User login
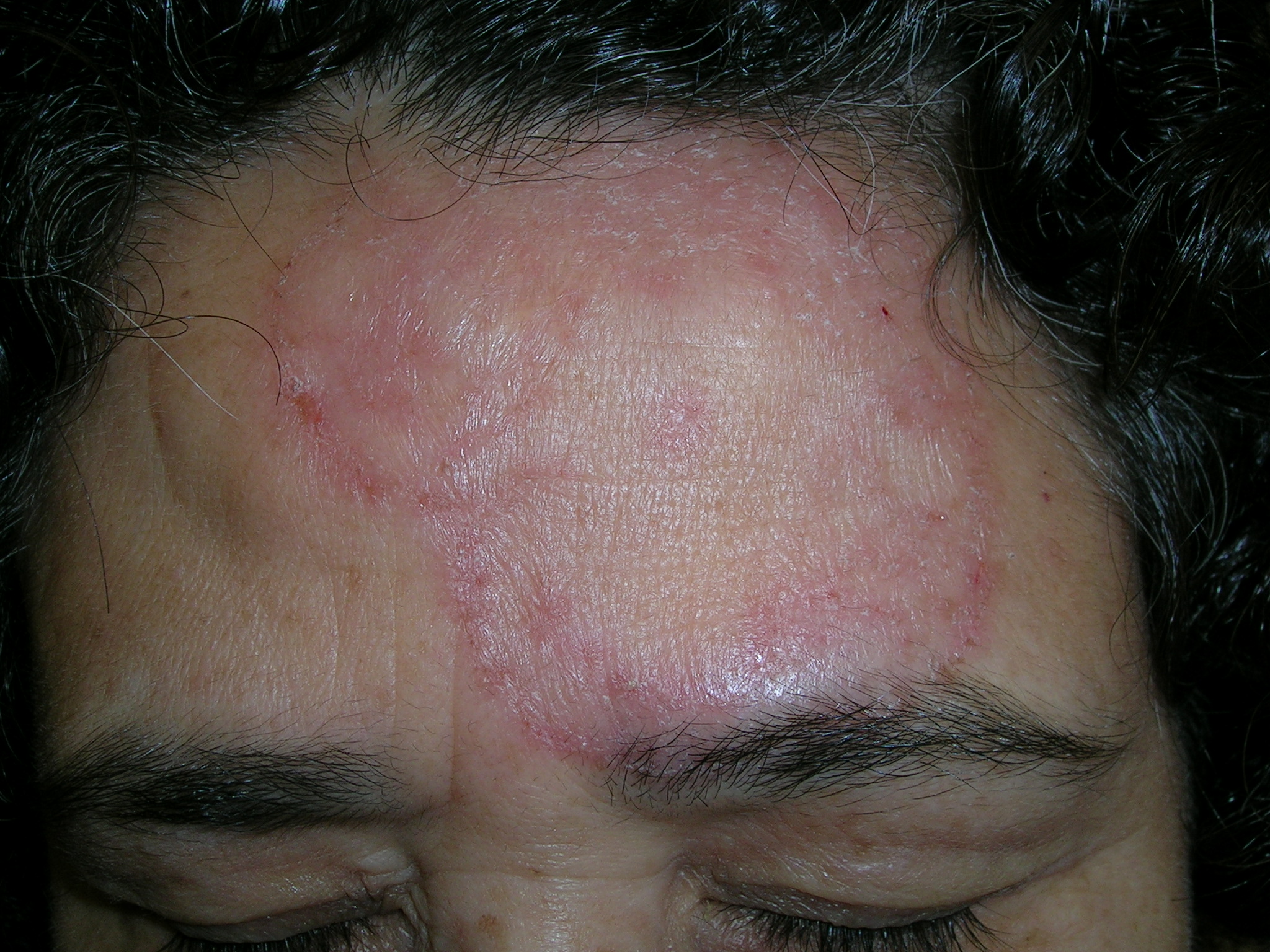
Red pruritic area on forehead
| 
|
We diagnosed a fungal infection. The annular pattern on the face (FIGURE 1) was suggestive of a dermatophyte infection. The differential diagnosis included granuloma annulare and psoriasis.
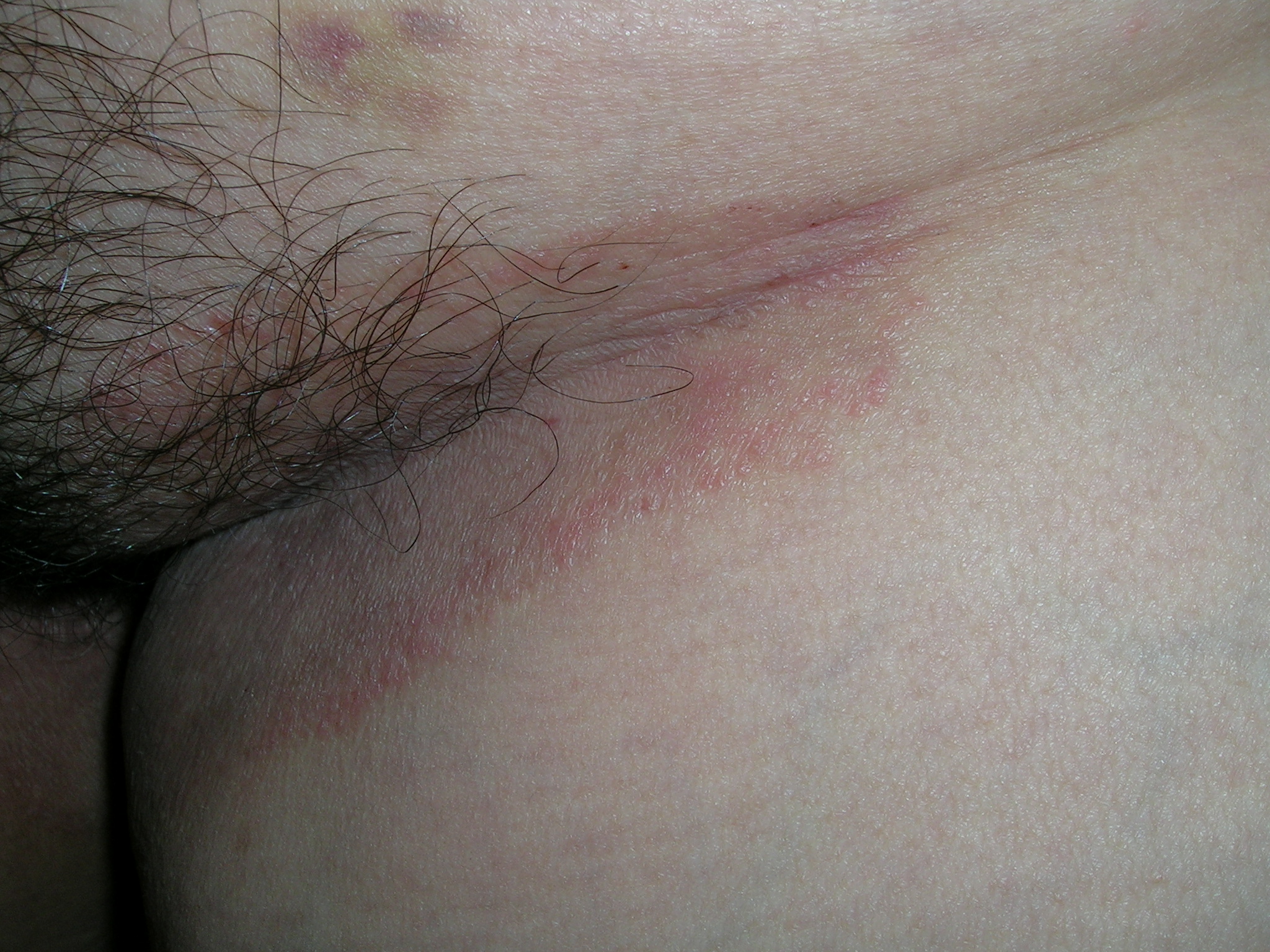
We looked at the patient’s feet, groin, and under the breasts for additional clues. This patient had what appeared to be fungal infections in all of these areas. The patient had severe tinea pedis in a moccasin distribution and tinea cruris (FIGURE 2). We performed a scraping for fungus from the groin infection and added a fungal stain with potassium hydroxide to the slide. Branching hyphae were seen under the microscope. The patient was treated with an oral antifungal agent (terbinafine, 250 mg daily) for 1 month. Her fungal infection cleared from all sites.
This case was adapted from: Usatine R. Cutaneous fungal infections: overview. In: Usatine R, Smith M, Mayeaux EJ, Chumley H, Tysinger J, eds. The Color Atlas of Family Medicine. New York, NY: McGraw-Hill; 2009:539-544.
To learn more about The Color Atlas of Family Medicine, see:
* http://www.amazon.com/Color-Atlas-Family-Medicine/dp/0071474641
|
|
|
We diagnosed a fungal infection. The annular pattern on the face (FIGURE 1) was suggestive of a dermatophyte infection. The differential diagnosis included granuloma annulare and psoriasis.
We looked at the patient’s feet, groin, and under the breasts for additional clues. This patient had what appeared to be fungal infections in all of these areas. The patient had severe tinea pedis in a moccasin distribution and tinea cruris (FIGURE 2). We performed a scraping for fungus from the groin infection and added a fungal stain with potassium hydroxide to the slide. Branching hyphae were seen under the microscope. The patient was treated with an oral antifungal agent (terbinafine, 250 mg daily) for 1 month. Her fungal infection cleared from all sites.
This case was adapted from: Usatine R. Cutaneous fungal infections: overview. In: Usatine R, Smith M, Mayeaux EJ, Chumley H, Tysinger J, eds. The Color Atlas of Family Medicine. New York, NY: McGraw-Hill; 2009:539-544.
To learn more about The Color Atlas of Family Medicine, see:
* http://www.amazon.com/Color-Atlas-Family-Medicine/dp/0071474641
|
|
|
We diagnosed a fungal infection. The annular pattern on the face (FIGURE 1) was suggestive of a dermatophyte infection. The differential diagnosis included granuloma annulare and psoriasis.
We looked at the patient’s feet, groin, and under the breasts for additional clues. This patient had what appeared to be fungal infections in all of these areas. The patient had severe tinea pedis in a moccasin distribution and tinea cruris (FIGURE 2). We performed a scraping for fungus from the groin infection and added a fungal stain with potassium hydroxide to the slide. Branching hyphae were seen under the microscope. The patient was treated with an oral antifungal agent (terbinafine, 250 mg daily) for 1 month. Her fungal infection cleared from all sites.
This case was adapted from: Usatine R. Cutaneous fungal infections: overview. In: Usatine R, Smith M, Mayeaux EJ, Chumley H, Tysinger J, eds. The Color Atlas of Family Medicine. New York, NY: McGraw-Hill; 2009:539-544.
To learn more about The Color Atlas of Family Medicine, see:
* http://www.amazon.com/Color-Atlas-Family-Medicine/dp/0071474641
Worsening low back pain
A 44-year-old African American man went to his primary care provider complaining of 5/10 dull lower back pain, which he attributed to a recent fall. He reported no radiation of the pain, lower extremity weakness or numbness, or any bowel/ bladder incontinence. Further review of systems and past medical history was unremarkable. He was initially treated with nonsteroidal anti-inflammatory agents and physical therapy. Approximately 2 months later he returned with similar persistent lower back pain complaints; an opiate was added to his pain control regimen. The following week he went to the emergency department (ED), where he reported worsening pain.
In the ED, he was afebrile with normal vital signs. He had a benign musculoskeletal and abdominal exam, and his neurological exam was without any focal deficits. Blood tests revealed a hemoglobin level of 9.8 g/dL with a hematocrit of 27.9%, serum blood urea nitrogen/creatinine ratio of 143/11.0 mg/dL, potassium of 6.5 mmol/L, calcium of 11.8 mg/dL, and phosphorus of 8.5 mg/dL. Urine protein was elevated at 88 mg/dL.
A stat CT of the patient’s abdomen and pelvis revealed no signs of obstructive uropathy to explain his acute renal failure. A sagittal CT reconstruction in bone windows of the initial ED radiological workup was revealing (FIGURE 1).
FIGURE 1
Sagittal CT reconstruction of thoracolumbar spine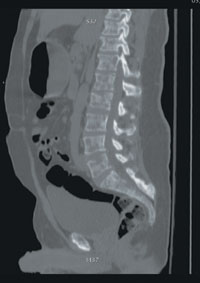
What is your diagnosis?
How would you manage this condition?
Dx: Multiple myeloma
The sagittal CT reconstruction in bone windows of the thoracolumbar spine (FIGURE 1) revealed multiple lucent foci throughout the osseous structures, with an anterior compression deformity of the L2 vertebral body. A subsequent skeletal survey showed a diffuse salt and pepper pattern affecting most of the osseous structures, with additional lytic lesions in the calvarium and extremities.
Following inpatient admission, the patient underwent a bone marrow biopsy that showed 90% marrow plasma cells (FIGURE 2). Serum protein electrophoresis (SPEP) and immunofixation electrophoresis revealed an elevated monoclonal protein of 7.52 g/dL IgG kappa, prompting us to diagnose multiple myeloma.
FIGURE 2
Bone marrow smear reveals 90% marrow plasma cells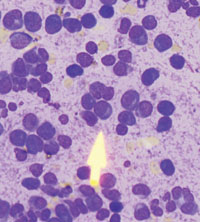
Patients are typically much older
Multiple myeloma is a malignant proliferation of plasma cells derived from a single clone and accounts for 13% of all hematologic malignancies in Caucasians and 33% in African Americans. As with most hematopoeitic malignancies, the incidence of multiple myeloma increases with age, with the median age at diagnosis estimated at 69 years. In addition, 47% of multiple myeloma patients are >70 years of age, and 75% are >60.1
Clinical findings in multiple myeloma vary from asymptomatic patients whose disease is discovered incidentally to patients presenting with life-threatening symptoms. In a recent study of more than 1000 newly diagnosed patients, the most common presenting symptoms were bone pain (58%), fatigue (32%), and weight loss (24%).2 Tumor cells, tumor products (ie, monoclonal immunoglobulin protein), and host response to both elements account for other focal and systemic symptoms, including bone fracture, anemia, renal failure, vascular manifestations of hyperviscosity, hypercalcemia, and increased susceptibility to infection.
This presentation, in conjunction with radiological evidence of lytic bone lesions, increased total serum protein concentration, and a monoclonal protein in the urine or serum, constitutes the hallmark findings of multiple myeloma.
Working group keys in on 3 criteria
The International Myeloma Working Group has agreed on 3 simplified criteria for diagnosis of symptomatic multiple myeloma.3 These include the presence of (1) clonal bone marrow plasma cells or plasmacytoma, (2) an M-protein in serum of unspecified concentration, and (3) tissue- or organ-related impairment, such as renal insufficiency or anemia.
Our patient easily met all 3. His bone marrow biopsy displayed 90% plasma cell cellularity. Additionally, his SPEP exhibited markedly increased IgG immunoglobulin, and immunofixation data suggested IgG type kappa monoclonal gammopathy. These findings, in combination with our patient’s blood chemistry abnormalities, marked proteinuria, and imaging findings, confirmed the diagnosis of multiple myeloma.
Rule out MGUS
The differential diagnosis for multiple myeloma includes monoclonal gammopathy of undetermined significance (MGUS). The characteristic findings of MGUS include:
- absence of symptoms,
- M-protein component (either IgG, IgA, or IgM) <3 g/dL,
- <10% plasma cells in the marrow,
- absence of lytic lesions, and
- no signs of anemia, hypercalcemia, or renal insufficiency.
Essentially, MGUS is a milder form of myeloma with a more indolent course. However, MGUS does carry a 1% annual risk of progression to frank myeloma. An additional concern for patients with multiple myeloma is progression to plasma cell leukemia (PCL). The prognosis for PCL is poor, and the diagnosis is made when the absolute plasma cell count exceeds 2000/mcL. The rate of occurrence of PCL as a progression of multiple myeloma is 1% to 4%.4
In addition to MGUS, the differential diagnosis for multiple myeloma includes tuberculosis, sarcoidosis, and metastatic disease.
A 2-pronged Tx approach
There has been a paradigm shift in the treatment of multiple myeloma in the past decade, and while it appears to be incurable with current approaches, considerable progress has been made.5 Median survival prior to 1997 was nearly 2.5 years; it is now nearly 4 years for patients diagnosed in the last decade.5
Treatment of multiple myeloma generally consists of systemic chemotherapy to control progression and supportive care to prevent serious complications. The standard treatment has traditionally consisted of intermittent pulses of an alkylating agent and prednisone administered for 4 to 7 days every 4 to 6 weeks.
Complications requiring supportive therapy include infections (eg, in the urinary tract), pneumonia, hypercalcemia, and renal failure. Of note, hypercalcemia and renal failure may be alleviated with adequate hydration. If necessary, more aggressive management with dialysis may be initiated.
Other considerations include the administration of allopurinol during chemotherapy, which may help control hyperuricemia from tumor lysis. Transfusions may be required for anemic patients, and plasmapheresis may be indicated to treat hyperviscosity syndrome.
Our patient improves
In the hospital our patient began 4 days on dexamethasone, a 7-day regimen of plasmapheresis, and dialysis. He was also started on a proteasome inhibitor (bortezomib), a newer antineoplastic agent approved for the treatment of multiple myeloma.6 The patient responded well and was discharged home with hematology-oncology follow-up, where he remains clinically improved after treatment with a combination of bortezomib and dexamethasone.
CORRESPONDENCE
Vincent Timpone, MD, Department of Radiology, David Grant United States Air Force Medical Center, Travis Air Force Base, CA 94535; [email protected]
1. Zulian GB, Babare R, Zagonel V. Multiple myeloma. Crit Rev Oncol Hematol. 1998;27:165-167.
2. Kyle RA, Gertz MA. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78:21-33.
3. International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003;121:749-757.
4. Blade J, Kyle RA. Nonsecretory myeloma, immunoglobulin D myeloma, and plasma cell leukemia. Hematol Oncol Clin North Am. 1999;13:1259-1272.
5. Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111:2516-2520.
6. Jagannath S, Durie BG, Wolf J, et al. Bortezomib therapy alone and in combination with dexamethasone for previously untreated symptomatic multiple myeloma. Br J Haematol. 2005;129:776-783.
