User login
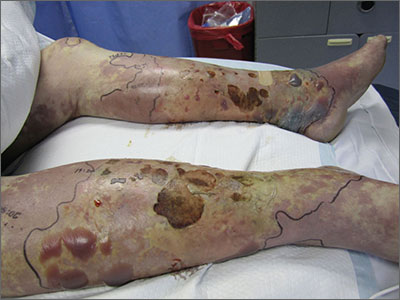
Violaceous bullae on legs

The FP suspected a Vibrio vulnificus infection secondary to ingesting the raw oysters, especially because the patient had a history of liver disease. V vulnificus grew out of the patient’s blood cultures, confirming the diagnosis.
V vulnificus is a free-living bacterium that is found in warm saltwater, such as in the Gulf of Mexico. This patient had been visiting the Gulf Coast when he ate the raw oysters. V vulnificus becomes concentrated in filter-feeding shellfish such as oysters.
Eating raw oysters can lead to overwhelming infections from V vulnificus, especially in those with liver disease, lymphoma, leukemia, and diabetes. The mortality rate in people with primary V vulnificus sepsis exceeds 40%.1
Unfortunately, the patient’s liver disease predisposed him to a more serious infection. Despite appropriate use of systemic antibiotics and supportive care, the patient died from sepsis.
1. Falcon LM, Pham L. Images in clinical medicine. Hemorrhagic cellulitis after consumption of raw oysters. N Engl J Med. 2005;353:1604.
Photo courtesy of Donna Nguyen, MD. Text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Cellulitis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:693-697.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP suspected a Vibrio vulnificus infection secondary to ingesting the raw oysters, especially because the patient had a history of liver disease. V vulnificus grew out of the patient’s blood cultures, confirming the diagnosis.
V vulnificus is a free-living bacterium that is found in warm saltwater, such as in the Gulf of Mexico. This patient had been visiting the Gulf Coast when he ate the raw oysters. V vulnificus becomes concentrated in filter-feeding shellfish such as oysters.
Eating raw oysters can lead to overwhelming infections from V vulnificus, especially in those with liver disease, lymphoma, leukemia, and diabetes. The mortality rate in people with primary V vulnificus sepsis exceeds 40%.1
Unfortunately, the patient’s liver disease predisposed him to a more serious infection. Despite appropriate use of systemic antibiotics and supportive care, the patient died from sepsis.
1. Falcon LM, Pham L. Images in clinical medicine. Hemorrhagic cellulitis after consumption of raw oysters. N Engl J Med. 2005;353:1604.
Photo courtesy of Donna Nguyen, MD. Text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Cellulitis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:693-697.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP suspected a Vibrio vulnificus infection secondary to ingesting the raw oysters, especially because the patient had a history of liver disease. V vulnificus grew out of the patient’s blood cultures, confirming the diagnosis.
V vulnificus is a free-living bacterium that is found in warm saltwater, such as in the Gulf of Mexico. This patient had been visiting the Gulf Coast when he ate the raw oysters. V vulnificus becomes concentrated in filter-feeding shellfish such as oysters.
Eating raw oysters can lead to overwhelming infections from V vulnificus, especially in those with liver disease, lymphoma, leukemia, and diabetes. The mortality rate in people with primary V vulnificus sepsis exceeds 40%.1
Unfortunately, the patient’s liver disease predisposed him to a more serious infection. Despite appropriate use of systemic antibiotics and supportive care, the patient died from sepsis.
1. Falcon LM, Pham L. Images in clinical medicine. Hemorrhagic cellulitis after consumption of raw oysters. N Engl J Med. 2005;353:1604.
Photo courtesy of Donna Nguyen, MD. Text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Cellulitis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:693-697.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
Cosmeceuticals for managing acne: more useful than you might think
ORLANDO – The increasingly popular role of cosmeceuticals in treating acne has created some confusion among both dermatologists and their patients as to what’s really effective and worth recommending.
This was the focus of a presentation at the Orlando Dermatology Aesthetic and Clinical Conference by Dr. Hilary E. Baldwin, who reviewed the cosmeceuticals most likely to make a clinical impact on patients with acne.

While there are no definitive data that prove that cosmeceuticals are the most effective means of managing acne, “sometimes cosmeceuticals may actually be helpful as adjunctive therapy,” said Dr. Baldwin, vice chair of dermatology at the State University of New York at Brooklyn. “Compared to prescription medications, I think these are just a drop in the bucket, but they’re a drop in the right direction.”
The main benefit of using cosmeceuticals for acne is to improve the barrier function of the skin. With increasing evidence that acne either causes or is caused by barrier defects, cosmeceuticals can be used, at the very least, as “extraordinarily well-made moisturizers,” according to Dr. Baldwin. In addition, because moisturizers are anti-inflammatory, they can improve the tolerability of other topical treatments dermatologists recommend to their patients, both prescription and over-the-counter.
For reducing Propionibacterium acnes, consider tea tree oil and lily leaf oil, both of which have a small but promising amount of clinical data behind them. For tea tree oil, Dr. Baldwin referred to a randomized study of 124 patients, which compared 5% tea tree oil gel with 5% benzoyl peroxide for treatment of mild to moderate acne (Med J Aust. 1990 Oct 15;153[8]:455-8). The study found that although the onset of action was slower for tea tree oil, overall it had a significant effect in improving acne in the patients, by reducing the number of inflamed and non-inflamed lesions.
“Both of them worked, but benzoyl peroxide was statistically better,” Dr. Baldwin said. “There were fewer side effects in the tea tree oil group, with less people complaining about skin discomfort.”
There are less data regarding lily leaf extract, however, with only one study she said was worth mentioning: a 4-week trial comparing lily leaf extract and 5% benzoyl peroxide that was “so complicated, and had so many arms, that they ended up having only 4-5 patients in each arm, so I don’t think they can conclude anything,” she remarked.
For management of acne-related inflammation, there is good evidence to suggest botanicals are an effective treatment. A double-blind, randomized, 12-week study coauthored by Dr. Baldwin found that in a cohort of 80 patients, benzoyl peroxide and salicylic acid were more effective when combined with botanical extracts than when used on their own (Semin Cutan Med Surg. 2015 Sep;34[5S]:S82-S85).
Furthermore, explained Dr. Baldwin, “evidence suggests that patients were also using [the botanical extract treatment] more because there was a preference for that,” indicating the increasing desire for more natural, cosmeceutical approaches to treating skin ailments by the general public.
“[Cosmeceuticals] appeal to this increasingly mature and demanding acne patient population,” she said. “[Patients] have a preference for a natural approach to skin disease, they believe that strengthening the host is more important than killing a pathogen, they think [cosmeceuticals] have less of a potential for side effects, and it also gives [patients] a sense of control, which attenuates some of the psychological sequelae of acne.
Dr. Baldwin also recommended oatmeal-based cosmeceuticals for their potential benefit in barrier repair, licorice-based cosmeceuticals for their ability to reduce both postinflammatory hyperpigmentation and post inflammatory erythema, and niacinamide. Niacinamide has been shown to reduce postinflammatory hyperpigmentation when used with other treatment options.
Dr. Baldwin emphasized, however, that cosmeceuticals should always be considered as a supplement to other, ongoing treatments, not the main treatment for acne.
She did not report any relevant financial disclosures.
ORLANDO – The increasingly popular role of cosmeceuticals in treating acne has created some confusion among both dermatologists and their patients as to what’s really effective and worth recommending.
This was the focus of a presentation at the Orlando Dermatology Aesthetic and Clinical Conference by Dr. Hilary E. Baldwin, who reviewed the cosmeceuticals most likely to make a clinical impact on patients with acne.
While there are no definitive data that prove that cosmeceuticals are the most effective means of managing acne, “sometimes cosmeceuticals may actually be helpful as adjunctive therapy,” said Dr. Baldwin, vice chair of dermatology at the State University of New York at Brooklyn. “Compared to prescription medications, I think these are just a drop in the bucket, but they’re a drop in the right direction.”
The main benefit of using cosmeceuticals for acne is to improve the barrier function of the skin. With increasing evidence that acne either causes or is caused by barrier defects, cosmeceuticals can be used, at the very least, as “extraordinarily well-made moisturizers,” according to Dr. Baldwin. In addition, because moisturizers are anti-inflammatory, they can improve the tolerability of other topical treatments dermatologists recommend to their patients, both prescription and over-the-counter.
For reducing Propionibacterium acnes, consider tea tree oil and lily leaf oil, both of which have a small but promising amount of clinical data behind them. For tea tree oil, Dr. Baldwin referred to a randomized study of 124 patients, which compared 5% tea tree oil gel with 5% benzoyl peroxide for treatment of mild to moderate acne (Med J Aust. 1990 Oct 15;153[8]:455-8). The study found that although the onset of action was slower for tea tree oil, overall it had a significant effect in improving acne in the patients, by reducing the number of inflamed and non-inflamed lesions.
“Both of them worked, but benzoyl peroxide was statistically better,” Dr. Baldwin said. “There were fewer side effects in the tea tree oil group, with less people complaining about skin discomfort.”
There are less data regarding lily leaf extract, however, with only one study she said was worth mentioning: a 4-week trial comparing lily leaf extract and 5% benzoyl peroxide that was “so complicated, and had so many arms, that they ended up having only 4-5 patients in each arm, so I don’t think they can conclude anything,” she remarked.
For management of acne-related inflammation, there is good evidence to suggest botanicals are an effective treatment. A double-blind, randomized, 12-week study coauthored by Dr. Baldwin found that in a cohort of 80 patients, benzoyl peroxide and salicylic acid were more effective when combined with botanical extracts than when used on their own (Semin Cutan Med Surg. 2015 Sep;34[5S]:S82-S85).
Furthermore, explained Dr. Baldwin, “evidence suggests that patients were also using [the botanical extract treatment] more because there was a preference for that,” indicating the increasing desire for more natural, cosmeceutical approaches to treating skin ailments by the general public.
“[Cosmeceuticals] appeal to this increasingly mature and demanding acne patient population,” she said. “[Patients] have a preference for a natural approach to skin disease, they believe that strengthening the host is more important than killing a pathogen, they think [cosmeceuticals] have less of a potential for side effects, and it also gives [patients] a sense of control, which attenuates some of the psychological sequelae of acne.
Dr. Baldwin also recommended oatmeal-based cosmeceuticals for their potential benefit in barrier repair, licorice-based cosmeceuticals for their ability to reduce both postinflammatory hyperpigmentation and post inflammatory erythema, and niacinamide. Niacinamide has been shown to reduce postinflammatory hyperpigmentation when used with other treatment options.
Dr. Baldwin emphasized, however, that cosmeceuticals should always be considered as a supplement to other, ongoing treatments, not the main treatment for acne.
She did not report any relevant financial disclosures.
ORLANDO – The increasingly popular role of cosmeceuticals in treating acne has created some confusion among both dermatologists and their patients as to what’s really effective and worth recommending.
This was the focus of a presentation at the Orlando Dermatology Aesthetic and Clinical Conference by Dr. Hilary E. Baldwin, who reviewed the cosmeceuticals most likely to make a clinical impact on patients with acne.
While there are no definitive data that prove that cosmeceuticals are the most effective means of managing acne, “sometimes cosmeceuticals may actually be helpful as adjunctive therapy,” said Dr. Baldwin, vice chair of dermatology at the State University of New York at Brooklyn. “Compared to prescription medications, I think these are just a drop in the bucket, but they’re a drop in the right direction.”
The main benefit of using cosmeceuticals for acne is to improve the barrier function of the skin. With increasing evidence that acne either causes or is caused by barrier defects, cosmeceuticals can be used, at the very least, as “extraordinarily well-made moisturizers,” according to Dr. Baldwin. In addition, because moisturizers are anti-inflammatory, they can improve the tolerability of other topical treatments dermatologists recommend to their patients, both prescription and over-the-counter.
For reducing Propionibacterium acnes, consider tea tree oil and lily leaf oil, both of which have a small but promising amount of clinical data behind them. For tea tree oil, Dr. Baldwin referred to a randomized study of 124 patients, which compared 5% tea tree oil gel with 5% benzoyl peroxide for treatment of mild to moderate acne (Med J Aust. 1990 Oct 15;153[8]:455-8). The study found that although the onset of action was slower for tea tree oil, overall it had a significant effect in improving acne in the patients, by reducing the number of inflamed and non-inflamed lesions.
“Both of them worked, but benzoyl peroxide was statistically better,” Dr. Baldwin said. “There were fewer side effects in the tea tree oil group, with less people complaining about skin discomfort.”
There are less data regarding lily leaf extract, however, with only one study she said was worth mentioning: a 4-week trial comparing lily leaf extract and 5% benzoyl peroxide that was “so complicated, and had so many arms, that they ended up having only 4-5 patients in each arm, so I don’t think they can conclude anything,” she remarked.
For management of acne-related inflammation, there is good evidence to suggest botanicals are an effective treatment. A double-blind, randomized, 12-week study coauthored by Dr. Baldwin found that in a cohort of 80 patients, benzoyl peroxide and salicylic acid were more effective when combined with botanical extracts than when used on their own (Semin Cutan Med Surg. 2015 Sep;34[5S]:S82-S85).
Furthermore, explained Dr. Baldwin, “evidence suggests that patients were also using [the botanical extract treatment] more because there was a preference for that,” indicating the increasing desire for more natural, cosmeceutical approaches to treating skin ailments by the general public.
“[Cosmeceuticals] appeal to this increasingly mature and demanding acne patient population,” she said. “[Patients] have a preference for a natural approach to skin disease, they believe that strengthening the host is more important than killing a pathogen, they think [cosmeceuticals] have less of a potential for side effects, and it also gives [patients] a sense of control, which attenuates some of the psychological sequelae of acne.
Dr. Baldwin also recommended oatmeal-based cosmeceuticals for their potential benefit in barrier repair, licorice-based cosmeceuticals for their ability to reduce both postinflammatory hyperpigmentation and post inflammatory erythema, and niacinamide. Niacinamide has been shown to reduce postinflammatory hyperpigmentation when used with other treatment options.
Dr. Baldwin emphasized, however, that cosmeceuticals should always be considered as a supplement to other, ongoing treatments, not the main treatment for acne.
She did not report any relevant financial disclosures.
AT THE ODAC CONFERENCE

Heightened emphasis on sex-specific cardiovascular risk factors
SNOWMASS, COLO. – Achieving continued reductions in cardiovascular deaths in U.S. women will require that physicians make greater use of sex-specific risk factors that aren’t incorporated in the ACC/AHA atherosclerotic cardiovascular disease risk score, Dr. Jennifer H. Mieres asserted at the Annual Cardiovascular Conference at Snowmass.
In the 13-year period beginning in 2000, with the launch of a national initiative to boost the research focus on cardiovascular disease in women, the annual number of women dying from cardiovascular disease has dropped by roughly 30%. That’s a steeper decline than in men. One of the keys to further reductions in women is more widespread physician evaluation of sex-specific risk factors – such as a history of elevated blood pressure in pregnancy, polycystic ovarian syndrome, or radiation therapy for breast cancer – as part of routine cardiovascular risk assessment in women, said Dr. Mieres, senior vice president office of community and public health at Hofstra Northwell in Hempstead, N.Y.

