitis may not require systemic antibiotics to clear local infections.2,4
Take steps to prevent spread of the disease
Early diagnosis of CA-MRSA infection is important to prevent the spread of this highly virulent disease in the community. It’s important to cover the infected area with sterile gauze and instruct the patient to avoid direct skin-to-skin contact with others, not share sports equipment or personal items, and decontaminate linens and surfaces.4,6
Stress the importance of frequent hand washing and using alcohol-based hand rubs to reduce transmission. Adjuvant topical therapy may be useful: intra-nasal mupirocin 2% ointment to prevent colonization and a persistent carrier state is prudent. Bathing with antimicrobial povidone-iodine or chlorhexidine gluconate is often recommended as an adjunctive treatment for skin and soft tissue infections, colonization, or both.14
Mupirocin 2% for our patient
We instructed our patient to continue the initial medications and to apply mupirocin 2% ointment to the nares twice a day for 1 week to eliminate the possibility of a staphylococcal carrier state.
At follow-up 2 weeks later, only a few excoriated papules and some post-inflammatory erythema remained. The topical clindamycin lotion and the doxycycline were continued for an additional 30 days, at which time the patient was clear and treatment was discontinued.
1. Elston DM. Epidemiology and prevention of skin and soft tissue infections. Cutis 2004;73:S3-7.
2. Moran GJ, Krishnadasan A, Gorwitz RJ, Fosheim GE, McDougal LK, et al. Methicillin-resistant S. aureus. infections among patients in the emergency department. N Engl J Med 2006;355:666-674.
3. Cohen PR, Kurzrock R. Community-acquired methicillin-resistant Staphylococcus aureus skin infection: An emerging clinical problem. J Am Acad Dermatol 2004;50:277-280.
4. Romero DV, Treston J, O’Sullivan AL. Hand-to-hand combat: Preventing MRSA infection. Adv Skin Wound Care 2006;19:328-333.
5. Moran GJ, Amii RN, Abrahamian FM, Talan DA. Methicillin-resistant Staphylococcus aureus in community-acquired skin infections. Emerg Infec Dis 2005;11:928-930.
6. Cohen PR, Grossman ME. Management of cutaneous lesions associated with an emerging epidemic: Community-acquired methicillin-resistant Staphylococcus aureus skin infections. J Am Acad Dermatol 2004;51:132-135.
7. Naimi TS, LeDell KH, Como-Sabetti K, Borchardt SM, Boxrud DJ, et al. Comparison of community-and health care-associated methicillin-resistant Staphylococcus aureus infection. JAMA 2003;290:2976-2984.
8. Vandenesch F, Naimi T, Enright MC, et al. Community-acquired methicillin-resistant Staphylococcus aureus carrying Panton-Valentine leukocidin genes: worldwide emergence. Emerg Infect Dis 2003;9:978-984.
9. Iyer S, Jones DH. Community-acquired methicillin-resistant Staphylococcus aureus skin infection: A retrospective analysis of clinical presentation and treatment of a local outbreak. J Am Acad Dermatol 2004;50:854-858.
10. Cohen PR. Community-acquired methicillin-resistant Staphylococcus aureus skin infection presenting as a periumbilical folliculitis. Cutis. 2006;77:229-231.
11. Bahrain M, Vasiliades M, Wolff M, Younus F. Five cases of bacterial endocarditis after furunculosis and the ongoing saga of community-acquired methicillin-resistant Staphylococcus aureus infections. Scand J Infect Dis 2006;38:702-707.
12. Herold B, Immergluck L, Maranan M, et al. Community-acquired methicillin-resistant Staphylococcus aureus in children with no identified predisposing risk. JAMA 1998;279:593-598.
13. Siberry GK, Tekle T, Carroll K, Dick J. Failure of clindamycin treatment of methicillin-resistant Staphylococcus aureus expressing inducible clindamycin resistance in vitro. Clin Infect Dis 2003;27:1257-1260.
14. Boyce JM. MRSA patient: proven methods to treat colonization and infection. J Hosp Infect 2001;48:S9-14.
A healthy 29-year-old woman presented to the clinic with pustules and boil-like lesions in the axillary areas, on the left forearm, and on the right thigh (FIGURE). She indicated that she’d had the lesions for 4 weeks. She said that she had not had any recent infections, and there was no evidence of immune compromise in her history.
We incised a pustule in the right axilla and expressed the pus. We sent the sample out for bacterial culture and sensitivity testing.
We immediately considered bacterial folliculitis and hidradenitis suppurativa as part of the differential. The acute nature of the process led us to favor a diagnosis of bacterial folliculitis.
FIGURE
Papules and papulopustules on right axilla
Would you favor this diagnosis?
What would you choose for initial empiric antibiotic treatment?
Diagnosis: MRSA folliculitis
We began empiric systemic antibiotic therapy with doxycycline 100 mg daily and topical treatment with clindamycin 1% lotion twice a day. The bacterial culture came back positive for methicillin-resistant Staphylococcus aureus (MRSA) and the organism proved to be tetracycline sensitive.
Staphylococci species have long been the most common causes of skin and soft tissue infections.1 A recent study in 11 major US cities identified MRSA as the most common cause of acute purulent skin and soft tissue infections in patients presenting to the emergency department.2 This rate, however, varied from 15% to 74% in various cities.2 Today, clinicians must consider MRSA as a potential causative agent whenever they treat a patient with a skin and soft tissue infection.
Community-acquired MRSA: Don’t expect typical risk factors
Community-acquired MRSA (CA-MRSA) cases differ from hospital-acquired MRSA (HA-MRSA) cases demographically, microbiologically, and clinically. CA-MRSA infections typically occur in young, healthy individuals without traditional MRSA risk factors such as recent hospitalization, residence in a long-term care facility, or prior antibiotic use.3,4
CA-MRSA outbreaks have been associated with participation in team sports, living in prison, dormitory, or group home settings, IV drug use, sharing personal items, and men who have sex with men.4,5 However, the prevalence of MRSA infections in patients without any recognized risk factors, like our patient, is increasing.6
CA-MRSA is genetically distinct from HA-MRSA in that it contains the Panton-Valentine leukocidin, an important virulence factor, and carries the type IV or type V SCCmec cassette, which resides at a different methicillin-resistant locus.7,8 CA-MRSA also does not demonstrate the multi-drug resistance typical of HA-MRSA, and these infections tend to be susceptible to most non-beta-lactam antibiotics.6
Is it MRSA or another pathogen?
