User login
Despite responses, imetelstat won’t move forward in ET
Although trial results suggest imetelstat is effective against essential thrombocythemia (ET), the telomerase inhibitor is only being developed to treat myelofibrosis (MF).
Imetelstat produced “rapid and durable” responses in a phase 2 trial of ET patients, but the drug also produced side effects that caused the US Food and Drug Administration (FDA) to place a full clinical hold on the drug.
The hold was lifted last November, but the company developing imetelstat decided not to pursue the drug as a treatment for ET or polycythemia vera.
Development continues for MF, however, and results of the pilot study of imetelstat in MF appear in NEJM.
Results from the trial of imetelstat in ET have been published in NEJM as well. Both studies were funded by Geron Corporation, the company developing imetelstat.
The ET trial included 18 patients with a median age of 59.5 (range, 21-83). All patients had received one or more prior treatments, including hydroxyurea (n=17, 94%), anagrelide (n=13, 72%), and interferon (n=4, 22%). Half of patients (n=9) were resistant to at least 1 prior therapy, and 78% (n=14) had experienced unacceptable side effects from a previous therapy.
The patients received imetelstat at an initial dose of 7.5 or 9.4 mg per kilogram of body weight intravenously once a week. Treatment continued until patients attained a platelet count of approximately 250,000 to 300,000 per cubic millimeter.
Responses
All 18 patients experienced hematologic responses, and 16 (89%) had a complete hematologic response. At a median follow-up of 17 months, 10 patients were still receiving treatment, and the median duration of response had not been reached (range, 5 to 30 months).
“[I]metelstat had a clinically significant effect on disease burden in ET patients,” said study investigator David Snyder, MD, of City of Hope in Duarte, California.
“This study was a first look at what happens when you treat ET patients with a drug that has a totally novel mechanism of action.”
Seven of the 8 patients (88%) who were positive for the JAK2 V617F mutation had a molecular response. All of the patients with CALR (n=5) or MPL (n=2) mutations saw a reduction in mutant allele burden, ranging from 15% to 66%.
“The molecular responses suggest that imetelstat may have broad activity across hematologic myeloid malignancies, which warrants further clinical study in other myeloproliferative neoplasms,” said investigator Gabriela M. Baerlocher, MD, of the University of Bern in Switzerland.
Toxicity, clinical hold, and discontinuation
The most common adverse events (≥50%) in this trial were fatigue (83%), diarrhea (78%), nausea (72%), dizziness (61%), increased alanine aminotransferase (ALT, 56%), increased aspartate aminotransferase (AST, 56%), constipation (50%), cough (50%), epistaxis (50%), and headache (50%).
Grade 3/4 adverse events included decreased neutrophil count (22%), neutropenia (22%), anemia (11%), syncope (11%), headache (11%), upper respiratory tract infection (6%), decreased white cell count (6%), myalgia (6%), hypokalemia (6%), fatigue (6%), cellulitis (6%), increased ALT (6%), increased AST (6%), and epistaxis (6%).
All 18 patients had at least one increase in grade, from baseline, in a liver-function value. Seventeen patients had an elevation in ALT, 17 in AST, 15 in alkaline phosphatase, and 8 in total bilirubin. Fourteen patients had persistent abnormalities (≥ 6 weeks), all of which were grade 1 or 2 in severity.
It was these liver-function abnormalities and the potential risk of chronic liver injury that prompted the FDA to place a clinical hold on imetelstat. The agency was concerned about whether the effects were reversible.
It turned out that, in many cases, abnormalities resolved after patients permanently discontinued treatment (as a result of the clinical hold). Sixteen of 17 patients with elevated ALT experienced a resolution, as did 12 of 17 patients with elevated AST, 9 of 15 with elevated alkaline phosphatase, and 7 of 8 with elevated bilirubin.
Of the 14 patients who had persistent abnormalities, 11 had their values reversed to normal or baseline values after stopping imetelstat. The median time to resolution after treatment discontinuation was 12 weeks.
Still, Geron Corporation decided not to develop imetelstat for patients with ET or polycythemia vera. And the FDA said the proposed clinical development plan for the drug, which is focused on MF and other high-risk myeloid disorders, was acceptable. So the clinical hold was lifted. ![]()
Although trial results suggest imetelstat is effective against essential thrombocythemia (ET), the telomerase inhibitor is only being developed to treat myelofibrosis (MF).
Imetelstat produced “rapid and durable” responses in a phase 2 trial of ET patients, but the drug also produced side effects that caused the US Food and Drug Administration (FDA) to place a full clinical hold on the drug.
The hold was lifted last November, but the company developing imetelstat decided not to pursue the drug as a treatment for ET or polycythemia vera.
Development continues for MF, however, and results of the pilot study of imetelstat in MF appear in NEJM.
Results from the trial of imetelstat in ET have been published in NEJM as well. Both studies were funded by Geron Corporation, the company developing imetelstat.
The ET trial included 18 patients with a median age of 59.5 (range, 21-83). All patients had received one or more prior treatments, including hydroxyurea (n=17, 94%), anagrelide (n=13, 72%), and interferon (n=4, 22%). Half of patients (n=9) were resistant to at least 1 prior therapy, and 78% (n=14) had experienced unacceptable side effects from a previous therapy.
The patients received imetelstat at an initial dose of 7.5 or 9.4 mg per kilogram of body weight intravenously once a week. Treatment continued until patients attained a platelet count of approximately 250,000 to 300,000 per cubic millimeter.
Responses
All 18 patients experienced hematologic responses, and 16 (89%) had a complete hematologic response. At a median follow-up of 17 months, 10 patients were still receiving treatment, and the median duration of response had not been reached (range, 5 to 30 months).
“[I]metelstat had a clinically significant effect on disease burden in ET patients,” said study investigator David Snyder, MD, of City of Hope in Duarte, California.
“This study was a first look at what happens when you treat ET patients with a drug that has a totally novel mechanism of action.”
Seven of the 8 patients (88%) who were positive for the JAK2 V617F mutation had a molecular response. All of the patients with CALR (n=5) or MPL (n=2) mutations saw a reduction in mutant allele burden, ranging from 15% to 66%.
“The molecular responses suggest that imetelstat may have broad activity across hematologic myeloid malignancies, which warrants further clinical study in other myeloproliferative neoplasms,” said investigator Gabriela M. Baerlocher, MD, of the University of Bern in Switzerland.
Toxicity, clinical hold, and discontinuation
The most common adverse events (≥50%) in this trial were fatigue (83%), diarrhea (78%), nausea (72%), dizziness (61%), increased alanine aminotransferase (ALT, 56%), increased aspartate aminotransferase (AST, 56%), constipation (50%), cough (50%), epistaxis (50%), and headache (50%).
Grade 3/4 adverse events included decreased neutrophil count (22%), neutropenia (22%), anemia (11%), syncope (11%), headache (11%), upper respiratory tract infection (6%), decreased white cell count (6%), myalgia (6%), hypokalemia (6%), fatigue (6%), cellulitis (6%), increased ALT (6%), increased AST (6%), and epistaxis (6%).
All 18 patients had at least one increase in grade, from baseline, in a liver-function value. Seventeen patients had an elevation in ALT, 17 in AST, 15 in alkaline phosphatase, and 8 in total bilirubin. Fourteen patients had persistent abnormalities (≥ 6 weeks), all of which were grade 1 or 2 in severity.
It was these liver-function abnormalities and the potential risk of chronic liver injury that prompted the FDA to place a clinical hold on imetelstat. The agency was concerned about whether the effects were reversible.
It turned out that, in many cases, abnormalities resolved after patients permanently discontinued treatment (as a result of the clinical hold). Sixteen of 17 patients with elevated ALT experienced a resolution, as did 12 of 17 patients with elevated AST, 9 of 15 with elevated alkaline phosphatase, and 7 of 8 with elevated bilirubin.
Of the 14 patients who had persistent abnormalities, 11 had their values reversed to normal or baseline values after stopping imetelstat. The median time to resolution after treatment discontinuation was 12 weeks.
Still, Geron Corporation decided not to develop imetelstat for patients with ET or polycythemia vera. And the FDA said the proposed clinical development plan for the drug, which is focused on MF and other high-risk myeloid disorders, was acceptable. So the clinical hold was lifted. ![]()
Although trial results suggest imetelstat is effective against essential thrombocythemia (ET), the telomerase inhibitor is only being developed to treat myelofibrosis (MF).
Imetelstat produced “rapid and durable” responses in a phase 2 trial of ET patients, but the drug also produced side effects that caused the US Food and Drug Administration (FDA) to place a full clinical hold on the drug.
The hold was lifted last November, but the company developing imetelstat decided not to pursue the drug as a treatment for ET or polycythemia vera.
Development continues for MF, however, and results of the pilot study of imetelstat in MF appear in NEJM.
Results from the trial of imetelstat in ET have been published in NEJM as well. Both studies were funded by Geron Corporation, the company developing imetelstat.
The ET trial included 18 patients with a median age of 59.5 (range, 21-83). All patients had received one or more prior treatments, including hydroxyurea (n=17, 94%), anagrelide (n=13, 72%), and interferon (n=4, 22%). Half of patients (n=9) were resistant to at least 1 prior therapy, and 78% (n=14) had experienced unacceptable side effects from a previous therapy.
The patients received imetelstat at an initial dose of 7.5 or 9.4 mg per kilogram of body weight intravenously once a week. Treatment continued until patients attained a platelet count of approximately 250,000 to 300,000 per cubic millimeter.
Responses
All 18 patients experienced hematologic responses, and 16 (89%) had a complete hematologic response. At a median follow-up of 17 months, 10 patients were still receiving treatment, and the median duration of response had not been reached (range, 5 to 30 months).
“[I]metelstat had a clinically significant effect on disease burden in ET patients,” said study investigator David Snyder, MD, of City of Hope in Duarte, California.
“This study was a first look at what happens when you treat ET patients with a drug that has a totally novel mechanism of action.”
Seven of the 8 patients (88%) who were positive for the JAK2 V617F mutation had a molecular response. All of the patients with CALR (n=5) or MPL (n=2) mutations saw a reduction in mutant allele burden, ranging from 15% to 66%.
“The molecular responses suggest that imetelstat may have broad activity across hematologic myeloid malignancies, which warrants further clinical study in other myeloproliferative neoplasms,” said investigator Gabriela M. Baerlocher, MD, of the University of Bern in Switzerland.
Toxicity, clinical hold, and discontinuation
The most common adverse events (≥50%) in this trial were fatigue (83%), diarrhea (78%), nausea (72%), dizziness (61%), increased alanine aminotransferase (ALT, 56%), increased aspartate aminotransferase (AST, 56%), constipation (50%), cough (50%), epistaxis (50%), and headache (50%).
Grade 3/4 adverse events included decreased neutrophil count (22%), neutropenia (22%), anemia (11%), syncope (11%), headache (11%), upper respiratory tract infection (6%), decreased white cell count (6%), myalgia (6%), hypokalemia (6%), fatigue (6%), cellulitis (6%), increased ALT (6%), increased AST (6%), and epistaxis (6%).
All 18 patients had at least one increase in grade, from baseline, in a liver-function value. Seventeen patients had an elevation in ALT, 17 in AST, 15 in alkaline phosphatase, and 8 in total bilirubin. Fourteen patients had persistent abnormalities (≥ 6 weeks), all of which were grade 1 or 2 in severity.
It was these liver-function abnormalities and the potential risk of chronic liver injury that prompted the FDA to place a clinical hold on imetelstat. The agency was concerned about whether the effects were reversible.
It turned out that, in many cases, abnormalities resolved after patients permanently discontinued treatment (as a result of the clinical hold). Sixteen of 17 patients with elevated ALT experienced a resolution, as did 12 of 17 patients with elevated AST, 9 of 15 with elevated alkaline phosphatase, and 7 of 8 with elevated bilirubin.
Of the 14 patients who had persistent abnormalities, 11 had their values reversed to normal or baseline values after stopping imetelstat. The median time to resolution after treatment discontinuation was 12 weeks.
Still, Geron Corporation decided not to develop imetelstat for patients with ET or polycythemia vera. And the FDA said the proposed clinical development plan for the drug, which is focused on MF and other high-risk myeloid disorders, was acceptable. So the clinical hold was lifted. ![]()
Imetelstat in MF: More research needed
The telomerase inhibitor imetelstat has exhibited unique activity in a pilot study of patients with intermediate- or high-risk myelofibrosis (MF), but more research is needed, according to investigators.
Imetelstat produced complete and partial responses in a minority of patients and reversed bone marrow fibrosis in complete responders.
However, imetelstat also prompted severe myelosuppression and liver-test abnormalities. And most patients ultimately discontinued treatment.
The investigators therefore concluded that additional research is needed to establish the most effective dosing of the drug, clarify its mechanism of action, and address concerns about toxicity.
Ayalew Tefferi, MD, of the Mayo Clinic in Rochester, Minnesota, and his colleagues reported the results of this trial in NEJM.
A phase 2 trial of imetelstat in patients with essential thrombocythemia was published in NEJM simultaneously. Both trials were sponsored by Geron Corporation, the company developing imetelstat.
The MF study included 33 patients, 18 with primary MF, 10 with post-polycythemia vera MF, and 5 with post-essential thrombocythemia MF. About 52% had high-risk disease, and the rest had intermediate-2-risk MF.
The patients had a median age of 67. About 79% of patients had received prior therapy, and 48% had received a JAK inhibitor. Thirty-nine percent of patients were dependent on red cell transfusions, 64% had constitutional symptoms, 70% had palpable splenomegaly, and 55% had an abnormal karyotype, including 18% with an unfavorable karyotype.
Imetelstat was administered as a 2-hour intravenous infusion, at a starting dose of 9.4 mg per kilogram of body weight. There were 2 dosing schedules: (1) once every 3 weeks or (2) weekly for 4 weeks, followed by once every 3 weeks.
Responses
“We observed that imetelstat was active and induced morphologic and molecular remissions in some patients with myelofibrosis,” Dr Tefferi said. “We also observed that imetelstat demonstrated selective anticlonal activity, inhibiting the growth of cancer cells, which we had not previously documented with other drugs.”
Overall, 21% of patients (7/33) experienced a complete response (n=4) or partial response (n=3) to treatment. The median duration of complete response was 18 months (range, 13-20+), and the median duration of partial response was 10 months (range, 7-10+).
“Some patients treated with imetelstat have reverted back to normal bone marrow,” Dr Tefferi noted. “Typically, myelofibrosis is characterized by marrow scarring, and, although patients may derive symptomatic relief from other treatments, such as ruxolitinib, they usually do not revert back to normal bone marrow.”
Bone marrow fibrosis was reversed in all 4 patients with a complete response. A molecular response was documented in 3 of these patients.
Mutations and telomere length
“We noted a difference in response rates, especially in complete remission rates, in patients with and without certain specific gene mutations, such as ASXL1, SF3B1, and U2AF1,” Dr Tefferi noted. “This underscores the need for laboratory correlative studies in future clinical trials.”
Responses occurred in 27% of patients with a JAK2 mutation and 0% of patients without a JAK2 mutation (P=0.30). Alternatively, responses occurred in 32% of patients without an ASXL1 mutation and 0% of patients with an ASXL1 mutation (P=0.07).
The rate of complete response was 38% among patients with a mutation in SF3B1 or U2AF1, compared to 4% among patients without either mutation (P=0.04).
The investigators also found that responses were not correlated with baseline telomere length.
