User login
How gynecologic procedures and pharmacologic treatments can affect the uterus
Transvaginal ultrasound: We are gaining a better understanding of its clinical applications

Steven R. Goldstein, MD
In my first book I coined the phrase "sonomicroscopy." We are seeing things with transvaginal ultrasonography (TVUS) that you could not see with your naked eye even if you could it hold it at arms length and squint at it. For instance, cardiac activity can be seen easily within an embryo of 4 mm at 47 days since the last menstrual period. If there were any possible way to hold this 4-mm embryo in your hand, you would not appreciate cardiac pulsations contained within it! This is one of the beauties, and yet potential foibles, of TVUS.
In this excellent pictorial article, Michelle Stalnaker Ozcan, MD, and Andrew M. Kaunitz, MD, have done an outstanding job of turning this low-power "sonomicroscope" into the uterus to better understand a number of unique yet important clinical applications of TVUS.
Tamoxifen is known to cause a slight but statistically significant increase in endometrial cancer. In 1994, I first described an unusual ultrasound appearance in the uterus of patients receiving tamoxifen, which was being misinterpreted as "endometrial thickening," and resulted in many unnecessary biopsies and dilation and curettage procedures.1 This type of uterine change has been seen in other selective estrogen-receptor modulators as well.2,3 In this article, Drs. Ozcan and Kaunitz correctly point out that such an ultrasound pattern does not necessitate any intervention in the absence of bleeding.
Another common question I am often asked is, "How do we handle the patient whose status is post-endometrial ablation and presents with staining?" The scarring shown in the figures that follow make any kind of meaningful evaluation extremely difficult.
There has been an epidemic of cesarean scar pregnancies when a subsequent gestation implants in the cesarean scar defect.4 Perhaps the time has come when all patients with a previous cesarean delivery should have their lower uterine segment scanned to look for such a defect as shown in the pictures that follow. If we are not yet ready for that, at least early TVUS scans in subsequent pregnancies, in my opinion, should be employed to make an early diagnosis of such cases that are the precursors of morbidly adherent placenta, a potentially life-threatening situation that appears to be increasing in frequency.
Finally, look to obgmanagement.com for next month's web-exclusive look at outstanding images of patients who have undergone transcervical sterilization.
Dr. Goldstein is Professor, Department of Obstetrics and Gynecology, New York University School of Medicine, Director, Gynecologic Ultrasound, and Co-Director, Bone Densitometry, New York University Medical Center. He also serves on the OBG Management Board of Editors.
Dr. Goldstein reports that he has an equipment loan from Philips, and is past President of the American Institute of Ultrasound in Medicine.
References
- Goldstein SR. Unusual ultrasonographic appearance of the uterus in patients receiving tamoxifen. Am J Obstet Gynecol. 1994;170(2):447–451.
- Goldstein SR, Neven P, Cummings S, et al. Postmenopausal evaluation and risk reduction with lasofoxifene (PEARL) trial: 5-year gynecological outcomes. Menopause. 2011;18(1):17–22.
- Goldstein SR, Nanavati N. Adverse events that are associated with the selective estrogen receptor modulator levormeloxifene in an aborted phase III osteoporosis treatment study. Am J Obstet Gynecol. 2002;187(3):521–527.
- Timor-Tritsch IE, Monteagudo A. Unforeseen consequences of the increasing rate of cesarean deliveries: early placenta accreta and cesarean scar pregnancy. A review. Am J Obstet Gynecol. 2012;207(1):14–29.
New technology, minimally invasive surgical procedures, and medications continue to change how physicians manage specific medical issues. Many procedures and medications used by gynecologists can cause characteristic findings on sonography. These findings can guide subsequent counseling and management decisions and are important to accurately interpret on imaging. Among these conditions are Asherman syndrome, postendometrial ablation uterine damage, cesarean scar defect, and altered endometrium as a result of tamoxifen use. In this article, we provide 2 dimensional and 3 dimensional sono‑graphic images of uterine presentations of these 4 conditions.
Asherman syndromeCharacterized by variable scarring, or intrauterine adhesions, inside the uterine cavity following endometrial trauma due to surgical procedures, Asherman syndrome can cause menstrual changes and infertility. Should pregnancy occur in the setting of Asherman syndrome, placental abnormalities may result.1 Intrauterine adhesions can follow many surgical procedures, including curettage (diagnostic or for missed/elective abortion or retained products of conception), cesarean delivery, and hysteroscopic myomectomy. They may even occur after spontaneous abortion without curettage. Rates of Asherman syndrome are highest after procedures that tend to cause the most intrauterine inflammation, including2:
- curettage after septic abortion
- late curettage after retained products of conception
- hysteroscopy with multiple myomectomies.
In severe cases Asherman syndrome can result in complete obliteration of the uterine cavity.3
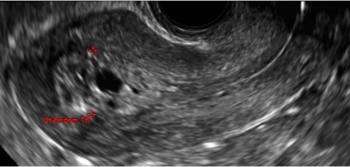
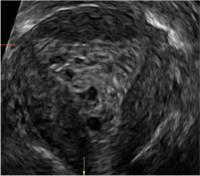
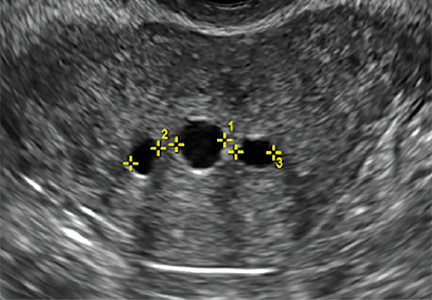
Clinicians should be cognizant of the appearance of Asherman syndrome on imaging because patients reporting menstrual abnormalities, pelvic pain (FIGURE 1), infertility, and other symptoms may exhibit intrauterine lesions on sonohysterography, or sometimes unenhanced sonography if endometrial fluid/blood is present. Depending on symptoms and patient reproductive plans, treatment may be indicated.2
| FIGURE 1 Asherman syndrome | ||||
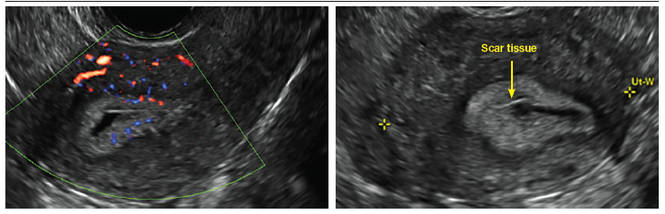
|
Postablation endometrial destruction
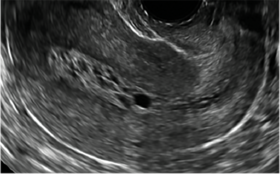
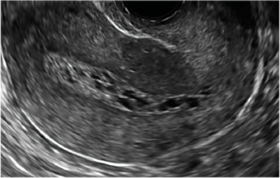
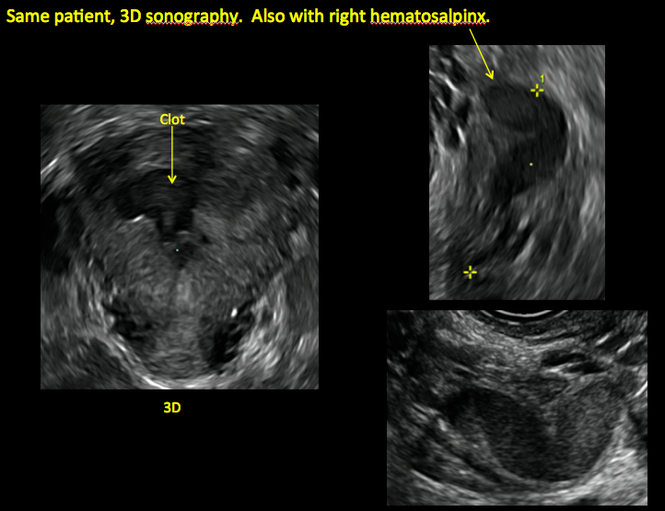
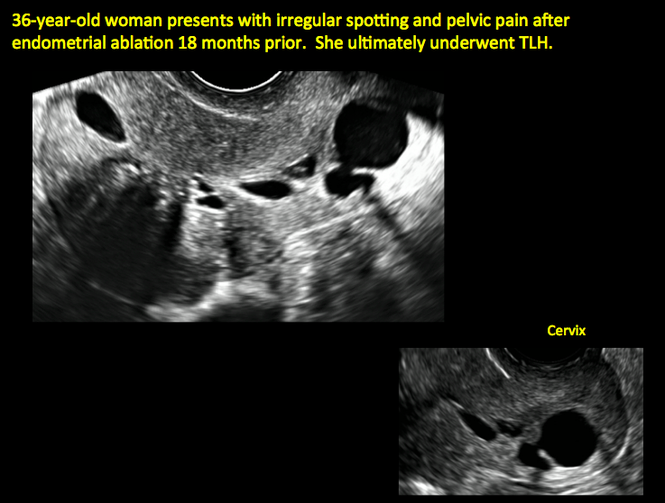
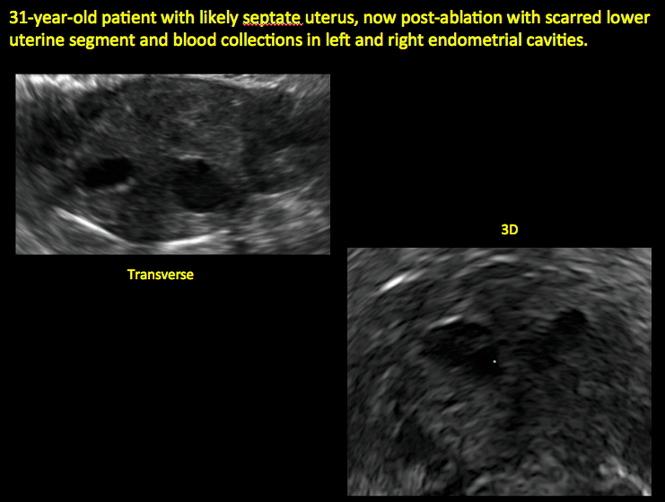
Surgical destruction of the endometrium to the level of the basalis has been associated with the formation of intrauterine adhesions (FIGURE 2) as well as pockets of hematometra (FIGURE 3). In a large Cochrane systematic review, the reported rate of hematometra was 0.9% following non− resectoscopic ablation and 2.4% following resectoscopic ablation.4
FIGURE 2 Intrauterine changes postablation | ||||
| ||||
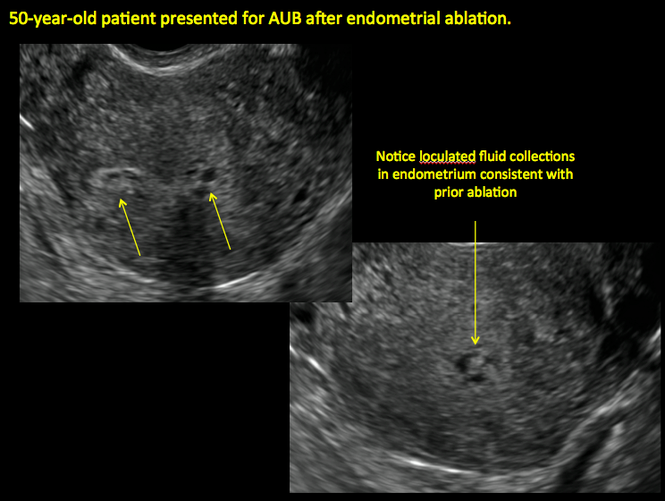
| Loculated fluid collections in the endometrium on transverse (A), sagittal (B), and 3 dimensional images (C) of a 41-year-old patient who presented with dysmenorrhea 3 years after an endometrial ablation procedure. The patient ultimately underwent transvaginal hysterectomy. | ||||
| ||||
| ||||
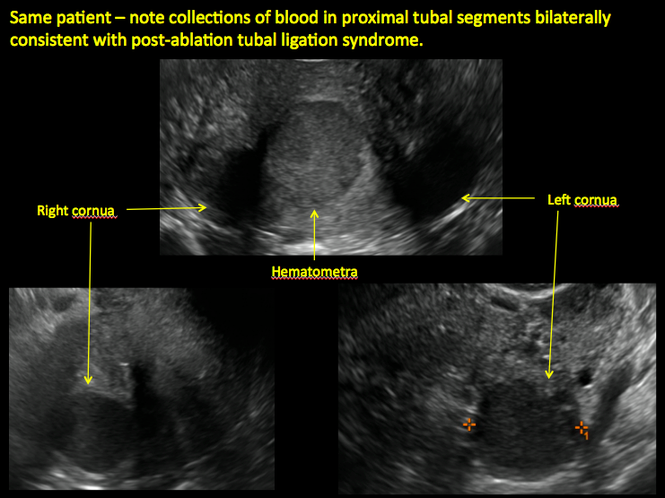
| 2 dimensional sonograms of a 40-year-old patient with a history of bilateral tubal ligation who presented for severe cyclic pelvic pain postablation. |
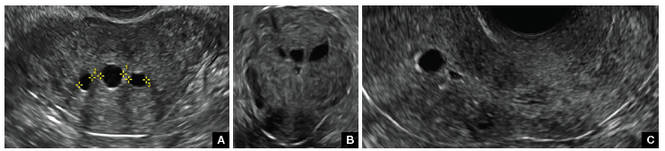
Postablation tubal sterilization syndrome—cyclic cramping with or without vaginal bleeding—occurs in up to 10% of previously sterilized women who undergo endometrial ablation.4 The syndrome is thought to be caused by bleeding from active endometrium trapped at the uterine cornua by intrauterine adhesions postablation.
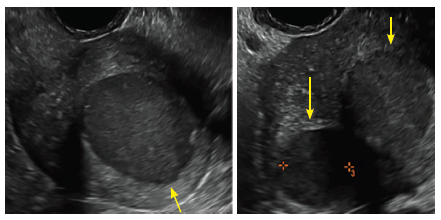
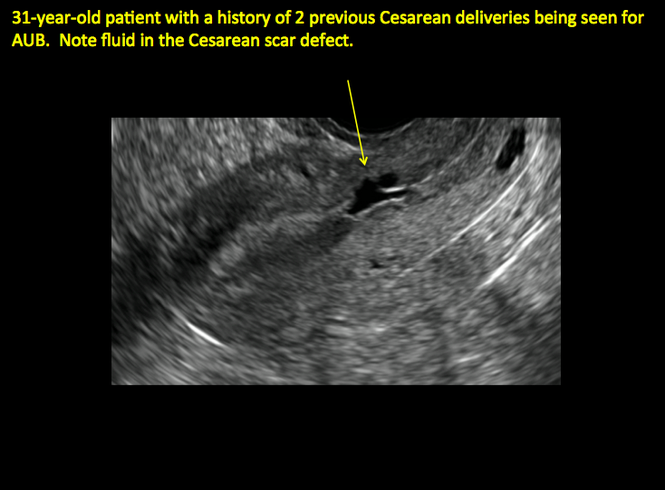
FIGURE 4 Cesarean scar defect with 1 previous cesarean delivery | ||||
| ||||
| Unenhanced sonogram in a 41-year-old patient. Myometrial notch is seen at both the endometrial surface and the serosal surface. | ||||
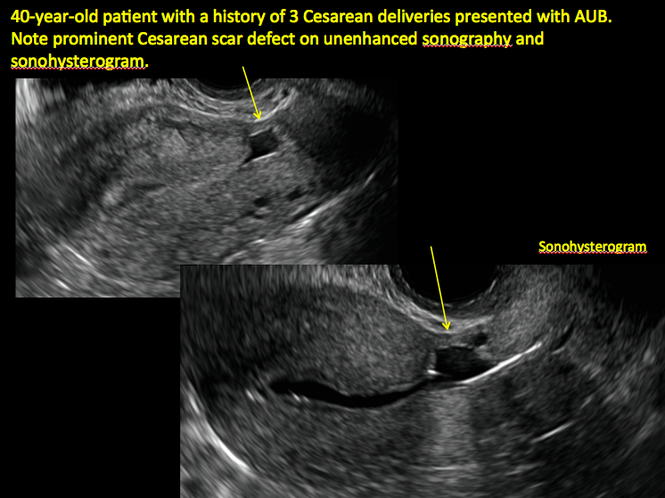
| ||||
| ||||
| Unenhanced sonogram (A) and sonohysterogram (B) in a 40-year-old patient. |
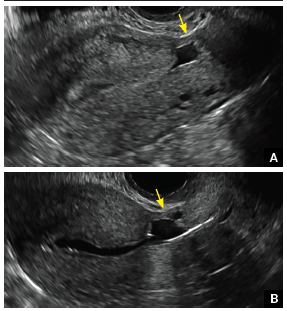
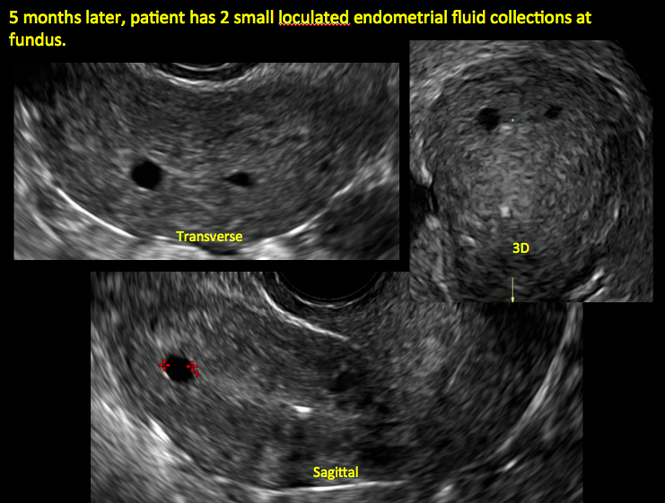
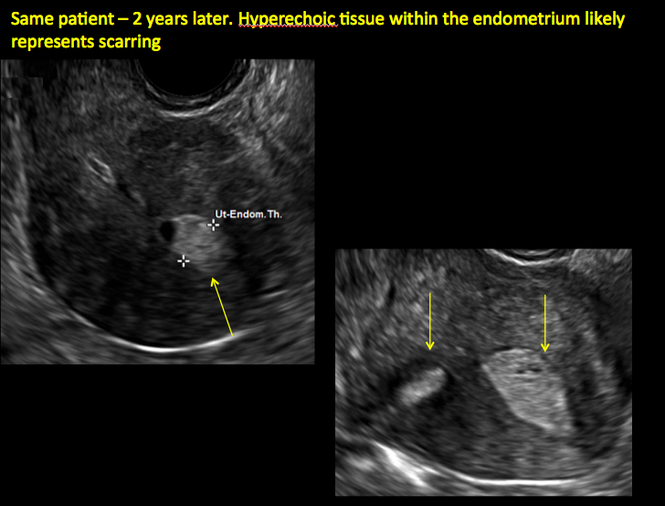
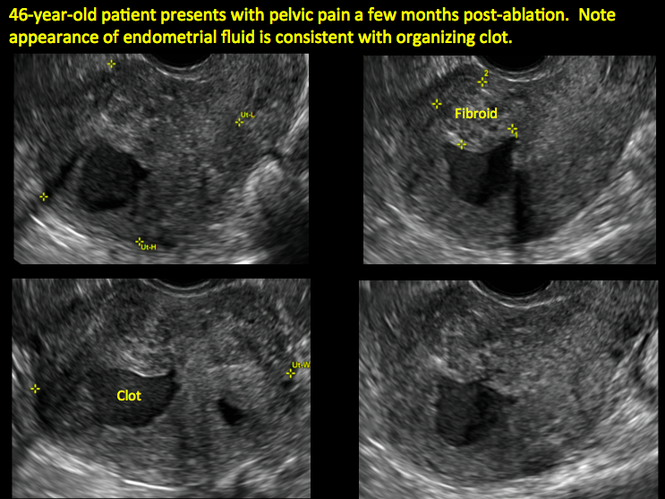
In patients with postablation tubal sterilization syndrome, imaging can reveal loculated endometrial fluid collections, hyperechoic foci/scarring, and a poorly defined endomyometrial interface. See ADDITIONAL CASES-Postablation at the bottom of this article for additional imaging case presentations.
Cesarean scar defect on imaging
In 1961, Poidevin first described the lower uterine segment myometrial notch or “niche,” now known as cesarean scar defect, as a wedge-shaped defect in the myometrium of women who had undergone cesarean delivery. He noted that it appeared after a 6-month healing period.5
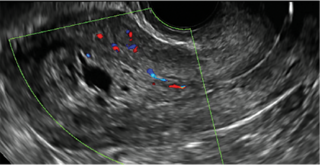
Using sonography with Doppler to view the defect, it appears relatively avascular and may decrease in size over time (FIGURES 4 and 5). Studies now are focusing on sonographic measurement of the cesarean scar defect as a clinical predictor of outcome for future pregnancies, as uterine rupture and abnormal placentation, including cesarean scar ectopics, can be associated with it.6-8
See ADDITIONAL CASES-Cesarean scar defect at the bottom of this article for 2 imaging case presentations.
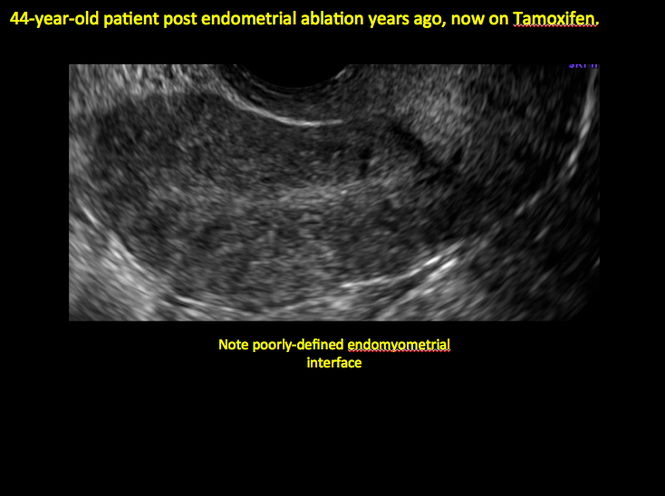
Endometrial changes with tamoxifen use
Tamoxifen use causes changes in the endometrium that on sonography can appear concerning for endometrial cancer. These changes include endometrial thickening and hyperechogenicity as well as cystic and heterogenous areas.9
Despite this imaging presentation, endometrial changes on sonography in the setting of tamoxifen use have been shown to be a poor predictor of actual endometrial pathology. In a study by Gerber and colleagues, the endometrial thickness in patients taking tamoxifen increased from a mean of 3.5 mm pretreatment to a mean of 9.2 mm after 3-year treatment.9 Using a cutoff value of 10 mm for abnormal endometrial thickness, screening transvaginal ultrasonography (TVUS) resulted in a high false-positive rate and iatrogenic morbidity. Endometrial cancer was detected in only 0.4% of patients (1 case), atrophy in 73%, polyps in 4%, and hyperplasia in 2%.9
Thus, routine screening sonographic assessment of the endometrium in asymptomatic women taking tamoxifen is not recommended. For women presenting with abnormal bleeding or other concerns, however, TVUS is appropriate (CASES 1 and 2).