When considering the differential diagnosis of MRSA folliculitis, consider methicillin-susceptible staphylococci species as equally likely pathogens. Gram-negative bacteria, including Pseudomonas aeruginosa, Malassezia furfur, and Candida species are less common, but notable causes of folliculitis. Other causes include eosinophilic folliculitis, non-bacterial/irritant folliculitis, pseudofolliculitis (chronic in-grown hairs), hidradenitis suppurativa, and acne vulgaris.
Use a low threshold for obtaining cultures CA-MRSA sometimes presents as an edematous, tender abscess, an expanding cellulitis, or both.9,10 These presentations, however, are neither sensitive nor specific. Cutaneous infection may also present as erythematous papules, nodules, pustules, crusted plaques, and infrequently suppurative folliculitis.3,10 Therefore, the clinician should have a low threshold to perform bacterial culture and sensitivity studies whenever a skin and soft tissue infection is suspected.
The potential complications of CA-MRSA infection, including pneumonia, sepsis, and endocarditis, can be avoided or minimized with early diagnosis and initiation of appropriate treatment.4,11,12
Consider community factors
In the past, folliculitis and other localized skin infections were traditionally caused by methicillin-susceptible Staphylococcus aureus and treated with beta-lactam antibiotics. In communities with high prevalence of CA-MRSA skin and soft tissue infections, beta-lactam antibiotics like cephalexin or dicloxacillin may no longer be appropriate.2,5 Therefore, let the prevalence of CA-MRSA in your community help guide your initial antibiotic choice while recognizing that empiric antibiotic therapy may be adjusted after cultures are available.
Good initial choices for CA-MRSA skin and soft tissue infections are trim-ethoprim/sulfamethoxazole with or without rifampin, clindamycin, gentamicin, and tetracycline,2,4,5,7 but inducible clindamycin resistance has been reported.13 if your community has a high prevalence of CA-MRSA, you may decide to begin empiric therapy that provides MRSA coverage.2 In addition, incision and drainage is necessary for adequate treatment of furuncles, carbuncles, and abscesses.5 Healthy, afebrile, immunocompetent patients without cellulitis may not require systemic antibiotics to clear local infections.2,4
Take steps to prevent spread of the disease
Early diagnosis of CA-MRSA infection is important to prevent the spread of this highly virulent disease in the community. It’s important to cover the infected area with sterile gauze and instruct the patient to avoid direct skin-to-skin contact with others, not share sports equipment or personal items, and decontaminate linens and surfaces.4,6
Stress the importance of frequent hand washing and using alcohol-based hand rubs to reduce transmission. Adjuvant topical therapy may be useful: intra-nasal mupirocin 2% ointment to prevent colonization and a persistent carrier state is prudent. Bathing with antimicrobial povidone-iodine or chlorhexidine gluconate is often recommended as an adjunctive treatment for skin and soft tissue infections, colonization, or both.14
Mupirocin 2% for our patient
We instructed our patient to continue the initial medications and to apply mupirocin 2% ointment to the nares twice a day for 1 week to eliminate the possibility of a staphylococcal carrier state.
At follow-up 2 weeks later, only a few excoriated papules and some post-inflammatory erythema remained. The topical clindamycin lotion and the doxycycline were continued for an additional 30 days, at which time the patient was clear and treatment was discontinued.
A healthy 29-year-old woman presented to the clinic with pustules and boil-like lesions in the axillary areas, on the left forearm, and on the right thigh (FIGURE). She indicated that she’d had the lesions for 4 weeks. She said that she had not had any recent infections, and there was no evidence of immune compromise in her history.
We incised a pustule in the right axilla and expressed the pus. We sent the sample out for bacterial culture and sensitivity testing.
We immediately considered bacterial folliculitis and hidradenitis suppurativa as part of the differential. The acute nature of the process led us to favor a diagnosis of bacterial folliculitis.
FIGURE
Papules and papulopustules on right axilla
Would you favor this diagnosis?
What would you choose for initial empiric antibiotic treatment?
Diagnosis: MRSA folliculitis
We began empiric systemic antibiotic therapy with doxycycline 100 mg daily and topical treatment with clindamycin 1% lotion twice a day. The bacterial culture came back positive for methicillin-resistant Staphylococcus aureus (MRSA) and the organism proved to be tetracycline sensitive.
Staphylococci species have long been the most common causes of skin and soft tissue infections.1 A recent study in 11 major US cities identified MRSA as the most common cause of acute purulent skin and soft tissue infections in patients presenting to the emergency department.2 This rate, however, varied from 15% to 74% in various cities.2 Today, clinicians must consider MRSA as a potential causative agent whenever they treat a patient with a skin and soft tissue infection.
Community-acquired MRSA: Don’t expect typical risk factors
Community-acquired MRSA (CA-MRSA) cases differ from hospital-acquired MRSA (HA-MRSA) cases demographically, microbiologically, and clinically. CA-MRSA infections typically occur in young, healthy individuals without traditional MRSA risk factors such as recent hospitalization, residence in a long-term care facility, or prior antibiotic use.3,4
CA-MRSA outbreaks have been associated with participation in team sports, living in prison, dormitory, or group home settings, IV drug use, sharing personal items, and men who have sex with men.4,5 However, the prevalence of MRSA infections in patients without any recognized risk factors, like our patient, is increasing.6
CA-MRSA is genetically distinct from HA-MRSA in that it contains the Panton-Valentine leukocidin, an important virulence factor, and carries the type IV or type V SCCmec cassette, which resides at a different methicillin-resistant locus.7,8 CA-MRSA also does not demonstrate the multi-drug resistance typical of HA-MRSA, and these infections tend to be susceptible to most non-beta-lactam antibiotics.6
Is it MRSA or another pathogen?
When considering the differential diagnosis of MRSA folliculitis, consider methicillin-susceptible staphylococci species as equally likely pathogens. Gram-negative bacteria, including Pseudomonas aeruginosa, Malassezia furfur, and Candida species are less common, but notable causes of folliculitis. Other causes include eosinophilic folliculitis, non-bacterial/irritant folliculitis, pseudofolliculitis (chronic in-grown hairs), hidradenitis suppurativa, and acne vulgaris.
Use a low threshold for obtaining cultures CA-MRSA sometimes presents as an edematous, tender abscess, an expanding cellulitis, or both.9,10 These presentations, however, are neither sensitive nor specific. Cutaneous infection may also present as erythematous papules, nodules, pustules, crusted plaques, and infrequently suppurative folliculitis.3,10 Therefore, the clinician should have a low threshold to perform bacterial culture and sensitivity studies whenever a skin and soft tissue infection is suspected.
