User login
ESC: Atrial fibrillation accelerates brain atrophy
LONDON – Atrial fibrillation in the elderly general population was independently associated with accelerated losses of brain volume and cognitive function in a major longitudinal study.
These findings from the population-based Age, Gene/Environment Susceptibility–Reykjavik Study (AGES-Reykjavik) have the potential to change the management of atrial fibrillation (AF), Dr. David O. Arnar said at the annual congress of the European Society of Cardiology.

“I think these data potentially suggest it’s better for the brain to remain in sinus rhythm than to pursue rate control in AF. We also have other studies, postablation studies, that show doing an ablation procedure to restore sinus rhythm delays the onset of cognitive dysfunction. So I think we have more and more data that are suggesting AF may be bad for the brain in more ways than just causing cerebral infarcts. That possibility needs to be considered as an endpoint in future studies of treatment strategies,” asserted Dr. Arnar, a cardiologist at Landspítali–The National University Hospital of Iceland, Reykjavik.
The AGES-Reykjavik Study is an ongoing project designed to investigate the genetic and environmental factors that contribute to clinical and subclinical diseases in older-age individuals.
The new data Dr. Arnar presented are an outgrowth of an earlier report from AGES-Reykjavik, which concluded that AF was associated with smaller brain volume and diminished cognitive performance independent of cerebral infarcts. The observed deficits were smallest in subjects with no history of AF, larger in those with paroxysmal AF, and largest of all in participants with persistent/permanent AF (Stroke. 2013 Apr;44[4]:1020-5). However, this was a cross-sectional analysis, which by definition doesn’t permit drawing conclusions regarding cause and effect.
That earlier report was the impetus for the new study featuring a mean of 5.2 years of longitudinal follow-up. The study included 2,472 elderly, nondemented subjects with a mean baseline age of 76 years who underwent brain MRIs and structured cognitive function testing, with repeated assessments roughly a half-decade later.
A total of 121 subjects had ECG-confirmed AF or a history of AF at entry. Another 132 developed new-onset AF during follow-up. Since the participants with prevalent or incident AF had significantly higher levels of cardiovascular risk factors, alcohol consumption, history of cerebral infarcts, and other potential confounders, extensive multivariate statistical adjustments were required in analyzing the data, according to the cardiologist.
During the follow-up period, the AF-free subjects experienced a mean 1.8% reduction in gray matter volume, compared with a 2.7% decrease in individuals with prevalent AF and a 3.88% reduction in those with incident AF. All differences were statistically significant.
Loss of white matter volume over time followed a similar pattern: a mean loss of 5.35% in the no-AF group, compared with a 5.5% drop in those with prevalent AF and a 6.56% decrease in individuals with incident AF.
The volume of white matter lesions rose by 31.6% in the elderly no-AF group, 26.9% in those with prevalent AF, and 43.5% in subjects with new-onset AF during follow-up.
“It surprised us that the changes were most pronounced in those with incident AF rather than prevalent AF,” Dr. Arnar confessed. “How do we explain that? Well, I don’t know, but you wonder if the effect of AF on the brain could be most pronounced initially and then as AF goes on, an adaptation process occurs so that the rate of change in the brain becomes less pronounced as the AF becomes more chronic.”
Turning to the results of cognitive function testing, a composite measure of processing speed declined over time by 10% in the no-AF group, 12.7% with prevalent AF, and 13.9% with incident AF. All differences were statistically significant.
The rate of decline in executive function was 8% in the no-AF subjects, 10.2% with prevalent AF, and 11.8% with incident AF.
Similarly, scores on memory testing dropped by 9.3% in the no-AF group, 9.9% with prevalent AF, and 11.9% with incident AF.
The mechanism by which AF accelerates brain aging is unknown. Dr. Arnar strongly suspects it is multifactorial, with candidate processes including altered autonomic regulation of blood flow, microemboli causing brain atrophy, and most assuredly AF-induced diminution of cerebral blood flow.
He presented cerebral blood flow data obtained via phase contrast MRI on 2,125 study participants. Those with no history of AF averaged a total cerebral blood flow of 540 mL/min. Subjects with a history of AF who were in sinus rhythm at the time of the brain scan averaged 520 mL/min. And subjects in AF when they were scanned averaged less than 480 mL/min.
Dr. Arnar also presented preliminary results from an ongoing brain perfusion imaging study conducted in AF patients before and after direct current cardioversion. Among 17 patients who responded to cardioversion by going into sinus rhythm and staying there for at least 10 weeks until their follow-up MRI, total cerebral blood flow improved by a mean of 70 mL/min from a precardioversion figure of 557 mL/min. Both white and gray matter perfusion improved by a mean of 16%.
In contrast, the 10 patients who remained in AF despite the cardioversion attempt showed no improvement in any of these three endpoints over the 10 weeks.
Audience members wondered whether being on warfarin or beta blocker therapy affected the rate of brain volume loss or cognitive function. Not in the cross-sectional study published in Stroke, Dr. Arnar replied. However, those analyses have yet to be conducted in the new longitudinal study.
The AGES-Reykjavik Study is funded by the U.S. National Institutes of Health, Icelandic Heart Association, and the Icelandic Parliament. Dr. Arnar reported having no financial conflicts.
LONDON – Atrial fibrillation in the elderly general population was independently associated with accelerated losses of brain volume and cognitive function in a major longitudinal study.
These findings from the population-based Age, Gene/Environment Susceptibility–Reykjavik Study (AGES-Reykjavik) have the potential to change the management of atrial fibrillation (AF), Dr. David O. Arnar said at the annual congress of the European Society of Cardiology.
“I think these data potentially suggest it’s better for the brain to remain in sinus rhythm than to pursue rate control in AF. We also have other studies, postablation studies, that show doing an ablation procedure to restore sinus rhythm delays the onset of cognitive dysfunction. So I think we have more and more data that are suggesting AF may be bad for the brain in more ways than just causing cerebral infarcts. That possibility needs to be considered as an endpoint in future studies of treatment strategies,” asserted Dr. Arnar, a cardiologist at Landspítali–The National University Hospital of Iceland, Reykjavik.
The AGES-Reykjavik Study is an ongoing project designed to investigate the genetic and environmental factors that contribute to clinical and subclinical diseases in older-age individuals.
The new data Dr. Arnar presented are an outgrowth of an earlier report from AGES-Reykjavik, which concluded that AF was associated with smaller brain volume and diminished cognitive performance independent of cerebral infarcts. The observed deficits were smallest in subjects with no history of AF, larger in those with paroxysmal AF, and largest of all in participants with persistent/permanent AF (Stroke. 2013 Apr;44[4]:1020-5). However, this was a cross-sectional analysis, which by definition doesn’t permit drawing conclusions regarding cause and effect.
That earlier report was the impetus for the new study featuring a mean of 5.2 years of longitudinal follow-up. The study included 2,472 elderly, nondemented subjects with a mean baseline age of 76 years who underwent brain MRIs and structured cognitive function testing, with repeated assessments roughly a half-decade later.
A total of 121 subjects had ECG-confirmed AF or a history of AF at entry. Another 132 developed new-onset AF during follow-up. Since the participants with prevalent or incident AF had significantly higher levels of cardiovascular risk factors, alcohol consumption, history of cerebral infarcts, and other potential confounders, extensive multivariate statistical adjustments were required in analyzing the data, according to the cardiologist.
During the follow-up period, the AF-free subjects experienced a mean 1.8% reduction in gray matter volume, compared with a 2.7% decrease in individuals with prevalent AF and a 3.88% reduction in those with incident AF. All differences were statistically significant.
Loss of white matter volume over time followed a similar pattern: a mean loss of 5.35% in the no-AF group, compared with a 5.5% drop in those with prevalent AF and a 6.56% decrease in individuals with incident AF.
The volume of white matter lesions rose by 31.6% in the elderly no-AF group, 26.9% in those with prevalent AF, and 43.5% in subjects with new-onset AF during follow-up.
“It surprised us that the changes were most pronounced in those with incident AF rather than prevalent AF,” Dr. Arnar confessed. “How do we explain that? Well, I don’t know, but you wonder if the effect of AF on the brain could be most pronounced initially and then as AF goes on, an adaptation process occurs so that the rate of change in the brain becomes less pronounced as the AF becomes more chronic.”
Turning to the results of cognitive function testing, a composite measure of processing speed declined over time by 10% in the no-AF group, 12.7% with prevalent AF, and 13.9% with incident AF. All differences were statistically significant.
The rate of decline in executive function was 8% in the no-AF subjects, 10.2% with prevalent AF, and 11.8% with incident AF.
Similarly, scores on memory testing dropped by 9.3% in the no-AF group, 9.9% with prevalent AF, and 11.9% with incident AF.
The mechanism by which AF accelerates brain aging is unknown. Dr. Arnar strongly suspects it is multifactorial, with candidate processes including altered autonomic regulation of blood flow, microemboli causing brain atrophy, and most assuredly AF-induced diminution of cerebral blood flow.
He presented cerebral blood flow data obtained via phase contrast MRI on 2,125 study participants. Those with no history of AF averaged a total cerebral blood flow of 540 mL/min. Subjects with a history of AF who were in sinus rhythm at the time of the brain scan averaged 520 mL/min. And subjects in AF when they were scanned averaged less than 480 mL/min.
Dr. Arnar also presented preliminary results from an ongoing brain perfusion imaging study conducted in AF patients before and after direct current cardioversion. Among 17 patients who responded to cardioversion by going into sinus rhythm and staying there for at least 10 weeks until their follow-up MRI, total cerebral blood flow improved by a mean of 70 mL/min from a precardioversion figure of 557 mL/min. Both white and gray matter perfusion improved by a mean of 16%.
In contrast, the 10 patients who remained in AF despite the cardioversion attempt showed no improvement in any of these three endpoints over the 10 weeks.
Audience members wondered whether being on warfarin or beta blocker therapy affected the rate of brain volume loss or cognitive function. Not in the cross-sectional study published in Stroke, Dr. Arnar replied. However, those analyses have yet to be conducted in the new longitudinal study.
The AGES-Reykjavik Study is funded by the U.S. National Institutes of Health, Icelandic Heart Association, and the Icelandic Parliament. Dr. Arnar reported having no financial conflicts.
LONDON – Atrial fibrillation in the elderly general population was independently associated with accelerated losses of brain volume and cognitive function in a major longitudinal study.
These findings from the population-based Age, Gene/Environment Susceptibility–Reykjavik Study (AGES-Reykjavik) have the potential to change the management of atrial fibrillation (AF), Dr. David O. Arnar said at the annual congress of the European Society of Cardiology.
“I think these data potentially suggest it’s better for the brain to remain in sinus rhythm than to pursue rate control in AF. We also have other studies, postablation studies, that show doing an ablation procedure to restore sinus rhythm delays the onset of cognitive dysfunction. So I think we have more and more data that are suggesting AF may be bad for the brain in more ways than just causing cerebral infarcts. That possibility needs to be considered as an endpoint in future studies of treatment strategies,” asserted Dr. Arnar, a cardiologist at Landspítali–The National University Hospital of Iceland, Reykjavik.
