User login
Sharing Notes for Better Doctor-Patient Communication
Excellent communication between physicians and patients is a crucial element of hospital quality, but it’s also an ongoing challenge for many institutions. One physician wondered whether letting patients read their physicians’ notes could help.
“I wanted to find new methods to improve patient understanding of their medical care plan,” says Craig Weinert, MD, MPH, medical director for adult inpatient services at the University of Minnesota Medical Center and author of “Giving Doctors’ Daily Progress Notes to Hospitalized Patients and Families to Improve Patient Experience” in the American Journal of Medical Quality. “It seemed logical to me that giving patients access to the same information that all the other members of the healthcare team were reading would improve communication. This is the overall hypothesis of the Open Notes movement.”
Another reason Dr. Weinert pursued the study: In his clinical job as an intensivist, he encounters frequent disagreements with patients’ families regarding prognosis and goals of care.
“No one has figured out how to increase the alignment of prognosis between the family and the medical team,” Dr. Weinert says. “I think having the families read the doctors’ notes, where the issues with poor-prognosis multi-organ failure are repeatedly spelled out, might help families more quickly grasp the futility of continuing care.”
During the study, hospitalized patients or family members on six wards of a university hospital received a printed copy of their medical team’s daily progress notes. Surveys afterward showed 74% to 86% of patients and family members responded favorably. Physicians were mostly satisfied, too.
“Most doctors, at the end of the study, thought that Open Notes went better than they had predicted,” Dr. Weinert says.
Complete transparency of medical records is the future of medicine, he says. It’s what patients want, “especially the younger generation.”
“Over the next 10 years,” he says, “I predict ... all [electronic medical record] vendors will have electronic portals that allow clinic and hospitalized patients access to almost everything in the EMR.”
Reference
1. Weinert C. Giving doctors’ daily progress notes to hospitalized patients and families to improve patient experience. Am J Med Qual. 2015. doi:10.1177/1062860615610424.
Excellent communication between physicians and patients is a crucial element of hospital quality, but it’s also an ongoing challenge for many institutions. One physician wondered whether letting patients read their physicians’ notes could help.
“I wanted to find new methods to improve patient understanding of their medical care plan,” says Craig Weinert, MD, MPH, medical director for adult inpatient services at the University of Minnesota Medical Center and author of “Giving Doctors’ Daily Progress Notes to Hospitalized Patients and Families to Improve Patient Experience” in the American Journal of Medical Quality. “It seemed logical to me that giving patients access to the same information that all the other members of the healthcare team were reading would improve communication. This is the overall hypothesis of the Open Notes movement.”
Another reason Dr. Weinert pursued the study: In his clinical job as an intensivist, he encounters frequent disagreements with patients’ families regarding prognosis and goals of care.
“No one has figured out how to increase the alignment of prognosis between the family and the medical team,” Dr. Weinert says. “I think having the families read the doctors’ notes, where the issues with poor-prognosis multi-organ failure are repeatedly spelled out, might help families more quickly grasp the futility of continuing care.”
During the study, hospitalized patients or family members on six wards of a university hospital received a printed copy of their medical team’s daily progress notes. Surveys afterward showed 74% to 86% of patients and family members responded favorably. Physicians were mostly satisfied, too.
“Most doctors, at the end of the study, thought that Open Notes went better than they had predicted,” Dr. Weinert says.
Complete transparency of medical records is the future of medicine, he says. It’s what patients want, “especially the younger generation.”
“Over the next 10 years,” he says, “I predict ... all [electronic medical record] vendors will have electronic portals that allow clinic and hospitalized patients access to almost everything in the EMR.”
Reference
1. Weinert C. Giving doctors’ daily progress notes to hospitalized patients and families to improve patient experience. Am J Med Qual. 2015. doi:10.1177/1062860615610424.
Excellent communication between physicians and patients is a crucial element of hospital quality, but it’s also an ongoing challenge for many institutions. One physician wondered whether letting patients read their physicians’ notes could help.
“I wanted to find new methods to improve patient understanding of their medical care plan,” says Craig Weinert, MD, MPH, medical director for adult inpatient services at the University of Minnesota Medical Center and author of “Giving Doctors’ Daily Progress Notes to Hospitalized Patients and Families to Improve Patient Experience” in the American Journal of Medical Quality. “It seemed logical to me that giving patients access to the same information that all the other members of the healthcare team were reading would improve communication. This is the overall hypothesis of the Open Notes movement.”
Another reason Dr. Weinert pursued the study: In his clinical job as an intensivist, he encounters frequent disagreements with patients’ families regarding prognosis and goals of care.
“No one has figured out how to increase the alignment of prognosis between the family and the medical team,” Dr. Weinert says. “I think having the families read the doctors’ notes, where the issues with poor-prognosis multi-organ failure are repeatedly spelled out, might help families more quickly grasp the futility of continuing care.”
During the study, hospitalized patients or family members on six wards of a university hospital received a printed copy of their medical team’s daily progress notes. Surveys afterward showed 74% to 86% of patients and family members responded favorably. Physicians were mostly satisfied, too.
“Most doctors, at the end of the study, thought that Open Notes went better than they had predicted,” Dr. Weinert says.
Complete transparency of medical records is the future of medicine, he says. It’s what patients want, “especially the younger generation.”
“Over the next 10 years,” he says, “I predict ... all [electronic medical record] vendors will have electronic portals that allow clinic and hospitalized patients access to almost everything in the EMR.”
Reference
1. Weinert C. Giving doctors’ daily progress notes to hospitalized patients and families to improve patient experience. Am J Med Qual. 2015. doi:10.1177/1062860615610424.
Elderly Patients Hospitalized For Cancer are More likely to Have Complications Afterward Compared to the Middle-Aged
(Reuters Health) - Elderly patients hospitalized for cancer surgery are more likely to have complications afterward compared to the middle-aged, particularly when they have several other health problems, a U.S. study suggests.
Overall, almost one in 10 adults age 55 and older had at least one post-operative issue like delirium, dehydration, falls, fractures, pressure ulcers or unusual weight loss, the study of nearly 1 million cancer surgery patients found.
These setbacks were even more common when patients were at least 65 years old, had two or more other serious health problems in addition to malignancies, or had surgeries for tumors of the digestive system or nearby organs.
But the odds were worst for people over 75 - about 46 percent of them had at least one complication, compared with 22 percent of adults aged 55 to 64.
"With the population aging, it's becoming increasingly important to consider not only the survival benefits of cancer surgery but the impact on functionality, vitality and quality of life," said lead study author Dr. Hung-Jui Tan, a researcher in urologic oncology at the University of California, Los Angeles.
While the events studied here are specific to the initial hospitalization, they can carry potential long-term ramifications," Tan added by email.
To see how age influences the risk of post-operative complications, Tan and colleagues reviewed hospital admission records for a nationwide sample of 940,000 adults age 55 and older who had cancer surgery from 2009 to 2011.
Compared with patients who were under age 65, those who were 65 to 74 years old were 23 percent more likely to have complications, while the over-75 group had 66 percent higher odds, researchers report in the Journal of Clinical Oncology.
Complications were most likely when patients were having surgery for cancers of the bladder, ovary, colon, rectum, pancreas or stomach.
After suffering post-operative setbacks, patients were also more likely to have further complications during their hospital stay, to remain in the hospital longer and to have more costly care. They were also more likely to die in the hospital and less likely to be discharged to home.
One limitation of the study is its reliance on administrative claims data, which is designed for billing purposes and might not always reflect the nuances of patients' medical conditions, the authors note. In addition, it's possible that some complications may have resulted from conditions patients had before they arrived at the hospital for cancer surgery.
The study can't prove that advanced age directly causes post-operative problems. But the findings suggest doctors and patients should consider these potential risks when deciding the best course of treatment, Tan said.
Patients should also understand that not all complications are equally devastating to quality of life. Dehydration and weight loss, for example, are nutritional problems that might be treated with fluids, noted Dr. Siri Rostoft, a geriatric medicine researcher at Oslo University Hospital in Norway.
"Cancer is often a lethal disease if left untreated that causes conditions such as bleeding, obstruction of the intestines, and pain," Rostoft, who wasn't involved in the study, said by email. "Not treating patients may be worse for their quality of life than operating."
Still, the findings add to a growing body of data on post-operative complications that may help doctors and patients decide if the potential benefits of surgery outweigh the possible risks, Dr. Steven Cunningham, a researcher at Saint Agnes Hospital and Cancer Institute in Baltimore who wasn't involved in the study, said by email.
Complications in the study were more likely at non-teaching hospitals and facilities that did fewer cancer surgeries, a factor that patients should also consider when they have a choice about where to go for surgery, noted Dr. Kwok-Leung Cheung, a researcher at the University of Nottingham in the U.K. and member of the surgical task force for the International Society of Geriatric Oncology.
Knowing when not to operate also matters, Cheung, who wasn't involved in the study, added by email.
"The surgeon should seriously consider the intensity of surgery, which has been identified as one of the important factors with post operative problems," Cheung added. "The use of minimally invasive techniques including laparoscopic and robotic surgery should be considered wherever appropriate."
(Reuters Health) - Elderly patients hospitalized for cancer surgery are more likely to have complications afterward compared to the middle-aged, particularly when they have several other health problems, a U.S. study suggests.
Overall, almost one in 10 adults age 55 and older had at least one post-operative issue like delirium, dehydration, falls, fractures, pressure ulcers or unusual weight loss, the study of nearly 1 million cancer surgery patients found.
These setbacks were even more common when patients were at least 65 years old, had two or more other serious health problems in addition to malignancies, or had surgeries for tumors of the digestive system or nearby organs.
But the odds were worst for people over 75 - about 46 percent of them had at least one complication, compared with 22 percent of adults aged 55 to 64.
"With the population aging, it's becoming increasingly important to consider not only the survival benefits of cancer surgery but the impact on functionality, vitality and quality of life," said lead study author Dr. Hung-Jui Tan, a researcher in urologic oncology at the University of California, Los Angeles.
While the events studied here are specific to the initial hospitalization, they can carry potential long-term ramifications," Tan added by email.
To see how age influences the risk of post-operative complications, Tan and colleagues reviewed hospital admission records for a nationwide sample of 940,000 adults age 55 and older who had cancer surgery from 2009 to 2011.
Compared with patients who were under age 65, those who were 65 to 74 years old were 23 percent more likely to have complications, while the over-75 group had 66 percent higher odds, researchers report in the Journal of Clinical Oncology.
Complications were most likely when patients were having surgery for cancers of the bladder, ovary, colon, rectum, pancreas or stomach.
After suffering post-operative setbacks, patients were also more likely to have further complications during their hospital stay, to remain in the hospital longer and to have more costly care. They were also more likely to die in the hospital and less likely to be discharged to home.
One limitation of the study is its reliance on administrative claims data, which is designed for billing purposes and might not always reflect the nuances of patients' medical conditions, the authors note. In addition, it's possible that some complications may have resulted from conditions patients had before they arrived at the hospital for cancer surgery.
The study can't prove that advanced age directly causes post-operative problems. But the findings suggest doctors and patients should consider these potential risks when deciding the best course of treatment, Tan said.
Patients should also understand that not all complications are equally devastating to quality of life. Dehydration and weight loss, for example, are nutritional problems that might be treated with fluids, noted Dr. Siri Rostoft, a geriatric medicine researcher at Oslo University Hospital in Norway.
"Cancer is often a lethal disease if left untreated that causes conditions such as bleeding, obstruction of the intestines, and pain," Rostoft, who wasn't involved in the study, said by email. "Not treating patients may be worse for their quality of life than operating."
Still, the findings add to a growing body of data on post-operative complications that may help doctors and patients decide if the potential benefits of surgery outweigh the possible risks, Dr. Steven Cunningham, a researcher at Saint Agnes Hospital and Cancer Institute in Baltimore who wasn't involved in the study, said by email.