A 44-year-old African American man went to his primary care provider complaining of 5/10 dull lower back pain, which he attributed to a recent fall. He reported no radiation of the pain, lower extremity weakness or numbness, or any bowel/ bladder incontinence. Further review of systems and past medical history was unremarkable. He was initially treated with nonsteroidal anti-inflammatory agents and physical therapy. Approximately 2 months later he returned with similar persistent lower back pain complaints; an opiate was added to his pain control regimen. The following week he went to the emergency department (ED), where he reported worsening pain.
In the ED, he was afebrile with normal vital signs. He had a benign musculoskeletal and abdominal exam, and his neurological exam was without any focal deficits. Blood tests revealed a hemoglobin level of 9.8 g/dL with a hematocrit of 27.9%, serum blood urea nitrogen/creatinine ratio of 143/11.0 mg/dL, potassium of 6.5 mmol/L, calcium of 11.8 mg/dL, and phosphorus of 8.5 mg/dL. Urine protein was elevated at 88 mg/dL.
A stat CT of the patient’s abdomen and pelvis revealed no signs of obstructive uropathy to explain his acute renal failure. A sagittal CT reconstruction in bone windows of the initial ED radiological workup was revealing (FIGURE 1).
FIGURE 1
Sagittal CT reconstruction of thoracolumbar spine
What is your diagnosis?
How would you manage this condition?
Dx: Multiple myeloma
The sagittal CT reconstruction in bone windows of the thoracolumbar spine (FIGURE 1) revealed multiple lucent foci throughout the osseous structures, with an anterior compression deformity of the L2 vertebral body. A subsequent skeletal survey showed a diffuse salt and pepper pattern affecting most of the osseous structures, with additional lytic lesions in the calvarium and extremities.
Following inpatient admission, the patient underwent a bone marrow biopsy that showed 90% marrow plasma cells (FIGURE 2). Serum protein electrophoresis (SPEP) and immunofixation electrophoresis revealed an elevated monoclonal protein of 7.52 g/dL IgG kappa, prompting us to diagnose multiple myeloma.
FIGURE 2
Bone marrow smear reveals 90% marrow plasma cells
Patients are typically much older
Multiple myeloma is a malignant proliferation of plasma cells derived from a single clone and accounts for 13% of all hematologic malignancies in Caucasians and 33% in African Americans. As with most hematopoeitic malignancies, the incidence of multiple myeloma increases with age, with the median age at diagnosis estimated at 69 years. In addition, 47% of multiple myeloma patients are >70 years of age, and 75% are >60.1
Clinical findings in multiple myeloma vary from asymptomatic patients whose disease is discovered incidentally to patients presenting with life-threatening symptoms. In a recent study of more than 1000 newly diagnosed patients, the most common presenting symptoms were bone pain (58%), fatigue (32%), and weight loss (24%).2 Tumor cells, tumor products (ie, monoclonal immunoglobulin protein), and host response to both elements account for other focal and systemic symptoms, including bone fracture, anemia, renal failure, vascular manifestations of hyperviscosity, hypercalcemia, and increased susceptibility to infection.
This presentation, in conjunction with radiological evidence of lytic bone lesions, increased total serum protein concentration, and a monoclonal protein in the urine or serum, constitutes the hallmark findings of multiple myeloma.
Working group keys in on 3 criteria
The International Myeloma Working Group has agreed on 3 simplified criteria for diagnosis of symptomatic multiple myeloma.3 These include the presence of (1) clonal bone marrow plasma cells or plasmacytoma, (2) an M-protein in serum of unspecified concentration, and (3) tissue- or organ-related impairment, such as renal insufficiency or anemia.
Our patient easily met all 3. His bone marrow biopsy displayed 90% plasma cell cellularity. Additionally, his SPEP exhibited markedly increased IgG immunoglobulin, and immunofixation data suggested IgG type kappa monoclonal gammopathy. These findings, in combination with our patient’s blood chemistry abnormalities, marked proteinuria, and imaging findings, confirmed the diagnosis of multiple myeloma.
Rule out MGUS
The differential diagnosis for multiple myeloma includes monoclonal gammopathy of undetermined significance (MGUS). The characteristic findings of MGUS include:
- absence of symptoms,
- M-protein component (either IgG, IgA, or IgM) <3 g/dL,
- <10% plasma cells in the marrow,
- absence of lytic lesions, and
- no signs of anemia, hypercalcemia, or renal insufficiency.
Essentially, MGUS is a milder form of myeloma with a more indolent course. However, MGUS does carry a 1% annual risk of progression to frank myeloma. An additional concern for patients with multiple myeloma is progression to plasma cell leukemia (PCL). The prognosis for PCL is poor, and the diagnosis is made when the absolute plasma cell count exceeds 2000/mcL. The rate of occurrence of PCL as a progression of multiple myeloma is 1% to 4%.4
In addition to MGUS, the differential diagnosis for multiple myeloma includes tuberculosis, sarcoidosis, and metastatic disease.
A 2-pronged Tx approach
There has been a paradigm shift in the treatment of multiple myeloma in the past decade, and while it appears to be incurable with current approaches, considerable progress has been made.5 Median survival prior to 1997 was nearly 2.5 years; it is now nearly 4 years for patients diagnosed in the last decade.5
Treatment of multiple myeloma generally consists of systemic chemotherapy to control progression and supportive care to prevent serious complications. The standard treatment has traditionally consisted of intermittent pulses of an alkylating agent and prednisone administered for 4 to 7 days every 4 to 6 weeks.
Complications requiring supportive therapy include infections (eg, in the urinary tract), pneumonia, hypercalcemia, and renal failure. Of note, hypercalcemia and renal failure may be alleviated with adequate hydration. If necessary, more aggressive management with dialysis may be initiated.
Other considerations include the administration of allopurinol during chemotherapy, which may help control hyperuricemia from tumor lysis. Transfusions may be required for anemic patients, and plasmapheresis may be indicated to treat hyperviscosity syndrome.
Our patient improves
In the hospital our patient began 4 days on dexamethasone, a 7-day regimen of plasmapheresis, and dialysis. He was also started on a proteasome inhibitor (bortezomib), a newer antineoplastic agent approved for the treatment of multiple myeloma.6 The patient responded well and was discharged home with hematology-oncology follow-up, where he remains clinically improved after treatment with a combination of bortezomib and dexamethasone.
CORRESPONDENCE
Vincent Timpone, MD, Department of Radiology, David Grant United States Air Force Medical Center, Travis Air Force Base, CA 94535; [email protected]
A 44-year-old African American man went to his primary care provider complaining of 5/10 dull lower back pain, which he attributed to a recent fall. He reported no radiation of the pain, lower extremity weakness or numbness, or any bowel/ bladder incontinence. Further review of systems and past medical history was unremarkable. He was initially treated with nonsteroidal anti-inflammatory agents and physical therapy. Approximately 2 months later he returned with similar persistent lower back pain complaints; an opiate was added to his pain control regimen. The following week he went to the emergency department (ED), where he reported worsening pain.
In the ED, he was afebrile with normal vital signs. He had a benign musculoskeletal and abdominal exam, and his neurological exam was without any focal deficits. Blood tests revealed a hemoglobin level of 9.8 g/dL with a hematocrit of 27.9%, serum blood urea nitrogen/creatinine ratio of 143/11.0 mg/dL, potassium of 6.5 mmol/L, calcium of 11.8 mg/dL, and phosphorus of 8.5 mg/dL. Urine protein was elevated at 88 mg/dL.
A stat CT of the patient’s abdomen and pelvis revealed no signs of obstructive uropathy to explain his acute renal failure. A sagittal CT reconstruction in bone windows of the initial ED radiological workup was revealing (FIGURE 1).
FIGURE 1
Sagittal CT reconstruction of thoracolumbar spine
What is your diagnosis?
How would you manage this condition?
Dx: Multiple myeloma
The sagittal CT reconstruction in bone windows of the thoracolumbar spine (FIGURE 1) revealed multiple lucent foci throughout the osseous structures, with an anterior compression deformity of the L2 vertebral body. A subsequent skeletal survey showed a diffuse salt and pepper pattern affecting most of the osseous structures, with additional lytic lesions in the calvarium and extremities.
Following inpatient admission, the patient underwent a bone marrow biopsy that showed 90% marrow plasma cells (FIGURE 2). Serum protein electrophoresis (SPEP) and immunofixation electrophoresis revealed an elevated monoclonal protein of 7.52 g/dL IgG kappa, prompting us to diagnose multiple myeloma.
FIGURE 2
Bone marrow smear reveals 90% marrow plasma cells
Patients are typically much older
Multiple myeloma is a malignant proliferation of plasma cells derived from a single clone and accounts for 13% of all hematologic malignancies in Caucasians and 33% in African Americans. As with most hematopoeitic malignancies, the incidence of multiple myeloma increases with age, with the median age at diagnosis estimated at 69 years. In addition, 47% of multiple myeloma patients are >70 years of age, and 75% are >60.1
Clinical findings in multiple myeloma vary from asymptomatic patients whose disease is discovered incidentally to patients presenting with life-threatening symptoms. In a recent study of more than 1000 newly diagnosed patients, the most common presenting symptoms were bone pain (58%), fatigue (32%), and weight loss (24%).2 Tumor cells, tumor products (ie, monoclonal immunoglobulin protein), and host response to both elements account for other focal and systemic symptoms, including bone fracture, anemia, renal failure, vascular manifestations of hyperviscosity, hypercalcemia, and increased susceptibility to infection.
This presentation, in conjunction with radiological evidence of lytic bone lesions, increased total serum protein concentration, and a monoclonal protein in the urine or serum, constitutes the hallmark findings of multiple myeloma.
Working group keys in on 3 criteria
The International Myeloma Working Group has agreed on 3 simplified criteria for diagnosis of symptomatic multiple myeloma.3 These include the presence of (1) clonal bone marrow plasma cells or plasmacytoma, (2) an M-protein in serum of unspecified concentration, and (3) tissue- or organ-related impairment, such as renal insufficiency or anemia.
Our patient easily met all 3. His bone marrow biopsy displayed 90% plasma cell cellularity. Additionally, his SPEP exhibited markedly increased IgG immunoglobulin, and immunofixation data suggested IgG type kappa monoclonal gammopathy. These findings, in combination with our patient’s blood chemistry abnormalities, marked proteinuria, and imaging findings, confirmed the diagnosis of multiple myeloma.
Rule out MGUS
The differential diagnosis for multiple myeloma includes monoclonal gammopathy of undetermined significance (MGUS). The characteristic findings of MGUS include:
- absence of symptoms,
- M-protein component (either IgG, IgA, or IgM) <3 g/dL,
- <10% plasma cells in the marrow,
- absence of lytic lesions, and
- no signs of anemia, hypercalcemia, or renal insufficiency.
Essentially, MGUS is a milder form of myeloma with a more indolent course. However, MGUS does carry a 1% annual risk of progression to frank myeloma. An additional concern for patients with multiple myeloma is progression to plasma cell leukemia (PCL). The prognosis for PCL is poor, and the diagnosis is made when the absolute plasma cell count exceeds 2000/mcL. The rate of occurrence of PCL as a progression of multiple myeloma is 1% to 4%.4
In addition to MGUS, the differential diagnosis for multiple myeloma includes tuberculosis, sarcoidosis, and metastatic disease.
A 2-pronged Tx approach
There has been a paradigm shift in the treatment of multiple myeloma in the past decade, and while it appears to be incurable with current approaches, considerable progress has been made.5 Median survival prior to 1997 was nearly 2.5 years; it is now nearly 4 years for patients diagnosed in the last decade.5
Treatment of multiple myeloma generally consists of systemic chemotherapy to control progression and supportive care to prevent serious complications. The standard treatment has traditionally consisted of intermittent pulses of an alkylating agent and prednisone administered for 4 to 7 days every 4 to 6 weeks.
Complications requiring supportive therapy include infections (eg, in the urinary tract), pneumonia, hypercalcemia, and renal failure. Of note, hypercalcemia and renal failure may be alleviated with adequate hydration. If necessary, more aggressive management with dialysis may be initiated.
Other considerations include the administration of allopurinol during chemotherapy, which may help control hyperuricemia from tumor lysis. Transfusions may be required for anemic patients, and plasmapheresis may be indicated to treat hyperviscosity syndrome.
Our patient improves
In the hospital our patient began 4 days on dexamethasone, a 7-day regimen of plasmapheresis, and dialysis. He was also started on a proteasome inhibitor (bortezomib), a newer antineoplastic agent approved for the treatment of multiple myeloma.6 The patient responded well and was discharged home with hematology-oncology follow-up, where he remains clinically improved after treatment with a combination of bortezomib and dexamethasone.
CORRESPONDENCE
Vincent Timpone, MD, Department of Radiology, David Grant United States Air Force Medical Center, Travis Air Force Base, CA 94535; [email protected]
1. Zulian GB, Babare R, Zagonel V. Multiple myeloma. Crit Rev Oncol Hematol. 1998;27:165-167.
2. Kyle RA, Gertz MA. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78:21-33.
3. International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003;121:749-757.
4. Blade J, Kyle RA. Nonsecretory myeloma, immunoglobulin D myeloma, and plasma cell leukemia. Hematol Oncol Clin North Am. 1999;13:1259-1272.
5. Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111:2516-2520.
6. Jagannath S, Durie BG, Wolf J, et al. Bortezomib therapy alone and in combination with dexamethasone for previously untreated symptomatic multiple myeloma. Br J Haematol. 2005;129:776-783.
1. Zulian GB, Babare R, Zagonel V. Multiple myeloma. Crit Rev Oncol Hematol. 1998;27:165-167.
2. Kyle RA, Gertz MA. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78:21-33.
3. International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003;121:749-757.
4. Blade J, Kyle RA. Nonsecretory myeloma, immunoglobulin D myeloma, and plasma cell leukemia. Hematol Oncol Clin North Am. 1999;13:1259-1272.
5. Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111:2516-2520.
6. Jagannath S, Durie BG, Wolf J, et al. Bortezomib therapy alone and in combination with dexamethasone for previously untreated symptomatic multiple myeloma. Br J Haematol. 2005;129:776-783.
Flat lesions on thigh
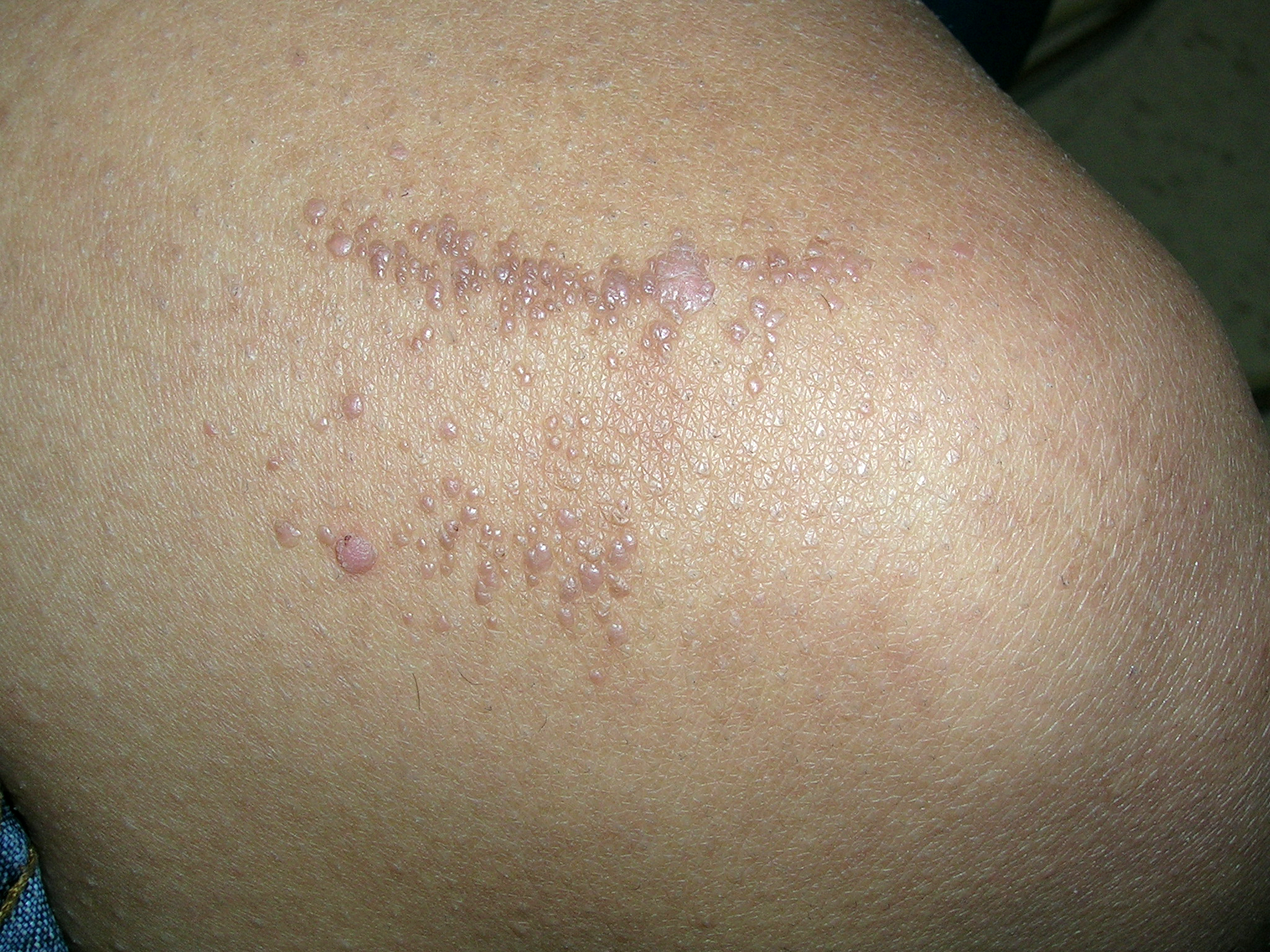
This patient was diagnosed with flat warts, based on their typical appearance and location. The differential diagnosis included lichen planus and seborrheic keratoses. Lichen planus is bilateral and the most common sites are the ankles, wrists, and back. Seborrheic keratoses are typically found in clusters on the face and trunk, so this would have been an unusual location for a cluster of these lesions.