The AGES-Reykjavik Study is an ongoing project designed to investigate the genetic and environmental factors that contribute to clinical and subclinical diseases in older-age individuals.
The new data Dr. Arnar presented are an outgrowth of an earlier report from AGES-Reykjavik, which concluded that AF was associated with smaller brain volume and diminished cognitive performance independent of cerebral infarcts. The observed deficits were smallest in subjects with no history of AF, larger in those with paroxysmal AF, and largest of all in participants with persistent/permanent AF (Stroke. 2013 Apr;44[4]:1020-5). However, this was a cross-sectional analysis, which by definition doesn’t permit drawing conclusions regarding cause and effect.
That earlier report was the impetus for the new study featuring a mean of 5.2 years of longitudinal follow-up. The study included 2,472 elderly, nondemented subjects with a mean baseline age of 76 years who underwent brain MRIs and structured cognitive function testing, with repeated assessments roughly a half-decade later.
A total of 121 subjects had ECG-confirmed AF or a history of AF at entry. Another 132 developed new-onset AF during follow-up. Since the participants with prevalent or incident AF had significantly higher levels of cardiovascular risk factors, alcohol consumption, history of cerebral infarcts, and other potential confounders, extensive multivariate statistical adjustments were required in analyzing the data, according to the cardiologist.
During the follow-up period, the AF-free subjects experienced a mean 1.8% reduction in gray matter volume, compared with a 2.7% decrease in individuals with prevalent AF and a 3.88% reduction in those with incident AF. All differences were statistically significant.
Loss of white matter volume over time followed a similar pattern: a mean loss of 5.35% in the no-AF group, compared with a 5.5% drop in those with prevalent AF and a 6.56% decrease in individuals with incident AF.
The volume of white matter lesions rose by 31.6% in the elderly no-AF group, 26.9% in those with prevalent AF, and 43.5% in subjects with new-onset AF during follow-up.
“It surprised us that the changes were most pronounced in those with incident AF rather than prevalent AF,” Dr. Arnar confessed. “How do we explain that? Well, I don’t know, but you wonder if the effect of AF on the brain could be most pronounced initially and then as AF goes on, an adaptation process occurs so that the rate of change in the brain becomes less pronounced as the AF becomes more chronic.”
Turning to the results of cognitive function testing, a composite measure of processing speed declined over time by 10% in the no-AF group, 12.7% with prevalent AF, and 13.9% with incident AF. All differences were statistically significant.
The rate of decline in executive function was 8% in the no-AF subjects, 10.2% with prevalent AF, and 11.8% with incident AF.
Similarly, scores on memory testing dropped by 9.3% in the no-AF group, 9.9% with prevalent AF, and 11.9% with incident AF.
The mechanism by which AF accelerates brain aging is unknown. Dr. Arnar strongly suspects it is multifactorial, with candidate processes including altered autonomic regulation of blood flow, microemboli causing brain atrophy, and most assuredly AF-induced diminution of cerebral blood flow.
He presented cerebral blood flow data obtained via phase contrast MRI on 2,125 study participants. Those with no history of AF averaged a total cerebral blood flow of 540 mL/min. Subjects with a history of AF who were in sinus rhythm at the time of the brain scan averaged 520 mL/min. And subjects in AF when they were scanned averaged less than 480 mL/min.
Dr. Arnar also presented preliminary results from an ongoing brain perfusion imaging study conducted in AF patients before and after direct current cardioversion. Among 17 patients who responded to cardioversion by going into sinus rhythm and staying there for at least 10 weeks until their follow-up MRI, total cerebral blood flow improved by a mean of 70 mL/min from a precardioversion figure of 557 mL/min. Both white and gray matter perfusion improved by a mean of 16%.
In contrast, the 10 patients who remained in AF despite the cardioversion attempt showed no improvement in any of these three endpoints over the 10 weeks.
Audience members wondered whether being on warfarin or beta blocker therapy affected the rate of brain volume loss or cognitive function. Not in the cross-sectional study published in Stroke, Dr. Arnar replied. However, those analyses have yet to be conducted in the new longitudinal study.
The AGES-Reykjavik Study is funded by the U.S. National Institutes of Health, Icelandic Heart Association, and the Icelandic Parliament. Dr. Arnar reported having no financial conflicts.
AT THE ESC CONGRESS 2015
Key clinical point: Atrial fibrillation appears to accelerate brain aging in the elderly independent of cerebral embolism.
Major finding: Both prevalent and incident atrial fibrillation were associated with accelerated loss of gray matter and total brain volume as well as key aspects of cognitive function.
Data source: This population-based study included 2,472 nondemented participants with a mean baseline age of 76 years who were prospectively followed with brain scans and structured cognitive function tests for a mean of 5.2 years.
Disclosures: The AGES-Reykjavik Study is funded by the U.S. National Institutes of Health, Icelandic Heart Association, and the Icelandic Parliament. The presenter reported having no financial conflicts.

The Eyes Have It, and It Itches Like Crazy
For several months, a 69-year-old woman has had a rash around her eyes. It is terribly symptomatic, burning and itching with or without treatment (attempts at which have encompassed moisturizers, petroleum jelly, topical vitamin E oil, and most recently, application of triple-antibiotic cream three times a day). She finally requests referral to dermatology from her primary care provider.
When the rash manifested, she reports, she made some alterations to her routine, eliminating or changing the type of makeup, soap, cleanser, and laundry detergent she used. None of these changes helped.
Even before the distressing symptoms started, a friend had suggested the patient might have an eye problem. She consulted an ophthalmologist, who prescribed eye drops (the patient doesn’t recall any details); these only produced more burning and itching around her eyes.
The patient’s history is significant for atopy, with a childhood history of seasonal allergies, asthma, and eczema.
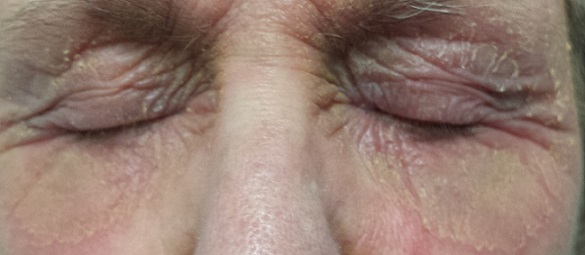
EXAMINATION
There is marked erythema and scaling in the bilateral periocular areas that spills onto both upper and lower lids. Very little edema is seen. The eyes themselves are free of changes.
What is the diagnosis?
DISCUSSION
For many patients, any problem that manifests close to the eye is deemed an “eye problem,” even when the eye itself is uninvolved. Eyelid dermatitis is an extremely common complaint, and this patient’s history is quite typical: The worse the problem gets, the more attempts the patient makes to relieve symptoms.
When this patient presented to dermatology, she was applying six different products (all OTC) to the affected areas. None helped, and in fact, most seemed to worsen the problem. Even if one had helped, she would never have known which. But desperation drives patients to do irrational things, especially when the problem is out in the open for the whole world to see.
Virtually every patient I’ve seen with eyelid dermatitis has (like this patient) already stopped using makeup and changed or discontinued use of laundry detergent and other products. These are almost never the problem; if any of them were, the effects would not be so sharply limited to the periocular area.
Rather, the sharp margins of this condition suggested irritant contact dermatitis. It often appears in this context: The periocular skin is unique in that it’s the thinnest in the female body. This means it is easily traumatized by rubbing and scratching and can be quite permeable to various contactants. This is especially true in atopic patients, whose skin is not only thin and dry but also overreactive to insult.
Though we may never know the full story, I suspect this patient had a more modest case of eczema on her eyelids until she began to apply product after product. One of them—triple-antibiotic cream—is a notorious topical sensitizer. In one sense, this patient is a victim of overattention to the problem.
I advised her to stop use of all the contactants and prescribed hydrocortisone 2.5% ointment for twice-daily application. I also gave her a prescription for a two-week taper of prednisone. When she returned for follow-up three weeks later, the rash was completely resolved.
Other conditions that can cause eyelid dermatitis include seborrhea and psoriasis.
TAKE-HOME LEARNING POINTS
• Eyelid dermatitis, an extremely common complaint, is rarely seen in men and is almost never caused by makeup, soap, or shampoo.
• Eyelid dermatitis is not an eye problem—rather, it is a skin problem that happens to occur near the eye.
• The differential for eyelid dermatitis includes atopic dermatitis, seborrhea and psoriasis.
• Stopping use of all contactant products (except prescription medications) is necessary.
• Class 5 or 6 steroids, especially hydrocortisone 2.5% in ointment form, are useful. In severe cases, a tapering course of oral glucocorticoids (prednisone) is extremely helpful.
For several months, a 69-year-old woman has had a rash around her eyes. It is terribly symptomatic, burning and itching with or without treatment (attempts at which have encompassed moisturizers, petroleum jelly, topical vitamin E oil, and most recently, application of triple-antibiotic cream three times a day). She finally requests referral to dermatology from her primary care provider.
When the rash manifested, she reports, she made some alterations to her routine, eliminating or changing the type of makeup, soap, cleanser, and laundry detergent she used. None of these changes helped.
Even before the distressing symptoms started, a friend had suggested the patient might have an eye problem. She consulted an ophthalmologist, who prescribed eye drops (the patient doesn’t recall any details); these only produced more burning and itching around her eyes.
The patient’s history is significant for atopy, with a childhood history of seasonal allergies, asthma, and eczema.
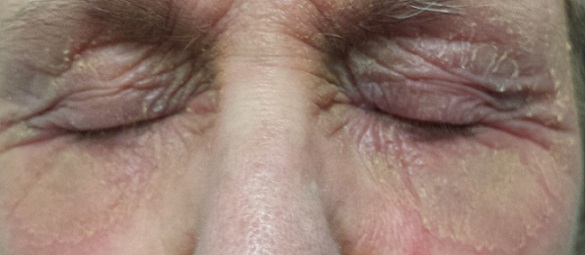
EXAMINATION
There is marked erythema and scaling in the bilateral periocular areas that spills onto both upper and lower lids. Very little edema is seen. The eyes themselves are free of changes.
What is the diagnosis?
DISCUSSION
For many patients, any problem that manifests close to the eye is deemed an “eye problem,” even when the eye itself is uninvolved. Eyelid dermatitis is an extremely common complaint, and this patient’s history is quite typical: The worse the problem gets, the more attempts the patient makes to relieve symptoms.
When this patient presented to dermatology, she was applying six different products (all OTC) to the affected areas. None helped, and in fact, most seemed to worsen the problem. Even if one had helped, she would never have known which. But desperation drives patients to do irrational things, especially when the problem is out in the open for the whole world to see.
Virtually every patient I’ve seen with eyelid dermatitis has (like this patient) already stopped using makeup and changed or discontinued use of laundry detergent and other products. These are almost never the problem; if any of them were, the effects would not be so sharply limited to the periocular area.