Complications in the study were more likely at non-teaching hospitals and facilities that did fewer cancer surgeries, a factor that patients should also consider when they have a choice about where to go for surgery, noted Dr. Kwok-Leung Cheung, a researcher at the University of Nottingham in the U.K. and member of the surgical task force for the International Society of Geriatric Oncology.
Knowing when not to operate also matters, Cheung, who wasn't involved in the study, added by email.
"The surgeon should seriously consider the intensity of surgery, which has been identified as one of the important factors with post operative problems," Cheung added. "The use of minimally invasive techniques including laparoscopic and robotic surgery should be considered wherever appropriate."
(Reuters Health) - Elderly patients hospitalized for cancer surgery are more likely to have complications afterward compared to the middle-aged, particularly when they have several other health problems, a U.S. study suggests.
Overall, almost one in 10 adults age 55 and older had at least one post-operative issue like delirium, dehydration, falls, fractures, pressure ulcers or unusual weight loss, the study of nearly 1 million cancer surgery patients found.
These setbacks were even more common when patients were at least 65 years old, had two or more other serious health problems in addition to malignancies, or had surgeries for tumors of the digestive system or nearby organs.
But the odds were worst for people over 75 - about 46 percent of them had at least one complication, compared with 22 percent of adults aged 55 to 64.
"With the population aging, it's becoming increasingly important to consider not only the survival benefits of cancer surgery but the impact on functionality, vitality and quality of life," said lead study author Dr. Hung-Jui Tan, a researcher in urologic oncology at the University of California, Los Angeles.
While the events studied here are specific to the initial hospitalization, they can carry potential long-term ramifications," Tan added by email.
To see how age influences the risk of post-operative complications, Tan and colleagues reviewed hospital admission records for a nationwide sample of 940,000 adults age 55 and older who had cancer surgery from 2009 to 2011.
Compared with patients who were under age 65, those who were 65 to 74 years old were 23 percent more likely to have complications, while the over-75 group had 66 percent higher odds, researchers report in the Journal of Clinical Oncology.
Complications were most likely when patients were having surgery for cancers of the bladder, ovary, colon, rectum, pancreas or stomach.
After suffering post-operative setbacks, patients were also more likely to have further complications during their hospital stay, to remain in the hospital longer and to have more costly care. They were also more likely to die in the hospital and less likely to be discharged to home.
One limitation of the study is its reliance on administrative claims data, which is designed for billing purposes and might not always reflect the nuances of patients' medical conditions, the authors note. In addition, it's possible that some complications may have resulted from conditions patients had before they arrived at the hospital for cancer surgery.
The study can't prove that advanced age directly causes post-operative problems. But the findings suggest doctors and patients should consider these potential risks when deciding the best course of treatment, Tan said.
Patients should also understand that not all complications are equally devastating to quality of life. Dehydration and weight loss, for example, are nutritional problems that might be treated with fluids, noted Dr. Siri Rostoft, a geriatric medicine researcher at Oslo University Hospital in Norway.
"Cancer is often a lethal disease if left untreated that causes conditions such as bleeding, obstruction of the intestines, and pain," Rostoft, who wasn't involved in the study, said by email. "Not treating patients may be worse for their quality of life than operating."
Still, the findings add to a growing body of data on post-operative complications that may help doctors and patients decide if the potential benefits of surgery outweigh the possible risks, Dr. Steven Cunningham, a researcher at Saint Agnes Hospital and Cancer Institute in Baltimore who wasn't involved in the study, said by email.
Complications in the study were more likely at non-teaching hospitals and facilities that did fewer cancer surgeries, a factor that patients should also consider when they have a choice about where to go for surgery, noted Dr. Kwok-Leung Cheung, a researcher at the University of Nottingham in the U.K. and member of the surgical task force for the International Society of Geriatric Oncology.
Knowing when not to operate also matters, Cheung, who wasn't involved in the study, added by email.
"The surgeon should seriously consider the intensity of surgery, which has been identified as one of the important factors with post operative problems," Cheung added. "The use of minimally invasive techniques including laparoscopic and robotic surgery should be considered wherever appropriate."
Team identifies potential target for T-ALL therapy

New research suggests T-cell acute lymphoblastic leukemia (T-ALL) cells use the tricarboxylic acid (TCA) cycle to support their growth and survival.
Investigators say the findings could aid the development of therapeutics that can kill T-ALL cells by targeting an enzyme that exists in the TCA cycle—dihydrolipoamide S-succinyltransferase (DLST).
The team described this research in the journal Leukemia.
“Researchers have wrongly assumed that cancer cells do not use the TCA cycle to support their growth,” said study author Hui Feng, MD, PhD, of Boston University Medical Center in Massachusetts.
“Our new findings provide solid evidence that these cancer cells depend on the TCA cycle for their survival. Additionally, we demonstrated the importance of DLST in T-cell leukemia development and have identified a targetable enzyme for T-cell leukemia treatment.”
For this study, the investigators set out to examine the mechanisms underlying MYC-mediated tumorigenesis in T-ALL.
They used a zebrafish model of MYC-induced T-ALL to screen for genes that contribute to disease onset. The results suggested the TCA-cycle enzyme DLST is an important contributor to T-ALL development.
And experiments showed that heterozygous inactivation of DLST significantly delayed disease onset in the zebrafish, apparently without affecting the development of the fish.
Further analysis revealed that inhibiting the activity of DLST could effectively kill human T-ALL cells. Specifically, RNAi knockdown of DLST decreased cell viability and induced apoptosis in human T-ALL cell
lines.
The investigators found that knockdown of DLST disrupted the TCA cycle in the human T-ALL cells. But adding succinate, the downstream TCA-cycle intermediate, to the cells rescued defects in cell viability caused by DLST knockdown.
The investigators said the therapeutic benefit of DLST inhibition may extend to cancers other than T-ALL as well. ![]()
New research suggests T-cell acute lymphoblastic leukemia (T-ALL) cells use the tricarboxylic acid (TCA) cycle to support their growth and survival.
Investigators say the findings could aid the development of therapeutics that can kill T-ALL cells by targeting an enzyme that exists in the TCA cycle—dihydrolipoamide S-succinyltransferase (DLST).
The team described this research in the journal Leukemia.
“Researchers have wrongly assumed that cancer cells do not use the TCA cycle to support their growth,” said study author Hui Feng, MD, PhD, of Boston University Medical Center in Massachusetts.
“Our new findings provide solid evidence that these cancer cells depend on the TCA cycle for their survival. Additionally, we demonstrated the importance of DLST in T-cell leukemia development and have identified a targetable enzyme for T-cell leukemia treatment.”
For this study, the investigators set out to examine the mechanisms underlying MYC-mediated tumorigenesis in T-ALL.
They used a zebrafish model of MYC-induced T-ALL to screen for genes that contribute to disease onset. The results suggested the TCA-cycle enzyme DLST is an important contributor to T-ALL development.
And experiments showed that heterozygous inactivation of DLST significantly delayed disease onset in the zebrafish, apparently without affecting the development of the fish.
Further analysis revealed that inhibiting the activity of DLST could effectively kill human T-ALL cells. Specifically, RNAi knockdown of DLST decreased cell viability and induced apoptosis in human T-ALL cell
lines.
The investigators found that knockdown of DLST disrupted the TCA cycle in the human T-ALL cells. But adding succinate, the downstream TCA-cycle intermediate, to the cells rescued defects in cell viability caused by DLST knockdown.
The investigators said the therapeutic benefit of DLST inhibition may extend to cancers other than T-ALL as well. ![]()
New research suggests T-cell acute lymphoblastic leukemia (T-ALL) cells use the tricarboxylic acid (TCA) cycle to support their growth and survival.
Investigators say the findings could aid the development of therapeutics that can kill T-ALL cells by targeting an enzyme that exists in the TCA cycle—dihydrolipoamide S-succinyltransferase (DLST).
The team described this research in the journal Leukemia.
“Researchers have wrongly assumed that cancer cells do not use the TCA cycle to support their growth,” said study author Hui Feng, MD, PhD, of Boston University Medical Center in Massachusetts.
“Our new findings provide solid evidence that these cancer cells depend on the TCA cycle for their survival. Additionally, we demonstrated the importance of DLST in T-cell leukemia development and have identified a targetable enzyme for T-cell leukemia treatment.”
For this study, the investigators set out to examine the mechanisms underlying MYC-mediated tumorigenesis in T-ALL.
They used a zebrafish model of MYC-induced T-ALL to screen for genes that contribute to disease onset. The results suggested the TCA-cycle enzyme DLST is an important contributor to T-ALL development.
And experiments showed that heterozygous inactivation of DLST significantly delayed disease onset in the zebrafish, apparently without affecting the development of the fish.
Further analysis revealed that inhibiting the activity of DLST could effectively kill human T-ALL cells. Specifically, RNAi knockdown of DLST decreased cell viability and induced apoptosis in human T-ALL cell
lines.
The investigators found that knockdown of DLST disrupted the TCA cycle in the human T-ALL cells. But adding succinate, the downstream TCA-cycle intermediate, to the cells rescued defects in cell viability caused by DLST knockdown.
The investigators said the therapeutic benefit of DLST inhibition may extend to cancers other than T-ALL as well. ![]()
Orphan designation recommended for BTK inhibitor

The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) is recommending orphan designation for the second-generation BTK inhibitor acalabrutinib (ACP-196) for 3 indications.
The COMP is recommending the drug receive orphan designation as a treatment for chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL), mantle cell lymphoma, and Waldenström’s macroglobulinemia.
The COMP adopts an opinion on orphan drug designation, and that opinion is submitted to the European Commission (EC) for endorsement.
To be granted orphan designation by the EC, a medicine must be intended for the treatment, prevention, or diagnosis of a disease that is life-threatening and has a prevalence of up to 5 in 10,000 in the European Union. Additionally, the medicine must aim to provide significant benefit to those affected by the condition.
Orphan designation provides companies with development and market exclusivity incentives for designated compounds and medicines.
About acalabrutinib
Acalabrutinib is under development by AstraZeneca and Acerta Pharma BV. The drug is currently being evaluated in trials of patients with CLL/SLL, mantle cell lymphoma, Waldentröm’s macroglobulinemia, and a range of other hematologic malignancies and solid tumor cancers.
Data from a phase 1/2 trial of acalabrutinib in CLL were presented at the 2015 ASH Annual Meeting and simultaneously published in NEJM.
The researchers reported on 61 patients with relapsed CLL who had a median age of 62 (range, 44-84) and a median of 3 prior therapies (range, 1-13).
Patients enrolled in the phase 1 portion of the study received escalating doses of acalabrutinib, with a maximum dose of 400 mg once daily. Patients involved in the phase 2 portion of the study were treated with a 100 mg dose twice daily.
At a median follow-up of 14.3 months (range, 0.5 to 20), 53 patients were still receiving treatment.
The most common adverse events of all grades (occurring in at least 20% of patients) were headache (43%), diarrhea (39%), increased weight (26%), pyrexia (23%), upper respiratory tract infection (23%), fatigue (21%), peripheral edema (21%), hypertension (20%), and nausea (20%).
Grade 3/4 adverse events included diarrhea (2%), increased weight (2%), pyrexia (3%), fatigue (3%), hypertension (7%), and arthralgia (2%).
The overall response rate among the 60 evaluable patients was 95%. This included partial responses in 85% of patients and partial responses with lymphocytosis in 10%. The rate of stable disease was 5%.
The researchers noted that responses occurred in all dosing cohorts, and the response rate increased over time. ![]()
The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) is recommending orphan designation for the second-generation BTK inhibitor acalabrutinib (ACP-196) for 3 indications.
The COMP is recommending the drug receive orphan designation as a treatment for chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL), mantle cell lymphoma, and Waldenström’s macroglobulinemia.
