User login
Swollen, painful leg
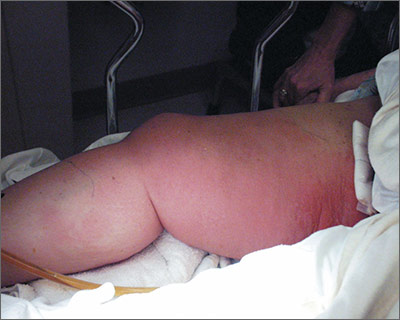
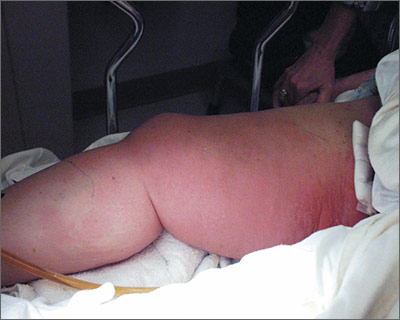
The ED physician suspected necrotizing fasciitis (NF), a rapidly progressive infection of the deep fascia with necrosis of the subcutaneous tissues. It usually occurs after surgery or trauma. Patients have erythema and pain disproportionate to the physical findings.
This patient had type I NF, a polymicrobial infection with aerobic and anaerobic bacteria. It is frequently caused by enteric Gram-negative pathogens including Enterobacteriaceae organisms and Bacteroides. It can also occur with Gram-positive organisms such as non–group A streptococci and Peptostreptococcus.
Risk factors for type I NF include diabetes, severe peripheral vascular disease, obesity, alcoholism, cirrhosis, intravenous drug use, decubitus ulcers, poor nutritional status, surgery, and penetrating trauma. Early recognition based on signs and symptoms is potentially lifesaving. Although lab tests and imaging studies can confirm one’s clinical impression, rapid treatment with antibiotics and surgery are crucial to improving survival.
The patient in this case was taken to the operating room for debridement of her NF. Broad-spectrum antibiotics were started, but the infection continued to quickly advance. The patient died the following day. Her wound culture later grew Escherichia coli, Proteus vulgaris, Corynebacterium, Enterococcus, Staphylococcus sp., and Peptostreptococcus.
Adapted from: Dufel S, Martino M. Simple cellulitis or a more serious infection? J Fam Pract. 2006;55:396-400.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The ED physician suspected necrotizing fasciitis (NF), a rapidly progressive infection of the deep fascia with necrosis of the subcutaneous tissues. It usually occurs after surgery or trauma. Patients have erythema and pain disproportionate to the physical findings.
This patient had type I NF, a polymicrobial infection with aerobic and anaerobic bacteria. It is frequently caused by enteric Gram-negative pathogens including Enterobacteriaceae organisms and Bacteroides. It can also occur with Gram-positive organisms such as non–group A streptococci and Peptostreptococcus.
Risk factors for type I NF include diabetes, severe peripheral vascular disease, obesity, alcoholism, cirrhosis, intravenous drug use, decubitus ulcers, poor nutritional status, surgery, and penetrating trauma. Early recognition based on signs and symptoms is potentially lifesaving. Although lab tests and imaging studies can confirm one’s clinical impression, rapid treatment with antibiotics and surgery are crucial to improving survival.
The patient in this case was taken to the operating room for debridement of her NF. Broad-spectrum antibiotics were started, but the infection continued to quickly advance. The patient died the following day. Her wound culture later grew Escherichia coli, Proteus vulgaris, Corynebacterium, Enterococcus, Staphylococcus sp., and Peptostreptococcus.
Adapted from: Dufel S, Martino M. Simple cellulitis or a more serious infection? J Fam Pract. 2006;55:396-400.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The ED physician suspected necrotizing fasciitis (NF), a rapidly progressive infection of the deep fascia with necrosis of the subcutaneous tissues. It usually occurs after surgery or trauma. Patients have erythema and pain disproportionate to the physical findings.
This patient had type I NF, a polymicrobial infection with aerobic and anaerobic bacteria. It is frequently caused by enteric Gram-negative pathogens including Enterobacteriaceae organisms and Bacteroides. It can also occur with Gram-positive organisms such as non–group A streptococci and Peptostreptococcus.
Risk factors for type I NF include diabetes, severe peripheral vascular disease, obesity, alcoholism, cirrhosis, intravenous drug use, decubitus ulcers, poor nutritional status, surgery, and penetrating trauma. Early recognition based on signs and symptoms is potentially lifesaving. Although lab tests and imaging studies can confirm one’s clinical impression, rapid treatment with antibiotics and surgery are crucial to improving survival.
The patient in this case was taken to the operating room for debridement of her NF. Broad-spectrum antibiotics were started, but the infection continued to quickly advance. The patient died the following day. Her wound culture later grew Escherichia coli, Proteus vulgaris, Corynebacterium, Enterococcus, Staphylococcus sp., and Peptostreptococcus.
Adapted from: Dufel S, Martino M. Simple cellulitis or a more serious infection? J Fam Pract. 2006;55:396-400.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
Swollen neck

The physician recognized that his patient had a neck abscess that was likely related to a dental abscess (odontogenic abscess). The patient’s alcoholism most likely weakened his ability to fight the infection, causing it to become potentially life-threatening. Risk factors for abscess formation include intravenous drug abuse, alcoholism, homelessness, dental disease, contact sports, and incarceration.
The physician transferred the patient to the local emergency department for hospitalization under the care of the ear, nose, and throat (ENT) service. The ENT physicians drained the abscess in the operating room without any complications. They cultured the abscess and started the patient on appropriate antibiotics, including penicillin G for dental aerobes and anaerobes.
The hospital team observed the patient for signs of alcohol withdrawal, but there were no complications because the patient hadn’t been drinking for the 5 days prior to hospitalization. Social Services was consulted and the patient was discharged to a respite bed in a local shelter that also had an alcohol and drug rehabilitation program. Arrangements were made for dental work in a charity dental clinic.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Abscess. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill;2013:698-701.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The physician recognized that his patient had a neck abscess that was likely related to a dental abscess (odontogenic abscess). The patient’s alcoholism most likely weakened his ability to fight the infection, causing it to become potentially life-threatening. Risk factors for abscess formation include intravenous drug abuse, alcoholism, homelessness, dental disease, contact sports, and incarceration.
The physician transferred the patient to the local emergency department for hospitalization under the care of the ear, nose, and throat (ENT) service. The ENT physicians drained the abscess in the operating room without any complications. They cultured the abscess and started the patient on appropriate antibiotics, including penicillin G for dental aerobes and anaerobes.
The hospital team observed the patient for signs of alcohol withdrawal, but there were no complications because the patient hadn’t been drinking for the 5 days prior to hospitalization. Social Services was consulted and the patient was discharged to a respite bed in a local shelter that also had an alcohol and drug rehabilitation program. Arrangements were made for dental work in a charity dental clinic.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Abscess. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill;2013:698-701.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The physician recognized that his patient had a neck abscess that was likely related to a dental abscess (odontogenic abscess). The patient’s alcoholism most likely weakened his ability to fight the infection, causing it to become potentially life-threatening. Risk factors for abscess formation include intravenous drug abuse, alcoholism, homelessness, dental disease, contact sports, and incarceration.
The physician transferred the patient to the local emergency department for hospitalization under the care of the ear, nose, and throat (ENT) service. The ENT physicians drained the abscess in the operating room without any complications. They cultured the abscess and started the patient on appropriate antibiotics, including penicillin G for dental aerobes and anaerobes.
The hospital team observed the patient for signs of alcohol withdrawal, but there were no complications because the patient hadn’t been drinking for the 5 days prior to hospitalization. Social Services was consulted and the patient was discharged to a respite bed in a local shelter that also had an alcohol and drug rehabilitation program. Arrangements were made for dental work in a charity dental clinic.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Abscess. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill;2013:698-701.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com

ECT may improve course of bipolar disorder
Electroconvulsive therapy increased illness-free intervals and reduced manic and depressive episodes in bipolar patients, reported Dr. Gian Paolo Minnai of the psychiatry unit at San Martino Hospital in Oristano, Italy, and coauthors.
In a retrospective study of 41 patients with bipolar disorder, the investigators analyzed the number of episodes and admissions 5 years before and after ECT treatment. The duration of free intervals before and after ECT was also studied.

The results showed “significantly longer” free intervals after treatment (13.2 ± 9.0 months before ECT to 25.1 ± 19.1 months after treatment [t = 3.8; P less than .0001]), Dr. Minnai and colleagues reported. In addition, the analysis found “significant reductions” in the number of manic and depressive episodes (5.9 ± 3.0 before ECT to 1.0 ± 1.7 after treatment [t = 9.3; P less than .0001]) and admissions (2.2 ± 1.3 before ECT to 0.2 ± 0.5 after treatment [t = 9.4; P less than .0001]).
The study suggests that “it is plausible that ECT, along with suspending antidepressant treatment, might carry intrinsic stabilizing effect on the course of [bipolar disorder],” the authors concluded.
No financial conflicts of interest were disclosed.
Read the full article in the Journal of Affective Disorders (May 2016;195:180-4).
Electroconvulsive therapy increased illness-free intervals and reduced manic and depressive episodes in bipolar patients, reported Dr. Gian Paolo Minnai of the psychiatry unit at San Martino Hospital in Oristano, Italy, and coauthors.
In a retrospective study of 41 patients with bipolar disorder, the investigators analyzed the number of episodes and admissions 5 years before and after ECT treatment. The duration of free intervals before and after ECT was also studied.
The results showed “significantly longer” free intervals after treatment (13.2 ± 9.0 months before ECT to 25.1 ± 19.1 months after treatment [t = 3.8; P less than .0001]), Dr. Minnai and colleagues reported. In addition, the analysis found “significant reductions” in the number of manic and depressive episodes (5.9 ± 3.0 before ECT to 1.0 ± 1.7 after treatment [t = 9.3; P less than .0001]) and admissions (2.2 ± 1.3 before ECT to 0.2 ± 0.5 after treatment [t = 9.4; P less than .0001]).
The study suggests that “it is plausible that ECT, along with suspending antidepressant treatment, might carry intrinsic stabilizing effect on the course of [bipolar disorder],” the authors concluded.
No financial conflicts of interest were disclosed.
Read the full article in the Journal of Affective Disorders (May 2016;195:180-4).
Electroconvulsive therapy increased illness-free intervals and reduced manic and depressive episodes in bipolar patients, reported Dr. Gian Paolo Minnai of the psychiatry unit at San Martino Hospital in Oristano, Italy, and coauthors.
In a retrospective study of 41 patients with bipolar disorder, the investigators analyzed the number of episodes and admissions 5 years before and after ECT treatment. The duration of free intervals before and after ECT was also studied.
The results showed “significantly longer” free intervals after treatment (13.2 ± 9.0 months before ECT to 25.1 ± 19.1 months after treatment [t = 3.8; P less than .0001]), Dr. Minnai and colleagues reported. In addition, the analysis found “significant reductions” in the number of manic and depressive episodes (5.9 ± 3.0 before ECT to 1.0 ± 1.7 after treatment [t = 9.3; P less than .0001]) and admissions (2.2 ± 1.3 before ECT to 0.2 ± 0.5 after treatment [t = 9.4; P less than .0001]).
The study suggests that “it is plausible that ECT, along with suspending antidepressant treatment, might carry intrinsic stabilizing effect on the course of [bipolar disorder],” the authors concluded.
No financial conflicts of interest were disclosed.
Read the full article in the Journal of Affective Disorders (May 2016;195:180-4).

