User login
Brown Macule on the Waist
The best diagnosis is:
a. granular cell tumor
b. intradermal nevus
c. Langerhans cell disease
d. mastocytosis
e. multicentric reticulohistiocytosis
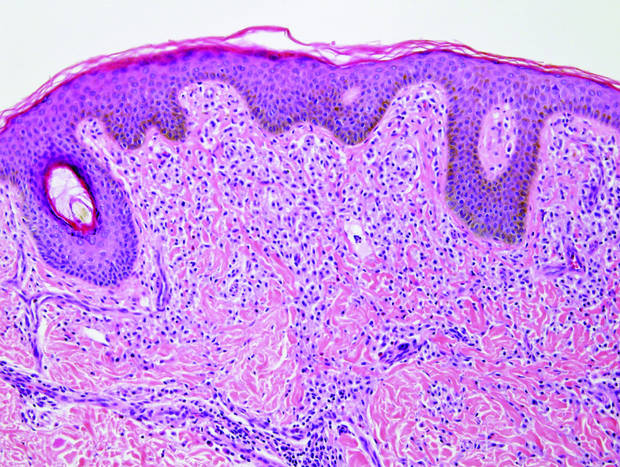
|
| Monomorphic cell infiltrate in the upper dermis (H&E, original magnification ×100). |
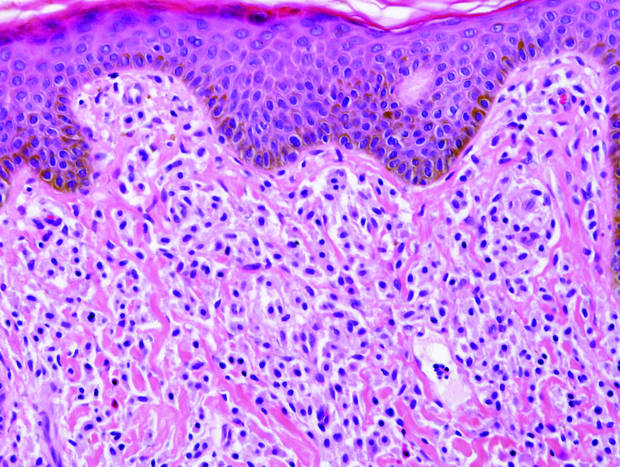
|
| A closer view reveals cuboidal or spindle cells with basal hyperpigmentation (H&E, original magnification ×200). |
Continue to the next page for the diagnosis >>
Mastocytosis
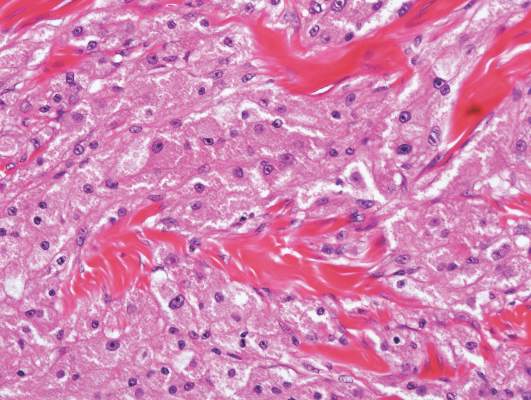
Mastocytosis is a clonal proliferation of mast cells in the skin and various systems of the body including the bone marrow, liver, lymph nodes, and gastrointestinal tract.1,2 Mast cell proliferation is closely associated with germline and acquired activating KIT mutations.3-5 Adult-onset mastocytosis is likely to involve several organs, whereas pediatric mastocytosis usually affects only the skin and is self-limiting. Patients with profound mast cell infiltration in the skin or other organs are likely to have attacks of flushing, palpitation, or diarrhea resulting from the degranulation of mast cells and release of histamine.6,7 In a majority of patients with advanced systemic mastocytosis, mast cells are positive for the Ki-1 antigen (CD30), whereas in most patients with indolent systemic mastocytosis, only a few mast cells are positive for CD30.8 Recently, CD30 was reported as a new drug target in patients with CD30+ advanced systemic mastocytosis.9 Because the skin frequently is involved and easily accessible in comparison with other organs, skin biopsy often is performed to establish a diagnosis of mastocytosis. Cutaneous mastocytosis comprises urticaria pigmentosa, solitary mastocytoma, diffuse cutaneous mastocytosis, and telangiectasia macularis eruptiva perstans; approximately 80% of all cases have urticaria pigmentosa.10-12 In cutaneous mastocytosis, skin biopsy typically shows monomorphous mast cell infiltrate mostly in the upper third of the dermis. The density of mast cells varies according to the clinical variant. For example, a lesion of telangiectasia macularis eruptiva perstans has only a perivascular mast cell infiltrate, whereas a solitary mastocytoma has sheets of mast cells in the dermis, sometimes extending into the subcutis. A skin biopsy of the brown macule on the waist showed a number of cuboidal or spindle mast cells in the upper dermis with occasional eosinophils. These mast cells are monomorphous, and no mitotic figures, necrotic cells, or atypical cells are seen. Mast cells have metachromatic granules in the cytoplasm, which can be seen with toluidine blue or Giemsa stain. CD117 (c-kit) also is positive. Mast cells in urticaria pigmentosa easily may be mistaken for nevus cells. Hyperpigmentation of the basal layer, a characteristic feature seen in urticaria pigmentosa, also may erroneously suggest a diagnosis of a melanocytic nevus.
Granular cell tumors predominantly affect the oral cavity, but the skin also can be involved. It comprises a fascicular infiltrate of large and polygonal cells with characteristic eosinophilic granular cytoplasm in the dermis (Figure 1).13 Cell membranes are not always distinct. Although the nuclei usually are small and centrally located, irregular and plump nuclei with distinct nucleoli also may be seen. The overlying epidermis tends to be hyperplastic. Granular cell tumor is considered a group of lesions of varying histogenesis. Cases in which tumors originated from a neural crest–derived peripheral nerve–related cell as well as a Schwann cell have been reported.14,15 The origin of granular cell tumors is controversial.
|
| Figure 1. Granular cell tumor showing fascicles of large and polygonal cells with characteristic eosinophilic granular cytoplasm in the dermis (H&E, original magnification ×200). |

|
| Figure 2. Intradermal nevus showing nests with melanin in the uppermost area of the lesion and neurotized nevus cells in the lower part (H&E, original magnification ×100). Pseudovascular spaces are seen on the right side. |
Intradermal nevus usually has nests and cords of nevus cells in the upper dermis. The uppermost melanocytes often contain a moderate amount of melanin, whereas nevus cells in the mid and lower dermis usually do not contain melanin (Figure 2). Shrinkage during tissue processing maycause clefts between nevus cells, resulting in pseudovascular spaces.16 The deeper dermis may have a neuroid appearance with spindle-shaped cells and Meissner corpuscle–like structures.17
Although Langerhans cell disease was formerly known as Langerhans cell histiocytosis and subdivided into several clinical subtypes, including Letterer-Siwe disease, Hand-Schüller-Christian disease, and eosinophilic granuloma, these clinical subtypes commonly overlapped. Langerhans cell disease is now used as a terminology that encompasses all subtypes.18,19 Langerhans cell disease is characterized by a proliferation of Langerhans cells with a variable mixture of other inflammatory cells. The constituent cells are large and ovoid with a distinct folded or lobulated, often kidney-shaped nucleus.20 Langerhans cells usually infiltrate the upper dermis and occasionally the epidermis (Figure 3). CD1a, HLA-DR, S-100 protein, and langerin are positive in Langerhans cells.21

|
| Figure 3. Langerhans cell disease showing an infiltrate of large and ovoid Langerhans cells with a distinct folded or lobulated, often kidney-shaped nucleus in the upper dermis and epidermis (H&E, original magnification ×200). |

|
| Figure 4. Multicentric reticulohistiocytosis showing a mixture of mononuclear and multinucleate histiocytes with abundant eosinophilic and finely granular cytoplasm (H&E, original magnification ×200). |
Multicentric reticulohistiocytosis is characterized by a combination of papulonodular cutaneous lesions and severe arthropathy.22 An irregular mixture of mononuclear and multinucleate histiocytes showing abundant eosinophilic and finely granular cytoplasm, often with a ground-glass appearance, is seen along with lymphocytic infiltration (Figure 4).23 A few giant cells may be seen in early lesions; older lesions more commonly have giant cells and fibrosis.
1. Arock M, Valent P. Pathogenesis, classification and treatment of mastocytosis: state of the art in 2010 and future perspectives. Expert Rev Hematol. 2010;3:497-516.
2. Pardanani A. Systemic mastocytosis in adults: 2013 update on diagnosis, risk stratification, and management. Am J Hematol. 2013;88:612-624.
3. Orfao A, Garcia-Montero AC, Sanchez L, et al. Recent advances in the understanding of mastocytosis: the role of KIT mutations. Br J Haematol. 2007;138:12-30.
4. Yanagihori H, Oyama N, Nakamura K, et al. c-KIT mutations in patients with childhood-onset mastocytosis and genotype-phenotype correlation. J Mol Diagn. 2005;7:252-257.
5. Bodemer C, Hermine O, Palmérini F, et al. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol. 2010;130:804-815.
6. Kettelhut BV, Metcalfe DD. Pediatric mastocytosis. Ann Allergy. 1994;73:197-202; quiz 202-207.
7. Longley J, Duffy TP, Kohn S. The mast cell and mast cell disease. J Am Acad Dermatol. 1995;32:545-561; quiz 562-564.
8. Sotlar K, Cerny-Reiterer S, Petat-Dutter K, et al. Aberrant expression of CD30 in neoplastic mast cells in high-grade mastocytosis. Mod Pathol. 2011;24:585-595.
9. Blatt K, Cerny-Reiterer S, Schwaab J, et al. Identification of the Ki-1 antigen (CD30) as a novel therapeutic target in systemic mastocytosis [published online October 20, 2015]. Blood. 2015;126:2832-2841.
10. Kiszewski AE, Duran-Mckinster C, Orozco-Covarrubias L, et al. Cutaneous mastocytosis in children: a clinical analysis of 71 cases. J Eur Acad Dermatol Venereol. 2004;18:285-290.
11. Akoglu G, Erkin G, Cakir B, et al. Cutaneous mastocytosis: demographic aspects and clinical features of 55 patients. J Eur Acad Dermatol Venereol. 2006;20:969-973.
12. Sarkany RP, Monk BE, Handfield-Jones SE. Telangiectasia macularis eruptiva perstans: a case report and review of the literature. Clin Exp Dermatol. 1998;23:38-39.
13. Lack EE, Worsham GF, Callihan MD, et al. Granular cell tumor: a clinicopathologic study of 110 patients. J Surg Oncol. 1980;13:301-316.
14. Buley ID, Gatter KC, Kelly PM, et al. Granular cell tumours revisited. an immunohistological and ultrastructural study. Histopathology. 1988;12:263-274.
15. Penneys NS, Adachi K, Ziegels-Weissman J, et al. Granular cell tumors of the skin contain myelin basic protein. Arch Pathol Lab Med. 1983;107:302-303.
16. Modlin RL, Gottlieb B, Taylor C, et al. Identification of cells lining pseudovascular spaces of benign pigmented nevi. Am J Dermatopathol. 1984;(suppl 6):25-29.
17. Fullen DR, Reed JA, Finnerty B, et al. S100A6 preferentially labels type C nevus cells and nevic corpuscles: additional support for Schwannian differentiation of intradermal nevi. J Cutan Pathol. 2001;28:393-399.
18. Newman B, Hu W, Nigro K, et al. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol. 2007;56:302-316.
19. Weedon D. Cutaneous infiltrates—non-lymphoid. In: Weedon D, ed. Weedon’s Skin Pathology. 3rd ed. Amsterdam, Netherlands: Elsevier; 2010:937-970.
20. Harrist TJ, Bhan AK, Murphy GF, et al. Histiocytosis-X: in situ characterization of cutaneous infiltrates with monoclonal antibodies. Am J Clin Pathol. 1983;79:294-300.
21. Lau SK, Chu PG, Weiss LM. Immunohistochemical expression of langerin in Langerhans cell histiocytosis and non-Langerhans cell histiocytic disorders. Am J Surg Pathol. 2008;32:615-619.
22. Lesher JL Jr, Allen BS. Multicentric reticulohistiocytosis. J Am Acad Dermatol. 1984;11:713-723.
23. Heathcote JG, Guenther LC, Wallace AC. Multicentric reticulohistiocytosis: a report of a case and a review of the pathology. Pathology. 1985;17:601-608.
The best diagnosis is:
a. granular cell tumor
b. intradermal nevus
c. Langerhans cell disease
d. mastocytosis
e. multicentric reticulohistiocytosis
|
|
| Monomorphic cell infiltrate in the upper dermis (H&E, original magnification ×100). |
|
|
| A closer view reveals cuboidal or spindle cells with basal hyperpigmentation (H&E, original magnification ×200). |
Continue to the next page for the diagnosis >>
Mastocytosis
Mastocytosis is a clonal proliferation of mast cells in the skin and various systems of the body including the bone marrow, liver, lymph nodes, and gastrointestinal tract.1,2 Mast cell proliferation is closely associated with germline and acquired activating KIT mutations.3-5 Adult-onset mastocytosis is likely to involve several organs, whereas pediatric mastocytosis usually affects only the skin and is self-limiting. Patients with profound mast cell infiltration in the skin or other organs are likely to have attacks of flushing, palpitation, or diarrhea resulting from the degranulation of mast cells and release of histamine.6,7 In a majority of patients with advanced systemic mastocytosis, mast cells are positive for the Ki-1 antigen (CD30), whereas in most patients with indolent systemic mastocytosis, only a few mast cells are positive for CD30.8 Recently, CD30 was reported as a new drug target in patients with CD30+ advanced systemic mastocytosis.9 Because the skin frequently is involved and easily accessible in comparison with other organs, skin biopsy often is performed to establish a diagnosis of mastocytosis. Cutaneous mastocytosis comprises urticaria pigmentosa, solitary mastocytoma, diffuse cutaneous mastocytosis, and telangiectasia macularis eruptiva perstans; approximately 80% of all cases have urticaria pigmentosa.10-12 In cutaneous mastocytosis, skin biopsy typically shows monomorphous mast cell infiltrate mostly in the upper third of the dermis. The density of mast cells varies according to the clinical variant. For example, a lesion of telangiectasia macularis eruptiva perstans has only a perivascular mast cell infiltrate, whereas a solitary mastocytoma has sheets of mast cells in the dermis, sometimes extending into the subcutis. A skin biopsy of the brown macule on the waist showed a number of cuboidal or spindle mast cells in the upper dermis with occasional eosinophils. These mast cells are monomorphous, and no mitotic figures, necrotic cells, or atypical cells are seen. Mast cells have metachromatic granules in the cytoplasm, which can be seen with toluidine blue or Giemsa stain. CD117 (c-kit) also is positive. Mast cells in urticaria pigmentosa easily may be mistaken for nevus cells. Hyperpigmentation of the basal layer, a characteristic feature seen in urticaria pigmentosa, also may erroneously suggest a diagnosis of a melanocytic nevus.