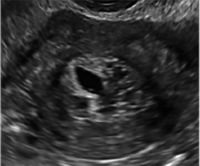
| CASE 1 Endometrial polyps identified with tamoxifen use | ||||
| A 56-year-old patient with a history of breast cancer presently taking tamoxifen presented with postmenopausal bleeding. Endometrial biopsy results revealed endometrial polyps. | ||||
| 
| 
| ||
| CASE 2 Benign endometrial changes with tamoxifen use | ||||
| A 50-year-old patient with a history of breast cancer currently taking tamoxifen presented with abnormal uterine bleeding. Endometrial biopsy results indicated benign endometrial changes. | ||||
| 
| 
| ||
ADDITIONAL CASES - Postablation

ADDITIONAL CASES - Cesarean scar defect
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Engelbrechtsen L, Langhoff-Roos J, Kjer JJ, Istre O. Placenta accreta: adherent placenta due to Asherman syndrome. Clin Case Rep. 2015;3(3):175−178.
- Conforti A, Alviggi C, Mollo A, De Placido G, Magos A. The management of Asherman syndrome: a review of literature. Reprod Biol Endocrinol. 2013;11:118.
- Song D, Xia E, Xiao Y, Li TC, Huang X, Liu Y. Management of false passage created during hysteroscopic adhesiolysis for Asherman’s syndrome. J Obstet Gynaecol. 2016;36(1):87−92.
- Lethaby A, Hickey M, Garry R, Penninx J. Endometrial resection/ablation techniques for heavy menstrual bleeding. Cochrane Database Syst Rev. 2009;4:CD001501.
- Poidevin LO. The value of hysterography in the prediction of cesarean section wound defects. Am J Obstet Gynecol. 1961;81:67−71.
- Naji O, Abdallah Y, Bij De Vaate AJ, et al. Standardized approach for imaging and measuring Cesarean section scars using ultrasonography. Ultrasound Obstet Gynecol. 2012;39(3):252−259.
- Kok N, Wiersma IC, Opmeer BC, et al. Sonographic measurement of lower uterine segment thickness to predict uterine rupture during a trial of labor in women with previous Cesarean section: a meta-analysis. Ultrasound Obstet Gynecol. 2013;42(2):132−139.
- Nezhat C, Grace L, Soliemannjad R, Razavi GM, Nezhat A. Cesarean scar defect: What is it and how should it be treated? OBG Manag. 2016;28(4):32, 34, 36, 38–39, 53.
- Gerber B, Krause A, Müller H, et al. Effects of adjuvant tamoxifen on the endometrium in postmenopausal women with breast cancer: a prospective long-term study using transvaginal ultrasound. J Clin Oncol. 2000; 18(20):3464–3667.

Dr. Ozcan is Assistant Professor and Co-Program Director, Obstetrics and Gynecology Residency, Department of Obstetrics and Gynecology, at the University of Central Florida College of Medicine−Orlando.

Dr. Kaunitz is University of Florida Research Foundation Professor and Associate Chairman, Department of Obstetrics and Gynecology, University of Florida College of Medicine–Jacksonville. Dr. Kaunitz serves as Medical Director and directs Menopause and Gynecologic Ultrasound Services at UF Women’s Health Specialists–Emerson. He serves on the OBG Management Board of Editors.
The authors report no financial relationships relevant to this article.
Dr. Ozcan is Assistant Professor and Co-Program Director, Obstetrics and Gynecology Residency, Department of Obstetrics and Gynecology, at the University of Central Florida College of Medicine−Orlando.
Dr. Kaunitz is University of Florida Research Foundation Professor and Associate Chairman, Department of Obstetrics and Gynecology, University of Florida College of Medicine–Jacksonville. Dr. Kaunitz serves as Medical Director and directs Menopause and Gynecologic Ultrasound Services at UF Women’s Health Specialists–Emerson. He serves on the OBG Management Board of Editors.
The authors report no financial relationships relevant to this article.
Dr. Ozcan is Assistant Professor and Co-Program Director, Obstetrics and Gynecology Residency, Department of Obstetrics and Gynecology, at the University of Central Florida College of Medicine−Orlando.
Dr. Kaunitz is University of Florida Research Foundation Professor and Associate Chairman, Department of Obstetrics and Gynecology, University of Florida College of Medicine–Jacksonville. Dr. Kaunitz serves as Medical Director and directs Menopause and Gynecologic Ultrasound Services at UF Women’s Health Specialists–Emerson. He serves on the OBG Management Board of Editors.
The authors report no financial relationships relevant to this article.
Transvaginal ultrasound: We are gaining a better understanding of its clinical applications
Steven R. Goldstein, MD
In my first book I coined the phrase "sonomicroscopy." We are seeing things with transvaginal ultrasonography (TVUS) that you could not see with your naked eye even if you could it hold it at arms length and squint at it. For instance, cardiac activity can be seen easily within an embryo of 4 mm at 47 days since the last menstrual period. If there were any possible way to hold this 4-mm embryo in your hand, you would not appreciate cardiac pulsations contained within it! This is one of the beauties, and yet potential foibles, of TVUS.
In this excellent pictorial article, Michelle Stalnaker Ozcan, MD, and Andrew M. Kaunitz, MD, have done an outstanding job of turning this low-power "sonomicroscope" into the uterus to better understand a number of unique yet important clinical applications of TVUS.
Tamoxifen is known to cause a slight but statistically significant increase in endometrial cancer. In 1994, I first described an unusual ultrasound appearance in the uterus of patients receiving tamoxifen, which was being misinterpreted as "endometrial thickening," and resulted in many unnecessary biopsies and dilation and curettage procedures.1 This type of uterine change has been seen in other selective estrogen-receptor modulators as well.2,3 In this article, Drs. Ozcan and Kaunitz correctly point out that such an ultrasound pattern does not necessitate any intervention in the absence of bleeding.
Another common question I am often asked is, "How do we handle the patient whose status is post-endometrial ablation and presents with staining?" The scarring shown in the figures that follow make any kind of meaningful evaluation extremely difficult.
There has been an epidemic of cesarean scar pregnancies when a subsequent gestation implants in the cesarean scar defect.4 Perhaps the time has come when all patients with a previous cesarean delivery should have their lower uterine segment scanned to look for such a defect as shown in the pictures that follow. If we are not yet ready for that, at least early TVUS scans in subsequent pregnancies, in my opinion, should be employed to make an early diagnosis of such cases that are the precursors of morbidly adherent placenta, a potentially life-threatening situation that appears to be increasing in frequency.
Finally, look to obgmanagement.com for next month's web-exclusive look at outstanding images of patients who have undergone transcervical sterilization.
Dr. Goldstein is Professor, Department of Obstetrics and Gynecology, New York University School of Medicine, Director, Gynecologic Ultrasound, and Co-Director, Bone Densitometry, New York University Medical Center. He also serves on the OBG Management Board of Editors.
Dr. Goldstein reports that he has an equipment loan from Philips, and is past President of the American Institute of Ultrasound in Medicine.
References
- Goldstein SR. Unusual ultrasonographic appearance of the uterus in patients receiving tamoxifen. Am J Obstet Gynecol. 1994;170(2):447–451.
- Goldstein SR, Neven P, Cummings S, et al. Postmenopausal evaluation and risk reduction with lasofoxifene (PEARL) trial: 5-year gynecological outcomes. Menopause. 2011;18(1):17–22.
- Goldstein SR, Nanavati N. Adverse events that are associated with the selective estrogen receptor modulator levormeloxifene in an aborted phase III osteoporosis treatment study. Am J Obstet Gynecol. 2002;187(3):521–527.
- Timor-Tritsch IE, Monteagudo A. Unforeseen consequences of the increasing rate of cesarean deliveries: early placenta accreta and cesarean scar pregnancy. A review. Am J Obstet Gynecol. 2012;207(1):14–29.
New technology, minimally invasive surgical procedures, and medications continue to change how physicians manage specific medical issues. Many procedures and medications used by gynecologists can cause characteristic findings on sonography. These findings can guide subsequent counseling and management decisions and are important to accurately interpret on imaging. Among these conditions are Asherman syndrome, postendometrial ablation uterine damage, cesarean scar defect, and altered endometrium as a result of tamoxifen use. In this article, we provide 2 dimensional and 3 dimensional sono‑graphic images of uterine presentations of these 4 conditions.
Asherman syndromeCharacterized by variable scarring, or intrauterine adhesions, inside the uterine cavity following endometrial trauma due to surgical procedures, Asherman syndrome can cause menstrual changes and infertility. Should pregnancy occur in the setting of Asherman syndrome, placental abnormalities may result.1 Intrauterine adhesions can follow many surgical procedures, including curettage (diagnostic or for missed/elective abortion or retained products of conception), cesarean delivery, and hysteroscopic myomectomy. They may even occur after spontaneous abortion without curettage. Rates of Asherman syndrome are highest after procedures that tend to cause the most intrauterine inflammation, including2:
- curettage after septic abortion
- late curettage after retained products of conception
- hysteroscopy with multiple myomectomies.
In severe cases Asherman syndrome can result in complete obliteration of the uterine cavity.3
Clinicians should be cognizant of the appearance of Asherman syndrome on imaging because patients reporting menstrual abnormalities, pelvic pain (FIGURE 1), infertility, and other symptoms may exhibit intrauterine lesions on sonohysterography, or sometimes unenhanced sonography if endometrial fluid/blood is present. Depending on symptoms and patient reproductive plans, treatment may be indicated.2
| FIGURE 1 Asherman syndrome | ||||
|
|
Postablation endometrial destruction
Surgical destruction of the endometrium to the level of the basalis has been associated with the formation of intrauterine adhesions (FIGURE 2) as well as pockets of hematometra (FIGURE 3). In a large Cochrane systematic review, the reported rate of hematometra was 0.9% following non− resectoscopic ablation and 2.4% following resectoscopic ablation.4
FIGURE 2 Intrauterine changes postablation | ||||
| ||||
| Loculated fluid collections in the endometrium on transverse (A), sagittal (B), and 3 dimensional images (C) of a 41-year-old patient who presented with dysmenorrhea 3 years after an endometrial ablation procedure. The patient ultimately underwent transvaginal hysterectomy. | ||||
| ||||
| ||||
| 2 dimensional sonograms of a 40-year-old patient with a history of bilateral tubal ligation who presented for severe cyclic pelvic pain postablation. |
Postablation tubal sterilization syndrome—cyclic cramping with or without vaginal bleeding—occurs in up to 10% of previously sterilized women who undergo endometrial ablation.4 The syndrome is thought to be caused by bleeding from active endometrium trapped at the uterine cornua by intrauterine adhesions postablation.
FIGURE 4 Cesarean scar defect with 1 previous cesarean delivery | ||||
| ||||
| Unenhanced sonogram in a 41-year-old patient. Myometrial notch is seen at both the endometrial surface and the serosal surface. | ||||
| ||||
| ||||
| Unenhanced sonogram (A) and sonohysterogram (B) in a 40-year-old patient. |
In patients with postablation tubal sterilization syndrome, imaging can reveal loculated endometrial fluid collections, hyperechoic foci/scarring, and a poorly defined endomyometrial interface. See ADDITIONAL CASES-Postablation at the bottom of this article for additional imaging case presentations.
Cesarean scar defect on imaging
In 1961, Poidevin first described the lower uterine segment myometrial notch or “niche,” now known as cesarean scar defect, as a wedge-shaped defect in the myometrium of women who had undergone cesarean delivery. He noted that it appeared after a 6-month healing period.5
Using sonography with Doppler to view the defect, it appears relatively avascular and may decrease in size over time (FIGURES 4 and 5). Studies now are focusing on sonographic measurement of the cesarean scar defect as a clinical predictor of outcome for future pregnancies, as uterine rupture and abnormal placentation, including cesarean scar ectopics, can be associated with it.6-8
See ADDITIONAL CASES-Cesarean scar defect at the bottom of this article for 2 imaging case presentations.
Endometrial changes with tamoxifen use
Tamoxifen use causes changes in the endometrium that on sonography can appear concerning for endometrial cancer. These changes include endometrial thickening and hyperechogenicity as well as cystic and heterogenous areas.9
Despite this imaging presentation, endometrial changes on sonography in the setting of tamoxifen use have been shown to be a poor predictor of actual endometrial pathology. In a study by Gerber and colleagues, the endometrial thickness in patients taking tamoxifen increased from a mean of 3.5 mm pretreatment to a mean of 9.2 mm after 3-year treatment.9 Using a cutoff value of 10 mm for abnormal endometrial thickness, screening transvaginal ultrasonography (TVUS) resulted in a high false-positive rate and iatrogenic morbidity. Endometrial cancer was detected in only 0.4% of patients (1 case), atrophy in 73%, polyps in 4%, and hyperplasia in 2%.9
Thus, routine screening sonographic assessment of the endometrium in asymptomatic women taking tamoxifen is not recommended. For women presenting with abnormal bleeding or other concerns, however, TVUS is appropriate (CASES 1 and 2).
| CASE 1 Endometrial polyps identified with tamoxifen use | ||||
| A 56-year-old patient with a history of breast cancer presently taking tamoxifen presented with postmenopausal bleeding. Endometrial biopsy results revealed endometrial polyps. | ||||
|
|
|
| ||
| CASE 2 Benign endometrial changes with tamoxifen use | ||||
| A 50-year-old patient with a history of breast cancer currently taking tamoxifen presented with abnormal uterine bleeding. Endometrial biopsy results indicated benign endometrial changes. | ||||
|
|
|
| ||
ADDITIONAL CASES - Postablation
ADDITIONAL CASES - Cesarean scar defect
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Transvaginal ultrasound: We are gaining a better understanding of its clinical applications
Steven R. Goldstein, MD
In my first book I coined the phrase "sonomicroscopy." We are seeing things with transvaginal ultrasonography (TVUS) that you could not see with your naked eye even if you could it hold it at arms length and squint at it. For instance, cardiac activity can be seen easily within an embryo of 4 mm at 47 days since the last menstrual period. If there were any possible way to hold this 4-mm embryo in your hand, you would not appreciate cardiac pulsations contained within it! This is one of the beauties, and yet potential foibles, of TVUS.
In this excellent pictorial article, Michelle Stalnaker Ozcan, MD, and Andrew M. Kaunitz, MD, have done an outstanding job of turning this low-power "sonomicroscope" into the uterus to better understand a number of unique yet important clinical applications of TVUS.
Tamoxifen is known to cause a slight but statistically significant increase in endometrial cancer. In 1994, I first described an unusual ultrasound appearance in the uterus of patients receiving tamoxifen, which was being misinterpreted as "endometrial thickening," and resulted in many unnecessary biopsies and dilation and curettage procedures.1 This type of uterine change has been seen in other selective estrogen-receptor modulators as well.2,3 In this article, Drs. Ozcan and Kaunitz correctly point out that such an ultrasound pattern does not necessitate any intervention in the absence of bleeding.
Another common question I am often asked is, "How do we handle the patient whose status is post-endometrial ablation and presents with staining?" The scarring shown in the figures that follow make any kind of meaningful evaluation extremely difficult.
There has been an epidemic of cesarean scar pregnancies when a subsequent gestation implants in the cesarean scar defect.4 Perhaps the time has come when all patients with a previous cesarean delivery should have their lower uterine segment scanned to look for such a defect as shown in the pictures that follow. If we are not yet ready for that, at least early TVUS scans in subsequent pregnancies, in my opinion, should be employed to make an early diagnosis of such cases that are the precursors of morbidly adherent placenta, a potentially life-threatening situation that appears to be increasing in frequency.
Finally, look to obgmanagement.com for next month's web-exclusive look at outstanding images of patients who have undergone transcervical sterilization.
Dr. Goldstein is Professor, Department of Obstetrics and Gynecology, New York University School of Medicine, Director, Gynecologic Ultrasound, and Co-Director, Bone Densitometry, New York University Medical Center. He also serves on the OBG Management Board of Editors.
Dr. Goldstein reports that he has an equipment loan from Philips, and is past President of the American Institute of Ultrasound in Medicine.
References
- Goldstein SR. Unusual ultrasonographic appearance of the uterus in patients receiving tamoxifen. Am J Obstet Gynecol. 1994;170(2):447–451.
- Goldstein SR, Neven P, Cummings S, et al. Postmenopausal evaluation and risk reduction with lasofoxifene (PEARL) trial: 5-year gynecological outcomes. Menopause. 2011;18(1):17–22.
- Goldstein SR, Nanavati N. Adverse events that are associated with the selective estrogen receptor modulator levormeloxifene in an aborted phase III osteoporosis treatment study. Am J Obstet Gynecol. 2002;187(3):521–527.
- Timor-Tritsch IE, Monteagudo A. Unforeseen consequences of the increasing rate of cesarean deliveries: early placenta accreta and cesarean scar pregnancy. A review. Am J Obstet Gynecol. 2012;207(1):14–29.
New technology, minimally invasive surgical procedures, and medications continue to change how physicians manage specific medical issues. Many procedures and medications used by gynecologists can cause characteristic findings on sonography. These findings can guide subsequent counseling and management decisions and are important to accurately interpret on imaging. Among these conditions are Asherman syndrome, postendometrial ablation uterine damage, cesarean scar defect, and altered endometrium as a result of tamoxifen use. In this article, we provide 2 dimensional and 3 dimensional sono‑graphic images of uterine presentations of these 4 conditions.
Asherman syndromeCharacterized by variable scarring, or intrauterine adhesions, inside the uterine cavity following endometrial trauma due to surgical procedures, Asherman syndrome can cause menstrual changes and infertility. Should pregnancy occur in the setting of Asherman syndrome, placental abnormalities may result.1 Intrauterine adhesions can follow many surgical procedures, including curettage (diagnostic or for missed/elective abortion or retained products of conception), cesarean delivery, and hysteroscopic myomectomy. They may even occur after spontaneous abortion without curettage. Rates of Asherman syndrome are highest after procedures that tend to cause the most intrauterine inflammation, including2:
- curettage after septic abortion
- late curettage after retained products of conception
- hysteroscopy with multiple myomectomies.
In severe cases Asherman syndrome can result in complete obliteration of the uterine cavity.3
Clinicians should be cognizant of the appearance of Asherman syndrome on imaging because patients reporting menstrual abnormalities, pelvic pain (FIGURE 1), infertility, and other symptoms may exhibit intrauterine lesions on sonohysterography, or sometimes unenhanced sonography if endometrial fluid/blood is present. Depending on symptoms and patient reproductive plans, treatment may be indicated.2
| FIGURE 1 Asherman syndrome | ||||
|
|
Postablation endometrial destruction
Surgical destruction of the endometrium to the level of the basalis has been associated with the formation of intrauterine adhesions (FIGURE 2) as well as pockets of hematometra (FIGURE 3). In a large Cochrane systematic review, the reported rate of hematometra was 0.9% following non− resectoscopic ablation and 2.4% following resectoscopic ablation.4
FIGURE 2 Intrauterine changes postablation | ||||
| ||||
| Loculated fluid collections in the endometrium on transverse (A), sagittal (B), and 3 dimensional images (C) of a 41-year-old patient who presented with dysmenorrhea 3 years after an endometrial ablation procedure. The patient ultimately underwent transvaginal hysterectomy. | ||||
| ||||
| ||||
| 2 dimensional sonograms of a 40-year-old patient with a history of bilateral tubal ligation who presented for severe cyclic pelvic pain postablation. |
Postablation tubal sterilization syndrome—cyclic cramping with or without vaginal bleeding—occurs in up to 10% of previously sterilized women who undergo endometrial ablation.4 The syndrome is thought to be caused by bleeding from active endometrium trapped at the uterine cornua by intrauterine adhesions postablation.
FIGURE 4 Cesarean scar defect with 1 previous cesarean delivery | ||||
| ||||
| Unenhanced sonogram in a 41-year-old patient. Myometrial notch is seen at both the endometrial surface and the serosal surface. | ||||
| ||||
| ||||
| Unenhanced sonogram (A) and sonohysterogram (B) in a 40-year-old patient. |
In patients with postablation tubal sterilization syndrome, imaging can reveal loculated endometrial fluid collections, hyperechoic foci/scarring, and a poorly defined endomyometrial interface. See ADDITIONAL CASES-Postablation at the bottom of this article for additional imaging case presentations.
Cesarean scar defect on imaging
In 1961, Poidevin first described the lower uterine segment myometrial notch or “niche,” now known as cesarean scar defect, as a wedge-shaped defect in the myometrium of women who had undergone cesarean delivery. He noted that it appeared after a 6-month healing period.5
Using sonography with Doppler to view the defect, it appears relatively avascular and may decrease in size over time (FIGURES 4 and 5). Studies now are focusing on sonographic measurement of the cesarean scar defect as a clinical predictor of outcome for future pregnancies, as uterine rupture and abnormal placentation, including cesarean scar ectopics, can be associated with it.6-8
See ADDITIONAL CASES-Cesarean scar defect at the bottom of this article for 2 imaging case presentations.
