User login
CRT in Patients with Heart Failure Without LBBB May Harm
NEW YORK (Reuters Health) - Cardiac resynchronization therapy (CRT) in patients with heart failure (HF) without left bundle branch block (LBBB) may not help and might even harm, according to an international group of investigators.
As Dr. Yitschak Biton told Reuters Health by email, "Our findings suggest that patients without LBBB electrocardiogram (ECG) morphology are not likely to benefit from CRT implantation and a subgroup of patients with short QRS duration might even be at higher risk for mortality."
In a January 28 online paper in Circulation: Heart Failure, Dr. Biton, of the University of Rochester Medical Center, New York, and colleagues note that the efficacy of CRT is well established in patients with both mild and moderate to severe HF symptoms. However, data on non-LBBB patients "are more limited and conflicting."
To investigate, the team examined data on 537 such patients with mild HF taking part in a larger study. At seven years, the cumulative probability of HF hospitalization or death was 45% in those randomized to an implantable cardioverter-defibrillator (ICD) and 56% in those given CRT with a defibrillator (CRT-D).
Multivariable-adjusted subgroup analysis by QRS duration showed that patients from the lower quartile (134 ms or less) had a 2.4-fold greater risk of HF hospitalization or death with CRT-D versus those with ICD-only therapy.
However, the effect of CRT-D in patients from the upper quartiles group (QRS greater than 134 ms) was neutral (hazard ratio 0.97).
In a further analysis based on PR interval, patients with prolonged QRS (more than 134 ms) and prolonged PR (at least 230 ms) were protected with CRT-D (HR 0.31). The association was neutral with prolonged QRS and shorter PR.
"Overall," the researchers conclude, "patients with mild HF but without left bundle branch block morphology did not derive clinical benefit with CRT-D during long-term follow-up. Relatively shorter QRS was associated with a significantly increased risk with CRT-D relative to implantable cardioverter-defibrillator only."
"This information should be taken into account when CRT therapy is considered in this subgroup of patients," Dr. Biton told Reuters Health.
Boston Scientific Corporation funded the clinical trial this research is based on. Five coauthors reported disclosures.
NEW YORK (Reuters Health) - Cardiac resynchronization therapy (CRT) in patients with heart failure (HF) without left bundle branch block (LBBB) may not help and might even harm, according to an international group of investigators.
As Dr. Yitschak Biton told Reuters Health by email, "Our findings suggest that patients without LBBB electrocardiogram (ECG) morphology are not likely to benefit from CRT implantation and a subgroup of patients with short QRS duration might even be at higher risk for mortality."
In a January 28 online paper in Circulation: Heart Failure, Dr. Biton, of the University of Rochester Medical Center, New York, and colleagues note that the efficacy of CRT is well established in patients with both mild and moderate to severe HF symptoms. However, data on non-LBBB patients "are more limited and conflicting."
To investigate, the team examined data on 537 such patients with mild HF taking part in a larger study. At seven years, the cumulative probability of HF hospitalization or death was 45% in those randomized to an implantable cardioverter-defibrillator (ICD) and 56% in those given CRT with a defibrillator (CRT-D).
Multivariable-adjusted subgroup analysis by QRS duration showed that patients from the lower quartile (134 ms or less) had a 2.4-fold greater risk of HF hospitalization or death with CRT-D versus those with ICD-only therapy.
However, the effect of CRT-D in patients from the upper quartiles group (QRS greater than 134 ms) was neutral (hazard ratio 0.97).
In a further analysis based on PR interval, patients with prolonged QRS (more than 134 ms) and prolonged PR (at least 230 ms) were protected with CRT-D (HR 0.31). The association was neutral with prolonged QRS and shorter PR.
"Overall," the researchers conclude, "patients with mild HF but without left bundle branch block morphology did not derive clinical benefit with CRT-D during long-term follow-up. Relatively shorter QRS was associated with a significantly increased risk with CRT-D relative to implantable cardioverter-defibrillator only."
"This information should be taken into account when CRT therapy is considered in this subgroup of patients," Dr. Biton told Reuters Health.
Boston Scientific Corporation funded the clinical trial this research is based on. Five coauthors reported disclosures.
NEW YORK (Reuters Health) - Cardiac resynchronization therapy (CRT) in patients with heart failure (HF) without left bundle branch block (LBBB) may not help and might even harm, according to an international group of investigators.
As Dr. Yitschak Biton told Reuters Health by email, "Our findings suggest that patients without LBBB electrocardiogram (ECG) morphology are not likely to benefit from CRT implantation and a subgroup of patients with short QRS duration might even be at higher risk for mortality."
In a January 28 online paper in Circulation: Heart Failure, Dr. Biton, of the University of Rochester Medical Center, New York, and colleagues note that the efficacy of CRT is well established in patients with both mild and moderate to severe HF symptoms. However, data on non-LBBB patients "are more limited and conflicting."
To investigate, the team examined data on 537 such patients with mild HF taking part in a larger study. At seven years, the cumulative probability of HF hospitalization or death was 45% in those randomized to an implantable cardioverter-defibrillator (ICD) and 56% in those given CRT with a defibrillator (CRT-D).
Multivariable-adjusted subgroup analysis by QRS duration showed that patients from the lower quartile (134 ms or less) had a 2.4-fold greater risk of HF hospitalization or death with CRT-D versus those with ICD-only therapy.
However, the effect of CRT-D in patients from the upper quartiles group (QRS greater than 134 ms) was neutral (hazard ratio 0.97).
In a further analysis based on PR interval, patients with prolonged QRS (more than 134 ms) and prolonged PR (at least 230 ms) were protected with CRT-D (HR 0.31). The association was neutral with prolonged QRS and shorter PR.
"Overall," the researchers conclude, "patients with mild HF but without left bundle branch block morphology did not derive clinical benefit with CRT-D during long-term follow-up. Relatively shorter QRS was associated with a significantly increased risk with CRT-D relative to implantable cardioverter-defibrillator only."
"This information should be taken into account when CRT therapy is considered in this subgroup of patients," Dr. Biton told Reuters Health.
Boston Scientific Corporation funded the clinical trial this research is based on. Five coauthors reported disclosures.
Gene therapy granted orphan designation for hemophilia A
Image courtesy of NHLBI
The US Food and Drug Administration (FDA) has granted orphan designation to BMN 270, an investigational gene therapy, for the treatment of patients with hemophilia A.
BMN 270 is an adeno-associated virus-factor VIII (FVIII) vector designed to restore FVIII plasma concentrations in these patients.
The FDA grants orphan designation to drugs that are intended to treat diseases or conditions affecting fewer than 200,000 patients in the US.
The designation provides the drug’s sponsor with various development incentives, including opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, and 7 years of US market exclusivity if the drug is approved.
BMN 270 is under development by BioMarin Pharmaceutical Inc.
BioMarin is conducting a phase 1/2 study to evaluate the safety and efficacy of BMN 270 in up to 12 patients with severe hemophilia A.
Researchers are assessing the safety of a single infusion of BMN 270 and the change in FVIII expression level from baseline to 16 weeks after infusion.
The group is also assessing the impact of BMN 270 on the frequency of FVIII replacement therapy, the number of bleeding episodes requiring treatment, and any potential immune responses.
Patients will be monitored for safety and durability of effect for 5 years. BioMarin plans to provide an update on this trial in April. ![]()
Image courtesy of NHLBI
The US Food and Drug Administration (FDA) has granted orphan designation to BMN 270, an investigational gene therapy, for the treatment of patients with hemophilia A.
BMN 270 is an adeno-associated virus-factor VIII (FVIII) vector designed to restore FVIII plasma concentrations in these patients.
The FDA grants orphan designation to drugs that are intended to treat diseases or conditions affecting fewer than 200,000 patients in the US.
The designation provides the drug’s sponsor with various development incentives, including opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, and 7 years of US market exclusivity if the drug is approved.
BMN 270 is under development by BioMarin Pharmaceutical Inc.
BioMarin is conducting a phase 1/2 study to evaluate the safety and efficacy of BMN 270 in up to 12 patients with severe hemophilia A.
Researchers are assessing the safety of a single infusion of BMN 270 and the change in FVIII expression level from baseline to 16 weeks after infusion.
The group is also assessing the impact of BMN 270 on the frequency of FVIII replacement therapy, the number of bleeding episodes requiring treatment, and any potential immune responses.
Patients will be monitored for safety and durability of effect for 5 years. BioMarin plans to provide an update on this trial in April. ![]()
Image courtesy of NHLBI
The US Food and Drug Administration (FDA) has granted orphan designation to BMN 270, an investigational gene therapy, for the treatment of patients with hemophilia A.
BMN 270 is an adeno-associated virus-factor VIII (FVIII) vector designed to restore FVIII plasma concentrations in these patients.
The FDA grants orphan designation to drugs that are intended to treat diseases or conditions affecting fewer than 200,000 patients in the US.
The designation provides the drug’s sponsor with various development incentives, including opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, and 7 years of US market exclusivity if the drug is approved.
BMN 270 is under development by BioMarin Pharmaceutical Inc.
BioMarin is conducting a phase 1/2 study to evaluate the safety and efficacy of BMN 270 in up to 12 patients with severe hemophilia A.
Researchers are assessing the safety of a single infusion of BMN 270 and the change in FVIII expression level from baseline to 16 weeks after infusion.
The group is also assessing the impact of BMN 270 on the frequency of FVIII replacement therapy, the number of bleeding episodes requiring treatment, and any potential immune responses.
Patients will be monitored for safety and durability of effect for 5 years. BioMarin plans to provide an update on this trial in April. ![]()
Bundle can decrease CLABSI incidence
Staphylococcus infection
Photo by Bill Branson
A central catheter maintenance bundle can decrease the incidence of central line-associated bloodstream infections (CLABSIs), according to a study published in the American Journal of Critical Care.
A team from the healthcare company Select Medical developed and implemented the bundle at 30 long-term acute care hospitals.
The team used infection prevention guidelines from the Centers for Disease Control and Prevention as the core of the bundle, with mandatory use of alcohol-based central catheter caps and chlorhexidine gluconate dressings.
Ongoing education of clinical staff about the protocol and a checklist to track compliance were also key elements of the initiative.
At each hospital, staff nurses who demonstrated competency in the care of central catheters monitored implementation of the bundle for the initial 6 months of the study.
Researchers reviewed the medical records of 6660 patients discharged during the 14 months prior to the study and 6559 patients discharged after implementation of the bundle. Patient days and central catheter days before and after the bundle was implemented were comparable.
Six months after the bundle was implemented, the CLABSI standardized infection rate had dropped 29%. The rate was 1.28 in the 6 months before the bundle was implemented and 0.96 six months after implementation.
There was a mean reduction of 4.5 CLABSIs per hospital for 14 months after the bundle was implemented.
“Our results encourage the development and implementation of similar bundles as effective infection reduction strategies in [long-term acute care hospitals],” said study author Antony Grigonis, PhD, vice-president of quality and healthcare analytics at Select Medical.
“Preventing these infections can help reduce complications and the length of stay for other patients. This infection reduction could also translate to a savings of approximately $3.7 million annually for the 30 long-term acute care hospitals studied.”
Select Medical, which is based in Mechanicsburg, Pennsylvania, owns long-term acute care and inpatient rehabilitation hospitals, as well as occupational health and physical therapy clinics. ![]()
Staphylococcus infection
Photo by Bill Branson
A central catheter maintenance bundle can decrease the incidence of central line-associated bloodstream infections (CLABSIs), according to a study published in the American Journal of Critical Care.
A team from the healthcare company Select Medical developed and implemented the bundle at 30 long-term acute care hospitals.
The team used infection prevention guidelines from the Centers for Disease Control and Prevention as the core of the bundle, with mandatory use of alcohol-based central catheter caps and chlorhexidine gluconate dressings.
Ongoing education of clinical staff about the protocol and a checklist to track compliance were also key elements of the initiative.
At each hospital, staff nurses who demonstrated competency in the care of central catheters monitored implementation of the bundle for the initial 6 months of the study.
Researchers reviewed the medical records of 6660 patients discharged during the 14 months prior to the study and 6559 patients discharged after implementation of the bundle. Patient days and central catheter days before and after the bundle was implemented were comparable.
Six months after the bundle was implemented, the CLABSI standardized infection rate had dropped 29%. The rate was 1.28 in the 6 months before the bundle was implemented and 0.96 six months after implementation.
There was a mean reduction of 4.5 CLABSIs per hospital for 14 months after the bundle was implemented.
“Our results encourage the development and implementation of similar bundles as effective infection reduction strategies in [long-term acute care hospitals],” said study author Antony Grigonis, PhD, vice-president of quality and healthcare analytics at Select Medical.
“Preventing these infections can help reduce complications and the length of stay for other patients. This infection reduction could also translate to a savings of approximately $3.7 million annually for the 30 long-term acute care hospitals studied.”
Select Medical, which is based in Mechanicsburg, Pennsylvania, owns long-term acute care and inpatient rehabilitation hospitals, as well as occupational health and physical therapy clinics. ![]()
Staphylococcus infection
Photo by Bill Branson
A central catheter maintenance bundle can decrease the incidence of central line-associated bloodstream infections (CLABSIs), according to a study published in the American Journal of Critical Care.
A team from the healthcare company Select Medical developed and implemented the bundle at 30 long-term acute care hospitals.
The team used infection prevention guidelines from the Centers for Disease Control and Prevention as the core of the bundle, with mandatory use of alcohol-based central catheter caps and chlorhexidine gluconate dressings.
Ongoing education of clinical staff about the protocol and a checklist to track compliance were also key elements of the initiative.
At each hospital, staff nurses who demonstrated competency in the care of central catheters monitored implementation of the bundle for the initial 6 months of the study.
Researchers reviewed the medical records of 6660 patients discharged during the 14 months prior to the study and 6559 patients discharged after implementation of the bundle. Patient days and central catheter days before and after the bundle was implemented were comparable.
Six months after the bundle was implemented, the CLABSI standardized infection rate had dropped 29%. The rate was 1.28 in the 6 months before the bundle was implemented and 0.96 six months after implementation.
There was a mean reduction of 4.5 CLABSIs per hospital for 14 months after the bundle was implemented.
“Our results encourage the development and implementation of similar bundles as effective infection reduction strategies in [long-term acute care hospitals],” said study author Antony Grigonis, PhD, vice-president of quality and healthcare analytics at Select Medical.
“Preventing these infections can help reduce complications and the length of stay for other patients. This infection reduction could also translate to a savings of approximately $3.7 million annually for the 30 long-term acute care hospitals studied.”
Select Medical, which is based in Mechanicsburg, Pennsylvania, owns long-term acute care and inpatient rehabilitation hospitals, as well as occupational health and physical therapy clinics. ![]()
A Perfect Storm: Patterns of care
Editor’s Note: This is the third installment of a five-part monthly series that will discuss the pathologic, genomic, and health system factors that contribute to the racial survival disparity in breast cancer. The series, which is adapted from an article that originally appeared in CA: A Cancer Journal for Clinicians1, a journal of the American Cancer Society, will also review exciting and innovative interventions to close the survival gap. This month’s column reviews patterns of care – the second element in the perfect storm.