The potential complications of CA-MRSA infection, including pneumonia, sepsis, and endocarditis, can be avoided or minimized with early diagnosis and initiation of appropriate treatment.4,11,12
Consider community factors
In the past, folliculitis and other localized skin infections were traditionally caused by methicillin-susceptible Staphylococcus aureus and treated with beta-lactam antibiotics. In communities with high prevalence of CA-MRSA skin and soft tissue infections, beta-lactam antibiotics like cephalexin or dicloxacillin may no longer be appropriate.2,5 Therefore, let the prevalence of CA-MRSA in your community help guide your initial antibiotic choice while recognizing that empiric antibiotic therapy may be adjusted after cultures are available.
Good initial choices for CA-MRSA skin and soft tissue infections are trim-ethoprim/sulfamethoxazole with or without rifampin, clindamycin, gentamicin, and tetracycline,2,4,5,7 but inducible clindamycin resistance has been reported.13 if your community has a high prevalence of CA-MRSA, you may decide to begin empiric therapy that provides MRSA coverage.2 In addition, incision and drainage is necessary for adequate treatment of furuncles, carbuncles, and abscesses.5 Healthy, afebrile, immunocompetent patients without cellulitis may not require systemic antibiotics to clear local infections.2,4
Take steps to prevent spread of the disease
Early diagnosis of CA-MRSA infection is important to prevent the spread of this highly virulent disease in the community. It’s important to cover the infected area with sterile gauze and instruct the patient to avoid direct skin-to-skin contact with others, not share sports equipment or personal items, and decontaminate linens and surfaces.4,6
Stress the importance of frequent hand washing and using alcohol-based hand rubs to reduce transmission. Adjuvant topical therapy may be useful: intra-nasal mupirocin 2% ointment to prevent colonization and a persistent carrier state is prudent. Bathing with antimicrobial povidone-iodine or chlorhexidine gluconate is often recommended as an adjunctive treatment for skin and soft tissue infections, colonization, or both.14
Mupirocin 2% for our patient
We instructed our patient to continue the initial medications and to apply mupirocin 2% ointment to the nares twice a day for 1 week to eliminate the possibility of a staphylococcal carrier state.
At follow-up 2 weeks later, only a few excoriated papules and some post-inflammatory erythema remained. The topical clindamycin lotion and the doxycycline were continued for an additional 30 days, at which time the patient was clear and treatment was discontinued.
1. Elston DM. Epidemiology and prevention of skin and soft tissue infections. Cutis 2004;73:S3-7.
2. Moran GJ, Krishnadasan A, Gorwitz RJ, Fosheim GE, McDougal LK, et al. Methicillin-resistant S. aureus. infections among patients in the emergency department. N Engl J Med 2006;355:666-674.
3. Cohen PR, Kurzrock R. Community-acquired methicillin-resistant Staphylococcus aureus skin infection: An emerging clinical problem. J Am Acad Dermatol 2004;50:277-280.
4. Romero DV, Treston J, O’Sullivan AL. Hand-to-hand combat: Preventing MRSA infection. Adv Skin Wound Care 2006;19:328-333.
5. Moran GJ, Amii RN, Abrahamian FM, Talan DA. Methicillin-resistant Staphylococcus aureus in community-acquired skin infections. Emerg Infec Dis 2005;11:928-930.
6. Cohen PR, Grossman ME. Management of cutaneous lesions associated with an emerging epidemic: Community-acquired methicillin-resistant Staphylococcus aureus skin infections. J Am Acad Dermatol 2004;51:132-135.
7. Naimi TS, LeDell KH, Como-Sabetti K, Borchardt SM, Boxrud DJ, et al. Comparison of community-and health care-associated methicillin-resistant Staphylococcus aureus infection. JAMA 2003;290:2976-2984.
8. Vandenesch F, Naimi T, Enright MC, et al. Community-acquired methicillin-resistant Staphylococcus aureus carrying Panton-Valentine leukocidin genes: worldwide emergence. Emerg Infect Dis 2003;9:978-984.
9. Iyer S, Jones DH. Community-acquired methicillin-resistant Staphylococcus aureus skin infection: A retrospective analysis of clinical presentation and treatment of a local outbreak. J Am Acad Dermatol 2004;50:854-858.
10. Cohen PR. Community-acquired methicillin-resistant Staphylococcus aureus skin infection presenting as a periumbilical folliculitis. Cutis. 2006;77:229-231.
11. Bahrain M, Vasiliades M, Wolff M, Younus F. Five cases of bacterial endocarditis after furunculosis and the ongoing saga of community-acquired methicillin-resistant Staphylococcus aureus infections. Scand J Infect Dis 2006;38:702-707.
12. Herold B, Immergluck L, Maranan M, et al. Community-acquired methicillin-resistant Staphylococcus aureus in children with no identified predisposing risk. JAMA 1998;279:593-598.
13. Siberry GK, Tekle T, Carroll K, Dick J. Failure of clindamycin treatment of methicillin-resistant Staphylococcus aureus expressing inducible clindamycin resistance in vitro. Clin Infect Dis 2003;27:1257-1260.
14. Boyce JM. MRSA patient: proven methods to treat colonization and infection. J Hosp Infect 2001;48:S9-14.
1. Elston DM. Epidemiology and prevention of skin and soft tissue infections. Cutis 2004;73:S3-7.
2. Moran GJ, Krishnadasan A, Gorwitz RJ, Fosheim GE, McDougal LK, et al. Methicillin-resistant S. aureus. infections among patients in the emergency department. N Engl J Med 2006;355:666-674.
3. Cohen PR, Kurzrock R. Community-acquired methicillin-resistant Staphylococcus aureus skin infection: An emerging clinical problem. J Am Acad Dermatol 2004;50:277-280.
4. Romero DV, Treston J, O’Sullivan AL. Hand-to-hand combat: Preventing MRSA infection. Adv Skin Wound Care 2006;19:328-333.
5. Moran GJ, Amii RN, Abrahamian FM, Talan DA. Methicillin-resistant Staphylococcus aureus in community-acquired skin infections. Emerg Infec Dis 2005;11:928-930.
6. Cohen PR, Grossman ME. Management of cutaneous lesions associated with an emerging epidemic: Community-acquired methicillin-resistant Staphylococcus aureus skin infections. J Am Acad Dermatol 2004;51:132-135.
7. Naimi TS, LeDell KH, Como-Sabetti K, Borchardt SM, Boxrud DJ, et al. Comparison of community-and health care-associated methicillin-resistant Staphylococcus aureus infection. JAMA 2003;290:2976-2984.
