User login
Secukinumab improves treatment outcomes and inhibits structural damage in PsA
Key clinical point: The achievement of stringent treatment goals with secukinumab led to clinically meaningful benefits in physical function in patients with psoriatic arthritis (PsA), with secukinumab showing a protective effect on radiographic progression.
Major finding: Overall, over 2 years, a relatively small percentage of patients receiving secukinumab achieved sustained remission (REM) according to very low disease activity (LDA; 19%-24%) or Disease Activity index for PsA (DAPSA) REM (30%-36%), with those achieving DAPSA LDA+REM or DAPSA REM showing numerically greater improvement in physical function. A higher proportion of secukinumab-treated patients were non-structural progressors at 2 years irrespective of achieving sustained LDA/REM.
Study details: This was a retrospective analysis from the FUTURE 5 study including 996 patients with active PsA who were randomly assigned to receive secukinumab or placebo.
Disclosures: This study was funded by Novartis Pharma AG. Two authors declared being employees of or owning stocks/shares in Novartis, and several authors reported ties with various sources, including Novartis.
Source: Coates LC et al. Secukinumab improves physical function and quality of life and inhibits structural damage in patients with PsA with sustained remission or low disease activity: Results from the 2-year phase 3 FUTURE 5 study. RMD Open. 2023;9(2):e002939 (Apr 24). Doi: 10.1136/rmdopen-2022-002939
Key clinical point: The achievement of stringent treatment goals with secukinumab led to clinically meaningful benefits in physical function in patients with psoriatic arthritis (PsA), with secukinumab showing a protective effect on radiographic progression.
Major finding: Overall, over 2 years, a relatively small percentage of patients receiving secukinumab achieved sustained remission (REM) according to very low disease activity (LDA; 19%-24%) or Disease Activity index for PsA (DAPSA) REM (30%-36%), with those achieving DAPSA LDA+REM or DAPSA REM showing numerically greater improvement in physical function. A higher proportion of secukinumab-treated patients were non-structural progressors at 2 years irrespective of achieving sustained LDA/REM.
Study details: This was a retrospective analysis from the FUTURE 5 study including 996 patients with active PsA who were randomly assigned to receive secukinumab or placebo.
Disclosures: This study was funded by Novartis Pharma AG. Two authors declared being employees of or owning stocks/shares in Novartis, and several authors reported ties with various sources, including Novartis.
Source: Coates LC et al. Secukinumab improves physical function and quality of life and inhibits structural damage in patients with PsA with sustained remission or low disease activity: Results from the 2-year phase 3 FUTURE 5 study. RMD Open. 2023;9(2):e002939 (Apr 24). Doi: 10.1136/rmdopen-2022-002939
Key clinical point: The achievement of stringent treatment goals with secukinumab led to clinically meaningful benefits in physical function in patients with psoriatic arthritis (PsA), with secukinumab showing a protective effect on radiographic progression.
Major finding: Overall, over 2 years, a relatively small percentage of patients receiving secukinumab achieved sustained remission (REM) according to very low disease activity (LDA; 19%-24%) or Disease Activity index for PsA (DAPSA) REM (30%-36%), with those achieving DAPSA LDA+REM or DAPSA REM showing numerically greater improvement in physical function. A higher proportion of secukinumab-treated patients were non-structural progressors at 2 years irrespective of achieving sustained LDA/REM.
Study details: This was a retrospective analysis from the FUTURE 5 study including 996 patients with active PsA who were randomly assigned to receive secukinumab or placebo.
Disclosures: This study was funded by Novartis Pharma AG. Two authors declared being employees of or owning stocks/shares in Novartis, and several authors reported ties with various sources, including Novartis.
Source: Coates LC et al. Secukinumab improves physical function and quality of life and inhibits structural damage in patients with PsA with sustained remission or low disease activity: Results from the 2-year phase 3 FUTURE 5 study. RMD Open. 2023;9(2):e002939 (Apr 24). Doi: 10.1136/rmdopen-2022-002939
PARP inhibitors and breast cancer: Questions remain about wider use
oncologists explained at the European Society for Medical Oncology Breast Cancer annual congress.
For now, the drugs are only approved in high-risk germline BRCA mutation (gBRCAmut) early breast cancer, oncologist Kevin Punie, MD, of Saint Augustine Hospital in Wilrijk, Belgium, said during a session at the meeting. Combining the drugs with chemotherapy “has not yet demonstrated significant benefits, and this is irrespective whether platinum was part of the chemotherapy backbone.”
PARP is a kind of enzyme that repairs damaged DNA in cells, especially cancerous ones. PARP inhibitors block the enzyme, potentially leading more cancer cells to die, the Dana-Farber Cancer Institute states.
In a separate presentation during the same session, oncologist Andrew Tutt, MBChB, PhD, noted that a study he led – a phase 3, double-blinded, randomized 2021 trial – found that patients with BRCA1- or BRCA2-mutated breast cancer who took the PARP inhibitor olarapib (Lynparza) versus placebo had improved outcomes on several measures, including 3-year invasive disease-free survival (85.9% vs. 77.1%, P < .001). However, the study noted that “olaparib had limited effects on global patient-reported quality of life.”
Dr. Tutt, of the Institute of Cancer Research, London, and Kings College London, said 57% of patients who took olarapib suffered nausea versus 24% of those who took placebo, and fatigue and anemia were also more common in the olarapib group. Anemia can be severe and lead to transfusions in some cases.
As Dr. Punie explained, there are many reasons to consider combining PARP inhibitors with other treatments such as chemotherapy, immunotherapy, and radiotherapy. The combinations may have synergetic effects, and they could have potential in both the neoadjuvant and adjuvant settings.
The combination of the PARP inhibitor olaparib and endocrine therapy is now approved by the European Medicines Agency for the adjuvant treatment of certain patients with germline BRCA1/2 mutations who have HER2-negative, high-risk early breast cancer, Dr. Punie noted.
The 2021 study led by Dr. Tutt reported that treatment or safety differences were found in those who received both olaparib and endocrine therapy versus those who only received olarapib.
So far, Dr. Punie said, “we not yet have enough clinical evidence to say that there’s really synergy between PNP inhibitors and other anticancer therapies.” According to the National Institutes of Health, medical synergy “describes the interaction of two or more drugs when their combined effect is greater than the sum of the effects seen when each drug is given alone.”
In regard to chemotherapy, it makes sense that PARP inhibitors would be helpful in combination, Dr. Punie said. DNA damage to cancer cells accumulates during chemotherapy, he said, and they’re more depending on PARP for repair.
Study results so far have been mixed. A 2022 study, for example, found that adding the experimental PARP inhibitor veliparib to the chemotherapy regimen carboplatin-paclitaxel didn’t improve outcomes, he said. A similar study examining the addition of olaparib to carboplatin-paclitaxel is ongoing.
As for combining radiotherapy and PARP inhibitors, Dr. Punie said that preclinical findings are promising, and research is underway. There’s also ongoing research into combining PARP inhibitors with immunotherapy.
Off-label use of olaparib with immunotherapy or sequential treatment may be appropriate in the setting of adjuvant gBRCAmut triple-negative breast cancer with residual disease, he said.
During his presentation, Dr. Tutt called for researchers to investigate the use of PARP inhibitors in the de-escalation of treatment in lower-risk gBRCAmut disease.
“Clearly, some patients require chemotherapy, and we know patients respond very well to neoadjuvant chemotherapy if they have a BRCA mutation, but we don’t yet know who we can de-escalate in,” he said.
He also highlighted the need to reduce anemia in patients on PARP inhibitors, “particularly if we’re moving into lower-risk populations or possibly considering prevention trials.
“The study of PARP inhibitor resistance ... is now urgent, so that we can address it,” he said.
Dr. Punie disclosed financial relationships with AstraZeneca, Eli Lilly, Exact Sciences, Focus Patient, Medscape, MSD, Mundi Pharma, Need, Novartis, Pierre Fabre, Pfizer, F. Hoffmann–La Roche, Sanofi, Seagen, and PharmaMar. Dr. Tutt disclosed financial relationships with Artios, Gilead, MD Anderson, Merck KGaA, Pfizer, Vertex, AstraZeneca, EM Partners, Medscape Education, CRUK, Inbiomotion, Myriad Genetics, and Breast Cancer Now.
oncologists explained at the European Society for Medical Oncology Breast Cancer annual congress.
For now, the drugs are only approved in high-risk germline BRCA mutation (gBRCAmut) early breast cancer, oncologist Kevin Punie, MD, of Saint Augustine Hospital in Wilrijk, Belgium, said during a session at the meeting. Combining the drugs with chemotherapy “has not yet demonstrated significant benefits, and this is irrespective whether platinum was part of the chemotherapy backbone.”
PARP is a kind of enzyme that repairs damaged DNA in cells, especially cancerous ones. PARP inhibitors block the enzyme, potentially leading more cancer cells to die, the Dana-Farber Cancer Institute states.
In a separate presentation during the same session, oncologist Andrew Tutt, MBChB, PhD, noted that a study he led – a phase 3, double-blinded, randomized 2021 trial – found that patients with BRCA1- or BRCA2-mutated breast cancer who took the PARP inhibitor olarapib (Lynparza) versus placebo had improved outcomes on several measures, including 3-year invasive disease-free survival (85.9% vs. 77.1%, P < .001). However, the study noted that “olaparib had limited effects on global patient-reported quality of life.”
Dr. Tutt, of the Institute of Cancer Research, London, and Kings College London, said 57% of patients who took olarapib suffered nausea versus 24% of those who took placebo, and fatigue and anemia were also more common in the olarapib group. Anemia can be severe and lead to transfusions in some cases.
As Dr. Punie explained, there are many reasons to consider combining PARP inhibitors with other treatments such as chemotherapy, immunotherapy, and radiotherapy. The combinations may have synergetic effects, and they could have potential in both the neoadjuvant and adjuvant settings.
The combination of the PARP inhibitor olaparib and endocrine therapy is now approved by the European Medicines Agency for the adjuvant treatment of certain patients with germline BRCA1/2 mutations who have HER2-negative, high-risk early breast cancer, Dr. Punie noted.
The 2021 study led by Dr. Tutt reported that treatment or safety differences were found in those who received both olaparib and endocrine therapy versus those who only received olarapib.
So far, Dr. Punie said, “we not yet have enough clinical evidence to say that there’s really synergy between PNP inhibitors and other anticancer therapies.” According to the National Institutes of Health, medical synergy “describes the interaction of two or more drugs when their combined effect is greater than the sum of the effects seen when each drug is given alone.”
In regard to chemotherapy, it makes sense that PARP inhibitors would be helpful in combination, Dr. Punie said. DNA damage to cancer cells accumulates during chemotherapy, he said, and they’re more depending on PARP for repair.
Study results so far have been mixed. A 2022 study, for example, found that adding the experimental PARP inhibitor veliparib to the chemotherapy regimen carboplatin-paclitaxel didn’t improve outcomes, he said. A similar study examining the addition of olaparib to carboplatin-paclitaxel is ongoing.
As for combining radiotherapy and PARP inhibitors, Dr. Punie said that preclinical findings are promising, and research is underway. There’s also ongoing research into combining PARP inhibitors with immunotherapy.
Off-label use of olaparib with immunotherapy or sequential treatment may be appropriate in the setting of adjuvant gBRCAmut triple-negative breast cancer with residual disease, he said.
During his presentation, Dr. Tutt called for researchers to investigate the use of PARP inhibitors in the de-escalation of treatment in lower-risk gBRCAmut disease.
“Clearly, some patients require chemotherapy, and we know patients respond very well to neoadjuvant chemotherapy if they have a BRCA mutation, but we don’t yet know who we can de-escalate in,” he said.
He also highlighted the need to reduce anemia in patients on PARP inhibitors, “particularly if we’re moving into lower-risk populations or possibly considering prevention trials.
“The study of PARP inhibitor resistance ... is now urgent, so that we can address it,” he said.
Dr. Punie disclosed financial relationships with AstraZeneca, Eli Lilly, Exact Sciences, Focus Patient, Medscape, MSD, Mundi Pharma, Need, Novartis, Pierre Fabre, Pfizer, F. Hoffmann–La Roche, Sanofi, Seagen, and PharmaMar. Dr. Tutt disclosed financial relationships with Artios, Gilead, MD Anderson, Merck KGaA, Pfizer, Vertex, AstraZeneca, EM Partners, Medscape Education, CRUK, Inbiomotion, Myriad Genetics, and Breast Cancer Now.
oncologists explained at the European Society for Medical Oncology Breast Cancer annual congress.
For now, the drugs are only approved in high-risk germline BRCA mutation (gBRCAmut) early breast cancer, oncologist Kevin Punie, MD, of Saint Augustine Hospital in Wilrijk, Belgium, said during a session at the meeting. Combining the drugs with chemotherapy “has not yet demonstrated significant benefits, and this is irrespective whether platinum was part of the chemotherapy backbone.”
PARP is a kind of enzyme that repairs damaged DNA in cells, especially cancerous ones. PARP inhibitors block the enzyme, potentially leading more cancer cells to die, the Dana-Farber Cancer Institute states.
In a separate presentation during the same session, oncologist Andrew Tutt, MBChB, PhD, noted that a study he led – a phase 3, double-blinded, randomized 2021 trial – found that patients with BRCA1- or BRCA2-mutated breast cancer who took the PARP inhibitor olarapib (Lynparza) versus placebo had improved outcomes on several measures, including 3-year invasive disease-free survival (85.9% vs. 77.1%, P < .001). However, the study noted that “olaparib had limited effects on global patient-reported quality of life.”
Dr. Tutt, of the Institute of Cancer Research, London, and Kings College London, said 57% of patients who took olarapib suffered nausea versus 24% of those who took placebo, and fatigue and anemia were also more common in the olarapib group. Anemia can be severe and lead to transfusions in some cases.
As Dr. Punie explained, there are many reasons to consider combining PARP inhibitors with other treatments such as chemotherapy, immunotherapy, and radiotherapy. The combinations may have synergetic effects, and they could have potential in both the neoadjuvant and adjuvant settings.