Toxicity
Dr Tefferi and his colleagues said the most clinically significant side effect of imetelstat was myelosuppression. It was the primary reason for a protocol-mandated dose reduction that occurred in 22 patients (67%).
Another “notable” side effect was the elevation of liver-enzyme levels. The investigators observed treatment-emergent (though not necessarily related) increases from baseline in total bilirubin (49%), alkaline phosphatase (58%), aspartate aminotransferase (58%), and alanine aminotransferase (27%).
None of these abnormalities were linked to clinically overt liver damage, and most patients ultimately saw their values return to baseline levels.
Adverse events that were considered at least possibly related to treatment and occurred in 3 or more patients included thrombocytopenia (45% grade 3/4), anemia (39% overall, 30% grade 3), neutropenia (27% grade 3/4), aspartate aminotransferase elevation (27% grade 1), alkaline phosphatase elevation (21% grade 1/2), elevation in total bilirubin (12% grade 1/2), infusion-related reactions (12% grade 1/2), diarrhea (9% grade 1/2), and epistaxis (9% grade 1/2).
Treatment discontinuation
At the data-cutoff date (December 5, 2014), 76% of patients had discontinued imetelstat (n=25). For all patients, the median duration of treatment was 8.6 months (range, 1.4 to 21.7).
Patients stopped treatment due to insufficient response (n=16), disease progression or relapse after response (n=3), death during the treatment period (n=2), adverse events (n=2), financial constraints (n=1), and pre-existing atrial fibrillation (n=1).
Both patients who discontinued imetelstat due to adverse events had persistent thrombocytopenia. Of the 2 deaths, 1 was considered treatment-related. That patient died of intracranial hemorrhage that was attributed to drug-induced, grade 4 thrombocytopenia after weekly dosing. The non-treatment-related death was the result of an upper gastrointestinal hemorrhage. ![]()
The telomerase inhibitor imetelstat has exhibited unique activity in a pilot study of patients with intermediate- or high-risk myelofibrosis (MF), but more research is needed, according to investigators.
Imetelstat produced complete and partial responses in a minority of patients and reversed bone marrow fibrosis in complete responders.
However, imetelstat also prompted severe myelosuppression and liver-test abnormalities. And most patients ultimately discontinued treatment.
The investigators therefore concluded that additional research is needed to establish the most effective dosing of the drug, clarify its mechanism of action, and address concerns about toxicity.
Ayalew Tefferi, MD, of the Mayo Clinic in Rochester, Minnesota, and his colleagues reported the results of this trial in NEJM.
A phase 2 trial of imetelstat in patients with essential thrombocythemia was published in NEJM simultaneously. Both trials were sponsored by Geron Corporation, the company developing imetelstat.
The MF study included 33 patients, 18 with primary MF, 10 with post-polycythemia vera MF, and 5 with post-essential thrombocythemia MF. About 52% had high-risk disease, and the rest had intermediate-2-risk MF.
The patients had a median age of 67. About 79% of patients had received prior therapy, and 48% had received a JAK inhibitor. Thirty-nine percent of patients were dependent on red cell transfusions, 64% had constitutional symptoms, 70% had palpable splenomegaly, and 55% had an abnormal karyotype, including 18% with an unfavorable karyotype.
Imetelstat was administered as a 2-hour intravenous infusion, at a starting dose of 9.4 mg per kilogram of body weight. There were 2 dosing schedules: (1) once every 3 weeks or (2) weekly for 4 weeks, followed by once every 3 weeks.
Responses
“We observed that imetelstat was active and induced morphologic and molecular remissions in some patients with myelofibrosis,” Dr Tefferi said. “We also observed that imetelstat demonstrated selective anticlonal activity, inhibiting the growth of cancer cells, which we had not previously documented with other drugs.”
Overall, 21% of patients (7/33) experienced a complete response (n=4) or partial response (n=3) to treatment. The median duration of complete response was 18 months (range, 13-20+), and the median duration of partial response was 10 months (range, 7-10+).
“Some patients treated with imetelstat have reverted back to normal bone marrow,” Dr Tefferi noted. “Typically, myelofibrosis is characterized by marrow scarring, and, although patients may derive symptomatic relief from other treatments, such as ruxolitinib, they usually do not revert back to normal bone marrow.”
Bone marrow fibrosis was reversed in all 4 patients with a complete response. A molecular response was documented in 3 of these patients.
Mutations and telomere length
“We noted a difference in response rates, especially in complete remission rates, in patients with and without certain specific gene mutations, such as ASXL1, SF3B1, and U2AF1,” Dr Tefferi noted. “This underscores the need for laboratory correlative studies in future clinical trials.”
Responses occurred in 27% of patients with a JAK2 mutation and 0% of patients without a JAK2 mutation (P=0.30). Alternatively, responses occurred in 32% of patients without an ASXL1 mutation and 0% of patients with an ASXL1 mutation (P=0.07).
The rate of complete response was 38% among patients with a mutation in SF3B1 or U2AF1, compared to 4% among patients without either mutation (P=0.04).
The investigators also found that responses were not correlated with baseline telomere length.
Toxicity
Dr Tefferi and his colleagues said the most clinically significant side effect of imetelstat was myelosuppression. It was the primary reason for a protocol-mandated dose reduction that occurred in 22 patients (67%).
Another “notable” side effect was the elevation of liver-enzyme levels. The investigators observed treatment-emergent (though not necessarily related) increases from baseline in total bilirubin (49%), alkaline phosphatase (58%), aspartate aminotransferase (58%), and alanine aminotransferase (27%).
None of these abnormalities were linked to clinically overt liver damage, and most patients ultimately saw their values return to baseline levels.
Adverse events that were considered at least possibly related to treatment and occurred in 3 or more patients included thrombocytopenia (45% grade 3/4), anemia (39% overall, 30% grade 3), neutropenia (27% grade 3/4), aspartate aminotransferase elevation (27% grade 1), alkaline phosphatase elevation (21% grade 1/2), elevation in total bilirubin (12% grade 1/2), infusion-related reactions (12% grade 1/2), diarrhea (9% grade 1/2), and epistaxis (9% grade 1/2).
Treatment discontinuation
At the data-cutoff date (December 5, 2014), 76% of patients had discontinued imetelstat (n=25). For all patients, the median duration of treatment was 8.6 months (range, 1.4 to 21.7).
Patients stopped treatment due to insufficient response (n=16), disease progression or relapse after response (n=3), death during the treatment period (n=2), adverse events (n=2), financial constraints (n=1), and pre-existing atrial fibrillation (n=1).
Both patients who discontinued imetelstat due to adverse events had persistent thrombocytopenia. Of the 2 deaths, 1 was considered treatment-related. That patient died of intracranial hemorrhage that was attributed to drug-induced, grade 4 thrombocytopenia after weekly dosing. The non-treatment-related death was the result of an upper gastrointestinal hemorrhage. ![]()
The telomerase inhibitor imetelstat has exhibited unique activity in a pilot study of patients with intermediate- or high-risk myelofibrosis (MF), but more research is needed, according to investigators.
Imetelstat produced complete and partial responses in a minority of patients and reversed bone marrow fibrosis in complete responders.
However, imetelstat also prompted severe myelosuppression and liver-test abnormalities. And most patients ultimately discontinued treatment.
The investigators therefore concluded that additional research is needed to establish the most effective dosing of the drug, clarify its mechanism of action, and address concerns about toxicity.
Ayalew Tefferi, MD, of the Mayo Clinic in Rochester, Minnesota, and his colleagues reported the results of this trial in NEJM.
A phase 2 trial of imetelstat in patients with essential thrombocythemia was published in NEJM simultaneously. Both trials were sponsored by Geron Corporation, the company developing imetelstat.
The MF study included 33 patients, 18 with primary MF, 10 with post-polycythemia vera MF, and 5 with post-essential thrombocythemia MF. About 52% had high-risk disease, and the rest had intermediate-2-risk MF.
The patients had a median age of 67. About 79% of patients had received prior therapy, and 48% had received a JAK inhibitor. Thirty-nine percent of patients were dependent on red cell transfusions, 64% had constitutional symptoms, 70% had palpable splenomegaly, and 55% had an abnormal karyotype, including 18% with an unfavorable karyotype.
Imetelstat was administered as a 2-hour intravenous infusion, at a starting dose of 9.4 mg per kilogram of body weight. There were 2 dosing schedules: (1) once every 3 weeks or (2) weekly for 4 weeks, followed by once every 3 weeks.
Responses
“We observed that imetelstat was active and induced morphologic and molecular remissions in some patients with myelofibrosis,” Dr Tefferi said. “We also observed that imetelstat demonstrated selective anticlonal activity, inhibiting the growth of cancer cells, which we had not previously documented with other drugs.”
Overall, 21% of patients (7/33) experienced a complete response (n=4) or partial response (n=3) to treatment. The median duration of complete response was 18 months (range, 13-20+), and the median duration of partial response was 10 months (range, 7-10+).
“Some patients treated with imetelstat have reverted back to normal bone marrow,” Dr Tefferi noted. “Typically, myelofibrosis is characterized by marrow scarring, and, although patients may derive symptomatic relief from other treatments, such as ruxolitinib, they usually do not revert back to normal bone marrow.”
Bone marrow fibrosis was reversed in all 4 patients with a complete response. A molecular response was documented in 3 of these patients.
Mutations and telomere length
“We noted a difference in response rates, especially in complete remission rates, in patients with and without certain specific gene mutations, such as ASXL1, SF3B1, and U2AF1,” Dr Tefferi noted. “This underscores the need for laboratory correlative studies in future clinical trials.”
Responses occurred in 27% of patients with a JAK2 mutation and 0% of patients without a JAK2 mutation (P=0.30). Alternatively, responses occurred in 32% of patients without an ASXL1 mutation and 0% of patients with an ASXL1 mutation (P=0.07).
The rate of complete response was 38% among patients with a mutation in SF3B1 or U2AF1, compared to 4% among patients without either mutation (P=0.04).
The investigators also found that responses were not correlated with baseline telomere length.
Toxicity
Dr Tefferi and his colleagues said the most clinically significant side effect of imetelstat was myelosuppression. It was the primary reason for a protocol-mandated dose reduction that occurred in 22 patients (67%).
Another “notable” side effect was the elevation of liver-enzyme levels. The investigators observed treatment-emergent (though not necessarily related) increases from baseline in total bilirubin (49%), alkaline phosphatase (58%), aspartate aminotransferase (58%), and alanine aminotransferase (27%).
None of these abnormalities were linked to clinically overt liver damage, and most patients ultimately saw their values return to baseline levels.
Adverse events that were considered at least possibly related to treatment and occurred in 3 or more patients included thrombocytopenia (45% grade 3/4), anemia (39% overall, 30% grade 3), neutropenia (27% grade 3/4), aspartate aminotransferase elevation (27% grade 1), alkaline phosphatase elevation (21% grade 1/2), elevation in total bilirubin (12% grade 1/2), infusion-related reactions (12% grade 1/2), diarrhea (9% grade 1/2), and epistaxis (9% grade 1/2).
Treatment discontinuation
At the data-cutoff date (December 5, 2014), 76% of patients had discontinued imetelstat (n=25). For all patients, the median duration of treatment was 8.6 months (range, 1.4 to 21.7).
Patients stopped treatment due to insufficient response (n=16), disease progression or relapse after response (n=3), death during the treatment period (n=2), adverse events (n=2), financial constraints (n=1), and pre-existing atrial fibrillation (n=1).
Both patients who discontinued imetelstat due to adverse events had persistent thrombocytopenia. Of the 2 deaths, 1 was considered treatment-related. That patient died of intracranial hemorrhage that was attributed to drug-induced, grade 4 thrombocytopenia after weekly dosing. The non-treatment-related death was the result of an upper gastrointestinal hemorrhage. ![]()
First biosimilar launched in US
© Sandoz Inc. 2015
The leukocyte growth factor Zarxio (filgrastim-sndz), the first biosimilar product to gain approval from the US Food and Drug Administration (FDA), is now available in the US.
Zarxio was approved by the FDA on March 6. The product, made by Sandoz, Inc., is biosimilar to Amgen Inc.’s Neupogen, which was originally licensed in 1991.
Zarxio is marketed as Zarzio outside the US. The biosimilar is available in more than 60 countries worldwide.
In the US, Zarxio is approved for the same indications as Neupogen. So Zarxio can be prescribed for the following 5 indications.
Patients with cancer receiving myelosuppressive chemotherapy: to decrease the incidence of infection, as manifested by febrile neutropenia, in patients with nonmyeloid malignancies receiving myelosuppressive anticancer drugs associated with a significant incidence of severe neutropenia with fever.
Patients with acute myeloid leukemia receiving induction or consolidation chemotherapy: to reduce the time to neutrophil recovery and the duration of fever, following induction or consolidation chemotherapy.
Patients with cancer undergoing bone marrow transplant: to reduce the duration of neutropenia and neutropenia-related clinical sequelae—eg, febrile neutropenia—in patients with nonmyeloid malignancies undergoing myeloablative chemotherapy followed by bone marrow transplant.
Patients undergoing autologous peripheral blood progenitor cell collection and therapy: for the mobilization of autologous hematopoietic progenitor cells into the peripheral blood for collection by leukapheresis.
Patients with severe chronic neutropenia: for chronic administration to reduce the incidence and duration of sequelae of neutropenia—eg, fever, infections, oropharyngeal ulcers—in symptomatic patients with congenital neutropenia, cyclic neutropenia, or idiopathic neutropenia.
PIONEER trial
The FDA’s approval of Zarxio was based on data showing that Zarxio is highly similar to Neupogen, with no clinically meaningful differences between the products.
The head-to-head PIONEER study was the final piece of evidence the FDA used to approve Zarxio as biosimilar to Neupogen. Results of the trial were presented at ASH 2014.
Zarxio and Neupogen both produced the expected reduction in the duration of severe neutropenia in breast cancer patients undergoing myelosuppressive chemotherapy—1.17 ± 1.11 and 1.20 ±1.02 days, respectively.
The mean time to absolute neutrophil count recovery in cycle 1 was also similar—1.8 ± 0.97 days in the Zarxio arm and 1.7 ± 0.81 days in the Neupogen arm. No immunogenicity or antibodies against rhG-CSF were detected throughout the study.
The researchers said there were no obvious differences between Zarxio and Neupogen with regard to treatment-emergent adverse events.
The most common side effects observed with Zarxio are aching bones/muscles and redness, swelling, or itching at the injection site. Serious side effects may include spleen rupture; serious allergic reactions that may cause rash, shortness of breath, wheezing and/or swelling around the mouth and eyes; fast pulse and sweating; and acute respiratory distress syndrome.
For more details on Zarxio, see the full prescribing information or visit www.zarxio.com. ![]()
© Sandoz Inc. 2015
The leukocyte growth factor Zarxio (filgrastim-sndz), the first biosimilar product to gain approval from the US Food and Drug Administration (FDA), is now available in the US.
Zarxio was approved by the FDA on March 6. The product, made by Sandoz, Inc., is biosimilar to Amgen Inc.’s Neupogen, which was originally licensed in 1991.
Zarxio is marketed as Zarzio outside the US. The biosimilar is available in more than 60 countries worldwide.
In the US, Zarxio is approved for the same indications as Neupogen. So Zarxio can be prescribed for the following 5 indications.
Patients with cancer receiving myelosuppressive chemotherapy: to decrease the incidence of infection, as manifested by febrile neutropenia, in patients with nonmyeloid malignancies receiving myelosuppressive anticancer drugs associated with a significant incidence of severe neutropenia with fever.