Rather, the sharp margins of this condition suggested irritant contact dermatitis. It often appears in this context: The periocular skin is unique in that it’s the thinnest in the female body. This means it is easily traumatized by rubbing and scratching and can be quite permeable to various contactants. This is especially true in atopic patients, whose skin is not only thin and dry but also overreactive to insult.
Though we may never know the full story, I suspect this patient had a more modest case of eczema on her eyelids until she began to apply product after product. One of them—triple-antibiotic cream—is a notorious topical sensitizer. In one sense, this patient is a victim of overattention to the problem.
I advised her to stop use of all the contactants and prescribed hydrocortisone 2.5% ointment for twice-daily application. I also gave her a prescription for a two-week taper of prednisone. When she returned for follow-up three weeks later, the rash was completely resolved.
Other conditions that can cause eyelid dermatitis include seborrhea and psoriasis.
TAKE-HOME LEARNING POINTS
• Eyelid dermatitis, an extremely common complaint, is rarely seen in men and is almost never caused by makeup, soap, or shampoo.
• Eyelid dermatitis is not an eye problem—rather, it is a skin problem that happens to occur near the eye.
• The differential for eyelid dermatitis includes atopic dermatitis, seborrhea and psoriasis.
• Stopping use of all contactant products (except prescription medications) is necessary.
• Class 5 or 6 steroids, especially hydrocortisone 2.5% in ointment form, are useful. In severe cases, a tapering course of oral glucocorticoids (prednisone) is extremely helpful.
For several months, a 69-year-old woman has had a rash around her eyes. It is terribly symptomatic, burning and itching with or without treatment (attempts at which have encompassed moisturizers, petroleum jelly, topical vitamin E oil, and most recently, application of triple-antibiotic cream three times a day). She finally requests referral to dermatology from her primary care provider.
When the rash manifested, she reports, she made some alterations to her routine, eliminating or changing the type of makeup, soap, cleanser, and laundry detergent she used. None of these changes helped.
Even before the distressing symptoms started, a friend had suggested the patient might have an eye problem. She consulted an ophthalmologist, who prescribed eye drops (the patient doesn’t recall any details); these only produced more burning and itching around her eyes.
The patient’s history is significant for atopy, with a childhood history of seasonal allergies, asthma, and eczema.
EXAMINATION
There is marked erythema and scaling in the bilateral periocular areas that spills onto both upper and lower lids. Very little edema is seen. The eyes themselves are free of changes.
What is the diagnosis?
DISCUSSION
For many patients, any problem that manifests close to the eye is deemed an “eye problem,” even when the eye itself is uninvolved. Eyelid dermatitis is an extremely common complaint, and this patient’s history is quite typical: The worse the problem gets, the more attempts the patient makes to relieve symptoms.
When this patient presented to dermatology, she was applying six different products (all OTC) to the affected areas. None helped, and in fact, most seemed to worsen the problem. Even if one had helped, she would never have known which. But desperation drives patients to do irrational things, especially when the problem is out in the open for the whole world to see.
Virtually every patient I’ve seen with eyelid dermatitis has (like this patient) already stopped using makeup and changed or discontinued use of laundry detergent and other products. These are almost never the problem; if any of them were, the effects would not be so sharply limited to the periocular area.
Rather, the sharp margins of this condition suggested irritant contact dermatitis. It often appears in this context: The periocular skin is unique in that it’s the thinnest in the female body. This means it is easily traumatized by rubbing and scratching and can be quite permeable to various contactants. This is especially true in atopic patients, whose skin is not only thin and dry but also overreactive to insult.
Though we may never know the full story, I suspect this patient had a more modest case of eczema on her eyelids until she began to apply product after product. One of them—triple-antibiotic cream—is a notorious topical sensitizer. In one sense, this patient is a victim of overattention to the problem.
I advised her to stop use of all the contactants and prescribed hydrocortisone 2.5% ointment for twice-daily application. I also gave her a prescription for a two-week taper of prednisone. When she returned for follow-up three weeks later, the rash was completely resolved.
Other conditions that can cause eyelid dermatitis include seborrhea and psoriasis.
TAKE-HOME LEARNING POINTS
• Eyelid dermatitis, an extremely common complaint, is rarely seen in men and is almost never caused by makeup, soap, or shampoo.
• Eyelid dermatitis is not an eye problem—rather, it is a skin problem that happens to occur near the eye.
• The differential for eyelid dermatitis includes atopic dermatitis, seborrhea and psoriasis.
• Stopping use of all contactant products (except prescription medications) is necessary.
• Class 5 or 6 steroids, especially hydrocortisone 2.5% in ointment form, are useful. In severe cases, a tapering course of oral glucocorticoids (prednisone) is extremely helpful.
Commentary to "CDC Will Soon Issue Guidelines for the Prevention of Surgical Site Infection"
Analyzing the Guidelines: It Can't All Be Level I
The demand for total joint arthroplasty continues to rise, resulting in a steady increase in the number of primary total hip and knee replacements every year. Unfortunately, as these numbers rise, so will the number of periprosthetic joint infections (PJIs). The economic burden and patient morbidity associated with PJI has resulted in the creation of multiple orthopedic societies and committees focused on formulating “best practice” guidelines in order to reduce the rates of PJI as much as possible.
The new guidelines for surgical site infection (SSI) prevention by the Centers for Disease Control and Prevention (CDC) recently forced the orthopedic community to critically analyze the current literature. Dr. Javad Parvizi’s editorial elegantly notes that many areas of infection prevention and treatment are not well evaluated, and many of our day-to-day practices are based on low levels of evidence. Level I studies continue to be a costly and time-consuming challenge due to the already very low SSI rate, and, in order to show an improvement in this rate, thousands of patients are required for study. This makes a multicenter approach necessary to ensure adequate power, and a multicenter study often requires significant resources and funding outlets. These requirements have resulted in many of our practice recommendations being based on retrospective reviews, which have inherent methodological limitations. The retrospective nature of these studies lacks the experimental design necessary to confidently make treatment recommendations; however, they do allow us to look at what strategies have been tried, and in essence, how well they worked. Although level III and IV studies do not allow us to compare treatments head to head, they do give us some insights into viable treatment strategies and should not be completely disregarded. The results of retrospective studies allow us to design prospective experiments based on what we have observed as successful treatment modalities in particular patient cohorts.
An alternative approach for evaluating new and existing treatment strategies is through basic science translational research. Future advancements in PJI diagnosis and treatment will likely be founded upon translational research efforts from clinician scientists testing treatment protocols both on the benchtop and in animal models. The most glaring knowledge gaps in PJI should be identified through the combined efforts of the CDC, the Musculoskeletal Infection Society, the American Academy of Orthopaedic Surgeons, and the Orthopaedic Research Society. Coordinated efforts should be made and strategies executed to systematically fund translational projects that answer these questions. Translational studies will be able to safely and methodically evaluate new and even established treatment protocols for PJI in a cost-effective manner.
We have made great strides in the prevention and treatment of PJI over the past 2 decades. When working together as a cohesive profession, we will undoubtedly continue to advance our knowledge base and improve treatment recommendations for our patients.
Analyzing the Guidelines: It Can't All Be Level I
The demand for total joint arthroplasty continues to rise, resulting in a steady increase in the number of primary total hip and knee replacements every year. Unfortunately, as these numbers rise, so will the number of periprosthetic joint infections (PJIs). The economic burden and patient morbidity associated with PJI has resulted in the creation of multiple orthopedic societies and committees focused on formulating “best practice” guidelines in order to reduce the rates of PJI as much as possible.
The new guidelines for surgical site infection (SSI) prevention by the Centers for Disease Control and Prevention (CDC) recently forced the orthopedic community to critically analyze the current literature. Dr. Javad Parvizi’s editorial elegantly notes that many areas of infection prevention and treatment are not well evaluated, and many of our day-to-day practices are based on low levels of evidence. Level I studies continue to be a costly and time-consuming challenge due to the already very low SSI rate, and, in order to show an improvement in this rate, thousands of patients are required for study. This makes a multicenter approach necessary to ensure adequate power, and a multicenter study often requires significant resources and funding outlets. These requirements have resulted in many of our practice recommendations being based on retrospective reviews, which have inherent methodological limitations. The retrospective nature of these studies lacks the experimental design necessary to confidently make treatment recommendations; however, they do allow us to look at what strategies have been tried, and in essence, how well they worked. Although level III and IV studies do not allow us to compare treatments head to head, they do give us some insights into viable treatment strategies and should not be completely disregarded. The results of retrospective studies allow us to design prospective experiments based on what we have observed as successful treatment modalities in particular patient cohorts.
An alternative approach for evaluating new and existing treatment strategies is through basic science translational research. Future advancements in PJI diagnosis and treatment will likely be founded upon translational research efforts from clinician scientists testing treatment protocols both on the benchtop and in animal models. The most glaring knowledge gaps in PJI should be identified through the combined efforts of the CDC, the Musculoskeletal Infection Society, the American Academy of Orthopaedic Surgeons, and the Orthopaedic Research Society. Coordinated efforts should be made and strategies executed to systematically fund translational projects that answer these questions. Translational studies will be able to safely and methodically evaluate new and even established treatment protocols for PJI in a cost-effective manner.
We have made great strides in the prevention and treatment of PJI over the past 2 decades. When working together as a cohesive profession, we will undoubtedly continue to advance our knowledge base and improve treatment recommendations for our patients.
Analyzing the Guidelines: It Can't All Be Level I
The demand for total joint arthroplasty continues to rise, resulting in a steady increase in the number of primary total hip and knee replacements every year. Unfortunately, as these numbers rise, so will the number of periprosthetic joint infections (PJIs). The economic burden and patient morbidity associated with PJI has resulted in the creation of multiple orthopedic societies and committees focused on formulating “best practice” guidelines in order to reduce the rates of PJI as much as possible.
The new guidelines for surgical site infection (SSI) prevention by the Centers for Disease Control and Prevention (CDC) recently forced the orthopedic community to critically analyze the current literature. Dr. Javad Parvizi’s editorial elegantly notes that many areas of infection prevention and treatment are not well evaluated, and many of our day-to-day practices are based on low levels of evidence. Level I studies continue to be a costly and time-consuming challenge due to the already very low SSI rate, and, in order to show an improvement in this rate, thousands of patients are required for study. This makes a multicenter approach necessary to ensure adequate power, and a multicenter study often requires significant resources and funding outlets. These requirements have resulted in many of our practice recommendations being based on retrospective reviews, which have inherent methodological limitations. The retrospective nature of these studies lacks the experimental design necessary to confidently make treatment recommendations; however, they do allow us to look at what strategies have been tried, and in essence, how well they worked. Although level III and IV studies do not allow us to compare treatments head to head, they do give us some insights into viable treatment strategies and should not be completely disregarded. The results of retrospective studies allow us to design prospective experiments based on what we have observed as successful treatment modalities in particular patient cohorts.
