User login
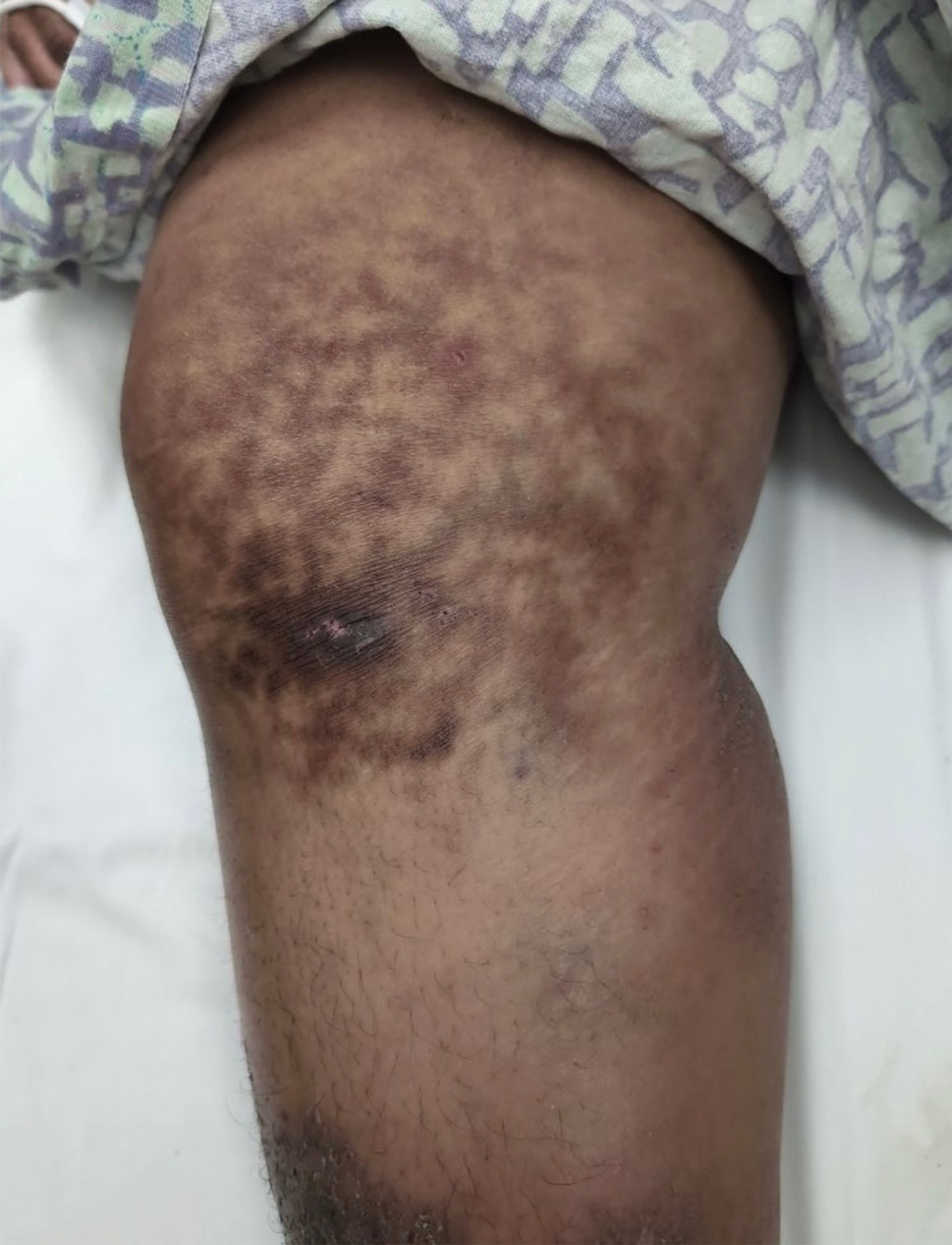
Reticulated Hyperpigmentation on the Knee and Thigh
Reticulated Hyperpigmentation on the Knee and Thigh
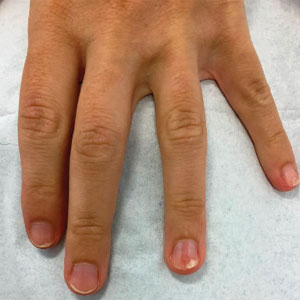
The patient was diagnosed with erythema ab igne based on characteristic skin findings on physical examination along with a convincing history of chronic localized heat exposure. Erythema ab igne manifests as a persistent reticulated, erythematous, or hyperpigmented rash at sites of chronic heat exposure.1 Commonplace items that emit heat such as electric heaters, car heaters, heating pads, hot water bottles, and, in our case, laptops also emit infrared radiation, which can lead to changes in the skin with long-term exposure.2 Because exposure to these sources often is limited to one area of the body, erythema ab igne usually manifests locally, as exemplified in this case. Chronic heat exposure and infrared radiation from these sources are thought to induce hyperthermia below the threshold for a thermal burn, and the cutaneous findings correspond with the dermal venous plexus.3
Diagnosis of erythema ab igne primarily is made clinically based on characteristic skin findings and exposure history. Relevant history may include occupations with prolonged heat exposure, such as baking, silversmithing, or foundry work. Heat exposure also may result from cultural practices such as cupping with moxibustion.4 Additionally, repeated use of heating pads or hot water bottles for pain relief by patients diagnosed with chronic pain or an underlying illness may contribute to development of erythema ab igne.1,4
Biopsy was not needed for diagnosis of this patient, but if the presentation is equivocal and history of potential exposures is unclear, a biopsy may be taken. A hematoxylin and eosin stain would reveal dilation of small vascular channels in the superficial dermis, contributing to the classic reticulated appearance. Biopsy findings also would reveal either an interface dermatitis or pigment incontinence containing melanin-laden macrophages correlating to either the erythema or hyperpigmentation, respectively.4
The prognosis for erythema ab igne is excellent, especially if diagnosed early. Treatment involves removal of the inciting heat source.1 The discoloration may resolve within a few months to years or may persist. If the hyperpigmentation is persistent, patients may consider laser treatments or lightening agents such as topical hydroquinone or topical tretinoin.4 However, if undiagnosed, patients may be at risk for development of a cutaneous malignancy, such as squamous cell carcinoma, Merkel cell carcinoma, poorly differentiated carcinoma, or cutaneous marginal zone lymphoma.2,4 Malignant transformation has been reported to occur decades after the initial skin eruption, although the risk is rare5; however, due to this risk, patients with erythema ab igne should be followed regularly and screened for new lesions in the affected areas.
- Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162:77-78.
- Miller K, Hunt R, Chu J, et al. Erythema ab igne. Dermatol Online J. 2011;17:28.
- Kesty K, Feldman SR. Erythema ab igne: evolving technology, evolving presentation. Dermatol Online J. 2014;20:13030.
- Harview CL, Krenitsky A. Erythema ab igne: a clinical review. Cutis. 2023;111:E33-E38. doi:10.12788/cutis.0771
- Wipf AJ, Brown MR. Malignant transformation of erythema ab igne. JAAD Case Rep. 2022;26:85-87. doi:10.1016/j.jdcr.2022.06.018
The patient was diagnosed with erythema ab igne based on characteristic skin findings on physical examination along with a convincing history of chronic localized heat exposure. Erythema ab igne manifests as a persistent reticulated, erythematous, or hyperpigmented rash at sites of chronic heat exposure.1 Commonplace items that emit heat such as electric heaters, car heaters, heating pads, hot water bottles, and, in our case, laptops also emit infrared radiation, which can lead to changes in the skin with long-term exposure.2 Because exposure to these sources often is limited to one area of the body, erythema ab igne usually manifests locally, as exemplified in this case. Chronic heat exposure and infrared radiation from these sources are thought to induce hyperthermia below the threshold for a thermal burn, and the cutaneous findings correspond with the dermal venous plexus.3
Diagnosis of erythema ab igne primarily is made clinically based on characteristic skin findings and exposure history. Relevant history may include occupations with prolonged heat exposure, such as baking, silversmithing, or foundry work. Heat exposure also may result from cultural practices such as cupping with moxibustion.4 Additionally, repeated use of heating pads or hot water bottles for pain relief by patients diagnosed with chronic pain or an underlying illness may contribute to development of erythema ab igne.1,4
Biopsy was not needed for diagnosis of this patient, but if the presentation is equivocal and history of potential exposures is unclear, a biopsy may be taken. A hematoxylin and eosin stain would reveal dilation of small vascular channels in the superficial dermis, contributing to the classic reticulated appearance. Biopsy findings also would reveal either an interface dermatitis or pigment incontinence containing melanin-laden macrophages correlating to either the erythema or hyperpigmentation, respectively.4
The prognosis for erythema ab igne is excellent, especially if diagnosed early. Treatment involves removal of the inciting heat source.1 The discoloration may resolve within a few months to years or may persist. If the hyperpigmentation is persistent, patients may consider laser treatments or lightening agents such as topical hydroquinone or topical tretinoin.4 However, if undiagnosed, patients may be at risk for development of a cutaneous malignancy, such as squamous cell carcinoma, Merkel cell carcinoma, poorly differentiated carcinoma, or cutaneous marginal zone lymphoma.2,4 Malignant transformation has been reported to occur decades after the initial skin eruption, although the risk is rare5; however, due to this risk, patients with erythema ab igne should be followed regularly and screened for new lesions in the affected areas.
The patient was diagnosed with erythema ab igne based on characteristic skin findings on physical examination along with a convincing history of chronic localized heat exposure. Erythema ab igne manifests as a persistent reticulated, erythematous, or hyperpigmented rash at sites of chronic heat exposure.1 Commonplace items that emit heat such as electric heaters, car heaters, heating pads, hot water bottles, and, in our case, laptops also emit infrared radiation, which can lead to changes in the skin with long-term exposure.2 Because exposure to these sources often is limited to one area of the body, erythema ab igne usually manifests locally, as exemplified in this case. Chronic heat exposure and infrared radiation from these sources are thought to induce hyperthermia below the threshold for a thermal burn, and the cutaneous findings correspond with the dermal venous plexus.3
Diagnosis of erythema ab igne primarily is made clinically based on characteristic skin findings and exposure history. Relevant history may include occupations with prolonged heat exposure, such as baking, silversmithing, or foundry work. Heat exposure also may result from cultural practices such as cupping with moxibustion.4 Additionally, repeated use of heating pads or hot water bottles for pain relief by patients diagnosed with chronic pain or an underlying illness may contribute to development of erythema ab igne.1,4
Biopsy was not needed for diagnosis of this patient, but if the presentation is equivocal and history of potential exposures is unclear, a biopsy may be taken. A hematoxylin and eosin stain would reveal dilation of small vascular channels in the superficial dermis, contributing to the classic reticulated appearance. Biopsy findings also would reveal either an interface dermatitis or pigment incontinence containing melanin-laden macrophages correlating to either the erythema or hyperpigmentation, respectively.4
The prognosis for erythema ab igne is excellent, especially if diagnosed early. Treatment involves removal of the inciting heat source.1 The discoloration may resolve within a few months to years or may persist. If the hyperpigmentation is persistent, patients may consider laser treatments or lightening agents such as topical hydroquinone or topical tretinoin.4 However, if undiagnosed, patients may be at risk for development of a cutaneous malignancy, such as squamous cell carcinoma, Merkel cell carcinoma, poorly differentiated carcinoma, or cutaneous marginal zone lymphoma.2,4 Malignant transformation has been reported to occur decades after the initial skin eruption, although the risk is rare5; however, due to this risk, patients with erythema ab igne should be followed regularly and screened for new lesions in the affected areas.
- Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162:77-78.
- Miller K, Hunt R, Chu J, et al. Erythema ab igne. Dermatol Online J. 2011;17:28.
- Kesty K, Feldman SR. Erythema ab igne: evolving technology, evolving presentation. Dermatol Online J. 2014;20:13030.
- Harview CL, Krenitsky A. Erythema ab igne: a clinical review. Cutis. 2023;111:E33-E38. doi:10.12788/cutis.0771
- Wipf AJ, Brown MR. Malignant transformation of erythema ab igne. JAAD Case Rep. 2022;26:85-87. doi:10.1016/j.jdcr.2022.06.018
- Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162:77-78.
- Miller K, Hunt R, Chu J, et al. Erythema ab igne. Dermatol Online J. 2011;17:28.
- Kesty K, Feldman SR. Erythema ab igne: evolving technology, evolving presentation. Dermatol Online J. 2014;20:13030.
- Harview CL, Krenitsky A. Erythema ab igne: a clinical review. Cutis. 2023;111:E33-E38. doi:10.12788/cutis.0771
- Wipf AJ, Brown MR. Malignant transformation of erythema ab igne. JAAD Case Rep. 2022;26:85-87. doi:10.1016/j.jdcr.2022.06.018
Reticulated Hyperpigmentation on the Knee and Thigh
Reticulated Hyperpigmentation on the Knee and Thigh
A 25-year-old woman with an unremarkable medical history presented to the dermatology clinic for evaluation of a persistent rash on the right knee and distal thigh of several months’ duration. The patient noted that the rash had been asymptomatic, and she denied any history of trauma to the area. She reported that she worked as a teacher and had repeatedly stayed up late using her laptop for months. Rather than use a desk, she often would work sitting with her laptop in her lap.

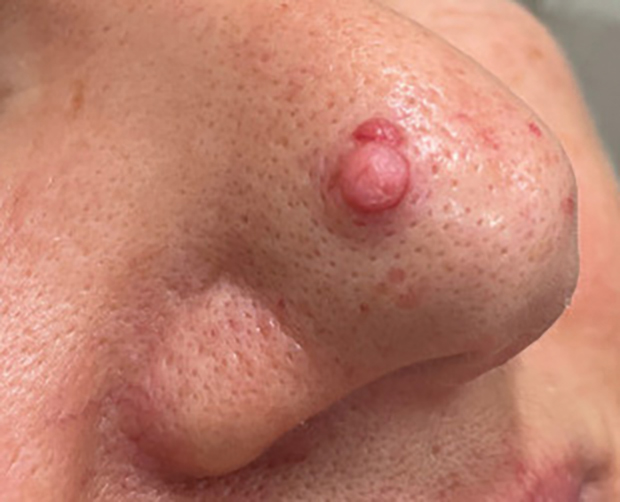
Flesh-Colored Lesion on the Ear
Flesh-Colored Lesion on the Ear
THE DIAGNOSIS: Gouty Tophus
The lesion was excised and sent for histopathologic examination (eFigures 1 and 2), revealing aggregates of feathery, amorphous, pale-pink material, which confirmed the diagnosis of gouty tophus. The surgical site was left to heal by secondary intention. Upon further evaluation, the patient reported recurrent monoarticular joint pain in the ankles and feet, and laboratory workup revealed elevated serum uric acid. He was advised to follow up with his primary care physician to discuss systemic treatment options for gout.
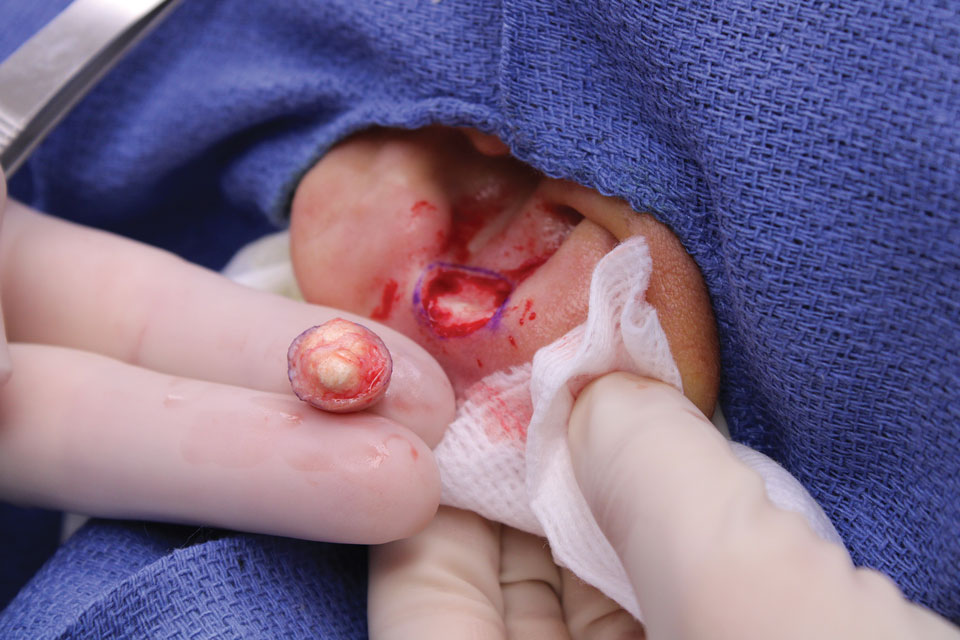
Gout is an inflammatory arthritis characterized by the deposition of monosodium urate monohydrate crystals in the joints, soft tissue, and bone due to elevated serum uric acid. Uric acid is the final product of purine metabolism, and serum levels may be elevated due to excess production or underexcretion. Multiple genetic, environmental, and metabolic factors influence these processes.1 Collections of monosodium urate crystals may develop intra- or extra-articularly, the latter of which are known as gouty tophi. These nodules have a classic chalklike consistency and typically are seen in patients with untreated gout starting approximately 10 years after the first flare. The most common locations for subcutaneous gouty tophi are acral sites (eg, fingertips, ears) as well as the wrists, knees, and elbows (olecranon bursae). Rarely, gouty panniculitis also may develop.2
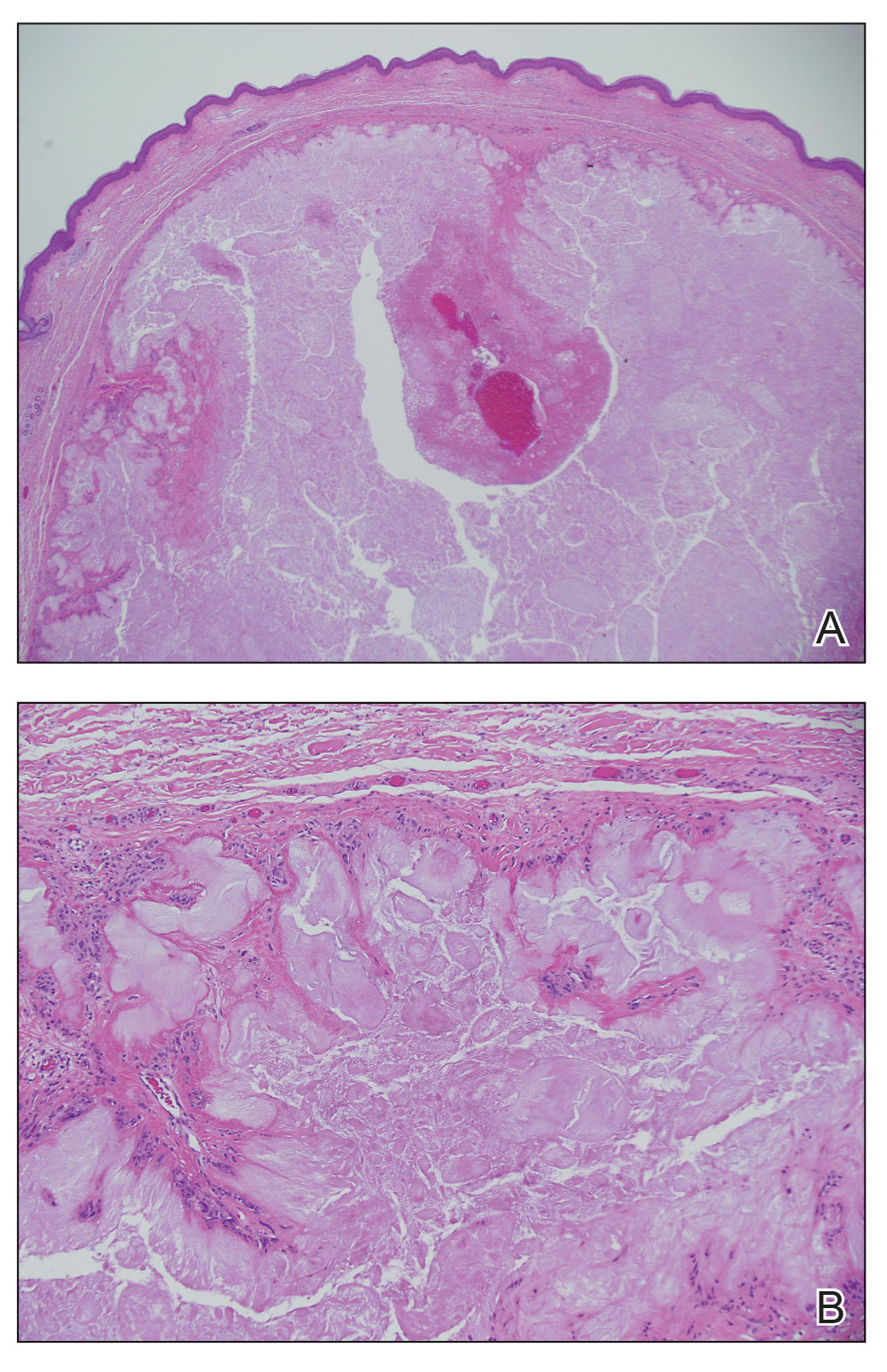
Histopathology of gouty tophi reveals nodular aggregates of acellular, amorphous, pale-pink material surrounded by palisading histiocytes and multinucleated giant cells. The presence of needlelike monosodium urate crystals, which display negative birefringence, is diagnostic. Unfortunately, these crystals are destroyed in routine formalin processing.3
There are limited data regarding treatment of gouty tophi. Urate-lowering systemic medications such as pegloticase may be beneficial, but more data are needed.4 We pursued surgical excision in our case for definitive diagnosis; however, it is not a common treatment for gouty tophi. Typically, urate-lowering therapy is utilized to resolve or shrink lesions over time.5
The differential diagnosis for gouty tophi includes epidermal inclusion cyst (EIC), the most common type of cutaneous cyst. Though EICs can manifest anywhere on the body, they are not as common on the ears as gouty tophi. Epidermal inclusion cysts clinically manifest as soft subcutaneous nodules, and a central punctum often is noted. These lesions are derived from the follicular infundibulum and histologically are characterized by a cystic cavity lined by a stratified squamous epithelium with a granular layer. The cavity contains loose laminated keratin material.6
Pseudocyst of the auricle is a benign cystic swelling of the pinna that can develop spontaneously but most often manifests following trauma to the area, which is believed to separate the tissue planes in the cartilage, allowing fluid to accumulate. This lesion typically is asymptomatic, though some patients report mild tenderness.7 Histology shows a cystic structure within the cartilage without an epithelial lining, and a perivascular inflammatory response often is observed.8
Pilomatricoma, also known as pilomatrixoma, is a benign tumor derived from the hair follicle matrix that manifests as a firm, slow-growing, painless subcutaneous nodule. It most often is found on the head and neck, commonly in the periauricular area.9 Though rare, it has been found on the auricle and external auditory canal.10 Histologically, pilomatricomas are well-defined tumors containing internal trabeculae. They contain populations of basaloid and ghost cells and often calcify, sometimes with resultant bone formation.9
Dermoid cysts are benign tumors that develop along lines of embryonic closure and often are diagnosed at birth or in early childhood. They most commonly manifest on the head and neck, typically in the supraorbital area. Rarely, they have been reported on the ear.6 Dermoid cysts may resemble EICs clinically and histopathologically, except that the cyst wall contains mature adnexal structures such as hair follicles and sebaceous glands.
- Dalbeth N, Merriman TR, Stamp LK. Gout. Lancet. 2016;388:2039-2052. doi:10.1016/S0140-6736(16)00346-9
- Gaviria JL, Ortega VG, Gaona J, et al. Unusual dermatological manifestations of gout: review of literature and a case report. Plast Reconstr Surg Glob Open. 2015;3:E445. doi:10.1097/GOX.0000000000000420
- Towiwat P, Chhana A, Dalbeth N. The anatomical pathology of gout: a systematic literature review. BMC Musculoskelet Disord. 2019;20:140. doi:10.1186/s12891-019-2519-y
- Sriranganathan MK, Vinik O, Pardo Pardo J, et al. Interventions for tophi in gout. Cochrane Database Syst Rev. 2021;8:CD010069. doi:10.1002/14651858.CD010069.pub3
- Evidence review for surgical excision of tophi. Gout: diagnosis and management. National Institute for Health and Care Excellence (NICE). June 2022. Accessed October 8, 2025. https://www.ncbi.nlm.nih.gov/books/NBK583526/
- Cho Y, Lee DH. Clinical characteristics of idiopathic epidermoid and dermoid cysts of the ear. J Audiol Otol. 2017;21:77-80. doi:10.7874 /jao.2017.21.2.77
- Ballan A, Zogheib S, Hanna C, et al. Auricular pseudocysts: a systematic review of the literature. Int J Dermatol. 2022;61:109-117. doi:10.1111/ijd.15816
- Lim CM, Goh YH, Chao SS, et al. Pseudocyst of the auricle: a histologic perspective. Laryngoscope. 2004;114:1281-1284. doi:10.1097/00005537-200407000-00026
- Jones CD, Ho W, Robertson BF, et al. Pilomatrixoma: a comprehensive review of the literature. Am J Dermatopathol. 2018; 40:631-641. doi:10.1097/DAD.0000000000001118
- McInerney NJ, Nae A, Brennan S, et al. Pilomatricoma of the external auditory canal. Royal College of Surgeons in Ireland. 2023. doi:10.1016/j.xocr.2023.10053
THE DIAGNOSIS: Gouty Tophus
The lesion was excised and sent for histopathologic examination (eFigures 1 and 2), revealing aggregates of feathery, amorphous, pale-pink material, which confirmed the diagnosis of gouty tophus. The surgical site was left to heal by secondary intention. Upon further evaluation, the patient reported recurrent monoarticular joint pain in the ankles and feet, and laboratory workup revealed elevated serum uric acid. He was advised to follow up with his primary care physician to discuss systemic treatment options for gout.
Gout is an inflammatory arthritis characterized by the deposition of monosodium urate monohydrate crystals in the joints, soft tissue, and bone due to elevated serum uric acid. Uric acid is the final product of purine metabolism, and serum levels may be elevated due to excess production or underexcretion. Multiple genetic, environmental, and metabolic factors influence these processes.1 Collections of monosodium urate crystals may develop intra- or extra-articularly, the latter of which are known as gouty tophi. These nodules have a classic chalklike consistency and typically are seen in patients with untreated gout starting approximately 10 years after the first flare. The most common locations for subcutaneous gouty tophi are acral sites (eg, fingertips, ears) as well as the wrists, knees, and elbows (olecranon bursae). Rarely, gouty panniculitis also may develop.2
Histopathology of gouty tophi reveals nodular aggregates of acellular, amorphous, pale-pink material surrounded by palisading histiocytes and multinucleated giant cells. The presence of needlelike monosodium urate crystals, which display negative birefringence, is diagnostic. Unfortunately, these crystals are destroyed in routine formalin processing.3
There are limited data regarding treatment of gouty tophi. Urate-lowering systemic medications such as pegloticase may be beneficial, but more data are needed.4 We pursued surgical excision in our case for definitive diagnosis; however, it is not a common treatment for gouty tophi. Typically, urate-lowering therapy is utilized to resolve or shrink lesions over time.5
The differential diagnosis for gouty tophi includes epidermal inclusion cyst (EIC), the most common type of cutaneous cyst. Though EICs can manifest anywhere on the body, they are not as common on the ears as gouty tophi. Epidermal inclusion cysts clinically manifest as soft subcutaneous nodules, and a central punctum often is noted. These lesions are derived from the follicular infundibulum and histologically are characterized by a cystic cavity lined by a stratified squamous epithelium with a granular layer. The cavity contains loose laminated keratin material.6
Pseudocyst of the auricle is a benign cystic swelling of the pinna that can develop spontaneously but most often manifests following trauma to the area, which is believed to separate the tissue planes in the cartilage, allowing fluid to accumulate. This lesion typically is asymptomatic, though some patients report mild tenderness.7 Histology shows a cystic structure within the cartilage without an epithelial lining, and a perivascular inflammatory response often is observed.8
Pilomatricoma, also known as pilomatrixoma, is a benign tumor derived from the hair follicle matrix that manifests as a firm, slow-growing, painless subcutaneous nodule. It most often is found on the head and neck, commonly in the periauricular area.9 Though rare, it has been found on the auricle and external auditory canal.10 Histologically, pilomatricomas are well-defined tumors containing internal trabeculae. They contain populations of basaloid and ghost cells and often calcify, sometimes with resultant bone formation.9
Dermoid cysts are benign tumors that develop along lines of embryonic closure and often are diagnosed at birth or in early childhood. They most commonly manifest on the head and neck, typically in the supraorbital area. Rarely, they have been reported on the ear.6 Dermoid cysts may resemble EICs clinically and histopathologically, except that the cyst wall contains mature adnexal structures such as hair follicles and sebaceous glands.
THE DIAGNOSIS: Gouty Tophus
The lesion was excised and sent for histopathologic examination (eFigures 1 and 2), revealing aggregates of feathery, amorphous, pale-pink material, which confirmed the diagnosis of gouty tophus. The surgical site was left to heal by secondary intention. Upon further evaluation, the patient reported recurrent monoarticular joint pain in the ankles and feet, and laboratory workup revealed elevated serum uric acid. He was advised to follow up with his primary care physician to discuss systemic treatment options for gout.
Gout is an inflammatory arthritis characterized by the deposition of monosodium urate monohydrate crystals in the joints, soft tissue, and bone due to elevated serum uric acid. Uric acid is the final product of purine metabolism, and serum levels may be elevated due to excess production or underexcretion. Multiple genetic, environmental, and metabolic factors influence these processes.1 Collections of monosodium urate crystals may develop intra- or extra-articularly, the latter of which are known as gouty tophi. These nodules have a classic chalklike consistency and typically are seen in patients with untreated gout starting approximately 10 years after the first flare. The most common locations for subcutaneous gouty tophi are acral sites (eg, fingertips, ears) as well as the wrists, knees, and elbows (olecranon bursae). Rarely, gouty panniculitis also may develop.2
Histopathology of gouty tophi reveals nodular aggregates of acellular, amorphous, pale-pink material surrounded by palisading histiocytes and multinucleated giant cells. The presence of needlelike monosodium urate crystals, which display negative birefringence, is diagnostic. Unfortunately, these crystals are destroyed in routine formalin processing.3
There are limited data regarding treatment of gouty tophi. Urate-lowering systemic medications such as pegloticase may be beneficial, but more data are needed.4 We pursued surgical excision in our case for definitive diagnosis; however, it is not a common treatment for gouty tophi. Typically, urate-lowering therapy is utilized to resolve or shrink lesions over time.5
The differential diagnosis for gouty tophi includes epidermal inclusion cyst (EIC), the most common type of cutaneous cyst. Though EICs can manifest anywhere on the body, they are not as common on the ears as gouty tophi. Epidermal inclusion cysts clinically manifest as soft subcutaneous nodules, and a central punctum often is noted. These lesions are derived from the follicular infundibulum and histologically are characterized by a cystic cavity lined by a stratified squamous epithelium with a granular layer. The cavity contains loose laminated keratin material.6
Pseudocyst of the auricle is a benign cystic swelling of the pinna that can develop spontaneously but most often manifests following trauma to the area, which is believed to separate the tissue planes in the cartilage, allowing fluid to accumulate. This lesion typically is asymptomatic, though some patients report mild tenderness.7 Histology shows a cystic structure within the cartilage without an epithelial lining, and a perivascular inflammatory response often is observed.8
Pilomatricoma, also known as pilomatrixoma, is a benign tumor derived from the hair follicle matrix that manifests as a firm, slow-growing, painless subcutaneous nodule. It most often is found on the head and neck, commonly in the periauricular area.9 Though rare, it has been found on the auricle and external auditory canal.10 Histologically, pilomatricomas are well-defined tumors containing internal trabeculae. They contain populations of basaloid and ghost cells and often calcify, sometimes with resultant bone formation.9
Dermoid cysts are benign tumors that develop along lines of embryonic closure and often are diagnosed at birth or in early childhood. They most commonly manifest on the head and neck, typically in the supraorbital area. Rarely, they have been reported on the ear.6 Dermoid cysts may resemble EICs clinically and histopathologically, except that the cyst wall contains mature adnexal structures such as hair follicles and sebaceous glands.