Patients with acute myeloid leukemia receiving induction or consolidation chemotherapy: to reduce the time to neutrophil recovery and the duration of fever, following induction or consolidation chemotherapy.
Patients with cancer undergoing bone marrow transplant: to reduce the duration of neutropenia and neutropenia-related clinical sequelae—eg, febrile neutropenia—in patients with nonmyeloid malignancies undergoing myeloablative chemotherapy followed by bone marrow transplant.
Patients undergoing autologous peripheral blood progenitor cell collection and therapy: for the mobilization of autologous hematopoietic progenitor cells into the peripheral blood for collection by leukapheresis.
Patients with severe chronic neutropenia: for chronic administration to reduce the incidence and duration of sequelae of neutropenia—eg, fever, infections, oropharyngeal ulcers—in symptomatic patients with congenital neutropenia, cyclic neutropenia, or idiopathic neutropenia.
PIONEER trial
The FDA’s approval of Zarxio was based on data showing that Zarxio is highly similar to Neupogen, with no clinically meaningful differences between the products.
The head-to-head PIONEER study was the final piece of evidence the FDA used to approve Zarxio as biosimilar to Neupogen. Results of the trial were presented at ASH 2014.
Zarxio and Neupogen both produced the expected reduction in the duration of severe neutropenia in breast cancer patients undergoing myelosuppressive chemotherapy—1.17 ± 1.11 and 1.20 ±1.02 days, respectively.
The mean time to absolute neutrophil count recovery in cycle 1 was also similar—1.8 ± 0.97 days in the Zarxio arm and 1.7 ± 0.81 days in the Neupogen arm. No immunogenicity or antibodies against rhG-CSF were detected throughout the study.
The researchers said there were no obvious differences between Zarxio and Neupogen with regard to treatment-emergent adverse events.
The most common side effects observed with Zarxio are aching bones/muscles and redness, swelling, or itching at the injection site. Serious side effects may include spleen rupture; serious allergic reactions that may cause rash, shortness of breath, wheezing and/or swelling around the mouth and eyes; fast pulse and sweating; and acute respiratory distress syndrome.
For more details on Zarxio, see the full prescribing information or visit www.zarxio.com. ![]()
© Sandoz Inc. 2015
The leukocyte growth factor Zarxio (filgrastim-sndz), the first biosimilar product to gain approval from the US Food and Drug Administration (FDA), is now available in the US.
Zarxio was approved by the FDA on March 6. The product, made by Sandoz, Inc., is biosimilar to Amgen Inc.’s Neupogen, which was originally licensed in 1991.
Zarxio is marketed as Zarzio outside the US. The biosimilar is available in more than 60 countries worldwide.
In the US, Zarxio is approved for the same indications as Neupogen. So Zarxio can be prescribed for the following 5 indications.
Patients with cancer receiving myelosuppressive chemotherapy: to decrease the incidence of infection, as manifested by febrile neutropenia, in patients with nonmyeloid malignancies receiving myelosuppressive anticancer drugs associated with a significant incidence of severe neutropenia with fever.
Patients with acute myeloid leukemia receiving induction or consolidation chemotherapy: to reduce the time to neutrophil recovery and the duration of fever, following induction or consolidation chemotherapy.
Patients with cancer undergoing bone marrow transplant: to reduce the duration of neutropenia and neutropenia-related clinical sequelae—eg, febrile neutropenia—in patients with nonmyeloid malignancies undergoing myeloablative chemotherapy followed by bone marrow transplant.
Patients undergoing autologous peripheral blood progenitor cell collection and therapy: for the mobilization of autologous hematopoietic progenitor cells into the peripheral blood for collection by leukapheresis.
Patients with severe chronic neutropenia: for chronic administration to reduce the incidence and duration of sequelae of neutropenia—eg, fever, infections, oropharyngeal ulcers—in symptomatic patients with congenital neutropenia, cyclic neutropenia, or idiopathic neutropenia.
PIONEER trial
The FDA’s approval of Zarxio was based on data showing that Zarxio is highly similar to Neupogen, with no clinically meaningful differences between the products.
The head-to-head PIONEER study was the final piece of evidence the FDA used to approve Zarxio as biosimilar to Neupogen. Results of the trial were presented at ASH 2014.
Zarxio and Neupogen both produced the expected reduction in the duration of severe neutropenia in breast cancer patients undergoing myelosuppressive chemotherapy—1.17 ± 1.11 and 1.20 ±1.02 days, respectively.
The mean time to absolute neutrophil count recovery in cycle 1 was also similar—1.8 ± 0.97 days in the Zarxio arm and 1.7 ± 0.81 days in the Neupogen arm. No immunogenicity or antibodies against rhG-CSF were detected throughout the study.
The researchers said there were no obvious differences between Zarxio and Neupogen with regard to treatment-emergent adverse events.
The most common side effects observed with Zarxio are aching bones/muscles and redness, swelling, or itching at the injection site. Serious side effects may include spleen rupture; serious allergic reactions that may cause rash, shortness of breath, wheezing and/or swelling around the mouth and eyes; fast pulse and sweating; and acute respiratory distress syndrome.
For more details on Zarxio, see the full prescribing information or visit www.zarxio.com. ![]()
Letter to the Editor
We greatly appreciate the thoughtful points made by Dr. Kerman regarding our recently published study evaluating the association of hospitalist continuity on adverse events (AEs).[1] We agree that a 7‐on/7‐off staffing model may limit discontinuity relative to models using shorter rotations lengths. Many hospital medicine programs use a 7‐on/7‐off model to optimize continuity. Longer rotation lengths are uncommon, as they may lead to fatigue and negatively affect physician work‐life balance. Shorter rotation lengths do exist, and we acknowledge that a study in a setting with greater fragmentation may have detected an effect.
We respectfully disagree with Dr. Kerman's concern that our methods for AE detection and confirmation may have been insensitive. We did not rely on incident reports, as these systems suffer from under‐reporting and often represent only a fraction of true AEs. We used a modified version of the classic 2‐stage method to identify and confirm AEs.[2] In the first stage, we used computerized screens, based on criteria from the Harvard Medical Practice Study and Institute for Healthcare Improvement global trigger tool, to identify potential AEs.[3, 4, 5] A research nurse created narrative summaries of potential AEs. A physician researcher then reviewed the narrative summaries to confirm whether an AE was truly present. This time‐consuming method is much more sensitive and specific than other options for patient safety measurement, including administrative data analyses and incident reporting systems.[6, 7]
With respect to other outcomes that may be affected by hospitalist continuity, we recently published a separate study showing that lower inpatient physician continuity was significantly associated with modest increases in hospital costs.[8] We found no association between continuity and patient satisfaction, but were likely underpowered to detect one. Interestingly, some of the models in our study suggested a slightly reduced risk of readmission with lower continuity. We were surprised by this finding and hypothesized that countervailing forces may be at play during handoffs of care from 1 hospitalist to another. Transitions of care introduce the opportunity for critical information to be lost, but they also introduce the potential for patient reassessment. A hospitalist newly taking over care from another may not be anchored to the initial diagnostic impressions and management plan established by the first. Of course, the potential benefit of a reassessment could only occur if the new hospitalist has time to perform one. At extremely high patient volumes, this theoretical benefit is unlikely to exist.
We did not include length of stay (LOS) as an outcome because hospitalist continuity and LOS are interdependent. Although discontinuity may lead to longer LOS, longer LOS definitely increases the probability of discontinuity. Thus, we controlled for LOS in our statistical models to isolate the effect of continuity. The study by Epstein and colleagues did not take into account the interdependence between LOS and hospitalist continuity.[9] Observational studies are not ideal for determining the effect of continuity on LOS. The Combing Incentives and Continuity Leading to Efficiency (CICLE) study by Chandra and colleagues was a pre‐post evaluation of a hospitalist staffing model specifically designed to improve continuity.[10] In the CICLE model, physicians work in a 4‐day rotation. On day 1, physicians exclusively admit patients. On day 2, physicians care for patients admitted on day 1 and accept patients admitted overnight. On days 3 and 4, physicians continue to care for patients received on days 1 and 2, but receive no additional patients. The remaining patients are transitioned to the next physician entering the cycle at the end of day 4. Chandra and colleagues found a 7.5% reduction in LOS and an 8.5% reduction in charges. Interestingly, they also found a 13.5% increase in readmissions that did not achieve statistical significance (P=0.08). The CICLE study suggests continuity does affect LOS, but is limited in that it did not account for a potential preexisting trend toward lower LOS.
Dr. Kerman presents data showing that it takes longer for a physician to care for a patient who is new to him or her than for a patient who is previously known. This finding has face validity. However, as we have suggested, the extra time spent by the oncoming physician may have both advantages and disadvantages. The disadvantages include time‐consuming cognitive work for the physician and the potential for information loss affecting patient care. The potential advantage is a second physician reassessing the diagnosis and management decisions established by the first, potentially correcting errors and optimizing care.
Ultimately, more research is needed to illuminate the effect of hospitalist continuity on patient outcomes. For now, we feel that hospital medicine group leaders need not institute lengthy rotations or staffing models that prioritize continuity above all other factors, as continuity appears to have little impact on patient outcomes.
- , , , et al. The effect of hospitalist discontinuity on adverse events. J Hosp Med. 2015;10(3):147–151.
- , , , et al. Comparison of manual abstraction to data warehouse facilitated abstraction to identify hosptial adverse events. BMJ Qual Saf. 2013;22(2):130–138.
- , . IHI global trigger tool for measuring adverse events: IHI innovation series white paper. Cambridge, MA: Institute for Healthcare Improvement; 2007.
- , BA, , et al. A study of medical injury and medical malpractice. N Engl J Med. 1989;321(7):480–484.
- , , , et al. Incidence and types of adverse events and negligent care in Utah and Colorado. Med Care. 2000;38(3):261–271.
- . The elephant of patient safety: what you see depends on how you look. Jt Comm J Qual Patient Saf. 2010;36(9):399–401.
- , . Measuring errors and adverse events in health care. J Gen Intern Med. 2003;18(1):61–67.
- , , , et al. The impact of hospitalist discontinuity on hospital cost, readmissions, and patient satisfaction. J Gen Intern Med. 2014;29(7):1004–1008.
- , , , , . The impact of fragmentation of hospitalist care on length of stay. J Hosp Med. 2010;5(6):335–338.
- , , . The Creating Incentives and Continuity Leading to Efficiency staffing model: a quality improvement initiative in hospital medicine. Mayo Clin Proc. 2012;87(4):364–371.
We greatly appreciate the thoughtful points made by Dr. Kerman regarding our recently published study evaluating the association of hospitalist continuity on adverse events (AEs).[1] We agree that a 7‐on/7‐off staffing model may limit discontinuity relative to models using shorter rotations lengths. Many hospital medicine programs use a 7‐on/7‐off model to optimize continuity. Longer rotation lengths are uncommon, as they may lead to fatigue and negatively affect physician work‐life balance. Shorter rotation lengths do exist, and we acknowledge that a study in a setting with greater fragmentation may have detected an effect.
We respectfully disagree with Dr. Kerman's concern that our methods for AE detection and confirmation may have been insensitive. We did not rely on incident reports, as these systems suffer from under‐reporting and often represent only a fraction of true AEs. We used a modified version of the classic 2‐stage method to identify and confirm AEs.[2] In the first stage, we used computerized screens, based on criteria from the Harvard Medical Practice Study and Institute for Healthcare Improvement global trigger tool, to identify potential AEs.[3, 4, 5] A research nurse created narrative summaries of potential AEs. A physician researcher then reviewed the narrative summaries to confirm whether an AE was truly present. This time‐consuming method is much more sensitive and specific than other options for patient safety measurement, including administrative data analyses and incident reporting systems.[6, 7]
With respect to other outcomes that may be affected by hospitalist continuity, we recently published a separate study showing that lower inpatient physician continuity was significantly associated with modest increases in hospital costs.[8] We found no association between continuity and patient satisfaction, but were likely underpowered to detect one. Interestingly, some of the models in our study suggested a slightly reduced risk of readmission with lower continuity. We were surprised by this finding and hypothesized that countervailing forces may be at play during handoffs of care from 1 hospitalist to another. Transitions of care introduce the opportunity for critical information to be lost, but they also introduce the potential for patient reassessment. A hospitalist newly taking over care from another may not be anchored to the initial diagnostic impressions and management plan established by the first. Of course, the potential benefit of a reassessment could only occur if the new hospitalist has time to perform one. At extremely high patient volumes, this theoretical benefit is unlikely to exist.
We did not include length of stay (LOS) as an outcome because hospitalist continuity and LOS are interdependent. Although discontinuity may lead to longer LOS, longer LOS definitely increases the probability of discontinuity. Thus, we controlled for LOS in our statistical models to isolate the effect of continuity. The study by Epstein and colleagues did not take into account the interdependence between LOS and hospitalist continuity.[9] Observational studies are not ideal for determining the effect of continuity on LOS. The Combing Incentives and Continuity Leading to Efficiency (CICLE) study by Chandra and colleagues was a pre‐post evaluation of a hospitalist staffing model specifically designed to improve continuity.[10] In the CICLE model, physicians work in a 4‐day rotation. On day 1, physicians exclusively admit patients. On day 2, physicians care for patients admitted on day 1 and accept patients admitted overnight. On days 3 and 4, physicians continue to care for patients received on days 1 and 2, but receive no additional patients. The remaining patients are transitioned to the next physician entering the cycle at the end of day 4. Chandra and colleagues found a 7.5% reduction in LOS and an 8.5% reduction in charges. Interestingly, they also found a 13.5% increase in readmissions that did not achieve statistical significance (P=0.08). The CICLE study suggests continuity does affect LOS, but is limited in that it did not account for a potential preexisting trend toward lower LOS.
Dr. Kerman presents data showing that it takes longer for a physician to care for a patient who is new to him or her than for a patient who is previously known. This finding has face validity. However, as we have suggested, the extra time spent by the oncoming physician may have both advantages and disadvantages. The disadvantages include time‐consuming cognitive work for the physician and the potential for information loss affecting patient care. The potential advantage is a second physician reassessing the diagnosis and management decisions established by the first, potentially correcting errors and optimizing care.
Ultimately, more research is needed to illuminate the effect of hospitalist continuity on patient outcomes. For now, we feel that hospital medicine group leaders need not institute lengthy rotations or staffing models that prioritize continuity above all other factors, as continuity appears to have little impact on patient outcomes.
We greatly appreciate the thoughtful points made by Dr. Kerman regarding our recently published study evaluating the association of hospitalist continuity on adverse events (AEs).[1] We agree that a 7‐on/7‐off staffing model may limit discontinuity relative to models using shorter rotations lengths. Many hospital medicine programs use a 7‐on/7‐off model to optimize continuity. Longer rotation lengths are uncommon, as they may lead to fatigue and negatively affect physician work‐life balance. Shorter rotation lengths do exist, and we acknowledge that a study in a setting with greater fragmentation may have detected an effect.