Flat warts may be treated with salicylates, 5-fluorouracil, imiquimod, cryotherapy, or tretinoin. The clinician discussed the treatment choices with the patient, and she chose to have initial cryotherapy followed by topical, over-the-counter salicylic acid.
This case was adapted from: Mayeaux EJ. Flat warts. In: Usatine R, Smith M, Mayeaux EJ, Chumley H, Tysinger J, eds. The Color Atlas of Family Medicine. New York, NY: McGraw-Hill; 2009:527-529.
To learn more about The Color Atlas of Family Medicine, see:
* http://www.amazon.com/Color-Atlas-Family-Medicine/dp/0071474641
This patient was diagnosed with flat warts, based on their typical appearance and location. The differential diagnosis included lichen planus and seborrheic keratoses. Lichen planus is bilateral and the most common sites are the ankles, wrists, and back. Seborrheic keratoses are typically found in clusters on the face and trunk, so this would have been an unusual location for a cluster of these lesions.
Flat warts may be treated with salicylates, 5-fluorouracil, imiquimod, cryotherapy, or tretinoin. The clinician discussed the treatment choices with the patient, and she chose to have initial cryotherapy followed by topical, over-the-counter salicylic acid.
This case was adapted from: Mayeaux EJ. Flat warts. In: Usatine R, Smith M, Mayeaux EJ, Chumley H, Tysinger J, eds. The Color Atlas of Family Medicine. New York, NY: McGraw-Hill; 2009:527-529.
To learn more about The Color Atlas of Family Medicine, see:
* http://www.amazon.com/Color-Atlas-Family-Medicine/dp/0071474641
This patient was diagnosed with flat warts, based on their typical appearance and location. The differential diagnosis included lichen planus and seborrheic keratoses. Lichen planus is bilateral and the most common sites are the ankles, wrists, and back. Seborrheic keratoses are typically found in clusters on the face and trunk, so this would have been an unusual location for a cluster of these lesions.
Flat warts may be treated with salicylates, 5-fluorouracil, imiquimod, cryotherapy, or tretinoin. The clinician discussed the treatment choices with the patient, and she chose to have initial cryotherapy followed by topical, over-the-counter salicylic acid.
This case was adapted from: Mayeaux EJ. Flat warts. In: Usatine R, Smith M, Mayeaux EJ, Chumley H, Tysinger J, eds. The Color Atlas of Family Medicine. New York, NY: McGraw-Hill; 2009:527-529.
To learn more about The Color Atlas of Family Medicine, see:
* http://www.amazon.com/Color-Atlas-Family-Medicine/dp/0071474641
Rash on back
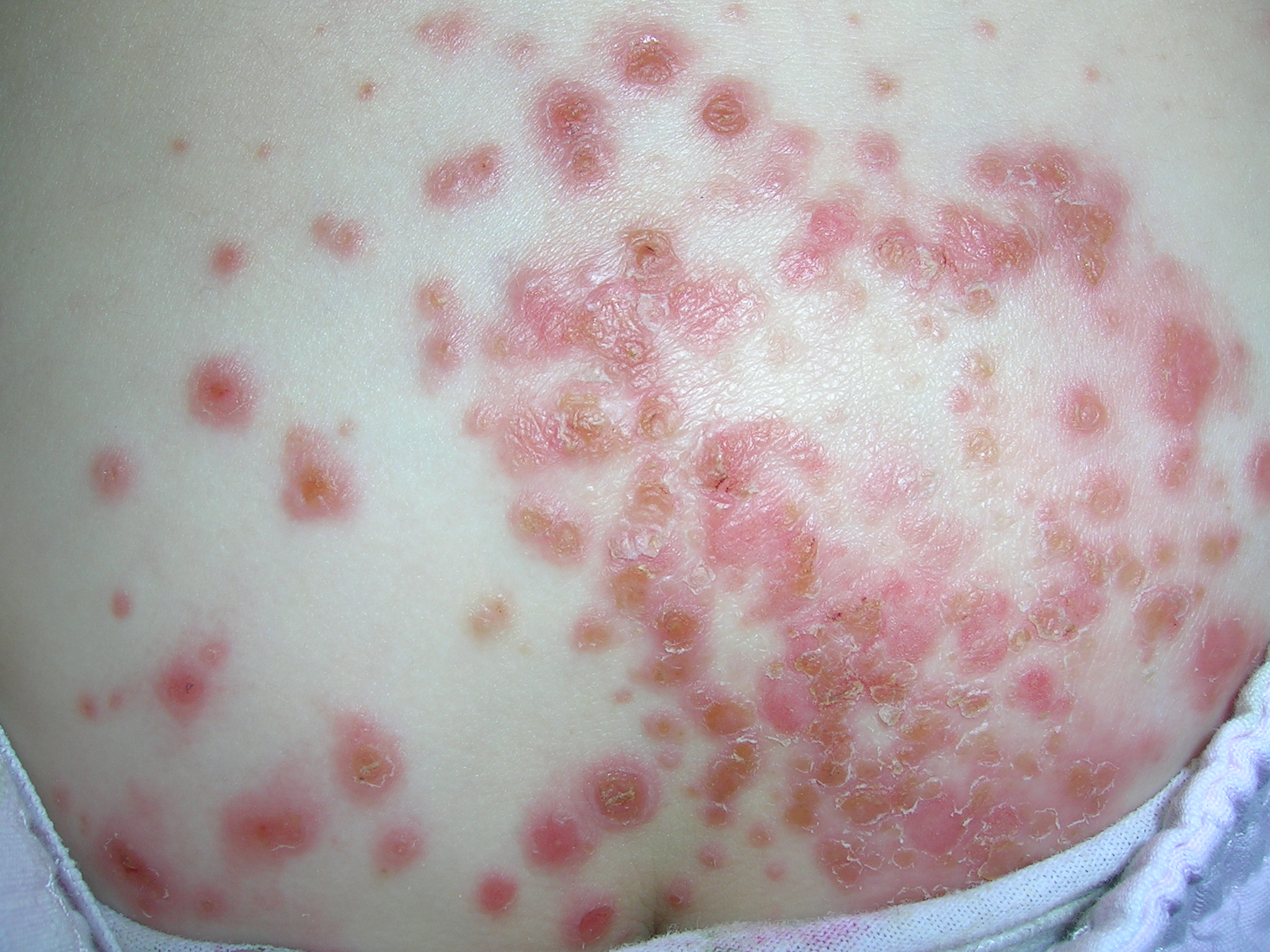
This patient had impetigo. The honey crusted erythematous lesions are typical for this superficial bacterial skin infection. Impetigo is caused by Staphylococcus aureus and/or group A beta-hemolytic Streptococcus (GABHS). The differential diagnosis for this case included herpes simplex, tinea corporis, and superinfected atopic dermatitis.
There is good evidence that topical mupirocin is equally or more effective than oral treatment for people with limited impetigo. Mupirocin also covers methicillin-resistant Staphylococcus aureus. Extensive impetigo should be treated for at least 7 days with antibiotics that cover group A beta-hemolytic Streptococcus and S aureus.
We treated our patient with oral cephalexin 35 mg/kg/day and the impetigo was resolved at a 2-week follow-up visit. The culture grew out S aureus sensitive to cephalosporins.
This case was adapted from: Usatine R. Impetigo. In: Usatine R, Smith M, Mayeaux EJ, Chumley H, Tysinger J, eds. The Color Atlas of Family Medicine. New York, NY: McGraw-Hill; 2009:461-465.
To learn more about The Color Atlas of Family Medicine, see:
* http://www.amazon.com/Color-Atlas-Family-Medicine/dp/0071474641
This patient had impetigo. The honey crusted erythematous lesions are typical for this superficial bacterial skin infection. Impetigo is caused by Staphylococcus aureus and/or group A beta-hemolytic Streptococcus (GABHS). The differential diagnosis for this case included herpes simplex, tinea corporis, and superinfected atopic dermatitis.
There is good evidence that topical mupirocin is equally or more effective than oral treatment for people with limited impetigo. Mupirocin also covers methicillin-resistant Staphylococcus aureus. Extensive impetigo should be treated for at least 7 days with antibiotics that cover group A beta-hemolytic Streptococcus and S aureus.
We treated our patient with oral cephalexin 35 mg/kg/day and the impetigo was resolved at a 2-week follow-up visit. The culture grew out S aureus sensitive to cephalosporins.
This case was adapted from: Usatine R. Impetigo. In: Usatine R, Smith M, Mayeaux EJ, Chumley H, Tysinger J, eds. The Color Atlas of Family Medicine. New York, NY: McGraw-Hill; 2009:461-465.
To learn more about The Color Atlas of Family Medicine, see:
* http://www.amazon.com/Color-Atlas-Family-Medicine/dp/0071474641
This patient had impetigo. The honey crusted erythematous lesions are typical for this superficial bacterial skin infection. Impetigo is caused by Staphylococcus aureus and/or group A beta-hemolytic Streptococcus (GABHS). The differential diagnosis for this case included herpes simplex, tinea corporis, and superinfected atopic dermatitis.
There is good evidence that topical mupirocin is equally or more effective than oral treatment for people with limited impetigo. Mupirocin also covers methicillin-resistant Staphylococcus aureus. Extensive impetigo should be treated for at least 7 days with antibiotics that cover group A beta-hemolytic Streptococcus and S aureus.
We treated our patient with oral cephalexin 35 mg/kg/day and the impetigo was resolved at a 2-week follow-up visit. The culture grew out S aureus sensitive to cephalosporins.
This case was adapted from: Usatine R. Impetigo. In: Usatine R, Smith M, Mayeaux EJ, Chumley H, Tysinger J, eds. The Color Atlas of Family Medicine. New York, NY: McGraw-Hill; 2009:461-465.
To learn more about The Color Atlas of Family Medicine, see:
* http://www.amazon.com/Color-Atlas-Family-Medicine/dp/0071474641
Painful sore on finger
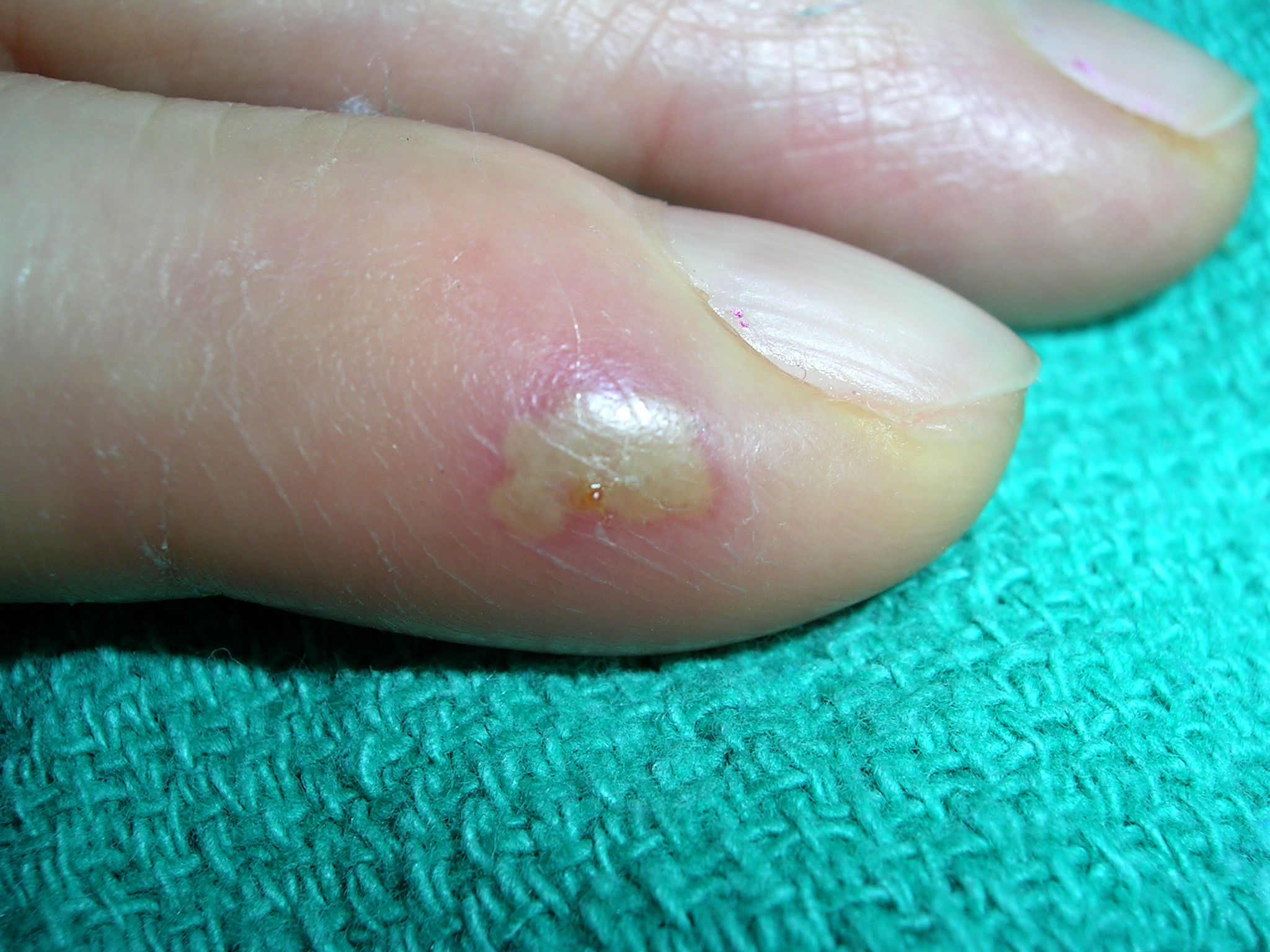
The diagnosis
The patient had herpetic whitlow—a herpes simplex virus (HSV) infection of the finger. Other conditions that could look like this include paronychia (infection of the nail fold) from a bacterial infection, or a felon, which is a bacterial infection of the pulp of the distal finger.
Herpetic whitlow used to be a common infection among dental hygienists. Now the use of gloves and universal precautions has made this less likely. Our patient had no idea where she picked up this infection, but was eager to receive treatment.
The physician pierced the pustule with a #11 blade and cultures were sent for bacteria and HSV. The patient was started on acyclovir since her physician felt HSV was the most likely cause of the infection. She had pain relief within a day and the lesion was fully clear in 2 weeks.
This case was adapted from: Mayeaux EJ. Herpes simplex. In: Usatine R, Smith M, Mayeaux EJ, Chumley H, Tysinger J, eds. The Color Atlas of Family Medicine. New York, NY: McGraw-Hill; 2009:513-517.
To learn more about The Color Atlas of Family Medicine, see:
* http://www.amazon.com/Color-Atlas-Family-Medicine/dp/0071474641
The diagnosis
The patient had herpetic whitlow—a herpes simplex virus (HSV) infection of the finger. Other conditions that could look like this include paronychia (infection of the nail fold) from a bacterial infection, or a felon, which is a bacterial infection of the pulp of the distal finger.
Herpetic whitlow used to be a common infection among dental hygienists. Now the use of gloves and universal precautions has made this less likely. Our patient had no idea where she picked up this infection, but was eager to receive treatment.
The physician pierced the pustule with a #11 blade and cultures were sent for bacteria and HSV. The patient was started on acyclovir since her physician felt HSV was the most likely cause of the infection. She had pain relief within a day and the lesion was fully clear in 2 weeks.
This case was adapted from: Mayeaux EJ. Herpes simplex. In: Usatine R, Smith M, Mayeaux EJ, Chumley H, Tysinger J, eds. The Color Atlas of Family Medicine. New York, NY: McGraw-Hill; 2009:513-517.
To learn more about The Color Atlas of Family Medicine, see:
* http://www.amazon.com/Color-Atlas-Family-Medicine/dp/0071474641
The diagnosis
The patient had herpetic whitlow—a herpes simplex virus (HSV) infection of the finger. Other conditions that could look like this include paronychia (infection of the nail fold) from a bacterial infection, or a felon, which is a bacterial infection of the pulp of the distal finger.
Herpetic whitlow used to be a common infection among dental hygienists. Now the use of gloves and universal precautions has made this less likely. Our patient had no idea where she picked up this infection, but was eager to receive treatment.
The physician pierced the pustule with a #11 blade and cultures were sent for bacteria and HSV. The patient was started on acyclovir since her physician felt HSV was the most likely cause of the infection. She had pain relief within a day and the lesion was fully clear in 2 weeks.
This case was adapted from: Mayeaux EJ. Herpes simplex. In: Usatine R, Smith M, Mayeaux EJ, Chumley H, Tysinger J, eds. The Color Atlas of Family Medicine. New York, NY: McGraw-Hill; 2009:513-517.
To learn more about The Color Atlas of Family Medicine, see:
* http://www.amazon.com/Color-Atlas-Family-Medicine/dp/0071474641
A newborn with peeling skin
Our patient, a Hispanic baby boy, was born at 37 weeks’ gestation after induction of labor. He was delivered vaginally without complication. The newborn’s Apgar scores were 8 and 9, and he weighed 2.24 kg.
The child was born with cracked and peeling skin on his face, chest, hands, and feet. The skin on his face and chest had a taut, cellophane-like appearance. He had fine stubble on his scalp and no eyebrows or eyelashes (FIGURE 1A AND 1B).
The mother’s medical history and serology were unremarkable. Her prior obstetric history included a female infant who had died at 3 months of age of pneumonia, a skin infection, and dehydration. The mother indicated that the deceased child had “fish scale disease.”
FIGURE 1
Cracked skin, absence of eyebrows and eyelashes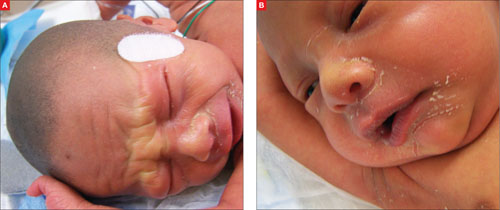
What is your diagnosis?
How would you manage this condition?
Diagnosis: Congenital ichthyosis
Our patient had the congenital form of ichthyosis—a disorder that is sometimes referred to as fish scale disease. Congenital ichthyosis is a phenotypic expression of several different genotypes, and presents with varying degrees of severity. It is fairly common, occurring in 1 in 250 to 300 people.1,2 An extremely rare acquired form of ichthyosis may appear in adults, usually as a result of systemic disease or a medication reaction.
