User login
Metronidazole and alcohol
A 32-year-old man develops diarrhea after receiving amoxicillin/clavulanate to treat an infection following a dog bite. He is diagnosed with Clostridium difficile and prescribed a 10-day course of metronidazole. He has no other medical problems. He will be the best man at his brother’s wedding tomorrow. What advice should you give him about alcohol use at the reception?
A. Do not take metronidazole the day of the wedding if you will be drinking alcohol.
B. Take metronidazole, do not drink alcohol.
C. It’s okay to drink alcohol.
For years, we have advised patients to not use alcohol if they are taking metronidazole because of concern for a disulfiram-like reaction between alcohol and metronidazole. This has been a standard warning given by physicians and appears as a contraindication in the prescribing information. It has been well accepted as a true, proven reaction.
Is it true?
As early as the 1960s, case reports and an uncontrolled study suggested that combining metronidazole with alcohol produced a disulfiram-like reaction, with case reports of severe reactions, including death.1, 2, 3 This was initially considered an area that might be therapeutic in the treatment of alcoholism, but several studies showed no benefit.4, 5
Caroline S. Williams and Dr. Kevin R. Woodcock reviewed the case reports for evidence of proof of a true interaction between metronidazole and ethanol.6 The case reports referenced textbooks to substantiate the interaction, but they did not present clear evidence of an interaction as the cause of elevated acetaldehyde levels.
Researchers have shown in a rat model that metronidazole can increase intracolonic, but not blood, acetaldehyde levels in rats that have received a combination of ethanol and metronidazole.7 Metronidazole did not have any inhibitory effect on hepatic or colonic alcohol dehydrogenase or aldehyde dehydrogenase. What was found was that rats treated with metronidazole had increased growth of Enterobacteriaceae, an alcohol dehydrogenase–containing aerobe, which could be the cause of the higher intracolonic acetaldehyde levels.
Jukka-Pekka Visapää and his colleagues studied the effect of coadministration of metronidazole and ethanol in young, healthy male volunteers.8 The study was a placebo-controlled, randomized trial. The study was small, with 12 participants. One-half of the study participants received metronidazole three times a day for 5 days; the other half received placebo. All participants then received ethanol 0.4g/kg, with blood testing being done every 20 minutes for the next 4 hours. Blood was tested for ethanol concentrations and for acetaldehyde levels. The study participants also had blood pressure, pulse, skin temperature, and symptoms monitored during the study.
There was no difference in blood acetaldehyde levels, vital signs, or symptoms between patients who received metronidazole or placebo. None of the subjects in the study had any measurable symptoms.
Metronidazole has many side effects, including nausea, vomiting, headache, dizziness, and seizures. These symptoms have a great deal of overlap with the symptoms of alcohol-disulfiram interaction. It has been assumed in early case reports that metronidazole caused a similar interaction with alcohol and raised acetaldehyde levels by interfering with aldehyde dehydrogenase.
Animal models and the human study do not show this to be the case. It is possible that metronidazole side effects alone were the cause of the symptoms in case reports. The one human study done was on healthy male volunteers, so projecting the results to a population with liver disease or other serious illness is a bit of a stretch. I think that if a problem exists with alcohol and metronidazole, it is uncommon and unlikely to occur in healthy individuals.
So, what would I advise the patient in the case about whether he can drink alcohol? I think that the risk would be minimal and that it would be safe for him to drink alcohol.
References
1. Br J Clin Pract. 1985 Jul;39(7):292-3.
2. Psychiatr Neurol. 1966;152:395-401.
3. Am J Forensic Med Pathol. 1996 Dec;17(4):343-6.
4. Q J Stud Alcohol. 1972 Sep;33: 734-40.
5. Q J Stud Ethanol. 1969 Mar;30: 140-51.
6. Ann Pharmacother. 2000 Feb;34(2):255-7.
7. Alcohol Clin Exp Res. 2000 Apr;24(4):570-5.
8. Ann Pharmacother. 2002 Jun;36(6):971-4.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and he serves as third-year medical student clerkship director at the University of Washington. Contact Dr. Paauw at [email protected].
A 32-year-old man develops diarrhea after receiving amoxicillin/clavulanate to treat an infection following a dog bite. He is diagnosed with Clostridium difficile and prescribed a 10-day course of metronidazole. He has no other medical problems. He will be the best man at his brother’s wedding tomorrow. What advice should you give him about alcohol use at the reception?
A. Do not take metronidazole the day of the wedding if you will be drinking alcohol.
B. Take metronidazole, do not drink alcohol.
C. It’s okay to drink alcohol.
For years, we have advised patients to not use alcohol if they are taking metronidazole because of concern for a disulfiram-like reaction between alcohol and metronidazole. This has been a standard warning given by physicians and appears as a contraindication in the prescribing information. It has been well accepted as a true, proven reaction.
Is it true?
As early as the 1960s, case reports and an uncontrolled study suggested that combining metronidazole with alcohol produced a disulfiram-like reaction, with case reports of severe reactions, including death.1, 2, 3 This was initially considered an area that might be therapeutic in the treatment of alcoholism, but several studies showed no benefit.4, 5
Caroline S. Williams and Dr. Kevin R. Woodcock reviewed the case reports for evidence of proof of a true interaction between metronidazole and ethanol.6 The case reports referenced textbooks to substantiate the interaction, but they did not present clear evidence of an interaction as the cause of elevated acetaldehyde levels.
Researchers have shown in a rat model that metronidazole can increase intracolonic, but not blood, acetaldehyde levels in rats that have received a combination of ethanol and metronidazole.7 Metronidazole did not have any inhibitory effect on hepatic or colonic alcohol dehydrogenase or aldehyde dehydrogenase. What was found was that rats treated with metronidazole had increased growth of Enterobacteriaceae, an alcohol dehydrogenase–containing aerobe, which could be the cause of the higher intracolonic acetaldehyde levels.
Jukka-Pekka Visapää and his colleagues studied the effect of coadministration of metronidazole and ethanol in young, healthy male volunteers.8 The study was a placebo-controlled, randomized trial. The study was small, with 12 participants. One-half of the study participants received metronidazole three times a day for 5 days; the other half received placebo. All participants then received ethanol 0.4g/kg, with blood testing being done every 20 minutes for the next 4 hours. Blood was tested for ethanol concentrations and for acetaldehyde levels. The study participants also had blood pressure, pulse, skin temperature, and symptoms monitored during the study.
There was no difference in blood acetaldehyde levels, vital signs, or symptoms between patients who received metronidazole or placebo. None of the subjects in the study had any measurable symptoms.
Metronidazole has many side effects, including nausea, vomiting, headache, dizziness, and seizures. These symptoms have a great deal of overlap with the symptoms of alcohol-disulfiram interaction. It has been assumed in early case reports that metronidazole caused a similar interaction with alcohol and raised acetaldehyde levels by interfering with aldehyde dehydrogenase.
Animal models and the human study do not show this to be the case. It is possible that metronidazole side effects alone were the cause of the symptoms in case reports. The one human study done was on healthy male volunteers, so projecting the results to a population with liver disease or other serious illness is a bit of a stretch. I think that if a problem exists with alcohol and metronidazole, it is uncommon and unlikely to occur in healthy individuals.
So, what would I advise the patient in the case about whether he can drink alcohol? I think that the risk would be minimal and that it would be safe for him to drink alcohol.
References
1. Br J Clin Pract. 1985 Jul;39(7):292-3.
2. Psychiatr Neurol. 1966;152:395-401.
3. Am J Forensic Med Pathol. 1996 Dec;17(4):343-6.
4. Q J Stud Alcohol. 1972 Sep;33: 734-40.
5. Q J Stud Ethanol. 1969 Mar;30: 140-51.
6. Ann Pharmacother. 2000 Feb;34(2):255-7.
7. Alcohol Clin Exp Res. 2000 Apr;24(4):570-5.
8. Ann Pharmacother. 2002 Jun;36(6):971-4.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and he serves as third-year medical student clerkship director at the University of Washington. Contact Dr. Paauw at [email protected].
A 32-year-old man develops diarrhea after receiving amoxicillin/clavulanate to treat an infection following a dog bite. He is diagnosed with Clostridium difficile and prescribed a 10-day course of metronidazole. He has no other medical problems. He will be the best man at his brother’s wedding tomorrow. What advice should you give him about alcohol use at the reception?
A. Do not take metronidazole the day of the wedding if you will be drinking alcohol.
B. Take metronidazole, do not drink alcohol.
C. It’s okay to drink alcohol.
For years, we have advised patients to not use alcohol if they are taking metronidazole because of concern for a disulfiram-like reaction between alcohol and metronidazole. This has been a standard warning given by physicians and appears as a contraindication in the prescribing information. It has been well accepted as a true, proven reaction.
Is it true?
As early as the 1960s, case reports and an uncontrolled study suggested that combining metronidazole with alcohol produced a disulfiram-like reaction, with case reports of severe reactions, including death.1, 2, 3 This was initially considered an area that might be therapeutic in the treatment of alcoholism, but several studies showed no benefit.4, 5
Caroline S. Williams and Dr. Kevin R. Woodcock reviewed the case reports for evidence of proof of a true interaction between metronidazole and ethanol.6 The case reports referenced textbooks to substantiate the interaction, but they did not present clear evidence of an interaction as the cause of elevated acetaldehyde levels.
Researchers have shown in a rat model that metronidazole can increase intracolonic, but not blood, acetaldehyde levels in rats that have received a combination of ethanol and metronidazole.7 Metronidazole did not have any inhibitory effect on hepatic or colonic alcohol dehydrogenase or aldehyde dehydrogenase. What was found was that rats treated with metronidazole had increased growth of Enterobacteriaceae, an alcohol dehydrogenase–containing aerobe, which could be the cause of the higher intracolonic acetaldehyde levels.
Jukka-Pekka Visapää and his colleagues studied the effect of coadministration of metronidazole and ethanol in young, healthy male volunteers.8 The study was a placebo-controlled, randomized trial. The study was small, with 12 participants. One-half of the study participants received metronidazole three times a day for 5 days; the other half received placebo. All participants then received ethanol 0.4g/kg, with blood testing being done every 20 minutes for the next 4 hours. Blood was tested for ethanol concentrations and for acetaldehyde levels. The study participants also had blood pressure, pulse, skin temperature, and symptoms monitored during the study.
There was no difference in blood acetaldehyde levels, vital signs, or symptoms between patients who received metronidazole or placebo. None of the subjects in the study had any measurable symptoms.
Metronidazole has many side effects, including nausea, vomiting, headache, dizziness, and seizures. These symptoms have a great deal of overlap with the symptoms of alcohol-disulfiram interaction. It has been assumed in early case reports that metronidazole caused a similar interaction with alcohol and raised acetaldehyde levels by interfering with aldehyde dehydrogenase.
Animal models and the human study do not show this to be the case. It is possible that metronidazole side effects alone were the cause of the symptoms in case reports. The one human study done was on healthy male volunteers, so projecting the results to a population with liver disease or other serious illness is a bit of a stretch. I think that if a problem exists with alcohol and metronidazole, it is uncommon and unlikely to occur in healthy individuals.
So, what would I advise the patient in the case about whether he can drink alcohol? I think that the risk would be minimal and that it would be safe for him to drink alcohol.
References
1. Br J Clin Pract. 1985 Jul;39(7):292-3.
2. Psychiatr Neurol. 1966;152:395-401.
3. Am J Forensic Med Pathol. 1996 Dec;17(4):343-6.
4. Q J Stud Alcohol. 1972 Sep;33: 734-40.
5. Q J Stud Ethanol. 1969 Mar;30: 140-51.
6. Ann Pharmacother. 2000 Feb;34(2):255-7.
7. Alcohol Clin Exp Res. 2000 Apr;24(4):570-5.
8. Ann Pharmacother. 2002 Jun;36(6):971-4.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and he serves as third-year medical student clerkship director at the University of Washington. Contact Dr. Paauw at [email protected].

E-cigarette Smokers Less Exposed to Carbon Monoxide
NEW YORK—Smokers who switch to e-cigarettes - even if only some of the time - may dramatically reduce their exposure to air pollutants including carbon monoxide and acrolein, a British study suggests.
Researchers gave e-cigarettes to 40 smokers who said they wanted to quit. After four weeks, the 16 participants using only e-cigarettes had about an 80% drop in exposure both to carbon monoxide and to acrolein, a harmful breakdown product that is also in some e-cigarettes' vapor. Acrolein is known to irritate exposed tissues and can destroy cilia.
The 17 participants who swapped some regular cigarettes for the electronic version had a 52% decline in carbon monoxide exposure and a 60% decline for acrolein, according to a report online September 3 in Cancer Prevention Research.
To get the most benefit from switching to e-cigarettes, smokers need to completely give up traditional cigarettes, lead study author Dr. Hayden McRobbie, of the Wolfson Institute of Preventive Medicine at Queen Mary University of London, said by email.
"Smokers may get some encouragement from the finding that there is some potential health benefit as soon as they start the process," Dr. McRobbie said.
While tobacco control advocates fear that e-cigarettes may give rise to a new generation of nicotine addicts who eventually transition to conventional cigarettes, the current study adds to a small but growing body of evidence suggesting the devices might benefit the health of people who already smoke.
An international analysis of published research by the Cochrane Review in December concluded the devices could help smokers quit but said much of the existing research on e-cigarettes was thin.
Even though the current study points to another potential benefit of e-cigarettes, more evidence is still needed from longer and larger trials before scientists can draw firm conclusions about any safety advantages, Dr. Nancy Rigotti, director of tobacco research at Massachusetts General Hospital in Boston, said by email.
"It is exactly the type of incremental, careful work that is needed but it is not yet a definitive study," Rigotti, who wasn't involved in the study, said.
Study participants were typically in their 40s and had attempted to quit at least twice before joining the trial. All of them were offered the same type of e-cigarette and encouraged to completely abandon traditional cigarettes.
Researchers measured carbon monoxide in participants' breath one week before switching to e-cigarettes, on the day they switched, and again four weeks later. They followed the same schedule for testing urine for exposure to acrolein.
A limitation of the study, the authors acknowledged, is that it only included people with a desire to quit smoking, making it possible the results would be different for smokers with no intention of quitting. It's also possible that the specific model of e-cigarette used in the study might not be representative of other devices.
Still, the findings suggest smokers should be told e-cigarettes may curb their exposure to toxic chemicals, Dr. Riccardo Polosa, head of the tobacco research center at the University of Catania in Italy, said by email.
"This study adds to the evidence that e-cigarettes are much less harmful compared to conventional cigarettes," said Polosa, who wasn't involved in the study.
NEW YORK—Smokers who switch to e-cigarettes - even if only some of the time - may dramatically reduce their exposure to air pollutants including carbon monoxide and acrolein, a British study suggests.
Researchers gave e-cigarettes to 40 smokers who said they wanted to quit. After four weeks, the 16 participants using only e-cigarettes had about an 80% drop in exposure both to carbon monoxide and to acrolein, a harmful breakdown product that is also in some e-cigarettes' vapor. Acrolein is known to irritate exposed tissues and can destroy cilia.
The 17 participants who swapped some regular cigarettes for the electronic version had a 52% decline in carbon monoxide exposure and a 60% decline for acrolein, according to a report online September 3 in Cancer Prevention Research.
To get the most benefit from switching to e-cigarettes, smokers need to completely give up traditional cigarettes, lead study author Dr. Hayden McRobbie, of the Wolfson Institute of Preventive Medicine at Queen Mary University of London, said by email.
"Smokers may get some encouragement from the finding that there is some potential health benefit as soon as they start the process," Dr. McRobbie said.
While tobacco control advocates fear that e-cigarettes may give rise to a new generation of nicotine addicts who eventually transition to conventional cigarettes, the current study adds to a small but growing body of evidence suggesting the devices might benefit the health of people who already smoke.
An international analysis of published research by the Cochrane Review in December concluded the devices could help smokers quit but said much of the existing research on e-cigarettes was thin.
Even though the current study points to another potential benefit of e-cigarettes, more evidence is still needed from longer and larger trials before scientists can draw firm conclusions about any safety advantages, Dr. Nancy Rigotti, director of tobacco research at Massachusetts General Hospital in Boston, said by email.
"It is exactly the type of incremental, careful work that is needed but it is not yet a definitive study," Rigotti, who wasn't involved in the study, said.
Study participants were typically in their 40s and had attempted to quit at least twice before joining the trial. All of them were offered the same type of e-cigarette and encouraged to completely abandon traditional cigarettes.
Researchers measured carbon monoxide in participants' breath one week before switching to e-cigarettes, on the day they switched, and again four weeks later. They followed the same schedule for testing urine for exposure to acrolein.
A limitation of the study, the authors acknowledged, is that it only included people with a desire to quit smoking, making it possible the results would be different for smokers with no intention of quitting. It's also possible that the specific model of e-cigarette used in the study might not be representative of other devices.
Still, the findings suggest smokers should be told e-cigarettes may curb their exposure to toxic chemicals, Dr. Riccardo Polosa, head of the tobacco research center at the University of Catania in Italy, said by email.
"This study adds to the evidence that e-cigarettes are much less harmful compared to conventional cigarettes," said Polosa, who wasn't involved in the study.
NEW YORK—Smokers who switch to e-cigarettes - even if only some of the time - may dramatically reduce their exposure to air pollutants including carbon monoxide and acrolein, a British study suggests.
Researchers gave e-cigarettes to 40 smokers who said they wanted to quit. After four weeks, the 16 participants using only e-cigarettes had about an 80% drop in exposure both to carbon monoxide and to acrolein, a harmful breakdown product that is also in some e-cigarettes' vapor. Acrolein is known to irritate exposed tissues and can destroy cilia.
The 17 participants who swapped some regular cigarettes for the electronic version had a 52% decline in carbon monoxide exposure and a 60% decline for acrolein, according to a report online September 3 in Cancer Prevention Research.
To get the most benefit from switching to e-cigarettes, smokers need to completely give up traditional cigarettes, lead study author Dr. Hayden McRobbie, of the Wolfson Institute of Preventive Medicine at Queen Mary University of London, said by email.
"Smokers may get some encouragement from the finding that there is some potential health benefit as soon as they start the process," Dr. McRobbie said.
While tobacco control advocates fear that e-cigarettes may give rise to a new generation of nicotine addicts who eventually transition to conventional cigarettes, the current study adds to a small but growing body of evidence suggesting the devices might benefit the health of people who already smoke.
An international analysis of published research by the Cochrane Review in December concluded the devices could help smokers quit but said much of the existing research on e-cigarettes was thin.
Even though the current study points to another potential benefit of e-cigarettes, more evidence is still needed from longer and larger trials before scientists can draw firm conclusions about any safety advantages, Dr. Nancy Rigotti, director of tobacco research at Massachusetts General Hospital in Boston, said by email.
"It is exactly the type of incremental, careful work that is needed but it is not yet a definitive study," Rigotti, who wasn't involved in the study, said.
Study participants were typically in their 40s and had attempted to quit at least twice before joining the trial. All of them were offered the same type of e-cigarette and encouraged to completely abandon traditional cigarettes.
Researchers measured carbon monoxide in participants' breath one week before switching to e-cigarettes, on the day they switched, and again four weeks later. They followed the same schedule for testing urine for exposure to acrolein.
A limitation of the study, the authors acknowledged, is that it only included people with a desire to quit smoking, making it possible the results would be different for smokers with no intention of quitting. It's also possible that the specific model of e-cigarette used in the study might not be representative of other devices.
Still, the findings suggest smokers should be told e-cigarettes may curb their exposure to toxic chemicals, Dr. Riccardo Polosa, head of the tobacco research center at the University of Catania in Italy, said by email.
"This study adds to the evidence that e-cigarettes are much less harmful compared to conventional cigarettes," said Polosa, who wasn't involved in the study.
Social factors may impact survival in AML

Photo by Rhoda Baer
A new study indicates that certain social factors may impact survival in adults with acute myelogenous leukemia (AML) who are under 65.
The research showed associations between patient survival and insurance status, marital status, and county-level income.
“We believe these 3 factors indicate lack of material and social support preventing young patients from successfully walking the long and difficult road towards a cure,” said Uma Borate, MD, of the University of Alabama at Birmingham.
To conduct this study, Dr Borate and her colleagues analyzed data on 5541 patients, ages 19 to 64, who were diagnosed with AML between 2007 and 2011.
The team reported their findings in Cancer.
Multivariable analysis showed that AML subtype, age, and sex were independently associated with patients’ survival. And the non-biological factors independently associated with survival were insurance status, marital status, and county-level median household income.
Specifically, there was a significantly increased risk of premature death among patients who were uninsured (P=0.005) or Medicaid beneficiaries (P<0.001), compared to patients with private insurance.
Single (P<0.001) or divorced (P=0.011) patients had a significantly higher risk of premature death than married patients. But there was no significant difference between married and widowed patients (P=0.206).
And patients who lived in areas with lower income—the lowest 3 of 5 income groups—had a significantly increased risk of premature death.
Compared to patients in the fifth income quintile ($58.3K-$79.9K), there was an increased risk of death in the first quintile ($16.2K-$38.8K, P=0.001), second quintile ($38.8K-$42.2K, P<0.001), and third quintile ($42.2K-$47.9K, P<0.001).
Early and late mortality
The researchers wanted to determine if the impact of non-biological factors on survival was related to early mortality (a possible surrogate for access to care or late presentation) or late mortality (a possible surrogate for access to post-remission therapy and hematopoietic stem cell transplant).
So they conducted an exploratory analysis of factors influencing the risk of death within the first 2 months of diagnosis.
Being a Medicaid beneficiary (P=0.01) or uninsured (P<0.001) was independently associated with an increased risk of death within the first 2 months.
The same was true for patients belonging to the first income quartile (P=0.001), second quartile (P=0.003), third quartile (P=0.02), and fourth quartile (P=0.028).
On the other hand, there was no significant difference in early death according to marital status.
The researchers also performed a landmark survival analysis including only patients who survived at least 2 months from diagnosis.
In this analysis, marital status (P<0.001), insurance status (P=0.001), and income (P=0.021) were all independent predictors of survival.
Implications
“As physicians, we often emphasize more of the biology of the cancer, especially with the recent focus on personalized medicine,” said study author Luciano Jose Costa, MD, PhD, also of the University of Alabama at Birmingham.
“But we need to pay the same attention to resources available to our patients, as this greatly impacts their chances to survive leukemia.”
The researchers believe this will be especially important as the US transitions to a healthcare system that ties physician and hospital payments to patient outcomes.
“Taking from the results of this study, factors that have nothing to do with quality of care need to be accounted for when comparing predicted with actual outcomes,” Dr Borate said. “Otherwise, we will create a disincentive for hospitals and doctors to care for less privileged patients.” ![]()
Photo by Rhoda Baer
A new study indicates that certain social factors may impact survival in adults with acute myelogenous leukemia (AML) who are under 65.
The research showed associations between patient survival and insurance status, marital status, and county-level income.
“We believe these 3 factors indicate lack of material and social support preventing young patients from successfully walking the long and difficult road towards a cure,” said Uma Borate, MD, of the University of Alabama at Birmingham.
To conduct this study, Dr Borate and her colleagues analyzed data on 5541 patients, ages 19 to 64, who were diagnosed with AML between 2007 and 2011.
The team reported their findings in Cancer.
Multivariable analysis showed that AML subtype, age, and sex were independently associated with patients’ survival. And the non-biological factors independently associated with survival were insurance status, marital status, and county-level median household income.
Specifically, there was a significantly increased risk of premature death among patients who were uninsured (P=0.005) or Medicaid beneficiaries (P<0.001), compared to patients with private insurance.
Single (P<0.001) or divorced (P=0.011) patients had a significantly higher risk of premature death than married patients. But there was no significant difference between married and widowed patients (P=0.206).
And patients who lived in areas with lower income—the lowest 3 of 5 income groups—had a significantly increased risk of premature death.
Compared to patients in the fifth income quintile ($58.3K-$79.9K), there was an increased risk of death in the first quintile ($16.2K-$38.8K, P=0.001), second quintile ($38.8K-$42.2K, P<0.001), and third quintile ($42.2K-$47.9K, P<0.001).
Early and late mortality
The researchers wanted to determine if the impact of non-biological factors on survival was related to early mortality (a possible surrogate for access to care or late presentation) or late mortality (a possible surrogate for access to post-remission therapy and hematopoietic stem cell transplant).
So they conducted an exploratory analysis of factors influencing the risk of death within the first 2 months of diagnosis.
Being a Medicaid beneficiary (P=0.01) or uninsured (P<0.001) was independently associated with an increased risk of death within the first 2 months.
The same was true for patients belonging to the first income quartile (P=0.001), second quartile (P=0.003), third quartile (P=0.02), and fourth quartile (P=0.028).
On the other hand, there was no significant difference in early death according to marital status.
The researchers also performed a landmark survival analysis including only patients who survived at least 2 months from diagnosis.
In this analysis, marital status (P<0.001), insurance status (P=0.001), and income (P=0.021) were all independent predictors of survival.