We respectfully disagree with Dr. Kerman's concern that our methods for AE detection and confirmation may have been insensitive. We did not rely on incident reports, as these systems suffer from under‐reporting and often represent only a fraction of true AEs. We used a modified version of the classic 2‐stage method to identify and confirm AEs.[2] In the first stage, we used computerized screens, based on criteria from the Harvard Medical Practice Study and Institute for Healthcare Improvement global trigger tool, to identify potential AEs.[3, 4, 5] A research nurse created narrative summaries of potential AEs. A physician researcher then reviewed the narrative summaries to confirm whether an AE was truly present. This time‐consuming method is much more sensitive and specific than other options for patient safety measurement, including administrative data analyses and incident reporting systems.[6, 7]
With respect to other outcomes that may be affected by hospitalist continuity, we recently published a separate study showing that lower inpatient physician continuity was significantly associated with modest increases in hospital costs.[8] We found no association between continuity and patient satisfaction, but were likely underpowered to detect one. Interestingly, some of the models in our study suggested a slightly reduced risk of readmission with lower continuity. We were surprised by this finding and hypothesized that countervailing forces may be at play during handoffs of care from 1 hospitalist to another. Transitions of care introduce the opportunity for critical information to be lost, but they also introduce the potential for patient reassessment. A hospitalist newly taking over care from another may not be anchored to the initial diagnostic impressions and management plan established by the first. Of course, the potential benefit of a reassessment could only occur if the new hospitalist has time to perform one. At extremely high patient volumes, this theoretical benefit is unlikely to exist.
We did not include length of stay (LOS) as an outcome because hospitalist continuity and LOS are interdependent. Although discontinuity may lead to longer LOS, longer LOS definitely increases the probability of discontinuity. Thus, we controlled for LOS in our statistical models to isolate the effect of continuity. The study by Epstein and colleagues did not take into account the interdependence between LOS and hospitalist continuity.[9] Observational studies are not ideal for determining the effect of continuity on LOS. The Combing Incentives and Continuity Leading to Efficiency (CICLE) study by Chandra and colleagues was a pre‐post evaluation of a hospitalist staffing model specifically designed to improve continuity.[10] In the CICLE model, physicians work in a 4‐day rotation. On day 1, physicians exclusively admit patients. On day 2, physicians care for patients admitted on day 1 and accept patients admitted overnight. On days 3 and 4, physicians continue to care for patients received on days 1 and 2, but receive no additional patients. The remaining patients are transitioned to the next physician entering the cycle at the end of day 4. Chandra and colleagues found a 7.5% reduction in LOS and an 8.5% reduction in charges. Interestingly, they also found a 13.5% increase in readmissions that did not achieve statistical significance (P=0.08). The CICLE study suggests continuity does affect LOS, but is limited in that it did not account for a potential preexisting trend toward lower LOS.
Dr. Kerman presents data showing that it takes longer for a physician to care for a patient who is new to him or her than for a patient who is previously known. This finding has face validity. However, as we have suggested, the extra time spent by the oncoming physician may have both advantages and disadvantages. The disadvantages include time‐consuming cognitive work for the physician and the potential for information loss affecting patient care. The potential advantage is a second physician reassessing the diagnosis and management decisions established by the first, potentially correcting errors and optimizing care.
Ultimately, more research is needed to illuminate the effect of hospitalist continuity on patient outcomes. For now, we feel that hospital medicine group leaders need not institute lengthy rotations or staffing models that prioritize continuity above all other factors, as continuity appears to have little impact on patient outcomes.
- , , , et al. The effect of hospitalist discontinuity on adverse events. J Hosp Med. 2015;10(3):147–151.
- , , , et al. Comparison of manual abstraction to data warehouse facilitated abstraction to identify hosptial adverse events. BMJ Qual Saf. 2013;22(2):130–138.
- , . IHI global trigger tool for measuring adverse events: IHI innovation series white paper. Cambridge, MA: Institute for Healthcare Improvement; 2007.
- , BA, , et al. A study of medical injury and medical malpractice. N Engl J Med. 1989;321(7):480–484.
- , , , et al. Incidence and types of adverse events and negligent care in Utah and Colorado. Med Care. 2000;38(3):261–271.
- . The elephant of patient safety: what you see depends on how you look. Jt Comm J Qual Patient Saf. 2010;36(9):399–401.
- , . Measuring errors and adverse events in health care. J Gen Intern Med. 2003;18(1):61–67.
- , , , et al. The impact of hospitalist discontinuity on hospital cost, readmissions, and patient satisfaction. J Gen Intern Med. 2014;29(7):1004–1008.
- , , , , . The impact of fragmentation of hospitalist care on length of stay. J Hosp Med. 2010;5(6):335–338.
- , , . The Creating Incentives and Continuity Leading to Efficiency staffing model: a quality improvement initiative in hospital medicine. Mayo Clin Proc. 2012;87(4):364–371.
- , , , et al. The effect of hospitalist discontinuity on adverse events. J Hosp Med. 2015;10(3):147–151.
- , , , et al. Comparison of manual abstraction to data warehouse facilitated abstraction to identify hosptial adverse events. BMJ Qual Saf. 2013;22(2):130–138.
- , . IHI global trigger tool for measuring adverse events: IHI innovation series white paper. Cambridge, MA: Institute for Healthcare Improvement; 2007.
- , BA, , et al. A study of medical injury and medical malpractice. N Engl J Med. 1989;321(7):480–484.
- , , , et al. Incidence and types of adverse events and negligent care in Utah and Colorado. Med Care. 2000;38(3):261–271.
- . The elephant of patient safety: what you see depends on how you look. Jt Comm J Qual Patient Saf. 2010;36(9):399–401.
- , . Measuring errors and adverse events in health care. J Gen Intern Med. 2003;18(1):61–67.
- , , , et al. The impact of hospitalist discontinuity on hospital cost, readmissions, and patient satisfaction. J Gen Intern Med. 2014;29(7):1004–1008.
- , , , , . The impact of fragmentation of hospitalist care on length of stay. J Hosp Med. 2010;5(6):335–338.
- , , . The Creating Incentives and Continuity Leading to Efficiency staffing model: a quality improvement initiative in hospital medicine. Mayo Clin Proc. 2012;87(4):364–371.
Letter to the Editor
Congratulations once again to Dr. Kevin O'Leary and his team at Northwestern Memorial Hospital for adding yet another thoughtful piece of research to the debate around continuity of care and team dynamics with their study The Effect of Hospitalist Continuity on Adverse Events published in the March 2015 issue of the Journal of Hospital Medicine.[1] However, I believe it would be unfortunate for their current negative study on frequency of adverse events to cause us to lose sight of the potential centrality of continuity of care to overall quality and efficiency and the need for further research.
I would like to add a perspective from our institutions experience, where fragmentation of care is common, with a focus on effects of continuity on work productivity. I would also like to comment in the context of the bigger scheme, where I believe there is a great deal of evidence that continuity of care is highly desirable for multiple reasons. Continuity in the inpatient setting has been shown to have effects on: (1) provider satisfaction, (2) length of stay, (3) efficiency, safety/medical errors, and (4) cost of care, patient satisfaction and readmission rates.[2]
For example, a study by Chandra et al.[3] showed improved continuity using the Creating Incentives and Continuity Leading to Efficiency model, which decreased LOS and reduced mean total charges by 20%. There is also evidence from the outpatient setting that greater continuity has been associated with better hypertensive control, lower risk of hospitalization, fewer emergency department visits, higher patient satisfaction, and higher physician satisfaction.[4, 5, 6, 7, 8]
Not captured in any of the literature I am aware of to date is the effect on work productivity of care fragmentation. According to data from an unpublished time study at our institution, each change of service to a new provider required an average of 12 extra minutes for rounding on each new patient to be evaluated, which included both time spent studying the chart as well as extra time spent reassessing the patient at the bedside. When we restructured our service to improve continuity by increasing the number of patients admitted and followed by the same provider (from 14% to 31%), we reduced the number of providers per stay from 2.4 to 2.1, and reduced the number of annual handoffs by 3600, and found that a total of 900 hours, or 0.45 full‐time equivalents (FTE) per year, were saved for our program of approximately 30 FTEs.
Although the study by O'Leary et al. is an important contribution to the literature, more research needs to be done on the effects of fragmentation and the benefits of continuity. Although safety and adverse events are among the most important indicators to look at, they also may represent a weak and hard to pick up signal without a multicentered and statistically high‐powered study. Extending the study of continuity's effects, including efficiency, safety, costs, provider and patient satisfaction, and readmissions, is well worth further effort.
- , , , et al. The effect of hospitalist discontinuity on adverse events. J Hosp Med. 2015;10(3):147–151.
- , , , , . The impact of fragmentation of hospitalist care on length of stay. J Hosp Med. 2010;5(6):335–338.
- , , . The Creating Incentives and Continuity Leading to Efficiency staffing model: a quality improvement initiative in hospital medicine. Mayo Clin Proc. 2012;87:364–371.
- , , , et al. The impact of hospitalist discontinuity on hospital cost, readmissions, and patient satisfaction. J Gen Intern Med. 2014;29(7):1004–1008.
- , , , . The association between continuity of care and outcomes: a systematic and critical review. J Eval Clin Pract. 2010;16:947–956.
- , . Interpersonal continuity of care and patient satisfaction: a critical review. Ann Fam Med. 2004;2:445–451.
- , , , . Continuity of care in a family practice residency program. Impact on physician satisfaction. J Fam Pract. 1990;31:69–73.
- , . Interpersonal continuity of care and care outcomes: a critical review. Ann Fam Med. 2005;3:159–166.
Congratulations once again to Dr. Kevin O'Leary and his team at Northwestern Memorial Hospital for adding yet another thoughtful piece of research to the debate around continuity of care and team dynamics with their study The Effect of Hospitalist Continuity on Adverse Events published in the March 2015 issue of the Journal of Hospital Medicine.[1] However, I believe it would be unfortunate for their current negative study on frequency of adverse events to cause us to lose sight of the potential centrality of continuity of care to overall quality and efficiency and the need for further research.
I would like to add a perspective from our institutions experience, where fragmentation of care is common, with a focus on effects of continuity on work productivity. I would also like to comment in the context of the bigger scheme, where I believe there is a great deal of evidence that continuity of care is highly desirable for multiple reasons. Continuity in the inpatient setting has been shown to have effects on: (1) provider satisfaction, (2) length of stay, (3) efficiency, safety/medical errors, and (4) cost of care, patient satisfaction and readmission rates.[2]
For example, a study by Chandra et al.[3] showed improved continuity using the Creating Incentives and Continuity Leading to Efficiency model, which decreased LOS and reduced mean total charges by 20%. There is also evidence from the outpatient setting that greater continuity has been associated with better hypertensive control, lower risk of hospitalization, fewer emergency department visits, higher patient satisfaction, and higher physician satisfaction.[4, 5, 6, 7, 8]
Not captured in any of the literature I am aware of to date is the effect on work productivity of care fragmentation. According to data from an unpublished time study at our institution, each change of service to a new provider required an average of 12 extra minutes for rounding on each new patient to be evaluated, which included both time spent studying the chart as well as extra time spent reassessing the patient at the bedside. When we restructured our service to improve continuity by increasing the number of patients admitted and followed by the same provider (from 14% to 31%), we reduced the number of providers per stay from 2.4 to 2.1, and reduced the number of annual handoffs by 3600, and found that a total of 900 hours, or 0.45 full‐time equivalents (FTE) per year, were saved for our program of approximately 30 FTEs.
Although the study by O'Leary et al. is an important contribution to the literature, more research needs to be done on the effects of fragmentation and the benefits of continuity. Although safety and adverse events are among the most important indicators to look at, they also may represent a weak and hard to pick up signal without a multicentered and statistically high‐powered study. Extending the study of continuity's effects, including efficiency, safety, costs, provider and patient satisfaction, and readmissions, is well worth further effort.
Congratulations once again to Dr. Kevin O'Leary and his team at Northwestern Memorial Hospital for adding yet another thoughtful piece of research to the debate around continuity of care and team dynamics with their study The Effect of Hospitalist Continuity on Adverse Events published in the March 2015 issue of the Journal of Hospital Medicine.[1] However, I believe it would be unfortunate for their current negative study on frequency of adverse events to cause us to lose sight of the potential centrality of continuity of care to overall quality and efficiency and the need for further research.
I would like to add a perspective from our institutions experience, where fragmentation of care is common, with a focus on effects of continuity on work productivity. I would also like to comment in the context of the bigger scheme, where I believe there is a great deal of evidence that continuity of care is highly desirable for multiple reasons. Continuity in the inpatient setting has been shown to have effects on: (1) provider satisfaction, (2) length of stay, (3) efficiency, safety/medical errors, and (4) cost of care, patient satisfaction and readmission rates.[2]
For example, a study by Chandra et al.[3] showed improved continuity using the Creating Incentives and Continuity Leading to Efficiency model, which decreased LOS and reduced mean total charges by 20%. There is also evidence from the outpatient setting that greater continuity has been associated with better hypertensive control, lower risk of hospitalization, fewer emergency department visits, higher patient satisfaction, and higher physician satisfaction.[4, 5, 6, 7, 8]
Not captured in any of the literature I am aware of to date is the effect on work productivity of care fragmentation. According to data from an unpublished time study at our institution, each change of service to a new provider required an average of 12 extra minutes for rounding on each new patient to be evaluated, which included both time spent studying the chart as well as extra time spent reassessing the patient at the bedside. When we restructured our service to improve continuity by increasing the number of patients admitted and followed by the same provider (from 14% to 31%), we reduced the number of providers per stay from 2.4 to 2.1, and reduced the number of annual handoffs by 3600, and found that a total of 900 hours, or 0.45 full‐time equivalents (FTE) per year, were saved for our program of approximately 30 FTEs.
Although the study by O'Leary et al. is an important contribution to the literature, more research needs to be done on the effects of fragmentation and the benefits of continuity. Although safety and adverse events are among the most important indicators to look at, they also may represent a weak and hard to pick up signal without a multicentered and statistically high‐powered study. Extending the study of continuity's effects, including efficiency, safety, costs, provider and patient satisfaction, and readmissions, is well worth further effort.
- , , , et al. The effect of hospitalist discontinuity on adverse events. J Hosp Med. 2015;10(3):147–151.
- , , , , . The impact of fragmentation of hospitalist care on length of stay. J Hosp Med. 2010;5(6):335–338.
- , , . The Creating Incentives and Continuity Leading to Efficiency staffing model: a quality improvement initiative in hospital medicine. Mayo Clin Proc. 2012;87:364–371.
- , , , et al. The impact of hospitalist discontinuity on hospital cost, readmissions, and patient satisfaction. J Gen Intern Med. 2014;29(7):1004–1008.
- , , , . The association between continuity of care and outcomes: a systematic and critical review. J Eval Clin Pract. 2010;16:947–956.
- , . Interpersonal continuity of care and patient satisfaction: a critical review. Ann Fam Med. 2004;2:445–451.
- , , , . Continuity of care in a family practice residency program. Impact on physician satisfaction. J Fam Pract. 1990;31:69–73.
- , . Interpersonal continuity of care and care outcomes: a critical review. Ann Fam Med. 2005;3:159–166.
- , , , et al. The effect of hospitalist discontinuity on adverse events. J Hosp Med. 2015;10(3):147–151.
- , , , , . The impact of fragmentation of hospitalist care on length of stay. J Hosp Med. 2010;5(6):335–338.
- , , . The Creating Incentives and Continuity Leading to Efficiency staffing model: a quality improvement initiative in hospital medicine. Mayo Clin Proc. 2012;87:364–371.
- , , , et al. The impact of hospitalist discontinuity on hospital cost, readmissions, and patient satisfaction. J Gen Intern Med. 2014;29(7):1004–1008.
- , , , . The association between continuity of care and outcomes: a systematic and critical review. J Eval Clin Pract. 2010;16:947–956.
- , . Interpersonal continuity of care and patient satisfaction: a critical review. Ann Fam Med. 2004;2:445–451.
- , , , . Continuity of care in a family practice residency program. Impact on physician satisfaction. J Fam Pract. 1990;31:69–73.