Practice pathway addresses problem behaviors for children with ASD
A new practice pathway provides primary care providers with evidence-based steps in assessing and addressing irritability and problem behavior in patients at least 3 years old who have been diagnosed with an autism spectrum disorder (ASD).
“As the medical professionals whom children with ASD will probably encounter first and most frequently, pediatric primary care providers need a breadth of knowledge to enable them to identify contributing factors to irritability and problem behavior, decide when and how to initiate treatment, and judge when to refer to specialists,” reported Dr. Kelly McGuire of Columbia University Medical Center and New York State Psychiatric Institute, and her associates (Pediatrics. 2016 Feb;137 Suppl 2:S136-48). “Therefore, this practice pathway can be viewed more as a comprehensive rather than an exhaustive recommendation for [primary care physicians], who must weigh their expertise and resources when addressing the range of issues that a child with ASD and irritability and problem behavior may present.”

The Irritability Workgroup comprised eight child psychiatrists, a developmental pediatrician, and a behavioral psychologist who met regularly to define irritability and problem behavior, and then to review the evidence base on assessing and treating factors that contribute to irritability and problem behavior. They grouped these factors into five domains: “co-occurring medical conditions, lack of functional communication, psychosocial stressors, maladaptive reinforcement patterns, and co-occurring psychiatric conditions.”
Then the group developed a consensus in determining each step in the practice pathway and when it should occur, along with opportunities for a primary care provider to refer the patient to a specialist or to collaborate with school and community providers. The 10 steps in the practice pathway are:
1. Assess the patient for irritability and problem behavior based on the presence of tantrums, meltdowns or rages; property destruction; aggression toward others; and self-injury.
2. Assess the safety of the patient and others in the home, enlisting home-based crisis services or considering hospitalization if safety is threatened.
3. Review the patient’s medical, developmental, communication, environmental, and psychiatric history before and after the problem behavior. Include information on the caretaker’s characteristics, any loss of skills by patient, and any interference occurring with functioning or relationships.
4. Prioritize the behaviors to address based on safety, severity, and impact on daily life.
5. Consider all contributing factors to the problem behavior, including medical problems, difficulties functionally communicating, psychosocial stressors, maladaptive reinforcement patterns (reducing inadvertent triggers), and comorbid psychiatric disorders.
6. Consider possible medication treatments, such as N-acetylcysteine, clonidine, risperidone or aripiprazole, depending on circumstances.
7. Develop an individualized treatment and safety plan.
8. Implement and monitor the treatment plan, setting clear and measurable treatment goals.
9. Follow up at 3 months to assess whether symptoms are continuing and a reassessment is needed.
10. Reevaluate every 3 months.
The article provides a useful and extensive checklist with a variety of specific things to consider when implementing the 10 steps.
“No other provider in the patient’s life combines the medical expertise and first-hand knowledge of the individual patient’s health and development” than the primary care provider, the authors wrote. “The practice pathway is most likely to be efficient and effective in generating a treatment plan if it is systematically followed and the specific combination of individual contributing factors is identified for each patient.”
The research was conducted through the Autism Speaks Autism Treatment Network and funded by the U.S. Department of Health and Human Services with the Autism Intervention Research Network on Physical Health and by Marilyn and James Simons Family Giving. Dr. Jeremy Veenstra-VanderWeele has received research funding from Seaside Therapeutics, Roche, Novartis, Forest, Sunovion, and SynapDx and has consulted with Roche, Novartis and SynapDx.
A new practice pathway provides primary care providers with evidence-based steps in assessing and addressing irritability and problem behavior in patients at least 3 years old who have been diagnosed with an autism spectrum disorder (ASD).
“As the medical professionals whom children with ASD will probably encounter first and most frequently, pediatric primary care providers need a breadth of knowledge to enable them to identify contributing factors to irritability and problem behavior, decide when and how to initiate treatment, and judge when to refer to specialists,” reported Dr. Kelly McGuire of Columbia University Medical Center and New York State Psychiatric Institute, and her associates (Pediatrics. 2016 Feb;137 Suppl 2:S136-48). “Therefore, this practice pathway can be viewed more as a comprehensive rather than an exhaustive recommendation for [primary care physicians], who must weigh their expertise and resources when addressing the range of issues that a child with ASD and irritability and problem behavior may present.”
The Irritability Workgroup comprised eight child psychiatrists, a developmental pediatrician, and a behavioral psychologist who met regularly to define irritability and problem behavior, and then to review the evidence base on assessing and treating factors that contribute to irritability and problem behavior. They grouped these factors into five domains: “co-occurring medical conditions, lack of functional communication, psychosocial stressors, maladaptive reinforcement patterns, and co-occurring psychiatric conditions.”
Then the group developed a consensus in determining each step in the practice pathway and when it should occur, along with opportunities for a primary care provider to refer the patient to a specialist or to collaborate with school and community providers. The 10 steps in the practice pathway are:
1. Assess the patient for irritability and problem behavior based on the presence of tantrums, meltdowns or rages; property destruction; aggression toward others; and self-injury.
2. Assess the safety of the patient and others in the home, enlisting home-based crisis services or considering hospitalization if safety is threatened.
3. Review the patient’s medical, developmental, communication, environmental, and psychiatric history before and after the problem behavior. Include information on the caretaker’s characteristics, any loss of skills by patient, and any interference occurring with functioning or relationships.
4. Prioritize the behaviors to address based on safety, severity, and impact on daily life.
5. Consider all contributing factors to the problem behavior, including medical problems, difficulties functionally communicating, psychosocial stressors, maladaptive reinforcement patterns (reducing inadvertent triggers), and comorbid psychiatric disorders.
6. Consider possible medication treatments, such as N-acetylcysteine, clonidine, risperidone or aripiprazole, depending on circumstances.
7. Develop an individualized treatment and safety plan.
8. Implement and monitor the treatment plan, setting clear and measurable treatment goals.
9. Follow up at 3 months to assess whether symptoms are continuing and a reassessment is needed.
10. Reevaluate every 3 months.
The article provides a useful and extensive checklist with a variety of specific things to consider when implementing the 10 steps.
“No other provider in the patient’s life combines the medical expertise and first-hand knowledge of the individual patient’s health and development” than the primary care provider, the authors wrote. “The practice pathway is most likely to be efficient and effective in generating a treatment plan if it is systematically followed and the specific combination of individual contributing factors is identified for each patient.”
The research was conducted through the Autism Speaks Autism Treatment Network and funded by the U.S. Department of Health and Human Services with the Autism Intervention Research Network on Physical Health and by Marilyn and James Simons Family Giving. Dr. Jeremy Veenstra-VanderWeele has received research funding from Seaside Therapeutics, Roche, Novartis, Forest, Sunovion, and SynapDx and has consulted with Roche, Novartis and SynapDx.
A new practice pathway provides primary care providers with evidence-based steps in assessing and addressing irritability and problem behavior in patients at least 3 years old who have been diagnosed with an autism spectrum disorder (ASD).
“As the medical professionals whom children with ASD will probably encounter first and most frequently, pediatric primary care providers need a breadth of knowledge to enable them to identify contributing factors to irritability and problem behavior, decide when and how to initiate treatment, and judge when to refer to specialists,” reported Dr. Kelly McGuire of Columbia University Medical Center and New York State Psychiatric Institute, and her associates (Pediatrics. 2016 Feb;137 Suppl 2:S136-48). “Therefore, this practice pathway can be viewed more as a comprehensive rather than an exhaustive recommendation for [primary care physicians], who must weigh their expertise and resources when addressing the range of issues that a child with ASD and irritability and problem behavior may present.”
The Irritability Workgroup comprised eight child psychiatrists, a developmental pediatrician, and a behavioral psychologist who met regularly to define irritability and problem behavior, and then to review the evidence base on assessing and treating factors that contribute to irritability and problem behavior. They grouped these factors into five domains: “co-occurring medical conditions, lack of functional communication, psychosocial stressors, maladaptive reinforcement patterns, and co-occurring psychiatric conditions.”
Then the group developed a consensus in determining each step in the practice pathway and when it should occur, along with opportunities for a primary care provider to refer the patient to a specialist or to collaborate with school and community providers. The 10 steps in the practice pathway are:
1. Assess the patient for irritability and problem behavior based on the presence of tantrums, meltdowns or rages; property destruction; aggression toward others; and self-injury.
2. Assess the safety of the patient and others in the home, enlisting home-based crisis services or considering hospitalization if safety is threatened.
3. Review the patient’s medical, developmental, communication, environmental, and psychiatric history before and after the problem behavior. Include information on the caretaker’s characteristics, any loss of skills by patient, and any interference occurring with functioning or relationships.
4. Prioritize the behaviors to address based on safety, severity, and impact on daily life.
5. Consider all contributing factors to the problem behavior, including medical problems, difficulties functionally communicating, psychosocial stressors, maladaptive reinforcement patterns (reducing inadvertent triggers), and comorbid psychiatric disorders.
6. Consider possible medication treatments, such as N-acetylcysteine, clonidine, risperidone or aripiprazole, depending on circumstances.
7. Develop an individualized treatment and safety plan.
8. Implement and monitor the treatment plan, setting clear and measurable treatment goals.
9. Follow up at 3 months to assess whether symptoms are continuing and a reassessment is needed.
10. Reevaluate every 3 months.
The article provides a useful and extensive checklist with a variety of specific things to consider when implementing the 10 steps.
“No other provider in the patient’s life combines the medical expertise and first-hand knowledge of the individual patient’s health and development” than the primary care provider, the authors wrote. “The practice pathway is most likely to be efficient and effective in generating a treatment plan if it is systematically followed and the specific combination of individual contributing factors is identified for each patient.”
The research was conducted through the Autism Speaks Autism Treatment Network and funded by the U.S. Department of Health and Human Services with the Autism Intervention Research Network on Physical Health and by Marilyn and James Simons Family Giving. Dr. Jeremy Veenstra-VanderWeele has received research funding from Seaside Therapeutics, Roche, Novartis, Forest, Sunovion, and SynapDx and has consulted with Roche, Novartis and SynapDx.
FROM PEDIATRICS
Key clinical point: A 10-step practice pathway helps primary care providers address irritability and problem behavior in children with ASD.
Major finding: Ten steps in the pathway include assessment of problems, identification of contributing factors, and formulation of a plan to address the underlying factors and the behavior.
Data source: The pathway is based on a consensus by a 10-member multidisciplinary work group charged with identifying available evidence on effective ways to assess and address irritability and problem behavior in children with ASD.
Disclosures: The research was conducted through the Autism Speaks Autism Treatment Network and funded by the U.S. Department of Health and Human Services with the Autism Intervention Research Network on Physical Health and by Marilyn and James Simons Family Giving. Dr. Jeremy Veenstra-VanderWeele has received research funding from Seaside Therapeutics, Roche, Novartis, Forest, Sunovion and SynapDx and has consulted with Roche, Novartis and SynapDx.

Safety of bioresorbable stents does not match that of metal stents
Bioresorbable vascular scaffold stents are improving rapidly but they are still associated with a higher risk of complications compared with drug-eluting metal stents, according to a meta-analysis of published studies presented at Cardiovascular Research Technologies 2016.
“Bioresorbable stents are clearly an attractive strategy, but our data suggest that physicians and patients should remain aware of the risks,” reported Dr. Alok Saurav of Creighton University Medical Center, Omaha, Neb.