Granular cell tumors predominantly affect the oral cavity, but the skin also can be involved. It comprises a fascicular infiltrate of large and polygonal cells with characteristic eosinophilic granular cytoplasm in the dermis (Figure 1).13 Cell membranes are not always distinct. Although the nuclei usually are small and centrally located, irregular and plump nuclei with distinct nucleoli also may be seen. The overlying epidermis tends to be hyperplastic. Granular cell tumor is considered a group of lesions of varying histogenesis. Cases in which tumors originated from a neural crest–derived peripheral nerve–related cell as well as a Schwann cell have been reported.14,15 The origin of granular cell tumors is controversial.
|
|
| Figure 1. Granular cell tumor showing fascicles of large and polygonal cells with characteristic eosinophilic granular cytoplasm in the dermis (H&E, original magnification ×200). |
|
|
| Figure 2. Intradermal nevus showing nests with melanin in the uppermost area of the lesion and neurotized nevus cells in the lower part (H&E, original magnification ×100). Pseudovascular spaces are seen on the right side. |
Intradermal nevus usually has nests and cords of nevus cells in the upper dermis. The uppermost melanocytes often contain a moderate amount of melanin, whereas nevus cells in the mid and lower dermis usually do not contain melanin (Figure 2). Shrinkage during tissue processing maycause clefts between nevus cells, resulting in pseudovascular spaces.16 The deeper dermis may have a neuroid appearance with spindle-shaped cells and Meissner corpuscle–like structures.17
Although Langerhans cell disease was formerly known as Langerhans cell histiocytosis and subdivided into several clinical subtypes, including Letterer-Siwe disease, Hand-Schüller-Christian disease, and eosinophilic granuloma, these clinical subtypes commonly overlapped. Langerhans cell disease is now used as a terminology that encompasses all subtypes.18,19 Langerhans cell disease is characterized by a proliferation of Langerhans cells with a variable mixture of other inflammatory cells. The constituent cells are large and ovoid with a distinct folded or lobulated, often kidney-shaped nucleus.20 Langerhans cells usually infiltrate the upper dermis and occasionally the epidermis (Figure 3). CD1a, HLA-DR, S-100 protein, and langerin are positive in Langerhans cells.21
|
|
| Figure 3. Langerhans cell disease showing an infiltrate of large and ovoid Langerhans cells with a distinct folded or lobulated, often kidney-shaped nucleus in the upper dermis and epidermis (H&E, original magnification ×200). |
|
|
| Figure 4. Multicentric reticulohistiocytosis showing a mixture of mononuclear and multinucleate histiocytes with abundant eosinophilic and finely granular cytoplasm (H&E, original magnification ×200). |
Multicentric reticulohistiocytosis is characterized by a combination of papulonodular cutaneous lesions and severe arthropathy.22 An irregular mixture of mononuclear and multinucleate histiocytes showing abundant eosinophilic and finely granular cytoplasm, often with a ground-glass appearance, is seen along with lymphocytic infiltration (Figure 4).23 A few giant cells may be seen in early lesions; older lesions more commonly have giant cells and fibrosis.
The best diagnosis is:
a. granular cell tumor
b. intradermal nevus
c. Langerhans cell disease
d. mastocytosis
e. multicentric reticulohistiocytosis
|
|
| Monomorphic cell infiltrate in the upper dermis (H&E, original magnification ×100). |
|
|
| A closer view reveals cuboidal or spindle cells with basal hyperpigmentation (H&E, original magnification ×200). |
Continue to the next page for the diagnosis >>
Mastocytosis
Mastocytosis is a clonal proliferation of mast cells in the skin and various systems of the body including the bone marrow, liver, lymph nodes, and gastrointestinal tract.1,2 Mast cell proliferation is closely associated with germline and acquired activating KIT mutations.3-5 Adult-onset mastocytosis is likely to involve several organs, whereas pediatric mastocytosis usually affects only the skin and is self-limiting. Patients with profound mast cell infiltration in the skin or other organs are likely to have attacks of flushing, palpitation, or diarrhea resulting from the degranulation of mast cells and release of histamine.6,7 In a majority of patients with advanced systemic mastocytosis, mast cells are positive for the Ki-1 antigen (CD30), whereas in most patients with indolent systemic mastocytosis, only a few mast cells are positive for CD30.8 Recently, CD30 was reported as a new drug target in patients with CD30+ advanced systemic mastocytosis.9 Because the skin frequently is involved and easily accessible in comparison with other organs, skin biopsy often is performed to establish a diagnosis of mastocytosis. Cutaneous mastocytosis comprises urticaria pigmentosa, solitary mastocytoma, diffuse cutaneous mastocytosis, and telangiectasia macularis eruptiva perstans; approximately 80% of all cases have urticaria pigmentosa.10-12 In cutaneous mastocytosis, skin biopsy typically shows monomorphous mast cell infiltrate mostly in the upper third of the dermis. The density of mast cells varies according to the clinical variant. For example, a lesion of telangiectasia macularis eruptiva perstans has only a perivascular mast cell infiltrate, whereas a solitary mastocytoma has sheets of mast cells in the dermis, sometimes extending into the subcutis. A skin biopsy of the brown macule on the waist showed a number of cuboidal or spindle mast cells in the upper dermis with occasional eosinophils. These mast cells are monomorphous, and no mitotic figures, necrotic cells, or atypical cells are seen. Mast cells have metachromatic granules in the cytoplasm, which can be seen with toluidine blue or Giemsa stain. CD117 (c-kit) also is positive. Mast cells in urticaria pigmentosa easily may be mistaken for nevus cells. Hyperpigmentation of the basal layer, a characteristic feature seen in urticaria pigmentosa, also may erroneously suggest a diagnosis of a melanocytic nevus.
Granular cell tumors predominantly affect the oral cavity, but the skin also can be involved. It comprises a fascicular infiltrate of large and polygonal cells with characteristic eosinophilic granular cytoplasm in the dermis (Figure 1).13 Cell membranes are not always distinct. Although the nuclei usually are small and centrally located, irregular and plump nuclei with distinct nucleoli also may be seen. The overlying epidermis tends to be hyperplastic. Granular cell tumor is considered a group of lesions of varying histogenesis. Cases in which tumors originated from a neural crest–derived peripheral nerve–related cell as well as a Schwann cell have been reported.14,15 The origin of granular cell tumors is controversial.
|
|
| Figure 1. Granular cell tumor showing fascicles of large and polygonal cells with characteristic eosinophilic granular cytoplasm in the dermis (H&E, original magnification ×200). |
|
|
| Figure 2. Intradermal nevus showing nests with melanin in the uppermost area of the lesion and neurotized nevus cells in the lower part (H&E, original magnification ×100). Pseudovascular spaces are seen on the right side. |
Intradermal nevus usually has nests and cords of nevus cells in the upper dermis. The uppermost melanocytes often contain a moderate amount of melanin, whereas nevus cells in the mid and lower dermis usually do not contain melanin (Figure 2). Shrinkage during tissue processing maycause clefts between nevus cells, resulting in pseudovascular spaces.16 The deeper dermis may have a neuroid appearance with spindle-shaped cells and Meissner corpuscle–like structures.17
Although Langerhans cell disease was formerly known as Langerhans cell histiocytosis and subdivided into several clinical subtypes, including Letterer-Siwe disease, Hand-Schüller-Christian disease, and eosinophilic granuloma, these clinical subtypes commonly overlapped. Langerhans cell disease is now used as a terminology that encompasses all subtypes.18,19 Langerhans cell disease is characterized by a proliferation of Langerhans cells with a variable mixture of other inflammatory cells. The constituent cells are large and ovoid with a distinct folded or lobulated, often kidney-shaped nucleus.20 Langerhans cells usually infiltrate the upper dermis and occasionally the epidermis (Figure 3). CD1a, HLA-DR, S-100 protein, and langerin are positive in Langerhans cells.21
|
|
| Figure 3. Langerhans cell disease showing an infiltrate of large and ovoid Langerhans cells with a distinct folded or lobulated, often kidney-shaped nucleus in the upper dermis and epidermis (H&E, original magnification ×200). |
|
|
| Figure 4. Multicentric reticulohistiocytosis showing a mixture of mononuclear and multinucleate histiocytes with abundant eosinophilic and finely granular cytoplasm (H&E, original magnification ×200). |
Multicentric reticulohistiocytosis is characterized by a combination of papulonodular cutaneous lesions and severe arthropathy.22 An irregular mixture of mononuclear and multinucleate histiocytes showing abundant eosinophilic and finely granular cytoplasm, often with a ground-glass appearance, is seen along with lymphocytic infiltration (Figure 4).23 A few giant cells may be seen in early lesions; older lesions more commonly have giant cells and fibrosis.
1. Arock M, Valent P. Pathogenesis, classification and treatment of mastocytosis: state of the art in 2010 and future perspectives. Expert Rev Hematol. 2010;3:497-516.
2. Pardanani A. Systemic mastocytosis in adults: 2013 update on diagnosis, risk stratification, and management. Am J Hematol. 2013;88:612-624.
3. Orfao A, Garcia-Montero AC, Sanchez L, et al. Recent advances in the understanding of mastocytosis: the role of KIT mutations. Br J Haematol. 2007;138:12-30.
4. Yanagihori H, Oyama N, Nakamura K, et al. c-KIT mutations in patients with childhood-onset mastocytosis and genotype-phenotype correlation. J Mol Diagn. 2005;7:252-257.
5. Bodemer C, Hermine O, Palmérini F, et al. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol. 2010;130:804-815.
6. Kettelhut BV, Metcalfe DD. Pediatric mastocytosis. Ann Allergy. 1994;73:197-202; quiz 202-207.
7. Longley J, Duffy TP, Kohn S. The mast cell and mast cell disease. J Am Acad Dermatol. 1995;32:545-561; quiz 562-564.
8. Sotlar K, Cerny-Reiterer S, Petat-Dutter K, et al. Aberrant expression of CD30 in neoplastic mast cells in high-grade mastocytosis. Mod Pathol. 2011;24:585-595.
9. Blatt K, Cerny-Reiterer S, Schwaab J, et al. Identification of the Ki-1 antigen (CD30) as a novel therapeutic target in systemic mastocytosis [published online October 20, 2015]. Blood. 2015;126:2832-2841.
10. Kiszewski AE, Duran-Mckinster C, Orozco-Covarrubias L, et al. Cutaneous mastocytosis in children: a clinical analysis of 71 cases. J Eur Acad Dermatol Venereol. 2004;18:285-290.
11. Akoglu G, Erkin G, Cakir B, et al. Cutaneous mastocytosis: demographic aspects and clinical features of 55 patients. J Eur Acad Dermatol Venereol. 2006;20:969-973.
12. Sarkany RP, Monk BE, Handfield-Jones SE. Telangiectasia macularis eruptiva perstans: a case report and review of the literature. Clin Exp Dermatol. 1998;23:38-39.
13. Lack EE, Worsham GF, Callihan MD, et al. Granular cell tumor: a clinicopathologic study of 110 patients. J Surg Oncol. 1980;13:301-316.
14. Buley ID, Gatter KC, Kelly PM, et al. Granular cell tumours revisited. an immunohistological and ultrastructural study. Histopathology. 1988;12:263-274.
15. Penneys NS, Adachi K, Ziegels-Weissman J, et al. Granular cell tumors of the skin contain myelin basic protein. Arch Pathol Lab Med. 1983;107:302-303.
16. Modlin RL, Gottlieb B, Taylor C, et al. Identification of cells lining pseudovascular spaces of benign pigmented nevi. Am J Dermatopathol. 1984;(suppl 6):25-29.
17. Fullen DR, Reed JA, Finnerty B, et al. S100A6 preferentially labels type C nevus cells and nevic corpuscles: additional support for Schwannian differentiation of intradermal nevi. J Cutan Pathol. 2001;28:393-399.
18. Newman B, Hu W, Nigro K, et al. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol. 2007;56:302-316.
19. Weedon D. Cutaneous infiltrates—non-lymphoid. In: Weedon D, ed. Weedon’s Skin Pathology. 3rd ed. Amsterdam, Netherlands: Elsevier; 2010:937-970.
20. Harrist TJ, Bhan AK, Murphy GF, et al. Histiocytosis-X: in situ characterization of cutaneous infiltrates with monoclonal antibodies. Am J Clin Pathol. 1983;79:294-300.
21. Lau SK, Chu PG, Weiss LM. Immunohistochemical expression of langerin in Langerhans cell histiocytosis and non-Langerhans cell histiocytic disorders. Am J Surg Pathol. 2008;32:615-619.
22. Lesher JL Jr, Allen BS. Multicentric reticulohistiocytosis. J Am Acad Dermatol. 1984;11:713-723.
23. Heathcote JG, Guenther LC, Wallace AC. Multicentric reticulohistiocytosis: a report of a case and a review of the pathology. Pathology. 1985;17:601-608.
1. Arock M, Valent P. Pathogenesis, classification and treatment of mastocytosis: state of the art in 2010 and future perspectives. Expert Rev Hematol. 2010;3:497-516.
2. Pardanani A. Systemic mastocytosis in adults: 2013 update on diagnosis, risk stratification, and management. Am J Hematol. 2013;88:612-624.
3. Orfao A, Garcia-Montero AC, Sanchez L, et al. Recent advances in the understanding of mastocytosis: the role of KIT mutations. Br J Haematol. 2007;138:12-30.
4. Yanagihori H, Oyama N, Nakamura K, et al. c-KIT mutations in patients with childhood-onset mastocytosis and genotype-phenotype correlation. J Mol Diagn. 2005;7:252-257.
5. Bodemer C, Hermine O, Palmérini F, et al. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol. 2010;130:804-815.
6. Kettelhut BV, Metcalfe DD. Pediatric mastocytosis. Ann Allergy. 1994;73:197-202; quiz 202-207.
7. Longley J, Duffy TP, Kohn S. The mast cell and mast cell disease. J Am Acad Dermatol. 1995;32:545-561; quiz 562-564.
8. Sotlar K, Cerny-Reiterer S, Petat-Dutter K, et al. Aberrant expression of CD30 in neoplastic mast cells in high-grade mastocytosis. Mod Pathol. 2011;24:585-595.
9. Blatt K, Cerny-Reiterer S, Schwaab J, et al. Identification of the Ki-1 antigen (CD30) as a novel therapeutic target in systemic mastocytosis [published online October 20, 2015]. Blood. 2015;126:2832-2841.
10. Kiszewski AE, Duran-Mckinster C, Orozco-Covarrubias L, et al. Cutaneous mastocytosis in children: a clinical analysis of 71 cases. J Eur Acad Dermatol Venereol. 2004;18:285-290.
11. Akoglu G, Erkin G, Cakir B, et al. Cutaneous mastocytosis: demographic aspects and clinical features of 55 patients. J Eur Acad Dermatol Venereol. 2006;20:969-973.
12. Sarkany RP, Monk BE, Handfield-Jones SE. Telangiectasia macularis eruptiva perstans: a case report and review of the literature. Clin Exp Dermatol. 1998;23:38-39.
13. Lack EE, Worsham GF, Callihan MD, et al. Granular cell tumor: a clinicopathologic study of 110 patients. J Surg Oncol. 1980;13:301-316.
14. Buley ID, Gatter KC, Kelly PM, et al. Granular cell tumours revisited. an immunohistological and ultrastructural study. Histopathology. 1988;12:263-274.
15. Penneys NS, Adachi K, Ziegels-Weissman J, et al. Granular cell tumors of the skin contain myelin basic protein. Arch Pathol Lab Med. 1983;107:302-303.
16. Modlin RL, Gottlieb B, Taylor C, et al. Identification of cells lining pseudovascular spaces of benign pigmented nevi. Am J Dermatopathol. 1984;(suppl 6):25-29.
17. Fullen DR, Reed JA, Finnerty B, et al. S100A6 preferentially labels type C nevus cells and nevic corpuscles: additional support for Schwannian differentiation of intradermal nevi. J Cutan Pathol. 2001;28:393-399.
18. Newman B, Hu W, Nigro K, et al. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol. 2007;56:302-316.
19. Weedon D. Cutaneous infiltrates—non-lymphoid. In: Weedon D, ed. Weedon’s Skin Pathology. 3rd ed. Amsterdam, Netherlands: Elsevier; 2010:937-970.
20. Harrist TJ, Bhan AK, Murphy GF, et al. Histiocytosis-X: in situ characterization of cutaneous infiltrates with monoclonal antibodies. Am J Clin Pathol. 1983;79:294-300.
21. Lau SK, Chu PG, Weiss LM. Immunohistochemical expression of langerin in Langerhans cell histiocytosis and non-Langerhans cell histiocytic disorders. Am J Surg Pathol. 2008;32:615-619.
22. Lesher JL Jr, Allen BS. Multicentric reticulohistiocytosis. J Am Acad Dermatol. 1984;11:713-723.
23. Heathcote JG, Guenther LC, Wallace AC. Multicentric reticulohistiocytosis: a report of a case and a review of the pathology. Pathology. 1985;17:601-608.
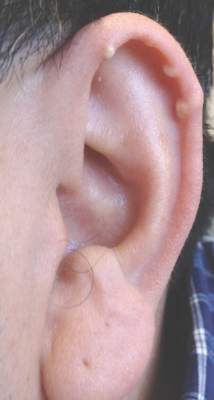
Multiple Superficial White Nodules on the Bilateral Helical Rims
The Diagnosis: Bilateral Auricular Tophaceous Gout
Histopathologic evaluation with hematoxylin and eosin staining demonstrated clusters of abundant granular amorphous material within the subcutaneous tissue (Figure 1). The overlying epidermis and dermis were unremarkable. The granular amorphous material demonstrated numerous monosodium urate crystals under polarized light (Figure 2). At a return visit following the biopsy results, the patient reported a history of a single episode of monoarticular gouty arthritis involving the right hallux approximately 6 months after the onset of the skin lesions. With the added clinical history and the biopsy results, his serum uric acid level was obtained and was found to be elevated at 9.2 mg/dL (reference range, 3.5–8 mg/dL).