8. Vandenesch F, Naimi T, Enright MC, et al. Community-acquired methicillin-resistant Staphylococcus aureus carrying Panton-Valentine leukocidin genes: worldwide emergence. Emerg Infect Dis 2003;9:978-984.
9. Iyer S, Jones DH. Community-acquired methicillin-resistant Staphylococcus aureus skin infection: A retrospective analysis of clinical presentation and treatment of a local outbreak. J Am Acad Dermatol 2004;50:854-858.
10. Cohen PR. Community-acquired methicillin-resistant Staphylococcus aureus skin infection presenting as a periumbilical folliculitis. Cutis. 2006;77:229-231.
11. Bahrain M, Vasiliades M, Wolff M, Younus F. Five cases of bacterial endocarditis after furunculosis and the ongoing saga of community-acquired methicillin-resistant Staphylococcus aureus infections. Scand J Infect Dis 2006;38:702-707.
12. Herold B, Immergluck L, Maranan M, et al. Community-acquired methicillin-resistant Staphylococcus aureus in children with no identified predisposing risk. JAMA 1998;279:593-598.
13. Siberry GK, Tekle T, Carroll K, Dick J. Failure of clindamycin treatment of methicillin-resistant Staphylococcus aureus expressing inducible clindamycin resistance in vitro. Clin Infect Dis 2003;27:1257-1260.
14. Boyce JM. MRSA patient: proven methods to treat colonization and infection. J Hosp Infect 2001;48:S9-14.
Pruritic blisters on legs and feet
A 23-year-old active duty US Navy sailor came to our medical center for treatment of 1–2 cm tense pruritic blisters on his dorsal feet, calves, and anterior lower extremities (FIGURE 1). He told us that several days earlier, he had noticed ill-defined “itchy red bumps” on both legs. He denied fever, night sweats, malaise, trauma, insect bites, contact with animals or plants, or recent illnesses. He did say, however, that he’d recently done outdoor fieldwork with the Marine Corps in southern California.
His medical history was unremarkable, and he was not taking any prescribed or over-the-counter medications or supplements. He had no family history of blistering or other autoimmune disorders.
On examination, we noticed clear fluid-filled vesicles and bullae on non-erythematous, non-urticarial bases that were haphazardly distributed on both legs and dorsal feet. Agminated or herpetiform configurations were not present. Ruptured bullae left erythematous to beefy red eroded bases, and there were numerous smaller red papules with vesicular surface changes. All of the lesions were below the knees; there was complete sparing of the trunk, upper extremities, intertriginous skin, head, and neck.
The remainder of the physical examination was unremarkable, and there was no lymphadenopathy. Complete blood count and chemistries were within normal limits, and the patient’s HIV status was negative.
We performed lesional and perilesional punch biopsies. The lesional biopsy demonstrated subepidermal blistering with a predominantly eosinophilic infiltrate in all layers of the dermis and within the blister. Direct immunofluorescence (DIF) was performed on the perilesional biopsy and was negative for IgA, IgG, IgM, C3, and fibrinogen. Gram stain, potassium hydroxide (KOH) prep, and wound culture were also negative.
FIGURE 1
Tense pruritic blisters
What is your diagnosis?
Diagnosis: Bullous arthropod bite reaction
Bullous arthropod bite reaction (BABR) occurs in sensitized individuals as a delayed hypersensitivity immune reaction to insect saliva.1 Patients typically present with grouped localized pruritic or asymptomatic blistering2 and otherwise appear well. Unless secondarily infected, the blisters are non-erythematous and non-purulent, and develop within hours to days of the bite.
Lesion location is key. The distribution and location of the lesions will tip you off to a BABR diagnosis. The lesions in BABR are usually grouped and localized to a specific area of the body, depending on the causative arthropod and the circumstances leading to the bites. For example:
- Lesions caused by Cheyletiella mites are typically found on the forearms, anterior thighs, and lower abdomen after an infested pet sits on an individual’s lap.2,3
- Blisters caused by flea bites are isolated to the lower extremities.4 (We suspect that flea bites were the culprit in our patient’s case.)
- Lesions caused by Cimex lectularius, more commonly known as bedbugs, may be found on the entire body and tend to occur in groups of 3.5
Insect bite? What insect bite?
Most patients will only complain of pruritus and will tell you that they don’t recall having had any insect bites.6 That said, the distribution of the lesions, lack of systemic illness, and otherwise unremarkable physical exam are sufficient for diagnosis.
Occasionally, a punch biopsy with DIF may be necessary to rule out more serious bullous disorders. In BABR, you may see both subepidermal and intraepidermal blistering, with perivascular and interstitial eosinophilic and lymphocytic infiltrates. Blisters separated by strands of keratinocytes create a characteristic multilocular appearance. Unlike autoimmune blistering disorders, DIF is negative in BABR.7 Gram stain, Tzanck smear, bacterial culture, and KOH prep may also provide additional information if infection is a concern.
For certain patients, the DX may be less clear-cut
Similar bullous lesions following insect bites have been reported in patients with HIV,8 chronic lymphocytic leukemia,9-12 EBV-associated Natural Killer leukemia/ lymphoma,13 and mantle cell lymphoma.14 There is ongoing debate as to whether the vesicular lesions in these patients truly represent an exaggerated response to an arthropod bite or mimic an insect-like bite reaction.10,12
Nevertheless, when you suspect a patient has BABR, be aware of its association with both hematologic malignancies and HIV. Appropriate evaluation, such as HIV screening and complete blood count, should be performed.
A condition that mimics contact dermatitis
Clinically and histologically, BABR can mimic the following:
- Contact dermatitis. With contact dermatitis, the blistering is more likely to appear in streaks or in a linear fashion.15 Lesions will be painful, as well as pruritic, and occur following direct contact with a plant or chemical allergen.
- Drug-induced pemphigoid. The patient’s history will increase your suspicion of drug-induced pemphigoid. Patients may be taking sulfur-containing drugs (furosemide), antibiotics (penicillins, fluoroquinolones), antihypertensives (ACE inhibitors, calcium channel blockers), neuroleptics, or sulfasalazine.7,16,17 The eruption is usually more generalized than BABR, and may involve the mucous membranes.
- Fungal infections. These infections will typically occur on the palms and soles. Infiltrate is typically neutrophilic, but can be eosinophilic.18
- Bullous scabies. Patients will have severe pruritus. Burrows and lesions can typically be found on moist areas (periumbilical, intertriginous skin).19,20
- Bullous pemphigoid. This is more commonly seen in elderly patients with comorbid conditions. Onset of blistering is gradual, and occurs predominantly on flexural skin.