Implications
“As physicians, we often emphasize more of the biology of the cancer, especially with the recent focus on personalized medicine,” said study author Luciano Jose Costa, MD, PhD, also of the University of Alabama at Birmingham.
“But we need to pay the same attention to resources available to our patients, as this greatly impacts their chances to survive leukemia.”
The researchers believe this will be especially important as the US transitions to a healthcare system that ties physician and hospital payments to patient outcomes.
“Taking from the results of this study, factors that have nothing to do with quality of care need to be accounted for when comparing predicted with actual outcomes,” Dr Borate said. “Otherwise, we will create a disincentive for hospitals and doctors to care for less privileged patients.” ![]()
Photo by Rhoda Baer
A new study indicates that certain social factors may impact survival in adults with acute myelogenous leukemia (AML) who are under 65.
The research showed associations between patient survival and insurance status, marital status, and county-level income.
“We believe these 3 factors indicate lack of material and social support preventing young patients from successfully walking the long and difficult road towards a cure,” said Uma Borate, MD, of the University of Alabama at Birmingham.
To conduct this study, Dr Borate and her colleagues analyzed data on 5541 patients, ages 19 to 64, who were diagnosed with AML between 2007 and 2011.
The team reported their findings in Cancer.
Multivariable analysis showed that AML subtype, age, and sex were independently associated with patients’ survival. And the non-biological factors independently associated with survival were insurance status, marital status, and county-level median household income.
Specifically, there was a significantly increased risk of premature death among patients who were uninsured (P=0.005) or Medicaid beneficiaries (P<0.001), compared to patients with private insurance.
Single (P<0.001) or divorced (P=0.011) patients had a significantly higher risk of premature death than married patients. But there was no significant difference between married and widowed patients (P=0.206).
And patients who lived in areas with lower income—the lowest 3 of 5 income groups—had a significantly increased risk of premature death.
Compared to patients in the fifth income quintile ($58.3K-$79.9K), there was an increased risk of death in the first quintile ($16.2K-$38.8K, P=0.001), second quintile ($38.8K-$42.2K, P<0.001), and third quintile ($42.2K-$47.9K, P<0.001).
Early and late mortality
The researchers wanted to determine if the impact of non-biological factors on survival was related to early mortality (a possible surrogate for access to care or late presentation) or late mortality (a possible surrogate for access to post-remission therapy and hematopoietic stem cell transplant).
So they conducted an exploratory analysis of factors influencing the risk of death within the first 2 months of diagnosis.
Being a Medicaid beneficiary (P=0.01) or uninsured (P<0.001) was independently associated with an increased risk of death within the first 2 months.
The same was true for patients belonging to the first income quartile (P=0.001), second quartile (P=0.003), third quartile (P=0.02), and fourth quartile (P=0.028).
On the other hand, there was no significant difference in early death according to marital status.
The researchers also performed a landmark survival analysis including only patients who survived at least 2 months from diagnosis.
In this analysis, marital status (P<0.001), insurance status (P=0.001), and income (P=0.021) were all independent predictors of survival.
Implications
“As physicians, we often emphasize more of the biology of the cancer, especially with the recent focus on personalized medicine,” said study author Luciano Jose Costa, MD, PhD, also of the University of Alabama at Birmingham.
“But we need to pay the same attention to resources available to our patients, as this greatly impacts their chances to survive leukemia.”
The researchers believe this will be especially important as the US transitions to a healthcare system that ties physician and hospital payments to patient outcomes.
“Taking from the results of this study, factors that have nothing to do with quality of care need to be accounted for when comparing predicted with actual outcomes,” Dr Borate said. “Otherwise, we will create a disincentive for hospitals and doctors to care for less privileged patients.” ![]()
EMA recommends orphan designation for LJPC-401
red blood cells
Image by Graham Beards
The European Medicines Agency’s (EMA’s) Committee for Orphan Medicinal Products (COMP) has adopted a positive opinion recommending that LJPC-401, a novel formulation of hepcidin, receive orphan designation to treat chronic iron overload requiring chelation therapy.
Chronic iron overload occurs in patients suffering from beta thalassemia, sickle cell disease, and hereditary hemochromatosis.
The COMP’s opinion, which is subject to review and approval by the European Commission, may include all or a subset of these conditions.
About LJPC-401
LJPC-401 is a novel formulation of hepcidin, an endogenous peptide hormone that is the body’s naturally occurring regulator of iron absorption and distribution. Hepcidin prevents excessive iron accumulation in tissues, such as the liver and heart, where it can cause significant damage and even result in death.
La Jolla Pharmaceutical Company is developing LJPC-401 for the treatment of iron overload occurring as a results of hereditary hemochromatosis, beta thalassemia, and sickle cell disease.
LJPC-401 has been shown to be effective in reducing serum iron in preclinical testing, according to La Jolla. The company said it expects to release preliminary results from a phase 1 trial of LJPC-401 by the end of this year.
About orphan designation
The EMA’s COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for endorsement.
In the European Union, orphan designation is granted to therapies intended to treat a life-threatening or chronically debilitating condition that affects no more than 5 in 10,000 persons and where no satisfactory treatment is available.
Companies that obtain orphan designation for a drug benefit from a number of incentives, including protocol assistance, a type of scientific advice specific for designated orphan medicines, and 10 years of market exclusivity if the medicine is approved. Fee reductions are also available, depending on the status of the sponsor and the type of service required. ![]()
red blood cells
Image by Graham Beards
The European Medicines Agency’s (EMA’s) Committee for Orphan Medicinal Products (COMP) has adopted a positive opinion recommending that LJPC-401, a novel formulation of hepcidin, receive orphan designation to treat chronic iron overload requiring chelation therapy.
Chronic iron overload occurs in patients suffering from beta thalassemia, sickle cell disease, and hereditary hemochromatosis.
The COMP’s opinion, which is subject to review and approval by the European Commission, may include all or a subset of these conditions.
About LJPC-401
LJPC-401 is a novel formulation of hepcidin, an endogenous peptide hormone that is the body’s naturally occurring regulator of iron absorption and distribution. Hepcidin prevents excessive iron accumulation in tissues, such as the liver and heart, where it can cause significant damage and even result in death.
La Jolla Pharmaceutical Company is developing LJPC-401 for the treatment of iron overload occurring as a results of hereditary hemochromatosis, beta thalassemia, and sickle cell disease.
LJPC-401 has been shown to be effective in reducing serum iron in preclinical testing, according to La Jolla. The company said it expects to release preliminary results from a phase 1 trial of LJPC-401 by the end of this year.
About orphan designation
The EMA’s COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for endorsement.
In the European Union, orphan designation is granted to therapies intended to treat a life-threatening or chronically debilitating condition that affects no more than 5 in 10,000 persons and where no satisfactory treatment is available.
Companies that obtain orphan designation for a drug benefit from a number of incentives, including protocol assistance, a type of scientific advice specific for designated orphan medicines, and 10 years of market exclusivity if the medicine is approved. Fee reductions are also available, depending on the status of the sponsor and the type of service required. ![]()
red blood cells
Image by Graham Beards
The European Medicines Agency’s (EMA’s) Committee for Orphan Medicinal Products (COMP) has adopted a positive opinion recommending that LJPC-401, a novel formulation of hepcidin, receive orphan designation to treat chronic iron overload requiring chelation therapy.
Chronic iron overload occurs in patients suffering from beta thalassemia, sickle cell disease, and hereditary hemochromatosis.
The COMP’s opinion, which is subject to review and approval by the European Commission, may include all or a subset of these conditions.
About LJPC-401
LJPC-401 is a novel formulation of hepcidin, an endogenous peptide hormone that is the body’s naturally occurring regulator of iron absorption and distribution. Hepcidin prevents excessive iron accumulation in tissues, such as the liver and heart, where it can cause significant damage and even result in death.
La Jolla Pharmaceutical Company is developing LJPC-401 for the treatment of iron overload occurring as a results of hereditary hemochromatosis, beta thalassemia, and sickle cell disease.
LJPC-401 has been shown to be effective in reducing serum iron in preclinical testing, according to La Jolla. The company said it expects to release preliminary results from a phase 1 trial of LJPC-401 by the end of this year.
About orphan designation
The EMA’s COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for endorsement.
In the European Union, orphan designation is granted to therapies intended to treat a life-threatening or chronically debilitating condition that affects no more than 5 in 10,000 persons and where no satisfactory treatment is available.
Companies that obtain orphan designation for a drug benefit from a number of incentives, including protocol assistance, a type of scientific advice specific for designated orphan medicines, and 10 years of market exclusivity if the medicine is approved. Fee reductions are also available, depending on the status of the sponsor and the type of service required. ![]()
Communication key to helping kids after disasters
Posttraumatic stress disorder can be hard to spot in kids after natural or manmade disasters.
They may not understand that intrusive thoughts, panic attacks, and other symptoms are problems that can be addressed, and are unlikely to mention them.
As a result, parents, teachers, and others often underestimate children’s distress levels and overestimate their resilience. One way around the problem is to ask children how they’re doing, and probe for signs of trouble. It helps to let them know that PTSD and adjustment problems are normal after a frightening event, and to teach them how to anticipate and cope with PTSD triggers.
That’s just a small fraction of the useful advice in new guidance from the American Academy of Pediatrics on the psychosocial support of children and families after disasters, published online Sept. 14 (Pediatrics. 2015 Sept. 14. doi:10.1542/peds.2015-2861).
“Children are particularly vulnerable to the effects of disasters and other traumatic events because of a lack of experience, skills, and resources to be able to independently meet their developmental, socioemotional, mental, and behavioral health needs,” said the authors, led by Dr. David Schonfeld of St. Christopher’s Hospital for Children, and Thomas Demaria, Ph.D., of Long Island (N.Y.) University.
Mental health triage should come right after medical stabilization. Dissociative symptoms; extreme confusion or inability to concentrate or make even simple decisions; intense fear, anxiety, panic, helplessness, or horror; depression at the time of the event; uncontrollable and intense grief; suicidal ideation; and marked somatization are among the warning signs that kids are in trouble.
Psychiatric medications to blunt such reactions are usually the wrong call. “Children need to develop an understanding of the event and learn to express and cope with their reactions.” If medication does seem necessary, its best to let an expert in childhood trauma make the decision, the authors said.
Dismissing children’s concerns is a mistake. “In reality, if children feel worried, then they are worried. Telling them that they should not be worried is usually ineffective.” It’s also a mistake to avoid talking about grief for fear of making it worse. Children’s “distress is caused by the reaction to the death itself, rather than any question or invitation to talk. Talking may provide some relief if not coerced. Avoiding discussion is rarely helpful and often isolates children at a time when they are most in need of support and assistance,” they said.
Simple, basic facts about the event – as long as they’re not graphic or overwhelming – will help children make sense of what they’ve been through, and reassurance that things will eventually be okay can be healing. Kids also have to know that the situation isn’t their fault, and how to cope with it.
Parents can share how they’re upset about losing their home, for instance, but then discuss how talking to another trusted adult, getting exercise, meditating, and helping others makes them feel better. Pediatricians can boost spirits by saying something like “the tornado created a big mess, but we are pulling together as a community” or “living in a shelter with all the other children in the neighborhood must have been a real adventure,” the authors said.
Having children contribute to food drives or draw hopeful pictures for victims in the hospital can help them regain a sense of control and usefulness. Resuming their routines as soon as possible will also help bring back a sense of normalcy.
Bereavement counseling is in order when children are struggling with the loss of a loved one, and cognitive behavioral therapy for kids with PTSD.
Posttraumatic stress disorder can be hard to spot in kids after natural or manmade disasters.
They may not understand that intrusive thoughts, panic attacks, and other symptoms are problems that can be addressed, and are unlikely to mention them.
As a result, parents, teachers, and others often underestimate children’s distress levels and overestimate their resilience. One way around the problem is to ask children how they’re doing, and probe for signs of trouble. It helps to let them know that PTSD and adjustment problems are normal after a frightening event, and to teach them how to anticipate and cope with PTSD triggers.
That’s just a small fraction of the useful advice in new guidance from the American Academy of Pediatrics on the psychosocial support of children and families after disasters, published online Sept. 14 (Pediatrics. 2015 Sept. 14. doi:10.1542/peds.2015-2861).
“Children are particularly vulnerable to the effects of disasters and other traumatic events because of a lack of experience, skills, and resources to be able to independently meet their developmental, socioemotional, mental, and behavioral health needs,” said the authors, led by Dr. David Schonfeld of St. Christopher’s Hospital for Children, and Thomas Demaria, Ph.D., of Long Island (N.Y.) University.
Mental health triage should come right after medical stabilization. Dissociative symptoms; extreme confusion or inability to concentrate or make even simple decisions; intense fear, anxiety, panic, helplessness, or horror; depression at the time of the event; uncontrollable and intense grief; suicidal ideation; and marked somatization are among the warning signs that kids are in trouble.
Psychiatric medications to blunt such reactions are usually the wrong call. “Children need to develop an understanding of the event and learn to express and cope with their reactions.” If medication does seem necessary, its best to let an expert in childhood trauma make the decision, the authors said.
Dismissing children’s concerns is a mistake. “In reality, if children feel worried, then they are worried. Telling them that they should not be worried is usually ineffective.” It’s also a mistake to avoid talking about grief for fear of making it worse. Children’s “distress is caused by the reaction to the death itself, rather than any question or invitation to talk. Talking may provide some relief if not coerced. Avoiding discussion is rarely helpful and often isolates children at a time when they are most in need of support and assistance,” they said.
Simple, basic facts about the event – as long as they’re not graphic or overwhelming – will help children make sense of what they’ve been through, and reassurance that things will eventually be okay can be healing. Kids also have to know that the situation isn’t their fault, and how to cope with it.
Parents can share how they’re upset about losing their home, for instance, but then discuss how talking to another trusted adult, getting exercise, meditating, and helping others makes them feel better. Pediatricians can boost spirits by saying something like “the tornado created a big mess, but we are pulling together as a community” or “living in a shelter with all the other children in the neighborhood must have been a real adventure,” the authors said.
Having children contribute to food drives or draw hopeful pictures for victims in the hospital can help them regain a sense of control and usefulness. Resuming their routines as soon as possible will also help bring back a sense of normalcy.
Bereavement counseling is in order when children are struggling with the loss of a loved one, and cognitive behavioral therapy for kids with PTSD.
Posttraumatic stress disorder can be hard to spot in kids after natural or manmade disasters.
They may not understand that intrusive thoughts, panic attacks, and other symptoms are problems that can be addressed, and are unlikely to mention them.
As a result, parents, teachers, and others often underestimate children’s distress levels and overestimate their resilience. One way around the problem is to ask children how they’re doing, and probe for signs of trouble. It helps to let them know that PTSD and adjustment problems are normal after a frightening event, and to teach them how to anticipate and cope with PTSD triggers.
That’s just a small fraction of the useful advice in new guidance from the American Academy of Pediatrics on the psychosocial support of children and families after disasters, published online Sept. 14 (Pediatrics. 2015 Sept. 14. doi:10.1542/peds.2015-2861).
“Children are particularly vulnerable to the effects of disasters and other traumatic events because of a lack of experience, skills, and resources to be able to independently meet their developmental, socioemotional, mental, and behavioral health needs,” said the authors, led by Dr. David Schonfeld of St. Christopher’s Hospital for Children, and Thomas Demaria, Ph.D., of Long Island (N.Y.) University.
Mental health triage should come right after medical stabilization. Dissociative symptoms; extreme confusion or inability to concentrate or make even simple decisions; intense fear, anxiety, panic, helplessness, or horror; depression at the time of the event; uncontrollable and intense grief; suicidal ideation; and marked somatization are among the warning signs that kids are in trouble.
Psychiatric medications to blunt such reactions are usually the wrong call. “Children need to develop an understanding of the event and learn to express and cope with their reactions.” If medication does seem necessary, its best to let an expert in childhood trauma make the decision, the authors said.
Dismissing children’s concerns is a mistake. “In reality, if children feel worried, then they are worried. Telling them that they should not be worried is usually ineffective.” It’s also a mistake to avoid talking about grief for fear of making it worse. Children’s “distress is caused by the reaction to the death itself, rather than any question or invitation to talk. Talking may provide some relief if not coerced. Avoiding discussion is rarely helpful and often isolates children at a time when they are most in need of support and assistance,” they said.
Simple, basic facts about the event – as long as they’re not graphic or overwhelming – will help children make sense of what they’ve been through, and reassurance that things will eventually be okay can be healing. Kids also have to know that the situation isn’t their fault, and how to cope with it.
Parents can share how they’re upset about losing their home, for instance, but then discuss how talking to another trusted adult, getting exercise, meditating, and helping others makes them feel better. Pediatricians can boost spirits by saying something like “the tornado created a big mess, but we are pulling together as a community” or “living in a shelter with all the other children in the neighborhood must have been a real adventure,” the authors said.
Having children contribute to food drives or draw hopeful pictures for victims in the hospital can help them regain a sense of control and usefulness. Resuming their routines as soon as possible will also help bring back a sense of normalcy.
Bereavement counseling is in order when children are struggling with the loss of a loved one, and cognitive behavioral therapy for kids with PTSD.
FROM PEDIATRICS
Product News: 09 2015
Cyclosporine A
Immune Pharmaceuticals acquires a nanoparticle topical formulation of cyclosporine A for the treatment of psoriasis, atopic dermatitis, pemphigus vulgaris, and other severe inflammatory dermatoses. Researchers have incorporated cyclosporine A into biodegradable nanocapsules and have developed stable topical formulations that are able to achieve therapeutic cyclosporine A levels in the targeted skin layers. This topical treatment provides a potential alternative to oral cyclosporine A and other topical immunosuppressive drugs. For more information, visit www.immunepharmaceuticals.com.
Picato Gel Warning
The US Food and Drug Administration (FDA) issues a warning about reports of severe allergic reactions and herpes zoster associated with the use of Picato Gel (ingenol mebutate) for the treatment of actinic keratosis. The allergic reaction may include throat tightness, difficulty breathing, feeling faint, or swelling of the lips or tongue. The FDA received reports of cases involving severe eye injuries associated with Picato Gel not being used according to the instructions for use on the label. Changes to the labeling have been requested by the FDA to warn about these new safety risks. Patients should not use Picato Gel for a longer period or on an area of skin larger than instructed on the drug label. Patients also should avoid application in, near, and around the mouth, lips, and eye area. For more information, visit www.fda.gov/MedWatch.
Ximino Extended-Release Capsules
Sun Pharmaceutical Industries Ltd announces US Food and Drug Administration approval of the Supplemental New Drug Application for Ximino (minocycline hydrochloride) extended-release capsules (45, 90, and 135 mg). Ximino extended-release capsules are indicated for the treatment of inflammatory lesions of nonnodular moderate to severe acne vulgaris in patients 12 years and older. Ximino extended-release capsules are expected to be available for patients during the fourth quarter of 2015. For more information, visit www.sunpharma.com.
If you would like your product included in Product News, please e-mail a press release to the Editorial Office at [email protected].
Cyclosporine A
Immune Pharmaceuticals acquires a nanoparticle topical formulation of cyclosporine A for the treatment of psoriasis, atopic dermatitis, pemphigus vulgaris, and other severe inflammatory dermatoses. Researchers have incorporated cyclosporine A into biodegradable nanocapsules and have developed stable topical formulations that are able to achieve therapeutic cyclosporine A levels in the targeted skin layers. This topical treatment provides a potential alternative to oral cyclosporine A and other topical immunosuppressive drugs. For more information, visit www.immunepharmaceuticals.com.
Picato Gel Warning
The US Food and Drug Administration (FDA) issues a warning about reports of severe allergic reactions and herpes zoster associated with the use of Picato Gel (ingenol mebutate) for the treatment of actinic keratosis. The allergic reaction may include throat tightness, difficulty breathing, feeling faint, or swelling of the lips or tongue. The FDA received reports of cases involving severe eye injuries associated with Picato Gel not being used according to the instructions for use on the label. Changes to the labeling have been requested by the FDA to warn about these new safety risks. Patients should not use Picato Gel for a longer period or on an area of skin larger than instructed on the drug label. Patients also should avoid application in, near, and around the mouth, lips, and eye area. For more information, visit www.fda.gov/MedWatch.
Ximino Extended-Release Capsules
Sun Pharmaceutical Industries Ltd announces US Food and Drug Administration approval of the Supplemental New Drug Application for Ximino (minocycline hydrochloride) extended-release capsules (45, 90, and 135 mg). Ximino extended-release capsules are indicated for the treatment of inflammatory lesions of nonnodular moderate to severe acne vulgaris in patients 12 years and older. Ximino extended-release capsules are expected to be available for patients during the fourth quarter of 2015. For more information, visit www.sunpharma.com.
If you would like your product included in Product News, please e-mail a press release to the Editorial Office at [email protected].
Cyclosporine A
Immune Pharmaceuticals acquires a nanoparticle topical formulation of cyclosporine A for the treatment of psoriasis, atopic dermatitis, pemphigus vulgaris, and other severe inflammatory dermatoses. Researchers have incorporated cyclosporine A into biodegradable nanocapsules and have developed stable topical formulations that are able to achieve therapeutic cyclosporine A levels in the targeted skin layers. This topical treatment provides a potential alternative to oral cyclosporine A and other topical immunosuppressive drugs. For more information, visit www.immunepharmaceuticals.com.
Picato Gel Warning
The US Food and Drug Administration (FDA) issues a warning about reports of severe allergic reactions and herpes zoster associated with the use of Picato Gel (ingenol mebutate) for the treatment of actinic keratosis. The allergic reaction may include throat tightness, difficulty breathing, feeling faint, or swelling of the lips or tongue. The FDA received reports of cases involving severe eye injuries associated with Picato Gel not being used according to the instructions for use on the label. Changes to the labeling have been requested by the FDA to warn about these new safety risks. Patients should not use Picato Gel for a longer period or on an area of skin larger than instructed on the drug label. Patients also should avoid application in, near, and around the mouth, lips, and eye area. For more information, visit www.fda.gov/MedWatch.
Ximino Extended-Release Capsules
Sun Pharmaceutical Industries Ltd announces US Food and Drug Administration approval of the Supplemental New Drug Application for Ximino (minocycline hydrochloride) extended-release capsules (45, 90, and 135 mg). Ximino extended-release capsules are indicated for the treatment of inflammatory lesions of nonnodular moderate to severe acne vulgaris in patients 12 years and older. Ximino extended-release capsules are expected to be available for patients during the fourth quarter of 2015. For more information, visit www.sunpharma.com.
If you would like your product included in Product News, please e-mail a press release to the Editorial Office at [email protected].

Erratum (Cutis. 2015;95:47-51)
Due to a submission error, the article “Reduced Degree of Irritation During a Second Cycle of Ingenol Mebutate Gel 0.015% for the Treatment of Actinic Keratosis” (Cutis. 2015;95:47-51) contained the incorrect scale for local skin reactions (LSRs). The text in the Methods should have stated:
Using standardized photographic guides, 6 individual LSRs—erythema, flaking/scaling, crusting, swelling, vesiculation/pustulation, and erosion/ulceration—were assessed on a scale of 0 (none) to 4 (severe), with higher numbers indicating more severe reactions.
The staff of Cutis® makes every possible effort to ensure accuracy in its articles and apologizes for the mistake.
Due to a submission error, the article “Reduced Degree of Irritation During a Second Cycle of Ingenol Mebutate Gel 0.015% for the Treatment of Actinic Keratosis” (Cutis. 2015;95:47-51) contained the incorrect scale for local skin reactions (LSRs). The text in the Methods should have stated:
Using standardized photographic guides, 6 individual LSRs—erythema, flaking/scaling, crusting, swelling, vesiculation/pustulation, and erosion/ulceration—were assessed on a scale of 0 (none) to 4 (severe), with higher numbers indicating more severe reactions.
The staff of Cutis® makes every possible effort to ensure accuracy in its articles and apologizes for the mistake.
Due to a submission error, the article “Reduced Degree of Irritation During a Second Cycle of Ingenol Mebutate Gel 0.015% for the Treatment of Actinic Keratosis” (Cutis. 2015;95:47-51) contained the incorrect scale for local skin reactions (LSRs). The text in the Methods should have stated:
Using standardized photographic guides, 6 individual LSRs—erythema, flaking/scaling, crusting, swelling, vesiculation/pustulation, and erosion/ulceration—were assessed on a scale of 0 (none) to 4 (severe), with higher numbers indicating more severe reactions.
The staff of Cutis® makes every possible effort to ensure accuracy in its articles and apologizes for the mistake.
