User login
Dusky plaque on the knee
A progressively growing lesion on the left knee prompted a 35-year-old woman to visit our clinic. She reported that about 3 months earlier, she had developed a small ulceration on the knee following a fall. With local wound care, the ulceration healed with a scar. The scar, however, continued to grow and she developed distinct papules outside the original scar.
FIGURE
Scar develops into a dusky plaque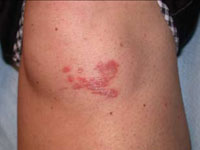
The scar subsequently became raised with violaceous discoloration. The patient reported having no history of excessive scarring or keloid forming after skin surgery or trauma. There was neither a personal nor family history of inflammatory or infectious granulomatous diseases.
On physical examination, there was an erythematous to dusky plaque with well-defined irregular borders. There were also discrete papules on the anterior and medial aspect of the knee. The plaque measured approximately 2.8 cm by 3.8 cm. There were no tender nodules on the shins, nor was lymphadenopathy present. A routine chest x-ray was normal.
To support our clinical diagnosis, we took a 4-mm punch biopsy from the center of the plaque. The histologic examination revealed changes in the dermis termed noncaseating “naked” granulomas.
What is your diagnosis?
How would you manage this condition?
Diagnosis: Scar sarcoidosis
Sarcoidosis is a systemic granulomatous disease that may affect any organ system, and therefore may present with various clinical manifestations.1 Sarcoidosis can be an incidental finding on chest x-ray or be discovered in patients that present with respiratory or constitutional symptoms.2
Cutaneous sarcoidosis occurs in up to one third of patients with systemic sarcoidosis.2 The classic skin lesions are “erythema nodosum”—an acute, nodular, erythematous eruption that usually is limited to the shins, and “lupus pernio”—red-to-purple or violaceous indurated nodules affecting the nose, cheeks, ears, and lips. There are other uncommon skin presentations of sarcoidosis ranging from scattered papules and annular lesions to erythrodermic skin manifestations.3
In scar sarcoidosis, there is spontaneous development of livid or reddish-brown plaques on scars that were previously atrophic for the most part. Scar sarcoidosis may be caused by:4-6
- venipuncture
- tuberculin skin tests
- herpes zoster
- tattoos
- cosmetic fillers such as hyaluronic acid injection.
Infection and other factors may be at work
Although the precise cause of sarcoidosis remains unknown, various infectious, noninfectious, environmental, and genetic factors may be at work. Researchers have theorized that immune dysregulation may be involved. Contact with a persistent antigen that is poorly cleared by the immune system may lead to T lymphocytes and mononuclear phagocytes accumulating in the granulomas of sarcoidosis.7 Researchers have proposed that inoculation of foreign matter from minor trauma may be one type of pathogenic mechanism in cutaneous sarcoid.8
Granuloma annulare is part of the differential
A wide range of diseases comprise the differential diagnosis of sarcoidosis. These diseases include:9
- Granuloma annulare. It is also a granulomatous skin disease, but it appears as single or multiple rings.
- Rheumatoid nodules. These usually appear in the context of a diagnosis of rheumatoid arthritis.
- Granulomatous mycosis fungoides. This type of cutaneous lymphoma has many clinical forms, including granuloma formation.
- Syringoma. On inspection, you’ll see small, firm adnexal benign tumors that usually appear around the upper cheeks and lower eyelids
- Xanthelasma. These are benign, yellow macules, papules, or plaques that tend to appear on the eyelids. Patients with xanthelasma often have a lipid disorder.
- Lichen planus. This is a very pruritic skin involvement with pink to violaceous papules and plaques. It may present in different locations, but the most common areas are the wrists and ankles.
- Granulomatous rosacea. This is a variant of rosacea characterized by uniform papules on the face.
Clinical findings, biopsy clinch the diagnosis
The diagnosis of sarcoidosis is made by a combination of clinical and histologic findings.1
- Clinical findings. Cutaneous involvement is either “specific” or “nonspecific.”
- With specific cutaneous involvement, which our patient had, you’ll see typical noncaseating granulomas, with no evidence of infection or a foreign body. It may be disfiguring, but it’s almost always nontender and it is rarely ulcerative.
- With nonspecific cutaneous involvement, you’ll see erythema nodosum lesions, especially on the legs. The serum angiotensin-converting enzyme (ACE) level is elevated in many of these patients.
Histologic findings. Skin biopsy demonstrating noncaseating granulomas provides definitive evidence of skin involvement. Typical sarcoid lesions are characterized by circumscribed granulomas of epithelioid cells with little or no necrosis. (The term “naked” granuloma refers to the absence, or small number, of surrounding lymphocytes.)
Other granulomatous diseases, such as berylliosis and tuberculosis, must be excluded since they often present the same way as scar sarcoidosis.7
Steroids control symptoms, slow disease progression
Topical, intralesional, and systemic corticosteroids are used to treat scar sarcoidosis, as are systemic medications such as chloroquine10 and allopurinol.11 Corticosteroids (local and systemic) are effective in controlling all sarcoid symptoms; they also slow disease progression.1
For localized skin involvement, intralesional corticosteroids are typically more effective than topical steroids. Systemic corticosteroids are reserved for widespread, progressive lesions or those that impair function.1,12,13 A starting dose of 1 mg/kg of prednisone is appropriate.
In general, the prognosis of cutaneous sarcoidosis is good.2 The course is variable, ranging from self-limited acute episodes to a chronic debilitating disease that may result in death.2 Spontaneous remissions occur in nearly two thirds of patients, but 10% to 30% have a more chronic or progressive course.1,2,13
Our patient responds to treatment
Our patient declined intralesional corticosteroid injections, so we started her on potent topical corticosteroid tapes (Cordran). She had significant improvement 6 weeks later.
Correspondence
Amor Khachemoune, MD, CWS, 450 Clarkson Avenue Box 46, Brooklyn, NY 11203; [email protected]
1. Howard A, White CR, Jr. Non-infectious granulomas. In: Bolognia JL, Jorizzo, JL, Rapini RP, eds. Dermatology. London: Mosby; 2003:1455-1460.
2. Giuffrida TJ, Kerdel FA. Sarcoidosis. Dermatol Clin 2002;20:435-447.
3. Okamoto H. Cutaneous sarcoidosis [in Japanese]. Nippon Rinsho 2002;60:1801-1806.
4. Barrazza V. Post-herpes zoster scar sarcoidosis. Acta Derm Venereol 1999;79:495.-
5. Dal Sacco D, Cozzani E, Parodi A, Rebora A. Scar sarcoidosis after hyaluronic acid injection. Int J Dermatol 2005;44:411-412.
6. Antonovich DD, Callen JP. Development of sarcoidosis in cosmetic tattoos. Arch Dermatol 2005;141:869-872.
7. Gal AA, Koss MN. The pathology of sarcoidosis. Curr Opin Pulm Med 2002;8:445-451.
8. Marcoval J, Mañà J, Moreno A, Gallego I, Fortuño Y, Peyrí J. Foreign bodies in granulomatous cutaneous lesions of patients with systemic sarcoidosis. Arch Dermatol 2001;137:427-430.
9. Katta R. Cutaneous sarcoidosis: a dermatologic masquerader. Am Fam Physician 2002;65:1581-1584.
10. Wallace DJ. The use of chloroquine and hydroxychloroquine for non-infectious conditions other than rheumatoid arthritis or lupus: a critical review. Lupus 1996;5 suppl 1:s59-64.
11. Bregnhoej A, Jemec GB. Low-dose allopurinol in the treatment of cutaneous sarcoidosis: response in four of seven patients. J Dermatolog Treat 2005;16:125-127.
12. Wu JJ, Schiff KR. Sarcoidosis. Am Fam Physician 2004;70:312-322.
13. Ahmed I, Harshad SR. Subcutaneous sarcoidosis: is it a specific subset of cutaneous sarcoidosis frequently associated with systemic disease? J Am Acad Dermatol 2006;54:55-60.
A progressively growing lesion on the left knee prompted a 35-year-old woman to visit our clinic. She reported that about 3 months earlier, she had developed a small ulceration on the knee following a fall. With local wound care, the ulceration healed with a scar. The scar, however, continued to grow and she developed distinct papules outside the original scar.
FIGURE
Scar develops into a dusky plaque
The scar subsequently became raised with violaceous discoloration. The patient reported having no history of excessive scarring or keloid forming after skin surgery or trauma. There was neither a personal nor family history of inflammatory or infectious granulomatous diseases.
On physical examination, there was an erythematous to dusky plaque with well-defined irregular borders. There were also discrete papules on the anterior and medial aspect of the knee. The plaque measured approximately 2.8 cm by 3.8 cm. There were no tender nodules on the shins, nor was lymphadenopathy present. A routine chest x-ray was normal.
To support our clinical diagnosis, we took a 4-mm punch biopsy from the center of the plaque. The histologic examination revealed changes in the dermis termed noncaseating “naked” granulomas.
What is your diagnosis?
How would you manage this condition?
Diagnosis: Scar sarcoidosis
Sarcoidosis is a systemic granulomatous disease that may affect any organ system, and therefore may present with various clinical manifestations.1 Sarcoidosis can be an incidental finding on chest x-ray or be discovered in patients that present with respiratory or constitutional symptoms.2
Cutaneous sarcoidosis occurs in up to one third of patients with systemic sarcoidosis.2 The classic skin lesions are “erythema nodosum”—an acute, nodular, erythematous eruption that usually is limited to the shins, and “lupus pernio”—red-to-purple or violaceous indurated nodules affecting the nose, cheeks, ears, and lips. There are other uncommon skin presentations of sarcoidosis ranging from scattered papules and annular lesions to erythrodermic skin manifestations.3
In scar sarcoidosis, there is spontaneous development of livid or reddish-brown plaques on scars that were previously atrophic for the most part. Scar sarcoidosis may be caused by:4-6
- venipuncture
- tuberculin skin tests
- herpes zoster
- tattoos
- cosmetic fillers such as hyaluronic acid injection.
Infection and other factors may be at work
Although the precise cause of sarcoidosis remains unknown, various infectious, noninfectious, environmental, and genetic factors may be at work. Researchers have theorized that immune dysregulation may be involved. Contact with a persistent antigen that is poorly cleared by the immune system may lead to T lymphocytes and mononuclear phagocytes accumulating in the granulomas of sarcoidosis.7 Researchers have proposed that inoculation of foreign matter from minor trauma may be one type of pathogenic mechanism in cutaneous sarcoid.8
Granuloma annulare is part of the differential
A wide range of diseases comprise the differential diagnosis of sarcoidosis. These diseases include:9
- Granuloma annulare. It is also a granulomatous skin disease, but it appears as single or multiple rings.
- Rheumatoid nodules. These usually appear in the context of a diagnosis of rheumatoid arthritis.
- Granulomatous mycosis fungoides. This type of cutaneous lymphoma has many clinical forms, including granuloma formation.
- Syringoma. On inspection, you’ll see small, firm adnexal benign tumors that usually appear around the upper cheeks and lower eyelids
- Xanthelasma. These are benign, yellow macules, papules, or plaques that tend to appear on the eyelids. Patients with xanthelasma often have a lipid disorder.
- Lichen planus. This is a very pruritic skin involvement with pink to violaceous papules and plaques. It may present in different locations, but the most common areas are the wrists and ankles.
- Granulomatous rosacea. This is a variant of rosacea characterized by uniform papules on the face.
Clinical findings, biopsy clinch the diagnosis
The diagnosis of sarcoidosis is made by a combination of clinical and histologic findings.1
- Clinical findings. Cutaneous involvement is either “specific” or “nonspecific.”
- With specific cutaneous involvement, which our patient had, you’ll see typical noncaseating granulomas, with no evidence of infection or a foreign body. It may be disfiguring, but it’s almost always nontender and it is rarely ulcerative.
- With nonspecific cutaneous involvement, you’ll see erythema nodosum lesions, especially on the legs. The serum angiotensin-converting enzyme (ACE) level is elevated in many of these patients.
Histologic findings. Skin biopsy demonstrating noncaseating granulomas provides definitive evidence of skin involvement. Typical sarcoid lesions are characterized by circumscribed granulomas of epithelioid cells with little or no necrosis. (The term “naked” granuloma refers to the absence, or small number, of surrounding lymphocytes.)
Other granulomatous diseases, such as berylliosis and tuberculosis, must be excluded since they often present the same way as scar sarcoidosis.7
Steroids control symptoms, slow disease progression
Topical, intralesional, and systemic corticosteroids are used to treat scar sarcoidosis, as are systemic medications such as chloroquine10 and allopurinol.11 Corticosteroids (local and systemic) are effective in controlling all sarcoid symptoms; they also slow disease progression.1
For localized skin involvement, intralesional corticosteroids are typically more effective than topical steroids. Systemic corticosteroids are reserved for widespread, progressive lesions or those that impair function.1,12,13 A starting dose of 1 mg/kg of prednisone is appropriate.
In general, the prognosis of cutaneous sarcoidosis is good.2 The course is variable, ranging from self-limited acute episodes to a chronic debilitating disease that may result in death.2 Spontaneous remissions occur in nearly two thirds of patients, but 10% to 30% have a more chronic or progressive course.1,2,13
Our patient responds to treatment
Our patient declined intralesional corticosteroid injections, so we started her on potent topical corticosteroid tapes (Cordran). She had significant improvement 6 weeks later.
Correspondence
Amor Khachemoune, MD, CWS, 450 Clarkson Avenue Box 46, Brooklyn, NY 11203; [email protected]
A progressively growing lesion on the left knee prompted a 35-year-old woman to visit our clinic. She reported that about 3 months earlier, she had developed a small ulceration on the knee following a fall. With local wound care, the ulceration healed with a scar. The scar, however, continued to grow and she developed distinct papules outside the original scar.
FIGURE
Scar develops into a dusky plaque
The scar subsequently became raised with violaceous discoloration. The patient reported having no history of excessive scarring or keloid forming after skin surgery or trauma. There was neither a personal nor family history of inflammatory or infectious granulomatous diseases.
On physical examination, there was an erythematous to dusky plaque with well-defined irregular borders. There were also discrete papules on the anterior and medial aspect of the knee. The plaque measured approximately 2.8 cm by 3.8 cm. There were no tender nodules on the shins, nor was lymphadenopathy present. A routine chest x-ray was normal.
To support our clinical diagnosis, we took a 4-mm punch biopsy from the center of the plaque. The histologic examination revealed changes in the dermis termed noncaseating “naked” granulomas.
What is your diagnosis?
How would you manage this condition?
Diagnosis: Scar sarcoidosis
Sarcoidosis is a systemic granulomatous disease that may affect any organ system, and therefore may present with various clinical manifestations.1 Sarcoidosis can be an incidental finding on chest x-ray or be discovered in patients that present with respiratory or constitutional symptoms.2
Cutaneous sarcoidosis occurs in up to one third of patients with systemic sarcoidosis.2 The classic skin lesions are “erythema nodosum”—an acute, nodular, erythematous eruption that usually is limited to the shins, and “lupus pernio”—red-to-purple or violaceous indurated nodules affecting the nose, cheeks, ears, and lips. There are other uncommon skin presentations of sarcoidosis ranging from scattered papules and annular lesions to erythrodermic skin manifestations.3
In scar sarcoidosis, there is spontaneous development of livid or reddish-brown plaques on scars that were previously atrophic for the most part. Scar sarcoidosis may be caused by:4-6
- venipuncture
- tuberculin skin tests
- herpes zoster
- tattoos
- cosmetic fillers such as hyaluronic acid injection.
Infection and other factors may be at work
Although the precise cause of sarcoidosis remains unknown, various infectious, noninfectious, environmental, and genetic factors may be at work. Researchers have theorized that immune dysregulation may be involved. Contact with a persistent antigen that is poorly cleared by the immune system may lead to T lymphocytes and mononuclear phagocytes accumulating in the granulomas of sarcoidosis.7 Researchers have proposed that inoculation of foreign matter from minor trauma may be one type of pathogenic mechanism in cutaneous sarcoid.8
Granuloma annulare is part of the differential
A wide range of diseases comprise the differential diagnosis of sarcoidosis. These diseases include:9
- Granuloma annulare. It is also a granulomatous skin disease, but it appears as single or multiple rings.
- Rheumatoid nodules. These usually appear in the context of a diagnosis of rheumatoid arthritis.
- Granulomatous mycosis fungoides. This type of cutaneous lymphoma has many clinical forms, including granuloma formation.
- Syringoma. On inspection, you’ll see small, firm adnexal benign tumors that usually appear around the upper cheeks and lower eyelids
- Xanthelasma. These are benign, yellow macules, papules, or plaques that tend to appear on the eyelids. Patients with xanthelasma often have a lipid disorder.
- Lichen planus. This is a very pruritic skin involvement with pink to violaceous papules and plaques. It may present in different locations, but the most common areas are the wrists and ankles.
- Granulomatous rosacea. This is a variant of rosacea characterized by uniform papules on the face.
Clinical findings, biopsy clinch the diagnosis
The diagnosis of sarcoidosis is made by a combination of clinical and histologic findings.1
- Clinical findings. Cutaneous involvement is either “specific” or “nonspecific.”
- With specific cutaneous involvement, which our patient had, you’ll see typical noncaseating granulomas, with no evidence of infection or a foreign body. It may be disfiguring, but it’s almost always nontender and it is rarely ulcerative.
- With nonspecific cutaneous involvement, you’ll see erythema nodosum lesions, especially on the legs. The serum angiotensin-converting enzyme (ACE) level is elevated in many of these patients.
Histologic findings. Skin biopsy demonstrating noncaseating granulomas provides definitive evidence of skin involvement. Typical sarcoid lesions are characterized by circumscribed granulomas of epithelioid cells with little or no necrosis. (The term “naked” granuloma refers to the absence, or small number, of surrounding lymphocytes.)
Other granulomatous diseases, such as berylliosis and tuberculosis, must be excluded since they often present the same way as scar sarcoidosis.7
Steroids control symptoms, slow disease progression
Topical, intralesional, and systemic corticosteroids are used to treat scar sarcoidosis, as are systemic medications such as chloroquine10 and allopurinol.11 Corticosteroids (local and systemic) are effective in controlling all sarcoid symptoms; they also slow disease progression.1
For localized skin involvement, intralesional corticosteroids are typically more effective than topical steroids. Systemic corticosteroids are reserved for widespread, progressive lesions or those that impair function.1,12,13 A starting dose of 1 mg/kg of prednisone is appropriate.
In general, the prognosis of cutaneous sarcoidosis is good.2 The course is variable, ranging from self-limited acute episodes to a chronic debilitating disease that may result in death.2 Spontaneous remissions occur in nearly two thirds of patients, but 10% to 30% have a more chronic or progressive course.1,2,13
Our patient responds to treatment
Our patient declined intralesional corticosteroid injections, so we started her on potent topical corticosteroid tapes (Cordran). She had significant improvement 6 weeks later.
Correspondence
Amor Khachemoune, MD, CWS, 450 Clarkson Avenue Box 46, Brooklyn, NY 11203; [email protected]
1. Howard A, White CR, Jr. Non-infectious granulomas. In: Bolognia JL, Jorizzo, JL, Rapini RP, eds. Dermatology. London: Mosby; 2003:1455-1460.
2. Giuffrida TJ, Kerdel FA. Sarcoidosis. Dermatol Clin 2002;20:435-447.
3. Okamoto H. Cutaneous sarcoidosis [in Japanese]. Nippon Rinsho 2002;60:1801-1806.
4. Barrazza V. Post-herpes zoster scar sarcoidosis. Acta Derm Venereol 1999;79:495.-
5. Dal Sacco D, Cozzani E, Parodi A, Rebora A. Scar sarcoidosis after hyaluronic acid injection. Int J Dermatol 2005;44:411-412.
6. Antonovich DD, Callen JP. Development of sarcoidosis in cosmetic tattoos. Arch Dermatol 2005;141:869-872.
7. Gal AA, Koss MN. The pathology of sarcoidosis. Curr Opin Pulm Med 2002;8:445-451.
8. Marcoval J, Mañà J, Moreno A, Gallego I, Fortuño Y, Peyrí J. Foreign bodies in granulomatous cutaneous lesions of patients with systemic sarcoidosis. Arch Dermatol 2001;137:427-430.
9. Katta R. Cutaneous sarcoidosis: a dermatologic masquerader. Am Fam Physician 2002;65:1581-1584.
10. Wallace DJ. The use of chloroquine and hydroxychloroquine for non-infectious conditions other than rheumatoid arthritis or lupus: a critical review. Lupus 1996;5 suppl 1:s59-64.
11. Bregnhoej A, Jemec GB. Low-dose allopurinol in the treatment of cutaneous sarcoidosis: response in four of seven patients. J Dermatolog Treat 2005;16:125-127.
12. Wu JJ, Schiff KR. Sarcoidosis. Am Fam Physician 2004;70:312-322.
13. Ahmed I, Harshad SR. Subcutaneous sarcoidosis: is it a specific subset of cutaneous sarcoidosis frequently associated with systemic disease? J Am Acad Dermatol 2006;54:55-60.
1. Howard A, White CR, Jr. Non-infectious granulomas. In: Bolognia JL, Jorizzo, JL, Rapini RP, eds. Dermatology. London: Mosby; 2003:1455-1460.
2. Giuffrida TJ, Kerdel FA. Sarcoidosis. Dermatol Clin 2002;20:435-447.
3. Okamoto H. Cutaneous sarcoidosis [in Japanese]. Nippon Rinsho 2002;60:1801-1806.
4. Barrazza V. Post-herpes zoster scar sarcoidosis. Acta Derm Venereol 1999;79:495.-
5. Dal Sacco D, Cozzani E, Parodi A, Rebora A. Scar sarcoidosis after hyaluronic acid injection. Int J Dermatol 2005;44:411-412.
6. Antonovich DD, Callen JP. Development of sarcoidosis in cosmetic tattoos. Arch Dermatol 2005;141:869-872.
7. Gal AA, Koss MN. The pathology of sarcoidosis. Curr Opin Pulm Med 2002;8:445-451.
8. Marcoval J, Mañà J, Moreno A, Gallego I, Fortuño Y, Peyrí J. Foreign bodies in granulomatous cutaneous lesions of patients with systemic sarcoidosis. Arch Dermatol 2001;137:427-430.
9. Katta R. Cutaneous sarcoidosis: a dermatologic masquerader. Am Fam Physician 2002;65:1581-1584.
10. Wallace DJ. The use of chloroquine and hydroxychloroquine for non-infectious conditions other than rheumatoid arthritis or lupus: a critical review. Lupus 1996;5 suppl 1:s59-64.
11. Bregnhoej A, Jemec GB. Low-dose allopurinol in the treatment of cutaneous sarcoidosis: response in four of seven patients. J Dermatolog Treat 2005;16:125-127.
12. Wu JJ, Schiff KR. Sarcoidosis. Am Fam Physician 2004;70:312-322.
13. Ahmed I, Harshad SR. Subcutaneous sarcoidosis: is it a specific subset of cutaneous sarcoidosis frequently associated with systemic disease? J Am Acad Dermatol 2006;54:55-60.
Lesion on the hard palate
A 58-year-old patient came into the office complaining of sinus congestion with sinus pressure, sore throat, and postnasal drip that had been getting progressively worse over the past week. He had tried over-the-counter decongestants, but his symptoms were not resolving. He told the physician’s assistant (DR) that he’d had sinus infections in the past and that he thought he had one now.
When asked about his general health, the patient indicated that he vaguely remembered being told years ago that his cholesterol was elevated, but at the time, declined any medical treatment. He indicated that he hadn’t had a physical examination in years. He said that he only went to the doctor when he was “sick.”
The patient was a cigarette smoker and had smoked a pack a day for 35 years. He said that while he smoked mostly cigarettes, he would also have an occasional cigar. He said that he had never smoked tobacco through a pipe. He also indicated that he drank 3 to 4 beers daily.
While inspecting his pharynx during the physical examination, DR noted a lesion on the posterior hard palate that extended to the soft palate (FIGURE 1). It appeared macular and erythematous, and had an eroded surface. The borders of the lesion were diffuse and irregular, measuring approximately 2 cm Ø 3 cm. During palpation, the lesion was mildly painful, but there was no firmness along the soft palate, buccal mucosa, or tongue. The patient had poor oral hygiene, multiple missing and carious teeth, and a removable partial denture. All other mucosal surfaces were within normal limits.
As the patient suspected, he did have a sinus infection. He left the office with a prescription for an antibiotic. He also left with a referral to an oral maxillofacial surgeon for further evaluation of the lesion.
FIGURE 1
Lesion on the hard palate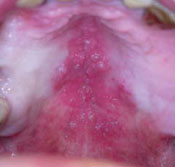
What is your diagnosis?
How would you manage this condition?
Diagnosis: Perianal streptococcal dermatitis
The oral maxillofacial surgeon (TAC) concurred with the previous examination findings. He was concerned by the appearance of the lesion and the history of tobacco use and performed a biopsy of the lesion. The pathology report revealed that the patient had nicotine stomatitis with a concomitant candidiasis infection.
Nicotine stomatitis is a common mucosal keratosis caused by smoking tobacco. It is localized to the hard palate of the mouth, but can extend to the soft palate. The mucosa of the hard palate becomes white and thickened due to hyperkeratosis of the tissue.1 Thin red lines of normal mucosa can be seen throughout the lesion. Red dots or papules can appear in the center of the lesion, which represent irritated salivary glands with inflamed duct openings (FIGURE 2).2
Our patient’s lesion had an atypical presentation. Classically the lesions are whiter, but in this case, the patient’s lesion was more erythematous. The reason: A super-infection of candidiasis caused an atrophic form of the condition.
FIGURE 2
Classic presentation of nicotine stomatitis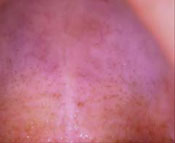
Differential Dx includes squamous cell carcinoma
The differential diagnosis in a case of nicotine stomatitis includes:
- irritation from dental appliances
- trauma from hot liquids
- squamous cell carcinoma
- atrophic candidiasis.
ill-fitting dentures can cause erythematous lesions along the hard palate and gingiva. The erythema from dentures is usually uniform in color without ulceration and follows the outline or shape of the oral appliance. In contrast, nicotine stomatitis will typically present on exposed areas of the palate and not follow any specific pattern. Most lesions caused by irritations or trauma resolve within 2 weeks of removal of the offending agent.
In the case of burns from food or liquids, the patient will tell you that he drank or ate something hot, and he’ll have a red lesion on the tongue or roof of his mouth.
Squamous cell carcinoma of the hard palate is part of the differential diagnosis because smoking increases a patient’s risk of this form of oral cancer. Squamous cell carcinoma of the palate accounts for 5% to 15% of intraoral carcinomas, depending on whether it is on the hard or soft palate.1 The lesions are typically red or red/white in color and have ulcerated and/or necrotic surfaces. The lesions can become exophytic if left untreated. In comparison, nicotine stomatitis does not ulcerate unless there is a concomitant disease process, which should prompt the clinician to perform further diagnostics tests.
Atrophic candidiasis is also part of the differential, and in the case of our patient, it was a concomitant infection. Candidiasis has a variable presentation, but typically presents with plaques in the oral cavity—commonly referred to as thrush. In the case of atrophic candidiasis, the lesion is usually raised and erythematous, giving a red velvety appearance of the oral mucosa.1 It is caused by the invasion of the candidal organism into the mucosal surface. Oral candidiasis presents in patients who are immunocompromised, taking long-term corticosteroids, and using antibiotics. It is also seen in infants.2 Our patient likely developed candidiasis because of his poor oral hygiene.
Pipe smoking is the usual red flag
Nicotine stomatitis is commonly seen in middle-age men who have a history of tobacco use.2 It is most commonly seen in pipe smokers, but can occur in cigar and cigarette smokers, as well. The intense heat in the oral cavity generated by smoking causes changes in the oral mucosa—typically on the hard palate. The stem of the pipe increases the amount of heat directed at the hard palate, resulting in a higher incidence of nicotine stomatitis in pipe smokers.3,4 The severity and extent of the lesion is directly proportional to the frequency of tobacco inhalation. Interestingly, the chemicals in the tobacco are not responsible for the mucosal changes, therefore there is no pre-malignant potential.