The diagnosis of congenital ichthyosis is made based on skin findings. Skin biopsy and genetic testing are typically not necessary for diagnosis.
Congenital ichthyosis is suspected in newborns who are either collodion babies (as was our patient) or who have harlequin ichthyosis.3
Collodion babies appear to be encased in a cellophane-like membrane at birth. The surface of the skin of the collodion baby usually appears taut and shiny. An absence of eyebrows, eyelashes, and scalp hair is common in these newborns; scarring alopecia can occur.4
The skin undergoes a variable degree of cracking and fissuring. Affected newborns may demonstrate ectropion (everted eyelids), eclabium (mouth held open by taut skin), and contracture of the fingers and joints.4,5 Degree of skin involvement at birth does not necessarily correlate with later disease severity.6
The collodion baby usually has a transglutaminase 1 gene mutation, which can cause either lamellar ichthyosis or congenital ichthyosiform erythroderma.7 (Absence of transglutaminase causes failed cross-linking of the proteins in the keratinocyte cellular envelope.)
Harlequin ichthyosis is a severe and usually lethal form of congenital ichthyosis, caused by a mutation in the ABCA12 gene. The product of this gene acts as a lipid transporter in epidermal keratinocytes. It is crucial for the correct formation of intercellular lipid layers in the stratum corneum, and its absence causes defective lipid transport and a loss of the skin lipid barrier.8
Affected infants have thick, armorlike plates of skin with deep moist fissures, along with severe ectropion and eclabium.4,5 Scaling and fissuring occur on the scalp, but hair is usually present.5
Differential diagnosis includes scalded skin syndrome
The differential for an infant with peeling skin includes staphylococcal scalded skin syndrome, physiologic desquamation, and infantile seborrheic dermatitis.
Staphylococcal scalded skin syndrome is a blistering skin disease induced by exfoliative toxins of Staphylococcus aureus. Toxins enter the skin, most often from the circulation, and disrupt intercellular linkages in the epidermis.9 Patients have generalized erythema, fever, and skin tenderness followed by the formation of large bullae, which rupture with slight pressure (Nikolsky sign). Rupture of these bullae results in extensive areas of denuded skin. These lesions do not scar because epidermal disruption occurs superficially.10 There is no hair loss.
Diagnosis is primarily clinical, but is supported by bacterial culture results. Sepsis can be a comorbid condition.
Physiologic desquamation is a common benign condition of full-term and post-date neonatal skin. Fine, diffuse scaling and peeling typically begin on the second day of life and last a few days. There is no hair loss or shiny membrane formation.11
Infantile seborrheic dermatitis, or “cradle cap,” is a common condition characterized by erythema and greasy white-to-yellowish scales on the scalp, forehead, eyebrows, cheeks, paranasal and nasolabial folds, retroauricular area, chest, and axillae.12 This condition usually develops in the first 3 to 4 weeks of life, but immunocompromised neonates often have generalized scaling and desquamation at birth.13 While hair loss is not caused by the primary process, aggressive scale removal during treatment can cause secondary hair loss. The etiology is unknown, but some evidence points to an abnormal host response to the yeast Malassezia.12
Management focuses on fluids and emollients
Management of congenital ichthyosis involves adequate pulmonary support, as the scaling and/or plate formation may cause a restrictive ventilatory defect; the use of humidified incubators; fluid and electrolyte replacement; emollients and wet compresses to maintain skin moisture and prevent further scaling or tautness of the membrane; ophthalmologic consultation to manage ectropion; and the treatment of infection.4,5 Oral retinoids are used in select cases.14
The prognosis varies with disease severity, but complications include infection, temperature instability, and dehydration secondary to skin barrier dysfunction. Most collodion babies survive to adulthood while few, if any, babies affected with harlequin ichthyosis survive the neonatal period. Ongoing research in corrective gene transfer provides hope for future therapy.15
Our patient required a humidified incubator
Following a consult with our hospital neonatologist, we closely monitored our patient’s volume status and electrolytes, placed him in a humidified incubator, and ensured that he was treated with emollient creams. Our patient was also treated for neonatal jaundice.
We obtained a consultation with a genetics counselor, who agreed with our diagnosis and ordered appropriate genetic testing. The patient has since been lost to follow-up.
Correspondence
T. Aaron Zeller, MD, 155 Academy Avenue, Greenwood, SC 29646; [email protected]
1. Schwartz RA. Ichthyosis vulgaris, hereditary and acquired. Available at: http://emedicine.medscape.com/article/1112753-overview. Accessed May 2, 2009.
2. Foundation for Ichthyosis & Related Skin Types FIRST). About ichthyosis. Available at: http://www.scalyskin.org/column.cfm?ColumnID=13. Accessed May 8, 2009.
3. Bale SJ, Richard G. Autosomal recessive congenital ichthyosis. Gene Reviews. December 11, 2008. Available at: http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=li-ar. Accessed April 29, 2009.
4. Shwayder T, Akland T. Neonatal skin barrier: structure, function, and disorders. Dermatol Ther. 2005;18:87-103.
5. Akiyama M. Severe congenital ichthyosis of the neonate. Int J Dermatol. 1998;37:722-728.
6. Moss C. Genetic skin disorders. Semin Neonatol. 2000;5:311-320.
7. Parmentier L, Blanchet-Bardon C, Nguyen S, et al. Autosomal recessive lamellar ichthyosis: identification of a new mutation in transglutaminase 1 and evidence for genetic heterogeneity. Hum Mol Genet. 1995;4:1391-1395.
8. Akiyama M. Pathomechanisms of harlequin ichthyosis and ABCA transporters in human diseases. Arch Dermatol. 2006;142:914-918.
9. Ladhani S, Evans RW. Staphylococcal scalded skin syndrome. Arch Dis Child. 1998;78:85-88.
10. Farrell AM. Staphylococcal scalded-skin syndrome. Lancet. 1999;354:880-881.
11. Wallach D. Diagnosis of common, benign neonatal dermatoses. Clin Dermatol. 2003;21:264-268.
12. Elish D, Silverberg NB. Infantile seborrheic dermatitis. Cutis. 2006;77:297-300.
13. Kim HJ, Lim YS, Choi HY, et al. Generalized seborrheic dermatitis in an immunodeficient newborn. Cutis. 2001;67:52-54.
14. Lacour M, Mehta-Nikhar B, Atherton DJ, et al. An appraisal of acitretin therapy in children with inherited disorders of keratinization. Br J Dermatol. 1996;134:1023-1029.
15. Akiyama M, Sugiyama-Nakagirl Y, Sakai K. Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J Clin Invest. 2005;115:1777-1784.
Our patient, a Hispanic baby boy, was born at 37 weeks’ gestation after induction of labor. He was delivered vaginally without complication. The newborn’s Apgar scores were 8 and 9, and he weighed 2.24 kg.
The child was born with cracked and peeling skin on his face, chest, hands, and feet. The skin on his face and chest had a taut, cellophane-like appearance. He had fine stubble on his scalp and no eyebrows or eyelashes (FIGURE 1A AND 1B).
The mother’s medical history and serology were unremarkable. Her prior obstetric history included a female infant who had died at 3 months of age of pneumonia, a skin infection, and dehydration. The mother indicated that the deceased child had “fish scale disease.”
FIGURE 1
Cracked skin, absence of eyebrows and eyelashes
What is your diagnosis?
How would you manage this condition?
Diagnosis: Congenital ichthyosis
Our patient had the congenital form of ichthyosis—a disorder that is sometimes referred to as fish scale disease. Congenital ichthyosis is a phenotypic expression of several different genotypes, and presents with varying degrees of severity. It is fairly common, occurring in 1 in 250 to 300 people.1,2 An extremely rare acquired form of ichthyosis may appear in adults, usually as a result of systemic disease or a medication reaction.
The diagnosis of congenital ichthyosis is made based on skin findings. Skin biopsy and genetic testing are typically not necessary for diagnosis.
Congenital ichthyosis is suspected in newborns who are either collodion babies (as was our patient) or who have harlequin ichthyosis.3
Collodion babies appear to be encased in a cellophane-like membrane at birth. The surface of the skin of the collodion baby usually appears taut and shiny. An absence of eyebrows, eyelashes, and scalp hair is common in these newborns; scarring alopecia can occur.4
The skin undergoes a variable degree of cracking and fissuring. Affected newborns may demonstrate ectropion (everted eyelids), eclabium (mouth held open by taut skin), and contracture of the fingers and joints.4,5 Degree of skin involvement at birth does not necessarily correlate with later disease severity.6
The collodion baby usually has a transglutaminase 1 gene mutation, which can cause either lamellar ichthyosis or congenital ichthyosiform erythroderma.7 (Absence of transglutaminase causes failed cross-linking of the proteins in the keratinocyte cellular envelope.)
Harlequin ichthyosis is a severe and usually lethal form of congenital ichthyosis, caused by a mutation in the ABCA12 gene. The product of this gene acts as a lipid transporter in epidermal keratinocytes. It is crucial for the correct formation of intercellular lipid layers in the stratum corneum, and its absence causes defective lipid transport and a loss of the skin lipid barrier.8
Affected infants have thick, armorlike plates of skin with deep moist fissures, along with severe ectropion and eclabium.4,5 Scaling and fissuring occur on the scalp, but hair is usually present.5
Differential diagnosis includes scalded skin syndrome
The differential for an infant with peeling skin includes staphylococcal scalded skin syndrome, physiologic desquamation, and infantile seborrheic dermatitis.
Staphylococcal scalded skin syndrome is a blistering skin disease induced by exfoliative toxins of Staphylococcus aureus. Toxins enter the skin, most often from the circulation, and disrupt intercellular linkages in the epidermis.9 Patients have generalized erythema, fever, and skin tenderness followed by the formation of large bullae, which rupture with slight pressure (Nikolsky sign). Rupture of these bullae results in extensive areas of denuded skin. These lesions do not scar because epidermal disruption occurs superficially.10 There is no hair loss.
Diagnosis is primarily clinical, but is supported by bacterial culture results. Sepsis can be a comorbid condition.
Physiologic desquamation is a common benign condition of full-term and post-date neonatal skin. Fine, diffuse scaling and peeling typically begin on the second day of life and last a few days. There is no hair loss or shiny membrane formation.11
Infantile seborrheic dermatitis, or “cradle cap,” is a common condition characterized by erythema and greasy white-to-yellowish scales on the scalp, forehead, eyebrows, cheeks, paranasal and nasolabial folds, retroauricular area, chest, and axillae.12 This condition usually develops in the first 3 to 4 weeks of life, but immunocompromised neonates often have generalized scaling and desquamation at birth.13 While hair loss is not caused by the primary process, aggressive scale removal during treatment can cause secondary hair loss. The etiology is unknown, but some evidence points to an abnormal host response to the yeast Malassezia.12
Management focuses on fluids and emollients
Management of congenital ichthyosis involves adequate pulmonary support, as the scaling and/or plate formation may cause a restrictive ventilatory defect; the use of humidified incubators; fluid and electrolyte replacement; emollients and wet compresses to maintain skin moisture and prevent further scaling or tautness of the membrane; ophthalmologic consultation to manage ectropion; and the treatment of infection.4,5 Oral retinoids are used in select cases.14
The prognosis varies with disease severity, but complications include infection, temperature instability, and dehydration secondary to skin barrier dysfunction. Most collodion babies survive to adulthood while few, if any, babies affected with harlequin ichthyosis survive the neonatal period. Ongoing research in corrective gene transfer provides hope for future therapy.15
Our patient required a humidified incubator
Following a consult with our hospital neonatologist, we closely monitored our patient’s volume status and electrolytes, placed him in a humidified incubator, and ensured that he was treated with emollient creams. Our patient was also treated for neonatal jaundice.
We obtained a consultation with a genetics counselor, who agreed with our diagnosis and ordered appropriate genetic testing. The patient has since been lost to follow-up.
Correspondence
T. Aaron Zeller, MD, 155 Academy Avenue, Greenwood, SC 29646; [email protected]
Our patient, a Hispanic baby boy, was born at 37 weeks’ gestation after induction of labor. He was delivered vaginally without complication. The newborn’s Apgar scores were 8 and 9, and he weighed 2.24 kg.
The child was born with cracked and peeling skin on his face, chest, hands, and feet. The skin on his face and chest had a taut, cellophane-like appearance. He had fine stubble on his scalp and no eyebrows or eyelashes (FIGURE 1A AND 1B).
The mother’s medical history and serology were unremarkable. Her prior obstetric history included a female infant who had died at 3 months of age of pneumonia, a skin infection, and dehydration. The mother indicated that the deceased child had “fish scale disease.”
FIGURE 1
Cracked skin, absence of eyebrows and eyelashes
What is your diagnosis?
How would you manage this condition?
Diagnosis: Congenital ichthyosis
Our patient had the congenital form of ichthyosis—a disorder that is sometimes referred to as fish scale disease. Congenital ichthyosis is a phenotypic expression of several different genotypes, and presents with varying degrees of severity. It is fairly common, occurring in 1 in 250 to 300 people.1,2 An extremely rare acquired form of ichthyosis may appear in adults, usually as a result of systemic disease or a medication reaction.
The diagnosis of congenital ichthyosis is made based on skin findings. Skin biopsy and genetic testing are typically not necessary for diagnosis.
Congenital ichthyosis is suspected in newborns who are either collodion babies (as was our patient) or who have harlequin ichthyosis.3
Collodion babies appear to be encased in a cellophane-like membrane at birth. The surface of the skin of the collodion baby usually appears taut and shiny. An absence of eyebrows, eyelashes, and scalp hair is common in these newborns; scarring alopecia can occur.4
The skin undergoes a variable degree of cracking and fissuring. Affected newborns may demonstrate ectropion (everted eyelids), eclabium (mouth held open by taut skin), and contracture of the fingers and joints.4,5 Degree of skin involvement at birth does not necessarily correlate with later disease severity.6
The collodion baby usually has a transglutaminase 1 gene mutation, which can cause either lamellar ichthyosis or congenital ichthyosiform erythroderma.7 (Absence of transglutaminase causes failed cross-linking of the proteins in the keratinocyte cellular envelope.)
Harlequin ichthyosis is a severe and usually lethal form of congenital ichthyosis, caused by a mutation in the ABCA12 gene. The product of this gene acts as a lipid transporter in epidermal keratinocytes. It is crucial for the correct formation of intercellular lipid layers in the stratum corneum, and its absence causes defective lipid transport and a loss of the skin lipid barrier.8
Affected infants have thick, armorlike plates of skin with deep moist fissures, along with severe ectropion and eclabium.4,5 Scaling and fissuring occur on the scalp, but hair is usually present.5
Differential diagnosis includes scalded skin syndrome
The differential for an infant with peeling skin includes staphylococcal scalded skin syndrome, physiologic desquamation, and infantile seborrheic dermatitis.
Staphylococcal scalded skin syndrome is a blistering skin disease induced by exfoliative toxins of Staphylococcus aureus. Toxins enter the skin, most often from the circulation, and disrupt intercellular linkages in the epidermis.9 Patients have generalized erythema, fever, and skin tenderness followed by the formation of large bullae, which rupture with slight pressure (Nikolsky sign). Rupture of these bullae results in extensive areas of denuded skin. These lesions do not scar because epidermal disruption occurs superficially.10 There is no hair loss.
Diagnosis is primarily clinical, but is supported by bacterial culture results. Sepsis can be a comorbid condition.
Physiologic desquamation is a common benign condition of full-term and post-date neonatal skin. Fine, diffuse scaling and peeling typically begin on the second day of life and last a few days. There is no hair loss or shiny membrane formation.11
Infantile seborrheic dermatitis, or “cradle cap,” is a common condition characterized by erythema and greasy white-to-yellowish scales on the scalp, forehead, eyebrows, cheeks, paranasal and nasolabial folds, retroauricular area, chest, and axillae.12 This condition usually develops in the first 3 to 4 weeks of life, but immunocompromised neonates often have generalized scaling and desquamation at birth.13 While hair loss is not caused by the primary process, aggressive scale removal during treatment can cause secondary hair loss. The etiology is unknown, but some evidence points to an abnormal host response to the yeast Malassezia.12
Management focuses on fluids and emollients
Management of congenital ichthyosis involves adequate pulmonary support, as the scaling and/or plate formation may cause a restrictive ventilatory defect; the use of humidified incubators; fluid and electrolyte replacement; emollients and wet compresses to maintain skin moisture and prevent further scaling or tautness of the membrane; ophthalmologic consultation to manage ectropion; and the treatment of infection.4,5 Oral retinoids are used in select cases.14
The prognosis varies with disease severity, but complications include infection, temperature instability, and dehydration secondary to skin barrier dysfunction. Most collodion babies survive to adulthood while few, if any, babies affected with harlequin ichthyosis survive the neonatal period. Ongoing research in corrective gene transfer provides hope for future therapy.15
Our patient required a humidified incubator
Following a consult with our hospital neonatologist, we closely monitored our patient’s volume status and electrolytes, placed him in a humidified incubator, and ensured that he was treated with emollient creams. Our patient was also treated for neonatal jaundice.
We obtained a consultation with a genetics counselor, who agreed with our diagnosis and ordered appropriate genetic testing. The patient has since been lost to follow-up.
Correspondence
T. Aaron Zeller, MD, 155 Academy Avenue, Greenwood, SC 29646; [email protected]
1. Schwartz RA. Ichthyosis vulgaris, hereditary and acquired. Available at: http://emedicine.medscape.com/article/1112753-overview. Accessed May 2, 2009.
2. Foundation for Ichthyosis & Related Skin Types FIRST). About ichthyosis. Available at: http://www.scalyskin.org/column.cfm?ColumnID=13. Accessed May 8, 2009.
3. Bale SJ, Richard G. Autosomal recessive congenital ichthyosis. Gene Reviews. December 11, 2008. Available at: http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=li-ar. Accessed April 29, 2009.
