User login
Giant Cell Tumor of Soft Tissue
Exome sequencing not always accurate, presenter says
Credit: Jeremy L. Grisham
MILAN, ITALY—New research suggests exome sequencing does not always produce high-quality results with regard to subsets of genes.
The American College of Medical Genetics and Genomics (ACMG) recommends physicians inform patients of clinically actionable genetic findings in the course of clinical exome testing.
Specifically, mutations in 56 specific genes with known clinical importance should be reported, even when they are incidental to the patient’s medical condition.
However, an analysis of 44 exome datasets from 4 different testing kits showed that they missed a high proportion of clinically relevant regions in the 56 ACMG genes.
“At least 1 gene in each exome method was missing more than 40% of disease-causing genetic variants, and we found that the worst-performing method missed more than 90% of such variants in four of the 56 genes,” said Eric Londin, PhD, of Thomas Jefferson University in Philadelphia, Pennsylvania.
He and his colleagues presented these findings at the European Society of Human Genetics Conference 2014 (abstract C07.6).
A central question, according to the researchers, is not how often a clinical diagnosis can be made using exome sequencing, but how often it is missed. And this study suggests there is a high false-negative rate using existing sequencing kits.
“Our concern is that when a clinical exome analysis does not report a disease-causing genetic variant, it may be that the location of that variant has not been analyzed, rather than the patient’s DNA being free of a disease-causing variant,” Dr Londin said.
“Depending on the method and the laboratory, a significant fraction (more than 10%) of the exome may be untested, and this raises concerns as to how results are being communicated to patients and their families.”
A total of 17,774 disease-causing genetic variants are annotated in the Human Gene Mutation Database for the 56 genes mentioned in the ACMG recommendations. The researchers examined the coverage of the exome datasets for the locations where the 17,774 disease-causing variants can occur.
Although the exome datasets are comparable in quality to other published clinical and research exome data sets, the coverage of the disease-causing locations was very heterogeneous and often poor.
The researchers believe that clinical labs that implement the ACMG reporting guidelines should recognize the substantial possibility of reporting false negative results.
The team said one potential improvement would be to have clinical exome sequencing use methods designed to provide a maximum yield of all clinically relevant genes.
“Many of the currently used exome kits are designed to provide a very broad dataset including genomic features that do not yet have a well-established clinical association,” Dr Londin said.
“There is a need to develop new kits and methods which provide adequate and reliable coverage of genes with known disease associations. If adequate performance cannot be obtained across the exome, then further use of targeted disease-specific panels of genes should be explored.”
The researchers also found that exome datasets generated from low amounts of sequence data (fewer than 6 gigabases) performed much worse than datasets that were generated from higher amounts of sequence data (more than 10 gigabases).
This finding is consistent with previous studies showing that exome methods do not have a linear relationship between sequence-generated and nucleotide coverage. Instead, a minimum threshold of sequencing data needs to be met before optimum nucleotide coverage is obtained.
“Current consensus and regulatory guidelines do not prescribe a minimum data requirement for clinical exome tests,” Dr Londin said. “The result is that when a causative variant cannot be identified, it does not necessarily imply that the variant is not present, rather that there may be a technical issue with the exome technology used.”
“In other words, a clinical ‘whole-exome’ study may not be ‘wholesome’ in coverage. Patients and their families should be made aware of this problem and of the implications of the genomic findings of clinical exome sequencing in its current state.” ![]()
Credit: Jeremy L. Grisham
MILAN, ITALY—New research suggests exome sequencing does not always produce high-quality results with regard to subsets of genes.
The American College of Medical Genetics and Genomics (ACMG) recommends physicians inform patients of clinically actionable genetic findings in the course of clinical exome testing.
Specifically, mutations in 56 specific genes with known clinical importance should be reported, even when they are incidental to the patient’s medical condition.
However, an analysis of 44 exome datasets from 4 different testing kits showed that they missed a high proportion of clinically relevant regions in the 56 ACMG genes.
“At least 1 gene in each exome method was missing more than 40% of disease-causing genetic variants, and we found that the worst-performing method missed more than 90% of such variants in four of the 56 genes,” said Eric Londin, PhD, of Thomas Jefferson University in Philadelphia, Pennsylvania.
He and his colleagues presented these findings at the European Society of Human Genetics Conference 2014 (abstract C07.6).
A central question, according to the researchers, is not how often a clinical diagnosis can be made using exome sequencing, but how often it is missed. And this study suggests there is a high false-negative rate using existing sequencing kits.
“Our concern is that when a clinical exome analysis does not report a disease-causing genetic variant, it may be that the location of that variant has not been analyzed, rather than the patient’s DNA being free of a disease-causing variant,” Dr Londin said.
“Depending on the method and the laboratory, a significant fraction (more than 10%) of the exome may be untested, and this raises concerns as to how results are being communicated to patients and their families.”
A total of 17,774 disease-causing genetic variants are annotated in the Human Gene Mutation Database for the 56 genes mentioned in the ACMG recommendations. The researchers examined the coverage of the exome datasets for the locations where the 17,774 disease-causing variants can occur.
Although the exome datasets are comparable in quality to other published clinical and research exome data sets, the coverage of the disease-causing locations was very heterogeneous and often poor.
The researchers believe that clinical labs that implement the ACMG reporting guidelines should recognize the substantial possibility of reporting false negative results.
The team said one potential improvement would be to have clinical exome sequencing use methods designed to provide a maximum yield of all clinically relevant genes.
“Many of the currently used exome kits are designed to provide a very broad dataset including genomic features that do not yet have a well-established clinical association,” Dr Londin said.
“There is a need to develop new kits and methods which provide adequate and reliable coverage of genes with known disease associations. If adequate performance cannot be obtained across the exome, then further use of targeted disease-specific panels of genes should be explored.”
The researchers also found that exome datasets generated from low amounts of sequence data (fewer than 6 gigabases) performed much worse than datasets that were generated from higher amounts of sequence data (more than 10 gigabases).
This finding is consistent with previous studies showing that exome methods do not have a linear relationship between sequence-generated and nucleotide coverage. Instead, a minimum threshold of sequencing data needs to be met before optimum nucleotide coverage is obtained.
“Current consensus and regulatory guidelines do not prescribe a minimum data requirement for clinical exome tests,” Dr Londin said. “The result is that when a causative variant cannot be identified, it does not necessarily imply that the variant is not present, rather that there may be a technical issue with the exome technology used.”
“In other words, a clinical ‘whole-exome’ study may not be ‘wholesome’ in coverage. Patients and their families should be made aware of this problem and of the implications of the genomic findings of clinical exome sequencing in its current state.” ![]()
Credit: Jeremy L. Grisham
MILAN, ITALY—New research suggests exome sequencing does not always produce high-quality results with regard to subsets of genes.
The American College of Medical Genetics and Genomics (ACMG) recommends physicians inform patients of clinically actionable genetic findings in the course of clinical exome testing.
Specifically, mutations in 56 specific genes with known clinical importance should be reported, even when they are incidental to the patient’s medical condition.
However, an analysis of 44 exome datasets from 4 different testing kits showed that they missed a high proportion of clinically relevant regions in the 56 ACMG genes.
“At least 1 gene in each exome method was missing more than 40% of disease-causing genetic variants, and we found that the worst-performing method missed more than 90% of such variants in four of the 56 genes,” said Eric Londin, PhD, of Thomas Jefferson University in Philadelphia, Pennsylvania.
He and his colleagues presented these findings at the European Society of Human Genetics Conference 2014 (abstract C07.6).
A central question, according to the researchers, is not how often a clinical diagnosis can be made using exome sequencing, but how often it is missed. And this study suggests there is a high false-negative rate using existing sequencing kits.
“Our concern is that when a clinical exome analysis does not report a disease-causing genetic variant, it may be that the location of that variant has not been analyzed, rather than the patient’s DNA being free of a disease-causing variant,” Dr Londin said.
“Depending on the method and the laboratory, a significant fraction (more than 10%) of the exome may be untested, and this raises concerns as to how results are being communicated to patients and their families.”
A total of 17,774 disease-causing genetic variants are annotated in the Human Gene Mutation Database for the 56 genes mentioned in the ACMG recommendations. The researchers examined the coverage of the exome datasets for the locations where the 17,774 disease-causing variants can occur.
Although the exome datasets are comparable in quality to other published clinical and research exome data sets, the coverage of the disease-causing locations was very heterogeneous and often poor.
The researchers believe that clinical labs that implement the ACMG reporting guidelines should recognize the substantial possibility of reporting false negative results.
The team said one potential improvement would be to have clinical exome sequencing use methods designed to provide a maximum yield of all clinically relevant genes.
“Many of the currently used exome kits are designed to provide a very broad dataset including genomic features that do not yet have a well-established clinical association,” Dr Londin said.
“There is a need to develop new kits and methods which provide adequate and reliable coverage of genes with known disease associations. If adequate performance cannot be obtained across the exome, then further use of targeted disease-specific panels of genes should be explored.”
The researchers also found that exome datasets generated from low amounts of sequence data (fewer than 6 gigabases) performed much worse than datasets that were generated from higher amounts of sequence data (more than 10 gigabases).
This finding is consistent with previous studies showing that exome methods do not have a linear relationship between sequence-generated and nucleotide coverage. Instead, a minimum threshold of sequencing data needs to be met before optimum nucleotide coverage is obtained.
“Current consensus and regulatory guidelines do not prescribe a minimum data requirement for clinical exome tests,” Dr Londin said. “The result is that when a causative variant cannot be identified, it does not necessarily imply that the variant is not present, rather that there may be a technical issue with the exome technology used.”
“In other words, a clinical ‘whole-exome’ study may not be ‘wholesome’ in coverage. Patients and their families should be made aware of this problem and of the implications of the genomic findings of clinical exome sequencing in its current state.” ![]()
Why Hospitalists Should Heed Choosing Wisely Recommendations

By now, most hospitalists are at least familiar with the Choosing Wisely campaign, which has been widely published and embraced by numerous medical societies, including the Society of Hospital Medicine.1 This campaign was conceived in 2009 by the National Physicians Alliance, which developed simple lists for three primary care specialties—internal medicine, pediatrics, and family medicine—to help them become more effective in utilizing specific resources.
The effort was first published in the Archives of Internal Medicine in 2011 by the “Good Stewardship Working Group,” which outlined the five most overutilized types of care by the three groups, including items such as routinely ordering complete blood counts or electrocardiograms, prescribing brand name versus generic statin drugs, and prescribing antibiotics for pediatric pharyngitis. From this small list alone, they found incredible variability among primary care practices, with utilization of these services ranging from 1% to 56% and resulting in an estimated annual cost of $6.8 billion. Although this first pilot found simple reductions in utilization can have a powerful impact on cost, the group estimated that this overutilization in primary care is only a very small fraction of overutilization cost in the U.S. As such, they called upon other specialties outside of primary care to identify their own sets of targets to reduce unnecessary utilization of low-value services.
Many specialty groups heeded this call to action, which resulted in the Choosing Wisely campaign, launched in April 2012. In just two short years, this simple effort has expanded to published recommendations about resource use in more than 60 specialty societies.2 Like the original primary care list, most recommendations have focused on overutilization of diagnostic testing (imaging, cardiac testing, labs, pathology) and medication use. Later this year, the campaign will expand to include non-physician provider organizations, including the American Dental Association, the American Physical Therapy Association, and the American Academy of Nursing.
The Next Phase
The program has evolved from asking specialty groups to develop consensus and abide by their lists to targeting patients and their families so that they can understand and abide by those same lists. In fact, one of the major aims of the campaign is empowering patients to insist on care that is evidence based, necessary, not duplicative, and more beneficial than harmful. To do this, Consumer Reports has partnered up with the Choosing Wisely campaign to develop patient-friendly educational materials and with multiple consumer groups to help these materials reach their target audience. Major funding for the project has been provided by the American Board of Internal Medicine (ABIM) Foundation and the Robert Wood Johnson Foundation (RWJF). So far, they have awarded 21 projects.
These grants have been awarded to medical societies (see “SHM Choosing Wisely Case Study Competition,” p. 4), regional health organizations, and consumer advocate groups. Many of the tactics will include educational campaigns to teach practitioners about the content of the recommendations, programs aimed to enhance physician communication skills geared toward practicing physicians, other educational campaigns geared toward patients and families, and the establishment of a learning network to assist practices in quickly and effectively learning from one another how to implement the various recommendations.3
The three major assets of the Choosing Wisely campaign are:
- It attacks a core issue within the medical industry: Healthcare costs are higher here than in any other industrialized nation in the world, without clear evidence of higher quality to justify that cost;
- The lists are created by those who are responsible for most of the spending; and
- The campaign is spending resources to get information to the patients and their families so that there will be bilateral exchange and acceptance of the recommendations.
Without widespread patient education, overutilization will likely continue; a recent survey sponsored by the Choosing Wisely campaign found about half of physicians admitted they would order a test they know is unnecessary if the patient is insistent.4
Variable Outcomes
While there are many reasons to celebrate the success of the campaign, there is some concern that the Choosing Wisely campaign may have unintended consequences. Although a major driver in the success of the program is the fact that the lists have been created and endorsed by physician societies, a sort of “self-governance,” with no influence or impact from payers, critics of the program note the variability that each list has on the actual practice or revenue of the physician groups enacting the lists.
For example, a recent New England Journal of Medicine (NEJM) essay notes that the list produced by the American Academy of Orthopedic Surgeons does not include procedures that are high volume and variably valuable (such as knee arthoplasty) but does include over-the-counter medication use and low-volume procedures (such as needle lavage for knee osteoarthritis).5 Some societies list specialty services that need to be curbed but neglect to mention their own.
And, although the campaign specifically states on its website that the “recommendations should not be used to establish coverage decisions or exclusions,” some are legitimately concerned that these Choosing Wisely lists might very well be used by payers and/or quality reporting bodies to determine payments. This is undeniably tempting: How can practitioners argue against public display and reimbursement schemes being tightly tethered to their performance on metrics that they themselves have deemed unnecessary? As the NEJM editorial summarizes, these efforts should be embraced as long as there is thoughtful discussion about inclusion criteria, exclusion criteria, and measurement beforehand.5
In Sum
Despite concerns, the impact of the Choosing Wisely campaign has been widespread and impressive. The full extent to which this will have an impact on utilization and healthcare cost remains to be seen, but this yeoman’s attempt to reduce waste by providers is long overdue. Whether the program will be used for unintended purposes, such as public reporting, financial penalties, or incentives for performance, is still unknown, but physician groups should be paying close attention to the lists that we can impact, and we should pledge to be good stewards of the finite healthcare resources available to our patients.
Dr. Scheurer is a hospitalist and chief quality officer at the Medical University of South Carolina in Charleston. She is physician editor of The Hospitalist. Email her at [email protected].
