User login
Clonazepam dosing
Dr. Scott Freeman’s useful discussion of targeting acute risk factors in suicidal patients (“Suicide assessment: Targeting acute risk factors,” Current Psychiatry, January 2012, p. 52-57) ends by resolving the clinical vignette with a summary of hospital treatment. Apart from failing to indicate any psychotherapeutic inroads, Dr. Freeman seems to support prescribing clonazepam, 0.5 mg twice daily and 1 mg at bedtime. Clonazepam apparently “worked” by alleviating the patient’s anxiety and insomnia, but defied any pharmacologic rationale insofar as clonazepam has a slow onset and long half-life, making 3 doses per day irrational. This treatment strategy also risks problems of cumulative excess in the long run after discharge.
Aggressive pharmacotherapy may be the hallmark of modern acute hospital treatment, but surely it should incorporate careful understanding of specific medications’ pharmacodynamics, especially when relying on benzodiazepines. Needless to say, beginning a psychological process in the hospital also appears to have been shortchanged.
Sara Hartley, MD
Lecturer, Clinical Skills Program
University of California,
Berkeley-University of California,
San Francisco Joint Medical Program
Berkeley, CA
Dr. Freeman responds
I appreciate Dr. Hartley’s interest in my article. Although I agree with her that psychotherapy is an integral part of any treatment plan, the clinical vignette was used only to emphasize the need to aggressively and quickly start antidepressant and, more importantly, anxiolytic pharmacologic treatment in acutely suicidal patients with severe anxiety and depression.
With regard to clonazepam’s pharmacokinetics, although it does have a long half-life, it is only weakly lipophilic compared with other long-acting benzodiazepines such as diazepam. In fact, clonazepam has been shown to be less lipophilic than lorazepam,1 meaning it has a much smaller volume of distribution and less accumulation in peripheral adipose tissue. Therefore, one would not be concerned about significant drug accumulation leading to unexpected toxicity with a less lipophilic agent such as clonazepam.
I do not agree that dosing clonazepam 3 times a day, especially in an acute crisis, is “irrational,” as Dr. Hartley suggests. According to the package insert, although clonazepam is recommended to be administered twice daily for panic disorder, it can be given 3 times a day for seizure disorders.2
Scott A. Freeman, MD
Medical Director
Schizophrenia and Bipolar Disorder Inpatient Unit
McLean Hospital
Belmont, MA
Dr. Scott Freeman’s useful discussion of targeting acute risk factors in suicidal patients (“Suicide assessment: Targeting acute risk factors,” Current Psychiatry, January 2012, p. 52-57) ends by resolving the clinical vignette with a summary of hospital treatment. Apart from failing to indicate any psychotherapeutic inroads, Dr. Freeman seems to support prescribing clonazepam, 0.5 mg twice daily and 1 mg at bedtime. Clonazepam apparently “worked” by alleviating the patient’s anxiety and insomnia, but defied any pharmacologic rationale insofar as clonazepam has a slow onset and long half-life, making 3 doses per day irrational. This treatment strategy also risks problems of cumulative excess in the long run after discharge.
Aggressive pharmacotherapy may be the hallmark of modern acute hospital treatment, but surely it should incorporate careful understanding of specific medications’ pharmacodynamics, especially when relying on benzodiazepines. Needless to say, beginning a psychological process in the hospital also appears to have been shortchanged.
Sara Hartley, MD
Lecturer, Clinical Skills Program
University of California,
Berkeley-University of California,
San Francisco Joint Medical Program
Berkeley, CA
Dr. Freeman responds
I appreciate Dr. Hartley’s interest in my article. Although I agree with her that psychotherapy is an integral part of any treatment plan, the clinical vignette was used only to emphasize the need to aggressively and quickly start antidepressant and, more importantly, anxiolytic pharmacologic treatment in acutely suicidal patients with severe anxiety and depression.
With regard to clonazepam’s pharmacokinetics, although it does have a long half-life, it is only weakly lipophilic compared with other long-acting benzodiazepines such as diazepam. In fact, clonazepam has been shown to be less lipophilic than lorazepam,1 meaning it has a much smaller volume of distribution and less accumulation in peripheral adipose tissue. Therefore, one would not be concerned about significant drug accumulation leading to unexpected toxicity with a less lipophilic agent such as clonazepam.
I do not agree that dosing clonazepam 3 times a day, especially in an acute crisis, is “irrational,” as Dr. Hartley suggests. According to the package insert, although clonazepam is recommended to be administered twice daily for panic disorder, it can be given 3 times a day for seizure disorders.2
Scott A. Freeman, MD
Medical Director
Schizophrenia and Bipolar Disorder Inpatient Unit
McLean Hospital
Belmont, MA
Dr. Scott Freeman’s useful discussion of targeting acute risk factors in suicidal patients (“Suicide assessment: Targeting acute risk factors,” Current Psychiatry, January 2012, p. 52-57) ends by resolving the clinical vignette with a summary of hospital treatment. Apart from failing to indicate any psychotherapeutic inroads, Dr. Freeman seems to support prescribing clonazepam, 0.5 mg twice daily and 1 mg at bedtime. Clonazepam apparently “worked” by alleviating the patient’s anxiety and insomnia, but defied any pharmacologic rationale insofar as clonazepam has a slow onset and long half-life, making 3 doses per day irrational. This treatment strategy also risks problems of cumulative excess in the long run after discharge.
Aggressive pharmacotherapy may be the hallmark of modern acute hospital treatment, but surely it should incorporate careful understanding of specific medications’ pharmacodynamics, especially when relying on benzodiazepines. Needless to say, beginning a psychological process in the hospital also appears to have been shortchanged.
Sara Hartley, MD
Lecturer, Clinical Skills Program
University of California,
Berkeley-University of California,
San Francisco Joint Medical Program
Berkeley, CA
Dr. Freeman responds
I appreciate Dr. Hartley’s interest in my article. Although I agree with her that psychotherapy is an integral part of any treatment plan, the clinical vignette was used only to emphasize the need to aggressively and quickly start antidepressant and, more importantly, anxiolytic pharmacologic treatment in acutely suicidal patients with severe anxiety and depression.
With regard to clonazepam’s pharmacokinetics, although it does have a long half-life, it is only weakly lipophilic compared with other long-acting benzodiazepines such as diazepam. In fact, clonazepam has been shown to be less lipophilic than lorazepam,1 meaning it has a much smaller volume of distribution and less accumulation in peripheral adipose tissue. Therefore, one would not be concerned about significant drug accumulation leading to unexpected toxicity with a less lipophilic agent such as clonazepam.
I do not agree that dosing clonazepam 3 times a day, especially in an acute crisis, is “irrational,” as Dr. Hartley suggests. According to the package insert, although clonazepam is recommended to be administered twice daily for panic disorder, it can be given 3 times a day for seizure disorders.2
Scott A. Freeman, MD
Medical Director
Schizophrenia and Bipolar Disorder Inpatient Unit
McLean Hospital
Belmont, MA
Your surgical toolbox should include topical hemostatic agents—here is why
Vessel-sealing devices and hemostatic adjuvants are expanding the surgical armamentarium. These products provide a spectrum of alternatives that can serve you and your surgical patient well when traditional techniques for obtaining hemostasis fail to provide a satisfactory result. (Keep in mind, however, that technology is no substitute for excellent technique!)
In this article, we highlight three common scenarios in which topical hemostatic agents may be useful during gynecologic surgery. In addition, in the sidebar, five surgeons describe the hemostatic products they rely on most often—and tell why.

Following hysterectomy, persistent oozing along the anterior vaginal margin, distal to the cuff and adjacent to the site of bladder mobilization, may be managed with the aid of a topical hemostatic agent—in this case, a fibrin sealant.
When the site of bleeding is difficult to reach
CASE 1: Oozing at the site of bladder mobilization
You perform total hysterectomy in a 44-year-old woman who has uterine fibroids. After the procedure, you notice persistent oozing along the anterior vaginal margin, distal to the cuff and adjacent to where the bladder was mobilized.
How do you manage the oozing?
Wide mobilization of the bladder is a vital step in the safe performance of hysterectomy. Adhesions may complicate the process if the patient has had previous abdominal surgery, infection, or inflammation. Following mobilization of the bladder and removal of the uterus, bleeding may be visible along the adventitia of the posterior bladder wall or along the anterior surface of the vagina, distal to the cuff, as it is in this case (see the illustration).
Judicious application of an energy source is an option, but thermal injury to the bladder is a concern. A good alternative is proper placement of a hemostatic suture, but it can sometimes be difficult to avoid incorporating the bladder or injuring or obstructing the nearby ureter.
In this case, the location of the bleeding deep in the operative field poses a challenge, because of limited exposure and the proximity of the bladder and ureters. Virtually any hemostatic agent would work well in this circumstance (TABLE). For example, a flowable agent or fibrin sealant could be thoroughly applied to the area during a minimally invasive or open procedure and would naturally conform to the irregularities in the tissue, particularly the junction between the vagina and bladder flap.
A pliable product such as Surgicel Nu-Knit or Fibrillar would also work well in these circumstances, although successful application during laparoscopy may depend on the size of the trocar. For example, Nu-Knit would require trimming to a size suitable for passage through a trocar, made easier by moistening with saline. The weave of Fibrillar makes it more challenging to pass, intact, through a trocar; rolling the material into a cylindrical shape may reduce its diameter and allow it to pass more easily.
CASE 1: Resolved
You apply a fibrin sealant to the site of bleeding, and the oozing abates. Once complete hemostasis is ensured, you conclude the surgery and transfer the patient to recovery, where she does well.
Profiles in hemostasis: Strengths and weaknesses of topical agent
| Agent (brands) | Composition | Forms available | Mechanism of action | Advantages | Caveats | Duration | Relative cost* |
|---|---|---|---|---|---|---|---|
| Physical agents | |||||||
| Gelatin matrix (Gelfoam, Gelfilm, Surgifoam) | Porcine- derived collagen | Sponge, film, powder | Provides physical matrix for clot formation | Non-antigenic; neutral pH; may be used with thrombin | Material expansion may cause compression; Not for use in closed spaces or near nerve structures | 4–6 weeks | $ |
| Oxidized regenerated cellulose (Surgicel Fibrillar, Surgicel Nu-Knit) | Wood pulp | Mesh or packed fibers | Provides physical matrix for clot formation; acidic pH causes hemolysis and local clot formation | Pliable, easy to place through laparoscope; acidic pH has antimicrobial effect | Works best in a dry field. Acidic pH inactivates biologic agents, such as thrombin, and may increase inflammation. Avoid using excess material. | 2–4 weeks | $ |
| Microfibrillar collagen (Avitene, Instat, Helitene Helistat) | Bovine-derived collagen | Powder, non-woven sheet, sponge | Absorbable acid salt. Provides physical scaffold for platelet activation and clot initiation. | Sheet form may be passed through laparoscope; minimal expansion | Rare allergic reactions reported; may contribute to granuloma formation | 8–12 weeks | $$ |
| Biologically active agents | |||||||
| Topical thrombin (Thrombin-JMI, Recothrom, Evithrom, rh Thrombin) | Bovine, human, or recombinant | Liquid | Promotes conversion of fibrinogen to fibrin | May be combined effectively with physical agents of neutral pH; recombinant human thrombin will be available in the near future | Risk of blood-borne infection with non-recombinant human thrombin; risk of anaphylaxis and antibody formation with bovine thrombin | N/A | $$ |
| Hemostatic matrix (Floseal, Surgiflo) | Thrombin plus gelatin | Foam | Gelatin granules provide expansion and compression while thrombin initiates clot formation | May be used in areas of small arterial bleeding | Requires contact with blood | 6–8 weeks | $$$ |
| Fibrin sealants (Evicel, Tisseel, Crosseal) | Human | Liquid | Combination of fibrinogen and thrombin causes cleavage of fibrinogen to fibrin and resultant clot initiation | Fast-acting; hemostatic and adhesive properties; works well for diffusely oozing surfaces | Contraindicated in patients who have a history of anaphylactic reaction to serum-derived products or IgA deficiency | 10–14 days | $$$ |
| * Median cost for use in one case Key: $=inexpensive; $$=moderately expensive; $$$=expensive | |||||||
Controlling bleeding without injuring underlying tissue
CASE 2: After adhesiolysis, bleeding at multiple sites
You perform adnexectomy on a 47-year-old woman who has a large (7 to 8 cm), benign ovarian mass. As you operate, you discover that the lesion is adherent to the sigmoid mesentery and the posterior aspect of the uterus; it is also adherent to the pelvic sidewall, directly along the course of the ureter. Although you are able to release the various adhesive attachments, persistent bleeding is noted at multiple pinpoint areas along the mesentery, uterine serosa, and pelvic sidewall, even after the application of direct pressure.
What do you do next?
Although cautery can be used liberally on the uterus, its application to mesentery carries a risk of injury to the mesenteric vessels and bowel wall. Caution is advised when you are attempting to control bleeding on the peritoneum overlying the ureter, whether you are using suture ligature or an energy source. Ideally, you should identify the ureter using a retroperitoneal approach and mobilize it laterally before employing any of these techniques.
There are several potential approaches to the bleeding described in Case 2, all of them involving hemostatic adjuvants. The first decision you need to make, however, is whether to address each region separately or all sites in unison. If you opt to address them together—either during an open procedure or laparoscopy—a fibrin sealant (e.g., Evicel, Tisseel) is one option. It can be applied using a dripping technique or aerosolization, either of which allows for broad application of a thin film of the agent. The limitation of this approach is the volume of agent required to resolve the bleeding, with a potential need for multiple doses to completely coat the area.
Because fibrin sealants function independently of the patient’s coagulation cascade, they are particularly useful in the presence of disseminated intravascular coagulation (DIC) and other coagulopathies that might limit the effectiveness of preparations that require the patient’s own serum.
An alternative approach to Case 2 is to apply an oxidized regenerated cellulose (ORC) derivative directly to the affected areas. Various forms are available (e.g., Surgicel Fibrillar, Surgicel Nu-Knit). These ORC products can be cut and customized to the area in need of hemostasis, allowing each site to be addressed individually. These agents typically remain adherent after they are applied due to the nature of the interaction between the product, blood, and tissue.
A liquid or foam hemostatic agent (e.g., Surgiflo, Floseal, topical thrombin) could also be employed in this case, but application can be a challenge on a large area with a heterogeneous topography because of the tendency of such agents to migrate under the force of gravity, pooling away from the source of bleeding.
Is combining agents a good idea?
Although they are not typically approved for use in combination, sequential application of hemostatic agents may be considered when bleeding persists.
All hemostatic agents work best in combination with the application of pressure. It usually is advisable to use moist gauze for this purpose because it can be lifted away without significant adherence to the underlying hemostatic complex, avoiding clot
disruption.
CASE 2: Resolved
You opt to use an ORC product, customizing it to fit each bleeding site, and apply direct pressure. When hemostasis has been achieved at all sites, you complete the operation. The patient has an uneventful postoperative course.
Protect structures along the pelvic sidewall
CASE 3: When the application of pressure isn’t enough
While performing a left salpingo-oophorectomy for a 12-cm ovarian lesion, you use a retroperitoneal approach to identify the structures along the pelvic sidewall. During identification of the ureter, you encounter bleeding from a small vessel in the adjacent fatty areolar tissue. After a period of observation, during which you apply pressure to the area of concern, bleeding persists.
What hemostatic agent do you employ to stop it?
The careful application of steady pressure is often enough to safely control bleeding in the area of the pelvic sidewall. In the event that pressure alone fails to resolve the bleeding, however, it is critical to choose a remedy that avoids injuring the ureter, iliac vessels, and infundibulopelvic ligament. Wide exposure of the space may allow for direct identification of the point of bleeding and precise application of cautery, a hemoclip, or a tie. When this approach is not feasible, other solutions must be sought.
When traditional hemostatic techniques fail in delicate anatomic sites, such as the periureteral area, hemostatic agents are an effective option that can minimize the risk of injury to surrounding vital structures. The contour of the space calls for a product that can intercalate, such as a foam, sealant, or Surgicel Fibrillar. Direct, precise application to the point of bleeding is critical, and the “bunching up” of a more rigid and bulky agent may limit its application to the area of concern. Use of a moist gauze to apply direct pressure after application of the agent will increase the likelihood of success.
CASE 3: Resolved
You decide to apply a foam hemostatic agent because of its ability to conform to the irregular space. You also continue to apply gentle pressure to the point of bleeding, using a moist gauze. Within minutes, hemostasis is achieved. You are then able to finish the operation.
Other variables to consider
As these three cases illustrate, the use of hemostatic agents to control surgical bleeding requires an individualized approach. The site and amount of bleeding, as well as the patient’s hemodynamic and coagulation status, are key variables to be considered when selecting an agent.
For instance, because of their components, fibrin sealants can function independently of the patient’s coagulation status. ORC products provide a matrix that facilitates platelet aggregration and may be less effective when anti-platelet agents have been used.
It is also appropriate for the surgeon to be familiar with the relative cost of the agents available at his or her institution. In particular, when several agents may be equally effective in a particular set of circumstances, cost may be the determining factor.
Availability of these agents varies from one institution to the next; as a result, it can be challenging to maintain familiarity with all of the products in the marketplace. Having access to a diverse, readily available set of “go to” agents is critical to ensure rapid application in a clinical setting.
The surgeon’s preference also is important, particularly in regard to the ease of preparation and handling. Some agents may not be as suitable for minimally invasive procedures (see TABLE). For others, special laparoscopic applicators are available.
When using a hemostatic agent, it pays to consider the duration of its effect in the surgical site. Both the quantity of the agent that is applied and characteristics of the local operative site influence how quickly the agent degrades. Keep this in mind when imaging studies are planned for the early postoperative period. An ORC preparation, for example, may appear with small pockets of air that resemble an abscess. Effective communication with the radiology team is critical to avoid the misinterpretation of findings.
Curious to discover the preferences and practices of surgeons likely to utilize topical hemostatic agents, OBG Management polled several experienced and expert surgeons, including members of the journal’s Board of Editors and Virtual Board of Editors. Their diverse responses offer a snapshot of gynecologic surgical practice in 2012—but all agree that hemostatic products are no substitute for sound surgical technique.
JANELLE YATES, SENIOR EDITOR





We want to hear from you! Tell us what you think.
Recommended reading
Achneck HE, Sileshi B, Jamiolkowski RM, et al. A comprehensive review of topical hemostatic agents: efficacy and recommendations for use. Ann Surg. 2010;251(2):217-228.
Chapman WC, Singla N, Genyk Y, et al. A phase 3, randomized, double-blind comparative study of the efficacy and safety of topical recombinant human thrombin and bovine thrombin in surgical hemostasis. J Am Coll Surg. 2007;205(2):256-265.
Holub Z, Jabor A. Laparoscopic management of bleeding after laparoscopic or vaginal hysterectomy. JSLS. 2004;8(3):235-238.
Sharma JB, Malhotra M. Laparoscopic oxidized cellulose (Surgicel) application for small uterine perforations. Int J Gynaecol Obstet. 2003;83(3):271-275.
Sharma JB, Malhotra M. Topical oxidized cellulose for tubal hemorrhage hemostasis during laparoscopic sterilization. Int J Gynaecol Obstet. 2003;82(2):221-222.

Lisa A. dos Santos, MD
Dr. dos Santos is Attending Physician, Division of Gynecologic Oncology, at North Shore University Hospital–Long Island Jewish Medical Center in Manhasset, NY, and Assistant Professor of Obstetrics and Gynecology at Hofstra North Shore—LIJ School of Medicine in Hempstead, NY.

Andrew W. Menzin, MD
Dr. Menzin is Vice Chairman for Academic Affairs and Associate Chief of Gynecologic Oncology at North Shore University Hospital–Long Island Jewish Medical Center in Manhasset, NY.
Dr. dos Santos reports no financial relationships relevant to this article. Dr. Menzin reports that he is a consultant to Ethicon Biosurgery.
Lisa A. dos Santos, MD
Dr. dos Santos is Attending Physician, Division of Gynecologic Oncology, at North Shore University Hospital–Long Island Jewish Medical Center in Manhasset, NY, and Assistant Professor of Obstetrics and Gynecology at Hofstra North Shore—LIJ School of Medicine in Hempstead, NY.
Andrew W. Menzin, MD
Dr. Menzin is Vice Chairman for Academic Affairs and Associate Chief of Gynecologic Oncology at North Shore University Hospital–Long Island Jewish Medical Center in Manhasset, NY.
Dr. dos Santos reports no financial relationships relevant to this article. Dr. Menzin reports that he is a consultant to Ethicon Biosurgery.
Lisa A. dos Santos, MD
Dr. dos Santos is Attending Physician, Division of Gynecologic Oncology, at North Shore University Hospital–Long Island Jewish Medical Center in Manhasset, NY, and Assistant Professor of Obstetrics and Gynecology at Hofstra North Shore—LIJ School of Medicine in Hempstead, NY.
Andrew W. Menzin, MD
Dr. Menzin is Vice Chairman for Academic Affairs and Associate Chief of Gynecologic Oncology at North Shore University Hospital–Long Island Jewish Medical Center in Manhasset, NY.
Dr. dos Santos reports no financial relationships relevant to this article. Dr. Menzin reports that he is a consultant to Ethicon Biosurgery.
Vessel-sealing devices and hemostatic adjuvants are expanding the surgical armamentarium. These products provide a spectrum of alternatives that can serve you and your surgical patient well when traditional techniques for obtaining hemostasis fail to provide a satisfactory result. (Keep in mind, however, that technology is no substitute for excellent technique!)
In this article, we highlight three common scenarios in which topical hemostatic agents may be useful during gynecologic surgery. In addition, in the sidebar, five surgeons describe the hemostatic products they rely on most often—and tell why.
Following hysterectomy, persistent oozing along the anterior vaginal margin, distal to the cuff and adjacent to the site of bladder mobilization, may be managed with the aid of a topical hemostatic agent—in this case, a fibrin sealant.
When the site of bleeding is difficult to reach
CASE 1: Oozing at the site of bladder mobilization
You perform total hysterectomy in a 44-year-old woman who has uterine fibroids. After the procedure, you notice persistent oozing along the anterior vaginal margin, distal to the cuff and adjacent to where the bladder was mobilized.
How do you manage the oozing?
Wide mobilization of the bladder is a vital step in the safe performance of hysterectomy. Adhesions may complicate the process if the patient has had previous abdominal surgery, infection, or inflammation. Following mobilization of the bladder and removal of the uterus, bleeding may be visible along the adventitia of the posterior bladder wall or along the anterior surface of the vagina, distal to the cuff, as it is in this case (see the illustration).
Judicious application of an energy source is an option, but thermal injury to the bladder is a concern. A good alternative is proper placement of a hemostatic suture, but it can sometimes be difficult to avoid incorporating the bladder or injuring or obstructing the nearby ureter.
In this case, the location of the bleeding deep in the operative field poses a challenge, because of limited exposure and the proximity of the bladder and ureters. Virtually any hemostatic agent would work well in this circumstance (TABLE). For example, a flowable agent or fibrin sealant could be thoroughly applied to the area during a minimally invasive or open procedure and would naturally conform to the irregularities in the tissue, particularly the junction between the vagina and bladder flap.
A pliable product such as Surgicel Nu-Knit or Fibrillar would also work well in these circumstances, although successful application during laparoscopy may depend on the size of the trocar. For example, Nu-Knit would require trimming to a size suitable for passage through a trocar, made easier by moistening with saline. The weave of Fibrillar makes it more challenging to pass, intact, through a trocar; rolling the material into a cylindrical shape may reduce its diameter and allow it to pass more easily.
CASE 1: Resolved
You apply a fibrin sealant to the site of bleeding, and the oozing abates. Once complete hemostasis is ensured, you conclude the surgery and transfer the patient to recovery, where she does well.
Profiles in hemostasis: Strengths and weaknesses of topical agent
| Agent (brands) | Composition | Forms available | Mechanism of action | Advantages | Caveats | Duration | Relative cost* |
|---|---|---|---|---|---|---|---|
| Physical agents | |||||||
| Gelatin matrix (Gelfoam, Gelfilm, Surgifoam) | Porcine- derived collagen | Sponge, film, powder | Provides physical matrix for clot formation | Non-antigenic; neutral pH; may be used with thrombin | Material expansion may cause compression; Not for use in closed spaces or near nerve structures | 4–6 weeks | $ |
| Oxidized regenerated cellulose (Surgicel Fibrillar, Surgicel Nu-Knit) | Wood pulp | Mesh or packed fibers | Provides physical matrix for clot formation; acidic pH causes hemolysis and local clot formation | Pliable, easy to place through laparoscope; acidic pH has antimicrobial effect | Works best in a dry field. Acidic pH inactivates biologic agents, such as thrombin, and may increase inflammation. Avoid using excess material. | 2–4 weeks | $ |
| Microfibrillar collagen (Avitene, Instat, Helitene Helistat) | Bovine-derived collagen | Powder, non-woven sheet, sponge | Absorbable acid salt. Provides physical scaffold for platelet activation and clot initiation. | Sheet form may be passed through laparoscope; minimal expansion | Rare allergic reactions reported; may contribute to granuloma formation | 8–12 weeks | $$ |
| Biologically active agents | |||||||
| Topical thrombin (Thrombin-JMI, Recothrom, Evithrom, rh Thrombin) | Bovine, human, or recombinant | Liquid | Promotes conversion of fibrinogen to fibrin | May be combined effectively with physical agents of neutral pH; recombinant human thrombin will be available in the near future | Risk of blood-borne infection with non-recombinant human thrombin; risk of anaphylaxis and antibody formation with bovine thrombin | N/A | $$ |
| Hemostatic matrix (Floseal, Surgiflo) | Thrombin plus gelatin | Foam | Gelatin granules provide expansion and compression while thrombin initiates clot formation | May be used in areas of small arterial bleeding | Requires contact with blood | 6–8 weeks | $$$ |
| Fibrin sealants (Evicel, Tisseel, Crosseal) | Human | Liquid | Combination of fibrinogen and thrombin causes cleavage of fibrinogen to fibrin and resultant clot initiation | Fast-acting; hemostatic and adhesive properties; works well for diffusely oozing surfaces | Contraindicated in patients who have a history of anaphylactic reaction to serum-derived products or IgA deficiency | 10–14 days | $$$ |
| * Median cost for use in one case Key: $=inexpensive; $$=moderately expensive; $$$=expensive | |||||||
Controlling bleeding without injuring underlying tissue
CASE 2: After adhesiolysis, bleeding at multiple sites
You perform adnexectomy on a 47-year-old woman who has a large (7 to 8 cm), benign ovarian mass. As you operate, you discover that the lesion is adherent to the sigmoid mesentery and the posterior aspect of the uterus; it is also adherent to the pelvic sidewall, directly along the course of the ureter. Although you are able to release the various adhesive attachments, persistent bleeding is noted at multiple pinpoint areas along the mesentery, uterine serosa, and pelvic sidewall, even after the application of direct pressure.
What do you do next?
Although cautery can be used liberally on the uterus, its application to mesentery carries a risk of injury to the mesenteric vessels and bowel wall. Caution is advised when you are attempting to control bleeding on the peritoneum overlying the ureter, whether you are using suture ligature or an energy source. Ideally, you should identify the ureter using a retroperitoneal approach and mobilize it laterally before employing any of these techniques.
There are several potential approaches to the bleeding described in Case 2, all of them involving hemostatic adjuvants. The first decision you need to make, however, is whether to address each region separately or all sites in unison. If you opt to address them together—either during an open procedure or laparoscopy—a fibrin sealant (e.g., Evicel, Tisseel) is one option. It can be applied using a dripping technique or aerosolization, either of which allows for broad application of a thin film of the agent. The limitation of this approach is the volume of agent required to resolve the bleeding, with a potential need for multiple doses to completely coat the area.
Because fibrin sealants function independently of the patient’s coagulation cascade, they are particularly useful in the presence of disseminated intravascular coagulation (DIC) and other coagulopathies that might limit the effectiveness of preparations that require the patient’s own serum.
An alternative approach to Case 2 is to apply an oxidized regenerated cellulose (ORC) derivative directly to the affected areas. Various forms are available (e.g., Surgicel Fibrillar, Surgicel Nu-Knit). These ORC products can be cut and customized to the area in need of hemostasis, allowing each site to be addressed individually. These agents typically remain adherent after they are applied due to the nature of the interaction between the product, blood, and tissue.
A liquid or foam hemostatic agent (e.g., Surgiflo, Floseal, topical thrombin) could also be employed in this case, but application can be a challenge on a large area with a heterogeneous topography because of the tendency of such agents to migrate under the force of gravity, pooling away from the source of bleeding.
Is combining agents a good idea?
Although they are not typically approved for use in combination, sequential application of hemostatic agents may be considered when bleeding persists.
All hemostatic agents work best in combination with the application of pressure. It usually is advisable to use moist gauze for this purpose because it can be lifted away without significant adherence to the underlying hemostatic complex, avoiding clot
disruption.
CASE 2: Resolved
You opt to use an ORC product, customizing it to fit each bleeding site, and apply direct pressure. When hemostasis has been achieved at all sites, you complete the operation. The patient has an uneventful postoperative course.
Protect structures along the pelvic sidewall
CASE 3: When the application of pressure isn’t enough
While performing a left salpingo-oophorectomy for a 12-cm ovarian lesion, you use a retroperitoneal approach to identify the structures along the pelvic sidewall. During identification of the ureter, you encounter bleeding from a small vessel in the adjacent fatty areolar tissue. After a period of observation, during which you apply pressure to the area of concern, bleeding persists.
What hemostatic agent do you employ to stop it?
The careful application of steady pressure is often enough to safely control bleeding in the area of the pelvic sidewall. In the event that pressure alone fails to resolve the bleeding, however, it is critical to choose a remedy that avoids injuring the ureter, iliac vessels, and infundibulopelvic ligament. Wide exposure of the space may allow for direct identification of the point of bleeding and precise application of cautery, a hemoclip, or a tie. When this approach is not feasible, other solutions must be sought.
When traditional hemostatic techniques fail in delicate anatomic sites, such as the periureteral area, hemostatic agents are an effective option that can minimize the risk of injury to surrounding vital structures. The contour of the space calls for a product that can intercalate, such as a foam, sealant, or Surgicel Fibrillar. Direct, precise application to the point of bleeding is critical, and the “bunching up” of a more rigid and bulky agent may limit its application to the area of concern. Use of a moist gauze to apply direct pressure after application of the agent will increase the likelihood of success.
CASE 3: Resolved
You decide to apply a foam hemostatic agent because of its ability to conform to the irregular space. You also continue to apply gentle pressure to the point of bleeding, using a moist gauze. Within minutes, hemostasis is achieved. You are then able to finish the operation.
Other variables to consider
As these three cases illustrate, the use of hemostatic agents to control surgical bleeding requires an individualized approach. The site and amount of bleeding, as well as the patient’s hemodynamic and coagulation status, are key variables to be considered when selecting an agent.
For instance, because of their components, fibrin sealants can function independently of the patient’s coagulation status. ORC products provide a matrix that facilitates platelet aggregration and may be less effective when anti-platelet agents have been used.
It is also appropriate for the surgeon to be familiar with the relative cost of the agents available at his or her institution. In particular, when several agents may be equally effective in a particular set of circumstances, cost may be the determining factor.
Availability of these agents varies from one institution to the next; as a result, it can be challenging to maintain familiarity with all of the products in the marketplace. Having access to a diverse, readily available set of “go to” agents is critical to ensure rapid application in a clinical setting.
The surgeon’s preference also is important, particularly in regard to the ease of preparation and handling. Some agents may not be as suitable for minimally invasive procedures (see TABLE). For others, special laparoscopic applicators are available.
When using a hemostatic agent, it pays to consider the duration of its effect in the surgical site. Both the quantity of the agent that is applied and characteristics of the local operative site influence how quickly the agent degrades. Keep this in mind when imaging studies are planned for the early postoperative period. An ORC preparation, for example, may appear with small pockets of air that resemble an abscess. Effective communication with the radiology team is critical to avoid the misinterpretation of findings.
Curious to discover the preferences and practices of surgeons likely to utilize topical hemostatic agents, OBG Management polled several experienced and expert surgeons, including members of the journal’s Board of Editors and Virtual Board of Editors. Their diverse responses offer a snapshot of gynecologic surgical practice in 2012—but all agree that hemostatic products are no substitute for sound surgical technique.
JANELLE YATES, SENIOR EDITOR
We want to hear from you! Tell us what you think.
Vessel-sealing devices and hemostatic adjuvants are expanding the surgical armamentarium. These products provide a spectrum of alternatives that can serve you and your surgical patient well when traditional techniques for obtaining hemostasis fail to provide a satisfactory result. (Keep in mind, however, that technology is no substitute for excellent technique!)
In this article, we highlight three common scenarios in which topical hemostatic agents may be useful during gynecologic surgery. In addition, in the sidebar, five surgeons describe the hemostatic products they rely on most often—and tell why.
Following hysterectomy, persistent oozing along the anterior vaginal margin, distal to the cuff and adjacent to the site of bladder mobilization, may be managed with the aid of a topical hemostatic agent—in this case, a fibrin sealant.
When the site of bleeding is difficult to reach
CASE 1: Oozing at the site of bladder mobilization
You perform total hysterectomy in a 44-year-old woman who has uterine fibroids. After the procedure, you notice persistent oozing along the anterior vaginal margin, distal to the cuff and adjacent to where the bladder was mobilized.