- Dalbeth N, Merriman TR, Stamp LK. Gout. Lancet. 2016;388:2039-2052. doi:10.1016/S0140-6736(16)00346-9
- Gaviria JL, Ortega VG, Gaona J, et al. Unusual dermatological manifestations of gout: review of literature and a case report. Plast Reconstr Surg Glob Open. 2015;3:E445. doi:10.1097/GOX.0000000000000420
- Towiwat P, Chhana A, Dalbeth N. The anatomical pathology of gout: a systematic literature review. BMC Musculoskelet Disord. 2019;20:140. doi:10.1186/s12891-019-2519-y
- Sriranganathan MK, Vinik O, Pardo Pardo J, et al. Interventions for tophi in gout. Cochrane Database Syst Rev. 2021;8:CD010069. doi:10.1002/14651858.CD010069.pub3
- Evidence review for surgical excision of tophi. Gout: diagnosis and management. National Institute for Health and Care Excellence (NICE). June 2022. Accessed October 8, 2025. https://www.ncbi.nlm.nih.gov/books/NBK583526/
- Cho Y, Lee DH. Clinical characteristics of idiopathic epidermoid and dermoid cysts of the ear. J Audiol Otol. 2017;21:77-80. doi:10.7874 /jao.2017.21.2.77
- Ballan A, Zogheib S, Hanna C, et al. Auricular pseudocysts: a systematic review of the literature. Int J Dermatol. 2022;61:109-117. doi:10.1111/ijd.15816
- Lim CM, Goh YH, Chao SS, et al. Pseudocyst of the auricle: a histologic perspective. Laryngoscope. 2004;114:1281-1284. doi:10.1097/00005537-200407000-00026
- Jones CD, Ho W, Robertson BF, et al. Pilomatrixoma: a comprehensive review of the literature. Am J Dermatopathol. 2018; 40:631-641. doi:10.1097/DAD.0000000000001118
- McInerney NJ, Nae A, Brennan S, et al. Pilomatricoma of the external auditory canal. Royal College of Surgeons in Ireland. 2023. doi:10.1016/j.xocr.2023.10053
- Dalbeth N, Merriman TR, Stamp LK. Gout. Lancet. 2016;388:2039-2052. doi:10.1016/S0140-6736(16)00346-9
- Gaviria JL, Ortega VG, Gaona J, et al. Unusual dermatological manifestations of gout: review of literature and a case report. Plast Reconstr Surg Glob Open. 2015;3:E445. doi:10.1097/GOX.0000000000000420
- Towiwat P, Chhana A, Dalbeth N. The anatomical pathology of gout: a systematic literature review. BMC Musculoskelet Disord. 2019;20:140. doi:10.1186/s12891-019-2519-y
- Sriranganathan MK, Vinik O, Pardo Pardo J, et al. Interventions for tophi in gout. Cochrane Database Syst Rev. 2021;8:CD010069. doi:10.1002/14651858.CD010069.pub3
- Evidence review for surgical excision of tophi. Gout: diagnosis and management. National Institute for Health and Care Excellence (NICE). June 2022. Accessed October 8, 2025. https://www.ncbi.nlm.nih.gov/books/NBK583526/
- Cho Y, Lee DH. Clinical characteristics of idiopathic epidermoid and dermoid cysts of the ear. J Audiol Otol. 2017;21:77-80. doi:10.7874 /jao.2017.21.2.77
- Ballan A, Zogheib S, Hanna C, et al. Auricular pseudocysts: a systematic review of the literature. Int J Dermatol. 2022;61:109-117. doi:10.1111/ijd.15816
- Lim CM, Goh YH, Chao SS, et al. Pseudocyst of the auricle: a histologic perspective. Laryngoscope. 2004;114:1281-1284. doi:10.1097/00005537-200407000-00026
- Jones CD, Ho W, Robertson BF, et al. Pilomatrixoma: a comprehensive review of the literature. Am J Dermatopathol. 2018; 40:631-641. doi:10.1097/DAD.0000000000001118
- McInerney NJ, Nae A, Brennan S, et al. Pilomatricoma of the external auditory canal. Royal College of Surgeons in Ireland. 2023. doi:10.1016/j.xocr.2023.10053
Flesh-Colored Lesion on the Ear
Flesh-Colored Lesion on the Ear
A 46-year-old man with a history of hypertension, hyperlipidemia, and type 2 diabetes presented to the dermatology clinic with a painless nodule on the left ear of 2 years’ duration. The patient denied any bleeding, drainage, or prior trauma to the area. He noted that the lesion had grown slowly over time. Physical examination revealed a 1.5×1.5-cm, flesh-colored, subcutaneous nodule with overlying telangiectasias on the left antihelix.
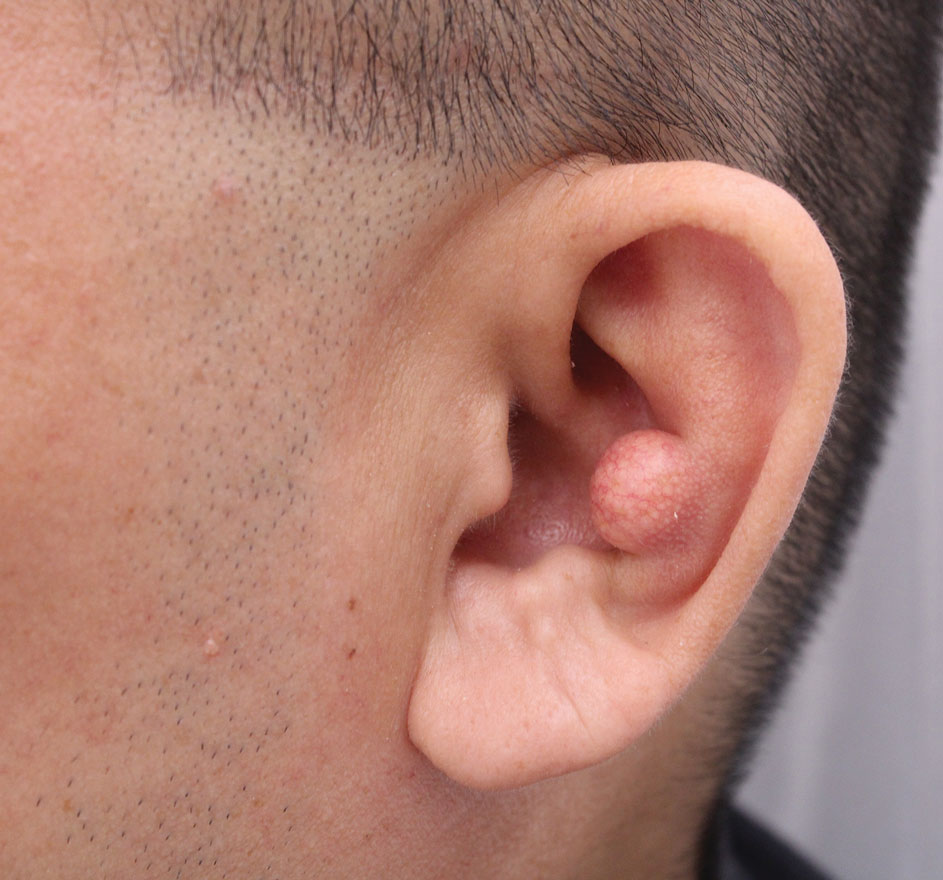

Longitudinal Erythronychia Manifesting With Pain and Cold Sensitivity
The Diagnosis: Glomangiomyoma
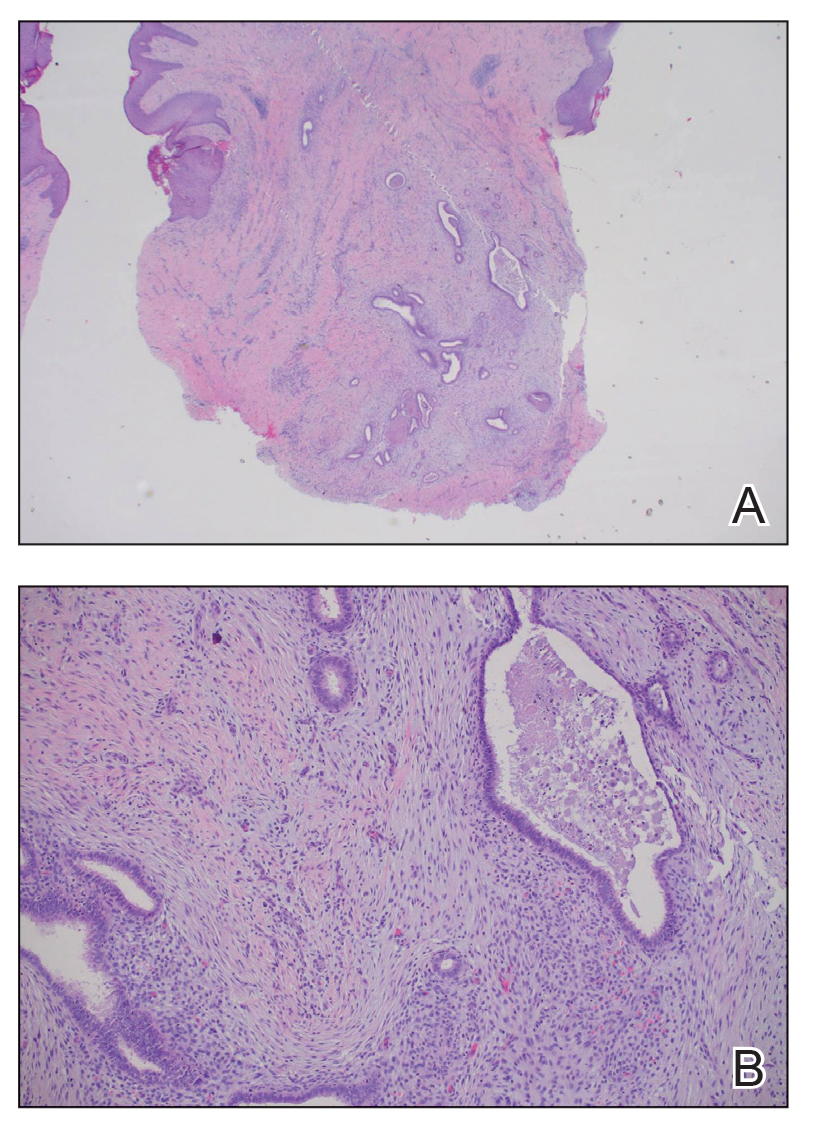
The nail unit excision specimen showed collections of cuboidal cells and spindled cells within the corium that were consistent with a diagnosis of a glomangiomyoma, a rare glomus tumor variant (Figure). Glomus tumors are benign neoplasms comprising glomus bodies, which are arteriovenous anastomoses involved in thermoregulation.1 They develop in areas densely populated by glomus bodies, including the fingers, toes, and subungual areas. Glomus tumors most commonly develop in middle-aged women.2 Clinically, they manifest with a characteristic triad of intense pain, point tenderness, and cold sensitivity and may appear as reddish-pink or blue macules under the nail plate and/or longitudinal erythronychia.2-6 The presence of multiple glomus tumors is associated with neurofibromatosis type 1.7
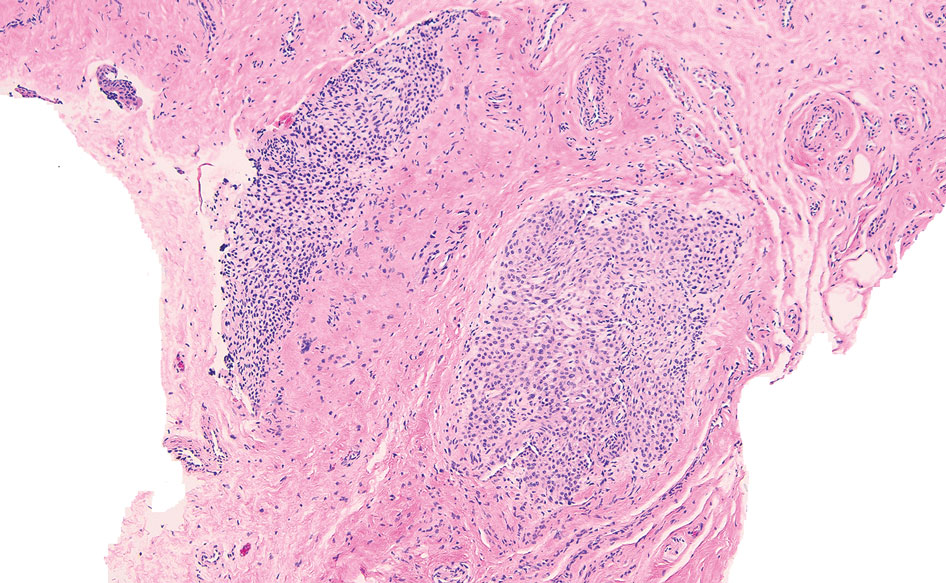
Advanced imaging including ultrasonography and magnetic resonance imaging (MRI) may help confirm the diagnosis but may not be cost effective, as excision with histopathology is needed to relieve symptoms and render a definitive diagnosis. Radiography is highly insensitive in identifying bone erosions associated with glomus tumors.8 With ultrasonography, glomus tumors appear hypoechoic; with Doppler ultrasonography, they appear hypervascular. With MRI, glomus tumors appear as well-defined nodular lesions with hypointense signal intensity on T1-weighted sequence and hyperintense signal intensity on T2-weighted sequence, with strong enhancement using gadolinium-based contrast.9,10 On histopathology, a glomus tumor appears as a nodular tumor with sheets of oval-nucleated cells arranged in multicellular layers surrounding blood vessels and are immunoreactive for α-smooth muscle actin, muscle-specific actin, and type IV collagen.11,12
There are several glomus tumor variants. The most common is a solid glomus tumor, which predominantly is composed of glomus cells, followed by glomangioma, which mainly is composed of blood vessels. Glomangiomyoma, which mostly is composed of smooth muscle cells, is the rarest variant.13
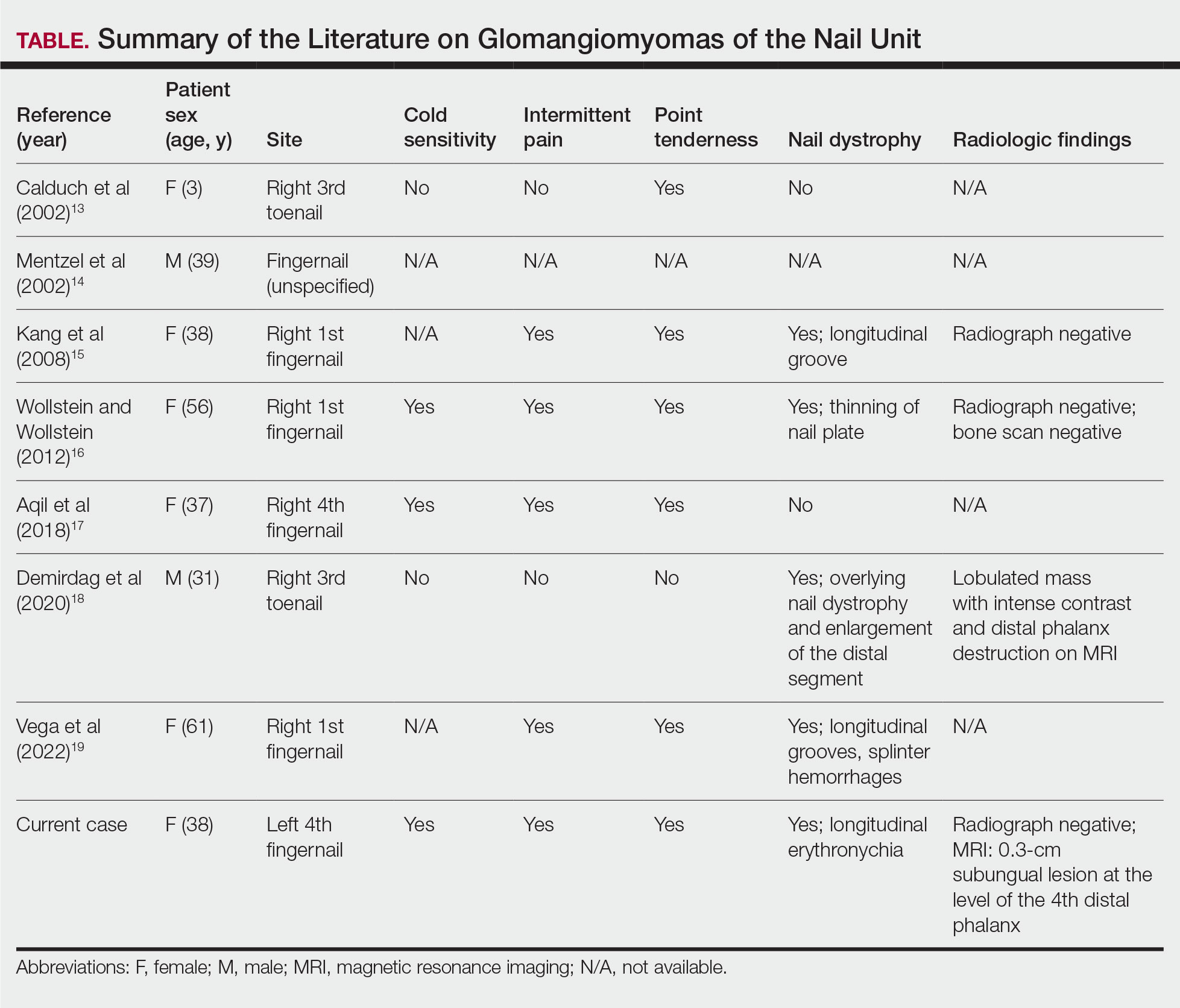
While glomus tumors are common in the subungual areas, it is an uncommon location for glomangiomyomas, which have been reported in the nail unit in only 7 prior case reports identified through searches of PubMed and Google Scholar using the terms glomangiomyoma, glomangiomyoma nail, and subungual glomangiomyoma (Table).13-19 Glomangiomyomas more commonly are described in solid organs, including the stomach, kidney, pancreas, and bladder.16 The mean age of patients with subungual glomangiomyomas, including our patient, was 40.4 years (range, 3-61 years), with the majority being female (75.0% [6/8]). Most patients presented with fingernail involvement (75.0% [6/8]), nail dystrophy (eg, nail plate thinning, longitudinal grooves, splinter hemorrhages, longitudinal erythronychia)(62.5% [5/8]), and intermittent pain and/or point tenderness in the affected nail (75.0% [6/8]).13-19 Notably, only our patient had longitudinal erythronychia as a clinical feature, and only one other case described MRI findings, which included a lobulated mass with intense contrast and distal phalanx destruction.18 One patient was a 3-year-old girl with a family history of generalized multiple glomangiomyomas. Although subungual glomangiomyoma was not confirmed on histopathology, the diagnosis in this patient was presumed based on her family history.13 On histopathology, glomangiomyomas are composed of oval-nucleated cells surrounding blood vessels. These oval-nucleated cells then gradually transition to smooth muscle cells.20
A myxoid cyst is composed of a pseudocyst, which lacks a cyst lining, and is a result of synovial fluid from the distal interphalangeal joint entering the pseudocyst space.2 It typically manifests with a longitudinal groove in the nail plate. A flesh-colored nodule may be appreciated between the cuticle and the distal interphalangeal joint.2 The depth of the longitudinal groove may vary depending on the volume of synovial fluid within the myxoid cyst.21 In a series of 35 cases of subungual myxoid cysts, none manifested with longitudinal erythronychia. Due to their composition, myxoid cysts can be distinguished easily from solid tumors of the nail unit via transillumination.22 Pain is a much less common with myxoid cysts vs glomus tumors, as the filling of the pseudocyst space with synovial fluid typically is gradual, allowing the surrounding tissue to accommodate and adapt over time.21 In equivocal cases, MRI or high-resolution ultrasonography may be used to distinguish myxoid cysts and glomus tumors.8 Histopathology shows accumulation of mucin in the dermis with surrounding fibrous stroma.23
Subungual neuromas are painful benign tumors that develop due to disorganized neural proliferation following disruption to peripheral nerves secondary to trauma or surgery. In 3 case reports, subungual neuromas manifested as painful subungual nodules, with proximal nail plate ridging, or onycholysis.24-26 Since neuromas have only rarely been described in the subungual region, reports of MRI and ultrasonography findings are unknown. Histopathology is needed to distinguish neuromas from glomus tumors. Histopathology shows an acapsular structure consisting of disorganized spindle-cell proliferation and nerve fibers arranged in a tangle of fascicles within fibrotic tissue.25 On immunochemistry, spindle cells typically are positive for cellular antigen protein S100.26
Leiomyomas are benign neoplasms derived from smooth muscle, typically localized to the uterus or gastrointestinal tract, and have been described rarely in the nail unit.27,28 It is hypothesized that subungual leiomyomas originate from the vascular smooth muscle in the subcutaneous layer of the nail unit.28 Like glomus tumors, leiomyomas of the subungual region often manifest with pain and longitudinal erythronychia.27-30 Subungual leiomyomas may be distinguished from glomus tumors via advanced imaging techniques, including ultrasonography and MRI. Cutaneous leiomyomas have been described with mild to moderate internal low flow vascularity on Doppler ultrasonography, while glomus tumors typically reveal high internal vascularity.28 Biopsy with histopathology is needed for definitive diagnosis. On histopathology, leiomyomas demonstrate bland-appearing spindle-shaped cells with elongated nuclei arranged in fascicles.27 They typically are positive for α-smooth muscle actin and caldesmon on immunostaining.
Eccrine spiradenomas are benign adnexal tumors likely of apocrine origin with limited case reports in the literature.31,32 Clinically, eccrine spiradenomas involving the nail unit may manifest with longitudinal nail splitting of the nail or as a papule on the proximal nail fold, with associated tenderness.31,32 In a report of a 50-year-old woman with a histopathologically confirmed eccrine spiradenoma manifesting with longitudinal splitting of the nail and pain in the proximal nail fold, the mass appeared hypoechoic on ultrasonography with increased intramass vascularity on Doppler, while MRI showed an intensely enhancing lesion.31 These imaging features, combined with a classically manifesting feature of pain, make eccrine spiradenomas difficult to distinguish from glomus tumors; therefore, histopathologic examination can provide a definitive diagnosis, and surgical excision is used for treatment.31 On histopathology, these tumors are well circumscribed and composed of both small dark basaloid cells with peripheral compact nuclei and larger cells with central pale nuclei, which may be arranged in tubules.31,32
- Gombos Z, Zhang PJ. Glomus tumor. Arch Pathol Lab Med. 2008;132: 1448-1452. doi:10.5858/2008-132-1448-gt
- Hare AQ, Rich P. Nail tumors. Dermatol Clin. 2021;39:281-292. doi:10.1016/j.det.2020.12.007
- Hazani R, Houle JM, Kasdan ML, et al. Glomus tumors of the hand. Eplasty. 2008;8:E48.
- Hwang JK, Lipner SR. Blue nail discoloration: literature review and diagnostic algorithms. Am J Clin Dermatol. 2023;24:419-441. doi:10.1007/s40257-023-00768-6
- Lipner SR, Scher RK. Longitudinal erythronychia of the fingernail. JAMA Dermatol. 2016;152:1271-1272. doi:10.1001/jamadermatol.2016.2747
- Jellinek NJ, Lipner SR. Longitudinal erythronychia: retrospective single-center study evaluating differential diagnosis and the likelihood of malignancy. Dermatol Surg. 2016;42:310-319. doi:10.1097 /DSS.0000000000000594
- Lipner SR, Scher RK. Subungual glomus tumors: underrecognized clinical findings in neurofibromatosis 1. J Am Acad Dermatol. 2021;84:E269. doi:10.1016/j.jaad.2020.08.129
- Dhami A, Vale SM, Richardson ML, et al. Comparing ultrasound with magnetic resonance imaging in the evaluation of subungual glomus tumors and subungual myxoid cysts. Skin Appendage Disord. 2023;9:262-267. doi:10.1159/000530397
- Baek HJ, Lee SJ, Cho KH, et al. Subungual tumors: clinicopathologic correlation with US and MR imaging findings. Radiographics. 2010;30:1621-1636. doi:10.1148/rg.306105514
- Patel T, Meena V, Meena P. Hand and foot glomus tumors: significance of MRI diagnosis followed by histopathological assessment. Cureus. 2022;14:E30038. doi:10.7759/cureus.30038
- Mravic M, LaChaud G, Nguyen A, et al. Clinical and histopathological diagnosis of glomus tumor: an institutional experience of 138 cases. Int J Surg Pathol. 2015;23:181-188. doi:10.1177/1066896914567330
- Folpe AL, Fanburg-Smith JC, Miettinen M, et al. Atypical and malignant glomus tumors: analysis of 52 cases, with a proposal for the reclassification of glomus tumors. Am J Surg Pathol. 2001;25:1-12. doi:10.1097/00000478-200101000-00001
- Calduch L, Monteagudo C, Martínez-Ruiz E, et al. Familial generalized multiple glomangiomyoma: report of a new family, with immunohistochemical and ultrastructural studies and review of the literature. Pediatr Dermatol. 2002;19:402-408. doi:10.1046/j.1525-1470.2002.00114.x
- Mentzel T, Hügel H, Kutzner H. CD34-positive glomus tumor: clinicopathologic and immunohistochemical analysis of six cases with myxoid stromal changes. J Cutan Pathol. 2002;29:421-425. doi:10.1034 /j.1600-0560.2002.290706.x
- Kang TW, Lee KH, Park CJ. A case of subungual glomangiomyoma with myxoid stromal change. Korean J Dermatol. 2008;46:550-553.
- Wollstein A, Wollstein R. Subungual glomangiomyoma—a case report. Hand Surg. 2012;17:271-273. doi:10.1142/S021881041272032X
- Aqil N, Gallouj S, Moustaide K, et al. Painful tumors in a patient with neurofibromatosis type 1: a case report. J Med Case Rep. 2018;12:319. doi:10.1186/s13256-018-1847-0
- Demirdag HG, Akay BN, Kirmizi A, et al. Subungual glomangiomyoma. J Am Podiatr Med Assoc. 2020;110:Article_13. doi:10.7547/19-051
- Vega SML, Ruiz SJA, Ramírez CS, et al. Subungual glomangiomyoma: a case report. Dermatol Cosmet Med Quir. 2022;20:258-262.
- Chalise S, Jha A, Neupane PR. Glomangiomyoma of uncertain malignant potential in the urinary bladder: a case report. JNMA J Nepal Med Assoc. 2021;59:719-722. doi:10.31729/jnma.5388
- de Berker D, Goettman S, Baran R. Subungual myxoid cysts: clinical manifestations and response to therapy. J Am Acad Dermatol. 2002;46:394-398. doi:10.1067/mjd.2002.119652
- Gupta MK, Lipner SR. Transillumination for improved diagnosis of digital myxoid cysts. Cutis. 2020;105:82.
- Fernandez-Flores A, Saeb-Lima M. Mucin as a diagnostic clue in dermatopathology. J Cutan Pathol. 2016;43:1005-1016. doi:10.1111/cup.12782
- Choi R, Kim SR, Glusac EJ, et al. Subungual neuroma masquerading as green nail syndrome. JAAD Case Rep. 2022;20:17-19. doi:10.1016 /j.jdcr.2021.11.025
- Rashid RM, Rashid RM, Thomas V. Subungal traumatic neuroma. J Am Acad Dermatol. 2010;63:E7-E8. doi:10.1016/j.jaad.2010.01.028
- Whitehouse HJ, Urwin R, Stables G. Traumatic subungual neuroma. Clin Exp Dermatol. 2018;43:65-66. doi:10.1111/ced.13247
- Lipner SR, Ko D, Husain S. Subungual leiyomyoma presenting as erythronychia: case report and review of the literature. J Drugs Dermatol. 2019;18:465-467.
- Taleb E, Saldías C, Gonzalez S, et al. Sonographic characteristics of leiomyomatous tumors of skin and nail: a case series. Dermatol Pract Concept. 2022;12:e2022082. doi:10.5826/dpc.1203a82
- Baran R, Requena L, Drapé JL. Subungual angioleiomyoma masquerading as a glomus tumour. Br J Dermatol. 2000;142:1239-1241. doi:10.1046/ j.1365-2133.2000.03560.x
- Watabe D, Sakurai E, Mori S, et al. Subungual angioleiomyoma. Indian J Dermatol Venereol Leprol. 2017;83:74-75. doi:10.4103/0378-6323 .185045
- Jha AK, Sinha R, Kumar A, et al. Spiradenoma causing longitudinal splitting of the nail. Clin Exp Dermatol. 2016;41:754-756. doi:10.1111 /ced.12886
- Leach BC, Graham BS. Papular lesion of the proximal nail fold. eccrine spiradenoma. Arch Dermatol. 2004;140:1003-1008. doi:10.1001 /archderm.140.8.1003-a
The Diagnosis: Glomangiomyoma
The nail unit excision specimen showed collections of cuboidal cells and spindled cells within the corium that were consistent with a diagnosis of a glomangiomyoma, a rare glomus tumor variant (Figure). Glomus tumors are benign neoplasms comprising glomus bodies, which are arteriovenous anastomoses involved in thermoregulation.1 They develop in areas densely populated by glomus bodies, including the fingers, toes, and subungual areas. Glomus tumors most commonly develop in middle-aged women.2 Clinically, they manifest with a characteristic triad of intense pain, point tenderness, and cold sensitivity and may appear as reddish-pink or blue macules under the nail plate and/or longitudinal erythronychia.2-6 The presence of multiple glomus tumors is associated with neurofibromatosis type 1.7
Advanced imaging including ultrasonography and magnetic resonance imaging (MRI) may help confirm the diagnosis but may not be cost effective, as excision with histopathology is needed to relieve symptoms and render a definitive diagnosis. Radiography is highly insensitive in identifying bone erosions associated with glomus tumors.8 With ultrasonography, glomus tumors appear hypoechoic; with Doppler ultrasonography, they appear hypervascular. With MRI, glomus tumors appear as well-defined nodular lesions with hypointense signal intensity on T1-weighted sequence and hyperintense signal intensity on T2-weighted sequence, with strong enhancement using gadolinium-based contrast.9,10 On histopathology, a glomus tumor appears as a nodular tumor with sheets of oval-nucleated cells arranged in multicellular layers surrounding blood vessels and are immunoreactive for α-smooth muscle actin, muscle-specific actin, and type IV collagen.11,12
There are several glomus tumor variants. The most common is a solid glomus tumor, which predominantly is composed of glomus cells, followed by glomangioma, which mainly is composed of blood vessels. Glomangiomyoma, which mostly is composed of smooth muscle cells, is the rarest variant.13
While glomus tumors are common in the subungual areas, it is an uncommon location for glomangiomyomas, which have been reported in the nail unit in only 7 prior case reports identified through searches of PubMed and Google Scholar using the terms glomangiomyoma, glomangiomyoma nail, and subungual glomangiomyoma (Table).13-19 Glomangiomyomas more commonly are described in solid organs, including the stomach, kidney, pancreas, and bladder.16 The mean age of patients with subungual glomangiomyomas, including our patient, was 40.4 years (range, 3-61 years), with the majority being female (75.0% [6/8]). Most patients presented with fingernail involvement (75.0% [6/8]), nail dystrophy (eg, nail plate thinning, longitudinal grooves, splinter hemorrhages, longitudinal erythronychia)(62.5% [5/8]), and intermittent pain and/or point tenderness in the affected nail (75.0% [6/8]).13-19 Notably, only our patient had longitudinal erythronychia as a clinical feature, and only one other case described MRI findings, which included a lobulated mass with intense contrast and distal phalanx destruction.18 One patient was a 3-year-old girl with a family history of generalized multiple glomangiomyomas. Although subungual glomangiomyoma was not confirmed on histopathology, the diagnosis in this patient was presumed based on her family history.13 On histopathology, glomangiomyomas are composed of oval-nucleated cells surrounding blood vessels. These oval-nucleated cells then gradually transition to smooth muscle cells.20
A myxoid cyst is composed of a pseudocyst, which lacks a cyst lining, and is a result of synovial fluid from the distal interphalangeal joint entering the pseudocyst space.2 It typically manifests with a longitudinal groove in the nail plate. A flesh-colored nodule may be appreciated between the cuticle and the distal interphalangeal joint.2 The depth of the longitudinal groove may vary depending on the volume of synovial fluid within the myxoid cyst.21 In a series of 35 cases of subungual myxoid cysts, none manifested with longitudinal erythronychia. Due to their composition, myxoid cysts can be distinguished easily from solid tumors of the nail unit via transillumination.22 Pain is a much less common with myxoid cysts vs glomus tumors, as the filling of the pseudocyst space with synovial fluid typically is gradual, allowing the surrounding tissue to accommodate and adapt over time.21 In equivocal cases, MRI or high-resolution ultrasonography may be used to distinguish myxoid cysts and glomus tumors.8 Histopathology shows accumulation of mucin in the dermis with surrounding fibrous stroma.23
Subungual neuromas are painful benign tumors that develop due to disorganized neural proliferation following disruption to peripheral nerves secondary to trauma or surgery. In 3 case reports, subungual neuromas manifested as painful subungual nodules, with proximal nail plate ridging, or onycholysis.24-26 Since neuromas have only rarely been described in the subungual region, reports of MRI and ultrasonography findings are unknown. Histopathology is needed to distinguish neuromas from glomus tumors. Histopathology shows an acapsular structure consisting of disorganized spindle-cell proliferation and nerve fibers arranged in a tangle of fascicles within fibrotic tissue.25 On immunochemistry, spindle cells typically are positive for cellular antigen protein S100.26
Leiomyomas are benign neoplasms derived from smooth muscle, typically localized to the uterus or gastrointestinal tract, and have been described rarely in the nail unit.27,28 It is hypothesized that subungual leiomyomas originate from the vascular smooth muscle in the subcutaneous layer of the nail unit.28 Like glomus tumors, leiomyomas of the subungual region often manifest with pain and longitudinal erythronychia.27-30 Subungual leiomyomas may be distinguished from glomus tumors via advanced imaging techniques, including ultrasonography and MRI. Cutaneous leiomyomas have been described with mild to moderate internal low flow vascularity on Doppler ultrasonography, while glomus tumors typically reveal high internal vascularity.28 Biopsy with histopathology is needed for definitive diagnosis. On histopathology, leiomyomas demonstrate bland-appearing spindle-shaped cells with elongated nuclei arranged in fascicles.27 They typically are positive for α-smooth muscle actin and caldesmon on immunostaining.