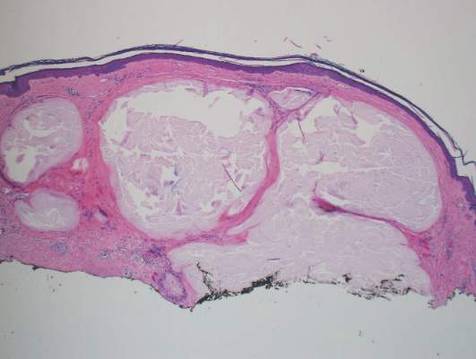
|
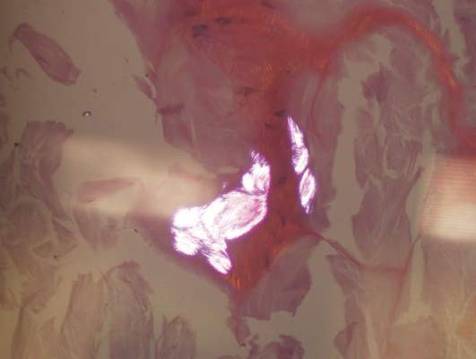
|
In our patient, the clinical differential diagnosis included calcium deposits, weathering nodules, and tophaceous gout. The differential diagnosis of auricular lesions is broad, and benign lesions may mimic cancerous entities such as basal cell carcinoma and squamous cell carcinoma.1 Therefore a detailed history, thorough physical examination, and tissue sampling are key to establishing the correct diagnosis. Our patient’s history of monoarticular gouty arthritis was only elucidated after a diagnosis of bilateral auricular tophaceous gout was made based on the biopsy results.
Subcutaneous tophi represent a chronic state of hyperuricemia and tend to manifest after long-standing polyarthritis and repeated acute gout attacks.2-5 These lesions develop in approximately 50% of gout patients and usually occur an average of 11.6 years after the onset of disease.2 There is a subset of individuals that are at higher risk for developing tophi, including elderly and female patients, diuretic and chronic nonsteroidal anti-inflammatory drug users, patients with a history of cyclosporine therapy, and patients with underlying chronic renal insufficiency.2,6,7 The most commonly affected tissues are those with poor blood supply and lower temperatures, such as the ear helix and first metacarpal joint.4 The auricle is the most common site of tophi on the head and neck. Tophi of the helices are generally asymptomatic and nontender; however, tophi can become large, inflamed, and ulcerated, causing pressure and discomfort.2 Combination treatment with dietary modification and antihyperuricemic therapy (eg, allopurinol) has been shown to reduce the size of lesions and prevent future tophi formation. However, these results may take months, warranting excision of large and symptomatic lesions.4,8
Our case is unusual in that the onset of the auricular lesions predated the articular gout by 6 months. Gouty tophi as the initial presentation of hyperuricemia is rare; however, tophi formation without concomitant arthritis has been reported.2,3,7,9 Wernick et al7 described 6 patients presenting with tophi before the onset of inflammatory arthritis that they attributed to changes in active inflammation by age (eg, elderly patients were more commonly immunosuppressed), chronic illnesses, and anti-inflammatory medications (eg, nonsteroidal anti-inflammatory drugs). Another possible explanation for this atypical presentation is misdiagnosis caused by other forms of arthritis (eg, rheumatoid arthritis, osteoarthritis) masking acute gout episodes. It also has been reported that monosodium urate crystals can be found in synovial fluid with no inflammation and therefore no symptoms.7
Tophi, although rare, may be the sole clinical manifestation of underlying gouty disease. It is important to be aware of this atypical presentation to prevent misdiagnosis and provide appropriate treatment.
- Dompmartin A. Nodules of the external ear [in French]. Ann Dermatol Venereol. 1999;126:261-266.
- Griffin G, Munns J, Fullen D, et al. Auricular tophi as the initial presentation of gout. Otolaryngol Head Neck Surg. 2009;141:153-154.
- Koley S, Salodkar A, Choudhary S, et al. Tophi as first manifestation of gout. Indian J Dermatol Venerol. 2010;76:393-393-396.
- Moriwaki Y. Tophaceous gout [in Japanese]. Nihon Rinsho. 2008;66:711-716.
- Eggebeen AT. Gout: an update. Am Fam Physician. 2007;76:801-808.
- Hollingworth P, Scott JT, Burry HC. Nonarticular gout: hyperuricemia and tophus formation without gouty arthritis. Arthritis Rheum. 1983;26:98-101.
- Wernick R, Winkler C, Campbell S. Tophi as the initial manifestation of gout. report of six cases and review of the literature. Arch Intern Med. 1992;152:873-876.
- Caldas CA, Fuller R. Excellent response to the clinical treatment of tophaceous gout. Clin Rheumatol. 2009;26:1553-1555.
- Iglesias A, Londono JC, Saaibi DL, et al. Gout nodulosis: widespread subcutaneous deposits without gout. Arthritis Care Res. 1996;9:74-77.
The Diagnosis: Bilateral Auricular Tophaceous Gout
Histopathologic evaluation with hematoxylin and eosin staining demonstrated clusters of abundant granular amorphous material within the subcutaneous tissue (Figure 1). The overlying epidermis and dermis were unremarkable. The granular amorphous material demonstrated numerous monosodium urate crystals under polarized light (Figure 2). At a return visit following the biopsy results, the patient reported a history of a single episode of monoarticular gouty arthritis involving the right hallux approximately 6 months after the onset of the skin lesions. With the added clinical history and the biopsy results, his serum uric acid level was obtained and was found to be elevated at 9.2 mg/dL (reference range, 3.5–8 mg/dL).
|
|
|
|
In our patient, the clinical differential diagnosis included calcium deposits, weathering nodules, and tophaceous gout. The differential diagnosis of auricular lesions is broad, and benign lesions may mimic cancerous entities such as basal cell carcinoma and squamous cell carcinoma.1 Therefore a detailed history, thorough physical examination, and tissue sampling are key to establishing the correct diagnosis. Our patient’s history of monoarticular gouty arthritis was only elucidated after a diagnosis of bilateral auricular tophaceous gout was made based on the biopsy results.
Subcutaneous tophi represent a chronic state of hyperuricemia and tend to manifest after long-standing polyarthritis and repeated acute gout attacks.2-5 These lesions develop in approximately 50% of gout patients and usually occur an average of 11.6 years after the onset of disease.2 There is a subset of individuals that are at higher risk for developing tophi, including elderly and female patients, diuretic and chronic nonsteroidal anti-inflammatory drug users, patients with a history of cyclosporine therapy, and patients with underlying chronic renal insufficiency.2,6,7 The most commonly affected tissues are those with poor blood supply and lower temperatures, such as the ear helix and first metacarpal joint.4 The auricle is the most common site of tophi on the head and neck. Tophi of the helices are generally asymptomatic and nontender; however, tophi can become large, inflamed, and ulcerated, causing pressure and discomfort.2 Combination treatment with dietary modification and antihyperuricemic therapy (eg, allopurinol) has been shown to reduce the size of lesions and prevent future tophi formation. However, these results may take months, warranting excision of large and symptomatic lesions.4,8
Our case is unusual in that the onset of the auricular lesions predated the articular gout by 6 months. Gouty tophi as the initial presentation of hyperuricemia is rare; however, tophi formation without concomitant arthritis has been reported.2,3,7,9 Wernick et al7 described 6 patients presenting with tophi before the onset of inflammatory arthritis that they attributed to changes in active inflammation by age (eg, elderly patients were more commonly immunosuppressed), chronic illnesses, and anti-inflammatory medications (eg, nonsteroidal anti-inflammatory drugs). Another possible explanation for this atypical presentation is misdiagnosis caused by other forms of arthritis (eg, rheumatoid arthritis, osteoarthritis) masking acute gout episodes. It also has been reported that monosodium urate crystals can be found in synovial fluid with no inflammation and therefore no symptoms.7
Tophi, although rare, may be the sole clinical manifestation of underlying gouty disease. It is important to be aware of this atypical presentation to prevent misdiagnosis and provide appropriate treatment.
The Diagnosis: Bilateral Auricular Tophaceous Gout
Histopathologic evaluation with hematoxylin and eosin staining demonstrated clusters of abundant granular amorphous material within the subcutaneous tissue (Figure 1). The overlying epidermis and dermis were unremarkable. The granular amorphous material demonstrated numerous monosodium urate crystals under polarized light (Figure 2). At a return visit following the biopsy results, the patient reported a history of a single episode of monoarticular gouty arthritis involving the right hallux approximately 6 months after the onset of the skin lesions. With the added clinical history and the biopsy results, his serum uric acid level was obtained and was found to be elevated at 9.2 mg/dL (reference range, 3.5–8 mg/dL).
|
|
|
|
In our patient, the clinical differential diagnosis included calcium deposits, weathering nodules, and tophaceous gout. The differential diagnosis of auricular lesions is broad, and benign lesions may mimic cancerous entities such as basal cell carcinoma and squamous cell carcinoma.1 Therefore a detailed history, thorough physical examination, and tissue sampling are key to establishing the correct diagnosis. Our patient’s history of monoarticular gouty arthritis was only elucidated after a diagnosis of bilateral auricular tophaceous gout was made based on the biopsy results.
Subcutaneous tophi represent a chronic state of hyperuricemia and tend to manifest after long-standing polyarthritis and repeated acute gout attacks.2-5 These lesions develop in approximately 50% of gout patients and usually occur an average of 11.6 years after the onset of disease.2 There is a subset of individuals that are at higher risk for developing tophi, including elderly and female patients, diuretic and chronic nonsteroidal anti-inflammatory drug users, patients with a history of cyclosporine therapy, and patients with underlying chronic renal insufficiency.2,6,7 The most commonly affected tissues are those with poor blood supply and lower temperatures, such as the ear helix and first metacarpal joint.4 The auricle is the most common site of tophi on the head and neck. Tophi of the helices are generally asymptomatic and nontender; however, tophi can become large, inflamed, and ulcerated, causing pressure and discomfort.2 Combination treatment with dietary modification and antihyperuricemic therapy (eg, allopurinol) has been shown to reduce the size of lesions and prevent future tophi formation. However, these results may take months, warranting excision of large and symptomatic lesions.4,8
Our case is unusual in that the onset of the auricular lesions predated the articular gout by 6 months. Gouty tophi as the initial presentation of hyperuricemia is rare; however, tophi formation without concomitant arthritis has been reported.2,3,7,9 Wernick et al7 described 6 patients presenting with tophi before the onset of inflammatory arthritis that they attributed to changes in active inflammation by age (eg, elderly patients were more commonly immunosuppressed), chronic illnesses, and anti-inflammatory medications (eg, nonsteroidal anti-inflammatory drugs). Another possible explanation for this atypical presentation is misdiagnosis caused by other forms of arthritis (eg, rheumatoid arthritis, osteoarthritis) masking acute gout episodes. It also has been reported that monosodium urate crystals can be found in synovial fluid with no inflammation and therefore no symptoms.7
Tophi, although rare, may be the sole clinical manifestation of underlying gouty disease. It is important to be aware of this atypical presentation to prevent misdiagnosis and provide appropriate treatment.
- Dompmartin A. Nodules of the external ear [in French]. Ann Dermatol Venereol. 1999;126:261-266.
- Griffin G, Munns J, Fullen D, et al. Auricular tophi as the initial presentation of gout. Otolaryngol Head Neck Surg. 2009;141:153-154.
- Koley S, Salodkar A, Choudhary S, et al. Tophi as first manifestation of gout. Indian J Dermatol Venerol. 2010;76:393-393-396.
- Moriwaki Y. Tophaceous gout [in Japanese]. Nihon Rinsho. 2008;66:711-716.
- Eggebeen AT. Gout: an update. Am Fam Physician. 2007;76:801-808.
- Hollingworth P, Scott JT, Burry HC. Nonarticular gout: hyperuricemia and tophus formation without gouty arthritis. Arthritis Rheum. 1983;26:98-101.
- Wernick R, Winkler C, Campbell S. Tophi as the initial manifestation of gout. report of six cases and review of the literature. Arch Intern Med. 1992;152:873-876.
- Caldas CA, Fuller R. Excellent response to the clinical treatment of tophaceous gout. Clin Rheumatol. 2009;26:1553-1555.
- Iglesias A, Londono JC, Saaibi DL, et al. Gout nodulosis: widespread subcutaneous deposits without gout. Arthritis Care Res. 1996;9:74-77.
- Dompmartin A. Nodules of the external ear [in French]. Ann Dermatol Venereol. 1999;126:261-266.
- Griffin G, Munns J, Fullen D, et al. Auricular tophi as the initial presentation of gout. Otolaryngol Head Neck Surg. 2009;141:153-154.
- Koley S, Salodkar A, Choudhary S, et al. Tophi as first manifestation of gout. Indian J Dermatol Venerol. 2010;76:393-393-396.
- Moriwaki Y. Tophaceous gout [in Japanese]. Nihon Rinsho. 2008;66:711-716.
- Eggebeen AT. Gout: an update. Am Fam Physician. 2007;76:801-808.
- Hollingworth P, Scott JT, Burry HC. Nonarticular gout: hyperuricemia and tophus formation without gouty arthritis. Arthritis Rheum. 1983;26:98-101.
- Wernick R, Winkler C, Campbell S. Tophi as the initial manifestation of gout. report of six cases and review of the literature. Arch Intern Med. 1992;152:873-876.
- Caldas CA, Fuller R. Excellent response to the clinical treatment of tophaceous gout. Clin Rheumatol. 2009;26:1553-1555.
- Iglesias A, Londono JC, Saaibi DL, et al. Gout nodulosis: widespread subcutaneous deposits without gout. Arthritis Care Res. 1996;9:74-77.
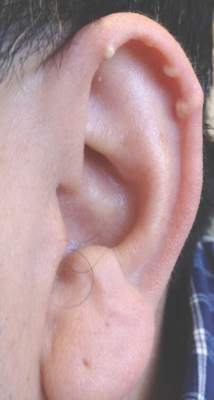
A 40-year-old man presented for evaluation of multiple small nodules on the bilateral auricles primarily involving the helices of 1 year’s duration. The lesions were nontender with no associated bleeding, burning, or pruritus. He denied any trauma to these sites and denied any systemic symptoms including fever, chills, joint pain, or weight loss. His medical history was remarkable for type 2 diabetes mellitus. He had no history of similar skin lesions or renal disease and denied any alcohol intake. He also denied taking any over-the-counter or prescription medications. Physical examination revealed several 1- to 4-mm superficial white dermal nodules located on the bilateral helical rims. The lesions were firm and well circumscribed and the surrounding skin showed mild erythema. Shave biopsies of the nodules were performed.
Judicious Use of Antibiotics in Dermatology

What does your patient need to know at the first visit? Does it apply to patients of all genders, ages, and races?
There are 3 scenarios in which antibiotics are used in dermatology. First, there is the treatment of a bona fide, verified skin infection, which may range from the relatively simple (impetigo) to the complex (botryomycosis) to the exotic (fish tank granuloma). The second scenario is antibiotic administration, often due to ancillary properties such as anti-inflammatory effects, in the management of noninfectious disorders, such as familial benign pemphigus or pityriasis lichenoides et varioliformis acuta. I try hard to avoid antibiotic use in these situations unless all else fails. The third scenario involves use of antibiotics at the patient’s request, usually associated with the phrase “just in case it’s infected.” In my opinion, this practice is completely ill advised.
Male and female patients of all ages, ethnic origins, and socioeconomic backgrounds are woefully uninformed regarding the promise and peril of antibiotics. I want patients to buy into the concept of good antibiotic stewardship. Thus, patients should understand that there must be a specific and justifiable reason for antibiotic use and that the recommended dose and duration of treatment should not be altered. In some situations, antibiotic therapy is intended to be of short duration, while in other situations, such therapy may be quite protracted. Patients also need to know at the outset of treatment when we plan to transition from a short-term, antibiotic-based modality to a long-term nonantibiotic maintenance regimen, which is especially true for acne and rosacea. I try to limit antibiotic use in these disorders to 3 months. Furthermore, patients should always be educated about the potential side effects associated with the particular antibiotic being prescribed. Hoarding and sharing leftover antibiotics should be strongly and explicitly discouraged.