The COMP adopts an opinion on orphan drug designation, and that opinion is submitted to the European Commission (EC) for endorsement.
To be granted orphan designation by the EC, a medicine must be intended for the treatment, prevention, or diagnosis of a disease that is life-threatening and has a prevalence of up to 5 in 10,000 in the European Union. Additionally, the medicine must aim to provide significant benefit to those affected by the condition.
Orphan designation provides companies with development and market exclusivity incentives for designated compounds and medicines.
About acalabrutinib
Acalabrutinib is under development by AstraZeneca and Acerta Pharma BV. The drug is currently being evaluated in trials of patients with CLL/SLL, mantle cell lymphoma, Waldentröm’s macroglobulinemia, and a range of other hematologic malignancies and solid tumor cancers.
Data from a phase 1/2 trial of acalabrutinib in CLL were presented at the 2015 ASH Annual Meeting and simultaneously published in NEJM.
The researchers reported on 61 patients with relapsed CLL who had a median age of 62 (range, 44-84) and a median of 3 prior therapies (range, 1-13).
Patients enrolled in the phase 1 portion of the study received escalating doses of acalabrutinib, with a maximum dose of 400 mg once daily. Patients involved in the phase 2 portion of the study were treated with a 100 mg dose twice daily.
At a median follow-up of 14.3 months (range, 0.5 to 20), 53 patients were still receiving treatment.
The most common adverse events of all grades (occurring in at least 20% of patients) were headache (43%), diarrhea (39%), increased weight (26%), pyrexia (23%), upper respiratory tract infection (23%), fatigue (21%), peripheral edema (21%), hypertension (20%), and nausea (20%).
Grade 3/4 adverse events included diarrhea (2%), increased weight (2%), pyrexia (3%), fatigue (3%), hypertension (7%), and arthralgia (2%).
The overall response rate among the 60 evaluable patients was 95%. This included partial responses in 85% of patients and partial responses with lymphocytosis in 10%. The rate of stable disease was 5%.
The researchers noted that responses occurred in all dosing cohorts, and the response rate increased over time. ![]()
The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) is recommending orphan designation for the second-generation BTK inhibitor acalabrutinib (ACP-196) for 3 indications.
The COMP is recommending the drug receive orphan designation as a treatment for chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL), mantle cell lymphoma, and Waldenström’s macroglobulinemia.
The COMP adopts an opinion on orphan drug designation, and that opinion is submitted to the European Commission (EC) for endorsement.
To be granted orphan designation by the EC, a medicine must be intended for the treatment, prevention, or diagnosis of a disease that is life-threatening and has a prevalence of up to 5 in 10,000 in the European Union. Additionally, the medicine must aim to provide significant benefit to those affected by the condition.
Orphan designation provides companies with development and market exclusivity incentives for designated compounds and medicines.
About acalabrutinib
Acalabrutinib is under development by AstraZeneca and Acerta Pharma BV. The drug is currently being evaluated in trials of patients with CLL/SLL, mantle cell lymphoma, Waldentröm’s macroglobulinemia, and a range of other hematologic malignancies and solid tumor cancers.
Data from a phase 1/2 trial of acalabrutinib in CLL were presented at the 2015 ASH Annual Meeting and simultaneously published in NEJM.
The researchers reported on 61 patients with relapsed CLL who had a median age of 62 (range, 44-84) and a median of 3 prior therapies (range, 1-13).
Patients enrolled in the phase 1 portion of the study received escalating doses of acalabrutinib, with a maximum dose of 400 mg once daily. Patients involved in the phase 2 portion of the study were treated with a 100 mg dose twice daily.
At a median follow-up of 14.3 months (range, 0.5 to 20), 53 patients were still receiving treatment.
The most common adverse events of all grades (occurring in at least 20% of patients) were headache (43%), diarrhea (39%), increased weight (26%), pyrexia (23%), upper respiratory tract infection (23%), fatigue (21%), peripheral edema (21%), hypertension (20%), and nausea (20%).
Grade 3/4 adverse events included diarrhea (2%), increased weight (2%), pyrexia (3%), fatigue (3%), hypertension (7%), and arthralgia (2%).
The overall response rate among the 60 evaluable patients was 95%. This included partial responses in 85% of patients and partial responses with lymphocytosis in 10%. The rate of stable disease was 5%.
The researchers noted that responses occurred in all dosing cohorts, and the response rate increased over time. ![]()
VIDEO: Novel tools measure disease progression in MS
NEW ORLEANS – The visual system is relevant and accessible for the study of multiple sclerosis and can aid in the measurement of neuronal and axonal injury.
Capturing primary neuronal loss in the afferent visual pathway was among the topics addressed during a session focused on novel methods for measuring disease progression in MS at a meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis.
In this video interview at the meeting, session chair Dr. Fiona Costello of the University of Calgary, Alta., discussed the presentation on the visual pathway, as well as presentations on the use of microRNA biomarkers and the use of MRI as an outcome measure in progressive MS.
“The gestalt is that the field is moving in a new direction; the field is looking for not only a better understanding of what causes disability in MS, but also more reliable, objective, accessible means of capturing the same thing that is relevant and meaningful to patients and their caretakers,” she said.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
NEW ORLEANS – The visual system is relevant and accessible for the study of multiple sclerosis and can aid in the measurement of neuronal and axonal injury.
Capturing primary neuronal loss in the afferent visual pathway was among the topics addressed during a session focused on novel methods for measuring disease progression in MS at a meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis.
In this video interview at the meeting, session chair Dr. Fiona Costello of the University of Calgary, Alta., discussed the presentation on the visual pathway, as well as presentations on the use of microRNA biomarkers and the use of MRI as an outcome measure in progressive MS.
“The gestalt is that the field is moving in a new direction; the field is looking for not only a better understanding of what causes disability in MS, but also more reliable, objective, accessible means of capturing the same thing that is relevant and meaningful to patients and their caretakers,” she said.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
NEW ORLEANS – The visual system is relevant and accessible for the study of multiple sclerosis and can aid in the measurement of neuronal and axonal injury.
Capturing primary neuronal loss in the afferent visual pathway was among the topics addressed during a session focused on novel methods for measuring disease progression in MS at a meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis.
In this video interview at the meeting, session chair Dr. Fiona Costello of the University of Calgary, Alta., discussed the presentation on the visual pathway, as well as presentations on the use of microRNA biomarkers and the use of MRI as an outcome measure in progressive MS.
“The gestalt is that the field is moving in a new direction; the field is looking for not only a better understanding of what causes disability in MS, but also more reliable, objective, accessible means of capturing the same thing that is relevant and meaningful to patients and their caretakers,” she said.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT ACTRIMS FORUM 2016

Medical scribes: How do their notes stack up?
ABSTRACT
Objective Medical scribes are increasingly employed to improve physician efficiency with regard to the electronic medical record (EMR). The impact of scribes on the quality of outpatient visit notes is not known. To assess the effect, we conducted a retrospective review of ambulatory progress notes written before and after 8 practice sites transitioned to the use of medical assistants as scribes.
Methods The Physician Documentation Quality Instrument 9 (PDQI-9) was used to compare the quality of outpatient progress notes written by medical assistant scribes with the quality of notes written by 18 primary care physicians working without a scribe. The notes pertained to diabetes encounters and same-day appointments and were written during the 3 to 6 months preceding the use of scribes (pre-scribe period) and the 3 to 6 months after scribes were employed (scribe period).
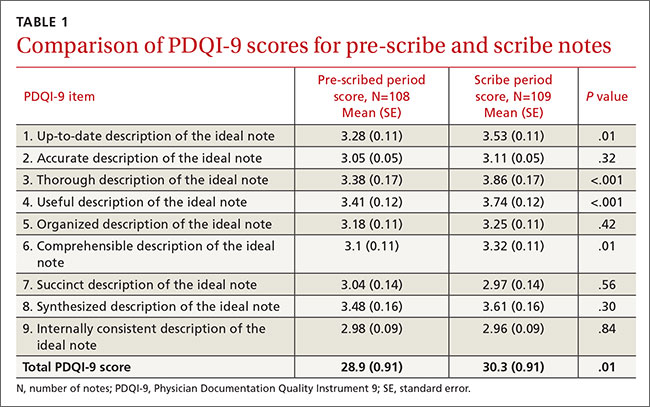
Results One hundred eight notes from the pre-scribe period and 109 from the scribe period were reviewed. Scribed notes were rated higher in overall quality than unscribed notes (mean total PDQI-9 score 30.3 for scribed notes vs 28.9 for nonscribed notes; P=.01) and more up-to-date, thorough, useful, and comprehensible. The differences were limited to diabetes encounters. For same-day appointments, scribed and nonscribed notes did not differ in quality. The total word count of all scribed and nonscribed notes was similar (mean words 618, standard deviation (SD) 273 for scribed notes vs 558 words, SD 289 for nonscribed notes; P=.12).
Conclusions In this retrospective review, ambulatory notes were of higher quality when medical assistants acted as scribes than when physicians wrote them alone, at least for diabetes visits. Our findings may not apply to professional scribes who are not part of the clinical care team. As the use of medical scribes expands, additional studies should examine the impact of scribes on other aspects of care quality.
Team-based models of primary care delivery may incorporate medical scribes to improve efficiency of electronic documentation.1-4 The employment of medical scribes has grown rapidly, and it is estimated that within several years there may be one scribe for every 9 physicians.3
Accurate documentation is important to providing high-quality patient care but can take a significant amount of time. Attending physicians have been estimated to spend as long as 52 minutes per day authoring notes.5 Medical scribes can help physicians improve the efficiency of electronic documentation6 and save time.2 Using scribes can also improve physician productivity7-10 and thereby potentially increase access to care. The impact of scribes on the quality of outpatient visit notes, however, is unknown.
A team-based care delivery model in our health system’s primary care clinics uses medical assistants to scribe notes during the outpatient encounter. We hypothesized that outpatient notes written by medical assistant scribes would be of similar quality to notes written by the same group of physicians without a scribe.
METHODS
Study design and sample
We conducted a retrospective review of ambulatory notes from 18 primary care physicians at 8 practice sites in our health system who had adopted a care model in which medical assistants act as scribes. Each physician works with 2 medical assistants. To train for the new model, the physician and medical assistants participated in 2 training sessions of 2 hours each and a half day of clinic observation and evaluation with a project manager.
Of the 18 primary care physicians included in this study, none had less than one year of experience in our health system. Tenure ranged from one to 24 years with a mean of 11.3 years.
For each participating provider, we requested all available outpatient progress notes with either an International Classification of Diseases, 9th revision (ICD-9) code for diabetes or a designation of “same day” for the 3 to 6 months preceding the use of scribes (pre-scribe period) and the 3 to 6 months after employing scribes (scribe period). We chose diabetes encounters as examples of notes addressing chronic disease management and same-day encounters as examples of problem-focused notes because these 2 types of encounters are common in outpatient primary care practice.
Note quality was evaluated using the Physician Documentation Quality Instrument 9 (PDQI-9), a validated instrument designed for this purpose, comprising 9 items rated subjectively on a 5-point Likert scale (1= not at all, 5= extremely). The items assess whether notes are up-to-date, accurate, thorough, useful, organized, comprehensible, succinct, synthesized, and internally consistent.11,12 The PDQI-9 has been applied previously in inpatient12 and outpatient settings.13
While the PDQI-9 is a validated tool, it relies on subjective ratings of note quality by the reviewer. To control for the subjective nature of the ratings, an experienced internist and an internal medicine resident coded 10 progress notes separately using the PDQI-9 and discussed the results. The process was repeated for a total of 20 notes, after which consensus was reached with >70% agreement on each attribute of the PDQI-9, suggesting that the resident’s ratings were reliable when compared with those of an experienced practicing physician.
The resident then evaluated a random sample of notes written by each physician for diabetes or same-day appointments in the pre-scribe and scribe periods. Word counts for the entire note were measured. The notes used to establish the reliability of the ratings were excluded from the analysis for this study.