Eccrine spiradenomas are benign adnexal tumors likely of apocrine origin with limited case reports in the literature.31,32 Clinically, eccrine spiradenomas involving the nail unit may manifest with longitudinal nail splitting of the nail or as a papule on the proximal nail fold, with associated tenderness.31,32 In a report of a 50-year-old woman with a histopathologically confirmed eccrine spiradenoma manifesting with longitudinal splitting of the nail and pain in the proximal nail fold, the mass appeared hypoechoic on ultrasonography with increased intramass vascularity on Doppler, while MRI showed an intensely enhancing lesion.31 These imaging features, combined with a classically manifesting feature of pain, make eccrine spiradenomas difficult to distinguish from glomus tumors; therefore, histopathologic examination can provide a definitive diagnosis, and surgical excision is used for treatment.31 On histopathology, these tumors are well circumscribed and composed of both small dark basaloid cells with peripheral compact nuclei and larger cells with central pale nuclei, which may be arranged in tubules.31,32
The Diagnosis: Glomangiomyoma
The nail unit excision specimen showed collections of cuboidal cells and spindled cells within the corium that were consistent with a diagnosis of a glomangiomyoma, a rare glomus tumor variant (Figure). Glomus tumors are benign neoplasms comprising glomus bodies, which are arteriovenous anastomoses involved in thermoregulation.1 They develop in areas densely populated by glomus bodies, including the fingers, toes, and subungual areas. Glomus tumors most commonly develop in middle-aged women.2 Clinically, they manifest with a characteristic triad of intense pain, point tenderness, and cold sensitivity and may appear as reddish-pink or blue macules under the nail plate and/or longitudinal erythronychia.2-6 The presence of multiple glomus tumors is associated with neurofibromatosis type 1.7
Advanced imaging including ultrasonography and magnetic resonance imaging (MRI) may help confirm the diagnosis but may not be cost effective, as excision with histopathology is needed to relieve symptoms and render a definitive diagnosis. Radiography is highly insensitive in identifying bone erosions associated with glomus tumors.8 With ultrasonography, glomus tumors appear hypoechoic; with Doppler ultrasonography, they appear hypervascular. With MRI, glomus tumors appear as well-defined nodular lesions with hypointense signal intensity on T1-weighted sequence and hyperintense signal intensity on T2-weighted sequence, with strong enhancement using gadolinium-based contrast.9,10 On histopathology, a glomus tumor appears as a nodular tumor with sheets of oval-nucleated cells arranged in multicellular layers surrounding blood vessels and are immunoreactive for α-smooth muscle actin, muscle-specific actin, and type IV collagen.11,12
There are several glomus tumor variants. The most common is a solid glomus tumor, which predominantly is composed of glomus cells, followed by glomangioma, which mainly is composed of blood vessels. Glomangiomyoma, which mostly is composed of smooth muscle cells, is the rarest variant.13
While glomus tumors are common in the subungual areas, it is an uncommon location for glomangiomyomas, which have been reported in the nail unit in only 7 prior case reports identified through searches of PubMed and Google Scholar using the terms glomangiomyoma, glomangiomyoma nail, and subungual glomangiomyoma (Table).13-19 Glomangiomyomas more commonly are described in solid organs, including the stomach, kidney, pancreas, and bladder.16 The mean age of patients with subungual glomangiomyomas, including our patient, was 40.4 years (range, 3-61 years), with the majority being female (75.0% [6/8]). Most patients presented with fingernail involvement (75.0% [6/8]), nail dystrophy (eg, nail plate thinning, longitudinal grooves, splinter hemorrhages, longitudinal erythronychia)(62.5% [5/8]), and intermittent pain and/or point tenderness in the affected nail (75.0% [6/8]).13-19 Notably, only our patient had longitudinal erythronychia as a clinical feature, and only one other case described MRI findings, which included a lobulated mass with intense contrast and distal phalanx destruction.18 One patient was a 3-year-old girl with a family history of generalized multiple glomangiomyomas. Although subungual glomangiomyoma was not confirmed on histopathology, the diagnosis in this patient was presumed based on her family history.13 On histopathology, glomangiomyomas are composed of oval-nucleated cells surrounding blood vessels. These oval-nucleated cells then gradually transition to smooth muscle cells.20
A myxoid cyst is composed of a pseudocyst, which lacks a cyst lining, and is a result of synovial fluid from the distal interphalangeal joint entering the pseudocyst space.2 It typically manifests with a longitudinal groove in the nail plate. A flesh-colored nodule may be appreciated between the cuticle and the distal interphalangeal joint.2 The depth of the longitudinal groove may vary depending on the volume of synovial fluid within the myxoid cyst.21 In a series of 35 cases of subungual myxoid cysts, none manifested with longitudinal erythronychia. Due to their composition, myxoid cysts can be distinguished easily from solid tumors of the nail unit via transillumination.22 Pain is a much less common with myxoid cysts vs glomus tumors, as the filling of the pseudocyst space with synovial fluid typically is gradual, allowing the surrounding tissue to accommodate and adapt over time.21 In equivocal cases, MRI or high-resolution ultrasonography may be used to distinguish myxoid cysts and glomus tumors.8 Histopathology shows accumulation of mucin in the dermis with surrounding fibrous stroma.23
Subungual neuromas are painful benign tumors that develop due to disorganized neural proliferation following disruption to peripheral nerves secondary to trauma or surgery. In 3 case reports, subungual neuromas manifested as painful subungual nodules, with proximal nail plate ridging, or onycholysis.24-26 Since neuromas have only rarely been described in the subungual region, reports of MRI and ultrasonography findings are unknown. Histopathology is needed to distinguish neuromas from glomus tumors. Histopathology shows an acapsular structure consisting of disorganized spindle-cell proliferation and nerve fibers arranged in a tangle of fascicles within fibrotic tissue.25 On immunochemistry, spindle cells typically are positive for cellular antigen protein S100.26
Leiomyomas are benign neoplasms derived from smooth muscle, typically localized to the uterus or gastrointestinal tract, and have been described rarely in the nail unit.27,28 It is hypothesized that subungual leiomyomas originate from the vascular smooth muscle in the subcutaneous layer of the nail unit.28 Like glomus tumors, leiomyomas of the subungual region often manifest with pain and longitudinal erythronychia.27-30 Subungual leiomyomas may be distinguished from glomus tumors via advanced imaging techniques, including ultrasonography and MRI. Cutaneous leiomyomas have been described with mild to moderate internal low flow vascularity on Doppler ultrasonography, while glomus tumors typically reveal high internal vascularity.28 Biopsy with histopathology is needed for definitive diagnosis. On histopathology, leiomyomas demonstrate bland-appearing spindle-shaped cells with elongated nuclei arranged in fascicles.27 They typically are positive for α-smooth muscle actin and caldesmon on immunostaining.
Eccrine spiradenomas are benign adnexal tumors likely of apocrine origin with limited case reports in the literature.31,32 Clinically, eccrine spiradenomas involving the nail unit may manifest with longitudinal nail splitting of the nail or as a papule on the proximal nail fold, with associated tenderness.31,32 In a report of a 50-year-old woman with a histopathologically confirmed eccrine spiradenoma manifesting with longitudinal splitting of the nail and pain in the proximal nail fold, the mass appeared hypoechoic on ultrasonography with increased intramass vascularity on Doppler, while MRI showed an intensely enhancing lesion.31 These imaging features, combined with a classically manifesting feature of pain, make eccrine spiradenomas difficult to distinguish from glomus tumors; therefore, histopathologic examination can provide a definitive diagnosis, and surgical excision is used for treatment.31 On histopathology, these tumors are well circumscribed and composed of both small dark basaloid cells with peripheral compact nuclei and larger cells with central pale nuclei, which may be arranged in tubules.31,32
- Gombos Z, Zhang PJ. Glomus tumor. Arch Pathol Lab Med. 2008;132: 1448-1452. doi:10.5858/2008-132-1448-gt
- Hare AQ, Rich P. Nail tumors. Dermatol Clin. 2021;39:281-292. doi:10.1016/j.det.2020.12.007
- Hazani R, Houle JM, Kasdan ML, et al. Glomus tumors of the hand. Eplasty. 2008;8:E48.
- Hwang JK, Lipner SR. Blue nail discoloration: literature review and diagnostic algorithms. Am J Clin Dermatol. 2023;24:419-441. doi:10.1007/s40257-023-00768-6
- Lipner SR, Scher RK. Longitudinal erythronychia of the fingernail. JAMA Dermatol. 2016;152:1271-1272. doi:10.1001/jamadermatol.2016.2747
- Jellinek NJ, Lipner SR. Longitudinal erythronychia: retrospective single-center study evaluating differential diagnosis and the likelihood of malignancy. Dermatol Surg. 2016;42:310-319. doi:10.1097 /DSS.0000000000000594
- Lipner SR, Scher RK. Subungual glomus tumors: underrecognized clinical findings in neurofibromatosis 1. J Am Acad Dermatol. 2021;84:E269. doi:10.1016/j.jaad.2020.08.129
- Dhami A, Vale SM, Richardson ML, et al. Comparing ultrasound with magnetic resonance imaging in the evaluation of subungual glomus tumors and subungual myxoid cysts. Skin Appendage Disord. 2023;9:262-267. doi:10.1159/000530397
- Baek HJ, Lee SJ, Cho KH, et al. Subungual tumors: clinicopathologic correlation with US and MR imaging findings. Radiographics. 2010;30:1621-1636. doi:10.1148/rg.306105514
- Patel T, Meena V, Meena P. Hand and foot glomus tumors: significance of MRI diagnosis followed by histopathological assessment. Cureus. 2022;14:E30038. doi:10.7759/cureus.30038
- Mravic M, LaChaud G, Nguyen A, et al. Clinical and histopathological diagnosis of glomus tumor: an institutional experience of 138 cases. Int J Surg Pathol. 2015;23:181-188. doi:10.1177/1066896914567330
- Folpe AL, Fanburg-Smith JC, Miettinen M, et al. Atypical and malignant glomus tumors: analysis of 52 cases, with a proposal for the reclassification of glomus tumors. Am J Surg Pathol. 2001;25:1-12. doi:10.1097/00000478-200101000-00001
- Calduch L, Monteagudo C, Martínez-Ruiz E, et al. Familial generalized multiple glomangiomyoma: report of a new family, with immunohistochemical and ultrastructural studies and review of the literature. Pediatr Dermatol. 2002;19:402-408. doi:10.1046/j.1525-1470.2002.00114.x
- Mentzel T, Hügel H, Kutzner H. CD34-positive glomus tumor: clinicopathologic and immunohistochemical analysis of six cases with myxoid stromal changes. J Cutan Pathol. 2002;29:421-425. doi:10.1034 /j.1600-0560.2002.290706.x
- Kang TW, Lee KH, Park CJ. A case of subungual glomangiomyoma with myxoid stromal change. Korean J Dermatol. 2008;46:550-553.
- Wollstein A, Wollstein R. Subungual glomangiomyoma—a case report. Hand Surg. 2012;17:271-273. doi:10.1142/S021881041272032X
- Aqil N, Gallouj S, Moustaide K, et al. Painful tumors in a patient with neurofibromatosis type 1: a case report. J Med Case Rep. 2018;12:319. doi:10.1186/s13256-018-1847-0
- Demirdag HG, Akay BN, Kirmizi A, et al. Subungual glomangiomyoma. J Am Podiatr Med Assoc. 2020;110:Article_13. doi:10.7547/19-051
- Vega SML, Ruiz SJA, Ramírez CS, et al. Subungual glomangiomyoma: a case report. Dermatol Cosmet Med Quir. 2022;20:258-262.
- Chalise S, Jha A, Neupane PR. Glomangiomyoma of uncertain malignant potential in the urinary bladder: a case report. JNMA J Nepal Med Assoc. 2021;59:719-722. doi:10.31729/jnma.5388
- de Berker D, Goettman S, Baran R. Subungual myxoid cysts: clinical manifestations and response to therapy. J Am Acad Dermatol. 2002;46:394-398. doi:10.1067/mjd.2002.119652
- Gupta MK, Lipner SR. Transillumination for improved diagnosis of digital myxoid cysts. Cutis. 2020;105:82.
- Fernandez-Flores A, Saeb-Lima M. Mucin as a diagnostic clue in dermatopathology. J Cutan Pathol. 2016;43:1005-1016. doi:10.1111/cup.12782
- Choi R, Kim SR, Glusac EJ, et al. Subungual neuroma masquerading as green nail syndrome. JAAD Case Rep. 2022;20:17-19. doi:10.1016 /j.jdcr.2021.11.025
- Rashid RM, Rashid RM, Thomas V. Subungal traumatic neuroma. J Am Acad Dermatol. 2010;63:E7-E8. doi:10.1016/j.jaad.2010.01.028
- Whitehouse HJ, Urwin R, Stables G. Traumatic subungual neuroma. Clin Exp Dermatol. 2018;43:65-66. doi:10.1111/ced.13247
- Lipner SR, Ko D, Husain S. Subungual leiyomyoma presenting as erythronychia: case report and review of the literature. J Drugs Dermatol. 2019;18:465-467.
- Taleb E, Saldías C, Gonzalez S, et al. Sonographic characteristics of leiomyomatous tumors of skin and nail: a case series. Dermatol Pract Concept. 2022;12:e2022082. doi:10.5826/dpc.1203a82
- Baran R, Requena L, Drapé JL. Subungual angioleiomyoma masquerading as a glomus tumour. Br J Dermatol. 2000;142:1239-1241. doi:10.1046/ j.1365-2133.2000.03560.x
- Watabe D, Sakurai E, Mori S, et al. Subungual angioleiomyoma. Indian J Dermatol Venereol Leprol. 2017;83:74-75. doi:10.4103/0378-6323 .185045
- Jha AK, Sinha R, Kumar A, et al. Spiradenoma causing longitudinal splitting of the nail. Clin Exp Dermatol. 2016;41:754-756. doi:10.1111 /ced.12886
- Leach BC, Graham BS. Papular lesion of the proximal nail fold. eccrine spiradenoma. Arch Dermatol. 2004;140:1003-1008. doi:10.1001 /archderm.140.8.1003-a
- Gombos Z, Zhang PJ. Glomus tumor. Arch Pathol Lab Med. 2008;132: 1448-1452. doi:10.5858/2008-132-1448-gt
- Hare AQ, Rich P. Nail tumors. Dermatol Clin. 2021;39:281-292. doi:10.1016/j.det.2020.12.007
- Hazani R, Houle JM, Kasdan ML, et al. Glomus tumors of the hand. Eplasty. 2008;8:E48.
- Hwang JK, Lipner SR. Blue nail discoloration: literature review and diagnostic algorithms. Am J Clin Dermatol. 2023;24:419-441. doi:10.1007/s40257-023-00768-6
- Lipner SR, Scher RK. Longitudinal erythronychia of the fingernail. JAMA Dermatol. 2016;152:1271-1272. doi:10.1001/jamadermatol.2016.2747
- Jellinek NJ, Lipner SR. Longitudinal erythronychia: retrospective single-center study evaluating differential diagnosis and the likelihood of malignancy. Dermatol Surg. 2016;42:310-319. doi:10.1097 /DSS.0000000000000594
- Lipner SR, Scher RK. Subungual glomus tumors: underrecognized clinical findings in neurofibromatosis 1. J Am Acad Dermatol. 2021;84:E269. doi:10.1016/j.jaad.2020.08.129
- Dhami A, Vale SM, Richardson ML, et al. Comparing ultrasound with magnetic resonance imaging in the evaluation of subungual glomus tumors and subungual myxoid cysts. Skin Appendage Disord. 2023;9:262-267. doi:10.1159/000530397
- Baek HJ, Lee SJ, Cho KH, et al. Subungual tumors: clinicopathologic correlation with US and MR imaging findings. Radiographics. 2010;30:1621-1636. doi:10.1148/rg.306105514
- Patel T, Meena V, Meena P. Hand and foot glomus tumors: significance of MRI diagnosis followed by histopathological assessment. Cureus. 2022;14:E30038. doi:10.7759/cureus.30038
- Mravic M, LaChaud G, Nguyen A, et al. Clinical and histopathological diagnosis of glomus tumor: an institutional experience of 138 cases. Int J Surg Pathol. 2015;23:181-188. doi:10.1177/1066896914567330
- Folpe AL, Fanburg-Smith JC, Miettinen M, et al. Atypical and malignant glomus tumors: analysis of 52 cases, with a proposal for the reclassification of glomus tumors. Am J Surg Pathol. 2001;25:1-12. doi:10.1097/00000478-200101000-00001
- Calduch L, Monteagudo C, Martínez-Ruiz E, et al. Familial generalized multiple glomangiomyoma: report of a new family, with immunohistochemical and ultrastructural studies and review of the literature. Pediatr Dermatol. 2002;19:402-408. doi:10.1046/j.1525-1470.2002.00114.x
- Mentzel T, Hügel H, Kutzner H. CD34-positive glomus tumor: clinicopathologic and immunohistochemical analysis of six cases with myxoid stromal changes. J Cutan Pathol. 2002;29:421-425. doi:10.1034 /j.1600-0560.2002.290706.x
- Kang TW, Lee KH, Park CJ. A case of subungual glomangiomyoma with myxoid stromal change. Korean J Dermatol. 2008;46:550-553.
- Wollstein A, Wollstein R. Subungual glomangiomyoma—a case report. Hand Surg. 2012;17:271-273. doi:10.1142/S021881041272032X
- Aqil N, Gallouj S, Moustaide K, et al. Painful tumors in a patient with neurofibromatosis type 1: a case report. J Med Case Rep. 2018;12:319. doi:10.1186/s13256-018-1847-0
- Demirdag HG, Akay BN, Kirmizi A, et al. Subungual glomangiomyoma. J Am Podiatr Med Assoc. 2020;110:Article_13. doi:10.7547/19-051
- Vega SML, Ruiz SJA, Ramírez CS, et al. Subungual glomangiomyoma: a case report. Dermatol Cosmet Med Quir. 2022;20:258-262.
- Chalise S, Jha A, Neupane PR. Glomangiomyoma of uncertain malignant potential in the urinary bladder: a case report. JNMA J Nepal Med Assoc. 2021;59:719-722. doi:10.31729/jnma.5388
- de Berker D, Goettman S, Baran R. Subungual myxoid cysts: clinical manifestations and response to therapy. J Am Acad Dermatol. 2002;46:394-398. doi:10.1067/mjd.2002.119652
- Gupta MK, Lipner SR. Transillumination for improved diagnosis of digital myxoid cysts. Cutis. 2020;105:82.
- Fernandez-Flores A, Saeb-Lima M. Mucin as a diagnostic clue in dermatopathology. J Cutan Pathol. 2016;43:1005-1016. doi:10.1111/cup.12782
- Choi R, Kim SR, Glusac EJ, et al. Subungual neuroma masquerading as green nail syndrome. JAAD Case Rep. 2022;20:17-19. doi:10.1016 /j.jdcr.2021.11.025
- Rashid RM, Rashid RM, Thomas V. Subungal traumatic neuroma. J Am Acad Dermatol. 2010;63:E7-E8. doi:10.1016/j.jaad.2010.01.028
- Whitehouse HJ, Urwin R, Stables G. Traumatic subungual neuroma. Clin Exp Dermatol. 2018;43:65-66. doi:10.1111/ced.13247
- Lipner SR, Ko D, Husain S. Subungual leiyomyoma presenting as erythronychia: case report and review of the literature. J Drugs Dermatol. 2019;18:465-467.
- Taleb E, Saldías C, Gonzalez S, et al. Sonographic characteristics of leiomyomatous tumors of skin and nail: a case series. Dermatol Pract Concept. 2022;12:e2022082. doi:10.5826/dpc.1203a82
- Baran R, Requena L, Drapé JL. Subungual angioleiomyoma masquerading as a glomus tumour. Br J Dermatol. 2000;142:1239-1241. doi:10.1046/ j.1365-2133.2000.03560.x
- Watabe D, Sakurai E, Mori S, et al. Subungual angioleiomyoma. Indian J Dermatol Venereol Leprol. 2017;83:74-75. doi:10.4103/0378-6323 .185045
- Jha AK, Sinha R, Kumar A, et al. Spiradenoma causing longitudinal splitting of the nail. Clin Exp Dermatol. 2016;41:754-756. doi:10.1111 /ced.12886
- Leach BC, Graham BS. Papular lesion of the proximal nail fold. eccrine spiradenoma. Arch Dermatol. 2004;140:1003-1008. doi:10.1001 /archderm.140.8.1003-a
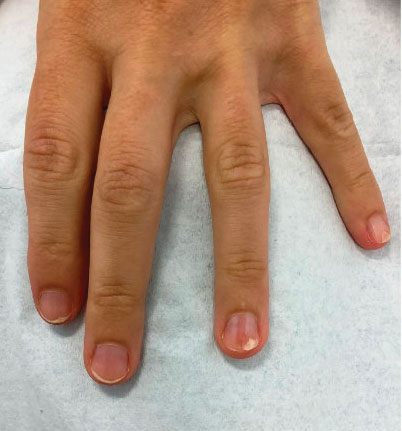
A 38-year-old woman presented to our nail specialty clinic with a red line and associated pain on the left fourth fingernail of 2 and 3 years’ duration, respectively. The patient described the pain as throbbing, with sensitivity to pressure and cold. She noted that the nail grew slowly and would sometimes split at the distal edge. She did not recall any discrete trauma to the digit or nail. The patient was right-handed, making the symptoms less likely to be due to overuse from daily activities. She had received no prior treatment for these symptoms.
The patient’s medical history included iron deficiency as well as acne and eczema. She had no personal or family history of skin cancer. Physical examination of the affected digit and nail revealed a longitudinal red line and distal onycholysis. With contact dermoscopy, the red line blanched. Pressure applied using a #11 scalpel blade elicited pinpoint tenderness (positive Love test), and application of an ice pack caused pain (positive cold test). A radiograph of the left hand was negative for bone erosions, and magnetic resonance imaging showed a 0.3-cm subungual lesion at the level of the fourth distal phalanx. An excision of the nail unit was performed.
Tender Nodule on the Umbilicus
Tender Nodule on the Umbilicus
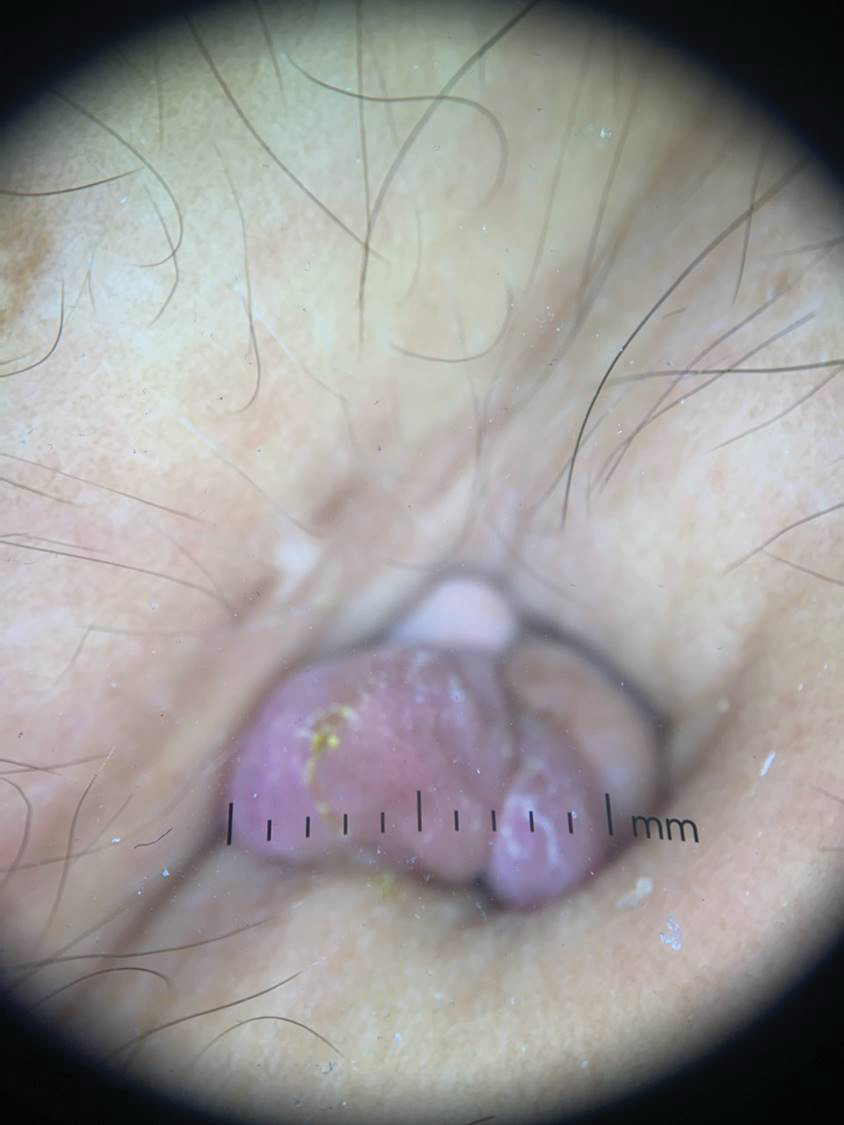
THE DIAGNOSIS: Villar Nodule
The biopsy revealed features consistent with cutaneous endometriosis in the setting of a painful, tender, multilobulated nodule with a cyclical bleeding pattern (Figure 1). The bleeding pattern of the nodule during menses and lack of surgical history supported the diagnosis of primary cutaneous endometriosis in our patient. She was diagnosed with endometriosis by gynecology, and her primary care physician started her on an oral contraceptive based on this diagnosis. She also was referred to gynecology and plastic surgery for a joint surgical consultation to remove the nodule. She initially decided to do a trial of the oral contraceptive but subsequently underwent umbilical endometrioma excision with neo-umbilicus creation with no evidence of recurrence.
Primary cutaneous endometriosis should be considered in young females who present with tender umbilical nodules. Endometriosis refers to the presence of an endometriumlike epithelium outside the endometrium and myometrium.1 The condition affects 10% to 15% of reproductive-aged (ie, 18-49 years) women in the United States and typically involves tissues within the pelvis, such as the ovaries, pouch of Douglas, or pelvic ligaments.2 Cutaneous endometriosis is the growth of endometrial tissue in the skin and is rare, accounting for less than 5.5% of cases of extrapelvic endometriosis worldwide, affecting primarily the umbilicus, abdominal wall, and vulva.3,4
The 2 main types of cutaneous endometriosis are primary (spontaneous) and secondary. Primary lesions develop in patients without prior surgical history, and secondary lesions occur within previous surgical incision sites, often scars from cesarean delivery.5 Less than 30% of cases of cutaneous endometriosis are primary disease.6 Primary cutaneous endometriosis of the umbilicus, known as Villar nodule, was first described in 1886.3,7 Up to 40% of patients with extrapelvic endometriosis worldwide presented with Villar nodules in a systematic literature review.6 The prevalence of these nodules is unknown, but the incidence is less than 1% of cases of extragenital endometriosis.4
There are 2 leading theories of primary cutaneous endometriosis pathogenesis. The first is the transportation theory, in which endometrial cells are transported outside the uterus via the lymphatic system.8 The second is the metaplasia theory, which proposed that endometrial cells develop in the coelomic mesothelium in the presence of high estrogen levels.8,9
Secondary cutaneous endometriosis, also known as scar endometriosis, is suspected to be caused by an iatrogenic implantation of endometrial cells at the scar of a prior surgical site.9 Although our patient had an existing umbilicus scar from a piercing, it was improbable for that to have been the nidus, as the keloid scar was superficial and did not have contact with the abdominal cavity for iatrogenic implantation. Clinical diagnosis for secondary cutaneous endometriosis often is made based on a triad of features: a nonmalignant abdominal mass, recurring pain and bleeding of the lesion with menses, and prior history of abdominal surgery.9,10 On clinical examination, these features typically manifest as a palpable subcutaneous mass that is black, blue, brown, or red. Often, the lesions enlarge and bleed during the menstrual cycle, causing pain, tenderness, or pruritus.3 Dermoscopic features of secondary cutaneous endometriosis are erythematous umbilical nodules with a homogeneous vascular pattern that appears red with a brownish hue (Figure 2).9,11 Dermoscopic features may vary with the hormone cycle; for example, the follicular phase (correlating with day 7 of menses) demonstrates polypoid projections, erythematous violaceous color, dark-brown spots, and active bleeding of the lesion.12 Clinical and dermoscopic examination are useful tools in this diagnosis.
Imaging such as ultrasonography, computed tomography, or magnetic resonance imaging may be useful in identifying abdominal endometriomas.8,13,14 Pelvic involvement of endometriosis was found in approximately 15% of patients in a case series,4 with concurrent primary umbilical endometriosis. Imaging studies may assist evaluation for fistula formation, presence of malignancies, and the extent of endometriosis within the abdominal cavity.
Histopathology is key to confirming cutaneous endometriosis and shows multiple bland-appearing glands of varying sizes with loose, concentric, edematous, or fibromyxoid stroma (Figure 1).3 Red blood cells sometimes are found with hemosiderin within the stroma. Immunohistochemical staining with estrogen receptors may aid in identifying the endometriumlike epithelial cells.13
Standard treatment involves surgical excision with 1-cm margins and umbilical preservation, which results in a recurrence rate of less than 10%.4,10 Medical therapy, such as aromatase inhibitors, progestogens, antiprogestogens, combined oral contraceptives, or gonadotropin-releasing hormone agonists or antagonists may help manage pain or reduce the size of the nodule.4,15 Simple observation also is a potential course for patients who decline treatment options.
Differential diagnoses include lobular capillary hemangioma, also known as pyogenic granuloma; Sister Mary Joseph nodule; umbilical hernia; and dermatofibrosarcoma protuberans. Lobular capillary hemangiomas commonly are acquired benign vascular proliferations of the skin that are friable and tend to ulcerate.16 These lesions typically grow rapidly and often are located on the face, lips, mucosae, and fingers. Histopathologic examination may show an exophytic lesion with lobules of proliferating capillaries within an edematous matrix, superficial ulceration, and an epithelial collarette.17 Treatment includes surgical excision, cauterization, laser treatments, sclerotherapy, injectable medications, and topical medications, but recurrence is possible with any of these interventions.18
Cutaneous metastasis of an internal solid organ cancer, commonly known as a Sister Mary Joseph nodule, typically manifests as an erythematous, irregularly shaped nodule that may protrude from the umbilicus.14 Gastrointestinal symptoms such as change in bowel habits or obstructive symptoms in the setting of a progressive malignancy are common.14 Clinical features include a firm fixed lesion, oozing, and ulceration.19 On dermoscopy, polymorphous vascular patterns, milky red structureless areas, and white lines typically are present.11 Although dermoscopic features may differentiate this entity from cutaneous endometriosis, tissue sampling and histologic examination are crucial diagnostic tools to identify malignant vs benign lesions.
An umbilical hernia is a protrusion of omentum, bowel, or other intra-abdominal organs in an abdominal wall defect. Clinical presentation includes a soft protrusion that may be reduced on palpation if nonstrangulated.20 Treatment includes watchful waiting or surgical repair. The reducibility and presence of an abdominal wall defect may point to this diagnosis. Imaging also may aid in the diagnosis if the history and physical examination are unclear.
Dermatofibrosarcoma protuberans is a slow-developing, low- to intermediate-grade, soft-tissue sarcoma that occurs in less than 0.1% of all cancers in the United States.21 Lesions often manifest as small, firm, slow-growing, painless, flesh-colored dermal plaques; subcutaneous thickening; or atrophic nonprotuberant lesions typically involving the trunk.21 Histopathologically, they are composed of uniform spindle-cell proliferation growing in a storiform pattern and subcutaneous fat trapping that has strong and diffuse CD34 immunoreactivity.21,22 Pathologic examination typically distinguishes this diagnosis from cutaneous endometriosis. Treatment includes tumor resection that may or may not involve radiotherapy and targeted therapy, as recurrence and metastases are possible.
Primary cutaneous endometriosis is a rare but important diagnosis for dermatologists to consider when evaluating umbilical nodules. Clinical features may include bleeding masses during menses in females of reproductive age. Dermoscopic examination aids in workup, and histopathologic testing can confirm the diagnosis and rule out malignancies. Surgical excision is the treatment of choice with a low rate of recurrence.
- International Working Group of AAGL, ESGE, ESHRE and WES; Tomassetti C, Johnson NP, et al. An international terminology for endometriosis, 2021. Hum Reprod Open. 2021;2021:hoab029. doi:10.1093/hropen/hoab029
- Batista M, Alves F, Cardoso J, et al. Cutaneous endometriosis: a differential diagnosis of umbilical nodule. Acta Med Port. 2020; 33:282-284. doi:10.20344/amp.10966
- Brown ME, Osswald S, Biediger T. Cutaneous endometriosis of the umbilicus (Villar’s nodule). Int J Womens Dermatol. 2020;6:214-215. doi:10.1016/j.ijwd.2020.01.001
- Bindra V, Sampurna S, Kade S, et al. Primary umbilical endometriosis - case series and review of clinical presentation, diagnosis and management. Int J Surg Case Rep. 2022;94:107134. doi:10.1016/j.ijscr.2022.107134
- Loh SH, Lew BL, Sim WY. Primary cutaneous endometriosis of umbilicus. Ann Dermatol. 2017;29:621-625. doi:10.5021/ad.2017.29.5.621
- Victory R, Diamond MP, Johns DA. Villar’s nodule: a case report and systematic literature review of endometriosis externa of the umbilicus. J Minim Invasive Gynecol. 2007;14:23-32. doi:10.1016/j.jmig.2006.07.01
- Van den Nouland D, Kaur M. Primary umbilical endometriosis: a case report. Facts Views Vis Obgyn. 2017;9:115-119.
- Machairiotis N, Stylianaki A, Dryllis G, et al. Extrapelvic endometriosis: a rare entity or an under diagnosed condition? Diagn Pathol. 2013;8:194. doi:10.1186/1746-1596-8-194
- Huang QF, Jiang B, Yang X, et al. Primary versus secondary cutaneous endometriosis: literature review and case study. Heliyon. 2023;9:E20094. doi:10.1016/j.heliyon.2023.e20094
- Gonzalez RH, Singh MS, Hamza SA. Cutaneous endometriosis: a case report and review of the literature. Am J Case Rep. 2021;22:E932493. doi:10.12659/AJCR.932493
- Buljan M, Arzberger E, Šitum M, et al. The use of dermoscopy in differentiating Sister Mary Joseph nodule and cutaneous endometriosis. Australas J Dermatol. 2019;60:E233-E235. doi:10.1111/ajd.12980
- Costa IM, Gomes CM, Morais OO, et al. Cutaneous endometriosis: dermoscopic findings related to phases of the female hormonal cycle. Int J Dermatol. 2014;53:E130-E132. doi:10.1111 /j.1365-4632.2012.05854.x
- Mohaghegh F, Hatami P, Rajabi P, et al. Coexistence of cutaneous endometriosis and ovarian endometrioma: a case report. J Med Case Rep. 2022;16:256. doi:10.1186/s13256-022-03483-8
- Raffi L, Suresh R, McCalmont TH, et al. Cutaneous endometriosis. Int J Womens Dermatol. 2019;5:384-386. doi:10.1016 /j.ijwd.2019.06.025
- Saunders PTK, Horne AW. Endometriosis: etiology, pathobiology, and therapeutic prospects. Cell. 2021;184:2807-2824. doi:10.1016 /j.cell.2021.04.041
- Habif TP. Clinical Dermatology a Color Guide to Diagnosis and Therapy. St. Louis, Mo. Elsevier; 2016.
- Patrice SJ, Wiss K, Mulliken JB. Pyogenic granuloma (lobular capillary hemangioma): a clinicopathologic study of 178 cases. Pediatr Dermatol. 1991;8:267-276. doi:10.1111/j.15251470.1991.tb00931.x
- Kaleeny JD, Janis JE. Pyogenic granuloma diagnosis and management: a practical review. Plast Reconstr Surg Glob Open. 2024;12:E6160. doi:10.1097/GOX.0000000000006160
- Ha DL, Yang MY, Shin JO, et al. Benign umbilical tumors resembling Sister Mary Joseph nodule. Clin Med Insights Oncol. 2021;15:1179554921995022. doi:10.1177/1179554921995022
- Lawrence PF, Smeds M, Jessica Beth O’connell. Essentials of General Surgery and Surgical Specialties. Wolters Kluwer Health; 2019.