The combination of the PARP inhibitor olaparib and endocrine therapy is now approved by the European Medicines Agency for the adjuvant treatment of certain patients with germline BRCA1/2 mutations who have HER2-negative, high-risk early breast cancer, Dr. Punie noted.
The 2021 study led by Dr. Tutt reported that treatment or safety differences were found in those who received both olaparib and endocrine therapy versus those who only received olarapib.
So far, Dr. Punie said, “we not yet have enough clinical evidence to say that there’s really synergy between PNP inhibitors and other anticancer therapies.” According to the National Institutes of Health, medical synergy “describes the interaction of two or more drugs when their combined effect is greater than the sum of the effects seen when each drug is given alone.”
In regard to chemotherapy, it makes sense that PARP inhibitors would be helpful in combination, Dr. Punie said. DNA damage to cancer cells accumulates during chemotherapy, he said, and they’re more depending on PARP for repair.
Study results so far have been mixed. A 2022 study, for example, found that adding the experimental PARP inhibitor veliparib to the chemotherapy regimen carboplatin-paclitaxel didn’t improve outcomes, he said. A similar study examining the addition of olaparib to carboplatin-paclitaxel is ongoing.
As for combining radiotherapy and PARP inhibitors, Dr. Punie said that preclinical findings are promising, and research is underway. There’s also ongoing research into combining PARP inhibitors with immunotherapy.
Off-label use of olaparib with immunotherapy or sequential treatment may be appropriate in the setting of adjuvant gBRCAmut triple-negative breast cancer with residual disease, he said.
During his presentation, Dr. Tutt called for researchers to investigate the use of PARP inhibitors in the de-escalation of treatment in lower-risk gBRCAmut disease.
“Clearly, some patients require chemotherapy, and we know patients respond very well to neoadjuvant chemotherapy if they have a BRCA mutation, but we don’t yet know who we can de-escalate in,” he said.
He also highlighted the need to reduce anemia in patients on PARP inhibitors, “particularly if we’re moving into lower-risk populations or possibly considering prevention trials.
“The study of PARP inhibitor resistance ... is now urgent, so that we can address it,” he said.
Dr. Punie disclosed financial relationships with AstraZeneca, Eli Lilly, Exact Sciences, Focus Patient, Medscape, MSD, Mundi Pharma, Need, Novartis, Pierre Fabre, Pfizer, F. Hoffmann–La Roche, Sanofi, Seagen, and PharmaMar. Dr. Tutt disclosed financial relationships with Artios, Gilead, MD Anderson, Merck KGaA, Pfizer, Vertex, AstraZeneca, EM Partners, Medscape Education, CRUK, Inbiomotion, Myriad Genetics, and Breast Cancer Now.
FROM ESMO BREAST CANCER 2023
Highlights in Multiple Sclerosis From AAN 2023
Conversion to multiple sclerosis (MS) and a novel drug to prevent it are among the MS highlights from the 2023 American Academy of Neurology (AAN) Annual Meeting, as reported by Dr Fred Lublin of the Icahn School of Medicine in New York.
Dr Lublin starts with three studies examining radiologic isolated syndrome (RIS), the first of which looked at MRI and spinal fluid factors associated with conversion to MS. The study indicated that less stringent criteria for RIS may have to be considered.
The second study, again using data obtained via MRI, showed that the presence of pragmatic rim lesions may be an indicator of more severe disease. The third study was a clinical trial of teriflunomide. This suggested that the drug reduced conversion rates to MS by almost two thirds compared with placebo.
Dr Lublin then turns to an investigation of the impact of assisted reproductive technologies on relapse risk in women with MS. Reassuringly, women who continued with disease-modifying therapy during reproductive assistance were less likely to relapse.
Finally, he discusses a study showing that women with MS who followed a Mediterranean diet had better cognition scores.
--
Fred Lublin, MD, Saunders Family Professor of Neurology; Director, The Corinne Goldsmith Dickinson Center for Multiple Sclerosis, Icahn School of Medicine at Mount Sinai, New York, NY
Fred D. Lublin, MD, has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for: Biogen; EMD Serono; Novartis; Teva; Actelion/Janssen; Sanofi/Genzyme; Acorda; Roche/Genentech; Viela Bio/Horizon
Serve(d) as a speaker or a member of a speakers bureau for: Sanofi; Biogen
Received research grant from: Biogen; Novartis; Actelion; Biogen; Sanofi; National Multiple Sclerosis Society; Brainstorm Cell Therapeutics; National Institutes of Health
Conversion to multiple sclerosis (MS) and a novel drug to prevent it are among the MS highlights from the 2023 American Academy of Neurology (AAN) Annual Meeting, as reported by Dr Fred Lublin of the Icahn School of Medicine in New York.
Dr Lublin starts with three studies examining radiologic isolated syndrome (RIS), the first of which looked at MRI and spinal fluid factors associated with conversion to MS. The study indicated that less stringent criteria for RIS may have to be considered.
The second study, again using data obtained via MRI, showed that the presence of pragmatic rim lesions may be an indicator of more severe disease. The third study was a clinical trial of teriflunomide. This suggested that the drug reduced conversion rates to MS by almost two thirds compared with placebo.
Dr Lublin then turns to an investigation of the impact of assisted reproductive technologies on relapse risk in women with MS. Reassuringly, women who continued with disease-modifying therapy during reproductive assistance were less likely to relapse.
Finally, he discusses a study showing that women with MS who followed a Mediterranean diet had better cognition scores.
--
Fred Lublin, MD, Saunders Family Professor of Neurology; Director, The Corinne Goldsmith Dickinson Center for Multiple Sclerosis, Icahn School of Medicine at Mount Sinai, New York, NY
Fred D. Lublin, MD, has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for: Biogen; EMD Serono; Novartis; Teva; Actelion/Janssen; Sanofi/Genzyme; Acorda; Roche/Genentech; Viela Bio/Horizon
Serve(d) as a speaker or a member of a speakers bureau for: Sanofi; Biogen
Received research grant from: Biogen; Novartis; Actelion; Biogen; Sanofi; National Multiple Sclerosis Society; Brainstorm Cell Therapeutics; National Institutes of Health
Conversion to multiple sclerosis (MS) and a novel drug to prevent it are among the MS highlights from the 2023 American Academy of Neurology (AAN) Annual Meeting, as reported by Dr Fred Lublin of the Icahn School of Medicine in New York.
Dr Lublin starts with three studies examining radiologic isolated syndrome (RIS), the first of which looked at MRI and spinal fluid factors associated with conversion to MS. The study indicated that less stringent criteria for RIS may have to be considered.
The second study, again using data obtained via MRI, showed that the presence of pragmatic rim lesions may be an indicator of more severe disease. The third study was a clinical trial of teriflunomide. This suggested that the drug reduced conversion rates to MS by almost two thirds compared with placebo.
Dr Lublin then turns to an investigation of the impact of assisted reproductive technologies on relapse risk in women with MS. Reassuringly, women who continued with disease-modifying therapy during reproductive assistance were less likely to relapse.
Finally, he discusses a study showing that women with MS who followed a Mediterranean diet had better cognition scores.
--
Fred Lublin, MD, Saunders Family Professor of Neurology; Director, The Corinne Goldsmith Dickinson Center for Multiple Sclerosis, Icahn School of Medicine at Mount Sinai, New York, NY
Fred D. Lublin, MD, has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for: Biogen; EMD Serono; Novartis; Teva; Actelion/Janssen; Sanofi/Genzyme; Acorda; Roche/Genentech; Viela Bio/Horizon
Serve(d) as a speaker or a member of a speakers bureau for: Sanofi; Biogen
Received research grant from: Biogen; Novartis; Actelion; Biogen; Sanofi; National Multiple Sclerosis Society; Brainstorm Cell Therapeutics; National Institutes of Health

Mailed HPV test kits boost cervical cancer screening
The self-sampling kits, which detect human papillomavirus (HPV), are available only for use in clinical trials, but the researchers hope that eventually these kits will be approved for use by the general public.
The researchers, from the University of North Carolina, explored use of these kits in the My Body, My Test-3 study, which was published online in The Lancet Public Health.
In a commentary published with the study, Runzhi Wang, MD, and Jennell Coleman, MD, MPH, both of Johns Hopkins University, Baltimore, said it “provides the required evidence that ... self-collected samples can be an effective strategy for hard-to-reach populations.”
The study involved 665 women (aged 25-64) in North Carolina who were either uninsured or enrolled in Medicaid or Medicare. The patients had low-income backgrounds and lived in urban areas. More than half self-reported as Black or Hispanic (55%), uninsured (78%) or unemployed (57%). None had a Pap smear in at least 4 years or a high-risk HPV test in the last 6 years.
Two-thirds of the women were mailed an HPV self-collection kit and received assistance with scheduling an in-person screening appointment. The kit included a Viba-Brush device, which is inserted into the vagina like a tampon to collect the sample.
The other third of women, the control group, only received scheduling assistance.
The team found that mailing the self-collection tests along with helping women book in-clinic appointments improved screening rates twofold, compared with just assisting patients to schedule an appointment.
Screening success among those who received the at-home collection kit was 72%, compared with 37% in the control group.
Of those who received the kits, 78% returned them. This is “impressive,” said Dr. Wang and Dr. Coleman, as previous studies have reported return rates of only 8%-20%.

About 23% of eligible women are overdue for cervical cancer screening by at least a year, according to the National Cancer Institute. Jennifer Smith, PhD, MPH, professor of epidemiology at the University of North Carolina at Chapel Hill and an author of the study, believes every woman deserves equal access to cervical screening.
“I think we really need to make efforts to increase cervical cancer screening among women who are overdue for screening by a year or more from the recommended guidelines,” Dr. Smith said. “We’ve proven along with the wide evidence both in the U.S. and globally that self-collection intervention works well and can motivate screening uptake by breaking down barriers for populations that have less access to care.”
“We’re hoping this research in combination with all of the extensive evidence on the positive performance of HPV self-collection will provide additional information to be considered by the FDA for approval of the kits for primary screening,” Dr. Smith said.

“Government approval of at-home HPV tests would have a huge impact,” said coauthor Noel Brewer, PhD, also of UNC Chapel Hill. “We could better reach those in rural areas where cervical cancer screening is hard to come by.”
Dr. Smith has received research grants, supply donations, and consultancies for Hologic and BD Diagnostics. Dr. Brewer, Dr. Wang, and Dr. Coleman reported no conflicts of interest.
A version of this article first appeared on WebMD.com.
The self-sampling kits, which detect human papillomavirus (HPV), are available only for use in clinical trials, but the researchers hope that eventually these kits will be approved for use by the general public.
The researchers, from the University of North Carolina, explored use of these kits in the My Body, My Test-3 study, which was published online in The Lancet Public Health.
In a commentary published with the study, Runzhi Wang, MD, and Jennell Coleman, MD, MPH, both of Johns Hopkins University, Baltimore, said it “provides the required evidence that ... self-collected samples can be an effective strategy for hard-to-reach populations.”
The study involved 665 women (aged 25-64) in North Carolina who were either uninsured or enrolled in Medicaid or Medicare. The patients had low-income backgrounds and lived in urban areas. More than half self-reported as Black or Hispanic (55%), uninsured (78%) or unemployed (57%). None had a Pap smear in at least 4 years or a high-risk HPV test in the last 6 years.
Two-thirds of the women were mailed an HPV self-collection kit and received assistance with scheduling an in-person screening appointment. The kit included a Viba-Brush device, which is inserted into the vagina like a tampon to collect the sample.
The other third of women, the control group, only received scheduling assistance.
The team found that mailing the self-collection tests along with helping women book in-clinic appointments improved screening rates twofold, compared with just assisting patients to schedule an appointment.
Screening success among those who received the at-home collection kit was 72%, compared with 37% in the control group.
Of those who received the kits, 78% returned them. This is “impressive,” said Dr. Wang and Dr. Coleman, as previous studies have reported return rates of only 8%-20%.
About 23% of eligible women are overdue for cervical cancer screening by at least a year, according to the National Cancer Institute. Jennifer Smith, PhD, MPH, professor of epidemiology at the University of North Carolina at Chapel Hill and an author of the study, believes every woman deserves equal access to cervical screening.
“I think we really need to make efforts to increase cervical cancer screening among women who are overdue for screening by a year or more from the recommended guidelines,” Dr. Smith said. “We’ve proven along with the wide evidence both in the U.S. and globally that self-collection intervention works well and can motivate screening uptake by breaking down barriers for populations that have less access to care.”
“We’re hoping this research in combination with all of the extensive evidence on the positive performance of HPV self-collection will provide additional information to be considered by the FDA for approval of the kits for primary screening,” Dr. Smith said.
“Government approval of at-home HPV tests would have a huge impact,” said coauthor Noel Brewer, PhD, also of UNC Chapel Hill. “We could better reach those in rural areas where cervical cancer screening is hard to come by.”
Dr. Smith has received research grants, supply donations, and consultancies for Hologic and BD Diagnostics. Dr. Brewer, Dr. Wang, and Dr. Coleman reported no conflicts of interest.
A version of this article first appeared on WebMD.com.
The self-sampling kits, which detect human papillomavirus (HPV), are available only for use in clinical trials, but the researchers hope that eventually these kits will be approved for use by the general public.
The researchers, from the University of North Carolina, explored use of these kits in the My Body, My Test-3 study, which was published online in The Lancet Public Health.
In a commentary published with the study, Runzhi Wang, MD, and Jennell Coleman, MD, MPH, both of Johns Hopkins University, Baltimore, said it “provides the required evidence that ... self-collected samples can be an effective strategy for hard-to-reach populations.”
The study involved 665 women (aged 25-64) in North Carolina who were either uninsured or enrolled in Medicaid or Medicare. The patients had low-income backgrounds and lived in urban areas. More than half self-reported as Black or Hispanic (55%), uninsured (78%) or unemployed (57%). None had a Pap smear in at least 4 years or a high-risk HPV test in the last 6 years.
Two-thirds of the women were mailed an HPV self-collection kit and received assistance with scheduling an in-person screening appointment. The kit included a Viba-Brush device, which is inserted into the vagina like a tampon to collect the sample.