An alternative approach for evaluating new and existing treatment strategies is through basic science translational research. Future advancements in PJI diagnosis and treatment will likely be founded upon translational research efforts from clinician scientists testing treatment protocols both on the benchtop and in animal models. The most glaring knowledge gaps in PJI should be identified through the combined efforts of the CDC, the Musculoskeletal Infection Society, the American Academy of Orthopaedic Surgeons, and the Orthopaedic Research Society. Coordinated efforts should be made and strategies executed to systematically fund translational projects that answer these questions. Translational studies will be able to safely and methodically evaluate new and even established treatment protocols for PJI in a cost-effective manner.
We have made great strides in the prevention and treatment of PJI over the past 2 decades. When working together as a cohesive profession, we will undoubtedly continue to advance our knowledge base and improve treatment recommendations for our patients.

Acne scars improved with topical epidermal growth factor serum
Topical synthetic epidermal growth factor serum moderately improved the appearance of atrophic acne scars in a small pilot study.
At the end of 12 weeks of twice-daily application, scar appearance improved from 2.875 to 2.38 points on a 5-point investigator global assessment scale. Mean Goodman and Baron acne scar grade fell from 3.00 to 2.75, with 3 representing moderate disease and 2 mild disease. Of eight pairs of before and after photographs given to a blinded investigator, posttreatment images were correctly identified in five. Two were assessed as 76%-100% improved, and three were assessed as 50%-75% improved (J Drugs Dermatol. 2015;14[9]:1005-1010).

The patients were an average of 38 years old, split about equally between the sexes, and racially diverse. They used a basic facial cleanser during the study, but were banned from using tretinoin and other topicals.
Previously studied topicals don’t do much for acne scars, so the usual go-to treatments are chemical peels, dermabrasion, resurfacing lasers, and percutaneous collagen needling. They all work in part by promoting collagen synthesis, but at the cost of pain and side effects. Epidermal growth factor (EGF) also promotes collagen synthesis, so the investigators thought it might help. The EGF used in the study – DNA Regeneration Serum, derived from barley – was supplied by its maker, DNA EGF Renewal in Los Angeles.
“The findings suggest EGF serum has the potential to be a modern, noninvasive treatment for an otherwise highly refractory condition. Whereas resurfacing procedures rely on skin injury to trigger [EGF] release, direct topical application offers the effects of EGF without the associated discomfort and recovery time,” said Dr. Ronald L. Moy of the University of Southern California, Los Angeles, and Rachel Seidel, a medical student at Georgetown University in Washington, D.C.
“All subjects in this study [also] noted improvements in skin texture, fine lines, and wrinkles, while the vast majority also saw a reduction in brown and age spots,” they said.
The investigators said they are interested next in seeing if topical EGF prevents scars in active acne. “We believe that, by counteracting collagen degradation during the course of the inflammatory response, significant tissue atrophy capable of causing visible scarring may be prevented,” they said.
Dr. Moy owns stock in DNA EGF Renewal and is the company’s scientific adviser.
Topical synthetic epidermal growth factor serum moderately improved the appearance of atrophic acne scars in a small pilot study.
At the end of 12 weeks of twice-daily application, scar appearance improved from 2.875 to 2.38 points on a 5-point investigator global assessment scale. Mean Goodman and Baron acne scar grade fell from 3.00 to 2.75, with 3 representing moderate disease and 2 mild disease. Of eight pairs of before and after photographs given to a blinded investigator, posttreatment images were correctly identified in five. Two were assessed as 76%-100% improved, and three were assessed as 50%-75% improved (J Drugs Dermatol. 2015;14[9]:1005-1010).
The patients were an average of 38 years old, split about equally between the sexes, and racially diverse. They used a basic facial cleanser during the study, but were banned from using tretinoin and other topicals.
Previously studied topicals don’t do much for acne scars, so the usual go-to treatments are chemical peels, dermabrasion, resurfacing lasers, and percutaneous collagen needling. They all work in part by promoting collagen synthesis, but at the cost of pain and side effects. Epidermal growth factor (EGF) also promotes collagen synthesis, so the investigators thought it might help. The EGF used in the study – DNA Regeneration Serum, derived from barley – was supplied by its maker, DNA EGF Renewal in Los Angeles.
“The findings suggest EGF serum has the potential to be a modern, noninvasive treatment for an otherwise highly refractory condition. Whereas resurfacing procedures rely on skin injury to trigger [EGF] release, direct topical application offers the effects of EGF without the associated discomfort and recovery time,” said Dr. Ronald L. Moy of the University of Southern California, Los Angeles, and Rachel Seidel, a medical student at Georgetown University in Washington, D.C.
“All subjects in this study [also] noted improvements in skin texture, fine lines, and wrinkles, while the vast majority also saw a reduction in brown and age spots,” they said.
The investigators said they are interested next in seeing if topical EGF prevents scars in active acne. “We believe that, by counteracting collagen degradation during the course of the inflammatory response, significant tissue atrophy capable of causing visible scarring may be prevented,” they said.
Dr. Moy owns stock in DNA EGF Renewal and is the company’s scientific adviser.
Topical synthetic epidermal growth factor serum moderately improved the appearance of atrophic acne scars in a small pilot study.
At the end of 12 weeks of twice-daily application, scar appearance improved from 2.875 to 2.38 points on a 5-point investigator global assessment scale. Mean Goodman and Baron acne scar grade fell from 3.00 to 2.75, with 3 representing moderate disease and 2 mild disease. Of eight pairs of before and after photographs given to a blinded investigator, posttreatment images were correctly identified in five. Two were assessed as 76%-100% improved, and three were assessed as 50%-75% improved (J Drugs Dermatol. 2015;14[9]:1005-1010).
The patients were an average of 38 years old, split about equally between the sexes, and racially diverse. They used a basic facial cleanser during the study, but were banned from using tretinoin and other topicals.
Previously studied topicals don’t do much for acne scars, so the usual go-to treatments are chemical peels, dermabrasion, resurfacing lasers, and percutaneous collagen needling. They all work in part by promoting collagen synthesis, but at the cost of pain and side effects. Epidermal growth factor (EGF) also promotes collagen synthesis, so the investigators thought it might help. The EGF used in the study – DNA Regeneration Serum, derived from barley – was supplied by its maker, DNA EGF Renewal in Los Angeles.
“The findings suggest EGF serum has the potential to be a modern, noninvasive treatment for an otherwise highly refractory condition. Whereas resurfacing procedures rely on skin injury to trigger [EGF] release, direct topical application offers the effects of EGF without the associated discomfort and recovery time,” said Dr. Ronald L. Moy of the University of Southern California, Los Angeles, and Rachel Seidel, a medical student at Georgetown University in Washington, D.C.
“All subjects in this study [also] noted improvements in skin texture, fine lines, and wrinkles, while the vast majority also saw a reduction in brown and age spots,” they said.
The investigators said they are interested next in seeing if topical EGF prevents scars in active acne. “We believe that, by counteracting collagen degradation during the course of the inflammatory response, significant tissue atrophy capable of causing visible scarring may be prevented,” they said.
Dr. Moy owns stock in DNA EGF Renewal and is the company’s scientific adviser.
FROM THE JOURNAL OF DRUGS AND DERMATOLOGY
Key clinical point: Topical synthetic epidermal growth factor serum may be a noninvasive way to improve the appearance of atrophic acne scars.
Major finding: Five of the 8 pairs of before and after photographs given to a blinded investigator were correctly identified as the posttreatment image.
Data source: A pilot study of eight patients with atrophic acne scars.
Disclosures: The EGF serum used in the study was supplied by its maker, DNA EGF Renewal. Dr. Moy owns stock in the company and is its scientific adviser.

Doctor, monitor thyself: The promise and perils of self-monitoring apps
I walked into my primary care doctor’s office the other day. I’m still young and healthy and a doctor, so making a doctor’s appointment is a rare event. As with most patients, it was symptoms that motivated me. I’m having a common, yet annoying problem: PVCs or premature ventricular contractions. I’ve had them on and off for a while, but now every time I push to my limit when exercising or double my espresso, they come back.
“Do you have them right now?” my doc asked me. “No. Just had them yesterday, though,” I replied. Dr. A is about my age and perhaps in even better shape than I am. He’s certainly smarter than I. Tall and athletic, he doesn’t wear a lab coat but is always immaculately dressed in a button-down shirt and light sweater. He walks from around his standing desk and hands me an iPhone cover. “Why don’t you try this?” Being the director of innovation, I recognized the device: It was a heart monitor. “Just download the app and track your EKG when you get symptoms,” he said.
I turned it over in my hands. It’s flimsier than I remembered from tech conferences. It’s even too small to fit on my iPhone 6+, although it doesn’t technically have to be on the phone to work. When I got home I downloaded the app and uploaded my first tracing. While right next to my phone, I gently touched the two sensors with my fingers. My tracing appeared on the screen. Wow, those are PQRS waves. (Indeed, I was a intern, too, once). The app requires that you submit the first recording for review before you can use it to verify that the tracing is normal.
The next morning, I hit the bike with everything I had, driving my heart rate to more than 170. (150 is working hard, 160 is painful, 170 is unsustainable for me. Sure enough, my PVCs returned later that day. Later that night, they were driving me crazy. I got out of bed and grabbed my phone. There, at 2 a.m., the glow of my iPhone lighting my bedroom, I could see my EKG: 1,2,3, PVC, 1,2,3, PVC. Wow! This is cool.
As the innovation director, most of the devices I review are from the viewpoint of a physician. This was different. I was clearly the patient in this story, and the device was meant to help me.
We talk about how digital medicine empowers our patients, and I suppose this is the idea. I now have access to diagnostic tools that ordinarily only my doctor would have. Yet, even though I clearly had PVCs this time, the app sends me back the same note as the first time I used it: “Normal EKG.” That’s true, technically. However, it’s easy for even a dermatologist to see that this tracing was different from the first.
I knew that quadrigeminy was a common and benign tracing, but if I wasn’t a physician (or hadn’t been trained by a great upper-level resident as an intern), then I might have been too anxious to fall back asleep.
Elizabeth Holmes in a recent Wall Street Journal article advocated for patients to be able to choose their own blood tests (and someday check their own blood, using her device, one presumes). Health care technology conferences abound with devices that promise to put the power of diagnostics in patients’ hands. But, as we all know, getting data is the easy part. It’s interpreting data – that’s why docs get the big bucks.
We also understand that often the best test is no test at all. If we randomly sampled EKGs from a population of everyone, we might find a few interesting tracings, most of which would have no meaningful consequences. Except if you’re a patient and your EKG has a funny blip on your at-home EKG device, or your iPhone dermatology app incorrectly reports a seborrheic keratosis as a possible melanoma. In such cases, these technologies have not empowered the user; rather, they’ve created needless anxiety, none of which existed before. The result is often more work for us physicians who must now spend time explaining why the patient’s finding is not important, and worse, might end up ordering more (real) tests to disconfirm what the at-home home test found.
Later, I brought my iPhone to my follow-up appointment and shared the tracings with my primary care doctor. “Looks like PVCs,” he confirmed, “and it looks normal.” But I already knew that.