How do you manage the oozing?
Wide mobilization of the bladder is a vital step in the safe performance of hysterectomy. Adhesions may complicate the process if the patient has had previous abdominal surgery, infection, or inflammation. Following mobilization of the bladder and removal of the uterus, bleeding may be visible along the adventitia of the posterior bladder wall or along the anterior surface of the vagina, distal to the cuff, as it is in this case (see the illustration).
Judicious application of an energy source is an option, but thermal injury to the bladder is a concern. A good alternative is proper placement of a hemostatic suture, but it can sometimes be difficult to avoid incorporating the bladder or injuring or obstructing the nearby ureter.
In this case, the location of the bleeding deep in the operative field poses a challenge, because of limited exposure and the proximity of the bladder and ureters. Virtually any hemostatic agent would work well in this circumstance (TABLE). For example, a flowable agent or fibrin sealant could be thoroughly applied to the area during a minimally invasive or open procedure and would naturally conform to the irregularities in the tissue, particularly the junction between the vagina and bladder flap.
A pliable product such as Surgicel Nu-Knit or Fibrillar would also work well in these circumstances, although successful application during laparoscopy may depend on the size of the trocar. For example, Nu-Knit would require trimming to a size suitable for passage through a trocar, made easier by moistening with saline. The weave of Fibrillar makes it more challenging to pass, intact, through a trocar; rolling the material into a cylindrical shape may reduce its diameter and allow it to pass more easily.
CASE 1: Resolved
You apply a fibrin sealant to the site of bleeding, and the oozing abates. Once complete hemostasis is ensured, you conclude the surgery and transfer the patient to recovery, where she does well.
Profiles in hemostasis: Strengths and weaknesses of topical agent
| Agent (brands) | Composition | Forms available | Mechanism of action | Advantages | Caveats | Duration | Relative cost* |
|---|---|---|---|---|---|---|---|
| Physical agents | |||||||
| Gelatin matrix (Gelfoam, Gelfilm, Surgifoam) | Porcine- derived collagen | Sponge, film, powder | Provides physical matrix for clot formation | Non-antigenic; neutral pH; may be used with thrombin | Material expansion may cause compression; Not for use in closed spaces or near nerve structures | 4–6 weeks | $ |
| Oxidized regenerated cellulose (Surgicel Fibrillar, Surgicel Nu-Knit) | Wood pulp | Mesh or packed fibers | Provides physical matrix for clot formation; acidic pH causes hemolysis and local clot formation | Pliable, easy to place through laparoscope; acidic pH has antimicrobial effect | Works best in a dry field. Acidic pH inactivates biologic agents, such as thrombin, and may increase inflammation. Avoid using excess material. | 2–4 weeks | $ |
| Microfibrillar collagen (Avitene, Instat, Helitene Helistat) | Bovine-derived collagen | Powder, non-woven sheet, sponge | Absorbable acid salt. Provides physical scaffold for platelet activation and clot initiation. | Sheet form may be passed through laparoscope; minimal expansion | Rare allergic reactions reported; may contribute to granuloma formation | 8–12 weeks | $$ |
| Biologically active agents | |||||||
| Topical thrombin (Thrombin-JMI, Recothrom, Evithrom, rh Thrombin) | Bovine, human, or recombinant | Liquid | Promotes conversion of fibrinogen to fibrin | May be combined effectively with physical agents of neutral pH; recombinant human thrombin will be available in the near future | Risk of blood-borne infection with non-recombinant human thrombin; risk of anaphylaxis and antibody formation with bovine thrombin | N/A | $$ |
| Hemostatic matrix (Floseal, Surgiflo) | Thrombin plus gelatin | Foam | Gelatin granules provide expansion and compression while thrombin initiates clot formation | May be used in areas of small arterial bleeding | Requires contact with blood | 6–8 weeks | $$$ |
| Fibrin sealants (Evicel, Tisseel, Crosseal) | Human | Liquid | Combination of fibrinogen and thrombin causes cleavage of fibrinogen to fibrin and resultant clot initiation | Fast-acting; hemostatic and adhesive properties; works well for diffusely oozing surfaces | Contraindicated in patients who have a history of anaphylactic reaction to serum-derived products or IgA deficiency | 10–14 days | $$$ |
| * Median cost for use in one case Key: $=inexpensive; $$=moderately expensive; $$$=expensive | |||||||
Controlling bleeding without injuring underlying tissue
CASE 2: After adhesiolysis, bleeding at multiple sites
You perform adnexectomy on a 47-year-old woman who has a large (7 to 8 cm), benign ovarian mass. As you operate, you discover that the lesion is adherent to the sigmoid mesentery and the posterior aspect of the uterus; it is also adherent to the pelvic sidewall, directly along the course of the ureter. Although you are able to release the various adhesive attachments, persistent bleeding is noted at multiple pinpoint areas along the mesentery, uterine serosa, and pelvic sidewall, even after the application of direct pressure.
What do you do next?
Although cautery can be used liberally on the uterus, its application to mesentery carries a risk of injury to the mesenteric vessels and bowel wall. Caution is advised when you are attempting to control bleeding on the peritoneum overlying the ureter, whether you are using suture ligature or an energy source. Ideally, you should identify the ureter using a retroperitoneal approach and mobilize it laterally before employing any of these techniques.
There are several potential approaches to the bleeding described in Case 2, all of them involving hemostatic adjuvants. The first decision you need to make, however, is whether to address each region separately or all sites in unison. If you opt to address them together—either during an open procedure or laparoscopy—a fibrin sealant (e.g., Evicel, Tisseel) is one option. It can be applied using a dripping technique or aerosolization, either of which allows for broad application of a thin film of the agent. The limitation of this approach is the volume of agent required to resolve the bleeding, with a potential need for multiple doses to completely coat the area.
Because fibrin sealants function independently of the patient’s coagulation cascade, they are particularly useful in the presence of disseminated intravascular coagulation (DIC) and other coagulopathies that might limit the effectiveness of preparations that require the patient’s own serum.
An alternative approach to Case 2 is to apply an oxidized regenerated cellulose (ORC) derivative directly to the affected areas. Various forms are available (e.g., Surgicel Fibrillar, Surgicel Nu-Knit). These ORC products can be cut and customized to the area in need of hemostasis, allowing each site to be addressed individually. These agents typically remain adherent after they are applied due to the nature of the interaction between the product, blood, and tissue.
A liquid or foam hemostatic agent (e.g., Surgiflo, Floseal, topical thrombin) could also be employed in this case, but application can be a challenge on a large area with a heterogeneous topography because of the tendency of such agents to migrate under the force of gravity, pooling away from the source of bleeding.
Is combining agents a good idea?
Although they are not typically approved for use in combination, sequential application of hemostatic agents may be considered when bleeding persists.
All hemostatic agents work best in combination with the application of pressure. It usually is advisable to use moist gauze for this purpose because it can be lifted away without significant adherence to the underlying hemostatic complex, avoiding clot
disruption.
CASE 2: Resolved
You opt to use an ORC product, customizing it to fit each bleeding site, and apply direct pressure. When hemostasis has been achieved at all sites, you complete the operation. The patient has an uneventful postoperative course.
Protect structures along the pelvic sidewall
CASE 3: When the application of pressure isn’t enough
While performing a left salpingo-oophorectomy for a 12-cm ovarian lesion, you use a retroperitoneal approach to identify the structures along the pelvic sidewall. During identification of the ureter, you encounter bleeding from a small vessel in the adjacent fatty areolar tissue. After a period of observation, during which you apply pressure to the area of concern, bleeding persists.
What hemostatic agent do you employ to stop it?
The careful application of steady pressure is often enough to safely control bleeding in the area of the pelvic sidewall. In the event that pressure alone fails to resolve the bleeding, however, it is critical to choose a remedy that avoids injuring the ureter, iliac vessels, and infundibulopelvic ligament. Wide exposure of the space may allow for direct identification of the point of bleeding and precise application of cautery, a hemoclip, or a tie. When this approach is not feasible, other solutions must be sought.
When traditional hemostatic techniques fail in delicate anatomic sites, such as the periureteral area, hemostatic agents are an effective option that can minimize the risk of injury to surrounding vital structures. The contour of the space calls for a product that can intercalate, such as a foam, sealant, or Surgicel Fibrillar. Direct, precise application to the point of bleeding is critical, and the “bunching up” of a more rigid and bulky agent may limit its application to the area of concern. Use of a moist gauze to apply direct pressure after application of the agent will increase the likelihood of success.
CASE 3: Resolved
You decide to apply a foam hemostatic agent because of its ability to conform to the irregular space. You also continue to apply gentle pressure to the point of bleeding, using a moist gauze. Within minutes, hemostasis is achieved. You are then able to finish the operation.
Other variables to consider
As these three cases illustrate, the use of hemostatic agents to control surgical bleeding requires an individualized approach. The site and amount of bleeding, as well as the patient’s hemodynamic and coagulation status, are key variables to be considered when selecting an agent.
For instance, because of their components, fibrin sealants can function independently of the patient’s coagulation status. ORC products provide a matrix that facilitates platelet aggregration and may be less effective when anti-platelet agents have been used.
It is also appropriate for the surgeon to be familiar with the relative cost of the agents available at his or her institution. In particular, when several agents may be equally effective in a particular set of circumstances, cost may be the determining factor.
Availability of these agents varies from one institution to the next; as a result, it can be challenging to maintain familiarity with all of the products in the marketplace. Having access to a diverse, readily available set of “go to” agents is critical to ensure rapid application in a clinical setting.
The surgeon’s preference also is important, particularly in regard to the ease of preparation and handling. Some agents may not be as suitable for minimally invasive procedures (see TABLE). For others, special laparoscopic applicators are available.
When using a hemostatic agent, it pays to consider the duration of its effect in the surgical site. Both the quantity of the agent that is applied and characteristics of the local operative site influence how quickly the agent degrades. Keep this in mind when imaging studies are planned for the early postoperative period. An ORC preparation, for example, may appear with small pockets of air that resemble an abscess. Effective communication with the radiology team is critical to avoid the misinterpretation of findings.
Curious to discover the preferences and practices of surgeons likely to utilize topical hemostatic agents, OBG Management polled several experienced and expert surgeons, including members of the journal’s Board of Editors and Virtual Board of Editors. Their diverse responses offer a snapshot of gynecologic surgical practice in 2012—but all agree that hemostatic products are no substitute for sound surgical technique.
JANELLE YATES, SENIOR EDITOR
We want to hear from you! Tell us what you think.
Recommended reading
Achneck HE, Sileshi B, Jamiolkowski RM, et al. A comprehensive review of topical hemostatic agents: efficacy and recommendations for use. Ann Surg. 2010;251(2):217-228.
Chapman WC, Singla N, Genyk Y, et al. A phase 3, randomized, double-blind comparative study of the efficacy and safety of topical recombinant human thrombin and bovine thrombin in surgical hemostasis. J Am Coll Surg. 2007;205(2):256-265.
Holub Z, Jabor A. Laparoscopic management of bleeding after laparoscopic or vaginal hysterectomy. JSLS. 2004;8(3):235-238.
Sharma JB, Malhotra M. Laparoscopic oxidized cellulose (Surgicel) application for small uterine perforations. Int J Gynaecol Obstet. 2003;83(3):271-275.
Sharma JB, Malhotra M. Topical oxidized cellulose for tubal hemorrhage hemostasis during laparoscopic sterilization. Int J Gynaecol Obstet. 2003;82(2):221-222.
Recommended reading
Achneck HE, Sileshi B, Jamiolkowski RM, et al. A comprehensive review of topical hemostatic agents: efficacy and recommendations for use. Ann Surg. 2010;251(2):217-228.
Chapman WC, Singla N, Genyk Y, et al. A phase 3, randomized, double-blind comparative study of the efficacy and safety of topical recombinant human thrombin and bovine thrombin in surgical hemostasis. J Am Coll Surg. 2007;205(2):256-265.
Holub Z, Jabor A. Laparoscopic management of bleeding after laparoscopic or vaginal hysterectomy. JSLS. 2004;8(3):235-238.
Sharma JB, Malhotra M. Laparoscopic oxidized cellulose (Surgicel) application for small uterine perforations. Int J Gynaecol Obstet. 2003;83(3):271-275.
Sharma JB, Malhotra M. Topical oxidized cellulose for tubal hemorrhage hemostasis during laparoscopic sterilization. Int J Gynaecol Obstet. 2003;82(2):221-222.
Clonazepam dosing
Dr. Scott Freeman’s useful discussion of targeting acute risk factors in suicidal patients (“Suicide assessment: Targeting acute risk factors,” Current Psychiatry, January 2012, p. 52-57) ends by resolving the clinical vignette with a summary of hospital treatment. Apart from failing to indicate any psychotherapeutic inroads, Dr. Freeman seems to support prescribing clonazepam, 0.5 mg twice daily and 1 mg at bedtime. Clonazepam apparently “worked” by alleviating the patient’s anxiety and insomnia, but defied any pharmacologic rationale insofar as clonazepam has a slow onset and long half-life, making 3 doses per day irrational. This treatment strategy also risks problems of cumulative excess in the long run after discharge.
Aggressive pharmacotherapy may be the hallmark of modern acute hospital treatment, but surely it should incorporate careful understanding of specific medications’ pharmacodynamics, especially when relying on benzodiazepines. Needless to say, beginning a psychological process in the hospital also appears to have been shortchanged.
Sara Hartley, MD
Lecturer, Clinical Skills Program
University of California,
Berkeley-University of California,
San Francisco Joint Medical Program
Berkeley, CA
Dr. Freeman responds
I appreciate Dr. Hartley’s interest in my article. Although I agree with her that psychotherapy is an integral part of any treatment plan, the clinical vignette was used only to emphasize the need to aggressively and quickly start antidepressant and, more importantly, anxiolytic pharmacologic treatment in acutely suicidal patients with severe anxiety and depression.
With regard to clonazepam’s pharmacokinetics, although it does have a long half-life, it is only weakly lipophilic compared with other long-acting benzodiazepines such as diazepam. In fact, clonazepam has been shown to be less lipophilic than lorazepam,1 meaning it has a much smaller volume of distribution and less accumulation in peripheral adipose tissue. Therefore, one would not be concerned about significant drug accumulation leading to unexpected toxicity with a less lipophilic agent such as clonazepam.
I do not agree that dosing clonazepam 3 times a day, especially in an acute crisis, is “irrational,” as Dr. Hartley suggests. According to the package insert, although clonazepam is recommended to be administered twice daily for panic disorder, it can be given 3 times a day for seizure disorders.2
Scott A. Freeman, MD
Medical Director
Schizophrenia and Bipolar Disorder Inpatient Unit
McLean Hospital
Belmont, MA
Dr. Scott Freeman’s useful discussion of targeting acute risk factors in suicidal patients (“Suicide assessment: Targeting acute risk factors,” Current Psychiatry, January 2012, p. 52-57) ends by resolving the clinical vignette with a summary of hospital treatment. Apart from failing to indicate any psychotherapeutic inroads, Dr. Freeman seems to support prescribing clonazepam, 0.5 mg twice daily and 1 mg at bedtime. Clonazepam apparently “worked” by alleviating the patient’s anxiety and insomnia, but defied any pharmacologic rationale insofar as clonazepam has a slow onset and long half-life, making 3 doses per day irrational. This treatment strategy also risks problems of cumulative excess in the long run after discharge.
Aggressive pharmacotherapy may be the hallmark of modern acute hospital treatment, but surely it should incorporate careful understanding of specific medications’ pharmacodynamics, especially when relying on benzodiazepines. Needless to say, beginning a psychological process in the hospital also appears to have been shortchanged.
Sara Hartley, MD
Lecturer, Clinical Skills Program
University of California,
Berkeley-University of California,
San Francisco Joint Medical Program
Berkeley, CA
Dr. Freeman responds
I appreciate Dr. Hartley’s interest in my article. Although I agree with her that psychotherapy is an integral part of any treatment plan, the clinical vignette was used only to emphasize the need to aggressively and quickly start antidepressant and, more importantly, anxiolytic pharmacologic treatment in acutely suicidal patients with severe anxiety and depression.
With regard to clonazepam’s pharmacokinetics, although it does have a long half-life, it is only weakly lipophilic compared with other long-acting benzodiazepines such as diazepam. In fact, clonazepam has been shown to be less lipophilic than lorazepam,1 meaning it has a much smaller volume of distribution and less accumulation in peripheral adipose tissue. Therefore, one would not be concerned about significant drug accumulation leading to unexpected toxicity with a less lipophilic agent such as clonazepam.
I do not agree that dosing clonazepam 3 times a day, especially in an acute crisis, is “irrational,” as Dr. Hartley suggests. According to the package insert, although clonazepam is recommended to be administered twice daily for panic disorder, it can be given 3 times a day for seizure disorders.2
Scott A. Freeman, MD
Medical Director
Schizophrenia and Bipolar Disorder Inpatient Unit
McLean Hospital
Belmont, MA
Dr. Scott Freeman’s useful discussion of targeting acute risk factors in suicidal patients (“Suicide assessment: Targeting acute risk factors,” Current Psychiatry, January 2012, p. 52-57) ends by resolving the clinical vignette with a summary of hospital treatment. Apart from failing to indicate any psychotherapeutic inroads, Dr. Freeman seems to support prescribing clonazepam, 0.5 mg twice daily and 1 mg at bedtime. Clonazepam apparently “worked” by alleviating the patient’s anxiety and insomnia, but defied any pharmacologic rationale insofar as clonazepam has a slow onset and long half-life, making 3 doses per day irrational. This treatment strategy also risks problems of cumulative excess in the long run after discharge.
Aggressive pharmacotherapy may be the hallmark of modern acute hospital treatment, but surely it should incorporate careful understanding of specific medications’ pharmacodynamics, especially when relying on benzodiazepines. Needless to say, beginning a psychological process in the hospital also appears to have been shortchanged.
Sara Hartley, MD
Lecturer, Clinical Skills Program
University of California,
Berkeley-University of California,
San Francisco Joint Medical Program
Berkeley, CA
Dr. Freeman responds
I appreciate Dr. Hartley’s interest in my article. Although I agree with her that psychotherapy is an integral part of any treatment plan, the clinical vignette was used only to emphasize the need to aggressively and quickly start antidepressant and, more importantly, anxiolytic pharmacologic treatment in acutely suicidal patients with severe anxiety and depression.
With regard to clonazepam’s pharmacokinetics, although it does have a long half-life, it is only weakly lipophilic compared with other long-acting benzodiazepines such as diazepam. In fact, clonazepam has been shown to be less lipophilic than lorazepam,1 meaning it has a much smaller volume of distribution and less accumulation in peripheral adipose tissue. Therefore, one would not be concerned about significant drug accumulation leading to unexpected toxicity with a less lipophilic agent such as clonazepam.
I do not agree that dosing clonazepam 3 times a day, especially in an acute crisis, is “irrational,” as Dr. Hartley suggests. According to the package insert, although clonazepam is recommended to be administered twice daily for panic disorder, it can be given 3 times a day for seizure disorders.2
Scott A. Freeman, MD
Medical Director
Schizophrenia and Bipolar Disorder Inpatient Unit
McLean Hospital
Belmont, MA
Using melatonin to reset the clock of hospitalized older patients
Helping hospitalized geriatric patients maintain an appropriate sleep-wake cycle can be a challenge. Older patients’ circadian rhythm may be affected by several factors—eg, obstructive sleep apnea and restless leg syndrome—that contribute to disrupted sleep and daytime fatigue. Some patients may have dementing illnesses that could dysregulate sleep. Many older patients experience delirium during hospitalization, of which sleep-wake cycle disturbances are a hallmark. Finally, geriatric patients’ natural sleep pattern often does not mimic a hospital’s typical schedule.
Sleep medication side effects
Medications used to promote sleep can cause side effects in geriatric patients. Benzodiazepine use by older adults is discouraged because these medications could cause falls or contribute to delirium. Non-benzodiazepine hypnotics such as zolpidem, zaleplon, and eszopiclone pose a similar risk. Medications containing diphenhydramine predispose patients to deliriogenic effects via their anticholinergic properties. Tricyclic antidepressants carry risks, such as delirium secondary to anticholinergic effects, orthostatic hypotension, falls from α-1 blockade, and cardiac arrythmias.
Atypical antipsychotics sometimes are used off-label to help initiate sleep, but they carry a “black-box” warning regarding sudden death from cardiovascular events in geriatric patients with dementia. Hydroxyzine and trazodone also are associated with side effects such as orthostatic hypotension and daytime sedation, and are not always effective.
Melatonin is a hormone secreted by the pineal gland in response to darkness, under the control of the suprachiasmatic nucleus (SCN), and is thought to promote sleep via synchronizing effects on the SCN.1 Melatonin is available as an over-the-counter dietary supplement and via prescription in dosages of 1 or 3 mg. The typical effective dose is 3 to 9 mg.1 Patients should take melatonin in the mid-evening, ideally between 7 pm and 8 pm, and effects become evident after a few days. Side effects are rare; the most common are headache and nausea. Daytime sedation and vivid dreams also have been reported. Melatonin can be used safely in conjunction with other sleep aids and its major drug-drug interactions involve enhancing the effects of other sedatives.2
We have found melatonin to be effective for treating sleep disturbances in older hospitalized patients. Its effectiveness may stem from the high incidence of dysregulated or calcified pineal glands in geriatric patients, which leads to a marked reduction in melatonin secretion.3 Recent evidence also suggests melatonin may reduce the incidence of delirium in older adults, and it has been proposed as a delirium treatment in post-operative and intensive care unit settings.4
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. de Jonghe A, Korevaar JC, van Munster BC, et al. Effectiveness of melatonin treatment on circadian rhythm disturbances in dementia. Are there implications for delirium? A systematic review. Int J Geriatr Psychiatry. 2010;25(12):1201-1208.
2. Werneke U, Turner T, Priebe S. Complementary medicines in psychiatry: review of effectiveness and safety. Br J Psychiatry. 2006;188:109-121.
3. Schmid HA. Decreased melatonin biosynthesis calcium flux, pineal gland calcification and aging: a hypothetical framework. Gerontology. 1993;39(4):189-199.
4. Al-Aama T, Brymer C, Gutmanis I, et al. Melatonin decreases delirium in elderly patients: a randomized, placebo-controlled trial. Int J Geriatr Psychiatry. 2011;26(7):687-694.
Helping hospitalized geriatric patients maintain an appropriate sleep-wake cycle can be a challenge. Older patients’ circadian rhythm may be affected by several factors—eg, obstructive sleep apnea and restless leg syndrome—that contribute to disrupted sleep and daytime fatigue. Some patients may have dementing illnesses that could dysregulate sleep. Many older patients experience delirium during hospitalization, of which sleep-wake cycle disturbances are a hallmark. Finally, geriatric patients’ natural sleep pattern often does not mimic a hospital’s typical schedule.
Sleep medication side effects
Medications used to promote sleep can cause side effects in geriatric patients. Benzodiazepine use by older adults is discouraged because these medications could cause falls or contribute to delirium. Non-benzodiazepine hypnotics such as zolpidem, zaleplon, and eszopiclone pose a similar risk. Medications containing diphenhydramine predispose patients to deliriogenic effects via their anticholinergic properties. Tricyclic antidepressants carry risks, such as delirium secondary to anticholinergic effects, orthostatic hypotension, falls from α-1 blockade, and cardiac arrythmias.
Atypical antipsychotics sometimes are used off-label to help initiate sleep, but they carry a “black-box” warning regarding sudden death from cardiovascular events in geriatric patients with dementia. Hydroxyzine and trazodone also are associated with side effects such as orthostatic hypotension and daytime sedation, and are not always effective.
Melatonin is a hormone secreted by the pineal gland in response to darkness, under the control of the suprachiasmatic nucleus (SCN), and is thought to promote sleep via synchronizing effects on the SCN.1 Melatonin is available as an over-the-counter dietary supplement and via prescription in dosages of 1 or 3 mg. The typical effective dose is 3 to 9 mg.1 Patients should take melatonin in the mid-evening, ideally between 7 pm and 8 pm, and effects become evident after a few days. Side effects are rare; the most common are headache and nausea. Daytime sedation and vivid dreams also have been reported. Melatonin can be used safely in conjunction with other sleep aids and its major drug-drug interactions involve enhancing the effects of other sedatives.2
We have found melatonin to be effective for treating sleep disturbances in older hospitalized patients. Its effectiveness may stem from the high incidence of dysregulated or calcified pineal glands in geriatric patients, which leads to a marked reduction in melatonin secretion.3 Recent evidence also suggests melatonin may reduce the incidence of delirium in older adults, and it has been proposed as a delirium treatment in post-operative and intensive care unit settings.4
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Helping hospitalized geriatric patients maintain an appropriate sleep-wake cycle can be a challenge. Older patients’ circadian rhythm may be affected by several factors—eg, obstructive sleep apnea and restless leg syndrome—that contribute to disrupted sleep and daytime fatigue. Some patients may have dementing illnesses that could dysregulate sleep. Many older patients experience delirium during hospitalization, of which sleep-wake cycle disturbances are a hallmark. Finally, geriatric patients’ natural sleep pattern often does not mimic a hospital’s typical schedule.
Sleep medication side effects
Medications used to promote sleep can cause side effects in geriatric patients. Benzodiazepine use by older adults is discouraged because these medications could cause falls or contribute to delirium. Non-benzodiazepine hypnotics such as zolpidem, zaleplon, and eszopiclone pose a similar risk. Medications containing diphenhydramine predispose patients to deliriogenic effects via their anticholinergic properties. Tricyclic antidepressants carry risks, such as delirium secondary to anticholinergic effects, orthostatic hypotension, falls from α-1 blockade, and cardiac arrythmias.
Atypical antipsychotics sometimes are used off-label to help initiate sleep, but they carry a “black-box” warning regarding sudden death from cardiovascular events in geriatric patients with dementia. Hydroxyzine and trazodone also are associated with side effects such as orthostatic hypotension and daytime sedation, and are not always effective.
Melatonin is a hormone secreted by the pineal gland in response to darkness, under the control of the suprachiasmatic nucleus (SCN), and is thought to promote sleep via synchronizing effects on the SCN.1 Melatonin is available as an over-the-counter dietary supplement and via prescription in dosages of 1 or 3 mg. The typical effective dose is 3 to 9 mg.1 Patients should take melatonin in the mid-evening, ideally between 7 pm and 8 pm, and effects become evident after a few days. Side effects are rare; the most common are headache and nausea. Daytime sedation and vivid dreams also have been reported. Melatonin can be used safely in conjunction with other sleep aids and its major drug-drug interactions involve enhancing the effects of other sedatives.2
We have found melatonin to be effective for treating sleep disturbances in older hospitalized patients. Its effectiveness may stem from the high incidence of dysregulated or calcified pineal glands in geriatric patients, which leads to a marked reduction in melatonin secretion.3 Recent evidence also suggests melatonin may reduce the incidence of delirium in older adults, and it has been proposed as a delirium treatment in post-operative and intensive care unit settings.4
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. de Jonghe A, Korevaar JC, van Munster BC, et al. Effectiveness of melatonin treatment on circadian rhythm disturbances in dementia. Are there implications for delirium? A systematic review. Int J Geriatr Psychiatry. 2010;25(12):1201-1208.
2. Werneke U, Turner T, Priebe S. Complementary medicines in psychiatry: review of effectiveness and safety. Br J Psychiatry. 2006;188:109-121.
3. Schmid HA. Decreased melatonin biosynthesis calcium flux, pineal gland calcification and aging: a hypothetical framework. Gerontology. 1993;39(4):189-199.
4. Al-Aama T, Brymer C, Gutmanis I, et al. Melatonin decreases delirium in elderly patients: a randomized, placebo-controlled trial. Int J Geriatr Psychiatry. 2011;26(7):687-694.
1. de Jonghe A, Korevaar JC, van Munster BC, et al. Effectiveness of melatonin treatment on circadian rhythm disturbances in dementia. Are there implications for delirium? A systematic review. Int J Geriatr Psychiatry. 2010;25(12):1201-1208.
2. Werneke U, Turner T, Priebe S. Complementary medicines in psychiatry: review of effectiveness and safety. Br J Psychiatry. 2006;188:109-121.
3. Schmid HA. Decreased melatonin biosynthesis calcium flux, pineal gland calcification and aging: a hypothetical framework. Gerontology. 1993;39(4):189-199.
4. Al-Aama T, Brymer C, Gutmanis I, et al. Melatonin decreases delirium in elderly patients: a randomized, placebo-controlled trial. Int J Geriatr Psychiatry. 2011;26(7):687-694.
Benzodiazepines: A versatile clinical tool
Since the discovery of chlordiazepoxide in the 1950s, benzodiazepines have revolutionized the treatment of anxiety and insomnia, largely because of their improved safety profile compared with barbiturates, formerly the preferred sedative-hypnotic.1 In addition to their anxiolytic and sedative-hypnotic effects, benzodiazepines exhibit anterograde amnesia, anticonvulsant, and muscle relaxant properties.1 Psychiatrists use benzodiazepines to treat anxiety and sleep disorders, acute agitation, alcohol withdrawal, catatonia, and psychotropic side effects such as akathisia. This article highlights the evidence for using benzodiazepines in anxiety and other disorders and why they generally should not be used for obsessive-compulsive disorder and posttraumatic stress disorder (Box 1).
Current evidence indicates little support for using benzodiazepines for obsessive-compulsive disorder (OCD). The American Psychiatric Association (APA) and the World Federation of Biological Psychiatry do not recommend benzodiazepines for treating OCD because of a lack of evidence for efficacy.a,b An earlier study suggested clonazepam monotherapy was effective for OCDc; however, a more recent study did not show a benefit on rate of response or degree of symptom improvement.d Augmentation strategies with benzodiazepines also do not appear to be beneficial for OCD management. A recent double-blind, placebo-controlled study failed to demonstrate faster symptom improvement by augmenting sertraline with clonazepam, although the study had a small sample size and high drop-out rate.e
Because benzodiazepines have negligible action on core posttraumatic stress disorder (PTSD) symptoms (re-experiencing, avoidance, and hyperarousal), selective serotonin reuptake inhibitors and other agents largely have supplanted them for PTSD treatment.f Use of benzodiazepines for PTSD is associated with withdrawal symptoms, more severe symptoms after discontinuation, and possible disinhibition, and may interfere with patients’ efforts to integrate trauma experiences. Although benzodiazepines may reduce distress associated with acute trauma, there is evidence—in clinical studies and animal models—that early benzodiazepine administration fails to prevent PTSD and may increase its incidence.g The International Consensus Group on Depression and Anxiety, the APA, and the British Association for Psychopharmacology all highlight the limited role, if any, for benzodiazepines in PTSD.h-j
References
- Bandelow B, Zohar J, Hollander E, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive-compulsive and post-traumatic stress disorders - first revision. World J Biol Psychiatry. 2008;9(4):248-312.
- American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder. Arlington, VA: American Psychiatric Publishing, Inc.; 2007.
- Hewlett WA, Vinogradov S, Agras WS. Clomipramine, clonazepam, and clonidine treatment of obsessive compulsive disorder. J Clin Psychopharmacol. 1992;12(6):420-430.
- Hollander E, Kaplan A, Stahl SM. A double-blind, placebo-controlled trial of clonazepam in obsessive-compulsive disorder. World J Biol Psychiatry. 2003;4(1):30-34.
- Crockett BA, Churchill E, Davidson JR. A double-blind combination study of clonazepam and sertraline in OCD. Ann Clin Psychiatry. 2004;16(3):127-132.
- Argyropoulos SV, Sandford JJ, Nutt DJ. The psychobiology of anxiolytic drugs. Part 2: pharmacological treatments of anxiety. Pharmacol Ther. 2000;88(3):213-227.
- Matar MA, Zohar J, Kaplan Z, et al. Alprazolam treatment immediately after stress exposure interferes with the normal HPA-stress response and increases vulnerability to subsequent stress in an animal model of PTSD. Eur Neuropsychopharmacol. 2009;19(4):283-295.
- Ballenger JC, Davidson JR, Lecrubier Y, et al. Consensus statement update on posttraumatic stress disorder from the international consensus group on depression and anxiety. J Clin Psychiatry. 2004;65(suppl 1):55-62.
- Ursano RJ, Bell C, Eth S, et al. Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Am J Psychiatry. 2004;161(11 suppl):3-31.
- Baldwin DS, Anderson IM, Nutt DJ, et al. Evidence-based guidelines for the pharmacological treatment of anxiety disorders: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2005;19(6):567-596.
Pharmacokinetic properties
Most benzodiazepines are considered to have similar efficacy; therefore, selection is based on pharmacokinetic considerations. Table 1 compares the indication, onset, and half-life of 12 commonly used benzodiazepines.2-6 Although Table 1 lists approximate equivalent doses, studies report inconsistent data. These are approximations only and should not be used independently to make therapy decisions.
