User login
When should lipid-lowering therapy be started in the hospitalized patient?
Case
A 52-year-old man with no medical history other than a transient ischemic attack (TIA) three months ago presents to the emergency department (ED) following multiple episodes of substernal (ST) chest pressure. He takes no medication. His electrocardiogram (ECG) revealed lateral ST segment depressions, and his cardiac biomarkers were elevated. He underwent cardiac catheterization, and a single drug-eluting stent was successfully placed to a culprit left circumflex lesion. He is now stable less than 24 hours following his initial presentation, without any evidence of heart failure. His providers prescribe aspirin, clopidogrel, metoprolol, and lisinopril. His fasting LDL level is 92 mg/dL.
What, if any, is the role for lipid-lowering therapy at this time?
Overview
Long-term therapy with HMG CoA reductase inhibitors (statins) has been shown through several large, randomized, controlled trials to reduce the risk for death, myocardial infarction (MI), and stroke in patients with established coronary disease. The most significant effects were evident after approximately two years of treatment.1,2,3,4
Subsequent trials have shown earlier and more significant reductions in the rates of recurrent ischemic cardiovascular events following acute coronary syndromes (ACS) when statins are administered early—within days of the initial event. This is a window of time in which most patients still are hospitalized.4,5,6,7
In addition to this data regarding statin use following ACS, a large, randomized, controlled trial demonstrated similar reductions in the incidence of strokes and cardiovascular events when high-dose atorvastatin was administered within one to six months following TIA or stroke in patients without established coronary disease.8 There is growing data supporting the hypothesis that statins have pleiotropic (non cholesterol-lowering), neuroprotective, properties that may improve patient outcomes following cerebrovascular events.9,10,11 There are ongoing trials investing the role of statins in the acute management of stroke.12,13
Hospitalists frequently manage patients in the stages immediately following ACS and stroke. Based on the large and evolving volume of data regarding the use of statins following these events, when and how should a statin be started in the hospital?
Review of the Data
Following Acute Coronary Syndrome: Death and recurrent ischemic events following ACS are most likely to occur in the early phase of recovery. Based on this observation and evidence supporting the early (in some cases within hours of administration) ‘pleiotropic’ or non-cholesterol lowering effects of statins, including improvement in endothelial function and decreases in platelet aggregation, thrombus deposition, and vascular inflammation, the MIRACL study was designed to answer the question of whether the initiation of treatment with a statin within 24 to 96 hours following ACS would reduce the occurrence of death and recurrent ischemia.4,7,14 Investigators randomized 3,086 patients within 24-96 hours (mean 63 hours) following admission for non-ST segment myocardial infarction (NSTEMI) or unstable angina (UA) to receive either atorvastatin 80 mg/d or placebo.
Investigators monitored patients for the primary end points of ischemic events (death, non-fatal MI, cardiac arrest with resuscitation, symptomatic myocardial ischemia with objective evidence) during a 16-week period. In the treatment arm, the risk of the primary combined end point was significantly reduced—relative risk (RR) 0.84; 95% confidence interval (CI), 0.70-1.00; p=0.048. (See Figure 1, pg. 39)
No significant differences were found between atorvastatin and placebo in the risk of death, non-fatal MI, or cardiac arrest with resuscitation. There was, however, a significantly lower risk of recurrent symptomatic myocardial ischemia with objective evidence requiring emergent re-hospitalization in the treatment arm (RR, 0.74; 95% CI, 0.57-0.95; p=0.02). The mean baseline LDL level in the treatment arm was 124 mg/dL, a value that may represent, in part, suppression of the LDL level in the setting of acute ACS. This is a phenomenon previously described in an analysis of the LUNAR trial.15
Suppression of LDL level after ACS appeared to be minimal, however, and is unlikely to be clinically significant. The benefits of atorvastatin in the MIRACL trial did not appear to depend on baseline LDL level—suggesting the decision to initiate statin therapy after ACS should not be influenced by LDL level at the time of the event.
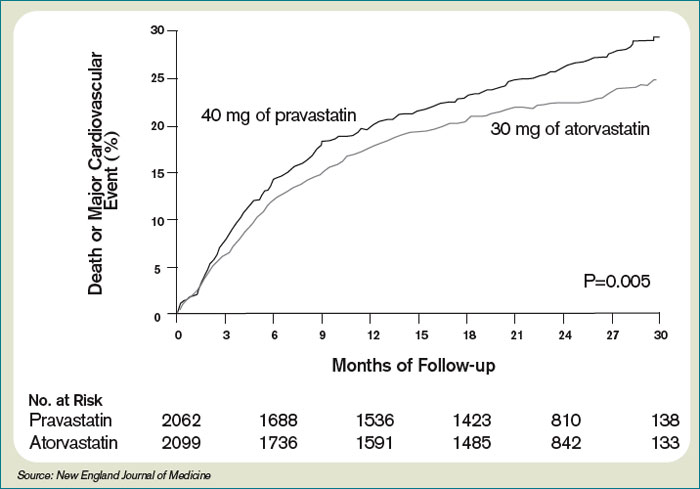
Only one dose of statin was used in the MIRACL trial, and the investigators commented they were unable to determine if a lower dose of atorvastatin or a gradual dose titration to a predetermined LDL target would have achieved similar benefits. The PROVE IT-TIMI 22 trial was designed to compare the reductions in death and major cardiovascular events following ACS between LDL lowering to approximately 100 mg/dL using 40 mg/d of pravastatin, and more intensive LDL lowering to approximately 70 mg/dL using 80 mg/d of atorvastatin.5 Investigators enrolled 4,162 patients for a median of seven days following ACS (STEMI, NSTEMI, or UA) to the two treatment arms. Investigators observed patients for a period of 18 to 36 months for the primary end points of death, MI, UA, revascularization, and stroke. The median LDL level at the time of enrollment was 106 mg/dL in both treatment arms. During follow up, the median LDL levels achieved were 95 mg/dL in the pravastatin group and 62 mg/dL in the atorvastatin group. After two years, a 16% reduction in the hazard ratio for any primary end point was seen favoring 80 mg/d of atorvastatin—p=0.005; 95% CI=5-26%. (See Figure 2, pg. 39) The benefit of high-dose atorvastatin was seen as early as 30 days after randomization and was sustained throughout the trial.
While the PROVE IT-TIMI 22 trial supported a specified dosing strategy for statin use following ACS, Phase Z of the A-to-Z trial was designed to evaluate the early initiation of intensive lipid lowering following ACS, as compared to a delayed and less-intensive strategy.5,6 Investigators randomized 4,497 patients (a mean of 3.7 days following either NSTEMI or STEMI) to receive either placebo for four months followed by simvastatin 20 mg/d or simvastatin 40 mg/d for one month followed by simvastatin 80 mg/d. They followed patients for 24 months for the primary end points of cardiovascular death, MI, readmission for ACS, or stroke. The primary end point occurred in 16.7% of the delayed, lower-intensity treatment group and in 14.4% of the early, higher-intensity treatment group (95% CI 0.76-1.04; p=0.14). Despite the lack of a significant difference in the composite primary end point between the two treatment arms, a significant reduction in the secondary end points of cardiovascular mortality (absolute risk reduction (ARR 1.3%; P=0.05) and congestive heart failure (ARR 1.3%; P=0.04) was evident favoring the early, intensive treatment strategy. These differences were not evident until at least four months after randomization. The A-to-Z trial investigators offered several possible explanations for the delay in evident clinical benefits in their trial when compared against the strong trend toward clinical benefit seen with 30 days following the early initiation of high-dose atorvastatin following ACS in the PROVE IT-TIMI 22 trial. In the PROVE IT trial, patients were enrolled an average of seven days after their index event, and as a result, 69% had undergone revascularization by this time. In the A-to-Z trial, patients were enrolled an average of three to four days earlier, and, therefore, were less likely to have undergone a revascularization procedure by the time of enrollment—and may have continued on with active thrombotic processes relatively less responsive to statin therapy.6 Another notable difference between PROVE IT and A-to-Z subjects was the C-reactive protein (CRP) concentrations in the A-to-Z subjects did not differ between treatment groups within 30 days despite significant differences in their LDL levels.16 This lack of a concurrent, pleiotropic, anti-inflammatory effect in the A-to-Z trial aggressive treatment arm may also have contributed to the delayed treatment effect.
In conclusion, the A-to-Z investigators suggest more intensive statin therapy (than the 40 mg Simvastatin in their intensive treatment arm) may be required to derive the most rapid and maximal clinical benefits during the highest risk period immediately following ACS.
Following stroke: Although there is more robust data supporting the benefits of early, intensive, statin therapy following ACS, there also is established and emerging data supporting similar treatment approaches following stroke.
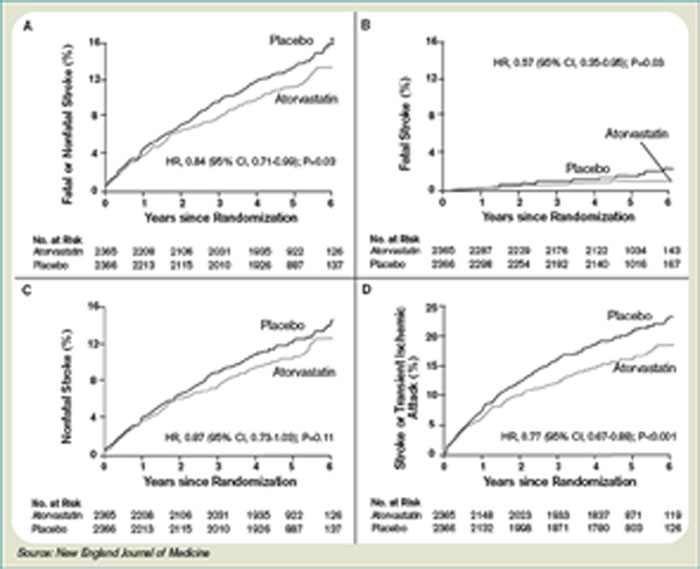
The Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) trial was designed to determine whether or not atorvastatin 80 mg daily would reduce the risk of stroke in patients without known coronary heart disease who had suffered a TIA or stroke within the preceding six months.8 Patients who experienced a hemorrhagic or ischemic TIA or stroke between one to six months before study entry were randomized to receive either atorvastatin 80 mg/d or placebo.
Investigators followed patients for a mean period of 4.9 years for the primary end point of time to non-fatal or fatal stroke. Secondary composite end points included stroke or TIA, and any coronary or peripheral arterial event, including death from cardiac causes, non-fatal MI, ACS, revascularization (coronary, carotid, peripheral), and death from any cause. No difference in mean baseline LDL levels was witnessed between the treatment and placebo arms (132.7 and 133.7 mg/dL, respectively). Atorvastatin was associated with a 16% relative reduction in the risk of stroke—hazard ratio, 0.84; 95% CI 0.71–0.99; p=0.03. This was found despite an increase in hemorrhagic stroke in the atorvastatin group—a finding that supports an epidemiologic association between low cholesterol levels and brain hemorrhage. The risk of cardiovascular events also was significantly reduced, however, no significant difference in overall mortality was observed between the two groups.
In conclusion, the authors recommend the initiation of high-dose atorvastatin “soon” after stroke or TIA. One can only conclude, based on these data, statin therapy should be initiated within six months of TIA or stroke, in accordance with the study design. There is retrospective data suggesting benefit to statin therapy initiated within four weeks following ischemic stroke, and there are prospective trials in process evaluating the potential benefits of statins initiated within 24 hours following ischemic stroke, however, no large, randomized, controlled trial can demonstrate the effect of statins when used as acute stroke therapy.9,12,13,17
Back to the Case
The patient described in our case has a history of TIA and experienced an acute coronary syndrome (NSTEMI) within the preceding 24 hours. He underwent a revascularization procedure (PCI with stent), and is on appropriate therapy, including dual anti-platelet therapy with aspirin and clopidogrel, a beta-blocker, and an angiotensin-converting enzyme inhibitor. Based on the data and conclusions of the MIRACL, PROVE IT-TIMI 22, and SPARCL trials, high-dose statin therapy with atorvastatin 80 mg/d should be initiated immediately in the patient in order to significantly reduce his risk of recurrent ischemic cardiovascular events and stroke following his acute coronary syndrome and TIA.
Bottom Line
Following ACS, high-dose statin therapy with 80 mg of atorvastatin per day should be initiated when the patient is still in the hospital, irrespective of baseline LDL level. Statin therapy should also strongly be considered for secondary stroke prevention in most patients with a history of stroke or transient ischemic attack. TH
Caleb Hale, MD, is a hospitalist at Beth Israel Deaconess Medical Center in Boston. Joseph Ming Wah Li is director of the hospital medicine program and associate chief, division of general medicine and primary care at Beth Israel Deaconess Medical Center in Boston, and assistant professor of Medicine at Harvard Medical School.
References
1. Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994;344:1383-1389.
2. Sacks RM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. N Engl J Med 1996;335:1001-1009.
3. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med 1998;339:1349-1357.
4. Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes. The MIRACL study: a randomized controlled trial. JAMA. 2001;285:1711-1718.
5. Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495-1504.
6. Lemos JA, Blazing MA, Wiviott SD, et al. Early intensive vs. a delayed conservative simvastatin strategy in patients with acute coronary syndromes. Phase Z of the A to Z trial. JAMA. 2004;292:1307-1316.
7. Waters D, Schwartz GG, Olsson AG. The myocardial ischemia reduction with acute cholesterol lowering (MIRACL) trial: a new frontier for statins? Curr Control Trials Cardiovasc Med. 2001;2;111-114.
8. The Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) Investigators. High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med. 2006;355:549-559.
9. Moonis M, Kane K, Schwiderski U, Sandage BW, Fisher M. HMG-CoA reductase inhibitors improve acute ischemic stroke outcome. Stroke. 2005;36:1298-1300.
10. Elkind MS, Flint AC, Sciacca RR, Sacco RL. Lipid-lowering agent use at ischemic stroke onset is associated with decreased mortality. Neurology. 2005;65:253-258.
11. Vaughan CJ, Delanty N. Neuroprotective properties of statins in cerebral ischemia and stroke. Stroke. 1999;30:1969-1973.
12. Elkind MS, Sacco RL, MacArthur RB, et al. The neuroprotection with statin therapy for acute recovery trial (NeuSTART): an adaptive design phase I dose-escalation study of high-dose lovastatin in acute ischemic stroke. Int J Stroke. 2008;3:210-218.
13. Montaner J, Chacon P, Krupinski J, et al. Simvastatin in the acute phase of ischemic stroke: a safety and efficacy pilot trial. Eur J of Neurol. 2008;15:82-90.
14. Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20-28.
15. Pitt B, Loscalzo J, Ycas J, Raichlen JS. Lipid levels after acute coronary syndromes. JACC. 2008;51:1440-1445.
16. Wiviott SD, de Lemos JA, Cannon CP, et al. A tale of two trials: a comparison of the post-acute coronary syndrome lipid-lowering trials of A to Z and PROVE IT-TIMI 22. Circulation. 2006;113:1406-1414.
17. Elking MS. Statins as acute-stroke treatment. Int J Stroke. 2006;1:224-225.
Case
A 52-year-old man with no medical history other than a transient ischemic attack (TIA) three months ago presents to the emergency department (ED) following multiple episodes of substernal (ST) chest pressure. He takes no medication. His electrocardiogram (ECG) revealed lateral ST segment depressions, and his cardiac biomarkers were elevated. He underwent cardiac catheterization, and a single drug-eluting stent was successfully placed to a culprit left circumflex lesion. He is now stable less than 24 hours following his initial presentation, without any evidence of heart failure. His providers prescribe aspirin, clopidogrel, metoprolol, and lisinopril. His fasting LDL level is 92 mg/dL.
What, if any, is the role for lipid-lowering therapy at this time?
Overview
Long-term therapy with HMG CoA reductase inhibitors (statins) has been shown through several large, randomized, controlled trials to reduce the risk for death, myocardial infarction (MI), and stroke in patients with established coronary disease. The most significant effects were evident after approximately two years of treatment.1,2,3,4
Subsequent trials have shown earlier and more significant reductions in the rates of recurrent ischemic cardiovascular events following acute coronary syndromes (ACS) when statins are administered early—within days of the initial event. This is a window of time in which most patients still are hospitalized.4,5,6,7
In addition to this data regarding statin use following ACS, a large, randomized, controlled trial demonstrated similar reductions in the incidence of strokes and cardiovascular events when high-dose atorvastatin was administered within one to six months following TIA or stroke in patients without established coronary disease.8 There is growing data supporting the hypothesis that statins have pleiotropic (non cholesterol-lowering), neuroprotective, properties that may improve patient outcomes following cerebrovascular events.9,10,11 There are ongoing trials investing the role of statins in the acute management of stroke.12,13
Hospitalists frequently manage patients in the stages immediately following ACS and stroke. Based on the large and evolving volume of data regarding the use of statins following these events, when and how should a statin be started in the hospital?
Review of the Data
Following Acute Coronary Syndrome: Death and recurrent ischemic events following ACS are most likely to occur in the early phase of recovery. Based on this observation and evidence supporting the early (in some cases within hours of administration) ‘pleiotropic’ or non-cholesterol lowering effects of statins, including improvement in endothelial function and decreases in platelet aggregation, thrombus deposition, and vascular inflammation, the MIRACL study was designed to answer the question of whether the initiation of treatment with a statin within 24 to 96 hours following ACS would reduce the occurrence of death and recurrent ischemia.4,7,14 Investigators randomized 3,086 patients within 24-96 hours (mean 63 hours) following admission for non-ST segment myocardial infarction (NSTEMI) or unstable angina (UA) to receive either atorvastatin 80 mg/d or placebo.
Investigators monitored patients for the primary end points of ischemic events (death, non-fatal MI, cardiac arrest with resuscitation, symptomatic myocardial ischemia with objective evidence) during a 16-week period. In the treatment arm, the risk of the primary combined end point was significantly reduced—relative risk (RR) 0.84; 95% confidence interval (CI), 0.70-1.00; p=0.048. (See Figure 1, pg. 39)
No significant differences were found between atorvastatin and placebo in the risk of death, non-fatal MI, or cardiac arrest with resuscitation. There was, however, a significantly lower risk of recurrent symptomatic myocardial ischemia with objective evidence requiring emergent re-hospitalization in the treatment arm (RR, 0.74; 95% CI, 0.57-0.95; p=0.02). The mean baseline LDL level in the treatment arm was 124 mg/dL, a value that may represent, in part, suppression of the LDL level in the setting of acute ACS. This is a phenomenon previously described in an analysis of the LUNAR trial.15
Suppression of LDL level after ACS appeared to be minimal, however, and is unlikely to be clinically significant. The benefits of atorvastatin in the MIRACL trial did not appear to depend on baseline LDL level—suggesting the decision to initiate statin therapy after ACS should not be influenced by LDL level at the time of the event.
Only one dose of statin was used in the MIRACL trial, and the investigators commented they were unable to determine if a lower dose of atorvastatin or a gradual dose titration to a predetermined LDL target would have achieved similar benefits. The PROVE IT-TIMI 22 trial was designed to compare the reductions in death and major cardiovascular events following ACS between LDL lowering to approximately 100 mg/dL using 40 mg/d of pravastatin, and more intensive LDL lowering to approximately 70 mg/dL using 80 mg/d of atorvastatin.5 Investigators enrolled 4,162 patients for a median of seven days following ACS (STEMI, NSTEMI, or UA) to the two treatment arms. Investigators observed patients for a period of 18 to 36 months for the primary end points of death, MI, UA, revascularization, and stroke. The median LDL level at the time of enrollment was 106 mg/dL in both treatment arms. During follow up, the median LDL levels achieved were 95 mg/dL in the pravastatin group and 62 mg/dL in the atorvastatin group. After two years, a 16% reduction in the hazard ratio for any primary end point was seen favoring 80 mg/d of atorvastatin—p=0.005; 95% CI=5-26%. (See Figure 2, pg. 39) The benefit of high-dose atorvastatin was seen as early as 30 days after randomization and was sustained throughout the trial.
While the PROVE IT-TIMI 22 trial supported a specified dosing strategy for statin use following ACS, Phase Z of the A-to-Z trial was designed to evaluate the early initiation of intensive lipid lowering following ACS, as compared to a delayed and less-intensive strategy.5,6 Investigators randomized 4,497 patients (a mean of 3.7 days following either NSTEMI or STEMI) to receive either placebo for four months followed by simvastatin 20 mg/d or simvastatin 40 mg/d for one month followed by simvastatin 80 mg/d. They followed patients for 24 months for the primary end points of cardiovascular death, MI, readmission for ACS, or stroke. The primary end point occurred in 16.7% of the delayed, lower-intensity treatment group and in 14.4% of the early, higher-intensity treatment group (95% CI 0.76-1.04; p=0.14). Despite the lack of a significant difference in the composite primary end point between the two treatment arms, a significant reduction in the secondary end points of cardiovascular mortality (absolute risk reduction (ARR 1.3%; P=0.05) and congestive heart failure (ARR 1.3%; P=0.04) was evident favoring the early, intensive treatment strategy. These differences were not evident until at least four months after randomization. The A-to-Z trial investigators offered several possible explanations for the delay in evident clinical benefits in their trial when compared against the strong trend toward clinical benefit seen with 30 days following the early initiation of high-dose atorvastatin following ACS in the PROVE IT-TIMI 22 trial. In the PROVE IT trial, patients were enrolled an average of seven days after their index event, and as a result, 69% had undergone revascularization by this time. In the A-to-Z trial, patients were enrolled an average of three to four days earlier, and, therefore, were less likely to have undergone a revascularization procedure by the time of enrollment—and may have continued on with active thrombotic processes relatively less responsive to statin therapy.6 Another notable difference between PROVE IT and A-to-Z subjects was the C-reactive protein (CRP) concentrations in the A-to-Z subjects did not differ between treatment groups within 30 days despite significant differences in their LDL levels.16 This lack of a concurrent, pleiotropic, anti-inflammatory effect in the A-to-Z trial aggressive treatment arm may also have contributed to the delayed treatment effect.
In conclusion, the A-to-Z investigators suggest more intensive statin therapy (than the 40 mg Simvastatin in their intensive treatment arm) may be required to derive the most rapid and maximal clinical benefits during the highest risk period immediately following ACS.
Following stroke: Although there is more robust data supporting the benefits of early, intensive, statin therapy following ACS, there also is established and emerging data supporting similar treatment approaches following stroke.
The Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) trial was designed to determine whether or not atorvastatin 80 mg daily would reduce the risk of stroke in patients without known coronary heart disease who had suffered a TIA or stroke within the preceding six months.8 Patients who experienced a hemorrhagic or ischemic TIA or stroke between one to six months before study entry were randomized to receive either atorvastatin 80 mg/d or placebo.
Investigators followed patients for a mean period of 4.9 years for the primary end point of time to non-fatal or fatal stroke. Secondary composite end points included stroke or TIA, and any coronary or peripheral arterial event, including death from cardiac causes, non-fatal MI, ACS, revascularization (coronary, carotid, peripheral), and death from any cause. No difference in mean baseline LDL levels was witnessed between the treatment and placebo arms (132.7 and 133.7 mg/dL, respectively). Atorvastatin was associated with a 16% relative reduction in the risk of stroke—hazard ratio, 0.84; 95% CI 0.71–0.99; p=0.03. This was found despite an increase in hemorrhagic stroke in the atorvastatin group—a finding that supports an epidemiologic association between low cholesterol levels and brain hemorrhage. The risk of cardiovascular events also was significantly reduced, however, no significant difference in overall mortality was observed between the two groups.
In conclusion, the authors recommend the initiation of high-dose atorvastatin “soon” after stroke or TIA. One can only conclude, based on these data, statin therapy should be initiated within six months of TIA or stroke, in accordance with the study design. There is retrospective data suggesting benefit to statin therapy initiated within four weeks following ischemic stroke, and there are prospective trials in process evaluating the potential benefits of statins initiated within 24 hours following ischemic stroke, however, no large, randomized, controlled trial can demonstrate the effect of statins when used as acute stroke therapy.9,12,13,17
Back to the Case
The patient described in our case has a history of TIA and experienced an acute coronary syndrome (NSTEMI) within the preceding 24 hours. He underwent a revascularization procedure (PCI with stent), and is on appropriate therapy, including dual anti-platelet therapy with aspirin and clopidogrel, a beta-blocker, and an angiotensin-converting enzyme inhibitor. Based on the data and conclusions of the MIRACL, PROVE IT-TIMI 22, and SPARCL trials, high-dose statin therapy with atorvastatin 80 mg/d should be initiated immediately in the patient in order to significantly reduce his risk of recurrent ischemic cardiovascular events and stroke following his acute coronary syndrome and TIA.
Bottom Line
Following ACS, high-dose statin therapy with 80 mg of atorvastatin per day should be initiated when the patient is still in the hospital, irrespective of baseline LDL level. Statin therapy should also strongly be considered for secondary stroke prevention in most patients with a history of stroke or transient ischemic attack. TH
Caleb Hale, MD, is a hospitalist at Beth Israel Deaconess Medical Center in Boston. Joseph Ming Wah Li is director of the hospital medicine program and associate chief, division of general medicine and primary care at Beth Israel Deaconess Medical Center in Boston, and assistant professor of Medicine at Harvard Medical School.
References
1. Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994;344:1383-1389.
2. Sacks RM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. N Engl J Med 1996;335:1001-1009.
3. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med 1998;339:1349-1357.
4. Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes. The MIRACL study: a randomized controlled trial. JAMA. 2001;285:1711-1718.
5. Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495-1504.
6. Lemos JA, Blazing MA, Wiviott SD, et al. Early intensive vs. a delayed conservative simvastatin strategy in patients with acute coronary syndromes. Phase Z of the A to Z trial. JAMA. 2004;292:1307-1316.
7. Waters D, Schwartz GG, Olsson AG. The myocardial ischemia reduction with acute cholesterol lowering (MIRACL) trial: a new frontier for statins? Curr Control Trials Cardiovasc Med. 2001;2;111-114.
8. The Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) Investigators. High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med. 2006;355:549-559.
9. Moonis M, Kane K, Schwiderski U, Sandage BW, Fisher M. HMG-CoA reductase inhibitors improve acute ischemic stroke outcome. Stroke. 2005;36:1298-1300.
10. Elkind MS, Flint AC, Sciacca RR, Sacco RL. Lipid-lowering agent use at ischemic stroke onset is associated with decreased mortality. Neurology. 2005;65:253-258.
11. Vaughan CJ, Delanty N. Neuroprotective properties of statins in cerebral ischemia and stroke. Stroke. 1999;30:1969-1973.
12. Elkind MS, Sacco RL, MacArthur RB, et al. The neuroprotection with statin therapy for acute recovery trial (NeuSTART): an adaptive design phase I dose-escalation study of high-dose lovastatin in acute ischemic stroke. Int J Stroke. 2008;3:210-218.
13. Montaner J, Chacon P, Krupinski J, et al. Simvastatin in the acute phase of ischemic stroke: a safety and efficacy pilot trial. Eur J of Neurol. 2008;15:82-90.
14. Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20-28.
15. Pitt B, Loscalzo J, Ycas J, Raichlen JS. Lipid levels after acute coronary syndromes. JACC. 2008;51:1440-1445.
16. Wiviott SD, de Lemos JA, Cannon CP, et al. A tale of two trials: a comparison of the post-acute coronary syndrome lipid-lowering trials of A to Z and PROVE IT-TIMI 22. Circulation. 2006;113:1406-1414.
17. Elking MS. Statins as acute-stroke treatment. Int J Stroke. 2006;1:224-225.
Case
A 52-year-old man with no medical history other than a transient ischemic attack (TIA) three months ago presents to the emergency department (ED) following multiple episodes of substernal (ST) chest pressure. He takes no medication. His electrocardiogram (ECG) revealed lateral ST segment depressions, and his cardiac biomarkers were elevated. He underwent cardiac catheterization, and a single drug-eluting stent was successfully placed to a culprit left circumflex lesion. He is now stable less than 24 hours following his initial presentation, without any evidence of heart failure. His providers prescribe aspirin, clopidogrel, metoprolol, and lisinopril. His fasting LDL level is 92 mg/dL.
What, if any, is the role for lipid-lowering therapy at this time?
Overview
Long-term therapy with HMG CoA reductase inhibitors (statins) has been shown through several large, randomized, controlled trials to reduce the risk for death, myocardial infarction (MI), and stroke in patients with established coronary disease. The most significant effects were evident after approximately two years of treatment.1,2,3,4
Subsequent trials have shown earlier and more significant reductions in the rates of recurrent ischemic cardiovascular events following acute coronary syndromes (ACS) when statins are administered early—within days of the initial event. This is a window of time in which most patients still are hospitalized.4,5,6,7
In addition to this data regarding statin use following ACS, a large, randomized, controlled trial demonstrated similar reductions in the incidence of strokes and cardiovascular events when high-dose atorvastatin was administered within one to six months following TIA or stroke in patients without established coronary disease.8 There is growing data supporting the hypothesis that statins have pleiotropic (non cholesterol-lowering), neuroprotective, properties that may improve patient outcomes following cerebrovascular events.9,10,11 There are ongoing trials investing the role of statins in the acute management of stroke.12,13
Hospitalists frequently manage patients in the stages immediately following ACS and stroke. Based on the large and evolving volume of data regarding the use of statins following these events, when and how should a statin be started in the hospital?
Review of the Data
Following Acute Coronary Syndrome: Death and recurrent ischemic events following ACS are most likely to occur in the early phase of recovery. Based on this observation and evidence supporting the early (in some cases within hours of administration) ‘pleiotropic’ or non-cholesterol lowering effects of statins, including improvement in endothelial function and decreases in platelet aggregation, thrombus deposition, and vascular inflammation, the MIRACL study was designed to answer the question of whether the initiation of treatment with a statin within 24 to 96 hours following ACS would reduce the occurrence of death and recurrent ischemia.4,7,14 Investigators randomized 3,086 patients within 24-96 hours (mean 63 hours) following admission for non-ST segment myocardial infarction (NSTEMI) or unstable angina (UA) to receive either atorvastatin 80 mg/d or placebo.
Investigators monitored patients for the primary end points of ischemic events (death, non-fatal MI, cardiac arrest with resuscitation, symptomatic myocardial ischemia with objective evidence) during a 16-week period. In the treatment arm, the risk of the primary combined end point was significantly reduced—relative risk (RR) 0.84; 95% confidence interval (CI), 0.70-1.00; p=0.048. (See Figure 1, pg. 39)
No significant differences were found between atorvastatin and placebo in the risk of death, non-fatal MI, or cardiac arrest with resuscitation. There was, however, a significantly lower risk of recurrent symptomatic myocardial ischemia with objective evidence requiring emergent re-hospitalization in the treatment arm (RR, 0.74; 95% CI, 0.57-0.95; p=0.02). The mean baseline LDL level in the treatment arm was 124 mg/dL, a value that may represent, in part, suppression of the LDL level in the setting of acute ACS. This is a phenomenon previously described in an analysis of the LUNAR trial.15
Suppression of LDL level after ACS appeared to be minimal, however, and is unlikely to be clinically significant. The benefits of atorvastatin in the MIRACL trial did not appear to depend on baseline LDL level—suggesting the decision to initiate statin therapy after ACS should not be influenced by LDL level at the time of the event.
Only one dose of statin was used in the MIRACL trial, and the investigators commented they were unable to determine if a lower dose of atorvastatin or a gradual dose titration to a predetermined LDL target would have achieved similar benefits. The PROVE IT-TIMI 22 trial was designed to compare the reductions in death and major cardiovascular events following ACS between LDL lowering to approximately 100 mg/dL using 40 mg/d of pravastatin, and more intensive LDL lowering to approximately 70 mg/dL using 80 mg/d of atorvastatin.5 Investigators enrolled 4,162 patients for a median of seven days following ACS (STEMI, NSTEMI, or UA) to the two treatment arms. Investigators observed patients for a period of 18 to 36 months for the primary end points of death, MI, UA, revascularization, and stroke. The median LDL level at the time of enrollment was 106 mg/dL in both treatment arms. During follow up, the median LDL levels achieved were 95 mg/dL in the pravastatin group and 62 mg/dL in the atorvastatin group. After two years, a 16% reduction in the hazard ratio for any primary end point was seen favoring 80 mg/d of atorvastatin—p=0.005; 95% CI=5-26%. (See Figure 2, pg. 39) The benefit of high-dose atorvastatin was seen as early as 30 days after randomization and was sustained throughout the trial.
While the PROVE IT-TIMI 22 trial supported a specified dosing strategy for statin use following ACS, Phase Z of the A-to-Z trial was designed to evaluate the early initiation of intensive lipid lowering following ACS, as compared to a delayed and less-intensive strategy.5,6 Investigators randomized 4,497 patients (a mean of 3.7 days following either NSTEMI or STEMI) to receive either placebo for four months followed by simvastatin 20 mg/d or simvastatin 40 mg/d for one month followed by simvastatin 80 mg/d. They followed patients for 24 months for the primary end points of cardiovascular death, MI, readmission for ACS, or stroke. The primary end point occurred in 16.7% of the delayed, lower-intensity treatment group and in 14.4% of the early, higher-intensity treatment group (95% CI 0.76-1.04; p=0.14). Despite the lack of a significant difference in the composite primary end point between the two treatment arms, a significant reduction in the secondary end points of cardiovascular mortality (absolute risk reduction (ARR 1.3%; P=0.05) and congestive heart failure (ARR 1.3%; P=0.04) was evident favoring the early, intensive treatment strategy. These differences were not evident until at least four months after randomization. The A-to-Z trial investigators offered several possible explanations for the delay in evident clinical benefits in their trial when compared against the strong trend toward clinical benefit seen with 30 days following the early initiation of high-dose atorvastatin following ACS in the PROVE IT-TIMI 22 trial. In the PROVE IT trial, patients were enrolled an average of seven days after their index event, and as a result, 69% had undergone revascularization by this time. In the A-to-Z trial, patients were enrolled an average of three to four days earlier, and, therefore, were less likely to have undergone a revascularization procedure by the time of enrollment—and may have continued on with active thrombotic processes relatively less responsive to statin therapy.6 Another notable difference between PROVE IT and A-to-Z subjects was the C-reactive protein (CRP) concentrations in the A-to-Z subjects did not differ between treatment groups within 30 days despite significant differences in their LDL levels.16 This lack of a concurrent, pleiotropic, anti-inflammatory effect in the A-to-Z trial aggressive treatment arm may also have contributed to the delayed treatment effect.