Data analysis
We used linear mixed-effects models to examine note quality measures by adjusting for possible correlations of notes from the same physician. Least-squares estimates were derived; the results were not adjusted for multiple comparisons.
RESULTS
One hundred eight notes from the pre-scribe period and 109 notes from the scribe period were reviewed. Compared with notes written by a physician alone, scribed notes were rated slightly higher in overall quality (mean total PDQI-9 score 30.3 for scribe notes vs 28.9 for pre-scribe notes; P=.01) and more up-to-date, thorough, useful, and comprehensible (TABLES 1 AND 2). The differences were limited to diabetes encounters. For same day appointments, scribed notes did not differ in quality from nonscribed notes (TABLE 2). Total word count did not vary significantly between all scribe and pre-scribe notes (mean words 618, SD 273 for scribed notes vs 558 words, SD 289 for nonscribed notes; P=.12).
DISCUSSION
In this retrospective review of ambulatory notes, progress notes written by medical assistant scribes were of higher quality than notes physicians wrote alone, at least for diabetes visits. Scribe and pre-scribe notes were of similar quality for problem-focused same-day visits. This is the first study of which we are aware that compares the quality of scribed notes with notes written by physicians.
Quality scribe notes can save physician time. The progress note is an important vehicle for describing care provided and transferring information among physicians caring for the same patient. Writing a note, however, adds a considerable amount of time to the physician’s workflow. Using a scribe can decrease the time burden of note writing, and if scribed notes are of similar or better quality, this practice innovation can allow the physician to focus more on clinical than clerical tasks.
Over-documentation is a possible concern. While implementation of the EMR may improve certain aspects of quality of care delivered14,15 and note quality,16 concern has been raised about over-documentation related to the connection between documentation and reimbursement.17 In our study, we found that physician notes and scribed notes for both diabetes and same-day encounters often used EMR-based note templates, which can lead to over-documentation.
In general, both physician and scribed notes were rated to be of average to low quality because none of the mean scores on the 9 individual components of the PDQI-9 reached 4.0. Scribed notes were not inaccurate and had word counts similar to physician notes.
Scribing has potential drawbacks—and benefits. Drawbacks to scribing have not been well-studied. It has been suggested that using scribes to work around the EMR may actually hinder its further advancement because scribing insulates physicians from the inefficiencies of current EMRs and will not spur demands for improvements.3 Inaccurate or poor-quality notes could represent another downside to scribing, although concern about the quality of notes has not been documented. Our results suggest the opposite may be true.
Note quality has not been associated with quality of care as assessed by clinical quality scores,13 but using scribes may improve the quality of care in other ways. For example, the EMR may negatively affect patient-physician communication,18,19 and freeing the physician from documentation may improve the interaction.8,20 Incorporating scribing into practice may also improve the physician experience,9,10,21,22 a possible benefit that we did not measure.
We also did not measure the cost of using a scribe to assist in EMR documentation compared with the cost of physician time spent in performing this task. If the scribe model were associated with cost savings through increased physician productivity, as well as improved physician experience, future EMR development might best focus on planned utilization by physician-scribe teams.
Study limitations. The study was conducted in a single health system, although at 8 different practice sites. The sites all used the same EMR, but templates used for documentation could be individualized by the physician and medical assistant team, so our findings may reflect variation in template design. Our analysis did adjust for possible correlations of notes from the same physician. The selection of note types in our study may make our results less generalizable to other encounter types. Our sample was not large enough to detect variations in note quality among different providers and scribes.
The ratings on the PDQI-9 may be subjective, and the reviewers were not blinded to whether a scribe was used to write the note. The differences in PDQI-9 scores were small. Although statistically significant, they may not significantly affect clinical practice. Our care model is unique in that scribes are active members of the clinical care team; the higher quality of scribed notes we found may not apply to professional scribes who are not part of the team.
Future research directions. In our study, medical assistants acting as scribes composed progress notes of similar or higher quality than physicians who wrote notes alone, although all notes were of generally average quality. As the use of scribes in medicine expands, additional studies should examine the impact of scribes on primary care workflow, quality and cost of care delivered, and quality of physician experience.
CORRESPONDENCE
Anita D. Misra-Hebert, MD, MPH, Center for Value-Based Care Research, Medicine Institute, 9500 Euclid Avenue, G10, Cleveland, OH 44195; [email protected].
1. Bodenheimer T, Willard-Grace R, Ghorob A. Expanding the roles of medical assistants: Who does what in primary care? JAMA Intern Med. 2014;174:1025-1026.
2. Reuben DB, Knudsen J, Senelick W, et al. The effect of a physician partner program on physician efficiency and patient satisfaction. JAMA Intern Med. 2014;174:1190-1193.
3. Gellert GA, Ramirea R, Webster S. The rise of the medical scribe industry: Implications for the advancement of electronic health records. JAMA. 2015;313:1315-1316.
4. Shultz CG, Holmstrom HL. The use of medical scribes in health care settings: a systematic review and future directions. J Am Board Fam Med. 2015;28:371-381.
5. Hripcsak G, Vawdrey DK, Fred MR, et al. Use of electronic clinical documentation: time spent and team interactions. J Am Med Inform Assoc. 2011;18:112-117.
6. Silverman L. Scribes Are Back, Helping Doctors Tackle Electronic Medical Records. NPR.org. Available at: www.npr.org/blogs/health/2014/04/21/303406306/scribes-are-back-helping-doctors-tackle-electronic-medical-records. Accessed April 23, 2014.
7. Arya R, Salovich DM, Ohman-Strickland P, et al. Impact of scribes on performance indicators in the emergency department. Acad Emerg Med. 2010;17:490-494.
8. Bank AJ, Obetz C, Konrardy A, et al. Impact of scribes on patient interaction, productivity, and revenue in a cardiology clinic: a prospective study. Clinicoecon Outcomes Res. 2013;5:399-406.
9. Bastani A, Shaqiri B, Palomba K, et al. An ED scribe program is able to improve throughput time and patient satisfaction. Am J Emerg Med. 2014;32:399-402.
10. Allen B, Banapoor B, Weeks EC, et al. An assessment of emergency department throughput and provider satisfaction after the implementation of a scribe program. Advances in Emergency Medicine. 2014;2014:e517319.
11. Stetson PD, Morrison FP, Bakken S, et al. Preliminary development of the Physician Documentation Quality Instrument. J Am Med Inform Assoc. 2008;15:534-541.
12. Stetson PD, Bakken S, Wrenn JO, et al. Assessing electronic note quality using the Physician Documentation Quality Instrument (PDQI-9). Appl Clin Inform. 2012;3:164-174.
13. Edwards ST, Neri PM, Volk LA, et al. Association of note quality and quality of care: a cross-sectional study. BMJ Qual Saf. 2013;23:406-413.
14. Schiff GD, Bates DW. Can electronic clinical documentation help prevent diagnostic errors? N Engl J Med. 2010;362:1066-1069.
15. Samal L, Wright A, Healey MJ, et al. Meaningful use and quality of care. JAMA Intern Med. 2014;174:997-998.
16. Burke HB, Sessums LL, Hoang A, et al. Electronic health records improve clinical note quality. J Am Med Inform Assoc. 2015;22:199-205.
17. Sheehy AM, Weissburg DJ, Dean SM. The role of copy-and-paste in the hospital electronic health record. JAMA Intern Med. 2014;174:1217-1218.
18. Shachak A, Hadas-Dayagi M, Ziv A, et al. Primary care physicians’ use of an electronic medical record system: a cognitive task analysis. J Gen Intern Med. 2009;24:341-348.
19. Shachak A, Reis S. The impact of electronic medical records on patient-doctor communication during consultation: a narrative literature review. J Eval Clin Pract. 2009;15:641-649.
20. Misra-Hebert AD, Rabovsky A, Yan C, et al. A team-based model of primary care delivery and physician-patient interaction. Am J Med. 2015;128:1025-1028.
21. Sinsky CA, Willard-Grace R, Schutzbank AM, et al. In search of joy in practice: a report of 23 high-functioning primary care practices. Ann Fam Med. 2013;11:272-278.
22. Koshy S, Feustel PJ, Hong M, et al. Scribes in an ambulatory urology practice: patient and physician satisfaction. J Urol. 2010;184:258-262.
ABSTRACT
Objective Medical scribes are increasingly employed to improve physician efficiency with regard to the electronic medical record (EMR). The impact of scribes on the quality of outpatient visit notes is not known. To assess the effect, we conducted a retrospective review of ambulatory progress notes written before and after 8 practice sites transitioned to the use of medical assistants as scribes.
Methods The Physician Documentation Quality Instrument 9 (PDQI-9) was used to compare the quality of outpatient progress notes written by medical assistant scribes with the quality of notes written by 18 primary care physicians working without a scribe. The notes pertained to diabetes encounters and same-day appointments and were written during the 3 to 6 months preceding the use of scribes (pre-scribe period) and the 3 to 6 months after scribes were employed (scribe period).
Results One hundred eight notes from the pre-scribe period and 109 from the scribe period were reviewed. Scribed notes were rated higher in overall quality than unscribed notes (mean total PDQI-9 score 30.3 for scribed notes vs 28.9 for nonscribed notes; P=.01) and more up-to-date, thorough, useful, and comprehensible. The differences were limited to diabetes encounters. For same-day appointments, scribed and nonscribed notes did not differ in quality. The total word count of all scribed and nonscribed notes was similar (mean words 618, standard deviation (SD) 273 for scribed notes vs 558 words, SD 289 for nonscribed notes; P=.12).
Conclusions In this retrospective review, ambulatory notes were of higher quality when medical assistants acted as scribes than when physicians wrote them alone, at least for diabetes visits. Our findings may not apply to professional scribes who are not part of the clinical care team. As the use of medical scribes expands, additional studies should examine the impact of scribes on other aspects of care quality.
Team-based models of primary care delivery may incorporate medical scribes to improve efficiency of electronic documentation.1-4 The employment of medical scribes has grown rapidly, and it is estimated that within several years there may be one scribe for every 9 physicians.3
Accurate documentation is important to providing high-quality patient care but can take a significant amount of time. Attending physicians have been estimated to spend as long as 52 minutes per day authoring notes.5 Medical scribes can help physicians improve the efficiency of electronic documentation6 and save time.2 Using scribes can also improve physician productivity7-10 and thereby potentially increase access to care. The impact of scribes on the quality of outpatient visit notes, however, is unknown.
A team-based care delivery model in our health system’s primary care clinics uses medical assistants to scribe notes during the outpatient encounter. We hypothesized that outpatient notes written by medical assistant scribes would be of similar quality to notes written by the same group of physicians without a scribe.
METHODS
Study design and sample
We conducted a retrospective review of ambulatory notes from 18 primary care physicians at 8 practice sites in our health system who had adopted a care model in which medical assistants act as scribes. Each physician works with 2 medical assistants. To train for the new model, the physician and medical assistants participated in 2 training sessions of 2 hours each and a half day of clinic observation and evaluation with a project manager.
Of the 18 primary care physicians included in this study, none had less than one year of experience in our health system. Tenure ranged from one to 24 years with a mean of 11.3 years.
For each participating provider, we requested all available outpatient progress notes with either an International Classification of Diseases, 9th revision (ICD-9) code for diabetes or a designation of “same day” for the 3 to 6 months preceding the use of scribes (pre-scribe period) and the 3 to 6 months after employing scribes (scribe period). We chose diabetes encounters as examples of notes addressing chronic disease management and same-day encounters as examples of problem-focused notes because these 2 types of encounters are common in outpatient primary care practice.