The first bioresorbable vascular scaffold (BVS) device, Synergy, was approved this past October, but this stent, despite bioresorbable struts, still has body parts that are not fully bioresorbable. However, several fully bioresorbable devices have reached late stages of testing and may receive regulatory approval this year.
In the meta-analysis, eight studies – five randomized trials, two studies with propensity matching, and an observational study –the primary goal was to compare BVS to drug eluting metal (DEM) stents for definite stent thrombosis. Secondary outcomes included subacute stent thrombosis within 30 days and within 1 year and cardiac death, all-cause death, MI, and ischemia-driven target vessel revascularization (TVR).
Despite the fact that the mean age and gender distribution was the same when the 2,760 patients receiving BVS stents were compared to the 2,212 receiving DEM stents, and both received comparable antiplatelet regimens after the stent was placed, there was an 80% greater relative risk for definite stent thrombosis in the BVS group. Although this difference fell short of statistical significance (P = .06), Dr. Saurav called it a “strong trend.”
Several of the adverse events that were analyzed as secondary outcomes in this study were less frequent with the BVS, such as cardiac death (relative risk, 0.83) and all-cause death (RR, 0.74), but the statistics did not suggest a trend, so Dr. Saurav characterized these outcomes as similar. MI was an exception. This was more frequent in those received a BVS stent (RR, 1.35; P = .049), and this reached significance.
Most of the studies included in this analysis were conducted with the everolimus-eluting Absorb BVS device, which many are predicting will be the first fully bioresorbable stent to receive regulatory approval.
It is notable that another meta-analysis including some of the same studies and published just weeks prior to the CRT meeting drew the same conclusion about the increased risk of stent thrombosis with BVS relative to DEM stents (Lancet 2016;387:537-44). This meta-analysis was restricted to six trials with 3,738 randomized patients. Unlike the meta-analysis presented at CRT, this study compared the two types of stents for both definite and probable stent thrombosis. For BVS relative to DEM stents, the relative risk for this outcome was 1.99 (P = .05).
“We think our restriction to definite stent thrombosis provides a stricter endpoint, but it’s notable that the results were relatively consistent,” Dr. Saurav reported.
Acknowledging that the increased risk of stent thrombosis appears to be modest for BVS relative to DEM stents, Dr. Saurav emphasized that these data should not discourage further development of bioresorbable stents, which are conceptually attractive.
“We cannot take these bioresorbable devices off the table,” he said. “But we do need more data to evaluate their risks relative to the conventional devices that are now available.”
The meeting was sponsored by the Cardiovascular Research Institute at Washington Hospital Center. Dr. Saurav reported no conflicts of interest.
Bioresorbable vascular scaffold stents are improving rapidly but they are still associated with a higher risk of complications compared with drug-eluting metal stents, according to a meta-analysis of published studies presented at Cardiovascular Research Technologies 2016.
“Bioresorbable stents are clearly an attractive strategy, but our data suggest that physicians and patients should remain aware of the risks,” reported Dr. Alok Saurav of Creighton University Medical Center, Omaha, Neb.
The first bioresorbable vascular scaffold (BVS) device, Synergy, was approved this past October, but this stent, despite bioresorbable struts, still has body parts that are not fully bioresorbable. However, several fully bioresorbable devices have reached late stages of testing and may receive regulatory approval this year.
In the meta-analysis, eight studies – five randomized trials, two studies with propensity matching, and an observational study –the primary goal was to compare BVS to drug eluting metal (DEM) stents for definite stent thrombosis. Secondary outcomes included subacute stent thrombosis within 30 days and within 1 year and cardiac death, all-cause death, MI, and ischemia-driven target vessel revascularization (TVR).
Despite the fact that the mean age and gender distribution was the same when the 2,760 patients receiving BVS stents were compared to the 2,212 receiving DEM stents, and both received comparable antiplatelet regimens after the stent was placed, there was an 80% greater relative risk for definite stent thrombosis in the BVS group. Although this difference fell short of statistical significance (P = .06), Dr. Saurav called it a “strong trend.”
Several of the adverse events that were analyzed as secondary outcomes in this study were less frequent with the BVS, such as cardiac death (relative risk, 0.83) and all-cause death (RR, 0.74), but the statistics did not suggest a trend, so Dr. Saurav characterized these outcomes as similar. MI was an exception. This was more frequent in those received a BVS stent (RR, 1.35; P = .049), and this reached significance.
Most of the studies included in this analysis were conducted with the everolimus-eluting Absorb BVS device, which many are predicting will be the first fully bioresorbable stent to receive regulatory approval.
It is notable that another meta-analysis including some of the same studies and published just weeks prior to the CRT meeting drew the same conclusion about the increased risk of stent thrombosis with BVS relative to DEM stents (Lancet 2016;387:537-44). This meta-analysis was restricted to six trials with 3,738 randomized patients. Unlike the meta-analysis presented at CRT, this study compared the two types of stents for both definite and probable stent thrombosis. For BVS relative to DEM stents, the relative risk for this outcome was 1.99 (P = .05).
“We think our restriction to definite stent thrombosis provides a stricter endpoint, but it’s notable that the results were relatively consistent,” Dr. Saurav reported.
Acknowledging that the increased risk of stent thrombosis appears to be modest for BVS relative to DEM stents, Dr. Saurav emphasized that these data should not discourage further development of bioresorbable stents, which are conceptually attractive.
“We cannot take these bioresorbable devices off the table,” he said. “But we do need more data to evaluate their risks relative to the conventional devices that are now available.”
The meeting was sponsored by the Cardiovascular Research Institute at Washington Hospital Center. Dr. Saurav reported no conflicts of interest.
Bioresorbable vascular scaffold stents are improving rapidly but they are still associated with a higher risk of complications compared with drug-eluting metal stents, according to a meta-analysis of published studies presented at Cardiovascular Research Technologies 2016.
“Bioresorbable stents are clearly an attractive strategy, but our data suggest that physicians and patients should remain aware of the risks,” reported Dr. Alok Saurav of Creighton University Medical Center, Omaha, Neb.
The first bioresorbable vascular scaffold (BVS) device, Synergy, was approved this past October, but this stent, despite bioresorbable struts, still has body parts that are not fully bioresorbable. However, several fully bioresorbable devices have reached late stages of testing and may receive regulatory approval this year.
In the meta-analysis, eight studies – five randomized trials, two studies with propensity matching, and an observational study –the primary goal was to compare BVS to drug eluting metal (DEM) stents for definite stent thrombosis. Secondary outcomes included subacute stent thrombosis within 30 days and within 1 year and cardiac death, all-cause death, MI, and ischemia-driven target vessel revascularization (TVR).
Despite the fact that the mean age and gender distribution was the same when the 2,760 patients receiving BVS stents were compared to the 2,212 receiving DEM stents, and both received comparable antiplatelet regimens after the stent was placed, there was an 80% greater relative risk for definite stent thrombosis in the BVS group. Although this difference fell short of statistical significance (P = .06), Dr. Saurav called it a “strong trend.”
Several of the adverse events that were analyzed as secondary outcomes in this study were less frequent with the BVS, such as cardiac death (relative risk, 0.83) and all-cause death (RR, 0.74), but the statistics did not suggest a trend, so Dr. Saurav characterized these outcomes as similar. MI was an exception. This was more frequent in those received a BVS stent (RR, 1.35; P = .049), and this reached significance.
Most of the studies included in this analysis were conducted with the everolimus-eluting Absorb BVS device, which many are predicting will be the first fully bioresorbable stent to receive regulatory approval.
It is notable that another meta-analysis including some of the same studies and published just weeks prior to the CRT meeting drew the same conclusion about the increased risk of stent thrombosis with BVS relative to DEM stents (Lancet 2016;387:537-44). This meta-analysis was restricted to six trials with 3,738 randomized patients. Unlike the meta-analysis presented at CRT, this study compared the two types of stents for both definite and probable stent thrombosis. For BVS relative to DEM stents, the relative risk for this outcome was 1.99 (P = .05).
“We think our restriction to definite stent thrombosis provides a stricter endpoint, but it’s notable that the results were relatively consistent,” Dr. Saurav reported.
Acknowledging that the increased risk of stent thrombosis appears to be modest for BVS relative to DEM stents, Dr. Saurav emphasized that these data should not discourage further development of bioresorbable stents, which are conceptually attractive.
“We cannot take these bioresorbable devices off the table,” he said. “But we do need more data to evaluate their risks relative to the conventional devices that are now available.”
The meeting was sponsored by the Cardiovascular Research Institute at Washington Hospital Center. Dr. Saurav reported no conflicts of interest.
AT CARDIOVASCULAR RESEARCH TECHNOLOGIES 2016
Key clinical point: Trial data suggest the risk of thrombosis and other adverse events remains higher with bioresorbable stents than with conventional drug-eluting metal stents.
Major finding: In a meta-analysis, the 80% increased risk of definite stent thrombosis for bioresorbable relative to metal stents fell just short of significance (P = .06) but the 35% increased risk of subsequent MI was significant (P = .049).
Data source: Meta-analysis of eight studies.
Disclosures: Dr. Saurav reported no conflicts of interest.

Limited posttreatment imaging suffices in HPV-positive oropharyngeal cancer
SCOTTSDALE, ARIZ. – Most patients who are treated for human papillomavirus (HPV)–positive oropharyngeal cancer can safely skip routine imaging after a negative 3-month posttreatment scan, suggest results of a retrospective cohort study reported at the Multidisciplinary Head and Neck Cancer Symposium.
Investigators led by Dr. Jessica M. Frakes of the H. Lee Moffitt Cancer Center in Tampa, Fla., studied 246 patients treated nonsurgically between 2006 and 2014 for nonmetastatic HPV-positive disease.