Finally, patients must be educated that taking shortcuts when prescribing antibiotics may lead to therapeutic failure, worsening disease, or serious long-term adverse consequences. For example, rational antibiotic use may require the added expense of an initial and/or subsequent test-of-cure culture and sensitivity. Is that swollen and tender hand following a cat bite due to Pasteurella multocida or methicillin-resistant Staphylococcus aureus? Is that new eruption in an atopic patient due to secondary impetigo or eczema herpeticum? Other laboratory testing also may be required, such as a follow-up serology after treating syphilis. Patients need to know why laboratory tests are being ordered and how the tests complement direct antibiotic intervention.
What are your go-to treatments? What are the side effects?
I am a fan of subantimicrobial-dose doxycycline for both rosacea (on label) and acne (off label). Studies have shown that neither quantitative nor qualitative changes occur in the cutaneous, oral, or gastrointestinal flora. Thus, I avoid contributing to the emerging global crisis of antimicrobial resistance. I am also a proponent of topical antibiotics whenever appropriate and reasonable. Mupirocin and retapamulin, for example, are quite effective for routine cases of impetigo. When incision and drainage alone are insufficient to resolve methicillin-resistant S aureus furunculosis, I prefer either trimethoprim-sulfamethoxazole or doxycycline. Of course, other specific oral and even parenteral antibiotics are appropriate for select disease states.
Although antibiotics generally are well tolerated, there are many possible side effects. Hypersensitivity reactions, ranging from self-limited fixed drug and pruritic maculopapular eruptions through acute urticaria to anaphylaxis, may occur with any antibiotic. Clostridium difficile–associated diarrhea also may occur in conjunction with the use of any antibacterial drug, especially those with a broad spectrum of activity. Nausea and headache are mild but common side effects of these agents. All tetracycline derivatives may be photosensitizers and may provoke intracranial hypertension. Minocycline may lead to hyperpigmentation of skin and teeth, vestibular disturbances (ie, dizziness, ataxia, vertigo, tinnitus) and rarely autoimmune hepatitis. Macrolide antibiotics have been linked to serious cardiotoxicity, and quinolone antibiotics have been linked to tendonitis/tendon rupture, cardiotoxicity, and insomnia. Many antibiotics can result in vaginal yeast infections. There is some evidence that prolonged antibiotic use may precipitate inflammatory bowel disease, especially in those who are genetically predisposed.
Finally, keep in mind that antibiotic administration changes the normal cutaneous flora, which may interfere with the normal antimicrobial and anti-inflammatory homeostatic roles played by resident skin microflora. Antibiotic administration also changes the gut flora and, in this manner, may help promote the development of resistant microbes.
How do you keep patients compliant with treatment?
The most important step to assure adherence is adequate pretreatment education. Whether short-term or long-term antibiotic treatment is anticipated, I always schedule a follow-up office visit in approximately 2 weeks to check on clinical progress and reinforce good habits. Younger patients benefit from periodic reminders using emails, text messages, and tweets.
What do you do if they refuse treatment?
In some instances, antibiotic phobia in patients can be totally accepted and alternative treatments explored. As an example, laser and light therapy, hormonal manipulation, zinc-based nutritional supplements, and intensive nonantibiotic topical combination drugs can supplant antibiotics for the management of acne.
What resources do you recommend to patients for more information?
There are some excellent resources online for patients such as “Using Antibiotics Wisely” and “Get Smart: Know When Antibiotics Work.”
Suggested Readings
Chon SY, Doan HQ, Mays RM. Antibiotic overuse and resistance in dermatology. Dermatol Ther. 2012;25:55-69.
Eichenfield LF, Del Rosso JQ, Mancini AJ, et al. Evolving perspectives on the etiology and pathogenesis of acne vulgaris. J Drugs Dermatol. 2015;14:263-272.
Gallo RL, Nakatsuji T. Microbial symbiosis with the innate immune defense system of the skin. J Invest Dermatol. 2011;131:1974-1980.
Gelband H, Miller-Petrie M, Pant S, et al. The State of the World’s Antibiotics, 2015. Washington, DC: Center for Disease Dynamics, Economics & Policy; 2015. http://cddep.org/publications/state_worlds_antibiotics_2015. Accessed February 11, 2016.
Get smart: know when antibiotics work. Centers for Disease Control and Prevention website. http://www.cdc.gov/getsmart/community/about/index.html. Updated April 17, 2015. Accessed February 11, 2016.
Harris AM, Hicks LA, Qaseem A. Appropriate antibiotic use for acute respiratory tract infection in adults: Advice for high-value care from the American College of Physicians and the Centers for Disease Control and Prevention [published online January 19, 2016]. Ann Intern Med. doi:10.7326/M15-1840.
Kirchner M, Mafura M, Hunt T, et al. Antimicrobial resistance characteristics and fitness of Gram-negative fecal bacteria from volunteers treated with minocycline or amoxicillin. Front Microbiol. 2014;5:722. doi:10.3389/fmicb.2014.00722.
Muhammad M, Rosen T. A controversial proposal: no more antibiotics for acne! Skin Therapy Lett. 2013;18:1-4.
Using antibiotics wisely. WedMD Medical Reference. http://www.webmd.com/a-to-z-guides/using-antibiotics-wisely-topic-overview. Updated November 14, 2014. Accessed February 11, 2016.
What does your patient need to know at the first visit? Does it apply to patients of all genders, ages, and races?
There are 3 scenarios in which antibiotics are used in dermatology. First, there is the treatment of a bona fide, verified skin infection, which may range from the relatively simple (impetigo) to the complex (botryomycosis) to the exotic (fish tank granuloma). The second scenario is antibiotic administration, often due to ancillary properties such as anti-inflammatory effects, in the management of noninfectious disorders, such as familial benign pemphigus or pityriasis lichenoides et varioliformis acuta. I try hard to avoid antibiotic use in these situations unless all else fails. The third scenario involves use of antibiotics at the patient’s request, usually associated with the phrase “just in case it’s infected.” In my opinion, this practice is completely ill advised.
Male and female patients of all ages, ethnic origins, and socioeconomic backgrounds are woefully uninformed regarding the promise and peril of antibiotics. I want patients to buy into the concept of good antibiotic stewardship. Thus, patients should understand that there must be a specific and justifiable reason for antibiotic use and that the recommended dose and duration of treatment should not be altered. In some situations, antibiotic therapy is intended to be of short duration, while in other situations, such therapy may be quite protracted. Patients also need to know at the outset of treatment when we plan to transition from a short-term, antibiotic-based modality to a long-term nonantibiotic maintenance regimen, which is especially true for acne and rosacea. I try to limit antibiotic use in these disorders to 3 months. Furthermore, patients should always be educated about the potential side effects associated with the particular antibiotic being prescribed. Hoarding and sharing leftover antibiotics should be strongly and explicitly discouraged.
Finally, patients must be educated that taking shortcuts when prescribing antibiotics may lead to therapeutic failure, worsening disease, or serious long-term adverse consequences. For example, rational antibiotic use may require the added expense of an initial and/or subsequent test-of-cure culture and sensitivity. Is that swollen and tender hand following a cat bite due to Pasteurella multocida or methicillin-resistant Staphylococcus aureus? Is that new eruption in an atopic patient due to secondary impetigo or eczema herpeticum? Other laboratory testing also may be required, such as a follow-up serology after treating syphilis. Patients need to know why laboratory tests are being ordered and how the tests complement direct antibiotic intervention.
What are your go-to treatments? What are the side effects?
I am a fan of subantimicrobial-dose doxycycline for both rosacea (on label) and acne (off label). Studies have shown that neither quantitative nor qualitative changes occur in the cutaneous, oral, or gastrointestinal flora. Thus, I avoid contributing to the emerging global crisis of antimicrobial resistance. I am also a proponent of topical antibiotics whenever appropriate and reasonable. Mupirocin and retapamulin, for example, are quite effective for routine cases of impetigo. When incision and drainage alone are insufficient to resolve methicillin-resistant S aureus furunculosis, I prefer either trimethoprim-sulfamethoxazole or doxycycline. Of course, other specific oral and even parenteral antibiotics are appropriate for select disease states.
Although antibiotics generally are well tolerated, there are many possible side effects. Hypersensitivity reactions, ranging from self-limited fixed drug and pruritic maculopapular eruptions through acute urticaria to anaphylaxis, may occur with any antibiotic. Clostridium difficile–associated diarrhea also may occur in conjunction with the use of any antibacterial drug, especially those with a broad spectrum of activity. Nausea and headache are mild but common side effects of these agents. All tetracycline derivatives may be photosensitizers and may provoke intracranial hypertension. Minocycline may lead to hyperpigmentation of skin and teeth, vestibular disturbances (ie, dizziness, ataxia, vertigo, tinnitus) and rarely autoimmune hepatitis. Macrolide antibiotics have been linked to serious cardiotoxicity, and quinolone antibiotics have been linked to tendonitis/tendon rupture, cardiotoxicity, and insomnia. Many antibiotics can result in vaginal yeast infections. There is some evidence that prolonged antibiotic use may precipitate inflammatory bowel disease, especially in those who are genetically predisposed.
Finally, keep in mind that antibiotic administration changes the normal cutaneous flora, which may interfere with the normal antimicrobial and anti-inflammatory homeostatic roles played by resident skin microflora. Antibiotic administration also changes the gut flora and, in this manner, may help promote the development of resistant microbes.
How do you keep patients compliant with treatment?
The most important step to assure adherence is adequate pretreatment education. Whether short-term or long-term antibiotic treatment is anticipated, I always schedule a follow-up office visit in approximately 2 weeks to check on clinical progress and reinforce good habits. Younger patients benefit from periodic reminders using emails, text messages, and tweets.
What do you do if they refuse treatment?
In some instances, antibiotic phobia in patients can be totally accepted and alternative treatments explored. As an example, laser and light therapy, hormonal manipulation, zinc-based nutritional supplements, and intensive nonantibiotic topical combination drugs can supplant antibiotics for the management of acne.
What resources do you recommend to patients for more information?
There are some excellent resources online for patients such as “Using Antibiotics Wisely” and “Get Smart: Know When Antibiotics Work.”
What does your patient need to know at the first visit? Does it apply to patients of all genders, ages, and races?
There are 3 scenarios in which antibiotics are used in dermatology. First, there is the treatment of a bona fide, verified skin infection, which may range from the relatively simple (impetigo) to the complex (botryomycosis) to the exotic (fish tank granuloma). The second scenario is antibiotic administration, often due to ancillary properties such as anti-inflammatory effects, in the management of noninfectious disorders, such as familial benign pemphigus or pityriasis lichenoides et varioliformis acuta. I try hard to avoid antibiotic use in these situations unless all else fails. The third scenario involves use of antibiotics at the patient’s request, usually associated with the phrase “just in case it’s infected.” In my opinion, this practice is completely ill advised.
Male and female patients of all ages, ethnic origins, and socioeconomic backgrounds are woefully uninformed regarding the promise and peril of antibiotics. I want patients to buy into the concept of good antibiotic stewardship. Thus, patients should understand that there must be a specific and justifiable reason for antibiotic use and that the recommended dose and duration of treatment should not be altered. In some situations, antibiotic therapy is intended to be of short duration, while in other situations, such therapy may be quite protracted. Patients also need to know at the outset of treatment when we plan to transition from a short-term, antibiotic-based modality to a long-term nonantibiotic maintenance regimen, which is especially true for acne and rosacea. I try to limit antibiotic use in these disorders to 3 months. Furthermore, patients should always be educated about the potential side effects associated with the particular antibiotic being prescribed. Hoarding and sharing leftover antibiotics should be strongly and explicitly discouraged.
Finally, patients must be educated that taking shortcuts when prescribing antibiotics may lead to therapeutic failure, worsening disease, or serious long-term adverse consequences. For example, rational antibiotic use may require the added expense of an initial and/or subsequent test-of-cure culture and sensitivity. Is that swollen and tender hand following a cat bite due to Pasteurella multocida or methicillin-resistant Staphylococcus aureus? Is that new eruption in an atopic patient due to secondary impetigo or eczema herpeticum? Other laboratory testing also may be required, such as a follow-up serology after treating syphilis. Patients need to know why laboratory tests are being ordered and how the tests complement direct antibiotic intervention.
What are your go-to treatments? What are the side effects?
I am a fan of subantimicrobial-dose doxycycline for both rosacea (on label) and acne (off label). Studies have shown that neither quantitative nor qualitative changes occur in the cutaneous, oral, or gastrointestinal flora. Thus, I avoid contributing to the emerging global crisis of antimicrobial resistance. I am also a proponent of topical antibiotics whenever appropriate and reasonable. Mupirocin and retapamulin, for example, are quite effective for routine cases of impetigo. When incision and drainage alone are insufficient to resolve methicillin-resistant S aureus furunculosis, I prefer either trimethoprim-sulfamethoxazole or doxycycline. Of course, other specific oral and even parenteral antibiotics are appropriate for select disease states.
Although antibiotics generally are well tolerated, there are many possible side effects. Hypersensitivity reactions, ranging from self-limited fixed drug and pruritic maculopapular eruptions through acute urticaria to anaphylaxis, may occur with any antibiotic. Clostridium difficile–associated diarrhea also may occur in conjunction with the use of any antibacterial drug, especially those with a broad spectrum of activity. Nausea and headache are mild but common side effects of these agents. All tetracycline derivatives may be photosensitizers and may provoke intracranial hypertension. Minocycline may lead to hyperpigmentation of skin and teeth, vestibular disturbances (ie, dizziness, ataxia, vertigo, tinnitus) and rarely autoimmune hepatitis. Macrolide antibiotics have been linked to serious cardiotoxicity, and quinolone antibiotics have been linked to tendonitis/tendon rupture, cardiotoxicity, and insomnia. Many antibiotics can result in vaginal yeast infections. There is some evidence that prolonged antibiotic use may precipitate inflammatory bowel disease, especially in those who are genetically predisposed.
Finally, keep in mind that antibiotic administration changes the normal cutaneous flora, which may interfere with the normal antimicrobial and anti-inflammatory homeostatic roles played by resident skin microflora. Antibiotic administration also changes the gut flora and, in this manner, may help promote the development of resistant microbes.
How do you keep patients compliant with treatment?
The most important step to assure adherence is adequate pretreatment education. Whether short-term or long-term antibiotic treatment is anticipated, I always schedule a follow-up office visit in approximately 2 weeks to check on clinical progress and reinforce good habits. Younger patients benefit from periodic reminders using emails, text messages, and tweets.
What do you do if they refuse treatment?
In some instances, antibiotic phobia in patients can be totally accepted and alternative treatments explored. As an example, laser and light therapy, hormonal manipulation, zinc-based nutritional supplements, and intensive nonantibiotic topical combination drugs can supplant antibiotics for the management of acne.
What resources do you recommend to patients for more information?
There are some excellent resources online for patients such as “Using Antibiotics Wisely” and “Get Smart: Know When Antibiotics Work.”
Suggested Readings
Chon SY, Doan HQ, Mays RM. Antibiotic overuse and resistance in dermatology. Dermatol Ther. 2012;25:55-69.
Eichenfield LF, Del Rosso JQ, Mancini AJ, et al. Evolving perspectives on the etiology and pathogenesis of acne vulgaris. J Drugs Dermatol. 2015;14:263-272.
Gallo RL, Nakatsuji T. Microbial symbiosis with the innate immune defense system of the skin. J Invest Dermatol. 2011;131:1974-1980.
Gelband H, Miller-Petrie M, Pant S, et al. The State of the World’s Antibiotics, 2015. Washington, DC: Center for Disease Dynamics, Economics & Policy; 2015. http://cddep.org/publications/state_worlds_antibiotics_2015. Accessed February 11, 2016.
Get smart: know when antibiotics work. Centers for Disease Control and Prevention website. http://www.cdc.gov/getsmart/community/about/index.html. Updated April 17, 2015. Accessed February 11, 2016.
Harris AM, Hicks LA, Qaseem A. Appropriate antibiotic use for acute respiratory tract infection in adults: Advice for high-value care from the American College of Physicians and the Centers for Disease Control and Prevention [published online January 19, 2016]. Ann Intern Med. doi:10.7326/M15-1840.
Kirchner M, Mafura M, Hunt T, et al. Antimicrobial resistance characteristics and fitness of Gram-negative fecal bacteria from volunteers treated with minocycline or amoxicillin. Front Microbiol. 2014;5:722. doi:10.3389/fmicb.2014.00722.