Hypertension in pregnancy as a harbinger of premature cardiovascular disease and other chronic diseases has been a topic of particularly fruitful research in the past few years.
“The ongoing hypothesis is that pregnancy is a sort of stress test. Pregnancy-related complications indicate an inability to adequately adapt to the physiologic stress of pregnancy and thus reveal the presence of underlying susceptibility to ischemic heart disease,” according to the cardiologist.
She cited a landmark prospective study of 10,314 women born in Northern Finland in 1966 and followed for an average of more than 39 years after a singleton pregnancy. The investigators showed that any elevation in blood pressure during pregnancy, including isolated systolic or diastolic hypertension that resolved during or shortly after pregnancy, was associated with increased future risks of various forms of cardiovascular disease.
For example, de novo gestational hypertension without proteinuria was associated with significantly increased risks of subsequent ischemic cerebrovascular disease, chronic kidney disease, diabetes, ischemic heart disease, acute MI, chronic hypertension, and heart failure. The MIs that occurred in Finns with a history of gestational hypertension were more serious, too, with an associated threefold greater risk of being fatal than MIs in women who had been normotensive in pregnancy (Circulation. 2013 Feb 12;127[6]:681-90).

New-onset isolated systolic or diastolic hypertension emerged during pregnancy in about 17% of the Finnish women. Roughly 30% of them had a cardiovascular event before their late 60s. This translated to a 14%-18% greater risk than in women who remained normotensive in pregnancy.
The highest risk of all in the Finnish study was seen in women with preeclampsia/eclampsia superimposed on a background of chronic hypertension. They had a 3.18-fold greater risk of subsequent MI than did women who were normotensive in pregnancy, a 3.32-fold increased risk of heart failure, and a 2.22-fold greater risk of developing diabetes.
In addition to the growing appreciation that it’s important to consider sex-specific cardiovascular risk factors, recent evidence shows that many of the traditional risk factors are stronger predictors of ischemic heart disease in women than men. These include diabetes, smoking, obesity, and hypertension, Dr. Mieres observed.
For example, a recent meta-analysis of 26 studies including more than 214,000 subjects concluded that women with type 1 diabetes had a 2.5-fold greater risk of incident coronary heart disease than did men with type 1 diabetes. The women with type 1 diabetes also had an 86% greater risk of fatal cardiovascular diseases, a 44% increase in the risk of fatal kidney disease, a 37% greater risk of stroke, and a 37% increase in all-cause mortality relative to type 1 diabetic men (Lancet Diabetes Endocrinol. 2015 Mar;3[3]:198-206).
A wealth of accumulating data indicates that type 2 diabetes, too, is a much stronger risk factor for cardiovascular diseases in women than in men. The evidence prompted a recent formal scientific statement to that effect by the American Heart Association (Circulation. 2015 Dec 22;132[25]:2424-47).
Dr. Mieres reported having no financial conflicts of interest regarding her presentation.
SNOWMASS, COLO. – Achieving continued reductions in cardiovascular deaths in U.S. women will require that physicians make greater use of sex-specific risk factors that aren’t incorporated in the ACC/AHA atherosclerotic cardiovascular disease risk score, Dr. Jennifer H. Mieres asserted at the Annual Cardiovascular Conference at Snowmass.
In the 13-year period beginning in 2000, with the launch of a national initiative to boost the research focus on cardiovascular disease in women, the annual number of women dying from cardiovascular disease has dropped by roughly 30%. That’s a steeper decline than in men. One of the keys to further reductions in women is more widespread physician evaluation of sex-specific risk factors – such as a history of elevated blood pressure in pregnancy, polycystic ovarian syndrome, or radiation therapy for breast cancer – as part of routine cardiovascular risk assessment in women, said Dr. Mieres, senior vice president office of community and public health at Hofstra Northwell in Hempstead, N.Y.
Hypertension in pregnancy as a harbinger of premature cardiovascular disease and other chronic diseases has been a topic of particularly fruitful research in the past few years.
“The ongoing hypothesis is that pregnancy is a sort of stress test. Pregnancy-related complications indicate an inability to adequately adapt to the physiologic stress of pregnancy and thus reveal the presence of underlying susceptibility to ischemic heart disease,” according to the cardiologist.
She cited a landmark prospective study of 10,314 women born in Northern Finland in 1966 and followed for an average of more than 39 years after a singleton pregnancy. The investigators showed that any elevation in blood pressure during pregnancy, including isolated systolic or diastolic hypertension that resolved during or shortly after pregnancy, was associated with increased future risks of various forms of cardiovascular disease.
For example, de novo gestational hypertension without proteinuria was associated with significantly increased risks of subsequent ischemic cerebrovascular disease, chronic kidney disease, diabetes, ischemic heart disease, acute MI, chronic hypertension, and heart failure. The MIs that occurred in Finns with a history of gestational hypertension were more serious, too, with an associated threefold greater risk of being fatal than MIs in women who had been normotensive in pregnancy (Circulation. 2013 Feb 12;127[6]:681-90).
New-onset isolated systolic or diastolic hypertension emerged during pregnancy in about 17% of the Finnish women. Roughly 30% of them had a cardiovascular event before their late 60s. This translated to a 14%-18% greater risk than in women who remained normotensive in pregnancy.
The highest risk of all in the Finnish study was seen in women with preeclampsia/eclampsia superimposed on a background of chronic hypertension. They had a 3.18-fold greater risk of subsequent MI than did women who were normotensive in pregnancy, a 3.32-fold increased risk of heart failure, and a 2.22-fold greater risk of developing diabetes.
In addition to the growing appreciation that it’s important to consider sex-specific cardiovascular risk factors, recent evidence shows that many of the traditional risk factors are stronger predictors of ischemic heart disease in women than men. These include diabetes, smoking, obesity, and hypertension, Dr. Mieres observed.
For example, a recent meta-analysis of 26 studies including more than 214,000 subjects concluded that women with type 1 diabetes had a 2.5-fold greater risk of incident coronary heart disease than did men with type 1 diabetes. The women with type 1 diabetes also had an 86% greater risk of fatal cardiovascular diseases, a 44% increase in the risk of fatal kidney disease, a 37% greater risk of stroke, and a 37% increase in all-cause mortality relative to type 1 diabetic men (Lancet Diabetes Endocrinol. 2015 Mar;3[3]:198-206).
A wealth of accumulating data indicates that type 2 diabetes, too, is a much stronger risk factor for cardiovascular diseases in women than in men. The evidence prompted a recent formal scientific statement to that effect by the American Heart Association (Circulation. 2015 Dec 22;132[25]:2424-47).
Dr. Mieres reported having no financial conflicts of interest regarding her presentation.
SNOWMASS, COLO. – Achieving continued reductions in cardiovascular deaths in U.S. women will require that physicians make greater use of sex-specific risk factors that aren’t incorporated in the ACC/AHA atherosclerotic cardiovascular disease risk score, Dr. Jennifer H. Mieres asserted at the Annual Cardiovascular Conference at Snowmass.
In the 13-year period beginning in 2000, with the launch of a national initiative to boost the research focus on cardiovascular disease in women, the annual number of women dying from cardiovascular disease has dropped by roughly 30%. That’s a steeper decline than in men. One of the keys to further reductions in women is more widespread physician evaluation of sex-specific risk factors – such as a history of elevated blood pressure in pregnancy, polycystic ovarian syndrome, or radiation therapy for breast cancer – as part of routine cardiovascular risk assessment in women, said Dr. Mieres, senior vice president office of community and public health at Hofstra Northwell in Hempstead, N.Y.
Hypertension in pregnancy as a harbinger of premature cardiovascular disease and other chronic diseases has been a topic of particularly fruitful research in the past few years.
“The ongoing hypothesis is that pregnancy is a sort of stress test. Pregnancy-related complications indicate an inability to adequately adapt to the physiologic stress of pregnancy and thus reveal the presence of underlying susceptibility to ischemic heart disease,” according to the cardiologist.
She cited a landmark prospective study of 10,314 women born in Northern Finland in 1966 and followed for an average of more than 39 years after a singleton pregnancy. The investigators showed that any elevation in blood pressure during pregnancy, including isolated systolic or diastolic hypertension that resolved during or shortly after pregnancy, was associated with increased future risks of various forms of cardiovascular disease.
For example, de novo gestational hypertension without proteinuria was associated with significantly increased risks of subsequent ischemic cerebrovascular disease, chronic kidney disease, diabetes, ischemic heart disease, acute MI, chronic hypertension, and heart failure. The MIs that occurred in Finns with a history of gestational hypertension were more serious, too, with an associated threefold greater risk of being fatal than MIs in women who had been normotensive in pregnancy (Circulation. 2013 Feb 12;127[6]:681-90).
New-onset isolated systolic or diastolic hypertension emerged during pregnancy in about 17% of the Finnish women. Roughly 30% of them had a cardiovascular event before their late 60s. This translated to a 14%-18% greater risk than in women who remained normotensive in pregnancy.
The highest risk of all in the Finnish study was seen in women with preeclampsia/eclampsia superimposed on a background of chronic hypertension. They had a 3.18-fold greater risk of subsequent MI than did women who were normotensive in pregnancy, a 3.32-fold increased risk of heart failure, and a 2.22-fold greater risk of developing diabetes.
In addition to the growing appreciation that it’s important to consider sex-specific cardiovascular risk factors, recent evidence shows that many of the traditional risk factors are stronger predictors of ischemic heart disease in women than men. These include diabetes, smoking, obesity, and hypertension, Dr. Mieres observed.
For example, a recent meta-analysis of 26 studies including more than 214,000 subjects concluded that women with type 1 diabetes had a 2.5-fold greater risk of incident coronary heart disease than did men with type 1 diabetes. The women with type 1 diabetes also had an 86% greater risk of fatal cardiovascular diseases, a 44% increase in the risk of fatal kidney disease, a 37% greater risk of stroke, and a 37% increase in all-cause mortality relative to type 1 diabetic men (Lancet Diabetes Endocrinol. 2015 Mar;3[3]:198-206).
A wealth of accumulating data indicates that type 2 diabetes, too, is a much stronger risk factor for cardiovascular diseases in women than in men. The evidence prompted a recent formal scientific statement to that effect by the American Heart Association (Circulation. 2015 Dec 22;132[25]:2424-47).
Dr. Mieres reported having no financial conflicts of interest regarding her presentation.
EXPERT ANALYSIS FROM THE CARDIOVASCULAR CONFERENCE AT SNOWMASS

PICCs Increase Risk for Upper- and Lower-Extremity DVT
Clinical question: Do peripherally inserted central catheters increase the risk for upper- and lower-extremity deep venous thromboses?
Bottom line: Although the association between peripherally inserted central catheters (PICCs) and upper-extremity deep venous thromboses (DVTs) was already known, this study shows that PICCs are also associated with a greater risk of lower-extremity DVTs, suggesting that PICC insertion in itself may be a trigger for thrombosis. (LOE = 2b)
Reference: Greene MT, Flander SA, Woller SC, Bernstein SJ, Chopra V. The association between PICC use and venous thromboembolism in upper and lower extremities. Am J Med 2015;128(9):986–993.
Study design: Cohort (retrospective)
Funding source: Industry
Setting: Inpatient (any location) with outpatient follow-up
Synopsis
Using a statewide registry as well as individual medical records, these investigators collected data for 76,242 hospitalized patients to examine the association between PICC placement and venous thromboembolism (VTE). Patients with a history of VTE, those undergoing surgery, those admitted to an intensive care unit, and those under observation were excluded. Patients were followed up for 90 days after index hospitalization to identify the development of symptomatic pulmonary emboli or upper- or lower-extremity proximal DVTs.
Overall, 5% of the cohort had PICCs present on admission or placed during the hospitalization. As compared with those without PICCs, patients with PICCs were more likely to be older than 70 years; have recent surgery or history of VTE; and have diabetes, inflammatory bowel disease, sepsis, or pneumonia. After adjusting for other risk factors for VTE, the presence of a PICC was not only associated with risk of upper-extremity DVT (hazard ratio [HR] = 10.49; 95% CI 7.79-14.11; P < .001), but also modestly associated with risk of lower-extremity DVT (HR = 1.48; 1.02-2.15; P = .038). The authors hypothesize that PICC line insertion may trigger a systemic thrombosis leading to DVTs in different locations, including the lower extremities. There was no significant association with pulmonary embolism.
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
Clinical question: Do peripherally inserted central catheters increase the risk for upper- and lower-extremity deep venous thromboses?
Bottom line: Although the association between peripherally inserted central catheters (PICCs) and upper-extremity deep venous thromboses (DVTs) was already known, this study shows that PICCs are also associated with a greater risk of lower-extremity DVTs, suggesting that PICC insertion in itself may be a trigger for thrombosis. (LOE = 2b)
Reference: Greene MT, Flander SA, Woller SC, Bernstein SJ, Chopra V. The association between PICC use and venous thromboembolism in upper and lower extremities. Am J Med 2015;128(9):986–993.
Study design: Cohort (retrospective)
Funding source: Industry
Setting: Inpatient (any location) with outpatient follow-up
Synopsis
Using a statewide registry as well as individual medical records, these investigators collected data for 76,242 hospitalized patients to examine the association between PICC placement and venous thromboembolism (VTE). Patients with a history of VTE, those undergoing surgery, those admitted to an intensive care unit, and those under observation were excluded. Patients were followed up for 90 days after index hospitalization to identify the development of symptomatic pulmonary emboli or upper- or lower-extremity proximal DVTs.
Overall, 5% of the cohort had PICCs present on admission or placed during the hospitalization. As compared with those without PICCs, patients with PICCs were more likely to be older than 70 years; have recent surgery or history of VTE; and have diabetes, inflammatory bowel disease, sepsis, or pneumonia. After adjusting for other risk factors for VTE, the presence of a PICC was not only associated with risk of upper-extremity DVT (hazard ratio [HR] = 10.49; 95% CI 7.79-14.11; P < .001), but also modestly associated with risk of lower-extremity DVT (HR = 1.48; 1.02-2.15; P = .038). The authors hypothesize that PICC line insertion may trigger a systemic thrombosis leading to DVTs in different locations, including the lower extremities. There was no significant association with pulmonary embolism.
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
Clinical question: Do peripherally inserted central catheters increase the risk for upper- and lower-extremity deep venous thromboses?
Bottom line: Although the association between peripherally inserted central catheters (PICCs) and upper-extremity deep venous thromboses (DVTs) was already known, this study shows that PICCs are also associated with a greater risk of lower-extremity DVTs, suggesting that PICC insertion in itself may be a trigger for thrombosis. (LOE = 2b)
Reference: Greene MT, Flander SA, Woller SC, Bernstein SJ, Chopra V. The association between PICC use and venous thromboembolism in upper and lower extremities. Am J Med 2015;128(9):986–993.
Study design: Cohort (retrospective)
Funding source: Industry
Setting: Inpatient (any location) with outpatient follow-up
Synopsis
Using a statewide registry as well as individual medical records, these investigators collected data for 76,242 hospitalized patients to examine the association between PICC placement and venous thromboembolism (VTE). Patients with a history of VTE, those undergoing surgery, those admitted to an intensive care unit, and those under observation were excluded. Patients were followed up for 90 days after index hospitalization to identify the development of symptomatic pulmonary emboli or upper- or lower-extremity proximal DVTs.