Mammography
Despite advances in breast cancer imaging technology, the mainstay of breast cancer screening has remained mammography. Chu et al.2 found that African American women have less early-stage disease in every age group for each hormone receptor status, and this raises the concern that mammography screening might be inadequate in this population. Although historically, African American women used mammography less than did white women, this difference has fortunately disappeared with time.3 According to results from the 2010 National Health Interview Survey, among women who were 40 years or older, 50.6% of non-Hispanic African Americans and 51.5% of non-Hispanic whites reported having had a mammogram within the past year.4
Although mammography uptake may be similar between these groups, there are still differences both in quality and in follow-up of abnormal imaging results. A study of mammography capacity and quality in a large urban setting found that the facilities that served predominantly minority women were more likely to be public institutions (31% vs. 0%) and less likely to be academic (27% vs. 71%), less likely to have digital mammography (18% vs. 71%), and less likely to have dedicated breast imaging specialists reading the films (23% vs. 87%). The authors concluded that the mammography process was broken, with quality differences in the manner in which the centers provided care and reported results.5

The accompanying graphic illustrates the disparities seen in breast cancer mammography and care for women in underserved communities on Chicago’s South Side. As the figure demonstrates, there are fewer mammography centers on the city’s South Side, with the concentration of breast cancer imaging and treatment resources localized in the more affluent communities of central and northern Chicago. A total of 300,000 women who were eligible for screening went unscreened because of improper management of resources.
Highlighting the importance of location in breast cancer care, Gehlert et al.6 asserted that ensuring that inner-city health facilities have up-to-date, well-maintained equipment and that mammographers have access to continuing training and opportunities for consultation should help reduce breast cancer mortality in African Americans.
With respect to follow-up of abnormal imaging results, a large retrospective cohort study of 6,722 women with abnormal mammogram results seen at a New York academic medical center from January 2002 through December 2002 found longer times to diagnostic follow-up for African American versus white women. The median number of days to diagnostic follow-up was 20 for African American patients versus 14 for white patients. In addition, racial disparities remained significant after the researchers controlled for age, Breast Imaging Reporting and Data System (BI-RADS) category, insurance status, provider practice location, and median household income. More important, in women with a BI-RADS classification of 4 or 5 – signifying a lesion seen on mammography that is either suspicious for or highly suggestive of malignancy, respectively – the median number of days to follow-up among those without same-day additional imaging was 26 for African Americans and 14 for whites (P < .05).7
Delays in treatment
A cascade of delays also has been documented in breast cancer care for African American women. Silber et al.8 investigated factors associated with differences in breast cancer outcomes in a large population-based study using Surveillance, Epidemiology, and End Results (SEER)-Medicare data. The mean time from diagnosis to treatment was 29.2 days for African Americans versus 22.5 days for whites (P < .001). The authors also found that African Americans were more likely to have very-long treatment delays. At least 6% of African Americans did not initiate treatment within the first 3 months of diagnosis, whereas only 3% of whites failed to start treatment (P < .001). Gwyn et al.9 also found potentially clinically significant treatment delays more often for African American women than for white women. The time from medical consultation to the initiation of treatment was longer than 3 months for 22.4% of African American women versus 14.3% of white women. Three months was chosen as a clinically significant time period, because Richards et al.10 demonstrated that a delay ≥ to 3 months affects survival. Thus, delays in the diagnosis and treatment of African American women are factors that worsen the survival gap.
Misuse of treatment
Once treatment is initiated, African Americans often receive inappropriate therapy, studies have demonstrated. In a prospective analysis of 957 patients in 101 oncology practices, Griggs et al.11 found more frequent use of non–guideline concordant adjuvant chemotherapy regimens in African American women. In a univariate analysis, African American patients were more likely than were whites to receive a nonstandard regimen (19% vs. 11%; P = .047). Although we will discuss further in this column whether guidelines based on clinical trials are appropriate for African American patients, the study demonstrates that these women are not uniformly receiving standard-of-care treatment.
Underuse of treatment
In addition to misuse of treatment, studies also have examined undertreatment of African American patients with breast cancer. One study investigated chemotherapy administration among African American patients with stage I-III breast cancer at 10 different treatment sites. Compared with white patients, African Americans received a lower dose proportion (actual vs. expected dose) and lower relative dose intensity.

The authors found that between-group differences in biological and medical characteristics, such as tolerance of therapy, comorbidities, and leukocyte counts, did not explain these variations in treatment. In fact, despite the association between lower leukocyte counts and African American ethnicity, there was no evidence that white blood cell levels accounted for the difference in dose proportion or relative dose intensity. Significantly, the authors discovered that more African Americans had chemotherapy dose reductions in the first cycle of treatment, perhaps indicating physician assumptions regarding African American patients’ ability to tolerate chemotherapy.12
Silber et al.8 also examined differences in the administration of chemotherapy between white and African American breast cancer patients. The authors found that 3.7% of African Americans received both an anthracycline and a taxane; that figure rose to 5.0% among whites who were matched to African Americans at presentation.
Bickell et al.13 explored further racial disparity in the underuse of adjuvant breast cancer treatment. The researchers examined the medical records of 677 women treated surgically for stage I or II breast cancer. The study defined underuse as omissions of radiotherapy after breast-conserving surgery, adjuvant chemotherapy after resection of hormone receptor–negative tumors ≥ 1 cm, or hormonal therapy for receptor-positive tumors ≥ 1 cm. Underuse of appropriate adjuvant treatment was found in 34% of African American patients versus 16% of white patients (P less than .001). There were racial disparities present in all three adjuvant therapies assessed.
Hormonal therapy has been shown effective in clinical trials for preventing breast cancer recurrence and death in women with early-stage breast cancer.14 The study by Bickell et al.13 documented underuse of this treatment in African American patients. Partridge et al.15 conducted the largest study of oral antineoplastic use outside of a clinical trial setting. Their study consisted of 2,378 primary breast cancer patients enrolled in New Jersey’s Medicaid or pharmaceutical assistance program; the main outcome was the number of days covered by filled tamoxifen prescriptions in the first year of therapy. The study found that nonwhite patients had significantly lower adherence rates than did whites. Although further investigation is needed to determine the drivers of this nonadherence in African American patients, medication cost has been proposed as a significant factor leading to underuse of these agents. Streeter et al.16 analyzed a nationally representative pharmacy claims database for oral antineoplastics and calculated abandonment rates for the initial claim. Not surprisingly, high cost sharing and low incomes were associated with a higher abandonment rate (P < .05). Despite being an important component of health equity research, treatment adherence has been identified by the Association of American Medical Colleges as a critically underrepresented area of disparities-focused health services research.17 More attention to this area is needed to understand the underuse of hormonal therapies in African American breast cancer patients.
The treatment strategies that have been shown to be delayed, underused, or misused in African American patients in the aforementioned studies have improved disease-free and overall survival in large randomized trials. Furthermore, diminished total dose and dose intensity of adjuvant chemotherapy both have been associated with lower breast cancer survival rates.18,19 These quality-of-care failures in breast cancer treatment for minority patients are thought to partially explain the survival disparity between African Americans and whites. It has been proposed that patients in both groups derive a similar benefit from systemic therapy when it is administered in accordance with their clinical and pathologic presentation,20 but that assumption becomes more nuanced when the clinical trial experience is reviewed.
Clinical trial experience
Dignam20 examined survival by race in several National Surgical Adjuvant Breast and Bowel Project trials. He found that the benefit from systemic adjuvant therapy for reductions in disease recurrence and mortality was comparable between African American and white patients. His survey of trials consistently indicated equivalent disease-free survival, but a mortality deficit for African Americans also was found consistently. Among African Americans, the excess risk of mortality was 21% for those who were lymph node–negative and 17% for those who were lymph node–positive. The excess mortality risk was thought to be attributable to greater mortality from noncancer causes among African American patients rather than a failure of African Americans to respond to breast cancer treatment.
In contrast to Dignam’s findings20, Hershman et al.21 assessed the association between race and treatment discontinuation/delay, white blood cell counts, and survival in women enrolled in the Southwest Oncology Group adjuvant breast cancer trials. The study found that African American women were significantly more likely to experience treatment discontinuation/delay than were white women (87% vs. 81%, respectively; P = .04). These delays were not accounted for by toxicities, which were experienced in similar proportions by race. African American women also were more likely to miss appointments (19% vs. 9%; P = .0002); perhaps, as Hassett and Griggs22 speculated, this finding speaks to economic barriers, including the inability to arrange alternate child care, miss work, or afford transportation to the clinic. Despite these barriers to care for African American patients, they still received the same mean relative dose intensity (87% vs. 86%).
In their survival analysis, Hershman et al.21 controlled for treatment-related factors such as dose reductions and delays, body surface area, baseline white blood cell counts, and other predictors of survival and still found that African Americans had worse disease-free and overall survival than did white women. The authors concluded that the study was “unable to demonstrate that any factor related to treatment quality or delivery contributed to racial differences in survival between the groups.”21 The study thus established two important findings related to the disparity gap. First, even in the controlled setting of a clinical trial, African American patients faced barriers to optimal treatment,22 and second, despite attempts to control for treatment quality and delivery, African American women still had worse outcomes. These findings suggest that tumor biology and genomics remain important.
In next month’s installment, we will discuss interventions aimed at closing the racial survival disparity in breast cancer. Eliminating racial disparities in cancer mortality through effective interventions has become an increasingly important imperative in federal, state, and community health care programs.
Other installments of this column can be found in the Related Content box.
1. Daly B, Olopade OI. A perfect storm: How tumor biology, genomics, and health care delivery patterns collide to create a racial survival disparity in breast cancer and proposed interventions for change. CA Cancer J Clin. 2015 May-Jun;65(3):221-38.
2. Chu KC, Lamar CA, Freeman HP. Racial disparities in breast carcinoma survival rates: Separating factors that affect diagnosis from factors that affect treatment. Cancer. 2003 Jun;97(11):2853-60.
3. DeLancey JO, Thun MJ, Jemal A, Ward EM. Recent trends in black-white disparities in cancer mortality. Cancer Epidemiol Biomarkers Prev. 2008 Nov;17(11):2908-12.
4. DeSantis C, Naishadham D, Jemal A. Cancer statistics for African Americans, 2013. CA Cancer J Clin. 2013 Nov;63(3):151-66.
5. Ansell D, Grabler P, Whitman S, et al. A community effort to reduce the black/white breast cancer mortality disparity in Chicago. Cancer Causes Control. 2009 Nov;20(9):1681-8.
6. Gehlert S, Sohmer D, Sacks T, Mininger C, McClintock M, Olopade O. Targeting health disparities: a model linking upstream determinants to downstream interventions. Health Aff (Millwood). 2008 Mar-Apr;27(2):339-49.
7. Press R, Carrasquillo O, Sciacca RR, Giardina EG. Racial/ethnic disparities in time to follow-up after an abnormal mammogram. J Womens Health (Larchmt). 2008 Jul;17(6):923-30.
8. Silber JH, Rosenbaum PR, Clark AS, et al. Characteristics associated with differences in survival among black and white women with breast cancer. JAMA. 2013 Jul;310(4):389-397.
9. Gwyn K, Bondy ML, Cohen DS, et al. Racial differences in diagnosis, treatment, and clinical delays in a population-based study of patients with newly diagnosed breast carcinoma. Cancer. 2004 Apr;100(8):1595-604.
10. Richards MA, Westcombe AM, Love SB, Littlejohns P, Ramirez AJ. Influence of delay on survival in patients with breast cancer: a systematic review. Lancet. 1999 Apr 3;353(9159):1119-26.
11. Griggs JJ, Culakova E, Sorbero ME, et al. Social and racial differences in selection of breast cancer adjuvant chemotherapy regimens. J Clin Oncol. 2007 Jun 20;25(18):2522-7.
12. Griggs JJ, Sorbero ME, Stark AT, Heininger SE, Dick AW. Racial disparity in the dose and dose intensity of breast cancer adjuvant chemotherapy. Breast Cancer Res Treat. 2003 Sep;81(1):21-31.
13. Bickell NA, Wang JJ, Oluwole S, et al. Missed opportunities: racial disparities in adjuvant breast cancer treatment. J Clin Oncol. 2006 Mar 20;24(9):1357-62. 14. Fisher B, Costantino J, Redmond C, et al. A randomized clinical trial evaluating tamoxifen in the treatment of patients with node-negative breast cancer who have estrogen-receptor-positive tumors. N Engl J Med. 1989 Feb 23;320(8):479-84.
15. Partridge AH, Wang PS, Winer EP, Avorn J. Nonadherence to adjuvant tamoxifen therapy in women with primary breast cancer. J Clin Oncol. 2003 Feb 15;21(4):602-6.
16. Streeter SB, Schwartzberg L, Husain N, Johnsrud M. Patient and plan characteristics affecting abandonment of oral oncolytic prescriptions. J Oncol Pract. 2011 Jul;7(3 Suppl):46s-51s.
17. Alberti PM KN, Sutton K, Johnson BH, Holve E. The state of health equity research: closing knowledge gaps to address inequities. ©2014 Association of American Medical Colleges. May not be reproduced or distributed without prior permission.
18. Wood WC, Budman DR, Korzun AH, et al. Dose and dose intensity of adjuvant chemotherapy for stage II, node-positive breast carcinoma. N Engl J Med. 1994 May 5;330(18):1253-9.
19. Budman DR, Berry DA, Cirrincione CT, et al. Dose and dose intensity as determinants of outcome in the adjuvant treatment of breast cancer. The Cancer and Leukemia Group B. J Natl Cancer Inst. 1998 Aug 19;90(16):1205-11.
20. Dignam JJ. Efficacy of systemic adjuvant therapy for breast cancer in African-American and Caucasian women. J Natl Cancer Inst Monogr. 2001(30):36-43.
21. Hershman DL, Unger JM, Barlow WE, et al. Treatment quality and outcomes of African American versus white breast cancer patients: retrospective analysis of Southwest Oncology studies S8814/S8897. J Clin Oncol. 2009 May;27(13):2157-62.
22. Hassett MJ, Griggs JJ. Disparities in breast cancer adjuvant chemotherapy: moving beyond yes or no. J Clin Oncol. 2009 May 1;27(13):2120-1.

Bobby Daly, MD, MBA, is the chief fellow in the section of hematology/oncology at the University of Chicago Medicine. His clinical focus is breast and thoracic oncology, and his research focus is health services. Specifically, Dr. Daly researches disparities in oncology care delivery, oncology health care utilization, aggressive end-of-life oncology care, and oncology payment models. He received his MD and MBA from Harvard Medical School and Harvard Business School, both in Boston, and a BA in Economics and History from Stanford (Calif.) University. He was the recipient of the Dean’s Award at Harvard Medical and Business Schools.

Olufunmilayo Olopade, MD, FACP, OON, is the Walter L. Palmer Distinguished Service Professor of Medicine and Human Genetics, and director, Center for Global Health at the University of Chicago. She is adopting emerging high throughput genomic and informatics strategies to identify genetic and nongenetic risk factors for breast cancer in order to implement precision health care in diverse populations. This innovative approach has the potential to improve the quality of care and reduce costs while saving more lives.
Disclosures: Dr. Olopade serves on the Medical Advisory Board for CancerIQ. Dr. Daly serves as a director of Quadrant Holdings Corporation and receives compensation from this entity. Frontline Medical Communications is a subsidiary of Quadrant Holdings Corporation.