References
- Choosing Wisely Campaign. Available at: http://www.choosingwisely.org. Accessed May 11, 2014.
- Choosing Wisely Consumer Partners. Available at: http://www.choosingwisely.org/partners. Access May 11, 2014.
- Choosing Wisely Grantees. Available at: http://www.choosingwisely.org/grantees. Accessed May 11, 2014.
- Choosing Wisely & Consumer Reports. Available at: http://consumerhealthchoices.org/wp-content/uploads/2014/03/ChoosingWiselyAndConsumerHealthChoices.pdf. Accessed May 11, 2014.
- Morden ME, Colla CH, Sequist TD, Rosenthal MB. Choosing Wisely—the politics and economics of labeling low-value services. Available at: http://www.nejm.org/doi/full/10.1056/NEJMp1314965. Accessed May 11, 2014.
By now, most hospitalists are at least familiar with the Choosing Wisely campaign, which has been widely published and embraced by numerous medical societies, including the Society of Hospital Medicine.1 This campaign was conceived in 2009 by the National Physicians Alliance, which developed simple lists for three primary care specialties—internal medicine, pediatrics, and family medicine—to help them become more effective in utilizing specific resources.
The effort was first published in the Archives of Internal Medicine in 2011 by the “Good Stewardship Working Group,” which outlined the five most overutilized types of care by the three groups, including items such as routinely ordering complete blood counts or electrocardiograms, prescribing brand name versus generic statin drugs, and prescribing antibiotics for pediatric pharyngitis. From this small list alone, they found incredible variability among primary care practices, with utilization of these services ranging from 1% to 56% and resulting in an estimated annual cost of $6.8 billion. Although this first pilot found simple reductions in utilization can have a powerful impact on cost, the group estimated that this overutilization in primary care is only a very small fraction of overutilization cost in the U.S. As such, they called upon other specialties outside of primary care to identify their own sets of targets to reduce unnecessary utilization of low-value services.
Many specialty groups heeded this call to action, which resulted in the Choosing Wisely campaign, launched in April 2012. In just two short years, this simple effort has expanded to published recommendations about resource use in more than 60 specialty societies.2 Like the original primary care list, most recommendations have focused on overutilization of diagnostic testing (imaging, cardiac testing, labs, pathology) and medication use. Later this year, the campaign will expand to include non-physician provider organizations, including the American Dental Association, the American Physical Therapy Association, and the American Academy of Nursing.
The Next Phase
The program has evolved from asking specialty groups to develop consensus and abide by their lists to targeting patients and their families so that they can understand and abide by those same lists. In fact, one of the major aims of the campaign is empowering patients to insist on care that is evidence based, necessary, not duplicative, and more beneficial than harmful. To do this, Consumer Reports has partnered up with the Choosing Wisely campaign to develop patient-friendly educational materials and with multiple consumer groups to help these materials reach their target audience. Major funding for the project has been provided by the American Board of Internal Medicine (ABIM) Foundation and the Robert Wood Johnson Foundation (RWJF). So far, they have awarded 21 projects.
These grants have been awarded to medical societies (see “SHM Choosing Wisely Case Study Competition,” p. 4), regional health organizations, and consumer advocate groups. Many of the tactics will include educational campaigns to teach practitioners about the content of the recommendations, programs aimed to enhance physician communication skills geared toward practicing physicians, other educational campaigns geared toward patients and families, and the establishment of a learning network to assist practices in quickly and effectively learning from one another how to implement the various recommendations.3
The three major assets of the Choosing Wisely campaign are:
- It attacks a core issue within the medical industry: Healthcare costs are higher here than in any other industrialized nation in the world, without clear evidence of higher quality to justify that cost;
- The lists are created by those who are responsible for most of the spending; and
- The campaign is spending resources to get information to the patients and their families so that there will be bilateral exchange and acceptance of the recommendations.
Without widespread patient education, overutilization will likely continue; a recent survey sponsored by the Choosing Wisely campaign found about half of physicians admitted they would order a test they know is unnecessary if the patient is insistent.4
Variable Outcomes
While there are many reasons to celebrate the success of the campaign, there is some concern that the Choosing Wisely campaign may have unintended consequences. Although a major driver in the success of the program is the fact that the lists have been created and endorsed by physician societies, a sort of “self-governance,” with no influence or impact from payers, critics of the program note the variability that each list has on the actual practice or revenue of the physician groups enacting the lists.
For example, a recent New England Journal of Medicine (NEJM) essay notes that the list produced by the American Academy of Orthopedic Surgeons does not include procedures that are high volume and variably valuable (such as knee arthoplasty) but does include over-the-counter medication use and low-volume procedures (such as needle lavage for knee osteoarthritis).5 Some societies list specialty services that need to be curbed but neglect to mention their own.
And, although the campaign specifically states on its website that the “recommendations should not be used to establish coverage decisions or exclusions,” some are legitimately concerned that these Choosing Wisely lists might very well be used by payers and/or quality reporting bodies to determine payments. This is undeniably tempting: How can practitioners argue against public display and reimbursement schemes being tightly tethered to their performance on metrics that they themselves have deemed unnecessary? As the NEJM editorial summarizes, these efforts should be embraced as long as there is thoughtful discussion about inclusion criteria, exclusion criteria, and measurement beforehand.5
In Sum
Despite concerns, the impact of the Choosing Wisely campaign has been widespread and impressive. The full extent to which this will have an impact on utilization and healthcare cost remains to be seen, but this yeoman’s attempt to reduce waste by providers is long overdue. Whether the program will be used for unintended purposes, such as public reporting, financial penalties, or incentives for performance, is still unknown, but physician groups should be paying close attention to the lists that we can impact, and we should pledge to be good stewards of the finite healthcare resources available to our patients.
Dr. Scheurer is a hospitalist and chief quality officer at the Medical University of South Carolina in Charleston. She is physician editor of The Hospitalist. Email her at [email protected].
References
- Choosing Wisely Campaign. Available at: http://www.choosingwisely.org. Accessed May 11, 2014.
- Choosing Wisely Consumer Partners. Available at: http://www.choosingwisely.org/partners. Access May 11, 2014.
- Choosing Wisely Grantees. Available at: http://www.choosingwisely.org/grantees. Accessed May 11, 2014.
- Choosing Wisely & Consumer Reports. Available at: http://consumerhealthchoices.org/wp-content/uploads/2014/03/ChoosingWiselyAndConsumerHealthChoices.pdf. Accessed May 11, 2014.
- Morden ME, Colla CH, Sequist TD, Rosenthal MB. Choosing Wisely—the politics and economics of labeling low-value services. Available at: http://www.nejm.org/doi/full/10.1056/NEJMp1314965. Accessed May 11, 2014.
By now, most hospitalists are at least familiar with the Choosing Wisely campaign, which has been widely published and embraced by numerous medical societies, including the Society of Hospital Medicine.1 This campaign was conceived in 2009 by the National Physicians Alliance, which developed simple lists for three primary care specialties—internal medicine, pediatrics, and family medicine—to help them become more effective in utilizing specific resources.
The effort was first published in the Archives of Internal Medicine in 2011 by the “Good Stewardship Working Group,” which outlined the five most overutilized types of care by the three groups, including items such as routinely ordering complete blood counts or electrocardiograms, prescribing brand name versus generic statin drugs, and prescribing antibiotics for pediatric pharyngitis. From this small list alone, they found incredible variability among primary care practices, with utilization of these services ranging from 1% to 56% and resulting in an estimated annual cost of $6.8 billion. Although this first pilot found simple reductions in utilization can have a powerful impact on cost, the group estimated that this overutilization in primary care is only a very small fraction of overutilization cost in the U.S. As such, they called upon other specialties outside of primary care to identify their own sets of targets to reduce unnecessary utilization of low-value services.
Many specialty groups heeded this call to action, which resulted in the Choosing Wisely campaign, launched in April 2012. In just two short years, this simple effort has expanded to published recommendations about resource use in more than 60 specialty societies.2 Like the original primary care list, most recommendations have focused on overutilization of diagnostic testing (imaging, cardiac testing, labs, pathology) and medication use. Later this year, the campaign will expand to include non-physician provider organizations, including the American Dental Association, the American Physical Therapy Association, and the American Academy of Nursing.
The Next Phase
The program has evolved from asking specialty groups to develop consensus and abide by their lists to targeting patients and their families so that they can understand and abide by those same lists. In fact, one of the major aims of the campaign is empowering patients to insist on care that is evidence based, necessary, not duplicative, and more beneficial than harmful. To do this, Consumer Reports has partnered up with the Choosing Wisely campaign to develop patient-friendly educational materials and with multiple consumer groups to help these materials reach their target audience. Major funding for the project has been provided by the American Board of Internal Medicine (ABIM) Foundation and the Robert Wood Johnson Foundation (RWJF). So far, they have awarded 21 projects.
These grants have been awarded to medical societies (see “SHM Choosing Wisely Case Study Competition,” p. 4), regional health organizations, and consumer advocate groups. Many of the tactics will include educational campaigns to teach practitioners about the content of the recommendations, programs aimed to enhance physician communication skills geared toward practicing physicians, other educational campaigns geared toward patients and families, and the establishment of a learning network to assist practices in quickly and effectively learning from one another how to implement the various recommendations.3
The three major assets of the Choosing Wisely campaign are:
- It attacks a core issue within the medical industry: Healthcare costs are higher here than in any other industrialized nation in the world, without clear evidence of higher quality to justify that cost;
- The lists are created by those who are responsible for most of the spending; and
- The campaign is spending resources to get information to the patients and their families so that there will be bilateral exchange and acceptance of the recommendations.
Without widespread patient education, overutilization will likely continue; a recent survey sponsored by the Choosing Wisely campaign found about half of physicians admitted they would order a test they know is unnecessary if the patient is insistent.4
Variable Outcomes
While there are many reasons to celebrate the success of the campaign, there is some concern that the Choosing Wisely campaign may have unintended consequences. Although a major driver in the success of the program is the fact that the lists have been created and endorsed by physician societies, a sort of “self-governance,” with no influence or impact from payers, critics of the program note the variability that each list has on the actual practice or revenue of the physician groups enacting the lists.
For example, a recent New England Journal of Medicine (NEJM) essay notes that the list produced by the American Academy of Orthopedic Surgeons does not include procedures that are high volume and variably valuable (such as knee arthoplasty) but does include over-the-counter medication use and low-volume procedures (such as needle lavage for knee osteoarthritis).5 Some societies list specialty services that need to be curbed but neglect to mention their own.
And, although the campaign specifically states on its website that the “recommendations should not be used to establish coverage decisions or exclusions,” some are legitimately concerned that these Choosing Wisely lists might very well be used by payers and/or quality reporting bodies to determine payments. This is undeniably tempting: How can practitioners argue against public display and reimbursement schemes being tightly tethered to their performance on metrics that they themselves have deemed unnecessary? As the NEJM editorial summarizes, these efforts should be embraced as long as there is thoughtful discussion about inclusion criteria, exclusion criteria, and measurement beforehand.5
In Sum
Despite concerns, the impact of the Choosing Wisely campaign has been widespread and impressive. The full extent to which this will have an impact on utilization and healthcare cost remains to be seen, but this yeoman’s attempt to reduce waste by providers is long overdue. Whether the program will be used for unintended purposes, such as public reporting, financial penalties, or incentives for performance, is still unknown, but physician groups should be paying close attention to the lists that we can impact, and we should pledge to be good stewards of the finite healthcare resources available to our patients.
Dr. Scheurer is a hospitalist and chief quality officer at the Medical University of South Carolina in Charleston. She is physician editor of The Hospitalist. Email her at [email protected].
References
- Choosing Wisely Campaign. Available at: http://www.choosingwisely.org. Accessed May 11, 2014.
- Choosing Wisely Consumer Partners. Available at: http://www.choosingwisely.org/partners. Access May 11, 2014.
- Choosing Wisely Grantees. Available at: http://www.choosingwisely.org/grantees. Accessed May 11, 2014.
- Choosing Wisely & Consumer Reports. Available at: http://consumerhealthchoices.org/wp-content/uploads/2014/03/ChoosingWiselyAndConsumerHealthChoices.pdf. Accessed May 11, 2014.
- Morden ME, Colla CH, Sequist TD, Rosenthal MB. Choosing Wisely—the politics and economics of labeling low-value services. Available at: http://www.nejm.org/doi/full/10.1056/NEJMp1314965. Accessed May 11, 2014.
How Generation X-Era Physicians Have Energized Hospital Medicine

“Each generation goes further than the generation preceding it, because it stands on the shoulders of that generation. You will have opportunities beyond anything we've ever known.”
—Former U.S. President Ronald Reagan
In the April 2014 issue of The Hospitalist, I began the tale of our specialty and the beginning of our evolution into a social movement. A social movement occurs when a large number of agents take coordinated action simultaneously.1 In the early days, managed care and other factors began driving doctors to try new, more efficient ways to practice, including early hospitalist practices, but usually these practices were one-offs and uncoordinated in their actions. Additionally, small numbers of doctors, perhaps a few hundred, focused their practice in the hospital. Drs. Wachter and Goldman published their “Sounding Board” article in the New England Journal of Medicine and, suddenly, hospital doctors across the country had a common identity: hospitalists.2 The specialty was poised for growth, but who would fill the need to come?
We should back up a few years to the dawn of the hospitalist movement in the late 1980s and early 1990s. For 20 years, Baby Boomers had been matriculating and graduating from medical school. The last Baby Boomers would graduate as the 1990s were just beginning. Baby Boomers were raised in the post-war era, largely by intact families with working fathers and stay-at-home mothers. They grew up in a competitive school environment—fueled by Sputnik—with a focus on success and working hard as the means to achieve that success. They were raised to be idealists and to question authority—remember Vietnam protests? These traits served the Baby Boomers well when managed care began to exert its pressure on physician practices. It was these physicians who figured out a new way to succeed in an altered landscape. It was either them or the big payers; their competitive nature was funneled into trying new, efficient practice models, to maintain income and control over their practices. Hospitalist systems were the most visible new practice model created in this era.
Fast forward a few years to the mid 1990s. A demographic shift was occurring. A new generation of Americans arrived on the scene of modern medicine—Generation X. The first Gen-X physicians graduated from medical school in 1991 and began moving into internship. They would graduate from residency in 1994, just as the early HM groups were starting to build a quiet but critical mass. This was a generation raised as latchkey kids in a time of rising divorce rates and working mothers. These kids were often home alone and grew up with more freedom and independence than any recent generation in history. Gen-X kids learned how to function on their own. Resourceful and self-reliant, they took on responsibility, but, conversely, did not appreciate being watched over. They liked to work at their own pace and valued work-life balance in a way that was foreign to the Baby Boomers. They weren’t born with keyboards in their cradles, but this was the first generation raised with technology—and they embraced it.