- , . Interpersonal continuity of care and care outcomes: a critical review. Ann Fam Med. 2005;3:159–166.
Improving Anticoagulation Transitions
Anticoagulants are among the prescriptions with the highest risk of an adverse drug event (ADE) after discharge, and many of these events are preventable.[1, 2, 3] In recognition of the high risk for adverse events, the Institute for Healthcare Improvement Map details several key features of a safe anticoagulation management program, including the recommendation during the transition period that clinicians ensure proper lab monitoring and establish capacity for follow‐up testing.[4]
Despite the potential for harm, most hospitals do not have a structured process for the transmission of vital information related to warfarin management from the inpatient to the ambulatory setting. Our aim was to develop a concise report containing the essential information regarding the patient's anticoagulation regimen, the Safe Transitions Anticoagulation Report (STAR), and a process to ensure the report can be readily accessed and utilized by ambulatory clinicians.
METHODS
We assembled an interdisciplinary team to develop a new report and workflow to ensure that information on inpatient warfarin management was transmitted to outpatient providers in a reliable and structured manner. Explicit goals were to maximize use of the electronic medical record (EMR) to autopopulate aspects of the new report and create a process that worked seamlessly into the workflow. The final items were selected based on the risk of harm if not conveyed and feasibility of incorporation through the EMR:
- Warfarin doses: the 7 warfarin doses immediately prior to discharge
- International normalized ratio (INR) values: the 7 INR values immediately prior to discharge
- Bridging anticoagulation: the low‐molecular‐weight heparin (LMWH) prescribed as a bridging anticoagulant, if any
The STAR resides in both the discharge summary and the after visit summary (AVS) for patients discharged on warfarin. At our institution, the AVS contains a medication list, discharge instructions, and appointments, and is automatically produced through our EMR. Our institution utilizes the Epic EMR (Epic Systems Corp., Verona, WI) in the hospital, ambulatory clinics, and faculty practices.
A process was developed where a structured table is automatically created (Figure 1). A field was added to our EMR's discharge summary template asking whether the patient is being discharged on warfarin. Answering yes produces a second question asking whether the patient is also being discharged on bridging anticoagulation with LMWH. The STAR is not inserted into the discharge summary if the clinician completing the discharge summary deletes the anticoagulation question. The STAR is automatically created for patients discharged on warfarin and inserted into the AVS by the EMR regardless of whether the discharge summary has been completed. Patients are instructed by their nurse to bring their AVS to their follow‐up appointments.
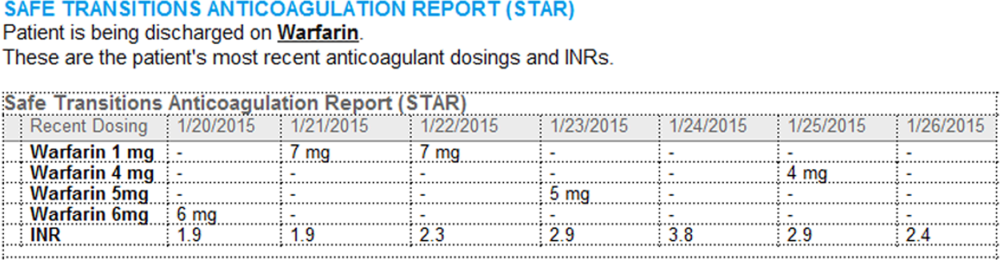
The STAR project team utilized plan‐do‐study‐act cycles to test small changes and make revisions. The workflow was piloted on 2 medical/surgical units from January 2014 through March 2014, and revised based on feedback from clinicians and nursing staff. The STAR initiative was fully implemented across our institution in April 2014.
The study was evaluated by the institutional review board of the Icahn School of Medicine at Mount Sinai, and full review was waived.
Outcomes
Our primary outcomes were the timeliness of laboratory monitoring and quality of anticoagulation management for patients with an established relationship at 1 of the main outpatient practices in our system. Our institution has an anticoagulation clinic for patients followed at the general medicine clinic. We defined an established relationship as having been seen in the same practice on at least 2 occasions in the 12 months prior to admission. The primary outcomes were the percentage of patients who had an INR measurement done and the percentage who had a therapeutic INR value within 10 days after discharge. As the 10‐day period is arbitrary, we also assessed these outcomes for the 3‐, 7‐, and 30‐day periods after discharge. The therapeutic range was defined for all patients as an INR of 2.0 to 3.0, as this is the target range for the large majority of patients on warfarin in our system. Outcomes during the intervention period were compared to baseline values during the preintervention period. For patients with multiple admissions, the first admission during each period was included.
Ambulatory Physician Survey
We surveyed ambulatory care physicians at the main practices for our health system. The survey assessed how often the STAR was viewed and incorporated into decision making, whether the report improved workflow, and whether ambulatory providers perceived that the report increased patient safety. The survey was distributed at the 6‐month interval during the intervention phase. The survey was disseminated electronically on 3 occasions, and a paper version was distributed on 1 occasion to housestaff and general medicine faculty.
Statistical Analysis
Comparisons for categorical data were performed using the 2 test. P values were based on 2‐tailed tests of significance, and a value 0.05 was considered significant.
RESULTS
The STAR was embedded in the discharge summary for 1370 (78.6%) discharges during the intervention period. A total of 505 patients in the preintervention period and 292 patients in the intervention period were established patients at an affiliated practice and comprised the study population. Demographics and indications for warfarin for the preintervention and intervention groups are listed in the Table 1.
| Preintervention Group, N=505 | Intervention Group, N=292 | P Value | |
|---|---|---|---|
| |||
| Age, y | 66.7 | 68.0 | 0.29 |
| Male gender, n (%) | 236 (46.7) | 153 (52.4) | 0.12 |
| Discharged on bridging agent, n (%) | 90 (17.8) | 36 (12.3) | 0.04 |
| Average length of stay, d | 7.1 | 7.6 | 0.46 |
| Newly prescribed warfarin, n (%) | 147 (29.1) | 62 (21.2) | 0.01 |
| INR 2.03.0 range at discharge, n (%) | 187 (37.0) | 137 (46.9) | 0.02 |
| Warfarin indication, n (%)* | |||
| Venous thromboembolism | 93 (18.4) | 39 (13.4) | |
| Atrial fibrillation | 204 (40.3) | 127 (43.5) | |
| Mechanical heart valve | 19 (3.8) | 17 (6.5) | |
| Prevention of thromboembolism | 142 (28.1) | 94 (32.2) | |
| Intracardiac thrombosis | 3 (0.5) | 6 (2.1) | |
| Thrombophilia | 4 (0.8) | 5 (1.7) | |
| Other | 19 (3.8) | 12 (4.1) | |
| No indication | 32 (6.3) | 1 (3.8) | |
The frequency of INR testing within 10 days of discharge was similar for the preintervention and intervention periods (41.4% and 47.6%, respectively, P=0.09). Similarly, the likelihood of having at least 1 therapeutic INR value within 10 days of discharge was not statistically different for the groups (17.0% and 21.2%, P=0.14). The pattern was similar for the 3‐, 7‐, and 30‐day periods; a higher percentage of the intervention group had INR testing and attained a therapeutic INR value, though for no time period did this reach statistical significance. This pattern was also found when limiting the analysis to patients discharged home rather than to a facility, patients on warfarin, prior to admission, and patients started on warfarin during the hospitalization.
A total of 87of 207 (42.0%) clinicians responded to the survey. Of respondents, 75% reported that they had seen 1 patient who had been discharged on warfarin since the STAR initiative had begun, 58% of whom reported having viewed the STAR. Most respondents who viewed the STAR found it to be helpful or very helpful in guiding warfarin management (67%), improving their workflow and efficiency (58%), and improving patient safety (77%). Approximately one‐third of respondents who had viewed the STAR (34%) reported that they selected a different dose than they would have chosen had the STAR not been available.
DISCUSSION
We developed a concise report that is seamlessly created and inserted into the discharge summary. Though the STAR was perceived as improving patient safety by ambulatory care providers, there was no impact on attaining a therapeutic INR after discharge. There are several possible explanations for a lack of benefit. Most notably, our intervention was comprised of a stand‐alone EMR‐based tool and focused on 1 component of the transitions process. Given the complexity of healthcare delivery and anticoagulant management, it is likely that broader interventions are required to improve clinical outcomes over the transition period. Potential targets of multifaceted approaches may include improving access to care, providing greater access to anticoagulation clinics, enhancing patient education, and promoting direct physician‐physician communication. Bundled interventions will likely need to include involvement of an interdisciplinary team, such as pharmacist involvement in the medication reconciliation process.
The transition period from the hospital to the outpatient setting has the potential to jeopardize patient safety if vital information is not reliably transmitted across venues.[5, 6, 7, 8] Forster and colleagues noted an 11% incidence of ADEs in the posthospitalization period, of which 60% were either preventable or ameliorable.[3] To decrease the risk to patient safety during the transitions period, the Transitions of Care Consensus policy statement by the Society of Hospital Medicine and other medical organizations called for incorporation of standard data transfer forms (templates and transmission protocols).[9] Despite the high risk and preventable nature of many of the events, few specific tools have been developed. As part of broader initiatives to improve the transitions process, the STAR may have the potential to be a means for health systems to improve the quality of the transition of care for patients on anticoagulants.
Our study has several limitations. First, it was performed at a single health system. It is unknown whether the EMR‐based report could be similarly employed at other systems. Second, our study was unable to assess clinical endpoints. Given the lack of effect on attaining a therapeutic INR, it is unlikely that downstream outcomes, such as thromboembolism, were impacted. Lastly, we were unable to examine whether our intervention improved the care of patients whose outpatient provider was external to our system.
The STAR is a concise tool developed to provide essential anticoagulant‐related information to ambulatory providers. Though the report was perceived as improving patient safety, our finding of a lack of impact on attaining a therapeutic INR after discharge suggests that the tool would need to be a component of a broader multifaceted intervention to impact clinical outcomes.
Disclosures
This project was funded by a grant from the Cardinal Health E3 Foundation. The authors report no conflicts of interest.
- , , , Medication errors: experience of the United States Pharmacopeia (USP) MEDMARX reporting system. J Clin Pharm. 2003;43:760–767.
- , , , , The incidence and severity of adverse events affecting patients after discharge from the hospital. Ann Intern Med. 2003;138:161–167.
- , , , , Adverse drug events occurring following hospital discharge. J Gen Int Med. 2005;204:317–323.
- IHI Improvement Map. Available at: http://app.ihi.org/imap/tool/#Process=54aa289b‐16fd‐4a64‐8329‐3941dfc565d1. Accessed February 20 2015.
- , , , Promoting effective transitions of care at hospital discharge: a review of key issues for hospitalists. J Hosp Med. 2007;2:314–323.
- , , , , , Deficits in communication and information transfer between hospital‐based and primary care physicians: implications for patient safety and continuity of care. JAMA. 2007;297:831–841.
- Transitions of care in patients receiving oral anticoagulants: general principles, procedures, and impact of new oral anticoagulants. J Cardiovasc Nurs. 2013;28:54–65.
- Care transitions in anticoagulation management for patients with atrial fibrillation: an emphasis on safety. Oschner J. 2013;13:419–427.
- , , , et al. Transitions of Care Consensus policy statement: American College of Physicians, Society of General Internal Medicine, Society of Hospital Medicine, American Geriatrics Society, American College Of Emergency Physicians, and Society for Academic Emergency Medicine. J Hosp Med. 2009;4:364–370.
Anticoagulants are among the prescriptions with the highest risk of an adverse drug event (ADE) after discharge, and many of these events are preventable.[1, 2, 3] In recognition of the high risk for adverse events, the Institute for Healthcare Improvement Map details several key features of a safe anticoagulation management program, including the recommendation during the transition period that clinicians ensure proper lab monitoring and establish capacity for follow‐up testing.[4]
Despite the potential for harm, most hospitals do not have a structured process for the transmission of vital information related to warfarin management from the inpatient to the ambulatory setting. Our aim was to develop a concise report containing the essential information regarding the patient's anticoagulation regimen, the Safe Transitions Anticoagulation Report (STAR), and a process to ensure the report can be readily accessed and utilized by ambulatory clinicians.
METHODS
We assembled an interdisciplinary team to develop a new report and workflow to ensure that information on inpatient warfarin management was transmitted to outpatient providers in a reliable and structured manner. Explicit goals were to maximize use of the electronic medical record (EMR) to autopopulate aspects of the new report and create a process that worked seamlessly into the workflow. The final items were selected based on the risk of harm if not conveyed and feasibility of incorporation through the EMR:
- Warfarin doses: the 7 warfarin doses immediately prior to discharge
- International normalized ratio (INR) values: the 7 INR values immediately prior to discharge
- Bridging anticoagulation: the low‐molecular‐weight heparin (LMWH) prescribed as a bridging anticoagulant, if any
The STAR resides in both the discharge summary and the after visit summary (AVS) for patients discharged on warfarin. At our institution, the AVS contains a medication list, discharge instructions, and appointments, and is automatically produced through our EMR. Our institution utilizes the Epic EMR (Epic Systems Corp., Verona, WI) in the hospital, ambulatory clinics, and faculty practices.
A process was developed where a structured table is automatically created (Figure 1). A field was added to our EMR's discharge summary template asking whether the patient is being discharged on warfarin. Answering yes produces a second question asking whether the patient is also being discharged on bridging anticoagulation with LMWH. The STAR is not inserted into the discharge summary if the clinician completing the discharge summary deletes the anticoagulation question. The STAR is automatically created for patients discharged on warfarin and inserted into the AVS by the EMR regardless of whether the discharge summary has been completed. Patients are instructed by their nurse to bring their AVS to their follow‐up appointments.
The STAR project team utilized plan‐do‐study‐act cycles to test small changes and make revisions. The workflow was piloted on 2 medical/surgical units from January 2014 through March 2014, and revised based on feedback from clinicians and nursing staff. The STAR initiative was fully implemented across our institution in April 2014.
The study was evaluated by the institutional review board of the Icahn School of Medicine at Mount Sinai, and full review was waived.
Outcomes
Our primary outcomes were the timeliness of laboratory monitoring and quality of anticoagulation management for patients with an established relationship at 1 of the main outpatient practices in our system. Our institution has an anticoagulation clinic for patients followed at the general medicine clinic. We defined an established relationship as having been seen in the same practice on at least 2 occasions in the 12 months prior to admission. The primary outcomes were the percentage of patients who had an INR measurement done and the percentage who had a therapeutic INR value within 10 days after discharge. As the 10‐day period is arbitrary, we also assessed these outcomes for the 3‐, 7‐, and 30‐day periods after discharge. The therapeutic range was defined for all patients as an INR of 2.0 to 3.0, as this is the target range for the large majority of patients on warfarin in our system. Outcomes during the intervention period were compared to baseline values during the preintervention period. For patients with multiple admissions, the first admission during each period was included.
Ambulatory Physician Survey
We surveyed ambulatory care physicians at the main practices for our health system. The survey assessed how often the STAR was viewed and incorporated into decision making, whether the report improved workflow, and whether ambulatory providers perceived that the report increased patient safety. The survey was distributed at the 6‐month interval during the intervention phase. The survey was disseminated electronically on 3 occasions, and a paper version was distributed on 1 occasion to housestaff and general medicine faculty.
Statistical Analysis
Comparisons for categorical data were performed using the 2 test. P values were based on 2‐tailed tests of significance, and a value 0.05 was considered significant.