Note quality was evaluated using the Physician Documentation Quality Instrument 9 (PDQI-9), a validated instrument designed for this purpose, comprising 9 items rated subjectively on a 5-point Likert scale (1= not at all, 5= extremely). The items assess whether notes are up-to-date, accurate, thorough, useful, organized, comprehensible, succinct, synthesized, and internally consistent.11,12 The PDQI-9 has been applied previously in inpatient12 and outpatient settings.13
While the PDQI-9 is a validated tool, it relies on subjective ratings of note quality by the reviewer. To control for the subjective nature of the ratings, an experienced internist and an internal medicine resident coded 10 progress notes separately using the PDQI-9 and discussed the results. The process was repeated for a total of 20 notes, after which consensus was reached with >70% agreement on each attribute of the PDQI-9, suggesting that the resident’s ratings were reliable when compared with those of an experienced practicing physician.
The resident then evaluated a random sample of notes written by each physician for diabetes or same-day appointments in the pre-scribe and scribe periods. Word counts for the entire note were measured. The notes used to establish the reliability of the ratings were excluded from the analysis for this study.
Data analysis
We used linear mixed-effects models to examine note quality measures by adjusting for possible correlations of notes from the same physician. Least-squares estimates were derived; the results were not adjusted for multiple comparisons.
RESULTS
One hundred eight notes from the pre-scribe period and 109 notes from the scribe period were reviewed. Compared with notes written by a physician alone, scribed notes were rated slightly higher in overall quality (mean total PDQI-9 score 30.3 for scribe notes vs 28.9 for pre-scribe notes; P=.01) and more up-to-date, thorough, useful, and comprehensible (TABLES 1 AND 2). The differences were limited to diabetes encounters. For same day appointments, scribed notes did not differ in quality from nonscribed notes (TABLE 2). Total word count did not vary significantly between all scribe and pre-scribe notes (mean words 618, SD 273 for scribed notes vs 558 words, SD 289 for nonscribed notes; P=.12).
DISCUSSION
In this retrospective review of ambulatory notes, progress notes written by medical assistant scribes were of higher quality than notes physicians wrote alone, at least for diabetes visits. Scribe and pre-scribe notes were of similar quality for problem-focused same-day visits. This is the first study of which we are aware that compares the quality of scribed notes with notes written by physicians.
Quality scribe notes can save physician time. The progress note is an important vehicle for describing care provided and transferring information among physicians caring for the same patient. Writing a note, however, adds a considerable amount of time to the physician’s workflow. Using a scribe can decrease the time burden of note writing, and if scribed notes are of similar or better quality, this practice innovation can allow the physician to focus more on clinical than clerical tasks.
Over-documentation is a possible concern. While implementation of the EMR may improve certain aspects of quality of care delivered14,15 and note quality,16 concern has been raised about over-documentation related to the connection between documentation and reimbursement.17 In our study, we found that physician notes and scribed notes for both diabetes and same-day encounters often used EMR-based note templates, which can lead to over-documentation.
In general, both physician and scribed notes were rated to be of average to low quality because none of the mean scores on the 9 individual components of the PDQI-9 reached 4.0. Scribed notes were not inaccurate and had word counts similar to physician notes.
Scribing has potential drawbacks—and benefits. Drawbacks to scribing have not been well-studied. It has been suggested that using scribes to work around the EMR may actually hinder its further advancement because scribing insulates physicians from the inefficiencies of current EMRs and will not spur demands for improvements.3 Inaccurate or poor-quality notes could represent another downside to scribing, although concern about the quality of notes has not been documented. Our results suggest the opposite may be true.
Note quality has not been associated with quality of care as assessed by clinical quality scores,13 but using scribes may improve the quality of care in other ways. For example, the EMR may negatively affect patient-physician communication,18,19 and freeing the physician from documentation may improve the interaction.8,20 Incorporating scribing into practice may also improve the physician experience,9,10,21,22 a possible benefit that we did not measure.
We also did not measure the cost of using a scribe to assist in EMR documentation compared with the cost of physician time spent in performing this task. If the scribe model were associated with cost savings through increased physician productivity, as well as improved physician experience, future EMR development might best focus on planned utilization by physician-scribe teams.
Study limitations. The study was conducted in a single health system, although at 8 different practice sites. The sites all used the same EMR, but templates used for documentation could be individualized by the physician and medical assistant team, so our findings may reflect variation in template design. Our analysis did adjust for possible correlations of notes from the same physician. The selection of note types in our study may make our results less generalizable to other encounter types. Our sample was not large enough to detect variations in note quality among different providers and scribes.
The ratings on the PDQI-9 may be subjective, and the reviewers were not blinded to whether a scribe was used to write the note. The differences in PDQI-9 scores were small. Although statistically significant, they may not significantly affect clinical practice. Our care model is unique in that scribes are active members of the clinical care team; the higher quality of scribed notes we found may not apply to professional scribes who are not part of the team.
Future research directions. In our study, medical assistants acting as scribes composed progress notes of similar or higher quality than physicians who wrote notes alone, although all notes were of generally average quality. As the use of scribes in medicine expands, additional studies should examine the impact of scribes on primary care workflow, quality and cost of care delivered, and quality of physician experience.
CORRESPONDENCE
Anita D. Misra-Hebert, MD, MPH, Center for Value-Based Care Research, Medicine Institute, 9500 Euclid Avenue, G10, Cleveland, OH 44195; [email protected].
ABSTRACT
Objective Medical scribes are increasingly employed to improve physician efficiency with regard to the electronic medical record (EMR). The impact of scribes on the quality of outpatient visit notes is not known. To assess the effect, we conducted a retrospective review of ambulatory progress notes written before and after 8 practice sites transitioned to the use of medical assistants as scribes.
Methods The Physician Documentation Quality Instrument 9 (PDQI-9) was used to compare the quality of outpatient progress notes written by medical assistant scribes with the quality of notes written by 18 primary care physicians working without a scribe. The notes pertained to diabetes encounters and same-day appointments and were written during the 3 to 6 months preceding the use of scribes (pre-scribe period) and the 3 to 6 months after scribes were employed (scribe period).
Results One hundred eight notes from the pre-scribe period and 109 from the scribe period were reviewed. Scribed notes were rated higher in overall quality than unscribed notes (mean total PDQI-9 score 30.3 for scribed notes vs 28.9 for nonscribed notes; P=.01) and more up-to-date, thorough, useful, and comprehensible. The differences were limited to diabetes encounters. For same-day appointments, scribed and nonscribed notes did not differ in quality. The total word count of all scribed and nonscribed notes was similar (mean words 618, standard deviation (SD) 273 for scribed notes vs 558 words, SD 289 for nonscribed notes; P=.12).
Conclusions In this retrospective review, ambulatory notes were of higher quality when medical assistants acted as scribes than when physicians wrote them alone, at least for diabetes visits. Our findings may not apply to professional scribes who are not part of the clinical care team. As the use of medical scribes expands, additional studies should examine the impact of scribes on other aspects of care quality.
Team-based models of primary care delivery may incorporate medical scribes to improve efficiency of electronic documentation.1-4 The employment of medical scribes has grown rapidly, and it is estimated that within several years there may be one scribe for every 9 physicians.3
Accurate documentation is important to providing high-quality patient care but can take a significant amount of time. Attending physicians have been estimated to spend as long as 52 minutes per day authoring notes.5 Medical scribes can help physicians improve the efficiency of electronic documentation6 and save time.2 Using scribes can also improve physician productivity7-10 and thereby potentially increase access to care. The impact of scribes on the quality of outpatient visit notes, however, is unknown.
A team-based care delivery model in our health system’s primary care clinics uses medical assistants to scribe notes during the outpatient encounter. We hypothesized that outpatient notes written by medical assistant scribes would be of similar quality to notes written by the same group of physicians without a scribe.
METHODS
Study design and sample
We conducted a retrospective review of ambulatory notes from 18 primary care physicians at 8 practice sites in our health system who had adopted a care model in which medical assistants act as scribes. Each physician works with 2 medical assistants. To train for the new model, the physician and medical assistants participated in 2 training sessions of 2 hours each and a half day of clinic observation and evaluation with a project manager.
Of the 18 primary care physicians included in this study, none had less than one year of experience in our health system. Tenure ranged from one to 24 years with a mean of 11.3 years.
For each participating provider, we requested all available outpatient progress notes with either an International Classification of Diseases, 9th revision (ICD-9) code for diabetes or a designation of “same day” for the 3 to 6 months preceding the use of scribes (pre-scribe period) and the 3 to 6 months after employing scribes (scribe period). We chose diabetes encounters as examples of notes addressing chronic disease management and same-day encounters as examples of problem-focused notes because these 2 types of encounters are common in outpatient primary care practice.
Note quality was evaluated using the Physician Documentation Quality Instrument 9 (PDQI-9), a validated instrument designed for this purpose, comprising 9 items rated subjectively on a 5-point Likert scale (1= not at all, 5= extremely). The items assess whether notes are up-to-date, accurate, thorough, useful, organized, comprehensible, succinct, synthesized, and internally consistent.11,12 The PDQI-9 has been applied previously in inpatient12 and outpatient settings.13
While the PDQI-9 is a validated tool, it relies on subjective ratings of note quality by the reviewer. To control for the subjective nature of the ratings, an experienced internist and an internal medicine resident coded 10 progress notes separately using the PDQI-9 and discussed the results. The process was repeated for a total of 20 notes, after which consensus was reached with >70% agreement on each attribute of the PDQI-9, suggesting that the resident’s ratings were reliable when compared with those of an experienced practicing physician.
The resident then evaluated a random sample of notes written by each physician for diabetes or same-day appointments in the pre-scribe and scribe periods. Word counts for the entire note were measured. The notes used to establish the reliability of the ratings were excluded from the analysis for this study.
Data analysis
We used linear mixed-effects models to examine note quality measures by adjusting for possible correlations of notes from the same physician. Least-squares estimates were derived; the results were not adjusted for multiple comparisons.
RESULTS
One hundred eight notes from the pre-scribe period and 109 notes from the scribe period were reviewed. Compared with notes written by a physician alone, scribed notes were rated slightly higher in overall quality (mean total PDQI-9 score 30.3 for scribe notes vs 28.9 for pre-scribe notes; P=.01) and more up-to-date, thorough, useful, and comprehensible (TABLES 1 AND 2). The differences were limited to diabetes encounters. For same day appointments, scribed notes did not differ in quality from nonscribed notes (TABLE 2). Total word count did not vary significantly between all scribe and pre-scribe notes (mean words 618, SD 273 for scribed notes vs 558 words, SD 289 for nonscribed notes; P=.12).
DISCUSSION
In this retrospective review of ambulatory notes, progress notes written by medical assistant scribes were of higher quality than notes physicians wrote alone, at least for diabetes visits. Scribe and pre-scribe notes were of similar quality for problem-focused same-day visits. This is the first study of which we are aware that compares the quality of scribed notes with notes written by physicians.
Quality scribe notes can save physician time. The progress note is an important vehicle for describing care provided and transferring information among physicians caring for the same patient. Writing a note, however, adds a considerable amount of time to the physician’s workflow. Using a scribe can decrease the time burden of note writing, and if scribed notes are of similar or better quality, this practice innovation can allow the physician to focus more on clinical than clerical tasks.
Over-documentation is a possible concern. While implementation of the EMR may improve certain aspects of quality of care delivered14,15 and note quality,16 concern has been raised about over-documentation related to the connection between documentation and reimbursement.17 In our study, we found that physician notes and scribed notes for both diabetes and same-day encounters often used EMR-based note templates, which can lead to over-documentation.
In general, both physician and scribed notes were rated to be of average to low quality because none of the mean scores on the 9 individual components of the PDQI-9 reached 4.0. Scribed notes were not inaccurate and had word counts similar to physician notes.
Scribing has potential drawbacks—and benefits. Drawbacks to scribing have not been well-studied. It has been suggested that using scribes to work around the EMR may actually hinder its further advancement because scribing insulates physicians from the inefficiencies of current EMRs and will not spur demands for improvements.3 Inaccurate or poor-quality notes could represent another downside to scribing, although concern about the quality of notes has not been documented. Our results suggest the opposite may be true.