Table 1
Oral benzodiazepines: Indications, onset, half-life, and equivalent doses
| Drug | FDA-approved indication(s) | Onset of action | Approximate half-life (hours) in healthy adults | Approximate equivalent dose (mg)a | Comments |
|---|---|---|---|---|---|
| Alprazolam | Anxiety disorders, panic disorder | Intermediate | 6.3 to 26.9 (IR), 10.7 to 15.8 (XR) | 0.5 | Increased risk for abuse because of greater lipid solubility |
| Chlordiazepoxide | Anxiety disorders, acute alcohol withdrawal, preoperative apprehension and anxiety | Intermediate | 24 to 48 | 10 | Risk for accumulation because of long-acting metabolites (desmethyldiazepam, oxazepam) |
| Clonazepam | Seizure disorders, panic disorder | Intermediate | 18 to 50 | 0.25 to 0.5 | Use caution in patients with liver disease |
| Clorazepate | Anxiety, seizures, acute alcohol withdrawal | Fast | 40 to 50 | 7.5 | Risk for accumulation because of long-acting metabolites (desmethyldiazepam, oxazepam) |
| Diazepam | Anxiety disorders, acute alcohol withdrawal, muscle spasms, convulsive disorders | Very fast | 20 to 100 | 5 | Risk for accumulation because of long-acting metabolites (temazepam, desmethyldiazepam, oxazepam). Increased risk for abuse because of quick onset |
| Estazolam | Insomnia | Intermediate | 10 to 24 | 0.3 to 2 | None |
| Flurazepam | Insomnia | Intermediate | 47 to 100 | 30 | Avoid in geriatric patients or patients with liver impairment |
| Lorazepam | Anxiety | Intermediate | 10 to 20 | 1 | Preferred for patients with liver impairment and geriatric patients |
| Oxazepam | Anxiety, acute alcohol withdrawal | Slow to intermediate | 5 to 20 | 30 | Preferred for patients with liver impairment and geriatric patients |
| Quazepam | Insomnia | Intermediate | 39 to 73 | 5 to 15 | Risk for accumulation because of long-acting metabolites (desmethyldiazepam, oxazepam) |
| Temazepam | Insomnia | Intermediate | 3.5 to 18.4 | 30 | Preferred for patients with liver impairment and geriatric patients |
| Triazolam | Insomnia | Fast | 1.5 to 5.5 | 0.25 | Lacks active metabolites |
| IR: immediate release; XR: extended release aInterpret with caution, conflicting data exist Source: References 2-6 | |||||
A diverse range of indications
Alcohol withdrawal. Benzodiazepines are the treatment of choice for alcohol withdrawal syndrome, particularly to prevent seizures.7 Research supports symptom-triggered therapy using the revised Clinical Institute Withdrawal Assessment for Alcohol. Benzodiazepines reduce CNS sympathetic hyperactivity to mitigate withdrawal from alcohol by decreasing tachycardia, tremor, insomnia, agitation, and anxiety. Furthermore, these agents provide prophylaxis against serious sequelae such as seizures and delirium.
Insomnia. The American Academy of Sleep Medicine considers benzodiazepine receptor agonists (BzRAs, which include benzodiazepines and non-benzodiazepines) and ramelteon first-line pharmacotherapy for primary insomnia.8 However, pharmacologic treatment should be short-term. Agents with short to intermediate half-lives and rapid onset, such as triazolam, can aid sleep initiation. Those with longer half-lives, such as temazepam, could address sleep maintenance. If a patient does not respond to the initial agent, try another medication within the same class, because patients may respond differently. Use lower starting doses in geriatric patients.9 Closely monitor for adverse effects, rebound insomnia, and potential abuse or tolerance. Identify comorbid conditions and medications that may impair sleep, and address them accordingly.
Psychological and behavioral treatments given over 4 to 8 weeks can yield stable sleep improvements for up to 2 years. If available, these interventions may be considered first-line for treating insomnia because of their lasting effects compared with BzRAs.10
Generalized anxiety disorder (GAD). Benzodiazepines effectively treat GAD because they work quickly and are well tolerated. However, there are better first-line treatment options when considering efficacy studies and dependence and tolerance concerns. One effect-size comparison of 21 double-blind, placebo-controlled trials showed that the efficacy of selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors (SNRIs), and pregabalin are comparable to benzodiazepines.11 Benzodiazepines can be used in the first 2 to 3 weeks after initiating antidepressants to alleviate and prevent worsening of anxiety that may occur at the start of antidepressant therapy. Recent treatment guidelines recommend benzodiazepines as a second-line treatment or for treatment-resistant GAD in patients who do not have a substance abuse history.12,13
Panic disorder. Efficacy of benzodiazepines for panic disorder is comparable to SSRIs, SNRIs, and tricyclic antidepressants (TCAs). SSRIs and SNRIs are considered first-line treatments for panic disorder because of their favorable side effect profile.14 In practice, benzodiazepines often are combined with SSRIs, SNRIs, or TCAs. A randomized controlled trial demonstrated that paroxetine and clonazepam (mean dose 1.6 mg/d at 5 weeks) resulted in a more rapid response compared with paroxetine alone, although this difference lasted only a few weeks.15 Furthermore, this study suggested that brief treatment with clonazepam followed by a taper is as effective as sustained treatment with paroxetine and clonazepam.15
There is a lack of high-quality data on combining cognitive-behavioral therapy (CBT) and benzodiazepines for panic disorder, although a Cochrane Review found that adding a benzodiazepine to CBT did not lead to a significant difference in response compared with psychotherapy alone.16 A recent randomized controlled trial demonstrated that tapering benzodiazepines combined with CBT was associated with successful discontinuation of the drug and prevented return of panic symptoms.17
Social anxiety. A meta-analysis found that for treating social anxiety, benzodiazepines have better efficacy than SSRIs, monoamine oxidase inhibitors, and anticonvulsants.18 Longer-acting benzodiazepines may be more effective than shorter-acting agents. One study of patients with social anxiety showed a 38% response rate for alprazolam vs 20% for placebo over 12 weeks, and a similar 10-week study demonstrated a 73% recovery rate with clonazepam vs 22% for placebo.19 In addition, studies have observed that patients can be maintained on clonazepam for up to 2 years without symptom relapse and will tolerate slow-taper discontinuation.18,20 Sedation and drowsiness can be lessened by limiting clonazepam doses to 2 to 3 mg/d.
Akathisia and tremor. Akathisia, a syndrome of motor restlessness and inner turmoil, is associated with antipsychotics but can occur with SSRIs. Reducing the dosage or switching to another, usually less potent agent often can relieve akathisia. When these remedies are not tenable, consider benzodiazepines along with other medications—including beta blockers and anticholinergic agents—with demonstrated efficacy in reducing akathisia symptoms. Lorazepam, diazepam, and clonazepam have demonstrated efficacy for relieving akathisia in comparison studies with placebo, propranolol, and diphenhydramine.21,22
Drug-induced postural tremor can occur with several psychotropics, including lithium, valproic acid, antidepressants, and antipsychotics. A tremor is considered mild if a patient can drink a glass of water with 1 hand without spilling and severe if holding a glass with 2 hands is difficult. Propranolol is most commonly prescribed for these tremors, but alprazolam and clonazepam have demonstrated efficacy, either as monotherapy or coadministered with a beta blocker.23
Acute agitation. Agitated patients often have acute psychosis and/or mania or dyscontrol secondary to axis II disorders.24 Patients may be paranoid, hostile, disruptive, and combative. Rapidly initiating medication can prevent the need for more restrictive measures, such as seclusion or restraint. Antipsychotics—especially high-potency agents such as haloperidol—and benzodiazepines, as monotherapy or in combination, are a mainstay treatment. Although treatment protocols favor atypical antipsychotics over typical antipsychotics, benzodiazepines are a viable option because of their anxiolytic and sedative effects. Advantages of benzodiazepine monotherapy include decreased extrapyramidal symptoms, greater patient acceptance/preference, and increased sedation compared with antipsychotics. Lorazepam, 1 to 2 mg intramuscularly (IM) or orally, is well tolerated because of its favorable drug-drug interaction profile and lack of significant cardiac side effects. Benzodiazepines can cause respiratory depression in patients with chronic lung disease and additive sedation secondary to opiates, other sedatives/hypnotics, or alcohol. Behavioral disinhibition is rare and is associated with preexisting CNS pathology or mental retardation.25 The IM olanzapine package insert warns against coadministering IM lorazepam because of additive cardiorespiratory depressive effects and excessive somnolence.26
Catatonia. The characteristic symptoms of catatonia are immobility, negativism, muteness, and failure to eat or drink. Benzodiazepines improve these symptoms in approximately 70% to 80% of catatonic patients with affective disorders. Response rates are lower in catatonia in patients with schizophrenia.27 If catatonia in a patient with psychosis is missed, giving antipsychotics before benzodiazepines may worsen catatonic symptoms or precipitate neuroleptic malignant syndrome in some cases. When you suspect a patient has catatonia, start with lorazepam, 1 to 2 mg IV or IM, and examine the patient for diminishing catatonic signs within 1 to 2 hours. If catatonia signs lessen, begin regularly scheduled lorazepam, with dosing varying by age—be more cautious in geriatric patients—and symptom severity. Titrate benzodiazepines for stuporous patients more slowly (eg, 1 mg 3 times a day as a starting dose) than for excited catatonic patients. Lorazepam can be increased gradually as tolerated; it is not unusual for patients to require up to 8 to 12 mg/d. Electroconvulsive therapy (ECT) is the treatment of choice when catatonic patients respond poorly or partially to high-dose benzodiazepines.28,29
Benzodiazepine reversal for ECT
Benzodiazepines have anticonvulsant properties that may interfere with the therapeutic efficacy of ECT.30 A multi-center study demonstrated that lorazepam (up to 4 mg/d as needed) in the 48 hours before the first ECT session was not associated with effects on seizure threshold or duration; however, larger lorazepam dosages were associated with briefer EEG seizure duration.31 Some patients may not tolerate withholding or tapering benzodiazepines in preparation for ECT. Studies investigating flumazenil for pre-ECT benzodiazepine reversal are lacking. One retrospective analysis showed that flumazenil administration immediately before and after ECT resulted in adequate seizures with no difference in clinical outcome compared with patients who were not receiving benzodiazepines or flumazenil.32
Tapering benzodiazepines
Slow discontinuation of benzodiazepines is recommended to avoid withdrawal symptoms, such as rebound anxiety, agitation, insomnia, or seizures, particularly when use exceeds 8 weeks. The onset of withdrawal symptoms varies, depending on the medication used. Withdrawal symptoms may appear in 1 to 2 days for agents with shorter half-lives, but may not appear until 3 to 7 days for agents with longer half-lives.33Table 2 lists recommended durations for tapering benzodiazepines.33,34 In general, decrease the total daily dose by 25% the first week, another 25% the second week, then 10% a week until discontinuation. When benzodiazepine use exceeds 1 year, a slower taper is recommended; for example, decrease 10% every 1 to 2 weeks. When 20% of the dosage remains, begin a 5% dose reduction every 2 to 4 weeks. Monitor patients for withdrawal symptoms or symptom exacerbation. If either occur, consider maintaining the current benzodiazepine dose or increasing the dose for 1 to 2 weeks or longer, if necessary, then continue to taper at a slower rate.34
Table 2
Recommendations for tapering benzodiazepines
| Duration of use | Recommended taper length | Comments |
| <6 to 8 weeks | Taper may not be required | Depending on clinical judgment and patient stability/preference, consider implementing a taper, particularly if using a high-dose benzodiazepine or an agent with a short or intermediate half-life, such as alprazolam or triazolam |
| 8 weeks to 6 months | Slowly over 2 to 3 weeks | Go slower during latter half of taper. Tapering will reduce, not eliminate, withdrawal symptoms. Patients should avoid alcohol and stimulants during benzodiazepine withdrawal |
| 6 months to 1 year | Slowly over 4 to 8 weeks | |
| >1 year | Slowly over 2 to 4 months | |
| Source: References 33,34 | ||
Risks of benzodiazepine use
For most indications, benzodiazepine therapy should be short-term.35 Use exceeding 2 to 4 weeks increases the risk for dependence and withdrawal. Tell patients to avoid alcohol while taking a benzodiazepine because this combination is potentially lethal. Benzodiazepines are commonly abused and abuse can lead to unintentional drug overdose. Benzodiazepines accounted for 37% of unintentional drug overdose deaths in West Virginia in 2006; in 46% of these cases, benzodiazepines were used for nonmedical purposes. Clinicians can help reduce the risk of diversion by limiting prescriptions to 30 days with no refills.36
Older patients taking benzodiazepines are at increased risk of falls and hip fractures.37 Lorazepam, oxazepam, and temazepam—agents with shorter half-lives that are not greatly affected by pharmacokinetic changes associated with aging—are preferred for these patients.34 Patients with dementia or other CNS-compromising conditions may become confused or delirious with regular benzodiazepine dosing. Educate patients to whom you prescribe benzodiazepines about the importance of gauging their level of sedation before driving or engaging in other tasks for which sedation could compromise their safety. Benzodiazepine use during pregnancy requires a careful discussion of risks and benefits (Box 2).38
Benzodiazepine use during pregnancy has been associated with cleft palate and urogenital and neurologic malformations in the fetus.38 Although data are conflicting—particularly among recent meta-analyses that fail to demonstrate an association—some experts advise against benzodiazepine use in the first trimester. Participate in shared decision making with your patients and educate them about the potential risks and benefits of benzodiazepine use during the first trimester and throughout pregnancy. After delivery, newborns may develop “floppy baby syndrome”—which is associated with lethargy, difficulty eating, and respiratory depression—or withdrawal.38 To minimize this risk, consider tapering the benzodiazepine as the patient approaches delivery.
Related Resources
- Substance Abuse and Mental Health Services Administration. www.samhsa.gov.
- National Institute on Drug Abuse resources for medical and health professionals. www.drugabuse.gov/medical-health-professionals.
- American Academy of Sleep Medicine. www.aasmnet.org.
Drug Brand Names
- Alprazolam • Xanax
- Chlordiazepoxide • Librium, Limbitrol
- Clonazepam • Klonopin
- Clorazepate • Tranxene
- Diazepam • Valium
- Diphenhydramine • Benadryl, others
- Estazolam • ProSom
- Flumazenil • Romazicon
- Flurazepam • Dalmane
- Haloperidol • Haldol
- Lithium • Lithobid
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Oxazepam • Serax
- Paroxetine • Paxil
- Pregabalin • Lyrica
- Propranolol • Inderal, InnoPran XL, others
- Quazepam • Doral
- Ramelteon • Rozerem
- Sertraline • Zoloft
- Temazepam • Restoril
- Triazolam • Halcion
- Valproic acid • Depakene, Stavzor, others
Disclosures
Drs. Bostwick and Yasugi report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Casher is a speaker for AstraZeneca and Sunovion Pharmaceuticals.
1. Mihic SJ, Harris RA. Hypnotics and sedatives. In: Brunton LL Chabner BA, Knollmann BC, eds. Goodman & Gilman’s the pharmacological basis of therapeutics. New York, NY: McGraw Hill and Company; 2011:457-480.
2. Facts and comparisons Web site. 2011 Wolters Kluwer Health Inc. http://online.factsandcomparisons.com. Accessed August 16, 2011.
3. DuPont RL, Greene W, Lydiard RB. Sedatives and hypnotics: pharmacology and epidemiology. In: Gold MS Hermann R, eds. UpToDate. http://www.uptodate.com/contents/sedatives-and-hypnotics-abuse-and-dependence-pharmacology-and-epidemiology. Accessed August 16, 2011.
4. U.S. Food and Drug Administration. Orange book: approved drug products with therapeutic equivalence evaluations. http://www.accessdata.fda.gov/scripts/cder/ob/default.cfm. Accessed August 16, 2011.
5. Chouinard G. Issues in the clinical use of benzodiazepines: potency withdrawal, and rebound. J Clin Psychiatry. 2004;65(suppl 5):7-12.
6. Shader RI, Greenblatt DJ. Can you provide a table of equivalencies for benzodiazepines and other marketed benzodiazepine receptor agonists? J Clin Psychopharmacol. 1997;17(4):331.-
7. Amato L, Minozzi S, Davoli M. Efficacy and safety of pharmacologic interventions for the treatment of the alcohol withdrawal syndrome. Cochrane Database Syst Rev. 2011;15(6):CD008537.-
8. Schutte-Rodin S, Broch L, Buysse D, et al. Clinical guideline for the evaluation and management of chronic insomnia in adults. J Clin Sleep Med. 2008;4(5):487-504.
9. Foral P, Dewan N, Malesker M. Insomnia: a therapeutic review for pharmacists. Consult Pharm. 2011;26(5):332-341.
10. Riemann D, Perlis ML. The treatments of chronic insomnia: a review of benzodiazepine receptor agonists and psychological and behavioral therapies. Sleep Med Rev. 2009;13(3):205-214.
11. Hidalgo RB, Tupler LA, Davidson JR. An effect-size analysis of pharmacologic treatments of generalized anxiety disorder. J Psychopharmacol. 2007;21(8):864-872.
12. Davidson JR, Zhang W, Connor KM, et al. A psychopharmacological treatment algorithm for generalised anxiety disorder (GAD). J Psychopharmacol. 2010;24(1):3-26.
13. Bandelow B, Zohar J, Hollander E, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive-compulsive and post-traumatic stress disorders - first revision. World J Biol Psychiatry. 2008;9(4):248-312.
14. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. 2nd ed. Arlington VA: American Psychiatric Publishing, Inc.; 2009.
15. Pollack MH, Simon NM, Worthington JJ, et al. Combined paroxetine and clonazepam treatment strategies compared to paroxetine monotherapy for panic disorder. J Psychopharmacol. 2003;17(3):276-282.
16. Watanabe N, Churchill R, Furukawa TA. Combined psychotherapy plus benzodiazepines for panic disorder. Cochrane Database Syst Rev. 2009;(1):CD005335.-
17. Otto MW, McHugh RK, Simon NM, et al. Efficacy of CBT for benzodiazepine discontinuation in patients with panic disorder: further evaluation. Behav Res Ther. 2010;48(8):720-727.
18. Davidson JR. Use of benzodiazepines in social anxiety disorder generalized anxiety disorder, and posttraumatic stress disorder. J Clin Psychiatry. 2004;65(suppl 5):29-33.
19. Argyropoulos SV, Sandford JJ, Nutt DJ. The psychobiology of anxiolytic drugs Part 2: pharmacological treatments of anxiety. Pharmacol Ther. 2000;88(3):213-227.
20. Connor KM, Davidson JR, Potts NL, et al. Discontinuation of clonazepam in the treatment of social phobia. J Clin Psychopharmacol. 1998;18(5):373-378.
21. Miller CH, Fleischhacker WW. Managing antipsychotic-induced acute and chronic akathisia. Drug Saf. 2000;22(1):73-81.
22. Rodnitzky RL. Drug-induced movement disorders. Clin Neuropharmacol. 2002;25(3):142-151.
23. Arbaizar B, Gómez-Acebo I, Llorca J. Postural induced tremor in psychiatry. Psychiatry Clin Neurosci. 2008;62(6):638-645.
24. Casher MI, Bess JD. Manual of inpatient psychiatry. New York NY: Cambridge University Press; 2010.
25. Battaglia J. Pharmacological management of acute agitation. Drugs. 2005;65(9):1207-1222.
26. Physicians’ desk reference. Montvale NJ: PDR Network, LLC; 2010.
27. Rosebush PI, Mazurek MF. Catatonia and its treatment. Schizophr Bull. 2010;36(2):239-242.
28. Ungvari GS, Kau LS, Wai-Kwong T, et al. The pharmacological treatment of catatonia: an overview. Eur Arch Psychiatry Clin Neurosci. 2001;251(suppl 1):I31-I34.
29. Fink M, Taylor MA. Catatonia: a clinician’s guide to diagnosis and treatment. New York NY: Cambridge University Press; 2003.
30. Naguib N, Koorn R. Interactions between psychotropics anaesthetics and electroconvulsive therapy: implications for drug choice and patient management. CNS Drugs. 2002;16(4):229-247.
31. Boylan LS, Haskett RF, Mulsant BH, et al. Determinants of seizure threshold in ECT: benzodiazepine use, anesthetic dosage, and other factors. J ECT. 2000;16(1):3-18.
32. Krystal AD, Watts BV, Weiner RD, et al. The use of flumazenil in the anxious and benzodiazepine-dependent ECT patient. J ECT. 1998;14(1):5-14.
33. Melton ST, Kirkwood CK. Anxiety disorders I: generalized anxiety panic, and social anxiety disorders. In: DiPiro JT, Talbert RL, Yee GC, et al, eds. Pharmacotherapy: a pathophysiologic approach. New York, NY: McGraw-Hill Companies; 2011:1209-1228.
34. Benzodiazepine toolkit. The Pharmacist’s Letter/Prescriber’s Letter. 2011;27(4):270406.-
35. Lader M. Benzodiazepines revisited – will we ever learn? Addiction. 2011;106(12):2086-2109.
36. Toblin RL, Paulozzi LJ, Logan JE, et al. Mental illness and psychotropic drug use among prescription drug overdose deaths: a medical examiner chart review. J Clin Psychiatry. 2010;71(4):491-496.
37. Ashton H. The diagnosis and management of benzodiazepine dependence. Curr Opin Psychiatry. 2005;18(3):249-255.
38. Menon SJ. Psychotropic medication during pregnancy and lactation. Arch Gynecol Obstet. 2008;277(1):1-13.
Since the discovery of chlordiazepoxide in the 1950s, benzodiazepines have revolutionized the treatment of anxiety and insomnia, largely because of their improved safety profile compared with barbiturates, formerly the preferred sedative-hypnotic.1 In addition to their anxiolytic and sedative-hypnotic effects, benzodiazepines exhibit anterograde amnesia, anticonvulsant, and muscle relaxant properties.1 Psychiatrists use benzodiazepines to treat anxiety and sleep disorders, acute agitation, alcohol withdrawal, catatonia, and psychotropic side effects such as akathisia. This article highlights the evidence for using benzodiazepines in anxiety and other disorders and why they generally should not be used for obsessive-compulsive disorder and posttraumatic stress disorder (Box 1).
Current evidence indicates little support for using benzodiazepines for obsessive-compulsive disorder (OCD). The American Psychiatric Association (APA) and the World Federation of Biological Psychiatry do not recommend benzodiazepines for treating OCD because of a lack of evidence for efficacy.a,b An earlier study suggested clonazepam monotherapy was effective for OCDc; however, a more recent study did not show a benefit on rate of response or degree of symptom improvement.d Augmentation strategies with benzodiazepines also do not appear to be beneficial for OCD management. A recent double-blind, placebo-controlled study failed to demonstrate faster symptom improvement by augmenting sertraline with clonazepam, although the study had a small sample size and high drop-out rate.e
Because benzodiazepines have negligible action on core posttraumatic stress disorder (PTSD) symptoms (re-experiencing, avoidance, and hyperarousal), selective serotonin reuptake inhibitors and other agents largely have supplanted them for PTSD treatment.f Use of benzodiazepines for PTSD is associated with withdrawal symptoms, more severe symptoms after discontinuation, and possible disinhibition, and may interfere with patients’ efforts to integrate trauma experiences. Although benzodiazepines may reduce distress associated with acute trauma, there is evidence—in clinical studies and animal models—that early benzodiazepine administration fails to prevent PTSD and may increase its incidence.g The International Consensus Group on Depression and Anxiety, the APA, and the British Association for Psychopharmacology all highlight the limited role, if any, for benzodiazepines in PTSD.h-j
References
- Bandelow B, Zohar J, Hollander E, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive-compulsive and post-traumatic stress disorders - first revision. World J Biol Psychiatry. 2008;9(4):248-312.
- American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder. Arlington, VA: American Psychiatric Publishing, Inc.; 2007.
- Hewlett WA, Vinogradov S, Agras WS. Clomipramine, clonazepam, and clonidine treatment of obsessive compulsive disorder. J Clin Psychopharmacol. 1992;12(6):420-430.
- Hollander E, Kaplan A, Stahl SM. A double-blind, placebo-controlled trial of clonazepam in obsessive-compulsive disorder. World J Biol Psychiatry. 2003;4(1):30-34.
- Crockett BA, Churchill E, Davidson JR. A double-blind combination study of clonazepam and sertraline in OCD. Ann Clin Psychiatry. 2004;16(3):127-132.
- Argyropoulos SV, Sandford JJ, Nutt DJ. The psychobiology of anxiolytic drugs. Part 2: pharmacological treatments of anxiety. Pharmacol Ther. 2000;88(3):213-227.
- Matar MA, Zohar J, Kaplan Z, et al. Alprazolam treatment immediately after stress exposure interferes with the normal HPA-stress response and increases vulnerability to subsequent stress in an animal model of PTSD. Eur Neuropsychopharmacol. 2009;19(4):283-295.
- Ballenger JC, Davidson JR, Lecrubier Y, et al. Consensus statement update on posttraumatic stress disorder from the international consensus group on depression and anxiety. J Clin Psychiatry. 2004;65(suppl 1):55-62.
- Ursano RJ, Bell C, Eth S, et al. Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Am J Psychiatry. 2004;161(11 suppl):3-31.
- Baldwin DS, Anderson IM, Nutt DJ, et al. Evidence-based guidelines for the pharmacological treatment of anxiety disorders: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2005;19(6):567-596.
Pharmacokinetic properties
Most benzodiazepines are considered to have similar efficacy; therefore, selection is based on pharmacokinetic considerations. Table 1 compares the indication, onset, and half-life of 12 commonly used benzodiazepines.2-6 Although Table 1 lists approximate equivalent doses, studies report inconsistent data. These are approximations only and should not be used independently to make therapy decisions.
Table 1
Oral benzodiazepines: Indications, onset, half-life, and equivalent doses
| Drug | FDA-approved indication(s) | Onset of action | Approximate half-life (hours) in healthy adults | Approximate equivalent dose (mg)a | Comments |
|---|---|---|---|---|---|
| Alprazolam | Anxiety disorders, panic disorder | Intermediate | 6.3 to 26.9 (IR), 10.7 to 15.8 (XR) | 0.5 | Increased risk for abuse because of greater lipid solubility |
| Chlordiazepoxide | Anxiety disorders, acute alcohol withdrawal, preoperative apprehension and anxiety | Intermediate | 24 to 48 | 10 | Risk for accumulation because of long-acting metabolites (desmethyldiazepam, oxazepam) |
| Clonazepam | Seizure disorders, panic disorder | Intermediate | 18 to 50 | 0.25 to 0.5 | Use caution in patients with liver disease |
| Clorazepate | Anxiety, seizures, acute alcohol withdrawal | Fast | 40 to 50 | 7.5 | Risk for accumulation because of long-acting metabolites (desmethyldiazepam, oxazepam) |
| Diazepam | Anxiety disorders, acute alcohol withdrawal, muscle spasms, convulsive disorders | Very fast | 20 to 100 | 5 | Risk for accumulation because of long-acting metabolites (temazepam, desmethyldiazepam, oxazepam). Increased risk for abuse because of quick onset |
| Estazolam | Insomnia | Intermediate | 10 to 24 | 0.3 to 2 | None |
| Flurazepam | Insomnia | Intermediate | 47 to 100 | 30 | Avoid in geriatric patients or patients with liver impairment |
| Lorazepam | Anxiety | Intermediate | 10 to 20 | 1 | Preferred for patients with liver impairment and geriatric patients |
| Oxazepam | Anxiety, acute alcohol withdrawal | Slow to intermediate | 5 to 20 | 30 | Preferred for patients with liver impairment and geriatric patients |
| Quazepam | Insomnia | Intermediate | 39 to 73 | 5 to 15 | Risk for accumulation because of long-acting metabolites (desmethyldiazepam, oxazepam) |
| Temazepam | Insomnia | Intermediate | 3.5 to 18.4 | 30 | Preferred for patients with liver impairment and geriatric patients |
| Triazolam | Insomnia | Fast | 1.5 to 5.5 | 0.25 | Lacks active metabolites |
| IR: immediate release; XR: extended release aInterpret with caution, conflicting data exist Source: References 2-6 | |||||
A diverse range of indications
Alcohol withdrawal. Benzodiazepines are the treatment of choice for alcohol withdrawal syndrome, particularly to prevent seizures.7 Research supports symptom-triggered therapy using the revised Clinical Institute Withdrawal Assessment for Alcohol. Benzodiazepines reduce CNS sympathetic hyperactivity to mitigate withdrawal from alcohol by decreasing tachycardia, tremor, insomnia, agitation, and anxiety. Furthermore, these agents provide prophylaxis against serious sequelae such as seizures and delirium.
Insomnia. The American Academy of Sleep Medicine considers benzodiazepine receptor agonists (BzRAs, which include benzodiazepines and non-benzodiazepines) and ramelteon first-line pharmacotherapy for primary insomnia.8 However, pharmacologic treatment should be short-term. Agents with short to intermediate half-lives and rapid onset, such as triazolam, can aid sleep initiation. Those with longer half-lives, such as temazepam, could address sleep maintenance. If a patient does not respond to the initial agent, try another medication within the same class, because patients may respond differently. Use lower starting doses in geriatric patients.9 Closely monitor for adverse effects, rebound insomnia, and potential abuse or tolerance. Identify comorbid conditions and medications that may impair sleep, and address them accordingly.
Psychological and behavioral treatments given over 4 to 8 weeks can yield stable sleep improvements for up to 2 years. If available, these interventions may be considered first-line for treating insomnia because of their lasting effects compared with BzRAs.10
Generalized anxiety disorder (GAD). Benzodiazepines effectively treat GAD because they work quickly and are well tolerated. However, there are better first-line treatment options when considering efficacy studies and dependence and tolerance concerns. One effect-size comparison of 21 double-blind, placebo-controlled trials showed that the efficacy of selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors (SNRIs), and pregabalin are comparable to benzodiazepines.11 Benzodiazepines can be used in the first 2 to 3 weeks after initiating antidepressants to alleviate and prevent worsening of anxiety that may occur at the start of antidepressant therapy. Recent treatment guidelines recommend benzodiazepines as a second-line treatment or for treatment-resistant GAD in patients who do not have a substance abuse history.12,13
Panic disorder. Efficacy of benzodiazepines for panic disorder is comparable to SSRIs, SNRIs, and tricyclic antidepressants (TCAs). SSRIs and SNRIs are considered first-line treatments for panic disorder because of their favorable side effect profile.14 In practice, benzodiazepines often are combined with SSRIs, SNRIs, or TCAs. A randomized controlled trial demonstrated that paroxetine and clonazepam (mean dose 1.6 mg/d at 5 weeks) resulted in a more rapid response compared with paroxetine alone, although this difference lasted only a few weeks.15 Furthermore, this study suggested that brief treatment with clonazepam followed by a taper is as effective as sustained treatment with paroxetine and clonazepam.15
There is a lack of high-quality data on combining cognitive-behavioral therapy (CBT) and benzodiazepines for panic disorder, although a Cochrane Review found that adding a benzodiazepine to CBT did not lead to a significant difference in response compared with psychotherapy alone.16 A recent randomized controlled trial demonstrated that tapering benzodiazepines combined with CBT was associated with successful discontinuation of the drug and prevented return of panic symptoms.17
Social anxiety. A meta-analysis found that for treating social anxiety, benzodiazepines have better efficacy than SSRIs, monoamine oxidase inhibitors, and anticonvulsants.18 Longer-acting benzodiazepines may be more effective than shorter-acting agents. One study of patients with social anxiety showed a 38% response rate for alprazolam vs 20% for placebo over 12 weeks, and a similar 10-week study demonstrated a 73% recovery rate with clonazepam vs 22% for placebo.19 In addition, studies have observed that patients can be maintained on clonazepam for up to 2 years without symptom relapse and will tolerate slow-taper discontinuation.18,20 Sedation and drowsiness can be lessened by limiting clonazepam doses to 2 to 3 mg/d.