As these Generation X’ers came out of residency looking for the right fit in a specialty, trying creative ways of doing things, and seeking balance in their lives, they saw the early hospitalist programs the Boomer pioneers had created and started to join. They saw in these early hospitalist programs all that they were looking for in a practice. In many programs, the first hospitalists were lonely souls—but lonely by choice, usually left to their own devices. Their partners in the clinic stopped coming to the hospital, and their administrative leaders focused on the engine of running the clinic and managing the capitated and non-capitated costs of care.
Hospitalist practices became bastions of independence and freedom. No longer were these physicians chained down to their small area in one hallway of the clinic, nor did they cling to the metaphor of the fast-moving production line. Hospitalists could roam the hospital at will, from inpatient unit to ED to ICU. Hospitalists could eat lunch when they wanted to! Gen-X physicians flocked to this model of independence that so aligned with their own inner desire to work at their own pace and in their own way.
These early hospitalist programs, still trying to find their way in a complicated and changing healthcare environment, necessitated continued resourcefulness. We saw creative approaches to scheduling that favored continuity (seven on/seven off), transitions to hospitalists as teaching attendings, and early attempts at night coverage. The transition from at-home call or coverage by residents to in-house shifts to nocturnists could fill its own column. The creative opportunities offered by these early practices strongly appealed to the Gen-X sensibilities and values.3
Lastly, work-life balance strongly resonated with Gen-X physicians. Many of the Boomer physicians of the time were content to stay in the clinic and “run faster” to keep up with the demands of managed care; however, the Boomers who migrated to the hospital and the early Gen-X physicians seemed to have a different mindset. They relished the opportunity to work in the new hybrid model. I say hybrid because it certainly wasn’t the ongoing continuity model that it originated from in primary care, but neither was it pure shift work like in the ED. It had the element of daily shifts—but clustered together in five to 14 day runs, often with an equal amount of time away from work. Additionally, nobody was taking work home—at least not until EMR. Work-life balance was the recruiting “pitch” during the late 90s and early 2000s.
So, after the creation of the field by Baby Boomers, the Gen-X doctors were the fuel needed to grow the specialty at a pace never before seen in medicine. Wachter and Goldman’s article opened the floodgates. Between 1997 and 2006, the number of hospitalists grew by 29%—not in total, but 29% per year!
Gen-X physicians latched onto the idea created and publicized by the Boomers, but the movement needed more to sustain, even accelerate, that early growth than just an interesting new idea for how to see hospital patients. What was the oxygen? What gave the HM social movement its purpose?
In my August column, I will explore what came next to propel HM from a new area of practice, an offshoot of primary care, into a full-fledged movement. It was that next thing that made our field “go viral.”
Dr. Kealey is SHM president and medical director of hospital specialties at HealthPartners Medical Group in St. Paul, Minn.
References
- Lancaster LC, Stillman D. When Generations Collide. Who They Are. Why They Clash. How to Solve the Generational Puzzle at Work. New York: HarperCollins; 2002.
- Wachter RM, Goldman L. The emerging role of “hospitalists” in the American health care system. New Engl J Med. 1996;335(7):514-517.
- Wachter R. Today’s NEJM hospitalist study: what’s the news? Available at: http://community.the-hospitalist.org/2009/03/13/today-s-nejm-hospitalist-study-what-s-the-news/. Accessed May 11, 2014.
“Each generation goes further than the generation preceding it, because it stands on the shoulders of that generation. You will have opportunities beyond anything we've ever known.”
—Former U.S. President Ronald Reagan
In the April 2014 issue of The Hospitalist, I began the tale of our specialty and the beginning of our evolution into a social movement. A social movement occurs when a large number of agents take coordinated action simultaneously.1 In the early days, managed care and other factors began driving doctors to try new, more efficient ways to practice, including early hospitalist practices, but usually these practices were one-offs and uncoordinated in their actions. Additionally, small numbers of doctors, perhaps a few hundred, focused their practice in the hospital. Drs. Wachter and Goldman published their “Sounding Board” article in the New England Journal of Medicine and, suddenly, hospital doctors across the country had a common identity: hospitalists.2 The specialty was poised for growth, but who would fill the need to come?
We should back up a few years to the dawn of the hospitalist movement in the late 1980s and early 1990s. For 20 years, Baby Boomers had been matriculating and graduating from medical school. The last Baby Boomers would graduate as the 1990s were just beginning. Baby Boomers were raised in the post-war era, largely by intact families with working fathers and stay-at-home mothers. They grew up in a competitive school environment—fueled by Sputnik—with a focus on success and working hard as the means to achieve that success. They were raised to be idealists and to question authority—remember Vietnam protests? These traits served the Baby Boomers well when managed care began to exert its pressure on physician practices. It was these physicians who figured out a new way to succeed in an altered landscape. It was either them or the big payers; their competitive nature was funneled into trying new, efficient practice models, to maintain income and control over their practices. Hospitalist systems were the most visible new practice model created in this era.
Fast forward a few years to the mid 1990s. A demographic shift was occurring. A new generation of Americans arrived on the scene of modern medicine—Generation X. The first Gen-X physicians graduated from medical school in 1991 and began moving into internship. They would graduate from residency in 1994, just as the early HM groups were starting to build a quiet but critical mass. This was a generation raised as latchkey kids in a time of rising divorce rates and working mothers. These kids were often home alone and grew up with more freedom and independence than any recent generation in history. Gen-X kids learned how to function on their own. Resourceful and self-reliant, they took on responsibility, but, conversely, did not appreciate being watched over. They liked to work at their own pace and valued work-life balance in a way that was foreign to the Baby Boomers. They weren’t born with keyboards in their cradles, but this was the first generation raised with technology—and they embraced it.
As these Generation X’ers came out of residency looking for the right fit in a specialty, trying creative ways of doing things, and seeking balance in their lives, they saw the early hospitalist programs the Boomer pioneers had created and started to join. They saw in these early hospitalist programs all that they were looking for in a practice. In many programs, the first hospitalists were lonely souls—but lonely by choice, usually left to their own devices. Their partners in the clinic stopped coming to the hospital, and their administrative leaders focused on the engine of running the clinic and managing the capitated and non-capitated costs of care.
Hospitalist practices became bastions of independence and freedom. No longer were these physicians chained down to their small area in one hallway of the clinic, nor did they cling to the metaphor of the fast-moving production line. Hospitalists could roam the hospital at will, from inpatient unit to ED to ICU. Hospitalists could eat lunch when they wanted to! Gen-X physicians flocked to this model of independence that so aligned with their own inner desire to work at their own pace and in their own way.
These early hospitalist programs, still trying to find their way in a complicated and changing healthcare environment, necessitated continued resourcefulness. We saw creative approaches to scheduling that favored continuity (seven on/seven off), transitions to hospitalists as teaching attendings, and early attempts at night coverage. The transition from at-home call or coverage by residents to in-house shifts to nocturnists could fill its own column. The creative opportunities offered by these early practices strongly appealed to the Gen-X sensibilities and values.3
Lastly, work-life balance strongly resonated with Gen-X physicians. Many of the Boomer physicians of the time were content to stay in the clinic and “run faster” to keep up with the demands of managed care; however, the Boomers who migrated to the hospital and the early Gen-X physicians seemed to have a different mindset. They relished the opportunity to work in the new hybrid model. I say hybrid because it certainly wasn’t the ongoing continuity model that it originated from in primary care, but neither was it pure shift work like in the ED. It had the element of daily shifts—but clustered together in five to 14 day runs, often with an equal amount of time away from work. Additionally, nobody was taking work home—at least not until EMR. Work-life balance was the recruiting “pitch” during the late 90s and early 2000s.
So, after the creation of the field by Baby Boomers, the Gen-X doctors were the fuel needed to grow the specialty at a pace never before seen in medicine. Wachter and Goldman’s article opened the floodgates. Between 1997 and 2006, the number of hospitalists grew by 29%—not in total, but 29% per year!
Gen-X physicians latched onto the idea created and publicized by the Boomers, but the movement needed more to sustain, even accelerate, that early growth than just an interesting new idea for how to see hospital patients. What was the oxygen? What gave the HM social movement its purpose?
In my August column, I will explore what came next to propel HM from a new area of practice, an offshoot of primary care, into a full-fledged movement. It was that next thing that made our field “go viral.”
Dr. Kealey is SHM president and medical director of hospital specialties at HealthPartners Medical Group in St. Paul, Minn.
References
- Lancaster LC, Stillman D. When Generations Collide. Who They Are. Why They Clash. How to Solve the Generational Puzzle at Work. New York: HarperCollins; 2002.
- Wachter RM, Goldman L. The emerging role of “hospitalists” in the American health care system. New Engl J Med. 1996;335(7):514-517.
- Wachter R. Today’s NEJM hospitalist study: what’s the news? Available at: http://community.the-hospitalist.org/2009/03/13/today-s-nejm-hospitalist-study-what-s-the-news/. Accessed May 11, 2014.
“Each generation goes further than the generation preceding it, because it stands on the shoulders of that generation. You will have opportunities beyond anything we've ever known.”
—Former U.S. President Ronald Reagan
In the April 2014 issue of The Hospitalist, I began the tale of our specialty and the beginning of our evolution into a social movement. A social movement occurs when a large number of agents take coordinated action simultaneously.1 In the early days, managed care and other factors began driving doctors to try new, more efficient ways to practice, including early hospitalist practices, but usually these practices were one-offs and uncoordinated in their actions. Additionally, small numbers of doctors, perhaps a few hundred, focused their practice in the hospital. Drs. Wachter and Goldman published their “Sounding Board” article in the New England Journal of Medicine and, suddenly, hospital doctors across the country had a common identity: hospitalists.2 The specialty was poised for growth, but who would fill the need to come?
We should back up a few years to the dawn of the hospitalist movement in the late 1980s and early 1990s. For 20 years, Baby Boomers had been matriculating and graduating from medical school. The last Baby Boomers would graduate as the 1990s were just beginning. Baby Boomers were raised in the post-war era, largely by intact families with working fathers and stay-at-home mothers. They grew up in a competitive school environment—fueled by Sputnik—with a focus on success and working hard as the means to achieve that success. They were raised to be idealists and to question authority—remember Vietnam protests? These traits served the Baby Boomers well when managed care began to exert its pressure on physician practices. It was these physicians who figured out a new way to succeed in an altered landscape. It was either them or the big payers; their competitive nature was funneled into trying new, efficient practice models, to maintain income and control over their practices. Hospitalist systems were the most visible new practice model created in this era.
Fast forward a few years to the mid 1990s. A demographic shift was occurring. A new generation of Americans arrived on the scene of modern medicine—Generation X. The first Gen-X physicians graduated from medical school in 1991 and began moving into internship. They would graduate from residency in 1994, just as the early HM groups were starting to build a quiet but critical mass. This was a generation raised as latchkey kids in a time of rising divorce rates and working mothers. These kids were often home alone and grew up with more freedom and independence than any recent generation in history. Gen-X kids learned how to function on their own. Resourceful and self-reliant, they took on responsibility, but, conversely, did not appreciate being watched over. They liked to work at their own pace and valued work-life balance in a way that was foreign to the Baby Boomers. They weren’t born with keyboards in their cradles, but this was the first generation raised with technology—and they embraced it.
As these Generation X’ers came out of residency looking for the right fit in a specialty, trying creative ways of doing things, and seeking balance in their lives, they saw the early hospitalist programs the Boomer pioneers had created and started to join. They saw in these early hospitalist programs all that they were looking for in a practice. In many programs, the first hospitalists were lonely souls—but lonely by choice, usually left to their own devices. Their partners in the clinic stopped coming to the hospital, and their administrative leaders focused on the engine of running the clinic and managing the capitated and non-capitated costs of care.
Hospitalist practices became bastions of independence and freedom. No longer were these physicians chained down to their small area in one hallway of the clinic, nor did they cling to the metaphor of the fast-moving production line. Hospitalists could roam the hospital at will, from inpatient unit to ED to ICU. Hospitalists could eat lunch when they wanted to! Gen-X physicians flocked to this model of independence that so aligned with their own inner desire to work at their own pace and in their own way.
These early hospitalist programs, still trying to find their way in a complicated and changing healthcare environment, necessitated continued resourcefulness. We saw creative approaches to scheduling that favored continuity (seven on/seven off), transitions to hospitalists as teaching attendings, and early attempts at night coverage. The transition from at-home call or coverage by residents to in-house shifts to nocturnists could fill its own column. The creative opportunities offered by these early practices strongly appealed to the Gen-X sensibilities and values.3
Lastly, work-life balance strongly resonated with Gen-X physicians. Many of the Boomer physicians of the time were content to stay in the clinic and “run faster” to keep up with the demands of managed care; however, the Boomers who migrated to the hospital and the early Gen-X physicians seemed to have a different mindset. They relished the opportunity to work in the new hybrid model. I say hybrid because it certainly wasn’t the ongoing continuity model that it originated from in primary care, but neither was it pure shift work like in the ED. It had the element of daily shifts—but clustered together in five to 14 day runs, often with an equal amount of time away from work. Additionally, nobody was taking work home—at least not until EMR. Work-life balance was the recruiting “pitch” during the late 90s and early 2000s.
So, after the creation of the field by Baby Boomers, the Gen-X doctors were the fuel needed to grow the specialty at a pace never before seen in medicine. Wachter and Goldman’s article opened the floodgates. Between 1997 and 2006, the number of hospitalists grew by 29%—not in total, but 29% per year!
Gen-X physicians latched onto the idea created and publicized by the Boomers, but the movement needed more to sustain, even accelerate, that early growth than just an interesting new idea for how to see hospital patients. What was the oxygen? What gave the HM social movement its purpose?
In my August column, I will explore what came next to propel HM from a new area of practice, an offshoot of primary care, into a full-fledged movement. It was that next thing that made our field “go viral.”
Dr. Kealey is SHM president and medical director of hospital specialties at HealthPartners Medical Group in St. Paul, Minn.
References
- Lancaster LC, Stillman D. When Generations Collide. Who They Are. Why They Clash. How to Solve the Generational Puzzle at Work. New York: HarperCollins; 2002.
- Wachter RM, Goldman L. The emerging role of “hospitalists” in the American health care system. New Engl J Med. 1996;335(7):514-517.
- Wachter R. Today’s NEJM hospitalist study: what’s the news? Available at: http://community.the-hospitalist.org/2009/03/13/today-s-nejm-hospitalist-study-what-s-the-news/. Accessed May 11, 2014.
American Board of Internal Medicine Foundation's Choosing Wisely Campaign Promotes Evidence-Based Patient Care

The American Board of Internal Medicine (ABIM) established the ABIM Foundation to advance professionalism in improving healthcare. The foundation initiated the Choosing Wisely campaign [www.choosingwisely.org] in April 2012 to promote conversations that help physicians guide patients in selecting care that is supported by evidence, not duplicative of other tests or procedures, not harmful, and truly necessary. In order to achieve this, national organizations representing medical specialists were asked to identify five common tests or procedures whose necessity should be questioned.