Dr. Benabio is a partner physician in the department of dermatology of the Southern California Permanente Group in San Diego, and a volunteer clinical assistant professor at the University of California, San Diego. Dr. Benabio is @dermdoc on Twitter.
I walked into my primary care doctor’s office the other day. I’m still young and healthy and a doctor, so making a doctor’s appointment is a rare event. As with most patients, it was symptoms that motivated me. I’m having a common, yet annoying problem: PVCs or premature ventricular contractions. I’ve had them on and off for a while, but now every time I push to my limit when exercising or double my espresso, they come back.
“Do you have them right now?” my doc asked me. “No. Just had them yesterday, though,” I replied. Dr. A is about my age and perhaps in even better shape than I am. He’s certainly smarter than I. Tall and athletic, he doesn’t wear a lab coat but is always immaculately dressed in a button-down shirt and light sweater. He walks from around his standing desk and hands me an iPhone cover. “Why don’t you try this?” Being the director of innovation, I recognized the device: It was a heart monitor. “Just download the app and track your EKG when you get symptoms,” he said.
I turned it over in my hands. It’s flimsier than I remembered from tech conferences. It’s even too small to fit on my iPhone 6+, although it doesn’t technically have to be on the phone to work. When I got home I downloaded the app and uploaded my first tracing. While right next to my phone, I gently touched the two sensors with my fingers. My tracing appeared on the screen. Wow, those are PQRS waves. (Indeed, I was a intern, too, once). The app requires that you submit the first recording for review before you can use it to verify that the tracing is normal.
The next morning, I hit the bike with everything I had, driving my heart rate to more than 170. (150 is working hard, 160 is painful, 170 is unsustainable for me. Sure enough, my PVCs returned later that day. Later that night, they were driving me crazy. I got out of bed and grabbed my phone. There, at 2 a.m., the glow of my iPhone lighting my bedroom, I could see my EKG: 1,2,3, PVC, 1,2,3, PVC. Wow! This is cool.
As the innovation director, most of the devices I review are from the viewpoint of a physician. This was different. I was clearly the patient in this story, and the device was meant to help me.
We talk about how digital medicine empowers our patients, and I suppose this is the idea. I now have access to diagnostic tools that ordinarily only my doctor would have. Yet, even though I clearly had PVCs this time, the app sends me back the same note as the first time I used it: “Normal EKG.” That’s true, technically. However, it’s easy for even a dermatologist to see that this tracing was different from the first.
I knew that quadrigeminy was a common and benign tracing, but if I wasn’t a physician (or hadn’t been trained by a great upper-level resident as an intern), then I might have been too anxious to fall back asleep.
Elizabeth Holmes in a recent Wall Street Journal article advocated for patients to be able to choose their own blood tests (and someday check their own blood, using her device, one presumes). Health care technology conferences abound with devices that promise to put the power of diagnostics in patients’ hands. But, as we all know, getting data is the easy part. It’s interpreting data – that’s why docs get the big bucks.
We also understand that often the best test is no test at all. If we randomly sampled EKGs from a population of everyone, we might find a few interesting tracings, most of which would have no meaningful consequences. Except if you’re a patient and your EKG has a funny blip on your at-home EKG device, or your iPhone dermatology app incorrectly reports a seborrheic keratosis as a possible melanoma. In such cases, these technologies have not empowered the user; rather, they’ve created needless anxiety, none of which existed before. The result is often more work for us physicians who must now spend time explaining why the patient’s finding is not important, and worse, might end up ordering more (real) tests to disconfirm what the at-home home test found.
Later, I brought my iPhone to my follow-up appointment and shared the tracings with my primary care doctor. “Looks like PVCs,” he confirmed, “and it looks normal.” But I already knew that.
Dr. Benabio is a partner physician in the department of dermatology of the Southern California Permanente Group in San Diego, and a volunteer clinical assistant professor at the University of California, San Diego. Dr. Benabio is @dermdoc on Twitter.
I walked into my primary care doctor’s office the other day. I’m still young and healthy and a doctor, so making a doctor’s appointment is a rare event. As with most patients, it was symptoms that motivated me. I’m having a common, yet annoying problem: PVCs or premature ventricular contractions. I’ve had them on and off for a while, but now every time I push to my limit when exercising or double my espresso, they come back.
“Do you have them right now?” my doc asked me. “No. Just had them yesterday, though,” I replied. Dr. A is about my age and perhaps in even better shape than I am. He’s certainly smarter than I. Tall and athletic, he doesn’t wear a lab coat but is always immaculately dressed in a button-down shirt and light sweater. He walks from around his standing desk and hands me an iPhone cover. “Why don’t you try this?” Being the director of innovation, I recognized the device: It was a heart monitor. “Just download the app and track your EKG when you get symptoms,” he said.
I turned it over in my hands. It’s flimsier than I remembered from tech conferences. It’s even too small to fit on my iPhone 6+, although it doesn’t technically have to be on the phone to work. When I got home I downloaded the app and uploaded my first tracing. While right next to my phone, I gently touched the two sensors with my fingers. My tracing appeared on the screen. Wow, those are PQRS waves. (Indeed, I was a intern, too, once). The app requires that you submit the first recording for review before you can use it to verify that the tracing is normal.
The next morning, I hit the bike with everything I had, driving my heart rate to more than 170. (150 is working hard, 160 is painful, 170 is unsustainable for me. Sure enough, my PVCs returned later that day. Later that night, they were driving me crazy. I got out of bed and grabbed my phone. There, at 2 a.m., the glow of my iPhone lighting my bedroom, I could see my EKG: 1,2,3, PVC, 1,2,3, PVC. Wow! This is cool.
As the innovation director, most of the devices I review are from the viewpoint of a physician. This was different. I was clearly the patient in this story, and the device was meant to help me.
We talk about how digital medicine empowers our patients, and I suppose this is the idea. I now have access to diagnostic tools that ordinarily only my doctor would have. Yet, even though I clearly had PVCs this time, the app sends me back the same note as the first time I used it: “Normal EKG.” That’s true, technically. However, it’s easy for even a dermatologist to see that this tracing was different from the first.
I knew that quadrigeminy was a common and benign tracing, but if I wasn’t a physician (or hadn’t been trained by a great upper-level resident as an intern), then I might have been too anxious to fall back asleep.
Elizabeth Holmes in a recent Wall Street Journal article advocated for patients to be able to choose their own blood tests (and someday check their own blood, using her device, one presumes). Health care technology conferences abound with devices that promise to put the power of diagnostics in patients’ hands. But, as we all know, getting data is the easy part. It’s interpreting data – that’s why docs get the big bucks.
We also understand that often the best test is no test at all. If we randomly sampled EKGs from a population of everyone, we might find a few interesting tracings, most of which would have no meaningful consequences. Except if you’re a patient and your EKG has a funny blip on your at-home EKG device, or your iPhone dermatology app incorrectly reports a seborrheic keratosis as a possible melanoma. In such cases, these technologies have not empowered the user; rather, they’ve created needless anxiety, none of which existed before. The result is often more work for us physicians who must now spend time explaining why the patient’s finding is not important, and worse, might end up ordering more (real) tests to disconfirm what the at-home home test found.
Later, I brought my iPhone to my follow-up appointment and shared the tracings with my primary care doctor. “Looks like PVCs,” he confirmed, “and it looks normal.” But I already knew that.
Dr. Benabio is a partner physician in the department of dermatology of the Southern California Permanente Group in San Diego, and a volunteer clinical assistant professor at the University of California, San Diego. Dr. Benabio is @dermdoc on Twitter.

Scientists describe new way to create etoposide

Scientists have reported a new way to produce the chemotherapeutic agent etoposide, and they believe this discovery could lead to a more stable supply of the drug.
Currently, producing etoposide requires isolating one of its precursors, (–)-podophyllotoxin, from the endangered, slow-growing, Himalayan Mayapple plant (Podophyllum hexandrum).
But researchers found they could generate the immediate precursor to etoposide—(–)-4’-desmethyl-epipodophyllotoxin—in a more easily accessible, faster-growing tobacco plant (Nicotiana benthamiana).
Elizabeth Sattely, PhD, of Stanford University in California, and her graduate student, Warren Lau, described this work in Science.
The pair noted that there are 4 known genes behind (–)-podophyllotoxin production, but the full recipe for this compound has eluded researchers, in part because of the Mayapple’s immense genome.
To tap into the Mayapple’s chemotherapeutic potential, Lau and Dr Sattely first focused on the 4 known genes—PLR, SDH, CYP719A23, and DIR. Then, they analyzed RNA sequencing data from the Mayapple to identify similar genes.
Next, the pair manipulated the tobacco plant to express multiple gene candidates at once and identified the resulting compounds in leaf tissue using mass spectrometry.
Dr Sattely and Lau identified 6 new genes—OMT3, CYP71CU1, OMT1, 2-ODD, CYP71BE54, and CYP82D61.
These genes, when expressed with the original 4, produce the immediate etoposide precursor (–)-4′-desmethyl-epipodophyllotoxin, which outperforms (–)-podophyllotoxin as a chemotherapy ingredient.
The researchers said this work has revealed a simpler and more direct route to etoposide that circumvents the semisynthetic epimerization and demethylation required to produce etoposide from (–)-podophyllotoxin.
However, Dr Sattely said the eventual goal is to use yeast instead of plants to produce etoposide. Yeast can be grown in large vats and may therefore provide a more stable source of drugs.
In addition, yeast provides the opportunity to modify genes to produce proteins with slightly different functions. And it may be possible to feed the yeast a slightly different starting product, thereby changing the chemical a molecular assembly line churns out.
These approaches could provide a way of tweaking existing drugs in an effort to improve them. ![]()
Scientists have reported a new way to produce the chemotherapeutic agent etoposide, and they believe this discovery could lead to a more stable supply of the drug.
Currently, producing etoposide requires isolating one of its precursors, (–)-podophyllotoxin, from the endangered, slow-growing, Himalayan Mayapple plant (Podophyllum hexandrum).
But researchers found they could generate the immediate precursor to etoposide—(–)-4’-desmethyl-epipodophyllotoxin—in a more easily accessible, faster-growing tobacco plant (Nicotiana benthamiana).
Elizabeth Sattely, PhD, of Stanford University in California, and her graduate student, Warren Lau, described this work in Science.
The pair noted that there are 4 known genes behind (–)-podophyllotoxin production, but the full recipe for this compound has eluded researchers, in part because of the Mayapple’s immense genome.
To tap into the Mayapple’s chemotherapeutic potential, Lau and Dr Sattely first focused on the 4 known genes—PLR, SDH, CYP719A23, and DIR. Then, they analyzed RNA sequencing data from the Mayapple to identify similar genes.
Next, the pair manipulated the tobacco plant to express multiple gene candidates at once and identified the resulting compounds in leaf tissue using mass spectrometry.
Dr Sattely and Lau identified 6 new genes—OMT3, CYP71CU1, OMT1, 2-ODD, CYP71BE54, and CYP82D61.
These genes, when expressed with the original 4, produce the immediate etoposide precursor (–)-4′-desmethyl-epipodophyllotoxin, which outperforms (–)-podophyllotoxin as a chemotherapy ingredient.