Published in conjunction with Susan G. Komen®.
Editor’s Note: This is the third installment of a five-part monthly series that will discuss the pathologic, genomic, and health system factors that contribute to the racial survival disparity in breast cancer. The series, which is adapted from an article that originally appeared in CA: A Cancer Journal for Clinicians1, a journal of the American Cancer Society, will also review exciting and innovative interventions to close the survival gap. This month’s column reviews patterns of care – the second element in the perfect storm.
Mammography
Despite advances in breast cancer imaging technology, the mainstay of breast cancer screening has remained mammography. Chu et al.2 found that African American women have less early-stage disease in every age group for each hormone receptor status, and this raises the concern that mammography screening might be inadequate in this population. Although historically, African American women used mammography less than did white women, this difference has fortunately disappeared with time.3 According to results from the 2010 National Health Interview Survey, among women who were 40 years or older, 50.6% of non-Hispanic African Americans and 51.5% of non-Hispanic whites reported having had a mammogram within the past year.4
Although mammography uptake may be similar between these groups, there are still differences both in quality and in follow-up of abnormal imaging results. A study of mammography capacity and quality in a large urban setting found that the facilities that served predominantly minority women were more likely to be public institutions (31% vs. 0%) and less likely to be academic (27% vs. 71%), less likely to have digital mammography (18% vs. 71%), and less likely to have dedicated breast imaging specialists reading the films (23% vs. 87%). The authors concluded that the mammography process was broken, with quality differences in the manner in which the centers provided care and reported results.5
The accompanying graphic illustrates the disparities seen in breast cancer mammography and care for women in underserved communities on Chicago’s South Side. As the figure demonstrates, there are fewer mammography centers on the city’s South Side, with the concentration of breast cancer imaging and treatment resources localized in the more affluent communities of central and northern Chicago. A total of 300,000 women who were eligible for screening went unscreened because of improper management of resources.
Highlighting the importance of location in breast cancer care, Gehlert et al.6 asserted that ensuring that inner-city health facilities have up-to-date, well-maintained equipment and that mammographers have access to continuing training and opportunities for consultation should help reduce breast cancer mortality in African Americans.
With respect to follow-up of abnormal imaging results, a large retrospective cohort study of 6,722 women with abnormal mammogram results seen at a New York academic medical center from January 2002 through December 2002 found longer times to diagnostic follow-up for African American versus white women. The median number of days to diagnostic follow-up was 20 for African American patients versus 14 for white patients. In addition, racial disparities remained significant after the researchers controlled for age, Breast Imaging Reporting and Data System (BI-RADS) category, insurance status, provider practice location, and median household income. More important, in women with a BI-RADS classification of 4 or 5 – signifying a lesion seen on mammography that is either suspicious for or highly suggestive of malignancy, respectively – the median number of days to follow-up among those without same-day additional imaging was 26 for African Americans and 14 for whites (P < .05).7
Delays in treatment
A cascade of delays also has been documented in breast cancer care for African American women. Silber et al.8 investigated factors associated with differences in breast cancer outcomes in a large population-based study using Surveillance, Epidemiology, and End Results (SEER)-Medicare data. The mean time from diagnosis to treatment was 29.2 days for African Americans versus 22.5 days for whites (P < .001). The authors also found that African Americans were more likely to have very-long treatment delays. At least 6% of African Americans did not initiate treatment within the first 3 months of diagnosis, whereas only 3% of whites failed to start treatment (P < .001). Gwyn et al.9 also found potentially clinically significant treatment delays more often for African American women than for white women. The time from medical consultation to the initiation of treatment was longer than 3 months for 22.4% of African American women versus 14.3% of white women. Three months was chosen as a clinically significant time period, because Richards et al.10 demonstrated that a delay ≥ to 3 months affects survival. Thus, delays in the diagnosis and treatment of African American women are factors that worsen the survival gap.
Misuse of treatment
Once treatment is initiated, African Americans often receive inappropriate therapy, studies have demonstrated. In a prospective analysis of 957 patients in 101 oncology practices, Griggs et al.11 found more frequent use of non–guideline concordant adjuvant chemotherapy regimens in African American women. In a univariate analysis, African American patients were more likely than were whites to receive a nonstandard regimen (19% vs. 11%; P = .047). Although we will discuss further in this column whether guidelines based on clinical trials are appropriate for African American patients, the study demonstrates that these women are not uniformly receiving standard-of-care treatment.
Underuse of treatment
In addition to misuse of treatment, studies also have examined undertreatment of African American patients with breast cancer. One study investigated chemotherapy administration among African American patients with stage I-III breast cancer at 10 different treatment sites. Compared with white patients, African Americans received a lower dose proportion (actual vs. expected dose) and lower relative dose intensity.
The authors found that between-group differences in biological and medical characteristics, such as tolerance of therapy, comorbidities, and leukocyte counts, did not explain these variations in treatment. In fact, despite the association between lower leukocyte counts and African American ethnicity, there was no evidence that white blood cell levels accounted for the difference in dose proportion or relative dose intensity. Significantly, the authors discovered that more African Americans had chemotherapy dose reductions in the first cycle of treatment, perhaps indicating physician assumptions regarding African American patients’ ability to tolerate chemotherapy.12
Silber et al.8 also examined differences in the administration of chemotherapy between white and African American breast cancer patients. The authors found that 3.7% of African Americans received both an anthracycline and a taxane; that figure rose to 5.0% among whites who were matched to African Americans at presentation.
Bickell et al.13 explored further racial disparity in the underuse of adjuvant breast cancer treatment. The researchers examined the medical records of 677 women treated surgically for stage I or II breast cancer. The study defined underuse as omissions of radiotherapy after breast-conserving surgery, adjuvant chemotherapy after resection of hormone receptor–negative tumors ≥ 1 cm, or hormonal therapy for receptor-positive tumors ≥ 1 cm. Underuse of appropriate adjuvant treatment was found in 34% of African American patients versus 16% of white patients (P less than .001). There were racial disparities present in all three adjuvant therapies assessed.
Hormonal therapy has been shown effective in clinical trials for preventing breast cancer recurrence and death in women with early-stage breast cancer.14 The study by Bickell et al.13 documented underuse of this treatment in African American patients. Partridge et al.15 conducted the largest study of oral antineoplastic use outside of a clinical trial setting. Their study consisted of 2,378 primary breast cancer patients enrolled in New Jersey’s Medicaid or pharmaceutical assistance program; the main outcome was the number of days covered by filled tamoxifen prescriptions in the first year of therapy. The study found that nonwhite patients had significantly lower adherence rates than did whites. Although further investigation is needed to determine the drivers of this nonadherence in African American patients, medication cost has been proposed as a significant factor leading to underuse of these agents. Streeter et al.16 analyzed a nationally representative pharmacy claims database for oral antineoplastics and calculated abandonment rates for the initial claim. Not surprisingly, high cost sharing and low incomes were associated with a higher abandonment rate (P < .05). Despite being an important component of health equity research, treatment adherence has been identified by the Association of American Medical Colleges as a critically underrepresented area of disparities-focused health services research.17 More attention to this area is needed to understand the underuse of hormonal therapies in African American breast cancer patients.
The treatment strategies that have been shown to be delayed, underused, or misused in African American patients in the aforementioned studies have improved disease-free and overall survival in large randomized trials. Furthermore, diminished total dose and dose intensity of adjuvant chemotherapy both have been associated with lower breast cancer survival rates.18,19 These quality-of-care failures in breast cancer treatment for minority patients are thought to partially explain the survival disparity between African Americans and whites. It has been proposed that patients in both groups derive a similar benefit from systemic therapy when it is administered in accordance with their clinical and pathologic presentation,20 but that assumption becomes more nuanced when the clinical trial experience is reviewed.
Clinical trial experience
Dignam20 examined survival by race in several National Surgical Adjuvant Breast and Bowel Project trials. He found that the benefit from systemic adjuvant therapy for reductions in disease recurrence and mortality was comparable between African American and white patients. His survey of trials consistently indicated equivalent disease-free survival, but a mortality deficit for African Americans also was found consistently. Among African Americans, the excess risk of mortality was 21% for those who were lymph node–negative and 17% for those who were lymph node–positive. The excess mortality risk was thought to be attributable to greater mortality from noncancer causes among African American patients rather than a failure of African Americans to respond to breast cancer treatment.
In contrast to Dignam’s findings20, Hershman et al.21 assessed the association between race and treatment discontinuation/delay, white blood cell counts, and survival in women enrolled in the Southwest Oncology Group adjuvant breast cancer trials. The study found that African American women were significantly more likely to experience treatment discontinuation/delay than were white women (87% vs. 81%, respectively; P = .04). These delays were not accounted for by toxicities, which were experienced in similar proportions by race. African American women also were more likely to miss appointments (19% vs. 9%; P = .0002); perhaps, as Hassett and Griggs22 speculated, this finding speaks to economic barriers, including the inability to arrange alternate child care, miss work, or afford transportation to the clinic. Despite these barriers to care for African American patients, they still received the same mean relative dose intensity (87% vs. 86%).
In their survival analysis, Hershman et al.21 controlled for treatment-related factors such as dose reductions and delays, body surface area, baseline white blood cell counts, and other predictors of survival and still found that African Americans had worse disease-free and overall survival than did white women. The authors concluded that the study was “unable to demonstrate that any factor related to treatment quality or delivery contributed to racial differences in survival between the groups.”21 The study thus established two important findings related to the disparity gap. First, even in the controlled setting of a clinical trial, African American patients faced barriers to optimal treatment,22 and second, despite attempts to control for treatment quality and delivery, African American women still had worse outcomes. These findings suggest that tumor biology and genomics remain important.
In next month’s installment, we will discuss interventions aimed at closing the racial survival disparity in breast cancer. Eliminating racial disparities in cancer mortality through effective interventions has become an increasingly important imperative in federal, state, and community health care programs.
Other installments of this column can be found in the Related Content box.
1. Daly B, Olopade OI. A perfect storm: How tumor biology, genomics, and health care delivery patterns collide to create a racial survival disparity in breast cancer and proposed interventions for change. CA Cancer J Clin. 2015 May-Jun;65(3):221-38.
2. Chu KC, Lamar CA, Freeman HP. Racial disparities in breast carcinoma survival rates: Separating factors that affect diagnosis from factors that affect treatment. Cancer. 2003 Jun;97(11):2853-60.
3. DeLancey JO, Thun MJ, Jemal A, Ward EM. Recent trends in black-white disparities in cancer mortality. Cancer Epidemiol Biomarkers Prev. 2008 Nov;17(11):2908-12.
4. DeSantis C, Naishadham D, Jemal A. Cancer statistics for African Americans, 2013. CA Cancer J Clin. 2013 Nov;63(3):151-66.
5. Ansell D, Grabler P, Whitman S, et al. A community effort to reduce the black/white breast cancer mortality disparity in Chicago. Cancer Causes Control. 2009 Nov;20(9):1681-8.
6. Gehlert S, Sohmer D, Sacks T, Mininger C, McClintock M, Olopade O. Targeting health disparities: a model linking upstream determinants to downstream interventions. Health Aff (Millwood). 2008 Mar-Apr;27(2):339-49.
7. Press R, Carrasquillo O, Sciacca RR, Giardina EG. Racial/ethnic disparities in time to follow-up after an abnormal mammogram. J Womens Health (Larchmt). 2008 Jul;17(6):923-30.
8. Silber JH, Rosenbaum PR, Clark AS, et al. Characteristics associated with differences in survival among black and white women with breast cancer. JAMA. 2013 Jul;310(4):389-397.
9. Gwyn K, Bondy ML, Cohen DS, et al. Racial differences in diagnosis, treatment, and clinical delays in a population-based study of patients with newly diagnosed breast carcinoma. Cancer. 2004 Apr;100(8):1595-604.
10. Richards MA, Westcombe AM, Love SB, Littlejohns P, Ramirez AJ. Influence of delay on survival in patients with breast cancer: a systematic review. Lancet. 1999 Apr 3;353(9159):1119-26.
11. Griggs JJ, Culakova E, Sorbero ME, et al. Social and racial differences in selection of breast cancer adjuvant chemotherapy regimens. J Clin Oncol. 2007 Jun 20;25(18):2522-7.
12. Griggs JJ, Sorbero ME, Stark AT, Heininger SE, Dick AW. Racial disparity in the dose and dose intensity of breast cancer adjuvant chemotherapy. Breast Cancer Res Treat. 2003 Sep;81(1):21-31.
13. Bickell NA, Wang JJ, Oluwole S, et al. Missed opportunities: racial disparities in adjuvant breast cancer treatment. J Clin Oncol. 2006 Mar 20;24(9):1357-62. 14. Fisher B, Costantino J, Redmond C, et al. A randomized clinical trial evaluating tamoxifen in the treatment of patients with node-negative breast cancer who have estrogen-receptor-positive tumors. N Engl J Med. 1989 Feb 23;320(8):479-84.
15. Partridge AH, Wang PS, Winer EP, Avorn J. Nonadherence to adjuvant tamoxifen therapy in women with primary breast cancer. J Clin Oncol. 2003 Feb 15;21(4):602-6.
16. Streeter SB, Schwartzberg L, Husain N, Johnsrud M. Patient and plan characteristics affecting abandonment of oral oncolytic prescriptions. J Oncol Pract. 2011 Jul;7(3 Suppl):46s-51s.
17. Alberti PM KN, Sutton K, Johnson BH, Holve E. The state of health equity research: closing knowledge gaps to address inequities. ©2014 Association of American Medical Colleges. May not be reproduced or distributed without prior permission.
18. Wood WC, Budman DR, Korzun AH, et al. Dose and dose intensity of adjuvant chemotherapy for stage II, node-positive breast carcinoma. N Engl J Med. 1994 May 5;330(18):1253-9.
19. Budman DR, Berry DA, Cirrincione CT, et al. Dose and dose intensity as determinants of outcome in the adjuvant treatment of breast cancer. The Cancer and Leukemia Group B. J Natl Cancer Inst. 1998 Aug 19;90(16):1205-11.
20. Dignam JJ. Efficacy of systemic adjuvant therapy for breast cancer in African-American and Caucasian women. J Natl Cancer Inst Monogr. 2001(30):36-43.
21. Hershman DL, Unger JM, Barlow WE, et al. Treatment quality and outcomes of African American versus white breast cancer patients: retrospective analysis of Southwest Oncology studies S8814/S8897. J Clin Oncol. 2009 May;27(13):2157-62.
22. Hassett MJ, Griggs JJ. Disparities in breast cancer adjuvant chemotherapy: moving beyond yes or no. J Clin Oncol. 2009 May 1;27(13):2120-1.
Bobby Daly, MD, MBA, is the chief fellow in the section of hematology/oncology at the University of Chicago Medicine. His clinical focus is breast and thoracic oncology, and his research focus is health services. Specifically, Dr. Daly researches disparities in oncology care delivery, oncology health care utilization, aggressive end-of-life oncology care, and oncology payment models. He received his MD and MBA from Harvard Medical School and Harvard Business School, both in Boston, and a BA in Economics and History from Stanford (Calif.) University. He was the recipient of the Dean’s Award at Harvard Medical and Business Schools.