John Bulger, DO, MBA, SFHM, chief quality officer at Geisinger Health System in Danville, Pa., chaired SHM’s Choosing Wisely recommendations committee. He says the “proximal concern over these tests may be the unnecessary cost of the test itself, [but] there are other unintended consequences.
“False positive or false negative results of unsupported testing may cause unwarranted emotional harm for the patient or may give a false sense of security,” he adds. “The latter may also be true for physicians who may fail to further investigate other ailments based on a previous false negative test. Tests ordered with little evidence tend to lead to more tests ordered with little evidence.”
To date, more than 60 specialty societies and 17 consumer groups have joined the Choosing Wisely effort, citing more than 300 potentially harmful tests and procedures that physicians should discuss with patients. New lists will be published throughout 2014.
—Karen Appold
The American Board of Internal Medicine (ABIM) established the ABIM Foundation to advance professionalism in improving healthcare. The foundation initiated the Choosing Wisely campaign [www.choosingwisely.org] in April 2012 to promote conversations that help physicians guide patients in selecting care that is supported by evidence, not duplicative of other tests or procedures, not harmful, and truly necessary. In order to achieve this, national organizations representing medical specialists were asked to identify five common tests or procedures whose necessity should be questioned.
John Bulger, DO, MBA, SFHM, chief quality officer at Geisinger Health System in Danville, Pa., chaired SHM’s Choosing Wisely recommendations committee. He says the “proximal concern over these tests may be the unnecessary cost of the test itself, [but] there are other unintended consequences.
“False positive or false negative results of unsupported testing may cause unwarranted emotional harm for the patient or may give a false sense of security,” he adds. “The latter may also be true for physicians who may fail to further investigate other ailments based on a previous false negative test. Tests ordered with little evidence tend to lead to more tests ordered with little evidence.”
To date, more than 60 specialty societies and 17 consumer groups have joined the Choosing Wisely effort, citing more than 300 potentially harmful tests and procedures that physicians should discuss with patients. New lists will be published throughout 2014.
—Karen Appold
The American Board of Internal Medicine (ABIM) established the ABIM Foundation to advance professionalism in improving healthcare. The foundation initiated the Choosing Wisely campaign [www.choosingwisely.org] in April 2012 to promote conversations that help physicians guide patients in selecting care that is supported by evidence, not duplicative of other tests or procedures, not harmful, and truly necessary. In order to achieve this, national organizations representing medical specialists were asked to identify five common tests or procedures whose necessity should be questioned.
John Bulger, DO, MBA, SFHM, chief quality officer at Geisinger Health System in Danville, Pa., chaired SHM’s Choosing Wisely recommendations committee. He says the “proximal concern over these tests may be the unnecessary cost of the test itself, [but] there are other unintended consequences.
“False positive or false negative results of unsupported testing may cause unwarranted emotional harm for the patient or may give a false sense of security,” he adds. “The latter may also be true for physicians who may fail to further investigate other ailments based on a previous false negative test. Tests ordered with little evidence tend to lead to more tests ordered with little evidence.”
To date, more than 60 specialty societies and 17 consumer groups have joined the Choosing Wisely effort, citing more than 300 potentially harmful tests and procedures that physicians should discuss with patients. New lists will be published throughout 2014.
—Karen Appold
When to Order Red Blood Cell Transfusion for Patients with Anemia
Background
Hospitalists commonly order red blood cell (RBC) transfusion as a therapy for patients with anemia resulting from a variety of clinical conditions. There has been lack of consensus on when to transfuse, because patients with anemia frequently have multiple co-morbidities, including coronary artery disease and congestive heart failure, which may influence their ability to tolerate a potentially ischemic state related to anemia or to accommodate volume fluctuations related to transfusion.
Furthermore, RBC transfusions are not without inherent risk. Life-threatening transfusion reactions occur in approximately seven per million transfused blood components, and transfusion-associated circulatory overload (TACO) can develop in one in 100 transfusions.1
Recently published guidelines provide recommendations for management of hemodynamically stable adults with anemia.
Guideline Update
The AABB published guidelines in the Annals of Internal Medicine in 2012 addressing RBC transfusion thresholds.1 The updated guideline makes a recommendation that clinicians utilize a restrictive transfusion strategy. Transfusion is strongly recommended for ICU patients with hemoglobin ≤7g/dL. In post-operative surgical patients and for post-operative patients with symptomatic anemia, transfusion is recommended for hemoglobin ≤8g/dL. The authors also made a weak recommendation to transfuse for hemoglobin ≤8g/dL or for symptoms in hospitalized hemodynamically stable patients with preexisting cardiovascular disease.
These recommendations draw from past literature, along with two more recent trials examining liberal or restrictive transfusion thresholds. The newer trials increased the total number of patients studied by nearly one third compared with prior reviews.2,3 The authors also incorporated recently published systematic reviews in their analysis.
Although the definition of a restrictive transfusion threshold varied across trials, including hemoglobin ≤7g/dL and ≤8g/dL, the authors used the pooled data to provide several recommendations in the new guideline. Of note, the pooled data was underpowered to detect up to a twofold increase in risk of myocardial infarction in patients in the restrictive strategy group.1
There were insufficient data for the authors to recommend for or against a restrictive transfusion strategy in patients with acute coronary syndrome, based on very low quality evidence.
Finally, the authors recommended that symptoms and hemoglobin level should both be used in determining transfusion criteria, based on low quality of evidence.
Analysis
The current AABB guidelines have two primary differences from earlier guidelines. First, the AABB authors used GRADE (Grading of Recommendations, Assessment, Development, and Evaluation) methodology to formalize evidence-based practice in their analysis of the literature. The authors purposely used the GRADE methodology to systematically evaluate the quality of the evidence base and explicitly state the strength of the recommendation for a particular transfusion threshold.4
Second, the AABB guidelines incorporated data from the more recently published FOCUS (Functional Outcomes in Cardiovascular patients Undergoing Surgical repair of hip fracture) and TRACS (Transfusion Requirements After Cardiac Surgery) trials, resulting in a stronger recommendation supporting the use of a restrictive transfusion strategy in non-ICU and post-operative patients. The findings of the FOCUS trial are especially applicable to hospitalists, because many patients who undergo hip fracture repair are directly cared for or are co-managed by hospitalists.
The current guidelines built upon previous guidelines that advocated a restrictive strategy (hemoglobin ≤7g/dL) in hemodynamically stable, critically ill adult patients.5 In general, restrictive transfusion strategy led to nearly 40% fewer patients receiving transfusion compared with the use of a liberal transfusion strategy.1 No additional harm to patients was evidenced in the restrictive transfusion group, though the trials were not designed to answer this question; moreover, there was no statistically significant difference in mortality or functional outcome between the two groups.
The authors of the current AABB guidelines recognized the importance of replicating the current findings in a more diverse patient population. An area where further study is indicated is in the use of specific transfusion thresholds in patients with acute coronary syndrome. These guidelines did not clarify whether or not there is a physiologic difference between use of different restrictive transfusion thresholds such as <8g/dL and <7g/dL.
The authors of the AABB guidelines also commented that performing a future trial to compare RBC transfusion for symptoms vs. hemoglobin “trigger” would be useful; however, they recognized that this may not be feasible due to the need to blind providers in the trial to hemoglobin values. Various society guidelines currently call for different transfusion thresholds or do not make a specific recommendation at all.1
Key Takeaways for Hospitalists
For the vast majority of medical patients, hospitalists can safely use a restrictive RBC transfusion threshold (≤7g/dL or ≤8g/dL), which can lead to a significant decrease in RBC transfusions without adversely affecting overall mortality.
Drs. Bortinger and Carbo are hospitalists at Beth Israel Deaconess Medical Center in Boston.
References
- Carson JL, Grossman BJ, Kleinman S, et al. Red blood cell transfusion: a clinical practice guideline from the AABB. Ann Inter Med. 2012;157(1):49-58.
- Carson AL, Terrin ML, Noveck H, et al. Liberal or restrictive transfusion in high-risk patients after hip surgery. N Engl J Med. 2011;367(26):2453-2462.
- Hajjar LA, Vincent JL, Galas FR, et al. Transfusion requirements after cardiac surgery: the TRACS randomized controlled trial. JAMA. 2010;304(14):1559-1567.
- Carson JL, Carless PA, Herbert PC. Transfusion threshold and other strategies for guiding allogenic red blood cell transfusion. Cochrane Database Syst Rev. 2012;CD002042.
- Napolitano LM, Kurek S, Luchette FA, et al. Clinical practice guideline: red blood cell transfusion in adult trauma and critical care. Crit Care Med. 2009;37(12):3124-3157.
Background
Hospitalists commonly order red blood cell (RBC) transfusion as a therapy for patients with anemia resulting from a variety of clinical conditions. There has been lack of consensus on when to transfuse, because patients with anemia frequently have multiple co-morbidities, including coronary artery disease and congestive heart failure, which may influence their ability to tolerate a potentially ischemic state related to anemia or to accommodate volume fluctuations related to transfusion.
Furthermore, RBC transfusions are not without inherent risk. Life-threatening transfusion reactions occur in approximately seven per million transfused blood components, and transfusion-associated circulatory overload (TACO) can develop in one in 100 transfusions.1
Recently published guidelines provide recommendations for management of hemodynamically stable adults with anemia.
Guideline Update
The AABB published guidelines in the Annals of Internal Medicine in 2012 addressing RBC transfusion thresholds.1 The updated guideline makes a recommendation that clinicians utilize a restrictive transfusion strategy. Transfusion is strongly recommended for ICU patients with hemoglobin ≤7g/dL. In post-operative surgical patients and for post-operative patients with symptomatic anemia, transfusion is recommended for hemoglobin ≤8g/dL. The authors also made a weak recommendation to transfuse for hemoglobin ≤8g/dL or for symptoms in hospitalized hemodynamically stable patients with preexisting cardiovascular disease.
These recommendations draw from past literature, along with two more recent trials examining liberal or restrictive transfusion thresholds. The newer trials increased the total number of patients studied by nearly one third compared with prior reviews.2,3 The authors also incorporated recently published systematic reviews in their analysis.
Although the definition of a restrictive transfusion threshold varied across trials, including hemoglobin ≤7g/dL and ≤8g/dL, the authors used the pooled data to provide several recommendations in the new guideline. Of note, the pooled data was underpowered to detect up to a twofold increase in risk of myocardial infarction in patients in the restrictive strategy group.1
There were insufficient data for the authors to recommend for or against a restrictive transfusion strategy in patients with acute coronary syndrome, based on very low quality evidence.
Finally, the authors recommended that symptoms and hemoglobin level should both be used in determining transfusion criteria, based on low quality of evidence.
Analysis
The current AABB guidelines have two primary differences from earlier guidelines. First, the AABB authors used GRADE (Grading of Recommendations, Assessment, Development, and Evaluation) methodology to formalize evidence-based practice in their analysis of the literature. The authors purposely used the GRADE methodology to systematically evaluate the quality of the evidence base and explicitly state the strength of the recommendation for a particular transfusion threshold.4
Second, the AABB guidelines incorporated data from the more recently published FOCUS (Functional Outcomes in Cardiovascular patients Undergoing Surgical repair of hip fracture) and TRACS (Transfusion Requirements After Cardiac Surgery) trials, resulting in a stronger recommendation supporting the use of a restrictive transfusion strategy in non-ICU and post-operative patients. The findings of the FOCUS trial are especially applicable to hospitalists, because many patients who undergo hip fracture repair are directly cared for or are co-managed by hospitalists.
The current guidelines built upon previous guidelines that advocated a restrictive strategy (hemoglobin ≤7g/dL) in hemodynamically stable, critically ill adult patients.5 In general, restrictive transfusion strategy led to nearly 40% fewer patients receiving transfusion compared with the use of a liberal transfusion strategy.1 No additional harm to patients was evidenced in the restrictive transfusion group, though the trials were not designed to answer this question; moreover, there was no statistically significant difference in mortality or functional outcome between the two groups.
The authors of the current AABB guidelines recognized the importance of replicating the current findings in a more diverse patient population. An area where further study is indicated is in the use of specific transfusion thresholds in patients with acute coronary syndrome. These guidelines did not clarify whether or not there is a physiologic difference between use of different restrictive transfusion thresholds such as <8g/dL and <7g/dL.
The authors of the AABB guidelines also commented that performing a future trial to compare RBC transfusion for symptoms vs. hemoglobin “trigger” would be useful; however, they recognized that this may not be feasible due to the need to blind providers in the trial to hemoglobin values. Various society guidelines currently call for different transfusion thresholds or do not make a specific recommendation at all.1
Key Takeaways for Hospitalists
For the vast majority of medical patients, hospitalists can safely use a restrictive RBC transfusion threshold (≤7g/dL or ≤8g/dL), which can lead to a significant decrease in RBC transfusions without adversely affecting overall mortality.
Drs. Bortinger and Carbo are hospitalists at Beth Israel Deaconess Medical Center in Boston.
References
- Carson JL, Grossman BJ, Kleinman S, et al. Red blood cell transfusion: a clinical practice guideline from the AABB. Ann Inter Med. 2012;157(1):49-58.
- Carson AL, Terrin ML, Noveck H, et al. Liberal or restrictive transfusion in high-risk patients after hip surgery. N Engl J Med. 2011;367(26):2453-2462.
- Hajjar LA, Vincent JL, Galas FR, et al. Transfusion requirements after cardiac surgery: the TRACS randomized controlled trial. JAMA. 2010;304(14):1559-1567.
- Carson JL, Carless PA, Herbert PC. Transfusion threshold and other strategies for guiding allogenic red blood cell transfusion. Cochrane Database Syst Rev. 2012;CD002042.
- Napolitano LM, Kurek S, Luchette FA, et al. Clinical practice guideline: red blood cell transfusion in adult trauma and critical care. Crit Care Med. 2009;37(12):3124-3157.
Background
Hospitalists commonly order red blood cell (RBC) transfusion as a therapy for patients with anemia resulting from a variety of clinical conditions. There has been lack of consensus on when to transfuse, because patients with anemia frequently have multiple co-morbidities, including coronary artery disease and congestive heart failure, which may influence their ability to tolerate a potentially ischemic state related to anemia or to accommodate volume fluctuations related to transfusion.
Furthermore, RBC transfusions are not without inherent risk. Life-threatening transfusion reactions occur in approximately seven per million transfused blood components, and transfusion-associated circulatory overload (TACO) can develop in one in 100 transfusions.1
Recently published guidelines provide recommendations for management of hemodynamically stable adults with anemia.