The risk of malignancy does, however, come into play if your patient does something called reverse smoking. Members of some Asian cultures practice this form of smoking, in which the lit end of the cigarette is placed in the mouth. This practice raises the risk of malignancy. Any patient with nicotine stomatitis who practices reverse smoking should have a biopsy of the lesion done.5
Our approach to the Dx differed from the norm
Typically, you’ll make the diagnosis of nicotine stomatitis clinically, since few lesions resemble its appearance.1 The atypical nature of our patient’s lesion, however, is what prompted a biopsy. You may also do a biopsy if there is any suspicion of cancer or if a lesion is still present after the patient stops smoking.
In our patient’s case, the pathology report revealed a thickened epithelial layer with no evidence of atypia or dysplasia. The minor salivary glands showed chronic inflammatory cells consistent with nicotine stomatitis. The area biopsied also contained a large amount of hyphae, consistent with candidiasis.
Treatment hinges on smoking cessation
Primary treatment for nicotine stomatitis is for the patient to stop smoking. Most lesions will resolve within several months of smoking cessation.1 The lesion is indicative of heavy tobacco use and other mucosal tissues of the oralpharyngeal tract may have similar damage. Therefore, the oropharyngeal cavity should be thoroughly evaluated for dysplastic or malignant lesions.
Our patient received counseling on tobacco cessation and oral hygiene practices. He also received clotrimazole troches for the Candida infection. Two weeks after he started taking the anti-fungal medication, and after he began smoking cessation efforts, the lesion significantly improved. The patient did not return for additional follow-up visits, so we do not know whether the lesion resolved completely.
Correspondence
Denise Rizzolo, PA-C, MS, 348 East Main street, First Floor, Somerville, NJ 08876; [email protected]
1. Mark RE, Stern D. Oral and Maxillofacial Pathology: A Rational for Diagnosis and Treatment. Carol stream, Ill: Quintessence Publishing Co, Inc; 2003: chap 7.
2. Regezi JA, Sciubba J, Jordan RCK. Oral Pathology: Clinical Pathologic Conditions. 4rd ed. Philadelphia, Pa: WB saunders Co; 2003.
3. Neville BW, Day TA. Oral cancer and precancerous lesions. CA Cancer J Clin 2002;52:195-215.
4. Taybos G. Oral changes associated with tobacco use. Am J Med Sci 2003;326:179-182.
5. Ramulu C, Raju MVS, Reddy CRRM. Nicotine stomatitis and its relation to carcinoma of the hard palate in reverse smokers of chuttas. J Dent Res 1973;52:711-718.
A 58-year-old patient came into the office complaining of sinus congestion with sinus pressure, sore throat, and postnasal drip that had been getting progressively worse over the past week. He had tried over-the-counter decongestants, but his symptoms were not resolving. He told the physician’s assistant (DR) that he’d had sinus infections in the past and that he thought he had one now.
When asked about his general health, the patient indicated that he vaguely remembered being told years ago that his cholesterol was elevated, but at the time, declined any medical treatment. He indicated that he hadn’t had a physical examination in years. He said that he only went to the doctor when he was “sick.”
The patient was a cigarette smoker and had smoked a pack a day for 35 years. He said that while he smoked mostly cigarettes, he would also have an occasional cigar. He said that he had never smoked tobacco through a pipe. He also indicated that he drank 3 to 4 beers daily.
While inspecting his pharynx during the physical examination, DR noted a lesion on the posterior hard palate that extended to the soft palate (FIGURE 1). It appeared macular and erythematous, and had an eroded surface. The borders of the lesion were diffuse and irregular, measuring approximately 2 cm Ø 3 cm. During palpation, the lesion was mildly painful, but there was no firmness along the soft palate, buccal mucosa, or tongue. The patient had poor oral hygiene, multiple missing and carious teeth, and a removable partial denture. All other mucosal surfaces were within normal limits.
As the patient suspected, he did have a sinus infection. He left the office with a prescription for an antibiotic. He also left with a referral to an oral maxillofacial surgeon for further evaluation of the lesion.
FIGURE 1
Lesion on the hard palate
What is your diagnosis?
How would you manage this condition?
Diagnosis: Perianal streptococcal dermatitis
The oral maxillofacial surgeon (TAC) concurred with the previous examination findings. He was concerned by the appearance of the lesion and the history of tobacco use and performed a biopsy of the lesion. The pathology report revealed that the patient had nicotine stomatitis with a concomitant candidiasis infection.
Nicotine stomatitis is a common mucosal keratosis caused by smoking tobacco. It is localized to the hard palate of the mouth, but can extend to the soft palate. The mucosa of the hard palate becomes white and thickened due to hyperkeratosis of the tissue.1 Thin red lines of normal mucosa can be seen throughout the lesion. Red dots or papules can appear in the center of the lesion, which represent irritated salivary glands with inflamed duct openings (FIGURE 2).2
Our patient’s lesion had an atypical presentation. Classically the lesions are whiter, but in this case, the patient’s lesion was more erythematous. The reason: A super-infection of candidiasis caused an atrophic form of the condition.
FIGURE 2
Classic presentation of nicotine stomatitis
Differential Dx includes squamous cell carcinoma
The differential diagnosis in a case of nicotine stomatitis includes:
- irritation from dental appliances
- trauma from hot liquids
- squamous cell carcinoma
- atrophic candidiasis.
ill-fitting dentures can cause erythematous lesions along the hard palate and gingiva. The erythema from dentures is usually uniform in color without ulceration and follows the outline or shape of the oral appliance. In contrast, nicotine stomatitis will typically present on exposed areas of the palate and not follow any specific pattern. Most lesions caused by irritations or trauma resolve within 2 weeks of removal of the offending agent.
In the case of burns from food or liquids, the patient will tell you that he drank or ate something hot, and he’ll have a red lesion on the tongue or roof of his mouth.
Squamous cell carcinoma of the hard palate is part of the differential diagnosis because smoking increases a patient’s risk of this form of oral cancer. Squamous cell carcinoma of the palate accounts for 5% to 15% of intraoral carcinomas, depending on whether it is on the hard or soft palate.1 The lesions are typically red or red/white in color and have ulcerated and/or necrotic surfaces. The lesions can become exophytic if left untreated. In comparison, nicotine stomatitis does not ulcerate unless there is a concomitant disease process, which should prompt the clinician to perform further diagnostics tests.
Atrophic candidiasis is also part of the differential, and in the case of our patient, it was a concomitant infection. Candidiasis has a variable presentation, but typically presents with plaques in the oral cavity—commonly referred to as thrush. In the case of atrophic candidiasis, the lesion is usually raised and erythematous, giving a red velvety appearance of the oral mucosa.1 It is caused by the invasion of the candidal organism into the mucosal surface. Oral candidiasis presents in patients who are immunocompromised, taking long-term corticosteroids, and using antibiotics. It is also seen in infants.2 Our patient likely developed candidiasis because of his poor oral hygiene.
Pipe smoking is the usual red flag
Nicotine stomatitis is commonly seen in middle-age men who have a history of tobacco use.2 It is most commonly seen in pipe smokers, but can occur in cigar and cigarette smokers, as well. The intense heat in the oral cavity generated by smoking causes changes in the oral mucosa—typically on the hard palate. The stem of the pipe increases the amount of heat directed at the hard palate, resulting in a higher incidence of nicotine stomatitis in pipe smokers.3,4 The severity and extent of the lesion is directly proportional to the frequency of tobacco inhalation. Interestingly, the chemicals in the tobacco are not responsible for the mucosal changes, therefore there is no pre-malignant potential.
The risk of malignancy does, however, come into play if your patient does something called reverse smoking. Members of some Asian cultures practice this form of smoking, in which the lit end of the cigarette is placed in the mouth. This practice raises the risk of malignancy. Any patient with nicotine stomatitis who practices reverse smoking should have a biopsy of the lesion done.5
Our approach to the Dx differed from the norm
Typically, you’ll make the diagnosis of nicotine stomatitis clinically, since few lesions resemble its appearance.1 The atypical nature of our patient’s lesion, however, is what prompted a biopsy. You may also do a biopsy if there is any suspicion of cancer or if a lesion is still present after the patient stops smoking.
In our patient’s case, the pathology report revealed a thickened epithelial layer with no evidence of atypia or dysplasia. The minor salivary glands showed chronic inflammatory cells consistent with nicotine stomatitis. The area biopsied also contained a large amount of hyphae, consistent with candidiasis.
Treatment hinges on smoking cessation
Primary treatment for nicotine stomatitis is for the patient to stop smoking. Most lesions will resolve within several months of smoking cessation.1 The lesion is indicative of heavy tobacco use and other mucosal tissues of the oralpharyngeal tract may have similar damage. Therefore, the oropharyngeal cavity should be thoroughly evaluated for dysplastic or malignant lesions.
Our patient received counseling on tobacco cessation and oral hygiene practices. He also received clotrimazole troches for the Candida infection. Two weeks after he started taking the anti-fungal medication, and after he began smoking cessation efforts, the lesion significantly improved. The patient did not return for additional follow-up visits, so we do not know whether the lesion resolved completely.
Correspondence
Denise Rizzolo, PA-C, MS, 348 East Main street, First Floor, Somerville, NJ 08876; [email protected]
A 58-year-old patient came into the office complaining of sinus congestion with sinus pressure, sore throat, and postnasal drip that had been getting progressively worse over the past week. He had tried over-the-counter decongestants, but his symptoms were not resolving. He told the physician’s assistant (DR) that he’d had sinus infections in the past and that he thought he had one now.
When asked about his general health, the patient indicated that he vaguely remembered being told years ago that his cholesterol was elevated, but at the time, declined any medical treatment. He indicated that he hadn’t had a physical examination in years. He said that he only went to the doctor when he was “sick.”
The patient was a cigarette smoker and had smoked a pack a day for 35 years. He said that while he smoked mostly cigarettes, he would also have an occasional cigar. He said that he had never smoked tobacco through a pipe. He also indicated that he drank 3 to 4 beers daily.
While inspecting his pharynx during the physical examination, DR noted a lesion on the posterior hard palate that extended to the soft palate (FIGURE 1). It appeared macular and erythematous, and had an eroded surface. The borders of the lesion were diffuse and irregular, measuring approximately 2 cm Ø 3 cm. During palpation, the lesion was mildly painful, but there was no firmness along the soft palate, buccal mucosa, or tongue. The patient had poor oral hygiene, multiple missing and carious teeth, and a removable partial denture. All other mucosal surfaces were within normal limits.
As the patient suspected, he did have a sinus infection. He left the office with a prescription for an antibiotic. He also left with a referral to an oral maxillofacial surgeon for further evaluation of the lesion.
FIGURE 1
Lesion on the hard palate
What is your diagnosis?
How would you manage this condition?
Diagnosis: Perianal streptococcal dermatitis
The oral maxillofacial surgeon (TAC) concurred with the previous examination findings. He was concerned by the appearance of the lesion and the history of tobacco use and performed a biopsy of the lesion. The pathology report revealed that the patient had nicotine stomatitis with a concomitant candidiasis infection.
Nicotine stomatitis is a common mucosal keratosis caused by smoking tobacco. It is localized to the hard palate of the mouth, but can extend to the soft palate. The mucosa of the hard palate becomes white and thickened due to hyperkeratosis of the tissue.1 Thin red lines of normal mucosa can be seen throughout the lesion. Red dots or papules can appear in the center of the lesion, which represent irritated salivary glands with inflamed duct openings (FIGURE 2).2
Our patient’s lesion had an atypical presentation. Classically the lesions are whiter, but in this case, the patient’s lesion was more erythematous. The reason: A super-infection of candidiasis caused an atrophic form of the condition.
FIGURE 2
Classic presentation of nicotine stomatitis
Differential Dx includes squamous cell carcinoma
The differential diagnosis in a case of nicotine stomatitis includes:
- irritation from dental appliances
- trauma from hot liquids
- squamous cell carcinoma
- atrophic candidiasis.
ill-fitting dentures can cause erythematous lesions along the hard palate and gingiva. The erythema from dentures is usually uniform in color without ulceration and follows the outline or shape of the oral appliance. In contrast, nicotine stomatitis will typically present on exposed areas of the palate and not follow any specific pattern. Most lesions caused by irritations or trauma resolve within 2 weeks of removal of the offending agent.
In the case of burns from food or liquids, the patient will tell you that he drank or ate something hot, and he’ll have a red lesion on the tongue or roof of his mouth.
Squamous cell carcinoma of the hard palate is part of the differential diagnosis because smoking increases a patient’s risk of this form of oral cancer. Squamous cell carcinoma of the palate accounts for 5% to 15% of intraoral carcinomas, depending on whether it is on the hard or soft palate.1 The lesions are typically red or red/white in color and have ulcerated and/or necrotic surfaces. The lesions can become exophytic if left untreated. In comparison, nicotine stomatitis does not ulcerate unless there is a concomitant disease process, which should prompt the clinician to perform further diagnostics tests.
Atrophic candidiasis is also part of the differential, and in the case of our patient, it was a concomitant infection. Candidiasis has a variable presentation, but typically presents with plaques in the oral cavity—commonly referred to as thrush. In the case of atrophic candidiasis, the lesion is usually raised and erythematous, giving a red velvety appearance of the oral mucosa.1 It is caused by the invasion of the candidal organism into the mucosal surface. Oral candidiasis presents in patients who are immunocompromised, taking long-term corticosteroids, and using antibiotics. It is also seen in infants.2 Our patient likely developed candidiasis because of his poor oral hygiene.
Pipe smoking is the usual red flag
Nicotine stomatitis is commonly seen in middle-age men who have a history of tobacco use.2 It is most commonly seen in pipe smokers, but can occur in cigar and cigarette smokers, as well. The intense heat in the oral cavity generated by smoking causes changes in the oral mucosa—typically on the hard palate. The stem of the pipe increases the amount of heat directed at the hard palate, resulting in a higher incidence of nicotine stomatitis in pipe smokers.3,4 The severity and extent of the lesion is directly proportional to the frequency of tobacco inhalation. Interestingly, the chemicals in the tobacco are not responsible for the mucosal changes, therefore there is no pre-malignant potential.
The risk of malignancy does, however, come into play if your patient does something called reverse smoking. Members of some Asian cultures practice this form of smoking, in which the lit end of the cigarette is placed in the mouth. This practice raises the risk of malignancy. Any patient with nicotine stomatitis who practices reverse smoking should have a biopsy of the lesion done.5
Our approach to the Dx differed from the norm
Typically, you’ll make the diagnosis of nicotine stomatitis clinically, since few lesions resemble its appearance.1 The atypical nature of our patient’s lesion, however, is what prompted a biopsy. You may also do a biopsy if there is any suspicion of cancer or if a lesion is still present after the patient stops smoking.
In our patient’s case, the pathology report revealed a thickened epithelial layer with no evidence of atypia or dysplasia. The minor salivary glands showed chronic inflammatory cells consistent with nicotine stomatitis. The area biopsied also contained a large amount of hyphae, consistent with candidiasis.
Treatment hinges on smoking cessation
Primary treatment for nicotine stomatitis is for the patient to stop smoking. Most lesions will resolve within several months of smoking cessation.1 The lesion is indicative of heavy tobacco use and other mucosal tissues of the oralpharyngeal tract may have similar damage. Therefore, the oropharyngeal cavity should be thoroughly evaluated for dysplastic or malignant lesions.
Our patient received counseling on tobacco cessation and oral hygiene practices. He also received clotrimazole troches for the Candida infection. Two weeks after he started taking the anti-fungal medication, and after he began smoking cessation efforts, the lesion significantly improved. The patient did not return for additional follow-up visits, so we do not know whether the lesion resolved completely.
Correspondence
Denise Rizzolo, PA-C, MS, 348 East Main street, First Floor, Somerville, NJ 08876; [email protected]
1. Mark RE, Stern D. Oral and Maxillofacial Pathology: A Rational for Diagnosis and Treatment. Carol stream, Ill: Quintessence Publishing Co, Inc; 2003: chap 7.
2. Regezi JA, Sciubba J, Jordan RCK. Oral Pathology: Clinical Pathologic Conditions. 4rd ed. Philadelphia, Pa: WB saunders Co; 2003.
3. Neville BW, Day TA. Oral cancer and precancerous lesions. CA Cancer J Clin 2002;52:195-215.
4. Taybos G. Oral changes associated with tobacco use. Am J Med Sci 2003;326:179-182.
5. Ramulu C, Raju MVS, Reddy CRRM. Nicotine stomatitis and its relation to carcinoma of the hard palate in reverse smokers of chuttas. J Dent Res 1973;52:711-718.
1. Mark RE, Stern D. Oral and Maxillofacial Pathology: A Rational for Diagnosis and Treatment. Carol stream, Ill: Quintessence Publishing Co, Inc; 2003: chap 7.
2. Regezi JA, Sciubba J, Jordan RCK. Oral Pathology: Clinical Pathologic Conditions. 4rd ed. Philadelphia, Pa: WB saunders Co; 2003.
3. Neville BW, Day TA. Oral cancer and precancerous lesions. CA Cancer J Clin 2002;52:195-215.
4. Taybos G. Oral changes associated with tobacco use. Am J Med Sci 2003;326:179-182.
5. Ramulu C, Raju MVS, Reddy CRRM. Nicotine stomatitis and its relation to carcinoma of the hard palate in reverse smokers of chuttas. J Dent Res 1973;52:711-718.
Itchy perianal erythema
A mother brought her 3½-year-old son into the office for a worsening rash in the perianal area (FIGURE 1). Mother and child had been in the office 2 weeks earlier for the rash, and the physician had prescribed topical clotrimazole cream for a presumed case of Candida infection. Despite using the cream as directed, the child’s rash worsened.
The child’s mother told the treating physician (SS) that his rash was painful and itchy. She also said he’d had a few blood-streaked stools.
The little boy had no significant medical history and was not taking any medications. The child otherwise felt well and was afebrile. An examination revealed sharply demarcated, bright red perianal erythema. The rash was moist, with a slight mucoid discharge. The area was not tender on palpation. The remainder of the little boy’s physical examination was unremarkable.
FIGURE 1
Perianal rash
What is your diagnosis?
How would you manage this condition?
Diagnosis: Perianal streptococcal dermatitis
A rapid strep test of the perianal lesion was positive, confirming the diagnosis of perianal streptococcal dermatitis (FIGURE 2). Perianal streptococcal dermatitis typically presents as a bright red, moist, sharply demarcated perianal rash. Itching, rectal pain, and blood-streaked stools are common.
Mucoid or serosanguinous oozing from the affected area can also occur. Less commonly, a patient may have perianal swelling, tenesmus, constipation, and anal fissures. Patients with perianal streptococcal dermatitis are afebrile and show no systemic signs of infection.
FIGURE 2
Rapid stress tests for the pharyngeal and perianal areas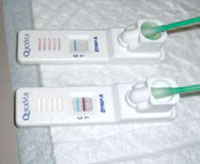
A common condition in children
Perianal streptococcal dermatitis occurs mostly in children between 6 months and 10 years of age, although cases in adults have been reported.1 The incidence in pediatric practices ranges from 1 in 200 to 1 in 2000, with a male to female ratio between 2:1 and 3:1.1 Transmission to family members and contacts in daycare settings have been reported.1,2
The culprit? group A strep
Group A beta-hemolytic streptococcus (GABHS) is the culprit with this form of dermatitis. While the condition was once referred to as “perianal cellulitis,” the indolent nature of the infection and the related itching lent support to the more common description of perianal streptococcal dermatitis.
Up to 92% of the cases of perianal streptococcal dermatitis involve positive pharyngeal cultures for GABHS, even in the absence of pharyngeal symptoms.3 This lends support to the theory that auto-inoculation is the cause for perianal or perineal disease.1 Asymptomatic perineal carriage of GABHS is rare in healthy people, but has been found in 6% of children with streptococcal pharyngitis.1
Rarely, group B or G beta-hemolytic streptococcus or Staphylococcus aureus is identified by culture as the cause of disease.
A condition that’s easy to mistake for candidiasis
The differential diagnosis of perianal streptococcal dermatitis includes candidiasis, diaper dermatitis, irritant dermatitis (such as trauma from heavy wiping), atopic dermatitis, allergic contact dermatitis, seborrheic dermatitis, pinworm infection, cellulitis, psoriasis, inflammatory bowel disease, histiocytosis, and sexual abuse. Patients are often initially misdiagnosed and come back to the office when treatment with topical steroids, topical antifungals, or oral regimens for pinworm infection fail.
Suspect perianal streptococcal dermatitis when the patient presents with a well-dermarcated, moist, bright red perianal rash with no satellite lesions. Also, consider this diagnosis when a perianal rash fails to respond to initial treatment as expected.
A rapid strep test or culture of the affected region helps to confirm perianal streptococcal dermatitis caused by GABHS.
Penicillin provides prompt improvement
Penicillin V or amoxicillin (40 mg/kg/ day divided into 3 oral doses daily) for 10 days is effective as a first-line treatment for perianal streptococcal dermatitis (strength of recommendation [SOR]: C).4
The amoxicillin suspension tastes better than the penicillin suspension, which may lead to better compliance in children.5
Topical mupirocin (Bactroban) 2% applied 3 times daily may also be effective. If a patient is allergic to penicillin, you may want to consider the macrolides (erythromycin, azithromycin, clarithromycin) or clindamycin.1
Once the patient takes his or her medication, clinical improvement is prompt; it often occurs within 24 hours.
Relapse is common. Clinical follow-up is indicated as relapse occurs in up to 39% of cases.6 Relapses usually respond to repeat courses with the same antibiotic. A prolonged treatment course (14 to 21 days) may increase cure rates in patients with relapse.1
No relapse for our young patient
Our young patient received a 10-day course of amoxicillin and his case of perianal streptococcal dermatitis resolved. He did not have a relapse.
Acknowledgments
The authors thank Robert Norman, MD for identifying and sharing this case.
Correspondence
Andrew D. Schechtman, MD, San Jose-O’Connor Hospital Family Medicine Residency, 455 O’Connor Dr.#210, San Jose, CA 95128; [email protected]
1. Herbst R. Perineal streptococcal dermatitis/disease: recognition and management. Am J Clin Dermatol 2003;4:555-560.
2. Brilliant LC. Perianal streptococcal dermatitis. Am Fam Physician 2000;61:391-397.
3. Mogielnicki NP, Schwartzman JD, Elliott JA. Perineal Group a streptococcal disease in a pediatric practice. Pediatrics 2000;106:276-280.
4. Barzilai A, Choen HA. Isolation of Group A Streptococci from children with perianal cellulitis and from their siblings. Pediatr Infect Dis J 1998;17:358-360.
5. Chan DS, Demers DM, Bass JW. Ann Pharmacother 1996;30:130-132.
6. Kokx NP, Comstock JA, Facklam RR. Streptococcal perianal disease in children. Pediatrics 1987;80:659-663.
A mother brought her 3½-year-old son into the office for a worsening rash in the perianal area (FIGURE 1). Mother and child had been in the office 2 weeks earlier for the rash, and the physician had prescribed topical clotrimazole cream for a presumed case of Candida infection. Despite using the cream as directed, the child’s rash worsened.
The child’s mother told the treating physician (SS) that his rash was painful and itchy. She also said he’d had a few blood-streaked stools.
The little boy had no significant medical history and was not taking any medications. The child otherwise felt well and was afebrile. An examination revealed sharply demarcated, bright red perianal erythema. The rash was moist, with a slight mucoid discharge. The area was not tender on palpation. The remainder of the little boy’s physical examination was unremarkable.
FIGURE 1
Perianal rash
What is your diagnosis?
How would you manage this condition?
Diagnosis: Perianal streptococcal dermatitis
A rapid strep test of the perianal lesion was positive, confirming the diagnosis of perianal streptococcal dermatitis (FIGURE 2). Perianal streptococcal dermatitis typically presents as a bright red, moist, sharply demarcated perianal rash. Itching, rectal pain, and blood-streaked stools are common.
Mucoid or serosanguinous oozing from the affected area can also occur. Less commonly, a patient may have perianal swelling, tenesmus, constipation, and anal fissures. Patients with perianal streptococcal dermatitis are afebrile and show no systemic signs of infection.
FIGURE 2
Rapid stress tests for the pharyngeal and perianal areas
A common condition in children
Perianal streptococcal dermatitis occurs mostly in children between 6 months and 10 years of age, although cases in adults have been reported.1 The incidence in pediatric practices ranges from 1 in 200 to 1 in 2000, with a male to female ratio between 2:1 and 3:1.1 Transmission to family members and contacts in daycare settings have been reported.1,2
The culprit? group A strep
Group A beta-hemolytic streptococcus (GABHS) is the culprit with this form of dermatitis. While the condition was once referred to as “perianal cellulitis,” the indolent nature of the infection and the related itching lent support to the more common description of perianal streptococcal dermatitis.
Up to 92% of the cases of perianal streptococcal dermatitis involve positive pharyngeal cultures for GABHS, even in the absence of pharyngeal symptoms.3 This lends support to the theory that auto-inoculation is the cause for perianal or perineal disease.1 Asymptomatic perineal carriage of GABHS is rare in healthy people, but has been found in 6% of children with streptococcal pharyngitis.1
Rarely, group B or G beta-hemolytic streptococcus or Staphylococcus aureus is identified by culture as the cause of disease.
A condition that’s easy to mistake for candidiasis
The differential diagnosis of perianal streptococcal dermatitis includes candidiasis, diaper dermatitis, irritant dermatitis (such as trauma from heavy wiping), atopic dermatitis, allergic contact dermatitis, seborrheic dermatitis, pinworm infection, cellulitis, psoriasis, inflammatory bowel disease, histiocytosis, and sexual abuse. Patients are often initially misdiagnosed and come back to the office when treatment with topical steroids, topical antifungals, or oral regimens for pinworm infection fail.
Suspect perianal streptococcal dermatitis when the patient presents with a well-dermarcated, moist, bright red perianal rash with no satellite lesions. Also, consider this diagnosis when a perianal rash fails to respond to initial treatment as expected.
A rapid strep test or culture of the affected region helps to confirm perianal streptococcal dermatitis caused by GABHS.
Penicillin provides prompt improvement
Penicillin V or amoxicillin (40 mg/kg/ day divided into 3 oral doses daily) for 10 days is effective as a first-line treatment for perianal streptococcal dermatitis (strength of recommendation [SOR]: C).4
The amoxicillin suspension tastes better than the penicillin suspension, which may lead to better compliance in children.5
Topical mupirocin (Bactroban) 2% applied 3 times daily may also be effective. If a patient is allergic to penicillin, you may want to consider the macrolides (erythromycin, azithromycin, clarithromycin) or clindamycin.1
Once the patient takes his or her medication, clinical improvement is prompt; it often occurs within 24 hours.
Relapse is common. Clinical follow-up is indicated as relapse occurs in up to 39% of cases.6 Relapses usually respond to repeat courses with the same antibiotic. A prolonged treatment course (14 to 21 days) may increase cure rates in patients with relapse.1
No relapse for our young patient
Our young patient received a 10-day course of amoxicillin and his case of perianal streptococcal dermatitis resolved. He did not have a relapse.
Acknowledgments
The authors thank Robert Norman, MD for identifying and sharing this case.
Correspondence
Andrew D. Schechtman, MD, San Jose-O’Connor Hospital Family Medicine Residency, 455 O’Connor Dr.#210, San Jose, CA 95128; [email protected]
A mother brought her 3½-year-old son into the office for a worsening rash in the perianal area (FIGURE 1). Mother and child had been in the office 2 weeks earlier for the rash, and the physician had prescribed topical clotrimazole cream for a presumed case of Candida infection. Despite using the cream as directed, the child’s rash worsened.
The child’s mother told the treating physician (SS) that his rash was painful and itchy. She also said he’d had a few blood-streaked stools.
The little boy had no significant medical history and was not taking any medications. The child otherwise felt well and was afebrile. An examination revealed sharply demarcated, bright red perianal erythema. The rash was moist, with a slight mucoid discharge. The area was not tender on palpation. The remainder of the little boy’s physical examination was unremarkable.