In conclusion, the A-to-Z investigators suggest more intensive statin therapy (than the 40 mg Simvastatin in their intensive treatment arm) may be required to derive the most rapid and maximal clinical benefits during the highest risk period immediately following ACS.
Following stroke: Although there is more robust data supporting the benefits of early, intensive, statin therapy following ACS, there also is established and emerging data supporting similar treatment approaches following stroke.
The Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) trial was designed to determine whether or not atorvastatin 80 mg daily would reduce the risk of stroke in patients without known coronary heart disease who had suffered a TIA or stroke within the preceding six months.8 Patients who experienced a hemorrhagic or ischemic TIA or stroke between one to six months before study entry were randomized to receive either atorvastatin 80 mg/d or placebo.
Investigators followed patients for a mean period of 4.9 years for the primary end point of time to non-fatal or fatal stroke. Secondary composite end points included stroke or TIA, and any coronary or peripheral arterial event, including death from cardiac causes, non-fatal MI, ACS, revascularization (coronary, carotid, peripheral), and death from any cause. No difference in mean baseline LDL levels was witnessed between the treatment and placebo arms (132.7 and 133.7 mg/dL, respectively). Atorvastatin was associated with a 16% relative reduction in the risk of stroke—hazard ratio, 0.84; 95% CI 0.71–0.99; p=0.03. This was found despite an increase in hemorrhagic stroke in the atorvastatin group—a finding that supports an epidemiologic association between low cholesterol levels and brain hemorrhage. The risk of cardiovascular events also was significantly reduced, however, no significant difference in overall mortality was observed between the two groups.
In conclusion, the authors recommend the initiation of high-dose atorvastatin “soon” after stroke or TIA. One can only conclude, based on these data, statin therapy should be initiated within six months of TIA or stroke, in accordance with the study design. There is retrospective data suggesting benefit to statin therapy initiated within four weeks following ischemic stroke, and there are prospective trials in process evaluating the potential benefits of statins initiated within 24 hours following ischemic stroke, however, no large, randomized, controlled trial can demonstrate the effect of statins when used as acute stroke therapy.9,12,13,17
Back to the Case
The patient described in our case has a history of TIA and experienced an acute coronary syndrome (NSTEMI) within the preceding 24 hours. He underwent a revascularization procedure (PCI with stent), and is on appropriate therapy, including dual anti-platelet therapy with aspirin and clopidogrel, a beta-blocker, and an angiotensin-converting enzyme inhibitor. Based on the data and conclusions of the MIRACL, PROVE IT-TIMI 22, and SPARCL trials, high-dose statin therapy with atorvastatin 80 mg/d should be initiated immediately in the patient in order to significantly reduce his risk of recurrent ischemic cardiovascular events and stroke following his acute coronary syndrome and TIA.
Bottom Line
Following ACS, high-dose statin therapy with 80 mg of atorvastatin per day should be initiated when the patient is still in the hospital, irrespective of baseline LDL level. Statin therapy should also strongly be considered for secondary stroke prevention in most patients with a history of stroke or transient ischemic attack. TH
Caleb Hale, MD, is a hospitalist at Beth Israel Deaconess Medical Center in Boston. Joseph Ming Wah Li is director of the hospital medicine program and associate chief, division of general medicine and primary care at Beth Israel Deaconess Medical Center in Boston, and assistant professor of Medicine at Harvard Medical School.
References
1. Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994;344:1383-1389.
2. Sacks RM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. N Engl J Med 1996;335:1001-1009.
3. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med 1998;339:1349-1357.
4. Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes. The MIRACL study: a randomized controlled trial. JAMA. 2001;285:1711-1718.
5. Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495-1504.
6. Lemos JA, Blazing MA, Wiviott SD, et al. Early intensive vs. a delayed conservative simvastatin strategy in patients with acute coronary syndromes. Phase Z of the A to Z trial. JAMA. 2004;292:1307-1316.
7. Waters D, Schwartz GG, Olsson AG. The myocardial ischemia reduction with acute cholesterol lowering (MIRACL) trial: a new frontier for statins? Curr Control Trials Cardiovasc Med. 2001;2;111-114.
8. The Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) Investigators. High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med. 2006;355:549-559.
9. Moonis M, Kane K, Schwiderski U, Sandage BW, Fisher M. HMG-CoA reductase inhibitors improve acute ischemic stroke outcome. Stroke. 2005;36:1298-1300.
10. Elkind MS, Flint AC, Sciacca RR, Sacco RL. Lipid-lowering agent use at ischemic stroke onset is associated with decreased mortality. Neurology. 2005;65:253-258.
11. Vaughan CJ, Delanty N. Neuroprotective properties of statins in cerebral ischemia and stroke. Stroke. 1999;30:1969-1973.
12. Elkind MS, Sacco RL, MacArthur RB, et al. The neuroprotection with statin therapy for acute recovery trial (NeuSTART): an adaptive design phase I dose-escalation study of high-dose lovastatin in acute ischemic stroke. Int J Stroke. 2008;3:210-218.
13. Montaner J, Chacon P, Krupinski J, et al. Simvastatin in the acute phase of ischemic stroke: a safety and efficacy pilot trial. Eur J of Neurol. 2008;15:82-90.
14. Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20-28.
15. Pitt B, Loscalzo J, Ycas J, Raichlen JS. Lipid levels after acute coronary syndromes. JACC. 2008;51:1440-1445.
16. Wiviott SD, de Lemos JA, Cannon CP, et al. A tale of two trials: a comparison of the post-acute coronary syndrome lipid-lowering trials of A to Z and PROVE IT-TIMI 22. Circulation. 2006;113:1406-1414.
17. Elking MS. Statins as acute-stroke treatment. Int J Stroke. 2006;1:224-225.
Cultural Considerations
Sharnjit Grewal, MD, a hospitalist at Mercy Medical Group in Sacramento, Calif., is familiar with what he calls “the double-take.” A Sikh born and raised in California, Dr. Grewal wears a traditional turban and full beard. When he walks into the room, some patient’s simply don’t know what to make of him, he admits.
“It’s confusing—even to my Hindu and Sikh patients,” Dr. Grewal says. “They sometimes say, ‘You talk like an American, you’re obviously from the West, but you follow a faith from the East. The line between religion and culture is obscured.”
Although the medical community stresses cultural awareness and sensitivity, Dr. Grewal’s experience highlights the fine line between religion and culture, and the barriers standing stand in the way of cultural awareness.
Today, hospitals experience shifting patient demographics and a growing number of languages and dialects observed in the United States today. Between 1990-2000, the foreign-born population in the U.S. increased by 57%, compared with a 9.3% increase for the native population and a 13% increase for the total U.S. population, according to the U.S. Census Bureau.
Break Down Walls
When hospitalists and patients share a culture or language, the result can be extremely positive. In fact, the Joint Commission report states some hospitals in the United States are working to increase racial and ethnic similarities between staff and patient populations.
Joseph Li, MD, a hospitalist at Beth Israel Deaconess Medical Center in Boston, frequently works with Cantonese-speaking patients referred to the hospital by the healthcare clinic in Boston’s Chinatown section. When he greets patients in their native tongue, Dr. Li says he can feel their comfort level rise; even though he speaks what he calls “5-year-old Cantonese.”
“There is an improved therapeutic relationship when doctors and patients share a language, culture, or belief,” Dr. Li says. “There’s a level of comfort that you are going to be understood and nothing will be lost in translation.”
A patient’s culture may drive decisions contradictory to traditional Western medicine, and hospitalists need to make the time to listen and respond. Recently, Dr. Grewal treated a dying, elderly Asian patient whose family insisted on administering an unknown, water-like fluid to cure the loved one. First, the family requested giving the fluid to the patient by mouth. Dr. Grewal denied the request, and told them the water would end up in the patient’s lungs because he was comatose and could not swallow. Then, the family asked if they could add it to the intravenous line. Again, Dr. Grewal denied the request, and told them water in an un-buffered solution could be harmful to red blood cells.
“It was frustrating for them,” Dr. Grewal says. “I told them, ‘It’s not that I don’t believe the water will cure him. Maybe it will or maybe it won’t. But from a medical standpoint, I know there will be complications and I just cannot do this.’ ”
Eventually, the family asked if a tube could be inserted into the patient’s stomach. When the request was denied, the family decided on comfort care for their loved one. Eventually, he passed away. The family, Dr. Grewal says, was grateful for the hospital staff’s care and effort, even though their requests to administer the fluid were denied.
Difficult Cases
Firm cultural beliefs may lead patients to resist treatment. Manish Patel, MD, a hospitalist and assistant professor with the division of General Internal Medicine at the University of Medicine and Dentistry of New Jersey-Robert Wood Johnson Medical School in New Brunswick, N.J., recalls working with an elderly member of the Indian community who refused to be transferred to a rehabilitation facility. Dr. Patel took time to speak to the patient and learned she came from a tradition that encouraged younger generations to care for the elderly. The patient interpreted her transfer to a rehabilitation facility as a sign her family was abandoning her, Dr. Patel says.
“Sometimes you have to probe to learn more,” Dr. Patel says. “Once we understood her fears, we were able to convey to her that this was a temporary situation and that her family could not provide her with the services that she needed at that point in time.”
Dr. Patel also interacts with Hispanic and Indian patients—many of whom revere doctors and defer to them for treatment decisions. In these situations, he uses the same approach as he does with patients who question his treatment recommendations.
“The patient may defer to you, but it’s important to empower the patient and give them all the information they need to make major choices in their healthcare.”
Information Pipeline
Hospitalists may prefer to be upfront about a patient’s condition and treatment, however, cultural norms sometimes dictate who receives information—and how much. For example, Scott Enderby, DO, a hospitalist at Alta Bates Summit Medical Center in Berkeley, Calif., says some Asian families prefer medical staff deliver bad news about the patient to them first. The family then decides what they will tell the patient, he says.
These situations create challenges and opportunities, Enderby says. Medical staff tries to establish a patient-centric care system, so it is important to continue appropriate communication with the patient. It also is important for healthcare providers to avoid putting the family in the middle and marginalizing the patient, he says. Healthcare teams can become frustrated when family members are at odds about decisions and options, and the patient is not involved at the family’s request, he says. In these cases, Dr. Enderby sees an opportunity to further engage the family, and, therefore, the patient.
“Often, when there are cultural and language barriers, a disengaged family can make caring for the patient very challenging,” Dr. Enderby says. “Having the family involved can help everyone feel more aligned with a treatment plan or strategy.”
For Alpesh Amin, MD, associate professor of medicine and vice chair for Clinical Affairs and Quality in the Department of Medicine at the University of California Irvine School of Medicine, being aware of a patient’s cultural values is critical to quality care. As executive director of the hospitalist program at the UCI Medical Center in Orange, Calif., Dr. Amin helped develop curriculum to train students on how to collect “values history” from patients, which includes asking questions about religion and culture. Students document their own values history, and then ask the same questions of a patient. Students discuss patient care and the importance of these histories during small group sessions.
“Knowing a patient's cultural information is just as important as knowing their sexual history or drug history,” Dr. Amin says. “It’s another piece of information that helps you get to know them as a whole. Their overall care is more comprehensive, if you have this knowledge.” TH
Gina Gotsill is a journalist based in California.
Sharnjit Grewal, MD, a hospitalist at Mercy Medical Group in Sacramento, Calif., is familiar with what he calls “the double-take.” A Sikh born and raised in California, Dr. Grewal wears a traditional turban and full beard. When he walks into the room, some patient’s simply don’t know what to make of him, he admits.
“It’s confusing—even to my Hindu and Sikh patients,” Dr. Grewal says. “They sometimes say, ‘You talk like an American, you’re obviously from the West, but you follow a faith from the East. The line between religion and culture is obscured.”
Although the medical community stresses cultural awareness and sensitivity, Dr. Grewal’s experience highlights the fine line between religion and culture, and the barriers standing stand in the way of cultural awareness.
Today, hospitals experience shifting patient demographics and a growing number of languages and dialects observed in the United States today. Between 1990-2000, the foreign-born population in the U.S. increased by 57%, compared with a 9.3% increase for the native population and a 13% increase for the total U.S. population, according to the U.S. Census Bureau.
Break Down Walls
When hospitalists and patients share a culture or language, the result can be extremely positive. In fact, the Joint Commission report states some hospitals in the United States are working to increase racial and ethnic similarities between staff and patient populations.
Joseph Li, MD, a hospitalist at Beth Israel Deaconess Medical Center in Boston, frequently works with Cantonese-speaking patients referred to the hospital by the healthcare clinic in Boston’s Chinatown section. When he greets patients in their native tongue, Dr. Li says he can feel their comfort level rise; even though he speaks what he calls “5-year-old Cantonese.”
“There is an improved therapeutic relationship when doctors and patients share a language, culture, or belief,” Dr. Li says. “There’s a level of comfort that you are going to be understood and nothing will be lost in translation.”
A patient’s culture may drive decisions contradictory to traditional Western medicine, and hospitalists need to make the time to listen and respond. Recently, Dr. Grewal treated a dying, elderly Asian patient whose family insisted on administering an unknown, water-like fluid to cure the loved one. First, the family requested giving the fluid to the patient by mouth. Dr. Grewal denied the request, and told them the water would end up in the patient’s lungs because he was comatose and could not swallow. Then, the family asked if they could add it to the intravenous line. Again, Dr. Grewal denied the request, and told them water in an un-buffered solution could be harmful to red blood cells.
“It was frustrating for them,” Dr. Grewal says. “I told them, ‘It’s not that I don’t believe the water will cure him. Maybe it will or maybe it won’t. But from a medical standpoint, I know there will be complications and I just cannot do this.’ ”
Eventually, the family asked if a tube could be inserted into the patient’s stomach. When the request was denied, the family decided on comfort care for their loved one. Eventually, he passed away. The family, Dr. Grewal says, was grateful for the hospital staff’s care and effort, even though their requests to administer the fluid were denied.
Difficult Cases
Firm cultural beliefs may lead patients to resist treatment. Manish Patel, MD, a hospitalist and assistant professor with the division of General Internal Medicine at the University of Medicine and Dentistry of New Jersey-Robert Wood Johnson Medical School in New Brunswick, N.J., recalls working with an elderly member of the Indian community who refused to be transferred to a rehabilitation facility. Dr. Patel took time to speak to the patient and learned she came from a tradition that encouraged younger generations to care for the elderly. The patient interpreted her transfer to a rehabilitation facility as a sign her family was abandoning her, Dr. Patel says.
“Sometimes you have to probe to learn more,” Dr. Patel says. “Once we understood her fears, we were able to convey to her that this was a temporary situation and that her family could not provide her with the services that she needed at that point in time.”
Dr. Patel also interacts with Hispanic and Indian patients—many of whom revere doctors and defer to them for treatment decisions. In these situations, he uses the same approach as he does with patients who question his treatment recommendations.
“The patient may defer to you, but it’s important to empower the patient and give them all the information they need to make major choices in their healthcare.”
Information Pipeline
Hospitalists may prefer to be upfront about a patient’s condition and treatment, however, cultural norms sometimes dictate who receives information—and how much. For example, Scott Enderby, DO, a hospitalist at Alta Bates Summit Medical Center in Berkeley, Calif., says some Asian families prefer medical staff deliver bad news about the patient to them first. The family then decides what they will tell the patient, he says.
These situations create challenges and opportunities, Enderby says. Medical staff tries to establish a patient-centric care system, so it is important to continue appropriate communication with the patient. It also is important for healthcare providers to avoid putting the family in the middle and marginalizing the patient, he says. Healthcare teams can become frustrated when family members are at odds about decisions and options, and the patient is not involved at the family’s request, he says. In these cases, Dr. Enderby sees an opportunity to further engage the family, and, therefore, the patient.
“Often, when there are cultural and language barriers, a disengaged family can make caring for the patient very challenging,” Dr. Enderby says. “Having the family involved can help everyone feel more aligned with a treatment plan or strategy.”
For Alpesh Amin, MD, associate professor of medicine and vice chair for Clinical Affairs and Quality in the Department of Medicine at the University of California Irvine School of Medicine, being aware of a patient’s cultural values is critical to quality care. As executive director of the hospitalist program at the UCI Medical Center in Orange, Calif., Dr. Amin helped develop curriculum to train students on how to collect “values history” from patients, which includes asking questions about religion and culture. Students document their own values history, and then ask the same questions of a patient. Students discuss patient care and the importance of these histories during small group sessions.
“Knowing a patient's cultural information is just as important as knowing their sexual history or drug history,” Dr. Amin says. “It’s another piece of information that helps you get to know them as a whole. Their overall care is more comprehensive, if you have this knowledge.” TH
Gina Gotsill is a journalist based in California.
Sharnjit Grewal, MD, a hospitalist at Mercy Medical Group in Sacramento, Calif., is familiar with what he calls “the double-take.” A Sikh born and raised in California, Dr. Grewal wears a traditional turban and full beard. When he walks into the room, some patient’s simply don’t know what to make of him, he admits.
“It’s confusing—even to my Hindu and Sikh patients,” Dr. Grewal says. “They sometimes say, ‘You talk like an American, you’re obviously from the West, but you follow a faith from the East. The line between religion and culture is obscured.”
Although the medical community stresses cultural awareness and sensitivity, Dr. Grewal’s experience highlights the fine line between religion and culture, and the barriers standing stand in the way of cultural awareness.
Today, hospitals experience shifting patient demographics and a growing number of languages and dialects observed in the United States today. Between 1990-2000, the foreign-born population in the U.S. increased by 57%, compared with a 9.3% increase for the native population and a 13% increase for the total U.S. population, according to the U.S. Census Bureau.
Break Down Walls
When hospitalists and patients share a culture or language, the result can be extremely positive. In fact, the Joint Commission report states some hospitals in the United States are working to increase racial and ethnic similarities between staff and patient populations.
Joseph Li, MD, a hospitalist at Beth Israel Deaconess Medical Center in Boston, frequently works with Cantonese-speaking patients referred to the hospital by the healthcare clinic in Boston’s Chinatown section. When he greets patients in their native tongue, Dr. Li says he can feel their comfort level rise; even though he speaks what he calls “5-year-old Cantonese.”
“There is an improved therapeutic relationship when doctors and patients share a language, culture, or belief,” Dr. Li says. “There’s a level of comfort that you are going to be understood and nothing will be lost in translation.”
A patient’s culture may drive decisions contradictory to traditional Western medicine, and hospitalists need to make the time to listen and respond. Recently, Dr. Grewal treated a dying, elderly Asian patient whose family insisted on administering an unknown, water-like fluid to cure the loved one. First, the family requested giving the fluid to the patient by mouth. Dr. Grewal denied the request, and told them the water would end up in the patient’s lungs because he was comatose and could not swallow. Then, the family asked if they could add it to the intravenous line. Again, Dr. Grewal denied the request, and told them water in an un-buffered solution could be harmful to red blood cells.
“It was frustrating for them,” Dr. Grewal says. “I told them, ‘It’s not that I don’t believe the water will cure him. Maybe it will or maybe it won’t. But from a medical standpoint, I know there will be complications and I just cannot do this.’ ”
Eventually, the family asked if a tube could be inserted into the patient’s stomach. When the request was denied, the family decided on comfort care for their loved one. Eventually, he passed away. The family, Dr. Grewal says, was grateful for the hospital staff’s care and effort, even though their requests to administer the fluid were denied.
Difficult Cases
Firm cultural beliefs may lead patients to resist treatment. Manish Patel, MD, a hospitalist and assistant professor with the division of General Internal Medicine at the University of Medicine and Dentistry of New Jersey-Robert Wood Johnson Medical School in New Brunswick, N.J., recalls working with an elderly member of the Indian community who refused to be transferred to a rehabilitation facility. Dr. Patel took time to speak to the patient and learned she came from a tradition that encouraged younger generations to care for the elderly. The patient interpreted her transfer to a rehabilitation facility as a sign her family was abandoning her, Dr. Patel says.
“Sometimes you have to probe to learn more,” Dr. Patel says. “Once we understood her fears, we were able to convey to her that this was a temporary situation and that her family could not provide her with the services that she needed at that point in time.”
Dr. Patel also interacts with Hispanic and Indian patients—many of whom revere doctors and defer to them for treatment decisions. In these situations, he uses the same approach as he does with patients who question his treatment recommendations.
“The patient may defer to you, but it’s important to empower the patient and give them all the information they need to make major choices in their healthcare.”
Information Pipeline
Hospitalists may prefer to be upfront about a patient’s condition and treatment, however, cultural norms sometimes dictate who receives information—and how much. For example, Scott Enderby, DO, a hospitalist at Alta Bates Summit Medical Center in Berkeley, Calif., says some Asian families prefer medical staff deliver bad news about the patient to them first. The family then decides what they will tell the patient, he says.
These situations create challenges and opportunities, Enderby says. Medical staff tries to establish a patient-centric care system, so it is important to continue appropriate communication with the patient. It also is important for healthcare providers to avoid putting the family in the middle and marginalizing the patient, he says. Healthcare teams can become frustrated when family members are at odds about decisions and options, and the patient is not involved at the family’s request, he says. In these cases, Dr. Enderby sees an opportunity to further engage the family, and, therefore, the patient.
“Often, when there are cultural and language barriers, a disengaged family can make caring for the patient very challenging,” Dr. Enderby says. “Having the family involved can help everyone feel more aligned with a treatment plan or strategy.”
For Alpesh Amin, MD, associate professor of medicine and vice chair for Clinical Affairs and Quality in the Department of Medicine at the University of California Irvine School of Medicine, being aware of a patient’s cultural values is critical to quality care. As executive director of the hospitalist program at the UCI Medical Center in Orange, Calif., Dr. Amin helped develop curriculum to train students on how to collect “values history” from patients, which includes asking questions about religion and culture. Students document their own values history, and then ask the same questions of a patient. Students discuss patient care and the importance of these histories during small group sessions.
“Knowing a patient's cultural information is just as important as knowing their sexual history or drug history,” Dr. Amin says. “It’s another piece of information that helps you get to know them as a whole. Their overall care is more comprehensive, if you have this knowledge.” TH
Gina Gotsill is a journalist based in California.
Industry Innovator Eyes HM Challenges Ahead
Brian Bossard, MD, was practicing as a hospitalist before he even knew what a hospitalist was. In 1993, Dr. Bossard, then a private practice internist, initiated a contract with Lincoln General Hospital in Lincoln, Neb. He agreed to care for hospital patients who didn’t have physicians. The hospital signed the contract—three years before HM pioneer Bob Wachter, MD, professor and chief of the division of hospital medicine at the University of California San Francisco, former SHM president and author of the blog “Wachter’s World” (www.wachtersworld.com), coined the term “hospitalist.”
Dr. Bossard, director and CEO of Inpatient Physician Associates in Lincoln, recently spoke with The Hospitalist about being at the forefront of the hospital medicine movement.
Q: How did you come to form your own hospitalist group?
—Brian Bossard, MD
A: [Starting in 1993], I was providing hospital medicine service while at the same time working in a private practice model. I took care of my own patients and also took care of all the assigned patients through the hospital. During that period, I started getting referrals from other physicians who wanted to turn their patients’ care over to me. It became clear after just a few years of doing that I was getting very busy and that there was a need for a more formal hospital medicine program. So, beginning in 1998, I started going to national hospital medicine meetings. I took my hospital administrator with me to the first meeting, and during the next four years developed an infrastructure for a mature hospital medicine program.
Q: What trends have you identified in HM since that time?
A: In the case of academic medicine models, hospital medicine developed because they needed to have a system to provide a cap for the residents—both in terms of number of hours they worked and the number of patients they saw. That was a new development and one that wasn’t in place when I went through training.
Private practice or community-based hospitals had physicians who were no longer interested in providing community call for taking care of patients that didn’t have physicians, or maybe didn’t have insurance. Community hospitals were finding that many physicians were opting out of that community call so they needed hospital medicine support to take care of those patients.
Q: What is the most significant change you’ve witnessed?
A: It’s become clear hospital medicine programs not only provide staffing to take care of those patients who otherwise wouldn’t be taken care of, but also provide a structure to take care of patients better. Probably the most positive and meaningful change since the mid-’90s is that hospital medicine programs are seen as quality drivers, efficiency drivers, and as a source of leadership within hospital policy making and decision making.
Q: What are your responsibilities as CEO of your group?
A: I run the business from top to bottom. Since I started the group in 2002, we’ve grown from just six physicians to 18 physicians and three nurse coordinators. So, I’ve had an opportunity during the last seven years to develop leadership roles within our group and delegate some activities to other leaders in the group. Where I once oversaw every little detail, I am now able to turn over some things to other, very talented group members. What I really focus on now is recruitment, the clinical aspects, public relations, and those sorts of things. But I never lose sight of the importance of developing data to drive our decisions, so I’m very involved in that, as well. As we add more and more physicians, I have to dedicate more time to management of the group. My clinical time goes down as the group grows.
Q: You mentioned that you collect data?
A: I work with the folks in the IT and Division Analysis departments in the hospital to identify what data we can get, what is important for me to know … so we can make decisions for the better of the group and the hospital. Some of that involves knowing what time of the day we have the highest admissions consults and what days of the week we’re busiest, and then organizing our schedule accordingly. It’s important to look at numbers and data, as opposed to going by when you feel you’re busy and when you’re not, because sometimes the feel is different from what is actually happening.
Q: What are the challenges facing your HMG?
A: Recruitment is a huge challenge. The growth of hospital medicine is much greater than anticipated even five years ago. Many programs are understaffed right now. That’s not because they don’t have financing, but because they don’t have physicians available to staff the slots. When I started my group, I was able to recruit a strong, core group of five physicians in six months. I don’t think there is any way you could do that now. That’s a trend that’s changed for the worst. I don’t think internal medicine is going to be able to support the need for care providers within hospital medicine programs.
Q: How should hospital medicine groups look to fill their vacancies?
A: I think opportunities will exist for well-trained and motivated family medicine physicians. Many more rural or community-based hospitals are turning to family physicians to staff programs. Typically, family physicians represent only 3% of hospital medicine program slots. I see that percentage increasing fairly significantly in the next five years. TH
Brian Bossard, MD, was practicing as a hospitalist before he even knew what a hospitalist was. In 1993, Dr. Bossard, then a private practice internist, initiated a contract with Lincoln General Hospital in Lincoln, Neb. He agreed to care for hospital patients who didn’t have physicians. The hospital signed the contract—three years before HM pioneer Bob Wachter, MD, professor and chief of the division of hospital medicine at the University of California San Francisco, former SHM president and author of the blog “Wachter’s World” (www.wachtersworld.com), coined the term “hospitalist.”
Dr. Bossard, director and CEO of Inpatient Physician Associates in Lincoln, recently spoke with The Hospitalist about being at the forefront of the hospital medicine movement.
Q: How did you come to form your own hospitalist group?
—Brian Bossard, MD
A: [Starting in 1993], I was providing hospital medicine service while at the same time working in a private practice model. I took care of my own patients and also took care of all the assigned patients through the hospital. During that period, I started getting referrals from other physicians who wanted to turn their patients’ care over to me. It became clear after just a few years of doing that I was getting very busy and that there was a need for a more formal hospital medicine program. So, beginning in 1998, I started going to national hospital medicine meetings. I took my hospital administrator with me to the first meeting, and during the next four years developed an infrastructure for a mature hospital medicine program.
Q: What trends have you identified in HM since that time?
A: In the case of academic medicine models, hospital medicine developed because they needed to have a system to provide a cap for the residents—both in terms of number of hours they worked and the number of patients they saw. That was a new development and one that wasn’t in place when I went through training.
Private practice or community-based hospitals had physicians who were no longer interested in providing community call for taking care of patients that didn’t have physicians, or maybe didn’t have insurance. Community hospitals were finding that many physicians were opting out of that community call so they needed hospital medicine support to take care of those patients.
Q: What is the most significant change you’ve witnessed?
A: It’s become clear hospital medicine programs not only provide staffing to take care of those patients who otherwise wouldn’t be taken care of, but also provide a structure to take care of patients better. Probably the most positive and meaningful change since the mid-’90s is that hospital medicine programs are seen as quality drivers, efficiency drivers, and as a source of leadership within hospital policy making and decision making.
Q: What are your responsibilities as CEO of your group?
A: I run the business from top to bottom. Since I started the group in 2002, we’ve grown from just six physicians to 18 physicians and three nurse coordinators. So, I’ve had an opportunity during the last seven years to develop leadership roles within our group and delegate some activities to other leaders in the group. Where I once oversaw every little detail, I am now able to turn over some things to other, very talented group members. What I really focus on now is recruitment, the clinical aspects, public relations, and those sorts of things. But I never lose sight of the importance of developing data to drive our decisions, so I’m very involved in that, as well. As we add more and more physicians, I have to dedicate more time to management of the group. My clinical time goes down as the group grows.
Q: You mentioned that you collect data?
A: I work with the folks in the IT and Division Analysis departments in the hospital to identify what data we can get, what is important for me to know … so we can make decisions for the better of the group and the hospital. Some of that involves knowing what time of the day we have the highest admissions consults and what days of the week we’re busiest, and then organizing our schedule accordingly. It’s important to look at numbers and data, as opposed to going by when you feel you’re busy and when you’re not, because sometimes the feel is different from what is actually happening.
Q: What are the challenges facing your HMG?
A: Recruitment is a huge challenge. The growth of hospital medicine is much greater than anticipated even five years ago. Many programs are understaffed right now. That’s not because they don’t have financing, but because they don’t have physicians available to staff the slots. When I started my group, I was able to recruit a strong, core group of five physicians in six months. I don’t think there is any way you could do that now. That’s a trend that’s changed for the worst. I don’t think internal medicine is going to be able to support the need for care providers within hospital medicine programs.
Q: How should hospital medicine groups look to fill their vacancies?
A: I think opportunities will exist for well-trained and motivated family medicine physicians. Many more rural or community-based hospitals are turning to family physicians to staff programs. Typically, family physicians represent only 3% of hospital medicine program slots. I see that percentage increasing fairly significantly in the next five years. TH
Brian Bossard, MD, was practicing as a hospitalist before he even knew what a hospitalist was. In 1993, Dr. Bossard, then a private practice internist, initiated a contract with Lincoln General Hospital in Lincoln, Neb. He agreed to care for hospital patients who didn’t have physicians. The hospital signed the contract—three years before HM pioneer Bob Wachter, MD, professor and chief of the division of hospital medicine at the University of California San Francisco, former SHM president and author of the blog “Wachter’s World” (www.wachtersworld.com), coined the term “hospitalist.”
Dr. Bossard, director and CEO of Inpatient Physician Associates in Lincoln, recently spoke with The Hospitalist about being at the forefront of the hospital medicine movement.
Q: How did you come to form your own hospitalist group?
—Brian Bossard, MD
A: [Starting in 1993], I was providing hospital medicine service while at the same time working in a private practice model. I took care of my own patients and also took care of all the assigned patients through the hospital. During that period, I started getting referrals from other physicians who wanted to turn their patients’ care over to me. It became clear after just a few years of doing that I was getting very busy and that there was a need for a more formal hospital medicine program. So, beginning in 1998, I started going to national hospital medicine meetings. I took my hospital administrator with me to the first meeting, and during the next four years developed an infrastructure for a mature hospital medicine program.
Q: What trends have you identified in HM since that time?
A: In the case of academic medicine models, hospital medicine developed because they needed to have a system to provide a cap for the residents—both in terms of number of hours they worked and the number of patients they saw. That was a new development and one that wasn’t in place when I went through training.
Private practice or community-based hospitals had physicians who were no longer interested in providing community call for taking care of patients that didn’t have physicians, or maybe didn’t have insurance. Community hospitals were finding that many physicians were opting out of that community call so they needed hospital medicine support to take care of those patients.
Q: What is the most significant change you’ve witnessed?
A: It’s become clear hospital medicine programs not only provide staffing to take care of those patients who otherwise wouldn’t be taken care of, but also provide a structure to take care of patients better. Probably the most positive and meaningful change since the mid-’90s is that hospital medicine programs are seen as quality drivers, efficiency drivers, and as a source of leadership within hospital policy making and decision making.
Q: What are your responsibilities as CEO of your group?
A: I run the business from top to bottom. Since I started the group in 2002, we’ve grown from just six physicians to 18 physicians and three nurse coordinators. So, I’ve had an opportunity during the last seven years to develop leadership roles within our group and delegate some activities to other leaders in the group. Where I once oversaw every little detail, I am now able to turn over some things to other, very talented group members. What I really focus on now is recruitment, the clinical aspects, public relations, and those sorts of things. But I never lose sight of the importance of developing data to drive our decisions, so I’m very involved in that, as well. As we add more and more physicians, I have to dedicate more time to management of the group. My clinical time goes down as the group grows.
Q: You mentioned that you collect data?
A: I work with the folks in the IT and Division Analysis departments in the hospital to identify what data we can get, what is important for me to know … so we can make decisions for the better of the group and the hospital. Some of that involves knowing what time of the day we have the highest admissions consults and what days of the week we’re busiest, and then organizing our schedule accordingly. It’s important to look at numbers and data, as opposed to going by when you feel you’re busy and when you’re not, because sometimes the feel is different from what is actually happening.
Q: What are the challenges facing your HMG?
A: Recruitment is a huge challenge. The growth of hospital medicine is much greater than anticipated even five years ago. Many programs are understaffed right now. That’s not because they don’t have financing, but because they don’t have physicians available to staff the slots. When I started my group, I was able to recruit a strong, core group of five physicians in six months. I don’t think there is any way you could do that now. That’s a trend that’s changed for the worst. I don’t think internal medicine is going to be able to support the need for care providers within hospital medicine programs.
Q: How should hospital medicine groups look to fill their vacancies?