- Hao X, Billings SD, Wu F, et al. Dermatofibrosarcoma protuberans: update on the diagnosis and treatment. J Clin Med. 2020;9:1752. doi:10.3390/jcm9061752
- Allen A, Ahn C, Sangüeza OP. Dermatofibrosarcoma protuberans. Dermatol Clin. 2019;37:483-488. doi:10.1016/j.det.2019.05.006
THE DIAGNOSIS: Villar Nodule
The biopsy revealed features consistent with cutaneous endometriosis in the setting of a painful, tender, multilobulated nodule with a cyclical bleeding pattern (Figure 1). The bleeding pattern of the nodule during menses and lack of surgical history supported the diagnosis of primary cutaneous endometriosis in our patient. She was diagnosed with endometriosis by gynecology, and her primary care physician started her on an oral contraceptive based on this diagnosis. She also was referred to gynecology and plastic surgery for a joint surgical consultation to remove the nodule. She initially decided to do a trial of the oral contraceptive but subsequently underwent umbilical endometrioma excision with neo-umbilicus creation with no evidence of recurrence.
Primary cutaneous endometriosis should be considered in young females who present with tender umbilical nodules. Endometriosis refers to the presence of an endometriumlike epithelium outside the endometrium and myometrium.1 The condition affects 10% to 15% of reproductive-aged (ie, 18-49 years) women in the United States and typically involves tissues within the pelvis, such as the ovaries, pouch of Douglas, or pelvic ligaments.2 Cutaneous endometriosis is the growth of endometrial tissue in the skin and is rare, accounting for less than 5.5% of cases of extrapelvic endometriosis worldwide, affecting primarily the umbilicus, abdominal wall, and vulva.3,4
The 2 main types of cutaneous endometriosis are primary (spontaneous) and secondary. Primary lesions develop in patients without prior surgical history, and secondary lesions occur within previous surgical incision sites, often scars from cesarean delivery.5 Less than 30% of cases of cutaneous endometriosis are primary disease.6 Primary cutaneous endometriosis of the umbilicus, known as Villar nodule, was first described in 1886.3,7 Up to 40% of patients with extrapelvic endometriosis worldwide presented with Villar nodules in a systematic literature review.6 The prevalence of these nodules is unknown, but the incidence is less than 1% of cases of extragenital endometriosis.4
There are 2 leading theories of primary cutaneous endometriosis pathogenesis. The first is the transportation theory, in which endometrial cells are transported outside the uterus via the lymphatic system.8 The second is the metaplasia theory, which proposed that endometrial cells develop in the coelomic mesothelium in the presence of high estrogen levels.8,9
Secondary cutaneous endometriosis, also known as scar endometriosis, is suspected to be caused by an iatrogenic implantation of endometrial cells at the scar of a prior surgical site.9 Although our patient had an existing umbilicus scar from a piercing, it was improbable for that to have been the nidus, as the keloid scar was superficial and did not have contact with the abdominal cavity for iatrogenic implantation. Clinical diagnosis for secondary cutaneous endometriosis often is made based on a triad of features: a nonmalignant abdominal mass, recurring pain and bleeding of the lesion with menses, and prior history of abdominal surgery.9,10 On clinical examination, these features typically manifest as a palpable subcutaneous mass that is black, blue, brown, or red. Often, the lesions enlarge and bleed during the menstrual cycle, causing pain, tenderness, or pruritus.3 Dermoscopic features of secondary cutaneous endometriosis are erythematous umbilical nodules with a homogeneous vascular pattern that appears red with a brownish hue (Figure 2).9,11 Dermoscopic features may vary with the hormone cycle; for example, the follicular phase (correlating with day 7 of menses) demonstrates polypoid projections, erythematous violaceous color, dark-brown spots, and active bleeding of the lesion.12 Clinical and dermoscopic examination are useful tools in this diagnosis.
Imaging such as ultrasonography, computed tomography, or magnetic resonance imaging may be useful in identifying abdominal endometriomas.8,13,14 Pelvic involvement of endometriosis was found in approximately 15% of patients in a case series,4 with concurrent primary umbilical endometriosis. Imaging studies may assist evaluation for fistula formation, presence of malignancies, and the extent of endometriosis within the abdominal cavity.
Histopathology is key to confirming cutaneous endometriosis and shows multiple bland-appearing glands of varying sizes with loose, concentric, edematous, or fibromyxoid stroma (Figure 1).3 Red blood cells sometimes are found with hemosiderin within the stroma. Immunohistochemical staining with estrogen receptors may aid in identifying the endometriumlike epithelial cells.13
Standard treatment involves surgical excision with 1-cm margins and umbilical preservation, which results in a recurrence rate of less than 10%.4,10 Medical therapy, such as aromatase inhibitors, progestogens, antiprogestogens, combined oral contraceptives, or gonadotropin-releasing hormone agonists or antagonists may help manage pain or reduce the size of the nodule.4,15 Simple observation also is a potential course for patients who decline treatment options.
Differential diagnoses include lobular capillary hemangioma, also known as pyogenic granuloma; Sister Mary Joseph nodule; umbilical hernia; and dermatofibrosarcoma protuberans. Lobular capillary hemangiomas commonly are acquired benign vascular proliferations of the skin that are friable and tend to ulcerate.16 These lesions typically grow rapidly and often are located on the face, lips, mucosae, and fingers. Histopathologic examination may show an exophytic lesion with lobules of proliferating capillaries within an edematous matrix, superficial ulceration, and an epithelial collarette.17 Treatment includes surgical excision, cauterization, laser treatments, sclerotherapy, injectable medications, and topical medications, but recurrence is possible with any of these interventions.18
Cutaneous metastasis of an internal solid organ cancer, commonly known as a Sister Mary Joseph nodule, typically manifests as an erythematous, irregularly shaped nodule that may protrude from the umbilicus.14 Gastrointestinal symptoms such as change in bowel habits or obstructive symptoms in the setting of a progressive malignancy are common.14 Clinical features include a firm fixed lesion, oozing, and ulceration.19 On dermoscopy, polymorphous vascular patterns, milky red structureless areas, and white lines typically are present.11 Although dermoscopic features may differentiate this entity from cutaneous endometriosis, tissue sampling and histologic examination are crucial diagnostic tools to identify malignant vs benign lesions.
An umbilical hernia is a protrusion of omentum, bowel, or other intra-abdominal organs in an abdominal wall defect. Clinical presentation includes a soft protrusion that may be reduced on palpation if nonstrangulated.20 Treatment includes watchful waiting or surgical repair. The reducibility and presence of an abdominal wall defect may point to this diagnosis. Imaging also may aid in the diagnosis if the history and physical examination are unclear.
Dermatofibrosarcoma protuberans is a slow-developing, low- to intermediate-grade, soft-tissue sarcoma that occurs in less than 0.1% of all cancers in the United States.21 Lesions often manifest as small, firm, slow-growing, painless, flesh-colored dermal plaques; subcutaneous thickening; or atrophic nonprotuberant lesions typically involving the trunk.21 Histopathologically, they are composed of uniform spindle-cell proliferation growing in a storiform pattern and subcutaneous fat trapping that has strong and diffuse CD34 immunoreactivity.21,22 Pathologic examination typically distinguishes this diagnosis from cutaneous endometriosis. Treatment includes tumor resection that may or may not involve radiotherapy and targeted therapy, as recurrence and metastases are possible.
Primary cutaneous endometriosis is a rare but important diagnosis for dermatologists to consider when evaluating umbilical nodules. Clinical features may include bleeding masses during menses in females of reproductive age. Dermoscopic examination aids in workup, and histopathologic testing can confirm the diagnosis and rule out malignancies. Surgical excision is the treatment of choice with a low rate of recurrence.
THE DIAGNOSIS: Villar Nodule
The biopsy revealed features consistent with cutaneous endometriosis in the setting of a painful, tender, multilobulated nodule with a cyclical bleeding pattern (Figure 1). The bleeding pattern of the nodule during menses and lack of surgical history supported the diagnosis of primary cutaneous endometriosis in our patient. She was diagnosed with endometriosis by gynecology, and her primary care physician started her on an oral contraceptive based on this diagnosis. She also was referred to gynecology and plastic surgery for a joint surgical consultation to remove the nodule. She initially decided to do a trial of the oral contraceptive but subsequently underwent umbilical endometrioma excision with neo-umbilicus creation with no evidence of recurrence.
Primary cutaneous endometriosis should be considered in young females who present with tender umbilical nodules. Endometriosis refers to the presence of an endometriumlike epithelium outside the endometrium and myometrium.1 The condition affects 10% to 15% of reproductive-aged (ie, 18-49 years) women in the United States and typically involves tissues within the pelvis, such as the ovaries, pouch of Douglas, or pelvic ligaments.2 Cutaneous endometriosis is the growth of endometrial tissue in the skin and is rare, accounting for less than 5.5% of cases of extrapelvic endometriosis worldwide, affecting primarily the umbilicus, abdominal wall, and vulva.3,4
The 2 main types of cutaneous endometriosis are primary (spontaneous) and secondary. Primary lesions develop in patients without prior surgical history, and secondary lesions occur within previous surgical incision sites, often scars from cesarean delivery.5 Less than 30% of cases of cutaneous endometriosis are primary disease.6 Primary cutaneous endometriosis of the umbilicus, known as Villar nodule, was first described in 1886.3,7 Up to 40% of patients with extrapelvic endometriosis worldwide presented with Villar nodules in a systematic literature review.6 The prevalence of these nodules is unknown, but the incidence is less than 1% of cases of extragenital endometriosis.4
There are 2 leading theories of primary cutaneous endometriosis pathogenesis. The first is the transportation theory, in which endometrial cells are transported outside the uterus via the lymphatic system.8 The second is the metaplasia theory, which proposed that endometrial cells develop in the coelomic mesothelium in the presence of high estrogen levels.8,9
Secondary cutaneous endometriosis, also known as scar endometriosis, is suspected to be caused by an iatrogenic implantation of endometrial cells at the scar of a prior surgical site.9 Although our patient had an existing umbilicus scar from a piercing, it was improbable for that to have been the nidus, as the keloid scar was superficial and did not have contact with the abdominal cavity for iatrogenic implantation. Clinical diagnosis for secondary cutaneous endometriosis often is made based on a triad of features: a nonmalignant abdominal mass, recurring pain and bleeding of the lesion with menses, and prior history of abdominal surgery.9,10 On clinical examination, these features typically manifest as a palpable subcutaneous mass that is black, blue, brown, or red. Often, the lesions enlarge and bleed during the menstrual cycle, causing pain, tenderness, or pruritus.3 Dermoscopic features of secondary cutaneous endometriosis are erythematous umbilical nodules with a homogeneous vascular pattern that appears red with a brownish hue (Figure 2).9,11 Dermoscopic features may vary with the hormone cycle; for example, the follicular phase (correlating with day 7 of menses) demonstrates polypoid projections, erythematous violaceous color, dark-brown spots, and active bleeding of the lesion.12 Clinical and dermoscopic examination are useful tools in this diagnosis.
Imaging such as ultrasonography, computed tomography, or magnetic resonance imaging may be useful in identifying abdominal endometriomas.8,13,14 Pelvic involvement of endometriosis was found in approximately 15% of patients in a case series,4 with concurrent primary umbilical endometriosis. Imaging studies may assist evaluation for fistula formation, presence of malignancies, and the extent of endometriosis within the abdominal cavity.
Histopathology is key to confirming cutaneous endometriosis and shows multiple bland-appearing glands of varying sizes with loose, concentric, edematous, or fibromyxoid stroma (Figure 1).3 Red blood cells sometimes are found with hemosiderin within the stroma. Immunohistochemical staining with estrogen receptors may aid in identifying the endometriumlike epithelial cells.13
Standard treatment involves surgical excision with 1-cm margins and umbilical preservation, which results in a recurrence rate of less than 10%.4,10 Medical therapy, such as aromatase inhibitors, progestogens, antiprogestogens, combined oral contraceptives, or gonadotropin-releasing hormone agonists or antagonists may help manage pain or reduce the size of the nodule.4,15 Simple observation also is a potential course for patients who decline treatment options.
Differential diagnoses include lobular capillary hemangioma, also known as pyogenic granuloma; Sister Mary Joseph nodule; umbilical hernia; and dermatofibrosarcoma protuberans. Lobular capillary hemangiomas commonly are acquired benign vascular proliferations of the skin that are friable and tend to ulcerate.16 These lesions typically grow rapidly and often are located on the face, lips, mucosae, and fingers. Histopathologic examination may show an exophytic lesion with lobules of proliferating capillaries within an edematous matrix, superficial ulceration, and an epithelial collarette.17 Treatment includes surgical excision, cauterization, laser treatments, sclerotherapy, injectable medications, and topical medications, but recurrence is possible with any of these interventions.18
Cutaneous metastasis of an internal solid organ cancer, commonly known as a Sister Mary Joseph nodule, typically manifests as an erythematous, irregularly shaped nodule that may protrude from the umbilicus.14 Gastrointestinal symptoms such as change in bowel habits or obstructive symptoms in the setting of a progressive malignancy are common.14 Clinical features include a firm fixed lesion, oozing, and ulceration.19 On dermoscopy, polymorphous vascular patterns, milky red structureless areas, and white lines typically are present.11 Although dermoscopic features may differentiate this entity from cutaneous endometriosis, tissue sampling and histologic examination are crucial diagnostic tools to identify malignant vs benign lesions.
An umbilical hernia is a protrusion of omentum, bowel, or other intra-abdominal organs in an abdominal wall defect. Clinical presentation includes a soft protrusion that may be reduced on palpation if nonstrangulated.20 Treatment includes watchful waiting or surgical repair. The reducibility and presence of an abdominal wall defect may point to this diagnosis. Imaging also may aid in the diagnosis if the history and physical examination are unclear.
Dermatofibrosarcoma protuberans is a slow-developing, low- to intermediate-grade, soft-tissue sarcoma that occurs in less than 0.1% of all cancers in the United States.21 Lesions often manifest as small, firm, slow-growing, painless, flesh-colored dermal plaques; subcutaneous thickening; or atrophic nonprotuberant lesions typically involving the trunk.21 Histopathologically, they are composed of uniform spindle-cell proliferation growing in a storiform pattern and subcutaneous fat trapping that has strong and diffuse CD34 immunoreactivity.21,22 Pathologic examination typically distinguishes this diagnosis from cutaneous endometriosis. Treatment includes tumor resection that may or may not involve radiotherapy and targeted therapy, as recurrence and metastases are possible.
Primary cutaneous endometriosis is a rare but important diagnosis for dermatologists to consider when evaluating umbilical nodules. Clinical features may include bleeding masses during menses in females of reproductive age. Dermoscopic examination aids in workup, and histopathologic testing can confirm the diagnosis and rule out malignancies. Surgical excision is the treatment of choice with a low rate of recurrence.
- International Working Group of AAGL, ESGE, ESHRE and WES; Tomassetti C, Johnson NP, et al. An international terminology for endometriosis, 2021. Hum Reprod Open. 2021;2021:hoab029. doi:10.1093/hropen/hoab029
- Batista M, Alves F, Cardoso J, et al. Cutaneous endometriosis: a differential diagnosis of umbilical nodule. Acta Med Port. 2020; 33:282-284. doi:10.20344/amp.10966
- Brown ME, Osswald S, Biediger T. Cutaneous endometriosis of the umbilicus (Villar’s nodule). Int J Womens Dermatol. 2020;6:214-215. doi:10.1016/j.ijwd.2020.01.001
- Bindra V, Sampurna S, Kade S, et al. Primary umbilical endometriosis - case series and review of clinical presentation, diagnosis and management. Int J Surg Case Rep. 2022;94:107134. doi:10.1016/j.ijscr.2022.107134
- Loh SH, Lew BL, Sim WY. Primary cutaneous endometriosis of umbilicus. Ann Dermatol. 2017;29:621-625. doi:10.5021/ad.2017.29.5.621
- Victory R, Diamond MP, Johns DA. Villar’s nodule: a case report and systematic literature review of endometriosis externa of the umbilicus. J Minim Invasive Gynecol. 2007;14:23-32. doi:10.1016/j.jmig.2006.07.01
- Van den Nouland D, Kaur M. Primary umbilical endometriosis: a case report. Facts Views Vis Obgyn. 2017;9:115-119.
- Machairiotis N, Stylianaki A, Dryllis G, et al. Extrapelvic endometriosis: a rare entity or an under diagnosed condition? Diagn Pathol. 2013;8:194. doi:10.1186/1746-1596-8-194
- Huang QF, Jiang B, Yang X, et al. Primary versus secondary cutaneous endometriosis: literature review and case study. Heliyon. 2023;9:E20094. doi:10.1016/j.heliyon.2023.e20094
- Gonzalez RH, Singh MS, Hamza SA. Cutaneous endometriosis: a case report and review of the literature. Am J Case Rep. 2021;22:E932493. doi:10.12659/AJCR.932493
- Buljan M, Arzberger E, Šitum M, et al. The use of dermoscopy in differentiating Sister Mary Joseph nodule and cutaneous endometriosis. Australas J Dermatol. 2019;60:E233-E235. doi:10.1111/ajd.12980
- Costa IM, Gomes CM, Morais OO, et al. Cutaneous endometriosis: dermoscopic findings related to phases of the female hormonal cycle. Int J Dermatol. 2014;53:E130-E132. doi:10.1111 /j.1365-4632.2012.05854.x
- Mohaghegh F, Hatami P, Rajabi P, et al. Coexistence of cutaneous endometriosis and ovarian endometrioma: a case report. J Med Case Rep. 2022;16:256. doi:10.1186/s13256-022-03483-8
- Raffi L, Suresh R, McCalmont TH, et al. Cutaneous endometriosis. Int J Womens Dermatol. 2019;5:384-386. doi:10.1016 /j.ijwd.2019.06.025
- Saunders PTK, Horne AW. Endometriosis: etiology, pathobiology, and therapeutic prospects. Cell. 2021;184:2807-2824. doi:10.1016 /j.cell.2021.04.041
- Habif TP. Clinical Dermatology a Color Guide to Diagnosis and Therapy. St. Louis, Mo. Elsevier; 2016.
- Patrice SJ, Wiss K, Mulliken JB. Pyogenic granuloma (lobular capillary hemangioma): a clinicopathologic study of 178 cases. Pediatr Dermatol. 1991;8:267-276. doi:10.1111/j.15251470.1991.tb00931.x
- Kaleeny JD, Janis JE. Pyogenic granuloma diagnosis and management: a practical review. Plast Reconstr Surg Glob Open. 2024;12:E6160. doi:10.1097/GOX.0000000000006160
- Ha DL, Yang MY, Shin JO, et al. Benign umbilical tumors resembling Sister Mary Joseph nodule. Clin Med Insights Oncol. 2021;15:1179554921995022. doi:10.1177/1179554921995022
- Lawrence PF, Smeds M, Jessica Beth O’connell. Essentials of General Surgery and Surgical Specialties. Wolters Kluwer Health; 2019.
- Hao X, Billings SD, Wu F, et al. Dermatofibrosarcoma protuberans: update on the diagnosis and treatment. J Clin Med. 2020;9:1752. doi:10.3390/jcm9061752
- Allen A, Ahn C, Sangüeza OP. Dermatofibrosarcoma protuberans. Dermatol Clin. 2019;37:483-488. doi:10.1016/j.det.2019.05.006
- International Working Group of AAGL, ESGE, ESHRE and WES; Tomassetti C, Johnson NP, et al. An international terminology for endometriosis, 2021. Hum Reprod Open. 2021;2021:hoab029. doi:10.1093/hropen/hoab029
- Batista M, Alves F, Cardoso J, et al. Cutaneous endometriosis: a differential diagnosis of umbilical nodule. Acta Med Port. 2020; 33:282-284. doi:10.20344/amp.10966
- Brown ME, Osswald S, Biediger T. Cutaneous endometriosis of the umbilicus (Villar’s nodule). Int J Womens Dermatol. 2020;6:214-215. doi:10.1016/j.ijwd.2020.01.001
- Bindra V, Sampurna S, Kade S, et al. Primary umbilical endometriosis - case series and review of clinical presentation, diagnosis and management. Int J Surg Case Rep. 2022;94:107134. doi:10.1016/j.ijscr.2022.107134
- Loh SH, Lew BL, Sim WY. Primary cutaneous endometriosis of umbilicus. Ann Dermatol. 2017;29:621-625. doi:10.5021/ad.2017.29.5.621
- Victory R, Diamond MP, Johns DA. Villar’s nodule: a case report and systematic literature review of endometriosis externa of the umbilicus. J Minim Invasive Gynecol. 2007;14:23-32. doi:10.1016/j.jmig.2006.07.01
- Van den Nouland D, Kaur M. Primary umbilical endometriosis: a case report. Facts Views Vis Obgyn. 2017;9:115-119.
- Machairiotis N, Stylianaki A, Dryllis G, et al. Extrapelvic endometriosis: a rare entity or an under diagnosed condition? Diagn Pathol. 2013;8:194. doi:10.1186/1746-1596-8-194
- Huang QF, Jiang B, Yang X, et al. Primary versus secondary cutaneous endometriosis: literature review and case study. Heliyon. 2023;9:E20094. doi:10.1016/j.heliyon.2023.e20094
- Gonzalez RH, Singh MS, Hamza SA. Cutaneous endometriosis: a case report and review of the literature. Am J Case Rep. 2021;22:E932493. doi:10.12659/AJCR.932493
- Buljan M, Arzberger E, Šitum M, et al. The use of dermoscopy in differentiating Sister Mary Joseph nodule and cutaneous endometriosis. Australas J Dermatol. 2019;60:E233-E235. doi:10.1111/ajd.12980
- Costa IM, Gomes CM, Morais OO, et al. Cutaneous endometriosis: dermoscopic findings related to phases of the female hormonal cycle. Int J Dermatol. 2014;53:E130-E132. doi:10.1111 /j.1365-4632.2012.05854.x
- Mohaghegh F, Hatami P, Rajabi P, et al. Coexistence of cutaneous endometriosis and ovarian endometrioma: a case report. J Med Case Rep. 2022;16:256. doi:10.1186/s13256-022-03483-8
- Raffi L, Suresh R, McCalmont TH, et al. Cutaneous endometriosis. Int J Womens Dermatol. 2019;5:384-386. doi:10.1016 /j.ijwd.2019.06.025
- Saunders PTK, Horne AW. Endometriosis: etiology, pathobiology, and therapeutic prospects. Cell. 2021;184:2807-2824. doi:10.1016 /j.cell.2021.04.041
- Habif TP. Clinical Dermatology a Color Guide to Diagnosis and Therapy. St. Louis, Mo. Elsevier; 2016.
- Patrice SJ, Wiss K, Mulliken JB. Pyogenic granuloma (lobular capillary hemangioma): a clinicopathologic study of 178 cases. Pediatr Dermatol. 1991;8:267-276. doi:10.1111/j.15251470.1991.tb00931.x
- Kaleeny JD, Janis JE. Pyogenic granuloma diagnosis and management: a practical review. Plast Reconstr Surg Glob Open. 2024;12:E6160. doi:10.1097/GOX.0000000000006160
- Ha DL, Yang MY, Shin JO, et al. Benign umbilical tumors resembling Sister Mary Joseph nodule. Clin Med Insights Oncol. 2021;15:1179554921995022. doi:10.1177/1179554921995022
- Lawrence PF, Smeds M, Jessica Beth O’connell. Essentials of General Surgery and Surgical Specialties. Wolters Kluwer Health; 2019.
- Hao X, Billings SD, Wu F, et al. Dermatofibrosarcoma protuberans: update on the diagnosis and treatment. J Clin Med. 2020;9:1752. doi:10.3390/jcm9061752
- Allen A, Ahn C, Sangüeza OP. Dermatofibrosarcoma protuberans. Dermatol Clin. 2019;37:483-488. doi:10.1016/j.det.2019.05.006
Tender Nodule on the Umbilicus
Tender Nodule on the Umbilicus
A 25-year-old woman was referred to the dermatology clinic by her primary care provider for evaluation of a tender nodule on the inferior umbilicus of 2 years' duration at the site of a preexisting keloid scar. The patient reported that the lesion caused occasional pain and tenderness. A few weeks prior to the current presentation, a dark-red bloody discharge developed at the superior aspect of the lesion that subsequently crusted over. The patient denied any use of oral contraceptives or history of abdominal surgery.
The original keloid scar had been treated successfully by an outside physician with intralesional steroid injections, and the patient was interested in a similar procedure for the current nodule. She also had a history of a hyperpigmented hypertrophic scar on the superior periumbilical area from a previous piercing that had resolved several years prior to presentation.
Physical examination of the lesion revealed a 1.2-cm, soft, tender, violaceous nodule with scant yellow crust along the superior surface of the umbilicus. There was no palpable abdominal wall defect, and the nodule was not reducible into the abdominal cavity. An interval history revealed bleeding of the lesion during the patient's menstrual cycle with persistent pain and tenderness. A punch biopsy was performed.
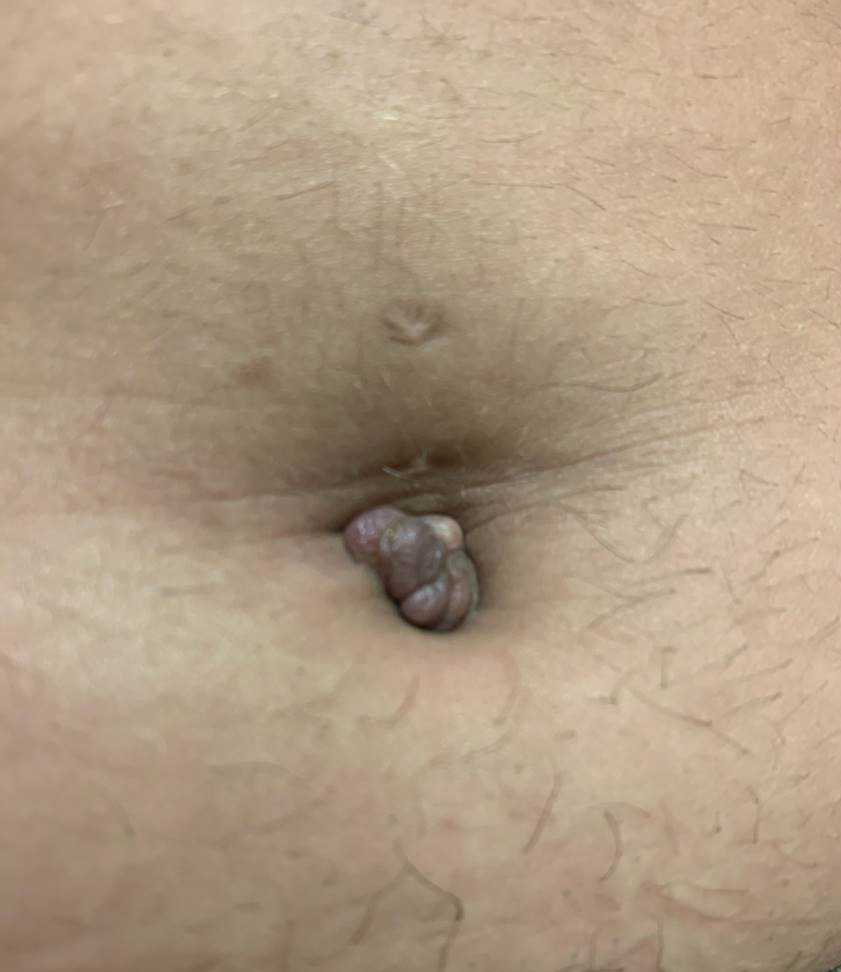

Diffuse Pruritic Keratotic Papules
Diffuse Pruritic Keratotic Papules
THE DIAGNOSIS: Reactive Perforating Collagenosis
Histopathology revealed invagination of the epidermis with hyperkeratosis; prominent epidermal hyperplasia; and a central basophilic plug of keratin, collagen, and inflammatory debris. Transepidermal elimination of bright eosinophilic altered collagen fibers was seen (Figure). The findings were consistent with a diagnosis of reactive perforating collagenosis (RPC).
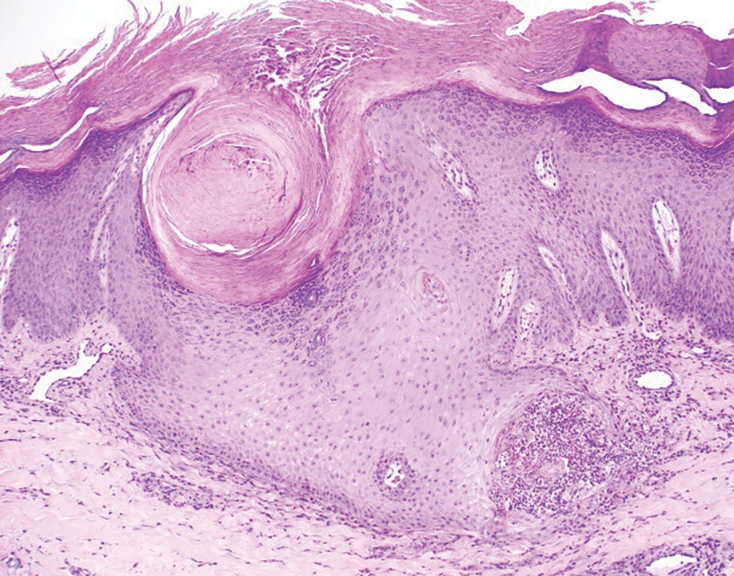
Reactive perforating collagenosis, a subtype of perforating dermatosis, is a rare skin condition in which altered collagen is eliminated through the epidermis.1 There are 2 forms of RPC: the inherited form, which is very rare and manifests in childhood, and the acquired form, which manifests in adulthood and is associated with systemic diseases, most notably diabetes and/or chronic renal failure, both of which our patient had been diagnosed with.1,2 The clinical presentation of RPC includes erythematous papules or nodules that evolve into umbilicated 4- to 10-mm craterlike ulcerations with a central keratotic plug. The lesions favor a linear distribution along the extensor surfaces of the arms and legs, trunk, and gluteal area. Involvement of the head, neck, and scalp has been reported less commonly, which makes our case particularly unique.3 Histopathologically, RPC is characterized by a cup-shaped depression of the epidermis with an overlying keratin plug containing inflammatory cells, keratinous debris, and collagen fibers. Vertically oriented collagen fibers are seen extruded through the epidermis.4,5
While the pathogenesis of RPC remains unknown, it is believed that superficial trauma due to chronic scratching results in transepithelial elimination of collagen. Due to the association of acquired RPC (ARPC) with diabetes, it also has been proposed that scratching can cause microtrauma and necrosis of the dermal structures, potentially due to diabetic microangiopathy.3 Additionally, RPC is associated with overexpression of transforming growth factor beta 3 in lesional skin, suggesting that transforming growth factor beta 3 is involved with tissue repair and extracellular remodeling in this condition.6
Treatment of ARPC should include the management of underlying disease. While no definitive treatment has been reported to date, topical corticosteroids, retinoids, keratolytics, emollients, antihistamines, narrow-band UVB phototherapy, and psoralen plus UVA phototherapy have been used with varying degrees of improvement. Typically, the lesions self-resolve within 6 to 8 weeks; however, they often recur and usually leave scarring with or without hyperpigmentation.2,7-10
Acquired RPC can be misdiagnosed initially, as it mimics several other conditions and commonly is associated with systemic diseases. While biopsy is necessary for diagnosis, if it cannot be performed or the results are indeterminate, dermoscopy can serve as a helpful diagnostic tool. The most common dermoscopic patterns seen in RPC include a yellow-brown structureless area in the center of the lesion with a peripheral surface crust and surrounding white rim—thought to represent epidermal invagination or keratinous debris. Additionally, inflammation with visible vessels both centrally and peripherally is represented by an outer pink circle on dermoscopy.5,11
The differential diagnoses for RPC include perforating folliculitis (PF), elastosis perforans serpiginosa (EPS), prurigo nodularis, and keratoacanthomas. The primary perforating dermatoses (PF, EPS, and RPC) are similarly characterized by elimination of altered dermal material through the epidermis. As these conditions manifest with similar features on clinical examination, differentiation is made by the type of epidermal damage and the features of elimination material, making histopathologic examination paramount for definitive diagnosis.
Perforating folliculitis manifests as erythematous, follicular papules with a small central keratotic core or a central hair. Histopathologically, PF reveals a widely dilated follicle containing keratin, necrotic debris, and degenerated inflammatory cells. Elastosis perforans serpiginosa manifests clinically as hyperkeratotic papules in serpiginous patterns rather than the linear pattern commonly seen with ARPC. Histopathologically, EPS reveals thickened elastic fibers, rather than collagen fibers as seen in ARPC, extruded through the epidermis. Prurigo nodularis manifests clinically as dome-shaped papules with possible excoriation and crusting. Histopathologic examination reveals epidermal hyperplasia and hyperkeratosis; however, the characteristic features of transepithelial elimination of collagen and invaginations of epidermis differentiate ARPC from prurigo nodularis.12,13 Keratoacanthomas manifest clinically as an eruption of small, round, pink papules that rapidly grow and evolve into 1- to 2-cm dome-shaped nodules with central keratinaceous plugs, mimicking a crateriform appearance. Histopathologic examination reveals a circumscribed proliferation of well-differentiated keratinocytes. Multilobular exophytic or endophytic cystlike invaginations of the epidermis also are noted. The expulsion of collagen from the epidermis is more consistent with ARPC.14
- Cohen RW, Auerbach R. Acquired reactive perforating collagenosis. J Am Acad Dermatol. 1989;20(2 pt 1):287-289. doi:10.1016/s0190 -9622(89)80059-3
- Bejjanki H, Siroy AE, Koratala A. Reactive perforating collagenosis in end-stage renal disease: not all that itches is uremic pruritis! Am J Med. 2019;132:E658-E660. doi:10.1016/j.amjmed.2019.03.015
- Gontijo JRV, Júnior FF, Pereira LB, et al. Trauma-induced acquired reactive perforating collagenosis. An Bras Dermatol. 2021;96:392-393. doi:10.1016/j.abd.2020.06.022
- Ambalathinkal JJ, Phiske MM, Someshwar SJ. Acquired reactive perforating collagenosis, a rare entity at uncommon site. Indian J Pathol Microbiol. 2022;65:895-897. doi:10.4103/ijpm.ijpm_333_21
- Ormerod E, Atwan A, Intzedy L, et al. Dermoscopy features of acquired reactive perforating collagenosis: a case series. Dermatol Pract Concept. 2018;8:303-305. doi:10.5826/dpc.0804a11
- Fei C, Wang Y, Gong Y, et al. Acquired reactive perforating collagenosis: a report of a typical case. Medicine (Baltimore). 2016;95:E4305. doi:10.1097/md.0000000000004305
- Bartling SJ, Naff JL, Canevari MM, et al. Pruritic rash in an elderly patient with uncontrolled diabetes mellitus. AACE Clin Case Rep. 2018;5:E146-E149. doi:10.4158/ACCR-2018-0388
- Kollipara H, Satya RS, Rao GR, et al. Acquired reactive perforating collagenosis: case series. Indian Dermatol Online J. 2023;14:72-76. doi:10.4103/idoj.idoj_373_22
- Wang C, Liu YH, Wang YX, et al. Acquired reactive perforating collagenosis. Chin Med J (Engl). 2020;133:2119-2120. doi:10.1097 /cm9.0000000000000906
- Harbaoui S, Litaiem N. Acquired perforating dermatosis. StatPearls [Internet]. Updated February 13, 2023. Accessed August 13, 2025. https://www.ncbi.nlm.nih.gov/books/NBK539715/
- Elmas ÖF, Kilitci A, Uyar B. Dermoscopic patterns of acquired reactive perforating collagenosis. Dermatol Pract Concept. 2021;11:E2020085. doi:10.5826/dpc.1101a85
- Patterson JW. The perforating disorders. J Am Acad Dermatol. 1984;10:561-581. doi:10.1016/s0190-9622(84)80259-5
- Huang AH, Williams KA, Kwatra SG. Prurigo nodularis: epidemiology and clinical features. J Am Acad Dermatol. 2020;83:1559-1565. doi:10.1016/j.jaad.2020.04.183
- Zito PM, Scharf R. Keratoacanthoma. StatPearls [Internet]. Updated August 8, 2023. Accessed August 13, 2025. https://www.ncbi.nlm.nih.gov/books/NBK499931/
THE DIAGNOSIS: Reactive Perforating Collagenosis
Histopathology revealed invagination of the epidermis with hyperkeratosis; prominent epidermal hyperplasia; and a central basophilic plug of keratin, collagen, and inflammatory debris. Transepidermal elimination of bright eosinophilic altered collagen fibers was seen (Figure). The findings were consistent with a diagnosis of reactive perforating collagenosis (RPC).