FIGURE 1
Perianal rash
What is your diagnosis?
How would you manage this condition?
Diagnosis: Perianal streptococcal dermatitis
A rapid strep test of the perianal lesion was positive, confirming the diagnosis of perianal streptococcal dermatitis (FIGURE 2). Perianal streptococcal dermatitis typically presents as a bright red, moist, sharply demarcated perianal rash. Itching, rectal pain, and blood-streaked stools are common.
Mucoid or serosanguinous oozing from the affected area can also occur. Less commonly, a patient may have perianal swelling, tenesmus, constipation, and anal fissures. Patients with perianal streptococcal dermatitis are afebrile and show no systemic signs of infection.
FIGURE 2
Rapid stress tests for the pharyngeal and perianal areas
A common condition in children
Perianal streptococcal dermatitis occurs mostly in children between 6 months and 10 years of age, although cases in adults have been reported.1 The incidence in pediatric practices ranges from 1 in 200 to 1 in 2000, with a male to female ratio between 2:1 and 3:1.1 Transmission to family members and contacts in daycare settings have been reported.1,2
The culprit? group A strep
Group A beta-hemolytic streptococcus (GABHS) is the culprit with this form of dermatitis. While the condition was once referred to as “perianal cellulitis,” the indolent nature of the infection and the related itching lent support to the more common description of perianal streptococcal dermatitis.
Up to 92% of the cases of perianal streptococcal dermatitis involve positive pharyngeal cultures for GABHS, even in the absence of pharyngeal symptoms.3 This lends support to the theory that auto-inoculation is the cause for perianal or perineal disease.1 Asymptomatic perineal carriage of GABHS is rare in healthy people, but has been found in 6% of children with streptococcal pharyngitis.1
Rarely, group B or G beta-hemolytic streptococcus or Staphylococcus aureus is identified by culture as the cause of disease.
A condition that’s easy to mistake for candidiasis
The differential diagnosis of perianal streptococcal dermatitis includes candidiasis, diaper dermatitis, irritant dermatitis (such as trauma from heavy wiping), atopic dermatitis, allergic contact dermatitis, seborrheic dermatitis, pinworm infection, cellulitis, psoriasis, inflammatory bowel disease, histiocytosis, and sexual abuse. Patients are often initially misdiagnosed and come back to the office when treatment with topical steroids, topical antifungals, or oral regimens for pinworm infection fail.
Suspect perianal streptococcal dermatitis when the patient presents with a well-dermarcated, moist, bright red perianal rash with no satellite lesions. Also, consider this diagnosis when a perianal rash fails to respond to initial treatment as expected.
A rapid strep test or culture of the affected region helps to confirm perianal streptococcal dermatitis caused by GABHS.
Penicillin provides prompt improvement
Penicillin V or amoxicillin (40 mg/kg/ day divided into 3 oral doses daily) for 10 days is effective as a first-line treatment for perianal streptococcal dermatitis (strength of recommendation [SOR]: C).4
The amoxicillin suspension tastes better than the penicillin suspension, which may lead to better compliance in children.5
Topical mupirocin (Bactroban) 2% applied 3 times daily may also be effective. If a patient is allergic to penicillin, you may want to consider the macrolides (erythromycin, azithromycin, clarithromycin) or clindamycin.1
Once the patient takes his or her medication, clinical improvement is prompt; it often occurs within 24 hours.
Relapse is common. Clinical follow-up is indicated as relapse occurs in up to 39% of cases.6 Relapses usually respond to repeat courses with the same antibiotic. A prolonged treatment course (14 to 21 days) may increase cure rates in patients with relapse.1
No relapse for our young patient
Our young patient received a 10-day course of amoxicillin and his case of perianal streptococcal dermatitis resolved. He did not have a relapse.
Acknowledgments
The authors thank Robert Norman, MD for identifying and sharing this case.
Correspondence
Andrew D. Schechtman, MD, San Jose-O’Connor Hospital Family Medicine Residency, 455 O’Connor Dr.#210, San Jose, CA 95128; [email protected]
1. Herbst R. Perineal streptococcal dermatitis/disease: recognition and management. Am J Clin Dermatol 2003;4:555-560.
2. Brilliant LC. Perianal streptococcal dermatitis. Am Fam Physician 2000;61:391-397.
3. Mogielnicki NP, Schwartzman JD, Elliott JA. Perineal Group a streptococcal disease in a pediatric practice. Pediatrics 2000;106:276-280.
4. Barzilai A, Choen HA. Isolation of Group A Streptococci from children with perianal cellulitis and from their siblings. Pediatr Infect Dis J 1998;17:358-360.
5. Chan DS, Demers DM, Bass JW. Ann Pharmacother 1996;30:130-132.
6. Kokx NP, Comstock JA, Facklam RR. Streptococcal perianal disease in children. Pediatrics 1987;80:659-663.
1. Herbst R. Perineal streptococcal dermatitis/disease: recognition and management. Am J Clin Dermatol 2003;4:555-560.
2. Brilliant LC. Perianal streptococcal dermatitis. Am Fam Physician 2000;61:391-397.
3. Mogielnicki NP, Schwartzman JD, Elliott JA. Perineal Group a streptococcal disease in a pediatric practice. Pediatrics 2000;106:276-280.
4. Barzilai A, Choen HA. Isolation of Group A Streptococci from children with perianal cellulitis and from their siblings. Pediatr Infect Dis J 1998;17:358-360.
5. Chan DS, Demers DM, Bass JW. Ann Pharmacother 1996;30:130-132.
6. Kokx NP, Comstock JA, Facklam RR. Streptococcal perianal disease in children. Pediatrics 1987;80:659-663.
Pruritus in pregnancy
A 32-year-old mother of 2 came into our facility during her 31st week of pregnancy and told us that she couldn’t stop itching. She said that her whole body was itchy and it got worse at night. She was unable to get a good night’s sleep. Up until this point, her pregnancy had been uncomplicated and she had no past history of medical problems.
An examination revealed excoriations—but no blistering—on her abdomen, chest, arms, and legs (FIGURES 1 AND 2). She had no jaundice or scleral icterus. The fundal height was 31 mm and the fetal heart tones were 150 bpm.
FIGURE 1 & 2
Excoriations on the abdomen and chest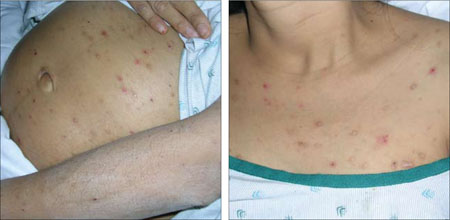
What is your diagnosis?
How would you manage this condition?
Diagnosis: Intrahepatic cholestasis of pregnancy
Intrahepatic cholestasis of pregnancy (ICP) is caused by maternal intrahepatic bile secretory dysfunction.1 The disorder, which is also referred to as obstetric cholestasis and pruritus gravidarum, has no primary skin lesions. Patients have generalized pruritus and secondary excoriations (FIGURE 3). In about 20% of cases, patients are also jaundiced.2
The sudden onset of generalized pruritus, which is the hallmark of ICP, starts during the late second (20%) or third (80%) trimester, followed by secondary skin lesions, namely linear excoriations and excoriated papules caused by scratching.3 These excoriations are typically localized on the extensor surfaces of the limbs, but may also be found on the abdomen and back. The itching may involve the palms and soles, as well. The severity of the skin lesions correlates with the duration and degree of pruritus.3
According to one study, ICP occurred in 0.5% of 3192 pregnancies. The disorder resolves after delivery, and recurs with subsequent pregnancies.4
ICP has been linked to fetal distress (20%–30%), stillbirths (1%–2%), and preterm delivery (20%–60%).3 Autopsies of the placenta have shown signs of acute anoxia. Fetal complications in ICP may be caused by decreased fetal elimination of toxic bile acids.3
Hormones, genes, and even the weather may play a role
Increased hormone production during pregnancy plays a role in ICP. Estrogen, which increases 100-fold during pregnancy, interferes with bile acid secretion across the basolateral and canalicular membrane of the hepatocyte. Particularly noteworthy is the fact that estriol-16 α-D-glucuronide, the estrogen metabolite that increases most during pregnancy, is cholestatic, according to animal studies.3 In addition, progesterone metabolites play an important part in the pathogenesis of ICP. Progesterone inhibits hepatic glucuronyl transferase, thereby reducing the clearance of estrogens and amplifying their effects.
Familial clustering and geographical variation indicate that there is a genetic predisposition for ICP. There is a high prevalence of ICP in Chile (14%), especially among Araucanian Indian women (24%), and in Bolivia.3 ICP patients may have a family history of cholelithiasis and a higher risk of gallstones. There is a family history of cholelithiasis in 50% of ICP cases.2
Exogenous factors have been implicated in the pathogenesis of ICP. There is a higher incidence of ICP during the winter. Low selenium levels have also been linked to ICP. This suggests that the environment and nutritional factors play a role.2
FIGURE 3
Excoriations down the legs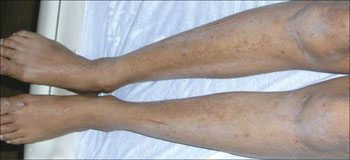
The differential Dx includes scabies
Itching has been reported to occur in 17% of pregnancies,2 so it is important to differentiate ICP from the conditions listed below.
- Pruritic urticarial papules and plaques of pregnancy—also known as polymorphic eruption of pregnancy—is a dermatosis of pregnancy. Unlike the excoriations of ICP, this condition involves papulovesicular or urticarial eruptions on the trunk and extremities. It is particularly pronounced around the abdominal striae, and is more common in nulliparous women.
- Pemphigoid gestationis, also known as herpes gestationis because of its appearance, is a bullous or blistering disease that is associated with pregnancy. It is often periumbilical and can also have target lesions, which are absent in ICP.
- Atopic eruption of pregnancy is a new term used to include previous non-specific diagnoses such as prurigo of pregnancy and pruritic folliculitis of pregnancy. Prurigo of pregnancy, which is also called Besnier’s prurigo gestationis, involves bite-like papules that resemble scabies. Pruritic folliculitis of pregnancy is characterized by red, follicle-based papules. These 2 conditions differ from ICP in that there is no cholestasis and liver studies are normal. (In ICP, there is an elevation in liver enzymes and serum bile acids.)
- Scabies infestation can occur during pregnancy. The mite burrows in the skin and produces severe itching between the fingers and in skin folds. Look for burrows and the typical distribution between the fingers, on the wrists, in the axilla, and around the waist. A positive scraping viewed under the microscope will show mites, eggs, and mite feces.
If you suspect ICP in a patient who is also jaundiced, you’ll also need to rule out several other conditions. These include:
- acute liver disease of pregnancy
- preeclampsia complicated by increased liver enzymes
- hyperemesis gravidarum
- viral hepatitis
- drug reaction
- obstructive biliary disease, such as a gallstone lodged in the common bile duct.
Order a blood chemistry or liver profile
If you suspect that your patient has ICP, start by ordering a blood chemistry or liver profile. If any of the liver tests are elevated, order a total bile acid level (which is the most sensitive indicator of ICP) and a hepatitis panel (or specific hepatitis tests based on the patient’s history of exposures and vaccinations). If there is laboratory evidence of cholestasis, a right upper quadrant ultrasound will help you to spot gallstones and evidence of obstruction.
In ICP, there will be mild abnormalities of the liver function tests, including transaminases, alkaline phosphatase, and bilirubin. Bilirubin may be mildly to moderately elevated (2–5 mg/dL). (Jaundice is seen only at the higher levels of bilirubin.) Our patient’s tests, for instance, revealed that her ALT and AST were both over 300; her total bilirubin was elevated at 2.1.
Serum levels of bile acid correlate with the severity of pruritus. Our patient’s bile acids were elevated and her hepatitis panel was negative. Her ultrasound showed gallstones, but we saw no obstruction. An ICP patient’s lipid profile may show mild elevations in total cholesterol and triglycerides, as was the case for our patient.
Malabsorption of fat may cause vitamin K deficiency resulting in a prolonged prothrombin time. Liver biopsy is unnecessary in suspected cases of ICP, but would show cholestatic changes such as dilated bile canaliculi, bile pigment in the parenchyma, and minimal inflammation.
Skin biopsy is not helpful in ICP
In suspected cases of ICP, skin biopsy will only reveal a spectrum of non-specific findings. It is, however, helpful if you suspect pemphigoid gestationis, since it will reveal subepidermal blisters. Similarly, biopsy for direct immunofluorescence is nonspecific in ICP, but helpful in pemphigoid gestationis.
Soothing baths can help, ursodiol is most effective
Mild cholestasis responds to symptomatic treatment with soothing baths, topical antipruritics, emollients, and primrose oil, among others. Antihistamines are rarely effective. Anion exchange resins, such as cholestyramine, can be helpful, too; they bind bile acids and decrease their enterohepatic circulation.2
Patients who do not respond to cholestyramine, or who cannot tolerate it, may be treated with ursodeoxycholic acid (ursodiol). The research indicates that ursodiol works faster than cholestyramine, has a more sustained effect on pruritus, and is more effective in improving the biochemical abnormalities of ICP (strength of recommendation [SOR]: A, based on good-quality patient-oriented evidence). Ursodiol is considered safe for both mother and fetus.5 For all of these reasons, ursodiol has replaced cholestyramine as the first-line agent for ICP.
Doses range from 1 g/day to high doses of 1.5 to 2.0 g/d.6 The dose is maintained until delivery. Davies et al5 suggest that the use of ursodiol can reduce fetal mortality associated with ICP (SOR: C, based on consensus, usual practice, opinion, disease-oriented evidence, case series).
Weekly non-stress tests beginning at the 34th week of gestation are advisable (SOR: C).2 Labor may need to be induced in the 38th week in mild cases of ICP, and in the 36th week in severe cases (SOR: C).2
Ursodiol for our patient, labor was induced
We treated our patient with oral ursodiol and topical 1% hydrocortisone cream. Her bile acids and transaminase levels dropped and her pruritus improved—though it did not completely resolve until after delivery. Our obstetrics department recommended weekly non-stress tests starting at the 34th week of gestation. The non-stress tests were reactive. Due to the severity of her condition, labor was induced at 36 weeks.
Our patient had a healthy baby girl without complications. After delivery, the itching went away completely and her skin began to heal from all of those excoriations. Our patient is planning an elective cholecystectomy in the coming months because she doesn’t want to take a chance that she might have problems with her gallstones in the future.
Disclosure
1. Galaria NA, Mercurio MG. Dermatoses of pregnancy. The Female Patient 2003. Available at: www.femalepatient.com/html/arc/sel/may03/028_05_024.asp. Accessed on October 8, 2007.
2. Kroumpouzos G. Intrahepatic cholestasis of pregnancy: what’s new. European Academy of Dermatology and Venereology 2002;16:316-318.
3. Ambros-Rudolph CM, Müllegger RR, Vaughan Jones SA, et al. The specific dermatoses of pregnancy revisited and reclassified: results of a retrospective two-center study on 505 pregnant patients. J Am Acad Dermatol 2006;54:395-404.
4. Odom R, James W. Andrews’Diseases of the Skin. 10th ed. Philadelphia, PA: WB Saunders Company; 2006.
5. Davies MH, da Silva RCMA, Jones SR, et al. Fetal mortality associated with cholestasis of pregnancy and the potential benefit of therapy with ursodeoxycholic acid. Gut 1995;37:580-584.
6. Mazzella G, Nicola R, Francesco A, et al. Ursodeoxycholic acid administration in patients with cholestasis of pregnancy: Effects on primary bile acids in babies and mothers. Hepatology 2001;33:504-508.
A 32-year-old mother of 2 came into our facility during her 31st week of pregnancy and told us that she couldn’t stop itching. She said that her whole body was itchy and it got worse at night. She was unable to get a good night’s sleep. Up until this point, her pregnancy had been uncomplicated and she had no past history of medical problems.
An examination revealed excoriations—but no blistering—on her abdomen, chest, arms, and legs (FIGURES 1 AND 2). She had no jaundice or scleral icterus. The fundal height was 31 mm and the fetal heart tones were 150 bpm.
FIGURE 1 & 2
Excoriations on the abdomen and chest
What is your diagnosis?
How would you manage this condition?
Diagnosis: Intrahepatic cholestasis of pregnancy
Intrahepatic cholestasis of pregnancy (ICP) is caused by maternal intrahepatic bile secretory dysfunction.1 The disorder, which is also referred to as obstetric cholestasis and pruritus gravidarum, has no primary skin lesions. Patients have generalized pruritus and secondary excoriations (FIGURE 3). In about 20% of cases, patients are also jaundiced.2
The sudden onset of generalized pruritus, which is the hallmark of ICP, starts during the late second (20%) or third (80%) trimester, followed by secondary skin lesions, namely linear excoriations and excoriated papules caused by scratching.3 These excoriations are typically localized on the extensor surfaces of the limbs, but may also be found on the abdomen and back. The itching may involve the palms and soles, as well. The severity of the skin lesions correlates with the duration and degree of pruritus.3
According to one study, ICP occurred in 0.5% of 3192 pregnancies. The disorder resolves after delivery, and recurs with subsequent pregnancies.4
ICP has been linked to fetal distress (20%–30%), stillbirths (1%–2%), and preterm delivery (20%–60%).3 Autopsies of the placenta have shown signs of acute anoxia. Fetal complications in ICP may be caused by decreased fetal elimination of toxic bile acids.3
Hormones, genes, and even the weather may play a role
Increased hormone production during pregnancy plays a role in ICP. Estrogen, which increases 100-fold during pregnancy, interferes with bile acid secretion across the basolateral and canalicular membrane of the hepatocyte. Particularly noteworthy is the fact that estriol-16 α-D-glucuronide, the estrogen metabolite that increases most during pregnancy, is cholestatic, according to animal studies.3 In addition, progesterone metabolites play an important part in the pathogenesis of ICP. Progesterone inhibits hepatic glucuronyl transferase, thereby reducing the clearance of estrogens and amplifying their effects.
Familial clustering and geographical variation indicate that there is a genetic predisposition for ICP. There is a high prevalence of ICP in Chile (14%), especially among Araucanian Indian women (24%), and in Bolivia.3 ICP patients may have a family history of cholelithiasis and a higher risk of gallstones. There is a family history of cholelithiasis in 50% of ICP cases.2
Exogenous factors have been implicated in the pathogenesis of ICP. There is a higher incidence of ICP during the winter. Low selenium levels have also been linked to ICP. This suggests that the environment and nutritional factors play a role.2
FIGURE 3
Excoriations down the legs
The differential Dx includes scabies
Itching has been reported to occur in 17% of pregnancies,2 so it is important to differentiate ICP from the conditions listed below.
- Pruritic urticarial papules and plaques of pregnancy—also known as polymorphic eruption of pregnancy—is a dermatosis of pregnancy. Unlike the excoriations of ICP, this condition involves papulovesicular or urticarial eruptions on the trunk and extremities. It is particularly pronounced around the abdominal striae, and is more common in nulliparous women.
- Pemphigoid gestationis, also known as herpes gestationis because of its appearance, is a bullous or blistering disease that is associated with pregnancy. It is often periumbilical and can also have target lesions, which are absent in ICP.
- Atopic eruption of pregnancy is a new term used to include previous non-specific diagnoses such as prurigo of pregnancy and pruritic folliculitis of pregnancy. Prurigo of pregnancy, which is also called Besnier’s prurigo gestationis, involves bite-like papules that resemble scabies. Pruritic folliculitis of pregnancy is characterized by red, follicle-based papules. These 2 conditions differ from ICP in that there is no cholestasis and liver studies are normal. (In ICP, there is an elevation in liver enzymes and serum bile acids.)
- Scabies infestation can occur during pregnancy. The mite burrows in the skin and produces severe itching between the fingers and in skin folds. Look for burrows and the typical distribution between the fingers, on the wrists, in the axilla, and around the waist. A positive scraping viewed under the microscope will show mites, eggs, and mite feces.
If you suspect ICP in a patient who is also jaundiced, you’ll also need to rule out several other conditions. These include:
- acute liver disease of pregnancy
- preeclampsia complicated by increased liver enzymes
- hyperemesis gravidarum
- viral hepatitis
- drug reaction
- obstructive biliary disease, such as a gallstone lodged in the common bile duct.
Order a blood chemistry or liver profile
If you suspect that your patient has ICP, start by ordering a blood chemistry or liver profile. If any of the liver tests are elevated, order a total bile acid level (which is the most sensitive indicator of ICP) and a hepatitis panel (or specific hepatitis tests based on the patient’s history of exposures and vaccinations). If there is laboratory evidence of cholestasis, a right upper quadrant ultrasound will help you to spot gallstones and evidence of obstruction.
In ICP, there will be mild abnormalities of the liver function tests, including transaminases, alkaline phosphatase, and bilirubin. Bilirubin may be mildly to moderately elevated (2–5 mg/dL). (Jaundice is seen only at the higher levels of bilirubin.) Our patient’s tests, for instance, revealed that her ALT and AST were both over 300; her total bilirubin was elevated at 2.1.
Serum levels of bile acid correlate with the severity of pruritus. Our patient’s bile acids were elevated and her hepatitis panel was negative. Her ultrasound showed gallstones, but we saw no obstruction. An ICP patient’s lipid profile may show mild elevations in total cholesterol and triglycerides, as was the case for our patient.
Malabsorption of fat may cause vitamin K deficiency resulting in a prolonged prothrombin time. Liver biopsy is unnecessary in suspected cases of ICP, but would show cholestatic changes such as dilated bile canaliculi, bile pigment in the parenchyma, and minimal inflammation.
Skin biopsy is not helpful in ICP
In suspected cases of ICP, skin biopsy will only reveal a spectrum of non-specific findings. It is, however, helpful if you suspect pemphigoid gestationis, since it will reveal subepidermal blisters. Similarly, biopsy for direct immunofluorescence is nonspecific in ICP, but helpful in pemphigoid gestationis.
Soothing baths can help, ursodiol is most effective
Mild cholestasis responds to symptomatic treatment with soothing baths, topical antipruritics, emollients, and primrose oil, among others. Antihistamines are rarely effective. Anion exchange resins, such as cholestyramine, can be helpful, too; they bind bile acids and decrease their enterohepatic circulation.2
Patients who do not respond to cholestyramine, or who cannot tolerate it, may be treated with ursodeoxycholic acid (ursodiol). The research indicates that ursodiol works faster than cholestyramine, has a more sustained effect on pruritus, and is more effective in improving the biochemical abnormalities of ICP (strength of recommendation [SOR]: A, based on good-quality patient-oriented evidence). Ursodiol is considered safe for both mother and fetus.5 For all of these reasons, ursodiol has replaced cholestyramine as the first-line agent for ICP.
Doses range from 1 g/day to high doses of 1.5 to 2.0 g/d.6 The dose is maintained until delivery. Davies et al5 suggest that the use of ursodiol can reduce fetal mortality associated with ICP (SOR: C, based on consensus, usual practice, opinion, disease-oriented evidence, case series).
Weekly non-stress tests beginning at the 34th week of gestation are advisable (SOR: C).2 Labor may need to be induced in the 38th week in mild cases of ICP, and in the 36th week in severe cases (SOR: C).2
Ursodiol for our patient, labor was induced
We treated our patient with oral ursodiol and topical 1% hydrocortisone cream. Her bile acids and transaminase levels dropped and her pruritus improved—though it did not completely resolve until after delivery. Our obstetrics department recommended weekly non-stress tests starting at the 34th week of gestation. The non-stress tests were reactive. Due to the severity of her condition, labor was induced at 36 weeks.
Our patient had a healthy baby girl without complications. After delivery, the itching went away completely and her skin began to heal from all of those excoriations. Our patient is planning an elective cholecystectomy in the coming months because she doesn’t want to take a chance that she might have problems with her gallstones in the future.
Disclosure
A 32-year-old mother of 2 came into our facility during her 31st week of pregnancy and told us that she couldn’t stop itching. She said that her whole body was itchy and it got worse at night. She was unable to get a good night’s sleep. Up until this point, her pregnancy had been uncomplicated and she had no past history of medical problems.
An examination revealed excoriations—but no blistering—on her abdomen, chest, arms, and legs (FIGURES 1 AND 2). She had no jaundice or scleral icterus. The fundal height was 31 mm and the fetal heart tones were 150 bpm.
FIGURE 1 & 2
Excoriations on the abdomen and chest
What is your diagnosis?
How would you manage this condition?
Diagnosis: Intrahepatic cholestasis of pregnancy
Intrahepatic cholestasis of pregnancy (ICP) is caused by maternal intrahepatic bile secretory dysfunction.1 The disorder, which is also referred to as obstetric cholestasis and pruritus gravidarum, has no primary skin lesions. Patients have generalized pruritus and secondary excoriations (FIGURE 3). In about 20% of cases, patients are also jaundiced.2
The sudden onset of generalized pruritus, which is the hallmark of ICP, starts during the late second (20%) or third (80%) trimester, followed by secondary skin lesions, namely linear excoriations and excoriated papules caused by scratching.3 These excoriations are typically localized on the extensor surfaces of the limbs, but may also be found on the abdomen and back. The itching may involve the palms and soles, as well. The severity of the skin lesions correlates with the duration and degree of pruritus.3
According to one study, ICP occurred in 0.5% of 3192 pregnancies. The disorder resolves after delivery, and recurs with subsequent pregnancies.4
ICP has been linked to fetal distress (20%–30%), stillbirths (1%–2%), and preterm delivery (20%–60%).3 Autopsies of the placenta have shown signs of acute anoxia. Fetal complications in ICP may be caused by decreased fetal elimination of toxic bile acids.3
Hormones, genes, and even the weather may play a role
Increased hormone production during pregnancy plays a role in ICP. Estrogen, which increases 100-fold during pregnancy, interferes with bile acid secretion across the basolateral and canalicular membrane of the hepatocyte. Particularly noteworthy is the fact that estriol-16 α-D-glucuronide, the estrogen metabolite that increases most during pregnancy, is cholestatic, according to animal studies.3 In addition, progesterone metabolites play an important part in the pathogenesis of ICP. Progesterone inhibits hepatic glucuronyl transferase, thereby reducing the clearance of estrogens and amplifying their effects.
Familial clustering and geographical variation indicate that there is a genetic predisposition for ICP. There is a high prevalence of ICP in Chile (14%), especially among Araucanian Indian women (24%), and in Bolivia.3 ICP patients may have a family history of cholelithiasis and a higher risk of gallstones. There is a family history of cholelithiasis in 50% of ICP cases.2
Exogenous factors have been implicated in the pathogenesis of ICP. There is a higher incidence of ICP during the winter. Low selenium levels have also been linked to ICP. This suggests that the environment and nutritional factors play a role.2
FIGURE 3
Excoriations down the legs
The differential Dx includes scabies
Itching has been reported to occur in 17% of pregnancies,2 so it is important to differentiate ICP from the conditions listed below.
- Pruritic urticarial papules and plaques of pregnancy—also known as polymorphic eruption of pregnancy—is a dermatosis of pregnancy. Unlike the excoriations of ICP, this condition involves papulovesicular or urticarial eruptions on the trunk and extremities. It is particularly pronounced around the abdominal striae, and is more common in nulliparous women.
- Pemphigoid gestationis, also known as herpes gestationis because of its appearance, is a bullous or blistering disease that is associated with pregnancy. It is often periumbilical and can also have target lesions, which are absent in ICP.
- Atopic eruption of pregnancy is a new term used to include previous non-specific diagnoses such as prurigo of pregnancy and pruritic folliculitis of pregnancy. Prurigo of pregnancy, which is also called Besnier’s prurigo gestationis, involves bite-like papules that resemble scabies. Pruritic folliculitis of pregnancy is characterized by red, follicle-based papules. These 2 conditions differ from ICP in that there is no cholestasis and liver studies are normal. (In ICP, there is an elevation in liver enzymes and serum bile acids.)