Note quality has not been associated with quality of care as assessed by clinical quality scores,13 but using scribes may improve the quality of care in other ways. For example, the EMR may negatively affect patient-physician communication,18,19 and freeing the physician from documentation may improve the interaction.8,20 Incorporating scribing into practice may also improve the physician experience,9,10,21,22 a possible benefit that we did not measure.
We also did not measure the cost of using a scribe to assist in EMR documentation compared with the cost of physician time spent in performing this task. If the scribe model were associated with cost savings through increased physician productivity, as well as improved physician experience, future EMR development might best focus on planned utilization by physician-scribe teams.
Study limitations. The study was conducted in a single health system, although at 8 different practice sites. The sites all used the same EMR, but templates used for documentation could be individualized by the physician and medical assistant team, so our findings may reflect variation in template design. Our analysis did adjust for possible correlations of notes from the same physician. The selection of note types in our study may make our results less generalizable to other encounter types. Our sample was not large enough to detect variations in note quality among different providers and scribes.
The ratings on the PDQI-9 may be subjective, and the reviewers were not blinded to whether a scribe was used to write the note. The differences in PDQI-9 scores were small. Although statistically significant, they may not significantly affect clinical practice. Our care model is unique in that scribes are active members of the clinical care team; the higher quality of scribed notes we found may not apply to professional scribes who are not part of the team.
Future research directions. In our study, medical assistants acting as scribes composed progress notes of similar or higher quality than physicians who wrote notes alone, although all notes were of generally average quality. As the use of scribes in medicine expands, additional studies should examine the impact of scribes on primary care workflow, quality and cost of care delivered, and quality of physician experience.
CORRESPONDENCE
Anita D. Misra-Hebert, MD, MPH, Center for Value-Based Care Research, Medicine Institute, 9500 Euclid Avenue, G10, Cleveland, OH 44195; [email protected].
1. Bodenheimer T, Willard-Grace R, Ghorob A. Expanding the roles of medical assistants: Who does what in primary care? JAMA Intern Med. 2014;174:1025-1026.
2. Reuben DB, Knudsen J, Senelick W, et al. The effect of a physician partner program on physician efficiency and patient satisfaction. JAMA Intern Med. 2014;174:1190-1193.
3. Gellert GA, Ramirea R, Webster S. The rise of the medical scribe industry: Implications for the advancement of electronic health records. JAMA. 2015;313:1315-1316.
4. Shultz CG, Holmstrom HL. The use of medical scribes in health care settings: a systematic review and future directions. J Am Board Fam Med. 2015;28:371-381.
5. Hripcsak G, Vawdrey DK, Fred MR, et al. Use of electronic clinical documentation: time spent and team interactions. J Am Med Inform Assoc. 2011;18:112-117.
6. Silverman L. Scribes Are Back, Helping Doctors Tackle Electronic Medical Records. NPR.org. Available at: www.npr.org/blogs/health/2014/04/21/303406306/scribes-are-back-helping-doctors-tackle-electronic-medical-records. Accessed April 23, 2014.
7. Arya R, Salovich DM, Ohman-Strickland P, et al. Impact of scribes on performance indicators in the emergency department. Acad Emerg Med. 2010;17:490-494.
8. Bank AJ, Obetz C, Konrardy A, et al. Impact of scribes on patient interaction, productivity, and revenue in a cardiology clinic: a prospective study. Clinicoecon Outcomes Res. 2013;5:399-406.
9. Bastani A, Shaqiri B, Palomba K, et al. An ED scribe program is able to improve throughput time and patient satisfaction. Am J Emerg Med. 2014;32:399-402.
10. Allen B, Banapoor B, Weeks EC, et al. An assessment of emergency department throughput and provider satisfaction after the implementation of a scribe program. Advances in Emergency Medicine. 2014;2014:e517319.
11. Stetson PD, Morrison FP, Bakken S, et al. Preliminary development of the Physician Documentation Quality Instrument. J Am Med Inform Assoc. 2008;15:534-541.
12. Stetson PD, Bakken S, Wrenn JO, et al. Assessing electronic note quality using the Physician Documentation Quality Instrument (PDQI-9). Appl Clin Inform. 2012;3:164-174.
13. Edwards ST, Neri PM, Volk LA, et al. Association of note quality and quality of care: a cross-sectional study. BMJ Qual Saf. 2013;23:406-413.
14. Schiff GD, Bates DW. Can electronic clinical documentation help prevent diagnostic errors? N Engl J Med. 2010;362:1066-1069.
15. Samal L, Wright A, Healey MJ, et al. Meaningful use and quality of care. JAMA Intern Med. 2014;174:997-998.
16. Burke HB, Sessums LL, Hoang A, et al. Electronic health records improve clinical note quality. J Am Med Inform Assoc. 2015;22:199-205.
17. Sheehy AM, Weissburg DJ, Dean SM. The role of copy-and-paste in the hospital electronic health record. JAMA Intern Med. 2014;174:1217-1218.
18. Shachak A, Hadas-Dayagi M, Ziv A, et al. Primary care physicians’ use of an electronic medical record system: a cognitive task analysis. J Gen Intern Med. 2009;24:341-348.
19. Shachak A, Reis S. The impact of electronic medical records on patient-doctor communication during consultation: a narrative literature review. J Eval Clin Pract. 2009;15:641-649.
20. Misra-Hebert AD, Rabovsky A, Yan C, et al. A team-based model of primary care delivery and physician-patient interaction. Am J Med. 2015;128:1025-1028.
21. Sinsky CA, Willard-Grace R, Schutzbank AM, et al. In search of joy in practice: a report of 23 high-functioning primary care practices. Ann Fam Med. 2013;11:272-278.
22. Koshy S, Feustel PJ, Hong M, et al. Scribes in an ambulatory urology practice: patient and physician satisfaction. J Urol. 2010;184:258-262.
1. Bodenheimer T, Willard-Grace R, Ghorob A. Expanding the roles of medical assistants: Who does what in primary care? JAMA Intern Med. 2014;174:1025-1026.
2. Reuben DB, Knudsen J, Senelick W, et al. The effect of a physician partner program on physician efficiency and patient satisfaction. JAMA Intern Med. 2014;174:1190-1193.
3. Gellert GA, Ramirea R, Webster S. The rise of the medical scribe industry: Implications for the advancement of electronic health records. JAMA. 2015;313:1315-1316.
4. Shultz CG, Holmstrom HL. The use of medical scribes in health care settings: a systematic review and future directions. J Am Board Fam Med. 2015;28:371-381.
5. Hripcsak G, Vawdrey DK, Fred MR, et al. Use of electronic clinical documentation: time spent and team interactions. J Am Med Inform Assoc. 2011;18:112-117.
6. Silverman L. Scribes Are Back, Helping Doctors Tackle Electronic Medical Records. NPR.org. Available at: www.npr.org/blogs/health/2014/04/21/303406306/scribes-are-back-helping-doctors-tackle-electronic-medical-records. Accessed April 23, 2014.
7. Arya R, Salovich DM, Ohman-Strickland P, et al. Impact of scribes on performance indicators in the emergency department. Acad Emerg Med. 2010;17:490-494.
8. Bank AJ, Obetz C, Konrardy A, et al. Impact of scribes on patient interaction, productivity, and revenue in a cardiology clinic: a prospective study. Clinicoecon Outcomes Res. 2013;5:399-406.
9. Bastani A, Shaqiri B, Palomba K, et al. An ED scribe program is able to improve throughput time and patient satisfaction. Am J Emerg Med. 2014;32:399-402.
10. Allen B, Banapoor B, Weeks EC, et al. An assessment of emergency department throughput and provider satisfaction after the implementation of a scribe program. Advances in Emergency Medicine. 2014;2014:e517319.
11. Stetson PD, Morrison FP, Bakken S, et al. Preliminary development of the Physician Documentation Quality Instrument. J Am Med Inform Assoc. 2008;15:534-541.
12. Stetson PD, Bakken S, Wrenn JO, et al. Assessing electronic note quality using the Physician Documentation Quality Instrument (PDQI-9). Appl Clin Inform. 2012;3:164-174.
13. Edwards ST, Neri PM, Volk LA, et al. Association of note quality and quality of care: a cross-sectional study. BMJ Qual Saf. 2013;23:406-413.
14. Schiff GD, Bates DW. Can electronic clinical documentation help prevent diagnostic errors? N Engl J Med. 2010;362:1066-1069.
15. Samal L, Wright A, Healey MJ, et al. Meaningful use and quality of care. JAMA Intern Med. 2014;174:997-998.
16. Burke HB, Sessums LL, Hoang A, et al. Electronic health records improve clinical note quality. J Am Med Inform Assoc. 2015;22:199-205.
17. Sheehy AM, Weissburg DJ, Dean SM. The role of copy-and-paste in the hospital electronic health record. JAMA Intern Med. 2014;174:1217-1218.
18. Shachak A, Hadas-Dayagi M, Ziv A, et al. Primary care physicians’ use of an electronic medical record system: a cognitive task analysis. J Gen Intern Med. 2009;24:341-348.
19. Shachak A, Reis S. The impact of electronic medical records on patient-doctor communication during consultation: a narrative literature review. J Eval Clin Pract. 2009;15:641-649.
20. Misra-Hebert AD, Rabovsky A, Yan C, et al. A team-based model of primary care delivery and physician-patient interaction. Am J Med. 2015;128:1025-1028.
21. Sinsky CA, Willard-Grace R, Schutzbank AM, et al. In search of joy in practice: a report of 23 high-functioning primary care practices. Ann Fam Med. 2013;11:272-278.
22. Koshy S, Feustel PJ, Hong M, et al. Scribes in an ambulatory urology practice: patient and physician satisfaction. J Urol. 2010;184:258-262.

Thrombocytopenia signals multiorgan system failure in acute liver failure
In patients with acute liver failure (ALF), decreasing platelet counts after hospital admission signaled systemic inflammation and a greater likelihood of systemic complications, such as high-grade hepatic encephalopathy, cardiovascular collapse, the need for liver transplant, and death, according to researchers.
Patients with systemic inflammatory response syndrome (SIRS) had significantly lower platelet counts at admission, compared with those without SIRS, and their platelet counts decreased dramatically (from 182 plus or minus 27 times 109/L on admission to 103 plus or minus 3.20 times 109/L on day 6) compared with stable platelet counts in patients without SIRS. For days 2-7 postadmission, lower platelet counts were associated with high-grade hepatic encephalopathy, as well as the need for vasopressor support and renal replacement therapy (P less than or equal to .001 for all).
“We hypothesize that the decrease in platelet count represents an integral event in the pathogenesis of the ALF syndrome rather than a nonspecific marker of suppressed bone marrow production. Further studies will be needed to prove the pivotal role of platelets in mediating the proinflammatory and prothrombotic features of the ALF syndrome,” wrote Dr. R. Todd Stravitz, professor of medicine, section of hepatology, Virginia Commonwealth University, Richmond, and his colleagues (Clin Gastroenterol Hepatol. 2016 Feb 25. doi: 10.1016/j.cgh.2015.09.029).
Results showed that SIRS and multiorgan system failure (MOSF) were more closely linked to a decrease in platelet counts than to an increase in the international normalized ratio (INR), a laboratory marker for liver injury. The INR was similar in patients with and without SIRS, in high-grade and low-grade hepatic encephalopathy, and in patients with and without requirements for vasopressor support and renal replacement therapy (indicators of MOSF). Given that both platelet counts and INR were associated with prognosis, the investigators suggest that these laboratory parameters may reflect different types of injury, with INR signaling primary liver injury and platelets reflecting the severity of systemic inflammation secondary to liver injury.