Muhammad M, Rosen T. A controversial proposal: no more antibiotics for acne! Skin Therapy Lett. 2013;18:1-4.
Using antibiotics wisely. WedMD Medical Reference. http://www.webmd.com/a-to-z-guides/using-antibiotics-wisely-topic-overview. Updated November 14, 2014. Accessed February 11, 2016.
Suggested Readings
Chon SY, Doan HQ, Mays RM. Antibiotic overuse and resistance in dermatology. Dermatol Ther. 2012;25:55-69.
Eichenfield LF, Del Rosso JQ, Mancini AJ, et al. Evolving perspectives on the etiology and pathogenesis of acne vulgaris. J Drugs Dermatol. 2015;14:263-272.
Gallo RL, Nakatsuji T. Microbial symbiosis with the innate immune defense system of the skin. J Invest Dermatol. 2011;131:1974-1980.
Gelband H, Miller-Petrie M, Pant S, et al. The State of the World’s Antibiotics, 2015. Washington, DC: Center for Disease Dynamics, Economics & Policy; 2015. http://cddep.org/publications/state_worlds_antibiotics_2015. Accessed February 11, 2016.
Get smart: know when antibiotics work. Centers for Disease Control and Prevention website. http://www.cdc.gov/getsmart/community/about/index.html. Updated April 17, 2015. Accessed February 11, 2016.
Harris AM, Hicks LA, Qaseem A. Appropriate antibiotic use for acute respiratory tract infection in adults: Advice for high-value care from the American College of Physicians and the Centers for Disease Control and Prevention [published online January 19, 2016]. Ann Intern Med. doi:10.7326/M15-1840.
Kirchner M, Mafura M, Hunt T, et al. Antimicrobial resistance characteristics and fitness of Gram-negative fecal bacteria from volunteers treated with minocycline or amoxicillin. Front Microbiol. 2014;5:722. doi:10.3389/fmicb.2014.00722.
Muhammad M, Rosen T. A controversial proposal: no more antibiotics for acne! Skin Therapy Lett. 2013;18:1-4.
Using antibiotics wisely. WedMD Medical Reference. http://www.webmd.com/a-to-z-guides/using-antibiotics-wisely-topic-overview. Updated November 14, 2014. Accessed February 11, 2016.

Non–Drug-Induced Pemphigus Foliaceus in a Patient With Rheumatoid Arthritis
To the Editor:
The term pemphigus describes a group of autoimmune blistering diseases that are histologically characterized by intraepidermal blisters caused by acantholysis. There are several types of pemphigus foliaceus, such as classic and endemic pemphigus foliaceus, pemphigus erythematosus, pemphigus herpetiformis, and drug-induced pemphigus foliaceus.1

|
| Figure 1. Multiple erosions and crusted lesions were present on the back. |

|
| Figure 2. Subcorneal bulla containing acantholytic keratinocytes and neutrophils (H&E, original magnification ×100). |
Drug-induced pemphigus foliaceus in patients treated with penicillamine for rheumatoid arthritis (RA) is well documented in the literature.2 An association between pemphigus foliaceus and RA without penicillamine therapy is rare. We present a case of a patient with a history of RA who developed this bullous disease.
A 67-year-old woman with a 15-year history of seropositive RA presented with widespread skin lesions of 4 weeks’ duration. Confluent scaly crusted erosions on an erythematous base were present on the back (Figure 1), chest, and abdomen. There was no alteration of the mucous membranes. Medical treatment consisted of methotrexate (10 mg weekly), folic acid (5 mg twice weekly), prednisolone (5 mg daily), and ketoprofen (50 mg daily). Routine blood analysis was unremarkable, except for a positive rheumatoid factor. Histologic examination showed a subcorneal bulla containing acantholytic keratinocytes and neutrophils. There was a mild lymphocytic and eosinophilic infiltrate in the papillary dermis (Figure 2).
Determination of anti-desmoglein 1 and 3 antibodies was performed by a commercial enzyme-linked immunosorbent assay. Desmoglein 1 antibodies were positive with titers of 30 U/mL (positive, ≥20 U/mL), whereas desmoglein 3 antibody was negative. Thus, a diagnosis of pemphigus foliaceus was established. The polymerase chain reaction ligation-based typing method and the nucleotide sequence was used to examine the protein drought-repressed 4 gene complex, DR4, which tested positive.
Based on a diagnosis of pemphigus foliaceus, the patient’s corticosteroid treatment was changed from 5 mg daily of prednisolone to 40 mg daily of methylprednisolone, leading to marked improvement of the cutaneous lesions. After tapering the steroid dosage over a period of 3 months, no relapse occurred.
Pemphigus foliaceus is a rare autoimmune blistering disease. It can be induced by drugs, such as penicillamine and captopril.2,3 Captopril, an angiotensin-converting enzyme inhibitor, is closely related to penicillamine structurally. Both drugs have highly active thiol groups capable of reducing disulfide bonds and inducing acantholysis.4 The drugs taken by our patient typically are not known to induce pemphigus foliaceus.
The association of pemphigus foliaceus with RA in the absence of penicillamine therapy was first described by Wilkinson et al.4 Since then, additional cases have been published.5,6 Pemphigus foliaceus also has been described with other autoimmune conditions such as autoimmune thyroid disease.7
Rheumatoid arthritis has been genetically linked to the HLA-DR4 gene complex, which also was found in our patient. Patients with pemphigus foliaceus and RA have an increased frequency of the class II major histocompatibility complex, serologically defined HLA-DR4, and HLA-DRw6 haplotypes.4 Therefore, we believe that the association of pemphigus foliaceus and RA in our patient might not be fortuitous.
1. Chams-Davatchi C, Valikhani M, Daneshpazhooh M, et al. Pemphigus: analysis of 1209 cases. Int J Dermatol. 2005;44:470-476.
2. Sugita K, Hirokawa H, Izu K, et al. D-penicillamine-induced pemphigus successfully treated with combination therapy of mizoribine and prednisolone. J Dermatolog Treat. 2004;15:214-217.
3. Kaplan RP, Potter TS, Fox JN. Drug-induced pemphigus related to angiotensin-converting enzyme inhibitors. J Am Acad Dermatol. 1992;26(2, pt 2):364-366.
4. Wilkinson SM, Smith AG, Davis MJ, et al. Rheumatoid arthritis: an association with pemphigus foliaceus. Acta Derm Venereol. 1992;72:289-291.
5. Sáez-de-Ocariz M, Granados J, Yamamoto-Furusho JK, et al. Rheumatoid arthritis associated with pemphigus foliaceus in a patient not taking penicillamine. Skinmed. 2007;6:252-254.
6. Gürcan HM, Ahmed RA. Analysis of current data on the use of methotrexate in the treatment of pemphigus and pemphigoid. Br J Dermatol. 2009;161:723-731.
7. Leshem YA, Katzenelson V, Yosipovitch G, et al. Autoimmune diseases in patients with pemphigus and their first-degree relatives. Int J Dermatol. 2011;50:827-831.
To the Editor:
The term pemphigus describes a group of autoimmune blistering diseases that are histologically characterized by intraepidermal blisters caused by acantholysis. There are several types of pemphigus foliaceus, such as classic and endemic pemphigus foliaceus, pemphigus erythematosus, pemphigus herpetiformis, and drug-induced pemphigus foliaceus.1
|
|
| Figure 1. Multiple erosions and crusted lesions were present on the back. |
|
|
| Figure 2. Subcorneal bulla containing acantholytic keratinocytes and neutrophils (H&E, original magnification ×100). |
Drug-induced pemphigus foliaceus in patients treated with penicillamine for rheumatoid arthritis (RA) is well documented in the literature.2 An association between pemphigus foliaceus and RA without penicillamine therapy is rare. We present a case of a patient with a history of RA who developed this bullous disease.
A 67-year-old woman with a 15-year history of seropositive RA presented with widespread skin lesions of 4 weeks’ duration. Confluent scaly crusted erosions on an erythematous base were present on the back (Figure 1), chest, and abdomen. There was no alteration of the mucous membranes. Medical treatment consisted of methotrexate (10 mg weekly), folic acid (5 mg twice weekly), prednisolone (5 mg daily), and ketoprofen (50 mg daily). Routine blood analysis was unremarkable, except for a positive rheumatoid factor. Histologic examination showed a subcorneal bulla containing acantholytic keratinocytes and neutrophils. There was a mild lymphocytic and eosinophilic infiltrate in the papillary dermis (Figure 2).
Determination of anti-desmoglein 1 and 3 antibodies was performed by a commercial enzyme-linked immunosorbent assay. Desmoglein 1 antibodies were positive with titers of 30 U/mL (positive, ≥20 U/mL), whereas desmoglein 3 antibody was negative. Thus, a diagnosis of pemphigus foliaceus was established. The polymerase chain reaction ligation-based typing method and the nucleotide sequence was used to examine the protein drought-repressed 4 gene complex, DR4, which tested positive.
Based on a diagnosis of pemphigus foliaceus, the patient’s corticosteroid treatment was changed from 5 mg daily of prednisolone to 40 mg daily of methylprednisolone, leading to marked improvement of the cutaneous lesions. After tapering the steroid dosage over a period of 3 months, no relapse occurred.
Pemphigus foliaceus is a rare autoimmune blistering disease. It can be induced by drugs, such as penicillamine and captopril.2,3 Captopril, an angiotensin-converting enzyme inhibitor, is closely related to penicillamine structurally. Both drugs have highly active thiol groups capable of reducing disulfide bonds and inducing acantholysis.4 The drugs taken by our patient typically are not known to induce pemphigus foliaceus.
The association of pemphigus foliaceus with RA in the absence of penicillamine therapy was first described by Wilkinson et al.4 Since then, additional cases have been published.5,6 Pemphigus foliaceus also has been described with other autoimmune conditions such as autoimmune thyroid disease.7
Rheumatoid arthritis has been genetically linked to the HLA-DR4 gene complex, which also was found in our patient. Patients with pemphigus foliaceus and RA have an increased frequency of the class II major histocompatibility complex, serologically defined HLA-DR4, and HLA-DRw6 haplotypes.4 Therefore, we believe that the association of pemphigus foliaceus and RA in our patient might not be fortuitous.
To the Editor:
The term pemphigus describes a group of autoimmune blistering diseases that are histologically characterized by intraepidermal blisters caused by acantholysis. There are several types of pemphigus foliaceus, such as classic and endemic pemphigus foliaceus, pemphigus erythematosus, pemphigus herpetiformis, and drug-induced pemphigus foliaceus.1
|
|
| Figure 1. Multiple erosions and crusted lesions were present on the back. |
|
|
| Figure 2. Subcorneal bulla containing acantholytic keratinocytes and neutrophils (H&E, original magnification ×100). |
Drug-induced pemphigus foliaceus in patients treated with penicillamine for rheumatoid arthritis (RA) is well documented in the literature.2 An association between pemphigus foliaceus and RA without penicillamine therapy is rare. We present a case of a patient with a history of RA who developed this bullous disease.
A 67-year-old woman with a 15-year history of seropositive RA presented with widespread skin lesions of 4 weeks’ duration. Confluent scaly crusted erosions on an erythematous base were present on the back (Figure 1), chest, and abdomen. There was no alteration of the mucous membranes. Medical treatment consisted of methotrexate (10 mg weekly), folic acid (5 mg twice weekly), prednisolone (5 mg daily), and ketoprofen (50 mg daily). Routine blood analysis was unremarkable, except for a positive rheumatoid factor. Histologic examination showed a subcorneal bulla containing acantholytic keratinocytes and neutrophils. There was a mild lymphocytic and eosinophilic infiltrate in the papillary dermis (Figure 2).
Determination of anti-desmoglein 1 and 3 antibodies was performed by a commercial enzyme-linked immunosorbent assay. Desmoglein 1 antibodies were positive with titers of 30 U/mL (positive, ≥20 U/mL), whereas desmoglein 3 antibody was negative. Thus, a diagnosis of pemphigus foliaceus was established. The polymerase chain reaction ligation-based typing method and the nucleotide sequence was used to examine the protein drought-repressed 4 gene complex, DR4, which tested positive.
Based on a diagnosis of pemphigus foliaceus, the patient’s corticosteroid treatment was changed from 5 mg daily of prednisolone to 40 mg daily of methylprednisolone, leading to marked improvement of the cutaneous lesions. After tapering the steroid dosage over a period of 3 months, no relapse occurred.
Pemphigus foliaceus is a rare autoimmune blistering disease. It can be induced by drugs, such as penicillamine and captopril.2,3 Captopril, an angiotensin-converting enzyme inhibitor, is closely related to penicillamine structurally. Both drugs have highly active thiol groups capable of reducing disulfide bonds and inducing acantholysis.4 The drugs taken by our patient typically are not known to induce pemphigus foliaceus.
The association of pemphigus foliaceus with RA in the absence of penicillamine therapy was first described by Wilkinson et al.4 Since then, additional cases have been published.5,6 Pemphigus foliaceus also has been described with other autoimmune conditions such as autoimmune thyroid disease.7
Rheumatoid arthritis has been genetically linked to the HLA-DR4 gene complex, which also was found in our patient. Patients with pemphigus foliaceus and RA have an increased frequency of the class II major histocompatibility complex, serologically defined HLA-DR4, and HLA-DRw6 haplotypes.4 Therefore, we believe that the association of pemphigus foliaceus and RA in our patient might not be fortuitous.
1. Chams-Davatchi C, Valikhani M, Daneshpazhooh M, et al. Pemphigus: analysis of 1209 cases. Int J Dermatol. 2005;44:470-476.
2. Sugita K, Hirokawa H, Izu K, et al. D-penicillamine-induced pemphigus successfully treated with combination therapy of mizoribine and prednisolone. J Dermatolog Treat. 2004;15:214-217.
3. Kaplan RP, Potter TS, Fox JN. Drug-induced pemphigus related to angiotensin-converting enzyme inhibitors. J Am Acad Dermatol. 1992;26(2, pt 2):364-366.
4. Wilkinson SM, Smith AG, Davis MJ, et al. Rheumatoid arthritis: an association with pemphigus foliaceus. Acta Derm Venereol. 1992;72:289-291.
5. Sáez-de-Ocariz M, Granados J, Yamamoto-Furusho JK, et al. Rheumatoid arthritis associated with pemphigus foliaceus in a patient not taking penicillamine. Skinmed. 2007;6:252-254.
6. Gürcan HM, Ahmed RA. Analysis of current data on the use of methotrexate in the treatment of pemphigus and pemphigoid. Br J Dermatol. 2009;161:723-731.
7. Leshem YA, Katzenelson V, Yosipovitch G, et al. Autoimmune diseases in patients with pemphigus and their first-degree relatives. Int J Dermatol. 2011;50:827-831.
1. Chams-Davatchi C, Valikhani M, Daneshpazhooh M, et al. Pemphigus: analysis of 1209 cases. Int J Dermatol. 2005;44:470-476.
2. Sugita K, Hirokawa H, Izu K, et al. D-penicillamine-induced pemphigus successfully treated with combination therapy of mizoribine and prednisolone. J Dermatolog Treat. 2004;15:214-217.
3. Kaplan RP, Potter TS, Fox JN. Drug-induced pemphigus related to angiotensin-converting enzyme inhibitors. J Am Acad Dermatol. 1992;26(2, pt 2):364-366.
4. Wilkinson SM, Smith AG, Davis MJ, et al. Rheumatoid arthritis: an association with pemphigus foliaceus. Acta Derm Venereol. 1992;72:289-291.
5. Sáez-de-Ocariz M, Granados J, Yamamoto-Furusho JK, et al. Rheumatoid arthritis associated with pemphigus foliaceus in a patient not taking penicillamine. Skinmed. 2007;6:252-254.
6. Gürcan HM, Ahmed RA. Analysis of current data on the use of methotrexate in the treatment of pemphigus and pemphigoid. Br J Dermatol. 2009;161:723-731.
7. Leshem YA, Katzenelson V, Yosipovitch G, et al. Autoimmune diseases in patients with pemphigus and their first-degree relatives. Int J Dermatol. 2011;50:827-831.
Practice Points
- Physicians should consider pemphigus foliaceus in the differential diagnosis in patients with rheumatoid arthritis and blistering eruptions.
- Appropriate analyses should be performed, including skin biopsy for histologic and immunohistochemical examination as well as search for circulating antibodies.