Overall, 5% of the cohort had PICCs present on admission or placed during the hospitalization. As compared with those without PICCs, patients with PICCs were more likely to be older than 70 years; have recent surgery or history of VTE; and have diabetes, inflammatory bowel disease, sepsis, or pneumonia. After adjusting for other risk factors for VTE, the presence of a PICC was not only associated with risk of upper-extremity DVT (hazard ratio [HR] = 10.49; 95% CI 7.79-14.11; P < .001), but also modestly associated with risk of lower-extremity DVT (HR = 1.48; 1.02-2.15; P = .038). The authors hypothesize that PICC line insertion may trigger a systemic thrombosis leading to DVTs in different locations, including the lower extremities. There was no significant association with pulmonary embolism.
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
NSAIDs Safe, Effective Option for Pleurodesis Pain
Clinical question: For pleurodesis, do nonsteroidal anti-inflammatory drugs and smaller chest tubes, as compared with opioids and larger tubes, provide better pain relief while maintaining efficacy of the procedure?
Bottom line: Although nonsteroidal anti-inflammatory drugs (NSAIDs) are not necessarily more effective than opiates for pain relief after pleurodesis, they should be considered a safe and effective analgesic option for these patients. NSAIDs do not lead to higher rates of pleurodesis failure. Using a smaller chest tube, on the other hand, does not provide a clinically significant pain benefit and may ultimately lead to a higher rate of pleurodesis failure. (LOE = 1b)
Reference: Rahman NM, Pepperell J, Rehal S, et al. Effect of opioids vs NSAIDs and larger vs smaller chest tube size on pain control and pleurodesis efficacy among patients with malignant pleural effusion. JAMA 2015;314(24):2614–2653.
Study design: Randomized controlled trial (nonblinded)
Funding source: Government
Allocation: Concealed
Setting: Inpatient (any location) with outpatient follow-up
Synopsis
Using concealed allocation, these investigators randomized 320 patients requiring pleurodesis for malignant pleural effusions to receive either NSAIDs or opioids and small or large (12F vs 24F) chest tubes. Patients who underwent thoracoscopy, which necessitates a 24F tube postprocedure, were randomized to receive either NSAIDs or opiates but were not included in the chest tube size analysis. Those who did not undergo thoracoscopy were randomized into both arms of the trial, either NSAIDs or opiates and either 12F or 24F chest tubes.
The NSAID groups received 800 mg ibuprofen 3 times daily as needed; the opiate groups received 10 mg to 20 mg oral morphine up to 4 times daily. The patients were not masked to any of the interventions. All patients also received scheduled 1g acetaminophen 4 times daily and intravenous morphine as needed for breakthrough pain. Pain was measured using a 100-mm visual analog scale. Pleurodesis failure was defined as requiring another pleural intervention within 3 months after randomization. Patients had similar baseline characteristics in all treatment groups, except for more men in the larger chest tube group.
Overall, there was no significant difference detected in mean pain scores while the chest tube was in place in the NSAID group as compared with the opiate group, but the NSAID group required more breakthrough intravenous morphine than the opiate group (38% vs 26%; P = .003). Patients in the smaller chest tube group reported less pain than the larger tube group (mean visual analog scale score = 22 mm vs 27 mm; P = .04). Although this finding was statistically significant, the absolute difference in pain scores was small and not necessarily clinically meaningful. For pleurodesis failure, the NSAID group was noninferior to the opiate group; however, the smaller chest tube group had a higher rate of pleurodesis failure and did not meet noninferiority criteria. Pain scores at 1 or 3 months, adverse events, and mortality did not differ for either of the comparisons.
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
Clinical question: For pleurodesis, do nonsteroidal anti-inflammatory drugs and smaller chest tubes, as compared with opioids and larger tubes, provide better pain relief while maintaining efficacy of the procedure?
Bottom line: Although nonsteroidal anti-inflammatory drugs (NSAIDs) are not necessarily more effective than opiates for pain relief after pleurodesis, they should be considered a safe and effective analgesic option for these patients. NSAIDs do not lead to higher rates of pleurodesis failure. Using a smaller chest tube, on the other hand, does not provide a clinically significant pain benefit and may ultimately lead to a higher rate of pleurodesis failure. (LOE = 1b)
Reference: Rahman NM, Pepperell J, Rehal S, et al. Effect of opioids vs NSAIDs and larger vs smaller chest tube size on pain control and pleurodesis efficacy among patients with malignant pleural effusion. JAMA 2015;314(24):2614–2653.
Study design: Randomized controlled trial (nonblinded)
Funding source: Government
Allocation: Concealed
Setting: Inpatient (any location) with outpatient follow-up
Synopsis
Using concealed allocation, these investigators randomized 320 patients requiring pleurodesis for malignant pleural effusions to receive either NSAIDs or opioids and small or large (12F vs 24F) chest tubes. Patients who underwent thoracoscopy, which necessitates a 24F tube postprocedure, were randomized to receive either NSAIDs or opiates but were not included in the chest tube size analysis. Those who did not undergo thoracoscopy were randomized into both arms of the trial, either NSAIDs or opiates and either 12F or 24F chest tubes.
The NSAID groups received 800 mg ibuprofen 3 times daily as needed; the opiate groups received 10 mg to 20 mg oral morphine up to 4 times daily. The patients were not masked to any of the interventions. All patients also received scheduled 1g acetaminophen 4 times daily and intravenous morphine as needed for breakthrough pain. Pain was measured using a 100-mm visual analog scale. Pleurodesis failure was defined as requiring another pleural intervention within 3 months after randomization. Patients had similar baseline characteristics in all treatment groups, except for more men in the larger chest tube group.
Overall, there was no significant difference detected in mean pain scores while the chest tube was in place in the NSAID group as compared with the opiate group, but the NSAID group required more breakthrough intravenous morphine than the opiate group (38% vs 26%; P = .003). Patients in the smaller chest tube group reported less pain than the larger tube group (mean visual analog scale score = 22 mm vs 27 mm; P = .04). Although this finding was statistically significant, the absolute difference in pain scores was small and not necessarily clinically meaningful. For pleurodesis failure, the NSAID group was noninferior to the opiate group; however, the smaller chest tube group had a higher rate of pleurodesis failure and did not meet noninferiority criteria. Pain scores at 1 or 3 months, adverse events, and mortality did not differ for either of the comparisons.
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
Clinical question: For pleurodesis, do nonsteroidal anti-inflammatory drugs and smaller chest tubes, as compared with opioids and larger tubes, provide better pain relief while maintaining efficacy of the procedure?
Bottom line: Although nonsteroidal anti-inflammatory drugs (NSAIDs) are not necessarily more effective than opiates for pain relief after pleurodesis, they should be considered a safe and effective analgesic option for these patients. NSAIDs do not lead to higher rates of pleurodesis failure. Using a smaller chest tube, on the other hand, does not provide a clinically significant pain benefit and may ultimately lead to a higher rate of pleurodesis failure. (LOE = 1b)
Reference: Rahman NM, Pepperell J, Rehal S, et al. Effect of opioids vs NSAIDs and larger vs smaller chest tube size on pain control and pleurodesis efficacy among patients with malignant pleural effusion. JAMA 2015;314(24):2614–2653.
Study design: Randomized controlled trial (nonblinded)
Funding source: Government
Allocation: Concealed
Setting: Inpatient (any location) with outpatient follow-up
Synopsis
Using concealed allocation, these investigators randomized 320 patients requiring pleurodesis for malignant pleural effusions to receive either NSAIDs or opioids and small or large (12F vs 24F) chest tubes. Patients who underwent thoracoscopy, which necessitates a 24F tube postprocedure, were randomized to receive either NSAIDs or opiates but were not included in the chest tube size analysis. Those who did not undergo thoracoscopy were randomized into both arms of the trial, either NSAIDs or opiates and either 12F or 24F chest tubes.
The NSAID groups received 800 mg ibuprofen 3 times daily as needed; the opiate groups received 10 mg to 20 mg oral morphine up to 4 times daily. The patients were not masked to any of the interventions. All patients also received scheduled 1g acetaminophen 4 times daily and intravenous morphine as needed for breakthrough pain. Pain was measured using a 100-mm visual analog scale. Pleurodesis failure was defined as requiring another pleural intervention within 3 months after randomization. Patients had similar baseline characteristics in all treatment groups, except for more men in the larger chest tube group.
Overall, there was no significant difference detected in mean pain scores while the chest tube was in place in the NSAID group as compared with the opiate group, but the NSAID group required more breakthrough intravenous morphine than the opiate group (38% vs 26%; P = .003). Patients in the smaller chest tube group reported less pain than the larger tube group (mean visual analog scale score = 22 mm vs 27 mm; P = .04). Although this finding was statistically significant, the absolute difference in pain scores was small and not necessarily clinically meaningful. For pleurodesis failure, the NSAID group was noninferior to the opiate group; however, the smaller chest tube group had a higher rate of pleurodesis failure and did not meet noninferiority criteria. Pain scores at 1 or 3 months, adverse events, and mortality did not differ for either of the comparisons.
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
CDC: Screen women for alcohol, birth control use
An estimated 3.3 million U.S. women aged 15-44 years risk conceiving children with fetal alcohol spectrum disorders by using alcohol but not birth control.
The finding has officials at the Centers for Disease Control and Prevention urging physicians to screen this group for concomitant drinking and nonuse of contraception. The data come from an analysis of 4,303 nonpregnant, nonsterile women ages 15-44 years from the 2011-2013 National Survey of Family Growth, conducted by the CDC (MMWR. 2016;65:1-7.).

“Alcohol can permanently harm a developing baby before a woman knows she is pregnant,” Dr. Anne Schuchat, the CDC’s principal deputy director, said during a media briefing on Feb. 2. “About half of all pregnancies in the United States are unplanned, and even if planned, most women won’t know they are pregnant for the first month or so, when they might still be drinking. The risk is real. Why take the chance?”
Fetal alcohol spectrum disorders (FASD) can include physical, behavioral, and intellectual disabilities that can last for a child’s lifetime. Dr, Schuchat said the CDC estimates that as many as 1 in 20 U.S. schoolchildren may have FASD. Currently, there are no data on what amounts of alcohol are safe for a woman to drink at any stage of pregnancy.
“Not drinking alcohol is one of the best things you can do to ensure the health of your baby,” Dr. Schuchat said.
For the study, a woman was considered at risk for an alcohol-exposed pregnancy during the past month if she was nonsterile and had sex with a nonsterile male, drank any alcohol, and did not use contraception in the past month. The CDC found the weighted prevalence of alcohol-exposed pregnancy risk among U.S. women aged 15-44 years was 7.3%.

During a 1-month period, approximately 3.3 million U.S. women were at risk for an alcohol-exposed pregnancy. The highest risk group – at 10.4% – were women aged 25-29 years. The lowest risk group were those aged 15-20 years, at 2.2%.
Neither race nor ethnicity were found to be risk factors, although the risk for an alcohol-exposed pregnancy was higher among married and cohabitating women at 11.7% and 13.6% respectively, compared with their single counterparts (2.3%).
The study also found that three-quarters of women who want to get pregnant as soon as possible do not stop drinking alcohol after discontinuing contraception.
Physicians and other health care providers should advise women who want to become pregnant to stop drinking alcohol as soon as they stop using birth control, Dr. Schuchat said.
She added that physicians should screen all adults for alcohol use, not just women, even though that is not currently standard practice. “We think it should be more common to do on a regular basis.” Dr. Schuchat said the federal government requires most health plans to cover alcohol screening without cost to the patient.
The CDC recommends that physicians:
• Screen all adult female patients for alcohol use annually.
• Advise women to cease all alcohol intake if there is any chance at all that she could be pregnant.
• Counsel, refer, and follow-up with patients who need additional support to not drink while pregnant.
• Use correct billing codes to be reimbursed for screening and counseling.
The American College of Obstetricians and Gynecologists, which recommends that women completely abstain from alcohol during pregnancy, praised the CDC’s guidance that physicians routinely screen women regarding their alcohol use.
Dr. Mark S. DeFrancesco, ACOG president, said the other important message from the CDC report is that physicians should counsel women about contraception use.
“As the CDC notes, roughly half of all pregnancies in the United States are unintended. In many cases of unintended pregnancy, women inadvertently expose their fetuses to alcohol and its teratogenic effects prior to discovering that they are pregnant,” he said in statement. “This is just another reason why it’s so important that health care providers counsel women about how to prevent unintended pregnancy through use of the contraceptive method that is right for them. There are many benefits to helping women become pregnant only when they are ready, and avoiding alcohol exposure is one of them.”
On Twitter @whitneymcknight
An estimated 3.3 million U.S. women aged 15-44 years risk conceiving children with fetal alcohol spectrum disorders by using alcohol but not birth control.
The finding has officials at the Centers for Disease Control and Prevention urging physicians to screen this group for concomitant drinking and nonuse of contraception. The data come from an analysis of 4,303 nonpregnant, nonsterile women ages 15-44 years from the 2011-2013 National Survey of Family Growth, conducted by the CDC (MMWR. 2016;65:1-7.).
“Alcohol can permanently harm a developing baby before a woman knows she is pregnant,” Dr. Anne Schuchat, the CDC’s principal deputy director, said during a media briefing on Feb. 2. “About half of all pregnancies in the United States are unplanned, and even if planned, most women won’t know they are pregnant for the first month or so, when they might still be drinking. The risk is real. Why take the chance?”
Fetal alcohol spectrum disorders (FASD) can include physical, behavioral, and intellectual disabilities that can last for a child’s lifetime. Dr, Schuchat said the CDC estimates that as many as 1 in 20 U.S. schoolchildren may have FASD. Currently, there are no data on what amounts of alcohol are safe for a woman to drink at any stage of pregnancy.
“Not drinking alcohol is one of the best things you can do to ensure the health of your baby,” Dr. Schuchat said.
For the study, a woman was considered at risk for an alcohol-exposed pregnancy during the past month if she was nonsterile and had sex with a nonsterile male, drank any alcohol, and did not use contraception in the past month. The CDC found the weighted prevalence of alcohol-exposed pregnancy risk among U.S. women aged 15-44 years was 7.3%.
During a 1-month period, approximately 3.3 million U.S. women were at risk for an alcohol-exposed pregnancy. The highest risk group – at 10.4% – were women aged 25-29 years. The lowest risk group were those aged 15-20 years, at 2.2%.
Neither race nor ethnicity were found to be risk factors, although the risk for an alcohol-exposed pregnancy was higher among married and cohabitating women at 11.7% and 13.6% respectively, compared with their single counterparts (2.3%).
The study also found that three-quarters of women who want to get pregnant as soon as possible do not stop drinking alcohol after discontinuing contraception.
Physicians and other health care providers should advise women who want to become pregnant to stop drinking alcohol as soon as they stop using birth control, Dr. Schuchat said.
She added that physicians should screen all adults for alcohol use, not just women, even though that is not currently standard practice. “We think it should be more common to do on a regular basis.” Dr. Schuchat said the federal government requires most health plans to cover alcohol screening without cost to the patient.
The CDC recommends that physicians:
• Screen all adult female patients for alcohol use annually.
• Advise women to cease all alcohol intake if there is any chance at all that she could be pregnant.
• Counsel, refer, and follow-up with patients who need additional support to not drink while pregnant.