RESULTS
The STAR was embedded in the discharge summary for 1370 (78.6%) discharges during the intervention period. A total of 505 patients in the preintervention period and 292 patients in the intervention period were established patients at an affiliated practice and comprised the study population. Demographics and indications for warfarin for the preintervention and intervention groups are listed in the Table 1.
| Preintervention Group, N=505 | Intervention Group, N=292 | P Value | |
|---|---|---|---|
| |||
| Age, y | 66.7 | 68.0 | 0.29 |
| Male gender, n (%) | 236 (46.7) | 153 (52.4) | 0.12 |
| Discharged on bridging agent, n (%) | 90 (17.8) | 36 (12.3) | 0.04 |
| Average length of stay, d | 7.1 | 7.6 | 0.46 |
| Newly prescribed warfarin, n (%) | 147 (29.1) | 62 (21.2) | 0.01 |
| INR 2.03.0 range at discharge, n (%) | 187 (37.0) | 137 (46.9) | 0.02 |
| Warfarin indication, n (%)* | |||
| Venous thromboembolism | 93 (18.4) | 39 (13.4) | |
| Atrial fibrillation | 204 (40.3) | 127 (43.5) | |
| Mechanical heart valve | 19 (3.8) | 17 (6.5) | |
| Prevention of thromboembolism | 142 (28.1) | 94 (32.2) | |
| Intracardiac thrombosis | 3 (0.5) | 6 (2.1) | |
| Thrombophilia | 4 (0.8) | 5 (1.7) | |
| Other | 19 (3.8) | 12 (4.1) | |
| No indication | 32 (6.3) | 1 (3.8) | |
The frequency of INR testing within 10 days of discharge was similar for the preintervention and intervention periods (41.4% and 47.6%, respectively, P=0.09). Similarly, the likelihood of having at least 1 therapeutic INR value within 10 days of discharge was not statistically different for the groups (17.0% and 21.2%, P=0.14). The pattern was similar for the 3‐, 7‐, and 30‐day periods; a higher percentage of the intervention group had INR testing and attained a therapeutic INR value, though for no time period did this reach statistical significance. This pattern was also found when limiting the analysis to patients discharged home rather than to a facility, patients on warfarin, prior to admission, and patients started on warfarin during the hospitalization.
A total of 87of 207 (42.0%) clinicians responded to the survey. Of respondents, 75% reported that they had seen 1 patient who had been discharged on warfarin since the STAR initiative had begun, 58% of whom reported having viewed the STAR. Most respondents who viewed the STAR found it to be helpful or very helpful in guiding warfarin management (67%), improving their workflow and efficiency (58%), and improving patient safety (77%). Approximately one‐third of respondents who had viewed the STAR (34%) reported that they selected a different dose than they would have chosen had the STAR not been available.
DISCUSSION
We developed a concise report that is seamlessly created and inserted into the discharge summary. Though the STAR was perceived as improving patient safety by ambulatory care providers, there was no impact on attaining a therapeutic INR after discharge. There are several possible explanations for a lack of benefit. Most notably, our intervention was comprised of a stand‐alone EMR‐based tool and focused on 1 component of the transitions process. Given the complexity of healthcare delivery and anticoagulant management, it is likely that broader interventions are required to improve clinical outcomes over the transition period. Potential targets of multifaceted approaches may include improving access to care, providing greater access to anticoagulation clinics, enhancing patient education, and promoting direct physician‐physician communication. Bundled interventions will likely need to include involvement of an interdisciplinary team, such as pharmacist involvement in the medication reconciliation process.
The transition period from the hospital to the outpatient setting has the potential to jeopardize patient safety if vital information is not reliably transmitted across venues.[5, 6, 7, 8] Forster and colleagues noted an 11% incidence of ADEs in the posthospitalization period, of which 60% were either preventable or ameliorable.[3] To decrease the risk to patient safety during the transitions period, the Transitions of Care Consensus policy statement by the Society of Hospital Medicine and other medical organizations called for incorporation of standard data transfer forms (templates and transmission protocols).[9] Despite the high risk and preventable nature of many of the events, few specific tools have been developed. As part of broader initiatives to improve the transitions process, the STAR may have the potential to be a means for health systems to improve the quality of the transition of care for patients on anticoagulants.
Our study has several limitations. First, it was performed at a single health system. It is unknown whether the EMR‐based report could be similarly employed at other systems. Second, our study was unable to assess clinical endpoints. Given the lack of effect on attaining a therapeutic INR, it is unlikely that downstream outcomes, such as thromboembolism, were impacted. Lastly, we were unable to examine whether our intervention improved the care of patients whose outpatient provider was external to our system.
The STAR is a concise tool developed to provide essential anticoagulant‐related information to ambulatory providers. Though the report was perceived as improving patient safety, our finding of a lack of impact on attaining a therapeutic INR after discharge suggests that the tool would need to be a component of a broader multifaceted intervention to impact clinical outcomes.
Disclosures
This project was funded by a grant from the Cardinal Health E3 Foundation. The authors report no conflicts of interest.
Anticoagulants are among the prescriptions with the highest risk of an adverse drug event (ADE) after discharge, and many of these events are preventable.[1, 2, 3] In recognition of the high risk for adverse events, the Institute for Healthcare Improvement Map details several key features of a safe anticoagulation management program, including the recommendation during the transition period that clinicians ensure proper lab monitoring and establish capacity for follow‐up testing.[4]
Despite the potential for harm, most hospitals do not have a structured process for the transmission of vital information related to warfarin management from the inpatient to the ambulatory setting. Our aim was to develop a concise report containing the essential information regarding the patient's anticoagulation regimen, the Safe Transitions Anticoagulation Report (STAR), and a process to ensure the report can be readily accessed and utilized by ambulatory clinicians.
METHODS
We assembled an interdisciplinary team to develop a new report and workflow to ensure that information on inpatient warfarin management was transmitted to outpatient providers in a reliable and structured manner. Explicit goals were to maximize use of the electronic medical record (EMR) to autopopulate aspects of the new report and create a process that worked seamlessly into the workflow. The final items were selected based on the risk of harm if not conveyed and feasibility of incorporation through the EMR:
- Warfarin doses: the 7 warfarin doses immediately prior to discharge
- International normalized ratio (INR) values: the 7 INR values immediately prior to discharge
- Bridging anticoagulation: the low‐molecular‐weight heparin (LMWH) prescribed as a bridging anticoagulant, if any
The STAR resides in both the discharge summary and the after visit summary (AVS) for patients discharged on warfarin. At our institution, the AVS contains a medication list, discharge instructions, and appointments, and is automatically produced through our EMR. Our institution utilizes the Epic EMR (Epic Systems Corp., Verona, WI) in the hospital, ambulatory clinics, and faculty practices.
A process was developed where a structured table is automatically created (Figure 1). A field was added to our EMR's discharge summary template asking whether the patient is being discharged on warfarin. Answering yes produces a second question asking whether the patient is also being discharged on bridging anticoagulation with LMWH. The STAR is not inserted into the discharge summary if the clinician completing the discharge summary deletes the anticoagulation question. The STAR is automatically created for patients discharged on warfarin and inserted into the AVS by the EMR regardless of whether the discharge summary has been completed. Patients are instructed by their nurse to bring their AVS to their follow‐up appointments.
The STAR project team utilized plan‐do‐study‐act cycles to test small changes and make revisions. The workflow was piloted on 2 medical/surgical units from January 2014 through March 2014, and revised based on feedback from clinicians and nursing staff. The STAR initiative was fully implemented across our institution in April 2014.
The study was evaluated by the institutional review board of the Icahn School of Medicine at Mount Sinai, and full review was waived.
Outcomes
Our primary outcomes were the timeliness of laboratory monitoring and quality of anticoagulation management for patients with an established relationship at 1 of the main outpatient practices in our system. Our institution has an anticoagulation clinic for patients followed at the general medicine clinic. We defined an established relationship as having been seen in the same practice on at least 2 occasions in the 12 months prior to admission. The primary outcomes were the percentage of patients who had an INR measurement done and the percentage who had a therapeutic INR value within 10 days after discharge. As the 10‐day period is arbitrary, we also assessed these outcomes for the 3‐, 7‐, and 30‐day periods after discharge. The therapeutic range was defined for all patients as an INR of 2.0 to 3.0, as this is the target range for the large majority of patients on warfarin in our system. Outcomes during the intervention period were compared to baseline values during the preintervention period. For patients with multiple admissions, the first admission during each period was included.
Ambulatory Physician Survey
We surveyed ambulatory care physicians at the main practices for our health system. The survey assessed how often the STAR was viewed and incorporated into decision making, whether the report improved workflow, and whether ambulatory providers perceived that the report increased patient safety. The survey was distributed at the 6‐month interval during the intervention phase. The survey was disseminated electronically on 3 occasions, and a paper version was distributed on 1 occasion to housestaff and general medicine faculty.
Statistical Analysis
Comparisons for categorical data were performed using the 2 test. P values were based on 2‐tailed tests of significance, and a value 0.05 was considered significant.
RESULTS
The STAR was embedded in the discharge summary for 1370 (78.6%) discharges during the intervention period. A total of 505 patients in the preintervention period and 292 patients in the intervention period were established patients at an affiliated practice and comprised the study population. Demographics and indications for warfarin for the preintervention and intervention groups are listed in the Table 1.
| Preintervention Group, N=505 | Intervention Group, N=292 | P Value | |
|---|---|---|---|
| |||
| Age, y | 66.7 | 68.0 | 0.29 |
| Male gender, n (%) | 236 (46.7) | 153 (52.4) | 0.12 |
| Discharged on bridging agent, n (%) | 90 (17.8) | 36 (12.3) | 0.04 |
| Average length of stay, d | 7.1 | 7.6 | 0.46 |
| Newly prescribed warfarin, n (%) | 147 (29.1) | 62 (21.2) | 0.01 |
| INR 2.03.0 range at discharge, n (%) | 187 (37.0) | 137 (46.9) | 0.02 |
| Warfarin indication, n (%)* | |||
| Venous thromboembolism | 93 (18.4) | 39 (13.4) | |
| Atrial fibrillation | 204 (40.3) | 127 (43.5) | |
| Mechanical heart valve | 19 (3.8) | 17 (6.5) | |
| Prevention of thromboembolism | 142 (28.1) | 94 (32.2) | |
| Intracardiac thrombosis | 3 (0.5) | 6 (2.1) | |
| Thrombophilia | 4 (0.8) | 5 (1.7) | |
| Other | 19 (3.8) | 12 (4.1) | |
| No indication | 32 (6.3) | 1 (3.8) | |
The frequency of INR testing within 10 days of discharge was similar for the preintervention and intervention periods (41.4% and 47.6%, respectively, P=0.09). Similarly, the likelihood of having at least 1 therapeutic INR value within 10 days of discharge was not statistically different for the groups (17.0% and 21.2%, P=0.14). The pattern was similar for the 3‐, 7‐, and 30‐day periods; a higher percentage of the intervention group had INR testing and attained a therapeutic INR value, though for no time period did this reach statistical significance. This pattern was also found when limiting the analysis to patients discharged home rather than to a facility, patients on warfarin, prior to admission, and patients started on warfarin during the hospitalization.
A total of 87of 207 (42.0%) clinicians responded to the survey. Of respondents, 75% reported that they had seen 1 patient who had been discharged on warfarin since the STAR initiative had begun, 58% of whom reported having viewed the STAR. Most respondents who viewed the STAR found it to be helpful or very helpful in guiding warfarin management (67%), improving their workflow and efficiency (58%), and improving patient safety (77%). Approximately one‐third of respondents who had viewed the STAR (34%) reported that they selected a different dose than they would have chosen had the STAR not been available.
DISCUSSION
We developed a concise report that is seamlessly created and inserted into the discharge summary. Though the STAR was perceived as improving patient safety by ambulatory care providers, there was no impact on attaining a therapeutic INR after discharge. There are several possible explanations for a lack of benefit. Most notably, our intervention was comprised of a stand‐alone EMR‐based tool and focused on 1 component of the transitions process. Given the complexity of healthcare delivery and anticoagulant management, it is likely that broader interventions are required to improve clinical outcomes over the transition period. Potential targets of multifaceted approaches may include improving access to care, providing greater access to anticoagulation clinics, enhancing patient education, and promoting direct physician‐physician communication. Bundled interventions will likely need to include involvement of an interdisciplinary team, such as pharmacist involvement in the medication reconciliation process.
The transition period from the hospital to the outpatient setting has the potential to jeopardize patient safety if vital information is not reliably transmitted across venues.[5, 6, 7, 8] Forster and colleagues noted an 11% incidence of ADEs in the posthospitalization period, of which 60% were either preventable or ameliorable.[3] To decrease the risk to patient safety during the transitions period, the Transitions of Care Consensus policy statement by the Society of Hospital Medicine and other medical organizations called for incorporation of standard data transfer forms (templates and transmission protocols).[9] Despite the high risk and preventable nature of many of the events, few specific tools have been developed. As part of broader initiatives to improve the transitions process, the STAR may have the potential to be a means for health systems to improve the quality of the transition of care for patients on anticoagulants.
Our study has several limitations. First, it was performed at a single health system. It is unknown whether the EMR‐based report could be similarly employed at other systems. Second, our study was unable to assess clinical endpoints. Given the lack of effect on attaining a therapeutic INR, it is unlikely that downstream outcomes, such as thromboembolism, were impacted. Lastly, we were unable to examine whether our intervention improved the care of patients whose outpatient provider was external to our system.
The STAR is a concise tool developed to provide essential anticoagulant‐related information to ambulatory providers. Though the report was perceived as improving patient safety, our finding of a lack of impact on attaining a therapeutic INR after discharge suggests that the tool would need to be a component of a broader multifaceted intervention to impact clinical outcomes.
Disclosures
This project was funded by a grant from the Cardinal Health E3 Foundation. The authors report no conflicts of interest.
- , , , Medication errors: experience of the United States Pharmacopeia (USP) MEDMARX reporting system. J Clin Pharm. 2003;43:760–767.
- , , , , The incidence and severity of adverse events affecting patients after discharge from the hospital. Ann Intern Med. 2003;138:161–167.
- , , , , Adverse drug events occurring following hospital discharge. J Gen Int Med. 2005;204:317–323.
- IHI Improvement Map. Available at: http://app.ihi.org/imap/tool/#Process=54aa289b‐16fd‐4a64‐8329‐3941dfc565d1. Accessed February 20 2015.
- , , , Promoting effective transitions of care at hospital discharge: a review of key issues for hospitalists. J Hosp Med. 2007;2:314–323.
- , , , , , Deficits in communication and information transfer between hospital‐based and primary care physicians: implications for patient safety and continuity of care. JAMA. 2007;297:831–841.
- Transitions of care in patients receiving oral anticoagulants: general principles, procedures, and impact of new oral anticoagulants. J Cardiovasc Nurs. 2013;28:54–65.
- Care transitions in anticoagulation management for patients with atrial fibrillation: an emphasis on safety. Oschner J. 2013;13:419–427.
- , , , et al. Transitions of Care Consensus policy statement: American College of Physicians, Society of General Internal Medicine, Society of Hospital Medicine, American Geriatrics Society, American College Of Emergency Physicians, and Society for Academic Emergency Medicine. J Hosp Med. 2009;4:364–370.
- , , , Medication errors: experience of the United States Pharmacopeia (USP) MEDMARX reporting system. J Clin Pharm. 2003;43:760–767.
- , , , , The incidence and severity of adverse events affecting patients after discharge from the hospital. Ann Intern Med. 2003;138:161–167.