A: I think opportunities will exist for well-trained and motivated family medicine physicians. Many more rural or community-based hospitals are turning to family physicians to staff programs. Typically, family physicians represent only 3% of hospital medicine program slots. I see that percentage increasing fairly significantly in the next five years. TH
The Bare Necessities
Medicare reimburses for procedures and services deemed “reasonable and necessary.” By statute, Medicare only may pay for items and services that are “reasonable and necessary for the diagnosis or treatment of illness or injury, or to improve the functioning of a malformed body member,” unless there is another statutory authorization for payment (e.g., colorectal cancer screening).1 Medical necessity is determined by evidence-based clinical standards of care, which guide the physician’s diagnostic and treatment process for certain patient populations, illnesses, or clinical circumstances.
National Coverage Determinations
The Centers for Medicare and Medicaid Services (CMS) develop national coverage determinations (NCDs) through an evidence-based process with opportunities for public participation. In some cases, CMS’ own research is supplemented by an outside technology assessment and/or consultation with the Medicare Evidence Development and Coverage Advisory Committee (MEDCAC).
All Medicare contractors must adhere to NCDs and cannot create additional limitations or guidelines. As an example, the NCD for pronouncement of death states an individual only is considered to have died as of the time he orshe is pronounced dead by a person who is legally authorized to make such a pronouncement, usually a physician; and medical services rendered up to and including pronouncement are considered reasonable and necessary.2 Further guidance authorizes physicians to report discharge day management codes (99238-99239) for the face-to-face pronouncement encounter.3 See the Medicare National Coverage Determination Manual (www.cms.hhs.gov/Manuals/ IOM/itemdetail.asp?filterType=none&filterByDID=-99&sortByDID=1&sort Order=ascending&itemID=CMS014961&intNumPerPage=10) for other applicable NCDs.
Local Coverage Determinations
In the absence of a national coverage policy, an item or service may be covered at the discretion of the Medicare contractors based on a local coverage determination (LCD).4
An LCD, as established by Section 522 of the Benefits Improvement and Protection Act (BIPA), is a decision made by a fiscal intermediary or carrier to cover a particular service on an intermediary-wide or carrier-wide basis, in accordance with Section 1862(a)(1)(A) of the Social Security Act (i.e., a determination as to whether the service is reasonable and necessary).5 LCDs may vary by state, causing an inconsistent approach to medical coverage. Non-Medicare payers do not have to follow federal guidelines, unless the member participates in a Medicare managed care plan. A list of Medicare contractor LCDs can be found at www.cms.hhs.gov/DeterminationProcess/04_LCDs.asp.
Certain payers develop coverage requirements for frequent or problematic procedures or services. Coverage requirements identify specific conditions (i.e., ICD-9-CM codes) for which the services or procedures are considered medically necessary. For example, echocardiography (99307) may not be considered medically necessary for a patient who presents with chest pain unless documentation also supports suspected acute myocardial ischemia and baseline electrocardiogram (ECG) is nondiagnostic; or in cases when the physician suspects aortic dissection.6
Medical Review Program
It is insufficient to develop billing compliance policies and standards without enforcement of these guidelines. In an effort to verify the appropriateness of claims and payment, CMS contracts with Medicare Administrative Contractors (MACs), Fiscal Intermediaries (FIs), and Program Safeguard Contractors (PSCs) to perform medical reviews. The goals of the Medical Review Program are reducing Medicare claims payment errors; decreased denials and increased timely payments; and increased educational opportunities.7
In order to determine which providers should be subject to medical review, contractors must analyze provider compliance with coverage and coding rules and take corrective action when necessary. The corrective action aims to modify behavior in need of change, collect overpayments, and deny improper payments.8 Several types of review exist:
- Prepayment review: The Medicare contractor requests medical records prior to payment;
- Postpayment review: The contractor requests medical records after payment has been received by the physician; this may result in upholding or reversing the initial payment determination;
- Probe review: The contractor requests medical records associated with 20 to 40 claims based upon provider-specific issues; and
- Comprehensive error rate testing (CERT) review: CMS measures the error rate and estimates improper claim payments by randomly selecting and reviewing a sample of claims for compliance.9
Prepayment reviews seem to be expanding as a response to the error rate for certain services. For example, high-level consultation services (99245 and 99255) have prompted review over the last several years to ensure documentation and medical necessity are appropriately supported and maintained. Hospitalists may have noticed a recent increase in prepayment record requests for subsequent hospital care (99232 or 99233) and discharge day management (99239) services. Responses to these and other record requests must be timely in order to prevent claim denial or repayment requests. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is on the faculty of SHM’s inpatient coding course.
References:
1. Exclusions from coverage and Medicare as a secondary payer. Social Security Online. www.ssa.gov/ OP_Home/ssact/title18/1862.htm. Updated October 28, 2008. Accessed October 15, 2008.
2. Centers for Medicare and Medicaid Services. Medicare national coverage determination manual: chapter 1, part 1, section 70.4. www.cms.hhs.gov/manuals/ downloads/ncd103c1_Part1.pdf. Accessed October 14, 2008.
3. Centers for Medicare and Medicaid Services. Transmittal 1460: Subsequent hospital visits and hospital discharge day management services (Codes 99231-99239). www.cms.hhs.gov/transmittals/downloads/R1460CP.pdf. Accessed October 14, 2008.
4. Centers for Medicare and Medicaid Services. Medicare coverage determination process: overview. www. cms.hhs.gov/DeterminationProcess/01_Overview.asp#TopOfPage. Updated August 5, 2008. Accessed October 15, 2008.
5. Centers for Medicare and Medicaid Services. Medicare coverage determination process: local coverage determinations. www.cms.hhs.gov/DeterminationProcess/ 04_LCDs.asp. Updated October 7, 2008. Accessed October 15, 2008.
6. Highmark Medicare Services. LCD L27536: transthoracic echocardiography. www.highmarkmedicareservices. com/policy/mac-ab/l27536-r3.html. Updated Septem-ber 23, 2008. Accessed October 16, 2008.
7. Centers for Medicare and Medicaid Services. The Medicare medical review program. www.cms. hhs.gov/MedicalReviewProcess/Downloads/mrfactsheet.pdf. Published September 2004. Accessed October 15, 2008.
8. Rudolph P, Shuren A. Dealing with Medicare. In: coding for chest medicine 2008. Northbrook, IL: Am Coll of Chest Physicians. 2008;23-35.
9. Centers for Medicare and Medicaid Services. Comprehensive error rate testing: overview. www. cms.hhs.gov/CERT/. Updated December 14, 2005. Accessed October 16, 2008.
10. Centers for Medicare and Medicaid Services. Medicare claims processing manual: chapter 12, section 30.6.10G. www.cms.hhs.gov/manuals/downloads/clm104c12.pdf. Updated July 9, 2008. Accessed October 16, 2008.
Medicare reimburses for procedures and services deemed “reasonable and necessary.” By statute, Medicare only may pay for items and services that are “reasonable and necessary for the diagnosis or treatment of illness or injury, or to improve the functioning of a malformed body member,” unless there is another statutory authorization for payment (e.g., colorectal cancer screening).1 Medical necessity is determined by evidence-based clinical standards of care, which guide the physician’s diagnostic and treatment process for certain patient populations, illnesses, or clinical circumstances.
National Coverage Determinations
The Centers for Medicare and Medicaid Services (CMS) develop national coverage determinations (NCDs) through an evidence-based process with opportunities for public participation. In some cases, CMS’ own research is supplemented by an outside technology assessment and/or consultation with the Medicare Evidence Development and Coverage Advisory Committee (MEDCAC).
All Medicare contractors must adhere to NCDs and cannot create additional limitations or guidelines. As an example, the NCD for pronouncement of death states an individual only is considered to have died as of the time he orshe is pronounced dead by a person who is legally authorized to make such a pronouncement, usually a physician; and medical services rendered up to and including pronouncement are considered reasonable and necessary.2 Further guidance authorizes physicians to report discharge day management codes (99238-99239) for the face-to-face pronouncement encounter.3 See the Medicare National Coverage Determination Manual (www.cms.hhs.gov/Manuals/ IOM/itemdetail.asp?filterType=none&filterByDID=-99&sortByDID=1&sort Order=ascending&itemID=CMS014961&intNumPerPage=10) for other applicable NCDs.
Local Coverage Determinations
In the absence of a national coverage policy, an item or service may be covered at the discretion of the Medicare contractors based on a local coverage determination (LCD).4
An LCD, as established by Section 522 of the Benefits Improvement and Protection Act (BIPA), is a decision made by a fiscal intermediary or carrier to cover a particular service on an intermediary-wide or carrier-wide basis, in accordance with Section 1862(a)(1)(A) of the Social Security Act (i.e., a determination as to whether the service is reasonable and necessary).5 LCDs may vary by state, causing an inconsistent approach to medical coverage. Non-Medicare payers do not have to follow federal guidelines, unless the member participates in a Medicare managed care plan. A list of Medicare contractor LCDs can be found at www.cms.hhs.gov/DeterminationProcess/04_LCDs.asp.
Certain payers develop coverage requirements for frequent or problematic procedures or services. Coverage requirements identify specific conditions (i.e., ICD-9-CM codes) for which the services or procedures are considered medically necessary. For example, echocardiography (99307) may not be considered medically necessary for a patient who presents with chest pain unless documentation also supports suspected acute myocardial ischemia and baseline electrocardiogram (ECG) is nondiagnostic; or in cases when the physician suspects aortic dissection.6
Medical Review Program
It is insufficient to develop billing compliance policies and standards without enforcement of these guidelines. In an effort to verify the appropriateness of claims and payment, CMS contracts with Medicare Administrative Contractors (MACs), Fiscal Intermediaries (FIs), and Program Safeguard Contractors (PSCs) to perform medical reviews. The goals of the Medical Review Program are reducing Medicare claims payment errors; decreased denials and increased timely payments; and increased educational opportunities.7
In order to determine which providers should be subject to medical review, contractors must analyze provider compliance with coverage and coding rules and take corrective action when necessary. The corrective action aims to modify behavior in need of change, collect overpayments, and deny improper payments.8 Several types of review exist:
- Prepayment review: The Medicare contractor requests medical records prior to payment;
- Postpayment review: The contractor requests medical records after payment has been received by the physician; this may result in upholding or reversing the initial payment determination;
- Probe review: The contractor requests medical records associated with 20 to 40 claims based upon provider-specific issues; and
- Comprehensive error rate testing (CERT) review: CMS measures the error rate and estimates improper claim payments by randomly selecting and reviewing a sample of claims for compliance.9
Prepayment reviews seem to be expanding as a response to the error rate for certain services. For example, high-level consultation services (99245 and 99255) have prompted review over the last several years to ensure documentation and medical necessity are appropriately supported and maintained. Hospitalists may have noticed a recent increase in prepayment record requests for subsequent hospital care (99232 or 99233) and discharge day management (99239) services. Responses to these and other record requests must be timely in order to prevent claim denial or repayment requests. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is on the faculty of SHM’s inpatient coding course.
References:
1. Exclusions from coverage and Medicare as a secondary payer. Social Security Online. www.ssa.gov/ OP_Home/ssact/title18/1862.htm. Updated October 28, 2008. Accessed October 15, 2008.
2. Centers for Medicare and Medicaid Services. Medicare national coverage determination manual: chapter 1, part 1, section 70.4. www.cms.hhs.gov/manuals/ downloads/ncd103c1_Part1.pdf. Accessed October 14, 2008.
3. Centers for Medicare and Medicaid Services. Transmittal 1460: Subsequent hospital visits and hospital discharge day management services (Codes 99231-99239). www.cms.hhs.gov/transmittals/downloads/R1460CP.pdf. Accessed October 14, 2008.
4. Centers for Medicare and Medicaid Services. Medicare coverage determination process: overview. www. cms.hhs.gov/DeterminationProcess/01_Overview.asp#TopOfPage. Updated August 5, 2008. Accessed October 15, 2008.
5. Centers for Medicare and Medicaid Services. Medicare coverage determination process: local coverage determinations. www.cms.hhs.gov/DeterminationProcess/ 04_LCDs.asp. Updated October 7, 2008. Accessed October 15, 2008.
6. Highmark Medicare Services. LCD L27536: transthoracic echocardiography. www.highmarkmedicareservices. com/policy/mac-ab/l27536-r3.html. Updated Septem-ber 23, 2008. Accessed October 16, 2008.
7. Centers for Medicare and Medicaid Services. The Medicare medical review program. www.cms. hhs.gov/MedicalReviewProcess/Downloads/mrfactsheet.pdf. Published September 2004. Accessed October 15, 2008.
8. Rudolph P, Shuren A. Dealing with Medicare. In: coding for chest medicine 2008. Northbrook, IL: Am Coll of Chest Physicians. 2008;23-35.
9. Centers for Medicare and Medicaid Services. Comprehensive error rate testing: overview. www. cms.hhs.gov/CERT/. Updated December 14, 2005. Accessed October 16, 2008.
10. Centers for Medicare and Medicaid Services. Medicare claims processing manual: chapter 12, section 30.6.10G. www.cms.hhs.gov/manuals/downloads/clm104c12.pdf. Updated July 9, 2008. Accessed October 16, 2008.
Medicare reimburses for procedures and services deemed “reasonable and necessary.” By statute, Medicare only may pay for items and services that are “reasonable and necessary for the diagnosis or treatment of illness or injury, or to improve the functioning of a malformed body member,” unless there is another statutory authorization for payment (e.g., colorectal cancer screening).1 Medical necessity is determined by evidence-based clinical standards of care, which guide the physician’s diagnostic and treatment process for certain patient populations, illnesses, or clinical circumstances.
National Coverage Determinations
The Centers for Medicare and Medicaid Services (CMS) develop national coverage determinations (NCDs) through an evidence-based process with opportunities for public participation. In some cases, CMS’ own research is supplemented by an outside technology assessment and/or consultation with the Medicare Evidence Development and Coverage Advisory Committee (MEDCAC).
All Medicare contractors must adhere to NCDs and cannot create additional limitations or guidelines. As an example, the NCD for pronouncement of death states an individual only is considered to have died as of the time he orshe is pronounced dead by a person who is legally authorized to make such a pronouncement, usually a physician; and medical services rendered up to and including pronouncement are considered reasonable and necessary.2 Further guidance authorizes physicians to report discharge day management codes (99238-99239) for the face-to-face pronouncement encounter.3 See the Medicare National Coverage Determination Manual (www.cms.hhs.gov/Manuals/ IOM/itemdetail.asp?filterType=none&filterByDID=-99&sortByDID=1&sort Order=ascending&itemID=CMS014961&intNumPerPage=10) for other applicable NCDs.
Local Coverage Determinations
In the absence of a national coverage policy, an item or service may be covered at the discretion of the Medicare contractors based on a local coverage determination (LCD).4
An LCD, as established by Section 522 of the Benefits Improvement and Protection Act (BIPA), is a decision made by a fiscal intermediary or carrier to cover a particular service on an intermediary-wide or carrier-wide basis, in accordance with Section 1862(a)(1)(A) of the Social Security Act (i.e., a determination as to whether the service is reasonable and necessary).5 LCDs may vary by state, causing an inconsistent approach to medical coverage. Non-Medicare payers do not have to follow federal guidelines, unless the member participates in a Medicare managed care plan. A list of Medicare contractor LCDs can be found at www.cms.hhs.gov/DeterminationProcess/04_LCDs.asp.
Certain payers develop coverage requirements for frequent or problematic procedures or services. Coverage requirements identify specific conditions (i.e., ICD-9-CM codes) for which the services or procedures are considered medically necessary. For example, echocardiography (99307) may not be considered medically necessary for a patient who presents with chest pain unless documentation also supports suspected acute myocardial ischemia and baseline electrocardiogram (ECG) is nondiagnostic; or in cases when the physician suspects aortic dissection.6
Medical Review Program
It is insufficient to develop billing compliance policies and standards without enforcement of these guidelines. In an effort to verify the appropriateness of claims and payment, CMS contracts with Medicare Administrative Contractors (MACs), Fiscal Intermediaries (FIs), and Program Safeguard Contractors (PSCs) to perform medical reviews. The goals of the Medical Review Program are reducing Medicare claims payment errors; decreased denials and increased timely payments; and increased educational opportunities.7
In order to determine which providers should be subject to medical review, contractors must analyze provider compliance with coverage and coding rules and take corrective action when necessary. The corrective action aims to modify behavior in need of change, collect overpayments, and deny improper payments.8 Several types of review exist:
- Prepayment review: The Medicare contractor requests medical records prior to payment;
- Postpayment review: The contractor requests medical records after payment has been received by the physician; this may result in upholding or reversing the initial payment determination;
- Probe review: The contractor requests medical records associated with 20 to 40 claims based upon provider-specific issues; and
- Comprehensive error rate testing (CERT) review: CMS measures the error rate and estimates improper claim payments by randomly selecting and reviewing a sample of claims for compliance.9
Prepayment reviews seem to be expanding as a response to the error rate for certain services. For example, high-level consultation services (99245 and 99255) have prompted review over the last several years to ensure documentation and medical necessity are appropriately supported and maintained. Hospitalists may have noticed a recent increase in prepayment record requests for subsequent hospital care (99232 or 99233) and discharge day management (99239) services. Responses to these and other record requests must be timely in order to prevent claim denial or repayment requests. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is on the faculty of SHM’s inpatient coding course.
References:
1. Exclusions from coverage and Medicare as a secondary payer. Social Security Online. www.ssa.gov/ OP_Home/ssact/title18/1862.htm. Updated October 28, 2008. Accessed October 15, 2008.
2. Centers for Medicare and Medicaid Services. Medicare national coverage determination manual: chapter 1, part 1, section 70.4. www.cms.hhs.gov/manuals/ downloads/ncd103c1_Part1.pdf. Accessed October 14, 2008.
3. Centers for Medicare and Medicaid Services. Transmittal 1460: Subsequent hospital visits and hospital discharge day management services (Codes 99231-99239). www.cms.hhs.gov/transmittals/downloads/R1460CP.pdf. Accessed October 14, 2008.
4. Centers for Medicare and Medicaid Services. Medicare coverage determination process: overview. www. cms.hhs.gov/DeterminationProcess/01_Overview.asp#TopOfPage. Updated August 5, 2008. Accessed October 15, 2008.
5. Centers for Medicare and Medicaid Services. Medicare coverage determination process: local coverage determinations. www.cms.hhs.gov/DeterminationProcess/ 04_LCDs.asp. Updated October 7, 2008. Accessed October 15, 2008.
6. Highmark Medicare Services. LCD L27536: transthoracic echocardiography. www.highmarkmedicareservices. com/policy/mac-ab/l27536-r3.html. Updated Septem-ber 23, 2008. Accessed October 16, 2008.
7. Centers for Medicare and Medicaid Services. The Medicare medical review program. www.cms. hhs.gov/MedicalReviewProcess/Downloads/mrfactsheet.pdf. Published September 2004. Accessed October 15, 2008.
8. Rudolph P, Shuren A. Dealing with Medicare. In: coding for chest medicine 2008. Northbrook, IL: Am Coll of Chest Physicians. 2008;23-35.
9. Centers for Medicare and Medicaid Services. Comprehensive error rate testing: overview. www. cms.hhs.gov/CERT/. Updated December 14, 2005. Accessed October 16, 2008.
10. Centers for Medicare and Medicaid Services. Medicare claims processing manual: chapter 12, section 30.6.10G. www.cms.hhs.gov/manuals/downloads/clm104c12.pdf. Updated July 9, 2008. Accessed October 16, 2008.
Grand Rounds: Man, 82, With New-Onset Headaches
An 82-year-old man presented to his primary care provider complaining of headaches for the past week. At the time of presentation, he reported persistent, nonthrobbing pain behind his right eye. Previously, he had experienced pain on the top and right side of his head.
The patient denied any recent visual changes. His last eye examination had taken place four weeks earlier. He was prescribed new eyeglasses, but he had not yet filled the prescription. He denied having symptoms of transient ischemic attack or stroke. He denied any nasal drainage, fever, or chills and reported no prior history of headaches. For the current headache, he had been taking acetaminophen intermittently and said it provided some relief.
The patient’s prior diagnoses included type 2 diabetes, hypertension, dyslipidemia, gout, metabolic syndrome, osteoarthritis, leg edema, and atrial fibrillation. His current medications were allopurinol, diltiazem, glipizide, hydrochlorothiazide, rosiglitazone, valsartan, vardenafil, and warfarin.
His most recent international normalized ratio (INR), measured five days earlier, was 3.34. Fifteen days earlier, however, his INR had been measured at 4.6.
The patient described himself as active, riding his bicycle 50 miles each week. He denied using tobacco but admitted to having “a couple of cocktails” before dinner each evening. He was a widower who lived alone. He owned an advertising company and was involved in its day-to-day operation.
On examination, the patient was alert and oriented. He had an irregularly irregular heart rate with a controlled ventricular response. Cranial nerves II through XII were intact. No papilledema was noted.
The patient was given a diagnosis of headaches of unknown etiology. He was told that he could continue using acetaminophen and was scheduled for head CT with and without contrast the following day.
CT revealed a 2.3-cm, right-sided subacute (mixed-density) subdural hematoma (SDH) with midline shift of 1.8 cm (see Figure 1). The patient’s provider was notified of the CT results, and the patient was sent directly from radiology to the emergency department. His INR was 2.7. The patient was given a partial dose of recombinant factor VIIa (rFVIIa), then emergently transferred to another facility for neurosurgical care.
Upon his arrival there, the patient was noted to be drowsy but oriented, without any focal neurologic deficits. The dose of rFVIIa was completed, and he was given 5 mg of vitamin K. He underwent an emergency craniotomy for clot evacuation. Intraoperatively, his INR was measured at 1.5, and he was given two units of fresh frozen plasma (FFP) to further reverse his coagulopathy.
Repeat head CT the following morning revealed nearly complete removal of the clot, with reexpansion of the brain (see Figure 2). The patient’s INR was 1.1. Additional doses of FFP or rFVIIa were deemed unnecessary. The patient recovered and was discharged from the hospital four days after his surgery. When he was seen at the clinic one month later, he had no neurologic deficits. Head CT was found stable with only a thin rim of residual subdural fluid noted (see Figure 3). He was followed as an outpatient with serial head CTs until all the subdural fluid completely resolved. At that time, he was allowed to restart warfarin.
Discussion
Use of anticoagulation therapy will become increasingly common as our population ages. While anticoagulants are important for preventing thromboembolic events that may result from use of mechanical heart valves, atrial fibrillation, and other conditions, their use is not without risk. The most significant and potentially lethal complication is hemorrhage.
Warfarin-Associated Hemorrhage
In patients who take warfarin, hemorrhage can occur in a variety of areas—most commonly, cerebral and gastrointestinal sites, the nose, the airways, the urinary tract, muscle, and skin.1,2 The site of hemorrhage that carries the highest risk of mortality and morbidity is cerebral.3-5 Among anticoagulated patients experiencing intracranial hemorrhage, a fourfold to fivefold increase in mortality has been reported.6 Among study patients who experienced intracranial hemorrhages while taking warfarin, only 14% were able to return to living independently.4
Excessive Anticoagulation
Recent studies have led to the conclusion that excessive anticoagulation, not anticoagulation targeting specific therapeutic levels, is associated with major bleeding events.7,8 In a review of 2,460 patients from 2000 to 2003 at Brigham and Women’s Hospital in Boston, Fanikos et al8 found that 83% of major bleeding events occurred in patients with an INR exceeding 3.0.
In addition, excessive anticoagulation has been associated with increased morbidity and mortality.5,9,10 Pieracci et al9 found that among patients who experienced a traumatic intracranial hemorrhage with an INR exceeding 3.5, the mortality rate was nearly 75%.
Intracranial Hemorrhage
Subdural hematoma is one of the most common types of intracranial hemorrhage. SDHs are classified based on radiographic findings and age. Acute SDHs are those less than three days old, subacute (mixed-density) SDHs are three to 20 days old, and chronic SDHs (CSDHs) are at least 21 days old.
Acute hemorrhages are more dense and appear white on CT, whereas CSDHs are hypodense and appear darker than the brain parenchyma. Subacute SDHs may have features of both acute SDHs and CSDHs or may appear isodense. While acute SDHs are often associated with trauma and are readily diagnosed, chronic and subacute SDHs present a greater diagnostic challenge. Clinically, subacute SDHs act like CSDHs and are treated similarly.11 For the purposes of this discussion, the case patient’s SDH will be considered a form of CSDH.
Pathophysiology of Chronic Subdural Hematomas
Chronic subdural hematomas form in a number of ways. Major causes are related to brain atrophy resulting from advanced age, alcoholism, brain injury, stroke, or other conditions.11 Atrophy of the brain causes the size of the subdural space to increase. This increased space causes the bridging veins between the cortical surface of the brain and the dura to become stretched and easily torn. As a result, seemingly minor trauma can easily lead to hemorrhage.
Over time, these small, acute hemorrhages in the subdural space may liquefy into CSDHs. Bleeding triggers an inflammatory response, and gradually, blood begins to break down, as with any bruise. Unlike most blood clots, however, blood in the subdural space is affected by fluid dynamics, fibrinolysis, and the formation of neomembranes.11,12 As a result, the blood may not be completely reabsorbed and may actually expand, causing patients to experience symptoms.
Potentially, SDHs can also be caused by subdural hygromas, low intracranial pressure, dehydration, or overdrainage of cerebrospinal fluid during lumbar puncture, spinal anesthesia, or shunting.13
Epidemiology
The annual incidence of CSDH is one to two cases per 100,000 persons. Incidence increases to seven cases per 100,000 among persons older than 70.13 The mortality rate for SDH is 31% to 36%.14,15 The mortality rate for CSDH is approximately 6%. For patients older than 60, the rate increases to 8.8%.16 Rates of morbidity (ie, severe disability or persistent vegetative state) associated with CSDHs have been reported at about 10%.16,17
Men are affected more commonly than are women (accounting for 61% to 70% of cases), and median ages between 71 and 78 have been reported.4,12,18,19
The risk factors for CSDH are listed in Table 1.4,10 SDHs frequently occur in the context of trauma, but they can occur spontaneously, especially in coagulopathic patients. Among patients with CSDHs who are taking warfarin, 45.5% to 52% deny recent experiences of trauma.4,14
Signs and Symptoms of Chronic Subdural Hematomas
The clinical onset of CSDH is insidious. Possible presenting symptoms are listed in Table 2.14,18,20,21 Frequently, the neurologic examination fails to reveal any focal deficits. Many of the symptoms are vague and nonspecific and may mimic those of other conditions that are common in the elderly, thus making diagnosis difficult. Despite clinical suspicion, the definitive diagnosis of SDH is based on CT results.
Reversing Warfarin-Induced Coagulopathy
In all patients with intracranial hemorrhages who are taking warfarin, the coagulopathy must be reversed. The agents commonly used to reverse the effects of warfarin include vitamin K, FFP, and rFVIIa.9,22-24 The choice of agents depends on the timing of intervention.
Vitamin K is commonly given to patients either intravenously or orally in combination with FFP and/or rFVIIa to promote the reversal of warfarin-induced coagulopathy. Vitamin K is seldom used alone, as its effects may not be seen for 24 hours or longer, and may not completely reverse the effects of warfarin.25
Another frequently used product is FFP. Unfortunately, FFP has been associated with complications such as fluid overload, infectious disease transmission, and anaphylaxis. Additionally, FFP too reverses coagulopathy very slowly. Boulis et al26 found that in patients given FFP with single-dose vitamin K, INR reduction averaged 0.18/hour. At this rate, it would take approximately 11 hours to correct an INR of 3.0 to the desired target of 1.0.
In contrast, rFVIIa, used off-label, has proved highly effective in rapidly reversing coagulopathy and allowing patients to safely undergo immediate surgical treatment.23,24 To its disadvantage, rFVIIa increases the risk of thromboembolism and is significantly more expensive than FFP. Compared with $105 for one unit of FFP, the cost of an 80-mcg/kg dose of rFVIIa for a patient weighing 80 kg is about $6,400.27
Factors Predicting Outcome for Subdural Hematomas
A number of factors determine post-SDH outcome. Rozzelle et al14 found that a Glasgow Coma Scale score below 7, age greater than 80, more acute hemorrhages, and hemorrhages requiring craniotomy rather than burr-hole drainage were associated with significantly higher mortality rates than when these factors were absent.
Other studies have revealed that patients with poor clinical status and larger hematomas with more midline shift are also prone to higher mortality rates.20,28 Merlicco et al29 found that younger, nonalcoholic patients without severe trauma whose hematomas were under high pressure had better chances for full recovery than other patients.
Patient Outcome
This case study illustrates the importance of patient education. The patient described here was aware of his excessive anticoagulation and told his provider that he was concerned about bleeding in the brain. Because the patient had been educated about the potential risks of warfarin therapy, he was able to alert his provider when he experienced symptoms of a possible complication. As a result, his condition was quickly diagnosed and treated, with an excellent outcome.
Conclusion
Intracranial hemorrhage is a serious and potentially life-threatening complication of warfarin therapy. CSDHs in particular are a significant cause of mortality and morbidity in older patients. The risk of death or disability increases in patients who are undergoing anticoagulation therapy. In addition, patients with an INR elevated above therapeutic levels face a significantly higher risk for major bleeding events. For this reason, it is important that anticoagulation be tightly controlled within the therapeutic range. It is equally important to educate patients and their families about anticoagulation’s potential risks and complications.
Making the diagnosis of CSDH can be difficult because its symptoms are so often nonspecific and a concomitant illness may be present. Thus, providers must maintain a low threshold for evaluating even minor patient complaints that may signal a complication of warfarin therapy. All too often, minor signs and symptoms go unrecognized, sometimes leading to devastating consequences.
Although many factors predict outcomes for CSDHs, the most important can be controlled by patients and their providers. If patients are well educated and providers listen to their patients, then early diagnosis of SDH can lead to early intervention and improved outcomes.
1. Pullicino P, Thompson JL. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med. 2003;348(3): 256-257.
2. Hurlen M, Abdelnoor M, Smith P, et al. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med. 2002;347(13):969-974.
3. DeSilvey DL. Clinical trials: advanced age, anticoagulation intensity, and risk for intracranial hemorrhage among patients taking warfarin for atrial fibrillation. Am J Geriatr Cardiol. 2005;14(2):98-99.
4. Fang MC, Chang Y, Hylek EM, et al. Advanced age, anticoagulation intensity, and risk for intracranial hemorrhage among patients taking warfarin for atrial fibrillation. Ann Intern Med. 2004;141(10):745-752.
5. Koo S, Kucher N, Nguyen PL, et al. The effect of excessive anticoagulation on mortality and morbidity in hospitalized patients with anticoagulant-related major hemorrhage. Arch Intern Med. 2004;164(14):1557-1560.
6. Mina AA, Knipfer JF, Park DY, et al. Intracranial complications of preinjury anticoagulation in trauma patients with head injury. J Trauma. 2002;53(4):668-672.
7. Pieracci FM, Eachempati SR, Shou J, et al. Degree of anticoagulation, but not warfarin use itself, predicts adverse outcomes after traumatic brain injury in elderly trauma patients. J Trauma. 2007;63(3):525-530.
8. Fanikos J, Grasso-Correnti N, Shah R, et al. Major bleeding complications in a specialized anticoagulation service. Am J Cardiol. 2005;96(4):595-598.
9. Pieracci FM, Eachempati SR, Shou J, et al. Use of long-term anticoagulation is associated with traumatic intracranial hemorrhage and subsequent mortality in elderly patients hospitalized after falls: analysis of the New York State Administrative Database. J Trauma. 2007;63(3):519-524.
10. Franko J, Kish KJ, O’Connell BG, et al. Advanced age and preinjury warfarin anticoagulation increase the risk of mortality after head trauma. J Trauma. 2006; 61(1):107-110.
11. Drapkin AJ. Chronic subdural hematoma: pathophysiological basis for treatment. Br J Neurosurg. 1991; 5(5):467-473.
12. Yamamoto H, Hirashima Y, Hamada H, et al. Independent predictors of recurrence of chronic subdural hematoma: results of multivariate analysis performed using a logistic regression model. J Neurosurg. 2003;98(6):1217-1221.
13. Iantosca MR, Simon RH. Chronic subdural hematoma in adult and elderly patients. Neurosurg Clin N Am. 2000;11(3):447-454.
14. Rozzelle CJ, Wofford JL, Branch CL. Predictors of hospital mortality in older patients with subdural hematoma. J Am Geriatr Soc. 1995;43(3):240-244.
15. Wintzen AR, Tijssen JG. Subdural hematoma and oral anticoagulant therapy. Arch Neurol. 1982;39(2): 69-72.
16. Ramachandran R, Hegde T. Chronic subdural hematomas: causes of morbidity and mortality. Surg Neurol. 2007;67(4):367-372.
17. Amirjamshidi A, Eftekhar B, Abouzari M, Rashidi A. The relationship between Glasgow coma/outcome scores and abnormal CT scan findings in chronic subdural hematoma. Clin Neurol Neurosurg. 2007;109(2): 152-157.
18. Lee JY, Ebel H, Ernestus RI, Klug N. Various surgical treatments of chronic subdural hematoma and outcome in 172 patients: is membranectomy necessary? Surg Neurol. 2004;61(6):523-527.
19. Gelabert-González M, Iglesias-Pais M, García-Allut A, Martínez-Rumbo R. Chronic subdural haematoma: surgical treatment and outcome in 1000 cases. Clin Neurol Neurosurg. 2005;107(3):223-229.
20. Mattle H, Kohler S, Huber P, et al. Anticoagulation-related intracranial extracerebral haemorrhage. J Neurol Neurosurg Psychiatry. 1989;52(7):829-837.
21. Sambasivan M. An overview of chronic subdural hematoma: experience with 2300 cases. Surg Neurol. 1997;47(5):418-422.
22. Lin J, Hanigan WC, Tarantino M, Wang J. The use of recombinant activated factor VII to reverse warfarin-induced anticoagulation in patients with hemorrhages in the central nervous system: preliminary findings. J Neurosurg. 2003;98(4):737-740.
23. Freeman WD, Brott TG, Barrett KM, et al. Recombinant factor VIIa for rapid reversal of warfarin anticoagulation in acute intracranial hemorrhage. Mayo Clin Proc. 2004;79(12):1495-1500.
24. Dager WE, King JH, Regalia RC, et al. Reversal of elevated international normalized ratios and bleeding with low-dose recombinant activated factor VII in patients receiving warfarin. Pharmacotherapy. 2006;26(8): 1091-1098.