Platelet counts varied according to outcome on day 21. Spontaneous survivors had higher mean platelet counts than patients who underwent liver transplant, and patients who died had the lowest platelet counts. Platelet counts in spontaneous survivors decreased from day 1 to 2, then subsequently recovered; platelets decreased progressively in patients who underwent liver transplant or died. The INR trend over time according to 21-day outcome was similar to that of platelet counts.
The retrospective study evaluated data from 1598 patients who enrolled in the ALF Study Group from 1998 to 2012. The mean age was 41 years, 76% were Caucasian, and 70% were female. Nearly one-half of participants (47%) had ALF due to acetaminophen overdose, 85% had at least one positive element of SIRS on admission, 32% required vasopressors, 33% required renal replacement therapy, and 50% developed high-grade (3 or 4) hepatic encephalopathy. In total, 47% of patients recovered without liver transplant, 24% underwent liver transplant, and 32% died.
The mechanism underlying development of thrombocytopenia in patients with ALF is poorly understood. Previous findings by the investigators showed that platelet-derived, prothrombotic microparticles increased in proportion to SIRS severity, and concentration of microparticles increased in parallel with laboratory markers of poor outcome. Current results suggest the converse to be true as well: platelet counts decreased in proportion to the severity of SIRS, the development of MOSF and poor outcome at 21 days.
The researchers proposed that deficiencies in the number of platelets or in liver-derived, prohemostatic coagulation factors may be compensated by systemic inflammation. Increased levels of platelet-derived microparticles in patients with ALF may overcompensate for thrombocytopenia due to a nearly 40-fold higher prothrombotic potential in tissue factor–dependent assays, compared with healthy control populations.
The findings point to the importance of platelet count in signaling impending complications in patients with ALF.
Dr. Stravitz and his coauthors reported having no disclosures.
In patients with acute liver failure (ALF), decreasing platelet counts after hospital admission signaled systemic inflammation and a greater likelihood of systemic complications, such as high-grade hepatic encephalopathy, cardiovascular collapse, the need for liver transplant, and death, according to researchers.
Patients with systemic inflammatory response syndrome (SIRS) had significantly lower platelet counts at admission, compared with those without SIRS, and their platelet counts decreased dramatically (from 182 plus or minus 27 times 109/L on admission to 103 plus or minus 3.20 times 109/L on day 6) compared with stable platelet counts in patients without SIRS. For days 2-7 postadmission, lower platelet counts were associated with high-grade hepatic encephalopathy, as well as the need for vasopressor support and renal replacement therapy (P less than or equal to .001 for all).
“We hypothesize that the decrease in platelet count represents an integral event in the pathogenesis of the ALF syndrome rather than a nonspecific marker of suppressed bone marrow production. Further studies will be needed to prove the pivotal role of platelets in mediating the proinflammatory and prothrombotic features of the ALF syndrome,” wrote Dr. R. Todd Stravitz, professor of medicine, section of hepatology, Virginia Commonwealth University, Richmond, and his colleagues (Clin Gastroenterol Hepatol. 2016 Feb 25. doi: 10.1016/j.cgh.2015.09.029).
Results showed that SIRS and multiorgan system failure (MOSF) were more closely linked to a decrease in platelet counts than to an increase in the international normalized ratio (INR), a laboratory marker for liver injury. The INR was similar in patients with and without SIRS, in high-grade and low-grade hepatic encephalopathy, and in patients with and without requirements for vasopressor support and renal replacement therapy (indicators of MOSF). Given that both platelet counts and INR were associated with prognosis, the investigators suggest that these laboratory parameters may reflect different types of injury, with INR signaling primary liver injury and platelets reflecting the severity of systemic inflammation secondary to liver injury.
Platelet counts varied according to outcome on day 21. Spontaneous survivors had higher mean platelet counts than patients who underwent liver transplant, and patients who died had the lowest platelet counts. Platelet counts in spontaneous survivors decreased from day 1 to 2, then subsequently recovered; platelets decreased progressively in patients who underwent liver transplant or died. The INR trend over time according to 21-day outcome was similar to that of platelet counts.
The retrospective study evaluated data from 1598 patients who enrolled in the ALF Study Group from 1998 to 2012. The mean age was 41 years, 76% were Caucasian, and 70% were female. Nearly one-half of participants (47%) had ALF due to acetaminophen overdose, 85% had at least one positive element of SIRS on admission, 32% required vasopressors, 33% required renal replacement therapy, and 50% developed high-grade (3 or 4) hepatic encephalopathy. In total, 47% of patients recovered without liver transplant, 24% underwent liver transplant, and 32% died.
The mechanism underlying development of thrombocytopenia in patients with ALF is poorly understood. Previous findings by the investigators showed that platelet-derived, prothrombotic microparticles increased in proportion to SIRS severity, and concentration of microparticles increased in parallel with laboratory markers of poor outcome. Current results suggest the converse to be true as well: platelet counts decreased in proportion to the severity of SIRS, the development of MOSF and poor outcome at 21 days.
The researchers proposed that deficiencies in the number of platelets or in liver-derived, prohemostatic coagulation factors may be compensated by systemic inflammation. Increased levels of platelet-derived microparticles in patients with ALF may overcompensate for thrombocytopenia due to a nearly 40-fold higher prothrombotic potential in tissue factor–dependent assays, compared with healthy control populations.
The findings point to the importance of platelet count in signaling impending complications in patients with ALF.
Dr. Stravitz and his coauthors reported having no disclosures.
In patients with acute liver failure (ALF), decreasing platelet counts after hospital admission signaled systemic inflammation and a greater likelihood of systemic complications, such as high-grade hepatic encephalopathy, cardiovascular collapse, the need for liver transplant, and death, according to researchers.
Patients with systemic inflammatory response syndrome (SIRS) had significantly lower platelet counts at admission, compared with those without SIRS, and their platelet counts decreased dramatically (from 182 plus or minus 27 times 109/L on admission to 103 plus or minus 3.20 times 109/L on day 6) compared with stable platelet counts in patients without SIRS. For days 2-7 postadmission, lower platelet counts were associated with high-grade hepatic encephalopathy, as well as the need for vasopressor support and renal replacement therapy (P less than or equal to .001 for all).
“We hypothesize that the decrease in platelet count represents an integral event in the pathogenesis of the ALF syndrome rather than a nonspecific marker of suppressed bone marrow production. Further studies will be needed to prove the pivotal role of platelets in mediating the proinflammatory and prothrombotic features of the ALF syndrome,” wrote Dr. R. Todd Stravitz, professor of medicine, section of hepatology, Virginia Commonwealth University, Richmond, and his colleagues (Clin Gastroenterol Hepatol. 2016 Feb 25. doi: 10.1016/j.cgh.2015.09.029).
Results showed that SIRS and multiorgan system failure (MOSF) were more closely linked to a decrease in platelet counts than to an increase in the international normalized ratio (INR), a laboratory marker for liver injury. The INR was similar in patients with and without SIRS, in high-grade and low-grade hepatic encephalopathy, and in patients with and without requirements for vasopressor support and renal replacement therapy (indicators of MOSF). Given that both platelet counts and INR were associated with prognosis, the investigators suggest that these laboratory parameters may reflect different types of injury, with INR signaling primary liver injury and platelets reflecting the severity of systemic inflammation secondary to liver injury.
Platelet counts varied according to outcome on day 21. Spontaneous survivors had higher mean platelet counts than patients who underwent liver transplant, and patients who died had the lowest platelet counts. Platelet counts in spontaneous survivors decreased from day 1 to 2, then subsequently recovered; platelets decreased progressively in patients who underwent liver transplant or died. The INR trend over time according to 21-day outcome was similar to that of platelet counts.
The retrospective study evaluated data from 1598 patients who enrolled in the ALF Study Group from 1998 to 2012. The mean age was 41 years, 76% were Caucasian, and 70% were female. Nearly one-half of participants (47%) had ALF due to acetaminophen overdose, 85% had at least one positive element of SIRS on admission, 32% required vasopressors, 33% required renal replacement therapy, and 50% developed high-grade (3 or 4) hepatic encephalopathy. In total, 47% of patients recovered without liver transplant, 24% underwent liver transplant, and 32% died.
The mechanism underlying development of thrombocytopenia in patients with ALF is poorly understood. Previous findings by the investigators showed that platelet-derived, prothrombotic microparticles increased in proportion to SIRS severity, and concentration of microparticles increased in parallel with laboratory markers of poor outcome. Current results suggest the converse to be true as well: platelet counts decreased in proportion to the severity of SIRS, the development of MOSF and poor outcome at 21 days.
The researchers proposed that deficiencies in the number of platelets or in liver-derived, prohemostatic coagulation factors may be compensated by systemic inflammation. Increased levels of platelet-derived microparticles in patients with ALF may overcompensate for thrombocytopenia due to a nearly 40-fold higher prothrombotic potential in tissue factor–dependent assays, compared with healthy control populations.
The findings point to the importance of platelet count in signaling impending complications in patients with ALF.
Dr. Stravitz and his coauthors reported having no disclosures.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Key clinical point: In patients with acute liver failure (ALF), decreasing platelet counts were associated with systemic inflammation and greater likelihood of serious systemic complications.
Major finding: Platelet counts decreased dramatically in patients with systemic inflammatory response syndrome (SIRS) (182 plus or minus 27 times 109/L on admission to 103 plus or minus 3.20 times 109/L on day 6) but remained stable in patients without SIRS; lower platelet counts were associated with high-grade hepatic encephalopathy, as well as the need for vasopressor support and renal replacement therapy (P less than or equal to .001 for all).
Data source: From 1998 to 2012 the ALF Study Group included 1,598 patients of mean age 41 years; 76% were Caucasian and 70% were female.
Disclosures: Dr. Stravitz and his coauthors reported having no disclosures.
2015-2016 flu vaccine 59% effective, CDC says
The overall rate of effectiveness for the 2015-2016 season’s influenza vaccine is 59%, according to preliminary data shared at a Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices meeting.
“This means that getting a flu vaccine this season reduced the risk of having to go to the doctor because of flu by nearly 60%,” Dr. Joseph Bresee – who heads the CDC’s Epidemiology and Prevention Branch – stated in a press release. “It’s good news and underscores the importance and the benefit of both annual and ongoing vaccination efforts this season.”

These findings are based on data collected by the U.S. Flu Vaccine Effectiveness Network between November 2, 2015, and February 12, 2016. A 59% effectiveness rate would be roughly in line with what previous years’ vaccines have rated when similar strains of influenza have been prevalent, according to the CDC.
Against the H1N1 strain, which was “responsible for most flu illness this season,” the influenza vaccine was 51% effective. Dr. Bresee noted that the CDC has received reports of unvaccinated individuals this season dying or falling seriously ill because of infection from the H1N1 strain.
The vaccine was 76% effective against all influenza B virus strains, and 79% effective against the B/Yamagata virus strains. There are not enough data at this time, however, to determine the vaccine’s effectiveness against B/Victoria or H3N2 strains.
The CDC cautions that, because the current influenza season is still weeks away from being over, the final rate of vaccine effectiveness for 2015-2016 may change once all the data are analyzed. On average, influenza seasons last 13 weeks.
“Flu activity this season started a bit later and has been lower so far than we’ve seen during the previous three seasons, but activity is still on the upswing and expected to continue for several weeks,” Dr. Bresee stated.
The overall rate of effectiveness for the 2015-2016 season’s influenza vaccine is 59%, according to preliminary data shared at a Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices meeting.
“This means that getting a flu vaccine this season reduced the risk of having to go to the doctor because of flu by nearly 60%,” Dr. Joseph Bresee – who heads the CDC’s Epidemiology and Prevention Branch – stated in a press release. “It’s good news and underscores the importance and the benefit of both annual and ongoing vaccination efforts this season.”
These findings are based on data collected by the U.S. Flu Vaccine Effectiveness Network between November 2, 2015, and February 12, 2016. A 59% effectiveness rate would be roughly in line with what previous years’ vaccines have rated when similar strains of influenza have been prevalent, according to the CDC.