Olufunmilayo Olopade, MD, FACP, OON, is the Walter L. Palmer Distinguished Service Professor of Medicine and Human Genetics, and director, Center for Global Health at the University of Chicago. She is adopting emerging high throughput genomic and informatics strategies to identify genetic and nongenetic risk factors for breast cancer in order to implement precision health care in diverse populations. This innovative approach has the potential to improve the quality of care and reduce costs while saving more lives.
Disclosures: Dr. Olopade serves on the Medical Advisory Board for CancerIQ. Dr. Daly serves as a director of Quadrant Holdings Corporation and receives compensation from this entity. Frontline Medical Communications is a subsidiary of Quadrant Holdings Corporation.
Published in conjunction with Susan G. Komen®.
Editor’s Note: This is the third installment of a five-part monthly series that will discuss the pathologic, genomic, and health system factors that contribute to the racial survival disparity in breast cancer. The series, which is adapted from an article that originally appeared in CA: A Cancer Journal for Clinicians1, a journal of the American Cancer Society, will also review exciting and innovative interventions to close the survival gap. This month’s column reviews patterns of care – the second element in the perfect storm.
Mammography
Despite advances in breast cancer imaging technology, the mainstay of breast cancer screening has remained mammography. Chu et al.2 found that African American women have less early-stage disease in every age group for each hormone receptor status, and this raises the concern that mammography screening might be inadequate in this population. Although historically, African American women used mammography less than did white women, this difference has fortunately disappeared with time.3 According to results from the 2010 National Health Interview Survey, among women who were 40 years or older, 50.6% of non-Hispanic African Americans and 51.5% of non-Hispanic whites reported having had a mammogram within the past year.4
Although mammography uptake may be similar between these groups, there are still differences both in quality and in follow-up of abnormal imaging results. A study of mammography capacity and quality in a large urban setting found that the facilities that served predominantly minority women were more likely to be public institutions (31% vs. 0%) and less likely to be academic (27% vs. 71%), less likely to have digital mammography (18% vs. 71%), and less likely to have dedicated breast imaging specialists reading the films (23% vs. 87%). The authors concluded that the mammography process was broken, with quality differences in the manner in which the centers provided care and reported results.5
The accompanying graphic illustrates the disparities seen in breast cancer mammography and care for women in underserved communities on Chicago’s South Side. As the figure demonstrates, there are fewer mammography centers on the city’s South Side, with the concentration of breast cancer imaging and treatment resources localized in the more affluent communities of central and northern Chicago. A total of 300,000 women who were eligible for screening went unscreened because of improper management of resources.
Highlighting the importance of location in breast cancer care, Gehlert et al.6 asserted that ensuring that inner-city health facilities have up-to-date, well-maintained equipment and that mammographers have access to continuing training and opportunities for consultation should help reduce breast cancer mortality in African Americans.
With respect to follow-up of abnormal imaging results, a large retrospective cohort study of 6,722 women with abnormal mammogram results seen at a New York academic medical center from January 2002 through December 2002 found longer times to diagnostic follow-up for African American versus white women. The median number of days to diagnostic follow-up was 20 for African American patients versus 14 for white patients. In addition, racial disparities remained significant after the researchers controlled for age, Breast Imaging Reporting and Data System (BI-RADS) category, insurance status, provider practice location, and median household income. More important, in women with a BI-RADS classification of 4 or 5 – signifying a lesion seen on mammography that is either suspicious for or highly suggestive of malignancy, respectively – the median number of days to follow-up among those without same-day additional imaging was 26 for African Americans and 14 for whites (P < .05).7
Delays in treatment
A cascade of delays also has been documented in breast cancer care for African American women. Silber et al.8 investigated factors associated with differences in breast cancer outcomes in a large population-based study using Surveillance, Epidemiology, and End Results (SEER)-Medicare data. The mean time from diagnosis to treatment was 29.2 days for African Americans versus 22.5 days for whites (P < .001). The authors also found that African Americans were more likely to have very-long treatment delays. At least 6% of African Americans did not initiate treatment within the first 3 months of diagnosis, whereas only 3% of whites failed to start treatment (P < .001). Gwyn et al.9 also found potentially clinically significant treatment delays more often for African American women than for white women. The time from medical consultation to the initiation of treatment was longer than 3 months for 22.4% of African American women versus 14.3% of white women. Three months was chosen as a clinically significant time period, because Richards et al.10 demonstrated that a delay ≥ to 3 months affects survival. Thus, delays in the diagnosis and treatment of African American women are factors that worsen the survival gap.
Misuse of treatment
Once treatment is initiated, African Americans often receive inappropriate therapy, studies have demonstrated. In a prospective analysis of 957 patients in 101 oncology practices, Griggs et al.11 found more frequent use of non–guideline concordant adjuvant chemotherapy regimens in African American women. In a univariate analysis, African American patients were more likely than were whites to receive a nonstandard regimen (19% vs. 11%; P = .047). Although we will discuss further in this column whether guidelines based on clinical trials are appropriate for African American patients, the study demonstrates that these women are not uniformly receiving standard-of-care treatment.
Underuse of treatment
In addition to misuse of treatment, studies also have examined undertreatment of African American patients with breast cancer. One study investigated chemotherapy administration among African American patients with stage I-III breast cancer at 10 different treatment sites. Compared with white patients, African Americans received a lower dose proportion (actual vs. expected dose) and lower relative dose intensity.
The authors found that between-group differences in biological and medical characteristics, such as tolerance of therapy, comorbidities, and leukocyte counts, did not explain these variations in treatment. In fact, despite the association between lower leukocyte counts and African American ethnicity, there was no evidence that white blood cell levels accounted for the difference in dose proportion or relative dose intensity. Significantly, the authors discovered that more African Americans had chemotherapy dose reductions in the first cycle of treatment, perhaps indicating physician assumptions regarding African American patients’ ability to tolerate chemotherapy.12
Silber et al.8 also examined differences in the administration of chemotherapy between white and African American breast cancer patients. The authors found that 3.7% of African Americans received both an anthracycline and a taxane; that figure rose to 5.0% among whites who were matched to African Americans at presentation.
Bickell et al.13 explored further racial disparity in the underuse of adjuvant breast cancer treatment. The researchers examined the medical records of 677 women treated surgically for stage I or II breast cancer. The study defined underuse as omissions of radiotherapy after breast-conserving surgery, adjuvant chemotherapy after resection of hormone receptor–negative tumors ≥ 1 cm, or hormonal therapy for receptor-positive tumors ≥ 1 cm. Underuse of appropriate adjuvant treatment was found in 34% of African American patients versus 16% of white patients (P less than .001). There were racial disparities present in all three adjuvant therapies assessed.
Hormonal therapy has been shown effective in clinical trials for preventing breast cancer recurrence and death in women with early-stage breast cancer.14 The study by Bickell et al.13 documented underuse of this treatment in African American patients. Partridge et al.15 conducted the largest study of oral antineoplastic use outside of a clinical trial setting. Their study consisted of 2,378 primary breast cancer patients enrolled in New Jersey’s Medicaid or pharmaceutical assistance program; the main outcome was the number of days covered by filled tamoxifen prescriptions in the first year of therapy. The study found that nonwhite patients had significantly lower adherence rates than did whites. Although further investigation is needed to determine the drivers of this nonadherence in African American patients, medication cost has been proposed as a significant factor leading to underuse of these agents. Streeter et al.16 analyzed a nationally representative pharmacy claims database for oral antineoplastics and calculated abandonment rates for the initial claim. Not surprisingly, high cost sharing and low incomes were associated with a higher abandonment rate (P < .05). Despite being an important component of health equity research, treatment adherence has been identified by the Association of American Medical Colleges as a critically underrepresented area of disparities-focused health services research.17 More attention to this area is needed to understand the underuse of hormonal therapies in African American breast cancer patients.
The treatment strategies that have been shown to be delayed, underused, or misused in African American patients in the aforementioned studies have improved disease-free and overall survival in large randomized trials. Furthermore, diminished total dose and dose intensity of adjuvant chemotherapy both have been associated with lower breast cancer survival rates.18,19 These quality-of-care failures in breast cancer treatment for minority patients are thought to partially explain the survival disparity between African Americans and whites. It has been proposed that patients in both groups derive a similar benefit from systemic therapy when it is administered in accordance with their clinical and pathologic presentation,20 but that assumption becomes more nuanced when the clinical trial experience is reviewed.
Clinical trial experience
Dignam20 examined survival by race in several National Surgical Adjuvant Breast and Bowel Project trials. He found that the benefit from systemic adjuvant therapy for reductions in disease recurrence and mortality was comparable between African American and white patients. His survey of trials consistently indicated equivalent disease-free survival, but a mortality deficit for African Americans also was found consistently. Among African Americans, the excess risk of mortality was 21% for those who were lymph node–negative and 17% for those who were lymph node–positive. The excess mortality risk was thought to be attributable to greater mortality from noncancer causes among African American patients rather than a failure of African Americans to respond to breast cancer treatment.
In contrast to Dignam’s findings20, Hershman et al.21 assessed the association between race and treatment discontinuation/delay, white blood cell counts, and survival in women enrolled in the Southwest Oncology Group adjuvant breast cancer trials. The study found that African American women were significantly more likely to experience treatment discontinuation/delay than were white women (87% vs. 81%, respectively; P = .04). These delays were not accounted for by toxicities, which were experienced in similar proportions by race. African American women also were more likely to miss appointments (19% vs. 9%; P = .0002); perhaps, as Hassett and Griggs22 speculated, this finding speaks to economic barriers, including the inability to arrange alternate child care, miss work, or afford transportation to the clinic. Despite these barriers to care for African American patients, they still received the same mean relative dose intensity (87% vs. 86%).
In their survival analysis, Hershman et al.21 controlled for treatment-related factors such as dose reductions and delays, body surface area, baseline white blood cell counts, and other predictors of survival and still found that African Americans had worse disease-free and overall survival than did white women. The authors concluded that the study was “unable to demonstrate that any factor related to treatment quality or delivery contributed to racial differences in survival between the groups.”21 The study thus established two important findings related to the disparity gap. First, even in the controlled setting of a clinical trial, African American patients faced barriers to optimal treatment,22 and second, despite attempts to control for treatment quality and delivery, African American women still had worse outcomes. These findings suggest that tumor biology and genomics remain important.
In next month’s installment, we will discuss interventions aimed at closing the racial survival disparity in breast cancer. Eliminating racial disparities in cancer mortality through effective interventions has become an increasingly important imperative in federal, state, and community health care programs.
Other installments of this column can be found in the Related Content box.
1. Daly B, Olopade OI. A perfect storm: How tumor biology, genomics, and health care delivery patterns collide to create a racial survival disparity in breast cancer and proposed interventions for change. CA Cancer J Clin. 2015 May-Jun;65(3):221-38.
2. Chu KC, Lamar CA, Freeman HP. Racial disparities in breast carcinoma survival rates: Separating factors that affect diagnosis from factors that affect treatment. Cancer. 2003 Jun;97(11):2853-60.
3. DeLancey JO, Thun MJ, Jemal A, Ward EM. Recent trends in black-white disparities in cancer mortality. Cancer Epidemiol Biomarkers Prev. 2008 Nov;17(11):2908-12.
4. DeSantis C, Naishadham D, Jemal A. Cancer statistics for African Americans, 2013. CA Cancer J Clin. 2013 Nov;63(3):151-66.
5. Ansell D, Grabler P, Whitman S, et al. A community effort to reduce the black/white breast cancer mortality disparity in Chicago. Cancer Causes Control. 2009 Nov;20(9):1681-8.
6. Gehlert S, Sohmer D, Sacks T, Mininger C, McClintock M, Olopade O. Targeting health disparities: a model linking upstream determinants to downstream interventions. Health Aff (Millwood). 2008 Mar-Apr;27(2):339-49.
7. Press R, Carrasquillo O, Sciacca RR, Giardina EG. Racial/ethnic disparities in time to follow-up after an abnormal mammogram. J Womens Health (Larchmt). 2008 Jul;17(6):923-30.
8. Silber JH, Rosenbaum PR, Clark AS, et al. Characteristics associated with differences in survival among black and white women with breast cancer. JAMA. 2013 Jul;310(4):389-397.
9. Gwyn K, Bondy ML, Cohen DS, et al. Racial differences in diagnosis, treatment, and clinical delays in a population-based study of patients with newly diagnosed breast carcinoma. Cancer. 2004 Apr;100(8):1595-604.
10. Richards MA, Westcombe AM, Love SB, Littlejohns P, Ramirez AJ. Influence of delay on survival in patients with breast cancer: a systematic review. Lancet. 1999 Apr 3;353(9159):1119-26.
11. Griggs JJ, Culakova E, Sorbero ME, et al. Social and racial differences in selection of breast cancer adjuvant chemotherapy regimens. J Clin Oncol. 2007 Jun 20;25(18):2522-7.
12. Griggs JJ, Sorbero ME, Stark AT, Heininger SE, Dick AW. Racial disparity in the dose and dose intensity of breast cancer adjuvant chemotherapy. Breast Cancer Res Treat. 2003 Sep;81(1):21-31.
13. Bickell NA, Wang JJ, Oluwole S, et al. Missed opportunities: racial disparities in adjuvant breast cancer treatment. J Clin Oncol. 2006 Mar 20;24(9):1357-62. 14. Fisher B, Costantino J, Redmond C, et al. A randomized clinical trial evaluating tamoxifen in the treatment of patients with node-negative breast cancer who have estrogen-receptor-positive tumors. N Engl J Med. 1989 Feb 23;320(8):479-84.
15. Partridge AH, Wang PS, Winer EP, Avorn J. Nonadherence to adjuvant tamoxifen therapy in women with primary breast cancer. J Clin Oncol. 2003 Feb 15;21(4):602-6.
16. Streeter SB, Schwartzberg L, Husain N, Johnsrud M. Patient and plan characteristics affecting abandonment of oral oncolytic prescriptions. J Oncol Pract. 2011 Jul;7(3 Suppl):46s-51s.
17. Alberti PM KN, Sutton K, Johnson BH, Holve E. The state of health equity research: closing knowledge gaps to address inequities. ©2014 Association of American Medical Colleges. May not be reproduced or distributed without prior permission.
18. Wood WC, Budman DR, Korzun AH, et al. Dose and dose intensity of adjuvant chemotherapy for stage II, node-positive breast carcinoma. N Engl J Med. 1994 May 5;330(18):1253-9.
19. Budman DR, Berry DA, Cirrincione CT, et al. Dose and dose intensity as determinants of outcome in the adjuvant treatment of breast cancer. The Cancer and Leukemia Group B. J Natl Cancer Inst. 1998 Aug 19;90(16):1205-11.
20. Dignam JJ. Efficacy of systemic adjuvant therapy for breast cancer in African-American and Caucasian women. J Natl Cancer Inst Monogr. 2001(30):36-43.
21. Hershman DL, Unger JM, Barlow WE, et al. Treatment quality and outcomes of African American versus white breast cancer patients: retrospective analysis of Southwest Oncology studies S8814/S8897. J Clin Oncol. 2009 May;27(13):2157-62.
22. Hassett MJ, Griggs JJ. Disparities in breast cancer adjuvant chemotherapy: moving beyond yes or no. J Clin Oncol. 2009 May 1;27(13):2120-1.