Guideline Update
The AABB published guidelines in the Annals of Internal Medicine in 2012 addressing RBC transfusion thresholds.1 The updated guideline makes a recommendation that clinicians utilize a restrictive transfusion strategy. Transfusion is strongly recommended for ICU patients with hemoglobin ≤7g/dL. In post-operative surgical patients and for post-operative patients with symptomatic anemia, transfusion is recommended for hemoglobin ≤8g/dL. The authors also made a weak recommendation to transfuse for hemoglobin ≤8g/dL or for symptoms in hospitalized hemodynamically stable patients with preexisting cardiovascular disease.
These recommendations draw from past literature, along with two more recent trials examining liberal or restrictive transfusion thresholds. The newer trials increased the total number of patients studied by nearly one third compared with prior reviews.2,3 The authors also incorporated recently published systematic reviews in their analysis.
Although the definition of a restrictive transfusion threshold varied across trials, including hemoglobin ≤7g/dL and ≤8g/dL, the authors used the pooled data to provide several recommendations in the new guideline. Of note, the pooled data was underpowered to detect up to a twofold increase in risk of myocardial infarction in patients in the restrictive strategy group.1
There were insufficient data for the authors to recommend for or against a restrictive transfusion strategy in patients with acute coronary syndrome, based on very low quality evidence.
Finally, the authors recommended that symptoms and hemoglobin level should both be used in determining transfusion criteria, based on low quality of evidence.
Analysis
The current AABB guidelines have two primary differences from earlier guidelines. First, the AABB authors used GRADE (Grading of Recommendations, Assessment, Development, and Evaluation) methodology to formalize evidence-based practice in their analysis of the literature. The authors purposely used the GRADE methodology to systematically evaluate the quality of the evidence base and explicitly state the strength of the recommendation for a particular transfusion threshold.4
Second, the AABB guidelines incorporated data from the more recently published FOCUS (Functional Outcomes in Cardiovascular patients Undergoing Surgical repair of hip fracture) and TRACS (Transfusion Requirements After Cardiac Surgery) trials, resulting in a stronger recommendation supporting the use of a restrictive transfusion strategy in non-ICU and post-operative patients. The findings of the FOCUS trial are especially applicable to hospitalists, because many patients who undergo hip fracture repair are directly cared for or are co-managed by hospitalists.
The current guidelines built upon previous guidelines that advocated a restrictive strategy (hemoglobin ≤7g/dL) in hemodynamically stable, critically ill adult patients.5 In general, restrictive transfusion strategy led to nearly 40% fewer patients receiving transfusion compared with the use of a liberal transfusion strategy.1 No additional harm to patients was evidenced in the restrictive transfusion group, though the trials were not designed to answer this question; moreover, there was no statistically significant difference in mortality or functional outcome between the two groups.
The authors of the current AABB guidelines recognized the importance of replicating the current findings in a more diverse patient population. An area where further study is indicated is in the use of specific transfusion thresholds in patients with acute coronary syndrome. These guidelines did not clarify whether or not there is a physiologic difference between use of different restrictive transfusion thresholds such as <8g/dL and <7g/dL.
The authors of the AABB guidelines also commented that performing a future trial to compare RBC transfusion for symptoms vs. hemoglobin “trigger” would be useful; however, they recognized that this may not be feasible due to the need to blind providers in the trial to hemoglobin values. Various society guidelines currently call for different transfusion thresholds or do not make a specific recommendation at all.1
Key Takeaways for Hospitalists
For the vast majority of medical patients, hospitalists can safely use a restrictive RBC transfusion threshold (≤7g/dL or ≤8g/dL), which can lead to a significant decrease in RBC transfusions without adversely affecting overall mortality.
Drs. Bortinger and Carbo are hospitalists at Beth Israel Deaconess Medical Center in Boston.
References
- Carson JL, Grossman BJ, Kleinman S, et al. Red blood cell transfusion: a clinical practice guideline from the AABB. Ann Inter Med. 2012;157(1):49-58.
- Carson AL, Terrin ML, Noveck H, et al. Liberal or restrictive transfusion in high-risk patients after hip surgery. N Engl J Med. 2011;367(26):2453-2462.
- Hajjar LA, Vincent JL, Galas FR, et al. Transfusion requirements after cardiac surgery: the TRACS randomized controlled trial. JAMA. 2010;304(14):1559-1567.
- Carson JL, Carless PA, Herbert PC. Transfusion threshold and other strategies for guiding allogenic red blood cell transfusion. Cochrane Database Syst Rev. 2012;CD002042.
- Napolitano LM, Kurek S, Luchette FA, et al. Clinical practice guideline: red blood cell transfusion in adult trauma and critical care. Crit Care Med. 2009;37(12):3124-3157.
Which Patients Should be Screened for Hepatitis C Virus Infection?
Case
A 65-year-old male with a history of a motor vehicle accident that required emergency surgery in 1982 is hospitalized for acute renal failure. He reports a distant history of IV heroin use and a brief incarceration. He does not currently use illicit drugs. He has no signs or symptoms of liver disease. Should this patient be screened for chronic hepatitis C virus (HCV) infection?
Brief Overview
HCV is a major public health concern in the United States and worldwide. It is estimated that more than 4.1 million people in the U.S. (1.6% prevalence) and more than 180 million worldwide (2.8% prevalence) are HCV antibody-positive.1,2 The acute infection is most often asymptomatic, and 80% to 100% of patients will remain HCV RNA-positive, 60% to 80% will have persistently elevated liver enzymes, and 16% will develop evidence of cirrhosis at 20 years after initial infection.3
A number of organizations in the United States have released HCV screening guidelines, including the CDC, the American Association for the Study of Liver Disease (AASLD), and the U.S. Preventive Services Task Force (USPSTF); however, despite these established recommendations, an estimated 50% of individuals with chronic HCV infection are unscreened and unaware of their infection status.4 Furthermore, in a recent study of one managed care network, even when one or more risk factors were present, only 29% of individuals underwent screening for HCV antibodies detection.5 The importance of detecting chronic HCV infection will have greater significance as newer and better-tolerated treatment options become available.6
Multiple organizations recommend screening for chronic HCV infection. This screening is recommended for patients with known risk factors and those in populations with a high prevalence of HCV infection.
Risk Factors and High-Prevalence Populations
IV or intranasal drug use. IV drug use is the main identifiable source of HCV infection in the U.S. It is estimated that 60% of new HCV infections occur in people who have injected drugs in the past six months.7 The prevalence of HCV antibodies in current IV drug users is between 72% and 96%.8 Intranasal cocaine use is also associated with a higher prevalence of HCV antibodies than the general population.8
Blood transfusion prior to July 1992. Testing of donor blood was not routinely done until 1990, and more sensitive testing was not implemented until July 1992.8 The prevalence of HCV antibodies in people who received blood transfusions prior to 1990 is 6%.8 Prior to 1990, the risk for transfusion-associated HCV infection was one in 526 units transfused.9 Since implementation of highly sensitive screening techniques, the risk of infection has dropped to less than one in 1.9 million units transfused.10
Clotting factors prior to 1987 or transplanted tissue prior to 1992. Individuals who have received clotting factors, other blood product transfusions, or transplanted tissue prior to 1987 are at an increased risk for developing HCV infection. For instance, individuals with hemophilia treated with clotting factors prior to 1987 had chronic HCV infection rates of up to 90%.8 In 1987, widespread use of protocols to inactivate HCV in clotting factors and other blood products was adopted.8 In addition, widespread screening of potential tissue donors and the use of HCV antibody-negative donors became routine.8
Alanine aminotransferase elevation. This can be considered screening or part of the diagnostic work-up of transaminitis. Regardless of the classification, this is a cohort of people with a high prevalence of HCV antibody. For individuals with one isolated alanine aminotransferase elevation, the prevalence is 3.2%.4 With two or more elevated aminotransferase results, the prevalence rises to 8.2%.4
Hemodialysis. Two major studies have estimated the prevalence of HCV antibody-positive in end-stage renal disease individuals on hemodialysis to be 7.8% and 10.4%.11,12 This prevalence can reach 64% at some dialysis centers.11 The risk of HCV infection has been associated with blood transfusions, longer duration of hemodialysis, and higher rates of HCV infection in the dialysis unit.13 With implementation of infection control practices in dialysis units, the incidence and prevalence of HCV infection are declining.13
Born in the U.S. between 1945 and 1965. The CDC and USPSTF recommend a one-time screening for HCV infection for people born in the U.S. between 1945 and 1965, regardless of the presence or absence of risk factors.6,14 This age group has an increased prevalence of HCV antibodies, at 3.25%.6
Human immunodeficiency virus (HIV). HCV has a prevalence of 30% in people infected with HIV.15 The rate of co-infection is likely secondary to shared routes of transmission. For example, 72.7% of HIV-infected individuals who used IV drugs had HCV antibodies, but only 3.5% of “low-risk” HIV-infected individuals had HCV antibodies.16
Born in a high prevalence country. In the U.S., a significant number of immigrants are from areas with a high endemic rate of HCV infection. High prevalence areas (greater than 3.5%) include Central Asia and East Asia, North Africa, and the Middle East.7 Of note, Egypt is thought to have the highest prevalence of chronic HCV infection in the world, with well over 10% of the population being antibody-positive.17 Although major guidelines do not currently recommend it, the high prevalence of chronic HCV infection in this population may warrant screening.
Other high-risk or high-prevalence populations. The prevalence of HCV infection in people who have had over 10 lifetime sexual partners (3% to 9%), those with a history of sexually transmitted disease (6%), men who have had sex with men (5%), and children born to HCV-infected mothers (5%) is increased compared with the general population.8 Incarcerated people in the U.S. have an HCV antibody prevalence of 16% to 41%.18 In addition, people who have sustained needle-stick injury or mucosal exposure, or those with potential exposures in unregulated tattoo or piercing salons, may also benefit from HCV antibody screening.14
Table 1 reviews HCV screening recommendations for the CDC, AASLD, and USPSTF.1,6,8,14
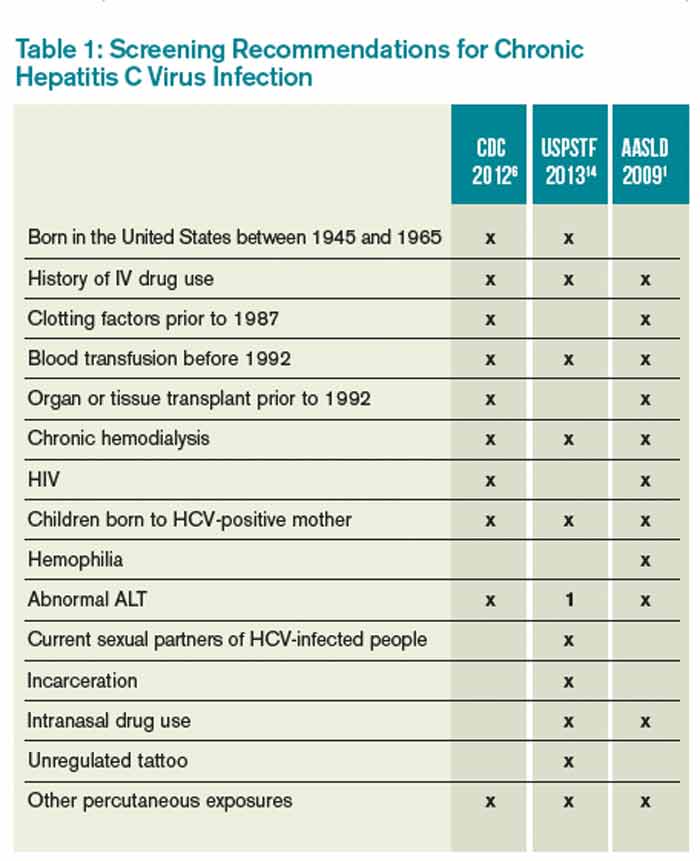
CDC=Centers for Disease Control and Prevention; USPSTF=U.S. Preventive Services Task Force; AASLD=American Association for the Study of Liver Disease; ALT=alanine aminotransferase; 1=considered diagnostic and not screening test
Screening Method
The most common initial screening test for the diagnosis of chronic HCV infection is the HCV antibody test. A positive antibody test should be followed by an HCV RNA test. In an individual with recent exposure, it takes between four and 10 weeks for the antibody to be detectable. HCV RNA testing can be positive as soon as two to three weeks after infection.8
Hospitalist Role in HCV Screening
None of the U.S.-based guidelines make recommendations on the preferred setting for HCV screening. According to the CDC, 60.4% of HCV screening was done in a physician office and 5.9% was done as a hospital inpatient.19 Traditionally, the PCP is responsible for screening for chronic diseases, including HCV infection; however, the current screening rate is insufficient, as 50% of people with chronic HCV infection remain unscreened.4
Given the insufficient rate of HCV screening at present, hospital medicine (HM) physicians have an opportunity to help improve this rate. Currently, there is no established standard of care for HCV screening in hospitalized patients. HM physicians could use the following strategies:
- Continue the current system and defer screening to outpatient providers;
- Offer screening to selected inpatients at high risk for chronic HCV infection; or
- Offer screening to all inpatients who meet screening criteria based on current guidelines.
Given the shortcomings of the current screening strategies, these authors would recommend widespread screening for chronic HCV infection in hospitalized people who meet screening criteria per current guidelines.
If HM physicians are to take an increased role in HCV screening, there are a number of important considerations. Because hospitalized patients have a limited length of stay, it would be unreasonable to expect HM physicians to test for HCV RNA viral load or genotype for all patients with a positive antibody test, because the duration of the inpatient stay may be shorter than the time it takes for these test results to return. These tests are often indicated after a positive HCV antibody test, however. Thus, communication of HCV antibody results to PCPs or other responsible providers is essential. If no follow-up is available or there are no responsible outpatient providers, HM physicians should continue with a limited screening strategy.
Back to the Case
This individual has multiple indications for chronic HCV infection screening. His risk factors include date of birth between 1945 and 1965, a history of IV drug use, and a history of incarceration. He also notes a history of emergency surgery, for which he may have received blood products prior to 1987. These factors significantly raise the likelihood of chronic HCV infection when compared with the general population. He was screened and found to be HCV antibody-positive. A follow-up HCV RNA viral load was also positive. He did not have any evidence of liver disease but did have a mild transaminitis. He has followed up as an outpatient with plans to start therapy.
Bottom Line
The current screening strategies for individuals with high prevalence of chronic HCV infection are insufficient. HM physicians have an opportunity to improve the rates of screening in this population.
Dr. Theisen-Toupal is an internist, Dr. Rosenthal is a clinical fellow in medicine, and Dr. Carbo is an assistant professor of medicine, all at Beth Israel Deaconess Medical Center in Boston. Dr. Li is an internist and associate professor of medicine at Harvard Medical School and director of the hospital medicine division at Beth Israel Deaconess Medical Center.