25. Denas G, Marzot F, Offelli P, et al. Effectiveness and safety of a management protocol to correct over-anticoagulation with oral vitamin K: a retrospective study of 1,043 cases. J Thromb Thrombolysis. 2008 Mar 13; [Epub ahead of print].
26. Boulis NM, Bobek MP, Schmaier A, Hoff JT. Use of factor IX complex in warfarin-related intracranial hemorrhage. Neurosurgery. 1999;45(5):1113-1118.
27. Kissela BM, Eckman MH. Cost effectiveness of recombinant factor VIIa for treatment of intracerebral hemorrhage. BMC Neurol. 2008;8:17.
28. Ernestus RI, Beldzinski P, Lanfermann H, Klug N. Chronic subdural hematoma: surgical treatment and outcome in 104 patients. Surg Neurol. 1997;48(3): 220-225.
29. Merlicco G, Pierangeli E, di Padova PL. Chronic subdural hematomas in adults: prognostic factors: analysis of 70 cases. Neurosurg Rev. 1995;18(4):247-251.
An 82-year-old man presented to his primary care provider complaining of headaches for the past week. At the time of presentation, he reported persistent, nonthrobbing pain behind his right eye. Previously, he had experienced pain on the top and right side of his head.
The patient denied any recent visual changes. His last eye examination had taken place four weeks earlier. He was prescribed new eyeglasses, but he had not yet filled the prescription. He denied having symptoms of transient ischemic attack or stroke. He denied any nasal drainage, fever, or chills and reported no prior history of headaches. For the current headache, he had been taking acetaminophen intermittently and said it provided some relief.
The patient’s prior diagnoses included type 2 diabetes, hypertension, dyslipidemia, gout, metabolic syndrome, osteoarthritis, leg edema, and atrial fibrillation. His current medications were allopurinol, diltiazem, glipizide, hydrochlorothiazide, rosiglitazone, valsartan, vardenafil, and warfarin.
His most recent international normalized ratio (INR), measured five days earlier, was 3.34. Fifteen days earlier, however, his INR had been measured at 4.6.
The patient described himself as active, riding his bicycle 50 miles each week. He denied using tobacco but admitted to having “a couple of cocktails” before dinner each evening. He was a widower who lived alone. He owned an advertising company and was involved in its day-to-day operation.
On examination, the patient was alert and oriented. He had an irregularly irregular heart rate with a controlled ventricular response. Cranial nerves II through XII were intact. No papilledema was noted.
The patient was given a diagnosis of headaches of unknown etiology. He was told that he could continue using acetaminophen and was scheduled for head CT with and without contrast the following day.
CT revealed a 2.3-cm, right-sided subacute (mixed-density) subdural hematoma (SDH) with midline shift of 1.8 cm (see Figure 1). The patient’s provider was notified of the CT results, and the patient was sent directly from radiology to the emergency department. His INR was 2.7. The patient was given a partial dose of recombinant factor VIIa (rFVIIa), then emergently transferred to another facility for neurosurgical care.
Upon his arrival there, the patient was noted to be drowsy but oriented, without any focal neurologic deficits. The dose of rFVIIa was completed, and he was given 5 mg of vitamin K. He underwent an emergency craniotomy for clot evacuation. Intraoperatively, his INR was measured at 1.5, and he was given two units of fresh frozen plasma (FFP) to further reverse his coagulopathy.
Repeat head CT the following morning revealed nearly complete removal of the clot, with reexpansion of the brain (see Figure 2). The patient’s INR was 1.1. Additional doses of FFP or rFVIIa were deemed unnecessary. The patient recovered and was discharged from the hospital four days after his surgery. When he was seen at the clinic one month later, he had no neurologic deficits. Head CT was found stable with only a thin rim of residual subdural fluid noted (see Figure 3). He was followed as an outpatient with serial head CTs until all the subdural fluid completely resolved. At that time, he was allowed to restart warfarin.
Discussion
Use of anticoagulation therapy will become increasingly common as our population ages. While anticoagulants are important for preventing thromboembolic events that may result from use of mechanical heart valves, atrial fibrillation, and other conditions, their use is not without risk. The most significant and potentially lethal complication is hemorrhage.
Warfarin-Associated Hemorrhage
In patients who take warfarin, hemorrhage can occur in a variety of areas—most commonly, cerebral and gastrointestinal sites, the nose, the airways, the urinary tract, muscle, and skin.1,2 The site of hemorrhage that carries the highest risk of mortality and morbidity is cerebral.3-5 Among anticoagulated patients experiencing intracranial hemorrhage, a fourfold to fivefold increase in mortality has been reported.6 Among study patients who experienced intracranial hemorrhages while taking warfarin, only 14% were able to return to living independently.4
Excessive Anticoagulation
Recent studies have led to the conclusion that excessive anticoagulation, not anticoagulation targeting specific therapeutic levels, is associated with major bleeding events.7,8 In a review of 2,460 patients from 2000 to 2003 at Brigham and Women’s Hospital in Boston, Fanikos et al8 found that 83% of major bleeding events occurred in patients with an INR exceeding 3.0.
In addition, excessive anticoagulation has been associated with increased morbidity and mortality.5,9,10 Pieracci et al9 found that among patients who experienced a traumatic intracranial hemorrhage with an INR exceeding 3.5, the mortality rate was nearly 75%.
Intracranial Hemorrhage
Subdural hematoma is one of the most common types of intracranial hemorrhage. SDHs are classified based on radiographic findings and age. Acute SDHs are those less than three days old, subacute (mixed-density) SDHs are three to 20 days old, and chronic SDHs (CSDHs) are at least 21 days old.
Acute hemorrhages are more dense and appear white on CT, whereas CSDHs are hypodense and appear darker than the brain parenchyma. Subacute SDHs may have features of both acute SDHs and CSDHs or may appear isodense. While acute SDHs are often associated with trauma and are readily diagnosed, chronic and subacute SDHs present a greater diagnostic challenge. Clinically, subacute SDHs act like CSDHs and are treated similarly.11 For the purposes of this discussion, the case patient’s SDH will be considered a form of CSDH.
Pathophysiology of Chronic Subdural Hematomas
Chronic subdural hematomas form in a number of ways. Major causes are related to brain atrophy resulting from advanced age, alcoholism, brain injury, stroke, or other conditions.11 Atrophy of the brain causes the size of the subdural space to increase. This increased space causes the bridging veins between the cortical surface of the brain and the dura to become stretched and easily torn. As a result, seemingly minor trauma can easily lead to hemorrhage.
Over time, these small, acute hemorrhages in the subdural space may liquefy into CSDHs. Bleeding triggers an inflammatory response, and gradually, blood begins to break down, as with any bruise. Unlike most blood clots, however, blood in the subdural space is affected by fluid dynamics, fibrinolysis, and the formation of neomembranes.11,12 As a result, the blood may not be completely reabsorbed and may actually expand, causing patients to experience symptoms.
Potentially, SDHs can also be caused by subdural hygromas, low intracranial pressure, dehydration, or overdrainage of cerebrospinal fluid during lumbar puncture, spinal anesthesia, or shunting.13
Epidemiology
The annual incidence of CSDH is one to two cases per 100,000 persons. Incidence increases to seven cases per 100,000 among persons older than 70.13 The mortality rate for SDH is 31% to 36%.14,15 The mortality rate for CSDH is approximately 6%. For patients older than 60, the rate increases to 8.8%.16 Rates of morbidity (ie, severe disability or persistent vegetative state) associated with CSDHs have been reported at about 10%.16,17
Men are affected more commonly than are women (accounting for 61% to 70% of cases), and median ages between 71 and 78 have been reported.4,12,18,19
The risk factors for CSDH are listed in Table 1.4,10 SDHs frequently occur in the context of trauma, but they can occur spontaneously, especially in coagulopathic patients. Among patients with CSDHs who are taking warfarin, 45.5% to 52% deny recent experiences of trauma.4,14
Signs and Symptoms of Chronic Subdural Hematomas
The clinical onset of CSDH is insidious. Possible presenting symptoms are listed in Table 2.14,18,20,21 Frequently, the neurologic examination fails to reveal any focal deficits. Many of the symptoms are vague and nonspecific and may mimic those of other conditions that are common in the elderly, thus making diagnosis difficult. Despite clinical suspicion, the definitive diagnosis of SDH is based on CT results.
Reversing Warfarin-Induced Coagulopathy
In all patients with intracranial hemorrhages who are taking warfarin, the coagulopathy must be reversed. The agents commonly used to reverse the effects of warfarin include vitamin K, FFP, and rFVIIa.9,22-24 The choice of agents depends on the timing of intervention.
Vitamin K is commonly given to patients either intravenously or orally in combination with FFP and/or rFVIIa to promote the reversal of warfarin-induced coagulopathy. Vitamin K is seldom used alone, as its effects may not be seen for 24 hours or longer, and may not completely reverse the effects of warfarin.25
Another frequently used product is FFP. Unfortunately, FFP has been associated with complications such as fluid overload, infectious disease transmission, and anaphylaxis. Additionally, FFP too reverses coagulopathy very slowly. Boulis et al26 found that in patients given FFP with single-dose vitamin K, INR reduction averaged 0.18/hour. At this rate, it would take approximately 11 hours to correct an INR of 3.0 to the desired target of 1.0.
In contrast, rFVIIa, used off-label, has proved highly effective in rapidly reversing coagulopathy and allowing patients to safely undergo immediate surgical treatment.23,24 To its disadvantage, rFVIIa increases the risk of thromboembolism and is significantly more expensive than FFP. Compared with $105 for one unit of FFP, the cost of an 80-mcg/kg dose of rFVIIa for a patient weighing 80 kg is about $6,400.27
Factors Predicting Outcome for Subdural Hematomas
A number of factors determine post-SDH outcome. Rozzelle et al14 found that a Glasgow Coma Scale score below 7, age greater than 80, more acute hemorrhages, and hemorrhages requiring craniotomy rather than burr-hole drainage were associated with significantly higher mortality rates than when these factors were absent.
Other studies have revealed that patients with poor clinical status and larger hematomas with more midline shift are also prone to higher mortality rates.20,28 Merlicco et al29 found that younger, nonalcoholic patients without severe trauma whose hematomas were under high pressure had better chances for full recovery than other patients.
Patient Outcome
This case study illustrates the importance of patient education. The patient described here was aware of his excessive anticoagulation and told his provider that he was concerned about bleeding in the brain. Because the patient had been educated about the potential risks of warfarin therapy, he was able to alert his provider when he experienced symptoms of a possible complication. As a result, his condition was quickly diagnosed and treated, with an excellent outcome.
Conclusion
Intracranial hemorrhage is a serious and potentially life-threatening complication of warfarin therapy. CSDHs in particular are a significant cause of mortality and morbidity in older patients. The risk of death or disability increases in patients who are undergoing anticoagulation therapy. In addition, patients with an INR elevated above therapeutic levels face a significantly higher risk for major bleeding events. For this reason, it is important that anticoagulation be tightly controlled within the therapeutic range. It is equally important to educate patients and their families about anticoagulation’s potential risks and complications.
Making the diagnosis of CSDH can be difficult because its symptoms are so often nonspecific and a concomitant illness may be present. Thus, providers must maintain a low threshold for evaluating even minor patient complaints that may signal a complication of warfarin therapy. All too often, minor signs and symptoms go unrecognized, sometimes leading to devastating consequences.
Although many factors predict outcomes for CSDHs, the most important can be controlled by patients and their providers. If patients are well educated and providers listen to their patients, then early diagnosis of SDH can lead to early intervention and improved outcomes.
An 82-year-old man presented to his primary care provider complaining of headaches for the past week. At the time of presentation, he reported persistent, nonthrobbing pain behind his right eye. Previously, he had experienced pain on the top and right side of his head.
The patient denied any recent visual changes. His last eye examination had taken place four weeks earlier. He was prescribed new eyeglasses, but he had not yet filled the prescription. He denied having symptoms of transient ischemic attack or stroke. He denied any nasal drainage, fever, or chills and reported no prior history of headaches. For the current headache, he had been taking acetaminophen intermittently and said it provided some relief.
The patient’s prior diagnoses included type 2 diabetes, hypertension, dyslipidemia, gout, metabolic syndrome, osteoarthritis, leg edema, and atrial fibrillation. His current medications were allopurinol, diltiazem, glipizide, hydrochlorothiazide, rosiglitazone, valsartan, vardenafil, and warfarin.
His most recent international normalized ratio (INR), measured five days earlier, was 3.34. Fifteen days earlier, however, his INR had been measured at 4.6.
The patient described himself as active, riding his bicycle 50 miles each week. He denied using tobacco but admitted to having “a couple of cocktails” before dinner each evening. He was a widower who lived alone. He owned an advertising company and was involved in its day-to-day operation.
On examination, the patient was alert and oriented. He had an irregularly irregular heart rate with a controlled ventricular response. Cranial nerves II through XII were intact. No papilledema was noted.
The patient was given a diagnosis of headaches of unknown etiology. He was told that he could continue using acetaminophen and was scheduled for head CT with and without contrast the following day.
CT revealed a 2.3-cm, right-sided subacute (mixed-density) subdural hematoma (SDH) with midline shift of 1.8 cm (see Figure 1). The patient’s provider was notified of the CT results, and the patient was sent directly from radiology to the emergency department. His INR was 2.7. The patient was given a partial dose of recombinant factor VIIa (rFVIIa), then emergently transferred to another facility for neurosurgical care.
Upon his arrival there, the patient was noted to be drowsy but oriented, without any focal neurologic deficits. The dose of rFVIIa was completed, and he was given 5 mg of vitamin K. He underwent an emergency craniotomy for clot evacuation. Intraoperatively, his INR was measured at 1.5, and he was given two units of fresh frozen plasma (FFP) to further reverse his coagulopathy.
Repeat head CT the following morning revealed nearly complete removal of the clot, with reexpansion of the brain (see Figure 2). The patient’s INR was 1.1. Additional doses of FFP or rFVIIa were deemed unnecessary. The patient recovered and was discharged from the hospital four days after his surgery. When he was seen at the clinic one month later, he had no neurologic deficits. Head CT was found stable with only a thin rim of residual subdural fluid noted (see Figure 3). He was followed as an outpatient with serial head CTs until all the subdural fluid completely resolved. At that time, he was allowed to restart warfarin.
Discussion
Use of anticoagulation therapy will become increasingly common as our population ages. While anticoagulants are important for preventing thromboembolic events that may result from use of mechanical heart valves, atrial fibrillation, and other conditions, their use is not without risk. The most significant and potentially lethal complication is hemorrhage.
Warfarin-Associated Hemorrhage
In patients who take warfarin, hemorrhage can occur in a variety of areas—most commonly, cerebral and gastrointestinal sites, the nose, the airways, the urinary tract, muscle, and skin.1,2 The site of hemorrhage that carries the highest risk of mortality and morbidity is cerebral.3-5 Among anticoagulated patients experiencing intracranial hemorrhage, a fourfold to fivefold increase in mortality has been reported.6 Among study patients who experienced intracranial hemorrhages while taking warfarin, only 14% were able to return to living independently.4
Excessive Anticoagulation
Recent studies have led to the conclusion that excessive anticoagulation, not anticoagulation targeting specific therapeutic levels, is associated with major bleeding events.7,8 In a review of 2,460 patients from 2000 to 2003 at Brigham and Women’s Hospital in Boston, Fanikos et al8 found that 83% of major bleeding events occurred in patients with an INR exceeding 3.0.
In addition, excessive anticoagulation has been associated with increased morbidity and mortality.5,9,10 Pieracci et al9 found that among patients who experienced a traumatic intracranial hemorrhage with an INR exceeding 3.5, the mortality rate was nearly 75%.
Intracranial Hemorrhage
Subdural hematoma is one of the most common types of intracranial hemorrhage. SDHs are classified based on radiographic findings and age. Acute SDHs are those less than three days old, subacute (mixed-density) SDHs are three to 20 days old, and chronic SDHs (CSDHs) are at least 21 days old.
Acute hemorrhages are more dense and appear white on CT, whereas CSDHs are hypodense and appear darker than the brain parenchyma. Subacute SDHs may have features of both acute SDHs and CSDHs or may appear isodense. While acute SDHs are often associated with trauma and are readily diagnosed, chronic and subacute SDHs present a greater diagnostic challenge. Clinically, subacute SDHs act like CSDHs and are treated similarly.11 For the purposes of this discussion, the case patient’s SDH will be considered a form of CSDH.
Pathophysiology of Chronic Subdural Hematomas
Chronic subdural hematomas form in a number of ways. Major causes are related to brain atrophy resulting from advanced age, alcoholism, brain injury, stroke, or other conditions.11 Atrophy of the brain causes the size of the subdural space to increase. This increased space causes the bridging veins between the cortical surface of the brain and the dura to become stretched and easily torn. As a result, seemingly minor trauma can easily lead to hemorrhage.
Over time, these small, acute hemorrhages in the subdural space may liquefy into CSDHs. Bleeding triggers an inflammatory response, and gradually, blood begins to break down, as with any bruise. Unlike most blood clots, however, blood in the subdural space is affected by fluid dynamics, fibrinolysis, and the formation of neomembranes.11,12 As a result, the blood may not be completely reabsorbed and may actually expand, causing patients to experience symptoms.
Potentially, SDHs can also be caused by subdural hygromas, low intracranial pressure, dehydration, or overdrainage of cerebrospinal fluid during lumbar puncture, spinal anesthesia, or shunting.13
Epidemiology
The annual incidence of CSDH is one to two cases per 100,000 persons. Incidence increases to seven cases per 100,000 among persons older than 70.13 The mortality rate for SDH is 31% to 36%.14,15 The mortality rate for CSDH is approximately 6%. For patients older than 60, the rate increases to 8.8%.16 Rates of morbidity (ie, severe disability or persistent vegetative state) associated with CSDHs have been reported at about 10%.16,17
Men are affected more commonly than are women (accounting for 61% to 70% of cases), and median ages between 71 and 78 have been reported.4,12,18,19
The risk factors for CSDH are listed in Table 1.4,10 SDHs frequently occur in the context of trauma, but they can occur spontaneously, especially in coagulopathic patients. Among patients with CSDHs who are taking warfarin, 45.5% to 52% deny recent experiences of trauma.4,14
Signs and Symptoms of Chronic Subdural Hematomas
The clinical onset of CSDH is insidious. Possible presenting symptoms are listed in Table 2.14,18,20,21 Frequently, the neurologic examination fails to reveal any focal deficits. Many of the symptoms are vague and nonspecific and may mimic those of other conditions that are common in the elderly, thus making diagnosis difficult. Despite clinical suspicion, the definitive diagnosis of SDH is based on CT results.
Reversing Warfarin-Induced Coagulopathy
In all patients with intracranial hemorrhages who are taking warfarin, the coagulopathy must be reversed. The agents commonly used to reverse the effects of warfarin include vitamin K, FFP, and rFVIIa.9,22-24 The choice of agents depends on the timing of intervention.
Vitamin K is commonly given to patients either intravenously or orally in combination with FFP and/or rFVIIa to promote the reversal of warfarin-induced coagulopathy. Vitamin K is seldom used alone, as its effects may not be seen for 24 hours or longer, and may not completely reverse the effects of warfarin.25
Another frequently used product is FFP. Unfortunately, FFP has been associated with complications such as fluid overload, infectious disease transmission, and anaphylaxis. Additionally, FFP too reverses coagulopathy very slowly. Boulis et al26 found that in patients given FFP with single-dose vitamin K, INR reduction averaged 0.18/hour. At this rate, it would take approximately 11 hours to correct an INR of 3.0 to the desired target of 1.0.
In contrast, rFVIIa, used off-label, has proved highly effective in rapidly reversing coagulopathy and allowing patients to safely undergo immediate surgical treatment.23,24 To its disadvantage, rFVIIa increases the risk of thromboembolism and is significantly more expensive than FFP. Compared with $105 for one unit of FFP, the cost of an 80-mcg/kg dose of rFVIIa for a patient weighing 80 kg is about $6,400.27
Factors Predicting Outcome for Subdural Hematomas
A number of factors determine post-SDH outcome. Rozzelle et al14 found that a Glasgow Coma Scale score below 7, age greater than 80, more acute hemorrhages, and hemorrhages requiring craniotomy rather than burr-hole drainage were associated with significantly higher mortality rates than when these factors were absent.
Other studies have revealed that patients with poor clinical status and larger hematomas with more midline shift are also prone to higher mortality rates.20,28 Merlicco et al29 found that younger, nonalcoholic patients without severe trauma whose hematomas were under high pressure had better chances for full recovery than other patients.
Patient Outcome
This case study illustrates the importance of patient education. The patient described here was aware of his excessive anticoagulation and told his provider that he was concerned about bleeding in the brain. Because the patient had been educated about the potential risks of warfarin therapy, he was able to alert his provider when he experienced symptoms of a possible complication. As a result, his condition was quickly diagnosed and treated, with an excellent outcome.
Conclusion
Intracranial hemorrhage is a serious and potentially life-threatening complication of warfarin therapy. CSDHs in particular are a significant cause of mortality and morbidity in older patients. The risk of death or disability increases in patients who are undergoing anticoagulation therapy. In addition, patients with an INR elevated above therapeutic levels face a significantly higher risk for major bleeding events. For this reason, it is important that anticoagulation be tightly controlled within the therapeutic range. It is equally important to educate patients and their families about anticoagulation’s potential risks and complications.
Making the diagnosis of CSDH can be difficult because its symptoms are so often nonspecific and a concomitant illness may be present. Thus, providers must maintain a low threshold for evaluating even minor patient complaints that may signal a complication of warfarin therapy. All too often, minor signs and symptoms go unrecognized, sometimes leading to devastating consequences.
Although many factors predict outcomes for CSDHs, the most important can be controlled by patients and their providers. If patients are well educated and providers listen to their patients, then early diagnosis of SDH can lead to early intervention and improved outcomes.
1. Pullicino P, Thompson JL. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med. 2003;348(3): 256-257.
2. Hurlen M, Abdelnoor M, Smith P, et al. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med. 2002;347(13):969-974.
3. DeSilvey DL. Clinical trials: advanced age, anticoagulation intensity, and risk for intracranial hemorrhage among patients taking warfarin for atrial fibrillation. Am J Geriatr Cardiol. 2005;14(2):98-99.
4. Fang MC, Chang Y, Hylek EM, et al. Advanced age, anticoagulation intensity, and risk for intracranial hemorrhage among patients taking warfarin for atrial fibrillation. Ann Intern Med. 2004;141(10):745-752.
5. Koo S, Kucher N, Nguyen PL, et al. The effect of excessive anticoagulation on mortality and morbidity in hospitalized patients with anticoagulant-related major hemorrhage. Arch Intern Med. 2004;164(14):1557-1560.
6. Mina AA, Knipfer JF, Park DY, et al. Intracranial complications of preinjury anticoagulation in trauma patients with head injury. J Trauma. 2002;53(4):668-672.
7. Pieracci FM, Eachempati SR, Shou J, et al. Degree of anticoagulation, but not warfarin use itself, predicts adverse outcomes after traumatic brain injury in elderly trauma patients. J Trauma. 2007;63(3):525-530.
8. Fanikos J, Grasso-Correnti N, Shah R, et al. Major bleeding complications in a specialized anticoagulation service. Am J Cardiol. 2005;96(4):595-598.
9. Pieracci FM, Eachempati SR, Shou J, et al. Use of long-term anticoagulation is associated with traumatic intracranial hemorrhage and subsequent mortality in elderly patients hospitalized after falls: analysis of the New York State Administrative Database. J Trauma. 2007;63(3):519-524.
10. Franko J, Kish KJ, O’Connell BG, et al. Advanced age and preinjury warfarin anticoagulation increase the risk of mortality after head trauma. J Trauma. 2006; 61(1):107-110.
11. Drapkin AJ. Chronic subdural hematoma: pathophysiological basis for treatment. Br J Neurosurg. 1991; 5(5):467-473.
12. Yamamoto H, Hirashima Y, Hamada H, et al. Independent predictors of recurrence of chronic subdural hematoma: results of multivariate analysis performed using a logistic regression model. J Neurosurg. 2003;98(6):1217-1221.
13. Iantosca MR, Simon RH. Chronic subdural hematoma in adult and elderly patients. Neurosurg Clin N Am. 2000;11(3):447-454.
14. Rozzelle CJ, Wofford JL, Branch CL. Predictors of hospital mortality in older patients with subdural hematoma. J Am Geriatr Soc. 1995;43(3):240-244.
15. Wintzen AR, Tijssen JG. Subdural hematoma and oral anticoagulant therapy. Arch Neurol. 1982;39(2): 69-72.
16. Ramachandran R, Hegde T. Chronic subdural hematomas: causes of morbidity and mortality. Surg Neurol. 2007;67(4):367-372.
17. Amirjamshidi A, Eftekhar B, Abouzari M, Rashidi A. The relationship between Glasgow coma/outcome scores and abnormal CT scan findings in chronic subdural hematoma. Clin Neurol Neurosurg. 2007;109(2): 152-157.
18. Lee JY, Ebel H, Ernestus RI, Klug N. Various surgical treatments of chronic subdural hematoma and outcome in 172 patients: is membranectomy necessary? Surg Neurol. 2004;61(6):523-527.
19. Gelabert-González M, Iglesias-Pais M, García-Allut A, Martínez-Rumbo R. Chronic subdural haematoma: surgical treatment and outcome in 1000 cases. Clin Neurol Neurosurg. 2005;107(3):223-229.
20. Mattle H, Kohler S, Huber P, et al. Anticoagulation-related intracranial extracerebral haemorrhage. J Neurol Neurosurg Psychiatry. 1989;52(7):829-837.
21. Sambasivan M. An overview of chronic subdural hematoma: experience with 2300 cases. Surg Neurol. 1997;47(5):418-422.
22. Lin J, Hanigan WC, Tarantino M, Wang J. The use of recombinant activated factor VII to reverse warfarin-induced anticoagulation in patients with hemorrhages in the central nervous system: preliminary findings. J Neurosurg. 2003;98(4):737-740.
23. Freeman WD, Brott TG, Barrett KM, et al. Recombinant factor VIIa for rapid reversal of warfarin anticoagulation in acute intracranial hemorrhage. Mayo Clin Proc. 2004;79(12):1495-1500.
24. Dager WE, King JH, Regalia RC, et al. Reversal of elevated international normalized ratios and bleeding with low-dose recombinant activated factor VII in patients receiving warfarin. Pharmacotherapy. 2006;26(8): 1091-1098.
25. Denas G, Marzot F, Offelli P, et al. Effectiveness and safety of a management protocol to correct over-anticoagulation with oral vitamin K: a retrospective study of 1,043 cases. J Thromb Thrombolysis. 2008 Mar 13; [Epub ahead of print].
26. Boulis NM, Bobek MP, Schmaier A, Hoff JT. Use of factor IX complex in warfarin-related intracranial hemorrhage. Neurosurgery. 1999;45(5):1113-1118.
27. Kissela BM, Eckman MH. Cost effectiveness of recombinant factor VIIa for treatment of intracerebral hemorrhage. BMC Neurol. 2008;8:17.
28. Ernestus RI, Beldzinski P, Lanfermann H, Klug N. Chronic subdural hematoma: surgical treatment and outcome in 104 patients. Surg Neurol. 1997;48(3): 220-225.
29. Merlicco G, Pierangeli E, di Padova PL. Chronic subdural hematomas in adults: prognostic factors: analysis of 70 cases. Neurosurg Rev. 1995;18(4):247-251.
1. Pullicino P, Thompson JL. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med. 2003;348(3): 256-257.
2. Hurlen M, Abdelnoor M, Smith P, et al. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med. 2002;347(13):969-974.
3. DeSilvey DL. Clinical trials: advanced age, anticoagulation intensity, and risk for intracranial hemorrhage among patients taking warfarin for atrial fibrillation. Am J Geriatr Cardiol. 2005;14(2):98-99.
4. Fang MC, Chang Y, Hylek EM, et al. Advanced age, anticoagulation intensity, and risk for intracranial hemorrhage among patients taking warfarin for atrial fibrillation. Ann Intern Med. 2004;141(10):745-752.
5. Koo S, Kucher N, Nguyen PL, et al. The effect of excessive anticoagulation on mortality and morbidity in hospitalized patients with anticoagulant-related major hemorrhage. Arch Intern Med. 2004;164(14):1557-1560.
6. Mina AA, Knipfer JF, Park DY, et al. Intracranial complications of preinjury anticoagulation in trauma patients with head injury. J Trauma. 2002;53(4):668-672.
7. Pieracci FM, Eachempati SR, Shou J, et al. Degree of anticoagulation, but not warfarin use itself, predicts adverse outcomes after traumatic brain injury in elderly trauma patients. J Trauma. 2007;63(3):525-530.
8. Fanikos J, Grasso-Correnti N, Shah R, et al. Major bleeding complications in a specialized anticoagulation service. Am J Cardiol. 2005;96(4):595-598.
9. Pieracci FM, Eachempati SR, Shou J, et al. Use of long-term anticoagulation is associated with traumatic intracranial hemorrhage and subsequent mortality in elderly patients hospitalized after falls: analysis of the New York State Administrative Database. J Trauma. 2007;63(3):519-524.
10. Franko J, Kish KJ, O’Connell BG, et al. Advanced age and preinjury warfarin anticoagulation increase the risk of mortality after head trauma. J Trauma. 2006; 61(1):107-110.
11. Drapkin AJ. Chronic subdural hematoma: pathophysiological basis for treatment. Br J Neurosurg. 1991; 5(5):467-473.
12. Yamamoto H, Hirashima Y, Hamada H, et al. Independent predictors of recurrence of chronic subdural hematoma: results of multivariate analysis performed using a logistic regression model. J Neurosurg. 2003;98(6):1217-1221.
13. Iantosca MR, Simon RH. Chronic subdural hematoma in adult and elderly patients. Neurosurg Clin N Am. 2000;11(3):447-454.
14. Rozzelle CJ, Wofford JL, Branch CL. Predictors of hospital mortality in older patients with subdural hematoma. J Am Geriatr Soc. 1995;43(3):240-244.
15. Wintzen AR, Tijssen JG. Subdural hematoma and oral anticoagulant therapy. Arch Neurol. 1982;39(2): 69-72.
16. Ramachandran R, Hegde T. Chronic subdural hematomas: causes of morbidity and mortality. Surg Neurol. 2007;67(4):367-372.
17. Amirjamshidi A, Eftekhar B, Abouzari M, Rashidi A. The relationship between Glasgow coma/outcome scores and abnormal CT scan findings in chronic subdural hematoma. Clin Neurol Neurosurg. 2007;109(2): 152-157.
18. Lee JY, Ebel H, Ernestus RI, Klug N. Various surgical treatments of chronic subdural hematoma and outcome in 172 patients: is membranectomy necessary? Surg Neurol. 2004;61(6):523-527.
19. Gelabert-González M, Iglesias-Pais M, García-Allut A, Martínez-Rumbo R. Chronic subdural haematoma: surgical treatment and outcome in 1000 cases. Clin Neurol Neurosurg. 2005;107(3):223-229.
20. Mattle H, Kohler S, Huber P, et al. Anticoagulation-related intracranial extracerebral haemorrhage. J Neurol Neurosurg Psychiatry. 1989;52(7):829-837.
21. Sambasivan M. An overview of chronic subdural hematoma: experience with 2300 cases. Surg Neurol. 1997;47(5):418-422.
22. Lin J, Hanigan WC, Tarantino M, Wang J. The use of recombinant activated factor VII to reverse warfarin-induced anticoagulation in patients with hemorrhages in the central nervous system: preliminary findings. J Neurosurg. 2003;98(4):737-740.
23. Freeman WD, Brott TG, Barrett KM, et al. Recombinant factor VIIa for rapid reversal of warfarin anticoagulation in acute intracranial hemorrhage. Mayo Clin Proc. 2004;79(12):1495-1500.
24. Dager WE, King JH, Regalia RC, et al. Reversal of elevated international normalized ratios and bleeding with low-dose recombinant activated factor VII in patients receiving warfarin. Pharmacotherapy. 2006;26(8): 1091-1098.
25. Denas G, Marzot F, Offelli P, et al. Effectiveness and safety of a management protocol to correct over-anticoagulation with oral vitamin K: a retrospective study of 1,043 cases. J Thromb Thrombolysis. 2008 Mar 13; [Epub ahead of print].
26. Boulis NM, Bobek MP, Schmaier A, Hoff JT. Use of factor IX complex in warfarin-related intracranial hemorrhage. Neurosurgery. 1999;45(5):1113-1118.
27. Kissela BM, Eckman MH. Cost effectiveness of recombinant factor VIIa for treatment of intracerebral hemorrhage. BMC Neurol. 2008;8:17.
28. Ernestus RI, Beldzinski P, Lanfermann H, Klug N. Chronic subdural hematoma: surgical treatment and outcome in 104 patients. Surg Neurol. 1997;48(3): 220-225.
29. Merlicco G, Pierangeli E, di Padova PL. Chronic subdural hematomas in adults: prognostic factors: analysis of 70 cases. Neurosurg Rev. 1995;18(4):247-251.
What's Eating You? Cutaneous Myiasis (Wohlfahrtia magnifica)
Dust mite control measures don’t help asthma patients
ILLUSTRATIVE CASE
The parents of a 10-year-old patient whom you recently diagnosed with asthma want to do everything they can to reduce his asthma symptoms. They are considering buying hypoallergenic mattress covers and an expensive air filtration system to decrease the levels of dust mite allergens in their home and want to know if you think that will help their son. What do you tell them?
We want to do everything we can to help our patients control their asthma symptoms, but when it comes to household dust mite control measures, this extensive Cochrane review confirms that interventions like mattress covers and air filtration don’t work, despite recent reviews and guidelines recommending them.
Dust mites (Dermatophagoides pteronyssinus) are one of the most common allergens that provoke asthma symptoms in children and adults.2 Dust mites live in warm, humid places and feed on human skin scales. The areas with the highest levels of household infestation are carpets, mattresses, pillows, drapes, upholstered furniture, and clothing.