Pemphigoid gestationis, erythema toxicum neonatorum, incontinentia pigmenti, and some pemphigus variants also have predominantly eosinophilic infiltration in the skin. These, however, are clinically distinct from BABR.
Focus on symptoms and prevention
BABR will resolve over time without aggressive intervention. Most patients are treated symptomatically with oral anti-histamines and topical steroids for pruritus.1 Prevention of further bites is important because of the risk of arthropod-transmitted diseases.21
Our patient couldn’t comfortably wear shoes
Our patient had extensive tense blistering on both legs that prevented him from comfortably wearing shoes (FIGURE 2). Using a #11 blade, we punctured all of the blisters at the most dependent portion of each lesion. We decompressed the lesions, but did not de-roof them so that the blistered skin could serve as a biological dressing. We applied topical mupirocin and wrapped both legs with a compressive dressing.
We gave the patient a 2-week tapering course of oral prednisone. At the 3-week follow-up, all of the blistered skin had completely healed with the exception of post-inflammatory hyperpigmentation. No new lesions developed. Our patient was well, with no recurrence of blistering, at his 6-month follow-up.
FIGURE 2
Wearing shoes was a problem
Correspondence
Kendall Lane, MD, Expeditionary Health Services Pacific, 3985 Cummings Rd, Suite 4, San Diego, CA 92136; [email protected]
1. North American arthropod envenomation and Parasitism. In: Auerbach PS, ed. Wilderness Medicine. 5th ed. Philadelphia, PA: Mosby; 2007;947–982.
2. Bjarke T, Hellgren L, Orstadius K. Cheyletiella parasitivorax dermatitis in man. Acta Derm Venereol 1973;53:217-224.
3. Shelley ED, Shelley WB. The diagnostic challenge of non-burrowing mite bites Cheyletiella yasguri. JAMA 1984;251:2690-2691.
4. Infestations and bites. In: Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Treatment. 4th ed. St Louis, Mo: Mosby; 2004;533–534.
5. Elston DM, Stockwell S. What’s eating you? Bed-bugs. Cutis 2000;65:262-264.
6. Steen C, Carbonaro P, Schwartz R. Arthropods in dermatology. J Am Acad Dermatol 2004;50:819-842.
7. The vesiculobullous reaction pattern. In: Weedon D. Skin Pathology. 2nd ed. Brisbane, Australia: Churchill Livingstone; 2002;129–191.
8. Ajithkumar K, Soshamma G, George B. Abnormal insect bite reactions: A manifestation of immunosuppression of HIV infection? Indian J Dermatol Venereol Leprol 2001;67:72-74.
9. Davis MD, Perniciaro C, Dahl PR, Randle HW, McEvoy MT, Leiferman KM. Exaggerated arthropod-bite lesions in patients with chronic lymphocytic leukemia: a clinical, histopathologic, and immunopathologic study of eight patients. J Am Acad Dermatol 1998;39:27-35.
10. Pendersen J, Carganello J, Van Der Weyden MB. Exaggerated reaction to insect bites in patients with chronic lymphocytic leukemia: clinical and histological findings. Pathology 1990;22:141-143.
11. Rosen LB, Frank BL, Rywlin AM. A characteristic vesticulobullous reaction in patients with chronic lymphocytic leukemia. J Am Acad Dermatol 1986;15:943-950.
12. Barzilai A, Shpiro D, Goldberg I, et al. Insect bite-like reaction in patients with hematologic malignant neoplasms. Arch Dermatol 1999;135:1503-1507.
13. Tokur Y, Ishihara S, Tagawa S, et al. Hypersensitivity to mosquito bites as the primary clinical manifestation of juvenile type of Epstein-Barr virus-associated natural killer cell leukemia/lymphoma. J Am Acad Dermatol 2001;45:569-578.
14. Khamaysi Z, Dodiuk-Gad RP, Weltfriend S, et al. Insect bite-like reaction associated with mantle cell lymphoma: clinicopathological, immunopathological, and molecular studies. Am J Dermatopathol 2005;27:290-295.
15. Allergic contact dermatitis. In: Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Treatment. 4th ed. St Louis, Mo: Mosby; 2004;84–97.
16. Kimyai-Asadi A, Usman A, Nousari HC. Ciprofloxacin-induced bullous pemphigoid. J Am Acad Dermatol 2000;42:847.-
17. Kashihara M, Danno K, Miyachi Y, Horiguchi Y, Imamura S. Bullous pemphigoid-like lesions induced by phenacetin. Report of a case and an immunopathologic study. Arch Dermatol 1984;120:1196-1199.
18. Mycoses and algal infections. In: Weedon D. Skin Pathology. 2nd ed. Brisbane, Australia: Churchill Livingstone; 2002: 663.
19. Nakamura E, Taniguchi H, Ohtaki N. A case of crusted scabies with a bullous pemphigoid-like eruption and nail involvement. J Dermatol 2006;33:196-201.
20. Ansarin H, Jalali MH, Mazloomi S, Soltani-Arabshahi R, Setarehshenas R. Scabies presenting with bullous pemphigoid-like lesions. Dermatol Online J 2006;12(1):19.-
21. Elston D. Prevention of arthropod-related disease. J Am Acad Dermatol 2004;947-954.
A 23-year-old active duty US Navy sailor came to our medical center for treatment of 1–2 cm tense pruritic blisters on his dorsal feet, calves, and anterior lower extremities (FIGURE 1). He told us that several days earlier, he had noticed ill-defined “itchy red bumps” on both legs. He denied fever, night sweats, malaise, trauma, insect bites, contact with animals or plants, or recent illnesses. He did say, however, that he’d recently done outdoor fieldwork with the Marine Corps in southern California.
His medical history was unremarkable, and he was not taking any prescribed or over-the-counter medications or supplements. He had no family history of blistering or other autoimmune disorders.
On examination, we noticed clear fluid-filled vesicles and bullae on non-erythematous, non-urticarial bases that were haphazardly distributed on both legs and dorsal feet. Agminated or herpetiform configurations were not present. Ruptured bullae left erythematous to beefy red eroded bases, and there were numerous smaller red papules with vesicular surface changes. All of the lesions were below the knees; there was complete sparing of the trunk, upper extremities, intertriginous skin, head, and neck.