Reactive perforating collagenosis, a subtype of perforating dermatosis, is a rare skin condition in which altered collagen is eliminated through the epidermis.1 There are 2 forms of RPC: the inherited form, which is very rare and manifests in childhood, and the acquired form, which manifests in adulthood and is associated with systemic diseases, most notably diabetes and/or chronic renal failure, both of which our patient had been diagnosed with.1,2 The clinical presentation of RPC includes erythematous papules or nodules that evolve into umbilicated 4- to 10-mm craterlike ulcerations with a central keratotic plug. The lesions favor a linear distribution along the extensor surfaces of the arms and legs, trunk, and gluteal area. Involvement of the head, neck, and scalp has been reported less commonly, which makes our case particularly unique.3 Histopathologically, RPC is characterized by a cup-shaped depression of the epidermis with an overlying keratin plug containing inflammatory cells, keratinous debris, and collagen fibers. Vertically oriented collagen fibers are seen extruded through the epidermis.4,5
While the pathogenesis of RPC remains unknown, it is believed that superficial trauma due to chronic scratching results in transepithelial elimination of collagen. Due to the association of acquired RPC (ARPC) with diabetes, it also has been proposed that scratching can cause microtrauma and necrosis of the dermal structures, potentially due to diabetic microangiopathy.3 Additionally, RPC is associated with overexpression of transforming growth factor beta 3 in lesional skin, suggesting that transforming growth factor beta 3 is involved with tissue repair and extracellular remodeling in this condition.6
Treatment of ARPC should include the management of underlying disease. While no definitive treatment has been reported to date, topical corticosteroids, retinoids, keratolytics, emollients, antihistamines, narrow-band UVB phototherapy, and psoralen plus UVA phototherapy have been used with varying degrees of improvement. Typically, the lesions self-resolve within 6 to 8 weeks; however, they often recur and usually leave scarring with or without hyperpigmentation.2,7-10
Acquired RPC can be misdiagnosed initially, as it mimics several other conditions and commonly is associated with systemic diseases. While biopsy is necessary for diagnosis, if it cannot be performed or the results are indeterminate, dermoscopy can serve as a helpful diagnostic tool. The most common dermoscopic patterns seen in RPC include a yellow-brown structureless area in the center of the lesion with a peripheral surface crust and surrounding white rim—thought to represent epidermal invagination or keratinous debris. Additionally, inflammation with visible vessels both centrally and peripherally is represented by an outer pink circle on dermoscopy.5,11
The differential diagnoses for RPC include perforating folliculitis (PF), elastosis perforans serpiginosa (EPS), prurigo nodularis, and keratoacanthomas. The primary perforating dermatoses (PF, EPS, and RPC) are similarly characterized by elimination of altered dermal material through the epidermis. As these conditions manifest with similar features on clinical examination, differentiation is made by the type of epidermal damage and the features of elimination material, making histopathologic examination paramount for definitive diagnosis.
Perforating folliculitis manifests as erythematous, follicular papules with a small central keratotic core or a central hair. Histopathologically, PF reveals a widely dilated follicle containing keratin, necrotic debris, and degenerated inflammatory cells. Elastosis perforans serpiginosa manifests clinically as hyperkeratotic papules in serpiginous patterns rather than the linear pattern commonly seen with ARPC. Histopathologically, EPS reveals thickened elastic fibers, rather than collagen fibers as seen in ARPC, extruded through the epidermis. Prurigo nodularis manifests clinically as dome-shaped papules with possible excoriation and crusting. Histopathologic examination reveals epidermal hyperplasia and hyperkeratosis; however, the characteristic features of transepithelial elimination of collagen and invaginations of epidermis differentiate ARPC from prurigo nodularis.12,13 Keratoacanthomas manifest clinically as an eruption of small, round, pink papules that rapidly grow and evolve into 1- to 2-cm dome-shaped nodules with central keratinaceous plugs, mimicking a crateriform appearance. Histopathologic examination reveals a circumscribed proliferation of well-differentiated keratinocytes. Multilobular exophytic or endophytic cystlike invaginations of the epidermis also are noted. The expulsion of collagen from the epidermis is more consistent with ARPC.14
THE DIAGNOSIS: Reactive Perforating Collagenosis
Histopathology revealed invagination of the epidermis with hyperkeratosis; prominent epidermal hyperplasia; and a central basophilic plug of keratin, collagen, and inflammatory debris. Transepidermal elimination of bright eosinophilic altered collagen fibers was seen (Figure). The findings were consistent with a diagnosis of reactive perforating collagenosis (RPC).
Reactive perforating collagenosis, a subtype of perforating dermatosis, is a rare skin condition in which altered collagen is eliminated through the epidermis.1 There are 2 forms of RPC: the inherited form, which is very rare and manifests in childhood, and the acquired form, which manifests in adulthood and is associated with systemic diseases, most notably diabetes and/or chronic renal failure, both of which our patient had been diagnosed with.1,2 The clinical presentation of RPC includes erythematous papules or nodules that evolve into umbilicated 4- to 10-mm craterlike ulcerations with a central keratotic plug. The lesions favor a linear distribution along the extensor surfaces of the arms and legs, trunk, and gluteal area. Involvement of the head, neck, and scalp has been reported less commonly, which makes our case particularly unique.3 Histopathologically, RPC is characterized by a cup-shaped depression of the epidermis with an overlying keratin plug containing inflammatory cells, keratinous debris, and collagen fibers. Vertically oriented collagen fibers are seen extruded through the epidermis.4,5
While the pathogenesis of RPC remains unknown, it is believed that superficial trauma due to chronic scratching results in transepithelial elimination of collagen. Due to the association of acquired RPC (ARPC) with diabetes, it also has been proposed that scratching can cause microtrauma and necrosis of the dermal structures, potentially due to diabetic microangiopathy.3 Additionally, RPC is associated with overexpression of transforming growth factor beta 3 in lesional skin, suggesting that transforming growth factor beta 3 is involved with tissue repair and extracellular remodeling in this condition.6
Treatment of ARPC should include the management of underlying disease. While no definitive treatment has been reported to date, topical corticosteroids, retinoids, keratolytics, emollients, antihistamines, narrow-band UVB phototherapy, and psoralen plus UVA phototherapy have been used with varying degrees of improvement. Typically, the lesions self-resolve within 6 to 8 weeks; however, they often recur and usually leave scarring with or without hyperpigmentation.2,7-10
Acquired RPC can be misdiagnosed initially, as it mimics several other conditions and commonly is associated with systemic diseases. While biopsy is necessary for diagnosis, if it cannot be performed or the results are indeterminate, dermoscopy can serve as a helpful diagnostic tool. The most common dermoscopic patterns seen in RPC include a yellow-brown structureless area in the center of the lesion with a peripheral surface crust and surrounding white rim—thought to represent epidermal invagination or keratinous debris. Additionally, inflammation with visible vessels both centrally and peripherally is represented by an outer pink circle on dermoscopy.5,11
The differential diagnoses for RPC include perforating folliculitis (PF), elastosis perforans serpiginosa (EPS), prurigo nodularis, and keratoacanthomas. The primary perforating dermatoses (PF, EPS, and RPC) are similarly characterized by elimination of altered dermal material through the epidermis. As these conditions manifest with similar features on clinical examination, differentiation is made by the type of epidermal damage and the features of elimination material, making histopathologic examination paramount for definitive diagnosis.
Perforating folliculitis manifests as erythematous, follicular papules with a small central keratotic core or a central hair. Histopathologically, PF reveals a widely dilated follicle containing keratin, necrotic debris, and degenerated inflammatory cells. Elastosis perforans serpiginosa manifests clinically as hyperkeratotic papules in serpiginous patterns rather than the linear pattern commonly seen with ARPC. Histopathologically, EPS reveals thickened elastic fibers, rather than collagen fibers as seen in ARPC, extruded through the epidermis. Prurigo nodularis manifests clinically as dome-shaped papules with possible excoriation and crusting. Histopathologic examination reveals epidermal hyperplasia and hyperkeratosis; however, the characteristic features of transepithelial elimination of collagen and invaginations of epidermis differentiate ARPC from prurigo nodularis.12,13 Keratoacanthomas manifest clinically as an eruption of small, round, pink papules that rapidly grow and evolve into 1- to 2-cm dome-shaped nodules with central keratinaceous plugs, mimicking a crateriform appearance. Histopathologic examination reveals a circumscribed proliferation of well-differentiated keratinocytes. Multilobular exophytic or endophytic cystlike invaginations of the epidermis also are noted. The expulsion of collagen from the epidermis is more consistent with ARPC.14
- Cohen RW, Auerbach R. Acquired reactive perforating collagenosis. J Am Acad Dermatol. 1989;20(2 pt 1):287-289. doi:10.1016/s0190 -9622(89)80059-3
- Bejjanki H, Siroy AE, Koratala A. Reactive perforating collagenosis in end-stage renal disease: not all that itches is uremic pruritis! Am J Med. 2019;132:E658-E660. doi:10.1016/j.amjmed.2019.03.015
- Gontijo JRV, Júnior FF, Pereira LB, et al. Trauma-induced acquired reactive perforating collagenosis. An Bras Dermatol. 2021;96:392-393. doi:10.1016/j.abd.2020.06.022
- Ambalathinkal JJ, Phiske MM, Someshwar SJ. Acquired reactive perforating collagenosis, a rare entity at uncommon site. Indian J Pathol Microbiol. 2022;65:895-897. doi:10.4103/ijpm.ijpm_333_21
- Ormerod E, Atwan A, Intzedy L, et al. Dermoscopy features of acquired reactive perforating collagenosis: a case series. Dermatol Pract Concept. 2018;8:303-305. doi:10.5826/dpc.0804a11
- Fei C, Wang Y, Gong Y, et al. Acquired reactive perforating collagenosis: a report of a typical case. Medicine (Baltimore). 2016;95:E4305. doi:10.1097/md.0000000000004305
- Bartling SJ, Naff JL, Canevari MM, et al. Pruritic rash in an elderly patient with uncontrolled diabetes mellitus. AACE Clin Case Rep. 2018;5:E146-E149. doi:10.4158/ACCR-2018-0388
- Kollipara H, Satya RS, Rao GR, et al. Acquired reactive perforating collagenosis: case series. Indian Dermatol Online J. 2023;14:72-76. doi:10.4103/idoj.idoj_373_22
- Wang C, Liu YH, Wang YX, et al. Acquired reactive perforating collagenosis. Chin Med J (Engl). 2020;133:2119-2120. doi:10.1097 /cm9.0000000000000906
- Harbaoui S, Litaiem N. Acquired perforating dermatosis. StatPearls [Internet]. Updated February 13, 2023. Accessed August 13, 2025. https://www.ncbi.nlm.nih.gov/books/NBK539715/
- Elmas ÖF, Kilitci A, Uyar B. Dermoscopic patterns of acquired reactive perforating collagenosis. Dermatol Pract Concept. 2021;11:E2020085. doi:10.5826/dpc.1101a85
- Patterson JW. The perforating disorders. J Am Acad Dermatol. 1984;10:561-581. doi:10.1016/s0190-9622(84)80259-5
- Huang AH, Williams KA, Kwatra SG. Prurigo nodularis: epidemiology and clinical features. J Am Acad Dermatol. 2020;83:1559-1565. doi:10.1016/j.jaad.2020.04.183
- Zito PM, Scharf R. Keratoacanthoma. StatPearls [Internet]. Updated August 8, 2023. Accessed August 13, 2025. https://www.ncbi.nlm.nih.gov/books/NBK499931/
- Cohen RW, Auerbach R. Acquired reactive perforating collagenosis. J Am Acad Dermatol. 1989;20(2 pt 1):287-289. doi:10.1016/s0190 -9622(89)80059-3
- Bejjanki H, Siroy AE, Koratala A. Reactive perforating collagenosis in end-stage renal disease: not all that itches is uremic pruritis! Am J Med. 2019;132:E658-E660. doi:10.1016/j.amjmed.2019.03.015
- Gontijo JRV, Júnior FF, Pereira LB, et al. Trauma-induced acquired reactive perforating collagenosis. An Bras Dermatol. 2021;96:392-393. doi:10.1016/j.abd.2020.06.022
- Ambalathinkal JJ, Phiske MM, Someshwar SJ. Acquired reactive perforating collagenosis, a rare entity at uncommon site. Indian J Pathol Microbiol. 2022;65:895-897. doi:10.4103/ijpm.ijpm_333_21
- Ormerod E, Atwan A, Intzedy L, et al. Dermoscopy features of acquired reactive perforating collagenosis: a case series. Dermatol Pract Concept. 2018;8:303-305. doi:10.5826/dpc.0804a11
- Fei C, Wang Y, Gong Y, et al. Acquired reactive perforating collagenosis: a report of a typical case. Medicine (Baltimore). 2016;95:E4305. doi:10.1097/md.0000000000004305
- Bartling SJ, Naff JL, Canevari MM, et al. Pruritic rash in an elderly patient with uncontrolled diabetes mellitus. AACE Clin Case Rep. 2018;5:E146-E149. doi:10.4158/ACCR-2018-0388
- Kollipara H, Satya RS, Rao GR, et al. Acquired reactive perforating collagenosis: case series. Indian Dermatol Online J. 2023;14:72-76. doi:10.4103/idoj.idoj_373_22
- Wang C, Liu YH, Wang YX, et al. Acquired reactive perforating collagenosis. Chin Med J (Engl). 2020;133:2119-2120. doi:10.1097 /cm9.0000000000000906
- Harbaoui S, Litaiem N. Acquired perforating dermatosis. StatPearls [Internet]. Updated February 13, 2023. Accessed August 13, 2025. https://www.ncbi.nlm.nih.gov/books/NBK539715/
- Elmas ÖF, Kilitci A, Uyar B. Dermoscopic patterns of acquired reactive perforating collagenosis. Dermatol Pract Concept. 2021;11:E2020085. doi:10.5826/dpc.1101a85
- Patterson JW. The perforating disorders. J Am Acad Dermatol. 1984;10:561-581. doi:10.1016/s0190-9622(84)80259-5
- Huang AH, Williams KA, Kwatra SG. Prurigo nodularis: epidemiology and clinical features. J Am Acad Dermatol. 2020;83:1559-1565. doi:10.1016/j.jaad.2020.04.183
- Zito PM, Scharf R. Keratoacanthoma. StatPearls [Internet]. Updated August 8, 2023. Accessed August 13, 2025. https://www.ncbi.nlm.nih.gov/books/NBK499931/
Diffuse Pruritic Keratotic Papules
Diffuse Pruritic Keratotic Papules
A 65-year-old woman presented to dermatology with an intensely pruritic rash on the arms, legs, neck, and face of several months’ duration. The patient reported scratching the lesions but denied any recent trauma to the affected areas. She previously had been evaluated by her primary care provider, who prescribed cephalexin with no improvement. Her medical history was remarkable for chronic renal failure on dialysis, diabetes, hypertension, and congestive heart failure. Physical examination of the skin revealed hard white cutaneous nodules distributed on the proximal posterior upper arms, bilateral proximal pretibial regions, right elbow, and left knee. Two shave biopsies from the right elbow and left knee were obtained for histopathology.

Painful Edematous Labial Erosions
Painful Edematous Labial Erosions
THE DIAGNOSIS: Kaposi Varicelliform Eruption
Genital erosions tested positive for herpes simplex virus (HSV) 2 via PCR, confirming a Kaposi varicelliform eruption (KVE) in a patient with mycosis fungoides. The medical team began antiviral therapy with intravenous (IV) acyclovir; however, susceptibility testing during the hospital admission confirmed acyclovir resistance, requiring a transition to cidofovir cream 1% and IV foscarnet.1 Subsequent concerns by the care team about chemical burns, dysuria, and renal impairment led to discontinuation of both the cidofovir and foscarnet, considerably narrowing the treatment options.1 The patient’s condition was complicated by polymicrobial bacteremia. Additionally, worsening acidosis and acute kidney injury required initiation of continuous renal replacement therapy.1 Considering these conditions, the patient was enrolled in a promising clinical trial for pritelivir, a novel antiviral medication; however, due to the development of oliguria and the progression of renal failure, this course of treatment had to be discontinued. Faced with potential viral encephalitis, the infectious disease team concluded that, despite previous adverse reactions, resumption of IV foscarnet treatment would present more benefits than risks, given the patient’s critical situation.1
Mycosis fungoides (MF) is a slowly progressive cutaneous T-cell lymphoma of CD4+ cells that primarily affects the skin. Clinically, it often is characterized by pruritic scaly patches or plaques with sharply demarcated borders, the enduring nature of which consistently poses a therapeutic challenge due to their noted resistance to preliminary lines of treatment. Presently, potential cures are limited to allogeneic stem cell transplantation and unilesional radiotherapy for advanced MF; however, no treatment has been found to notably improve survival rates.1 Mycosis fungoides can result in various complications including diffuse spread of a skin infection caused by HSV, known as KVE.1 Kaposi varicelliform eruption usually manifests clinically with painful skin vesicles that often are accompanied by systemic signs such as fever and malaise. The vesicles rapidly progress into pustules or erosions, predominantly affecting regions such as the head, neck, groin, and upper torso (Figure 1).2 Kaposi varicelliform eruption is considered a dermatologic emergency due to its potential to precipitate serious complications such as life-threatening secondary bacterial infection, HSV viremia, and multiorgan involvement; it also carries the risk of instigating ocular complications, such as keratitis, conjunctivitis, blepharitis, uveitis, and potential vision loss.2
Kaposi varicelliform eruption usually is diagnosed through clinical examination supported by polymerase chain reaction, viral culture, histopathology, HSV serology, and Tzanck smear.2 The differential diagnosis includes varicella, atypical varicella, herpes genitalis, herpes zoster, allergic or irritant contact dermatitis, or MF, which may result in painful skin ulcers.2-4 If an HSV superinfection is suspected, a polymerase chain reaction test ideally should be conducted within the first 72 hours of symptom onset.2 Herpes simplex virus infection may be reinforced by histologic features such as intraepidermal blistering, acantholysis, keratinocyte ballooning degeneration, and multinuclear giant cells with intranuclear inclusions. Given its severe nature, immediate empiric antiviral treatment for KVE is essential, even while awaiting confirmatory tests. The recommended treatment protocol involves acyclovir (400 mg orally 3 times daily or 10 mg/kg IV) or valacyclovir (500 mg orally twice daily), continued until KVE resolves.2
Herpes genitalis caused by HSV-2 is estimated to affect approximately 45 million adults in the United States.2 First-line treatment for HSV-2 includes acyclovir and its derivatives, which are viral nucleoside analogs that inhibit viral DNA polymerases.5,6 However, over the past 2 decades, increasing HSV resistance to acyclovir and its derivatives has been noted among immunocompromised patients.5,6 Second-line agents, such as IV foscarnet and cidofovir, require close laboratory monitoring for nephrotoxicity and are contraindicated in those with renal insufficiency, thus limiting their use.5 To combat acyclovir resistance, novel antivirals such as pritelivir are being developed. Pritelivir targets the HSV helicase-primase complex and has been shown to outperform acyclovir in in-vitro animal models.7 Due to its unique mechanism of action (Figure 2), pritelivir is effective against acyclovir-resistant HSV strains, and clinical trials suggest its serum half-life may allow for daily dosing. A phase 2 study showed pritelivir reduced viral shedding days, sped up genital lesion healing in adults infected with HSV-2, and exhibited a good safety profile.7 Our patient participated in ongoing open-label trials of pritelivir that aimed to assess its efficacy and safety in immunocompromised patients. Given the limited alternative treatments for acyclovir-resistant HSV-2, clinicians need to stay updated on antiviral agents under development.
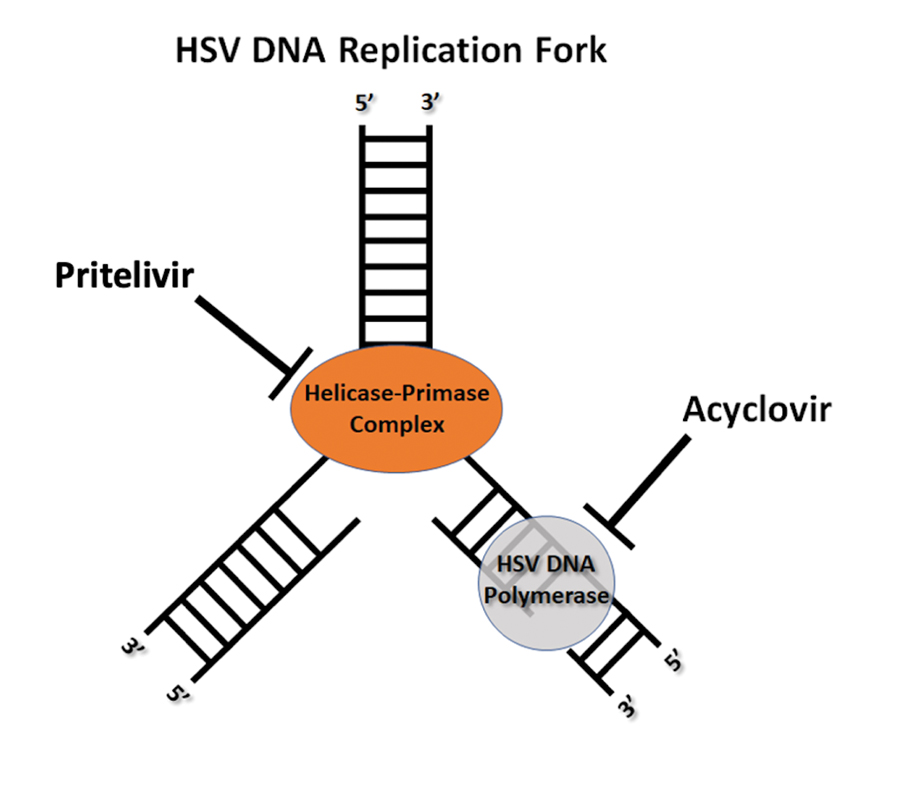
- García-Díaz N, Piris MÁ, Ortiz-Romero PL, et al. Mycosis fungoides and Sézary syndrome: an integrative review of the pathophysiology, molecular drivers, and targeted therapy. Cancers. 2021;13:1931. doi:10.3390/cancers13081931
- Baaniya B, Agrawal S. Kaposi varicelliform eruption in a patient with pemphigus vulgaris: a case report and review of the literature. Case Rep Dermatol Med. 2020;2020:6695342. doi:10.1155/2020/6695342
- Shin D, Lee MS, Kim DY, et al. Increased large unstained cells value in varicella patients: a valuable parameter to aid rapid diagnosis of varicella infection. J Dermatol. 2015;42:795-799. doi:10.1111
- Joshi A, Sah SP, Agrawal S. Kaposi’s varicelliform eruption or atypical chickenpox in a normal individual. Australas J Dermatol. 2000;41:126-127.
- Groves MJ. Genital herpes: a review. Am Fam Physician. 2016; 93:928-934.
- Fleming DT, Leone P, Esposito D, et al. Herpes virus type 2 infection and genital symptoms in primary care patients. Sex Transm Dis. 2006;33:416-421. doi:10.1097/01.olq.0000200578.86276.0b
- Poole CL, James SH. Antiviral therapies for herpesviruses: current agents and new directions. Clin Ther. 2018;40:1282-1298. doi:10.1016 /j.clinthera.2018.07.006.
THE DIAGNOSIS: Kaposi Varicelliform Eruption
Genital erosions tested positive for herpes simplex virus (HSV) 2 via PCR, confirming a Kaposi varicelliform eruption (KVE) in a patient with mycosis fungoides. The medical team began antiviral therapy with intravenous (IV) acyclovir; however, susceptibility testing during the hospital admission confirmed acyclovir resistance, requiring a transition to cidofovir cream 1% and IV foscarnet.1 Subsequent concerns by the care team about chemical burns, dysuria, and renal impairment led to discontinuation of both the cidofovir and foscarnet, considerably narrowing the treatment options.1 The patient’s condition was complicated by polymicrobial bacteremia. Additionally, worsening acidosis and acute kidney injury required initiation of continuous renal replacement therapy.1 Considering these conditions, the patient was enrolled in a promising clinical trial for pritelivir, a novel antiviral medication; however, due to the development of oliguria and the progression of renal failure, this course of treatment had to be discontinued. Faced with potential viral encephalitis, the infectious disease team concluded that, despite previous adverse reactions, resumption of IV foscarnet treatment would present more benefits than risks, given the patient’s critical situation.1
Mycosis fungoides (MF) is a slowly progressive cutaneous T-cell lymphoma of CD4+ cells that primarily affects the skin. Clinically, it often is characterized by pruritic scaly patches or plaques with sharply demarcated borders, the enduring nature of which consistently poses a therapeutic challenge due to their noted resistance to preliminary lines of treatment. Presently, potential cures are limited to allogeneic stem cell transplantation and unilesional radiotherapy for advanced MF; however, no treatment has been found to notably improve survival rates.1 Mycosis fungoides can result in various complications including diffuse spread of a skin infection caused by HSV, known as KVE.1 Kaposi varicelliform eruption usually manifests clinically with painful skin vesicles that often are accompanied by systemic signs such as fever and malaise. The vesicles rapidly progress into pustules or erosions, predominantly affecting regions such as the head, neck, groin, and upper torso (Figure 1).2 Kaposi varicelliform eruption is considered a dermatologic emergency due to its potential to precipitate serious complications such as life-threatening secondary bacterial infection, HSV viremia, and multiorgan involvement; it also carries the risk of instigating ocular complications, such as keratitis, conjunctivitis, blepharitis, uveitis, and potential vision loss.2
Kaposi varicelliform eruption usually is diagnosed through clinical examination supported by polymerase chain reaction, viral culture, histopathology, HSV serology, and Tzanck smear.2 The differential diagnosis includes varicella, atypical varicella, herpes genitalis, herpes zoster, allergic or irritant contact dermatitis, or MF, which may result in painful skin ulcers.2-4 If an HSV superinfection is suspected, a polymerase chain reaction test ideally should be conducted within the first 72 hours of symptom onset.2 Herpes simplex virus infection may be reinforced by histologic features such as intraepidermal blistering, acantholysis, keratinocyte ballooning degeneration, and multinuclear giant cells with intranuclear inclusions. Given its severe nature, immediate empiric antiviral treatment for KVE is essential, even while awaiting confirmatory tests. The recommended treatment protocol involves acyclovir (400 mg orally 3 times daily or 10 mg/kg IV) or valacyclovir (500 mg orally twice daily), continued until KVE resolves.2
Herpes genitalis caused by HSV-2 is estimated to affect approximately 45 million adults in the United States.2 First-line treatment for HSV-2 includes acyclovir and its derivatives, which are viral nucleoside analogs that inhibit viral DNA polymerases.5,6 However, over the past 2 decades, increasing HSV resistance to acyclovir and its derivatives has been noted among immunocompromised patients.5,6 Second-line agents, such as IV foscarnet and cidofovir, require close laboratory monitoring for nephrotoxicity and are contraindicated in those with renal insufficiency, thus limiting their use.5 To combat acyclovir resistance, novel antivirals such as pritelivir are being developed. Pritelivir targets the HSV helicase-primase complex and has been shown to outperform acyclovir in in-vitro animal models.7 Due to its unique mechanism of action (Figure 2), pritelivir is effective against acyclovir-resistant HSV strains, and clinical trials suggest its serum half-life may allow for daily dosing. A phase 2 study showed pritelivir reduced viral shedding days, sped up genital lesion healing in adults infected with HSV-2, and exhibited a good safety profile.7 Our patient participated in ongoing open-label trials of pritelivir that aimed to assess its efficacy and safety in immunocompromised patients. Given the limited alternative treatments for acyclovir-resistant HSV-2, clinicians need to stay updated on antiviral agents under development.
THE DIAGNOSIS: Kaposi Varicelliform Eruption
Genital erosions tested positive for herpes simplex virus (HSV) 2 via PCR, confirming a Kaposi varicelliform eruption (KVE) in a patient with mycosis fungoides. The medical team began antiviral therapy with intravenous (IV) acyclovir; however, susceptibility testing during the hospital admission confirmed acyclovir resistance, requiring a transition to cidofovir cream 1% and IV foscarnet.1 Subsequent concerns by the care team about chemical burns, dysuria, and renal impairment led to discontinuation of both the cidofovir and foscarnet, considerably narrowing the treatment options.1 The patient’s condition was complicated by polymicrobial bacteremia. Additionally, worsening acidosis and acute kidney injury required initiation of continuous renal replacement therapy.1 Considering these conditions, the patient was enrolled in a promising clinical trial for pritelivir, a novel antiviral medication; however, due to the development of oliguria and the progression of renal failure, this course of treatment had to be discontinued. Faced with potential viral encephalitis, the infectious disease team concluded that, despite previous adverse reactions, resumption of IV foscarnet treatment would present more benefits than risks, given the patient’s critical situation.1
Mycosis fungoides (MF) is a slowly progressive cutaneous T-cell lymphoma of CD4+ cells that primarily affects the skin. Clinically, it often is characterized by pruritic scaly patches or plaques with sharply demarcated borders, the enduring nature of which consistently poses a therapeutic challenge due to their noted resistance to preliminary lines of treatment. Presently, potential cures are limited to allogeneic stem cell transplantation and unilesional radiotherapy for advanced MF; however, no treatment has been found to notably improve survival rates.1 Mycosis fungoides can result in various complications including diffuse spread of a skin infection caused by HSV, known as KVE.1 Kaposi varicelliform eruption usually manifests clinically with painful skin vesicles that often are accompanied by systemic signs such as fever and malaise. The vesicles rapidly progress into pustules or erosions, predominantly affecting regions such as the head, neck, groin, and upper torso (Figure 1).2 Kaposi varicelliform eruption is considered a dermatologic emergency due to its potential to precipitate serious complications such as life-threatening secondary bacterial infection, HSV viremia, and multiorgan involvement; it also carries the risk of instigating ocular complications, such as keratitis, conjunctivitis, blepharitis, uveitis, and potential vision loss.2
Kaposi varicelliform eruption usually is diagnosed through clinical examination supported by polymerase chain reaction, viral culture, histopathology, HSV serology, and Tzanck smear.2 The differential diagnosis includes varicella, atypical varicella, herpes genitalis, herpes zoster, allergic or irritant contact dermatitis, or MF, which may result in painful skin ulcers.2-4 If an HSV superinfection is suspected, a polymerase chain reaction test ideally should be conducted within the first 72 hours of symptom onset.2 Herpes simplex virus infection may be reinforced by histologic features such as intraepidermal blistering, acantholysis, keratinocyte ballooning degeneration, and multinuclear giant cells with intranuclear inclusions. Given its severe nature, immediate empiric antiviral treatment for KVE is essential, even while awaiting confirmatory tests. The recommended treatment protocol involves acyclovir (400 mg orally 3 times daily or 10 mg/kg IV) or valacyclovir (500 mg orally twice daily), continued until KVE resolves.2
Herpes genitalis caused by HSV-2 is estimated to affect approximately 45 million adults in the United States.2 First-line treatment for HSV-2 includes acyclovir and its derivatives, which are viral nucleoside analogs that inhibit viral DNA polymerases.5,6 However, over the past 2 decades, increasing HSV resistance to acyclovir and its derivatives has been noted among immunocompromised patients.5,6 Second-line agents, such as IV foscarnet and cidofovir, require close laboratory monitoring for nephrotoxicity and are contraindicated in those with renal insufficiency, thus limiting their use.5 To combat acyclovir resistance, novel antivirals such as pritelivir are being developed. Pritelivir targets the HSV helicase-primase complex and has been shown to outperform acyclovir in in-vitro animal models.7 Due to its unique mechanism of action (Figure 2), pritelivir is effective against acyclovir-resistant HSV strains, and clinical trials suggest its serum half-life may allow for daily dosing. A phase 2 study showed pritelivir reduced viral shedding days, sped up genital lesion healing in adults infected with HSV-2, and exhibited a good safety profile.7 Our patient participated in ongoing open-label trials of pritelivir that aimed to assess its efficacy and safety in immunocompromised patients. Given the limited alternative treatments for acyclovir-resistant HSV-2, clinicians need to stay updated on antiviral agents under development.
- García-Díaz N, Piris MÁ, Ortiz-Romero PL, et al. Mycosis fungoides and Sézary syndrome: an integrative review of the pathophysiology, molecular drivers, and targeted therapy. Cancers. 2021;13:1931. doi:10.3390/cancers13081931
- Baaniya B, Agrawal S. Kaposi varicelliform eruption in a patient with pemphigus vulgaris: a case report and review of the literature. Case Rep Dermatol Med. 2020;2020:6695342. doi:10.1155/2020/6695342
- Shin D, Lee MS, Kim DY, et al. Increased large unstained cells value in varicella patients: a valuable parameter to aid rapid diagnosis of varicella infection. J Dermatol. 2015;42:795-799. doi:10.1111
- Joshi A, Sah SP, Agrawal S. Kaposi’s varicelliform eruption or atypical chickenpox in a normal individual. Australas J Dermatol. 2000;41:126-127.