4. Shwayder T, Akland T. Neonatal skin barrier: structure, function, and disorders. Dermatol Ther. 2005;18:87-103.
5. Akiyama M. Severe congenital ichthyosis of the neonate. Int J Dermatol. 1998;37:722-728.
6. Moss C. Genetic skin disorders. Semin Neonatol. 2000;5:311-320.
7. Parmentier L, Blanchet-Bardon C, Nguyen S, et al. Autosomal recessive lamellar ichthyosis: identification of a new mutation in transglutaminase 1 and evidence for genetic heterogeneity. Hum Mol Genet. 1995;4:1391-1395.
8. Akiyama M. Pathomechanisms of harlequin ichthyosis and ABCA transporters in human diseases. Arch Dermatol. 2006;142:914-918.
9. Ladhani S, Evans RW. Staphylococcal scalded skin syndrome. Arch Dis Child. 1998;78:85-88.
10. Farrell AM. Staphylococcal scalded-skin syndrome. Lancet. 1999;354:880-881.
11. Wallach D. Diagnosis of common, benign neonatal dermatoses. Clin Dermatol. 2003;21:264-268.
12. Elish D, Silverberg NB. Infantile seborrheic dermatitis. Cutis. 2006;77:297-300.
13. Kim HJ, Lim YS, Choi HY, et al. Generalized seborrheic dermatitis in an immunodeficient newborn. Cutis. 2001;67:52-54.
14. Lacour M, Mehta-Nikhar B, Atherton DJ, et al. An appraisal of acitretin therapy in children with inherited disorders of keratinization. Br J Dermatol. 1996;134:1023-1029.
15. Akiyama M, Sugiyama-Nakagirl Y, Sakai K. Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J Clin Invest. 2005;115:1777-1784.
1. Schwartz RA. Ichthyosis vulgaris, hereditary and acquired. Available at: http://emedicine.medscape.com/article/1112753-overview. Accessed May 2, 2009.
2. Foundation for Ichthyosis & Related Skin Types FIRST). About ichthyosis. Available at: http://www.scalyskin.org/column.cfm?ColumnID=13. Accessed May 8, 2009.
3. Bale SJ, Richard G. Autosomal recessive congenital ichthyosis. Gene Reviews. December 11, 2008. Available at: http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=li-ar. Accessed April 29, 2009.
4. Shwayder T, Akland T. Neonatal skin barrier: structure, function, and disorders. Dermatol Ther. 2005;18:87-103.
5. Akiyama M. Severe congenital ichthyosis of the neonate. Int J Dermatol. 1998;37:722-728.
6. Moss C. Genetic skin disorders. Semin Neonatol. 2000;5:311-320.
7. Parmentier L, Blanchet-Bardon C, Nguyen S, et al. Autosomal recessive lamellar ichthyosis: identification of a new mutation in transglutaminase 1 and evidence for genetic heterogeneity. Hum Mol Genet. 1995;4:1391-1395.
8. Akiyama M. Pathomechanisms of harlequin ichthyosis and ABCA transporters in human diseases. Arch Dermatol. 2006;142:914-918.
9. Ladhani S, Evans RW. Staphylococcal scalded skin syndrome. Arch Dis Child. 1998;78:85-88.
10. Farrell AM. Staphylococcal scalded-skin syndrome. Lancet. 1999;354:880-881.
11. Wallach D. Diagnosis of common, benign neonatal dermatoses. Clin Dermatol. 2003;21:264-268.
12. Elish D, Silverberg NB. Infantile seborrheic dermatitis. Cutis. 2006;77:297-300.
13. Kim HJ, Lim YS, Choi HY, et al. Generalized seborrheic dermatitis in an immunodeficient newborn. Cutis. 2001;67:52-54.
14. Lacour M, Mehta-Nikhar B, Atherton DJ, et al. An appraisal of acitretin therapy in children with inherited disorders of keratinization. Br J Dermatol. 1996;134:1023-1029.
15. Akiyama M, Sugiyama-Nakagirl Y, Sakai K. Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J Clin Invest. 2005;115:1777-1784.
Photo Rounds Friday
PHOTO ROUNDS FRIDAY
| Click to see full size image |
The diagnosis
This patient had a typical case of infantile acropustulosis, which is often misdiagnosed as scabies. This intensely pruritic, vesiculopustular disease of young children is rare, and typically begins in the second or third months of life, though it can begin when the patient is 10 months old. It occurs slightly more often in darker skinned patients and in boys, can be recurrent, and typically remits when the child is 6 to 36 months of age.
It’s unclear what causes acropustulosis, though some speculate that it is a persistent reaction to scabies (“postscabies syndrome”). Oral antihistamines may be helpful in controlling pruritus. Pramoxine (lotion or cream) may be used topically to control itching as it works by a different mechanism than antihistamine. Corticosteroids (topical and oral) are generally not effective.
We reassured the mother that the condition is not dangerous and would go away on its own. We told her to return with her son if the pustular eruption significantly worsened.
This case was adapted from: Shedd A, Usatine R. Pustular diseases of childhood. In: Usatine R, Smith M, Mayeaux EJ, Chumley H, Tysinger J, eds. The Color Atlas of Family Medicine. New York, NY: McGraw-Hill; 2009: 432-434.
To learn more about The Color Atlas of Family Medicine, see:
* http://www.amazon.com/Color-Atlas-Family-Medicine/dp/0071474641
* http://www.mhprofessional.com/product.php?isbn=0071474641
PHOTO ROUNDS FRIDAY
| Click to see full size image |
The diagnosis
This patient had a typical case of infantile acropustulosis, which is often misdiagnosed as scabies. This intensely pruritic, vesiculopustular disease of young children is rare, and typically begins in the second or third months of life, though it can begin when the patient is 10 months old. It occurs slightly more often in darker skinned patients and in boys, can be recurrent, and typically remits when the child is 6 to 36 months of age.
It’s unclear what causes acropustulosis, though some speculate that it is a persistent reaction to scabies (“postscabies syndrome”). Oral antihistamines may be helpful in controlling pruritus. Pramoxine (lotion or cream) may be used topically to control itching as it works by a different mechanism than antihistamine. Corticosteroids (topical and oral) are generally not effective.
We reassured the mother that the condition is not dangerous and would go away on its own. We told her to return with her son if the pustular eruption significantly worsened.
This case was adapted from: Shedd A, Usatine R. Pustular diseases of childhood. In: Usatine R, Smith M, Mayeaux EJ, Chumley H, Tysinger J, eds. The Color Atlas of Family Medicine. New York, NY: McGraw-Hill; 2009: 432-434.
To learn more about The Color Atlas of Family Medicine, see:
* http://www.amazon.com/Color-Atlas-Family-Medicine/dp/0071474641
* http://www.mhprofessional.com/product.php?isbn=0071474641
PHOTO ROUNDS FRIDAY
| Click to see full size image |
The diagnosis
This patient had a typical case of infantile acropustulosis, which is often misdiagnosed as scabies. This intensely pruritic, vesiculopustular disease of young children is rare, and typically begins in the second or third months of life, though it can begin when the patient is 10 months old. It occurs slightly more often in darker skinned patients and in boys, can be recurrent, and typically remits when the child is 6 to 36 months of age.
It’s unclear what causes acropustulosis, though some speculate that it is a persistent reaction to scabies (“postscabies syndrome”). Oral antihistamines may be helpful in controlling pruritus. Pramoxine (lotion or cream) may be used topically to control itching as it works by a different mechanism than antihistamine. Corticosteroids (topical and oral) are generally not effective.
We reassured the mother that the condition is not dangerous and would go away on its own. We told her to return with her son if the pustular eruption significantly worsened.
This case was adapted from: Shedd A, Usatine R. Pustular diseases of childhood. In: Usatine R, Smith M, Mayeaux EJ, Chumley H, Tysinger J, eds. The Color Atlas of Family Medicine. New York, NY: McGraw-Hill; 2009: 432-434.
To learn more about The Color Atlas of Family Medicine, see:
* http://www.amazon.com/Color-Atlas-Family-Medicine/dp/0071474641
* http://www.mhprofessional.com/product.php?isbn=0071474641
Hyperpigmented papules and plaques on chest
A 20-year-old black man came into our medical center with a mildly pruritic scaly rash affecting his neck and upper body for 2 weeks. Physical exam revealed well-demarcated, hyperpigmented hyperkeratotic papules coalescing to form large plaques on his central chest, back, and shoulders. He had a reticulated pattern on his shoulders and arms (FIGURE 1). His face, intertriginous skin, genitals, mucous membranes, and lower extremities were spared. The remainder of the physical exam was unremarkable.
Woods lamp and potassium hydroxide (KOH) preparation were negative. Labs, including fasting blood glucose and thyroid function test, were normal. Our patient denied any recent travel, fever, night sweats, or weight loss. He noted only that he used the weight benches at the gym. His medical and family histories were unremarkable, and he was not taking any medications or supplements.
FIGURE 1
Plaques on chest, reticulated pattern on arms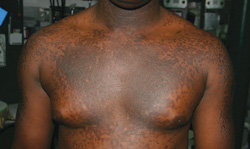
What is your diagnosis?
How would you manage this condition?
Dx: Confluent and reticulated papillomatosis
Confluent and reticulated papillomatosis of Gougerot and Carteaud (CRP) is a rare skin disorder characterized by benign blue-gray or brown hyperpigmented hyperkeratotic papules and plaques. The lesions initially occur on the trunk or central chest as 1- to 2-mm warty papules that become confluent to form plaques, spreading to the neck, abdomen, and upper extremities. Peripherally, the lesions are distributed in a reticular pattern. Although less common, CRP may be isolated to one part of the body, including the face and genitals; the mucous membranes are swpared.
With the exception of Japan, where a male predominance is seen, young women are more commonly affected.1 Patients are typically asymptomatic, or complain of mild pruritus and cosmetic concerns.
A disorder of keratinocyte maturation and differentiation?
The cause of CRP is unknown. Several theories have been entertained: an underlying endocrine disorder, a rare form of cutaneous amyloidosis, reaction to bacteria or fungus on the skin, and a keratinization abnormality.1 Most patients with CRP do not have an underlying endocrine disorder or any evidence of amyloidosis, making these theories less likely. KOH preparation for fungal elements is typically negative and patients do not respond to antifungal therapy.
A bacterial cause has been implicated because CRP responds to antibiotics, but many have argued that antibiotics are acting as an anti-inflammatory agent rather than an antibacterial medication.1 Others have suggested antibiotics may be acting to decrease epidermal proliferation by blocking protein and DNA synthesis and reducing keratinocyte production of cytokines.2
Overall, the most accepted theory is that it is a disorder of keratinocyte maturation and differentiation.1,3 Histological analysis, electron microscopy (EM), and immunohistochemical studies support this theory. Hyperkeratosis can be seen on histological examination. On EM, the stratum granulosum contains more lamellar granules and a larger transition cell layer, and immunochemical analysis reveals an increased expression of genetic markers associated with keratinization.1
The etiology of this keratinization abnormality is poorly understood.
CRP has been reported in several family members, prompting suggestions by some of a possible genetic component.4 Others have proposed staphylococcal enterotoxin B may promote certain immune factors causing abnormal keratinization.2
Is it CRP or tinea versicolor?
The differential for CRP includes:
- tinea or pityriasis versicolor
- tinea corporis
- seborrheic dermatitis
- keratosis follicularis (Darier disease)
- acanthosis nigricans
- macular amyloidosis.
Of all of these, however, CRP is most likely to be confused with tinea versicolor, as both are associated with hyper- or hypopigmented papules and plaques on the chest and back, as well as mild pruritus. In CRP, however, a Woods lamp and KOH will be negative, while with tinea versicolor, KOH will be positive and the Woods lamp may reveal yellow-green fluorescence.
If a patient’s KOH is negative and/ or the patient does not respond to treatment for tinea versicolor (which includes topical or oral antifungals), a trial of oral antibiotics for CRP may be reasonable. Response to an oral antibiotic, such as minocycline, will help to confirm the diagnosis. Although most patients with CRP do not have an endocrine disorder, it’s a good idea to keep this reported association in mind, and perform further testing, as needed.
Oral minocycline is the treatment of choice
The preferred treatment for CRP is oral minocycline (100 mg orally twice a day for 6 weeks).1,2 Oral azithromycin, erythromycin, clarithromycin, tetracycline, cefdinir,3 roxithromycin,5 doxycycline,2 and amoxicillin6 have also been used. Isotretinoin is an effective alternative to oral antibiotics, but clinicians often avoid it because of the adverse side effect profile.
With oral antibiotic therapy, the patient may completely clear and stay clear, or go on to have multiple recurrences or exacerbations. Topical retinoids have also been used with some success,4 but most reported cases have been successfully treated with oral antibiotics.1
Our patient required Tx for several months
We initially treated our patient with doxycycline (100 mg orally twice a day) for 1 month. The lesions cleared after 2 weeks and then recurred during week 4 of treatment. We discontinued the doxycycline, and started the patient on minocycline. The primary lesions resolved after 5 weeks of minocycline, though we noted residual post-inflammatory hyper-pigmentation (FIGURE 2). Our patient continued the medication for an additional 8 weeks. He was lost to follow-up.
FIGURE 2
Weeks later, hyperpigmentation remains
Correspondence
Kendall Lane, MD, LCDR, MC, United States Navy, Navy Medical Center San Diego, 34520 Bob Wilson Drive, Suite 300, San Diego, CA 92134
1. Schwartz R. Confluent and reticulated papillomatosis. Available at: http://www.emedicine.com/derm/topic82.htm. Accessed March 25, 2009.
2. Greenblatt D, Cintra M, Teixiera F, et al. Hyperpigmented plaques on a young man. J Am Acad Dermatol. 2007;56:896-898.
3. Scheinfeld N. Confluent and reticulated papillomatosis: a review of the literature. Am J Clin Dermatol. 2006;7:305-313.
4. Schwartzberg JB, Schwartzberg HA. Response of confluent and reticulate papillomatosis of Gougerot and Carteaud to topical tretinoin. Cutis. 2000;66:291-293.
5. Ito S, Hatamochi A, Yamazaki S. A case of confluent and reticulated papillomatosis that successfully responded to roxithromycin. J Dermatol. 2006;33:71-72.
6. Davis RF, Harman KE. Confluent and reticulated papillomatosis successfully treated with amoxicillin. Br J Dermatol. 2007;156:583-584.
A 20-year-old black man came into our medical center with a mildly pruritic scaly rash affecting his neck and upper body for 2 weeks. Physical exam revealed well-demarcated, hyperpigmented hyperkeratotic papules coalescing to form large plaques on his central chest, back, and shoulders. He had a reticulated pattern on his shoulders and arms (FIGURE 1). His face, intertriginous skin, genitals, mucous membranes, and lower extremities were spared. The remainder of the physical exam was unremarkable.
Woods lamp and potassium hydroxide (KOH) preparation were negative. Labs, including fasting blood glucose and thyroid function test, were normal. Our patient denied any recent travel, fever, night sweats, or weight loss. He noted only that he used the weight benches at the gym. His medical and family histories were unremarkable, and he was not taking any medications or supplements.
FIGURE 1
Plaques on chest, reticulated pattern on arms
What is your diagnosis?
How would you manage this condition?
Dx: Confluent and reticulated papillomatosis
Confluent and reticulated papillomatosis of Gougerot and Carteaud (CRP) is a rare skin disorder characterized by benign blue-gray or brown hyperpigmented hyperkeratotic papules and plaques. The lesions initially occur on the trunk or central chest as 1- to 2-mm warty papules that become confluent to form plaques, spreading to the neck, abdomen, and upper extremities. Peripherally, the lesions are distributed in a reticular pattern. Although less common, CRP may be isolated to one part of the body, including the face and genitals; the mucous membranes are swpared.
With the exception of Japan, where a male predominance is seen, young women are more commonly affected.1 Patients are typically asymptomatic, or complain of mild pruritus and cosmetic concerns.
A disorder of keratinocyte maturation and differentiation?
The cause of CRP is unknown. Several theories have been entertained: an underlying endocrine disorder, a rare form of cutaneous amyloidosis, reaction to bacteria or fungus on the skin, and a keratinization abnormality.1 Most patients with CRP do not have an underlying endocrine disorder or any evidence of amyloidosis, making these theories less likely. KOH preparation for fungal elements is typically negative and patients do not respond to antifungal therapy.
A bacterial cause has been implicated because CRP responds to antibiotics, but many have argued that antibiotics are acting as an anti-inflammatory agent rather than an antibacterial medication.1 Others have suggested antibiotics may be acting to decrease epidermal proliferation by blocking protein and DNA synthesis and reducing keratinocyte production of cytokines.2
Overall, the most accepted theory is that it is a disorder of keratinocyte maturation and differentiation.1,3 Histological analysis, electron microscopy (EM), and immunohistochemical studies support this theory. Hyperkeratosis can be seen on histological examination. On EM, the stratum granulosum contains more lamellar granules and a larger transition cell layer, and immunochemical analysis reveals an increased expression of genetic markers associated with keratinization.1
The etiology of this keratinization abnormality is poorly understood.
CRP has been reported in several family members, prompting suggestions by some of a possible genetic component.4 Others have proposed staphylococcal enterotoxin B may promote certain immune factors causing abnormal keratinization.2
Is it CRP or tinea versicolor?
The differential for CRP includes:
- tinea or pityriasis versicolor
- tinea corporis
- seborrheic dermatitis
- keratosis follicularis (Darier disease)
- acanthosis nigricans
- macular amyloidosis.
Of all of these, however, CRP is most likely to be confused with tinea versicolor, as both are associated with hyper- or hypopigmented papules and plaques on the chest and back, as well as mild pruritus. In CRP, however, a Woods lamp and KOH will be negative, while with tinea versicolor, KOH will be positive and the Woods lamp may reveal yellow-green fluorescence.
If a patient’s KOH is negative and/ or the patient does not respond to treatment for tinea versicolor (which includes topical or oral antifungals), a trial of oral antibiotics for CRP may be reasonable. Response to an oral antibiotic, such as minocycline, will help to confirm the diagnosis. Although most patients with CRP do not have an endocrine disorder, it’s a good idea to keep this reported association in mind, and perform further testing, as needed.
Oral minocycline is the treatment of choice
The preferred treatment for CRP is oral minocycline (100 mg orally twice a day for 6 weeks).1,2 Oral azithromycin, erythromycin, clarithromycin, tetracycline, cefdinir,3 roxithromycin,5 doxycycline,2 and amoxicillin6 have also been used. Isotretinoin is an effective alternative to oral antibiotics, but clinicians often avoid it because of the adverse side effect profile.