References
- Ghany MG, Strader DB, Thomas DL, Seeff LB; American Association for the Study of Liver Diseases. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49(4):1335-1374.
- Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology. 2013;57(4):1333-1342.
- Chopra S. Clinical manifestations and natural history of chronic hepatitis C virus infection. UpToDate. Available at: http://www.uptodate.com/contents/clinical-manifestations-and-natural-history-of-chronic-hepatitis-c-virus-infection. Accessed March 5, 2014.
- Spradling PR, Rupp L, Moorman AC, et al. Hepatitis B and C virus infection among 1.2 million people with access to care: factors associated with testing and infection prevalence. Clin Infect Dis. 2012;55(8):1047-1055.
- Roblin DW, Smith BD, Weinbaum CM, Sabin ME. HCV screening practices and prevalence in an MCO, 2000-2007. Am J Manag Care. 2011;17(8):548-555.
- Centers for Disease Control and Prevention. Recommendations for the identification of chronic hepatitis C virus infection among people born during 1945-1965. MMWR. August 17, 2012. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr6104a1.htm. Accessed March 5, 2014.
- Chopra S. Epidemiology and transmission of hepatitis C virus infection. UpToDate. Available at: http://www.uptodate.com/contents/epidemiology-and-transmission-of-hepatitis-c-virus-infection?source=search_result&search=%22Epidemiology+and+transmission+of+hepatitis+C+virus+infection%22&selectedTitle=1~150. Accessed March 5, 2014.
- Centers for Disease Control and Prevention. Recommendations for prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. MMWR. October 16, 1998. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/00055154.htm. Accessed March 5, 2014.
- Donahue JG, Muñoz A, Ness PM, et al. The declining risk of post-transfusion hepatitis C virus infection. N Engl J Med. 1992;327(6):369-373.
- Pomper GJ, Wu Y, Snyder EL. Risks of transfusion-transmitted infections: 2003. Curr Opin Hematol. 2003;10(6):412-418.
- Tokars JI, Miller ER, Alter MJ, Arduino MJ. National surveillance of dialysis associated diseases in the United States, 1995. ASAIO J. 1998;44(1):98-107.
- Finelli L, Miller JT, Tokars JI, Alter MJ, Arduino MJ. National surveillance of dialysis-associated diseases in the United States, 2002. Semin Dial. 2005;18(1):52-61.
- Natov S, Pereira BJG. Hepatitis C virus infection in patients on maintenance dialysis. UpToDate. Available at: http://www.uptodate.com/contents/hepatitis-c-virus-infection-in-patients-on-maintenance-dialysis?source=search_result&search=Hepatitis+C+virus+infection+in+patients+on+maintenance+dialysis.&selectedTitle=1~150. Accessed March 5, 2014.
- Moyer VA, U.S. Preventive Services Task Force. Screening for hepatitis C virus infection in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2013;159(5):349-357.
- Staples CT II, Rimland D, Dudas D. Hepatitis C in the HIV (human immunodeficiency virus) Atlanta V.A. (Veterans Affairs Medical Center) Cohort Study (HAVACS): the effect of coinfection on survival. Clin Infect Dis. 1999;29(1):150-154.
- Sherman KE, Rouster SD, Chung RT, Rajicic N. Hepatitis C virus prevalence among patients infected with human immunodeficiency virus: a cross-sectional analysis of the U.S. adult AIDS clinical trials group. Clin Infect Dis. 2002;34(6):831-837.
- Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the United States. Clin Infect Dis. 2012;55 Suppl 1:S10-15.
- Centers for Disease Control and Prevention. Prevention and control of infections with hepatitis viruses in correctional settings. MMWR. January 24, 2003. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5201a1.htm. Accessed March 5, 2014.
- Centers for Disease Control and Prevention. Locations and reasons for initial testing for hepatitis C infection—chronic hepatitis cohort study, United States, 2006-2010. MMWR. August 16, 2013. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6232a3.htm?s_cid=mm6232a3_w. Accessed March 5, 2014.
Case
A 65-year-old male with a history of a motor vehicle accident that required emergency surgery in 1982 is hospitalized for acute renal failure. He reports a distant history of IV heroin use and a brief incarceration. He does not currently use illicit drugs. He has no signs or symptoms of liver disease. Should this patient be screened for chronic hepatitis C virus (HCV) infection?
Brief Overview
HCV is a major public health concern in the United States and worldwide. It is estimated that more than 4.1 million people in the U.S. (1.6% prevalence) and more than 180 million worldwide (2.8% prevalence) are HCV antibody-positive.1,2 The acute infection is most often asymptomatic, and 80% to 100% of patients will remain HCV RNA-positive, 60% to 80% will have persistently elevated liver enzymes, and 16% will develop evidence of cirrhosis at 20 years after initial infection.3
A number of organizations in the United States have released HCV screening guidelines, including the CDC, the American Association for the Study of Liver Disease (AASLD), and the U.S. Preventive Services Task Force (USPSTF); however, despite these established recommendations, an estimated 50% of individuals with chronic HCV infection are unscreened and unaware of their infection status.4 Furthermore, in a recent study of one managed care network, even when one or more risk factors were present, only 29% of individuals underwent screening for HCV antibodies detection.5 The importance of detecting chronic HCV infection will have greater significance as newer and better-tolerated treatment options become available.6
Multiple organizations recommend screening for chronic HCV infection. This screening is recommended for patients with known risk factors and those in populations with a high prevalence of HCV infection.
Risk Factors and High-Prevalence Populations
IV or intranasal drug use. IV drug use is the main identifiable source of HCV infection in the U.S. It is estimated that 60% of new HCV infections occur in people who have injected drugs in the past six months.7 The prevalence of HCV antibodies in current IV drug users is between 72% and 96%.8 Intranasal cocaine use is also associated with a higher prevalence of HCV antibodies than the general population.8
Blood transfusion prior to July 1992. Testing of donor blood was not routinely done until 1990, and more sensitive testing was not implemented until July 1992.8 The prevalence of HCV antibodies in people who received blood transfusions prior to 1990 is 6%.8 Prior to 1990, the risk for transfusion-associated HCV infection was one in 526 units transfused.9 Since implementation of highly sensitive screening techniques, the risk of infection has dropped to less than one in 1.9 million units transfused.10
Clotting factors prior to 1987 or transplanted tissue prior to 1992. Individuals who have received clotting factors, other blood product transfusions, or transplanted tissue prior to 1987 are at an increased risk for developing HCV infection. For instance, individuals with hemophilia treated with clotting factors prior to 1987 had chronic HCV infection rates of up to 90%.8 In 1987, widespread use of protocols to inactivate HCV in clotting factors and other blood products was adopted.8 In addition, widespread screening of potential tissue donors and the use of HCV antibody-negative donors became routine.8
Alanine aminotransferase elevation. This can be considered screening or part of the diagnostic work-up of transaminitis. Regardless of the classification, this is a cohort of people with a high prevalence of HCV antibody. For individuals with one isolated alanine aminotransferase elevation, the prevalence is 3.2%.4 With two or more elevated aminotransferase results, the prevalence rises to 8.2%.4
Hemodialysis. Two major studies have estimated the prevalence of HCV antibody-positive in end-stage renal disease individuals on hemodialysis to be 7.8% and 10.4%.11,12 This prevalence can reach 64% at some dialysis centers.11 The risk of HCV infection has been associated with blood transfusions, longer duration of hemodialysis, and higher rates of HCV infection in the dialysis unit.13 With implementation of infection control practices in dialysis units, the incidence and prevalence of HCV infection are declining.13
Born in the U.S. between 1945 and 1965. The CDC and USPSTF recommend a one-time screening for HCV infection for people born in the U.S. between 1945 and 1965, regardless of the presence or absence of risk factors.6,14 This age group has an increased prevalence of HCV antibodies, at 3.25%.6
Human immunodeficiency virus (HIV). HCV has a prevalence of 30% in people infected with HIV.15 The rate of co-infection is likely secondary to shared routes of transmission. For example, 72.7% of HIV-infected individuals who used IV drugs had HCV antibodies, but only 3.5% of “low-risk” HIV-infected individuals had HCV antibodies.16
Born in a high prevalence country. In the U.S., a significant number of immigrants are from areas with a high endemic rate of HCV infection. High prevalence areas (greater than 3.5%) include Central Asia and East Asia, North Africa, and the Middle East.7 Of note, Egypt is thought to have the highest prevalence of chronic HCV infection in the world, with well over 10% of the population being antibody-positive.17 Although major guidelines do not currently recommend it, the high prevalence of chronic HCV infection in this population may warrant screening.
Other high-risk or high-prevalence populations. The prevalence of HCV infection in people who have had over 10 lifetime sexual partners (3% to 9%), those with a history of sexually transmitted disease (6%), men who have had sex with men (5%), and children born to HCV-infected mothers (5%) is increased compared with the general population.8 Incarcerated people in the U.S. have an HCV antibody prevalence of 16% to 41%.18 In addition, people who have sustained needle-stick injury or mucosal exposure, or those with potential exposures in unregulated tattoo or piercing salons, may also benefit from HCV antibody screening.14
Table 1 reviews HCV screening recommendations for the CDC, AASLD, and USPSTF.1,6,8,14
CDC=Centers for Disease Control and Prevention; USPSTF=U.S. Preventive Services Task Force; AASLD=American Association for the Study of Liver Disease; ALT=alanine aminotransferase; 1=considered diagnostic and not screening test
Screening Method
The most common initial screening test for the diagnosis of chronic HCV infection is the HCV antibody test. A positive antibody test should be followed by an HCV RNA test. In an individual with recent exposure, it takes between four and 10 weeks for the antibody to be detectable. HCV RNA testing can be positive as soon as two to three weeks after infection.8
Hospitalist Role in HCV Screening
None of the U.S.-based guidelines make recommendations on the preferred setting for HCV screening. According to the CDC, 60.4% of HCV screening was done in a physician office and 5.9% was done as a hospital inpatient.19 Traditionally, the PCP is responsible for screening for chronic diseases, including HCV infection; however, the current screening rate is insufficient, as 50% of people with chronic HCV infection remain unscreened.4
Given the insufficient rate of HCV screening at present, hospital medicine (HM) physicians have an opportunity to help improve this rate. Currently, there is no established standard of care for HCV screening in hospitalized patients. HM physicians could use the following strategies:
- Continue the current system and defer screening to outpatient providers;
- Offer screening to selected inpatients at high risk for chronic HCV infection; or
- Offer screening to all inpatients who meet screening criteria based on current guidelines.
Given the shortcomings of the current screening strategies, these authors would recommend widespread screening for chronic HCV infection in hospitalized people who meet screening criteria per current guidelines.
If HM physicians are to take an increased role in HCV screening, there are a number of important considerations. Because hospitalized patients have a limited length of stay, it would be unreasonable to expect HM physicians to test for HCV RNA viral load or genotype for all patients with a positive antibody test, because the duration of the inpatient stay may be shorter than the time it takes for these test results to return. These tests are often indicated after a positive HCV antibody test, however. Thus, communication of HCV antibody results to PCPs or other responsible providers is essential. If no follow-up is available or there are no responsible outpatient providers, HM physicians should continue with a limited screening strategy.
Back to the Case
This individual has multiple indications for chronic HCV infection screening. His risk factors include date of birth between 1945 and 1965, a history of IV drug use, and a history of incarceration. He also notes a history of emergency surgery, for which he may have received blood products prior to 1987. These factors significantly raise the likelihood of chronic HCV infection when compared with the general population. He was screened and found to be HCV antibody-positive. A follow-up HCV RNA viral load was also positive. He did not have any evidence of liver disease but did have a mild transaminitis. He has followed up as an outpatient with plans to start therapy.
Bottom Line
The current screening strategies for individuals with high prevalence of chronic HCV infection are insufficient. HM physicians have an opportunity to improve the rates of screening in this population.
Dr. Theisen-Toupal is an internist, Dr. Rosenthal is a clinical fellow in medicine, and Dr. Carbo is an assistant professor of medicine, all at Beth Israel Deaconess Medical Center in Boston. Dr. Li is an internist and associate professor of medicine at Harvard Medical School and director of the hospital medicine division at Beth Israel Deaconess Medical Center.
References
- Ghany MG, Strader DB, Thomas DL, Seeff LB; American Association for the Study of Liver Diseases. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49(4):1335-1374.
- Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology. 2013;57(4):1333-1342.
- Chopra S. Clinical manifestations and natural history of chronic hepatitis C virus infection. UpToDate. Available at: http://www.uptodate.com/contents/clinical-manifestations-and-natural-history-of-chronic-hepatitis-c-virus-infection. Accessed March 5, 2014.
- Spradling PR, Rupp L, Moorman AC, et al. Hepatitis B and C virus infection among 1.2 million people with access to care: factors associated with testing and infection prevalence. Clin Infect Dis. 2012;55(8):1047-1055.
- Roblin DW, Smith BD, Weinbaum CM, Sabin ME. HCV screening practices and prevalence in an MCO, 2000-2007. Am J Manag Care. 2011;17(8):548-555.
- Centers for Disease Control and Prevention. Recommendations for the identification of chronic hepatitis C virus infection among people born during 1945-1965. MMWR. August 17, 2012. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr6104a1.htm. Accessed March 5, 2014.
- Chopra S. Epidemiology and transmission of hepatitis C virus infection. UpToDate. Available at: http://www.uptodate.com/contents/epidemiology-and-transmission-of-hepatitis-c-virus-infection?source=search_result&search=%22Epidemiology+and+transmission+of+hepatitis+C+virus+infection%22&selectedTitle=1~150. Accessed March 5, 2014.
- Centers for Disease Control and Prevention. Recommendations for prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. MMWR. October 16, 1998. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/00055154.htm. Accessed March 5, 2014.
- Donahue JG, Muñoz A, Ness PM, et al. The declining risk of post-transfusion hepatitis C virus infection. N Engl J Med. 1992;327(6):369-373.
- Pomper GJ, Wu Y, Snyder EL. Risks of transfusion-transmitted infections: 2003. Curr Opin Hematol. 2003;10(6):412-418.
- Tokars JI, Miller ER, Alter MJ, Arduino MJ. National surveillance of dialysis associated diseases in the United States, 1995. ASAIO J. 1998;44(1):98-107.
- Finelli L, Miller JT, Tokars JI, Alter MJ, Arduino MJ. National surveillance of dialysis-associated diseases in the United States, 2002. Semin Dial. 2005;18(1):52-61.
- Natov S, Pereira BJG. Hepatitis C virus infection in patients on maintenance dialysis. UpToDate. Available at: http://www.uptodate.com/contents/hepatitis-c-virus-infection-in-patients-on-maintenance-dialysis?source=search_result&search=Hepatitis+C+virus+infection+in+patients+on+maintenance+dialysis.&selectedTitle=1~150. Accessed March 5, 2014.