Akathisia and tremor. Akathisia, a syndrome of motor restlessness and inner turmoil, is associated with antipsychotics but can occur with SSRIs. Reducing the dosage or switching to another, usually less potent agent often can relieve akathisia. When these remedies are not tenable, consider benzodiazepines along with other medications—including beta blockers and anticholinergic agents—with demonstrated efficacy in reducing akathisia symptoms. Lorazepam, diazepam, and clonazepam have demonstrated efficacy for relieving akathisia in comparison studies with placebo, propranolol, and diphenhydramine.21,22
Drug-induced postural tremor can occur with several psychotropics, including lithium, valproic acid, antidepressants, and antipsychotics. A tremor is considered mild if a patient can drink a glass of water with 1 hand without spilling and severe if holding a glass with 2 hands is difficult. Propranolol is most commonly prescribed for these tremors, but alprazolam and clonazepam have demonstrated efficacy, either as monotherapy or coadministered with a beta blocker.23
Acute agitation. Agitated patients often have acute psychosis and/or mania or dyscontrol secondary to axis II disorders.24 Patients may be paranoid, hostile, disruptive, and combative. Rapidly initiating medication can prevent the need for more restrictive measures, such as seclusion or restraint. Antipsychotics—especially high-potency agents such as haloperidol—and benzodiazepines, as monotherapy or in combination, are a mainstay treatment. Although treatment protocols favor atypical antipsychotics over typical antipsychotics, benzodiazepines are a viable option because of their anxiolytic and sedative effects. Advantages of benzodiazepine monotherapy include decreased extrapyramidal symptoms, greater patient acceptance/preference, and increased sedation compared with antipsychotics. Lorazepam, 1 to 2 mg intramuscularly (IM) or orally, is well tolerated because of its favorable drug-drug interaction profile and lack of significant cardiac side effects. Benzodiazepines can cause respiratory depression in patients with chronic lung disease and additive sedation secondary to opiates, other sedatives/hypnotics, or alcohol. Behavioral disinhibition is rare and is associated with preexisting CNS pathology or mental retardation.25 The IM olanzapine package insert warns against coadministering IM lorazepam because of additive cardiorespiratory depressive effects and excessive somnolence.26
Catatonia. The characteristic symptoms of catatonia are immobility, negativism, muteness, and failure to eat or drink. Benzodiazepines improve these symptoms in approximately 70% to 80% of catatonic patients with affective disorders. Response rates are lower in catatonia in patients with schizophrenia.27 If catatonia in a patient with psychosis is missed, giving antipsychotics before benzodiazepines may worsen catatonic symptoms or precipitate neuroleptic malignant syndrome in some cases. When you suspect a patient has catatonia, start with lorazepam, 1 to 2 mg IV or IM, and examine the patient for diminishing catatonic signs within 1 to 2 hours. If catatonia signs lessen, begin regularly scheduled lorazepam, with dosing varying by age—be more cautious in geriatric patients—and symptom severity. Titrate benzodiazepines for stuporous patients more slowly (eg, 1 mg 3 times a day as a starting dose) than for excited catatonic patients. Lorazepam can be increased gradually as tolerated; it is not unusual for patients to require up to 8 to 12 mg/d. Electroconvulsive therapy (ECT) is the treatment of choice when catatonic patients respond poorly or partially to high-dose benzodiazepines.28,29
Benzodiazepine reversal for ECT
Benzodiazepines have anticonvulsant properties that may interfere with the therapeutic efficacy of ECT.30 A multi-center study demonstrated that lorazepam (up to 4 mg/d as needed) in the 48 hours before the first ECT session was not associated with effects on seizure threshold or duration; however, larger lorazepam dosages were associated with briefer EEG seizure duration.31 Some patients may not tolerate withholding or tapering benzodiazepines in preparation for ECT. Studies investigating flumazenil for pre-ECT benzodiazepine reversal are lacking. One retrospective analysis showed that flumazenil administration immediately before and after ECT resulted in adequate seizures with no difference in clinical outcome compared with patients who were not receiving benzodiazepines or flumazenil.32
Tapering benzodiazepines
Slow discontinuation of benzodiazepines is recommended to avoid withdrawal symptoms, such as rebound anxiety, agitation, insomnia, or seizures, particularly when use exceeds 8 weeks. The onset of withdrawal symptoms varies, depending on the medication used. Withdrawal symptoms may appear in 1 to 2 days for agents with shorter half-lives, but may not appear until 3 to 7 days for agents with longer half-lives.33Table 2 lists recommended durations for tapering benzodiazepines.33,34 In general, decrease the total daily dose by 25% the first week, another 25% the second week, then 10% a week until discontinuation. When benzodiazepine use exceeds 1 year, a slower taper is recommended; for example, decrease 10% every 1 to 2 weeks. When 20% of the dosage remains, begin a 5% dose reduction every 2 to 4 weeks. Monitor patients for withdrawal symptoms or symptom exacerbation. If either occur, consider maintaining the current benzodiazepine dose or increasing the dose for 1 to 2 weeks or longer, if necessary, then continue to taper at a slower rate.34
Table 2
Recommendations for tapering benzodiazepines
| Duration of use | Recommended taper length | Comments |
| <6 to 8 weeks | Taper may not be required | Depending on clinical judgment and patient stability/preference, consider implementing a taper, particularly if using a high-dose benzodiazepine or an agent with a short or intermediate half-life, such as alprazolam or triazolam |
| 8 weeks to 6 months | Slowly over 2 to 3 weeks | Go slower during latter half of taper. Tapering will reduce, not eliminate, withdrawal symptoms. Patients should avoid alcohol and stimulants during benzodiazepine withdrawal |
| 6 months to 1 year | Slowly over 4 to 8 weeks | |
| >1 year | Slowly over 2 to 4 months | |
| Source: References 33,34 | ||
Risks of benzodiazepine use
For most indications, benzodiazepine therapy should be short-term.35 Use exceeding 2 to 4 weeks increases the risk for dependence and withdrawal. Tell patients to avoid alcohol while taking a benzodiazepine because this combination is potentially lethal. Benzodiazepines are commonly abused and abuse can lead to unintentional drug overdose. Benzodiazepines accounted for 37% of unintentional drug overdose deaths in West Virginia in 2006; in 46% of these cases, benzodiazepines were used for nonmedical purposes. Clinicians can help reduce the risk of diversion by limiting prescriptions to 30 days with no refills.36
Older patients taking benzodiazepines are at increased risk of falls and hip fractures.37 Lorazepam, oxazepam, and temazepam—agents with shorter half-lives that are not greatly affected by pharmacokinetic changes associated with aging—are preferred for these patients.34 Patients with dementia or other CNS-compromising conditions may become confused or delirious with regular benzodiazepine dosing. Educate patients to whom you prescribe benzodiazepines about the importance of gauging their level of sedation before driving or engaging in other tasks for which sedation could compromise their safety. Benzodiazepine use during pregnancy requires a careful discussion of risks and benefits (Box 2).38
Benzodiazepine use during pregnancy has been associated with cleft palate and urogenital and neurologic malformations in the fetus.38 Although data are conflicting—particularly among recent meta-analyses that fail to demonstrate an association—some experts advise against benzodiazepine use in the first trimester. Participate in shared decision making with your patients and educate them about the potential risks and benefits of benzodiazepine use during the first trimester and throughout pregnancy. After delivery, newborns may develop “floppy baby syndrome”—which is associated with lethargy, difficulty eating, and respiratory depression—or withdrawal.38 To minimize this risk, consider tapering the benzodiazepine as the patient approaches delivery.
Related Resources
- Substance Abuse and Mental Health Services Administration. www.samhsa.gov.
- National Institute on Drug Abuse resources for medical and health professionals. www.drugabuse.gov/medical-health-professionals.
- American Academy of Sleep Medicine. www.aasmnet.org.
Drug Brand Names
- Alprazolam • Xanax
- Chlordiazepoxide • Librium, Limbitrol
- Clonazepam • Klonopin
- Clorazepate • Tranxene
- Diazepam • Valium
- Diphenhydramine • Benadryl, others
- Estazolam • ProSom
- Flumazenil • Romazicon
- Flurazepam • Dalmane
- Haloperidol • Haldol
- Lithium • Lithobid
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Oxazepam • Serax
- Paroxetine • Paxil
- Pregabalin • Lyrica
- Propranolol • Inderal, InnoPran XL, others
- Quazepam • Doral
- Ramelteon • Rozerem
- Sertraline • Zoloft
- Temazepam • Restoril
- Triazolam • Halcion
- Valproic acid • Depakene, Stavzor, others
Disclosures
Drs. Bostwick and Yasugi report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Casher is a speaker for AstraZeneca and Sunovion Pharmaceuticals.
Since the discovery of chlordiazepoxide in the 1950s, benzodiazepines have revolutionized the treatment of anxiety and insomnia, largely because of their improved safety profile compared with barbiturates, formerly the preferred sedative-hypnotic.1 In addition to their anxiolytic and sedative-hypnotic effects, benzodiazepines exhibit anterograde amnesia, anticonvulsant, and muscle relaxant properties.1 Psychiatrists use benzodiazepines to treat anxiety and sleep disorders, acute agitation, alcohol withdrawal, catatonia, and psychotropic side effects such as akathisia. This article highlights the evidence for using benzodiazepines in anxiety and other disorders and why they generally should not be used for obsessive-compulsive disorder and posttraumatic stress disorder (Box 1).
Current evidence indicates little support for using benzodiazepines for obsessive-compulsive disorder (OCD). The American Psychiatric Association (APA) and the World Federation of Biological Psychiatry do not recommend benzodiazepines for treating OCD because of a lack of evidence for efficacy.a,b An earlier study suggested clonazepam monotherapy was effective for OCDc; however, a more recent study did not show a benefit on rate of response or degree of symptom improvement.d Augmentation strategies with benzodiazepines also do not appear to be beneficial for OCD management. A recent double-blind, placebo-controlled study failed to demonstrate faster symptom improvement by augmenting sertraline with clonazepam, although the study had a small sample size and high drop-out rate.e
Because benzodiazepines have negligible action on core posttraumatic stress disorder (PTSD) symptoms (re-experiencing, avoidance, and hyperarousal), selective serotonin reuptake inhibitors and other agents largely have supplanted them for PTSD treatment.f Use of benzodiazepines for PTSD is associated with withdrawal symptoms, more severe symptoms after discontinuation, and possible disinhibition, and may interfere with patients’ efforts to integrate trauma experiences. Although benzodiazepines may reduce distress associated with acute trauma, there is evidence—in clinical studies and animal models—that early benzodiazepine administration fails to prevent PTSD and may increase its incidence.g The International Consensus Group on Depression and Anxiety, the APA, and the British Association for Psychopharmacology all highlight the limited role, if any, for benzodiazepines in PTSD.h-j
References
- Bandelow B, Zohar J, Hollander E, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive-compulsive and post-traumatic stress disorders - first revision. World J Biol Psychiatry. 2008;9(4):248-312.
- American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder. Arlington, VA: American Psychiatric Publishing, Inc.; 2007.
- Hewlett WA, Vinogradov S, Agras WS. Clomipramine, clonazepam, and clonidine treatment of obsessive compulsive disorder. J Clin Psychopharmacol. 1992;12(6):420-430.
- Hollander E, Kaplan A, Stahl SM. A double-blind, placebo-controlled trial of clonazepam in obsessive-compulsive disorder. World J Biol Psychiatry. 2003;4(1):30-34.
- Crockett BA, Churchill E, Davidson JR. A double-blind combination study of clonazepam and sertraline in OCD. Ann Clin Psychiatry. 2004;16(3):127-132.
- Argyropoulos SV, Sandford JJ, Nutt DJ. The psychobiology of anxiolytic drugs. Part 2: pharmacological treatments of anxiety. Pharmacol Ther. 2000;88(3):213-227.
- Matar MA, Zohar J, Kaplan Z, et al. Alprazolam treatment immediately after stress exposure interferes with the normal HPA-stress response and increases vulnerability to subsequent stress in an animal model of PTSD. Eur Neuropsychopharmacol. 2009;19(4):283-295.
- Ballenger JC, Davidson JR, Lecrubier Y, et al. Consensus statement update on posttraumatic stress disorder from the international consensus group on depression and anxiety. J Clin Psychiatry. 2004;65(suppl 1):55-62.
- Ursano RJ, Bell C, Eth S, et al. Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Am J Psychiatry. 2004;161(11 suppl):3-31.
- Baldwin DS, Anderson IM, Nutt DJ, et al. Evidence-based guidelines for the pharmacological treatment of anxiety disorders: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2005;19(6):567-596.
Pharmacokinetic properties
Most benzodiazepines are considered to have similar efficacy; therefore, selection is based on pharmacokinetic considerations. Table 1 compares the indication, onset, and half-life of 12 commonly used benzodiazepines.2-6 Although Table 1 lists approximate equivalent doses, studies report inconsistent data. These are approximations only and should not be used independently to make therapy decisions.
Table 1
Oral benzodiazepines: Indications, onset, half-life, and equivalent doses
| Drug | FDA-approved indication(s) | Onset of action | Approximate half-life (hours) in healthy adults | Approximate equivalent dose (mg)a | Comments |
|---|---|---|---|---|---|
| Alprazolam | Anxiety disorders, panic disorder | Intermediate | 6.3 to 26.9 (IR), 10.7 to 15.8 (XR) | 0.5 | Increased risk for abuse because of greater lipid solubility |
| Chlordiazepoxide | Anxiety disorders, acute alcohol withdrawal, preoperative apprehension and anxiety | Intermediate | 24 to 48 | 10 | Risk for accumulation because of long-acting metabolites (desmethyldiazepam, oxazepam) |
| Clonazepam | Seizure disorders, panic disorder | Intermediate | 18 to 50 | 0.25 to 0.5 | Use caution in patients with liver disease |
| Clorazepate | Anxiety, seizures, acute alcohol withdrawal | Fast | 40 to 50 | 7.5 | Risk for accumulation because of long-acting metabolites (desmethyldiazepam, oxazepam) |
| Diazepam | Anxiety disorders, acute alcohol withdrawal, muscle spasms, convulsive disorders | Very fast | 20 to 100 | 5 | Risk for accumulation because of long-acting metabolites (temazepam, desmethyldiazepam, oxazepam). Increased risk for abuse because of quick onset |
| Estazolam | Insomnia | Intermediate | 10 to 24 | 0.3 to 2 | None |
| Flurazepam | Insomnia | Intermediate | 47 to 100 | 30 | Avoid in geriatric patients or patients with liver impairment |
| Lorazepam | Anxiety | Intermediate | 10 to 20 | 1 | Preferred for patients with liver impairment and geriatric patients |
| Oxazepam | Anxiety, acute alcohol withdrawal | Slow to intermediate | 5 to 20 | 30 | Preferred for patients with liver impairment and geriatric patients |
| Quazepam | Insomnia | Intermediate | 39 to 73 | 5 to 15 | Risk for accumulation because of long-acting metabolites (desmethyldiazepam, oxazepam) |
| Temazepam | Insomnia | Intermediate | 3.5 to 18.4 | 30 | Preferred for patients with liver impairment and geriatric patients |
| Triazolam | Insomnia | Fast | 1.5 to 5.5 | 0.25 | Lacks active metabolites |
| IR: immediate release; XR: extended release aInterpret with caution, conflicting data exist Source: References 2-6 | |||||
A diverse range of indications
Alcohol withdrawal. Benzodiazepines are the treatment of choice for alcohol withdrawal syndrome, particularly to prevent seizures.7 Research supports symptom-triggered therapy using the revised Clinical Institute Withdrawal Assessment for Alcohol. Benzodiazepines reduce CNS sympathetic hyperactivity to mitigate withdrawal from alcohol by decreasing tachycardia, tremor, insomnia, agitation, and anxiety. Furthermore, these agents provide prophylaxis against serious sequelae such as seizures and delirium.
Insomnia. The American Academy of Sleep Medicine considers benzodiazepine receptor agonists (BzRAs, which include benzodiazepines and non-benzodiazepines) and ramelteon first-line pharmacotherapy for primary insomnia.8 However, pharmacologic treatment should be short-term. Agents with short to intermediate half-lives and rapid onset, such as triazolam, can aid sleep initiation. Those with longer half-lives, such as temazepam, could address sleep maintenance. If a patient does not respond to the initial agent, try another medication within the same class, because patients may respond differently. Use lower starting doses in geriatric patients.9 Closely monitor for adverse effects, rebound insomnia, and potential abuse or tolerance. Identify comorbid conditions and medications that may impair sleep, and address them accordingly.
Psychological and behavioral treatments given over 4 to 8 weeks can yield stable sleep improvements for up to 2 years. If available, these interventions may be considered first-line for treating insomnia because of their lasting effects compared with BzRAs.10
Generalized anxiety disorder (GAD). Benzodiazepines effectively treat GAD because they work quickly and are well tolerated. However, there are better first-line treatment options when considering efficacy studies and dependence and tolerance concerns. One effect-size comparison of 21 double-blind, placebo-controlled trials showed that the efficacy of selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors (SNRIs), and pregabalin are comparable to benzodiazepines.11 Benzodiazepines can be used in the first 2 to 3 weeks after initiating antidepressants to alleviate and prevent worsening of anxiety that may occur at the start of antidepressant therapy. Recent treatment guidelines recommend benzodiazepines as a second-line treatment or for treatment-resistant GAD in patients who do not have a substance abuse history.12,13
Panic disorder. Efficacy of benzodiazepines for panic disorder is comparable to SSRIs, SNRIs, and tricyclic antidepressants (TCAs). SSRIs and SNRIs are considered first-line treatments for panic disorder because of their favorable side effect profile.14 In practice, benzodiazepines often are combined with SSRIs, SNRIs, or TCAs. A randomized controlled trial demonstrated that paroxetine and clonazepam (mean dose 1.6 mg/d at 5 weeks) resulted in a more rapid response compared with paroxetine alone, although this difference lasted only a few weeks.15 Furthermore, this study suggested that brief treatment with clonazepam followed by a taper is as effective as sustained treatment with paroxetine and clonazepam.15
There is a lack of high-quality data on combining cognitive-behavioral therapy (CBT) and benzodiazepines for panic disorder, although a Cochrane Review found that adding a benzodiazepine to CBT did not lead to a significant difference in response compared with psychotherapy alone.16 A recent randomized controlled trial demonstrated that tapering benzodiazepines combined with CBT was associated with successful discontinuation of the drug and prevented return of panic symptoms.17
Social anxiety. A meta-analysis found that for treating social anxiety, benzodiazepines have better efficacy than SSRIs, monoamine oxidase inhibitors, and anticonvulsants.18 Longer-acting benzodiazepines may be more effective than shorter-acting agents. One study of patients with social anxiety showed a 38% response rate for alprazolam vs 20% for placebo over 12 weeks, and a similar 10-week study demonstrated a 73% recovery rate with clonazepam vs 22% for placebo.19 In addition, studies have observed that patients can be maintained on clonazepam for up to 2 years without symptom relapse and will tolerate slow-taper discontinuation.18,20 Sedation and drowsiness can be lessened by limiting clonazepam doses to 2 to 3 mg/d.
Akathisia and tremor. Akathisia, a syndrome of motor restlessness and inner turmoil, is associated with antipsychotics but can occur with SSRIs. Reducing the dosage or switching to another, usually less potent agent often can relieve akathisia. When these remedies are not tenable, consider benzodiazepines along with other medications—including beta blockers and anticholinergic agents—with demonstrated efficacy in reducing akathisia symptoms. Lorazepam, diazepam, and clonazepam have demonstrated efficacy for relieving akathisia in comparison studies with placebo, propranolol, and diphenhydramine.21,22
Drug-induced postural tremor can occur with several psychotropics, including lithium, valproic acid, antidepressants, and antipsychotics. A tremor is considered mild if a patient can drink a glass of water with 1 hand without spilling and severe if holding a glass with 2 hands is difficult. Propranolol is most commonly prescribed for these tremors, but alprazolam and clonazepam have demonstrated efficacy, either as monotherapy or coadministered with a beta blocker.23
Acute agitation. Agitated patients often have acute psychosis and/or mania or dyscontrol secondary to axis II disorders.24 Patients may be paranoid, hostile, disruptive, and combative. Rapidly initiating medication can prevent the need for more restrictive measures, such as seclusion or restraint. Antipsychotics—especially high-potency agents such as haloperidol—and benzodiazepines, as monotherapy or in combination, are a mainstay treatment. Although treatment protocols favor atypical antipsychotics over typical antipsychotics, benzodiazepines are a viable option because of their anxiolytic and sedative effects. Advantages of benzodiazepine monotherapy include decreased extrapyramidal symptoms, greater patient acceptance/preference, and increased sedation compared with antipsychotics. Lorazepam, 1 to 2 mg intramuscularly (IM) or orally, is well tolerated because of its favorable drug-drug interaction profile and lack of significant cardiac side effects. Benzodiazepines can cause respiratory depression in patients with chronic lung disease and additive sedation secondary to opiates, other sedatives/hypnotics, or alcohol. Behavioral disinhibition is rare and is associated with preexisting CNS pathology or mental retardation.25 The IM olanzapine package insert warns against coadministering IM lorazepam because of additive cardiorespiratory depressive effects and excessive somnolence.26
Catatonia. The characteristic symptoms of catatonia are immobility, negativism, muteness, and failure to eat or drink. Benzodiazepines improve these symptoms in approximately 70% to 80% of catatonic patients with affective disorders. Response rates are lower in catatonia in patients with schizophrenia.27 If catatonia in a patient with psychosis is missed, giving antipsychotics before benzodiazepines may worsen catatonic symptoms or precipitate neuroleptic malignant syndrome in some cases. When you suspect a patient has catatonia, start with lorazepam, 1 to 2 mg IV or IM, and examine the patient for diminishing catatonic signs within 1 to 2 hours. If catatonia signs lessen, begin regularly scheduled lorazepam, with dosing varying by age—be more cautious in geriatric patients—and symptom severity. Titrate benzodiazepines for stuporous patients more slowly (eg, 1 mg 3 times a day as a starting dose) than for excited catatonic patients. Lorazepam can be increased gradually as tolerated; it is not unusual for patients to require up to 8 to 12 mg/d. Electroconvulsive therapy (ECT) is the treatment of choice when catatonic patients respond poorly or partially to high-dose benzodiazepines.28,29
Benzodiazepine reversal for ECT
Benzodiazepines have anticonvulsant properties that may interfere with the therapeutic efficacy of ECT.30 A multi-center study demonstrated that lorazepam (up to 4 mg/d as needed) in the 48 hours before the first ECT session was not associated with effects on seizure threshold or duration; however, larger lorazepam dosages were associated with briefer EEG seizure duration.31 Some patients may not tolerate withholding or tapering benzodiazepines in preparation for ECT. Studies investigating flumazenil for pre-ECT benzodiazepine reversal are lacking. One retrospective analysis showed that flumazenil administration immediately before and after ECT resulted in adequate seizures with no difference in clinical outcome compared with patients who were not receiving benzodiazepines or flumazenil.32
Tapering benzodiazepines
Slow discontinuation of benzodiazepines is recommended to avoid withdrawal symptoms, such as rebound anxiety, agitation, insomnia, or seizures, particularly when use exceeds 8 weeks. The onset of withdrawal symptoms varies, depending on the medication used. Withdrawal symptoms may appear in 1 to 2 days for agents with shorter half-lives, but may not appear until 3 to 7 days for agents with longer half-lives.33Table 2 lists recommended durations for tapering benzodiazepines.33,34 In general, decrease the total daily dose by 25% the first week, another 25% the second week, then 10% a week until discontinuation. When benzodiazepine use exceeds 1 year, a slower taper is recommended; for example, decrease 10% every 1 to 2 weeks. When 20% of the dosage remains, begin a 5% dose reduction every 2 to 4 weeks. Monitor patients for withdrawal symptoms or symptom exacerbation. If either occur, consider maintaining the current benzodiazepine dose or increasing the dose for 1 to 2 weeks or longer, if necessary, then continue to taper at a slower rate.34
Table 2
Recommendations for tapering benzodiazepines
| Duration of use | Recommended taper length | Comments |
| <6 to 8 weeks | Taper may not be required | Depending on clinical judgment and patient stability/preference, consider implementing a taper, particularly if using a high-dose benzodiazepine or an agent with a short or intermediate half-life, such as alprazolam or triazolam |
| 8 weeks to 6 months | Slowly over 2 to 3 weeks | Go slower during latter half of taper. Tapering will reduce, not eliminate, withdrawal symptoms. Patients should avoid alcohol and stimulants during benzodiazepine withdrawal |
| 6 months to 1 year | Slowly over 4 to 8 weeks | |
| >1 year | Slowly over 2 to 4 months | |
| Source: References 33,34 | ||
Risks of benzodiazepine use
For most indications, benzodiazepine therapy should be short-term.35 Use exceeding 2 to 4 weeks increases the risk for dependence and withdrawal. Tell patients to avoid alcohol while taking a benzodiazepine because this combination is potentially lethal. Benzodiazepines are commonly abused and abuse can lead to unintentional drug overdose. Benzodiazepines accounted for 37% of unintentional drug overdose deaths in West Virginia in 2006; in 46% of these cases, benzodiazepines were used for nonmedical purposes. Clinicians can help reduce the risk of diversion by limiting prescriptions to 30 days with no refills.36
Older patients taking benzodiazepines are at increased risk of falls and hip fractures.37 Lorazepam, oxazepam, and temazepam—agents with shorter half-lives that are not greatly affected by pharmacokinetic changes associated with aging—are preferred for these patients.34 Patients with dementia or other CNS-compromising conditions may become confused or delirious with regular benzodiazepine dosing. Educate patients to whom you prescribe benzodiazepines about the importance of gauging their level of sedation before driving or engaging in other tasks for which sedation could compromise their safety. Benzodiazepine use during pregnancy requires a careful discussion of risks and benefits (Box 2).38
Benzodiazepine use during pregnancy has been associated with cleft palate and urogenital and neurologic malformations in the fetus.38 Although data are conflicting—particularly among recent meta-analyses that fail to demonstrate an association—some experts advise against benzodiazepine use in the first trimester. Participate in shared decision making with your patients and educate them about the potential risks and benefits of benzodiazepine use during the first trimester and throughout pregnancy. After delivery, newborns may develop “floppy baby syndrome”—which is associated with lethargy, difficulty eating, and respiratory depression—or withdrawal.38 To minimize this risk, consider tapering the benzodiazepine as the patient approaches delivery.
Related Resources
- Substance Abuse and Mental Health Services Administration. www.samhsa.gov.
- National Institute on Drug Abuse resources for medical and health professionals. www.drugabuse.gov/medical-health-professionals.
- American Academy of Sleep Medicine. www.aasmnet.org.
Drug Brand Names
- Alprazolam • Xanax
- Chlordiazepoxide • Librium, Limbitrol
- Clonazepam • Klonopin
- Clorazepate • Tranxene
- Diazepam • Valium
- Diphenhydramine • Benadryl, others
- Estazolam • ProSom
- Flumazenil • Romazicon
- Flurazepam • Dalmane
- Haloperidol • Haldol
- Lithium • Lithobid
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Oxazepam • Serax
- Paroxetine • Paxil
- Pregabalin • Lyrica
- Propranolol • Inderal, InnoPran XL, others
- Quazepam • Doral
- Ramelteon • Rozerem
- Sertraline • Zoloft
- Temazepam • Restoril
- Triazolam • Halcion
- Valproic acid • Depakene, Stavzor, others
Disclosures
Drs. Bostwick and Yasugi report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Casher is a speaker for AstraZeneca and Sunovion Pharmaceuticals.
1. Mihic SJ, Harris RA. Hypnotics and sedatives. In: Brunton LL Chabner BA, Knollmann BC, eds. Goodman & Gilman’s the pharmacological basis of therapeutics. New York, NY: McGraw Hill and Company; 2011:457-480.
2. Facts and comparisons Web site. 2011 Wolters Kluwer Health Inc. http://online.factsandcomparisons.com. Accessed August 16, 2011.
3. DuPont RL, Greene W, Lydiard RB. Sedatives and hypnotics: pharmacology and epidemiology. In: Gold MS Hermann R, eds. UpToDate. http://www.uptodate.com/contents/sedatives-and-hypnotics-abuse-and-dependence-pharmacology-and-epidemiology. Accessed August 16, 2011.
4. U.S. Food and Drug Administration. Orange book: approved drug products with therapeutic equivalence evaluations. http://www.accessdata.fda.gov/scripts/cder/ob/default.cfm. Accessed August 16, 2011.
5. Chouinard G. Issues in the clinical use of benzodiazepines: potency withdrawal, and rebound. J Clin Psychiatry. 2004;65(suppl 5):7-12.
6. Shader RI, Greenblatt DJ. Can you provide a table of equivalencies for benzodiazepines and other marketed benzodiazepine receptor agonists? J Clin Psychopharmacol. 1997;17(4):331.-
7. Amato L, Minozzi S, Davoli M. Efficacy and safety of pharmacologic interventions for the treatment of the alcohol withdrawal syndrome. Cochrane Database Syst Rev. 2011;15(6):CD008537.-
8. Schutte-Rodin S, Broch L, Buysse D, et al. Clinical guideline for the evaluation and management of chronic insomnia in adults. J Clin Sleep Med. 2008;4(5):487-504.
9. Foral P, Dewan N, Malesker M. Insomnia: a therapeutic review for pharmacists. Consult Pharm. 2011;26(5):332-341.
10. Riemann D, Perlis ML. The treatments of chronic insomnia: a review of benzodiazepine receptor agonists and psychological and behavioral therapies. Sleep Med Rev. 2009;13(3):205-214.
11. Hidalgo RB, Tupler LA, Davidson JR. An effect-size analysis of pharmacologic treatments of generalized anxiety disorder. J Psychopharmacol. 2007;21(8):864-872.
12. Davidson JR, Zhang W, Connor KM, et al. A psychopharmacological treatment algorithm for generalised anxiety disorder (GAD). J Psychopharmacol. 2010;24(1):3-26.
13. Bandelow B, Zohar J, Hollander E, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive-compulsive and post-traumatic stress disorders - first revision. World J Biol Psychiatry. 2008;9(4):248-312.
14. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. 2nd ed. Arlington VA: American Psychiatric Publishing, Inc.; 2009.
15. Pollack MH, Simon NM, Worthington JJ, et al. Combined paroxetine and clonazepam treatment strategies compared to paroxetine monotherapy for panic disorder. J Psychopharmacol. 2003;17(3):276-282.
16. Watanabe N, Churchill R, Furukawa TA. Combined psychotherapy plus benzodiazepines for panic disorder. Cochrane Database Syst Rev. 2009;(1):CD005335.-
17. Otto MW, McHugh RK, Simon NM, et al. Efficacy of CBT for benzodiazepine discontinuation in patients with panic disorder: further evaluation. Behav Res Ther. 2010;48(8):720-727.
18. Davidson JR. Use of benzodiazepines in social anxiety disorder generalized anxiety disorder, and posttraumatic stress disorder. J Clin Psychiatry. 2004;65(suppl 5):29-33.
19. Argyropoulos SV, Sandford JJ, Nutt DJ. The psychobiology of anxiolytic drugs Part 2: pharmacological treatments of anxiety. Pharmacol Ther. 2000;88(3):213-227.
20. Connor KM, Davidson JR, Potts NL, et al. Discontinuation of clonazepam in the treatment of social phobia. J Clin Psychopharmacol. 1998;18(5):373-378.
21. Miller CH, Fleischhacker WW. Managing antipsychotic-induced acute and chronic akathisia. Drug Saf. 2000;22(1):73-81.
22. Rodnitzky RL. Drug-induced movement disorders. Clin Neuropharmacol. 2002;25(3):142-151.
23. Arbaizar B, Gómez-Acebo I, Llorca J. Postural induced tremor in psychiatry. Psychiatry Clin Neurosci. 2008;62(6):638-645.
24. Casher MI, Bess JD. Manual of inpatient psychiatry. New York NY: Cambridge University Press; 2010.
25. Battaglia J. Pharmacological management of acute agitation. Drugs. 2005;65(9):1207-1222.
26. Physicians’ desk reference. Montvale NJ: PDR Network, LLC; 2010.
27. Rosebush PI, Mazurek MF. Catatonia and its treatment. Schizophr Bull. 2010;36(2):239-242.
28. Ungvari GS, Kau LS, Wai-Kwong T, et al. The pharmacological treatment of catatonia: an overview. Eur Arch Psychiatry Clin Neurosci. 2001;251(suppl 1):I31-I34.
29. Fink M, Taylor MA. Catatonia: a clinician’s guide to diagnosis and treatment. New York NY: Cambridge University Press; 2003.
30. Naguib N, Koorn R. Interactions between psychotropics anaesthetics and electroconvulsive therapy: implications for drug choice and patient management. CNS Drugs. 2002;16(4):229-247.
31. Boylan LS, Haskett RF, Mulsant BH, et al. Determinants of seizure threshold in ECT: benzodiazepine use, anesthetic dosage, and other factors. J ECT. 2000;16(1):3-18.
32. Krystal AD, Watts BV, Weiner RD, et al. The use of flumazenil in the anxious and benzodiazepine-dependent ECT patient. J ECT. 1998;14(1):5-14.
33. Melton ST, Kirkwood CK. Anxiety disorders I: generalized anxiety panic, and social anxiety disorders. In: DiPiro JT, Talbert RL, Yee GC, et al, eds. Pharmacotherapy: a pathophysiologic approach. New York, NY: McGraw-Hill Companies; 2011:1209-1228.
34. Benzodiazepine toolkit. The Pharmacist’s Letter/Prescriber’s Letter. 2011;27(4):270406.-
35. Lader M. Benzodiazepines revisited – will we ever learn? Addiction. 2011;106(12):2086-2109.
36. Toblin RL, Paulozzi LJ, Logan JE, et al. Mental illness and psychotropic drug use among prescription drug overdose deaths: a medical examiner chart review. J Clin Psychiatry. 2010;71(4):491-496.
37. Ashton H. The diagnosis and management of benzodiazepine dependence. Curr Opin Psychiatry. 2005;18(3):249-255.
38. Menon SJ. Psychotropic medication during pregnancy and lactation. Arch Gynecol Obstet. 2008;277(1):1-13.
1. Mihic SJ, Harris RA. Hypnotics and sedatives. In: Brunton LL Chabner BA, Knollmann BC, eds. Goodman & Gilman’s the pharmacological basis of therapeutics. New York, NY: McGraw Hill and Company; 2011:457-480.
2. Facts and comparisons Web site. 2011 Wolters Kluwer Health Inc. http://online.factsandcomparisons.com. Accessed August 16, 2011.
3. DuPont RL, Greene W, Lydiard RB. Sedatives and hypnotics: pharmacology and epidemiology. In: Gold MS Hermann R, eds. UpToDate. http://www.uptodate.com/contents/sedatives-and-hypnotics-abuse-and-dependence-pharmacology-and-epidemiology. Accessed August 16, 2011.
4. U.S. Food and Drug Administration. Orange book: approved drug products with therapeutic equivalence evaluations. http://www.accessdata.fda.gov/scripts/cder/ob/default.cfm. Accessed August 16, 2011.
5. Chouinard G. Issues in the clinical use of benzodiazepines: potency withdrawal, and rebound. J Clin Psychiatry. 2004;65(suppl 5):7-12.