- Groves MJ. Genital herpes: a review. Am Fam Physician. 2016; 93:928-934.
- Fleming DT, Leone P, Esposito D, et al. Herpes virus type 2 infection and genital symptoms in primary care patients. Sex Transm Dis. 2006;33:416-421. doi:10.1097/01.olq.0000200578.86276.0b
- Poole CL, James SH. Antiviral therapies for herpesviruses: current agents and new directions. Clin Ther. 2018;40:1282-1298. doi:10.1016 /j.clinthera.2018.07.006.
- García-Díaz N, Piris MÁ, Ortiz-Romero PL, et al. Mycosis fungoides and Sézary syndrome: an integrative review of the pathophysiology, molecular drivers, and targeted therapy. Cancers. 2021;13:1931. doi:10.3390/cancers13081931
- Baaniya B, Agrawal S. Kaposi varicelliform eruption in a patient with pemphigus vulgaris: a case report and review of the literature. Case Rep Dermatol Med. 2020;2020:6695342. doi:10.1155/2020/6695342
- Shin D, Lee MS, Kim DY, et al. Increased large unstained cells value in varicella patients: a valuable parameter to aid rapid diagnosis of varicella infection. J Dermatol. 2015;42:795-799. doi:10.1111
- Joshi A, Sah SP, Agrawal S. Kaposi’s varicelliform eruption or atypical chickenpox in a normal individual. Australas J Dermatol. 2000;41:126-127.
- Groves MJ. Genital herpes: a review. Am Fam Physician. 2016; 93:928-934.
- Fleming DT, Leone P, Esposito D, et al. Herpes virus type 2 infection and genital symptoms in primary care patients. Sex Transm Dis. 2006;33:416-421. doi:10.1097/01.olq.0000200578.86276.0b
- Poole CL, James SH. Antiviral therapies for herpesviruses: current agents and new directions. Clin Ther. 2018;40:1282-1298. doi:10.1016 /j.clinthera.2018.07.006.
Painful Edematous Labial Erosions
Painful Edematous Labial Erosions
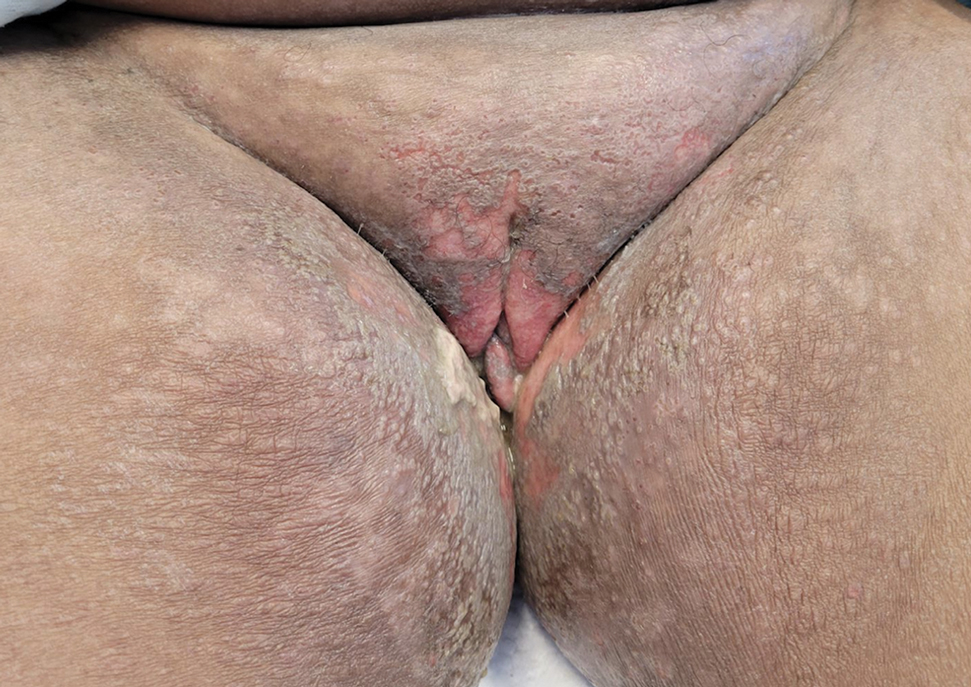
A 40-year-old woman presented to the emergency department with painful, well-defined, edematous labial erosions of several weeks’ duration. The patient reported a medical history of stage IIIA (T4N0M0B0) mycosis fungoides. She had been hospitalized 2 months prior for sepsis that was attributed to a polymicrobial bacteremia involving Acinetobacter baumannii and Staphylococcus epidermidis. During that admission, a polymerase chain reaction test conducted on a skin swab from a lesion on the labia majora confirmed the presence of herpes simplex virus type 2. At the current presentation, physical examination by dermatology also revealed discrete, coalescing, erythematous erosions on the buttocks, groin, and proximal thighs with a punched-out appearance. These areas also exhibited skin sloughing and were covered with a gray-brown exudate. No other mucosal surfaces were involved.

Generalized Erythematous Plaques and Pustules in a Pregnant Patient
Generalized Erythematous Plaques and Pustules in a Pregnant Patient
THE DIAGNOSIS: Impetigo Herpetiformis
Histopathology revealed epidermal acanthosis and spongiosis with overlying parakeratosis associated with subcorneal and intracorneal neutrophils, papillary dermal edema, and dermal mixed inflammation with neutrophils and eosinophils. Both direct immunofluorescence and periodic acid–Schiff studies were negative. Blood and pustule cultures were sterile and the skin flora were normal. Based on these findings, a diagnosis of impetigo herpetiformis (IH) was made. The condition improved with systemic and topical steroids, supportive care, and an intravenous infusion of infliximab 5 mg/kg. At 3 weeks’ follow-up, the patient demonstrated near-complete resolution and later delivered successfully at 40 weeks’ gestation without complications.
Impetigo herpetiformis, also known as pustular psoriasis of pregnancy, is an exceedingly rare gestational dermatosis that typically manifests in the third trimester and can be life-threatening for both the mother and fetus. The term was first used in 1872 to describe 5 pregnant women with extensive acute pustular eruptions, all in unstable condition; 4 (80%)of the cases resulted in maternal death, and all resulted in fetal death.1 Impetigo herpetiformis is characterized by pruritic and painful erythematous patches studded at the periphery with subcorneal pustules. Eruptions usually occur in the flexural areas and spread centrifugally, with extension of the lesions peripherally as the center erodes and crusts. Sparing of the face, palms, and soles is expected, and mucosal involvement is rare. Generalized involvement and exfoliation may occur in extreme cases.2 While IH typically manifests during the third trimester, it may occur any time throughout pregnancy or immediately postpartum.3 A few cases have been reported in the puerperium.2 Common symptoms include fever, chills, malaise, anorexia, nausea, vomiting, diarrhea, and arthralgias. Less common complications include hypoalbuminemia and severe hypocalcemia leading to tetany, seizures, and delirium.2,3 While maternal mortality is uncommon, fetal mortality often is a more pressing risk and is attributed to placental insufficiency.3,4 For this reason, early delivery commonly is considered in severe cases.
Whether IH is a separate entity or a variant of pustular psoriasis remains heavily debated. Although the histopathology of IH is identical to pustular psoriasis, the lack of a personal and family history of psoriasis, symptom resolution with delivery, and possible recurrence during successive pregnancies help differentiate IH from generalized pustular psoriasis.2,5 Earlier onset, diffuse involvement, faster progression, and recurrence in subsequent pregnancies all have been linked to a worse prognosis.4
The differential diagnosis for IH includes acute generalized exanthematous pustulosis, pemphigoid gestationis, dermatitis herpetiformis, and subcorneal pustular dermatosis. Acute generalized exanthematous pustulosis is an uncommon severe cutaneous drug reaction characterized by the sudden onset of numerous sterile pustules on erythematous skin within 48 hours of exposure. The most common offending medications include pristinamycin and beta-lactam antibiotics. A high fever, neutrophilic leukocytosis, and hypocalcemia often accompany acute generalized exanthematous pustulosis.6 Prompt diagnosis and withdrawal of the offending drug as well as supportive care and symptomatic treatment are crucial for disease management, as systemic symptoms and even organ involvement may occur.6
Pemphigoid gestationis, also known as gestational pemphigoid or herpes gestationis, is a rare autoimmune blistering disorder that primarily affects pregnant women. It typically manifests in the second or third trimester or shortly after delivery. Clinically, it manifests as an intensely pruritic polymorphic eruption of urticarial papules and plaques accompanied by vesicles and bullae and often is distributed on the abdomen and extends to other body regions. Although the exact etiology is unknown, pemphigoid gestationis is caused by autoantibodies targeting the BP180 and BP230 hemidesmosomal proteins.7 Treatment usually involves systemic corticosteroids and may require additional immunosuppressive therapy. In most cases, patients see resolution within 6 months of delivery.7
Dermatitis herpetiformis is a chronic autoimmune blistering skin disorder characterized by intensely pruritic, grouped vesicles and papules, often distributed symmetrically on extensor surfaces such as the elbows, knees, buttocks, and back. It is closely associated with celiac disease and is triggered by gluten ingestion in genetically predisposed individuals with human leukocyte antigen DQ2 and DQ8 haplotypes. Dermatitis herpetiformis is caused by deposition of IgA antibodies that target tissue transglutaminase 3 at the dermal papillae, leading to inflammation and blister formation. 8 Treatment typically involves a gluten-free diet and medications such as dapsone to alleviate symptoms and prevent recurrence.
Subcorneal pustular dermatosis, also known as Sneddon-Wilkinson disease, is a rare chronic relapsing pustular dermatosis characterized by sterile superficial pustules arranged in annular or circinate patterns on erythematous plaques. It predominantly affects middleaged women and often is associated with underlying conditions such as IgA gammopathy or monoclonal gammopathy of undetermined significance. The pathogenesis remains unclear, but immune dysregulation is thought to play a role. Some authors still question whether subcorneal pustular dermatosis is a distinct entity from pustular psoriasis.4,5,12 Dapsone is the preferred first-line treatment, with adjunct therapies including topical or systemic corticosteroids, other immunosuppressive agents, tumor necrosis factor inhibitors, and UV light therapy.9
Definitive management of IH is achieved through early delivery; however, systemic corticosteroids often are used in varying doses to control the disease and to extend the pregnancy period closer to term or until delivery is considered viable. Additional therapies that can be considered include infliximab, cyclosporine, and topical corticosteroids, in conjunction with fluid and electrolyte maintenance.2,4,10 If symptoms persist despite supportive care and pharmacologic intervention, induction of labor or termination of pregnancy may be indicated. In nonbreastfeeding postpartum mothers with persistent disease, therapies commonly used in generalized pustular psoriasis may be given.11
- Hebra F. Ueber einzelne wahrend Schwangerschaft, des wacherbette unde bei uterinal. Krankheiten der Frauen zu beobachtende Hautkrankheiten. Wien Med Wochenschr. 1872;48:1197-1202.
- Fouda UM, Fouda RM, Ammar HM, et al. Impetigo herpetiformis during the puerperium triggered by secondary hypoparathyroidism: a case report. Cases J. 2009;2:9338. doi:10.1186/1757-1626-2-9338
- Kroumpouzos G, Cohen LM. Dermatoses of pregnancy. J Am Acad Dermatol. 2001;45:1-22. doi:10.1067/mjd.2001.114595
- Liu J, Ali K, Lou H, et al. First-trimester impetigo herpetiformis leads to stillbirth: a case report. Dermatol Ther (Heidelb). 2022;12:1271-1279. doi:10.1007/s13555-022-00735-9
- Lotem M, Katzenelson V, Rotem A, et al. Impetigo herpetiformis: a variant of pustular psoriasis or a separate entity? J Am Acad Dermatol. 1989;20:338-41. doi:10.1016/s0190-9622(89)70042-6
- Stadler PC, Oschmann A, Kerl-French K, et al. Acute generalized exanthematous pustulosis: clinical characteristics, pathogenesis, and management. Dermatology. 2023;239:328-333. doi:10.1159/000529218
- Abdelhafez MMA, Ahmed KAM, Daud MNBM, et al. Pemphigoid gestationis and adverse pregnancy outcomes: a literature review. J Gynecol Obstet Hum Reprod. 2022;51:102370. doi:10.1016 /j.jogoh.2022.102370
- Reunala T, Hervonen K, Salmi T. Dermatitis herpetiformis: an update on diagnosis and management. Am J Clin Dermatol. 2021;22:329-338. doi:10.1007/s40257-020-00584-2
- Watts PJ, Khachemoune A. Subcorneal pustular dermatosis: a review of 30 years of progress. Am J Clin Dermatol. 2016;17:653-671. doi:10.1007 /s40257-016-0202-8
- Robinson A, Van Voorhees AS, Hsu S, et al. Treatment of pustular psoriasis: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol. 2012;67:279-288. doi:10.1016/j.jaad.2011.01.032
- Bukhari IA. Impetigo herpetiformis in a primigravida: successful treatment with etanercept. J Drugs Dermatol. 2004;3:449-451.
- Chang SE, Kim HH, Choi JH, et al. Impetigo herpetiformis followed by generalized pustular psoriasis: more evidence of same disease entity. Int J Dermatol. 2003;42(9):754-755.
THE DIAGNOSIS: Impetigo Herpetiformis
Histopathology revealed epidermal acanthosis and spongiosis with overlying parakeratosis associated with subcorneal and intracorneal neutrophils, papillary dermal edema, and dermal mixed inflammation with neutrophils and eosinophils. Both direct immunofluorescence and periodic acid–Schiff studies were negative. Blood and pustule cultures were sterile and the skin flora were normal. Based on these findings, a diagnosis of impetigo herpetiformis (IH) was made. The condition improved with systemic and topical steroids, supportive care, and an intravenous infusion of infliximab 5 mg/kg. At 3 weeks’ follow-up, the patient demonstrated near-complete resolution and later delivered successfully at 40 weeks’ gestation without complications.
Impetigo herpetiformis, also known as pustular psoriasis of pregnancy, is an exceedingly rare gestational dermatosis that typically manifests in the third trimester and can be life-threatening for both the mother and fetus. The term was first used in 1872 to describe 5 pregnant women with extensive acute pustular eruptions, all in unstable condition; 4 (80%)of the cases resulted in maternal death, and all resulted in fetal death.1 Impetigo herpetiformis is characterized by pruritic and painful erythematous patches studded at the periphery with subcorneal pustules. Eruptions usually occur in the flexural areas and spread centrifugally, with extension of the lesions peripherally as the center erodes and crusts. Sparing of the face, palms, and soles is expected, and mucosal involvement is rare. Generalized involvement and exfoliation may occur in extreme cases.2 While IH typically manifests during the third trimester, it may occur any time throughout pregnancy or immediately postpartum.3 A few cases have been reported in the puerperium.2 Common symptoms include fever, chills, malaise, anorexia, nausea, vomiting, diarrhea, and arthralgias. Less common complications include hypoalbuminemia and severe hypocalcemia leading to tetany, seizures, and delirium.2,3 While maternal mortality is uncommon, fetal mortality often is a more pressing risk and is attributed to placental insufficiency.3,4 For this reason, early delivery commonly is considered in severe cases.
Whether IH is a separate entity or a variant of pustular psoriasis remains heavily debated. Although the histopathology of IH is identical to pustular psoriasis, the lack of a personal and family history of psoriasis, symptom resolution with delivery, and possible recurrence during successive pregnancies help differentiate IH from generalized pustular psoriasis.2,5 Earlier onset, diffuse involvement, faster progression, and recurrence in subsequent pregnancies all have been linked to a worse prognosis.4
The differential diagnosis for IH includes acute generalized exanthematous pustulosis, pemphigoid gestationis, dermatitis herpetiformis, and subcorneal pustular dermatosis. Acute generalized exanthematous pustulosis is an uncommon severe cutaneous drug reaction characterized by the sudden onset of numerous sterile pustules on erythematous skin within 48 hours of exposure. The most common offending medications include pristinamycin and beta-lactam antibiotics. A high fever, neutrophilic leukocytosis, and hypocalcemia often accompany acute generalized exanthematous pustulosis.6 Prompt diagnosis and withdrawal of the offending drug as well as supportive care and symptomatic treatment are crucial for disease management, as systemic symptoms and even organ involvement may occur.6
Pemphigoid gestationis, also known as gestational pemphigoid or herpes gestationis, is a rare autoimmune blistering disorder that primarily affects pregnant women. It typically manifests in the second or third trimester or shortly after delivery. Clinically, it manifests as an intensely pruritic polymorphic eruption of urticarial papules and plaques accompanied by vesicles and bullae and often is distributed on the abdomen and extends to other body regions. Although the exact etiology is unknown, pemphigoid gestationis is caused by autoantibodies targeting the BP180 and BP230 hemidesmosomal proteins.7 Treatment usually involves systemic corticosteroids and may require additional immunosuppressive therapy. In most cases, patients see resolution within 6 months of delivery.7
Dermatitis herpetiformis is a chronic autoimmune blistering skin disorder characterized by intensely pruritic, grouped vesicles and papules, often distributed symmetrically on extensor surfaces such as the elbows, knees, buttocks, and back. It is closely associated with celiac disease and is triggered by gluten ingestion in genetically predisposed individuals with human leukocyte antigen DQ2 and DQ8 haplotypes. Dermatitis herpetiformis is caused by deposition of IgA antibodies that target tissue transglutaminase 3 at the dermal papillae, leading to inflammation and blister formation. 8 Treatment typically involves a gluten-free diet and medications such as dapsone to alleviate symptoms and prevent recurrence.
Subcorneal pustular dermatosis, also known as Sneddon-Wilkinson disease, is a rare chronic relapsing pustular dermatosis characterized by sterile superficial pustules arranged in annular or circinate patterns on erythematous plaques. It predominantly affects middleaged women and often is associated with underlying conditions such as IgA gammopathy or monoclonal gammopathy of undetermined significance. The pathogenesis remains unclear, but immune dysregulation is thought to play a role. Some authors still question whether subcorneal pustular dermatosis is a distinct entity from pustular psoriasis.4,5,12 Dapsone is the preferred first-line treatment, with adjunct therapies including topical or systemic corticosteroids, other immunosuppressive agents, tumor necrosis factor inhibitors, and UV light therapy.9
Definitive management of IH is achieved through early delivery; however, systemic corticosteroids often are used in varying doses to control the disease and to extend the pregnancy period closer to term or until delivery is considered viable. Additional therapies that can be considered include infliximab, cyclosporine, and topical corticosteroids, in conjunction with fluid and electrolyte maintenance.2,4,10 If symptoms persist despite supportive care and pharmacologic intervention, induction of labor or termination of pregnancy may be indicated. In nonbreastfeeding postpartum mothers with persistent disease, therapies commonly used in generalized pustular psoriasis may be given.11
THE DIAGNOSIS: Impetigo Herpetiformis
Histopathology revealed epidermal acanthosis and spongiosis with overlying parakeratosis associated with subcorneal and intracorneal neutrophils, papillary dermal edema, and dermal mixed inflammation with neutrophils and eosinophils. Both direct immunofluorescence and periodic acid–Schiff studies were negative. Blood and pustule cultures were sterile and the skin flora were normal. Based on these findings, a diagnosis of impetigo herpetiformis (IH) was made. The condition improved with systemic and topical steroids, supportive care, and an intravenous infusion of infliximab 5 mg/kg. At 3 weeks’ follow-up, the patient demonstrated near-complete resolution and later delivered successfully at 40 weeks’ gestation without complications.
Impetigo herpetiformis, also known as pustular psoriasis of pregnancy, is an exceedingly rare gestational dermatosis that typically manifests in the third trimester and can be life-threatening for both the mother and fetus. The term was first used in 1872 to describe 5 pregnant women with extensive acute pustular eruptions, all in unstable condition; 4 (80%)of the cases resulted in maternal death, and all resulted in fetal death.1 Impetigo herpetiformis is characterized by pruritic and painful erythematous patches studded at the periphery with subcorneal pustules. Eruptions usually occur in the flexural areas and spread centrifugally, with extension of the lesions peripherally as the center erodes and crusts. Sparing of the face, palms, and soles is expected, and mucosal involvement is rare. Generalized involvement and exfoliation may occur in extreme cases.2 While IH typically manifests during the third trimester, it may occur any time throughout pregnancy or immediately postpartum.3 A few cases have been reported in the puerperium.2 Common symptoms include fever, chills, malaise, anorexia, nausea, vomiting, diarrhea, and arthralgias. Less common complications include hypoalbuminemia and severe hypocalcemia leading to tetany, seizures, and delirium.2,3 While maternal mortality is uncommon, fetal mortality often is a more pressing risk and is attributed to placental insufficiency.3,4 For this reason, early delivery commonly is considered in severe cases.
Whether IH is a separate entity or a variant of pustular psoriasis remains heavily debated. Although the histopathology of IH is identical to pustular psoriasis, the lack of a personal and family history of psoriasis, symptom resolution with delivery, and possible recurrence during successive pregnancies help differentiate IH from generalized pustular psoriasis.2,5 Earlier onset, diffuse involvement, faster progression, and recurrence in subsequent pregnancies all have been linked to a worse prognosis.4
The differential diagnosis for IH includes acute generalized exanthematous pustulosis, pemphigoid gestationis, dermatitis herpetiformis, and subcorneal pustular dermatosis. Acute generalized exanthematous pustulosis is an uncommon severe cutaneous drug reaction characterized by the sudden onset of numerous sterile pustules on erythematous skin within 48 hours of exposure. The most common offending medications include pristinamycin and beta-lactam antibiotics. A high fever, neutrophilic leukocytosis, and hypocalcemia often accompany acute generalized exanthematous pustulosis.6 Prompt diagnosis and withdrawal of the offending drug as well as supportive care and symptomatic treatment are crucial for disease management, as systemic symptoms and even organ involvement may occur.6
Pemphigoid gestationis, also known as gestational pemphigoid or herpes gestationis, is a rare autoimmune blistering disorder that primarily affects pregnant women. It typically manifests in the second or third trimester or shortly after delivery. Clinically, it manifests as an intensely pruritic polymorphic eruption of urticarial papules and plaques accompanied by vesicles and bullae and often is distributed on the abdomen and extends to other body regions. Although the exact etiology is unknown, pemphigoid gestationis is caused by autoantibodies targeting the BP180 and BP230 hemidesmosomal proteins.7 Treatment usually involves systemic corticosteroids and may require additional immunosuppressive therapy. In most cases, patients see resolution within 6 months of delivery.7
Dermatitis herpetiformis is a chronic autoimmune blistering skin disorder characterized by intensely pruritic, grouped vesicles and papules, often distributed symmetrically on extensor surfaces such as the elbows, knees, buttocks, and back. It is closely associated with celiac disease and is triggered by gluten ingestion in genetically predisposed individuals with human leukocyte antigen DQ2 and DQ8 haplotypes. Dermatitis herpetiformis is caused by deposition of IgA antibodies that target tissue transglutaminase 3 at the dermal papillae, leading to inflammation and blister formation. 8 Treatment typically involves a gluten-free diet and medications such as dapsone to alleviate symptoms and prevent recurrence.
Subcorneal pustular dermatosis, also known as Sneddon-Wilkinson disease, is a rare chronic relapsing pustular dermatosis characterized by sterile superficial pustules arranged in annular or circinate patterns on erythematous plaques. It predominantly affects middleaged women and often is associated with underlying conditions such as IgA gammopathy or monoclonal gammopathy of undetermined significance. The pathogenesis remains unclear, but immune dysregulation is thought to play a role. Some authors still question whether subcorneal pustular dermatosis is a distinct entity from pustular psoriasis.4,5,12 Dapsone is the preferred first-line treatment, with adjunct therapies including topical or systemic corticosteroids, other immunosuppressive agents, tumor necrosis factor inhibitors, and UV light therapy.9
Definitive management of IH is achieved through early delivery; however, systemic corticosteroids often are used in varying doses to control the disease and to extend the pregnancy period closer to term or until delivery is considered viable. Additional therapies that can be considered include infliximab, cyclosporine, and topical corticosteroids, in conjunction with fluid and electrolyte maintenance.2,4,10 If symptoms persist despite supportive care and pharmacologic intervention, induction of labor or termination of pregnancy may be indicated. In nonbreastfeeding postpartum mothers with persistent disease, therapies commonly used in generalized pustular psoriasis may be given.11
- Hebra F. Ueber einzelne wahrend Schwangerschaft, des wacherbette unde bei uterinal. Krankheiten der Frauen zu beobachtende Hautkrankheiten. Wien Med Wochenschr. 1872;48:1197-1202.
- Fouda UM, Fouda RM, Ammar HM, et al. Impetigo herpetiformis during the puerperium triggered by secondary hypoparathyroidism: a case report. Cases J. 2009;2:9338. doi:10.1186/1757-1626-2-9338
- Kroumpouzos G, Cohen LM. Dermatoses of pregnancy. J Am Acad Dermatol. 2001;45:1-22. doi:10.1067/mjd.2001.114595
- Liu J, Ali K, Lou H, et al. First-trimester impetigo herpetiformis leads to stillbirth: a case report. Dermatol Ther (Heidelb). 2022;12:1271-1279. doi:10.1007/s13555-022-00735-9
- Lotem M, Katzenelson V, Rotem A, et al. Impetigo herpetiformis: a variant of pustular psoriasis or a separate entity? J Am Acad Dermatol. 1989;20:338-41. doi:10.1016/s0190-9622(89)70042-6
- Stadler PC, Oschmann A, Kerl-French K, et al. Acute generalized exanthematous pustulosis: clinical characteristics, pathogenesis, and management. Dermatology. 2023;239:328-333. doi:10.1159/000529218
- Abdelhafez MMA, Ahmed KAM, Daud MNBM, et al. Pemphigoid gestationis and adverse pregnancy outcomes: a literature review. J Gynecol Obstet Hum Reprod. 2022;51:102370. doi:10.1016 /j.jogoh.2022.102370
- Reunala T, Hervonen K, Salmi T. Dermatitis herpetiformis: an update on diagnosis and management. Am J Clin Dermatol. 2021;22:329-338. doi:10.1007/s40257-020-00584-2
- Watts PJ, Khachemoune A. Subcorneal pustular dermatosis: a review of 30 years of progress. Am J Clin Dermatol. 2016;17:653-671. doi:10.1007 /s40257-016-0202-8
- Robinson A, Van Voorhees AS, Hsu S, et al. Treatment of pustular psoriasis: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol. 2012;67:279-288. doi:10.1016/j.jaad.2011.01.032
- Bukhari IA. Impetigo herpetiformis in a primigravida: successful treatment with etanercept. J Drugs Dermatol. 2004;3:449-451.
- Chang SE, Kim HH, Choi JH, et al. Impetigo herpetiformis followed by generalized pustular psoriasis: more evidence of same disease entity. Int J Dermatol. 2003;42(9):754-755.
- Hebra F. Ueber einzelne wahrend Schwangerschaft, des wacherbette unde bei uterinal. Krankheiten der Frauen zu beobachtende Hautkrankheiten. Wien Med Wochenschr. 1872;48:1197-1202.
- Fouda UM, Fouda RM, Ammar HM, et al. Impetigo herpetiformis during the puerperium triggered by secondary hypoparathyroidism: a case report. Cases J. 2009;2:9338. doi:10.1186/1757-1626-2-9338
- Kroumpouzos G, Cohen LM. Dermatoses of pregnancy. J Am Acad Dermatol. 2001;45:1-22. doi:10.1067/mjd.2001.114595
- Liu J, Ali K, Lou H, et al. First-trimester impetigo herpetiformis leads to stillbirth: a case report. Dermatol Ther (Heidelb). 2022;12:1271-1279. doi:10.1007/s13555-022-00735-9
- Lotem M, Katzenelson V, Rotem A, et al. Impetigo herpetiformis: a variant of pustular psoriasis or a separate entity? J Am Acad Dermatol. 1989;20:338-41. doi:10.1016/s0190-9622(89)70042-6
- Stadler PC, Oschmann A, Kerl-French K, et al. Acute generalized exanthematous pustulosis: clinical characteristics, pathogenesis, and management. Dermatology. 2023;239:328-333. doi:10.1159/000529218
- Abdelhafez MMA, Ahmed KAM, Daud MNBM, et al. Pemphigoid gestationis and adverse pregnancy outcomes: a literature review. J Gynecol Obstet Hum Reprod. 2022;51:102370. doi:10.1016 /j.jogoh.2022.102370
- Reunala T, Hervonen K, Salmi T. Dermatitis herpetiformis: an update on diagnosis and management. Am J Clin Dermatol. 2021;22:329-338. doi:10.1007/s40257-020-00584-2
- Watts PJ, Khachemoune A. Subcorneal pustular dermatosis: a review of 30 years of progress. Am J Clin Dermatol. 2016;17:653-671. doi:10.1007 /s40257-016-0202-8
- Robinson A, Van Voorhees AS, Hsu S, et al. Treatment of pustular psoriasis: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol. 2012;67:279-288. doi:10.1016/j.jaad.2011.01.032
- Bukhari IA. Impetigo herpetiformis in a primigravida: successful treatment with etanercept. J Drugs Dermatol. 2004;3:449-451.
- Chang SE, Kim HH, Choi JH, et al. Impetigo herpetiformis followed by generalized pustular psoriasis: more evidence of same disease entity. Int J Dermatol. 2003;42(9):754-755.
Generalized Erythematous Plaques and Pustules in a Pregnant Patient
Generalized Erythematous Plaques and Pustules in a Pregnant Patient
A 17-year-old girl was admitted to the hospital at 19 weeks' gestation for a widespread eruption of erythematous plaques with pustules covering more than 60% of the body and signs of sepsis. The rash initially appeared as a few small spots on the upper chest and under the breasts 5 weeks prior to hospital admission with subsequent spread to the abdomen and groin. At admission, the patient had a mild fever and tachycardia. She reported a history of eczema, herpes simplex virus, and intertrigo. Physical examination performed by dermatology revealed generalized erythematous plaques with pustule-studded margins and overlying scale involving the neck, torso, arms, and legs favoring the flexural areas. There was no involvement of the face, eyes, oral mucosa, palms, soles, or nails. Laboratory testing revealed hypoalbuminemia (2.4 g/dL [reference range, 3.5-5.5 g/dL]) and elevated inflammatory markers, including leukocytosis (15.83×103μL [reference range, 4.50- 11.00×103/μL]), absolute neutrophil count (12.87×103/μL [reference range, 1.50-8.00×103/μL]), and erythrocyte sedimentation rate (124 mm/h [reference range, 0-20 mm/h]). A culture from an abdominal pustule grew 1 colony of taphylococcus epidermidis, a suspected contaminant. A biopsy from a lesion on the right chest was performed.

Painless Nodule on the Lower Eyelid
Painless Nodule on the Lower Eyelid
THE DIAGNOSIS: Idiopathic Facial Aseptic Granuloma
Histopathology showed a ruptured follicle, perifollicular granulomatous inflammation, and admixed multinucleated giant cells in the superficial dermis. The deeper tissue exhibited edema, histiocytic/granulomatous inflammation forming ill-defined loose granulomas, and a single neutrophilic microabscess (Figure). Stains for periodic acid-Schiff with diastase and acid-fast bacillus were negative for microorganisms. The clinical examination and pathology findings supported a diagnosis of idiopathic facial aseptic granuloma (IFAG).
First reported in 1999, IFAG was described using the French term pyodermite froide du visage, which translates to “cold pyoderma of the face”; however, it was renamed to represent its granulomatous characteristics and noninfectious etiology.1 The pathogenesis of IFAG is unknown, but the leading hypothesis is that it may be a type of childhood granulomatous rosacea, given its association with relapsing chalazions, papulopustular eruptions on the face, and facial flushing.2 Other hypotheses are that IFAG is idiopathic or a granulomatous response to an insect bite, minor trauma, or embryologic remnant.3
A rare condition arising in early childhood, IFAG manifests as a single or multiple, painless, erythematous or violaceous nodule(s) on the face, most often on the cheeks or eyelids.4 A thorough history and clinical examination often suffice for diagnosis. Dermoscopy may reveal white perifollicular halos and follicular plugs on an erythematous base with linear vessels.4 If diagnostic tests are performed, there are notable characteristic findings: ultrasonography often shows a well-circumscribed, hypoechoic, ovoid dermal lesion without calcifications. Bacterial, fungal, and mycobacterial cultures commonly are negative.4 On biopsy, histopathology may reveal granulomatous inflammation in the superficial and deep dermis, multinucleated giant cells, and surrounding lymphocytic, neutrophilic, and eosinophilic infiltration with no calcium deposits.3,5,6 Histopathology findings for IFAG and rosacea lesions are similar; both may demonstrate folliculitis, perifollicular granulomas, and admixed lymphohistiocytic inflammation.7
Differentiating IFAG from other dermatologic lesions can be challenging, as the differential includes benign neoplasms (eg, dermoid cyst, chalazion, pilomatricoma, xanthoma, xanthogranuloma2) and infectious etiologies such as bacterial pyoderma and mycobacterial, fungal, and parasitic infections (eg, cutaneous leishmaniasis). Pilomatricomas, although often seen on the face or extremities in young girls, more often are well circumscribed and located in the dermis. Ultrasonography of a pilomatricoma classically shows variable foci of calcification. Xanthoma and xanthogranuloma also were considered in our case since the lesion was subtly yellowish on examination. Similar to IFAG, these conditions may manifest as single or multiple lesions. Abnormalities in the patient’s blood lipid panel or family history may be needed to diagnose xanthoma. Biopsy of a juvenile xanthogranuloma would exhibit a dense dermal nodular proliferation of histiocytic cells with a smaller number of admixed lymphocytes, neutrophils, and eosinophils, in contrast to the multiple smaller loose epithelioid granulomas seen in IFAG. Additional diagnoses in the differential for IFAG include pyogenic granuloma, Spitz nevus, nodulocystic infantile acne, granulomatous rosacea, and hemangioma.1,3,9 In particular, granulomatous rosacea is challenging to differentiate from IFAG given the overlapping clinical findings. Multiple lesions, the presence of papules and pustules, and associated rosacea symptoms such as flushing suggest a diagnosis of granulomatous rosacea over IFAG.2
The prognosis for IFAG is excellent; most lesions self-resolve without treatment or procedural intervention within 1 year without scarring or relapse.3 Topical and oral antibiotic treatments such as metronidazole 0.75% gel or cream, oral erythromycin, oral clarithromycin, and doxycycline (in patients older than 8 years) have been used to treat IFAG with variable clinic responses.2,3,6,8 Persistent IFAG has been treated with surgical excision.3 Our patient was treated with a combination of gentamicin ointment 0.3% and tacrolimus ointment 0.3% and experienced approximately 50% improvement in the first month of treatment.