- Scabies infestation can occur during pregnancy. The mite burrows in the skin and produces severe itching between the fingers and in skin folds. Look for burrows and the typical distribution between the fingers, on the wrists, in the axilla, and around the waist. A positive scraping viewed under the microscope will show mites, eggs, and mite feces.
If you suspect ICP in a patient who is also jaundiced, you’ll also need to rule out several other conditions. These include:
- acute liver disease of pregnancy
- preeclampsia complicated by increased liver enzymes
- hyperemesis gravidarum
- viral hepatitis
- drug reaction
- obstructive biliary disease, such as a gallstone lodged in the common bile duct.
Order a blood chemistry or liver profile
If you suspect that your patient has ICP, start by ordering a blood chemistry or liver profile. If any of the liver tests are elevated, order a total bile acid level (which is the most sensitive indicator of ICP) and a hepatitis panel (or specific hepatitis tests based on the patient’s history of exposures and vaccinations). If there is laboratory evidence of cholestasis, a right upper quadrant ultrasound will help you to spot gallstones and evidence of obstruction.
In ICP, there will be mild abnormalities of the liver function tests, including transaminases, alkaline phosphatase, and bilirubin. Bilirubin may be mildly to moderately elevated (2–5 mg/dL). (Jaundice is seen only at the higher levels of bilirubin.) Our patient’s tests, for instance, revealed that her ALT and AST were both over 300; her total bilirubin was elevated at 2.1.
Serum levels of bile acid correlate with the severity of pruritus. Our patient’s bile acids were elevated and her hepatitis panel was negative. Her ultrasound showed gallstones, but we saw no obstruction. An ICP patient’s lipid profile may show mild elevations in total cholesterol and triglycerides, as was the case for our patient.
Malabsorption of fat may cause vitamin K deficiency resulting in a prolonged prothrombin time. Liver biopsy is unnecessary in suspected cases of ICP, but would show cholestatic changes such as dilated bile canaliculi, bile pigment in the parenchyma, and minimal inflammation.
Skin biopsy is not helpful in ICP
In suspected cases of ICP, skin biopsy will only reveal a spectrum of non-specific findings. It is, however, helpful if you suspect pemphigoid gestationis, since it will reveal subepidermal blisters. Similarly, biopsy for direct immunofluorescence is nonspecific in ICP, but helpful in pemphigoid gestationis.
Soothing baths can help, ursodiol is most effective
Mild cholestasis responds to symptomatic treatment with soothing baths, topical antipruritics, emollients, and primrose oil, among others. Antihistamines are rarely effective. Anion exchange resins, such as cholestyramine, can be helpful, too; they bind bile acids and decrease their enterohepatic circulation.2
Patients who do not respond to cholestyramine, or who cannot tolerate it, may be treated with ursodeoxycholic acid (ursodiol). The research indicates that ursodiol works faster than cholestyramine, has a more sustained effect on pruritus, and is more effective in improving the biochemical abnormalities of ICP (strength of recommendation [SOR]: A, based on good-quality patient-oriented evidence). Ursodiol is considered safe for both mother and fetus.5 For all of these reasons, ursodiol has replaced cholestyramine as the first-line agent for ICP.
Doses range from 1 g/day to high doses of 1.5 to 2.0 g/d.6 The dose is maintained until delivery. Davies et al5 suggest that the use of ursodiol can reduce fetal mortality associated with ICP (SOR: C, based on consensus, usual practice, opinion, disease-oriented evidence, case series).
Weekly non-stress tests beginning at the 34th week of gestation are advisable (SOR: C).2 Labor may need to be induced in the 38th week in mild cases of ICP, and in the 36th week in severe cases (SOR: C).2
Ursodiol for our patient, labor was induced
We treated our patient with oral ursodiol and topical 1% hydrocortisone cream. Her bile acids and transaminase levels dropped and her pruritus improved—though it did not completely resolve until after delivery. Our obstetrics department recommended weekly non-stress tests starting at the 34th week of gestation. The non-stress tests were reactive. Due to the severity of her condition, labor was induced at 36 weeks.
Our patient had a healthy baby girl without complications. After delivery, the itching went away completely and her skin began to heal from all of those excoriations. Our patient is planning an elective cholecystectomy in the coming months because she doesn’t want to take a chance that she might have problems with her gallstones in the future.
Disclosure
1. Galaria NA, Mercurio MG. Dermatoses of pregnancy. The Female Patient 2003. Available at: www.femalepatient.com/html/arc/sel/may03/028_05_024.asp. Accessed on October 8, 2007.
2. Kroumpouzos G. Intrahepatic cholestasis of pregnancy: what’s new. European Academy of Dermatology and Venereology 2002;16:316-318.
3. Ambros-Rudolph CM, Müllegger RR, Vaughan Jones SA, et al. The specific dermatoses of pregnancy revisited and reclassified: results of a retrospective two-center study on 505 pregnant patients. J Am Acad Dermatol 2006;54:395-404.
4. Odom R, James W. Andrews’Diseases of the Skin. 10th ed. Philadelphia, PA: WB Saunders Company; 2006.
5. Davies MH, da Silva RCMA, Jones SR, et al. Fetal mortality associated with cholestasis of pregnancy and the potential benefit of therapy with ursodeoxycholic acid. Gut 1995;37:580-584.
6. Mazzella G, Nicola R, Francesco A, et al. Ursodeoxycholic acid administration in patients with cholestasis of pregnancy: Effects on primary bile acids in babies and mothers. Hepatology 2001;33:504-508.
1. Galaria NA, Mercurio MG. Dermatoses of pregnancy. The Female Patient 2003. Available at: www.femalepatient.com/html/arc/sel/may03/028_05_024.asp. Accessed on October 8, 2007.
2. Kroumpouzos G. Intrahepatic cholestasis of pregnancy: what’s new. European Academy of Dermatology and Venereology 2002;16:316-318.
3. Ambros-Rudolph CM, Müllegger RR, Vaughan Jones SA, et al. The specific dermatoses of pregnancy revisited and reclassified: results of a retrospective two-center study on 505 pregnant patients. J Am Acad Dermatol 2006;54:395-404.
4. Odom R, James W. Andrews’Diseases of the Skin. 10th ed. Philadelphia, PA: WB Saunders Company; 2006.
5. Davies MH, da Silva RCMA, Jones SR, et al. Fetal mortality associated with cholestasis of pregnancy and the potential benefit of therapy with ursodeoxycholic acid. Gut 1995;37:580-584.
6. Mazzella G, Nicola R, Francesco A, et al. Ursodeoxycholic acid administration in patients with cholestasis of pregnancy: Effects on primary bile acids in babies and mothers. Hepatology 2001;33:504-508.
Acute onset of rash and oligoarthritis
A 29-year-old man sought treatment at our clinic for an extensive rash he’d developed the month before. The rash was on his scalp, umbilicus, glans penis, palms, and soles of his feet. He reported swelling in his left knee and his fourth toes bilaterally that was exacerbated by weight bearing. During the 2 days prior to his visit to the clinic, the patient said he’d had a fever and night sweats; he denied ocular symptoms, GI complaints, dysuria, or penile discharge.
When asked about his sexual history, the patient noted that he’d had unprotected intercourse with a woman a year earlier that resulted in pain on urination and resolved on its own. Other than a resolved case of oral thrush, the patient had a noncontributory past medical history, took no medications, and had no family history of psoriasis.
A physical exam revealed circinate, scaly, and erythematous plaques covering his entire scalp ( FIGURE 1 ). The patient’s conjunctiva and oropharynx were clear. His fingernails showed hyperkeratosis, subungual debris, and nail fold erythema, without pitting. He also had bilateral swelling of the distal interphalangeal joints of his index fingers.
The patient’s umbilicus had a scaly erythematous plaque, while there were confluent erythematous plaques in the groin area, and on the glans penis. There were also similar erythematous plaques in the axilla and inguinal folds; plaques on the lower extremities had a thicker layer of scale. The patient’s feet had crusted plaques on the plantar surface, hyperkeratotic nails with thick subungual debris, and swelling and tenderness of the fourth toes bilaterally ( FIGURE 2 ).
FIGURE 1
Circinate plaques
FIGURE 2
Hyperkeratotic nails, swollen toe
What is your diagnosis?
Diagnosis: Reiter’s syndrome
This young man had Reiter’s syndrome (RS), a form of reactive arthritis that comprises a small subset of cases within the larger family of rheumatoid factor- seronegative spondyloarthritides—conditions noted primarily for inflammation of the axial skeleton.1
Of historical interest is the fact that this diagnosis shares its name with the man who first described it, Hans Reiter, a Nazi physician who tested unapproved vaccines and performed experimental procedures on victims in concentration camps. The infamous legacy of Reiter’s name has led to the proposal that the syndrome be referred to by another, more descriptive name.2 For the sake of simplicity, we’ll refer to the syndrome by the abbreviation RS.
Look for elements of the classic triad
RS is notoriously inconsistent in its presentation. Only a third of patients will develop the “classic triad”—that is: peripheral arthritis lasting at least 1 month, urethritis (or cervicitis), and conjunctivitis. Nearly half of patients will have only a single element of the triad.3
Patients with RS will complain of generalized malaise and fever and will often describe dysuria with concomitant urethral discharge. If conjunctivitis is present, the patient will report reddened, sensitive eyes. Pain will often originate from axial bones, lower extremities (in an oligoarticular asymmetrical pattern), swollen digits, and the heels (from enthesopathy).
Skin manifestations are often very noticeable and include psoriasiform lesions ( FIGURE 3 ) on the palms, soles, and glans penis. Specifically, you’ll see keratoderma blenorrhagicum ( FIGURE 4 ), brown and red macules/papules with pustular or hyperkeratotic features, on the palmar and plantar surfaces. Erythematous and scaly lesions resembling psoriatic plaques often appear elsewhere on the body. On the uncircumcised penis, these shallow ulcerations have a micropustular, serpiginous border and are referred to as balanitis circinata. However, they may also appear psoriasiform in nature on circumcised men, as was the case with our patient.
Coincident findings include onycholysis and subungual hyperkeratosis, lesions mimicking migratory glossitis, and anterior uveitis.
FIGURE 3
Psoriasiform plaque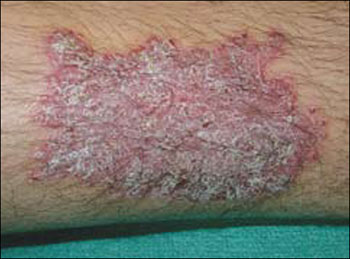
FIGURE 4
Keratoderma blenorrhagicum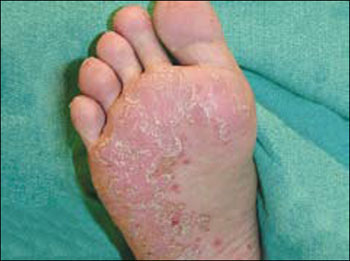
The typical patient? A young, white man
Patients with RS are almost always Caucasian males in their early twenties and are typically HLA-B27 positive. Seronegativity for this HLA factor may portend a less severe version of the syndrome. Individuals infected with HIV show increased incidence of developing RS.3
A microbial antigen is likely responsible for the initial activation of RS. This is followed by an immune reaction involving the joints, skin, and eyes. This theory is supported by the absence of auto- antibodies, the frequent association with HLA-B27, and the fact that patients with advanced AIDS experience the same severity of RS symptoms, despite depressed CD4+ T cell function.1
Bacteria trigger syndrome via 1 of 2 pathways
The bacteria that trigger RS typically enter the body through one of 2 pathways: the genitourinary tract or the gastrointestinal tract.
- The sexual transmission pathway involves infection with Chlamydia trachomatis or Ureaplasma urealyticum 1 to 4 weeks prior to development of urethritis and possibly conjunctivitis. The arthritic component follows later.
- The gastrointestinal pathway involves an enteric pathogen, such as Salmonella enteritidis, Yersinia enterocolitica, Campylobacter fetus, or Shigella flexneri that infects the host and follows the same time frame as noted earlier, though diarrhea rather than urethritis emerges as a chief complaint.4
Various forms of arthritis comprise the differential
A number of conditions must be ruled out before the RS diagnosis is considered definitive. The most likely imposters include:
- Gonococcal arthritis
- Rheumatoid arthritis
- Ankylosing spondylitis
- Psoriatic arthritis.
In addition, an attack of gouty arthritis, systemic lupus erythematosus, serum sickness, Behçet’s syndrome, rheumatic fever, Still’s disease, and HIV could also present in a similar fashion.
The lab work, detailed below, separates RS from the imposters.
Test blood and urine; check the ankles
Although there is no specific test for RS, several laboratory procedures are essential to honing in on the diagnosis. Hematological inquiry will confirm anemia, leukocytosis, thrombocytosis, and an elevated erythrocyte sedimentation rate (ESR). Though the urethral test may not be positive for a suspected organism, this procedure must be done to rule out gonococcal or chlamydial infection. This can now be done on a urine specimen rather than inserting a swab into the urethra. The urine is sent for a polymerase chain reaction (PCR) test rather than a culture. If enteritis was the preceding infection, a stool culture to elucidate potential pathogens is warranted.
You’ll also need to order serological tests for antinuclear antibodies (ANAs), rheumatoid factor (RF), and HIV. As you would expect, these tests will be abnormal for systemic lupus erythematosus, rheumatoid arthritis, and HIV respectively. Though these tests are often negative in RS patients, a strong association with HIV infection does exist.
Keep in mind, too, that you can differentiate gonococcal arthritis from RS based on historical features, as well as clinical features, including migratory polyarthritis with necrotic and pustular skin lesions. Patients with gonococcal arthritis will also have a positive gonococcal culture and rapid improvement with antibiotics.
If you order a biopsy, pathology is likely to find a variety of features in an RS patient, such as spongiform pustules, neutrophilic infiltrate in a perivascular pattern, and an epidermal hyperplasia that resembles psoriasis.3
Radiographic imaging for a suspected case of RS may reveal a number of signs that resemble psoriatic arthritis (pencilin-cup deformity, syndesmophytes, sacroiliitis), but enthesitis, particularly in the ankle joint region, should raise your index of suspicion for RS.6
Tx: Antibiotics, NSAIDs, and steroids
Antibiotic therapy for 3 months is indicated if a patient’s case of RS can be traced back to an infection. If a Chlamydia species is the offending organism, then doxycycline or tetracycline can be used7 (strength of recommendation [SOR]: B). If the infectious agent is unknown, then ciprofloxacin can offer broad-spectrum coverage8 (SOR: B).
Though few studies have evaluated the long-term effects of NSAID treatment on RS, a regular schedule of high doses for several weeks is appropriate for inflammation and pain management. It’s most effective when given early in the disease course5 (SOR: B).
Topical corticosteroids can be used on mucosal and skin lesions. For refractory disease, immunosuppressive agents such as sulfasalazine at 2000 mg/day9 (SOR: B) or a subcutaneous injection of etanercept at 25 mg twice weekly10 (SOR: B) offer relief.
Our patient’s treatment included an NSAID and corticosteroids
Because our patient’s syndrome involved a variety of systemic manifestations, we used several medications to cover all of his symptoms. We prescribed piroxicam 20 mg daily, clobetasol 0.05% ointment applied daily to legs and feet, triamcinolone 0.1% cream applied to scalp twice daily and genitals and armpits once daily, and acitretin 25 mg daily. We consulted Rheumatology to assess and treat his joint disease. We also consulted Ophthalmology to assess for potential ocular manifestations.
Though the patient did report a history of a sexually transmitted infection, it occurred long before his visit, and we were unable to identify an infectious agent. As a result, we did not start him on any antibiotics.
We instructed the patient to return in 2 weeks. Unfortunately, he was lost to follow-up. Patients with RS, though, typically make a full recovery from their symptoms. Some patients, however—10% to 20%—may go on to have a chronic, deforming arthritis.3
1. Winchester R. Reiter’s syndrome. In Freedberg IM, Eisen AZ, Wolf K, Austen KF, Goldsmith LA, Katz SI. Fitzpatrick’s Dermatology in General Medicine. 6th ed. New York, NY: McGraw-Hill; 2003:1769-1776.
2. James WD, Berger TG, Elston DM. Andrew’s Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, Pa: saunders elsevier; 2006.
3. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 5th ed. New York, NY: McGraw-Hill, 2005.
4. Colmegna I, Cuchacovich R, Espinoza LR. HLA-B27-associated reactive arthritis: pathogenetic and clinical considerations. Clin Microbiol Rev 2004;17:348-369.
5. Schachner LA, Hansen RC. Pediatric Dermatology. 2nd ed. New York, NY: churchill livingstone, Inc; 1995.
6. Gladman DD. Clinical aspects of the spondyloarthropathies. Am J Med Sci 1998;316:234-238.
7. Lauhio A, Leirisalo-Repo M, Lähdevirta J, Saikku P, Repo H. Double-blind, placebo-controlled study of three-month treatment with lymecycline in reactive arthritis, with special reference to chlamydia arthritis. Arthritis Rheum 1991;34:6-14.
8. Yli-Kerttula T, Luukkainen R, Yli-Kerttula U, et al. Effect of a three month course of ciprofloxacin on the late prognosis of reactive arthritis. Ann Rheum Dis 2003;62:880-884.
9. Clegg DO, Reda DJ, Weisman MH, et al. Comparison of sulfasalazine and placebo in the treatment of reactive arthritis (Reiter’s syndrome). A Department of Veterans Affairs Cooperative Study. Arthritis Rheum 1996;39:2021-2027.
10. Flagg SD, Meador R, Hsia E, Kitumnuaypong T, Schumacher HR, Jr. Decreased pain and synovial inflammation after etanercept therapy in patients with reactive and undifferentiated arthritis: an open-label trial. Arthritis Rheum 2005;53:613-617.
A 29-year-old man sought treatment at our clinic for an extensive rash he’d developed the month before. The rash was on his scalp, umbilicus, glans penis, palms, and soles of his feet. He reported swelling in his left knee and his fourth toes bilaterally that was exacerbated by weight bearing. During the 2 days prior to his visit to the clinic, the patient said he’d had a fever and night sweats; he denied ocular symptoms, GI complaints, dysuria, or penile discharge.
When asked about his sexual history, the patient noted that he’d had unprotected intercourse with a woman a year earlier that resulted in pain on urination and resolved on its own. Other than a resolved case of oral thrush, the patient had a noncontributory past medical history, took no medications, and had no family history of psoriasis.
A physical exam revealed circinate, scaly, and erythematous plaques covering his entire scalp ( FIGURE 1 ). The patient’s conjunctiva and oropharynx were clear. His fingernails showed hyperkeratosis, subungual debris, and nail fold erythema, without pitting. He also had bilateral swelling of the distal interphalangeal joints of his index fingers.
The patient’s umbilicus had a scaly erythematous plaque, while there were confluent erythematous plaques in the groin area, and on the glans penis. There were also similar erythematous plaques in the axilla and inguinal folds; plaques on the lower extremities had a thicker layer of scale. The patient’s feet had crusted plaques on the plantar surface, hyperkeratotic nails with thick subungual debris, and swelling and tenderness of the fourth toes bilaterally ( FIGURE 2 ).
FIGURE 1
Circinate plaques
FIGURE 2
Hyperkeratotic nails, swollen toe
What is your diagnosis?
Diagnosis: Reiter’s syndrome
This young man had Reiter’s syndrome (RS), a form of reactive arthritis that comprises a small subset of cases within the larger family of rheumatoid factor- seronegative spondyloarthritides—conditions noted primarily for inflammation of the axial skeleton.1
Of historical interest is the fact that this diagnosis shares its name with the man who first described it, Hans Reiter, a Nazi physician who tested unapproved vaccines and performed experimental procedures on victims in concentration camps. The infamous legacy of Reiter’s name has led to the proposal that the syndrome be referred to by another, more descriptive name.2 For the sake of simplicity, we’ll refer to the syndrome by the abbreviation RS.
Look for elements of the classic triad
RS is notoriously inconsistent in its presentation. Only a third of patients will develop the “classic triad”—that is: peripheral arthritis lasting at least 1 month, urethritis (or cervicitis), and conjunctivitis. Nearly half of patients will have only a single element of the triad.3
Patients with RS will complain of generalized malaise and fever and will often describe dysuria with concomitant urethral discharge. If conjunctivitis is present, the patient will report reddened, sensitive eyes. Pain will often originate from axial bones, lower extremities (in an oligoarticular asymmetrical pattern), swollen digits, and the heels (from enthesopathy).
Skin manifestations are often very noticeable and include psoriasiform lesions ( FIGURE 3 ) on the palms, soles, and glans penis. Specifically, you’ll see keratoderma blenorrhagicum ( FIGURE 4 ), brown and red macules/papules with pustular or hyperkeratotic features, on the palmar and plantar surfaces. Erythematous and scaly lesions resembling psoriatic plaques often appear elsewhere on the body. On the uncircumcised penis, these shallow ulcerations have a micropustular, serpiginous border and are referred to as balanitis circinata. However, they may also appear psoriasiform in nature on circumcised men, as was the case with our patient.
Coincident findings include onycholysis and subungual hyperkeratosis, lesions mimicking migratory glossitis, and anterior uveitis.
FIGURE 3
Psoriasiform plaque
FIGURE 4
Keratoderma blenorrhagicum
The typical patient? A young, white man
Patients with RS are almost always Caucasian males in their early twenties and are typically HLA-B27 positive. Seronegativity for this HLA factor may portend a less severe version of the syndrome. Individuals infected with HIV show increased incidence of developing RS.3
A microbial antigen is likely responsible for the initial activation of RS. This is followed by an immune reaction involving the joints, skin, and eyes. This theory is supported by the absence of auto- antibodies, the frequent association with HLA-B27, and the fact that patients with advanced AIDS experience the same severity of RS symptoms, despite depressed CD4+ T cell function.1
Bacteria trigger syndrome via 1 of 2 pathways
The bacteria that trigger RS typically enter the body through one of 2 pathways: the genitourinary tract or the gastrointestinal tract.
- The sexual transmission pathway involves infection with Chlamydia trachomatis or Ureaplasma urealyticum 1 to 4 weeks prior to development of urethritis and possibly conjunctivitis. The arthritic component follows later.
- The gastrointestinal pathway involves an enteric pathogen, such as Salmonella enteritidis, Yersinia enterocolitica, Campylobacter fetus, or Shigella flexneri that infects the host and follows the same time frame as noted earlier, though diarrhea rather than urethritis emerges as a chief complaint.4
Various forms of arthritis comprise the differential
A number of conditions must be ruled out before the RS diagnosis is considered definitive. The most likely imposters include:
- Gonococcal arthritis
- Rheumatoid arthritis
- Ankylosing spondylitis
- Psoriatic arthritis.
In addition, an attack of gouty arthritis, systemic lupus erythematosus, serum sickness, Behçet’s syndrome, rheumatic fever, Still’s disease, and HIV could also present in a similar fashion.
The lab work, detailed below, separates RS from the imposters.
Test blood and urine; check the ankles
Although there is no specific test for RS, several laboratory procedures are essential to honing in on the diagnosis. Hematological inquiry will confirm anemia, leukocytosis, thrombocytosis, and an elevated erythrocyte sedimentation rate (ESR). Though the urethral test may not be positive for a suspected organism, this procedure must be done to rule out gonococcal or chlamydial infection. This can now be done on a urine specimen rather than inserting a swab into the urethra. The urine is sent for a polymerase chain reaction (PCR) test rather than a culture. If enteritis was the preceding infection, a stool culture to elucidate potential pathogens is warranted.
You’ll also need to order serological tests for antinuclear antibodies (ANAs), rheumatoid factor (RF), and HIV. As you would expect, these tests will be abnormal for systemic lupus erythematosus, rheumatoid arthritis, and HIV respectively. Though these tests are often negative in RS patients, a strong association with HIV infection does exist.
Keep in mind, too, that you can differentiate gonococcal arthritis from RS based on historical features, as well as clinical features, including migratory polyarthritis with necrotic and pustular skin lesions. Patients with gonococcal arthritis will also have a positive gonococcal culture and rapid improvement with antibiotics.
If you order a biopsy, pathology is likely to find a variety of features in an RS patient, such as spongiform pustules, neutrophilic infiltrate in a perivascular pattern, and an epidermal hyperplasia that resembles psoriasis.3
Radiographic imaging for a suspected case of RS may reveal a number of signs that resemble psoriatic arthritis (pencilin-cup deformity, syndesmophytes, sacroiliitis), but enthesitis, particularly in the ankle joint region, should raise your index of suspicion for RS.6
Tx: Antibiotics, NSAIDs, and steroids
Antibiotic therapy for 3 months is indicated if a patient’s case of RS can be traced back to an infection. If a Chlamydia species is the offending organism, then doxycycline or tetracycline can be used7 (strength of recommendation [SOR]: B). If the infectious agent is unknown, then ciprofloxacin can offer broad-spectrum coverage8 (SOR: B).
Though few studies have evaluated the long-term effects of NSAID treatment on RS, a regular schedule of high doses for several weeks is appropriate for inflammation and pain management. It’s most effective when given early in the disease course5 (SOR: B).
Topical corticosteroids can be used on mucosal and skin lesions. For refractory disease, immunosuppressive agents such as sulfasalazine at 2000 mg/day9 (SOR: B) or a subcutaneous injection of etanercept at 25 mg twice weekly10 (SOR: B) offer relief.
Our patient’s treatment included an NSAID and corticosteroids
Because our patient’s syndrome involved a variety of systemic manifestations, we used several medications to cover all of his symptoms. We prescribed piroxicam 20 mg daily, clobetasol 0.05% ointment applied daily to legs and feet, triamcinolone 0.1% cream applied to scalp twice daily and genitals and armpits once daily, and acitretin 25 mg daily. We consulted Rheumatology to assess and treat his joint disease. We also consulted Ophthalmology to assess for potential ocular manifestations.
Though the patient did report a history of a sexually transmitted infection, it occurred long before his visit, and we were unable to identify an infectious agent. As a result, we did not start him on any antibiotics.
We instructed the patient to return in 2 weeks. Unfortunately, he was lost to follow-up. Patients with RS, though, typically make a full recovery from their symptoms. Some patients, however—10% to 20%—may go on to have a chronic, deforming arthritis.3
A 29-year-old man sought treatment at our clinic for an extensive rash he’d developed the month before. The rash was on his scalp, umbilicus, glans penis, palms, and soles of his feet. He reported swelling in his left knee and his fourth toes bilaterally that was exacerbated by weight bearing. During the 2 days prior to his visit to the clinic, the patient said he’d had a fever and night sweats; he denied ocular symptoms, GI complaints, dysuria, or penile discharge.
When asked about his sexual history, the patient noted that he’d had unprotected intercourse with a woman a year earlier that resulted in pain on urination and resolved on its own. Other than a resolved case of oral thrush, the patient had a noncontributory past medical history, took no medications, and had no family history of psoriasis.
A physical exam revealed circinate, scaly, and erythematous plaques covering his entire scalp ( FIGURE 1 ). The patient’s conjunctiva and oropharynx were clear. His fingernails showed hyperkeratosis, subungual debris, and nail fold erythema, without pitting. He also had bilateral swelling of the distal interphalangeal joints of his index fingers.
The patient’s umbilicus had a scaly erythematous plaque, while there were confluent erythematous plaques in the groin area, and on the glans penis. There were also similar erythematous plaques in the axilla and inguinal folds; plaques on the lower extremities had a thicker layer of scale. The patient’s feet had crusted plaques on the plantar surface, hyperkeratotic nails with thick subungual debris, and swelling and tenderness of the fourth toes bilaterally ( FIGURE 2 ).
FIGURE 1
Circinate plaques
FIGURE 2
Hyperkeratotic nails, swollen toe
What is your diagnosis?
Diagnosis: Reiter’s syndrome
This young man had Reiter’s syndrome (RS), a form of reactive arthritis that comprises a small subset of cases within the larger family of rheumatoid factor- seronegative spondyloarthritides—conditions noted primarily for inflammation of the axial skeleton.1
Of historical interest is the fact that this diagnosis shares its name with the man who first described it, Hans Reiter, a Nazi physician who tested unapproved vaccines and performed experimental procedures on victims in concentration camps. The infamous legacy of Reiter’s name has led to the proposal that the syndrome be referred to by another, more descriptive name.2 For the sake of simplicity, we’ll refer to the syndrome by the abbreviation RS.