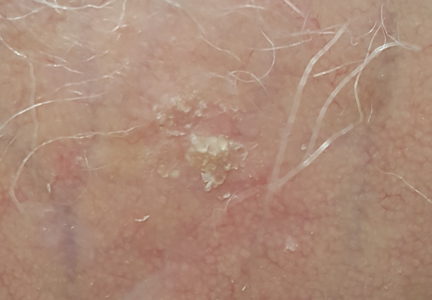
What Is Your Diagnosis? Idiopathic Guttate Hypomelanosis
The Diagnosis: Idiopathic Guttate Hypomelanosis
A biopsy of the largest lesion from the left leg superior to the lateral malleolus was performed. Histopathologic examination revealed solar elastosis, diminished number of focal melanocytes and pigment within keratinocytes compared to uninvolved skin, and presence of hyperkeratosis with flattening of rete ridges. The clinical presentation along with histopathologic analysis confirmed a diagnosis of idiopathic guttate hypomelanosis (IGH). The lesions were treated with short-exposure cryotherapy, which resulted in partial repigmentation after several treatments.
Idiopathic guttate hypomelanosis is a common but underreported condition in elderly patients that usually presents with small, discrete, asymptomatic, hypopigmented macules. The frequency of IGH increases with age.1 Frequency of the condition is much lower in patients aged 21 to 30 years and does not exceed 7%. Lesions of IGH have a predilection for sun-exposed areas such as the arms and legs but rarely can be seen on the face and trunk. Facial lesions of IGH are more frequently reported in women.1 The size of lesions can be up to 1.5 cm in diameter. The condition generally is self-limited, but some patients may express aesthetic concerns. Rare cases of IGH in children have been associated with prolonged sun exposure.2
The etiology of IGH is unknown but an association with sun exposure has been noted. Patients with IGH frequently show other signs of photoaging, such as numerous seborrheic keratoses, solar lentigines, xeroses, freckles, and actinic keratoses.1 Short-term exposure to UVB radiation and psoralen plus UVA therapy has been shown to cause IGH in patients with chronic diseases such as mycosis fungoides.3-5 One small study that examined renal transplant recipients determined an association between HLA-DQ3 antigens and IGH, whereas HLA-DR8 antigens were not identified in any patients with IGH, indicating it may have some advantage in preventing the development of IGH.6 Shin et al1 reported that IGH was prevalent among patients who regularly traumatized their skin by scrubbing.
Clinically, IGH should be differentiated from other conditions characterized by hypopigmentation, such as pityriasis alba, pityriasis versicolor, postinflammatory hypopigmentation, progressive macular hypomelanosis, and vitiligo. Aside from clinical examination, histopathologic studies are helpful in making a definitive diagnosis. The differential diagnosis of IGH is presented in the Table.

Histopathology of IGH lesions usually reveals slight atrophy of the epidermis with flattening of rete ridges and concomitant hyperkeratosis. A thickened stratum granulosum also has been noted in lesions of IGH.2 The diminished number of melanocytes and melanin pigment granules along with hyperkeratosis both appear to contribute to the hypopigmentation noted in IGH.7 Ultrastructural studies of lesions of IGH can confirm melanocytic degeneration and a decreased number of melanosomes in melanocytes and keratinocytes.2,8
There is no uniformly effective treatment of IGH. Topical application of tacrolimus and tretinoin have shown efficacy in repigmenting IGH lesions.8,9 Short-exposure cryotherapy with a duration of 3 to 5 seconds, localized chemical peels, and/or local dermabrasion can be helpful.10-12 CO2 lasers also have demonstrated promising results.13
- Shin MK, Jeong KH, Oh IH, et al. Clinical features of idiopathic guttate hypomelanosis in 646 subjects and association with other aspects of photoaging. Int J Dermatol. 2011;50:798-805.
- Kim SK, Kim EH, Kang HY, et al. Comprehensive understanding of idiopathic guttate hypomelanosis: clinical and histopathological correlation. Int J Dermatol. 2010;49:162-166.
- Friedland R, David M, Feinmesser M, et al. Idiopathic guttate hypomelanosis-like lesions in patients with mycosis fungoides: a new adverse effect of phototherapy. J Eur Acad Dermatol Venereol. 2010;24:1026-1030.
- Kaya TI, Yazici AC, Tursen U, et al. Idiopathic guttate hypomelanosis: idiopathic or ultraviolet induced? Photodermatol Photoimmunol Photomed. 2005;21:270-271.
- Loquai C, Metze D, Nashan D, et al. Confetti-like lesions with hyperkeratosis: a novel ultraviolet-induced hypomelanotic disorder? Br J Dermatol. 2005;153:190-193.
- Arrunategui A, Trujillo RA, Marulanda MP, et al. HLA-DQ3 is associated with idiopathic guttate hypomelanosis, whereas HLA-DR8 is not, in a group of renal transplant patients. Int J Dermatol. 2002;41:744-747.
- Wallace ML, Grichnik JM, Prieto VG, et al. Numbers and differentiation status of melanocytes in idiopathic guttate hypomelanosis. J Cutan Pathol. 1998;25:375-379.
- Ortonne JP, Perrot H. Idiopathic guttate hypomelanosis. ultrastructural study. Arch Dermatol. 1980;116:664-668.
- Rerknimitr P, Disphanurat W, Achariyakul M. Topical tacrolimus significantly promotes repigmentation in idiopathic guttate hypomelanosis: a double-blind, randomized, placebo-controlled study. J Eur Acad Dermatol Venereol. 2013;27:460-464.
- Pagnoni A, Kligman AM, Sadiq I, et al. Hypopigmented macules of photodamaged skin and their treatment with topical tretinoin. Acta Derm Venereol. 1999;79:305-310.
- Kumarasinghe SP. 3-5 second cryotherapy is effective in idiopathic guttate hypomelanosis. J Dermatol. 2004;31:457-459.
- Hexsel DM. Treatment of idiopathic guttate hypomelanosis by localized superficial dermabrasion. Dermatol Surg. 1999;25:917-918.
- Shin J, Kim M, Park SH, et al. The effect of fractional carbon dioxide lasers on idiopathic guttate hypomelanosis: a preliminary study. J Eur Acad Dermatol Venereol. 2013;27:e243-e246.
The Diagnosis: Idiopathic Guttate Hypomelanosis
A biopsy of the largest lesion from the left leg superior to the lateral malleolus was performed. Histopathologic examination revealed solar elastosis, diminished number of focal melanocytes and pigment within keratinocytes compared to uninvolved skin, and presence of hyperkeratosis with flattening of rete ridges. The clinical presentation along with histopathologic analysis confirmed a diagnosis of idiopathic guttate hypomelanosis (IGH). The lesions were treated with short-exposure cryotherapy, which resulted in partial repigmentation after several treatments.
Idiopathic guttate hypomelanosis is a common but underreported condition in elderly patients that usually presents with small, discrete, asymptomatic, hypopigmented macules. The frequency of IGH increases with age.1 Frequency of the condition is much lower in patients aged 21 to 30 years and does not exceed 7%. Lesions of IGH have a predilection for sun-exposed areas such as the arms and legs but rarely can be seen on the face and trunk. Facial lesions of IGH are more frequently reported in women.1 The size of lesions can be up to 1.5 cm in diameter. The condition generally is self-limited, but some patients may express aesthetic concerns. Rare cases of IGH in children have been associated with prolonged sun exposure.2
The etiology of IGH is unknown but an association with sun exposure has been noted. Patients with IGH frequently show other signs of photoaging, such as numerous seborrheic keratoses, solar lentigines, xeroses, freckles, and actinic keratoses.1 Short-term exposure to UVB radiation and psoralen plus UVA therapy has been shown to cause IGH in patients with chronic diseases such as mycosis fungoides.3-5 One small study that examined renal transplant recipients determined an association between HLA-DQ3 antigens and IGH, whereas HLA-DR8 antigens were not identified in any patients with IGH, indicating it may have some advantage in preventing the development of IGH.6 Shin et al1 reported that IGH was prevalent among patients who regularly traumatized their skin by scrubbing.
Clinically, IGH should be differentiated from other conditions characterized by hypopigmentation, such as pityriasis alba, pityriasis versicolor, postinflammatory hypopigmentation, progressive macular hypomelanosis, and vitiligo. Aside from clinical examination, histopathologic studies are helpful in making a definitive diagnosis. The differential diagnosis of IGH is presented in the Table.
Histopathology of IGH lesions usually reveals slight atrophy of the epidermis with flattening of rete ridges and concomitant hyperkeratosis. A thickened stratum granulosum also has been noted in lesions of IGH.2 The diminished number of melanocytes and melanin pigment granules along with hyperkeratosis both appear to contribute to the hypopigmentation noted in IGH.7 Ultrastructural studies of lesions of IGH can confirm melanocytic degeneration and a decreased number of melanosomes in melanocytes and keratinocytes.2,8
There is no uniformly effective treatment of IGH. Topical application of tacrolimus and tretinoin have shown efficacy in repigmenting IGH lesions.8,9 Short-exposure cryotherapy with a duration of 3 to 5 seconds, localized chemical peels, and/or local dermabrasion can be helpful.10-12 CO2 lasers also have demonstrated promising results.13
The Diagnosis: Idiopathic Guttate Hypomelanosis
A biopsy of the largest lesion from the left leg superior to the lateral malleolus was performed. Histopathologic examination revealed solar elastosis, diminished number of focal melanocytes and pigment within keratinocytes compared to uninvolved skin, and presence of hyperkeratosis with flattening of rete ridges. The clinical presentation along with histopathologic analysis confirmed a diagnosis of idiopathic guttate hypomelanosis (IGH). The lesions were treated with short-exposure cryotherapy, which resulted in partial repigmentation after several treatments.
Idiopathic guttate hypomelanosis is a common but underreported condition in elderly patients that usually presents with small, discrete, asymptomatic, hypopigmented macules. The frequency of IGH increases with age.1 Frequency of the condition is much lower in patients aged 21 to 30 years and does not exceed 7%. Lesions of IGH have a predilection for sun-exposed areas such as the arms and legs but rarely can be seen on the face and trunk. Facial lesions of IGH are more frequently reported in women.1 The size of lesions can be up to 1.5 cm in diameter. The condition generally is self-limited, but some patients may express aesthetic concerns. Rare cases of IGH in children have been associated with prolonged sun exposure.2
The etiology of IGH is unknown but an association with sun exposure has been noted. Patients with IGH frequently show other signs of photoaging, such as numerous seborrheic keratoses, solar lentigines, xeroses, freckles, and actinic keratoses.1 Short-term exposure to UVB radiation and psoralen plus UVA therapy has been shown to cause IGH in patients with chronic diseases such as mycosis fungoides.3-5 One small study that examined renal transplant recipients determined an association between HLA-DQ3 antigens and IGH, whereas HLA-DR8 antigens were not identified in any patients with IGH, indicating it may have some advantage in preventing the development of IGH.6 Shin et al1 reported that IGH was prevalent among patients who regularly traumatized their skin by scrubbing.
Clinically, IGH should be differentiated from other conditions characterized by hypopigmentation, such as pityriasis alba, pityriasis versicolor, postinflammatory hypopigmentation, progressive macular hypomelanosis, and vitiligo. Aside from clinical examination, histopathologic studies are helpful in making a definitive diagnosis. The differential diagnosis of IGH is presented in the Table.
Histopathology of IGH lesions usually reveals slight atrophy of the epidermis with flattening of rete ridges and concomitant hyperkeratosis. A thickened stratum granulosum also has been noted in lesions of IGH.2 The diminished number of melanocytes and melanin pigment granules along with hyperkeratosis both appear to contribute to the hypopigmentation noted in IGH.7 Ultrastructural studies of lesions of IGH can confirm melanocytic degeneration and a decreased number of melanosomes in melanocytes and keratinocytes.2,8
There is no uniformly effective treatment of IGH. Topical application of tacrolimus and tretinoin have shown efficacy in repigmenting IGH lesions.8,9 Short-exposure cryotherapy with a duration of 3 to 5 seconds, localized chemical peels, and/or local dermabrasion can be helpful.10-12 CO2 lasers also have demonstrated promising results.13
- Shin MK, Jeong KH, Oh IH, et al. Clinical features of idiopathic guttate hypomelanosis in 646 subjects and association with other aspects of photoaging. Int J Dermatol. 2011;50:798-805.
- Kim SK, Kim EH, Kang HY, et al. Comprehensive understanding of idiopathic guttate hypomelanosis: clinical and histopathological correlation. Int J Dermatol. 2010;49:162-166.
- Friedland R, David M, Feinmesser M, et al. Idiopathic guttate hypomelanosis-like lesions in patients with mycosis fungoides: a new adverse effect of phototherapy. J Eur Acad Dermatol Venereol. 2010;24:1026-1030.
- Kaya TI, Yazici AC, Tursen U, et al. Idiopathic guttate hypomelanosis: idiopathic or ultraviolet induced? Photodermatol Photoimmunol Photomed. 2005;21:270-271.
- Loquai C, Metze D, Nashan D, et al. Confetti-like lesions with hyperkeratosis: a novel ultraviolet-induced hypomelanotic disorder? Br J Dermatol. 2005;153:190-193.
- Arrunategui A, Trujillo RA, Marulanda MP, et al. HLA-DQ3 is associated with idiopathic guttate hypomelanosis, whereas HLA-DR8 is not, in a group of renal transplant patients. Int J Dermatol. 2002;41:744-747.
- Wallace ML, Grichnik JM, Prieto VG, et al. Numbers and differentiation status of melanocytes in idiopathic guttate hypomelanosis. J Cutan Pathol. 1998;25:375-379.
- Ortonne JP, Perrot H. Idiopathic guttate hypomelanosis. ultrastructural study. Arch Dermatol. 1980;116:664-668.
- Rerknimitr P, Disphanurat W, Achariyakul M. Topical tacrolimus significantly promotes repigmentation in idiopathic guttate hypomelanosis: a double-blind, randomized, placebo-controlled study. J Eur Acad Dermatol Venereol. 2013;27:460-464.
- Pagnoni A, Kligman AM, Sadiq I, et al. Hypopigmented macules of photodamaged skin and their treatment with topical tretinoin. Acta Derm Venereol. 1999;79:305-310.
- Kumarasinghe SP. 3-5 second cryotherapy is effective in idiopathic guttate hypomelanosis. J Dermatol. 2004;31:457-459.
- Hexsel DM. Treatment of idiopathic guttate hypomelanosis by localized superficial dermabrasion. Dermatol Surg. 1999;25:917-918.
- Shin J, Kim M, Park SH, et al. The effect of fractional carbon dioxide lasers on idiopathic guttate hypomelanosis: a preliminary study. J Eur Acad Dermatol Venereol. 2013;27:e243-e246.
- Shin MK, Jeong KH, Oh IH, et al. Clinical features of idiopathic guttate hypomelanosis in 646 subjects and association with other aspects of photoaging. Int J Dermatol. 2011;50:798-805.
- Kim SK, Kim EH, Kang HY, et al. Comprehensive understanding of idiopathic guttate hypomelanosis: clinical and histopathological correlation. Int J Dermatol. 2010;49:162-166.
- Friedland R, David M, Feinmesser M, et al. Idiopathic guttate hypomelanosis-like lesions in patients with mycosis fungoides: a new adverse effect of phototherapy. J Eur Acad Dermatol Venereol. 2010;24:1026-1030.
- Kaya TI, Yazici AC, Tursen U, et al. Idiopathic guttate hypomelanosis: idiopathic or ultraviolet induced? Photodermatol Photoimmunol Photomed. 2005;21:270-271.
- Loquai C, Metze D, Nashan D, et al. Confetti-like lesions with hyperkeratosis: a novel ultraviolet-induced hypomelanotic disorder? Br J Dermatol. 2005;153:190-193.
- Arrunategui A, Trujillo RA, Marulanda MP, et al. HLA-DQ3 is associated with idiopathic guttate hypomelanosis, whereas HLA-DR8 is not, in a group of renal transplant patients. Int J Dermatol. 2002;41:744-747.
- Wallace ML, Grichnik JM, Prieto VG, et al. Numbers and differentiation status of melanocytes in idiopathic guttate hypomelanosis. J Cutan Pathol. 1998;25:375-379.
- Ortonne JP, Perrot H. Idiopathic guttate hypomelanosis. ultrastructural study. Arch Dermatol. 1980;116:664-668.
- Rerknimitr P, Disphanurat W, Achariyakul M. Topical tacrolimus significantly promotes repigmentation in idiopathic guttate hypomelanosis: a double-blind, randomized, placebo-controlled study. J Eur Acad Dermatol Venereol. 2013;27:460-464.
- Pagnoni A, Kligman AM, Sadiq I, et al. Hypopigmented macules of photodamaged skin and their treatment with topical tretinoin. Acta Derm Venereol. 1999;79:305-310.
- Kumarasinghe SP. 3-5 second cryotherapy is effective in idiopathic guttate hypomelanosis. J Dermatol. 2004;31:457-459.
- Hexsel DM. Treatment of idiopathic guttate hypomelanosis by localized superficial dermabrasion. Dermatol Surg. 1999;25:917-918.
- Shin J, Kim M, Park SH, et al. The effect of fractional carbon dioxide lasers on idiopathic guttate hypomelanosis: a preliminary study. J Eur Acad Dermatol Venereol. 2013;27:e243-e246.
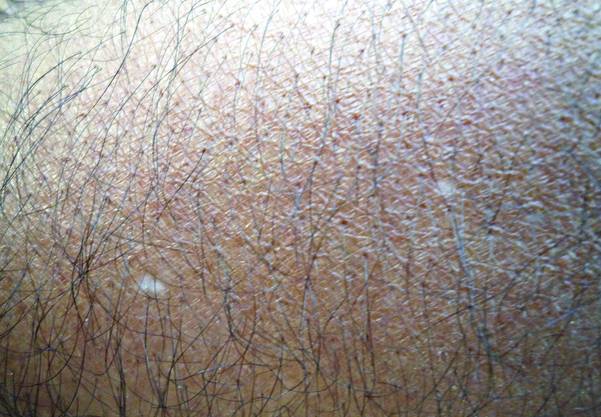
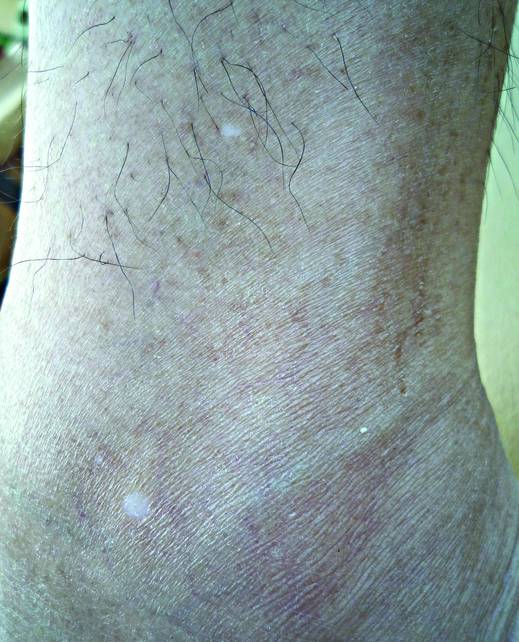
A 58-year-old man presented with disseminated, hypopigmented, asymptomatic lesions on the right arm (top) and left leg (bottom) that had been present for approximately 6 years. The patient reported that the lesions had become more visible and greater in number within the last year. Multiple circular hypopigmented macules of various sizes ranging from 1 to 3 mm in diameter were identified. No scaling was seen. Physical examination was otherwise unremarkable.
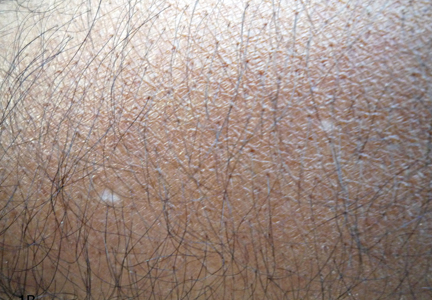
Syringoid Eccrine Carcinoma
Syringoid eccrine carcinoma is a rare malignant adnexal tumor with eccrine differentiation that histologically resembles a syringoma.1 Originally described as eccrine epithelioma by Freeman and Winklemann2 in 1969, syringoid eccrine carcinoma has been reported in the literature as eccrine carcinoma, eccrine syringomatous carcinoma, and sclerosing sweat duct carcinoma.3 Clinically, syringoid eccrine carcinoma most commonly presents as a tender plaque or nodule on the scalp, and histologic examination generally reveals a dermal-based lesion that rarely shows epidermal connection. It demonstrates syringomalike tadpole morphology (epithelial strands with lumen formation) composed of basaloid epithelium with uniform hyperchromatic nuclei (Figure 1). There usually is an infiltrative growth pattern to the subcutis (Figure 2 [left]) or skeletal muscle as well as remarkable perineural invasion (Figure 2 [right]). Mitotic activity is minimal to absent. The tumor cells of syringoid eccrine carcinoma typically show positive immuno-staining for high- and low-molecular-weight cytokeratin, while the lumina are highlighted by epithelial membrane antigen and carcinoembryonic antigen.4 However, immunohistochemistry often is not contributory in diagnosing primary eccrine carcinomas.
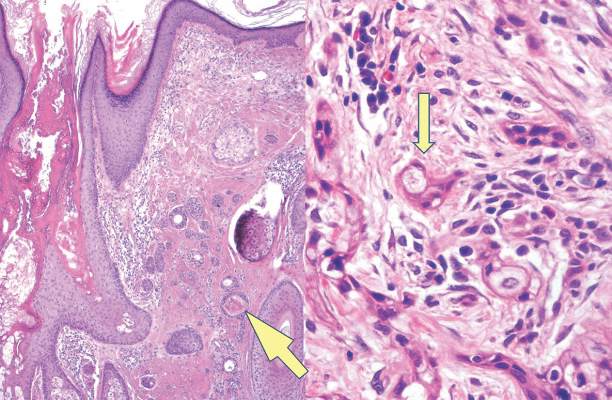
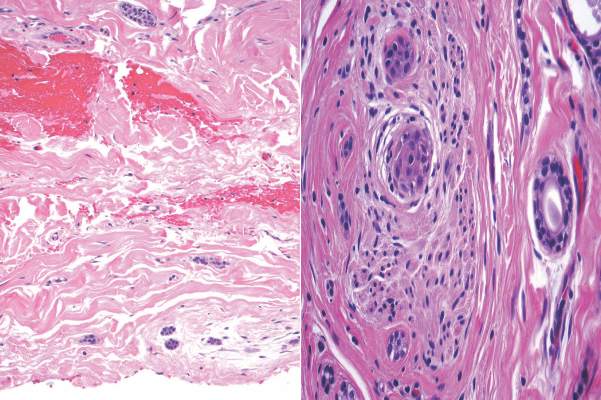
The differential diagnosis of syringoid eccrine carcinoma includes cutaneous adenoid cystic carcinoma, metastatic adenocarcinoma, sclerosing basal cell carcinoma, and syringoma. Cutaneous adenoid cystic carcinoma is a rare, slow-growing, flesh-colored tumor that consists of lobules, islands, and cords of basaloid cells with prominent cystic cribriforming (Figure 3). The tumor cells typically are small, cuboidal, and monomorphic. Metastatic adenoid cystic carcinoma, such as from a primary tumor of the salivary glands or breasts, must be excluded before rendering a diagnosis of primary cutaneous disease.
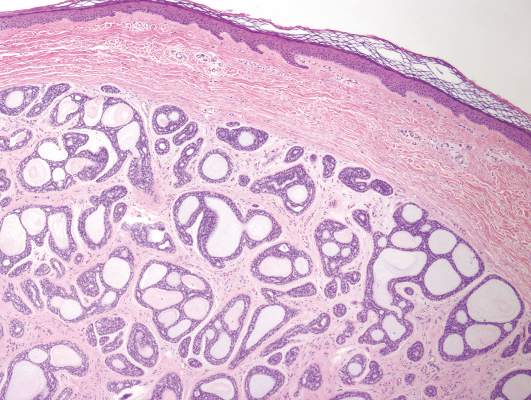
Metastatic adenocarcinoma of the skin usually presents in patients with a clinical history of preexisting disease. The breasts, colon, stomach, and ovaries are common origins of metastases. The histopathologic and immunohistochemical findings depend on the particular site of origin of the metastasis. Compared with primary eccrine carcinomas, metastatic adenocarcinomas of the skin generally are high-grade lesions with prominent atypia, mitosis, and necrosis (Figure 4).