• Use correct billing codes to be reimbursed for screening and counseling.
The American College of Obstetricians and Gynecologists, which recommends that women completely abstain from alcohol during pregnancy, praised the CDC’s guidance that physicians routinely screen women regarding their alcohol use.
Dr. Mark S. DeFrancesco, ACOG president, said the other important message from the CDC report is that physicians should counsel women about contraception use.
“As the CDC notes, roughly half of all pregnancies in the United States are unintended. In many cases of unintended pregnancy, women inadvertently expose their fetuses to alcohol and its teratogenic effects prior to discovering that they are pregnant,” he said in statement. “This is just another reason why it’s so important that health care providers counsel women about how to prevent unintended pregnancy through use of the contraceptive method that is right for them. There are many benefits to helping women become pregnant only when they are ready, and avoiding alcohol exposure is one of them.”
On Twitter @whitneymcknight
An estimated 3.3 million U.S. women aged 15-44 years risk conceiving children with fetal alcohol spectrum disorders by using alcohol but not birth control.
The finding has officials at the Centers for Disease Control and Prevention urging physicians to screen this group for concomitant drinking and nonuse of contraception. The data come from an analysis of 4,303 nonpregnant, nonsterile women ages 15-44 years from the 2011-2013 National Survey of Family Growth, conducted by the CDC (MMWR. 2016;65:1-7.).
“Alcohol can permanently harm a developing baby before a woman knows she is pregnant,” Dr. Anne Schuchat, the CDC’s principal deputy director, said during a media briefing on Feb. 2. “About half of all pregnancies in the United States are unplanned, and even if planned, most women won’t know they are pregnant for the first month or so, when they might still be drinking. The risk is real. Why take the chance?”
Fetal alcohol spectrum disorders (FASD) can include physical, behavioral, and intellectual disabilities that can last for a child’s lifetime. Dr, Schuchat said the CDC estimates that as many as 1 in 20 U.S. schoolchildren may have FASD. Currently, there are no data on what amounts of alcohol are safe for a woman to drink at any stage of pregnancy.
“Not drinking alcohol is one of the best things you can do to ensure the health of your baby,” Dr. Schuchat said.
For the study, a woman was considered at risk for an alcohol-exposed pregnancy during the past month if she was nonsterile and had sex with a nonsterile male, drank any alcohol, and did not use contraception in the past month. The CDC found the weighted prevalence of alcohol-exposed pregnancy risk among U.S. women aged 15-44 years was 7.3%.
During a 1-month period, approximately 3.3 million U.S. women were at risk for an alcohol-exposed pregnancy. The highest risk group – at 10.4% – were women aged 25-29 years. The lowest risk group were those aged 15-20 years, at 2.2%.
Neither race nor ethnicity were found to be risk factors, although the risk for an alcohol-exposed pregnancy was higher among married and cohabitating women at 11.7% and 13.6% respectively, compared with their single counterparts (2.3%).
The study also found that three-quarters of women who want to get pregnant as soon as possible do not stop drinking alcohol after discontinuing contraception.
Physicians and other health care providers should advise women who want to become pregnant to stop drinking alcohol as soon as they stop using birth control, Dr. Schuchat said.
She added that physicians should screen all adults for alcohol use, not just women, even though that is not currently standard practice. “We think it should be more common to do on a regular basis.” Dr. Schuchat said the federal government requires most health plans to cover alcohol screening without cost to the patient.
The CDC recommends that physicians:
• Screen all adult female patients for alcohol use annually.
• Advise women to cease all alcohol intake if there is any chance at all that she could be pregnant.
• Counsel, refer, and follow-up with patients who need additional support to not drink while pregnant.
• Use correct billing codes to be reimbursed for screening and counseling.
The American College of Obstetricians and Gynecologists, which recommends that women completely abstain from alcohol during pregnancy, praised the CDC’s guidance that physicians routinely screen women regarding their alcohol use.
Dr. Mark S. DeFrancesco, ACOG president, said the other important message from the CDC report is that physicians should counsel women about contraception use.
“As the CDC notes, roughly half of all pregnancies in the United States are unintended. In many cases of unintended pregnancy, women inadvertently expose their fetuses to alcohol and its teratogenic effects prior to discovering that they are pregnant,” he said in statement. “This is just another reason why it’s so important that health care providers counsel women about how to prevent unintended pregnancy through use of the contraceptive method that is right for them. There are many benefits to helping women become pregnant only when they are ready, and avoiding alcohol exposure is one of them.”
On Twitter @whitneymcknight
FROM THE MMWR
Key clinical point: The CDC advises physicians to screen women for alcohol use and provide contraception counseling.
Major finding: A total of 3.3 million women aged 15-44 years risk conceiving a child with FASD by using alcohol and having unprotected sex.
Data source: Data on 4,303 nonpregnant, nonsterile women aged 15-44 years from the 2011-2013 National Survey of Family Growth.
Disclosures: The researchers did not report having any financial disclosures.

Shorter hours, longer breaks for surgery residents not shown to improve patient outcomes
JACKSONVILLE, FLA. – Accreditation Council for Graduate Medical Education (ACGME) rules that shortened surgery resident shifts and expanded breaks didn’t improve patient safety or surgery resident well-being in a trial presented at the Association for Academic Surgery/Society of University Surgeons Academic Surgical Congress.
“This national, prospective, randomized trial showed that flexible, less-restrictive duty-hour policies for surgical residents were noninferior to standard ACGME duty-hour policies,” wrote Dr. Karl Bilimoria, associate surgery professor at Northwestern University, Chicago, and associates. The work was published simultaneously Feb. 2 in the New England Journal of Medicine (doi: 10.1056/NEJMoa1515724).

Recent ACGME residency reforms were meant to reduce fatigue-related errors, but there have been concerns that they have come at the cost of increased handoffs and reduced education.
To get a handle on the situation, the investigators randomized 59 teaching-hospital surgery programs to standard ACGME duty hours and 58 others to a freer approach in the 2014-2015 academic year. Residents weren’t allowed to work more than 80 hours per week in either group, but hospitals in the flexible-hour arm were allowed to push residents past ACGME policy, working first-year residents longer than 16 hours per shift and others more than 28 hours, with breaks of less than 14 hours after 24-hour shifts and less than 8-10 hours after shorter ones.
Among the 138,691 adult general surgery cases during the academic year, there was no increase in 30-day rates of postoperative deaths or serious complications in the flexible group (9.1% vs. 9.0% with standard policy, P = .92) or secondary postoperative outcomes, based on risk-adjusted data from the American College of Surgeons’ National Surgical Quality Improvement Program (ACS NSQIP).
The 4,330 residents in the study filled out a multiple-choice questionnaire midway through the project in January 2015. Those in the flexible group said they weren’t significantly unhappier with the quality of their education (11.0% vs. 10.7% in the standard group, P = .86) or well-being (14.9% and 12.0%, P = .10). The investigators didn’t report the lengths of shifts or breaks.
There were no significant differences in resident-reported perceptions of fatigue on personal or patient safety. Residents in the flexible group were less likely to report leaving an operation (7.0% vs. 13.2%, P less than .001) or handing off patients with active issues (32% vs. 46.3%, P less than .001).
Flexible duty-hour residents “noted numerous benefits with respect to nearly all aspects of patient safety, continuity of care, surgical training, and professionalism. However, residents reported that less-restrictive duty-hour policies had a negative effect on [their] time with family and friends, time for extracurricular activities, rest, and health. Importantly … residents’ satisfaction with overall well-being did not differ significantly between study groups,” Dr. Karl Bilimoria and associates concluded.
The investigators “did not specifically collect data on needle sticks and car accidents, because these are notoriously challenging outcomes to capture in surveys,” they noted.
In an interview, Dr. Bilimoria commented, “Increasingly over time we’ve had more regulations of duty hours, and with each set of regulations the surgical community became increasingly concerned about patient handoffs and continuity of care, so our focus was to identify those policies that we thought affected continuity of care and work with the ACGME to waive those for the centers that were in the flexible arm of the study.”
His comments on the impact on residents: “The residents very clearly noted that the flexible policy arm provided better continuity of care, allowed them to take care of their patients in a way that they wanted to and stay with their patients in the operating room and at times when their patients were unstable.”
When asked if the findings could be extrapolated to these smaller nonparticipating centers, Dr. Bilimoria responded, “We captured the majority of residents, and we’re working on an analysis now that seeks to understand what the generalizability would be to those nonparticipating programs. That will be fairly enlightening as well.”
“95% of eligible programs participated in the trial, showing overwhelming support from the community for bringing high level data to this question. There had never before been a randomized trial nationally on this topic and for understanding and testing the notion of flexibility. They saw a need for both of those things.”
ACGME paid for the work, along with the American Board of Surgery and the American College of Surgeons. Dr. Bilimoria and five other authors reported payments from ACGME and the other entities.
UPDATE: This story was updated 2/2/16
What do the results of [this] trial mean for ACGME policy on resident duty hours? The authors conclude, as will many surgeons, that surgical training programs should be afforded more flexibility in applying work-hour rules. This interpretation implicitly places the burden of proof on the ACGME. Thus, because the [trial] found no evidence that removing restrictions on resident shift length and time off between shifts was harmful to patients, programs should have more autonomy to train residents as they choose.
I reach a different conclusion. The [trial] effectively debunks concerns that patients will suffer as a result of increased handoffs and breaks in the continuity of care. Rather than backtrack on the ACGME duty-hour rules, surgical leaders should focus on developing safe, resilient health systems that do not depend on overworked resident physicians. They also should recognize the changing expectations of postmillennial learners. To many current residents and medical students, 80-hour (or even 72-hour) work weeks and 24-hour shifts probably seem long enough. Although few surgical residents would ever acknowledge this publicly, I’m sure that many love to hear, “We can take care of this case without you. Go home, see your family, and come in fresh tomorrow.”
Dr. John Birkmeyer is professor of surgery at the Geisel School of Medicine at Dartmouth in Hanover, N.H. He wasn’t involved in the study; his comments appeared in an editorial (N Eng J Med. 2016 Feb 2. doi: 10.1056/NEJMe1516572).
What do the results of [this] trial mean for ACGME policy on resident duty hours? The authors conclude, as will many surgeons, that surgical training programs should be afforded more flexibility in applying work-hour rules. This interpretation implicitly places the burden of proof on the ACGME. Thus, because the [trial] found no evidence that removing restrictions on resident shift length and time off between shifts was harmful to patients, programs should have more autonomy to train residents as they choose.
I reach a different conclusion. The [trial] effectively debunks concerns that patients will suffer as a result of increased handoffs and breaks in the continuity of care. Rather than backtrack on the ACGME duty-hour rules, surgical leaders should focus on developing safe, resilient health systems that do not depend on overworked resident physicians. They also should recognize the changing expectations of postmillennial learners. To many current residents and medical students, 80-hour (or even 72-hour) work weeks and 24-hour shifts probably seem long enough. Although few surgical residents would ever acknowledge this publicly, I’m sure that many love to hear, “We can take care of this case without you. Go home, see your family, and come in fresh tomorrow.”
Dr. John Birkmeyer is professor of surgery at the Geisel School of Medicine at Dartmouth in Hanover, N.H. He wasn’t involved in the study; his comments appeared in an editorial (N Eng J Med. 2016 Feb 2. doi: 10.1056/NEJMe1516572).
What do the results of [this] trial mean for ACGME policy on resident duty hours? The authors conclude, as will many surgeons, that surgical training programs should be afforded more flexibility in applying work-hour rules. This interpretation implicitly places the burden of proof on the ACGME. Thus, because the [trial] found no evidence that removing restrictions on resident shift length and time off between shifts was harmful to patients, programs should have more autonomy to train residents as they choose.
I reach a different conclusion. The [trial] effectively debunks concerns that patients will suffer as a result of increased handoffs and breaks in the continuity of care. Rather than backtrack on the ACGME duty-hour rules, surgical leaders should focus on developing safe, resilient health systems that do not depend on overworked resident physicians. They also should recognize the changing expectations of postmillennial learners. To many current residents and medical students, 80-hour (or even 72-hour) work weeks and 24-hour shifts probably seem long enough. Although few surgical residents would ever acknowledge this publicly, I’m sure that many love to hear, “We can take care of this case without you. Go home, see your family, and come in fresh tomorrow.”
Dr. John Birkmeyer is professor of surgery at the Geisel School of Medicine at Dartmouth in Hanover, N.H. He wasn’t involved in the study; his comments appeared in an editorial (N Eng J Med. 2016 Feb 2. doi: 10.1056/NEJMe1516572).
JACKSONVILLE, FLA. – Accreditation Council for Graduate Medical Education (ACGME) rules that shortened surgery resident shifts and expanded breaks didn’t improve patient safety or surgery resident well-being in a trial presented at the Association for Academic Surgery/Society of University Surgeons Academic Surgical Congress.
“This national, prospective, randomized trial showed that flexible, less-restrictive duty-hour policies for surgical residents were noninferior to standard ACGME duty-hour policies,” wrote Dr. Karl Bilimoria, associate surgery professor at Northwestern University, Chicago, and associates. The work was published simultaneously Feb. 2 in the New England Journal of Medicine (doi: 10.1056/NEJMoa1515724).
Recent ACGME residency reforms were meant to reduce fatigue-related errors, but there have been concerns that they have come at the cost of increased handoffs and reduced education.
To get a handle on the situation, the investigators randomized 59 teaching-hospital surgery programs to standard ACGME duty hours and 58 others to a freer approach in the 2014-2015 academic year. Residents weren’t allowed to work more than 80 hours per week in either group, but hospitals in the flexible-hour arm were allowed to push residents past ACGME policy, working first-year residents longer than 16 hours per shift and others more than 28 hours, with breaks of less than 14 hours after 24-hour shifts and less than 8-10 hours after shorter ones.
Among the 138,691 adult general surgery cases during the academic year, there was no increase in 30-day rates of postoperative deaths or serious complications in the flexible group (9.1% vs. 9.0% with standard policy, P = .92) or secondary postoperative outcomes, based on risk-adjusted data from the American College of Surgeons’ National Surgical Quality Improvement Program (ACS NSQIP).
The 4,330 residents in the study filled out a multiple-choice questionnaire midway through the project in January 2015. Those in the flexible group said they weren’t significantly unhappier with the quality of their education (11.0% vs. 10.7% in the standard group, P = .86) or well-being (14.9% and 12.0%, P = .10). The investigators didn’t report the lengths of shifts or breaks.
There were no significant differences in resident-reported perceptions of fatigue on personal or patient safety. Residents in the flexible group were less likely to report leaving an operation (7.0% vs. 13.2%, P less than .001) or handing off patients with active issues (32% vs. 46.3%, P less than .001).
Flexible duty-hour residents “noted numerous benefits with respect to nearly all aspects of patient safety, continuity of care, surgical training, and professionalism. However, residents reported that less-restrictive duty-hour policies had a negative effect on [their] time with family and friends, time for extracurricular activities, rest, and health. Importantly … residents’ satisfaction with overall well-being did not differ significantly between study groups,” Dr. Karl Bilimoria and associates concluded.
The investigators “did not specifically collect data on needle sticks and car accidents, because these are notoriously challenging outcomes to capture in surveys,” they noted.