With a median follow-up of 36 months, all local recurrences and the large majority of regional and distant recurrences were detected from symptoms, physical exam, and the 3-month posttreatment PET-CT imaging, according to data reported in a session and related press briefing.
“Routine imaging is not recommended after posttreatment imaging shows a complete response to treatment, unless the patient presents with symptoms or something else that would warrant imaging,” Dr. Frakes commented. “Follow-up should include history and physical examination with direct visualization.”
Currently, the National Comprehensive Cancer Network advises a one-size-fits-all approach to follow-up that does not consider tumor HPV status, she noted. But reducing surveillance imaging for HPV-positive patients would likely have considerable benefit in terms of less stress and anxiety (provided patients are educated about the safety of clinical follow-up) and lower financial burden for both the patient and the health care system as a whole.
Results additionally showed that the majority of recurrences occurred within the first 6 months, a pattern that was consistent whether or not patients had risk factors for recurrence. The 3-year rate of freedom from local failure exceeded 97%, and only 2% of patients had grade 3 or worse toxicity at their last follow-up.
“Our outcomes are excellent with low rates of permanent toxicity, and we think that this partly is due to the fact that they are treated by specialized multidisciplinary team,” Dr. Frakes commented at the meeting.
Press briefing moderator Dr. Christine Gourin of Johns Hopkins University in Baltimore, commented, “This study is one that’s dear to my heart because I think that we probably do too much posttreatment surveillance, and they are exactly right that the NCCN is fairly vague about when to perform imaging.”
“I can tell you that we have stopped routinely imaging patients after 3 months if the PET is negative, and it’s true that we do pick up recurrences more clinically than we do radiologically,” she added. “And of course the false positives are causing much morbidity.”
Introducing the study, Dr. Frakes commented, “Several retrospective and prospective trials have shown increased survivals and decreased toxicity in patients with HPV-associated oropharynx cancer. As the number of patients and survivors grows, so does the need to determine general time to recurrence and the most effective modes of recurrence detection, thereby guiding our standards for optimal follow-up care.”
All patients studied received definitive radiation therapy, and 85% of them also received concurrent chemotherapy.
The patients had a 3-month posttreatment PET-CT scan, plus physical exams every 3 months in the first year post treatment, every 4 months in the second year, every 6 months in the third through fifth years, and annually thereafter.
Results showed that the 3-year rate of local control was 97.8%, and 100% of the local failures were detected by physical exam, including direct visualization or flexible laryngoscopy.
The rate of regional control was 95.3%, and 89% of cases of regional failure were detected through symptoms or the 3-month posttreatment imaging. Risk factors for regional recurrence included involvement of five or more lymph nodes in the neck and involvement of level 4 (low neck) lymph nodes (P less than .05 for each).
The rate of freedom from distant metastases was 91%, and 71% of cases of distant metastases were detected through symptoms or the 3-month posttreatment imaging. Risk factors for distant metastases included tumor in the lymph nodes measuring greater than 6 cm, involvement of bilateral lymph nodes, involvement of five or more lymph nodes in the neck, and involvement of level 4 lymph nodes (P less than .05 for each).
Overall, 9% of patients experienced grade 3 or worse late toxicity (occurring at 3 months or thereafter), consisting of feeding/gastrostomy tube (G-tube) placement, necrosis or ulcer, and tracheostomy. However, these toxicities had resolved as of the last follow-up in most cases, with a final rate of toxicity of only 2%.
The center follows an aggressive approach to preventing and managing toxicity, noted Dr. Frakes.
“Even when the patients have their G-tube in place, we really do encourage p.o. [oral] intake as much as possible with pain medication. And I think that really does make a big difference for our patients,” she said. “They do meet with a speech pathologist and our nutritionist weekly when they are on treatment.” Patients are also allowed to have the G-tube removed by last follow-up, she added.
SCOTTSDALE, ARIZ. – Most patients who are treated for human papillomavirus (HPV)–positive oropharyngeal cancer can safely skip routine imaging after a negative 3-month posttreatment scan, suggest results of a retrospective cohort study reported at the Multidisciplinary Head and Neck Cancer Symposium.
Investigators led by Dr. Jessica M. Frakes of the H. Lee Moffitt Cancer Center in Tampa, Fla., studied 246 patients treated nonsurgically between 2006 and 2014 for nonmetastatic HPV-positive disease.
With a median follow-up of 36 months, all local recurrences and the large majority of regional and distant recurrences were detected from symptoms, physical exam, and the 3-month posttreatment PET-CT imaging, according to data reported in a session and related press briefing.
“Routine imaging is not recommended after posttreatment imaging shows a complete response to treatment, unless the patient presents with symptoms or something else that would warrant imaging,” Dr. Frakes commented. “Follow-up should include history and physical examination with direct visualization.”
Currently, the National Comprehensive Cancer Network advises a one-size-fits-all approach to follow-up that does not consider tumor HPV status, she noted. But reducing surveillance imaging for HPV-positive patients would likely have considerable benefit in terms of less stress and anxiety (provided patients are educated about the safety of clinical follow-up) and lower financial burden for both the patient and the health care system as a whole.
Results additionally showed that the majority of recurrences occurred within the first 6 months, a pattern that was consistent whether or not patients had risk factors for recurrence. The 3-year rate of freedom from local failure exceeded 97%, and only 2% of patients had grade 3 or worse toxicity at their last follow-up.
“Our outcomes are excellent with low rates of permanent toxicity, and we think that this partly is due to the fact that they are treated by specialized multidisciplinary team,” Dr. Frakes commented at the meeting.
Press briefing moderator Dr. Christine Gourin of Johns Hopkins University in Baltimore, commented, “This study is one that’s dear to my heart because I think that we probably do too much posttreatment surveillance, and they are exactly right that the NCCN is fairly vague about when to perform imaging.”
“I can tell you that we have stopped routinely imaging patients after 3 months if the PET is negative, and it’s true that we do pick up recurrences more clinically than we do radiologically,” she added. “And of course the false positives are causing much morbidity.”
Introducing the study, Dr. Frakes commented, “Several retrospective and prospective trials have shown increased survivals and decreased toxicity in patients with HPV-associated oropharynx cancer. As the number of patients and survivors grows, so does the need to determine general time to recurrence and the most effective modes of recurrence detection, thereby guiding our standards for optimal follow-up care.”
All patients studied received definitive radiation therapy, and 85% of them also received concurrent chemotherapy.
The patients had a 3-month posttreatment PET-CT scan, plus physical exams every 3 months in the first year post treatment, every 4 months in the second year, every 6 months in the third through fifth years, and annually thereafter.
Results showed that the 3-year rate of local control was 97.8%, and 100% of the local failures were detected by physical exam, including direct visualization or flexible laryngoscopy.
The rate of regional control was 95.3%, and 89% of cases of regional failure were detected through symptoms or the 3-month posttreatment imaging. Risk factors for regional recurrence included involvement of five or more lymph nodes in the neck and involvement of level 4 (low neck) lymph nodes (P less than .05 for each).
The rate of freedom from distant metastases was 91%, and 71% of cases of distant metastases were detected through symptoms or the 3-month posttreatment imaging. Risk factors for distant metastases included tumor in the lymph nodes measuring greater than 6 cm, involvement of bilateral lymph nodes, involvement of five or more lymph nodes in the neck, and involvement of level 4 lymph nodes (P less than .05 for each).
Overall, 9% of patients experienced grade 3 or worse late toxicity (occurring at 3 months or thereafter), consisting of feeding/gastrostomy tube (G-tube) placement, necrosis or ulcer, and tracheostomy. However, these toxicities had resolved as of the last follow-up in most cases, with a final rate of toxicity of only 2%.
The center follows an aggressive approach to preventing and managing toxicity, noted Dr. Frakes.
“Even when the patients have their G-tube in place, we really do encourage p.o. [oral] intake as much as possible with pain medication. And I think that really does make a big difference for our patients,” she said. “They do meet with a speech pathologist and our nutritionist weekly when they are on treatment.” Patients are also allowed to have the G-tube removed by last follow-up, she added.
SCOTTSDALE, ARIZ. – Most patients who are treated for human papillomavirus (HPV)–positive oropharyngeal cancer can safely skip routine imaging after a negative 3-month posttreatment scan, suggest results of a retrospective cohort study reported at the Multidisciplinary Head and Neck Cancer Symposium.
Investigators led by Dr. Jessica M. Frakes of the H. Lee Moffitt Cancer Center in Tampa, Fla., studied 246 patients treated nonsurgically between 2006 and 2014 for nonmetastatic HPV-positive disease.
With a median follow-up of 36 months, all local recurrences and the large majority of regional and distant recurrences were detected from symptoms, physical exam, and the 3-month posttreatment PET-CT imaging, according to data reported in a session and related press briefing.
“Routine imaging is not recommended after posttreatment imaging shows a complete response to treatment, unless the patient presents with symptoms or something else that would warrant imaging,” Dr. Frakes commented. “Follow-up should include history and physical examination with direct visualization.”
Currently, the National Comprehensive Cancer Network advises a one-size-fits-all approach to follow-up that does not consider tumor HPV status, she noted. But reducing surveillance imaging for HPV-positive patients would likely have considerable benefit in terms of less stress and anxiety (provided patients are educated about the safety of clinical follow-up) and lower financial burden for both the patient and the health care system as a whole.
Results additionally showed that the majority of recurrences occurred within the first 6 months, a pattern that was consistent whether or not patients had risk factors for recurrence. The 3-year rate of freedom from local failure exceeded 97%, and only 2% of patients had grade 3 or worse toxicity at their last follow-up.
“Our outcomes are excellent with low rates of permanent toxicity, and we think that this partly is due to the fact that they are treated by specialized multidisciplinary team,” Dr. Frakes commented at the meeting.
Press briefing moderator Dr. Christine Gourin of Johns Hopkins University in Baltimore, commented, “This study is one that’s dear to my heart because I think that we probably do too much posttreatment surveillance, and they are exactly right that the NCCN is fairly vague about when to perform imaging.”
“I can tell you that we have stopped routinely imaging patients after 3 months if the PET is negative, and it’s true that we do pick up recurrences more clinically than we do radiologically,” she added. “And of course the false positives are causing much morbidity.”
Introducing the study, Dr. Frakes commented, “Several retrospective and prospective trials have shown increased survivals and decreased toxicity in patients with HPV-associated oropharynx cancer. As the number of patients and survivors grows, so does the need to determine general time to recurrence and the most effective modes of recurrence detection, thereby guiding our standards for optimal follow-up care.”
All patients studied received definitive radiation therapy, and 85% of them also received concurrent chemotherapy.
The patients had a 3-month posttreatment PET-CT scan, plus physical exams every 3 months in the first year post treatment, every 4 months in the second year, every 6 months in the third through fifth years, and annually thereafter.
Results showed that the 3-year rate of local control was 97.8%, and 100% of the local failures were detected by physical exam, including direct visualization or flexible laryngoscopy.
The rate of regional control was 95.3%, and 89% of cases of regional failure were detected through symptoms or the 3-month posttreatment imaging. Risk factors for regional recurrence included involvement of five or more lymph nodes in the neck and involvement of level 4 (low neck) lymph nodes (P less than .05 for each).
The rate of freedom from distant metastases was 91%, and 71% of cases of distant metastases were detected through symptoms or the 3-month posttreatment imaging. Risk factors for distant metastases included tumor in the lymph nodes measuring greater than 6 cm, involvement of bilateral lymph nodes, involvement of five or more lymph nodes in the neck, and involvement of level 4 lymph nodes (P less than .05 for each).
Overall, 9% of patients experienced grade 3 or worse late toxicity (occurring at 3 months or thereafter), consisting of feeding/gastrostomy tube (G-tube) placement, necrosis or ulcer, and tracheostomy. However, these toxicities had resolved as of the last follow-up in most cases, with a final rate of toxicity of only 2%.
The center follows an aggressive approach to preventing and managing toxicity, noted Dr. Frakes.
“Even when the patients have their G-tube in place, we really do encourage p.o. [oral] intake as much as possible with pain medication. And I think that really does make a big difference for our patients,” she said. “They do meet with a speech pathologist and our nutritionist weekly when they are on treatment.” Patients are also allowed to have the G-tube removed by last follow-up, she added.
AT THE HEAD AND NECK CANCER SYMPOSIUM
Key clinical point: Most patients with HPV-positive oropharyngeal cancer do not need routine imaging after a negative 3-month posttreatment scan.
Major finding: Overall, 100%, 89%, and 71% of local, regional, and distant recurrences, respectively, were detected from symptoms, physical exam, and 3-month posttreatment imaging.
Data source: A retrospective cohort study of 246 patients treated for HPV-positive oropharyngeal cancer.
Disclosures: Dr. Frakes disclosed that she had no relevant conflicts of interest.

Childhood maltreatment tied to lifetime anxiety disorders in bipolar
Childhood maltreatment is associated with lifetime anxiety among people with bipolar disorder, Barbara Pavlova, Ph.D., and her associates reported.
The researchers recruited 174 adult outpatients with a diagnosis of bipolar disorder I or bipolar disorder II, of whom 29% had one anxiety disorder and 20% had two or more. More than half (56%) of the patients were female, and their median age was 42. The types of anxiety disorders among the patients ranged from generalized anxiety disorder (28%) to obsessive-compulsive disorder (4%).