The other third of women, the control group, only received scheduling assistance.
The team found that mailing the self-collection tests along with helping women book in-clinic appointments improved screening rates twofold, compared with just assisting patients to schedule an appointment.
Screening success among those who received the at-home collection kit was 72%, compared with 37% in the control group.
Of those who received the kits, 78% returned them. This is “impressive,” said Dr. Wang and Dr. Coleman, as previous studies have reported return rates of only 8%-20%.
About 23% of eligible women are overdue for cervical cancer screening by at least a year, according to the National Cancer Institute. Jennifer Smith, PhD, MPH, professor of epidemiology at the University of North Carolina at Chapel Hill and an author of the study, believes every woman deserves equal access to cervical screening.
“I think we really need to make efforts to increase cervical cancer screening among women who are overdue for screening by a year or more from the recommended guidelines,” Dr. Smith said. “We’ve proven along with the wide evidence both in the U.S. and globally that self-collection intervention works well and can motivate screening uptake by breaking down barriers for populations that have less access to care.”
“We’re hoping this research in combination with all of the extensive evidence on the positive performance of HPV self-collection will provide additional information to be considered by the FDA for approval of the kits for primary screening,” Dr. Smith said.
“Government approval of at-home HPV tests would have a huge impact,” said coauthor Noel Brewer, PhD, also of UNC Chapel Hill. “We could better reach those in rural areas where cervical cancer screening is hard to come by.”
Dr. Smith has received research grants, supply donations, and consultancies for Hologic and BD Diagnostics. Dr. Brewer, Dr. Wang, and Dr. Coleman reported no conflicts of interest.
A version of this article first appeared on WebMD.com.
FROM THE LANCET PUBLIC HEALTH
Expunging ‘penicillin allergy’: Your questions answered
Last month, I described a 28-year-old patient with a history of injection drug use who presented with pain in his left forearm. His history showed that, within the past 2 years, he’d been seen for cutaneous infections multiple times as an outpatient and in the emergency department. His records indicated that he was diagnosed with a penicillin allergy as a child when he developed a rash after receiving amoxicillin. I believed the next course of action should be to test for a penicillin allergy with an oral amoxicillin challenge.
Thank you for your excellent questions regarding this case. Great to hear the enthusiasm for testing for penicillin allergy!
One question focused on the course of action in the case of a mild or moderate IgE-mediated reaction after a single dose test with amoxicillin. Treatment for these reactions should include an antihistamine. I would reserve intravenous antihistamines for more severe cases, which also require treatment with a course of corticosteroids. However, the risk for a moderate to severe reaction to amoxicillin on retesting is quite low.
Clinicians need to exercise caution in the use of systemic corticosteroids. These drugs can be lifesaving, but even short courses of corticosteroids are associated with potentially serious adverse events. In a review of adverse events associated with short-course systemic corticosteroids among children, the rate of vomiting was 5.4%; behavioral change, 4.7%; and sleep disturbance, 4.3%. One child died after contracting herpes zoster, more than one-third of children developed elevated blood pressure, and 81.1% had evidence of suppression of the hypothalamic-pituitary-adrenal axis.
Among adults, short courses of systemic corticosteroids are associated with acute increases in the risks for gastrointestinal bleeding and hypertension. Cumulative exposure to short courses of corticosteroids over time results in higher risks for obesity, type 2 diabetes, and osteoporosis.
Another question prompted by this young man’s case focused on the durability of IgE reactions against penicillin. The IgE response to penicillin does indeed wane over time; 80% of patients with a previous true penicillin allergy can tolerate the antibiotic after 10 years. Thus, about 95% of patients with a remote history of penicillin allergy are tolerant of penicillin, and testing can be performed using the algorithm described.
Clinicians should avoid applying current guidelines for the evaluation of patients with penicillin allergy to other common drug allergies. The overall prevalence of sulfonamide allergy is 3%-8%, and the vast majority of these reactions follow treatment with trimethoprim-sulfamethoxazole. Sulfa allergy is even more common among persons living with HIV infection. The natural history of sulfa allergy is not as well established as penicillin allergy. Allergy testing is encouraged in these cases. Graded oral challenge testing is best reserved for patients who are unlikely to have a true sulfa allergy based on their history.
A version of this article first appeared on Medscape.com.
Last month, I described a 28-year-old patient with a history of injection drug use who presented with pain in his left forearm. His history showed that, within the past 2 years, he’d been seen for cutaneous infections multiple times as an outpatient and in the emergency department. His records indicated that he was diagnosed with a penicillin allergy as a child when he developed a rash after receiving amoxicillin. I believed the next course of action should be to test for a penicillin allergy with an oral amoxicillin challenge.
Thank you for your excellent questions regarding this case. Great to hear the enthusiasm for testing for penicillin allergy!
One question focused on the course of action in the case of a mild or moderate IgE-mediated reaction after a single dose test with amoxicillin. Treatment for these reactions should include an antihistamine. I would reserve intravenous antihistamines for more severe cases, which also require treatment with a course of corticosteroids. However, the risk for a moderate to severe reaction to amoxicillin on retesting is quite low.
Clinicians need to exercise caution in the use of systemic corticosteroids. These drugs can be lifesaving, but even short courses of corticosteroids are associated with potentially serious adverse events. In a review of adverse events associated with short-course systemic corticosteroids among children, the rate of vomiting was 5.4%; behavioral change, 4.7%; and sleep disturbance, 4.3%. One child died after contracting herpes zoster, more than one-third of children developed elevated blood pressure, and 81.1% had evidence of suppression of the hypothalamic-pituitary-adrenal axis.
Among adults, short courses of systemic corticosteroids are associated with acute increases in the risks for gastrointestinal bleeding and hypertension. Cumulative exposure to short courses of corticosteroids over time results in higher risks for obesity, type 2 diabetes, and osteoporosis.
Another question prompted by this young man’s case focused on the durability of IgE reactions against penicillin. The IgE response to penicillin does indeed wane over time; 80% of patients with a previous true penicillin allergy can tolerate the antibiotic after 10 years. Thus, about 95% of patients with a remote history of penicillin allergy are tolerant of penicillin, and testing can be performed using the algorithm described.
Clinicians should avoid applying current guidelines for the evaluation of patients with penicillin allergy to other common drug allergies. The overall prevalence of sulfonamide allergy is 3%-8%, and the vast majority of these reactions follow treatment with trimethoprim-sulfamethoxazole. Sulfa allergy is even more common among persons living with HIV infection. The natural history of sulfa allergy is not as well established as penicillin allergy. Allergy testing is encouraged in these cases. Graded oral challenge testing is best reserved for patients who are unlikely to have a true sulfa allergy based on their history.
A version of this article first appeared on Medscape.com.
Last month, I described a 28-year-old patient with a history of injection drug use who presented with pain in his left forearm. His history showed that, within the past 2 years, he’d been seen for cutaneous infections multiple times as an outpatient and in the emergency department. His records indicated that he was diagnosed with a penicillin allergy as a child when he developed a rash after receiving amoxicillin. I believed the next course of action should be to test for a penicillin allergy with an oral amoxicillin challenge.
Thank you for your excellent questions regarding this case. Great to hear the enthusiasm for testing for penicillin allergy!
One question focused on the course of action in the case of a mild or moderate IgE-mediated reaction after a single dose test with amoxicillin. Treatment for these reactions should include an antihistamine. I would reserve intravenous antihistamines for more severe cases, which also require treatment with a course of corticosteroids. However, the risk for a moderate to severe reaction to amoxicillin on retesting is quite low.
Clinicians need to exercise caution in the use of systemic corticosteroids. These drugs can be lifesaving, but even short courses of corticosteroids are associated with potentially serious adverse events. In a review of adverse events associated with short-course systemic corticosteroids among children, the rate of vomiting was 5.4%; behavioral change, 4.7%; and sleep disturbance, 4.3%. One child died after contracting herpes zoster, more than one-third of children developed elevated blood pressure, and 81.1% had evidence of suppression of the hypothalamic-pituitary-adrenal axis.
Among adults, short courses of systemic corticosteroids are associated with acute increases in the risks for gastrointestinal bleeding and hypertension. Cumulative exposure to short courses of corticosteroids over time results in higher risks for obesity, type 2 diabetes, and osteoporosis.
Another question prompted by this young man’s case focused on the durability of IgE reactions against penicillin. The IgE response to penicillin does indeed wane over time; 80% of patients with a previous true penicillin allergy can tolerate the antibiotic after 10 years. Thus, about 95% of patients with a remote history of penicillin allergy are tolerant of penicillin, and testing can be performed using the algorithm described.
Clinicians should avoid applying current guidelines for the evaluation of patients with penicillin allergy to other common drug allergies. The overall prevalence of sulfonamide allergy is 3%-8%, and the vast majority of these reactions follow treatment with trimethoprim-sulfamethoxazole. Sulfa allergy is even more common among persons living with HIV infection. The natural history of sulfa allergy is not as well established as penicillin allergy. Allergy testing is encouraged in these cases. Graded oral challenge testing is best reserved for patients who are unlikely to have a true sulfa allergy based on their history.
A version of this article first appeared on Medscape.com.
Combination Therapies for Patients With Type 2 Diabetes
The treatment of type 2 diabetes (T2D) has traditionally followed a stepwise approach, beginning with metformin and continuing with the addition of other agents to achieve target glycemic control.
In this ReCAP, Dr Chika Anekwe, of the Massachusetts General Hospital Weight Center in Boston, Massachusetts, discusses categories for antidiabetic therapies, when to combine medications that have differing mechanisms, and the clinical benefits of combination therapy, such as a stronger response in the reduction of hemoglobin A1c as well as loss of body weight.
She also discusses tirzepatide, a dual therapy combining glucagon-like peptide-1 agonist and glucose-dependent insulinotropic polypeptide (GIP) agonist, which has shown clinically meaningful results for both blood sugar reduction and body weight reduction for patients with T2D.
--
Chika V. Anekwe, MD, MPH, Obesity Medicine Physician, Massachusetts General Hospital Weight Center, Boston, Massachusetts
Chika V. Anekwe, MD, MPH, has disclosed no relevant financial relationships.
The treatment of type 2 diabetes (T2D) has traditionally followed a stepwise approach, beginning with metformin and continuing with the addition of other agents to achieve target glycemic control.
In this ReCAP, Dr Chika Anekwe, of the Massachusetts General Hospital Weight Center in Boston, Massachusetts, discusses categories for antidiabetic therapies, when to combine medications that have differing mechanisms, and the clinical benefits of combination therapy, such as a stronger response in the reduction of hemoglobin A1c as well as loss of body weight.
She also discusses tirzepatide, a dual therapy combining glucagon-like peptide-1 agonist and glucose-dependent insulinotropic polypeptide (GIP) agonist, which has shown clinically meaningful results for both blood sugar reduction and body weight reduction for patients with T2D.
--
Chika V. Anekwe, MD, MPH, Obesity Medicine Physician, Massachusetts General Hospital Weight Center, Boston, Massachusetts
Chika V. Anekwe, MD, MPH, has disclosed no relevant financial relationships.
The treatment of type 2 diabetes (T2D) has traditionally followed a stepwise approach, beginning with metformin and continuing with the addition of other agents to achieve target glycemic control.
In this ReCAP, Dr Chika Anekwe, of the Massachusetts General Hospital Weight Center in Boston, Massachusetts, discusses categories for antidiabetic therapies, when to combine medications that have differing mechanisms, and the clinical benefits of combination therapy, such as a stronger response in the reduction of hemoglobin A1c as well as loss of body weight.
She also discusses tirzepatide, a dual therapy combining glucagon-like peptide-1 agonist and glucose-dependent insulinotropic polypeptide (GIP) agonist, which has shown clinically meaningful results for both blood sugar reduction and body weight reduction for patients with T2D.
--
Chika V. Anekwe, MD, MPH, Obesity Medicine Physician, Massachusetts General Hospital Weight Center, Boston, Massachusetts
Chika V. Anekwe, MD, MPH, has disclosed no relevant financial relationships.

Would you prescribe antenatal steroids to a pregnant patient at high risk for delivering at 22 weeks’ gestation?
For many decades, the limit of newborn viability was at approximately 24 weeks’ gestation. Recent advances in pregnancy and neonatal care suggest that the new limit of viability is 22 (22 weeks and 0 days to 22 weeks and 6 days) or 23 (23 weeks and 0 days to 23 weeks and 6 days) weeks of gestation. In addition, data from observational cohort studies indicate that for infants born at 22 and 23 weeks’ gestation, survival is dependent on a course of antenatal steroids administered prior to birth plus intensive respiratory and cardiovascular support at delivery and in the neonatal intensive care unit (NICU).
Antenatal steroids: Critical for survival at 22 and 23 weeks of gestation
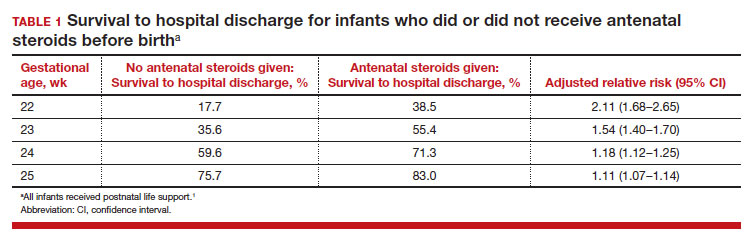
Most studies of birth outcomes at 22 and 23 weeks’ gestation rely on observational cohorts where unmeasured differences among the maternal-fetal dyads that received or did not receive a specific treatment confounds the interpretation of the data. However, data from multiple large observational cohorts suggest that between 22 and 24 weeks of gestation, completion of a course of antenatal steroids will optimize infant outcomes. Particularly noteworthy was the observation that the incremental survival benefit of antenatal steroids was greatest at 22 and 23 weeks’ gestation (TABLE 1).1 Similar results have been reported by Rossi and colleagues (TABLE 2).2

The importance of a completed course of antenatal steroids before birth was confirmed in another cohort study of 431 infants born in 2016 to 2019 at 22 weeks and 0 days’ to 23 weeks and 6 days’ gestation.3 Survival to discharge occurred in 53.9% of infants who received a full course of antenatal steroids before birth and 35.5% among those who did not receive antenatal steroids..3 Survival to discharge without major neonatal morbidities was 26.9% in those who received a full course of antenatal steroids and 10% among those who did not. In this cohort, major neonatal morbidities included severe intracranial hemorrhage, cystic periventricular leukomalacia, severe bronchopulmonary dysplasia, surgical necrotizing enterocolitis, or severe retinopathy of prematurity requiring treatment.