6. Shader RI, Greenblatt DJ. Can you provide a table of equivalencies for benzodiazepines and other marketed benzodiazepine receptor agonists? J Clin Psychopharmacol. 1997;17(4):331.-
7. Amato L, Minozzi S, Davoli M. Efficacy and safety of pharmacologic interventions for the treatment of the alcohol withdrawal syndrome. Cochrane Database Syst Rev. 2011;15(6):CD008537.-
8. Schutte-Rodin S, Broch L, Buysse D, et al. Clinical guideline for the evaluation and management of chronic insomnia in adults. J Clin Sleep Med. 2008;4(5):487-504.
9. Foral P, Dewan N, Malesker M. Insomnia: a therapeutic review for pharmacists. Consult Pharm. 2011;26(5):332-341.
10. Riemann D, Perlis ML. The treatments of chronic insomnia: a review of benzodiazepine receptor agonists and psychological and behavioral therapies. Sleep Med Rev. 2009;13(3):205-214.
11. Hidalgo RB, Tupler LA, Davidson JR. An effect-size analysis of pharmacologic treatments of generalized anxiety disorder. J Psychopharmacol. 2007;21(8):864-872.
12. Davidson JR, Zhang W, Connor KM, et al. A psychopharmacological treatment algorithm for generalised anxiety disorder (GAD). J Psychopharmacol. 2010;24(1):3-26.
13. Bandelow B, Zohar J, Hollander E, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive-compulsive and post-traumatic stress disorders - first revision. World J Biol Psychiatry. 2008;9(4):248-312.
14. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. 2nd ed. Arlington VA: American Psychiatric Publishing, Inc.; 2009.
15. Pollack MH, Simon NM, Worthington JJ, et al. Combined paroxetine and clonazepam treatment strategies compared to paroxetine monotherapy for panic disorder. J Psychopharmacol. 2003;17(3):276-282.
16. Watanabe N, Churchill R, Furukawa TA. Combined psychotherapy plus benzodiazepines for panic disorder. Cochrane Database Syst Rev. 2009;(1):CD005335.-
17. Otto MW, McHugh RK, Simon NM, et al. Efficacy of CBT for benzodiazepine discontinuation in patients with panic disorder: further evaluation. Behav Res Ther. 2010;48(8):720-727.
18. Davidson JR. Use of benzodiazepines in social anxiety disorder generalized anxiety disorder, and posttraumatic stress disorder. J Clin Psychiatry. 2004;65(suppl 5):29-33.
19. Argyropoulos SV, Sandford JJ, Nutt DJ. The psychobiology of anxiolytic drugs Part 2: pharmacological treatments of anxiety. Pharmacol Ther. 2000;88(3):213-227.
20. Connor KM, Davidson JR, Potts NL, et al. Discontinuation of clonazepam in the treatment of social phobia. J Clin Psychopharmacol. 1998;18(5):373-378.
21. Miller CH, Fleischhacker WW. Managing antipsychotic-induced acute and chronic akathisia. Drug Saf. 2000;22(1):73-81.
22. Rodnitzky RL. Drug-induced movement disorders. Clin Neuropharmacol. 2002;25(3):142-151.
23. Arbaizar B, Gómez-Acebo I, Llorca J. Postural induced tremor in psychiatry. Psychiatry Clin Neurosci. 2008;62(6):638-645.
24. Casher MI, Bess JD. Manual of inpatient psychiatry. New York NY: Cambridge University Press; 2010.
25. Battaglia J. Pharmacological management of acute agitation. Drugs. 2005;65(9):1207-1222.
26. Physicians’ desk reference. Montvale NJ: PDR Network, LLC; 2010.
27. Rosebush PI, Mazurek MF. Catatonia and its treatment. Schizophr Bull. 2010;36(2):239-242.
28. Ungvari GS, Kau LS, Wai-Kwong T, et al. The pharmacological treatment of catatonia: an overview. Eur Arch Psychiatry Clin Neurosci. 2001;251(suppl 1):I31-I34.
29. Fink M, Taylor MA. Catatonia: a clinician’s guide to diagnosis and treatment. New York NY: Cambridge University Press; 2003.
30. Naguib N, Koorn R. Interactions between psychotropics anaesthetics and electroconvulsive therapy: implications for drug choice and patient management. CNS Drugs. 2002;16(4):229-247.
31. Boylan LS, Haskett RF, Mulsant BH, et al. Determinants of seizure threshold in ECT: benzodiazepine use, anesthetic dosage, and other factors. J ECT. 2000;16(1):3-18.
32. Krystal AD, Watts BV, Weiner RD, et al. The use of flumazenil in the anxious and benzodiazepine-dependent ECT patient. J ECT. 1998;14(1):5-14.
33. Melton ST, Kirkwood CK. Anxiety disorders I: generalized anxiety panic, and social anxiety disorders. In: DiPiro JT, Talbert RL, Yee GC, et al, eds. Pharmacotherapy: a pathophysiologic approach. New York, NY: McGraw-Hill Companies; 2011:1209-1228.
34. Benzodiazepine toolkit. The Pharmacist’s Letter/Prescriber’s Letter. 2011;27(4):270406.-
35. Lader M. Benzodiazepines revisited – will we ever learn? Addiction. 2011;106(12):2086-2109.
36. Toblin RL, Paulozzi LJ, Logan JE, et al. Mental illness and psychotropic drug use among prescription drug overdose deaths: a medical examiner chart review. J Clin Psychiatry. 2010;71(4):491-496.
37. Ashton H. The diagnosis and management of benzodiazepine dependence. Curr Opin Psychiatry. 2005;18(3):249-255.
38. Menon SJ. Psychotropic medication during pregnancy and lactation. Arch Gynecol Obstet. 2008;277(1):1-13.
Bedside visit comes too late . . . Unrecognized spinal infection leads to paralysis . . .
Bedside visit comes too late
A 22-YEAR-OLD MAN underwent a liver biopsy after being admitted to the hospital a week earlier with fever, chills, diarrhea, and general malaise. A number of specialists had seen him in the hospital because of abnormal laboratory studies, increasing fever, and a maculopapular rash over his trunk and face.
After the biopsy, the patient was dizzy and diaphoretic. His attending physician ordered hemoglobin and hematocrit levels, which were lower than earlier that day. Repeat testing showed a further decrease, prompting the physician to order 2 units of red blood cells.
Typing and cross-matching delayed the transfusion for several hours. Before it could be started, the patient was found unresponsive. When the attending physician came to the bedside, the patient had no palpable pulse. A code was called, but resuscitation efforts failed.
An autopsy found a small hole in the liver and 3500 mL of blood in the peritoneal cavity, as well as hepatitis with zonal and submassive necrosis, hemoperitoneum, and hypertrophy of the heart. An HIV test performed before the biopsy eventually came back positive.
PLAINTIFF’S CLAIM The attending physician and nurses were negligent in failing to respond to signs and symptoms of internal bleeding, including falling hematocrit and hemoglobin levels. The attending physician, who was at the hospital when the patient’s condition deteriorated, should have gone to the bedside and taken steps to prevent his death.
THE DEFENSE The patient had been stable overnight; a bedside exam was unnecessary.
VERDICT $1,815,658 Texas verdict.
COMMENT Considering the many demands on clinicians’ time, it’s easy to postpone a face-to-face evaluation of a patient after a procedure. In this case, such a delay cost more than $1.8 million. A laboratory test or nurses’ notes are sometimes inadequate substitutes for a physician’s evaluation.
Failure to investigate suspicious symptoms ends badly
A MAN WITH SIGNS AND SYMPTOMS SUGGESTIVE OF AORTIC ANEURYSM/DISSECTION—including chest pain, pericardial effusion, aortic regurgitation, and aortic dilatation—saw his physician, but the doctor didn’t order any tests, such as computed tomography (CT) with contrast, magnetic resonance imaging (MRI), or transesophageal echocardiogram (TEE).
Two weeks later, the 43-year-old patient returned to the physician, who noted left ventricular hypertrophy with pericardial effusion and mild aortic loop dilatation. Once again, the doctor didn’t order tests to rule out aneurysm/dissection.
Three weeks after the second office visit, the patient collapsed and was taken by ambulance to a hospital, where he was pronounced dead. An autopsy indicated that the cause of death was cardiac tamponade resulting from an undiagnosed aortic dissection.
PLAINTIFF’S CLAIM The physician should have ordered a CT scan with contrast, an MRI, or a TEE, any of which would have confirmed an aortic aneurysm/dissection, mandating immediate admission to a hospital for surgery.
THE DEFENSE No information about the defense is available.
VERDICT $1 million Maryland settlement.
COMMENT Although many common conditions will resolve spontaneously, it’s hard to imagine temporizing in a patient with chest pain and presumed aortic dissection.
Unrecognized spinal infection leads to paralysis
A 355-LB MAN WITH DIABETES AND SPINAL DISC DISEASE experienced a sharp pain between his shoulder blades after playing golf, followed by constant back pain radiating to his chest. He went to the emergency department (ED) the next day and was admitted to the hospital to rule out a heart attack.
During a week in the hospital, the patient was seen by several doctors and diagnosed with pneumonia and excessive myoglobin levels. A computed tomography (CT) scan of the thorax and abdomen showing fluid buildup in the lining around the lungs led to the pneumonia diagnosis. No definitive spinal view was available, however, because of a mixup between a secretary and a radiology technician.
When the patient saw the hospital attending physician (at the family practice group where she was a partner) after discharge from the hospital, he complained of shooting pain down his spine. The doctor prescribed muscle relaxants. Soon afterward, the patient developed difficulty walking and reported no bowel movements for 13 days.
Almost 2 weeks after discharge from the hospital, the patient broke his ankle. He told the paramedics who responded that he felt numb from his nipples to his feet. He was taken to a community hospital, where a doctor ordered another CT scan. The radiologist who read the scan failed to identify the serious spinal infection it indicated.
The patient was transferred back to the original hospital. No doctor saw him for 8 hours after transfer, by which time he was paralyzed from the chest down.
PLAINTIFF’S CLAIM The fluid buildup on the first CT scan was caused not by pneumonia but by an infection in the spinal discs that had spread to the vertebrae and surrounding tissue.
THE DEFENSE The attending physician denied at trial that the patient had told her about the shooting pains down his spine during the posthospitalization visit.
VERDICT $4.75 million Illinois verdict, preceded by more than $2.7 million in settlements with some of the doctors involved and the community hospital.
COMMENT Careful follow-up of ED visits and coordinated care are essential to avoid large verdicts such as this one.
Bedside visit comes too late
A 22-YEAR-OLD MAN underwent a liver biopsy after being admitted to the hospital a week earlier with fever, chills, diarrhea, and general malaise. A number of specialists had seen him in the hospital because of abnormal laboratory studies, increasing fever, and a maculopapular rash over his trunk and face.
After the biopsy, the patient was dizzy and diaphoretic. His attending physician ordered hemoglobin and hematocrit levels, which were lower than earlier that day. Repeat testing showed a further decrease, prompting the physician to order 2 units of red blood cells.
Typing and cross-matching delayed the transfusion for several hours. Before it could be started, the patient was found unresponsive. When the attending physician came to the bedside, the patient had no palpable pulse. A code was called, but resuscitation efforts failed.
An autopsy found a small hole in the liver and 3500 mL of blood in the peritoneal cavity, as well as hepatitis with zonal and submassive necrosis, hemoperitoneum, and hypertrophy of the heart. An HIV test performed before the biopsy eventually came back positive.
PLAINTIFF’S CLAIM The attending physician and nurses were negligent in failing to respond to signs and symptoms of internal bleeding, including falling hematocrit and hemoglobin levels. The attending physician, who was at the hospital when the patient’s condition deteriorated, should have gone to the bedside and taken steps to prevent his death.
THE DEFENSE The patient had been stable overnight; a bedside exam was unnecessary.
VERDICT $1,815,658 Texas verdict.
COMMENT Considering the many demands on clinicians’ time, it’s easy to postpone a face-to-face evaluation of a patient after a procedure. In this case, such a delay cost more than $1.8 million. A laboratory test or nurses’ notes are sometimes inadequate substitutes for a physician’s evaluation.
Failure to investigate suspicious symptoms ends badly
A MAN WITH SIGNS AND SYMPTOMS SUGGESTIVE OF AORTIC ANEURYSM/DISSECTION—including chest pain, pericardial effusion, aortic regurgitation, and aortic dilatation—saw his physician, but the doctor didn’t order any tests, such as computed tomography (CT) with contrast, magnetic resonance imaging (MRI), or transesophageal echocardiogram (TEE).
Two weeks later, the 43-year-old patient returned to the physician, who noted left ventricular hypertrophy with pericardial effusion and mild aortic loop dilatation. Once again, the doctor didn’t order tests to rule out aneurysm/dissection.
Three weeks after the second office visit, the patient collapsed and was taken by ambulance to a hospital, where he was pronounced dead. An autopsy indicated that the cause of death was cardiac tamponade resulting from an undiagnosed aortic dissection.
PLAINTIFF’S CLAIM The physician should have ordered a CT scan with contrast, an MRI, or a TEE, any of which would have confirmed an aortic aneurysm/dissection, mandating immediate admission to a hospital for surgery.
THE DEFENSE No information about the defense is available.
VERDICT $1 million Maryland settlement.
COMMENT Although many common conditions will resolve spontaneously, it’s hard to imagine temporizing in a patient with chest pain and presumed aortic dissection.
Unrecognized spinal infection leads to paralysis
A 355-LB MAN WITH DIABETES AND SPINAL DISC DISEASE experienced a sharp pain between his shoulder blades after playing golf, followed by constant back pain radiating to his chest. He went to the emergency department (ED) the next day and was admitted to the hospital to rule out a heart attack.
During a week in the hospital, the patient was seen by several doctors and diagnosed with pneumonia and excessive myoglobin levels. A computed tomography (CT) scan of the thorax and abdomen showing fluid buildup in the lining around the lungs led to the pneumonia diagnosis. No definitive spinal view was available, however, because of a mixup between a secretary and a radiology technician.
When the patient saw the hospital attending physician (at the family practice group where she was a partner) after discharge from the hospital, he complained of shooting pain down his spine. The doctor prescribed muscle relaxants. Soon afterward, the patient developed difficulty walking and reported no bowel movements for 13 days.
Almost 2 weeks after discharge from the hospital, the patient broke his ankle. He told the paramedics who responded that he felt numb from his nipples to his feet. He was taken to a community hospital, where a doctor ordered another CT scan. The radiologist who read the scan failed to identify the serious spinal infection it indicated.
The patient was transferred back to the original hospital. No doctor saw him for 8 hours after transfer, by which time he was paralyzed from the chest down.
PLAINTIFF’S CLAIM The fluid buildup on the first CT scan was caused not by pneumonia but by an infection in the spinal discs that had spread to the vertebrae and surrounding tissue.
THE DEFENSE The attending physician denied at trial that the patient had told her about the shooting pains down his spine during the posthospitalization visit.
VERDICT $4.75 million Illinois verdict, preceded by more than $2.7 million in settlements with some of the doctors involved and the community hospital.
COMMENT Careful follow-up of ED visits and coordinated care are essential to avoid large verdicts such as this one.
Bedside visit comes too late
A 22-YEAR-OLD MAN underwent a liver biopsy after being admitted to the hospital a week earlier with fever, chills, diarrhea, and general malaise. A number of specialists had seen him in the hospital because of abnormal laboratory studies, increasing fever, and a maculopapular rash over his trunk and face.
After the biopsy, the patient was dizzy and diaphoretic. His attending physician ordered hemoglobin and hematocrit levels, which were lower than earlier that day. Repeat testing showed a further decrease, prompting the physician to order 2 units of red blood cells.
Typing and cross-matching delayed the transfusion for several hours. Before it could be started, the patient was found unresponsive. When the attending physician came to the bedside, the patient had no palpable pulse. A code was called, but resuscitation efforts failed.
An autopsy found a small hole in the liver and 3500 mL of blood in the peritoneal cavity, as well as hepatitis with zonal and submassive necrosis, hemoperitoneum, and hypertrophy of the heart. An HIV test performed before the biopsy eventually came back positive.
PLAINTIFF’S CLAIM The attending physician and nurses were negligent in failing to respond to signs and symptoms of internal bleeding, including falling hematocrit and hemoglobin levels. The attending physician, who was at the hospital when the patient’s condition deteriorated, should have gone to the bedside and taken steps to prevent his death.
THE DEFENSE The patient had been stable overnight; a bedside exam was unnecessary.
VERDICT $1,815,658 Texas verdict.
COMMENT Considering the many demands on clinicians’ time, it’s easy to postpone a face-to-face evaluation of a patient after a procedure. In this case, such a delay cost more than $1.8 million. A laboratory test or nurses’ notes are sometimes inadequate substitutes for a physician’s evaluation.
Failure to investigate suspicious symptoms ends badly
A MAN WITH SIGNS AND SYMPTOMS SUGGESTIVE OF AORTIC ANEURYSM/DISSECTION—including chest pain, pericardial effusion, aortic regurgitation, and aortic dilatation—saw his physician, but the doctor didn’t order any tests, such as computed tomography (CT) with contrast, magnetic resonance imaging (MRI), or transesophageal echocardiogram (TEE).
Two weeks later, the 43-year-old patient returned to the physician, who noted left ventricular hypertrophy with pericardial effusion and mild aortic loop dilatation. Once again, the doctor didn’t order tests to rule out aneurysm/dissection.
Three weeks after the second office visit, the patient collapsed and was taken by ambulance to a hospital, where he was pronounced dead. An autopsy indicated that the cause of death was cardiac tamponade resulting from an undiagnosed aortic dissection.
PLAINTIFF’S CLAIM The physician should have ordered a CT scan with contrast, an MRI, or a TEE, any of which would have confirmed an aortic aneurysm/dissection, mandating immediate admission to a hospital for surgery.
THE DEFENSE No information about the defense is available.
VERDICT $1 million Maryland settlement.
COMMENT Although many common conditions will resolve spontaneously, it’s hard to imagine temporizing in a patient with chest pain and presumed aortic dissection.
Unrecognized spinal infection leads to paralysis
A 355-LB MAN WITH DIABETES AND SPINAL DISC DISEASE experienced a sharp pain between his shoulder blades after playing golf, followed by constant back pain radiating to his chest. He went to the emergency department (ED) the next day and was admitted to the hospital to rule out a heart attack.
During a week in the hospital, the patient was seen by several doctors and diagnosed with pneumonia and excessive myoglobin levels. A computed tomography (CT) scan of the thorax and abdomen showing fluid buildup in the lining around the lungs led to the pneumonia diagnosis. No definitive spinal view was available, however, because of a mixup between a secretary and a radiology technician.
When the patient saw the hospital attending physician (at the family practice group where she was a partner) after discharge from the hospital, he complained of shooting pain down his spine. The doctor prescribed muscle relaxants. Soon afterward, the patient developed difficulty walking and reported no bowel movements for 13 days.
Almost 2 weeks after discharge from the hospital, the patient broke his ankle. He told the paramedics who responded that he felt numb from his nipples to his feet. He was taken to a community hospital, where a doctor ordered another CT scan. The radiologist who read the scan failed to identify the serious spinal infection it indicated.
The patient was transferred back to the original hospital. No doctor saw him for 8 hours after transfer, by which time he was paralyzed from the chest down.
PLAINTIFF’S CLAIM The fluid buildup on the first CT scan was caused not by pneumonia but by an infection in the spinal discs that had spread to the vertebrae and surrounding tissue.
THE DEFENSE The attending physician denied at trial that the patient had told her about the shooting pains down his spine during the posthospitalization visit.
VERDICT $4.75 million Illinois verdict, preceded by more than $2.7 million in settlements with some of the doctors involved and the community hospital.
COMMENT Careful follow-up of ED visits and coordinated care are essential to avoid large verdicts such as this one.
What’s best for IBS?
Recommend antispasmodics or antidepressants for patients with irritable bowel syndrome (IBS) and explain that, while fiber may have other benefits, it is unlikely to relieve IBS symptoms.1
STRENGTH OF RECOMMENDATION
A: Based on a meta-analysis.
Ruepert L, Quartero AO, deWit NJ, et al. Bulking agents, antispasmodics and antidepressants for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2011;(8):CD003460.
ILLUSTRATIVE CASE
A 25-year-old woman who has been your patient for several years has intermittent bouts of abdominal pain, constipation, gas, and bloating. You believe she can benefit from treatment for IBS. What should you recommend?
IBS is the most common functional disorder of the gastrointestinal (GI) tract, affecting approximately 15% of the US population2 and accounting for annual health care costs of roughly $30 billion.3 The primary symptoms are bloating, gas, and abdominal pain that often improves immediately after a bowel movement. Patients may have intermittent diarrhea and constipation, as well.
IBS may be related to “brain-gut dysfunction”
The etiology of IBS is unclear, but many agree that a combination of abnormal GI motility, visceral hypersensitivity, and “brain-gut dysfunction”—the inability of the brain to send signals that turn down pain produced in the GI tract—are contributing factors. Although IBS is not life threatening, it has a significant personal, social, and psychological impact. Despite its high prevalence and impact, only a limited number of large studies have assessed the effectiveness of various treatments.
STUDY SUMMARY: Antispasmodics, antidepressants offer relief—fiber does not
The Cochrane review included 56 randomized controlled trials (RCTs) comparing the efficacy of bulking agents (fiber supplements), anti-spasmodics, or antidepressants with placebo for the treatment of IBS. Twelve RCTs (n=621) focused on bulking agents, 29 (n=2333) on antispasmodics, and 15 (n=922) on antidepressants. Inclusion criteria included age (>12 years) and an IBS diagnosis. The outcomes analyzed were improvement in abdominal pain, global health assessments, and IBS symptom scores. Adverse effects were not evaluated.
Bulking agents. In studies ranging from 4 to 16 weeks, bulking agents were found to have no significant effect on abdominal pain (4 studies; standardized mean difference [SMD], 0.03; 95% confidence interval [CI], -0.34 to 0.40; P=.87) or global functioning (11 studies; risk ratio [RR]=1.11; 95% CI, 0.91-1.35; P=.32). Nor was there an improvement in IBS symptom score (3 studies; SMD=0.00; 95% CI, -0.43 to 0.43; P=1.00).
Antispasmodics. Assessed in RCTs ranging from one week to 6 months, antispasmodics significantly improved abdominal pain (RR=1.3; 95% CI, 1.1-1.55; P<.001; number needed to treat [NNT]=7); global functioning (RR=1.5; 95% CI, 1.2-1.8; P<.0001; NNT=5), and IBS symptom score (RR=1.9; 95% CI, 1.3-2.8; P<.01; NNT=3). Ten different antispasmodic agents were studied; in subgroup analyses, 5 of them— cimetropium/dicyclomine, peppermint oil, pinaverium, and trimebutine—were found to have statistically significant benefits.
Antidepressants. In studies of both tricyclics and selective serotonin reuptake inhibitors (SSRIs), antidepressants were found to have a significant effect on improving abdominal pain (RR=1.5; 95% CI, 1.0-2.1; P<.03; NNT=5), global functioning (RR=1.6; 95% CI, 1.2-2; P<.001; NNT=4), and IBS symptom score (RR=2.0; 95% CI, 1.3-3.0; P<.001; NNT=4). Subgroup analyses found statistically significant benefits in global functioning for SSRIs, and in abdominal pain and symptom scores for tricyclics.
WHAT’S NEW: More evidence against fiber for IBS symptoms
This Cochrane review confirms earlier findings—that both antispasmodics and antidepressants are effective treatments for IBS, but bulking agents are not. This is an important finding because dietary fiber adjustment is still among the first recommendations made by leading organizations like the American Gastroenterological Association and the World Gastroenterology Organisation.4,5
CAVEATS: Limitations of studies included in the meta-analysis
Adverse effects of antispasmodics and antidepressants, which may limit compliance and treatment efficacy, were not addressed by the Cochrane reviewers. The total number of participants in trials of bulking agents was much smaller than that of the other treatments, so it is possible that clinically meaningful improvements were missed due to inadequate statistical power. In addition, the duration of interventions was highly variable, ranging from one to 4 months for bulking agents and antidepressants and from one week to 6 months for antispasmodics.
It is also important to note that 8 of the 12 studies of bulking agents were conducted in GI clinics. (The settings in which the other 4 studies were conducted is unclear.) Given the possibility that patients referred to GI clinics have already tried and failed to respond to fiber (and thus, that those who do respond to fiber are not given referrals), it may be reasonable for family physicians to recommend a trial of bulking agents for patients with IBS and to monitor them for symptom improvement.
CHALLENGES TO IMPLEMENTATION: Patients may favor fiber
Patients with IBS may be reluctant to take antidepressants or antispasmodics, due to concern about adverse effects (eg, headache, insomnia, nervousness, dry mouth, and constipation) or because of a preference for what they see as a more “natural” remedy. It may be helpful to explain that while fiber may have some health benefits, such as lowering cholesterol,6 antispasmodics and antidepressants have been found to improve IBS symptoms but thus far, fiber has not.
Acknowledgement
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National center for Research Resources, a clinical Translational Science Award to the University of chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National center for Research Resources or the National Institutes of Health.
1. Ruepert L, Quartero AO, deWit NJ, et al. Bulking agents, antispasmodics and antidepressants for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2011;(8):CD003460.-
2. Saito YA, Schoenfeld P, Locke GR 3rd. The epidemiology of irritable bowel syndrome in North America: a systematic review. Am J Gastroenterol. 2002;97:1910-1915.
3. Hulisz D. The burden of illness of irritable bowel syndrome: current challenges and hope for the future. J Manag Care Pharm. 2004;10:299-309.
4. American Gastroenterological Association. IBS: A patient’s guide to living with irritable bowel syndrome. Available at: http://www.gastro.org/patient-center/digestive-conditions/irritable-bowel-syndrome. Accessed March 21, 2012.
5. World Gastroenterology Organisation.WGO practice guideline— irritable bowel syndrome: a global perspective. 2009. Available at: http://www.worldgastroenterology.org/irritable-bowel-syndrome.html. Accessed March 16, 2012.
6. Gunness P, Gidley MJ. Mechanisms underlying the cholesterol-lowering properties of soluble dietary fibre polysaccharides. Food Funct. 2010;1:149-155.
Recommend antispasmodics or antidepressants for patients with irritable bowel syndrome (IBS) and explain that, while fiber may have other benefits, it is unlikely to relieve IBS symptoms.1
STRENGTH OF RECOMMENDATION
A: Based on a meta-analysis.
Ruepert L, Quartero AO, deWit NJ, et al. Bulking agents, antispasmodics and antidepressants for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2011;(8):CD003460.
ILLUSTRATIVE CASE
A 25-year-old woman who has been your patient for several years has intermittent bouts of abdominal pain, constipation, gas, and bloating. You believe she can benefit from treatment for IBS. What should you recommend?
IBS is the most common functional disorder of the gastrointestinal (GI) tract, affecting approximately 15% of the US population2 and accounting for annual health care costs of roughly $30 billion.3 The primary symptoms are bloating, gas, and abdominal pain that often improves immediately after a bowel movement. Patients may have intermittent diarrhea and constipation, as well.
IBS may be related to “brain-gut dysfunction”
The etiology of IBS is unclear, but many agree that a combination of abnormal GI motility, visceral hypersensitivity, and “brain-gut dysfunction”—the inability of the brain to send signals that turn down pain produced in the GI tract—are contributing factors. Although IBS is not life threatening, it has a significant personal, social, and psychological impact. Despite its high prevalence and impact, only a limited number of large studies have assessed the effectiveness of various treatments.
STUDY SUMMARY: Antispasmodics, antidepressants offer relief—fiber does not
The Cochrane review included 56 randomized controlled trials (RCTs) comparing the efficacy of bulking agents (fiber supplements), anti-spasmodics, or antidepressants with placebo for the treatment of IBS. Twelve RCTs (n=621) focused on bulking agents, 29 (n=2333) on antispasmodics, and 15 (n=922) on antidepressants. Inclusion criteria included age (>12 years) and an IBS diagnosis. The outcomes analyzed were improvement in abdominal pain, global health assessments, and IBS symptom scores. Adverse effects were not evaluated.
Bulking agents. In studies ranging from 4 to 16 weeks, bulking agents were found to have no significant effect on abdominal pain (4 studies; standardized mean difference [SMD], 0.03; 95% confidence interval [CI], -0.34 to 0.40; P=.87) or global functioning (11 studies; risk ratio [RR]=1.11; 95% CI, 0.91-1.35; P=.32). Nor was there an improvement in IBS symptom score (3 studies; SMD=0.00; 95% CI, -0.43 to 0.43; P=1.00).
Antispasmodics. Assessed in RCTs ranging from one week to 6 months, antispasmodics significantly improved abdominal pain (RR=1.3; 95% CI, 1.1-1.55; P<.001; number needed to treat [NNT]=7); global functioning (RR=1.5; 95% CI, 1.2-1.8; P<.0001; NNT=5), and IBS symptom score (RR=1.9; 95% CI, 1.3-2.8; P<.01; NNT=3). Ten different antispasmodic agents were studied; in subgroup analyses, 5 of them— cimetropium/dicyclomine, peppermint oil, pinaverium, and trimebutine—were found to have statistically significant benefits.
Antidepressants. In studies of both tricyclics and selective serotonin reuptake inhibitors (SSRIs), antidepressants were found to have a significant effect on improving abdominal pain (RR=1.5; 95% CI, 1.0-2.1; P<.03; NNT=5), global functioning (RR=1.6; 95% CI, 1.2-2; P<.001; NNT=4), and IBS symptom score (RR=2.0; 95% CI, 1.3-3.0; P<.001; NNT=4). Subgroup analyses found statistically significant benefits in global functioning for SSRIs, and in abdominal pain and symptom scores for tricyclics.
WHAT’S NEW: More evidence against fiber for IBS symptoms
This Cochrane review confirms earlier findings—that both antispasmodics and antidepressants are effective treatments for IBS, but bulking agents are not. This is an important finding because dietary fiber adjustment is still among the first recommendations made by leading organizations like the American Gastroenterological Association and the World Gastroenterology Organisation.4,5
CAVEATS: Limitations of studies included in the meta-analysis
Adverse effects of antispasmodics and antidepressants, which may limit compliance and treatment efficacy, were not addressed by the Cochrane reviewers. The total number of participants in trials of bulking agents was much smaller than that of the other treatments, so it is possible that clinically meaningful improvements were missed due to inadequate statistical power. In addition, the duration of interventions was highly variable, ranging from one to 4 months for bulking agents and antidepressants and from one week to 6 months for antispasmodics.
It is also important to note that 8 of the 12 studies of bulking agents were conducted in GI clinics. (The settings in which the other 4 studies were conducted is unclear.) Given the possibility that patients referred to GI clinics have already tried and failed to respond to fiber (and thus, that those who do respond to fiber are not given referrals), it may be reasonable for family physicians to recommend a trial of bulking agents for patients with IBS and to monitor them for symptom improvement.
CHALLENGES TO IMPLEMENTATION: Patients may favor fiber
Patients with IBS may be reluctant to take antidepressants or antispasmodics, due to concern about adverse effects (eg, headache, insomnia, nervousness, dry mouth, and constipation) or because of a preference for what they see as a more “natural” remedy. It may be helpful to explain that while fiber may have some health benefits, such as lowering cholesterol,6 antispasmodics and antidepressants have been found to improve IBS symptoms but thus far, fiber has not.
Acknowledgement
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National center for Research Resources, a clinical Translational Science Award to the University of chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National center for Research Resources or the National Institutes of Health.
Recommend antispasmodics or antidepressants for patients with irritable bowel syndrome (IBS) and explain that, while fiber may have other benefits, it is unlikely to relieve IBS symptoms.1
STRENGTH OF RECOMMENDATION
A: Based on a meta-analysis.
Ruepert L, Quartero AO, deWit NJ, et al. Bulking agents, antispasmodics and antidepressants for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2011;(8):CD003460.
ILLUSTRATIVE CASE
A 25-year-old woman who has been your patient for several years has intermittent bouts of abdominal pain, constipation, gas, and bloating. You believe she can benefit from treatment for IBS. What should you recommend?
IBS is the most common functional disorder of the gastrointestinal (GI) tract, affecting approximately 15% of the US population2 and accounting for annual health care costs of roughly $30 billion.3 The primary symptoms are bloating, gas, and abdominal pain that often improves immediately after a bowel movement. Patients may have intermittent diarrhea and constipation, as well.
IBS may be related to “brain-gut dysfunction”
The etiology of IBS is unclear, but many agree that a combination of abnormal GI motility, visceral hypersensitivity, and “brain-gut dysfunction”—the inability of the brain to send signals that turn down pain produced in the GI tract—are contributing factors. Although IBS is not life threatening, it has a significant personal, social, and psychological impact. Despite its high prevalence and impact, only a limited number of large studies have assessed the effectiveness of various treatments.
STUDY SUMMARY: Antispasmodics, antidepressants offer relief—fiber does not
The Cochrane review included 56 randomized controlled trials (RCTs) comparing the efficacy of bulking agents (fiber supplements), anti-spasmodics, or antidepressants with placebo for the treatment of IBS. Twelve RCTs (n=621) focused on bulking agents, 29 (n=2333) on antispasmodics, and 15 (n=922) on antidepressants. Inclusion criteria included age (>12 years) and an IBS diagnosis. The outcomes analyzed were improvement in abdominal pain, global health assessments, and IBS symptom scores. Adverse effects were not evaluated.