Guidelines still encourage mattress cover use
The National Asthma Education and Prevention Program (NAEPP) 2007 guidelines recommend using allergen-impermeable mattress and pillow covers and washing sheets and blankets in hot water. They also recommend “considering” reducing indoor humidity, removing bedroom carpets, and washing stuffed toys weekly. The NAEPP Expert Panel cites many studies to support these recommendations.3
The National Environmental Education and Training Foundation (NEETF) 2005 guidelines recommend additional measures to reduce dust mite exposure including vacuuming using a high-efficiency particulate air (HEPA) filter, removing draperies, and considering using a portable air cleaner with a HEPA filter.4
STUDY SUMMARY: 54 trials, but no support for dust mite measures
This Cochrane systematic review included 54 randomized trials that assessed the effects of physical and/or chemical interventions to reduce exposure to house dust mite antigens in the homes of patients with mite-sensitive asthma. These studies included a total of 3002 pediatric and adult asthma patients (9 - 628 patients analyzed per trial) with mite sensitization confirmed by skin testing or IgE serum assays.
Thirty-six studies tested physical interventions, including mattress covers, vacuum cleaning, heating, ventilation, freezing, washing, air filtration, and ionizers. Ten used chemical interventions to kill dust mites; 8 used a combination of physical and chemical methods. Control groups received either placebo or no treatment.
Outcomes studied. The authors extracted data for the following outcomes: subjective well-being, asthma symptom scores, use of medication, days of sick leave from school or work, number of unscheduled visits to a physician or hospital, forced expiratory volume in 1 second (FEV1), peak expiratory flow rate (PEFR), and provocative concentration that causes a 20% fall in FEV1 (PC20). Length of the intervention and follow-up ranged from 2 weeks to 2 years.
Quality of studies. According to modern standards for randomized trials, the quality of many of the 54 studies was not optimal, especially in the descriptions of randomization and the reporting of outcomes. The method of randomization and concealment of allocation was rarely described. Eleven trial reports did not contain any usable data for the meta-analysis because of the way data were reported, and there was significant potential for reporting bias in favor of a treatment effect in the studies included. Mite reduction was successful in 17 trials, unsuccessful in 24 trials, and not reported in 13 trials.
Interventions didn’t help. There were no differences between the intervention and control groups for any of the outcomes. The percentage of patients who improved after the experimental interventions was not significantly different from the percentage of patients in the control groups (relative risk [RR]=1.01; 95% confidence interval [CI], 0.80-1.27; data based on 7 trials). There was no difference in medication usage (data from 10 trials), FEV1 (data from 14 trials), morning PEFR (data from 23 trials), or PC 20 (data from 14 trials) between the intervention and control groups ( TABLE ).1
TABLE
Dust mite control measures didn’t improve these outcomes
| OUTCOME | STANDARDIZED MEAN DIFFERENCE* (95% CI) |
|---|---|
| Medication usage | -0.06 (-0.18 to 0.07) |
| FEV1 | 0.11 (-0.05 to 0.28) |
| Morning PEFR | 0.00 (-1.0 to 0.10) |
| PC 20 | 0.05 (-0.13 to 0.22) |
| CI, confidence interval; FEV1, forced expiratory volume in 1 second; PC20, provocative concentration that causes a 20% fall in FEV1; PEFR, peak expiratory flow rate. | |
| *Standardized mean difference is a common way to combine results of different studies for comparison purposes. If the 95% CI crosses 0, there is no effect of the intervention compared with the control. | |
WHAT’S NEW?: Nothing is new, yet this will be “news” to many
This Cochrane review includes 5 additional trials that have been conducted since the last Cochrane review of this topic in 2004. However, the 2004 review reported the same conclusion—that interventions to reduce house dust mite exposure in asthma patients are ineffective—as did 3 other Cochrane reviews on the same topic beginning in 1998.5-8
So why are the guidelines out of step? Schmidt and Gøtzsche (one of the authors of the Cochrane review) conducted a systematic review of narrative review articles in 2005 to answer this question. They found 70 review articles, 90% of which recommended physical methods to reduce exposure to house dust mites. They discovered that although these review articles included references to support their recommendations of dust mite control measures, the reviews showed significant bias in favor of positive studies and highlighted the results of low-quality studies, including non-randomized studies that had been excluded from the Cochrane reviews.9
CAVEATS: Duration of studies not long enough?
We know that extreme measures to reduce exposure to dust mite allergen, such as relocating to a high altitude or prolonged hospitalization, can reduce asthma symptoms,10,11 but these are clearly not practical solutions for most patients with dust mite-sensitive asthma. When it comes to this Cochrane review, some might argue that many of the interventions included were not of sufficient duration and did not sufficiently reduce the level of house mite allergen to improve asthma symptoms.
However, the subgroups of trials with long treatment duration (1-2 years) and successful mite reduction (determined by different methods, including mite counts and measured antigen levels in dust samples) also failed to show a significant difference between intervention and control groups.1
Tweak the approach? Most dust mite-sensitive asthma patients are sensitive to other allergens, so perhaps multifaceted interventions that target multiple allergens would be more effective.12 But until these potential interventions are supported by stronger evidence, we should not recommend them to our patients.
CHALLENGES TO IMPLEMENTATION: Swimming against the tide is never easy
Although the evidence to date indicates that interventions to reduce home dust mite exposure are ineffective, there are hundreds of products—including mattress and pillow covers ($10-$100), ionizers ($100-$200), and air filtration systems ($500-$800)—that are being marketed to patients with asthma. In addition, patient education handouts from sources such as the American Academy of Family Physicians, the American Academy of Pediatrics, and UpToDate recommend implementing dust mite control measures to reduce dust mite allergen exposure.13-15
We need to start educating our asthma patients properly so they can spend their time, energy, and money on interventions, such as medications, that work—and not on interventions that make no difference.
Acknowledgements
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
This study was selected and evaluated using FPIN’s Priority Updates from the Research Literature (PURL) Surveillance System methodology. The criteria and findings leading to the selection of this study as a PURL can be accessed at www.jfponline.com/purls.
1. Gotzsche PC, Johansen HK. House dust mite control measures for asthma. Cochrane Database Syst Rev. 2008;(2):CD001187.-
2. German JA, Harper MB. Environmental control of allergic diseases. Am Fam Physician. 2002;66:421-426.
3. National Asthma Education and Prevention Program (NAEPP). Control of environmental factors and comorbid conditions that affect asthma. In: Expert panel report 3: guidelines for the diagnosis and management of asthma. Bethesda, Md: National Heart, Lung, and Blood Institute; 2007.
4. National Environmental Education & Training Foundation (NEETF). Environmental management of pediatric asthma. Guidelines for health care providers. Washington, DC: National Environmental Education & Training Foundation (NEETF); 2005.
5. Gøtzsche PC, Hammarquist C, Burr M. House dust mite control measures in the management of asthma: meta-analysis. BMJ. 1998;317:1105-1110.
6. Hammarquist C, Burr ML, Gotzsche PC. House dust mite control measures for asthma. Cochrane Database Syst Rev. 2000;(2):CD001187.-
7. Gøtzsche PC, Johansen HK, Burr ML, Hammarquist C. House dust mite control measures for asthma. Cochrane Database Syst Rev. 2001;(3):CD001187.-
8. Gøtzsche PC, Johansen HK, Schmidt LM, Burr ML. House dust mite control measures for asthma. Cochrane Database Syst Rev. 2004;(4):CD001187.-
9. Schmidt LM, Gøtzsche PC. Of mites and men: reference bias in narrative review articles: a systematic review. J Fam Pract. 2005;54:334-338.
10. Platts-Mills TA, Tovey ER, Mitchell EB, Moszoro H, Nock P, Wilkins SR. Reduction of bronchial hyperreactivity during prolonged allergen avoidance. Lancet 1982;2:675-678.
11. Grootendorst DC, Dahlen SE. Benefits of high altitude allergen avoidance in atopic adolescents with moderate to severe asthma, over and above treatment with high dose inhaled steroids. Clin Exp Allergy. 2001;31:400-408.
12. Morgan WJ, Crain EF, Gruchalla RS, et al. Results of a home-based environmental intervention among urban children with asthma. N Engl J Med. 2004;351:1068-1080.
13. American Academy of Family Physicians. Dust mites in the home [patient handout]. Available at: http://familydoctor.org/online/famdocen/home/common/asthma/triggers/683.html. Accessed October 23, 2008.
14. American Academy of Pediatrics. Non-pharmacologic approaches to asthma management [patient handout]. Available at: http://www.aap.org/sections/allergy/nonrxchild.pdf. Accessed October 23, 2008.
15. Bailey W. Patient information: Trigger avoidance in asthma. UpToDate [online database]. Version 16.2. Waltham, Mass: UpToDate; 2008.
ILLUSTRATIVE CASE
The parents of a 10-year-old patient whom you recently diagnosed with asthma want to do everything they can to reduce his asthma symptoms. They are considering buying hypoallergenic mattress covers and an expensive air filtration system to decrease the levels of dust mite allergens in their home and want to know if you think that will help their son. What do you tell them?
We want to do everything we can to help our patients control their asthma symptoms, but when it comes to household dust mite control measures, this extensive Cochrane review confirms that interventions like mattress covers and air filtration don’t work, despite recent reviews and guidelines recommending them.
Dust mites (Dermatophagoides pteronyssinus) are one of the most common allergens that provoke asthma symptoms in children and adults.2 Dust mites live in warm, humid places and feed on human skin scales. The areas with the highest levels of household infestation are carpets, mattresses, pillows, drapes, upholstered furniture, and clothing.
Guidelines still encourage mattress cover use
The National Asthma Education and Prevention Program (NAEPP) 2007 guidelines recommend using allergen-impermeable mattress and pillow covers and washing sheets and blankets in hot water. They also recommend “considering” reducing indoor humidity, removing bedroom carpets, and washing stuffed toys weekly. The NAEPP Expert Panel cites many studies to support these recommendations.3
The National Environmental Education and Training Foundation (NEETF) 2005 guidelines recommend additional measures to reduce dust mite exposure including vacuuming using a high-efficiency particulate air (HEPA) filter, removing draperies, and considering using a portable air cleaner with a HEPA filter.4
STUDY SUMMARY: 54 trials, but no support for dust mite measures
This Cochrane systematic review included 54 randomized trials that assessed the effects of physical and/or chemical interventions to reduce exposure to house dust mite antigens in the homes of patients with mite-sensitive asthma. These studies included a total of 3002 pediatric and adult asthma patients (9 - 628 patients analyzed per trial) with mite sensitization confirmed by skin testing or IgE serum assays.
Thirty-six studies tested physical interventions, including mattress covers, vacuum cleaning, heating, ventilation, freezing, washing, air filtration, and ionizers. Ten used chemical interventions to kill dust mites; 8 used a combination of physical and chemical methods. Control groups received either placebo or no treatment.
Outcomes studied. The authors extracted data for the following outcomes: subjective well-being, asthma symptom scores, use of medication, days of sick leave from school or work, number of unscheduled visits to a physician or hospital, forced expiratory volume in 1 second (FEV1), peak expiratory flow rate (PEFR), and provocative concentration that causes a 20% fall in FEV1 (PC20). Length of the intervention and follow-up ranged from 2 weeks to 2 years.
Quality of studies. According to modern standards for randomized trials, the quality of many of the 54 studies was not optimal, especially in the descriptions of randomization and the reporting of outcomes. The method of randomization and concealment of allocation was rarely described. Eleven trial reports did not contain any usable data for the meta-analysis because of the way data were reported, and there was significant potential for reporting bias in favor of a treatment effect in the studies included. Mite reduction was successful in 17 trials, unsuccessful in 24 trials, and not reported in 13 trials.
Interventions didn’t help. There were no differences between the intervention and control groups for any of the outcomes. The percentage of patients who improved after the experimental interventions was not significantly different from the percentage of patients in the control groups (relative risk [RR]=1.01; 95% confidence interval [CI], 0.80-1.27; data based on 7 trials). There was no difference in medication usage (data from 10 trials), FEV1 (data from 14 trials), morning PEFR (data from 23 trials), or PC 20 (data from 14 trials) between the intervention and control groups ( TABLE ).1
TABLE
Dust mite control measures didn’t improve these outcomes
| OUTCOME | STANDARDIZED MEAN DIFFERENCE* (95% CI) |
|---|---|
| Medication usage | -0.06 (-0.18 to 0.07) |
| FEV1 | 0.11 (-0.05 to 0.28) |
| Morning PEFR | 0.00 (-1.0 to 0.10) |
| PC 20 | 0.05 (-0.13 to 0.22) |
| CI, confidence interval; FEV1, forced expiratory volume in 1 second; PC20, provocative concentration that causes a 20% fall in FEV1; PEFR, peak expiratory flow rate. | |
| *Standardized mean difference is a common way to combine results of different studies for comparison purposes. If the 95% CI crosses 0, there is no effect of the intervention compared with the control. | |
WHAT’S NEW?: Nothing is new, yet this will be “news” to many
This Cochrane review includes 5 additional trials that have been conducted since the last Cochrane review of this topic in 2004. However, the 2004 review reported the same conclusion—that interventions to reduce house dust mite exposure in asthma patients are ineffective—as did 3 other Cochrane reviews on the same topic beginning in 1998.5-8
So why are the guidelines out of step? Schmidt and Gøtzsche (one of the authors of the Cochrane review) conducted a systematic review of narrative review articles in 2005 to answer this question. They found 70 review articles, 90% of which recommended physical methods to reduce exposure to house dust mites. They discovered that although these review articles included references to support their recommendations of dust mite control measures, the reviews showed significant bias in favor of positive studies and highlighted the results of low-quality studies, including non-randomized studies that had been excluded from the Cochrane reviews.9
CAVEATS: Duration of studies not long enough?
We know that extreme measures to reduce exposure to dust mite allergen, such as relocating to a high altitude or prolonged hospitalization, can reduce asthma symptoms,10,11 but these are clearly not practical solutions for most patients with dust mite-sensitive asthma. When it comes to this Cochrane review, some might argue that many of the interventions included were not of sufficient duration and did not sufficiently reduce the level of house mite allergen to improve asthma symptoms.
However, the subgroups of trials with long treatment duration (1-2 years) and successful mite reduction (determined by different methods, including mite counts and measured antigen levels in dust samples) also failed to show a significant difference between intervention and control groups.1
Tweak the approach? Most dust mite-sensitive asthma patients are sensitive to other allergens, so perhaps multifaceted interventions that target multiple allergens would be more effective.12 But until these potential interventions are supported by stronger evidence, we should not recommend them to our patients.
CHALLENGES TO IMPLEMENTATION: Swimming against the tide is never easy
Although the evidence to date indicates that interventions to reduce home dust mite exposure are ineffective, there are hundreds of products—including mattress and pillow covers ($10-$100), ionizers ($100-$200), and air filtration systems ($500-$800)—that are being marketed to patients with asthma. In addition, patient education handouts from sources such as the American Academy of Family Physicians, the American Academy of Pediatrics, and UpToDate recommend implementing dust mite control measures to reduce dust mite allergen exposure.13-15
We need to start educating our asthma patients properly so they can spend their time, energy, and money on interventions, such as medications, that work—and not on interventions that make no difference.
Acknowledgements
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
This study was selected and evaluated using FPIN’s Priority Updates from the Research Literature (PURL) Surveillance System methodology. The criteria and findings leading to the selection of this study as a PURL can be accessed at www.jfponline.com/purls.
ILLUSTRATIVE CASE
The parents of a 10-year-old patient whom you recently diagnosed with asthma want to do everything they can to reduce his asthma symptoms. They are considering buying hypoallergenic mattress covers and an expensive air filtration system to decrease the levels of dust mite allergens in their home and want to know if you think that will help their son. What do you tell them?
We want to do everything we can to help our patients control their asthma symptoms, but when it comes to household dust mite control measures, this extensive Cochrane review confirms that interventions like mattress covers and air filtration don’t work, despite recent reviews and guidelines recommending them.
Dust mites (Dermatophagoides pteronyssinus) are one of the most common allergens that provoke asthma symptoms in children and adults.2 Dust mites live in warm, humid places and feed on human skin scales. The areas with the highest levels of household infestation are carpets, mattresses, pillows, drapes, upholstered furniture, and clothing.
Guidelines still encourage mattress cover use
The National Asthma Education and Prevention Program (NAEPP) 2007 guidelines recommend using allergen-impermeable mattress and pillow covers and washing sheets and blankets in hot water. They also recommend “considering” reducing indoor humidity, removing bedroom carpets, and washing stuffed toys weekly. The NAEPP Expert Panel cites many studies to support these recommendations.3
The National Environmental Education and Training Foundation (NEETF) 2005 guidelines recommend additional measures to reduce dust mite exposure including vacuuming using a high-efficiency particulate air (HEPA) filter, removing draperies, and considering using a portable air cleaner with a HEPA filter.4
STUDY SUMMARY: 54 trials, but no support for dust mite measures
This Cochrane systematic review included 54 randomized trials that assessed the effects of physical and/or chemical interventions to reduce exposure to house dust mite antigens in the homes of patients with mite-sensitive asthma. These studies included a total of 3002 pediatric and adult asthma patients (9 - 628 patients analyzed per trial) with mite sensitization confirmed by skin testing or IgE serum assays.
Thirty-six studies tested physical interventions, including mattress covers, vacuum cleaning, heating, ventilation, freezing, washing, air filtration, and ionizers. Ten used chemical interventions to kill dust mites; 8 used a combination of physical and chemical methods. Control groups received either placebo or no treatment.
Outcomes studied. The authors extracted data for the following outcomes: subjective well-being, asthma symptom scores, use of medication, days of sick leave from school or work, number of unscheduled visits to a physician or hospital, forced expiratory volume in 1 second (FEV1), peak expiratory flow rate (PEFR), and provocative concentration that causes a 20% fall in FEV1 (PC20). Length of the intervention and follow-up ranged from 2 weeks to 2 years.
Quality of studies. According to modern standards for randomized trials, the quality of many of the 54 studies was not optimal, especially in the descriptions of randomization and the reporting of outcomes. The method of randomization and concealment of allocation was rarely described. Eleven trial reports did not contain any usable data for the meta-analysis because of the way data were reported, and there was significant potential for reporting bias in favor of a treatment effect in the studies included. Mite reduction was successful in 17 trials, unsuccessful in 24 trials, and not reported in 13 trials.
Interventions didn’t help. There were no differences between the intervention and control groups for any of the outcomes. The percentage of patients who improved after the experimental interventions was not significantly different from the percentage of patients in the control groups (relative risk [RR]=1.01; 95% confidence interval [CI], 0.80-1.27; data based on 7 trials). There was no difference in medication usage (data from 10 trials), FEV1 (data from 14 trials), morning PEFR (data from 23 trials), or PC 20 (data from 14 trials) between the intervention and control groups ( TABLE ).1
TABLE
Dust mite control measures didn’t improve these outcomes
| OUTCOME | STANDARDIZED MEAN DIFFERENCE* (95% CI) |
|---|---|
| Medication usage | -0.06 (-0.18 to 0.07) |
| FEV1 | 0.11 (-0.05 to 0.28) |
| Morning PEFR | 0.00 (-1.0 to 0.10) |
| PC 20 | 0.05 (-0.13 to 0.22) |
| CI, confidence interval; FEV1, forced expiratory volume in 1 second; PC20, provocative concentration that causes a 20% fall in FEV1; PEFR, peak expiratory flow rate. | |
| *Standardized mean difference is a common way to combine results of different studies for comparison purposes. If the 95% CI crosses 0, there is no effect of the intervention compared with the control. | |
WHAT’S NEW?: Nothing is new, yet this will be “news” to many
This Cochrane review includes 5 additional trials that have been conducted since the last Cochrane review of this topic in 2004. However, the 2004 review reported the same conclusion—that interventions to reduce house dust mite exposure in asthma patients are ineffective—as did 3 other Cochrane reviews on the same topic beginning in 1998.5-8
So why are the guidelines out of step? Schmidt and Gøtzsche (one of the authors of the Cochrane review) conducted a systematic review of narrative review articles in 2005 to answer this question. They found 70 review articles, 90% of which recommended physical methods to reduce exposure to house dust mites. They discovered that although these review articles included references to support their recommendations of dust mite control measures, the reviews showed significant bias in favor of positive studies and highlighted the results of low-quality studies, including non-randomized studies that had been excluded from the Cochrane reviews.9
CAVEATS: Duration of studies not long enough?
We know that extreme measures to reduce exposure to dust mite allergen, such as relocating to a high altitude or prolonged hospitalization, can reduce asthma symptoms,10,11 but these are clearly not practical solutions for most patients with dust mite-sensitive asthma. When it comes to this Cochrane review, some might argue that many of the interventions included were not of sufficient duration and did not sufficiently reduce the level of house mite allergen to improve asthma symptoms.
However, the subgroups of trials with long treatment duration (1-2 years) and successful mite reduction (determined by different methods, including mite counts and measured antigen levels in dust samples) also failed to show a significant difference between intervention and control groups.1
Tweak the approach? Most dust mite-sensitive asthma patients are sensitive to other allergens, so perhaps multifaceted interventions that target multiple allergens would be more effective.12 But until these potential interventions are supported by stronger evidence, we should not recommend them to our patients.
CHALLENGES TO IMPLEMENTATION: Swimming against the tide is never easy
Although the evidence to date indicates that interventions to reduce home dust mite exposure are ineffective, there are hundreds of products—including mattress and pillow covers ($10-$100), ionizers ($100-$200), and air filtration systems ($500-$800)—that are being marketed to patients with asthma. In addition, patient education handouts from sources such as the American Academy of Family Physicians, the American Academy of Pediatrics, and UpToDate recommend implementing dust mite control measures to reduce dust mite allergen exposure.13-15
We need to start educating our asthma patients properly so they can spend their time, energy, and money on interventions, such as medications, that work—and not on interventions that make no difference.
Acknowledgements
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
This study was selected and evaluated using FPIN’s Priority Updates from the Research Literature (PURL) Surveillance System methodology. The criteria and findings leading to the selection of this study as a PURL can be accessed at www.jfponline.com/purls.
1. Gotzsche PC, Johansen HK. House dust mite control measures for asthma. Cochrane Database Syst Rev. 2008;(2):CD001187.-
2. German JA, Harper MB. Environmental control of allergic diseases. Am Fam Physician. 2002;66:421-426.
3. National Asthma Education and Prevention Program (NAEPP). Control of environmental factors and comorbid conditions that affect asthma. In: Expert panel report 3: guidelines for the diagnosis and management of asthma. Bethesda, Md: National Heart, Lung, and Blood Institute; 2007.
4. National Environmental Education & Training Foundation (NEETF). Environmental management of pediatric asthma. Guidelines for health care providers. Washington, DC: National Environmental Education & Training Foundation (NEETF); 2005.
5. Gøtzsche PC, Hammarquist C, Burr M. House dust mite control measures in the management of asthma: meta-analysis. BMJ. 1998;317:1105-1110.
6. Hammarquist C, Burr ML, Gotzsche PC. House dust mite control measures for asthma. Cochrane Database Syst Rev. 2000;(2):CD001187.-
7. Gøtzsche PC, Johansen HK, Burr ML, Hammarquist C. House dust mite control measures for asthma. Cochrane Database Syst Rev. 2001;(3):CD001187.-
8. Gøtzsche PC, Johansen HK, Schmidt LM, Burr ML. House dust mite control measures for asthma. Cochrane Database Syst Rev. 2004;(4):CD001187.-
9. Schmidt LM, Gøtzsche PC. Of mites and men: reference bias in narrative review articles: a systematic review. J Fam Pract. 2005;54:334-338.
10. Platts-Mills TA, Tovey ER, Mitchell EB, Moszoro H, Nock P, Wilkins SR. Reduction of bronchial hyperreactivity during prolonged allergen avoidance. Lancet 1982;2:675-678.
11. Grootendorst DC, Dahlen SE. Benefits of high altitude allergen avoidance in atopic adolescents with moderate to severe asthma, over and above treatment with high dose inhaled steroids. Clin Exp Allergy. 2001;31:400-408.
12. Morgan WJ, Crain EF, Gruchalla RS, et al. Results of a home-based environmental intervention among urban children with asthma. N Engl J Med. 2004;351:1068-1080.
13. American Academy of Family Physicians. Dust mites in the home [patient handout]. Available at: http://familydoctor.org/online/famdocen/home/common/asthma/triggers/683.html. Accessed October 23, 2008.
14. American Academy of Pediatrics. Non-pharmacologic approaches to asthma management [patient handout]. Available at: http://www.aap.org/sections/allergy/nonrxchild.pdf. Accessed October 23, 2008.
15. Bailey W. Patient information: Trigger avoidance in asthma. UpToDate [online database]. Version 16.2. Waltham, Mass: UpToDate; 2008.
1. Gotzsche PC, Johansen HK. House dust mite control measures for asthma. Cochrane Database Syst Rev. 2008;(2):CD001187.-
2. German JA, Harper MB. Environmental control of allergic diseases. Am Fam Physician. 2002;66:421-426.
3. National Asthma Education and Prevention Program (NAEPP). Control of environmental factors and comorbid conditions that affect asthma. In: Expert panel report 3: guidelines for the diagnosis and management of asthma. Bethesda, Md: National Heart, Lung, and Blood Institute; 2007.
4. National Environmental Education & Training Foundation (NEETF). Environmental management of pediatric asthma. Guidelines for health care providers. Washington, DC: National Environmental Education & Training Foundation (NEETF); 2005.
5. Gøtzsche PC, Hammarquist C, Burr M. House dust mite control measures in the management of asthma: meta-analysis. BMJ. 1998;317:1105-1110.
6. Hammarquist C, Burr ML, Gotzsche PC. House dust mite control measures for asthma. Cochrane Database Syst Rev. 2000;(2):CD001187.-
7. Gøtzsche PC, Johansen HK, Burr ML, Hammarquist C. House dust mite control measures for asthma. Cochrane Database Syst Rev. 2001;(3):CD001187.-
8. Gøtzsche PC, Johansen HK, Schmidt LM, Burr ML. House dust mite control measures for asthma. Cochrane Database Syst Rev. 2004;(4):CD001187.-
9. Schmidt LM, Gøtzsche PC. Of mites and men: reference bias in narrative review articles: a systematic review. J Fam Pract. 2005;54:334-338.
10. Platts-Mills TA, Tovey ER, Mitchell EB, Moszoro H, Nock P, Wilkins SR. Reduction of bronchial hyperreactivity during prolonged allergen avoidance. Lancet 1982;2:675-678.
11. Grootendorst DC, Dahlen SE. Benefits of high altitude allergen avoidance in atopic adolescents with moderate to severe asthma, over and above treatment with high dose inhaled steroids. Clin Exp Allergy. 2001;31:400-408.
12. Morgan WJ, Crain EF, Gruchalla RS, et al. Results of a home-based environmental intervention among urban children with asthma. N Engl J Med. 2004;351:1068-1080.
13. American Academy of Family Physicians. Dust mites in the home [patient handout]. Available at: http://familydoctor.org/online/famdocen/home/common/asthma/triggers/683.html. Accessed October 23, 2008.
14. American Academy of Pediatrics. Non-pharmacologic approaches to asthma management [patient handout]. Available at: http://www.aap.org/sections/allergy/nonrxchild.pdf. Accessed October 23, 2008.
15. Bailey W. Patient information: Trigger avoidance in asthma. UpToDate [online database]. Version 16.2. Waltham, Mass: UpToDate; 2008.
Copyright © 2008 The Family Physicians Inquiries Network.
All rights reserved.
Unsedated colonoscopy: Time to revisit this option?
- Consider recommending unsedated colonoscopy to patients who have issues with cost, concerns about sedation, or are unable to get an escort or avoid work following the procedure.
- Explore resources in your area that offer unsedated colonoscopy.
Background Access to potentially life-saving screening colonoscopy is limited by the high cost of sedation. We explored the practicability of having supervised trainees perform unsedated colonoscopies.
Method A nursing shortage at our Veterans Administration gastroenterology training program necessitated discontinuing sedated colonoscopy. We offered the procedure without sedation to restore local access to screening colonoscopy.
Results From September 2002 to June 2005, 145 of 483 patients accepted the unsedated option. The procedure was done by second-year gastroenterology (GI) fellows who had performed about 100 sedated colonoscopies in their first year of training. Cecal intubation was achieved in 81% of 138 wellpurged patients without obstructive lesions. Implementation obviated the need for 2 registered nurses, the escort requirement, and postprocedure activity restriction. It also eliminated sedation-related complications.
Conclusion This report confirms the feasibility of unsedated colonoscopy performed by supervised trainees. The unsedated option minimizes direct and indirect costs of colonoscopy. Describing unsedated screening colonoscopy to patients as a “sedation risk–free” procedure encouraged them to consider the benefits. We recommend that future studies test primary care providers’ willingness to inform patients of the feasibility of this nonstandard option, and perhaps reshape the practice of colonoscopy for colorectal cancer screening.
Monitored sedation given for colonoscopy is a measure meant to ensure patient safety,1 but its high cost limits access to the potentially life-saving screening procedure.2,3 Unsedated colonoscopy is an option, but a controversial one, raising issues both pro4-6 and con.7-11 In the United States, gastroenterologists perform unsedated colonoscopy both for unescorted patients (~2% of all screening colonos-copies)12,13 and individuals who simply prefer to avoid sedation (~6%).7 Family physicians, too, perform unsedated colonoscopy in both rural and urban settings.14-17
Would it be feasible to make training in unsedated colonoscopy more readily available to providers, and thereby reduce costs and inconvenience for patients? We took advantage of a changing environment at our Veterans Administration program to explore this question.
Method
A nursing shortage in our VA academic GI program necessitated discontinuing sedated colonoscopy. We reviewed the literature on unsedated colonoscopy and found that it is a feasible alternative performed elsewhere.4-7,12,14,16 Our attending staff discussed the options with patients and obtained informed consent18 using the following general message:
“Sedated colonoscopy is usual practice. Even though the risks of sedation are very small, nurses are required to monitor patients continuously. Because of a nursing shortage, we must send you to another VA facility 15 miles away for sedated colonoscopy. You must have an escort, as you will not be allowed to drive after sedation. One of the medicines they administer will make you forget the discomfort you may have experienced, as well as the discussions after the examination.
“Alternatively, you may choose unsedated colonoscopy, which is practiced in the United States and many other countries. Because no medicines are used, there are no medication-induced complications. An escort is not required, and there is no activity restriction afterwards.
“You will feel air in the colon and the endoscope being pushed around inside you. The colonoscopist will talk to you throughout the examination. When you begin to experience discomfort, the colonoscopist will remove air inside the colon or straighten the loops in the colonoscope to minimize the discomfort before it becomes severe. If discomfort does become severe, you and the colonoscopist can agree to stop the advancement of the colonoscope. Complications related to taking biopsies or removing polyps are similar in sedated and unsedated procedures.”
Supervised trainees (second-year GI fellows who had performed about 100 sedated colonoscopies in their first year of training), assisted by a licensed vocational nurse, performed the procedures using appropriate techniques.5-7
The Institution Review Board of the Veterans Affairs Greater Los Angeles Healthcare System (VAGLAHS) approved our review of the patient data for publication.
Results
From September 2002 to June 2005, 145 of 483 patients accepted the unsedated option. The number of patients choosing this option increased in each successive academic year (31, 50, and 64), as did the wait-time in days (27±4, 46±5, 72±6) (mean±standard error of the mean [SEM]). Seven patients (4%) had poor bowel preparation or obstructing lesions limiting completion. Among the 138 well-purged patients, we achieved cecal intubation in 112 (81%). Discomfort limited completion in the remaining 19%.
Other than transient vasovagal reactions in 2 patients, no complications occurred. Patients with incomplete examinations due to discomfort underwent sedated colonoscopy or barium enema or received no further assessment, depending on the initial findings. Those who subsequently underwent sedated colonoscopy (10%) had to be purged again, escorted, and comply with activity restriction.
Colonoscopy was developed as an unsedated procedure.19,20 Discomfort experienced by some patients during sigmoid intubation led to the use of medications,20,21 which are now administered routinely.22 Interestingly, competency in unsedated colonoscopy is not required of GI fellows.23 Indeed, until recently,18 teaching GI trainees unsedated colonoscopy was deemed impractical.7 Family physicians, on the other hand, have long practiced and taught unsedated colonoscopy,14-17,24 although the actual number of family physicians performing colonoscopy is fairly small.25 One reason so few family physicians offer the service—estimated at 3.7% of the specialty—may be the intensive and costly education required.16 Even more difficult has been gaining privileges to perform the sedated procedure,26 given the training requirements set forth by GI professional societies.27
Many patients would opt for unsedated colonoscopy. The favorable reception of unsedated colonoscopy in our study is evident in the increasing number of veterans each year who opted for the procedure despite the lengthening wait-time (which was due to increased demand rather than decreased availability of endoscopists). In the course of our project, we found that the terms “unsedated,” “no sedation,” or “without sedation” tended to convey the negative connotation that relief of discomfort and induced amnesia are withheld.8,9 The term “sedation risk–free”28 emphasized the benefits of no sedation.
Cost factors favoring unsedated colonoscopy. Sedated colonoscopies performed by family physicians have offered substantial health care savings.29,30 It is intuitively obvious that the unsedated option in the hands of those with the necessary skills14-16,24 would be even less costly. Our unsedated colonoscopy project reduced direct costs, which included the cost of having 2 registered nurses on hand. Indirect costs to patients31 were also minimized by avoiding the need for an escort or activity restriction. Moreover, there were no sedation-related complications.32,33
An estimated 40 million healthy Americans are eligible for colorectal cancer screening.34 Primary care providers play a pivotal role in counseling many of these patients, who may find the indirect cost savings of unsedated colonoscopy performed by that same provider appealing.