The American College of Obstetricians and Gynecologists (ACOG) recommends against antenatal steroids prior to 22 weeks and 0 days gestation.4 However, some neonatologists might recommend that antenatal steroids be given starting at 21 weeks and 5 days of gestation if birth is anticipated in the 22nd week of gestation and the patient prefers aggressive treatment of the newborn.
Active respiratory and cardiovascular support improves newborn outcomes
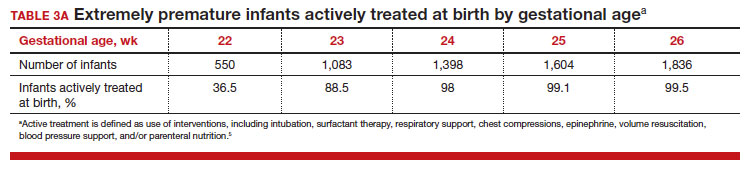
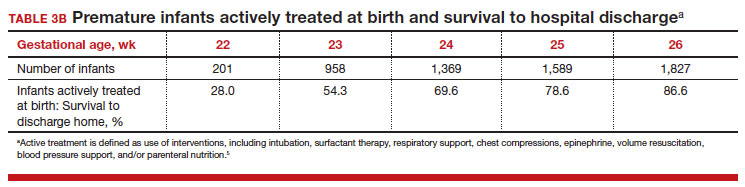
To maximize survival, infants born at 22 and 23 weeks’ gestation always require intensive active treatment at birth and in the following days in the NICU. Active treatment may include respiratory support, surfactant treatment, pressors, closure of a patent ductus arteriosus, transfusion of red blood cells, and parenteral nutrition. In one observational cohort study, active treatment at birth was not routinely provided at 22 and 23 weeks’ gestation but was routinely provided at later gestational ages (TABLE 3A).5 Not surprisingly, active treatment, especially at early gestational ages, is associated with improved survival to discharge. For example, at 22 weeks’ gestation, survival to discharge in infants who received or did not receive intensive active treatment was 28% and 0%, respectively.5 However, specific clinical characteristics of the pregnant patient and newborn may have influenced which infants were actively treated, confounding interpretation of the observation. In this cohort of extremely premature newborns, survival to hospital discharge increased substantially between 22 weeks and 26 weeks of gestational age (TABLE 3B).5
Many of the surviving infants needed chronic support treatment. Among surviving infants born at 22 weeks and 26 weeks, chronic support treatments were being used by 22.6% and 10.6% of infants, respectively, 2 years after birth.5 For surviving infants born at 22 weeks, the specific chronic support treatments included gastrostomy or feeding tube (19.4%), oxygen (9.7%), pulse oximeter (9.7%), and/or tracheostomy (3.2%). For surviving infants born at 26 weeks’ gestation, the specific chronic support treatments included gastrotomy or feeding tube (8.5%), pulse oximeter (4.4%), oxygen (3.2%), tracheostomy (2.3%), an apnea monitor (1.5%), and/or ventilator or continuous positive airway pressure (1.1%).5
Continue to: Evolving improvement in infant outcomes...
Evolving improvement in infant outcomes
In 1963, Jacqueline Bouvier Kennedy went into preterm labor at 34 weeks of gestation and delivered her son Patrick at a community facility. Due to severe respiratory distress syndrome, Patrick was transferred to the Boston Children’s Hospital, and he died shortly thereafter.6 Sixty years later, due to advances in obstetric and neonatal care, death from respiratory distress syndrome at 34 weeks of gestation is uncommon in the United States.
Infant outcomes following birth at 22 and 23 weeks’ gestation continue to improve. An observational cohort study from Sweden reported that at 22 weeks’ gestation, the percentage of live-born infants who survived to 1-year post birth in 2004 to 2007 and 2014 to 2016 was 10% and 30%, respectively.7 Similarly, at 23 weeks’ gestation, the percentage of live-born infants who survived to 1-year post birth in 2004 to 2007 and 2014 to 2016 was 52% and 61%, respectively.7 However, most of the surviving infants in this cohort had one or more major neonatal morbidities, including intraventricular hemorrhage grade 3 or 4; periventricular leukomalacia; necrotizing enterocolitis; retinopathy of prematurity grade 3, 4, or 5; or severe bronchopulmonary dysplasia.7
In a cohort of infants born in Japan at 22 to 24 weeks of gestation, there was a notable decrease in major neurodisability at 3 years of age for births occurring in 2 epochs, 2003 to 2007 and 2008 to 2012.8 When comparing outcomes in 2003 to 2007 versus 2008 to 2012, the change in rate of various major complications included the following: cerebral palsy (15.9% vs 9.5%), visual impairment (13.6% vs 4.4%), blindness (4.8% vs 1.3%), and hearing impairment (2.6% vs 1.0%). In contrast, the rate of cognitive impairment, defined as less than 70% of standard test performance for chronological age, was similar in the 2 time periods (36.5% and 37.9%, respectively).8 Based on data reported between 2000 and 2020, a systematic review and meta-analysis by Backes and colleagues concluded that there has been substantial improvement in the survival of infants born at 22 weeks of gestation.9
The small baby unit
A feature of modern medicine is the relentless evolution of new clinical subspecialties and sub-subspecialties. NICUs evolved from newborn nurseries to serve the needs of the most severely ill newborns, with care provided by a cadre of highly trained subspecialized neonatologists and neonatal nurses. A new era is dawning, with some NICUs developing a sub-subspecialized small baby unit to care for infants born between 22 and 26 weeks of gestation. These units often are staffed by clinicians with a specific interest in optimizing the care of extremely preterm infants, providing continuity of care over a long hospitalization.10 The benefits of a small baby unit may include:
- relentless standardization and adherence to the best intensive care practices
- daily use of checklists
- strict adherence to central line care
- timely extubation and transition to continuous positive airway pressure
- adherence to breastfeeding guidelines
- limiting the number of clinicians responsible for the patient
- promotion of kangaroo care
- avoidance of noxious stimuli.10,11
Continue to: Ethical and clinical issues...
Ethical and clinical issues
Providing clinical care to infants born at the edge of viability is challenging and raises many ethical and clinical concerns.12,13 For an infant born at the edge of viability, clinicians and parents do not want to initiate a care process that improves survival but results in an extremely poor quality of life. At the same time, clinicians and parents do not want to withhold care that could help an extremely premature newborn survive and thrive. Consequently, the counseling process is complex and requires coordination between the obstetrical and neonatology disciplines, involving physicians and nurses from both. A primary consideration in deciding to institute active treatment at birth is the preference of the pregnant patient and the patient’s trusted family members. A thorough discussion of these issues is beyond the scope of this editorial. ACOG provides detailed advice about the approach to counseling patients who face the possibility of a periviable birth.14
To help standardize the counseling process, institutions may find it helpful to recommend that clinicians consistently use a calculator to provide newborn outcome data to patients. The National Institute of Child Health and Human Development’s Extremely Preterm Birth Outcomes calculator uses the following inputs:
- gestational age
- estimated birth weight
- sex
- singleton/multiple gestation
- antenatal steroid treatment.
It also provides the following outputs as percentages:
- survival with active treatment at birth
- survival without active treatment at birth
- profound neurodevelopmental impairment
- moderate to severe neurodevelopmental impairment
- blindness
- deafness
- moderate to severe cerebral palsy
- cognitive developmental delay.15
A full assessment of all known clinical factors should influence the interpretation of the output from the clinical calculator. An alternativeis to use data from the Vermont Oxford Network. NICUs with sufficient clinical volume may prefer to use their own outcome data in the counseling process.
Institutions and clinical teams may improve the consistency of the counseling process by identifying criteria for 3 main treatment options:
- clinical situations where active treatment at birth is not generally offered (eg, <22 weeks’ gestation)
- clinical situations where active treatment at birth is almost always routinely provided (eg, >25 weeks’ gestation)
- clinical situations where patient preferences are especially important in guiding the use of active treatment.
Most institutions do not routinely offer active treatment of the newborn at a gestational age of less than 22 weeks and 0 days. Instead, comfort care often is provided for these newborns. Most institutions routinely provide active treatment at birth beginning at 24 or 25 weeks’ gestation unless unique risk factors or comorbidities warrant not providing active treatment (TABLE 3A). Some professional societies recommend setting a threshold for recommending active treatment at birth. For example, the British Association of Perinatal Medicine recommends that if there is 50% or higher probability of survival without severe disability, active treatment at birth should be considered because it is in the best interest of the newborn.16 In the hours and days following birth, the clinical course of the newborn greatly influences the treatment plan and care goals. After the initial resuscitation, if the clinical condition of an extremely preterm infant worsens and the prognosis is grim, a pivot to palliative care may be considered.
Final thoughts
Periviability is the earliest stage of fetal development where there is a reasonable chance, but not a high likelihood, of survival outside the womb. For decades, the threshold for periviability was approximately 24 weeks of gestation. With current obstetrical and neonatal practice, the new threshold for periviability is 22 to 23 weeks of gestation, but death prior to hospital discharge occurs in approximately half of these newborns. For the survivors, lifelong neurodevelopmental complications and pulmonary disease are common. Obstetricians play a key role in counseling patients who are at risk of giving birth before 24 weeks of gestation. Given the challenges faced by an infant born at 22 and 23 weeks’ gestation, pregnant patients and trusted family members should approach the decision to actively resuscitate the newborn with caution. However, if the clinical team, patient, and trusted family members agree to pursue active treatment, completion of a course of antenatal steroids and appropriate respiratory and cardiovascular support at birth are key to improving long-term outcomes. ●
- Ehret DEY, Edwards EM, Greenberg LT, et al. Association of antenatal steroid exposure with survival among infants receiving postnatal life support at 22 to 25 weeks gestation. JAMA Network Open. 2018;E183235.
- Rossi RM, DeFranco EA, Hall ES. Association of antenatal corticosteroid exposure and infant survival at 22 and 23 weeks [published online November 28, 2021]. Am J Perinatol. doi:10.1055/s-004-1740062
- Chawla S, Wyckoff MH, Rysavy MA, et al. Association of antenatal steroid exposure at 21 to 22 weeks of gestation with neonatal survival and survival without morbidities. JAMA Network Open. 2022;5:E2233331.
- Use of antenatal corticosteroids at 22 weeks of gestation. ACOG website. Published September 2021. Accessed April 10, 2023. https://www.acog .org/clinical/clinical-guidance/practice -advisory/articles/2021/09/use-of-antenatal -corticosteroids-at-22-weeks-of-gestation
- Bell EF, Hintz SR, Hansen NI, et al. Mortality, in-hospital morbidity, care practices and 2-year outcomes for extremely preterm infants in the US, 2013-2018. JAMA. 2022;327:248-263.
- The tragic death of Patrick, JFK and Jackie’s newborn son, in 1963. Irish Central website. Published November 6, 2022. Accessed April 10, 2023. https://www.irishcentral.com/roots/history /tragic-death-patrick-kennedy-jfk-jackie
- Norman M, Hallberg B, Abrahamsson T, et al. Association between year of birth and 1-year survival among extremely preterm infants in Sweden during 2004-2007 and 2014-2016. JAMA. 2019;32:1188-1199.
- Kono Y, Yonemoto N, Nakanishi H, et al. Changes in survival and neurodevelopmental outcomes of infants born at <25 weeks gestation: a retrospective observational study in tertiary centres in Japan. BMJ Paediatrics Open. 2018;2:E000211.
- Backes CH, Rivera BK, Pavlek L, et al. Proactive neonatal treatment at 22 weeks of gestation: a systematic review and meta-analysis. Am J Obstet Gynecol. 2021;224:158-174.
- Morris M, Cleary JP, Soliman A. Small baby unit improves quality and outcomes in extremely low birth weight infants. Pediatrics. 2015;136:E1007-E1015.
- Fathi O, Nelin LD, Shephard EG, et al. Development of a small baby unit to improve outcomes for the extremely premature infant. J Perinatology. 2002;42:157-164.
- Lantos JD. Ethical issues in treatment of babies born at 22 weeks of gestation. Arch Dis Child. 2021;106:1155-1157.
- Shinwell ES. Ethics of birth at the limit of viability: the risky business of prediction. Neonatology. 2015;107:317-320.
- American College of Obstetricians and Gynecologists; Society for Maternal-Fetal Medicine. Obstetric Care Consensus No 6. periviable birth. Obstet Gynecol. 2017;E187-E199.
- Extremely preterm birth outcomes tool. NICHD website. Updated March 2, 2020. Accessed April 10, 2023. https://www.nichd.nih.gov/research /supported/EPBO/use#
- Mactier H, Bates SE, Johnston T, et al. Perinatal management of extreme preterm birth before 27 weeks of gestation: a framework for practice. Arch Dis Child Fetal Neonatal Ed. 2020;105:F232-F239.

Robert L. Barbieri, MD
Editor in Chief, OBG Management
Chair Emeritus, Department of Obstetrics and Gynecology
Brigham and Women’s Hospital
Kate Macy Ladd Distinguished Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Boston, Massachusetts
The author reports no conflict of interest related to this article.
Robert L. Barbieri, MD
Editor in Chief, OBG Management
Chair Emeritus, Department of Obstetrics and Gynecology
Brigham and Women’s Hospital
Kate Macy Ladd Distinguished Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Boston, Massachusetts
The author reports no conflict of interest related to this article.
Robert L. Barbieri, MD
Editor in Chief, OBG Management
Chair Emeritus, Department of Obstetrics and Gynecology
Brigham and Women’s Hospital
Kate Macy Ladd Distinguished Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Boston, Massachusetts
The author reports no conflict of interest related to this article.