Bobby Daly, MD, MBA, is the chief fellow in the section of hematology/oncology at the University of Chicago Medicine. His clinical focus is breast and thoracic oncology, and his research focus is health services. Specifically, Dr. Daly researches disparities in oncology care delivery, oncology health care utilization, aggressive end-of-life oncology care, and oncology payment models. He received his MD and MBA from Harvard Medical School and Harvard Business School, both in Boston, and a BA in Economics and History from Stanford (Calif.) University. He was the recipient of the Dean’s Award at Harvard Medical and Business Schools.
Olufunmilayo Olopade, MD, FACP, OON, is the Walter L. Palmer Distinguished Service Professor of Medicine and Human Genetics, and director, Center for Global Health at the University of Chicago. She is adopting emerging high throughput genomic and informatics strategies to identify genetic and nongenetic risk factors for breast cancer in order to implement precision health care in diverse populations. This innovative approach has the potential to improve the quality of care and reduce costs while saving more lives.
Disclosures: Dr. Olopade serves on the Medical Advisory Board for CancerIQ. Dr. Daly serves as a director of Quadrant Holdings Corporation and receives compensation from this entity. Frontline Medical Communications is a subsidiary of Quadrant Holdings Corporation.
Published in conjunction with Susan G. Komen®.
An Eruption While on Total Parenteral Nutrition
The Diagnosis: Acquired Acrodermatitis Enteropathica
Acquired acrodermatitis enteropathica (AAE) is a rare disorder caused by severe zinc deficiency. Although acrodermatitis enteropathica is an autosomal-recessive disorder that typically manifests in infancy, AAE also can result from poor zinc intake, impaired absorption, or accelerated losses. There are reports of AAE in patients with zinc-deficient diets,1 eating disorders,2 bariatric and other gastrointestinal surgeries,3 malabsorptive diseases,4 and nephrotic syndrome.5
Zinc plays an important role in DNA and RNA synthesis, reactive oxygen species attenuation, and energy metabolism, allowing for proper wound healing, skin differentiation, and proliferation.6 Zinc is found in most foods, but animal protein contains higher concentrations (Table).7 Approximately 85% of zinc is stored in muscles and bones, with only a small amount of accessible zinc available in the liver. Liver stores can be depleted as quickly as 1 week.8 Total parenteral nutrition without trace element supplementation can quickly predispose patients to AAE.
|
|
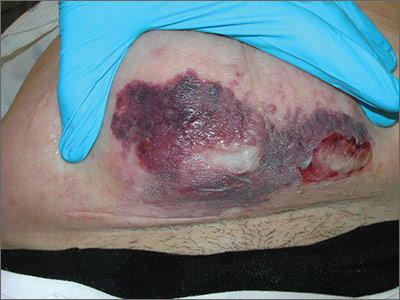
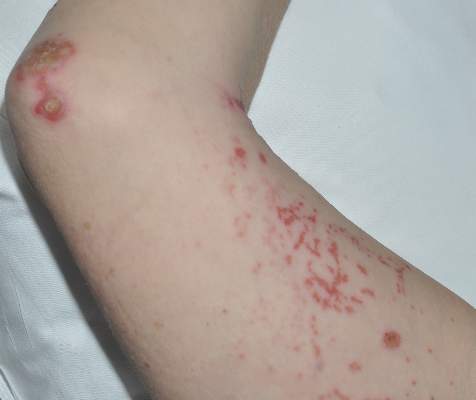
Diagnosis of this condition requires triangulation of clinical presentation, histopathology examination, and laboratory findings. Acrodermatitis enteropathica typically is characterized by dermatitis, diarrhea, and epidermal appendage findings. In its early stages, the dermatitis often manifests with angular cheilitis and paronychia.9 Patients then develop erythema, erosions, and occasionally vesicles or psoriasiform plaques in periorificial, perineal, and acral sites (Figure 1). Epidermal appendage effects include generalized alopecia and thinning nails with white transverse ridges. Although dermatologic and gastrointestinal manifestations are the most obvious, severe AAE may cause other symptoms, including mental slowing, hypogonadism, and impaired immune function.9
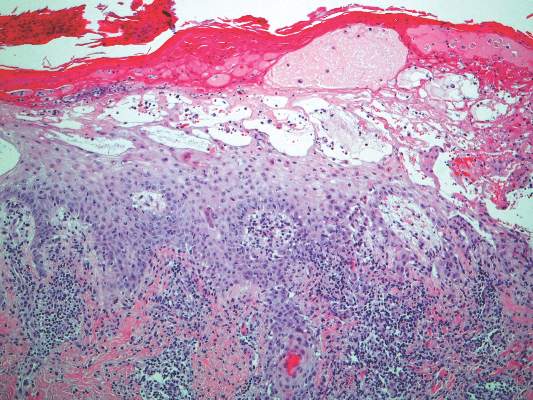
Histopathology of AAE skin lesions is similar to other nutritional deficiencies. Early changes are more specific to deficiency dermatitis and include cytoplasmic pallor and ballooning degeneration of keratinocytes in the stratum spinosum and granulosum.9 Necrolysis results in confluent keratinocyte necrosis developing into subcorneal bulla. Later in the disease course, the presentation becomes psoriasiform with keratinocyte dyskeratosis and confluent parakeratosis10 (Figure 2). Dermal edema with dilated tortuous vessels and a neutrophilic infiltrate may be present throughout disease progression.
Common laboratory abnormalities used to confirm zinc deficiency are decreased plasma zinc and alkaline phosphatase levels. Plasma zinc levels should be drawn after fasting because zinc levels decrease after food intake.9 Concurrent albumin levels should be drawn to correct for low levels caused by hypoalbuminemia. Acquired acrodermatitis enteropathica has been seen in patients with only mildly decreased plasma zinc levels or even zinc levels within reference range.11 Alkaline phosphatase metalloenzyme synthesis requires zinc and a decreased level suggests zinc deficiency even with a plasma zinc level within reference range. Alkaline phosphatase levels usually can be ascertained in a matter of hours, while the zinc levels take much longer to result.

Acquired acrodermatitis enteropathica is treated with oral elemental zinc supplementation at 1 to 2 mg/kg daily.12 Diarrhea typically resolves within 24 hours, but skin lesions heal in 1 to 2 weeks or longer. Although there is no consensus on when to discontinue zinc replacement therapy, therapy generally is not lifelong. Once the patient is zinc replete and the inciting factor has resolved, patients can discontinue supplementation without risk for recurrence.
Trace elements had not been added to our patient’s total parenteral nutrition prior to admission. Basic nutrition laboratory results and zinc levels returned markedly low: 14 μg/dL (reference range, 60–120 μg/dL). Alkaline phosphatase, a zinc-dependent protein, also was low at 12 U/L (reference range, 40–150 U/L). We added trace elements and vitamins and began empiric zinc replacement with 440 mg oral zinc sulfate daily (100 mg elemental zinc). Cephalexin was prescribed for impetiginized skin lesions. The patient noted skin improvement after 3 days on zinc replacement therapy.
- Saritha M, Gupta D, Chandrashekar L, et al. Acquired zinc deficiency in an adult female. Indian J Dermatol. 2012;57:492-494.
- Kim ST, Kang JS, Baek JW, et al. Acrodermatitis enteropathica with anorexia nervosa. J Dermatol. 2010;37:726-729.
- Bae-Harboe YS, Solky A, Masterpol KS. A case of acquired zinc deficiency. Dermatol Online J. 2012;18:1.
- Krasovec M, Frenk E. Acrodermatitis enteropathica secondary to Crohn’s disease. Dermatol Basel Switz. 1996;193:361-363.
- Reichel M, Mauro TM, Ziboh VA, et al. Acrodermatitis enteropathica in a patient with the acquired immunodeficiency syndrome. Arch Dermatol. 1992;128:415-417.
- Perafan-Riveros C, Franca LFS, Alves ACF, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- National Nutrient Database for Standard Reference, Release 28. United States Department of Agriculture, Agricultural Research Service website. http://ndb.nal.usda.gov/ndb/nutrients/report/nutrientsfrm?max=25&offset=0&totCount=0&nutrient1=309&nutrient2=&nutrient3=&subset=0&fg=&sort=f&measureby=m. Accessed December 14, 2015.
- McPherson RA, Pincus MR. Henry’s Clinical Diagnosis and Management by Laboratory Methods. 22nd ed. Philadelphia, PA: Saunders Elsevier; 2011.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Macdonald JB, Connolly SM, DiCaudo DJ. Think zinc deficiency: acquired acrodermatitis enteropathica due to poor diet and common medications. Arch Dermatol. 2012;148:961-963.
- Kumar P, Lal NR, Mondal A, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
The Diagnosis: Acquired Acrodermatitis Enteropathica
Acquired acrodermatitis enteropathica (AAE) is a rare disorder caused by severe zinc deficiency. Although acrodermatitis enteropathica is an autosomal-recessive disorder that typically manifests in infancy, AAE also can result from poor zinc intake, impaired absorption, or accelerated losses. There are reports of AAE in patients with zinc-deficient diets,1 eating disorders,2 bariatric and other gastrointestinal surgeries,3 malabsorptive diseases,4 and nephrotic syndrome.5
Zinc plays an important role in DNA and RNA synthesis, reactive oxygen species attenuation, and energy metabolism, allowing for proper wound healing, skin differentiation, and proliferation.6 Zinc is found in most foods, but animal protein contains higher concentrations (Table).7 Approximately 85% of zinc is stored in muscles and bones, with only a small amount of accessible zinc available in the liver. Liver stores can be depleted as quickly as 1 week.8 Total parenteral nutrition without trace element supplementation can quickly predispose patients to AAE.
|
|
|
|
Diagnosis of this condition requires triangulation of clinical presentation, histopathology examination, and laboratory findings. Acrodermatitis enteropathica typically is characterized by dermatitis, diarrhea, and epidermal appendage findings. In its early stages, the dermatitis often manifests with angular cheilitis and paronychia.9 Patients then develop erythema, erosions, and occasionally vesicles or psoriasiform plaques in periorificial, perineal, and acral sites (Figure 1). Epidermal appendage effects include generalized alopecia and thinning nails with white transverse ridges. Although dermatologic and gastrointestinal manifestations are the most obvious, severe AAE may cause other symptoms, including mental slowing, hypogonadism, and impaired immune function.9
Histopathology of AAE skin lesions is similar to other nutritional deficiencies. Early changes are more specific to deficiency dermatitis and include cytoplasmic pallor and ballooning degeneration of keratinocytes in the stratum spinosum and granulosum.9 Necrolysis results in confluent keratinocyte necrosis developing into subcorneal bulla. Later in the disease course, the presentation becomes psoriasiform with keratinocyte dyskeratosis and confluent parakeratosis10 (Figure 2). Dermal edema with dilated tortuous vessels and a neutrophilic infiltrate may be present throughout disease progression.
Common laboratory abnormalities used to confirm zinc deficiency are decreased plasma zinc and alkaline phosphatase levels. Plasma zinc levels should be drawn after fasting because zinc levels decrease after food intake.9 Concurrent albumin levels should be drawn to correct for low levels caused by hypoalbuminemia. Acquired acrodermatitis enteropathica has been seen in patients with only mildly decreased plasma zinc levels or even zinc levels within reference range.11 Alkaline phosphatase metalloenzyme synthesis requires zinc and a decreased level suggests zinc deficiency even with a plasma zinc level within reference range. Alkaline phosphatase levels usually can be ascertained in a matter of hours, while the zinc levels take much longer to result.
Acquired acrodermatitis enteropathica is treated with oral elemental zinc supplementation at 1 to 2 mg/kg daily.12 Diarrhea typically resolves within 24 hours, but skin lesions heal in 1 to 2 weeks or longer. Although there is no consensus on when to discontinue zinc replacement therapy, therapy generally is not lifelong. Once the patient is zinc replete and the inciting factor has resolved, patients can discontinue supplementation without risk for recurrence.
Trace elements had not been added to our patient’s total parenteral nutrition prior to admission. Basic nutrition laboratory results and zinc levels returned markedly low: 14 μg/dL (reference range, 60–120 μg/dL). Alkaline phosphatase, a zinc-dependent protein, also was low at 12 U/L (reference range, 40–150 U/L). We added trace elements and vitamins and began empiric zinc replacement with 440 mg oral zinc sulfate daily (100 mg elemental zinc). Cephalexin was prescribed for impetiginized skin lesions. The patient noted skin improvement after 3 days on zinc replacement therapy.
The Diagnosis: Acquired Acrodermatitis Enteropathica
Acquired acrodermatitis enteropathica (AAE) is a rare disorder caused by severe zinc deficiency. Although acrodermatitis enteropathica is an autosomal-recessive disorder that typically manifests in infancy, AAE also can result from poor zinc intake, impaired absorption, or accelerated losses. There are reports of AAE in patients with zinc-deficient diets,1 eating disorders,2 bariatric and other gastrointestinal surgeries,3 malabsorptive diseases,4 and nephrotic syndrome.5
Zinc plays an important role in DNA and RNA synthesis, reactive oxygen species attenuation, and energy metabolism, allowing for proper wound healing, skin differentiation, and proliferation.6 Zinc is found in most foods, but animal protein contains higher concentrations (Table).7 Approximately 85% of zinc is stored in muscles and bones, with only a small amount of accessible zinc available in the liver. Liver stores can be depleted as quickly as 1 week.8 Total parenteral nutrition without trace element supplementation can quickly predispose patients to AAE.
|
|
|
|
Diagnosis of this condition requires triangulation of clinical presentation, histopathology examination, and laboratory findings. Acrodermatitis enteropathica typically is characterized by dermatitis, diarrhea, and epidermal appendage findings. In its early stages, the dermatitis often manifests with angular cheilitis and paronychia.9 Patients then develop erythema, erosions, and occasionally vesicles or psoriasiform plaques in periorificial, perineal, and acral sites (Figure 1). Epidermal appendage effects include generalized alopecia and thinning nails with white transverse ridges. Although dermatologic and gastrointestinal manifestations are the most obvious, severe AAE may cause other symptoms, including mental slowing, hypogonadism, and impaired immune function.9
Histopathology of AAE skin lesions is similar to other nutritional deficiencies. Early changes are more specific to deficiency dermatitis and include cytoplasmic pallor and ballooning degeneration of keratinocytes in the stratum spinosum and granulosum.9 Necrolysis results in confluent keratinocyte necrosis developing into subcorneal bulla. Later in the disease course, the presentation becomes psoriasiform with keratinocyte dyskeratosis and confluent parakeratosis10 (Figure 2). Dermal edema with dilated tortuous vessels and a neutrophilic infiltrate may be present throughout disease progression.
Common laboratory abnormalities used to confirm zinc deficiency are decreased plasma zinc and alkaline phosphatase levels. Plasma zinc levels should be drawn after fasting because zinc levels decrease after food intake.9 Concurrent albumin levels should be drawn to correct for low levels caused by hypoalbuminemia. Acquired acrodermatitis enteropathica has been seen in patients with only mildly decreased plasma zinc levels or even zinc levels within reference range.11 Alkaline phosphatase metalloenzyme synthesis requires zinc and a decreased level suggests zinc deficiency even with a plasma zinc level within reference range. Alkaline phosphatase levels usually can be ascertained in a matter of hours, while the zinc levels take much longer to result.