A logical transition from flexible sigmoidoscopy. An unsedated colonoscopy is very similar to an extended flexible sigmoidoscopy.14-17,35 In patients who can tolerate a flexible sigmoidoscopy well, extended flexible sigmoidoscopy can reach the cecum >70% of the time.36 To enhance the cecal intubation rate among unsedated veterans, we developed (subsequent to the findings reported here) a novel method of water infusion in lieu of air insufflation during insertion of the colonoscope.37 This measure improved the cecal intubation rate from 76% to 97%.38 For family physicians who perform flexible sigmoidoscopy, it is worth considering performing extended flexible sigmoidoscopy or unsedated colonoscopy using this water infusion method37,38 or other methods to minimize discomfort in unsedated patients.39
Limitations of our study, and opportunities. Our report is based on uncontrolled, nonrandomized observational data. Nevertheless, it affirms the feasibility of unsedated colonoscopy performed by supervised trainees, as previously reported by a family practice training program.16 It also underscores the benefits of the unsedated option on direct and indirect costs.
Since only 3.7% of family physicians in a recent survey reported performing colonoscopy,25 it is uncertain whether primary care providers would voluntarily inform patients about the unsedated option. In select settings, gastroenterologists are willing to provide unsedated colonoscopy.6,7,12,13,18 A reasonable hypothesis to test is that primary care providers informing patients about unsedated colonoscopy could reshape the future practice of screening colonoscopy in family medicine and gastroenterology.
Funding/Support
Supported in part by vA medical research Funds and an American Society for Gastrointestinal endoscopy career Development Award (FWl 1985).
Correspondence
Felix W. Leung, MD, Division of Gastroenterology (111G), Sepulveda Ambulatory Care Center, Veterans Affairs Greater los Angeles Healthcare System, 16111 Plummer Street, North Hills, CA 91343; .
1. Comprehensive Accreditation Manual for Hospitals; The Official Handbook. Oakbrook Terrace, III: Joint Commission on Accreditation of Healthcare Organizations; 2005:160-223.
2. Lieberman DA, Weiss DG, Bond JH, et al. Use of colonoscopy to screen asymptomatic adults for colorectal cancer. N Engl J Med. 2000;343:162-168.
3. El-Serag HB, Petersen L, Hampel H, et al. The use of screening colonoscopy for patients cared for by the Department of Veterans Affairs. Arch Intern Med. 2006;166:2202-2208.
4. Herman FN. Avoidance of sedation during total colonoscopy. Dis Colon Rectum. 1990;33:70-72.
5. Cataldo PA. Colonoscopy without sedation: a viable alternative. Dis Colon Rectum. 1996;39:257-261.
6. Hoffman MS, Butler TW, Shaver T. Colonoscopy without sedation. J Clin Gastroenterol. 1998;26:279-282.
7. Rex DK, Imperiale TF, Portish V. Patients willing to try colonoscopy without sedation: associated clinical factors and results of a randomized controlled trial. Gastrointest Endosc. 1999;49:554-559.
8. Levenson D. Health quality organization criticizes colonoscopies given without pain medication. Rept Med Guidelines Outcomes Res. 2001;12:9-12.
9. Leo RA. Unsedated endoscopy: you don’t get a medal for it! South Med J. 2004;97:797-798.
10. Faulx AL, Vela S, Das A, et al. The changing landscape of practice patterns regarding unsedated endoscopy and propofol use: a national Web survey. Gastrointest Endosc. 2005;62:9-15.
11. Rex DK, Khalfan HK. Sedation and the technical performance of colonoscopy. Gastrointest Endosc Clin N Am. 2005;15:661-672.
12. Nelson DB, MCQuaid KR, Bond JH, et al. Procedural success and complications of large-scale screening colonoscopy. Gastrointest Endosc. 2002;55:307-314.
13. Aslinia F, Uradomo L, Steele A, et al. Quality assessment of colonoscopic cecal intubation: an analysis of 6 years of continuous practice at a University Hospital. Am J Gastroenterol. 2006;101:721-731.
14. Hopper W, Kyker KA, Rodney WM. Colonoscopy by a family physician: a 9-year experience of 1048 procedures. J Fam Pract. 1996;43:561 -566.
15. Carr KW, Worthington JM, Rodney WM, et al. Advancing from flexible sigmoidoscopy to colonoscopy in rural family practice. Tenn Med. 1998;91:21-26.
16. Rodney WM, Dabov G, Orientale E, et al. Sedation associated with a more complete colonoscopy. J Fam Pract. 1993;36:394-400.
17. Knox L, Hahn RG, Lane C. A comparison of unsedated colonoscopy and flexible sigmoidoscopy in the family medicine setting: an LA Net study. J Am Board Fam Med. 2007;20:444-450.
18. Leung FW, Aharonian HS, Guth PH, et al. Involvement of trainees in routine unsedated colonoscopy—review of pilot experience. Gastrointest Endosc. 2008;67:718-722.
19. Wolff WI, Shinya H. Colonofiberoscopy. JAMA. 1971;217:1509-1512.
20. Williams C, Teague RH. Colonoscopy. Gut. 1973;14:990-1003.
21. Waye JD. Colonoscopy. Surg Clin North Am. 1972;52:1013-1024.
22. National Cancer Institute Web Site. Available at: http://www.cancer.gov/cancertopics/types/colon-and-rectal. Accessed January 23, 2008.
23. Gastroenterology required curriculum. Chicago, III: Accreditation council on Graduate medical education; 2005.
24. Edwards JK, Norris TE. Colonoscopy in rural communities: can family physicians perform the procedure with safe and efficacious results? J Am Board Fam Pract. 2004;17:353-358.
25. Wilkins T, Wagner P, Thomas A, et al. Attitudes toward performance of endoscopic colon cancer screening by family physicians. Fam Med. 2007;39:578-584.
26. Frank M. American Academy of Family Physicians response to American college of Gastroenterology’s letter and legal opinion. Sent to hospital administrators on December 12, 2005.
27. Faigel D, Baron T, Lewis B, et al. Ensuring competence in endoscopy. Prepared by the ASGe Taskforce on ensuring competence in endoscopy and American college of Gastroenterology executive and Practice management committees, 2005: 1-36.
28. Leung FW. Should minimization of cue be an option as a quality indicator in colonoscopy performed for colorectal cancer screening? Gastroin-test Endosc. 2008;67:579-580.
29. Short MW, Kelly KM, Runser lA. Colonoscopy by a family physician: a case series demonstrating health care savings. Mil Med. 2007;172:1089-1092.
30. Perry RE, Christensen JB, Christensen MA, et al. Office colonoscopy—a safe procedure in selected patients. Dis Colon Rectum. 1989;32:1031-1033.
31. Glied S. Estimating the indirect cost of illness: an assessment of the forgone earnings approach. Am J Public Health. 1996;86:1723-1728.
32. Arrowsmith JB, Gerstman BB, Fleischer DE, et al. Results from the American Society for Gastrointestinal endoscopy/uS Food and Drug Administration collaborative study on complication rates and drug use during gastrointestinal endoscopy. Gastroin-test Endosc. 1991;37:421-427.
33. Sharma VK, Nguyen CC, Crowell MD, et al. A national study of cardiopulmonary unplanned events after GI endoscopy. Gastrointest Endosc. 2007;66:27-34.
34. Seeff LC, Manninen DL, Dong FB, et al. Is there endoscopic capacity to provide colorectal cancer screening to the unscreened population in the united States? Gastroenterology. 2004;127:1661-1669.
35. Dervin JV. Feasibility of 105-cm flexible sigmoidoscopy in family practice. J Fam Pract. 1986;23:341-344.
36. Lee JG, Lum D, Urayama S, et al. Unsedated extended flexible sigmoidoscopy for colorectal cancer screening: a pilot study. Aliment Pharmacol Ther. 2006;23:945-951.
37. Leung JW, Mann S, Leung FW. Option of screening colonoscopy without sedation—a pilot study in united States veterans. Aliment Pharmacol Ther. 2007;26:627-631.
38. Leung FW, Aharonian HS, Leung JW, et al. Impact of a novel water method on scheduled unsedated colonoscopy in U.S. veterans. Gastrointest Endosc. In press.
39. Leung FW. Methods of reducing discomfort during colonoscopy. Dig Dis Sci. 2008;53:1462-1467.
- Consider recommending unsedated colonoscopy to patients who have issues with cost, concerns about sedation, or are unable to get an escort or avoid work following the procedure.
- Explore resources in your area that offer unsedated colonoscopy.
Background Access to potentially life-saving screening colonoscopy is limited by the high cost of sedation. We explored the practicability of having supervised trainees perform unsedated colonoscopies.
Method A nursing shortage at our Veterans Administration gastroenterology training program necessitated discontinuing sedated colonoscopy. We offered the procedure without sedation to restore local access to screening colonoscopy.
Results From September 2002 to June 2005, 145 of 483 patients accepted the unsedated option. The procedure was done by second-year gastroenterology (GI) fellows who had performed about 100 sedated colonoscopies in their first year of training. Cecal intubation was achieved in 81% of 138 wellpurged patients without obstructive lesions. Implementation obviated the need for 2 registered nurses, the escort requirement, and postprocedure activity restriction. It also eliminated sedation-related complications.
Conclusion This report confirms the feasibility of unsedated colonoscopy performed by supervised trainees. The unsedated option minimizes direct and indirect costs of colonoscopy. Describing unsedated screening colonoscopy to patients as a “sedation risk–free” procedure encouraged them to consider the benefits. We recommend that future studies test primary care providers’ willingness to inform patients of the feasibility of this nonstandard option, and perhaps reshape the practice of colonoscopy for colorectal cancer screening.
Monitored sedation given for colonoscopy is a measure meant to ensure patient safety,1 but its high cost limits access to the potentially life-saving screening procedure.2,3 Unsedated colonoscopy is an option, but a controversial one, raising issues both pro4-6 and con.7-11 In the United States, gastroenterologists perform unsedated colonoscopy both for unescorted patients (~2% of all screening colonos-copies)12,13 and individuals who simply prefer to avoid sedation (~6%).7 Family physicians, too, perform unsedated colonoscopy in both rural and urban settings.14-17
Would it be feasible to make training in unsedated colonoscopy more readily available to providers, and thereby reduce costs and inconvenience for patients? We took advantage of a changing environment at our Veterans Administration program to explore this question.
Method
A nursing shortage in our VA academic GI program necessitated discontinuing sedated colonoscopy. We reviewed the literature on unsedated colonoscopy and found that it is a feasible alternative performed elsewhere.4-7,12,14,16 Our attending staff discussed the options with patients and obtained informed consent18 using the following general message:
“Sedated colonoscopy is usual practice. Even though the risks of sedation are very small, nurses are required to monitor patients continuously. Because of a nursing shortage, we must send you to another VA facility 15 miles away for sedated colonoscopy. You must have an escort, as you will not be allowed to drive after sedation. One of the medicines they administer will make you forget the discomfort you may have experienced, as well as the discussions after the examination.
“Alternatively, you may choose unsedated colonoscopy, which is practiced in the United States and many other countries. Because no medicines are used, there are no medication-induced complications. An escort is not required, and there is no activity restriction afterwards.
“You will feel air in the colon and the endoscope being pushed around inside you. The colonoscopist will talk to you throughout the examination. When you begin to experience discomfort, the colonoscopist will remove air inside the colon or straighten the loops in the colonoscope to minimize the discomfort before it becomes severe. If discomfort does become severe, you and the colonoscopist can agree to stop the advancement of the colonoscope. Complications related to taking biopsies or removing polyps are similar in sedated and unsedated procedures.”
Supervised trainees (second-year GI fellows who had performed about 100 sedated colonoscopies in their first year of training), assisted by a licensed vocational nurse, performed the procedures using appropriate techniques.5-7
The Institution Review Board of the Veterans Affairs Greater Los Angeles Healthcare System (VAGLAHS) approved our review of the patient data for publication.
Results
From September 2002 to June 2005, 145 of 483 patients accepted the unsedated option. The number of patients choosing this option increased in each successive academic year (31, 50, and 64), as did the wait-time in days (27±4, 46±5, 72±6) (mean±standard error of the mean [SEM]). Seven patients (4%) had poor bowel preparation or obstructing lesions limiting completion. Among the 138 well-purged patients, we achieved cecal intubation in 112 (81%). Discomfort limited completion in the remaining 19%.
Other than transient vasovagal reactions in 2 patients, no complications occurred. Patients with incomplete examinations due to discomfort underwent sedated colonoscopy or barium enema or received no further assessment, depending on the initial findings. Those who subsequently underwent sedated colonoscopy (10%) had to be purged again, escorted, and comply with activity restriction.
Colonoscopy was developed as an unsedated procedure.19,20 Discomfort experienced by some patients during sigmoid intubation led to the use of medications,20,21 which are now administered routinely.22 Interestingly, competency in unsedated colonoscopy is not required of GI fellows.23 Indeed, until recently,18 teaching GI trainees unsedated colonoscopy was deemed impractical.7 Family physicians, on the other hand, have long practiced and taught unsedated colonoscopy,14-17,24 although the actual number of family physicians performing colonoscopy is fairly small.25 One reason so few family physicians offer the service—estimated at 3.7% of the specialty—may be the intensive and costly education required.16 Even more difficult has been gaining privileges to perform the sedated procedure,26 given the training requirements set forth by GI professional societies.27
Many patients would opt for unsedated colonoscopy. The favorable reception of unsedated colonoscopy in our study is evident in the increasing number of veterans each year who opted for the procedure despite the lengthening wait-time (which was due to increased demand rather than decreased availability of endoscopists). In the course of our project, we found that the terms “unsedated,” “no sedation,” or “without sedation” tended to convey the negative connotation that relief of discomfort and induced amnesia are withheld.8,9 The term “sedation risk–free”28 emphasized the benefits of no sedation.
Cost factors favoring unsedated colonoscopy. Sedated colonoscopies performed by family physicians have offered substantial health care savings.29,30 It is intuitively obvious that the unsedated option in the hands of those with the necessary skills14-16,24 would be even less costly. Our unsedated colonoscopy project reduced direct costs, which included the cost of having 2 registered nurses on hand. Indirect costs to patients31 were also minimized by avoiding the need for an escort or activity restriction. Moreover, there were no sedation-related complications.32,33
An estimated 40 million healthy Americans are eligible for colorectal cancer screening.34 Primary care providers play a pivotal role in counseling many of these patients, who may find the indirect cost savings of unsedated colonoscopy performed by that same provider appealing.
A logical transition from flexible sigmoidoscopy. An unsedated colonoscopy is very similar to an extended flexible sigmoidoscopy.14-17,35 In patients who can tolerate a flexible sigmoidoscopy well, extended flexible sigmoidoscopy can reach the cecum >70% of the time.36 To enhance the cecal intubation rate among unsedated veterans, we developed (subsequent to the findings reported here) a novel method of water infusion in lieu of air insufflation during insertion of the colonoscope.37 This measure improved the cecal intubation rate from 76% to 97%.38 For family physicians who perform flexible sigmoidoscopy, it is worth considering performing extended flexible sigmoidoscopy or unsedated colonoscopy using this water infusion method37,38 or other methods to minimize discomfort in unsedated patients.39
Limitations of our study, and opportunities. Our report is based on uncontrolled, nonrandomized observational data. Nevertheless, it affirms the feasibility of unsedated colonoscopy performed by supervised trainees, as previously reported by a family practice training program.16 It also underscores the benefits of the unsedated option on direct and indirect costs.
Since only 3.7% of family physicians in a recent survey reported performing colonoscopy,25 it is uncertain whether primary care providers would voluntarily inform patients about the unsedated option. In select settings, gastroenterologists are willing to provide unsedated colonoscopy.6,7,12,13,18 A reasonable hypothesis to test is that primary care providers informing patients about unsedated colonoscopy could reshape the future practice of screening colonoscopy in family medicine and gastroenterology.
Funding/Support
Supported in part by vA medical research Funds and an American Society for Gastrointestinal endoscopy career Development Award (FWl 1985).
Correspondence
Felix W. Leung, MD, Division of Gastroenterology (111G), Sepulveda Ambulatory Care Center, Veterans Affairs Greater los Angeles Healthcare System, 16111 Plummer Street, North Hills, CA 91343; .
- Consider recommending unsedated colonoscopy to patients who have issues with cost, concerns about sedation, or are unable to get an escort or avoid work following the procedure.
- Explore resources in your area that offer unsedated colonoscopy.
Background Access to potentially life-saving screening colonoscopy is limited by the high cost of sedation. We explored the practicability of having supervised trainees perform unsedated colonoscopies.
Method A nursing shortage at our Veterans Administration gastroenterology training program necessitated discontinuing sedated colonoscopy. We offered the procedure without sedation to restore local access to screening colonoscopy.
Results From September 2002 to June 2005, 145 of 483 patients accepted the unsedated option. The procedure was done by second-year gastroenterology (GI) fellows who had performed about 100 sedated colonoscopies in their first year of training. Cecal intubation was achieved in 81% of 138 wellpurged patients without obstructive lesions. Implementation obviated the need for 2 registered nurses, the escort requirement, and postprocedure activity restriction. It also eliminated sedation-related complications.
Conclusion This report confirms the feasibility of unsedated colonoscopy performed by supervised trainees. The unsedated option minimizes direct and indirect costs of colonoscopy. Describing unsedated screening colonoscopy to patients as a “sedation risk–free” procedure encouraged them to consider the benefits. We recommend that future studies test primary care providers’ willingness to inform patients of the feasibility of this nonstandard option, and perhaps reshape the practice of colonoscopy for colorectal cancer screening.
Monitored sedation given for colonoscopy is a measure meant to ensure patient safety,1 but its high cost limits access to the potentially life-saving screening procedure.2,3 Unsedated colonoscopy is an option, but a controversial one, raising issues both pro4-6 and con.7-11 In the United States, gastroenterologists perform unsedated colonoscopy both for unescorted patients (~2% of all screening colonos-copies)12,13 and individuals who simply prefer to avoid sedation (~6%).7 Family physicians, too, perform unsedated colonoscopy in both rural and urban settings.14-17
Would it be feasible to make training in unsedated colonoscopy more readily available to providers, and thereby reduce costs and inconvenience for patients? We took advantage of a changing environment at our Veterans Administration program to explore this question.
Method
A nursing shortage in our VA academic GI program necessitated discontinuing sedated colonoscopy. We reviewed the literature on unsedated colonoscopy and found that it is a feasible alternative performed elsewhere.4-7,12,14,16 Our attending staff discussed the options with patients and obtained informed consent18 using the following general message:
“Sedated colonoscopy is usual practice. Even though the risks of sedation are very small, nurses are required to monitor patients continuously. Because of a nursing shortage, we must send you to another VA facility 15 miles away for sedated colonoscopy. You must have an escort, as you will not be allowed to drive after sedation. One of the medicines they administer will make you forget the discomfort you may have experienced, as well as the discussions after the examination.
“Alternatively, you may choose unsedated colonoscopy, which is practiced in the United States and many other countries. Because no medicines are used, there are no medication-induced complications. An escort is not required, and there is no activity restriction afterwards.
“You will feel air in the colon and the endoscope being pushed around inside you. The colonoscopist will talk to you throughout the examination. When you begin to experience discomfort, the colonoscopist will remove air inside the colon or straighten the loops in the colonoscope to minimize the discomfort before it becomes severe. If discomfort does become severe, you and the colonoscopist can agree to stop the advancement of the colonoscope. Complications related to taking biopsies or removing polyps are similar in sedated and unsedated procedures.”
Supervised trainees (second-year GI fellows who had performed about 100 sedated colonoscopies in their first year of training), assisted by a licensed vocational nurse, performed the procedures using appropriate techniques.5-7
The Institution Review Board of the Veterans Affairs Greater Los Angeles Healthcare System (VAGLAHS) approved our review of the patient data for publication.
Results
From September 2002 to June 2005, 145 of 483 patients accepted the unsedated option. The number of patients choosing this option increased in each successive academic year (31, 50, and 64), as did the wait-time in days (27±4, 46±5, 72±6) (mean±standard error of the mean [SEM]). Seven patients (4%) had poor bowel preparation or obstructing lesions limiting completion. Among the 138 well-purged patients, we achieved cecal intubation in 112 (81%). Discomfort limited completion in the remaining 19%.
Other than transient vasovagal reactions in 2 patients, no complications occurred. Patients with incomplete examinations due to discomfort underwent sedated colonoscopy or barium enema or received no further assessment, depending on the initial findings. Those who subsequently underwent sedated colonoscopy (10%) had to be purged again, escorted, and comply with activity restriction.
Colonoscopy was developed as an unsedated procedure.19,20 Discomfort experienced by some patients during sigmoid intubation led to the use of medications,20,21 which are now administered routinely.22 Interestingly, competency in unsedated colonoscopy is not required of GI fellows.23 Indeed, until recently,18 teaching GI trainees unsedated colonoscopy was deemed impractical.7 Family physicians, on the other hand, have long practiced and taught unsedated colonoscopy,14-17,24 although the actual number of family physicians performing colonoscopy is fairly small.25 One reason so few family physicians offer the service—estimated at 3.7% of the specialty—may be the intensive and costly education required.16 Even more difficult has been gaining privileges to perform the sedated procedure,26 given the training requirements set forth by GI professional societies.27
Many patients would opt for unsedated colonoscopy. The favorable reception of unsedated colonoscopy in our study is evident in the increasing number of veterans each year who opted for the procedure despite the lengthening wait-time (which was due to increased demand rather than decreased availability of endoscopists). In the course of our project, we found that the terms “unsedated,” “no sedation,” or “without sedation” tended to convey the negative connotation that relief of discomfort and induced amnesia are withheld.8,9 The term “sedation risk–free”28 emphasized the benefits of no sedation.
Cost factors favoring unsedated colonoscopy. Sedated colonoscopies performed by family physicians have offered substantial health care savings.29,30 It is intuitively obvious that the unsedated option in the hands of those with the necessary skills14-16,24 would be even less costly. Our unsedated colonoscopy project reduced direct costs, which included the cost of having 2 registered nurses on hand. Indirect costs to patients31 were also minimized by avoiding the need for an escort or activity restriction. Moreover, there were no sedation-related complications.32,33
An estimated 40 million healthy Americans are eligible for colorectal cancer screening.34 Primary care providers play a pivotal role in counseling many of these patients, who may find the indirect cost savings of unsedated colonoscopy performed by that same provider appealing.
A logical transition from flexible sigmoidoscopy. An unsedated colonoscopy is very similar to an extended flexible sigmoidoscopy.14-17,35 In patients who can tolerate a flexible sigmoidoscopy well, extended flexible sigmoidoscopy can reach the cecum >70% of the time.36 To enhance the cecal intubation rate among unsedated veterans, we developed (subsequent to the findings reported here) a novel method of water infusion in lieu of air insufflation during insertion of the colonoscope.37 This measure improved the cecal intubation rate from 76% to 97%.38 For family physicians who perform flexible sigmoidoscopy, it is worth considering performing extended flexible sigmoidoscopy or unsedated colonoscopy using this water infusion method37,38 or other methods to minimize discomfort in unsedated patients.39
Limitations of our study, and opportunities. Our report is based on uncontrolled, nonrandomized observational data. Nevertheless, it affirms the feasibility of unsedated colonoscopy performed by supervised trainees, as previously reported by a family practice training program.16 It also underscores the benefits of the unsedated option on direct and indirect costs.
Since only 3.7% of family physicians in a recent survey reported performing colonoscopy,25 it is uncertain whether primary care providers would voluntarily inform patients about the unsedated option. In select settings, gastroenterologists are willing to provide unsedated colonoscopy.6,7,12,13,18 A reasonable hypothesis to test is that primary care providers informing patients about unsedated colonoscopy could reshape the future practice of screening colonoscopy in family medicine and gastroenterology.
Funding/Support
Supported in part by vA medical research Funds and an American Society for Gastrointestinal endoscopy career Development Award (FWl 1985).
Correspondence
Felix W. Leung, MD, Division of Gastroenterology (111G), Sepulveda Ambulatory Care Center, Veterans Affairs Greater los Angeles Healthcare System, 16111 Plummer Street, North Hills, CA 91343; .
1. Comprehensive Accreditation Manual for Hospitals; The Official Handbook. Oakbrook Terrace, III: Joint Commission on Accreditation of Healthcare Organizations; 2005:160-223.
2. Lieberman DA, Weiss DG, Bond JH, et al. Use of colonoscopy to screen asymptomatic adults for colorectal cancer. N Engl J Med. 2000;343:162-168.
3. El-Serag HB, Petersen L, Hampel H, et al. The use of screening colonoscopy for patients cared for by the Department of Veterans Affairs. Arch Intern Med. 2006;166:2202-2208.
4. Herman FN. Avoidance of sedation during total colonoscopy. Dis Colon Rectum. 1990;33:70-72.
5. Cataldo PA. Colonoscopy without sedation: a viable alternative. Dis Colon Rectum. 1996;39:257-261.
6. Hoffman MS, Butler TW, Shaver T. Colonoscopy without sedation. J Clin Gastroenterol. 1998;26:279-282.
7. Rex DK, Imperiale TF, Portish V. Patients willing to try colonoscopy without sedation: associated clinical factors and results of a randomized controlled trial. Gastrointest Endosc. 1999;49:554-559.
8. Levenson D. Health quality organization criticizes colonoscopies given without pain medication. Rept Med Guidelines Outcomes Res. 2001;12:9-12.
9. Leo RA. Unsedated endoscopy: you don’t get a medal for it! South Med J. 2004;97:797-798.
10. Faulx AL, Vela S, Das A, et al. The changing landscape of practice patterns regarding unsedated endoscopy and propofol use: a national Web survey. Gastrointest Endosc. 2005;62:9-15.
11. Rex DK, Khalfan HK. Sedation and the technical performance of colonoscopy. Gastrointest Endosc Clin N Am. 2005;15:661-672.
12. Nelson DB, MCQuaid KR, Bond JH, et al. Procedural success and complications of large-scale screening colonoscopy. Gastrointest Endosc. 2002;55:307-314.
13. Aslinia F, Uradomo L, Steele A, et al. Quality assessment of colonoscopic cecal intubation: an analysis of 6 years of continuous practice at a University Hospital. Am J Gastroenterol. 2006;101:721-731.
14. Hopper W, Kyker KA, Rodney WM. Colonoscopy by a family physician: a 9-year experience of 1048 procedures. J Fam Pract. 1996;43:561 -566.
15. Carr KW, Worthington JM, Rodney WM, et al. Advancing from flexible sigmoidoscopy to colonoscopy in rural family practice. Tenn Med. 1998;91:21-26.
16. Rodney WM, Dabov G, Orientale E, et al. Sedation associated with a more complete colonoscopy. J Fam Pract. 1993;36:394-400.
17. Knox L, Hahn RG, Lane C. A comparison of unsedated colonoscopy and flexible sigmoidoscopy in the family medicine setting: an LA Net study. J Am Board Fam Med. 2007;20:444-450.
18. Leung FW, Aharonian HS, Guth PH, et al. Involvement of trainees in routine unsedated colonoscopy—review of pilot experience. Gastrointest Endosc. 2008;67:718-722.
19. Wolff WI, Shinya H. Colonofiberoscopy. JAMA. 1971;217:1509-1512.
20. Williams C, Teague RH. Colonoscopy. Gut. 1973;14:990-1003.
21. Waye JD. Colonoscopy. Surg Clin North Am. 1972;52:1013-1024.
22. National Cancer Institute Web Site. Available at: http://www.cancer.gov/cancertopics/types/colon-and-rectal. Accessed January 23, 2008.
23. Gastroenterology required curriculum. Chicago, III: Accreditation council on Graduate medical education; 2005.
24. Edwards JK, Norris TE. Colonoscopy in rural communities: can family physicians perform the procedure with safe and efficacious results? J Am Board Fam Pract. 2004;17:353-358.
25. Wilkins T, Wagner P, Thomas A, et al. Attitudes toward performance of endoscopic colon cancer screening by family physicians. Fam Med. 2007;39:578-584.
26. Frank M. American Academy of Family Physicians response to American college of Gastroenterology’s letter and legal opinion. Sent to hospital administrators on December 12, 2005.
27. Faigel D, Baron T, Lewis B, et al. Ensuring competence in endoscopy. Prepared by the ASGe Taskforce on ensuring competence in endoscopy and American college of Gastroenterology executive and Practice management committees, 2005: 1-36.
28. Leung FW. Should minimization of cue be an option as a quality indicator in colonoscopy performed for colorectal cancer screening? Gastroin-test Endosc. 2008;67:579-580.
29. Short MW, Kelly KM, Runser lA. Colonoscopy by a family physician: a case series demonstrating health care savings. Mil Med. 2007;172:1089-1092.
30. Perry RE, Christensen JB, Christensen MA, et al. Office colonoscopy—a safe procedure in selected patients. Dis Colon Rectum. 1989;32:1031-1033.
31. Glied S. Estimating the indirect cost of illness: an assessment of the forgone earnings approach. Am J Public Health. 1996;86:1723-1728.
32. Arrowsmith JB, Gerstman BB, Fleischer DE, et al. Results from the American Society for Gastrointestinal endoscopy/uS Food and Drug Administration collaborative study on complication rates and drug use during gastrointestinal endoscopy. Gastroin-test Endosc. 1991;37:421-427.
33. Sharma VK, Nguyen CC, Crowell MD, et al. A national study of cardiopulmonary unplanned events after GI endoscopy. Gastrointest Endosc. 2007;66:27-34.
34. Seeff LC, Manninen DL, Dong FB, et al. Is there endoscopic capacity to provide colorectal cancer screening to the unscreened population in the united States? Gastroenterology. 2004;127:1661-1669.
35. Dervin JV. Feasibility of 105-cm flexible sigmoidoscopy in family practice. J Fam Pract. 1986;23:341-344.
36. Lee JG, Lum D, Urayama S, et al. Unsedated extended flexible sigmoidoscopy for colorectal cancer screening: a pilot study. Aliment Pharmacol Ther. 2006;23:945-951.
37. Leung JW, Mann S, Leung FW. Option of screening colonoscopy without sedation—a pilot study in united States veterans. Aliment Pharmacol Ther. 2007;26:627-631.
38. Leung FW, Aharonian HS, Leung JW, et al. Impact of a novel water method on scheduled unsedated colonoscopy in U.S. veterans. Gastrointest Endosc. In press.
39. Leung FW. Methods of reducing discomfort during colonoscopy. Dig Dis Sci. 2008;53:1462-1467.
1. Comprehensive Accreditation Manual for Hospitals; The Official Handbook. Oakbrook Terrace, III: Joint Commission on Accreditation of Healthcare Organizations; 2005:160-223.
2. Lieberman DA, Weiss DG, Bond JH, et al. Use of colonoscopy to screen asymptomatic adults for colorectal cancer. N Engl J Med. 2000;343:162-168.
3. El-Serag HB, Petersen L, Hampel H, et al. The use of screening colonoscopy for patients cared for by the Department of Veterans Affairs. Arch Intern Med. 2006;166:2202-2208.
4. Herman FN. Avoidance of sedation during total colonoscopy. Dis Colon Rectum. 1990;33:70-72.
5. Cataldo PA. Colonoscopy without sedation: a viable alternative. Dis Colon Rectum. 1996;39:257-261.
6. Hoffman MS, Butler TW, Shaver T. Colonoscopy without sedation. J Clin Gastroenterol. 1998;26:279-282.
7. Rex DK, Imperiale TF, Portish V. Patients willing to try colonoscopy without sedation: associated clinical factors and results of a randomized controlled trial. Gastrointest Endosc. 1999;49:554-559.
8. Levenson D. Health quality organization criticizes colonoscopies given without pain medication. Rept Med Guidelines Outcomes Res. 2001;12:9-12.
9. Leo RA. Unsedated endoscopy: you don’t get a medal for it! South Med J. 2004;97:797-798.
10. Faulx AL, Vela S, Das A, et al. The changing landscape of practice patterns regarding unsedated endoscopy and propofol use: a national Web survey. Gastrointest Endosc. 2005;62:9-15.
11. Rex DK, Khalfan HK. Sedation and the technical performance of colonoscopy. Gastrointest Endosc Clin N Am. 2005;15:661-672.
12. Nelson DB, MCQuaid KR, Bond JH, et al. Procedural success and complications of large-scale screening colonoscopy. Gastrointest Endosc. 2002;55:307-314.
13. Aslinia F, Uradomo L, Steele A, et al. Quality assessment of colonoscopic cecal intubation: an analysis of 6 years of continuous practice at a University Hospital. Am J Gastroenterol. 2006;101:721-731.
14. Hopper W, Kyker KA, Rodney WM. Colonoscopy by a family physician: a 9-year experience of 1048 procedures. J Fam Pract. 1996;43:561 -566.
15. Carr KW, Worthington JM, Rodney WM, et al. Advancing from flexible sigmoidoscopy to colonoscopy in rural family practice. Tenn Med. 1998;91:21-26.
16. Rodney WM, Dabov G, Orientale E, et al. Sedation associated with a more complete colonoscopy. J Fam Pract. 1993;36:394-400.
17. Knox L, Hahn RG, Lane C. A comparison of unsedated colonoscopy and flexible sigmoidoscopy in the family medicine setting: an LA Net study. J Am Board Fam Med. 2007;20:444-450.
18. Leung FW, Aharonian HS, Guth PH, et al. Involvement of trainees in routine unsedated colonoscopy—review of pilot experience. Gastrointest Endosc. 2008;67:718-722.
19. Wolff WI, Shinya H. Colonofiberoscopy. JAMA. 1971;217:1509-1512.
20. Williams C, Teague RH. Colonoscopy. Gut. 1973;14:990-1003.
21. Waye JD. Colonoscopy. Surg Clin North Am. 1972;52:1013-1024.
22. National Cancer Institute Web Site. Available at: http://www.cancer.gov/cancertopics/types/colon-and-rectal. Accessed January 23, 2008.
23. Gastroenterology required curriculum. Chicago, III: Accreditation council on Graduate medical education; 2005.
24. Edwards JK, Norris TE. Colonoscopy in rural communities: can family physicians perform the procedure with safe and efficacious results? J Am Board Fam Pract. 2004;17:353-358.
25. Wilkins T, Wagner P, Thomas A, et al. Attitudes toward performance of endoscopic colon cancer screening by family physicians. Fam Med. 2007;39:578-584.
26. Frank M. American Academy of Family Physicians response to American college of Gastroenterology’s letter and legal opinion. Sent to hospital administrators on December 12, 2005.