Endometrial changes with tamoxifen use
Tamoxifen use causes changes in the endometrium that on sonography can appear concerning for endometrial cancer. These changes include endometrial thickening and hyperechogenicity as well as cystic and heterogenous areas.9
Despite this imaging presentation, endometrial changes on sonography in the setting of tamoxifen use have been shown to be a poor predictor of actual endometrial pathology. In a study by Gerber and colleagues, the endometrial thickness in patients taking tamoxifen increased from a mean of 3.5 mm pretreatment to a mean of 9.2 mm after 3-year treatment.9 Using a cutoff value of 10 mm for abnormal endometrial thickness, screening transvaginal ultrasonography (TVUS) resulted in a high false-positive rate and iatrogenic morbidity. Endometrial cancer was detected in only 0.4% of patients (1 case), atrophy in 73%, polyps in 4%, and hyperplasia in 2%.9
Thus, routine screening sonographic assessment of the endometrium in asymptomatic women taking tamoxifen is not recommended. For women presenting with abnormal bleeding or other concerns, however, TVUS is appropriate (CASES 1 and 2).
| CASE 1 Endometrial polyps identified with tamoxifen use | ||||
| A 56-year-old patient with a history of breast cancer presently taking tamoxifen presented with postmenopausal bleeding. Endometrial biopsy results revealed endometrial polyps. | ||||
|
|
|
| ||
| CASE 2 Benign endometrial changes with tamoxifen use | ||||
| A 50-year-old patient with a history of breast cancer currently taking tamoxifen presented with abnormal uterine bleeding. Endometrial biopsy results indicated benign endometrial changes. | ||||
|
|
|
| ||
ADDITIONAL CASES - Postablation
ADDITIONAL CASES - Cesarean scar defect
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Engelbrechtsen L, Langhoff-Roos J, Kjer JJ, Istre O. Placenta accreta: adherent placenta due to Asherman syndrome. Clin Case Rep. 2015;3(3):175−178.
- Conforti A, Alviggi C, Mollo A, De Placido G, Magos A. The management of Asherman syndrome: a review of literature. Reprod Biol Endocrinol. 2013;11:118.
- Song D, Xia E, Xiao Y, Li TC, Huang X, Liu Y. Management of false passage created during hysteroscopic adhesiolysis for Asherman’s syndrome. J Obstet Gynaecol. 2016;36(1):87−92.
- Lethaby A, Hickey M, Garry R, Penninx J. Endometrial resection/ablation techniques for heavy menstrual bleeding. Cochrane Database Syst Rev. 2009;4:CD001501.
- Poidevin LO. The value of hysterography in the prediction of cesarean section wound defects. Am J Obstet Gynecol. 1961;81:67−71.
- Naji O, Abdallah Y, Bij De Vaate AJ, et al. Standardized approach for imaging and measuring Cesarean section scars using ultrasonography. Ultrasound Obstet Gynecol. 2012;39(3):252−259.
- Kok N, Wiersma IC, Opmeer BC, et al. Sonographic measurement of lower uterine segment thickness to predict uterine rupture during a trial of labor in women with previous Cesarean section: a meta-analysis. Ultrasound Obstet Gynecol. 2013;42(2):132−139.
- Nezhat C, Grace L, Soliemannjad R, Razavi GM, Nezhat A. Cesarean scar defect: What is it and how should it be treated? OBG Manag. 2016;28(4):32, 34, 36, 38–39, 53.
- Gerber B, Krause A, Müller H, et al. Effects of adjuvant tamoxifen on the endometrium in postmenopausal women with breast cancer: a prospective long-term study using transvaginal ultrasound. J Clin Oncol. 2000; 18(20):3464–3667.
- Engelbrechtsen L, Langhoff-Roos J, Kjer JJ, Istre O. Placenta accreta: adherent placenta due to Asherman syndrome. Clin Case Rep. 2015;3(3):175−178.
- Conforti A, Alviggi C, Mollo A, De Placido G, Magos A. The management of Asherman syndrome: a review of literature. Reprod Biol Endocrinol. 2013;11:118.
- Song D, Xia E, Xiao Y, Li TC, Huang X, Liu Y. Management of false passage created during hysteroscopic adhesiolysis for Asherman’s syndrome. J Obstet Gynaecol. 2016;36(1):87−92.
- Lethaby A, Hickey M, Garry R, Penninx J. Endometrial resection/ablation techniques for heavy menstrual bleeding. Cochrane Database Syst Rev. 2009;4:CD001501.
- Poidevin LO. The value of hysterography in the prediction of cesarean section wound defects. Am J Obstet Gynecol. 1961;81:67−71.
- Naji O, Abdallah Y, Bij De Vaate AJ, et al. Standardized approach for imaging and measuring Cesarean section scars using ultrasonography. Ultrasound Obstet Gynecol. 2012;39(3):252−259.
- Kok N, Wiersma IC, Opmeer BC, et al. Sonographic measurement of lower uterine segment thickness to predict uterine rupture during a trial of labor in women with previous Cesarean section: a meta-analysis. Ultrasound Obstet Gynecol. 2013;42(2):132−139.
- Nezhat C, Grace L, Soliemannjad R, Razavi GM, Nezhat A. Cesarean scar defect: What is it and how should it be treated? OBG Manag. 2016;28(4):32, 34, 36, 38–39, 53.
- Gerber B, Krause A, Müller H, et al. Effects of adjuvant tamoxifen on the endometrium in postmenopausal women with breast cancer: a prospective long-term study using transvaginal ultrasound. J Clin Oncol. 2000; 18(20):3464–3667.
In this Article
- Foreword by Steven R. Goldstein, MD
- Uterine changes postablation
- Endometrial changes with tamoxifen use
The Effect of Orthopedic Advertising and Self-Promotion on a Naïve Population
In 1975, the American Medical Association (AMA) lifted the professional ban on physician advertising after a successful Federal Trade Commission suit.1 Since then, there has been a marked increase in the number of physicians marketing themselves directly to patients and consumers. With the pervasive nature of the Internet, never before has it been so easy and inexpensive to effectively communicate with a targeted population of people and influence their behavior. Few would dispute the role of advertising on consumer choices when used to sell products and services, change behavior, and educate consumers across all types of industries and professions. Thus, it is reasonable to hypothesize that the nature and content of a surgeon’s web presence could significantly affect patients’ decision-making and their impression of the orthopedic surgery profession.
There is a lack of consensus among physician organizations regarding physician advertising. For example, the American Association of Physicians and Surgeons (AAPS) takes an ethical stand on physician self-promotion. Their position states “The physician should not solicit patients. Professional reputation is the major source of patient referrals. The physician should be circumspect and restrained in dealing with the communication media, always avoiding self-aggrandizement.2” In contrast, the AMA has a less defined stance on physician self-promotion. With the exception of conflicts of interest and privacy guidelines, the AMA has few recommendations regarding the content of physician websites. The organization’s position states “There are no restrictions on advertising by physicians except those that can be specifically justified to protect the public from deceptive practices. …Nothing in this opinion is intended to discourage or to limit advertising and representations which are not false or deceptive.3” This guideline emphasizes accuracy of health-related information, but does not limit physician self-promotion or self-aggrandizement. The American Academy of Orthopaedic Surgeons (AAOS) holds a similar position. In their position statement on advertising by orthopedic surgeons, they encourage advertising and competition as “ethical and acceptable” as long as they are representing services in a “clear and accurate manner.”4 Furthermore, the AAOS also states that “An orthopaedic surgeon shall not use photographs, images, endorsements and/or statements in a false or misleading manner that communicate a degree of relief, safety, effectiveness, or benefits from orthopaedic care that are not representative of results attained by that orthopaedic surgeon.”4 The surgeon is responsible for his/her advertising materials and the content and claims therein, and is generally policed by peers through a complaint process with the AAOS.
Notably, up to 75% of Americans use the Internet for health-related information and this number is likely to increase.5Patients who utilize the Internet must choose from a vast array of search results for medical information from credible resources. Which sources are to be believed and relied upon? This depends on the health literacy among the general population. Inadequate health literacy is defined as “limited ability to obtain, process, and understand basic health information and services needed to make appropriate health decisions and follow instructions for treatment.”6 Patients have different levels of health literacy often unknown to even the most well-intentioned healthcare professional. It is often difficult to provide appropriate and meaningful information at a level that is most beneficial to the patient. It is estimated that 89 million people in the US have insufficient health literacy to understand treatments or preventive care.7 Certainly, with this information in mind, the orthopedic surgeon must consider his/her audience, and the potential for a fiduciary responsibility when preparing Internet content.
A tangible example of marketing results is the increasing popularity of robotic surgery over the last decade.8 Hospitals routinely advertise the availability of robotic surgery at their institution through various means, including roadside billboards. Despite limited evidence supporting a benefit of robotic surgery beyond less expensive conventional laparoscopic surgery, patients are increasingly seeking robotic surgery.8 With society’s increasing infatuation with technology, this is likely based on the presumption that robotic surgery is better and safer than conventional methods. It is likely that marketing pressure is at least partly responsible for the widespread adoption of robotic-assisted surgery and words used in marketing highlighting novelty have an important influence on patient preference.8
Orthopedic surgery, with its large proportion of elective surgeries, offers a unique venue to study differences in patient perceptions. Preoperative evaluations in orthopedics are often performed after an assessment of a surgeon’s reputation, which offers the patient an ability to choose their surgeon within their community.
We pondered how different promotional styles would affect potential patients’ perceptions. Would people believe that a self-promoting physician was more competent? Could fellow doctors “see through” the self-promotion of their peers? Based on the premise that advertising and self-promotion are undertaken because they are effective, we hypothesized that nonphysician patients perceive self-promoting orthopedic surgeons more favorably compared to members of the medical community.
Although numerous anonymous physician review sites exist, our analysis focused on surgeon self-promotion through personal websites or web pages. Within these sources, there exists a wide array of information and methods that physicians utilize to present themselves. Some physicians merely post their educational background and qualifications. This appears most often when the physician is associated with an academic institution and their profile is part of an institution’s website. Others post extensive self-promoting statements about technical skill and innovations in clinical practice. They sometimes include information regarding charity donations, level of community involvement, and practice philosophy.
Materials and Methods
Categorization of Surgeon Websites and Ratings
Surgeon websites were selected from the 5 largest population centers in the United States. Analysis was undertaken to categorize the self-promotion content of each selected website using an objective scale to quantitatively assess the number of times that physicians referred to themselves in a positive manner. A thorough search of the literature did not reveal any validated questionnaire or assessment tool usable for this purpose. Five blinded raters were asked to count the number of positive self-directed remarks made by the author of each website. Websites were ranked based on the number of such statements. No rater was exposed to any styling or graphical information from any website. Only textual statements were used for the purposes of this study. All statements were printed on paper and evaluated without the use of a computer to prevent any searching or contamination of the subject or rater pool.
Websites were considered as self-promoting (using language that promotes the physician beyond the use of basic facts), or non-self-promoting(presenting little beyond basic biographical information) based on the presence of many (more than 5) or few (less than 5) self-promoting statements. The breakpoint of 5 self-promoting statements served to highlight a clear transition between the 2 general types of websites and provided a good demarcation between self-promoters and non-self-promoters. This distinction allowed for the choosing of contrasting websites, which could directly probe the question in our hypothesis about the effect of such websites on naïve or surgeon-peer respondents.
Each website was judged independently by 5 blinded raters. Inter-rater reliability scores were then calculated using Fleiss’ Kappa to assess reliability of the categorization of self-promoter or non-self-promoter. This value was calculated to be k = .80, 95% confidence interval (0.58-1.01), which is suggestive of a “substantial level of agreement.”9 Websites categorized as non-self-promoting contained a mean number of self-promoting statements of less than 2 (0-1.8) as judged by the 5 raters. By contrast, websites categorized as self-promoting had a mean number of self-promoting statements of 6.4 or higher (6.4-22.6). When the self-promoting websites and the non-self-promoting websites were compared, they were significantly different in the number of self-promoting statements t (43) = 7.90, P < .001, with self-promoting websites having significantly more self-promoting statements than non-self-promoting websites.
Surveys and Respondents
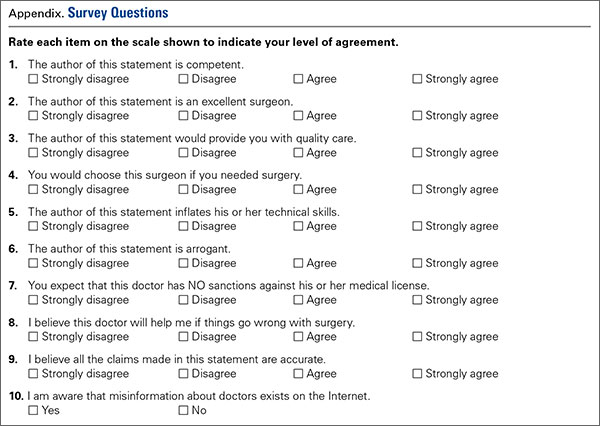
Next, a survey of 10 questions of interest was developed. A thorough literature search revealed no validated measure or survey to measure the effects of surgeon or physician self-promotion. We developed a 10-question survey to prove the impressions and allow for assessment of differences between respondent groups to measure the effect of promotion. The questions (see Appendix for survey questions) included a forced Likert rating system. Each response occurs and is presented on a scale from 0 to 3 (0 = Strongly Disagree, 1 = Disagree, 2 = Agree, and 3 = Strongly Agree).
Respondents were true volunteers recruited from 2 groups that were termed “surgeon-peers” and “naïve subjects.” Surgeon-peers were board-certified orthopedic surgeons (N = 21, all with medical doctorates). Demographic breakdown of the surgeon-peers revealed them to be reflective of the general population of orthopedic surgeons (71.4% male, 28.6% female, 90.2% Caucasian, 4.8% African American, and 4.8% Asian, all with professional degrees). Naïve subjects (N = 24, average age 41 years) were selected based on the criterion of having no affiliation with a healthcare system and no history of interaction with an orthopedic surgery or surgery in general. The demographic breakdown of naïve subjects was 45.8% male, 54.2% female, 79.1% Caucasian, 16.7% African American, and 4.2% Asian. Half of the naïve respondents had a Bachelor’s degree, 17% had a Master’s degree, 4% had a professional degree, and 29% had a high school diploma. No volunteer, in either group, received any form of inducement or reward for participation so as not to skew any responses in favor of physicians.
All participants were asked to read each surgeon’s statements and then complete a survey for each statement. Volunteers were not informed of a surgeon’s calculated level of self-promotion, and they were presented the survey questions in random order. Survey completion required unreimbursed time of approximately 1 to 2 hours.
Statistical Methods
The data compiled was then analyzed with SAS/STAT Software (SAS Institute Inc.) and a LR Type III analysis using the GENMOD procedure. The method of analysis and presentation of data focuses on the relationship between respondents perceptions between the surgeon-peer and naïve subject groups. The P values presented are the significance of the testing of interactions comparing the difference between surgeon-peers and naïve subjects, and the differences in their responses to each question for self-promoters and non-self-promoters. Surgeon-peers answer questions differently based on their assessment of a self-promoter or non-self-promoter website. It is this difference that is compared to the analogous difference for naïve subjects and statistically evaluated. The LR statistic for type III analysis tests if the differences are significantly different, ie, if the difference between the 2 subject groups is statistically significant. All statistical methods were performed by a qualified statistician who helped guide the design of this study.
Results
Each respondent was asked if they were aware that misinformation about doctors exists on the Internet. Half of the naïve subjects affirmed awareness of this whereas the other half were unaware. All surgeon-peers were aware of the presence of misinformation regarding physicians on the Internet.
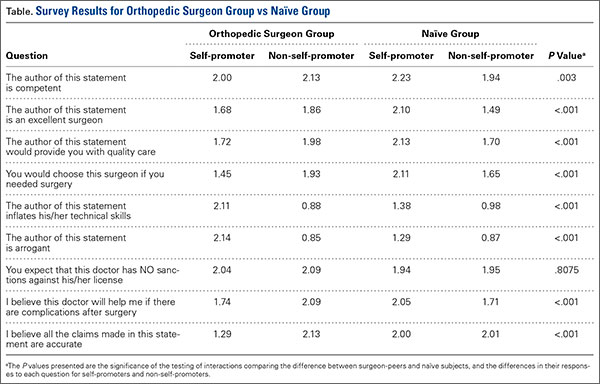
The results of the comparisons are shown in the Table. The columns show the average response to each question for self-promoters and non-self-promoters grouped by either surgeon-peer or naïve subject. In judging the overall accuracy of statements made on the Internet, naïve subjects found no difference between self-promoters and non-self-promoters, whereas surgeon-peers judged the difference to be large and significant in favor of non-self-promoting surgeons. Surgeon-peers generally rated non-self-promoters with significantly more positive Likert scores, indicating improved “competence”, “excellence”, and “better quality of care” when compared to naïve respondents (Table). The direction and magnitude of the difference was also striking, with the naïve respondents favoring self-promoters on all of these questions. This held true for the choice of orthopedic surgeon, where naïve responders favored self-promoters and surgeon-peers favored non-self-promoters. Moreover, naïve subjects believed that self-promoters would be significantly more likely to help them in the event of a complication, whereas surgeon-peers believed the opposite. Even when the direction of difference was the same in both groups, statistically significant differences in the responses were evident, as was the case when respondents were asked “Did the surgeon inflate his/her technical skills?” or “Did the author of this statement seem arrogant?” Both groups favored self-promoters for these questions, but the differences were larger among surgeon-peers, indicating that naïve subjects were somewhat less sensitive to the differences between promoters and non-self-promoters. There was no difference between surgeon-peers and naïve subjects in their expectations of sanctions against self-promoters’ licenses when compared to non-self-promoters, which was the only question to fail to garner a significant difference between respondents.
Discussion
This study explores the differences in the perceptions of physician websites between board-certified orthopedic surgeons and naïve individuals. These websites contain varying amounts of information presented in numerous ways. While we did not poll the website authors regarding their intent, the purpose of a website seems naturally to communicate believable information to the public. The information provided ranges widely from basic facts regarding education and contact information to statements regarding technical skills, reputation, television appearances, and the friendly nature of the office staff.
Our results suggest that board-certified orthopedic surgeons, peers of the writers of these websites, tend to view self-promoting surgeons more negatively than do their nonphysician counterparts. These findings support our hypothesis that self-promoting surgeons are perceived more favorably by the naïve, nonphysician population.
At first glance, our results suggest that the mere absence of a surgeon from the medium may affect the patient’s choice, because 50% of our naïve respondents indicated that they would use the Internet to choose a doctor. Interestingly, both the surgeon-peer group and naïve subjects were equally aware that misinformation exists on the Internet. However, when reviewing the websites, naïve subjects were significantly more likely to view self-promoters as more competent, more excellent, and more likely to provide quality care, and were more likely to choose the self-promoter if they needed surgery compared to the surgeon-peer group. The naïve group viewed self-promoters as less likely to inflate their technical skills but more likely to be arrogant. They viewed self-promoters as more likely to help if things went wrong and more likely to make accurate statements compared to the surgeon-peer group. This suggests that patients with little experience are more likely to choose a self-promoting physician than one who does not self-promote for reasons that cannot be proven true or false in the confines of a website. Further study is needed to see if perceptions based on web content translate into actual changes in healthcare choices.
This study had several limitations. Though statistically sound, the sample size of 45 people was small and should likely be expanded in further investigations to allow for analysis of demographics and socioeconomic factors. The study focused only on the text content of websites and purposely removed the influences of the other potential content mentioned previously. While a biography serves as an introduction, further research is needed to determine how initial perceptions affect future perceptions throughout the course of the patient-physician relationship. The small number of Internet biographies used cannot represent the vast array of information that could be displayed in numerous ways, but was necessary given the length of time donated by each uncompensated subject (1-2 hours). To minimize complexity, we purposefully ignored websites in the middle, somewhere in the continuum between self-promoting and non-self-promoting. Instead we selected websites that would be stark in their self-promotion to allow for the assessment of our hypothesis. Finally, this study was not designed to address economic implications of promotional advertising. The goal of much advertising is to generate revenue, and in the case of orthopedic surgery, one goal is likely attracting more patients, but this effect is beyond the scope of the current study. Given the elective nature of many orthopedic surgery procedures, the effect of promotional websites on a person’s decision to have surgery or not is an important topic for future study.
Taken together, the data suggests a profound influence of the content of the Internet website in the impressions made on different groups of people. These facts, although profound in their influence and unregulated by the medical profession, present both great opportunities and liabilities. The opportunities arise from the professional community to help guide what surgeons do to generate interest on the Internet. The liabilities arise on consideration of the consequences of self-promotion in the setting of real world surgical complications.
1. Tomycz ND. A profession selling out: lamenting the paradigm shift in physician advertising. J Med Ethics. 2006;32(1):26-28.
2. The principles of medical ethics of the Association of American Physicians and Surgeons. Association of American Physicians and Surgeons Web site. http://www.aapsonline.org/index.php/principles_of_medical_ethics. Accessed September 20, 2013.
3. Opinion 5.027 – Use of health-related online sites. American Medical Association Web site. http://www.ama-assn.org/ama/pub/physician-resources/medical-ethics/code-medical-ethics/opinion5027.page. Accessed September 10, 2013.
4. Standards of professionalism. Advertising by orthopaedic surgeons. Adopted April 18, 2007. American Academy of Orthopaedic Surgeons Web site. http://www.aaos.org/cc_files/aaosorg/member/profcomp/advertisingbyos.pdf. Accessed May 6, 2016.
5. Mostaghimi A, Crotty BH, Landon BE. The availability and nature of physician information on the internet. J Gen Intern Med. 2010;25(11):1152-1156.
6. Ad Hoc Committee on Health Literacy for the Council on Scientific Affairs, American Medical Association. Health Literacy: Report of the Council on Scientific Affairs. JAMA. 1999;281(6):552-557. doi:10.1001/jama.281.6.552.
7. Leroy G, Endicott JE, Mouradi O, Kauchak D, Just ML. Improving perceived and actual text difficulty for health information consumers using semi-automated methods. AMIA Annu Symp Proc. 2012;2012:522–531.
8. Dixon PR, Grant RC, Urbach DR. The impact of promotional language on patient preference for innovative procedures. J Am Coll Surg. 2013;217(3):S100.
9. Landis JR, Koch GG. A one-way components of variance model for categorical data. Biometrics. 1977;33(4):671–679.
In 1975, the American Medical Association (AMA) lifted the professional ban on physician advertising after a successful Federal Trade Commission suit.1 Since then, there has been a marked increase in the number of physicians marketing themselves directly to patients and consumers. With the pervasive nature of the Internet, never before has it been so easy and inexpensive to effectively communicate with a targeted population of people and influence their behavior. Few would dispute the role of advertising on consumer choices when used to sell products and services, change behavior, and educate consumers across all types of industries and professions. Thus, it is reasonable to hypothesize that the nature and content of a surgeon’s web presence could significantly affect patients’ decision-making and their impression of the orthopedic surgery profession.