Look for elements of the classic triad
RS is notoriously inconsistent in its presentation. Only a third of patients will develop the “classic triad”—that is: peripheral arthritis lasting at least 1 month, urethritis (or cervicitis), and conjunctivitis. Nearly half of patients will have only a single element of the triad.3
Patients with RS will complain of generalized malaise and fever and will often describe dysuria with concomitant urethral discharge. If conjunctivitis is present, the patient will report reddened, sensitive eyes. Pain will often originate from axial bones, lower extremities (in an oligoarticular asymmetrical pattern), swollen digits, and the heels (from enthesopathy).
Skin manifestations are often very noticeable and include psoriasiform lesions ( FIGURE 3 ) on the palms, soles, and glans penis. Specifically, you’ll see keratoderma blenorrhagicum ( FIGURE 4 ), brown and red macules/papules with pustular or hyperkeratotic features, on the palmar and plantar surfaces. Erythematous and scaly lesions resembling psoriatic plaques often appear elsewhere on the body. On the uncircumcised penis, these shallow ulcerations have a micropustular, serpiginous border and are referred to as balanitis circinata. However, they may also appear psoriasiform in nature on circumcised men, as was the case with our patient.
Coincident findings include onycholysis and subungual hyperkeratosis, lesions mimicking migratory glossitis, and anterior uveitis.
FIGURE 3
Psoriasiform plaque
FIGURE 4
Keratoderma blenorrhagicum
The typical patient? A young, white man
Patients with RS are almost always Caucasian males in their early twenties and are typically HLA-B27 positive. Seronegativity for this HLA factor may portend a less severe version of the syndrome. Individuals infected with HIV show increased incidence of developing RS.3
A microbial antigen is likely responsible for the initial activation of RS. This is followed by an immune reaction involving the joints, skin, and eyes. This theory is supported by the absence of auto- antibodies, the frequent association with HLA-B27, and the fact that patients with advanced AIDS experience the same severity of RS symptoms, despite depressed CD4+ T cell function.1
Bacteria trigger syndrome via 1 of 2 pathways
The bacteria that trigger RS typically enter the body through one of 2 pathways: the genitourinary tract or the gastrointestinal tract.
- The sexual transmission pathway involves infection with Chlamydia trachomatis or Ureaplasma urealyticum 1 to 4 weeks prior to development of urethritis and possibly conjunctivitis. The arthritic component follows later.
- The gastrointestinal pathway involves an enteric pathogen, such as Salmonella enteritidis, Yersinia enterocolitica, Campylobacter fetus, or Shigella flexneri that infects the host and follows the same time frame as noted earlier, though diarrhea rather than urethritis emerges as a chief complaint.4
Various forms of arthritis comprise the differential
A number of conditions must be ruled out before the RS diagnosis is considered definitive. The most likely imposters include:
- Gonococcal arthritis
- Rheumatoid arthritis
- Ankylosing spondylitis
- Psoriatic arthritis.
In addition, an attack of gouty arthritis, systemic lupus erythematosus, serum sickness, Behçet’s syndrome, rheumatic fever, Still’s disease, and HIV could also present in a similar fashion.
The lab work, detailed below, separates RS from the imposters.
Test blood and urine; check the ankles
Although there is no specific test for RS, several laboratory procedures are essential to honing in on the diagnosis. Hematological inquiry will confirm anemia, leukocytosis, thrombocytosis, and an elevated erythrocyte sedimentation rate (ESR). Though the urethral test may not be positive for a suspected organism, this procedure must be done to rule out gonococcal or chlamydial infection. This can now be done on a urine specimen rather than inserting a swab into the urethra. The urine is sent for a polymerase chain reaction (PCR) test rather than a culture. If enteritis was the preceding infection, a stool culture to elucidate potential pathogens is warranted.
You’ll also need to order serological tests for antinuclear antibodies (ANAs), rheumatoid factor (RF), and HIV. As you would expect, these tests will be abnormal for systemic lupus erythematosus, rheumatoid arthritis, and HIV respectively. Though these tests are often negative in RS patients, a strong association with HIV infection does exist.
Keep in mind, too, that you can differentiate gonococcal arthritis from RS based on historical features, as well as clinical features, including migratory polyarthritis with necrotic and pustular skin lesions. Patients with gonococcal arthritis will also have a positive gonococcal culture and rapid improvement with antibiotics.
If you order a biopsy, pathology is likely to find a variety of features in an RS patient, such as spongiform pustules, neutrophilic infiltrate in a perivascular pattern, and an epidermal hyperplasia that resembles psoriasis.3
Radiographic imaging for a suspected case of RS may reveal a number of signs that resemble psoriatic arthritis (pencilin-cup deformity, syndesmophytes, sacroiliitis), but enthesitis, particularly in the ankle joint region, should raise your index of suspicion for RS.6
Tx: Antibiotics, NSAIDs, and steroids
Antibiotic therapy for 3 months is indicated if a patient’s case of RS can be traced back to an infection. If a Chlamydia species is the offending organism, then doxycycline or tetracycline can be used7 (strength of recommendation [SOR]: B). If the infectious agent is unknown, then ciprofloxacin can offer broad-spectrum coverage8 (SOR: B).
Though few studies have evaluated the long-term effects of NSAID treatment on RS, a regular schedule of high doses for several weeks is appropriate for inflammation and pain management. It’s most effective when given early in the disease course5 (SOR: B).
Topical corticosteroids can be used on mucosal and skin lesions. For refractory disease, immunosuppressive agents such as sulfasalazine at 2000 mg/day9 (SOR: B) or a subcutaneous injection of etanercept at 25 mg twice weekly10 (SOR: B) offer relief.
Our patient’s treatment included an NSAID and corticosteroids
Because our patient’s syndrome involved a variety of systemic manifestations, we used several medications to cover all of his symptoms. We prescribed piroxicam 20 mg daily, clobetasol 0.05% ointment applied daily to legs and feet, triamcinolone 0.1% cream applied to scalp twice daily and genitals and armpits once daily, and acitretin 25 mg daily. We consulted Rheumatology to assess and treat his joint disease. We also consulted Ophthalmology to assess for potential ocular manifestations.
Though the patient did report a history of a sexually transmitted infection, it occurred long before his visit, and we were unable to identify an infectious agent. As a result, we did not start him on any antibiotics.
We instructed the patient to return in 2 weeks. Unfortunately, he was lost to follow-up. Patients with RS, though, typically make a full recovery from their symptoms. Some patients, however—10% to 20%—may go on to have a chronic, deforming arthritis.3
1. Winchester R. Reiter’s syndrome. In Freedberg IM, Eisen AZ, Wolf K, Austen KF, Goldsmith LA, Katz SI. Fitzpatrick’s Dermatology in General Medicine. 6th ed. New York, NY: McGraw-Hill; 2003:1769-1776.
2. James WD, Berger TG, Elston DM. Andrew’s Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, Pa: saunders elsevier; 2006.
3. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 5th ed. New York, NY: McGraw-Hill, 2005.
4. Colmegna I, Cuchacovich R, Espinoza LR. HLA-B27-associated reactive arthritis: pathogenetic and clinical considerations. Clin Microbiol Rev 2004;17:348-369.
5. Schachner LA, Hansen RC. Pediatric Dermatology. 2nd ed. New York, NY: churchill livingstone, Inc; 1995.
6. Gladman DD. Clinical aspects of the spondyloarthropathies. Am J Med Sci 1998;316:234-238.
7. Lauhio A, Leirisalo-Repo M, Lähdevirta J, Saikku P, Repo H. Double-blind, placebo-controlled study of three-month treatment with lymecycline in reactive arthritis, with special reference to chlamydia arthritis. Arthritis Rheum 1991;34:6-14.
8. Yli-Kerttula T, Luukkainen R, Yli-Kerttula U, et al. Effect of a three month course of ciprofloxacin on the late prognosis of reactive arthritis. Ann Rheum Dis 2003;62:880-884.
9. Clegg DO, Reda DJ, Weisman MH, et al. Comparison of sulfasalazine and placebo in the treatment of reactive arthritis (Reiter’s syndrome). A Department of Veterans Affairs Cooperative Study. Arthritis Rheum 1996;39:2021-2027.
10. Flagg SD, Meador R, Hsia E, Kitumnuaypong T, Schumacher HR, Jr. Decreased pain and synovial inflammation after etanercept therapy in patients with reactive and undifferentiated arthritis: an open-label trial. Arthritis Rheum 2005;53:613-617.
1. Winchester R. Reiter’s syndrome. In Freedberg IM, Eisen AZ, Wolf K, Austen KF, Goldsmith LA, Katz SI. Fitzpatrick’s Dermatology in General Medicine. 6th ed. New York, NY: McGraw-Hill; 2003:1769-1776.
2. James WD, Berger TG, Elston DM. Andrew’s Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, Pa: saunders elsevier; 2006.
3. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 5th ed. New York, NY: McGraw-Hill, 2005.
4. Colmegna I, Cuchacovich R, Espinoza LR. HLA-B27-associated reactive arthritis: pathogenetic and clinical considerations. Clin Microbiol Rev 2004;17:348-369.
5. Schachner LA, Hansen RC. Pediatric Dermatology. 2nd ed. New York, NY: churchill livingstone, Inc; 1995.
6. Gladman DD. Clinical aspects of the spondyloarthropathies. Am J Med Sci 1998;316:234-238.
7. Lauhio A, Leirisalo-Repo M, Lähdevirta J, Saikku P, Repo H. Double-blind, placebo-controlled study of three-month treatment with lymecycline in reactive arthritis, with special reference to chlamydia arthritis. Arthritis Rheum 1991;34:6-14.
8. Yli-Kerttula T, Luukkainen R, Yli-Kerttula U, et al. Effect of a three month course of ciprofloxacin on the late prognosis of reactive arthritis. Ann Rheum Dis 2003;62:880-884.
9. Clegg DO, Reda DJ, Weisman MH, et al. Comparison of sulfasalazine and placebo in the treatment of reactive arthritis (Reiter’s syndrome). A Department of Veterans Affairs Cooperative Study. Arthritis Rheum 1996;39:2021-2027.
10. Flagg SD, Meador R, Hsia E, Kitumnuaypong T, Schumacher HR, Jr. Decreased pain and synovial inflammation after etanercept therapy in patients with reactive and undifferentiated arthritis: an open-label trial. Arthritis Rheum 2005;53:613-617.
Confluent annular lesion on the dorsum of the hand
A 48-year-old man came to the office with a pruritic lesion that had been on the dorsum of his hand for 2 months. He said that the lesion began as 2 flesh-colored papules that had coalesced to form a larger lesion.
He denied any recent trauma, foreign travel, insect bites, disseminated rashes, or systemic symptoms associated with the appearance of the lesion. His medical history was unremarkable, and he indicated that he’d otherwise been feeling well.
His physical exam was significant for a 6 cm, violaceous annular confluent plaque with a firm, slightly raised border (FIGURE 1). There was no visible scale.
FIGURE 1
Violaceous lesion on the hand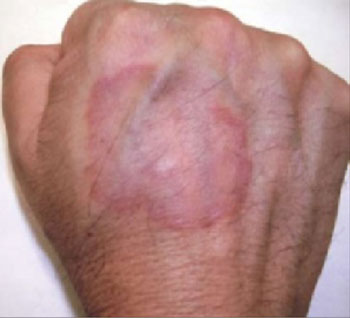
What is your diagnosis?
How would you manage this condition?
Diagnosis: Granuloma annulare
Granuloma annulare is a benign inflammatory skin condition classically described as annular papules and plaques. The lesions typically range from skin-colored to violaceous in appearance. There is no racial predilection for this condition.
There are 4 variants of granuloma annulare:
- The localized form—which our patient had—accounts for approximately 75% of cases.1 Lesions are typically found on the lateral or dorsal surfaces of the hands and feet. Most cases of localized granuloma annulare occur in young adult women.2
- The generalized (disseminated) form involves a number of lesions, and thus, is more widespread (FIGURE 2). Lesions tend to appear on the extremities and the trunk.
- The perforating form is rare, and occurs in both localized and generalized granuloma annulare. Papules may develop into lesions that exude a thick and creamy or clear and viscous fluid.
- The subcutaneous form presents as asymptomatic solitary lesions or in clusters that are most commonly found on the lower extremities—often on pretibial areas. The lesions with this form of granuloma annulare are deeper than the localized form, so there is more swelling, and less surface definition.
Anecdotal reports link condition to diabetes
Although the cause of granuloma annulare is unknown, it has been linked with trauma, thyroid disease, viral infection, malignancy, and diabetes mellitus. Some reports suggest that the lesions are the result of a delayed type hypersensitivity reaction. Of note: Our patient had type 2 diabetes mellitus, something we only discovered while doing a work-up for his lesion. Though largely based on anecdote and case reports, retrospective studies have supported this association.3,4
More recently however, a small case-control study failed to reveal any significant correlation between the 2.5 At this time, there is no indication to screen for diabetes in an otherwise asymptomatic patient.
FIGURE 2
Generalized granuloma annulare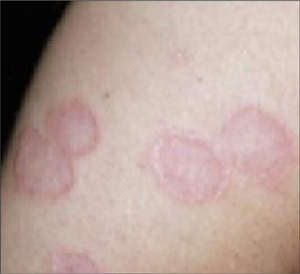
Differential Dx includes ringworm, Lyme disease
Localized granuloma annulare must be differentiated from the most common annular lesion, tinea corporis, as well as other annular lesions, including necrobiosis lipoidica and erythema migrans.
- Tinea corporis, or ringworm, is a superficial fungal infection of the skin. Similar to granuloma annulare, patients present with a gradually enlarging annular, well-demarcated papular lesion. Ringworm can be distinguished from granuloma annulare by noting the presence of scale, and by performing a potassium hydroxide slide preparation, which would reveal septate hyphae.
- Necrobiosis lipoidica classically presents as annular violaceous plaques on the anterior legs, but may appear on the arms, hands, feet, or scalp. As the lesion expands, the advancing border becomes red and the central area typically develops a waxy yellow surface, with prominent telangiectasias. This lesion is generally flat or atrophic, and does not have the same raised border as is seen in granuloma annulare.
- Erythema migrans is the characteristic rash of early localized Lyme disease. This classic “bull’s-eye” rash is an annular lesion with concentric redness and expanding central clearing. Erythema migrans typically appears 7 to 10 days after a patient has been bitten by an infected tick. The classic red lesion starts as a small papule at the site of inoculation, while the expanding ring remains flat and lacks the papular appearance of granuloma annulare.
While uncommon, cutaneous sarcoidosis should also be considered in the differential diagnosis. Cutaneous manifestations occur in up to 20% of patients with sarcoidosis.2 The most common presentation is a maculopapular eruption involving the face. Lesions may also be nodular or plaque-like. Biopsy is necessary for diagnosis.
Clinical presentation typically clinches it
Diagnosis of granuloma annulare is often made by its characteristic clinical presentation. If the diagnosis is unclear, a skin biopsy may be needed. Histologic exam will reveal histiocytes in surrounding dermal tissue with increased mucin deposition.
Cosmetic concerns?
Localized lesions that are asymptomatic are often left to resolve spontaneously. If there are cosmetic concerns, or if there is significant pruritus, treatment options include intralesional steroid injection into the raised border with triamcinolone, occlusion therapy with clobetasol propionate, or liquid nitrogen therapy.
One small study of 31 patients with localized granuloma annulare showed resolution after 1 treatment with liquid nitrogen in 81% of patients.6 Topical steroids alone do not produce significant results.5 Other agents, including UV light and systemic medications, are available for the generalized form, however, none are curative and relapses are common.
No treatment for lesion; Metformin for diabetes
Our patient chose to have no treatment for his granuloma annulare, but we did put him on metformin for his diabetes. At a 3-month follow-up visit, our patient’s lesion was unchanged in appearance.
Although the disease course is variable, 50% of patients with localized granuloma annulare will see spontaneous resolution within 2 years without scarring.7
1. Nopper A, Markus R, Esterly N. When it’s not ring-worm: annular lesions of childhood. Pediatr Ann. 1998;27:136-148.
2. Habif TP. Clinical Dermatology. 4th ed. St louis, mo: mosby, Inc; 2004.
3. Studer EM, Calza AM, Saurat JH. Precipitating factors and associated diseases in 84 patients with granuloma annulare: a retrospective study. Dermatology. 1996;193:364-368.
4. Muhlemann MF. Localized granuloma annulare is associated with insulin-dependent diabetes mellitus. Br J Dermatol. 1984;111:325-329.
5. Nebesio CL, Lewis C, Chuang TY. Lack of an association between granuloma annulare and type 2 diabetes mellitus. Br J Dermatol. 2002;146:122-124.
6. Blume-peytavi U, Zouboulis CH, Jacobi H, Scholz A, Bisson S, Orfanos CE. Successful outcome of cryosurgery in patients with granuloma annulare. Br J Dermatol. 1994;130:494-497.
7. Smith MD, Downie JB, DiCostanzo D. Granuloma annulare. Int J Dermatol. 1997;36:326-333.
A 48-year-old man came to the office with a pruritic lesion that had been on the dorsum of his hand for 2 months. He said that the lesion began as 2 flesh-colored papules that had coalesced to form a larger lesion.
He denied any recent trauma, foreign travel, insect bites, disseminated rashes, or systemic symptoms associated with the appearance of the lesion. His medical history was unremarkable, and he indicated that he’d otherwise been feeling well.
His physical exam was significant for a 6 cm, violaceous annular confluent plaque with a firm, slightly raised border (FIGURE 1). There was no visible scale.
FIGURE 1
Violaceous lesion on the hand
What is your diagnosis?
How would you manage this condition?
Diagnosis: Granuloma annulare
Granuloma annulare is a benign inflammatory skin condition classically described as annular papules and plaques. The lesions typically range from skin-colored to violaceous in appearance. There is no racial predilection for this condition.
There are 4 variants of granuloma annulare:
- The localized form—which our patient had—accounts for approximately 75% of cases.1 Lesions are typically found on the lateral or dorsal surfaces of the hands and feet. Most cases of localized granuloma annulare occur in young adult women.2
- The generalized (disseminated) form involves a number of lesions, and thus, is more widespread (FIGURE 2). Lesions tend to appear on the extremities and the trunk.
- The perforating form is rare, and occurs in both localized and generalized granuloma annulare. Papules may develop into lesions that exude a thick and creamy or clear and viscous fluid.
- The subcutaneous form presents as asymptomatic solitary lesions or in clusters that are most commonly found on the lower extremities—often on pretibial areas. The lesions with this form of granuloma annulare are deeper than the localized form, so there is more swelling, and less surface definition.
Anecdotal reports link condition to diabetes
Although the cause of granuloma annulare is unknown, it has been linked with trauma, thyroid disease, viral infection, malignancy, and diabetes mellitus. Some reports suggest that the lesions are the result of a delayed type hypersensitivity reaction. Of note: Our patient had type 2 diabetes mellitus, something we only discovered while doing a work-up for his lesion. Though largely based on anecdote and case reports, retrospective studies have supported this association.3,4
More recently however, a small case-control study failed to reveal any significant correlation between the 2.5 At this time, there is no indication to screen for diabetes in an otherwise asymptomatic patient.
FIGURE 2
Generalized granuloma annulare
Differential Dx includes ringworm, Lyme disease
Localized granuloma annulare must be differentiated from the most common annular lesion, tinea corporis, as well as other annular lesions, including necrobiosis lipoidica and erythema migrans.
- Tinea corporis, or ringworm, is a superficial fungal infection of the skin. Similar to granuloma annulare, patients present with a gradually enlarging annular, well-demarcated papular lesion. Ringworm can be distinguished from granuloma annulare by noting the presence of scale, and by performing a potassium hydroxide slide preparation, which would reveal septate hyphae.
- Necrobiosis lipoidica classically presents as annular violaceous plaques on the anterior legs, but may appear on the arms, hands, feet, or scalp. As the lesion expands, the advancing border becomes red and the central area typically develops a waxy yellow surface, with prominent telangiectasias. This lesion is generally flat or atrophic, and does not have the same raised border as is seen in granuloma annulare.
- Erythema migrans is the characteristic rash of early localized Lyme disease. This classic “bull’s-eye” rash is an annular lesion with concentric redness and expanding central clearing. Erythema migrans typically appears 7 to 10 days after a patient has been bitten by an infected tick. The classic red lesion starts as a small papule at the site of inoculation, while the expanding ring remains flat and lacks the papular appearance of granuloma annulare.
While uncommon, cutaneous sarcoidosis should also be considered in the differential diagnosis. Cutaneous manifestations occur in up to 20% of patients with sarcoidosis.2 The most common presentation is a maculopapular eruption involving the face. Lesions may also be nodular or plaque-like. Biopsy is necessary for diagnosis.
Clinical presentation typically clinches it
Diagnosis of granuloma annulare is often made by its characteristic clinical presentation. If the diagnosis is unclear, a skin biopsy may be needed. Histologic exam will reveal histiocytes in surrounding dermal tissue with increased mucin deposition.
Cosmetic concerns?
Localized lesions that are asymptomatic are often left to resolve spontaneously. If there are cosmetic concerns, or if there is significant pruritus, treatment options include intralesional steroid injection into the raised border with triamcinolone, occlusion therapy with clobetasol propionate, or liquid nitrogen therapy.
One small study of 31 patients with localized granuloma annulare showed resolution after 1 treatment with liquid nitrogen in 81% of patients.6 Topical steroids alone do not produce significant results.5 Other agents, including UV light and systemic medications, are available for the generalized form, however, none are curative and relapses are common.
No treatment for lesion; Metformin for diabetes
Our patient chose to have no treatment for his granuloma annulare, but we did put him on metformin for his diabetes. At a 3-month follow-up visit, our patient’s lesion was unchanged in appearance.
Although the disease course is variable, 50% of patients with localized granuloma annulare will see spontaneous resolution within 2 years without scarring.7
A 48-year-old man came to the office with a pruritic lesion that had been on the dorsum of his hand for 2 months. He said that the lesion began as 2 flesh-colored papules that had coalesced to form a larger lesion.
He denied any recent trauma, foreign travel, insect bites, disseminated rashes, or systemic symptoms associated with the appearance of the lesion. His medical history was unremarkable, and he indicated that he’d otherwise been feeling well.
His physical exam was significant for a 6 cm, violaceous annular confluent plaque with a firm, slightly raised border (FIGURE 1). There was no visible scale.
FIGURE 1
Violaceous lesion on the hand
What is your diagnosis?
How would you manage this condition?
Diagnosis: Granuloma annulare
Granuloma annulare is a benign inflammatory skin condition classically described as annular papules and plaques. The lesions typically range from skin-colored to violaceous in appearance. There is no racial predilection for this condition.
There are 4 variants of granuloma annulare:
- The localized form—which our patient had—accounts for approximately 75% of cases.1 Lesions are typically found on the lateral or dorsal surfaces of the hands and feet. Most cases of localized granuloma annulare occur in young adult women.2
- The generalized (disseminated) form involves a number of lesions, and thus, is more widespread (FIGURE 2). Lesions tend to appear on the extremities and the trunk.
- The perforating form is rare, and occurs in both localized and generalized granuloma annulare. Papules may develop into lesions that exude a thick and creamy or clear and viscous fluid.
- The subcutaneous form presents as asymptomatic solitary lesions or in clusters that are most commonly found on the lower extremities—often on pretibial areas. The lesions with this form of granuloma annulare are deeper than the localized form, so there is more swelling, and less surface definition.
Anecdotal reports link condition to diabetes
Although the cause of granuloma annulare is unknown, it has been linked with trauma, thyroid disease, viral infection, malignancy, and diabetes mellitus. Some reports suggest that the lesions are the result of a delayed type hypersensitivity reaction. Of note: Our patient had type 2 diabetes mellitus, something we only discovered while doing a work-up for his lesion. Though largely based on anecdote and case reports, retrospective studies have supported this association.3,4
More recently however, a small case-control study failed to reveal any significant correlation between the 2.5 At this time, there is no indication to screen for diabetes in an otherwise asymptomatic patient.
FIGURE 2
Generalized granuloma annulare
Differential Dx includes ringworm, Lyme disease
Localized granuloma annulare must be differentiated from the most common annular lesion, tinea corporis, as well as other annular lesions, including necrobiosis lipoidica and erythema migrans.
- Tinea corporis, or ringworm, is a superficial fungal infection of the skin. Similar to granuloma annulare, patients present with a gradually enlarging annular, well-demarcated papular lesion. Ringworm can be distinguished from granuloma annulare by noting the presence of scale, and by performing a potassium hydroxide slide preparation, which would reveal septate hyphae.
- Necrobiosis lipoidica classically presents as annular violaceous plaques on the anterior legs, but may appear on the arms, hands, feet, or scalp. As the lesion expands, the advancing border becomes red and the central area typically develops a waxy yellow surface, with prominent telangiectasias. This lesion is generally flat or atrophic, and does not have the same raised border as is seen in granuloma annulare.
- Erythema migrans is the characteristic rash of early localized Lyme disease. This classic “bull’s-eye” rash is an annular lesion with concentric redness and expanding central clearing. Erythema migrans typically appears 7 to 10 days after a patient has been bitten by an infected tick. The classic red lesion starts as a small papule at the site of inoculation, while the expanding ring remains flat and lacks the papular appearance of granuloma annulare.
While uncommon, cutaneous sarcoidosis should also be considered in the differential diagnosis. Cutaneous manifestations occur in up to 20% of patients with sarcoidosis.2 The most common presentation is a maculopapular eruption involving the face. Lesions may also be nodular or plaque-like. Biopsy is necessary for diagnosis.
Clinical presentation typically clinches it
Diagnosis of granuloma annulare is often made by its characteristic clinical presentation. If the diagnosis is unclear, a skin biopsy may be needed. Histologic exam will reveal histiocytes in surrounding dermal tissue with increased mucin deposition.
Cosmetic concerns?
Localized lesions that are asymptomatic are often left to resolve spontaneously. If there are cosmetic concerns, or if there is significant pruritus, treatment options include intralesional steroid injection into the raised border with triamcinolone, occlusion therapy with clobetasol propionate, or liquid nitrogen therapy.
One small study of 31 patients with localized granuloma annulare showed resolution after 1 treatment with liquid nitrogen in 81% of patients.6 Topical steroids alone do not produce significant results.5 Other agents, including UV light and systemic medications, are available for the generalized form, however, none are curative and relapses are common.
No treatment for lesion; Metformin for diabetes
Our patient chose to have no treatment for his granuloma annulare, but we did put him on metformin for his diabetes. At a 3-month follow-up visit, our patient’s lesion was unchanged in appearance.
Although the disease course is variable, 50% of patients with localized granuloma annulare will see spontaneous resolution within 2 years without scarring.7
1. Nopper A, Markus R, Esterly N. When it’s not ring-worm: annular lesions of childhood. Pediatr Ann. 1998;27:136-148.
2. Habif TP. Clinical Dermatology. 4th ed. St louis, mo: mosby, Inc; 2004.
3. Studer EM, Calza AM, Saurat JH. Precipitating factors and associated diseases in 84 patients with granuloma annulare: a retrospective study. Dermatology. 1996;193:364-368.
4. Muhlemann MF. Localized granuloma annulare is associated with insulin-dependent diabetes mellitus. Br J Dermatol. 1984;111:325-329.
5. Nebesio CL, Lewis C, Chuang TY. Lack of an association between granuloma annulare and type 2 diabetes mellitus. Br J Dermatol. 2002;146:122-124.
6. Blume-peytavi U, Zouboulis CH, Jacobi H, Scholz A, Bisson S, Orfanos CE. Successful outcome of cryosurgery in patients with granuloma annulare. Br J Dermatol. 1994;130:494-497.