VIDEO – Survey: Isolation, rejection is real for acne patients
WASHINGTON – Acne patients are telling the truth when they describe feeling isolated, rejected, and stigmatized – and now there are data to prove it.
Dr. Alexa B. Kimball, professor of dermatology at Harvard Medical School, Boston, found that almost 70% of people surveyed believe that those with acne are unattractive and hesitate to be seen with them. Her survey of 56 people also found that they harbor fears that acne is infectious and can be transmitted, that it’s caused by poor hygiene and diet.
“The widespread misconceptions about acne contribute to negative perceptions, which can affect patients’ quality of life and social interaction,” Dr. Kimball said. “When our patients describe these feelings, they are describing their real, day-to-day life experiences.”
See more of her comments on treating patients with acne in this video.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
WASHINGTON – Acne patients are telling the truth when they describe feeling isolated, rejected, and stigmatized – and now there are data to prove it.
Dr. Alexa B. Kimball, professor of dermatology at Harvard Medical School, Boston, found that almost 70% of people surveyed believe that those with acne are unattractive and hesitate to be seen with them. Her survey of 56 people also found that they harbor fears that acne is infectious and can be transmitted, that it’s caused by poor hygiene and diet.
“The widespread misconceptions about acne contribute to negative perceptions, which can affect patients’ quality of life and social interaction,” Dr. Kimball said. “When our patients describe these feelings, they are describing their real, day-to-day life experiences.”
See more of her comments on treating patients with acne in this video.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
WASHINGTON – Acne patients are telling the truth when they describe feeling isolated, rejected, and stigmatized – and now there are data to prove it.
Dr. Alexa B. Kimball, professor of dermatology at Harvard Medical School, Boston, found that almost 70% of people surveyed believe that those with acne are unattractive and hesitate to be seen with them. Her survey of 56 people also found that they harbor fears that acne is infectious and can be transmitted, that it’s caused by poor hygiene and diet.
“The widespread misconceptions about acne contribute to negative perceptions, which can affect patients’ quality of life and social interaction,” Dr. Kimball said. “When our patients describe these feelings, they are describing their real, day-to-day life experiences.”
See more of her comments on treating patients with acne in this video.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT AAD 16

New SHM Members – March 2016
D. Holt, Arkansas
S. Bhansali, MD, California
E. Bhansin, BS, DO, California
A. Bogart, MD, California
D. Donald, MD, California
S. Evans, FNP, California
R. Godbout, California
E. Gustafson, MD, California
M. Hasan, MD, California
M. Kantor, MD, California
M. McClellan, California
A. Milin, MD, California
E. Nguyen, California
J. Young, BSN, RN, California
M. Hicks, MD, Colorado
J. Tyler, DO, Colorado
P. Sharma, MHA, Connecticut
J. Meyer, DO, Florida
M. Prabhu, ACMPE, Florida
K. Slazinski, MD, MA, Florida
P. Zipper, Florida
S. Gayle, MD, Georgia
N. Palmer, DO, Georgia
R. Schaefer, MD, Hawaii
E. Diehl, MD, Iowa
C. Kuehn, MD, Iowa
A. Buchwach, MD, Illinois
F. Evangelista, MD, Illinois
S. Kurup, Illinois
J. Shanahan, Illinois
U. Tekin, MD, Illinois
C. Dennis, Massachusetts
D. Gewanter, Massachusetts
S. Master, MD, Massachusetts
C. Mathew, MD, Massachusetts
M. Sakr, MD, Massachusetts
M. Mason, PA-C, Maryland
M. Kowalczyk, ACNP, Minnesota
M. Doose, MD, Minnesota
F. Abualrub, Missouri
R. Adkison, DO, Missouri
S. Katukoori, MD, Missouri
A. Persaud, PA-C, Missouri
L. Gerstle, NP, Montana
S. Brown, MD, North Carolina
S. Okorie, New Jersey
P. Mathew, MD, New Mexico
A. Turney, DO, Nevada
J. Chester, MD, New York
C. Flynn, DO, New York
M. Hoefer, PMGR, New York
M. Islam, MD, New York
C. Karno, MD, New York
S. Mir, New York
P. Nadkarni, MD, New York
A. Rana, New York
G. Rubinfeld, New York
A. Black, MBA, Ohio
R. Cartabuke, MHA, Ohio
P. He, DO, Ohio
R. Rivero, DO, Ohio
K. Welch, Ohio
S. Parker, APRN-BC, Oklahoma
T. Basra, MD, MBBS, Oregon
B. Baxter, Oregon
S. Mehta, MD, Oregon
V. Karper, ACNP, Pennsylvania
D. Messner, MSN, NP, Pennsylvania
M. Scoulos-Hanson, Pennsylvania
K. Willoughby, MD, Pennsylvania
R. Yazdanfar, MD, Pennsylvania
J. Hennessey, CCFP, Canada
J. Brown, MD, South Carolina
K. Medlin, RN, South Carolina
K. Kays, Tennessee
M. Begum, MD, Texas
S. Blinchevsky, MD, Texas
C. Ciborowski, Texas
K. Gupta, MD, Texas
H. Lam, MD, Texas
A. Owens, Texas
K. Salciccioli, BSE, Texas
D. Lundberg, Wisconsin
H. Peto, Wisconsin
B. Quinn, MD, Wisconsin
N. Ros, PhD, Spain
S. Takeuchi, MD, Japan
D. Holt, Arkansas
S. Bhansali, MD, California
E. Bhansin, BS, DO, California
A. Bogart, MD, California
D. Donald, MD, California
S. Evans, FNP, California
R. Godbout, California
E. Gustafson, MD, California
M. Hasan, MD, California
M. Kantor, MD, California
M. McClellan, California
A. Milin, MD, California
E. Nguyen, California
J. Young, BSN, RN, California
M. Hicks, MD, Colorado
J. Tyler, DO, Colorado
P. Sharma, MHA, Connecticut
J. Meyer, DO, Florida
M. Prabhu, ACMPE, Florida
K. Slazinski, MD, MA, Florida
P. Zipper, Florida
S. Gayle, MD, Georgia
N. Palmer, DO, Georgia
R. Schaefer, MD, Hawaii
E. Diehl, MD, Iowa
C. Kuehn, MD, Iowa
A. Buchwach, MD, Illinois
F. Evangelista, MD, Illinois
S. Kurup, Illinois
J. Shanahan, Illinois
U. Tekin, MD, Illinois
C. Dennis, Massachusetts
D. Gewanter, Massachusetts
S. Master, MD, Massachusetts
C. Mathew, MD, Massachusetts
M. Sakr, MD, Massachusetts
M. Mason, PA-C, Maryland
M. Kowalczyk, ACNP, Minnesota
M. Doose, MD, Minnesota
F. Abualrub, Missouri
R. Adkison, DO, Missouri
S. Katukoori, MD, Missouri
A. Persaud, PA-C, Missouri
L. Gerstle, NP, Montana
S. Brown, MD, North Carolina
S. Okorie, New Jersey
P. Mathew, MD, New Mexico
A. Turney, DO, Nevada
J. Chester, MD, New York
C. Flynn, DO, New York
M. Hoefer, PMGR, New York
M. Islam, MD, New York
C. Karno, MD, New York
S. Mir, New York
P. Nadkarni, MD, New York
A. Rana, New York
G. Rubinfeld, New York
A. Black, MBA, Ohio
R. Cartabuke, MHA, Ohio
P. He, DO, Ohio
R. Rivero, DO, Ohio
K. Welch, Ohio
S. Parker, APRN-BC, Oklahoma
T. Basra, MD, MBBS, Oregon
B. Baxter, Oregon
S. Mehta, MD, Oregon
V. Karper, ACNP, Pennsylvania
D. Messner, MSN, NP, Pennsylvania
M. Scoulos-Hanson, Pennsylvania
K. Willoughby, MD, Pennsylvania
R. Yazdanfar, MD, Pennsylvania
J. Hennessey, CCFP, Canada
J. Brown, MD, South Carolina
K. Medlin, RN, South Carolina
K. Kays, Tennessee
M. Begum, MD, Texas
S. Blinchevsky, MD, Texas
C. Ciborowski, Texas
K. Gupta, MD, Texas
H. Lam, MD, Texas
A. Owens, Texas
K. Salciccioli, BSE, Texas
D. Lundberg, Wisconsin
H. Peto, Wisconsin
B. Quinn, MD, Wisconsin
N. Ros, PhD, Spain
S. Takeuchi, MD, Japan
D. Holt, Arkansas
S. Bhansali, MD, California
E. Bhansin, BS, DO, California
A. Bogart, MD, California
D. Donald, MD, California
S. Evans, FNP, California
R. Godbout, California
E. Gustafson, MD, California
M. Hasan, MD, California
M. Kantor, MD, California
M. McClellan, California
A. Milin, MD, California
E. Nguyen, California
J. Young, BSN, RN, California
M. Hicks, MD, Colorado
J. Tyler, DO, Colorado
P. Sharma, MHA, Connecticut
J. Meyer, DO, Florida
M. Prabhu, ACMPE, Florida
K. Slazinski, MD, MA, Florida
P. Zipper, Florida
S. Gayle, MD, Georgia
N. Palmer, DO, Georgia
R. Schaefer, MD, Hawaii
E. Diehl, MD, Iowa
C. Kuehn, MD, Iowa
A. Buchwach, MD, Illinois
F. Evangelista, MD, Illinois
S. Kurup, Illinois
J. Shanahan, Illinois
U. Tekin, MD, Illinois
C. Dennis, Massachusetts
D. Gewanter, Massachusetts
S. Master, MD, Massachusetts
C. Mathew, MD, Massachusetts
M. Sakr, MD, Massachusetts
M. Mason, PA-C, Maryland
M. Kowalczyk, ACNP, Minnesota
M. Doose, MD, Minnesota
F. Abualrub, Missouri
R. Adkison, DO, Missouri
S. Katukoori, MD, Missouri
A. Persaud, PA-C, Missouri
L. Gerstle, NP, Montana
S. Brown, MD, North Carolina
S. Okorie, New Jersey
P. Mathew, MD, New Mexico
A. Turney, DO, Nevada
J. Chester, MD, New York
C. Flynn, DO, New York
M. Hoefer, PMGR, New York
M. Islam, MD, New York
C. Karno, MD, New York
S. Mir, New York
P. Nadkarni, MD, New York
A. Rana, New York
G. Rubinfeld, New York
A. Black, MBA, Ohio
R. Cartabuke, MHA, Ohio
P. He, DO, Ohio
R. Rivero, DO, Ohio
K. Welch, Ohio
S. Parker, APRN-BC, Oklahoma
T. Basra, MD, MBBS, Oregon
B. Baxter, Oregon
S. Mehta, MD, Oregon
V. Karper, ACNP, Pennsylvania
D. Messner, MSN, NP, Pennsylvania
M. Scoulos-Hanson, Pennsylvania
K. Willoughby, MD, Pennsylvania
R. Yazdanfar, MD, Pennsylvania
J. Hennessey, CCFP, Canada
J. Brown, MD, South Carolina
K. Medlin, RN, South Carolina
K. Kays, Tennessee
M. Begum, MD, Texas
S. Blinchevsky, MD, Texas
C. Ciborowski, Texas
K. Gupta, MD, Texas
H. Lam, MD, Texas
A. Owens, Texas
K. Salciccioli, BSE, Texas
D. Lundberg, Wisconsin
H. Peto, Wisconsin
B. Quinn, MD, Wisconsin
N. Ros, PhD, Spain
S. Takeuchi, MD, Japan
FDA approves long-acting hemophilia B therapy
The US Food and Drug Administration (FDA) has approved Idelvion (Coagulation Factor IX [Recombinant], Albumin Fusion Protein) for the treatment of hemophilia B.
The product—a fusion protein linking recombinant coagulation factor IX with recombinant albumin—is intended for use in children and adults for routine prophylaxis to prevent or reduce the frequency of bleeding episodes, for on-demand control and prevention of bleeding episodes, and for the perioperative management of bleeding.
Appropriate patients age 12 and older can go up to 14 days between Idelvion infusions. This dosing interval has been achieved while maintaining high levels of factor IX activity—above 5% over 14 days at 75 IU/kg.
Idelvion is the first FDA-approved recombinant factor IX therapy that can extend the dosing interval up to 14 days.
Idelvion is expected to be available in the US later this month. The product is being developed by CSL Behring. For more details on the drug, see the full prescribing information.
Phase 3 trial
The FDA approved Idelvion based on results of the PROLONG-9FP clinical development program. PROLONG-9FP includes phase 1, 2, and 3 studies evaluating the safety and efficacy of Idelvion in adults and children (ages 1 to 61) with hemophilia B.
Data from the phase 3 study were recently published in Blood. This study included 63 previously treated male patients with severe hemophilia B. They had a mean age of 33 (range, 12 to 61).
The patients were divided into 2 groups. Group 1 (n=40) received routine prophylaxis with Idelvion once every 7 days for 26 weeks, followed by a 7-, 10- or 14-day prophylaxis regimen for a mean of 50, 38, or 51 weeks, respectively.
Group 2 received on-demand treatment with Idelvion for bleeding episodes for 26 weeks (n=23) and then switched to a 7-day prophylaxis regimen for a mean of 45 weeks (n=19).
The median annualized bleeding rate (ABR) was 2.0 in the prophylaxis arm (group 1) and 23.5 in the on-demand treatment arm (group 2). The median spontaneous ABRs were 0.0 and 17.0, respectively.
For patients in group 2, there was a significant reduction in median ABRs when patients switched from on-demand treatment to prophylaxis—19.22 and 1.58, respectively (P<0.0001). And there was a significant reduction in median spontaneous ABRs—15.43 and 0.00, respectively (P<0.0001).
Overall, 98.6% of bleeding episodes were treated successfully, including 93.6% that were treated with a single injection of Idelvion.
None of the patients developed inhibitors or experienced thromboembolic events, anaphylaxis, or life-threatening adverse events (AEs).
There were 347 treatment-emergent AEs reported in 54 (85.7%) patients. The most common were nasopharyngitis (25.4%), headache (23.8%), arthralgia (4.3%), and influenza (11.1%).
Eleven mild/moderate AEs in 5 patients (7.9%) were considered possibly related to Idelvion. Two patients discontinued treatment due to AEs—1 with hypersensitivity and 1 with headache. ![]()
The US Food and Drug Administration (FDA) has approved Idelvion (Coagulation Factor IX [Recombinant], Albumin Fusion Protein) for the treatment of hemophilia B.
The product—a fusion protein linking recombinant coagulation factor IX with recombinant albumin—is intended for use in children and adults for routine prophylaxis to prevent or reduce the frequency of bleeding episodes, for on-demand control and prevention of bleeding episodes, and for the perioperative management of bleeding.
Appropriate patients age 12 and older can go up to 14 days between Idelvion infusions. This dosing interval has been achieved while maintaining high levels of factor IX activity—above 5% over 14 days at 75 IU/kg.
Idelvion is the first FDA-approved recombinant factor IX therapy that can extend the dosing interval up to 14 days.
Idelvion is expected to be available in the US later this month. The product is being developed by CSL Behring. For more details on the drug, see the full prescribing information.
Phase 3 trial
The FDA approved Idelvion based on results of the PROLONG-9FP clinical development program. PROLONG-9FP includes phase 1, 2, and 3 studies evaluating the safety and efficacy of Idelvion in adults and children (ages 1 to 61) with hemophilia B.
Data from the phase 3 study were recently published in Blood. This study included 63 previously treated male patients with severe hemophilia B. They had a mean age of 33 (range, 12 to 61).
The patients were divided into 2 groups. Group 1 (n=40) received routine prophylaxis with Idelvion once every 7 days for 26 weeks, followed by a 7-, 10- or 14-day prophylaxis regimen for a mean of 50, 38, or 51 weeks, respectively.
Group 2 received on-demand treatment with Idelvion for bleeding episodes for 26 weeks (n=23) and then switched to a 7-day prophylaxis regimen for a mean of 45 weeks (n=19).
The median annualized bleeding rate (ABR) was 2.0 in the prophylaxis arm (group 1) and 23.5 in the on-demand treatment arm (group 2). The median spontaneous ABRs were 0.0 and 17.0, respectively.
For patients in group 2, there was a significant reduction in median ABRs when patients switched from on-demand treatment to prophylaxis—19.22 and 1.58, respectively (P<0.0001). And there was a significant reduction in median spontaneous ABRs—15.43 and 0.00, respectively (P<0.0001).