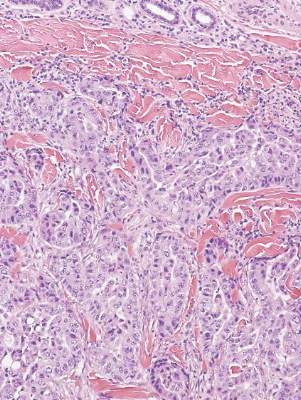
Sclerosing basal cell carcinoma shows basaloid tumor cells with deep infiltration. Unlike syringoid eccrine carcinoma, basal cell carcinoma is an epidermal tumor that does not have true lumen formation. Furthermore, other variants of basal cell carcinoma, including nodular, micronodular, or superficial multicentric tumors, often coexist with the sclerosing variant in the same lesion and constitute a useful diagnostic clue (Figure 5). Staining for epithelial membrane antigen may be useful in identifying the absence of lumen formation, and Ber-EP4 highlights the epidermal origin of the lesion.5
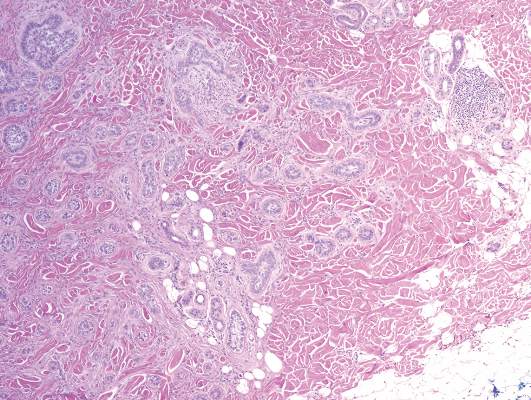
Syringomas most commonly present as multiple small flesh-colored papules on the eyelids. On histology, syringomas present as small superficial dermal lesions composed of small ducts that may form tadpolelike structures in a fibrotic stroma (Figure 6). The ducts are lined by benign cuboidal cells. In contrast to syringoid eccrine carcinomas, syringomas usually present as multiple lesions that are microscopically superficial without perineural involvement.
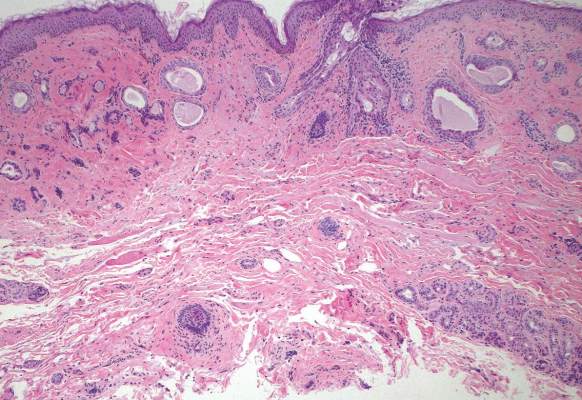
1. Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
2. Freeman RG, Winklemann RK. Basal cell tumor with eccrine differentiations (eccrine epithelioma). Arch Dermatol. 1969;100:234-242.
3. Nishizawa A, Nakanishi Y, Sasajima Y, et al. Syringoid carcinoma with apparently aggressive transformation: case report and review of the literature. Int J Dermatol. 2006;45:1218-1221.
4. Urso C, Bondi R, Paglierani M, et al. Carcinomas of sweat glands: report of 60 cases. Arch Pathol Lab Med. 2001;125:498-505.
5. Cassarino D. Diagnostic Pathology: Neoplastic Dermatopathology. Salt Lake City, UT: Amirsys Publishing Inc; 2012.
Syringoid eccrine carcinoma is a rare malignant adnexal tumor with eccrine differentiation that histologically resembles a syringoma.1 Originally described as eccrine epithelioma by Freeman and Winklemann2 in 1969, syringoid eccrine carcinoma has been reported in the literature as eccrine carcinoma, eccrine syringomatous carcinoma, and sclerosing sweat duct carcinoma.3 Clinically, syringoid eccrine carcinoma most commonly presents as a tender plaque or nodule on the scalp, and histologic examination generally reveals a dermal-based lesion that rarely shows epidermal connection. It demonstrates syringomalike tadpole morphology (epithelial strands with lumen formation) composed of basaloid epithelium with uniform hyperchromatic nuclei (Figure 1). There usually is an infiltrative growth pattern to the subcutis (Figure 2 [left]) or skeletal muscle as well as remarkable perineural invasion (Figure 2 [right]). Mitotic activity is minimal to absent. The tumor cells of syringoid eccrine carcinoma typically show positive immuno-staining for high- and low-molecular-weight cytokeratin, while the lumina are highlighted by epithelial membrane antigen and carcinoembryonic antigen.4 However, immunohistochemistry often is not contributory in diagnosing primary eccrine carcinomas.
The differential diagnosis of syringoid eccrine carcinoma includes cutaneous adenoid cystic carcinoma, metastatic adenocarcinoma, sclerosing basal cell carcinoma, and syringoma. Cutaneous adenoid cystic carcinoma is a rare, slow-growing, flesh-colored tumor that consists of lobules, islands, and cords of basaloid cells with prominent cystic cribriforming (Figure 3). The tumor cells typically are small, cuboidal, and monomorphic. Metastatic adenoid cystic carcinoma, such as from a primary tumor of the salivary glands or breasts, must be excluded before rendering a diagnosis of primary cutaneous disease.
Metastatic adenocarcinoma of the skin usually presents in patients with a clinical history of preexisting disease. The breasts, colon, stomach, and ovaries are common origins of metastases. The histopathologic and immunohistochemical findings depend on the particular site of origin of the metastasis. Compared with primary eccrine carcinomas, metastatic adenocarcinomas of the skin generally are high-grade lesions with prominent atypia, mitosis, and necrosis (Figure 4).
Sclerosing basal cell carcinoma shows basaloid tumor cells with deep infiltration. Unlike syringoid eccrine carcinoma, basal cell carcinoma is an epidermal tumor that does not have true lumen formation. Furthermore, other variants of basal cell carcinoma, including nodular, micronodular, or superficial multicentric tumors, often coexist with the sclerosing variant in the same lesion and constitute a useful diagnostic clue (Figure 5). Staining for epithelial membrane antigen may be useful in identifying the absence of lumen formation, and Ber-EP4 highlights the epidermal origin of the lesion.5
Syringomas most commonly present as multiple small flesh-colored papules on the eyelids. On histology, syringomas present as small superficial dermal lesions composed of small ducts that may form tadpolelike structures in a fibrotic stroma (Figure 6). The ducts are lined by benign cuboidal cells. In contrast to syringoid eccrine carcinomas, syringomas usually present as multiple lesions that are microscopically superficial without perineural involvement.
Syringoid eccrine carcinoma is a rare malignant adnexal tumor with eccrine differentiation that histologically resembles a syringoma.1 Originally described as eccrine epithelioma by Freeman and Winklemann2 in 1969, syringoid eccrine carcinoma has been reported in the literature as eccrine carcinoma, eccrine syringomatous carcinoma, and sclerosing sweat duct carcinoma.3 Clinically, syringoid eccrine carcinoma most commonly presents as a tender plaque or nodule on the scalp, and histologic examination generally reveals a dermal-based lesion that rarely shows epidermal connection. It demonstrates syringomalike tadpole morphology (epithelial strands with lumen formation) composed of basaloid epithelium with uniform hyperchromatic nuclei (Figure 1). There usually is an infiltrative growth pattern to the subcutis (Figure 2 [left]) or skeletal muscle as well as remarkable perineural invasion (Figure 2 [right]). Mitotic activity is minimal to absent. The tumor cells of syringoid eccrine carcinoma typically show positive immuno-staining for high- and low-molecular-weight cytokeratin, while the lumina are highlighted by epithelial membrane antigen and carcinoembryonic antigen.4 However, immunohistochemistry often is not contributory in diagnosing primary eccrine carcinomas.
The differential diagnosis of syringoid eccrine carcinoma includes cutaneous adenoid cystic carcinoma, metastatic adenocarcinoma, sclerosing basal cell carcinoma, and syringoma. Cutaneous adenoid cystic carcinoma is a rare, slow-growing, flesh-colored tumor that consists of lobules, islands, and cords of basaloid cells with prominent cystic cribriforming (Figure 3). The tumor cells typically are small, cuboidal, and monomorphic. Metastatic adenoid cystic carcinoma, such as from a primary tumor of the salivary glands or breasts, must be excluded before rendering a diagnosis of primary cutaneous disease.
Metastatic adenocarcinoma of the skin usually presents in patients with a clinical history of preexisting disease. The breasts, colon, stomach, and ovaries are common origins of metastases. The histopathologic and immunohistochemical findings depend on the particular site of origin of the metastasis. Compared with primary eccrine carcinomas, metastatic adenocarcinomas of the skin generally are high-grade lesions with prominent atypia, mitosis, and necrosis (Figure 4).
Sclerosing basal cell carcinoma shows basaloid tumor cells with deep infiltration. Unlike syringoid eccrine carcinoma, basal cell carcinoma is an epidermal tumor that does not have true lumen formation. Furthermore, other variants of basal cell carcinoma, including nodular, micronodular, or superficial multicentric tumors, often coexist with the sclerosing variant in the same lesion and constitute a useful diagnostic clue (Figure 5). Staining for epithelial membrane antigen may be useful in identifying the absence of lumen formation, and Ber-EP4 highlights the epidermal origin of the lesion.5
Syringomas most commonly present as multiple small flesh-colored papules on the eyelids. On histology, syringomas present as small superficial dermal lesions composed of small ducts that may form tadpolelike structures in a fibrotic stroma (Figure 6). The ducts are lined by benign cuboidal cells. In contrast to syringoid eccrine carcinomas, syringomas usually present as multiple lesions that are microscopically superficial without perineural involvement.
1. Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
2. Freeman RG, Winklemann RK. Basal cell tumor with eccrine differentiations (eccrine epithelioma). Arch Dermatol. 1969;100:234-242.
3. Nishizawa A, Nakanishi Y, Sasajima Y, et al. Syringoid carcinoma with apparently aggressive transformation: case report and review of the literature. Int J Dermatol. 2006;45:1218-1221.
4. Urso C, Bondi R, Paglierani M, et al. Carcinomas of sweat glands: report of 60 cases. Arch Pathol Lab Med. 2001;125:498-505.
5. Cassarino D. Diagnostic Pathology: Neoplastic Dermatopathology. Salt Lake City, UT: Amirsys Publishing Inc; 2012.
1. Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
2. Freeman RG, Winklemann RK. Basal cell tumor with eccrine differentiations (eccrine epithelioma). Arch Dermatol. 1969;100:234-242.
3. Nishizawa A, Nakanishi Y, Sasajima Y, et al. Syringoid carcinoma with apparently aggressive transformation: case report and review of the literature. Int J Dermatol. 2006;45:1218-1221.
4. Urso C, Bondi R, Paglierani M, et al. Carcinomas of sweat glands: report of 60 cases. Arch Pathol Lab Med. 2001;125:498-505.
5. Cassarino D. Diagnostic Pathology: Neoplastic Dermatopathology. Salt Lake City, UT: Amirsys Publishing Inc; 2012.
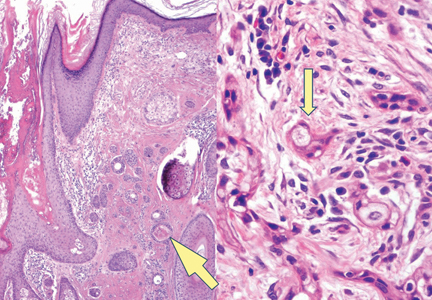
Therapies for Actinic Keratosis With a Focus on Cosmetic Outcomes
Actinic keratosis (AK), also referred to as solar keratosis or senile keratosis, is an intraepidermal proliferation of dysplastic keratinocytes that develops in response to chronic exposure to UV radiation. Actinic keratoses are among the most commonly encountered lesions seen by dermatologists, and it has been estimated that 60% of predisposed individuals older than 40 years have at least one AK.1,2 Prevalence is notably higher in light-skinned individuals and increases with age, presumably from higher cumulative sun exposure and decreased effectiveness of the immune system.1,3 It remains a point of contention as to whether or not AKs actually represent squamous cell carcinoma (SCC) in situ, but the potential for progression to invasive disease has been well demonstrated, as the majority of SCCs develop from preexisting AKs.4-6 The risk for progression to invasive disease for an individual AK has been estimated to range from 0.025% to 16% per year, with an average of approximately 8% in immunocompetent patients.7
The clinical morphology of AK can vary widely, but the most common presentation is an erythematous scaly macule, papule, or plaque on sun-exposed skin. The skin surrounding AKs typically shows evidence of solar damage with deep wrinkling, mottled pigmentation, scattered telangiectases, purpura, or xerosis (Figure). A variety of clinical variants with unique presentations exist, including atrophic, hypertrophic, acantholytic, lichenoid, bowenoid, and pigmented subtypes. Because more than 80% of AKs occur on highly visible areas such as the head, neck, back of the hands, and forearms, AKs can have an obvious detrimental effect on cosmetic appearance. Studies also have shown a strong association between AKs and decreased overall quality of life (QOL).3,8,9

Because of the risk for AK progression to invasive cancer along with its negative impact on cosmesis and QOL, clinicians generally opt to treat AKs. Numerous different treatment options exist, including topical medications, procedural modalities, and light-based therapies. Here, we review the efficacy of the most commonly utilized treatments and discuss the relevant cosmetic considerations and outcomes.
Topical Treatments
5-Fluorouracil
5-Fluorouracil (5-FU) is a US Food and Drug Administration (FDA)–approved, topically applied pyrimidine analogue that inhibits thymidylate synthase. The resulting suppression of DNA and RNA synthesis induces cell death with a preference for mitotically active cells.10 5-Fluorouracil has been used for more than 50 years as a treatment of AK and its efficacy is well established. A systematic review of 5 randomized controlled studies of topical 5-FU reported an average of 49% of 423 patients achieving complete lesion clearance with 5-FU cream 5% applied once or twice daily for up to 7 weeks.11 Some notable drawbacks of 5-FU, however, are application-site erythema, blistering, pruritus, necrosis, erosion, and pain. These effects often lead to premature cessation of therapy, but newer formulations of 5-FU cream 0.5% have shown good efficacy with better tolerability.12 A randomized, double-blind, multicenter, parallel-group study of 177 patients using 5-FU cream 0.5% once daily for either 1, 2, or 4 weeks demonstrated significant (P<.001) efficacy over vehicle gel in all treatment arms.13 The most effective therapy was 4 weeks of treatment, which achieved a mean 91.7% reduction in lesion count as assessed 1 month after cessation of therapy. The primary adverse effect (AE) reported in this trial was mild to moderate facial irritation, which generally resolved within 18 to 21 days after treatment cessation.13 Overall, 5-FU is a highly effective therapy for treating AKs that also can improve signs of photoaging, but patients should be aware of cosmetically unappealing effects that generally occur throughout therapy and during the immediate posttreatment period.14
Chemical Peels
Chemical peels traditionally employ acidic compounds to strip away outer layers of skin to variable depths depending on the concentration of the agent being applied. For treatment of AK, trichloroacetic acid (TCA) is a commonly employed cauterant that has shown efficacy comparable to topical 5-FU as well as ablative CO2 laser resurfacing.15 Trichloroacetic acid peels also are a convenient therapy, as good results can be achieved after a single treatment session. A split-face study of 15 patients treated with either a single application of 35% TCA and Jessner solution or twice-daily application of 5-FU cream 5% for 3 weeks demonstrated a reduction in 75% of visible AKs in both treatment arms over a 1-year follow-up period.16 Although 80% of patients self-reported considerable cosmetic improvement with both therapies, patient preference was reported to be in favor of the TCA peel, given its quick results and relatively mild side effects as compared to 5-FU. Treatment with chemical peels will result in temporary erythema and mild desquamation that usually resolves within 2 weeks; however, there are cases in which erythema has been reported to persist for several months.16 Adverse effects such as permanent scarring or pigmentation changes rarely are seen with TCA concentrations less than 45%.17 Caution should be used in patients with a history of herpes simplex virus, keloids, postinflammatory hyperpigmentation, radiation exposure, immunosuppression, and those unable or unwilling to use sunscreen and avoid sun exposure in the immediate posttreatment period.
Diclofenac Sodium
Diclofenac sodium (DFS) is an FDA-approved topical, nonsteroidal,
anti-inflammatory drug whose mechanism of action in the treatment of AK is thought to involve inhibition of the cyclooxygenase 2 enzyme.18 The resulting reduction of prostaglandins is believed to inhibit tumor angiogenesis, induce apoptosis, and inhibit cell differentiation.19-22 In a multicenter, double-blind, placebo-controlled study of 195 patients, application of DFS 3% in hyaluronan gel 2.5% twice daily for 60 days showed significant (P<.05) efficacy over placebo in achieving complete resolution of target lesions during a 30-day follow-up period (31% vs 10%). Furthermore, qualitative patient assessment of complete global improvement also was significantly (P<.05) higher in the active treatment group as compared to placebo (31% vs 10%).23 Additional studies of DFS 3% in hyaluronan gel 2.5% applied twice daily for 90 days have shown even higher rates of success, with complete resolution of target lesions in 40% to 58% of cases.24,25 This therapy also has been reported to substantially improve QOL following treatment completion.26 The most frequently cited AEs include pruritus, rash, dry skin, erythema, and application-site reactions. Overall, DFS is a
well-tolerated therapy with efficacy comparable to that of 5-FU but with a lower incidence of AEs
and higher patient satisfaction as determined in
2 head-to-head studies.27,28
ImiquimodImiquimod (IMQ) is an FDA-approved topical agent that functions as an immune response modifier via agonism of toll-like receptor 7.18 The resulting cytokine production and release enhances the innate and acquired immune responses leading to anticancer activity.29 The efficacy of IMQ for treatment of AK has been demonstrated in numerous well-designed clinical trials. A
meta-analysis of 5 randomized, double-blind trials including 1293 patients treated with IMQ cream 5%
2 to 3 times per week for 12 to 16 weeks reported complete clearance of AKs in 50% of patients treated with IMQ as compared to 5% of patients treated with vehicle.30 The most frequently reported AEs with this therapy include erythema, scabbing, flaking, and erosion. These effects generally resolve following cessation of treatment, and therapy is considered to be well tolerated; however, there are case reports of IMQ triggering or exacerbating existing inflammatory conditions.31 Imiquimod cream also is approved at 2.5% and 3.75% concentrations, which have demonstrated significant (P<.001) efficacy over placebo and a reduced incidence of AEs; complete clearance rates have been reported as 30.6% and 35.6%, respectively.32 Notably, a study comparing 75 patients randomized to either IMQ cream 5%
3 times per week for 4 weeks, 1 or 2 courses of cryosurgery, or 5-FU ointment 5% twice daily for 4 weeks reported that IMQ achieved significantly (P<.01) superior sustained clearance rates during a 12-month follow-up period over cryosurgery and 5-FU
(73% vs 4% vs 33%).33 Additionally, cosmetic outcomes as determined by both participants and investigators were reported as excellent at 12 months posttreatment in more than 80% of participants treated with IMQ. These excellent, long-lasting cosmetic outcomes also were determined to be significantly (P<.0001) superior to the cosmetic outcomes of 5-FU and cryotherapy, which both reported excellent outcomes in less than 10% of cases.33
Ingenol MebutateIngenol mebutate (IM) is a macrocyclic diterpene ester derived from the Euphorbia peplus plant that is FDA approved for the treatment of AK.1 Ingenol mebutate’s mechanism of action is thought to involve induction of cell death via disruption of the plasma membrane and mitochondria in addition to production of an inflammatory response, which produces tumor-specific antibodies and a large influx of neutrophils.34,35 The overall evidence for the efficacy of IM is strong. A combined analysis of 4 multicenter, randomized, double-blind studies of 1005 participants reported that IM gel 0.015% applied once daily for 3 days to the face or scalp was significantly superior (P<.001) to placebo in achieving complete clearance as assessed 54 days after completion of therapy (42.2% vs 3.7%) and that IM gel 0.05% applied once daily for 2 days to the trunk or extremities also was significantly superior (P<.001) to placebo in achieving complete clearance as determined 55 days after completion of therapy (34.1% vs 4.7%).36 A follow-up report to this study indicated that IM also appears to achieve long-lasting effects with an overall 87% decrease in total AKs at 12 months follow-up in both trial groups.37 Additionally, it has been recently reported that treatment with IM in these trials was associated with significantly higher overall treatment satisfaction (P<.001) and improved QOL (P<.001) as compared to vehicle.38 Cosmetic outcomes of IM therapy have been assessed in a trial analyzing the efficacy of IM gel 0.025% for 3 days or IM gel 0.05% for 2 or 3 days on nonfacial AKs. This study reported significantly (P<.0001) higher patient satisfaction with the cosmetic outcome at 8 weeks after therapy as compared to vehicle.34 Studies performed in mice have demonstrated that IM is able to promote collagen matrix turnover and impose dermal elasticity, which may contribute to these good cosmetic outcomes.39 The most common AEs of IM therapy are erythema, crusting, and flaking; these effects generally occur 3 to 8 days after starting treatment. These effects, however, generally are short lived and resolve within 2 weeks of treatment cessation when IM is applied to the face or scalp or 4 weeks when applied to the trunk or extremities.40 Overall, IM is a useful therapeutic option given its relatively short treatment course as compared to other topically applied agents, as well as its lasting efficacy, mild AEs, and good cosmetic outcomes.
Procedural Modalities
Surgical Procedures
Surgical approaches for the treatment of AK include excision, curettage with or without electrodesiccation, and dermabrasion. In the past, these modalities were used with greater frequency, but the advent of effective topical medications with lower risks of AEs has largely reduced their use.41 Excision may still be indicated in cases where SCC is suspected, and curettage can be used for treatment of thicker hypertrophic AKs.42 Although these approaches have not been evaluated in clinical trials, they are generally effective but require the use of local anesthetics and come with substantial risk for infection, permanent scarring, and hypopigmentation. Dermabrasion employs the use of a motorized device equipped with an abrasive material to physically remove superficial layers of the skin. Studies are limited, but this method has been reported as an effective treatment in a retrospective review of 23 participants in which 96% remained free of AKs at 1 year, 83% at 2 years, 64% at 4 years, and 54% at 5 years posttherapy.43 Notably, one split-face study of 40 participants treated with dermabrasion followed by 25% TCA on one side and either Jessner solution and 35% TCA or dermabrasion alone on the other side reported that the combination of dermabrasion with 25% TCA consistently produced excellent cosmetic results with nearly complete eradication of AKs.44 In general, however, cosmetic outcomes with dermabrasion are variable, as the technique is highly operator dependent and treatment is associated with notable discomfort as well as risk for scarring and permanent pigmentation alteration.
Cryotherapy
Cryotherapy remains one of the most commonly utilized treatments of AK and involves the delivery of liquid nitrogen via a spray device or a cotton tip applicator to rapidly freeze cells, thus causing cellular destruction via ice crystal formation and protein denaturation.45 Efficacy with this technique has been reported to be as high as 98.8% at 12 months follow-up, but more recent studies cite lower rates of success.46 A prospective multicenter study of 90 participants with 421 AKs on the face or scalp treated with a single freeze-thaw cycle of liquid nitrogen reported an overall complete response rate of 67.2% at 3 months posttherapy. Additionally, higher complete response rates were associated with longer freeze times, and cosmetic outcomes were reported as good to excellent in 94% of complete response lesions.47 Similar results were reported in an open-label, prospective, randomized, controlled clinical trial of 200 participants with 543 AKs, which compared a single freeze-thaw cycle with liquid nitrogen to a single session of CO2 laser ablation in the treatment of isolated AKs of the face and scalp.48 At 3 months posttherapy, complete clearance was observed in 71.6% of participants treated with cryotherapy and in 65.3% of participants treated with laser ablation (P=.532). At 12 months posttherapy, participants who originally showed complete response at 3 months were assessed for relapse. Complete clearance was preserved in 72.6% of participants treated with cryotherapy versus 21.9% of participants treated with laser ablation (P<.0001), and cosmetic outcomes were reported by participants as good or excellent at 3 months follow-up in more than 93% of participants for both treatment arms.48 Possible AEs of cryotherapy include pain during treatment, blister formation with possible hemorrhage, infection, scarring, and permanent pigmentary changes.47,48 Notably, the risk for hypopigmentation increases with longer freezing times, thus requiring clinicians to consider the balance between improved efficacy and reduced cosmetic outcomes.47
Light-Based Therapies
Laser Therapy
Ablative laser resurfacing with either the CO2 or erbium-doped:YAG (Er:YAG) laser utilizes light of specific wavelengths to selectively induce thermolysis and destruction of the epidermal layer. Both lasers have been studied as treatments of AK, but there is a lack of large, well-designed studies. In one small study of 14 participants treated with 1 to 2 passes of the CO2 laser, complete clearance was reported in all cases without any recurrences during a follow-up period of 6 to 24 months. Additionally, all participants in this study reported satisfaction with the cosmetic outcome.49 The CO2 laser also has demonstrated efficacy comparable to that of the TCA peel and 5-FU therapy in a prospective randomized trial of 34 patients with facial or scalp AKs who received either CO2 laser with 2 passes, 30% TCA peel, or 5-FU cream 5% twice daily for 3 weeks.15 Reduction in mean AK counts at 3 months posttherapy was significantly (P<.03) higher in all treatment arms as compared to the control group (92% for CO2 laser, 89% for TCA peel, and 83% for 5-FU cream). No significant (P=.31) difference in outcomes was noted among the different treatment arms.15 Similar results were reported for the Er:YAG laser in a small prospective study of 5 participants treated with 2 to 3 passes with the Er:YAG laser in which reduction in mean AK counts was reported as ranging from 86% to 96% at 3 months posttherapy.50 The Er:YAG laser in combination with the CO2 laser has shown notable long-term efficacy in achieving higher lesion clearance rates and sustained complete clearance rates over treatment with topical 5-FU.51 In a prospective randomized study of 55 par-ticipants with multiple AKs on the face or scalp, participants were assigned to receive either combination laser ablation with the Er:YAG and CO2 lasers down to the level of the papillary dermis or 5-FU cream 5% applied twice daily for 2 to 7 weeks until an appropriate clinical inflammatory response was achieved. At 12 months follow-up, the laser treatment group achieved significantly (P=.048) higher mean lesion clearance rates (91.1%) as compared to the 5-FU arm (76.6%) and significantly (P=.003) higher sustained complete clearance rates (59.3%) as compared to 5-FU (29.2%). The proportion of participants with an improvement in photoaging score at 12 months follow-up approached statistical significance (P=.07), with 74% of the laser-treated group showing improvement as compared to 43% of the 5-FU–treated group. Long-term, cosmetically unappealing side effects such as erythema and hypopigmentation occurred notably more often in the laser-treated group as compared to the 5-FU group.51 In summary, ablative lasers appear to be a highly effective therapy for AK but at the cost of increased risk for AEs such as permanent pigmentary changes, prolonged erythema lasting up to several months, and scarring.50,52-55
Fractional photothermolysis is a relatively new advancement in the field of laser therapy that has received FDA approval for the treatment of AK.56 This treatment works by creating multiple noncontiguous microscopic columns of thermal injury while sparing adjacent zones of viable tissue.57 Although there are limited studies involving the use of such lasers in the treatment of AK, initial findings suggest that 1927-nm thulium lasers may be more effective than 1550-nm erbium lasers in achieving lesion clearance. A trial of 14 participants who received 5 laser treatments with a 1550-nm fractionated erbium-doped fiber laser reported an average reduction in AK counts of 66.2% at 3 months follow-up and a 55.6% reduction at 6 months follow-up. A participant-determined marked or very significant improvement of lesions was reported in 83% of participants at 1 month posttreatment but only in 44% of participants at 6 months posttreatment.58 A similar trial of 24 participants treated with up to 4 treatment sessions of the fractionated 1927-nm thulium laser reported an 87.3% reduction in number of AKs at 3 months follow-up and an 86.6% reduction at 6 months follow-up.56 The primary advantage of fractional laser therapy is a faster recovery period generally lasting only 2 or 3 days as compared to 2 weeks or more with traditional ablative lasers, thus limiting the amount of time a patient must tolerate cosmetically unappealing erythema.59,60 The quick recovery time has been attributed to the fractional laser’s ability to preserve the stratum corneum and skin barrier, which also helps reduce the risk for other AEs such as scarring and infection.56,59-61 Additional studies are needed to better assess the true efficacy of fractional laser therapy, but treatment with the fractional 1927-nm thulium laser appears to be a promising and well-tolerated therapeutic option for treatment of AK with similar efficacy to traditional ablative lasers but with a lower risk of AEs.