7. Smith MD, Downie JB, DiCostanzo D. Granuloma annulare. Int J Dermatol. 1997;36:326-333.
1. Nopper A, Markus R, Esterly N. When it’s not ring-worm: annular lesions of childhood. Pediatr Ann. 1998;27:136-148.
2. Habif TP. Clinical Dermatology. 4th ed. St louis, mo: mosby, Inc; 2004.
3. Studer EM, Calza AM, Saurat JH. Precipitating factors and associated diseases in 84 patients with granuloma annulare: a retrospective study. Dermatology. 1996;193:364-368.
4. Muhlemann MF. Localized granuloma annulare is associated with insulin-dependent diabetes mellitus. Br J Dermatol. 1984;111:325-329.
5. Nebesio CL, Lewis C, Chuang TY. Lack of an association between granuloma annulare and type 2 diabetes mellitus. Br J Dermatol. 2002;146:122-124.
6. Blume-peytavi U, Zouboulis CH, Jacobi H, Scholz A, Bisson S, Orfanos CE. Successful outcome of cryosurgery in patients with granuloma annulare. Br J Dermatol. 1994;130:494-497.
7. Smith MD, Downie JB, DiCostanzo D. Granuloma annulare. Int J Dermatol. 1997;36:326-333.
A bright red pruritic rash on the forearms
A 50-year-old woman presented to the clinic with an intensely pruritic rash that had come on suddenly and extended over the dorsal aspect of both arms. She said that she had not begun taking any new medicines, and had no recent exposures to any new chemicals. She did, however, note that she’d recently spent some time in the sun.
Her history included schizoaffective disorder, bipolar disorder, reactive airways disease, type 2 diabetes mellitus, and hypertension.
She was taking a number of medications including trihexyphenidyl, flu- phenazine, mirtazapine, ibuprofen, propranolol, acetaminophen, albuterol/ipratropium inhaled, and triamcinolone inhaled. She reported being allergic to lithium, erythromycin, and haloperidol.
Her skin exam showed erythematous plaques over the dorsum of both forearms (Figure 1). There were no lesions on her left forearm where she had been wearing a watch, and there were no lesions on her face or lower extremities. Her vital signs were normal.
We ordered a punch biopsy.
FIGURE 1
Plaque on forearms, except under watchband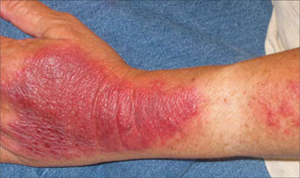
What is your diagnosis?
Diagnosis: Polymorphous light eruption
Biopsy findings
The biopsy showed extensive spongiosis and edema of the dermis with a deep lymphohistiocytic infiltrate, consistent with polymorphous light eruption—an idiopathic, delayed type hypersensitivity to UVA and UVB light.1-3
Characteristics
Polymorphous light eruption may affect up to 10% of the population, with a predilection for females.3,4 The prevalence increases in northern latitudes. All skin types may be affected, but those with Fitzpatrick skin types I or II are most frequently affected.4
Polymorphous light eruption may appear spontaneously at any age and may manifest as plaques (as was the case with our patient), papules, or rarely, vesicles (Figure 2). It tends to present early in the spring and summer, with the first significant UV exposure of the year. The rash typically develops 1 to 4 days after sun exposure, and with time, remits spontaneously. Occasionally, though, it will last as long as there is significant sun exposure.
FIGURE 2
Polymorphous light eruption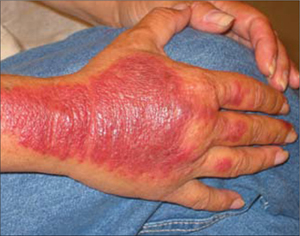
A condition that mimics a phototoxic drug reaction
The differential diagnosis for polymorphous light eruption includes phototoxic drug reaction, systemic lupus erythematosus, and porphyria cutanea tarda.
A phototoxic drug reaction is a non-immunologic reaction that manifests 2 to 6 hours after exposure to sunlight.1-3 It can be difficult to differentiate from polymorphous light eruption without a biopsy. A phototoxic drug reaction can be likened to sunburn, with a mild form causing slight erythema and a severe form causing vesicles or bullae.
Our patient certainly used medicines known to cause phototoxic reactions (ibuprofen, fluphenazine). However, she’d had no reactions with prior sun exposure. The significant dermal edema (hence the plaque) without any vesicles or bullae made a phototoxic drug reaction diagnosis less likely. The biopsy clarified the issue.
Systemic lupus erythematosus
SLE was also a possibility with our patient. Sunlight can precipitate a lupus rash, so the photodistribution made lupus plausible. Its abrupt onset and marked pruritus spoke against it, though, as did the negative serum antinuclear antibody test, which had been ordered.
Porphyria cutanea tarda
This disorder, which can also be precipitated by sunlight, was also a possibility with our patient. It tends to present with vesicles or bullae in sun-exposed areas, especially the dorsum of the hands.2 The bullae generally have no surrounding erythema, which made it an unlikely diagnosis for our patient. A diagnosis of porphyria cutanea tarda hinges on increased porphyrin levels, measured in a 24-hour urine collection.
Steroids, antihistamines, sunblock, and long sleeves
Treatment for polymorphous light eruption includes topical steroids and antihistamines.1-3 Patients can attempt to prevent future episodes by applying broad-spectrum (UVA and UVB coverage) sunblock and wearing long-sleeved garments when going out in the sun.
Desensitization with phototherapy is often necessary. UVA, UVB, and PUVA have all proven beneficial.5
Hydroxychloroquine. Recalcitrant cases may require hydroxychloroquine during the summer months.6
Nonadherence hinders our patient’s recovery
We started our patient on oral antihistamines and topical steroids and recommended that she avoid direct sunlight.
Our patient, however, didn’t avoid sun exposure, and when we saw her on follow-up, her pruritis had improved, but the lesions were essentially unchanged.
We subsequently lost our patient to follow-up.
1. James WD, Berger TG, Elston DA. Andrews’ Diseases of the Skin, Clinical Dermatology. 10th ed. Philadelphia, Pa: W.B. Saunders; 2006.
2. Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 4th ed. Philadelphia, Pa: Mosby; 2004.
3. Fitzpatrick TB, Johnson RA, Wolff K, Suurmond D. Color Atlas and Synopsis of Clinical Dermatology, Common and Serious Diseases. 4th ed. New York, NY: McGraw Hill; 2001.
4. Fesq H, Ring J, Abeck D. Management of polymorphous light eruption. Clinical course, pathogenesis, diagnosis and intervention. Am J Clin Dermatol 2003;4:399-406.
5. Murphy GM, Logan RA, Lovell CR, Morris RW, Hawk JL, Magnus IA. Prophylactic PUVA and UVB therapy in polymorphic light eruption—a controlled trial. Br J Dermatol 1987;116:531-538.
6. Murphy GM, Hawk JLM, Magnus IA. Hydroxychloroquine in polymorphic light eruption: a controlled trial with drug and visual sensitivity monitoring. Br J Dermatol 1987;116:379-386.
A 50-year-old woman presented to the clinic with an intensely pruritic rash that had come on suddenly and extended over the dorsal aspect of both arms. She said that she had not begun taking any new medicines, and had no recent exposures to any new chemicals. She did, however, note that she’d recently spent some time in the sun.
Her history included schizoaffective disorder, bipolar disorder, reactive airways disease, type 2 diabetes mellitus, and hypertension.
She was taking a number of medications including trihexyphenidyl, flu- phenazine, mirtazapine, ibuprofen, propranolol, acetaminophen, albuterol/ipratropium inhaled, and triamcinolone inhaled. She reported being allergic to lithium, erythromycin, and haloperidol.
Her skin exam showed erythematous plaques over the dorsum of both forearms (Figure 1). There were no lesions on her left forearm where she had been wearing a watch, and there were no lesions on her face or lower extremities. Her vital signs were normal.
We ordered a punch biopsy.
FIGURE 1
Plaque on forearms, except under watchband
What is your diagnosis?
Diagnosis: Polymorphous light eruption
Biopsy findings
The biopsy showed extensive spongiosis and edema of the dermis with a deep lymphohistiocytic infiltrate, consistent with polymorphous light eruption—an idiopathic, delayed type hypersensitivity to UVA and UVB light.1-3
Characteristics
Polymorphous light eruption may affect up to 10% of the population, with a predilection for females.3,4 The prevalence increases in northern latitudes. All skin types may be affected, but those with Fitzpatrick skin types I or II are most frequently affected.4
Polymorphous light eruption may appear spontaneously at any age and may manifest as plaques (as was the case with our patient), papules, or rarely, vesicles (Figure 2). It tends to present early in the spring and summer, with the first significant UV exposure of the year. The rash typically develops 1 to 4 days after sun exposure, and with time, remits spontaneously. Occasionally, though, it will last as long as there is significant sun exposure.
FIGURE 2
Polymorphous light eruption
A condition that mimics a phototoxic drug reaction
The differential diagnosis for polymorphous light eruption includes phototoxic drug reaction, systemic lupus erythematosus, and porphyria cutanea tarda.
A phototoxic drug reaction is a non-immunologic reaction that manifests 2 to 6 hours after exposure to sunlight.1-3 It can be difficult to differentiate from polymorphous light eruption without a biopsy. A phototoxic drug reaction can be likened to sunburn, with a mild form causing slight erythema and a severe form causing vesicles or bullae.
Our patient certainly used medicines known to cause phototoxic reactions (ibuprofen, fluphenazine). However, she’d had no reactions with prior sun exposure. The significant dermal edema (hence the plaque) without any vesicles or bullae made a phototoxic drug reaction diagnosis less likely. The biopsy clarified the issue.
Systemic lupus erythematosus
SLE was also a possibility with our patient. Sunlight can precipitate a lupus rash, so the photodistribution made lupus plausible. Its abrupt onset and marked pruritus spoke against it, though, as did the negative serum antinuclear antibody test, which had been ordered.
Porphyria cutanea tarda
This disorder, which can also be precipitated by sunlight, was also a possibility with our patient. It tends to present with vesicles or bullae in sun-exposed areas, especially the dorsum of the hands.2 The bullae generally have no surrounding erythema, which made it an unlikely diagnosis for our patient. A diagnosis of porphyria cutanea tarda hinges on increased porphyrin levels, measured in a 24-hour urine collection.
Steroids, antihistamines, sunblock, and long sleeves
Treatment for polymorphous light eruption includes topical steroids and antihistamines.1-3 Patients can attempt to prevent future episodes by applying broad-spectrum (UVA and UVB coverage) sunblock and wearing long-sleeved garments when going out in the sun.
Desensitization with phototherapy is often necessary. UVA, UVB, and PUVA have all proven beneficial.5
Hydroxychloroquine. Recalcitrant cases may require hydroxychloroquine during the summer months.6
Nonadherence hinders our patient’s recovery
We started our patient on oral antihistamines and topical steroids and recommended that she avoid direct sunlight.
Our patient, however, didn’t avoid sun exposure, and when we saw her on follow-up, her pruritis had improved, but the lesions were essentially unchanged.
We subsequently lost our patient to follow-up.
A 50-year-old woman presented to the clinic with an intensely pruritic rash that had come on suddenly and extended over the dorsal aspect of both arms. She said that she had not begun taking any new medicines, and had no recent exposures to any new chemicals. She did, however, note that she’d recently spent some time in the sun.
Her history included schizoaffective disorder, bipolar disorder, reactive airways disease, type 2 diabetes mellitus, and hypertension.
She was taking a number of medications including trihexyphenidyl, flu- phenazine, mirtazapine, ibuprofen, propranolol, acetaminophen, albuterol/ipratropium inhaled, and triamcinolone inhaled. She reported being allergic to lithium, erythromycin, and haloperidol.
Her skin exam showed erythematous plaques over the dorsum of both forearms (Figure 1). There were no lesions on her left forearm where she had been wearing a watch, and there were no lesions on her face or lower extremities. Her vital signs were normal.
We ordered a punch biopsy.
FIGURE 1
Plaque on forearms, except under watchband
What is your diagnosis?
Diagnosis: Polymorphous light eruption
Biopsy findings
The biopsy showed extensive spongiosis and edema of the dermis with a deep lymphohistiocytic infiltrate, consistent with polymorphous light eruption—an idiopathic, delayed type hypersensitivity to UVA and UVB light.1-3
Characteristics
Polymorphous light eruption may affect up to 10% of the population, with a predilection for females.3,4 The prevalence increases in northern latitudes. All skin types may be affected, but those with Fitzpatrick skin types I or II are most frequently affected.4
Polymorphous light eruption may appear spontaneously at any age and may manifest as plaques (as was the case with our patient), papules, or rarely, vesicles (Figure 2). It tends to present early in the spring and summer, with the first significant UV exposure of the year. The rash typically develops 1 to 4 days after sun exposure, and with time, remits spontaneously. Occasionally, though, it will last as long as there is significant sun exposure.
FIGURE 2
Polymorphous light eruption
A condition that mimics a phototoxic drug reaction
The differential diagnosis for polymorphous light eruption includes phototoxic drug reaction, systemic lupus erythematosus, and porphyria cutanea tarda.
A phototoxic drug reaction is a non-immunologic reaction that manifests 2 to 6 hours after exposure to sunlight.1-3 It can be difficult to differentiate from polymorphous light eruption without a biopsy. A phototoxic drug reaction can be likened to sunburn, with a mild form causing slight erythema and a severe form causing vesicles or bullae.
Our patient certainly used medicines known to cause phototoxic reactions (ibuprofen, fluphenazine). However, she’d had no reactions with prior sun exposure. The significant dermal edema (hence the plaque) without any vesicles or bullae made a phototoxic drug reaction diagnosis less likely. The biopsy clarified the issue.
Systemic lupus erythematosus
SLE was also a possibility with our patient. Sunlight can precipitate a lupus rash, so the photodistribution made lupus plausible. Its abrupt onset and marked pruritus spoke against it, though, as did the negative serum antinuclear antibody test, which had been ordered.
Porphyria cutanea tarda
This disorder, which can also be precipitated by sunlight, was also a possibility with our patient. It tends to present with vesicles or bullae in sun-exposed areas, especially the dorsum of the hands.2 The bullae generally have no surrounding erythema, which made it an unlikely diagnosis for our patient. A diagnosis of porphyria cutanea tarda hinges on increased porphyrin levels, measured in a 24-hour urine collection.
Steroids, antihistamines, sunblock, and long sleeves
Treatment for polymorphous light eruption includes topical steroids and antihistamines.1-3 Patients can attempt to prevent future episodes by applying broad-spectrum (UVA and UVB coverage) sunblock and wearing long-sleeved garments when going out in the sun.
Desensitization with phototherapy is often necessary. UVA, UVB, and PUVA have all proven beneficial.5
Hydroxychloroquine. Recalcitrant cases may require hydroxychloroquine during the summer months.6
Nonadherence hinders our patient’s recovery
We started our patient on oral antihistamines and topical steroids and recommended that she avoid direct sunlight.
Our patient, however, didn’t avoid sun exposure, and when we saw her on follow-up, her pruritis had improved, but the lesions were essentially unchanged.
We subsequently lost our patient to follow-up.
1. James WD, Berger TG, Elston DA. Andrews’ Diseases of the Skin, Clinical Dermatology. 10th ed. Philadelphia, Pa: W.B. Saunders; 2006.
2. Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 4th ed. Philadelphia, Pa: Mosby; 2004.
3. Fitzpatrick TB, Johnson RA, Wolff K, Suurmond D. Color Atlas and Synopsis of Clinical Dermatology, Common and Serious Diseases. 4th ed. New York, NY: McGraw Hill; 2001.
4. Fesq H, Ring J, Abeck D. Management of polymorphous light eruption. Clinical course, pathogenesis, diagnosis and intervention. Am J Clin Dermatol 2003;4:399-406.
5. Murphy GM, Logan RA, Lovell CR, Morris RW, Hawk JL, Magnus IA. Prophylactic PUVA and UVB therapy in polymorphic light eruption—a controlled trial. Br J Dermatol 1987;116:531-538.
6. Murphy GM, Hawk JLM, Magnus IA. Hydroxychloroquine in polymorphic light eruption: a controlled trial with drug and visual sensitivity monitoring. Br J Dermatol 1987;116:379-386.
1. James WD, Berger TG, Elston DA. Andrews’ Diseases of the Skin, Clinical Dermatology. 10th ed. Philadelphia, Pa: W.B. Saunders; 2006.
2. Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 4th ed. Philadelphia, Pa: Mosby; 2004.
3. Fitzpatrick TB, Johnson RA, Wolff K, Suurmond D. Color Atlas and Synopsis of Clinical Dermatology, Common and Serious Diseases. 4th ed. New York, NY: McGraw Hill; 2001.
4. Fesq H, Ring J, Abeck D. Management of polymorphous light eruption. Clinical course, pathogenesis, diagnosis and intervention. Am J Clin Dermatol 2003;4:399-406.
5. Murphy GM, Logan RA, Lovell CR, Morris RW, Hawk JL, Magnus IA. Prophylactic PUVA and UVB therapy in polymorphic light eruption—a controlled trial. Br J Dermatol 1987;116:531-538.
6. Murphy GM, Hawk JLM, Magnus IA. Hydroxychloroquine in polymorphic light eruption: a controlled trial with drug and visual sensitivity monitoring. Br J Dermatol 1987;116:379-386.
A young girl with blisters on her forehead
A 5-year-old girl came into the office complaining of severe burning and tingling sensation of her right forehead. She’d had fever, chills, myalgia, and a relentless headache for 3 days. The morning of her appointment, a few “bumps” and water-filled blisters began to appear on the right side of the forehead; the lesions then started to multiply and grow (FIGURE). The patient’s mother expressed concern over the rapid development of the lesions, which were accompanied by marked edema of the forehead and right eyelid. The mother indicated that no one else in the family was affected at the time of presentation.
The child appeared ill and pale at the time of presentation, but there was no history of immediate antecedent illness or any drug intake prior to the current eruption. Her growth and development were in the lower normal range. The child had a history of recurrent bacterial infections; she was not vaccinated for varicella. There was, however, a personal (and family) history of varicella when the child was about 3 years old.
FIGURE
Vesicles and bullae on forehead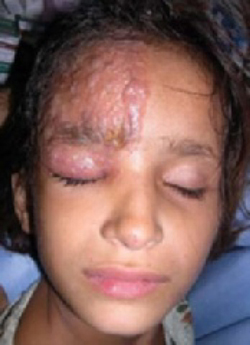
On physical examination, there were multiple vesicles and bullae that varied in size from 2 mm to 1 cm in diameter on the right side of the forehead; there were areas of dried yellowish serous exudates limited to the right eyebrow. The forehead and periorbital areas were edematous with underlying erythematous skin. The entire eruption appeared to be restricted to the right upper part of the face, extending from the eyelid and medial canthus up to the frontal scalp. On close examination, several vesicles and crusts were present on the right side of the nasal tip. Serology for HIV was negative.
What is your diagnosis?
How would you treat?
Diagnosis: Herpes zoster ophthalmicus
This case of herpes zoster ophthalmicus (HZO) was unusual—not because of the way the patient presented, but because of her age. This condition is rarely seen in children, though it is not uncommon in adults—specifically, patients who are 60 years of age and older.1-4
Herpes zoster ophthalmicus, which refers to the involvement of the ophthalmic branch of the trigeminal nerve or the fifth cranial nerve, usually manifests with a typical vesicular or bullous eruption of the forehead, often in unilateral fashion. The lesions of herpes zoster (shingles) are similar to those of varicella (chickenpox) but in herpes zoster, painful unilateral vesicular eruptions are usually limited to one dermatome. (If hematogenous dissemination occurs, more than 20 vesicles will form in skin areas away from the affected dermatome.1)
Patients typically present with fever, headache, and abnormal sensations that precede the development of cutaneous lesions by a few days. The eruptions may be pustular and hemorrhagic initially, and within 10 days evolve into crusts.5,6
Differential diagnosis: Pain sets HZO apart
In its classical presentation, HZO does not pose a diagnostic challenge for practitioners well-versed in skin disease management. The clinical differential diagnosis of herpes zoster will include other causes of blisters or vesicular eruptions of autoimmune etiologies, viral infections, or hypersensitivity reactions.
The most common blistering diseases that may be mistaken for zoster include herpes simplex, contact dermatitis, erythema multiforme, and cellulitis. The prodromal stage and the characteristics of pain usually set herpes zoster apart from the other diagnoses.
Beware of complications
Eye complications. HZO may result in paralytic ptosis, conjunctivitis, keratitis, cataracts, glaucoma, retinitis, and optic neuritis and atrophy.2,5,7
Herpetic lesions on the tip of the nose are believed to herald ocular involvement and may precede typical dermatomal eruption of HZO.2 Such cutaneous involvement along the distribution of the nasociliary nerve is called Hutchinson’s sign. It should prompt a complete ophthalmologic evaluation, as it did with our patient.
Neurologic complications. The most common complication of herpes zoster infection in general is postherpetic neuralgia (PHN), which results in severe pain that persists for months—or years—after the skin lesions have completely healed. Patients over 70 have a 70% chance of developing PHN.4,7
Neurological complications such as encephalitis, myelitis, and Guillain-Barré syndrome have also been reported.
Other complications. Other complications include secondary bacterial infections, pneumonitis, and polyradiculitis.
Management: Antivirals and pain meds
Patients with HZO have a 50% chance of having eye complications (iritis and keratitis) without antiviral treatment.2,5,6 Therefore, treatment is recommended for all HZO patients.
Antivirals ASAP
Start antiviral drugs within 72 hours of clinical presentation, and when new lesions are still appearing on the skin, to achieve optimal effect. Acyclovir, famciclovir, and valacyclovir are the antivirals of choice.6 The usual dosages for adults are: acyclovir (800 mg orally 5 times per day for 7–10 days), valacyclovir (1000 mg orally 3 times daily for 7 days) and famciclovir (500 mg orally 3 times daily for 7 days). The suggested dose of acyclovir for children is 10 to 20 mg/kg/dose qid for 5 days; not to exceed 800 mg per day.2,5,6
Preventing postherpetic neuralgia
The role of systemic corticosteroids in the prevention of PHN, decreasing duration and severity of the acute symptoms in the initial days of herpes zoster infection, remains controversial.8
Immunosuppressed individuals may be treated with acyclovir, interferon-alpha, and vidarabine. In this population the live, attenuated vaccine is safer and is preferred to the varicella zoster immunoglobulins (VZIG).1,6,8,9
PHN can be reduced by treating the patient within the first 24 hours of symptom onset. Pain usually resolves within 3 months in 50% of patients and within 1 year in 75% of patients.1,6,7
Pain therapies for PHN
Therapeutic approaches to the management of PHN include topical anesthetic creams (lidocaine plus prilocaine), capsaicin, and oral medications such as tricyclic antidepressants (amitriptyline and desipramine), carbamazepine, and gabapentin, as well as nerve blocks.6,7
Gloves and handwashing are key for caregivers
Individuals who have not had a confirmed varicella infection should avoid contact with those who have shingles unless a varicella zoster virus antibody is satisfactory and shows immunity.6,7
Gloves should be worn when touching the lesions or infectious drainage, and hands should be washed after glove removal.5-9
Patients with disseminated disease require hospitalization with airborne precautions, which include a negative air pressure room and ensuring that caregivers wear N95 respirators.
What put our patient at risk?
In the case of our young patient, low immunity may have played a role in her contracting HZO. She had a history of recurrent bacterial infections and suffered from generally poor health.
She was referred to an ophthalmologist, who did not find any dendritic corneal ulcer or other complications on weekly follow up. The patient was successfully managed with oral acyclovir 10 mg/kg/dose for 5 days, and she responded well to treatment. We also prescribed acyclovir eye ointment, and local wet compresses with good outcome.
Acknowledgments
The authors thank Dr Shahbaz Janjua for his assistance in the preparation of this manuscript.
Correspondence
Amor Khachemoune, MD, CWS, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016; [email protected].
1. Trent JT, Kirsner RS. Herpesvirus infections and herpetic wounds. Adv Skin Wound Care 2003;16:236-243.
2. Zaal MJ, Volker-Dieben HJ, D’Amaro J. Prognostic value of Hutchinson’s sign in acute herpes zoster ophthalmicus. Graefes Arch Clin Exp Ophthalmol 2003;241:187-191.
3. Binder NR, Holland GN, Hosea S, Silverberg ML. Herpes zoster ophthalmicus in an otherwisehealthy child. JAAPOS 2005;9:597-598.
4. Gebo KA, Kalyani R, Moore RD, Polydefkis MJ. The incidence of, risk factors for, and sequelae of herpes zoster among HIV patients in the highly active antiretroviral therapy era. JAIDS 2005;40:169-174.
5. Liesegang TJ. Herpes zoster virus infection. Curr Opin Ophthalmol 2004;15:531-536.
6. McCrary ML, Severson J, Tyring SK. Varicella zoster virus. J Am Acad Dermatol 1999;41:1-14.
7. Cohen JI, Brunell PA, Straus SE, Krause PR. Recent advances in varicella-zoster virus infection. Ann Intern Med 1999;130:922-932.
8. Santee JA. Corticosteroids for herpes zoster: what do they accomplish? Am J Clin Dermatol 2002;3:517-524.
9. Oxman MN, Levin MJ, Johnson GR, et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med 2005;352:2271-2284.
A 5-year-old girl came into the office complaining of severe burning and tingling sensation of her right forehead. She’d had fever, chills, myalgia, and a relentless headache for 3 days. The morning of her appointment, a few “bumps” and water-filled blisters began to appear on the right side of the forehead; the lesions then started to multiply and grow (FIGURE). The patient’s mother expressed concern over the rapid development of the lesions, which were accompanied by marked edema of the forehead and right eyelid. The mother indicated that no one else in the family was affected at the time of presentation.
The child appeared ill and pale at the time of presentation, but there was no history of immediate antecedent illness or any drug intake prior to the current eruption. Her growth and development were in the lower normal range. The child had a history of recurrent bacterial infections; she was not vaccinated for varicella. There was, however, a personal (and family) history of varicella when the child was about 3 years old.
FIGURE
Vesicles and bullae on forehead
On physical examination, there were multiple vesicles and bullae that varied in size from 2 mm to 1 cm in diameter on the right side of the forehead; there were areas of dried yellowish serous exudates limited to the right eyebrow. The forehead and periorbital areas were edematous with underlying erythematous skin. The entire eruption appeared to be restricted to the right upper part of the face, extending from the eyelid and medial canthus up to the frontal scalp. On close examination, several vesicles and crusts were present on the right side of the nasal tip. Serology for HIV was negative.
What is your diagnosis?
How would you treat?
Diagnosis: Herpes zoster ophthalmicus
This case of herpes zoster ophthalmicus (HZO) was unusual—not because of the way the patient presented, but because of her age. This condition is rarely seen in children, though it is not uncommon in adults—specifically, patients who are 60 years of age and older.1-4
Herpes zoster ophthalmicus, which refers to the involvement of the ophthalmic branch of the trigeminal nerve or the fifth cranial nerve, usually manifests with a typical vesicular or bullous eruption of the forehead, often in unilateral fashion. The lesions of herpes zoster (shingles) are similar to those of varicella (chickenpox) but in herpes zoster, painful unilateral vesicular eruptions are usually limited to one dermatome. (If hematogenous dissemination occurs, more than 20 vesicles will form in skin areas away from the affected dermatome.1)
Patients typically present with fever, headache, and abnormal sensations that precede the development of cutaneous lesions by a few days. The eruptions may be pustular and hemorrhagic initially, and within 10 days evolve into crusts.5,6
Differential diagnosis: Pain sets HZO apart
In its classical presentation, HZO does not pose a diagnostic challenge for practitioners well-versed in skin disease management. The clinical differential diagnosis of herpes zoster will include other causes of blisters or vesicular eruptions of autoimmune etiologies, viral infections, or hypersensitivity reactions.
The most common blistering diseases that may be mistaken for zoster include herpes simplex, contact dermatitis, erythema multiforme, and cellulitis. The prodromal stage and the characteristics of pain usually set herpes zoster apart from the other diagnoses.