- Moyer VA, U.S. Preventive Services Task Force. Screening for hepatitis C virus infection in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2013;159(5):349-357.
- Staples CT II, Rimland D, Dudas D. Hepatitis C in the HIV (human immunodeficiency virus) Atlanta V.A. (Veterans Affairs Medical Center) Cohort Study (HAVACS): the effect of coinfection on survival. Clin Infect Dis. 1999;29(1):150-154.
- Sherman KE, Rouster SD, Chung RT, Rajicic N. Hepatitis C virus prevalence among patients infected with human immunodeficiency virus: a cross-sectional analysis of the U.S. adult AIDS clinical trials group. Clin Infect Dis. 2002;34(6):831-837.
- Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the United States. Clin Infect Dis. 2012;55 Suppl 1:S10-15.
- Centers for Disease Control and Prevention. Prevention and control of infections with hepatitis viruses in correctional settings. MMWR. January 24, 2003. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5201a1.htm. Accessed March 5, 2014.
- Centers for Disease Control and Prevention. Locations and reasons for initial testing for hepatitis C infection—chronic hepatitis cohort study, United States, 2006-2010. MMWR. August 16, 2013. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6232a3.htm?s_cid=mm6232a3_w. Accessed March 5, 2014.
Case
A 65-year-old male with a history of a motor vehicle accident that required emergency surgery in 1982 is hospitalized for acute renal failure. He reports a distant history of IV heroin use and a brief incarceration. He does not currently use illicit drugs. He has no signs or symptoms of liver disease. Should this patient be screened for chronic hepatitis C virus (HCV) infection?
Brief Overview
HCV is a major public health concern in the United States and worldwide. It is estimated that more than 4.1 million people in the U.S. (1.6% prevalence) and more than 180 million worldwide (2.8% prevalence) are HCV antibody-positive.1,2 The acute infection is most often asymptomatic, and 80% to 100% of patients will remain HCV RNA-positive, 60% to 80% will have persistently elevated liver enzymes, and 16% will develop evidence of cirrhosis at 20 years after initial infection.3
A number of organizations in the United States have released HCV screening guidelines, including the CDC, the American Association for the Study of Liver Disease (AASLD), and the U.S. Preventive Services Task Force (USPSTF); however, despite these established recommendations, an estimated 50% of individuals with chronic HCV infection are unscreened and unaware of their infection status.4 Furthermore, in a recent study of one managed care network, even when one or more risk factors were present, only 29% of individuals underwent screening for HCV antibodies detection.5 The importance of detecting chronic HCV infection will have greater significance as newer and better-tolerated treatment options become available.6
Multiple organizations recommend screening for chronic HCV infection. This screening is recommended for patients with known risk factors and those in populations with a high prevalence of HCV infection.
Risk Factors and High-Prevalence Populations
IV or intranasal drug use. IV drug use is the main identifiable source of HCV infection in the U.S. It is estimated that 60% of new HCV infections occur in people who have injected drugs in the past six months.7 The prevalence of HCV antibodies in current IV drug users is between 72% and 96%.8 Intranasal cocaine use is also associated with a higher prevalence of HCV antibodies than the general population.8
Blood transfusion prior to July 1992. Testing of donor blood was not routinely done until 1990, and more sensitive testing was not implemented until July 1992.8 The prevalence of HCV antibodies in people who received blood transfusions prior to 1990 is 6%.8 Prior to 1990, the risk for transfusion-associated HCV infection was one in 526 units transfused.9 Since implementation of highly sensitive screening techniques, the risk of infection has dropped to less than one in 1.9 million units transfused.10
Clotting factors prior to 1987 or transplanted tissue prior to 1992. Individuals who have received clotting factors, other blood product transfusions, or transplanted tissue prior to 1987 are at an increased risk for developing HCV infection. For instance, individuals with hemophilia treated with clotting factors prior to 1987 had chronic HCV infection rates of up to 90%.8 In 1987, widespread use of protocols to inactivate HCV in clotting factors and other blood products was adopted.8 In addition, widespread screening of potential tissue donors and the use of HCV antibody-negative donors became routine.8
Alanine aminotransferase elevation. This can be considered screening or part of the diagnostic work-up of transaminitis. Regardless of the classification, this is a cohort of people with a high prevalence of HCV antibody. For individuals with one isolated alanine aminotransferase elevation, the prevalence is 3.2%.4 With two or more elevated aminotransferase results, the prevalence rises to 8.2%.4
Hemodialysis. Two major studies have estimated the prevalence of HCV antibody-positive in end-stage renal disease individuals on hemodialysis to be 7.8% and 10.4%.11,12 This prevalence can reach 64% at some dialysis centers.11 The risk of HCV infection has been associated with blood transfusions, longer duration of hemodialysis, and higher rates of HCV infection in the dialysis unit.13 With implementation of infection control practices in dialysis units, the incidence and prevalence of HCV infection are declining.13
Born in the U.S. between 1945 and 1965. The CDC and USPSTF recommend a one-time screening for HCV infection for people born in the U.S. between 1945 and 1965, regardless of the presence or absence of risk factors.6,14 This age group has an increased prevalence of HCV antibodies, at 3.25%.6
Human immunodeficiency virus (HIV). HCV has a prevalence of 30% in people infected with HIV.15 The rate of co-infection is likely secondary to shared routes of transmission. For example, 72.7% of HIV-infected individuals who used IV drugs had HCV antibodies, but only 3.5% of “low-risk” HIV-infected individuals had HCV antibodies.16
Born in a high prevalence country. In the U.S., a significant number of immigrants are from areas with a high endemic rate of HCV infection. High prevalence areas (greater than 3.5%) include Central Asia and East Asia, North Africa, and the Middle East.7 Of note, Egypt is thought to have the highest prevalence of chronic HCV infection in the world, with well over 10% of the population being antibody-positive.17 Although major guidelines do not currently recommend it, the high prevalence of chronic HCV infection in this population may warrant screening.
Other high-risk or high-prevalence populations. The prevalence of HCV infection in people who have had over 10 lifetime sexual partners (3% to 9%), those with a history of sexually transmitted disease (6%), men who have had sex with men (5%), and children born to HCV-infected mothers (5%) is increased compared with the general population.8 Incarcerated people in the U.S. have an HCV antibody prevalence of 16% to 41%.18 In addition, people who have sustained needle-stick injury or mucosal exposure, or those with potential exposures in unregulated tattoo or piercing salons, may also benefit from HCV antibody screening.14
Table 1 reviews HCV screening recommendations for the CDC, AASLD, and USPSTF.1,6,8,14
CDC=Centers for Disease Control and Prevention; USPSTF=U.S. Preventive Services Task Force; AASLD=American Association for the Study of Liver Disease; ALT=alanine aminotransferase; 1=considered diagnostic and not screening test
Screening Method
The most common initial screening test for the diagnosis of chronic HCV infection is the HCV antibody test. A positive antibody test should be followed by an HCV RNA test. In an individual with recent exposure, it takes between four and 10 weeks for the antibody to be detectable. HCV RNA testing can be positive as soon as two to three weeks after infection.8
Hospitalist Role in HCV Screening
None of the U.S.-based guidelines make recommendations on the preferred setting for HCV screening. According to the CDC, 60.4% of HCV screening was done in a physician office and 5.9% was done as a hospital inpatient.19 Traditionally, the PCP is responsible for screening for chronic diseases, including HCV infection; however, the current screening rate is insufficient, as 50% of people with chronic HCV infection remain unscreened.4
Given the insufficient rate of HCV screening at present, hospital medicine (HM) physicians have an opportunity to help improve this rate. Currently, there is no established standard of care for HCV screening in hospitalized patients. HM physicians could use the following strategies:
- Continue the current system and defer screening to outpatient providers;
- Offer screening to selected inpatients at high risk for chronic HCV infection; or
- Offer screening to all inpatients who meet screening criteria based on current guidelines.
Given the shortcomings of the current screening strategies, these authors would recommend widespread screening for chronic HCV infection in hospitalized people who meet screening criteria per current guidelines.
If HM physicians are to take an increased role in HCV screening, there are a number of important considerations. Because hospitalized patients have a limited length of stay, it would be unreasonable to expect HM physicians to test for HCV RNA viral load or genotype for all patients with a positive antibody test, because the duration of the inpatient stay may be shorter than the time it takes for these test results to return. These tests are often indicated after a positive HCV antibody test, however. Thus, communication of HCV antibody results to PCPs or other responsible providers is essential. If no follow-up is available or there are no responsible outpatient providers, HM physicians should continue with a limited screening strategy.
Back to the Case
This individual has multiple indications for chronic HCV infection screening. His risk factors include date of birth between 1945 and 1965, a history of IV drug use, and a history of incarceration. He also notes a history of emergency surgery, for which he may have received blood products prior to 1987. These factors significantly raise the likelihood of chronic HCV infection when compared with the general population. He was screened and found to be HCV antibody-positive. A follow-up HCV RNA viral load was also positive. He did not have any evidence of liver disease but did have a mild transaminitis. He has followed up as an outpatient with plans to start therapy.
Bottom Line
The current screening strategies for individuals with high prevalence of chronic HCV infection are insufficient. HM physicians have an opportunity to improve the rates of screening in this population.
Dr. Theisen-Toupal is an internist, Dr. Rosenthal is a clinical fellow in medicine, and Dr. Carbo is an assistant professor of medicine, all at Beth Israel Deaconess Medical Center in Boston. Dr. Li is an internist and associate professor of medicine at Harvard Medical School and director of the hospital medicine division at Beth Israel Deaconess Medical Center.
References
- Ghany MG, Strader DB, Thomas DL, Seeff LB; American Association for the Study of Liver Diseases. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49(4):1335-1374.
- Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology. 2013;57(4):1333-1342.
- Chopra S. Clinical manifestations and natural history of chronic hepatitis C virus infection. UpToDate. Available at: http://www.uptodate.com/contents/clinical-manifestations-and-natural-history-of-chronic-hepatitis-c-virus-infection. Accessed March 5, 2014.
- Spradling PR, Rupp L, Moorman AC, et al. Hepatitis B and C virus infection among 1.2 million people with access to care: factors associated with testing and infection prevalence. Clin Infect Dis. 2012;55(8):1047-1055.
- Roblin DW, Smith BD, Weinbaum CM, Sabin ME. HCV screening practices and prevalence in an MCO, 2000-2007. Am J Manag Care. 2011;17(8):548-555.
- Centers for Disease Control and Prevention. Recommendations for the identification of chronic hepatitis C virus infection among people born during 1945-1965. MMWR. August 17, 2012. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr6104a1.htm. Accessed March 5, 2014.
- Chopra S. Epidemiology and transmission of hepatitis C virus infection. UpToDate. Available at: http://www.uptodate.com/contents/epidemiology-and-transmission-of-hepatitis-c-virus-infection?source=search_result&search=%22Epidemiology+and+transmission+of+hepatitis+C+virus+infection%22&selectedTitle=1~150. Accessed March 5, 2014.
- Centers for Disease Control and Prevention. Recommendations for prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. MMWR. October 16, 1998. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/00055154.htm. Accessed March 5, 2014.
- Donahue JG, Muñoz A, Ness PM, et al. The declining risk of post-transfusion hepatitis C virus infection. N Engl J Med. 1992;327(6):369-373.
- Pomper GJ, Wu Y, Snyder EL. Risks of transfusion-transmitted infections: 2003. Curr Opin Hematol. 2003;10(6):412-418.
- Tokars JI, Miller ER, Alter MJ, Arduino MJ. National surveillance of dialysis associated diseases in the United States, 1995. ASAIO J. 1998;44(1):98-107.
- Finelli L, Miller JT, Tokars JI, Alter MJ, Arduino MJ. National surveillance of dialysis-associated diseases in the United States, 2002. Semin Dial. 2005;18(1):52-61.
- Natov S, Pereira BJG. Hepatitis C virus infection in patients on maintenance dialysis. UpToDate. Available at: http://www.uptodate.com/contents/hepatitis-c-virus-infection-in-patients-on-maintenance-dialysis?source=search_result&search=Hepatitis+C+virus+infection+in+patients+on+maintenance+dialysis.&selectedTitle=1~150. Accessed March 5, 2014.
- Moyer VA, U.S. Preventive Services Task Force. Screening for hepatitis C virus infection in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2013;159(5):349-357.
- Staples CT II, Rimland D, Dudas D. Hepatitis C in the HIV (human immunodeficiency virus) Atlanta V.A. (Veterans Affairs Medical Center) Cohort Study (HAVACS): the effect of coinfection on survival. Clin Infect Dis. 1999;29(1):150-154.
- Sherman KE, Rouster SD, Chung RT, Rajicic N. Hepatitis C virus prevalence among patients infected with human immunodeficiency virus: a cross-sectional analysis of the U.S. adult AIDS clinical trials group. Clin Infect Dis. 2002;34(6):831-837.
- Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the United States. Clin Infect Dis. 2012;55 Suppl 1:S10-15.
- Centers for Disease Control and Prevention. Prevention and control of infections with hepatitis viruses in correctional settings. MMWR. January 24, 2003. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5201a1.htm. Accessed March 5, 2014.
- Centers for Disease Control and Prevention. Locations and reasons for initial testing for hepatitis C infection—chronic hepatitis cohort study, United States, 2006-2010. MMWR. August 16, 2013. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6232a3.htm?s_cid=mm6232a3_w. Accessed March 5, 2014.
Inhaled Corticosteroids Increase Risk of Serious Pneumonia in Patients with COPD

Clinical question: Does the risk of pneumonia vary for different inhaled agents?
Background: Inhaled corticosteroids (ICS) are known to increase the risk of pneumonia in COPD patients; duration, dosage, and various agents were investigated, especially fluticasone and budesonide.
Study design: Nested, case-control analysis.
Setting: Quebec health insurance database for new users with COPD, 1990-2005, with follow-up through 2007.
Synopsis: Investigators analyzed 163,514 patients, including 20,344 patients with serious pneumonia; current use of ICS was associated with a 69% increase in the rate of serious pneumonia (RR 1.69; 95% CI 1.63-1.75). The increased risk was sustained with long-term use but declined gradually to zero at six months after stopping ICS. The risk of serious pneumonia was higher with fluticasone (RR 2.01; 95% CI 1.93-2.10) than budesonide (RR 1.17; 95% CI 1.09-1.26).
Bottom line: Fluticasone was associated with an increased risk of pneumonia in COPD patients, consistent with earlier clinical trials, but the risk with budesonide was much lower.
Citation: Suissa S, Patenaude V, Lapi F, Ernst P. Inhaled corticosteroids in COPD and the risk of serious pneumonia. Thorax. 2013;68(11):1029-1036.
Clinical question: Does the risk of pneumonia vary for different inhaled agents?