Bulking agents. In studies ranging from 4 to 16 weeks, bulking agents were found to have no significant effect on abdominal pain (4 studies; standardized mean difference [SMD], 0.03; 95% confidence interval [CI], -0.34 to 0.40; P=.87) or global functioning (11 studies; risk ratio [RR]=1.11; 95% CI, 0.91-1.35; P=.32). Nor was there an improvement in IBS symptom score (3 studies; SMD=0.00; 95% CI, -0.43 to 0.43; P=1.00).
Antispasmodics. Assessed in RCTs ranging from one week to 6 months, antispasmodics significantly improved abdominal pain (RR=1.3; 95% CI, 1.1-1.55; P<.001; number needed to treat [NNT]=7); global functioning (RR=1.5; 95% CI, 1.2-1.8; P<.0001; NNT=5), and IBS symptom score (RR=1.9; 95% CI, 1.3-2.8; P<.01; NNT=3). Ten different antispasmodic agents were studied; in subgroup analyses, 5 of them— cimetropium/dicyclomine, peppermint oil, pinaverium, and trimebutine—were found to have statistically significant benefits.
Antidepressants. In studies of both tricyclics and selective serotonin reuptake inhibitors (SSRIs), antidepressants were found to have a significant effect on improving abdominal pain (RR=1.5; 95% CI, 1.0-2.1; P<.03; NNT=5), global functioning (RR=1.6; 95% CI, 1.2-2; P<.001; NNT=4), and IBS symptom score (RR=2.0; 95% CI, 1.3-3.0; P<.001; NNT=4). Subgroup analyses found statistically significant benefits in global functioning for SSRIs, and in abdominal pain and symptom scores for tricyclics.
WHAT’S NEW: More evidence against fiber for IBS symptoms
This Cochrane review confirms earlier findings—that both antispasmodics and antidepressants are effective treatments for IBS, but bulking agents are not. This is an important finding because dietary fiber adjustment is still among the first recommendations made by leading organizations like the American Gastroenterological Association and the World Gastroenterology Organisation.4,5
CAVEATS: Limitations of studies included in the meta-analysis
Adverse effects of antispasmodics and antidepressants, which may limit compliance and treatment efficacy, were not addressed by the Cochrane reviewers. The total number of participants in trials of bulking agents was much smaller than that of the other treatments, so it is possible that clinically meaningful improvements were missed due to inadequate statistical power. In addition, the duration of interventions was highly variable, ranging from one to 4 months for bulking agents and antidepressants and from one week to 6 months for antispasmodics.
It is also important to note that 8 of the 12 studies of bulking agents were conducted in GI clinics. (The settings in which the other 4 studies were conducted is unclear.) Given the possibility that patients referred to GI clinics have already tried and failed to respond to fiber (and thus, that those who do respond to fiber are not given referrals), it may be reasonable for family physicians to recommend a trial of bulking agents for patients with IBS and to monitor them for symptom improvement.
CHALLENGES TO IMPLEMENTATION: Patients may favor fiber
Patients with IBS may be reluctant to take antidepressants or antispasmodics, due to concern about adverse effects (eg, headache, insomnia, nervousness, dry mouth, and constipation) or because of a preference for what they see as a more “natural” remedy. It may be helpful to explain that while fiber may have some health benefits, such as lowering cholesterol,6 antispasmodics and antidepressants have been found to improve IBS symptoms but thus far, fiber has not.
Acknowledgement
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National center for Research Resources, a clinical Translational Science Award to the University of chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National center for Research Resources or the National Institutes of Health.
1. Ruepert L, Quartero AO, deWit NJ, et al. Bulking agents, antispasmodics and antidepressants for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2011;(8):CD003460.-
2. Saito YA, Schoenfeld P, Locke GR 3rd. The epidemiology of irritable bowel syndrome in North America: a systematic review. Am J Gastroenterol. 2002;97:1910-1915.
3. Hulisz D. The burden of illness of irritable bowel syndrome: current challenges and hope for the future. J Manag Care Pharm. 2004;10:299-309.
4. American Gastroenterological Association. IBS: A patient’s guide to living with irritable bowel syndrome. Available at: http://www.gastro.org/patient-center/digestive-conditions/irritable-bowel-syndrome. Accessed March 21, 2012.
5. World Gastroenterology Organisation.WGO practice guideline— irritable bowel syndrome: a global perspective. 2009. Available at: http://www.worldgastroenterology.org/irritable-bowel-syndrome.html. Accessed March 16, 2012.
6. Gunness P, Gidley MJ. Mechanisms underlying the cholesterol-lowering properties of soluble dietary fibre polysaccharides. Food Funct. 2010;1:149-155.
1. Ruepert L, Quartero AO, deWit NJ, et al. Bulking agents, antispasmodics and antidepressants for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2011;(8):CD003460.-
2. Saito YA, Schoenfeld P, Locke GR 3rd. The epidemiology of irritable bowel syndrome in North America: a systematic review. Am J Gastroenterol. 2002;97:1910-1915.
3. Hulisz D. The burden of illness of irritable bowel syndrome: current challenges and hope for the future. J Manag Care Pharm. 2004;10:299-309.
4. American Gastroenterological Association. IBS: A patient’s guide to living with irritable bowel syndrome. Available at: http://www.gastro.org/patient-center/digestive-conditions/irritable-bowel-syndrome. Accessed March 21, 2012.
5. World Gastroenterology Organisation.WGO practice guideline— irritable bowel syndrome: a global perspective. 2009. Available at: http://www.worldgastroenterology.org/irritable-bowel-syndrome.html. Accessed March 16, 2012.
6. Gunness P, Gidley MJ. Mechanisms underlying the cholesterol-lowering properties of soluble dietary fibre polysaccharides. Food Funct. 2010;1:149-155.
Copyright ©2012 The Family Physicians Inquiries Network. All rights reserved.
How to recognize a patient who’s high on “bath salts”
• Include cathinone use in the differential diagnosis for any patient exhibiting paranoid psychotic behavior or hallucinatory delirium. C
• Keep in mind that cathinone effects can mimic the “excited delirium” attributed to cocaine, methamphetamine, PCP, and Ecstasy. C
• Consider using benzodiazepines to control agitation, or low-dose antipsychotics to treat hallucinations. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 31-year-old construction worker with a history of intermittent cocaine use was brought to the emergency department (ED) by the police. He was handcuffed and appeared confused and frightened. The patient’s wife had phoned the police after he began running through a field in pursuit of perceived invaders of their home. The wife reported that a few hours earlier, the patient had begun to hallucinate and had become very fearful after getting high.
His heart rate was 126, blood pressure 136/96 mm Hg, and temperature 99.6°F. During the initial exam, the patient became agitated, attempted to assault a nurse, and tried to leave the ED before being subdued. A urine screen for drugs was negative for cocaine. His creatine phosphokinase was 850 U/L, creatinine 2.32 mg/dL, and blood urea nitrogen 27 mg/dL.
The health care team learned that the drug he’d been snorting earlier that day—and the day before—was “bath salts.”
This patient was one of the 30 that we’ve seen at our university hospital over the past year.
Since early 2010, EDs, psychiatric facilities, and poison control centers have seen a surge in the number of patients abusing novel synthetic stimulants—cathinones—that had once been sold in convenience stores and tobacco shops and often labeled innocuously as “bath salts” or “plant food.” Sales have largely gone underground, sold by those trafficking in methamphetamine and cocaine. These products are also available for online purchase and may be sold under such provocative names as “Cloud Nine” or “Rave.”1 In 2010, poison control centers received 304 calls related to the use of these substances; in 2011, the number was 6138.2
Cathinone: An emerging recreational drug
For centuries the peoples of East Africa and the Arabian Peninsula have used the leaves of the indigenous khat plant (Catha edulis) for its amphetamine-like properties.3 Its active ingredient, cathinone, is a central nervous system stimulant that inhibits dopamine reuptake.4 In 2005, extracts from the plant were imported to Israel as “Hagigat” and promoted as a stimulant or aphrodisiac. These products were banned by the Israeli government in 2008 following documented cases of cardiovascular and neurologic sequelae.5
The growing problem of synthetic cathinone analogs in the United States. In 2008, synthetic analogs of cathinone were first identified in an analysis of drugs seized in the United States from individuals suffering psychological reactions to their use.1 Two such substances, 4-methylmethcathinone (mephedrone) and 3,4-methylenedioxypyrovalerone (MDPV), have since circulated worldwide, publicized by information on the Internet. Although the packets sold as “bath salts” clearly state that the contents are not for human consumption, Web sites promote the chemicals as “legal highs.”6
While these substances were being banned in many Western European countries, their use rapidly increased throughout the United States and elsewhere, often as an alternative to cocaine. Increases in the number of reports to poison control centers throughout the United States provide evidence of the increasing use of these drugs, despite legislation outlawing possession and sale in many states.7 In September 2011, the US Drug Enforcement Agency, using its emergency scheduling authority, made possession and sale of MDPV and mephedrone illegal throughout the United States.8
Who’s using bath salts? A review of calls to 2 poison control centers involving 236 patients over a 7-month period ending in February 2011 suggests that users of cathinones are primarily male (78%) and young (modal age 26).7 Many users of cathinones do not regularly use other drugs recreationally, and they believe the open sale of these substances implies low risk.7 However, one reported series from a hospital in Michigan indicated that 69% of users presenting to the ED had acknowledged past use of illicit drugs.9
What the drugs look like. Mephedrone and MDPV are supplied as white powders packaged in small packets of 500 mg and sell for about $25. Most users take the drug by nasal insufflation, although there is an alarming trend toward intravenous use.7 The intended effects in using these stimulants are improved attention and energy, as well as euphoria. Doses of about 25 mg produce these effects in most individuals and last for 2 to 3 hours, leading some users to compulsively re-dose to maintain the effects.
Use of bath salts leads to paranoid delusions, violent behavior
Both mephedrone and MDPV are strong inhibitors of dopamine reuptake in areas of the brain regulating reward and motivation.10,11 With prolonged exposure, the resultant stimulant effect of dopamine in reward centers of the brain moves a user from recreational pleasure seeking to addictive use just to maintain normal function.12 This transition occurs in a matter of days or weeks in some individuals, and we have seen multiple readmissions for paranoid psychotic reactions shortly after discharge from hospitalization.
Multiple serious complications of use have been described. The mainstream media have reported bizarre suicides and some homicides.13,14 Our clinic has reported on a unique hallucinatory delirium after use of MDPV, resulting in paranoid delusions and violent behavior in response to vivid hallucinations.15 Other patients suffer prolonged anxiety and panic reactions or depressive symptoms with suicidal ideation.16
Cardiovascular and other sequelae
About half of patients presenting to hospital EDs have cardiovascular complications such as tachycardia, chest pain, and hypertension from the sympathomimetic effects of these agents.9,16 There have also been reports of rhabdomyolysis and renal failure requiring intensive medical treatment.7,9,16
Taken together, mental status changes and physiologic reactions are similar to the “excited delirium” attributed to cocaine, methamphetamine, phencyclidine (PCP), and methylenedioxymethamphetamine (Ecstasy), all drugs that act on central monoamines.17–20 There have also been reports of death after the use of these drugs.21
Cathinones do not show up on routine drug screens
A routine urine screen for synthetic cathinones is not available, although a specific test arranged through commercial laboratories is available for cases when use is suspected. According to a written communication from A. Macher, MD, in 2011 (manuscript in preparation), urine drug screens for PCP using the immunoassay method may yield a false-positive result in the presence of MDPV. Simply asking patients whether they’ve been using these products often elicits an honest answer.
Treatment is largely supportive
Management guidance based on clinical trials is lacking. Cardiovascular complications require usual treatment.9,16 Serious psychiatric reactions may necessitate hospitalization to assure patient safety, particularly with evidence suggesting the potential to act on paranoid delusions or suicidal ideation. Benzodiazepines may be needed to control agitation, and low-dose antipsychotics, such as risperidone 1 mg, can aid in treating hallucinations.9,15
The hallucinatory psychosis seen with these substances is best characterized as a toxic delirium.15 Aggressive use of antipsychotic agents is not advised, given the risk of treatment-related morbidity in patients with a history of repeated stimulant use.22 Many patients presenting with acute delirium may require restraints. These procedures should be used with caution to minimize muscle tissue damage; patients should be monitored frequently for hyperthermia, dehydration, and rhabdomyolysis.16
Nothing is known about the long-term effects of these drugs, although substances with similar actions are associated with long-term cognitive and memory deficits after repeated use.4
CORRESPONDENCE Thomas M. Penders, MD, LFAPA, Department of Psychiatry, Brody School of Medicine, East Carolina University, 600 Moye Boulevard, Greenville, NC 27834; [email protected]
1. National Drug Intelligence Center. Synthetic Cathinones (Bath Salts): An Emerging Domestic Threat. Washington, DC: Department of Justice; July 2011. Publication no. 2011-S0787-004.
2. American Association of Poison Control Centers. Bath salts data, updated February 8, 2012. Available at: http://www.aapcc.org/dnn/Portals/0/Bath%20Salts%20Data%20for%20Website%202.8.2012.pdf. Accessed March 2, 2012.
3. Kalix P. Catha edulis, a plant that has amphetamine effects. Pharm World Sci. 1996;18:69-73.
4. Hadlock GC, Webb KM, McFadden LM, et al. 4-Methylmethcathinone (mephedrone): neuropharmacologic effects of a designer stimulant of abuse. J Pharmacol Exp Ther. 2011;339:530-536.
5. Bentur Y, Bloom-Krasik A, Raikhlin-Eisenkraft B. Illicit cathinone (“Hagigat”) poisoning. Clin Toxicol (Phila). 2008;46:206-210.
6. Winstock AR, Mitcheson LR, Deluca P, et al. Mephedrone, new kid for the chop? Addiction. 2010;106:154-161.
7. Spiller HA, Ryan ML, Weston RG, et al. Clinical experience with and analytic confirmation of “bath salts” and “legal highs” (synthetic cathinones) in the United States. Clin Toxicol (Phila). 2011;49:499-505.
8. DeNoon DJ. “Bath salts” used to get high are now illegal. WebMD. Available at: http://www.webmd.com/mental-health/news/20110908/bath-salts-used-to-get-high-are-now-illegal. Accessed October 10, 2011.
9. Centers for Disease Control and Prevention. Emergency department visits after use of a drug sold as “bath salts”—Michigan, November 13, 2010 – March 31, 2011. MMWR Morb Mortal Wkly Rep. 2011;60:624-627.
10. Kelly JP. Cathinone derivatives: a review of the chemistry, pharmacology and toxicology. Drug Test Anal. 2011;3:439-453.
11. Advisory Council on the Misuse of Drugs (ACMD). ACMD report on the consideration of the cathinones. London, UK: Home Office; March 31, 2010. Available at: http://www.homeoffice.gov.uk/publications/drugs/acmd1/acmd-cathinodes-report-2010. Accessed October 10, 2011.
12. Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217-238.
13. Lewis B. Man commits suicide after using bath salts. WNDU.com: 2011. Available at: http://www.wndu.com/hometop/headlines/Man_commits_suicide_after_doing_bath_salts_123520219.html. Accessed October 9, 2011.
14. Goodnough A, Zezima K. An alarming new stimulant, legal in many states. The New York Times; July 16, 2011. Available at: http://www.nytimes.com/2011/07/17/us/17salts.html?pagewanted=all. Accessed October 12, 2011.
15. Penders T, Gestring R. Hallucinatory delirium following use of MDPV (“bath salts”). Gen Hosp Psychiatry. 2011;33:525-526.
16. Ross EA, Watson M, Goldberger B. “Bath salts” intoxication. N Engl J Med. 2011;365:967-968.
17. Ruttenber AJ, McAnally HB, Wetli CV. Cocaine-associated rhabdomyolysis and excited delirium: different stages of the same syndrome. Am J Forensic Med Pathol. 1999;20:120-127.
18. Richards JR, Johnson EB, Stark RW, et al. Methamphetamine abuse and rhabdomyolysis in the ED: a 5-year study. Am J Emerg Med. 1999;17:681-685.
19. Henry JA, Jeffreys KJ, Dawling S. Toxicity and deaths from 3,4 methylenedioxymethamphetamine (“ecstasy”). Lancet. 1992;340:384-387.
20. Lahmeyer HW, Stock PG. Phencyclidine intoxication, physical restraint, and acute renal failure: case report. J Clin Psychiatry. 1983;44:184-185.
21. Murray BL, Murphy CM, Beuhler MC. Death following recreational use of designer drug “Bath Salts” containing 3,4, methylenedioxypyrovalerone (MDPV). J Med Toxicol. 2012;8:69-75.
22. Akpaffiong MJ, Ruiz P. Neuroleptic malignant syndrome: a complication of neuroleptics and cocaine abuse. Psychiatr Q. 1991;62:299-309.
• Include cathinone use in the differential diagnosis for any patient exhibiting paranoid psychotic behavior or hallucinatory delirium. C
• Keep in mind that cathinone effects can mimic the “excited delirium” attributed to cocaine, methamphetamine, PCP, and Ecstasy. C
• Consider using benzodiazepines to control agitation, or low-dose antipsychotics to treat hallucinations. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 31-year-old construction worker with a history of intermittent cocaine use was brought to the emergency department (ED) by the police. He was handcuffed and appeared confused and frightened. The patient’s wife had phoned the police after he began running through a field in pursuit of perceived invaders of their home. The wife reported that a few hours earlier, the patient had begun to hallucinate and had become very fearful after getting high.
His heart rate was 126, blood pressure 136/96 mm Hg, and temperature 99.6°F. During the initial exam, the patient became agitated, attempted to assault a nurse, and tried to leave the ED before being subdued. A urine screen for drugs was negative for cocaine. His creatine phosphokinase was 850 U/L, creatinine 2.32 mg/dL, and blood urea nitrogen 27 mg/dL.
The health care team learned that the drug he’d been snorting earlier that day—and the day before—was “bath salts.”
This patient was one of the 30 that we’ve seen at our university hospital over the past year.
Since early 2010, EDs, psychiatric facilities, and poison control centers have seen a surge in the number of patients abusing novel synthetic stimulants—cathinones—that had once been sold in convenience stores and tobacco shops and often labeled innocuously as “bath salts” or “plant food.” Sales have largely gone underground, sold by those trafficking in methamphetamine and cocaine. These products are also available for online purchase and may be sold under such provocative names as “Cloud Nine” or “Rave.”1 In 2010, poison control centers received 304 calls related to the use of these substances; in 2011, the number was 6138.2
Cathinone: An emerging recreational drug
For centuries the peoples of East Africa and the Arabian Peninsula have used the leaves of the indigenous khat plant (Catha edulis) for its amphetamine-like properties.3 Its active ingredient, cathinone, is a central nervous system stimulant that inhibits dopamine reuptake.4 In 2005, extracts from the plant were imported to Israel as “Hagigat” and promoted as a stimulant or aphrodisiac. These products were banned by the Israeli government in 2008 following documented cases of cardiovascular and neurologic sequelae.5
The growing problem of synthetic cathinone analogs in the United States. In 2008, synthetic analogs of cathinone were first identified in an analysis of drugs seized in the United States from individuals suffering psychological reactions to their use.1 Two such substances, 4-methylmethcathinone (mephedrone) and 3,4-methylenedioxypyrovalerone (MDPV), have since circulated worldwide, publicized by information on the Internet. Although the packets sold as “bath salts” clearly state that the contents are not for human consumption, Web sites promote the chemicals as “legal highs.”6
While these substances were being banned in many Western European countries, their use rapidly increased throughout the United States and elsewhere, often as an alternative to cocaine. Increases in the number of reports to poison control centers throughout the United States provide evidence of the increasing use of these drugs, despite legislation outlawing possession and sale in many states.7 In September 2011, the US Drug Enforcement Agency, using its emergency scheduling authority, made possession and sale of MDPV and mephedrone illegal throughout the United States.8
Who’s using bath salts? A review of calls to 2 poison control centers involving 236 patients over a 7-month period ending in February 2011 suggests that users of cathinones are primarily male (78%) and young (modal age 26).7 Many users of cathinones do not regularly use other drugs recreationally, and they believe the open sale of these substances implies low risk.7 However, one reported series from a hospital in Michigan indicated that 69% of users presenting to the ED had acknowledged past use of illicit drugs.9
What the drugs look like. Mephedrone and MDPV are supplied as white powders packaged in small packets of 500 mg and sell for about $25. Most users take the drug by nasal insufflation, although there is an alarming trend toward intravenous use.7 The intended effects in using these stimulants are improved attention and energy, as well as euphoria. Doses of about 25 mg produce these effects in most individuals and last for 2 to 3 hours, leading some users to compulsively re-dose to maintain the effects.
Use of bath salts leads to paranoid delusions, violent behavior
Both mephedrone and MDPV are strong inhibitors of dopamine reuptake in areas of the brain regulating reward and motivation.10,11 With prolonged exposure, the resultant stimulant effect of dopamine in reward centers of the brain moves a user from recreational pleasure seeking to addictive use just to maintain normal function.12 This transition occurs in a matter of days or weeks in some individuals, and we have seen multiple readmissions for paranoid psychotic reactions shortly after discharge from hospitalization.
Multiple serious complications of use have been described. The mainstream media have reported bizarre suicides and some homicides.13,14 Our clinic has reported on a unique hallucinatory delirium after use of MDPV, resulting in paranoid delusions and violent behavior in response to vivid hallucinations.15 Other patients suffer prolonged anxiety and panic reactions or depressive symptoms with suicidal ideation.16
Cardiovascular and other sequelae
About half of patients presenting to hospital EDs have cardiovascular complications such as tachycardia, chest pain, and hypertension from the sympathomimetic effects of these agents.9,16 There have also been reports of rhabdomyolysis and renal failure requiring intensive medical treatment.7,9,16
Taken together, mental status changes and physiologic reactions are similar to the “excited delirium” attributed to cocaine, methamphetamine, phencyclidine (PCP), and methylenedioxymethamphetamine (Ecstasy), all drugs that act on central monoamines.17–20 There have also been reports of death after the use of these drugs.21
Cathinones do not show up on routine drug screens
A routine urine screen for synthetic cathinones is not available, although a specific test arranged through commercial laboratories is available for cases when use is suspected. According to a written communication from A. Macher, MD, in 2011 (manuscript in preparation), urine drug screens for PCP using the immunoassay method may yield a false-positive result in the presence of MDPV. Simply asking patients whether they’ve been using these products often elicits an honest answer.
Treatment is largely supportive
Management guidance based on clinical trials is lacking. Cardiovascular complications require usual treatment.9,16 Serious psychiatric reactions may necessitate hospitalization to assure patient safety, particularly with evidence suggesting the potential to act on paranoid delusions or suicidal ideation. Benzodiazepines may be needed to control agitation, and low-dose antipsychotics, such as risperidone 1 mg, can aid in treating hallucinations.9,15
The hallucinatory psychosis seen with these substances is best characterized as a toxic delirium.15 Aggressive use of antipsychotic agents is not advised, given the risk of treatment-related morbidity in patients with a history of repeated stimulant use.22 Many patients presenting with acute delirium may require restraints. These procedures should be used with caution to minimize muscle tissue damage; patients should be monitored frequently for hyperthermia, dehydration, and rhabdomyolysis.16
Nothing is known about the long-term effects of these drugs, although substances with similar actions are associated with long-term cognitive and memory deficits after repeated use.4
CORRESPONDENCE Thomas M. Penders, MD, LFAPA, Department of Psychiatry, Brody School of Medicine, East Carolina University, 600 Moye Boulevard, Greenville, NC 27834; [email protected]
• Include cathinone use in the differential diagnosis for any patient exhibiting paranoid psychotic behavior or hallucinatory delirium. C
• Keep in mind that cathinone effects can mimic the “excited delirium” attributed to cocaine, methamphetamine, PCP, and Ecstasy. C
• Consider using benzodiazepines to control agitation, or low-dose antipsychotics to treat hallucinations. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 31-year-old construction worker with a history of intermittent cocaine use was brought to the emergency department (ED) by the police. He was handcuffed and appeared confused and frightened. The patient’s wife had phoned the police after he began running through a field in pursuit of perceived invaders of their home. The wife reported that a few hours earlier, the patient had begun to hallucinate and had become very fearful after getting high.
His heart rate was 126, blood pressure 136/96 mm Hg, and temperature 99.6°F. During the initial exam, the patient became agitated, attempted to assault a nurse, and tried to leave the ED before being subdued. A urine screen for drugs was negative for cocaine. His creatine phosphokinase was 850 U/L, creatinine 2.32 mg/dL, and blood urea nitrogen 27 mg/dL.
The health care team learned that the drug he’d been snorting earlier that day—and the day before—was “bath salts.”
This patient was one of the 30 that we’ve seen at our university hospital over the past year.
Since early 2010, EDs, psychiatric facilities, and poison control centers have seen a surge in the number of patients abusing novel synthetic stimulants—cathinones—that had once been sold in convenience stores and tobacco shops and often labeled innocuously as “bath salts” or “plant food.” Sales have largely gone underground, sold by those trafficking in methamphetamine and cocaine. These products are also available for online purchase and may be sold under such provocative names as “Cloud Nine” or “Rave.”1 In 2010, poison control centers received 304 calls related to the use of these substances; in 2011, the number was 6138.2
Cathinone: An emerging recreational drug
For centuries the peoples of East Africa and the Arabian Peninsula have used the leaves of the indigenous khat plant (Catha edulis) for its amphetamine-like properties.3 Its active ingredient, cathinone, is a central nervous system stimulant that inhibits dopamine reuptake.4 In 2005, extracts from the plant were imported to Israel as “Hagigat” and promoted as a stimulant or aphrodisiac. These products were banned by the Israeli government in 2008 following documented cases of cardiovascular and neurologic sequelae.5
The growing problem of synthetic cathinone analogs in the United States. In 2008, synthetic analogs of cathinone were first identified in an analysis of drugs seized in the United States from individuals suffering psychological reactions to their use.1 Two such substances, 4-methylmethcathinone (mephedrone) and 3,4-methylenedioxypyrovalerone (MDPV), have since circulated worldwide, publicized by information on the Internet. Although the packets sold as “bath salts” clearly state that the contents are not for human consumption, Web sites promote the chemicals as “legal highs.”6
While these substances were being banned in many Western European countries, their use rapidly increased throughout the United States and elsewhere, often as an alternative to cocaine. Increases in the number of reports to poison control centers throughout the United States provide evidence of the increasing use of these drugs, despite legislation outlawing possession and sale in many states.7 In September 2011, the US Drug Enforcement Agency, using its emergency scheduling authority, made possession and sale of MDPV and mephedrone illegal throughout the United States.8
Who’s using bath salts? A review of calls to 2 poison control centers involving 236 patients over a 7-month period ending in February 2011 suggests that users of cathinones are primarily male (78%) and young (modal age 26).7 Many users of cathinones do not regularly use other drugs recreationally, and they believe the open sale of these substances implies low risk.7 However, one reported series from a hospital in Michigan indicated that 69% of users presenting to the ED had acknowledged past use of illicit drugs.9
What the drugs look like. Mephedrone and MDPV are supplied as white powders packaged in small packets of 500 mg and sell for about $25. Most users take the drug by nasal insufflation, although there is an alarming trend toward intravenous use.7 The intended effects in using these stimulants are improved attention and energy, as well as euphoria. Doses of about 25 mg produce these effects in most individuals and last for 2 to 3 hours, leading some users to compulsively re-dose to maintain the effects.
Use of bath salts leads to paranoid delusions, violent behavior
Both mephedrone and MDPV are strong inhibitors of dopamine reuptake in areas of the brain regulating reward and motivation.10,11 With prolonged exposure, the resultant stimulant effect of dopamine in reward centers of the brain moves a user from recreational pleasure seeking to addictive use just to maintain normal function.12 This transition occurs in a matter of days or weeks in some individuals, and we have seen multiple readmissions for paranoid psychotic reactions shortly after discharge from hospitalization.
Multiple serious complications of use have been described. The mainstream media have reported bizarre suicides and some homicides.13,14 Our clinic has reported on a unique hallucinatory delirium after use of MDPV, resulting in paranoid delusions and violent behavior in response to vivid hallucinations.15 Other patients suffer prolonged anxiety and panic reactions or depressive symptoms with suicidal ideation.16
Cardiovascular and other sequelae
About half of patients presenting to hospital EDs have cardiovascular complications such as tachycardia, chest pain, and hypertension from the sympathomimetic effects of these agents.9,16 There have also been reports of rhabdomyolysis and renal failure requiring intensive medical treatment.7,9,16
Taken together, mental status changes and physiologic reactions are similar to the “excited delirium” attributed to cocaine, methamphetamine, phencyclidine (PCP), and methylenedioxymethamphetamine (Ecstasy), all drugs that act on central monoamines.17–20 There have also been reports of death after the use of these drugs.21
Cathinones do not show up on routine drug screens
A routine urine screen for synthetic cathinones is not available, although a specific test arranged through commercial laboratories is available for cases when use is suspected. According to a written communication from A. Macher, MD, in 2011 (manuscript in preparation), urine drug screens for PCP using the immunoassay method may yield a false-positive result in the presence of MDPV. Simply asking patients whether they’ve been using these products often elicits an honest answer.
Treatment is largely supportive
Management guidance based on clinical trials is lacking. Cardiovascular complications require usual treatment.9,16 Serious psychiatric reactions may necessitate hospitalization to assure patient safety, particularly with evidence suggesting the potential to act on paranoid delusions or suicidal ideation. Benzodiazepines may be needed to control agitation, and low-dose antipsychotics, such as risperidone 1 mg, can aid in treating hallucinations.9,15
The hallucinatory psychosis seen with these substances is best characterized as a toxic delirium.15 Aggressive use of antipsychotic agents is not advised, given the risk of treatment-related morbidity in patients with a history of repeated stimulant use.22 Many patients presenting with acute delirium may require restraints. These procedures should be used with caution to minimize muscle tissue damage; patients should be monitored frequently for hyperthermia, dehydration, and rhabdomyolysis.16
Nothing is known about the long-term effects of these drugs, although substances with similar actions are associated with long-term cognitive and memory deficits after repeated use.4
CORRESPONDENCE Thomas M. Penders, MD, LFAPA, Department of Psychiatry, Brody School of Medicine, East Carolina University, 600 Moye Boulevard, Greenville, NC 27834; [email protected]
1. National Drug Intelligence Center. Synthetic Cathinones (Bath Salts): An Emerging Domestic Threat. Washington, DC: Department of Justice; July 2011. Publication no. 2011-S0787-004.
2. American Association of Poison Control Centers. Bath salts data, updated February 8, 2012. Available at: http://www.aapcc.org/dnn/Portals/0/Bath%20Salts%20Data%20for%20Website%202.8.2012.pdf. Accessed March 2, 2012.
3. Kalix P. Catha edulis, a plant that has amphetamine effects. Pharm World Sci. 1996;18:69-73.
4. Hadlock GC, Webb KM, McFadden LM, et al. 4-Methylmethcathinone (mephedrone): neuropharmacologic effects of a designer stimulant of abuse. J Pharmacol Exp Ther. 2011;339:530-536.
5. Bentur Y, Bloom-Krasik A, Raikhlin-Eisenkraft B. Illicit cathinone (“Hagigat”) poisoning. Clin Toxicol (Phila). 2008;46:206-210.
6. Winstock AR, Mitcheson LR, Deluca P, et al. Mephedrone, new kid for the chop? Addiction. 2010;106:154-161.
7. Spiller HA, Ryan ML, Weston RG, et al. Clinical experience with and analytic confirmation of “bath salts” and “legal highs” (synthetic cathinones) in the United States. Clin Toxicol (Phila). 2011;49:499-505.
8. DeNoon DJ. “Bath salts” used to get high are now illegal. WebMD. Available at: http://www.webmd.com/mental-health/news/20110908/bath-salts-used-to-get-high-are-now-illegal. Accessed October 10, 2011.
9. Centers for Disease Control and Prevention. Emergency department visits after use of a drug sold as “bath salts”—Michigan, November 13, 2010 – March 31, 2011. MMWR Morb Mortal Wkly Rep. 2011;60:624-627.
10. Kelly JP. Cathinone derivatives: a review of the chemistry, pharmacology and toxicology. Drug Test Anal. 2011;3:439-453.
11. Advisory Council on the Misuse of Drugs (ACMD). ACMD report on the consideration of the cathinones. London, UK: Home Office; March 31, 2010. Available at: http://www.homeoffice.gov.uk/publications/drugs/acmd1/acmd-cathinodes-report-2010. Accessed October 10, 2011.
12. Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217-238.
13. Lewis B. Man commits suicide after using bath salts. WNDU.com: 2011. Available at: http://www.wndu.com/hometop/headlines/Man_commits_suicide_after_doing_bath_salts_123520219.html. Accessed October 9, 2011.
14. Goodnough A, Zezima K. An alarming new stimulant, legal in many states. The New York Times; July 16, 2011. Available at: http://www.nytimes.com/2011/07/17/us/17salts.html?pagewanted=all. Accessed October 12, 2011.
15. Penders T, Gestring R. Hallucinatory delirium following use of MDPV (“bath salts”). Gen Hosp Psychiatry. 2011;33:525-526.
16. Ross EA, Watson M, Goldberger B. “Bath salts” intoxication. N Engl J Med. 2011;365:967-968.
17. Ruttenber AJ, McAnally HB, Wetli CV. Cocaine-associated rhabdomyolysis and excited delirium: different stages of the same syndrome. Am J Forensic Med Pathol. 1999;20:120-127.