Overall, 98.6% of bleeding episodes were treated successfully, including 93.6% that were treated with a single injection of Idelvion.
None of the patients developed inhibitors or experienced thromboembolic events, anaphylaxis, or life-threatening adverse events (AEs).
There were 347 treatment-emergent AEs reported in 54 (85.7%) patients. The most common were nasopharyngitis (25.4%), headache (23.8%), arthralgia (4.3%), and influenza (11.1%).
Eleven mild/moderate AEs in 5 patients (7.9%) were considered possibly related to Idelvion. Two patients discontinued treatment due to AEs—1 with hypersensitivity and 1 with headache. ![]()
The US Food and Drug Administration (FDA) has approved Idelvion (Coagulation Factor IX [Recombinant], Albumin Fusion Protein) for the treatment of hemophilia B.
The product—a fusion protein linking recombinant coagulation factor IX with recombinant albumin—is intended for use in children and adults for routine prophylaxis to prevent or reduce the frequency of bleeding episodes, for on-demand control and prevention of bleeding episodes, and for the perioperative management of bleeding.
Appropriate patients age 12 and older can go up to 14 days between Idelvion infusions. This dosing interval has been achieved while maintaining high levels of factor IX activity—above 5% over 14 days at 75 IU/kg.
Idelvion is the first FDA-approved recombinant factor IX therapy that can extend the dosing interval up to 14 days.
Idelvion is expected to be available in the US later this month. The product is being developed by CSL Behring. For more details on the drug, see the full prescribing information.
Phase 3 trial
The FDA approved Idelvion based on results of the PROLONG-9FP clinical development program. PROLONG-9FP includes phase 1, 2, and 3 studies evaluating the safety and efficacy of Idelvion in adults and children (ages 1 to 61) with hemophilia B.
Data from the phase 3 study were recently published in Blood. This study included 63 previously treated male patients with severe hemophilia B. They had a mean age of 33 (range, 12 to 61).
The patients were divided into 2 groups. Group 1 (n=40) received routine prophylaxis with Idelvion once every 7 days for 26 weeks, followed by a 7-, 10- or 14-day prophylaxis regimen for a mean of 50, 38, or 51 weeks, respectively.
Group 2 received on-demand treatment with Idelvion for bleeding episodes for 26 weeks (n=23) and then switched to a 7-day prophylaxis regimen for a mean of 45 weeks (n=19).
The median annualized bleeding rate (ABR) was 2.0 in the prophylaxis arm (group 1) and 23.5 in the on-demand treatment arm (group 2). The median spontaneous ABRs were 0.0 and 17.0, respectively.
For patients in group 2, there was a significant reduction in median ABRs when patients switched from on-demand treatment to prophylaxis—19.22 and 1.58, respectively (P<0.0001). And there was a significant reduction in median spontaneous ABRs—15.43 and 0.00, respectively (P<0.0001).
Overall, 98.6% of bleeding episodes were treated successfully, including 93.6% that were treated with a single injection of Idelvion.
None of the patients developed inhibitors or experienced thromboembolic events, anaphylaxis, or life-threatening adverse events (AEs).
There were 347 treatment-emergent AEs reported in 54 (85.7%) patients. The most common were nasopharyngitis (25.4%), headache (23.8%), arthralgia (4.3%), and influenza (11.1%).
Eleven mild/moderate AEs in 5 patients (7.9%) were considered possibly related to Idelvion. Two patients discontinued treatment due to AEs—1 with hypersensitivity and 1 with headache. ![]()
U.S. flu activity falls for first time since early January
Influenza-like illness (ILI) activity in the 2015-2016 U.S. flu season declined for the first time since early January, according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for ILI was reported at 3.2% for the week ending Feb. 20, but the CDC has adjusted that figure to 3.3%, which makes the 3.2% reported for this most recent week (week 20 of the season, ending Feb. 27, 2016) a decrease from the week before.
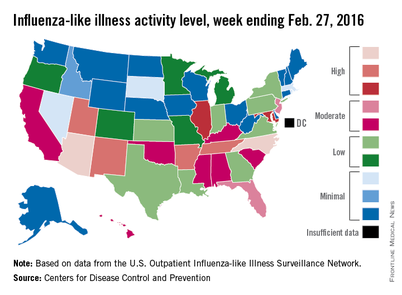
Despite that drop, two states were at level 10 on the CDC’s 1-10 scale of ILI activity for the first time this season. Arizona had already reached level 10, and joining it there last week was North Carolina, moving up from level 8 the week before. Other states in the “high” range of activity were Arkansas, New Mexico, Tennessee, and Utah at level 9, and Illinois and Maryland at level 8, the CDC reported March 4. Puerto Rico, which had been at level 10 for several weeks, moved down to level 8.
States in the “moderate” range of activity for the week ending Feb. 27 were Florida and New Jersey at level 7 and Alabama, California, Hawaii, Kentucky, Mississippi, Oklahoma, and South Carolina at level 6. Altogether, there were 35 states at level 2 or higher, according to data from the CDC’s Outpatient Influenza-like Illness Surveillance Network.
Four pediatric ILI-related deaths were reported to the CDC during week 20, but three actually occurred during week 19. There have been 18 ILI-related deaths so far during the 2015-2016 season, the CDC said.
Influenza-like illness (ILI) activity in the 2015-2016 U.S. flu season declined for the first time since early January, according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for ILI was reported at 3.2% for the week ending Feb. 20, but the CDC has adjusted that figure to 3.3%, which makes the 3.2% reported for this most recent week (week 20 of the season, ending Feb. 27, 2016) a decrease from the week before.
Despite that drop, two states were at level 10 on the CDC’s 1-10 scale of ILI activity for the first time this season. Arizona had already reached level 10, and joining it there last week was North Carolina, moving up from level 8 the week before. Other states in the “high” range of activity were Arkansas, New Mexico, Tennessee, and Utah at level 9, and Illinois and Maryland at level 8, the CDC reported March 4. Puerto Rico, which had been at level 10 for several weeks, moved down to level 8.
States in the “moderate” range of activity for the week ending Feb. 27 were Florida and New Jersey at level 7 and Alabama, California, Hawaii, Kentucky, Mississippi, Oklahoma, and South Carolina at level 6. Altogether, there were 35 states at level 2 or higher, according to data from the CDC’s Outpatient Influenza-like Illness Surveillance Network.
Four pediatric ILI-related deaths were reported to the CDC during week 20, but three actually occurred during week 19. There have been 18 ILI-related deaths so far during the 2015-2016 season, the CDC said.
Influenza-like illness (ILI) activity in the 2015-2016 U.S. flu season declined for the first time since early January, according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for ILI was reported at 3.2% for the week ending Feb. 20, but the CDC has adjusted that figure to 3.3%, which makes the 3.2% reported for this most recent week (week 20 of the season, ending Feb. 27, 2016) a decrease from the week before.
Despite that drop, two states were at level 10 on the CDC’s 1-10 scale of ILI activity for the first time this season. Arizona had already reached level 10, and joining it there last week was North Carolina, moving up from level 8 the week before. Other states in the “high” range of activity were Arkansas, New Mexico, Tennessee, and Utah at level 9, and Illinois and Maryland at level 8, the CDC reported March 4. Puerto Rico, which had been at level 10 for several weeks, moved down to level 8.
States in the “moderate” range of activity for the week ending Feb. 27 were Florida and New Jersey at level 7 and Alabama, California, Hawaii, Kentucky, Mississippi, Oklahoma, and South Carolina at level 6. Altogether, there were 35 states at level 2 or higher, according to data from the CDC’s Outpatient Influenza-like Illness Surveillance Network.
Four pediatric ILI-related deaths were reported to the CDC during week 20, but three actually occurred during week 19. There have been 18 ILI-related deaths so far during the 2015-2016 season, the CDC said.
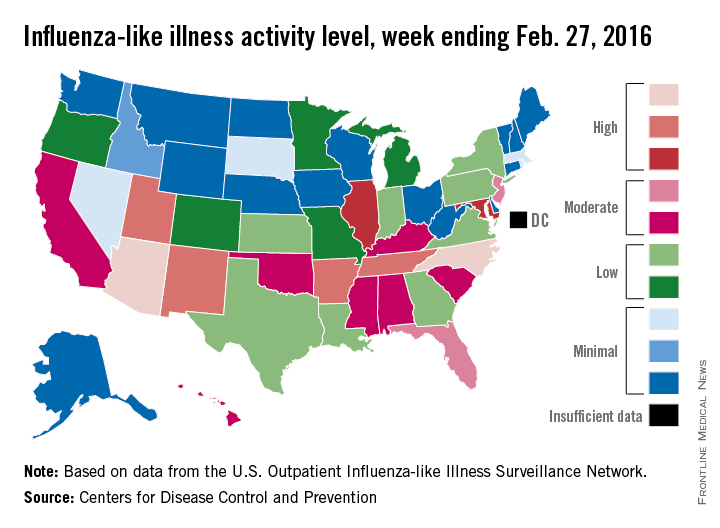
Guidelines: Combine topical, oral therapy for most effective acne treatment
WASHINGTON – Monotherapy is not recommended in treating moderate-severe acne, and antibiotics should always be coupled with topical therapy, according to the latest guidelines from the American Academy of Dermatology.
And although it may be hard – even nearly impossible – to discontinue antibiotics completely, patients should be reevaluated every 3-4 months to determine whether reducing the dosage may be possible while maintaining effectiveness, the document says.
AAD published the guideline on Feb 17. At the academy’s annual meeting, a panel met to discuss its practical application.
Topical therapy
Benzoyl peroxide is a first-line agent that not only effectively fights Propionibacterium acnes, but also discourages the development of antibiotic resistance. Topical antibiotics also decrease P. acnes populations and exert a mild anti-inflammatory effect; however, monotherapy with a topical antibiotic is strongly discouraged. These should be used in combination with another agent such as a retinoid, benzoyl peroxide, adapalene, azelaic acid, or dapsone. This approach decreases the chance of antibiotic resistance, attacks the acne on several fronts, and provides for a maintenance transition.
Systemic antibiotics
Tetracycline-class antibiotics are still the best option for moderate-severe acne. A Cochrane review found that minocycline and doxycycline are equally effective (Cochrane Skin Group Nov 2011. doi: 10.1002/14651858.CD002086.pub2).
The incidence of adverse events associated with each is low, although minocycline may be marginally more troublesome. Low doses seem to be as effective as traditional doses, but pulsed therapy is inadequate. To prevent antibiotic resistance, limit both dose and length of therapy as much as possible. This can best be accomplished by adding a topical agent – either benzoyl peroxide or a retinoid – to the regimen.
“This is critical,” Dr. Jonette E. Keri of the University of Miami said at the meeting. “When antibiotics are eventually discontinued, the retinoid will fulfill the need for maintenance therapy.”
Hormonal agents
Four combination oral contraceptives are Food and Drug Administration–approved for acne treatment. Each of them decreases androgens by interrupting the pathway of testosterone production. There are no data suggesting that one is better than the other; patient preferences and their individual clinical picture should drive choice. Because of the cardiovascular risks associated with these combination OCs, they should not be prescribed for anyone with a personal or family history of clotting disorders or thromboembolic events. Smoking should also be a contraindication.
Oral contraceptives can be tried alone or as part of a comprehensive treatment regimen, including one containing antibiotics. Rifampin and griseofulvin are the only antibiotics known to decrease the contraceptive effect of the medications.
The tincture of time is an important part of this therapy, said Dr. Diane M. Thiboutot, professor of dermatology at Pennsylvania State University, Hershey. “You can’t rush it. It may take three cycles to see any real improvement in acne, and patients should be aware of this.”
Isotretinoin
Oral isotretinoin is a highly effective treatment for severe, recalcitrant acne. It decreases sebum production, acne lesion count, and scarring. Despite concerns about depression and suicidality, isotretinoin treatment can actually improve mood in most patients, said Dr. Megha M. Tollefson of the Mayo Clinic, Rochester, Minn.
“A very well-done Swedish study published in 2010 in BMJ found a slightly increased risk of suicide in the first 6 months after treatment started, but that risk was already rising before treatment started, so it could [be unrelated] to the drug,” she said. “And, in those who got isotretinoin, the [suicide] rate after that was actually decreased, compared to the general population.”
Female patients need education on isotretinoin’s teratogenic potential. After discussions, they should sign the SMART or iPLEDGE agreements about using effective birth control while taking the drug. Unfortunately, Dr. Tollefson said, “We continue to see hundreds of isotretinoin-exposed pregnancies each year.”
A recent study found that up to 30% of women did not comply with the birth control measures they agreed to while taking the drug (J Am Acad Dermatol. 2011 Oct. doi: 10.1016/j.jaad.2013.08.034).
The link between isotretinoin and inflammatory bowel disease is not well founded, Dr. Tollefson said. Studies have been contradictory, and most evidence is based on case report and association studies. There is, however, some evidence suggesting an innate connection between acne and inflammatory bowel disease, she noted.
Diet
Emerging evidence suggests that high glycemic diets may be associated with acne, but these studies are small. However, those randomized to a low glycemic index diet showed decreased sebum production and inflammation.
A small case-control study in 2012 suggested a link between milk and acne. AAD makes no recommendation based on this. Milk remains an important source of calcium and vitamin D for Americans, especially children, the panel said.
Dr. Tollefson had no financial disclosures. Dr. Keri said she has been a consultant for Hoffmann-LaRoche.
WASHINGTON – Monotherapy is not recommended in treating moderate-severe acne, and antibiotics should always be coupled with topical therapy, according to the latest guidelines from the American Academy of Dermatology.
And although it may be hard – even nearly impossible – to discontinue antibiotics completely, patients should be reevaluated every 3-4 months to determine whether reducing the dosage may be possible while maintaining effectiveness, the document says.
AAD published the guideline on Feb 17. At the academy’s annual meeting, a panel met to discuss its practical application.
Topical therapy
Benzoyl peroxide is a first-line agent that not only effectively fights Propionibacterium acnes, but also discourages the development of antibiotic resistance. Topical antibiotics also decrease P. acnes populations and exert a mild anti-inflammatory effect; however, monotherapy with a topical antibiotic is strongly discouraged. These should be used in combination with another agent such as a retinoid, benzoyl peroxide, adapalene, azelaic acid, or dapsone. This approach decreases the chance of antibiotic resistance, attacks the acne on several fronts, and provides for a maintenance transition.
Systemic antibiotics
Tetracycline-class antibiotics are still the best option for moderate-severe acne. A Cochrane review found that minocycline and doxycycline are equally effective (Cochrane Skin Group Nov 2011. doi: 10.1002/14651858.CD002086.pub2).
The incidence of adverse events associated with each is low, although minocycline may be marginally more troublesome. Low doses seem to be as effective as traditional doses, but pulsed therapy is inadequate. To prevent antibiotic resistance, limit both dose and length of therapy as much as possible. This can best be accomplished by adding a topical agent – either benzoyl peroxide or a retinoid – to the regimen.
“This is critical,” Dr. Jonette E. Keri of the University of Miami said at the meeting. “When antibiotics are eventually discontinued, the retinoid will fulfill the need for maintenance therapy.”
Hormonal agents
Four combination oral contraceptives are Food and Drug Administration–approved for acne treatment. Each of them decreases androgens by interrupting the pathway of testosterone production. There are no data suggesting that one is better than the other; patient preferences and their individual clinical picture should drive choice. Because of the cardiovascular risks associated with these combination OCs, they should not be prescribed for anyone with a personal or family history of clotting disorders or thromboembolic events. Smoking should also be a contraindication.
Oral contraceptives can be tried alone or as part of a comprehensive treatment regimen, including one containing antibiotics. Rifampin and griseofulvin are the only antibiotics known to decrease the contraceptive effect of the medications.
The tincture of time is an important part of this therapy, said Dr. Diane M. Thiboutot, professor of dermatology at Pennsylvania State University, Hershey. “You can’t rush it. It may take three cycles to see any real improvement in acne, and patients should be aware of this.”
Isotretinoin
Oral isotretinoin is a highly effective treatment for severe, recalcitrant acne. It decreases sebum production, acne lesion count, and scarring. Despite concerns about depression and suicidality, isotretinoin treatment can actually improve mood in most patients, said Dr. Megha M. Tollefson of the Mayo Clinic, Rochester, Minn.
“A very well-done Swedish study published in 2010 in BMJ found a slightly increased risk of suicide in the first 6 months after treatment started, but that risk was already rising before treatment started, so it could [be unrelated] to the drug,” she said. “And, in those who got isotretinoin, the [suicide] rate after that was actually decreased, compared to the general population.”