The researchers said this work has revealed a simpler and more direct route to etoposide that circumvents the semisynthetic epimerization and demethylation required to produce etoposide from (–)-podophyllotoxin.
However, Dr Sattely said the eventual goal is to use yeast instead of plants to produce etoposide. Yeast can be grown in large vats and may therefore provide a more stable source of drugs.
In addition, yeast provides the opportunity to modify genes to produce proteins with slightly different functions. And it may be possible to feed the yeast a slightly different starting product, thereby changing the chemical a molecular assembly line churns out.
These approaches could provide a way of tweaking existing drugs in an effort to improve them. ![]()
Scientists have reported a new way to produce the chemotherapeutic agent etoposide, and they believe this discovery could lead to a more stable supply of the drug.
Currently, producing etoposide requires isolating one of its precursors, (–)-podophyllotoxin, from the endangered, slow-growing, Himalayan Mayapple plant (Podophyllum hexandrum).
But researchers found they could generate the immediate precursor to etoposide—(–)-4’-desmethyl-epipodophyllotoxin—in a more easily accessible, faster-growing tobacco plant (Nicotiana benthamiana).
Elizabeth Sattely, PhD, of Stanford University in California, and her graduate student, Warren Lau, described this work in Science.
The pair noted that there are 4 known genes behind (–)-podophyllotoxin production, but the full recipe for this compound has eluded researchers, in part because of the Mayapple’s immense genome.
To tap into the Mayapple’s chemotherapeutic potential, Lau and Dr Sattely first focused on the 4 known genes—PLR, SDH, CYP719A23, and DIR. Then, they analyzed RNA sequencing data from the Mayapple to identify similar genes.
Next, the pair manipulated the tobacco plant to express multiple gene candidates at once and identified the resulting compounds in leaf tissue using mass spectrometry.
Dr Sattely and Lau identified 6 new genes—OMT3, CYP71CU1, OMT1, 2-ODD, CYP71BE54, and CYP82D61.
These genes, when expressed with the original 4, produce the immediate etoposide precursor (–)-4′-desmethyl-epipodophyllotoxin, which outperforms (–)-podophyllotoxin as a chemotherapy ingredient.
The researchers said this work has revealed a simpler and more direct route to etoposide that circumvents the semisynthetic epimerization and demethylation required to produce etoposide from (–)-podophyllotoxin.
However, Dr Sattely said the eventual goal is to use yeast instead of plants to produce etoposide. Yeast can be grown in large vats and may therefore provide a more stable source of drugs.
In addition, yeast provides the opportunity to modify genes to produce proteins with slightly different functions. And it may be possible to feed the yeast a slightly different starting product, thereby changing the chemical a molecular assembly line churns out.
These approaches could provide a way of tweaking existing drugs in an effort to improve them. ![]()
T-cell exhaustion may be therapeutic target for AML

Image courtesy of NIAID
New research has revealed a population of T cells associated with relapse of acute myeloid leukemia (AML) after allogeneic stem cell transplant (allo-SCT).
Patients experienced an increase in this cell population before their relapse was diagnosed clinically.
The cells also appear to be markers of T-cell exhaustion, which suggests therapies targeting T-cell exhaustion might be able to treat or prevent AML relapse after
allo-SCT.
Hong Zheng, MD, PhD, of Penn State University College of Medicine in Hershey, Pennsylvania, and her colleagues conducted this research and reported the results in Blood Cancer Journal.
The investigators noted that T cells are major players in the graft-vs-leukemia effect, but persistent antigen stimulation can lead to T-cell exhaustion.
To determine whether T-cell exhaustion is involved in relapse after allo-SCT, the team performed phenotypic and functional studies on T cells from the peripheral blood of AML transplant recipients.
Dr Zheng and her colleagues discovered that patients who relapsed after allo-SCT had significantly elevated levels of PD-1hiTIM-3+ T cells.
These T cells produced fewer cytokines—interleukin 2, tumor necrosis factor-α, and interferon-γ—than normal T cells, which is a sign of T-cell exhaustion.
In addition, while other T cells mostly consisted of all 4 T-cell subsets, PD-1hiTIM-3+ T cells had no naive T cells (CCR7+CD45RA+) and significantly decreased levels of terminally differentiated effector T cells (CCR7-CD45RA+).
The investigators said this suggests PD-1hiTIM-3+ cells are phenotypically antigen-experienced T cells that have lost functional subsets, which is consistent with a state of T-cell exhaustion.
The team believes their findings could pave the way for new treatments for AML patients undergoing allo-SCT.
The PD-1 inhibitor nivolumab has been shown to block T-cell exhaustion in patients with solid tumors. So Dr Zheng is planning a clinical trial to see if such treatment could work for AML patients as well.
She also believes that PD-1hiTIM-3+ T cells could be used to diagnose relapses earlier than is currently possible.
“Two months before we were able to clinically diagnose relapse in these patients, we found these markers elevated in them,” Dr Zheng said. “If we can have an early diagnostic marker, we will potentially be able to improve clinical outcome significantly.”
Dr Zheng is currently investigating the trigger for T-cell exhaustion in transplant recipients with AML.
“The hypothesis is that when you have chronic antigen stimulation, the T cell can get exhausted,” she said. “We think that residual leukemia in our patients is the chronic stimulator that causes this.” ![]()
Image courtesy of NIAID
New research has revealed a population of T cells associated with relapse of acute myeloid leukemia (AML) after allogeneic stem cell transplant (allo-SCT).
Patients experienced an increase in this cell population before their relapse was diagnosed clinically.
The cells also appear to be markers of T-cell exhaustion, which suggests therapies targeting T-cell exhaustion might be able to treat or prevent AML relapse after
allo-SCT.
Hong Zheng, MD, PhD, of Penn State University College of Medicine in Hershey, Pennsylvania, and her colleagues conducted this research and reported the results in Blood Cancer Journal.
The investigators noted that T cells are major players in the graft-vs-leukemia effect, but persistent antigen stimulation can lead to T-cell exhaustion.
To determine whether T-cell exhaustion is involved in relapse after allo-SCT, the team performed phenotypic and functional studies on T cells from the peripheral blood of AML transplant recipients.
Dr Zheng and her colleagues discovered that patients who relapsed after allo-SCT had significantly elevated levels of PD-1hiTIM-3+ T cells.
These T cells produced fewer cytokines—interleukin 2, tumor necrosis factor-α, and interferon-γ—than normal T cells, which is a sign of T-cell exhaustion.
In addition, while other T cells mostly consisted of all 4 T-cell subsets, PD-1hiTIM-3+ T cells had no naive T cells (CCR7+CD45RA+) and significantly decreased levels of terminally differentiated effector T cells (CCR7-CD45RA+).
The investigators said this suggests PD-1hiTIM-3+ cells are phenotypically antigen-experienced T cells that have lost functional subsets, which is consistent with a state of T-cell exhaustion.
The team believes their findings could pave the way for new treatments for AML patients undergoing allo-SCT.
The PD-1 inhibitor nivolumab has been shown to block T-cell exhaustion in patients with solid tumors. So Dr Zheng is planning a clinical trial to see if such treatment could work for AML patients as well.
She also believes that PD-1hiTIM-3+ T cells could be used to diagnose relapses earlier than is currently possible.
“Two months before we were able to clinically diagnose relapse in these patients, we found these markers elevated in them,” Dr Zheng said. “If we can have an early diagnostic marker, we will potentially be able to improve clinical outcome significantly.”
Dr Zheng is currently investigating the trigger for T-cell exhaustion in transplant recipients with AML.
“The hypothesis is that when you have chronic antigen stimulation, the T cell can get exhausted,” she said. “We think that residual leukemia in our patients is the chronic stimulator that causes this.” ![]()
Image courtesy of NIAID
New research has revealed a population of T cells associated with relapse of acute myeloid leukemia (AML) after allogeneic stem cell transplant (allo-SCT).
Patients experienced an increase in this cell population before their relapse was diagnosed clinically.
The cells also appear to be markers of T-cell exhaustion, which suggests therapies targeting T-cell exhaustion might be able to treat or prevent AML relapse after
allo-SCT.
Hong Zheng, MD, PhD, of Penn State University College of Medicine in Hershey, Pennsylvania, and her colleagues conducted this research and reported the results in Blood Cancer Journal.
The investigators noted that T cells are major players in the graft-vs-leukemia effect, but persistent antigen stimulation can lead to T-cell exhaustion.
To determine whether T-cell exhaustion is involved in relapse after allo-SCT, the team performed phenotypic and functional studies on T cells from the peripheral blood of AML transplant recipients.
Dr Zheng and her colleagues discovered that patients who relapsed after allo-SCT had significantly elevated levels of PD-1hiTIM-3+ T cells.
These T cells produced fewer cytokines—interleukin 2, tumor necrosis factor-α, and interferon-γ—than normal T cells, which is a sign of T-cell exhaustion.
In addition, while other T cells mostly consisted of all 4 T-cell subsets, PD-1hiTIM-3+ T cells had no naive T cells (CCR7+CD45RA+) and significantly decreased levels of terminally differentiated effector T cells (CCR7-CD45RA+).
The investigators said this suggests PD-1hiTIM-3+ cells are phenotypically antigen-experienced T cells that have lost functional subsets, which is consistent with a state of T-cell exhaustion.
The team believes their findings could pave the way for new treatments for AML patients undergoing allo-SCT.
The PD-1 inhibitor nivolumab has been shown to block T-cell exhaustion in patients with solid tumors. So Dr Zheng is planning a clinical trial to see if such treatment could work for AML patients as well.
She also believes that PD-1hiTIM-3+ T cells could be used to diagnose relapses earlier than is currently possible.
“Two months before we were able to clinically diagnose relapse in these patients, we found these markers elevated in them,” Dr Zheng said. “If we can have an early diagnostic marker, we will potentially be able to improve clinical outcome significantly.”
Dr Zheng is currently investigating the trigger for T-cell exhaustion in transplant recipients with AML.
“The hypothesis is that when you have chronic antigen stimulation, the T cell can get exhausted,” she said. “We think that residual leukemia in our patients is the chronic stimulator that causes this.” ![]()
Online tool may aid study of immune system

Photo by Darren Baker
Researchers say they have designed an online tool that can predict the role of proteins and genes involved in immunological diseases and processes.
The tool uses information compiled from 38,088 public experiments to predict new immune pathway interactions, mechanisms, and disease-associated genes.
Details on this publicly available tool, known as ImmuNet, were recently published in Immunity.
“This new tool unlocks the insight contained in big data, the world’s biomedical research output, to help understand immunological mechanisms and diseases,” said Stuart Sealfon, MD, of Mount Sinai Health System in New York, New York.
“The goal of ImmuNet is to accelerate the understanding of immune pathways and genes, ultimately leading to the development of improved treatment for diseases with an immunological component.”
ImmuNet enables immunology researchers without special computational training to use the statistical techniques of Bayesian data integration and machine learning algorithms to “interrogate” this compendium of public data.