With oral antibiotic therapy, the patient may completely clear and stay clear, or go on to have multiple recurrences or exacerbations. Topical retinoids have also been used with some success,4 but most reported cases have been successfully treated with oral antibiotics.1
Our patient required Tx for several months
We initially treated our patient with doxycycline (100 mg orally twice a day) for 1 month. The lesions cleared after 2 weeks and then recurred during week 4 of treatment. We discontinued the doxycycline, and started the patient on minocycline. The primary lesions resolved after 5 weeks of minocycline, though we noted residual post-inflammatory hyper-pigmentation (FIGURE 2). Our patient continued the medication for an additional 8 weeks. He was lost to follow-up.
FIGURE 2
Weeks later, hyperpigmentation remains
Correspondence
Kendall Lane, MD, LCDR, MC, United States Navy, Navy Medical Center San Diego, 34520 Bob Wilson Drive, Suite 300, San Diego, CA 92134
A 20-year-old black man came into our medical center with a mildly pruritic scaly rash affecting his neck and upper body for 2 weeks. Physical exam revealed well-demarcated, hyperpigmented hyperkeratotic papules coalescing to form large plaques on his central chest, back, and shoulders. He had a reticulated pattern on his shoulders and arms (FIGURE 1). His face, intertriginous skin, genitals, mucous membranes, and lower extremities were spared. The remainder of the physical exam was unremarkable.
Woods lamp and potassium hydroxide (KOH) preparation were negative. Labs, including fasting blood glucose and thyroid function test, were normal. Our patient denied any recent travel, fever, night sweats, or weight loss. He noted only that he used the weight benches at the gym. His medical and family histories were unremarkable, and he was not taking any medications or supplements.
FIGURE 1
Plaques on chest, reticulated pattern on arms
What is your diagnosis?
How would you manage this condition?
Dx: Confluent and reticulated papillomatosis
Confluent and reticulated papillomatosis of Gougerot and Carteaud (CRP) is a rare skin disorder characterized by benign blue-gray or brown hyperpigmented hyperkeratotic papules and plaques. The lesions initially occur on the trunk or central chest as 1- to 2-mm warty papules that become confluent to form plaques, spreading to the neck, abdomen, and upper extremities. Peripherally, the lesions are distributed in a reticular pattern. Although less common, CRP may be isolated to one part of the body, including the face and genitals; the mucous membranes are swpared.
With the exception of Japan, where a male predominance is seen, young women are more commonly affected.1 Patients are typically asymptomatic, or complain of mild pruritus and cosmetic concerns.
A disorder of keratinocyte maturation and differentiation?
The cause of CRP is unknown. Several theories have been entertained: an underlying endocrine disorder, a rare form of cutaneous amyloidosis, reaction to bacteria or fungus on the skin, and a keratinization abnormality.1 Most patients with CRP do not have an underlying endocrine disorder or any evidence of amyloidosis, making these theories less likely. KOH preparation for fungal elements is typically negative and patients do not respond to antifungal therapy.
A bacterial cause has been implicated because CRP responds to antibiotics, but many have argued that antibiotics are acting as an anti-inflammatory agent rather than an antibacterial medication.1 Others have suggested antibiotics may be acting to decrease epidermal proliferation by blocking protein and DNA synthesis and reducing keratinocyte production of cytokines.2
Overall, the most accepted theory is that it is a disorder of keratinocyte maturation and differentiation.1,3 Histological analysis, electron microscopy (EM), and immunohistochemical studies support this theory. Hyperkeratosis can be seen on histological examination. On EM, the stratum granulosum contains more lamellar granules and a larger transition cell layer, and immunochemical analysis reveals an increased expression of genetic markers associated with keratinization.1
The etiology of this keratinization abnormality is poorly understood.
CRP has been reported in several family members, prompting suggestions by some of a possible genetic component.4 Others have proposed staphylococcal enterotoxin B may promote certain immune factors causing abnormal keratinization.2
Is it CRP or tinea versicolor?
The differential for CRP includes:
- tinea or pityriasis versicolor
- tinea corporis
- seborrheic dermatitis
- keratosis follicularis (Darier disease)
- acanthosis nigricans
- macular amyloidosis.
Of all of these, however, CRP is most likely to be confused with tinea versicolor, as both are associated with hyper- or hypopigmented papules and plaques on the chest and back, as well as mild pruritus. In CRP, however, a Woods lamp and KOH will be negative, while with tinea versicolor, KOH will be positive and the Woods lamp may reveal yellow-green fluorescence.
If a patient’s KOH is negative and/ or the patient does not respond to treatment for tinea versicolor (which includes topical or oral antifungals), a trial of oral antibiotics for CRP may be reasonable. Response to an oral antibiotic, such as minocycline, will help to confirm the diagnosis. Although most patients with CRP do not have an endocrine disorder, it’s a good idea to keep this reported association in mind, and perform further testing, as needed.
Oral minocycline is the treatment of choice
The preferred treatment for CRP is oral minocycline (100 mg orally twice a day for 6 weeks).1,2 Oral azithromycin, erythromycin, clarithromycin, tetracycline, cefdinir,3 roxithromycin,5 doxycycline,2 and amoxicillin6 have also been used. Isotretinoin is an effective alternative to oral antibiotics, but clinicians often avoid it because of the adverse side effect profile.
With oral antibiotic therapy, the patient may completely clear and stay clear, or go on to have multiple recurrences or exacerbations. Topical retinoids have also been used with some success,4 but most reported cases have been successfully treated with oral antibiotics.1
Our patient required Tx for several months
We initially treated our patient with doxycycline (100 mg orally twice a day) for 1 month. The lesions cleared after 2 weeks and then recurred during week 4 of treatment. We discontinued the doxycycline, and started the patient on minocycline. The primary lesions resolved after 5 weeks of minocycline, though we noted residual post-inflammatory hyper-pigmentation (FIGURE 2). Our patient continued the medication for an additional 8 weeks. He was lost to follow-up.
FIGURE 2
Weeks later, hyperpigmentation remains
Correspondence
Kendall Lane, MD, LCDR, MC, United States Navy, Navy Medical Center San Diego, 34520 Bob Wilson Drive, Suite 300, San Diego, CA 92134
1. Schwartz R. Confluent and reticulated papillomatosis. Available at: http://www.emedicine.com/derm/topic82.htm. Accessed March 25, 2009.
2. Greenblatt D, Cintra M, Teixiera F, et al. Hyperpigmented plaques on a young man. J Am Acad Dermatol. 2007;56:896-898.
3. Scheinfeld N. Confluent and reticulated papillomatosis: a review of the literature. Am J Clin Dermatol. 2006;7:305-313.
4. Schwartzberg JB, Schwartzberg HA. Response of confluent and reticulate papillomatosis of Gougerot and Carteaud to topical tretinoin. Cutis. 2000;66:291-293.
5. Ito S, Hatamochi A, Yamazaki S. A case of confluent and reticulated papillomatosis that successfully responded to roxithromycin. J Dermatol. 2006;33:71-72.
6. Davis RF, Harman KE. Confluent and reticulated papillomatosis successfully treated with amoxicillin. Br J Dermatol. 2007;156:583-584.
1. Schwartz R. Confluent and reticulated papillomatosis. Available at: http://www.emedicine.com/derm/topic82.htm. Accessed March 25, 2009.
2. Greenblatt D, Cintra M, Teixiera F, et al. Hyperpigmented plaques on a young man. J Am Acad Dermatol. 2007;56:896-898.
3. Scheinfeld N. Confluent and reticulated papillomatosis: a review of the literature. Am J Clin Dermatol. 2006;7:305-313.
4. Schwartzberg JB, Schwartzberg HA. Response of confluent and reticulate papillomatosis of Gougerot and Carteaud to topical tretinoin. Cutis. 2000;66:291-293.
5. Ito S, Hatamochi A, Yamazaki S. A case of confluent and reticulated papillomatosis that successfully responded to roxithromycin. J Dermatol. 2006;33:71-72.
6. Davis RF, Harman KE. Confluent and reticulated papillomatosis successfully treated with amoxicillin. Br J Dermatol. 2007;156:583-584.
Warty papule and scaling around finger
A 35-year-old Caucasian man was referred to our clinic for treatment of a nonhealing “wart-like” growth on his left index finger. He said that the lesion had been there for at least 2 years and complained of extensive periungual erythema and scaling of the same finger. The patient was immunocompetent and denied trauma, chronic nail infection, arsenic exposure, or radiation to this particular finger. On several occasions, liquid nitrogen cryotherapy had been used on the growth, without improvement.
Physical examination revealed a well-demarcated erythematous patch with scaling involving the medial and proximal periungual areas of the left index finger (FIGURE). There was also a distinct, rough-surfaced 5×4 mm papule with scales, crusts, and small fissures in the middle of the patch. In addition, there was onychodystrophy with keratotic debris on the medial aspect of the same finger. Our attempt to scrape this papule failed and was painful for the patient.
The other nails were normal. Review of the patient’s systems, family history, and personal history were otherwise unremarkable. We biopsied the lesion.
FIGURE
Erythematous patch and rough-surfaced papule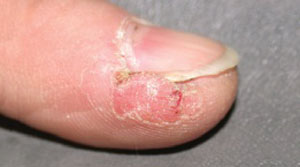
What is your diagnosis?
How would you manage this condition?
Dx: Bowen’s disease of the nail
Bowen’s disease (BD), a form of intraepidermal (in situ) squamous cell carcinoma (SCC), may affect the skin but also the nail unit. It presents as periungual or subungual verrucous plaques, erosions, and ulcerations, with nail discoloration, dystrophy, or onycholysis.
Our patient was younger than the norm. BD of the nail—which typically involves the fingers—occurs in both men and women, but is most common in men in their 50s. It often presents with verrucous, scaly, crusting, erythematous, or fissuring lesions that may involve any portion of the nail apparatus with associated onycholysis, nail dystrophy, or longitudinal erythronychia or melanonychia (red or black longitudinal nail streak).1-6 BD of the nail may involve more than one digit on multiple extremities, simultaneously or sequentially.5 Initial BD of the nail may spread to periungual or subungual areas or vice versa. Bleeding, ulceration, or a nodule may signal the development of invasive SCC.3,6
Radiation and HPV linked to BD of the nail
While the etiology of this condition is unclear, trauma, chronic paronychia, ionizing radiation, infectious agents, ultraviolet exposure, arsenic or pesticide exposure, and immunosuppression are a few proposed predisposing factors.1,3,4,6
Human papilloma virus (HPV) subtypes 16, 34, and 35 have been identified in BD and SCC lesions. These subtypes with oncogenic potential may play a role in the development of BD and invasive SCC of the nail and other cutaneous regions.
In cases where there has been HPV-16 infection in a digit and the genital region, researchers have suggested autoinoculation as a transmission mode from the anogenital area to the digit, or vice versa.6-8
The pathogenesis behind polydactylous BD of the nail has been linked to factors such as trauma, radiation, and immunosuppression (eg, post-transplant, oncological, and HIV patients).5,8
Differential includes onychomycosis, eczema
The differential for BD of the nail includes onychomycosis, paronychia, verruca vulgaris, eczema, pyogenic granuloma, glomus tumor, and verrucous tuberculosis. The differential also includes: subungual exostosis, onychomatricoma, amelanotic malignant melanoma, keratoacanthoma, fibrokeratoma, and gouty tophus.
Irregular borders and the presence of a scaly patch with papules should raise your suspicion of BD. Biopsy is necessary to confirm your suspicions. In fact, all chronic and recalcitrant lesions of the nail apparatus should be biopsied to rule out BD.9
Biopsy with care
The matrix is a germinating portion of the nail and requires special care, because damage to it may permanently affect nail formation and function. Proper anesthesia and hemostasis are also key, given that the nail apparatus is very vascular and well innervated.9
The histopathology of BD lesions is characterized by hyperkeratosis; parakeratosis; loss of orderly maturation, polarity, and a granular layer; and keratinocytic atypia involving the entire acanthotic epithelial layer. The atypia and dyskeratosis are confined to the epidermis. However, some microscopic specimens of BD may simultaneously demonstrate features of invasive SCC in other areas of the lesion. Microinvasion is common in long-term BD with reports of invasive carcinoma in approximately 15% of cases.4-6,9
Consider CO2 laser therapy, Mohs surgery
Various therapeutic modalities have been used for BD of the nail, including electrodesiccation and curettage, 5% fluorouracil cream (Efudex), cryosurgery, and radiotherapy. Most of these treatments have not been successful and are associated with high recurrence rates.10-12
Simple excision of nail bed and matrix is successful in small and localized BD lesions. CO2 laser therapy for periungual BD has been reported to have up to an 80% cure rate, with less scarring and contractures when compared with surgical excision.10 Recently, imiquimod (Aldara 5% cream) has been used with success for BD of the nail—especially recurrent disease.11
Mohs surgery is considered the best treatment approach, with cure rates of up to 96%.12 Mohs surgery allows for adequate depth of tumor resection, great preservation of normal digit function, and excellent cosmetic results, with healing by secondary intention. Although the 5-year recurrence rate after Mohs surgery is small (about 3%), it is still a good idea to follow the patient closely to assess for a potential relapse.
If invasive SCC is suspected, the patient’s regional lymph nodes should be evaluated for possible metastasis. Radiologic study of the digit(s) to assess for bone invasion should also be considered. Amputation of the affected digit, although a drastic measure, is an option if there is evidence of bone involvement.13
Our patient’s course. After discussing the different treatment options with our patient, we referred him to plastic surgery for wide excision. He was subsequently lost to follow up.
Correspondence
Amor Khachemoune, MD, CWS, Assistant Professor, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016; [email protected]
1. Fleckman P, Allan C. Surgical anatomy of the nail unit. Dermatol Surg. 2001;27:257-260.
2. Baran R, Perrin C. Longitudinal erythronychia with distal subungual keratosis: onychopapilloma of the nail bed and Bowen’s disease. Br J Dermatol. 2000;143:132-135.
3. Kaiser JF, Proctor-Shipman L. Squamous cell carcinoma in situ (Bowen’s disease) mimicking subungual verruca vulgaris. J Fam Pract. 1994;39:384-387.
4. Ongenae K, Kerckhove MV, Naeyaert J. Bowen’s disease of the nail. Dermatology. 2002;204:348-350.
5. Koch A, Schonlebe J, Haroske G, et al. Polydactylous Bowen’s disease. J Eur Acad Dermatol Venereol. 2003;17:213-215.
6. Ratner D. Recurrent squamous cell carcinoma in situ of the finger. SKINmed. 2003;2:251-252.
7. McHugh RW, Hazen P, Eliezri YD, et al. Metastatic periungual squamous cell carcinoma: detection of human papillomavirus type 35 RNA in the digital tumor and axillary lymph node metastases. J Am Acad Dermatol. 1996;34:1080-1082.
8. Moy RL, Eliezri YD, Nuovo GJ, et al. Human papilloma type 16 DNA in periungual squamous cell carcinomas. JAMA. 1989;261:2669-2673.
9. Rich P. Nail biopsy: indications and methods. Dermatol Surg. 2001;27:229-234.
10. Gordon KB, Garden JM, Robinson JK. Bowen’s disease of the distal digit. Outcome of treatment with carbon dioxide laser vaporization. Dermatol Surg. 1996;22:723-728.
11. Laffitte E, Saurat JH. Recurrent Bowen’s disease of the nail: treatment by topical imiquimod (Aldara). Ann Dermatol Venereol. 2003;130:211-213.
12. Goldminz D, Bennett RG. Mohs micrographic surgery of the nail unit. J Dermatol Surg Oncol. 1992;18:721-726.
13. Peterson SR, Layton EG, Joseph AK. Squamous cell carcinoma of the nail unit with evidence of bony involvement: a multidisciplinary approach to resection and reconstruction. Dermatol Surg. 2004;30:218-221.
A 35-year-old Caucasian man was referred to our clinic for treatment of a nonhealing “wart-like” growth on his left index finger. He said that the lesion had been there for at least 2 years and complained of extensive periungual erythema and scaling of the same finger. The patient was immunocompetent and denied trauma, chronic nail infection, arsenic exposure, or radiation to this particular finger. On several occasions, liquid nitrogen cryotherapy had been used on the growth, without improvement.
Physical examination revealed a well-demarcated erythematous patch with scaling involving the medial and proximal periungual areas of the left index finger (FIGURE). There was also a distinct, rough-surfaced 5×4 mm papule with scales, crusts, and small fissures in the middle of the patch. In addition, there was onychodystrophy with keratotic debris on the medial aspect of the same finger. Our attempt to scrape this papule failed and was painful for the patient.
The other nails were normal. Review of the patient’s systems, family history, and personal history were otherwise unremarkable. We biopsied the lesion.
FIGURE
Erythematous patch and rough-surfaced papule
What is your diagnosis?
How would you manage this condition?
Dx: Bowen’s disease of the nail
Bowen’s disease (BD), a form of intraepidermal (in situ) squamous cell carcinoma (SCC), may affect the skin but also the nail unit. It presents as periungual or subungual verrucous plaques, erosions, and ulcerations, with nail discoloration, dystrophy, or onycholysis.
Our patient was younger than the norm. BD of the nail—which typically involves the fingers—occurs in both men and women, but is most common in men in their 50s. It often presents with verrucous, scaly, crusting, erythematous, or fissuring lesions that may involve any portion of the nail apparatus with associated onycholysis, nail dystrophy, or longitudinal erythronychia or melanonychia (red or black longitudinal nail streak).1-6 BD of the nail may involve more than one digit on multiple extremities, simultaneously or sequentially.5 Initial BD of the nail may spread to periungual or subungual areas or vice versa. Bleeding, ulceration, or a nodule may signal the development of invasive SCC.3,6
Radiation and HPV linked to BD of the nail
While the etiology of this condition is unclear, trauma, chronic paronychia, ionizing radiation, infectious agents, ultraviolet exposure, arsenic or pesticide exposure, and immunosuppression are a few proposed predisposing factors.1,3,4,6
Human papilloma virus (HPV) subtypes 16, 34, and 35 have been identified in BD and SCC lesions. These subtypes with oncogenic potential may play a role in the development of BD and invasive SCC of the nail and other cutaneous regions.