Photodynamic TherapyPhotodynamic therapy (PDT) is an FDA-approved treatment that involves the use of a topical photosensitizing agent such as 5-aminolevulinic acid (ALA) or methyl aminovulinate (MAL) before exposure to an activating light source to generate reactive oxygen species that lead to cell death.62-65 Multiple PDT regimens with varying combinations of photosensitizers, incubation time, and light sources have been studied, but a 2012 Cochrane review determined that treatment with conventional formulations of MAL and ALA with either blue- or red-light PDT were similarly efficacious for treatment of individual AKs as compared to vehicle with blue- or red-light PDT. One exception was that longer incubation time (ie, 4 hours) with ALA resulted in better results than shorter incubation times (ie, 0.5, 1, 2 hours) with ALA.66
Standard PDT treatment with MAL also has consistently demonstrated superior efficacy in achieving complete clearance rates in addition to superior cosmetic outcomes over treatment with either cryotherapy, DFS, or 5-FU.67-73 Three studies in particular noted an excellent or good investigator-determined cosmetic outcome in 96% to 98% of participants treated with MAL-PDT.69,71,74 Photodynamic therapy with ALA also has been reported as superior over CO2 laser ablation for AK reduction as well as both patient and investigator overall satisfaction.75
More recently, several methods of improving photosensitizer delivery have been studied, which have demonstrated remarkable efficacy at achieving lesion clearance over standard cream formulations or application routines. One such method involves the use of gentle heating to increase photosensitizer uptake. In a split-extremity study of 20 participants who were treated with 20% ALA under occlusion for 1 hour with one side heated to 38.8°C, the heated side demonstrated significant (P<.0001) efficacy at achieving higher median clearance rates over control when evaluated at 2 and 6 months posttherapy.76 Notably, occlusion of ALA in itself during the incubation period also has been demonstrated to significantly (P<.0001) improve clearance rates.77 Another method involves the use of a new nanoemulsion-based formulation of ALA gel, known as BF-200 ALA, which has demonstrated remarkable efficacy over standard MAL cream and placebo in a long-term follow-up analysis of 2 prospective, randomized, controlled trials.78 In a similar vein, 3 prospective randomized trials with a minimum follow-up time of 3 months demonstrated that MAL-PDT in combination with fractional ablative laser pretreatment has significant (P<.02 in all trials) efficacy over MAL-PDT without pretreatment in achieving complete AK clearance. Although the cosmetic outcomes were good or excellent in 87% to 100% of patients, they were not significantly different from stand-alone MAL-PDT treatment in any of the trials.79-81 However, pretreatment with microneedling in MAL-PDT has been shown to achieve superior cosmetic outcomes over MAL-PDT without microneedling, according to one small split-face study of 10 participants.82
Overall, PDT is an excellent therapeutic option that is able to provide efficacious clearance of AKs as well as superior cosmetic outcomes. Common AEs of PDT include burning, itching, and stinging during therapy, but pain intensity decreases dramatically upon termination of illumination, with cessation of most symptoms by 12 hours posttherapy.73 Permanent pigmentation changes have been reported to occasionally occur following PDT therapy.81
Conclusion
When determining which therapy to use in a patient, clinicians must take into account a variety of factors such as patient preference, cost of treatment, availability, tolerance for AEs, and the need for field therapy. Although all therapies discussed within this article are effective and reasonable treatment choices, patients who are particularly concerned about cosmetic outcomes would most likely benefit from either IMQ or PDT, as the data for cosmetic outcomes with these therapies are the strongest. Combination or sequential treatments may be required in some cases and all patients should be monitored for lesion recurrence regardless of treatment choice. A summary of the therapies and key studies discussed here is available in the PDF.
- Lebwohl M. Actinic keratosis: epidemiology and progression to squamous cell carcinoma. Br J Dermatol. 2003;149(suppl 66):31-33.
- Drake LA, Ceilley RI, Cornelison RL, et al. Guidelines of care for actinic keratoses. Committee on Guidelines of Care. J Am Acad Dermatol. 1995;32:95-98.
- Salasche SJ. Epidemiology of actinic keratoses and squamous cell carcinoma. J Am Acad Dermatol. 2000;42(1, pt 2):4-7.
- Ackerman AB, Mones JM. Solar (actinic) keratosis is squamous cell carcinoma. Br J Dermatol. 2006;155:9-22.
- Anwar J, Wrone DA, Kimyai-Asadi A, et al. The development of actinic keratosis into invasive squamous cell carcinoma: evidence and evolving classification schemes. Clin Dermatol. 2004;22:189-196.
- Criscione VD, Weinstock MA, Naylor MF, et al. Actinic keratoses: natural history and risk of malignant transformation in the Veterans Affairs Topical Tretinoin Chemoprevention Trial. Cancer. 2009;115:2523-2530.
- Glogau RG. The risk of progression to invasive disease. J Am Acad Dermatol. 2000;42(1, pt 2):23-24.
- Esmann S, Jemec GB. Management of actinic keratosis patients: a qualitative study. J Dermatolog Treat. 2007;18:53-58.
- Weinstock MA, Lee KC, Chren MM, et al. Quality of life in the actinic neoplasia syndrome: the VA Topical Tretinoin Chemoprevention (VATTC) trial. J Am Acad Dermatol. 2009;61:207-215.
- Berman B, Villa AM, Ramirez CC. Mechanisms of action of new treatment modalities for actinic keratosis. J Drugs Dermatol. 2006;5:167-173.
- Askew DA, Mickan SM, Soyer HP. Effectiveness of 5-fluorouracil treatment for actinic keratosis: a systematic review of randomized controlled trials. Int J Dermatol. 2009;46:452-463.
- Levy S, Furst K, Chern W. A pharmacokinetic evaluation of 0.5% and 5% fluorouracil topical cream in patients with actinic keratosis. Clin Ther. 2001;23:908-920.
- Jorizzo J, Stewart D, Bucko A, et al. Randomized trial evaluating a new 0.5% fluorouracil formulation demonstrates efficacy after 1-, 2-, or 4-week treatment in patients with actinic keratosis. Cutis. 2002;70:335-359.
- Sachs DL, Kang S, Hammerberg C, et al. Topical fluorouracil for actinic keratoses and photoaging: a clinical and molecular analysis. Arch Dermatol. 2009;145:659-666.
- Hantash BM, Stewart DB, Cooper ZA, et al. Facial resurfacing for nonmelanoma skin cancer prophylaxis. Arch Dermatol. 2006;142:976-982.
- Lawrence N, Cox SE, Cockerell CJ, et al. A comparison of the efficacy and safety of Jessner’s solution and 35% trichloroacetic acid vs 5% fluorouracil in the treatment of widespread facial actinic keratoses. Arch Dermatol. 1995;131:176-181.
- Monheit GD. The Jessner’s + TCA peel: a medium-depth chemical peel. J Dermatol Surg Oncol. 1989;15:945-950.
- Hemmi H, Kaisho T, Takeuchi O, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196-200.
- Adamson DJ, Frew D, Tatoud R, et al. Diclofenac antagonizes peroxisome proliferator-activated receptor-gamma signaling. Mol Pharmacol. 2002;61:7-12.
- Alam CA, Seed MP, Willoughby DA. Angiostasis and vascular regression in chronic granulomatous inflammation induced by diclofenac in combination with hyaluronan in mice. J Pharm Pharmacol. 1995;47:407-411.
- Lu X, Xie W, Reed D, et al. Nonsteroidal antiinflammatory drugs cause apoptosis and induce cyclooxygenases in chicken embryo fibroblasts. Proc Natl Acad Sci USA. 1995;92:7961-7965.
- Seed MP, Brown JR, Freemantle CN, et al. The inhibition of colon-26 adenocarcinoma development and angiogenesis by topical diclofenac in 2.5% hyaluronan. Cancer Res. 1997;57:1625-1629.
- Rivers JK, Arlette J, Shear N, et al. Topical treatment of actinic keratoses with 3.0% diclofenac in 2.5% hyaluronan gel. Br J Dermatol. 2002;146:94-100.
- Wolf JE, Taylor JR, Tschen E, et al. Topical 3.0% diclo-fenac in 2.5% hyaluronan gel in the treatment of actinic keratoses. Int J Dermatol. 2001;40:709-713.
- Nelson C, Rigel D, Smith S, et al. Phase IV, open-label assessment of the treatment of actinic keratosis with 3.0% diclofenac sodium topical gel (Solaraze). J Drugs Dermatol. 2004;3:401-407.
- Pflugfelder A, Welter AK, Leiter U, et al. Open label randomized study comparing 3 months vs. 6 months treatment of actinic keratoses with 3% diclofenac in 2.5% hyaluronic acid gel: a trial of the German Dermatologic Cooperative Oncology Group. J Eur Acad Dermatol Venereol. 2012;26:48-53.
- Smith SR, Morhenn VB, Piacquadio DJ. Bilateral comparison of the efficacy and tolerability of 3% diclofenac sodium gel and 5% 5-fluorouracil cream in the treatment of actinic keratoses of the face and scalp. J Drugs Dermatol. 2006;5:156-159.
- Segatto MM, Dornelles SI, Silveira VB, et al. Comparative study of actinic keratosis treatment with 3% diclo- fenac sodium and 5% 5-fluorouracil. An Bras Dermatol. 2013;88:732-738.
- Vidal D. Topical imiquimod: mechanism of action and clinical applications. Mini Rev Med Chem. 2006;6:499-503.
- Hadley G, Derry S, Moore RA. Imiquimod for actinic keratosis: systematic review and meta-analysis. J Invest Dermatol. 2006;126:1251-1255.
- Caperton C, Berman B. Safety, efficacy, and patient acceptability of imiquimod for topical treatment of actinic keratoses. Clin Cosmet Investig Dermatol. 2011;4:35-40.
- Swanson N, Smith CC, Kaur M, et al. Imiquimod 2.5% and 3.75% for the treatment of actinic keratoses: two phase 3, multicenter, randomized, double-blind, placebo-controlled studies. J Drugs Dermatol. 2014;13:166-169.
- Krawtchenko N, Roewert-Huber J, Ulrich M, et al. A randomised study of topical 5% imiquimod vs. topical 5-fluorouracil vs. cryosurgery in immunocompetent patients with actinic keratoses: a comparison of clinical and histological outcomes including 1-year follow-up. Br J Dermatol. 2007;157(suppl 2):34-40.
- Anderson L, Schmieder GJ, Werschler WP, et al. Randomized, double-blind, double-dummy, vehicle-controlled study of ingenol mebutate gel 0.025% and 0.05% for actinic keratosis. J Am Acad Dermatol. 2009;60:934-943.
- Ogbourne SM, Suhrbier A, Jones B, et al. Antitumor activity of 3-ingenyl angelate: plasma membrane and mitochondrial disruption and necrotic cell death. Cancer Res. 2004;64:2833-2839.
- Lebwohl M, Swanson N, Anderson LL, et al. Ingenol mebutate gel for actinic keratosis. N Engl J Med. 2012;366:1010-1019.
- Lebwohl M, Shumack S, Stein-Gold L, et al. Long-term follow-up study of ingenol mebutate gel for the treatment of actinic keratoses. JAMA Dermatol. 2013;149:666-670.
- Augustin M, Tu JH, Knudsen KM, et al. Ingenol mebutate gel for actinic keratosis: the link between quality of life, treatment satisfaction, and clinical outcomes. J Am Acad Dermatol. 2015;72:816-821.
- Kane-Maguire N, Moseley R, Cozzi S, et al. Modulation of fibroblast phenotype and extracellular matrix composition by ingenol mebutate may be associated with scar resolution and improved dermal cosmesis. J Am Acad Dermatol. 2012;66:AB218.
- Martin G, Swanson N. Clinical findings using ingenol mebutate gel to treat actinic keratoses. J Am Acad Dermatol. 2013;68(1, suppl 1):S39-S48.
- Feldman SR, Fleischer AB, Williford PM, et al. Destructive procedures are the standard of care for treatment of actinic keratoses. J Am Acad Dermatol. 1999;40:43-47.
- Berlin JM. Current and emerging treatment strategies for the treatment of actinic keratosis. Clin Cosmet Investig Dermatol. 2010;3:119-126.
- Coleman WP, Yarborough JM, Mandy SH. Dermabrasion for prophylaxis and treatment of actinic keratoses. Dermatol Surg. 1996;22:17-21.
- Cooley JE, Casey DL, Kauffman CL. Manual resurfacing and trichloroacetic acid for the treatment of patients with widespread actinic damage. clinical and histologic observations. Dermatol Surg. 1997;23:373-379.
- Goldberg LH, Kaplan B, Vergilis-Kalner I, et al. Liquid nitrogen: temperature control in the treatment of actinic keratosis. Dermatol Surg. 2010;36:1956-1961.
- Lubritz RR, Smolewski SA. Cryosurgery cure rate of actinic keratoses. J Am Acad Dermatol. 1982;7:631-632.
- Thai KE, Fergin P, Freeman M, et al. A prospective study of the use of cryosurgery for the treatment of actinic keratoses. Int J Dermatol. 2004;43:687-692.
- Zane C, Facchinetti E, Rossi MT, et al. Cryotherapy is preferable to ablative CO2 laser for the treatment of isolated actinic keratoses of the face and scalp: a randomized clinical trial. Br J Dermatol. 2014;170:1114-1121.
- Trimas SJ, Ellis DA, Metz RD. The carbon dioxide laser. an alternative for the treatment of actinically damaged skin. Dermatol Surg. 1997;23:885-889.
- Jiang SB, Levine VJ, Nehal KS, et al. Er:YAG laser for the treatment of actinic keratoses. Dermatol Surg. 2000;26:437-440.
- Ostertag JU, Quaedvlieg PJ, Van der geer S, et al. A clinical comparison and long-term follow-up of topical 5-fluorouracil versus laser resurfacing in the treatment of widespread actinic keratoses. Lasers Surg Med. 2006;38:731-739.
- Iyer S, Friedli A, Bowes L, et al. Full face laser resurfacing: therapy and prophylaxis for actinic keratoses and non-melanoma skin cancer. Lasers Surg Med. 2004;34:114-119.
- Rubin MG. A peeler’s thoughts on skin improvement with chemical peels and laser resurfacing. Clin Plast Surg. 1997;24:407-409.
- Riggs K, Keller M, Humphreys TR. Ablative laser resurfacing: high-energy pulsed carbon dioxide and erbium:yttrium-aluminum-garnet. Clin Dermatol. 2007;25:462-473.
- Adrian RM. Pulsed carbon dioxide and long pulse 10-ms erbium-YAG laser resurfacing: a comparative clinical and histological study. J Cutan Laser Ther. 1999;1:197-202.
- Weiss ET, Brauer JA, Anolik R, et al. 1927-nm fractional resurfacing of facial actinic keratoses: a promising new therapeutic option. J Am Acad Dermatol. 2013; 68:98-102.
- Manstein D, Herron GS, Sink RK, et al. Fractional photothermolysis: a new concept for cutaneous remodeling using microscopic patterns of thermal injury. Lasers Surg Med. 2004;34:426-438.
- Katz TM, Goldberg LH, Marquez D, et al. Nonablative fractional photothermolysis for facial actinic keratoses: 6-month follow-up with histologic evaluation. J Am Acad Dermatol. 2011;65:349-356.
- Prens SP, De Vries K, Neumann HA, et al. Non-ablative fractional resurfacing in combination with topical tretinoin cream as a field treatment modality for multiple actinic keratosis: a pilot study and a review of other field treatment modalities. J Dermatolog Treat. 2013;24:227-231.
- Alexiades-Armenakas MR, Dover JS, Arndt KA. The spectrum of laser skin resurfacing: nonablative, fractional, and ablative laser resurfacing. J Am Acad Dermatol. 2008;58:719-737.
- Tannous Z. Fractional resurfacing. Clin Dermatol. 2007;25:480-486.
- Gold MH. Continuing medical education article-skin treatment: photodynamic therapy: indications and treatment. Aesthet Surg J. 2008;28:545-552.
- Juarranz A, Jaén P, Sanz-Rodríguez F, et al. Photodynamic therapy of cancer. basic principles and applications. Clin Transl Oncol. 2008;10:148-154.
- Juzeniene A, Peng Q, Moan J. Milestones in the development of photodynamic therapy and fluorescence diagnosis. Photochem Photobiol Sci. 2007;6:1234-1245.
- Moan J, Berg K. The photodegradation of porphyrins in cells can be used to estimate the lifetime of singlet oxygen. Photochem Photobiol. 1991;53:549-553.
- Gupta AK, Paquet M, Villanueva E, et al. Interventions for actinic keratoses. Cochrane Database Syst Rev. 2012;12:CD004415.
- Patel G, Armstrong AW, Eisen DB. Efficacy of photodynamic therapy vs other interventions in randomized clinical trials for the treatment of actinic keratoses: a systematic review and meta-analysis. JAMA Dermatol. 2014;150:1281-1288.
- Kaufmann R, Spelman L, Weightman W, et al. Multicentre intraindividual randomized trial of topical methyl aminolaevulinate-photodynamic therapy vs. cryotherapy for multiple actinic keratoses on the extremities. Br J Dermatol. 2008;158:994-999.
- Freeman M, Vinciullo C, Francis D, et al. A comparison of photodynamic therapy using topical methyl aminolevulinate (Metvix) with single cycle cryotherapy in patients with actinic keratosis: a prospective, randomized study. J Dermatolog Treat. 2003;14:99-106.
- Morton C, Campbell S, Gupta G, et al. Intraindividual, right-left comparison of topical methyl aminolaevulinate-photodynamic therapy and cryotherapy in subjects with actinic keratoses: a multicentre, randomized controlled study. Br J Dermatol. 2006;155:1029-1036.
- Pariser DM, Lowe NJ, Stewart DM, et al. Photodynamic therapy with topical methyl aminolevulinate for actinic keratosis: results of a prospective randomized multicenter trial. J Am Acad Dermatol. 2003;48:227-232.
- Zane C, Facchinetti E, Rossi MT, et al. A randomized clinical trial of photodynamic therapy with methyl aminolaevulinate vs. diclofenac 3% plus hyaluronic acid gel for the treatment of multiple actinic keratoses of the face and scalp. Br J Dermatol. 2014;170:1143-1150.
- Perrett CM, McGregor JM, Warwick J, et al. Treatment of post-transplant premalignant skin disease: a randomized intrapatient comparative study of 5-fluorouracil cream and topical photodynamic therapy. Br J Dermatol. 2007;156:320-328.
- Szeimies RM, Karrer S, Radakovic-Fijan S, et al. Photodynamic therapy using topical methyl 5-aminolevulinate compared with cryotherapy for actinic keratosis: a prospective, randomized study. J Am Acad Dermatol. 2002; 47:258-262.
- Scola N, Terras S, Georgas D, et al. A randomized, half-side comparative study of aminolaevulinate photodynamic therapy vs. CO(2) laser ablation in immunocompetent patients with multiple actinic keratoses. Br J Dermatol. 2012;167:1366-1373.
- Willey A, Anderson RR, Sakamoto FH. Temperature-modulated photodynamic therapy for the treatment of actinic keratosis on the extremities: a pilot study. Dermatol Surg. 2014;40:1094-1102.
- Pariser DM. Management of Actinic Keratoses: Treatment Selection and Optimizing Outcomes. Presented at: Winter Clinical Dermatology Conference Hawaii; January 18, 2015; Kaanapali, HI.
- Dirschka T, Radny P, Dominicus R, et al. Long-term (6 and 12 months) follow-up of two prospective, randomized, controlled phase III trials of photodynamic therapy with BF-200 ALA and methyl aminolaevulinate for the treatment of actinic keratosis. Br J Dermatol. 2013;168:825-836.
- Choi SH, Kim KH, Song KH. Efficacy of ablative fractional laser-assisted photodynamic therapy with short-incubation time for the treatment of facial and scalp actinic keratosis: 12-month follow-up results of a randomized, prospective, comparative trial. J Eur Acad Dermatol Venereol. 2015;29:1598-1605.
- Ko DY, Jeon SY, Kim KH, et al. Fractional erbium:YAG laser-assisted photodynamic therapy for facial actinic keratoses: a randomized, comparative, prospective study. J Eur Acad Dermatol Venereol. 2014;28:1529-1539.
- Togsverd-Ho K, Haak CS, Thaysen-Petersen D, et al. Intensified photodynamic therapy of actinic keratoses with fractional CO2 laser: a randomized clinical trial. Br J Dermatol. 2012;166:1262-1269.
- Torezan L, Chaves Y, Niwa A, et al. A pilot split-face study comparing conventional methyl aminolevulinate-photodynamic therapy (PDT) with microneedling-assisted PDT on actinically damaged skin. Dermatol Surg. 2013;39:1197-1201.
Actinic keratosis (AK), also referred to as solar keratosis or senile keratosis, is an intraepidermal proliferation of dysplastic keratinocytes that develops in response to chronic exposure to UV radiation. Actinic keratoses are among the most commonly encountered lesions seen by dermatologists, and it has been estimated that 60% of predisposed individuals older than 40 years have at least one AK.1,2 Prevalence is notably higher in light-skinned individuals and increases with age, presumably from higher cumulative sun exposure and decreased effectiveness of the immune system.1,3 It remains a point of contention as to whether or not AKs actually represent squamous cell carcinoma (SCC) in situ, but the potential for progression to invasive disease has been well demonstrated, as the majority of SCCs develop from preexisting AKs.4-6 The risk for progression to invasive disease for an individual AK has been estimated to range from 0.025% to 16% per year, with an average of approximately 8% in immunocompetent patients.7
The clinical morphology of AK can vary widely, but the most common presentation is an erythematous scaly macule, papule, or plaque on sun-exposed skin. The skin surrounding AKs typically shows evidence of solar damage with deep wrinkling, mottled pigmentation, scattered telangiectases, purpura, or xerosis (Figure). A variety of clinical variants with unique presentations exist, including atrophic, hypertrophic, acantholytic, lichenoid, bowenoid, and pigmented subtypes. Because more than 80% of AKs occur on highly visible areas such as the head, neck, back of the hands, and forearms, AKs can have an obvious detrimental effect on cosmetic appearance. Studies also have shown a strong association between AKs and decreased overall quality of life (QOL).3,8,9
Because of the risk for AK progression to invasive cancer along with its negative impact on cosmesis and QOL, clinicians generally opt to treat AKs. Numerous different treatment options exist, including topical medications, procedural modalities, and light-based therapies. Here, we review the efficacy of the most commonly utilized treatments and discuss the relevant cosmetic considerations and outcomes.