“We expect the applicability of ImmuNet to wide-ranging areas of immunology will grow with the incorporation of continually increasing public big data,” said Olga Troyanskaya, PhD, of Princeton University in New Jersey.
“By enabling immune researchers from diverse backgrounds to leverage these valuable and heterogeneous data collections, ImmuNet has the potential to accelerate discovery in immunology.” ![]()
Photo by Darren Baker
Researchers say they have designed an online tool that can predict the role of proteins and genes involved in immunological diseases and processes.
The tool uses information compiled from 38,088 public experiments to predict new immune pathway interactions, mechanisms, and disease-associated genes.
Details on this publicly available tool, known as ImmuNet, were recently published in Immunity.
“This new tool unlocks the insight contained in big data, the world’s biomedical research output, to help understand immunological mechanisms and diseases,” said Stuart Sealfon, MD, of Mount Sinai Health System in New York, New York.
“The goal of ImmuNet is to accelerate the understanding of immune pathways and genes, ultimately leading to the development of improved treatment for diseases with an immunological component.”
ImmuNet enables immunology researchers without special computational training to use the statistical techniques of Bayesian data integration and machine learning algorithms to “interrogate” this compendium of public data.
“We expect the applicability of ImmuNet to wide-ranging areas of immunology will grow with the incorporation of continually increasing public big data,” said Olga Troyanskaya, PhD, of Princeton University in New Jersey.
“By enabling immune researchers from diverse backgrounds to leverage these valuable and heterogeneous data collections, ImmuNet has the potential to accelerate discovery in immunology.” ![]()
Photo by Darren Baker
Researchers say they have designed an online tool that can predict the role of proteins and genes involved in immunological diseases and processes.
The tool uses information compiled from 38,088 public experiments to predict new immune pathway interactions, mechanisms, and disease-associated genes.
Details on this publicly available tool, known as ImmuNet, were recently published in Immunity.
“This new tool unlocks the insight contained in big data, the world’s biomedical research output, to help understand immunological mechanisms and diseases,” said Stuart Sealfon, MD, of Mount Sinai Health System in New York, New York.
“The goal of ImmuNet is to accelerate the understanding of immune pathways and genes, ultimately leading to the development of improved treatment for diseases with an immunological component.”
ImmuNet enables immunology researchers without special computational training to use the statistical techniques of Bayesian data integration and machine learning algorithms to “interrogate” this compendium of public data.
“We expect the applicability of ImmuNet to wide-ranging areas of immunology will grow with the incorporation of continually increasing public big data,” said Olga Troyanskaya, PhD, of Princeton University in New Jersey.
“By enabling immune researchers from diverse backgrounds to leverage these valuable and heterogeneous data collections, ImmuNet has the potential to accelerate discovery in immunology.” ![]()
Drug granted orphan designation for AML

Image by Lance Liotta
The investigational therapy SL-401 has received orphan designation to treat acute myeloid leukemia (AML) in the European Union (EU).
SL-401 targets the interleukin-3 receptor (IL-3R). It is comprised of human IL-3 coupled to a truncated diphtheria toxin payload that inhibits protein synthesis.
SL-401 is under development to treat a range of hematologic malignancies, as IL-3R is overexpressed on cancer stem cells in multiple hematologic malignancies.
SL-401 has orphan designation from the US Food and Drug Administration (FDA) for the treatment of AML and blastic plasmacytoid dendritic cell neoplasm (BPDCN).
SL-401 research
At ASH 2012 (abstract 3625), researchers reported results with SL-401 in a study of patients with AML, BPDCN, and myelodysplastic syndromes (MDS).
At that time, the study had enrolled 80 patients, including 59 with relapsed or refractory AML, 11 with de novo AML unfit for chemotherapy, 7 with high-risk MDS, and 3 with relapsed/refractory BPDCN.
Patients received a single cycle of SL-401 as a 15-minute intravenous infusion in 1 of 2 dosing regimens to determine the maximum tolerated dose (MTD) and assess antitumor activity.
With regimen A, 45 patients received doses ranging from 4 to 12.5 μg/kg every other day for up to 6 doses. With regimen B, 35 patients received doses ranging from 7.1 to 22.1 μg/kg daily for up to 5 doses.
Of the 59 patients with relapsed/refractory AML, 2 achieved complete responses (CRs), 5 had partial responses (PRs), and 8 had minor responses (MRs). One CR lasted more than 8 months, and the other lasted more than 25 months.
Of the 11 patients with AML who were not candidates for chemotherapy, 2 had PRs and 1 had an MR. Among the 7 patients with high-risk MDS, there was 1 PR and 1 MR.
And among the 3 patients with BPDCN, there were 2 CRs. One CR lasted more than 2 months, and the other lasted more than 4 months.
The MTD was not achieved with regimen A, but the MTD for regimen B was 16.6 μg/kg/day. The dose-limiting toxicities were a gastrointestinal bleed (n=1), transaminase and creatinine kinase elevations (n=1), and capillary leak syndrome (n=3). There was no evidence of treatment-related bone marrow suppression.
Last year, researchers reported additional results in BPDCN patients (Frankel et al, Blood 2014).
Eleven BPDCN patients received a single course of SL-401 (at 12.5 μg/kg intravenously over 15 minutes) daily for up to 5 doses. Three patients who had initial responses to SL-401 received a second course while in relapse.
Seven of 9 evaluable (78%) patients responded to a single course of SL-401. There were 5 CRs and 2 PRs. The median duration of responses was 5 months (range, 1-20+ months).
The most common adverse events were transient and included fever, chills, hypotension, edema, hypoalbuminemia, thrombocytopenia, and transaminasemia.
Three multicenter clinical trials of SL-401 are currently open in the following indications:
- BPDCN and relapsed/refractory AML
- AML patients in first complete remission with minimal residual disease
- Four types of advanced, high-risk myeloproliferative neoplasms, including systemic mastocytosis, advanced symptomatic hypereosinophilic disorder, myelofibrosis, and chronic myelomonocytic leukemia.
Additional SL-401 studies are planned for patients with myeloma, lymphomas, and other leukemias.
About orphan designation
In the EU, orphan designation is granted to therapies intended to treat a life-threatening or chronically debilitating condition that affects no more than 5 in 10,000 persons and where no satisfactory treatment is available.
Companies that obtain orphan designation for a drug in the EU benefit from a number of incentives, including protocol assistance, a type of scientific advice specific for designated orphan medicines, and 10 years of market exclusivity once the medicine is on the market. Fee reductions are also available, depending on the status of the sponsor and the type of service required.
The FDA grants orphan designation to drugs that are intended to treat diseases or conditions affecting fewer than 200,000 patients in the US.
In the US, orphan designation provides the sponsor of a drug with various development incentives, including opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, and 7 years of US market exclusivity if the drug is approved. ![]()
Image by Lance Liotta
The investigational therapy SL-401 has received orphan designation to treat acute myeloid leukemia (AML) in the European Union (EU).
SL-401 targets the interleukin-3 receptor (IL-3R). It is comprised of human IL-3 coupled to a truncated diphtheria toxin payload that inhibits protein synthesis.
SL-401 is under development to treat a range of hematologic malignancies, as IL-3R is overexpressed on cancer stem cells in multiple hematologic malignancies.
SL-401 has orphan designation from the US Food and Drug Administration (FDA) for the treatment of AML and blastic plasmacytoid dendritic cell neoplasm (BPDCN).
SL-401 research
At ASH 2012 (abstract 3625), researchers reported results with SL-401 in a study of patients with AML, BPDCN, and myelodysplastic syndromes (MDS).
At that time, the study had enrolled 80 patients, including 59 with relapsed or refractory AML, 11 with de novo AML unfit for chemotherapy, 7 with high-risk MDS, and 3 with relapsed/refractory BPDCN.
Patients received a single cycle of SL-401 as a 15-minute intravenous infusion in 1 of 2 dosing regimens to determine the maximum tolerated dose (MTD) and assess antitumor activity.
With regimen A, 45 patients received doses ranging from 4 to 12.5 μg/kg every other day for up to 6 doses. With regimen B, 35 patients received doses ranging from 7.1 to 22.1 μg/kg daily for up to 5 doses.
Of the 59 patients with relapsed/refractory AML, 2 achieved complete responses (CRs), 5 had partial responses (PRs), and 8 had minor responses (MRs). One CR lasted more than 8 months, and the other lasted more than 25 months.
Of the 11 patients with AML who were not candidates for chemotherapy, 2 had PRs and 1 had an MR. Among the 7 patients with high-risk MDS, there was 1 PR and 1 MR.
And among the 3 patients with BPDCN, there were 2 CRs. One CR lasted more than 2 months, and the other lasted more than 4 months.
The MTD was not achieved with regimen A, but the MTD for regimen B was 16.6 μg/kg/day. The dose-limiting toxicities were a gastrointestinal bleed (n=1), transaminase and creatinine kinase elevations (n=1), and capillary leak syndrome (n=3). There was no evidence of treatment-related bone marrow suppression.
Last year, researchers reported additional results in BPDCN patients (Frankel et al, Blood 2014).
Eleven BPDCN patients received a single course of SL-401 (at 12.5 μg/kg intravenously over 15 minutes) daily for up to 5 doses. Three patients who had initial responses to SL-401 received a second course while in relapse.
Seven of 9 evaluable (78%) patients responded to a single course of SL-401. There were 5 CRs and 2 PRs. The median duration of responses was 5 months (range, 1-20+ months).
The most common adverse events were transient and included fever, chills, hypotension, edema, hypoalbuminemia, thrombocytopenia, and transaminasemia.
Three multicenter clinical trials of SL-401 are currently open in the following indications:
- BPDCN and relapsed/refractory AML
- AML patients in first complete remission with minimal residual disease
- Four types of advanced, high-risk myeloproliferative neoplasms, including systemic mastocytosis, advanced symptomatic hypereosinophilic disorder, myelofibrosis, and chronic myelomonocytic leukemia.
Additional SL-401 studies are planned for patients with myeloma, lymphomas, and other leukemias.
About orphan designation
In the EU, orphan designation is granted to therapies intended to treat a life-threatening or chronically debilitating condition that affects no more than 5 in 10,000 persons and where no satisfactory treatment is available.
Companies that obtain orphan designation for a drug in the EU benefit from a number of incentives, including protocol assistance, a type of scientific advice specific for designated orphan medicines, and 10 years of market exclusivity once the medicine is on the market. Fee reductions are also available, depending on the status of the sponsor and the type of service required.
The FDA grants orphan designation to drugs that are intended to treat diseases or conditions affecting fewer than 200,000 patients in the US.
In the US, orphan designation provides the sponsor of a drug with various development incentives, including opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, and 7 years of US market exclusivity if the drug is approved. ![]()
Image by Lance Liotta
The investigational therapy SL-401 has received orphan designation to treat acute myeloid leukemia (AML) in the European Union (EU).
SL-401 targets the interleukin-3 receptor (IL-3R). It is comprised of human IL-3 coupled to a truncated diphtheria toxin payload that inhibits protein synthesis.