27. Faigel D, Baron T, Lewis B, et al. Ensuring competence in endoscopy. Prepared by the ASGe Taskforce on ensuring competence in endoscopy and American college of Gastroenterology executive and Practice management committees, 2005: 1-36.
28. Leung FW. Should minimization of cue be an option as a quality indicator in colonoscopy performed for colorectal cancer screening? Gastroin-test Endosc. 2008;67:579-580.
29. Short MW, Kelly KM, Runser lA. Colonoscopy by a family physician: a case series demonstrating health care savings. Mil Med. 2007;172:1089-1092.
30. Perry RE, Christensen JB, Christensen MA, et al. Office colonoscopy—a safe procedure in selected patients. Dis Colon Rectum. 1989;32:1031-1033.
31. Glied S. Estimating the indirect cost of illness: an assessment of the forgone earnings approach. Am J Public Health. 1996;86:1723-1728.
32. Arrowsmith JB, Gerstman BB, Fleischer DE, et al. Results from the American Society for Gastrointestinal endoscopy/uS Food and Drug Administration collaborative study on complication rates and drug use during gastrointestinal endoscopy. Gastroin-test Endosc. 1991;37:421-427.
33. Sharma VK, Nguyen CC, Crowell MD, et al. A national study of cardiopulmonary unplanned events after GI endoscopy. Gastrointest Endosc. 2007;66:27-34.
34. Seeff LC, Manninen DL, Dong FB, et al. Is there endoscopic capacity to provide colorectal cancer screening to the unscreened population in the united States? Gastroenterology. 2004;127:1661-1669.
35. Dervin JV. Feasibility of 105-cm flexible sigmoidoscopy in family practice. J Fam Pract. 1986;23:341-344.
36. Lee JG, Lum D, Urayama S, et al. Unsedated extended flexible sigmoidoscopy for colorectal cancer screening: a pilot study. Aliment Pharmacol Ther. 2006;23:945-951.
37. Leung JW, Mann S, Leung FW. Option of screening colonoscopy without sedation—a pilot study in united States veterans. Aliment Pharmacol Ther. 2007;26:627-631.
38. Leung FW, Aharonian HS, Leung JW, et al. Impact of a novel water method on scheduled unsedated colonoscopy in U.S. veterans. Gastrointest Endosc. In press.
39. Leung FW. Methods of reducing discomfort during colonoscopy. Dig Dis Sci. 2008;53:1462-1467.
Driving with dementia: How to assess safety behind the wheel
Mr. D, age 75, presents to your office with a 5-year history of gradually declining memory. His wife reports he is having difficulty with word finding, managing his finances, and shopping, and he needs supervision when using the stove. Nonetheless, he enjoys playing golf and drives himself to the golf course 3 times a week. He is compliant with his chronic medical therapy for hypertension, hypercholesterolemia, and asthma.
Patients with dementia who continue to drive pose a potential danger on the road, worry their families, and present challenges to clinicians. Most people would agree that patients with moderate or severe dementia should not drive, but a careful evaluation is required to assess whether a patient such as Mr. D with mild dementia remains fit to drive.
This article explores how dementia exacerbates age-related changes in driving ability and discusses how to assess driving in patients with dementia. Our goal is to help clinicians sort through data from in-office physical and cognitive assessments, family caregivers/informants’ reports, and (when available) on-road testing. We also discuss:
- guidelines for assessing older drivers that can help balance patients’ need for autonomy with public safety
- strategies for discussing driving cessation with patients and their families.
Driving: A privilege, not a right
Driving is central to older adults’ autonomy, and >75% of persons age ≥75 rely on driving as their primary mode of transportation.1 Driving cessation in this population has been associated with a 3-fold decrease in out-of-home activity2 and a 2.5-fold increase in depressive symptoms.3 Nonetheless, some 4.5 million Americans have Alzheimer’s disease (AD),4 and dementia poses a substantial risk to safe driving.
Although driving must be sacrificed when personal and public safety is at risk, most physicians perceive an uncomfortable conflict of interest between patient confidentiality and public safety.5 Assessing driving safety of patients with dementia can undermine the doctor-patient relationship and pose hardships for patients.
Mr. D has a 5-year history of memory problems that affect his daily functioning, yet he continues to drive. A longitudinal study of persons with dementia found that among the 29% who were driving at baseline, more than one-half were still behind the wheel 2 years later.6
Age and driving safety. Even in the absence of dementia, driving ability declines with aging (Tables 1 and 2).7,8 Older persons may self-regulate and restrict their driving to shorter distances, with fewer trips at night, on high-speed roads, or in unfamiliar situations. Their driving is rarely aggressive and they are unlikely to speed, but they may drive more slowly than other traffic.7,8 Although the overall rate of motor vehicle collisions declines with age:
- the rate of collisions per mile driven increases after age 659
- drivers age >65 have the highest fatality rate per mile driven among adults age ≥25.10
A dementia diagnosis is not sufficient to withdraw driving privileges, according to American Medical Association (AMA)/National Highway Traffic Safety Administration (NHTSA) guidelines. These recommend that you base decisions on the individual’s driving ability, and—when you have concerns—factor in a focused medical assessment and formal assessment of driving skills.10
Table 1
Age-related changes that may affect driving fitness
| Decreased physical capabilities, including declining muscle tone, flexibility, and reaction time |
| Decreased hearing and visual acuity |
| Increased fragility, resulting in longer time to heal should injuries occur |
| Increased medication use with possible side effect of drowsiness |
| Source: References 7,8 |
Table 2
Older drivers’ common traffic violations leading to crashes*
| Failure to obey traffic signals, including stop signs and red lights |
| Unsafe left turns (driver may inaccurately judge speed of oncoming vehicle) |
| Inappropriate turns (such as difficulty judging distance from oncoming cars, wide or narrow turns, or not timing the turn correctly with traffic lights) |
| Unsafe passing |
| Failure to yield |
| * These errors often lead to multivehicle accidents Source: References 7,8 |
CASE CONTINUED: Cognitive deficits quantified
You perform a Mini-Mental State Examination (MMSE). Mr. D scores 24/30, losing 1 point for orientation, 2 points for attention, 2 points for recall, and 1 point for copying. This score, along with his history, indicates mild dementia, although he claims he is a safe driver. On further cognitive testing, Mr. D completes the Trails A test in 90 seconds and Trails B test in 250 seconds (well below 1.5 standard deviations of the norm for his age and education).11 On the clock-drawing task, he drew a poorly organized clock, with unequal spaces between numbers and hands pointing to “10” and “11” instead of properly indicating “10 after 11.”
Mr. D and his wife live in a rural area, 5 miles from the nearest grocery store. His wife never drove, and she relies on him for weekly shopping trips and to drive her to her bridge club. She denies any problems with his driving but states, “Other drivers have become so aggressive; they’re always honking at him.” Their daughter denies that Mr. D has driving problems but admits that for the last 2 years she has refused to allow her child to ride in his car.
Focused in-office assessment
Information to assess driving ability can come from the patient, family caregiver/informant, and clinical judgment. Patients with dementia are notoriously inaccurate in self-reported driving ability, either for lack of insight or as a testament to the importance of driving to their autonomy. Caregivers often are more accurate in describing a patient’s driving, but other agendas may color their responses.
In a study of patients with very mild or mild AD, 94% reported themselves as safe drivers, whereas on-road driving instructors rated <50% of drivers in these groups as safe. Caregivers were better able to classify driving performance, but 36% of their ratings were incorrect.12
Cognitive assessment. To assess older drivers’ cognition, AMA/NHTSA’s Guide to Assessing and Counseling Older Drivers recommends the Trail-Making Test, Part B and the clock-drawing test.10 The Canadian Medical Association suggests the MMSE.13 Both guides say that abnormalities in these tests indicate a need for more detailed testing, including referral to specialized driving assessment and retesting at regular intervals (Algorithm). Retest patients with mild dementia at least every 6 months or sooner when dementia severity increases noticeably14 (Box 1).6,15
The MMSE is widely used to screen for cognitive impairment and identify dementia or delirium, but it is not a diagnostic tool or proxy driving test. A patient with dementia may produce a high MMSE score and yet be an unsafe driver. For example, well-educated patients or those with vascular or frontotemporal dementia may retain cognitive abilities as measured by the MMSE until later in the disease.
Considerable effort has been put into developing tools to help clinicians quickly and accurately differentiate safe from unsafe drivers by assessing cognition. Unfortunately, no consistent link has been found between cognitive test results and driving outcome measures. A systematic review of office-based predictors of fitness to drive in dementia found 5 studies showing an association between MMSE scores and driving and 5 studies showing no such association.16 Thus, although the AMA/NHTSA guide recommends the MMSE, Trails B, and clock-drawing tests, cognitive tests—including these—are not sufficient to assess driving ability.
Severity of dementia. International consensus groups have attempted to create guidelines for patients with dementia who drive. American, Canadian, and Australian groups suggest that a diagnosis of moderate to severe dementia precludes driving, and the driver’s licenses of persons with these conditions should be revoked.17
In general, AD is considered severe when the MMSE score is <10 or the patient becomes dependent on a caregiver for survival.18 AD of moderate severity is more difficult to define, but a Canadian consensus conference suggested a practical approach: Patients with AD would be considered to have moderate to severe dementia and should not drive when they cannot independently perform multiple instrumental activities of daily living or any of the basic activities of daily living.19
Some dementias may impair driving more quickly than AD does. For example, hallucinations may occur early in Lewy body dementia, as may impulsivity in frontotemporal dementia and motor impairment in vascular dementia.
Mrs. Y visits your office for a follow-up regarding mild Alzheimer’s disease (AD), which was diagnosed 2 years ago. She passed an on-road test 3 months ago and has an Mini-Mental State Examination score of 24/30. Over the last month she has become depressed, with insomnia and mild psychomotor retardation. She occasionally has hallucinations.
Behavioral and psychological symptoms such as agitation, aggression, hallucinations, apathy, depression, and anxiety are common neuropsychiatric sequelae of AD. Little is known about the risks these symptoms pose to road safety, but we recommend that clinicians strongly consider the potential for impaired driving.
In a longitudinal study, cognitive impairment and behavioral disturbances—especially agitation, apathy, and hallucinations—were strong predictors of driving cessation among patients with dementia.6 Furthermore, a case crossover study of patients with dementia found a 54% increase in risk of motor vehicle collisions associated with the use of psychotropic medications.15
Consider all aspects of the patient’s clinical status, including neuropsychiatric symptoms, psychotropic medications, comorbid medical conditions (including hearing and vision impairment), and concomitant therapy for medical conditions. Any could change a safe driver with mild dementia into an unsafe driver.
Algorithm: 3 options for drivers with dementia, based on in-office assessment
* Observe legislation or statutes that address reporting unsafe drivers to the department of motor vehicles or ministry of transportation
On-road driving tests
Because some individuals with mild dementia can drive safely for extended periods, international recommendations for assessing the driver with dementia emphasize on-road driving tests.10,13,20–22 American10 and Canadian guidelines13 suggest that a dementia diagnosis is not sufficient to withdraw licensure.
A formal driving assessment is necessary to establish road safety for patients with mild dementia except when the need for license withdrawal is evident, such as when the patient has:
- a history of major driving problems (such as crashes or driving the wrong way on a highway)
- significant contraindications to driving on the history or physical examination (such as severe inattention or psychosis).
Challenges of on-road testing. On-road tests may be the gold standard, but they are not without clinical problems.
Need to retest. Because almost all dementias are progressive and driving skills deteriorate over time, most guidelines recommend periodic retesting. For patients with dementia who pass on-road evaluations, limited evidence supports retesting every 6 months.14 Take an individual approach, however, because of the various rates at which the dementias progress.
Testing vs real world conditions. Structured on-road testing is not equivalent to unstructured real-world driving, in which the patient often must navigate without instruction or assistance.
Rural vs urban driving. Road tests conducted in urban areas assess skills associated with complex conditions and the need to respond quickly to crises. They might not assess as well rural driving, which requires sustained attention on monotonous roads.
Inaccessibility. Cost and lack of availability of on-road tests, particularly in rural areas, limit the number of patients whose performance can be evaluated.
CASE CONTINUED: Distressing results
Mr. D has a history of decline in cognition and function, objective cognitive difficulties, and a subtle history of driving problems. You refer him for a specialized on-road test, and the report indicates that he failed. Errors included wide turns, driving too slowly, getting caught in an intersection twice during red lights while attempting to turn left, driving on the shoulder, and failing to signal lane changes. You review the results with Mr. D and his wife and recommend that he cease driving immediately.
Mr. D is furious, and his wife is dismayed. He demands to know how he can continue to play golf, which is his only form of exercise and recreation. Will she have to give up her bridge club? How will they shop for food? They request permission to at least to drive to the grocery store during the daytime.
You explain that no system allows individuals to drive only at certain times, and for the sake of safety you cannot grant them special permission. You discuss alternatives, such as asking their daughter for assistance with grocery shopping and taking taxis or ride-sharing with friends who play golf and bridge.
Remain firm, but ease the blow
Driving cessation orders distress patients, families, and clinicians. A failed road test clearly indicates unsafe driving, and driving cessation is critical to public safety.
A review by Man-Son-Hing et al23 found that drivers with dementia performed worse than nondemented controls in all studies that examined driving performance (on-road, simulator, or caregiver report). Simulators showed problems such as off-road driving, deviation from posted speed, and more time to negotiate left turns.24
By comparison, only 1 of 3 studies using state crash records showed an increased risk of collisions in persons with dementia compared with controls.23 From a research perspective, however, studies that use state-reported collisions to assess driving risk are confounded by driving restrictions on persons with dementia.
Mr. D wants to continue driving with restrictions. No studies have shown reduced crash rates when drivers with dementia used compensatory strategies such as restrictions, retraining/education, having a passenger “co-pilot,” on-board navigation, or cognitive enhancers.23
If Mr. D had passed the road test, the situation would have been more ambiguous. Two studies have examined on-road driving performance over time in patients with early-stage dementia.25,26 Both studies followed drivers prospectively for 2 years, and those with mild dementia (vs very mild or no dementia) were most likely to show a decline in driving skills:
- All participants with mild dementia were rated as “not safe” by the end of 2 years by Duchek et al.25
- Median time to “failure” (or a rating of unsafe) was 324 days for drivers with mild dementia vs 605 days for those with very mild dementia, as reported by Ott et al.26
Mr. D’s passionate plea for reconsideration highlights the need for communities to develop alternate transportation for seniors whose driving becomes unsafe (Box 2).
Legal liability? Physicians often are concerned about legal responsibilities and risks involved in reporting unsafe drivers. Be aware of local statutes or legislations regarding mandatory reporting of patients you deem unsafe to drive.17 These laws usually protect physicians from lawsuits related to violating patient confidentiality. Civil lawsuits remain possible, however, if clinicians fail to report an unsafe driver who subsequently is involved in a motor vehicle collision.27
1. Meet with family first. Help them assume a positive and supportive role. Explain concretely and empathically your concern for the safety of the patient and others. Clearly outline your findings that the patient is not fit to drive, and explain that the law requires you to report the patient to the authorities.
Remind family members that the goal of driving assessment is to prevent a collision, and they carry some responsibility because they are aware of the potential risk of letting their family member continue to drive. If necessary, have family members witness a repeat performance by the patient on the most revealing test. Discuss the importance of finding alternate transportation to reduce the risk of isolation and depression that can follow driving cessation.
2. Meet with patient. Having the family present can be helpful, but ask them to assume a supportive role. Give the patient a positive role by recognizing that he or she has been a responsible driver, and part of this responsibility is to stop driving before an accident occurs. Acknowledge that it is normal to be unhappy upon learning that one’s driving privileges are being revoked.
Sometimes it helps to give the patient a prescription in their name that says, “Do not drive.” Families who receive a copy may find this very helpful, too, for reminding the patient later about what you said.
If your patient argues with your position, remain firm and do not argue. Indicate that you have made notes on the meeting and are notifying the authorities about the patient’s unsafe driving. You can add that your chart could be subpoenaed and the patient may be legally liable and financially responsible should he or she continue to drive and have a collision.
3. Talk about transportation options. Family members could share driving responsibilities. Taxi rides can cost less than maintaining a car if the patient drives <4,000 km (2,500 miles) per year. Suggest that patients or families find volunteer drivers or contact helpful taxi drivers a day before an outing is planned.
4. If patient refuses to comply, meet with the family again and encourage them to remove the patient’s opportunity to drive (confiscate the keys, disable the car, or remove the car altogether).
Provide a written statement to the patient and family outlining why the patient can no longer drive. Indicate that it is your legal responsibility to report unsafe drivers, and you intend to notify the authorities regarding the patient’s driving status. If the patient remains noncompliant, continue to encourage family to remove the opportunity to drive.
Related resources
- Physician’s guide to assessing and counseling older drivers. American Medical Association and the National Highway Traffic Safety Administration, 2003. www.nhtsa.dot.gov/people/injury/olddrive/OlderDriversBook.
- Determining medical fitness to operate motor vehicles. Canadian Medical Association. 2006. www.cma.ca/index.cfm/ci_id/18223/la_id/1.htm.
- Canadian Driving Research Initiative for Vehicular Safety in the Elderly (CanDRIVE). www.candrive.ca.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Rapoport receives grant/research support from the Canadian Institute of Health Research and the Ontario Neurotrauma Foundation.
1. Stowell-Ritter A, Straight A, Evans EL. Understanding senior transportation: report and analysis of a survey of consumers age 50+. Washington, DC: American Association of Retired Persons; 2002.
2. Marottoli RA, de Leon CFM, Glass TA, et al. Consequences of driving cessation: decreased out-of-home activity levels. J Gerontol B Psychol Sci Soc Sci 2000;55(6):S334-40.
3. Marottoli RA, Mendes de Leon CF, Glass TA, et al. Driving cessation and increased depressive symptoms: prospective evidence from the New Haven EPESE. Established Populations for Epidemiologic Studies of the Elderly. J Am Geriatr Soc 1997;45(2):202-6.
4. Hebert LE, Scherr PA, Bienias JL, et al. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 2003;60(8):1119-22.
5. Jang RW, Man-Son-Hing M, Molnar FJ, et al. Family physicians’ attitudes and practices regarding assessments of medical fitness to drive in older persons. J Gen Intern Med 2007;22(4):531-43.
6. Herrmann N, Rapoport MJ, Sambrook R, et al. Predictors of driving cessation in mild-to-moderate dementia. CMAJ 2006;175(6):591-5.
7. Hedlund J. Countermeasures that work: a highway safety countermeasure guide for state highway safety offices. Washington, DC: National Highway Traffic Safety Administration; 2006.
8. Dobbs B. Medical conditions and driving: a review of the literature (1960-2000). Washington, DC: National Highway Traffic Safety Administration; 2005. Available at: http://www.nhtsa.dot.gov/people/injury/research/Medical_Condition_Driving/pages/TRD.html. Accessed September 29, 2008.
9. Li G, Braver ER, Chen LH. Fragility versus excessive crash involvement as determinants of high death rates per vehicle-mile of travel among older drivers. Accid Anal Prev 2003;35(2):227-35.
10. Physician’s guide to assessing and counseling older drivers. Washington, DC: National Highway Traffic Safety Administration; 2003. Available at: http://www.nhtsa.dot.gov/people/injury/olddrive/OlderDriversBook. Accessed September 29, 2008.
11. Yeudall LT, Reddon JR, Gill DM, et al. Normative data for the Halstead-Reitan neuropsychological tests stratified by age and sex. J Clin Psychol 1987;43(3):346-67.
12. Brown LB, Ott BR, Papandonatos GD, et al. Prediction of on-road driving performance in patients with early Alzheimer’s disease. J Am Geriatr Soc 2005;53(1):94-8.
13. Determining medical fitness to operate motor vehicles. CMA driver’s guide. 7th ed. Ottawa, Ontario, Canada: Canadian Medical Association; 2006.
14. Molnar FJ, Patel A, Marshall SC, et al. Systematic review of the optimal frequency of follow-up in persons with mild dementia who continue to drive. Alzheimer Dis Assoc Disord 2006;20(4):295-7.
15. Rapoport MJ, Herrmann N, Molnar FJ, et al. Psychotropic medications and motor vehicle collisions in patients with dementia (letter). J Am Geriatr Soc 2008;56(10):1968-70.
16. Molnar FJ, Patel A, Marshall SC, et al. Clinical utility of office-based cognitive predictors of fitness to drive in persons with dementia: a systematic review. J Am Geriatr Soc 2006;54(12):1809-24.
17. Rapoport MJ, Herrmann N, Molnar FJ, et al. Sharing the responsibility for assessing the risk of the driver with dementia. CMAJ. 2007;177(6):599-601.
18. Herrmann N, Gauthier S, Lysy PG. Clinical practice guidelines for severe Alzheimer’s disease. Alzheimers Dement 2007;3(4):385-97.
19. Hogan DB, Bailey P, Carswell A, et al. Management of mild to moderate Alzheimer’s disease and dementia. Alzheimers Dement 2007;3(4):355-84.
19. Assessing fitness to drive for commercial and private vehicle drivers. Sydney, Australia: National Library of Australia; 2006. Available at: http://www.austroads.com.au/aftd/index.html. Accessed November 4, 2008.
21. Medical aspects of fitness to drive. A guide for medical practitioners. Wellington, New Zealand: Land Transport Safety Authority; 2002. Available at: http://www.transfund.govt.nz/licensing/docs/ltsa-medical-aspects.pdf. Accessed September 29, 2008.
22. At a glance guide to the current medical standards of fitness to drive. Swansea, UK: Drivers Medical Group, Driver and Vehicle Licensing Agency; 2008. Available at: http://www.dvla.gov.uk/medical/ataglance.aspx. Accessed September 29, 2008.
23. Man-Son-Hing M, Marshall SC, Molnar FJ, Wilson KG. Systematic review of driving risk and the efficacy of compensatory strategies in persons with dementia. J Am Geriatr Soc 2007;55(6):878-84.
24. Cox DJ, Quillian WC, Thorndike FP, et al. Evaluating driving performance of outpatients with Alzheimer disease. J Am Board Fam Pract 1998;11(4):264-71.
25. Duchek JM, Carr DB, Hunt L, et al. Longitudinal driving performance in early-stage dementia of the Alzheimer type. J Am Geriatr Soc 2003;51(10):1342-7.
26. Ott BR, Heindel WC, Papandonatos GD, et al. A longitudinal study of drivers with Alzheimer disease. Neurology 2008;70(14):1171-8.
27. Molnar FJ, Byszewski AM, Marshall SC, Man-Son-Hing M. In-office evaluation of medical fitness to drive: practical approaches for assessing older people. Can Fam Physician 2005;51:372-9.
Mr. D, age 75, presents to your office with a 5-year history of gradually declining memory. His wife reports he is having difficulty with word finding, managing his finances, and shopping, and he needs supervision when using the stove. Nonetheless, he enjoys playing golf and drives himself to the golf course 3 times a week. He is compliant with his chronic medical therapy for hypertension, hypercholesterolemia, and asthma.
Patients with dementia who continue to drive pose a potential danger on the road, worry their families, and present challenges to clinicians. Most people would agree that patients with moderate or severe dementia should not drive, but a careful evaluation is required to assess whether a patient such as Mr. D with mild dementia remains fit to drive.
This article explores how dementia exacerbates age-related changes in driving ability and discusses how to assess driving in patients with dementia. Our goal is to help clinicians sort through data from in-office physical and cognitive assessments, family caregivers/informants’ reports, and (when available) on-road testing. We also discuss:
- guidelines for assessing older drivers that can help balance patients’ need for autonomy with public safety
- strategies for discussing driving cessation with patients and their families.
Driving: A privilege, not a right
Driving is central to older adults’ autonomy, and >75% of persons age ≥75 rely on driving as their primary mode of transportation.1 Driving cessation in this population has been associated with a 3-fold decrease in out-of-home activity2 and a 2.5-fold increase in depressive symptoms.3 Nonetheless, some 4.5 million Americans have Alzheimer’s disease (AD),4 and dementia poses a substantial risk to safe driving.
Although driving must be sacrificed when personal and public safety is at risk, most physicians perceive an uncomfortable conflict of interest between patient confidentiality and public safety.5 Assessing driving safety of patients with dementia can undermine the doctor-patient relationship and pose hardships for patients.
Mr. D has a 5-year history of memory problems that affect his daily functioning, yet he continues to drive. A longitudinal study of persons with dementia found that among the 29% who were driving at baseline, more than one-half were still behind the wheel 2 years later.6
Age and driving safety. Even in the absence of dementia, driving ability declines with aging (Tables 1 and 2).7,8 Older persons may self-regulate and restrict their driving to shorter distances, with fewer trips at night, on high-speed roads, or in unfamiliar situations. Their driving is rarely aggressive and they are unlikely to speed, but they may drive more slowly than other traffic.7,8 Although the overall rate of motor vehicle collisions declines with age:
- the rate of collisions per mile driven increases after age 659
- drivers age >65 have the highest fatality rate per mile driven among adults age ≥25.10
A dementia diagnosis is not sufficient to withdraw driving privileges, according to American Medical Association (AMA)/National Highway Traffic Safety Administration (NHTSA) guidelines. These recommend that you base decisions on the individual’s driving ability, and—when you have concerns—factor in a focused medical assessment and formal assessment of driving skills.10
Table 1
Age-related changes that may affect driving fitness
| Decreased physical capabilities, including declining muscle tone, flexibility, and reaction time |
| Decreased hearing and visual acuity |
| Increased fragility, resulting in longer time to heal should injuries occur |
| Increased medication use with possible side effect of drowsiness |
| Source: References 7,8 |
Table 2
Older drivers’ common traffic violations leading to crashes*
| Failure to obey traffic signals, including stop signs and red lights |
| Unsafe left turns (driver may inaccurately judge speed of oncoming vehicle) |
| Inappropriate turns (such as difficulty judging distance from oncoming cars, wide or narrow turns, or not timing the turn correctly with traffic lights) |
| Unsafe passing |
| Failure to yield |
| * These errors often lead to multivehicle accidents Source: References 7,8 |
CASE CONTINUED: Cognitive deficits quantified
You perform a Mini-Mental State Examination (MMSE). Mr. D scores 24/30, losing 1 point for orientation, 2 points for attention, 2 points for recall, and 1 point for copying. This score, along with his history, indicates mild dementia, although he claims he is a safe driver. On further cognitive testing, Mr. D completes the Trails A test in 90 seconds and Trails B test in 250 seconds (well below 1.5 standard deviations of the norm for his age and education).11 On the clock-drawing task, he drew a poorly organized clock, with unequal spaces between numbers and hands pointing to “10” and “11” instead of properly indicating “10 after 11.”
Mr. D and his wife live in a rural area, 5 miles from the nearest grocery store. His wife never drove, and she relies on him for weekly shopping trips and to drive her to her bridge club. She denies any problems with his driving but states, “Other drivers have become so aggressive; they’re always honking at him.” Their daughter denies that Mr. D has driving problems but admits that for the last 2 years she has refused to allow her child to ride in his car.
Focused in-office assessment
Information to assess driving ability can come from the patient, family caregiver/informant, and clinical judgment. Patients with dementia are notoriously inaccurate in self-reported driving ability, either for lack of insight or as a testament to the importance of driving to their autonomy. Caregivers often are more accurate in describing a patient’s driving, but other agendas may color their responses.
In a study of patients with very mild or mild AD, 94% reported themselves as safe drivers, whereas on-road driving instructors rated <50% of drivers in these groups as safe. Caregivers were better able to classify driving performance, but 36% of their ratings were incorrect.12
Cognitive assessment. To assess older drivers’ cognition, AMA/NHTSA’s Guide to Assessing and Counseling Older Drivers recommends the Trail-Making Test, Part B and the clock-drawing test.10 The Canadian Medical Association suggests the MMSE.13 Both guides say that abnormalities in these tests indicate a need for more detailed testing, including referral to specialized driving assessment and retesting at regular intervals (Algorithm). Retest patients with mild dementia at least every 6 months or sooner when dementia severity increases noticeably14 (Box 1).6,15
The MMSE is widely used to screen for cognitive impairment and identify dementia or delirium, but it is not a diagnostic tool or proxy driving test. A patient with dementia may produce a high MMSE score and yet be an unsafe driver. For example, well-educated patients or those with vascular or frontotemporal dementia may retain cognitive abilities as measured by the MMSE until later in the disease.
Considerable effort has been put into developing tools to help clinicians quickly and accurately differentiate safe from unsafe drivers by assessing cognition. Unfortunately, no consistent link has been found between cognitive test results and driving outcome measures. A systematic review of office-based predictors of fitness to drive in dementia found 5 studies showing an association between MMSE scores and driving and 5 studies showing no such association.16 Thus, although the AMA/NHTSA guide recommends the MMSE, Trails B, and clock-drawing tests, cognitive tests—including these—are not sufficient to assess driving ability.
Severity of dementia. International consensus groups have attempted to create guidelines for patients with dementia who drive. American, Canadian, and Australian groups suggest that a diagnosis of moderate to severe dementia precludes driving, and the driver’s licenses of persons with these conditions should be revoked.17
In general, AD is considered severe when the MMSE score is <10 or the patient becomes dependent on a caregiver for survival.18 AD of moderate severity is more difficult to define, but a Canadian consensus conference suggested a practical approach: Patients with AD would be considered to have moderate to severe dementia and should not drive when they cannot independently perform multiple instrumental activities of daily living or any of the basic activities of daily living.19
Some dementias may impair driving more quickly than AD does. For example, hallucinations may occur early in Lewy body dementia, as may impulsivity in frontotemporal dementia and motor impairment in vascular dementia.
Mrs. Y visits your office for a follow-up regarding mild Alzheimer’s disease (AD), which was diagnosed 2 years ago. She passed an on-road test 3 months ago and has an Mini-Mental State Examination score of 24/30. Over the last month she has become depressed, with insomnia and mild psychomotor retardation. She occasionally has hallucinations.
Behavioral and psychological symptoms such as agitation, aggression, hallucinations, apathy, depression, and anxiety are common neuropsychiatric sequelae of AD. Little is known about the risks these symptoms pose to road safety, but we recommend that clinicians strongly consider the potential for impaired driving.
In a longitudinal study, cognitive impairment and behavioral disturbances—especially agitation, apathy, and hallucinations—were strong predictors of driving cessation among patients with dementia.6 Furthermore, a case crossover study of patients with dementia found a 54% increase in risk of motor vehicle collisions associated with the use of psychotropic medications.15
Consider all aspects of the patient’s clinical status, including neuropsychiatric symptoms, psychotropic medications, comorbid medical conditions (including hearing and vision impairment), and concomitant therapy for medical conditions. Any could change a safe driver with mild dementia into an unsafe driver.
Algorithm: 3 options for drivers with dementia, based on in-office assessment
* Observe legislation or statutes that address reporting unsafe drivers to the department of motor vehicles or ministry of transportation
On-road driving tests
Because some individuals with mild dementia can drive safely for extended periods, international recommendations for assessing the driver with dementia emphasize on-road driving tests.10,13,20–22 American10 and Canadian guidelines13 suggest that a dementia diagnosis is not sufficient to withdraw licensure.
A formal driving assessment is necessary to establish road safety for patients with mild dementia except when the need for license withdrawal is evident, such as when the patient has:
- a history of major driving problems (such as crashes or driving the wrong way on a highway)
- significant contraindications to driving on the history or physical examination (such as severe inattention or psychosis).
Challenges of on-road testing. On-road tests may be the gold standard, but they are not without clinical problems.
Need to retest. Because almost all dementias are progressive and driving skills deteriorate over time, most guidelines recommend periodic retesting. For patients with dementia who pass on-road evaluations, limited evidence supports retesting every 6 months.14 Take an individual approach, however, because of the various rates at which the dementias progress.
Testing vs real world conditions. Structured on-road testing is not equivalent to unstructured real-world driving, in which the patient often must navigate without instruction or assistance.
Rural vs urban driving. Road tests conducted in urban areas assess skills associated with complex conditions and the need to respond quickly to crises. They might not assess as well rural driving, which requires sustained attention on monotonous roads.
Inaccessibility. Cost and lack of availability of on-road tests, particularly in rural areas, limit the number of patients whose performance can be evaluated.
CASE CONTINUED: Distressing results
Mr. D has a history of decline in cognition and function, objective cognitive difficulties, and a subtle history of driving problems. You refer him for a specialized on-road test, and the report indicates that he failed. Errors included wide turns, driving too slowly, getting caught in an intersection twice during red lights while attempting to turn left, driving on the shoulder, and failing to signal lane changes. You review the results with Mr. D and his wife and recommend that he cease driving immediately.
Mr. D is furious, and his wife is dismayed. He demands to know how he can continue to play golf, which is his only form of exercise and recreation. Will she have to give up her bridge club? How will they shop for food? They request permission to at least to drive to the grocery store during the daytime.
You explain that no system allows individuals to drive only at certain times, and for the sake of safety you cannot grant them special permission. You discuss alternatives, such as asking their daughter for assistance with grocery shopping and taking taxis or ride-sharing with friends who play golf and bridge.
Remain firm, but ease the blow
Driving cessation orders distress patients, families, and clinicians. A failed road test clearly indicates unsafe driving, and driving cessation is critical to public safety.
A review by Man-Son-Hing et al23 found that drivers with dementia performed worse than nondemented controls in all studies that examined driving performance (on-road, simulator, or caregiver report). Simulators showed problems such as off-road driving, deviation from posted speed, and more time to negotiate left turns.24
By comparison, only 1 of 3 studies using state crash records showed an increased risk of collisions in persons with dementia compared with controls.23 From a research perspective, however, studies that use state-reported collisions to assess driving risk are confounded by driving restrictions on persons with dementia.
Mr. D wants to continue driving with restrictions. No studies have shown reduced crash rates when drivers with dementia used compensatory strategies such as restrictions, retraining/education, having a passenger “co-pilot,” on-board navigation, or cognitive enhancers.23
If Mr. D had passed the road test, the situation would have been more ambiguous. Two studies have examined on-road driving performance over time in patients with early-stage dementia.25,26 Both studies followed drivers prospectively for 2 years, and those with mild dementia (vs very mild or no dementia) were most likely to show a decline in driving skills:
- All participants with mild dementia were rated as “not safe” by the end of 2 years by Duchek et al.25
- Median time to “failure” (or a rating of unsafe) was 324 days for drivers with mild dementia vs 605 days for those with very mild dementia, as reported by Ott et al.26
Mr. D’s passionate plea for reconsideration highlights the need for communities to develop alternate transportation for seniors whose driving becomes unsafe (Box 2).