Acquired acrodermatitis enteropathica is treated with oral elemental zinc supplementation at 1 to 2 mg/kg daily.12 Diarrhea typically resolves within 24 hours, but skin lesions heal in 1 to 2 weeks or longer. Although there is no consensus on when to discontinue zinc replacement therapy, therapy generally is not lifelong. Once the patient is zinc replete and the inciting factor has resolved, patients can discontinue supplementation without risk for recurrence.
Trace elements had not been added to our patient’s total parenteral nutrition prior to admission. Basic nutrition laboratory results and zinc levels returned markedly low: 14 μg/dL (reference range, 60–120 μg/dL). Alkaline phosphatase, a zinc-dependent protein, also was low at 12 U/L (reference range, 40–150 U/L). We added trace elements and vitamins and began empiric zinc replacement with 440 mg oral zinc sulfate daily (100 mg elemental zinc). Cephalexin was prescribed for impetiginized skin lesions. The patient noted skin improvement after 3 days on zinc replacement therapy.
- Saritha M, Gupta D, Chandrashekar L, et al. Acquired zinc deficiency in an adult female. Indian J Dermatol. 2012;57:492-494.
- Kim ST, Kang JS, Baek JW, et al. Acrodermatitis enteropathica with anorexia nervosa. J Dermatol. 2010;37:726-729.
- Bae-Harboe YS, Solky A, Masterpol KS. A case of acquired zinc deficiency. Dermatol Online J. 2012;18:1.
- Krasovec M, Frenk E. Acrodermatitis enteropathica secondary to Crohn’s disease. Dermatol Basel Switz. 1996;193:361-363.
- Reichel M, Mauro TM, Ziboh VA, et al. Acrodermatitis enteropathica in a patient with the acquired immunodeficiency syndrome. Arch Dermatol. 1992;128:415-417.
- Perafan-Riveros C, Franca LFS, Alves ACF, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- National Nutrient Database for Standard Reference, Release 28. United States Department of Agriculture, Agricultural Research Service website. http://ndb.nal.usda.gov/ndb/nutrients/report/nutrientsfrm?max=25&offset=0&totCount=0&nutrient1=309&nutrient2=&nutrient3=&subset=0&fg=&sort=f&measureby=m. Accessed December 14, 2015.
- McPherson RA, Pincus MR. Henry’s Clinical Diagnosis and Management by Laboratory Methods. 22nd ed. Philadelphia, PA: Saunders Elsevier; 2011.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Macdonald JB, Connolly SM, DiCaudo DJ. Think zinc deficiency: acquired acrodermatitis enteropathica due to poor diet and common medications. Arch Dermatol. 2012;148:961-963.
- Kumar P, Lal NR, Mondal A, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Saritha M, Gupta D, Chandrashekar L, et al. Acquired zinc deficiency in an adult female. Indian J Dermatol. 2012;57:492-494.
- Kim ST, Kang JS, Baek JW, et al. Acrodermatitis enteropathica with anorexia nervosa. J Dermatol. 2010;37:726-729.
- Bae-Harboe YS, Solky A, Masterpol KS. A case of acquired zinc deficiency. Dermatol Online J. 2012;18:1.
- Krasovec M, Frenk E. Acrodermatitis enteropathica secondary to Crohn’s disease. Dermatol Basel Switz. 1996;193:361-363.
- Reichel M, Mauro TM, Ziboh VA, et al. Acrodermatitis enteropathica in a patient with the acquired immunodeficiency syndrome. Arch Dermatol. 1992;128:415-417.
- Perafan-Riveros C, Franca LFS, Alves ACF, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- National Nutrient Database for Standard Reference, Release 28. United States Department of Agriculture, Agricultural Research Service website. http://ndb.nal.usda.gov/ndb/nutrients/report/nutrientsfrm?max=25&offset=0&totCount=0&nutrient1=309&nutrient2=&nutrient3=&subset=0&fg=&sort=f&measureby=m. Accessed December 14, 2015.
- McPherson RA, Pincus MR. Henry’s Clinical Diagnosis and Management by Laboratory Methods. 22nd ed. Philadelphia, PA: Saunders Elsevier; 2011.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Macdonald JB, Connolly SM, DiCaudo DJ. Think zinc deficiency: acquired acrodermatitis enteropathica due to poor diet and common medications. Arch Dermatol. 2012;148:961-963.
- Kumar P, Lal NR, Mondal A, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
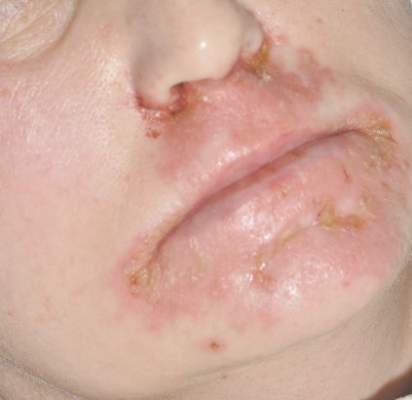
A 47-year-old woman with a history of bulimia and gastroparesis who had been on total parenteral nutrition for 8 weeks presented with a painful, perioral, perineal, and acral eruption of 7 weeks’ duration. Additionally, she had experienced diarrhea, vomiting, and a 13.5-kg weight loss in the last 4 months. Physical examination revealed perioral and perineal, well-demarcated, erythematous, scaly plaques with yellow crusting. She had edematous crusted erosions on the bilateral palms and soles and psoriasiform plaques along the right arm and flank. Punch biopsies (4 mm) from the right inguinal fold and right elbow were obtained.
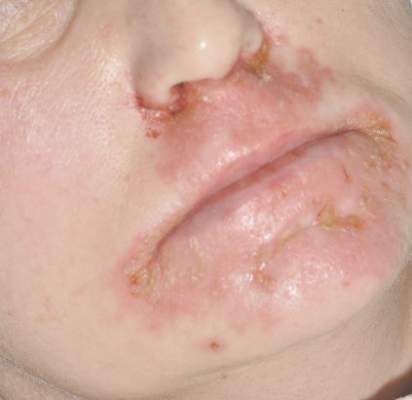
High coffee consumption linked to decreased risk for MS
High coffee consumption decreased the odds of developing multiple sclerosis (MS) in two separate case-control studies conducted by Dr. Anna K. Hedström of the Karolinska Institute, Stockholm, and her associates.
The investigators analyzed coffee consumption across certain ages among 1,620 cases and 2,788 controls in the Swedish Epidemiological Investigation of Multiple Sclerosis (EIMS) and 1,159 cases and 1,172 controls in the Kaiser Permanente Medical Care Plan, Northern California Region (KPNC).

In EIMS, the adjusted odds ratio (OR) was 0.70 (95% confidence interval, 0.49-0.99; P = .04) among participants who drank six or more cups of coffee (greater than 900 mL) daily at the index year, which was defined as the year of the initial appearance of symptoms indicative of MS. The corresponding OR for those who reported high coffee consumption at 5 and 10 years before the study was 0.72 and 0.71, respectively, but neither comparison reached statistical significance.
In KPNC, those who consumed four or more cups of coffee (more than 948 mL in this study) daily were significantly less likely to develop MS than were those who never drank coffee (OR, 0.69; 95% CI, 0.50-0.96; P = .05). And the cohorts who drank four or more cups of coffee daily at least 5 years prior to the study were associated with significantly reduced odds of MS (OR, 0.64).
The combined results of the two studies in a meta-analysis found a significant, 29% reduction in the likelihood of developing MS among the highest drinkers of coffee (greater than 900 mL daily in EIMS and greater than 948 mL in KPNC). The investigators adjusted all the analyses for many demographic and environmental risk factors for MS, including age, gender, residential area, ancestry, smoking habits, exposure to passive smoking, sun exposure habits, and body mass index at age 20 years.
No evidence was found for associations between increased amounts of tea or soda intake and MS.
“Further studies are required to establish if it is in fact caffeine, or if there is another molecule in coffee underlying the findings, to longitudinally assess the association between consumption of coffee and disease activity in MS, and to evaluate the mechanisms by which coffee may be acting, which could thus lead to new therapeutic targets,” the researchers concluded.
Find the full study in the Journal of Neurology, Neurosurgery & Psychiatry (doi: 10.1136/jnnp-2015-312176).
High coffee consumption decreased the odds of developing multiple sclerosis (MS) in two separate case-control studies conducted by Dr. Anna K. Hedström of the Karolinska Institute, Stockholm, and her associates.
The investigators analyzed coffee consumption across certain ages among 1,620 cases and 2,788 controls in the Swedish Epidemiological Investigation of Multiple Sclerosis (EIMS) and 1,159 cases and 1,172 controls in the Kaiser Permanente Medical Care Plan, Northern California Region (KPNC).
In EIMS, the adjusted odds ratio (OR) was 0.70 (95% confidence interval, 0.49-0.99; P = .04) among participants who drank six or more cups of coffee (greater than 900 mL) daily at the index year, which was defined as the year of the initial appearance of symptoms indicative of MS. The corresponding OR for those who reported high coffee consumption at 5 and 10 years before the study was 0.72 and 0.71, respectively, but neither comparison reached statistical significance.
In KPNC, those who consumed four or more cups of coffee (more than 948 mL in this study) daily were significantly less likely to develop MS than were those who never drank coffee (OR, 0.69; 95% CI, 0.50-0.96; P = .05). And the cohorts who drank four or more cups of coffee daily at least 5 years prior to the study were associated with significantly reduced odds of MS (OR, 0.64).
The combined results of the two studies in a meta-analysis found a significant, 29% reduction in the likelihood of developing MS among the highest drinkers of coffee (greater than 900 mL daily in EIMS and greater than 948 mL in KPNC). The investigators adjusted all the analyses for many demographic and environmental risk factors for MS, including age, gender, residential area, ancestry, smoking habits, exposure to passive smoking, sun exposure habits, and body mass index at age 20 years.
No evidence was found for associations between increased amounts of tea or soda intake and MS.
“Further studies are required to establish if it is in fact caffeine, or if there is another molecule in coffee underlying the findings, to longitudinally assess the association between consumption of coffee and disease activity in MS, and to evaluate the mechanisms by which coffee may be acting, which could thus lead to new therapeutic targets,” the researchers concluded.
Find the full study in the Journal of Neurology, Neurosurgery & Psychiatry (doi: 10.1136/jnnp-2015-312176).
High coffee consumption decreased the odds of developing multiple sclerosis (MS) in two separate case-control studies conducted by Dr. Anna K. Hedström of the Karolinska Institute, Stockholm, and her associates.
The investigators analyzed coffee consumption across certain ages among 1,620 cases and 2,788 controls in the Swedish Epidemiological Investigation of Multiple Sclerosis (EIMS) and 1,159 cases and 1,172 controls in the Kaiser Permanente Medical Care Plan, Northern California Region (KPNC).
In EIMS, the adjusted odds ratio (OR) was 0.70 (95% confidence interval, 0.49-0.99; P = .04) among participants who drank six or more cups of coffee (greater than 900 mL) daily at the index year, which was defined as the year of the initial appearance of symptoms indicative of MS. The corresponding OR for those who reported high coffee consumption at 5 and 10 years before the study was 0.72 and 0.71, respectively, but neither comparison reached statistical significance.
In KPNC, those who consumed four or more cups of coffee (more than 948 mL in this study) daily were significantly less likely to develop MS than were those who never drank coffee (OR, 0.69; 95% CI, 0.50-0.96; P = .05). And the cohorts who drank four or more cups of coffee daily at least 5 years prior to the study were associated with significantly reduced odds of MS (OR, 0.64).
The combined results of the two studies in a meta-analysis found a significant, 29% reduction in the likelihood of developing MS among the highest drinkers of coffee (greater than 900 mL daily in EIMS and greater than 948 mL in KPNC). The investigators adjusted all the analyses for many demographic and environmental risk factors for MS, including age, gender, residential area, ancestry, smoking habits, exposure to passive smoking, sun exposure habits, and body mass index at age 20 years.
No evidence was found for associations between increased amounts of tea or soda intake and MS.
“Further studies are required to establish if it is in fact caffeine, or if there is another molecule in coffee underlying the findings, to longitudinally assess the association between consumption of coffee and disease activity in MS, and to evaluate the mechanisms by which coffee may be acting, which could thus lead to new therapeutic targets,” the researchers concluded.
Find the full study in the Journal of Neurology, Neurosurgery & Psychiatry (doi: 10.1136/jnnp-2015-312176).
FROM JOURNAL OF NEUROLOGY, NEUROSURGERY & PSYCHIATRY

Age alone shouldn’t preclude use of chemo in older adults with head and neck cancer
SCOTTSDALE, ARIZ. – Oncologists should consider not only age, but also comorbidities and disease extent when deciding whether to offer concurrent chemoradiation to older adults with locally advanced head and neck cancer, suggests a cohort study using data from the National Cancer Data Base.
The study of 4,042 patients aged 71 years or older found that adding chemotherapy to radiation therapy (RT) reduced the risk of death by at least one-fourth overall, according to results reported in a poster session and related press briefing at the Multidisciplinary Head and Neck Cancer Symposium.
In further analysis, benefit was limited to those who were aged 81 years or younger with low comorbidity and more advanced disease.

“Does age matter? The answer to that question is yes and no,” commented senior author Dr. Sana Karam of the University of Colorado at Denver, Aurora. “The physician needs to use his or her clinical judgment.”
“Don’t just look at the age of the patient,” she advised. “In this day and age where patients are healthier and living longer, give them the benefit of curative intent with the addition of chemo. Assess the patient clinically. If they are not healthy; their comorbidity score, KPS, ECOG, whatever you are using in your practice, is poor; or if they have earlier-stage disease, early T, no bulky nodes, then it’s okay to just give RT alone. But the addition of chemotherapy can improve survival dramatically” for other patients.
The new findings are likely to temper those of the pivotal MACH-NC (Meta-Analysis of Chemotherapy in Head and Neck Cancers). That analysis found little to no survival benefit from adding concurrent chemotherapy to radiation therapy in patients aged 71 years or older, but only 4% of the included patients fell into this age-group.
“So it was very underpowered, but yet, it has set our clinical practice guidelines,” Dr. Karam noted at the meeting cosponsored by the American Society for Radiation Oncology and the American Society of Clinical Oncology. “And we know from many of our clinical trials this is a patient population that’s generally heavily underrepresented on clinical trials.”
Press briefing moderator Dr. Christine Gourin of Johns Hopkins University, Baltimore, commented, “Your data are very important because we all know the MACH-NC meta-analysis is used by our colleagues in Europe to support not using chemotherapy in elderly patients,” she added. “And in fact your data suggest it’s really not age, but comorbidity” that should be considered.
Dr. Gourin and colleagues performed a similar analysis, but instead used the Surveillance, Epidemiology, and End Results (SEER) Medicare database. Their results suggested that the impact of adding concurrent chemotherapy to radiation depended on the site: Patients with oropharynx cancer benefited, but those with larynx cancer actually fared worse because of late toxicity.
“Did you find any difference when you drilled down between larynx and hypopharynx and oropharynx?” she asked.