18. Richards JR, Johnson EB, Stark RW, et al. Methamphetamine abuse and rhabdomyolysis in the ED: a 5-year study. Am J Emerg Med. 1999;17:681-685.
19. Henry JA, Jeffreys KJ, Dawling S. Toxicity and deaths from 3,4 methylenedioxymethamphetamine (“ecstasy”). Lancet. 1992;340:384-387.
20. Lahmeyer HW, Stock PG. Phencyclidine intoxication, physical restraint, and acute renal failure: case report. J Clin Psychiatry. 1983;44:184-185.
21. Murray BL, Murphy CM, Beuhler MC. Death following recreational use of designer drug “Bath Salts” containing 3,4, methylenedioxypyrovalerone (MDPV). J Med Toxicol. 2012;8:69-75.
22. Akpaffiong MJ, Ruiz P. Neuroleptic malignant syndrome: a complication of neuroleptics and cocaine abuse. Psychiatr Q. 1991;62:299-309.
1. National Drug Intelligence Center. Synthetic Cathinones (Bath Salts): An Emerging Domestic Threat. Washington, DC: Department of Justice; July 2011. Publication no. 2011-S0787-004.
2. American Association of Poison Control Centers. Bath salts data, updated February 8, 2012. Available at: http://www.aapcc.org/dnn/Portals/0/Bath%20Salts%20Data%20for%20Website%202.8.2012.pdf. Accessed March 2, 2012.
3. Kalix P. Catha edulis, a plant that has amphetamine effects. Pharm World Sci. 1996;18:69-73.
4. Hadlock GC, Webb KM, McFadden LM, et al. 4-Methylmethcathinone (mephedrone): neuropharmacologic effects of a designer stimulant of abuse. J Pharmacol Exp Ther. 2011;339:530-536.
5. Bentur Y, Bloom-Krasik A, Raikhlin-Eisenkraft B. Illicit cathinone (“Hagigat”) poisoning. Clin Toxicol (Phila). 2008;46:206-210.
6. Winstock AR, Mitcheson LR, Deluca P, et al. Mephedrone, new kid for the chop? Addiction. 2010;106:154-161.
7. Spiller HA, Ryan ML, Weston RG, et al. Clinical experience with and analytic confirmation of “bath salts” and “legal highs” (synthetic cathinones) in the United States. Clin Toxicol (Phila). 2011;49:499-505.
8. DeNoon DJ. “Bath salts” used to get high are now illegal. WebMD. Available at: http://www.webmd.com/mental-health/news/20110908/bath-salts-used-to-get-high-are-now-illegal. Accessed October 10, 2011.
9. Centers for Disease Control and Prevention. Emergency department visits after use of a drug sold as “bath salts”—Michigan, November 13, 2010 – March 31, 2011. MMWR Morb Mortal Wkly Rep. 2011;60:624-627.
10. Kelly JP. Cathinone derivatives: a review of the chemistry, pharmacology and toxicology. Drug Test Anal. 2011;3:439-453.
11. Advisory Council on the Misuse of Drugs (ACMD). ACMD report on the consideration of the cathinones. London, UK: Home Office; March 31, 2010. Available at: http://www.homeoffice.gov.uk/publications/drugs/acmd1/acmd-cathinodes-report-2010. Accessed October 10, 2011.
12. Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217-238.
13. Lewis B. Man commits suicide after using bath salts. WNDU.com: 2011. Available at: http://www.wndu.com/hometop/headlines/Man_commits_suicide_after_doing_bath_salts_123520219.html. Accessed October 9, 2011.
14. Goodnough A, Zezima K. An alarming new stimulant, legal in many states. The New York Times; July 16, 2011. Available at: http://www.nytimes.com/2011/07/17/us/17salts.html?pagewanted=all. Accessed October 12, 2011.
15. Penders T, Gestring R. Hallucinatory delirium following use of MDPV (“bath salts”). Gen Hosp Psychiatry. 2011;33:525-526.
16. Ross EA, Watson M, Goldberger B. “Bath salts” intoxication. N Engl J Med. 2011;365:967-968.
17. Ruttenber AJ, McAnally HB, Wetli CV. Cocaine-associated rhabdomyolysis and excited delirium: different stages of the same syndrome. Am J Forensic Med Pathol. 1999;20:120-127.
18. Richards JR, Johnson EB, Stark RW, et al. Methamphetamine abuse and rhabdomyolysis in the ED: a 5-year study. Am J Emerg Med. 1999;17:681-685.
19. Henry JA, Jeffreys KJ, Dawling S. Toxicity and deaths from 3,4 methylenedioxymethamphetamine (“ecstasy”). Lancet. 1992;340:384-387.
20. Lahmeyer HW, Stock PG. Phencyclidine intoxication, physical restraint, and acute renal failure: case report. J Clin Psychiatry. 1983;44:184-185.
21. Murray BL, Murphy CM, Beuhler MC. Death following recreational use of designer drug “Bath Salts” containing 3,4, methylenedioxypyrovalerone (MDPV). J Med Toxicol. 2012;8:69-75.
22. Akpaffiong MJ, Ruiz P. Neuroleptic malignant syndrome: a complication of neuroleptics and cocaine abuse. Psychiatr Q. 1991;62:299-309.
Not just a sprain: 4 foot and ankle injuries you may be missing
• Treat a nondisplaced shaft fracture of the fifth metatarsal conservatively, with 6 to 8 weeks of immobilization with a protective orthosis. B
• Suspect a navicular fracture in patients who describe a gradual onset of vague, dorsal midfoot pain associated with athletic activity. C
• Order magnetic resonance imaging when you suspect osteochondritis dissecans, as radiographs are insensitive for identifying these lesions. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Ankle sprain, one of the more common injuries that primary care physicians evaluate, is usually managed with conservative treatment. Not uncommonly, however, lateral ankle sprain is diagnosed without consideration of a broader differential diagnosis.
Contributing to the problem is the fact that the clinical presentation of some fractures and tendon injuries is similar to that of a routine sprain. In some cases, the mechanism of injury—sprains are usually caused by excessive inversion of the ankle on a plantar-flexed foot—is similar, as well. What’s more, radiographs are often omitted or misinterpreted.
In the pages that follow, we highlight 4 commonly misdiagnosed injuries: fifth metatarsal fractures, navicular fractures, talar dome lesions, and peroneal tendon injuries. These injuries should be included in the differential diagnosis of an acute ankle injury—or a subacute foot or ankle injury that fails to respond as expected. Prompt recognition and appropriate treatment result in optimal outcomes. When foot and ankle fractures and tendon injuries are misdiagnosed (or simply missed) and do not receive adequate treatment, long-term morbidity, including frequent reinjury and disability, may result.1
Are x-rays needed? Turn to the Ottawa rules
Ankle sprains represent a disruption in a ligament supporting a joint, and result in pain, edema, and ecchymosis, and often affect a patient’s ability to bear weight. While uncomplicated sprains generally heal with conservative treatment, other common foot and ankle injuries may require a different approach.
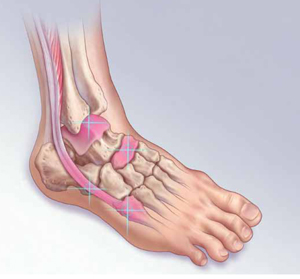
The Ottawa foot and ankle rules are an evidence-based guide to the use of initial radiographs after acute ankle injury (TABLE 1).2-4 Pain—near the malleoli (for the ankle) or in the midfoot—is the key criterion, but x-rays are recommended only if at least one other specified criterion is also met. With a sensitivity of nearly 100%, the rules have been shown to reliably exclude, and diagnose, ankle and midfoot fractures in children >5 years and adults.2,5
Table 1
Ottawa ankle and foot rules2-4
| Ankle |
X-rays are required only if the patient has pain near the malleolus and one or more of the following:
|
| Foot |
X-rays are required only if the patient has pain in the midfoot and one or more of the following:
|
Fifth metatarsal fractures are easily missed
The mechanism of injury for a fifth metatarsal fracture is often similar to that of a lateral ankle sprain. In addition, isolated ankle radiographs may not adequately evaluate the fifth metatarsal, which increases the risk of misdiagnosis.6
3 types of fifth metatarsal fractures
Fifth metatarsal fractures involve one of the following:
- an avulsion fracture, caused by the pull of the plantar aponeurosis and the peroneus brevis tendon at the tuberosity of the bone
- a Jones fracture, at the base of the fourth and fifth metatarsal (FIGURE 1)
- a shaft fracture, distal to the fifth metatarsal joint in the proximal diaphysis.6-8
FIGURE 1
Jones fractures heal slowly
This 50-year-old patient presented with pain and swelling in the ankle and lateral foot shortly after an inversion ankle injury. A radiograph (A) taken at that time reveals a Jones fracture. The second radiograph (B) was taken 6 weeks later, after continued immobilization with no weight-bearing. Three months after the injury (C), the patient was clinically asymptomatic.
While avulsion fractures are generally the result of an inversion ankle injury, Jones fractures are usually caused by a large adductive force applied to the forefoot on a plantar-flexed ankle.6 Shaft fractures, also known as diaphyseal stress fractures, are overuse injuries from chronic overload, usually after a sudden increase in running or walking.9
Patients with fifth metatarsal fractures typically have tenderness with palpation over the area of injury, with edema and ecchymosis when the injury is acute. Evidence-based guidelines recommend x-rays of the foot, including anteroposterior (AP), lateral, and oblique views.2-4 One study supports the use of an additional x-ray—an AP view of the ankle, including the base of the fifth metatarsal—if clinical suspicion is high and initial radiographs are negative or inconclusive.10
Shaft fractures may not be seen on x-rays in the first 3 weeks, but a periosteal reaction or linear lucency near the symptomatic area may be noticeable on radiographs taken at a later date.11 If this overuse injury seems likely but does not show up on the initial x-rays, however, magnetic resonance imaging (MRI) or a technetium bone scan can reliably identify a stress fracture.9
How to treat, when to refer
Treatment of fifth metatarsal fractures range from conservative to surgical, depending on the type (and extent) of injury (TABLE 2).1,5,6,12-14
TABLE 2
Nondisplaced avulsion fractures can be treated conservatively, with relative immobilization. In one prospective study, the use of a stiff-soled shoe, with weight-bearing as tolerated, was associated with excellent long-term outcomes.11 Orthopedic referral for probable reduction and fixation is indicated for avulsion fractures that are comminuted or >2 mm displaced, or have >30% involvement of the cubometatarsal joint.15,16
Jones fractures are known for prolonged healing and nonunion, as well as a high rate of complications. If the fracture is nondisplaced, start with conservative treatment, consisting of nonweight-bearing immobilization for 6 to 8 weeks, with additional immobilization dependent on radiographs. One randomized controlled trial of patients with Jones fractures showed a relatively high failure rate (44%) with casting; patients for whom casting was successful still had a median time to bony union of 15 weeks.17 Specialty consultation may be needed when there is fracture displacement, absence of bony union, or high clinical concern.6,17
Is your patient an athlete? Surgical fixation is favored for injured athletes with Jones fractures because failure rates are lower and both clinical union and return to play are shorter.18,19 In a case series involving 23 athletic patients with Jones fractures, the success rate for immediate surgical screw fixation approached 100% within 6 to 8 weeks.18
Nondisplaced shaft fractures may be treated conservatively, with 6 to 8 weeks of immobilization with a protective orthosis. An orthopedic referral is recommended for patients whose fractures have >3 mm displacement or >10 degree angulation.15
Navicular fractures are overuse injuries
The navicular is predisposed to stress injury because the central third of the bone is relatively avascular. In addition, the navicular is the area of greatest stress and impingement between the talus and cuneiform bones during repetitive foot strikes.12,20 Navicular fractures occur predominantly in track and field athletes.12
Patients presenting with a navicular stress fracture often report a gradual onset of vague dorsal midfoot pain associated with their workout.17 Examination typically reveals tenderness on palpation over the dorsal aspect of the navicular; passive eversion and active inversion may be painful, but edema and ecchymosis are usually absent.21
When pain is elicited by palpation of the navicular, radiographs are recommended.2,6 X-rays have a relatively low sensitivity (33%), however, for detecting acute navicular stress fractures. If initial radiographs are negative but there is a high clinical suspicion, advanced studies—with either MRI or a technetium bone scan—are recommended for a definitive diagnosis.12,22 While both are highly sensitive for navicular stress fractures, MRI provides greater specificity and anatomic detail.23
Most navicular fractures are nondisplaced
Nondisplaced navicular fractures can be treated conservatively, with nonweight-bearing immobilization for 6 to 8 weeks followed by progressive activity.24 Prospective studies have found that conservative treatment has a high success rate, with athletes usually able to return to play within 6 months.22,24,25 If tenderness remains after 6 to 8 weeks of immobilization, treatment choices are continued immobilization with no weight-bearing or orthopedic referral.26
Referral is indicated for navicular fractures that are comminuted or displaced, or involve more than one bone cortex.26 Surgical screw fixation may be recommended for navicular stress fractures in selected athletes because of its high success rate—and likelihood of an earlier return to play.27
Talar injuries are characterized by persistent pain
Injuries to the talus commonly occur at the same time as ankle sprains and may cause persistent pain, even after the sprain has healed.28 Evidence suggests that up to 90% of residual pain is related to an underlying cartilage injury.29,30 Most talar injuries are associated with the disruption of the cartilage overlaying the talar dome, which may lead to osteochondritis dissecans.29 Subtle talus fractures are also a concern after an acute ankle injury.
Osteochondral lesions are associated with a dull ankle pain deep in a location with a prior ankle injury; in some cases, the pain will become chronic.31 Physical exam findings typically include ankle joint effusion with localized tenderness around the joint.31
Ankle radiographs are insensitive for identifying osteochondral lesions, and MRI is recommended for evaluating suspected lesions.29,31 Treatment varies, depending on symptoms and severity. Patients with minimal symptoms may be treated conservatively; however, high failure rates have been reported.32 Surgical treatment depends on the size and site of the lesion and the degree of cartilage injury, and surgical consultation is recommended.31
Fractures of the talar dome (FIGURE 2) may be either medial or lateral and are often the result of inversion ankle injuries.14 History and clinical findings vary depending on the type of fracture.
FIGURE 2
Talar dome injuries often result from inversion ankle injuries
As with osteochondral lesions, ankle radiographs may fail to identify talus fractures. Computed tomography (CT) should be used to evaluate acute fractures of the talus, as CT scan is better able to define displacement, size, and intra-articular involvement.33 Talar fractures may be managed conservatively with immobilization and nonweight-bearing for 4 to 6 weeks, but specialty consultation should be considered.14,33
A tarsal coalition—an incomplete, congenital separation of the bones, occasionally involving the talus and the calcaneus—can also be a cause of persistent pain after a sprain.28 Physical exam typically demonstrates decreased range of motion in the subtalar or transverse tarsal joint. Radiographs may identify the coalition, but MRI or CT scan provides optimal visualization. Immobilization for 6 weeks is the recommended initial treatment, but if that fails, surgical excision or fusion may be necessary.
Peroneal tendon injuries may cause ankle instability
Peroneal tendon injuries, which include strains, subluxation, dislocation, and tears of one or both of the peroneal tendons, are often caused by ankle inversion similar to that of an uncomplicated sprain. Subsequent ankle instability may result from untreated peroneal tendon injuries.34 Peroneal tendon subluxation accounts for a very small number (0.3%-0.5%) of traumatic ankle injuries.35
Peroneal tendon injuries often occur during sports that involve frequent lateral movement or cutting—eg, football, basketball, and soccer—and are often caused by sudden dorsiflexion of the inverted foot, with coincident contraction of the peroneal muscles.36,37 This mechanism can disrupt the superior peroneal retinaculum, leading to recurrent subluxation or dislocation and subsequent ankle instability.36,38 Chronic subluxation can also result in longitudinal tears of the peroneal tendons, especially of the peroneus brevis.36,38,39
Patients with peroneal tendon injuries may report a “pop” at the time of injury. Pain is typically located posterior to the lateral malleolus, and recurrent subluxation is often described as a “snapping” around the lateral ankle during athletic activities.37,38 Instability is common in patients with subacute or chronic peroneal tendon injuries, especially on uneven surfaces.38
Acute peroneal tendon injuries cause posterolateral ankle pain, swelling, and weakness; exam findings include tenderness along the course of the peroneal tendons with associated edema.37 Subluxation or dislocation of the peroneal brevis tendon may be confirmed by placing the foot in plantar flexion and inversion and asking the patient to forcibly dorsiflex and evert the injured ankle.
Plain radiographs are usually normal in an isolated injury to the peroneal tendons. A fracture of the posterolateral margin of the fibula is a rare finding but indicates disruption of the peroneal retinaculum.36 MRI provides the best imaging for peroneal tendons and the stabilizing retinaculum, although a CT scan can provide detailed bony anatomy when subtle fractures are suspected or additional evaluation is needed.
Subluxation or dislocation indicate a need for surgery
Conservative management is recommended for peroneal tendon strains, but surgical treatment is increasingly recommended for subluxation or dislocation, especially if the problem is recurrent.36,37 Conservative treatment consists of short-term immobilization with a walking boot or brace, followed by physical therapy to improve strength and motion. Surgical treatment of subluxation and dislocation by stabilizing the peroneal tendons within the peroneal groove has been shown to provide lasting stability and improvement.37,38,40,41
CORRESPONDENCE Scott Hall, MD, University of Nevada-Reno, Brigham Building 316, Reno, NV 89557; [email protected]
1. van Rijn RM, van Os AG, Bernsen RM, et al. What is the clinical course of acute ankle sprains? A systematic literature review. Am J Med. 2008;121:324-331.
2. Nugent PJ. Ottawa ankle rules accurately assess injuries and reduce reliance on radiographs. J Fam Pract. 2004;53:785-788.
3. Stiell IG, McKnight RD, Greenberg GH, et al. Implementation of the Ottawa ankle rules. JAMA. 1994;271:827-832.
4. Stiell IG, Greenberg GH, McKnight RD, et al. Decision rules for the use of radiography in acute ankle injuries. JAMA. 1993;269:1127-1132.
5. Judd DB, Kim DH. Foot fractures frequently misdiagnosed as ankle sprains. Am Fam Physician. 2002;66:785-795.
6. Den Hartog BD. Fracture of the proximal fifth metatarsal. J Am Acad Orthop Surg. 2009;17:458-464.
7. Torg JS, Balduini FC, Zelko RR, et al. Fractures of the base of the fifth metatarsal distal to the tuberosity. J Bone Joint Surg Am. 1984;66:209-214.
8. Dameron TB. Fractures and anatomical variations of the proximal portion of the fifth metatarsal. J Bone Joint Surg Am. 1975;57:788-792.
9. Boden BP, Oshbahr DC, Jimenez C. Low-risk stress fractures. Am J Sports Med. 2001;29:100-113.
10. Pao DG. Avulsion fracture of the base of the fifth metatarsal not seen on conventional radiography of the foot: the need for an additional projection. Am J Roentgenol. 2000;175:549-552.
11. Egol K. Avulsion fractures of the fifth metatarsal base: a prospective outcome study. Foot Ankle Int. 2007;28:581-583.
12. Jones MH, Amendola AS. Navicular stress fractures. Clin Sports Med. 2006;25:151-158.
13. Fitch KD, Blackwell JB, Gilmour WN. Operation for non-union of stress fracture of the tarsal navicular. J Bone Joint Surg Br. 1989;71:105-110.
14. Judd DB, Kim DH. Foot fractures frequently misdiagnosed as ankle sprains. Am Fam Physician. 2002;66:785-794.
15. Zwitser EW, Breederveld BS. Fractures of the fifth metatarsal; diagnosis and treatment. Injury. 2010;41:555-562.
16. Koslowsky TC, Gausepohl T, Mader K, et al. Treatment of displaced proximal fifth metatarsal fractures using a new one-step fixation technique. J Trauma. 2010;68:122-125.
17. Mologne TS. Early screw fixation versus casting in the treatment of acute Jones fractures. Am J Sports Med. 2005;33:970-975.
18. Porter DA, Duncan M, Meyer SJF. Fifth metatarsal Jones fracture fixation with a 4.5-mm cannulated stainless steel screw in the competitive and recreational athlete. Am J Sports Med. 2005;33:726-733.
19. Vu D, McDiarmid T, Brown M. What is the most effective management of acute fractures of the base of the fifth metatarsal? J Fam Pract. 2006;55:713-717.
20. Monteleone GP. Stress fractures in the athlete. Orthop Clin North Am. 1995;26:423-432.
21. Torg JS, Pavlov H, Cooley LH, et al. Stress fractures of the tarsal navicular. A retrospective review of twenty-one cases. J Bone Joint Surg Am. 1982;64:700-712.
22. Khan KM, Fuller PJ, Brukner PD, et al. Outcome of conservative and surgical management of navicular stress fracture in athletes. Eighty-six cases proven with computerized tomography. Am J Sports Med. 1992;20:657-666.
23. Sizensky JA, Marks RM. Imaging of the navicular. Foot Ankle Clin. 2004;9:181-209.
24. Torg JS, Moyer J, Gaughan JP, et al. Management of tarsal navicular stress fractures: conservative versus surgical treatment: a meta-analysis. Am J Sports Med. 2010;38:1048-1053.
25. Bojanic I. Conservative treatment of stress fractures of the tarsal navicular in athletes. Rev Chir Orthop Reparatrice Appar Mot. 1997;83:133-138.
26. Ostlie DK, Simons SM. Tarsal navicular stress fracture in a young athlete: case report with clinical, radiologic, and pathophysiologic correlations. J Am Board Fam Pract. 2001;14:381-385.
27. Towne LC, Blazina ME, Cozen LN. Fatigue fracture of the tarsal navicular. J Bone Joint Surg Am. 1970;52:376-378.
28. Strauss JE, Fornberg JA, Lippert FG. Chronic lateral ankle instability and associated conditions: a rationale for treatment. Foot Ankle Int. 2007;28:1041-1044.
29. Schachter AK, Chen AL, Reddy PD, et al. Osteochondral lesions of the talus. J Am Acad Orthop Surg. 2005;13:152-158.
30. Taga I, Shino K, Inoue M, et al. Articular cartilage lesion in ankles with lateral ligament injury. Am J Sport Med. 1993;21:120-127.
31. O’Loughlin PF, Heyworth BE, Kennedy JG. Current concepts in the diagnosis and treatment of osteochondral lesions of the ankle. Am J Sports Med. 2010;38:392-404.
32. Shearer C, Loomer R, Clement D. Nonoperatively managed stage 5 osteochondral talar lesions. Foot Ankle Int. 2002;23:651-654.
33. Haverstock BD. Foot and ankle imaging in the athlete. Clin Podiatr Med Surg. 2008;25:249-262.
34. Geppert M, Sobel M, Bohne W. Lateral ankle instability as a cause of superior peroneal retinacular laxity: an anatomic and biomechanical study of cadaveric feet. Foot Ankle. 1993;14:330-334.
35. Butler BW, Lanthier J, Wertheimer SJ. Subluxing peroneals: a review of the literature and case report. J Foot Ankle Surg. 1993;32:134-139.
36. Roth JA, Taylor WC, Whalen J. Peroneal tendon subluxation: the other lateral ankle injury. Br J Sports Med. 2010;44:1047-1053.
37. Maffulli N, Ferran NA, Oliva F, et al. Recurrent subluxation of the peroneal tendons. Am J Sports Med. 2006;34:986-992.
38. Mason RB, Henderson JP. Traumatic peroneal tendon instability. Am J Sports Med. 1996;24:652-658.
39. Brodsky J, Krause J. Peroneus brevis tendon tears: pathophysiology, surgical reconstruction, and clinical results. Foot Ankle Int. 1998;19:271-279.
40. Marten MA, Noyez JF, Mulier JC. Recurrent dislocation of the peroneal tendons. Results of rerouting the tendons under the calcaneofibular ligament. Am J Sports Med. 1986;14:148-150.
41. Escalas F, Figueras JM, Merino JA. Dislocation of the peroneal tendons. Long-term results of surgical treatment. J Bone Joint Surg Am. 1980;62:451-453.
• Treat a nondisplaced shaft fracture of the fifth metatarsal conservatively, with 6 to 8 weeks of immobilization with a protective orthosis. B
• Suspect a navicular fracture in patients who describe a gradual onset of vague, dorsal midfoot pain associated with athletic activity. C
• Order magnetic resonance imaging when you suspect osteochondritis dissecans, as radiographs are insensitive for identifying these lesions. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Ankle sprain, one of the more common injuries that primary care physicians evaluate, is usually managed with conservative treatment. Not uncommonly, however, lateral ankle sprain is diagnosed without consideration of a broader differential diagnosis.
Contributing to the problem is the fact that the clinical presentation of some fractures and tendon injuries is similar to that of a routine sprain. In some cases, the mechanism of injury—sprains are usually caused by excessive inversion of the ankle on a plantar-flexed foot—is similar, as well. What’s more, radiographs are often omitted or misinterpreted.
In the pages that follow, we highlight 4 commonly misdiagnosed injuries: fifth metatarsal fractures, navicular fractures, talar dome lesions, and peroneal tendon injuries. These injuries should be included in the differential diagnosis of an acute ankle injury—or a subacute foot or ankle injury that fails to respond as expected. Prompt recognition and appropriate treatment result in optimal outcomes. When foot and ankle fractures and tendon injuries are misdiagnosed (or simply missed) and do not receive adequate treatment, long-term morbidity, including frequent reinjury and disability, may result.1
Are x-rays needed? Turn to the Ottawa rules
Ankle sprains represent a disruption in a ligament supporting a joint, and result in pain, edema, and ecchymosis, and often affect a patient’s ability to bear weight. While uncomplicated sprains generally heal with conservative treatment, other common foot and ankle injuries may require a different approach.
The Ottawa foot and ankle rules are an evidence-based guide to the use of initial radiographs after acute ankle injury (TABLE 1).2-4 Pain—near the malleoli (for the ankle) or in the midfoot—is the key criterion, but x-rays are recommended only if at least one other specified criterion is also met. With a sensitivity of nearly 100%, the rules have been shown to reliably exclude, and diagnose, ankle and midfoot fractures in children >5 years and adults.2,5
Table 1
Ottawa ankle and foot rules2-4
| Ankle |
X-rays are required only if the patient has pain near the malleolus and one or more of the following:
|
| Foot |
X-rays are required only if the patient has pain in the midfoot and one or more of the following:
|
Fifth metatarsal fractures are easily missed
The mechanism of injury for a fifth metatarsal fracture is often similar to that of a lateral ankle sprain. In addition, isolated ankle radiographs may not adequately evaluate the fifth metatarsal, which increases the risk of misdiagnosis.6
3 types of fifth metatarsal fractures
Fifth metatarsal fractures involve one of the following:
- an avulsion fracture, caused by the pull of the plantar aponeurosis and the peroneus brevis tendon at the tuberosity of the bone
- a Jones fracture, at the base of the fourth and fifth metatarsal (FIGURE 1)
- a shaft fracture, distal to the fifth metatarsal joint in the proximal diaphysis.6-8
FIGURE 1
Jones fractures heal slowly
This 50-year-old patient presented with pain and swelling in the ankle and lateral foot shortly after an inversion ankle injury. A radiograph (A) taken at that time reveals a Jones fracture. The second radiograph (B) was taken 6 weeks later, after continued immobilization with no weight-bearing. Three months after the injury (C), the patient was clinically asymptomatic.
While avulsion fractures are generally the result of an inversion ankle injury, Jones fractures are usually caused by a large adductive force applied to the forefoot on a plantar-flexed ankle.6 Shaft fractures, also known as diaphyseal stress fractures, are overuse injuries from chronic overload, usually after a sudden increase in running or walking.9
Patients with fifth metatarsal fractures typically have tenderness with palpation over the area of injury, with edema and ecchymosis when the injury is acute. Evidence-based guidelines recommend x-rays of the foot, including anteroposterior (AP), lateral, and oblique views.2-4 One study supports the use of an additional x-ray—an AP view of the ankle, including the base of the fifth metatarsal—if clinical suspicion is high and initial radiographs are negative or inconclusive.10
Shaft fractures may not be seen on x-rays in the first 3 weeks, but a periosteal reaction or linear lucency near the symptomatic area may be noticeable on radiographs taken at a later date.11 If this overuse injury seems likely but does not show up on the initial x-rays, however, magnetic resonance imaging (MRI) or a technetium bone scan can reliably identify a stress fracture.9
How to treat, when to refer
Treatment of fifth metatarsal fractures range from conservative to surgical, depending on the type (and extent) of injury (TABLE 2).1,5,6,12-14
TABLE 2
Nondisplaced avulsion fractures can be treated conservatively, with relative immobilization. In one prospective study, the use of a stiff-soled shoe, with weight-bearing as tolerated, was associated with excellent long-term outcomes.11 Orthopedic referral for probable reduction and fixation is indicated for avulsion fractures that are comminuted or >2 mm displaced, or have >30% involvement of the cubometatarsal joint.15,16
Jones fractures are known for prolonged healing and nonunion, as well as a high rate of complications. If the fracture is nondisplaced, start with conservative treatment, consisting of nonweight-bearing immobilization for 6 to 8 weeks, with additional immobilization dependent on radiographs. One randomized controlled trial of patients with Jones fractures showed a relatively high failure rate (44%) with casting; patients for whom casting was successful still had a median time to bony union of 15 weeks.17 Specialty consultation may be needed when there is fracture displacement, absence of bony union, or high clinical concern.6,17
Is your patient an athlete? Surgical fixation is favored for injured athletes with Jones fractures because failure rates are lower and both clinical union and return to play are shorter.18,19 In a case series involving 23 athletic patients with Jones fractures, the success rate for immediate surgical screw fixation approached 100% within 6 to 8 weeks.18
Nondisplaced shaft fractures may be treated conservatively, with 6 to 8 weeks of immobilization with a protective orthosis. An orthopedic referral is recommended for patients whose fractures have >3 mm displacement or >10 degree angulation.15
Navicular fractures are overuse injuries
The navicular is predisposed to stress injury because the central third of the bone is relatively avascular. In addition, the navicular is the area of greatest stress and impingement between the talus and cuneiform bones during repetitive foot strikes.12,20 Navicular fractures occur predominantly in track and field athletes.12
Patients presenting with a navicular stress fracture often report a gradual onset of vague dorsal midfoot pain associated with their workout.17 Examination typically reveals tenderness on palpation over the dorsal aspect of the navicular; passive eversion and active inversion may be painful, but edema and ecchymosis are usually absent.21
When pain is elicited by palpation of the navicular, radiographs are recommended.2,6 X-rays have a relatively low sensitivity (33%), however, for detecting acute navicular stress fractures. If initial radiographs are negative but there is a high clinical suspicion, advanced studies—with either MRI or a technetium bone scan—are recommended for a definitive diagnosis.12,22 While both are highly sensitive for navicular stress fractures, MRI provides greater specificity and anatomic detail.23
Most navicular fractures are nondisplaced
Nondisplaced navicular fractures can be treated conservatively, with nonweight-bearing immobilization for 6 to 8 weeks followed by progressive activity.24 Prospective studies have found that conservative treatment has a high success rate, with athletes usually able to return to play within 6 months.22,24,25 If tenderness remains after 6 to 8 weeks of immobilization, treatment choices are continued immobilization with no weight-bearing or orthopedic referral.26
Referral is indicated for navicular fractures that are comminuted or displaced, or involve more than one bone cortex.26 Surgical screw fixation may be recommended for navicular stress fractures in selected athletes because of its high success rate—and likelihood of an earlier return to play.27
Talar injuries are characterized by persistent pain
Injuries to the talus commonly occur at the same time as ankle sprains and may cause persistent pain, even after the sprain has healed.28 Evidence suggests that up to 90% of residual pain is related to an underlying cartilage injury.29,30 Most talar injuries are associated with the disruption of the cartilage overlaying the talar dome, which may lead to osteochondritis dissecans.29 Subtle talus fractures are also a concern after an acute ankle injury.
Osteochondral lesions are associated with a dull ankle pain deep in a location with a prior ankle injury; in some cases, the pain will become chronic.31 Physical exam findings typically include ankle joint effusion with localized tenderness around the joint.31
Ankle radiographs are insensitive for identifying osteochondral lesions, and MRI is recommended for evaluating suspected lesions.29,31 Treatment varies, depending on symptoms and severity. Patients with minimal symptoms may be treated conservatively; however, high failure rates have been reported.32 Surgical treatment depends on the size and site of the lesion and the degree of cartilage injury, and surgical consultation is recommended.31
Fractures of the talar dome (FIGURE 2) may be either medial or lateral and are often the result of inversion ankle injuries.14 History and clinical findings vary depending on the type of fracture.
FIGURE 2
Talar dome injuries often result from inversion ankle injuries
As with osteochondral lesions, ankle radiographs may fail to identify talus fractures. Computed tomography (CT) should be used to evaluate acute fractures of the talus, as CT scan is better able to define displacement, size, and intra-articular involvement.33 Talar fractures may be managed conservatively with immobilization and nonweight-bearing for 4 to 6 weeks, but specialty consultation should be considered.14,33
A tarsal coalition—an incomplete, congenital separation of the bones, occasionally involving the talus and the calcaneus—can also be a cause of persistent pain after a sprain.28 Physical exam typically demonstrates decreased range of motion in the subtalar or transverse tarsal joint. Radiographs may identify the coalition, but MRI or CT scan provides optimal visualization. Immobilization for 6 weeks is the recommended initial treatment, but if that fails, surgical excision or fusion may be necessary.