Topical Treatments
5-Fluorouracil
5-Fluorouracil (5-FU) is a US Food and Drug Administration (FDA)–approved, topically applied pyrimidine analogue that inhibits thymidylate synthase. The resulting suppression of DNA and RNA synthesis induces cell death with a preference for mitotically active cells.10 5-Fluorouracil has been used for more than 50 years as a treatment of AK and its efficacy is well established. A systematic review of 5 randomized controlled studies of topical 5-FU reported an average of 49% of 423 patients achieving complete lesion clearance with 5-FU cream 5% applied once or twice daily for up to 7 weeks.11 Some notable drawbacks of 5-FU, however, are application-site erythema, blistering, pruritus, necrosis, erosion, and pain. These effects often lead to premature cessation of therapy, but newer formulations of 5-FU cream 0.5% have shown good efficacy with better tolerability.12 A randomized, double-blind, multicenter, parallel-group study of 177 patients using 5-FU cream 0.5% once daily for either 1, 2, or 4 weeks demonstrated significant (P<.001) efficacy over vehicle gel in all treatment arms.13 The most effective therapy was 4 weeks of treatment, which achieved a mean 91.7% reduction in lesion count as assessed 1 month after cessation of therapy. The primary adverse effect (AE) reported in this trial was mild to moderate facial irritation, which generally resolved within 18 to 21 days after treatment cessation.13 Overall, 5-FU is a highly effective therapy for treating AKs that also can improve signs of photoaging, but patients should be aware of cosmetically unappealing effects that generally occur throughout therapy and during the immediate posttreatment period.14
Chemical Peels
Chemical peels traditionally employ acidic compounds to strip away outer layers of skin to variable depths depending on the concentration of the agent being applied. For treatment of AK, trichloroacetic acid (TCA) is a commonly employed cauterant that has shown efficacy comparable to topical 5-FU as well as ablative CO2 laser resurfacing.15 Trichloroacetic acid peels also are a convenient therapy, as good results can be achieved after a single treatment session. A split-face study of 15 patients treated with either a single application of 35% TCA and Jessner solution or twice-daily application of 5-FU cream 5% for 3 weeks demonstrated a reduction in 75% of visible AKs in both treatment arms over a 1-year follow-up period.16 Although 80% of patients self-reported considerable cosmetic improvement with both therapies, patient preference was reported to be in favor of the TCA peel, given its quick results and relatively mild side effects as compared to 5-FU. Treatment with chemical peels will result in temporary erythema and mild desquamation that usually resolves within 2 weeks; however, there are cases in which erythema has been reported to persist for several months.16 Adverse effects such as permanent scarring or pigmentation changes rarely are seen with TCA concentrations less than 45%.17 Caution should be used in patients with a history of herpes simplex virus, keloids, postinflammatory hyperpigmentation, radiation exposure, immunosuppression, and those unable or unwilling to use sunscreen and avoid sun exposure in the immediate posttreatment period.
Diclofenac Sodium
Diclofenac sodium (DFS) is an FDA-approved topical, nonsteroidal,
anti-inflammatory drug whose mechanism of action in the treatment of AK is thought to involve inhibition of the cyclooxygenase 2 enzyme.18 The resulting reduction of prostaglandins is believed to inhibit tumor angiogenesis, induce apoptosis, and inhibit cell differentiation.19-22 In a multicenter, double-blind, placebo-controlled study of 195 patients, application of DFS 3% in hyaluronan gel 2.5% twice daily for 60 days showed significant (P<.05) efficacy over placebo in achieving complete resolution of target lesions during a 30-day follow-up period (31% vs 10%). Furthermore, qualitative patient assessment of complete global improvement also was significantly (P<.05) higher in the active treatment group as compared to placebo (31% vs 10%).23 Additional studies of DFS 3% in hyaluronan gel 2.5% applied twice daily for 90 days have shown even higher rates of success, with complete resolution of target lesions in 40% to 58% of cases.24,25 This therapy also has been reported to substantially improve QOL following treatment completion.26 The most frequently cited AEs include pruritus, rash, dry skin, erythema, and application-site reactions. Overall, DFS is a
well-tolerated therapy with efficacy comparable to that of 5-FU but with a lower incidence of AEs
and higher patient satisfaction as determined in
2 head-to-head studies.27,28
ImiquimodImiquimod (IMQ) is an FDA-approved topical agent that functions as an immune response modifier via agonism of toll-like receptor 7.18 The resulting cytokine production and release enhances the innate and acquired immune responses leading to anticancer activity.29 The efficacy of IMQ for treatment of AK has been demonstrated in numerous well-designed clinical trials. A
meta-analysis of 5 randomized, double-blind trials including 1293 patients treated with IMQ cream 5%
2 to 3 times per week for 12 to 16 weeks reported complete clearance of AKs in 50% of patients treated with IMQ as compared to 5% of patients treated with vehicle.30 The most frequently reported AEs with this therapy include erythema, scabbing, flaking, and erosion. These effects generally resolve following cessation of treatment, and therapy is considered to be well tolerated; however, there are case reports of IMQ triggering or exacerbating existing inflammatory conditions.31 Imiquimod cream also is approved at 2.5% and 3.75% concentrations, which have demonstrated significant (P<.001) efficacy over placebo and a reduced incidence of AEs; complete clearance rates have been reported as 30.6% and 35.6%, respectively.32 Notably, a study comparing 75 patients randomized to either IMQ cream 5%
3 times per week for 4 weeks, 1 or 2 courses of cryosurgery, or 5-FU ointment 5% twice daily for 4 weeks reported that IMQ achieved significantly (P<.01) superior sustained clearance rates during a 12-month follow-up period over cryosurgery and 5-FU
(73% vs 4% vs 33%).33 Additionally, cosmetic outcomes as determined by both participants and investigators were reported as excellent at 12 months posttreatment in more than 80% of participants treated with IMQ. These excellent, long-lasting cosmetic outcomes also were determined to be significantly (P<.0001) superior to the cosmetic outcomes of 5-FU and cryotherapy, which both reported excellent outcomes in less than 10% of cases.33
Ingenol MebutateIngenol mebutate (IM) is a macrocyclic diterpene ester derived from the Euphorbia peplus plant that is FDA approved for the treatment of AK.1 Ingenol mebutate’s mechanism of action is thought to involve induction of cell death via disruption of the plasma membrane and mitochondria in addition to production of an inflammatory response, which produces tumor-specific antibodies and a large influx of neutrophils.34,35 The overall evidence for the efficacy of IM is strong. A combined analysis of 4 multicenter, randomized, double-blind studies of 1005 participants reported that IM gel 0.015% applied once daily for 3 days to the face or scalp was significantly superior (P<.001) to placebo in achieving complete clearance as assessed 54 days after completion of therapy (42.2% vs 3.7%) and that IM gel 0.05% applied once daily for 2 days to the trunk or extremities also was significantly superior (P<.001) to placebo in achieving complete clearance as determined 55 days after completion of therapy (34.1% vs 4.7%).36 A follow-up report to this study indicated that IM also appears to achieve long-lasting effects with an overall 87% decrease in total AKs at 12 months follow-up in both trial groups.37 Additionally, it has been recently reported that treatment with IM in these trials was associated with significantly higher overall treatment satisfaction (P<.001) and improved QOL (P<.001) as compared to vehicle.38 Cosmetic outcomes of IM therapy have been assessed in a trial analyzing the efficacy of IM gel 0.025% for 3 days or IM gel 0.05% for 2 or 3 days on nonfacial AKs. This study reported significantly (P<.0001) higher patient satisfaction with the cosmetic outcome at 8 weeks after therapy as compared to vehicle.34 Studies performed in mice have demonstrated that IM is able to promote collagen matrix turnover and impose dermal elasticity, which may contribute to these good cosmetic outcomes.39 The most common AEs of IM therapy are erythema, crusting, and flaking; these effects generally occur 3 to 8 days after starting treatment. These effects, however, generally are short lived and resolve within 2 weeks of treatment cessation when IM is applied to the face or scalp or 4 weeks when applied to the trunk or extremities.40 Overall, IM is a useful therapeutic option given its relatively short treatment course as compared to other topically applied agents, as well as its lasting efficacy, mild AEs, and good cosmetic outcomes.
Procedural Modalities
Surgical Procedures
Surgical approaches for the treatment of AK include excision, curettage with or without electrodesiccation, and dermabrasion. In the past, these modalities were used with greater frequency, but the advent of effective topical medications with lower risks of AEs has largely reduced their use.41 Excision may still be indicated in cases where SCC is suspected, and curettage can be used for treatment of thicker hypertrophic AKs.42 Although these approaches have not been evaluated in clinical trials, they are generally effective but require the use of local anesthetics and come with substantial risk for infection, permanent scarring, and hypopigmentation. Dermabrasion employs the use of a motorized device equipped with an abrasive material to physically remove superficial layers of the skin. Studies are limited, but this method has been reported as an effective treatment in a retrospective review of 23 participants in which 96% remained free of AKs at 1 year, 83% at 2 years, 64% at 4 years, and 54% at 5 years posttherapy.43 Notably, one split-face study of 40 participants treated with dermabrasion followed by 25% TCA on one side and either Jessner solution and 35% TCA or dermabrasion alone on the other side reported that the combination of dermabrasion with 25% TCA consistently produced excellent cosmetic results with nearly complete eradication of AKs.44 In general, however, cosmetic outcomes with dermabrasion are variable, as the technique is highly operator dependent and treatment is associated with notable discomfort as well as risk for scarring and permanent pigmentation alteration.
Cryotherapy
Cryotherapy remains one of the most commonly utilized treatments of AK and involves the delivery of liquid nitrogen via a spray device or a cotton tip applicator to rapidly freeze cells, thus causing cellular destruction via ice crystal formation and protein denaturation.45 Efficacy with this technique has been reported to be as high as 98.8% at 12 months follow-up, but more recent studies cite lower rates of success.46 A prospective multicenter study of 90 participants with 421 AKs on the face or scalp treated with a single freeze-thaw cycle of liquid nitrogen reported an overall complete response rate of 67.2% at 3 months posttherapy. Additionally, higher complete response rates were associated with longer freeze times, and cosmetic outcomes were reported as good to excellent in 94% of complete response lesions.47 Similar results were reported in an open-label, prospective, randomized, controlled clinical trial of 200 participants with 543 AKs, which compared a single freeze-thaw cycle with liquid nitrogen to a single session of CO2 laser ablation in the treatment of isolated AKs of the face and scalp.48 At 3 months posttherapy, complete clearance was observed in 71.6% of participants treated with cryotherapy and in 65.3% of participants treated with laser ablation (P=.532). At 12 months posttherapy, participants who originally showed complete response at 3 months were assessed for relapse. Complete clearance was preserved in 72.6% of participants treated with cryotherapy versus 21.9% of participants treated with laser ablation (P<.0001), and cosmetic outcomes were reported by participants as good or excellent at 3 months follow-up in more than 93% of participants for both treatment arms.48 Possible AEs of cryotherapy include pain during treatment, blister formation with possible hemorrhage, infection, scarring, and permanent pigmentary changes.47,48 Notably, the risk for hypopigmentation increases with longer freezing times, thus requiring clinicians to consider the balance between improved efficacy and reduced cosmetic outcomes.47
Light-Based Therapies
Laser Therapy
Ablative laser resurfacing with either the CO2 or erbium-doped:YAG (Er:YAG) laser utilizes light of specific wavelengths to selectively induce thermolysis and destruction of the epidermal layer. Both lasers have been studied as treatments of AK, but there is a lack of large, well-designed studies. In one small study of 14 participants treated with 1 to 2 passes of the CO2 laser, complete clearance was reported in all cases without any recurrences during a follow-up period of 6 to 24 months. Additionally, all participants in this study reported satisfaction with the cosmetic outcome.49 The CO2 laser also has demonstrated efficacy comparable to that of the TCA peel and 5-FU therapy in a prospective randomized trial of 34 patients with facial or scalp AKs who received either CO2 laser with 2 passes, 30% TCA peel, or 5-FU cream 5% twice daily for 3 weeks.15 Reduction in mean AK counts at 3 months posttherapy was significantly (P<.03) higher in all treatment arms as compared to the control group (92% for CO2 laser, 89% for TCA peel, and 83% for 5-FU cream). No significant (P=.31) difference in outcomes was noted among the different treatment arms.15 Similar results were reported for the Er:YAG laser in a small prospective study of 5 participants treated with 2 to 3 passes with the Er:YAG laser in which reduction in mean AK counts was reported as ranging from 86% to 96% at 3 months posttherapy.50 The Er:YAG laser in combination with the CO2 laser has shown notable long-term efficacy in achieving higher lesion clearance rates and sustained complete clearance rates over treatment with topical 5-FU.51 In a prospective randomized study of 55 par-ticipants with multiple AKs on the face or scalp, participants were assigned to receive either combination laser ablation with the Er:YAG and CO2 lasers down to the level of the papillary dermis or 5-FU cream 5% applied twice daily for 2 to 7 weeks until an appropriate clinical inflammatory response was achieved. At 12 months follow-up, the laser treatment group achieved significantly (P=.048) higher mean lesion clearance rates (91.1%) as compared to the 5-FU arm (76.6%) and significantly (P=.003) higher sustained complete clearance rates (59.3%) as compared to 5-FU (29.2%). The proportion of participants with an improvement in photoaging score at 12 months follow-up approached statistical significance (P=.07), with 74% of the laser-treated group showing improvement as compared to 43% of the 5-FU–treated group. Long-term, cosmetically unappealing side effects such as erythema and hypopigmentation occurred notably more often in the laser-treated group as compared to the 5-FU group.51 In summary, ablative lasers appear to be a highly effective therapy for AK but at the cost of increased risk for AEs such as permanent pigmentary changes, prolonged erythema lasting up to several months, and scarring.50,52-55
Fractional photothermolysis is a relatively new advancement in the field of laser therapy that has received FDA approval for the treatment of AK.56 This treatment works by creating multiple noncontiguous microscopic columns of thermal injury while sparing adjacent zones of viable tissue.57 Although there are limited studies involving the use of such lasers in the treatment of AK, initial findings suggest that 1927-nm thulium lasers may be more effective than 1550-nm erbium lasers in achieving lesion clearance. A trial of 14 participants who received 5 laser treatments with a 1550-nm fractionated erbium-doped fiber laser reported an average reduction in AK counts of 66.2% at 3 months follow-up and a 55.6% reduction at 6 months follow-up. A participant-determined marked or very significant improvement of lesions was reported in 83% of participants at 1 month posttreatment but only in 44% of participants at 6 months posttreatment.58 A similar trial of 24 participants treated with up to 4 treatment sessions of the fractionated 1927-nm thulium laser reported an 87.3% reduction in number of AKs at 3 months follow-up and an 86.6% reduction at 6 months follow-up.56 The primary advantage of fractional laser therapy is a faster recovery period generally lasting only 2 or 3 days as compared to 2 weeks or more with traditional ablative lasers, thus limiting the amount of time a patient must tolerate cosmetically unappealing erythema.59,60 The quick recovery time has been attributed to the fractional laser’s ability to preserve the stratum corneum and skin barrier, which also helps reduce the risk for other AEs such as scarring and infection.56,59-61 Additional studies are needed to better assess the true efficacy of fractional laser therapy, but treatment with the fractional 1927-nm thulium laser appears to be a promising and well-tolerated therapeutic option for treatment of AK with similar efficacy to traditional ablative lasers but with a lower risk of AEs.
Photodynamic TherapyPhotodynamic therapy (PDT) is an FDA-approved treatment that involves the use of a topical photosensitizing agent such as 5-aminolevulinic acid (ALA) or methyl aminovulinate (MAL) before exposure to an activating light source to generate reactive oxygen species that lead to cell death.62-65 Multiple PDT regimens with varying combinations of photosensitizers, incubation time, and light sources have been studied, but a 2012 Cochrane review determined that treatment with conventional formulations of MAL and ALA with either blue- or red-light PDT were similarly efficacious for treatment of individual AKs as compared to vehicle with blue- or red-light PDT. One exception was that longer incubation time (ie, 4 hours) with ALA resulted in better results than shorter incubation times (ie, 0.5, 1, 2 hours) with ALA.66
Standard PDT treatment with MAL also has consistently demonstrated superior efficacy in achieving complete clearance rates in addition to superior cosmetic outcomes over treatment with either cryotherapy, DFS, or 5-FU.67-73 Three studies in particular noted an excellent or good investigator-determined cosmetic outcome in 96% to 98% of participants treated with MAL-PDT.69,71,74 Photodynamic therapy with ALA also has been reported as superior over CO2 laser ablation for AK reduction as well as both patient and investigator overall satisfaction.75
More recently, several methods of improving photosensitizer delivery have been studied, which have demonstrated remarkable efficacy at achieving lesion clearance over standard cream formulations or application routines. One such method involves the use of gentle heating to increase photosensitizer uptake. In a split-extremity study of 20 participants who were treated with 20% ALA under occlusion for 1 hour with one side heated to 38.8°C, the heated side demonstrated significant (P<.0001) efficacy at achieving higher median clearance rates over control when evaluated at 2 and 6 months posttherapy.76 Notably, occlusion of ALA in itself during the incubation period also has been demonstrated to significantly (P<.0001) improve clearance rates.77 Another method involves the use of a new nanoemulsion-based formulation of ALA gel, known as BF-200 ALA, which has demonstrated remarkable efficacy over standard MAL cream and placebo in a long-term follow-up analysis of 2 prospective, randomized, controlled trials.78 In a similar vein, 3 prospective randomized trials with a minimum follow-up time of 3 months demonstrated that MAL-PDT in combination with fractional ablative laser pretreatment has significant (P<.02 in all trials) efficacy over MAL-PDT without pretreatment in achieving complete AK clearance. Although the cosmetic outcomes were good or excellent in 87% to 100% of patients, they were not significantly different from stand-alone MAL-PDT treatment in any of the trials.79-81 However, pretreatment with microneedling in MAL-PDT has been shown to achieve superior cosmetic outcomes over MAL-PDT without microneedling, according to one small split-face study of 10 participants.82
Overall, PDT is an excellent therapeutic option that is able to provide efficacious clearance of AKs as well as superior cosmetic outcomes. Common AEs of PDT include burning, itching, and stinging during therapy, but pain intensity decreases dramatically upon termination of illumination, with cessation of most symptoms by 12 hours posttherapy.73 Permanent pigmentation changes have been reported to occasionally occur following PDT therapy.81
Conclusion
When determining which therapy to use in a patient, clinicians must take into account a variety of factors such as patient preference, cost of treatment, availability, tolerance for AEs, and the need for field therapy. Although all therapies discussed within this article are effective and reasonable treatment choices, patients who are particularly concerned about cosmetic outcomes would most likely benefit from either IMQ or PDT, as the data for cosmetic outcomes with these therapies are the strongest. Combination or sequential treatments may be required in some cases and all patients should be monitored for lesion recurrence regardless of treatment choice. A summary of the therapies and key studies discussed here is available in the PDF.
Actinic keratosis (AK), also referred to as solar keratosis or senile keratosis, is an intraepidermal proliferation of dysplastic keratinocytes that develops in response to chronic exposure to UV radiation. Actinic keratoses are among the most commonly encountered lesions seen by dermatologists, and it has been estimated that 60% of predisposed individuals older than 40 years have at least one AK.1,2 Prevalence is notably higher in light-skinned individuals and increases with age, presumably from higher cumulative sun exposure and decreased effectiveness of the immune system.1,3 It remains a point of contention as to whether or not AKs actually represent squamous cell carcinoma (SCC) in situ, but the potential for progression to invasive disease has been well demonstrated, as the majority of SCCs develop from preexisting AKs.4-6 The risk for progression to invasive disease for an individual AK has been estimated to range from 0.025% to 16% per year, with an average of approximately 8% in immunocompetent patients.7
The clinical morphology of AK can vary widely, but the most common presentation is an erythematous scaly macule, papule, or plaque on sun-exposed skin. The skin surrounding AKs typically shows evidence of solar damage with deep wrinkling, mottled pigmentation, scattered telangiectases, purpura, or xerosis (Figure). A variety of clinical variants with unique presentations exist, including atrophic, hypertrophic, acantholytic, lichenoid, bowenoid, and pigmented subtypes. Because more than 80% of AKs occur on highly visible areas such as the head, neck, back of the hands, and forearms, AKs can have an obvious detrimental effect on cosmetic appearance. Studies also have shown a strong association between AKs and decreased overall quality of life (QOL).3,8,9
Because of the risk for AK progression to invasive cancer along with its negative impact on cosmesis and QOL, clinicians generally opt to treat AKs. Numerous different treatment options exist, including topical medications, procedural modalities, and light-based therapies. Here, we review the efficacy of the most commonly utilized treatments and discuss the relevant cosmetic considerations and outcomes.
Topical Treatments
5-Fluorouracil
5-Fluorouracil (5-FU) is a US Food and Drug Administration (FDA)–approved, topically applied pyrimidine analogue that inhibits thymidylate synthase. The resulting suppression of DNA and RNA synthesis induces cell death with a preference for mitotically active cells.10 5-Fluorouracil has been used for more than 50 years as a treatment of AK and its efficacy is well established. A systematic review of 5 randomized controlled studies of topical 5-FU reported an average of 49% of 423 patients achieving complete lesion clearance with 5-FU cream 5% applied once or twice daily for up to 7 weeks.11 Some notable drawbacks of 5-FU, however, are application-site erythema, blistering, pruritus, necrosis, erosion, and pain. These effects often lead to premature cessation of therapy, but newer formulations of 5-FU cream 0.5% have shown good efficacy with better tolerability.12 A randomized, double-blind, multicenter, parallel-group study of 177 patients using 5-FU cream 0.5% once daily for either 1, 2, or 4 weeks demonstrated significant (P<.001) efficacy over vehicle gel in all treatment arms.13 The most effective therapy was 4 weeks of treatment, which achieved a mean 91.7% reduction in lesion count as assessed 1 month after cessation of therapy. The primary adverse effect (AE) reported in this trial was mild to moderate facial irritation, which generally resolved within 18 to 21 days after treatment cessation.13 Overall, 5-FU is a highly effective therapy for treating AKs that also can improve signs of photoaging, but patients should be aware of cosmetically unappealing effects that generally occur throughout therapy and during the immediate posttreatment period.14
Chemical Peels
Chemical peels traditionally employ acidic compounds to strip away outer layers of skin to variable depths depending on the concentration of the agent being applied. For treatment of AK, trichloroacetic acid (TCA) is a commonly employed cauterant that has shown efficacy comparable to topical 5-FU as well as ablative CO2 laser resurfacing.15 Trichloroacetic acid peels also are a convenient therapy, as good results can be achieved after a single treatment session. A split-face study of 15 patients treated with either a single application of 35% TCA and Jessner solution or twice-daily application of 5-FU cream 5% for 3 weeks demonstrated a reduction in 75% of visible AKs in both treatment arms over a 1-year follow-up period.16 Although 80% of patients self-reported considerable cosmetic improvement with both therapies, patient preference was reported to be in favor of the TCA peel, given its quick results and relatively mild side effects as compared to 5-FU. Treatment with chemical peels will result in temporary erythema and mild desquamation that usually resolves within 2 weeks; however, there are cases in which erythema has been reported to persist for several months.16 Adverse effects such as permanent scarring or pigmentation changes rarely are seen with TCA concentrations less than 45%.17 Caution should be used in patients with a history of herpes simplex virus, keloids, postinflammatory hyperpigmentation, radiation exposure, immunosuppression, and those unable or unwilling to use sunscreen and avoid sun exposure in the immediate posttreatment period.