- , , , , Adverse drug events occurring following hospital discharge. J Gen Int Med. 2005;204:317–323.
- IHI Improvement Map. Available at: http://app.ihi.org/imap/tool/#Process=54aa289b‐16fd‐4a64‐8329‐3941dfc565d1. Accessed February 20 2015.
- , , , Promoting effective transitions of care at hospital discharge: a review of key issues for hospitalists. J Hosp Med. 2007;2:314–323.
- , , , , , Deficits in communication and information transfer between hospital‐based and primary care physicians: implications for patient safety and continuity of care. JAMA. 2007;297:831–841.
- Transitions of care in patients receiving oral anticoagulants: general principles, procedures, and impact of new oral anticoagulants. J Cardiovasc Nurs. 2013;28:54–65.
- Care transitions in anticoagulation management for patients with atrial fibrillation: an emphasis on safety. Oschner J. 2013;13:419–427.
- , , , et al. Transitions of Care Consensus policy statement: American College of Physicians, Society of General Internal Medicine, Society of Hospital Medicine, American Geriatrics Society, American College Of Emergency Physicians, and Society for Academic Emergency Medicine. J Hosp Med. 2009;4:364–370.
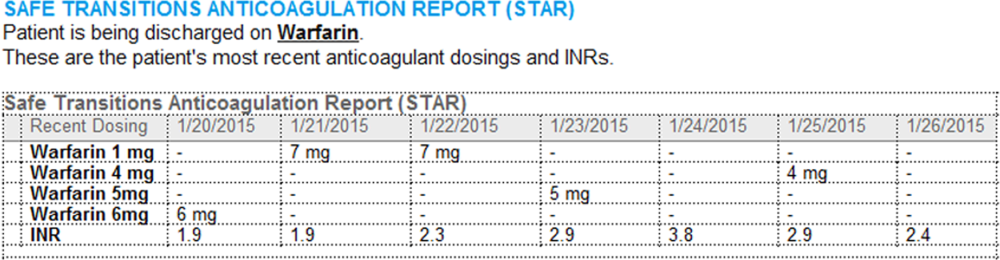
Increased Mortality With Perioperative Beta-Blockade in Low-Risk Patients Undergoing Noncardiac Surgery
Clinical question: Does the use of perioperative beta-blockers affect the outcomes in patients undergoing noncardiac surgery?
Bottom line: Determining the presence or absence of cardiac risk factors is important when deciding whether to use beta-blockers during the perioperative period for patients undergoing noncardiac surgery. This study shows that although perioperative beta-blockade may benefit patients with high cardiac risk, it increases short-term mortality in those with no cardiac risk factors. (LOE = 2b)
Reference: Friedell ML, Van Way CW, Freyberg RW, Almenoff PL. Beta-blockade and operative mortality in noncardiac surgery: harmful or helpful. JAMA Surg. 2015;150(7):658-663.
Study design: Cohort (retrospective)
Funding source: Unknown/not stated
Allocation: Uncertain
Setting: Inpatient (any location) with outpatient follow-up
Synopsis: Using data collected from the Veterans Health Administration, these investigators identified more than 325,000 patients hospitalized for surgery. The use of perioperative beta-blockers in this cohort was determined by using pharmacy data. It was unclear whether the beta-blocker was a new medication or a continuation of a home medication and the study did not measure if the beta-blocker was given preoperatively or postoperatively.
Each patient was assigned a cardiac risk score (1 point each for the presence of renal failure, coronary artery disease, diabetes, and abdominal/thoracic surgery) and grouped into 1 of 3 categories: 0 risk factors, 1 to 2 risk factors, and 3 to 4 risk factors. The results showed that the effect of the beta-blockers on mortality varied according to the presence of cardiac risk factors in patients undergoing noncardiac surgery (n = 314,114). In an adjusted analysis, patients with no cardiac risk factors who received beta-blockers had increased 30-day mortality compared with those who did not receive beta-blockers (odds ratio [OR] 1.19, 95% CI 1.06-1.35).
The opposite was true for patients with 3 to 4 cardiac risk factors: Those who received beta-blockers were less likely to die than those who did not receive them (OR 0.63, 95% CI 0.43-0.93). For patients with 1 to 2 risk factors, there was a nonsignificant reduction in mortality with the use of beta-blockers. For the minority of the cohort who actually underwent cardiac surgery (n = 12,375), there was no significant interaction seen between the number of cardiac risk factors and the use of beta-blockers on mortality. Of note, more than 90% of patients in this study population were men, thus these findings may not be generalizable to women.
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
Clinical question: Does the use of perioperative beta-blockers affect the outcomes in patients undergoing noncardiac surgery?
Bottom line: Determining the presence or absence of cardiac risk factors is important when deciding whether to use beta-blockers during the perioperative period for patients undergoing noncardiac surgery. This study shows that although perioperative beta-blockade may benefit patients with high cardiac risk, it increases short-term mortality in those with no cardiac risk factors. (LOE = 2b)
Reference: Friedell ML, Van Way CW, Freyberg RW, Almenoff PL. Beta-blockade and operative mortality in noncardiac surgery: harmful or helpful. JAMA Surg. 2015;150(7):658-663.
Study design: Cohort (retrospective)
Funding source: Unknown/not stated
Allocation: Uncertain
Setting: Inpatient (any location) with outpatient follow-up
Synopsis: Using data collected from the Veterans Health Administration, these investigators identified more than 325,000 patients hospitalized for surgery. The use of perioperative beta-blockers in this cohort was determined by using pharmacy data. It was unclear whether the beta-blocker was a new medication or a continuation of a home medication and the study did not measure if the beta-blocker was given preoperatively or postoperatively.
Each patient was assigned a cardiac risk score (1 point each for the presence of renal failure, coronary artery disease, diabetes, and abdominal/thoracic surgery) and grouped into 1 of 3 categories: 0 risk factors, 1 to 2 risk factors, and 3 to 4 risk factors. The results showed that the effect of the beta-blockers on mortality varied according to the presence of cardiac risk factors in patients undergoing noncardiac surgery (n = 314,114). In an adjusted analysis, patients with no cardiac risk factors who received beta-blockers had increased 30-day mortality compared with those who did not receive beta-blockers (odds ratio [OR] 1.19, 95% CI 1.06-1.35).
The opposite was true for patients with 3 to 4 cardiac risk factors: Those who received beta-blockers were less likely to die than those who did not receive them (OR 0.63, 95% CI 0.43-0.93). For patients with 1 to 2 risk factors, there was a nonsignificant reduction in mortality with the use of beta-blockers. For the minority of the cohort who actually underwent cardiac surgery (n = 12,375), there was no significant interaction seen between the number of cardiac risk factors and the use of beta-blockers on mortality. Of note, more than 90% of patients in this study population were men, thus these findings may not be generalizable to women.
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
Clinical question: Does the use of perioperative beta-blockers affect the outcomes in patients undergoing noncardiac surgery?
Bottom line: Determining the presence or absence of cardiac risk factors is important when deciding whether to use beta-blockers during the perioperative period for patients undergoing noncardiac surgery. This study shows that although perioperative beta-blockade may benefit patients with high cardiac risk, it increases short-term mortality in those with no cardiac risk factors. (LOE = 2b)
Reference: Friedell ML, Van Way CW, Freyberg RW, Almenoff PL. Beta-blockade and operative mortality in noncardiac surgery: harmful or helpful. JAMA Surg. 2015;150(7):658-663.
Study design: Cohort (retrospective)
Funding source: Unknown/not stated
Allocation: Uncertain
Setting: Inpatient (any location) with outpatient follow-up
Synopsis: Using data collected from the Veterans Health Administration, these investigators identified more than 325,000 patients hospitalized for surgery. The use of perioperative beta-blockers in this cohort was determined by using pharmacy data. It was unclear whether the beta-blocker was a new medication or a continuation of a home medication and the study did not measure if the beta-blocker was given preoperatively or postoperatively.
Each patient was assigned a cardiac risk score (1 point each for the presence of renal failure, coronary artery disease, diabetes, and abdominal/thoracic surgery) and grouped into 1 of 3 categories: 0 risk factors, 1 to 2 risk factors, and 3 to 4 risk factors. The results showed that the effect of the beta-blockers on mortality varied according to the presence of cardiac risk factors in patients undergoing noncardiac surgery (n = 314,114). In an adjusted analysis, patients with no cardiac risk factors who received beta-blockers had increased 30-day mortality compared with those who did not receive beta-blockers (odds ratio [OR] 1.19, 95% CI 1.06-1.35).
The opposite was true for patients with 3 to 4 cardiac risk factors: Those who received beta-blockers were less likely to die than those who did not receive them (OR 0.63, 95% CI 0.43-0.93). For patients with 1 to 2 risk factors, there was a nonsignificant reduction in mortality with the use of beta-blockers. For the minority of the cohort who actually underwent cardiac surgery (n = 12,375), there was no significant interaction seen between the number of cardiac risk factors and the use of beta-blockers on mortality. Of note, more than 90% of patients in this study population were men, thus these findings may not be generalizable to women.
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
Head for oral contraceptives to target women’s acne
NEW YORK – Almost all women with acne will have at least a fair response to therapy with oral contraceptive pills.
Most should experience at least a 50% reduction in lesions, Dr. Bethanee Schlosser said at the American Academy of Dermatology summer meeting.
“From baseline, you are generally speaking about a 50% decrease in inflammatory and noninflammatory lesions and total lesion count,” said Dr. Schlosser of Northwestern University, Chicago. “The important thing, though, is that you have to tell a patient this is not an overnight thing. You have to wait at least three cycles before you make any kind of judgment on whether it’s working.”
The improvement will be seen on all affected areas, not just the face, she said.
“This is important. It’s not just facial acne that’s hormonally sensitive. For us to say it’s just the facial distribution that’s hormonally sensitive is ridiculous. We all know as dermatologists that all acne is androgen driven, and it’s all hormonally sensitive.”
Oral contraceptives can be used alone, as Dr. Schlosser usually initiates treatment, or they can be used in conjunction with spironolactone or antibiotics. Three OCs are approved by the FDA for the treatment of acne.
“I often get asked which OC is the best,” she said. “Just because the FDA approved some for acne doesn’t mean they are better. It means the company had the studies done and basically paid for this labeling indication.”
A 2012 Cochrane review examined 31 studies that compared different OCs to placebo and to each other. The investigators found that all the OCs were consistently more effective than placebo (Cochrane Database Syst Rev. doi: 10.1002/14651858.CD004425.pub6). The head-to-head comparisons produced conflicting results with no clear advantage of one formulation over another.
“I would say use what you are comfortable with,” Dr. Schlosser said.
Some personal and family history and health screenings are necessary before prescribing OCs, although leading women’s health associations, as well as the FDA have said there’s no need for a pelvic exam and Pap smear. “You do have to make sure they are not pregnant, hypertensive, or at risk for stroke or heart disease.”
Spironolactone is usually prescribed at 100-150 mg/day and rarely up to 200 mg/day, she noted. It can be added to an OC regimen if the patient has not adequately responded to monotherapy. It can also be combined with drospirenone, an antibiotic, or with both OCs and antibiotics.
Since spironolactone is a diuretic, women should be monitored for increased thirst and urination, and signs of hypokalemia (lethargy, muscle cramps, dizziness, and increased heart rate). In utero exposure can cause feminization of a male fetus, so reliable contraception is a must.
The drug does carry a boxed warning, as it was carcinogenic in rat studies – but only when given at 50-100 times the usual human dose.
Dr. Schlosser disclosed that she is an investigator with Galderma and Allergan.
On Twitter @Alz_Gal
NEW YORK – Almost all women with acne will have at least a fair response to therapy with oral contraceptive pills.
Most should experience at least a 50% reduction in lesions, Dr. Bethanee Schlosser said at the American Academy of Dermatology summer meeting.
“From baseline, you are generally speaking about a 50% decrease in inflammatory and noninflammatory lesions and total lesion count,” said Dr. Schlosser of Northwestern University, Chicago. “The important thing, though, is that you have to tell a patient this is not an overnight thing. You have to wait at least three cycles before you make any kind of judgment on whether it’s working.”
The improvement will be seen on all affected areas, not just the face, she said.
“This is important. It’s not just facial acne that’s hormonally sensitive. For us to say it’s just the facial distribution that’s hormonally sensitive is ridiculous. We all know as dermatologists that all acne is androgen driven, and it’s all hormonally sensitive.”
Oral contraceptives can be used alone, as Dr. Schlosser usually initiates treatment, or they can be used in conjunction with spironolactone or antibiotics. Three OCs are approved by the FDA for the treatment of acne.
“I often get asked which OC is the best,” she said. “Just because the FDA approved some for acne doesn’t mean they are better. It means the company had the studies done and basically paid for this labeling indication.”
A 2012 Cochrane review examined 31 studies that compared different OCs to placebo and to each other. The investigators found that all the OCs were consistently more effective than placebo (Cochrane Database Syst Rev. doi: 10.1002/14651858.CD004425.pub6). The head-to-head comparisons produced conflicting results with no clear advantage of one formulation over another.
“I would say use what you are comfortable with,” Dr. Schlosser said.
Some personal and family history and health screenings are necessary before prescribing OCs, although leading women’s health associations, as well as the FDA have said there’s no need for a pelvic exam and Pap smear. “You do have to make sure they are not pregnant, hypertensive, or at risk for stroke or heart disease.”
Spironolactone is usually prescribed at 100-150 mg/day and rarely up to 200 mg/day, she noted. It can be added to an OC regimen if the patient has not adequately responded to monotherapy. It can also be combined with drospirenone, an antibiotic, or with both OCs and antibiotics.
Since spironolactone is a diuretic, women should be monitored for increased thirst and urination, and signs of hypokalemia (lethargy, muscle cramps, dizziness, and increased heart rate). In utero exposure can cause feminization of a male fetus, so reliable contraception is a must.
The drug does carry a boxed warning, as it was carcinogenic in rat studies – but only when given at 50-100 times the usual human dose.
Dr. Schlosser disclosed that she is an investigator with Galderma and Allergan.
On Twitter @Alz_Gal
NEW YORK – Almost all women with acne will have at least a fair response to therapy with oral contraceptive pills.
Most should experience at least a 50% reduction in lesions, Dr. Bethanee Schlosser said at the American Academy of Dermatology summer meeting.
“From baseline, you are generally speaking about a 50% decrease in inflammatory and noninflammatory lesions and total lesion count,” said Dr. Schlosser of Northwestern University, Chicago. “The important thing, though, is that you have to tell a patient this is not an overnight thing. You have to wait at least three cycles before you make any kind of judgment on whether it’s working.”
The improvement will be seen on all affected areas, not just the face, she said.
“This is important. It’s not just facial acne that’s hormonally sensitive. For us to say it’s just the facial distribution that’s hormonally sensitive is ridiculous. We all know as dermatologists that all acne is androgen driven, and it’s all hormonally sensitive.”
Oral contraceptives can be used alone, as Dr. Schlosser usually initiates treatment, or they can be used in conjunction with spironolactone or antibiotics. Three OCs are approved by the FDA for the treatment of acne.
“I often get asked which OC is the best,” she said. “Just because the FDA approved some for acne doesn’t mean they are better. It means the company had the studies done and basically paid for this labeling indication.”
A 2012 Cochrane review examined 31 studies that compared different OCs to placebo and to each other. The investigators found that all the OCs were consistently more effective than placebo (Cochrane Database Syst Rev. doi: 10.1002/14651858.CD004425.pub6). The head-to-head comparisons produced conflicting results with no clear advantage of one formulation over another.