Dr. Pavlova and her associates assessed the patients’ history of maltreatment in childhood using the Childhood Trauma Questionnaire (CTQ), a 28-item self-report measure that asks about emotional, physical, and sexual abuse and about emotional and physical neglect. Anxiety disorders were assessed using the Mini-International Neuropsychiatric Interview (MINI), wrote Dr. Pavlova of the psychiatry department at Dalhousie University, Halifax, N.S.
They found that childhood maltreatment, indexed by higher CTQ total scores, was linked to a higher number of lifetime anxiety disorders (odds ratio, 1.5; 95% confidence interval, 1.01-2.14; P = .04). In addition, panic disorder was most strongly tied to childhood maltreatment (OR, 2.27; 95% CI, 1.28-4.02; P = .01).
The results suggest “that bipolar disorder with comorbid anxiety constitutes an [etiologic] subtype shaped to a greater extent by early environment,” the investigators wrote.
Read the full study here: (J Affect Dis. 2016 Mar 1;192:22-7).
On Twittter @ginalhenderson
Childhood maltreatment is associated with lifetime anxiety among people with bipolar disorder, Barbara Pavlova, Ph.D., and her associates reported.
The researchers recruited 174 adult outpatients with a diagnosis of bipolar disorder I or bipolar disorder II, of whom 29% had one anxiety disorder and 20% had two or more. More than half (56%) of the patients were female, and their median age was 42. The types of anxiety disorders among the patients ranged from generalized anxiety disorder (28%) to obsessive-compulsive disorder (4%).
Dr. Pavlova and her associates assessed the patients’ history of maltreatment in childhood using the Childhood Trauma Questionnaire (CTQ), a 28-item self-report measure that asks about emotional, physical, and sexual abuse and about emotional and physical neglect. Anxiety disorders were assessed using the Mini-International Neuropsychiatric Interview (MINI), wrote Dr. Pavlova of the psychiatry department at Dalhousie University, Halifax, N.S.
They found that childhood maltreatment, indexed by higher CTQ total scores, was linked to a higher number of lifetime anxiety disorders (odds ratio, 1.5; 95% confidence interval, 1.01-2.14; P = .04). In addition, panic disorder was most strongly tied to childhood maltreatment (OR, 2.27; 95% CI, 1.28-4.02; P = .01).
The results suggest “that bipolar disorder with comorbid anxiety constitutes an [etiologic] subtype shaped to a greater extent by early environment,” the investigators wrote.
Read the full study here: (J Affect Dis. 2016 Mar 1;192:22-7).
On Twittter @ginalhenderson
Childhood maltreatment is associated with lifetime anxiety among people with bipolar disorder, Barbara Pavlova, Ph.D., and her associates reported.
The researchers recruited 174 adult outpatients with a diagnosis of bipolar disorder I or bipolar disorder II, of whom 29% had one anxiety disorder and 20% had two or more. More than half (56%) of the patients were female, and their median age was 42. The types of anxiety disorders among the patients ranged from generalized anxiety disorder (28%) to obsessive-compulsive disorder (4%).
Dr. Pavlova and her associates assessed the patients’ history of maltreatment in childhood using the Childhood Trauma Questionnaire (CTQ), a 28-item self-report measure that asks about emotional, physical, and sexual abuse and about emotional and physical neglect. Anxiety disorders were assessed using the Mini-International Neuropsychiatric Interview (MINI), wrote Dr. Pavlova of the psychiatry department at Dalhousie University, Halifax, N.S.
They found that childhood maltreatment, indexed by higher CTQ total scores, was linked to a higher number of lifetime anxiety disorders (odds ratio, 1.5; 95% confidence interval, 1.01-2.14; P = .04). In addition, panic disorder was most strongly tied to childhood maltreatment (OR, 2.27; 95% CI, 1.28-4.02; P = .01).
The results suggest “that bipolar disorder with comorbid anxiety constitutes an [etiologic] subtype shaped to a greater extent by early environment,” the investigators wrote.
Read the full study here: (J Affect Dis. 2016 Mar 1;192:22-7).
On Twittter @ginalhenderson
FROM THE JOURNAL OF AFFECTIVE DISORDERS

What Matters: What’s the magic behind successful bariatric patients?
A fair number of my patients have had or are undergoing bariatric surgery. Disconcertingly, a not insignificant number of them are regaining the weight after surgery. Weight regain will occur in 20% of patients undergoing bariatric surgery after initial weight loss.
When this occurs, not only do we have a patient with an altered gut putting them at risk for nutritional deficiencies if we are not fastidious in our follow-up, but they are discouraged and overweight again.

Add this to the concern that bariatric surgery has been associated with an increase in suicides (2.33-3.63 per 1000 patient-years), and we may have some cause for alarm.
So, what predicts success – and can we facilitate it?
Several factors have been shown to predict successful weight loss after bariatric surgery. An “active coping style” (that is, planning vs. denial) and adherence to follow-up after bariatric surgery have both been shown to be associated with a higher percentage of excess weight loss. Interestingly, psychological burden and motivation have not been associated with weight loss.
In a recent article, Lori Liebl, Ph.D., and her colleagues conducted a qualitative study of the experiences of adults who successfully maintained weight loss after bariatric surgery (J Clin Nurs. 2016 Feb 23. doi: 10.1111/jocn.13129). Success was defined as 50% or more of the excessive weight loss 24 months after bariatric surgery.
The voice of the successful bariatric patient is an interesting and important one. Several themes were identified: 1) taking life back (“I did it for myself”); 2) a new lease on life (“There are things I can do now that I am not exhausted”); 3) the importance of social support; 4) avoiding the negative (terminating unhealthy relationships in which “food is love”); 5) the void (food addiction and sense of loss); 6) fighting food demons; 7) finding the happy weight; and 8) a ripple effect (that is, if you don’t eat it, the rest of family doesn’t, either).
I was left wondering how I can best help my patients using this information.
First, I think the themes can mature our empathy for the struggles that these patients face, and perhaps help us combat bias. Second, I think this knowledge can inform early discussions around what sorts of things need to be lined up for after the procedure, such as social support.
Finally, I think the themes can be universalized and help us counsel patients who may be struggling with weight, but who are otherwise not candidates for bariatric surgery.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article.
A fair number of my patients have had or are undergoing bariatric surgery. Disconcertingly, a not insignificant number of them are regaining the weight after surgery. Weight regain will occur in 20% of patients undergoing bariatric surgery after initial weight loss.
When this occurs, not only do we have a patient with an altered gut putting them at risk for nutritional deficiencies if we are not fastidious in our follow-up, but they are discouraged and overweight again.
Add this to the concern that bariatric surgery has been associated with an increase in suicides (2.33-3.63 per 1000 patient-years), and we may have some cause for alarm.
So, what predicts success – and can we facilitate it?
Several factors have been shown to predict successful weight loss after bariatric surgery. An “active coping style” (that is, planning vs. denial) and adherence to follow-up after bariatric surgery have both been shown to be associated with a higher percentage of excess weight loss. Interestingly, psychological burden and motivation have not been associated with weight loss.
In a recent article, Lori Liebl, Ph.D., and her colleagues conducted a qualitative study of the experiences of adults who successfully maintained weight loss after bariatric surgery (J Clin Nurs. 2016 Feb 23. doi: 10.1111/jocn.13129). Success was defined as 50% or more of the excessive weight loss 24 months after bariatric surgery.
The voice of the successful bariatric patient is an interesting and important one. Several themes were identified: 1) taking life back (“I did it for myself”); 2) a new lease on life (“There are things I can do now that I am not exhausted”); 3) the importance of social support; 4) avoiding the negative (terminating unhealthy relationships in which “food is love”); 5) the void (food addiction and sense of loss); 6) fighting food demons; 7) finding the happy weight; and 8) a ripple effect (that is, if you don’t eat it, the rest of family doesn’t, either).
I was left wondering how I can best help my patients using this information.
First, I think the themes can mature our empathy for the struggles that these patients face, and perhaps help us combat bias. Second, I think this knowledge can inform early discussions around what sorts of things need to be lined up for after the procedure, such as social support.
Finally, I think the themes can be universalized and help us counsel patients who may be struggling with weight, but who are otherwise not candidates for bariatric surgery.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article.
A fair number of my patients have had or are undergoing bariatric surgery. Disconcertingly, a not insignificant number of them are regaining the weight after surgery. Weight regain will occur in 20% of patients undergoing bariatric surgery after initial weight loss.
When this occurs, not only do we have a patient with an altered gut putting them at risk for nutritional deficiencies if we are not fastidious in our follow-up, but they are discouraged and overweight again.
Add this to the concern that bariatric surgery has been associated with an increase in suicides (2.33-3.63 per 1000 patient-years), and we may have some cause for alarm.
So, what predicts success – and can we facilitate it?
Several factors have been shown to predict successful weight loss after bariatric surgery. An “active coping style” (that is, planning vs. denial) and adherence to follow-up after bariatric surgery have both been shown to be associated with a higher percentage of excess weight loss. Interestingly, psychological burden and motivation have not been associated with weight loss.
In a recent article, Lori Liebl, Ph.D., and her colleagues conducted a qualitative study of the experiences of adults who successfully maintained weight loss after bariatric surgery (J Clin Nurs. 2016 Feb 23. doi: 10.1111/jocn.13129). Success was defined as 50% or more of the excessive weight loss 24 months after bariatric surgery.
The voice of the successful bariatric patient is an interesting and important one. Several themes were identified: 1) taking life back (“I did it for myself”); 2) a new lease on life (“There are things I can do now that I am not exhausted”); 3) the importance of social support; 4) avoiding the negative (terminating unhealthy relationships in which “food is love”); 5) the void (food addiction and sense of loss); 6) fighting food demons; 7) finding the happy weight; and 8) a ripple effect (that is, if you don’t eat it, the rest of family doesn’t, either).
I was left wondering how I can best help my patients using this information.
First, I think the themes can mature our empathy for the struggles that these patients face, and perhaps help us combat bias. Second, I think this knowledge can inform early discussions around what sorts of things need to be lined up for after the procedure, such as social support.
Finally, I think the themes can be universalized and help us counsel patients who may be struggling with weight, but who are otherwise not candidates for bariatric surgery.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article.