Diclofenac Sodium
Diclofenac sodium (DFS) is an FDA-approved topical, nonsteroidal,
anti-inflammatory drug whose mechanism of action in the treatment of AK is thought to involve inhibition of the cyclooxygenase 2 enzyme.18 The resulting reduction of prostaglandins is believed to inhibit tumor angiogenesis, induce apoptosis, and inhibit cell differentiation.19-22 In a multicenter, double-blind, placebo-controlled study of 195 patients, application of DFS 3% in hyaluronan gel 2.5% twice daily for 60 days showed significant (P<.05) efficacy over placebo in achieving complete resolution of target lesions during a 30-day follow-up period (31% vs 10%). Furthermore, qualitative patient assessment of complete global improvement also was significantly (P<.05) higher in the active treatment group as compared to placebo (31% vs 10%).23 Additional studies of DFS 3% in hyaluronan gel 2.5% applied twice daily for 90 days have shown even higher rates of success, with complete resolution of target lesions in 40% to 58% of cases.24,25 This therapy also has been reported to substantially improve QOL following treatment completion.26 The most frequently cited AEs include pruritus, rash, dry skin, erythema, and application-site reactions. Overall, DFS is a
well-tolerated therapy with efficacy comparable to that of 5-FU but with a lower incidence of AEs
and higher patient satisfaction as determined in
2 head-to-head studies.27,28
ImiquimodImiquimod (IMQ) is an FDA-approved topical agent that functions as an immune response modifier via agonism of toll-like receptor 7.18 The resulting cytokine production and release enhances the innate and acquired immune responses leading to anticancer activity.29 The efficacy of IMQ for treatment of AK has been demonstrated in numerous well-designed clinical trials. A
meta-analysis of 5 randomized, double-blind trials including 1293 patients treated with IMQ cream 5%
2 to 3 times per week for 12 to 16 weeks reported complete clearance of AKs in 50% of patients treated with IMQ as compared to 5% of patients treated with vehicle.30 The most frequently reported AEs with this therapy include erythema, scabbing, flaking, and erosion. These effects generally resolve following cessation of treatment, and therapy is considered to be well tolerated; however, there are case reports of IMQ triggering or exacerbating existing inflammatory conditions.31 Imiquimod cream also is approved at 2.5% and 3.75% concentrations, which have demonstrated significant (P<.001) efficacy over placebo and a reduced incidence of AEs; complete clearance rates have been reported as 30.6% and 35.6%, respectively.32 Notably, a study comparing 75 patients randomized to either IMQ cream 5%
3 times per week for 4 weeks, 1 or 2 courses of cryosurgery, or 5-FU ointment 5% twice daily for 4 weeks reported that IMQ achieved significantly (P<.01) superior sustained clearance rates during a 12-month follow-up period over cryosurgery and 5-FU
(73% vs 4% vs 33%).33 Additionally, cosmetic outcomes as determined by both participants and investigators were reported as excellent at 12 months posttreatment in more than 80% of participants treated with IMQ. These excellent, long-lasting cosmetic outcomes also were determined to be significantly (P<.0001) superior to the cosmetic outcomes of 5-FU and cryotherapy, which both reported excellent outcomes in less than 10% of cases.33
Ingenol MebutateIngenol mebutate (IM) is a macrocyclic diterpene ester derived from the Euphorbia peplus plant that is FDA approved for the treatment of AK.1 Ingenol mebutate’s mechanism of action is thought to involve induction of cell death via disruption of the plasma membrane and mitochondria in addition to production of an inflammatory response, which produces tumor-specific antibodies and a large influx of neutrophils.34,35 The overall evidence for the efficacy of IM is strong. A combined analysis of 4 multicenter, randomized, double-blind studies of 1005 participants reported that IM gel 0.015% applied once daily for 3 days to the face or scalp was significantly superior (P<.001) to placebo in achieving complete clearance as assessed 54 days after completion of therapy (42.2% vs 3.7%) and that IM gel 0.05% applied once daily for 2 days to the trunk or extremities also was significantly superior (P<.001) to placebo in achieving complete clearance as determined 55 days after completion of therapy (34.1% vs 4.7%).36 A follow-up report to this study indicated that IM also appears to achieve long-lasting effects with an overall 87% decrease in total AKs at 12 months follow-up in both trial groups.37 Additionally, it has been recently reported that treatment with IM in these trials was associated with significantly higher overall treatment satisfaction (P<.001) and improved QOL (P<.001) as compared to vehicle.38 Cosmetic outcomes of IM therapy have been assessed in a trial analyzing the efficacy of IM gel 0.025% for 3 days or IM gel 0.05% for 2 or 3 days on nonfacial AKs. This study reported significantly (P<.0001) higher patient satisfaction with the cosmetic outcome at 8 weeks after therapy as compared to vehicle.34 Studies performed in mice have demonstrated that IM is able to promote collagen matrix turnover and impose dermal elasticity, which may contribute to these good cosmetic outcomes.39 The most common AEs of IM therapy are erythema, crusting, and flaking; these effects generally occur 3 to 8 days after starting treatment. These effects, however, generally are short lived and resolve within 2 weeks of treatment cessation when IM is applied to the face or scalp or 4 weeks when applied to the trunk or extremities.40 Overall, IM is a useful therapeutic option given its relatively short treatment course as compared to other topically applied agents, as well as its lasting efficacy, mild AEs, and good cosmetic outcomes.
Procedural Modalities
Surgical Procedures
Surgical approaches for the treatment of AK include excision, curettage with or without electrodesiccation, and dermabrasion. In the past, these modalities were used with greater frequency, but the advent of effective topical medications with lower risks of AEs has largely reduced their use.41 Excision may still be indicated in cases where SCC is suspected, and curettage can be used for treatment of thicker hypertrophic AKs.42 Although these approaches have not been evaluated in clinical trials, they are generally effective but require the use of local anesthetics and come with substantial risk for infection, permanent scarring, and hypopigmentation. Dermabrasion employs the use of a motorized device equipped with an abrasive material to physically remove superficial layers of the skin. Studies are limited, but this method has been reported as an effective treatment in a retrospective review of 23 participants in which 96% remained free of AKs at 1 year, 83% at 2 years, 64% at 4 years, and 54% at 5 years posttherapy.43 Notably, one split-face study of 40 participants treated with dermabrasion followed by 25% TCA on one side and either Jessner solution and 35% TCA or dermabrasion alone on the other side reported that the combination of dermabrasion with 25% TCA consistently produced excellent cosmetic results with nearly complete eradication of AKs.44 In general, however, cosmetic outcomes with dermabrasion are variable, as the technique is highly operator dependent and treatment is associated with notable discomfort as well as risk for scarring and permanent pigmentation alteration.
Cryotherapy
Cryotherapy remains one of the most commonly utilized treatments of AK and involves the delivery of liquid nitrogen via a spray device or a cotton tip applicator to rapidly freeze cells, thus causing cellular destruction via ice crystal formation and protein denaturation.45 Efficacy with this technique has been reported to be as high as 98.8% at 12 months follow-up, but more recent studies cite lower rates of success.46 A prospective multicenter study of 90 participants with 421 AKs on the face or scalp treated with a single freeze-thaw cycle of liquid nitrogen reported an overall complete response rate of 67.2% at 3 months posttherapy. Additionally, higher complete response rates were associated with longer freeze times, and cosmetic outcomes were reported as good to excellent in 94% of complete response lesions.47 Similar results were reported in an open-label, prospective, randomized, controlled clinical trial of 200 participants with 543 AKs, which compared a single freeze-thaw cycle with liquid nitrogen to a single session of CO2 laser ablation in the treatment of isolated AKs of the face and scalp.48 At 3 months posttherapy, complete clearance was observed in 71.6% of participants treated with cryotherapy and in 65.3% of participants treated with laser ablation (P=.532). At 12 months posttherapy, participants who originally showed complete response at 3 months were assessed for relapse. Complete clearance was preserved in 72.6% of participants treated with cryotherapy versus 21.9% of participants treated with laser ablation (P<.0001), and cosmetic outcomes were reported by participants as good or excellent at 3 months follow-up in more than 93% of participants for both treatment arms.48 Possible AEs of cryotherapy include pain during treatment, blister formation with possible hemorrhage, infection, scarring, and permanent pigmentary changes.47,48 Notably, the risk for hypopigmentation increases with longer freezing times, thus requiring clinicians to consider the balance between improved efficacy and reduced cosmetic outcomes.47
Light-Based Therapies
Laser Therapy
Ablative laser resurfacing with either the CO2 or erbium-doped:YAG (Er:YAG) laser utilizes light of specific wavelengths to selectively induce thermolysis and destruction of the epidermal layer. Both lasers have been studied as treatments of AK, but there is a lack of large, well-designed studies. In one small study of 14 participants treated with 1 to 2 passes of the CO2 laser, complete clearance was reported in all cases without any recurrences during a follow-up period of 6 to 24 months. Additionally, all participants in this study reported satisfaction with the cosmetic outcome.49 The CO2 laser also has demonstrated efficacy comparable to that of the TCA peel and 5-FU therapy in a prospective randomized trial of 34 patients with facial or scalp AKs who received either CO2 laser with 2 passes, 30% TCA peel, or 5-FU cream 5% twice daily for 3 weeks.15 Reduction in mean AK counts at 3 months posttherapy was significantly (P<.03) higher in all treatment arms as compared to the control group (92% for CO2 laser, 89% for TCA peel, and 83% for 5-FU cream). No significant (P=.31) difference in outcomes was noted among the different treatment arms.15 Similar results were reported for the Er:YAG laser in a small prospective study of 5 participants treated with 2 to 3 passes with the Er:YAG laser in which reduction in mean AK counts was reported as ranging from 86% to 96% at 3 months posttherapy.50 The Er:YAG laser in combination with the CO2 laser has shown notable long-term efficacy in achieving higher lesion clearance rates and sustained complete clearance rates over treatment with topical 5-FU.51 In a prospective randomized study of 55 par-ticipants with multiple AKs on the face or scalp, participants were assigned to receive either combination laser ablation with the Er:YAG and CO2 lasers down to the level of the papillary dermis or 5-FU cream 5% applied twice daily for 2 to 7 weeks until an appropriate clinical inflammatory response was achieved. At 12 months follow-up, the laser treatment group achieved significantly (P=.048) higher mean lesion clearance rates (91.1%) as compared to the 5-FU arm (76.6%) and significantly (P=.003) higher sustained complete clearance rates (59.3%) as compared to 5-FU (29.2%). The proportion of participants with an improvement in photoaging score at 12 months follow-up approached statistical significance (P=.07), with 74% of the laser-treated group showing improvement as compared to 43% of the 5-FU–treated group. Long-term, cosmetically unappealing side effects such as erythema and hypopigmentation occurred notably more often in the laser-treated group as compared to the 5-FU group.51 In summary, ablative lasers appear to be a highly effective therapy for AK but at the cost of increased risk for AEs such as permanent pigmentary changes, prolonged erythema lasting up to several months, and scarring.50,52-55
Fractional photothermolysis is a relatively new advancement in the field of laser therapy that has received FDA approval for the treatment of AK.56 This treatment works by creating multiple noncontiguous microscopic columns of thermal injury while sparing adjacent zones of viable tissue.57 Although there are limited studies involving the use of such lasers in the treatment of AK, initial findings suggest that 1927-nm thulium lasers may be more effective than 1550-nm erbium lasers in achieving lesion clearance. A trial of 14 participants who received 5 laser treatments with a 1550-nm fractionated erbium-doped fiber laser reported an average reduction in AK counts of 66.2% at 3 months follow-up and a 55.6% reduction at 6 months follow-up. A participant-determined marked or very significant improvement of lesions was reported in 83% of participants at 1 month posttreatment but only in 44% of participants at 6 months posttreatment.58 A similar trial of 24 participants treated with up to 4 treatment sessions of the fractionated 1927-nm thulium laser reported an 87.3% reduction in number of AKs at 3 months follow-up and an 86.6% reduction at 6 months follow-up.56 The primary advantage of fractional laser therapy is a faster recovery period generally lasting only 2 or 3 days as compared to 2 weeks or more with traditional ablative lasers, thus limiting the amount of time a patient must tolerate cosmetically unappealing erythema.59,60 The quick recovery time has been attributed to the fractional laser’s ability to preserve the stratum corneum and skin barrier, which also helps reduce the risk for other AEs such as scarring and infection.56,59-61 Additional studies are needed to better assess the true efficacy of fractional laser therapy, but treatment with the fractional 1927-nm thulium laser appears to be a promising and well-tolerated therapeutic option for treatment of AK with similar efficacy to traditional ablative lasers but with a lower risk of AEs.
Photodynamic TherapyPhotodynamic therapy (PDT) is an FDA-approved treatment that involves the use of a topical photosensitizing agent such as 5-aminolevulinic acid (ALA) or methyl aminovulinate (MAL) before exposure to an activating light source to generate reactive oxygen species that lead to cell death.62-65 Multiple PDT regimens with varying combinations of photosensitizers, incubation time, and light sources have been studied, but a 2012 Cochrane review determined that treatment with conventional formulations of MAL and ALA with either blue- or red-light PDT were similarly efficacious for treatment of individual AKs as compared to vehicle with blue- or red-light PDT. One exception was that longer incubation time (ie, 4 hours) with ALA resulted in better results than shorter incubation times (ie, 0.5, 1, 2 hours) with ALA.66
Standard PDT treatment with MAL also has consistently demonstrated superior efficacy in achieving complete clearance rates in addition to superior cosmetic outcomes over treatment with either cryotherapy, DFS, or 5-FU.67-73 Three studies in particular noted an excellent or good investigator-determined cosmetic outcome in 96% to 98% of participants treated with MAL-PDT.69,71,74 Photodynamic therapy with ALA also has been reported as superior over CO2 laser ablation for AK reduction as well as both patient and investigator overall satisfaction.75
More recently, several methods of improving photosensitizer delivery have been studied, which have demonstrated remarkable efficacy at achieving lesion clearance over standard cream formulations or application routines. One such method involves the use of gentle heating to increase photosensitizer uptake. In a split-extremity study of 20 participants who were treated with 20% ALA under occlusion for 1 hour with one side heated to 38.8°C, the heated side demonstrated significant (P<.0001) efficacy at achieving higher median clearance rates over control when evaluated at 2 and 6 months posttherapy.76 Notably, occlusion of ALA in itself during the incubation period also has been demonstrated to significantly (P<.0001) improve clearance rates.77 Another method involves the use of a new nanoemulsion-based formulation of ALA gel, known as BF-200 ALA, which has demonstrated remarkable efficacy over standard MAL cream and placebo in a long-term follow-up analysis of 2 prospective, randomized, controlled trials.78 In a similar vein, 3 prospective randomized trials with a minimum follow-up time of 3 months demonstrated that MAL-PDT in combination with fractional ablative laser pretreatment has significant (P<.02 in all trials) efficacy over MAL-PDT without pretreatment in achieving complete AK clearance. Although the cosmetic outcomes were good or excellent in 87% to 100% of patients, they were not significantly different from stand-alone MAL-PDT treatment in any of the trials.79-81 However, pretreatment with microneedling in MAL-PDT has been shown to achieve superior cosmetic outcomes over MAL-PDT without microneedling, according to one small split-face study of 10 participants.82
Overall, PDT is an excellent therapeutic option that is able to provide efficacious clearance of AKs as well as superior cosmetic outcomes. Common AEs of PDT include burning, itching, and stinging during therapy, but pain intensity decreases dramatically upon termination of illumination, with cessation of most symptoms by 12 hours posttherapy.73 Permanent pigmentation changes have been reported to occasionally occur following PDT therapy.81
Conclusion
When determining which therapy to use in a patient, clinicians must take into account a variety of factors such as patient preference, cost of treatment, availability, tolerance for AEs, and the need for field therapy. Although all therapies discussed within this article are effective and reasonable treatment choices, patients who are particularly concerned about cosmetic outcomes would most likely benefit from either IMQ or PDT, as the data for cosmetic outcomes with these therapies are the strongest. Combination or sequential treatments may be required in some cases and all patients should be monitored for lesion recurrence regardless of treatment choice. A summary of the therapies and key studies discussed here is available in the PDF.
- Lebwohl M. Actinic keratosis: epidemiology and progression to squamous cell carcinoma. Br J Dermatol. 2003;149(suppl 66):31-33.
- Drake LA, Ceilley RI, Cornelison RL, et al. Guidelines of care for actinic keratoses. Committee on Guidelines of Care. J Am Acad Dermatol. 1995;32:95-98.
- Salasche SJ. Epidemiology of actinic keratoses and squamous cell carcinoma. J Am Acad Dermatol. 2000;42(1, pt 2):4-7.
- Ackerman AB, Mones JM. Solar (actinic) keratosis is squamous cell carcinoma. Br J Dermatol. 2006;155:9-22.
- Anwar J, Wrone DA, Kimyai-Asadi A, et al. The development of actinic keratosis into invasive squamous cell carcinoma: evidence and evolving classification schemes. Clin Dermatol. 2004;22:189-196.
- Criscione VD, Weinstock MA, Naylor MF, et al. Actinic keratoses: natural history and risk of malignant transformation in the Veterans Affairs Topical Tretinoin Chemoprevention Trial. Cancer. 2009;115:2523-2530.
- Glogau RG. The risk of progression to invasive disease. J Am Acad Dermatol. 2000;42(1, pt 2):23-24.
- Esmann S, Jemec GB. Management of actinic keratosis patients: a qualitative study. J Dermatolog Treat. 2007;18:53-58.
- Weinstock MA, Lee KC, Chren MM, et al. Quality of life in the actinic neoplasia syndrome: the VA Topical Tretinoin Chemoprevention (VATTC) trial. J Am Acad Dermatol. 2009;61:207-215.
- Berman B, Villa AM, Ramirez CC. Mechanisms of action of new treatment modalities for actinic keratosis. J Drugs Dermatol. 2006;5:167-173.
- Askew DA, Mickan SM, Soyer HP. Effectiveness of 5-fluorouracil treatment for actinic keratosis: a systematic review of randomized controlled trials. Int J Dermatol. 2009;46:452-463.
- Levy S, Furst K, Chern W. A pharmacokinetic evaluation of 0.5% and 5% fluorouracil topical cream in patients with actinic keratosis. Clin Ther. 2001;23:908-920.
- Jorizzo J, Stewart D, Bucko A, et al. Randomized trial evaluating a new 0.5% fluorouracil formulation demonstrates efficacy after 1-, 2-, or 4-week treatment in patients with actinic keratosis. Cutis. 2002;70:335-359.
- Sachs DL, Kang S, Hammerberg C, et al. Topical fluorouracil for actinic keratoses and photoaging: a clinical and molecular analysis. Arch Dermatol. 2009;145:659-666.
- Hantash BM, Stewart DB, Cooper ZA, et al. Facial resurfacing for nonmelanoma skin cancer prophylaxis. Arch Dermatol. 2006;142:976-982.
- Lawrence N, Cox SE, Cockerell CJ, et al. A comparison of the efficacy and safety of Jessner’s solution and 35% trichloroacetic acid vs 5% fluorouracil in the treatment of widespread facial actinic keratoses. Arch Dermatol. 1995;131:176-181.
- Monheit GD. The Jessner’s + TCA peel: a medium-depth chemical peel. J Dermatol Surg Oncol. 1989;15:945-950.
- Hemmi H, Kaisho T, Takeuchi O, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196-200.
- Adamson DJ, Frew D, Tatoud R, et al. Diclofenac antagonizes peroxisome proliferator-activated receptor-gamma signaling. Mol Pharmacol. 2002;61:7-12.
- Alam CA, Seed MP, Willoughby DA. Angiostasis and vascular regression in chronic granulomatous inflammation induced by diclofenac in combination with hyaluronan in mice. J Pharm Pharmacol. 1995;47:407-411.
- Lu X, Xie W, Reed D, et al. Nonsteroidal antiinflammatory drugs cause apoptosis and induce cyclooxygenases in chicken embryo fibroblasts. Proc Natl Acad Sci USA. 1995;92:7961-7965.
- Seed MP, Brown JR, Freemantle CN, et al. The inhibition of colon-26 adenocarcinoma development and angiogenesis by topical diclofenac in 2.5% hyaluronan. Cancer Res. 1997;57:1625-1629.
- Rivers JK, Arlette J, Shear N, et al. Topical treatment of actinic keratoses with 3.0% diclofenac in 2.5% hyaluronan gel. Br J Dermatol. 2002;146:94-100.
- Wolf JE, Taylor JR, Tschen E, et al. Topical 3.0% diclo-fenac in 2.5% hyaluronan gel in the treatment of actinic keratoses. Int J Dermatol. 2001;40:709-713.
- Nelson C, Rigel D, Smith S, et al. Phase IV, open-label assessment of the treatment of actinic keratosis with 3.0% diclofenac sodium topical gel (Solaraze). J Drugs Dermatol. 2004;3:401-407.
- Pflugfelder A, Welter AK, Leiter U, et al. Open label randomized study comparing 3 months vs. 6 months treatment of actinic keratoses with 3% diclofenac in 2.5% hyaluronic acid gel: a trial of the German Dermatologic Cooperative Oncology Group. J Eur Acad Dermatol Venereol. 2012;26:48-53.
- Smith SR, Morhenn VB, Piacquadio DJ. Bilateral comparison of the efficacy and tolerability of 3% diclofenac sodium gel and 5% 5-fluorouracil cream in the treatment of actinic keratoses of the face and scalp. J Drugs Dermatol. 2006;5:156-159.
- Segatto MM, Dornelles SI, Silveira VB, et al. Comparative study of actinic keratosis treatment with 3% diclo- fenac sodium and 5% 5-fluorouracil. An Bras Dermatol. 2013;88:732-738.
- Vidal D. Topical imiquimod: mechanism of action and clinical applications. Mini Rev Med Chem. 2006;6:499-503.
- Hadley G, Derry S, Moore RA. Imiquimod for actinic keratosis: systematic review and meta-analysis. J Invest Dermatol. 2006;126:1251-1255.
- Caperton C, Berman B. Safety, efficacy, and patient acceptability of imiquimod for topical treatment of actinic keratoses. Clin Cosmet Investig Dermatol. 2011;4:35-40.
- Swanson N, Smith CC, Kaur M, et al. Imiquimod 2.5% and 3.75% for the treatment of actinic keratoses: two phase 3, multicenter, randomized, double-blind, placebo-controlled studies. J Drugs Dermatol. 2014;13:166-169.
- Krawtchenko N, Roewert-Huber J, Ulrich M, et al. A randomised study of topical 5% imiquimod vs. topical 5-fluorouracil vs. cryosurgery in immunocompetent patients with actinic keratoses: a comparison of clinical and histological outcomes including 1-year follow-up. Br J Dermatol. 2007;157(suppl 2):34-40.
- Anderson L, Schmieder GJ, Werschler WP, et al. Randomized, double-blind, double-dummy, vehicle-controlled study of ingenol mebutate gel 0.025% and 0.05% for actinic keratosis. J Am Acad Dermatol. 2009;60:934-943.
- Ogbourne SM, Suhrbier A, Jones B, et al. Antitumor activity of 3-ingenyl angelate: plasma membrane and mitochondrial disruption and necrotic cell death. Cancer Res. 2004;64:2833-2839.
- Lebwohl M, Swanson N, Anderson LL, et al. Ingenol mebutate gel for actinic keratosis. N Engl J Med. 2012;366:1010-1019.
- Lebwohl M, Shumack S, Stein-Gold L, et al. Long-term follow-up study of ingenol mebutate gel for the treatment of actinic keratoses. JAMA Dermatol. 2013;149:666-670.
- Augustin M, Tu JH, Knudsen KM, et al. Ingenol mebutate gel for actinic keratosis: the link between quality of life, treatment satisfaction, and clinical outcomes. J Am Acad Dermatol. 2015;72:816-821.
- Kane-Maguire N, Moseley R, Cozzi S, et al. Modulation of fibroblast phenotype and extracellular matrix composition by ingenol mebutate may be associated with scar resolution and improved dermal cosmesis. J Am Acad Dermatol. 2012;66:AB218.
- Martin G, Swanson N. Clinical findings using ingenol mebutate gel to treat actinic keratoses. J Am Acad Dermatol. 2013;68(1, suppl 1):S39-S48.
- Feldman SR, Fleischer AB, Williford PM, et al. Destructive procedures are the standard of care for treatment of actinic keratoses. J Am Acad Dermatol. 1999;40:43-47.
- Berlin JM. Current and emerging treatment strategies for the treatment of actinic keratosis. Clin Cosmet Investig Dermatol. 2010;3:119-126.
- Coleman WP, Yarborough JM, Mandy SH. Dermabrasion for prophylaxis and treatment of actinic keratoses. Dermatol Surg. 1996;22:17-21.
- Cooley JE, Casey DL, Kauffman CL. Manual resurfacing and trichloroacetic acid for the treatment of patients with widespread actinic damage. clinical and histologic observations. Dermatol Surg. 1997;23:373-379.
- Goldberg LH, Kaplan B, Vergilis-Kalner I, et al. Liquid nitrogen: temperature control in the treatment of actinic keratosis. Dermatol Surg. 2010;36:1956-1961.
- Lubritz RR, Smolewski SA. Cryosurgery cure rate of actinic keratoses. J Am Acad Dermatol. 1982;7:631-632.
- Thai KE, Fergin P, Freeman M, et al. A prospective study of the use of cryosurgery for the treatment of actinic keratoses. Int J Dermatol. 2004;43:687-692.
- Zane C, Facchinetti E, Rossi MT, et al. Cryotherapy is preferable to ablative CO2 laser for the treatment of isolated actinic keratoses of the face and scalp: a randomized clinical trial. Br J Dermatol. 2014;170:1114-1121.