“I would say use what you are comfortable with,” Dr. Schlosser said.
Some personal and family history and health screenings are necessary before prescribing OCs, although leading women’s health associations, as well as the FDA have said there’s no need for a pelvic exam and Pap smear. “You do have to make sure they are not pregnant, hypertensive, or at risk for stroke or heart disease.”
Spironolactone is usually prescribed at 100-150 mg/day and rarely up to 200 mg/day, she noted. It can be added to an OC regimen if the patient has not adequately responded to monotherapy. It can also be combined with drospirenone, an antibiotic, or with both OCs and antibiotics.
Since spironolactone is a diuretic, women should be monitored for increased thirst and urination, and signs of hypokalemia (lethargy, muscle cramps, dizziness, and increased heart rate). In utero exposure can cause feminization of a male fetus, so reliable contraception is a must.
The drug does carry a boxed warning, as it was carcinogenic in rat studies – but only when given at 50-100 times the usual human dose.
Dr. Schlosser disclosed that she is an investigator with Galderma and Allergan.
On Twitter @Alz_Gal
EXPERT ANALYSIS FROM THE AAD SUMMER ACADEMY 2015

Wells Score Not Helpful in Hospitalized Patients With Suspected DVT
Clinical question: How useful is the Wells score for risk-stratifying hospitalized patients with suspected deep vein thrombosis?
Bottom line: The Wells score is not helpful in the inpatient setting to predict the presence or absence of deep vein thrombosis (DVT). Based on this study, if a hospitalized patient has a low Wells score, the risk of having DVT is still relatively high (6%). If a patient has a moderate or high score, however, the risk of having DVT is fairly low (10% to 16%). In all 3 categories, a patient would need further testing with ultrasound to evaluate for DVT. (LOE = 2b)
Reference: Silveira PC, Ip IK, Goldhaber SZ, Piazza G, Benson CB, Khorasani R. Performance of Wells score for deep vein thrombosis in the inpatient setting. JAMA Intern Med. 2015;175(7):1112-1117.
Study design: Diagnostic test evaluation
Funding source: Unknown/not stated
Allocation: Uncertain
Setting: Inpatient (any location)
Synopsis: The Wells score has been previously validated to risk-stratify outpatients with suspected DVT but its utility in the inpatient setting is unknown. These investigators evaluated 1135 hospitalized patients with suspected DVT who underwent a lower extremity ultrasound study in the hospital. When ordering these studies, clinicians were required to enter information regarding clinical predictors in order to calculate an individual patient's Wells score. The patients were divided into 3 Wells score categories that determined their pre-test probability for DVT (low risk = 0 or lower, moderate risk = 1 or 2, high risk = 3 or higher). Baseline characteristics for the patients in the study showed that 71% were recently bedridden or had recent major surgery and almost 40% had active cancer.
Overall, 12% of patients in the study had proximal DVT confirmed by a lower extremity ultrasound study. When classified by Wells score categories, the incidence of proximal DVT was 5.9%, 9.5%, and 16.4% in low, moderate, and high pre-test probability groups, respectively. The area under the receiving operating characteristics curve for the Wells score as a diagnostic test was 0.60.
This indicates that the ability of the Wells score to discriminate between the presence and absence of DVT in hospitalized patients was only slightly better than chance. The authors postulate that the reason for this is that hospitalized patients are inherently different from outpatients: they have a higher prevalence of immobilization and/or have active cancer; they receive routine DVT prophylaxis; and they are more likely to have other comorbidities that increase DVT risk, such as heart failure and chronic obstructive pulmonary disease — risk factors that are not accounted for in the Wells score calculation. As such, the Wells score is less meaningful in this population.
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
Clinical question: How useful is the Wells score for risk-stratifying hospitalized patients with suspected deep vein thrombosis?
Bottom line: The Wells score is not helpful in the inpatient setting to predict the presence or absence of deep vein thrombosis (DVT). Based on this study, if a hospitalized patient has a low Wells score, the risk of having DVT is still relatively high (6%). If a patient has a moderate or high score, however, the risk of having DVT is fairly low (10% to 16%). In all 3 categories, a patient would need further testing with ultrasound to evaluate for DVT. (LOE = 2b)
Reference: Silveira PC, Ip IK, Goldhaber SZ, Piazza G, Benson CB, Khorasani R. Performance of Wells score for deep vein thrombosis in the inpatient setting. JAMA Intern Med. 2015;175(7):1112-1117.
Study design: Diagnostic test evaluation
Funding source: Unknown/not stated
Allocation: Uncertain
Setting: Inpatient (any location)
Synopsis: The Wells score has been previously validated to risk-stratify outpatients with suspected DVT but its utility in the inpatient setting is unknown. These investigators evaluated 1135 hospitalized patients with suspected DVT who underwent a lower extremity ultrasound study in the hospital. When ordering these studies, clinicians were required to enter information regarding clinical predictors in order to calculate an individual patient's Wells score. The patients were divided into 3 Wells score categories that determined their pre-test probability for DVT (low risk = 0 or lower, moderate risk = 1 or 2, high risk = 3 or higher). Baseline characteristics for the patients in the study showed that 71% were recently bedridden or had recent major surgery and almost 40% had active cancer.
Overall, 12% of patients in the study had proximal DVT confirmed by a lower extremity ultrasound study. When classified by Wells score categories, the incidence of proximal DVT was 5.9%, 9.5%, and 16.4% in low, moderate, and high pre-test probability groups, respectively. The area under the receiving operating characteristics curve for the Wells score as a diagnostic test was 0.60.
This indicates that the ability of the Wells score to discriminate between the presence and absence of DVT in hospitalized patients was only slightly better than chance. The authors postulate that the reason for this is that hospitalized patients are inherently different from outpatients: they have a higher prevalence of immobilization and/or have active cancer; they receive routine DVT prophylaxis; and they are more likely to have other comorbidities that increase DVT risk, such as heart failure and chronic obstructive pulmonary disease — risk factors that are not accounted for in the Wells score calculation. As such, the Wells score is less meaningful in this population.
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
Clinical question: How useful is the Wells score for risk-stratifying hospitalized patients with suspected deep vein thrombosis?
Bottom line: The Wells score is not helpful in the inpatient setting to predict the presence or absence of deep vein thrombosis (DVT). Based on this study, if a hospitalized patient has a low Wells score, the risk of having DVT is still relatively high (6%). If a patient has a moderate or high score, however, the risk of having DVT is fairly low (10% to 16%). In all 3 categories, a patient would need further testing with ultrasound to evaluate for DVT. (LOE = 2b)
Reference: Silveira PC, Ip IK, Goldhaber SZ, Piazza G, Benson CB, Khorasani R. Performance of Wells score for deep vein thrombosis in the inpatient setting. JAMA Intern Med. 2015;175(7):1112-1117.
Study design: Diagnostic test evaluation
Funding source: Unknown/not stated
Allocation: Uncertain
Setting: Inpatient (any location)
Synopsis: The Wells score has been previously validated to risk-stratify outpatients with suspected DVT but its utility in the inpatient setting is unknown. These investigators evaluated 1135 hospitalized patients with suspected DVT who underwent a lower extremity ultrasound study in the hospital. When ordering these studies, clinicians were required to enter information regarding clinical predictors in order to calculate an individual patient's Wells score. The patients were divided into 3 Wells score categories that determined their pre-test probability for DVT (low risk = 0 or lower, moderate risk = 1 or 2, high risk = 3 or higher). Baseline characteristics for the patients in the study showed that 71% were recently bedridden or had recent major surgery and almost 40% had active cancer.
Overall, 12% of patients in the study had proximal DVT confirmed by a lower extremity ultrasound study. When classified by Wells score categories, the incidence of proximal DVT was 5.9%, 9.5%, and 16.4% in low, moderate, and high pre-test probability groups, respectively. The area under the receiving operating characteristics curve for the Wells score as a diagnostic test was 0.60.
This indicates that the ability of the Wells score to discriminate between the presence and absence of DVT in hospitalized patients was only slightly better than chance. The authors postulate that the reason for this is that hospitalized patients are inherently different from outpatients: they have a higher prevalence of immobilization and/or have active cancer; they receive routine DVT prophylaxis; and they are more likely to have other comorbidities that increase DVT risk, such as heart failure and chronic obstructive pulmonary disease — risk factors that are not accounted for in the Wells score calculation. As such, the Wells score is less meaningful in this population.
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
Total Knee Replacement Is Effective in Patients With Rheumatoid Arthritis
According to a new study, total knee arthroplasty is highly effective in reducing clinically relevant knee pain to a greater extent than other subjective health-related quality-of-life indicies in patients with rheumatoid arthritis, although this improvement is less marked as compared to outcomes in patients with osteoarthritis. These study findings were published online ahead of print July 20 in Arthritis & Rheumatology.
The study included patients with rheumatologist-diagnosed arthritis undergoing primary total knee arthroplasty during 1999 to 2012. Indices of pain knee, and health-related quality of life were obtained in 3 consecutive 6-month intervals: preoperative, perioperative, and postoperative. Descriptive statistics and 1-way analysis of variance were used to compare total knee arthroplasty outcomes by diagnosis. Effect sizes and standardized response means were calculated between baseline and recovery.

Of the participating 18,897 patients, 834 people with rheumatoid arthritis, and 315 people with osteoarthritis had undergone index total knee arthroplasty at mean ages 65 and 68. Post total knee arthroplasty, significant improvements were observed for most domains of pain, function, and health-related quality of life within both disease groups, with greater impact in osteoarthritis. Based on the standardized response means, the maximum improvement was shown in index knee pain.
The Health Assessment Questionnaire II and the Short Form 36 physical component summary were the most responsive health-related quality of life indices in detecting post-total knee arthroplasty improvement in rheumatoid arthritis. A diagnosis of rheumatoid arthritis, lower income, and preoperative anxiety were independently associated with a lower degree of improvement in index knee pain following total knee arthroplasty.
Senior author Kaleb D. Michaud, PhD, Associate Professor in the Division of Rheumatology and Immunology at the University of Nebraska Medical Center in Omaha, and colleagues said that total knee replacement can serve as a “time machine” by which patients can return to a less disabled lifestyle, before the arthritic process catches up.
“A new knee can give osteoarthritis patients 10 to 20 years of painless use, whereas rheumatoid arthritis continues to affect the joint soon afterward,” the researchers said. “It’s an important and effective treatment, but patients with rheumatoid arthritis shouldn’t expect the same, often dramatic results experienced by their osteoarthritis counterparts,” Dr. Michaud said.
Suggested Reading
Dusad A, Pedro S, Mikuls TR, et al. Impact of total knee arthroplasty as assessed using patient-reported pain and health-related quality of life indices: rheumatoid arthritis versus osteoarthritis. Arthritis Rheumatol. 2015 July 20 [Epub ahead of print].
According to a new study, total knee arthroplasty is highly effective in reducing clinically relevant knee pain to a greater extent than other subjective health-related quality-of-life indicies in patients with rheumatoid arthritis, although this improvement is less marked as compared to outcomes in patients with osteoarthritis. These study findings were published online ahead of print July 20 in Arthritis & Rheumatology.
The study included patients with rheumatologist-diagnosed arthritis undergoing primary total knee arthroplasty during 1999 to 2012. Indices of pain knee, and health-related quality of life were obtained in 3 consecutive 6-month intervals: preoperative, perioperative, and postoperative. Descriptive statistics and 1-way analysis of variance were used to compare total knee arthroplasty outcomes by diagnosis. Effect sizes and standardized response means were calculated between baseline and recovery.
Of the participating 18,897 patients, 834 people with rheumatoid arthritis, and 315 people with osteoarthritis had undergone index total knee arthroplasty at mean ages 65 and 68. Post total knee arthroplasty, significant improvements were observed for most domains of pain, function, and health-related quality of life within both disease groups, with greater impact in osteoarthritis. Based on the standardized response means, the maximum improvement was shown in index knee pain.
The Health Assessment Questionnaire II and the Short Form 36 physical component summary were the most responsive health-related quality of life indices in detecting post-total knee arthroplasty improvement in rheumatoid arthritis. A diagnosis of rheumatoid arthritis, lower income, and preoperative anxiety were independently associated with a lower degree of improvement in index knee pain following total knee arthroplasty.
Senior author Kaleb D. Michaud, PhD, Associate Professor in the Division of Rheumatology and Immunology at the University of Nebraska Medical Center in Omaha, and colleagues said that total knee replacement can serve as a “time machine” by which patients can return to a less disabled lifestyle, before the arthritic process catches up.
“A new knee can give osteoarthritis patients 10 to 20 years of painless use, whereas rheumatoid arthritis continues to affect the joint soon afterward,” the researchers said. “It’s an important and effective treatment, but patients with rheumatoid arthritis shouldn’t expect the same, often dramatic results experienced by their osteoarthritis counterparts,” Dr. Michaud said.
According to a new study, total knee arthroplasty is highly effective in reducing clinically relevant knee pain to a greater extent than other subjective health-related quality-of-life indicies in patients with rheumatoid arthritis, although this improvement is less marked as compared to outcomes in patients with osteoarthritis. These study findings were published online ahead of print July 20 in Arthritis & Rheumatology.
The study included patients with rheumatologist-diagnosed arthritis undergoing primary total knee arthroplasty during 1999 to 2012. Indices of pain knee, and health-related quality of life were obtained in 3 consecutive 6-month intervals: preoperative, perioperative, and postoperative. Descriptive statistics and 1-way analysis of variance were used to compare total knee arthroplasty outcomes by diagnosis. Effect sizes and standardized response means were calculated between baseline and recovery.
Of the participating 18,897 patients, 834 people with rheumatoid arthritis, and 315 people with osteoarthritis had undergone index total knee arthroplasty at mean ages 65 and 68. Post total knee arthroplasty, significant improvements were observed for most domains of pain, function, and health-related quality of life within both disease groups, with greater impact in osteoarthritis. Based on the standardized response means, the maximum improvement was shown in index knee pain.
The Health Assessment Questionnaire II and the Short Form 36 physical component summary were the most responsive health-related quality of life indices in detecting post-total knee arthroplasty improvement in rheumatoid arthritis. A diagnosis of rheumatoid arthritis, lower income, and preoperative anxiety were independently associated with a lower degree of improvement in index knee pain following total knee arthroplasty.
Senior author Kaleb D. Michaud, PhD, Associate Professor in the Division of Rheumatology and Immunology at the University of Nebraska Medical Center in Omaha, and colleagues said that total knee replacement can serve as a “time machine” by which patients can return to a less disabled lifestyle, before the arthritic process catches up.
“A new knee can give osteoarthritis patients 10 to 20 years of painless use, whereas rheumatoid arthritis continues to affect the joint soon afterward,” the researchers said. “It’s an important and effective treatment, but patients with rheumatoid arthritis shouldn’t expect the same, often dramatic results experienced by their osteoarthritis counterparts,” Dr. Michaud said.
Suggested Reading
Dusad A, Pedro S, Mikuls TR, et al. Impact of total knee arthroplasty as assessed using patient-reported pain and health-related quality of life indices: rheumatoid arthritis versus osteoarthritis. Arthritis Rheumatol. 2015 July 20 [Epub ahead of print].
Suggested Reading
Dusad A, Pedro S, Mikuls TR, et al. Impact of total knee arthroplasty as assessed using patient-reported pain and health-related quality of life indices: rheumatoid arthritis versus osteoarthritis. Arthritis Rheumatol. 2015 July 20 [Epub ahead of print].