The remainder of the physical examination was unremarkable, and there was no lymphadenopathy. Complete blood count and chemistries were within normal limits, and the patient’s HIV status was negative.
We performed lesional and perilesional punch biopsies. The lesional biopsy demonstrated subepidermal blistering with a predominantly eosinophilic infiltrate in all layers of the dermis and within the blister. Direct immunofluorescence (DIF) was performed on the perilesional biopsy and was negative for IgA, IgG, IgM, C3, and fibrinogen. Gram stain, potassium hydroxide (KOH) prep, and wound culture were also negative.
FIGURE 1
Tense pruritic blisters
What is your diagnosis?
Diagnosis: Bullous arthropod bite reaction
Bullous arthropod bite reaction (BABR) occurs in sensitized individuals as a delayed hypersensitivity immune reaction to insect saliva.1 Patients typically present with grouped localized pruritic or asymptomatic blistering2 and otherwise appear well. Unless secondarily infected, the blisters are non-erythematous and non-purulent, and develop within hours to days of the bite.
Lesion location is key. The distribution and location of the lesions will tip you off to a BABR diagnosis. The lesions in BABR are usually grouped and localized to a specific area of the body, depending on the causative arthropod and the circumstances leading to the bites. For example:
- Lesions caused by Cheyletiella mites are typically found on the forearms, anterior thighs, and lower abdomen after an infested pet sits on an individual’s lap.2,3
- Blisters caused by flea bites are isolated to the lower extremities.4 (We suspect that flea bites were the culprit in our patient’s case.)
- Lesions caused by Cimex lectularius, more commonly known as bedbugs, may be found on the entire body and tend to occur in groups of 3.5
Insect bite? What insect bite?
Most patients will only complain of pruritus and will tell you that they don’t recall having had any insect bites.6 That said, the distribution of the lesions, lack of systemic illness, and otherwise unremarkable physical exam are sufficient for diagnosis.
Occasionally, a punch biopsy with DIF may be necessary to rule out more serious bullous disorders. In BABR, you may see both subepidermal and intraepidermal blistering, with perivascular and interstitial eosinophilic and lymphocytic infiltrates. Blisters separated by strands of keratinocytes create a characteristic multilocular appearance. Unlike autoimmune blistering disorders, DIF is negative in BABR.7 Gram stain, Tzanck smear, bacterial culture, and KOH prep may also provide additional information if infection is a concern.
For certain patients, the DX may be less clear-cut
Similar bullous lesions following insect bites have been reported in patients with HIV,8 chronic lymphocytic leukemia,9-12 EBV-associated Natural Killer leukemia/ lymphoma,13 and mantle cell lymphoma.14 There is ongoing debate as to whether the vesicular lesions in these patients truly represent an exaggerated response to an arthropod bite or mimic an insect-like bite reaction.10,12
Nevertheless, when you suspect a patient has BABR, be aware of its association with both hematologic malignancies and HIV. Appropriate evaluation, such as HIV screening and complete blood count, should be performed.
A condition that mimics contact dermatitis
Clinically and histologically, BABR can mimic the following:
- Contact dermatitis. With contact dermatitis, the blistering is more likely to appear in streaks or in a linear fashion.15 Lesions will be painful, as well as pruritic, and occur following direct contact with a plant or chemical allergen.
- Drug-induced pemphigoid. The patient’s history will increase your suspicion of drug-induced pemphigoid. Patients may be taking sulfur-containing drugs (furosemide), antibiotics (penicillins, fluoroquinolones), antihypertensives (ACE inhibitors, calcium channel blockers), neuroleptics, or sulfasalazine.7,16,17 The eruption is usually more generalized than BABR, and may involve the mucous membranes.
- Fungal infections. These infections will typically occur on the palms and soles. Infiltrate is typically neutrophilic, but can be eosinophilic.18
- Bullous scabies. Patients will have severe pruritus. Burrows and lesions can typically be found on moist areas (periumbilical, intertriginous skin).19,20
- Bullous pemphigoid. This is more commonly seen in elderly patients with comorbid conditions. Onset of blistering is gradual, and occurs predominantly on flexural skin.
Pemphigoid gestationis, erythema toxicum neonatorum, incontinentia pigmenti, and some pemphigus variants also have predominantly eosinophilic infiltration in the skin. These, however, are clinically distinct from BABR.
Focus on symptoms and prevention
BABR will resolve over time without aggressive intervention. Most patients are treated symptomatically with oral anti-histamines and topical steroids for pruritus.1 Prevention of further bites is important because of the risk of arthropod-transmitted diseases.21
Our patient couldn’t comfortably wear shoes
Our patient had extensive tense blistering on both legs that prevented him from comfortably wearing shoes (FIGURE 2). Using a #11 blade, we punctured all of the blisters at the most dependent portion of each lesion. We decompressed the lesions, but did not de-roof them so that the blistered skin could serve as a biological dressing. We applied topical mupirocin and wrapped both legs with a compressive dressing.
We gave the patient a 2-week tapering course of oral prednisone. At the 3-week follow-up, all of the blistered skin had completely healed with the exception of post-inflammatory hyperpigmentation. No new lesions developed. Our patient was well, with no recurrence of blistering, at his 6-month follow-up.
FIGURE 2
Wearing shoes was a problem
Correspondence
Kendall Lane, MD, Expeditionary Health Services Pacific, 3985 Cummings Rd, Suite 4, San Diego, CA 92136; [email protected]
A 23-year-old active duty US Navy sailor came to our medical center for treatment of 1–2 cm tense pruritic blisters on his dorsal feet, calves, and anterior lower extremities (FIGURE 1). He told us that several days earlier, he had noticed ill-defined “itchy red bumps” on both legs. He denied fever, night sweats, malaise, trauma, insect bites, contact with animals or plants, or recent illnesses. He did say, however, that he’d recently done outdoor fieldwork with the Marine Corps in southern California.
His medical history was unremarkable, and he was not taking any prescribed or over-the-counter medications or supplements. He had no family history of blistering or other autoimmune disorders.
On examination, we noticed clear fluid-filled vesicles and bullae on non-erythematous, non-urticarial bases that were haphazardly distributed on both legs and dorsal feet. Agminated or herpetiform configurations were not present. Ruptured bullae left erythematous to beefy red eroded bases, and there were numerous smaller red papules with vesicular surface changes. All of the lesions were below the knees; there was complete sparing of the trunk, upper extremities, intertriginous skin, head, and neck.
The remainder of the physical examination was unremarkable, and there was no lymphadenopathy. Complete blood count and chemistries were within normal limits, and the patient’s HIV status was negative.