In an interview, Dr. Bilimoria commented, “Increasingly over time we’ve had more regulations of duty hours, and with each set of regulations the surgical community became increasingly concerned about patient handoffs and continuity of care, so our focus was to identify those policies that we thought affected continuity of care and work with the ACGME to waive those for the centers that were in the flexible arm of the study.”
His comments on the impact on residents: “The residents very clearly noted that the flexible policy arm provided better continuity of care, allowed them to take care of their patients in a way that they wanted to and stay with their patients in the operating room and at times when their patients were unstable.”
When asked if the findings could be extrapolated to these smaller nonparticipating centers, Dr. Bilimoria responded, “We captured the majority of residents, and we’re working on an analysis now that seeks to understand what the generalizability would be to those nonparticipating programs. That will be fairly enlightening as well.”
“95% of eligible programs participated in the trial, showing overwhelming support from the community for bringing high level data to this question. There had never before been a randomized trial nationally on this topic and for understanding and testing the notion of flexibility. They saw a need for both of those things.”
ACGME paid for the work, along with the American Board of Surgery and the American College of Surgeons. Dr. Bilimoria and five other authors reported payments from ACGME and the other entities.
UPDATE: This story was updated 2/2/16
JACKSONVILLE, FLA. – Accreditation Council for Graduate Medical Education (ACGME) rules that shortened surgery resident shifts and expanded breaks didn’t improve patient safety or surgery resident well-being in a trial presented at the Association for Academic Surgery/Society of University Surgeons Academic Surgical Congress.
“This national, prospective, randomized trial showed that flexible, less-restrictive duty-hour policies for surgical residents were noninferior to standard ACGME duty-hour policies,” wrote Dr. Karl Bilimoria, associate surgery professor at Northwestern University, Chicago, and associates. The work was published simultaneously Feb. 2 in the New England Journal of Medicine (doi: 10.1056/NEJMoa1515724).
Recent ACGME residency reforms were meant to reduce fatigue-related errors, but there have been concerns that they have come at the cost of increased handoffs and reduced education.
To get a handle on the situation, the investigators randomized 59 teaching-hospital surgery programs to standard ACGME duty hours and 58 others to a freer approach in the 2014-2015 academic year. Residents weren’t allowed to work more than 80 hours per week in either group, but hospitals in the flexible-hour arm were allowed to push residents past ACGME policy, working first-year residents longer than 16 hours per shift and others more than 28 hours, with breaks of less than 14 hours after 24-hour shifts and less than 8-10 hours after shorter ones.
Among the 138,691 adult general surgery cases during the academic year, there was no increase in 30-day rates of postoperative deaths or serious complications in the flexible group (9.1% vs. 9.0% with standard policy, P = .92) or secondary postoperative outcomes, based on risk-adjusted data from the American College of Surgeons’ National Surgical Quality Improvement Program (ACS NSQIP).
The 4,330 residents in the study filled out a multiple-choice questionnaire midway through the project in January 2015. Those in the flexible group said they weren’t significantly unhappier with the quality of their education (11.0% vs. 10.7% in the standard group, P = .86) or well-being (14.9% and 12.0%, P = .10). The investigators didn’t report the lengths of shifts or breaks.
There were no significant differences in resident-reported perceptions of fatigue on personal or patient safety. Residents in the flexible group were less likely to report leaving an operation (7.0% vs. 13.2%, P less than .001) or handing off patients with active issues (32% vs. 46.3%, P less than .001).
Flexible duty-hour residents “noted numerous benefits with respect to nearly all aspects of patient safety, continuity of care, surgical training, and professionalism. However, residents reported that less-restrictive duty-hour policies had a negative effect on [their] time with family and friends, time for extracurricular activities, rest, and health. Importantly … residents’ satisfaction with overall well-being did not differ significantly between study groups,” Dr. Karl Bilimoria and associates concluded.
The investigators “did not specifically collect data on needle sticks and car accidents, because these are notoriously challenging outcomes to capture in surveys,” they noted.
In an interview, Dr. Bilimoria commented, “Increasingly over time we’ve had more regulations of duty hours, and with each set of regulations the surgical community became increasingly concerned about patient handoffs and continuity of care, so our focus was to identify those policies that we thought affected continuity of care and work with the ACGME to waive those for the centers that were in the flexible arm of the study.”
His comments on the impact on residents: “The residents very clearly noted that the flexible policy arm provided better continuity of care, allowed them to take care of their patients in a way that they wanted to and stay with their patients in the operating room and at times when their patients were unstable.”
When asked if the findings could be extrapolated to these smaller nonparticipating centers, Dr. Bilimoria responded, “We captured the majority of residents, and we’re working on an analysis now that seeks to understand what the generalizability would be to those nonparticipating programs. That will be fairly enlightening as well.”
“95% of eligible programs participated in the trial, showing overwhelming support from the community for bringing high level data to this question. There had never before been a randomized trial nationally on this topic and for understanding and testing the notion of flexibility. They saw a need for both of those things.”
ACGME paid for the work, along with the American Board of Surgery and the American College of Surgeons. Dr. Bilimoria and five other authors reported payments from ACGME and the other entities.
UPDATE: This story was updated 2/2/16
AT THE ACADEMIC SURGICAL CONGRESS
Key clinical point: Patient outcome were not affected by the duty hours of general surgery residents.
Major finding: There was no increase in 30-day rates of postoperative deaths or serious complications when residents exceeded Accreditation Council for Graduate Medical Education (ACGME) hours (9.1% vs. 9.0% for residents not going beyond ACGME policy, P = .92).
Data source: 1-year randomized trial of 117 general surgery residency programs in the United States.
Disclosures: ACGME paid for the work, along with the American Board of Surgery and the American College of Surgeons. The lead author and five others reported payments from those groups.

New and Noteworthy Information—February 2016
Herpes zoster is associated with a short-term increased risk of stroke, and preventing infection may prevent this increased risk, according to a study published online ahead of print December 9, 2015, in Mayo Clinic Proceedings. In a community cohort study, researchers compared the risk of stroke and myocardial infarction at four time points in 4,862 adults with and without herpes zoster. People with herpes zoster had more risk or confounding factors for myocardial infarction and stroke, suggesting that they had worse health status overall. People with herpes zoster were at increased risk for stroke at three months after infection, compared with those without a history of herpes zoster. Herpes zoster was not associated with an increased risk of stroke or myocardial infarction at any point beyond three months.
Supplementation with 10,400 IU of vitamin D3 daily is safe and well-tolerated in patients with multiple sclerosis (MS), according to a study published online ahead of print December 30, 2015, in Neurology. Supplementation also may mitigate patients’ hyperactive immune response. In a double-blind, single-center study, 40 patients with relapsing-remitting MS were randomized to receive 10,400 IU or 800 IU of vitamin D3 daily for six months. Blood tests were performed at baseline and three and six months. In the high-dose group, researchers found a reduction in the proportion of interleukin-17+CD4+ T cells, CD161+CD4+ T cells, and effector memory CD4+ T cells, with a concomitant increase in the proportion of central memory CD4+ T cells and naive CD4+ T cells. These effects were not observed in the low-dose group.
Weight loss is associated with rapid progression of Parkinson’s disease, according to a study published online ahead of print January 11 in JAMA Neurology. Researchers analyzed data for 1,673 participants in the National Institute of Neurological Disorders and Stroke Exploratory Trials in PD Long-term Study-1. Of this cohort, 158 people lost weight, whereas 233 gained weight. After adjusting for covariates, researchers found that mean motor score increased by 1.48 more points per visit among people who lost weight than among people whose weight was stable. Mean motor score decreased by 0.51 points per visit for people who gained weight, relative to participants with stable weight. The observed difference in survival between the three BMI groups was not a significant outcome after data were adjusted for covariates.
Distal flow status is associated with risk for subsequent stroke in patients with symptomatic atherosclerotic vertebrobasilar occlusive disease, according to a study published online ahead of print December 21, 2015, in JAMA Neurology. Researchers conducted a prospective, blinded, longitudinal cohort study of 82 patients with recent vertebrobasilar transient ischemic attack or stroke and 50% or more atherosclerotic stenosis or occlusion in vertebral or basilar arteries. Distal flow status was low in 18 of the 72 participants included in the analysis and was significantly associated with risk for a subsequent vertebrobasilar stroke. The 12- and 24-month event-free survival rates were 78% and 70%, respectively, in the low-flow group, compared with 96% and 87%, respectively, in the normal-flow group. Hazard ratio for stroke was 11.55 among people with low distal flow.
The FDA has approved incobotulinumtoxinA for the treatment of upper limb spasticity in adult patients. The approval is based on the results of a randomized, multicenter, placebo-controlled trial. Treatment with incobotulinumtoxinA for adult upper limb spasticity resulted in statistically and clinically significant improvements in muscle tone. The product’s safety and efficacy were evaluated in multiple phase III clinical studies that included more than 400 patients. The safety profile for this indication is similar to that observed for other indications. FDA first approved incobotulinumtoxinA in August 2010 for the treatment of adults with cervical dystonia and blepharospasm. The most common adverse reactions include seizure, nasopharyngitis, dry mouth, and upper respiratory tract infection. Merz Pharma Group, headquartered in Raleigh, North Carolina, markets the product under the name Xeomin.
Cannabidiol may reduce seizure frequency and have an adequate safety profile in children and young adults with highly treatment-resistant epilepsy, according to a study published online ahead of print December 23, 2015, in Lancet Neurology. Patients received 2 to 5 mg/kg/day of oral cannabidiol, and the dose was increased until intolerance or to a maximum dose of 25 mg/kg/day or 50 mg/kg/day. Adverse events included somnolence, decreased appetite, diarrhea, fatigue, and convulsion. Five patients discontinued treatment because of an adverse event. Serious adverse events were reported in 48 patients, including one death regarded as unrelated to the study drug. Twenty patients had severe adverse events possibly related to cannabidiol use, the most common being status epilepticus. The median reduction in monthly motor seizures was 36.5%.
For every hour of reperfusion delay, the benefit of intra-arterial treatment for ischemic stroke decreases and the absolute risk difference for a good outcome is reduced by 6%, according to a study published online ahead of print December 21, 2015, in JAMA Neurology. The Multicenter Randomized Clinical Trial of Endovascular Treatment of Acute Ischemic Stroke in the Netherlands (MR CLEAN) compared intra-arterial treatment with no intra-arterial treatment in 500 patients. The median time to treatment was 260 minutes. Median time from treatment to reperfusion was 340 minutes. The researchers found an interaction between time from treatment to reperfusion and treatment effect, but not between time to treatment and treatment effect. The adjusted risk difference was 25.9% when reperfusion was achieved at three hours, 18.8% at four hours, and 6.7% at six hours.
Anticholinergic drugs are not associated with impaired cognitive performance among patients with Parkinson’s disease, according to a study published October 2, 2015, in Journal of Parkinson’s Disease. Using data from the Incidence of Cognitive Impairment in Cohorts with Longitudinal Evaluation—Parkinson’s Disease study, the researchers studied 195 patients with Parkinson’s disease and 84 controls. Patients’ detailed medication history, including over-the-counter drugs, was evaluated using the Anticholinergic Drug Scale (ADS). Each drug’s anticholinergic activity was classified on a scale from 0 to 3. Follow-up lasted 18 months. The investigators found no differences in global cognition, attention, memory, or executive function between patients with Parkinson’s disease who used anticholinergic drugs and those who did not. The proportion of patients with mild cognitive impairment was similar in both groups.
Anxiety symptoms are associated with an increased risk of dementia, according to a study published online ahead of print November 6, 2015, in Alzheimer’s & Dementia. The study included 1,082 fraternal and identical twins without dementia. Participants completed an assessment of anxiety symptoms in 1984 and were followed for 28 years. The twins also completed in-person tests every three years, answered questionnaires, and were screened for dementia throughout the study. Baseline anxiety score, independent of depressive symptoms, was significantly associated with incident dementia over follow-up. There was a 48% increased risk of dementia for people who had experienced high anxiety at any time, compared with those who had not. In co-twin analyses, the association between anxiety symptoms and dementia was greater for dizygotic, compared with monozygotic twins.
Common variants of MS4A6A and ABCA7 are associated with atrophy in cortical and hippocampal regions of the brain, according to a study published online ahead of print November 5, 2015, in Neurobiology of Aging. Researchers studied the relationship between the top 10 genetic variants associated with Alzheimer’s disease risk, excluding APOE, with cortical and hippocampal atrophy. They performed 1.5-T MRI to measure brain size and conducted genetic analyses for 50 cognitively normal participants and 98 participants with mild cognitive impairment. After explicit matching of cortical and hippocampal morphology, investigators computed in 3D the cortical thickness and hippocampal radial distance measures for each participant. MS4A6A rs610932 and ABCA7 rs3764650 had significant associations with cortical and hippocampal atrophy. The study may be the first to report the effect of these variants on neurodegeneration.
Anemia is associated with an increased risk of mild cognitive impairment (MCI), independent of traditional cardiovascular risk factors, according to a study published November 21, 2015, in Journal of Alzheimer’s Disease. Researchers examined 4,033 participants in a cohort study with available hemoglobin data and complete cognitive assessments. Participants’ age ranged between 50 and 80, and they were assessed between 2000 and 2003. Participants with anemia (ie, hemoglobin level less than 13 g/dl in men and less than 12 g/dl in women) had poorer cognitive performance in verbal memory and executive function, compared with people without anemia. The fully adjusted odds ratios for MCI, amnestic MCI, and nonamnestic MCI in anemic versus nonanemic participants were 1.92, 1.96, and 1.88, respectively.
By manipulating the WNT pathway, researchers efficiently differentiated human pluripotent stem cells to cells resembling central serotonin neurons, according to a study published in the January issue of Nature Biotechnology. For their investigation, the researchers used stem cells derived from embryos and stem cells derived from adult cells. The resulting serotonin neurons resembled those located in the rhombomeric segments 2-3 of the rostral raphe. They expressed a series of molecules essential for serotonergic development, including tryptophan hydroxylase 2. The cells also exhibited typical electrophysiologic properties and released serotonin in an activity-dependent manner. When treated with tramadol and escitalopram oxalate, the serotonin neurons released or took up serotonin in a dose- and time-dependent manner. These cells may help researchers to evaluate drug candidates, according to the investigators.
Vascular and Lewy body pathologies and vascular risk factors modify the risk of psychosis in Alzheimer’s disease, according to a study published November 30, 2015, in Journal of Alzheimer’s Disease. Researchers reviewed a group of patients with clinically diagnosed Alzheimer’s disease who had neuropathology data, as well as a group of neuropathologically definite cases of Alzheimer’s disease. They investigated the relationships between psychosis and clinical variables, neuropathologic correlates, and vascular risk factors. In all, 1,073 participants were included in this study. A total of 34% of clinically diagnosed patients and 37% of neuropathologically definite cases had psychotic symptoms during their illness. Overall, Lewy body pathology, subcortical arteriosclerotic leukoencephalopathy, and vascular risk factors, including a history of hypertension and diabetes, were associated with the development of psychosis.