For many decades, the limit of newborn viability was at approximately 24 weeks’ gestation. Recent advances in pregnancy and neonatal care suggest that the new limit of viability is 22 (22 weeks and 0 days to 22 weeks and 6 days) or 23 (23 weeks and 0 days to 23 weeks and 6 days) weeks of gestation. In addition, data from observational cohort studies indicate that for infants born at 22 and 23 weeks’ gestation, survival is dependent on a course of antenatal steroids administered prior to birth plus intensive respiratory and cardiovascular support at delivery and in the neonatal intensive care unit (NICU).
Antenatal steroids: Critical for survival at 22 and 23 weeks of gestation
Most studies of birth outcomes at 22 and 23 weeks’ gestation rely on observational cohorts where unmeasured differences among the maternal-fetal dyads that received or did not receive a specific treatment confounds the interpretation of the data. However, data from multiple large observational cohorts suggest that between 22 and 24 weeks of gestation, completion of a course of antenatal steroids will optimize infant outcomes. Particularly noteworthy was the observation that the incremental survival benefit of antenatal steroids was greatest at 22 and 23 weeks’ gestation (TABLE 1).1 Similar results have been reported by Rossi and colleagues (TABLE 2).2
The importance of a completed course of antenatal steroids before birth was confirmed in another cohort study of 431 infants born in 2016 to 2019 at 22 weeks and 0 days’ to 23 weeks and 6 days’ gestation.3 Survival to discharge occurred in 53.9% of infants who received a full course of antenatal steroids before birth and 35.5% among those who did not receive antenatal steroids..3 Survival to discharge without major neonatal morbidities was 26.9% in those who received a full course of antenatal steroids and 10% among those who did not. In this cohort, major neonatal morbidities included severe intracranial hemorrhage, cystic periventricular leukomalacia, severe bronchopulmonary dysplasia, surgical necrotizing enterocolitis, or severe retinopathy of prematurity requiring treatment.
The American College of Obstetricians and Gynecologists (ACOG) recommends against antenatal steroids prior to 22 weeks and 0 days gestation.4 However, some neonatologists might recommend that antenatal steroids be given starting at 21 weeks and 5 days of gestation if birth is anticipated in the 22nd week of gestation and the patient prefers aggressive treatment of the newborn.
Active respiratory and cardiovascular support improves newborn outcomes
To maximize survival, infants born at 22 and 23 weeks’ gestation always require intensive active treatment at birth and in the following days in the NICU. Active treatment may include respiratory support, surfactant treatment, pressors, closure of a patent ductus arteriosus, transfusion of red blood cells, and parenteral nutrition. In one observational cohort study, active treatment at birth was not routinely provided at 22 and 23 weeks’ gestation but was routinely provided at later gestational ages (TABLE 3A).5 Not surprisingly, active treatment, especially at early gestational ages, is associated with improved survival to discharge. For example, at 22 weeks’ gestation, survival to discharge in infants who received or did not receive intensive active treatment was 28% and 0%, respectively.5 However, specific clinical characteristics of the pregnant patient and newborn may have influenced which infants were actively treated, confounding interpretation of the observation. In this cohort of extremely premature newborns, survival to hospital discharge increased substantially between 22 weeks and 26 weeks of gestational age (TABLE 3B).5
Many of the surviving infants needed chronic support treatment. Among surviving infants born at 22 weeks and 26 weeks, chronic support treatments were being used by 22.6% and 10.6% of infants, respectively, 2 years after birth.5 For surviving infants born at 22 weeks, the specific chronic support treatments included gastrostomy or feeding tube (19.4%), oxygen (9.7%), pulse oximeter (9.7%), and/or tracheostomy (3.2%). For surviving infants born at 26 weeks’ gestation, the specific chronic support treatments included gastrotomy or feeding tube (8.5%), pulse oximeter (4.4%), oxygen (3.2%), tracheostomy (2.3%), an apnea monitor (1.5%), and/or ventilator or continuous positive airway pressure (1.1%).5
Continue to: Evolving improvement in infant outcomes...
Evolving improvement in infant outcomes
In 1963, Jacqueline Bouvier Kennedy went into preterm labor at 34 weeks of gestation and delivered her son Patrick at a community facility. Due to severe respiratory distress syndrome, Patrick was transferred to the Boston Children’s Hospital, and he died shortly thereafter.6 Sixty years later, due to advances in obstetric and neonatal care, death from respiratory distress syndrome at 34 weeks of gestation is uncommon in the United States.
Infant outcomes following birth at 22 and 23 weeks’ gestation continue to improve. An observational cohort study from Sweden reported that at 22 weeks’ gestation, the percentage of live-born infants who survived to 1-year post birth in 2004 to 2007 and 2014 to 2016 was 10% and 30%, respectively.7 Similarly, at 23 weeks’ gestation, the percentage of live-born infants who survived to 1-year post birth in 2004 to 2007 and 2014 to 2016 was 52% and 61%, respectively.7 However, most of the surviving infants in this cohort had one or more major neonatal morbidities, including intraventricular hemorrhage grade 3 or 4; periventricular leukomalacia; necrotizing enterocolitis; retinopathy of prematurity grade 3, 4, or 5; or severe bronchopulmonary dysplasia.7
In a cohort of infants born in Japan at 22 to 24 weeks of gestation, there was a notable decrease in major neurodisability at 3 years of age for births occurring in 2 epochs, 2003 to 2007 and 2008 to 2012.8 When comparing outcomes in 2003 to 2007 versus 2008 to 2012, the change in rate of various major complications included the following: cerebral palsy (15.9% vs 9.5%), visual impairment (13.6% vs 4.4%), blindness (4.8% vs 1.3%), and hearing impairment (2.6% vs 1.0%). In contrast, the rate of cognitive impairment, defined as less than 70% of standard test performance for chronological age, was similar in the 2 time periods (36.5% and 37.9%, respectively).8 Based on data reported between 2000 and 2020, a systematic review and meta-analysis by Backes and colleagues concluded that there has been substantial improvement in the survival of infants born at 22 weeks of gestation.9
The small baby unit
A feature of modern medicine is the relentless evolution of new clinical subspecialties and sub-subspecialties. NICUs evolved from newborn nurseries to serve the needs of the most severely ill newborns, with care provided by a cadre of highly trained subspecialized neonatologists and neonatal nurses. A new era is dawning, with some NICUs developing a sub-subspecialized small baby unit to care for infants born between 22 and 26 weeks of gestation. These units often are staffed by clinicians with a specific interest in optimizing the care of extremely preterm infants, providing continuity of care over a long hospitalization.10 The benefits of a small baby unit may include:
- relentless standardization and adherence to the best intensive care practices
- daily use of checklists
- strict adherence to central line care
- timely extubation and transition to continuous positive airway pressure
- adherence to breastfeeding guidelines
- limiting the number of clinicians responsible for the patient
- promotion of kangaroo care
- avoidance of noxious stimuli.10,11
Continue to: Ethical and clinical issues...
Ethical and clinical issues
Providing clinical care to infants born at the edge of viability is challenging and raises many ethical and clinical concerns.12,13 For an infant born at the edge of viability, clinicians and parents do not want to initiate a care process that improves survival but results in an extremely poor quality of life. At the same time, clinicians and parents do not want to withhold care that could help an extremely premature newborn survive and thrive. Consequently, the counseling process is complex and requires coordination between the obstetrical and neonatology disciplines, involving physicians and nurses from both. A primary consideration in deciding to institute active treatment at birth is the preference of the pregnant patient and the patient’s trusted family members. A thorough discussion of these issues is beyond the scope of this editorial. ACOG provides detailed advice about the approach to counseling patients who face the possibility of a periviable birth.14
To help standardize the counseling process, institutions may find it helpful to recommend that clinicians consistently use a calculator to provide newborn outcome data to patients. The National Institute of Child Health and Human Development’s Extremely Preterm Birth Outcomes calculator uses the following inputs:
- gestational age
- estimated birth weight
- sex
- singleton/multiple gestation
- antenatal steroid treatment.
It also provides the following outputs as percentages:
- survival with active treatment at birth
- survival without active treatment at birth
- profound neurodevelopmental impairment
- moderate to severe neurodevelopmental impairment
- blindness
- deafness
- moderate to severe cerebral palsy
- cognitive developmental delay.15
A full assessment of all known clinical factors should influence the interpretation of the output from the clinical calculator. An alternativeis to use data from the Vermont Oxford Network. NICUs with sufficient clinical volume may prefer to use their own outcome data in the counseling process.
Institutions and clinical teams may improve the consistency of the counseling process by identifying criteria for 3 main treatment options:
- clinical situations where active treatment at birth is not generally offered (eg, <22 weeks’ gestation)
- clinical situations where active treatment at birth is almost always routinely provided (eg, >25 weeks’ gestation)
- clinical situations where patient preferences are especially important in guiding the use of active treatment.
Most institutions do not routinely offer active treatment of the newborn at a gestational age of less than 22 weeks and 0 days. Instead, comfort care often is provided for these newborns. Most institutions routinely provide active treatment at birth beginning at 24 or 25 weeks’ gestation unless unique risk factors or comorbidities warrant not providing active treatment (TABLE 3A). Some professional societies recommend setting a threshold for recommending active treatment at birth. For example, the British Association of Perinatal Medicine recommends that if there is 50% or higher probability of survival without severe disability, active treatment at birth should be considered because it is in the best interest of the newborn.16 In the hours and days following birth, the clinical course of the newborn greatly influences the treatment plan and care goals. After the initial resuscitation, if the clinical condition of an extremely preterm infant worsens and the prognosis is grim, a pivot to palliative care may be considered.
Final thoughts
Periviability is the earliest stage of fetal development where there is a reasonable chance, but not a high likelihood, of survival outside the womb. For decades, the threshold for periviability was approximately 24 weeks of gestation. With current obstetrical and neonatal practice, the new threshold for periviability is 22 to 23 weeks of gestation, but death prior to hospital discharge occurs in approximately half of these newborns. For the survivors, lifelong neurodevelopmental complications and pulmonary disease are common. Obstetricians play a key role in counseling patients who are at risk of giving birth before 24 weeks of gestation. Given the challenges faced by an infant born at 22 and 23 weeks’ gestation, pregnant patients and trusted family members should approach the decision to actively resuscitate the newborn with caution. However, if the clinical team, patient, and trusted family members agree to pursue active treatment, completion of a course of antenatal steroids and appropriate respiratory and cardiovascular support at birth are key to improving long-term outcomes. ●
For many decades, the limit of newborn viability was at approximately 24 weeks’ gestation. Recent advances in pregnancy and neonatal care suggest that the new limit of viability is 22 (22 weeks and 0 days to 22 weeks and 6 days) or 23 (23 weeks and 0 days to 23 weeks and 6 days) weeks of gestation. In addition, data from observational cohort studies indicate that for infants born at 22 and 23 weeks’ gestation, survival is dependent on a course of antenatal steroids administered prior to birth plus intensive respiratory and cardiovascular support at delivery and in the neonatal intensive care unit (NICU).
Antenatal steroids: Critical for survival at 22 and 23 weeks of gestation
Most studies of birth outcomes at 22 and 23 weeks’ gestation rely on observational cohorts where unmeasured differences among the maternal-fetal dyads that received or did not receive a specific treatment confounds the interpretation of the data. However, data from multiple large observational cohorts suggest that between 22 and 24 weeks of gestation, completion of a course of antenatal steroids will optimize infant outcomes. Particularly noteworthy was the observation that the incremental survival benefit of antenatal steroids was greatest at 22 and 23 weeks’ gestation (TABLE 1).1 Similar results have been reported by Rossi and colleagues (TABLE 2).2
The importance of a completed course of antenatal steroids before birth was confirmed in another cohort study of 431 infants born in 2016 to 2019 at 22 weeks and 0 days’ to 23 weeks and 6 days’ gestation.3 Survival to discharge occurred in 53.9% of infants who received a full course of antenatal steroids before birth and 35.5% among those who did not receive antenatal steroids..3 Survival to discharge without major neonatal morbidities was 26.9% in those who received a full course of antenatal steroids and 10% among those who did not. In this cohort, major neonatal morbidities included severe intracranial hemorrhage, cystic periventricular leukomalacia, severe bronchopulmonary dysplasia, surgical necrotizing enterocolitis, or severe retinopathy of prematurity requiring treatment.
The American College of Obstetricians and Gynecologists (ACOG) recommends against antenatal steroids prior to 22 weeks and 0 days gestation.4 However, some neonatologists might recommend that antenatal steroids be given starting at 21 weeks and 5 days of gestation if birth is anticipated in the 22nd week of gestation and the patient prefers aggressive treatment of the newborn.
Active respiratory and cardiovascular support improves newborn outcomes
To maximize survival, infants born at 22 and 23 weeks’ gestation always require intensive active treatment at birth and in the following days in the NICU. Active treatment may include respiratory support, surfactant treatment, pressors, closure of a patent ductus arteriosus, transfusion of red blood cells, and parenteral nutrition. In one observational cohort study, active treatment at birth was not routinely provided at 22 and 23 weeks’ gestation but was routinely provided at later gestational ages (TABLE 3A).5 Not surprisingly, active treatment, especially at early gestational ages, is associated with improved survival to discharge. For example, at 22 weeks’ gestation, survival to discharge in infants who received or did not receive intensive active treatment was 28% and 0%, respectively.5 However, specific clinical characteristics of the pregnant patient and newborn may have influenced which infants were actively treated, confounding interpretation of the observation. In this cohort of extremely premature newborns, survival to hospital discharge increased substantially between 22 weeks and 26 weeks of gestational age (TABLE 3B).5
Many of the surviving infants needed chronic support treatment. Among surviving infants born at 22 weeks and 26 weeks, chronic support treatments were being used by 22.6% and 10.6% of infants, respectively, 2 years after birth.5 For surviving infants born at 22 weeks, the specific chronic support treatments included gastrostomy or feeding tube (19.4%), oxygen (9.7%), pulse oximeter (9.7%), and/or tracheostomy (3.2%). For surviving infants born at 26 weeks’ gestation, the specific chronic support treatments included gastrotomy or feeding tube (8.5%), pulse oximeter (4.4%), oxygen (3.2%), tracheostomy (2.3%), an apnea monitor (1.5%), and/or ventilator or continuous positive airway pressure (1.1%).5
Continue to: Evolving improvement in infant outcomes...