There is a lack of consensus among physician organizations regarding physician advertising. For example, the American Association of Physicians and Surgeons (AAPS) takes an ethical stand on physician self-promotion. Their position states “The physician should not solicit patients. Professional reputation is the major source of patient referrals. The physician should be circumspect and restrained in dealing with the communication media, always avoiding self-aggrandizement.2” In contrast, the AMA has a less defined stance on physician self-promotion. With the exception of conflicts of interest and privacy guidelines, the AMA has few recommendations regarding the content of physician websites. The organization’s position states “There are no restrictions on advertising by physicians except those that can be specifically justified to protect the public from deceptive practices. …Nothing in this opinion is intended to discourage or to limit advertising and representations which are not false or deceptive.3” This guideline emphasizes accuracy of health-related information, but does not limit physician self-promotion or self-aggrandizement. The American Academy of Orthopaedic Surgeons (AAOS) holds a similar position. In their position statement on advertising by orthopedic surgeons, they encourage advertising and competition as “ethical and acceptable” as long as they are representing services in a “clear and accurate manner.”4 Furthermore, the AAOS also states that “An orthopaedic surgeon shall not use photographs, images, endorsements and/or statements in a false or misleading manner that communicate a degree of relief, safety, effectiveness, or benefits from orthopaedic care that are not representative of results attained by that orthopaedic surgeon.”4 The surgeon is responsible for his/her advertising materials and the content and claims therein, and is generally policed by peers through a complaint process with the AAOS.
Notably, up to 75% of Americans use the Internet for health-related information and this number is likely to increase.5Patients who utilize the Internet must choose from a vast array of search results for medical information from credible resources. Which sources are to be believed and relied upon? This depends on the health literacy among the general population. Inadequate health literacy is defined as “limited ability to obtain, process, and understand basic health information and services needed to make appropriate health decisions and follow instructions for treatment.”6 Patients have different levels of health literacy often unknown to even the most well-intentioned healthcare professional. It is often difficult to provide appropriate and meaningful information at a level that is most beneficial to the patient. It is estimated that 89 million people in the US have insufficient health literacy to understand treatments or preventive care.7 Certainly, with this information in mind, the orthopedic surgeon must consider his/her audience, and the potential for a fiduciary responsibility when preparing Internet content.
A tangible example of marketing results is the increasing popularity of robotic surgery over the last decade.8 Hospitals routinely advertise the availability of robotic surgery at their institution through various means, including roadside billboards. Despite limited evidence supporting a benefit of robotic surgery beyond less expensive conventional laparoscopic surgery, patients are increasingly seeking robotic surgery.8 With society’s increasing infatuation with technology, this is likely based on the presumption that robotic surgery is better and safer than conventional methods. It is likely that marketing pressure is at least partly responsible for the widespread adoption of robotic-assisted surgery and words used in marketing highlighting novelty have an important influence on patient preference.8
Orthopedic surgery, with its large proportion of elective surgeries, offers a unique venue to study differences in patient perceptions. Preoperative evaluations in orthopedics are often performed after an assessment of a surgeon’s reputation, which offers the patient an ability to choose their surgeon within their community.
We pondered how different promotional styles would affect potential patients’ perceptions. Would people believe that a self-promoting physician was more competent? Could fellow doctors “see through” the self-promotion of their peers? Based on the premise that advertising and self-promotion are undertaken because they are effective, we hypothesized that nonphysician patients perceive self-promoting orthopedic surgeons more favorably compared to members of the medical community.
Although numerous anonymous physician review sites exist, our analysis focused on surgeon self-promotion through personal websites or web pages. Within these sources, there exists a wide array of information and methods that physicians utilize to present themselves. Some physicians merely post their educational background and qualifications. This appears most often when the physician is associated with an academic institution and their profile is part of an institution’s website. Others post extensive self-promoting statements about technical skill and innovations in clinical practice. They sometimes include information regarding charity donations, level of community involvement, and practice philosophy.
Materials and Methods
Categorization of Surgeon Websites and Ratings
Surgeon websites were selected from the 5 largest population centers in the United States. Analysis was undertaken to categorize the self-promotion content of each selected website using an objective scale to quantitatively assess the number of times that physicians referred to themselves in a positive manner. A thorough search of the literature did not reveal any validated questionnaire or assessment tool usable for this purpose. Five blinded raters were asked to count the number of positive self-directed remarks made by the author of each website. Websites were ranked based on the number of such statements. No rater was exposed to any styling or graphical information from any website. Only textual statements were used for the purposes of this study. All statements were printed on paper and evaluated without the use of a computer to prevent any searching or contamination of the subject or rater pool.
Websites were considered as self-promoting (using language that promotes the physician beyond the use of basic facts), or non-self-promoting(presenting little beyond basic biographical information) based on the presence of many (more than 5) or few (less than 5) self-promoting statements. The breakpoint of 5 self-promoting statements served to highlight a clear transition between the 2 general types of websites and provided a good demarcation between self-promoters and non-self-promoters. This distinction allowed for the choosing of contrasting websites, which could directly probe the question in our hypothesis about the effect of such websites on naïve or surgeon-peer respondents.
Each website was judged independently by 5 blinded raters. Inter-rater reliability scores were then calculated using Fleiss’ Kappa to assess reliability of the categorization of self-promoter or non-self-promoter. This value was calculated to be k = .80, 95% confidence interval (0.58-1.01), which is suggestive of a “substantial level of agreement.”9 Websites categorized as non-self-promoting contained a mean number of self-promoting statements of less than 2 (0-1.8) as judged by the 5 raters. By contrast, websites categorized as self-promoting had a mean number of self-promoting statements of 6.4 or higher (6.4-22.6). When the self-promoting websites and the non-self-promoting websites were compared, they were significantly different in the number of self-promoting statements t (43) = 7.90, P < .001, with self-promoting websites having significantly more self-promoting statements than non-self-promoting websites.
Surveys and Respondents
Next, a survey of 10 questions of interest was developed. A thorough literature search revealed no validated measure or survey to measure the effects of surgeon or physician self-promotion. We developed a 10-question survey to prove the impressions and allow for assessment of differences between respondent groups to measure the effect of promotion. The questions (see Appendix for survey questions) included a forced Likert rating system. Each response occurs and is presented on a scale from 0 to 3 (0 = Strongly Disagree, 1 = Disagree, 2 = Agree, and 3 = Strongly Agree).
Respondents were true volunteers recruited from 2 groups that were termed “surgeon-peers” and “naïve subjects.” Surgeon-peers were board-certified orthopedic surgeons (N = 21, all with medical doctorates). Demographic breakdown of the surgeon-peers revealed them to be reflective of the general population of orthopedic surgeons (71.4% male, 28.6% female, 90.2% Caucasian, 4.8% African American, and 4.8% Asian, all with professional degrees). Naïve subjects (N = 24, average age 41 years) were selected based on the criterion of having no affiliation with a healthcare system and no history of interaction with an orthopedic surgery or surgery in general. The demographic breakdown of naïve subjects was 45.8% male, 54.2% female, 79.1% Caucasian, 16.7% African American, and 4.2% Asian. Half of the naïve respondents had a Bachelor’s degree, 17% had a Master’s degree, 4% had a professional degree, and 29% had a high school diploma. No volunteer, in either group, received any form of inducement or reward for participation so as not to skew any responses in favor of physicians.
All participants were asked to read each surgeon’s statements and then complete a survey for each statement. Volunteers were not informed of a surgeon’s calculated level of self-promotion, and they were presented the survey questions in random order. Survey completion required unreimbursed time of approximately 1 to 2 hours.
Statistical Methods
The data compiled was then analyzed with SAS/STAT Software (SAS Institute Inc.) and a LR Type III analysis using the GENMOD procedure. The method of analysis and presentation of data focuses on the relationship between respondents perceptions between the surgeon-peer and naïve subject groups. The P values presented are the significance of the testing of interactions comparing the difference between surgeon-peers and naïve subjects, and the differences in their responses to each question for self-promoters and non-self-promoters. Surgeon-peers answer questions differently based on their assessment of a self-promoter or non-self-promoter website. It is this difference that is compared to the analogous difference for naïve subjects and statistically evaluated. The LR statistic for type III analysis tests if the differences are significantly different, ie, if the difference between the 2 subject groups is statistically significant. All statistical methods were performed by a qualified statistician who helped guide the design of this study.
Results
Each respondent was asked if they were aware that misinformation about doctors exists on the Internet. Half of the naïve subjects affirmed awareness of this whereas the other half were unaware. All surgeon-peers were aware of the presence of misinformation regarding physicians on the Internet.
The results of the comparisons are shown in the Table. The columns show the average response to each question for self-promoters and non-self-promoters grouped by either surgeon-peer or naïve subject. In judging the overall accuracy of statements made on the Internet, naïve subjects found no difference between self-promoters and non-self-promoters, whereas surgeon-peers judged the difference to be large and significant in favor of non-self-promoting surgeons. Surgeon-peers generally rated non-self-promoters with significantly more positive Likert scores, indicating improved “competence”, “excellence”, and “better quality of care” when compared to naïve respondents (Table). The direction and magnitude of the difference was also striking, with the naïve respondents favoring self-promoters on all of these questions. This held true for the choice of orthopedic surgeon, where naïve responders favored self-promoters and surgeon-peers favored non-self-promoters. Moreover, naïve subjects believed that self-promoters would be significantly more likely to help them in the event of a complication, whereas surgeon-peers believed the opposite. Even when the direction of difference was the same in both groups, statistically significant differences in the responses were evident, as was the case when respondents were asked “Did the surgeon inflate his/her technical skills?” or “Did the author of this statement seem arrogant?” Both groups favored self-promoters for these questions, but the differences were larger among surgeon-peers, indicating that naïve subjects were somewhat less sensitive to the differences between promoters and non-self-promoters. There was no difference between surgeon-peers and naïve subjects in their expectations of sanctions against self-promoters’ licenses when compared to non-self-promoters, which was the only question to fail to garner a significant difference between respondents.
Discussion
This study explores the differences in the perceptions of physician websites between board-certified orthopedic surgeons and naïve individuals. These websites contain varying amounts of information presented in numerous ways. While we did not poll the website authors regarding their intent, the purpose of a website seems naturally to communicate believable information to the public. The information provided ranges widely from basic facts regarding education and contact information to statements regarding technical skills, reputation, television appearances, and the friendly nature of the office staff.
Our results suggest that board-certified orthopedic surgeons, peers of the writers of these websites, tend to view self-promoting surgeons more negatively than do their nonphysician counterparts. These findings support our hypothesis that self-promoting surgeons are perceived more favorably by the naïve, nonphysician population.
At first glance, our results suggest that the mere absence of a surgeon from the medium may affect the patient’s choice, because 50% of our naïve respondents indicated that they would use the Internet to choose a doctor. Interestingly, both the surgeon-peer group and naïve subjects were equally aware that misinformation exists on the Internet. However, when reviewing the websites, naïve subjects were significantly more likely to view self-promoters as more competent, more excellent, and more likely to provide quality care, and were more likely to choose the self-promoter if they needed surgery compared to the surgeon-peer group. The naïve group viewed self-promoters as less likely to inflate their technical skills but more likely to be arrogant. They viewed self-promoters as more likely to help if things went wrong and more likely to make accurate statements compared to the surgeon-peer group. This suggests that patients with little experience are more likely to choose a self-promoting physician than one who does not self-promote for reasons that cannot be proven true or false in the confines of a website. Further study is needed to see if perceptions based on web content translate into actual changes in healthcare choices.
This study had several limitations. Though statistically sound, the sample size of 45 people was small and should likely be expanded in further investigations to allow for analysis of demographics and socioeconomic factors. The study focused only on the text content of websites and purposely removed the influences of the other potential content mentioned previously. While a biography serves as an introduction, further research is needed to determine how initial perceptions affect future perceptions throughout the course of the patient-physician relationship. The small number of Internet biographies used cannot represent the vast array of information that could be displayed in numerous ways, but was necessary given the length of time donated by each uncompensated subject (1-2 hours). To minimize complexity, we purposefully ignored websites in the middle, somewhere in the continuum between self-promoting and non-self-promoting. Instead we selected websites that would be stark in their self-promotion to allow for the assessment of our hypothesis. Finally, this study was not designed to address economic implications of promotional advertising. The goal of much advertising is to generate revenue, and in the case of orthopedic surgery, one goal is likely attracting more patients, but this effect is beyond the scope of the current study. Given the elective nature of many orthopedic surgery procedures, the effect of promotional websites on a person’s decision to have surgery or not is an important topic for future study.
Taken together, the data suggests a profound influence of the content of the Internet website in the impressions made on different groups of people. These facts, although profound in their influence and unregulated by the medical profession, present both great opportunities and liabilities. The opportunities arise from the professional community to help guide what surgeons do to generate interest on the Internet. The liabilities arise on consideration of the consequences of self-promotion in the setting of real world surgical complications.
In 1975, the American Medical Association (AMA) lifted the professional ban on physician advertising after a successful Federal Trade Commission suit.1 Since then, there has been a marked increase in the number of physicians marketing themselves directly to patients and consumers. With the pervasive nature of the Internet, never before has it been so easy and inexpensive to effectively communicate with a targeted population of people and influence their behavior. Few would dispute the role of advertising on consumer choices when used to sell products and services, change behavior, and educate consumers across all types of industries and professions. Thus, it is reasonable to hypothesize that the nature and content of a surgeon’s web presence could significantly affect patients’ decision-making and their impression of the orthopedic surgery profession.
There is a lack of consensus among physician organizations regarding physician advertising. For example, the American Association of Physicians and Surgeons (AAPS) takes an ethical stand on physician self-promotion. Their position states “The physician should not solicit patients. Professional reputation is the major source of patient referrals. The physician should be circumspect and restrained in dealing with the communication media, always avoiding self-aggrandizement.2” In contrast, the AMA has a less defined stance on physician self-promotion. With the exception of conflicts of interest and privacy guidelines, the AMA has few recommendations regarding the content of physician websites. The organization’s position states “There are no restrictions on advertising by physicians except those that can be specifically justified to protect the public from deceptive practices. …Nothing in this opinion is intended to discourage or to limit advertising and representations which are not false or deceptive.3” This guideline emphasizes accuracy of health-related information, but does not limit physician self-promotion or self-aggrandizement. The American Academy of Orthopaedic Surgeons (AAOS) holds a similar position. In their position statement on advertising by orthopedic surgeons, they encourage advertising and competition as “ethical and acceptable” as long as they are representing services in a “clear and accurate manner.”4 Furthermore, the AAOS also states that “An orthopaedic surgeon shall not use photographs, images, endorsements and/or statements in a false or misleading manner that communicate a degree of relief, safety, effectiveness, or benefits from orthopaedic care that are not representative of results attained by that orthopaedic surgeon.”4 The surgeon is responsible for his/her advertising materials and the content and claims therein, and is generally policed by peers through a complaint process with the AAOS.
Notably, up to 75% of Americans use the Internet for health-related information and this number is likely to increase.5Patients who utilize the Internet must choose from a vast array of search results for medical information from credible resources. Which sources are to be believed and relied upon? This depends on the health literacy among the general population. Inadequate health literacy is defined as “limited ability to obtain, process, and understand basic health information and services needed to make appropriate health decisions and follow instructions for treatment.”6 Patients have different levels of health literacy often unknown to even the most well-intentioned healthcare professional. It is often difficult to provide appropriate and meaningful information at a level that is most beneficial to the patient. It is estimated that 89 million people in the US have insufficient health literacy to understand treatments or preventive care.7 Certainly, with this information in mind, the orthopedic surgeon must consider his/her audience, and the potential for a fiduciary responsibility when preparing Internet content.
A tangible example of marketing results is the increasing popularity of robotic surgery over the last decade.8 Hospitals routinely advertise the availability of robotic surgery at their institution through various means, including roadside billboards. Despite limited evidence supporting a benefit of robotic surgery beyond less expensive conventional laparoscopic surgery, patients are increasingly seeking robotic surgery.8 With society’s increasing infatuation with technology, this is likely based on the presumption that robotic surgery is better and safer than conventional methods. It is likely that marketing pressure is at least partly responsible for the widespread adoption of robotic-assisted surgery and words used in marketing highlighting novelty have an important influence on patient preference.8
Orthopedic surgery, with its large proportion of elective surgeries, offers a unique venue to study differences in patient perceptions. Preoperative evaluations in orthopedics are often performed after an assessment of a surgeon’s reputation, which offers the patient an ability to choose their surgeon within their community.
We pondered how different promotional styles would affect potential patients’ perceptions. Would people believe that a self-promoting physician was more competent? Could fellow doctors “see through” the self-promotion of their peers? Based on the premise that advertising and self-promotion are undertaken because they are effective, we hypothesized that nonphysician patients perceive self-promoting orthopedic surgeons more favorably compared to members of the medical community.
Although numerous anonymous physician review sites exist, our analysis focused on surgeon self-promotion through personal websites or web pages. Within these sources, there exists a wide array of information and methods that physicians utilize to present themselves. Some physicians merely post their educational background and qualifications. This appears most often when the physician is associated with an academic institution and their profile is part of an institution’s website. Others post extensive self-promoting statements about technical skill and innovations in clinical practice. They sometimes include information regarding charity donations, level of community involvement, and practice philosophy.
Materials and Methods
Categorization of Surgeon Websites and Ratings
Surgeon websites were selected from the 5 largest population centers in the United States. Analysis was undertaken to categorize the self-promotion content of each selected website using an objective scale to quantitatively assess the number of times that physicians referred to themselves in a positive manner. A thorough search of the literature did not reveal any validated questionnaire or assessment tool usable for this purpose. Five blinded raters were asked to count the number of positive self-directed remarks made by the author of each website. Websites were ranked based on the number of such statements. No rater was exposed to any styling or graphical information from any website. Only textual statements were used for the purposes of this study. All statements were printed on paper and evaluated without the use of a computer to prevent any searching or contamination of the subject or rater pool.
Websites were considered as self-promoting (using language that promotes the physician beyond the use of basic facts), or non-self-promoting(presenting little beyond basic biographical information) based on the presence of many (more than 5) or few (less than 5) self-promoting statements. The breakpoint of 5 self-promoting statements served to highlight a clear transition between the 2 general types of websites and provided a good demarcation between self-promoters and non-self-promoters. This distinction allowed for the choosing of contrasting websites, which could directly probe the question in our hypothesis about the effect of such websites on naïve or surgeon-peer respondents.
Each website was judged independently by 5 blinded raters. Inter-rater reliability scores were then calculated using Fleiss’ Kappa to assess reliability of the categorization of self-promoter or non-self-promoter. This value was calculated to be k = .80, 95% confidence interval (0.58-1.01), which is suggestive of a “substantial level of agreement.”9 Websites categorized as non-self-promoting contained a mean number of self-promoting statements of less than 2 (0-1.8) as judged by the 5 raters. By contrast, websites categorized as self-promoting had a mean number of self-promoting statements of 6.4 or higher (6.4-22.6). When the self-promoting websites and the non-self-promoting websites were compared, they were significantly different in the number of self-promoting statements t (43) = 7.90, P < .001, with self-promoting websites having significantly more self-promoting statements than non-self-promoting websites.
Surveys and Respondents
Next, a survey of 10 questions of interest was developed. A thorough literature search revealed no validated measure or survey to measure the effects of surgeon or physician self-promotion. We developed a 10-question survey to prove the impressions and allow for assessment of differences between respondent groups to measure the effect of promotion. The questions (see Appendix for survey questions) included a forced Likert rating system. Each response occurs and is presented on a scale from 0 to 3 (0 = Strongly Disagree, 1 = Disagree, 2 = Agree, and 3 = Strongly Agree).
Respondents were true volunteers recruited from 2 groups that were termed “surgeon-peers” and “naïve subjects.” Surgeon-peers were board-certified orthopedic surgeons (N = 21, all with medical doctorates). Demographic breakdown of the surgeon-peers revealed them to be reflective of the general population of orthopedic surgeons (71.4% male, 28.6% female, 90.2% Caucasian, 4.8% African American, and 4.8% Asian, all with professional degrees). Naïve subjects (N = 24, average age 41 years) were selected based on the criterion of having no affiliation with a healthcare system and no history of interaction with an orthopedic surgery or surgery in general. The demographic breakdown of naïve subjects was 45.8% male, 54.2% female, 79.1% Caucasian, 16.7% African American, and 4.2% Asian. Half of the naïve respondents had a Bachelor’s degree, 17% had a Master’s degree, 4% had a professional degree, and 29% had a high school diploma. No volunteer, in either group, received any form of inducement or reward for participation so as not to skew any responses in favor of physicians.
All participants were asked to read each surgeon’s statements and then complete a survey for each statement. Volunteers were not informed of a surgeon’s calculated level of self-promotion, and they were presented the survey questions in random order. Survey completion required unreimbursed time of approximately 1 to 2 hours.
Statistical Methods
The data compiled was then analyzed with SAS/STAT Software (SAS Institute Inc.) and a LR Type III analysis using the GENMOD procedure. The method of analysis and presentation of data focuses on the relationship between respondents perceptions between the surgeon-peer and naïve subject groups. The P values presented are the significance of the testing of interactions comparing the difference between surgeon-peers and naïve subjects, and the differences in their responses to each question for self-promoters and non-self-promoters. Surgeon-peers answer questions differently based on their assessment of a self-promoter or non-self-promoter website. It is this difference that is compared to the analogous difference for naïve subjects and statistically evaluated. The LR statistic for type III analysis tests if the differences are significantly different, ie, if the difference between the 2 subject groups is statistically significant. All statistical methods were performed by a qualified statistician who helped guide the design of this study.
Results
Each respondent was asked if they were aware that misinformation about doctors exists on the Internet. Half of the naïve subjects affirmed awareness of this whereas the other half were unaware. All surgeon-peers were aware of the presence of misinformation regarding physicians on the Internet.