$1.8 Billion Wasted With Single Dose Chemotherapy Vials
The use of single dose vials for chemotherapy leads to significant wastage and unnecessary expense, according to a study published in BMJ. Researchers, led by Dr. Peter B. Bach, director of the Center for Health Policy and Outcomes at Memorial Sloan Kettering Cancer Center, examined the top 20 cancer drugs based on sales projections for 2016 that are packaged in single dose vials and dosed by body size, which often results in leftover and unused medication. According to the authors, as much as 10% of the drugs were not used. However, hospital systems, including the VA and the DoD, pay for the entire dose, “making wasted drug a source of unnecessary spending,” the researchers note. The authors estimate the cost for this waste could reach $1.8 billion.
Related:FDA Approves Rescue Drug for Chemotherapy Overdose
Currently, safety standards from the U.S. Pharmacopeial Convention only permit sharing if a leftover drug is used within 6 hours, and only in specialized pharmacies. “Policy makers should also revisit the current FDA guidance on the appropriate packaging of infused drugs in single dose vials and encourage the FDA, Centers for Drug Control and Prevention, Centers for Medicare and Medicaid Services, and US Pharmacopeial Convention to reconcile their views on vial contents and vial sharing,” the authors urged. “Such steps could lead to savings for our health care system without sacrificing health outcomes. Opportunities to eradicate waste of this kind are rare.”.
Related: DoD Proposed 2017 Budget Include Cost Hikes For Military Retirees
To measure the waste the researchers estimated Medicare claims records to determine how frequently vial sharing occurred. They then calculated the most efficient way to combine available vial sizes to achieve the lowest FDA-approved dose in a representative sample of the US population derived from the National Health and Nutrition Examination Survey with adjustments for the cancer patient population.
Related: The Cost of Oncology Drugs: A Pharmacy Perspective, Part I
To reduce waste, the authors suggest that manufacturers be required to package drugs in quantities that allow better matching with required doses or enable virtual return of leftover drug.
The use of single dose vials for chemotherapy leads to significant wastage and unnecessary expense, according to a study published in BMJ. Researchers, led by Dr. Peter B. Bach, director of the Center for Health Policy and Outcomes at Memorial Sloan Kettering Cancer Center, examined the top 20 cancer drugs based on sales projections for 2016 that are packaged in single dose vials and dosed by body size, which often results in leftover and unused medication. According to the authors, as much as 10% of the drugs were not used. However, hospital systems, including the VA and the DoD, pay for the entire dose, “making wasted drug a source of unnecessary spending,” the researchers note. The authors estimate the cost for this waste could reach $1.8 billion.
Related:FDA Approves Rescue Drug for Chemotherapy Overdose
Currently, safety standards from the U.S. Pharmacopeial Convention only permit sharing if a leftover drug is used within 6 hours, and only in specialized pharmacies. “Policy makers should also revisit the current FDA guidance on the appropriate packaging of infused drugs in single dose vials and encourage the FDA, Centers for Drug Control and Prevention, Centers for Medicare and Medicaid Services, and US Pharmacopeial Convention to reconcile their views on vial contents and vial sharing,” the authors urged. “Such steps could lead to savings for our health care system without sacrificing health outcomes. Opportunities to eradicate waste of this kind are rare.”.
Related: DoD Proposed 2017 Budget Include Cost Hikes For Military Retirees
To measure the waste the researchers estimated Medicare claims records to determine how frequently vial sharing occurred. They then calculated the most efficient way to combine available vial sizes to achieve the lowest FDA-approved dose in a representative sample of the US population derived from the National Health and Nutrition Examination Survey with adjustments for the cancer patient population.
Related: The Cost of Oncology Drugs: A Pharmacy Perspective, Part I
To reduce waste, the authors suggest that manufacturers be required to package drugs in quantities that allow better matching with required doses or enable virtual return of leftover drug.
The use of single dose vials for chemotherapy leads to significant wastage and unnecessary expense, according to a study published in BMJ. Researchers, led by Dr. Peter B. Bach, director of the Center for Health Policy and Outcomes at Memorial Sloan Kettering Cancer Center, examined the top 20 cancer drugs based on sales projections for 2016 that are packaged in single dose vials and dosed by body size, which often results in leftover and unused medication. According to the authors, as much as 10% of the drugs were not used. However, hospital systems, including the VA and the DoD, pay for the entire dose, “making wasted drug a source of unnecessary spending,” the researchers note. The authors estimate the cost for this waste could reach $1.8 billion.
Related:FDA Approves Rescue Drug for Chemotherapy Overdose
Currently, safety standards from the U.S. Pharmacopeial Convention only permit sharing if a leftover drug is used within 6 hours, and only in specialized pharmacies. “Policy makers should also revisit the current FDA guidance on the appropriate packaging of infused drugs in single dose vials and encourage the FDA, Centers for Drug Control and Prevention, Centers for Medicare and Medicaid Services, and US Pharmacopeial Convention to reconcile their views on vial contents and vial sharing,” the authors urged. “Such steps could lead to savings for our health care system without sacrificing health outcomes. Opportunities to eradicate waste of this kind are rare.”.
Related: DoD Proposed 2017 Budget Include Cost Hikes For Military Retirees
To measure the waste the researchers estimated Medicare claims records to determine how frequently vial sharing occurred. They then calculated the most efficient way to combine available vial sizes to achieve the lowest FDA-approved dose in a representative sample of the US population derived from the National Health and Nutrition Examination Survey with adjustments for the cancer patient population.
Related: The Cost of Oncology Drugs: A Pharmacy Perspective, Part I
To reduce waste, the authors suggest that manufacturers be required to package drugs in quantities that allow better matching with required doses or enable virtual return of leftover drug.

Intranasal Drug Delivery Bypasses the Blood–Brain Barrier
LAS VEGAS—The nasal mucosa in the upper third of the nasal cavity provides a direct pathway from the external environment to the brain and, according to William H. Frey II, PhD, that pathway can be used to noninvasively deliver therapeutics into the brain. This pathway effectively bypasses the blood–brain barrier and avoids the systemic exposure and side effects associated with therapeutics that enter the bloodstream. At the 19th Annual Meeting of the North American Neuromodulation Society, Dr. Frey presented an in-depth look at intranasal delivery of therapeutics to the brain.