Evolving improvement in infant outcomes
In 1963, Jacqueline Bouvier Kennedy went into preterm labor at 34 weeks of gestation and delivered her son Patrick at a community facility. Due to severe respiratory distress syndrome, Patrick was transferred to the Boston Children’s Hospital, and he died shortly thereafter.6 Sixty years later, due to advances in obstetric and neonatal care, death from respiratory distress syndrome at 34 weeks of gestation is uncommon in the United States.
Infant outcomes following birth at 22 and 23 weeks’ gestation continue to improve. An observational cohort study from Sweden reported that at 22 weeks’ gestation, the percentage of live-born infants who survived to 1-year post birth in 2004 to 2007 and 2014 to 2016 was 10% and 30%, respectively.7 Similarly, at 23 weeks’ gestation, the percentage of live-born infants who survived to 1-year post birth in 2004 to 2007 and 2014 to 2016 was 52% and 61%, respectively.7 However, most of the surviving infants in this cohort had one or more major neonatal morbidities, including intraventricular hemorrhage grade 3 or 4; periventricular leukomalacia; necrotizing enterocolitis; retinopathy of prematurity grade 3, 4, or 5; or severe bronchopulmonary dysplasia.7
In a cohort of infants born in Japan at 22 to 24 weeks of gestation, there was a notable decrease in major neurodisability at 3 years of age for births occurring in 2 epochs, 2003 to 2007 and 2008 to 2012.8 When comparing outcomes in 2003 to 2007 versus 2008 to 2012, the change in rate of various major complications included the following: cerebral palsy (15.9% vs 9.5%), visual impairment (13.6% vs 4.4%), blindness (4.8% vs 1.3%), and hearing impairment (2.6% vs 1.0%). In contrast, the rate of cognitive impairment, defined as less than 70% of standard test performance for chronological age, was similar in the 2 time periods (36.5% and 37.9%, respectively).8 Based on data reported between 2000 and 2020, a systematic review and meta-analysis by Backes and colleagues concluded that there has been substantial improvement in the survival of infants born at 22 weeks of gestation.9
The small baby unit
A feature of modern medicine is the relentless evolution of new clinical subspecialties and sub-subspecialties. NICUs evolved from newborn nurseries to serve the needs of the most severely ill newborns, with care provided by a cadre of highly trained subspecialized neonatologists and neonatal nurses. A new era is dawning, with some NICUs developing a sub-subspecialized small baby unit to care for infants born between 22 and 26 weeks of gestation. These units often are staffed by clinicians with a specific interest in optimizing the care of extremely preterm infants, providing continuity of care over a long hospitalization.10 The benefits of a small baby unit may include:
- relentless standardization and adherence to the best intensive care practices
- daily use of checklists
- strict adherence to central line care
- timely extubation and transition to continuous positive airway pressure
- adherence to breastfeeding guidelines
- limiting the number of clinicians responsible for the patient
- promotion of kangaroo care
- avoidance of noxious stimuli.10,11
Continue to: Ethical and clinical issues...
Ethical and clinical issues
Providing clinical care to infants born at the edge of viability is challenging and raises many ethical and clinical concerns.12,13 For an infant born at the edge of viability, clinicians and parents do not want to initiate a care process that improves survival but results in an extremely poor quality of life. At the same time, clinicians and parents do not want to withhold care that could help an extremely premature newborn survive and thrive. Consequently, the counseling process is complex and requires coordination between the obstetrical and neonatology disciplines, involving physicians and nurses from both. A primary consideration in deciding to institute active treatment at birth is the preference of the pregnant patient and the patient’s trusted family members. A thorough discussion of these issues is beyond the scope of this editorial. ACOG provides detailed advice about the approach to counseling patients who face the possibility of a periviable birth.14
To help standardize the counseling process, institutions may find it helpful to recommend that clinicians consistently use a calculator to provide newborn outcome data to patients. The National Institute of Child Health and Human Development’s Extremely Preterm Birth Outcomes calculator uses the following inputs:
- gestational age
- estimated birth weight
- sex
- singleton/multiple gestation
- antenatal steroid treatment.
It also provides the following outputs as percentages:
- survival with active treatment at birth
- survival without active treatment at birth
- profound neurodevelopmental impairment
- moderate to severe neurodevelopmental impairment
- blindness
- deafness
- moderate to severe cerebral palsy
- cognitive developmental delay.15
A full assessment of all known clinical factors should influence the interpretation of the output from the clinical calculator. An alternativeis to use data from the Vermont Oxford Network. NICUs with sufficient clinical volume may prefer to use their own outcome data in the counseling process.
Institutions and clinical teams may improve the consistency of the counseling process by identifying criteria for 3 main treatment options:
- clinical situations where active treatment at birth is not generally offered (eg, <22 weeks’ gestation)
- clinical situations where active treatment at birth is almost always routinely provided (eg, >25 weeks’ gestation)
- clinical situations where patient preferences are especially important in guiding the use of active treatment.
Most institutions do not routinely offer active treatment of the newborn at a gestational age of less than 22 weeks and 0 days. Instead, comfort care often is provided for these newborns. Most institutions routinely provide active treatment at birth beginning at 24 or 25 weeks’ gestation unless unique risk factors or comorbidities warrant not providing active treatment (TABLE 3A). Some professional societies recommend setting a threshold for recommending active treatment at birth. For example, the British Association of Perinatal Medicine recommends that if there is 50% or higher probability of survival without severe disability, active treatment at birth should be considered because it is in the best interest of the newborn.16 In the hours and days following birth, the clinical course of the newborn greatly influences the treatment plan and care goals. After the initial resuscitation, if the clinical condition of an extremely preterm infant worsens and the prognosis is grim, a pivot to palliative care may be considered.
Final thoughts
Periviability is the earliest stage of fetal development where there is a reasonable chance, but not a high likelihood, of survival outside the womb. For decades, the threshold for periviability was approximately 24 weeks of gestation. With current obstetrical and neonatal practice, the new threshold for periviability is 22 to 23 weeks of gestation, but death prior to hospital discharge occurs in approximately half of these newborns. For the survivors, lifelong neurodevelopmental complications and pulmonary disease are common. Obstetricians play a key role in counseling patients who are at risk of giving birth before 24 weeks of gestation. Given the challenges faced by an infant born at 22 and 23 weeks’ gestation, pregnant patients and trusted family members should approach the decision to actively resuscitate the newborn with caution. However, if the clinical team, patient, and trusted family members agree to pursue active treatment, completion of a course of antenatal steroids and appropriate respiratory and cardiovascular support at birth are key to improving long-term outcomes. ●
- Ehret DEY, Edwards EM, Greenberg LT, et al. Association of antenatal steroid exposure with survival among infants receiving postnatal life support at 22 to 25 weeks gestation. JAMA Network Open. 2018;E183235.
- Rossi RM, DeFranco EA, Hall ES. Association of antenatal corticosteroid exposure and infant survival at 22 and 23 weeks [published online November 28, 2021]. Am J Perinatol. doi:10.1055/s-004-1740062
- Chawla S, Wyckoff MH, Rysavy MA, et al. Association of antenatal steroid exposure at 21 to 22 weeks of gestation with neonatal survival and survival without morbidities. JAMA Network Open. 2022;5:E2233331.
- Use of antenatal corticosteroids at 22 weeks of gestation. ACOG website. Published September 2021. Accessed April 10, 2023. https://www.acog .org/clinical/clinical-guidance/practice -advisory/articles/2021/09/use-of-antenatal -corticosteroids-at-22-weeks-of-gestation
- Bell EF, Hintz SR, Hansen NI, et al. Mortality, in-hospital morbidity, care practices and 2-year outcomes for extremely preterm infants in the US, 2013-2018. JAMA. 2022;327:248-263.
- The tragic death of Patrick, JFK and Jackie’s newborn son, in 1963. Irish Central website. Published November 6, 2022. Accessed April 10, 2023. https://www.irishcentral.com/roots/history /tragic-death-patrick-kennedy-jfk-jackie
- Norman M, Hallberg B, Abrahamsson T, et al. Association between year of birth and 1-year survival among extremely preterm infants in Sweden during 2004-2007 and 2014-2016. JAMA. 2019;32:1188-1199.
- Kono Y, Yonemoto N, Nakanishi H, et al. Changes in survival and neurodevelopmental outcomes of infants born at <25 weeks gestation: a retrospective observational study in tertiary centres in Japan. BMJ Paediatrics Open. 2018;2:E000211.
- Backes CH, Rivera BK, Pavlek L, et al. Proactive neonatal treatment at 22 weeks of gestation: a systematic review and meta-analysis. Am J Obstet Gynecol. 2021;224:158-174.
- Morris M, Cleary JP, Soliman A. Small baby unit improves quality and outcomes in extremely low birth weight infants. Pediatrics. 2015;136:E1007-E1015.
- Fathi O, Nelin LD, Shephard EG, et al. Development of a small baby unit to improve outcomes for the extremely premature infant. J Perinatology. 2002;42:157-164.
- Lantos JD. Ethical issues in treatment of babies born at 22 weeks of gestation. Arch Dis Child. 2021;106:1155-1157.
- Shinwell ES. Ethics of birth at the limit of viability: the risky business of prediction. Neonatology. 2015;107:317-320.
- American College of Obstetricians and Gynecologists; Society for Maternal-Fetal Medicine. Obstetric Care Consensus No 6. periviable birth. Obstet Gynecol. 2017;E187-E199.
- Extremely preterm birth outcomes tool. NICHD website. Updated March 2, 2020. Accessed April 10, 2023. https://www.nichd.nih.gov/research /supported/EPBO/use#
- Mactier H, Bates SE, Johnston T, et al. Perinatal management of extreme preterm birth before 27 weeks of gestation: a framework for practice. Arch Dis Child Fetal Neonatal Ed. 2020;105:F232-F239.
- Ehret DEY, Edwards EM, Greenberg LT, et al. Association of antenatal steroid exposure with survival among infants receiving postnatal life support at 22 to 25 weeks gestation. JAMA Network Open. 2018;E183235.
- Rossi RM, DeFranco EA, Hall ES. Association of antenatal corticosteroid exposure and infant survival at 22 and 23 weeks [published online November 28, 2021]. Am J Perinatol. doi:10.1055/s-004-1740062
- Chawla S, Wyckoff MH, Rysavy MA, et al. Association of antenatal steroid exposure at 21 to 22 weeks of gestation with neonatal survival and survival without morbidities. JAMA Network Open. 2022;5:E2233331.
- Use of antenatal corticosteroids at 22 weeks of gestation. ACOG website. Published September 2021. Accessed April 10, 2023. https://www.acog .org/clinical/clinical-guidance/practice -advisory/articles/2021/09/use-of-antenatal -corticosteroids-at-22-weeks-of-gestation
- Bell EF, Hintz SR, Hansen NI, et al. Mortality, in-hospital morbidity, care practices and 2-year outcomes for extremely preterm infants in the US, 2013-2018. JAMA. 2022;327:248-263.
- The tragic death of Patrick, JFK and Jackie’s newborn son, in 1963. Irish Central website. Published November 6, 2022. Accessed April 10, 2023. https://www.irishcentral.com/roots/history /tragic-death-patrick-kennedy-jfk-jackie
- Norman M, Hallberg B, Abrahamsson T, et al. Association between year of birth and 1-year survival among extremely preterm infants in Sweden during 2004-2007 and 2014-2016. JAMA. 2019;32:1188-1199.
- Kono Y, Yonemoto N, Nakanishi H, et al. Changes in survival and neurodevelopmental outcomes of infants born at <25 weeks gestation: a retrospective observational study in tertiary centres in Japan. BMJ Paediatrics Open. 2018;2:E000211.
- Backes CH, Rivera BK, Pavlek L, et al. Proactive neonatal treatment at 22 weeks of gestation: a systematic review and meta-analysis. Am J Obstet Gynecol. 2021;224:158-174.
- Morris M, Cleary JP, Soliman A. Small baby unit improves quality and outcomes in extremely low birth weight infants. Pediatrics. 2015;136:E1007-E1015.
- Fathi O, Nelin LD, Shephard EG, et al. Development of a small baby unit to improve outcomes for the extremely premature infant. J Perinatology. 2002;42:157-164.
- Lantos JD. Ethical issues in treatment of babies born at 22 weeks of gestation. Arch Dis Child. 2021;106:1155-1157.
- Shinwell ES. Ethics of birth at the limit of viability: the risky business of prediction. Neonatology. 2015;107:317-320.
- American College of Obstetricians and Gynecologists; Society for Maternal-Fetal Medicine. Obstetric Care Consensus No 6. periviable birth. Obstet Gynecol. 2017;E187-E199.
- Extremely preterm birth outcomes tool. NICHD website. Updated March 2, 2020. Accessed April 10, 2023. https://www.nichd.nih.gov/research /supported/EPBO/use#
- Mactier H, Bates SE, Johnston T, et al. Perinatal management of extreme preterm birth before 27 weeks of gestation: a framework for practice. Arch Dis Child Fetal Neonatal Ed. 2020;105:F232-F239.
News & Perspectives from Ob.Gyn. News
DRUGS, PREGNANCY, AND LACTATION
Canadian Task Force recommendation on screening for postpartum depression misses the mark
Director, Ammon-Pinizzotto Center for Women’s Mental Health at Massachusetts General Hospital, Boston, Massachusetts.
Postpartum/perinatal depression (PPD) remains the most common complication in modern obstetrics, with a prevalence of 10%-15% based on multiple studies over the last 2 decades. Over those same 2 decades, there has been growing interest and motivation across the country—from small community hospitals to major academic centers—to promote screening. Such screening is integrated into obstetrical practices, typically using the Edinburgh Postnatal Depression Scale (EPDS), the most widely used validated screen for PPD globally.
As mentioned in previous columns, the U.S. Preventive Services Task Force recommended screening for PPD in 2016, which includes screening women at highest risk, and both acutely treating and preventing PPD.