SL-401 is under development to treat a range of hematologic malignancies, as IL-3R is overexpressed on cancer stem cells in multiple hematologic malignancies.
SL-401 has orphan designation from the US Food and Drug Administration (FDA) for the treatment of AML and blastic plasmacytoid dendritic cell neoplasm (BPDCN).
SL-401 research
At ASH 2012 (abstract 3625), researchers reported results with SL-401 in a study of patients with AML, BPDCN, and myelodysplastic syndromes (MDS).
At that time, the study had enrolled 80 patients, including 59 with relapsed or refractory AML, 11 with de novo AML unfit for chemotherapy, 7 with high-risk MDS, and 3 with relapsed/refractory BPDCN.
Patients received a single cycle of SL-401 as a 15-minute intravenous infusion in 1 of 2 dosing regimens to determine the maximum tolerated dose (MTD) and assess antitumor activity.
With regimen A, 45 patients received doses ranging from 4 to 12.5 μg/kg every other day for up to 6 doses. With regimen B, 35 patients received doses ranging from 7.1 to 22.1 μg/kg daily for up to 5 doses.
Of the 59 patients with relapsed/refractory AML, 2 achieved complete responses (CRs), 5 had partial responses (PRs), and 8 had minor responses (MRs). One CR lasted more than 8 months, and the other lasted more than 25 months.
Of the 11 patients with AML who were not candidates for chemotherapy, 2 had PRs and 1 had an MR. Among the 7 patients with high-risk MDS, there was 1 PR and 1 MR.
And among the 3 patients with BPDCN, there were 2 CRs. One CR lasted more than 2 months, and the other lasted more than 4 months.
The MTD was not achieved with regimen A, but the MTD for regimen B was 16.6 μg/kg/day. The dose-limiting toxicities were a gastrointestinal bleed (n=1), transaminase and creatinine kinase elevations (n=1), and capillary leak syndrome (n=3). There was no evidence of treatment-related bone marrow suppression.
Last year, researchers reported additional results in BPDCN patients (Frankel et al, Blood 2014).
Eleven BPDCN patients received a single course of SL-401 (at 12.5 μg/kg intravenously over 15 minutes) daily for up to 5 doses. Three patients who had initial responses to SL-401 received a second course while in relapse.
Seven of 9 evaluable (78%) patients responded to a single course of SL-401. There were 5 CRs and 2 PRs. The median duration of responses was 5 months (range, 1-20+ months).
The most common adverse events were transient and included fever, chills, hypotension, edema, hypoalbuminemia, thrombocytopenia, and transaminasemia.
Three multicenter clinical trials of SL-401 are currently open in the following indications:
- BPDCN and relapsed/refractory AML
- AML patients in first complete remission with minimal residual disease
- Four types of advanced, high-risk myeloproliferative neoplasms, including systemic mastocytosis, advanced symptomatic hypereosinophilic disorder, myelofibrosis, and chronic myelomonocytic leukemia.
Additional SL-401 studies are planned for patients with myeloma, lymphomas, and other leukemias.
About orphan designation
In the EU, orphan designation is granted to therapies intended to treat a life-threatening or chronically debilitating condition that affects no more than 5 in 10,000 persons and where no satisfactory treatment is available.
Companies that obtain orphan designation for a drug in the EU benefit from a number of incentives, including protocol assistance, a type of scientific advice specific for designated orphan medicines, and 10 years of market exclusivity once the medicine is on the market. Fee reductions are also available, depending on the status of the sponsor and the type of service required.
The FDA grants orphan designation to drugs that are intended to treat diseases or conditions affecting fewer than 200,000 patients in the US.
In the US, orphan designation provides the sponsor of a drug with various development incentives, including opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, and 7 years of US market exclusivity if the drug is approved. ![]()
Death and dying
Learning about death and dying in residency is perhaps one of the hardest things about being a doctor. ... This list might make it a tiny bit easier.
1. The Hippocratic oath challenges us to be teachers not only to students, but also to our patients – especially to our dying patients. Remember you are teachers.
2. Nurses spend the most time with your patients; utilize them and learn from them. Nurses will make you better teachers and physicians.
3. When a patient is dying, hold that person’s hand if family isn’t around. Touch your patient.
4. Accompany your residents, fellows, or attending physicians to deliver bad news.
5. Ask to debrief after a patient dies.

6. While discussing treatment or prognosis with families, pay attention to the emotional data in the room. Tears should prompt tissues or hand holding. A wrinkled forehead or fearful face warrants a pause in conversation for the family to absorb the content. Tears don’t mean to hurry.
7. Attempt to understand your patient’s expectations. In other words, learn what the family’s goals are for treatment.
8. When a patient dies, reflect upon it in moderation. Don’t just move on to the next task. It’s okay to feel and to grieve … you are human.
9. When families declare DNR [do not resuscitate], don’t ask them repeatedly if they are sure. Also, make certain that they know they can change their minds at any time. Respect a patient’s values and wishes.
10. Take the time to have the difficult conversations early in diagnosis so families know what to expect. This empowers decisions that are proactive as opposed to reactive. Be proactive in your role as a teacher.
11. When discussing options, talk about what you CAN do before you talk about what you CAN’T do.
12. Withdrawing treatment and forgoing treatment are the same thing ethically and legally, but not emotionally.
13. When your patient is actively dying, see it out. Stay with the family, even if it’s time to go home.
14. When the family prays, be present.
15. Palliative care is neither hospice nor social work.
16. Ask your dying patients if they are afraid of anything – if the moment presents itself. Answer questions about death or find someone who can answer their questions.
17. No patient should die in pain. The doctrine of double effect allows a caregiver to provide medication for pain even though it may hasten death.
18. For the dying child, ask the family where they want their child to die. Don’t assume the family wants the death of their child to be at home or in the hospital. Ask.
19. Transitioning from treatment-oriented management to comfort care does not ever mean “there is nothing we can do.”
20. Lastly, and most importantly, I leave you with a question that should be in the back of our minds regarding quality of life and treatment options: Just because we can, should we?
Dr. Morvant is a second-year pediatric resident at Monroe Carell Jr. Children’s Hospital at Vanderbilt, Nashville, Tenn. E-mail her at [email protected].
Learning about death and dying in residency is perhaps one of the hardest things about being a doctor. ... This list might make it a tiny bit easier.
1. The Hippocratic oath challenges us to be teachers not only to students, but also to our patients – especially to our dying patients. Remember you are teachers.
2. Nurses spend the most time with your patients; utilize them and learn from them. Nurses will make you better teachers and physicians.
3. When a patient is dying, hold that person’s hand if family isn’t around. Touch your patient.
4. Accompany your residents, fellows, or attending physicians to deliver bad news.
5. Ask to debrief after a patient dies.
6. While discussing treatment or prognosis with families, pay attention to the emotional data in the room. Tears should prompt tissues or hand holding. A wrinkled forehead or fearful face warrants a pause in conversation for the family to absorb the content. Tears don’t mean to hurry.
7. Attempt to understand your patient’s expectations. In other words, learn what the family’s goals are for treatment.
8. When a patient dies, reflect upon it in moderation. Don’t just move on to the next task. It’s okay to feel and to grieve … you are human.
9. When families declare DNR [do not resuscitate], don’t ask them repeatedly if they are sure. Also, make certain that they know they can change their minds at any time. Respect a patient’s values and wishes.
10. Take the time to have the difficult conversations early in diagnosis so families know what to expect. This empowers decisions that are proactive as opposed to reactive. Be proactive in your role as a teacher.
11. When discussing options, talk about what you CAN do before you talk about what you CAN’T do.
12. Withdrawing treatment and forgoing treatment are the same thing ethically and legally, but not emotionally.
13. When your patient is actively dying, see it out. Stay with the family, even if it’s time to go home.
14. When the family prays, be present.
15. Palliative care is neither hospice nor social work.
16. Ask your dying patients if they are afraid of anything – if the moment presents itself. Answer questions about death or find someone who can answer their questions.
17. No patient should die in pain. The doctrine of double effect allows a caregiver to provide medication for pain even though it may hasten death.
18. For the dying child, ask the family where they want their child to die. Don’t assume the family wants the death of their child to be at home or in the hospital. Ask.
19. Transitioning from treatment-oriented management to comfort care does not ever mean “there is nothing we can do.”
20. Lastly, and most importantly, I leave you with a question that should be in the back of our minds regarding quality of life and treatment options: Just because we can, should we?
Dr. Morvant is a second-year pediatric resident at Monroe Carell Jr. Children’s Hospital at Vanderbilt, Nashville, Tenn. E-mail her at [email protected].
Learning about death and dying in residency is perhaps one of the hardest things about being a doctor. ... This list might make it a tiny bit easier.
1. The Hippocratic oath challenges us to be teachers not only to students, but also to our patients – especially to our dying patients. Remember you are teachers.
2. Nurses spend the most time with your patients; utilize them and learn from them. Nurses will make you better teachers and physicians.
3. When a patient is dying, hold that person’s hand if family isn’t around. Touch your patient.
4. Accompany your residents, fellows, or attending physicians to deliver bad news.
5. Ask to debrief after a patient dies.
6. While discussing treatment or prognosis with families, pay attention to the emotional data in the room. Tears should prompt tissues or hand holding. A wrinkled forehead or fearful face warrants a pause in conversation for the family to absorb the content. Tears don’t mean to hurry.
7. Attempt to understand your patient’s expectations. In other words, learn what the family’s goals are for treatment.
8. When a patient dies, reflect upon it in moderation. Don’t just move on to the next task. It’s okay to feel and to grieve … you are human.
9. When families declare DNR [do not resuscitate], don’t ask them repeatedly if they are sure. Also, make certain that they know they can change their minds at any time. Respect a patient’s values and wishes.
10. Take the time to have the difficult conversations early in diagnosis so families know what to expect. This empowers decisions that are proactive as opposed to reactive. Be proactive in your role as a teacher.
11. When discussing options, talk about what you CAN do before you talk about what you CAN’T do.
12. Withdrawing treatment and forgoing treatment are the same thing ethically and legally, but not emotionally.
13. When your patient is actively dying, see it out. Stay with the family, even if it’s time to go home.
14. When the family prays, be present.
15. Palliative care is neither hospice nor social work.
16. Ask your dying patients if they are afraid of anything – if the moment presents itself. Answer questions about death or find someone who can answer their questions.
17. No patient should die in pain. The doctrine of double effect allows a caregiver to provide medication for pain even though it may hasten death.
18. For the dying child, ask the family where they want their child to die. Don’t assume the family wants the death of their child to be at home or in the hospital. Ask.
19. Transitioning from treatment-oriented management to comfort care does not ever mean “there is nothing we can do.”
20. Lastly, and most importantly, I leave you with a question that should be in the back of our minds regarding quality of life and treatment options: Just because we can, should we?
Dr. Morvant is a second-year pediatric resident at Monroe Carell Jr. Children’s Hospital at Vanderbilt, Nashville, Tenn. E-mail her at [email protected].