- Roul S, Léauté-Labrèze C, Boralevi F, et al. Idiopathic aseptic facial granuloma (pyodermite froide du visage): a pediatric entity? Arch Dermatol. 2001;137:1253-1255.
- Prey S, Ezzedine K, Mazereeuw-Hautier J, et al. IFAG and childhood rosacea: a possible link? Pediatr Dermatol. 2013;30:429-432. doi:10.1111/pde.12137
- Boralevi F, Léauté-Labrèze C, Lepreux S, et al. Idiopathic facial aseptic granuloma: a multicentre prospective study of 30 cases. Br J Dermatol. 2007;156:705-708. doi:10.1111/j.1365-2133.2006.07741.x
- Lobato-Berezo A, Montoro-Romero S, Pujol RM, et al. Dermoscopic features of idiopathic facial aseptic granuloma. Pediatr Dermatol. 2018;35:E308-E309. doi:10.1111/pde.13582
- González Rodríguez AJ, Jordá Cuevas E. Idiopathic facial aseptic granuloma. Clin Exp Dermatol. 2015;40:298-300. doi:10.1111/ced.12535
- Orion C, Sfecci A, Tisseau L, et al. Idiopathic facial aseptic granuloma in a 13-year-old boy dramatically improved with oral doxycycline and topical metronidazole: evidence for a link with childhood rosacea. Case Rep Dermatol. 2016;8:197-201. doi:10.1159/000447624
- Neri I, Raone B, Dondi A, et al. Should idiopathic facial aseptic granuloma be considered granulomatous rosacea? report of three pediatric cases. Pediatr Dermatol. 2013;30:109-111. doi:10.1111 /j.1525-1470.2011.01689.x
- Miconi F, Principi N, Cassiani L, et al. A cheek nodule in a child: be aware of idiopathic facial aseptic granuloma and its differential diagnosis. Int J Environ Res Public Health. 2019;16:2471. doi:10.3390/ijerph16142471
- Baroni A, Russo T, Faccenda F, et al. Idiopathic facial aseptic granuloma in a child: a possible expression of childhood rosacea. Pediatr Dermatol. 2013;30:394-395. doi:10.1111/j.1525-1470.2012.01805.x
THE DIAGNOSIS: Idiopathic Facial Aseptic Granuloma
Histopathology showed a ruptured follicle, perifollicular granulomatous inflammation, and admixed multinucleated giant cells in the superficial dermis. The deeper tissue exhibited edema, histiocytic/granulomatous inflammation forming ill-defined loose granulomas, and a single neutrophilic microabscess (Figure). Stains for periodic acid-Schiff with diastase and acid-fast bacillus were negative for microorganisms. The clinical examination and pathology findings supported a diagnosis of idiopathic facial aseptic granuloma (IFAG).
First reported in 1999, IFAG was described using the French term pyodermite froide du visage, which translates to “cold pyoderma of the face”; however, it was renamed to represent its granulomatous characteristics and noninfectious etiology.1 The pathogenesis of IFAG is unknown, but the leading hypothesis is that it may be a type of childhood granulomatous rosacea, given its association with relapsing chalazions, papulopustular eruptions on the face, and facial flushing.2 Other hypotheses are that IFAG is idiopathic or a granulomatous response to an insect bite, minor trauma, or embryologic remnant.3
A rare condition arising in early childhood, IFAG manifests as a single or multiple, painless, erythematous or violaceous nodule(s) on the face, most often on the cheeks or eyelids.4 A thorough history and clinical examination often suffice for diagnosis. Dermoscopy may reveal white perifollicular halos and follicular plugs on an erythematous base with linear vessels.4 If diagnostic tests are performed, there are notable characteristic findings: ultrasonography often shows a well-circumscribed, hypoechoic, ovoid dermal lesion without calcifications. Bacterial, fungal, and mycobacterial cultures commonly are negative.4 On biopsy, histopathology may reveal granulomatous inflammation in the superficial and deep dermis, multinucleated giant cells, and surrounding lymphocytic, neutrophilic, and eosinophilic infiltration with no calcium deposits.3,5,6 Histopathology findings for IFAG and rosacea lesions are similar; both may demonstrate folliculitis, perifollicular granulomas, and admixed lymphohistiocytic inflammation.7
Differentiating IFAG from other dermatologic lesions can be challenging, as the differential includes benign neoplasms (eg, dermoid cyst, chalazion, pilomatricoma, xanthoma, xanthogranuloma2) and infectious etiologies such as bacterial pyoderma and mycobacterial, fungal, and parasitic infections (eg, cutaneous leishmaniasis). Pilomatricomas, although often seen on the face or extremities in young girls, more often are well circumscribed and located in the dermis. Ultrasonography of a pilomatricoma classically shows variable foci of calcification. Xanthoma and xanthogranuloma also were considered in our case since the lesion was subtly yellowish on examination. Similar to IFAG, these conditions may manifest as single or multiple lesions. Abnormalities in the patient’s blood lipid panel or family history may be needed to diagnose xanthoma. Biopsy of a juvenile xanthogranuloma would exhibit a dense dermal nodular proliferation of histiocytic cells with a smaller number of admixed lymphocytes, neutrophils, and eosinophils, in contrast to the multiple smaller loose epithelioid granulomas seen in IFAG. Additional diagnoses in the differential for IFAG include pyogenic granuloma, Spitz nevus, nodulocystic infantile acne, granulomatous rosacea, and hemangioma.1,3,9 In particular, granulomatous rosacea is challenging to differentiate from IFAG given the overlapping clinical findings. Multiple lesions, the presence of papules and pustules, and associated rosacea symptoms such as flushing suggest a diagnosis of granulomatous rosacea over IFAG.2
The prognosis for IFAG is excellent; most lesions self-resolve without treatment or procedural intervention within 1 year without scarring or relapse.3 Topical and oral antibiotic treatments such as metronidazole 0.75% gel or cream, oral erythromycin, oral clarithromycin, and doxycycline (in patients older than 8 years) have been used to treat IFAG with variable clinic responses.2,3,6,8 Persistent IFAG has been treated with surgical excision.3 Our patient was treated with a combination of gentamicin ointment 0.3% and tacrolimus ointment 0.3% and experienced approximately 50% improvement in the first month of treatment.
THE DIAGNOSIS: Idiopathic Facial Aseptic Granuloma
Histopathology showed a ruptured follicle, perifollicular granulomatous inflammation, and admixed multinucleated giant cells in the superficial dermis. The deeper tissue exhibited edema, histiocytic/granulomatous inflammation forming ill-defined loose granulomas, and a single neutrophilic microabscess (Figure). Stains for periodic acid-Schiff with diastase and acid-fast bacillus were negative for microorganisms. The clinical examination and pathology findings supported a diagnosis of idiopathic facial aseptic granuloma (IFAG).
First reported in 1999, IFAG was described using the French term pyodermite froide du visage, which translates to “cold pyoderma of the face”; however, it was renamed to represent its granulomatous characteristics and noninfectious etiology.1 The pathogenesis of IFAG is unknown, but the leading hypothesis is that it may be a type of childhood granulomatous rosacea, given its association with relapsing chalazions, papulopustular eruptions on the face, and facial flushing.2 Other hypotheses are that IFAG is idiopathic or a granulomatous response to an insect bite, minor trauma, or embryologic remnant.3
A rare condition arising in early childhood, IFAG manifests as a single or multiple, painless, erythematous or violaceous nodule(s) on the face, most often on the cheeks or eyelids.4 A thorough history and clinical examination often suffice for diagnosis. Dermoscopy may reveal white perifollicular halos and follicular plugs on an erythematous base with linear vessels.4 If diagnostic tests are performed, there are notable characteristic findings: ultrasonography often shows a well-circumscribed, hypoechoic, ovoid dermal lesion without calcifications. Bacterial, fungal, and mycobacterial cultures commonly are negative.4 On biopsy, histopathology may reveal granulomatous inflammation in the superficial and deep dermis, multinucleated giant cells, and surrounding lymphocytic, neutrophilic, and eosinophilic infiltration with no calcium deposits.3,5,6 Histopathology findings for IFAG and rosacea lesions are similar; both may demonstrate folliculitis, perifollicular granulomas, and admixed lymphohistiocytic inflammation.7
Differentiating IFAG from other dermatologic lesions can be challenging, as the differential includes benign neoplasms (eg, dermoid cyst, chalazion, pilomatricoma, xanthoma, xanthogranuloma2) and infectious etiologies such as bacterial pyoderma and mycobacterial, fungal, and parasitic infections (eg, cutaneous leishmaniasis). Pilomatricomas, although often seen on the face or extremities in young girls, more often are well circumscribed and located in the dermis. Ultrasonography of a pilomatricoma classically shows variable foci of calcification. Xanthoma and xanthogranuloma also were considered in our case since the lesion was subtly yellowish on examination. Similar to IFAG, these conditions may manifest as single or multiple lesions. Abnormalities in the patient’s blood lipid panel or family history may be needed to diagnose xanthoma. Biopsy of a juvenile xanthogranuloma would exhibit a dense dermal nodular proliferation of histiocytic cells with a smaller number of admixed lymphocytes, neutrophils, and eosinophils, in contrast to the multiple smaller loose epithelioid granulomas seen in IFAG. Additional diagnoses in the differential for IFAG include pyogenic granuloma, Spitz nevus, nodulocystic infantile acne, granulomatous rosacea, and hemangioma.1,3,9 In particular, granulomatous rosacea is challenging to differentiate from IFAG given the overlapping clinical findings. Multiple lesions, the presence of papules and pustules, and associated rosacea symptoms such as flushing suggest a diagnosis of granulomatous rosacea over IFAG.2
The prognosis for IFAG is excellent; most lesions self-resolve without treatment or procedural intervention within 1 year without scarring or relapse.3 Topical and oral antibiotic treatments such as metronidazole 0.75% gel or cream, oral erythromycin, oral clarithromycin, and doxycycline (in patients older than 8 years) have been used to treat IFAG with variable clinic responses.2,3,6,8 Persistent IFAG has been treated with surgical excision.3 Our patient was treated with a combination of gentamicin ointment 0.3% and tacrolimus ointment 0.3% and experienced approximately 50% improvement in the first month of treatment.
- Roul S, Léauté-Labrèze C, Boralevi F, et al. Idiopathic aseptic facial granuloma (pyodermite froide du visage): a pediatric entity? Arch Dermatol. 2001;137:1253-1255.
- Prey S, Ezzedine K, Mazereeuw-Hautier J, et al. IFAG and childhood rosacea: a possible link? Pediatr Dermatol. 2013;30:429-432. doi:10.1111/pde.12137
- Boralevi F, Léauté-Labrèze C, Lepreux S, et al. Idiopathic facial aseptic granuloma: a multicentre prospective study of 30 cases. Br J Dermatol. 2007;156:705-708. doi:10.1111/j.1365-2133.2006.07741.x
- Lobato-Berezo A, Montoro-Romero S, Pujol RM, et al. Dermoscopic features of idiopathic facial aseptic granuloma. Pediatr Dermatol. 2018;35:E308-E309. doi:10.1111/pde.13582
- González Rodríguez AJ, Jordá Cuevas E. Idiopathic facial aseptic granuloma. Clin Exp Dermatol. 2015;40:298-300. doi:10.1111/ced.12535
- Orion C, Sfecci A, Tisseau L, et al. Idiopathic facial aseptic granuloma in a 13-year-old boy dramatically improved with oral doxycycline and topical metronidazole: evidence for a link with childhood rosacea. Case Rep Dermatol. 2016;8:197-201. doi:10.1159/000447624
- Neri I, Raone B, Dondi A, et al. Should idiopathic facial aseptic granuloma be considered granulomatous rosacea? report of three pediatric cases. Pediatr Dermatol. 2013;30:109-111. doi:10.1111 /j.1525-1470.2011.01689.x
- Miconi F, Principi N, Cassiani L, et al. A cheek nodule in a child: be aware of idiopathic facial aseptic granuloma and its differential diagnosis. Int J Environ Res Public Health. 2019;16:2471. doi:10.3390/ijerph16142471
- Baroni A, Russo T, Faccenda F, et al. Idiopathic facial aseptic granuloma in a child: a possible expression of childhood rosacea. Pediatr Dermatol. 2013;30:394-395. doi:10.1111/j.1525-1470.2012.01805.x
- Roul S, Léauté-Labrèze C, Boralevi F, et al. Idiopathic aseptic facial granuloma (pyodermite froide du visage): a pediatric entity? Arch Dermatol. 2001;137:1253-1255.
- Prey S, Ezzedine K, Mazereeuw-Hautier J, et al. IFAG and childhood rosacea: a possible link? Pediatr Dermatol. 2013;30:429-432. doi:10.1111/pde.12137
- Boralevi F, Léauté-Labrèze C, Lepreux S, et al. Idiopathic facial aseptic granuloma: a multicentre prospective study of 30 cases. Br J Dermatol. 2007;156:705-708. doi:10.1111/j.1365-2133.2006.07741.x
- Lobato-Berezo A, Montoro-Romero S, Pujol RM, et al. Dermoscopic features of idiopathic facial aseptic granuloma. Pediatr Dermatol. 2018;35:E308-E309. doi:10.1111/pde.13582
- González Rodríguez AJ, Jordá Cuevas E. Idiopathic facial aseptic granuloma. Clin Exp Dermatol. 2015;40:298-300. doi:10.1111/ced.12535
- Orion C, Sfecci A, Tisseau L, et al. Idiopathic facial aseptic granuloma in a 13-year-old boy dramatically improved with oral doxycycline and topical metronidazole: evidence for a link with childhood rosacea. Case Rep Dermatol. 2016;8:197-201. doi:10.1159/000447624
- Neri I, Raone B, Dondi A, et al. Should idiopathic facial aseptic granuloma be considered granulomatous rosacea? report of three pediatric cases. Pediatr Dermatol. 2013;30:109-111. doi:10.1111 /j.1525-1470.2011.01689.x
- Miconi F, Principi N, Cassiani L, et al. A cheek nodule in a child: be aware of idiopathic facial aseptic granuloma and its differential diagnosis. Int J Environ Res Public Health. 2019;16:2471. doi:10.3390/ijerph16142471
- Baroni A, Russo T, Faccenda F, et al. Idiopathic facial aseptic granuloma in a child: a possible expression of childhood rosacea. Pediatr Dermatol. 2013;30:394-395. doi:10.1111/j.1525-1470.2012.01805.x
Painless Nodule on the Lower Eyelid
Painless Nodule on the Lower Eyelid
A 4-year-old girl presented to the dermatology clinic with a painless, red to golden-yellowish nodule on the right lower eyelid of 4 months’ duration. The patient had no history of skin disease and was otherwise healthy. Physical examination revealed a single 1-cm, soft, erythematous and yellowish plaque on the right lower eyelid that was subtly fluctuant on palpation. She had no associated systemic symptoms or lymphadenopathy. A punch biopsy of the lesion was performed.

Reddish Nodule on the Left Shoulder
Reddish Nodule on the Left Shoulder
THE DIAGNOSIS: Atypical Fibroxanthoma
Given the appearance of the nodule and the absence of features of a keloid scar, a soft-tissue or adnexal tumor was suspected. Histology revealed a thin epidermis with loss of rete ridges and a Grenz zone. There was a nodular uncircumscribed dermal proliferation of spindle cells forming interweaving fascicles with elongated ovoid nuclei and prominent nucleoli (Figure). There was moderate cellular and nuclear atypia, and no necrosis was observed. The spindle cells stained positive for CD10 and negative for AE1/AE3, cytokeratin 5/6, S100, melanoma triple marker, Factor XIII 1, ERG, CD31, CD34, desmin, and smooth muscle actin; ERG, CD31, CD34, and SMA highlighted small vessels within the tumor. The histologic diagnosis was an atypical spindle cell tumor favoring atypical fibroxanthoma (AFX). The excisional biopsy margins were clear.
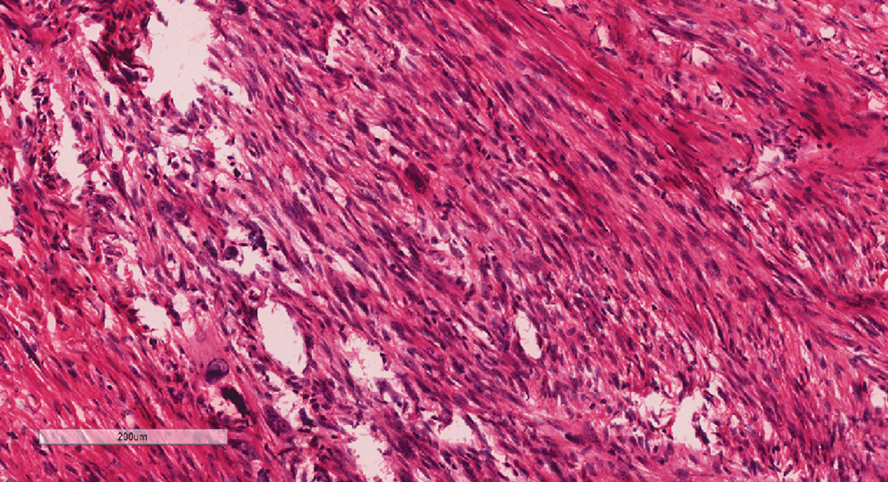
The patient was referred to surgical oncology to consider re-excision of margins after the diagnosis was made. A chest radiograph was clear, and magnetic resonance imaging showed mild skin thickening and image enhancement at the left shoulder—possibly a postsurgical change—with no nodularity suggesting a residual or recurrent tumor. Surgical oncology determined that the patient did not require further excision and placed him on regular follow-up every 2 to 3 months for the next 2 years.
uncertain origin that is considered to be on a spectrum with the more aggressive pleomorphic dermal sarcoma (PDS); it can be distinguished from PDS by histologic features such as nerve or vessel invasion.1 Both entities share oncogenes (eg, tumor protein 53 gene mutations) and are histologically and immunohistochemically similar. Atypical fibroxanthoma largely is viewed as an intermediate-risk tumor that is locally aggressive but rarely metastasizes, with a reported local recurrence rate of 5% to 11% and metastasis risk of 1% to 2%. Conversely, PDS is a more aggressive diagnosis with a high risk for local recurrence and metastasis (7%-69% and 4%-20%, respectively).1
Atypical fibroxanthomas may mimic other entities, both clinically and histologically. It commonly manifests as a flesh-colored to erythematous, sometimes ulcerated nodule on sun-exposed skin in elderly patients, leading to a broad range of clinical differential diagnoses, including other primary cutaneous malignancies (eg, squamous cell carcinoma, amelanotic melanoma), cutaneous sarcomas (eg, dermatofibrosarcoma protuberans), adnexal and other tumors (eg, pleomorphic fibroma, pilomatricoma), cutaneous metastases, and even keloid scars. As the differentials can look clinically similar, a skin biopsy may be necessary to confirm the diagnosis.
Histologically, AFX tends to show an undifferentiated pleomorphic spindle cell morphology. Notably, histology can be highly variable, with other reported histologic patterns including keloidlike, pleomorphic, epithelioid, rhabdoid, clear-cell, foamy cell, granular cell, bizarre cell, pseudoangiomatous, inflammatory, and osteoclast-rich patterns.2 Thus, the histologic differential diagnosis also is broad, and AFX primarily is a diagnosis of exclusion without specific immunohistochemical markers that serve to exclude other diagnoses. For example, AFX tends to stain positive for CD10 and CD68, though these are not specific markers for AFX. Furthermore, although certain histologic markers may commonly be more positive in AFX than PDS (eg, CD74 stains positive in 20% of AFXs and only 1% of PDSs), this is not reliable enough to be diagnostic.3 As such, AFX is distinguished from PDS primarily by histologic features such as subcutaneous tissue invasion, vascular or perineural invasion, necrosis, or local invasion/ metastases.1 Given the rarity of both tumors, no established management guidelines exist, although excision (wide local excision or Mohs micrographic surgery) usually is recommended, with some authors suggesting margins of 1 cm for AFX and 2 cm to 3 cm for PDS.1
This atypical case of AFX arising in non–sun-exposed skin in a young man raises questions about whether unknown genetic factors or possibly prior immunosuppression could have contributed to the development of the tumor. A thorough history and physical examination can provide valuable clues for biopsy, including ongoing growth, absence of known prior trauma or acne at the site, and clinical appearance, such as the reddish, solitary, dome-shaped lesion in our patient.
- Ørholt M, Abebe K, Rasmussen LE, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: local recurrence and metastasis in a nationwide population-based cohort of 1118 patients. J Am Acad Dermatol. 2023;89:1177-1184. doi:10.1016/j.jaad.2023.08.050
- Agaimy A. The many faces of atypical fibroxanthoma. Semin Diagn Pathol. 2023;40:306-312. doi:10.1053/j.semdp.2023.06.001
- Rapini RP. Practical Dermatopathology. 3rd ed. Elsevier Health Sciences; 2021.
THE DIAGNOSIS: Atypical Fibroxanthoma
Given the appearance of the nodule and the absence of features of a keloid scar, a soft-tissue or adnexal tumor was suspected. Histology revealed a thin epidermis with loss of rete ridges and a Grenz zone. There was a nodular uncircumscribed dermal proliferation of spindle cells forming interweaving fascicles with elongated ovoid nuclei and prominent nucleoli (Figure). There was moderate cellular and nuclear atypia, and no necrosis was observed. The spindle cells stained positive for CD10 and negative for AE1/AE3, cytokeratin 5/6, S100, melanoma triple marker, Factor XIII 1, ERG, CD31, CD34, desmin, and smooth muscle actin; ERG, CD31, CD34, and SMA highlighted small vessels within the tumor. The histologic diagnosis was an atypical spindle cell tumor favoring atypical fibroxanthoma (AFX). The excisional biopsy margins were clear.
The patient was referred to surgical oncology to consider re-excision of margins after the diagnosis was made. A chest radiograph was clear, and magnetic resonance imaging showed mild skin thickening and image enhancement at the left shoulder—possibly a postsurgical change—with no nodularity suggesting a residual or recurrent tumor. Surgical oncology determined that the patient did not require further excision and placed him on regular follow-up every 2 to 3 months for the next 2 years.
uncertain origin that is considered to be on a spectrum with the more aggressive pleomorphic dermal sarcoma (PDS); it can be distinguished from PDS by histologic features such as nerve or vessel invasion.1 Both entities share oncogenes (eg, tumor protein 53 gene mutations) and are histologically and immunohistochemically similar. Atypical fibroxanthoma largely is viewed as an intermediate-risk tumor that is locally aggressive but rarely metastasizes, with a reported local recurrence rate of 5% to 11% and metastasis risk of 1% to 2%. Conversely, PDS is a more aggressive diagnosis with a high risk for local recurrence and metastasis (7%-69% and 4%-20%, respectively).1
Atypical fibroxanthomas may mimic other entities, both clinically and histologically. It commonly manifests as a flesh-colored to erythematous, sometimes ulcerated nodule on sun-exposed skin in elderly patients, leading to a broad range of clinical differential diagnoses, including other primary cutaneous malignancies (eg, squamous cell carcinoma, amelanotic melanoma), cutaneous sarcomas (eg, dermatofibrosarcoma protuberans), adnexal and other tumors (eg, pleomorphic fibroma, pilomatricoma), cutaneous metastases, and even keloid scars. As the differentials can look clinically similar, a skin biopsy may be necessary to confirm the diagnosis.
Histologically, AFX tends to show an undifferentiated pleomorphic spindle cell morphology. Notably, histology can be highly variable, with other reported histologic patterns including keloidlike, pleomorphic, epithelioid, rhabdoid, clear-cell, foamy cell, granular cell, bizarre cell, pseudoangiomatous, inflammatory, and osteoclast-rich patterns.2 Thus, the histologic differential diagnosis also is broad, and AFX primarily is a diagnosis of exclusion without specific immunohistochemical markers that serve to exclude other diagnoses. For example, AFX tends to stain positive for CD10 and CD68, though these are not specific markers for AFX. Furthermore, although certain histologic markers may commonly be more positive in AFX than PDS (eg, CD74 stains positive in 20% of AFXs and only 1% of PDSs), this is not reliable enough to be diagnostic.3 As such, AFX is distinguished from PDS primarily by histologic features such as subcutaneous tissue invasion, vascular or perineural invasion, necrosis, or local invasion/ metastases.1 Given the rarity of both tumors, no established management guidelines exist, although excision (wide local excision or Mohs micrographic surgery) usually is recommended, with some authors suggesting margins of 1 cm for AFX and 2 cm to 3 cm for PDS.1
This atypical case of AFX arising in non–sun-exposed skin in a young man raises questions about whether unknown genetic factors or possibly prior immunosuppression could have contributed to the development of the tumor. A thorough history and physical examination can provide valuable clues for biopsy, including ongoing growth, absence of known prior trauma or acne at the site, and clinical appearance, such as the reddish, solitary, dome-shaped lesion in our patient.
THE DIAGNOSIS: Atypical Fibroxanthoma
Given the appearance of the nodule and the absence of features of a keloid scar, a soft-tissue or adnexal tumor was suspected. Histology revealed a thin epidermis with loss of rete ridges and a Grenz zone. There was a nodular uncircumscribed dermal proliferation of spindle cells forming interweaving fascicles with elongated ovoid nuclei and prominent nucleoli (Figure). There was moderate cellular and nuclear atypia, and no necrosis was observed. The spindle cells stained positive for CD10 and negative for AE1/AE3, cytokeratin 5/6, S100, melanoma triple marker, Factor XIII 1, ERG, CD31, CD34, desmin, and smooth muscle actin; ERG, CD31, CD34, and SMA highlighted small vessels within the tumor. The histologic diagnosis was an atypical spindle cell tumor favoring atypical fibroxanthoma (AFX). The excisional biopsy margins were clear.
The patient was referred to surgical oncology to consider re-excision of margins after the diagnosis was made. A chest radiograph was clear, and magnetic resonance imaging showed mild skin thickening and image enhancement at the left shoulder—possibly a postsurgical change—with no nodularity suggesting a residual or recurrent tumor. Surgical oncology determined that the patient did not require further excision and placed him on regular follow-up every 2 to 3 months for the next 2 years.
uncertain origin that is considered to be on a spectrum with the more aggressive pleomorphic dermal sarcoma (PDS); it can be distinguished from PDS by histologic features such as nerve or vessel invasion.1 Both entities share oncogenes (eg, tumor protein 53 gene mutations) and are histologically and immunohistochemically similar. Atypical fibroxanthoma largely is viewed as an intermediate-risk tumor that is locally aggressive but rarely metastasizes, with a reported local recurrence rate of 5% to 11% and metastasis risk of 1% to 2%. Conversely, PDS is a more aggressive diagnosis with a high risk for local recurrence and metastasis (7%-69% and 4%-20%, respectively).1
Atypical fibroxanthomas may mimic other entities, both clinically and histologically. It commonly manifests as a flesh-colored to erythematous, sometimes ulcerated nodule on sun-exposed skin in elderly patients, leading to a broad range of clinical differential diagnoses, including other primary cutaneous malignancies (eg, squamous cell carcinoma, amelanotic melanoma), cutaneous sarcomas (eg, dermatofibrosarcoma protuberans), adnexal and other tumors (eg, pleomorphic fibroma, pilomatricoma), cutaneous metastases, and even keloid scars. As the differentials can look clinically similar, a skin biopsy may be necessary to confirm the diagnosis.
Histologically, AFX tends to show an undifferentiated pleomorphic spindle cell morphology. Notably, histology can be highly variable, with other reported histologic patterns including keloidlike, pleomorphic, epithelioid, rhabdoid, clear-cell, foamy cell, granular cell, bizarre cell, pseudoangiomatous, inflammatory, and osteoclast-rich patterns.2 Thus, the histologic differential diagnosis also is broad, and AFX primarily is a diagnosis of exclusion without specific immunohistochemical markers that serve to exclude other diagnoses. For example, AFX tends to stain positive for CD10 and CD68, though these are not specific markers for AFX. Furthermore, although certain histologic markers may commonly be more positive in AFX than PDS (eg, CD74 stains positive in 20% of AFXs and only 1% of PDSs), this is not reliable enough to be diagnostic.3 As such, AFX is distinguished from PDS primarily by histologic features such as subcutaneous tissue invasion, vascular or perineural invasion, necrosis, or local invasion/ metastases.1 Given the rarity of both tumors, no established management guidelines exist, although excision (wide local excision or Mohs micrographic surgery) usually is recommended, with some authors suggesting margins of 1 cm for AFX and 2 cm to 3 cm for PDS.1
This atypical case of AFX arising in non–sun-exposed skin in a young man raises questions about whether unknown genetic factors or possibly prior immunosuppression could have contributed to the development of the tumor. A thorough history and physical examination can provide valuable clues for biopsy, including ongoing growth, absence of known prior trauma or acne at the site, and clinical appearance, such as the reddish, solitary, dome-shaped lesion in our patient.
- Ørholt M, Abebe K, Rasmussen LE, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: local recurrence and metastasis in a nationwide population-based cohort of 1118 patients. J Am Acad Dermatol. 2023;89:1177-1184. doi:10.1016/j.jaad.2023.08.050
- Agaimy A. The many faces of atypical fibroxanthoma. Semin Diagn Pathol. 2023;40:306-312. doi:10.1053/j.semdp.2023.06.001
- Rapini RP. Practical Dermatopathology. 3rd ed. Elsevier Health Sciences; 2021.
- Ørholt M, Abebe K, Rasmussen LE, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: local recurrence and metastasis in a nationwide population-based cohort of 1118 patients. J Am Acad Dermatol. 2023;89:1177-1184. doi:10.1016/j.jaad.2023.08.050
- Agaimy A. The many faces of atypical fibroxanthoma. Semin Diagn Pathol. 2023;40:306-312. doi:10.1053/j.semdp.2023.06.001
- Rapini RP. Practical Dermatopathology. 3rd ed. Elsevier Health Sciences; 2021.
Reddish Nodule on the Left Shoulder
Reddish Nodule on the Left Shoulder
A 20-year-old man presented to the dermatology clinic for evaluation of a slow-growing nodule on the left shoulder of 1 year’s duration. The patient reported a history of eczema since childhood, which had been treated by an external physician with cyclosporine and methotrexate; however, exact treatment records were unavailable as the patient had been treated at another institution. The eczema had been well controlled over the past year on topical steroids alone. The nodule was asymptomatic, and the patient denied any history of trauma or acne at the affected site. He also denied any family history of similar nodules or other notable skin findings. Physical examination revealed a well circumscribed, 15×12-mm, firm, flesh-colored to reddish nodule on the left shoulder with a slightly whitish center. An excisional biopsy was performed.
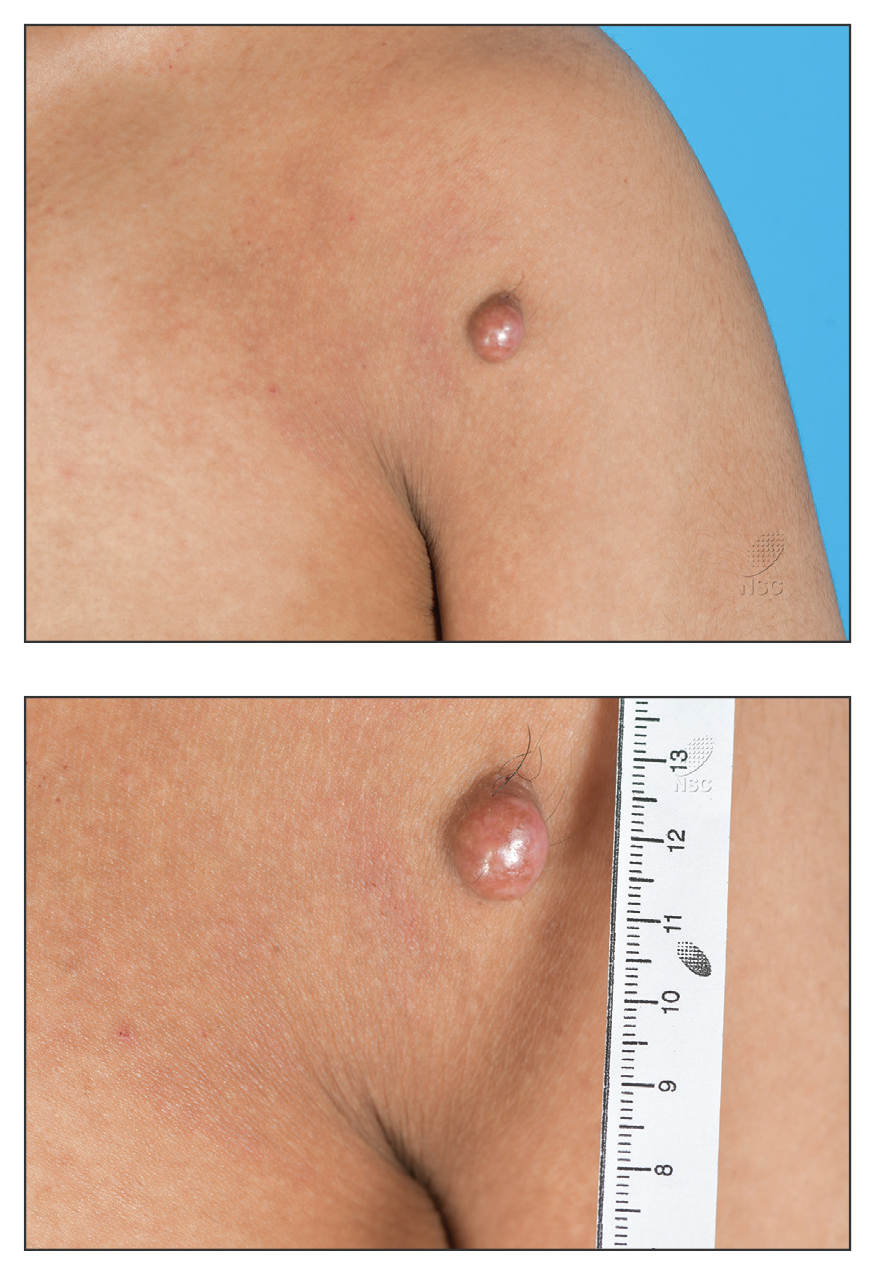

Pedunculated Pink Papule on the Nose
THE DIAGNOSIS: Pedunculated Lipofibroma
Histopathology confirmed a pedunculated/polypoid lesion with intradermal lobules of adipocytes/mature adipose tissue admixed with connective tissue bundles and vascular ectasias. Overlying epidermal acanthosis with slight papillomatosis and hyperkeratosis was present (Figure 1). Masson trichrome staining highlighted admixed collagen bundles (Figure 2). Verhoeff–van Gieson staining showed marked reduction in elastic fibers (Figure 3). Immunostaining was negative for smooth muscle actin and desmin. A diagnosis of pedunculated lipofibroma on the nose was made based on both clinical and histopathologic findings.