The results of the comparisons are shown in the Table. The columns show the average response to each question for self-promoters and non-self-promoters grouped by either surgeon-peer or naïve subject. In judging the overall accuracy of statements made on the Internet, naïve subjects found no difference between self-promoters and non-self-promoters, whereas surgeon-peers judged the difference to be large and significant in favor of non-self-promoting surgeons. Surgeon-peers generally rated non-self-promoters with significantly more positive Likert scores, indicating improved “competence”, “excellence”, and “better quality of care” when compared to naïve respondents (Table). The direction and magnitude of the difference was also striking, with the naïve respondents favoring self-promoters on all of these questions. This held true for the choice of orthopedic surgeon, where naïve responders favored self-promoters and surgeon-peers favored non-self-promoters. Moreover, naïve subjects believed that self-promoters would be significantly more likely to help them in the event of a complication, whereas surgeon-peers believed the opposite. Even when the direction of difference was the same in both groups, statistically significant differences in the responses were evident, as was the case when respondents were asked “Did the surgeon inflate his/her technical skills?” or “Did the author of this statement seem arrogant?” Both groups favored self-promoters for these questions, but the differences were larger among surgeon-peers, indicating that naïve subjects were somewhat less sensitive to the differences between promoters and non-self-promoters. There was no difference between surgeon-peers and naïve subjects in their expectations of sanctions against self-promoters’ licenses when compared to non-self-promoters, which was the only question to fail to garner a significant difference between respondents.
Discussion
This study explores the differences in the perceptions of physician websites between board-certified orthopedic surgeons and naïve individuals. These websites contain varying amounts of information presented in numerous ways. While we did not poll the website authors regarding their intent, the purpose of a website seems naturally to communicate believable information to the public. The information provided ranges widely from basic facts regarding education and contact information to statements regarding technical skills, reputation, television appearances, and the friendly nature of the office staff.
Our results suggest that board-certified orthopedic surgeons, peers of the writers of these websites, tend to view self-promoting surgeons more negatively than do their nonphysician counterparts. These findings support our hypothesis that self-promoting surgeons are perceived more favorably by the naïve, nonphysician population.
At first glance, our results suggest that the mere absence of a surgeon from the medium may affect the patient’s choice, because 50% of our naïve respondents indicated that they would use the Internet to choose a doctor. Interestingly, both the surgeon-peer group and naïve subjects were equally aware that misinformation exists on the Internet. However, when reviewing the websites, naïve subjects were significantly more likely to view self-promoters as more competent, more excellent, and more likely to provide quality care, and were more likely to choose the self-promoter if they needed surgery compared to the surgeon-peer group. The naïve group viewed self-promoters as less likely to inflate their technical skills but more likely to be arrogant. They viewed self-promoters as more likely to help if things went wrong and more likely to make accurate statements compared to the surgeon-peer group. This suggests that patients with little experience are more likely to choose a self-promoting physician than one who does not self-promote for reasons that cannot be proven true or false in the confines of a website. Further study is needed to see if perceptions based on web content translate into actual changes in healthcare choices.
This study had several limitations. Though statistically sound, the sample size of 45 people was small and should likely be expanded in further investigations to allow for analysis of demographics and socioeconomic factors. The study focused only on the text content of websites and purposely removed the influences of the other potential content mentioned previously. While a biography serves as an introduction, further research is needed to determine how initial perceptions affect future perceptions throughout the course of the patient-physician relationship. The small number of Internet biographies used cannot represent the vast array of information that could be displayed in numerous ways, but was necessary given the length of time donated by each uncompensated subject (1-2 hours). To minimize complexity, we purposefully ignored websites in the middle, somewhere in the continuum between self-promoting and non-self-promoting. Instead we selected websites that would be stark in their self-promotion to allow for the assessment of our hypothesis. Finally, this study was not designed to address economic implications of promotional advertising. The goal of much advertising is to generate revenue, and in the case of orthopedic surgery, one goal is likely attracting more patients, but this effect is beyond the scope of the current study. Given the elective nature of many orthopedic surgery procedures, the effect of promotional websites on a person’s decision to have surgery or not is an important topic for future study.
Taken together, the data suggests a profound influence of the content of the Internet website in the impressions made on different groups of people. These facts, although profound in their influence and unregulated by the medical profession, present both great opportunities and liabilities. The opportunities arise from the professional community to help guide what surgeons do to generate interest on the Internet. The liabilities arise on consideration of the consequences of self-promotion in the setting of real world surgical complications.
1. Tomycz ND. A profession selling out: lamenting the paradigm shift in physician advertising. J Med Ethics. 2006;32(1):26-28.
2. The principles of medical ethics of the Association of American Physicians and Surgeons. Association of American Physicians and Surgeons Web site. http://www.aapsonline.org/index.php/principles_of_medical_ethics. Accessed September 20, 2013.
3. Opinion 5.027 – Use of health-related online sites. American Medical Association Web site. http://www.ama-assn.org/ama/pub/physician-resources/medical-ethics/code-medical-ethics/opinion5027.page. Accessed September 10, 2013.
4. Standards of professionalism. Advertising by orthopaedic surgeons. Adopted April 18, 2007. American Academy of Orthopaedic Surgeons Web site. http://www.aaos.org/cc_files/aaosorg/member/profcomp/advertisingbyos.pdf. Accessed May 6, 2016.
5. Mostaghimi A, Crotty BH, Landon BE. The availability and nature of physician information on the internet. J Gen Intern Med. 2010;25(11):1152-1156.
6. Ad Hoc Committee on Health Literacy for the Council on Scientific Affairs, American Medical Association. Health Literacy: Report of the Council on Scientific Affairs. JAMA. 1999;281(6):552-557. doi:10.1001/jama.281.6.552.
7. Leroy G, Endicott JE, Mouradi O, Kauchak D, Just ML. Improving perceived and actual text difficulty for health information consumers using semi-automated methods. AMIA Annu Symp Proc. 2012;2012:522–531.
8. Dixon PR, Grant RC, Urbach DR. The impact of promotional language on patient preference for innovative procedures. J Am Coll Surg. 2013;217(3):S100.
9. Landis JR, Koch GG. A one-way components of variance model for categorical data. Biometrics. 1977;33(4):671–679.
1. Tomycz ND. A profession selling out: lamenting the paradigm shift in physician advertising. J Med Ethics. 2006;32(1):26-28.
2. The principles of medical ethics of the Association of American Physicians and Surgeons. Association of American Physicians and Surgeons Web site. http://www.aapsonline.org/index.php/principles_of_medical_ethics. Accessed September 20, 2013.
3. Opinion 5.027 – Use of health-related online sites. American Medical Association Web site. http://www.ama-assn.org/ama/pub/physician-resources/medical-ethics/code-medical-ethics/opinion5027.page. Accessed September 10, 2013.
4. Standards of professionalism. Advertising by orthopaedic surgeons. Adopted April 18, 2007. American Academy of Orthopaedic Surgeons Web site. http://www.aaos.org/cc_files/aaosorg/member/profcomp/advertisingbyos.pdf. Accessed May 6, 2016.
5. Mostaghimi A, Crotty BH, Landon BE. The availability and nature of physician information on the internet. J Gen Intern Med. 2010;25(11):1152-1156.
6. Ad Hoc Committee on Health Literacy for the Council on Scientific Affairs, American Medical Association. Health Literacy: Report of the Council on Scientific Affairs. JAMA. 1999;281(6):552-557. doi:10.1001/jama.281.6.552.
7. Leroy G, Endicott JE, Mouradi O, Kauchak D, Just ML. Improving perceived and actual text difficulty for health information consumers using semi-automated methods. AMIA Annu Symp Proc. 2012;2012:522–531.
8. Dixon PR, Grant RC, Urbach DR. The impact of promotional language on patient preference for innovative procedures. J Am Coll Surg. 2013;217(3):S100.
9. Landis JR, Koch GG. A one-way components of variance model for categorical data. Biometrics. 1977;33(4):671–679.
Fewer adults with psychological distress getting mental health care
The percentage of adults with psychological distress who saw or spoke with a mental health professional has gone down every year since 2012, according to the National Center for Heath Statistics.
In 2012, almost 42% of adults aged 18-64 years who reported experiencing serious psychological distress in the previous 30 days had seen or spoken with a mental health professional in the previous 12 months. By 2015 (January through September), the percentage was down to 34.2%, and it dropped each of the 2 years between, with the trend significant at P less than .05, the NCHS reported.
For adults without psychological distress, contacts with mental health professionals were consistently around 7% for all 4 years, the NCHS noted.
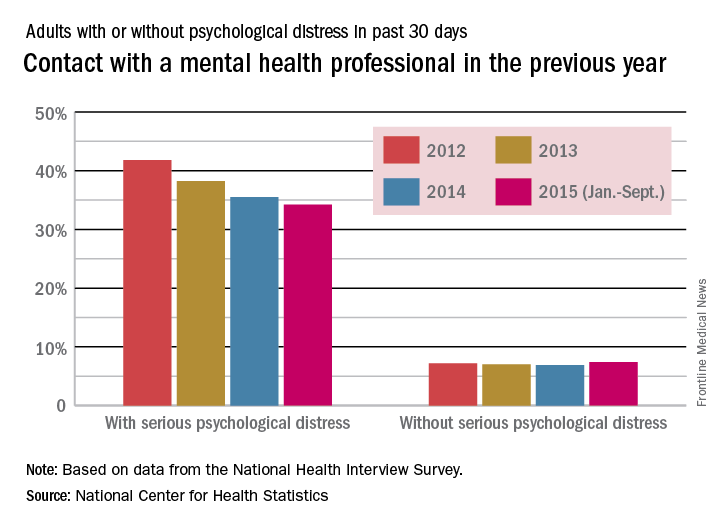
In the first 9 months of 2015, 3.8% of all adults aged 18-64 years experienced serious psychological distress, compared with 3.4% in 2014, 4% in 2013, and 3.2% in 2012, according to data from the National Health Interview Survey.
The percentage of adults with psychological distress who saw or spoke with a mental health professional has gone down every year since 2012, according to the National Center for Heath Statistics.
In 2012, almost 42% of adults aged 18-64 years who reported experiencing serious psychological distress in the previous 30 days had seen or spoken with a mental health professional in the previous 12 months. By 2015 (January through September), the percentage was down to 34.2%, and it dropped each of the 2 years between, with the trend significant at P less than .05, the NCHS reported.
For adults without psychological distress, contacts with mental health professionals were consistently around 7% for all 4 years, the NCHS noted.
In the first 9 months of 2015, 3.8% of all adults aged 18-64 years experienced serious psychological distress, compared with 3.4% in 2014, 4% in 2013, and 3.2% in 2012, according to data from the National Health Interview Survey.
The percentage of adults with psychological distress who saw or spoke with a mental health professional has gone down every year since 2012, according to the National Center for Heath Statistics.
In 2012, almost 42% of adults aged 18-64 years who reported experiencing serious psychological distress in the previous 30 days had seen or spoken with a mental health professional in the previous 12 months. By 2015 (January through September), the percentage was down to 34.2%, and it dropped each of the 2 years between, with the trend significant at P less than .05, the NCHS reported.
For adults without psychological distress, contacts with mental health professionals were consistently around 7% for all 4 years, the NCHS noted.
In the first 9 months of 2015, 3.8% of all adults aged 18-64 years experienced serious psychological distress, compared with 3.4% in 2014, 4% in 2013, and 3.2% in 2012, according to data from the National Health Interview Survey.
High spondyloarthritis risk found in first-degree relatives of ankylosing spondylitis patients
As many as one-third of healthy first-degree relatives of individuals with ankylosing spondylitis may meet the criteria for spondyloarthritis, according to results from a prospective cohort study.
Dr. Maureen C. Turina of the Amsterdam Rheumatology and Immunology Center at the University of Amsterdam and her coauthors enrolled 51 first-degree relatives of individuals with HLA-B27–positive ankylosing spondylitis in the study to see whether any of these otherwise healthy individuals showed early signs of the disease. They found that 17 (33%) of these first-degree relatives fulfilled any of the spondyloarthritis (SpA) classification criteria at baseline – a higher rate than reported in previous studies – and 7 (14%) met both the Assessment of Spondyloarthritis International Society (ASAS) criteria and European Spondyloarthropathy Study Group (ESSG) criteria.
The authors suggested that the higher rate, compared with previous studies, could be due to channeling bias, as individuals with clinical symptoms may have been more willing to participate in the study.
However, 4 of the 38 individuals who did not fulfill either of these criteria for SpA still showed imaging abnormalities – including syndesmophytes on the cervical spine and bone marrow edema in the sacroiliac joint.
The authors suggested these imaging abnormalities may represent a subclinical phase of SpA (Arthritis Rheumatol. 2016 May 23. doi: 10.1002/art.39766).
The participants who met the ASAS and/or ESSG classification criteria for SpA had more axial, entheseal, and joint pain, compared with those who did not meet the criteria. All of these patients also reported back pain, compared with 35% of those who did not fulfill the criteria.
However, the individuals meeting the SpA criteria had low overall disease scores, and did not show any increase in inflammatory serum markers.
“Importantly, some key features of SpA including the presence of peripheral disease or extra-articular manifestations and increased inflammatory parameters were only rarely observed in FDRs [first-degree relatives] and were also not different in those fulfilling the ASAS axSpA and/or ESSG classification criteria versus those who did not,” the authors reported.
“Future follow-up will learn if and which of these FDRs will evolve into the clinically established phase of SpA.”
When specifically asked, more than half (57%) of the first-degree relatives said they had back pain, and 40% said they had experienced or were experiencing arthralgia.
“The fact that these FDRs were not investigated for or diagnosed with axial SpA before inclusion in the study may be related either to the fact that the back pain symptoms were relatively mild or to ignorance of general physicians for these alarm symptoms,” the authors wrote.
One person said they had past or present peripheral arthritis but none reported enthesitis or dactylitis.
While none of the subjects had arthritis, 16% reported at least one tender joint, and 22% had a modified Schober score of less than 4.5 cm, with 4% showing a chest expansion of less than 3.6 cm.
Researchers also examined whether HLA-B27 status had any impact on the first-degree relatives, as previous studies had suggested that SpA is more likely to present in HLA-B27–positive first-degree relatives.
However they found no differences between the HLA-B27–positive and HLA-B27–negative groups in terms of demographics, symptom history, disease activity, clinical examination, or laboratory or imaging data; a finding the authors described as “intriguing.”
“However, if confirmed in a larger sample set, further follow-up of these FDRs will allow us to determine if FDRs showing signs and symptoms of SpA will evolve to more active and severe disease, independently of HLA-B27 status or, alternatively, if the presence of HLA-B27 may promote exacerbation and persistence of subclinical pathology.”
The study was supported by the Dutch Arthritis Foundation. No conflicts of interest were declared.
As many as one-third of healthy first-degree relatives of individuals with ankylosing spondylitis may meet the criteria for spondyloarthritis, according to results from a prospective cohort study.
Dr. Maureen C. Turina of the Amsterdam Rheumatology and Immunology Center at the University of Amsterdam and her coauthors enrolled 51 first-degree relatives of individuals with HLA-B27–positive ankylosing spondylitis in the study to see whether any of these otherwise healthy individuals showed early signs of the disease. They found that 17 (33%) of these first-degree relatives fulfilled any of the spondyloarthritis (SpA) classification criteria at baseline – a higher rate than reported in previous studies – and 7 (14%) met both the Assessment of Spondyloarthritis International Society (ASAS) criteria and European Spondyloarthropathy Study Group (ESSG) criteria.
The authors suggested that the higher rate, compared with previous studies, could be due to channeling bias, as individuals with clinical symptoms may have been more willing to participate in the study.
However, 4 of the 38 individuals who did not fulfill either of these criteria for SpA still showed imaging abnormalities – including syndesmophytes on the cervical spine and bone marrow edema in the sacroiliac joint.
The authors suggested these imaging abnormalities may represent a subclinical phase of SpA (Arthritis Rheumatol. 2016 May 23. doi: 10.1002/art.39766).
The participants who met the ASAS and/or ESSG classification criteria for SpA had more axial, entheseal, and joint pain, compared with those who did not meet the criteria. All of these patients also reported back pain, compared with 35% of those who did not fulfill the criteria.
However, the individuals meeting the SpA criteria had low overall disease scores, and did not show any increase in inflammatory serum markers.
“Importantly, some key features of SpA including the presence of peripheral disease or extra-articular manifestations and increased inflammatory parameters were only rarely observed in FDRs [first-degree relatives] and were also not different in those fulfilling the ASAS axSpA and/or ESSG classification criteria versus those who did not,” the authors reported.
“Future follow-up will learn if and which of these FDRs will evolve into the clinically established phase of SpA.”
When specifically asked, more than half (57%) of the first-degree relatives said they had back pain, and 40% said they had experienced or were experiencing arthralgia.
“The fact that these FDRs were not investigated for or diagnosed with axial SpA before inclusion in the study may be related either to the fact that the back pain symptoms were relatively mild or to ignorance of general physicians for these alarm symptoms,” the authors wrote.
One person said they had past or present peripheral arthritis but none reported enthesitis or dactylitis.
While none of the subjects had arthritis, 16% reported at least one tender joint, and 22% had a modified Schober score of less than 4.5 cm, with 4% showing a chest expansion of less than 3.6 cm.
Researchers also examined whether HLA-B27 status had any impact on the first-degree relatives, as previous studies had suggested that SpA is more likely to present in HLA-B27–positive first-degree relatives.
However they found no differences between the HLA-B27–positive and HLA-B27–negative groups in terms of demographics, symptom history, disease activity, clinical examination, or laboratory or imaging data; a finding the authors described as “intriguing.”
“However, if confirmed in a larger sample set, further follow-up of these FDRs will allow us to determine if FDRs showing signs and symptoms of SpA will evolve to more active and severe disease, independently of HLA-B27 status or, alternatively, if the presence of HLA-B27 may promote exacerbation and persistence of subclinical pathology.”
The study was supported by the Dutch Arthritis Foundation. No conflicts of interest were declared.
As many as one-third of healthy first-degree relatives of individuals with ankylosing spondylitis may meet the criteria for spondyloarthritis, according to results from a prospective cohort study.
Dr. Maureen C. Turina of the Amsterdam Rheumatology and Immunology Center at the University of Amsterdam and her coauthors enrolled 51 first-degree relatives of individuals with HLA-B27–positive ankylosing spondylitis in the study to see whether any of these otherwise healthy individuals showed early signs of the disease. They found that 17 (33%) of these first-degree relatives fulfilled any of the spondyloarthritis (SpA) classification criteria at baseline – a higher rate than reported in previous studies – and 7 (14%) met both the Assessment of Spondyloarthritis International Society (ASAS) criteria and European Spondyloarthropathy Study Group (ESSG) criteria.
The authors suggested that the higher rate, compared with previous studies, could be due to channeling bias, as individuals with clinical symptoms may have been more willing to participate in the study.
However, 4 of the 38 individuals who did not fulfill either of these criteria for SpA still showed imaging abnormalities – including syndesmophytes on the cervical spine and bone marrow edema in the sacroiliac joint.
The authors suggested these imaging abnormalities may represent a subclinical phase of SpA (Arthritis Rheumatol. 2016 May 23. doi: 10.1002/art.39766).
The participants who met the ASAS and/or ESSG classification criteria for SpA had more axial, entheseal, and joint pain, compared with those who did not meet the criteria. All of these patients also reported back pain, compared with 35% of those who did not fulfill the criteria.
However, the individuals meeting the SpA criteria had low overall disease scores, and did not show any increase in inflammatory serum markers.
“Importantly, some key features of SpA including the presence of peripheral disease or extra-articular manifestations and increased inflammatory parameters were only rarely observed in FDRs [first-degree relatives] and were also not different in those fulfilling the ASAS axSpA and/or ESSG classification criteria versus those who did not,” the authors reported.
“Future follow-up will learn if and which of these FDRs will evolve into the clinically established phase of SpA.”
When specifically asked, more than half (57%) of the first-degree relatives said they had back pain, and 40% said they had experienced or were experiencing arthralgia.
“The fact that these FDRs were not investigated for or diagnosed with axial SpA before inclusion in the study may be related either to the fact that the back pain symptoms were relatively mild or to ignorance of general physicians for these alarm symptoms,” the authors wrote.
One person said they had past or present peripheral arthritis but none reported enthesitis or dactylitis.
While none of the subjects had arthritis, 16% reported at least one tender joint, and 22% had a modified Schober score of less than 4.5 cm, with 4% showing a chest expansion of less than 3.6 cm.
Researchers also examined whether HLA-B27 status had any impact on the first-degree relatives, as previous studies had suggested that SpA is more likely to present in HLA-B27–positive first-degree relatives.
However they found no differences between the HLA-B27–positive and HLA-B27–negative groups in terms of demographics, symptom history, disease activity, clinical examination, or laboratory or imaging data; a finding the authors described as “intriguing.”
“However, if confirmed in a larger sample set, further follow-up of these FDRs will allow us to determine if FDRs showing signs and symptoms of SpA will evolve to more active and severe disease, independently of HLA-B27 status or, alternatively, if the presence of HLA-B27 may promote exacerbation and persistence of subclinical pathology.”
The study was supported by the Dutch Arthritis Foundation. No conflicts of interest were declared.
FROM ARTHRITIS & RHEUMATOLOGY
Key clinical point: Healthy first-degree relatives of individuals with ankylosing spondylitis may have signs of preclinical spondyloarthritis.
Major finding: One-third of first-degree relatives of individuals with ankylosing spondylitis meet one of two sets of criteria for spondyloarthritis.
Data source: Prospective cohort study of 51 first-degree relatives of individuals with HLA-B27–positive ankylosing spondylitis.
Disclosures: The study was supported by the Dutch Arthritis Foundation. No conflicts of interest were declared.
Less education tied to more anxiety
Adults with less than a high school education are more than twice as likely as are those with high school degrees to have reported an anxiety disorder in the past year, according to the National Survey on Drug Use and Health’s 2008 to 2012 Mental Health Surveillance Study (MHSS), published June 2.
The data identified anxiety disorders in 13% of non–high school graduates vs. 5% of high school graduates. Adults with at least a college degree had the lowest rates of past year anxiety (4.3%). The MHSS estimated that approximately 13 million adults in the United States had at least one anxiety disorder within the past year.
“Although the MHSS results cannot be used to determine whether anxiety stopped people from finishing high school, having an anxiety disorder can lower the odds of graduating from high school and the odds of attending college,” the researchers wrote. The findings emphasize the need to support people with anxiety to help them remain in school and succeed, they added.