In cases where there has been HPV-16 infection in a digit and the genital region, researchers have suggested autoinoculation as a transmission mode from the anogenital area to the digit, or vice versa.6-8
The pathogenesis behind polydactylous BD of the nail has been linked to factors such as trauma, radiation, and immunosuppression (eg, post-transplant, oncological, and HIV patients).5,8
Differential includes onychomycosis, eczema
The differential for BD of the nail includes onychomycosis, paronychia, verruca vulgaris, eczema, pyogenic granuloma, glomus tumor, and verrucous tuberculosis. The differential also includes: subungual exostosis, onychomatricoma, amelanotic malignant melanoma, keratoacanthoma, fibrokeratoma, and gouty tophus.
Irregular borders and the presence of a scaly patch with papules should raise your suspicion of BD. Biopsy is necessary to confirm your suspicions. In fact, all chronic and recalcitrant lesions of the nail apparatus should be biopsied to rule out BD.9
Biopsy with care
The matrix is a germinating portion of the nail and requires special care, because damage to it may permanently affect nail formation and function. Proper anesthesia and hemostasis are also key, given that the nail apparatus is very vascular and well innervated.9
The histopathology of BD lesions is characterized by hyperkeratosis; parakeratosis; loss of orderly maturation, polarity, and a granular layer; and keratinocytic atypia involving the entire acanthotic epithelial layer. The atypia and dyskeratosis are confined to the epidermis. However, some microscopic specimens of BD may simultaneously demonstrate features of invasive SCC in other areas of the lesion. Microinvasion is common in long-term BD with reports of invasive carcinoma in approximately 15% of cases.4-6,9
Consider CO2 laser therapy, Mohs surgery
Various therapeutic modalities have been used for BD of the nail, including electrodesiccation and curettage, 5% fluorouracil cream (Efudex), cryosurgery, and radiotherapy. Most of these treatments have not been successful and are associated with high recurrence rates.10-12
Simple excision of nail bed and matrix is successful in small and localized BD lesions. CO2 laser therapy for periungual BD has been reported to have up to an 80% cure rate, with less scarring and contractures when compared with surgical excision.10 Recently, imiquimod (Aldara 5% cream) has been used with success for BD of the nail—especially recurrent disease.11
Mohs surgery is considered the best treatment approach, with cure rates of up to 96%.12 Mohs surgery allows for adequate depth of tumor resection, great preservation of normal digit function, and excellent cosmetic results, with healing by secondary intention. Although the 5-year recurrence rate after Mohs surgery is small (about 3%), it is still a good idea to follow the patient closely to assess for a potential relapse.
If invasive SCC is suspected, the patient’s regional lymph nodes should be evaluated for possible metastasis. Radiologic study of the digit(s) to assess for bone invasion should also be considered. Amputation of the affected digit, although a drastic measure, is an option if there is evidence of bone involvement.13
Our patient’s course. After discussing the different treatment options with our patient, we referred him to plastic surgery for wide excision. He was subsequently lost to follow up.
Correspondence
Amor Khachemoune, MD, CWS, Assistant Professor, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016; [email protected]
A 35-year-old Caucasian man was referred to our clinic for treatment of a nonhealing “wart-like” growth on his left index finger. He said that the lesion had been there for at least 2 years and complained of extensive periungual erythema and scaling of the same finger. The patient was immunocompetent and denied trauma, chronic nail infection, arsenic exposure, or radiation to this particular finger. On several occasions, liquid nitrogen cryotherapy had been used on the growth, without improvement.
Physical examination revealed a well-demarcated erythematous patch with scaling involving the medial and proximal periungual areas of the left index finger (FIGURE). There was also a distinct, rough-surfaced 5×4 mm papule with scales, crusts, and small fissures in the middle of the patch. In addition, there was onychodystrophy with keratotic debris on the medial aspect of the same finger. Our attempt to scrape this papule failed and was painful for the patient.
The other nails were normal. Review of the patient’s systems, family history, and personal history were otherwise unremarkable. We biopsied the lesion.
FIGURE
Erythematous patch and rough-surfaced papule
What is your diagnosis?
How would you manage this condition?
Dx: Bowen’s disease of the nail
Bowen’s disease (BD), a form of intraepidermal (in situ) squamous cell carcinoma (SCC), may affect the skin but also the nail unit. It presents as periungual or subungual verrucous plaques, erosions, and ulcerations, with nail discoloration, dystrophy, or onycholysis.
Our patient was younger than the norm. BD of the nail—which typically involves the fingers—occurs in both men and women, but is most common in men in their 50s. It often presents with verrucous, scaly, crusting, erythematous, or fissuring lesions that may involve any portion of the nail apparatus with associated onycholysis, nail dystrophy, or longitudinal erythronychia or melanonychia (red or black longitudinal nail streak).1-6 BD of the nail may involve more than one digit on multiple extremities, simultaneously or sequentially.5 Initial BD of the nail may spread to periungual or subungual areas or vice versa. Bleeding, ulceration, or a nodule may signal the development of invasive SCC.3,6
Radiation and HPV linked to BD of the nail
While the etiology of this condition is unclear, trauma, chronic paronychia, ionizing radiation, infectious agents, ultraviolet exposure, arsenic or pesticide exposure, and immunosuppression are a few proposed predisposing factors.1,3,4,6
Human papilloma virus (HPV) subtypes 16, 34, and 35 have been identified in BD and SCC lesions. These subtypes with oncogenic potential may play a role in the development of BD and invasive SCC of the nail and other cutaneous regions.
In cases where there has been HPV-16 infection in a digit and the genital region, researchers have suggested autoinoculation as a transmission mode from the anogenital area to the digit, or vice versa.6-8
The pathogenesis behind polydactylous BD of the nail has been linked to factors such as trauma, radiation, and immunosuppression (eg, post-transplant, oncological, and HIV patients).5,8
Differential includes onychomycosis, eczema
The differential for BD of the nail includes onychomycosis, paronychia, verruca vulgaris, eczema, pyogenic granuloma, glomus tumor, and verrucous tuberculosis. The differential also includes: subungual exostosis, onychomatricoma, amelanotic malignant melanoma, keratoacanthoma, fibrokeratoma, and gouty tophus.
Irregular borders and the presence of a scaly patch with papules should raise your suspicion of BD. Biopsy is necessary to confirm your suspicions. In fact, all chronic and recalcitrant lesions of the nail apparatus should be biopsied to rule out BD.9
Biopsy with care
The matrix is a germinating portion of the nail and requires special care, because damage to it may permanently affect nail formation and function. Proper anesthesia and hemostasis are also key, given that the nail apparatus is very vascular and well innervated.9
The histopathology of BD lesions is characterized by hyperkeratosis; parakeratosis; loss of orderly maturation, polarity, and a granular layer; and keratinocytic atypia involving the entire acanthotic epithelial layer. The atypia and dyskeratosis are confined to the epidermis. However, some microscopic specimens of BD may simultaneously demonstrate features of invasive SCC in other areas of the lesion. Microinvasion is common in long-term BD with reports of invasive carcinoma in approximately 15% of cases.4-6,9
Consider CO2 laser therapy, Mohs surgery
Various therapeutic modalities have been used for BD of the nail, including electrodesiccation and curettage, 5% fluorouracil cream (Efudex), cryosurgery, and radiotherapy. Most of these treatments have not been successful and are associated with high recurrence rates.10-12
Simple excision of nail bed and matrix is successful in small and localized BD lesions. CO2 laser therapy for periungual BD has been reported to have up to an 80% cure rate, with less scarring and contractures when compared with surgical excision.10 Recently, imiquimod (Aldara 5% cream) has been used with success for BD of the nail—especially recurrent disease.11
Mohs surgery is considered the best treatment approach, with cure rates of up to 96%.12 Mohs surgery allows for adequate depth of tumor resection, great preservation of normal digit function, and excellent cosmetic results, with healing by secondary intention. Although the 5-year recurrence rate after Mohs surgery is small (about 3%), it is still a good idea to follow the patient closely to assess for a potential relapse.
If invasive SCC is suspected, the patient’s regional lymph nodes should be evaluated for possible metastasis. Radiologic study of the digit(s) to assess for bone invasion should also be considered. Amputation of the affected digit, although a drastic measure, is an option if there is evidence of bone involvement.13
Our patient’s course. After discussing the different treatment options with our patient, we referred him to plastic surgery for wide excision. He was subsequently lost to follow up.
Correspondence
Amor Khachemoune, MD, CWS, Assistant Professor, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016; [email protected]
1. Fleckman P, Allan C. Surgical anatomy of the nail unit. Dermatol Surg. 2001;27:257-260.
2. Baran R, Perrin C. Longitudinal erythronychia with distal subungual keratosis: onychopapilloma of the nail bed and Bowen’s disease. Br J Dermatol. 2000;143:132-135.
3. Kaiser JF, Proctor-Shipman L. Squamous cell carcinoma in situ (Bowen’s disease) mimicking subungual verruca vulgaris. J Fam Pract. 1994;39:384-387.
4. Ongenae K, Kerckhove MV, Naeyaert J. Bowen’s disease of the nail. Dermatology. 2002;204:348-350.
5. Koch A, Schonlebe J, Haroske G, et al. Polydactylous Bowen’s disease. J Eur Acad Dermatol Venereol. 2003;17:213-215.
6. Ratner D. Recurrent squamous cell carcinoma in situ of the finger. SKINmed. 2003;2:251-252.
7. McHugh RW, Hazen P, Eliezri YD, et al. Metastatic periungual squamous cell carcinoma: detection of human papillomavirus type 35 RNA in the digital tumor and axillary lymph node metastases. J Am Acad Dermatol. 1996;34:1080-1082.
8. Moy RL, Eliezri YD, Nuovo GJ, et al. Human papilloma type 16 DNA in periungual squamous cell carcinomas. JAMA. 1989;261:2669-2673.
9. Rich P. Nail biopsy: indications and methods. Dermatol Surg. 2001;27:229-234.
10. Gordon KB, Garden JM, Robinson JK. Bowen’s disease of the distal digit. Outcome of treatment with carbon dioxide laser vaporization. Dermatol Surg. 1996;22:723-728.
11. Laffitte E, Saurat JH. Recurrent Bowen’s disease of the nail: treatment by topical imiquimod (Aldara). Ann Dermatol Venereol. 2003;130:211-213.
12. Goldminz D, Bennett RG. Mohs micrographic surgery of the nail unit. J Dermatol Surg Oncol. 1992;18:721-726.
13. Peterson SR, Layton EG, Joseph AK. Squamous cell carcinoma of the nail unit with evidence of bony involvement: a multidisciplinary approach to resection and reconstruction. Dermatol Surg. 2004;30:218-221.
1. Fleckman P, Allan C. Surgical anatomy of the nail unit. Dermatol Surg. 2001;27:257-260.
2. Baran R, Perrin C. Longitudinal erythronychia with distal subungual keratosis: onychopapilloma of the nail bed and Bowen’s disease. Br J Dermatol. 2000;143:132-135.
3. Kaiser JF, Proctor-Shipman L. Squamous cell carcinoma in situ (Bowen’s disease) mimicking subungual verruca vulgaris. J Fam Pract. 1994;39:384-387.
4. Ongenae K, Kerckhove MV, Naeyaert J. Bowen’s disease of the nail. Dermatology. 2002;204:348-350.
5. Koch A, Schonlebe J, Haroske G, et al. Polydactylous Bowen’s disease. J Eur Acad Dermatol Venereol. 2003;17:213-215.
6. Ratner D. Recurrent squamous cell carcinoma in situ of the finger. SKINmed. 2003;2:251-252.
7. McHugh RW, Hazen P, Eliezri YD, et al. Metastatic periungual squamous cell carcinoma: detection of human papillomavirus type 35 RNA in the digital tumor and axillary lymph node metastases. J Am Acad Dermatol. 1996;34:1080-1082.
8. Moy RL, Eliezri YD, Nuovo GJ, et al. Human papilloma type 16 DNA in periungual squamous cell carcinomas. JAMA. 1989;261:2669-2673.
9. Rich P. Nail biopsy: indications and methods. Dermatol Surg. 2001;27:229-234.
10. Gordon KB, Garden JM, Robinson JK. Bowen’s disease of the distal digit. Outcome of treatment with carbon dioxide laser vaporization. Dermatol Surg. 1996;22:723-728.
11. Laffitte E, Saurat JH. Recurrent Bowen’s disease of the nail: treatment by topical imiquimod (Aldara). Ann Dermatol Venereol. 2003;130:211-213.
12. Goldminz D, Bennett RG. Mohs micrographic surgery of the nail unit. J Dermatol Surg Oncol. 1992;18:721-726.
13. Peterson SR, Layton EG, Joseph AK. Squamous cell carcinoma of the nail unit with evidence of bony involvement: a multidisciplinary approach to resection and reconstruction. Dermatol Surg. 2004;30:218-221.
Bilateral leg edema and difficulty swallowing
A 70-year-old man came to our medical center for treatment of painful bilateral leg swelling that had gotten progressively worse over the past week. He had no significant past medical or surgical history, took an aspirin daily, did not smoke tobacco or drink alcohol, and had not taken any trips recently. He denied any chest pain, dyspnea, or orthopnea, but indicated that he’d been having difficulty swallowing food for the past month.
The patient had a cachectic appearance, diminished breath sounds and dullness to percussion over the right middle and lower lung fields, and pitting edema up to the knees bilaterally.
Lab studies showed a normal brain natriuretic peptide of 16 pg/mL, normocytic anemia (hemoglobin, 9.5 g/dL; hematocrit, 29.6%; and a mean corpuscular volume of 82.6 fL), potassium of 2.4 mEq/L, calcium of 5.4 mg/dL, and albumin of 1.4 g/dL. An electrocardiogram (EKG) revealed a bifascicular heart block, which was not new based on older EKGs. A plain chest radiograph was performed (FIGURES 1A AND 1B).
FIGURE 1
Chest x-rays reveal large mass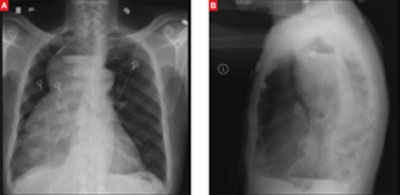
What is your diagnosis?
Diagnosis: Achalasia
Achalasia is characterized by an incomplete relaxation of the lower esophageal sphincter (LES) accompanied by aperistalsis of the esophageal body. The etiology of primary achalasia is unknown, but involves the degeneration of inhibitory neurons within the myenteric plexus responsible for LES relaxation and esophageal peristalsis.
Many potential causes. Infectious, inflammatory, autoimmune, and genetic causes have all been proposed. Achalasia has a prevalence of less than 1 in 10,000; its incidence is 0.3 to 1 per 100,000 per year, with peaks in the third and seventh decades of life. Men and women are affected equally.1
Conversely, pseudoachalasia—or secondary achalasia—has a known cause that either destroys the neurons of LES relaxation or has a mass effect that limits LES relaxation. Its most common cause is malignancy involving the gastroesophageal junction. Other causes include Chagas disease, an infection by the parasite Trypanosoma cruzi that affects the myenteric plexus, and amyloidosis.2
A disorder that goes undiagnosed for years
In primary achalasia symptom onset is insidious, with the disorder typically going undiagnosed for several years. Patients experience a gradual dysphagia for solids, progressing to liquids. Over time, patients adopt particular behaviors to aid the transit of food boluses down the esophagus and through the contracted LES, including eating slowly, stretching, moving side-to-side, or walking after meals.1 Regurgitation of food, chest pain, weight loss, heartburn, and difficulty belching are also common complaints.3 A more rapid onset and progression of symptoms (less than 6 months) is suggestive of pseudoachalasia and cancer.
Left untreated, primary achalasia leads to a progressively dilating esophagus with increased risk of aspiration, perforation, malnutrition, weight loss, and esophageal cancer.3 This case is remarkable for how far the patient’s disease progressed before he presented with signs and symptoms more indicative of secondary malnutrition than the primary disease.
Differential Dx includes hiatal hernia
The differential diagnosis for achalasia includes hiatal hernia, right middle lobe pneumonia, empyema, and lung abscess. Most patients with hiatal hernias are asymptomatic, and the diagnosis is made incidentally on chest x-ray as a retrocardiac air-fluid level or on upper gastrointestinal studies. Right middle lobe pneumonia would appear on chest x-ray as a well-demarcated opacity within the confines of that lobe, but would not have an air-fluid level. Empyema and lung abscess are complications of pneumonia, and patients present with persistent fever, cough, dyspnea, and malaise. With an empyema, chest x-ray reveals a pleural effusion. A lung abscess is an intrapulmonary cavitary lesion with an air-fluid level, most commonly due to aspiration pneumonia.
This patient’s chest x-ray showed a mass outside the lung tissue and pleural space that had an air-fluid level, was within the mediastinum, and displaced the trachea anteriorly, all suggestive of a dilated esophagus filled with undigested food unable to pass the LES.
Esophageal manometry clinches the diagnosis
Esophageal manometry is the gold standard for diagnosis. In achalasia, manometry shows poor peristalsis of the esophageal body and a constricted LES that does not relax sufficiently with swallowing.3 Manometry cannot, however, reliably distinguish primary achalasia from pseudoachalasia.
Barium swallow studies and endoscopy can assist in the diagnosis and help rule out pseudoachalasia. Timed barium studies can show a lack of peristalsis, an air-fluid level at the top of a barium column retained within the dilated esophagus, and the narrowing of the distal esophagus into the pathognomonic “bird’s beak” of primary achalasia.4 Endoscopy can visualize a mass beyond the LES, but cannot adequately assess esophageal peristalsis or LES relaxation.1
In our patient’s initial workup, a CT (FIGURE 2), barium study (FIGURE 3), and endoscopy were all used to rule out an obstructive lesion.