“We found that the overall survival benefit was regardless of the subsite,” Dr. Karam replied, noting that the patient populations in the two cohorts differed somewhat. “Unfortunately, we don’t have clear-cut variables for toxicity, but what we did look at is time to completion of RT, and we found that patients who did get concurrent chemoradiation had a longer time to completion of RT, suggesting perhaps more treatment breaks maybe. … But still, despite the treatment breaks, even after controlling for that, we still found an overall survival advantage, regardless of the subsite.”
At her institution, patients are already being treated with a tailored approach, Dr. Gourin commented. “We have young patients who are so sick that they are not great candidates for chemotherapy, and then we have old patients who are healthier than I am who are great candidates for chemotherapy. So I would say that we have been doing what Dr. Karam suggests, which is looking at age not as a number, but rather comorbidity and the overall functional status of the patient.”
“That’s why I really liked your study: It’s great to see that in writing, because I know that colleagues in other countries that I talk to about health care reform and how to cut costs, they actually use the MACH-NC to define who gets treated and who doesn’t,” she added.
For their analysis, Dr. Karam and colleagues identified patients in the database given a diagnosis of locally advanced cancer of the oropharynx, larynx, or hypopharynx between 1998 and 2011 who were treated with radiation therapy.
Overall, 53% received chemotherapy concurrently, defined as starting it in the 14 days before or 14 days after initiation of the radiation therapy, according to Dr. Karam.
The specific agents given could not be ascertained, she acknowledged. “Unfortunately, the NCDB does not give us data in regard to the type of chemotherapy, and they only started collecting cetuximab data in 2013.”
With a median follow-up of 19 months, the unadjusted 5-year overall survival rate was 15.2% with radiation therapy alone and 30.3% with concurrent chemoradiation (hazard ratio, 0.59; P less than .001). Benefit fell only slightly after multivariate adjustment (HR, 0.63; P less than .001).
Findings were similar in a propensity-matched analysis, which showed an 18.1% survival with radiation therapy alone versus 26.4% with concurrent chemoradiation (HR, 0.73; P less than .001).
On recursive partitioning analysis, chemoradiation was associated with better survival among patients 81 years of age or younger who had low comorbidity based on Charlson-Deyo score and either T1-2,N2-3 disease or T3-4,N0-3 disease.
There was no survival benefit in patients older than age 81. And among patients aged 71-80, there was no benefit for those having less advanced disease (stage T1-2,N1) and low comorbidity, or having more advanced disease (T3-4,N1+ disease) and high comorbidity.
SCOTTSDALE, ARIZ. – Oncologists should consider not only age, but also comorbidities and disease extent when deciding whether to offer concurrent chemoradiation to older adults with locally advanced head and neck cancer, suggests a cohort study using data from the National Cancer Data Base.
The study of 4,042 patients aged 71 years or older found that adding chemotherapy to radiation therapy (RT) reduced the risk of death by at least one-fourth overall, according to results reported in a poster session and related press briefing at the Multidisciplinary Head and Neck Cancer Symposium.
In further analysis, benefit was limited to those who were aged 81 years or younger with low comorbidity and more advanced disease.
“Does age matter? The answer to that question is yes and no,” commented senior author Dr. Sana Karam of the University of Colorado at Denver, Aurora. “The physician needs to use his or her clinical judgment.”
“Don’t just look at the age of the patient,” she advised. “In this day and age where patients are healthier and living longer, give them the benefit of curative intent with the addition of chemo. Assess the patient clinically. If they are not healthy; their comorbidity score, KPS, ECOG, whatever you are using in your practice, is poor; or if they have earlier-stage disease, early T, no bulky nodes, then it’s okay to just give RT alone. But the addition of chemotherapy can improve survival dramatically” for other patients.
The new findings are likely to temper those of the pivotal MACH-NC (Meta-Analysis of Chemotherapy in Head and Neck Cancers). That analysis found little to no survival benefit from adding concurrent chemotherapy to radiation therapy in patients aged 71 years or older, but only 4% of the included patients fell into this age-group.
“So it was very underpowered, but yet, it has set our clinical practice guidelines,” Dr. Karam noted at the meeting cosponsored by the American Society for Radiation Oncology and the American Society of Clinical Oncology. “And we know from many of our clinical trials this is a patient population that’s generally heavily underrepresented on clinical trials.”
Press briefing moderator Dr. Christine Gourin of Johns Hopkins University, Baltimore, commented, “Your data are very important because we all know the MACH-NC meta-analysis is used by our colleagues in Europe to support not using chemotherapy in elderly patients,” she added. “And in fact your data suggest it’s really not age, but comorbidity” that should be considered.
Dr. Gourin and colleagues performed a similar analysis, but instead used the Surveillance, Epidemiology, and End Results (SEER) Medicare database. Their results suggested that the impact of adding concurrent chemotherapy to radiation depended on the site: Patients with oropharynx cancer benefited, but those with larynx cancer actually fared worse because of late toxicity.
“Did you find any difference when you drilled down between larynx and hypopharynx and oropharynx?” she asked.
“We found that the overall survival benefit was regardless of the subsite,” Dr. Karam replied, noting that the patient populations in the two cohorts differed somewhat. “Unfortunately, we don’t have clear-cut variables for toxicity, but what we did look at is time to completion of RT, and we found that patients who did get concurrent chemoradiation had a longer time to completion of RT, suggesting perhaps more treatment breaks maybe. … But still, despite the treatment breaks, even after controlling for that, we still found an overall survival advantage, regardless of the subsite.”
At her institution, patients are already being treated with a tailored approach, Dr. Gourin commented. “We have young patients who are so sick that they are not great candidates for chemotherapy, and then we have old patients who are healthier than I am who are great candidates for chemotherapy. So I would say that we have been doing what Dr. Karam suggests, which is looking at age not as a number, but rather comorbidity and the overall functional status of the patient.”
“That’s why I really liked your study: It’s great to see that in writing, because I know that colleagues in other countries that I talk to about health care reform and how to cut costs, they actually use the MACH-NC to define who gets treated and who doesn’t,” she added.
For their analysis, Dr. Karam and colleagues identified patients in the database given a diagnosis of locally advanced cancer of the oropharynx, larynx, or hypopharynx between 1998 and 2011 who were treated with radiation therapy.
Overall, 53% received chemotherapy concurrently, defined as starting it in the 14 days before or 14 days after initiation of the radiation therapy, according to Dr. Karam.
The specific agents given could not be ascertained, she acknowledged. “Unfortunately, the NCDB does not give us data in regard to the type of chemotherapy, and they only started collecting cetuximab data in 2013.”
With a median follow-up of 19 months, the unadjusted 5-year overall survival rate was 15.2% with radiation therapy alone and 30.3% with concurrent chemoradiation (hazard ratio, 0.59; P less than .001). Benefit fell only slightly after multivariate adjustment (HR, 0.63; P less than .001).
Findings were similar in a propensity-matched analysis, which showed an 18.1% survival with radiation therapy alone versus 26.4% with concurrent chemoradiation (HR, 0.73; P less than .001).
On recursive partitioning analysis, chemoradiation was associated with better survival among patients 81 years of age or younger who had low comorbidity based on Charlson-Deyo score and either T1-2,N2-3 disease or T3-4,N0-3 disease.
There was no survival benefit in patients older than age 81. And among patients aged 71-80, there was no benefit for those having less advanced disease (stage T1-2,N1) and low comorbidity, or having more advanced disease (T3-4,N1+ disease) and high comorbidity.
SCOTTSDALE, ARIZ. – Oncologists should consider not only age, but also comorbidities and disease extent when deciding whether to offer concurrent chemoradiation to older adults with locally advanced head and neck cancer, suggests a cohort study using data from the National Cancer Data Base.
The study of 4,042 patients aged 71 years or older found that adding chemotherapy to radiation therapy (RT) reduced the risk of death by at least one-fourth overall, according to results reported in a poster session and related press briefing at the Multidisciplinary Head and Neck Cancer Symposium.
In further analysis, benefit was limited to those who were aged 81 years or younger with low comorbidity and more advanced disease.
“Does age matter? The answer to that question is yes and no,” commented senior author Dr. Sana Karam of the University of Colorado at Denver, Aurora. “The physician needs to use his or her clinical judgment.”
“Don’t just look at the age of the patient,” she advised. “In this day and age where patients are healthier and living longer, give them the benefit of curative intent with the addition of chemo. Assess the patient clinically. If they are not healthy; their comorbidity score, KPS, ECOG, whatever you are using in your practice, is poor; or if they have earlier-stage disease, early T, no bulky nodes, then it’s okay to just give RT alone. But the addition of chemotherapy can improve survival dramatically” for other patients.
The new findings are likely to temper those of the pivotal MACH-NC (Meta-Analysis of Chemotherapy in Head and Neck Cancers). That analysis found little to no survival benefit from adding concurrent chemotherapy to radiation therapy in patients aged 71 years or older, but only 4% of the included patients fell into this age-group.
“So it was very underpowered, but yet, it has set our clinical practice guidelines,” Dr. Karam noted at the meeting cosponsored by the American Society for Radiation Oncology and the American Society of Clinical Oncology. “And we know from many of our clinical trials this is a patient population that’s generally heavily underrepresented on clinical trials.”
Press briefing moderator Dr. Christine Gourin of Johns Hopkins University, Baltimore, commented, “Your data are very important because we all know the MACH-NC meta-analysis is used by our colleagues in Europe to support not using chemotherapy in elderly patients,” she added. “And in fact your data suggest it’s really not age, but comorbidity” that should be considered.
Dr. Gourin and colleagues performed a similar analysis, but instead used the Surveillance, Epidemiology, and End Results (SEER) Medicare database. Their results suggested that the impact of adding concurrent chemotherapy to radiation depended on the site: Patients with oropharynx cancer benefited, but those with larynx cancer actually fared worse because of late toxicity.
“Did you find any difference when you drilled down between larynx and hypopharynx and oropharynx?” she asked.
“We found that the overall survival benefit was regardless of the subsite,” Dr. Karam replied, noting that the patient populations in the two cohorts differed somewhat. “Unfortunately, we don’t have clear-cut variables for toxicity, but what we did look at is time to completion of RT, and we found that patients who did get concurrent chemoradiation had a longer time to completion of RT, suggesting perhaps more treatment breaks maybe. … But still, despite the treatment breaks, even after controlling for that, we still found an overall survival advantage, regardless of the subsite.”
At her institution, patients are already being treated with a tailored approach, Dr. Gourin commented. “We have young patients who are so sick that they are not great candidates for chemotherapy, and then we have old patients who are healthier than I am who are great candidates for chemotherapy. So I would say that we have been doing what Dr. Karam suggests, which is looking at age not as a number, but rather comorbidity and the overall functional status of the patient.”
“That’s why I really liked your study: It’s great to see that in writing, because I know that colleagues in other countries that I talk to about health care reform and how to cut costs, they actually use the MACH-NC to define who gets treated and who doesn’t,” she added.
For their analysis, Dr. Karam and colleagues identified patients in the database given a diagnosis of locally advanced cancer of the oropharynx, larynx, or hypopharynx between 1998 and 2011 who were treated with radiation therapy.
Overall, 53% received chemotherapy concurrently, defined as starting it in the 14 days before or 14 days after initiation of the radiation therapy, according to Dr. Karam.
The specific agents given could not be ascertained, she acknowledged. “Unfortunately, the NCDB does not give us data in regard to the type of chemotherapy, and they only started collecting cetuximab data in 2013.”
With a median follow-up of 19 months, the unadjusted 5-year overall survival rate was 15.2% with radiation therapy alone and 30.3% with concurrent chemoradiation (hazard ratio, 0.59; P less than .001). Benefit fell only slightly after multivariate adjustment (HR, 0.63; P less than .001).
Findings were similar in a propensity-matched analysis, which showed an 18.1% survival with radiation therapy alone versus 26.4% with concurrent chemoradiation (HR, 0.73; P less than .001).
On recursive partitioning analysis, chemoradiation was associated with better survival among patients 81 years of age or younger who had low comorbidity based on Charlson-Deyo score and either T1-2,N2-3 disease or T3-4,N0-3 disease.
There was no survival benefit in patients older than age 81. And among patients aged 71-80, there was no benefit for those having less advanced disease (stage T1-2,N1) and low comorbidity, or having more advanced disease (T3-4,N1+ disease) and high comorbidity.
AT THE HEAD AND NECK CANCER SYMPOSIUM
Key clinical point: Age alone is not a reason to withhold concurrent chemoradiation for head and neck cancer.
Major finding: Addition of concurrent chemotherapy improved overall survival for patients aged 81 years or younger who had low comorbidity and more advanced disease.
Data source: A retrospective cohort study of 4,042 patients aged 71 years or older with locally advanced head and neck cancer given radiation therapy (National Cancer Data Base).
Disclosures: Dr. Karam disclosed that she had no relevant conflicts of interest.

Antibiotic-resistant infections remain a persistent threat
One in every seven infections in acute care hospitals related to catheters and surgeries was caused by antibiotic-resistant bacteria. In long-term acute care hospitals, that number increased to one in four.
Those are key findings from a study published March 3 in the Centers for Disease Control and Prevention’s Morbidity and Mortality Weekly Report that is the first to combine national data on antibiotic-resistant (AR) bacteria threats with progress on health care–associated infections (HAIs).
“Antibiotic resistance threatens to return us to a time when a simple infection could kill,” CDC Director Thomas Frieden said during a March 3 telebriefing. “The more people who get infected with resistant bacteria, the more people who suffer complications, the more who, tragically, may die from preventable infections. On any given day about one in 25 hospitalized patients has at least one health care–associated infection that they didn’t come in with. No one should get sick when they’re trying to get well.”
For the study, researchers led by Dr. Clifford McDonald of the CDC’s Division of Healthcare Quality Promotion, collected data on specific infections that were reported to the National Healthcare Safety Network in 2014 by approximately 4,000 short-term acute care hospitals, 501 long-term acute care hospitals, and 1,135 inpatient rehabilitation facilities in all 50 states (MMWR. 2016 Mar 3. doi: 10.15585/mmwr.mm6509e1er). Next, they determined the proportions of AR pathogens and HAIs caused by any of six resistant bacteria highlighted by the CDC in 2013 as urgent or serious threats: CRE (carbapenem-resistant Enterobacteriaceae), MRSA (methicillin-resistant Staphylococcus aureus), ESBL-producing Enterobacteriaceae (extended-spectrum beta-lactamases), VRE (vancomycin-resistant enterococci), multidrug-resistant pseudomonas, and multidrug-resistant Acinetobacter.
The researchers found that, compared with historical data from 5-8 years earlier, central line–associated bloodstream infections decreased by 50% and surgical site infections (SSIs) by 17% in 2014.

“There is encouraging news here,” Dr. Frieden said. “Doctors, nurses, hospitals, health care systems and other partners have made progress preventing some health care–associated infections.” However, the study found that one in six remaining central line-associated bloodstream infections were caused by urgent or serious antibiotic-resistant bacteria, while one in seven remaining surgical site infections were caused by urgent or serious antibiotic-resistant bacteria.
While catheter-associated urinary tract infections appear unchanged from baseline, there have been recent decreases, according to the study. In addition, C. difficile infections in hospitals decreased 8% between 2011 and 2014.
Dr. McDonald and his associates determined that in 2014, one in seven infections in acute care hospitals related to catheters and surgeries was caused by one of the six antibiotic-resistance threat bacteria, “which is deeply concerning,” Dr. Frieden said. That number increased to one in four infections in long-term acute care hospitals, a proportion that he characterized as “chilling.”