Peroneal tendon injuries may cause ankle instability
Peroneal tendon injuries, which include strains, subluxation, dislocation, and tears of one or both of the peroneal tendons, are often caused by ankle inversion similar to that of an uncomplicated sprain. Subsequent ankle instability may result from untreated peroneal tendon injuries.34 Peroneal tendon subluxation accounts for a very small number (0.3%-0.5%) of traumatic ankle injuries.35
Peroneal tendon injuries often occur during sports that involve frequent lateral movement or cutting—eg, football, basketball, and soccer—and are often caused by sudden dorsiflexion of the inverted foot, with coincident contraction of the peroneal muscles.36,37 This mechanism can disrupt the superior peroneal retinaculum, leading to recurrent subluxation or dislocation and subsequent ankle instability.36,38 Chronic subluxation can also result in longitudinal tears of the peroneal tendons, especially of the peroneus brevis.36,38,39
Patients with peroneal tendon injuries may report a “pop” at the time of injury. Pain is typically located posterior to the lateral malleolus, and recurrent subluxation is often described as a “snapping” around the lateral ankle during athletic activities.37,38 Instability is common in patients with subacute or chronic peroneal tendon injuries, especially on uneven surfaces.38
Acute peroneal tendon injuries cause posterolateral ankle pain, swelling, and weakness; exam findings include tenderness along the course of the peroneal tendons with associated edema.37 Subluxation or dislocation of the peroneal brevis tendon may be confirmed by placing the foot in plantar flexion and inversion and asking the patient to forcibly dorsiflex and evert the injured ankle.
Plain radiographs are usually normal in an isolated injury to the peroneal tendons. A fracture of the posterolateral margin of the fibula is a rare finding but indicates disruption of the peroneal retinaculum.36 MRI provides the best imaging for peroneal tendons and the stabilizing retinaculum, although a CT scan can provide detailed bony anatomy when subtle fractures are suspected or additional evaluation is needed.
Subluxation or dislocation indicate a need for surgery
Conservative management is recommended for peroneal tendon strains, but surgical treatment is increasingly recommended for subluxation or dislocation, especially if the problem is recurrent.36,37 Conservative treatment consists of short-term immobilization with a walking boot or brace, followed by physical therapy to improve strength and motion. Surgical treatment of subluxation and dislocation by stabilizing the peroneal tendons within the peroneal groove has been shown to provide lasting stability and improvement.37,38,40,41
CORRESPONDENCE Scott Hall, MD, University of Nevada-Reno, Brigham Building 316, Reno, NV 89557; [email protected]
• Treat a nondisplaced shaft fracture of the fifth metatarsal conservatively, with 6 to 8 weeks of immobilization with a protective orthosis. B
• Suspect a navicular fracture in patients who describe a gradual onset of vague, dorsal midfoot pain associated with athletic activity. C
• Order magnetic resonance imaging when you suspect osteochondritis dissecans, as radiographs are insensitive for identifying these lesions. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Ankle sprain, one of the more common injuries that primary care physicians evaluate, is usually managed with conservative treatment. Not uncommonly, however, lateral ankle sprain is diagnosed without consideration of a broader differential diagnosis.
Contributing to the problem is the fact that the clinical presentation of some fractures and tendon injuries is similar to that of a routine sprain. In some cases, the mechanism of injury—sprains are usually caused by excessive inversion of the ankle on a plantar-flexed foot—is similar, as well. What’s more, radiographs are often omitted or misinterpreted.
In the pages that follow, we highlight 4 commonly misdiagnosed injuries: fifth metatarsal fractures, navicular fractures, talar dome lesions, and peroneal tendon injuries. These injuries should be included in the differential diagnosis of an acute ankle injury—or a subacute foot or ankle injury that fails to respond as expected. Prompt recognition and appropriate treatment result in optimal outcomes. When foot and ankle fractures and tendon injuries are misdiagnosed (or simply missed) and do not receive adequate treatment, long-term morbidity, including frequent reinjury and disability, may result.1
Are x-rays needed? Turn to the Ottawa rules
Ankle sprains represent a disruption in a ligament supporting a joint, and result in pain, edema, and ecchymosis, and often affect a patient’s ability to bear weight. While uncomplicated sprains generally heal with conservative treatment, other common foot and ankle injuries may require a different approach.
The Ottawa foot and ankle rules are an evidence-based guide to the use of initial radiographs after acute ankle injury (TABLE 1).2-4 Pain—near the malleoli (for the ankle) or in the midfoot—is the key criterion, but x-rays are recommended only if at least one other specified criterion is also met. With a sensitivity of nearly 100%, the rules have been shown to reliably exclude, and diagnose, ankle and midfoot fractures in children >5 years and adults.2,5
Table 1
Ottawa ankle and foot rules2-4
| Ankle |
X-rays are required only if the patient has pain near the malleolus and one or more of the following:
|
| Foot |
X-rays are required only if the patient has pain in the midfoot and one or more of the following:
|
Fifth metatarsal fractures are easily missed
The mechanism of injury for a fifth metatarsal fracture is often similar to that of a lateral ankle sprain. In addition, isolated ankle radiographs may not adequately evaluate the fifth metatarsal, which increases the risk of misdiagnosis.6
3 types of fifth metatarsal fractures
Fifth metatarsal fractures involve one of the following:
- an avulsion fracture, caused by the pull of the plantar aponeurosis and the peroneus brevis tendon at the tuberosity of the bone
- a Jones fracture, at the base of the fourth and fifth metatarsal (FIGURE 1)
- a shaft fracture, distal to the fifth metatarsal joint in the proximal diaphysis.6-8
FIGURE 1
Jones fractures heal slowly
This 50-year-old patient presented with pain and swelling in the ankle and lateral foot shortly after an inversion ankle injury. A radiograph (A) taken at that time reveals a Jones fracture. The second radiograph (B) was taken 6 weeks later, after continued immobilization with no weight-bearing. Three months after the injury (C), the patient was clinically asymptomatic.
While avulsion fractures are generally the result of an inversion ankle injury, Jones fractures are usually caused by a large adductive force applied to the forefoot on a plantar-flexed ankle.6 Shaft fractures, also known as diaphyseal stress fractures, are overuse injuries from chronic overload, usually after a sudden increase in running or walking.9
Patients with fifth metatarsal fractures typically have tenderness with palpation over the area of injury, with edema and ecchymosis when the injury is acute. Evidence-based guidelines recommend x-rays of the foot, including anteroposterior (AP), lateral, and oblique views.2-4 One study supports the use of an additional x-ray—an AP view of the ankle, including the base of the fifth metatarsal—if clinical suspicion is high and initial radiographs are negative or inconclusive.10
Shaft fractures may not be seen on x-rays in the first 3 weeks, but a periosteal reaction or linear lucency near the symptomatic area may be noticeable on radiographs taken at a later date.11 If this overuse injury seems likely but does not show up on the initial x-rays, however, magnetic resonance imaging (MRI) or a technetium bone scan can reliably identify a stress fracture.9
How to treat, when to refer
Treatment of fifth metatarsal fractures range from conservative to surgical, depending on the type (and extent) of injury (TABLE 2).1,5,6,12-14
TABLE 2
Nondisplaced avulsion fractures can be treated conservatively, with relative immobilization. In one prospective study, the use of a stiff-soled shoe, with weight-bearing as tolerated, was associated with excellent long-term outcomes.11 Orthopedic referral for probable reduction and fixation is indicated for avulsion fractures that are comminuted or >2 mm displaced, or have >30% involvement of the cubometatarsal joint.15,16
Jones fractures are known for prolonged healing and nonunion, as well as a high rate of complications. If the fracture is nondisplaced, start with conservative treatment, consisting of nonweight-bearing immobilization for 6 to 8 weeks, with additional immobilization dependent on radiographs. One randomized controlled trial of patients with Jones fractures showed a relatively high failure rate (44%) with casting; patients for whom casting was successful still had a median time to bony union of 15 weeks.17 Specialty consultation may be needed when there is fracture displacement, absence of bony union, or high clinical concern.6,17
Is your patient an athlete? Surgical fixation is favored for injured athletes with Jones fractures because failure rates are lower and both clinical union and return to play are shorter.18,19 In a case series involving 23 athletic patients with Jones fractures, the success rate for immediate surgical screw fixation approached 100% within 6 to 8 weeks.18
Nondisplaced shaft fractures may be treated conservatively, with 6 to 8 weeks of immobilization with a protective orthosis. An orthopedic referral is recommended for patients whose fractures have >3 mm displacement or >10 degree angulation.15
Navicular fractures are overuse injuries
The navicular is predisposed to stress injury because the central third of the bone is relatively avascular. In addition, the navicular is the area of greatest stress and impingement between the talus and cuneiform bones during repetitive foot strikes.12,20 Navicular fractures occur predominantly in track and field athletes.12
Patients presenting with a navicular stress fracture often report a gradual onset of vague dorsal midfoot pain associated with their workout.17 Examination typically reveals tenderness on palpation over the dorsal aspect of the navicular; passive eversion and active inversion may be painful, but edema and ecchymosis are usually absent.21
When pain is elicited by palpation of the navicular, radiographs are recommended.2,6 X-rays have a relatively low sensitivity (33%), however, for detecting acute navicular stress fractures. If initial radiographs are negative but there is a high clinical suspicion, advanced studies—with either MRI or a technetium bone scan—are recommended for a definitive diagnosis.12,22 While both are highly sensitive for navicular stress fractures, MRI provides greater specificity and anatomic detail.23
Most navicular fractures are nondisplaced
Nondisplaced navicular fractures can be treated conservatively, with nonweight-bearing immobilization for 6 to 8 weeks followed by progressive activity.24 Prospective studies have found that conservative treatment has a high success rate, with athletes usually able to return to play within 6 months.22,24,25 If tenderness remains after 6 to 8 weeks of immobilization, treatment choices are continued immobilization with no weight-bearing or orthopedic referral.26
Referral is indicated for navicular fractures that are comminuted or displaced, or involve more than one bone cortex.26 Surgical screw fixation may be recommended for navicular stress fractures in selected athletes because of its high success rate—and likelihood of an earlier return to play.27
Talar injuries are characterized by persistent pain
Injuries to the talus commonly occur at the same time as ankle sprains and may cause persistent pain, even after the sprain has healed.28 Evidence suggests that up to 90% of residual pain is related to an underlying cartilage injury.29,30 Most talar injuries are associated with the disruption of the cartilage overlaying the talar dome, which may lead to osteochondritis dissecans.29 Subtle talus fractures are also a concern after an acute ankle injury.
Osteochondral lesions are associated with a dull ankle pain deep in a location with a prior ankle injury; in some cases, the pain will become chronic.31 Physical exam findings typically include ankle joint effusion with localized tenderness around the joint.31
Ankle radiographs are insensitive for identifying osteochondral lesions, and MRI is recommended for evaluating suspected lesions.29,31 Treatment varies, depending on symptoms and severity. Patients with minimal symptoms may be treated conservatively; however, high failure rates have been reported.32 Surgical treatment depends on the size and site of the lesion and the degree of cartilage injury, and surgical consultation is recommended.31
Fractures of the talar dome (FIGURE 2) may be either medial or lateral and are often the result of inversion ankle injuries.14 History and clinical findings vary depending on the type of fracture.
FIGURE 2
Talar dome injuries often result from inversion ankle injuries
As with osteochondral lesions, ankle radiographs may fail to identify talus fractures. Computed tomography (CT) should be used to evaluate acute fractures of the talus, as CT scan is better able to define displacement, size, and intra-articular involvement.33 Talar fractures may be managed conservatively with immobilization and nonweight-bearing for 4 to 6 weeks, but specialty consultation should be considered.14,33
A tarsal coalition—an incomplete, congenital separation of the bones, occasionally involving the talus and the calcaneus—can also be a cause of persistent pain after a sprain.28 Physical exam typically demonstrates decreased range of motion in the subtalar or transverse tarsal joint. Radiographs may identify the coalition, but MRI or CT scan provides optimal visualization. Immobilization for 6 weeks is the recommended initial treatment, but if that fails, surgical excision or fusion may be necessary.
Peroneal tendon injuries may cause ankle instability
Peroneal tendon injuries, which include strains, subluxation, dislocation, and tears of one or both of the peroneal tendons, are often caused by ankle inversion similar to that of an uncomplicated sprain. Subsequent ankle instability may result from untreated peroneal tendon injuries.34 Peroneal tendon subluxation accounts for a very small number (0.3%-0.5%) of traumatic ankle injuries.35
Peroneal tendon injuries often occur during sports that involve frequent lateral movement or cutting—eg, football, basketball, and soccer—and are often caused by sudden dorsiflexion of the inverted foot, with coincident contraction of the peroneal muscles.36,37 This mechanism can disrupt the superior peroneal retinaculum, leading to recurrent subluxation or dislocation and subsequent ankle instability.36,38 Chronic subluxation can also result in longitudinal tears of the peroneal tendons, especially of the peroneus brevis.36,38,39
Patients with peroneal tendon injuries may report a “pop” at the time of injury. Pain is typically located posterior to the lateral malleolus, and recurrent subluxation is often described as a “snapping” around the lateral ankle during athletic activities.37,38 Instability is common in patients with subacute or chronic peroneal tendon injuries, especially on uneven surfaces.38
Acute peroneal tendon injuries cause posterolateral ankle pain, swelling, and weakness; exam findings include tenderness along the course of the peroneal tendons with associated edema.37 Subluxation or dislocation of the peroneal brevis tendon may be confirmed by placing the foot in plantar flexion and inversion and asking the patient to forcibly dorsiflex and evert the injured ankle.
Plain radiographs are usually normal in an isolated injury to the peroneal tendons. A fracture of the posterolateral margin of the fibula is a rare finding but indicates disruption of the peroneal retinaculum.36 MRI provides the best imaging for peroneal tendons and the stabilizing retinaculum, although a CT scan can provide detailed bony anatomy when subtle fractures are suspected or additional evaluation is needed.
Subluxation or dislocation indicate a need for surgery
Conservative management is recommended for peroneal tendon strains, but surgical treatment is increasingly recommended for subluxation or dislocation, especially if the problem is recurrent.36,37 Conservative treatment consists of short-term immobilization with a walking boot or brace, followed by physical therapy to improve strength and motion. Surgical treatment of subluxation and dislocation by stabilizing the peroneal tendons within the peroneal groove has been shown to provide lasting stability and improvement.37,38,40,41
CORRESPONDENCE Scott Hall, MD, University of Nevada-Reno, Brigham Building 316, Reno, NV 89557; [email protected]
1. van Rijn RM, van Os AG, Bernsen RM, et al. What is the clinical course of acute ankle sprains? A systematic literature review. Am J Med. 2008;121:324-331.
2. Nugent PJ. Ottawa ankle rules accurately assess injuries and reduce reliance on radiographs. J Fam Pract. 2004;53:785-788.
3. Stiell IG, McKnight RD, Greenberg GH, et al. Implementation of the Ottawa ankle rules. JAMA. 1994;271:827-832.
4. Stiell IG, Greenberg GH, McKnight RD, et al. Decision rules for the use of radiography in acute ankle injuries. JAMA. 1993;269:1127-1132.
5. Judd DB, Kim DH. Foot fractures frequently misdiagnosed as ankle sprains. Am Fam Physician. 2002;66:785-795.
6. Den Hartog BD. Fracture of the proximal fifth metatarsal. J Am Acad Orthop Surg. 2009;17:458-464.
7. Torg JS, Balduini FC, Zelko RR, et al. Fractures of the base of the fifth metatarsal distal to the tuberosity. J Bone Joint Surg Am. 1984;66:209-214.
8. Dameron TB. Fractures and anatomical variations of the proximal portion of the fifth metatarsal. J Bone Joint Surg Am. 1975;57:788-792.
9. Boden BP, Oshbahr DC, Jimenez C. Low-risk stress fractures. Am J Sports Med. 2001;29:100-113.
10. Pao DG. Avulsion fracture of the base of the fifth metatarsal not seen on conventional radiography of the foot: the need for an additional projection. Am J Roentgenol. 2000;175:549-552.
11. Egol K. Avulsion fractures of the fifth metatarsal base: a prospective outcome study. Foot Ankle Int. 2007;28:581-583.
12. Jones MH, Amendola AS. Navicular stress fractures. Clin Sports Med. 2006;25:151-158.
13. Fitch KD, Blackwell JB, Gilmour WN. Operation for non-union of stress fracture of the tarsal navicular. J Bone Joint Surg Br. 1989;71:105-110.
14. Judd DB, Kim DH. Foot fractures frequently misdiagnosed as ankle sprains. Am Fam Physician. 2002;66:785-794.
15. Zwitser EW, Breederveld BS. Fractures of the fifth metatarsal; diagnosis and treatment. Injury. 2010;41:555-562.
16. Koslowsky TC, Gausepohl T, Mader K, et al. Treatment of displaced proximal fifth metatarsal fractures using a new one-step fixation technique. J Trauma. 2010;68:122-125.
17. Mologne TS. Early screw fixation versus casting in the treatment of acute Jones fractures. Am J Sports Med. 2005;33:970-975.
18. Porter DA, Duncan M, Meyer SJF. Fifth metatarsal Jones fracture fixation with a 4.5-mm cannulated stainless steel screw in the competitive and recreational athlete. Am J Sports Med. 2005;33:726-733.
19. Vu D, McDiarmid T, Brown M. What is the most effective management of acute fractures of the base of the fifth metatarsal? J Fam Pract. 2006;55:713-717.
20. Monteleone GP. Stress fractures in the athlete. Orthop Clin North Am. 1995;26:423-432.
21. Torg JS, Pavlov H, Cooley LH, et al. Stress fractures of the tarsal navicular. A retrospective review of twenty-one cases. J Bone Joint Surg Am. 1982;64:700-712.
22. Khan KM, Fuller PJ, Brukner PD, et al. Outcome of conservative and surgical management of navicular stress fracture in athletes. Eighty-six cases proven with computerized tomography. Am J Sports Med. 1992;20:657-666.
23. Sizensky JA, Marks RM. Imaging of the navicular. Foot Ankle Clin. 2004;9:181-209.
24. Torg JS, Moyer J, Gaughan JP, et al. Management of tarsal navicular stress fractures: conservative versus surgical treatment: a meta-analysis. Am J Sports Med. 2010;38:1048-1053.
25. Bojanic I. Conservative treatment of stress fractures of the tarsal navicular in athletes. Rev Chir Orthop Reparatrice Appar Mot. 1997;83:133-138.
26. Ostlie DK, Simons SM. Tarsal navicular stress fracture in a young athlete: case report with clinical, radiologic, and pathophysiologic correlations. J Am Board Fam Pract. 2001;14:381-385.
27. Towne LC, Blazina ME, Cozen LN. Fatigue fracture of the tarsal navicular. J Bone Joint Surg Am. 1970;52:376-378.
28. Strauss JE, Fornberg JA, Lippert FG. Chronic lateral ankle instability and associated conditions: a rationale for treatment. Foot Ankle Int. 2007;28:1041-1044.
29. Schachter AK, Chen AL, Reddy PD, et al. Osteochondral lesions of the talus. J Am Acad Orthop Surg. 2005;13:152-158.
30. Taga I, Shino K, Inoue M, et al. Articular cartilage lesion in ankles with lateral ligament injury. Am J Sport Med. 1993;21:120-127.
31. O’Loughlin PF, Heyworth BE, Kennedy JG. Current concepts in the diagnosis and treatment of osteochondral lesions of the ankle. Am J Sports Med. 2010;38:392-404.
32. Shearer C, Loomer R, Clement D. Nonoperatively managed stage 5 osteochondral talar lesions. Foot Ankle Int. 2002;23:651-654.
33. Haverstock BD. Foot and ankle imaging in the athlete. Clin Podiatr Med Surg. 2008;25:249-262.
34. Geppert M, Sobel M, Bohne W. Lateral ankle instability as a cause of superior peroneal retinacular laxity: an anatomic and biomechanical study of cadaveric feet. Foot Ankle. 1993;14:330-334.
35. Butler BW, Lanthier J, Wertheimer SJ. Subluxing peroneals: a review of the literature and case report. J Foot Ankle Surg. 1993;32:134-139.
36. Roth JA, Taylor WC, Whalen J. Peroneal tendon subluxation: the other lateral ankle injury. Br J Sports Med. 2010;44:1047-1053.
37. Maffulli N, Ferran NA, Oliva F, et al. Recurrent subluxation of the peroneal tendons. Am J Sports Med. 2006;34:986-992.
38. Mason RB, Henderson JP. Traumatic peroneal tendon instability. Am J Sports Med. 1996;24:652-658.
39. Brodsky J, Krause J. Peroneus brevis tendon tears: pathophysiology, surgical reconstruction, and clinical results. Foot Ankle Int. 1998;19:271-279.
40. Marten MA, Noyez JF, Mulier JC. Recurrent dislocation of the peroneal tendons. Results of rerouting the tendons under the calcaneofibular ligament. Am J Sports Med. 1986;14:148-150.
41. Escalas F, Figueras JM, Merino JA. Dislocation of the peroneal tendons. Long-term results of surgical treatment. J Bone Joint Surg Am. 1980;62:451-453.
1. van Rijn RM, van Os AG, Bernsen RM, et al. What is the clinical course of acute ankle sprains? A systematic literature review. Am J Med. 2008;121:324-331.
2. Nugent PJ. Ottawa ankle rules accurately assess injuries and reduce reliance on radiographs. J Fam Pract. 2004;53:785-788.
3. Stiell IG, McKnight RD, Greenberg GH, et al. Implementation of the Ottawa ankle rules. JAMA. 1994;271:827-832.
4. Stiell IG, Greenberg GH, McKnight RD, et al. Decision rules for the use of radiography in acute ankle injuries. JAMA. 1993;269:1127-1132.
5. Judd DB, Kim DH. Foot fractures frequently misdiagnosed as ankle sprains. Am Fam Physician. 2002;66:785-795.
6. Den Hartog BD. Fracture of the proximal fifth metatarsal. J Am Acad Orthop Surg. 2009;17:458-464.
7. Torg JS, Balduini FC, Zelko RR, et al. Fractures of the base of the fifth metatarsal distal to the tuberosity. J Bone Joint Surg Am. 1984;66:209-214.
8. Dameron TB. Fractures and anatomical variations of the proximal portion of the fifth metatarsal. J Bone Joint Surg Am. 1975;57:788-792.
9. Boden BP, Oshbahr DC, Jimenez C. Low-risk stress fractures. Am J Sports Med. 2001;29:100-113.
10. Pao DG. Avulsion fracture of the base of the fifth metatarsal not seen on conventional radiography of the foot: the need for an additional projection. Am J Roentgenol. 2000;175:549-552.
11. Egol K. Avulsion fractures of the fifth metatarsal base: a prospective outcome study. Foot Ankle Int. 2007;28:581-583.
12. Jones MH, Amendola AS. Navicular stress fractures. Clin Sports Med. 2006;25:151-158.
13. Fitch KD, Blackwell JB, Gilmour WN. Operation for non-union of stress fracture of the tarsal navicular. J Bone Joint Surg Br. 1989;71:105-110.
14. Judd DB, Kim DH. Foot fractures frequently misdiagnosed as ankle sprains. Am Fam Physician. 2002;66:785-794.
15. Zwitser EW, Breederveld BS. Fractures of the fifth metatarsal; diagnosis and treatment. Injury. 2010;41:555-562.
16. Koslowsky TC, Gausepohl T, Mader K, et al. Treatment of displaced proximal fifth metatarsal fractures using a new one-step fixation technique. J Trauma. 2010;68:122-125.
17. Mologne TS. Early screw fixation versus casting in the treatment of acute Jones fractures. Am J Sports Med. 2005;33:970-975.
18. Porter DA, Duncan M, Meyer SJF. Fifth metatarsal Jones fracture fixation with a 4.5-mm cannulated stainless steel screw in the competitive and recreational athlete. Am J Sports Med. 2005;33:726-733.
19. Vu D, McDiarmid T, Brown M. What is the most effective management of acute fractures of the base of the fifth metatarsal? J Fam Pract. 2006;55:713-717.
20. Monteleone GP. Stress fractures in the athlete. Orthop Clin North Am. 1995;26:423-432.
21. Torg JS, Pavlov H, Cooley LH, et al. Stress fractures of the tarsal navicular. A retrospective review of twenty-one cases. J Bone Joint Surg Am. 1982;64:700-712.
22. Khan KM, Fuller PJ, Brukner PD, et al. Outcome of conservative and surgical management of navicular stress fracture in athletes. Eighty-six cases proven with computerized tomography. Am J Sports Med. 1992;20:657-666.
23. Sizensky JA, Marks RM. Imaging of the navicular. Foot Ankle Clin. 2004;9:181-209.
24. Torg JS, Moyer J, Gaughan JP, et al. Management of tarsal navicular stress fractures: conservative versus surgical treatment: a meta-analysis. Am J Sports Med. 2010;38:1048-1053.
25. Bojanic I. Conservative treatment of stress fractures of the tarsal navicular in athletes. Rev Chir Orthop Reparatrice Appar Mot. 1997;83:133-138.
26. Ostlie DK, Simons SM. Tarsal navicular stress fracture in a young athlete: case report with clinical, radiologic, and pathophysiologic correlations. J Am Board Fam Pract. 2001;14:381-385.
27. Towne LC, Blazina ME, Cozen LN. Fatigue fracture of the tarsal navicular. J Bone Joint Surg Am. 1970;52:376-378.
28. Strauss JE, Fornberg JA, Lippert FG. Chronic lateral ankle instability and associated conditions: a rationale for treatment. Foot Ankle Int. 2007;28:1041-1044.
29. Schachter AK, Chen AL, Reddy PD, et al. Osteochondral lesions of the talus. J Am Acad Orthop Surg. 2005;13:152-158.
30. Taga I, Shino K, Inoue M, et al. Articular cartilage lesion in ankles with lateral ligament injury. Am J Sport Med. 1993;21:120-127.
31. O’Loughlin PF, Heyworth BE, Kennedy JG. Current concepts in the diagnosis and treatment of osteochondral lesions of the ankle. Am J Sports Med. 2010;38:392-404.
32. Shearer C, Loomer R, Clement D. Nonoperatively managed stage 5 osteochondral talar lesions. Foot Ankle Int. 2002;23:651-654.
33. Haverstock BD. Foot and ankle imaging in the athlete. Clin Podiatr Med Surg. 2008;25:249-262.
34. Geppert M, Sobel M, Bohne W. Lateral ankle instability as a cause of superior peroneal retinacular laxity: an anatomic and biomechanical study of cadaveric feet. Foot Ankle. 1993;14:330-334.
35. Butler BW, Lanthier J, Wertheimer SJ. Subluxing peroneals: a review of the literature and case report. J Foot Ankle Surg. 1993;32:134-139.
36. Roth JA, Taylor WC, Whalen J. Peroneal tendon subluxation: the other lateral ankle injury. Br J Sports Med. 2010;44:1047-1053.
37. Maffulli N, Ferran NA, Oliva F, et al. Recurrent subluxation of the peroneal tendons. Am J Sports Med. 2006;34:986-992.
38. Mason RB, Henderson JP. Traumatic peroneal tendon instability. Am J Sports Med. 1996;24:652-658.
39. Brodsky J, Krause J. Peroneus brevis tendon tears: pathophysiology, surgical reconstruction, and clinical results. Foot Ankle Int. 1998;19:271-279.
40. Marten MA, Noyez JF, Mulier JC. Recurrent dislocation of the peroneal tendons. Results of rerouting the tendons under the calcaneofibular ligament. Am J Sports Med. 1986;14:148-150.
41. Escalas F, Figueras JM, Merino JA. Dislocation of the peroneal tendons. Long-term results of surgical treatment. J Bone Joint Surg Am. 1980;62:451-453.
Depression and Quality of Life in Cancer Patients Undergoing Chemotherapy: Relation between the Zung Self-Rating Depression Scale and Functional Assessment of Cancer Therapy-General
Objective: Although depression is prevalent among cancer patients, it remains underdiagnosed and undertreated. Quality of life is an important outcome in cancer patients and can be measured by questionnaires such as the Functional Assessment of Cancer Therapy-General version (FACT-G). The purpose of our study was to establish whether or not a group of items in FACT-G could be used as a screening tool for depression as well as for assessing quality of life.
Methods: A total of 62 chemotherapy patients (median age, 62 years [range, 22-81 years]; 55% women) completed Zung Self-Rating Depression Scale (ZSDS) and FACT-G questionnaires. Patients with ZSDS scores of 40 or more underwent clinical interviews for major depression. Pearson’s correlation was used to examine the relationship between the ZSDS and FACT-G scores. FACT-G score results were then analyzed to evaluate if subsets of the FACT-G can be used as a screening tool for major depression.
Results: In all, 30 of 62 patients (48%) had ZSDS scores less than 40 and were ruled out for major depression, and 30 of the 32 patients with ZSDS scores greater or equal to 40 participated in clinical interviews. Of those who were interviewed, 7 patients (23%) were confirmed to have major depression. Overall, the prevalence of major depression was 7 of 60 patients (12%; 95% CI: 5%-23%). The ZSDS and FACT-G scores had strong correlation (r -0.75). The composite score of six statements in FACT-G were found to have sensitivity of 100% and specificity of 81% in predicting major depression, using a cut-off value of 12 (range, 0-24). The six statements were, I have a lack of energy; I feel sad; I feel nervous; I am able to enjoy life; I am sleeping well; and I am enjoying the things I usually do for fun.
Conclusions: The prevalence of major depression among all participants was 12%. The ZSDS score and FACT-G score had strong correlation; the subsets of FACT-G may be useful as a screening tool for depression.
*For a PDF of the full article, click on the link to the left of this introduction.
Objective: Although depression is prevalent among cancer patients, it remains underdiagnosed and undertreated. Quality of life is an important outcome in cancer patients and can be measured by questionnaires such as the Functional Assessment of Cancer Therapy-General version (FACT-G). The purpose of our study was to establish whether or not a group of items in FACT-G could be used as a screening tool for depression as well as for assessing quality of life.
Methods: A total of 62 chemotherapy patients (median age, 62 years [range, 22-81 years]; 55% women) completed Zung Self-Rating Depression Scale (ZSDS) and FACT-G questionnaires. Patients with ZSDS scores of 40 or more underwent clinical interviews for major depression. Pearson’s correlation was used to examine the relationship between the ZSDS and FACT-G scores. FACT-G score results were then analyzed to evaluate if subsets of the FACT-G can be used as a screening tool for major depression.
Results: In all, 30 of 62 patients (48%) had ZSDS scores less than 40 and were ruled out for major depression, and 30 of the 32 patients with ZSDS scores greater or equal to 40 participated in clinical interviews. Of those who were interviewed, 7 patients (23%) were confirmed to have major depression. Overall, the prevalence of major depression was 7 of 60 patients (12%; 95% CI: 5%-23%). The ZSDS and FACT-G scores had strong correlation (r -0.75). The composite score of six statements in FACT-G were found to have sensitivity of 100% and specificity of 81% in predicting major depression, using a cut-off value of 12 (range, 0-24). The six statements were, I have a lack of energy; I feel sad; I feel nervous; I am able to enjoy life; I am sleeping well; and I am enjoying the things I usually do for fun.
Conclusions: The prevalence of major depression among all participants was 12%. The ZSDS score and FACT-G score had strong correlation; the subsets of FACT-G may be useful as a screening tool for depression.
*For a PDF of the full article, click on the link to the left of this introduction.
Objective: Although depression is prevalent among cancer patients, it remains underdiagnosed and undertreated. Quality of life is an important outcome in cancer patients and can be measured by questionnaires such as the Functional Assessment of Cancer Therapy-General version (FACT-G). The purpose of our study was to establish whether or not a group of items in FACT-G could be used as a screening tool for depression as well as for assessing quality of life.
Methods: A total of 62 chemotherapy patients (median age, 62 years [range, 22-81 years]; 55% women) completed Zung Self-Rating Depression Scale (ZSDS) and FACT-G questionnaires. Patients with ZSDS scores of 40 or more underwent clinical interviews for major depression. Pearson’s correlation was used to examine the relationship between the ZSDS and FACT-G scores. FACT-G score results were then analyzed to evaluate if subsets of the FACT-G can be used as a screening tool for major depression.
Results: In all, 30 of 62 patients (48%) had ZSDS scores less than 40 and were ruled out for major depression, and 30 of the 32 patients with ZSDS scores greater or equal to 40 participated in clinical interviews. Of those who were interviewed, 7 patients (23%) were confirmed to have major depression. Overall, the prevalence of major depression was 7 of 60 patients (12%; 95% CI: 5%-23%). The ZSDS and FACT-G scores had strong correlation (r -0.75). The composite score of six statements in FACT-G were found to have sensitivity of 100% and specificity of 81% in predicting major depression, using a cut-off value of 12 (range, 0-24). The six statements were, I have a lack of energy; I feel sad; I feel nervous; I am able to enjoy life; I am sleeping well; and I am enjoying the things I usually do for fun.
Conclusions: The prevalence of major depression among all participants was 12%. The ZSDS score and FACT-G score had strong correlation; the subsets of FACT-G may be useful as a screening tool for depression.
*For a PDF of the full article, click on the link to the left of this introduction.