- Trimas SJ, Ellis DA, Metz RD. The carbon dioxide laser. an alternative for the treatment of actinically damaged skin. Dermatol Surg. 1997;23:885-889.
- Jiang SB, Levine VJ, Nehal KS, et al. Er:YAG laser for the treatment of actinic keratoses. Dermatol Surg. 2000;26:437-440.
- Ostertag JU, Quaedvlieg PJ, Van der geer S, et al. A clinical comparison and long-term follow-up of topical 5-fluorouracil versus laser resurfacing in the treatment of widespread actinic keratoses. Lasers Surg Med. 2006;38:731-739.
- Iyer S, Friedli A, Bowes L, et al. Full face laser resurfacing: therapy and prophylaxis for actinic keratoses and non-melanoma skin cancer. Lasers Surg Med. 2004;34:114-119.
- Rubin MG. A peeler’s thoughts on skin improvement with chemical peels and laser resurfacing. Clin Plast Surg. 1997;24:407-409.
- Riggs K, Keller M, Humphreys TR. Ablative laser resurfacing: high-energy pulsed carbon dioxide and erbium:yttrium-aluminum-garnet. Clin Dermatol. 2007;25:462-473.
- Adrian RM. Pulsed carbon dioxide and long pulse 10-ms erbium-YAG laser resurfacing: a comparative clinical and histological study. J Cutan Laser Ther. 1999;1:197-202.
- Weiss ET, Brauer JA, Anolik R, et al. 1927-nm fractional resurfacing of facial actinic keratoses: a promising new therapeutic option. J Am Acad Dermatol. 2013; 68:98-102.
- Manstein D, Herron GS, Sink RK, et al. Fractional photothermolysis: a new concept for cutaneous remodeling using microscopic patterns of thermal injury. Lasers Surg Med. 2004;34:426-438.
- Katz TM, Goldberg LH, Marquez D, et al. Nonablative fractional photothermolysis for facial actinic keratoses: 6-month follow-up with histologic evaluation. J Am Acad Dermatol. 2011;65:349-356.
- Prens SP, De Vries K, Neumann HA, et al. Non-ablative fractional resurfacing in combination with topical tretinoin cream as a field treatment modality for multiple actinic keratosis: a pilot study and a review of other field treatment modalities. J Dermatolog Treat. 2013;24:227-231.
- Alexiades-Armenakas MR, Dover JS, Arndt KA. The spectrum of laser skin resurfacing: nonablative, fractional, and ablative laser resurfacing. J Am Acad Dermatol. 2008;58:719-737.
- Tannous Z. Fractional resurfacing. Clin Dermatol. 2007;25:480-486.
- Gold MH. Continuing medical education article-skin treatment: photodynamic therapy: indications and treatment. Aesthet Surg J. 2008;28:545-552.
- Juarranz A, Jaén P, Sanz-Rodríguez F, et al. Photodynamic therapy of cancer. basic principles and applications. Clin Transl Oncol. 2008;10:148-154.
- Juzeniene A, Peng Q, Moan J. Milestones in the development of photodynamic therapy and fluorescence diagnosis. Photochem Photobiol Sci. 2007;6:1234-1245.
- Moan J, Berg K. The photodegradation of porphyrins in cells can be used to estimate the lifetime of singlet oxygen. Photochem Photobiol. 1991;53:549-553.
- Gupta AK, Paquet M, Villanueva E, et al. Interventions for actinic keratoses. Cochrane Database Syst Rev. 2012;12:CD004415.
- Patel G, Armstrong AW, Eisen DB. Efficacy of photodynamic therapy vs other interventions in randomized clinical trials for the treatment of actinic keratoses: a systematic review and meta-analysis. JAMA Dermatol. 2014;150:1281-1288.
- Kaufmann R, Spelman L, Weightman W, et al. Multicentre intraindividual randomized trial of topical methyl aminolaevulinate-photodynamic therapy vs. cryotherapy for multiple actinic keratoses on the extremities. Br J Dermatol. 2008;158:994-999.
- Freeman M, Vinciullo C, Francis D, et al. A comparison of photodynamic therapy using topical methyl aminolevulinate (Metvix) with single cycle cryotherapy in patients with actinic keratosis: a prospective, randomized study. J Dermatolog Treat. 2003;14:99-106.
- Morton C, Campbell S, Gupta G, et al. Intraindividual, right-left comparison of topical methyl aminolaevulinate-photodynamic therapy and cryotherapy in subjects with actinic keratoses: a multicentre, randomized controlled study. Br J Dermatol. 2006;155:1029-1036.
- Pariser DM, Lowe NJ, Stewart DM, et al. Photodynamic therapy with topical methyl aminolevulinate for actinic keratosis: results of a prospective randomized multicenter trial. J Am Acad Dermatol. 2003;48:227-232.
- Zane C, Facchinetti E, Rossi MT, et al. A randomized clinical trial of photodynamic therapy with methyl aminolaevulinate vs. diclofenac 3% plus hyaluronic acid gel for the treatment of multiple actinic keratoses of the face and scalp. Br J Dermatol. 2014;170:1143-1150.
- Perrett CM, McGregor JM, Warwick J, et al. Treatment of post-transplant premalignant skin disease: a randomized intrapatient comparative study of 5-fluorouracil cream and topical photodynamic therapy. Br J Dermatol. 2007;156:320-328.
- Szeimies RM, Karrer S, Radakovic-Fijan S, et al. Photodynamic therapy using topical methyl 5-aminolevulinate compared with cryotherapy for actinic keratosis: a prospective, randomized study. J Am Acad Dermatol. 2002; 47:258-262.
- Scola N, Terras S, Georgas D, et al. A randomized, half-side comparative study of aminolaevulinate photodynamic therapy vs. CO(2) laser ablation in immunocompetent patients with multiple actinic keratoses. Br J Dermatol. 2012;167:1366-1373.
- Willey A, Anderson RR, Sakamoto FH. Temperature-modulated photodynamic therapy for the treatment of actinic keratosis on the extremities: a pilot study. Dermatol Surg. 2014;40:1094-1102.
- Pariser DM. Management of Actinic Keratoses: Treatment Selection and Optimizing Outcomes. Presented at: Winter Clinical Dermatology Conference Hawaii; January 18, 2015; Kaanapali, HI.
- Dirschka T, Radny P, Dominicus R, et al. Long-term (6 and 12 months) follow-up of two prospective, randomized, controlled phase III trials of photodynamic therapy with BF-200 ALA and methyl aminolaevulinate for the treatment of actinic keratosis. Br J Dermatol. 2013;168:825-836.
- Choi SH, Kim KH, Song KH. Efficacy of ablative fractional laser-assisted photodynamic therapy with short-incubation time for the treatment of facial and scalp actinic keratosis: 12-month follow-up results of a randomized, prospective, comparative trial. J Eur Acad Dermatol Venereol. 2015;29:1598-1605.
- Ko DY, Jeon SY, Kim KH, et al. Fractional erbium:YAG laser-assisted photodynamic therapy for facial actinic keratoses: a randomized, comparative, prospective study. J Eur Acad Dermatol Venereol. 2014;28:1529-1539.
- Togsverd-Ho K, Haak CS, Thaysen-Petersen D, et al. Intensified photodynamic therapy of actinic keratoses with fractional CO2 laser: a randomized clinical trial. Br J Dermatol. 2012;166:1262-1269.
- Torezan L, Chaves Y, Niwa A, et al. A pilot split-face study comparing conventional methyl aminolevulinate-photodynamic therapy (PDT) with microneedling-assisted PDT on actinically damaged skin. Dermatol Surg. 2013;39:1197-1201.
- Lebwohl M. Actinic keratosis: epidemiology and progression to squamous cell carcinoma. Br J Dermatol. 2003;149(suppl 66):31-33.
- Drake LA, Ceilley RI, Cornelison RL, et al. Guidelines of care for actinic keratoses. Committee on Guidelines of Care. J Am Acad Dermatol. 1995;32:95-98.
- Salasche SJ. Epidemiology of actinic keratoses and squamous cell carcinoma. J Am Acad Dermatol. 2000;42(1, pt 2):4-7.
- Ackerman AB, Mones JM. Solar (actinic) keratosis is squamous cell carcinoma. Br J Dermatol. 2006;155:9-22.
- Anwar J, Wrone DA, Kimyai-Asadi A, et al. The development of actinic keratosis into invasive squamous cell carcinoma: evidence and evolving classification schemes. Clin Dermatol. 2004;22:189-196.
- Criscione VD, Weinstock MA, Naylor MF, et al. Actinic keratoses: natural history and risk of malignant transformation in the Veterans Affairs Topical Tretinoin Chemoprevention Trial. Cancer. 2009;115:2523-2530.
- Glogau RG. The risk of progression to invasive disease. J Am Acad Dermatol. 2000;42(1, pt 2):23-24.
- Esmann S, Jemec GB. Management of actinic keratosis patients: a qualitative study. J Dermatolog Treat. 2007;18:53-58.
- Weinstock MA, Lee KC, Chren MM, et al. Quality of life in the actinic neoplasia syndrome: the VA Topical Tretinoin Chemoprevention (VATTC) trial. J Am Acad Dermatol. 2009;61:207-215.
- Berman B, Villa AM, Ramirez CC. Mechanisms of action of new treatment modalities for actinic keratosis. J Drugs Dermatol. 2006;5:167-173.
- Askew DA, Mickan SM, Soyer HP. Effectiveness of 5-fluorouracil treatment for actinic keratosis: a systematic review of randomized controlled trials. Int J Dermatol. 2009;46:452-463.
- Levy S, Furst K, Chern W. A pharmacokinetic evaluation of 0.5% and 5% fluorouracil topical cream in patients with actinic keratosis. Clin Ther. 2001;23:908-920.
- Jorizzo J, Stewart D, Bucko A, et al. Randomized trial evaluating a new 0.5% fluorouracil formulation demonstrates efficacy after 1-, 2-, or 4-week treatment in patients with actinic keratosis. Cutis. 2002;70:335-359.
- Sachs DL, Kang S, Hammerberg C, et al. Topical fluorouracil for actinic keratoses and photoaging: a clinical and molecular analysis. Arch Dermatol. 2009;145:659-666.
- Hantash BM, Stewart DB, Cooper ZA, et al. Facial resurfacing for nonmelanoma skin cancer prophylaxis. Arch Dermatol. 2006;142:976-982.
- Lawrence N, Cox SE, Cockerell CJ, et al. A comparison of the efficacy and safety of Jessner’s solution and 35% trichloroacetic acid vs 5% fluorouracil in the treatment of widespread facial actinic keratoses. Arch Dermatol. 1995;131:176-181.
- Monheit GD. The Jessner’s + TCA peel: a medium-depth chemical peel. J Dermatol Surg Oncol. 1989;15:945-950.
- Hemmi H, Kaisho T, Takeuchi O, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196-200.
- Adamson DJ, Frew D, Tatoud R, et al. Diclofenac antagonizes peroxisome proliferator-activated receptor-gamma signaling. Mol Pharmacol. 2002;61:7-12.
- Alam CA, Seed MP, Willoughby DA. Angiostasis and vascular regression in chronic granulomatous inflammation induced by diclofenac in combination with hyaluronan in mice. J Pharm Pharmacol. 1995;47:407-411.
- Lu X, Xie W, Reed D, et al. Nonsteroidal antiinflammatory drugs cause apoptosis and induce cyclooxygenases in chicken embryo fibroblasts. Proc Natl Acad Sci USA. 1995;92:7961-7965.
- Seed MP, Brown JR, Freemantle CN, et al. The inhibition of colon-26 adenocarcinoma development and angiogenesis by topical diclofenac in 2.5% hyaluronan. Cancer Res. 1997;57:1625-1629.
- Rivers JK, Arlette J, Shear N, et al. Topical treatment of actinic keratoses with 3.0% diclofenac in 2.5% hyaluronan gel. Br J Dermatol. 2002;146:94-100.
- Wolf JE, Taylor JR, Tschen E, et al. Topical 3.0% diclo-fenac in 2.5% hyaluronan gel in the treatment of actinic keratoses. Int J Dermatol. 2001;40:709-713.
- Nelson C, Rigel D, Smith S, et al. Phase IV, open-label assessment of the treatment of actinic keratosis with 3.0% diclofenac sodium topical gel (Solaraze). J Drugs Dermatol. 2004;3:401-407.
- Pflugfelder A, Welter AK, Leiter U, et al. Open label randomized study comparing 3 months vs. 6 months treatment of actinic keratoses with 3% diclofenac in 2.5% hyaluronic acid gel: a trial of the German Dermatologic Cooperative Oncology Group. J Eur Acad Dermatol Venereol. 2012;26:48-53.
- Smith SR, Morhenn VB, Piacquadio DJ. Bilateral comparison of the efficacy and tolerability of 3% diclofenac sodium gel and 5% 5-fluorouracil cream in the treatment of actinic keratoses of the face and scalp. J Drugs Dermatol. 2006;5:156-159.
- Segatto MM, Dornelles SI, Silveira VB, et al. Comparative study of actinic keratosis treatment with 3% diclo- fenac sodium and 5% 5-fluorouracil. An Bras Dermatol. 2013;88:732-738.
- Vidal D. Topical imiquimod: mechanism of action and clinical applications. Mini Rev Med Chem. 2006;6:499-503.
- Hadley G, Derry S, Moore RA. Imiquimod for actinic keratosis: systematic review and meta-analysis. J Invest Dermatol. 2006;126:1251-1255.
- Caperton C, Berman B. Safety, efficacy, and patient acceptability of imiquimod for topical treatment of actinic keratoses. Clin Cosmet Investig Dermatol. 2011;4:35-40.
- Swanson N, Smith CC, Kaur M, et al. Imiquimod 2.5% and 3.75% for the treatment of actinic keratoses: two phase 3, multicenter, randomized, double-blind, placebo-controlled studies. J Drugs Dermatol. 2014;13:166-169.
- Krawtchenko N, Roewert-Huber J, Ulrich M, et al. A randomised study of topical 5% imiquimod vs. topical 5-fluorouracil vs. cryosurgery in immunocompetent patients with actinic keratoses: a comparison of clinical and histological outcomes including 1-year follow-up. Br J Dermatol. 2007;157(suppl 2):34-40.
- Anderson L, Schmieder GJ, Werschler WP, et al. Randomized, double-blind, double-dummy, vehicle-controlled study of ingenol mebutate gel 0.025% and 0.05% for actinic keratosis. J Am Acad Dermatol. 2009;60:934-943.
- Ogbourne SM, Suhrbier A, Jones B, et al. Antitumor activity of 3-ingenyl angelate: plasma membrane and mitochondrial disruption and necrotic cell death. Cancer Res. 2004;64:2833-2839.
- Lebwohl M, Swanson N, Anderson LL, et al. Ingenol mebutate gel for actinic keratosis. N Engl J Med. 2012;366:1010-1019.
- Lebwohl M, Shumack S, Stein-Gold L, et al. Long-term follow-up study of ingenol mebutate gel for the treatment of actinic keratoses. JAMA Dermatol. 2013;149:666-670.
- Augustin M, Tu JH, Knudsen KM, et al. Ingenol mebutate gel for actinic keratosis: the link between quality of life, treatment satisfaction, and clinical outcomes. J Am Acad Dermatol. 2015;72:816-821.
- Kane-Maguire N, Moseley R, Cozzi S, et al. Modulation of fibroblast phenotype and extracellular matrix composition by ingenol mebutate may be associated with scar resolution and improved dermal cosmesis. J Am Acad Dermatol. 2012;66:AB218.
- Martin G, Swanson N. Clinical findings using ingenol mebutate gel to treat actinic keratoses. J Am Acad Dermatol. 2013;68(1, suppl 1):S39-S48.
- Feldman SR, Fleischer AB, Williford PM, et al. Destructive procedures are the standard of care for treatment of actinic keratoses. J Am Acad Dermatol. 1999;40:43-47.
- Berlin JM. Current and emerging treatment strategies for the treatment of actinic keratosis. Clin Cosmet Investig Dermatol. 2010;3:119-126.
- Coleman WP, Yarborough JM, Mandy SH. Dermabrasion for prophylaxis and treatment of actinic keratoses. Dermatol Surg. 1996;22:17-21.
- Cooley JE, Casey DL, Kauffman CL. Manual resurfacing and trichloroacetic acid for the treatment of patients with widespread actinic damage. clinical and histologic observations. Dermatol Surg. 1997;23:373-379.
- Goldberg LH, Kaplan B, Vergilis-Kalner I, et al. Liquid nitrogen: temperature control in the treatment of actinic keratosis. Dermatol Surg. 2010;36:1956-1961.
- Lubritz RR, Smolewski SA. Cryosurgery cure rate of actinic keratoses. J Am Acad Dermatol. 1982;7:631-632.
- Thai KE, Fergin P, Freeman M, et al. A prospective study of the use of cryosurgery for the treatment of actinic keratoses. Int J Dermatol. 2004;43:687-692.
- Zane C, Facchinetti E, Rossi MT, et al. Cryotherapy is preferable to ablative CO2 laser for the treatment of isolated actinic keratoses of the face and scalp: a randomized clinical trial. Br J Dermatol. 2014;170:1114-1121.
- Trimas SJ, Ellis DA, Metz RD. The carbon dioxide laser. an alternative for the treatment of actinically damaged skin. Dermatol Surg. 1997;23:885-889.
- Jiang SB, Levine VJ, Nehal KS, et al. Er:YAG laser for the treatment of actinic keratoses. Dermatol Surg. 2000;26:437-440.
- Ostertag JU, Quaedvlieg PJ, Van der geer S, et al. A clinical comparison and long-term follow-up of topical 5-fluorouracil versus laser resurfacing in the treatment of widespread actinic keratoses. Lasers Surg Med. 2006;38:731-739.
- Iyer S, Friedli A, Bowes L, et al. Full face laser resurfacing: therapy and prophylaxis for actinic keratoses and non-melanoma skin cancer. Lasers Surg Med. 2004;34:114-119.
- Rubin MG. A peeler’s thoughts on skin improvement with chemical peels and laser resurfacing. Clin Plast Surg. 1997;24:407-409.
- Riggs K, Keller M, Humphreys TR. Ablative laser resurfacing: high-energy pulsed carbon dioxide and erbium:yttrium-aluminum-garnet. Clin Dermatol. 2007;25:462-473.
- Adrian RM. Pulsed carbon dioxide and long pulse 10-ms erbium-YAG laser resurfacing: a comparative clinical and histological study. J Cutan Laser Ther. 1999;1:197-202.
- Weiss ET, Brauer JA, Anolik R, et al. 1927-nm fractional resurfacing of facial actinic keratoses: a promising new therapeutic option. J Am Acad Dermatol. 2013; 68:98-102.
- Manstein D, Herron GS, Sink RK, et al. Fractional photothermolysis: a new concept for cutaneous remodeling using microscopic patterns of thermal injury. Lasers Surg Med. 2004;34:426-438.
- Katz TM, Goldberg LH, Marquez D, et al. Nonablative fractional photothermolysis for facial actinic keratoses: 6-month follow-up with histologic evaluation. J Am Acad Dermatol. 2011;65:349-356.
- Prens SP, De Vries K, Neumann HA, et al. Non-ablative fractional resurfacing in combination with topical tretinoin cream as a field treatment modality for multiple actinic keratosis: a pilot study and a review of other field treatment modalities. J Dermatolog Treat. 2013;24:227-231.
- Alexiades-Armenakas MR, Dover JS, Arndt KA. The spectrum of laser skin resurfacing: nonablative, fractional, and ablative laser resurfacing. J Am Acad Dermatol. 2008;58:719-737.
- Tannous Z. Fractional resurfacing. Clin Dermatol. 2007;25:480-486.
- Gold MH. Continuing medical education article-skin treatment: photodynamic therapy: indications and treatment. Aesthet Surg J. 2008;28:545-552.
- Juarranz A, Jaén P, Sanz-Rodríguez F, et al. Photodynamic therapy of cancer. basic principles and applications. Clin Transl Oncol. 2008;10:148-154.
- Juzeniene A, Peng Q, Moan J. Milestones in the development of photodynamic therapy and fluorescence diagnosis. Photochem Photobiol Sci. 2007;6:1234-1245.
- Moan J, Berg K. The photodegradation of porphyrins in cells can be used to estimate the lifetime of singlet oxygen. Photochem Photobiol. 1991;53:549-553.
- Gupta AK, Paquet M, Villanueva E, et al. Interventions for actinic keratoses. Cochrane Database Syst Rev. 2012;12:CD004415.
- Patel G, Armstrong AW, Eisen DB. Efficacy of photodynamic therapy vs other interventions in randomized clinical trials for the treatment of actinic keratoses: a systematic review and meta-analysis. JAMA Dermatol. 2014;150:1281-1288.
- Kaufmann R, Spelman L, Weightman W, et al. Multicentre intraindividual randomized trial of topical methyl aminolaevulinate-photodynamic therapy vs. cryotherapy for multiple actinic keratoses on the extremities. Br J Dermatol. 2008;158:994-999.
- Freeman M, Vinciullo C, Francis D, et al. A comparison of photodynamic therapy using topical methyl aminolevulinate (Metvix) with single cycle cryotherapy in patients with actinic keratosis: a prospective, randomized study. J Dermatolog Treat. 2003;14:99-106.
- Morton C, Campbell S, Gupta G, et al. Intraindividual, right-left comparison of topical methyl aminolaevulinate-photodynamic therapy and cryotherapy in subjects with actinic keratoses: a multicentre, randomized controlled study. Br J Dermatol. 2006;155:1029-1036.
- Pariser DM, Lowe NJ, Stewart DM, et al. Photodynamic therapy with topical methyl aminolevulinate for actinic keratosis: results of a prospective randomized multicenter trial. J Am Acad Dermatol. 2003;48:227-232.
- Zane C, Facchinetti E, Rossi MT, et al. A randomized clinical trial of photodynamic therapy with methyl aminolaevulinate vs. diclofenac 3% plus hyaluronic acid gel for the treatment of multiple actinic keratoses of the face and scalp. Br J Dermatol. 2014;170:1143-1150.
- Perrett CM, McGregor JM, Warwick J, et al. Treatment of post-transplant premalignant skin disease: a randomized intrapatient comparative study of 5-fluorouracil cream and topical photodynamic therapy. Br J Dermatol. 2007;156:320-328.
- Szeimies RM, Karrer S, Radakovic-Fijan S, et al. Photodynamic therapy using topical methyl 5-aminolevulinate compared with cryotherapy for actinic keratosis: a prospective, randomized study. J Am Acad Dermatol. 2002; 47:258-262.
- Scola N, Terras S, Georgas D, et al. A randomized, half-side comparative study of aminolaevulinate photodynamic therapy vs. CO(2) laser ablation in immunocompetent patients with multiple actinic keratoses. Br J Dermatol. 2012;167:1366-1373.
- Willey A, Anderson RR, Sakamoto FH. Temperature-modulated photodynamic therapy for the treatment of actinic keratosis on the extremities: a pilot study. Dermatol Surg. 2014;40:1094-1102.
- Pariser DM. Management of Actinic Keratoses: Treatment Selection and Optimizing Outcomes. Presented at: Winter Clinical Dermatology Conference Hawaii; January 18, 2015; Kaanapali, HI.
- Dirschka T, Radny P, Dominicus R, et al. Long-term (6 and 12 months) follow-up of two prospective, randomized, controlled phase III trials of photodynamic therapy with BF-200 ALA and methyl aminolaevulinate for the treatment of actinic keratosis. Br J Dermatol. 2013;168:825-836.
- Choi SH, Kim KH, Song KH. Efficacy of ablative fractional laser-assisted photodynamic therapy with short-incubation time for the treatment of facial and scalp actinic keratosis: 12-month follow-up results of a randomized, prospective, comparative trial. J Eur Acad Dermatol Venereol. 2015;29:1598-1605.
- Ko DY, Jeon SY, Kim KH, et al. Fractional erbium:YAG laser-assisted photodynamic therapy for facial actinic keratoses: a randomized, comparative, prospective study. J Eur Acad Dermatol Venereol. 2014;28:1529-1539.
- Togsverd-Ho K, Haak CS, Thaysen-Petersen D, et al. Intensified photodynamic therapy of actinic keratoses with fractional CO2 laser: a randomized clinical trial. Br J Dermatol. 2012;166:1262-1269.
- Torezan L, Chaves Y, Niwa A, et al. A pilot split-face study comparing conventional methyl aminolevulinate-photodynamic therapy (PDT) with microneedling-assisted PDT on actinically damaged skin. Dermatol Surg. 2013;39:1197-1201.
Practice Points
- In addition to their risk for progression to malignancy, actinic keratoses (AKs) can have negative impacts on cosmetic appearance and quality of life.
- A variety of topical medications, procedural modalities, and light-based therapies are available for treatment of AKs, which offer varying degrees of efficacy for clearance of lesions and cosmetic outcomes. Based on the current data, imiquimod and photodynamic therapy are the treatments most likely to provide an excellent cosmetic outcome.