Beware of complications
Eye complications. HZO may result in paralytic ptosis, conjunctivitis, keratitis, cataracts, glaucoma, retinitis, and optic neuritis and atrophy.2,5,7
Herpetic lesions on the tip of the nose are believed to herald ocular involvement and may precede typical dermatomal eruption of HZO.2 Such cutaneous involvement along the distribution of the nasociliary nerve is called Hutchinson’s sign. It should prompt a complete ophthalmologic evaluation, as it did with our patient.
Neurologic complications. The most common complication of herpes zoster infection in general is postherpetic neuralgia (PHN), which results in severe pain that persists for months—or years—after the skin lesions have completely healed. Patients over 70 have a 70% chance of developing PHN.4,7
Neurological complications such as encephalitis, myelitis, and Guillain-Barré syndrome have also been reported.
Other complications. Other complications include secondary bacterial infections, pneumonitis, and polyradiculitis.
Management: Antivirals and pain meds
Patients with HZO have a 50% chance of having eye complications (iritis and keratitis) without antiviral treatment.2,5,6 Therefore, treatment is recommended for all HZO patients.
Antivirals ASAP
Start antiviral drugs within 72 hours of clinical presentation, and when new lesions are still appearing on the skin, to achieve optimal effect. Acyclovir, famciclovir, and valacyclovir are the antivirals of choice.6 The usual dosages for adults are: acyclovir (800 mg orally 5 times per day for 7–10 days), valacyclovir (1000 mg orally 3 times daily for 7 days) and famciclovir (500 mg orally 3 times daily for 7 days). The suggested dose of acyclovir for children is 10 to 20 mg/kg/dose qid for 5 days; not to exceed 800 mg per day.2,5,6
Preventing postherpetic neuralgia
The role of systemic corticosteroids in the prevention of PHN, decreasing duration and severity of the acute symptoms in the initial days of herpes zoster infection, remains controversial.8
Immunosuppressed individuals may be treated with acyclovir, interferon-alpha, and vidarabine. In this population the live, attenuated vaccine is safer and is preferred to the varicella zoster immunoglobulins (VZIG).1,6,8,9
PHN can be reduced by treating the patient within the first 24 hours of symptom onset. Pain usually resolves within 3 months in 50% of patients and within 1 year in 75% of patients.1,6,7
Pain therapies for PHN
Therapeutic approaches to the management of PHN include topical anesthetic creams (lidocaine plus prilocaine), capsaicin, and oral medications such as tricyclic antidepressants (amitriptyline and desipramine), carbamazepine, and gabapentin, as well as nerve blocks.6,7
Gloves and handwashing are key for caregivers
Individuals who have not had a confirmed varicella infection should avoid contact with those who have shingles unless a varicella zoster virus antibody is satisfactory and shows immunity.6,7
Gloves should be worn when touching the lesions or infectious drainage, and hands should be washed after glove removal.5-9
Patients with disseminated disease require hospitalization with airborne precautions, which include a negative air pressure room and ensuring that caregivers wear N95 respirators.
What put our patient at risk?
In the case of our young patient, low immunity may have played a role in her contracting HZO. She had a history of recurrent bacterial infections and suffered from generally poor health.
She was referred to an ophthalmologist, who did not find any dendritic corneal ulcer or other complications on weekly follow up. The patient was successfully managed with oral acyclovir 10 mg/kg/dose for 5 days, and she responded well to treatment. We also prescribed acyclovir eye ointment, and local wet compresses with good outcome.
Acknowledgments
The authors thank Dr Shahbaz Janjua for his assistance in the preparation of this manuscript.
Correspondence
Amor Khachemoune, MD, CWS, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016; [email protected].
A 5-year-old girl came into the office complaining of severe burning and tingling sensation of her right forehead. She’d had fever, chills, myalgia, and a relentless headache for 3 days. The morning of her appointment, a few “bumps” and water-filled blisters began to appear on the right side of the forehead; the lesions then started to multiply and grow (FIGURE). The patient’s mother expressed concern over the rapid development of the lesions, which were accompanied by marked edema of the forehead and right eyelid. The mother indicated that no one else in the family was affected at the time of presentation.
The child appeared ill and pale at the time of presentation, but there was no history of immediate antecedent illness or any drug intake prior to the current eruption. Her growth and development were in the lower normal range. The child had a history of recurrent bacterial infections; she was not vaccinated for varicella. There was, however, a personal (and family) history of varicella when the child was about 3 years old.
FIGURE
Vesicles and bullae on forehead
On physical examination, there were multiple vesicles and bullae that varied in size from 2 mm to 1 cm in diameter on the right side of the forehead; there were areas of dried yellowish serous exudates limited to the right eyebrow. The forehead and periorbital areas were edematous with underlying erythematous skin. The entire eruption appeared to be restricted to the right upper part of the face, extending from the eyelid and medial canthus up to the frontal scalp. On close examination, several vesicles and crusts were present on the right side of the nasal tip. Serology for HIV was negative.
What is your diagnosis?
How would you treat?
Diagnosis: Herpes zoster ophthalmicus
This case of herpes zoster ophthalmicus (HZO) was unusual—not because of the way the patient presented, but because of her age. This condition is rarely seen in children, though it is not uncommon in adults—specifically, patients who are 60 years of age and older.1-4
Herpes zoster ophthalmicus, which refers to the involvement of the ophthalmic branch of the trigeminal nerve or the fifth cranial nerve, usually manifests with a typical vesicular or bullous eruption of the forehead, often in unilateral fashion. The lesions of herpes zoster (shingles) are similar to those of varicella (chickenpox) but in herpes zoster, painful unilateral vesicular eruptions are usually limited to one dermatome. (If hematogenous dissemination occurs, more than 20 vesicles will form in skin areas away from the affected dermatome.1)
Patients typically present with fever, headache, and abnormal sensations that precede the development of cutaneous lesions by a few days. The eruptions may be pustular and hemorrhagic initially, and within 10 days evolve into crusts.5,6
Differential diagnosis: Pain sets HZO apart
In its classical presentation, HZO does not pose a diagnostic challenge for practitioners well-versed in skin disease management. The clinical differential diagnosis of herpes zoster will include other causes of blisters or vesicular eruptions of autoimmune etiologies, viral infections, or hypersensitivity reactions.
The most common blistering diseases that may be mistaken for zoster include herpes simplex, contact dermatitis, erythema multiforme, and cellulitis. The prodromal stage and the characteristics of pain usually set herpes zoster apart from the other diagnoses.
Beware of complications
Eye complications. HZO may result in paralytic ptosis, conjunctivitis, keratitis, cataracts, glaucoma, retinitis, and optic neuritis and atrophy.2,5,7
Herpetic lesions on the tip of the nose are believed to herald ocular involvement and may precede typical dermatomal eruption of HZO.2 Such cutaneous involvement along the distribution of the nasociliary nerve is called Hutchinson’s sign. It should prompt a complete ophthalmologic evaluation, as it did with our patient.
Neurologic complications. The most common complication of herpes zoster infection in general is postherpetic neuralgia (PHN), which results in severe pain that persists for months—or years—after the skin lesions have completely healed. Patients over 70 have a 70% chance of developing PHN.4,7
Neurological complications such as encephalitis, myelitis, and Guillain-Barré syndrome have also been reported.
Other complications. Other complications include secondary bacterial infections, pneumonitis, and polyradiculitis.
Management: Antivirals and pain meds
Patients with HZO have a 50% chance of having eye complications (iritis and keratitis) without antiviral treatment.2,5,6 Therefore, treatment is recommended for all HZO patients.
Antivirals ASAP
Start antiviral drugs within 72 hours of clinical presentation, and when new lesions are still appearing on the skin, to achieve optimal effect. Acyclovir, famciclovir, and valacyclovir are the antivirals of choice.6 The usual dosages for adults are: acyclovir (800 mg orally 5 times per day for 7–10 days), valacyclovir (1000 mg orally 3 times daily for 7 days) and famciclovir (500 mg orally 3 times daily for 7 days). The suggested dose of acyclovir for children is 10 to 20 mg/kg/dose qid for 5 days; not to exceed 800 mg per day.2,5,6
Preventing postherpetic neuralgia
The role of systemic corticosteroids in the prevention of PHN, decreasing duration and severity of the acute symptoms in the initial days of herpes zoster infection, remains controversial.8
Immunosuppressed individuals may be treated with acyclovir, interferon-alpha, and vidarabine. In this population the live, attenuated vaccine is safer and is preferred to the varicella zoster immunoglobulins (VZIG).1,6,8,9
PHN can be reduced by treating the patient within the first 24 hours of symptom onset. Pain usually resolves within 3 months in 50% of patients and within 1 year in 75% of patients.1,6,7
Pain therapies for PHN
Therapeutic approaches to the management of PHN include topical anesthetic creams (lidocaine plus prilocaine), capsaicin, and oral medications such as tricyclic antidepressants (amitriptyline and desipramine), carbamazepine, and gabapentin, as well as nerve blocks.6,7
Gloves and handwashing are key for caregivers
Individuals who have not had a confirmed varicella infection should avoid contact with those who have shingles unless a varicella zoster virus antibody is satisfactory and shows immunity.6,7
Gloves should be worn when touching the lesions or infectious drainage, and hands should be washed after glove removal.5-9
Patients with disseminated disease require hospitalization with airborne precautions, which include a negative air pressure room and ensuring that caregivers wear N95 respirators.
What put our patient at risk?
In the case of our young patient, low immunity may have played a role in her contracting HZO. She had a history of recurrent bacterial infections and suffered from generally poor health.
She was referred to an ophthalmologist, who did not find any dendritic corneal ulcer or other complications on weekly follow up. The patient was successfully managed with oral acyclovir 10 mg/kg/dose for 5 days, and she responded well to treatment. We also prescribed acyclovir eye ointment, and local wet compresses with good outcome.
Acknowledgments
The authors thank Dr Shahbaz Janjua for his assistance in the preparation of this manuscript.
Correspondence
Amor Khachemoune, MD, CWS, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016; [email protected].
1. Trent JT, Kirsner RS. Herpesvirus infections and herpetic wounds. Adv Skin Wound Care 2003;16:236-243.
2. Zaal MJ, Volker-Dieben HJ, D’Amaro J. Prognostic value of Hutchinson’s sign in acute herpes zoster ophthalmicus. Graefes Arch Clin Exp Ophthalmol 2003;241:187-191.
3. Binder NR, Holland GN, Hosea S, Silverberg ML. Herpes zoster ophthalmicus in an otherwisehealthy child. JAAPOS 2005;9:597-598.
4. Gebo KA, Kalyani R, Moore RD, Polydefkis MJ. The incidence of, risk factors for, and sequelae of herpes zoster among HIV patients in the highly active antiretroviral therapy era. JAIDS 2005;40:169-174.
5. Liesegang TJ. Herpes zoster virus infection. Curr Opin Ophthalmol 2004;15:531-536.
6. McCrary ML, Severson J, Tyring SK. Varicella zoster virus. J Am Acad Dermatol 1999;41:1-14.
7. Cohen JI, Brunell PA, Straus SE, Krause PR. Recent advances in varicella-zoster virus infection. Ann Intern Med 1999;130:922-932.
8. Santee JA. Corticosteroids for herpes zoster: what do they accomplish? Am J Clin Dermatol 2002;3:517-524.
9. Oxman MN, Levin MJ, Johnson GR, et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med 2005;352:2271-2284.
1. Trent JT, Kirsner RS. Herpesvirus infections and herpetic wounds. Adv Skin Wound Care 2003;16:236-243.
2. Zaal MJ, Volker-Dieben HJ, D’Amaro J. Prognostic value of Hutchinson’s sign in acute herpes zoster ophthalmicus. Graefes Arch Clin Exp Ophthalmol 2003;241:187-191.
3. Binder NR, Holland GN, Hosea S, Silverberg ML. Herpes zoster ophthalmicus in an otherwisehealthy child. JAAPOS 2005;9:597-598.
4. Gebo KA, Kalyani R, Moore RD, Polydefkis MJ. The incidence of, risk factors for, and sequelae of herpes zoster among HIV patients in the highly active antiretroviral therapy era. JAIDS 2005;40:169-174.
5. Liesegang TJ. Herpes zoster virus infection. Curr Opin Ophthalmol 2004;15:531-536.
6. McCrary ML, Severson J, Tyring SK. Varicella zoster virus. J Am Acad Dermatol 1999;41:1-14.
7. Cohen JI, Brunell PA, Straus SE, Krause PR. Recent advances in varicella-zoster virus infection. Ann Intern Med 1999;130:922-932.
8. Santee JA. Corticosteroids for herpes zoster: what do they accomplish? Am J Clin Dermatol 2002;3:517-524.
9. Oxman MN, Levin MJ, Johnson GR, et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med 2005;352:2271-2284.
Hurricane Katrina evacuee develops a persistent rash
Seven months after Hurricane Katrina, a 52-year-old African American woman who was evacuated from New Orleans came to our office with a hypopigmented rash on both her upper thighs and arms. Immediately following the 2005 hurricane, she was forced to wade through the polluted waters of New Orleans for many hours before being rescued by boat. Four days passed before she had access to a shower. It was after this shower that she first noticed a single erythematous spot the size of a silver dollar on her left thigh, that several weeks later faded to hypopigmented macules and plaques. Over time, the rash spread to both thighs and arms (FIGURE 1 AND 2. She did not have any itching, pain, bleeding, fever, chills, weight changes, or gastrointestinal symptoms after the rash appeared.
The patient reported that she was married and had worked as a chef for 20 years. She smoked cigarettes, drank alcohol occasionally, and was obese. She had synovitis of her left ankle, which led to surgery. She had no known drug allergies and was taking ibuprofen. Her mother, age 79, had glucose intolerance; her father, age 82, had a renal cell carcinoma removed.
Due to a confluence of situational, economic, and medical problems, she did not seek care for this rash until April 2006. At that time, a punch biopsy revealed the findings in FIGURE 3. Physical exam revealed her skin to have symmetrically distributed hypopigmented macules and plaques to all 4 extremities. She had no lymphadenopathy.
FIGURE 1
Rash on patients’ legs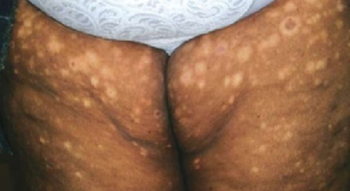
FIGURE 2
Rash on right arm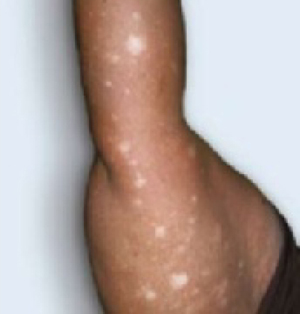
FIGURE 3
Hematoxylin/eosin stain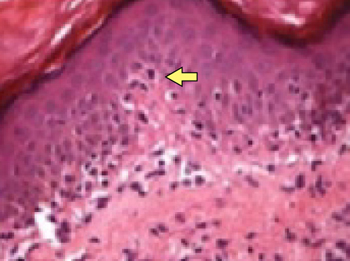
What is the diagnosis?
Were Katrina’s flood waters to blame?
Dx: T-cell lymphoma—timing is coincidental
The biopsy revealed cutaneous T-cell lymphoma (CTCL)—specifically a type known as mycosis fungoides, named after the mushroom-like skin tumors seen in severe cases. CTCL is a malignant lymphoma of helper T-cells that have an affinity for the skin.
Normal life expectancy likely if diagnosed early
In about 10% of cases, the T-cells spread via the lymphatic system to metastasize to the liver, lung, and bone marrow, but more often remain confined to the skin and lymph nodes, and most patients diagnosed early have a normal life expectancy.1
The disease is rare, with about 1000 new cases per year in the US,1 and is more common in African Americans,2 although the photos in dermatological atlases overwhelmingly show Caucasians. CTCL presents in numerous ways, but patients usually have a long course of persistent rash that is often pruritic and usually erythematous or hyperpigmented.
No one knows what causes CTCL, but current theories point to exposure to environmental hazards such as Agent Orange.2 And while the number of environmental hazards in the floodwaters of Katrina were vast, we have no scientific evidence that an environmental exposure was to blame. In fact, while we cared for a number of Katrina evacuees—many of whom had skin infections—this was the only case of mycosis fungoides.
It’s likely that the start of the visible mycosis fungoides lesions after the flood was purely coincidental.
Diagnosis hinges on palpation and biopsy
To diagnose CTCL, you need to palpate for enlarged lymph nodes and perform a full-thickness punch biopsy of the lesion. If the biopsy is negative and the rash persists, take another biopsy—results can be falsely negative at the beginning stages of the disease.
There are no guidelines on which studies should be used for staging CTCL (TABLE), but some sources recommend that if lymph node involvement is suspected on physical exam, lymph node biopsies should be done in addition to a chest radiograph.3 In more advanced stages, consider a computed tomography (CT) scan of the abdomen and pelvis. A recent study concluded that CT and positron-emission tomography (PET) scans used together were more sensitive in staging, but this may not be cost-effective.4
Late-stage mycosis fungoides is usually associated with immunocompromise. Therefore, HIV testing should be performed in all patients with CTCL. Other important laboratory studies include complete blood count—with differential—as well as peripheral smear looking for Sézary cells, lactic dehydrogenase (LDH), liver function tests, and uric acid.
TABLE
Staging for cutaneous T-cell lymphoma based on the Tumor, Node, Metastasis (TNM) system
| STAGE | TNM STAGING | RECOMMENDED TREATMENTS |
|---|---|---|
| IA | T1, N0, M0: Patch or plaque <10% body surface area | Topical high-potency steroids, PUVA, topical nitrogen |
| IB | T2, N0, M0: Patch or plaque >10% body surface area | mustard, carmustine, or bexarotene |
| IIA | T1–2, N1, M0: Patch or plaque with palpable but pathologically normal lymph node | Same as Stage I; if refractory, use total skin electron beam therapy |
| IIB | T3, N0–1, M0: Tumor/nodule | |
| IIIA | T4, N0, M0:Generalized erythroderma | |
| IIIB | T4, N1, M0: Erythroderma and palpable but pathologically normal lymph node | Chemotherapy or photophoresis, refer to medical oncologist, radiation oncologist, and dermatologist |
| IVA | T1–4, N2–3, M0: Pathological lymph node | |
| IVB | T1–4, N0–3, M1: Visceral (M1) or blood involvement | |
| T: 0–4=indicates size or direct extent of the primary tumor | ||
| N: 0=tumor cells absent from regional lymph nodes; 1=tumor cells spread to closest or small number of regional lymph nodes; 3=tumor cells spread to most distant or numerous regional lymph nodes | ||
| M: 0=no distant metastasis; 1=metastasis to distant organs (beyond regional lymph nodes) | ||
A disease that’s easy to mistake for vitiligo
The hypopigmented spots of CTCL look so much like vitiligo, it is frightening to think how easy it would be to miss the diagnosis. Complicating matters: All vitiligo does not need a biopsy to confirm the clinical impression.
So what made this case suspicious enough to warrant a biopsy? First, the hypopigmented spots on our patient were not a typical distribution for vitiligo, which has a predilection for the hands and face. Second, our patient had a hypopigmented patch that had a dark, slightly raised plaque within it, which also would not be typical for vitiligo. (See the patient’s left upper thigh, just below the inguinal area, in FIGURE 1.) Finally, our patient had a rapid onset of multiple discrete macules that did not coalesce into larger hypopigmented areas; in vitiligo you would expect otherwise.
The differential diagnosis also includes idiopathic guttate hypomelanosis, a benign condition that can cause multiple small hypopigmented macules. The size of immunoglobulin H macules, however, is tiny compared with what you’ll see with CTCL. The absence of scales makes eczema, psoriasis, or tinea corporis very unlikely.
Treatment starts with topical steroids
This is a rare disease that lacks the data needed to support an evidence-based approach to treatment. Topical steroids are recommended for stage I when the disease is local to the skin (see TABLE for recommended treatments of other stages). Ultraviolet light (PUVA and UVB) is also used; a recent study suggests the PUVA is a good treatment alternative for stages IA and IB (SOR: C).1
Our patient’s course
Initially, we prescribed a high-potency generic steroid (clobetasol 0.05% cream) for this patient, to be used twice daily to the affected areas.
The patient reported no improvement with this approach, while she awaited her appointments with dermatology and oncology specialists.
Her blood tests were essentially normal, including a negative HIV test.
She is currently receiving narrowband UVB treatment twice weekly.
1. Pinter-Brown LC. Mycosis fungoides. Emedicine, 8 September 2006. Available at: www.emedicine.com/Med/topic1541.htm. Accessed on May 8, 2007.
2. James WD, Berger T, Elston T. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, Pa: Saunders Elsevier; 2006:727–740.
3. Gettler SL, Fung MA. Efficacy of treatments for mycosis fungoides and sézary syndrome: nationwide survey responses. Dermatol Online J 2005;11:6.-
4. Tsai EY, Taur A, Espinosa L, et al. Staging accuracy in mycosis fungoides and sezary syndrome using integrated positron emission tomography and computed tomography. Arch Dermatol 2006;142:577-584.
Seven months after Hurricane Katrina, a 52-year-old African American woman who was evacuated from New Orleans came to our office with a hypopigmented rash on both her upper thighs and arms. Immediately following the 2005 hurricane, she was forced to wade through the polluted waters of New Orleans for many hours before being rescued by boat. Four days passed before she had access to a shower. It was after this shower that she first noticed a single erythematous spot the size of a silver dollar on her left thigh, that several weeks later faded to hypopigmented macules and plaques. Over time, the rash spread to both thighs and arms (FIGURE 1 AND 2. She did not have any itching, pain, bleeding, fever, chills, weight changes, or gastrointestinal symptoms after the rash appeared.
The patient reported that she was married and had worked as a chef for 20 years. She smoked cigarettes, drank alcohol occasionally, and was obese. She had synovitis of her left ankle, which led to surgery. She had no known drug allergies and was taking ibuprofen. Her mother, age 79, had glucose intolerance; her father, age 82, had a renal cell carcinoma removed.
Due to a confluence of situational, economic, and medical problems, she did not seek care for this rash until April 2006. At that time, a punch biopsy revealed the findings in FIGURE 3. Physical exam revealed her skin to have symmetrically distributed hypopigmented macules and plaques to all 4 extremities. She had no lymphadenopathy.
FIGURE 1
Rash on patients’ legs
FIGURE 2
Rash on right arm
FIGURE 3
Hematoxylin/eosin stain
What is the diagnosis?
Were Katrina’s flood waters to blame?
Dx: T-cell lymphoma—timing is coincidental
The biopsy revealed cutaneous T-cell lymphoma (CTCL)—specifically a type known as mycosis fungoides, named after the mushroom-like skin tumors seen in severe cases. CTCL is a malignant lymphoma of helper T-cells that have an affinity for the skin.
Normal life expectancy likely if diagnosed early
In about 10% of cases, the T-cells spread via the lymphatic system to metastasize to the liver, lung, and bone marrow, but more often remain confined to the skin and lymph nodes, and most patients diagnosed early have a normal life expectancy.1
The disease is rare, with about 1000 new cases per year in the US,1 and is more common in African Americans,2 although the photos in dermatological atlases overwhelmingly show Caucasians. CTCL presents in numerous ways, but patients usually have a long course of persistent rash that is often pruritic and usually erythematous or hyperpigmented.
No one knows what causes CTCL, but current theories point to exposure to environmental hazards such as Agent Orange.2 And while the number of environmental hazards in the floodwaters of Katrina were vast, we have no scientific evidence that an environmental exposure was to blame. In fact, while we cared for a number of Katrina evacuees—many of whom had skin infections—this was the only case of mycosis fungoides.
It’s likely that the start of the visible mycosis fungoides lesions after the flood was purely coincidental.
Diagnosis hinges on palpation and biopsy
To diagnose CTCL, you need to palpate for enlarged lymph nodes and perform a full-thickness punch biopsy of the lesion. If the biopsy is negative and the rash persists, take another biopsy—results can be falsely negative at the beginning stages of the disease.
There are no guidelines on which studies should be used for staging CTCL (TABLE), but some sources recommend that if lymph node involvement is suspected on physical exam, lymph node biopsies should be done in addition to a chest radiograph.3 In more advanced stages, consider a computed tomography (CT) scan of the abdomen and pelvis. A recent study concluded that CT and positron-emission tomography (PET) scans used together were more sensitive in staging, but this may not be cost-effective.4
Late-stage mycosis fungoides is usually associated with immunocompromise. Therefore, HIV testing should be performed in all patients with CTCL. Other important laboratory studies include complete blood count—with differential—as well as peripheral smear looking for Sézary cells, lactic dehydrogenase (LDH), liver function tests, and uric acid.
TABLE
Staging for cutaneous T-cell lymphoma based on the Tumor, Node, Metastasis (TNM) system
| STAGE | TNM STAGING | RECOMMENDED TREATMENTS |
|---|---|---|
| IA | T1, N0, M0: Patch or plaque <10% body surface area | Topical high-potency steroids, PUVA, topical nitrogen |
| IB | T2, N0, M0: Patch or plaque >10% body surface area | mustard, carmustine, or bexarotene |
| IIA | T1–2, N1, M0: Patch or plaque with palpable but pathologically normal lymph node | Same as Stage I; if refractory, use total skin electron beam therapy |
| IIB | T3, N0–1, M0: Tumor/nodule | |
| IIIA | T4, N0, M0:Generalized erythroderma | |
| IIIB | T4, N1, M0: Erythroderma and palpable but pathologically normal lymph node | Chemotherapy or photophoresis, refer to medical oncologist, radiation oncologist, and dermatologist |
| IVA | T1–4, N2–3, M0: Pathological lymph node | |
| IVB | T1–4, N0–3, M1: Visceral (M1) or blood involvement | |
| T: 0–4=indicates size or direct extent of the primary tumor | ||
| N: 0=tumor cells absent from regional lymph nodes; 1=tumor cells spread to closest or small number of regional lymph nodes; 3=tumor cells spread to most distant or numerous regional lymph nodes | ||
| M: 0=no distant metastasis; 1=metastasis to distant organs (beyond regional lymph nodes) | ||
A disease that’s easy to mistake for vitiligo
The hypopigmented spots of CTCL look so much like vitiligo, it is frightening to think how easy it would be to miss the diagnosis. Complicating matters: All vitiligo does not need a biopsy to confirm the clinical impression.
So what made this case suspicious enough to warrant a biopsy? First, the hypopigmented spots on our patient were not a typical distribution for vitiligo, which has a predilection for the hands and face. Second, our patient had a hypopigmented patch that had a dark, slightly raised plaque within it, which also would not be typical for vitiligo. (See the patient’s left upper thigh, just below the inguinal area, in FIGURE 1.) Finally, our patient had a rapid onset of multiple discrete macules that did not coalesce into larger hypopigmented areas; in vitiligo you would expect otherwise.
The differential diagnosis also includes idiopathic guttate hypomelanosis, a benign condition that can cause multiple small hypopigmented macules. The size of immunoglobulin H macules, however, is tiny compared with what you’ll see with CTCL. The absence of scales makes eczema, psoriasis, or tinea corporis very unlikely.
Treatment starts with topical steroids
This is a rare disease that lacks the data needed to support an evidence-based approach to treatment. Topical steroids are recommended for stage I when the disease is local to the skin (see TABLE for recommended treatments of other stages). Ultraviolet light (PUVA and UVB) is also used; a recent study suggests the PUVA is a good treatment alternative for stages IA and IB (SOR: C).1
Our patient’s course
Initially, we prescribed a high-potency generic steroid (clobetasol 0.05% cream) for this patient, to be used twice daily to the affected areas.
The patient reported no improvement with this approach, while she awaited her appointments with dermatology and oncology specialists.
Her blood tests were essentially normal, including a negative HIV test.
She is currently receiving narrowband UVB treatment twice weekly.
Seven months after Hurricane Katrina, a 52-year-old African American woman who was evacuated from New Orleans came to our office with a hypopigmented rash on both her upper thighs and arms. Immediately following the 2005 hurricane, she was forced to wade through the polluted waters of New Orleans for many hours before being rescued by boat. Four days passed before she had access to a shower. It was after this shower that she first noticed a single erythematous spot the size of a silver dollar on her left thigh, that several weeks later faded to hypopigmented macules and plaques. Over time, the rash spread to both thighs and arms (FIGURE 1 AND 2. She did not have any itching, pain, bleeding, fever, chills, weight changes, or gastrointestinal symptoms after the rash appeared.