—Kimberly Williams
Herpes zoster is associated with a short-term increased risk of stroke, and preventing infection may prevent this increased risk, according to a study published online ahead of print December 9, 2015, in Mayo Clinic Proceedings. In a community cohort study, researchers compared the risk of stroke and myocardial infarction at four time points in 4,862 adults with and without herpes zoster. People with herpes zoster had more risk or confounding factors for myocardial infarction and stroke, suggesting that they had worse health status overall. People with herpes zoster were at increased risk for stroke at three months after infection, compared with those without a history of herpes zoster. Herpes zoster was not associated with an increased risk of stroke or myocardial infarction at any point beyond three months.
Supplementation with 10,400 IU of vitamin D3 daily is safe and well-tolerated in patients with multiple sclerosis (MS), according to a study published online ahead of print December 30, 2015, in Neurology. Supplementation also may mitigate patients’ hyperactive immune response. In a double-blind, single-center study, 40 patients with relapsing-remitting MS were randomized to receive 10,400 IU or 800 IU of vitamin D3 daily for six months. Blood tests were performed at baseline and three and six months. In the high-dose group, researchers found a reduction in the proportion of interleukin-17+CD4+ T cells, CD161+CD4+ T cells, and effector memory CD4+ T cells, with a concomitant increase in the proportion of central memory CD4+ T cells and naive CD4+ T cells. These effects were not observed in the low-dose group.
Weight loss is associated with rapid progression of Parkinson’s disease, according to a study published online ahead of print January 11 in JAMA Neurology. Researchers analyzed data for 1,673 participants in the National Institute of Neurological Disorders and Stroke Exploratory Trials in PD Long-term Study-1. Of this cohort, 158 people lost weight, whereas 233 gained weight. After adjusting for covariates, researchers found that mean motor score increased by 1.48 more points per visit among people who lost weight than among people whose weight was stable. Mean motor score decreased by 0.51 points per visit for people who gained weight, relative to participants with stable weight. The observed difference in survival between the three BMI groups was not a significant outcome after data were adjusted for covariates.
Distal flow status is associated with risk for subsequent stroke in patients with symptomatic atherosclerotic vertebrobasilar occlusive disease, according to a study published online ahead of print December 21, 2015, in JAMA Neurology. Researchers conducted a prospective, blinded, longitudinal cohort study of 82 patients with recent vertebrobasilar transient ischemic attack or stroke and 50% or more atherosclerotic stenosis or occlusion in vertebral or basilar arteries. Distal flow status was low in 18 of the 72 participants included in the analysis and was significantly associated with risk for a subsequent vertebrobasilar stroke. The 12- and 24-month event-free survival rates were 78% and 70%, respectively, in the low-flow group, compared with 96% and 87%, respectively, in the normal-flow group. Hazard ratio for stroke was 11.55 among people with low distal flow.
The FDA has approved incobotulinumtoxinA for the treatment of upper limb spasticity in adult patients. The approval is based on the results of a randomized, multicenter, placebo-controlled trial. Treatment with incobotulinumtoxinA for adult upper limb spasticity resulted in statistically and clinically significant improvements in muscle tone. The product’s safety and efficacy were evaluated in multiple phase III clinical studies that included more than 400 patients. The safety profile for this indication is similar to that observed for other indications. FDA first approved incobotulinumtoxinA in August 2010 for the treatment of adults with cervical dystonia and blepharospasm. The most common adverse reactions include seizure, nasopharyngitis, dry mouth, and upper respiratory tract infection. Merz Pharma Group, headquartered in Raleigh, North Carolina, markets the product under the name Xeomin.
Cannabidiol may reduce seizure frequency and have an adequate safety profile in children and young adults with highly treatment-resistant epilepsy, according to a study published online ahead of print December 23, 2015, in Lancet Neurology. Patients received 2 to 5 mg/kg/day of oral cannabidiol, and the dose was increased until intolerance or to a maximum dose of 25 mg/kg/day or 50 mg/kg/day. Adverse events included somnolence, decreased appetite, diarrhea, fatigue, and convulsion. Five patients discontinued treatment because of an adverse event. Serious adverse events were reported in 48 patients, including one death regarded as unrelated to the study drug. Twenty patients had severe adverse events possibly related to cannabidiol use, the most common being status epilepticus. The median reduction in monthly motor seizures was 36.5%.
For every hour of reperfusion delay, the benefit of intra-arterial treatment for ischemic stroke decreases and the absolute risk difference for a good outcome is reduced by 6%, according to a study published online ahead of print December 21, 2015, in JAMA Neurology. The Multicenter Randomized Clinical Trial of Endovascular Treatment of Acute Ischemic Stroke in the Netherlands (MR CLEAN) compared intra-arterial treatment with no intra-arterial treatment in 500 patients. The median time to treatment was 260 minutes. Median time from treatment to reperfusion was 340 minutes. The researchers found an interaction between time from treatment to reperfusion and treatment effect, but not between time to treatment and treatment effect. The adjusted risk difference was 25.9% when reperfusion was achieved at three hours, 18.8% at four hours, and 6.7% at six hours.
Anticholinergic drugs are not associated with impaired cognitive performance among patients with Parkinson’s disease, according to a study published October 2, 2015, in Journal of Parkinson’s Disease. Using data from the Incidence of Cognitive Impairment in Cohorts with Longitudinal Evaluation—Parkinson’s Disease study, the researchers studied 195 patients with Parkinson’s disease and 84 controls. Patients’ detailed medication history, including over-the-counter drugs, was evaluated using the Anticholinergic Drug Scale (ADS). Each drug’s anticholinergic activity was classified on a scale from 0 to 3. Follow-up lasted 18 months. The investigators found no differences in global cognition, attention, memory, or executive function between patients with Parkinson’s disease who used anticholinergic drugs and those who did not. The proportion of patients with mild cognitive impairment was similar in both groups.
Anxiety symptoms are associated with an increased risk of dementia, according to a study published online ahead of print November 6, 2015, in Alzheimer’s & Dementia. The study included 1,082 fraternal and identical twins without dementia. Participants completed an assessment of anxiety symptoms in 1984 and were followed for 28 years. The twins also completed in-person tests every three years, answered questionnaires, and were screened for dementia throughout the study. Baseline anxiety score, independent of depressive symptoms, was significantly associated with incident dementia over follow-up. There was a 48% increased risk of dementia for people who had experienced high anxiety at any time, compared with those who had not. In co-twin analyses, the association between anxiety symptoms and dementia was greater for dizygotic, compared with monozygotic twins.
Common variants of MS4A6A and ABCA7 are associated with atrophy in cortical and hippocampal regions of the brain, according to a study published online ahead of print November 5, 2015, in Neurobiology of Aging. Researchers studied the relationship between the top 10 genetic variants associated with Alzheimer’s disease risk, excluding APOE, with cortical and hippocampal atrophy. They performed 1.5-T MRI to measure brain size and conducted genetic analyses for 50 cognitively normal participants and 98 participants with mild cognitive impairment. After explicit matching of cortical and hippocampal morphology, investigators computed in 3D the cortical thickness and hippocampal radial distance measures for each participant. MS4A6A rs610932 and ABCA7 rs3764650 had significant associations with cortical and hippocampal atrophy. The study may be the first to report the effect of these variants on neurodegeneration.
Anemia is associated with an increased risk of mild cognitive impairment (MCI), independent of traditional cardiovascular risk factors, according to a study published November 21, 2015, in Journal of Alzheimer’s Disease. Researchers examined 4,033 participants in a cohort study with available hemoglobin data and complete cognitive assessments. Participants’ age ranged between 50 and 80, and they were assessed between 2000 and 2003. Participants with anemia (ie, hemoglobin level less than 13 g/dl in men and less than 12 g/dl in women) had poorer cognitive performance in verbal memory and executive function, compared with people without anemia. The fully adjusted odds ratios for MCI, amnestic MCI, and nonamnestic MCI in anemic versus nonanemic participants were 1.92, 1.96, and 1.88, respectively.
By manipulating the WNT pathway, researchers efficiently differentiated human pluripotent stem cells to cells resembling central serotonin neurons, according to a study published in the January issue of Nature Biotechnology. For their investigation, the researchers used stem cells derived from embryos and stem cells derived from adult cells. The resulting serotonin neurons resembled those located in the rhombomeric segments 2-3 of the rostral raphe. They expressed a series of molecules essential for serotonergic development, including tryptophan hydroxylase 2. The cells also exhibited typical electrophysiologic properties and released serotonin in an activity-dependent manner. When treated with tramadol and escitalopram oxalate, the serotonin neurons released or took up serotonin in a dose- and time-dependent manner. These cells may help researchers to evaluate drug candidates, according to the investigators.
Vascular and Lewy body pathologies and vascular risk factors modify the risk of psychosis in Alzheimer’s disease, according to a study published November 30, 2015, in Journal of Alzheimer’s Disease. Researchers reviewed a group of patients with clinically diagnosed Alzheimer’s disease who had neuropathology data, as well as a group of neuropathologically definite cases of Alzheimer’s disease. They investigated the relationships between psychosis and clinical variables, neuropathologic correlates, and vascular risk factors. In all, 1,073 participants were included in this study. A total of 34% of clinically diagnosed patients and 37% of neuropathologically definite cases had psychotic symptoms during their illness. Overall, Lewy body pathology, subcortical arteriosclerotic leukoencephalopathy, and vascular risk factors, including a history of hypertension and diabetes, were associated with the development of psychosis.
—Kimberly Williams
Herpes zoster is associated with a short-term increased risk of stroke, and preventing infection may prevent this increased risk, according to a study published online ahead of print December 9, 2015, in Mayo Clinic Proceedings. In a community cohort study, researchers compared the risk of stroke and myocardial infarction at four time points in 4,862 adults with and without herpes zoster. People with herpes zoster had more risk or confounding factors for myocardial infarction and stroke, suggesting that they had worse health status overall. People with herpes zoster were at increased risk for stroke at three months after infection, compared with those without a history of herpes zoster. Herpes zoster was not associated with an increased risk of stroke or myocardial infarction at any point beyond three months.
Supplementation with 10,400 IU of vitamin D3 daily is safe and well-tolerated in patients with multiple sclerosis (MS), according to a study published online ahead of print December 30, 2015, in Neurology. Supplementation also may mitigate patients’ hyperactive immune response. In a double-blind, single-center study, 40 patients with relapsing-remitting MS were randomized to receive 10,400 IU or 800 IU of vitamin D3 daily for six months. Blood tests were performed at baseline and three and six months. In the high-dose group, researchers found a reduction in the proportion of interleukin-17+CD4+ T cells, CD161+CD4+ T cells, and effector memory CD4+ T cells, with a concomitant increase in the proportion of central memory CD4+ T cells and naive CD4+ T cells. These effects were not observed in the low-dose group.
Weight loss is associated with rapid progression of Parkinson’s disease, according to a study published online ahead of print January 11 in JAMA Neurology. Researchers analyzed data for 1,673 participants in the National Institute of Neurological Disorders and Stroke Exploratory Trials in PD Long-term Study-1. Of this cohort, 158 people lost weight, whereas 233 gained weight. After adjusting for covariates, researchers found that mean motor score increased by 1.48 more points per visit among people who lost weight than among people whose weight was stable. Mean motor score decreased by 0.51 points per visit for people who gained weight, relative to participants with stable weight. The observed difference in survival between the three BMI groups was not a significant outcome after data were adjusted for covariates.
Distal flow status is associated with risk for subsequent stroke in patients with symptomatic atherosclerotic vertebrobasilar occlusive disease, according to a study published online ahead of print December 21, 2015, in JAMA Neurology. Researchers conducted a prospective, blinded, longitudinal cohort study of 82 patients with recent vertebrobasilar transient ischemic attack or stroke and 50% or more atherosclerotic stenosis or occlusion in vertebral or basilar arteries. Distal flow status was low in 18 of the 72 participants included in the analysis and was significantly associated with risk for a subsequent vertebrobasilar stroke. The 12- and 24-month event-free survival rates were 78% and 70%, respectively, in the low-flow group, compared with 96% and 87%, respectively, in the normal-flow group. Hazard ratio for stroke was 11.55 among people with low distal flow.
The FDA has approved incobotulinumtoxinA for the treatment of upper limb spasticity in adult patients. The approval is based on the results of a randomized, multicenter, placebo-controlled trial. Treatment with incobotulinumtoxinA for adult upper limb spasticity resulted in statistically and clinically significant improvements in muscle tone. The product’s safety and efficacy were evaluated in multiple phase III clinical studies that included more than 400 patients. The safety profile for this indication is similar to that observed for other indications. FDA first approved incobotulinumtoxinA in August 2010 for the treatment of adults with cervical dystonia and blepharospasm. The most common adverse reactions include seizure, nasopharyngitis, dry mouth, and upper respiratory tract infection. Merz Pharma Group, headquartered in Raleigh, North Carolina, markets the product under the name Xeomin.
Cannabidiol may reduce seizure frequency and have an adequate safety profile in children and young adults with highly treatment-resistant epilepsy, according to a study published online ahead of print December 23, 2015, in Lancet Neurology. Patients received 2 to 5 mg/kg/day of oral cannabidiol, and the dose was increased until intolerance or to a maximum dose of 25 mg/kg/day or 50 mg/kg/day. Adverse events included somnolence, decreased appetite, diarrhea, fatigue, and convulsion. Five patients discontinued treatment because of an adverse event. Serious adverse events were reported in 48 patients, including one death regarded as unrelated to the study drug. Twenty patients had severe adverse events possibly related to cannabidiol use, the most common being status epilepticus. The median reduction in monthly motor seizures was 36.5%.
For every hour of reperfusion delay, the benefit of intra-arterial treatment for ischemic stroke decreases and the absolute risk difference for a good outcome is reduced by 6%, according to a study published online ahead of print December 21, 2015, in JAMA Neurology. The Multicenter Randomized Clinical Trial of Endovascular Treatment of Acute Ischemic Stroke in the Netherlands (MR CLEAN) compared intra-arterial treatment with no intra-arterial treatment in 500 patients. The median time to treatment was 260 minutes. Median time from treatment to reperfusion was 340 minutes. The researchers found an interaction between time from treatment to reperfusion and treatment effect, but not between time to treatment and treatment effect. The adjusted risk difference was 25.9% when reperfusion was achieved at three hours, 18.8% at four hours, and 6.7% at six hours.
Anticholinergic drugs are not associated with impaired cognitive performance among patients with Parkinson’s disease, according to a study published October 2, 2015, in Journal of Parkinson’s Disease. Using data from the Incidence of Cognitive Impairment in Cohorts with Longitudinal Evaluation—Parkinson’s Disease study, the researchers studied 195 patients with Parkinson’s disease and 84 controls. Patients’ detailed medication history, including over-the-counter drugs, was evaluated using the Anticholinergic Drug Scale (ADS). Each drug’s anticholinergic activity was classified on a scale from 0 to 3. Follow-up lasted 18 months. The investigators found no differences in global cognition, attention, memory, or executive function between patients with Parkinson’s disease who used anticholinergic drugs and those who did not. The proportion of patients with mild cognitive impairment was similar in both groups.
Anxiety symptoms are associated with an increased risk of dementia, according to a study published online ahead of print November 6, 2015, in Alzheimer’s & Dementia. The study included 1,082 fraternal and identical twins without dementia. Participants completed an assessment of anxiety symptoms in 1984 and were followed for 28 years. The twins also completed in-person tests every three years, answered questionnaires, and were screened for dementia throughout the study. Baseline anxiety score, independent of depressive symptoms, was significantly associated with incident dementia over follow-up. There was a 48% increased risk of dementia for people who had experienced high anxiety at any time, compared with those who had not. In co-twin analyses, the association between anxiety symptoms and dementia was greater for dizygotic, compared with monozygotic twins.