Since then, screening women for a common clinical problem like PPD has been widely adopted by clinicians representing a broad spectrum of interdisciplinary care. Providers who are engaged in the treatment of postpartum women—obstetricians, psychiatrists, doulas, lactation consultants, facilitators of postpartum support groups, and advocacy groups among others—are included.
An open question and one of great concern recently to our group and others has been what happens after screening. It is clear that identification of PPD per se is not necessarily a challenge, and we have multiple effective treatments from antidepressants to mindfulness-based cognitive therapy to cognitive-behavioral interventions. There is also a growing number of digital applications aimed at mitigation of depressive symptoms in women with postpartum major depressive disorder. One unanswered question is how to engage women after identification of PPD and how to facilitate access to care in a way that maximizes the likelihood that women who actually are suffering from PPD get adequate treatment.
The “perinatal treatment cascade” refers to the majority of women who, on the other side of identification of PPD, fail to receive adequate treatment and continue to have persistent depression. This is perhaps the greatest challenge to the field and to clinicians—how do we, on the other side of screening, see that these women get access to care and get well?
https://www.mdedge.com/obgyn/drugs-pregnancy-lactation
GENDER-AFFIRMING GYNECOLOGY
Caring for the aging transgender patient
Ob.Gyn. and fellowship-trained gender-affirming surgeon in West Reading, Pennsylvania.
The elderly transgender population is rapidly expanding and remains significantly overlooked. Although emerging evidence provides some guidance for medical and surgical treatment for transgender youth, there is still a paucity of research directed at the management of gender-diverse elders.
To a large extent, the challenges that transgender elders face are no different from those experienced by the general elder population. Irrespective of gender identity, patients begin to undergo cognitive and physical changes, encounter difficulties with activities of daily living, suffer the loss of social networks and friends, and face end-of-life issues. Attributes that contribute to successful aging in the general population include good health, social engagement and support, and having a positive outlook on life. Yet, stigma surrounding gender identity and sexual orientation continues to negatively affect elder transgender people.
https://www.mdedge.com/obgyn/gender-affirming-gynecology
Continue to: LATEST NEWS...
LATEST NEWS
Study: Prenatal supplements fail to meet nutrient needs
Although drugstore shelves might suggest otherwise,affordable dietary supplements that provide critical nutrients in appropriate doses for pregnant women are virtually nonexistent, researchers have found.
In a new study published in the American Journal of Clinical Nutrition, investigators observed what many physicians have long suspected: Most prenatal vitamins and other supplements do not adequately make up the difference of what food-based intake of nutrients leave lacking. Despite patients believing they are getting everything they need with their product purchase, they fall short of guideline-recommended requirements.
“There is no magic pill,” said Katherine A. Sauder, PhD, an associate professor of pediatrics at the University of Colorado Anschutz Medical Campus, Aurora, and lead author of the study. “There is no easy answer here.”
The researchers analyzed 24-hour dietary intake data from 2,450 study participants across five states from 2007 to 2019. Dr. Sauder and colleagues focused on six of the more than 20 key nutrients recommended for pregnant people and determined the target dose for vitamin A, vitamin D, folate, calcium, iron, and omega-3 fatty acids.
The researchers tested more than 20,500 dietary supplements, of which 421 were prenatal products. Only 69 products—three prenatal—included all six nutrients. Just seven products —two prenatal—contained target doses for five nutrients. Only one product, which was not marketed as prenatal, contained target doses for all six nutrients but required seven tablets a serving and cost patients approximately $200 a month.
SARS-CoV-2 crosses placenta and infects brains of two infants: ‘This is a first’
Researchers have found for the first time that COVID infection has crossed the placenta and caused brain damage in two newborns, according to a study published online today in Pediatrics.
One of the infants died at 13 months and the other remained in hospice care at time of manuscript submission.
Lead author Merline Benny, MD,with the division of neonatology, department of pediatrics at University of Miami, and colleagues briefed reporters today ahead of the release.
“This is a first,” said senior author Shahnaz Duara, MD, medical director of the Neonatal Intensive Care Unit at Holtz Children’s Hospital, Miami, explaining it is the first study to confirm cross-placental SARS-CoV-2 transmission leading to brain injury in a newborn.
Both infants negative for the virus at birth
The two infants were admitted in the early days of the pandemic in the Delta wave to the neonatal ICU at Holtz Children’s Hospital at University of Miami/Jackson Memorial Medical Center.
Both infants tested negative for the virus at birth, but had significantly elevated SARS-CoV-2 antibodies in their blood, indicating that either antibodies crossed the placenta, or the virus crossed and the immune response was the baby’s.
Dr. Benny explained that the researchers have seen, to this point, more than 700 mother/infant pairs in whom the mother tested positive for COVID in Jackson hospital.
Most who tested positive for COVID were asymptomatic and most of the mothers and infants left the hospital without complications.
“However, (these) two babies had a very unusual clinical picture,” Dr. Benny said.
Those infants were born to mothers who became COVID positive in the second trimester and delivered a few weeks later.
Perinatal HIV nearly eradicated in U.S.
Rates of perinatal HIV have dropped so much that the disease is effectively eliminated in the United States, with less than 1 baby for every 100,000 live births having the virus, a new study released by researchers at the Centers for Disease Control and Prevention finds.
The report marks significant progress on the U.S. government’s goal to eradicate perinatal HIV, an immune-weakening and potentially deadly virus that is passed from mother to baby during pregnancy. Just 32 children in the country were diagnosed in 2019, compared with twice as many in 2010, according to the CDC.
Mothers who are HIV positive can prevent transmission of the infection by receiving antiretroviral therapy, according to Monica Gandhi, MD, MPH, a professor of medicine at University of California, San Francisco’s division of HIV, infectious disease and global medicine.
Dr. Gandhi said she could recall only one case of perinatal HIV in the San Francisco area over the last decade.
DRUGS, PREGNANCY, AND LACTATION
Canadian Task Force recommendation on screening for postpartum depression misses the mark
Director, Ammon-Pinizzotto Center for Women’s Mental Health at Massachusetts General Hospital, Boston, Massachusetts.
Postpartum/perinatal depression (PPD) remains the most common complication in modern obstetrics, with a prevalence of 10%-15% based on multiple studies over the last 2 decades. Over those same 2 decades, there has been growing interest and motivation across the country—from small community hospitals to major academic centers—to promote screening. Such screening is integrated into obstetrical practices, typically using the Edinburgh Postnatal Depression Scale (EPDS), the most widely used validated screen for PPD globally.
As mentioned in previous columns, the U.S. Preventive Services Task Force recommended screening for PPD in 2016, which includes screening women at highest risk, and both acutely treating and preventing PPD.
Since then, screening women for a common clinical problem like PPD has been widely adopted by clinicians representing a broad spectrum of interdisciplinary care. Providers who are engaged in the treatment of postpartum women—obstetricians, psychiatrists, doulas, lactation consultants, facilitators of postpartum support groups, and advocacy groups among others—are included.
An open question and one of great concern recently to our group and others has been what happens after screening. It is clear that identification of PPD per se is not necessarily a challenge, and we have multiple effective treatments from antidepressants to mindfulness-based cognitive therapy to cognitive-behavioral interventions. There is also a growing number of digital applications aimed at mitigation of depressive symptoms in women with postpartum major depressive disorder. One unanswered question is how to engage women after identification of PPD and how to facilitate access to care in a way that maximizes the likelihood that women who actually are suffering from PPD get adequate treatment.
The “perinatal treatment cascade” refers to the majority of women who, on the other side of identification of PPD, fail to receive adequate treatment and continue to have persistent depression. This is perhaps the greatest challenge to the field and to clinicians—how do we, on the other side of screening, see that these women get access to care and get well?
https://www.mdedge.com/obgyn/drugs-pregnancy-lactation
GENDER-AFFIRMING GYNECOLOGY
Caring for the aging transgender patient
Ob.Gyn. and fellowship-trained gender-affirming surgeon in West Reading, Pennsylvania.
The elderly transgender population is rapidly expanding and remains significantly overlooked. Although emerging evidence provides some guidance for medical and surgical treatment for transgender youth, there is still a paucity of research directed at the management of gender-diverse elders.
To a large extent, the challenges that transgender elders face are no different from those experienced by the general elder population. Irrespective of gender identity, patients begin to undergo cognitive and physical changes, encounter difficulties with activities of daily living, suffer the loss of social networks and friends, and face end-of-life issues. Attributes that contribute to successful aging in the general population include good health, social engagement and support, and having a positive outlook on life. Yet, stigma surrounding gender identity and sexual orientation continues to negatively affect elder transgender people.
https://www.mdedge.com/obgyn/gender-affirming-gynecology
Continue to: LATEST NEWS...
LATEST NEWS
Study: Prenatal supplements fail to meet nutrient needs
Although drugstore shelves might suggest otherwise,affordable dietary supplements that provide critical nutrients in appropriate doses for pregnant women are virtually nonexistent, researchers have found.
In a new study published in the American Journal of Clinical Nutrition, investigators observed what many physicians have long suspected: Most prenatal vitamins and other supplements do not adequately make up the difference of what food-based intake of nutrients leave lacking. Despite patients believing they are getting everything they need with their product purchase, they fall short of guideline-recommended requirements.
“There is no magic pill,” said Katherine A. Sauder, PhD, an associate professor of pediatrics at the University of Colorado Anschutz Medical Campus, Aurora, and lead author of the study. “There is no easy answer here.”
The researchers analyzed 24-hour dietary intake data from 2,450 study participants across five states from 2007 to 2019. Dr. Sauder and colleagues focused on six of the more than 20 key nutrients recommended for pregnant people and determined the target dose for vitamin A, vitamin D, folate, calcium, iron, and omega-3 fatty acids.
The researchers tested more than 20,500 dietary supplements, of which 421 were prenatal products. Only 69 products—three prenatal—included all six nutrients. Just seven products —two prenatal—contained target doses for five nutrients. Only one product, which was not marketed as prenatal, contained target doses for all six nutrients but required seven tablets a serving and cost patients approximately $200 a month.
SARS-CoV-2 crosses placenta and infects brains of two infants: ‘This is a first’
Researchers have found for the first time that COVID infection has crossed the placenta and caused brain damage in two newborns, according to a study published online today in Pediatrics.
One of the infants died at 13 months and the other remained in hospice care at time of manuscript submission.
Lead author Merline Benny, MD,with the division of neonatology, department of pediatrics at University of Miami, and colleagues briefed reporters today ahead of the release.
“This is a first,” said senior author Shahnaz Duara, MD, medical director of the Neonatal Intensive Care Unit at Holtz Children’s Hospital, Miami, explaining it is the first study to confirm cross-placental SARS-CoV-2 transmission leading to brain injury in a newborn.
Both infants negative for the virus at birth
The two infants were admitted in the early days of the pandemic in the Delta wave to the neonatal ICU at Holtz Children’s Hospital at University of Miami/Jackson Memorial Medical Center.
Both infants tested negative for the virus at birth, but had significantly elevated SARS-CoV-2 antibodies in their blood, indicating that either antibodies crossed the placenta, or the virus crossed and the immune response was the baby’s.
Dr. Benny explained that the researchers have seen, to this point, more than 700 mother/infant pairs in whom the mother tested positive for COVID in Jackson hospital.
Most who tested positive for COVID were asymptomatic and most of the mothers and infants left the hospital without complications.
“However, (these) two babies had a very unusual clinical picture,” Dr. Benny said.
Those infants were born to mothers who became COVID positive in the second trimester and delivered a few weeks later.
Perinatal HIV nearly eradicated in U.S.
Rates of perinatal HIV have dropped so much that the disease is effectively eliminated in the United States, with less than 1 baby for every 100,000 live births having the virus, a new study released by researchers at the Centers for Disease Control and Prevention finds.
The report marks significant progress on the U.S. government’s goal to eradicate perinatal HIV, an immune-weakening and potentially deadly virus that is passed from mother to baby during pregnancy. Just 32 children in the country were diagnosed in 2019, compared with twice as many in 2010, according to the CDC.
Mothers who are HIV positive can prevent transmission of the infection by receiving antiretroviral therapy, according to Monica Gandhi, MD, MPH, a professor of medicine at University of California, San Francisco’s division of HIV, infectious disease and global medicine.
Dr. Gandhi said she could recall only one case of perinatal HIV in the San Francisco area over the last decade.
DRUGS, PREGNANCY, AND LACTATION
Canadian Task Force recommendation on screening for postpartum depression misses the mark
Director, Ammon-Pinizzotto Center for Women’s Mental Health at Massachusetts General Hospital, Boston, Massachusetts.
Postpartum/perinatal depression (PPD) remains the most common complication in modern obstetrics, with a prevalence of 10%-15% based on multiple studies over the last 2 decades. Over those same 2 decades, there has been growing interest and motivation across the country—from small community hospitals to major academic centers—to promote screening. Such screening is integrated into obstetrical practices, typically using the Edinburgh Postnatal Depression Scale (EPDS), the most widely used validated screen for PPD globally.
As mentioned in previous columns, the U.S. Preventive Services Task Force recommended screening for PPD in 2016, which includes screening women at highest risk, and both acutely treating and preventing PPD.
Since then, screening women for a common clinical problem like PPD has been widely adopted by clinicians representing a broad spectrum of interdisciplinary care. Providers who are engaged in the treatment of postpartum women—obstetricians, psychiatrists, doulas, lactation consultants, facilitators of postpartum support groups, and advocacy groups among others—are included.
An open question and one of great concern recently to our group and others has been what happens after screening. It is clear that identification of PPD per se is not necessarily a challenge, and we have multiple effective treatments from antidepressants to mindfulness-based cognitive therapy to cognitive-behavioral interventions. There is also a growing number of digital applications aimed at mitigation of depressive symptoms in women with postpartum major depressive disorder. One unanswered question is how to engage women after identification of PPD and how to facilitate access to care in a way that maximizes the likelihood that women who actually are suffering from PPD get adequate treatment.