The patient reported that she was married and had worked as a chef for 20 years. She smoked cigarettes, drank alcohol occasionally, and was obese. She had synovitis of her left ankle, which led to surgery. She had no known drug allergies and was taking ibuprofen. Her mother, age 79, had glucose intolerance; her father, age 82, had a renal cell carcinoma removed.
Due to a confluence of situational, economic, and medical problems, she did not seek care for this rash until April 2006. At that time, a punch biopsy revealed the findings in FIGURE 3. Physical exam revealed her skin to have symmetrically distributed hypopigmented macules and plaques to all 4 extremities. She had no lymphadenopathy.
FIGURE 1
Rash on patients’ legs
FIGURE 2
Rash on right arm
FIGURE 3
Hematoxylin/eosin stain
What is the diagnosis?
Were Katrina’s flood waters to blame?
Dx: T-cell lymphoma—timing is coincidental
The biopsy revealed cutaneous T-cell lymphoma (CTCL)—specifically a type known as mycosis fungoides, named after the mushroom-like skin tumors seen in severe cases. CTCL is a malignant lymphoma of helper T-cells that have an affinity for the skin.
Normal life expectancy likely if diagnosed early
In about 10% of cases, the T-cells spread via the lymphatic system to metastasize to the liver, lung, and bone marrow, but more often remain confined to the skin and lymph nodes, and most patients diagnosed early have a normal life expectancy.1
The disease is rare, with about 1000 new cases per year in the US,1 and is more common in African Americans,2 although the photos in dermatological atlases overwhelmingly show Caucasians. CTCL presents in numerous ways, but patients usually have a long course of persistent rash that is often pruritic and usually erythematous or hyperpigmented.
No one knows what causes CTCL, but current theories point to exposure to environmental hazards such as Agent Orange.2 And while the number of environmental hazards in the floodwaters of Katrina were vast, we have no scientific evidence that an environmental exposure was to blame. In fact, while we cared for a number of Katrina evacuees—many of whom had skin infections—this was the only case of mycosis fungoides.
It’s likely that the start of the visible mycosis fungoides lesions after the flood was purely coincidental.
Diagnosis hinges on palpation and biopsy
To diagnose CTCL, you need to palpate for enlarged lymph nodes and perform a full-thickness punch biopsy of the lesion. If the biopsy is negative and the rash persists, take another biopsy—results can be falsely negative at the beginning stages of the disease.
There are no guidelines on which studies should be used for staging CTCL (TABLE), but some sources recommend that if lymph node involvement is suspected on physical exam, lymph node biopsies should be done in addition to a chest radiograph.3 In more advanced stages, consider a computed tomography (CT) scan of the abdomen and pelvis. A recent study concluded that CT and positron-emission tomography (PET) scans used together were more sensitive in staging, but this may not be cost-effective.4
Late-stage mycosis fungoides is usually associated with immunocompromise. Therefore, HIV testing should be performed in all patients with CTCL. Other important laboratory studies include complete blood count—with differential—as well as peripheral smear looking for Sézary cells, lactic dehydrogenase (LDH), liver function tests, and uric acid.
TABLE
Staging for cutaneous T-cell lymphoma based on the Tumor, Node, Metastasis (TNM) system
| STAGE | TNM STAGING | RECOMMENDED TREATMENTS |
|---|---|---|
| IA | T1, N0, M0: Patch or plaque <10% body surface area | Topical high-potency steroids, PUVA, topical nitrogen |
| IB | T2, N0, M0: Patch or plaque >10% body surface area | mustard, carmustine, or bexarotene |
| IIA | T1–2, N1, M0: Patch or plaque with palpable but pathologically normal lymph node | Same as Stage I; if refractory, use total skin electron beam therapy |
| IIB | T3, N0–1, M0: Tumor/nodule | |
| IIIA | T4, N0, M0:Generalized erythroderma | |
| IIIB | T4, N1, M0: Erythroderma and palpable but pathologically normal lymph node | Chemotherapy or photophoresis, refer to medical oncologist, radiation oncologist, and dermatologist |
| IVA | T1–4, N2–3, M0: Pathological lymph node | |
| IVB | T1–4, N0–3, M1: Visceral (M1) or blood involvement | |
| T: 0–4=indicates size or direct extent of the primary tumor | ||
| N: 0=tumor cells absent from regional lymph nodes; 1=tumor cells spread to closest or small number of regional lymph nodes; 3=tumor cells spread to most distant or numerous regional lymph nodes | ||
| M: 0=no distant metastasis; 1=metastasis to distant organs (beyond regional lymph nodes) | ||
A disease that’s easy to mistake for vitiligo
The hypopigmented spots of CTCL look so much like vitiligo, it is frightening to think how easy it would be to miss the diagnosis. Complicating matters: All vitiligo does not need a biopsy to confirm the clinical impression.
So what made this case suspicious enough to warrant a biopsy? First, the hypopigmented spots on our patient were not a typical distribution for vitiligo, which has a predilection for the hands and face. Second, our patient had a hypopigmented patch that had a dark, slightly raised plaque within it, which also would not be typical for vitiligo. (See the patient’s left upper thigh, just below the inguinal area, in FIGURE 1.) Finally, our patient had a rapid onset of multiple discrete macules that did not coalesce into larger hypopigmented areas; in vitiligo you would expect otherwise.
The differential diagnosis also includes idiopathic guttate hypomelanosis, a benign condition that can cause multiple small hypopigmented macules. The size of immunoglobulin H macules, however, is tiny compared with what you’ll see with CTCL. The absence of scales makes eczema, psoriasis, or tinea corporis very unlikely.
Treatment starts with topical steroids
This is a rare disease that lacks the data needed to support an evidence-based approach to treatment. Topical steroids are recommended for stage I when the disease is local to the skin (see TABLE for recommended treatments of other stages). Ultraviolet light (PUVA and UVB) is also used; a recent study suggests the PUVA is a good treatment alternative for stages IA and IB (SOR: C).1
Our patient’s course
Initially, we prescribed a high-potency generic steroid (clobetasol 0.05% cream) for this patient, to be used twice daily to the affected areas.
The patient reported no improvement with this approach, while she awaited her appointments with dermatology and oncology specialists.
Her blood tests were essentially normal, including a negative HIV test.
She is currently receiving narrowband UVB treatment twice weekly.
1. Pinter-Brown LC. Mycosis fungoides. Emedicine, 8 September 2006. Available at: www.emedicine.com/Med/topic1541.htm. Accessed on May 8, 2007.
2. James WD, Berger T, Elston T. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, Pa: Saunders Elsevier; 2006:727–740.
3. Gettler SL, Fung MA. Efficacy of treatments for mycosis fungoides and sézary syndrome: nationwide survey responses. Dermatol Online J 2005;11:6.-
4. Tsai EY, Taur A, Espinosa L, et al. Staging accuracy in mycosis fungoides and sezary syndrome using integrated positron emission tomography and computed tomography. Arch Dermatol 2006;142:577-584.
1. Pinter-Brown LC. Mycosis fungoides. Emedicine, 8 September 2006. Available at: www.emedicine.com/Med/topic1541.htm. Accessed on May 8, 2007.
2. James WD, Berger T, Elston T. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, Pa: Saunders Elsevier; 2006:727–740.
3. Gettler SL, Fung MA. Efficacy of treatments for mycosis fungoides and sézary syndrome: nationwide survey responses. Dermatol Online J 2005;11:6.-
4. Tsai EY, Taur A, Espinosa L, et al. Staging accuracy in mycosis fungoides and sezary syndrome using integrated positron emission tomography and computed tomography. Arch Dermatol 2006;142:577-584.
5 cases, 1 cause of irritated eyes
Irritated and watery eyes. Mild erythema of the nasal bulbar conjunctiva. Photophobia. Blurred vision. These were just some of the signs and symptoms that prompted the following 5 patients to seek treatment. Though the specifics of their cases varied, their diagnosis was the same.
CASE 1 A 35-year-old man presented with a foreign-body sensation and tearing of his right eye that had lasted for a few days. The eye showed mild erythema of the nasal bulbar conjunctiva and linear corneal abrasions.
CASE 2 A 23-year-old woman came in complaining of an irritated and watery eye that had been bothering her for the past 24 hours. She had normal visual acuity, bulbar conjunctival injection, and linear corneal abrasions.
CASE 3 A 28-year-old man presented with irritation, photophobia, and redness of his left eye that had been bothering him for the last 2 weeks. He had been treated with a topical antibiotic, but showed no improvement.
CASE 4 A 15-year-old girl came in complaining of irritation of the left eye over the last month. She was seen by an ophthalmologist, who attributed her symptoms to exposure keratopathy due to lagophthalmos—inability to close, or poor closure of, the eyelids (FIGURE). He treated her with different lubricants and antibiotics, without improvement.
CASE 5 A 15-year-old boy came in complaining of blurred vision in his right eye. His ophthalmic history was significant for corneal abrasion following a vague history of trauma to that eye a month ago. His best corrected visual acuity in that eye was 20/60.
FIGURE
Eye irritation attributed to exposure keratopathy
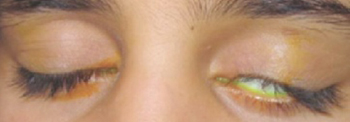
What is your diagnosis?
Diagnosis: Misplaced eyelash
The life cycle of eyelashes is usually an asymptomatic process. Sometimes, though, lashes can find their way into unusual anatomical sites, as occurred in the 5 cases described on the previous page. Here, broken down by the location in which the eyelash was found, is a discussion of the trouble a misplaced eyelash can cause.
Punctum: Upper is more common than lower
Once an eyelash is shed into the external ocular surface, it can cause foreign body sensation, leading to reflex tearing that will carry it away to the lacus lacrimalis. At this point, it comes in close contact with the punctum and can be propelled by the lids or sucked into the canaliculus in the blink cycle.
If it finds its way into the punctum, the barbs on the hair prevent it from being expelled. They can obstruct the canaliculi, causing epiphora, or incite inflammation and possibly infection, causing canaliculitis or dacryocystitis.1
Eyelashes are often reported to enter the upper and lower punctum, though Nagashima and Kido found that cilia in upper punctum was 3 times more frequent than cilia in the lower punctum.2
In CASE 1, involving mild erythema of the nasal bulbar conjunctiva and linear corneal abrasions, the 35-year-old patient had an eyelash protruding from the upper punctum. The rest of the exam was unremarkable. His physician removed the eyelash mechanically, without resistance. This relieved his symptoms and he was given a prophylactic topical antibiotic.
In CASE 2, the 23-year old woman with bulbar conjunctival injection and linear corneal abrasions, there was an eyelash projecting out of the lower punctum. The eyelash was removed easily and she, too, was treated with topical antibiotic ointment.
CASE 1 Upper punctum
Eyelash protruding from the upper punctum.
CASE 2 Lower punctum
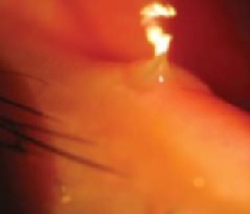
Meibomian gland: An uncommon spot
Cilia in the meibomian gland is unusual. Clinicians who suspect this is the cause of a patient’s discomfort will need to differentiate it from acquired distichiasis, which is metaplastic eyelash growth that results from chronic lid inflammation. Careful examination, and the absence of resistance on removal, differentiate the 2 conditions.
In CASE 3, involving the 28-year-old man with eye irritation and photosensitivity, an examination showed an eyelash protruding from a meibomian gland orifice 2 mm lateral to the lower punctum. He also had corneal punctate epithelial erosions and mild ciliary flush. His physician removed the eyelash with forceps without resistance. He was given a topical antibiotic.
In CASE 4, involving the 15-year-old pictured on page 365 who had been unsuccessfully treated with different lubricants and antibiotics for exposure keratopathy, an examination revealed shortening of the upper and lower eyelids of the affected eye and abnormal upper eye lid contour causing 4 to 5 mm of lagophthalmos. She had a positive Bell’s phenomenon and normal tear break-up time.
Further examination of the upper eyelid margin, pictured at far right, showed a cilium projecting out of a meibomian gland orifice toward the ocular surface with corneal punctuate erosions. The eyelash was removed smoothly with forceps without resistance, and her symptoms resolved within a few days.
CASE 3 Meibomian gland near lower punctum
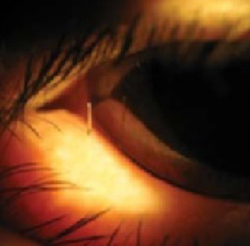
Eyelash protruding from a meibomian gland orifice 2 mm lateral to the lower punctum.
CASE 4 Meibomian gland
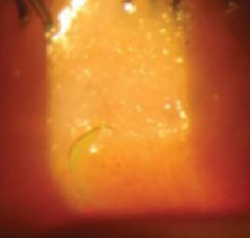
Closer examination of patient shown on page 365 revealed an eyelash projecting out of a meibomian gland orifice toward the ocular surface with corneal punctuate erosions.
Corneal stroma: Suspect trauma
Spontaneously or after trauma, loose eyelashes can penetrate the anterior ocular surface layers by their pointed tip.3,4 These dislodged eyelashes can cause no symptoms at all, or cause foreign body sensation, reflex tearing, and localized erythema mimicking other ocular conditions. Moreover, eyelashes can be washed out into the lower drainage system where they can cause tear stagnation and act as a nidus for dacryolith.5
Considering the history of trauma in CASE 5, we believe the cilium was mechanically embedded on the cornea at the time of injury and got buried as the surface reepithelialized. Examination revealed a corneal opacity containing a horizontally lying cilium. The patient was referred to a cornea specialist for further management.
CASE 5 Corneal opacity

When to suspect a misplaced eyelash
It’s important to examine the eyelid margin carefully in patients with nonspecific eye symptoms, as misplaced eyelashes can be easily overlooked and treated inappropriately. Moreover even in the presence of an obvious pathology, be sure to look for associated findings, especially when that pathology cannot fully explain the symptoms. That was especially important in Case 4, with the rare occurrence of exposure keratopathy in a young patient.
CORRESPONDENCE
Kimia Ziahosseini, MD, Flat 5, 25 Higher Hill Gate, Stockport, Cheshire SK1 3ED United Kingdom. [email protected]
1. Duke-Elder S. System of Ophthalmology. Vol 13. London: Henry Kimpton; 1974.
2. Nagashima K, Kido R. Relative roles of upper and lower lacrimal canaliculi in normal tear drainage. Jpn J Ophthalmol 1984;28:259-262.
3. Taneja S, Arora R, Yadava U. Fingernail trauma causing corneal laceration and intraocular cilia. Arch Ophthalmol 1998;116:530-531.
4. Oh KT, Oh KT, Singerman LJ. An eyelash in the vitreous cavity without apparent etiology. Ophthal Surg Lasers 1996;27:243-245.
5. Rosen WJ, Rose GE. Intranasal passage of dacryoliths. Br J Ophthalmol 2000;84:799-800.
Irritated and watery eyes. Mild erythema of the nasal bulbar conjunctiva. Photophobia. Blurred vision. These were just some of the signs and symptoms that prompted the following 5 patients to seek treatment. Though the specifics of their cases varied, their diagnosis was the same.
CASE 1 A 35-year-old man presented with a foreign-body sensation and tearing of his right eye that had lasted for a few days. The eye showed mild erythema of the nasal bulbar conjunctiva and linear corneal abrasions.
CASE 2 A 23-year-old woman came in complaining of an irritated and watery eye that had been bothering her for the past 24 hours. She had normal visual acuity, bulbar conjunctival injection, and linear corneal abrasions.
CASE 3 A 28-year-old man presented with irritation, photophobia, and redness of his left eye that had been bothering him for the last 2 weeks. He had been treated with a topical antibiotic, but showed no improvement.
CASE 4 A 15-year-old girl came in complaining of irritation of the left eye over the last month. She was seen by an ophthalmologist, who attributed her symptoms to exposure keratopathy due to lagophthalmos—inability to close, or poor closure of, the eyelids (FIGURE). He treated her with different lubricants and antibiotics, without improvement.
CASE 5 A 15-year-old boy came in complaining of blurred vision in his right eye. His ophthalmic history was significant for corneal abrasion following a vague history of trauma to that eye a month ago. His best corrected visual acuity in that eye was 20/60.
FIGURE
Eye irritation attributed to exposure keratopathy
What is your diagnosis?
Diagnosis: Misplaced eyelash
The life cycle of eyelashes is usually an asymptomatic process. Sometimes, though, lashes can find their way into unusual anatomical sites, as occurred in the 5 cases described on the previous page. Here, broken down by the location in which the eyelash was found, is a discussion of the trouble a misplaced eyelash can cause.
Punctum: Upper is more common than lower
Once an eyelash is shed into the external ocular surface, it can cause foreign body sensation, leading to reflex tearing that will carry it away to the lacus lacrimalis. At this point, it comes in close contact with the punctum and can be propelled by the lids or sucked into the canaliculus in the blink cycle.
If it finds its way into the punctum, the barbs on the hair prevent it from being expelled. They can obstruct the canaliculi, causing epiphora, or incite inflammation and possibly infection, causing canaliculitis or dacryocystitis.1
Eyelashes are often reported to enter the upper and lower punctum, though Nagashima and Kido found that cilia in upper punctum was 3 times more frequent than cilia in the lower punctum.2
In CASE 1, involving mild erythema of the nasal bulbar conjunctiva and linear corneal abrasions, the 35-year-old patient had an eyelash protruding from the upper punctum. The rest of the exam was unremarkable. His physician removed the eyelash mechanically, without resistance. This relieved his symptoms and he was given a prophylactic topical antibiotic.
In CASE 2, the 23-year old woman with bulbar conjunctival injection and linear corneal abrasions, there was an eyelash projecting out of the lower punctum. The eyelash was removed easily and she, too, was treated with topical antibiotic ointment.
CASE 1 Upper punctum
Eyelash protruding from the upper punctum.
CASE 2 Lower punctum
Meibomian gland: An uncommon spot
Cilia in the meibomian gland is unusual. Clinicians who suspect this is the cause of a patient’s discomfort will need to differentiate it from acquired distichiasis, which is metaplastic eyelash growth that results from chronic lid inflammation. Careful examination, and the absence of resistance on removal, differentiate the 2 conditions.
In CASE 3, involving the 28-year-old man with eye irritation and photosensitivity, an examination showed an eyelash protruding from a meibomian gland orifice 2 mm lateral to the lower punctum. He also had corneal punctate epithelial erosions and mild ciliary flush. His physician removed the eyelash with forceps without resistance. He was given a topical antibiotic.
In CASE 4, involving the 15-year-old pictured on page 365 who had been unsuccessfully treated with different lubricants and antibiotics for exposure keratopathy, an examination revealed shortening of the upper and lower eyelids of the affected eye and abnormal upper eye lid contour causing 4 to 5 mm of lagophthalmos. She had a positive Bell’s phenomenon and normal tear break-up time.
Further examination of the upper eyelid margin, pictured at far right, showed a cilium projecting out of a meibomian gland orifice toward the ocular surface with corneal punctuate erosions. The eyelash was removed smoothly with forceps without resistance, and her symptoms resolved within a few days.
CASE 3 Meibomian gland near lower punctum
Eyelash protruding from a meibomian gland orifice 2 mm lateral to the lower punctum.
CASE 4 Meibomian gland
Closer examination of patient shown on page 365 revealed an eyelash projecting out of a meibomian gland orifice toward the ocular surface with corneal punctuate erosions.
Corneal stroma: Suspect trauma
Spontaneously or after trauma, loose eyelashes can penetrate the anterior ocular surface layers by their pointed tip.3,4 These dislodged eyelashes can cause no symptoms at all, or cause foreign body sensation, reflex tearing, and localized erythema mimicking other ocular conditions. Moreover, eyelashes can be washed out into the lower drainage system where they can cause tear stagnation and act as a nidus for dacryolith.5
Considering the history of trauma in CASE 5, we believe the cilium was mechanically embedded on the cornea at the time of injury and got buried as the surface reepithelialized. Examination revealed a corneal opacity containing a horizontally lying cilium. The patient was referred to a cornea specialist for further management.
CASE 5 Corneal opacity
When to suspect a misplaced eyelash
It’s important to examine the eyelid margin carefully in patients with nonspecific eye symptoms, as misplaced eyelashes can be easily overlooked and treated inappropriately. Moreover even in the presence of an obvious pathology, be sure to look for associated findings, especially when that pathology cannot fully explain the symptoms. That was especially important in Case 4, with the rare occurrence of exposure keratopathy in a young patient.
CORRESPONDENCE
Kimia Ziahosseini, MD, Flat 5, 25 Higher Hill Gate, Stockport, Cheshire SK1 3ED United Kingdom. [email protected]
Irritated and watery eyes. Mild erythema of the nasal bulbar conjunctiva. Photophobia. Blurred vision. These were just some of the signs and symptoms that prompted the following 5 patients to seek treatment. Though the specifics of their cases varied, their diagnosis was the same.
CASE 1 A 35-year-old man presented with a foreign-body sensation and tearing of his right eye that had lasted for a few days. The eye showed mild erythema of the nasal bulbar conjunctiva and linear corneal abrasions.
CASE 2 A 23-year-old woman came in complaining of an irritated and watery eye that had been bothering her for the past 24 hours. She had normal visual acuity, bulbar conjunctival injection, and linear corneal abrasions.
CASE 3 A 28-year-old man presented with irritation, photophobia, and redness of his left eye that had been bothering him for the last 2 weeks. He had been treated with a topical antibiotic, but showed no improvement.
CASE 4 A 15-year-old girl came in complaining of irritation of the left eye over the last month. She was seen by an ophthalmologist, who attributed her symptoms to exposure keratopathy due to lagophthalmos—inability to close, or poor closure of, the eyelids (FIGURE). He treated her with different lubricants and antibiotics, without improvement.
CASE 5 A 15-year-old boy came in complaining of blurred vision in his right eye. His ophthalmic history was significant for corneal abrasion following a vague history of trauma to that eye a month ago. His best corrected visual acuity in that eye was 20/60.
FIGURE
Eye irritation attributed to exposure keratopathy
What is your diagnosis?
Diagnosis: Misplaced eyelash
The life cycle of eyelashes is usually an asymptomatic process. Sometimes, though, lashes can find their way into unusual anatomical sites, as occurred in the 5 cases described on the previous page. Here, broken down by the location in which the eyelash was found, is a discussion of the trouble a misplaced eyelash can cause.
Punctum: Upper is more common than lower
Once an eyelash is shed into the external ocular surface, it can cause foreign body sensation, leading to reflex tearing that will carry it away to the lacus lacrimalis. At this point, it comes in close contact with the punctum and can be propelled by the lids or sucked into the canaliculus in the blink cycle.
If it finds its way into the punctum, the barbs on the hair prevent it from being expelled. They can obstruct the canaliculi, causing epiphora, or incite inflammation and possibly infection, causing canaliculitis or dacryocystitis.1
Eyelashes are often reported to enter the upper and lower punctum, though Nagashima and Kido found that cilia in upper punctum was 3 times more frequent than cilia in the lower punctum.2
In CASE 1, involving mild erythema of the nasal bulbar conjunctiva and linear corneal abrasions, the 35-year-old patient had an eyelash protruding from the upper punctum. The rest of the exam was unremarkable. His physician removed the eyelash mechanically, without resistance. This relieved his symptoms and he was given a prophylactic topical antibiotic.
In CASE 2, the 23-year old woman with bulbar conjunctival injection and linear corneal abrasions, there was an eyelash projecting out of the lower punctum. The eyelash was removed easily and she, too, was treated with topical antibiotic ointment.
CASE 1 Upper punctum
Eyelash protruding from the upper punctum.
CASE 2 Lower punctum
Meibomian gland: An uncommon spot
Cilia in the meibomian gland is unusual. Clinicians who suspect this is the cause of a patient’s discomfort will need to differentiate it from acquired distichiasis, which is metaplastic eyelash growth that results from chronic lid inflammation. Careful examination, and the absence of resistance on removal, differentiate the 2 conditions.
In CASE 3, involving the 28-year-old man with eye irritation and photosensitivity, an examination showed an eyelash protruding from a meibomian gland orifice 2 mm lateral to the lower punctum. He also had corneal punctate epithelial erosions and mild ciliary flush. His physician removed the eyelash with forceps without resistance. He was given a topical antibiotic.
In CASE 4, involving the 15-year-old pictured on page 365 who had been unsuccessfully treated with different lubricants and antibiotics for exposure keratopathy, an examination revealed shortening of the upper and lower eyelids of the affected eye and abnormal upper eye lid contour causing 4 to 5 mm of lagophthalmos. She had a positive Bell’s phenomenon and normal tear break-up time.
Further examination of the upper eyelid margin, pictured at far right, showed a cilium projecting out of a meibomian gland orifice toward the ocular surface with corneal punctuate erosions. The eyelash was removed smoothly with forceps without resistance, and her symptoms resolved within a few days.
CASE 3 Meibomian gland near lower punctum
Eyelash protruding from a meibomian gland orifice 2 mm lateral to the lower punctum.
CASE 4 Meibomian gland
Closer examination of patient shown on page 365 revealed an eyelash projecting out of a meibomian gland orifice toward the ocular surface with corneal punctuate erosions.
Corneal stroma: Suspect trauma
Spontaneously or after trauma, loose eyelashes can penetrate the anterior ocular surface layers by their pointed tip.3,4 These dislodged eyelashes can cause no symptoms at all, or cause foreign body sensation, reflex tearing, and localized erythema mimicking other ocular conditions. Moreover, eyelashes can be washed out into the lower drainage system where they can cause tear stagnation and act as a nidus for dacryolith.5
Considering the history of trauma in CASE 5, we believe the cilium was mechanically embedded on the cornea at the time of injury and got buried as the surface reepithelialized. Examination revealed a corneal opacity containing a horizontally lying cilium. The patient was referred to a cornea specialist for further management.
CASE 5 Corneal opacity
When to suspect a misplaced eyelash
It’s important to examine the eyelid margin carefully in patients with nonspecific eye symptoms, as misplaced eyelashes can be easily overlooked and treated inappropriately. Moreover even in the presence of an obvious pathology, be sure to look for associated findings, especially when that pathology cannot fully explain the symptoms. That was especially important in Case 4, with the rare occurrence of exposure keratopathy in a young patient.
CORRESPONDENCE
Kimia Ziahosseini, MD, Flat 5, 25 Higher Hill Gate, Stockport, Cheshire SK1 3ED United Kingdom. [email protected]
1. Duke-Elder S. System of Ophthalmology. Vol 13. London: Henry Kimpton; 1974.
2. Nagashima K, Kido R. Relative roles of upper and lower lacrimal canaliculi in normal tear drainage. Jpn J Ophthalmol 1984;28:259-262.
3. Taneja S, Arora R, Yadava U. Fingernail trauma causing corneal laceration and intraocular cilia. Arch Ophthalmol 1998;116:530-531.
4. Oh KT, Oh KT, Singerman LJ. An eyelash in the vitreous cavity without apparent etiology. Ophthal Surg Lasers 1996;27:243-245.
5. Rosen WJ, Rose GE. Intranasal passage of dacryoliths. Br J Ophthalmol 2000;84:799-800.
1. Duke-Elder S. System of Ophthalmology. Vol 13. London: Henry Kimpton; 1974.
2. Nagashima K, Kido R. Relative roles of upper and lower lacrimal canaliculi in normal tear drainage. Jpn J Ophthalmol 1984;28:259-262.
3. Taneja S, Arora R, Yadava U. Fingernail trauma causing corneal laceration and intraocular cilia. Arch Ophthalmol 1998;116:530-531.
4. Oh KT, Oh KT, Singerman LJ. An eyelash in the vitreous cavity without apparent etiology. Ophthal Surg Lasers 1996;27:243-245.
5. Rosen WJ, Rose GE. Intranasal passage of dacryoliths. Br J Ophthalmol 2000;84:799-800.