Background: Inhaled corticosteroids (ICS) are known to increase the risk of pneumonia in COPD patients; duration, dosage, and various agents were investigated, especially fluticasone and budesonide.
Study design: Nested, case-control analysis.
Setting: Quebec health insurance database for new users with COPD, 1990-2005, with follow-up through 2007.
Synopsis: Investigators analyzed 163,514 patients, including 20,344 patients with serious pneumonia; current use of ICS was associated with a 69% increase in the rate of serious pneumonia (RR 1.69; 95% CI 1.63-1.75). The increased risk was sustained with long-term use but declined gradually to zero at six months after stopping ICS. The risk of serious pneumonia was higher with fluticasone (RR 2.01; 95% CI 1.93-2.10) than budesonide (RR 1.17; 95% CI 1.09-1.26).
Bottom line: Fluticasone was associated with an increased risk of pneumonia in COPD patients, consistent with earlier clinical trials, but the risk with budesonide was much lower.
Citation: Suissa S, Patenaude V, Lapi F, Ernst P. Inhaled corticosteroids in COPD and the risk of serious pneumonia. Thorax. 2013;68(11):1029-1036.
Clinical question: Does the risk of pneumonia vary for different inhaled agents?
Background: Inhaled corticosteroids (ICS) are known to increase the risk of pneumonia in COPD patients; duration, dosage, and various agents were investigated, especially fluticasone and budesonide.
Study design: Nested, case-control analysis.
Setting: Quebec health insurance database for new users with COPD, 1990-2005, with follow-up through 2007.
Synopsis: Investigators analyzed 163,514 patients, including 20,344 patients with serious pneumonia; current use of ICS was associated with a 69% increase in the rate of serious pneumonia (RR 1.69; 95% CI 1.63-1.75). The increased risk was sustained with long-term use but declined gradually to zero at six months after stopping ICS. The risk of serious pneumonia was higher with fluticasone (RR 2.01; 95% CI 1.93-2.10) than budesonide (RR 1.17; 95% CI 1.09-1.26).
Bottom line: Fluticasone was associated with an increased risk of pneumonia in COPD patients, consistent with earlier clinical trials, but the risk with budesonide was much lower.
Citation: Suissa S, Patenaude V, Lapi F, Ernst P. Inhaled corticosteroids in COPD and the risk of serious pneumonia. Thorax. 2013;68(11):1029-1036.
Ambulatory Patients with COPD Exacerbations Can Be Managed Without Antibiotics in the Absence of Increased Sputum Purulence, Elevated C-Reactive Protein
Clinical question: Which criteria identify ambulatory patients with exacerbations of mild to moderate COPD who do not need antibiotics?
Background: The Anthonisen criteria (increased dyspnea, sputum volume, sputum purulence) are commonly used to identify which patients with COPD exacerbations would benefit from antibiotics. These criteria, however, were derived in patients with severe COPD. It is unknown whether these criteria are predictive in patients with mild to moderate COPD.
Study design: Multivariate logistic regression analysis of placebo group of a double-blinded RCT.
Setting: Multicenter, ambulatory, primary care clinics in Spain.
Synopsis: The original RCT enrolled 310 ambulatory patients with exacerbations of mild to moderate COPD and tested the efficacy of amoxicillin/clavulanate. Clinical failure without antibiotics was 19.9% compared to 9.5% with antibiotics (P=0.022). Here they analyzed the 152 patients from the placebo group to identify factors associated with increased risk of clinical failure. Only increased sputum purulence (OR 6.1, CI 1.5-25; P=0.005) or C-reactive protein (CRP) >40 mg/L (OR 13.4, CI 4.5-38.8, P<0.001) were independently associated with increased risk of failure. Presence of both predicted a 63.7% failure without antibiotics.
The study did not define “increased sputum purulence,” but this is similar to real-life clinical practice. Placebo effect cannot be ruled out, but correlation of the objective measures with the clinical assessments suggests that the clinical assessments were accurate. The study did not have a protocol for administering co-medications such as steroids and inhalers. Despite these limitations, the criteria of increased sputum purulence and CRP >40 mg/L identified COPD patients likely to have a clinical failure without antibiotics.
Bottom line: Patients with exacerbations of mild to moderate COPD who do not have increased sputum purulence or CRP >40 mg/L can be safely managed without antibiotics.
Citation: Maravitlles M, Moravas A, Hernandez S, Bayona C, Llor C. Is it possible to identify exacerbations of mild to moderate COPD that do not require antibiotic treatment? Chest. 2013;144(5):1571-1577.
Clinical question: Which criteria identify ambulatory patients with exacerbations of mild to moderate COPD who do not need antibiotics?
Background: The Anthonisen criteria (increased dyspnea, sputum volume, sputum purulence) are commonly used to identify which patients with COPD exacerbations would benefit from antibiotics. These criteria, however, were derived in patients with severe COPD. It is unknown whether these criteria are predictive in patients with mild to moderate COPD.
Study design: Multivariate logistic regression analysis of placebo group of a double-blinded RCT.
Setting: Multicenter, ambulatory, primary care clinics in Spain.
Synopsis: The original RCT enrolled 310 ambulatory patients with exacerbations of mild to moderate COPD and tested the efficacy of amoxicillin/clavulanate. Clinical failure without antibiotics was 19.9% compared to 9.5% with antibiotics (P=0.022). Here they analyzed the 152 patients from the placebo group to identify factors associated with increased risk of clinical failure. Only increased sputum purulence (OR 6.1, CI 1.5-25; P=0.005) or C-reactive protein (CRP) >40 mg/L (OR 13.4, CI 4.5-38.8, P<0.001) were independently associated with increased risk of failure. Presence of both predicted a 63.7% failure without antibiotics.
The study did not define “increased sputum purulence,” but this is similar to real-life clinical practice. Placebo effect cannot be ruled out, but correlation of the objective measures with the clinical assessments suggests that the clinical assessments were accurate. The study did not have a protocol for administering co-medications such as steroids and inhalers. Despite these limitations, the criteria of increased sputum purulence and CRP >40 mg/L identified COPD patients likely to have a clinical failure without antibiotics.
Bottom line: Patients with exacerbations of mild to moderate COPD who do not have increased sputum purulence or CRP >40 mg/L can be safely managed without antibiotics.
Citation: Maravitlles M, Moravas A, Hernandez S, Bayona C, Llor C. Is it possible to identify exacerbations of mild to moderate COPD that do not require antibiotic treatment? Chest. 2013;144(5):1571-1577.
Clinical question: Which criteria identify ambulatory patients with exacerbations of mild to moderate COPD who do not need antibiotics?
Background: The Anthonisen criteria (increased dyspnea, sputum volume, sputum purulence) are commonly used to identify which patients with COPD exacerbations would benefit from antibiotics. These criteria, however, were derived in patients with severe COPD. It is unknown whether these criteria are predictive in patients with mild to moderate COPD.
Study design: Multivariate logistic regression analysis of placebo group of a double-blinded RCT.
Setting: Multicenter, ambulatory, primary care clinics in Spain.
Synopsis: The original RCT enrolled 310 ambulatory patients with exacerbations of mild to moderate COPD and tested the efficacy of amoxicillin/clavulanate. Clinical failure without antibiotics was 19.9% compared to 9.5% with antibiotics (P=0.022). Here they analyzed the 152 patients from the placebo group to identify factors associated with increased risk of clinical failure. Only increased sputum purulence (OR 6.1, CI 1.5-25; P=0.005) or C-reactive protein (CRP) >40 mg/L (OR 13.4, CI 4.5-38.8, P<0.001) were independently associated with increased risk of failure. Presence of both predicted a 63.7% failure without antibiotics.
The study did not define “increased sputum purulence,” but this is similar to real-life clinical practice. Placebo effect cannot be ruled out, but correlation of the objective measures with the clinical assessments suggests that the clinical assessments were accurate. The study did not have a protocol for administering co-medications such as steroids and inhalers. Despite these limitations, the criteria of increased sputum purulence and CRP >40 mg/L identified COPD patients likely to have a clinical failure without antibiotics.
Bottom line: Patients with exacerbations of mild to moderate COPD who do not have increased sputum purulence or CRP >40 mg/L can be safely managed without antibiotics.
Citation: Maravitlles M, Moravas A, Hernandez S, Bayona C, Llor C. Is it possible to identify exacerbations of mild to moderate COPD that do not require antibiotic treatment? Chest. 2013;144(5):1571-1577.
Pre-Operative Angiotensin Axis Blockade Increases Risk of Hypotension, Acute Kidney Injury with Major Orthopedic Surgery
Clinical question: Do patients receiving pre-operative angiotensin axis blockade (AAB) prior to elective major orthopedic surgery have an increased risk of peri-operative hypotension and acute kidney injury (AKI)?
Background: Patients with pre-operative AAB from angiotensin-converting enzyme inhibitors or angiotensin receptor blockers have an increased incidence of peri-operative hypotension. Patients undergoing cardiothoracic and vascular surgery with pre-operative AAB have increased incidence of post-operative AKI; however, there is scant literature evaluating the hypotensive and renal effects of pre-operative AAB prior to elective major orthopedic surgery.
Study design: Retrospective, cohort study.
Setting: Academic medical center.
Synopsis: Retrospective review of 922 patients undergoing spinal fusion, total knee arthroplasty, or total hip arthroplasty in one academic medical center in 2010 found that 37% received pre-operative AAB. Post-induction hypotension (systolic blood pressure ≤80 mm Hg for five minutes) was significantly higher in patients receiving AAB (12.2% vs. 6.7%; odds ratio [OR] 1.93, P=0.005). Post-operative AKI was significantly higher in patients receiving AAB (8.3% vs. 1.7%; OR 5.40, P<0.001), remaining significant after adjusting for intra-operative hypotension (OR 2.60, P=0.042). Developing AKI resulted in a significantly higher mean length of stay (5.76 vs. 3.28 days, P<0.001) but no difference in two-year mortality.
The findings suggest an association exists between pre-operative angiotensin-converting enzyme inhibitors/ARB, hypotension, and AKI following major orthopedic surgeries but does not demonstrate causality. A prospective, multi-center, randomized trial is needed to confirm that holding pre-operative AAB would decrease the incidence of AKI in patients undergoing major orthopedic procedures under general anesthesia.
Bottom line: Patients who underwent elective major orthopedic surgery who received pre-operative AAB therapy had an associated increased risk of post-induction hypotension and post-operative AKI, resulting in a greater hospital length of stay.
Citation: Nielson E, Hennrikus E, Lehman E, Mets B. Angiotensin axis blockade, hypotension, and acute kidney injury in elective major orthopedic surgery. J Hosp Med. 2014;9(5):283-288.
Clinical question: Do patients receiving pre-operative angiotensin axis blockade (AAB) prior to elective major orthopedic surgery have an increased risk of peri-operative hypotension and acute kidney injury (AKI)?
Background: Patients with pre-operative AAB from angiotensin-converting enzyme inhibitors or angiotensin receptor blockers have an increased incidence of peri-operative hypotension. Patients undergoing cardiothoracic and vascular surgery with pre-operative AAB have increased incidence of post-operative AKI; however, there is scant literature evaluating the hypotensive and renal effects of pre-operative AAB prior to elective major orthopedic surgery.
Study design: Retrospective, cohort study.
Setting: Academic medical center.
Synopsis: Retrospective review of 922 patients undergoing spinal fusion, total knee arthroplasty, or total hip arthroplasty in one academic medical center in 2010 found that 37% received pre-operative AAB. Post-induction hypotension (systolic blood pressure ≤80 mm Hg for five minutes) was significantly higher in patients receiving AAB (12.2% vs. 6.7%; odds ratio [OR] 1.93, P=0.005). Post-operative AKI was significantly higher in patients receiving AAB (8.3% vs. 1.7%; OR 5.40, P<0.001), remaining significant after adjusting for intra-operative hypotension (OR 2.60, P=0.042). Developing AKI resulted in a significantly higher mean length of stay (5.76 vs. 3.28 days, P<0.001) but no difference in two-year mortality.
The findings suggest an association exists between pre-operative angiotensin-converting enzyme inhibitors/ARB, hypotension, and AKI following major orthopedic surgeries but does not demonstrate causality. A prospective, multi-center, randomized trial is needed to confirm that holding pre-operative AAB would decrease the incidence of AKI in patients undergoing major orthopedic procedures under general anesthesia.
Bottom line: Patients who underwent elective major orthopedic surgery who received pre-operative AAB therapy had an associated increased risk of post-induction hypotension and post-operative AKI, resulting in a greater hospital length of stay.
Citation: Nielson E, Hennrikus E, Lehman E, Mets B. Angiotensin axis blockade, hypotension, and acute kidney injury in elective major orthopedic surgery. J Hosp Med. 2014;9(5):283-288.
Clinical question: Do patients receiving pre-operative angiotensin axis blockade (AAB) prior to elective major orthopedic surgery have an increased risk of peri-operative hypotension and acute kidney injury (AKI)?
Background: Patients with pre-operative AAB from angiotensin-converting enzyme inhibitors or angiotensin receptor blockers have an increased incidence of peri-operative hypotension. Patients undergoing cardiothoracic and vascular surgery with pre-operative AAB have increased incidence of post-operative AKI; however, there is scant literature evaluating the hypotensive and renal effects of pre-operative AAB prior to elective major orthopedic surgery.
Study design: Retrospective, cohort study.
Setting: Academic medical center.
Synopsis: Retrospective review of 922 patients undergoing spinal fusion, total knee arthroplasty, or total hip arthroplasty in one academic medical center in 2010 found that 37% received pre-operative AAB. Post-induction hypotension (systolic blood pressure ≤80 mm Hg for five minutes) was significantly higher in patients receiving AAB (12.2% vs. 6.7%; odds ratio [OR] 1.93, P=0.005). Post-operative AKI was significantly higher in patients receiving AAB (8.3% vs. 1.7%; OR 5.40, P<0.001), remaining significant after adjusting for intra-operative hypotension (OR 2.60, P=0.042). Developing AKI resulted in a significantly higher mean length of stay (5.76 vs. 3.28 days, P<0.001) but no difference in two-year mortality.
The findings suggest an association exists between pre-operative angiotensin-converting enzyme inhibitors/ARB, hypotension, and AKI following major orthopedic surgeries but does not demonstrate causality. A prospective, multi-center, randomized trial is needed to confirm that holding pre-operative AAB would decrease the incidence of AKI in patients undergoing major orthopedic procedures under general anesthesia.
Bottom line: Patients who underwent elective major orthopedic surgery who received pre-operative AAB therapy had an associated increased risk of post-induction hypotension and post-operative AKI, resulting in a greater hospital length of stay.
Citation: Nielson E, Hennrikus E, Lehman E, Mets B. Angiotensin axis blockade, hypotension, and acute kidney injury in elective major orthopedic surgery. J Hosp Med. 2014;9(5):283-288.