“We have learned from experience that therapeutics sprayed into the nose or even given as nose drops can travel extracellularly and paracellularly along the olfactory axon bundles and along the trigeminal nerve pathway from the nose to the brain,” said Dr. Frey, who is Founder and Codirector of the Alzheimer’s Research Center at Regions Hospital and Senior Director of HealthPartners Neuroscience Research in St. Paul.
Therapeutics that can be delivered intranasally include proteins like insulin, small molecules, charged molecules, oligonucleotides, therapeutic cells like stem cells and Treg cells, nanoparticles, and microparticles. “You do not have to modify your drug or therapeutic in any way in order to do this, but this method only works for really potent therapeutics that are active in the picomolar, nanomolar, or very low micromolar concentration range,” Dr. Frey said.
This technique is being investigated in various disorders. “Most of the studies have been done in animal models, but the Alzheimer’s work has also been done in humans,” Dr. Frey said.
The Neuroanatomy of Intranasal Delivery
The cribriform plate of the skull separates the upper part of the nasal cavity from the brain. The primary olfactory nerves are located in the roof of the nasal cavity under the cribriform plate and include the olfactory sensory neurons and odorant receptors. Sniffing brings molecules into the nose, thus allowing them to bind to odorant receptors and send a signal. Intranasal delivery of therapeutics involves spraying therapeutics into the upper part of the nasal cavity to enable them to follow these olfactory axon bundles directly into the brain through foramena in the cribriform plate. Once across the cribriform plate, the therapeutics penetrate the subarachnoid space and enter the perivascular spaces of the brain’s blood vessels.
When the heart pumps, a corresponding pulsation in the cerebrovasculature creates a perivascular pumping mechanism that moves the therapeutics throughout the brain. “They are near the blood vessels, but on the brain side of the blood–brain barrier throughout the brain,” Dr. Frey explained. Drugs also follow the trigeminal nerves that innervate the entire nasal mucosa and follow the trigeminal neural pathway through the trigeminal ganglion and into the brain and upper spinal cord.
“[This method] results in rapid delivery—within 10 minutes in mice, rats, and monkeys—to the brain and upper spinal cord,” Dr. Frey said. In humans, intranasal neuropeptides reach the CSF within 10 minutes.
Stroke
Preclinical studies have examined intranasal therapy for stroke. Researchers gave rats a stroke by occluding the middle cerebral artery. Two hours of occlusion were followed by reperfusion. Ten minutes after the reperfusion was initiated, investigators administered nose drops containing insulin-like growth factor 1—a 7,600-Da neurotrophic protein naturally found in humans. Compared with controls, rats that received 150 mg of this peptide intranasally had an infarct volume or amount of brain damage that was reduced by 63%. Benefit was also seen when treatment was delayed for two or four hours.
Brain Tumors
A different intranasal treatment uses GRN163, a polynucleotide that inhibits the enzyme telomerase. Telomerase is expressed highly in brain tumors and is required for the brain tumor cells to keep dividing. Investigators tagged the negatively charged large polynucleotide with a fluorescent label and administered it. They observed that GRN163 accumulated in the brain tumor over a period of four hours but did not accumulate in the normal brain. After 24 hours, GRN163 was completely cleared from the brain. Survival time was doubled following treatment with the intranasal polynucleotide.
Neurodegenerative Disease
Iron accumulates abnormally in the brain in all of the neurodegenerative disorders. “Obviously, our bodies need iron, but the abnormal accumulation of free iron is damaging because it is a strong promoter of free-radical damage,” Dr. Frey said. Data also indicate that the key receptor for memory, the human brain muscarinic cholinergic receptor, is rapidly inactivated by free iron or free heme, which are present at increased levels in the brains of people with Alzheimer’s disease. “We have a potent iron chelator, deferoxamine mesylate, that has been around for about 40 years. It has a high affinity for iron and it is a generic drug. It has been used to treat beta thalassemia, sickle cell anemia, and various conditions where too much iron is accumulated in the blood. When given intramuscularly over a period of two years to patients with Alzheimer’s disease, it reduced their cognitive decline by 50%. That’s a far bigger benefit than any drug on the market today for Alzheimer’s disease,” Dr. Frey noted. But there were significant side effects at the injection site, and deferoxamine does not cross the blood–brain barrier well. “Consequently, we’ve been developing and have patented intranasal deferoxamine to treat Parkinson’s disease, Alzheimer’s disease, stroke, and traumatic brain injury,” Dr. Frey said.
“We’ve shown that intranasal deferoxamine protects dopamine brain cells and improves movement in animals with Parkinson’s disease… We’ve shown that just a few nose drops of deferoxamine given before or after a stroke reduce brain damage in rats by 55%. We’ve shown that even in normal mice, it improves memory when given intranasally. And it also improves or reduces memory loss in Alzheimer transgenic mice.”
Alzheimer’s Disease
Fludeoxyglucose (18F) PET scans reveal adequate uptake and utilization of glucose, the main energy source for brain cells, in the brains of healthy elderly controls. But the brains of patients with Alzheimer’s disease do not take up glucose normally, and their brain cells consequently have less energy. “A number of areas of the brain require insulin to take up glucose, and the hippocampus is one of those areas,” Dr. Frey explained. “Insulin signaling is reduced in the brains of patients with Alzheimer’s disease, causing what some have called type 3 diabetes, or diabetes of the brain, which leaves these brain cells starved for energy and not able to function normally.”
Dr. Frey obtained several patents on the direct intranasal delivery of therapeutics, including insulin, to the brain, and various clinical trials have been conducted. “Four trials in patients with Alzheimer’s disease and five trials in normal, healthy adults have demonstrated improved memory following intranasal insulin treatment, with no change in the blood levels of insulin or glucose,” Dr. Frey reported.
In one of the first trials, a single intranasal insulin treatment improved verbal memory for individuals with Alzheimer’s disease within 15 minutes. In a three-week trial, intranasal insulin enhanced memory, compared with placebo, and significantly improved attention and functional status in patients with Alzheimer’s disease. However, patients who carried the APOE ε4 gene allele were not improved with intranasal regular insulin. “Only long-acting insulin detemir, given intranasally, has been shown to improve memory in patients who have the APOE ε4 gene allele,”Dr. Frey said.
The longest completed trial lasted for four months and showed improved memory and function in patients who were given insulin twice per day in a nasal spray. It also showed that the treatment reduced the loss of glucose uptake and utilization in key brain regions, as seen in PET scans. A new six-month treatment trial is now underway at the HealthPartners Center for Memory & Aging in St. Paul.
Mechanism of Action
One open question is whether intranasal insulin only provides symptomatic treatment (ie, improved memory and functioning in patients with Alzheimer’s disease) or also has the potential to change the underlying disease process. “We know it can provide energy to prevent brain cells from degenerating and allow the cells to produce materials to replace worn-out parts,” said Dr. Frey. “That result has been shown in humans using P-31 MRI. After administration of intranasal insulin, levels of brain cell adenosine triphosphate (ATP) and brain cell phosphocreatine increase significantly. We know that insulin, after it causes signaling, and glucose uptake occurs, causes the production of insulin-degrading enzyme to reduce the insulin signal so that the next time the signal comes in, it can be easily detected. That turns out to be the enzyme that degrades beta amyloid, which accumulates abnormally in the brains of individuals with Alzheimer disease. So, if you don’t have insulin signaling, you don’t make insulin-degrading enzyme, and you accumulate beta amyloid. Insulin also inhibits glycogen synthase kinase 3 beta that phosphorylates tau to form Alzheimer neurofibrillary tangles. Insulin is also needed to maintain synaptic density, so it is possible that if humans were given intranasal insulin at the first sign of an insulin-signaling deficiency or a decrease in glucose uptake in the brain, it might be possible to delay, or maybe even prevent, the onset of this disease,” Dr. Frey said.
Stem Cells
Dr. Frey and research collaborators in Germany, including Lusine Danielyan MD, discovered and patented that intranasal stem cells bypass the blood–brain barrier to reach the brain and treat Parkinson’s disease in rats. Adult bone marrow–derived stem cells have anti-inflammatory and neurotrophic properties. “After cell treatment, the proinflammatory cytokines in this inflammatory brain disease go down to normal levels. Our study showed highly significant improvement, compared with placebo, in motor function or movement,” Dr. Frey said.
Researchers in the Netherlands demonstrated that intranasal adult stem cells treat neonatal ischemia and neonatal brain damage. Other researchers at Emory University reported treatment of stroke with adult stem cells from bone marrow. Swedish researchers reported that intranasal Treg cells treat multiple sclerosis (MS). “These studies are all in animals,” Dr. Frey noted. Other researchers reported that neuronal stem cells induce recovery and remyelination in an animal model of MS. Brain tumors have also been treated in animals with intranasal stem cells. Recently, intranasal stem cell therapy was also reported to improve motor function and reduce lesion size in spinal cord injury in animals. “Noninvasive intranasal delivery can target therapeutics to the brain while reducing systemic exposure to facilitate the treatment of brain disorders,” said Dr. Frey.
—Glenn S. Williams
Suggested Reading
Danielyan L, Schäfer R, von Ameln-Mayerhofer A, et al. Therapeutic efficacy of intranasally delivered mesenchymal stem cells in a rat model of Parkinson’s disease. Rejuvenation Res. 2011;14(1):3-16.
Danielyan L, Beer-Hammer S, Stolzing A, et al. Intranasal delivery of bone marrow-derived mesenchymal stem cells, macrophages, and microglia to the brain in mouse models of Alzheimer’s and Parkinson’s disease. Cell Transplant. 2014;23(suppl 1):S123-S139.
Frey WH 2nd. Noninvasive intranasal stem cells bypass the blood-brain barrier to target the brain to treat Parkinson’s disease, stroke, MS, brain tumors, cerebral ischemia, Alzheimer’s and other CNS disorders. J Nat Sci. 2015;1(1):e23.
Lochhead JJ, Wolak DJ, Pizzo ME, Thorne RG. Rapid transport within cerebral perivascular spaces underlies widespread tracer distribution in the brain after intranasal administration. J Cereb Blood Flow Metab. 2015;35(3):371-381.
LAS VEGAS—The nasal mucosa in the upper third of the nasal cavity provides a direct pathway from the external environment to the brain and, according to William H. Frey II, PhD, that pathway can be used to noninvasively deliver therapeutics into the brain. This pathway effectively bypasses the blood–brain barrier and avoids the systemic exposure and side effects associated with therapeutics that enter the bloodstream. At the 19th Annual Meeting of the North American Neuromodulation Society, Dr. Frey presented an in-depth look at intranasal delivery of therapeutics to the brain.
“We have learned from experience that therapeutics sprayed into the nose or even given as nose drops can travel extracellularly and paracellularly along the olfactory axon bundles and along the trigeminal nerve pathway from the nose to the brain,” said Dr. Frey, who is Founder and Codirector of the Alzheimer’s Research Center at Regions Hospital and Senior Director of HealthPartners Neuroscience Research in St. Paul.
Therapeutics that can be delivered intranasally include proteins like insulin, small molecules, charged molecules, oligonucleotides, therapeutic cells like stem cells and Treg cells, nanoparticles, and microparticles. “You do not have to modify your drug or therapeutic in any way in order to do this, but this method only works for really potent therapeutics that are active in the picomolar, nanomolar, or very low micromolar concentration range,” Dr. Frey said.
This technique is being investigated in various disorders. “Most of the studies have been done in animal models, but the Alzheimer’s work has also been done in humans,” Dr. Frey said.
The Neuroanatomy of Intranasal Delivery
The cribriform plate of the skull separates the upper part of the nasal cavity from the brain. The primary olfactory nerves are located in the roof of the nasal cavity under the cribriform plate and include the olfactory sensory neurons and odorant receptors. Sniffing brings molecules into the nose, thus allowing them to bind to odorant receptors and send a signal. Intranasal delivery of therapeutics involves spraying therapeutics into the upper part of the nasal cavity to enable them to follow these olfactory axon bundles directly into the brain through foramena in the cribriform plate. Once across the cribriform plate, the therapeutics penetrate the subarachnoid space and enter the perivascular spaces of the brain’s blood vessels.
When the heart pumps, a corresponding pulsation in the cerebrovasculature creates a perivascular pumping mechanism that moves the therapeutics throughout the brain. “They are near the blood vessels, but on the brain side of the blood–brain barrier throughout the brain,” Dr. Frey explained. Drugs also follow the trigeminal nerves that innervate the entire nasal mucosa and follow the trigeminal neural pathway through the trigeminal ganglion and into the brain and upper spinal cord.
“[This method] results in rapid delivery—within 10 minutes in mice, rats, and monkeys—to the brain and upper spinal cord,” Dr. Frey said. In humans, intranasal neuropeptides reach the CSF within 10 minutes.
Stroke
Preclinical studies have examined intranasal therapy for stroke. Researchers gave rats a stroke by occluding the middle cerebral artery. Two hours of occlusion were followed by reperfusion. Ten minutes after the reperfusion was initiated, investigators administered nose drops containing insulin-like growth factor 1—a 7,600-Da neurotrophic protein naturally found in humans. Compared with controls, rats that received 150 mg of this peptide intranasally had an infarct volume or amount of brain damage that was reduced by 63%. Benefit was also seen when treatment was delayed for two or four hours.
Brain Tumors
A different intranasal treatment uses GRN163, a polynucleotide that inhibits the enzyme telomerase. Telomerase is expressed highly in brain tumors and is required for the brain tumor cells to keep dividing. Investigators tagged the negatively charged large polynucleotide with a fluorescent label and administered it. They observed that GRN163 accumulated in the brain tumor over a period of four hours but did not accumulate in the normal brain. After 24 hours, GRN163 was completely cleared from the brain. Survival time was doubled following treatment with the intranasal polynucleotide.
Neurodegenerative Disease
Iron accumulates abnormally in the brain in all of the neurodegenerative disorders. “Obviously, our bodies need iron, but the abnormal accumulation of free iron is damaging because it is a strong promoter of free-radical damage,” Dr. Frey said. Data also indicate that the key receptor for memory, the human brain muscarinic cholinergic receptor, is rapidly inactivated by free iron or free heme, which are present at increased levels in the brains of people with Alzheimer’s disease. “We have a potent iron chelator, deferoxamine mesylate, that has been around for about 40 years. It has a high affinity for iron and it is a generic drug. It has been used to treat beta thalassemia, sickle cell anemia, and various conditions where too much iron is accumulated in the blood. When given intramuscularly over a period of two years to patients with Alzheimer’s disease, it reduced their cognitive decline by 50%. That’s a far bigger benefit than any drug on the market today for Alzheimer’s disease,” Dr. Frey noted. But there were significant side effects at the injection site, and deferoxamine does not cross the blood–brain barrier well. “Consequently, we’ve been developing and have patented intranasal deferoxamine to treat Parkinson’s disease, Alzheimer’s disease, stroke, and traumatic brain injury,” Dr. Frey said.
“We’ve shown that intranasal deferoxamine protects dopamine brain cells and improves movement in animals with Parkinson’s disease… We’ve shown that just a few nose drops of deferoxamine given before or after a stroke reduce brain damage in rats by 55%. We’ve shown that even in normal mice, it improves memory when given intranasally. And it also improves or reduces memory loss in Alzheimer transgenic mice.”
Alzheimer’s Disease