The “perinatal treatment cascade” refers to the majority of women who, on the other side of identification of PPD, fail to receive adequate treatment and continue to have persistent depression. This is perhaps the greatest challenge to the field and to clinicians—how do we, on the other side of screening, see that these women get access to care and get well?
https://www.mdedge.com/obgyn/drugs-pregnancy-lactation
GENDER-AFFIRMING GYNECOLOGY
Caring for the aging transgender patient
Ob.Gyn. and fellowship-trained gender-affirming surgeon in West Reading, Pennsylvania.
The elderly transgender population is rapidly expanding and remains significantly overlooked. Although emerging evidence provides some guidance for medical and surgical treatment for transgender youth, there is still a paucity of research directed at the management of gender-diverse elders.
To a large extent, the challenges that transgender elders face are no different from those experienced by the general elder population. Irrespective of gender identity, patients begin to undergo cognitive and physical changes, encounter difficulties with activities of daily living, suffer the loss of social networks and friends, and face end-of-life issues. Attributes that contribute to successful aging in the general population include good health, social engagement and support, and having a positive outlook on life. Yet, stigma surrounding gender identity and sexual orientation continues to negatively affect elder transgender people.
https://www.mdedge.com/obgyn/gender-affirming-gynecology
Continue to: LATEST NEWS...
LATEST NEWS
Study: Prenatal supplements fail to meet nutrient needs
Although drugstore shelves might suggest otherwise,affordable dietary supplements that provide critical nutrients in appropriate doses for pregnant women are virtually nonexistent, researchers have found.
In a new study published in the American Journal of Clinical Nutrition, investigators observed what many physicians have long suspected: Most prenatal vitamins and other supplements do not adequately make up the difference of what food-based intake of nutrients leave lacking. Despite patients believing they are getting everything they need with their product purchase, they fall short of guideline-recommended requirements.
“There is no magic pill,” said Katherine A. Sauder, PhD, an associate professor of pediatrics at the University of Colorado Anschutz Medical Campus, Aurora, and lead author of the study. “There is no easy answer here.”
The researchers analyzed 24-hour dietary intake data from 2,450 study participants across five states from 2007 to 2019. Dr. Sauder and colleagues focused on six of the more than 20 key nutrients recommended for pregnant people and determined the target dose for vitamin A, vitamin D, folate, calcium, iron, and omega-3 fatty acids.
The researchers tested more than 20,500 dietary supplements, of which 421 were prenatal products. Only 69 products—three prenatal—included all six nutrients. Just seven products —two prenatal—contained target doses for five nutrients. Only one product, which was not marketed as prenatal, contained target doses for all six nutrients but required seven tablets a serving and cost patients approximately $200 a month.
SARS-CoV-2 crosses placenta and infects brains of two infants: ‘This is a first’
Researchers have found for the first time that COVID infection has crossed the placenta and caused brain damage in two newborns, according to a study published online today in Pediatrics.
One of the infants died at 13 months and the other remained in hospice care at time of manuscript submission.
Lead author Merline Benny, MD,with the division of neonatology, department of pediatrics at University of Miami, and colleagues briefed reporters today ahead of the release.
“This is a first,” said senior author Shahnaz Duara, MD, medical director of the Neonatal Intensive Care Unit at Holtz Children’s Hospital, Miami, explaining it is the first study to confirm cross-placental SARS-CoV-2 transmission leading to brain injury in a newborn.
Both infants negative for the virus at birth
The two infants were admitted in the early days of the pandemic in the Delta wave to the neonatal ICU at Holtz Children’s Hospital at University of Miami/Jackson Memorial Medical Center.
Both infants tested negative for the virus at birth, but had significantly elevated SARS-CoV-2 antibodies in their blood, indicating that either antibodies crossed the placenta, or the virus crossed and the immune response was the baby’s.
Dr. Benny explained that the researchers have seen, to this point, more than 700 mother/infant pairs in whom the mother tested positive for COVID in Jackson hospital.
Most who tested positive for COVID were asymptomatic and most of the mothers and infants left the hospital without complications.
“However, (these) two babies had a very unusual clinical picture,” Dr. Benny said.
Those infants were born to mothers who became COVID positive in the second trimester and delivered a few weeks later.
Perinatal HIV nearly eradicated in U.S.
Rates of perinatal HIV have dropped so much that the disease is effectively eliminated in the United States, with less than 1 baby for every 100,000 live births having the virus, a new study released by researchers at the Centers for Disease Control and Prevention finds.
The report marks significant progress on the U.S. government’s goal to eradicate perinatal HIV, an immune-weakening and potentially deadly virus that is passed from mother to baby during pregnancy. Just 32 children in the country were diagnosed in 2019, compared with twice as many in 2010, according to the CDC.
Mothers who are HIV positive can prevent transmission of the infection by receiving antiretroviral therapy, according to Monica Gandhi, MD, MPH, a professor of medicine at University of California, San Francisco’s division of HIV, infectious disease and global medicine.
Dr. Gandhi said she could recall only one case of perinatal HIV in the San Francisco area over the last decade.
Investigational drug peresolimab shows efficacy in patients with RA
(RA), according to results from a phase 2 clinical trial.
After 12 weeks, patients receiving peresolimab 700 mg saw a greater improvement in the primary endpoint of change in Disease Activity Score for 28 joints based on C-reactive protein (DAS28-CRP), compared with placebo.
“These results provide evidence that stimulation of the PD-1 receptor has potential efficacy in the treatment of rheumatoid arthritis,” said the authors, led by Jay Tuttle, PhD, of Eli Lilly and Company. The study was published in the New England Journal of Medicine.
A total of 98 patients with treatment-resistant, moderate to severe RA were enrolled in the double-blind, placebo-controlled trial. All patients had previously experienced treatment failure with biologic, targeted synthetic, or conventional synthetic disease-modifying antirheumatic drugs. Patients were randomized to receive 700 mg of peresolimab (49 patients), 300 mg of peresolimab (25 patients), or placebo (24 patients) intravenously once every 4 weeks.
Only patients taking peresolimab 700 mg had a significantly greater change in DAS28-CRP scores after 12 weeks, compared with placebo. In secondary outcomes, 71% of the 700-mg group experienced an improvement of at least 20% in American College of Rheumatology response criteria (ACR20), as compared with 42% in the placebo group. There was no difference between the placebo and peresolimab groups in ACR50 or ACR70 responses.
The safety profiles were similar across all three groups, although the 700-mg peresolimab group had numerically more adverse events (n = 14) than the 300-mg peresolimab group (n = 8) and the placebo group (n = 9). There were no severe adverse events reported during the study period. The authors noted that larger and longer studies are necessary to understand the safety of peresolimab.
“Careful evaluation of the effect of peresolimab on the risk of cancer will be important given the efficacy of PD-1 inhibitors in oncologic disease,” the authors wrote.
Eli Lilly funded the research. Researchers disclosed financial relationships with AbbVie, Eli Lilly, Janssen, Novartis, Pfizer, and several other pharmaceutical companies.
(RA), according to results from a phase 2 clinical trial.
After 12 weeks, patients receiving peresolimab 700 mg saw a greater improvement in the primary endpoint of change in Disease Activity Score for 28 joints based on C-reactive protein (DAS28-CRP), compared with placebo.
“These results provide evidence that stimulation of the PD-1 receptor has potential efficacy in the treatment of rheumatoid arthritis,” said the authors, led by Jay Tuttle, PhD, of Eli Lilly and Company. The study was published in the New England Journal of Medicine.
A total of 98 patients with treatment-resistant, moderate to severe RA were enrolled in the double-blind, placebo-controlled trial. All patients had previously experienced treatment failure with biologic, targeted synthetic, or conventional synthetic disease-modifying antirheumatic drugs. Patients were randomized to receive 700 mg of peresolimab (49 patients), 300 mg of peresolimab (25 patients), or placebo (24 patients) intravenously once every 4 weeks.
Only patients taking peresolimab 700 mg had a significantly greater change in DAS28-CRP scores after 12 weeks, compared with placebo. In secondary outcomes, 71% of the 700-mg group experienced an improvement of at least 20% in American College of Rheumatology response criteria (ACR20), as compared with 42% in the placebo group. There was no difference between the placebo and peresolimab groups in ACR50 or ACR70 responses.
The safety profiles were similar across all three groups, although the 700-mg peresolimab group had numerically more adverse events (n = 14) than the 300-mg peresolimab group (n = 8) and the placebo group (n = 9). There were no severe adverse events reported during the study period. The authors noted that larger and longer studies are necessary to understand the safety of peresolimab.
“Careful evaluation of the effect of peresolimab on the risk of cancer will be important given the efficacy of PD-1 inhibitors in oncologic disease,” the authors wrote.
Eli Lilly funded the research. Researchers disclosed financial relationships with AbbVie, Eli Lilly, Janssen, Novartis, Pfizer, and several other pharmaceutical companies.
(RA), according to results from a phase 2 clinical trial.
After 12 weeks, patients receiving peresolimab 700 mg saw a greater improvement in the primary endpoint of change in Disease Activity Score for 28 joints based on C-reactive protein (DAS28-CRP), compared with placebo.
“These results provide evidence that stimulation of the PD-1 receptor has potential efficacy in the treatment of rheumatoid arthritis,” said the authors, led by Jay Tuttle, PhD, of Eli Lilly and Company. The study was published in the New England Journal of Medicine.
A total of 98 patients with treatment-resistant, moderate to severe RA were enrolled in the double-blind, placebo-controlled trial. All patients had previously experienced treatment failure with biologic, targeted synthetic, or conventional synthetic disease-modifying antirheumatic drugs. Patients were randomized to receive 700 mg of peresolimab (49 patients), 300 mg of peresolimab (25 patients), or placebo (24 patients) intravenously once every 4 weeks.
Only patients taking peresolimab 700 mg had a significantly greater change in DAS28-CRP scores after 12 weeks, compared with placebo. In secondary outcomes, 71% of the 700-mg group experienced an improvement of at least 20% in American College of Rheumatology response criteria (ACR20), as compared with 42% in the placebo group. There was no difference between the placebo and peresolimab groups in ACR50 or ACR70 responses.
The safety profiles were similar across all three groups, although the 700-mg peresolimab group had numerically more adverse events (n = 14) than the 300-mg peresolimab group (n = 8) and the placebo group (n = 9). There were no severe adverse events reported during the study period. The authors noted that larger and longer studies are necessary to understand the safety of peresolimab.
“Careful evaluation of the effect of peresolimab on the risk of cancer will be important given the efficacy of PD-1 inhibitors in oncologic disease,” the authors wrote.
Eli Lilly funded the research. Researchers disclosed financial relationships with AbbVie, Eli Lilly, Janssen, Novartis, Pfizer, and several other pharmaceutical companies.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Independent risk factors-based referral tool may help identify PsA in psoriasis
Key clinical point: A referral tool based on five independent risk factors for concomitant psoriatic arthritis (PsA) among patients with psoriasis may help dermatologists to identify patients with psoriasis who could benefit from a rheumatologist referral.
Major finding: The predictive variables for concomitant PsA among patients with psoriasis that were included in the referral tool were treatment history with conventional systemics (P = .04) and biologics/small molecule inhibitors (P = .01), patient-reported history of joint pain without trauma (P = .02), swollen joints (P < .001), and sausage-like swollen digits (P = .01). The referral tool had an area under curve of 0.82.
Study details: This study analyzed the data of 303 patients with psoriasis from the prospective observational DAPPER study who had visited the dermatology outpatient clinic.
Disclosures: This study was funded by a PhD grant from Radboud University Medical Center/Sint Maartenskliniek, Netherlands, and local structural research funding of the Sint Maartenskliniek. Several authors reported financial and non-financial ties with various sources.
Source: van Hal TW et al. Development of a new referral tool to identify psoriasis patients with concomitant psoriatic arthritis: Results of the prospective DAPPER cohort. Acta Derm Venereol. 2023 (Apr 27). Doi: 10.2340/actadv.v103.5269
Key clinical point: A referral tool based on five independent risk factors for concomitant psoriatic arthritis (PsA) among patients with psoriasis may help dermatologists to identify patients with psoriasis who could benefit from a rheumatologist referral.
Major finding: The predictive variables for concomitant PsA among patients with psoriasis that were included in the referral tool were treatment history with conventional systemics (P = .04) and biologics/small molecule inhibitors (P = .01), patient-reported history of joint pain without trauma (P = .02), swollen joints (P < .001), and sausage-like swollen digits (P = .01). The referral tool had an area under curve of 0.82.
Study details: This study analyzed the data of 303 patients with psoriasis from the prospective observational DAPPER study who had visited the dermatology outpatient clinic.
Disclosures: This study was funded by a PhD grant from Radboud University Medical Center/Sint Maartenskliniek, Netherlands, and local structural research funding of the Sint Maartenskliniek. Several authors reported financial and non-financial ties with various sources.
Source: van Hal TW et al. Development of a new referral tool to identify psoriasis patients with concomitant psoriatic arthritis: Results of the prospective DAPPER cohort. Acta Derm Venereol. 2023 (Apr 27). Doi: 10.2340/actadv.v103.5269
Key clinical point: A referral tool based on five independent risk factors for concomitant psoriatic arthritis (PsA) among patients with psoriasis may help dermatologists to identify patients with psoriasis who could benefit from a rheumatologist referral.
Major finding: The predictive variables for concomitant PsA among patients with psoriasis that were included in the referral tool were treatment history with conventional systemics (P = .04) and biologics/small molecule inhibitors (P = .01), patient-reported history of joint pain without trauma (P = .02), swollen joints (P < .001), and sausage-like swollen digits (P = .01). The referral tool had an area under curve of 0.82.
Study details: This study analyzed the data of 303 patients with psoriasis from the prospective observational DAPPER study who had visited the dermatology outpatient clinic.
Disclosures: This study was funded by a PhD grant from Radboud University Medical Center/Sint Maartenskliniek, Netherlands, and local structural research funding of the Sint Maartenskliniek. Several authors reported financial and non-financial ties with various sources.
Source: van Hal TW et al. Development of a new referral tool to identify psoriasis patients with concomitant psoriatic arthritis: Results of the prospective DAPPER cohort. Acta Derm Venereol. 2023 (Apr 27). Doi: 10.2340/actadv.v103.5269