The findings were published in the Center for Behavioral Health Statistics and Quality Report. Read the full study here.
Adults with less than a high school education are more than twice as likely as are those with high school degrees to have reported an anxiety disorder in the past year, according to the National Survey on Drug Use and Health’s 2008 to 2012 Mental Health Surveillance Study (MHSS), published June 2.
The data identified anxiety disorders in 13% of non–high school graduates vs. 5% of high school graduates. Adults with at least a college degree had the lowest rates of past year anxiety (4.3%). The MHSS estimated that approximately 13 million adults in the United States had at least one anxiety disorder within the past year.
“Although the MHSS results cannot be used to determine whether anxiety stopped people from finishing high school, having an anxiety disorder can lower the odds of graduating from high school and the odds of attending college,” the researchers wrote. The findings emphasize the need to support people with anxiety to help them remain in school and succeed, they added.
The findings were published in the Center for Behavioral Health Statistics and Quality Report. Read the full study here.
Adults with less than a high school education are more than twice as likely as are those with high school degrees to have reported an anxiety disorder in the past year, according to the National Survey on Drug Use and Health’s 2008 to 2012 Mental Health Surveillance Study (MHSS), published June 2.
The data identified anxiety disorders in 13% of non–high school graduates vs. 5% of high school graduates. Adults with at least a college degree had the lowest rates of past year anxiety (4.3%). The MHSS estimated that approximately 13 million adults in the United States had at least one anxiety disorder within the past year.
“Although the MHSS results cannot be used to determine whether anxiety stopped people from finishing high school, having an anxiety disorder can lower the odds of graduating from high school and the odds of attending college,” the researchers wrote. The findings emphasize the need to support people with anxiety to help them remain in school and succeed, they added.
The findings were published in the Center for Behavioral Health Statistics and Quality Report. Read the full study here.
FROM THE CBHSQ REPORT
Necrolytic Migratory Erythema With Recalcitrant Dermatitis as the Only Presenting Symptom
To the Editor:
A 52-year-old man presented with recalcitrant dermatitis of 6 years’ duration. He was otherwise in excellent health. On initial presentation, physical examination revealed symmetrical, erythematous, blanching plaques with areas of erosions and overlying hemorrhagic crust on the eyebrows, scalp, back, dorsal aspects of the hands, axillae, abdomen (Figure), buttocks, groin, scrotum, pubis, and lower legs. Some areas showed slight necrosis. He denied any fevers, chills, night sweats, cough, chest pain, shortness of breath, dizziness, lightheadedness, weight loss, or appetite change.
Throughout the disease course the patient had visited numerous dermatologists seeking treatment. He had response to higher doses of oral prednisone (80 mg taper), but the condition would recur at the end of an extended taper. Treatment with narrowband UVB, mycophenolate mofetil, methotrexate, acitretin, topical clobetasol, and topical pimecrolimus provided no relief. Eventually he was placed on azathioprine 100 mg twice daily, which led to near-complete resolution. Outbreaks continued every few months and required courses of prednisone.
Multiple biopsies over the years revealed subacute spongiotic or psoriasiform dermatitis. At multiple visits it was noted that during flares there were areas of crusting and mild necrosis, which led to an extensive biochemical investigation. The glucagon level was markedly elevated at 630 ng/L (reference range, 40–130 ng/L), as was insulin at 71 μIU/mL (reference range, 6–27 μIU/mL). Complete blood cell counts over the disease course showed mild normochromic normocytic anemia. The abnormal laboratory findings led to computed tomography of the abdomen, which revealed a mass in the body of the pancreas measuring 3×3.8 cm. After computed tomography, the patient underwent a laparoscopic distal pancreatectomy and splenectomy. Histologic examination revealed a well-differentiated pancreatic endocrine tumor (glucagonoma) confined to the pancreas. After the surgery, the patient’s rash resolved within a few days and he discontinued all medications.
Diagnosis of glucagonomas often is delayed due to their rarity and lack of classical signs and symptoms. The distribution of the lesions seen in necrolytic migratory erythema (NME) usually involves the inguinal crease, perineum, lower extremities, buttocks, and other intertriginous areas.1 Our patient had involvement in the typical distribution but also had involvement of the scalp, face, and upper body. The typical histology for NME is crusted psoriasiform dermatitis with a tendency for the upper epidermis to have necrosis and a vacuolated pale epidermis.2 Our patient’s histologic findings were less specific showing epidermal spongiosis with a scant lymphocytic infiltrate and at times acanthosis. The lack of classical skin findings and histology delayed diagnosis. In more than 50% of patients, metastasis has already occurred by the time the patient is diagnosed.3 Treatment is aimed at complete removal of the pancreatic tumor, which typically leads to a rapid improvement in symptoms. For patients unable to undergo surgery, chemotherapy agents and octreotide are used; unfortunately, symptoms may persist.4 The response to azathioprine in our patient suggests it is a possible alternate therapy for those with persistent NME.
This patient highlights the difficulty of diagnosing a glucagonoma when the only clinical manifestation may be NME. Moreover, skin biopsies that can sometimes be diagnostic may be nonspecific. This patient also shows a potential benefit of azathioprine in the treatment of NME.
- Shi W, Liao W, Mei X, et al. Necrolytic migratory erythema associated with glucagonoma syndrome [published online June 7, 2010]. J Clin Oncol. 2010;28:e329-e331.
- Rapini RP. Practical Dermatopathology. London, England: Elsevier Mosby; 2005.
- Oberg K, Eriksson B. Endocrine tumors of the pancreas. Best Pract Res Clin Gastroenterol. 2005;19:753-781.
- Wermers RA, Fatourechi V, Wynne AG, et al. The glucagonoma syndrome: clinical and pathologic features in 21 patients. Medicine (Baltimore). 1996;72:53-63.
To the Editor:
A 52-year-old man presented with recalcitrant dermatitis of 6 years’ duration. He was otherwise in excellent health. On initial presentation, physical examination revealed symmetrical, erythematous, blanching plaques with areas of erosions and overlying hemorrhagic crust on the eyebrows, scalp, back, dorsal aspects of the hands, axillae, abdomen (Figure), buttocks, groin, scrotum, pubis, and lower legs. Some areas showed slight necrosis. He denied any fevers, chills, night sweats, cough, chest pain, shortness of breath, dizziness, lightheadedness, weight loss, or appetite change.
Throughout the disease course the patient had visited numerous dermatologists seeking treatment. He had response to higher doses of oral prednisone (80 mg taper), but the condition would recur at the end of an extended taper. Treatment with narrowband UVB, mycophenolate mofetil, methotrexate, acitretin, topical clobetasol, and topical pimecrolimus provided no relief. Eventually he was placed on azathioprine 100 mg twice daily, which led to near-complete resolution. Outbreaks continued every few months and required courses of prednisone.
Multiple biopsies over the years revealed subacute spongiotic or psoriasiform dermatitis. At multiple visits it was noted that during flares there were areas of crusting and mild necrosis, which led to an extensive biochemical investigation. The glucagon level was markedly elevated at 630 ng/L (reference range, 40–130 ng/L), as was insulin at 71 μIU/mL (reference range, 6–27 μIU/mL). Complete blood cell counts over the disease course showed mild normochromic normocytic anemia. The abnormal laboratory findings led to computed tomography of the abdomen, which revealed a mass in the body of the pancreas measuring 3×3.8 cm. After computed tomography, the patient underwent a laparoscopic distal pancreatectomy and splenectomy. Histologic examination revealed a well-differentiated pancreatic endocrine tumor (glucagonoma) confined to the pancreas. After the surgery, the patient’s rash resolved within a few days and he discontinued all medications.
Diagnosis of glucagonomas often is delayed due to their rarity and lack of classical signs and symptoms. The distribution of the lesions seen in necrolytic migratory erythema (NME) usually involves the inguinal crease, perineum, lower extremities, buttocks, and other intertriginous areas.1 Our patient had involvement in the typical distribution but also had involvement of the scalp, face, and upper body. The typical histology for NME is crusted psoriasiform dermatitis with a tendency for the upper epidermis to have necrosis and a vacuolated pale epidermis.2 Our patient’s histologic findings were less specific showing epidermal spongiosis with a scant lymphocytic infiltrate and at times acanthosis. The lack of classical skin findings and histology delayed diagnosis. In more than 50% of patients, metastasis has already occurred by the time the patient is diagnosed.3 Treatment is aimed at complete removal of the pancreatic tumor, which typically leads to a rapid improvement in symptoms. For patients unable to undergo surgery, chemotherapy agents and octreotide are used; unfortunately, symptoms may persist.4 The response to azathioprine in our patient suggests it is a possible alternate therapy for those with persistent NME.
This patient highlights the difficulty of diagnosing a glucagonoma when the only clinical manifestation may be NME. Moreover, skin biopsies that can sometimes be diagnostic may be nonspecific. This patient also shows a potential benefit of azathioprine in the treatment of NME.
To the Editor:
A 52-year-old man presented with recalcitrant dermatitis of 6 years’ duration. He was otherwise in excellent health. On initial presentation, physical examination revealed symmetrical, erythematous, blanching plaques with areas of erosions and overlying hemorrhagic crust on the eyebrows, scalp, back, dorsal aspects of the hands, axillae, abdomen (Figure), buttocks, groin, scrotum, pubis, and lower legs. Some areas showed slight necrosis. He denied any fevers, chills, night sweats, cough, chest pain, shortness of breath, dizziness, lightheadedness, weight loss, or appetite change.
Throughout the disease course the patient had visited numerous dermatologists seeking treatment. He had response to higher doses of oral prednisone (80 mg taper), but the condition would recur at the end of an extended taper. Treatment with narrowband UVB, mycophenolate mofetil, methotrexate, acitretin, topical clobetasol, and topical pimecrolimus provided no relief. Eventually he was placed on azathioprine 100 mg twice daily, which led to near-complete resolution. Outbreaks continued every few months and required courses of prednisone.
Multiple biopsies over the years revealed subacute spongiotic or psoriasiform dermatitis. At multiple visits it was noted that during flares there were areas of crusting and mild necrosis, which led to an extensive biochemical investigation. The glucagon level was markedly elevated at 630 ng/L (reference range, 40–130 ng/L), as was insulin at 71 μIU/mL (reference range, 6–27 μIU/mL). Complete blood cell counts over the disease course showed mild normochromic normocytic anemia. The abnormal laboratory findings led to computed tomography of the abdomen, which revealed a mass in the body of the pancreas measuring 3×3.8 cm. After computed tomography, the patient underwent a laparoscopic distal pancreatectomy and splenectomy. Histologic examination revealed a well-differentiated pancreatic endocrine tumor (glucagonoma) confined to the pancreas. After the surgery, the patient’s rash resolved within a few days and he discontinued all medications.
Diagnosis of glucagonomas often is delayed due to their rarity and lack of classical signs and symptoms. The distribution of the lesions seen in necrolytic migratory erythema (NME) usually involves the inguinal crease, perineum, lower extremities, buttocks, and other intertriginous areas.1 Our patient had involvement in the typical distribution but also had involvement of the scalp, face, and upper body. The typical histology for NME is crusted psoriasiform dermatitis with a tendency for the upper epidermis to have necrosis and a vacuolated pale epidermis.2 Our patient’s histologic findings were less specific showing epidermal spongiosis with a scant lymphocytic infiltrate and at times acanthosis. The lack of classical skin findings and histology delayed diagnosis. In more than 50% of patients, metastasis has already occurred by the time the patient is diagnosed.3 Treatment is aimed at complete removal of the pancreatic tumor, which typically leads to a rapid improvement in symptoms. For patients unable to undergo surgery, chemotherapy agents and octreotide are used; unfortunately, symptoms may persist.4 The response to azathioprine in our patient suggests it is a possible alternate therapy for those with persistent NME.
This patient highlights the difficulty of diagnosing a glucagonoma when the only clinical manifestation may be NME. Moreover, skin biopsies that can sometimes be diagnostic may be nonspecific. This patient also shows a potential benefit of azathioprine in the treatment of NME.
- Shi W, Liao W, Mei X, et al. Necrolytic migratory erythema associated with glucagonoma syndrome [published online June 7, 2010]. J Clin Oncol. 2010;28:e329-e331.
- Rapini RP. Practical Dermatopathology. London, England: Elsevier Mosby; 2005.
- Oberg K, Eriksson B. Endocrine tumors of the pancreas. Best Pract Res Clin Gastroenterol. 2005;19:753-781.
- Wermers RA, Fatourechi V, Wynne AG, et al. The glucagonoma syndrome: clinical and pathologic features in 21 patients. Medicine (Baltimore). 1996;72:53-63.
- Shi W, Liao W, Mei X, et al. Necrolytic migratory erythema associated with glucagonoma syndrome [published online June 7, 2010]. J Clin Oncol. 2010;28:e329-e331.
- Rapini RP. Practical Dermatopathology. London, England: Elsevier Mosby; 2005.
- Oberg K, Eriksson B. Endocrine tumors of the pancreas. Best Pract Res Clin Gastroenterol. 2005;19:753-781.
- Wermers RA, Fatourechi V, Wynne AG, et al. The glucagonoma syndrome: clinical and pathologic features in 21 patients. Medicine (Baltimore). 1996;72:53-63.
Practice Points
- Recalcitrant dermatitis may be a symptom of internal malignancy.
- Glucagon levels are helpful in identifying glucagonomas of the pancreas.
- Although surgical excision is the preferred treatment of glucagonomas, azathioprine also can control dermatitis associated with necrolytic migratory erythema.
Sell skin care products to protect your patients
The ethics behind selling skin care products to patients has been hotly debated within the field of cosmetic dermatology for several decades. In 15 years of practice, I have come to the conclusion that patients want you to and need you to because otherwise they are easily taken advantage of. Other physicians are doing it but we – the dermatologists – are the most qualified to offer skin care advice. This article will discuss the reasons that you need to get over the ethical dilemma and offer skin care to your patients.

Using the correct skin care regimen for the face and body will improve outcomes
Whether a patient suffers from acne, rosacea, melasma, psoriasis, eczema, contact dermatitis, or even tinea versicolor, using the proper skin care regimen will improve outcomes by affecting the skin barrier, pH, hydration level, and function of the keratinocytes and fibroblasts. In fact, every personal care product that touches the skin has an impact on skin health. For example, if a patient uses a detergent-laden bar soap, the skin barrier will be impaired, which can cause them to react to allergens and irritants. Personal care products can affect the skin pH; this is shown to play a role in Malassezia colonization in atopic dermatitis patients (J Clin Med. 2015;4[6]:1217-28). As dermatologists, we know better than anyone that daily use of SPF improves skin health and lowers the risk of postinflammatory pigmentation. We all agree that patients should cleanse the skin and apply a SPF every day. Giving them guidance about which to choose is very important.
Giving the patient exact instructions will lead to improved compliance
Why should recommending skin care products be perceived differently than prescribing a prescription medication? We should prescribe to our patients in writing the exact skin care regimen they should use for their face or body to ensure that they understand the directions. I have been surprised by patients who have said, “I did not know I was supposed to wash my cleanser off,” or “I wash my face with hand soap.” We can help them by educating them and giving them specific instructions. Improved education and communication results in increased compliance. When you do surgery on a wound, you probably tell them to apply a topical antibiotic ointment, but do you direct them to what cleanser to use or tell them which SPF to use on the stitched wound? Providing written instructions for all dermatologic disorders and postprocedure care is necessary to improve compliance and outcomes.
Combine cosmeceuticals, prescription medications, and medical procedures
You (unlike the cosmetic counter salesperson) have the ability to combine cosmeceuticals with prescription medications and medical procedures. In fact, selling your patients the right skin care products to use after a procedure saves them a trip to the store and ensures that they use the correct products. Of course it makes sense that patients getting toxins and fillers should use a retinoid to improve skin aging; however, many general dermatologic diseases would improve with the proper skin care. For example, do you use biologics for psoriasis? Using the proper skin care to regulate skin pH and improve the skin barrier may help prevent colonization of yeast, fungus, and bacteria. The same is true for atopic patients. Do you use liquid nitrogen? Studies show that using a retinoid before a procedure speeds healing. Skin care goes way beyond wrinkles and dark circles under the eyes, so if you are not prescribing the patient an exact regimen, you are not maximizing outcomes.
I don’t have time to talk to my patients about skin care
The missing piece is that most of us don’t have the time to spend discussing skin care. This is where using a standardized scientific methodology is crucial. I developed and use a skin typing methodology in my office and have seen improved physician/patient relationships and increased patient satisfaction resulting in a significant amount of referrals. We also have noted decreased call backs and fewer adverse events from products because the patients have a better understanding of how to properly apply the cosmeceuticals and prescription products. The best part is, it does not add any time onto the patient visit when standardized methodologies are properly adopted.
What if I still do not feel comfortable profiting from the sale of skin care products?
First you need to realize that time is money and you are saving the patient the cost in time it would have taken them to go to a store, park, and shop for the correct product. I have seen data presented from several companies that show that patients usually spend a large amount of money on skin care products after they see their dermatologist. Without guidance, they will likely buy the incorrect products. If they buy the wrong product, you save them the hassle of having to make another office visit and the aggravation of the side effects from the incorrect product. These are often of poor quality or not appropriate for their skin issues. Counterfeit products are rampant on the Internet and many new companies tout worthless products with stem cells and other nonsense. Only you can help your patients make sure that money is spent on the proper products.
Conclusion
Do you really want someone else giving your patients skin care advice? Your patients deserve to have someone with your insights, knowledge, compassion, and honesty help them achieve optimal skin health through use of the proper cosmeceuticals and prescription medications. It is up to you and your staff to save your patients from falling prey to persuasive salespeople with no scientific knowledge or concern for long-term skin health.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera Biopharmaceuticals, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Topix Pharmaceuticals, and Unilever. Dr. Baumann also developed and owns the Baumann Skin Type Solution skin typing systems and related products.
The ethics behind selling skin care products to patients has been hotly debated within the field of cosmetic dermatology for several decades. In 15 years of practice, I have come to the conclusion that patients want you to and need you to because otherwise they are easily taken advantage of. Other physicians are doing it but we – the dermatologists – are the most qualified to offer skin care advice. This article will discuss the reasons that you need to get over the ethical dilemma and offer skin care to your patients.
Using the correct skin care regimen for the face and body will improve outcomes
Whether a patient suffers from acne, rosacea, melasma, psoriasis, eczema, contact dermatitis, or even tinea versicolor, using the proper skin care regimen will improve outcomes by affecting the skin barrier, pH, hydration level, and function of the keratinocytes and fibroblasts. In fact, every personal care product that touches the skin has an impact on skin health. For example, if a patient uses a detergent-laden bar soap, the skin barrier will be impaired, which can cause them to react to allergens and irritants. Personal care products can affect the skin pH; this is shown to play a role in Malassezia colonization in atopic dermatitis patients (J Clin Med. 2015;4[6]:1217-28). As dermatologists, we know better than anyone that daily use of SPF improves skin health and lowers the risk of postinflammatory pigmentation. We all agree that patients should cleanse the skin and apply a SPF every day. Giving them guidance about which to choose is very important.
Giving the patient exact instructions will lead to improved compliance
Why should recommending skin care products be perceived differently than prescribing a prescription medication? We should prescribe to our patients in writing the exact skin care regimen they should use for their face or body to ensure that they understand the directions. I have been surprised by patients who have said, “I did not know I was supposed to wash my cleanser off,” or “I wash my face with hand soap.” We can help them by educating them and giving them specific instructions. Improved education and communication results in increased compliance. When you do surgery on a wound, you probably tell them to apply a topical antibiotic ointment, but do you direct them to what cleanser to use or tell them which SPF to use on the stitched wound? Providing written instructions for all dermatologic disorders and postprocedure care is necessary to improve compliance and outcomes.
Combine cosmeceuticals, prescription medications, and medical procedures
You (unlike the cosmetic counter salesperson) have the ability to combine cosmeceuticals with prescription medications and medical procedures. In fact, selling your patients the right skin care products to use after a procedure saves them a trip to the store and ensures that they use the correct products. Of course it makes sense that patients getting toxins and fillers should use a retinoid to improve skin aging; however, many general dermatologic diseases would improve with the proper skin care. For example, do you use biologics for psoriasis? Using the proper skin care to regulate skin pH and improve the skin barrier may help prevent colonization of yeast, fungus, and bacteria. The same is true for atopic patients. Do you use liquid nitrogen? Studies show that using a retinoid before a procedure speeds healing. Skin care goes way beyond wrinkles and dark circles under the eyes, so if you are not prescribing the patient an exact regimen, you are not maximizing outcomes.
I don’t have time to talk to my patients about skin care
The missing piece is that most of us don’t have the time to spend discussing skin care. This is where using a standardized scientific methodology is crucial. I developed and use a skin typing methodology in my office and have seen improved physician/patient relationships and increased patient satisfaction resulting in a significant amount of referrals. We also have noted decreased call backs and fewer adverse events from products because the patients have a better understanding of how to properly apply the cosmeceuticals and prescription products. The best part is, it does not add any time onto the patient visit when standardized methodologies are properly adopted.
What if I still do not feel comfortable profiting from the sale of skin care products?
First you need to realize that time is money and you are saving the patient the cost in time it would have taken them to go to a store, park, and shop for the correct product. I have seen data presented from several companies that show that patients usually spend a large amount of money on skin care products after they see their dermatologist. Without guidance, they will likely buy the incorrect products. If they buy the wrong product, you save them the hassle of having to make another office visit and the aggravation of the side effects from the incorrect product. These are often of poor quality or not appropriate for their skin issues. Counterfeit products are rampant on the Internet and many new companies tout worthless products with stem cells and other nonsense. Only you can help your patients make sure that money is spent on the proper products.
Conclusion
Do you really want someone else giving your patients skin care advice? Your patients deserve to have someone with your insights, knowledge, compassion, and honesty help them achieve optimal skin health through use of the proper cosmeceuticals and prescription medications. It is up to you and your staff to save your patients from falling prey to persuasive salespeople with no scientific knowledge or concern for long-term skin health.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera Biopharmaceuticals, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Topix Pharmaceuticals, and Unilever. Dr. Baumann also developed and owns the Baumann Skin Type Solution skin typing systems and related products.