Common variants of MS4A6A and ABCA7 are associated with atrophy in cortical and hippocampal regions of the brain, according to a study published online ahead of print November 5, 2015, in Neurobiology of Aging. Researchers studied the relationship between the top 10 genetic variants associated with Alzheimer’s disease risk, excluding APOE, with cortical and hippocampal atrophy. They performed 1.5-T MRI to measure brain size and conducted genetic analyses for 50 cognitively normal participants and 98 participants with mild cognitive impairment. After explicit matching of cortical and hippocampal morphology, investigators computed in 3D the cortical thickness and hippocampal radial distance measures for each participant. MS4A6A rs610932 and ABCA7 rs3764650 had significant associations with cortical and hippocampal atrophy. The study may be the first to report the effect of these variants on neurodegeneration.
Anemia is associated with an increased risk of mild cognitive impairment (MCI), independent of traditional cardiovascular risk factors, according to a study published November 21, 2015, in Journal of Alzheimer’s Disease. Researchers examined 4,033 participants in a cohort study with available hemoglobin data and complete cognitive assessments. Participants’ age ranged between 50 and 80, and they were assessed between 2000 and 2003. Participants with anemia (ie, hemoglobin level less than 13 g/dl in men and less than 12 g/dl in women) had poorer cognitive performance in verbal memory and executive function, compared with people without anemia. The fully adjusted odds ratios for MCI, amnestic MCI, and nonamnestic MCI in anemic versus nonanemic participants were 1.92, 1.96, and 1.88, respectively.
By manipulating the WNT pathway, researchers efficiently differentiated human pluripotent stem cells to cells resembling central serotonin neurons, according to a study published in the January issue of Nature Biotechnology. For their investigation, the researchers used stem cells derived from embryos and stem cells derived from adult cells. The resulting serotonin neurons resembled those located in the rhombomeric segments 2-3 of the rostral raphe. They expressed a series of molecules essential for serotonergic development, including tryptophan hydroxylase 2. The cells also exhibited typical electrophysiologic properties and released serotonin in an activity-dependent manner. When treated with tramadol and escitalopram oxalate, the serotonin neurons released or took up serotonin in a dose- and time-dependent manner. These cells may help researchers to evaluate drug candidates, according to the investigators.
Vascular and Lewy body pathologies and vascular risk factors modify the risk of psychosis in Alzheimer’s disease, according to a study published November 30, 2015, in Journal of Alzheimer’s Disease. Researchers reviewed a group of patients with clinically diagnosed Alzheimer’s disease who had neuropathology data, as well as a group of neuropathologically definite cases of Alzheimer’s disease. They investigated the relationships between psychosis and clinical variables, neuropathologic correlates, and vascular risk factors. In all, 1,073 participants were included in this study. A total of 34% of clinically diagnosed patients and 37% of neuropathologically definite cases had psychotic symptoms during their illness. Overall, Lewy body pathology, subcortical arteriosclerotic leukoencephalopathy, and vascular risk factors, including a history of hypertension and diabetes, were associated with the development of psychosis.
—Kimberly Williams

Molecular Biomarkers May Predict Conversion to MS
BARCELONA—CSF levels of chitinase 3-like-1 protein and of the light subunit of neurofilaments predict conversion to multiple sclerosis (MS) and the development of neurologic disability in patients with clinically isolated syndrome (CIS), according to an overview presented at the 31st Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS). Ongoing research will indicate whether combinations of CSF biomarkers provide more accurate prognoses than individual biomarkers do.
Clinical manifestations, disease course, radiologic findings, histopathologic characteristics of lesions, and response to treatment vary greatly among patients with MS. The heterogeneity of the disease creates a need for reliable biomarkers that may improve diagnosis, stratification, prediction of disease course, identification of new therapies, and development of personalized therapy, said Manuel Comabella, MD, Head of the Laboratory of the Clinical Neuroimmunology Unit at the MS Center of Catalonia in Barcelona.
Neurofilament Proteins
Previous studies indicate that CSF oligoclonal bands and the 14-3-3 protein individually predict conversion from CIS to MS. Newer research indicates that the levels of neurofilament proteins in CSF can help neurologists quantify axonal damage. Neurofilaments comprise heavy, medium, and light subunits, and neurofilaments’ axonal diameter is influenced by the degree to which they are phosphorylated. Pathologic processes that cause axonal damage release the neurofilament proteins into the CSF, where they can be detected.
Teunissen et al observed that CSF levels of the light subunit of the neurofilaments were significantly higher in patients with CIS who converted to clinically definite MS, compared with patients with CIS who did not convert. More recently, Modvig and colleagues found that levels of the light subunit of the neurofilaments predicted long-term disability, as measured by the MS severity scale and the nine-hole peg test, in patients with optic neuritis after more than three years of follow-up.
Levels of the light subunit of the neurofilaments also may predict disease progression in patients with MS. Investigators found that conversion from relapsing-remitting MS to secondary progressive MS was more likely among patients with high levels of the light subunit of the neurofilaments, compared with patients with intermediate or undetectable levels. Also, case reviews indicate that common MS therapies such as natalizumab or fingolimod modify CSF levels of the light subunit of the neurofilaments, “which suggests that they can be used as a biomarker to monitor the response to therapies and … the neuroprotective effects of treatments,” said Dr. Comabella.
Chitinase 3-Like-1 Protein
Investigators also have examined chitinase 3-like-1 protein as a potential biomarker. The protein results from strong proinflammatory cytokines such as TNF-α and IL-1β. Serum and plasma levels of the protein usually are elevated in people with disorders characterized by chronic inflammation.
Dr. Comabella and colleagues conducted a study to identify CSF biomarkers associated with conversion from CIS to MS. The investigators classified patients into two groups. One group included patients with CIS who had a normal MRI and were positive for IgG oligoclonal bands at baseline, and who converted to clinically definite MS during follow-up. The other group included patients with CIS who had a normal MRI at baseline and after five years of follow-up, were negative for IgG oligoclonal bands, and did not convert to MS during follow-up.
The investigators applied a mass-spectrometry-based proteomic approach to identify proteins that were present in different quantities in the CSF of patients who converted to MS, compared with those who did not. They found that CSF levels of chitinase 3-like-1 protein were significantly higher in patients who converted to MS, compared with those who did not. CSF levels of the protein also correlated with MRI abnormalities at baseline and with disability progression during follow-up. In addition, high levels of chitinase 3-like-1 protein were associated with a shorter time to MS.
These results prompted Dr. Comabella’s group to begin a study evaluating chitinase 3-like-1 protein as a prognostic biomarker for conversion to MS. The investigators examined more than 800 CSF samples from patients with CIS. They used multivariable Cox regression models to investigate the association between CSF chitinase 3-like-1 protein levels and the time to MS, based on Poser or 2005 McDonald criteria, and also based on the time to reach an Expanded Disability Status Scale score of 3.
CSF levels of chitinase 3-like-1 protein were a risk factor for conversion to MS, independent of other predictors such as IgG oligoclonal bands and MRI. Chitinase 3-like-1 protein levels were the only significant independent risk factor associated with the development of neurologic disability, with a hazard ratio of approximately 4. “A chitinase level of 170 ng/mL was the best cutoff that allowed us to classify protein levels into high and low, and 44% of the patients had protein levels above this cutoff,” said Dr. Comabella. Levels higher than 170 ng/mL were associated with a shorter time to MS and with faster development of disability.
Dr. Comabella’s results are consistent with those of other recent investigations of the protein. Using a similar proteomic approach, Hinsinger et al identified chitinase 3-like-1 protein as one of the best predictors of conversion to MS. They identified 189 ng/ml as the cutoff that best classified protein levels as high or low. Levels above the cutoff were associated with shorter time to MS, based on the 2005 McDonald criteria. Also, Modvig’s study of patients with optic neuritis found that chitinase 3-like-1 protein, MRI, and age together were the best predictor of clinically definite MS. The protein also predicted long-term cognitive impairment in that study.
Do Biomarker Combinations Improve Predictions?
Combinations of biomarkers may improve prognostic predictions for patients with CIS, compared with individual biomarkers, said Dr. Comabella. He and his colleagues are investigating the predictive value of the combination of chitinase 3-like-1 protein, dipeptidase, and semaphorin 7A. Data suggest that this combination is better at distinguishing between patients with CIS who convert to MS and those who do not, compared with each biomarker considered individually.
Dr. Comabella’s group also is investigating the potential neurotoxic effect of chitinase 3-like-1 protein. They are adding the protein to primary cultures of neurons at the concentrations above and below the cutoff of 170 ng/ml. Preliminary data suggest that the protein is neurotoxic.
—Erik Greb
Suggested Reading
Comabella M, Fernández M, Martin R, et al. Cerebrospinal fluid chitinase 3-like 1 levels are associated with conversion to multiple sclerosis. Brain. 2010;133(Pt 4):1082-1093.
Hinsinger G, Galéotti N, Nabholz N, et al. Chitinase 3-like proteins as diagnostic and prognostic biomarkers of multiple sclerosis. Mult Scler. 2015;21(10):1251-1261.
Modvig S, Degn M, Roed H, et al. Cerebrospinal fluid levels of chitinase 3-like 1 and neurofilament light chain predict multiple sclerosis development and disability after optic neuritis. Mult Scler. 2015;21(14):1761-1770.
BARCELONA—CSF levels of chitinase 3-like-1 protein and of the light subunit of neurofilaments predict conversion to multiple sclerosis (MS) and the development of neurologic disability in patients with clinically isolated syndrome (CIS), according to an overview presented at the 31st Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS). Ongoing research will indicate whether combinations of CSF biomarkers provide more accurate prognoses than individual biomarkers do.
Clinical manifestations, disease course, radiologic findings, histopathologic characteristics of lesions, and response to treatment vary greatly among patients with MS. The heterogeneity of the disease creates a need for reliable biomarkers that may improve diagnosis, stratification, prediction of disease course, identification of new therapies, and development of personalized therapy, said Manuel Comabella, MD, Head of the Laboratory of the Clinical Neuroimmunology Unit at the MS Center of Catalonia in Barcelona.
Neurofilament Proteins
Previous studies indicate that CSF oligoclonal bands and the 14-3-3 protein individually predict conversion from CIS to MS. Newer research indicates that the levels of neurofilament proteins in CSF can help neurologists quantify axonal damage. Neurofilaments comprise heavy, medium, and light subunits, and neurofilaments’ axonal diameter is influenced by the degree to which they are phosphorylated. Pathologic processes that cause axonal damage release the neurofilament proteins into the CSF, where they can be detected.
Teunissen et al observed that CSF levels of the light subunit of the neurofilaments were significantly higher in patients with CIS who converted to clinically definite MS, compared with patients with CIS who did not convert. More recently, Modvig and colleagues found that levels of the light subunit of the neurofilaments predicted long-term disability, as measured by the MS severity scale and the nine-hole peg test, in patients with optic neuritis after more than three years of follow-up.
Levels of the light subunit of the neurofilaments also may predict disease progression in patients with MS. Investigators found that conversion from relapsing-remitting MS to secondary progressive MS was more likely among patients with high levels of the light subunit of the neurofilaments, compared with patients with intermediate or undetectable levels. Also, case reviews indicate that common MS therapies such as natalizumab or fingolimod modify CSF levels of the light subunit of the neurofilaments, “which suggests that they can be used as a biomarker to monitor the response to therapies and … the neuroprotective effects of treatments,” said Dr. Comabella.
Chitinase 3-Like-1 Protein
Investigators also have examined chitinase 3-like-1 protein as a potential biomarker. The protein results from strong proinflammatory cytokines such as TNF-α and IL-1β. Serum and plasma levels of the protein usually are elevated in people with disorders characterized by chronic inflammation.
Dr. Comabella and colleagues conducted a study to identify CSF biomarkers associated with conversion from CIS to MS. The investigators classified patients into two groups. One group included patients with CIS who had a normal MRI and were positive for IgG oligoclonal bands at baseline, and who converted to clinically definite MS during follow-up. The other group included patients with CIS who had a normal MRI at baseline and after five years of follow-up, were negative for IgG oligoclonal bands, and did not convert to MS during follow-up.
The investigators applied a mass-spectrometry-based proteomic approach to identify proteins that were present in different quantities in the CSF of patients who converted to MS, compared with those who did not. They found that CSF levels of chitinase 3-like-1 protein were significantly higher in patients who converted to MS, compared with those who did not. CSF levels of the protein also correlated with MRI abnormalities at baseline and with disability progression during follow-up. In addition, high levels of chitinase 3-like-1 protein were associated with a shorter time to MS.
These results prompted Dr. Comabella’s group to begin a study evaluating chitinase 3-like-1 protein as a prognostic biomarker for conversion to MS. The investigators examined more than 800 CSF samples from patients with CIS. They used multivariable Cox regression models to investigate the association between CSF chitinase 3-like-1 protein levels and the time to MS, based on Poser or 2005 McDonald criteria, and also based on the time to reach an Expanded Disability Status Scale score of 3.
CSF levels of chitinase 3-like-1 protein were a risk factor for conversion to MS, independent of other predictors such as IgG oligoclonal bands and MRI. Chitinase 3-like-1 protein levels were the only significant independent risk factor associated with the development of neurologic disability, with a hazard ratio of approximately 4. “A chitinase level of 170 ng/mL was the best cutoff that allowed us to classify protein levels into high and low, and 44% of the patients had protein levels above this cutoff,” said Dr. Comabella. Levels higher than 170 ng/mL were associated with a shorter time to MS and with faster development of disability.
Dr. Comabella’s results are consistent with those of other recent investigations of the protein. Using a similar proteomic approach, Hinsinger et al identified chitinase 3-like-1 protein as one of the best predictors of conversion to MS. They identified 189 ng/ml as the cutoff that best classified protein levels as high or low. Levels above the cutoff were associated with shorter time to MS, based on the 2005 McDonald criteria. Also, Modvig’s study of patients with optic neuritis found that chitinase 3-like-1 protein, MRI, and age together were the best predictor of clinically definite MS. The protein also predicted long-term cognitive impairment in that study.
Do Biomarker Combinations Improve Predictions?
Combinations of biomarkers may improve prognostic predictions for patients with CIS, compared with individual biomarkers, said Dr. Comabella. He and his colleagues are investigating the predictive value of the combination of chitinase 3-like-1 protein, dipeptidase, and semaphorin 7A. Data suggest that this combination is better at distinguishing between patients with CIS who convert to MS and those who do not, compared with each biomarker considered individually.
Dr. Comabella’s group also is investigating the potential neurotoxic effect of chitinase 3-like-1 protein. They are adding the protein to primary cultures of neurons at the concentrations above and below the cutoff of 170 ng/ml. Preliminary data suggest that the protein is neurotoxic.
—Erik Greb
BARCELONA—CSF levels of chitinase 3-like-1 protein and of the light subunit of neurofilaments predict conversion to multiple sclerosis (MS) and the development of neurologic disability in patients with clinically isolated syndrome (CIS), according to an overview presented at the 31st Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS). Ongoing research will indicate whether combinations of CSF biomarkers provide more accurate prognoses than individual biomarkers do.