The CDC recommends three strategies that doctors, nurses, and other health care providers should take with every patient, to prevent HAIs and stop the spread of antibiotic resistance:
• Prevent the spread of bacteria between patients. Dr. Peter Pronovost, who participated in the telebriefing, said that he and his associates at Johns Hopkins University in Baltimore “do this by practicing good hand hygiene techniques by wearing sterile equipment when inserting lines.”
• Prevent surgery-related infections and/or placement of a catheter. “Check catheters frequently and remove them when you no longer need them,” advised Dr. Pronovost, director of the Armstrong Institute for Patient Safety and Quality at Johns Hopkins. “Ask if you actually need them before you even place them.”
• Improve antibiotic use through stewardship. This means using “the right antibiotics for the right duration,” Dr. Pronovost said. “Antibiotics could be lifesaving and are necessary for critically ill patients, especially those with septic shock. But these antibiotics need to be adjusted based on lab results and new information about the organisms that are causing these infections. Forty-eight hours after antibiotics are initiated, take a ‘time out.’ Perform a brief but focused assessment to determine if antibiotic therapy is still needed, or if it should be refined. A common mistake we make is to continue vancomycin when there is no presence of MRSA. We often tell our staff at Johns Hopkins, ‘if it doesn’t grow, let it go.’ ”
Dr. Frieden concluded his remarks by noting that physicians and other clinicians on the front lines “need support of their facility leadership,” to prevent HAIs. “Health care facilities, CEOs, and administrators are a major part of the solution. It’s important that they make a priority of infection prevention, sepsis prevention, and antibiotic stewardship. Know your facility’s data and target prevention efforts to ensure improvements in patient safety.”
One in every seven infections in acute care hospitals related to catheters and surgeries was caused by antibiotic-resistant bacteria. In long-term acute care hospitals, that number increased to one in four.
Those are key findings from a study published March 3 in the Centers for Disease Control and Prevention’s Morbidity and Mortality Weekly Report that is the first to combine national data on antibiotic-resistant (AR) bacteria threats with progress on health care–associated infections (HAIs).
“Antibiotic resistance threatens to return us to a time when a simple infection could kill,” CDC Director Thomas Frieden said during a March 3 telebriefing. “The more people who get infected with resistant bacteria, the more people who suffer complications, the more who, tragically, may die from preventable infections. On any given day about one in 25 hospitalized patients has at least one health care–associated infection that they didn’t come in with. No one should get sick when they’re trying to get well.”
For the study, researchers led by Dr. Clifford McDonald of the CDC’s Division of Healthcare Quality Promotion, collected data on specific infections that were reported to the National Healthcare Safety Network in 2014 by approximately 4,000 short-term acute care hospitals, 501 long-term acute care hospitals, and 1,135 inpatient rehabilitation facilities in all 50 states (MMWR. 2016 Mar 3. doi: 10.15585/mmwr.mm6509e1er). Next, they determined the proportions of AR pathogens and HAIs caused by any of six resistant bacteria highlighted by the CDC in 2013 as urgent or serious threats: CRE (carbapenem-resistant Enterobacteriaceae), MRSA (methicillin-resistant Staphylococcus aureus), ESBL-producing Enterobacteriaceae (extended-spectrum beta-lactamases), VRE (vancomycin-resistant enterococci), multidrug-resistant pseudomonas, and multidrug-resistant Acinetobacter.
The researchers found that, compared with historical data from 5-8 years earlier, central line–associated bloodstream infections decreased by 50% and surgical site infections (SSIs) by 17% in 2014.
“There is encouraging news here,” Dr. Frieden said. “Doctors, nurses, hospitals, health care systems and other partners have made progress preventing some health care–associated infections.” However, the study found that one in six remaining central line-associated bloodstream infections were caused by urgent or serious antibiotic-resistant bacteria, while one in seven remaining surgical site infections were caused by urgent or serious antibiotic-resistant bacteria.
While catheter-associated urinary tract infections appear unchanged from baseline, there have been recent decreases, according to the study. In addition, C. difficile infections in hospitals decreased 8% between 2011 and 2014.
Dr. McDonald and his associates determined that in 2014, one in seven infections in acute care hospitals related to catheters and surgeries was caused by one of the six antibiotic-resistance threat bacteria, “which is deeply concerning,” Dr. Frieden said. That number increased to one in four infections in long-term acute care hospitals, a proportion that he characterized as “chilling.”
The CDC recommends three strategies that doctors, nurses, and other health care providers should take with every patient, to prevent HAIs and stop the spread of antibiotic resistance:
• Prevent the spread of bacteria between patients. Dr. Peter Pronovost, who participated in the telebriefing, said that he and his associates at Johns Hopkins University in Baltimore “do this by practicing good hand hygiene techniques by wearing sterile equipment when inserting lines.”
• Prevent surgery-related infections and/or placement of a catheter. “Check catheters frequently and remove them when you no longer need them,” advised Dr. Pronovost, director of the Armstrong Institute for Patient Safety and Quality at Johns Hopkins. “Ask if you actually need them before you even place them.”
• Improve antibiotic use through stewardship. This means using “the right antibiotics for the right duration,” Dr. Pronovost said. “Antibiotics could be lifesaving and are necessary for critically ill patients, especially those with septic shock. But these antibiotics need to be adjusted based on lab results and new information about the organisms that are causing these infections. Forty-eight hours after antibiotics are initiated, take a ‘time out.’ Perform a brief but focused assessment to determine if antibiotic therapy is still needed, or if it should be refined. A common mistake we make is to continue vancomycin when there is no presence of MRSA. We often tell our staff at Johns Hopkins, ‘if it doesn’t grow, let it go.’ ”
Dr. Frieden concluded his remarks by noting that physicians and other clinicians on the front lines “need support of their facility leadership,” to prevent HAIs. “Health care facilities, CEOs, and administrators are a major part of the solution. It’s important that they make a priority of infection prevention, sepsis prevention, and antibiotic stewardship. Know your facility’s data and target prevention efforts to ensure improvements in patient safety.”
One in every seven infections in acute care hospitals related to catheters and surgeries was caused by antibiotic-resistant bacteria. In long-term acute care hospitals, that number increased to one in four.
Those are key findings from a study published March 3 in the Centers for Disease Control and Prevention’s Morbidity and Mortality Weekly Report that is the first to combine national data on antibiotic-resistant (AR) bacteria threats with progress on health care–associated infections (HAIs).
“Antibiotic resistance threatens to return us to a time when a simple infection could kill,” CDC Director Thomas Frieden said during a March 3 telebriefing. “The more people who get infected with resistant bacteria, the more people who suffer complications, the more who, tragically, may die from preventable infections. On any given day about one in 25 hospitalized patients has at least one health care–associated infection that they didn’t come in with. No one should get sick when they’re trying to get well.”
For the study, researchers led by Dr. Clifford McDonald of the CDC’s Division of Healthcare Quality Promotion, collected data on specific infections that were reported to the National Healthcare Safety Network in 2014 by approximately 4,000 short-term acute care hospitals, 501 long-term acute care hospitals, and 1,135 inpatient rehabilitation facilities in all 50 states (MMWR. 2016 Mar 3. doi: 10.15585/mmwr.mm6509e1er). Next, they determined the proportions of AR pathogens and HAIs caused by any of six resistant bacteria highlighted by the CDC in 2013 as urgent or serious threats: CRE (carbapenem-resistant Enterobacteriaceae), MRSA (methicillin-resistant Staphylococcus aureus), ESBL-producing Enterobacteriaceae (extended-spectrum beta-lactamases), VRE (vancomycin-resistant enterococci), multidrug-resistant pseudomonas, and multidrug-resistant Acinetobacter.
The researchers found that, compared with historical data from 5-8 years earlier, central line–associated bloodstream infections decreased by 50% and surgical site infections (SSIs) by 17% in 2014.
“There is encouraging news here,” Dr. Frieden said. “Doctors, nurses, hospitals, health care systems and other partners have made progress preventing some health care–associated infections.” However, the study found that one in six remaining central line-associated bloodstream infections were caused by urgent or serious antibiotic-resistant bacteria, while one in seven remaining surgical site infections were caused by urgent or serious antibiotic-resistant bacteria.
While catheter-associated urinary tract infections appear unchanged from baseline, there have been recent decreases, according to the study. In addition, C. difficile infections in hospitals decreased 8% between 2011 and 2014.
Dr. McDonald and his associates determined that in 2014, one in seven infections in acute care hospitals related to catheters and surgeries was caused by one of the six antibiotic-resistance threat bacteria, “which is deeply concerning,” Dr. Frieden said. That number increased to one in four infections in long-term acute care hospitals, a proportion that he characterized as “chilling.”
The CDC recommends three strategies that doctors, nurses, and other health care providers should take with every patient, to prevent HAIs and stop the spread of antibiotic resistance:
• Prevent the spread of bacteria between patients. Dr. Peter Pronovost, who participated in the telebriefing, said that he and his associates at Johns Hopkins University in Baltimore “do this by practicing good hand hygiene techniques by wearing sterile equipment when inserting lines.”
• Prevent surgery-related infections and/or placement of a catheter. “Check catheters frequently and remove them when you no longer need them,” advised Dr. Pronovost, director of the Armstrong Institute for Patient Safety and Quality at Johns Hopkins. “Ask if you actually need them before you even place them.”
• Improve antibiotic use through stewardship. This means using “the right antibiotics for the right duration,” Dr. Pronovost said. “Antibiotics could be lifesaving and are necessary for critically ill patients, especially those with septic shock. But these antibiotics need to be adjusted based on lab results and new information about the organisms that are causing these infections. Forty-eight hours after antibiotics are initiated, take a ‘time out.’ Perform a brief but focused assessment to determine if antibiotic therapy is still needed, or if it should be refined. A common mistake we make is to continue vancomycin when there is no presence of MRSA. We often tell our staff at Johns Hopkins, ‘if it doesn’t grow, let it go.’ ”
Dr. Frieden concluded his remarks by noting that physicians and other clinicians on the front lines “need support of their facility leadership,” to prevent HAIs. “Health care facilities, CEOs, and administrators are a major part of the solution. It’s important that they make a priority of infection prevention, sepsis prevention, and antibiotic stewardship. Know your facility’s data and target prevention efforts to ensure improvements in patient safety.”
FROM MMWR
Haseeb Rahman, MD
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Painful area above cesarean scar
The ED physician suspected necrotizing fasciitis (NF) and immediately called for a surgical consult. This patient had type II NF, which can be caused by common skin organisms, such as Streptococcus pyogenes and Staphylococcus aureus.
Surgical debridement is the primary therapeutic modality. If the first debridement occurs within 24 hours of the onset of symptoms, there is a significantly improved chance of survival. Extensive, definitive debridement should be the goal with the first surgery. This may require amputation of an extremity to control the disease. Surgical debridement is repeated until all infected devitalized tissue is removed.
Antibiotics are the main adjunctive therapy to surgery. Broad-spectrum empiric antibiotics should be started immediately when NF is suspected and should include coverage of Gram-positive, Gram-negative, and anaerobic organisms. Antimicrobial therapy must be directed at the known or suspected pathogens and used in appropriate doses until repeated operative procedures are no longer needed. Empiric vancomycin should be considered while awaiting culture results to cover for the increasing incidence of methicillin-resistant Staphylococcus aureus (MRSA). Aggressive fluid resuscitation is often necessary because of massive capillary leak syndrome.
In this case, the patient was started on empiric broad-spectrum intravenous antibiotics to cover Gram-positive, Gram-negative, and anaerobic organisms. The surgical team then rapidly admitted the patient to the operating room (OR) for debridement. From the OR, she was transferred to the surgical intensive care unit where she received ongoing supportive and postoperative care. The intraoperative tissue cultures grew out MRSA.
The patient in this case was fortunate. She survived because of the speed with which she received a proper diagnosis and treatment.
Photo courtesy of Michael Babcock, MD. Text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R, Franklin J. Necrotizing fasciitis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:702-706.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The ED physician suspected necrotizing fasciitis (NF) and immediately called for a surgical consult. This patient had type II NF, which can be caused by common skin organisms, such as Streptococcus pyogenes and Staphylococcus aureus.
Surgical debridement is the primary therapeutic modality. If the first debridement occurs within 24 hours of the onset of symptoms, there is a significantly improved chance of survival. Extensive, definitive debridement should be the goal with the first surgery. This may require amputation of an extremity to control the disease. Surgical debridement is repeated until all infected devitalized tissue is removed.
Antibiotics are the main adjunctive therapy to surgery. Broad-spectrum empiric antibiotics should be started immediately when NF is suspected and should include coverage of Gram-positive, Gram-negative, and anaerobic organisms. Antimicrobial therapy must be directed at the known or suspected pathogens and used in appropriate doses until repeated operative procedures are no longer needed. Empiric vancomycin should be considered while awaiting culture results to cover for the increasing incidence of methicillin-resistant Staphylococcus aureus (MRSA). Aggressive fluid resuscitation is often necessary because of massive capillary leak syndrome.
In this case, the patient was started on empiric broad-spectrum intravenous antibiotics to cover Gram-positive, Gram-negative, and anaerobic organisms. The surgical team then rapidly admitted the patient to the operating room (OR) for debridement. From the OR, she was transferred to the surgical intensive care unit where she received ongoing supportive and postoperative care. The intraoperative tissue cultures grew out MRSA.
The patient in this case was fortunate. She survived because of the speed with which she received a proper diagnosis and treatment.
Photo courtesy of Michael Babcock, MD. Text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R, Franklin J. Necrotizing fasciitis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:702-706.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The ED physician suspected necrotizing fasciitis (NF) and immediately called for a surgical consult. This patient had type II NF, which can be caused by common skin organisms, such as Streptococcus pyogenes and Staphylococcus aureus.
Surgical debridement is the primary therapeutic modality. If the first debridement occurs within 24 hours of the onset of symptoms, there is a significantly improved chance of survival. Extensive, definitive debridement should be the goal with the first surgery. This may require amputation of an extremity to control the disease. Surgical debridement is repeated until all infected devitalized tissue is removed.
Antibiotics are the main adjunctive therapy to surgery. Broad-spectrum empiric antibiotics should be started immediately when NF is suspected and should include coverage of Gram-positive, Gram-negative, and anaerobic organisms. Antimicrobial therapy must be directed at the known or suspected pathogens and used in appropriate doses until repeated operative procedures are no longer needed. Empiric vancomycin should be considered while awaiting culture results to cover for the increasing incidence of methicillin-resistant Staphylococcus aureus (MRSA). Aggressive fluid resuscitation is often necessary because of massive capillary leak syndrome.
In this case, the patient was started on empiric broad-spectrum intravenous antibiotics to cover Gram-positive, Gram-negative, and anaerobic organisms. The surgical team then rapidly admitted the patient to the operating room (OR) for debridement. From the OR, she was transferred to the surgical intensive care unit where she received ongoing supportive and postoperative care. The intraoperative tissue cultures grew out MRSA.
The patient in this case was fortunate. She survived because of the speed with which she received a proper diagnosis and treatment.
Photo courtesy of Michael Babcock, MD. Text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R, Franklin J. Necrotizing fasciitis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:702-706.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com