Legal liability? Physicians often are concerned about legal responsibilities and risks involved in reporting unsafe drivers. Be aware of local statutes or legislations regarding mandatory reporting of patients you deem unsafe to drive.17 These laws usually protect physicians from lawsuits related to violating patient confidentiality. Civil lawsuits remain possible, however, if clinicians fail to report an unsafe driver who subsequently is involved in a motor vehicle collision.27
1. Meet with family first. Help them assume a positive and supportive role. Explain concretely and empathically your concern for the safety of the patient and others. Clearly outline your findings that the patient is not fit to drive, and explain that the law requires you to report the patient to the authorities.
Remind family members that the goal of driving assessment is to prevent a collision, and they carry some responsibility because they are aware of the potential risk of letting their family member continue to drive. If necessary, have family members witness a repeat performance by the patient on the most revealing test. Discuss the importance of finding alternate transportation to reduce the risk of isolation and depression that can follow driving cessation.
2. Meet with patient. Having the family present can be helpful, but ask them to assume a supportive role. Give the patient a positive role by recognizing that he or she has been a responsible driver, and part of this responsibility is to stop driving before an accident occurs. Acknowledge that it is normal to be unhappy upon learning that one’s driving privileges are being revoked.
Sometimes it helps to give the patient a prescription in their name that says, “Do not drive.” Families who receive a copy may find this very helpful, too, for reminding the patient later about what you said.
If your patient argues with your position, remain firm and do not argue. Indicate that you have made notes on the meeting and are notifying the authorities about the patient’s unsafe driving. You can add that your chart could be subpoenaed and the patient may be legally liable and financially responsible should he or she continue to drive and have a collision.
3. Talk about transportation options. Family members could share driving responsibilities. Taxi rides can cost less than maintaining a car if the patient drives <4,000 km (2,500 miles) per year. Suggest that patients or families find volunteer drivers or contact helpful taxi drivers a day before an outing is planned.
4. If patient refuses to comply, meet with the family again and encourage them to remove the patient’s opportunity to drive (confiscate the keys, disable the car, or remove the car altogether).
Provide a written statement to the patient and family outlining why the patient can no longer drive. Indicate that it is your legal responsibility to report unsafe drivers, and you intend to notify the authorities regarding the patient’s driving status. If the patient remains noncompliant, continue to encourage family to remove the opportunity to drive.
Related resources
- Physician’s guide to assessing and counseling older drivers. American Medical Association and the National Highway Traffic Safety Administration, 2003. www.nhtsa.dot.gov/people/injury/olddrive/OlderDriversBook.
- Determining medical fitness to operate motor vehicles. Canadian Medical Association. 2006. www.cma.ca/index.cfm/ci_id/18223/la_id/1.htm.
- Canadian Driving Research Initiative for Vehicular Safety in the Elderly (CanDRIVE). www.candrive.ca.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Rapoport receives grant/research support from the Canadian Institute of Health Research and the Ontario Neurotrauma Foundation.
Mr. D, age 75, presents to your office with a 5-year history of gradually declining memory. His wife reports he is having difficulty with word finding, managing his finances, and shopping, and he needs supervision when using the stove. Nonetheless, he enjoys playing golf and drives himself to the golf course 3 times a week. He is compliant with his chronic medical therapy for hypertension, hypercholesterolemia, and asthma.
Patients with dementia who continue to drive pose a potential danger on the road, worry their families, and present challenges to clinicians. Most people would agree that patients with moderate or severe dementia should not drive, but a careful evaluation is required to assess whether a patient such as Mr. D with mild dementia remains fit to drive.
This article explores how dementia exacerbates age-related changes in driving ability and discusses how to assess driving in patients with dementia. Our goal is to help clinicians sort through data from in-office physical and cognitive assessments, family caregivers/informants’ reports, and (when available) on-road testing. We also discuss:
- guidelines for assessing older drivers that can help balance patients’ need for autonomy with public safety
- strategies for discussing driving cessation with patients and their families.
Driving: A privilege, not a right
Driving is central to older adults’ autonomy, and >75% of persons age ≥75 rely on driving as their primary mode of transportation.1 Driving cessation in this population has been associated with a 3-fold decrease in out-of-home activity2 and a 2.5-fold increase in depressive symptoms.3 Nonetheless, some 4.5 million Americans have Alzheimer’s disease (AD),4 and dementia poses a substantial risk to safe driving.
Although driving must be sacrificed when personal and public safety is at risk, most physicians perceive an uncomfortable conflict of interest between patient confidentiality and public safety.5 Assessing driving safety of patients with dementia can undermine the doctor-patient relationship and pose hardships for patients.
Mr. D has a 5-year history of memory problems that affect his daily functioning, yet he continues to drive. A longitudinal study of persons with dementia found that among the 29% who were driving at baseline, more than one-half were still behind the wheel 2 years later.6
Age and driving safety. Even in the absence of dementia, driving ability declines with aging (Tables 1 and 2).7,8 Older persons may self-regulate and restrict their driving to shorter distances, with fewer trips at night, on high-speed roads, or in unfamiliar situations. Their driving is rarely aggressive and they are unlikely to speed, but they may drive more slowly than other traffic.7,8 Although the overall rate of motor vehicle collisions declines with age:
- the rate of collisions per mile driven increases after age 659
- drivers age >65 have the highest fatality rate per mile driven among adults age ≥25.10
A dementia diagnosis is not sufficient to withdraw driving privileges, according to American Medical Association (AMA)/National Highway Traffic Safety Administration (NHTSA) guidelines. These recommend that you base decisions on the individual’s driving ability, and—when you have concerns—factor in a focused medical assessment and formal assessment of driving skills.10
Table 1
Age-related changes that may affect driving fitness
| Decreased physical capabilities, including declining muscle tone, flexibility, and reaction time |
| Decreased hearing and visual acuity |
| Increased fragility, resulting in longer time to heal should injuries occur |
| Increased medication use with possible side effect of drowsiness |
| Source: References 7,8 |
Table 2
Older drivers’ common traffic violations leading to crashes*
| Failure to obey traffic signals, including stop signs and red lights |
| Unsafe left turns (driver may inaccurately judge speed of oncoming vehicle) |
| Inappropriate turns (such as difficulty judging distance from oncoming cars, wide or narrow turns, or not timing the turn correctly with traffic lights) |
| Unsafe passing |
| Failure to yield |
| * These errors often lead to multivehicle accidents Source: References 7,8 |
CASE CONTINUED: Cognitive deficits quantified
You perform a Mini-Mental State Examination (MMSE). Mr. D scores 24/30, losing 1 point for orientation, 2 points for attention, 2 points for recall, and 1 point for copying. This score, along with his history, indicates mild dementia, although he claims he is a safe driver. On further cognitive testing, Mr. D completes the Trails A test in 90 seconds and Trails B test in 250 seconds (well below 1.5 standard deviations of the norm for his age and education).11 On the clock-drawing task, he drew a poorly organized clock, with unequal spaces between numbers and hands pointing to “10” and “11” instead of properly indicating “10 after 11.”
Mr. D and his wife live in a rural area, 5 miles from the nearest grocery store. His wife never drove, and she relies on him for weekly shopping trips and to drive her to her bridge club. She denies any problems with his driving but states, “Other drivers have become so aggressive; they’re always honking at him.” Their daughter denies that Mr. D has driving problems but admits that for the last 2 years she has refused to allow her child to ride in his car.
Focused in-office assessment
Information to assess driving ability can come from the patient, family caregiver/informant, and clinical judgment. Patients with dementia are notoriously inaccurate in self-reported driving ability, either for lack of insight or as a testament to the importance of driving to their autonomy. Caregivers often are more accurate in describing a patient’s driving, but other agendas may color their responses.
In a study of patients with very mild or mild AD, 94% reported themselves as safe drivers, whereas on-road driving instructors rated <50% of drivers in these groups as safe. Caregivers were better able to classify driving performance, but 36% of their ratings were incorrect.12
Cognitive assessment. To assess older drivers’ cognition, AMA/NHTSA’s Guide to Assessing and Counseling Older Drivers recommends the Trail-Making Test, Part B and the clock-drawing test.10 The Canadian Medical Association suggests the MMSE.13 Both guides say that abnormalities in these tests indicate a need for more detailed testing, including referral to specialized driving assessment and retesting at regular intervals (Algorithm). Retest patients with mild dementia at least every 6 months or sooner when dementia severity increases noticeably14 (Box 1).6,15
The MMSE is widely used to screen for cognitive impairment and identify dementia or delirium, but it is not a diagnostic tool or proxy driving test. A patient with dementia may produce a high MMSE score and yet be an unsafe driver. For example, well-educated patients or those with vascular or frontotemporal dementia may retain cognitive abilities as measured by the MMSE until later in the disease.
Considerable effort has been put into developing tools to help clinicians quickly and accurately differentiate safe from unsafe drivers by assessing cognition. Unfortunately, no consistent link has been found between cognitive test results and driving outcome measures. A systematic review of office-based predictors of fitness to drive in dementia found 5 studies showing an association between MMSE scores and driving and 5 studies showing no such association.16 Thus, although the AMA/NHTSA guide recommends the MMSE, Trails B, and clock-drawing tests, cognitive tests—including these—are not sufficient to assess driving ability.
Severity of dementia. International consensus groups have attempted to create guidelines for patients with dementia who drive. American, Canadian, and Australian groups suggest that a diagnosis of moderate to severe dementia precludes driving, and the driver’s licenses of persons with these conditions should be revoked.17
In general, AD is considered severe when the MMSE score is <10 or the patient becomes dependent on a caregiver for survival.18 AD of moderate severity is more difficult to define, but a Canadian consensus conference suggested a practical approach: Patients with AD would be considered to have moderate to severe dementia and should not drive when they cannot independently perform multiple instrumental activities of daily living or any of the basic activities of daily living.19
Some dementias may impair driving more quickly than AD does. For example, hallucinations may occur early in Lewy body dementia, as may impulsivity in frontotemporal dementia and motor impairment in vascular dementia.
Mrs. Y visits your office for a follow-up regarding mild Alzheimer’s disease (AD), which was diagnosed 2 years ago. She passed an on-road test 3 months ago and has an Mini-Mental State Examination score of 24/30. Over the last month she has become depressed, with insomnia and mild psychomotor retardation. She occasionally has hallucinations.
Behavioral and psychological symptoms such as agitation, aggression, hallucinations, apathy, depression, and anxiety are common neuropsychiatric sequelae of AD. Little is known about the risks these symptoms pose to road safety, but we recommend that clinicians strongly consider the potential for impaired driving.
In a longitudinal study, cognitive impairment and behavioral disturbances—especially agitation, apathy, and hallucinations—were strong predictors of driving cessation among patients with dementia.6 Furthermore, a case crossover study of patients with dementia found a 54% increase in risk of motor vehicle collisions associated with the use of psychotropic medications.15
Consider all aspects of the patient’s clinical status, including neuropsychiatric symptoms, psychotropic medications, comorbid medical conditions (including hearing and vision impairment), and concomitant therapy for medical conditions. Any could change a safe driver with mild dementia into an unsafe driver.
Algorithm: 3 options for drivers with dementia, based on in-office assessment
* Observe legislation or statutes that address reporting unsafe drivers to the department of motor vehicles or ministry of transportation
On-road driving tests
Because some individuals with mild dementia can drive safely for extended periods, international recommendations for assessing the driver with dementia emphasize on-road driving tests.10,13,20–22 American10 and Canadian guidelines13 suggest that a dementia diagnosis is not sufficient to withdraw licensure.
A formal driving assessment is necessary to establish road safety for patients with mild dementia except when the need for license withdrawal is evident, such as when the patient has:
- a history of major driving problems (such as crashes or driving the wrong way on a highway)
- significant contraindications to driving on the history or physical examination (such as severe inattention or psychosis).
Challenges of on-road testing. On-road tests may be the gold standard, but they are not without clinical problems.
Need to retest. Because almost all dementias are progressive and driving skills deteriorate over time, most guidelines recommend periodic retesting. For patients with dementia who pass on-road evaluations, limited evidence supports retesting every 6 months.14 Take an individual approach, however, because of the various rates at which the dementias progress.
Testing vs real world conditions. Structured on-road testing is not equivalent to unstructured real-world driving, in which the patient often must navigate without instruction or assistance.
Rural vs urban driving. Road tests conducted in urban areas assess skills associated with complex conditions and the need to respond quickly to crises. They might not assess as well rural driving, which requires sustained attention on monotonous roads.
Inaccessibility. Cost and lack of availability of on-road tests, particularly in rural areas, limit the number of patients whose performance can be evaluated.
CASE CONTINUED: Distressing results
Mr. D has a history of decline in cognition and function, objective cognitive difficulties, and a subtle history of driving problems. You refer him for a specialized on-road test, and the report indicates that he failed. Errors included wide turns, driving too slowly, getting caught in an intersection twice during red lights while attempting to turn left, driving on the shoulder, and failing to signal lane changes. You review the results with Mr. D and his wife and recommend that he cease driving immediately.
Mr. D is furious, and his wife is dismayed. He demands to know how he can continue to play golf, which is his only form of exercise and recreation. Will she have to give up her bridge club? How will they shop for food? They request permission to at least to drive to the grocery store during the daytime.
You explain that no system allows individuals to drive only at certain times, and for the sake of safety you cannot grant them special permission. You discuss alternatives, such as asking their daughter for assistance with grocery shopping and taking taxis or ride-sharing with friends who play golf and bridge.
Remain firm, but ease the blow
Driving cessation orders distress patients, families, and clinicians. A failed road test clearly indicates unsafe driving, and driving cessation is critical to public safety.
A review by Man-Son-Hing et al23 found that drivers with dementia performed worse than nondemented controls in all studies that examined driving performance (on-road, simulator, or caregiver report). Simulators showed problems such as off-road driving, deviation from posted speed, and more time to negotiate left turns.24
By comparison, only 1 of 3 studies using state crash records showed an increased risk of collisions in persons with dementia compared with controls.23 From a research perspective, however, studies that use state-reported collisions to assess driving risk are confounded by driving restrictions on persons with dementia.
Mr. D wants to continue driving with restrictions. No studies have shown reduced crash rates when drivers with dementia used compensatory strategies such as restrictions, retraining/education, having a passenger “co-pilot,” on-board navigation, or cognitive enhancers.23
If Mr. D had passed the road test, the situation would have been more ambiguous. Two studies have examined on-road driving performance over time in patients with early-stage dementia.25,26 Both studies followed drivers prospectively for 2 years, and those with mild dementia (vs very mild or no dementia) were most likely to show a decline in driving skills:
- All participants with mild dementia were rated as “not safe” by the end of 2 years by Duchek et al.25
- Median time to “failure” (or a rating of unsafe) was 324 days for drivers with mild dementia vs 605 days for those with very mild dementia, as reported by Ott et al.26
Mr. D’s passionate plea for reconsideration highlights the need for communities to develop alternate transportation for seniors whose driving becomes unsafe (Box 2).
Legal liability? Physicians often are concerned about legal responsibilities and risks involved in reporting unsafe drivers. Be aware of local statutes or legislations regarding mandatory reporting of patients you deem unsafe to drive.17 These laws usually protect physicians from lawsuits related to violating patient confidentiality. Civil lawsuits remain possible, however, if clinicians fail to report an unsafe driver who subsequently is involved in a motor vehicle collision.27
1. Meet with family first. Help them assume a positive and supportive role. Explain concretely and empathically your concern for the safety of the patient and others. Clearly outline your findings that the patient is not fit to drive, and explain that the law requires you to report the patient to the authorities.
Remind family members that the goal of driving assessment is to prevent a collision, and they carry some responsibility because they are aware of the potential risk of letting their family member continue to drive. If necessary, have family members witness a repeat performance by the patient on the most revealing test. Discuss the importance of finding alternate transportation to reduce the risk of isolation and depression that can follow driving cessation.
2. Meet with patient. Having the family present can be helpful, but ask them to assume a supportive role. Give the patient a positive role by recognizing that he or she has been a responsible driver, and part of this responsibility is to stop driving before an accident occurs. Acknowledge that it is normal to be unhappy upon learning that one’s driving privileges are being revoked.
Sometimes it helps to give the patient a prescription in their name that says, “Do not drive.” Families who receive a copy may find this very helpful, too, for reminding the patient later about what you said.
If your patient argues with your position, remain firm and do not argue. Indicate that you have made notes on the meeting and are notifying the authorities about the patient’s unsafe driving. You can add that your chart could be subpoenaed and the patient may be legally liable and financially responsible should he or she continue to drive and have a collision.
3. Talk about transportation options. Family members could share driving responsibilities. Taxi rides can cost less than maintaining a car if the patient drives <4,000 km (2,500 miles) per year. Suggest that patients or families find volunteer drivers or contact helpful taxi drivers a day before an outing is planned.
4. If patient refuses to comply, meet with the family again and encourage them to remove the patient’s opportunity to drive (confiscate the keys, disable the car, or remove the car altogether).
Provide a written statement to the patient and family outlining why the patient can no longer drive. Indicate that it is your legal responsibility to report unsafe drivers, and you intend to notify the authorities regarding the patient’s driving status. If the patient remains noncompliant, continue to encourage family to remove the opportunity to drive.
Related resources
- Physician’s guide to assessing and counseling older drivers. American Medical Association and the National Highway Traffic Safety Administration, 2003. www.nhtsa.dot.gov/people/injury/olddrive/OlderDriversBook.
- Determining medical fitness to operate motor vehicles. Canadian Medical Association. 2006. www.cma.ca/index.cfm/ci_id/18223/la_id/1.htm.
- Canadian Driving Research Initiative for Vehicular Safety in the Elderly (CanDRIVE). www.candrive.ca.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Rapoport receives grant/research support from the Canadian Institute of Health Research and the Ontario Neurotrauma Foundation.
1. Stowell-Ritter A, Straight A, Evans EL. Understanding senior transportation: report and analysis of a survey of consumers age 50+. Washington, DC: American Association of Retired Persons; 2002.
2. Marottoli RA, de Leon CFM, Glass TA, et al. Consequences of driving cessation: decreased out-of-home activity levels. J Gerontol B Psychol Sci Soc Sci 2000;55(6):S334-40.
3. Marottoli RA, Mendes de Leon CF, Glass TA, et al. Driving cessation and increased depressive symptoms: prospective evidence from the New Haven EPESE. Established Populations for Epidemiologic Studies of the Elderly. J Am Geriatr Soc 1997;45(2):202-6.
4. Hebert LE, Scherr PA, Bienias JL, et al. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 2003;60(8):1119-22.
5. Jang RW, Man-Son-Hing M, Molnar FJ, et al. Family physicians’ attitudes and practices regarding assessments of medical fitness to drive in older persons. J Gen Intern Med 2007;22(4):531-43.
6. Herrmann N, Rapoport MJ, Sambrook R, et al. Predictors of driving cessation in mild-to-moderate dementia. CMAJ 2006;175(6):591-5.
7. Hedlund J. Countermeasures that work: a highway safety countermeasure guide for state highway safety offices. Washington, DC: National Highway Traffic Safety Administration; 2006.
8. Dobbs B. Medical conditions and driving: a review of the literature (1960-2000). Washington, DC: National Highway Traffic Safety Administration; 2005. Available at: http://www.nhtsa.dot.gov/people/injury/research/Medical_Condition_Driving/pages/TRD.html. Accessed September 29, 2008.
9. Li G, Braver ER, Chen LH. Fragility versus excessive crash involvement as determinants of high death rates per vehicle-mile of travel among older drivers. Accid Anal Prev 2003;35(2):227-35.
10. Physician’s guide to assessing and counseling older drivers. Washington, DC: National Highway Traffic Safety Administration; 2003. Available at: http://www.nhtsa.dot.gov/people/injury/olddrive/OlderDriversBook. Accessed September 29, 2008.
11. Yeudall LT, Reddon JR, Gill DM, et al. Normative data for the Halstead-Reitan neuropsychological tests stratified by age and sex. J Clin Psychol 1987;43(3):346-67.
12. Brown LB, Ott BR, Papandonatos GD, et al. Prediction of on-road driving performance in patients with early Alzheimer’s disease. J Am Geriatr Soc 2005;53(1):94-8.
13. Determining medical fitness to operate motor vehicles. CMA driver’s guide. 7th ed. Ottawa, Ontario, Canada: Canadian Medical Association; 2006.
14. Molnar FJ, Patel A, Marshall SC, et al. Systematic review of the optimal frequency of follow-up in persons with mild dementia who continue to drive. Alzheimer Dis Assoc Disord 2006;20(4):295-7.
15. Rapoport MJ, Herrmann N, Molnar FJ, et al. Psychotropic medications and motor vehicle collisions in patients with dementia (letter). J Am Geriatr Soc 2008;56(10):1968-70.
16. Molnar FJ, Patel A, Marshall SC, et al. Clinical utility of office-based cognitive predictors of fitness to drive in persons with dementia: a systematic review. J Am Geriatr Soc 2006;54(12):1809-24.
17. Rapoport MJ, Herrmann N, Molnar FJ, et al. Sharing the responsibility for assessing the risk of the driver with dementia. CMAJ. 2007;177(6):599-601.
18. Herrmann N, Gauthier S, Lysy PG. Clinical practice guidelines for severe Alzheimer’s disease. Alzheimers Dement 2007;3(4):385-97.
19. Hogan DB, Bailey P, Carswell A, et al. Management of mild to moderate Alzheimer’s disease and dementia. Alzheimers Dement 2007;3(4):355-84.
19. Assessing fitness to drive for commercial and private vehicle drivers. Sydney, Australia: National Library of Australia; 2006. Available at: http://www.austroads.com.au/aftd/index.html. Accessed November 4, 2008.
21. Medical aspects of fitness to drive. A guide for medical practitioners. Wellington, New Zealand: Land Transport Safety Authority; 2002. Available at: http://www.transfund.govt.nz/licensing/docs/ltsa-medical-aspects.pdf. Accessed September 29, 2008.
22. At a glance guide to the current medical standards of fitness to drive. Swansea, UK: Drivers Medical Group, Driver and Vehicle Licensing Agency; 2008. Available at: http://www.dvla.gov.uk/medical/ataglance.aspx. Accessed September 29, 2008.
23. Man-Son-Hing M, Marshall SC, Molnar FJ, Wilson KG. Systematic review of driving risk and the efficacy of compensatory strategies in persons with dementia. J Am Geriatr Soc 2007;55(6):878-84.
24. Cox DJ, Quillian WC, Thorndike FP, et al. Evaluating driving performance of outpatients with Alzheimer disease. J Am Board Fam Pract 1998;11(4):264-71.
25. Duchek JM, Carr DB, Hunt L, et al. Longitudinal driving performance in early-stage dementia of the Alzheimer type. J Am Geriatr Soc 2003;51(10):1342-7.
26. Ott BR, Heindel WC, Papandonatos GD, et al. A longitudinal study of drivers with Alzheimer disease. Neurology 2008;70(14):1171-8.
27. Molnar FJ, Byszewski AM, Marshall SC, Man-Son-Hing M. In-office evaluation of medical fitness to drive: practical approaches for assessing older people. Can Fam Physician 2005;51:372-9.
1. Stowell-Ritter A, Straight A, Evans EL. Understanding senior transportation: report and analysis of a survey of consumers age 50+. Washington, DC: American Association of Retired Persons; 2002.
2. Marottoli RA, de Leon CFM, Glass TA, et al. Consequences of driving cessation: decreased out-of-home activity levels. J Gerontol B Psychol Sci Soc Sci 2000;55(6):S334-40.
3. Marottoli RA, Mendes de Leon CF, Glass TA, et al. Driving cessation and increased depressive symptoms: prospective evidence from the New Haven EPESE. Established Populations for Epidemiologic Studies of the Elderly. J Am Geriatr Soc 1997;45(2):202-6.
4. Hebert LE, Scherr PA, Bienias JL, et al. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 2003;60(8):1119-22.
5. Jang RW, Man-Son-Hing M, Molnar FJ, et al. Family physicians’ attitudes and practices regarding assessments of medical fitness to drive in older persons. J Gen Intern Med 2007;22(4):531-43.
6. Herrmann N, Rapoport MJ, Sambrook R, et al. Predictors of driving cessation in mild-to-moderate dementia. CMAJ 2006;175(6):591-5.
7. Hedlund J. Countermeasures that work: a highway safety countermeasure guide for state highway safety offices. Washington, DC: National Highway Traffic Safety Administration; 2006.
8. Dobbs B. Medical conditions and driving: a review of the literature (1960-2000). Washington, DC: National Highway Traffic Safety Administration; 2005. Available at: http://www.nhtsa.dot.gov/people/injury/research/Medical_Condition_Driving/pages/TRD.html. Accessed September 29, 2008.
9. Li G, Braver ER, Chen LH. Fragility versus excessive crash involvement as determinants of high death rates per vehicle-mile of travel among older drivers. Accid Anal Prev 2003;35(2):227-35.
10. Physician’s guide to assessing and counseling older drivers. Washington, DC: National Highway Traffic Safety Administration; 2003. Available at: http://www.nhtsa.dot.gov/people/injury/olddrive/OlderDriversBook. Accessed September 29, 2008.
11. Yeudall LT, Reddon JR, Gill DM, et al. Normative data for the Halstead-Reitan neuropsychological tests stratified by age and sex. J Clin Psychol 1987;43(3):346-67.
12. Brown LB, Ott BR, Papandonatos GD, et al. Prediction of on-road driving performance in patients with early Alzheimer’s disease. J Am Geriatr Soc 2005;53(1):94-8.
13. Determining medical fitness to operate motor vehicles. CMA driver’s guide. 7th ed. Ottawa, Ontario, Canada: Canadian Medical Association; 2006.
14. Molnar FJ, Patel A, Marshall SC, et al. Systematic review of the optimal frequency of follow-up in persons with mild dementia who continue to drive. Alzheimer Dis Assoc Disord 2006;20(4):295-7.
15. Rapoport MJ, Herrmann N, Molnar FJ, et al. Psychotropic medications and motor vehicle collisions in patients with dementia (letter). J Am Geriatr Soc 2008;56(10):1968-70.
16. Molnar FJ, Patel A, Marshall SC, et al. Clinical utility of office-based cognitive predictors of fitness to drive in persons with dementia: a systematic review. J Am Geriatr Soc 2006;54(12):1809-24.
17. Rapoport MJ, Herrmann N, Molnar FJ, et al. Sharing the responsibility for assessing the risk of the driver with dementia. CMAJ. 2007;177(6):599-601.
18. Herrmann N, Gauthier S, Lysy PG. Clinical practice guidelines for severe Alzheimer’s disease. Alzheimers Dement 2007;3(4):385-97.
19. Hogan DB, Bailey P, Carswell A, et al. Management of mild to moderate Alzheimer’s disease and dementia. Alzheimers Dement 2007;3(4):355-84.
19. Assessing fitness to drive for commercial and private vehicle drivers. Sydney, Australia: National Library of Australia; 2006. Available at: http://www.austroads.com.au/aftd/index.html. Accessed November 4, 2008.
21. Medical aspects of fitness to drive. A guide for medical practitioners. Wellington, New Zealand: Land Transport Safety Authority; 2002. Available at: http://www.transfund.govt.nz/licensing/docs/ltsa-medical-aspects.pdf. Accessed September 29, 2008.
22. At a glance guide to the current medical standards of fitness to drive. Swansea, UK: Drivers Medical Group, Driver and Vehicle Licensing Agency; 2008. Available at: http://www.dvla.gov.uk/medical/ataglance.aspx. Accessed September 29, 2008.
23. Man-Son-Hing M, Marshall SC, Molnar FJ, Wilson KG. Systematic review of driving risk and the efficacy of compensatory strategies in persons with dementia. J Am Geriatr Soc 2007;55(6):878-84.
24. Cox DJ, Quillian WC, Thorndike FP, et al. Evaluating driving performance of outpatients with Alzheimer disease. J Am Board Fam Pract 1998;11(4):264-71.
25. Duchek JM, Carr DB, Hunt L, et al. Longitudinal driving performance in early-stage dementia of the Alzheimer type. J Am Geriatr Soc 2003;51(10):1342-7.
26. Ott BR, Heindel WC, Papandonatos GD, et al. A longitudinal study of drivers with Alzheimer disease. Neurology 2008;70(14):1171-8.
27. Molnar FJ, Byszewski AM, Marshall SC, Man-Son-Hing M. In-office evaluation of medical fitness to drive: practical approaches for assessing older people. Can Fam Physician 2005;51:372-9.
Novel agent more effective than standard therapy in NHL
Preliminary results of a phase 3 study indicate that pixantrone is more effective than standard chemotherapy in patients with advanced, relapsed, aggressive non-Hodgkin lymphoma (NHL).
The phase 3 EXTEND PIX301 trial enrolled 140 NHL patients from 130 sites in 17 countries. Patients had received 2 or more prior therapies and were sensitive to anthracycline treatment.
They were randomized to receive either pixantrone or another single-agent drug currently used in this patient population and selected by a physician. The trial assessed patients’ complete remission or unconfirmed complete remission rate, overall survival, and progression-free survival.
Twenty percent of patients who received pixantrone achieved either a confirmed or unconfirmed complete remission, compared to 5.7% of patients on standard chemotherapy. Eleven percent of pixantrone patients’ remissions were confirmed, whereas none of the standard chemotherapy remissions were.
The overall response rate was 37.1% with pixantrone and 14.3% for patients on standard chemotherapy. Response rates were determined by an independent assessment panel that was blinded to treatment assignments.
Complete safety information for this study is not yet available. However, the study was monitored on an ongoing basis by an independent Data Safety Monitoring Committee, and no serious concerns were raised. The most common serious toxicities (> 5%) observed in previous trials of pixantrone include grade 3 and 4 neutropenia and febrile neutropenia.
Seventy-four percent of patients enrolled in this study discontinued therapy due to disease progression or death, the majority of which were in the standard chemotherapy control arm.
This study was funded by Cell Therapeutics, Inc., the company developing pixantrone.
Cell Therapeutics says it plans to submit complete study data for presentation at a major scientific conference. The organization also plans to request a pre-New Drug Application meeting with the FDA and expects to begin submission of a rolling New Drug Application to the FDA in early 2009.
Pixantrone is an antitumor agent that contains an aza-anthracenedione molecular structure, which differentiates it from anthracycline chemotherapy agents. ![]()
Preliminary results of a phase 3 study indicate that pixantrone is more effective than standard chemotherapy in patients with advanced, relapsed, aggressive non-Hodgkin lymphoma (NHL).
The phase 3 EXTEND PIX301 trial enrolled 140 NHL patients from 130 sites in 17 countries. Patients had received 2 or more prior therapies and were sensitive to anthracycline treatment.
They were randomized to receive either pixantrone or another single-agent drug currently used in this patient population and selected by a physician. The trial assessed patients’ complete remission or unconfirmed complete remission rate, overall survival, and progression-free survival.
Twenty percent of patients who received pixantrone achieved either a confirmed or unconfirmed complete remission, compared to 5.7% of patients on standard chemotherapy. Eleven percent of pixantrone patients’ remissions were confirmed, whereas none of the standard chemotherapy remissions were.
The overall response rate was 37.1% with pixantrone and 14.3% for patients on standard chemotherapy. Response rates were determined by an independent assessment panel that was blinded to treatment assignments.
Complete safety information for this study is not yet available. However, the study was monitored on an ongoing basis by an independent Data Safety Monitoring Committee, and no serious concerns were raised. The most common serious toxicities (> 5%) observed in previous trials of pixantrone include grade 3 and 4 neutropenia and febrile neutropenia.
Seventy-four percent of patients enrolled in this study discontinued therapy due to disease progression or death, the majority of which were in the standard chemotherapy control arm.
This study was funded by Cell Therapeutics, Inc., the company developing pixantrone.
Cell Therapeutics says it plans to submit complete study data for presentation at a major scientific conference. The organization also plans to request a pre-New Drug Application meeting with the FDA and expects to begin submission of a rolling New Drug Application to the FDA in early 2009.
Pixantrone is an antitumor agent that contains an aza-anthracenedione molecular structure, which differentiates it from anthracycline chemotherapy agents. ![]()
Preliminary results of a phase 3 study indicate that pixantrone is more effective than standard chemotherapy in patients with advanced, relapsed, aggressive non-Hodgkin lymphoma (NHL).
The phase 3 EXTEND PIX301 trial enrolled 140 NHL patients from 130 sites in 17 countries. Patients had received 2 or more prior therapies and were sensitive to anthracycline treatment.
They were randomized to receive either pixantrone or another single-agent drug currently used in this patient population and selected by a physician. The trial assessed patients’ complete remission or unconfirmed complete remission rate, overall survival, and progression-free survival.
Twenty percent of patients who received pixantrone achieved either a confirmed or unconfirmed complete remission, compared to 5.7% of patients on standard chemotherapy. Eleven percent of pixantrone patients’ remissions were confirmed, whereas none of the standard chemotherapy remissions were.
The overall response rate was 37.1% with pixantrone and 14.3% for patients on standard chemotherapy. Response rates were determined by an independent assessment panel that was blinded to treatment assignments.
Complete safety information for this study is not yet available. However, the study was monitored on an ongoing basis by an independent Data Safety Monitoring Committee, and no serious concerns were raised. The most common serious toxicities (> 5%) observed in previous trials of pixantrone include grade 3 and 4 neutropenia and febrile neutropenia.
Seventy-four percent of patients enrolled in this study discontinued therapy due to disease progression or death, the majority of which were in the standard chemotherapy control arm.
This study was funded by Cell Therapeutics, Inc., the company developing pixantrone.
Cell Therapeutics says it plans to submit complete study data for presentation at a major scientific conference. The organization also plans to request a pre-New Drug Application meeting with the FDA and expects to begin submission of a rolling New Drug Application to the FDA in early 2009.
Pixantrone is an antitumor agent that contains an aza-anthracenedione molecular structure, which differentiates it from anthracycline chemotherapy agents. ![]()