FIGURE 2
Massively distended esophagus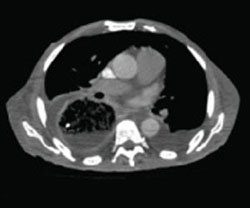
FIGURE 3
The “bird’s beak” of achalasia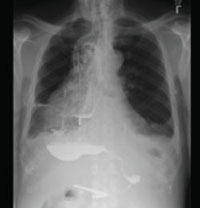
Treatment targets the constricted LES
The goal of treatment in achalasia is to help food move through the constricted LES. Pharmacologic therapy, used to decrease LES resting tone, has limited benefit and becomes less effective as the disease progresses. Calcium channel blockers, particularly nifedipine, and nitrates are the most commonly used agents.5
Pneumatic dilation is the most effective nonsurgical treatment of achalasia. It involves the controlled rupture of LES muscle fibers to dilate the sphincter and allow gravity to move food from the esophagus into the stomach.6 Symptom improvement lasts for 60% of patients at 1 year, 50% at 5 years, and 36% at 10 years. Repeated dilations are possible and sometimes necessary, but have diminishing success rates. The most worrisome complication of pneumatic dilation is esophageal rupture, which occurs 2% of the time.5
Botulinum toxin injection is another accepted nonsurgical therapy and is recommended for patients who would like to avoid more invasive treatment. When injected into the LES, the toxin causes temporary atrophy of the neurons responsible for LES contraction. After the procedure, 70% of patients report improvement in symptoms, with half in remission at 6 months and one-third at 1 year. Patients may require repeat injections every 3 to 4 months, but the toxin’s effectiveness diminishes with each injection.6
The laparoscopic Heller myotomy involves cutting the LES muscle, making it incompetent and allowing food boluses to pass through the LES. Ninety percent of patients experience symptom relief after surgery,5 with 10-year remission rates of 67% to 85%.1 Comparisons to pneumatic dilation do not show significant differences in outcomes over time, although most studies favor myotomy over dilation.
Surgical treatment is indicated for patients who are younger than 40 years of age, those who have failed repeated nonsurgical therapies, and those at high risk of esophageal perforation.5
Patient’s course highlights Tx risks
Due to the severity of the patient’s achalasia, a Heller myotomy was performed. During the postoperative period he had several complications. He ultimately died from an uncontrollable hemorrhage after an esophageal perforation, highlighting the seriousness of this disease and the risks inherent to its treatment.
Correspondence
Drew C. Baird, MD, Department of Family and Community Medicine, D.D. Eisenhower Army Medical Center, 300 Hospital Road, Fort Gordon, GA 30905.
1. Pohl D, Tutuian R. Achalasia: an overview of diagnosis and treatment. J Gastrointestin Liver Dis. 2007;16:297-303.
2. Spechler SJ, Castell DO. Classification of oesophageal motility abnormalities. Gut. 2001;49:145-151.
3. Farrokhi F, Vaezi MF. Idiopathic (primary) achalasia. Orphanet J Rare Dis. 2007;2:38.-
4. Levine MS, Rubesin SE. Diseases of the esophagus: diagnosis with esophagography. Radiology. 2005;237:414-427.
5. Roberts KE, Duffy AJ, Bell RL. Controversies in the treatment of gastroesophageal reflux and achalasia. World J Gastroenterol. 2006;12:3155-3161.
6. Annese V, Bassotti G. Non-surgical treatment of esophageal achalasia. World J Gastroenterol. 2006;12:5763-5766.
A 70-year-old man came to our medical center for treatment of painful bilateral leg swelling that had gotten progressively worse over the past week. He had no significant past medical or surgical history, took an aspirin daily, did not smoke tobacco or drink alcohol, and had not taken any trips recently. He denied any chest pain, dyspnea, or orthopnea, but indicated that he’d been having difficulty swallowing food for the past month.
The patient had a cachectic appearance, diminished breath sounds and dullness to percussion over the right middle and lower lung fields, and pitting edema up to the knees bilaterally.
Lab studies showed a normal brain natriuretic peptide of 16 pg/mL, normocytic anemia (hemoglobin, 9.5 g/dL; hematocrit, 29.6%; and a mean corpuscular volume of 82.6 fL), potassium of 2.4 mEq/L, calcium of 5.4 mg/dL, and albumin of 1.4 g/dL. An electrocardiogram (EKG) revealed a bifascicular heart block, which was not new based on older EKGs. A plain chest radiograph was performed (FIGURES 1A AND 1B).
FIGURE 1
Chest x-rays reveal large mass
What is your diagnosis?
Diagnosis: Achalasia
Achalasia is characterized by an incomplete relaxation of the lower esophageal sphincter (LES) accompanied by aperistalsis of the esophageal body. The etiology of primary achalasia is unknown, but involves the degeneration of inhibitory neurons within the myenteric plexus responsible for LES relaxation and esophageal peristalsis.
Many potential causes. Infectious, inflammatory, autoimmune, and genetic causes have all been proposed. Achalasia has a prevalence of less than 1 in 10,000; its incidence is 0.3 to 1 per 100,000 per year, with peaks in the third and seventh decades of life. Men and women are affected equally.1
Conversely, pseudoachalasia—or secondary achalasia—has a known cause that either destroys the neurons of LES relaxation or has a mass effect that limits LES relaxation. Its most common cause is malignancy involving the gastroesophageal junction. Other causes include Chagas disease, an infection by the parasite Trypanosoma cruzi that affects the myenteric plexus, and amyloidosis.2
A disorder that goes undiagnosed for years
In primary achalasia symptom onset is insidious, with the disorder typically going undiagnosed for several years. Patients experience a gradual dysphagia for solids, progressing to liquids. Over time, patients adopt particular behaviors to aid the transit of food boluses down the esophagus and through the contracted LES, including eating slowly, stretching, moving side-to-side, or walking after meals.1 Regurgitation of food, chest pain, weight loss, heartburn, and difficulty belching are also common complaints.3 A more rapid onset and progression of symptoms (less than 6 months) is suggestive of pseudoachalasia and cancer.
Left untreated, primary achalasia leads to a progressively dilating esophagus with increased risk of aspiration, perforation, malnutrition, weight loss, and esophageal cancer.3 This case is remarkable for how far the patient’s disease progressed before he presented with signs and symptoms more indicative of secondary malnutrition than the primary disease.
Differential Dx includes hiatal hernia
The differential diagnosis for achalasia includes hiatal hernia, right middle lobe pneumonia, empyema, and lung abscess. Most patients with hiatal hernias are asymptomatic, and the diagnosis is made incidentally on chest x-ray as a retrocardiac air-fluid level or on upper gastrointestinal studies. Right middle lobe pneumonia would appear on chest x-ray as a well-demarcated opacity within the confines of that lobe, but would not have an air-fluid level. Empyema and lung abscess are complications of pneumonia, and patients present with persistent fever, cough, dyspnea, and malaise. With an empyema, chest x-ray reveals a pleural effusion. A lung abscess is an intrapulmonary cavitary lesion with an air-fluid level, most commonly due to aspiration pneumonia.
This patient’s chest x-ray showed a mass outside the lung tissue and pleural space that had an air-fluid level, was within the mediastinum, and displaced the trachea anteriorly, all suggestive of a dilated esophagus filled with undigested food unable to pass the LES.
Esophageal manometry clinches the diagnosis
Esophageal manometry is the gold standard for diagnosis. In achalasia, manometry shows poor peristalsis of the esophageal body and a constricted LES that does not relax sufficiently with swallowing.3 Manometry cannot, however, reliably distinguish primary achalasia from pseudoachalasia.
Barium swallow studies and endoscopy can assist in the diagnosis and help rule out pseudoachalasia. Timed barium studies can show a lack of peristalsis, an air-fluid level at the top of a barium column retained within the dilated esophagus, and the narrowing of the distal esophagus into the pathognomonic “bird’s beak” of primary achalasia.4 Endoscopy can visualize a mass beyond the LES, but cannot adequately assess esophageal peristalsis or LES relaxation.1
In our patient’s initial workup, a CT (FIGURE 2), barium study (FIGURE 3), and endoscopy were all used to rule out an obstructive lesion.
FIGURE 2
Massively distended esophagus
FIGURE 3
The “bird’s beak” of achalasia
Treatment targets the constricted LES
The goal of treatment in achalasia is to help food move through the constricted LES. Pharmacologic therapy, used to decrease LES resting tone, has limited benefit and becomes less effective as the disease progresses. Calcium channel blockers, particularly nifedipine, and nitrates are the most commonly used agents.5
Pneumatic dilation is the most effective nonsurgical treatment of achalasia. It involves the controlled rupture of LES muscle fibers to dilate the sphincter and allow gravity to move food from the esophagus into the stomach.6 Symptom improvement lasts for 60% of patients at 1 year, 50% at 5 years, and 36% at 10 years. Repeated dilations are possible and sometimes necessary, but have diminishing success rates. The most worrisome complication of pneumatic dilation is esophageal rupture, which occurs 2% of the time.5
Botulinum toxin injection is another accepted nonsurgical therapy and is recommended for patients who would like to avoid more invasive treatment. When injected into the LES, the toxin causes temporary atrophy of the neurons responsible for LES contraction. After the procedure, 70% of patients report improvement in symptoms, with half in remission at 6 months and one-third at 1 year. Patients may require repeat injections every 3 to 4 months, but the toxin’s effectiveness diminishes with each injection.6
The laparoscopic Heller myotomy involves cutting the LES muscle, making it incompetent and allowing food boluses to pass through the LES. Ninety percent of patients experience symptom relief after surgery,5 with 10-year remission rates of 67% to 85%.1 Comparisons to pneumatic dilation do not show significant differences in outcomes over time, although most studies favor myotomy over dilation.
Surgical treatment is indicated for patients who are younger than 40 years of age, those who have failed repeated nonsurgical therapies, and those at high risk of esophageal perforation.5
Patient’s course highlights Tx risks
Due to the severity of the patient’s achalasia, a Heller myotomy was performed. During the postoperative period he had several complications. He ultimately died from an uncontrollable hemorrhage after an esophageal perforation, highlighting the seriousness of this disease and the risks inherent to its treatment.
Correspondence
Drew C. Baird, MD, Department of Family and Community Medicine, D.D. Eisenhower Army Medical Center, 300 Hospital Road, Fort Gordon, GA 30905.
A 70-year-old man came to our medical center for treatment of painful bilateral leg swelling that had gotten progressively worse over the past week. He had no significant past medical or surgical history, took an aspirin daily, did not smoke tobacco or drink alcohol, and had not taken any trips recently. He denied any chest pain, dyspnea, or orthopnea, but indicated that he’d been having difficulty swallowing food for the past month.
The patient had a cachectic appearance, diminished breath sounds and dullness to percussion over the right middle and lower lung fields, and pitting edema up to the knees bilaterally.
Lab studies showed a normal brain natriuretic peptide of 16 pg/mL, normocytic anemia (hemoglobin, 9.5 g/dL; hematocrit, 29.6%; and a mean corpuscular volume of 82.6 fL), potassium of 2.4 mEq/L, calcium of 5.4 mg/dL, and albumin of 1.4 g/dL. An electrocardiogram (EKG) revealed a bifascicular heart block, which was not new based on older EKGs. A plain chest radiograph was performed (FIGURES 1A AND 1B).
FIGURE 1
Chest x-rays reveal large mass
What is your diagnosis?
Diagnosis: Achalasia
Achalasia is characterized by an incomplete relaxation of the lower esophageal sphincter (LES) accompanied by aperistalsis of the esophageal body. The etiology of primary achalasia is unknown, but involves the degeneration of inhibitory neurons within the myenteric plexus responsible for LES relaxation and esophageal peristalsis.
Many potential causes. Infectious, inflammatory, autoimmune, and genetic causes have all been proposed. Achalasia has a prevalence of less than 1 in 10,000; its incidence is 0.3 to 1 per 100,000 per year, with peaks in the third and seventh decades of life. Men and women are affected equally.1
Conversely, pseudoachalasia—or secondary achalasia—has a known cause that either destroys the neurons of LES relaxation or has a mass effect that limits LES relaxation. Its most common cause is malignancy involving the gastroesophageal junction. Other causes include Chagas disease, an infection by the parasite Trypanosoma cruzi that affects the myenteric plexus, and amyloidosis.2
A disorder that goes undiagnosed for years
In primary achalasia symptom onset is insidious, with the disorder typically going undiagnosed for several years. Patients experience a gradual dysphagia for solids, progressing to liquids. Over time, patients adopt particular behaviors to aid the transit of food boluses down the esophagus and through the contracted LES, including eating slowly, stretching, moving side-to-side, or walking after meals.1 Regurgitation of food, chest pain, weight loss, heartburn, and difficulty belching are also common complaints.3 A more rapid onset and progression of symptoms (less than 6 months) is suggestive of pseudoachalasia and cancer.
Left untreated, primary achalasia leads to a progressively dilating esophagus with increased risk of aspiration, perforation, malnutrition, weight loss, and esophageal cancer.3 This case is remarkable for how far the patient’s disease progressed before he presented with signs and symptoms more indicative of secondary malnutrition than the primary disease.
Differential Dx includes hiatal hernia
The differential diagnosis for achalasia includes hiatal hernia, right middle lobe pneumonia, empyema, and lung abscess. Most patients with hiatal hernias are asymptomatic, and the diagnosis is made incidentally on chest x-ray as a retrocardiac air-fluid level or on upper gastrointestinal studies. Right middle lobe pneumonia would appear on chest x-ray as a well-demarcated opacity within the confines of that lobe, but would not have an air-fluid level. Empyema and lung abscess are complications of pneumonia, and patients present with persistent fever, cough, dyspnea, and malaise. With an empyema, chest x-ray reveals a pleural effusion. A lung abscess is an intrapulmonary cavitary lesion with an air-fluid level, most commonly due to aspiration pneumonia.
This patient’s chest x-ray showed a mass outside the lung tissue and pleural space that had an air-fluid level, was within the mediastinum, and displaced the trachea anteriorly, all suggestive of a dilated esophagus filled with undigested food unable to pass the LES.
Esophageal manometry clinches the diagnosis
Esophageal manometry is the gold standard for diagnosis. In achalasia, manometry shows poor peristalsis of the esophageal body and a constricted LES that does not relax sufficiently with swallowing.3 Manometry cannot, however, reliably distinguish primary achalasia from pseudoachalasia.
Barium swallow studies and endoscopy can assist in the diagnosis and help rule out pseudoachalasia. Timed barium studies can show a lack of peristalsis, an air-fluid level at the top of a barium column retained within the dilated esophagus, and the narrowing of the distal esophagus into the pathognomonic “bird’s beak” of primary achalasia.4 Endoscopy can visualize a mass beyond the LES, but cannot adequately assess esophageal peristalsis or LES relaxation.1
In our patient’s initial workup, a CT (FIGURE 2), barium study (FIGURE 3), and endoscopy were all used to rule out an obstructive lesion.
FIGURE 2
Massively distended esophagus
FIGURE 3
The “bird’s beak” of achalasia
Treatment targets the constricted LES
The goal of treatment in achalasia is to help food move through the constricted LES. Pharmacologic therapy, used to decrease LES resting tone, has limited benefit and becomes less effective as the disease progresses. Calcium channel blockers, particularly nifedipine, and nitrates are the most commonly used agents.5
Pneumatic dilation is the most effective nonsurgical treatment of achalasia. It involves the controlled rupture of LES muscle fibers to dilate the sphincter and allow gravity to move food from the esophagus into the stomach.6 Symptom improvement lasts for 60% of patients at 1 year, 50% at 5 years, and 36% at 10 years. Repeated dilations are possible and sometimes necessary, but have diminishing success rates. The most worrisome complication of pneumatic dilation is esophageal rupture, which occurs 2% of the time.5
Botulinum toxin injection is another accepted nonsurgical therapy and is recommended for patients who would like to avoid more invasive treatment. When injected into the LES, the toxin causes temporary atrophy of the neurons responsible for LES contraction. After the procedure, 70% of patients report improvement in symptoms, with half in remission at 6 months and one-third at 1 year. Patients may require repeat injections every 3 to 4 months, but the toxin’s effectiveness diminishes with each injection.6
The laparoscopic Heller myotomy involves cutting the LES muscle, making it incompetent and allowing food boluses to pass through the LES. Ninety percent of patients experience symptom relief after surgery,5 with 10-year remission rates of 67% to 85%.1 Comparisons to pneumatic dilation do not show significant differences in outcomes over time, although most studies favor myotomy over dilation.
Surgical treatment is indicated for patients who are younger than 40 years of age, those who have failed repeated nonsurgical therapies, and those at high risk of esophageal perforation.5
Patient’s course highlights Tx risks
Due to the severity of the patient’s achalasia, a Heller myotomy was performed. During the postoperative period he had several complications. He ultimately died from an uncontrollable hemorrhage after an esophageal perforation, highlighting the seriousness of this disease and the risks inherent to its treatment.
Correspondence
Drew C. Baird, MD, Department of Family and Community Medicine, D.D. Eisenhower Army Medical Center, 300 Hospital Road, Fort Gordon, GA 30905.
1. Pohl D, Tutuian R. Achalasia: an overview of diagnosis and treatment. J Gastrointestin Liver Dis. 2007;16:297-303.
2. Spechler SJ, Castell DO. Classification of oesophageal motility abnormalities. Gut. 2001;49:145-151.
3. Farrokhi F, Vaezi MF. Idiopathic (primary) achalasia. Orphanet J Rare Dis. 2007;2:38.-
4. Levine MS, Rubesin SE. Diseases of the esophagus: diagnosis with esophagography. Radiology. 2005;237:414-427.
5. Roberts KE, Duffy AJ, Bell RL. Controversies in the treatment of gastroesophageal reflux and achalasia. World J Gastroenterol. 2006;12:3155-3161.
6. Annese V, Bassotti G. Non-surgical treatment of esophageal achalasia. World J Gastroenterol. 2006;12:5763-5766.
1. Pohl D, Tutuian R. Achalasia: an overview of diagnosis and treatment. J Gastrointestin Liver Dis. 2007;16:297-303.
2. Spechler SJ, Castell DO. Classification of oesophageal motility abnormalities. Gut. 2001;49:145-151.
3. Farrokhi F, Vaezi MF. Idiopathic (primary) achalasia. Orphanet J Rare Dis. 2007;2:38.-
4. Levine MS, Rubesin SE. Diseases of the esophagus: diagnosis with esophagography. Radiology. 2005;237:414-427.
5. Roberts KE, Duffy AJ, Bell RL. Controversies in the treatment of gastroesophageal reflux and achalasia. World J Gastroenterol. 2006;12:3155-3161.
6. Annese V, Bassotti G. Non-surgical treatment of esophageal achalasia. World J Gastroenterol. 2006;12:5763-5766.