We performed lesional and perilesional punch biopsies. The lesional biopsy demonstrated subepidermal blistering with a predominantly eosinophilic infiltrate in all layers of the dermis and within the blister. Direct immunofluorescence (DIF) was performed on the perilesional biopsy and was negative for IgA, IgG, IgM, C3, and fibrinogen. Gram stain, potassium hydroxide (KOH) prep, and wound culture were also negative.
FIGURE 1
Tense pruritic blisters
What is your diagnosis?
Diagnosis: Bullous arthropod bite reaction
Bullous arthropod bite reaction (BABR) occurs in sensitized individuals as a delayed hypersensitivity immune reaction to insect saliva.1 Patients typically present with grouped localized pruritic or asymptomatic blistering2 and otherwise appear well. Unless secondarily infected, the blisters are non-erythematous and non-purulent, and develop within hours to days of the bite.
Lesion location is key. The distribution and location of the lesions will tip you off to a BABR diagnosis. The lesions in BABR are usually grouped and localized to a specific area of the body, depending on the causative arthropod and the circumstances leading to the bites. For example:
- Lesions caused by Cheyletiella mites are typically found on the forearms, anterior thighs, and lower abdomen after an infested pet sits on an individual’s lap.2,3
- Blisters caused by flea bites are isolated to the lower extremities.4 (We suspect that flea bites were the culprit in our patient’s case.)
- Lesions caused by Cimex lectularius, more commonly known as bedbugs, may be found on the entire body and tend to occur in groups of 3.5
Insect bite? What insect bite?
Most patients will only complain of pruritus and will tell you that they don’t recall having had any insect bites.6 That said, the distribution of the lesions, lack of systemic illness, and otherwise unremarkable physical exam are sufficient for diagnosis.
Occasionally, a punch biopsy with DIF may be necessary to rule out more serious bullous disorders. In BABR, you may see both subepidermal and intraepidermal blistering, with perivascular and interstitial eosinophilic and lymphocytic infiltrates. Blisters separated by strands of keratinocytes create a characteristic multilocular appearance. Unlike autoimmune blistering disorders, DIF is negative in BABR.7 Gram stain, Tzanck smear, bacterial culture, and KOH prep may also provide additional information if infection is a concern.
For certain patients, the DX may be less clear-cut
Similar bullous lesions following insect bites have been reported in patients with HIV,8 chronic lymphocytic leukemia,9-12 EBV-associated Natural Killer leukemia/ lymphoma,13 and mantle cell lymphoma.14 There is ongoing debate as to whether the vesicular lesions in these patients truly represent an exaggerated response to an arthropod bite or mimic an insect-like bite reaction.10,12
Nevertheless, when you suspect a patient has BABR, be aware of its association with both hematologic malignancies and HIV. Appropriate evaluation, such as HIV screening and complete blood count, should be performed.
A condition that mimics contact dermatitis
Clinically and histologically, BABR can mimic the following:
- Contact dermatitis. With contact dermatitis, the blistering is more likely to appear in streaks or in a linear fashion.15 Lesions will be painful, as well as pruritic, and occur following direct contact with a plant or chemical allergen.
- Drug-induced pemphigoid. The patient’s history will increase your suspicion of drug-induced pemphigoid. Patients may be taking sulfur-containing drugs (furosemide), antibiotics (penicillins, fluoroquinolones), antihypertensives (ACE inhibitors, calcium channel blockers), neuroleptics, or sulfasalazine.7,16,17 The eruption is usually more generalized than BABR, and may involve the mucous membranes.
- Fungal infections. These infections will typically occur on the palms and soles. Infiltrate is typically neutrophilic, but can be eosinophilic.18
- Bullous scabies. Patients will have severe pruritus. Burrows and lesions can typically be found on moist areas (periumbilical, intertriginous skin).19,20
- Bullous pemphigoid. This is more commonly seen in elderly patients with comorbid conditions. Onset of blistering is gradual, and occurs predominantly on flexural skin.
Pemphigoid gestationis, erythema toxicum neonatorum, incontinentia pigmenti, and some pemphigus variants also have predominantly eosinophilic infiltration in the skin. These, however, are clinically distinct from BABR.
Focus on symptoms and prevention
BABR will resolve over time without aggressive intervention. Most patients are treated symptomatically with oral anti-histamines and topical steroids for pruritus.1 Prevention of further bites is important because of the risk of arthropod-transmitted diseases.21
Our patient couldn’t comfortably wear shoes
Our patient had extensive tense blistering on both legs that prevented him from comfortably wearing shoes (FIGURE 2). Using a #11 blade, we punctured all of the blisters at the most dependent portion of each lesion. We decompressed the lesions, but did not de-roof them so that the blistered skin could serve as a biological dressing. We applied topical mupirocin and wrapped both legs with a compressive dressing.
We gave the patient a 2-week tapering course of oral prednisone. At the 3-week follow-up, all of the blistered skin had completely healed with the exception of post-inflammatory hyperpigmentation. No new lesions developed. Our patient was well, with no recurrence of blistering, at his 6-month follow-up.
FIGURE 2
Wearing shoes was a problem
Correspondence
Kendall Lane, MD, Expeditionary Health Services Pacific, 3985 Cummings Rd, Suite 4, San Diego, CA 92136; [email protected]
1. North American arthropod envenomation and Parasitism. In: Auerbach PS, ed. Wilderness Medicine. 5th ed. Philadelphia, PA: Mosby; 2007;947–982.
2. Bjarke T, Hellgren L, Orstadius K. Cheyletiella parasitivorax dermatitis in man. Acta Derm Venereol 1973;53:217-224.
3. Shelley ED, Shelley WB. The diagnostic challenge of non-burrowing mite bites Cheyletiella yasguri. JAMA 1984;251:2690-2691.
4. Infestations and bites. In: Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Treatment. 4th ed. St Louis, Mo: Mosby; 2004;533–534.
5. Elston DM, Stockwell S. What’s eating you? Bed-bugs. Cutis 2000;65:262-264.
6. Steen C, Carbonaro P, Schwartz R. Arthropods in dermatology. J Am Acad Dermatol 2004;50:819-842.
7. The vesiculobullous reaction pattern. In: Weedon D. Skin Pathology. 2nd ed. Brisbane, Australia: Churchill Livingstone; 2002;129–191.
8. Ajithkumar K, Soshamma G, George B. Abnormal insect bite reactions: A manifestation of immunosuppression of HIV infection? Indian J Dermatol Venereol Leprol 2001;67:72-74.