Clinical manifestations, disease course, radiologic findings, histopathologic characteristics of lesions, and response to treatment vary greatly among patients with MS. The heterogeneity of the disease creates a need for reliable biomarkers that may improve diagnosis, stratification, prediction of disease course, identification of new therapies, and development of personalized therapy, said Manuel Comabella, MD, Head of the Laboratory of the Clinical Neuroimmunology Unit at the MS Center of Catalonia in Barcelona.
Neurofilament Proteins
Previous studies indicate that CSF oligoclonal bands and the 14-3-3 protein individually predict conversion from CIS to MS. Newer research indicates that the levels of neurofilament proteins in CSF can help neurologists quantify axonal damage. Neurofilaments comprise heavy, medium, and light subunits, and neurofilaments’ axonal diameter is influenced by the degree to which they are phosphorylated. Pathologic processes that cause axonal damage release the neurofilament proteins into the CSF, where they can be detected.
Teunissen et al observed that CSF levels of the light subunit of the neurofilaments were significantly higher in patients with CIS who converted to clinically definite MS, compared with patients with CIS who did not convert. More recently, Modvig and colleagues found that levels of the light subunit of the neurofilaments predicted long-term disability, as measured by the MS severity scale and the nine-hole peg test, in patients with optic neuritis after more than three years of follow-up.
Levels of the light subunit of the neurofilaments also may predict disease progression in patients with MS. Investigators found that conversion from relapsing-remitting MS to secondary progressive MS was more likely among patients with high levels of the light subunit of the neurofilaments, compared with patients with intermediate or undetectable levels. Also, case reviews indicate that common MS therapies such as natalizumab or fingolimod modify CSF levels of the light subunit of the neurofilaments, “which suggests that they can be used as a biomarker to monitor the response to therapies and … the neuroprotective effects of treatments,” said Dr. Comabella.
Chitinase 3-Like-1 Protein
Investigators also have examined chitinase 3-like-1 protein as a potential biomarker. The protein results from strong proinflammatory cytokines such as TNF-α and IL-1β. Serum and plasma levels of the protein usually are elevated in people with disorders characterized by chronic inflammation.
Dr. Comabella and colleagues conducted a study to identify CSF biomarkers associated with conversion from CIS to MS. The investigators classified patients into two groups. One group included patients with CIS who had a normal MRI and were positive for IgG oligoclonal bands at baseline, and who converted to clinically definite MS during follow-up. The other group included patients with CIS who had a normal MRI at baseline and after five years of follow-up, were negative for IgG oligoclonal bands, and did not convert to MS during follow-up.
The investigators applied a mass-spectrometry-based proteomic approach to identify proteins that were present in different quantities in the CSF of patients who converted to MS, compared with those who did not. They found that CSF levels of chitinase 3-like-1 protein were significantly higher in patients who converted to MS, compared with those who did not. CSF levels of the protein also correlated with MRI abnormalities at baseline and with disability progression during follow-up. In addition, high levels of chitinase 3-like-1 protein were associated with a shorter time to MS.
These results prompted Dr. Comabella’s group to begin a study evaluating chitinase 3-like-1 protein as a prognostic biomarker for conversion to MS. The investigators examined more than 800 CSF samples from patients with CIS. They used multivariable Cox regression models to investigate the association between CSF chitinase 3-like-1 protein levels and the time to MS, based on Poser or 2005 McDonald criteria, and also based on the time to reach an Expanded Disability Status Scale score of 3.
CSF levels of chitinase 3-like-1 protein were a risk factor for conversion to MS, independent of other predictors such as IgG oligoclonal bands and MRI. Chitinase 3-like-1 protein levels were the only significant independent risk factor associated with the development of neurologic disability, with a hazard ratio of approximately 4. “A chitinase level of 170 ng/mL was the best cutoff that allowed us to classify protein levels into high and low, and 44% of the patients had protein levels above this cutoff,” said Dr. Comabella. Levels higher than 170 ng/mL were associated with a shorter time to MS and with faster development of disability.
Dr. Comabella’s results are consistent with those of other recent investigations of the protein. Using a similar proteomic approach, Hinsinger et al identified chitinase 3-like-1 protein as one of the best predictors of conversion to MS. They identified 189 ng/ml as the cutoff that best classified protein levels as high or low. Levels above the cutoff were associated with shorter time to MS, based on the 2005 McDonald criteria. Also, Modvig’s study of patients with optic neuritis found that chitinase 3-like-1 protein, MRI, and age together were the best predictor of clinically definite MS. The protein also predicted long-term cognitive impairment in that study.
Do Biomarker Combinations Improve Predictions?
Combinations of biomarkers may improve prognostic predictions for patients with CIS, compared with individual biomarkers, said Dr. Comabella. He and his colleagues are investigating the predictive value of the combination of chitinase 3-like-1 protein, dipeptidase, and semaphorin 7A. Data suggest that this combination is better at distinguishing between patients with CIS who convert to MS and those who do not, compared with each biomarker considered individually.
Dr. Comabella’s group also is investigating the potential neurotoxic effect of chitinase 3-like-1 protein. They are adding the protein to primary cultures of neurons at the concentrations above and below the cutoff of 170 ng/ml. Preliminary data suggest that the protein is neurotoxic.
—Erik Greb
Suggested Reading
Comabella M, Fernández M, Martin R, et al. Cerebrospinal fluid chitinase 3-like 1 levels are associated with conversion to multiple sclerosis. Brain. 2010;133(Pt 4):1082-1093.
Hinsinger G, Galéotti N, Nabholz N, et al. Chitinase 3-like proteins as diagnostic and prognostic biomarkers of multiple sclerosis. Mult Scler. 2015;21(10):1251-1261.
Modvig S, Degn M, Roed H, et al. Cerebrospinal fluid levels of chitinase 3-like 1 and neurofilament light chain predict multiple sclerosis development and disability after optic neuritis. Mult Scler. 2015;21(14):1761-1770.
Suggested Reading
Comabella M, Fernández M, Martin R, et al. Cerebrospinal fluid chitinase 3-like 1 levels are associated with conversion to multiple sclerosis. Brain. 2010;133(Pt 4):1082-1093.
Hinsinger G, Galéotti N, Nabholz N, et al. Chitinase 3-like proteins as diagnostic and prognostic biomarkers of multiple sclerosis. Mult Scler. 2015;21(10):1251-1261.
Modvig S, Degn M, Roed H, et al. Cerebrospinal fluid levels of chitinase 3-like 1 and neurofilament light chain predict multiple sclerosis development and disability after optic neuritis. Mult Scler. 2015;21(14):1761-1770.
Electronic Screen Exposure Is Associated With Migraine in Young Adults
Too much time in front of electronic screens can increase the risk of headaches and migraine in young adults, according to research published online ahead of print December 2, 2015, in Cephalalgia.
Previous studies have observed associations between screen time exposure and headaches in children between ages 10 and 12 and adolescents. Data also have found an association between screen time exposure and low-back and shoulder pain in adolescents. “This [information] had led to speculation that the high amount of screen time exposure among students of higher education institutions may be correlated with the high prevalence of headache and migraine observed in this population,” said Ilaria Montagni, PhD, a research fellow at the University of Bordeaux in Talence, France.
Dr. Montagni and her coinvestigators studied 4,927 individuals in France, all of whom were age 18 or older and part of the Internet-based Students Health Research Enterprise (i-Share) project cohort, which is an ongoing, prospective, population-based study of students at French-speaking universities and institutions of higher education. The mean age of the students was 20.8, and 75.5% were female.
Subjects completed self-reported surveys on the average amount of time they spend in front of screens during the following five activities: computer or tablet work, playing video games on a computer or tablet, Internet surfing on a computer or tablet, watching videos on a computer or tablet, and using a smartphone. All questions were scored using a five-point scale. A score of 0 indicated never, 1 indicated less than 30 minutes, 2 indicated 30 minutes to two hours, 3 indicated four to eight hours, and 5 indicated eight hours or more. Scores from the surveys were divided into quartiles of very low, low, high, and very high screen-time exposure.
Surveys also asked if participants had experienced any headaches that had lasted several hours in the previous 12 months. Participants who answered negatively were classified as “no headache,” while those who answered positively were asked a series of follow-up questions related to symptom type and severity, sensitivity to light or sound, nausea, vomiting, and disruption of daily routines. To establish a classification of migraine, the investigators used the “probable migraine” category of the International Classification of Headache Disorders, third edition. With these data, the investigators used multinomial logistic regression models to calculate odds ratios of any relationship between screen time exposure and the presence and severity of headaches.
Of the 4,927 participants, 2,773 (56.3%) reported no headaches. In all, 710 (14.4%) participants reported a nonmigraine headache, 791 (16.1%) reported migraine without aura, and 653 (13.3%) reported migraine with aura. In comparisons against very low screen time exposure, very high exposure increased the likelihood of having migraine by 37%, which resulted in a statistically significant 50% greater odds for migraine without aura, but not for migraine with aura.
“Students reporting very high screen time exposure were more likely to be male, to be older, to have higher BMI, and to consume cannabis [and] were also more likely to report nonmigraine headache or migraine,” the authors noted. Furthermore, higher exposure to screens was a significant indicator of recurrent headaches in adolescent males, and the same indicator was seen in adolescent females who spent more time on the computer and in front of the TV.
—Deepak Chitnis
Suggested Reading
Montagni I, Guichard E, Carpenet C, et al. Screen time exposure and reporting of headaches in young adults: A cross-sectional study. Cephalalgia. 2015 Dec 2 [Epub ahead of print].
Too much time in front of electronic screens can increase the risk of headaches and migraine in young adults, according to research published online ahead of print December 2, 2015, in Cephalalgia.
Previous studies have observed associations between screen time exposure and headaches in children between ages 10 and 12 and adolescents. Data also have found an association between screen time exposure and low-back and shoulder pain in adolescents. “This [information] had led to speculation that the high amount of screen time exposure among students of higher education institutions may be correlated with the high prevalence of headache and migraine observed in this population,” said Ilaria Montagni, PhD, a research fellow at the University of Bordeaux in Talence, France.
Dr. Montagni and her coinvestigators studied 4,927 individuals in France, all of whom were age 18 or older and part of the Internet-based Students Health Research Enterprise (i-Share) project cohort, which is an ongoing, prospective, population-based study of students at French-speaking universities and institutions of higher education. The mean age of the students was 20.8, and 75.5% were female.
Subjects completed self-reported surveys on the average amount of time they spend in front of screens during the following five activities: computer or tablet work, playing video games on a computer or tablet, Internet surfing on a computer or tablet, watching videos on a computer or tablet, and using a smartphone. All questions were scored using a five-point scale. A score of 0 indicated never, 1 indicated less than 30 minutes, 2 indicated 30 minutes to two hours, 3 indicated four to eight hours, and 5 indicated eight hours or more. Scores from the surveys were divided into quartiles of very low, low, high, and very high screen-time exposure.
Surveys also asked if participants had experienced any headaches that had lasted several hours in the previous 12 months. Participants who answered negatively were classified as “no headache,” while those who answered positively were asked a series of follow-up questions related to symptom type and severity, sensitivity to light or sound, nausea, vomiting, and disruption of daily routines. To establish a classification of migraine, the investigators used the “probable migraine” category of the International Classification of Headache Disorders, third edition. With these data, the investigators used multinomial logistic regression models to calculate odds ratios of any relationship between screen time exposure and the presence and severity of headaches.
Of the 4,927 participants, 2,773 (56.3%) reported no headaches. In all, 710 (14.4%) participants reported a nonmigraine headache, 791 (16.1%) reported migraine without aura, and 653 (13.3%) reported migraine with aura. In comparisons against very low screen time exposure, very high exposure increased the likelihood of having migraine by 37%, which resulted in a statistically significant 50% greater odds for migraine without aura, but not for migraine with aura.
“Students reporting very high screen time exposure were more likely to be male, to be older, to have higher BMI, and to consume cannabis [and] were also more likely to report nonmigraine headache or migraine,” the authors noted. Furthermore, higher exposure to screens was a significant indicator of recurrent headaches in adolescent males, and the same indicator was seen in adolescent females who spent more time on the computer and in front of the TV.
—Deepak Chitnis
Too much time in front of electronic screens can increase the risk of headaches and migraine in young adults, according to research published online ahead of print December 2, 2015, in Cephalalgia.
Previous studies have observed associations between screen time exposure and headaches in children between ages 10 and 12 and adolescents. Data also have found an association between screen time exposure and low-back and shoulder pain in adolescents. “This [information] had led to speculation that the high amount of screen time exposure among students of higher education institutions may be correlated with the high prevalence of headache and migraine observed in this population,” said Ilaria Montagni, PhD, a research fellow at the University of Bordeaux in Talence, France.
Dr. Montagni and her coinvestigators studied 4,927 individuals in France, all of whom were age 18 or older and part of the Internet-based Students Health Research Enterprise (i-Share) project cohort, which is an ongoing, prospective, population-based study of students at French-speaking universities and institutions of higher education. The mean age of the students was 20.8, and 75.5% were female.
Subjects completed self-reported surveys on the average amount of time they spend in front of screens during the following five activities: computer or tablet work, playing video games on a computer or tablet, Internet surfing on a computer or tablet, watching videos on a computer or tablet, and using a smartphone. All questions were scored using a five-point scale. A score of 0 indicated never, 1 indicated less than 30 minutes, 2 indicated 30 minutes to two hours, 3 indicated four to eight hours, and 5 indicated eight hours or more. Scores from the surveys were divided into quartiles of very low, low, high, and very high screen-time exposure.
Surveys also asked if participants had experienced any headaches that had lasted several hours in the previous 12 months. Participants who answered negatively were classified as “no headache,” while those who answered positively were asked a series of follow-up questions related to symptom type and severity, sensitivity to light or sound, nausea, vomiting, and disruption of daily routines. To establish a classification of migraine, the investigators used the “probable migraine” category of the International Classification of Headache Disorders, third edition. With these data, the investigators used multinomial logistic regression models to calculate odds ratios of any relationship between screen time exposure and the presence and severity of headaches.
Of the 4,927 participants, 2,773 (56.3%) reported no headaches. In all, 710 (14.4%) participants reported a nonmigraine headache, 791 (16.1%) reported migraine without aura, and 653 (13.3%) reported migraine with aura. In comparisons against very low screen time exposure, very high exposure increased the likelihood of having migraine by 37%, which resulted in a statistically significant 50% greater odds for migraine without aura, but not for migraine with aura.
“Students reporting very high screen time exposure were more likely to be male, to be older, to have higher BMI, and to consume cannabis [and] were also more likely to report nonmigraine headache or migraine,” the authors noted. Furthermore, higher exposure to screens was a significant indicator of recurrent headaches in adolescent males, and the same indicator was seen in adolescent females who spent more time on the computer and in front of the TV.
—Deepak Chitnis
Suggested Reading
Montagni I, Guichard E, Carpenet C, et al. Screen time exposure and reporting of headaches in young adults: A cross-sectional study. Cephalalgia. 2015 Dec 2 [Epub ahead of print].
Suggested Reading
Montagni I, Guichard E, Carpenet C, et al. Screen time exposure and reporting of headaches in young adults: A cross-sectional study. Cephalalgia. 2015 Dec 2 [Epub ahead of print].