Female patients need education on isotretinoin’s teratogenic potential. After discussions, they should sign the SMART or iPLEDGE agreements about using effective birth control while taking the drug. Unfortunately, Dr. Tollefson said, “We continue to see hundreds of isotretinoin-exposed pregnancies each year.”
A recent study found that up to 30% of women did not comply with the birth control measures they agreed to while taking the drug (J Am Acad Dermatol. 2011 Oct. doi: 10.1016/j.jaad.2013.08.034).
The link between isotretinoin and inflammatory bowel disease is not well founded, Dr. Tollefson said. Studies have been contradictory, and most evidence is based on case report and association studies. There is, however, some evidence suggesting an innate connection between acne and inflammatory bowel disease, she noted.
Diet
Emerging evidence suggests that high glycemic diets may be associated with acne, but these studies are small. However, those randomized to a low glycemic index diet showed decreased sebum production and inflammation.
A small case-control study in 2012 suggested a link between milk and acne. AAD makes no recommendation based on this. Milk remains an important source of calcium and vitamin D for Americans, especially children, the panel said.
Dr. Tollefson had no financial disclosures. Dr. Keri said she has been a consultant for Hoffmann-LaRoche.
WASHINGTON – Monotherapy is not recommended in treating moderate-severe acne, and antibiotics should always be coupled with topical therapy, according to the latest guidelines from the American Academy of Dermatology.
And although it may be hard – even nearly impossible – to discontinue antibiotics completely, patients should be reevaluated every 3-4 months to determine whether reducing the dosage may be possible while maintaining effectiveness, the document says.
AAD published the guideline on Feb 17. At the academy’s annual meeting, a panel met to discuss its practical application.
Topical therapy
Benzoyl peroxide is a first-line agent that not only effectively fights Propionibacterium acnes, but also discourages the development of antibiotic resistance. Topical antibiotics also decrease P. acnes populations and exert a mild anti-inflammatory effect; however, monotherapy with a topical antibiotic is strongly discouraged. These should be used in combination with another agent such as a retinoid, benzoyl peroxide, adapalene, azelaic acid, or dapsone. This approach decreases the chance of antibiotic resistance, attacks the acne on several fronts, and provides for a maintenance transition.
Systemic antibiotics
Tetracycline-class antibiotics are still the best option for moderate-severe acne. A Cochrane review found that minocycline and doxycycline are equally effective (Cochrane Skin Group Nov 2011. doi: 10.1002/14651858.CD002086.pub2).
The incidence of adverse events associated with each is low, although minocycline may be marginally more troublesome. Low doses seem to be as effective as traditional doses, but pulsed therapy is inadequate. To prevent antibiotic resistance, limit both dose and length of therapy as much as possible. This can best be accomplished by adding a topical agent – either benzoyl peroxide or a retinoid – to the regimen.
“This is critical,” Dr. Jonette E. Keri of the University of Miami said at the meeting. “When antibiotics are eventually discontinued, the retinoid will fulfill the need for maintenance therapy.”
Hormonal agents
Four combination oral contraceptives are Food and Drug Administration–approved for acne treatment. Each of them decreases androgens by interrupting the pathway of testosterone production. There are no data suggesting that one is better than the other; patient preferences and their individual clinical picture should drive choice. Because of the cardiovascular risks associated with these combination OCs, they should not be prescribed for anyone with a personal or family history of clotting disorders or thromboembolic events. Smoking should also be a contraindication.
Oral contraceptives can be tried alone or as part of a comprehensive treatment regimen, including one containing antibiotics. Rifampin and griseofulvin are the only antibiotics known to decrease the contraceptive effect of the medications.
The tincture of time is an important part of this therapy, said Dr. Diane M. Thiboutot, professor of dermatology at Pennsylvania State University, Hershey. “You can’t rush it. It may take three cycles to see any real improvement in acne, and patients should be aware of this.”
Isotretinoin
Oral isotretinoin is a highly effective treatment for severe, recalcitrant acne. It decreases sebum production, acne lesion count, and scarring. Despite concerns about depression and suicidality, isotretinoin treatment can actually improve mood in most patients, said Dr. Megha M. Tollefson of the Mayo Clinic, Rochester, Minn.
“A very well-done Swedish study published in 2010 in BMJ found a slightly increased risk of suicide in the first 6 months after treatment started, but that risk was already rising before treatment started, so it could [be unrelated] to the drug,” she said. “And, in those who got isotretinoin, the [suicide] rate after that was actually decreased, compared to the general population.”
Female patients need education on isotretinoin’s teratogenic potential. After discussions, they should sign the SMART or iPLEDGE agreements about using effective birth control while taking the drug. Unfortunately, Dr. Tollefson said, “We continue to see hundreds of isotretinoin-exposed pregnancies each year.”
A recent study found that up to 30% of women did not comply with the birth control measures they agreed to while taking the drug (J Am Acad Dermatol. 2011 Oct. doi: 10.1016/j.jaad.2013.08.034).
The link between isotretinoin and inflammatory bowel disease is not well founded, Dr. Tollefson said. Studies have been contradictory, and most evidence is based on case report and association studies. There is, however, some evidence suggesting an innate connection between acne and inflammatory bowel disease, she noted.
Diet
Emerging evidence suggests that high glycemic diets may be associated with acne, but these studies are small. However, those randomized to a low glycemic index diet showed decreased sebum production and inflammation.
A small case-control study in 2012 suggested a link between milk and acne. AAD makes no recommendation based on this. Milk remains an important source of calcium and vitamin D for Americans, especially children, the panel said.
Dr. Tollefson had no financial disclosures. Dr. Keri said she has been a consultant for Hoffmann-LaRoche.
AT AAD 2016
FDA approves ibrutinib as first-line CLL therapy

Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has approved the BTK inhibitor ibrutinib (Imbruvica) as a first-line treatment for patients with chronic lymphocytic leukemia (CLL).
This means ibrutinib is now FDA-approved to treat CLL patients regardless of their treatment history, including patients with 17p deletion.
Ibrutinib is also FDA-approved to treat Waldenström’s macroglobulinemia, and the drug was granted accelerated approval to treat patients with mantle cell lymphoma who have received at least 1 prior therapy.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc. For more details on the drug, see the full prescribing information, available at imbruvica.com.
RESONATE-2 trial
The latest FDA approval for ibrutinib is based on results from the phase 3 RESONATE-2 trial (PCYC-1115), which were presented at the 2015 ASH Annual Meeting and simultaneously published in NEJM.
RESONATE-2 enrolled 269 treatment-naïve patients with CLL or small lymphocytic lymphoma who were 65 or older.
Patients were randomized to receive ibrutinib (n=136) at 420 mg once a day until progression or unacceptable toxicity, or chlorambucil (n=133) on days 1 and 15 of each 28-day cycle for up to 12 cycles. The starting dose for chlorambucil in cycle 1 was 0.5 mg/kg and was increased based on tolerability in cycle 2 by increments of 0.1 mg/kg to a maximum of 0.8 mg/kg.
The primary endpoint of the study was progression-free survival (PFS), as assessed by an independent review committee (IRC) according to the International Workshop on Chronic Lymphocytic Leukemia (iWCLL) 2008 criteria, with modification for treatment-related lymphocytosis.
Key secondary endpoints included overall response rate (based on the same iWCLL criteria), overall survival (OS), and safety.
Ibrutinib significantly prolonged PFS, as determined by the IRC, reducing the risk of progression or death by 84% compared to chlorambucil. The hazard ratio was 0.16 (P<0.001). The median PFS was not reached in the ibrutinib arm but was 18.9 months for the chlorambucil arm.
Ibrutinib significantly prolonged OS as well, although the median OS was not reached in either treatment arm. The OS rate at 24 months was 98% with ibrutinib and 85% with chlorambucil. The relative risk of death with ibrutinib was 84% lower than that with chlorambucil. The hazard ratio was 0.16 (P=0.001).
Ibrutinib was associated with a significantly higher IRC-assessed overall response rate compared to chlorambucil—82% and 35%, respectively (P<0.0001). Five patients (4%) in the ibrutinib arm achieved a complete response, as did 2 patients (2%) in the chlorambucil arm.
The median duration of treatment was 17.4 months in the ibrutinib arm and 7.1 months in the chlorambucil arm.
The most common adverse events of any grade—in the ibrutinib and chlorambucil arms, respectively—were diarrhea (42% and 17%), fatigue (30% and 38%), cough (22% and 15%), nausea (22% and 39%), peripheral edema (19% and 9%), dry eye (17% and 5%), arthralgia (16% and 7%), neutropenia (16% and 23%), and vomiting (13% and 20%).
Adverse events of grade 3 or higher—in the ibrutinib and chlorambucil arms, respectively—were neutropenia (10% and 18%), anemia (6% and 8%), hypertension (4% and 0%), pneumonia (4% and 2%), diarrhea (4% and 0%), maculopapular rash (3% and 2%), decreased platelet count (3% and 1%), abdominal pain (3% and 1%), hyponatremia (3% and 0%), thrombocytopenia (2% and 6%), febrile neutropenia (2% and 2%), upper respiratory tract infection (2% and 2%), pleural effusion (2% and 1%), cellulitis (2% and 0%), fatigue (1% and 5%), syncope (1% and 2%), and hemolytic anemia (0% and 2%). ![]()
Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has approved the BTK inhibitor ibrutinib (Imbruvica) as a first-line treatment for patients with chronic lymphocytic leukemia (CLL).
This means ibrutinib is now FDA-approved to treat CLL patients regardless of their treatment history, including patients with 17p deletion.
Ibrutinib is also FDA-approved to treat Waldenström’s macroglobulinemia, and the drug was granted accelerated approval to treat patients with mantle cell lymphoma who have received at least 1 prior therapy.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc. For more details on the drug, see the full prescribing information, available at imbruvica.com.
RESONATE-2 trial
The latest FDA approval for ibrutinib is based on results from the phase 3 RESONATE-2 trial (PCYC-1115), which were presented at the 2015 ASH Annual Meeting and simultaneously published in NEJM.
RESONATE-2 enrolled 269 treatment-naïve patients with CLL or small lymphocytic lymphoma who were 65 or older.
Patients were randomized to receive ibrutinib (n=136) at 420 mg once a day until progression or unacceptable toxicity, or chlorambucil (n=133) on days 1 and 15 of each 28-day cycle for up to 12 cycles. The starting dose for chlorambucil in cycle 1 was 0.5 mg/kg and was increased based on tolerability in cycle 2 by increments of 0.1 mg/kg to a maximum of 0.8 mg/kg.
The primary endpoint of the study was progression-free survival (PFS), as assessed by an independent review committee (IRC) according to the International Workshop on Chronic Lymphocytic Leukemia (iWCLL) 2008 criteria, with modification for treatment-related lymphocytosis.
Key secondary endpoints included overall response rate (based on the same iWCLL criteria), overall survival (OS), and safety.
Ibrutinib significantly prolonged PFS, as determined by the IRC, reducing the risk of progression or death by 84% compared to chlorambucil. The hazard ratio was 0.16 (P<0.001). The median PFS was not reached in the ibrutinib arm but was 18.9 months for the chlorambucil arm.
Ibrutinib significantly prolonged OS as well, although the median OS was not reached in either treatment arm. The OS rate at 24 months was 98% with ibrutinib and 85% with chlorambucil. The relative risk of death with ibrutinib was 84% lower than that with chlorambucil. The hazard ratio was 0.16 (P=0.001).
Ibrutinib was associated with a significantly higher IRC-assessed overall response rate compared to chlorambucil—82% and 35%, respectively (P<0.0001). Five patients (4%) in the ibrutinib arm achieved a complete response, as did 2 patients (2%) in the chlorambucil arm.
The median duration of treatment was 17.4 months in the ibrutinib arm and 7.1 months in the chlorambucil arm.
The most common adverse events of any grade—in the ibrutinib and chlorambucil arms, respectively—were diarrhea (42% and 17%), fatigue (30% and 38%), cough (22% and 15%), nausea (22% and 39%), peripheral edema (19% and 9%), dry eye (17% and 5%), arthralgia (16% and 7%), neutropenia (16% and 23%), and vomiting (13% and 20%).
Adverse events of grade 3 or higher—in the ibrutinib and chlorambucil arms, respectively—were neutropenia (10% and 18%), anemia (6% and 8%), hypertension (4% and 0%), pneumonia (4% and 2%), diarrhea (4% and 0%), maculopapular rash (3% and 2%), decreased platelet count (3% and 1%), abdominal pain (3% and 1%), hyponatremia (3% and 0%), thrombocytopenia (2% and 6%), febrile neutropenia (2% and 2%), upper respiratory tract infection (2% and 2%), pleural effusion (2% and 1%), cellulitis (2% and 0%), fatigue (1% and 5%), syncope (1% and 2%), and hemolytic anemia (0% and 2%). ![]()
Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has approved the BTK inhibitor ibrutinib (Imbruvica) as a first-line treatment for patients with chronic lymphocytic leukemia (CLL).
This means ibrutinib is now FDA-approved to treat CLL patients regardless of their treatment history, including patients with 17p deletion.
Ibrutinib is also FDA-approved to treat Waldenström’s macroglobulinemia, and the drug was granted accelerated approval to treat patients with mantle cell lymphoma who have received at least 1 prior therapy.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc. For more details on the drug, see the full prescribing information, available at imbruvica.com.
RESONATE-2 trial
The latest FDA approval for ibrutinib is based on results from the phase 3 RESONATE-2 trial (PCYC-1115), which were presented at the 2015 ASH Annual Meeting and simultaneously published in NEJM.
RESONATE-2 enrolled 269 treatment-naïve patients with CLL or small lymphocytic lymphoma who were 65 or older.
Patients were randomized to receive ibrutinib (n=136) at 420 mg once a day until progression or unacceptable toxicity, or chlorambucil (n=133) on days 1 and 15 of each 28-day cycle for up to 12 cycles. The starting dose for chlorambucil in cycle 1 was 0.5 mg/kg and was increased based on tolerability in cycle 2 by increments of 0.1 mg/kg to a maximum of 0.8 mg/kg.
The primary endpoint of the study was progression-free survival (PFS), as assessed by an independent review committee (IRC) according to the International Workshop on Chronic Lymphocytic Leukemia (iWCLL) 2008 criteria, with modification for treatment-related lymphocytosis.
Key secondary endpoints included overall response rate (based on the same iWCLL criteria), overall survival (OS), and safety.
Ibrutinib significantly prolonged PFS, as determined by the IRC, reducing the risk of progression or death by 84% compared to chlorambucil. The hazard ratio was 0.16 (P<0.001). The median PFS was not reached in the ibrutinib arm but was 18.9 months for the chlorambucil arm.
Ibrutinib significantly prolonged OS as well, although the median OS was not reached in either treatment arm. The OS rate at 24 months was 98% with ibrutinib and 85% with chlorambucil. The relative risk of death with ibrutinib was 84% lower than that with chlorambucil. The hazard ratio was 0.16 (P=0.001).
Ibrutinib was associated with a significantly higher IRC-assessed overall response rate compared to chlorambucil—82% and 35%, respectively (P<0.0001). Five patients (4%) in the ibrutinib arm achieved a complete response, as did 2 patients (2%) in the chlorambucil arm.
The median duration of treatment was 17.4 months in the ibrutinib arm and 7.1 months in the chlorambucil arm.
The most common adverse events of any grade—in the ibrutinib and chlorambucil arms, respectively—were diarrhea (42% and 17%), fatigue (30% and 38%), cough (22% and 15%), nausea (22% and 39%), peripheral edema (19% and 9%), dry eye (17% and 5%), arthralgia (16% and 7%), neutropenia (16% and 23%), and vomiting (13% and 20%).
Adverse events of grade 3 or higher—in the ibrutinib and chlorambucil arms, respectively—were neutropenia (10% and 18%), anemia (6% and 8%), hypertension (4% and 0%), pneumonia (4% and 2%), diarrhea (4% and 0%), maculopapular rash (3% and 2%), decreased platelet count (3% and 1%), abdominal pain (3% and 1%), hyponatremia (3% and 0%), thrombocytopenia (2% and 6%), febrile neutropenia (2% and 2%), upper respiratory tract infection (2% and 2%), pleural effusion (2% and 1%), cellulitis (2% and 0%), fatigue (1% and 5%), syncope (1% and 2%), and hemolytic anemia (0% and 2%). ![]()