9. Davis MD, Perniciaro C, Dahl PR, Randle HW, McEvoy MT, Leiferman KM. Exaggerated arthropod-bite lesions in patients with chronic lymphocytic leukemia: a clinical, histopathologic, and immunopathologic study of eight patients. J Am Acad Dermatol 1998;39:27-35.
10. Pendersen J, Carganello J, Van Der Weyden MB. Exaggerated reaction to insect bites in patients with chronic lymphocytic leukemia: clinical and histological findings. Pathology 1990;22:141-143.
11. Rosen LB, Frank BL, Rywlin AM. A characteristic vesticulobullous reaction in patients with chronic lymphocytic leukemia. J Am Acad Dermatol 1986;15:943-950.
12. Barzilai A, Shpiro D, Goldberg I, et al. Insect bite-like reaction in patients with hematologic malignant neoplasms. Arch Dermatol 1999;135:1503-1507.
13. Tokur Y, Ishihara S, Tagawa S, et al. Hypersensitivity to mosquito bites as the primary clinical manifestation of juvenile type of Epstein-Barr virus-associated natural killer cell leukemia/lymphoma. J Am Acad Dermatol 2001;45:569-578.
14. Khamaysi Z, Dodiuk-Gad RP, Weltfriend S, et al. Insect bite-like reaction associated with mantle cell lymphoma: clinicopathological, immunopathological, and molecular studies. Am J Dermatopathol 2005;27:290-295.
15. Allergic contact dermatitis. In: Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Treatment. 4th ed. St Louis, Mo: Mosby; 2004;84–97.
16. Kimyai-Asadi A, Usman A, Nousari HC. Ciprofloxacin-induced bullous pemphigoid. J Am Acad Dermatol 2000;42:847.-
17. Kashihara M, Danno K, Miyachi Y, Horiguchi Y, Imamura S. Bullous pemphigoid-like lesions induced by phenacetin. Report of a case and an immunopathologic study. Arch Dermatol 1984;120:1196-1199.
18. Mycoses and algal infections. In: Weedon D. Skin Pathology. 2nd ed. Brisbane, Australia: Churchill Livingstone; 2002: 663.
19. Nakamura E, Taniguchi H, Ohtaki N. A case of crusted scabies with a bullous pemphigoid-like eruption and nail involvement. J Dermatol 2006;33:196-201.
20. Ansarin H, Jalali MH, Mazloomi S, Soltani-Arabshahi R, Setarehshenas R. Scabies presenting with bullous pemphigoid-like lesions. Dermatol Online J 2006;12(1):19.-
21. Elston D. Prevention of arthropod-related disease. J Am Acad Dermatol 2004;947-954.
1. North American arthropod envenomation and Parasitism. In: Auerbach PS, ed. Wilderness Medicine. 5th ed. Philadelphia, PA: Mosby; 2007;947–982.
2. Bjarke T, Hellgren L, Orstadius K. Cheyletiella parasitivorax dermatitis in man. Acta Derm Venereol 1973;53:217-224.
3. Shelley ED, Shelley WB. The diagnostic challenge of non-burrowing mite bites Cheyletiella yasguri. JAMA 1984;251:2690-2691.
4. Infestations and bites. In: Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Treatment. 4th ed. St Louis, Mo: Mosby; 2004;533–534.
5. Elston DM, Stockwell S. What’s eating you? Bed-bugs. Cutis 2000;65:262-264.
6. Steen C, Carbonaro P, Schwartz R. Arthropods in dermatology. J Am Acad Dermatol 2004;50:819-842.
7. The vesiculobullous reaction pattern. In: Weedon D. Skin Pathology. 2nd ed. Brisbane, Australia: Churchill Livingstone; 2002;129–191.
8. Ajithkumar K, Soshamma G, George B. Abnormal insect bite reactions: A manifestation of immunosuppression of HIV infection? Indian J Dermatol Venereol Leprol 2001;67:72-74.
9. Davis MD, Perniciaro C, Dahl PR, Randle HW, McEvoy MT, Leiferman KM. Exaggerated arthropod-bite lesions in patients with chronic lymphocytic leukemia: a clinical, histopathologic, and immunopathologic study of eight patients. J Am Acad Dermatol 1998;39:27-35.
10. Pendersen J, Carganello J, Van Der Weyden MB. Exaggerated reaction to insect bites in patients with chronic lymphocytic leukemia: clinical and histological findings. Pathology 1990;22:141-143.
11. Rosen LB, Frank BL, Rywlin AM. A characteristic vesticulobullous reaction in patients with chronic lymphocytic leukemia. J Am Acad Dermatol 1986;15:943-950.
12. Barzilai A, Shpiro D, Goldberg I, et al. Insect bite-like reaction in patients with hematologic malignant neoplasms. Arch Dermatol 1999;135:1503-1507.
13. Tokur Y, Ishihara S, Tagawa S, et al. Hypersensitivity to mosquito bites as the primary clinical manifestation of juvenile type of Epstein-Barr virus-associated natural killer cell leukemia/lymphoma. J Am Acad Dermatol 2001;45:569-578.
14. Khamaysi Z, Dodiuk-Gad RP, Weltfriend S, et al. Insect bite-like reaction associated with mantle cell lymphoma: clinicopathological, immunopathological, and molecular studies. Am J Dermatopathol 2005;27:290-295.
15. Allergic contact dermatitis. In: Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Treatment. 4th ed. St Louis, Mo: Mosby; 2004;84–97.
16. Kimyai-Asadi A, Usman A, Nousari HC. Ciprofloxacin-induced bullous pemphigoid. J Am Acad Dermatol 2000;42:847.-
17. Kashihara M, Danno K, Miyachi Y, Horiguchi Y, Imamura S. Bullous pemphigoid-like lesions induced by phenacetin. Report of a case and an immunopathologic study. Arch Dermatol 1984;120:1196-1199.
18. Mycoses and algal infections. In: Weedon D. Skin Pathology. 2nd ed. Brisbane, Australia: Churchill Livingstone; 2002: 663.
19. Nakamura E, Taniguchi H, Ohtaki N. A case of crusted scabies with a bullous pemphigoid-like eruption and nail involvement. J Dermatol 2006;33:196-201.
20. Ansarin H, Jalali MH, Mazloomi S, Soltani-Arabshahi R, Setarehshenas R. Scabies presenting with bullous pemphigoid-like lesions. Dermatol Online J 2006;12(1):19.-
21. Elston D. Prevention of arthropod-related disease. J Am Acad Dermatol 2004;947-954.