The ethics behind selling skin care products to patients has been hotly debated within the field of cosmetic dermatology for several decades. In 15 years of practice, I have come to the conclusion that patients want you to and need you to because otherwise they are easily taken advantage of. Other physicians are doing it but we – the dermatologists – are the most qualified to offer skin care advice. This article will discuss the reasons that you need to get over the ethical dilemma and offer skin care to your patients.
Using the correct skin care regimen for the face and body will improve outcomes
Whether a patient suffers from acne, rosacea, melasma, psoriasis, eczema, contact dermatitis, or even tinea versicolor, using the proper skin care regimen will improve outcomes by affecting the skin barrier, pH, hydration level, and function of the keratinocytes and fibroblasts. In fact, every personal care product that touches the skin has an impact on skin health. For example, if a patient uses a detergent-laden bar soap, the skin barrier will be impaired, which can cause them to react to allergens and irritants. Personal care products can affect the skin pH; this is shown to play a role in Malassezia colonization in atopic dermatitis patients (J Clin Med. 2015;4[6]:1217-28). As dermatologists, we know better than anyone that daily use of SPF improves skin health and lowers the risk of postinflammatory pigmentation. We all agree that patients should cleanse the skin and apply a SPF every day. Giving them guidance about which to choose is very important.
Giving the patient exact instructions will lead to improved compliance
Why should recommending skin care products be perceived differently than prescribing a prescription medication? We should prescribe to our patients in writing the exact skin care regimen they should use for their face or body to ensure that they understand the directions. I have been surprised by patients who have said, “I did not know I was supposed to wash my cleanser off,” or “I wash my face with hand soap.” We can help them by educating them and giving them specific instructions. Improved education and communication results in increased compliance. When you do surgery on a wound, you probably tell them to apply a topical antibiotic ointment, but do you direct them to what cleanser to use or tell them which SPF to use on the stitched wound? Providing written instructions for all dermatologic disorders and postprocedure care is necessary to improve compliance and outcomes.
Combine cosmeceuticals, prescription medications, and medical procedures
You (unlike the cosmetic counter salesperson) have the ability to combine cosmeceuticals with prescription medications and medical procedures. In fact, selling your patients the right skin care products to use after a procedure saves them a trip to the store and ensures that they use the correct products. Of course it makes sense that patients getting toxins and fillers should use a retinoid to improve skin aging; however, many general dermatologic diseases would improve with the proper skin care. For example, do you use biologics for psoriasis? Using the proper skin care to regulate skin pH and improve the skin barrier may help prevent colonization of yeast, fungus, and bacteria. The same is true for atopic patients. Do you use liquid nitrogen? Studies show that using a retinoid before a procedure speeds healing. Skin care goes way beyond wrinkles and dark circles under the eyes, so if you are not prescribing the patient an exact regimen, you are not maximizing outcomes.
I don’t have time to talk to my patients about skin care
The missing piece is that most of us don’t have the time to spend discussing skin care. This is where using a standardized scientific methodology is crucial. I developed and use a skin typing methodology in my office and have seen improved physician/patient relationships and increased patient satisfaction resulting in a significant amount of referrals. We also have noted decreased call backs and fewer adverse events from products because the patients have a better understanding of how to properly apply the cosmeceuticals and prescription products. The best part is, it does not add any time onto the patient visit when standardized methodologies are properly adopted.
What if I still do not feel comfortable profiting from the sale of skin care products?
First you need to realize that time is money and you are saving the patient the cost in time it would have taken them to go to a store, park, and shop for the correct product. I have seen data presented from several companies that show that patients usually spend a large amount of money on skin care products after they see their dermatologist. Without guidance, they will likely buy the incorrect products. If they buy the wrong product, you save them the hassle of having to make another office visit and the aggravation of the side effects from the incorrect product. These are often of poor quality or not appropriate for their skin issues. Counterfeit products are rampant on the Internet and many new companies tout worthless products with stem cells and other nonsense. Only you can help your patients make sure that money is spent on the proper products.
Conclusion
Do you really want someone else giving your patients skin care advice? Your patients deserve to have someone with your insights, knowledge, compassion, and honesty help them achieve optimal skin health through use of the proper cosmeceuticals and prescription medications. It is up to you and your staff to save your patients from falling prey to persuasive salespeople with no scientific knowledge or concern for long-term skin health.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera Biopharmaceuticals, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Topix Pharmaceuticals, and Unilever. Dr. Baumann also developed and owns the Baumann Skin Type Solution skin typing systems and related products.
• Most skin care products that patients buy are not appropriate for their skin issues.
• Dermatologists have the most knowledge and insights to prescribe skin care.
• Giving specific skin care instructions helps improve communication.
• Increased communication improves outcomes.

Targeting vagal activity could improve breast cancer survival
ATLANTA – Vagal activity predicts survival in patients with metastatic or recurrent breast cancer, a study showed.
The findings are intriguing, given that vagal activity is modifiable, according to Dr. David Spiegel, Willson Professor of Psychiatry and Behavioral Sciences and director of the center on stress and health at Stanford (Calif.) University.

The study, conducted by Dr. Spiegel and his colleagues, is one of several that together are beginning to elucidate the connections among sleep, stress, and vagal tone, and the effects these factors have on cancer outcomes. For example, in one earlier study of metastatic breast cancer patients, the group demonstrated that good sleep efficiency predicted longer survival (Sleep. 2014 May 1;37[5]:837-42).
“There’s something about sleep that we think has an effect on disease progression,” Dr. Spiegel said at the annual meeting of the American Psychiatric Association.
In another study, the team showed that breast cancer patients who slept better at night had better vagal tone the following morning.
“We all kind of know that a problem that has been keeping you from getting to sleep or worrying you a lot the night before suddenly seems more soluble in the morning after you’ve had a good night’s sleep. You’re better able to self-soothe in the morning,” he said, noting the importance of this evidence that “sleep improves vagal tone.”
Heart rate variability is a good measure of vagal tone, vagal activity, and the ability to self-soothe, he explained, noting that heart rate variability also predicts longer survival with cardiac disease; it seems to reduce the risk of fatal arrhythmias, and also predicts recovery from myocardial infarction.
Others have suggested that it might have an effect on cancer, and there seems to be a link between vagal activity and inflammatory processes, Dr. Spiegel said.
“There is reason to think that poor heart rate variability might be associated with cancer progression as well, and that’s what we wanted to study in a group of metastatic cancer patients,” he said.
Dr. Spiegel and his colleagues measured high-frequency heart rate variability (HF-HRV), which appears to be the best measure of parasympathetic tone, has been associated with longer survival in humans and animals, and is related to immune system functioning.
“We hypothesized that higher heart rate variability would predict longer survival in patients with MRBC [metastatic or recurrent breast cancer],” he said.
In 87 patients with metastases to bone, skin, or viscera who underwent a variety of stress measures, including a 5-minute resting baseline electrocardiogram, 43 had higher HF-HRV, and 44 had lower HF-HRV. Higher baseline HF-HRV did, indeed, predict significantly longer survival (hazard ratio, 0.75).
“The main hypothesis was confirmed – that patients with better vagal tone, higher high-frequency heart rate variability had significantly longer survival over the ensuing 7 years, compared with the patients who had poorer heart rate variability, poorer vagal tone,” he said (Psychosom Med. 2015;77[4]:346-55).
Visceral metastasis status and baseline heart rate both were related to HF-HRV and survival, and the combination of HF-HRV and heart rate further improved survival prediction (HR, 0.64), he noted.
“This is basically coactivation of higher parasympathetic and lower sympathetic activity related to longer survival,” he explained.
Reconstructive surgery, the presence of visceral metastases, and sleep efficiency each were found to be associated with heart rate variability; thus several analyses were conducted “to try to disentangle these relationships and determine what the major variables were that predicted survival,” Dr. Spiegel said.
“It turns out that heart rate variability and visceral metastases were significantly related, and heart rate variability did not predict survival,” he said, explaining that those with visceral metastases (and therefore, cancer with a much poorer prognosis) died sooner, but heart rate variability didn’t make much of a difference. “Where we saw the heart rate variability effect was among those with better prognosis.”
A combination measure of high heart rate variability (“a pretty pure measure of vagal activity, not sympathetic activity”) and low heart rate (“more driven by the sympathetic adrenal-medullary system”) is an even stronger predictor of overall survival, he said.
This suggests that autonomic nervous system variables play a strong role in predicting overall cancer survival, Dr. Spiegel said, noting that depression was not a confounder.
“This is important, because we have, in other studies, found that depression is associated with lower heart rate variability, as you might expect,” he said. In fact, depression has been found to predict shorter survival in cancer patients over a period of 10 years. In an earlier study, cancer patients with worsening depression in the first year died sooner than those with depression that improved during the first year (J Clin Oncol. 2011;29[4]:413-20).
“The median survival difference was about 2 years, so this is not a trivial difference in overall survival,” he said, stressing that the finding is based on more chronic and severe depression.
Other studies have demonstrated relationships between circadian rhythm disruption and cancer survival. In one such study involving patients with metastatic colorectal cancer, circadian rhythm/rest activity cycle (more activity during the day, more rest at night) was associated with better quality of life and predicted survival.
“This has been shown now in several cancers, and it’s clear that a combination of higher activity and better sleep predict longer survival with different kinds of cancers,” Dr. Spiegel said, explaining that a hallmark of a healthy hypothalamic-pituitary-adrenal axis is good diurnal variation of cortisol with high levels in the morning and declining levels throughout the day.
Women with metastatic breast cancer and poorer survival tended to have flat or increasing levels of cortisol throughout the day, he said, adding that the same was true in a study of patients with lung cancer, and that there is evidence that those among them with flatter, more abnormal cortisol patterns throughout the day also have shorter survival.
Animal studies suggest that cortisol might directly suppress the activity of tumor suppressor genes, he explained, noting that there is also increasing evidence of autonomic dysregulation effects on inflammatory processes associated with tumor growth.
“Some basic research on this in animal models shows that if you block adrenergic arousal, you can block the growth of blood vessels from tumors. This has led some people to look at the use of antiadrenergic drugs like the beta-blocker propranolol, and it turned out – to everyone’s surprise – that breast cancer patients who happened to be on beta-blockers for hypertension actually lived longer than those who didn’t,” he said.
This adds to the growing evidence that dysregulation in the sympathetic and parasympathetic systems have effects on survival, he said.
A look at another factor related to sleep disruption – bedtime misalignment – showed that patients who adhere to their preferred sleep pattern, and who are therefore sleeping better, had a difference in disease-free interval; those whose bedtime was misaligned had a shorter time between diagnosis and disease recurrence (Chronobiol Int. 2014 Mar 31[2]214-21).
“Disease-free interval is a very strong predictor of ultimate overall survival, so there seems to be another relationship here between circadian disruption and disease progression in breast cancer,” Dr. Spiegel said.
Taken together, these findings demonstrate a strong association between vagal activity and survival in patients with metastatic or recurrent breast cancer, extending the known predictive window of HF-HRV beyond palliative care to cancer, Dr. Spiegel said.
“Vagal activity can be altered through behavioral, pharmacological, and surgical interventions and thus may be a promising target for increasing survival in patients with metastatic cancer,” he said.
Dr. Spiegel’s studies were funded by the National Cancer Institute and the National Institute on Aging.
ATLANTA – Vagal activity predicts survival in patients with metastatic or recurrent breast cancer, a study showed.
The findings are intriguing, given that vagal activity is modifiable, according to Dr. David Spiegel, Willson Professor of Psychiatry and Behavioral Sciences and director of the center on stress and health at Stanford (Calif.) University.
The study, conducted by Dr. Spiegel and his colleagues, is one of several that together are beginning to elucidate the connections among sleep, stress, and vagal tone, and the effects these factors have on cancer outcomes. For example, in one earlier study of metastatic breast cancer patients, the group demonstrated that good sleep efficiency predicted longer survival (Sleep. 2014 May 1;37[5]:837-42).
“There’s something about sleep that we think has an effect on disease progression,” Dr. Spiegel said at the annual meeting of the American Psychiatric Association.
In another study, the team showed that breast cancer patients who slept better at night had better vagal tone the following morning.
“We all kind of know that a problem that has been keeping you from getting to sleep or worrying you a lot the night before suddenly seems more soluble in the morning after you’ve had a good night’s sleep. You’re better able to self-soothe in the morning,” he said, noting the importance of this evidence that “sleep improves vagal tone.”
Heart rate variability is a good measure of vagal tone, vagal activity, and the ability to self-soothe, he explained, noting that heart rate variability also predicts longer survival with cardiac disease; it seems to reduce the risk of fatal arrhythmias, and also predicts recovery from myocardial infarction.
Others have suggested that it might have an effect on cancer, and there seems to be a link between vagal activity and inflammatory processes, Dr. Spiegel said.
“There is reason to think that poor heart rate variability might be associated with cancer progression as well, and that’s what we wanted to study in a group of metastatic cancer patients,” he said.
Dr. Spiegel and his colleagues measured high-frequency heart rate variability (HF-HRV), which appears to be the best measure of parasympathetic tone, has been associated with longer survival in humans and animals, and is related to immune system functioning.
“We hypothesized that higher heart rate variability would predict longer survival in patients with MRBC [metastatic or recurrent breast cancer],” he said.
In 87 patients with metastases to bone, skin, or viscera who underwent a variety of stress measures, including a 5-minute resting baseline electrocardiogram, 43 had higher HF-HRV, and 44 had lower HF-HRV. Higher baseline HF-HRV did, indeed, predict significantly longer survival (hazard ratio, 0.75).
“The main hypothesis was confirmed – that patients with better vagal tone, higher high-frequency heart rate variability had significantly longer survival over the ensuing 7 years, compared with the patients who had poorer heart rate variability, poorer vagal tone,” he said (Psychosom Med. 2015;77[4]:346-55).
Visceral metastasis status and baseline heart rate both were related to HF-HRV and survival, and the combination of HF-HRV and heart rate further improved survival prediction (HR, 0.64), he noted.
“This is basically coactivation of higher parasympathetic and lower sympathetic activity related to longer survival,” he explained.
Reconstructive surgery, the presence of visceral metastases, and sleep efficiency each were found to be associated with heart rate variability; thus several analyses were conducted “to try to disentangle these relationships and determine what the major variables were that predicted survival,” Dr. Spiegel said.
“It turns out that heart rate variability and visceral metastases were significantly related, and heart rate variability did not predict survival,” he said, explaining that those with visceral metastases (and therefore, cancer with a much poorer prognosis) died sooner, but heart rate variability didn’t make much of a difference. “Where we saw the heart rate variability effect was among those with better prognosis.”
A combination measure of high heart rate variability (“a pretty pure measure of vagal activity, not sympathetic activity”) and low heart rate (“more driven by the sympathetic adrenal-medullary system”) is an even stronger predictor of overall survival, he said.
This suggests that autonomic nervous system variables play a strong role in predicting overall cancer survival, Dr. Spiegel said, noting that depression was not a confounder.
“This is important, because we have, in other studies, found that depression is associated with lower heart rate variability, as you might expect,” he said. In fact, depression has been found to predict shorter survival in cancer patients over a period of 10 years. In an earlier study, cancer patients with worsening depression in the first year died sooner than those with depression that improved during the first year (J Clin Oncol. 2011;29[4]:413-20).
“The median survival difference was about 2 years, so this is not a trivial difference in overall survival,” he said, stressing that the finding is based on more chronic and severe depression.
Other studies have demonstrated relationships between circadian rhythm disruption and cancer survival. In one such study involving patients with metastatic colorectal cancer, circadian rhythm/rest activity cycle (more activity during the day, more rest at night) was associated with better quality of life and predicted survival.
“This has been shown now in several cancers, and it’s clear that a combination of higher activity and better sleep predict longer survival with different kinds of cancers,” Dr. Spiegel said, explaining that a hallmark of a healthy hypothalamic-pituitary-adrenal axis is good diurnal variation of cortisol with high levels in the morning and declining levels throughout the day.
Women with metastatic breast cancer and poorer survival tended to have flat or increasing levels of cortisol throughout the day, he said, adding that the same was true in a study of patients with lung cancer, and that there is evidence that those among them with flatter, more abnormal cortisol patterns throughout the day also have shorter survival.
Animal studies suggest that cortisol might directly suppress the activity of tumor suppressor genes, he explained, noting that there is also increasing evidence of autonomic dysregulation effects on inflammatory processes associated with tumor growth.
“Some basic research on this in animal models shows that if you block adrenergic arousal, you can block the growth of blood vessels from tumors. This has led some people to look at the use of antiadrenergic drugs like the beta-blocker propranolol, and it turned out – to everyone’s surprise – that breast cancer patients who happened to be on beta-blockers for hypertension actually lived longer than those who didn’t,” he said.
This adds to the growing evidence that dysregulation in the sympathetic and parasympathetic systems have effects on survival, he said.
A look at another factor related to sleep disruption – bedtime misalignment – showed that patients who adhere to their preferred sleep pattern, and who are therefore sleeping better, had a difference in disease-free interval; those whose bedtime was misaligned had a shorter time between diagnosis and disease recurrence (Chronobiol Int. 2014 Mar 31[2]214-21).
“Disease-free interval is a very strong predictor of ultimate overall survival, so there seems to be another relationship here between circadian disruption and disease progression in breast cancer,” Dr. Spiegel said.
Taken together, these findings demonstrate a strong association between vagal activity and survival in patients with metastatic or recurrent breast cancer, extending the known predictive window of HF-HRV beyond palliative care to cancer, Dr. Spiegel said.
“Vagal activity can be altered through behavioral, pharmacological, and surgical interventions and thus may be a promising target for increasing survival in patients with metastatic cancer,” he said.
Dr. Spiegel’s studies were funded by the National Cancer Institute and the National Institute on Aging.
ATLANTA – Vagal activity predicts survival in patients with metastatic or recurrent breast cancer, a study showed.
The findings are intriguing, given that vagal activity is modifiable, according to Dr. David Spiegel, Willson Professor of Psychiatry and Behavioral Sciences and director of the center on stress and health at Stanford (Calif.) University.
The study, conducted by Dr. Spiegel and his colleagues, is one of several that together are beginning to elucidate the connections among sleep, stress, and vagal tone, and the effects these factors have on cancer outcomes. For example, in one earlier study of metastatic breast cancer patients, the group demonstrated that good sleep efficiency predicted longer survival (Sleep. 2014 May 1;37[5]:837-42).
“There’s something about sleep that we think has an effect on disease progression,” Dr. Spiegel said at the annual meeting of the American Psychiatric Association.
In another study, the team showed that breast cancer patients who slept better at night had better vagal tone the following morning.
“We all kind of know that a problem that has been keeping you from getting to sleep or worrying you a lot the night before suddenly seems more soluble in the morning after you’ve had a good night’s sleep. You’re better able to self-soothe in the morning,” he said, noting the importance of this evidence that “sleep improves vagal tone.”
Heart rate variability is a good measure of vagal tone, vagal activity, and the ability to self-soothe, he explained, noting that heart rate variability also predicts longer survival with cardiac disease; it seems to reduce the risk of fatal arrhythmias, and also predicts recovery from myocardial infarction.
Others have suggested that it might have an effect on cancer, and there seems to be a link between vagal activity and inflammatory processes, Dr. Spiegel said.
“There is reason to think that poor heart rate variability might be associated with cancer progression as well, and that’s what we wanted to study in a group of metastatic cancer patients,” he said.
Dr. Spiegel and his colleagues measured high-frequency heart rate variability (HF-HRV), which appears to be the best measure of parasympathetic tone, has been associated with longer survival in humans and animals, and is related to immune system functioning.
“We hypothesized that higher heart rate variability would predict longer survival in patients with MRBC [metastatic or recurrent breast cancer],” he said.
In 87 patients with metastases to bone, skin, or viscera who underwent a variety of stress measures, including a 5-minute resting baseline electrocardiogram, 43 had higher HF-HRV, and 44 had lower HF-HRV. Higher baseline HF-HRV did, indeed, predict significantly longer survival (hazard ratio, 0.75).
“The main hypothesis was confirmed – that patients with better vagal tone, higher high-frequency heart rate variability had significantly longer survival over the ensuing 7 years, compared with the patients who had poorer heart rate variability, poorer vagal tone,” he said (Psychosom Med. 2015;77[4]:346-55).
Visceral metastasis status and baseline heart rate both were related to HF-HRV and survival, and the combination of HF-HRV and heart rate further improved survival prediction (HR, 0.64), he noted.
“This is basically coactivation of higher parasympathetic and lower sympathetic activity related to longer survival,” he explained.
Reconstructive surgery, the presence of visceral metastases, and sleep efficiency each were found to be associated with heart rate variability; thus several analyses were conducted “to try to disentangle these relationships and determine what the major variables were that predicted survival,” Dr. Spiegel said.
“It turns out that heart rate variability and visceral metastases were significantly related, and heart rate variability did not predict survival,” he said, explaining that those with visceral metastases (and therefore, cancer with a much poorer prognosis) died sooner, but heart rate variability didn’t make much of a difference. “Where we saw the heart rate variability effect was among those with better prognosis.”
A combination measure of high heart rate variability (“a pretty pure measure of vagal activity, not sympathetic activity”) and low heart rate (“more driven by the sympathetic adrenal-medullary system”) is an even stronger predictor of overall survival, he said.
This suggests that autonomic nervous system variables play a strong role in predicting overall cancer survival, Dr. Spiegel said, noting that depression was not a confounder.
“This is important, because we have, in other studies, found that depression is associated with lower heart rate variability, as you might expect,” he said. In fact, depression has been found to predict shorter survival in cancer patients over a period of 10 years. In an earlier study, cancer patients with worsening depression in the first year died sooner than those with depression that improved during the first year (J Clin Oncol. 2011;29[4]:413-20).
“The median survival difference was about 2 years, so this is not a trivial difference in overall survival,” he said, stressing that the finding is based on more chronic and severe depression.
Other studies have demonstrated relationships between circadian rhythm disruption and cancer survival. In one such study involving patients with metastatic colorectal cancer, circadian rhythm/rest activity cycle (more activity during the day, more rest at night) was associated with better quality of life and predicted survival.