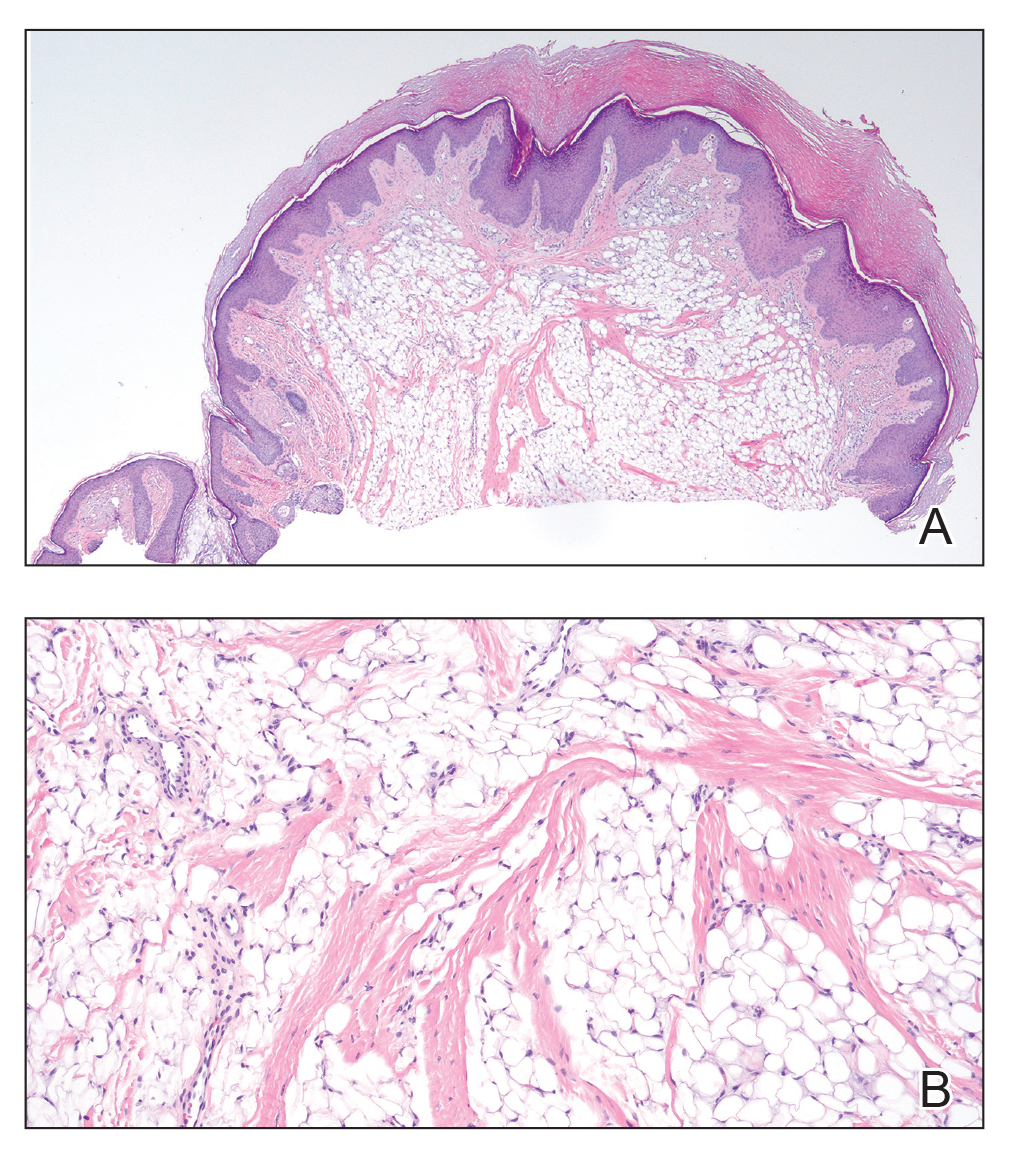
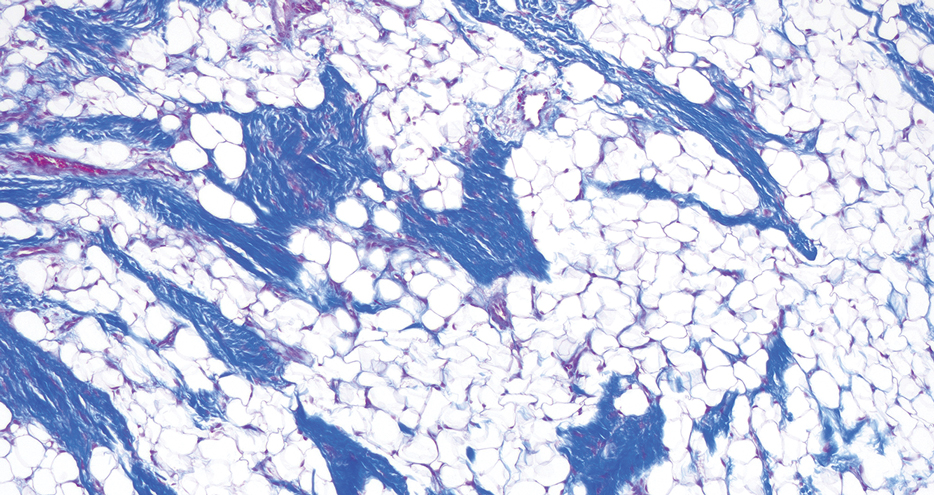
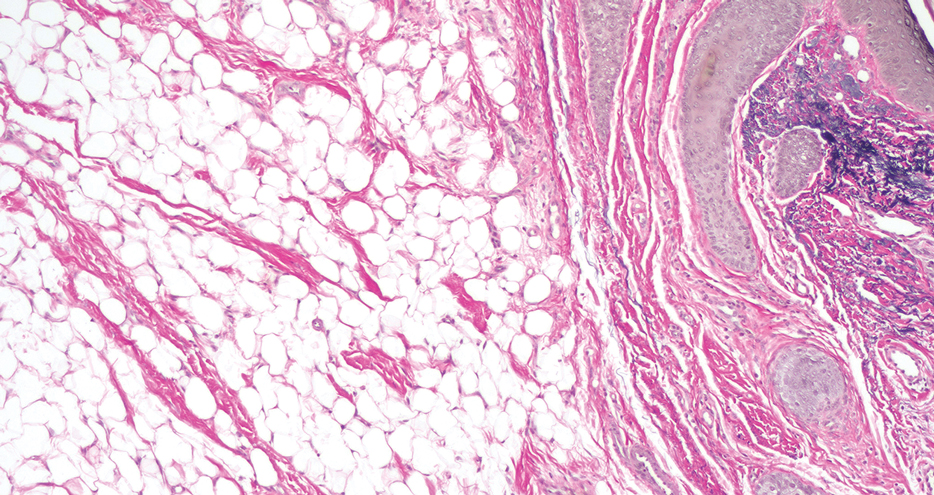
Pedunculated lipofibroma (or solitary lipofibroma) is the solitary form of nevus lipomatosus cutaneous superficialis (NLCS).7 First described by Hoffmann and Zurhelle1 in 1921, NLCS is an uncommon benign hamartomatous cutaneous lesion/connective tissue nevus that also has a classic multiple form.1-13 The etiology of NLCS remains unclear, but several theories have been proposed to explain its pathogenesis, including deposition of adipocytes secondary to degenerative changes in dermal connective tissue, focal/local heterotopic development of adipose tissue, and derivation from differentiating lipoblasts (preadipose tissue) originating from precursor vascular or perivascular cells.2-13
Pedunculated lipofibroma usually develops during the third to sixth decades of life and manifests as a single cutaneous lesion with a smooth surface, often on a non–pelvic girdle location.7-13 No particular predilection sites are noted, with lesions reported on the arm, axilla, back, upper thigh, knee, and sole.5,12 There are rare reports of this type of NLCS on the ear, scalp, forehead, or eyelid.7-11
In the classic form of NLCS, multiple cutaneous lesions are present at birth or develop within the first 2 to 3 decades of life.2-6 Lesions consist of soft, nontender, pedunculated, flesh-colored or yellowish papules and nodules with a verrucoid or cerebriform surface that may later coalesce to form plaques.2-6 Predilection sites include the pelvic girdle, buttocks, sacral and coccygeal regions, and upper posterior thighs, with a linear or zosteriform pattern of distribution.2-6 Rarely, the classic form can arise in elderly patients and/or at an atypical anatomic location (eg, clitoris,3 shoulder,5 thorax,5 abdomen5) and can demonstrate extension of lesions across the midline.4 Rare cases of classic NLCS on the scalp2 and face3-6 have been reported, including lesions localized to the nose3 and chin4 and others extending from the right mandible to the neck5 and right lower lip to the submandibular/posteriorateral cervical region.6 In some cases, lesions clinically resemble plane xanthoma4 and localized scleroderma.6
Adotama et al13 proposed a set of clinical features to differentiate classic NLCS, pedunculated lipofibroma (solitary NLCS), and fibroepithelial polyp with adipocytes (distinguished by their furrowed surface, hyperpigmentation, and anatomic predilection for the neck and axilla). Lesions are asymptomatic in both forms of NLCS.2-13 Family history or predominant sex involvement have not been reported in either clinical type.2-13 Reported associations with NLCS include a number of endocrinologic conditions including diabetes.7 Other coexisting skin findings can include café-au-lait macules, leukodermic (white) spots, overlying hypertrichosis, comedolike alterations, angiokeratoma, hemangioma, and folliculosebaceous cystic hamartoma.4 None of these were evident in our patient.
Lesions from both types of NLCS are indistinguishable on histopathology, characterized by the presence of a central core of ectopic mature adipocytes in the papillary/reticular dermis.2-13 Additional light microscopic features (some seen in our case) have been described, including thickened collagen bundles, reduction of elastic fibers, increased numbers of fibroblasts and/or mast cells, increased (small-vessel) vascularity, focal mucin deposition/myxoid degeneration, a mild perivascular lymphocytic infiltrate, attenuation of adnexal structures, and abnormalities of the epidermis (eg, surface ulceration).2-13
Prior to biopsy, the differential diagnosis in our patient included angiofibroma, pyogenic granuloma, and basal cell carcinoma given the exophytic, pink, papular appearance of the lesion; however, the histopathologic differential diagnosis included angiofibroma, angiomyolipoma, lymphangioma, nevus sebaceus, and spindle cell lipoma (SCL). In angiofibroma, a dermal proliferation of stellate fibroblasts, dilated blood vessels, and collagenous stroma are seen. Cutaneous angiomyolipoma demonstrates smooth muscle bundles in addition to thickened blood vessels and variable proportions of mature adipocytes. Lymphangioma is characterized by dilated lymph channels lined by flat endothelial cells. Nevus sebaceus shows superficial immature and abnormally formed pilosebaceous units, with epidermal papillomatosis.
Rare cases of SCL on the nose have been described.14 Similar to pedunculated lipofibroma, reported examples demonstrate mature univacuolar adipocytes with thick collagen fibers and bland uniform spindle cells. Unlike the lesion seen in our patient, nasal SCL may be clinically mobile and typically is localized to the subcutaneous tissue, although dermal tumors also occur.14 Variably reported histopathologic findings in nasal SCL include circumscription/encapsulation, spindle cells arranged in short fascicles with nuclear palisading, a myxoid/mucinous interstitial matrix, and/or multinucleated giant cells—all light microscopic features that were not identified in our case; however, variable proportions of adipocytic, fibrous, and myxoid components among reported examples of SCL on the nose14 can make distinction from pedunculated lipofibroma difficult, as both are benign lipomatous tumor variants.
Clinically, pedunculated lipofibroma may be confused with more common benign cutaneous lesions and must be distinguished from other fibrolipomatous lesions on the nose. Specifically, the differential diagnosis includes benign cutaneous papillomas such as acrochordon, angiofibroma, melanocytic nevi, neurofibroma, nevus sebaceus, lymphangioma, and eccrine poroma.7-13 These all can be readily excluded on histopathology. Pedunculated lipofibroma on the nose, as in our patient, must be distinguished from fibrolipoma15 and dendritic myxofibrolipoma.16 Fibrolipoma is a subcutaneous proliferation of mature adipose tissue and fibrous tissue and comprises 1.6% of all facial lipomas reported worldwide.15 Dendritic myxofibrolipoma is a recently described benign soft-tissue tumor characterized by an admixture of mature adipose tissue, spindle and stellate cells, and an abundant myxoid stroma with prominent collagenization.16
Treatment of pedunculated lipofibroma on the nose is not indicated except for cosmetic reasons, in which case simple surgical excision would be considered satisfactory. Following biopsy, no further treatment was pursued in our patient.
- Hoffmann E, Zurhelle E. Uber einen naevus lipomatodes cutaneous superficialis der linken Glutaalgegend. Arch Derm Syph. 1921;130:327-333.
- Chanoki M, Isukos S, Suzuki S, et al. Nevus lipomatosus cutaneus superficialis of the scalp. Cutis. 1989;43:143-144.
- Sáez Rodríguez M, Rodríguez-Martin M, Carnerero A, et al. Naevus lipomatosus cutaneous superficialis on the nose. J Eur Acad Dermatol Venereol. 2005;19:751-752.
- Hassab-El-Naby HMM, Rageh MA. Adult-onset nevus lipomatosus cutaneous superficialis mimicking plane xanthoma. J Clin Aesthet Dermatol. 2022;15:10-11.
- Park HJ, Park CJ, Yi JY, et al. Nevus lipomatosus superficialis on the face. Int J Dermatol. 1997;36:435-437.
- Ioannidou DJ, Stefanidou MP, Panayiotides JG, et al. Nevus lipomatosus cutaneous superficialis (Hoffman-Zurhelle) with localized scleroderma like appearance. Int J Dermatol. 2001;40:54-57.
- Nogita T, Wong TY, Hidano A, et al. Pedunculated lipofibroma. a clinicopathologic study of thirty-two cases supporting a simplified nomenclature. J Am Acad Dermatol. 1994;31(2 pt 1):235-240.
- Sawada Y. Solitary nevus lipomatosus superficialis on the forehead. Ann Plast Surg. 1986;16:356-358.
- Knoth W. Uber Naevus lipomatosus cutaneus superficialis Hoffmann-Zurhelle und uber Naevus naevocellularis partim lipomatodes. Dermatologica. 1962;125:161.
- Weitzner S. Solitary naevus lipomatosus cutaneus superficialis of scalp. Arch Dermatol. 1968;97:540-542.
- Kaw P, Carlson A, Meyer DR. Nevus lipomatosus (pedunculated lipofibroma) of the eyelid. Ophthalmic Plast Reconstr Surg. 2005;21:74-76.
- Vano-Galvan S, Moreno C, Vano-Galvan E, et al. Solitary naevus lipomatosus cutaneous superficialis on the sole. Eur J Dermatol. 2008;18:353-354.
- Adotama P, Hutson SD, Rieder EA, et al. Revisiting solitary pedunculated lipofibromas. Am J Clin Pathol. 2021;156:954-957.
- Kubin ME, Lantto U, Lindgren O, et al. A rare, recurrent spindle cell lipoma of the nose. Acta Derm Venereol. 2021;101:adv00571.
- Jung SN, Shin JW, Kwon H, et al. Fibrolipoma of the tip of the nose. J Craniofac Surg. 2009;20:555-556.
- Han XC, Zheng LQ, Shang XL. Dendritic fibromyxolipoma on the nasal tip in an old patient. Int J Clin Exp Pathol. 2014;7:7064-7067.
THE DIAGNOSIS: Pedunculated Lipofibroma
Histopathology confirmed a pedunculated/polypoid lesion with intradermal lobules of adipocytes/mature adipose tissue admixed with connective tissue bundles and vascular ectasias. Overlying epidermal acanthosis with slight papillomatosis and hyperkeratosis was present (Figure 1). Masson trichrome staining highlighted admixed collagen bundles (Figure 2). Verhoeff–van Gieson staining showed marked reduction in elastic fibers (Figure 3). Immunostaining was negative for smooth muscle actin and desmin. A diagnosis of pedunculated lipofibroma on the nose was made based on both clinical and histopathologic findings.
Pedunculated lipofibroma (or solitary lipofibroma) is the solitary form of nevus lipomatosus cutaneous superficialis (NLCS).7 First described by Hoffmann and Zurhelle1 in 1921, NLCS is an uncommon benign hamartomatous cutaneous lesion/connective tissue nevus that also has a classic multiple form.1-13 The etiology of NLCS remains unclear, but several theories have been proposed to explain its pathogenesis, including deposition of adipocytes secondary to degenerative changes in dermal connective tissue, focal/local heterotopic development of adipose tissue, and derivation from differentiating lipoblasts (preadipose tissue) originating from precursor vascular or perivascular cells.2-13
Pedunculated lipofibroma usually develops during the third to sixth decades of life and manifests as a single cutaneous lesion with a smooth surface, often on a non–pelvic girdle location.7-13 No particular predilection sites are noted, with lesions reported on the arm, axilla, back, upper thigh, knee, and sole.5,12 There are rare reports of this type of NLCS on the ear, scalp, forehead, or eyelid.7-11
In the classic form of NLCS, multiple cutaneous lesions are present at birth or develop within the first 2 to 3 decades of life.2-6 Lesions consist of soft, nontender, pedunculated, flesh-colored or yellowish papules and nodules with a verrucoid or cerebriform surface that may later coalesce to form plaques.2-6 Predilection sites include the pelvic girdle, buttocks, sacral and coccygeal regions, and upper posterior thighs, with a linear or zosteriform pattern of distribution.2-6 Rarely, the classic form can arise in elderly patients and/or at an atypical anatomic location (eg, clitoris,3 shoulder,5 thorax,5 abdomen5) and can demonstrate extension of lesions across the midline.4 Rare cases of classic NLCS on the scalp2 and face3-6 have been reported, including lesions localized to the nose3 and chin4 and others extending from the right mandible to the neck5 and right lower lip to the submandibular/posteriorateral cervical region.6 In some cases, lesions clinically resemble plane xanthoma4 and localized scleroderma.6
Adotama et al13 proposed a set of clinical features to differentiate classic NLCS, pedunculated lipofibroma (solitary NLCS), and fibroepithelial polyp with adipocytes (distinguished by their furrowed surface, hyperpigmentation, and anatomic predilection for the neck and axilla). Lesions are asymptomatic in both forms of NLCS.2-13 Family history or predominant sex involvement have not been reported in either clinical type.2-13 Reported associations with NLCS include a number of endocrinologic conditions including diabetes.7 Other coexisting skin findings can include café-au-lait macules, leukodermic (white) spots, overlying hypertrichosis, comedolike alterations, angiokeratoma, hemangioma, and folliculosebaceous cystic hamartoma.4 None of these were evident in our patient.
Lesions from both types of NLCS are indistinguishable on histopathology, characterized by the presence of a central core of ectopic mature adipocytes in the papillary/reticular dermis.2-13 Additional light microscopic features (some seen in our case) have been described, including thickened collagen bundles, reduction of elastic fibers, increased numbers of fibroblasts and/or mast cells, increased (small-vessel) vascularity, focal mucin deposition/myxoid degeneration, a mild perivascular lymphocytic infiltrate, attenuation of adnexal structures, and abnormalities of the epidermis (eg, surface ulceration).2-13
Prior to biopsy, the differential diagnosis in our patient included angiofibroma, pyogenic granuloma, and basal cell carcinoma given the exophytic, pink, papular appearance of the lesion; however, the histopathologic differential diagnosis included angiofibroma, angiomyolipoma, lymphangioma, nevus sebaceus, and spindle cell lipoma (SCL). In angiofibroma, a dermal proliferation of stellate fibroblasts, dilated blood vessels, and collagenous stroma are seen. Cutaneous angiomyolipoma demonstrates smooth muscle bundles in addition to thickened blood vessels and variable proportions of mature adipocytes. Lymphangioma is characterized by dilated lymph channels lined by flat endothelial cells. Nevus sebaceus shows superficial immature and abnormally formed pilosebaceous units, with epidermal papillomatosis.
Rare cases of SCL on the nose have been described.14 Similar to pedunculated lipofibroma, reported examples demonstrate mature univacuolar adipocytes with thick collagen fibers and bland uniform spindle cells. Unlike the lesion seen in our patient, nasal SCL may be clinically mobile and typically is localized to the subcutaneous tissue, although dermal tumors also occur.14 Variably reported histopathologic findings in nasal SCL include circumscription/encapsulation, spindle cells arranged in short fascicles with nuclear palisading, a myxoid/mucinous interstitial matrix, and/or multinucleated giant cells—all light microscopic features that were not identified in our case; however, variable proportions of adipocytic, fibrous, and myxoid components among reported examples of SCL on the nose14 can make distinction from pedunculated lipofibroma difficult, as both are benign lipomatous tumor variants.
Clinically, pedunculated lipofibroma may be confused with more common benign cutaneous lesions and must be distinguished from other fibrolipomatous lesions on the nose. Specifically, the differential diagnosis includes benign cutaneous papillomas such as acrochordon, angiofibroma, melanocytic nevi, neurofibroma, nevus sebaceus, lymphangioma, and eccrine poroma.7-13 These all can be readily excluded on histopathology. Pedunculated lipofibroma on the nose, as in our patient, must be distinguished from fibrolipoma15 and dendritic myxofibrolipoma.16 Fibrolipoma is a subcutaneous proliferation of mature adipose tissue and fibrous tissue and comprises 1.6% of all facial lipomas reported worldwide.15 Dendritic myxofibrolipoma is a recently described benign soft-tissue tumor characterized by an admixture of mature adipose tissue, spindle and stellate cells, and an abundant myxoid stroma with prominent collagenization.16
Treatment of pedunculated lipofibroma on the nose is not indicated except for cosmetic reasons, in which case simple surgical excision would be considered satisfactory. Following biopsy, no further treatment was pursued in our patient.
THE DIAGNOSIS: Pedunculated Lipofibroma
Histopathology confirmed a pedunculated/polypoid lesion with intradermal lobules of adipocytes/mature adipose tissue admixed with connective tissue bundles and vascular ectasias. Overlying epidermal acanthosis with slight papillomatosis and hyperkeratosis was present (Figure 1). Masson trichrome staining highlighted admixed collagen bundles (Figure 2). Verhoeff–van Gieson staining showed marked reduction in elastic fibers (Figure 3). Immunostaining was negative for smooth muscle actin and desmin. A diagnosis of pedunculated lipofibroma on the nose was made based on both clinical and histopathologic findings.
Pedunculated lipofibroma (or solitary lipofibroma) is the solitary form of nevus lipomatosus cutaneous superficialis (NLCS).7 First described by Hoffmann and Zurhelle1 in 1921, NLCS is an uncommon benign hamartomatous cutaneous lesion/connective tissue nevus that also has a classic multiple form.1-13 The etiology of NLCS remains unclear, but several theories have been proposed to explain its pathogenesis, including deposition of adipocytes secondary to degenerative changes in dermal connective tissue, focal/local heterotopic development of adipose tissue, and derivation from differentiating lipoblasts (preadipose tissue) originating from precursor vascular or perivascular cells.2-13
Pedunculated lipofibroma usually develops during the third to sixth decades of life and manifests as a single cutaneous lesion with a smooth surface, often on a non–pelvic girdle location.7-13 No particular predilection sites are noted, with lesions reported on the arm, axilla, back, upper thigh, knee, and sole.5,12 There are rare reports of this type of NLCS on the ear, scalp, forehead, or eyelid.7-11
In the classic form of NLCS, multiple cutaneous lesions are present at birth or develop within the first 2 to 3 decades of life.2-6 Lesions consist of soft, nontender, pedunculated, flesh-colored or yellowish papules and nodules with a verrucoid or cerebriform surface that may later coalesce to form plaques.2-6 Predilection sites include the pelvic girdle, buttocks, sacral and coccygeal regions, and upper posterior thighs, with a linear or zosteriform pattern of distribution.2-6 Rarely, the classic form can arise in elderly patients and/or at an atypical anatomic location (eg, clitoris,3 shoulder,5 thorax,5 abdomen5) and can demonstrate extension of lesions across the midline.4 Rare cases of classic NLCS on the scalp2 and face3-6 have been reported, including lesions localized to the nose3 and chin4 and others extending from the right mandible to the neck5 and right lower lip to the submandibular/posteriorateral cervical region.6 In some cases, lesions clinically resemble plane xanthoma4 and localized scleroderma.6
Adotama et al13 proposed a set of clinical features to differentiate classic NLCS, pedunculated lipofibroma (solitary NLCS), and fibroepithelial polyp with adipocytes (distinguished by their furrowed surface, hyperpigmentation, and anatomic predilection for the neck and axilla). Lesions are asymptomatic in both forms of NLCS.2-13 Family history or predominant sex involvement have not been reported in either clinical type.2-13 Reported associations with NLCS include a number of endocrinologic conditions including diabetes.7 Other coexisting skin findings can include café-au-lait macules, leukodermic (white) spots, overlying hypertrichosis, comedolike alterations, angiokeratoma, hemangioma, and folliculosebaceous cystic hamartoma.4 None of these were evident in our patient.
Lesions from both types of NLCS are indistinguishable on histopathology, characterized by the presence of a central core of ectopic mature adipocytes in the papillary/reticular dermis.2-13 Additional light microscopic features (some seen in our case) have been described, including thickened collagen bundles, reduction of elastic fibers, increased numbers of fibroblasts and/or mast cells, increased (small-vessel) vascularity, focal mucin deposition/myxoid degeneration, a mild perivascular lymphocytic infiltrate, attenuation of adnexal structures, and abnormalities of the epidermis (eg, surface ulceration).2-13
Prior to biopsy, the differential diagnosis in our patient included angiofibroma, pyogenic granuloma, and basal cell carcinoma given the exophytic, pink, papular appearance of the lesion; however, the histopathologic differential diagnosis included angiofibroma, angiomyolipoma, lymphangioma, nevus sebaceus, and spindle cell lipoma (SCL). In angiofibroma, a dermal proliferation of stellate fibroblasts, dilated blood vessels, and collagenous stroma are seen. Cutaneous angiomyolipoma demonstrates smooth muscle bundles in addition to thickened blood vessels and variable proportions of mature adipocytes. Lymphangioma is characterized by dilated lymph channels lined by flat endothelial cells. Nevus sebaceus shows superficial immature and abnormally formed pilosebaceous units, with epidermal papillomatosis.
Rare cases of SCL on the nose have been described.14 Similar to pedunculated lipofibroma, reported examples demonstrate mature univacuolar adipocytes with thick collagen fibers and bland uniform spindle cells. Unlike the lesion seen in our patient, nasal SCL may be clinically mobile and typically is localized to the subcutaneous tissue, although dermal tumors also occur.14 Variably reported histopathologic findings in nasal SCL include circumscription/encapsulation, spindle cells arranged in short fascicles with nuclear palisading, a myxoid/mucinous interstitial matrix, and/or multinucleated giant cells—all light microscopic features that were not identified in our case; however, variable proportions of adipocytic, fibrous, and myxoid components among reported examples of SCL on the nose14 can make distinction from pedunculated lipofibroma difficult, as both are benign lipomatous tumor variants.
Clinically, pedunculated lipofibroma may be confused with more common benign cutaneous lesions and must be distinguished from other fibrolipomatous lesions on the nose. Specifically, the differential diagnosis includes benign cutaneous papillomas such as acrochordon, angiofibroma, melanocytic nevi, neurofibroma, nevus sebaceus, lymphangioma, and eccrine poroma.7-13 These all can be readily excluded on histopathology. Pedunculated lipofibroma on the nose, as in our patient, must be distinguished from fibrolipoma15 and dendritic myxofibrolipoma.16 Fibrolipoma is a subcutaneous proliferation of mature adipose tissue and fibrous tissue and comprises 1.6% of all facial lipomas reported worldwide.15 Dendritic myxofibrolipoma is a recently described benign soft-tissue tumor characterized by an admixture of mature adipose tissue, spindle and stellate cells, and an abundant myxoid stroma with prominent collagenization.16
Treatment of pedunculated lipofibroma on the nose is not indicated except for cosmetic reasons, in which case simple surgical excision would be considered satisfactory. Following biopsy, no further treatment was pursued in our patient.
- Hoffmann E, Zurhelle E. Uber einen naevus lipomatodes cutaneous superficialis der linken Glutaalgegend. Arch Derm Syph. 1921;130:327-333.
- Chanoki M, Isukos S, Suzuki S, et al. Nevus lipomatosus cutaneus superficialis of the scalp. Cutis. 1989;43:143-144.
- Sáez Rodríguez M, Rodríguez-Martin M, Carnerero A, et al. Naevus lipomatosus cutaneous superficialis on the nose. J Eur Acad Dermatol Venereol. 2005;19:751-752.
- Hassab-El-Naby HMM, Rageh MA. Adult-onset nevus lipomatosus cutaneous superficialis mimicking plane xanthoma. J Clin Aesthet Dermatol. 2022;15:10-11.
- Park HJ, Park CJ, Yi JY, et al. Nevus lipomatosus superficialis on the face. Int J Dermatol. 1997;36:435-437.
- Ioannidou DJ, Stefanidou MP, Panayiotides JG, et al. Nevus lipomatosus cutaneous superficialis (Hoffman-Zurhelle) with localized scleroderma like appearance. Int J Dermatol. 2001;40:54-57.
- Nogita T, Wong TY, Hidano A, et al. Pedunculated lipofibroma. a clinicopathologic study of thirty-two cases supporting a simplified nomenclature. J Am Acad Dermatol. 1994;31(2 pt 1):235-240.
- Sawada Y. Solitary nevus lipomatosus superficialis on the forehead. Ann Plast Surg. 1986;16:356-358.
- Knoth W. Uber Naevus lipomatosus cutaneus superficialis Hoffmann-Zurhelle und uber Naevus naevocellularis partim lipomatodes. Dermatologica. 1962;125:161.
- Weitzner S. Solitary naevus lipomatosus cutaneus superficialis of scalp. Arch Dermatol. 1968;97:540-542.
- Kaw P, Carlson A, Meyer DR. Nevus lipomatosus (pedunculated lipofibroma) of the eyelid. Ophthalmic Plast Reconstr Surg. 2005;21:74-76.
- Vano-Galvan S, Moreno C, Vano-Galvan E, et al. Solitary naevus lipomatosus cutaneous superficialis on the sole. Eur J Dermatol. 2008;18:353-354.
- Adotama P, Hutson SD, Rieder EA, et al. Revisiting solitary pedunculated lipofibromas. Am J Clin Pathol. 2021;156:954-957.
- Kubin ME, Lantto U, Lindgren O, et al. A rare, recurrent spindle cell lipoma of the nose. Acta Derm Venereol. 2021;101:adv00571.
- Jung SN, Shin JW, Kwon H, et al. Fibrolipoma of the tip of the nose. J Craniofac Surg. 2009;20:555-556.
- Han XC, Zheng LQ, Shang XL. Dendritic fibromyxolipoma on the nasal tip in an old patient. Int J Clin Exp Pathol. 2014;7:7064-7067.
- Hoffmann E, Zurhelle E. Uber einen naevus lipomatodes cutaneous superficialis der linken Glutaalgegend. Arch Derm Syph. 1921;130:327-333.
- Chanoki M, Isukos S, Suzuki S, et al. Nevus lipomatosus cutaneus superficialis of the scalp. Cutis. 1989;43:143-144.
- Sáez Rodríguez M, Rodríguez-Martin M, Carnerero A, et al. Naevus lipomatosus cutaneous superficialis on the nose. J Eur Acad Dermatol Venereol. 2005;19:751-752.
- Hassab-El-Naby HMM, Rageh MA. Adult-onset nevus lipomatosus cutaneous superficialis mimicking plane xanthoma. J Clin Aesthet Dermatol. 2022;15:10-11.
- Park HJ, Park CJ, Yi JY, et al. Nevus lipomatosus superficialis on the face. Int J Dermatol. 1997;36:435-437.
- Ioannidou DJ, Stefanidou MP, Panayiotides JG, et al. Nevus lipomatosus cutaneous superficialis (Hoffman-Zurhelle) with localized scleroderma like appearance. Int J Dermatol. 2001;40:54-57.
- Nogita T, Wong TY, Hidano A, et al. Pedunculated lipofibroma. a clinicopathologic study of thirty-two cases supporting a simplified nomenclature. J Am Acad Dermatol. 1994;31(2 pt 1):235-240.
- Sawada Y. Solitary nevus lipomatosus superficialis on the forehead. Ann Plast Surg. 1986;16:356-358.
- Knoth W. Uber Naevus lipomatosus cutaneus superficialis Hoffmann-Zurhelle und uber Naevus naevocellularis partim lipomatodes. Dermatologica. 1962;125:161.
- Weitzner S. Solitary naevus lipomatosus cutaneus superficialis of scalp. Arch Dermatol. 1968;97:540-542.
- Kaw P, Carlson A, Meyer DR. Nevus lipomatosus (pedunculated lipofibroma) of the eyelid. Ophthalmic Plast Reconstr Surg. 2005;21:74-76.
- Vano-Galvan S, Moreno C, Vano-Galvan E, et al. Solitary naevus lipomatosus cutaneous superficialis on the sole. Eur J Dermatol. 2008;18:353-354.
- Adotama P, Hutson SD, Rieder EA, et al. Revisiting solitary pedunculated lipofibromas. Am J Clin Pathol. 2021;156:954-957.
- Kubin ME, Lantto U, Lindgren O, et al. A rare, recurrent spindle cell lipoma of the nose. Acta Derm Venereol. 2021;101:adv00571.
- Jung SN, Shin JW, Kwon H, et al. Fibrolipoma of the tip of the nose. J Craniofac Surg. 2009;20:555-556.
- Han XC, Zheng LQ, Shang XL. Dendritic fibromyxolipoma on the nasal tip in an old patient. Int J Clin Exp Pathol. 2014;7:7064-7067.
A 60-year-old woman presented to the dermatology department with a 6-mm, firm, pink, nonulcerated, nonmobile papule on the right nasal side wall of 1 year’s duration. It had grown slowly and was asymptomatic with no tenderness or bleeding. No other skin lesions were noted on physical examination, and her medical history was otherwise unremarkable. A shave biopsy was performed.