Against the H1N1 strain, which was “responsible for most flu illness this season,” the influenza vaccine was 51% effective. Dr. Bresee noted that the CDC has received reports of unvaccinated individuals this season dying or falling seriously ill because of infection from the H1N1 strain.
The vaccine was 76% effective against all influenza B virus strains, and 79% effective against the B/Yamagata virus strains. There are not enough data at this time, however, to determine the vaccine’s effectiveness against B/Victoria or H3N2 strains.
The CDC cautions that, because the current influenza season is still weeks away from being over, the final rate of vaccine effectiveness for 2015-2016 may change once all the data are analyzed. On average, influenza seasons last 13 weeks.
“Flu activity this season started a bit later and has been lower so far than we’ve seen during the previous three seasons, but activity is still on the upswing and expected to continue for several weeks,” Dr. Bresee stated.
The overall rate of effectiveness for the 2015-2016 season’s influenza vaccine is 59%, according to preliminary data shared at a Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices meeting.
“This means that getting a flu vaccine this season reduced the risk of having to go to the doctor because of flu by nearly 60%,” Dr. Joseph Bresee – who heads the CDC’s Epidemiology and Prevention Branch – stated in a press release. “It’s good news and underscores the importance and the benefit of both annual and ongoing vaccination efforts this season.”
These findings are based on data collected by the U.S. Flu Vaccine Effectiveness Network between November 2, 2015, and February 12, 2016. A 59% effectiveness rate would be roughly in line with what previous years’ vaccines have rated when similar strains of influenza have been prevalent, according to the CDC.
Against the H1N1 strain, which was “responsible for most flu illness this season,” the influenza vaccine was 51% effective. Dr. Bresee noted that the CDC has received reports of unvaccinated individuals this season dying or falling seriously ill because of infection from the H1N1 strain.
The vaccine was 76% effective against all influenza B virus strains, and 79% effective against the B/Yamagata virus strains. There are not enough data at this time, however, to determine the vaccine’s effectiveness against B/Victoria or H3N2 strains.
The CDC cautions that, because the current influenza season is still weeks away from being over, the final rate of vaccine effectiveness for 2015-2016 may change once all the data are analyzed. On average, influenza seasons last 13 weeks.
“Flu activity this season started a bit later and has been lower so far than we’ve seen during the previous three seasons, but activity is still on the upswing and expected to continue for several weeks,” Dr. Bresee stated.
FROM AN ACIP MEETING

Allergic Contact Dermatitis, Part 4

Practice Questions
1. The most common allergen of hand dermatitis in hairdressers can cross-react with which of the following allergens?
a. benzocaine
b. para-aminobenzoic acid
c. procaine
d. sulfanomides
e. all of the above
2. Patients with a documented allergy to quaternium-15 should avoid all of the following ingredients except:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. imidazolidinyl urea
e. paraben mix
3. Which of the following is a screening agent for hydrocortisone allergy?
a. budesonide
b. clobetasol
c. desoximetasone
d. paraben mix
e. tixocortol pivalate
4. This allergen often is found in black synthetic henna tattoos:
a. paraben mix
b. potassium dichromate
c. PPD
d. quaternium-15
e. thimerosol
5. A patient with a documented allergy to paraben mix also should avoid the following agent:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. PPD
e. thiuram mix
Practice Question Answers
1. The most common allergen of hand dermatitis in hairdressers can cross-react with which of the following allergens?
a. benzocaine
b. para-aminobenzoic acid
c. procaine
d. sulfanomides
e. all of the above
2. Patients with a documented allergy to quaternium-15 should avoid all of the following ingredients except:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. imidazolidinyl urea
e. paraben mix
3. Which of the following is a screening agent for hydrocortisone allergy?
a. budesonide
b. clobetasol
c. desoximetasone
d. paraben mix
e. tixocortol pivalate
4. This allergen often is found in black synthetic henna tattoos:
a. paraben mix
b. potassium dichromate
c. PPD
d. quaternium-15
e. thimerosol
5. A patient with a documented allergy to paraben mix also should avoid the following agent:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. PPD
e. thiuram mix
Practice Questions
1. The most common allergen of hand dermatitis in hairdressers can cross-react with which of the following allergens?
a. benzocaine
b. para-aminobenzoic acid
c. procaine
d. sulfanomides
e. all of the above
2. Patients with a documented allergy to quaternium-15 should avoid all of the following ingredients except:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. imidazolidinyl urea
e. paraben mix
3. Which of the following is a screening agent for hydrocortisone allergy?
a. budesonide
b. clobetasol
c. desoximetasone
d. paraben mix
e. tixocortol pivalate
4. This allergen often is found in black synthetic henna tattoos:
a. paraben mix
b. potassium dichromate
c. PPD
d. quaternium-15
e. thimerosol
5. A patient with a documented allergy to paraben mix also should avoid the following agent:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. PPD
e. thiuram mix
Practice Question Answers
1. The most common allergen of hand dermatitis in hairdressers can cross-react with which of the following allergens?
a. benzocaine
b. para-aminobenzoic acid
c. procaine
d. sulfanomides
e. all of the above
2. Patients with a documented allergy to quaternium-15 should avoid all of the following ingredients except:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. imidazolidinyl urea
e. paraben mix
3. Which of the following is a screening agent for hydrocortisone allergy?
a. budesonide
b. clobetasol
c. desoximetasone
d. paraben mix
e. tixocortol pivalate
4. This allergen often is found in black synthetic henna tattoos:
a. paraben mix
b. potassium dichromate
c. PPD
d. quaternium-15
e. thimerosol
5. A patient with a documented allergy to paraben mix also should avoid the following agent:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. PPD
e. thiuram mix
Practice Questions
1. The most common allergen of hand dermatitis in hairdressers can cross-react with which of the following allergens?
a. benzocaine
b. para-aminobenzoic acid
c. procaine
d. sulfanomides
e. all of the above
2. Patients with a documented allergy to quaternium-15 should avoid all of the following ingredients except:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. imidazolidinyl urea
e. paraben mix
3. Which of the following is a screening agent for hydrocortisone allergy?
a. budesonide
b. clobetasol
c. desoximetasone
d. paraben mix
e. tixocortol pivalate
4. This allergen often is found in black synthetic henna tattoos:
a. paraben mix
b. potassium dichromate
c. PPD
d. quaternium-15
e. thimerosol
5. A patient with a documented allergy to paraben mix also should avoid the following agent:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. PPD
e. thiuram mix
Practice Question Answers
1. The most common allergen of hand dermatitis in hairdressers can cross-react with which of the following allergens?
a. benzocaine
b. para-aminobenzoic acid
c. procaine
d. sulfanomides
e. all of the above
2. Patients with a documented allergy to quaternium-15 should avoid all of the following ingredients except:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. imidazolidinyl urea
e. paraben mix
3. Which of the following is a screening agent for hydrocortisone allergy?
a. budesonide
b. clobetasol
c. desoximetasone
d. paraben mix
e. tixocortol pivalate
4. This allergen often is found in black synthetic henna tattoos:
a. paraben mix
b. potassium dichromate
c. PPD
d. quaternium-15
e. thimerosol
5. A patient with a documented allergy to paraben mix also should avoid the following agent:
a. bronopol
b. diazolidinyl urea
c. DMDM hydantoin
d. PPD
e. thiuram mix

ACIP recommends LAIV as an option for all people with egg allergies
Live attenuated influenza vaccine (LAIV) is likely to be an option for all individuals with egg allergies, regardless of allergy severity, because the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices voted to approve proposed amendments to the existing recommendations regarding administration of LAIV in individuals with egg allergies.
With 11 members voting “yes” and three choosing to abstain, the committee agreed to leave sections 1, 2, and 3 as they currently are, with a slight change to section 1 so that it will now include the latter half of recommendations that were previously placed under section 4. Therefore, section 1 of the recommendations for influenza vaccination of persons with egg allergy – which already states that “Regardless of a recipient’s allergy history, all vaccines should be administered in settings in which personnel and equipment for rapid recognition and treatment of anaphylaxis are available” – will now also include text stating that the vaccine should be administered in a medical setting and supervised by a health care provider with “experience in the recognition and management of severe allergic conditions,” or that such a medical professional should be “immediately available.”
Sections 2 and 3 will remain the same. Section 2 states that a previous allergic reaction to the influenza vaccine is a contraindication to ever receiving the vaccine again in the future. Section 3 states that individuals with egg allergy who have only experienced hives due to eggs still should be administered the influenza vaccine.
The committee voted to strike section 5 of the existing recommendations, which calls for a 30-minute postvaccination observation period. However, the committee still advises that health care providers monitor all patients, particularly adolescents, for 15 minutes immediately after vaccination, and that individuals should be seated or lying down “to decrease the risk for injury should syncopy occur.”
The CDC generally follows ACIP’s recommendations.
Live attenuated influenza vaccine (LAIV) is likely to be an option for all individuals with egg allergies, regardless of allergy severity, because the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices voted to approve proposed amendments to the existing recommendations regarding administration of LAIV in individuals with egg allergies.
With 11 members voting “yes” and three choosing to abstain, the committee agreed to leave sections 1, 2, and 3 as they currently are, with a slight change to section 1 so that it will now include the latter half of recommendations that were previously placed under section 4. Therefore, section 1 of the recommendations for influenza vaccination of persons with egg allergy – which already states that “Regardless of a recipient’s allergy history, all vaccines should be administered in settings in which personnel and equipment for rapid recognition and treatment of anaphylaxis are available” – will now also include text stating that the vaccine should be administered in a medical setting and supervised by a health care provider with “experience in the recognition and management of severe allergic conditions,” or that such a medical professional should be “immediately available.”
Sections 2 and 3 will remain the same. Section 2 states that a previous allergic reaction to the influenza vaccine is a contraindication to ever receiving the vaccine again in the future. Section 3 states that individuals with egg allergy who have only experienced hives due to eggs still should be administered the influenza vaccine.
The committee voted to strike section 5 of the existing recommendations, which calls for a 30-minute postvaccination observation period. However, the committee still advises that health care providers monitor all patients, particularly adolescents, for 15 minutes immediately after vaccination, and that individuals should be seated or lying down “to decrease the risk for injury should syncopy occur.”
The CDC generally follows ACIP’s recommendations.
Live attenuated influenza vaccine (LAIV) is likely to be an option for all individuals with egg allergies, regardless of allergy severity, because the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices voted to approve proposed amendments to the existing recommendations regarding administration of LAIV in individuals with egg allergies.
With 11 members voting “yes” and three choosing to abstain, the committee agreed to leave sections 1, 2, and 3 as they currently are, with a slight change to section 1 so that it will now include the latter half of recommendations that were previously placed under section 4. Therefore, section 1 of the recommendations for influenza vaccination of persons with egg allergy – which already states that “Regardless of a recipient’s allergy history, all vaccines should be administered in settings in which personnel and equipment for rapid recognition and treatment of anaphylaxis are available” – will now also include text stating that the vaccine should be administered in a medical setting and supervised by a health care provider with “experience in the recognition and management of severe allergic conditions,” or that such a medical professional should be “immediately available.”
Sections 2 and 3 will remain the same. Section 2 states that a previous allergic reaction to the influenza vaccine is a contraindication to ever receiving the vaccine again in the future. Section 3 states that individuals with egg allergy who have only experienced hives due to eggs still should be administered the influenza vaccine.
The committee voted to strike section 5 of the existing recommendations, which calls for a 30-minute postvaccination observation period. However, the committee still advises that health care providers monitor all patients, particularly adolescents, for 15 minutes immediately after vaccination, and that individuals should be seated or lying down “to decrease the risk for injury should syncopy occur.”
The CDC generally follows ACIP’s recommendations.
FROM AN ACIP MEETING