Fludeoxyglucose (18F) PET scans reveal adequate uptake and utilization of glucose, the main energy source for brain cells, in the brains of healthy elderly controls. But the brains of patients with Alzheimer’s disease do not take up glucose normally, and their brain cells consequently have less energy. “A number of areas of the brain require insulin to take up glucose, and the hippocampus is one of those areas,” Dr. Frey explained. “Insulin signaling is reduced in the brains of patients with Alzheimer’s disease, causing what some have called type 3 diabetes, or diabetes of the brain, which leaves these brain cells starved for energy and not able to function normally.”
Dr. Frey obtained several patents on the direct intranasal delivery of therapeutics, including insulin, to the brain, and various clinical trials have been conducted. “Four trials in patients with Alzheimer’s disease and five trials in normal, healthy adults have demonstrated improved memory following intranasal insulin treatment, with no change in the blood levels of insulin or glucose,” Dr. Frey reported.
In one of the first trials, a single intranasal insulin treatment improved verbal memory for individuals with Alzheimer’s disease within 15 minutes. In a three-week trial, intranasal insulin enhanced memory, compared with placebo, and significantly improved attention and functional status in patients with Alzheimer’s disease. However, patients who carried the APOE ε4 gene allele were not improved with intranasal regular insulin. “Only long-acting insulin detemir, given intranasally, has been shown to improve memory in patients who have the APOE ε4 gene allele,”Dr. Frey said.
The longest completed trial lasted for four months and showed improved memory and function in patients who were given insulin twice per day in a nasal spray. It also showed that the treatment reduced the loss of glucose uptake and utilization in key brain regions, as seen in PET scans. A new six-month treatment trial is now underway at the HealthPartners Center for Memory & Aging in St. Paul.
Mechanism of Action
One open question is whether intranasal insulin only provides symptomatic treatment (ie, improved memory and functioning in patients with Alzheimer’s disease) or also has the potential to change the underlying disease process. “We know it can provide energy to prevent brain cells from degenerating and allow the cells to produce materials to replace worn-out parts,” said Dr. Frey. “That result has been shown in humans using P-31 MRI. After administration of intranasal insulin, levels of brain cell adenosine triphosphate (ATP) and brain cell phosphocreatine increase significantly. We know that insulin, after it causes signaling, and glucose uptake occurs, causes the production of insulin-degrading enzyme to reduce the insulin signal so that the next time the signal comes in, it can be easily detected. That turns out to be the enzyme that degrades beta amyloid, which accumulates abnormally in the brains of individuals with Alzheimer disease. So, if you don’t have insulin signaling, you don’t make insulin-degrading enzyme, and you accumulate beta amyloid. Insulin also inhibits glycogen synthase kinase 3 beta that phosphorylates tau to form Alzheimer neurofibrillary tangles. Insulin is also needed to maintain synaptic density, so it is possible that if humans were given intranasal insulin at the first sign of an insulin-signaling deficiency or a decrease in glucose uptake in the brain, it might be possible to delay, or maybe even prevent, the onset of this disease,” Dr. Frey said.
Stem Cells
Dr. Frey and research collaborators in Germany, including Lusine Danielyan MD, discovered and patented that intranasal stem cells bypass the blood–brain barrier to reach the brain and treat Parkinson’s disease in rats. Adult bone marrow–derived stem cells have anti-inflammatory and neurotrophic properties. “After cell treatment, the proinflammatory cytokines in this inflammatory brain disease go down to normal levels. Our study showed highly significant improvement, compared with placebo, in motor function or movement,” Dr. Frey said.
Researchers in the Netherlands demonstrated that intranasal adult stem cells treat neonatal ischemia and neonatal brain damage. Other researchers at Emory University reported treatment of stroke with adult stem cells from bone marrow. Swedish researchers reported that intranasal Treg cells treat multiple sclerosis (MS). “These studies are all in animals,” Dr. Frey noted. Other researchers reported that neuronal stem cells induce recovery and remyelination in an animal model of MS. Brain tumors have also been treated in animals with intranasal stem cells. Recently, intranasal stem cell therapy was also reported to improve motor function and reduce lesion size in spinal cord injury in animals. “Noninvasive intranasal delivery can target therapeutics to the brain while reducing systemic exposure to facilitate the treatment of brain disorders,” said Dr. Frey.
—Glenn S. Williams
LAS VEGAS—The nasal mucosa in the upper third of the nasal cavity provides a direct pathway from the external environment to the brain and, according to William H. Frey II, PhD, that pathway can be used to noninvasively deliver therapeutics into the brain. This pathway effectively bypasses the blood–brain barrier and avoids the systemic exposure and side effects associated with therapeutics that enter the bloodstream. At the 19th Annual Meeting of the North American Neuromodulation Society, Dr. Frey presented an in-depth look at intranasal delivery of therapeutics to the brain.
“We have learned from experience that therapeutics sprayed into the nose or even given as nose drops can travel extracellularly and paracellularly along the olfactory axon bundles and along the trigeminal nerve pathway from the nose to the brain,” said Dr. Frey, who is Founder and Codirector of the Alzheimer’s Research Center at Regions Hospital and Senior Director of HealthPartners Neuroscience Research in St. Paul.
Therapeutics that can be delivered intranasally include proteins like insulin, small molecules, charged molecules, oligonucleotides, therapeutic cells like stem cells and Treg cells, nanoparticles, and microparticles. “You do not have to modify your drug or therapeutic in any way in order to do this, but this method only works for really potent therapeutics that are active in the picomolar, nanomolar, or very low micromolar concentration range,” Dr. Frey said.
This technique is being investigated in various disorders. “Most of the studies have been done in animal models, but the Alzheimer’s work has also been done in humans,” Dr. Frey said.
The Neuroanatomy of Intranasal Delivery
The cribriform plate of the skull separates the upper part of the nasal cavity from the brain. The primary olfactory nerves are located in the roof of the nasal cavity under the cribriform plate and include the olfactory sensory neurons and odorant receptors. Sniffing brings molecules into the nose, thus allowing them to bind to odorant receptors and send a signal. Intranasal delivery of therapeutics involves spraying therapeutics into the upper part of the nasal cavity to enable them to follow these olfactory axon bundles directly into the brain through foramena in the cribriform plate. Once across the cribriform plate, the therapeutics penetrate the subarachnoid space and enter the perivascular spaces of the brain’s blood vessels.
When the heart pumps, a corresponding pulsation in the cerebrovasculature creates a perivascular pumping mechanism that moves the therapeutics throughout the brain. “They are near the blood vessels, but on the brain side of the blood–brain barrier throughout the brain,” Dr. Frey explained. Drugs also follow the trigeminal nerves that innervate the entire nasal mucosa and follow the trigeminal neural pathway through the trigeminal ganglion and into the brain and upper spinal cord.
“[This method] results in rapid delivery—within 10 minutes in mice, rats, and monkeys—to the brain and upper spinal cord,” Dr. Frey said. In humans, intranasal neuropeptides reach the CSF within 10 minutes.
Stroke
Preclinical studies have examined intranasal therapy for stroke. Researchers gave rats a stroke by occluding the middle cerebral artery. Two hours of occlusion were followed by reperfusion. Ten minutes after the reperfusion was initiated, investigators administered nose drops containing insulin-like growth factor 1—a 7,600-Da neurotrophic protein naturally found in humans. Compared with controls, rats that received 150 mg of this peptide intranasally had an infarct volume or amount of brain damage that was reduced by 63%. Benefit was also seen when treatment was delayed for two or four hours.
Brain Tumors
A different intranasal treatment uses GRN163, a polynucleotide that inhibits the enzyme telomerase. Telomerase is expressed highly in brain tumors and is required for the brain tumor cells to keep dividing. Investigators tagged the negatively charged large polynucleotide with a fluorescent label and administered it. They observed that GRN163 accumulated in the brain tumor over a period of four hours but did not accumulate in the normal brain. After 24 hours, GRN163 was completely cleared from the brain. Survival time was doubled following treatment with the intranasal polynucleotide.
Neurodegenerative Disease
Iron accumulates abnormally in the brain in all of the neurodegenerative disorders. “Obviously, our bodies need iron, but the abnormal accumulation of free iron is damaging because it is a strong promoter of free-radical damage,” Dr. Frey said. Data also indicate that the key receptor for memory, the human brain muscarinic cholinergic receptor, is rapidly inactivated by free iron or free heme, which are present at increased levels in the brains of people with Alzheimer’s disease. “We have a potent iron chelator, deferoxamine mesylate, that has been around for about 40 years. It has a high affinity for iron and it is a generic drug. It has been used to treat beta thalassemia, sickle cell anemia, and various conditions where too much iron is accumulated in the blood. When given intramuscularly over a period of two years to patients with Alzheimer’s disease, it reduced their cognitive decline by 50%. That’s a far bigger benefit than any drug on the market today for Alzheimer’s disease,” Dr. Frey noted. But there were significant side effects at the injection site, and deferoxamine does not cross the blood–brain barrier well. “Consequently, we’ve been developing and have patented intranasal deferoxamine to treat Parkinson’s disease, Alzheimer’s disease, stroke, and traumatic brain injury,” Dr. Frey said.
“We’ve shown that intranasal deferoxamine protects dopamine brain cells and improves movement in animals with Parkinson’s disease… We’ve shown that just a few nose drops of deferoxamine given before or after a stroke reduce brain damage in rats by 55%. We’ve shown that even in normal mice, it improves memory when given intranasally. And it also improves or reduces memory loss in Alzheimer transgenic mice.”
Alzheimer’s Disease
Fludeoxyglucose (18F) PET scans reveal adequate uptake and utilization of glucose, the main energy source for brain cells, in the brains of healthy elderly controls. But the brains of patients with Alzheimer’s disease do not take up glucose normally, and their brain cells consequently have less energy. “A number of areas of the brain require insulin to take up glucose, and the hippocampus is one of those areas,” Dr. Frey explained. “Insulin signaling is reduced in the brains of patients with Alzheimer’s disease, causing what some have called type 3 diabetes, or diabetes of the brain, which leaves these brain cells starved for energy and not able to function normally.”
Dr. Frey obtained several patents on the direct intranasal delivery of therapeutics, including insulin, to the brain, and various clinical trials have been conducted. “Four trials in patients with Alzheimer’s disease and five trials in normal, healthy adults have demonstrated improved memory following intranasal insulin treatment, with no change in the blood levels of insulin or glucose,” Dr. Frey reported.
In one of the first trials, a single intranasal insulin treatment improved verbal memory for individuals with Alzheimer’s disease within 15 minutes. In a three-week trial, intranasal insulin enhanced memory, compared with placebo, and significantly improved attention and functional status in patients with Alzheimer’s disease. However, patients who carried the APOE ε4 gene allele were not improved with intranasal regular insulin. “Only long-acting insulin detemir, given intranasally, has been shown to improve memory in patients who have the APOE ε4 gene allele,”Dr. Frey said.
The longest completed trial lasted for four months and showed improved memory and function in patients who were given insulin twice per day in a nasal spray. It also showed that the treatment reduced the loss of glucose uptake and utilization in key brain regions, as seen in PET scans. A new six-month treatment trial is now underway at the HealthPartners Center for Memory & Aging in St. Paul.
Mechanism of Action
One open question is whether intranasal insulin only provides symptomatic treatment (ie, improved memory and functioning in patients with Alzheimer’s disease) or also has the potential to change the underlying disease process. “We know it can provide energy to prevent brain cells from degenerating and allow the cells to produce materials to replace worn-out parts,” said Dr. Frey. “That result has been shown in humans using P-31 MRI. After administration of intranasal insulin, levels of brain cell adenosine triphosphate (ATP) and brain cell phosphocreatine increase significantly. We know that insulin, after it causes signaling, and glucose uptake occurs, causes the production of insulin-degrading enzyme to reduce the insulin signal so that the next time the signal comes in, it can be easily detected. That turns out to be the enzyme that degrades beta amyloid, which accumulates abnormally in the brains of individuals with Alzheimer disease. So, if you don’t have insulin signaling, you don’t make insulin-degrading enzyme, and you accumulate beta amyloid. Insulin also inhibits glycogen synthase kinase 3 beta that phosphorylates tau to form Alzheimer neurofibrillary tangles. Insulin is also needed to maintain synaptic density, so it is possible that if humans were given intranasal insulin at the first sign of an insulin-signaling deficiency or a decrease in glucose uptake in the brain, it might be possible to delay, or maybe even prevent, the onset of this disease,” Dr. Frey said.
Stem Cells
Dr. Frey and research collaborators in Germany, including Lusine Danielyan MD, discovered and patented that intranasal stem cells bypass the blood–brain barrier to reach the brain and treat Parkinson’s disease in rats. Adult bone marrow–derived stem cells have anti-inflammatory and neurotrophic properties. “After cell treatment, the proinflammatory cytokines in this inflammatory brain disease go down to normal levels. Our study showed highly significant improvement, compared with placebo, in motor function or movement,” Dr. Frey said.
Researchers in the Netherlands demonstrated that intranasal adult stem cells treat neonatal ischemia and neonatal brain damage. Other researchers at Emory University reported treatment of stroke with adult stem cells from bone marrow. Swedish researchers reported that intranasal Treg cells treat multiple sclerosis (MS). “These studies are all in animals,” Dr. Frey noted. Other researchers reported that neuronal stem cells induce recovery and remyelination in an animal model of MS. Brain tumors have also been treated in animals with intranasal stem cells. Recently, intranasal stem cell therapy was also reported to improve motor function and reduce lesion size in spinal cord injury in animals. “Noninvasive intranasal delivery can target therapeutics to the brain while reducing systemic exposure to facilitate the treatment of brain disorders,” said Dr. Frey.
—Glenn S. Williams
Suggested Reading
Danielyan L, Schäfer R, von Ameln-Mayerhofer A, et al. Therapeutic efficacy of intranasally delivered mesenchymal stem cells in a rat model of Parkinson’s disease. Rejuvenation Res. 2011;14(1):3-16.
Danielyan L, Beer-Hammer S, Stolzing A, et al. Intranasal delivery of bone marrow-derived mesenchymal stem cells, macrophages, and microglia to the brain in mouse models of Alzheimer’s and Parkinson’s disease. Cell Transplant. 2014;23(suppl 1):S123-S139.
Frey WH 2nd. Noninvasive intranasal stem cells bypass the blood-brain barrier to target the brain to treat Parkinson’s disease, stroke, MS, brain tumors, cerebral ischemia, Alzheimer’s and other CNS disorders. J Nat Sci. 2015;1(1):e23.
Lochhead JJ, Wolak DJ, Pizzo ME, Thorne RG. Rapid transport within cerebral perivascular spaces underlies widespread tracer distribution in the brain after intranasal administration. J Cereb Blood Flow Metab. 2015;35(3):371-381.
Suggested Reading
Danielyan L, Schäfer R, von Ameln-Mayerhofer A, et al. Therapeutic efficacy of intranasally delivered mesenchymal stem cells in a rat model of Parkinson’s disease. Rejuvenation Res. 2011;14(1):3-16.
Danielyan L, Beer-Hammer S, Stolzing A, et al. Intranasal delivery of bone marrow-derived mesenchymal stem cells, macrophages, and microglia to the brain in mouse models of Alzheimer’s and Parkinson’s disease. Cell Transplant. 2014;23(suppl 1):S123-S139.
Frey WH 2nd. Noninvasive intranasal stem cells bypass the blood-brain barrier to target the brain to treat Parkinson’s disease, stroke, MS, brain tumors, cerebral ischemia, Alzheimer’s and other CNS disorders. J Nat Sci. 2015;1(1):e23.
Lochhead JJ, Wolak DJ, Pizzo ME, Thorne RG. Rapid transport within cerebral perivascular spaces underlies widespread tracer distribution in the brain after intranasal administration. J Cereb Blood Flow Metab. 2015;35(3):371-381.