“This has been shown now in several cancers, and it’s clear that a combination of higher activity and better sleep predict longer survival with different kinds of cancers,” Dr. Spiegel said, explaining that a hallmark of a healthy hypothalamic-pituitary-adrenal axis is good diurnal variation of cortisol with high levels in the morning and declining levels throughout the day.
Women with metastatic breast cancer and poorer survival tended to have flat or increasing levels of cortisol throughout the day, he said, adding that the same was true in a study of patients with lung cancer, and that there is evidence that those among them with flatter, more abnormal cortisol patterns throughout the day also have shorter survival.
Animal studies suggest that cortisol might directly suppress the activity of tumor suppressor genes, he explained, noting that there is also increasing evidence of autonomic dysregulation effects on inflammatory processes associated with tumor growth.
“Some basic research on this in animal models shows that if you block adrenergic arousal, you can block the growth of blood vessels from tumors. This has led some people to look at the use of antiadrenergic drugs like the beta-blocker propranolol, and it turned out – to everyone’s surprise – that breast cancer patients who happened to be on beta-blockers for hypertension actually lived longer than those who didn’t,” he said.
This adds to the growing evidence that dysregulation in the sympathetic and parasympathetic systems have effects on survival, he said.
A look at another factor related to sleep disruption – bedtime misalignment – showed that patients who adhere to their preferred sleep pattern, and who are therefore sleeping better, had a difference in disease-free interval; those whose bedtime was misaligned had a shorter time between diagnosis and disease recurrence (Chronobiol Int. 2014 Mar 31[2]214-21).
“Disease-free interval is a very strong predictor of ultimate overall survival, so there seems to be another relationship here between circadian disruption and disease progression in breast cancer,” Dr. Spiegel said.
Taken together, these findings demonstrate a strong association between vagal activity and survival in patients with metastatic or recurrent breast cancer, extending the known predictive window of HF-HRV beyond palliative care to cancer, Dr. Spiegel said.
“Vagal activity can be altered through behavioral, pharmacological, and surgical interventions and thus may be a promising target for increasing survival in patients with metastatic cancer,” he said.
Dr. Spiegel’s studies were funded by the National Cancer Institute and the National Institute on Aging.
AT THE APA ANNUAL MEETING
Key clinical point: Vagal activity predicted survival in patients with metastatic or recurrent breast cancer.
Major finding: Higher baseline HF-HRV predicted significantly longer survival (hazard ratio, 0.75).
Data source: A study of 87 patients with metastatic or recurrent breast cancer.
Disclosures: Dr. Spiegel’s studies were funded by the National Cancer Institute and the National Institute on Aging.

Book offers even-handed, scholarly treatment of AA
Several years ago, the British psychoanalyst Enid Balint suggested that people might not necessarily emerge from a group experience less neurotic or psychotic, but invariably, they were more mature.1 Her suggestion is consistent with my clinical experience. Some of the most admirable and mature patients with whom I have worked have been individuals who participated regularly in 12-step meetings as part of their treatment and recovery.
In this book, “What is Alcoholics Anonymous? A Path From Addiction to Recovery,” (Oxford University Press, 2016), Dr. Marc Galanter, a distinguished scholar and clinician, provides a basis to understand how this occurs. He has devoted a better part of his career to the study and treatment of addictive disorders with a special interest in how 12-step groups curtail the unbridled drinking of alcoholic individuals and stimulate a process for their growth and recovery.

Despite the volumes written about AA, Dr. Galanter maintains that there are few if any scholarly accounts that explain how it works, and how it benefits those who attend. He believes that this book will help the alcoholic who wonders whether AA is for him or her, as it will guide stymied family members and friends who wonder what help to offer, as well as health professionals who need a “coherent and objective sense of what the fellowship is about.”
Dr. Galanter invites the reader to witness the changes he and others have observed that occur in the lives of alcoholics with their encounters and immersion in AA. Allowance is made that not all who try the program benefit or continue, but for those who do, the change and help of AA, as Dr. Galanter repeatedly provides compelling data and touching examples, are transformative.
He offers that an informed appreciation of what AA is about, and most importantly, guides alcoholic individuals to understand how AA can help them, as well as assist their family and friends.

The book also provides health professionals and the public an awareness of essential aspects of the program that meet the needs of alcoholic individuals. Dr. Galanter benefits the reader by providing a brief background on the beginnings of AA, how it governs itself, and the different pathways by which its participants achieve recovery. Helpful chapters addressing controversies such as the God concept and whether alcoholism is a disease are balanced and illuminating, as are the chapters that review the 12 steps and the process of engagement.
He also provides a balanced explanation of the spiritual elements of the program, the different form they take, and how the program helps those who can draw on those elements. In addition, Dr. Galanter reviews the evidence that AA changes the brain, and finally, he concludes with a scholarly consideration and formulation on the effectiveness of AA. His even-handed, scholarly assessment of these issues is refreshing and welcome, given recent polarizations and controversies about the effectiveness AA.
As Dr. Galanter and others see and understand the culture, it is a bottom-up, democratic one in which the guides, and wisdom for sobriety and recovery for its members are garnered from each other and not from authorities on high. To his credit, Dr. Galanter avoids doctrinaire views on rules for participation, such as number and frequency of meetings (in contrast to rigid and strident rules advocated by some members), but describes flexible and alternative ways individuals participate and benefit. Dr. Galanter brings the reader to this experience with his own erudite and investigative accounts, but just as compelling, through the absorbing words and experiences of the many who have experienced the help and wisdom of the program.
I find little to criticize about this book on the merits. It is lucid and well written, and it will be instructive for all who take it up. Dr. Galanter clearly succeeds in getting to the main audiences he targets, namely those who wonder whether AA is for them, for family and friends who worry about those with the condition, and for clinicians who want guidance in how and why the program works. But ultimately, the book will profit anyone who wants a better insight into how AA achieves its successes.
References
1. Int J Psychoanal. 1972;53[1]61-5.
Dr. Khantzian is a professor of psychiatry, Harvard Medical School, Boston, and past president of the American Academy of Addiction Psychiatry.
Several years ago, the British psychoanalyst Enid Balint suggested that people might not necessarily emerge from a group experience less neurotic or psychotic, but invariably, they were more mature.1 Her suggestion is consistent with my clinical experience. Some of the most admirable and mature patients with whom I have worked have been individuals who participated regularly in 12-step meetings as part of their treatment and recovery.
In this book, “What is Alcoholics Anonymous? A Path From Addiction to Recovery,” (Oxford University Press, 2016), Dr. Marc Galanter, a distinguished scholar and clinician, provides a basis to understand how this occurs. He has devoted a better part of his career to the study and treatment of addictive disorders with a special interest in how 12-step groups curtail the unbridled drinking of alcoholic individuals and stimulate a process for their growth and recovery.
Despite the volumes written about AA, Dr. Galanter maintains that there are few if any scholarly accounts that explain how it works, and how it benefits those who attend. He believes that this book will help the alcoholic who wonders whether AA is for him or her, as it will guide stymied family members and friends who wonder what help to offer, as well as health professionals who need a “coherent and objective sense of what the fellowship is about.”
Dr. Galanter invites the reader to witness the changes he and others have observed that occur in the lives of alcoholics with their encounters and immersion in AA. Allowance is made that not all who try the program benefit or continue, but for those who do, the change and help of AA, as Dr. Galanter repeatedly provides compelling data and touching examples, are transformative.
He offers that an informed appreciation of what AA is about, and most importantly, guides alcoholic individuals to understand how AA can help them, as well as assist their family and friends.
The book also provides health professionals and the public an awareness of essential aspects of the program that meet the needs of alcoholic individuals. Dr. Galanter benefits the reader by providing a brief background on the beginnings of AA, how it governs itself, and the different pathways by which its participants achieve recovery. Helpful chapters addressing controversies such as the God concept and whether alcoholism is a disease are balanced and illuminating, as are the chapters that review the 12 steps and the process of engagement.
He also provides a balanced explanation of the spiritual elements of the program, the different form they take, and how the program helps those who can draw on those elements. In addition, Dr. Galanter reviews the evidence that AA changes the brain, and finally, he concludes with a scholarly consideration and formulation on the effectiveness of AA. His even-handed, scholarly assessment of these issues is refreshing and welcome, given recent polarizations and controversies about the effectiveness AA.
As Dr. Galanter and others see and understand the culture, it is a bottom-up, democratic one in which the guides, and wisdom for sobriety and recovery for its members are garnered from each other and not from authorities on high. To his credit, Dr. Galanter avoids doctrinaire views on rules for participation, such as number and frequency of meetings (in contrast to rigid and strident rules advocated by some members), but describes flexible and alternative ways individuals participate and benefit. Dr. Galanter brings the reader to this experience with his own erudite and investigative accounts, but just as compelling, through the absorbing words and experiences of the many who have experienced the help and wisdom of the program.
I find little to criticize about this book on the merits. It is lucid and well written, and it will be instructive for all who take it up. Dr. Galanter clearly succeeds in getting to the main audiences he targets, namely those who wonder whether AA is for them, for family and friends who worry about those with the condition, and for clinicians who want guidance in how and why the program works. But ultimately, the book will profit anyone who wants a better insight into how AA achieves its successes.
References
1. Int J Psychoanal. 1972;53[1]61-5.
Dr. Khantzian is a professor of psychiatry, Harvard Medical School, Boston, and past president of the American Academy of Addiction Psychiatry.
Several years ago, the British psychoanalyst Enid Balint suggested that people might not necessarily emerge from a group experience less neurotic or psychotic, but invariably, they were more mature.1 Her suggestion is consistent with my clinical experience. Some of the most admirable and mature patients with whom I have worked have been individuals who participated regularly in 12-step meetings as part of their treatment and recovery.
In this book, “What is Alcoholics Anonymous? A Path From Addiction to Recovery,” (Oxford University Press, 2016), Dr. Marc Galanter, a distinguished scholar and clinician, provides a basis to understand how this occurs. He has devoted a better part of his career to the study and treatment of addictive disorders with a special interest in how 12-step groups curtail the unbridled drinking of alcoholic individuals and stimulate a process for their growth and recovery.
Despite the volumes written about AA, Dr. Galanter maintains that there are few if any scholarly accounts that explain how it works, and how it benefits those who attend. He believes that this book will help the alcoholic who wonders whether AA is for him or her, as it will guide stymied family members and friends who wonder what help to offer, as well as health professionals who need a “coherent and objective sense of what the fellowship is about.”
Dr. Galanter invites the reader to witness the changes he and others have observed that occur in the lives of alcoholics with their encounters and immersion in AA. Allowance is made that not all who try the program benefit or continue, but for those who do, the change and help of AA, as Dr. Galanter repeatedly provides compelling data and touching examples, are transformative.
He offers that an informed appreciation of what AA is about, and most importantly, guides alcoholic individuals to understand how AA can help them, as well as assist their family and friends.
The book also provides health professionals and the public an awareness of essential aspects of the program that meet the needs of alcoholic individuals. Dr. Galanter benefits the reader by providing a brief background on the beginnings of AA, how it governs itself, and the different pathways by which its participants achieve recovery. Helpful chapters addressing controversies such as the God concept and whether alcoholism is a disease are balanced and illuminating, as are the chapters that review the 12 steps and the process of engagement.
He also provides a balanced explanation of the spiritual elements of the program, the different form they take, and how the program helps those who can draw on those elements. In addition, Dr. Galanter reviews the evidence that AA changes the brain, and finally, he concludes with a scholarly consideration and formulation on the effectiveness of AA. His even-handed, scholarly assessment of these issues is refreshing and welcome, given recent polarizations and controversies about the effectiveness AA.
As Dr. Galanter and others see and understand the culture, it is a bottom-up, democratic one in which the guides, and wisdom for sobriety and recovery for its members are garnered from each other and not from authorities on high. To his credit, Dr. Galanter avoids doctrinaire views on rules for participation, such as number and frequency of meetings (in contrast to rigid and strident rules advocated by some members), but describes flexible and alternative ways individuals participate and benefit. Dr. Galanter brings the reader to this experience with his own erudite and investigative accounts, but just as compelling, through the absorbing words and experiences of the many who have experienced the help and wisdom of the program.
I find little to criticize about this book on the merits. It is lucid and well written, and it will be instructive for all who take it up. Dr. Galanter clearly succeeds in getting to the main audiences he targets, namely those who wonder whether AA is for them, for family and friends who worry about those with the condition, and for clinicians who want guidance in how and why the program works. But ultimately, the book will profit anyone who wants a better insight into how AA achieves its successes.
References
1. Int J Psychoanal. 1972;53[1]61-5.
Dr. Khantzian is a professor of psychiatry, Harvard Medical School, Boston, and past president of the American Academy of Addiction Psychiatry.

Hungry and obese
Does it seem strange to you that while on one hand we hear from multiple sources that a troubling number of adults and children are going hungry, but on the other hand data from the Centers for Disease Control and Prevention indicate that self reports of obesity by adults in 2014 in different ethnic groups ranged from 27% to 38% and approximately 17% of children and adolescents aged 2-19 years are obese per 2011-2012 data?
One might guess that this situation is simply that too many Americans can afford to overeat and their numbers overwhelm the data from a smaller segment of the population who are underweight because they can’t afford to feed themselves.
But this isn’t the case at all, because the obesity rates in our poorest counties are nearly 12% above the national median (“Obesity: The New Hunger” by Robert Paarlberg, Ph.D., The Wall Street Journal, May 10, 2016). So the question is why do we have so many overweight adults and children if we also have a hunger problem? The answer is very complicated and even more complicated because of how we define hunger.
While obesity is relatively easy to measure with a scale and a tape measure, hunger is a perception that is difficult, if not impossible, to quantify. Possibly in an attempt to create clarity for people like me who are confused by the coexistence of hunger and obesity, there has been a trend toward replacing “hunger” with the more techno-sounding buzz words, “food insecurity.”
According to Dr. Paarlberg, an adjunct professor of public policy at Harvard University, Cambridge, Mass., and author of “The United States of Excess: Gluttony and the Dark Side of American Exceptionalism” (New York, N.Y.: Oxford University Press, 2015), the United States Department of Agriculture calculates our national food insecurity quotient by way of an annual survey of a sample of households. Family members are asked questions such as whether “they had failed to eat or worried about running out of food for lack of money at any time in the previous 12 months.”

There are many reasons why a survey respondent might be concerned that he or she wouldn’t have enough to eat on a given day. It could have been poor planning on the part of the head of the household or a consequence of family chaos. And we have to assume that in some cases, it is simply because there wasn’t enough money to buy food that day. If it were only a matter of money, the solution would be easy. We could simply provide economically challenged families with more money to buy more food, but that is already being done through programs such as the Supplemental Nutrition Assistance Program (often referred to as food stamps or SNAP). But the coexistence of obesity and hunger suggests to me that more food isn’t the answer.
Part of the problem is that “food” is too broadly defined. Some foods are more likely to contribute to obesity than others, and some foods satiate more quickly than others. While some restrictions have been built into the SNAP program to encourage participants to eat a healthier diet, the fact that soda and candy can be bought with food stamps is a serious error that must be corrected. It may be time to take a harder look at tightening other guidelines to make the subsidized diet healthier.
Unfortunately, the last step in the process occurs in the home. A diet that discourages obesity often includes fresh fruits and vegetables that can be expensive and may not be appealing to a family accustomed to calorie-dense foods. And a healthy diet often requires preparation skills and time, both of which economically challenged families may not have.
All of this makes me wonder whether we should stop worrying so much about hunger in America and shift the focus of our nutritional support programs more toward obesity prevention. Of course, that is easy to say for someone like myself who is lean and can always find something in the refrigerator to eat. But let’s remember that while starvation and obesity can kill, hunger doesn’t.
The problem is that “hunger” and the less emotionally charged term “food insecurity” are potent motivators for legislators who control the funding of our critical nutritional support programs. It still makes sense politically to continue to talk about eliminating hunger. But we need to craft our programs so that they address the larger problem of obesity.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.”
Does it seem strange to you that while on one hand we hear from multiple sources that a troubling number of adults and children are going hungry, but on the other hand data from the Centers for Disease Control and Prevention indicate that self reports of obesity by adults in 2014 in different ethnic groups ranged from 27% to 38% and approximately 17% of children and adolescents aged 2-19 years are obese per 2011-2012 data?
One might guess that this situation is simply that too many Americans can afford to overeat and their numbers overwhelm the data from a smaller segment of the population who are underweight because they can’t afford to feed themselves.
But this isn’t the case at all, because the obesity rates in our poorest counties are nearly 12% above the national median (“Obesity: The New Hunger” by Robert Paarlberg, Ph.D., The Wall Street Journal, May 10, 2016). So the question is why do we have so many overweight adults and children if we also have a hunger problem? The answer is very complicated and even more complicated because of how we define hunger.
While obesity is relatively easy to measure with a scale and a tape measure, hunger is a perception that is difficult, if not impossible, to quantify. Possibly in an attempt to create clarity for people like me who are confused by the coexistence of hunger and obesity, there has been a trend toward replacing “hunger” with the more techno-sounding buzz words, “food insecurity.”
According to Dr. Paarlberg, an adjunct professor of public policy at Harvard University, Cambridge, Mass., and author of “The United States of Excess: Gluttony and the Dark Side of American Exceptionalism” (New York, N.Y.: Oxford University Press, 2015), the United States Department of Agriculture calculates our national food insecurity quotient by way of an annual survey of a sample of households. Family members are asked questions such as whether “they had failed to eat or worried about running out of food for lack of money at any time in the previous 12 months.”
There are many reasons why a survey respondent might be concerned that he or she wouldn’t have enough to eat on a given day. It could have been poor planning on the part of the head of the household or a consequence of family chaos. And we have to assume that in some cases, it is simply because there wasn’t enough money to buy food that day. If it were only a matter of money, the solution would be easy. We could simply provide economically challenged families with more money to buy more food, but that is already being done through programs such as the Supplemental Nutrition Assistance Program (often referred to as food stamps or SNAP). But the coexistence of obesity and hunger suggests to me that more food isn’t the answer.
Part of the problem is that “food” is too broadly defined. Some foods are more likely to contribute to obesity than others, and some foods satiate more quickly than others. While some restrictions have been built into the SNAP program to encourage participants to eat a healthier diet, the fact that soda and candy can be bought with food stamps is a serious error that must be corrected. It may be time to take a harder look at tightening other guidelines to make the subsidized diet healthier.
Unfortunately, the last step in the process occurs in the home. A diet that discourages obesity often includes fresh fruits and vegetables that can be expensive and may not be appealing to a family accustomed to calorie-dense foods. And a healthy diet often requires preparation skills and time, both of which economically challenged families may not have.
All of this makes me wonder whether we should stop worrying so much about hunger in America and shift the focus of our nutritional support programs more toward obesity prevention. Of course, that is easy to say for someone like myself who is lean and can always find something in the refrigerator to eat. But let’s remember that while starvation and obesity can kill, hunger doesn’t.
The problem is that “hunger” and the less emotionally charged term “food insecurity” are potent motivators for legislators who control the funding of our critical nutritional support programs. It still makes sense politically to continue to talk about eliminating hunger. But we need to craft our programs so that they address the larger problem of obesity.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.”
Does it seem strange to you that while on one hand we hear from multiple sources that a troubling number of adults and children are going hungry, but on the other hand data from the Centers for Disease Control and Prevention indicate that self reports of obesity by adults in 2014 in different ethnic groups ranged from 27% to 38% and approximately 17% of children and adolescents aged 2-19 years are obese per 2011-2012 data?
One might guess that this situation is simply that too many Americans can afford to overeat and their numbers overwhelm the data from a smaller segment of the population who are underweight because they can’t afford to feed themselves.
But this isn’t the case at all, because the obesity rates in our poorest counties are nearly 12% above the national median (“Obesity: The New Hunger” by Robert Paarlberg, Ph.D., The Wall Street Journal, May 10, 2016). So the question is why do we have so many overweight adults and children if we also have a hunger problem? The answer is very complicated and even more complicated because of how we define hunger.
While obesity is relatively easy to measure with a scale and a tape measure, hunger is a perception that is difficult, if not impossible, to quantify. Possibly in an attempt to create clarity for people like me who are confused by the coexistence of hunger and obesity, there has been a trend toward replacing “hunger” with the more techno-sounding buzz words, “food insecurity.”
According to Dr. Paarlberg, an adjunct professor of public policy at Harvard University, Cambridge, Mass., and author of “The United States of Excess: Gluttony and the Dark Side of American Exceptionalism” (New York, N.Y.: Oxford University Press, 2015), the United States Department of Agriculture calculates our national food insecurity quotient by way of an annual survey of a sample of households. Family members are asked questions such as whether “they had failed to eat or worried about running out of food for lack of money at any time in the previous 12 months.”
There are many reasons why a survey respondent might be concerned that he or she wouldn’t have enough to eat on a given day. It could have been poor planning on the part of the head of the household or a consequence of family chaos. And we have to assume that in some cases, it is simply because there wasn’t enough money to buy food that day. If it were only a matter of money, the solution would be easy. We could simply provide economically challenged families with more money to buy more food, but that is already being done through programs such as the Supplemental Nutrition Assistance Program (often referred to as food stamps or SNAP). But the coexistence of obesity and hunger suggests to me that more food isn’t the answer.
Part of the problem is that “food” is too broadly defined. Some foods are more likely to contribute to obesity than others, and some foods satiate more quickly than others. While some restrictions have been built into the SNAP program to encourage participants to eat a healthier diet, the fact that soda and candy can be bought with food stamps is a serious error that must be corrected. It may be time to take a harder look at tightening other guidelines to make the subsidized diet healthier.
Unfortunately, the last step in the process occurs in the home. A diet that discourages obesity often includes fresh fruits and vegetables that can be expensive and may not be appealing to a family accustomed to calorie-dense foods. And a healthy diet often requires preparation skills and time, both of which economically challenged families may not have.
All of this makes me wonder whether we should stop worrying so much about hunger in America and shift the focus of our nutritional support programs more toward obesity prevention. Of course, that is easy to say for someone like myself who is lean and can always find something in the refrigerator to eat. But let’s remember that while starvation and obesity can kill, hunger doesn’t.
The problem is that “hunger” and the less emotionally charged term “food insecurity” are potent motivators for legislators who control the funding of our critical nutritional support programs. It still makes sense politically to continue to talk about eliminating hunger. But we need to craft our programs so that they address the larger problem of obesity.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.”
